WO2016194743A1 - 変性ポリエチレン樹脂組成物 - Google Patents
変性ポリエチレン樹脂組成物 Download PDFInfo
- Publication number
- WO2016194743A1 WO2016194743A1 PCT/JP2016/065515 JP2016065515W WO2016194743A1 WO 2016194743 A1 WO2016194743 A1 WO 2016194743A1 JP 2016065515 W JP2016065515 W JP 2016065515W WO 2016194743 A1 WO2016194743 A1 WO 2016194743A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- resin composition
- polyethylene resin
- modified polyethylene
- mass
- molecular weight
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/26—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L59/00—Compositions of polyacetals; Compositions of derivatives of polyacetals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L77/00—Compositions of polyamides obtained by reactions forming a carboxylic amide link in the main chain; Compositions of derivatives of such polymers
Definitions
- the present invention relates to a modified polyethylene resin composition capable of dramatically improving the wear resistance of various thermoplastic resins.
- Engineering tools represented by polyamide resin, polyacetal resin, polyester resin, polyphenylene sulfide resin, polycarbonate resin, etc. have excellent heat resistance, oil resistance, moldability, rigidity, toughness, etc. It is widely used as various functional parts such as general industrial parts, machine parts, electronic parts, automotive interior / exterior parts, engine room interior parts, automotive electrical parts and the like.
- Patent Document 1 the sliding properties of polyacetal, polyester, etc. are improved by using ultrahigh molecular weight polyethylene having an intrinsic viscosity [ ⁇ ] of 6 dl / g or more and polyethylene having an intrinsic viscosity [ ⁇ ] of 0.1 to 5 dl / g. Examples of use as agents have been proposed.
- Patent Document 2 discloses ultra-high molecular weight polyethylene having an intrinsic viscosity [ ⁇ ] of 6 dl / g or more and an intrinsic viscosity [ ⁇ ] of 0.00 as a sliding property improving agent blended in polyamide, polyacetal, polyester, and the like.
- a sliding property improver comprising 1 to 5 dl / g of polyethylene, wherein the ultrahigh molecular weight polyethylene and / or polyethylene is modified with an unsaturated carboxylic acid.
- an object of the present invention is to provide a polyethylene resin composition capable of dramatically improving the wear resistance (particularly the wear resistance in a high temperature environment) of various resins such as polyamide, polyacetal, polyester, and polycarbonate.
- the present inventors have found that a modified polyethylene resin composition in which a modified polyethylene having a different molecular weight is finely dispersed and a sea-island structure is formed.
- the present inventors have found that specific physical properties are improved due to the interaction between the resin and the modifying group (functional group) in the case of using the above composition, and have completed the present invention. That is, the present invention is specified by the following [1] to [12].
- [1] It has a sea-island structure and has no more than one island phase with a major axis particle diameter of 100 ⁇ m or more in a 300 ⁇ m square field of view with an optical microscope. b) There are 3 or less island phases with a major axis particle diameter of 30 ⁇ m or more and less than 100 ⁇ m in a 300 ⁇ m square field of view, c) There are 4 or more island phases with a major axis particle diameter of 10 ⁇ m or more and less than 30 ⁇ m in a 300 ⁇ m square field of view, d) The intrinsic viscosity [ ⁇ ] at 135 ° C.
- a polyethylene resin composition (B) produced by a method including the following steps and having an intrinsic viscosity [ ⁇ ] measured in decalin at 135 ° C. in the range of 1.5 to 15 dL / g is modified.
- II Low molecular weight having intrinsic viscosity [ ⁇ ] in the range of 0.3 to 1.0 dL / g To the process of producing high molecular weight polyethylene
- a resin composition comprising the modified polyethylene resin composition (A) according to [1] and another resin (E). [8] 50 to 95 parts by mass of the polyamide resin (E1) and 5 to 50 parts by mass of the modified polyethylene resin composition (A) according to [1] (of the polyamide resin (E1) and the modified polyethylene resin composition (A) [7]
- the resin composition according to [7] comprising: 100 parts by mass in total.
- the resin composition according to [7] comprising: 100 parts by mass in total.
- a modified polyethylene resin composition having an excellent balance between moldability and wear resistance can be provided.
- the modified polyethylene resin composition of the present invention is used for various resins such as polyamide, polyacetal, polyester, polycarbonate and the like, for example, the abrasion resistance (particularly, the abrasion resistance under a high temperature environment) is remarkably improved. be able to.
- the modified polyethylene resin composition (A) of the present invention has a sea-island structure, and in a 300 ⁇ m square field of view with an optical microscope, there are 1 or less, preferably 0 island phases having a long axis particle diameter of 100 ⁇ m or more. 3 or less, preferably 2 or less island phases with an axial particle diameter of 30 ⁇ m or more and less than 100 ⁇ m, and 4 or more, preferably 10 or more, 1150 or less island phases with a major axis particle diameter of 10 ⁇ m or more and less than 30 ⁇ m More preferably, it is 20 or more and 300 or less.
- the island and sea phase characteristics are expressed in a well-balanced manner by the sea-island structure in which island phases are finely dispersed.
- the island phase and the sea phase is polyethylene having a high molecular weight, but it is particularly preferable that the island phase has a higher molecular weight.
- the island phase is ultra high molecular weight polyethylene and the sea phase is low molecular weight or high molecular weight polyethylene, it is easy to satisfy the requirement f) described later, and the coefficient of dynamic friction decreases when a composition with another resin is used. The sliding property tends to be improved.
- the number of these island phases can be observed and measured using a commercially available optical microscope as shown in the examples described later. That is, the strand section of the modified polyethylene resin composition (A) is observed with a crossed Nicol using a commercially available optical microscope, and the major axis particle diameter and the number of island phases in a 300 ⁇ m square field of view may be measured.
- modified polyethylene resin composition (A) The modified polyethylene resin composition (A) of the present invention can be obtained, for example, by graft-modifying a polyethylene resin composition (B) composed of ultrahigh molecular weight polyethylene and low or high molecular weight polyethylene and introducing a functional group.
- the functional group used for modification is selected from the group consisting of a carboxyl group, an amino group, a hydroxyl group and a silanol group. Among these, a carboxyl group and a hydroxyl group are preferable, and a carboxyl group is most preferable.
- Examples of the structural unit containing the functional group include a structure derived from a compound such as an unsaturated carboxylic acid or a derivative thereof, a hydroxyl group-containing ethylenically unsaturated compound, an amino group-containing ethylenically unsaturated compound, or a vinyl group-containing organosilicon compound. Units are listed. Of these, unsaturated carboxylic acids or derivatives thereof, and hydroxyl group-containing ethylenically unsaturated compounds are preferred, and unsaturated carboxylic acids or derivatives thereof are most preferred.
- Examples of the unsaturated carboxylic acid or derivative thereof include unsaturated compounds having at least one carboxylic acid group, esters of a compound having a carboxylic acid group and an alkyl alcohol, unsaturated compounds having at least one carboxylic anhydride group, and the like. it can.
- Examples of the unsaturated group include a vinyl group, a vinylene group, and an unsaturated cyclic hydrocarbon group. As these compounds, known compounds can be used, and there is no particular limitation.
- the compound include acrylic acid, maleic acid, fumaric acid, tetrahydrophthalic acid, itaconic acid, citraconic acid, crotonic acid, isocrotonic acid, nadic acid [trademark] (endocis-bicyclo [2.2.1] hept Unsaturated carboxylic acids such as -5-ene-2,3-dicarboxylic acid); or derivatives thereof such as acid halides, amides, imides, anhydrides, esters and the like.
- Specific examples of such derivatives include maleyl chloride, maleimide, maleic anhydride, citraconic anhydride, monomethyl maleate, dimethyl maleate, glycidyl maleate and the like.
- These unsaturated carboxylic acids and / or derivatives thereof can be used alone or in combination of two or more.
- maleic anhydride and acrylic acid are preferable, and maleic anhydride is particularly preferable in terms of high reactivity.
- the content of the structural unit containing the functional group is 0.01 to 10% by mass, preferably 0.1 to 5% by mass, more preferably 0.2 to 3% by mass.
- Graft modification of the polyethylene resin composition (B) can be accomplished, for example, by dissolving the polyethylene resin composition (B) in an organic solvent and then providing a structural unit containing a functional group (such as maleic anhydride) and a radical initiator. Or the like is added to the solution, and the reaction is preferably performed at a temperature of 70 to 200 ° C., more preferably 80 to 190 ° C., preferably for 0.5 to 15 hours, more preferably for 1 to 10 hours.
- the modified polyethylene resin composition (A) is produced by reacting the polyethylene resin composition (B) with a compound for imparting a structural unit containing a functional group without solvent using an extruder or the like. You can also. This reaction is usually carried out at a temperature not lower than the melting point of the polyethylene resin composition (B), specifically at a temperature of 160 to 330 ° C., usually for 0.5 to 10 minutes.
- the kneading time is preferably 5 minutes or less, more preferably 3 minutes or less, whereby the difference in the amount of modification of the sea-island phase due to maleic anhydride or the like is increased, and the performance is easily exhibited.
- radical initiators used for graft modification include organic peroxides or azo compounds.
- the radical initiator can be used as it is mixed with the polyethylene resin composition (B) and the compound for imparting a structural unit containing a functional group, but it can also be used after being dissolved in a small amount of an organic solvent. it can. Any organic solvent that can dissolve the radical initiator can be used without particular limitation.
- a reducing substance may be used for graft modification.
- the graft amount of a compound for imparting a structural unit containing a functional group can be improved.
- the modification rate of the modified polyethylene resin composition (A) thus obtained is obtained by measuring the ratio of the intensity ratio of the peak of the functional group and the CH 2 group by IR analysis as shown in the examples described later. It is done.
- the ratio of the intensity ratio of the functional group peak to the CH 2 group peak in the IR of the sea phase (IRS) and the ratio of the intensity ratio of the functional group peak to the CH 2 group peak in the IR of the island phase (IRI) Ratio (IRS / IRI) exceeds 1.0.
- the lower limit is more preferably 1.005, particularly preferably 1.01, most preferably 1.03
- the upper limit is more preferably 10, particularly preferably 5, most preferably 3. .
- the modified polyethylene resin composition (A) is a composition with a resin such as polyamide (engineering plastic).
- the low molecular weight or high molecular weight polyethylene part is strongly compatible with the resin by the interaction with the modifying group (functional group) and follows the deformation of the resin, and the ultra high molecular weight polyethylene part is resistant to deformation stress. It is considered that a molded product having excellent wear resistance can be obtained because it exhibits the inherent properties.
- the intrinsic viscosity [ ⁇ ] of the modified polyethylene resin composition (A) at 135 ° C. in decalin is 1.5 to 15 dL / g, preferably 1.8 to 12 dL / g, more preferably 2.0 to 10 dL / g. Is in range. By having such an intrinsic viscosity [ ⁇ ], the balance of physical properties is excellent. Further, the intrinsic viscosity [ ⁇ ] of the polyethylene resin composition (B) before modification is usually the same intrinsic viscosity [ ⁇ ] as that of the polyethylene resin composition (A) after modification.
- the island phase and the sea phase is high molecular weight polyethylene.
- the island phase has a higher molecular weight.
- polyethylene having a high molecular weight is an ultrahigh molecular weight polyethylene having an intrinsic viscosity [ ⁇ ] in a decalin solvent at 135 ° C. of preferably 20 to 40 dL / g
- a polyethylene having a lower molecular weight is Low molecular weight or high molecular weight polyethylene having an intrinsic viscosity [ ⁇ ] of preferably in the range of 0.3 to 1.0 dL / g is preferable.
- the unique properties of both are expressed in a balanced manner, and as a result, a composition having an excellent balance of physical properties can be obtained.
- the polyethylene resin composition (B) before modification is preferably 5 to 45 masses of ultrahigh molecular weight polyethylene (C) with respect to 100 mass parts of the total amount of ultrahigh molecular weight polyethylene (C) and low or high molecular weight polyethylene (D). Parts, more preferably 5 to 30 parts by weight, particularly preferably 5 to 28 parts by weight, most preferably 8 to 25 parts by weight, and preferably low to high molecular weight polyethylene (D) 55 to 95 parts by weight, more
- the content is preferably 70 to 95 parts by mass, particularly preferably 72 to 95 parts by mass, and most preferably 75 to 92 parts by mass, and preferably satisfies the following requirements (i) and (ii).
- the density of the polyethylene resin composition (B) is 955 to 980 kg / m 3 (measured in accordance with ASTM D1505), preferably 957 to 970 kg / m 3 , more preferably 960 to 969 kg / m m is 3.
- the intrinsic viscosity [ ⁇ ] of the polyethylene resin composition (B) measured in a decalin solvent at 135 ° C. is 1.5 to 15 dl / g, preferably 1.8 to 12 dl / g, more preferably 2 to 10 dl / g.
- the polyethylene resin composition (B) contains an ultra high molecular weight polyethylene (C) and a low molecular weight or high molecular weight polyethylene (D) in the above proportions, so that a modified polyethylene resin composition having excellent wear resistance and moldability.
- a product (A) is obtained.
- the resulting modified polyethylene resin composition (A) Since the polyethylene resin composition (B) has a density within the above range, the resulting modified polyethylene resin composition (A) has an excellent balance of wear resistance, rigidity, and impact resistance. Moreover, by having the intrinsic viscosity [ ⁇ ] within the above range, the resulting resin composition is excellent in wear resistance, impact strength, appearance, moldability, and the like.
- Ultra high molecular weight polyethylene (C) is, for example, a homopolymer of ethylene, or ethylene and propylene, 1-butene, 1-pentene, 1-hexene, 1-octene, 1-decene, 1-dodecene, 4-methyl.
- a copolymer with an ⁇ -olefin such as -1-pentene or 3-methyl-1-pentene.
- the ultrahigh molecular weight polyethylene (C) contained in the polyethylene resin composition (B) before modification has an intrinsic viscosity [ ⁇ ] in a decalin solvent at 135 ° C. of preferably 20 to 40 dL / g, more preferably 25 to 35 dL / g.
- an intrinsic viscosity [ ⁇ ] in a decalin solvent at 135 ° C. of preferably 20 to 40 dL / g, more preferably 25 to 35 dL / g.
- the low molecular weight to high molecular weight polyethylene (D) is, for example, a homopolymer of ethylene or a copolymer of ethylene and ⁇ -olefin.
- the ⁇ -olefin constituting the copolymer of ethylene and ⁇ -olefin is a linear or branched ⁇ -olefin having 3 to 20 carbon atoms, specifically, propylene, 1-butene, 1- Pentene, 3-methyl-1-butene, 1-hexene, 4-methyl-1-pentene, 3-methyl-1-pentene, 3,4-dimethyl-1-pentene, 4-methyl-1-hexene, 3- Ethyl-1-pentene, 3-ethyl-4-methyl-1-pentene, 3,4-dimethyl-1-hexene, 4-methyl-1-heptene, 3,4-dimethyl-1-heptene, 1-octene, Examples include 1-decene, 1-dodecene, 1-tetradecene, 1-hexadecene, 1-octadecene, 1-eicocene and the like.
- the low molecular weight to high molecular weight polyethylene (D) is preferably an ethylene / ⁇ -olefin copolymer composed mainly of ethylene, more preferably having an ethylene content of 60 mol% or more, and an ethylene content. Is more preferably 80 mol% or more, and most preferably an ethylene homopolymer.
- the low molecular weight to high molecular weight polyethylene (D) has an intrinsic viscosity [ ⁇ ] at 135 ° C. in decalin of preferably 0.3 to 1.0 dL / g, more preferably 0.5 to 0.8 dL / g. .
- the low molecular weight to high molecular weight polyethylene (D) preferably has a density of 953 to 980 kg / m 3 (measured in accordance with ASTM D1505), more preferably 953 to 970 kg / m 3 , still more preferably 954 to 969 kg / m 3 .
- a low molecular weight or high molecular weight polyethylene having a density of less than 953 kg / m 3 it is not preferable from the viewpoint of wear resistance of the resulting resin composition.
- the method (2) is preferable from the point that a more finely dispersed sea-island structure can be formed as compared with the method (1). Furthermore, in the method (2), it is particularly preferable to perform the step (I) of producing ultrahigh molecular weight polyethylene as the first step from the viewpoint of the polymerization treatment operation and the control of physical properties of the produced polyethylene. At this time, various olefins described in the items of low molecular weight to high molecular weight polyethylene (D) and ultrahigh molecular weight polyethylene (C) can be used without limitation as olefins such as ethylene used for polymerization.
- the modified polyethylene resin composition obtained from the blend amount ratio or the polymerization amount ratio of the ultrahigh molecular weight polyethylene (C) and the low molecular weight or high molecular weight polyethylene (D) In some cases, it can be determined whether the sea phase or the island phase of (A) is derived from ultra-high molecular weight polyethylene (C) or low molecular weight or high molecular weight polyethylene (D).
- the modified polyethylene resin composition (A) of the present invention has an intrinsic viscosity [ ⁇ ] measured in decalin at 135 ° C. produced by a method including the following steps in a range of 1.5 to 15 dL / g. It is preferably obtained by modifying a certain polyethylene resin composition (B).
- II) Intrinsic viscosity [ ⁇ ] in the range of 0.3 to 1.0 dL / g Process for producing a certain low molecular weight or high molecular weight polyethylene (D)
- a more finely dispersed sea-island structure is formed by the multistage polymerization in which the step (I) is first performed and the step (II) is subsequently performed in the same batch as compared with the method of mixing two types of polyethylene. Furthermore, as compared with the continuous production method (non-batch type), a composition having more uniform characteristics can be obtained stably.
- ultra-high molecular weight polyethylene is uniformly finely dispersed and bonded, it has an excellent balance of properties such as wear resistance, self-lubrication, impact strength, chemical resistance, appearance and moldability, and especially wear resistance, appearance and molding.
- a polyethylene resin composition having an excellent balance between properties and flexibility can be obtained.
- the inner phase is usually low molecular weight or high molecular weight polyethylene (D) and the outer phase is ultra high molecular weight polyethylene (C).
- Polyethylene particles having a core-shell particle structure are obtained. Further, by thermally melting and modifying these particles, a modified polyethylene resin composition (A) in which the island phase is derived from ultra-high molecular weight polyethylene (C) and the sea phase is derived from low molecular weight to high molecular weight polyethylene (D) is obtained. can get.
- polyethylene particles having a core-shell particle structure are subjected to solid phase modification using an electron beam or the like, so that the sea phase is derived from ultrahigh molecular weight polyethylene (C) and the island phase is low or high molecular weight polyethylene (D).
- any catalyst can be used without particular limitation as long as it can produce ultra high molecular weight polyethylene, low molecular weight or high molecular weight polyethylene and the like.
- Specific examples include a Ziegler-Natta catalyst made of titanium tetrachloride or titanium trichloride, a carrier-supported solid titanium catalyst in which titanium is supported on a carrier such as magnesium, a metallocene catalyst, and a post metallocene catalyst.
- a preferred embodiment for obtaining the modified polyethylene resin composition (A) of the present invention is to use a solid titanium catalyst component having a particle diameter of about several micrometers to about 25 ⁇ m and having a tendency of narrow particle size distribution, Preferable examples thereof include a liquid magnesium compound, a liquid titanium compound, and an electron donor as required, as exemplified by JP-A-56-811 and JP-A-58-83006.
- the embodiment of the catalyst containing the solid titanium catalyst component produced in this manner is preferred.
- Resin (E) which can improve characteristics, such as a sliding characteristic, using the modified polyethylene resin composition (A) of this invention is thermoplastic resins, such as engineering plastics, for example.
- resin (E) include the following (E1) to (E6).
- Polyamide resin (E1) examples include hexamethylene diamine, decamethylene diamine, dodecamethylene diamine, 2,2,4- or 2,4,4-trimethylhexamethylene diamine, 1,3- or 1,4-bis.
- Aliphatic diamines such as (aminomethyl) cyclohexane, bis (p-aminocyclohexylmethane), m- or p-xylylenediamine, diamines such as alicyclic diamine or aromatic diamine, and adipic acid, suberic acid, sebacine Polyamide obtained by polycondensation with dicarboxylic acids such as acids, cyclohexanedicarboxylic acid, terephthalic acid, isophthalic acid and the like, dicarboxylic acids such as alicyclic dicarboxylic acid, aromatic dicarboxylic acid, ⁇ -aminocaproic acid, 11-aminoundecane Polyamides obtained by condensation of aminocarboxylic acids such as acids De, .epsilon.-caprolactam, .omega.
- this polyamide resin (E1) include nylon 6, nylon 66, nylon 6110, nylon 9, nylon 11, nylon 12, nylon 6/66, nylon 66/610, nylon 6/11, aromatic nylon, nylon 6T, nylon 9T, nylon 10T, MXD6 and the like.
- the polyacetal resin (E2) is, for example, a thermoplastic resin having an oxymethylene group as a main structural unit, and includes a polyacetal homopolymer or polyoxymethylene, and a polyacetal copolymer containing an oxymethylene unit and a comonomer unit.
- a polyacetal homopolymer and a polyacetal copolymer can be used.
- polyacetal resin (E2) examples include polyacetal homopolymers (for example, trade name “Dellin” manufactured by DuPont, USA, trade name “Tenac” manufactured by Asahi Kasei Kogyo Co., Ltd.), and polyacetal copolymers (eg, polyplastics). Company name "Duracon”).
- polyester resin (E3) examples include aliphatic glycols such as ethylene glycol, propylene glycol, 1,4-butanediol, neopentyl glycol and hexamethylene glycol, alicyclic glycols such as cyclohexanedimethanol, and aromatics such as bisphenol.
- Dihydroxy compounds such as terephthalic acid, isophthalic acid, 2,6-naphthalene dicarboxylic acid, oxalic acid, succinic acid, adipic acid, sebacic acid, undecadicarboxylic acid, hexahydro It is a crystalline thermoplastic resin formed from an alicyclic dicarboxylic acid such as terephthalic acid or two or more dicarboxylic acids selected from these. As long as this polyester resin (E3) exhibits thermoplasticity, it may be modified with a small amount of a trihydroxy or higher polyhydroxy compound such as triol or tricarboxylic acid, or polycarboxylic acid.
- polyester resin (E3) examples include polyethylene terephthalate, polybutylene terephthalate, polyethylene isophthalate / terephthalate copolymer, and the like. Of these, polybutylene terephthalate (PBT) is preferable.
- Polyphenylene sulfide resin (E4) The type and molecular weight of the polyphenylene sulfide resin (E4) are not particularly limited. For example, those obtained by a polymerization method such as a linear type, a crosslinked type, and a semi-crosslinked type can be used.
- the polycarbonate resin (E5) is not particularly limited. Specific examples thereof include, for example, a polymer synthesized from aromatic dioxy compounds (bisphenol), particularly bisphenol A, among high molecular weight polymers having a carbonic acid ester bond in the molecular chain.
- the polyolefin resin (E6) is not particularly limited as long as it is different from the modified polyethylene resin composition (A). Specific examples thereof include polyethylene, polypropylene, polybutene, poly-4-1-methylpentene and the like.
- composition ratio of resin (E) and modified polyethylene resin composition (A) When a resin composition having improved properties such as sliding properties of the resin (E) is obtained by using the modified polyethylene resin composition (A) of the present invention, the preferred composition ratio varies depending on the type of the resin (E). .
- the content of the resin (E) is preferably 50 to 98 parts by mass, more preferably 55 to 95 parts by mass
- the content of the modified polyethylene resin composition (A) is particularly preferably 2 to 50 parts by mass, more preferably 5 to 45 parts by mass, and particularly preferably 5 to 30 parts by mass. is there.
- any one of (E1) to (E6) may be used alone, or a plurality thereof may be used in combination at any ratio. However, it is preferable to use either one alone.
- the total amount of the polyamide resin (E1) and the modified polyethylene resin composition (A) is 100 parts by mass, and the polyamide (
- the content of E1) is preferably 50 to 95 parts by mass, more preferably 80 to 90 parts by mass, and the content of the modified polyethylene resin composition (A) is preferably 5 to 50 parts by mass, more preferably 10 to 20 parts by mass.
- the total amount of the polyacetal resin (E2) and the modified polyethylene resin composition (A) is 100 parts by mass, and the polyacetal resin
- the content of (E2) is preferably 80 to 95 parts by mass, more preferably 85 to 95 parts by mass
- the content of the modified polyethylene resin composition (A) is preferably 5 to 20 parts by mass, more preferably Is 5 to 15 parts by mass.
- the wear resistance is particularly excellent.
- the total amount of the polyester resin (E3) and the modified polyethylene resin composition (A) is 100 parts by mass, and the polyester resin is used.
- the content of (E3) is preferably 50 to 95 parts by mass, more preferably 65 to 95 parts by mass, and the content of the modified polyethylene resin composition (A) is preferably 5 to 50 parts by mass, more preferably Is 5 to 35 parts by mass.
- Examples of the method for preparing the resin composition containing the resin (E) and the modified polyethylene resin composition (A) include a method of melt-kneading the resin (E) and the modified polyethylene resin (A).
- the resin (E) and the modified polyethylene resin (A) may be kneaded at a temperature at which both are melted, and the temperature is not particularly limited. Usually, it may be melt-kneaded at a temperature of 200 to 350 ° C., preferably 200 to 310 ° C. Kneading can be performed using a single screw extruder, a twin screw extruder, a Banbury mixer, or the like.
- the molded product of the present invention is a molded product obtained by using a resin composition comprising the modified polyethylene resin composition (A) and the other resin (E) described above in part or in whole.
- This resin composition can be molded by various conventionally known methods. Specifically, this resin composition can be used, for example, by injection molding, profile extrusion molding, pipe molding, tube molding, dissimilar molded coating molding, injection blow molding, direct blow molding, T-die sheet. Alternatively, it can be formed into a container shape, a tray shape, a sheet shape, a rod shape, a film shape, or a coating of various shaped articles by a forming method such as a film forming method, an inflation film forming method, or a press forming method.
- the molded body obtained by the above molding method can be widely used for conventionally known applications.
- it has an excellent balance of properties such as wear resistance, self-lubricating properties, impact strength, and thin-wall molding.
- Applications that require these properties include, for example, steel pipes, electric wires, and automobile coating door rails (lamination).
- the modified polyethylene resin composition (A) of the present invention can be widely used for conventionally known polyethylene applications in addition to improving the slidability of various resins. Since it has an excellent balance of properties such as wear resistance and moldability, the applications that require these are, for example, steel pipes, electric wires, automotive sliding door rails and other metal coatings (laminates), pressure rubber hoses, and automotive doors.
- Gaskets clean room door gaskets, automotive glass run channels, automotive weather strips and other rubber coatings (laminates), hoppers, chutes and other linings, gears, bearings, rollers, tape reels, various guide rails and elevator rail guides, It can use suitably for uses, such as sliding materials, such as various protective liner materials.
- the modified polyethylene resin composition (A) of the present invention, and the polyethylene resin composition (B) and the resin (E) that are the raw materials of the modified polyethylene resin (A) are not limited to the range that does not impair the effects of the present invention. These resins or polymers and / or additives for resins can be optionally added.
- additives for resins include nucleating agents, anti-blocking agents, slip agents, pigments, dyes, fillers (fillers), lubricants, plasticizers, mold release agents, antioxidants, flame retardants, ultraviolet absorbers, Antibacterial agent, surfactant, antistatic agent, weather resistance stabilizer, heat resistance stabilizer, anti-slip agent, foaming agent, crystallization aid, anti-fogging agent, (transparent) nucleating agent, anti-aging agent, hydrochloric acid absorbent, impact Examples thereof include an improver, a crosslinking agent, a co-crosslinking agent, a crosslinking aid, an adhesive, a softener, a processing aid, a petroleum resin, a wax, an olefin oil, and a silicone oil. These additives can be used singly or in appropriate combination of two or more.
- thermoplastic resins (F) can be widely used.
- the amount of the resin or polymer added is preferably 0.1 to 30% by mass with respect to the total mass of the fat composition.
- thermoplastic resin (F) The thermoplastic resin (F) is not particularly limited. Examples of the thermoplastic resin (F) include the following resins.
- Thermoplastic polyolefin resin for example, low density, medium density, high density polyethylene, high pressure low density polyethylene, isotactic polypropylene, syndiotactic polypropylene, poly 1-butene, poly 4-methyl-1-pentene, poly 3 -Methyl-1-pentene, poly-3-methyl-1-butene, ethylene / ⁇ -olefin copolymer, propylene / ⁇ -olefin copolymer, 1-butene / ⁇ -olefin copolymer, cyclic olefin copolymer , Chlorinated polyolefins, and modified polyolefin resins obtained by modifying these olefin resins,
- Thermoplastic polyamide resin for example, aliphatic polyamide (nylon 6, nylon 11, nylon 12, nylon 66, nylon 610, nylon 612),
- Thermoplastic polyester resin for example, polyethylene terephthalate, polybutylene
- polypropylene examples include isotactic polypropylene and syndiotactic polypropylene as described above.
- the isotactic polypropylene may be a homopolypropylene, a propylene / ⁇ -olefin having 2 to 20 carbon atoms (excluding propylene) random copolymer, or a propylene block copolymer. .
- Poly-4-methyl-1-pentene is a homopolymer of 4-methyl-1-pentene or a 4-methyl-1-pentene / ⁇ -olefin copolymer.
- ethylene, propylene, 1-butene, 1-hexene, 1-octene can be used as the ⁇ -olefin copolymerized with 4-methyl-1-pentene.
- polyethylene low density polyethylene, medium density polyethylene, high density polyethylene, and high pressure method low density polyethylene, which are produced by a conventionally known method as described above, can be used.
- Polybutene is a homopolymer of 1-butene or a copolymer of 1-butene and olefins excluding 1-butene.
- the olefin include those described above, and these olefins can be used alone or in combination of two or more.
- the copolymer include 1-butene / ethylene random copolymer, 1-butene / propylene random copolymer, 1-butene / methylpentene copolymer, 1-butene / methylbutene copolymer, 1-butene / Examples include propylene / ethylene copolymers.
- the 1-butene content is preferably 50 mol% or more, more preferably 70 mol% or more, and particularly preferably 85% or more. .
- the modified polyolefin resin can be obtained by graft-modifying the above-described polyolefin resin with an ethylenically unsaturated bond-containing monomer using an organic peroxide.
- functional groups possessed by the modified polyolefin include halogen atoms, carboxyl groups, acid anhydride groups, epoxy groups, hydroxyl groups, amino groups, amide groups, imide groups, ester groups, alkoxysilane groups, acid halide groups, and nitrile groups. Is mentioned.
- rosin-based resins include natural rosin, polymerized rosin, modified rosin and rosin derivatives modified with maleic acid, fumaric acid, (meth) acrylic acid, and the like.
- examples of the rosin derivative include the above-mentioned natural rosin, polymerized rosin or esterified product of modified rosin, phenol-modified product and esterified product thereof.
- hydrogenated substances can also be mentioned.
- terpene resin examples include resins made of ⁇ -pinene, ⁇ -pinene, limonene, dipentene, terpene phenol, terpene alcohol, terpene aldehyde, etc., and ⁇ -pinene, ⁇ -pinene, limonene, dipentene, etc.
- An aromatic modified terpene resin obtained by polymerizing an aromatic monomer may also be used.
- these hydrogenated substances can also be mentioned.
- Examples of the petroleum resin include an aliphatic petroleum resin whose main raw material is a C5 fraction of tar naphtha, an aromatic petroleum resin whose main raw material is a C9 fraction, and a copolymer petroleum resin thereof. That is, C5 petroleum resin (resin obtained by polymerizing C5 fraction of naphtha cracked oil), C9 petroleum resin (resin obtained by polymerizing C9 fraction of naphtha cracked oil), C5C9 copolymerized petroleum resin (C5 fraction of naphtha cracked oil) A co-polymer of styrene and indene, coumarone and other dicyclopentadiene, p-tertiarybutylphenol, and the like. Examples thereof include alkylphenol resins represented by condensates of acetylene, xylene resins obtained by reacting o-xylene, p-xylene or m-xylene with formalin.
- the rosin resin, terpene resin and petroleum resin are preferably hydrogenated derivatives because they are excellent in weather resistance and discoloration resistance.
- the softening point of the resin by the ring and ball method is preferably in the range of 40 to 180 ° C.
- the number average molecular weight (Mn) molecular weight measured by GPC of the resin is preferably in the range of about 100 to 10,000.
- Commercial products can also be used as rosin resins, terpene resins, and petroleum resins.
- thermoplastic resin (F) a commercial item can also be used as each above-mentioned thermoplastic resin (F).
- One of these thermoplastic resins (F) can be used alone, or two or more can be used in combination.
- a known nucleating agent can be used as the nucleating agent in order to further improve the moldability of the olefin polymer, that is, to increase the crystallization temperature and increase the crystallization speed.
- the blending amount of the nucleating agent is not particularly limited, but is preferably 0.1 to 1 part by mass with respect to 100 parts by mass of the olefin polymer.
- the nucleating agent can be appropriately added during polymerization, after polymerization, or during molding.
- antiblocking agent known antiblocking agents can be used. Specifically, fine powder silica, fine powder aluminum oxide, fine powder clay, powdered or liquid silicon resin, tetrafluoroethylene resin, fine powder cross-linked resin, for example, cross-linked acrylic, methacrylic resin powder, etc. it can. Of these, fine powder silica and crosslinked acrylic and methacrylic resin powders are preferred.
- the pigment examples include inorganic contents (titanium oxide, iron oxide, chromium oxide, cadmium sulfide, etc.) and organic pigments (azo lake, thioindigo, phthalocyanine, anthraquinone).
- the dye examples include azo series, anthraquinone series, and triphenylmethane series. The addition amount of these pigments and dyes is not particularly limited, but is generally 5% by mass or less, preferably 0.1 to 3% by mass with respect to the total mass of the resin composition.
- filler glass fiber, carbon fiber, silica fiber, metal (stainless steel, aluminum, titanium, copper, etc.) fiber, carbon black, silica, glass beads, silicate (calcium silicate, talc, clay, etc.), Examples include metal oxides (iron oxide, titanium oxide, alumina, etc.), metal carbonates (calcium sulfate, barium sulfate, etc.) and various metal (magnesium, silicon, aluminum, titanium, copper, etc.) powders, mica, and glass flakes. . These fillers may be used alone or in combination of two or more.
- lubricant examples include wax (carnauba wax etc.), higher fatty acid (eg stearic acid), higher alcohol (stearyl alcohol etc.), higher fatty acid amide (eg stearic acid amide) and the like.
- Plasticizers include aromatic carboxylic acid esters (such as dibutyl phthalate), aliphatic carboxylic acid esters (such as methyl acetyl ricinoleate), aliphatic dialcolic acid esters (such as adipic acid-propylene glycol polyester), and aliphatic tricarboxylic acids.
- aromatic carboxylic acid esters such as dibutyl phthalate
- aliphatic carboxylic acid esters such as methyl acetyl ricinoleate
- aliphatic dialcolic acid esters such as adipic acid-propylene glycol polyester
- aliphatic tricarboxylic acids include esters (such as triethyl citrate), phosphoric acid triesters (such as triphenyl phosphate), epoxy fatty acid esters (such as epoxybutyl stearate), and petroleum resins.
- Release agents include higher fatty acid lower (C1-4) alcohol esters (butyl stearate, etc.), fatty acid (C4-30) polyhydric alcohol esters (hardened castor oil, etc.), fatty acid glycol esters, liquid paraffin, etc. Is mentioned.
- antioxidants known antioxidants can be used. Specifically, phenol-based (2,6-di-t-butyl-4-methylphenol, etc.), polycyclic phenol-based (2,2′-methylenebis (4-methyl-6-t-butylphenol, etc.), phosphorus System (tetrakis (2,4-di-t-butylphenyl) -4,4-biphenylene diphosphonate, etc.), sulfur system (dilauryl thiodipropionate, etc.), amine system (N, N-diisopropyl-p- Phenylenediamine, etc.), lactone-based antioxidants, and the like.
- flame retardants examples include organic flame retardants (nitrogen-containing, sulfur-containing, silicon-containing, phosphorus-containing, etc.) and inorganic flame retardants (antimony trioxide, magnesium hydroxide, zinc borate, red phosphorus, etc.). Can be mentioned.
- UV absorber examples include benzotriazole, benzophenone, salicylic acid, and acrylate.
- Antibacterial agents include quaternary ammonium salts, pyridine compounds, organic acids, organic acid esters, halogenated phenols, organic iodine, and the like.
- Nonionic surfactants include nonionic, anionic, cationic or amphoteric surfactants.
- Nonionic surfactants include polyethylene glycol type nonionic surfactants such as higher alcohol ethylene oxide adducts, fatty acid ethylene oxide adducts, higher alkylamine ethylene oxide adducts, polypropylene glycol ethylene oxide adducts, fatty acid esters of polyethylene oxide and glycerin.
- Polyanhydric alcohol type nonionic surfactants such as fatty acid ester of pentaerythritol, fatty acid ester of sorbit or sorbitan, alkyl ether of polyhydric alcohol, aliphatic amide of alkanolamine, etc.
- sulfate salts such as alkali metal salts of higher fatty acids, sulfonates such as alkylbenzene sulfonates, alkyl sulfonates, paraffin sulfonates, Include a phosphoric acid ester salts such as grade alcohol phosphate ester salt, the cationic surfactants, such as quaternary ammonium salts such as alkyl trimethyl ammonium salts.
- amphoteric surfactants include amino acid-type double-sided surfactants such as higher alkylaminopropionates, betaine-type amphoteric surfactants such as higher alkyldimethylbetaine and higher alkyl hydroxyethylbetaine.
- antistatic agent examples include the above-mentioned surfactants, fatty acid esters, and polymer type antistatic agents.
- fatty acid esters examples include esters of stearic acid and oleic acid
- polymer antistatic agents include polyether ester amides.
- additives such as the above fillers, lubricants, plasticizers, mold release agents, antioxidants, flame retardants, ultraviolet absorbers, antibacterial agents, surfactants, antistatic agents, and the like impairs the purpose of the present invention.
- it is preferably 0.1 to 30% by mass with respect to the total mass of the resin composition.
- Polarization is adjusted so that the island phase particles can be easily observed by applying polarized light, and the sea-island structure is photographed at three places at a magnification of 200 times, and a 300 ⁇ m square is randomly determined from the photographed field of view.
- the number of island phases having a major axis diameter and a predetermined major axis particle diameter was determined.
- the average ratio of 3 times the measured sea phase and island phase each CH 2 group peak intensity (1470 cm -1) and a carboxyl group peak intensity (sum of the intensities of 1710 cm -1 and 1790 cm -1) by calculating the, IRS , IRI, and IRS / IRI were determined.
- a method of calculating the peak intensities in a state where the interference fringe and noise to obtain a free spectrum for CH 2 group peak intensity, starting from the inflection point in the range from 1400 cm -1 to 1410 cm -1, from 1510 cm -1 1520 cm The inflection point in the range of -1 was determined as the end point, and the peak intensity was measured from the area.
- peak intensity at around 1710 cm -1 of the carboxyl group peak intensity determined from 1670 cm -1 starting from the inflection point in the range of 1680 cm -1, and ending the inflection point in the range from 1745 cm -1 to 1755 cm -1
- peak intensity at around 1790 cm -1 is determined from 1760 cm -1 starting from the inflection point in the range of 1770 cm -1, and ending the inflection point in the range from 1810 cm -1 to 1820 cm -1, the peak intensity from their area was measured.
- melt flow rate (MFR) was measured under the condition of a load of 2.16 kg under the temperature conditions of 190 ° C. and 230 ° C., 235 ° C. or 280 ° C.
- ⁇ Tensile test> Based on ISO-527-1, 2, the shape of the test piece was a multipurpose test piece, the tensile speed was 50 mm / min, and the tensile yield point stress (yield point strength) and elongation at break were determined.
- ⁇ Dispersion diameter> About the resin composition containing the modified polyethylene resin composition (A) and the resin (E), using a multi-purpose test piece prepared for measuring physical properties, a cutting cross section is prepared with a cryo ultra microtome perpendicular to the MD direction, and carbon Vapor deposition was performed to obtain an observation specimen. As an observation method, the core layer of the observation specimen end view was observed using an electron microscope (JFM-7001F, manufactured by JEOL Ltd.), and the dispersion diameter was measured.
- JFM-7001F electron microscope
- Example 1-1 ⁇ Preparation of solid titanium catalyst component> An anhydrous magnesium chloride 95.2 g, decane 398.4 g and 2-ethylhexyl alcohol 306 g were heated and reacted at a temperature of 140 ° C. for 6 hours to obtain a homogeneous solution, and then 17.6 g of ethyl benzoate was added to the solution. The mixture was further stirred and mixed at 130 ° C. for 1 hour.
- the solid titanium catalyst component prepared by the above operation was stored as a decant slurry.
- the solid titanium catalyst component When analyzed by the ICP method, the solid titanium catalyst component contained 3.5% by mass of the Ti component.
- the average particle diameter of the catalyst particles measured with a laser diffraction scattering method particle size distribution analyzer manufactured by Beckman Coulter was 7 ⁇ m, and the maximum particle diameter was 18 ⁇ m.
- the white solid sample (1) had an intrinsic viscosity [ ⁇ ] of 28 dl / g. Further, the content of the ultrahigh molecular weight polyethylene produced in the first stage is 19.0% by mass from the supply amount of ethylene. The intrinsic viscosity of the polymer produced in the second stage was 0.5 dl / g as estimated from the following formula.
- Modified Polyethylene Resin Composition (A-1) 100 parts by weight of the polyethylene resin composition (B-1) obtained above, 0.8 parts by weight of maleic anhydride, and 0.07 parts by weight of an organic peroxide [manufactured by NOF Corporation, trade name Perhexin-25B] , And the resulting mixture is melt-grafted with a 100 mm ⁇ twin screw extruder set at 270 ° C. in a kneading time of about 1 minute and 30 seconds, whereby a modified polyethylene resin composition (A- 1) was obtained.
- the amount of maleic anhydride grafted in the graft-modified polyethylene composition was measured by IR analysis and found to be 0.8% by mass.
- a clear sea-island structure was observed with an optical microscope.
- the island phase is considered to be the first stage polymer, and the sea phase is considered to be the second stage polymer.
- the other analysis results are shown in Table 1.
- modified polyethylene resin composition (A-2) The polyethylene resin composition (B-2) obtained above was graft-modified in the same manner as in the production of the ethylene polymer (A-1) to obtain a modified polyethylene resin composition (A-2).
- this modified polyethylene resin composition (A-2) a clear sea-island structure was not observed. That is, it was considered that the first and second stage polymers were compatible.
- the analysis results are shown in Table 1.
- Examples 1 to 3 Comparative Example 1
- a polyamide resin (E1) PA6 manufactured by Toray Industries, Inc., Amilan (registered trademark) CM1007
- a modified polyethylene resin composition (A-1) in the amounts shown in Table 2, 43 mm ⁇ vent type twin screw Pellets were produced by melt mixing in an extruder at a cylinder temperature of 200 to 240 ° C.
- An injection-molded test piece was prepared using the pellets thus obtained, and the performance described above was evaluated.
- PA6 alone was also evaluated.
- polyester resin (E3) polybutylene terephthalate (PBT) [manufactured by Polyplastics Co., Ltd., DURANEX (registered trademark) 500FP] and a modified polyethylene resin composition (A-1) were used in the amounts shown in Table 3.
- the resin composition pellets were manufactured by melt mixing at a cylinder temperature of 230 to 250 ° C. using a 43 mm ⁇ vented twin screw extruder. An injection-molded test piece was prepared using the pellets thus obtained, and the performance described above was evaluated. In addition, evaluation with a single PBT was also performed.
- the modified polyethylene resin composition of the present invention is excellent in the balance of properties such as wear resistance and moldability, for example, a metal coating such as a steel pipe, an electric wire, and an automobile slide door rail (for example) is required.
- a metal coating such as a steel pipe, an electric wire, and an automobile slide door rail (for example) is required.
- Lamination pressure-resistant rubber hose, automotive door gasket, clean room door gasket, automotive glass run channel, automotive weather strip and other rubber coatings (lamination), hopper, chute, etc. lining, gear, bearing, roller, tape reel It is very useful for sliding materials such as various guide rails, elevator rail guides and various protective liner materials.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
a)海島構造を有し、光学顕微鏡での300μm角視野中に長軸粒子径100μm以上の島相が1個以下、b)長軸粒子径30μm以上100μm未満の島相が3個以下、c)長軸粒子径10μm以上30μm未満の島相が4個以上であり、d)デカリン中135℃での極限粘度[η]が1.5~15dL/gの範囲にあり、e)カルボキシル基、アミノ基、水酸基及びシラノール基からなる群より選ばれる官能基を含む構造単位を0.01~10質量%含み、f)海相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRS)と島相のIRにおける同比(IRI)との比(IRS/IRI)が1.0を超える変性ポリエチレン樹脂組成物が開示される。
Description
本発明は、各種熱可塑性樹脂の耐摩耗性を飛躍的に向上させうる変性ポリエチレン樹脂組成物に関する。
ポリアミド樹脂、ポリアセタール樹脂、ポリエステル樹脂、ポリフェニレンスルフィド樹脂、ポリカーボネート樹脂などに代表されるエンジニアリングプラスチックは、優れた耐熱性、耐油性、成形性、剛性、強靭性などの特徴を有しているため電動工具、一般工業部品、機械部品、電子部品、自動車内外装部品、エンジンルーム内部品、自動車電装部品などの種々の機能部品として広く利用されている。
しかしながら、それらの樹脂は、金属材料などに比較すると、摺動特性である摩擦係数が高く、摺動時の発熱により、樹脂自体の溶融、凝着による摩耗が大きく、摺動特性が不充分であった。このため、ポリオレフィン成分などの添加物を配合させて、摺動特性を向上させることが試みられている。
たとえば、特許文献1には、ポリアセタール、ポリエステルなどに極限粘度[η]が6dl/g以上の超高分子量ポリエチレンと、極限粘度[η]が0.1~5dl/gのポリエチレンを摺動特性改良剤として使用する例が提案されている。
また、特許文献2には、ポリアミド、ポリアセタール、ポリエステルなどに配合される摺動特性改良剤として、極限粘度[η]が6dl/g以上の超高分子量ポリエチレンと、極限粘度[η]が0.1~5dl/gのポリエチレンとを含み、かつ前記超高分子量ポリエチレンおよび/またはポリエチレンが不飽和カルボン酸で変性されてなる摺動特性改良剤が例示されている。
本発明者らの検討によれば、特許文献1、2に開示された摺動特性改良剤では摺動特性の改良効果が不十分な場合が想定され、さらに高い耐摩耗性、低摩擦係数を達成することが望ましかった。また、摩擦時の発熱を抑制したい要望があった。さらには、100℃程度の比較的高温環境におかれた場合の耐摩耗性が不足していることがわかった。
すなわち本発明の目的は、ポリアミド、ポリアセタール、ポリエステル、ポリカーボネート等の各種樹脂の耐摩耗性(特に高温環境下の耐摩耗性)を飛躍的に向上させうるポリエチレン樹脂組成物を提供することにある。
本発明者は、鋭意検討した結果、分子量の異なる変性ポリエチレンが微分散して海島構造が形成された変性ポリエチレン樹脂組成物において、海相と島相の変性率が異なることによって、他の樹脂との組成物にした場合の当該樹脂と変性基(官能基)との相互作用などに起因して特定の物性が向上することを見出し、本発明を完成するに至った。すなわち本発明は、以下の[1]~[12]により特定される。
[1]a) 海島構造を有し、光学顕微鏡での300μm角視野中に長軸粒子径100μm以上の島相が1個以下であり、
b) 300μm角視野中に長軸粒子径30μm以上、100μm未満の島相が3個以下であり、
c) 300μm角視野中に長軸粒子径10μm以上、30μm未満の島相が4個以上であり、
d) デカリン中135℃での極限粘度[η]が1.5~15dL/gの範囲にあり、
e) カルボキシル基、アミノ基、水酸基及びシラノール基からなる群より選ばれる官能基を含む構造単位を0.01~10質量%含み、
f) 海相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRS)と島相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRI)との比(IRS/IRI)が1.0を超える変性ポリエチレン樹脂組成物(A)。
b) 300μm角視野中に長軸粒子径30μm以上、100μm未満の島相が3個以下であり、
c) 300μm角視野中に長軸粒子径10μm以上、30μm未満の島相が4個以上であり、
d) デカリン中135℃での極限粘度[η]が1.5~15dL/gの範囲にあり、
e) カルボキシル基、アミノ基、水酸基及びシラノール基からなる群より選ばれる官能基を含む構造単位を0.01~10質量%含み、
f) 海相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRS)と島相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRI)との比(IRS/IRI)が1.0を超える変性ポリエチレン樹脂組成物(A)。
[2]e)の官能基がカルボキシル基である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[3]e)の構造単位が無水マレイン酸由来の構造単位である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[4]f)の比(IRS/IRI)が1.0を超えて5以下である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[5]f)の比(IRS/IRI)が1.001以上3以下である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[6]下記の工程を含む方法で製造された、135℃のデカリン中で測定される極限粘度[η]が1.5~15dL/gの範囲にあるポリエチレン樹脂組成物(B)を変性して得られる[1]に記載の変性ポリエチレン樹脂組成物(A)。
(I)極限粘度[η]が20~40dL/gの範囲にある超高分子量ポリエチレンを製造する工程
(II)極限粘度[η]が0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレンを製造する工程
[3]e)の構造単位が無水マレイン酸由来の構造単位である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[4]f)の比(IRS/IRI)が1.0を超えて5以下である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[5]f)の比(IRS/IRI)が1.001以上3以下である[1]に記載の変性ポリエチレン樹脂組成物(A)。
[6]下記の工程を含む方法で製造された、135℃のデカリン中で測定される極限粘度[η]が1.5~15dL/gの範囲にあるポリエチレン樹脂組成物(B)を変性して得られる[1]に記載の変性ポリエチレン樹脂組成物(A)。
(I)極限粘度[η]が20~40dL/gの範囲にある超高分子量ポリエチレンを製造する工程
(II)極限粘度[η]が0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレンを製造する工程
[7][1]に記載の変性ポリエチレン樹脂組成物(A)と、他の樹脂(E)とを含んでなる樹脂組成物。
[8]ポリアミド樹脂(E1)50~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリアミド樹脂(E1)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[9]ポリアセタール樹脂(E2)80~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~20質量部(ポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[10]ポリエステル樹脂(E3)50~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリエステル樹脂(E3)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[8]ポリアミド樹脂(E1)50~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリアミド樹脂(E1)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[9]ポリアセタール樹脂(E2)80~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~20質量部(ポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[10]ポリエステル樹脂(E3)50~95質量部と、[1]に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリエステル樹脂(E3)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる[7]に記載の樹脂組成物。
[11][7]に記載の樹脂組成物をその一部または全部に使用してなる成形体。
[12]摺動部材である、[11]に記載の成形体。
[12]摺動部材である、[11]に記載の成形体。
本発明によれば、成形性と耐摩耗性のバランスに優れた変性ポリエチレン樹脂組成物を提供することができる。特に、ポリアミド、ポリアセタール、ポリエステル、ポリカーボネート等の各種樹脂に対して、本発明の変性ポリエチレン樹脂組成物を用いると、例えば耐摩耗性(特に高温環境下の耐摩耗性)などを飛躍的に向上させることができる。
[海島構造]
本発明の変性ポリエチレン樹脂組成物(A)は海島構造を有し、光学顕微鏡での300μm角視野中に、長軸粒子径100μm以上の島相が1個以下、好ましくは0個であり、長軸粒子径30μm以上、100μm未満の島相が3個以下、好ましくは2個以下であり、長軸粒子径10μm以上、30μm未満の島相が4個以上、好ましくは10個以上、1150個以下、より好ましくは20以上、300個以下である。
本発明の変性ポリエチレン樹脂組成物(A)は海島構造を有し、光学顕微鏡での300μm角視野中に、長軸粒子径100μm以上の島相が1個以下、好ましくは0個であり、長軸粒子径30μm以上、100μm未満の島相が3個以下、好ましくは2個以下であり、長軸粒子径10μm以上、30μm未満の島相が4個以上、好ましくは10個以上、1150個以下、より好ましくは20以上、300個以下である。
このような島相が微分散した海島構造によって、島相と海相の各特性がバランス良く発現する。本発明においては、島相と海相のどちらが分子量の高いポリエチレンであるかは限定されないが、特に島相の分子量の方が高いことが好ましい。例えば、島相が超高分子量ポリエチレンであり、海相が低分子量ないし高分子量ポリエチレンである場合、後述する要件f)を満たしやすく、他の樹脂との組成物にしたときに動摩擦係数が低下し摺動性が向上する傾向にある。
これら島相の個数は、後述する実施例に示すように市販の光学顕微鏡を用いて観察・測定できる。すなわち、変性ポリエチレン樹脂組成物(A)のストランド切片を市販の光学顕微鏡を用いてクロスニコルで観察し、300μm角視野中の島相の長軸粒子径と個数を測定すれば良い。
[変性ポリエチレン樹脂組成物(A)]
本発明の変性ポリエチレン樹脂組成物(A)は、例えば、超高分子量ポリエチレンと低分子量ないし高分子量ポリエチレンからなるポリエチレン樹脂組成物(B)をグラフト変性して官能基を導入することにより得られる。
本発明の変性ポリエチレン樹脂組成物(A)は、例えば、超高分子量ポリエチレンと低分子量ないし高分子量ポリエチレンからなるポリエチレン樹脂組成物(B)をグラフト変性して官能基を導入することにより得られる。
変性に用いる官能基は、カルボキシル基、アミノ基、水酸基及びシラノール基からなる群より選ばれる。中でも、カルボキシル基、水酸基が好ましく、カルボキシル基が最も好ましい。
前記官能基を含む構造単位としては、例えば、不飽和カルボン酸又はその誘導体、水酸基含有エチレン性不飽和化合物、アミノ基含有エチレン性不飽和化合物、ビニル基含有有機ケイ素化合物などの化合物に由来する構造単位が挙げられる。これらのうち、不飽和カルボン酸又はその誘導体、水酸基含有エチレン性不飽和化合物が好ましく、不飽和カルボン酸又はその誘導体が最も好ましい。
不飽和カルボン酸又はその誘導体としては、カルボン酸基を1以上有する不飽和化合物、カルボン酸基を有する化合物とアルキルアルコールとのエステル、無水カルボン酸基を1以上有する不飽和化合物等を挙げることができる。不飽和基としては、ビニル基、ビニレン基、不飽和環状炭化水素基などを挙げることができる。これらの化合物は公知のものが使用でき、特に制限はない。具体的な化合物としては、例えばアクリル酸、マレイン酸、フマル酸、テトラヒドロフタル酸、イタコン酸、シトラコン酸、クロトン酸、イソクロトン酸、ナジック酸〔商標〕(エンドシス-ビシクロ[2.2.1]ヘプト-5-エン-2,3-ジカルボン酸)等の不飽和カルボン酸;又はその誘導体、例えば酸ハライド、アミド、イミド、無水物、エステル等が挙げられる。かかる誘導体の具体例としては、例えば塩化マレニル、マレイミド、無水マレイン酸、無水シトラコン酸、マレイン酸モノメチル、マレイン酸ジメチル、グリシジルマレエート等が挙げられる。これらの不飽和カルボン酸及び/又はその誘導体は、1種単独で使用することもできるし、2種以上を組み合せて使用することもできる。これらの中でも、無水マレイン酸、アクリル酸が好ましく、無水マレイン酸が反応性が高い点で特に好ましい。
前記官能基を含む構造単位の含有量は、0.01~10質量%、好ましくは0.1~5質量%、より好ましくは0.2~3質量%である。
ポリエチレン樹脂組成物(B)のグラフト変性は、例えばポリエチレン樹脂組成物(B)を有機溶媒に溶解し、次いで官能基を含む構造単位を付与する為の化合物(無水マレイン酸など)及びラジカル開始剤などを溶液に加え、好ましくは70~200℃、より好ましくは80~190℃の温度で、好ましくは0.5~15時間、より好ましくは1~10時間反応させることにより行うことができる。また、押出機などを用いて、無溶媒で、ポリエチレン樹脂組成物(B)と官能基を含む構造単位を付与する為の化合物とを反応させて、変性ポリエチレン樹脂組成物(A)を製造することもできる。この反応は、通常ポリエチレン樹脂組成物(B)の融点以上、具体的には160~330℃の温度で、通常0.5~10分間行なわれることが望ましい。
特に、好ましくは300℃以下、より好ましくは280℃以下で混練することで、海島相の無水マレイン酸等による変性量の差が大きくなり、性能を発揮しやすい。さらに混練時間については、好ましくは5分以下、より好ましくは3分以下で混練することで、海島相の無水マレイン酸等による変性量の差が大きくなり、性能を発揮しやすい。
グラフト変性に用いられるラジカル開始剤としては、有機過酸化物あるいはアゾ化合物などが挙げられる。ラジカル開始剤は、ポリエチレン樹脂組成物(B)及び官能基を含む構造単位を付与する為の化合物にそのまま混合して使用することもできるが、少量の有機溶媒に溶解してから使用することもできる。この有機溶媒としては、ラジカル開始剤を溶解し得る有機溶媒であれば特に限定することなく用いることができる。
グラフト変性させる際には、還元性物質を用いてもよい。還元性物質を用いると、官能基を含む構造単位を付与する為の化合物のグラフト量を向上させることができる。
このようにして得られる変性ポリエチレン樹脂組成物(A)の変性率は、後述する実施例に示すようにIR分析により官能基とCH2基のピークの強度比の比を測定することによりを求められる。本発明においては、海相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRS)と島相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRI)との比(IRS/IRI)が1.0を超える。好ましくは1.001以上100以下、下限はより好ましくは1.005、特に好ましくは1.01、最も好ましくは1.03、上限はより好ましくは10、特に好ましくは5、最も好ましくは3である。本発明においては、このように海相と島相の変性率が異なることによって、他の樹脂との組成物にした場合の当該樹脂と変性基(官能基)との相互作用などに起因して特定の物性が向上する。例えば、島相が超高分子量ポリエチレンであり、海相が低分子量ないし高分子量ポリエチレンである場合は、変性ポリエチレン樹脂組成物(A)をポリアミドなどの樹脂(エンジニアリングプラスチック)との組成物にすると、低分子量ないし高分子量ポリエチレン部分は変性基(官能基)との相互作用によって当該樹脂により強固に相溶し、当該樹脂の変形に追随すると共に、超高分子量ポリエチレン部分は変形のストレスに抗して本来有する特性を発揮するため耐摩耗性に優れる成形品を得ることができると考えられる。
変性ポリエチレン樹脂組成物(A)のデカリン中135℃での極限粘度[η]は1.5~15dL/g、好ましくは1.8~12dL/g、より好ましくは2.0~10dL/gの範囲にある。このような極限粘度[η]であることにより、各物性のバランスに優れたものとなる。また変性前のポリエチレン樹脂組成物(B)の極限粘度[η]は、通常、変性後のポリエチレン樹脂組成物(A)と同様の極限粘度[η]である。
先に述べた通り、本発明においては島相と海相のどちらが分子量の高いポリエチレンであるか限定されない。ただし、特に島相の分子量の方が高いことが好ましい。特に、分子量が高いポリエチレンは、135℃のデカリン溶媒中での極限粘度[η]が好ましくは20~40dL/gの範囲にある超高分子量ポリエチレンであり、それよりも分子量が低いポリエチレンは、その極限粘度[η]が好ましくは0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレンであることが好ましい。この態様により、両者が各々有する固有の特性がバランス良く発現し、その結果として、各物性のバランスに優れた組成物が得られる。
[変性前のポリエチレン樹脂組成物(B)]
ポリエチレン樹脂組成物(B)は、超高分子量ポリエチレン(C)と低分子量ないし高分子量ポリエチレン(D)の合計量100質量部に対して、超高分子量ポリエチレン(C)を好ましくは5~45質量部、より好ましくは5~30質量部、特に好ましくは5~28質量部、もっとも好ましくは8~25質量部含有し、低分子量ないし高分子量ポリエチレン(D)を好ましくは55~95質量部、より好ましくは70~95質量部、特に好ましくは72~95質量部、もっとも好ましくは75~92質量部含有し、かつ、以下の要件(i),(ii)を満たすことが好ましい。
ポリエチレン樹脂組成物(B)は、超高分子量ポリエチレン(C)と低分子量ないし高分子量ポリエチレン(D)の合計量100質量部に対して、超高分子量ポリエチレン(C)を好ましくは5~45質量部、より好ましくは5~30質量部、特に好ましくは5~28質量部、もっとも好ましくは8~25質量部含有し、低分子量ないし高分子量ポリエチレン(D)を好ましくは55~95質量部、より好ましくは70~95質量部、特に好ましくは72~95質量部、もっとも好ましくは75~92質量部含有し、かつ、以下の要件(i),(ii)を満たすことが好ましい。
(i)ポリエチレン樹脂組成物(B)の密度が、955~980kg/m3(ASTM D1505に準拠して測定される)であり、好ましくは957~970kg/m3、より好ましくは960~969kg/m3である。
(ii)ポリエチレン樹脂組成物(B)の135℃のデカリン溶媒中で測定される極限粘度[η]が1.5~15dl/gであり、好ましくは1.8~12dl/g、より好ましくは2~10dl/gである。
ポリエチレン樹脂組成物(B)が、超高分子量ポリエチレン(C)と、低分子量ないし高分子量ポリエチレン(D)とを上記の割合で含むことにより、耐摩耗性と成形性に優れた変性ポリエチレン樹脂組成物(A)が得られる。
ポリエチレン樹脂組成物(B)は、密度が上記範囲であることにより、得られる変性ポリエチレン樹脂組成物(A)の耐摩耗性と剛性、耐衝撃性のバランスに優れる。また、上記の範囲内の極限粘度[η]を有することにより、得られる樹脂組成物の耐摩耗性、衝撃強度、外観および成形性などに優れる。
以下、上記態様のポリエチレン樹脂組成物(B)における超高分子量ポリエチレン(C)と、低分子量ないし高分子量ポリエチレン(D)について説明する。
[超高分子量ポリエチレン(C)]
超高分子量ポリエチレン(C)は、例えば、エチレンの単独重合体、又は、エチレンとプロピレン、1-ブテン、1-ペンテン、1-ヘキセン、1-オクテン、1-デセン、1-ドデセン、4-メチル-1-ペンテンもしくは3-メチル-1-ペンテンなどのα-オレフィンとの共重合体である。中でも、エチレンの単独重合体、又はエチレンと前記のα-オレフィンとの共重合体であって、エチレンを主成分として構成される共重合体を使用することが好ましく、エチレンの単独重合体であることが特に好ましい。
超高分子量ポリエチレン(C)は、例えば、エチレンの単独重合体、又は、エチレンとプロピレン、1-ブテン、1-ペンテン、1-ヘキセン、1-オクテン、1-デセン、1-ドデセン、4-メチル-1-ペンテンもしくは3-メチル-1-ペンテンなどのα-オレフィンとの共重合体である。中でも、エチレンの単独重合体、又はエチレンと前記のα-オレフィンとの共重合体であって、エチレンを主成分として構成される共重合体を使用することが好ましく、エチレンの単独重合体であることが特に好ましい。
変性前のポリエチレン樹脂組成物(B)に含まれる超高分子量ポリエチレン(C)は、135℃のデカリン溶媒中での極限粘度[η]が、好ましくは20~40dL/g、より好ましくは25~35dL/gである。このような極限粘度[η]の超高分子量ポリエチレンを使用することにより、耐摩耗性等の特性が向上する。
[低分子量ないし高分子量ポリエチレン(D)]
低分子量ないし高分子量ポリエチレン(D)は、例えば、エチレンの単独重合体、又は、エチレンとα-オレフィンの共重合体である。エチレンとα-オレフィンの共重合体を構成するα-オレフィンとしては、炭素原子数3~20の直鎖状又は分岐状のα-オレフィンであり、具体的にはプロピレン、1-ブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセン、4-メチル-1-ペンテン、3-メチル-1-ペンテン、3,4-ジメチル-1-ペンテン、4-メチル-1-ヘキセン、3-エチル-1-ペンテン、3-エチル-4-メチル-1-ペンテン、3,4-ジメチル-1-ヘキセン、4-メチル-1-ヘプテン、3,4-ジメチル-1-ヘプテン、1-オクテン、1-デセン、1-ドデセン、1-テトラデセン、1-ヘキサデセン、1-オクタデセン、1-エイコセンなどが挙げられる。
低分子量ないし高分子量ポリエチレン(D)は、例えば、エチレンの単独重合体、又は、エチレンとα-オレフィンの共重合体である。エチレンとα-オレフィンの共重合体を構成するα-オレフィンとしては、炭素原子数3~20の直鎖状又は分岐状のα-オレフィンであり、具体的にはプロピレン、1-ブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセン、4-メチル-1-ペンテン、3-メチル-1-ペンテン、3,4-ジメチル-1-ペンテン、4-メチル-1-ヘキセン、3-エチル-1-ペンテン、3-エチル-4-メチル-1-ペンテン、3,4-ジメチル-1-ヘキセン、4-メチル-1-ヘプテン、3,4-ジメチル-1-ヘプテン、1-オクテン、1-デセン、1-ドデセン、1-テトラデセン、1-ヘキサデセン、1-オクタデセン、1-エイコセンなどが挙げられる。
これらのうち、プロピレン、1-ブテンが好ましく用いられる。なお、低分子量ないし高分子量ポリエチレン(D)は、エチレンを主成分として構成されるエチレン・α-オレフィン共重合体であることが好ましく、エチレン含量が60mol%以上であることがより好ましく、エチレン含量が80mol%以上であることがさらに好ましく、最も好ましくはエチレン単独重合体である。
低分子量ないし高分子量ポリエチレン(D)は、デカリン中135℃での極限粘度[η]が、好ましくは0.3~1.0dL/g、より好ましくは0.5~0.8dL/gである。このような極限粘度[η]の低分子量ないし高分子量ポリエチレンを使用することにより、成形性等の特性が向上する。
低分子量ないし高分子量ポリエチレン(D)は、密度が好ましくは953~980kg/m3(ASTM D1505に準拠して測定される)であり、より好ましくは953~970kg/m3、さらに好ましくは954~969kg/m3である。密度が953kg/m3未満の低分子量ないし高分子量ポリエチレンを用いた場合は、得られる樹脂組成物の耐摩耗性の点で好ましくない。
[変性前のポリエチレン樹脂組成物(B)の製造方法]
変性前のポリエチレン樹脂組成物(B)の製造方法について特に制限はない。その製造方法の代表的な態様として、以下の(1)及び(2)を例示することができる。
変性前のポリエチレン樹脂組成物(B)の製造方法について特に制限はない。その製造方法の代表的な態様として、以下の(1)及び(2)を例示することができる。
(1)超高分子量ポリエチレン(C)と低分子量ないし高分子量ポリエチレン(D)とをそれぞれ、予め公知のオレフィン重合用触媒の存在下で製造したものをブレンドすることにより製造する方法。
(2)公知のオレフィン重合用触媒の存在下、超高分子量ポリエチレン(C)を製造する工程(I)と、低分子量ないし高分子量ポリエチレン(D)を製造する工程(II)の少なくとも2段階の工程を含んで構成される多段重合法により製造する方法。
特に上記(2)の方法は、上記(1)の方法に比べて、より微分散した海島構造を形成できる点などから好適である。さらに、上記(2)の方法において、特に第1工程として超高分子量ポリエチレンを生成させる工程(I)を行うのが、重合処理操作及び生成ポリエチレンの物性の制御の点などから好適である。なお、この際、重合に用いるエチレンなどのオレフィンとして、前記低分子量ないし高分子量ポリエチレン(D)及び超高分子量ポリエチレン(C)の項目において記載した各種オレフィンを制限無く用いることができる。
上記(1)及び(2)いずれの製造方法においても、超高分子量ポリエチレン(C)と低分子量ないし高分子量ポリエチレン(D)とのブレンド量比あるいは重合量比から、得られる変性ポリエチレン樹脂組成物(A)の海相および島相のいずれが超高分子量ポリエチレン(C)あるいは低分子量ないし高分子量ポリエチレン(D)に由来するものであるかを判断できる場合もある。
特に本発明の変性ポリエチレン樹脂組成物(A)は、下記の工程を含む方法で製造された、135℃のデカリン中で測定される極限粘度[η]が1.5~15dL/gの範囲にあるポリエチレン樹脂組成物(B)を変性して得られるものであることが好ましい。
(I)極限粘度[η]が20~40dL/gの範囲にある超高分子量ポリエチレン(C)を製造する工程
(II)極限粘度[η]が0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレン(D)を製造する工程
(I)極限粘度[η]が20~40dL/gの範囲にある超高分子量ポリエチレン(C)を製造する工程
(II)極限粘度[η]が0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレン(D)を製造する工程
例えばまず上記(I)の工程を行い、引き続き上記(II)の工程を同バッチ内で行う多段階重合により、二種のポリエチレンを混合する方法と比べ、より微分散した海島構造を形成することができ、さらに連続生産法(非バッチ式)と比べ、より均一な特性を有する組成物を安定して得ることができる。特に、超高分子量ポリエチレンが均一に微分散して結合すると耐摩耗性、自己潤滑性、衝撃強度、耐薬品性、外観及び成形性などの特性のバランスに優れ、とりわけ耐摩耗性、外観と成形性と柔軟性のバランスに優れたポリエチレン樹脂組成物が得られる。
なお、上記(I)の工程を行い、引き続き上記(II)の工程を行うと、通常、内相が低分子量ないし高分子量ポリエチレン(D)であり、外相が超高分子量ポリエチレン(C)であるコアシェル粒子構造のポリエチレン粒子が得られる。さらにこの粒子を熱溶融および変性することにより、島相が超高分子量ポリエチレン(C)に由来し、海相が低分子量ないし高分子量ポリエチレン(D)に由来する変性ポリエチレン樹脂組成物(A)が得られる。一方でこのコアシェル粒子構造のポリエチレン粒子に対し、電子線などを用いて固相変性させることで、海相が超高分子量ポリエチレン(C)に由来し、島相が低分子量ないし高分子量ポリエチレン(D)に由来する変性ポリエチレン樹脂組成物(A)が得られる場合もある。
ポリエチレン等の重合に用いるオレフィン重合用触媒としては、超高分子量ポリエチレン及び低分子量ないし高分子量ポリエチレンなどを製造することができるものであれば、特に制限無く用いることができる。具体的には、四塩化チタン又は三塩化チタンからなるチーグラー・ナッタ触媒、チタンをマグネシウム等の担体に担持した担体担持型固体状チタン触媒、メタロセン触媒や、ポストメタロセン触媒が挙げられる。
本発明の変性ポリエチレン樹脂組成物(A)を得るのに好ましい態様としては、数マイクロメートル~25μm程度の粒子径で、且つ粒度分布が狭い傾向を有する固体状チタン触媒成分を用いることであり、その好適な例としては、特開昭56-811号公報、特開昭58-83006号公報などを代表例とする液状のマグネシウム化合物と液状のチタン化合物と必要に応じて電子供与体とを用いて製造される固体状チタン触媒成分を含む触媒の態様が好ましい。
[樹脂(E)]
本発明の変性ポリエチレン樹脂組成物(A)を用いて摺動特性などの特性を向上させることができる樹脂(E)は、例えばエンジニアリングプラスチック等の熱可塑性樹脂である。樹脂(E)として、具体的には次の(E1)~(E6)が例示される。
(E1)ポリアミド樹脂、
(E2)ポリアセタール樹脂、
(E3)ポリエステル樹脂、
(E4)ポリフェニレンスルフィド樹脂、
(E5)ポリカーボネート樹脂、
(E6)ポリオレフィン樹脂
本発明の変性ポリエチレン樹脂組成物(A)を用いて摺動特性などの特性を向上させることができる樹脂(E)は、例えばエンジニアリングプラスチック等の熱可塑性樹脂である。樹脂(E)として、具体的には次の(E1)~(E6)が例示される。
(E1)ポリアミド樹脂、
(E2)ポリアセタール樹脂、
(E3)ポリエステル樹脂、
(E4)ポリフェニレンスルフィド樹脂、
(E5)ポリカーボネート樹脂、
(E6)ポリオレフィン樹脂
[ポリアミド樹脂(E1)]
ポリアミド樹脂(E1)としては、例えば、ヘキサメチレンジアミン、デカメチレンジアミン、ドデカメチレンジアミン、2,2,4-又は2,4,4-トリメチルヘキサメチレンジアミン、1,3-又は1,4-ビス(アミノメチル)シクロヘキサン、ビス(p-アミノシクロヘキシルメタン)、m-又はp-キシリレンジアミン等の脂肪族ジアミン、脂環式ジアミン又は芳香族ジアミンなどのジアミン類と、アジピン酸、スベリン酸、セバシン酸、シクロヘキサンジカルボン酸、テレフタル酸、イソフタル酸等の脂肪族ジカルボン酸、脂環式ジカルボン酸、芳香族ジカルボン酸などのジカルボン酸類との重縮合によって得られるポリアミド、ε-アミノカプロン酸、11-アミノウンデカン酸等のアミノカルボン酸の縮合によって得られるポリアミド、ε-カプロラクタム、ω-ラウロラクタム等のラクタムから得られるポリアミド、あるいはこれらの成分からなる共重合ポリアミド、さらにはこれらのポリアミドの混合物などが挙げられる。このポリアミド樹脂(E1)の具体例としては、ナイロン6、ナイロン66、ナイロン6110、ナイロン9、ナイロン11、ナイロン12、ナイロン6/66、ナイロン66/610、ナイロン6/11、芳香族ナイロン、ナイロン6T、ナイロン9T、ナイロン10T、MXD6等が挙げられる。
ポリアミド樹脂(E1)としては、例えば、ヘキサメチレンジアミン、デカメチレンジアミン、ドデカメチレンジアミン、2,2,4-又は2,4,4-トリメチルヘキサメチレンジアミン、1,3-又は1,4-ビス(アミノメチル)シクロヘキサン、ビス(p-アミノシクロヘキシルメタン)、m-又はp-キシリレンジアミン等の脂肪族ジアミン、脂環式ジアミン又は芳香族ジアミンなどのジアミン類と、アジピン酸、スベリン酸、セバシン酸、シクロヘキサンジカルボン酸、テレフタル酸、イソフタル酸等の脂肪族ジカルボン酸、脂環式ジカルボン酸、芳香族ジカルボン酸などのジカルボン酸類との重縮合によって得られるポリアミド、ε-アミノカプロン酸、11-アミノウンデカン酸等のアミノカルボン酸の縮合によって得られるポリアミド、ε-カプロラクタム、ω-ラウロラクタム等のラクタムから得られるポリアミド、あるいはこれらの成分からなる共重合ポリアミド、さらにはこれらのポリアミドの混合物などが挙げられる。このポリアミド樹脂(E1)の具体例としては、ナイロン6、ナイロン66、ナイロン6110、ナイロン9、ナイロン11、ナイロン12、ナイロン6/66、ナイロン66/610、ナイロン6/11、芳香族ナイロン、ナイロン6T、ナイロン9T、ナイロン10T、MXD6等が挙げられる。
[ポリアセタール樹脂(E2)]
ポリアセタール樹脂(E2)としては、例えば、オキシメチレン基を主たる構成単位とする熱可塑性樹脂であり、ポリアセタールホモポリマー又はポリオキシメチレンと、オキシメチレン単位とコモノマー単位とを含有するポリアセタールコポリマーが含まれる。本発明においては、ポリアセタールホモポリマー、ポリアセタールコポリマーのいずれも使用することが可能である。このポリアセタール樹脂(E2)の具体例としては、ポリアセタールホモポリマー(例えば米国デュポン社製の商品名「デルリン」、旭化成工業社製の商品名「テナック」など)、また、ポリアセタールコポリマー(例えばポリプラスチックス社製の商品名「ジュラコン」など)が挙げられる。
ポリアセタール樹脂(E2)としては、例えば、オキシメチレン基を主たる構成単位とする熱可塑性樹脂であり、ポリアセタールホモポリマー又はポリオキシメチレンと、オキシメチレン単位とコモノマー単位とを含有するポリアセタールコポリマーが含まれる。本発明においては、ポリアセタールホモポリマー、ポリアセタールコポリマーのいずれも使用することが可能である。このポリアセタール樹脂(E2)の具体例としては、ポリアセタールホモポリマー(例えば米国デュポン社製の商品名「デルリン」、旭化成工業社製の商品名「テナック」など)、また、ポリアセタールコポリマー(例えばポリプラスチックス社製の商品名「ジュラコン」など)が挙げられる。
[ポリエステル樹脂(E3)]
ポリエステル樹脂(E3)としては、例えば、エチレングリコール、プロピレングリコール、1,4-ブタンジオール、ネオペンチルグリコール、ヘキサメチレングリコール等の脂肪族グリコール、シクロヘキサンジメタノール等の脂環式グリコール、ビスフェノール等の芳香族ジヒドロキシ化合物と、テレフタル酸、イソフタル酸、2,6-ナフタリンジカルボン酸等の芳香族ジカルボン酸、シュウ酸、コハク酸、アジピン酸、セバシン酸、ウンデカジカルボン酸等の脂肪族ジカルボン酸、ヘキサヒドロテレフタル酸等の脂環式ジカルボン酸、あるいはこれらから選ばれる2種以上のジカルボン酸とから形成される結晶性の熱可塑性樹脂である。このポリエステル樹脂(E3)は、熱可塑性を示す限り、少量のトリオールやトリカルボン酸等の3価以上のポリヒドロキシ化合物やポリカルボン酸などで変性されていてもよい。このポリエステル樹脂(E3)の具体例として、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンイソフタレート・テレフタレート共重合体等が挙げられる。なかでも、ポリブチレンテレフタレート(PBT)が好ましい。
ポリエステル樹脂(E3)としては、例えば、エチレングリコール、プロピレングリコール、1,4-ブタンジオール、ネオペンチルグリコール、ヘキサメチレングリコール等の脂肪族グリコール、シクロヘキサンジメタノール等の脂環式グリコール、ビスフェノール等の芳香族ジヒドロキシ化合物と、テレフタル酸、イソフタル酸、2,6-ナフタリンジカルボン酸等の芳香族ジカルボン酸、シュウ酸、コハク酸、アジピン酸、セバシン酸、ウンデカジカルボン酸等の脂肪族ジカルボン酸、ヘキサヒドロテレフタル酸等の脂環式ジカルボン酸、あるいはこれらから選ばれる2種以上のジカルボン酸とから形成される結晶性の熱可塑性樹脂である。このポリエステル樹脂(E3)は、熱可塑性を示す限り、少量のトリオールやトリカルボン酸等の3価以上のポリヒドロキシ化合物やポリカルボン酸などで変性されていてもよい。このポリエステル樹脂(E3)の具体例として、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエチレンイソフタレート・テレフタレート共重合体等が挙げられる。なかでも、ポリブチレンテレフタレート(PBT)が好ましい。
[ポリフェニレンスルフィド樹脂(E4)]
ポリフェニレンスルフィド樹脂(E4)は、その種類、分子量等とくに限定されない。例えば、直鎖型、架橋型、半架橋型等の重合方法により得られたものを用いることができる。
ポリフェニレンスルフィド樹脂(E4)は、その種類、分子量等とくに限定されない。例えば、直鎖型、架橋型、半架橋型等の重合方法により得られたものを用いることができる。
[ポリカーボネート樹脂(E5)]
ポリカーボネート樹脂(E5)としては、特に限定されるものではない。その具体例としては、例えば、炭酸エステル結合を分子鎖中に有する高分子量重合体の中でも、芳香族ジオキシ化合物(ビスフェノール)、特にビスフェノールAを原料として合成される高分子重合体が挙げられる。
ポリカーボネート樹脂(E5)としては、特に限定されるものではない。その具体例としては、例えば、炭酸エステル結合を分子鎖中に有する高分子量重合体の中でも、芳香族ジオキシ化合物(ビスフェノール)、特にビスフェノールAを原料として合成される高分子重合体が挙げられる。
[ポリオレフィン樹脂(E6)]
ポリオレフィン樹脂(E6)としては、変性ポリエチレン樹脂組成物(A)と異なるものであればとくに限定されるものではない。その具体例としては、例えば、ポリエチレン、ポリプロピレン、ポリブテン、ポリ-4-1-メチルペンテン等が挙げられる。
ポリオレフィン樹脂(E6)としては、変性ポリエチレン樹脂組成物(A)と異なるものであればとくに限定されるものではない。その具体例としては、例えば、ポリエチレン、ポリプロピレン、ポリブテン、ポリ-4-1-メチルペンテン等が挙げられる。
[樹脂(E)と変性ポリエチレン樹脂組成物(A)の組成比]
本発明の変性ポリエチレン樹脂組成物(A)を用いることによって樹脂(E)の摺動特性等の特性が向上した樹脂組成物を得る場合、その好適な組成比は樹脂(E)の種類によって異なる。例えば、樹脂(E)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部として、樹脂(E)の含有量は、好ましくは50~98質量部、より好ましくは55~95質量部、特に好ましくは70~95質量部であり、変性ポリエチレン樹脂組成物(A)の含有量は、好ましくは2~50質量部、より好ましくは5~45質量部、特に好ましくは5~30質量部である。樹脂(E)の含有量が多過ぎると摺動特性が十分に得られないおそれがあり、少な過ぎると樹脂(E)の本来持つ機械的特性が損なわれるおそれがある。樹脂(E)は(E1)~(E6)の何れかを単独で用いてもよいし、複数を任意の比率で組み合わせて用いることもできる。ただし、何れかを単独で用いることが好ましい。
本発明の変性ポリエチレン樹脂組成物(A)を用いることによって樹脂(E)の摺動特性等の特性が向上した樹脂組成物を得る場合、その好適な組成比は樹脂(E)の種類によって異なる。例えば、樹脂(E)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部として、樹脂(E)の含有量は、好ましくは50~98質量部、より好ましくは55~95質量部、特に好ましくは70~95質量部であり、変性ポリエチレン樹脂組成物(A)の含有量は、好ましくは2~50質量部、より好ましくは5~45質量部、特に好ましくは5~30質量部である。樹脂(E)の含有量が多過ぎると摺動特性が十分に得られないおそれがあり、少な過ぎると樹脂(E)の本来持つ機械的特性が損なわれるおそれがある。樹脂(E)は(E1)~(E6)の何れかを単独で用いてもよいし、複数を任意の比率で組み合わせて用いることもできる。ただし、何れかを単独で用いることが好ましい。
特に、ポリアミド樹脂(E1)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の場合は、ポリアミド樹脂(E1)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部として、ポリアミド(E1)の含有量は、好ましくは50~95質量部、より好ましくは80~90質量部であり、変性ポリエチレン樹脂組成物(A)の含有量は、好ましくは5~50質量部、より好ましくは10~20質量部である。これら範囲にあるとき、摩擦発熱MAX温度の抑制効果が特に高くなる。
また、ポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の場合は、ポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部として、ポリアセタール樹脂(E2)の含有量は、好ましくは80~95質量部、より好ましくは85~95質量部であり、変性ポリエチレン樹脂組成物(A)の含有量は、好ましくは5~20質量部、より好ましくは5~15質量部である。これら範囲にあるとき、耐摩耗性が特に優れる。その理由については明らかではないが、おそらく、樹脂組成物におけるポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)とが相構造をなし、それらの界面から摩耗が起こりうるところ、上記範囲の比率において界面の形状等が最適となって耐摩耗性が特に優れるものと推察される。
また、ポリエステル樹脂(E3)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の場合は、ポリエステル樹脂(E3)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部として、ポリエステル樹脂(E3)の含有量は、好ましくは50~95質量部、より好ましくは65~95質量部であり、変性ポリエチレン樹脂組成物(A)の含有量は、好ましくは5~50質量部、より好ましくは5~35質量部である。
[樹脂(E)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の調製方法]
樹脂(E)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の調製方法としては、例えば、樹脂(E)と、変性ポリエチレン樹脂(A)とを溶融混練する方法が挙げられる。この方法においては、樹脂(E)と、変性ポリエチレン樹脂(A)のいずれもが溶融する温度で混練すればよく、その温度は特に限定されない。通常は、200~350℃、好ましくは200~310℃の温度で溶融混練すればよい。混練は、一軸押出機、二軸押出機、バンバリミキサー等を使用して行うことができる。
樹脂(E)と変性ポリエチレン樹脂組成物(A)を含む樹脂組成物の調製方法としては、例えば、樹脂(E)と、変性ポリエチレン樹脂(A)とを溶融混練する方法が挙げられる。この方法においては、樹脂(E)と、変性ポリエチレン樹脂(A)のいずれもが溶融する温度で混練すればよく、その温度は特に限定されない。通常は、200~350℃、好ましくは200~310℃の温度で溶融混練すればよい。混練は、一軸押出機、二軸押出機、バンバリミキサー等を使用して行うことができる。
[樹脂組成物の摺動特性]
エンジニアリングプラスチック等の樹脂(E)単独あるいは変性超高分子量ポリエチレン単独の場合は高温における摩耗特性に劣るが、樹脂(E)と本発明の変性ポリエチレン樹脂組成物(A)を併用した場合は高温における摩耗特性が予想外に向上するという効果が得られる。これは、変性率が比較的高い海相が樹脂(E)により強固に相溶し、たとえ海相が変形し易いものであっても樹脂(E)によってその変形が抑制され、その結果、島相の物性(特に摺動特性など)を発揮し易い状態を作り出しているものと考えられる。
[成形体]
エンジニアリングプラスチック等の樹脂(E)単独あるいは変性超高分子量ポリエチレン単独の場合は高温における摩耗特性に劣るが、樹脂(E)と本発明の変性ポリエチレン樹脂組成物(A)を併用した場合は高温における摩耗特性が予想外に向上するという効果が得られる。これは、変性率が比較的高い海相が樹脂(E)により強固に相溶し、たとえ海相が変形し易いものであっても樹脂(E)によってその変形が抑制され、その結果、島相の物性(特に摺動特性など)を発揮し易い状態を作り出しているものと考えられる。
[成形体]
本発明の成形体は、以上説明した変性ポリエチレン樹脂組成物(A)と他の樹脂(E)とを含んでなる樹脂組成物をその一部または全部に使用してなる成形体である。この樹脂組成物は、従来種々公知の方法により成形できる。具体的には、この樹脂組成物を、例えば、射出成形法、異形押出成形法、パイプ成形法、チューブ成形法、異種成形体の被覆成形法、インジェクションブロー成形法、ダイレクトブロー成形法、Tダイシートまたはフィルム成形法、インフレーションフィルム成形法、プレス成形法などの成形方法により、容器状、トレー状、シート状、棒状、フィルム状または各種成形体の被覆などに成形することができる。
上記成形方法で得られた成形体は、従来公知の用途に広く使用できる。特に耐摩耗性、自己潤滑性、衝撃強度、薄肉成形などの特性のバランスに優れているので、これらが要求される用途として、例えば、鋼管、電線、自動車スライドドアレールなどの金属の被覆(積層)、耐圧ゴムホース、自動車ドア用ガスケット、クリーンルームドア用ガスケット、自動車グラスランチャンネル、自動車ウエザストリップなどの各種ゴムの被覆(積層)、ホッパー、シュートなどのライニング用、ギアー、軸受、ブッシュ、ローラー、テープリール、各種ガイドレールやエレベーターレールガイド、各種保護ライナー材などの摺動部材などに使用される。
[変性ポリエチレン樹脂組成物(A)のその他の用途]
本発明の変性ポリエチレン樹脂組成物(A)は、各種樹脂の摺動性を向上させる以外にも、従来公知のポリエチレン用途に広く使用できる。特に耐摩耗性、成形性などの特性のバランスに優れているので、これらが要求される用途として、例えば、鋼管、電線、自動車スライドドアレールなどの金属の被覆(積層)、耐圧ゴムホース、自動車ドア用ガスケット、クリーンルームドア用ガスケット、自動車グラスランチャンネル、自動車ウエザストリップなどの各種ゴムの被覆(積層)、ホッパー、シュートなどのライニング用、ギアー、軸受、ローラー、テープリール、各種ガイドレールやエレベーターレールガイド、各種保護ライナー材などの摺動材などの用途に好適に用いることができる。
本発明の変性ポリエチレン樹脂組成物(A)は、各種樹脂の摺動性を向上させる以外にも、従来公知のポリエチレン用途に広く使用できる。特に耐摩耗性、成形性などの特性のバランスに優れているので、これらが要求される用途として、例えば、鋼管、電線、自動車スライドドアレールなどの金属の被覆(積層)、耐圧ゴムホース、自動車ドア用ガスケット、クリーンルームドア用ガスケット、自動車グラスランチャンネル、自動車ウエザストリップなどの各種ゴムの被覆(積層)、ホッパー、シュートなどのライニング用、ギアー、軸受、ローラー、テープリール、各種ガイドレールやエレベーターレールガイド、各種保護ライナー材などの摺動材などの用途に好適に用いることができる。
[その他の成分]
本発明の変性ポリエチレン樹脂組成物(A)、および、変性ポリエチレン樹脂(A)の原料となるポリエチレン樹脂組成物(B)、および樹脂(E)には、本発明の効果を阻害しない範囲で他の樹脂あるいは重合体および/または樹脂用添加剤を任意に添加することができる。
本発明の変性ポリエチレン樹脂組成物(A)、および、変性ポリエチレン樹脂(A)の原料となるポリエチレン樹脂組成物(B)、および樹脂(E)には、本発明の効果を阻害しない範囲で他の樹脂あるいは重合体および/または樹脂用添加剤を任意に添加することができる。
かかる樹脂用添加剤としては、例えば、核剤、アンチブロッキング剤、スリップ剤、顔料、染料、充填剤(フィラー)、滑剤、可塑剤、離型剤、酸化防止剤、難燃剤、紫外線吸収剤、抗菌剤、界面活性剤、帯電防止剤、耐候安定剤、耐熱安定剤、スリップ防止剤、発泡剤、結晶化助剤、防曇剤、(透明)核剤、老化防止剤、塩酸吸収剤、衝撃改良剤、架橋剤、共架橋剤、架橋助剤、粘着剤、軟化剤、加工助剤、石油樹脂、ワックス、オレフィン系オイル、シリコーンオイルなどが挙げられる。これらの添加剤は、1種単独でも、適宜2種以上を組み合わせても用いることができる。
添加する他の樹脂あるいは重合体としては、下記の熱可塑性樹脂(F)を広く用いることができる。これら樹脂あるいは重合体の添加量は脂組成物の総質量に対して、0.1~30質量%であることが好ましい。
[熱可塑性樹脂(F)]
熱可塑性樹脂(F)は特に制限されない。熱可塑性樹脂(F)としては、例えば、以下の樹脂が挙げられる。
熱可塑性樹脂(F)は特に制限されない。熱可塑性樹脂(F)としては、例えば、以下の樹脂が挙げられる。
熱可塑性ポリオレフィン系樹脂;例えば、低密度、中密度、高密度ポリエチレン、高圧法低密度ポリエチレン、アイソタクティックポリプロピレン、シンジオタクティックポリプロピレン、ポリ1-ブテン、ポリ4-メチル-1-ペンテン、ポリ3-メチル-1-ペンテン、ポリ3-メチル-1-ブテン、エチレン・α-オレフィン共重合体、プロピレン・α-オレフィン共重合体、1-ブテン・α-オレフィン共重合体、環状オレフィン共重合体、塩素化ポリオレフィン、およびこれらのオレフィン系樹脂を変性した変性ポリオレフィン樹脂、
熱可塑性ポリアミド系樹脂;例えば、脂肪族ポリアミド(ナイロン6、ナイロン11、ナイロン12、ナイロン66、ナイロン610、ナイロン612)、
熱可塑性ポリエステル系樹脂;例えば、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステル系エラストマー、
熱可塑性ビニル芳香族系樹脂;例えば、ポリスチレン、ABS樹脂、AS樹脂、スチレン系エラストマー(スチレン・ブタジエン・スチレンブロックポリマー、スチレン・イソプレン・スチレンブロックポリマー、スチレン・イソブチレン・スチレンブロックポリマー、前述の水素添加物)、
熱可塑性ポリウレタン;塩化ビニル樹脂;塩化ビニリデン樹脂;アクリル樹脂;エチレン・酢酸ビニル共重合体;エチレン・メタクリル酸アクリレート共重合体;アイオノマー;エチレン・ビニルアルコール共重合体;ポリビニルアルコール;フッ素系樹脂ポリカーボネート;ポリアセタール;ポリフェニレンオキシド;ポリフェニレンサルファイドポリイミド;ポリアリレート;ポリスルホン;ポリエーテルスルホン;ロジン系樹脂;テルペン系樹脂および石油樹脂;
共重合体ゴム;例えば、エチレン・α-オレフィン・ジエン共重合体、プロピレン・α-オレフィン・ジエン共重合体、1-ブテン・α-オレフィン・ジエン共重合体、ポリブタジエンゴム、ポリイソプレンゴム、ネオプレンゴム、ニトリルゴム、ブチルゴム、ポリイソブチレンゴム、天然ゴム、シリコーンゴム。
熱可塑性ポリアミド系樹脂;例えば、脂肪族ポリアミド(ナイロン6、ナイロン11、ナイロン12、ナイロン66、ナイロン610、ナイロン612)、
熱可塑性ポリエステル系樹脂;例えば、ポリエチレンテレフタレート、ポリブチレンテレフタレート、ポリエステル系エラストマー、
熱可塑性ビニル芳香族系樹脂;例えば、ポリスチレン、ABS樹脂、AS樹脂、スチレン系エラストマー(スチレン・ブタジエン・スチレンブロックポリマー、スチレン・イソプレン・スチレンブロックポリマー、スチレン・イソブチレン・スチレンブロックポリマー、前述の水素添加物)、
熱可塑性ポリウレタン;塩化ビニル樹脂;塩化ビニリデン樹脂;アクリル樹脂;エチレン・酢酸ビニル共重合体;エチレン・メタクリル酸アクリレート共重合体;アイオノマー;エチレン・ビニルアルコール共重合体;ポリビニルアルコール;フッ素系樹脂ポリカーボネート;ポリアセタール;ポリフェニレンオキシド;ポリフェニレンサルファイドポリイミド;ポリアリレート;ポリスルホン;ポリエーテルスルホン;ロジン系樹脂;テルペン系樹脂および石油樹脂;
共重合体ゴム;例えば、エチレン・α-オレフィン・ジエン共重合体、プロピレン・α-オレフィン・ジエン共重合体、1-ブテン・α-オレフィン・ジエン共重合体、ポリブタジエンゴム、ポリイソプレンゴム、ネオプレンゴム、ニトリルゴム、ブチルゴム、ポリイソブチレンゴム、天然ゴム、シリコーンゴム。
ポリプロピレンとしては、上記の通りアイソタクティックポリプロピレンとシンジオタクティックポリプロピレンが挙げられる。アイソタクティックポリプロピレンは、ホモポリプロピレンであっても、プロピレン・炭素原子数2~20のα-オレフィン(ただしプロピレンを除く)ランダム共重合体であっても、プロピレンブロック共重合体であってもよい。
ポリ4-メチル-1-ペンテンは、4-メチル-1-ペンテンの単独重合体、または4-メチル-1-ペンテン・α-オレフィン共重合体である。4-メチル-1-ペンテン・α-オレフィンランダム共重合体の場合、4-メチル-1-ペンテンと共重合するα-オレフィンとしては、エチレン、プロピレン、1-ブテン、1-ヘキセン、1-オクテン、1-デセン、1-ドデセン、1-テトラデセン、1-ヘキサデセン、1-オクタデセン、1-エイコセンなどの炭素原子数2~20 、好ましくは6~20のα-オレフィンが挙げられる。これらは、1種単独で、あるいは2種以上組み合せて用いることができる。
ポリエチレンとしては、上記の通り従来公知の手法で製造されている、低密度ポリエチレン、中密度ポリエチレン、高密度ポリエチレン、高圧法低密度ポリエチレンを使用することが出来る。
ポリブテンとしては、1-ブテンのホモポリマー、あるいは1-ブテンと、1-ブテンを除くオレフィンとの共重合体である。オレフィンは、上記のものが挙げられ、これらのオレフィンは、単独で、または2種以上組み合せて用いることができる。共重合体として、例えば、1-ブテン・エチレンランダム共重合体、1-ブテン・プロピレンランダム共重合体、1-ブテン・メチルペンテン共重合体、1-ブテン・メチルブテン共重合体、1-ブテン・プロピレン・エチレン共重合体などが挙げられる。このような共重合体において、耐熱性の点から、1-ブテン含有量が50モル%以上であることが好ましく、70モル%以上であることが更に好ましく、85%以上であることが特に好ましい。
変性ポリオレフィン樹脂は、上述したポリオレフィン樹脂にエチレン性不飽和結合含有モノマーを、有機過酸化物を用いてグラフト変性することにより得ることができる。変性ポリオレフィンが有する官能基の種類としては、ハロゲン原子、カルボキシル基、酸無水物基、エポキシ基、水酸基、アミノ基、アミド基、イミド基、エステル基、アルコキシシラン基、酸ハライド基およびニトリル基等が挙げられる。
ロジン系樹脂としては、天然ロジン、重合ロジン、マレイン酸、フマル酸、(メタ)アクリル酸などで変性した変性ロジン、ロジン誘導体が挙げられる。また、このロジン誘導体としては、前記の天然ロジン、重合ロジンまたは変性ロジンのエステル化物、フェノール変性物およびそのエステル化物などが挙げられる。さらに、これらの水素添加物も挙げることができる。
テルペン系樹脂としては、α-ピネン、β-ピネン、リモネン、ジペンテン、テルペンフェノール、テルペンアルコール、テルペンアルデヒドなどからなる樹脂が挙げられ、α-ピネン、β-ピネン、リモネン、ジペンテンなどにスチレンなどの芳香族モノマーを重合させた芳香族変性のテルペン系樹脂なども挙げられる。また、これらの水素添加物も挙げることができる。
石油樹脂としては、たとえば、タールナフサのC5留分を主原料とする脂肪族系石油樹脂、C9留分を主原料とする芳香族系石油樹脂およびそれらの共重合石油樹脂が挙げられる。すなわち、C5系石油樹脂(ナフサ分解油のC5留分を重合した樹脂)、C9系石油樹脂(ナフサ分解油のC9留分を重合した樹脂)、C5C9共重合石油樹脂(ナフサ分解油のC5留分とC9留分とを共重合した樹脂)が挙げられ、タールナフサ留分のスチレン類、インデン類、クマロン、その他ジシクロペンタジエンなどを含有しているクマロンインデン系樹脂、p-ターシャリブチルフェノールとアセチレンの縮合物に代表されるアルキルフェノール類樹脂、ο-キシレン、p-キシレンまたはm-キシレンをホルマリンと反応させてなるキシレン系樹脂なども挙げられる。
また、ロジン系樹脂、テルペン系樹脂および石油樹脂は、耐候性および耐変色性に優れるために水素添加誘導体が好ましい。前記樹脂の環球法による軟化点は、40~180℃の範囲にあることが好ましい。また、前記樹脂のGPCにより測定される数平均分子量(Mn)分子量は100~10,000程度の範囲にあることが好ましい。ロジン系樹脂、テルペン系樹脂、石油樹脂として市販品を使用することもできる。
また以上の各熱可塑性樹脂(F)として、市販品を使用することもできる。これらの熱可塑性樹脂(F)の中から1種単独で使用することもできるし、2種以上を組み合せて使用することもできる。
[添加剤]
先に例示列挙した各添加剤のうち、核剤としては、オレフィン重合体の成形性をさらに改善させる、すなわち結晶化温度を高め結晶化速度を速めるために公知の核剤が使用可能である。具体的には、ジベンジリデンソルビトール系核剤、リン酸エステル塩系核剤、ロジン系核剤、安息香酸金属塩系核剤、フッ素化ポリエチレン、2,2-メチレンビス(4,6-ジ-t-ブチルフェニル)リン酸ナトリウム、ピメリン酸やその塩、2,6-ナフタレン酸ジカルボン酸ジシクロヘキシルアミド等が挙げられる。核剤の配合量は、特に限定されないが、上記オレフィン重合体100質量部に対して、好ましくは0.1~1質量部である。核剤は、重合中、重合後、あるいは成形加工時など適宜添加が可能である。
先に例示列挙した各添加剤のうち、核剤としては、オレフィン重合体の成形性をさらに改善させる、すなわち結晶化温度を高め結晶化速度を速めるために公知の核剤が使用可能である。具体的には、ジベンジリデンソルビトール系核剤、リン酸エステル塩系核剤、ロジン系核剤、安息香酸金属塩系核剤、フッ素化ポリエチレン、2,2-メチレンビス(4,6-ジ-t-ブチルフェニル)リン酸ナトリウム、ピメリン酸やその塩、2,6-ナフタレン酸ジカルボン酸ジシクロヘキシルアミド等が挙げられる。核剤の配合量は、特に限定されないが、上記オレフィン重合体100質量部に対して、好ましくは0.1~1質量部である。核剤は、重合中、重合後、あるいは成形加工時など適宜添加が可能である。
アンチブロッキング剤としては、公知のアンチブロッキング剤が使用可能である。具体的には、微粉末シリカ、微粉末酸化アルミニウム、微粉末クレー、粉末状もしくは液状のシリコン樹脂、テトラフロロエチレン樹脂、微粉末架橋樹脂、例えば架橋されたアクリル、メタクリル樹脂粉末等をあげることができる。これらのうちでは、微粉末シリカおよび架橋されたアクリル、メタクリル樹脂粉末が好ましい。
顔料としては、無機含量(酸化チタン、酸化鉄、酸化クロム、硫化カドミウム等)、有機顔料(アゾレーキ系、チオインジゴ系、フタロシアニン系、アントラキノン系)が挙げられる。染料としてはアゾ系、アントラキノン系、トリフェニルメタン系等が挙げられる。これら顔料および染料の添加量は、特に限定されないが、樹脂組成物の総質量に対して、合計で、通常5質量%以下、好ましくは0.1~3質量%である。
充填剤(フィラー)としては、ガラス繊維、炭素繊維、シリカ繊維、金属(ステンレス、アルミニウム、チタン、銅等)繊維、カーボンブラック、シリカ、ガラスビーズ、珪酸塩(珪酸カルシウム、タルク、クレー等)、金属酸化物(酸化鉄、酸化チタン、アルミナ等)、金属の炭酸塩(硫酸カルシウム、硫酸バリウム)および各種金属(マグネシウム、珪素、アルミニウム、チタン、銅等)粉末、マイカ、ガラスフレーク等が挙げられる。これらの充填剤は1種単独または2種以上の併用いずれでもよい。
滑剤としては、ワックス(カルナバロウワックス等)、高級脂肪酸(ステアリン酸等)、高級アルコール(ステアリルアルコール等)、高級脂肪酸アミド(ステアリン酸アミド等)等が挙げられる。
可塑剤としては、芳香族カルボン酸エステル(フタル酸ジブチル等)、脂肪族カルボン酸エステル(メチルアセチルリシノレート等)、脂肪族ジアルボン酸エステル(アジピン酸-プロピレングリコール系ポリエステル等)、脂肪族トリカルボン酸エステル(クエン酸トリエチル等)、リン酸トリエステル(リン酸トリフェニル等)、エポキシ脂肪酸エステル(ステアリン酸エポキシブチル等)、石油樹脂等が挙げられる。
離型剤としては、高級脂肪酸の低級(C1~4)アルコールエステル(ステアリン酸ブチル等)、脂肪酸(C4~30)の多価アルコールエステル(硬化ヒマシ油等)、脂肪酸のグリコールエステル、流動パラフィン等が挙げられる。
酸化防止剤としては、公知の酸化防止剤が使用可能である。具体的には、フェノール系(2,6-ジ-t-ブチル-4-メチルフェノール等)、多環フェノール系(2,2’-メチレンビス(4-メチル-6-t-ブチルフェノール等)、リン系(テトラキス(2,4-ジ-t-ブチルフェニル)-4,4-ビフェニレンジホスフォネート等)、イオウ系(チオジプロピオン酸ジラウリル等)、アミン系(N,N-ジイソプロピル-p-フェニレンジアミン等)、ラクトン系の酸化防止剤等が挙げられ、これらを数種類組み合わせても使用できる。
難燃剤としては、有機系難燃剤(含窒素系、含硫黄系、含珪素系、含リン系等)、無機系難燃剤(三酸化アンチモン、水酸化マグネシウム、ホウ酸亜鉛、赤リン等)が挙げられる。
紫外線吸収剤としては、ベンゾトリアゾール系、ベンゾフェノン系、サリチル酸系、アクリレート系等が挙げられる。
抗菌剤としては、4級アンモニウム塩、ピリジン系化合物、有機酸、有機酸エステル、ハロゲン化フェノール、有機ヨウ素等が挙げられる。
界面活性剤としては非イオン性、アニオン性、カチオン性または両性の界面活性剤を挙げることができる。非イオン性界面活性剤としては、高級アルコールエチレンオキシド付加物、脂肪酸エチレンオキシド付加物、高級アルキルアミンエチレンオキシド付加物、ポリプロピレングリコールエチレンオキシド付加物等のポリエチレングリコール型非イオン界面活性剤、ポリエチレンオキシド、グリセリンの脂肪酸エステル、ペンタエリスリトールの脂肪酸エステル、ソルビットもしくはソルビタンの脂肪酸エステル、多価アルコールのアルキルエーテル、アルカノールアミンの脂肪族アミド等の多価アルコール型非イオン性界面活性剤などが挙げられ、アニオン性界面活性剤としては、例えば、高級脂肪酸のアルカリ金属塩等の硫酸エステル塩、アルキルベンゼンスルホン酸塩、アルキルスルホン酸塩、パラフィンスルホン酸塩等のスルホン酸塩、高級アルコールリン酸エステル塩等のリン酸エステル塩などが挙げられ、カチオン性界面活性剤としては、アルキルトリメチルアンモニウム塩等の第4級アンモニウム塩などが挙げられる。両性界面活性剤としては、高級アルキルアミノプロピオン酸塩等のアミノ酸型両面界面活性剤、高級アルキルジメチルベタイン、高級アルキル時ヒドロキシエチルベタイン等のベタイン型両性界面活性剤などが挙げられる。
帯電防止剤としては、上記の界面活性剤、脂肪酸エステル、高分子型帯電防止剤が挙げられる。脂肪酸エステルとしてはステアリン酸やオレイン酸のエステルなどが挙げられ、高分子型帯電防止剤としてはポリエーテルエステルアミドが挙げられる。
上記充填剤、滑剤、可塑剤、離型剤、酸化防止剤、難燃剤、紫外線吸収剤、抗菌剤、界面活性剤、帯電防止剤などの各種添加剤の添加量は、本発明の目的を損なわない範囲内で用途に応じて、特に限定されないが、樹脂組成物の総質量に対して、それぞれ、0.1~30質量%であることが好ましい。
以下、実施例に基づいて本発明をさらに具体的に説明するが、本発明はこれらの実施例に限定されるものではない。本発明の実施例および比較例における測定方法は以下の通りである。
<島相の観察方法と長軸粒子径の算出方法>
変性ポリエチレン樹脂組成物(A)を用いて、JIS K7210に準拠して、190℃、10kg荷重でMFRを測定する際のストランドを作製し、ストランド方向に対して垂直にクライオウルトラミクロトームにて約5μm厚の切片を作製し観察検体とした。観察方法としては、光学顕微鏡(ニコン社製、製品名LVI0OPOL)を用いて、クロスニコルで観察した。偏光をかけることで島相の粒子が観察しやすいように偏光を調整し、海島構造を200倍の倍率で3か所撮影し、撮影した視野中から300μm角を無作為に定めて、島相長軸方向の径と、所定の長軸粒子径を有する島相の各個数を求めた。
変性ポリエチレン樹脂組成物(A)を用いて、JIS K7210に準拠して、190℃、10kg荷重でMFRを測定する際のストランドを作製し、ストランド方向に対して垂直にクライオウルトラミクロトームにて約5μm厚の切片を作製し観察検体とした。観察方法としては、光学顕微鏡(ニコン社製、製品名LVI0OPOL)を用いて、クロスニコルで観察した。偏光をかけることで島相の粒子が観察しやすいように偏光を調整し、海島構造を200倍の倍率で3か所撮影し、撮影した視野中から300μm角を無作為に定めて、島相長軸方向の径と、所定の長軸粒子径を有する島相の各個数を求めた。
<IRS、IRI、及びIRS/IRIの算出方法>
上述で得られた観察検体を用いて、島相については10μm以上30μm以下の粒子で、それぞれが50μm以上離れた粒子から無作為に3個選定して、また海相については、各選定した10μm以上30μm以下の粒子それぞれの外周から、10μm以上、30μm以下の範囲内の海相を選定して、選定したそれぞれの島相、海相をFT-IR顕微システム(VARIAN製、製品名FTS-7000/UMA-600)を用いて、顕微透過イメージング法によって3回測定した。3回測定した海相と島相それぞれのCH2基ピーク強度(1470cm-1)とカルボキシル基ピーク強度(1710cm-1と1790cm-1の強度の和)の比の平均を算出することで、IRS、IRI、及びIRS/IRIを求めた。ピーク強度の算出方法としては、干渉縞やノイズが無いスペクトルを得た状態で、CH2基ピーク強度については、1400cm-1から1410cm-1の範囲の変曲点を始点、1510cm-1から1520cm-1の範囲の変曲点を終点と定め、それらの面積からピーク強度を測定した。また、カルボキシル基ピーク強度の1710cm-1付近のピーク強度は、1670cm-1から1680cm-1の範囲の変曲点を始点、1745cm-1から1755cm-1の範囲の変曲点を終点と定め、1790cm-1付近のピーク強度は、1760cm-1から1770cm-1の範囲の変曲点を始点、1810cm-1から1820cm-1の範囲の変曲点を終点と定めて、それらの面積からピーク強度を測定した。
上述で得られた観察検体を用いて、島相については10μm以上30μm以下の粒子で、それぞれが50μm以上離れた粒子から無作為に3個選定して、また海相については、各選定した10μm以上30μm以下の粒子それぞれの外周から、10μm以上、30μm以下の範囲内の海相を選定して、選定したそれぞれの島相、海相をFT-IR顕微システム(VARIAN製、製品名FTS-7000/UMA-600)を用いて、顕微透過イメージング法によって3回測定した。3回測定した海相と島相それぞれのCH2基ピーク強度(1470cm-1)とカルボキシル基ピーク強度(1710cm-1と1790cm-1の強度の和)の比の平均を算出することで、IRS、IRI、及びIRS/IRIを求めた。ピーク強度の算出方法としては、干渉縞やノイズが無いスペクトルを得た状態で、CH2基ピーク強度については、1400cm-1から1410cm-1の範囲の変曲点を始点、1510cm-1から1520cm-1の範囲の変曲点を終点と定め、それらの面積からピーク強度を測定した。また、カルボキシル基ピーク強度の1710cm-1付近のピーク強度は、1670cm-1から1680cm-1の範囲の変曲点を始点、1745cm-1から1755cm-1の範囲の変曲点を終点と定め、1790cm-1付近のピーク強度は、1760cm-1から1770cm-1の範囲の変曲点を始点、1810cm-1から1820cm-1の範囲の変曲点を終点と定めて、それらの面積からピーク強度を測定した。
<極限粘度[η]>
デカリン溶媒を用いて、135℃で極限粘度[η]を測定した。すなわち、サンプルの造粒ペレット(約20mg)をデカリン溶媒(15mL)に溶解し、135℃のオイルバス中で比粘度ηspを測定した。このデカリン溶液にデカリン溶媒(5mL)を追加して希釈した後、前記と同様に比粘度ηspを測定した。この希釈操作をさらに2回繰り返し、サンプルの濃度(C)を0に外挿したときのηsp/Cの値をオレフィン重合体の極限粘度[η]とした。
極限粘度[η]=lim(ηsp/C) (C→0)
デカリン溶媒を用いて、135℃で極限粘度[η]を測定した。すなわち、サンプルの造粒ペレット(約20mg)をデカリン溶媒(15mL)に溶解し、135℃のオイルバス中で比粘度ηspを測定した。このデカリン溶液にデカリン溶媒(5mL)を追加して希釈した後、前記と同様に比粘度ηspを測定した。この希釈操作をさらに2回繰り返し、サンプルの濃度(C)を0に外挿したときのηsp/Cの値をオレフィン重合体の極限粘度[η]とした。
極限粘度[η]=lim(ηsp/C) (C→0)
<密度>
ASTM D1505に準拠して密度を測定した。
ASTM D1505に準拠して密度を測定した。
<MFR>
JIS K7210に準拠して、190℃と230℃、235℃又は280℃の温度条件で、2.16kg荷重の条件下でメルトフローレート(MFR)を測定した。
JIS K7210に準拠して、190℃と230℃、235℃又は280℃の温度条件で、2.16kg荷重の条件下でメルトフローレート(MFR)を測定した。
<引張試験>
ISO-527-1,2に準拠して、試験片形状を多目的試験片及び引張速度を50mm/minとし、引張降伏点応力(降伏点強度)及び破断点伸びを求めた。
ISO-527-1,2に準拠して、試験片形状を多目的試験片及び引張速度を50mm/minとし、引張降伏点応力(降伏点強度)及び破断点伸びを求めた。
<曲げ試験>
ISO-178に準拠して、多目的試験片を用いて、曲げスパン48.0mm、試験速度5.0mm/minとし、曲げ強度、曲げ弾性率を求めた。
ISO-178に準拠して、多目的試験片を用いて、曲げスパン48.0mm、試験速度5.0mm/minとし、曲げ強度、曲げ弾性率を求めた。
<シャルピー衝撃強度>
ISO-179に準拠して、ノッチ付き多目的試験片を用いてシャルピー衝撃強度を測定した。
ISO-179に準拠して、ノッチ付き多目的試験片を用いてシャルピー衝撃強度を測定した。
<摩擦係数、比摩耗量及び摩擦発熱MAX温度>
JIS K7218「プラスチックの滑り摩耗試験A法」に準拠して、松原式摩擦摩耗試験機を使用して動摩擦係数および比摩耗量を測定した。試験条件は、相手材:S45C、速度:50cm/秒、距離:3km、荷重:15kg(動摩擦係数)または2.5kg(比摩耗量)、測定環境温度:23℃とした。また測定環境温度を50℃、100℃、150℃、170℃としたこと以外は同じ条件で、高温下における動摩擦係数および比摩耗量を測定した。摩擦発熱MAX温度の測定は、相手材であるS45Cに温度測定用の熱電対を設置して、試験中の温度を測定し、試験中の温度上昇のMAXの値を算出した。
JIS K7218「プラスチックの滑り摩耗試験A法」に準拠して、松原式摩擦摩耗試験機を使用して動摩擦係数および比摩耗量を測定した。試験条件は、相手材:S45C、速度:50cm/秒、距離:3km、荷重:15kg(動摩擦係数)または2.5kg(比摩耗量)、測定環境温度:23℃とした。また測定環境温度を50℃、100℃、150℃、170℃としたこと以外は同じ条件で、高温下における動摩擦係数および比摩耗量を測定した。摩擦発熱MAX温度の測定は、相手材であるS45Cに温度測定用の熱電対を設置して、試験中の温度を測定し、試験中の温度上昇のMAXの値を算出した。
<限界PV値>
ステップワイズ法〔JIS K7218(SUSリング/樹脂シート)〕により評価した。具体的には、摺動速度:0.2m/s、試験荷重:0.25~25MPa(0.25MPa毎ステップ)、試験温度:23℃として、試験荷重による樹脂の摩耗による融着、変形による摩擦係数上昇、発熱温度上昇までの試験荷重と摺動速度から限界PV値を算出した。
ステップワイズ法〔JIS K7218(SUSリング/樹脂シート)〕により評価した。具体的には、摺動速度:0.2m/s、試験荷重:0.25~25MPa(0.25MPa毎ステップ)、試験温度:23℃として、試験荷重による樹脂の摩耗による融着、変形による摩擦係数上昇、発熱温度上昇までの試験荷重と摺動速度から限界PV値を算出した。
<分散径>
変性ポリエチレン樹脂組成物(A)と樹脂(E)を含む樹脂組成物について、物性測定用に作製した多目的試験片を用いて、MD方向に垂直にクライオウルトラミクロトームにて切削断面を作製し、カーボン蒸着を施して観察検体とした。観察方法としては観察検体エンドビューのコア層を、電子顕微鏡(日本電子社製、JFM-7001F)を用いて観察し、分散径を測定した。
変性ポリエチレン樹脂組成物(A)と樹脂(E)を含む樹脂組成物について、物性測定用に作製した多目的試験片を用いて、MD方向に垂直にクライオウルトラミクロトームにて切削断面を作製し、カーボン蒸着を施して観察検体とした。観察方法としては観察検体エンドビューのコア層を、電子顕微鏡(日本電子社製、JFM-7001F)を用いて観察し、分散径を測定した。
[実施例1-1]
<固体状チタン触媒成分の調製>
無水塩化マグネシウム95.2g、デカン398.4gおよび2-エチルヘキシルアルコ-ル306gを温度140℃で6時間加熱反応させて均一溶液とした後、この溶液中に安息香酸エチル17.6gを添加し、更に130℃にて1時間攪拌混合を行なった。
<固体状チタン触媒成分の調製>
無水塩化マグネシウム95.2g、デカン398.4gおよび2-エチルヘキシルアルコ-ル306gを温度140℃で6時間加熱反応させて均一溶液とした後、この溶液中に安息香酸エチル17.6gを添加し、更に130℃にて1時間攪拌混合を行なった。
このようにして得られた均一溶液を室温まで冷却した後、この均一溶液50mlを0℃に保持した四塩化チタン200ml中に一定の撹拌速度で攪拌しつつ1時間にわたって全量滴下装入した。装入終了後、この混合液の温度を2.5時間かけて80℃に昇温し、80℃になったところで混合液中に安息香酸エチル2.35gを添加し、2時間同温度にて攪拌下保持した。2時間の反応終了後、熱濾過にて固体部を採取し、この固体部を100mlの四塩化チタンにて再懸濁させた後、90℃で2時間、加熱反応を行った。反応終了後、再び熱濾過にて固体部を採取し、温度90℃のデカンおよびヘキサンで洗液中に遊離のチタン化合物が検出されなくなるまで充分洗浄した。以上の操作によって調製した固体状チタン触媒成分はデカンスラリ-として保存した。
ICP法で分析したところ、固体状チタン触媒成分中、Ti成分が3.5質量%含まれていた。ベックマン・コールター社製レーザー回折散乱法粒度分布測定装置で測定した触媒粒子の平均粒径は7μm、最大粒径は18μmであった。
<ポリエチレン樹脂組成物(B-1)の重合>
充分に窒素置換された攪拌機付24Lのオートクレーブに12Lの精製n-デカンを添加した後、トリエチルアルミニウムをアルミニウム換算で14ミリモル、上記固体状チタン触媒成分をチタン換算で0.3ミリモル加え、十分に撹拌しながら45℃まで昇温しつつ、4.2L/分の速度でエチレンを供給して重合を開始した。オートクレーブの内圧は6kg/cm2・Gに保持した。重合温度は45~46℃に維持した。エチレンを880L供給した時点でエチレンの供給を一旦停止し、内圧が3kg/cm2・Gとなるまで温度を一定に保持した後、速やかに常圧まで脱圧した。(この段階で、得られたスラリーを少量サンプリングし、デカンとヘキサンとで洗浄して白色固体サンプル(1)を得た。)次いで水素を41リットル導入し、温度を85℃に上げつつエチレンを11.6L/分の速度で供給しながら2段目の重合を開始した。全圧を6.4kg/cm2・G、温度は85℃に保持した。
充分に窒素置換された攪拌機付24Lのオートクレーブに12Lの精製n-デカンを添加した後、トリエチルアルミニウムをアルミニウム換算で14ミリモル、上記固体状チタン触媒成分をチタン換算で0.3ミリモル加え、十分に撹拌しながら45℃まで昇温しつつ、4.2L/分の速度でエチレンを供給して重合を開始した。オートクレーブの内圧は6kg/cm2・Gに保持した。重合温度は45~46℃に維持した。エチレンを880L供給した時点でエチレンの供給を一旦停止し、内圧が3kg/cm2・Gとなるまで温度を一定に保持した後、速やかに常圧まで脱圧した。(この段階で、得られたスラリーを少量サンプリングし、デカンとヘキサンとで洗浄して白色固体サンプル(1)を得た。)次いで水素を41リットル導入し、温度を85℃に上げつつエチレンを11.6L/分の速度で供給しながら2段目の重合を開始した。全圧を6.4kg/cm2・G、温度は85℃に保持した。
エチレンを3800L供給したところで、エチレンの供給を停止し、内圧が3kg/cm2・Gになるまで温度は85℃に保持し、その後、常圧、常温まで脱圧、冷却し、重合終了とした。
重合終了後、得られたスラリーから固体状白色固体を分離し、デカン、ヘキサンで洗浄した後、これを80℃で減圧乾燥した。得られた白色固体(ポリエチレン樹脂組成物(B-1))の密度は967kg/cm3、極限粘度[η]は5.73dl/gであった。
一方、白色個体サンプル(1)は、極限粘度[η]が28dl/gであった。また、エチレンの供給量より、第1段目で製造した超高分子量ポリエチレンの含有量は19.0質量%である。第2段目で生成した重合体の極限粘度は下記式より推算すると、0.5dl/gであった。
[η]all=[η]A×wtA+[η]B×wtB
[η]all:ポリマー全体(ポリエチレン樹脂組成物)の極限粘度(dl/g)
[η]A:超高分子量ポリエチレンの極限粘度(dl/g)
wtA:超高分子量ポリエチレンの含有量(質量%)
[η]B:低分子量ないし高分子量ポリエチレンの極限粘度(dl/g)
wtB:低分子量ないし高分子量ポリエチレンの含有量(質量%)
[η]all=[η]A×wtA+[η]B×wtB
[η]all:ポリマー全体(ポリエチレン樹脂組成物)の極限粘度(dl/g)
[η]A:超高分子量ポリエチレンの極限粘度(dl/g)
wtA:超高分子量ポリエチレンの含有量(質量%)
[η]B:低分子量ないし高分子量ポリエチレンの極限粘度(dl/g)
wtB:低分子量ないし高分子量ポリエチレンの含有量(質量%)
<変性ポリエチレン樹脂組成物(A-1)の製造>
上記で得たポリエチレン樹脂組成物(B-1)100質量部、無水マレイン酸0.8質量部、および有機過酸化物[日本油脂(株)製、商品名パーヘキシン-25B]0.07質量部、をヘキシェルミキサーで混合し、得られた混合物を270℃に設定した100mmφの二軸押出機で、混練時間1分30秒程で溶融グラフト変性することによって、変性ポリエチレン樹脂組成物(A-1)を得た。このグラフト変性ポリエチレン組成物の無水マレイン酸グラフト量をIR分析で測定したところ、0.8質量%であった。また、光学顕微鏡により明確な海島構造が観察された。その島相は1段目の重合体、海相は2段目の重合体と考えられる。その他の分析結果を表1に示す。
上記で得たポリエチレン樹脂組成物(B-1)100質量部、無水マレイン酸0.8質量部、および有機過酸化物[日本油脂(株)製、商品名パーヘキシン-25B]0.07質量部、をヘキシェルミキサーで混合し、得られた混合物を270℃に設定した100mmφの二軸押出機で、混練時間1分30秒程で溶融グラフト変性することによって、変性ポリエチレン樹脂組成物(A-1)を得た。このグラフト変性ポリエチレン組成物の無水マレイン酸グラフト量をIR分析で測定したところ、0.8質量%であった。また、光学顕微鏡により明確な海島構造が観察された。その島相は1段目の重合体、海相は2段目の重合体と考えられる。その他の分析結果を表1に示す。
[比較例1-1]
<ポリエチレン樹脂組成物(B-2)の重合>
実施例1の重合方法において、第1段目で得られる重合体の極限粘度が7dl/g、1段目と2段目の重合体量比が50/50となるように重合条件を調整して、極限粘度3.75dl/g、密度が966kg/m3の白色固体(ポリエチレン樹脂組成物(B-2))を得た。
<ポリエチレン樹脂組成物(B-2)の重合>
実施例1の重合方法において、第1段目で得られる重合体の極限粘度が7dl/g、1段目と2段目の重合体量比が50/50となるように重合条件を調整して、極限粘度3.75dl/g、密度が966kg/m3の白色固体(ポリエチレン樹脂組成物(B-2))を得た。
<変性ポリエチレン樹脂組成物(A-2)の製造>
上記で得たポリエチレン樹脂組成物(B-2)を、エチレン系重合体(A-1)の製造と同様にしてグラフト変性して変性ポリエチレン樹脂組成物(A-2)を得た。なお、この変性ポリエチレン樹脂組成物(A-2)では明確な海島構造は観察されなかった。すなわち、1段目と2段目の重合体が相溶していると考えられた。分析結果を表1に示す。
上記で得たポリエチレン樹脂組成物(B-2)を、エチレン系重合体(A-1)の製造と同様にしてグラフト変性して変性ポリエチレン樹脂組成物(A-2)を得た。なお、この変性ポリエチレン樹脂組成物(A-2)では明確な海島構造は観察されなかった。すなわち、1段目と2段目の重合体が相溶していると考えられた。分析結果を表1に示す。
[比較例1-2]
<変性ポリエチレン樹脂組成物(A-3)の製造>
特開平4-351647号公報の実施例と同様の変性ポリエチレン樹脂組成物を得た。すなわち、極限粘度[η]21dl/gの超高分子量ポリエチレン25質量%と、極限粘度1.5dl/gのポリエチレン75質量%からなるポリエチレン樹脂組成物をグラフト変性し、無水マレイン酸含有率(変性率)1質量%の変性ポリエチレン樹脂組成物(A-3)を得た。なお、グラフト変性はエチレン系重合体(A-1)の製造と同様にして行った。この変性ポリエチレン樹脂組成物(A-3)には海島構造が観察されたが、その島相のほとんどは長軸粒子径が100μm以上の粗大なものであった。したがって、上述のIRI等の測定(長軸粒子径が10μm以上30μm以下の島相が測定対象)は行わなかった。それ以外の分析結果を表1に示す。
<変性ポリエチレン樹脂組成物(A-3)の製造>
特開平4-351647号公報の実施例と同様の変性ポリエチレン樹脂組成物を得た。すなわち、極限粘度[η]21dl/gの超高分子量ポリエチレン25質量%と、極限粘度1.5dl/gのポリエチレン75質量%からなるポリエチレン樹脂組成物をグラフト変性し、無水マレイン酸含有率(変性率)1質量%の変性ポリエチレン樹脂組成物(A-3)を得た。なお、グラフト変性はエチレン系重合体(A-1)の製造と同様にして行った。この変性ポリエチレン樹脂組成物(A-3)には海島構造が観察されたが、その島相のほとんどは長軸粒子径が100μm以上の粗大なものであった。したがって、上述のIRI等の測定(長軸粒子径が10μm以上30μm以下の島相が測定対象)は行わなかった。それ以外の分析結果を表1に示す。
[実施例1~3、比較例1]
ポリアミド樹脂(E1)としてPA6(東レ(株)製、アミラン(登録商標)CM1007)と、変性ポリエチレン樹脂組成物(A-1)を表2に示す量使用して、43mmφのベント式二軸スクリュー押出機により200~240℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PA6単体での評価も行った。
ポリアミド樹脂(E1)としてPA6(東レ(株)製、アミラン(登録商標)CM1007)と、変性ポリエチレン樹脂組成物(A-1)を表2に示す量使用して、43mmφのベント式二軸スクリュー押出機により200~240℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PA6単体での評価も行った。
[比較例2]
ポリエチレン樹脂組成物(B-1)を43mmφのベント式二軸スクリュー押出機により200~240℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。
ポリエチレン樹脂組成物(B-1)を43mmφのベント式二軸スクリュー押出機により200~240℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。
[比較例3~5]
表2に示す組成比で同様の評価を行った。
表2に示す組成比で同様の評価を行った。
[実施例4~6、比較例6]
ポリアミド樹脂(E1)としてPA66(デュポン製、ザイテル(登録商標)101L)と、変性ポリエチレン樹脂組成物(A-1)を表2に示す量使用して、43mmφのベント式二軸スクリュー押出機により240~260℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PA66単体での評価も行った。
ポリアミド樹脂(E1)としてPA66(デュポン製、ザイテル(登録商標)101L)と、変性ポリエチレン樹脂組成物(A-1)を表2に示す量使用して、43mmφのベント式二軸スクリュー押出機により240~260℃のシリンダー温度条件で溶融混合してペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PA66単体での評価も行った。
[比較例7~9]
表2に示す組成比で同様の評価を行った。
表2に示す組成比で同様の評価を行った。
[実施例7~9、比較例10]
ポリアセタール樹脂(E2)としてポリオキシメチレン(POM)[ポリプラスチックス株式会社製、DURACON(登録商標)M90-44]と、変性ポリエチレン樹脂組成物(A-1)を表3に示す量使用して、43mmφのベント式二軸スクリュー押出機により180~200℃のシリンダー温度条件で溶融混合して樹脂組成物のペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、POM単体での評価も行った。
ポリアセタール樹脂(E2)としてポリオキシメチレン(POM)[ポリプラスチックス株式会社製、DURACON(登録商標)M90-44]と、変性ポリエチレン樹脂組成物(A-1)を表3に示す量使用して、43mmφのベント式二軸スクリュー押出機により180~200℃のシリンダー温度条件で溶融混合して樹脂組成物のペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、POM単体での評価も行った。
[比較例11~13]
表3に示す組成比で同様の評価を行った。
表3に示す組成比で同様の評価を行った。
[実施例10~12、比較例14]
ポリエステル樹脂(E3)としてポリブチレンテレフタラート(PBT)[ポリプラスチックス株式会社製、ジュラネックス(登録商標)500FP]と、変性ポリエチレン樹脂組成物(A-1)を表3に示す量使用して、43mmφのベント式二軸スクリュー押出機により230~250℃のシリンダー温度条件で溶融混合して樹脂組成物のペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PBT単体での評価も行った。
ポリエステル樹脂(E3)としてポリブチレンテレフタラート(PBT)[ポリプラスチックス株式会社製、ジュラネックス(登録商標)500FP]と、変性ポリエチレン樹脂組成物(A-1)を表3に示す量使用して、43mmφのベント式二軸スクリュー押出機により230~250℃のシリンダー温度条件で溶融混合して樹脂組成物のペレットを製造した。こうして得られたペレットを用いて射出成型試験片を作製し、上記した性能を評価した。また、PBT単体での評価も行った。
[比較例15~17]
表3に示す組成比で同様の評価を行った。
表3に示す組成比で同様の評価を行った。


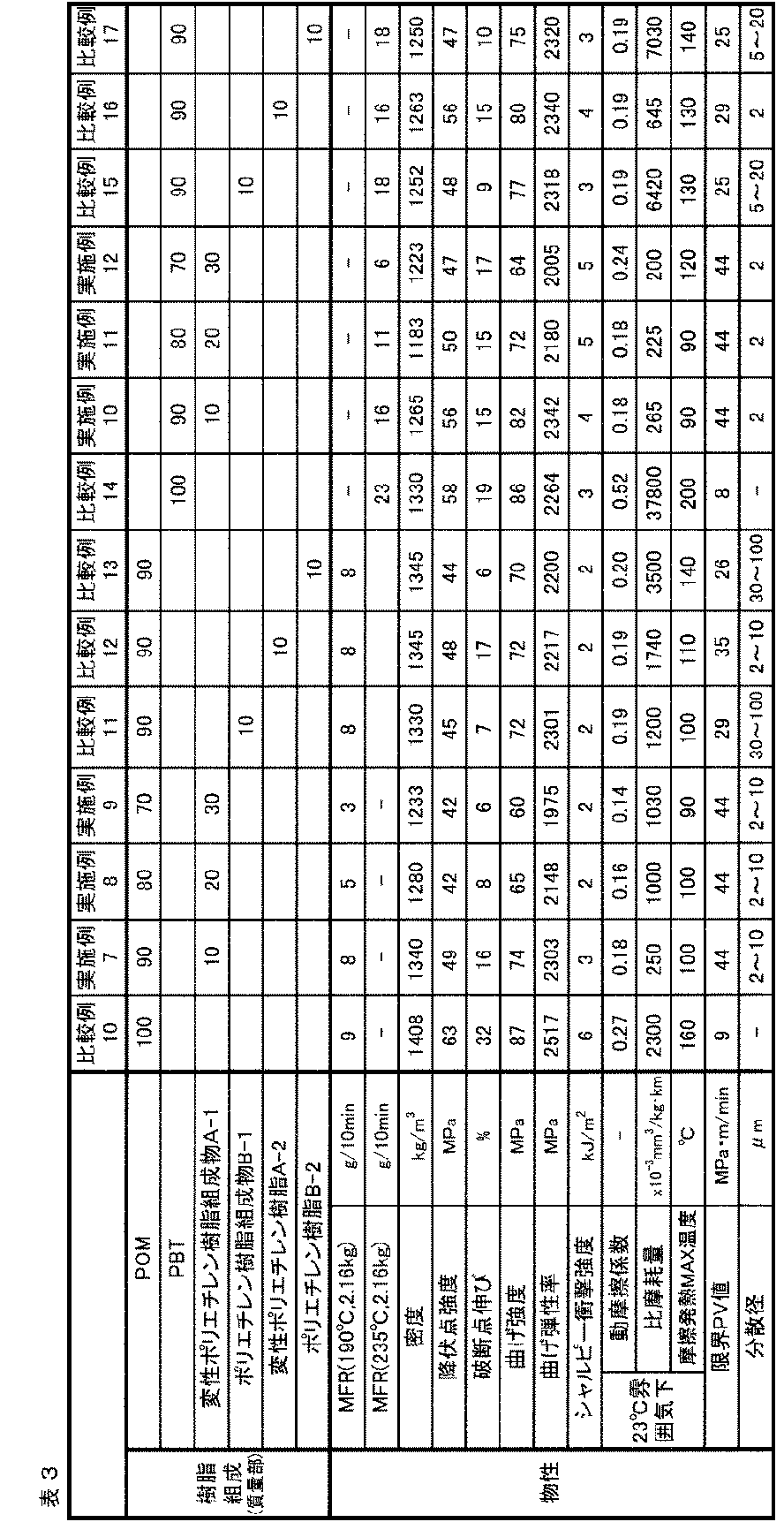
本発明の変性ポリエチレン樹脂組成物は、耐摩耗性、成形性などの特性のバランスに優れているので、これらが要求される用途として、例えば、鋼管、電線、自動車スライドドアレールなどの金属の被覆(積層)、耐圧ゴムホース、自動車ドア用ガスケット、クリーンルームドア用ガスケット、自動車グラスランチャンネル、自動車ウエザストリップなどの各種ゴムの被覆(積層)、ホッパー、シュートなどのライニング用、ギアー、軸受、ローラー、テープリール、各種ガイドレールやエレベーターレールガイド、各種保護ライナー材などの摺動材などに非常に有用である。
Claims (12)
- a) 海島構造を有し、光学顕微鏡での300μm角視野中に長軸粒子径100μm以上の島相が1個以下であり、
b) 300μm角視野中に長軸粒子径30μm以上、100μm未満の島相が3個以下であり、
c) 300μm角視野中に長軸粒子径10μm以上、30μm未満の島相が4個以上であり、
d) デカリン中135℃での極限粘度[η]が1.5~15dL/gの範囲にあり、
e) カルボキシル基、アミノ基、水酸基及びシラノール基からなる群より選ばれる官能基を含む構造単位を0.01~10質量%含み、
f) 海相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRS)と島相のIRにおける前記官能基ピークとCH2基ピークの強度比の比(IRI)との比(IRS/IRI)が1.0を超える変性ポリエチレン樹脂組成物。 - e)の官能基がカルボキシル基である請求項1記載の変性ポリエチレン樹脂組成物。
- e)の構造単位が無水マレイン酸由来の構造単位である請求項1に記載の変性ポリエチレン樹脂組成物。
- f)の比(IRS/IRI)が1.0を超えて5以下である請求項1に記載の変性ポリエチレン樹脂組成物。
- f)の比(IRS/IRI)が1.001以上3以下である請求項1に記載の変性ポリエチレン樹脂組成物。
- 下記の工程を含む方法で製造された、135℃のデカリン中で測定される極限粘度[η]が1.5~15dL/gの範囲にあるポリエチレン樹脂組成物を変性して得られる請求項1に記載の変性ポリエチレン樹脂組成物。
(I)極限粘度[η]が20~40dL/gの範囲にある超高分子量ポリエチレンを製造する工程
(II)極限粘度[η]が0.3~1.0dL/gの範囲にある低分子量ないし高分子量ポリエチレンを製造する工程 - 請求項1に記載の変性ポリエチレン樹脂組成物(A)と、他の樹脂(E)とを含んでなる樹脂組成物。
- ポリアミド樹脂(E1)50~95質量部と、請求項1に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリアミド樹脂(E1)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる請求項7に記載の樹脂組成物。
- ポリアセタール樹脂(E2)80~95質量部と、請求項1に記載の変性ポリエチレン樹脂組成物(A)5~20質量部(ポリアセタール樹脂(E2)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる請求項7に記載の樹脂組成物。
- ポリエステル樹脂(E3)50~95質量部と、請求項1に記載の変性ポリエチレン樹脂組成物(A)5~50質量部(ポリエステル樹脂(E3)と変性ポリエチレン樹脂組成物(A)の合計量を100質量部とする)を含んでなる請求項7に記載の樹脂組成物。
- 請求項7に記載の樹脂組成物をその一部または全部に使用してなる成形体。
- 摺動部材である、請求項11に記載の成形体。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015109267A JP2018119018A (ja) | 2015-05-29 | 2015-05-29 | 変性ポリエチレン樹脂組成物 |
| JP2015-109267 | 2015-05-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016194743A1 true WO2016194743A1 (ja) | 2016-12-08 |
Family
ID=57441473
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/065515 WO2016194743A1 (ja) | 2015-05-29 | 2016-05-25 | 変性ポリエチレン樹脂組成物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2018119018A (ja) |
| WO (1) | WO2016194743A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018193436A (ja) * | 2017-05-15 | 2018-12-06 | バンドー化学株式会社 | 樹脂部材 |
| JP2019143066A (ja) * | 2018-02-22 | 2019-08-29 | 三井化学株式会社 | 4−メチル−1−ペンテン系樹脂組成物およびその成形体 |
| CN113597447A (zh) * | 2019-03-29 | 2021-11-02 | 古河电气工业株式会社 | 绝缘性树脂组合物及其制造方法、绝缘带及其制造方法、绝缘层形成方法、以及电力电缆及其制造方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7261636B2 (ja) * | 2019-03-28 | 2023-04-20 | 三井化学株式会社 | 樹脂組成物および成形体 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63175069A (ja) * | 1987-01-14 | 1988-07-19 | Mitsui Petrochem Ind Ltd | 合成樹脂の低摩擦性改良用添加剤 |
| JPH04351647A (ja) * | 1991-05-28 | 1992-12-07 | Mitsui Petrochem Ind Ltd | 摺動特性改良剤 |
| JP2001059042A (ja) * | 1999-08-24 | 2001-03-06 | Mitsui Chemicals Inc | ポリオレフィン組成物 |
| JP2001279093A (ja) * | 2000-03-31 | 2001-10-10 | Mitsui Chemicals Inc | ポリアミド樹脂組成物 |
-
2015
- 2015-05-29 JP JP2015109267A patent/JP2018119018A/ja active Pending
-
2016
- 2016-05-25 WO PCT/JP2016/065515 patent/WO2016194743A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63175069A (ja) * | 1987-01-14 | 1988-07-19 | Mitsui Petrochem Ind Ltd | 合成樹脂の低摩擦性改良用添加剤 |
| JPH04351647A (ja) * | 1991-05-28 | 1992-12-07 | Mitsui Petrochem Ind Ltd | 摺動特性改良剤 |
| JP2001059042A (ja) * | 1999-08-24 | 2001-03-06 | Mitsui Chemicals Inc | ポリオレフィン組成物 |
| JP2001279093A (ja) * | 2000-03-31 | 2001-10-10 | Mitsui Chemicals Inc | ポリアミド樹脂組成物 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018193436A (ja) * | 2017-05-15 | 2018-12-06 | バンドー化学株式会社 | 樹脂部材 |
| JP2019143066A (ja) * | 2018-02-22 | 2019-08-29 | 三井化学株式会社 | 4−メチル−1−ペンテン系樹脂組成物およびその成形体 |
| JP7088689B2 (ja) | 2018-02-22 | 2022-06-21 | 三井化学株式会社 | 4-メチル-1-ペンテン系樹脂組成物およびその成形体 |
| CN113597447A (zh) * | 2019-03-29 | 2021-11-02 | 古河电气工业株式会社 | 绝缘性树脂组合物及其制造方法、绝缘带及其制造方法、绝缘层形成方法、以及电力电缆及其制造方法 |
| CN113597447B (zh) * | 2019-03-29 | 2024-03-05 | 古河电气工业株式会社 | 绝缘性树脂组合物及其制造方法、绝缘带及其制造方法、绝缘层形成方法、以及电力电缆及其制造方法 |
| US11990253B2 (en) | 2019-03-29 | 2024-05-21 | Furukawa Electric Co., Ltd. | Insulating resin composition and production method therefor, insulating tape and production method therefor, insulating layer formation method, and power cable and production method therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2018119018A (ja) | 2018-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100244882B1 (ko) | 폴리카보네이트/폴리올레핀계 수지 조성물,그의 제조방법 및 용도 | |
| CN101622299B (zh) | 对油的作用具有改善的耐受性的柔性热塑性组合物和该组合物的用途 | |
| JP4173444B2 (ja) | ポリエチレン樹脂組成物 | |
| WO2016194743A1 (ja) | 変性ポリエチレン樹脂組成物 | |
| US11680165B2 (en) | Polyamide resin composition with high fluidity | |
| US20120015202A1 (en) | Thermoplastic Elastomer Compositions, Articles Made Therefrom, and Methods for Making Such Articles | |
| JP5351108B2 (ja) | ポリエチレン樹脂組成物 | |
| EP1554337B2 (en) | Scratch and mar resistant soft ethylene elastomer compositions | |
| JP5584594B2 (ja) | ポリプロピレン系樹脂組成物 | |
| Bragaglia et al. | Compatibilization of an immiscible blend of EPDM and POM with an Ionomer | |
| JP2015117362A (ja) | 低分子量ポリオレフィンの製造法 | |
| JP6243729B2 (ja) | ポリプロピレン系樹脂組成物 | |
| JP6857529B2 (ja) | ポリプロピレン樹脂組成物および成形体 | |
| JP7088689B2 (ja) | 4-メチル-1-ペンテン系樹脂組成物およびその成形体 | |
| JP6523030B2 (ja) | ポリエチレン樹脂組成物 | |
| JP7456793B2 (ja) | 樹脂組成物および積層体 | |
| JP7398989B2 (ja) | 樹脂組成物および積層体の製造方法 | |
| JP7261636B2 (ja) | 樹脂組成物および成形体 | |
| JP7362342B2 (ja) | フィラー含有ポリプロピレン系樹脂組成物及び自動車外装用部品 | |
| JP2022098911A (ja) | オレフィン系重合体組成物およびその用途 | |
| CA2503153C (en) | Scratch and mar resistant soft ethylene elastomer compositions | |
| JP2014189710A (ja) | 樹脂組成物及びそれを含む成形体 | |
| JP2023041362A (ja) | 成形品および摺動部品 | |
| US20230250265A1 (en) | Polypropylene resin composition | |
| JP2018024752A (ja) | 樹脂組成物、及びこれから得られる成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16803178 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16803178 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |