WO2016098801A1 - 触媒の調製方法 - Google Patents
触媒の調製方法 Download PDFInfo
- Publication number
- WO2016098801A1 WO2016098801A1 PCT/JP2015/085175 JP2015085175W WO2016098801A1 WO 2016098801 A1 WO2016098801 A1 WO 2016098801A1 JP 2015085175 W JP2015085175 W JP 2015085175W WO 2016098801 A1 WO2016098801 A1 WO 2016098801A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- less
- preparing
- aqueous solution
- catalyst according
- Prior art date
Links
- 239000003054 catalyst Substances 0.000 title claims abstract description 178
- 238000000034 method Methods 0.000 title claims abstract description 45
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 70
- 239000007864 aqueous solution Substances 0.000 claims abstract description 51
- 239000006185 dispersion Substances 0.000 claims abstract description 31
- 238000004519 manufacturing process Methods 0.000 claims abstract description 21
- 238000002360 preparation method Methods 0.000 claims description 52
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 31
- 229910052797 bismuth Inorganic materials 0.000 claims description 25
- 150000005215 alkyl ethers Chemical class 0.000 claims description 24
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 20
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 20
- 239000001301 oxygen Substances 0.000 claims description 20
- 229910052760 oxygen Inorganic materials 0.000 claims description 20
- 229910052697 platinum Inorganic materials 0.000 claims description 20
- 239000002253 acid Substances 0.000 claims description 16
- 125000004432 carbon atom Chemical group C* 0.000 claims description 10
- 239000000203 mixture Substances 0.000 claims description 10
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims description 9
- 125000001931 aliphatic group Chemical group 0.000 claims description 9
- 150000002500 ions Chemical class 0.000 claims description 9
- 229910017604 nitric acid Inorganic materials 0.000 claims description 9
- FBXVOTBTGXARNA-UHFFFAOYSA-N bismuth;trinitrate;pentahydrate Chemical compound O.O.O.O.O.[Bi+3].[O-][N+]([O-])=O.[O-][N+]([O-])=O.[O-][N+]([O-])=O FBXVOTBTGXARNA-UHFFFAOYSA-N 0.000 claims description 7
- 125000005529 alkyleneoxy group Chemical group 0.000 claims description 6
- 238000002186 photoelectron spectrum Methods 0.000 claims description 5
- 229940036348 bismuth carbonate Drugs 0.000 claims description 3
- 229910000416 bismuth oxide Inorganic materials 0.000 claims description 3
- TYIXMATWDRGMPF-UHFFFAOYSA-N dibismuth;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Bi+3].[Bi+3] TYIXMATWDRGMPF-UHFFFAOYSA-N 0.000 claims description 3
- GMZOPRQQINFLPQ-UHFFFAOYSA-H dibismuth;tricarbonate Chemical compound [Bi+3].[Bi+3].[O-]C([O-])=O.[O-]C([O-])=O.[O-]C([O-])=O GMZOPRQQINFLPQ-UHFFFAOYSA-H 0.000 claims description 3
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 3
- 229940049676 bismuth hydroxide Drugs 0.000 claims 1
- 229940036359 bismuth oxide Drugs 0.000 claims 1
- TZSXPYWRDWEXHG-UHFFFAOYSA-K bismuth;trihydroxide Chemical compound [OH-].[OH-].[OH-].[Bi+3] TZSXPYWRDWEXHG-UHFFFAOYSA-K 0.000 claims 1
- 229910052751 metal Inorganic materials 0.000 abstract description 41
- 239000002184 metal Substances 0.000 abstract description 41
- 238000010828 elution Methods 0.000 abstract description 32
- 238000007254 oxidation reaction Methods 0.000 abstract description 18
- 239000007791 liquid phase Substances 0.000 abstract description 16
- 239000003513 alkali Substances 0.000 abstract description 7
- 229910052799 carbon Inorganic materials 0.000 abstract description 6
- 230000003647 oxidation Effects 0.000 abstract description 6
- 230000003197 catalytic effect Effects 0.000 abstract description 3
- 150000002739 metals Chemical class 0.000 abstract description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 75
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 60
- 239000007788 liquid Substances 0.000 description 48
- 239000007789 gas Substances 0.000 description 37
- -1 carboxyl compound Chemical class 0.000 description 29
- 238000006243 chemical reaction Methods 0.000 description 28
- 238000001914 filtration Methods 0.000 description 24
- 238000003756 stirring Methods 0.000 description 23
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 22
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 22
- 239000000243 solution Substances 0.000 description 22
- 238000005406 washing Methods 0.000 description 22
- 230000000694 effects Effects 0.000 description 19
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 17
- 230000000052 comparative effect Effects 0.000 description 16
- 239000001257 hydrogen Substances 0.000 description 13
- 229910052739 hydrogen Inorganic materials 0.000 description 13
- 230000009467 reduction Effects 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 12
- 238000001035 drying Methods 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 238000005259 measurement Methods 0.000 description 11
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 125000000217 alkyl group Chemical group 0.000 description 9
- 239000003638 chemical reducing agent Substances 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 238000006386 neutralization reaction Methods 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 230000009257 reactivity Effects 0.000 description 8
- 239000002994 raw material Substances 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- 238000004833 X-ray photoelectron spectroscopy Methods 0.000 description 6
- 239000012752 auxiliary agent Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 238000004817 gas chromatography Methods 0.000 description 5
- 239000000523 sample Substances 0.000 description 5
- 238000003828 vacuum filtration Methods 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 230000002776 aggregation Effects 0.000 description 4
- 125000003342 alkenyl group Chemical group 0.000 description 4
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 4
- 238000004140 cleaning Methods 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- 150000007524 organic acids Chemical class 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000012086 standard solution Substances 0.000 description 4
- 229910015902 Bi 2 O 3 Inorganic materials 0.000 description 3
- 239000004809 Teflon Substances 0.000 description 3
- 229920006362 Teflon® Polymers 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- QZPSXPBJTPJTSZ-UHFFFAOYSA-N aqua regia Chemical compound Cl.O[N+]([O-])=O QZPSXPBJTPJTSZ-UHFFFAOYSA-N 0.000 description 3
- 238000004364 calculation method Methods 0.000 description 3
- 150000002440 hydroxy compounds Chemical class 0.000 description 3
- 239000011261 inert gas Substances 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 230000001590 oxidative effect Effects 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 239000011148 porous material Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- OOCCDEMITAIZTP-QPJJXVBHSA-N (E)-cinnamyl alcohol Chemical compound OC\C=C\C1=CC=CC=C1 OOCCDEMITAIZTP-QPJJXVBHSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 2
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 235000011054 acetic acid Nutrition 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 150000001621 bismuth Chemical class 0.000 description 2
- IAQAJTTVJUUIQJ-UHFFFAOYSA-N bismuth;trihydrate Chemical compound O.O.O.[Bi] IAQAJTTVJUUIQJ-UHFFFAOYSA-N 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 239000012295 chemical reaction liquid Substances 0.000 description 2
- 235000015165 citric acid Nutrition 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 229910001882 dioxygen Inorganic materials 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 229910000510 noble metal Inorganic materials 0.000 description 2
- SJWFXCIHNDVPSH-UHFFFAOYSA-N octan-2-ol Chemical compound CCCCCCC(C)O SJWFXCIHNDVPSH-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 230000002572 peristaltic effect Effects 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 238000011085 pressure filtration Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000012488 sample solution Substances 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- WAPNOHKVXSQRPX-UHFFFAOYSA-N 1-phenylethanol Chemical compound CC(O)C1=CC=CC=C1 WAPNOHKVXSQRPX-UHFFFAOYSA-N 0.000 description 1
- PRWVJCOHFSWPFN-UHFFFAOYSA-N C(C)(=O)O.[N+](=O)([O-])[O-].[Bi+3].[N+](=O)([O-])[O-].[N+](=O)([O-])[O-] Chemical compound C(C)(=O)O.[N+](=O)([O-])[O-].[Bi+3].[N+](=O)([O-])[O-].[N+](=O)([O-])[O-] PRWVJCOHFSWPFN-UHFFFAOYSA-N 0.000 description 1
- 235000013162 Cocos nucifera Nutrition 0.000 description 1
- 244000060011 Cocos nucifera Species 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical group [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229910018879 Pt—Pd Inorganic materials 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- OOCCDEMITAIZTP-UHFFFAOYSA-N allylic benzylic alcohol Natural products OCC=CC1=CC=CC=C1 OOCCDEMITAIZTP-UHFFFAOYSA-N 0.000 description 1
- XPNGNIFUDRPBFJ-UHFFFAOYSA-N alpha-methylbenzylalcohol Natural products CC1=CC=CC=C1CO XPNGNIFUDRPBFJ-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 238000004380 ashing Methods 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- RXPAJWPEYBDXOG-UHFFFAOYSA-N hydron;methyl 4-methoxypyridine-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1=CC(OC)=CC=N1 RXPAJWPEYBDXOG-UHFFFAOYSA-N 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052745 lead Inorganic materials 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 239000010815 organic waste Substances 0.000 description 1
- 238000001139 pH measurement Methods 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 229910052573 porcelain Inorganic materials 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 229910021642 ultra pure water Inorganic materials 0.000 description 1
- 239000012498 ultrapure water Substances 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/04—Mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/18—Carbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/644—Arsenic, antimony or bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/644—Arsenic, antimony or bismuth
- B01J23/6447—Bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/20—Catalysts, in general, characterised by their form or physical properties characterised by their non-solid state
- B01J35/23—Catalysts, in general, characterised by their form or physical properties characterised by their non-solid state in a colloidal state
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J35/00—Catalysts, in general, characterised by their form or physical properties
- B01J35/30—Catalysts, in general, characterised by their form or physical properties characterised by their physical properties
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/009—Preparation by separation, e.g. by filtration, decantation, screening
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/02—Impregnation, coating or precipitation
- B01J37/03—Precipitation; Co-precipitation
- B01J37/031—Precipitation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/16—Reducing
- B01J37/18—Reducing with gases containing free hydrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/23—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of oxygen-containing groups to carboxyl groups
- C07C51/235—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of oxygen-containing groups to carboxyl groups of —CHO groups or primary alcohol groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C59/00—Compounds having carboxyl groups bound to acyclic carbon atoms and containing any of the groups OH, O—metal, —CHO, keto, ether, groups, groups, or groups
- C07C59/125—Saturated compounds having only one carboxyl group and containing ether groups, groups, groups, or groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B61/00—Other general methods
Definitions
- the present invention relates to a method for preparing a catalyst. More specifically, the present invention relates to a method for preparing an oxidation reaction catalyst that can be suitably used for producing a carboxyl compound or a ketone compound by oxidizing a hydroxy compound such as alcohol or polyoxyalkylene alkyl ether or an aldehyde compound.
- Pt which is a noble metal catalyst
- Bi a hydroxy compound or an aldehyde compound
- a hydroxy compound or an aldehyde compound is catalytically oxidized and converted into a corresponding carboxyl compound or ketone compound.
- a catalyst in which Bi is supported by dropping a nitric acid aqueous solution of bismuth nitrate into a dispersion of a commercially available Pt / C catalyst is prepared, and this is used for 2-substitution.
- JP-A-62-228093 describes that when Bi is supported on Pd / C, the activity is higher than when Pd is supported on Bi / C.
- it does not provide knowledge that can sufficiently cope with the above-mentioned problems, and in the same manner as in JP-A-11-279110, it is necessary to use a catalyst and an alkali together in the reaction.
- An object of the present invention is to provide a method for preparing a highly durable catalyst having high activity and suppressing elution of a catalyst metal when performing a liquid phase oxidation reaction without using an alkali.
- the inventor of the present invention has completed the present invention as a result of continuing research on a method for preparing a Pt—Bi / C catalyst in order to solve the problems of the Pt—Bi / C catalyst in the liquid phase oxidation reaction as described above.
- the present invention is a method for preparing a catalyst having the following step 1, step 2 and step 3.
- Step 1 A step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon
- Step 2 A step of preparing an aqueous solution containing Bi in an ionic state
- Step 3 An aqueous solution obtained in Step 2 is obtained in Step 1.
- Step 1 A step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon
- Step 2 A step of preparing an aqueous solution containing Bi in an ionic state
- Step 3 An aqueous solution obtained in Step 2 is obtained in Step 1.
- Step 1 A step of preparing an aqueous disper
- a highly durable catalyst with high activity and suppressed elution of catalytic metal can be obtained.
- the method of manufacturing an oxide with high efficiency using such a catalyst is provided.
- FIG. 1 is a photoelectron spectrum of Bi4f orbit
- spectrum 1 shows the result of measuring the catalyst prepared in Example 1 in Example 7
- spectrum 2 is measured in Comparative Example 3 of the catalyst prepared in Comparative Example 1. Results are shown.
- the present invention is a method for preparing a catalyst having the following step 1, step 2 and step 3.
- Step 1 A step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon
- Step 2 A step of preparing an aqueous solution containing Bi in an ionic state
- Step 3 An aqueous solution obtained in Step 2 is obtained in Step 1.
- Step 1 A step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon
- Step 2 A step of preparing an aqueous solution containing Bi in an ionic state
- Step 3 An aqueous solution obtained in Step 2 is obtained in Step 1.
- Step 1 A step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon
- Step 3 A step of preparing an aqueous solution containing Bi in an ionic state
- Step 3 An aqueous solution obtained in Step 2 is obtained in Step 1.
- Bi ions can be brought into contact with Pt more evenly. Therefore, since the Pt—Bi complex can be formed efficiently, the activity is improved, and metal elution during the liquid phase oxidation reaction can be suppressed. However, this action is an estimate and does not limit the scope of the present invention.
- Step 1 is a step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon (hereinafter also referred to as a Pt / C catalyst).
- the activated carbon is not particularly limited, and any type of activated carbon can be used as long as it can adsorb and carry Pt.
- Examples of the activated carbon include vegetable activated carbon such as coconut shell activated carbon, mineral activated carbon such as coal-based activated carbon, pulp waste liquid, synthetic resin, and activated carbon of organic waste. There are no particular restrictions on the activation method, pore distribution, shape, and the like.
- the particle size of Pt supported on the activated carbon is not particularly limited, but is preferably 20 nm or less, more preferably 15 nm or less, and further preferably 10 nm or less, from the viewpoint of enhancing dispersibility and reaction activity. It may be above.
- the supported amount of Pt metal in the catalyst solid content is preferably 0.1% or more, more preferably 1% or more, still more preferably 3% or more, and even more preferably 5% or more from the viewpoint of reactivity. From the viewpoint of enhancing the properties, it is preferably 20% or less, more preferably 15% or less, and still more preferably 10% or less.
- the Pt / C catalyst can be prepared by a known impregnation method or precipitation precipitation method, but a commercially available product can also be used.
- the aqueous dispersion is prepared by adding a Pt / C catalyst to ion-exchanged water, distilled water, pure water, and the like, and stirring.
- the concentration of the Pt / C catalyst in the dispersion is preferably 4% or more, more preferably 5% or more, still more preferably 6% or more, and even more preferably 7% or more, from the viewpoint of efficiency in the subsequent step. From the viewpoint of operability, it is preferably 12% or less, more preferably 9% or less, and still more preferably 8% or less.
- the preparation temperature is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, more preferably 20 ° C. or higher from the viewpoint of economy, and preferably 60 ° C. or lower, from the viewpoint of suppressing Pt denaturation and aggregation. Is 50 ° C. or lower, more preferably 40 ° C. or lower, still more preferably 30 ° C. or lower, and still more preferably 25 ° C. or lower.
- Step 2 is a step of preparing an aqueous solution containing Bi in an ionic state (hereinafter also referred to as Bi aqueous solution).
- the ion source of Bi ions needs to be Bi species in which Bi salt can be dissolved in water or acidic aqueous solution.
- Bi species in which Bi salt can be dissolved in water or acidic aqueous solution.
- bismuth nitrate pentahydrate Bi (NO 3 ) 3 .5H 2 O is preferable.
- Bismuth oxide Bi 2 O 3
- bismuth carbonate ((BiO) 2 CO 3 )
- bismuth hydroxide (Bi (OH) 3 ) more preferably bismuth nitrate pentahydrate It is.
- the Bi aqueous solution preferably contains an acid from the viewpoint of dissolving the Bi salt.
- the acid used may be either an inorganic acid or an organic acid.
- the organic acid is preferably an acid having a carboxyl group, more preferably one or more selected from acetic acid, formic acid, citric acid and oxalic acid from the viewpoint of suppressing metal elution from the catalyst prepared during the liquid phase oxidation reaction, Acetic acid is preferred.
- the inorganic acid is preferably at least one selected from nitric acid, hydrochloric acid, phosphoric acid and sulfuric acid, more preferably nitric acid, from the viewpoint of suppressing metal elution from the catalyst prepared during the liquid phase oxidation reaction.
- the amount of the acid to be used is preferably 1% or more, more preferably 1.5% or more in the Bi aqueous solution from the viewpoint of dissolving the Bi salt, and preferably 5% or less, more preferably from the economical viewpoint. 4% or less, more preferably 3% or less.
- the amount of Bi in the Bi aqueous solution is preferably 0.0001M or more, more preferably 0.0005M or more, and further preferably 0.001M or more. To preferably 0.1M or less, more preferably 0.05M or less, still more preferably 0.03M or less, still more preferably 0.02M or less, and still more preferably 0.01M or less.
- the pH of the Bi aqueous solution is preferably 3.8 or less, more preferably 3.6 or less, even more preferably 3.4 or less, preferably 1.0 or more, from the viewpoint of suppressing metal elution from the prepared catalyst. More preferably, it is 1.5 or more, More preferably, it is 2.0 or more, More preferably, it is 2.5 or more.
- the temperature for preparing the Bi aqueous solution is not particularly limited, but is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, further preferably 20 ° C. or higher, preferably 60 ° C. or lower, from the viewpoint of economy. More preferably, it is 50 ° C. or less, more preferably 40 ° C. or less, still more preferably 30 ° C. or less, and still more preferably 25 ° C. or less.
- the Bi aqueous solution obtained in step 1 can be treated with a reducing agent or reducing gas that can be used to reduce Pt in the next step 3.
- the treatment with the reducing agent can be performed by adding formalin, sodium borohydride, or the like to the obtained aqueous dispersion or Bi aqueous solution.
- the treatment with the reducing gas can be performed by circulating a reducing gas such as hydrogen or carbon monoxide through the aqueous dispersion or the Bi aqueous solution to form a reducing atmosphere.
- a reducing gas such as hydrogen or carbon monoxide
- the treatment temperature with the reducing agent or reducing gas is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, and still more preferably 20 ° C. or higher, from the viewpoint of economy. From the viewpoint of suppressing Pt denaturation and aggregation, Preferably it is 60 degrees C or less, More preferably, it is 50 degrees C or less, More preferably, it is 40 degrees C or less, More preferably, it is 30 degrees C or less, More preferably, it is 25 degrees C or less.
- the supply amount of the reducing gas to be circulated is an amount that is largely excessive with respect to the catalyst from the viewpoint of sufficiently performing the treatment with the reducing gas, preferably It is 60 mL / min or more, More preferably, it is 80 mL / min or more, From a viewpoint of economical efficiency, Preferably it is 500 mL / min or less, More preferably, it is 300 mL / min or less, More preferably, it is 150 mL / min or less.
- the treatment time with the reducing agent or reducing gas is preferably 10 minutes or more, more preferably 15 minutes or more, and even more preferably 20 minutes or more from the viewpoint of sufficiently performing the treatment with the reducing agent or the reducing gas. From the viewpoint, it is preferably 2 hours or less, more preferably 1 hour or less, and further preferably 30 minutes or less.
- Step 3 is a step of adding the aqueous dispersion obtained in Step 1 to the Bi aqueous solution obtained in Step 2.
- a Pt—Bi / C catalyst complex is efficiently formed by adding an aqueous dispersion of a Pt / C catalyst to an aqueous Bi solution.
- the addition may be any of continuous addition, divided addition, and batch addition, but continuous addition or divided addition is preferred, and continuous divided addition such as dropwise addition is more preferred.
- the addition rate is preferably 1 mL / min or more, more preferably 3 mL / min or more as an aqueous dispersion of a Pt / C catalyst.
- the aqueous dispersion of the Pt / C catalyst is preferably 10 mL / min or less, more preferably 5 mL / min or less. Is speed.
- the time required for the addition is not particularly limited, but is preferably 15 minutes or more, more preferably 30 minutes or more, and still more preferably from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst. Is 1 hour or more, more preferably 2 hours or more, and from the viewpoint of productivity, it is preferably 10 hours or less, more preferably 7 hours or less, still more preferably 5 hours or less, and even more preferably 3 hours or less.
- the temperature of the Bi aqueous solution that is added or dripped in Step 3 is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, and more preferably, from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst. Is 20 ° C. or higher, and preferably 60 ° C. or lower, more preferably 50 ° C. or lower, still more preferably 40 ° C. or lower, even more preferably 30 ° C. or lower, and still more preferably, from the viewpoint of suppressing Pt modification and aggregation. It is 25 degrees C or less.
- reduction treatment is preferably performed in step 3.
- the reduction treatment in step 3 can be performed on either or both of the liquid to be added or dropped and the liquid to be added or dropped. From the viewpoint of operability, the reduction treatment is preferably performed on the liquid to be added or dropped.
- the reduction treatment in step 3 is preferably performed in a reducing atmosphere from the viewpoint of suppressing metal elution from the prepared catalyst.
- the reduction treatment in step 3 is performed by flowing a reducing gas such as hydrogen or carbon monoxide, more preferably hydrogen gas.
- the preferable ranges of the reducing gas supply amount when the reducing treatment temperature and the reducing gas are circulated are used for the reducing gas and the reducing gas or the treating temperature by the reducing gas prior to Step 3, respectively. This is the same as the preferable range of the supply amount.
- the reduction treatment in step 3 is preferably performed until the addition of the aqueous dispersion of Pt / C catalyst is completed.
- step 4 a liquid containing the catalyst obtained in step 3 and a reducing aid are mixed, and Pt and Bi are supported on activated carbon (that is, the above Pt—Bi / C catalyst). And a step of obtaining a liquid containing a reducing aid. This step is preferable from the viewpoint of suppressing metal elution from the prepared catalyst.
- the reducing auxiliary agent maintains the reduced state of the catalyst in which Pt and Bi are supported on activated carbon, and a reducing organic solvent such as isopropyl alcohol (IPA) can be used.
- IPA isopropyl alcohol
- the amount of the reducing aid used is preferably 0.1 parts by mass or more, more preferably 0.1 parts by mass with respect to 100 parts by mass of the liquid containing the catalyst obtained in Step 3. 5 mass parts or more, More preferably, it is 1.0 mass part or more, From a viewpoint of economical efficiency, Preferably it is 10 mass parts or less, More preferably, it is 5 mass parts or less, More preferably, it is 2 mass parts or less.
- the method for preparing a catalyst of the present invention can include, as step 5, a step of washing a catalyst in which Pt and Bi in the solution obtained in step 4 are supported on activated carbon with a washing solution. This step is preferred from the viewpoint of the activity of the prepared catalyst and the suppression of metal elution from the prepared catalyst.
- the cleaning liquid used in step 5 is preferably a liquid containing at least one selected from water and the above-mentioned reducing aid.
- it is a liquid containing one or more of the above-mentioned reducing aids and water, more preferably an IPA aqueous solution.
- step 4 it is preferable to filter the liquid obtained in step 4 before washing and filter the catalyst in which Pt and Bi are supported on activated carbon. Filtration can be performed by either vacuum filtration or pressure filtration.
- the method for preparing a catalyst of the present invention can include, as step 6, a step of drying the catalyst having Pt and Bi washed in step 5 supported on activated carbon. This step is preferable from the viewpoint of handling.
- Drying is performed under the flow of an inert gas such as nitrogen gas.
- the catalyst preparation method of the present invention it is preferable not to perform neutralization after Step 3 from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst.
- the pH of the liquid containing the catalyst obtained in step 3 is preferably 3.8 or less, more preferably 3.6, from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst.
- it is further preferably 3.4 or less, preferably 1.0 or more, more preferably 1.5 or more, further preferably 2.0 or more, and still more preferably 2.5 or more.
- the pH of the filtrate obtained by filtering the liquid obtained in Step 4 is preferably 3.8 from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst. Or less, more preferably 3.6 or less, still more preferably 3.4 or less, preferably 1.0 or more, more preferably 1.5 or more, still more preferably 2.0 or more, and even more preferably 2.5. That's it.
- the pH of the filtrate after washing is preferably 3.8 or less, more preferably from the viewpoint of improving the activity of the prepared catalyst and suppressing metal elution from the prepared catalyst. Is 3.6 or less, more preferably 3.4 or less, preferably 1.0 or more, more preferably 1.5 or more, still more preferably 2.0 or more, and even more preferably 2.5 or more.
- the amount of Bi supported in the solid content of the Pt—Bi / C catalyst is preferably 0.01% or more, more preferably 0.5% or more, preferably from the viewpoint of improving the productivity of the product of the oxidation reaction. It is 10% or less, more preferably 5% or less, further preferably 3.5% or less, and still more preferably 1.5% or less.
- the mass ratio (atomic ratio) Bi / Pt of Bi to Pt in the Pt—Bi / C catalyst is preferably 0.05 or more, more preferably 0.1 or more, from the viewpoint of improving the productivity of the product of the oxidation reaction. Yes, preferably 1.0 or less, more preferably 0.6 or less, still more preferably 0.3 or less, and still more preferably 0.2 or less.
- the catalyst in which Pt and Bi of the present invention are supported on activated carbon has a binding energy in the Bi4f orbital photoelectron spectrum measured by XPS.
- the binding energy value of the peak top in the range of 162 to 155 eV is preferably 158.5 eV or less, more preferably 158.2 eV or less, still more preferably 158.0 eV or less, preferably 157.0 eV or more, more preferably 157 .2 eV or more, more preferably 157.5 eV or more.
- the catalyst of the present invention is suitably used for a reaction for producing a carboxyl compound or a ketone compound by oxidizing a hydroxy compound or an aldehyde compound, for example, for a reaction for producing a corresponding carboxyl compound by liquid phase oxidation of a polyoxyalkylene alkyl ether.
- the present invention provides an oxide of alcohol that deoxygenates the alcohol or polyoxyalkylene alkyl ether by supplying oxygen to the composition containing the alcohol or polyoxyalkylene alkyl ether and water in the presence of the catalyst.
- the present invention also relates to a method for producing an oxide of a polyoxyalkylene alkyl ether (hereinafter also referred to as the oxide).
- a step of obtaining the catalyst a step of supplying oxygen to a composition containing an alcohol or polyoxyalkylene alkyl ether and water in the presence of the catalyst, and dehydrogenating and oxidizing the alcohol or polyoxyalkylene alkyl ether. It has a manufacturing method of the oxide concerned.
- the alcohol or polyoxyalkylene alkyl ether is one or more kinds represented by the following general formula (1) or general formula (2).
- R 1 O—H (1)
- R 1 is an aliphatic hydrocarbon group having 2 to 40 carbon atoms.
- R 2 represents a hydrocarbon group having 2 to 40 carbon atoms
- AO represents an alkyleneoxy group having 2 to 4 carbon atoms
- n represents the number of added moles of the alkyleneoxy group, It is an integer of 30 or less.
- R 1 is preferably a linear or branched primary or secondary aliphatic hydrocarbon group, more preferably a linear or branched primary or secondary alkyl group, or An alkenyl group, more preferably a linear primary or secondary alkyl group.
- the carbon number of R 1 is not particularly limited, but may be 6 or more, 8 or more, 10 or more, or 12 or more, and is preferably 36 or less, more preferably 22 or less, from the viewpoint of reactivity. More preferably, it is 18 or less, More preferably, it is 14 or less.
- R 2 is preferably an aliphatic hydrocarbon group, more preferably a linear or branched primary or secondary aliphatic hydrocarbon group, still more preferably a linear or branched chain, A primary or secondary alkyl group or alkenyl group, more preferably a linear primary or secondary alkyl group.
- the carbon number of R 2 is not particularly limited, but may be 6 or more, 8 or more, 10 or more, or 12 or more, and is preferably 36 or less, more preferably 22 or less, from the viewpoint of reactivity. More preferably, it is 18 or less, More preferably, it is 14 or less.
- the mass ratio of the alcohol or polyoxyalkylene alkyl ether used as the raw material to the water used for the reaction is the oxidation of the alcohol or polyoxyalkylene alkyl ether. From the viewpoint of improving the productivity of the product and suppressing the increase in viscosity of the liquid phase, it is preferably 60/40 or more, more preferably 70/30 or more, still more preferably 75/25 or more, preferably 95/5 or less, more preferably Is 90/10 or less, more preferably 85/15 or less.
- the supply of oxygen to the liquid phase which is the composition or the reaction liquid thereof, can be performed by circulating an oxygen-containing gas in the liquid phase.
- oxygen-containing gas include mixed gases containing oxygen such as oxygen gas and air.
- the gas used in combination with oxygen is preferably an inert gas such as helium, argon, or nitrogen from the viewpoint of not affecting the activity.
- the oxygen concentration in the oxygen-containing gas is preferably 10% by volume or more, more preferably 50% by volume or more, still more preferably 70% by volume or more, and still more preferably 90% by volume or more. , Still more preferably substantially 100% by volume, even more preferably 100% by volume.
- Such production can be carried out in a continuous, batch or semi-batch manner.
- the reaction temperature is preferably 50 ° C. or higher, more preferably 60 ° C. or higher from the viewpoint of reactivity, and preferably 100 ° C. or lower, more preferably 90 ° C. or lower, more preferably 80 ° C. or lower, from the viewpoint of equipment load. It is.
- the reaction may be performed under normal pressure or under pressure.
- the reaction pressure is an absolute pressure from the viewpoint of reactivity, preferably 0.09 MPa or more, more preferably 0.10 MPa or more, and from the viewpoint of equipment load, preferably 0.5 MPa or less, more preferably 0.2 MPa. Hereinafter, it is more preferably 0.11 MPa or less.
- the amount of the catalyst used can be arbitrarily selected within the range where a practical reaction rate can be obtained according to the reaction temperature or reaction pressure, but when the reaction is carried out batchwise, from the viewpoint of reactivity, polyoxyalkylene alkyl ether
- the mass of the Pt metal with respect to 100 parts by mass is preferably 0.1 parts by mass or more, more preferably 1 part by mass or more, further preferably 3 parts by mass or more, and still more preferably 6 parts by mass or more. Therefore, it is preferably 20 parts by mass or less, more preferably 15 parts by mass or less, and still more preferably 10 parts by mass or less.
- Step 1 Step of preparing an aqueous dispersion of a catalyst in which Pt is supported on activated carbon (Pt / C catalyst)
- Step 2 Step of preparing an aqueous solution (Bi aqueous solution) containing Bi in an ionic state
- Step 3 In Step 2 The step of adding the aqueous dispersion obtained in step 1 to the obtained aqueous solution
- ⁇ 2> The preparation method according to ⁇ 1>, wherein the particle diameter of Pt is preferably 20 nm or less, more preferably 15 nm or less, still more preferably 10 nm or less, and may be 1 nm or more.
- the supported amount of Pt metal in the catalyst solid content is preferably 0.1% or more, more preferably 1% or more, still more preferably 3% or more, still more preferably 5% or more, preferably 20%.
- the preparation method according to ⁇ 1> or ⁇ 2> which is more preferably 15% or less, and still more preferably 10% or less.
- the concentration of the Pt / C catalyst in the aqueous dispersion is preferably 4% or more, more preferably 5% or more, still more preferably 6% or more, still more preferably 7% or more, preferably 12% or less.
- the preparation temperature in step 1 is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, further preferably 20 ° C. or higher, preferably 60 ° C. or lower, more preferably 50 ° C. or lower, more preferably 40 ° C. or lower. More preferably, the preparation method according to any one of ⁇ 1> to ⁇ 4>, which is 30 ° C. or less, more preferably 25 ° C. or less.
- An ion source of Bi ions is preferably bismuth nitrate pentahydrate (Bi (NO 3 ) 3 .5H 2 O), bismuth oxide (Bi 2 O 3 ), bismuth carbonate ((BiO) 2 CO 3 ). And bismuth hydroxide (Bi (OH) 3 ), more preferably bismuth nitrate pentahydrate, the preparation method according to any one of ⁇ 1> to ⁇ 5>.
- ⁇ 7> The preparation method according to any one of ⁇ 1> to ⁇ 6>, wherein the aqueous solution obtained in step 2 contains an acid.
- the acid is an inorganic acid or an organic acid, preferably an organic acid, more preferably an acid having a carboxyl group, more preferably one or more selected from acetic acid, formic acid, citric acid and oxalic acid,
- the acid is an inorganic acid, preferably one or more selected from nitric acid, hydrochloric acid, phosphoric acid, and sulfuric acid, and more preferably nitric acid.
- the amount of acid used in the Bi aqueous solution is preferably 1% or more, more preferably 1.5% or more, preferably 5% or less, more preferably 4% or less, and even more preferably 3% or less.
- the Bi content in the Bi aqueous solution is preferably 0.0001M or more, more preferably 0.0005M or more, still more preferably 0.001M or more, preferably 0.1M or less, more preferably 0.05M.
- the pH of the ⁇ 13> Bi aqueous solution is preferably 3.8 or less, more preferably 3.6 or less, still more preferably 3.4 or less, preferably 1.0 or more, more preferably 1.5 or more, and still more preferably
- the temperature at which the ⁇ 14> Bi aqueous solution is prepared is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, further preferably 20 ° C. or higher, preferably 60 ° C. or lower, more preferably 50 ° C. or lower, and still more preferably 40
- ⁇ 15> Either or both of the aqueous dispersion obtained in Step 1 and the Bi aqueous solution obtained in Step 2, preferably the liquid which is added or dropped in Step 3, ie, the Bi aqueous solution obtained in Step 2, is a step.
- the treatment temperature with a reducing agent or reducing gas is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, still more preferably 20 ° C. or higher, preferably 60 ° C. or lower, more preferably 50 ° C. or lower, still more preferably. Is 40 ° C. or lower, more preferably 30 ° C. or lower, and still more preferably 25 ° C. or lower.
- the treatment time with the reducing agent or reducing gas is preferably 10 minutes or more, more preferably 15 minutes or more, still more preferably 20 minutes or more, preferably 2 hours or less, more preferably 1 hour or less, even more preferably. Is 30 minutes or less, The preparation method as described in ⁇ 15> or ⁇ 16>.
- ⁇ 18> The preparation method according to any one of ⁇ 15> to ⁇ 17>, wherein the treatment with the reducing agent or the reducing gas is performed by flowing a reducing gas, preferably hydrogen gas.
- the supply amount of the reducing gas is preferably 60 mL / min or more, more preferably 80 mL / min or more, further preferably 120 mL / min or more, preferably 500 mL / min or less, more preferably 300 mL / min or less.
- the addition in step 3 is continuous addition, divided addition, batch addition, or continuous divided addition, preferably continuous addition, divided addition, or continuous divided addition, and more preferably dropwise.
- the addition rate is preferably 1 mL / min or more, more preferably 3 mL / min or more, preferably 10 mL / min or less, more preferably 5 mL / min or less as an aqueous dispersion of the Pt / C catalyst.
- the time required for ⁇ 22> addition is preferably 15 minutes or more, more preferably 30 minutes or more, further preferably 1 hour or more, still more preferably 2 hours or more, preferably 10 hours or less, more preferably 7 hours.
- the temperature of the Bi aqueous solution subjected to addition in Step 3 is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, further preferably 20 ° C. or higher, preferably 60 ° C. or lower, more preferably 50 ° C. or lower.
- the reduction treatment is preferably performed in the step 3, and the reduction treatment in the step 3 can be performed on either or both of the liquid to be added or dropped and the liquid to be added or dropped, preferably added or dropped.
- the reduction treatment temperature in ⁇ 26> step 3 is preferably 10 ° C. or higher, more preferably 15 ° C. or higher, further preferably 20 ° C. or higher, preferably 60 ° C. or lower, more preferably 50 ° C. or lower, still more preferably 40 ° C.
- the preparation method according to ⁇ 24> or ⁇ 25> more preferably 30 ° C. or less, and further preferably 25 ° C. or less.
- ⁇ 27> The preparation method according to any one of ⁇ 24> to ⁇ 26>, wherein the reduction treatment in step 3 is performed by flowing a reducing gas, preferably hydrogen gas.
- the supply amount of the reducing gas in step 3 is preferably 60 mL / min or more, more preferably 80 mL / min or more, still more preferably 120 mL / min or more, preferably 500 mL / min or less, more preferably 300 mL. / Min or less, more preferably 150 mL / min or less, The preparation method as described in ⁇ 27>.
- the pH of the liquid containing the catalyst obtained in step 3 is preferably 3.8 or less, more preferably 3.6 or less, still more preferably 3.4 or less, and preferably 1.0 or more, more preferably
- ⁇ 31> A step of obtaining a liquid containing a catalyst (Pt-Bi / C catalyst) in which Pt and Bi are supported on activated carbon and a reducing auxiliary agent by mixing the liquid containing the catalyst obtained in step 3 and a reducing auxiliary agent.
- the reduction auxiliary agent is preferably an organic solvent having reducibility, and more preferably isopropyl alcohol (IPA).
- the amount of ⁇ 33> reducing aid used is preferably 0.1 parts by mass or more, more preferably 0.5 parts by mass or more, and further preferably 1 with respect to 100 parts by mass of the liquid containing the catalyst obtained in Step 3.
- the preparation method according to ⁇ 31> or ⁇ 32> which is 0.0 part by mass or more, preferably 10 parts by mass or less, more preferably 5 parts by mass or less, and further preferably 2 parts by mass or less.
- the pH of the filtrate obtained by filtering the liquid obtained in step 4 is preferably 3.8 or less, more preferably 3.6 or less, still more preferably 3.4 or less, and preferably 1.0 or more, more preferably
- ⁇ 35> The preparation method according to any one of ⁇ 31> to ⁇ 34>, further comprising a step 5 of washing the catalyst obtained by supporting Pt and Bi in the liquid obtained in the step 4 on activated carbon with a washing solution.
- the cleaning liquid is preferably a liquid containing one or more selected from water and a reducing auxiliary agent, more preferably a liquid containing one or more reducing auxiliary agents and water, and still more preferably an IPA aqueous solution ⁇ 35>.
- the pH of the filtrate after washing in step 5 is preferably 3.8 or less, more preferably 3.6 or less, still more preferably 3.4 or less, preferably 1.0 or more, more preferably 1.
- the supported amount of Bi in the solid content of the Pt—Bi / C catalyst is preferably 0.01% or more, more preferably 0.5% or more, preferably 10% or less, more preferably 5% or less, The catalyst according to ⁇ 41>, further preferably 3.5% or less, and still more preferably 1.5% or less.
- the mass ratio (atomic ratio) Bi / Pt of Bi to Pt in the Pt—Bi / C catalyst is preferably 0.05 or more, more preferably 0.1 or more, preferably 1.0 or less, more preferably The catalyst according to ⁇ 41> or ⁇ 42>, which is 0.6 or less, more preferably 0.3 or less, and still more preferably 0.2 or less.
- the peak top binding energy value in the range of binding energy 162 to 155 eV is preferably 158.5 eV or less, more preferably 158.2 eV or less, and further preferably 158.0 eV.
- a catalyst in which Pt and Bi are supported on activated carbon which is below, preferably 157.0 eV or more, more preferably 157.2 eV or more, and further preferably 157.5 eV or more.
- oxygen is supplied to the composition containing an alcohol or a polyoxyalkylene alkyl ether and water, and the alcohol or the polyoxyalkylene alkyl is supplied.
- R 1 O—H (1)
- R 1 is an aliphatic hydrocarbon group having 2 to 40 carbon atoms.
- R 2 O— (AO) n —H (2)
- R represents a hydrocarbon group having 2 to 40 carbon atoms
- AO represents an alkyleneoxy group having 2 to 4 carbon atoms
- n represents the number of added moles of the alkyleneoxy group, and 1 or more. It is an integer of 30 or less.
- R 1 is preferably a linear or branched primary or secondary aliphatic hydrocarbon group, more preferably a linear or branched primary or secondary alkyl group or alkenyl group, more preferably The method for producing an oxide according to ⁇ 46>, which is a linear primary or secondary alkyl group.
- the carbon number of R 1 is 6 or more, 8 or more, 10 or more, or 12 or more, preferably 36 or less, more preferably 22 or less, still more preferably 18 or less, still more preferably 14 or less, ⁇ 46> Or the manufacturing method of the oxide as described in ⁇ 47>.
- R 2 is preferably an aliphatic hydrocarbon group, more preferably a linear or branched primary or secondary aliphatic hydrocarbon group, still more preferably a linear or branched primary or secondary chain.
- the number of carbon atoms of R 2 is 6 or more, 8 or more, 10 or more or 12 or more, preferably 36 or less, more preferably 22 or less, more preferably 18 or less, even more preferably 14 or less, ⁇ 46>
- the mass ratio of ⁇ 51> alcohol or polyoxyalkylene alkyl ether to water used in the reaction is preferably 60/40 or more, more preferably 70/30 or more, further preferably 75/25 or more, preferably 95.
- ⁇ 52> Supply of oxygen to the liquid phase which is the composition or a reaction liquid thereof is performed by circulating an oxygen-containing gas in the liquid phase, and the oxygen-containing gas is preferably a mixed gas such as oxygen gas or air.
- the oxygen concentration in the oxygen-containing gas is preferably 10% by volume or more, more preferably 50% by volume or more, still more preferably 70% by volume or more, still more preferably 90% by volume or more, and still more preferably substantially.
- the method for producing an oxide according to ⁇ 52> which is 100% by volume, more preferably 100% by volume.
- the reaction temperature is preferably 50 ° C. or higher, more preferably 60 ° C. or higher, preferably 100 ° C. or lower, more preferably 90 ° C. or lower, and still more preferably 80 ° C. or lower.
- the manufacturing method of the oxide in any one.
- the reaction pressure is preferably 0.09 MPa or more, more preferably 0.10 MPa or more, preferably 0.5 MPa or less, more preferably 0.2 MPa or less, and further preferably 0.11 MPa or less ⁇ 45>
- the amount of the catalyst used is preferably 0.1 parts by mass or more, more preferably 1 part by mass or more, further preferably 3 parts by mass or more, more preferably as the mass of the Pt metal with respect to 100 parts by mass of the polyoxyalkylene alkyl ether.
- the temperature condition is 20 ° C.
- the stirring condition is 200 rpm
- the pressure condition is normal pressure.
- pH test paper “roll type UNIV” manufactured by ADVANTEC was used for pH measurement.
- Step 1 Preparation of Pt / C slurry (hereinafter referred to as liquid A): A 1 L beaker was charged with 40 g of 10% Pt / C (Evonik Japan, Pt particle size 14 nm, water content 59.2%) and 500 g of ion-exchanged water, and a “Fine stirrer was used to prevent precipitation of the solid at the bottom of the beaker. F-202 "(manufactured by Tokyo Glass Instrument Co., Ltd.) was used for stirring at about 100 rpm.
- Step 2 Preparation of an acidic aqueous solution containing bismuth (hereinafter referred to as liquid B): Into a 2 L separable flask equipped with a mechanical stirrer “Teflon (registered trademark) stirring blade crescent shape” (manufactured by ASONE, stirring blade width 75 mm ⁇ height 20 mm ⁇ thickness 4 mm), bismuth nitrate pentahydrate ( (Wako Pure Chemical Industries, Ltd.) 0.38 g and 2% acetic acid aqueous solution 808 mL were charged. The Bi concentration in a 2% aqueous acetic acid solution is 0.001M.
- Step 3 the liquid B in the separable flask that was dropped in Step 3 was passed through hydrogen at 86-129 mL / min for 20 minutes while stirring with a mechanical stirrer at 200 rpm. Replacement was performed. Thereafter, 2 mL of acetic acid was added under stirring and hydrogen flow. The pH of the solution after addition of acetic acid was 3.
- Step 3 While continuing the hydrogen flow and stirring to the liquid B in the separable flask, the liquid A in the beaker was dropped into the liquid B in the separable flask using a peristaltic pump. The dropping was carried out while stirring the liquid B while flowing hydrogen through the liquid B under the conditions of 86 to 129 mL / min.
- the dropping speed of dropping liquid A, dropping time, and temperature of liquid B receiving dropping were as shown in Table 1.
- the pH of the solution after dropping was 3.
- IPA isopropyl alcohol
- Aging and IPA addition were performed while continuing the hydrogen flow and stirring to the liquid B.
- vacuum filtration was performed while stopping hydrogen and flowing nitrogen. This filtration is performed by placing a membrane filter “PTFE membrane filter T020A142C” (ADVANTEC, pore size 0.2 ⁇ m) on a filter holder “glass type KGS-90” (manufactured by ADVANTEC) for vacuum filtration, and adding the solution after addition of IPA.
- the solid was separated by flowing in a suction environment.
- the pH of the filtrate was 3.
- the filtered solid was returned to the separable flask, and a 1% IPA aqueous solution (300 mL) was further added, followed by stirring and washing for 30 minutes. Thereafter, filtration was performed in the same manner as above, and the solid obtained by filtration was dried in about 1 hour to obtain a 10% Pt-1% Bi / C catalyst.
- the operations of stirring and washing, filtration and drying were performed under a nitrogen flow.
- the pH of the filtrate obtained by filtration after washing and before drying was 3-4.
- Step 1 Preparation of Pt / C slurry (hereinafter referred to as liquid A): A 2 L separable flask equipped with a mechanical stirrer “Teflon (registered trademark) stirring blade crescent moon” (manufactured by ASONE, stirring blade width 75 mm ⁇ height 20 mm ⁇ thickness 4 mm) was added with 10% Pt / C (Evonik). 40 g of water (59.2%, manufactured by Japan) and 500 g of ion-exchanged water were charged.
- Step 2 Preparation of an acidic aqueous solution containing bismuth (hereinafter referred to as liquid B): In a 1 L beaker, 0.38 g of bismuth nitrate pentahydrate (manufactured by Wako Pure Chemical Industries, Ltd.) and 808 mL of 2% acetic acid aqueous solution were charged. The Bi concentration in a 2% aqueous acetic acid solution is 0.001M. In order to completely dissolve the bismuth salt, ultrasonic waves were applied for 5 minutes under the condition of a frequency of 38 kHz using an ultrasonic cleaner “Fine ultrasonic cleaner FU-9H” (manufactured by Tokyo Glass Instrument Co., Ltd.).
- Step 3 the liquid A in the separable flask that was dropped in Step 3 was passed through hydrogen at 86-129 mL / min for 20 minutes while stirring with a mechanical stirrer at 200 rpm. Replacement was performed. Thereafter, 2 mL of acetic acid was added under stirring and hydrogen flow. The pH of the solution after addition of acetic acid was 3.
- Step 3 While continuing the hydrogen flow and stirring to the liquid A in the separable flask, the liquid B in the beaker was dropped into the liquid A in the separable flask using a peristaltic pump.
- the dropping speed of dropping liquid B, dropping time, and temperature of liquid A receiving dropping were as shown in Table 1.
- the pH of the solution after dropping was 4.
- IPA isopropyl alcohol
- vacuum filtration was performed while stopping hydrogen and flowing nitrogen. This filtration is performed by placing a membrane filter “PTFE membrane filter T020A142C” (ADVANTEC, pore size 0.2 ⁇ m) on a filter holder “glass type KGS-90” (manufactured by ADVANTEC) for vacuum filtration, and adding the solution after addition of IPA. The solid was separated by flowing in a suction environment. The pH of the filtrate was 3-4.
- the filtered solid was returned to the separable flask, and a 1% IPA aqueous solution (300 mL) was further added, followed by stirring and washing for 30 minutes. Thereafter, filtration was performed in the same manner as above, and the solid obtained by filtration was dried in about 1 hour to obtain a 10% Pt-1% Bi / C catalyst.
- the operations of stirring and washing, filtration and drying were performed under a nitrogen flow.
- the pH of the filtrate obtained by filtration after washing and before drying was 3-4.
- Example 2 A catalyst was obtained by carrying out the same steps as in Example 1 except that a Pt / C catalyst having a Pt loading of 5% was used and neutralization, washing and filtration were carried out after addition of IPA and filtration. The filtration was performed by the method described in Example 1. Moreover, neutralization and washing
- the solid separated by filtration was returned to the separable flask, added with 300 mL of 0.05M NaHCO 3 aqueous solution, and neutralized by stirring for 30 minutes. Thereafter, filtration was performed, the solid was returned to the separable flask, and 300 mL of ion-exchanged water was further added, and the mixture was stirred and washed for 30 minutes. Neutralization and washing operations were performed under nitrogen flow. The pH of the filtrate immediately after neutralization and after washing was 7.
- Comparative Example 2 A catalyst was obtained by carrying out the same steps as in Comparative Example 1 except that a Pt / C catalyst having a Pt loading of 5% was used, and that neutralization, washing and filtration were performed after addition of IPA and filtration. The filtration was performed by the method described in Comparative Example 1. Moreover, neutralization and washing
- FIG. 1 A catalyst was obtained by carrying out the same steps as in Comparative Example 1 except that a Pt / C catalyst having a Pt loading of 5% was used, and that neutralization, washing and filtration were performed after addition of IPA and filtration. The filtration was performed by the method described in Comparative Example 1. Moreover, neutralization and washing
- Example 1 When determining the catalyst performance, determine the amount of ion-exchanged water charged. Therefore, it is necessary to measure the water content in the catalyst.
- the water content in the catalysts obtained in Example 1, Comparative Example 1, Example 2 and Comparative Example 2 was measured by the following method. The obtained results are shown in Tables 1 and 2.
- ether carboxylate was produced by the following method, and the amount of metal elution from the catalyst and the yield of ether carboxylate were measured. .
- the measurement results are shown in Tables 1 and 2.
- the solution was promptly filtered at 70 ° C. and separated from the catalyst.
- the pressure filtration was performed while the filter was heated to 70 ° C. in advance, and the solution after completion of the reaction (70 ° C.) was poured into the filter while injecting nitrogen at 4 kgf / cm 2 .
- ICP analysis was performed under the following measurement conditions to measure the elution amounts of Pt and Bi from the catalyst.
- Reagents Hydrochloric acid For atomic absorption analysis, manufactured by Kanto Chemical Co., Ltd.
- Nitric acid For atomic absorption analysis, manufactured by Kanto Chemical Co., Ltd.
- Sulfuric acid For precision analysis, manufactured by Wako Pure Chemical Industries, Ltd.
- Platinum standard solution Standard solution for atomic absorption analysis 1000 mg / L, manufactured by Kanto Chemical Co., Ltd.
- Bismuth standard solution standard solution for atomic absorption analysis 1000 mg / L, manufactured by Kanto Chemical Co., Ltd.
- the filter residue is ashed together with the filter paper (sulfuric acid is added appropriately during the ashing step to completely ash
- Calibration curve solution preparation A calibration curve solution of 0.1 to 2.0 mg / L was prepared using a standard solution for atomic absorption analysis (Pt and Bi: 1000 mg / L). Aqua regia was added to each solution so as to be about the same as the sample (about 8%).
- ICP measurement conditions Analysis device iCAP 6500 Duo manufactured by Thermo Fisher Scientific Wavelength: Pt 214.423nm, Bi 223.061nm RF power: 1150W Coolant gas flow rate: 12L / min Nebulizer flow rate: 0.70 L / min Auxiliary gas: 0.5 L / min Pump flow rate: 50rpm
- Examples 3-5 A catalyst was obtained by carrying out the process in the same manner as in Example 1 except that the conditions described in Table 3 were used. Moreover, the ether carboxylate was manufactured by the same method as Example 1, and the metal elution amount from a catalyst and the yield of ether carboxylate were measured. Table 3 shows the measurement results.
- Example 6 A catalyst was prepared by carrying out the steps in the same manner as in Example 1. Moreover, ether carboxylate was manufactured by the same method as Example 1 except having made reaction time into 14 hours, and the metal elution amount from a catalyst and the yield of ether carboxylate were measured. Table 3 shows the measurement results. The metal elution amount was measured in the same manner as in Example 1 except for the following (1) and (2).
- (1) In the sample solution preparation (1) of Example 1, instead of using a magnetic crucible to heat-dissolve the sample with acid, the sample was treated with acid by a microwave method using a dedicated sealed container.
- ICP measurement an ICP mass spectrometer ELAN DRC II manufactured by PerkinElmer, Inc. was used.
- Example 7 Using the catalyst produced in Example 1, XPS (X-ray Photoelectron Spectroscopy) measurement was performed by the following method. The result is shown in FIG. The peak top binding energy value in the range of the binding energy 162 to 155 eV was 157.8 eV.
- XPS X-ray Photoelectron Spectroscopy
- Comparative Example 3 XPS measurement was performed in the same manner as in Example 7 except that the catalyst produced in Comparative Example 1 was used.
- the photoelectron spectrum of the Bi4f orbit is shown in FIG.
- the peak top binding energy value in the range of binding energy 162 to 155 eV was 158.7 eV.
- the catalyst prepared in Example 1 has a smaller binding energy value at the peak top than that of the catalyst prepared in Comparative Example 1, and thus it is presumed that Bi is uniformly supported on Pt. And as a result of carrying Bi uniformly on Pt, the catalyst prepared in Example 1 is considered to have improved activity and suppressed metal elution.
- ⁇ XPS measurement method> An analysis sample was prepared by spreading the catalyst on a carbon double-sided tape bonded to a copper plate.
- the equipment and conditions used for the analysis are as follows.
- PHI Quantera SXM (ULVAC-PHI Inc.) -X-ray source: Monochromatic AlK ⁇ 1486.6 eV, 25W, 15kV ⁇ Beam diameter: 100 ⁇ m ⁇ Measurement range: 500 ⁇ 500 ⁇ m 2 ⁇ Pass energy: 280.0 eV (survey) 112.0 eV (narrow) ⁇ Step: 1.00 eV (survey) 0.20 eV (narrow) ⁇ Charging correction: Neutralizer and Ar + irradiation ⁇ Photoelectron extraction angle: 45 ° ⁇ Detection elements: C1s (5), O1s (10), Na1s (20), Pt4f (30), Bi4f (30) ⁇ Correction of bond energy value was performed at C1s 284.8 eV derived from carbon CH
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Dispersion Chemistry (AREA)
Abstract
【課題】アルカリを併用せずに液相酸化反応を行う場合に、活性が高く、触媒金属の溶出が抑制された耐久性の高い触媒の調製方法、及びその触媒を用いて酸化物を高効率で製造する方法の提供。 下記の工程1、工程2及び工程3を有する触媒の調製方法。 工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程 工程2:Biをイオンの状態で含む水溶液を調製する工程 工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
Description
本発明は触媒の調製方法に関する。より詳細には、アルコールやポリオキシアルキレンアルキルエーテルのようなヒドロキシ化合物、又はアルデヒド化合物を酸化してカルボキシル化合物又はケトン化合物を製造する際に好適に用いることができる酸化反応触媒の調製方法に関する。
背景技術
背景技術
従来より、貴金属触媒であるPtに助触媒としてBiを組み合わせて用い、ヒドロキシ化合物やアルデヒド化合物を接触酸化し、相当するカルボキシル化合物やケトン化合物に変換する方法が知られている。例えば、特開平11-279110号公報には、市販のPt/C触媒の分散液に硝酸ビスマス塩の硝酸水溶液を滴下してBiを担持させた触媒を調製し、これを用いて、2-置換-1,3-プロパンジオールを酸素含有ガスと接触反応させて、2-置換-3-ヒドロキシプロピオン酸類や2-置換マロン酸類を製造する方法が開示されている。また、Journal of Catalysis 225(2004)p.138-146には、Pt/Al2O3に硝酸ビスマス塩の酢酸水溶液を滴下してBiを担持させて触媒を調製し、これを用いて、1-フェニルエタノール、2-オクタノール、シンナミルアルコールを液相酸化する方法が開示されている。特開昭62-228093号公報には、グルコースからグルコネートを得る反応において、触媒としてPd/Cを用い、促進剤としてBiを担持させることが記載されている。担持はPd/Cの懸濁液に促進剤溶液を混合することによって行われる。なおPtは白金を意味し、Biはビスマスを意味し、Pdはパラジウムを意味し、Cは活性炭を意味する。
発明の概要
発明の概要
従来のPt-Bi/C触媒を酸化反応に使用すると、反応初期から中期にかけては反応が円滑に進行するが、特に反応末期になると反応速度が著しく低下するか、或いは反応が未完結のまま停止してしまうという現象が観測される。また、触媒から反応後の溶液へと、担持されている金属が溶出して耐久性が劣化してしまうという現象も観測される。これらの現象は、被酸化物の構造によって程度に差はあるものの、Biを担持させた貴金属触媒を用いる酸化反応一般において観測される。
特開平11-279110号公報では2-置換-1,3-プロパンジオールから2-置換-3-ヒドロキシプロピオン酸、2-アルキルマロン酸を得る反応において、第2金属成分としてBiを担持させたPt-Bi/C触媒だけではなく、PdやPbを担持させたPt-Pd/C触媒やPt-Pb/C触媒を用いることも記載されている。しかしながら、収率が低い。また特開平11-279110号公報の反応では全て、触媒以外にアルカリを添加して選択性を向上させることを必要としている。
また、特開昭62-228093号公報では、Pd/C上にBiを担持させると、Bi/C上にPdを担持させる場合よりも活性が高いことが記載されている。しかし、上記の問題点に十分に対処しうる知見をもたらすものではなく、また特開平11-279110号公報の場合と同様、反応に際しては触媒とアルカリの併用が必要である。
さらに、Journal of Catalysis 225(2004)p.138-146ではヒドロキシル基を有する化合物の液相酸化において上記のようにPt-Bi/Al2O3触媒を用いている。しかし、アルカリを併用せずに反応を行うと活性及び金属溶出いずれの点でも満足できるものではない。
本発明の課題は、アルカリを併用せずに液相酸化反応を行う場合に、活性が高く、また触媒金属の溶出が抑制された耐久性の高い触媒の調製方法を提供することである。
本発明者は、上記の如き液相酸化反応におけるPt-Bi/C触媒の問題点を解決すべく、Pt-Bi/C触媒の調製法について研究を続けた結果、本発明を完成した。
本発明は、下記の工程1、工程2及び工程3を有する触媒の調製方法である。
工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
本発明は、下記の工程1、工程2及び工程3を有する触媒の調製方法である。
工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
本発明によれば、アルカリを併用せずに液相酸化反応を行う場合に、活性が高く、また触媒金属の溶出が抑制された耐久性の高い触媒が得られる。またかかる触媒を用いて酸化物を高効率で製造する方法が提供される。
本発明は、下記の工程1、工程2及び工程3を有する触媒の調製方法である。
工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
工程1で調製した、活性炭にPtを担持させた触媒の水分散液を、工程2で調製したBiをイオンの状態で含む水溶液に添加することにより、アルカリを併用せずに液相酸化反応を行う場合に、活性が高く、また触媒金属の溶出が抑制された耐久性の高い触媒を調製することができる。その理由は定かではないが以下のように考える。活性炭にPtを担持させた触媒の水分散液を、Biイオンを含む水溶液に添加すると、PtとBiを共沈させる場合や、Biイオンを含む水溶液を、活性炭にPtを担持させた触媒の水分散液に添加したり供給したりする場合と比較して、Biイオンをより均等にPtに接触させることができる。そのため、Pt-Bi複合体を効率良く形成させることができるため、活性が向上し、また液相酸化反応中の金属溶出を抑制することができる。しかしながらかかる作用は推定であり、本発明の範囲を制限するものではない。
以下の記載において、特に断らない限り、「%」は「質量%」を示す。
工程1は、活性炭にPtを担持させた触媒(以下、Pt/C触媒ともいう)の水分散液を調製する工程である。
活性炭には特に制限はなく、Ptを吸着し担持しうるものであれば、どのような種類の活性炭であっても使用することができる。活性炭の例としては、ヤシ殻活性炭等の植物質の活性炭、石炭系活性炭等の鉱物質の活性炭、パルプ廃液、合成樹脂、有機性廃棄物の活性炭などを挙げることができる。また賦活方法や細孔分布、形状などにも特に制限はない。
活性炭に担持されるPtの粒径に特に制限はないが、分散性と反応活性を高める観点から、好ましくは20nm以下、より好ましくは15nm以下、さらに好ましくは10nm以下であり、また、例えば、1nm以上であってよい。
触媒固形分中のPt金属の担持量は、反応性の観点から好ましくは0.1%以上、より好ましくは1%以上、さらに好ましくは3%以上、よりさらに好ましくは5%以上であり、分散性を高める観点から、好ましくは20%以下、より好ましくは15%以下、さらに好ましくは10%以下である。またPt/C触媒は公知の含浸法や析出沈殿法によって調製可能であるが、市販品を用いることもできる。
水分散液の調製は、Pt/C触媒をイオン交換水、蒸留水、純水などに添加し、撹拌することによって行われる。
分散液中のPt/C触媒の濃度は後工程での効率の観点から、好ましくは4%以上、より好ましくは5%以上、さらに好ましくは6%以上、よりさらに好ましくは7%以上であり、操作性の観点から、好ましくは12%以下、より好ましくは9%以下、さらに好ましくは8%以下である。
調製温度は、経済性の観点から、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、Ptの変性や凝集を抑制する観点から好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である。
工程2は、Biをイオンの状態で含む水溶液(以下、Bi水溶液ともよぶ)を調製する工程である。
Biイオンのイオン源は、Bi塩が水や酸性水溶液に溶解可能なBi種であることが必要であり、この観点から好ましくは硝酸ビスマス五水和物(Bi(NO3)3・5H2O)、酸化ビスマス(Bi2O3)、炭酸ビスマス((BiO)2CO3)及び水酸化ビスマス(Bi(OH)3)から選ばれる少なくとも1種であり、より好ましくは硝酸ビスマス五水和物である。
Bi水溶液は、Bi塩を溶解する観点から、好ましくは酸を含有する。使用する酸としては、無機酸と有機酸のいずれでもよい。有機酸としては、液相酸化反応時に調製した触媒からの金属溶出を抑制する観点から好ましくはカルボキシル基を有する酸、より好ましくは酢酸、ギ酸、クエン酸及びシュウ酸から選ばれる1種以上、さらに好ましくは酢酸である。無機酸としては、液相酸化反応時に調製した触媒からの金属溶出を抑制する観点から好ましくは硝酸、塩酸、リン酸及び硫酸から選ばれる1種以上、より好ましくは硝酸である。
使用する酸の量は、Bi塩を溶解する観点から、Bi水溶液中、好ましくは1%以上、より好ましくは1.5%以上であり、経済性の観点から好ましくは5%以下、より好ましくは4%以下、さらに好ましくは3%以下である。
調製した触媒からの金属溶出を抑制する観点から、Bi水溶液中のBiの配合量は好ましくは0.0001M以上、より好ましくは0.0005M以上、さらに好ましくは0.001M以上であり、同様の観点から好ましくは0.1M以下、より好ましくは0.05M以下、さらに好ましくは0.03M以下、よりさらに好ましくは0.02M以下、よりさらに好ましくは0.01M以下である。
Bi水溶液のpHは、調製した触媒からの金属溶出を抑制する観点から、好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である。
Bi水溶液を調製する温度は、特に制限されるものではないが、経済性の観点から、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である。
工程3に先立って、工程1で得られた水分散液及び工程2で得られたBi水溶液のいずれか一方又は両方、操作性の観点から好ましくは工程3において添加又は滴下を受ける液すなわち工程2で得られたBi水溶液は、調製した触媒からの金属溶出を抑制する観点から、次の工程3でPtを還元処理するために使用することのできる還元剤や還元性ガスにより処理をすることが好ましい。還元剤による処理は、ホルマリンやソジウムボロハイドライドなどを得られた水分散液やBi水溶液に添加することによって行うことができる。また、還元性ガスによる処理は、水素や一酸化炭素などの還元性ガスを水分散液やBi水溶液に流通させて還元雰囲気としたりすることによって行うことができる。好ましくは還元性ガスを流通させ、より好ましくは水素ガスを流通させる。
還元剤や還元性ガスによる処理温度は、経済性の観点から、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、Ptの変性や凝集を抑制する観点から、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である。
また、還元性ガスを流通させて処理を行う場合、流通させる還元性ガスの供給量は、還元性ガスによる処理を十分に行う観点から、触媒に対して大過剰となる量であり、好ましくは60mL/分以上、より好ましくは80mL/分以上であり、経済性の観点から好ましくは500mL/分以下、より好ましくは、300mL/分以下、さらに好ましくは150mL/分以下である。
還元剤や還元性ガスによる処理時間は、還元剤や還元性ガスによる処理を十分に行う観点から好ましくは10分以上、より好ましくは15分以上、さらに好ましくは20分以上であり、生産性の観点から好ましくは2時間以下、より好ましくは1時間以下、さらに好ましくは30分以下である。
工程3は、工程2で得られたBi水溶液に、工程1で得られた水分散液を添加する工程である。
Bi水溶液にPt/C触媒の水分散液を添加することによって、Pt-Bi/C触媒の複合体が効率良く形成されると考えられる。添加は連続添加、分割添加、一括添加のいずれでもよいが、連続添加又は分割添加が好ましく、滴下等の連続的な分割添加がより好ましい。
連続添加又は滴下等の連続的な分割添加の場合、生産性の観点から、添加速度は好ましくはPt/C触媒の水分散液として1mL/分以上、より好ましくは3mL/分以上となる速度であり、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、好ましくはPt/C触媒の水分散液として10mL/分以下、より好ましくは5mL/分以下となる速度である。
また、添加に要する時間は、特に制限はないが、調製した触媒の活性を向上させ調製した触媒からの金属溶出を抑制する観点から、好ましくは15分以上、より好ましくは30分以上、さらに好ましくは1時間以上、よりさらに好ましくは2時間以上であり、生産性の観点から好ましくは10時間以下、より好ましくは7時間以下、さらに好ましくは5時間以下、よりさらに好ましくは3時間以下である。
工程3において添加又は滴下を受けるBi水溶液の温度は、調製した触媒の活性を向上させ調製した触媒からの金属溶出を抑制する観点から、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、Ptの変性や凝集を抑制する観点から、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である。
調製した触媒からの金属溶出を抑制する観点から、工程3に際して好ましくは還元処理を行う。工程3の還元処理は、添加又は滴下する液及び添加又は滴下を受ける液のいずれか一方又は両方に行うことができるが、操作性の観点から、好ましくは添加又は滴下を受ける液に行う。工程3の還元処理は、調製した触媒からの金属溶出を抑制する観点から、好ましくは工程3を還元雰囲気下に行う。好ましくは工程3の還元処理は水素や一酸化炭素などの還元性ガス、より好ましくは水素ガスの流通によって行われる。還元処理温度及び還元性ガスを流通させて還元処理を行う場合の還元性ガスの供給量の好ましい範囲は、それぞれ、工程3に先立って行う還元剤や還元性ガスによる処理温度及び還元性ガスの供給量の好ましい範囲と同様である。工程3の還元処理は好ましくはPt/C触媒の水分散液の添加が終了するまで行われる。
本発明の触媒の調製方法は工程4として、工程3で得た触媒を含有する液と還元補助剤を混合し、PtとBiを活性炭に担持させた触媒(即ち上記Pt-Bi/C触媒)及び還元補助剤を含有する液を得る工程を有することができる。この工程は、調製した触媒からの金属溶出を抑制する観点から好ましい。
還元補助剤はPtとBiを活性炭に担持させた触媒の還元状態を維持するものであり、還元性を有する有機溶媒、例えばイソプロピルアルコール(IPA)を用いることができる。
調製した触媒からの金属溶出を抑制する観点から還元補助剤の使用量は、工程3で得た触媒を含有する液100質量部に対して好ましくは0.1質量部以上、より好ましくは0.5質量部以上、さらに好ましくは1.0質量部以上であり、経済性の観点から好ましくは10質量部以下、より好ましくは5質量部以下、さらに好ましくは2質量部以下である。
本発明の触媒の調製方法は工程5として、工程4で得た液中のPtとBiを活性炭に担持させた触媒を洗浄液で洗浄する工程を有することができる。この工程は、調製した触媒の活性の観点及び調製した触媒からの金属溶出を抑制する観点から好ましい。
工程5で使用する洗浄液は、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、好ましくは水及び上記還元補助剤から選ばれる1種以上を含む液、より好ましくは上記還元補助剤の1種以上及び水を含有する液、さらに好ましくはIPA水溶液である。
洗浄を効率的に行う観点から、洗浄前に工程4で得た液を濾過し、PtとBiを活性炭に担持させた触媒を濾別することが好ましい。濾過は減圧濾過、加圧濾過のいずれでも行うことができる。
本発明の触媒の調製方法は工程6として、工程5で洗浄したPtとBiを活性炭に担持させた触媒を乾燥する工程を有することができる。この工程は、取扱の観点から好ましい。
乾燥は例えば窒素ガス等の不活性ガスの流通下に行われる。
本発明の触媒の調製方法では、工程3の後、調製した触媒の活性を向上させる観点及び調製した触媒から金属溶出を抑制する観点から中和処理を行わないことが好ましい。
工程3で得た触媒を含有する液のpHは、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である。
また、工程4を行う場合、工程4で得た液を濾過した濾液のpHは、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である。
また、工程5の洗浄を行う場合、洗浄後の濾液のpHは、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である。
Pt-Bi/C触媒固形分中のBiの担持量は、酸化反応の生成物の生産性向上の観点から、好ましくは0.01%以上、より好ましくは0.5%以上であり、好ましくは10%以下、より好ましくは5%以下、さらに好ましくは3.5%以下、よりさらに好ましくは1.5%以下である。
Pt-Bi/C触媒におけるBiのPtに対する質量比(原子比)Bi/Ptは、酸化反応の生成物の生産性向上の観点から、好ましくは0.05以上、より好ましくは0.1以上であり、好ましくは1.0以下、より好ましくは0.6以下、さらに好ましくは0.3以下、よりさらに好ましくは0.2以下である。
本発明のPtとBiを活性炭に担持させた触媒は、調製した触媒の活性を向上させる観点及び調製した触媒からの金属溶出を抑制する観点から、XPSにより測定したBi4f軌道の光電子スペクトルにおいて結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値が、好ましくは158.5eV以下、より好ましくは158.2eV以下、更に好ましくは158.0eV以下であり、好ましくは157.0eV以上、より好ましくは157.2eV以上、さらに好ましくは157.5eV以上である。
本発明の触媒は、ヒドロキシ化合物又はアルデヒド化合物を酸化してカルボキシル化合物又はケトン化合物を製造する反応、例えば、ポリオキシアルキレンアルキルエーテルを液相酸化して対応するカルボキシル化合物を製造する反応に好適に使用される。即ち本発明は、上記触媒の存在下、アルコール又はポリオキシアルキレンアルキルエーテルと水を含有する組成物に酸素を供給して、前記アルコール又はポリオキシアルキレンアルキルエーテルを脱水素酸化する、アルコールの酸化物又はポリオキシアルキレンアルキルエーテルの酸化物(以下、当該酸化物ともいう)の製造方法にも関する。すなわち、上記触媒を得る工程、該触媒の存在下、アルコール又はポリオキシアルキレンアルキルエーテルと水を含有する組成物に酸素を供給して、前記アルコール又はポリオキシアルキレンアルキルエーテルを脱水素酸化する工程を有する、当該酸化物の製造方法に関する。
好ましくはアルコール又はポリオキシアルキレンアルキルエーテルは、下記一般式(1)又は一般式(2)で表される1種又は2種以上である。
R1O-H (1)
但し、一般式(1)においてR1は炭素数2以上40以下の脂肪族炭化水素基である。
但し、一般式(1)においてR1は炭素数2以上40以下の脂肪族炭化水素基である。
R2O-(AO)n-H (2)
但し、一般式(2)においてR2は炭素数2以上40以下の炭化水素基、AOは炭素数2以上4以下のアルキレンオキシ基を表し、nはアルキレンオキシ基の付加モル数であり、1以上30以下の整数である。
但し、一般式(2)においてR2は炭素数2以上40以下の炭化水素基、AOは炭素数2以上4以下のアルキレンオキシ基を表し、nはアルキレンオキシ基の付加モル数であり、1以上30以下の整数である。
R1は、反応性の観点から、好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、より好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、さらに好ましくは直鎖の、1級若しくは2級のアルキル基である。
R1の炭素数は、特に制限されるものではないが、6以上、8以上、10以上又は12以上であってもよく、反応性の観点から、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である。
R2は、反応性の観点から、好ましくは脂肪族炭化水素基、より好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、さらに好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、よりさらに好ましくは直鎖の、1級若しくは2級のアルキル基である。
R2の炭素数は、特に制限されるものではないが、6以上、8以上、10以上又は12以上であってもよく、反応性の観点から、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である。
反応に使用する水に対する、原料として使用するアルコール又はポリオキシアルキレンアルキルエーテルの質量比(原料として使用するアルコール又はポリオキシアルキレンアルキルエーテル/水)は、アルコールの酸化物又はポリオキシアルキレンアルキルエーテルの酸化物の生産性向上及び液相の粘度増加抑制の観点から、好ましくは60/40以上、より好ましくは70/30以上、さらに好ましくは75/25以上であり、好ましくは95/5以下、より好ましくは90/10以下、さらに好ましくは85/15以下である。
前記組成物又はその反応液である液相への酸素の供給は、酸素含有ガスを液相に流通させることによって実施できる。酸素含有ガスとしては、酸素ガス、空気などの酸素を含有する混合ガスが挙げられる。
酸素を含有する混合ガスを用いる場合、酸素と併用するガスは、活性に影響を与えない観点から、好ましくはヘリウム、アルゴン、窒素などの不活性ガスである。
酸素含有ガス中の酸素濃度は、当該酸化物の生産性の観点から、好ましくは10体積%以上、より好ましくは50体積%以上、さらに好ましくは70体積%以上、よりさらに好ましくは90体積%以上、よりさらに好ましくは実質的に100体積%、よりさらに好ましくは100体積%である。
かかる製造は連続式、回分式又は半回分式で行うことができる。
反応温度は、反応性の観点から、好ましくは50℃以上、より好ましくは60℃以上であり、設備負荷の観点から、好ましくは100℃以下、より好ましくは90℃以下、さらに好ましくは80℃以下である。
反応は常圧下で行っても、加圧下で行ってもよい。反応圧力は、反応性の観点から、絶対圧力で、好ましくは0.09MPa以上、より好ましくは0.10MPa以上であり、設備負荷の観点から、好ましくは0.5MPa以下、より好ましくは0.2MPa以下、さらに好ましくは0.11MPa以下である。
触媒の使用量は反応温度あるいは反応圧力に応じ、実用的な反応速度が得られる範囲内において任意に選択できるが、反応を回分式で行う場合は、反応性の観点から、ポリオキシアルキレンアルキルエーテル100質量部に対するPt金属の質量として、好ましくは0.1質量部以上、より好ましくは1質量部以上、さらに好ましくは3質量部以上、よりさらに好ましくは6質量部以上であり、経済性の観点から、好ましくは20質量部以下、より好ましくは15質量部以下、さらに好ましくは10質量部以下である。
以下では本発明の好適な実施態様について述べる。
<1>下記の工程1、工程2及び工程3を有する触媒の調製方法。
工程1:活性炭にPtを担持させた触媒(Pt/C触媒)の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液(Bi水溶液)を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
<1>下記の工程1、工程2及び工程3を有する触媒の調製方法。
工程1:活性炭にPtを担持させた触媒(Pt/C触媒)の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液(Bi水溶液)を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程
<2>Ptの粒径が好ましくは20nm以下、より好ましくは15nm以下、さらに好ましくは10nm以下であり、また、1nm以上であってよい<1>に記載の調製方法。
<3>触媒固形分中のPt金属の担持量が好ましくは0.1%以上、より好ましくは1%以上、さらに好ましくは3%以上、よりさらに好ましくは5%以上であり、好ましくは20%以下、より好ましくは15%以下、さらに好ましくは10%以下である<1>又は<2>に記載の調製方法。
<4>水分散液中のPt/C触媒の濃度が好ましくは4%以上、より好ましくは5%以上、さらに好ましくは6%以上、よりさらに好ましくは7%以上であり、好ましくは12%以下、より好ましくは9%以下、さらに好ましくは8%以下である<1>~<3>の何れか1に記載の調製方法。
<5>工程1の調製温度が好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である<1>~<4>の何れか1に記載の調製方法。
<6>Biイオンのイオン源が、好ましくは硝酸ビスマス五水和物(Bi(NO3)3・5H2O)、酸化ビスマス(Bi2O3)、炭酸ビスマス((BiO)2CO3)及び水酸化ビスマス(Bi(OH)3)から選ばれる少なくとも1種であり、より好ましくは硝酸ビスマス五水和物である、<1>~<5>の何れか1に記載の調製方法。
<7>工程2で得られた水溶液が酸を含有する<1>~<6>の何れか1に記載の調製方法。
<8>酸が無機酸又は有機酸であり、好ましくは有機酸であり、より好ましくはカルボキシル基を有する酸であり、さらに好ましくは酢酸、ギ酸、クエン酸及びシュウ酸から選ばれる1種以上、よりさらに好ましくは酢酸である<7>に記載の調製方法。
<9>酸が無機酸であり、好ましくは硝酸、塩酸、リン酸及び硫酸から選ばれる1種以上であり、より好ましくは硝酸である<7>に記載の調製方法。
<10>酸が酢酸及び硝酸から選ばれる1種である、<7>に記載の触媒の調製方法。
<11>使用する酸の量が、Bi水溶液中、好ましくは1%以上、より好ましくは1.5%以上であり、好ましくは5%以下、より好ましくは4%以下、さらに好ましくは3%以下である、<7>~<10>の何れか1に記載の調製方法。
<12>Bi水溶液中のBiの配合量が好ましくは0.0001M以上、より好ましくは0.0005M以上、さらに好ましくは0.001M以上であり、好ましくは0.1M以下、より好ましくは0.05M以下、さらに好ましくは0.03M以下、よりさらに好ましくは0.02M以下、よりさらに好ましくは0.01M以下である<1>~<11>の何れか1に記載の調製方法。
<13>Bi水溶液のpHが好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である<1>~<12>の何れか1に記載の調製方法。
<14>Bi水溶液を調製する温度が、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である、<1>~<13>の何れか1に記載の調製方法。
<15>工程1で得られた水分散液及び工程2で得られたBi水溶液のいずれか一方又は両方、好ましくは工程3において添加又は滴下を受ける液すなわち工程2で得られたBi水溶液が工程3に先立って還元剤や還元性ガスにより処理される<1>~<14>の何れか1に記載の調製方法。
<16>還元剤や還元性ガスによる処理温度が好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である<15>に記載の調製方法。
<17>還元剤や還元性ガスによる処理時間が好ましくは10分以上、より好ましくは15分以上、さらに好ましくは20分以上であり、好ましくは2時間以下、より好ましくは1時間以下、さらに好ましくは30分以下である<15>又は<16>に記載の調製方法。
<18>還元剤や還元性ガスによる処理が還元性ガス、好ましくは水素ガスの流通によって行われる<15>~<17>の何れか1に記載の調製方法。
<19>還元性ガスの供給量が好ましくは60mL/分以上、より好ましくは80mL/分以上、さらに好ましくは120mL/分以上であり、好ましくは500mL/分以下、より好ましくは、300mL/分以下、さらに好ましくは150mL/分以下である<18>に記載の調製方法。
<20>工程3の添加が連続添加、分割添加、一括添加、又は連続的な分割添加であり、好ましくは連続添加、分割添加、又は連続的な分割添加であり、より好ましくは滴下である<1>~<19>の何れか1に記載の調製方法。
<21>添加速度がPt/C触媒の水分散液として好ましくは1mL/分以上、より好ましくは3mL/分以上であり、好ましくは10mL/分以下、より好ましくは5mL/分以下である<1>~<20>の何れか1に記載の調製方法。
<22>添加に要する時間が、好ましくは15分以上、より好ましくは30分以上、さらに好ましくは1時間以上、よりさらに好ましくは2時間以上であり、好ましくは10時間以下、より好ましくは7時間以下、さらに好ましくは5時間以下、よりさらに好ましくは3時間以下である<1>~<21>の何れか1に記載の調製方法。
<23>工程3において添加を受けるBi水溶液の温度が、好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である、<1>~<22>の何れか1に記載の調製方法。
<24>工程3に際して好ましくは還元処理を行い、工程3の還元処理は、添加又は滴下する液及び添加又は滴下を受ける液のいずれか一方又は両方に行うことができるが、好ましくは添加又は滴下を受ける液に行い、工程3の還元処理は好ましくは工程3を還元雰囲気下に行う、<1>~<23>の何れか1に記載の調製方法。
<25>工程3の還元処理が好ましくはPt/C触媒の水分散液の添加が終了するまで行われる、<24>に記載の調製方法。
<26>工程3の還元処理温度が好ましくは10℃以上、より好ましくは15℃以上、さらに好ましくは20℃以上であり、好ましくは60℃以下、より好ましくは50℃以下、さらに好ましくは40℃以下、よりさらに好ましくは30℃以下、よりさらに好ましくは25℃以下である<24>又は<25>に記載の調製方法。
<27>工程3の還元処理が還元性ガス、好ましくは水素ガスの流通によって行われる<24>~<26>の何れか1に記載の調製方法。
<28>工程3の還元性ガスの供給量が好ましくは60mL/分以上、より好ましくは80mL/分以上、さらに好ましくは120mL/分以上であり、好ましくは500mL/分以下、より好ましくは、300mL/分以下、さらに好ましくは150mL/分以下である<27>に記載の調製方法。
<29>工程3の後に中和処理を行わない<1>~<28>の何れか1に記載の調製方法。
<30>工程3で得た触媒を含有する液のpHが好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である<1>~<29>の何れか1に記載の調製方法。
<31>工程3で得た触媒を含有する液と還元補助剤を混合し、PtとBiを活性炭に担持させた触媒(Pt-Bi/C触媒)及び還元補助剤を含有する液を得る工程4を有する、<1>~<30>の何れか1に記載の調製方法。
<32>還元補助剤が好ましくは還元性を有する有機溶媒であり、より好ましくはイソプロピルアルコール(IPA)である<31>に記載の調製方法。
<33>還元補助剤の使用量が、工程3で得た触媒を含有する液100質量部に対して好ましくは0.1質量部以上、より好ましくは0.5質量部以上、さらに好ましくは1.0質量部以上であり、好ましくは10質量部以下、より好ましくは5質量部以下、さらに好ましくは2質量部以下である<31>又は<32>に記載の調製方法。
<34>工程4で得た液を濾過した濾液のpHが好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である<31>~<33>の何れか1に記載の調製方法。
<35>工程4で得た液中のPtとBiを活性炭に担持させた触媒を洗浄液で洗浄する工程5を有する<31>~<34>の何れか1に記載の調製方法。
<36>洗浄液が、好ましくは水及び還元補助剤から選ばれる1種以上を含む液、より好ましくは還元補助剤の1種以上及び水を含有する液、さらに好ましくはIPA水溶液である<35>に記載の調製方法。
<37>洗浄前に工程4で得た液を濾過し、PtとBiを活性炭に担持させた触媒を濾別する<35>又は<36>に記載の調製方法。
<38>工程5の洗浄後の濾液のpHが好ましくは3.8以下、より好ましくは3.6以下、さらに好ましくは3.4以下であり、好ましくは1.0以上、より好ましくは1.5以上、さらに好ましくは2.0以上、よりさらに好ましくは2.5以上である<35>~<37>の何れか1に記載の調製方法。
<39>工程5で洗浄したPtとBiを活性炭に担持させた触媒を乾燥する工程6を有する<35>~<38>の何れか1に記載の調製方法。
<40>乾燥が好ましくは不活性ガス、より好ましくは窒素ガスの流通下に行われる<39>に記載の調製方法。
<41><1>~<40>の何れか1に記載の方法により得られたPt-Bi/C触媒。
<42>Pt-Bi/C触媒固形分中のBiの担持量が好ましくは0.01%以上、より好ましくは0.5%以上であり、好ましくは10%以下、より好ましくは5%以下、さらに好ましくは3.5%以下、よりさらに好ましくは1.5%以下である<41>に記載の触媒。
<43>Pt-Bi/C触媒におけるBiのPtに対する質量比(原子比)Bi/Ptが好ましくは0.05以上、より好ましくは0.1以上であり、好ましくは1.0以下、より好ましくは0.6以下、さらに好ましくは0.3以下、よりさらに好ましくは0.2以下である<41>又は<42>に記載の触媒。
<44>XPSにより測定したBi4f軌道の光電子スペクトルにおいて結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値が、好ましくは158.5eV以下、より好ましくは158.2eV以下、更に好ましくは158.0eV以下であり、好ましくは157.0eV以上、より好ましくは157.2eV以上、さらに好ましくは157.5eV以上である、PtとBiを活性炭に担持させた触媒。
<45><41>~<44>の何れか1に記載の触媒の存在下、アルコール又はポリオキシアルキレンアルキルエーテルと水を含有する組成物に酸素を供給して、前記アルコール又はポリオキシアルキレンアルキルエーテルを脱水素酸化する、アルコールの酸化物又はポリオキシアルキレンアルキルエーテルの酸化物の製造方法。
<46>アルコール又はポリオキシアルキレンアルキルエーテルが下記一般式(1)又は一般式(2)で表される1種又は2種以上である<45>に記載のアルコールの酸化物又はポリオキシアルキレンアルキルエーテルの酸化物の製造方法。
R1O-H (1)
但し、一般式(1)においてR1は炭素数2以上40以下の脂肪族炭化水素基である。
R2O-(AO)n-H (2)
但し、一般式(2)においてRは炭素数2以上40以下の炭化水素基、AOは炭素数2以上4以下のアルキレンオキシ基を表し、nはアルキレンオキシ基の付加モル数であり、1以上30以下の整数である。
R1O-H (1)
但し、一般式(1)においてR1は炭素数2以上40以下の脂肪族炭化水素基である。
R2O-(AO)n-H (2)
但し、一般式(2)においてRは炭素数2以上40以下の炭化水素基、AOは炭素数2以上4以下のアルキレンオキシ基を表し、nはアルキレンオキシ基の付加モル数であり、1以上30以下の整数である。
<47>
R1が、好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、より好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、さらに好ましくは直鎖の、1級若しくは2級のアルキル基である、<46>に記載の酸化物の製造方法。
R1が、好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、より好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、さらに好ましくは直鎖の、1級若しくは2級のアルキル基である、<46>に記載の酸化物の製造方法。
<48>
R1の炭素数が、6以上、8以上、10以上又は12以上であり、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である、<46>又は<47>に記載の酸化物の製造方法。
R1の炭素数が、6以上、8以上、10以上又は12以上であり、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である、<46>又は<47>に記載の酸化物の製造方法。
<49>
R2が、好ましくは脂肪族炭化水素基、より好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、さらに好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、よりさらに好ましくは直鎖の、1級若しくは2級のアルキル基である、<46>~<48>の何れか1に記載の酸化物の製造方法。
R2が、好ましくは脂肪族炭化水素基、より好ましくは直鎖又は分岐鎖の、1級又は2級の脂肪族炭化水素基、さらに好ましくは直鎖若しくは分岐鎖の、1級若しくは2級のアルキル基又はアルケニル基、よりさらに好ましくは直鎖の、1級若しくは2級のアルキル基である、<46>~<48>の何れか1に記載の酸化物の製造方法。
<50>
R2の炭素数が、6以上、8以上、10以上又は12以上であり、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である、<46>~<49>の何れか1に記載の酸化物の製造方法。
R2の炭素数が、6以上、8以上、10以上又は12以上であり、好ましくは36以下、より好ましくは22以下、さらに好ましくは18以下、よりさらに好ましくは14以下である、<46>~<49>の何れか1に記載の酸化物の製造方法。
<51>アルコール又はポリオキシアルキレンアルキルエーテルの、反応に使用する水に対する質量比が、好ましくは60/40以上、より好ましくは70/30以上、さらに好ましくは75/25以上であり、好ましくは95/5以下、より好ましくは90/10以下、さらに好ましくは85/15以下である<45>~<50>の何れか1に記載の酸化物の製造方法。
<52>前記組成物又はその反応液である液相への酸素の供給が、酸素含有ガスを液相に流通させることによって実施され、酸素含有ガスが好ましくは酸素ガス又は空気などの混合ガスである<45>~<51>の何れか1に記載の酸化物の製造方法。
<53>酸素含有ガス中の酸素濃度が好ましくは10体積%以上、より好ましくは50体積%以上、さらに好ましくは70体積%以上、よりさらに好ましくは90体積%以上、よりさらに好ましくは実質的に100体積%、よりさらに好ましくは100体積%である<52>に記載の酸化物の製造方法。
<54>反応温度が好ましくは50℃以上、より好ましくは60℃以上であり、好ましくは100℃以下、より好ましくは90℃以下、さらに好ましくは80℃以下である<45>~<53>の何れか1に記載の酸化物の製造方法。
<55>反応圧力が好ましくは0.09MPa以上、より好ましくは0.10MPa以上であり、好ましくは0.5MPa以下、より好ましくは0.2MPa以下、さらに好ましくは0.11MPa以下である<45>~<54>の何れか1に記載の酸化物の製造方法。
<56>触媒の使用量が、ポリオキシアルキレンアルキルエーテル100質量部に対するPt金属の質量として好ましくは0.1質量部以上、より好ましくは1質量部以上、さらに好ましくは3質量部以上、よりさらに好ましくは6質量部以上であり、好ましくは20質量部以下、より好ましくは15質量部以下、さらに好ましくは10質量部以下である<45>~<55>の何れか1に記載の酸化物の製造方法。
<57><41>~<44>の何れか1に記載の触媒のアルコール又はポリオキシアルキレンアルキルエーテルの脱水素酸化触媒としての使用。
実施例
実施例
以下の実施例及び比較例において、特に断らない限り、温度条件は20℃、撹拌条件は200rpm、圧力条件は常圧である。また、pHの測定にはADVANTEC社製pH試験紙「ロールタイプ UNIV」を用いた。
実施例1
<10%Pt-1%Bi/C触媒の調製>
工程1:Pt/Cスラリー(以下、液Aという)の調製:
1Lのビーカーに、10% Pt/C(Evonik Japan社製、Pt粒径14nm、水分含量59.2%)40gとイオン交換水500gを仕込み、固体がビーカーの底に沈殿しないように「FineスターラーF-202」(東京硝子器械株式会社製)を用いて約100rpmの条件にて撹拌した。
<10%Pt-1%Bi/C触媒の調製>
工程1:Pt/Cスラリー(以下、液Aという)の調製:
1Lのビーカーに、10% Pt/C(Evonik Japan社製、Pt粒径14nm、水分含量59.2%)40gとイオン交換水500gを仕込み、固体がビーカーの底に沈殿しないように「FineスターラーF-202」(東京硝子器械株式会社製)を用いて約100rpmの条件にて撹拌した。
工程2:ビスマスを含む酸性水溶液(以下、液Bという)の調製:
メカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた2Lのセパラブルフラスコに、硝酸ビスマス五水和物(和光純薬工業株式会社製)0.38gと2%酢酸水溶液808mLを仕込んだ。2%酢酸水溶液中のBi濃度は0.001Mである。ビスマス塩を完全に溶解させるため、超音波洗浄機「Fine超音波洗浄器FU-9H」(東京硝子器械株式会社製)で周波数38kHzの条件下にて5分間超音波をあてた。
メカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた2Lのセパラブルフラスコに、硝酸ビスマス五水和物(和光純薬工業株式会社製)0.38gと2%酢酸水溶液808mLを仕込んだ。2%酢酸水溶液中のBi濃度は0.001Mである。ビスマス塩を完全に溶解させるため、超音波洗浄機「Fine超音波洗浄器FU-9H」(東京硝子器械株式会社製)で周波数38kHzの条件下にて5分間超音波をあてた。
工程3に先立って、工程3で滴下を受けるセパラブルフラスコ内の液Bを、200rpmの条件下にてメカニカルスターラーで攪拌しながら20分間水素を86-129mL/分の条件にて流通させて水素置換を行った。
その後、撹拌及び水素流通下で酢酸を2mL添加した。酢酸添加後の溶液のpHは3であった。
その後、撹拌及び水素流通下で酢酸を2mL添加した。酢酸添加後の溶液のpHは3であった。
工程3:
セパラブルフラスコ内の液Bへの水素流通及び撹拌を継続しながら、ぜん動ポンプを用いてビーカー内の液Aを、セパラブルフラスコ内の液Bへ滴下した。滴下は、86-129mL/分の条件にて水素を液Bに流通しながら、液Bを撹拌しながら行った。滴下する液Aの滴下速度、滴下時間及び滴下を受ける液Bの温度は表1記載の通りであった。また、滴下後の溶液のpHは3であった。
セパラブルフラスコ内の液Bへの水素流通及び撹拌を継続しながら、ぜん動ポンプを用いてビーカー内の液Aを、セパラブルフラスコ内の液Bへ滴下した。滴下は、86-129mL/分の条件にて水素を液Bに流通しながら、液Bを撹拌しながら行った。滴下する液Aの滴下速度、滴下時間及び滴下を受ける液Bの温度は表1記載の通りであった。また、滴下後の溶液のpHは3であった。
滴下終了後、5分間熟成し、金属の還元状態を維持するためにイソプロピルアルコール(以下IPA)18mLを添加した。熟成及びIPA添加は液Bへの水素流通及び撹拌を継続しながら行った。IPA添加後、水素を止めて窒素を流通させながら減圧濾過を行った。この濾過は、減圧濾過用フィルターホルダー「ガラスタイプ KGS-90」(ADVANTEC社製)にメンブレンフィルター「PTFEメンブレンフィルターT020A142C」(ADVANTEC社製、孔サイズ0.2μm)をのせ、IPA添加後の溶液を流して吸引環境下に行い、固体を分離した。濾液のpHは3であった。
濾過後の固体はセパラブルフラスコに戻して、さらに1%IPA水溶液(300mL)を添加し、30分間撹拌洗浄した。その後、上記と同様にして濾過を行い、濾過で得られた固体を1時間程度で乾燥させ、10%Pt-1%Bi/C触媒を得た。尚、撹拌洗浄、濾過、乾燥の操作は窒素流通下で行った。また、洗浄後、乾燥前に行った濾過で得られた濾液のpHは3~4であった。
比較例1
<10%Pt-1%Bi/C触媒の調製2>
工程1:Pt/Cスラリー(以下、液Aという)の調製:
メカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた2Lのセパラブルフラスコに、10% Pt/C(Evonik Japan社製、水分含量59.2%)40gとイオン交換水500gを仕込んだ。
<10%Pt-1%Bi/C触媒の調製2>
工程1:Pt/Cスラリー(以下、液Aという)の調製:
メカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた2Lのセパラブルフラスコに、10% Pt/C(Evonik Japan社製、水分含量59.2%)40gとイオン交換水500gを仕込んだ。
工程2:ビスマスを含む酸性水溶液(以下、液Bという)の調製:
1Lのビーカーに、硝酸ビスマス五水和物(和光純薬工業株式会社製)0.38gと2%酢酸水溶液808mLを仕込んだ。2%酢酸水溶液中のBi濃度は0.001Mである。ビスマス塩を完全に溶解させるため、超音波洗浄機「Fine超音波洗浄器FU-9H」(東京硝子器械株式会社製)で周波数38kHzの条件下にて5分間超音波をあてた。
1Lのビーカーに、硝酸ビスマス五水和物(和光純薬工業株式会社製)0.38gと2%酢酸水溶液808mLを仕込んだ。2%酢酸水溶液中のBi濃度は0.001Mである。ビスマス塩を完全に溶解させるため、超音波洗浄機「Fine超音波洗浄器FU-9H」(東京硝子器械株式会社製)で周波数38kHzの条件下にて5分間超音波をあてた。
工程3に先立って、工程3で滴下を受けるセパラブルフラスコ内の液Aを、200rpmの条件下にてメカニカルスターラーで攪拌しながら20分間水素を86-129mL/分の条件にて流通させて水素置換を行った。その後、撹拌及び水素流通下で酢酸を2mL添加した。酢酸添加後の溶液のpHは3であった。
工程3:
セパラブルフラスコ内の液Aへの水素流通及び撹拌を継続しながら、ぜん動ポンプを用いてビーカー内の液Bを、セパラブルフラスコ内の液Aへ滴下した。滴下する液Bの滴下速度、滴下時間及び滴下を受ける液Aの温度は表1記載の通りであった。また、滴下後の溶液のpHは4であった。
セパラブルフラスコ内の液Aへの水素流通及び撹拌を継続しながら、ぜん動ポンプを用いてビーカー内の液Bを、セパラブルフラスコ内の液Aへ滴下した。滴下する液Bの滴下速度、滴下時間及び滴下を受ける液Aの温度は表1記載の通りであった。また、滴下後の溶液のpHは4であった。
滴下終了後、水素流通及び撹拌を継続しながら5分間熟成し、金属の還元状態を維持するためにイソプロピルアルコール(以下IPA)18mLを添加した。IPA添加後、水素を止めて窒素を流通させながら減圧濾過を行った。この濾過は、減圧濾過用フィルターホルダー「ガラスタイプ KGS-90」(ADVANTEC社製)にメンブレンフィルター「PTFEメンブレンフィルターT020A142C」(ADVANTEC社製、孔サイズ0.2μm)をのせ、IPA添加後の溶液を流して吸引環境下に行い、固体を分離した。濾液のpHは3~4であった。
濾過後の固体はセパラブルフラスコに戻して、さらに1%IPA水溶液(300mL)を添加し、30分間撹拌洗浄した。その後、上記と同様にして濾過を行い、濾過で得られた固体を1時間程度で乾燥させ、10%Pt-1%Bi/C触媒を得た。尚、撹拌洗浄、濾過、乾燥の操作は窒素流通下で行った。また、洗浄後、乾燥前に行った濾過で得られた濾液のpHは3~4であった。
実施例2
Pt担持量が5%のPt/C触媒を用い、また、IPA添加、濾過後に、中和、洗浄及び濾過をした以外は、実施例1と同様に工程を行って触媒を得た。なお、濾過は実施例1に記載の方法で行った。また、中和及び洗浄は次のように行った。
Pt担持量が5%のPt/C触媒を用い、また、IPA添加、濾過後に、中和、洗浄及び濾過をした以外は、実施例1と同様に工程を行って触媒を得た。なお、濾過は実施例1に記載の方法で行った。また、中和及び洗浄は次のように行った。
IPA添加、濾過後、濾別された固体をセパラブルフラスコに戻して0.05M NaHCO3水溶液300mLを添加し、30分間撹拌して、中和した。その後、濾過を行い、固体をセパラブルフラスコに戻して、さらにイオン交換水300mLを添加し、30分間撹拌し洗浄した。中和及び洗浄の操作は窒素流通下で行った。また、中和直後及び洗浄後の濾液のpHは7であった。
比較例2
Pt担持量が5%のPt/C触媒を用い、また、IPA添加、濾過後に、中和、洗浄及び濾過をした以外は、比較例1と同様の工程を行って触媒を得た。なお、濾過は比較例1に記載の方法で行った。また、中和及び洗浄は実施例2と同様の工程を行った。
Pt担持量が5%のPt/C触媒を用い、また、IPA添加、濾過後に、中和、洗浄及び濾過をした以外は、比較例1と同様の工程を行って触媒を得た。なお、濾過は比較例1に記載の方法で行った。また、中和及び洗浄は実施例2と同様の工程を行った。
触媒の性能評価を行う際に、イオン交換水の仕込み量を決める。そのために触媒中の含水率の測定が必要である。実施例1、比較例1、実施例2及び比較例2で得た触媒中の含水率を下記の方法で測定した。得られた結果を表1及び表2に記載する。
<触媒中の含水率の測定方法>
触媒中の含水率は、次のように触媒を乾燥して乾燥前後の触媒の重量を測定して求める。
触媒3g程度をシャーレ「フラットシャーレFS-90B」(VIDREX社製)に入れて重量を測る。その後、「VACUUM DRYING OVEN DRR420DA」(ADVANTEC社製)を使用して-80kPa~-100kPaまで減圧し、70℃で4時間減圧乾燥を行い、再び重量を測定する。重量減少分は触媒に含まれている水分であると仮定し、次式により含水率を求める。
(触媒中の含水率%)=100×(重量減少分g)/(乾燥前の触媒重量g)
触媒中の含水率は、次のように触媒を乾燥して乾燥前後の触媒の重量を測定して求める。
触媒3g程度をシャーレ「フラットシャーレFS-90B」(VIDREX社製)に入れて重量を測る。その後、「VACUUM DRYING OVEN DRR420DA」(ADVANTEC社製)を使用して-80kPa~-100kPaまで減圧し、70℃で4時間減圧乾燥を行い、再び重量を測定する。重量減少分は触媒に含まれている水分であると仮定し、次式により含水率を求める。
(触媒中の含水率%)=100×(重量減少分g)/(乾燥前の触媒重量g)
実施例1、比較例1、実施例2及び比較例2で得た触媒を用いて、以下の方法でエーテルカルボキシレートを製造し、触媒からの金属溶出量及びエーテルカルボキシレートの収率を測定した。測定結果を表1及び表2に示す。
<エーテルカルボキシレートの製造>
還流管、pHメーター「デジタルpHコントローラーFD-02」(東京硝子器械株式会社製)、溶存酸素濃度計「InPro6850i/12/220」(METTLER TOLEDO社製)及びメカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた500mLの7つ口フラスコに、原料であるポリオキシエチレンアルキルエーテル(ラウリルアルコールにエチレンオキシドを平均3.6mol付加したもの)265g、触媒を原料100質量部に対して触媒中のPt及びBiの金属量の合計が、実施例1と比較例1では6.4質量部、実施例2と比較例2では3.2質量部となる量、及びイオン交換水を下記の計算方法で得られる水仕込み量g、仕込んだ。
還流管、pHメーター「デジタルpHコントローラーFD-02」(東京硝子器械株式会社製)、溶存酸素濃度計「InPro6850i/12/220」(METTLER TOLEDO社製)及びメカニカルスターラー「テフロン(登録商標)製撹拌羽根三日月形」(ASONE社製、撹拌翼の幅75mm×高さ20mm×厚さ4mm)を備えた500mLの7つ口フラスコに、原料であるポリオキシエチレンアルキルエーテル(ラウリルアルコールにエチレンオキシドを平均3.6mol付加したもの)265g、触媒を原料100質量部に対して触媒中のPt及びBiの金属量の合計が、実施例1と比較例1では6.4質量部、実施例2と比較例2では3.2質量部となる量、及びイオン交換水を下記の計算方法で得られる水仕込み量g、仕込んだ。
<水仕込み量の計算方法>
原料に対する水の量が80.9:19.1となるようにして、次のように計算する。
(水仕込み量g)=191×(原料仕込み量g)/809-(触媒中の含水率%)×(触媒仕込み量g)
原料に対する水の量が80.9:19.1となるようにして、次のように計算する。
(水仕込み量g)=191×(原料仕込み量g)/809-(触媒中の含水率%)×(触媒仕込み量g)
上記7つ口フラスコ中に仕込んだ原料、触媒及び水を450rpmの条件下にてメカニカルスターラーで攪拌しながら窒素流通下70℃に昇温し、70℃に達して15分間は引き続き窒素を流通させた。その後酸素に切り替えて90mL/分の条件下にて8時間流通し、反応させ、対応するエーテルカルボキシレートを得た。
反応終了後、溶液を速やかに70℃で加圧濾過して触媒と分離した。加圧濾過は、濾過器をあらかじめ70℃に加温しておき、そこへ反応終了後の溶液(70℃)を流し込んで窒素を4kgf/cm2で圧入しながら行った。
この濾液を用いて触媒からの金属溶出量及びエーテルカルボキシレートの収率を下記の方法で求めた。
<金属溶出量>
下記の測定条件にてICP分析によりを行い、触媒からのPt及びBiの溶出量を測定した。
試薬
塩酸:原子吸光分析用、関東化学株式会社製
硝酸:原子吸光分析用、関東化学株式会社製
硫酸:精密分析用、和光純薬工業株式会社製
プラチナ標準液:原子吸光分析用標準液1000mg/L、関東化学株式会社製
ビスマス標準液:原子吸光分析用標準液1000mg/L、関東化学株式会社製
超純水 :ミリQ水、ミリポア社製
下記の測定条件にてICP分析によりを行い、触媒からのPt及びBiの溶出量を測定した。
試薬
塩酸:原子吸光分析用、関東化学株式会社製
硝酸:原子吸光分析用、関東化学株式会社製
硫酸:精密分析用、和光純薬工業株式会社製
プラチナ標準液:原子吸光分析用標準液1000mg/L、関東化学株式会社製
ビスマス標準液:原子吸光分析用標準液1000mg/L、関東化学株式会社製
超純水 :ミリQ水、ミリポア社製
試料溶液調製:
(1)試料0.1gを磁器るつぼに採取しヒーターで焼成後、王水(塩酸:硝酸=3:1)8mLを添加し、加熱溶解する。冷却後、濾過し、超純水で100mLにメスアップする。
(2)濾過残渣を濾紙ごと灰化し(途中で硫酸を適宜添加し550℃で完全に灰化)、(1)と同様に王水で溶解し、冷却後、濾過し、メスアップする。
(3)(1)で調製した液と(2)で調製した液のBi量及びPt量を測定して、それぞれ、合算する。
(4)(2)の濾過残渣は蛍光X線で定性分析を行い、Pt、Biが残っていないことを確認する。
(1)試料0.1gを磁器るつぼに採取しヒーターで焼成後、王水(塩酸:硝酸=3:1)8mLを添加し、加熱溶解する。冷却後、濾過し、超純水で100mLにメスアップする。
(2)濾過残渣を濾紙ごと灰化し(途中で硫酸を適宜添加し550℃で完全に灰化)、(1)と同様に王水で溶解し、冷却後、濾過し、メスアップする。
(3)(1)で調製した液と(2)で調製した液のBi量及びPt量を測定して、それぞれ、合算する。
(4)(2)の濾過残渣は蛍光X線で定性分析を行い、Pt、Biが残っていないことを確認する。
検量線溶液調製:
原子吸光分析用標準液(Pt及びBi:1000mg/L)を用いて、0.1~2.0mg/Lの検量線溶液を調製した。それぞれの溶液には試料と同程度(約8%)となるように王水を添加した。
原子吸光分析用標準液(Pt及びBi:1000mg/L)を用いて、0.1~2.0mg/Lの検量線溶液を調製した。それぞれの溶液には試料と同程度(約8%)となるように王水を添加した。
ICP測定条件
分析装置:サーモフィッシャーサイエンティフィック社製 iCAP 6500Duo
波長:Pt 214.423nm、Bi 223.061nm
RFパワー:1150W
クーラントガス流量:12L/min
ネブライザー流量:0.70L/min
補助ガス:0.5L/min
ポンプ流量:50rpm
分析装置:サーモフィッシャーサイエンティフィック社製 iCAP 6500Duo
波長:Pt 214.423nm、Bi 223.061nm
RFパワー:1150W
クーラントガス流量:12L/min
ネブライザー流量:0.70L/min
補助ガス:0.5L/min
ポンプ流量:50rpm
<エーテルカルボキシレート収率>
下記の測定条件にて、ガスクロマトグラフィー(GC)分析により、原料、中間体であるアルデヒド及びエーテルカルボキシレートのピーク面積を求めた。これら3成分のピーク面積の合計に対するエーテルカルボキシレートのピーク面積の割合をエーテルカルボキシレートの収率として求め、百分率で示した。
下記の測定条件にて、ガスクロマトグラフィー(GC)分析により、原料、中間体であるアルデヒド及びエーテルカルボキシレートのピーク面積を求めた。これら3成分のピーク面積の合計に対するエーテルカルボキシレートのピーク面積の割合をエーテルカルボキシレートの収率として求め、百分率で示した。
GC分析に使用する溶液の調製:
(1)濾液0.22gをスクリュー管に測り取り、イオン交換水と飽和食塩水をそれぞれ3mLずつ添加し、さらにジエチルエーテル10mLを添加し分層する。
(2)そのエーテル層2.5mLをスクリュー管に測り取ってジアゾメタンでメチルエステル化する。
(3)窒素を流通させてジアゾメタンを留去する。
(4)溶液を1.5 mLに濃縮してGC分析に使用する溶液とする。
(1)濾液0.22gをスクリュー管に測り取り、イオン交換水と飽和食塩水をそれぞれ3mLずつ添加し、さらにジエチルエーテル10mLを添加し分層する。
(2)そのエーテル層2.5mLをスクリュー管に測り取ってジアゾメタンでメチルエステル化する。
(3)窒素を流通させてジアゾメタンを留去する。
(4)溶液を1.5 mLに濃縮してGC分析に使用する溶液とする。
GC測定条件
装置:Agilent Technologies 19091A-102E(Agilent Technologies社製)
カラム:Ultra1 Methyl Siloxane(25.0m×200μm×0.33μm)
Injection temp.325℃
Detector temp.300℃
装置:Agilent Technologies 19091A-102E(Agilent Technologies社製)
カラム:Ultra1 Methyl Siloxane(25.0m×200μm×0.33μm)
Injection temp.325℃
Detector temp.300℃
温度プログラム
Initial temp.100℃
Initial time.5min.
Increasing rate 5℃/min.
Final temp.300℃
Final time.45min.
Injection volume 1.0μL
Split ratio 25:1
Total flow rate 28.1mL/min.(He)
Initial temp.100℃
Initial time.5min.
Increasing rate 5℃/min.
Final temp.300℃
Final time.45min.
Injection volume 1.0μL
Split ratio 25:1
Total flow rate 28.1mL/min.(He)
以上の結果を表1及び表2に示す。


実施例3~5
それぞれ表3に記載した条件に変えた以外は、実施例1と同様に工程を行って触媒を得た。また、実施例1と同様の方法でエーテルカルボキシレートを製造し、触媒からの金属溶出量及びエーテルカルボキシレートの収率を測定した。測定結果を表3に示す。
それぞれ表3に記載した条件に変えた以外は、実施例1と同様に工程を行って触媒を得た。また、実施例1と同様の方法でエーテルカルボキシレートを製造し、触媒からの金属溶出量及びエーテルカルボキシレートの収率を測定した。測定結果を表3に示す。
実施例6
実施例1と同様に工程を行って触媒を調製した。また、反応時間を14時間とした以外は実施例1と同様の方法でエーテルカルボキシレートを製造し、触媒からの金属溶出量及びエーテルカルボキシレートの収率を測定した。測定結果を表3に示す。なお、金属溶出量の測定は、下記の(1)および(2)以外は実施例1と同様に行った。
(1)実施例1の試料溶液調製(1)において、磁性るつぼを用いて試料を酸で加熱溶解する代わりに、専用の密閉容器を用いてマイクロウェーブ法にて試料を酸で処理した。
(2)ICP測定において、PerkinElmer, Inc.製のICP質量分析装置ELAN DRC IIを用いた。
実施例1と同様に工程を行って触媒を調製した。また、反応時間を14時間とした以外は実施例1と同様の方法でエーテルカルボキシレートを製造し、触媒からの金属溶出量及びエーテルカルボキシレートの収率を測定した。測定結果を表3に示す。なお、金属溶出量の測定は、下記の(1)および(2)以外は実施例1と同様に行った。
(1)実施例1の試料溶液調製(1)において、磁性るつぼを用いて試料を酸で加熱溶解する代わりに、専用の密閉容器を用いてマイクロウェーブ法にて試料を酸で処理した。
(2)ICP測定において、PerkinElmer, Inc.製のICP質量分析装置ELAN DRC IIを用いた。
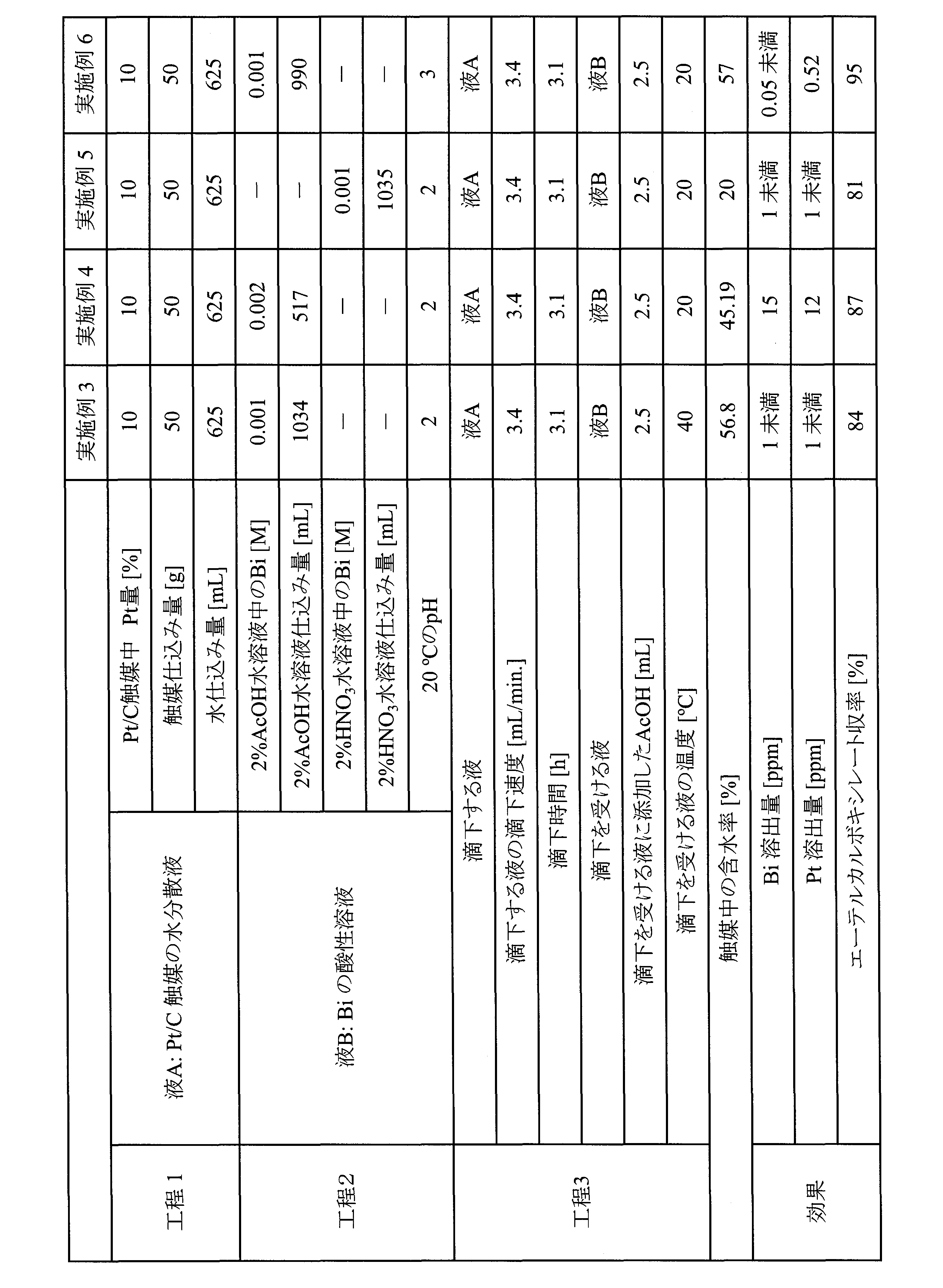
実施例7
実施例1で製造した触媒を用い、下記の方法でXPS(X-ray Photoelectron Spectroscopy)測定を行った。その結果を図1に示す。
結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値は157.8eVであった。
実施例1で製造した触媒を用い、下記の方法でXPS(X-ray Photoelectron Spectroscopy)測定を行った。その結果を図1に示す。
結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値は157.8eVであった。
比較例3
比較例1で製造した触媒を用いた以外は実施例7と同様にXPS測定を行った。そのBi4f軌道の光電子スペクトルを図1に示す。
結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値は158.7 eVであった。
比較例1で製造した触媒を用いた以外は実施例7と同様にXPS測定を行った。そのBi4f軌道の光電子スペクトルを図1に示す。
結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値は158.7 eVであった。
Pt上にBiが担持されている場合、Biは近傍に存在するPtから電子を受け取ることにより結合エネルギーが低下すると考えられる。すなわち、Biが凝集することなく、Pt上に均一に担持されるほど、前記ピークトップの結合エネルギー値は低下すると考えられる。
上記の通り、実施例1で調製した触媒は、比較例1で調製した触媒よりも、前記ピークトップの結合エネルギー値が小さいので、Pt上でBiが均一に担持されていると推測される。そして、Pt上でBiが均一に担持された結果、実施例1で調整した触媒は、活性が向上し、かつ金属溶出が抑制されたと考えられる。
<XPS測定方法>
触媒を銅プレートに接着したカーボン両面テープ上に散布したものを分析サンプルとした。分析に使用した装置と条件は以下の通りである。
触媒を銅プレートに接着したカーボン両面テープ上に散布したものを分析サンプルとした。分析に使用した装置と条件は以下の通りである。
分析に使用した装置と測定条件
・装置:PHI Quantera SXM (ULVAC-PHI Inc.)
・X線源:単色化AlKα 1486.6 eV, 25W, 15kV
・ビーム径:100μm
・測定範囲:500×500μm2
・Pass energy:280.0 eV (survey) 112.0 eV (narrow)
・Step:1.00 eV (survey) 0.20 eV (narrow)
・帯電補正:NeutralizerおよびAr+照射
・光電子取り出し角度:45°
・検出元素:C1s (5), O1s (10), Na1s (20), Pt4f (30), Bi4f (30)
・結合エネルギー値の補正は、炭素のCHに由来するC1s 284.8 eVで行った
・装置:PHI Quantera SXM (ULVAC-PHI Inc.)
・X線源:単色化AlKα 1486.6 eV, 25W, 15kV
・ビーム径:100μm
・測定範囲:500×500μm2
・Pass energy:280.0 eV (survey) 112.0 eV (narrow)
・Step:1.00 eV (survey) 0.20 eV (narrow)
・帯電補正:NeutralizerおよびAr+照射
・光電子取り出し角度:45°
・検出元素:C1s (5), O1s (10), Na1s (20), Pt4f (30), Bi4f (30)
・結合エネルギー値の補正は、炭素のCHに由来するC1s 284.8 eVで行った
Claims (16)
- 下記の工程1、工程2及び工程3を有する触媒の調製方法。
工程1:活性炭にPtを担持させた触媒の水分散液を調製する工程
工程2:Biをイオンの状態で含む水溶液を調製する工程
工程3:工程2で得られた水溶液に、工程1で得られた水分散液を添加する工程 - 工程3を還元雰囲気下に行う、請求項1の調製方法。
- 工程2で得られた水溶液が酸を含有する、請求項1又は2記載の触媒の調製方法。
- 酸が酢酸及び硝酸から選ばれる1種である、請求項3に記載の触媒の調製方法。
- 工程3における添加が連続添加又は分割添加で行われる、請求項1~4の何れか1項に記載の触媒の調製方法。
- 工程3において、添加を受けるBi水溶液の温度が10℃以上60℃以下である、請求項5記載の触媒の調製方法。
- 工程3において、添加速度が活性炭にPtを担持させた触媒の水分散液として1mL/分以上10mL/分以下である、請求項5又は6記載の触媒の調製方法。
- 工程3において、添加に要する時間が15分以上10時間以下である、請求項5~7の何れか1項に記載の触媒の調製方法。
- 水分散液中の活性炭にPtを担持させた触媒の濃度が、4質量%以上12質量%以下である、請求項1~8の何れか1項に記載の触媒の調製方法。
- 使用する酸の量が、Bi水溶液中、1質量%以上5質量%以下である、請求項3又は3を引用する請求項4~9の何れか1項に記載の触媒の調製方法。
- Bi水溶液中のBiの配合量が、0.0001M以上0.1M以下である、請求項1~10の何れか1項に記載の触媒の調製方法。
- Biイオンのイオン源が、硝酸ビスマス五水和物、酸化ビスマス、炭酸ビスマス及び水酸化ビスマスから選ばれる少なくとも1種である、請求項1~11の何れか1項に記載の触媒の調製方法。
- 請求項1~12の何れか1項に記載の調製方法により得られた触媒。
- XPSにより測定したBi4f軌道の光電子スペクトルにおいて結合エネルギー162~155eVの範囲のピークトップの結合エネルギー値が158.5eV以下、157.0eV以上である、PtとBiを活性炭に担持させた触媒。
- 請求項13又は14に記載の触媒の存在下、アルコール又はポリオキシアルキレンアルキルエーテルと水を含有する組成物に酸素を供給して、前記アルコール又はポリオキシアルキレンアルキルエーテルを脱水素酸化する、アルコールの酸化物又はポリオキシアルキレンアルキルエーテルの酸化物の製造方法。
- アルコール又はポリオキシアルキレンアルキルエーテルが下記一般式(1)又は一般式(2)で表される1種又は2種以上である、請求項15に記載の酸化物の製造方法。
R1O-H (1)
但し、一般式(1)においてR1は炭素数2以上40以下の脂肪族炭化水素基である。
R2O-(AO)n-H (2)
但し、一般式(2)においてR2は炭素数2以上40以下の炭化水素基、AOは炭素数2以上4以下のアルキレンオキシ基を表し、nはアルキレンオキシ基の付加モル数であり、1以上30以下の整数である。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201580066748.4A CN106999917A (zh) | 2014-12-18 | 2015-12-16 | 催化剂的调制方法 |
| US15/532,929 US10058857B2 (en) | 2014-12-18 | 2015-12-16 | Method for preparing catalyst |
| EP15870003.9A EP3235564A4 (en) | 2014-12-18 | 2015-12-16 | Method for preparing catalyst |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-255846 | 2014-12-18 | ||
| JP2014255846 | 2014-12-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016098801A1 true WO2016098801A1 (ja) | 2016-06-23 |
Family
ID=56126686
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/085175 WO2016098801A1 (ja) | 2014-12-18 | 2015-12-16 | 触媒の調製方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10058857B2 (ja) |
| EP (1) | EP3235564A4 (ja) |
| JP (1) | JP5957592B2 (ja) |
| CN (1) | CN106999917A (ja) |
| WO (1) | WO2016098801A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022196790A1 (ja) * | 2021-03-19 | 2022-09-22 | 花王株式会社 | 酸化物の製造方法、及びPt/Bi複合触媒の製造方法 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7448153B2 (ja) * | 2020-09-08 | 2024-03-12 | 株式会社クラレ | 触媒の製造方法、及び触媒、並びに3-ヒドロキシ-3-メチルブタン酸の製造方法 |
| CN115772143B (zh) * | 2021-09-08 | 2024-06-11 | 中国石油化工股份有限公司 | 一种制备2,5-呋喃二甲酸的方法 |
| CN115806537B (zh) * | 2021-09-13 | 2024-09-24 | 中国石油化工股份有限公司 | 一种制备糠酸的方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0272137A (ja) * | 1988-07-09 | 1990-03-12 | Degussa Ag | グルコン酸またはそのアルカリ金属塩の製造方法 |
| JPH04305538A (ja) * | 1991-03-06 | 1992-10-28 | A G Technol Kk | 含水素クロロメタン類の製造法 |
| JPH07173099A (ja) * | 1993-12-17 | 1995-07-11 | Mitsui Toatsu Chem Inc | グリオキシル酸の製造方法 |
| JPH09512286A (ja) * | 1995-02-20 | 1997-12-09 | ローヌ−プーラン・シミ | 置換4−ヒドロキシベンズアルデヒドの製造方法 |
| JPH10158227A (ja) * | 1996-12-03 | 1998-06-16 | Mitsui Chem Inc | N,n−ジメチルホルムアミドの製造方法 |
| JPH11279110A (ja) * | 1998-03-25 | 1999-10-12 | Mitsubishi Rayon Co Ltd | 2−置換カルボン酸類の製造法 |
| JP2002504427A (ja) * | 1998-02-25 | 2002-02-12 | モンサント カンパニー | 深く還元された酸化触媒および液相酸化反応を触媒するためのその使用 |
| JP2012000595A (ja) * | 2010-06-18 | 2012-01-05 | Ict:Kk | 排ガス浄化用触媒、その製造方法およびそれを用いた排ガス浄化方法 |
| WO2012169585A1 (ja) * | 2011-06-10 | 2012-12-13 | 花王株式会社 | ポリオキシエチレンアルキルエーテル酢酸の製造方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2597474B1 (fr) | 1986-01-30 | 1988-09-23 | Roquette Freres | Procede d'oxydation d'aldoses, catalyseur mis en oeuvre et produits ainsi obtenus. |
| US7737296B2 (en) * | 2005-08-08 | 2010-06-15 | Nippoh Chemicals Co., Ltd. | Method for producing 2-hydroxyester compound |
| JP5511369B2 (ja) * | 2009-12-28 | 2014-06-04 | 花王株式会社 | カルボン酸の製造方法 |
-
2015
- 2015-12-16 EP EP15870003.9A patent/EP3235564A4/en active Pending
- 2015-12-16 US US15/532,929 patent/US10058857B2/en active Active
- 2015-12-16 JP JP2015244919A patent/JP5957592B2/ja active Active
- 2015-12-16 CN CN201580066748.4A patent/CN106999917A/zh active Pending
- 2015-12-16 WO PCT/JP2015/085175 patent/WO2016098801A1/ja active Application Filing
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0272137A (ja) * | 1988-07-09 | 1990-03-12 | Degussa Ag | グルコン酸またはそのアルカリ金属塩の製造方法 |
| JPH04305538A (ja) * | 1991-03-06 | 1992-10-28 | A G Technol Kk | 含水素クロロメタン類の製造法 |
| JPH07173099A (ja) * | 1993-12-17 | 1995-07-11 | Mitsui Toatsu Chem Inc | グリオキシル酸の製造方法 |
| JPH09512286A (ja) * | 1995-02-20 | 1997-12-09 | ローヌ−プーラン・シミ | 置換4−ヒドロキシベンズアルデヒドの製造方法 |
| JPH10158227A (ja) * | 1996-12-03 | 1998-06-16 | Mitsui Chem Inc | N,n−ジメチルホルムアミドの製造方法 |
| JP2002504427A (ja) * | 1998-02-25 | 2002-02-12 | モンサント カンパニー | 深く還元された酸化触媒および液相酸化反応を触媒するためのその使用 |
| JPH11279110A (ja) * | 1998-03-25 | 1999-10-12 | Mitsubishi Rayon Co Ltd | 2−置換カルボン酸類の製造法 |
| JP2012000595A (ja) * | 2010-06-18 | 2012-01-05 | Ict:Kk | 排ガス浄化用触媒、その製造方法およびそれを用いた排ガス浄化方法 |
| WO2012169585A1 (ja) * | 2011-06-10 | 2012-12-13 | 花王株式会社 | ポリオキシエチレンアルキルエーテル酢酸の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3235564A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022196790A1 (ja) * | 2021-03-19 | 2022-09-22 | 花王株式会社 | 酸化物の製造方法、及びPt/Bi複合触媒の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016120484A (ja) | 2016-07-07 |
| JP5957592B2 (ja) | 2016-07-27 |
| US20170341070A1 (en) | 2017-11-30 |
| EP3235564A4 (en) | 2018-07-25 |
| US10058857B2 (en) | 2018-08-28 |
| CN106999917A (zh) | 2017-08-01 |
| EP3235564A1 (en) | 2017-10-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Kawawaki et al. | Creation of High‐Performance Heterogeneous Photocatalysts by Controlling Ligand Desorption and Particle Size of Gold Nanocluster | |
| JP5957592B2 (ja) | 触媒の調製方法 | |
| Liu et al. | Highly active iridium–rhenium catalyst condensed on silica support for hydrogenolysis of glycerol to 1, 3-propanediol | |
| Yu et al. | Reduced graphene oxide supported Au nanoparticles as an efficient catalyst for aerobic oxidation of benzyl alcohol | |
| Nie et al. | CO oxidation catalyzed by oxide-supported Au25 (SR) 18 nanoclusters and identification of perimeter sites as active centers | |
| Okumura et al. | A career in catalysis: Masatake Haruta | |
| Villa et al. | Selective oxidation of glycerol under acidic conditions using gold catalysts | |
| Wang et al. | MnO2 nanorod supported gold nanoparticles with enhanced activity for solvent-free aerobic alcohol oxidation | |
| Grass et al. | Colloidally synthesized monodisperse Rh nanoparticles supported on SBA-15 for size-and pretreatment-dependent studies of CO oxidation | |
| JP6017777B2 (ja) | アンモニア製造用触媒組成物の製造方法及びアンモニア製造方法 | |
| Lei et al. | Controllable decoration of palladium sub-nanoclusters on reduced graphene oxide with superior catalytic performance in selective oxidation of alcohols | |
| Kawawaki et al. | Supported,∼ 1-nm-Sized platinum clusters: controlled preparation and enhanced catalytic activity | |
| Oh et al. | Coverage of capping ligands determining the selectivity of multi-carbon products and morphological evolution of Cu nanocatalysts in electrochemical reduction of CO 2 | |
| JP2007203284A (ja) | パラジウム担持触媒及びその製造方法 | |
| JP2017001037A (ja) | アンモニア製造用触媒組成物、アンモニア製造用触媒組成物の製造方法及びアンモニア製造方法 | |
| RU2446009C1 (ru) | Способ приготовления платино-рутениевых электрокатализаторов | |
| JP2013034937A (ja) | 白金微粒子の製造方法、白金微粒子担持触媒の製造方法、及び浄化用触媒 | |
| WO2019172273A1 (ja) | 電極触媒 | |
| JP5062993B2 (ja) | 担持型触媒の製造およびその使用 | |
| JP5479939B2 (ja) | α,β−不飽和カルボン酸の製造方法 | |
| Redina et al. | High-performance Pt/CeO2-ZrO2 catalysts for selective hydrogenation of α, β-unsaturated aldehydes to unsaturated alcohols under mild reaction conditions:“Giant” hydrogen spillover behind the activity enhancement | |
| Mallat et al. | Partial oxidation of cinnamyl alcohol on bimetallic catalysts of improved resistance to self-poisoning | |
| JP5247081B2 (ja) | α,β−不飽和カルボン酸の製造方法 | |
| KR101933710B1 (ko) | 산화막 형성에 의해 갈바닉 치환반응성을 조절한 AuPd 나노입자를 포함하는 산화환원용 전극촉매 및 그의 제조방법 | |
| Campbell et al. | Alcohol Oxidations Using Reduced Polyoxovanadates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15870003 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015870003 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15532929 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |