WO2015181059A1 - Multiparticles safeguarded against ethanolic dose-dumping - Google Patents
Multiparticles safeguarded against ethanolic dose-dumping Download PDFInfo
- Publication number
- WO2015181059A1 WO2015181059A1 PCT/EP2015/061343 EP2015061343W WO2015181059A1 WO 2015181059 A1 WO2015181059 A1 WO 2015181059A1 EP 2015061343 W EP2015061343 W EP 2015061343W WO 2015181059 A1 WO2015181059 A1 WO 2015181059A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dosage form
- active ingredient
- pharmacologically active
- heteropolysaccharide
- alkyl cellulose
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1652—Polysaccharides, e.g. alginate, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/137—Arylalkylamines, e.g. amphetamine, epinephrine, salbutamol, ephedrine or methadone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/485—Morphinan derivatives, e.g. morphine, codeine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
Definitions
- the invention relates to an oral pharmaceutical dosage form providing resistance against dose dumping in aqueous ethanol and comprising a pharmacologically active ingredient, preferably an opioid, embedded in a matrix material,
- the matrix material comprises an alkyl cellulose, preferably ethyl cellulose, and a heteropolysaccharide, preferably xanthan gum; and
- the relative weight ratio of heteropolysaccharide to alkyl cellulose is within the range of from 1 : 20 to 20 : 1 ;
- the content of the alkyl cellulose is at least 10 wt.-%, relative to the total weight of the dosage form;
- the content of the alkyl cellulose in the dosage form is higher than the content of the heteropolysaccharide in the dosage form.
- a large number of pharmacologically active substances have a potential for being intentionally or unintentionally abused or misused, i.e. they can be used to produce effects which are not consistent with their intended use.
- opioids which exhibit an excellent efficacy in controlling severe to extremely severe pain are frequently abused to induce euphoric states similar to being intoxicated.
- active substances which have a psychotropic effect are abused accordingly.
- the corresponding pharmaceutical dosage forms such as pharmaceutical dosage forms or capsules can be taken with alcohol (oral abuse).
- oral abuse Alternatively, the dosage forms are crushed, for example ground by the abuser, the active substance is extracted from the thus obtained powder using a preferably aqueous liquid and after being optionally filtered through cotton wool or cellulose wadding, the resultant solution is administered parenterally, in particular intravenously.
- This type of dosage results in an even faster diffusion of the active substance compared to the oral abuse, with the result desired by the abuser, namely the kick.
- This kick or these intoxication-like, euphoric states are also reached if the powdered pharmaceutical dosage form is administered nasally, i.e. is sniffed.
- Dosage forms containing active ingredients having a high solubility in water usually have a high susceptibility to ethanolic dose dumping.
- aversive agents and/or antagonists have been proposed to incorporate in pharmaceutical dosage forms so that they only produce their aversive and/or antagonizing effects when the pharmaceutical dosage forms are tampered with.
- aversive agents and/or antagonists have been proposed to incorporate in pharmaceutical dosage forms in a manner so that they only produce their aversive and/or antagonizing effects when the pharmaceutical dosage forms are tampered with.
- the presence of such aversive agents is principally not desirable and there is a need to provide sufficient tamper -resistance without relying on aversive agents and/or antagonists.
- Another concept to prevent abuse relies on the mechanical properties of the pharmaceutical dosage forms, particularly an increased breaking strength (resistance to crushing).
- the major advantage of such pharmaceutical dosage forms is that comminuting, particularly pulverization, by conventional means, such as grinding in a mortar or fracturing by means of a hammer, is impossible or at least substantially impeded.
- the pulverization, necessary for abuse, of the pharmaceutical dosage forms by the means usually available to a potential abuser is prevented or at least complicated.
- Such pharmaceutical dosage forms are useful for avoiding drug abuse of the pharmacologically active ingredient contained therein, as they may not be powdered by conventional means and thus, cannot be administered in powdered form, e.g. nasally.
- tamper-resistant pharmaceutical dosage forms it can be referred to, e.g., WO 2005/016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097, WO 2006/082099, and WO2009/092601.
- monolithic dosage forms some formulation concepts are known which provide to some degree a controlled release of the drug substance even in ethanolic media. Further, however, monolithic dosage forms are not suitable for all patient groups, as they are required to be swallowed intact. Due to the big size of such formulations this is not possible for patients having difficulties in swallowing as e.g. the elderly and children. These patients have a high risk of choking on monolithic dosage forms. Pulverization of these dosage forms on the other hand solves the choking hazard, but endangers the patients by releasing a potentially toxic dose of the drug substance. The swallowing issue can be overcome by the use of multiparticulate dosage forms, e.g.
- MUPS multiple unit pellet system
- MUPS multiple unit pellet system
- Working examples of monolithic dosage forms contain hydrophilic polymer matrices, wherein control of drug release is achieved by a long diffusion way within the formulation.
- long diffusion ways do not exist due to the small size of the individual particles.
- a common technique to overcome this problem is the application of a functional barrier coating on top of the individual particle, e.g. ethylcellulose for diffusion control.
- ethylcellulose is alcohol soluble, these formulation approaches are not resistant against ethanolic dose dumping.
- US 2008/0085304 discloses robust sustained release formulations, solid dosage forms comprising robust sustained release formulations, and methods for making and using these formulations and solid dosage forms are provided.
- Robustness of the sustained release formulation is related to the particle size of the hydrophilic gum.
- Sustained release formulations resist dose-dumping when ingested with alcohol.
- the formulations are useful for treating a patient suffering from a condition, e.g., pain.
- the formulations comprise at least one drug.
- the drug is an opioid, e.g., oxymorphone.
- WO 2009/034541 relates to a solid dosage form for the controlled release of trimetazidine suitable for once-daily dosing, in which the dosage form exhibits a controlled in vitro release of trimetazidine in phosphate buffer at pH 6.8 of not less than about 75% after 16 hours when measured using USP Apparatus I at 100 rpm, thereby decreasing the incidence and severity of burst release or dose dumping.
- WO 2013/084059 relates to a pharmaceutical dosage form comprising a mixture in the form of an extended release matrix formulation, the mixture comprising at least: (1) at least one poly(s-caprolactone), and (2) at least one polyethylene oxide, and (3) at least one active agent.
- the dosage form is said to be tamper resistant and to provide extended release of the active agent.
- poly(s-caprolactone) is not a pharmacopeial excipient for oral use according to the Ph. Eur. and the USP, respectively.
- WO 2012/166474 relates to a solid dose form comprising a film coating composition encapsulating a core, wherein the core comprises an active ingredient comprising at least one of a pharmaceutical, veterinary, or nutraceutical active ingredient; the film coating composition comprises ethylcellulose and guar gum; and the guar gum is present in an amount greater than 5 wt% based on the weight of the guar gum and ethylcellulose.
- the solid dose form is said to provide controlled release of the active ingredient and to be ethanol resistant.
- Extended release tablets comprising a lipid matrix containing glyceryl (di)behenate (commercially available as Compritol ® 888 ATO) in which the active ingredient is embedded are said to not being susceptible to alcohol- related dose dumping.
- the drug substance is said to be released from the dosage form by diffusion, thereby leaving behind an in principle structurally intact tablet matrix.
- the remaining "washed-out" lipid tablet will remain visible in human stool after excretion. This observation (“ghosting”) is known to lead to increased complaints by patients and a reduced patient compliance.
- mechanical manipulation of the tablet e.g. dividing it to allow easier swallowing, leads to an accelerated drug release due to reduced diffusion ways eventually resulting in higher plasma concentrations of the drug substance including toxic levels.
- a first aspect of the invention relates to an oral pharmaceutical dosage form providing resistance against dose dumping in aqueous ethanol and comprising a pharmacologically active ingredient, preferably an opioid, embedded in a matrix material,
- the matrix material comprises an alkyl cellulose, preferably ethyl cellulose, and a heteropolysaccharide, preferably xanthan gum; and
- the relative weight ratio of heteropolysaccharide to alkyl cellulose is within the range of from 1 : 20 to 20 : 1 ;
- the content of the alkyl cellulose is at least 10 wt.-%, relative to the total weight of the dosage form;
- the content of the alkyl cellulose in the dosage form is higher than the content of the heteropolysaccharide in the dosage form.
- an oral pharmaceutical dosage form comprising a pharmacologically active ingredient, preferably an opioid, an alkyl cellulose and a heteropolysaccharide can be prepared, wherein the dosage form exhibits tamper resistance, especially in terms of resistance against dose-dumping of the pharmacologically active ingredient in aqueous ethanol.
- the content of the pharmacologically active ingredient, preferably the opioid in the dosage form and in the particles, respectively, can be optimized in order to provide the best compromise between tamper-resistance, disintegration time and drug release, drug load, processability (especially pharmaceutical dosage formability) and patient compliance.
- the dosage forms provide a retarded release when the release medium additionally contains ethanol compared to the release in aqueous medium not containing ethanol. This result was completely unexpected because of the good solubility of alkyl celluloses, especially ethylcellulose in ethanol.
- the term "pharmaceutical dosage form” and “dosage form”, respectively, refers to a pharmaceutical entity that comprises a pharmacologically active ingredient, preferably an opioid, and which is actually administered to, or taken by, a patient. It may be compressed or molded in its manufacture, and it may be of almost any size, shape, weight, and color.
- the dosage form is preferably solid or semisolid.
- dosage forms according to the invention include, but are not limited to, tablets, capsules, pills, granules, pellets, sachets and effervescent, powders, and the like.
- the dosage form is a filled capsule or a tablet.
- the capsule can be a hard or soft gelatin capsule.
- the dosage form according to the invention is particulate.
- the dosage form is preferably comprises a multitude of particles or granules.
- An advantage of particulate dosage forms is that the particles may be mixed in different amounts to thereby produce dosage forms of different strengths.
- the dosage form according to the invention can be regarded as a MUPS formulation (multiple unit pellet system).
- the dosage form according to the invention contains all ingredients in a dense compact unit which in comparison to capsules has a comparatively high density.
- the dosage forms according to the invention preferably comprise subunits having different morphology and properties, namely drug-containing particles and an outer matrix material, wherein the particles form a discontinuous phase within the outer matrix material.
- the constituents of the outer matrix material are preferably different from the constituents of the drug-containing particles.
- the outer matrix material neither contains a pharmacologically active ingredient nor an alkyl cellulose nor a heteropolysaccharide.
- the particles typically have mechanical properties that differ from the mechanical properties of the outer matrix material.
- the particles can preferably be visualized by conventional means such as solid state nuclear magnetic resonance spectroscopy, raster electron microscopy, terahertz spectroscopy and the like.
- the dosage form according to the invention is monolithic.
- monolithic preferably means that the dosage form is formed or composed of material without joints or seams or consists of or constitutes a single unit.
- the dosage form according to the invention has preferably a total weight in the range of 0.01 to 1.5 g, more preferably in the range of 0.05 to 1.2 g, still more preferably in the range of 0.1 g to 1.0 g, yet more preferably in the range of 0.2 g to 0.9 g, and most preferably in the range of 0.3 g to 0.8 g.
- the total weight of the dosage form is within the range of 350+300 mg, more preferably 350+250 mg, still more preferably 350+200 mg, yet more preferably 350+150 mg, most preferably 350+100 mg, and in particular 350+50 mg.
- the total weight of the dosage form is within the range of 500+450 mg, more preferably 500+300 mg, still more preferably 500+200 mg, yet more preferably 500+150 mg, most preferably 500+100 mg, and in particular 500+50 mg.
- the total weight of the dosage form is within the range of 600+450 mg, more preferably 600+300 mg, still more preferably 600+200 mg, yet more preferably 600+150 mg, most preferably 600+100 mg, and in particular 600+50 mg.
- the dosage form according to the invention is a filled capsule.
- Dosage forms of this embodiment preferably have a lengthwise extension (longitudinal extension) of about 4 mm to about 30 mm, more preferably about 6 mm to about 25 mm, most preferably about 8 mm to about 23 mm, and in particular about 10 mm to about 20 mm; and an internal diameter in the range of about 1 mm to about 20 mm, more preferably about 3 mm to about 17 mm, most preferably about 5 mm to about 15 mm, an in particular about 7 mm to about 13 mm.
- the dosage form according to the invention is a round dosage form.
- Dosage forms of this embodiment preferably have a diameter in the range of about 1 mm to about 30 mm, more preferably about 2 mm to about 25 mm, most preferably about 5 mm to about 23 mm, and in particular about 7 mm to about 13 mm; and a thickness in the range of about 1.0 mm to about 12 mm, more preferably about 2.0 mm to about 10 mm, most preferably about 3.0 mm to about 9.0 mm, and in particular about 4.0 mm to about 8.0 mm.
- the dosage form according to the invention is an oblong dosage form.
- Dosage forms of this embodiment preferably have a lengthwise extension (longitudinal extension) of about 1 mm to about 30 mm, more preferably about 2 mm to about 25 mm, most preferably about 5 mm to about 23 mm, and in particular about 7 mm to about 20 mm; a width in the range of about 1 mm to about 30 mm, more preferably about 2 mm to about 25 mm, most preferably about 5 mm to about 23 mm, and in particular about 7 mm to about 13 mm; and a thickness in the range of about 1.0 mm to about 12 mm, more preferably about 2.0 mm to about 10 mm, most preferably about 3.0 mm to about 9.0 mm, and in particular about 4.0 mm to about 8.0 mm.
- the dosage form according to the invention when it is monolithic, it preferably has an extension in any direction of at least 2.0 mm, more preferably at least 2.5 mm, still more preferably at least 3.0 mm, yet more preferably at least 3.5 mm, even more preferably at least 4.0 mm, most preferably at least 4.5 mm and in particular at least 5.0 mm.
- the dosage form or the particles if the dosage form is in a particulate form may optionally comprise a coating, e.g. a cosmetic coating.
- the coating is preferably applied after formation of the pharmaceutical dosage form.
- the coating may be applied prior to or after the curing process.
- an alkyl cellulose such as ethyl cellulose and/or a heteropolysaccharide such as xanthan gum or guar gum, are preferably not contained in a coating which may be applied to the dosage form and the particles, respectively.
- the dosage form is not coated and/or when the dosage form is particulate, the particles are not coated.
- the dosage forms according to the invention are film coated with conventional film coating compositions.
- Suitable coating materials are commercially available, e.g. under the trademarks Opadry ® and Eudragit ® .
- suitable materials include cellulose esters and cellulose ethers, such as methylcellulose (MC), hydroxypropylmethylcellulose (HPMC), hydroxypropylcellulose (HPC), hydroxyethylcellulose (HEC), sodium carboxymethylcellulose (Na-CMC), poly(meth)acrylates, such as aminoalkylmethacrylate copolymers, methacrylic acid methylmethacrylate copolymers, methacrylic acid methylmethacrylate copolymers; vinyl polymers, such as polyvinylpyrrolidone, polyvinyl alcohol, polyvinylacetate; and natural film formers.
- MC methylcellulose
- HPMC hydroxypropylmethylcellulose
- HPC hydroxypropylcellulose
- HEC hydroxyethylcellulose
- Na-CMC sodium carboxymethylcellulose
- poly(meth)acrylates such as aminoalkylmethacrylate copolymers, methacrylic acid methylmethacrylate copolymers, methacrylic acid methylmeth
- the coating can be resistant to gastric juices and dissolve as a function of the pH value of the release environment. By means of this coating, it is possible to ensure that the dosage form according to the invention passes through the stomach undissolved and the active compound is only released in the intestines.
- the coating which is resistant to gastric juices preferably dissolves at a pH value of between 5 and 7.5.
- the coating can also be applied e.g. to improve the aesthetic impression and/or the taste of the dosage forms and the ease with which they can be swallowed. Coating the dosage forms according to the invention can also serve other purposes, e.g. improving stability and shelf-life.
- Suitable coating formulations comprise a film forming polymer such as, for example, polyvinyl alcohol or hydroxypropyl methylcellulose, e.g. hypromellose, a plasticizer such as, for example, a glycol, e.g. propylene glycol or polyethylene glycol, an opacifier, such as, for example, titanium dioxide, and a film smoothener, such as, for example, talc.
- Suitable coating solvents are water as well as organic solvents.
- Coated pharmaceutical dosage forms according to the invention are preferably prepared by first making the cores and subsequently coating said cores using conventional techniques, such as coating in a coating pan.
- the term "particle” refers to a discrete mass of material that is solid, e.g. at 20°C or at room temperature or ambient temperature.
- a particle is solid at 20 C.
- the particles are monoliths.
- the pharmacologically active ingredient which is preferably an opioid
- the alkyl cellulose which is preferably ethyl cellulose
- the heteropolysaccharide which is preferably xanthan gum
- the pharmacologically active ingredient is preferably an opioid
- the alkyl cellulose which is preferably ethyl cellulose
- the heteropolysaccharide which is preferably xanthan gum
- the pharmacologically active ingredient which is preferably an opioid
- the alkyl cellulose which is preferably ethyl cellulose
- the heteropolysaccharide which is preferably xanthan gum
- the dosage form When the dosage form is particulate, it preferably comprises a multitude i.e. plurality of particles containing pharmacologically active ingredient (drug-containing particles) and may optionally further comprise particles not containing any pharmacologically active ingredient (drug-free particles).
- drug-containing particles pharmacologically active ingredient
- drug-free particles particles not containing any pharmacologically active ingredient
- all particles are drug-containing particles.
- the particles are not film coated.
- the dosage form preferably comprises at least 2, more preferably at least 4, still more preferably at least 6, yet more preferably at least 8, even more preferably at least 10, most preferably at least 15 and in particular at least 20 or at least 100 or at least 1000 drug-containing particles.
- the dosage form preferably comprises at most 10, more preferably at most 9, still more preferably at most 8, yet more preferably at most 7, even more preferably at most 6, most preferably at most 5, and in particular at most 4 or 3 or 2 drug-containing particles.
- the particles are preferably of macroscopic size, typically the average diameter is within the range of from 100 ⁇ to 5,000 ⁇ , preferably 200 ⁇ to 4,000 ⁇ , more preferably 300 ⁇ to 3,000 ⁇ , still more preferably 400 ⁇ to 2,000 ⁇ , most preferably 500 ⁇ to 1,500 ⁇ , and in particular 500 ⁇ to 1,000 ⁇ .
- the particles in the dosage form have an average particle size of at least 50 ⁇ , more preferably at least 100 ⁇ , still more preferably at least 150 ⁇ or at least 200 ⁇ , yet more preferably at least 250 ⁇ or at least 300 ⁇ , most preferably at least 400 ⁇ or at least 500 ⁇ , and in particular at least 550 ⁇ or at least 600 ⁇ .
- the particles in the dosage form have an average particle size of at least 700 ⁇ , more preferably at least 800 ⁇ and most preferably at least 900 ⁇ .
- the dosage forms according to the invention comprise particles as a discontinuous phase, i.e. the particles form a discontinuous phase in an outer matrix material which in turn preferably forms a continuous phase.
- discontinuous means that not each and every particle is in intimate contact with another particle but that the particles are at least partially separated from one another by the outer matrix material in which the particles are embedded.
- the particles preferably do not form a single coherent mass within the dosage forms according to the invention.
- the dosage form according to the invention when the dosage form according to the invention is particulate, the dosage form does not contain an outer matrix material.
- the dosage form preferably is a filled capsule.
- the content of the particles in the dosage forms according to the invention is at most 95 wt.-%, more preferably at most 90 wt.-%, still more preferably at most 85 wt.-%, yet more preferably at most 80 wt.-%, most preferably at most 75 wt.-% and in particular at most 70 wt.-%, based on the total weight of the dosage forms.
- the content of the particles in the dosage forms according to the invention is at least 10 wt.-%, at least 15 wt.-%, at least 20 wt.-% or at least 25 wt.-%; more preferably at least 30 wt.-%, at least 35 wt.-%, at least 40 wt.-% or at least 45 wt.-%; most preferably at least 50 wt.-%, at least 55 wt.-%, at least 60 wt.-% or at least 65 wt.-%; and in particular at least 70 wt.-%, at least 75 wt.-%, at least 80 wt.- % or at least 85 wt.-%; based on the total weight of the dosage form.
- the shape of the particles is not particularly limited.
- the particles are manufactured by granulation, preferably wet, dry or fluid bed granulation.
- the particles preferably have an irregular shape.
- the particles preferably have a particle size in the range of from 300 ⁇ to 5 mm, more preferably 400 ⁇ to 4 mm, still more preferably 500 ⁇ to 3 mm, yet more preferably 600 ⁇ to 2 mm, most preferably 700 ⁇ to 1.5 mm and in particular 850 ⁇ to 1.25 mm.
- the dosage form according to the invention is particulate and when the particles are manufactured by granulation, preferably the dosage form is a filled capsule.
- the particles are manufactured by hot-melt extrusion.
- the particles preferably are generally cylindrical in shape.
- the diameter of such particles is therefore the diameter of their circular cross section.
- the cylindrical shape is caused by the extrusion process according to which the diameter of the circular cross section is a function of the extrusion die and the length of the cylinders is a function of the cutting length according to which the extruded strand of material is cut into pieces of preferably more or less predetermined length.
- Preferred particles manufactured by hot-melt extrusion have an average length and average diameter of about 1,000 ⁇ or less.
- the "length" of particles is the dimension of the particles that is parallel to the direction of extrusion.
- the minimum average length of the particles is determined by the cutting step and may be, e.g. 4.0 mm, 3.0 mm, 2.0 mm, 2.5 mm, 2.0 mm, 1.5 mm, 1.0 mm, 0.9 mm, 0.8 mm, 0.7 mm, 0.6 mm, 0.5 mm, 0.4 mm, 0.3 mm or 0.2 mm.
- the "diameter" of particles is the largest dimension that is perpendicular to the direction of extrusion.
- the particles preferably have an average diameter in the range of 200 to 1500 ⁇ , more preferably 400 to 800 ⁇ , still more preferably 450 to 700 ⁇ , yet more preferably 500 to 650 ⁇ , e.g. about 500 to 600 ⁇ .
- the particles when they have been manufactured by hot-melt extrusion, they have an average length in the range of 500 to 5000 ⁇ , more preferably 750 to 4600 ⁇ , still more preferably 1000 to 4200 ⁇ , yet more preferably 1250 to 3800 ⁇ , even more preferably 1500 to 3400 ⁇ , most preferably 1750 to 3200 ⁇ and in particular 2000 to 3000 ⁇ .
- particles manufactured by hot-melt extrusion have an average length in the range of 200 to 1000 ⁇ , more preferably 400 to 800 ⁇ , still more preferably 450 to 700 ⁇ , yet more preferably 500 to 650 ⁇ , e.g. about 500 to 600 ⁇ .
- the size of particles may be determined by any conventional procedure known in the art, e.g. laser light scattering, sieve analysis, light microscopy or image analysis.
- the plurality of particles that is contained in the dosage form according to the invention has an arithmetic average weight, in the following referred to as "aaw", wherein at least 70%, more preferably at least 75%, still more preferably at least 80%, yet more preferably at least 85%, most preferably at least 90% and in particular at least 95% of the individual particles contained in said plurality of particles has an individual weight within the range of aaw+30%, more preferably aaw+25%, still more preferably aaw+20%, yet more preferably aaw+15%, most preferably aaw+10%, and in particular aaw+5%.
- aaw arithmetic average weight
- the dosage form according to the invention contains a plurality of 100 particles and aaw of said plurality of particles is 1.00 mg, at least 75 individual particles (i.e. 75%) have an individual weight within the range of from 0.70 to 1.30 mg (1.00 mg +30%).
- the particles each have a weight of less than 20 mg, more preferably less than 18 mg, still more preferably less than 16 mg, yet more preferably less than 14 mg, even more preferably less than 12 mg or less than 10 mg, most preferably less than 8 mg, and in particular less than 6 or 4 mg.
- all individual particles each preferably have a weight of from 1 to 19 mg, more preferably 1.5 to 15 mg, still more preferably 2.0 to 12 mg, yet more preferably 2.2 to 10 mg, even more preferably 2.5 to 8 mg, most preferably 2.8 to 6 mg and in particular 3 to 5 mg.
- the particles, more preferably the drug-containing particles each have a weight of 20 mg or more.
- all individual particles preferably each have a weight of at least 30 mg, more preferably at least 40 mg, still more preferably at least 50 mg, most preferably at least 60 mg and in particular at least 100 mg.
- all individual particles each have a weight of from 20 to 1000 mg, more preferably 30 to 800 mg, still more preferably 40 to 600 mg, yet more preferably 50 to 400 mg, even more preferably 60 to 200 mg, most preferably 70 to 150 mg and in particular 80 to 120 mg.
- the particles of the dosage form, more preferably the drug-containing particles of the dosage form preferably each have an extension in any given direction of at least 2.0 mm or 3.0 mm and have a weight of at least 20 mg.
- the particles When the dosage form is particulate, the particles may be e.g. loosely contained in a capsule, or the particles may be incorporated into an outer matrix material. From a macroscopic perspective, the outer matrix material preferably forms a continuous phase in which the particles are embedded as discontinuous phase.
- the outer matrix material is preferably a homogenous coherent mass, preferably a homogeneous mixture of solid constituents, in which the particles are embedded thereby spatially separating the particles from one another. While it is possible that the surfaces of particles are in contact or at least in very close proximity with one another, the plurality of particles preferably cannot be regarded as a single continuous coherent mass within the dosage form.
- the dosage form according to the invention preferably comprises the particles as volume element(s) of a first type in which the pharmacologically active ingredient, the alkyl cellulose and the heteropolysaccharide are contained, and the outer matrix material as volume element of a second type differing from the material that forms the particles, preferably containing neither pharmacologically active ingredient, nor alkyl cellulose, nor heteropolysaccharide.
- the relative weight ratio of particles to outer matrix material is not particularly limited.
- said relative weight ratio is within the range of 1 : 1.00+0.75, more preferably 1 : 1.00+0.50, still more preferably 1 : 1.00+0.40, yet more preferably 1 : 1.00+0.30, most preferably 1 : 1.00+0.20, and in particular 1 : 1.00+0.10.
- the content of the outer matrix material is at least 2.5 wt.-%, at least 5 wt.-%, at least 10 wt.-%, at least 15 wt.-%, at least 20 wt.-%, at least 25 wt.-%, at least 30 wt.-%, at least 35 wt.-% or at least 40 wt.-%; more preferably at least 45 wt.-% or at least 50 wt.-%; still more preferably at least 55 wt.-% or at least 60 wt.-%; yet more preferably at least 65 wt.-% or at least 70 wt.-%; most preferably at least 75 wt.-% or at least 80 wt.-%; and in particular at least 85 wt.-% or at least 90 wt.-%; based on the total weight of the dosage form.
- the content of the outer matrix material is at most 90 wt.-% or at most 85 wt.-%; more preferably at most 80 wt.-% or at most 75 wt.-%; still more preferably at most 70 wt.-% or at most 65 wt.-%; yet more preferably at most 60 wt.-% or at most 55 wt.-%; most preferably at most 50 wt.-% or at most 45 wt.-%; and in particular at most 40 wt.-% or at most 35 wt.-%; based on the total weight of the dosage form.
- the outer matrix material is a mixture, preferably a homogeneous mixture of at least two different constituents, more preferably of at least three different constituents.
- all constituents of the outer matrix material are homogeneously distributed in the continuous phase that is formed by the outer matrix material.
- the outer matrix material is also provided in particulate form, i.e. in the course of the manufacture of the dosage forms according to the invention, the constituents of the outer matrix material are preferably processed into particles, subsequently mixed with the particles that contain the pharmacologically active ingredient, which is preferably an opioid, the alkyl cellulose and the heteropolysaccharide, and then compressed into the dosage forms.
- the pharmacologically active ingredient which is preferably an opioid, the alkyl cellulose and the heteropolysaccharide
- the average size of the particles of the outer matrix material is within the range of +60%, more preferably +50%, still more preferably +40%, yet more preferably +30%, most preferably +20%, and in particular +10% of the average size of the particles that contain the pharmacologically active ingredient, which is preferably an opioid, the alkyl cellulose and the heteropolysaccharide.
- the particles of the outer matrix material can be manufactured by conventional methods for the preparation of aggregates and agglomerates from powder mixtures such as granulating and compacting.
- the mixture of all constituents of the outer matrix material is blended and pre- compacted thereby yielding a pre-compacted outer matrix material.
- the outer matrix material preferably does not contain any pharmacologically active ingredient.
- the outer matrix material comprises a filler or a binder.
- filler/binder refers to any excipient that is suitable as filler, binder or both.
- the outer matrix material preferably comprises a filler/binder.
- Preferred fillers are selected from the group consisting of silicium dioxide (e.g. Aerosil ® ), microcrystalline cellulose (e.g. Avicel ® , Elcema ® , Emocel ® , ExCel ® , Vitacell ® ); cellulose ether (e.g. Natrosol ® , Klucel ® , Methocel ® , Blanose ® , Pharmacoat ® , Viscontran ® ); mannitol; dextrines; dextrose; calciumhydrogen phosphate (e.g. Emcompress ® ); maltodextrine (e.g. Emdex ® ); lactose (e.g.
- silicium dioxide e.g. Aerosil ®
- microcrystalline cellulose e.g. Avicel ® , Elcema ® , Emocel ® , ExCel ® , Vitacell ®
- cellulose ether e.g
- Fast-Flow Lactose ® ; Ludipress ® ' Pharmaceutical dosage formtose ® , Zeparox ® ); polyvinylpyrrolidone (PVP) (e.g. Kollidone ® , Polyplasdone ® , Polydone ® ); saccharose (e.g. Nu-Tab ® , Sugar Tab ® ); magnesium salts (e.g. MgC0 3 , MgO, MgSi(3 ⁇ 4); starches and pretreated starches (e.g. Prejel ® , Primotab ® ET, Starch ® 1500).
- the outer matrix material comprises a glidant such as silicium dioxide.
- the content of the filler/binder or mixture of fillers binders in the outer matrix material is within the range of from 1 to 99 wt.-%, more preferably 25 to 90 wt.-%, based on the total weight of outer matrix material.
- the filler/binder is contained in the outer matrix material but not in the drug-containing particles of the dosage form according to the invention.
- the outer matrix material comprises a diluent or lubricant, preferably selected from the group consisting of calcium stearate; magnesium stearate; glycerol monobehenate (e.g. Compritol ® ); Myvatex ® ; Precirol ® ; Precirol ® Ato5; sodium stearylfumarate (e.g. Pruv ® ); and talcum.
- a diluent or lubricant preferably selected from the group consisting of calcium stearate; magnesium stearate; glycerol monobehenate (e.g. Compritol ® ); Myvatex ® ; Precirol ® ; Precirol ® Ato5; sodium stearylfumarate (e.g. Pruv ® ); and talcum.
- Magnesium stearate is particularly preferred.
- the content of the lubricant in the outer matrix material is at most 10.0 wt.-%, more preferably at most 7.5 wt.-%, still more preferably at most 5.0 wt.-%, yet more preferably at most 2.0 wt.-%, even more preferably at most 1.0 wt.-%, and most preferably at most 0.5 wt.-%, based on the total weight of the outer matrix material and based on the total weight of the dosage form.
- the outer matrix material comprises a combination of filler/binder and lubricant.
- the outer matrix material of the dosage forms according to the invention may additionally contain other excipients that are conventional in the art, e.g. diluents, binders, granulating aids, colorants, flavor additives, glidants, wet-regulating agents and disintegrants.
- excipients that are conventional in the art, e.g. diluents, binders, granulating aids, colorants, flavor additives, glidants, wet-regulating agents and disintegrants.
- diluents e.g. diluents, binders, granulating aids, colorants, flavor additives, glidants, wet-regulating agents and disintegrants.
- the pharmacologically active ingredient preferably an opioid
- the matrix material preferably dispersed in the matrix material.
- matrix preferably refers to the matrix material comprising the embedded pharmacologically active ingredient and the term “matrix material” refers to a preferably homogeneous, intimate mixture of the alkyl cellulose, the heteropolysaccharide and optionally present excipients.
- the pharmacologically active ingredient more preferably the opioid is embedded in a matrix material consisting of an alkyl cellulose, a heteropolysaccharide and optional excipients approved for oral use according to the Ph. Eur. and the USP, respectively.
- the matrix comprising the alkyl cellulose and the heteropolysaccharide provides resistance against dose dumping in aqueous ethanol.
- the dosage form provides prolonged release of the pharmacologically active ingredient.
- the matrix comprising the alkyl cellulose and the heteropolysaccharide provides prolonged release of the pharmacologically active ingredient embedded therein.
- the matrix provides resistance against dose dumping in aqueous ethanol and/or the matrix provides prolonged release of the pharmacologically active ingredient, preferably the opioid.
- the particles preferably comprise the matrix material and at least a portion of the total amount of the pharmacologically active ingredient that is contained in the pharmaceutical dosage form.
- the particles comprise the total amount of the pharmacologically active ingredient that is contained in the dosage form.
- the pharmacologically active ingredient, the alkyl cellulose and the heteropolysaccharide are intimately homogeneously distributed within the particles so that the particles do not contain any segments where either pharmacologically active ingredient is present in the absence of the alkyl cellulose and/or the heteropolysaccharide or the alkyl cellulose is present in the absence of the pharmacologically active ingredient and/or the heteropolysaccharide or the heteropolysaccharide is present in the absence of the pharmacologically active ingredient and/or the alkyl cellulose.
- the dosage form according to the invention can be regarded as a MUPS formulation which preferably comprises drug-containing particles and an outer matrix material
- the outer matrix material is not a constituent of the matrix material and, thus, is to be distinguished from the matrix material of the dosage form according to the invention.
- the matrix material in which the pharmacologically active ingredient, preferably the opioid is embedded preferably forms the body of the dosage form.
- the pharmacologically active ingredient, the alkyl cellulose and the heteropolysaccharide are intimately homogeneously distributed within the monolithic dosage form so that the monolithic dosage form does not contain any segments where either pharmacologically active ingredient is present in the absence of the alkyl cellulose and/or the heteropolysaccharide or the alkyl cellulose is present in the absence of the pharmacologically active ingredient and/or the heteropolysaccharide or the heteropolysaccharide is present in the absence of the pharmacologically active ingredient and/or the alkyl cellulose.
- the relative weight ratio of the pharmacologically active ingredient, preferably the opioid to the matrix material is in the range of from 1 : 1 to 1 : 50, more preferably 1 : 1.5 to 1 : 45, still more preferably 1 : 2 to 1 : 40, even more preferably 1 : 2.5 to 1 : 35, yet more preferably 1 : 3 to 1 : 30, most preferably 1 : 3.5 to 1 : 25, and in particular 1 : 4 to 1 : 20.
- the total content of the matrix material is at least 35 wt.-%, more preferably at least 40 wt.-%, still more preferably at least 45 wt.-%, even more preferably at least 50 wt.-%, yet more preferably at least 55 wt.-%, most preferably at least 60 wt.-%, and in particular at least 65 wt.-%, relative to the total weight of the dosage form.
- the total content of the matrix material is at most 95 wt.-%, more preferably at most 90 wt.-%, still more preferably at most 85 wt.-%, most preferably at most 80 wt.-%, and in particular at most 75 wt.-%, relative to the total weight of the dosage form.
- the total content of the matrix material is within the range of from 35 to 95 wt.-%, more preferably 45 to 85 wt.-%, most preferably 55 to 80 wt.-%, and in particular 65 to 75 wt.-%, relative to the total weight of the dosage form.
- the total content of alkyl cellulose and heteropolysaccharide is at least 50 wt.-%, more preferably at least 60 wt.-%, still more preferably at least 70 wt.-%, even more preferably at least 80 wt.-%, yet more preferably at least 90 wt.-%, most preferably at least 95 wt.-%, and in particular at least 99.999 wt.-%, relative to the total weight of the matrix material.
- the total content of alkyl cellulose and heteropolysaccharide is at most 99.999 wt.-%, more preferably at most 99 wt.-%, still more preferably at most 97 wt.-%, most preferably at most 95 wt.-%, and in particular at most 93 wt.-%, relative to the total weight of the matrix material.
- the total content of alkyl cellulose and heteropolysaccharide is within the range of from 50 to 99.999 wt.-%, more preferably 60 to 99.999 wt.-%, still more preferably 70 to 99.999 wt.-%, most preferably 80 to 99.999 wt.-%, and in particular 90 to 99.999 wt.-%, relative to the total weight of the matrix material.
- the total content of alkyl cellulose and heteropolysaccharide is at least 35 wt.-%, preferably at least 40 wt.-% or at least 45 wt.-% or at least 50 wt.-%, more preferably at least 55 wt.-%, still more preferably at least 65 wt.-%, even more preferably at least 70 wt.-%, yet more preferably at least 75 wt.-%, most preferably at least 80 wt.-%, and in particular at least 85 wt.-%, relative to the total weight of the dosage form.
- the total content of alkyl cellulose and heteropolysaccharide is at most 99 wt.-%, more preferably at most 97 wt.-%, still more preferably at most 95 wt.-%, even more preferably at most 93 wt.-%, most preferably at most 91 wt.-%, and in particular at most 90 wt.-%, relative to the total weight of the dosage form.
- the total content of alkyl cellulose and heteropolysaccharide is within the range of from 35 to 99 wt.- %, more preferably 45 to 97 wt.-%, still more preferably 55 to 95 wt.-%, even more preferably 65 to 93 wt.-%, most preferably 75 to 91 wt.-%, and in particular 85 to 90 wt.-%, relative to the total weight of the dosage form.
- the relative weight ratio of heteropolysaccharide to alkyl cellulose is within the range of from 1 : 20 to 20 : 1, preferably 1 : 19 to 15 to : 1, more preferably 1 : 18 to 10 : 1, still more preferably 1 : 18 to 7 : 1 or 1 : 14 to 7 : 1, even more preferably 1 : 18 to 4 : 1 or 1 : 12 to 4 : 1, yet more preferably 1 : 18 to 2 : 1 or 1 : 11 to 2 : 1, most preferably 1 : 18 to 1 : 1 or 1 : 10 to 1 : 1, and in particular 1 : 18 to 1 : 4 or 1 : 8 to 1 : 4.
- the relative weight ratio of heteropolysaccharide to alkyl cellulose is within the range of from 1 : 18 to 2 : 1.
- the content of the alkyl cellulose in the dosage form is higher than the content of the heteropolysaccharide in the dosage form.
- the dosage form comprises a matrix material which in turn comprises an alkyl cellulose.
- the dosage form and the matrix material respectively, contains only one alkyl cellulose.
- the dosage form and the matrix material respectively, contains a mixture of two or more alkyl celluloses.
- Preferred alkyl celluloses are selected from Ci_6-alkyl celluloses, more preferably unsubstituted Ci_6-alkyl celluloses, i.e. Ci_6-alkyl celluloses wherein the Ci_6-alkyl moiety is not substituted.
- the alkyl cellulose has a solution viscosity within the range of from 1 mPa- s to 150 mPa- s, more preferably 1 mPa- s to 7 mPa- s, or 5 mPa- s to 10 mPa- s, or 7 mPa- s to 13 mPa- s, or 15 mPa- s to 25 mPa- s, or 38 mPa- s to 52 mPa- s, or 60 mPa- s to 140 mPa- s, measured in a 5 wt.-% solution of 80 wt.-% toluene and 20 wt.-% ethanol at 25°C in an Ubbelohde viscosimeter.
- the alkyl cellulose has a solution viscosity within the range of from 70 mPa- s to 130 mPa- s, more preferably 80 mPa- s to 120 mPa- s and most preferably 90 mPa- s to 110 mPa- s, measured in a 5 wt.-% solution of 80 wt.-% toluene and 20 wt.-% ethanol at 25°C in an Ubbelohde viscosimeter.
- the alkyl cellulose has an alkoxyl content of from 10 wt.-% to 80 wt.-%, more preferably 20 wt.-% to 70 wt.-%, still more preferably 22 wt.-% to 40 wt.-% or 40 wt.-% to 60 wt.-%, most preferably 24 wt.-% to 35 wt.-% or 44 wt.-% to 51 wt.-%, and in particular 26 wt.-% to 33 wt.-% or 48 wt.-% to 49.5 wt.-%.
- the alkyl cellulose is selected from the group consisting of ethyl cellulose, hydroxyethyl cellulose, ethylmethyl cellulose, hydroxyethyl methyl cellulose, ethylhydroxy ethyl cellulose, methyl cellulose, carboxymethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, hydroxybutyl methyl cellulose, and carboxymethyl hydroxyethyl cellulose.
- Preferred alkyl celluloses are ethyl cellulose, methyl cellulose and ethylmethyl cellulose. In a particularly preferred embodiment, the alkyl cellulose is ethyl cellulose.
- the alkyl cellulose is ethyl cellulose having an ethoxyl content of from 40 wt.-% to 60 wt.-%, more preferably 44 wt.-% to 51 wt.-%, most preferably 48 wt.-% to 49.5 wt.-%.
- the alkyl cellulose is ethyl cellulose having a solution viscosity within the range of from 70 mPa- s to 130 mPa- s, more preferably 80 mPa- s to 120 mPa- s and most preferably 90 mPa- s to 110 mPa- s, measured in a 5 wt.-% solution of 80 wt.-% toluene and 20 wt.-% ethanol at 25°C in an Ubbelohde viscosimeter.
- the alkyl cellulose is ethyl cellulose, having
- an ethoxyl content of from 40 wt.-% to 60 wt.-%;
- a solution viscosity within the range of from 70 mPa- s to 130 mPa- s, measured in a 5 wt.-% solution of 80 wt.-% toluene and 20 wt.-% ethanol at 25°C in an Ubbelohde viscosimeter.
- Preferred commercially available alkyl celluloses include ETHOCEL Polymers, in particular ETHOCEL Standard 100 Premium, ETHOCEL Standard 4 Premium, ETHOCEL Standard 7 Premium, ETHOCEL Standard 10 Premium, ETHOCEL Standard 20 Premium and ETHOCEL Standard 45 Premium.
- the content of the alkyl cellulose in the matrix material is preferably at least 20 wt.-%, more preferably at least 30 wt.-%, still more preferably at least 40 wt.-%, even more preferably at least 50 wt.-%, yet more preferably at least 60 wt.-%, most preferably at least 70 wt.-%, and in particular at least 71 wt.-%, relative to the total weight of the matrix material.
- the content of the alkyl cellulose in the matrix material is preferably at most 95 wt.- , more preferably at most 94 wt.- , still more preferably at most 93 wt.- , even more preferably at most 92 wt.- , most preferably at most 91 wt.- , and in particular at most 90 wt.- , relative to the total weight of the matrix material.
- the content of the alkyl cellulose in the matrix material is in the range of from 20 to 95 wt.-%, more preferably 30 to 94 wt.-%, still more preferably 40 to 93 wt.-%, even more preferably 50 to 92 wt.-%, most preferably 60 to 91 wt.-%, and in particular 70 to 90 wt.-% or 75 to 90 wt.-%, relative to the total weight of the matrix material.
- the content of the alkyl cellulose is at least 10 wt.-%, more preferably at least 20 w - %, most preferably at least 30 wt.-%, and in particular at least 40 wt.-%, relative to the total weight of the dosage form.
- the content of the alkyl cellulose is at least 45 wt.-%, more preferably at least 50 wt.-%, still more preferably at least 55 wt.-%, most preferably at least 60 wt.-%, and in particular at least 63 wt.-%, relative to the total weight of the dosage form.
- the content of the alkyl cellulose is at most 95 wt.-%, more preferably at most 93 wt.-%, still more preferably at most 91 wt.-%, even more preferably at most 89 wt.-%, most preferably at most 87 wt.-%, and in particular at most 86 wt.-%, relative to the total weight of the dosage form.
- the content of the alkyl cellulose is within the range of from 10 to 95 wt.-%, more preferably 25 to 93 wt.-%, still more preferably 35 to 91 wt.-%, even more preferably 45 to 89 wt.-%, most preferably 55 to 87 wt.- %, and in particular 63 to 86 wt.-%, relative to the total weight of the dosage form.
- the alkyl cellulose is ethyl cellulose which content is within the range of from 63 to 86 wt.-%, relative to the total weight of the dosage form.
- the amount of the alkyl cellulose which is contained in the dosage form is within the range of from 50 to 600 mg, more preferably 100 to 575 mg, still more preferably 150 to 550 mg, yet more preferably 200 to 525 mg, even more preferably 250 to 500 mg, most preferably 270 to 475 mg, and in particular 290 to 450 mg.
- the relative weight ratio of the pharmacologically active ingredient, preferably the opioid to the alkyl cellulose is in the range of from 1 : 30 to 10 : 1, more preferably 1 : 25 to 7 : 1, still more preferably 1 : 22 to 4 : 1, yet more preferably 1 : 20 to 1 : 1, most preferably 1 : 18 to 1 : 3, and in particular 1 : 17 to 1 : 5.
- the dosage form according to the invention contains a matrix material comprising a heteropolysaccharide.
- the dosage form and the matrix material respectively, contains only one heteropolysaccharide.
- the dosage form and the matrix material respectively, contains a mixture of two or more heteropolysaccharides.
- Heteropolysaccharides are polysaccharides which are based on two or more different monosaccharides.
- the heteropolysaccharide may be acidic or neutral.
- the term "acidic heteropolysaccharide” also includes any derivative of acidic heteropolysaccharides, such as e.g. salts, esters and amides.
- the heteropolysaccharide is acidic and preferably selected from the group consisting of xanthan gum, agar, alginic acid, sodium alginate, propylene glycol alginate, gum arabic, ⁇ -carrageenan, ⁇ - carrageenan, i-carrageenan, fucoidan, fucogalactan (GFS), gellan gum, gum ghatti, gum karaya, pectin, psyllium seed gum, gum tragacanth, welan gum, their corresponding salts and mixtures thereof.
- xanthan gum agar, alginic acid, sodium alginate, propylene glycol alginate, gum arabic, ⁇ -carrageenan, ⁇ - carrageenan, i-carrageenan, fucoidan, fucogalactan (GFS), gellan gum, gum ghatti, gum karaya, pectin, psyllium seed gum, gum tragacanth, welan
- the heteropolysaccharide is neutral and preferably selected from the group consisting of chitin, chitosan, curdlan, dextran, guar gum, inulin, ivory nut mannan, konjac glucomannan, laminaran, larch arabinogalactan, locust bean gum, pullulan, scleroglucan, tamarind gum, tara gum, their derivatives and mixtures thereof.
- the heteropolysaccharide is selected from the group consisting of xanthan gum, guar gum, alginic acid, sodium alginate, carrageenans, locust bean gum, and mixtures thereof.
- the heteropolysaccharide is xanthan gum or guar gum. Particularly preferably, the heteropolysaccharide is xanthan gum.
- Preferred commercially available heteropolysaccharides include Xanthan Gum Type 602.
- the dosage form contains a singly type of a heteropolysaccharide, preferably only xanthan gum, but no additional heteropolysaccharide.
- the dosage form does not comprise a combination of xanthan gum and locust bean gum.
- the alkyl cellulose is ethyl cellulose
- heteropolysaccharide is xanthan gum.
- the content of the heteropolysaccharide in the matrix material is preferably at least 1 wt.-%, more preferably at least 3 wt.-%, still more preferably at least 5 wt.-%, even more preferably at least 7 wt.-%, yet more preferably at least 9 wt.-%, most preferably at least 10 wt.-%, and in particular at least 11 wt.-%, relative to the total weight of the matrix material.
- the content of the heteropolysaccharide in the matrix material is preferably at least 11 wt.-%, more preferably at least 13 wt.-%, still more preferably at least 15 wt.-%, even more preferably at least 17 wt.-%, yet more preferably at least 19 wt.-%, most preferably at least 21 wt.-%, and in particular at least 23 wt.-% or at least 25 wt.-%, relative to the total weight of the matrix material.
- the content of the heteropolysaccharide in the matrix material is preferably at most 90 wt.-%, more preferably at most 80 wt.-%, still more preferably at most 70 wt.-%, even more preferably at most 60 wt.-%, yet more preferably at most 50 wt.- , most preferably at most 40 wt.- , and in particular at most 30 wt.-% or at most 29 wt.- , relative to the total weight of the matrix material.
- the content of the heteropolysaccharide in the matrix material is in the range of from 1 to 90 wt.-%, more preferably 3 to 80 wt.-%, still more preferably 5 to 70 wt.-%, even more preferably 7 to 60 wt.-%, yet more preferably 8 to 50 wt.-%, most preferably 9 to 40 wt.-%, and in particular 10 to 30 wt.-% or 11 to 29 wt.-%, relative to the total weight of the matrix material.
- the content of the heteropolysaccharide is below 80 wt.-%, more preferably below 70 wt.-%, still more preferably below 65 wt.-%, most preferably below 55 wt.-%, and in particular below 50 wt.-%, relative to the total weight of the pharmaceutical dosage form.
- the content of the heteropolysaccharide is below 45 wt.-%, more preferably below 40 wt.-%, still more preferably below 35 wt.-%, most preferably below 30 wt.-%, and in particular below 28 wt.-%, relative to the total weight of the dosage form.
- the content of the heteropolysaccharide is above 1 wt.-%, more preferably above 3 wt.-%, still more preferably above 5 wt.-%, most preferably above 7 wt.-% or above 10 wt.-%, and in particular above 9 wt.-% or above 15 wt.-% or above 20 wt.-%, relative to the total weight of the dosage form.
- the content of the heteropolysaccharide is within the range of from 2 to 80 wt.-%, more preferably 3 to 70 wt.-%, still more preferably 4 to 60 wt.-%, yet more preferably 5 to 50 wt.-%, even more preferably 6 to 40 wt.-%, most preferably 7 to 30 wt.-%, and in particular 8 to 28 wt.-%, relative to the total weight of the dosage form.
- the heteropolysaccharide is xanthan gum which content is within the range of from 8 to 28 wt.-%, relative to the total weight of the dosage form.
- the amount of the heteropolysaccharide which is contained in the dosage form is within the range of from 5 to 300 mg, more preferably 15 to 250 mg, still more preferably 20 to 200 mg, yet more preferably 25 to 180 mg, even more preferably 30 to 160 mg, most preferably 35 to 140 mg, and in particular 40 to 130 mg.
- the relative weight ratio of the pharmacologically active ingredient, preferably the opioid to the heteropolysaccharide is in the range of from 1 : 10 to 10 : 1, more preferably 1 : 9 to 9 : 1, still more preferably 1 : 7 to 7 : 1, yet more preferably 1 : 5 to 5 : 1, most preferably 1 : 3 to 3 : 1, and in particular 1 : 2.5 to 2.5 : 1.
- the dosage form contains only one pharmacologically active ingredient, preferably one opioid. In another preferred embodiment, the dosage form contains a combination of two or more pharmacologically active ingredients.
- the pharmacologically active ingredient is soluble in water.
- the pharmacologically active ingredient is selected from ATC class [N], more preferably [N02] according to the WHO.
- the pharmacologically active ingredient is an opioid.
- opioid shall refer to any opioid as well as any physiologically acceptable salt thereof.
- the dosage form comprises an opioid or a physiologically acceptable salt thereof.
- Opioids are active ingredients with potential for being abused and potential for dose dumping in ethanol.
- opioids are divided into natural opium alkaloids, phenylpiperidine derivatives, diphenylpropylamine derivatives, benzomorphan derivatives, oripavine derivatives, morphinan derivatives and others.
- the pharmacologically active ingredient is selected from the group consisting of morphine, hydromorphone, nicomorphine, oxycodone, oxymorphone, dihydrocodeine, ketobemidone, pethidine, fenantyl, dextromoramide, piritramide, dextropropoxyphene, bezitramide, pentazocine, phenazocine, buprenorphine, butorphanol, nalbuphine, tilidine, tramadol, dezocine, meptazinol, tapentadol, and the physiologically acceptable salts thereof.
- the pharmacologically active ingredient is selected from the group consisting of tramadol, tapentadol, faxeladol and axomadol.
- the pharmacologically active ingredient is selected from the group consisting of oxycodone, oxymorphone, hydrocodone, hydromorphone, tramadol, tapentadol, morphine, buprenorphine and the physiologically acceptable salts thereof.
- the pharmacologically active ingredient is selected from the group consisting of 1, 1 -(3-dimethylamino-3-phenylpentamethylene)-6-fluoro- 1 ,3,4,9-tetrahydropyrano[3,4-b]indole, particularly its hemicitrate; 1 , 1 -[3-dimethylamino-3-(2-thienyl)pentamethylene]- 1 ,3,4,9-tetrahydropyrano[3,4- bjindole, particularly its citrate; and l, l-[3-dimethylamino-3-(2-thienyl)pentamethylene]-l,3,4,9-tetrahydro- pyrano[3,4-b]-6-fluoroindole, particularly its hemicitrate.
- These compounds are known from, e.g., WO 2004/043967, WO 2005/066183.
- the pharmacologically active ingredient is selected from the following compounds: alfentanil, allyl- prodine, alphaprodine, apocodeine, axomadol, bemidone, benzylmorphine, bezitramide, buprenorphine, butorphanol, carfentanil, clonitazene, cocaine, codeine, cyclorphan, cyprenorphine, desomorphine, dextromoramide, dextropropoxyphene, dezocine, diampromide, diamorphone, dihydrocodeine, dihydromorphine, dihydromorphone, dimenoxadol, dimephetamol, dimethylthiambutene, dioxaphetylbutyrate, dipipanone, eptazocine, ethoheptazine, ethylmethylthiambutene, ethylmorphine, etonitazene,
- the pharmacologically active ingredient is selected from the group consisting of DPI-125, M6G (CE-04-410), ADL-5859, CR-665, NRP290 and sebacoyl dinalbuphine ester.
- the pharmacologically active ingredient is selected from the group consisting of rabeprazole, fentanyl, risedronate, nifedipine, amphetamine salts, everolimus, alprazolam, lovastatin, Zolpidem, dalfampridine, cyclobenzaprine, bupropion, mesalamine, tipranavir, donepezil, diclofenac, aspirin, sulfasalazine, morphine, dutasteride, clarithromycin, praziquantel, bisacodyl, ibandronate, verapamil, nicardipine, diltiazem, doxazosin, cefuroxime, mycophenolate, activated charcoal, ciprofloxacin, docusate, colestipol, methylphenidate, nicotine, carvedilol, pancrelipase, indinavir, duloxetine, cyclo
- the pharmacologically active ingredient preferably the opioid may be present in form of a physiologically acceptable salt, e.g. physiologically acceptable acid addition salt.
- Physiologically acceptable acid addition salts comprise the acid addition salt forms which can conveniently be obtained by treating the base form of the pharmacologically active ingredient, preferably the opioid with appropriate organic and inorganic acids.
- Pharmacologically active ingredients preferably opioids containing an acidic proton may be converted into their non-toxic metal or amine addition salt forms by treatment with appropriate organic and inorganic bases.
- the term addition salt also comprises the hydrates and solvent addition forms which the active ingredients are able to form. Examples of such forms are e.g. hydrates, alcoholates and the like.
- the content of the pharmacologically active ingredient, preferably the opioid in the dosage form and in the particles, respectively, can be optimized in order to provide the best compromise between tamper-resistance, disintegration time and drug release, drug load, processability (especially pharmaceutical dosage formability) and patient compliance.
- the pharmacologically active ingredient preferably the opioid is present in the dosage form in a therapeutically effective amount.
- the amount that constitutes a therapeutically effective amount varies according to the pharmacologically active ingredients being used, the condition being treated, the severity of said condition, the patient being treated, and the frequency of administration.
- the content of the pharmacologically active ingredient in the dosage form is not limited.
- the dose of the pharmacologically active ingredient, preferably the opioid which is adapted for administration preferably is in the range of 0.1 mg to 500 mg, more preferably in the range of 1.0 mg to 400 mg, even more preferably in the range of 5.0 mg to 300 mg, and most preferably in the range of 10 mg to 250 mg.
- the total amount of the pharmacologically active ingredient, preferably the opioid that is contained in the dosage form is within the range of from 0.01 to 200 mg, more preferably 0.1 to 190 mg, still more preferably 1.0 to 180 mg, yet more preferably 1.5 to 160 mg, most preferably 2.0 to 100 mg and in particular 2.5 to 80 mg.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of from 0.01 to 80 wt.-%, more preferably 0.1 to 50 wt.-%, still more preferably 1 to 35 wt.-%, based on the total weight of the dosage form.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of from 5.0+4.5 wt.-%, or 10+9.0 wt.-%, or 15+14 wt.-%, or 20+19 wt.-%, or 25+24 wt.-%; more preferably 5.0+4.0 wt.-%, or 10+8.0 wt.-%, or 15+12 wt.-%, or 20+19 wt.-%, or 25+24 wt.-%; still more preferably 5.0+3.5 wt.-%, or 10+7.0 wt.-%, or 15+10 wt.-%, or 20+17 wt.-%, or 25+21 wt.-%; yet more preferably 5.0+3.0 wt.-%, or 10+6.0 wt.-%, or 15+8.0 wt.-%, or 20+15 wt.-%, or 25+18
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 5+4 wt.-%, more preferably 5+3 wt.-%, still more preferably 5+2 wt.-%, most preferably 5+1 wt.-%, and in particular 5+0.5 wt.-%, either based on the total weight of the dosage form or, when the dosage form is particulate, based on the total weight of the particles that contain the pharmacologically active ingredient.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 10+9 wt.-%, more preferably 10+7 wt.-%, still more preferably 10+5 wt.-%, yet more preferably 10+3 wt.-%, most preferably 10+1 wt.-%, and in particular 10+0.5 wt.-%, either based on the total weight of the dosage form or, when the dosage form is particulate, based on the total weight of the particles that contain the pharmacologically active ingredient.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 15+14 wt.-%, more preferably 15+11 wt.-%, still more preferably 15+8 wt.-%, yet more preferably 15+5 wt.-%, most preferably 15+2 wt.-%, and in particular 15+0.5 wt.-%, either based on the total weight of the dosage form or, when the dosage form is particulate, based on the total weight of the particles that contain the pharmacologically active ingredient.
- an appropriate amount of pharmacologically active ingredient, preferably opioid to include in a dosage form may readily determine an appropriate amount of pharmacologically active ingredient, preferably opioid to include in a dosage form.
- the total amount of pharmacologically active ingredient, preferably opioid present in the dosage form is that sufficient to provide analgesia.
- the total amount of pharmacologically active ingredient, preferably opioid administered to a patient in a dose will vary depending on numerous factors including the nature of the pharmacologically active ingredient, the weight of the patient, the severity of the pain, the nature of other therapeutic agents being administered etc.
- the pharmacologically active ingredient preferably the opioid is contained in the dosage form in an amount of 7.5+5 mg, 10+5 mg, 15+5 mg, 20+5 mg, 25+5 mg, 30+5 mg, 35+5 mg, 40+5 mg, 45+5 mg, 50+5 mg, 55+5 mg, 60+5 mg, 65+5 mg, 70+5 mg, 75+5 mg, 80+5 mg, 85+5 mg, 90+5 mg, 95+5 mg, 100+5 mg, 110+5 mg, 120+5 mg, 130+5, 140+5 mg, 150+5 mg, 160+5 mg, 170+5 mg, 180+5 mg, 190+5 mg, 200+5 mg, 210+5 mg, 220+5 mg, 230+5 mg, 240+5 mg, 250+5 mg, 260+5 mg, 270+5 mg, 280+5 mg, 290+5 mg, or 300+5 mg.
- the pharmacologically active ingredient preferably the opioid is contained in the dosage form in an amount of 5+2.5 mg, 7.5+2.5 mg, 10+2.5 mg, 15+2.5 mg, 20+2.5 mg, 25+2.5 mg, 30+2.5 mg, 35+2.5 mg, 40+2.5 mg, 45+2.5 mg, 50+2.5 mg, 55+2.5 mg, 60+2.5 mg, 65+2.5 mg, 70+2.5 mg, 75+2.5 mg, 80+2.5 mg, 85+2.5 mg, 90+2.5 mg, 95+2.5 mg, 100+2.5 mg, 105+2.5 mg, 110+2.5 mg, 115+2.5 mg, 120+2.5 mg, 125+2.5 mg, 130+2.5 mg, 135+2.5 mg, 140+2.5 mg, 145+2.5 mg, 150+2.5 mg, 155+2.5 mg, 160+2.5 mg, 165+2.5 mg, 170+2.5 mg, 175+2.5 mg, 180+2.5 mg, 185+2.5 mg, 190+2.5 mg, 195+2.5 mg, 200+2.5 mg, 205+2.5 mg, 210+
- the pharmacologically active ingredient is tramadol, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 300 mg.
- the pharmacologically active ingredient is tramadol, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 10 to 500 mg.
- the pharmacologically active ingredient is oxycodone, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 1 to 80 mg.
- the pharmacologically active ingredient is oxycodone, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 2 to 320 mg.
- the pharmacologically active ingredient is oxymorphone, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 40 mg.
- the pharmacologically active ingredient is oxymorphone, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 10 to 80 mg.
- the pharmacologically active ingredient is tapentadol, preferably its HC1 salt, and the dosage form is adapted for administration once daily or twice daily.
- pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 25 to 250 mg.
- the pharmacologically active ingredient is hydromorphone, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 2 to 52 mg.
- the pharmacologically active ingredient is hydromorphone, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 4 to 104 mg.
- the pharmacologically active ingredient is hydrocodone, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 250 mg.
- the pharmacologically active ingredient is hydrocodone, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 250 mg.
- the pharmacologically active ingredient is morphine, preferably its HC1 or H 2 S0 4 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 250 mg.
- the pharmacologically active ingredient is morphine, preferably its HC1 or H 2 SO 4 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 5 to 250 mg.
- the pharmacologically active ingredient is buprenorphine, preferably its HC1 salt, and the dosage form is adapted for administration twice daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 1 to 12 mg.
- the pharmacologically active ingredient is buprenorphine, preferably its HC1 salt, and the dosage form is adapted for administration once daily.
- the pharmacologically active ingredient is preferably contained in the dosage form in a total amount of from 2 to 12 mg.
- the particles present in the dosage form according to the invention preferably comprise 1 to 75 wt.-% of the pharmacologically active ingredient, preferably the opioid, more preferably 2 to 60 wt.-% of the pharmacologically active ingredient, preferably the opioid, still more preferably 3 to 40 wt.-% of the pharmacologically active ingredient, preferably the opioid, most preferably 4 to 25 wt.-% of the pharmacologically active ingredient, preferably the opioid and in particular 4.5 to 17 wt.-% of the pharmacologically active ingredient, preferably the opioid, based on the total weight of a particle.
- the content of the pharmacologically active ingredient, preferably the opioid is preferably at least 1 wt.-%, more preferably at least 2 wt.-%, still more preferably at least 3 wt.-%, most preferably at least 4 wt.-% and in particular at least 5 wt.-%, based on the total weight of a particle.
- the content of the pharmacologically active ingredient, preferably the opioid is preferably at most 70 wt.-%, more preferably at most 65 wt.-%, still more preferably at most 50 wt.-%, yet more preferably at most 35 wt.-%, most preferably at most 20 wt.-%, based on the total weight of a particle.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 5+4 wt.-%, more preferably 5+3 wt.-%, still more preferably 5+2 wt.-%, most preferably 5+1 wt.-%, and in particular 5+0.5 wt.-%, based on the total weight of a particle.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 10+9 wt.-%, more preferably 10+7 wt.-%, still more preferably 10+5 wt.-%, yet more preferably 10+3 wt.-%, most preferably 10+1 wt.-%, and in particular 10+0.5 wt.-%, based on the total weight of a particle.
- the content of the pharmacologically active ingredient, preferably the opioid is within the range of 15+14 wt.-%, more preferably 15+11 wt.-%, still more preferably 15+8 wt.-%, yet more preferably 15+5 wt.-%, most preferably 15+2 wt.-%, and in particular 15+0.5 wt.-%, based on the total weight of a particle.
- the pharmacologically active ingredient preferably the opioid that is included in the preparation of the dosage forms according to the invention preferably has an average particle size of less than 500 microns, still more preferably less than 300 microns, yet more preferably less than 200 or 100 microns. There is no lower limit on the average particle size and it may be, for example, 50 microns.
- the particle size of the pharmacologically active ingredient may be determined by any technique conventional in the art, e.g. laser light scattering, sieve analysis, light microscopy or image analysis. When the dosage form is particulate, it is preferable that the largest dimension of the pharmacologically active ingredient particle be less than the size of the particles (e.g. less than the smallest dimension of the particles).
- the dosage form contains a combination of a pharmacologically active ingredient, preferably an opioid and a further pharmacologically active ingredient which is not an opioid.
- the dosage form does not contain any further pharmacologically active ingredient.
- Said further pharmacologically active ingredient is preferably selected from ATC classes [M01A], [M01C], [N02B] and [N02C] according to the WHO.
- the further pharmacologically active ingredient is selected from the group consisting of acetylsalicylic acid, aloxiprin, choline salicylate, sodium salicylate, salicylamide, salsalate, ethenzamide, morpholine salicylate, dipyrocetyl, benorilate, diflunisal, potassium salicylate, guacetisal, carbasalate calcium, imidazole salicylate, phenazone, metamizole sodium, aminophenazone, propyphenazone, nifenazone, paracetamol, phenacetin, bucetin, propacetamol, rimazolium, glafenine, floctafenine, viminol, nefopam, flupirtine, ziconotide, methoxyflurane, nabiximols, dihydroergotamine, ergotamine, methysergide, lisuride, flumedroxone
- the dosage form comprises a further pharmacologically active ingredient
- said further pharmacologically active ingredient preferably is present in the dosage form in a therapeutically effective amount.
- the amount that constitutes a therapeutically effective amount varies according to the further pharmacologically active ingredient being used, the condition being treated, the severity of said condition, the patient being treated, and the frequency of administration.
- the content of the further pharmacologically active ingredient in the dosage form is not limited.
- the dose of the further pharmacologically active ingredient which is adapted for administration preferably is in the range of 0.1 mg to 4 g.
- the matrix material and the dosage form may contain additional pharmaceutical excipients conventionally contained in pharmaceutical dosage forms in conventional amounts, such as antioxidants, preservatives, lubricants, plasticizer, fillers, binders, and the like.
- the matrix material and the dosage form respectively, only comprises excipients which are approved for oral use according to the Ph. Eur. and the USP, respectively. Therefore, in a preferred embodiment, the dosage form according to the present invention does not contain any compound which is not approved for oral use. More preferably, the dosage form does not contain poly(s-caprolactone).
- the dosage form does not contain a disintegrant.
- the matrix material and the dosage form respectively, further comprises an antioxidant.
- Suitable antioxidants include ascorbic acid, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), salts of ascorbic acid, monothioglycerol, phosphorous acid, vitamin C, vitamin E and the derivatives thereof, coniferyl benzoate, nordihydroguajaretic acid, gallus acid esters, sodium bisulfite, particularly preferably butylhydroxytoluene or butylhydroxyanisole and oc-tocopherol.
- BHA butylated hydroxyanisole
- BHT butylated hydroxytoluene
- the antioxidant is preferably present in quantities of 0.01 wt.-% to 10 wt.-%, more preferably of 0.03 wt.-% to 5 wt.-%, most preferably of 0.05 wt.-% to 2.5 wt.-%, based on the total weight of the dosage form and the matrix material, respectively.
- the matrix material and the dosage form respectively, further comprise an acid, preferably citric acid.
- the amount of acid is preferably in the range of 0.01 wt.-% to about 20 wt.-%, more preferably in the range of 0.02 wt.-% to about 10 wt.-%, and still more preferably in the range of 0.05 wt.-% to about 5 wt.-%, and most preferably in the range of 0.1 wt.-% to about 1.0 wt.-%, based on the total weight of the dosage form and the matrix material, respectively.
- the matrix material and the dosage form respectively, contain at least one lubricant.
- Especially preferred lubricants are selected from
- glycerol fatty acid esters such as mixtures of mono-, di- and triesters of glycerol and di- and monoesters of macrogols having molecular weights within the range of from 200 to 4000 g/mol, e.g., macrogolglycerolcaprylocaprate, macrogolglycerollaurate, macrogolglycerolococoate, macrogolglycerol- linoleate, macrogol-20-glycerolmonostearate, macrogol-6-glycerolcaprylocaprate, macrogolglycerololeate; macrogolglycerolstearate, macrogolglycerolhydroxystearate, and macrogolglycerolrizinoleate;
- macrogolglycerolcaprylocaprate e.g., macrogolglycerolcaprylocaprate, macrogolglycerollaurate, macrogolglycerolococoate, macrogolglycerol- linoleate, macrogol-20-g
- fatty alcohols that may be linear or branched, such as cetylalcohol, stearylalcohol, cetylstearyl alcohol, 2- octyldodecane- 1 -ol and 2-hexyldecane-l-ol; and
- the amount of the lubricant ranges from 0.01 wt.-% to about 10 wt.-%, more preferably in the range of 0.05 wt.-% to about 7.5 wt.-%, most preferably in the range of 0.1 wt.-% to about 5 wt.-%, and in particular in the range of 0.1 wt.-% to about 1 wt.-%, based on the total weight of the dosage form and the matrix material, respectively.
- the matrix material and the dosage form, respectively, further comprise a plasticizer.
- the plasticizer improves the matrix material.
- a preferred plasticizer is polyalkylene glycol, like polyethylene glycol, triacetin, fatty acids, fatty acid esters, waxes and/or microcrystalline waxes.
- Particularly preferred plasticizers are polyethylene glycols, such as PEG 6000.
- the content of the plasticizer is within the range of from 0.5 to 30 wt.-%, more preferably 1.0 to 25 wt.-%, still more preferably 2.5 wt.-% to 22.5 wt.-%, yet more preferably 5.0 wt.-% to 20 wt.-%, most preferably 6 to 20 wt.-% and in particular 7 wt.-% to 17.5 wt.-%, based on the total weight of the dosage form and the matrix material, respectively.
- Plasticizers can sometimes act as a lubricant, and lubricants can sometimes act as a plasticizer.
- the matrix material and the dosage form contain no antioxidant and/or no acid and/or no lubricant and/or no plasticizer. More preferably, the matrix material and the dosage form, respectively, contain no excipients.
- Preferred contents of the pharmacologically active ingredient, preferably the opioid, the alkyl cellulose, the heteropolysaccharide and excipients, relative to the total weight of the dosage form are summarized as embodiments A 1 to A 12 in the tables here below:
- the dosage form more preferably the matrix comprising the alkyl cellulose and the heteropolysaccharide provides prolonged release of the pharmacologically active ingredient, preferably the opioid.
- the dosage form, more preferably the matrix comprising the alkyl cellulose and the heteropolysaccharide provides immediate release of the pharmacologically active ingredient, preferably the opioid.
- Prolonged release is known to the skilled artisan.
- the term “prolonged release” preferably refers to a release rate of the pharmacologically active ingredient from the formulation that has been reduced over time in order to maintain therapeutic activity, to reduce toxic effects, or for some other therapeutic purpose such as reducing the dosing frequency.
- immediate release is known to the skilled artisan.
- immediate release preferably refers to a release rate of the pharmacologically active ingredient from the formulation that is comparatively fast and not retarded.
- the release of the pharmacologically active ingredient is preferably not controlled by erosion of the surface of the dosage form. If the dosage form according to the present invention is particulate, the release of the pharmacologically active ingredient is preferably neither controlled by erosion of the surface of the particles, nor by erosion of the surface of the dosage form.
- the dosage form provides prolonged release of the pharmacologically active ingredient, preferably the opioid.
- the matrix provides for a prolonged release of the pharmacologically active ingredient, preferably the opioid from dosage form.
- the dosage form has released after 30 minutes 0.1 to 75%, after 240 minutes 0.5 to 99%, after 480 minutes 1.0 to 100% and after 720 minutes 2.5 to 100% of the pharmacologically active ingredient, preferably the opioid.
- Suitable in vitro conditions are known to the skilled artisan. In this regard it can be referred to, e.g., the Eur. Ph.
- the release profile is measured under the following conditions: Paddle apparatus, 50 rpm, 37+5 °C, 900 mL 0.1 M HCl (pH 1.0) or simulated intestinal fluid pH 6.8 (phosphate buffer) or pH 4.5.
- the rotational speed of the paddle is increased to 75 rpm.
- the release profile is determined under the following conditions: basket method, 75 rpm, 37+5 °C, 900 mL 0.1 N HCl or 900 mL of SIF sp (pH 6.8) or 900 mL of 0.1 N HCl+40 vol.-% ethanol.
- Preferred release profiles R 1 to R 7 are summarized in the table here below [all data in wt.-% of released pharmacologically active ingredient] :
- the dosage form under in vitro conditions in 900 mL 0.1 N HCl (pH 1.0), using the paddle method according to Ph. Eur. at 50 rpm, after 1 h under physiological conditions, the dosage form has released at most 80%, more preferably at most 70%, most preferably at most 65% and in particular at most 60% of the pharmacologically active ingredient, preferably the opioid relative to the total amount of the pharmacologically active ingredient originally contained in the dosage form.
- the dosage form provides immediate release of the pharmacologically active ingredient, preferably the opioid.
- the matrix provides for an immediate release of the pharmacologically active ingredient, preferably the opioid from the dosage form.
- the dosage form has released after 15 minutes 20 to 90%, after 30 minutes 35 to 99%, after 45 minutes 50 to 99% and after 60 minutes more than 60% or more than 70% or more than 80% or more than 90% or more than 95% of the pharmacologically active ingredient, preferably the opioid.
- the release profile is measured under the following conditions: Paddle apparatus, 50 rpm, 37+5 °C, 900 mL 0.1 M HC1 (pH 1.0) or simulated intestinal fluid pH 6.8 (phosphate buffer) or pH 4.5.
- the rotational speed of the paddle is increased to 75 rpm.
- the release profile is determined under the following conditions: basket method, 75 rpm, 37+5 °C, 900 mL 0.1 N HC1 or 900 mL of SIF sp (pH 6.8) or 900 mL of 0.1 N HCl+40% ethanol.
- the dosage form according to the invention has a breaking strength of less than 300 N, more preferably less than 200 N, or, when the dosage form is particulate, the particles have a breaking strength of less than 300 N, more preferably less than 200 N.
- the dosage form preferably is particulate and in form of a filled capsule.
- the dosage form according to the invention has a breaking strength of at least 200 N, more preferably at least 300 N, or, when the dosage form is particulate, the particles have a breaking strength of at least 200 N, more preferably at least 300 N.
- the dosage form or, when it is particulate, the particles according to the invention which contain the pharmacologically active ingredient preferably have a breaking strength of at least 300 N, at least 400 N, or at least 500 N, preferably at least 600 N, more preferably at least 700 N, still more preferably at least 800 N, yet more preferably at least 1000 N, most preferably at least 1250 N and in particular at least 1500 N.
- the dosage form and the particles, respectively cannot be pulverized by the application of force with conventional means, such as for example a pestle and mortar, a hammer, a mallet or other usual means for pulverization, in particular devices developed for this purpose (dosage form crushers).
- pulverization means crumbling into small particles. Avoidance of pulverization virtually rules out oral or parenteral, in particular intravenous or nasal abuse.
- the "breaking strength" (resistance to crushing) of a dosage form and of a particle is known to the skilled person. In this regard it can be referred to, e.g., W.A. Ritschel, Die Tablette, 2. Auflage, Editio Cantor Verlag Aulendorf, 2002; H Liebermann et al., Pharmaceutical dosage forms: Pharmaceutical dosage forms, Vol. 2, Informa Healthcare; 2 edition, 1990; and Encyclopedia of Pharmaceutical Technology, Informa Healthcare; 1 edition.
- the breaking strength can be measured in accordance with the Eur. Ph. 5.0, 2.9.8 or 6.0, 2.09.08 "Resistance to Crushing of Pharmaceutical dosage forms".
- the particles may be subjected to the same or similar breaking strength test as the dosage form.
- the test is intended to determine, under defined conditions, the resistance to crushing of dosage forms and individual particles, respectively, measured by the force needed to disrupt them by crushing.
- the apparatus consists of 2 jaws facing each other, one of which moves towards the other.
- the flat surfaces of the jaws are perpendicular to the direction of movement.
- the crushing surfaces of the jaws are flat and larger than the zone of contact with the dosage form and individual particle, respectively.
- the apparatus is calibrated using a system with a precision of 1 Newton.
- the dosage form and particle, respectively, is placed between the jaws, taking into account, where applicable, the shape, the break-mark and the inscription; for each measurement the dosage form and particle, respectively, is oriented in the same way with respect to the direction of application of the force (and the direction of extension in which the breaking strength is to be measured).
- the measurement is carried out on 10 dosage forms and particles, respectively, taking care that all fragments have been removed before each determination.
- the result is expressed as the mean, minimum and maximum values of the forces measured, all expressed in Newton.
- breaking strength breaking force
- the breaking strength can alternatively be measured in accordance with the method described therein where it is stated that the breaking strength is the force required to cause a dosage form and particle, respectively, to fail (i.e., break) in a specific plane.
- the dosage forms and particles, respectively are generally placed between two platens, one of which moves to apply sufficient force to the dosage form and particle, respectively, to cause fracture.
- loading occurs across their diameter (sometimes referred to as diametral loading), and fracture occurs in the plane.
- the breaking force of dosage forms and particles, respectively is commonly called hardness in the pharmaceutical literature; however, the use of this term is misleading.
- hardness refers to the resistance of a surface to penetration or indentation by a small probe.
- crushing strength is also frequently used to describe the resistance of dosage forms and particle, respectively, to the application of a compressive load. Although this term describes the true nature of the test more accurately than does hardness, it implies that dosage forms and particles, respectively, are actually crushed during the test, which is often not the case.
- the breaking strength can be measured in accordance with WO 2008/107149, which can be regarded as a modification of the method described in the Eur. Ph.
- the dosage form and particle, respectively, is regarded as being broken if it is fractured into at least two separate pieces.
- the dosage form according to the invention provides tamper resistance in terms of resistance against dose- dumping in aqueous ethanol.
- the dosage form more preferably the matrix, further provides resistance against solvent extraction and/or resistance against grinding.
- the dosage form more preferably the matrix, provides tamper resistance. Tamper resistance preferably means that the dosage form and the matrix, respectively,
- (iii) preferably provides resistance against grinding.
- the dosage form and the matrix apart from exhibiting resistance (i), does not necessarily need to further exhibit resistances (ii) and (iii); but may preferably exhibit a combination thereof; namely a combination of only (i) and (ii); a combination of only (i) and (iii); or a combination of (i) and (ii) and (iii).
- the term "tamper-resistant” refers to dosage forms or segments that are resistant to conversion into a form suitable for misuse or abuse, particular for nasal and/or intravenous administration, by conventional means.
- the dosage form according to the invention provides resistance against dose dumping in aqueous ethanol.
- the matrix provides the dosage form with resistance against dose dumping in aqueous ethanol.
- the dosage form can be tested in vitro using 0.1 N HC1 with 40 vol.-% ethanol to evaluate alcohol extractability. Testing is preferably performed using standard procedures, e.g. USP Apparatus 1 (basket) or USP Apparatus 2 (paddle) at e.g. 50 rpm in e.g. 900 mL of media at 37°C, using a Perkin Elmer UV/VIS Spectrometer Lambda 20, UV at an appropriate wavelength for detection of the pharmacologically active ingredient present therein. Sample time points preferably include 0.5 and 1 hour.
- the in vitro release 0.1 N HCl / ethanol (40 vol.-%) is preferably not substantially accelerated compared to the in vitro release in 0.1 N HCl.
- substantially means that at any given time point the in vitro release in 0.1 N HCl / ethanol (40 vol.-%) relatively deviates from the in vitro release in 0.1 N HCl by not more than +15%, more preferably not more than +10%, still more preferably not more than +8%, yet more preferably not more than +6%, even more preferably not more than +4%, most preferably not more than +2% and in particular not more than +1 % or not more than +0.5% or not more than +0.1 %.
- the in vitro release in 0.1 N HCl / ethanol (40 vol.-%) relatively deviates from the in vitro release in 0.1 N HCl by at least -0.01 %, more preferably at least -0.05%, still more preferably at least -0.1 %, most preferably at least -0.5% and in particular at least -1 %.
- the dosage form can be tested in vitro using ethanol / simulated gastric fluid of 0%, 20% and 40% to evaluate alcohol extractability. Testing is preferably performed using standard procedures, e.g. USP Apparatus 1 (basket) or USP Apparatus 2 (paddle) at e.g. 50 rpm in e.g. 900 mL of media at 37°C, using a Perkin Elmer UV/VIS Spectrometer Lambda 20, UV at an appropriate wavelength for detection of the pharmacologically active ingredient present therein. Sample time points preferably include 0.5 and 1 hour.
- the in vitro release in ethanol / simulated gastric fluid is preferably not substantially accelerated compared to the in vitro release in simulated gastric fluid.
- substantially means that at any given time point the in vitro release in ethanol / simulated gastric fluid (40 vol.-%) relatively deviates from the in vitro release in simulated gastric fluid by not more than +15%, more preferably not more than +10%, still more preferably not more than +8%, yet more preferably not more than +6%, even more preferably not more than +4%, most preferably not more than +2% and in particular not more than +1 %.
- the in vitro release in ethanol / simulated gastric fluid (40 vol.-%) compared to the in vitro release in simulated gastric fluid is observed.
- the in vitro release in ethanol / simulated gastric fluid (40 vol.-%) relatively deviates from the in vitro release in simulated gastric fluid by at least -0.01 %, more preferably at least -0.05%, still more preferably at least -0.1 %, most preferably at least -0.5% and in particular at least -1 %.
- the dosage form according to the invention preferably exhibits resistance against solvent extraction.
- the matrix provides the dosage form according to the invention with resistance against solvent extraction.
- the liquid part of the formulation that can be separated from the remainder by means of a syringe at room temperature is as little as possible, preferably it contains not more than 45 or 40 wt.-%, more preferably not more than 35 wt.-%, still more preferably not more than 30 wt.-%, yet more preferably not more than 25 wt.-%, even more preferably not more than 20 wt.-%, most preferably not more than 15 wt.-% and in particular not more than 10 wt.-% of the original content of the pharmacologically active ingredient, preferably the opioid.
- this property is tested by (i) dispensing a dosage form that is either intact or has been manually comminuted by means of two spoons in 5 ml of solvent, either purified water or aqueous ethanol (40 vol.%), (ii) allowing the dispersion to stand for 10 min at room temperature, (iii) drawing up the hot liquid into a syringe (needle 21G equipped with a cigarette filter), and (iv) determining the amount of the pharmacologically active ingredient contained in the liquid within the syringe.
- the dosage form exhibits resistance against grinding. In another preferred embodiment, the dosage form does not exhibit resistance against grinding.
- the dosage form according to the invention is particulate, wherein the particles are obtained by wet granulation, dry granulation or fluid bed granulation.
- a further aspect of the invention relates to a process for the production of an oral pharmaceutical dosage form as described herein comprising the steps of
- a pharmacologically active ingredient preferably an opioid; an alkyl cellulose; a heteropolysaccharide and optionally ethanol; and
- step (ii) granulating the mixture obtained in step (i).
- the process further comprises the steps of
- step (iii) screening the granules obtained in step (ii) through a sieve having a mesh size of preferably 1,000 ⁇ ;
- step (iv) drying the screened granules obtained in step (iii);
- step (v) filling the dried granules obtained in step (iv) in a capsule;
- the granules preferably obtained in any of steps (ii) to (iv), preferably in combination with an outer matrix material, are compressed into tablets.
- the dosage form or, when it is particulate, the particles that contain the pharmacologically active ingredient are preferably thermoformed, preferably by melt-extrusion, although also other methods of thermoforming may be useful, such as press-molding at elevated temperature or heating of compacts that were manufactured by conventional compression in a first step and then heated above the softening temperature of the matrix material, in a second step to form break resistant, hardened compacts, i.e. monolithic dosage forms or particles, respectively.
- thermoforming preferably means the forming or molding of a mass after, before or during the application of heat.
- thermoforming is performed by hot-melt extrusion.
- the dosage form when the dosage form is particulate and the particles are hot-melt extruded, the dosage form is a filled capsule.
- hot-melt extrusion is performed by means of a twin-screw-extruder.
- Melt extrusion preferably provides a melt-extruded strand that is preferably cut into monoliths, which are then optionally compressed and formed.
- compression is achieved by means of a die and a punch, preferably from a monolithic mass obtained by melt extrusion. If obtained via melt extrusion, the compressing step is preferably carried out with a monolithic mass exhibiting ambient temperature, that is, a temperature in the range from 20 to 25°C.
- the dosage forms and particles, respectively, are manufactured by thermoforming, they may be produced by different processes.
- Several suitable processes have already been described in the prior art. In this regard it can be referred to, e.g., WO 2005/016313, WO 2005/016314, WO 2005/063214, WO 2005/102286, WO 2006/002883, WO 2006/002884, WO 2006/002886, WO 2006/082097, and WO 2006/082099.
- the dosage form when it is in form of a tablet, it is prepared by compression.
- the particles as hereinbefore defined are preferably mixed, e.g. blended and/or granulated (e.g. wet granulated), with outer matrix material and the resulting mix (e.g. blend or granulate) is then compressed, preferably in molds, to form dosage forms.
- the particles herein described may be incorporated into a matrix using other processes, such as by melt granulation (e.g. using fatty alcohols and/or water-soluble waxes and/or water-insoluble waxes) or high shear granulation, followed by compression.
- the compression force is preferably within the range of from 5 to 15 kN.
- the compression force is preferably within the range of from 5 to 40 kN, in certain embodiments >25 kN, in other embodiments about 13 kN.
- Another aspect of the invention relates to a dosage form which is obtainable by any of the processes described above.
- the release profile, the pharmacologically active ingredient, the alkyl cellulose, the heteropolysaccharide and optionally present pharmaceutical excipients are stable upon storage, preferably upon storage at elevated temperature, e.g. 40°C, for 3 months in sealed containers.
- “stable" preferably means that when comparing the initial release profile with the release profile after storage, at any given time point the release profiles deviate from one another by not more than 20%, more preferably not more than 15%, still more preferably not more than 10%, yet more preferably not more than 7.5%, most preferably not more than 5.0% and in particular not more than 2.5%.
- the alkyl cellulose, the heteropolysaccharide, the optionally present further pharmacologically active ingredient and the optionally present pharmaceutical excipients "stable" preferably means that the dosage forms satisfy the requirements of EMEA concerning shelf- life of pharmaceutical products.
- the content of the pharmacologically active ingredient, preferably the opioid in the dosage form according to the invention amounts to at least 98.0%, more preferably at least 98.5%, still more preferably at least 99.0%, yet more preferably at least 99.2%, most preferably at least 99.4% and in particular at least 99.6%, of its original content before storage.
- the dosage form according to the invention contains no substances which irritate the nasal passages and/or pharynx, i.e. substances which, when administered via the nasal passages and/or pharynx, bring about a physical reaction which is either so unpleasant for the patient that he/she does not wish to or cannot continue administration, for example burning, or physiologically counteracts taking of the corresponding active compound, for example due to increased nasal secretion or sneezing.
- substances which irritate the nasal passages and/or pharynx are those which cause burning, itching, urge to sneeze, increased formation of secretions or a combination of at least two of these stimuli.
- Corresponding substances and the quantities thereof which are conventionally to be used are known to the person skilled in the art. Some of the substances which irritate the nasal passages and/or pharynx are accordingly based on one or more constituents or one or more plant parts of a hot substance drug.
- Corresponding hot substance drugs are known per se to the person skilled in the art and are described, for example, in "Pharmazeutician Biologie - Drogen und Strukturbericht" by Prof. Dr. Hildebert Wagner, 2nd., revised edition, Gustav Fischer Verlag, Stuttgart-New York, 1982, pages 82 et seq. The corresponding description is hereby introduced as a reference and is deemed to be part of the disclosure.
- the dosage form according to the invention furthermore preferably contains no antagonists for the pharmacologically active ingredient, preferably the opioid and the optionally present further pharmacologically active ingredient, preferably no antagonists against psychotropic substances, in particular no antagonists against opioids.
- Antagonists suitable for a given pharmacologically active ingredient are known to the person skilled in the art and may be present as such or in the form of corresponding derivatives, in particular esters or ethers, or in each case in the form of corresponding physiologically acceptable compounds, in particular in the form of the salts or solvates thereof.
- the dosage form according to the invention preferably contains no antagonists selected from among the group comprising naloxone, naltrexone, nalmefene, nalide, nalmexone, nalorphine or naluphine, in each case optionally in the form of a corresponding physiologically acceptable compound, in particular in the form of a base, a salt or solvate; and no neuroleptics, for example a compound selected from among the group comprising haloperidol, promethacine, fluphenazine, perphenazine, levomepromazine, thioridazine, perazine, chlorpromazine, chlorprothixine, zuclopenthixol, flupentixol, prothipendyl, zotepine, benperidol, pipamperone, melperone and bromperidol.
- no antagonists selected from among the group comprising naloxone, naltrex
- the dosage form according to the invention furthermore preferably contains no emetic.
- Emetics are known to the person skilled in the art and may be present as such or in the form of corresponding derivatives, in particular esters or ethers, or in each case in the form of corresponding physiologically acceptable compounds, in particular in the form of the salts or solvates thereof.
- the dosage form according to the invention preferably contains no emetic based on one or more constituents of ipecacuanha (ipecac) root, for example based on the constituent emetine, as are, for example, described in "Pharmazeutician Biologie - Drogen und Hä Kunststoffsstoffe" by Prof. Dr. Hildebert Wagner, 2nd, revised edition, Gustav Fischer Verlag, Stuttgart, New York, 1982.
- the corresponding literature description is hereby introduced as a reference and is deemed to be part of the disclosure.
- the dosage form according to the invention preferably also contains no apomorphine as an emetic.
- the dosage form according to the invention preferably also contains no bitter substance.
- bitter substances and the quantities effective for use may be found in US 2003/0064099 Al, the corresponding disclosure of which should be deemed to be the disclosure of the present application and is hereby introduced as a reference.
- bitter substances are aromatic oils, such as peppermint oil, eucalyptus oil, bitter almond oil, menthol, fruit aroma substances, aroma substances from lemons, oranges, limes, grapefruit or mixtures thereof, and/or denatonium benzoate.
- the dosage form according to the invention accordingly preferably contains neither substances which irritate the nasal passages and/or pharynx, nor antagonists for the pharmacologically active ingredient, nor emetics, nor bitter substances.
- the dosage form according to the invention is adapted for administration once daily, preferably orally. In another preferred embodiment, the dosage form according to the invention is adapted for administration twice daily, preferably orally. In still another preferred embodiment, the dosage form according to the invention is adapted for administration thrice daily, preferably orally. In yet another preferred embodiment, the dosage form according to the invention is adapted for administration more frequently than thrice daily, for example 4 times daily, 5 times daily, 6 times daily, 7 times daily or 8 times daily, in each case preferably orally.
- time intervals i.e., about every 12 hours, or different time intervals, e.g., 8 and 16 hours or 10 and 14 hours, between the individual administrations.
- thrice daily means equal or nearly equal time intervals, i.e., about every 8 hours, or different time intervals, e.g., 6, 6 and 12 hours; or 7, 7 and 10 hours, between the individual administrations.
- the dosage forms according to the invention may be used in medicine, e.g. as an analgesic.
- the dosage forms are therefore particularly suitable for the treatment or management of pain.
- a further aspect of the invention relates to the dosage form as described above for use in the treatment of pain.
- a further aspect of the invention relates to the use of a pharmacologically active ingredient, preferably an opioid for the manufacture of a dosage form as described above for treating pain.
- a further aspect of the invention relates to a method of treating pain comprising the administration of the dosage form as described above to a subject in need thereof.
- a further aspect according to the invention relates to the use of a dosage form as described above for avoiding or hindering the unintentional overdose of the pharmacologically active ingredient, preferably the opioid contained therein.
- the invention also relates to the use of a dosage form as described above for the prophylaxis and/or the treatment of a disorder, thereby preventing an overdose of the pharmacologically active ingredient, preferably the opioid, particularly due to dose dumping in aqueous ethanol.
- the dosage form is particulate and in form of a filled capsule, wherein the pharmacologically active ingredient is selected from the group consisting of oxycodone, oxymorphone, hydrocodone, hydromorphone, tramadol, tapentadol, morphine, buprenorphine and the physiologically acceptable salts thereof; and
- the alkyl cellulose is ethyl cellulose
- heteropolysaccharide is xanthan gum
- the relative weight ratio of xanthan gum to ethyl cellulose is within the range of from 1 : 18 to 1 : 1.
- the weight ratio of heteropolysaccharide to alkyl cellulose is within the range of from 1 : 18 to 1 : 1 ; and/or the total content of alkyl cellulose and heteropolysaccharide is at least 95 wt.-%, relative to the total weight of the matrix material, or the matrix material consists of the alkyl cellulose and the heteropolysaccharide; and/or
- the alkyl cellulose is selected from unsubstituted Ci_6-alkyl celluloses; and/or
- the alkyl cellulose is ethyl cellulose having an ethoxyl content of from 40 wt.-% to 60 wt.-%; and/or having a solution viscosity within the range of from 70 mPa- s to 130 mPa- s, measured in a 5 wt.-% solution of 80 wt.- % toluene and 20 wt.-% ethanol at 25°C in an Ubbelohde viscosimeter; and/or
- the content of the alkyl cellulose in the matrix material is at most 90 wt.-%, relative to the total weight of the matrix material;
- the content of the alkyl cellulose is within the range of from 45 to 89 wt.-%, relative to the total weight of the dosage form; and/or the heteropolysaccharide is selected from acidic heteropolysaccharides; and/or
- heteropolysaccharide is xanthan gum
- the content of the heteropolysaccharide in the matrix material is at least 10 wt.- , relative to the total weight of the matrix material;
- the content of the heteropolysaccharide is within the range of from 8 to 27 wt.- , relative to the total weight of the dosage form;
- the dosage form contains only one pharmacologically active ingredient selected from the group consisting of oxycodone, oxymorphone, hydrocodone, hydromorphone, tramadol, tapentadol, morphine, buprenorphine and the physiologically acceptable salts thereof; and/or
- the pharmacologically active ingredient preferably the opioid is present in the dosage form in a therapeutically effective amount; and/or
- the dosage form does not contain any further pharmacologically active ingredient;
- the dosage form has a breaking strength of less than 200 N, or, when the dosage form is particulate, the particles have a breaking strength of less than 200 N;
- the in vitro release of the pharmacologically active ingredient, preferably the opioid from the dosage form in 0.1 N HCl / ethanol (40 vol.-%) relatively deviates from the in vitro release in 0.1 N HCl by not more than +1 %; or by at least -1 %; and/or
- the dosage form provides prolonged release of the pharmacologically active ingredient, preferably the opioid; and/or
- the dosage form is particulate, wherein the particles have been manufactured by granulation; and/or the dosage form is a filled capsule; and/or
- the dosage form does not contain any outer matrix material.
- the xanthan gum was found to have a viscosity of 281 mPa- s, measured in a 1 % aqueous solution at a shear rate of 50 s "1 rotationally at 20°C after 1 minute equilibration using a 6 cm acrylic cone (1°), wherein the shear was ramped up linearly from 1 to 50 s "1 in 25 steps over 29 s.
- the release profiles were determined in 0.1 N HC1 with and without addition of 40% (v/v) ethanol in a USP Apparatus 2 (paddle) at 75 rpm in 600 mL of media at 37°C with a LabSwiss sinker, using a HPLC method.
- the mobile phase consisted of 1460 mL potassium dihydrogenphosphate buffer pH 2.7 wit 540 mL methanol with a flow rate of 2.5 mL/min.
- the stationary phase was a Supelcosil LC-8 DB 5 ⁇ 150*4.6mm chromatographic column conditioned at 35°C. Injected volume was 30 ⁇ L, detection was performed by UV absorption at a wavelength of 215 nm.
- the obtained release data were normalized in that always the highest value measured after quickly stirring for a longer time was taken as 100% value ("infinity value").
- Figure 1 shows the release profile of the capsules in 0.1 N HC1 and in a mixture of 0.1 N HC1 and 40% ethanol, respectively.
- Figure 2 shows the release profile of the capsules in 0.1 N HC1 and in a mixture of 0.1 N HC1 and 40% ethanol, respectively. The data are also summarized in Table 10.
- Figure 3 shows the release profile of the capsules in 0.1 N HC1 and in a mixture of 0.1 N HC1 and 40% ethanol, respectively.
- the data are also summarized in Table 10.
- Figure 4 shows the release profile of the capsules in 0.1 N HC1 and in a mixture of 0.1 N HC1 and 40% ethanol, respectively.
- the data are also summarized in Table 10.
- Figure 4 in both media the release of the active ingredient is distinctly prolonged and is further retarded by addition of ethanol.
- Figure 5 shows the release profile of the capsules in 0.1 N HCl and in a mixture of 0.1 N HCl and 40% ethanol, respectively.
- the data are also summarized in Table 10.
Abstract
Description











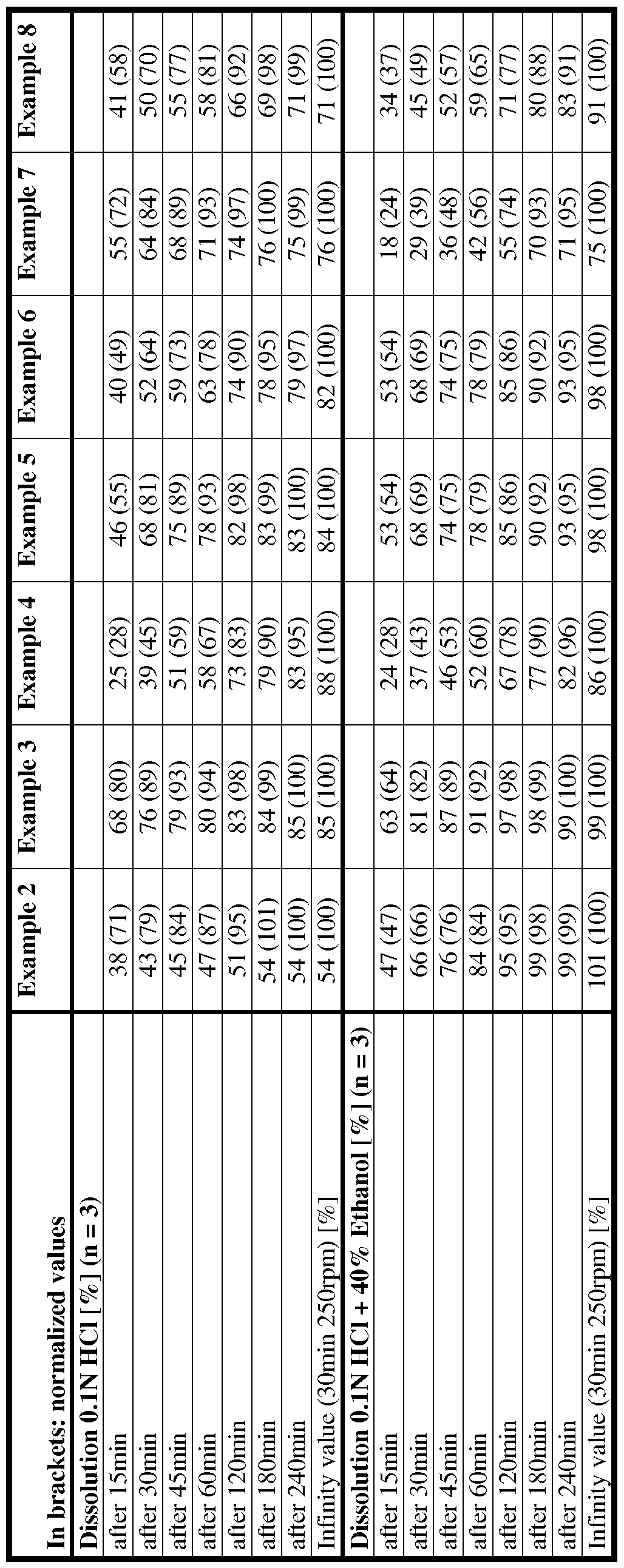
Claims
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2015266117A AU2015266117A1 (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping |
| CA2949422A CA2949422A1 (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping |
| MX2016015417A MX2016015417A (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping. |
| EA201692388A EA201692388A1 (en) | 2014-05-26 | 2015-05-22 | DOSAGE FORM AS PARTICLE MULTIPLE, PROTECTED AGAINST CALLED DOSE RESET BY ETHANOL |
| CN201580027660.1A CN106456550A (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping |
| EP15724622.4A EP3148512A1 (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping |
| JP2016569686A JP2017516789A (en) | 2014-05-26 | 2015-05-22 | Multiparticulates protected against ethanol overdose |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14169801 | 2014-05-26 | ||
| EP14169801.9 | 2014-05-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015181059A1 true WO2015181059A1 (en) | 2015-12-03 |
Family
ID=50774716
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2015/061343 WO2015181059A1 (en) | 2014-05-26 | 2015-05-22 | Multiparticles safeguarded against ethanolic dose-dumping |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9872835B2 (en) |
| EP (1) | EP3148512A1 (en) |
| JP (1) | JP2017516789A (en) |
| CN (1) | CN106456550A (en) |
| AU (1) | AU2015266117A1 (en) |
| CA (1) | CA2949422A1 (en) |
| EA (1) | EA201692388A1 (en) |
| MX (1) | MX2016015417A (en) |
| WO (1) | WO2015181059A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11077055B2 (en) | 2015-04-29 | 2021-08-03 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2938699A1 (en) | 2014-02-05 | 2015-08-13 | Kashiv Pharma Llc | Abuse-resistant drug formulations with built-in overdose protection |
| US20170266112A1 (en) | 2014-06-11 | 2017-09-21 | Massachusetts Institute Of Technology | Residence structures and related methods |
| AU2015274456B2 (en) | 2014-06-11 | 2020-03-26 | Massachusetts Institute Of Technology | Self-assembled residence devices and related methods |
| WO2017019393A1 (en) * | 2015-07-27 | 2017-02-02 | Dow Global Technologies Llc | Method to additive manufacture biocompatible material and articles made by the method |
| EP3364946A4 (en) | 2015-10-23 | 2019-06-26 | Lyndra, Inc. | Gastric residence systems for sustained release of therapeutic agents and methods of use thereof |
| US10076494B2 (en) | 2016-06-16 | 2018-09-18 | Dexcel Pharma Technologies Ltd. | Stable orally disintegrating pharmaceutical compositions |
| WO2018064630A1 (en) * | 2016-09-30 | 2018-04-05 | Lyndra, Inc. | Gastric residence systems for sustained delivery of adamantane-class drugs |
| US20180140536A1 (en) * | 2016-11-22 | 2018-05-24 | Epc Natural Products Co., Ltd. | Ivory nut powder and mannan from ivory nut |
| EP3473246A1 (en) | 2017-10-19 | 2019-04-24 | Capsugel Belgium NV | Immediate release abuse deterrent formulations |
| WO2020106735A1 (en) * | 2018-11-19 | 2020-05-28 | Jazz Pharmaceuticals Ireland Limited | Alcohol-resistant drug formulations |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080085304A1 (en) * | 2006-10-10 | 2008-04-10 | Penwest Pharmaceuticals Co. | Robust sustained release formulations |
| WO2009034541A2 (en) * | 2007-09-11 | 2009-03-19 | Ranbaxy Laboratories Limited | Controlled release pharmaceutical dosage forms of trimetazidine |
Family Cites Families (508)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA722109A (en) | 1965-11-23 | W. Mock Henry | Extrusion of ethylene oxide polymers | |
| CA229621A (en) | 1923-03-20 | Bowden Thomas | Toy vehicle | |
| US2524855A (en) | 1950-10-10 | Process for the manufacture of | ||
| US2806033A (en) | 1955-08-03 | 1957-09-10 | Lewenstein | Morphine derivative |
| US2987445A (en) | 1958-10-10 | 1961-06-06 | Rohm & Haas | Drug composition |
| US3370035A (en) | 1961-06-23 | 1968-02-20 | Takeda Chemical Industries Ltd | Stabilization of polyalkylene oxide |
| US3332950A (en) | 1963-03-23 | 1967-07-25 | Endo Lab | 14-hydroxydihydronormorphinone derivatives |
| GB1147210A (en) | 1965-06-30 | 1969-04-02 | Eastman Kodak Co | Improvements in or relating to vitamins |
| US3652589A (en) | 1967-07-27 | 1972-03-28 | Gruenenthal Chemie | 1-(m-substituted phenyl)-2-aminomethyl cyclohexanols |
| US3806603A (en) | 1969-10-13 | 1974-04-23 | W Gaunt | Pharmaceutical carriers of plasticized dried milled particles of hydrated cooked rice endosperm |
| DE2210071A1 (en) | 1971-03-09 | 1972-09-14 | PPG Industries Inc., Pittsburgh, Pa. (V.StA.) | Process for applying and curing a wide variety of coatings |
| US3865108A (en) | 1971-05-17 | 1975-02-11 | Ortho Pharma Corp | Expandable drug delivery device |
| US3966747A (en) | 1972-10-26 | 1976-06-29 | Bristol-Myers Company | 9-Hydroxy-6,7-benzomorphans |
| US4014965A (en) | 1972-11-24 | 1977-03-29 | The Dow Chemical Company | Process for scrapless forming of plastic articles |
| US3980766A (en) | 1973-08-13 | 1976-09-14 | West Laboratories, Inc. | Orally administered drug composition for therapy in the treatment of narcotic drug addiction |
| US3941865A (en) | 1973-12-10 | 1976-03-02 | Union Carbide Corporation | Extrusion of ethylene oxide resins |
| US4002173A (en) | 1974-07-23 | 1977-01-11 | International Paper Company | Diester crosslinked polyglucan hydrogels and reticulated sponges thereof |
| DE2530563C2 (en) | 1975-07-09 | 1986-07-24 | Bayer Ag, 5090 Leverkusen | Analgesic drugs with reduced potential for abuse |
| JPS603286B2 (en) | 1977-03-03 | 1985-01-26 | 日本化薬株式会社 | Constant-dissolution formulation |
| US4207893A (en) | 1977-08-29 | 1980-06-17 | Alza Corporation | Device using hydrophilic polymer for delivering drug to biological environment |
| US4175119A (en) | 1978-01-11 | 1979-11-20 | Porter Garry L | Composition and method to prevent accidental and intentional overdosage with psychoactive drugs |
| DE2822324C3 (en) | 1978-05-22 | 1981-02-26 | Basf Ag, 6700 Ludwigshafen | Manufacture of vitamin E dry powder |
| US4211681A (en) | 1978-08-16 | 1980-07-08 | Union Carbide Corporation | Poly(ethylene oxide) compositions |
| US4200704A (en) | 1978-09-28 | 1980-04-29 | Union Carbide Corporation | Controlled degradation of poly(ethylene oxide) |
| NO793297L (en) | 1978-10-19 | 1980-04-22 | Mallinckrodt Inc | PROCEDURE FOR THE MANUFACTURE OF OXYMORPHONE |
| US4215104A (en) | 1979-03-26 | 1980-07-29 | Mead Johnson & Company | Multi-fractionable tablet structure |
| US4258027A (en) | 1979-03-26 | 1981-03-24 | Mead Johnson & Company | Multi-fractionable tablet structure |
| CA1146866A (en) | 1979-07-05 | 1983-05-24 | Yamanouchi Pharmaceutical Co. Ltd. | Process for the production of sustained release pharmaceutical composition of solid medical material |
| US4353887A (en) | 1979-08-16 | 1982-10-12 | Ciba-Geigy Corporation | Divisible tablet having controlled and delayed release of the active substance |
| CH648754A5 (en) | 1979-08-16 | 1985-04-15 | Ciba Geigy Ag | Pharmaceutical slow release tablet |
| US4457933A (en) | 1980-01-24 | 1984-07-03 | Bristol-Myers Company | Prevention of analgesic abuse |
| JPS56169622A (en) | 1980-06-03 | 1981-12-26 | Kissei Pharmaceut Co Ltd | Method of making solid preparation from oily substance |
| DE3024416C2 (en) | 1980-06-28 | 1982-04-15 | Gödecke AG, 1000 Berlin | Process for the production of medicaments with sustained release of active substances |
| US4473640A (en) | 1982-06-03 | 1984-09-25 | Combie Joan D | Detection of morphine and its analogues using enzymatic hydrolysis |
| US4462941A (en) | 1982-06-10 | 1984-07-31 | The Regents Of The University Of California | Dynorphin amide analogs |
| US4427778A (en) | 1982-06-29 | 1984-01-24 | Biochem Technology, Inc. | Enzymatic preparation of particulate cellulose for tablet making |
| US4485211A (en) | 1982-09-15 | 1984-11-27 | The B. F. Goodrich Company | Poly(glycidyl ether)block copolymers and process for their preparation |
| US4427681A (en) | 1982-09-16 | 1984-01-24 | Richardson-Vicks, Inc. | Thixotropic compositions easily convertible to pourable liquids |
| US4529583A (en) | 1983-03-07 | 1985-07-16 | Clear Lake Development Group | Composition and method of immobilizing emetics and method of treating human beings with emetics |
| US4603143A (en) | 1983-05-02 | 1986-07-29 | Basf Corporation | Free-flowing, high density, fat soluble vitamin powders with improved stability |
| US4612008A (en) | 1983-05-11 | 1986-09-16 | Alza Corporation | Osmotic device with dual thermodynamic activity |
| US4783337A (en) | 1983-05-11 | 1988-11-08 | Alza Corporation | Osmotic system comprising plurality of members for dispensing drug |
| US5082668A (en) | 1983-05-11 | 1992-01-21 | Alza Corporation | Controlled-release system with constant pushing source |
| US4765989A (en) | 1983-05-11 | 1988-08-23 | Alza Corporation | Osmotic device for administering certain drugs |
| US4599342A (en) | 1984-01-16 | 1986-07-08 | The Procter & Gamble Company | Pharmaceutical products providing enhanced analgesia |
| US4629621A (en) | 1984-07-23 | 1986-12-16 | Zetachron, Inc. | Erodible matrix for sustained release bioactive composition |
| AU592065B2 (en) | 1984-10-09 | 1990-01-04 | Dow Chemical Company, The | Sustained release dosage form based on highly plasticized cellulose ether gels |
| GB8507779D0 (en) | 1985-03-26 | 1985-05-01 | Fujisawa Pharmaceutical Co | Drug carrier |
| DE3673789D1 (en) | 1985-06-24 | 1990-10-04 | Ici Australia Ltd | PACKABLE CAPSULES. |
| EP0227806B1 (en) | 1985-06-28 | 1989-08-30 | Carrington Laboratories, Inc. | Processes for preparation of aloe products, products produced thereby and compositions thereof |
| US4992279A (en) | 1985-07-03 | 1991-02-12 | Kraft General Foods, Inc. | Sweetness inhibitor |
| US4851521A (en) | 1985-07-08 | 1989-07-25 | Fidia, S.P.A. | Esters of hyaluronic acid |
| DE3689650T2 (en) | 1985-12-17 | 1994-05-26 | United States Surgical Corp | High molecular weight bioabsorbable polymers and implants thereof. |
| US5229164A (en) | 1985-12-19 | 1993-07-20 | Capsoid Pharma Gmbh | Process for producing individually dosed administration forms |
| US4711894A (en) | 1986-01-16 | 1987-12-08 | Henkel Corporation | Stabilized tocopherol in dry, particulate, free-flowing form |
| US5198226A (en) | 1986-01-30 | 1993-03-30 | Syntex (U.S.A.) Inc. | Long acting nicardipine hydrochloride formulation |
| US4940556A (en) | 1986-01-30 | 1990-07-10 | Syntex (U.S.A.) Inc. | Method of preparing long acting formulation |
| US4764378A (en) | 1986-02-10 | 1988-08-16 | Zetachron, Inc. | Buccal drug dosage form |
| EP0239973A3 (en) | 1986-03-31 | 1989-11-08 | Union Carbide Corporation | Catalyst and process for alkylene oxide polymerization |
| DE3612211A1 (en) | 1986-04-11 | 1987-10-15 | Basf Ag | CONTINUOUS TABLET METHOD |
| US4667013A (en) | 1986-05-02 | 1987-05-19 | Union Carbide Corporation | Process for alkylene oxide polymerization |
| US4713243A (en) | 1986-06-16 | 1987-12-15 | Johnson & Johnson Products, Inc. | Bioadhesive extruded film for intra-oral drug delivery and process |
| USRE33093E (en) | 1986-06-16 | 1989-10-17 | Johnson & Johnson Consumer Products, Inc. | Bioadhesive extruded film for intra-oral drug delivery and process |
| USRE34990E (en) | 1986-08-07 | 1995-07-04 | Ciba-Geigy Corporation | Oral therapeutic system having systemic action |
| CA1335748C (en) | 1986-09-25 | 1995-05-30 | Jeffrey Lawrence Finnan | Crosslinked gelatins |
| US5227157A (en) | 1986-10-14 | 1993-07-13 | Board Of Regents, The University Of Texas System | Delivery of therapeutic agents |
| JP2962731B2 (en) | 1986-11-10 | 1999-10-12 | バイオピュアー、コーポレーション | Ultra-pure semi-synthetic blood substitute |
| US4892889A (en) | 1986-11-18 | 1990-01-09 | Basf Corporation | Process for making a spray-dried, directly-compressible vitamin powder comprising unhydrolyzed gelatin |
| JPH0831303B2 (en) | 1986-12-01 | 1996-03-27 | オムロン株式会社 | Chip type fuse |
| DE3868077D1 (en) | 1987-01-14 | 1992-03-12 | Ciba Geigy Ag | THERAPEUTIC SYSTEM FOR HEAVY-SOLUBLE ACTIVE SUBSTANCES. |
| US4892778A (en) | 1987-05-27 | 1990-01-09 | Alza Corporation | Juxtaposed laminated arrangement |
| US5051261A (en) | 1987-11-24 | 1991-09-24 | Fmc Corporation | Method for preparing a solid sustained release form of a functionally active composition |
| KR900700071A (en) | 1987-12-17 | 1990-08-11 | 로버어트 에이 아미테이지 | Tri-scored Drug Tablets |
| DE3812567A1 (en) | 1988-04-15 | 1989-10-26 | Basf Ag | METHOD FOR PRODUCING PHARMACEUTICAL MIXTURES |
| US4954346A (en) | 1988-06-08 | 1990-09-04 | Ciba-Geigy Corporation | Orally administrable nifedipine solution in a solid light resistant dosage form |
| US4960814A (en) | 1988-06-13 | 1990-10-02 | Eastman Kodak Company | Water-dispersible polymeric compositions |
| US5350741A (en) | 1988-07-30 | 1994-09-27 | Kanji Takada | Enteric formulations of physiologically active peptides and proteins |
| JPH0249719A (en) | 1988-08-11 | 1990-02-20 | Dai Ichi Kogyo Seiyaku Co Ltd | Oil soluble-vitamin powder having readily water-dispersible and soluble performance |
| GB8820327D0 (en) | 1988-08-26 | 1988-09-28 | May & Baker Ltd | New compositions of matter |
| DE3830353A1 (en) | 1988-09-07 | 1990-03-15 | Basf Ag | METHOD FOR THE CONTINUOUS PRODUCTION OF SOLID PHARMACEUTICAL FORMS |
| US5004601A (en) | 1988-10-14 | 1991-04-02 | Zetachron, Inc. | Low-melting moldable pharmaceutical excipient and dosage forms prepared therewith |
| US5139790A (en) | 1988-10-14 | 1992-08-18 | Zetachron, Inc. | Low-melting moldable pharmaceutical excipient and dosage forms prepared therewith |
| US4957668A (en) | 1988-12-07 | 1990-09-18 | General Motors Corporation | Ultrasonic compacting and bonding particles |
| US5190760A (en) | 1989-07-08 | 1993-03-02 | Coopers Animal Health Limited | Solid pharmaceutical composition |
| US5169645A (en) | 1989-10-31 | 1992-12-08 | Duquesne University Of The Holy Ghost | Directly compressible granules having improved flow properties |
| US5200197A (en) | 1989-11-16 | 1993-04-06 | Alza Corporation | Contraceptive pill |
| GB8926612D0 (en) | 1989-11-24 | 1990-01-17 | Erba Farmitalia | Pharmaceutical compositions |
| EP0449775A3 (en) | 1990-03-29 | 1992-09-02 | Ciba-Geigy Ag | Polyether-polyester block copolymers and their use as dispersing agents |
| SU1759445A1 (en) | 1990-06-15 | 1992-09-07 | Ленинградский Технологический Институт Им.Ленсовета | Method of producing encapsulated hydrophobic substances |
| FR2664851B1 (en) | 1990-07-20 | 1992-10-16 | Oreal | METHOD OF COMPACTING A POWDER MIXTURE FOR OBTAINING A COMPACT ABSORBENT OR PARTIALLY DELITABLE PRODUCT AND PRODUCT OBTAINED BY THIS PROCESS. |
| EP0477135A1 (en) | 1990-09-07 | 1992-03-25 | Warner-Lambert Company | Chewable spheroidal coated microcapsules and methods for preparing same |
| US5126151A (en) | 1991-01-24 | 1992-06-30 | Warner-Lambert Company | Encapsulation matrix |
| US5273758A (en) | 1991-03-18 | 1993-12-28 | Sandoz Ltd. | Directly compressible polyethylene oxide vehicle for preparing therapeutic dosage forms |
| US5149538A (en) | 1991-06-14 | 1992-09-22 | Warner-Lambert Company | Misuse-resistive transdermal opioid dosage form |
| JP3073054B2 (en) | 1991-07-11 | 2000-08-07 | 住友精化株式会社 | Method for producing alkylene oxide polymer |
| US5496563A (en) | 1991-08-30 | 1996-03-05 | Showa Yakuhin Kako Co., Ltd. | Dry gel composition |
| WO1993006723A1 (en) | 1991-10-04 | 1993-04-15 | Olin Corporation | Fungicide tablet |
| KR100205276B1 (en) | 1991-10-04 | 1999-07-01 | 가마쿠라 아키오 | Sustained-release tablet |
| DE4138513A1 (en) | 1991-11-23 | 1993-05-27 | Basf Ag | SOLID PHARMACEUTICAL RETARD FORM |
| US5266331A (en) | 1991-11-27 | 1993-11-30 | Euroceltique, S.A. | Controlled release oxycodone compositions |
| EP0615438B1 (en) | 1991-12-05 | 1996-07-24 | Mallinckrodt Veterinary, Inc. | A carbohydrate glass matrix for the sustained release of a therapeutic agent |
| JP3722293B2 (en) | 1991-12-18 | 2005-11-30 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | Novel pharmaceutical solid dispersion |
| US5225417A (en) | 1992-01-21 | 1993-07-06 | G. D. Searle & Co. | Opioid agonist compounds |
| IL105553A (en) | 1992-05-06 | 1998-01-04 | Janssen Pharmaceutica Inc | Solid dosage form comprising a porous network of matrix forming material which disperses rapidly in water |
| ATE136459T1 (en) | 1992-05-22 | 1996-04-15 | Goedecke Ag | METHOD FOR PRODUCING SUSTAINED-RELEASE MEDICINAL PREPARATIONS |
| GB9217295D0 (en) | 1992-08-14 | 1992-09-30 | Wellcome Found | Controlled released tablets |
| DE4227385A1 (en) | 1992-08-19 | 1994-02-24 | Kali Chemie Pharma Gmbh | Pancreatin micropellets |
| DE4229085C2 (en) | 1992-09-01 | 1996-07-11 | Boehringer Mannheim Gmbh | Elongated, divisible tablet |
| KR100355130B1 (en) | 1992-09-18 | 2003-01-30 | 야마노우치세이야쿠 가부시키가이샤 | Hydrogel Sustained Release Tablet |
| US5472943A (en) | 1992-09-21 | 1995-12-05 | Albert Einstein College Of Medicine Of Yeshiva University, | Method of simultaneously enhancing analgesic potency and attenuating dependence liability caused by morphine and other opioid agonists |
| FI101039B (en) | 1992-10-09 | 1998-04-15 | Eeva Kristoffersson | Method for preparing medicated pellets |
| AU679937B2 (en) | 1992-11-18 | 1997-07-17 | Johnson & Johnson Consumer Products, Inc. | Extrudable compositions for topical or transdermal drug delivery |
| ATE183641T1 (en) | 1992-12-23 | 1999-09-15 | Saitec Srl | METHOD FOR PRODUCING MEDICINAL FORMS AND CONTROLLED RELEASE AND THE MEDICINAL FORMS OBTAINED |
| GB2273874A (en) | 1992-12-31 | 1994-07-06 | Pertti Olavi Toermaelae | Preparation of pharmaceuticals in a polymer matrix |
| US6071970A (en) | 1993-02-08 | 2000-06-06 | Nps Pharmaceuticals, Inc. | Compounds active at a novel site on receptor-operated calcium channels useful for treatment of neurological disorders and diseases |
| DE4309528C2 (en) | 1993-03-24 | 1998-05-20 | Doxa Gmbh | Casein film or film tube, process for their production and their use |
| IL119660A (en) | 1993-05-10 | 2002-09-12 | Euro Celtique Sa | Controlled release formulation comprising tramadol |
| IL109944A (en) | 1993-07-01 | 1998-12-06 | Euro Celtique Sa | Sustained release dosage unit forms containing morphine and a method of preparing these sustained release dosage unit forms |
| DE4329794C2 (en) | 1993-09-03 | 1997-09-18 | Gruenenthal Gmbh | Tramadol salt-containing drugs with delayed release |
| EP1442745A1 (en) | 1993-10-07 | 2004-08-04 | Euro-Celtique | Orally administrable opioid formulations having extended duration of effect |
| PT654263E (en) | 1993-11-23 | 2002-06-28 | Euro Celtique Sa | METHOD FOR THE PREPARATION OF A PROLONGED LIBERTYACAO COMPOSITION |
| KR100354702B1 (en) | 1993-11-23 | 2002-12-28 | 유로-셀티크 소시에떼 아노뉨 | Manufacturing method and sustained release composition of pharmaceutical composition |
| AU1266895A (en) | 1993-12-20 | 1995-07-10 | Procter & Gamble Company, The | Process for making laxatives containing dioctyl sulfosuccinate |
| IL112106A0 (en) | 1993-12-22 | 1995-03-15 | Ergo Science Inc | Accelerated release composition containing bromocriptine |
| GB9401894D0 (en) | 1994-02-01 | 1994-03-30 | Rhone Poulenc Rorer Ltd | New compositions of matter |
| EP0744941B1 (en) | 1994-02-16 | 2003-06-04 | Abbott Laboratories | Process for preparing fine particle pharmaceutical formulations |
| SE9503924D0 (en) | 1995-08-18 | 1995-11-07 | Astra Ab | Novel opioid peptides |
| US5458887A (en) | 1994-03-02 | 1995-10-17 | Andrx Pharmaceuticals, Inc. | Controlled release tablet formulation |
| DE4413350A1 (en) | 1994-04-18 | 1995-10-19 | Basf Ag | Retard matrix pellets and process for their production |
| ES2163504T5 (en) | 1994-05-06 | 2008-05-16 | Pfizer Inc. | DOSAGE FORMS OF CONTROLLED AZITROMYCIN RELEASE. |
| DE19509807A1 (en) | 1995-03-21 | 1996-09-26 | Basf Ag | Process for the preparation of active substance preparations in the form of a solid solution of the active substance in a polymer matrix, and active substance preparations produced using this method |
| AT403988B (en) | 1994-05-18 | 1998-07-27 | Lannacher Heilmittel | SOLID ORAL RETARDED PREPARATION |
| US5460826A (en) | 1994-06-27 | 1995-10-24 | Alza Corporation | Morphine therapy |
| DE4426245A1 (en) | 1994-07-23 | 1996-02-22 | Gruenenthal Gmbh | 1-phenyl-3-dimethylamino-propane compounds with pharmacological activity |
| IT1274879B (en) | 1994-08-03 | 1997-07-25 | Saitec Srl | APPARATUS AND METHOD FOR PREPARING SOLID PHARMACEUTICAL FORMS WITH CONTROLLED RELEASE OF THE ACTIVE INGREDIENT. |
| JP3285452B2 (en) | 1994-08-11 | 2002-05-27 | サンスター株式会社 | Toothpaste composition |
| US5837790A (en) | 1994-10-24 | 1998-11-17 | Amcol International Corporation | Precipitation polymerization process for producing an oil adsorbent polymer capable of entrapping solid particles and liquids and the product thereof |
| AUPM897594A0 (en) | 1994-10-25 | 1994-11-17 | Daratech Pty Ltd | Controlled release container |
| US5965161A (en) | 1994-11-04 | 1999-10-12 | Euro-Celtique, S.A. | Extruded multi-particulates |
| DE4446470A1 (en) | 1994-12-23 | 1996-06-27 | Basf Ag | Process for the production of dividable tablets |
| DE19504832A1 (en) | 1995-02-14 | 1996-08-22 | Basf Ag | Solid drug preparations |
| US5945125A (en) | 1995-02-28 | 1999-08-31 | Temple University | Controlled release tablet |
| US6117453A (en) | 1995-04-14 | 2000-09-12 | Pharma Pass | Solid compositions containing polyethylene oxide and an active ingredient |
| US6348469B1 (en) | 1995-04-14 | 2002-02-19 | Pharma Pass Llc | Solid compositions containing glipizide and polyethylene oxide |
| US5900425A (en) | 1995-05-02 | 1999-05-04 | Bayer Aktiengesellschaft | Pharmaceutical preparations having controlled release of active compound and processes for their preparation |
| DE19522899C1 (en) | 1995-06-23 | 1996-12-19 | Hexal Pharmaforschung Gmbh | Process for the continuous sintering of a granulate |
| US5759583A (en) | 1995-08-30 | 1998-06-02 | Syntex (U.S.A.) Inc. | Sustained release poly (lactic/glycolic) matrices |
| US6007843A (en) | 1995-09-29 | 1999-12-28 | Lam Pharmaceuticals Corp. | Sustained release delivery system |
| US5811126A (en) | 1995-10-02 | 1998-09-22 | Euro-Celtique, S.A. | Controlled release matrix for pharmaceuticals |
| DE19539361A1 (en) | 1995-10-23 | 1997-04-24 | Basf Ag | Process for the preparation of multilayer, solid pharmaceutical forms for oral or rectal administration |
| US5908850A (en) | 1995-12-04 | 1999-06-01 | Celgene Corporation | Method of treating attention deficit disorders with d-threo methylphenidate |
| US6355656B1 (en) | 1995-12-04 | 2002-03-12 | Celgene Corporation | Phenidate drug formulations having diminished abuse potential |
| DE19547766A1 (en) | 1995-12-20 | 1997-06-26 | Gruenenthal Gmbh | 1-phenyl-2-dimethylaminomethyl-cyclohexan-1-ol compounds as active pharmaceutical ingredients |
| WO1997033566A2 (en) | 1996-03-12 | 1997-09-18 | Alza Corporation | Composition and dosage form comprising opioid antagonist |
| US6461644B1 (en) | 1996-03-25 | 2002-10-08 | Richard R. Jackson | Anesthetizing plastics, drug delivery plastics, and related medical products, systems and methods |
| US20020114838A1 (en) | 1996-04-05 | 2002-08-22 | Ayer Atul D. | Uniform drug delivery therapy |
| US6096339A (en) | 1997-04-04 | 2000-08-01 | Alza Corporation | Dosage form, process of making and using same |
| SI0914158T2 (en) | 1996-04-05 | 2006-04-30 | Takeda Chemical Industries Ltd | Pharmaceutical combination containing a compound having angiotensin ii antagonistic activity and a compound which increases the insulin-sensitivity |
| US5817343A (en) | 1996-05-14 | 1998-10-06 | Alkermes, Inc. | Method for fabricating polymer-based controlled-release devices |
| DK1014941T3 (en) | 1996-06-26 | 2009-07-27 | Univ Texas | Extrudable pharmaceutical hot-melt formulation |
| US6093420A (en) * | 1996-07-08 | 2000-07-25 | Edward Mendell Co., Inc. | Sustained release matrix for high-dose insoluble drugs |
| DE19629753A1 (en) | 1996-07-23 | 1998-01-29 | Basf Ag | Process for the production of solid dosage forms |
| NL1003684C2 (en) | 1996-07-25 | 1998-01-28 | Weterings B V H | Device for dispensing a liquid. |
| DE19630236A1 (en) | 1996-07-26 | 1998-01-29 | Wolff Walsrode Ag | Biaxially stretched, biodegradable and compostable sausage casing |
| BE1010353A5 (en) | 1996-08-14 | 1998-06-02 | Boss Pharmaceuticals Ag | Method for manufacture of pharmaceutical products, device for such a method and pharmaceutical products obtained. |
| DK0947559T3 (en) | 1996-11-05 | 2005-02-14 | Novamont Spa | Biodegradable polymer compositions containing starch and a thermoplastic polymer |
| US5991799A (en) | 1996-12-20 | 1999-11-23 | Liberate Technologies | Information retrieval system using an internet multiplexer to focus user selection |
| DE19705538C1 (en) | 1997-02-14 | 1998-08-27 | Goedecke Ag | Process for the separation of active substances in solid pharmaceutical preparations |
| US5948787A (en) | 1997-02-28 | 1999-09-07 | Alza Corporation | Compositions containing opiate analgesics |
| DE19710008A1 (en) | 1997-03-12 | 1998-09-17 | Basf Ag | Solid, at least two-phase formulations of a sustained-release opioid analgesic |
| DE19710213A1 (en) | 1997-03-12 | 1998-09-17 | Basf Ag | Process for the manufacture of solid combination dosage forms |
| DE19710009A1 (en) | 1997-03-12 | 1998-09-24 | Knoll Ag | Multi-phase preparation forms containing active ingredients |
| US6139770A (en) | 1997-05-16 | 2000-10-31 | Chevron Chemical Company Llc | Photoinitiators and oxygen scavenging compositions |
| DE19721467A1 (en) | 1997-05-22 | 1998-11-26 | Basf Ag | Process for the preparation of small-scale preparations of biologically active substances |
| PT998271E (en) | 1997-06-06 | 2005-10-31 | Depomed Inc | FORMS OF ORAL DOSAGE OF DRUGS WITH GASTRIC RETENTION FOR THE CONTROLLED LIBERATION OF HIGHLY SOLUABLE DRUGS |
| US6635280B2 (en) | 1997-06-06 | 2003-10-21 | Depomed, Inc. | Extending the duration of drug release within the stomach during the fed mode |
| DE69834195T2 (en) | 1997-07-02 | 2007-03-29 | Euro-Celtique S.A. | STABILIZED TRAMADOL FORMULATIONS WITH DELAYED RELEASE |
| IE970588A1 (en) | 1997-08-01 | 2000-08-23 | Elan Corp Plc | Controlled release pharmaceutical compositions containing tiagabine |
| AU741599B2 (en) | 1997-09-10 | 2001-12-06 | Fram Group Ip Llc | Injection molding of structural zirconia-based materials by an aqueous process |
| US6009390A (en) | 1997-09-11 | 1999-12-28 | Lucent Technologies Inc. | Technique for selective use of Gaussian kernels and mixture component weights of tied-mixture hidden Markov models for speech recognition |
| PT1033975E (en) | 1997-11-28 | 2002-07-31 | Knoll Ag | PROCESS FOR THE PREPARATION OF BIOLOGICALLY ACTIVE NAO-CRYSTALLINE SUBSTANCES ISSUED OF DISSOLVENTS |
| DE19753534A1 (en) | 1997-12-03 | 1999-06-10 | Bayer Ag | Biodegradable thermoplastic polyester-amides with good mechanical properties for molding, film and fiber, useful for e.g. compostable refuse bag |
| JP2001525433A (en) | 1997-12-03 | 2001-12-11 | バイエル・アクチエンゲゼルシヤフト | Polyetheresteramides |
| US6375957B1 (en) | 1997-12-22 | 2002-04-23 | Euro-Celtique, S.A. | Opioid agonist/opioid antagonist/acetaminophen combinations |
| EP1041988A4 (en) | 1997-12-22 | 2002-03-13 | Euro Celtique Sa | A method of preventing abuse of opioid dosage forms |
| DE19800689C1 (en) | 1998-01-10 | 1999-07-15 | Deloro Stellite Gmbh | Shaped body made of a wear-resistant material |
| DE19800698A1 (en) | 1998-01-10 | 1999-07-15 | Bayer Ag | Biodegradable polyester amides with block-like polyester and polyamide segments |
| WO1999045067A1 (en) | 1998-03-05 | 1999-09-10 | Mitsui Chemicals, Inc. | Polylactic acid composition and film thereof |
| US6245357B1 (en) | 1998-03-06 | 2001-06-12 | Alza Corporation | Extended release dosage form |
| US6090411A (en) | 1998-03-09 | 2000-07-18 | Temple University | Monolithic tablet for controlled drug release |
| US6110500A (en) | 1998-03-25 | 2000-08-29 | Temple University | Coated tablet with long term parabolic and zero-order release kinetics |
| WO1999052135A1 (en) | 1998-04-02 | 1999-10-14 | Applied Materials, Inc. | Method for etching low k dielectrics |
| EP1067910B1 (en) | 1998-04-03 | 2004-05-26 | Egalet A/S | Controlled release composition |
| US5962488A (en) | 1998-04-08 | 1999-10-05 | Roberts Laboratories, Inc. | Stable pharmaceutical formulations for treating internal bowel syndrome containing isoxazole derivatives |
| DE19822979A1 (en) | 1998-05-25 | 1999-12-02 | Kalle Nalo Gmbh & Co Kg | Film with starch or starch derivatives and polyester urethanes and process for their production |
| US6333087B1 (en) | 1998-08-27 | 2001-12-25 | Chevron Chemical Company Llc | Oxygen scavenging packaging |
| DE19841244A1 (en) | 1998-09-09 | 2000-03-16 | Knoll Ag | Method and device for making tablets |
| US6268177B1 (en) | 1998-09-22 | 2001-07-31 | Smithkline Beecham Corporation | Isolated nucleic acid encoding nucleotide pyrophosphorylase |
| CN1327384A (en) | 1998-10-20 | 2001-12-19 | 韩国科学技术研究院 | Bioflavonoids as plasma high density lipoprotein level increasing agent |
| US6322819B1 (en) | 1998-10-21 | 2001-11-27 | Shire Laboratories, Inc. | Oral pulsed dose drug delivery system |
| US20060240105A1 (en) | 1998-11-02 | 2006-10-26 | Elan Corporation, Plc | Multiparticulate modified release composition |
| DE19855440A1 (en) | 1998-12-01 | 2000-06-08 | Basf Ag | Process for the production of solid dosage forms by melt extrusion |
| DE19856147A1 (en) | 1998-12-04 | 2000-06-08 | Knoll Ag | Divisible solid dosage forms and methods for their preparation |
| EP1005863A1 (en) | 1998-12-04 | 2000-06-07 | Synthelabo | Controlled-release dosage forms comprising a short acting hypnotic or a salt thereof |
| US6238697B1 (en) | 1998-12-21 | 2001-05-29 | Pharmalogix, Inc. | Methods and formulations for making bupropion hydrochloride tablets using direct compression |
| AU3469100A (en) | 1999-01-05 | 2000-07-24 | Copley Pharmaceutical Inc. | Sustained release formulation with reduced moisture sensitivity |
| US7374779B2 (en) | 1999-02-26 | 2008-05-20 | Lipocine, Inc. | Pharmaceutical formulations and systems for improved absorption and multistage release of active agents |
| US6375963B1 (en) | 1999-06-16 | 2002-04-23 | Michael A. Repka | Bioadhesive hot-melt extruded film for topical and mucosal adhesion applications and drug delivery and process for preparation thereof |
| JP2003522127A (en) | 1999-07-29 | 2003-07-22 | ロクセニ ラボラトリーズ インコーポレイテッド | Opioid sustained release formulation |
| US20030118641A1 (en) | 2000-07-27 | 2003-06-26 | Roxane Laboratories, Inc. | Abuse-resistant sustained-release opioid formulation |
| HU229295B1 (en) | 1999-08-04 | 2013-10-28 | Astellas Pharma Inc | Stable medicinal compositions for oral use |
| US6562375B1 (en) | 1999-08-04 | 2003-05-13 | Yamanouchi Pharmaceuticals, Co., Ltd. | Stable pharmaceutical composition for oral use |
| KR100345214B1 (en) | 1999-08-17 | 2002-07-25 | 이강춘 | The nasal transmucosal delivery of peptides conjugated with biocompatible polymers |
| DE19940944B4 (en) | 1999-08-31 | 2006-10-12 | Grünenthal GmbH | Retarded, oral, pharmaceutical dosage forms |
| HUP0203623A2 (en) | 1999-08-31 | 2003-02-28 | Grünenthal GmbH | Delayed-action form of administration containing tramadol saccharinate and its use |
| DE19940740A1 (en) | 1999-08-31 | 2001-03-01 | Gruenenthal Gmbh | Pharmaceutical salts |
| DE19960494A1 (en) | 1999-12-15 | 2001-06-21 | Knoll Ag | Device and method for producing solid active substance-containing forms |
| US6680070B1 (en) | 2000-01-18 | 2004-01-20 | Albemarle Corporation | Particulate blends and compacted products formed therefrom, and the preparation thereof |
| OA12215A (en) | 2000-02-08 | 2006-05-09 | Euro Celtique Sa | Tamper-resistant oral opioid agonist formulations. |
| US20020015730A1 (en) | 2000-03-09 | 2002-02-07 | Torsten Hoffmann | Pharmaceutical formulations and method for making |
| DE10015479A1 (en) | 2000-03-29 | 2001-10-11 | Basf Ag | Solid oral dosage forms with delayed release of active ingredient and high mechanical stability |
| US8012504B2 (en) | 2000-04-28 | 2011-09-06 | Reckitt Benckiser Inc. | Sustained release of guaifenesin combination drugs |
| US6419954B1 (en) | 2000-05-19 | 2002-07-16 | Yamanouchi Pharmaceutical Co., Ltd. | Tablets and methods for modified release of hydrophilic and other active agents |
| EP2357004B1 (en) | 2000-05-23 | 2016-10-12 | CeNeS Pharmaceuticals, Inc. | NRG-2 nucleic acid molecules, polypeptides and diagnostic and therapeutical methods |
| DE10029201A1 (en) | 2000-06-19 | 2001-12-20 | Basf Ag | Retarded release oral dosage form, obtained by granulating mixture containing active agent and polyvinyl acetate-polyvinyl pyrrolidone mixture below the melting temperature |
| US6488962B1 (en) | 2000-06-20 | 2002-12-03 | Depomed, Inc. | Tablet shapes to enhance gastric retention of swellable controlled-release oral dosage forms |
| US6607748B1 (en) | 2000-06-29 | 2003-08-19 | Vincent Lenaerts | Cross-linked high amylose starch for use in controlled-release pharmaceutical formulations and processes for its manufacture |
| DE10036400A1 (en) | 2000-07-26 | 2002-06-06 | Mitsubishi Polyester Film Gmbh | White, biaxially oriented polyester film |
| EP1363673A2 (en) | 2000-09-25 | 2003-11-26 | Pro-Pharmaceuticals, Inc. | Compositions for reducing side effects in chemotherapeutic treatments |
| NZ523686A (en) | 2000-09-27 | 2004-12-24 | Danisco | Antimicrobial agent |
| WO2002026928A1 (en) | 2000-09-28 | 2002-04-04 | The Dow Chemical Company | Polymer composite structures useful for controlled release systems |
| US6344215B1 (en) | 2000-10-27 | 2002-02-05 | Eurand America, Inc. | Methylphenidate modified release formulations |
| EP1337244A4 (en) | 2000-10-30 | 2006-01-11 | Euro Celtique Sa | Controlled release hydrocodone formulations |
| WO2002035991A2 (en) | 2000-10-30 | 2002-05-10 | The Board Of Regents, The University Of Texas System | Spherical particles produced by a hot-melt extrusion/spheronization process |
| DE10109763A1 (en) | 2001-02-28 | 2002-09-05 | Gruenenthal Gmbh | Pharmaceutical salts |
| JP2002265592A (en) | 2001-03-07 | 2002-09-18 | Sumitomo Seika Chem Co Ltd | Process for producing alkylene oxide polymer |
| WO2002071860A1 (en) | 2001-03-13 | 2002-09-19 | L.A. Dreyfus Co. | Gum base and gum manufacturing using particulated gum base ingredients |
| JP3967554B2 (en) | 2001-03-15 | 2007-08-29 | 株式会社ポッカコーポレーション | Flavonoid compound and method for producing the same |
| US20020132395A1 (en) | 2001-03-16 | 2002-09-19 | International Business Machines Corporation | Body contact in SOI devices by electrically weakening the oxide under the body |
| EP1241110A1 (en) | 2001-03-16 | 2002-09-18 | Pfizer Products Inc. | Dispensing unit for oxygen-sensitive drugs |
| US20020187192A1 (en) | 2001-04-30 | 2002-12-12 | Yatindra Joshi | Pharmaceutical composition which reduces or eliminates drug abuse potential |
| ATE328028T1 (en) | 2001-05-01 | 2006-06-15 | Union Carbide Chem Plastic | PHARMACEUTICAL COMPOSITION CONTAINING POLYALKYLENE OXIDES WITH REDUCED AMOUNTS OF FORMIC ACID AND FORMIC ACID DERIVATIVES |
| UA81224C2 (en) | 2001-05-02 | 2007-12-25 | Euro Celtic S A | Dosage form of oxycodone and use thereof |
| JP4522652B2 (en) | 2001-05-11 | 2010-08-11 | エンドー ファーマシューティカルズ, インコーポレイティド | Abuse prevention controlled release opioid dosage form |
| US6623754B2 (en) | 2001-05-21 | 2003-09-23 | Noveon Ip Holdings Corp. | Dosage form of N-acetyl cysteine |
| US7125561B2 (en) | 2001-05-22 | 2006-10-24 | Euro-Celtique S.A. | Compartmentalized dosage form |
| US20030064122A1 (en) | 2001-05-23 | 2003-04-03 | Endo Pharmaceuticals, Inc. | Abuse resistant pharmaceutical composition containing capsaicin |
| US7968119B2 (en) | 2001-06-26 | 2011-06-28 | Farrell John J | Tamper-proof narcotic delivery system |
| US20030008409A1 (en) | 2001-07-03 | 2003-01-09 | Spearman Steven R. | Method and apparatus for determining sunlight exposure |
| PL208484B1 (en) | 2001-07-06 | 2011-05-31 | Penwest Pharmaceuticals Company | Methods of making sustained release formulations of oxymorphone related applications |
| EP1406630A1 (en) | 2001-07-06 | 2004-04-14 | Endo Pharmaceuticals Inc. | Parenteral administration of 6-hydroxy-oxymorphone for use as an analgesic |
| US8329216B2 (en) | 2001-07-06 | 2012-12-11 | Endo Pharmaceuticals Inc. | Oxymorphone controlled release formulations |
| JP2003020517A (en) | 2001-07-10 | 2003-01-24 | Calp Corp | Resin composition for compound fiber |
| US7943173B2 (en) | 2001-07-18 | 2011-05-17 | Purdue Pharma L.P. | Pharmaceutical combinations of oxycodone and naloxone |
| US6883976B2 (en) | 2001-07-30 | 2005-04-26 | Seikoh Giken Co., Ltd. | Optical fiber ferrule assembly and optical module and optical connector using the same |
| AU2002324624A1 (en) | 2001-08-06 | 2003-02-24 | Euro-Celtique S.A. | Sequestered antagonist formulations |
| DE20220917U1 (en) | 2001-08-06 | 2004-08-19 | Euro-Celtique S.A. | Anti-abuse compositions for opioids |
| EP1414418A1 (en) | 2001-08-06 | 2004-05-06 | Euro-Celtique S.A. | Compositions and methods to prevent abuse of opioids |
| US20030044458A1 (en) | 2001-08-06 | 2003-03-06 | Curtis Wright | Oral dosage form comprising a therapeutic agent and an adverse-effect agent |
| US7141250B2 (en) | 2001-08-06 | 2006-11-28 | Euro-Celtique S.A. | Pharmaceutical formulation containing bittering agent |
| US7144587B2 (en) | 2001-08-06 | 2006-12-05 | Euro-Celtique S.A. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and bittering agent |
| US7332182B2 (en) | 2001-08-06 | 2008-02-19 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and irritant |
| WO2003015531A2 (en) | 2001-08-06 | 2003-02-27 | Thomas Gruber | Pharmaceutical formulation containing dye |
| US20030068375A1 (en) | 2001-08-06 | 2003-04-10 | Curtis Wright | Pharmaceutical formulation containing gelling agent |
| US7157103B2 (en) | 2001-08-06 | 2007-01-02 | Euro-Celtique S.A. | Pharmaceutical formulation containing irritant |
| US7842307B2 (en) | 2001-08-06 | 2010-11-30 | Purdue Pharma L.P. | Pharmaceutical formulation containing opioid agonist, opioid antagonist and gelling agent |
| US20030049272A1 (en) | 2001-08-30 | 2003-03-13 | Yatindra Joshi | Pharmaceutical composition which produces irritation |
| US20030068276A1 (en) | 2001-09-17 | 2003-04-10 | Lyn Hughes | Dosage forms |
| US20030092724A1 (en) | 2001-09-18 | 2003-05-15 | Huaihung Kao | Combination sustained release-immediate release oral dosage forms with an opioid analgesic and a non-opioid analgesic |
| EP1929998A3 (en) | 2001-09-21 | 2008-11-26 | Egalet A/S | Controlled release solid dispersions of carvedilol |
| WO2003024430A1 (en) | 2001-09-21 | 2003-03-27 | Egalet A/S | Morphine polymer release system |
| AU2002342755A1 (en) | 2001-09-26 | 2003-04-14 | Klaus-Jurgen Steffens | Method and device for producing granulates that comprise at least one pharmaceutical active substance |
| CA2459976A1 (en) | 2001-09-26 | 2003-04-03 | Penwest Pharmaceuticals Company | Opioid formulations having reduced potential for abuse |
| BR0212951A (en) | 2001-09-28 | 2004-10-26 | Mcneil Ppc Inc | Composite Dosage Forms |
| US6837696B2 (en) | 2001-09-28 | 2005-01-04 | Mcneil-Ppc, Inc. | Apparatus for manufacturing dosage forms |
| CA2460435A1 (en) | 2001-10-09 | 2003-04-17 | The Procter & Gamble Company | Aqueous compositions for treating a surface comprising a polymeric biguanide |
| US6592901B2 (en) | 2001-10-15 | 2003-07-15 | Hercules Incorporated | Highly compressible ethylcellulose for tableting |
| JP2003125706A (en) | 2001-10-23 | 2003-05-07 | Lion Corp | Mouth freshening preparation |
| PE20030527A1 (en) | 2001-10-24 | 2003-07-26 | Gruenenthal Chemie | DELAYED-RELEASE PHARMACEUTICAL FORMULATION CONTAINING 3- (3-DIMETHYLAMINO-1-ETHYL-2-METHYL-PROPYL) PHENOL OR A PHARMACEUTICALLY ACCEPTABLE SALT OF THE SAME AND ORAL TABLETS CONTAINING IT |
| US20030152622A1 (en) | 2001-10-25 | 2003-08-14 | Jenny Louie-Helm | Formulation of an erodible, gastric retentive oral diuretic |
| US20030104052A1 (en) | 2001-10-25 | 2003-06-05 | Bret Berner | Gastric retentive oral dosage form with restricted drug release in the lower gastrointestinal tract |
| US20030091630A1 (en) | 2001-10-25 | 2003-05-15 | Jenny Louie-Helm | Formulation of an erodible, gastric retentive oral dosage form using in vitro disintegration test data |
| TWI312285B (en) | 2001-10-25 | 2009-07-21 | Depomed Inc | Methods of treatment using a gastric retained gabapentin dosage |
| CA2409552A1 (en) | 2001-10-25 | 2003-04-25 | Depomed, Inc. | Gastric retentive oral dosage form with restricted drug release in the lower gastrointestinal tract |
| US6723340B2 (en) | 2001-10-25 | 2004-04-20 | Depomed, Inc. | Optimal polymer mixtures for gastric retentive tablets |
| JP4551089B2 (en) | 2001-10-29 | 2010-09-22 | マサチューセッツ インスティテュート オブ テクノロジー | System for producing sustained release dosage forms such as zero order release profile dosage forms produced by three-dimensional printing |
| WO2003039561A1 (en) | 2001-11-02 | 2003-05-15 | Elan Corporation, Plc | Pharmaceutical composition |
| US20040126428A1 (en) | 2001-11-02 | 2004-07-01 | Lyn Hughes | Pharmaceutical formulation including a resinate and an aversive agent |
| US20030175345A1 (en) | 2001-12-06 | 2003-09-18 | Hite Michael P. | Isoflavone composition for oral delivery |
| FR2833838B1 (en) | 2001-12-21 | 2005-09-16 | Ellipse Pharmaceuticals | METHOD FOR MANUFACTURING A TABLET INCLUDING A MORPHINIC ANALGESIC AND TABLET OBTAINED |
| AUPS044502A0 (en) | 2002-02-11 | 2002-03-07 | Commonwealth Scientific And Industrial Research Organisation | Novel catalysts and processes for their preparation |
| US20040033253A1 (en) | 2002-02-19 | 2004-02-19 | Ihor Shevchuk | Acyl opioid antagonists |
| US20030190343A1 (en) | 2002-03-05 | 2003-10-09 | Pfizer Inc. | Palatable pharmaceutical compositions for companion animals |
| US6572889B1 (en) | 2002-03-07 | 2003-06-03 | Noveon Ip Holdings Corp. | Controlled release solid dosage carbamazepine formulations |
| US6753009B2 (en) | 2002-03-13 | 2004-06-22 | Mcneil-Ppc, Inc. | Soft tablet containing high molecular weight polyethylene oxide |
| NZ535286A (en) | 2002-04-05 | 2007-07-27 | Euro Celtique Sa | Matrix for sustained, invariant and independent release of active compounds |
| DE10217232B4 (en) | 2002-04-18 | 2004-08-19 | Ticona Gmbh | Process for the production of filled granules from polyethylene of high or ultra-high molecular weight |
| US6960617B2 (en) | 2002-04-22 | 2005-11-01 | Purdue Research Foundation | Hydrogels having enhanced elasticity and mechanical strength properties |
| WO2003092648A1 (en) | 2002-04-29 | 2003-11-13 | Alza Corporation | Methods and dosage forms for controlled delivery of oxycodone |
| US20050106249A1 (en) | 2002-04-29 | 2005-05-19 | Stephen Hwang | Once-a-day, oral, controlled-release, oxycodone dosage forms |
| CA2486075A1 (en) | 2002-05-13 | 2003-11-20 | Endo Pharmaceuticals Inc. | Abuse-resistant opioid solid dosage form |
| DE10250083A1 (en) | 2002-06-17 | 2003-12-24 | Gruenenthal Gmbh | Dosage form protected against abuse |
| US7776314B2 (en) | 2002-06-17 | 2010-08-17 | Grunenthal Gmbh | Abuse-proofed dosage system |
| US20050175690A1 (en) | 2003-12-29 | 2005-08-11 | David Edgren | Novel drug compositions and dosage forms |
| WO2004004693A1 (en) | 2002-07-05 | 2004-01-15 | Collgegium Pharmaceutical | Abuse-deterrent pharmaceutical compositions of opiods and other drugs |
| US20040011806A1 (en) | 2002-07-17 | 2004-01-22 | Luciano Packaging Technologies, Inc. | Tablet filler device with star wheel |
| MY136318A (en) | 2002-07-25 | 2008-09-30 | Pharmacia Corp | Sustained-release tablet composition |
| US7388068B2 (en) | 2002-08-21 | 2008-06-17 | Clariant Produkte (Deutschland) Gmbh | Copolymers made of alkylene oxides and glycidyl ethers and use thereof as polymerizable emulsifiers |
| AU2003259336A1 (en) | 2002-08-21 | 2004-03-11 | Phoqus Pharmaceuticals Limited | Use of an aqueous solution of citric acid and a water-soluble sugar like lactitol as granulation liquid in the manufacture of tablets |
| US20040052844A1 (en) | 2002-09-16 | 2004-03-18 | Fang-Hsiung Hsiao | Time-controlled, sustained release, pharmaceutical composition containing water-soluble resins |
| DK1635830T3 (en) | 2002-09-17 | 2009-02-23 | Wyeth Corp | Granulation formulation of the rapamycin ester CCI-779 |
| ATE519474T1 (en) | 2002-09-20 | 2011-08-15 | Fmc Corp | COSMETIC COMPOSITION CONTAINING MICROCRYSTALLINE CELLULOSE |
| EP1555022B1 (en) | 2002-09-21 | 2008-02-20 | Shuyi Zhang | Sustained release formulation of acetaminophen and tramadol |
| WO2004026262A2 (en) | 2002-09-23 | 2004-04-01 | Verion, Inc. | Abuse-resistant pharmaceutical compositions |
| US20050186139A1 (en) | 2002-10-25 | 2005-08-25 | Gruenenthal Gmbh | Abuse-proofed dosage form |
| DE10250087A1 (en) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Dosage form protected against abuse |
| US20050191244A1 (en) | 2002-10-25 | 2005-09-01 | Gruenenthal Gmbh | Abuse-resistant pharmaceutical dosage form |
| DE10250084A1 (en) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Dosage form protected against abuse |
| DE10250088A1 (en) | 2002-10-25 | 2004-05-06 | Grünenthal GmbH | Dosage form protected against abuse |
| JP2006507277A (en) | 2002-10-25 | 2006-03-02 | ラボファーマ インコーポレイテッド | 24 hour sustained release tramadol formulation |
| DE10252667A1 (en) | 2002-11-11 | 2004-05-27 | Grünenthal GmbH | New spiro-((cyclohexane)-tetrahydropyrano-(3,4-b)-indole) derivatives, are ORL1 receptor ligands useful e.g. for treating anxiety, depression, epilepsy, senile dementia, withdrawal symptoms or especially pain |
| US20040091528A1 (en) | 2002-11-12 | 2004-05-13 | Yamanouchi Pharma Technologies, Inc. | Soluble drug extended release system |
| US20040185097A1 (en) | 2003-01-31 | 2004-09-23 | Glenmark Pharmaceuticals Ltd. | Controlled release modifying complex and pharmaceutical compositions thereof |
| US7442387B2 (en) | 2003-03-06 | 2008-10-28 | Astellas Pharma Inc. | Pharmaceutical composition for controlled release of active substances and manufacturing method thereof |
| WO2004082620A2 (en) | 2003-03-13 | 2004-09-30 | Controlled Chemicals, Inc. | Oxycodone conjugates with lower the abuse potential and extended duration of action |
| EP1974726B1 (en) | 2003-03-26 | 2010-01-13 | Egalet A/S | Matrix compositions for controlled delivery of drug substances |
| ES2570454T3 (en) | 2003-03-26 | 2016-05-18 | Egalet Ltd | Morphine controlled release system |
| AU2004232370B2 (en) | 2003-04-21 | 2009-12-10 | Euro-Celtique S.A. | Tamper resistant dosage form comprising co-extruded, adverse agent particles and process of making same |
| MY135852A (en) | 2003-04-21 | 2008-07-31 | Euro Celtique Sa | Pharmaceutical products |
| JP5048324B2 (en) | 2003-04-30 | 2012-10-17 | パーデュー、ファーマ、リミテッド、パートナーシップ | Tamper-resistant transdermal dosage form comprising an active substance element and a side-effect substance element distal to the active substance layer |
| US8906413B2 (en) | 2003-05-12 | 2014-12-09 | Supernus Pharmaceuticals, Inc. | Drug formulations having reduced abuse potential |
| CN1473562A (en) | 2003-06-27 | 2004-02-11 | 辉 刘 | Mouth cavity quick dissolving quick disintegrating freeze-dried tablet and its preparing method |
| US20050015730A1 (en) | 2003-07-14 | 2005-01-20 | Srimanth Gunturi | Systems, methods and computer program products for identifying tab order sequence of graphically represented elements |
| DE10336400A1 (en) | 2003-08-06 | 2005-03-24 | Grünenthal GmbH | Anti-abuse dosage form |
| US8075872B2 (en) | 2003-08-06 | 2011-12-13 | Gruenenthal Gmbh | Abuse-proofed dosage form |
| US20070048228A1 (en) | 2003-08-06 | 2007-03-01 | Elisabeth Arkenau-Maric | Abuse-proofed dosage form |
| DE102004032051A1 (en) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Process for the preparation of a secured against misuse, solid dosage form |
| US20070183980A1 (en) | 2003-08-06 | 2007-08-09 | Elisabeth Arkenau-Maric | Dosage form that is safeguarded from abuse |
| DE10361596A1 (en) | 2003-12-24 | 2005-09-29 | Grünenthal GmbH | Process for producing an anti-abuse dosage form |
| DE102005005446A1 (en) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Break-resistant dosage forms with sustained release |
| DE102004020220A1 (en) | 2004-04-22 | 2005-11-10 | Grünenthal GmbH | Process for the preparation of a secured against misuse, solid dosage form |
| RU2339365C2 (en) | 2003-08-06 | 2008-11-27 | Грюненталь Гмбх | Drug dosage form, protected from unintended application |
| US20050063214A1 (en) | 2003-09-22 | 2005-03-24 | Daisaburo Takashima | Semiconductor integrated circuit device |
| AU2004277898B2 (en) | 2003-09-25 | 2009-04-02 | Euro-Celtique S.A. | Pharmaceutical combinations of hydrocodone and naltrexone |
| AU2004277980A1 (en) | 2003-09-30 | 2005-04-14 | Alza Corporation | Osmotically driven active agent delivery device providing an ascending release profile |
| US20060172006A1 (en) | 2003-10-10 | 2006-08-03 | Vincent Lenaerts | Sustained-release tramadol formulations with 24-hour clinical efficacy |
| US20060009478A1 (en) | 2003-10-15 | 2006-01-12 | Nadav Friedmann | Methods for the treatment of back pain |
| CA2546691A1 (en) | 2003-10-29 | 2005-05-12 | Alza Corporation | Once-a-day, oral, controlled-release, oxycodone dosage forms |
| US7201920B2 (en) | 2003-11-26 | 2007-04-10 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of opioid containing dosage forms |
| BRPI0417348A (en) | 2003-12-04 | 2007-03-13 | Pfizer Prod Inc | spray gelatinization process using an extruder for preparing multiparticulate crystalline drug compositions preferably containing a poloxamer and a glyceride |
| DK1691892T3 (en) | 2003-12-09 | 2007-06-25 | Euro Celtique Sa | Resistant, coextruded dosage form containing an active agent and an antagonist, and process for preparing the same |
| WO2005060942A1 (en) | 2003-12-19 | 2005-07-07 | Aurobindo Pharma Ltd | Extended release pharmaceutical composition of metformin |
| DE10360792A1 (en) | 2003-12-23 | 2005-07-28 | Grünenthal GmbH | Spirocyclic cyclohexane derivatives |
| US20070196396A1 (en) | 2004-02-11 | 2007-08-23 | Rubicon Research Private Limited | Controlled release pharmaceutical compositions with improved bioavailability |
| TWI350762B (en) | 2004-02-12 | 2011-10-21 | Euro Celtique Sa | Particulates |
| GB0403100D0 (en) | 2004-02-12 | 2004-03-17 | Euro Celtique Sa | Particulates |
| GB0403098D0 (en) | 2004-02-12 | 2004-03-17 | Euro Celtique Sa | Extrusion |
| ME02661B (en) | 2004-02-23 | 2017-06-20 | Euro Celtique Sa | Abuse resistance opioid transdermal delivery device |
| TWI365880B (en) | 2004-03-30 | 2012-06-11 | Euro Celtique Sa | Process for preparing oxycodone hydrochloride having less than 25 ppm 14-hydroxycodeinone and oxycodone hydrochloride composition,pharmaceutical dosage form,sustained release oeal dosage form and pharmaceutically acceptable package having less than 25 pp |
| US20050220877A1 (en) | 2004-03-31 | 2005-10-06 | Patel Ashish A | Bilayer tablet comprising an antihistamine and a decongestant |
| DE102004019916A1 (en) | 2004-04-21 | 2005-11-17 | Grünenthal GmbH | Anti-abuse drug-containing patch |
| EP1740156B8 (en) | 2004-04-22 | 2012-07-11 | Grünenthal GmbH | Method for the production of an abuse-proof, solid form of administration |
| WO2005105036A1 (en) | 2004-04-28 | 2005-11-10 | Natco Pharma Limited | Controlled release mucoadhesive matrix formulation containing tolterodine and a process for its preparation |
| TWI356036B (en) | 2004-06-09 | 2012-01-11 | Smithkline Beecham Corp | Apparatus and method for pharmaceutical production |
| ATE368639T1 (en) | 2004-06-28 | 2007-08-15 | Gruenenthal Gmbh | CRYSTALLINE FORMS OF (-)-(1R,2R)-3-(3-DIMETHYLAMINO-1-ETHYL-2-METHYLPROPYL)-PHENOL HYDROCHLORIDE |
| ITMI20041317A1 (en) | 2004-06-30 | 2004-09-30 | Ibsa Inst Biochimique Sa | PHARMACEUTICAL FORMULATIONS FOR THE SAFE ADMINISTRATION OF DRUGS USED IN THE TREATMENT OF DRUG ADDICTION AND PROCEDURE FOR THEIR OBTAINING |
| DE102004032103A1 (en) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Anti-abuse, oral dosage form |
| PL1765303T5 (en) | 2004-07-01 | 2023-05-22 | Grünenthal GmbH | Oral dosage form safeguarded against abuse |
| EP1765298B1 (en) | 2004-07-01 | 2012-10-24 | Gruenenthal Gmbh | Method for producing a solid dosage form, which is safeguarded against abuse, while using a planetary gear extruder |
| AR053304A1 (en) | 2004-07-01 | 2007-05-02 | Gruenenthal Gmbh | PROTECTED ORAL PHARMACEUTICAL FORMS AGAINST ABUSE WITH CONTROLLED RELEASE OF (1R, 2R) -3- (3 DIMETHYLAMIN-1-ETIL-2METIL-PROPIL) PHENOL AND PROCEDURE FOR PRODUCTION. |
| DE102004032049A1 (en) | 2004-07-01 | 2006-01-19 | Grünenthal GmbH | Anti-abuse, oral dosage form |
| KR20060007225A (en) | 2004-07-19 | 2006-01-24 | 삼성전자주식회사 | Auto-stabilization method in control of driving image pickup device and reading memory and apparatus thereof |
| BRPI0513608A (en) | 2004-07-27 | 2008-05-13 | Unilever Nv | hair care composition, process for preparing a hair care composition and method for hair treatment |
| US20070077297A1 (en) | 2004-09-30 | 2007-04-05 | Scolr Pharma, Inc. | Modified release ibuprofen dosage form |
| US20060068009A1 (en) | 2004-09-30 | 2006-03-30 | Scolr Pharma, Inc. | Modified release ibuprofen dosage form |
| US20080152595A1 (en) | 2004-11-24 | 2008-06-26 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| US20070231268A1 (en) | 2004-11-24 | 2007-10-04 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| US20060177380A1 (en) | 2004-11-24 | 2006-08-10 | Acura Pharmaceuticals, Inc. | Methods and compositions for deterring abuse of orally administered pharmaceutical products |
| LT1849470T (en) | 2005-01-26 | 2017-07-25 | Taiho Pharmaceutical Co., Ltd. | Anticancer drug containing alpha, alpha, alpha-trifluorothymidine and thymidine phosphorylase inhibitor |
| EA015615B1 (en) | 2005-01-28 | 2011-10-31 | Еуро-Селтик С.А. | Alcohol resistant dosage forms |
| DE102005005449A1 (en) | 2005-02-04 | 2006-08-10 | Grünenthal GmbH | Process for producing an anti-abuse dosage form |
| WO2006084474A2 (en) | 2005-02-10 | 2006-08-17 | Lifecycle Pharma A/S | A stable pharmaceutical composition comprising a fixed dose combination of fenofibrate and an hmg-coa reductase inhibitor |
| US20060194759A1 (en) | 2005-02-25 | 2006-08-31 | Eidelson Stewart G | Topical compositions and methods for treating pain and inflammation |
| EP1695700A1 (en) | 2005-02-28 | 2006-08-30 | Euro-Celtique S.A. | Dosage form containing oxycodone and naloxone |
| RS51568B (en) | 2005-03-04 | 2011-08-31 | Euro-Celtique S.A. | Method of reducing alpha, beta- unsaturated ketones in opioid compositions |
| US7732427B2 (en) | 2005-03-31 | 2010-06-08 | University Of Delaware | Multifunctional and biologically active matrices from multicomponent polymeric solutions |
| CA2603649C (en) | 2005-04-08 | 2014-10-14 | Ozpharma Pty Ltd | Buccal delivery system |
| EP1881819A1 (en) | 2005-05-10 | 2008-01-30 | Novartis AG | Extrusion process for making compositions with poorly compressible therapeutic compounds |
| US20090274759A1 (en) | 2005-06-03 | 2009-11-05 | Egalet A/S | Solid pharmaceutical composition with a first fraction of a dispersion medium and a second fraction of a matrix, the latter being at least partially first exposed to gastrointestinal fluids |
| WO2007005716A2 (en) | 2005-06-30 | 2007-01-11 | Cinergen, Llc | Methods of treatment and compositions for use thereof |
| AU2006269225B2 (en) | 2005-07-07 | 2011-10-06 | Farnam Companies, Inc. | Sustained release pharmaceutical compositions for highly water soluble drugs |
| US8858993B2 (en) | 2005-07-25 | 2014-10-14 | Metrics, Inc. | Coated tablet with zero-order or near zero-order release kinetics |
| EP1909760A1 (en) | 2005-08-03 | 2008-04-16 | Eastman Chemical Company | Tocopheryl polyethylene glycol succinate powder and process for preparing same |
| CA2625055C (en) | 2005-10-14 | 2013-12-24 | The Kitasato Institute | Novel dihydropseudoerythromycin derivatives |
| US20070092573A1 (en) | 2005-10-24 | 2007-04-26 | Laxminarayan Joshi | Stabilized extended release pharmaceutical compositions comprising a beta-adrenoreceptor antagonist |
| PL116330U1 (en) | 2005-10-31 | 2007-04-02 | Alza Corp | Method for the reduction of alcohol provoked rapid increase in the released dose of the orally administered opioide with prolonged liberation |
| WO2008134071A1 (en) | 2007-04-26 | 2008-11-06 | Theraquest Biosciences, Inc. | Multimodal abuse resistant extended release formulations |
| US8329744B2 (en) | 2005-11-02 | 2012-12-11 | Relmada Therapeutics, Inc. | Methods of preventing the serotonin syndrome and compositions for use thereof |
| FR2892937B1 (en) | 2005-11-10 | 2013-04-05 | Flamel Tech Sa | MICROPARTICULAR ORAL PHARMACEUTICAL FORM ANTI-MEASURING |
| US8652529B2 (en) | 2005-11-10 | 2014-02-18 | Flamel Technologies | Anti-misuse microparticulate oral pharmaceutical form |
| BRPI0706753A2 (en) | 2006-01-21 | 2011-04-05 | Abbott Gmbh & Co Kg | dosage form and method for the delivery of drugs of abuse |
| US20090022798A1 (en) | 2007-07-20 | 2009-01-22 | Abbott Gmbh & Co. Kg | Formulations of nonopioid and confined opioid analgesics |
| US20100172989A1 (en) | 2006-01-21 | 2010-07-08 | Abbott Laboratories | Abuse resistant melt extruded formulation having reduced alcohol interaction |
| US20090317355A1 (en) | 2006-01-21 | 2009-12-24 | Abbott Gmbh & Co. Kg, | Abuse resistant melt extruded formulation having reduced alcohol interaction |
| EP1813276A1 (en) | 2006-01-27 | 2007-08-01 | Euro-Celtique S.A. | Tamper resistant dosage forms |
| FR2898056B1 (en) | 2006-03-01 | 2012-01-20 | Ethypharm Sa | SQUEEZE-RESISTANT TABLETS TO PREVENT UNLAWFUL MISUSE |
| WO2007103286A2 (en) | 2006-03-02 | 2007-09-13 | Spherics, Inc. | Rate-controlled bioadhesive oral dosage formulations |
| WO2007103105A2 (en) | 2006-03-02 | 2007-09-13 | Mallinckrodt Inc. | Processes for preparing morphinan-6-one products with low levels of alpha, beta-unsaturated ketone compounds |
| US20070224637A1 (en) | 2006-03-24 | 2007-09-27 | Mcauliffe Joseph C | Oxidative protection of lipid layer biosensors |
| CA2647809C (en) | 2006-03-24 | 2016-08-16 | Auxilium Pharmaceuticals, Inc. | Stabilized compositions containing alkaline labile drugs |
| US8465759B2 (en) | 2006-03-24 | 2013-06-18 | Auxilium Us Holdings, Llc | Process for the preparation of a hot-melt extruded laminate |
| US10960077B2 (en) | 2006-05-12 | 2021-03-30 | Intellipharmaceutics Corp. | Abuse and alcohol resistant drug composition |
| US9023400B2 (en) | 2006-05-24 | 2015-05-05 | Flamel Technologies | Prolonged-release multimicroparticulate oral pharmaceutical form |
| US20070292508A1 (en) | 2006-06-05 | 2007-12-20 | Balchem Corporation | Orally disintegrating dosage forms |
| US20080069891A1 (en) | 2006-09-15 | 2008-03-20 | Cima Labs, Inc. | Abuse resistant drug formulation |
| CN101091721A (en) | 2006-06-22 | 2007-12-26 | 孙明 | Method for preparing new type asshide |
| GB0613836D0 (en) | 2006-07-13 | 2006-08-23 | Univ Greenwich | New medical use of triazine derivatives |
| JP4029109B1 (en) | 2006-07-18 | 2008-01-09 | タマ生化学株式会社 | Complex powder of vitamin E and proline and method for producing the same |
| US20080075771A1 (en) | 2006-07-21 | 2008-03-27 | Vaughn Jason M | Hydrophilic opioid abuse deterrent delivery system using opioid antagonists |
| SA07280459B1 (en) | 2006-08-25 | 2011-07-20 | بيورديو فارما إل. بي. | Tamper Resistant Oral Pharmaceutical Dosage Forms Comprising an Opioid Analgesic |
| US8445018B2 (en) | 2006-09-15 | 2013-05-21 | Cima Labs Inc. | Abuse resistant drug formulation |
| US8187636B2 (en) | 2006-09-25 | 2012-05-29 | Atlantic Pharmaceuticals, Inc. | Dosage forms for tamper prone therapeutic agents |
| KR101400824B1 (en) | 2006-09-25 | 2014-05-29 | 후지필름 가부시키가이샤 | Resist composition, resin for use in the resist composition, compound for use in the synthesis of the resin, and pattern-forming method usign the resist composition |
| AU2006349402A1 (en) * | 2006-10-10 | 2008-04-17 | Penwest Pharmaceuticals Co. | Robust sustained release formulations |
| GB0624880D0 (en) | 2006-12-14 | 2007-01-24 | Johnson Matthey Plc | Improved method for making analgesics |
| DE102006062120A1 (en) | 2006-12-22 | 2008-06-26 | Grünenthal GmbH | Pharmaceutical composition for acne treatment |
| CA2930487A1 (en) | 2007-01-16 | 2008-07-24 | Egalet Ltd. | Use of i) a polyglycol and ii) an active drug substance for the preparation of a pharmaceutical composition for i)mitigating the risk of alcohol induced dose dumping and/or ii) reducing the risk of drug abuse |
| US20080181932A1 (en) | 2007-01-30 | 2008-07-31 | Drugtech Corporation | Compositions for oral delivery of pharmaceuticals |
| CN100579525C (en) | 2007-02-02 | 2010-01-13 | 东南大学 | Sustained release preparation of licardipine hydrochloride and its preparing process |
| CN101057849A (en) | 2007-02-27 | 2007-10-24 | 齐齐哈尔医学院 | Slow-releasing preparation containing metformin hydrochloride and glipizide and its preparation method |
| DE602008002073D1 (en) | 2007-03-02 | 2010-09-16 | Farnam Co Inc | WAX-LIKE MATERIAL-CONTAINING TABLETS WITH DELAYED RELEASE |
| DE102007011485A1 (en) | 2007-03-07 | 2008-09-11 | Grünenthal GmbH | Dosage form with more difficult abuse |
| EP1980245A1 (en) | 2007-04-11 | 2008-10-15 | Cephalon France | Bilayer lyophilized pharmaceutical compositions and methods of making and using same |
| US8951570B2 (en) | 2007-04-26 | 2015-02-10 | Sigmoid Pharma Limited | Manufacture of multiple minicapsules |
| WO2008142627A2 (en) | 2007-05-17 | 2008-11-27 | Ranbaxy Laboratories Limited | Multilayered modified release formulation comprising amoxicillin and clavulanate |
| US8202542B1 (en) | 2007-05-31 | 2012-06-19 | Tris Pharma | Abuse resistant opioid drug-ion exchange resin complexes having hybrid coatings |
| WO2008148798A2 (en) | 2007-06-04 | 2008-12-11 | Egalet A/S | Controlled release pharmaceutical compositions for prolonged effect |
| US20100035886A1 (en) | 2007-06-21 | 2010-02-11 | Veroscience, Llc | Parenteral formulations of dopamine agonists |
| JP2010532358A (en) | 2007-07-01 | 2010-10-07 | ピーター ハバウシ,ジョセフ | Formulation with chewable outer layer |
| KR20100055431A (en) | 2007-07-20 | 2010-05-26 | 애보트 게엠베하 운트 콤파니 카게 | Formulations of nonopioid and confined opioid analgesics |
| MX336861B (en) | 2007-09-13 | 2016-02-04 | Cima Labs Inc | Abuse resistant drug formulation. |
| WO2009051819A1 (en) | 2007-10-17 | 2009-04-23 | Axxia Pharmaceuticals, Llc | Polymeric drug delivery systems and thermoplastic extrusion processes for producing such systems |
| CN104958282B (en) | 2007-11-23 | 2018-05-29 | 格吕伦塔尔有限公司 | Tapentadol hydrochloride composition |
| JP2011506318A (en) | 2007-12-06 | 2011-03-03 | デュレクト コーポレーション | Oral pharmaceutical formulation |
| US20100280047A1 (en) | 2007-12-12 | 2010-11-04 | Basf Se | Salts of active ingredients with polymeric counter-ions |
| AU2008338207A1 (en) | 2007-12-17 | 2009-06-25 | Labopharm (Barbados) Limited | Misuse preventative, controlled release formulation |
| CA2713128C (en) | 2008-01-25 | 2016-04-05 | Gruenenthal Gmbh | Pharmaceutical dosage form |
| KR100970665B1 (en) | 2008-02-04 | 2010-07-15 | 삼일제약주식회사 | Sustained release tablet containing alfuzosin or its salt |
| CN101969931A (en) | 2008-03-05 | 2011-02-09 | 万能药生物有限公司 | Modified release pharmaceutical compositions comprising mycophenolate and processes thereof |
| US8372432B2 (en) | 2008-03-11 | 2013-02-12 | Depomed, Inc. | Gastric retentive extended-release dosage forms comprising combinations of a non-opioid analgesic and an opioid analgesic |
| EP2100598A1 (en) | 2008-03-13 | 2009-09-16 | Laboratorios Almirall, S.A. | Inhalation composition containing aclidinium for treatment of asthma and chronic obstructive pulmonary disease |
| WO2009135680A1 (en) | 2008-05-09 | 2009-11-12 | Grünenthal GmbH | Process for the preparation of an intermediate powder formulation and a final solid dosage form under usage of a spray congealing step |
| JP2011526928A (en) | 2008-07-03 | 2011-10-20 | ノバルティス アーゲー | Melt granulation method |
| US9192578B2 (en) | 2008-08-20 | 2015-11-24 | Board Of Regents, The University Of Texas System | Hot-melt extrusion of modified release multi-particulates |
| WO2010044842A1 (en) | 2008-10-16 | 2010-04-22 | University Of Tennessee Research Foundation | Tamper resistant oral dosage forms containing an embolizing agent |
| US20100104638A1 (en) | 2008-10-27 | 2010-04-29 | Wei-Guo Dai | Extended release oral acetaminophen/tramadol dosage form |
| TW201022253A (en) | 2008-11-14 | 2010-06-16 | Portola Pharm Inc | Solid composition for controlled release of ionizable active agents with poor aqueous solubility at low pH and methods of use thereof |
| BRPI0917608B8 (en) | 2008-12-12 | 2021-05-25 | Paladin Labs Inc | oral drug formulation for the reduction of potential abuse, process for manufacturing a drug formulation and its use |
| WO2010069050A1 (en) | 2008-12-16 | 2010-06-24 | Labopharm Inc. | Misuse preventative, controlled release formulation |
| EP2700400A1 (en) | 2009-01-26 | 2014-02-26 | Egalet Ltd. | Controlled release formulation with continuous efficacy |
| US8603526B2 (en) | 2009-02-06 | 2013-12-10 | Egalet Ltd. | Pharmaceutical compositions resistant to abuse |
| CN102355893A (en) | 2009-03-18 | 2012-02-15 | 赢创罗姆有限公司 | Controlled release pharmaceutical composition with resistance against influence of ethanol employing coating comprising neutral vinyl polymers and excipients |
| EP2246063A1 (en) | 2009-04-29 | 2010-11-03 | Ipsen Pharma S.A.S. | Sustained release formulations comprising GnRH analogues |
| GB0909680D0 (en) | 2009-06-05 | 2009-07-22 | Euro Celtique Sa | Dosage form |
| AU2010265213B2 (en) | 2009-06-24 | 2012-08-23 | Egalet Ltd. | Controlled release formulations |
| WO2011008298A2 (en) | 2009-07-16 | 2011-01-20 | Nectid, Inc. | Novel axomadol dosage forms |
| ES2534908T3 (en) | 2009-07-22 | 2015-04-30 | Grünenthal GmbH | Hot melt extruded controlled release dosage form |
| CN102639118B (en) | 2009-07-22 | 2015-07-29 | 格吕伦塔尔有限公司 | Oxidation-stabilized tamper resistant dosage form |
| EP2506838A1 (en) | 2009-12-01 | 2012-10-10 | Noven Pharmaceuticals, INC. | Transdermal testosterone device and delivery |
| CN102821757B (en) | 2010-02-03 | 2016-01-20 | 格吕伦塔尔有限公司 | By extrusion mechanism for powdery medicine compositions |
| WO2011123866A1 (en) | 2010-04-02 | 2011-10-06 | Alltranz Inc. | Abuse-deterrent transdermal formulations of opiate agonists and agonist-antagonists |
| GB201006200D0 (en) | 2010-04-14 | 2010-06-02 | Ayanda As | Composition |
| ES2689520T3 (en) | 2010-04-23 | 2018-11-14 | Kempharm, Inc. | Therapeutic formulation to reduce drug side effects |
| FR2960775A1 (en) | 2010-06-07 | 2011-12-09 | Ethypharm Sa | MICROGRANULES RESISTANT TO MISMATCH |
| PL2611426T3 (en) | 2010-09-02 | 2014-09-30 | Gruenenthal Gmbh | Tamper resistant dosage form comprising inorganic salt |
| JP5925778B2 (en) | 2010-09-02 | 2016-05-25 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | Tamper resistant dosage forms containing anionic polymers |
| MX2013002293A (en) | 2010-09-02 | 2013-05-09 | Gruenenthal Gmbh | Tamper resistant dosage form comprising an anionic polymer. |
| US20120202838A1 (en) | 2010-11-04 | 2012-08-09 | Abbott Laboratories | Drug formulations |
| US20120231083A1 (en) | 2010-11-18 | 2012-09-13 | The Board Of Trustees Of The University Of Illinois | Sustained release cannabinoid medicaments |
| GB201020895D0 (en) | 2010-12-09 | 2011-01-26 | Euro Celtique Sa | Dosage form |
| BR112013015939A2 (en) | 2010-12-23 | 2020-08-04 | Purdue Pharma L.P. | adulteration-resistant solid oral dosage forms |
| MX2013009492A (en) | 2011-02-17 | 2014-07-30 | Qrxpharma Ltd | Technology for preventing abuse of solid dosage forms. |
| CA2828631C (en) | 2011-03-04 | 2019-10-15 | Grunenthal Gmbh | Aqueous pharmaceutical formulation of tapentadol for oral administration |
| US8858963B1 (en) | 2011-05-17 | 2014-10-14 | Mallinckrodt Llc | Tamper resistant composition comprising hydrocodone and acetaminophen for rapid onset and extended duration of analgesia |
| JP6042880B2 (en) | 2011-06-01 | 2016-12-14 | エフ エム シー コーポレーションFmc Corporation | Controlled release solid dosage form |
| EP2726065A4 (en) | 2011-06-30 | 2014-11-26 | Neos Therapeutics Lp | Abuse resistant drug forms |
| RS56527B1 (en) | 2011-07-29 | 2018-02-28 | Gruenenthal Gmbh | Tamper-resistant tablet providing immediate drug release |
| DK2736495T3 (en) | 2011-07-29 | 2017-11-13 | Gruenenthal Gmbh | ABUSE RESPONSIBLE TABLE THAT PROVIDES IMMEDIATE RELEASE OF MEDICINE |
| FR2979242A1 (en) | 2011-08-29 | 2013-03-01 | Sanofi Sa | COMPRESSES AGAINST ABUSIVE USE, BASED ON PARACETAMOL AND OXYCODONE |
| EP2763664A2 (en) | 2011-10-06 | 2014-08-13 | Grünenthal GmbH | Tamper-resistant oral pharmaceutical dosage form comprising opioid agonist and opioid antagonist |
| AR088875A1 (en) | 2011-11-17 | 2014-07-16 | Gruenenthal Gmbh | ORAL PHARMACEUTICAL DOSE FORM FOR HANDLING PROOF |
| EP2787978B1 (en) | 2011-12-09 | 2016-09-21 | Purdue Pharma LP | Pharmaceutical dosage forms comprising poly(epsilon-caprolactone) and polyethylene oxide |
| EP2819656A1 (en) | 2012-02-28 | 2015-01-07 | Grünenthal GmbH | Tamper-resistant dosage form comprising pharmacologically active compound and anionic polymer |
| US20130225625A1 (en) | 2012-02-28 | 2013-08-29 | Grunenthal Gmbh | Tamper-resistant pharmaceutical dosage form comprising nonionic surfactant |
| PE20142320A1 (en) | 2012-03-02 | 2015-01-16 | Rhodes Pharmaceuticals Lp | IMMEDIATE RELEASE FORMULATIONS RESISTANT TO HANDLING |
| MX362357B (en) | 2012-04-18 | 2019-01-14 | Gruenenthal Gmbh | Tamper resistant and dose-dumping resistant pharmaceutical dosage form. |
| JP2015516406A (en) | 2012-05-11 | 2015-06-11 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | Thermoformed tamper-resistant pharmaceutical dosage forms containing zinc |
| US10064945B2 (en) | 2012-05-11 | 2018-09-04 | Gruenenthal Gmbh | Thermoformed, tamper-resistant pharmaceutical dosage form containing zinc |
| WO2014059512A1 (en) | 2012-10-15 | 2014-04-24 | Isa Odidi | Oral drug delivery formulations |
| AU2014273226B2 (en) | 2013-05-29 | 2019-06-27 | Grunenthal Gmbh | Tamper resistant dosage form with bimodal release profile |
| US10154966B2 (en) | 2013-05-29 | 2018-12-18 | Grünenthal GmbH | Tamper-resistant dosage form containing one or more particles |
| CA2817728A1 (en) | 2013-05-31 | 2014-11-30 | Pharmascience Inc. | Abuse deterrent immediate release formulation |
| JP6449871B2 (en) | 2013-07-12 | 2019-01-09 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | Anti-modified dosage form containing ethylene-vinyl acetate polymer |
| US9770514B2 (en) | 2013-09-03 | 2017-09-26 | ExxPharma Therapeutics LLC | Tamper-resistant pharmaceutical dosage forms |
| US20150118300A1 (en) | 2013-10-31 | 2015-04-30 | Cima Labs Inc. | Immediate Release Abuse-Deterrent Granulated Dosage Forms |
| US10744131B2 (en) | 2013-12-31 | 2020-08-18 | Kashiv Biosciences, Llc | Abuse-resistant drug formulations |
| KR102030891B1 (en) | 2014-02-12 | 2019-10-11 | 제넨테크, 인크. | Anti-jagged1 antibodies and methods of use |
| WO2016125001A1 (en) | 2015-02-05 | 2016-08-11 | Grey Orange Pte, Ltd. | Apparatus and method for navigation path compensation |
-
2015
- 2015-05-22 CN CN201580027660.1A patent/CN106456550A/en active Pending
- 2015-05-22 EA EA201692388A patent/EA201692388A1/en unknown
- 2015-05-22 WO PCT/EP2015/061343 patent/WO2015181059A1/en active Application Filing
- 2015-05-22 CA CA2949422A patent/CA2949422A1/en not_active Abandoned
- 2015-05-22 AU AU2015266117A patent/AU2015266117A1/en not_active Abandoned
- 2015-05-22 EP EP15724622.4A patent/EP3148512A1/en not_active Withdrawn
- 2015-05-22 JP JP2016569686A patent/JP2017516789A/en active Pending
- 2015-05-22 US US14/719,351 patent/US9872835B2/en not_active Expired - Fee Related
- 2015-05-22 MX MX2016015417A patent/MX2016015417A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080085304A1 (en) * | 2006-10-10 | 2008-04-10 | Penwest Pharmaceuticals Co. | Robust sustained release formulations |
| WO2009034541A2 (en) * | 2007-09-11 | 2009-03-19 | Ranbaxy Laboratories Limited | Controlled release pharmaceutical dosage forms of trimetazidine |
Non-Patent Citations (1)
| Title |
|---|
| ROSIAUX Y ET AL: "Ethanol-resistant ethylcellulose/guar gum coatings - Importance of formulation parame", EUROPEAN JOURNAL OF PHARMACEUTICS AND BIOPHARMACEUTICS, vol. 85, no. 3, 25 July 2013 (2013-07-25), pages 1250 - 1258, XP028791007, ISSN: 0939-6411, DOI: 10.1016/J.EJPB.2013.07.014 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11077055B2 (en) | 2015-04-29 | 2021-08-03 | Dexcel Pharma Technologies Ltd. | Orally disintegrating compositions |
Also Published As
| Publication number | Publication date |
|---|---|
| EA201692388A1 (en) | 2017-05-31 |
| CA2949422A1 (en) | 2015-12-03 |
| JP2017516789A (en) | 2017-06-22 |
| US20150335592A1 (en) | 2015-11-26 |
| AU2015266117A1 (en) | 2016-11-24 |
| CN106456550A (en) | 2017-02-22 |
| EP3148512A1 (en) | 2017-04-05 |
| MX2016015417A (en) | 2017-02-22 |
| US9872835B2 (en) | 2018-01-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10864164B2 (en) | Tamper-resistant tablet providing immediate drug release | |
| US9872835B2 (en) | Multiparticles safeguarded against ethanolic dose-dumping | |
| US9737490B2 (en) | Tamper resistant dosage form with bimodal release profile | |
| WO2014191397A1 (en) | Tamper-resistant dosage form containing one or more particles | |
| AU2012289764A1 (en) | Tamper-resistant tablet providing immediate drug release | |
| WO2013017242A1 (en) | Tamper-resistant tablet providing immediate drug release | |
| US20160310428A1 (en) | Tamper-resistant fixed dose combination providing fast release of two drugs from different particles | |
| US20160310437A1 (en) | Tamper-resistant fixed dose combination providing fast release of two drugs from particles | |
| US20160310486A1 (en) | Tamper-resistant fixed dose combination providing fast release of two drugs from particles and a matrix | |
| US20160310427A1 (en) | Tamper-resistant fixed dose combination providing fast release of two drugs from particles and a powder |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15724622 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| REEP | Request for entry into the european phase |
Ref document number: 2015724622 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015724622 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2949422 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2015266117 Country of ref document: AU Date of ref document: 20150522 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2016/015417 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2016569686 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112016027582 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201692388 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 112016027582 Country of ref document: BR Kind code of ref document: A2 Effective date: 20161124 |