WO2015137238A1 - 2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの結晶多形体およびその製造方法 - Google Patents
2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの結晶多形体およびその製造方法 Download PDFInfo
- Publication number
- WO2015137238A1 WO2015137238A1 PCT/JP2015/056555 JP2015056555W WO2015137238A1 WO 2015137238 A1 WO2015137238 A1 WO 2015137238A1 JP 2015056555 W JP2015056555 W JP 2015056555W WO 2015137238 A1 WO2015137238 A1 WO 2015137238A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- crystal
- hydroxyethoxy
- bis
- binaphthalene
- crystals
- Prior art date
Links
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C43/00—Ethers; Compounds having groups, groups or groups
- C07C43/02—Ethers
- C07C43/20—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring
- C07C43/23—Ethers having an ether-oxygen atom bound to a carbon atom of a six-membered aromatic ring containing hydroxy or O-metal groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/34—Separation; Purification; Stabilisation; Use of additives
- C07C41/40—Separation; Purification; Stabilisation; Use of additives by change of physical state, e.g. by crystallisation
Definitions
- the present invention is suitable as a monomer for forming a resin (optical resin) constituting an optical member typified by an optical lens or an optical film, and is a novel 2,2′-bis (2-
- the present invention relates to a crystalline polymorph of hydroxyethoxy) -1,1′-binaphthalene and a method for producing the same.
- Resin materials such as polycarbonate, polyester, polyacrylate, polyurethane, and epoxy using binaphthalenes such as 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene as raw materials have optical properties, heat resistance, etc.
- binaphthalenes such as 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene
- Patent Document 1 describes 1,1′-biphthalene in Synthesis Example 1 thereof. -2-Naphthol and an excess amount of ethylene carbonate are reacted in the presence of a potassium hydroxide catalyst, the resulting reaction product is dissolved in methyl isobutyl ketone, washed with water, and then methyl isobutyl ketone is removed to remove the resin. A method for obtaining a product is disclosed.
- Example 1 was reacted with 1,1′-bi-2-naphthol and an excess amount of ethylene carbonate in a toluene solvent in the presence of a potassium carbonate catalyst.
- the obtained reaction product is washed with a 1% aqueous sodium hydroxide solution and water, and then the solvent is removed under reduced pressure to obtain 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene.
- a method is disclosed. However, the methods described in these patent documents are methods in which the solvent is completely removed from a solution in which 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene is dissolved. Is a difficult method.
- Non-Patent Document 1 is extremely poor in productivity because the filterability and liquid breakage during crystal washing are poor, and as a result, the crystals before drying contain a large amount of solvent.
- An object of the present invention is to obtain 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene, which is suitably used as a raw material monomer for various resins including optical resins, as crystals. It is an object of the present invention to provide a production method which can be carried out commercially and crystals of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene which can be obtained industrially advantageously.
- the diffraction angle 2 ⁇ in the powder X-ray diffraction pattern by Cu—K ⁇ rays is 10.4 ⁇ 0.2 °, 12.1 ⁇ 0.2 °, 14.8 ⁇ 0.2 °, and 22.3 ⁇ 0.2 °. 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene crystal having a peak at.
- the diffraction angle 2 ⁇ in the powder X-ray diffraction pattern by Cu—K ⁇ rays is 10.0 ⁇ 0.2 °, 19.2 ⁇ 0.2 °, 19.7 ⁇ 0.2 ° and 22.2 ⁇ 0.2 °. 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene crystal having a peak at.
- the ratio Dmode / Dmedian between the mode diameter Dmode and the median diameter Dmedian is 2.0 or less
- the ratio L / W between the maximum crystal length L and the width W obtained from the optical micrograph is 1 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene crystal
- 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene crystals can be obtained by a method with high yield and good operability.
- the novel plate-like crystal or bulk crystal obtained by the present invention is different from the conventionally known needle-like crystal.
- a small amount of solvent is used when the crystal is precipitated.
- 2) -bis (2-hydroxyethoxy) -1,1'-binaphthalene crystals are efficiently crystallized with a small amount of solvent compared to the case of isolating known needle-like crystals.
- the filterability is good, and the amount of the solvent contained in the crystal before drying is small, so that the productivity can be improved along with the shortening of the filtration / drying time.
- the obtained plate-like or block crystal of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene is characterized by being less colored and having a higher purity than known acicular crystals. Therefore, it can be suitably used as a monomer for optical resins.
- FIG. 3 is a diagram showing a differential scanning calorimetry (DSC) curve of a crystal (polymorph A) obtained in Production Example 1.
- FIG. 4 is a diagram showing a differential scanning calorimetry (DSC) curve of the crystal (polymorph B) obtained in Example 3.
- FIG. 2 is a diagram showing a differential scanning calorimetry (DSC) curve of the crystal (polymorph C) obtained in Example 1.
- FIG. 2 is a diagram showing a powder X-ray diffraction pattern of a crystal (polymorph A) obtained in Production Example 1.
- FIG. 4 is a diagram showing a powder X-ray diffraction pattern of the crystal (polymorph B) obtained in Example 3.
- FIG. 1 is a diagram showing a powder X-ray diffraction pattern of a crystal (polymorph C) obtained in Example 1.
- FIG. 2 is a digital microscope photograph of the crystal (polymorph A) obtained in Production Example 1.
- FIG. 4 is a digital microscope photograph of the crystal (polymorph B) obtained in Example 3.
- FIG. 2 is a digital microscope photograph of the crystal (polymorph C) obtained in Example 1.
- FIG. 6 is a digital microscope photograph of a bulk crystal obtained in Example 6.
- 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene of the present invention has a crystal form different from that of conventionally known acicular crystals. There are two types of crystals. First, the plate crystal will be described in detail.
- the plate-like crystal of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene of the present invention is a novel of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene. It is a crystalline polymorph.
- the plate-like crystal in the present invention is a crystal having a small aspect ratio. Specifically, 2,2′-bis (2-hydroxyethoxy) -1,1′-photographed with an optical microscope (digital microscope). It is defined by the ratio L / W (aspect ratio) between the maximum crystal length L and the width W of the binaphthalene crystal.
- the maximum crystal length L here is defined as the length of the crystal taken from the crystal photograph taken with an optical microscope so that the length of the crystal is the longest, and the width W is the maximum crystal length.
- the angle is defined as the maximum length at an angle of 90 °.
- the maximum crystal length L and width W are average values calculated by measuring at least 30 crystals randomly selected from an optical micrograph.
- the plate-like crystal in the present invention has a value L / W (aspect ratio) of 1 to 8, preferably 1 to 5, obtained by dividing the maximum crystal length L defined above by the width W.
- L / W aspect ratio
- the width W of the plate crystal of the present invention is usually 3 ⁇ m or more, preferably 5 ⁇ m or more. When the width W of the crystal is smaller than 3 ⁇ m and the aspect ratio is larger than 8, a needle-like crystal is formed.
- the plate-like crystal in the present invention is a crystal form different from the crystal form of a conventionally known acicular crystal (referred to as polymorph A in the present application), the melting endotherm maximum by differential scanning calorimetry (DSC), and powder X-rays It has two novel crystal forms (referred to herein as polymorph B and polymorph C, respectively) distinguished by at least one feature of the diffraction angle 2 ⁇ in the diffraction pattern.
- polymorph B of the present invention has a melting endotherm maximum of 109-112 ° C. by differential scanning calorimetry
- polymorph C of the present invention has a melting endotherm of 106-108 ° C. by differential scanning calorimetry. is there.
- the maximum melting endotherm by differential scanning calorimetry in the present invention refers to the temperature at which the maximum endothermic peak is observed when differential scanning calorimetry is performed under the conditions described below. Note that the maximum melting endotherm exhibited by polymorph B and polymorph C of the present invention may fluctuate up and down due to several factors.
- Factors involved in such deviation include the heating rate of the sample when performing the analysis, the calibration standard used, the calibration method of the instrument, the relative humidity of the analysis environment and the chemical purity of the sample.
- the maximum melting endotherm observed for a given sample may vary from device to device, but generally will be within the range defined in this application if the device is similarly calibrated.
- Polymorph B of the present invention has a diffraction angle 2 ⁇ of 10.4 ⁇ 0.2 °, 12.1 ⁇ 0.2 °, 14.8 ⁇ 0.2 ° in a powder X-ray diffraction pattern by Cu—K ⁇ rays and It has a characteristic peak at 22.3 ⁇ 0.2 °.
- the diffraction angle 2 ⁇ has a maximum peak at 22.3 ⁇ 0.2 °.
- Polymorph C of the present invention has a diffraction angle 2 ⁇ of 10.0 ° ⁇ 0.2 °, 19.2 ° ⁇ 0.2 °, 19.7 ⁇ 0.2 in a powder X-ray diffraction pattern by Cu—K ⁇ rays. It has characteristic peaks at ° and 22.2 ⁇ 0.2 °. The diffraction angle 2 ⁇ has a maximum peak at 10.0 ° ⁇ 0.2 °.
- the plate-like crystal (polymorph B, polymorph C) of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene of the present invention is a racemate, but the effects of the present invention described above are manifested. Any one of the optically active substances may be contained in the range.
- the purity of the plate-like crystal of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene of the present invention is usually 90% or more, preferably 95% or more, more preferably 98% as described later. % Or more.
- the bulk crystal of 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene of the present invention is a crystal having a new shape and has at least the following two characteristics.
- the ratio Dmode / Dmedian between the mode diameter Dmode and the median diameter Dmedian is 2.0 or less.
- the ratio L / W between the maximum crystal length L and the width W determined based on the optical micrograph is 1-8.
- the mode diameter (Dmode) and the median diameter (Dmedian) in the present invention are numerical values obtained based on the particle size measurement by the laser diffraction method, and the mode diameter is the most frequent diameter indicating the highest frequency value. Yes, the median diameter indicates a 50% cumulative particle diameter in the cumulative particle size distribution.
- the mode diameter and the median diameter are measured by a method described later, and the ratio (Dmode / Dmedian) is calculated.
- the bulk crystal in the present invention has a ratio of mode diameter to median diameter (Dmode / Dmedian) obtained by the above-mentioned method of 2.0 or less, preferably 1.0 to 1.6. When it is higher than 2.0, the obtained crystal becomes a known acicular crystal, and the effect of the present invention described above is not exhibited.
- the bulk crystal has a ratio L / W (aspect ratio) between the maximum crystal length L and the width W obtained from an optical micrograph of 1 to 8, preferably 1 to 3.
- the width W of the bulk crystal of the present invention is usually 3 ⁇ m or more, preferably 5 ⁇ m or more.
- the width W of the crystal is smaller than 3 ⁇ m and the aspect ratio is larger than 8, a needle-like crystal is formed.
- the aspect ratio is measured by the same method as that for the plate crystal described above.
- the purity of the bulk crystals of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene of the present invention is usually 90% or higher, preferably 95% or higher, more preferably 98%, as described later. That's it.
- the bulk crystal of the present invention is produced by the method described later. Since polymorph B or polymorph C is transformed to polymorph A during production, the melting endotherm maximum by differential scanning calorimetry is 114 to 116 ° C.
- the powder X-ray diffraction pattern by Cu-K ⁇ ray shows the same pattern as that of polymorph A.
- the block crystal is less colored than the known acicular crystal and has a high purity, so that the crystal is excellent in industrial handling.
- the block crystal may include a plate crystal (polymorph B and / or polymorph C).
- the target 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene in the present invention is a compound in which 1 mol of 1,1′-bi-2-naphthol is reacted with 2 mol of ethylene carbonate.
- a compound obtained by reacting 1 mol of 1,1′-bi-2-naphthol and 1 mol of ethylene carbonate (hereinafter sometimes referred to as 1 mol adduct), 1,1′-bi- A compound in which 1 mol of 2-naphthol and 3 mol of ethylene carbonate have reacted (hereinafter sometimes referred to as a 3 mol adduct), 1 mol of 1,1′-bi-2-naphthols and 4 mol or more of ethylene carbonate have reacted
- a compound hereinafter sometimes referred to as an adduct of 4 mol or more
- a compound in which the target product is polymerized by 2 mol or more with a carbonate ester bond (hereinafter may be referred to as a polymer), etc. That.
- the reaction is preferably performed in the presence of an inert organic solvent.
- the amount of the inert organic solvent used is usually 0.1 to 4 times by weight, preferably 0.5 to 2 times by weight, relative to 1,1'-bi-2-naphthol.
- the inert organic solvent may be any solvent that does not inhibit the reaction, for example, aromatic hydrocarbons such as benzene, toluene, xylene, mesitylene, aliphatic hydrocarbons such as pentane, hexane, heptane, chlorobenzene, dichlorobenzene, etc.
- Aromatic hydrocarbons and halogenated aromatic hydrocarbons are preferred, and toluene and xylene are particularly preferred.
- the catalyst to be used may be either an alkali catalyst or an acid catalyst, but an alkali catalyst is preferred from the viewpoint that the reaction proceeds rapidly and impurities are reduced.
- alkali catalyst include potassium hydroxide, sodium hydroxide, barium hydroxide, magnesium oxide, sodium carbonate, potassium carbonate and the like. Of these, potassium hydroxide, sodium hydroxide, and potassium carbonate are preferred.
- an acid catalyst examples of usable acid catalysts include sulfuric acid, paratoluenesulfonic acid, methanesulfonic acid and the like.
- the amount of the catalyst used is usually 0.01 to 0.2 mol, preferably 0.05 to 0.2 mol, per 1 mol of 1,1'-bi-2-naphthol.
- the reaction temperature is usually 150 ° C. or lower, preferably 140 to 40 ° C., more preferably 130 to 70 ° C., particularly 120 to 90 ° C. If the reaction temperature is too high, it may cause a decrease in yield or a deterioration in hue due to an increase in side reaction products. If the reaction temperature is too low, the reaction may not proceed rapidly.
- the reaction can be carried out in the air, but it is preferably carried out in an inert gas atmosphere such as nitrogen or argon from the viewpoint of safety.
- the reaction can be followed by analytical means such as liquid chromatography.
- reaction mixture when the reaction mixture is in a slurry state, it is preferable to wash with an aqueous alkali solution and / or water in order to decompose and remove unreacted ethylene carbonate and polymer after dissolving by adding a solvent. Further, post-treatment operations such as dehydration, filtration, and adsorption treatment may be appropriately performed as necessary.
- the plate-like crystal of the present invention (polymorph B or C) has 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene dissolved in at least one solvent selected from aromatic hydrocarbons.
- This second crystal is a plate crystal (polymorph B or C).
- at least one solvent selected from aromatic hydrocarbons may be further used.
- a reaction containing 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene obtained by reaction of 1,1′-bi-2-naphthol and ethylene carbonate It is also preferred to use the liquid further.
- the reaction solution is used as at least part of the 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene used in the step of preparing the second solution.
- Polymorph C is obtained by precipitating crystals from the second solution at a temperature of 48 ° C. or lower, preferably 20 to 48 ° C., more preferably 30 to 45 ° C.
- a method for producing a mother liquor in the absence of the mother liquor used in the present invention (a step of obtaining a mother liquor in the method for producing 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene crystal of the present invention) is described in detail. Describe.
- the mother liquor crystallizes a reaction solution containing 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene obtained by the reaction of 1,1'-bi-2-naphthol and ethylene carbonate.
- the crystal and the mother liquor are obtained by solid-liquid separation. Therefore, crystallization conditions for obtaining the mother liquor will be described in detail.
- Examples of the at least one solvent selected from aromatic hydrocarbons used in the step of obtaining the mother liquor include benzene, toluene, xylene, mesitylene, chlorobenzene, dichlorobenzene, and the like. Toluene and xylene are preferable, and toluene is more preferable. These solvents can be used singly or as a mixture of two or more.
- crystallization is performed using a solvent not containing aromatic hydrocarbons to obtain a mother liquor, and crystallization is performed using the obtained mother liquor, the crystals obtained are needle-like crystals (polymorph A).
- the aromatic hydrocarbon in the solvent in the solution is at least 10% by weight, preferably 50% by weight or more.
- the amount of the solvent used is not limited so long as crystallization can be carried out. For example, 12 to 50% by weight with respect to 1 weight times 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene in the first solution. Use twice.
- the temperature during dissolution is usually 65 ° C. or higher and below the boiling point of the solvent, preferably 80 ° C. to 110 ° C.
- the dissolution time is usually 0.5 to 5 hours, preferably 1 to 3 hours with stirring.
- the cooling rate is usually 0.05 to 1 ° C. per minute, preferably 0.1 to 0.5 ° C. per minute, more preferably 0.1 to 0.3 ° C. per minute. If the cooling rate is too high, impurities are likely to be incorporated into the crystal, which causes a decrease in purity.
- the slurry liquid may be further cooled after crystal precipitation.
- the temperature at the end of cooling is usually ⁇ 10 to 40 ° C., preferably 0 to 30 ° C., more preferably 10 to 30 ° C.
- the mother liquor is obtained by solid-liquid separation of the slurry liquid containing the first crystal of 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene obtained as described above.
- the method of solid-liquid separation is not particularly limited, and examples thereof include a method using a filter and a method using a centrifuge.
- the first crystals of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene obtained by solid-liquid separation are used as various raw materials such as monomers for optical resins by performing an operation such as drying as necessary.
- step of preparing the second solution and the step of precipitating the second crystals (hereinafter, these may be collectively referred to as a step of reusing the mother liquor) will be described in detail.
- the second solution can also consist of at least a part of the mother liquor, but at least a part of the mother liquor and at least one solvent selected from aromatic hydrocarbons different from the aromatic hydrocarbons contained in the mother liquor It is preferable to prepare it.
- the amount of the mother liquor in the total of the mother liquor and the separate solvent is usually 10% by weight or more, preferably 25% by weight or more, more preferably 40% by weight or more, and particularly 50% by weight or more. If the amount of mother liquor contained is less than 10% by weight, polymorph C may not be obtained.
- the mother liquor may contain a solvent used for further washing the crystals separated during solid-liquid separation.
- the mother liquor to be reused may be a mother liquor separated from any of the crystalline forms of polymorph A, polymorph B, and polymorph C, but in order to obtain polymorph C stably, polymorph B or polymorph A mother liquor from which Form C has been produced and separated is preferred, and a mother liquor separated from Polymorph C is particularly preferred.
- mother liquor from reaction liquid containing 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene obtained by reaction of 1,1′-bi-2-naphthol and ethylene carbonate only
- the mother liquor obtained by crystallization of the first solution is a mother liquor separated from the polymorph A, and the mother liquor obtained by reusing and crystallization of the mother liquor is polymorph B or polymorph C.
- the mother liquor to be used may be contained when performing the step of precipitating the second crystal, and may be added immediately before the crystallization of the step of precipitating the second crystal, or may be added in the previous step. Good.
- the mother liquor used may be one that has never been used, or may be a mother liquor that has been used repeatedly. Further, mother liquors derived from different sources may be mixed and used.
- the at least one solvent selected from the aromatic hydrocarbons that can be used together with the mother liquor may be the same kind as that used in the process for obtaining the mother liquor described above or any other kind.
- the amount used is at least one total amount selected from aromatic hydrocarbons contained in the second solution (crystallization solution) before crystallization after addition of the mother liquor (aromatics contained in the mother liquor)
- the sum of the hydrocarbons and the newly added aromatic hydrocarbons) is 3 to 20 times the weight of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene in the second solution. It is preferably adjusted so that it is 5 to 12 times by weight.
- the temperature during dissolution is usually 65 ° C. or higher and below the boiling point of the solvent, preferably 80 ° C. to 110 ° C.
- the dissolution time is usually 0.5 to 5 hours, preferably 1 to 3 hours with stirring.
- the temperature at which the crystals are precipitated is 48 ° C. or lower, preferably 20 to 48 ° C., more preferably 30 to 45 ° C. When it is higher than 48 ° C., polymorph A or polymorph B is obtained. In particular, when the crystal precipitation temperature is higher than 60 ° C., it becomes a needle crystal (polymorph A) and the above-mentioned effects are not exhibited.
- the cooling rate before and after crystal precipitation is usually 0.05 to 1 ° C. per minute, preferably 0.1 to 0.5 ° C. per minute, in particular 0.1 to 0.3 ° C. per minute. . If the cooling rate is too slow, crystals tend to precipitate at a temperature higher than 60 ° C., and as a result, needle-like crystals are obtained. If the cooling rate is too high, impurities can easily be incorporated into the crystal and cause a decrease in purity.
- the slurry liquid may be further cooled after crystal precipitation.
- the temperature at the end of cooling is usually ⁇ 10 to 40 ° C., preferably 0 to 30 ° C., more preferably 10 to 30 ° C.
- the second crystals of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene contained in the slurry liquid are recovered by solid-liquid separation such as filtration and centrifugation.
- the obtained crystal may be washed using the solvent or the like used for the above crystallization, or may be dried.
- the crystal of 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene thus obtained is a plate-like crystal and is polymorph C. Its purity is usually 95% or more.
- the mother liquor separated by the solid-liquid separation can be reused as the mother liquor used in the step of preparing the second solution.
- Polymorph B is obtained by precipitating crystals at 50-60 ° C. from the second solution.
- the crystallization conditions and operation for obtaining the mother liquor and the crystallization conditions and operation in the step of reusing the mother liquor can be carried out under the same conditions as in the step of obtaining polymorph C except for the following points.
- the first difference is that the mother liquor to be reused may be a mother liquor separated from any of the crystal forms of polymorph A, polymorph B and polymorph C. In order to obtain polymorph B stably. Is preferably a mother liquor from which polymorph B or C is produced and separated, particularly preferably a mother liquor separated from polymorph B.
- the second difference is that the second crystal of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene needs to be precipitated at a temperature of 50 to 60 ° C. It is preferable to add.
- the crystal of 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene is preferably seeded at a temperature of 50 to 60 ° C., more preferably 52 to 58 ° C. during cooling.
- the crystals are precipitated at the same temperature, and more preferably, after the seed crystals are added, the mixture is stirred at a temperature of 50 ° C. or higher for a certain time (for example, 1 to 5 hours, preferably 1 to 3 hours).
- the 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene crystal used as a seed crystal may be any of polymorph A, polymorph B and polymorph C, but preferably polymorph B Or polymorph C, particularly preferably polymorph B.
- the amount of seed crystals added is usually 0.001 to 5% by weight with respect to 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene in the second solution (crystallization solution).
- the content is preferably 0.005 to 1% by weight, more preferably 0.01 to 0.5% by weight.
- the second crystals of 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene thus precipitated are recovered by solid-liquid separation such as filtration and centrifugation.
- the obtained crystal may be washed using the solvent or the like used for the above crystallization, or may be dried.
- the crystal of 2,2'-bis (2-hydroxyethoxy) -1,1'-binaphthalene thus obtained is a plate-like crystal and is polymorph B. Its purity is usually 95% or more.
- the mother liquor separated by the solid-liquid separation can be reused as the mother liquor used in the step of preparing the second solution.
- the bulk crystal of the present invention is a plate crystal (polymorph B and / or polymorph C) at 40 to 108 ° C., preferably 60 to 102 ° C., usually 6 hours or more, preferably 12 to 72 hours, more preferably 12 Obtained by heating for ⁇ 48 hours.
- the heating is preferably carried out under conditions such that the crystals are stirred in a container containing the crystals.
- a block crystal cannot be obtained, and when it heats at a temperature higher than 108 degreeC, since a plate-like crystal melts, a block crystal cannot be obtained.
- the acicular crystal (polymorph A) is heated to the above temperature range, it does not change into a lump crystal and remains as an acicular crystal.
- the plate crystal may contain the solvent used in the crystallization described above.
- the solvent content is 40% by weight or less, preferably 20% by weight or less. If the amount of the solvent contained in the plate-like crystal is more than 40% by weight, the crystal may be dissolved during heating to form a needle-like crystal.
- crystallization you may perform solvent removal operation, such as drying under reduced pressure, in parallel.
- DSC ⁇ Differential scanning calorimetry
- Apparatus Color difference meter (Nippon Denshoku Industries Co., Ltd. SE6000), Cell used: Optical path length 33 mm Quartz cell.
- Apparatus Digital microscope (manufactured by Keyence Corporation, VHX-1000), Measurement magnification of each photo: 600 times.
- ⁇ Ratio of mode diameter to median diameter (Dmode / Dmedian)> The mode diameter and the median diameter were measured under the following conditions, and the ratio of the mode diameter to the median diameter (Dmode / Dmedian) was calculated based on the obtained value.
- Measuring device SALD-2200 (laser diffraction particle size distribution measuring device) manufactured by Shimadzu Corporation, Measurement range: 1000 to 0.030 ⁇ m, Dispersing solvent: distilled water + neutral detergent, Dispersion method: ultrasonic dispersion.
- the obtained organic solvent layer was dehydrated using a Dean-Stark apparatus under reflux, and a toluene solution (crystallization) in which (RS) -2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene was dissolved. Solution). Thereafter, when the crystallization solution was cooled, crystals precipitated at a stretch at 63 ° C., making stirring difficult. Therefore, 1200 g of toluene was added at the same temperature to make it a slurry state containing crystals, and then it was stirred, and further cooled to 30 ° C.
- the crystal was further washed with 200 g of toluene to separate the crystal part and the mother liquor. This filtration washing operation took 40 minutes. Further, when a part of the obtained crystal was collected and analyzed, the solvent content in the crystal was 50% by weight, and the shape of the crystal was a needle-like crystal. The mother liquor separated by filtration was 2630 g. Next, the crystals obtained by filtration were dried to obtain 198 g of light yellow crystals of (RS) -2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene (yield 84.1). %, HPLC purity 99.1%, YI value: 11).
- the physical properties of the obtained crystal are as follows [DSC melting endotherm maximum: 116 ° C., powder X-ray diffraction pattern: polymorph A, crystal shape: needle crystal, aspect ratio: 68.0 (absolute width: 2 ⁇ m) , Ratio of mode diameter to median diameter (Dmode / Dmedian): 2.5].
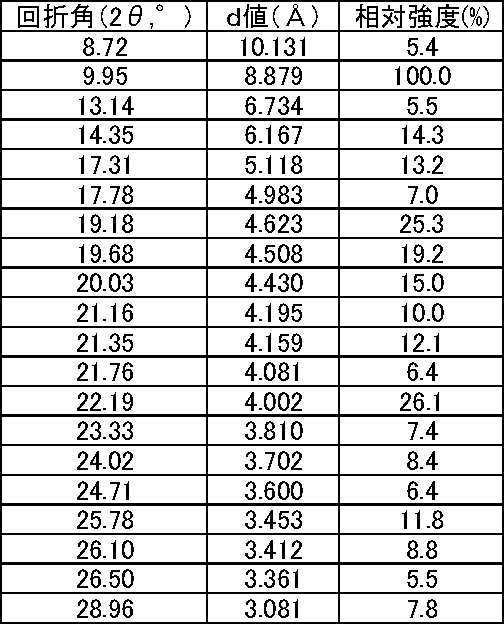
- the DSC analysis chart is shown in FIG. 1, the powder X-ray pattern is shown in FIG. 4, and the main peaks of powder X-rays (those having a relative intensity exceeding 5%) are listed in Table 1.
- a digital microscope photograph is shown in FIG.
- Example 1 In a glass reactor equipped with a stirrer, a cooler, and a thermometer, 180 g (0.629 mol) of (RS) -1,1′-bi-2-naphthol, 127 g (1.439 mol) of ethylene carbonate, potassium carbonate 9.0 g and 180 g of toluene were charged and stirred at 110 ° C. for 10 hours. After adding 200 g of toluene and 1100 g of the mother liquor obtained in Production Example 1 to this reaction product, the organic solvent layer was kept at 80 ° C. and washed with an aqueous sodium hydroxide solution. Next, this organic solvent layer was washed with water until the washing water became neutral.
- the obtained organic solvent layer was dehydrated under reflux using a Dean-Stark apparatus to obtain a crystallization solution, and cooled to 30 ° C. after 8 hours to precipitate crystals at 39 ° C.
- the slurry liquid thus obtained was filtered under the above conditions, and the crystals were further washed with 200 g of toluene to separate the crystal part and the mother liquor. This filtration washing operation took 10 minutes. Further, when a part of the obtained crystal was collected and analyzed, the solvent content in the crystal was 15% by weight and the crystal shape was a plate crystal.
- the mother liquor separated by filtration was 1603 g.
- the DSC analysis chart is shown in FIG. 3, the powder X-ray pattern is shown in FIG. 6, and the main peaks of powder X-rays (those having a relative intensity exceeding 5%) are listed in Table 3.
- Polymorph C has diffraction peaks characteristic of diffraction angles 2 ⁇ of 10.0 ⁇ 0.2 °, 19.2 ⁇ 0.2 °, 19.7 ⁇ 0.2 ° and 22.2 ⁇ 0.2 °. showed that.
- a digital microscope photograph is shown in FIG.
- Example 2 A crystallization solution was obtained in the same manner as in Example 1 except that 1100 g of the mother liquor obtained in Example 1 was added instead of 1100 g of the mother liquor obtained in Production Example 1. Crystals were precipitated at 36 ° C. by cooling the crystallization solution to 30 ° C. in 10 hours after the start of cooling. The slurry liquid thus obtained was filtered under the above conditions, and the crystals were further washed with 200 g of toluene to separate the crystal part and the mother liquor. This filtration washing operation took 10 minutes. Further, when a part of the obtained crystal was collected and analyzed, the solvent content in the crystal was 14.5% by weight, and the crystal shape was a plate crystal. The mother liquor separated by filtration was 1610 g.
- Example 3 In a glass reactor equipped with a stirrer, a cooler, and a thermometer, 180 g (0.629 mol) of (RS) -1,1′-bi-2-naphthol, 127 g (1.439 mol) of ethylene carbonate, potassium carbonate 9.0 g and 180 g of toluene were charged and stirred at 110 ° C. for 10 hours.
- the reaction product was diluted by adding 760 g of toluene and 410 g of the mother liquor obtained in Example 2, and the organic solvent layer was kept at 80 ° C. and washed with an aqueous sodium hydroxide solution. Next, this organic solvent layer was washed with water until the washing water became neutral.
- the obtained organic solvent layer was dehydrated under reflux using a Dean Stark apparatus to obtain a crystallization solution. After cooling this crystallization solution to 55 ° C. over 5 hours, at the same temperature, 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene (polymorph C) obtained in Example 2 was used. ) 0.085 g was added as a seed crystal, and the mixture was further stirred at the same temperature for 2 hours to precipitate crystals, and then cooled to 30 ° C. over 3 hours. After filtering this under the above-mentioned conditions, the crystal was further washed with 200 g of toluene to separate the crystal part and the mother liquor. This filtration washing operation took 10 minutes.
- the physical properties of the obtained crystal are as follows [DSC melting endotherm maximum: 110 ° C., powder X-ray diffraction pattern: polymorph B, crystal shape: plate crystal, aspect ratio: 1.7 (absolute value of width: 97 ⁇ m) , Ratio of mode diameter to median diameter (Dmode / Dmedian): 1.0].
- FIG. 2 shows a DSC analysis chart
- FIG. 5 shows a powder X-ray pattern
- Table 2 lists main peaks (having a relative intensity exceeding 5%) of the powder X-ray.
- Polymorph B has diffraction peaks characteristic of diffraction angles 2 ⁇ of 10.4 ⁇ 0.2 °, 12.1 ⁇ 0.2 °, 14.8 ⁇ 0.2 ° and 22.3 ⁇ 0.2 °. showed that.
- a digital microscope photograph is shown in FIG.
- Example 4 In a glass reactor equipped with a stirrer, a cooler, and a thermometer, 270 g (0.943 mol) of (RS) -1,1′-bi-2-naphthol, 190 g (2.158 mol) of ethylene carbonate, potassium carbonate 13.5 g and 270 g of toluene were charged and stirred at 110 ° C. for 10 hours.
- the reaction product was diluted with 1080 g of toluene and 1350 g of the mother liquor obtained in Example 3, and the organic solvent layer was kept at 80 ° C. and washed with an aqueous sodium hydroxide solution. Next, this organic solvent layer was washed with water until the washing water became neutral.
- the obtained organic solvent layer was dehydrated under reflux using a Dean Stark apparatus to obtain a crystallization solution.
- the crystallization solution was cooled to 55 ° C. over 5 hours, and then at the same temperature, 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene (polymorph B) obtained in Example 3 was used.
- polymorph B 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene obtained in Example 3 was used.
- 0.270 g was added as seed crystals, and further stirred at the same temperature for 2 hours to precipitate crystals, and then cooled to 30 ° C. over 4 hours. After filtering this under the above-mentioned filtration conditions, the crystals were further washed with 300 g of toluene to separate the crystal part and the mother liquor. This filtration washing operation took 15 minutes.
- the physical properties of the obtained crystal are as follows [DSC melting endotherm maximum: 110 ° C., powder X-ray diffraction pattern: polymorph B, crystal shape: plate crystal, aspect ratio: 1.9 (absolute value of width: 48 ⁇ m) , Ratio of mode diameter to median diameter (Dmode / Dmedian): 1.1].
- Example 5 In a glass reactor equipped with a stirrer, a cooler, and a thermometer, 180 g (0.629 mol) of (RS) -1,1′-bi-2-naphthol, 127 g (1.439 mol) of ethylene carbonate, potassium carbonate 9.0 g and xylene 180 g were charged and stirred at 110 ° C. for 10 hours.
- the reaction product was diluted by adding 670 g of xylene and 500 g of the mother liquor obtained in Example 4, and the organic solvent layer was kept at 80 ° C. and washed with an aqueous sodium hydroxide solution. Next, this organic solvent layer was washed with water until the washing water became neutral.
- the obtained organic solvent layer was dehydrated under reflux using a Dean Stark apparatus to obtain a crystallization solution. After cooling this crystallization solution to 55 ° C. over 5 hours, the 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene (polymorph B) obtained in Example 4 was used at the same temperature. 0.900 g was added as a seed crystal, and further stirred at the same temperature for 2 hours to precipitate a crystal, and then cooled to 30 ° C. over 4 hours. After filtering this under the above-mentioned filtration conditions, the crystal was further washed with 200 g of xylene to separate the crystal part and the mother liquor. This filtration washing operation took 10 minutes.
- the physical properties of the obtained crystal are as follows [DSC melting endotherm maximum: 110 ° C., powder X-ray diffraction pattern: polymorph B, crystal shape: plate crystal, aspect ratio: 1.8 (absolute value of width: 20 ⁇ m) , Ratio of mode diameter to median diameter (Dmode / Dmedian): 1.0].
- Example 6 50 g of bulk crystals were obtained by stirring the plate-like crystals (polymorph C) obtained in Example 1 for 36 hours at an external temperature of 95 ° C. with a rotary evaporator.
- the physical properties of the obtained bulk crystals were as follows [HPLC purity 99.7%, YI value: 3, DSC melting endotherm: 115 ° C. (powder X-ray diffraction pattern: polymorph A), crystal shape: bulk crystals, Aspect ratio: 1.8 (absolute value of width: 50 ⁇ m), mode diameter: 91.15 ⁇ m, median diameter: 74.63 ⁇ m, ratio of mode diameter to median diameter (Dmode / Dmedian): 1.2].
- a digital microscope photograph of the obtained bulk crystal is shown in FIG.
- the obtained organic solvent layer was concentrated to remove water and toluene, and then 540 g of acetone was added to the residue and dissolved by heating at 56 ° C. Next, when this solution was cooled to 30 ° C. in 3 hours after the start of cooling, crystals were precipitated at 35 ° C.
- the slurry liquid thus obtained was filtered under the above-mentioned filtration conditions to separate the crystal part and the mother liquor. This filtration operation took 30 minutes. Further, when a part of the obtained crystal was collected and analyzed, the solvent content in the crystal was 48% by weight and the crystal shape was a needle crystal.
- the obtained organic solvent layer was concentrated to remove water and toluene, and then 540 g of methanol was added to the residue and dissolved by heating at 64 ° C. Next, when this solution was cooled to 30 ° C. in 3 hours after the start of cooling, crystals were precipitated at 32 ° C.
- the obtained slurry was filtered under the above filtration conditions, and then separated into a crystal part and a mother liquor. This filtration operation took 30 minutes. Further, when a part of the obtained crystal was collected and analyzed, the solvent content in the crystal was 45% by weight, and the shape of the crystal was a needle crystal.
- the physical properties of the obtained crystal are as follows [DSC melting endotherm maximum: 116 ° C., powder X-ray diffraction pattern: polymorph A, crystal shape: needle crystal, aspect ratio: 70.0 (absolute value of width: 2 ⁇ m) Ratio of mode diameter to median diameter (Dmode / Dmedian): 2.1].
- This resinous product is a polycondensate obtained by reacting 1 mol of 1,1′-bi-2-naphthols with 3 mol of ethylene carbonate, and 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene. It was 2,2′-bis (2-hydroxyethoxy) -1,1′-binaphthalene containing a carbonate ester body and a salt polymerized by 2 mol by a carbonate ester bond. (HPLC purity 94.1%).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
所定の結晶形を有する2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレン結晶、および芳香族炭化水素から選ばれる少なくとも1種の溶媒中に2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンが溶解している第1溶液から2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの第1結晶を析出させた後、固液分離して母液を得る工程と、該母液の少なくとも一部を用いて2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンが溶解している第2溶液を調製する工程と、該第2溶液から、60℃以下の温度で2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの第2結晶を析出させる工程とを含む2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレン結晶の製造方法が提供される。
Description
本発明は、光学レンズや光学フィルムに代表される光学部材を構成する樹脂(光学樹脂)を形成するモノマーとして好適で、加工性、生産性に優れた新規な2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶多形体およびその製造方法に関する。
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンなどのビナフタレン類を原料モノマーとするポリカーボネート、ポリエステル、ポリアクリレート、ポリウレタン、エポキシなどの樹脂材料は、光学特性、耐熱性等に優れることから、近年、光学レンズや光学シートなどの新たな光学材料として注目されている。
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを得る方法として、特開2011-153248号公報(特許文献1)には、その合成例1に1,1’-ビ-2-ナフトールと過剰量のエチレンカーボネートとを水酸化カリウム触媒存在下に反応させ、得られた反応生成物をメチルイソブチルケトンに溶解し、水で洗浄した後、メチルイソブチルケトンを除去して樹脂状物を得る方法が開示されている。特開2010-018753号公報(特許文献2)には、その実施例1に1,1’-ビ-2-ナフトールと過剰量のエチレンカーボネートとを炭酸カリウム触媒存在下、トルエン溶媒中で反応させ、得られた反応生成物を1%水酸化ナトリウム水溶液および水で洗浄した後、減圧下で溶媒を除去して2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを得る方法が開示されている。しかし、これらの特許文献に記載される方法は、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解した溶液から溶媒を全量除去する方法であり、工業的な実施が困難な方法である。更には、1,1’-ビ-2-ナフトールと過剰量のエチレンカーボネートとの反応においては目的物の他に、1,1’-ビ-2-ナフトール1分子とエチレンカーボネート1分子が反応した化合物、1,1’-ビ-2-ナフトール1分子とエチレンカーボネート3分子以上が反応した化合物や目的物が炭酸エステル結合などにより2モル以上重合した化合物などが副生する。そのため、前記特許文献記載のように反応生成物から溶媒を留去し、その濃縮残として目的物を得る方法では、樹脂状の固まりとなり、純度や色相の不十分なものしか得られない。
一方、カラムクロマトグラフィーで得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶としては、融点が112~113℃のラセミ体結晶が知られている〔J.Org.Chem.,Vol.42,No.26,1977,4173-4184(非特許文献1)〕。そこで本発明者らがアルカリ触媒下、1,1’-ビ-2-ナフトールと過剰量のエチレンカーボネートとを反応させた後、反応生成物をトルエンに溶解させ定法により晶析を行った結果、当該文献記載と同様に融点112~113℃(示差走査熱分析の融解吸熱最大では114~116℃)の結晶を得た。本結晶の結晶形状は針状結晶であった。
しかしながら、前記晶析操作を行った際、結晶析出と同時に撹拌することができなくなった。そこで、撹拌可能な状態とするために多量の溶媒を追加し、析出した結晶の一部を再度溶解させ、撹拌を再開できる状態とする必要があった。また、濾過性や結晶洗浄時の液切れが悪く、その結果、乾燥前の結晶が多量の溶媒を含むため、非特許文献1記載の方法は、生産性が著しく悪いことが判明した。
J.Org.Chem.,Vol.42,No.26,1977,4173-4184
本発明の目的は、光学樹脂をはじめとする各種樹脂の原料モノマーとして好適に用いられる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを結晶として取得可能である、工業的に実施可能な製造方法、および工業的有利に取得可能な2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を提供することにある。
本発明者らは、前記の課題を解決すべく鋭意研究を重ねた結果、特定の晶析条件によれば、所定の結晶形を有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を析出させることが可能であり、これにより2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを当該結晶として工業的有利に得ることが可能であることを見出した。具体的には以下の発明を含む。
[1]
示差走査熱分析による融解吸熱最大が109~112℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
示差走査熱分析による融解吸熱最大が109~112℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[2]
Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.4±0.2°、12.1±0.2°、14.8±0.2°および22.3±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.4±0.2°、12.1±0.2°、14.8±0.2°および22.3±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[3]
示差走査熱分析による融解吸熱最大が106~108℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
示差走査熱分析による融解吸熱最大が106~108℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[4]
Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.0±0.2°、19.2±0.2°、19.7±0.2°および22.2±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.0±0.2°、19.2±0.2°、19.7±0.2°および22.2±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[5]
板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[6]
板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である[1]~[4]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である[1]~[4]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[7]
板状結晶の幅Wが3μm以上である[5]または[6]に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
板状結晶の幅Wが3μm以上である[5]または[6]に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[8]
塊状結晶であって、モード径Dmodeとメジアン径Dmedianとの比Dmode/Dmedianが2.0以下であり、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶
[9]
示差走査熱分析による融解吸熱最大が114~116℃である[8]に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
塊状結晶であって、モード径Dmodeとメジアン径Dmedianとの比Dmode/Dmedianが2.0以下であり、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶
[9]
示差走査熱分析による融解吸熱最大が114~116℃である[8]に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
[10]
芳香族炭化水素から選ばれる少なくとも1種の溶媒中に2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第1溶液から2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶を析出させた後、固液分離して母液を得る工程と、
前記母液の少なくとも一部を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第2溶液を調製する工程と、
前記第2溶液から、60℃以下の温度で2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶を析出させる工程と、
を含む[1]~[7]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶の製造方法。
芳香族炭化水素から選ばれる少なくとも1種の溶媒中に2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第1溶液から2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶を析出させた後、固液分離して母液を得る工程と、
前記母液の少なくとも一部を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第2溶液を調製する工程と、
前記第2溶液から、60℃以下の温度で2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶を析出させる工程と、
を含む[1]~[7]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶の製造方法。
[11]
前記第2溶液から、50~60℃の温度で前記第2結晶を析出させる[10]に記載の製造方法。
前記第2溶液から、50~60℃の温度で前記第2結晶を析出させる[10]に記載の製造方法。
[12]
前記第2溶液から、48℃以下の温度で前記第2結晶を析出させる[10]に記載の製造方法。
前記第2溶液から、48℃以下の温度で前記第2結晶を析出させる[10]に記載の製造方法。
[13]
前記母液を得る工程において、芳香族炭化水素から選ばれる少なくとも1種の溶媒中に溶解している2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが、1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得られたものである[10]~[12]のいずれかに記載の製造方法。
前記母液を得る工程において、芳香族炭化水素から選ばれる少なくとも1種の溶媒中に溶解している2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが、1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得られたものである[10]~[12]のいずれかに記載の製造方法。
[14]
[1]~[7]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を用意する工程と、
前記結晶を40~108℃に加熱する工程と、
を含む2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶の製造方法。
[1]~[7]のいずれかに記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を用意する工程と、
前記結晶を40~108℃に加熱する工程と、
を含む2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶の製造方法。
本発明によれば、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を高収率かつ操作性が良好な方法により得ることが可能となる。具体的には、本発明により得られる新規な板状結晶、あるいは塊状結晶は従来公知の針状結晶とは異なり、晶析にて結晶を単離する場合、結晶が析出した際に少量の溶媒で撹拌が可能であることから公知の針状結晶を単離する場合に比べ少量の溶媒で効率よく2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を晶析により得ることが可能であり、更には濾過性が良く、乾燥前の結晶に含まれる溶媒量が少ないため、濾過・乾燥時間の短縮に伴う生産性の改善が可能となる。
また得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状結晶あるいは塊状結晶は公知の針状結晶に比べ着色が少なく、高純度であるといった特徴を有しているため、光学樹脂用モノマーとして好適に用いることができる。
<本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンについて>
本発明における2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンは、従来公知の針状結晶とは異なる結晶形を有しており、その形状から、板状結晶と塊状結晶の二つに大別される。まずは板状結晶について詳述する。
本発明における2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンは、従来公知の針状結晶とは異なる結晶形を有しており、その形状から、板状結晶と塊状結晶の二つに大別される。まずは板状結晶について詳述する。
本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状結晶は、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの新規な結晶多形体である。本発明における板状結晶とは、縦横比の小さい結晶であり、具体的には、光学顕微鏡(デジタルマイクロスコープ)で撮影した2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶の最大結晶長さLと幅Wとの比L/W(アスペクト比)で定義される。ここでいう最大結晶長さLとは、光学顕微鏡で撮影された結晶写真から結晶の長さが最も長くなるようにとった結晶の長さで定義し、また幅Wとは最大結晶長さに対し90°の角度をなし、且つ最大の長さとなる長さで定義する。なお、最大結晶長さLと幅Wは光学顕微鏡写真から無作為に選択した少なくとも30個の結晶を測定し算出した平均値である。
本発明における板状結晶とは、上述のように定義された最大結晶長さLを幅Wで除した値L/W(アスペクト比)が1~8であり、好ましくは1~5である。アスペクト比が大きいと加工性、生産性が悪くなり、更には純度、色相等の品質が低下し得る。また、本発明の板状結晶の幅Wは、通常、3μm以上であり、好ましくは5μm以上である。結晶の幅Wが3μmより小さく、アスペクト比が8より大きいと針状結晶となる。
本発明における板状結晶は、従来公知の針状結晶(本願において多形体Aと称する)の結晶形とは異なる結晶形であり、示差走査熱分析(DSC)による融解吸熱最大、および粉末X線回折パターンにおける回折角2θの少なくとも1つの特徴により区別される新規な2種類の結晶形(本願においてそれぞれ多形体B、多形体Cと称する)を有する。
具体的には、本発明の多形体Bは、示差走査熱分析による融解吸熱最大が109~112℃であり、本発明の多形体Cは示差走査熱分析による融解吸熱最大が106~108℃である。本発明における示差走査熱分析による融解吸熱最大とは、後述する条件にて示差走査熱分析を実施した際、最大吸熱ピークが観測される温度のことをいう。なお、本発明の多形体Bおよび多形体Cの示す融解吸熱最大は、いくつかの要因により、上下に変動することがある。このような偏差に関与する要因としては、分析を実施する際の試料の加熱速度、使用される校正標準、機器の校正方法、分析環境の相対湿度および試料の化学的純度がある。与えられた試料について観察される融解吸熱最大は、装置毎に異なることがあるが、一般に、装置が同様に校正されていれば、本願に定義される範囲内となる。
本発明の多形体Bは、Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.4±0.2°、12.1±0.2°、14.8±0.2°および22.3±0.2°に特徴的なピークを有する。また、回折角2θが22.3±0.2°に最大ピークを有する。
本発明の多形体Cは、Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.0°±0.2°、19.2°±0.2°、19.7±0.2°および22.2±0.2°に特徴的なピークを有する。また、回折角2θが10.0°±0.2°に最大ピークを有する。
本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状結晶(多形体B、多形体C)はラセミ体であるが、上述した本願の効果が発現する範囲でいずれかの光学活性体が多く含まれていても良い。R体とS体の比率はR体/S体=45/55~55/45であることが好ましい。
本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状結晶の純度は、後述するHPLC純度が通常90%以上、好ましくは95%以上、より好ましくは98%以上である。
続いて本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶について詳述する。
本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶は新たな形状を有する結晶であり、少なくとも以下の二つの特徴を有している。
(1)モード径Dmodeとメジアン径Dmedianとの比Dmode/Dmedianが2.0以下である、
(2)光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である。
(2)光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である。
本発明におけるモード径(Dmode)とメジアン径(Dmedian)とは、レーザー回折法による粒度測定に基づき得られた数値であって、モード径とは、最も高い頻度値を示す最頻径のことであり、メジアン径とは累積粒度分布において、累積値50%粒子径のことを示す。なお、本発明においては、後述する方法にてモード径とメジアン径とを測定し、その比(Dmode/Dmedian)を算出する。
本発明における塊状結晶とは、上述の方法により得られたモード径とメジアン径との比(Dmode/Dmedian)が2.0以下であり、好ましくは1.0~1.6である。2.0より高い場合、得られる結晶は公知の針状結晶となり、上述した本願効果が発現しなくなる。
本発明における塊状結晶とは、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/W(アスペクト比)が1~8であり、好ましくは1~3である。アスペクト比が大きいと加工性、生産性が悪くなり、更には純度、色相等の品質が低下し得る。また、本発明の塊状結晶の幅Wは、通常、3μm以上であり、好ましくは5μm以上である。結晶の幅Wが3μmより小さく、アスペクト比が8より大きいと針状結晶となる。アスペクト比は、上述した板状結晶と同一の方法によって測定する。
本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶の純度は、後述するHPLC純度が通常90%以上、好ましくは95%以上、より好ましくは98%以上である。
本発明の塊状結晶は後述する方法にて製造されるが、製造中に多形体Bまたは多形体Cが多形体Aへ転移するため、示差走査熱分析による融解吸熱最大は114~116℃であり、Cu-Kα線による粉末X線回折パターンは多形体Aと同一のパターンを示す。しかしながら、前述の通り塊状結晶とすることにより公知の針状結晶と比べ着色が少なく、高純度であることから工業的な取扱いに優れた結晶となる。なお、塊状結晶は板状結晶(多形体Bおよび/または多形体C)を含んでいても良い。
<2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状および塊状結晶の製造方法について>
続いて上述した2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状および塊状結晶の製造方法について詳述する。本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンは、例えば1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得ることができる。以下、1,1’-ビ-2-ナフトールとエチレンカーボネートから2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを製造する方法について説明する。
続いて上述した2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの板状および塊状結晶の製造方法について詳述する。本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンは、例えば1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得ることができる。以下、1,1’-ビ-2-ナフトールとエチレンカーボネートから2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを製造する方法について説明する。
本発明において目的とする2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンは1,1’-ビ-2-ナフトール1モルとエチレンカーボネート2モルが反応した化合物であるが、この他に副反応物として1,1’-ビ-2-ナフトール1モルとエチレンカーボネート1モルが反応した化合物(以下1モル付加体と記載する場合がある)、1,1’-ビ-2-ナフトール1モルとエチレンカーボネート3モルが反応した化合物(以下3モル付加体と記載する場合がある)、1,1’-ビ-2-ナフトール類1モルとエチレンカーボネート4モル以上が反応した化合物(以下4モル以上付加体と記載する場合がある)、目的物が炭酸エステル結合で2モル以上重合した化合物(以下重合体と記載する場合がある)などが生成する。
1,1’-ビ-2-ナフトールとエチレンカーボネートの使用量は後述する溶媒を使用するか否かによっても異なるが、通常、1,1’-ビ-2-ナフトールとエチレンカーボネートの比率(モル比)が1,1’-ビ-2-ナフトール/エチレンカーボネート=1/1.9~1/2.4、好ましくは1/2.0~1/2.4、更に好ましくは1/2.0~1/2.3である。エチレンカーボネートの使用量が1/1.9より少ない場合、攪拌困難となり反応が進行しないか、反応が遅延する場合がある。反応が進行した場合においても未反応1,1’-ビ-2-ナフトールや1モル付加体などの副反応物が多く、収率や純度が低下する場合がある。エチレンカーボネートの使用量が1/2.4より多いと3モル付加体、4モル以上付加体や重合体などの副反応物の増加により、収率や純度が低下する場合がある。
溶媒は用いても用いなくてもよいが、好ましくは不活性有機溶媒共存下で反応する。不活性有機溶媒の使用量は通常、1,1’-ビ-2-ナフトールに対して0.1~4重量倍、好ましくは0.5~2重量倍である。
不活性有機溶媒とは、反応を阻害しないものであればよく、例えば、ベンゼン、トルエン、キシレン、メシチレンなどの芳香族炭化水素、ペンタン、ヘキサン、ヘプタンなどの脂肪族炭化水素、クロロベンゼン、ジクロロベンゼンなどのハロゲン化芳香族炭化水素、ジクロロメタン、1,2-ジクロロエタンなどのハロゲン化脂肪族炭化水素などが使用可能である。好ましくは芳香族炭化水素、ハロゲン化芳香族炭化水素であり、特にトルエン、キシレンが好ましい。
反応時、必要に応じ触媒を使用する。使用する触媒は、アルカリ触媒、酸触媒のいずれであってもよいが、反応の進行が速く、不純物が少なくなる点からアルカリ触媒が好ましい。アルカリ触媒としては、例えば水酸化カリウム、水酸化ナトリウム、水酸化バリウム、酸化マグネシウム、炭酸ナトリウム、炭酸カリウムなどが挙げられる。中でも水酸化カリウム、水酸化ナトリウム、炭酸カリウムが好ましい。酸触媒を使用する場合、使用可能な酸触媒として例えば硫酸、パラトルエンスルホン酸、メタンスルホン酸などが挙げられる。触媒の使用量は通常、1,1’-ビ-2-ナフトール1モルに対して0.01~0.2モル、好ましくは0.05~0.2モルである。
反応温度は通常、150℃以下、好ましくは140~40℃、更に好ましくは130~70℃、特に120~90℃である。反応温度が高すぎると副反応物の増加による収率低下や色相悪化の原因となる場合がある。反応温度が低すぎると反応が速やかに進行しない場合がある。
反応は大気下でも実施することができるが、安全性の観点から、窒素、アルゴンなどの不活性ガス雰囲気下で行うことが好ましい。反応は液体クロマトグラフィーなどの分析手段で追跡することができる。
反応後、反応混合物がスラリー状態の場合は溶媒を加えて溶解した後、未反応のエチレンカーボネートや重合体を分解、除去するためにアルカリ水溶液または/および水で洗浄を行うと良い。さらに必要に応じて脱水、濾過、吸着処理などの後処理操作を適宜施しても良い。
続いて、上記反応で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの晶析工程について詳述する。上述の方法にて得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを晶析することにより2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶が得られるが、この結晶は、後述する方法にて製造しない場合、公知の針状結晶となり、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を高収率かつ操作性が良好な方法により得ることができない。
本発明の板状結晶(多形体BまたはC)は、芳香族炭化水素から選ばれる少なくとも1種の溶媒中に2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第1溶液(晶析溶液)から2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶を析出させた後、固液分離して母液を得る工程と、該母液の少なくとも一部を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第2溶液を調製する工程と、該第2溶液から、60℃以下の温度で第2結晶を析出させることによって得られる。この第2結晶が板状結晶(多形体BまたはC)である。第2溶液の調製にあたって、芳香族炭化水素から選ばれる少なくとも1種の溶媒をさらに使用しても良い。また、第2溶液の調製にあたって、1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応で得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを含む反応液をさらに使用することも好ましい。この場合、第2溶液を調製する工程に使用する上記2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの少なくとも一部として当該反応液が使用される。
母液を含まず、芳香族炭化水素から選ばれる少なくとも1種を溶媒とする溶液から2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを晶析した場合や、芳香族炭化水素以外を溶媒とする溶液から晶析した場合等、本発明に従わない条件にて晶析し、60℃より高い温度で結晶を析出させた場合、得られる結晶は針状結晶(多形体A)となる。一方、本発明に従う条件にて晶析し、かつ結晶を析出させる温度が50~60℃である場合、板状結晶の中でも多形体Bが析出し、結晶を析出させる温度が48℃以下の場合、板状結晶の中でも多形体Cが析出する。以下、各多形体を得る条件を詳述することにより、本発明の板状結晶を得る条件を詳述する。
多形体Cを得る方法について説明する。多形体Cは、上記第2溶液から、48℃以下、好ましくは20~48℃、より好ましくは30~45℃の温度で結晶を析出させることによって得られる。
まず本発明に使用する母液がない場合の母液の製造方法(本発明の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶の製造方法における母液を得る工程)について詳述する。母液は上述の1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応で得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを含む反応液を晶析し、結晶と母液とを固液分離することによって得られる。そこで、母液を得るための晶析条件を詳述する。
母液を得る工程に用いられる芳香族炭化水素から選ばれる少なくとも1種の溶媒としては、例えば、ベンゼン、トルエン、キシレン、メシチレン、クロロベンゼン、ジクロロベンゼンなどが挙げられる。好ましくはトルエン、キシレン、さらに好ましくはトルエンである。これらの溶媒は1種、あるいは2種以上の混合物として用いる事ができる。芳香族炭化水素を含まない溶媒を用いて晶析し母液を得、得られた母液を使用して晶析した場合、得られる結晶は針状結晶(多形体A)となるため、上記第1溶液における溶媒中の芳香族炭化水素は少なくとも10重量%以上、好ましくは50重量%以上とする。溶媒の使用量は晶析が実施できる量であれば良く、第1溶液中の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン1重量倍に対し例えば12~50重量倍使用する。
第1溶液の晶析前に結晶を完全に加熱溶解させることが好ましい。溶解時の温度は通常65℃以上、溶媒の沸点以下、好ましくは80℃~110℃である。また、溶解時間は通常、撹拌下、0.5~5時間、好ましくは1~3時間である。
冷却速度は通常、毎分0.05~1℃、好ましくは、毎分0.1~0.5℃、より好ましくは、毎分0.1~0.3℃である。冷却速度が速すぎると結晶中に不純物を取り込みやすく純度低下の原因となり好ましくない。
結晶析出後、スラリー液をさらに冷却してもよい。冷却終点の温度は通常-10~40℃、好ましくは0~30℃、更に好ましくは10~30℃である。
上述のようにして得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶を含むスラリー液を固液分離して母液を得る。固液分離の方法は特に限定されず、例えば濾過機を用いる方法、遠心分離機を用いる方法等が例示される。固液分離により得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶は必要に応じ乾燥等の操作を行い光学樹脂用モノマー等各種原料として使用することも可能であるが、1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを含む反応液のみからなる第1溶液を晶析して得られる第1結晶は多形体Aであるので、この場合は、得られた母液と共に後述する条件で第1結晶を再晶析することが好ましい。
続いて、第2溶液を調製する工程および第2結晶を析出させる工程(以下、これらを総称して、母液を再使用する工程と称することもある)について詳述する。
第2溶液は、母液の少なくとも一部からなることもできるが、母液の少なくとも一部と、母液に含まれる芳香族炭化水素とは別の、芳香族炭化水素から選ばれる少なくとも1種の溶媒とを含んで調製されることが好ましい。この場合、母液と別途の上記溶媒との合計中の母液の量は、通常10重量%以上、好ましくは25重量%以上、より好ましくは40重量%以上、特に50重量%以上である。含まれる母液の量が10重量%より少ないと多形体Cが得られない場合がある。なお、母液には、固液分離時に分離した結晶を更に洗浄するために使用した溶媒が含まれていてもよい。再使用する母液は多形体A、多形体B、多形体Cのいずれの結晶形から分離された母液を用いても良いが、安定的に多形体Cを得るためには、多形体Bまたは多形体Cを生成させ分離した母液が好ましく、特に好ましくは多形体Cから分離された母液である。最初に得られる母液(1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンを含む反応液のみからなる第1溶液を晶析して得られる母液)は、多形体Aから分離される母液であるが、この母液を再使用し、晶析して得られる母液は、多形体Bまたは多形体Cから分離される母液となる。
使用する母液は第2結晶を析出させる工程を行う際に含まれていればよく、第2結晶を析出させる工程の晶析直前に添加しても良いし、さらに前の工程で添加されてもよい。使用する母液は一度も使用されていないものでも良く、繰り返し使用された母液であっても良い。また、異なる由来の母液を混合して使用しても良い。
母液とともに使用可能である芳香族炭化水素から選ばれる少なくとも1種の溶媒は、前述した母液を得るための工程で使用したものと同種、他種いずれでも良い。また、該溶媒を用いる場合の使用量は、母液添加後、晶析前の第2溶液(晶析溶液)に含まれる芳香族炭化水素から選ばれる少なくとも1種の総量(母液に含まれる芳香族炭化水素類と新たに加える芳香族炭化水素類の和)が、第2溶液中の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンに対して3~20重量倍となるよう調整されることが好ましく、5~12重量倍となるよう調整されることがより好ましい。これらの溶媒は新たに添加することも可能であるし、あるいは新たに添加することなく、反応または精製工程で用いた溶媒をそのまま用いても良い。
第2溶液の晶析前に結晶を完全に加熱溶解させることが好ましい。溶解時の温度は通常65℃以上、溶媒の沸点以下、好ましくは80℃~110℃である。また、溶解時間は通常、撹拌下、0.5~5時間、好ましくは1~3時間である。
結晶を析出させる温度は48℃以下、好ましくは20~48℃、より好ましくは30~45℃である。48℃より高い場合、多形体Aまたは多形体Bが得られる。特に結晶析出温度が60℃より高い場合、針状結晶(多形体A)となり上述した本願効果が発現しない。
結晶析出前および結晶析出後の冷却速度は通常、毎分0.05~1℃、好ましくは、毎分0.1~0.5℃、特に、毎分0.1~0.3℃である。冷却速度が遅すぎると60℃より高い温度で結晶が析出しやすく、その結果針状結晶が得られる。冷却速度が速すぎると結晶中に不純物を取り込みやすく純度低下の原因となり得る。
結晶析出後、スラリー液をさらに冷却してもよい。冷却終点の温度は通常-10~40℃、好ましくは0~30℃、更に好ましくは10~30℃である。
スラリー液に含まれる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶は、濾過、遠心分離等の固液分離により回収される。得られた結晶は、上記の晶析に用いた溶媒等を用いて洗浄してもよいし、乾燥してもよい。かくして得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶は、板状結晶であって多形体Cとなる。その純度は、通常95%以上である。上記固液分離によって分離された母液は、第2溶液を調製する工程で使用する母液として再利用することができる。
次に多形体Bを得る方法について説明する。多形体Bは、上記第2溶液から、50~60℃で結晶を析出させることによって得られる。
母液を得るための晶析条件および操作、ならびに母液を再使用する工程の晶析条件および操作については下記点を除き多形体Cを得る工程と同様の条件にて実施することができる。一つ目の相違点として、再使用する母液は多形体A、多形体B、多形体Cのいずれの結晶形から分離された母液を用いても良いが、安定的に多形体Bを得るためには、多形体Bまたは多形体Cを生成させ分離した母液が好ましく、特に好ましくは多形体Bから分離された母液である点である。
二つ目の相違点としては、50~60℃の温度で2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶を析出させる必要があり、そのために種晶を添加することが好ましい点である。具体的には、冷却の途中で2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を好ましくは50~60℃、より好ましくは52~58℃の温度で種晶として添加し、同温度で結晶を析出させ、更に好ましくは、種晶を添加した後は50℃以上の温度で一定時間(例えば1~5時間、好ましくは1~3時間)撹拌する。種晶として用いる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶は多形体A、多形体B、多形体Cのいずれの結晶形でもよいが、好ましくは多形体Bまたは多形体C、特に好ましくは多形体Bである。添加される種晶の量は、通常、第2溶液(晶析溶液)中の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンに対して0.001~5重量%、好ましくは0.005~1重量%、更に好ましくは0.01~0.5重量%である。
こうして析出させた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶は濾過、遠心分離等の固液分離により回収される。得られた結晶は、上記の晶析に用いた溶媒等を用いて洗浄してもよいし、乾燥してもよい。かくして得られる2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶は、板状結晶であって多形体Bとなる。その純度は、通常95%以上である。上記固液分離によって分離された母液は、第2溶液を調製する工程で使用する母液として再利用することができる。
続いて本発明の塊状結晶の製造方法について詳述する。本発明の塊状結晶は板状結晶(多形体Bおよび/または多形体C)を40~108℃、好ましくは60~102℃で、通常6時間以上、好ましくは12~72時間、より好ましくは12~48時間加熱することにより得られる。加熱は、結晶を入れた容器中で結晶が撹拌されるような条件で実施することが好ましい。なお、40℃より低い温度で加熱しても塊状結晶は得られず、108℃より高い温度で加熱した場合、板状結晶が溶融するため塊状結晶は得られない。また、針状結晶(多形体A)を上記温度範囲に加熱しても塊状結晶に変化せず、針状結晶のまま存在する。
塊状結晶を製造する際、板状結晶に上述した晶析で用いた溶媒が含まれていても良い。溶媒が含まれている場合、溶媒含量は40重量%以下、好ましくは20重量%以下とする。板状結晶に含まれる溶媒が40重量%より多いと加熱時に結晶が溶解し針状結晶となる場合がある。なお、結晶に溶媒が含まれている場合、減圧乾燥等の溶媒除去操作を並行して行っても良い。
以下に実施例および試験例を挙げて本発明を具体的に説明するが、本発明はこれに何ら限定されるものではない。なお、例中、各種測定は下記の方法で実施した。また、以下実施例、比較例における収率は特に断りのない限り、反応で使用した1,1’-ビ-2-ナフトールに対する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの有姿収率(得られた結晶が全て2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンであると仮定した場合の収率)であり、HPLC純度は下記条件で測定したHPLCの面積百分率である。
<HPLC純度>
装置 :島津製作所製 LC-2010A、
カラム:SUMIPAX ODS A-211(5μm、4.6mmφ×250mm)、
移動相:純水/アセトニトリル(アセトニトリル30%→100%)、
流量 :1.0ml/min、カラム温度:40℃、検出波長:UV 254nm。
装置 :島津製作所製 LC-2010A、
カラム:SUMIPAX ODS A-211(5μm、4.6mmφ×250mm)、
移動相:純水/アセトニトリル(アセトニトリル30%→100%)、
流量 :1.0ml/min、カラム温度:40℃、検出波長:UV 254nm。
<示差走査熱量測定(DSC)>
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶5mgをアルミパンに精密に秤取し、示差走査熱量計(エスアイアイ・ナノテクノロジー株式会社:DSC7020)を用い、酸化アルミニウムを対照として下記操作条件で測定した。
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶5mgをアルミパンに精密に秤取し、示差走査熱量計(エスアイアイ・ナノテクノロジー株式会社:DSC7020)を用い、酸化アルミニウムを対照として下記操作条件で測定した。
(操作条件)
昇温速度:10℃/min、
測定範囲:30-200℃、
雰囲気 :開放、窒素40ml/min。
昇温速度:10℃/min、
測定範囲:30-200℃、
雰囲気 :開放、窒素40ml/min。
<粉末X線回折>
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶150mgをガラス試験板の試料充填部に充填し、粉末X線回折装置(スペクトリス製:X’PertPRO)を用いて下記の条件で測定した。
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶150mgをガラス試験板の試料充填部に充填し、粉末X線回折装置(スペクトリス製:X’PertPRO)を用いて下記の条件で測定した。
X線源 :CuKα、
出力 :1.8kW(45kV-40mA)、
測定範囲 :2θ=5°~70°、
スキャン速度:2θ=2°/min、
スリット :DS=1°、マスク=15mm、RS=可変(0.1mm~)。
出力 :1.8kW(45kV-40mA)、
測定範囲 :2θ=5°~70°、
スキャン速度:2θ=2°/min、
スリット :DS=1°、マスク=15mm、RS=可変(0.1mm~)。
<YI値>
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶を、得られる溶液が28.6質量%となるようにN,N―ジメチルホルムアミドに溶解させ、以下の条件で得られたN,N―ジメチルホルムアミド溶液のYI値(黄変度)を測定した。
2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶を、得られる溶液が28.6質量%となるようにN,N―ジメチルホルムアミドに溶解させ、以下の条件で得られたN,N―ジメチルホルムアミド溶液のYI値(黄変度)を測定した。
装置 :色差計(日本電色工業社製,SE6000)、
使用セル:光路長33mm 石英セル。
使用セル:光路長33mm 石英セル。
<アスペクト比、絶対幅、結晶形状>
下記装置を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶の最大結晶長さLおよび幅Wを測定し、得られた最大結晶長さLを幅Wで除した値をアスペクト比とした。最大結晶長さLおよび幅Wの定義は上述のとおりであり、それぞれ、光学顕微鏡写真から無作為に選択した30個の平均値として求めた。
下記装置を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの各結晶の最大結晶長さLおよび幅Wを測定し、得られた最大結晶長さLを幅Wで除した値をアスペクト比とした。最大結晶長さLおよび幅Wの定義は上述のとおりであり、それぞれ、光学顕微鏡写真から無作為に選択した30個の平均値として求めた。
装置:デジタルマイクロスコープ(株式会社キーエンス製、VHX-1000)、
各写真の測定倍率:600倍。
各写真の測定倍率:600倍。
<モード径とメジアン径との比(Dmode/Dmedian)>
下記条件にてモード径とメジアン径を測定し、得られた値を基にモード径とメジアン径との比(Dmode/Dmedian)を算出した。
下記条件にてモード径とメジアン径を測定し、得られた値を基にモード径とメジアン径との比(Dmode/Dmedian)を算出した。
測定装置:島津製作所製 SALD-2200(レーザー回折粒度分布測定装置)、
測定範囲:1000~0.030μm、
分散溶媒:蒸留水+中性洗剤、
分散方法:超音波分散。
測定範囲:1000~0.030μm、
分散溶媒:蒸留水+中性洗剤、
分散方法:超音波分散。
<濾過条件>
各製造例、実施例、比較例における濾過・洗浄操作は下記条件にて実施した。
各製造例、実施例、比較例における濾過・洗浄操作は下記条件にて実施した。
濾過器:桐山製作所製 SU-95、
ろ紙:桐山製作所製 No.5C、
受器減圧度:4.67kPa。
ろ紙:桐山製作所製 No.5C、
受器減圧度:4.67kPa。
<製造例1(母液の製造)>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加えた後、有機溶媒層を80℃に保ちながら水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下で脱水し、(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解したトルエン溶液(晶析溶液)を得た。その後、晶析溶液を冷却したところ63℃で一気に結晶が析出し撹拌困難となった。そこで同温度でトルエン1200gを加え結晶を含むスラリー状態として撹拌可能な状態とした後、更に30℃まで冷却した。これを前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に40分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は50重量%であり、結晶の形状は針状結晶であった。また、濾過により分離された母液は2630gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶198gを得た(収率84.1%、HPLC純度99.1%、YI値:11)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:68.0(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.5]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加えた後、有機溶媒層を80℃に保ちながら水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下で脱水し、(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解したトルエン溶液(晶析溶液)を得た。その後、晶析溶液を冷却したところ63℃で一気に結晶が析出し撹拌困難となった。そこで同温度でトルエン1200gを加え結晶を含むスラリー状態として撹拌可能な状態とした後、更に30℃まで冷却した。これを前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に40分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は50重量%であり、結晶の形状は針状結晶であった。また、濾過により分離された母液は2630gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶198gを得た(収率84.1%、HPLC純度99.1%、YI値:11)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:68.0(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.5]。
DSC分析チャートを図1に、粉末X線のパターンを図4に、粉末X線の主なピーク(5%を超える相対強度を有するもの)を表1に列挙する。また、デジタルマイクロスコープ写真を図7に示す。
<実施例1>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン200gおよび製造例1で得られた母液1100gを加えた後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下で脱水し晶析溶液を得、8時間後に30℃となるように冷却することにより39℃で結晶を析出させた。こうして得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は15重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1603gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶211gを得た(収率89.5%、HPLC純度99.7%、YI値:4)。得られた結晶の物性は次の通り[DSC融解吸熱最大:107℃、粉末X線回折パターン:多形体C、結晶形状:板状結晶、アスペクト比:1.3(幅の絶対値:70μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン200gおよび製造例1で得られた母液1100gを加えた後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下で脱水し晶析溶液を得、8時間後に30℃となるように冷却することにより39℃で結晶を析出させた。こうして得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は15重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1603gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶211gを得た(収率89.5%、HPLC純度99.7%、YI値:4)。得られた結晶の物性は次の通り[DSC融解吸熱最大:107℃、粉末X線回折パターン:多形体C、結晶形状:板状結晶、アスペクト比:1.3(幅の絶対値:70μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
DSC分析チャートを図3に、粉末X線のパターンを図6に、粉末X線の主なピーク(5%を超える相対強度を有するもの)を表3に列挙する。多形体Cは、回折角2θが10.0±0.2°、19.2±0.2°、19.7±0.2°および22.2±0.2°に特徴的な回折ピークを示した。また、デジタルマイクロスコープ写真を図9に示す。
<実施例2>
製造例1で得られた母液1100gの代わりに、実施例1で得られた母液1100gを加えた以外は実施例1と同様に行い、晶析溶液を得た。この晶析溶液を冷却開始後10時間で30℃となるように冷却することにより、36℃で結晶を析出させた。こうして得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は14.5重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1610gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶212gを得た(収率90.0%、HPLC純度99.8%、YI値:3)。得られた結晶の物性は次の通り[DSC融解吸熱最大:108℃、粉末X線回折パターン:多形体C、結晶形状:板状結晶、アスペクト比:1.9(幅の絶対値:45μm)、モード径とメジアン径との比(Dmode/Dmedian):1.1]。
製造例1で得られた母液1100gの代わりに、実施例1で得られた母液1100gを加えた以外は実施例1と同様に行い、晶析溶液を得た。この晶析溶液を冷却開始後10時間で30℃となるように冷却することにより、36℃で結晶を析出させた。こうして得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は14.5重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1610gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶212gを得た(収率90.0%、HPLC純度99.8%、YI値:3)。得られた結晶の物性は次の通り[DSC融解吸熱最大:108℃、粉末X線回折パターン:多形体C、結晶形状:板状結晶、アスペクト比:1.9(幅の絶対値:45μm)、モード径とメジアン径との比(Dmode/Dmedian):1.1]。
<実施例3>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン760gおよび実施例2で得られた母液410gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例2で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体C)0.085gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、3時間かけて30℃まで冷却した。これを前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は14重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1510gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶211gを得た(収率89.7%、HPLC純度99.7%、YI値:2)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.7(幅の絶対値:97μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン760gおよび実施例2で得られた母液410gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例2で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体C)0.085gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、3時間かけて30℃まで冷却した。これを前述の条件で濾過した後、更に結晶をトルエン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は14重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1510gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶211gを得た(収率89.7%、HPLC純度99.7%、YI値:2)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.7(幅の絶対値:97μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
DSC分析チャートを図2に、粉末X線のパターンを図5に、粉末X線の主なピーク(5%を超える相対強度を有するもの)を表2に列挙する。多形体Bは、回折角2θが10.4±0.2°、12.1±0.2°、14.8±0.2°および22.3±0.2°に特徴的な回折ピークを示した。また、デジタルマイクロスコープ写真を図8に示す。
<実施例4>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール270g(0.943mol)、エチレンカーボネート190g(2.158mol)、炭酸カリウム13.5gおよびトルエン270gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1080gおよび実施例3で得られた母液1350gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例3で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体B)0.270gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、4時間かけて30℃まで冷却した。これを前述の濾過条件で濾過した後、更に結晶をトルエン300gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に15分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は15重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は2990gであった。次いで、濾過操作より得られた結晶部を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶315gを得た(収率89.2%、HPLC純度99.8%、YI値:2)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.9(幅の絶対値:48μm)、モード径とメジアン径との比(Dmode/Dmedian):1.1]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール270g(0.943mol)、エチレンカーボネート190g(2.158mol)、炭酸カリウム13.5gおよびトルエン270gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1080gおよび実施例3で得られた母液1350gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例3で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体B)0.270gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、4時間かけて30℃まで冷却した。これを前述の濾過条件で濾過した後、更に結晶をトルエン300gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に15分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は15重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は2990gであった。次いで、濾過操作より得られた結晶部を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶315gを得た(収率89.2%、HPLC純度99.8%、YI値:2)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.9(幅の絶対値:48μm)、モード径とメジアン径との比(Dmode/Dmedian):1.1]。
<実施例5>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびキシレン180gを仕込み、110℃で10時間撹拌した。この反応生成物にキシレン670gおよび実施例4で得られた母液500gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例4で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体B)0.900gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、4時間かけて30℃まで冷却した。これを前述の濾過条件で濾過した後、更に結晶をキシレン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は18重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1541gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶208gを得た(収率88.5%、HPLC純度99.8%、YI値:3)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.8(幅の絶対値:20μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびキシレン180gを仕込み、110℃で10時間撹拌した。この反応生成物にキシレン670gおよび実施例4で得られた母液500gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層をディーンスターク装置を用いて還流下脱水し晶析溶液を得た。この晶析溶液を5時間かけて55℃まで冷却した後、同温度で、実施例4で得られた2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン(多形体B)0.900gを種晶として添加し、更に同温度で2時間撹拌して結晶を析出させた後、4時間かけて30℃まで冷却した。これを前述の濾過条件で濾過した後、更に結晶をキシレン200gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に10分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は18重量%であり、結晶の形状は板状結晶であった。濾過により分離された母液は1541gであった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶208gを得た(収率88.5%、HPLC純度99.8%、YI値:3)。得られた結晶の物性は次の通り[DSC融解吸熱最大:110℃、粉末X線回折パターン:多形体B、結晶形状:板状結晶、アスペクト比:1.8(幅の絶対値:20μm)、モード径とメジアン径との比(Dmode/Dmedian):1.0]。
<実施例6>
実施例1で得られた板状結晶(多形体C)50gをロータリーエバポレーターにて外温95℃で36時間撹拌することにより、塊状結晶50g得た。得られた塊状結晶の物性は次の通り[HPLC純度99.7%、YI値:3、DSC融解吸熱最大:115℃、(粉末X線回折パターン:多形体A)、結晶形状:塊状結晶、アスペクト比:1.8(幅の絶対値:50μm)、モード径:91.15μm、メジアン径:74.63μm、モード径とメジアン径との比(Dmode/Dmedian):1.2]。得られた塊状結晶のデジタルマイクロスコープ写真を図10に示す。
実施例1で得られた板状結晶(多形体C)50gをロータリーエバポレーターにて外温95℃で36時間撹拌することにより、塊状結晶50g得た。得られた塊状結晶の物性は次の通り[HPLC純度99.7%、YI値:3、DSC融解吸熱最大:115℃、(粉末X線回折パターン:多形体A)、結晶形状:塊状結晶、アスペクト比:1.8(幅の絶対値:50μm)、モード径:91.15μm、メジアン径:74.63μm、モード径とメジアン径との比(Dmode/Dmedian):1.2]。得られた塊状結晶のデジタルマイクロスコープ写真を図10に示す。
<比較例1>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加え希釈した後、反応生成物を含む有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を濃縮し、水およびトルエンを除去した後、残渣にアセトン540g加え、56℃で加熱溶解した。次いでこの溶液を冷却開始後3時間で30℃となるように冷却したところ、35℃で結晶が析出した。こうして得られたスラリー液を前述の濾過条件で濾過し、結晶部と母液に分離した。この濾過操作に30分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は48重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶110gを得た(収率46.7%、HPLC純度99.5%、YI値:7)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:65(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加え希釈した後、反応生成物を含む有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を濃縮し、水およびトルエンを除去した後、残渣にアセトン540g加え、56℃で加熱溶解した。次いでこの溶液を冷却開始後3時間で30℃となるように冷却したところ、35℃で結晶が析出した。こうして得られたスラリー液を前述の濾過条件で濾過し、結晶部と母液に分離した。この濾過操作に30分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は48重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの白色結晶110gを得た(収率46.7%、HPLC純度99.5%、YI値:7)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:65(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
<比較例2>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を濃縮し水およびトルエンを除去した後、残渣にメタノール540g加え、64℃で加熱溶解した。次いでこの溶液を冷却開始後3時間で30℃となるように冷却したところ、32℃で結晶を析出した。得られたスラリー液を前述の濾過条件で濾過した後、結晶部と母液に分離した。この濾過操作に30分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は45重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶部を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶125gを得た(収率53%、HPLC純度99.3%、YI値:8)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:11(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.4]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応生成物にトルエン1300gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を濃縮し水およびトルエンを除去した後、残渣にメタノール540g加え、64℃で加熱溶解した。次いでこの溶液を冷却開始後3時間で30℃となるように冷却したところ、32℃で結晶を析出した。得られたスラリー液を前述の濾過条件で濾過した後、結晶部と母液に分離した。この濾過操作に30分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は45重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶部を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶125gを得た(収率53%、HPLC純度99.3%、YI値:8)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:11(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.4]。
<比較例3>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応混合物にトルエン1300gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を還流下で脱水した。これにトルエン400gおよびヘキサン800gを加えて冷却したところ65℃で一気に結晶が析出した。これを4時間かけて30℃まで冷却し、得られたスラリー液を前述の濾過条件で濾過した後、更に結晶をトルエン/ヘキサン=140g/60gの混合溶媒で洗浄して、溶媒を含む結晶と母液に分離した。この濾過操作に40分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は52重量%であり、結晶の形状は針状結晶であった。次いで固液分離により得られた溶媒を含む結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶197gを得た(収率83.9%、HPLC純度98.2%、YI値:13)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:17(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート127g(1.439mol)、炭酸カリウム9.0gおよびトルエン180gを仕込み、110℃で10時間撹拌した。この反応混合物にトルエン1300gを加え希釈した後、有機溶媒層を80℃に保ち、水酸化ナトリウ水溶液で洗浄した。次いでこの有機溶媒層を、洗浄水が中性となるまで水洗を行った。得られた有機溶媒層を還流下で脱水した。これにトルエン400gおよびヘキサン800gを加えて冷却したところ65℃で一気に結晶が析出した。これを4時間かけて30℃まで冷却し、得られたスラリー液を前述の濾過条件で濾過した後、更に結晶をトルエン/ヘキサン=140g/60gの混合溶媒で洗浄して、溶媒を含む結晶と母液に分離した。この濾過操作に40分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は52重量%であり、結晶の形状は針状結晶であった。次いで固液分離により得られた溶媒を含む結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶197gを得た(収率83.9%、HPLC純度98.2%、YI値:13)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:17(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
<比較例4>
攪拌器、冷却器、および温度計を備えたガラス製反応器に、製造例1で得た結晶100gおよびトルエン1200gを仕込み、110℃で1時間撹拌し完溶させた後、6時間で25℃になるように冷却したところ、52℃で結晶が析出した。更に25℃まで冷却した。得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン150gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に35分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は50重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶84.2gを得た(回収率84.2%、HPLC純度99.5%、YI値:10)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:70.0(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
攪拌器、冷却器、および温度計を備えたガラス製反応器に、製造例1で得た結晶100gおよびトルエン1200gを仕込み、110℃で1時間撹拌し完溶させた後、6時間で25℃になるように冷却したところ、52℃で結晶が析出した。更に25℃まで冷却した。得られたスラリー液を前述の条件で濾過した後、更に結晶をトルエン150gで洗浄して、結晶部と母液に分離した。この濾過洗浄操作に35分を要した。また、得られた結晶の一部を採取し分析したところ、結晶中の溶媒含量は50重量%であり、結晶の形状は針状結晶であった。次いで、濾過操作により得られた結晶を乾燥して(RS)-2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの淡黄色結晶84.2gを得た(回収率84.2%、HPLC純度99.5%、YI値:10)。得られた結晶の物性は次の通り[DSC融解吸熱最大:116℃、粉末X線回折パターン:多形体A、結晶形状:針状結晶、アスペクト比:70.0(幅の絶対値:2μm)、モード径とメジアン径との比(Dmode/Dmedian):2.1]。
<参考例1>
特開2011-153248号公報(上記特許文献1)合成例1に準じ、攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート132.9g(1.510mol)、48%水酸化カリウム5.0gを加え、170℃で4時間撹拌した。その後、メチルイソブチルケトン315gを加え溶解した後、水210gで2回水洗を行った。得られた有機層を150℃で濃縮し溶媒を除去したところ、淡黄色の樹脂状物220gが得られた。この樹脂状物は1,1’-ビ-2-ナフトール類1モルとエチレンカーボネート3モルが反応した重縮合体、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが炭酸エステル結合により2モル重合した炭酸エステル体および塩を含む2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンであった。(HPLC純度94.1%)。
特開2011-153248号公報(上記特許文献1)合成例1に準じ、攪拌器、冷却器、および温度計を備えたガラス製反応器に、(RS)-1,1’-ビ-2-ナフトール180g(0.629mol)、エチレンカーボネート132.9g(1.510mol)、48%水酸化カリウム5.0gを加え、170℃で4時間撹拌した。その後、メチルイソブチルケトン315gを加え溶解した後、水210gで2回水洗を行った。得られた有機層を150℃で濃縮し溶媒を除去したところ、淡黄色の樹脂状物220gが得られた。この樹脂状物は1,1’-ビ-2-ナフトール類1モルとエチレンカーボネート3モルが反応した重縮合体、2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが炭酸エステル結合により2モル重合した炭酸エステル体および塩を含む2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンであった。(HPLC純度94.1%)。


Claims (14)
- 示差走査熱分析による融解吸熱最大が109~112℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.4±0.2°、12.1±0.2°、14.8±0.2°および22.3±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 示差走査熱分析による融解吸熱最大が106~108℃である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- Cu-Kα線による粉末X線回折パターンにおける回折角2θが10.0±0.2°、19.2±0.2°、19.7±0.2°および22.2±0.2°にピークを有する2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 板状結晶であって、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である請求項1~4のいずれか1項に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 板状結晶の幅Wが3μm以上である請求項5または6に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 塊状結晶であって、モード径Dmodeとメジアン径Dmedianとの比Dmode/Dmedianが2.0以下であり、光学顕微鏡写真に基づき求めた最大結晶長さLと幅Wとの比L/Wが1~8である2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 示差走査熱分析による融解吸熱最大が114~116℃である請求項8に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶。
- 芳香族炭化水素から選ばれる少なくとも1種の溶媒中に2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第1溶液から2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第1結晶を析出させた後、固液分離して母液を得る工程と、
前記母液の少なくとも一部を用いて2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが溶解している第2溶液を調製する工程と、
前記第2溶液から、60℃以下の温度で2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの第2結晶を析出させる工程と、
を含む請求項1~7のいずれか1項に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレン結晶の製造方法。 - 前記第2溶液から、50~60℃の温度で前記第2結晶を析出させる請求項10に記載の製造方法。
- 前記第2溶液から、48℃以下の温度で前記第2結晶を析出させる請求項10に記載の製造方法。
- 前記母液を得る工程において、芳香族炭化水素から選ばれる少なくとも1種の溶媒中に溶解している2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンが、1,1’-ビ-2-ナフトールとエチレンカーボネートとの反応により得られたものである請求項10~12のいずれか1項に記載の製造方法。
- 請求項1~7のいずれか1項に記載の2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの結晶を用意する工程と、
前記結晶を40~108℃に加熱する工程と、
を含む2,2’-ビス(2-ヒドロキシエトキシ)-1,1’-ビナフタレンの塊状結晶の製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201580012293.8A CN106170469A (zh) | 2014-03-10 | 2015-03-05 | 2,2’‑双(2‑羟基乙氧基)‑1,1’‑联萘的结晶多形体及其制造方法 |
| KR1020167027866A KR102219152B1 (ko) | 2014-03-10 | 2015-03-05 | 2,2'-비스(2-하이드록시에톡시)-1,1'-비나프탈렌의 결정 다형체 및 그 제조 방법 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014-045921 | 2014-03-10 | ||
| JP2014045927 | 2014-03-10 | ||
| JP2014-045927 | 2014-03-10 | ||
| JP2014045921A JP6253099B2 (ja) | 2014-03-10 | 2014-03-10 | 2,2’−ビス(2−ヒドロキシエトキシ)−1,1’−ビナフタレンの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015137238A1 true WO2015137238A1 (ja) | 2015-09-17 |
Family
ID=54071684
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/056555 WO2015137238A1 (ja) | 2014-03-10 | 2015-03-05 | 2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの結晶多形体およびその製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| KR (1) | KR102219152B1 (ja) |
| CN (2) | CN106170469A (ja) |
| TW (1) | TWI596083B (ja) |
| WO (1) | WO2015137238A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015187098A (ja) * | 2014-03-10 | 2015-10-29 | 田岡化学工業株式会社 | 2,2’−ビス(2−ヒドロキシエトキシ)−1,1’−ビナフタレンの結晶多形体およびその製造方法 |
| JP2021006511A (ja) * | 2019-06-28 | 2021-01-21 | 帝人株式会社 | ビナフタレン骨格を有する化合物およびその結晶多形体、ならびにその製造方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN107176905B (zh) * | 2017-04-25 | 2020-06-26 | 江苏永星化工股份有限公司 | 高纯度(±)-2,2’-二-(2-羟基乙氧基)-1,1’-联萘的制备方法 |
| KR102509154B1 (ko) * | 2017-08-30 | 2023-03-10 | 미츠비시 가스 가가쿠 가부시키가이샤 | 폴리카보네이트 수지, 그 제조 방법, 및, 광학 렌즈 |
| JP7295692B2 (ja) * | 2018-05-31 | 2023-06-21 | 本州化学工業株式会社 | 2,2’-ビス(カルボキシメトキシ)-1,1’-ビナフチルの結晶体 |
| CN110483259A (zh) * | 2019-09-12 | 2019-11-22 | 江苏永星化工股份有限公司 | 一种(±)-2,2’-二-(2-羟基乙氧基)-1,1’-联萘晶体及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010018753A (ja) * | 2008-07-14 | 2010-01-28 | Nippon Kayaku Co Ltd | (メタ)アクリレート化合物及びそれを含有する活性エネルギー線硬化型樹脂組成物並びにその硬化物 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4001279A (en) * | 1973-03-29 | 1977-01-04 | The Regents Of The University Of California | Asymmetric macrocycles containing oxygen and binaphthyl ring members |
| JP5509880B2 (ja) * | 2010-01-28 | 2014-06-04 | Dic株式会社 | 硬化性樹脂組成物、その硬化物、及びプラスチックレンズ |
-
2015
- 2015-03-05 CN CN201580012293.8A patent/CN106170469A/zh active Pending
- 2015-03-05 CN CN201811413011.7A patent/CN109942386A/zh active Pending
- 2015-03-05 KR KR1020167027866A patent/KR102219152B1/ko active IP Right Grant
- 2015-03-05 WO PCT/JP2015/056555 patent/WO2015137238A1/ja active Application Filing
- 2015-03-10 TW TW104107641A patent/TWI596083B/zh active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010018753A (ja) * | 2008-07-14 | 2010-01-28 | Nippon Kayaku Co Ltd | (メタ)アクリレート化合物及びそれを含有する活性エネルギー線硬化型樹脂組成物並びにその硬化物 |
Non-Patent Citations (1)
| Title |
|---|
| KYBA, EVAN P. ET AL.: "Host-guest complexation. 7. The binaphthyl structural unit in host compounds", JOURNAL OF ORGANIC CHEMISTRY, vol. 42, no. 26, 1977, pages 4173 - 84 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015187098A (ja) * | 2014-03-10 | 2015-10-29 | 田岡化学工業株式会社 | 2,2’−ビス(2−ヒドロキシエトキシ)−1,1’−ビナフタレンの結晶多形体およびその製造方法 |
| JP2021006511A (ja) * | 2019-06-28 | 2021-01-21 | 帝人株式会社 | ビナフタレン骨格を有する化合物およびその結晶多形体、ならびにその製造方法 |
| JP7315386B2 (ja) | 2019-06-28 | 2023-07-26 | 帝人株式会社 | ビナフタレン骨格を有する化合物およびその結晶多形体、ならびにその製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN109942386A (zh) | 2019-06-28 |
| CN106170469A (zh) | 2016-11-30 |
| TW201617306A (zh) | 2016-05-16 |
| KR20160134716A (ko) | 2016-11-23 |
| TWI596083B (zh) | 2017-08-21 |
| KR102219152B1 (ko) | 2021-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6484465B2 (ja) | 2,2’−ビス(2−ヒドロキシエトキシ)−1,1’−ビナフタレンの結晶多形体およびその製造方法 | |
| WO2015137238A1 (ja) | 2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの結晶多形体およびその製造方法 | |
| JP6253099B2 (ja) | 2,2’−ビス(2−ヒドロキシエトキシ)−1,1’−ビナフタレンの製造方法 | |
| TWI503306B (zh) | 茀衍生物之結晶多形體 | |
| JP6083900B2 (ja) | ビナフタレン化合物の製造方法 | |
| KR102077352B1 (ko) | 9,9-비스(4-(2-히드록시에톡시)페닐)플루오렌의 제조방법, 그의 결정체 및 그의 결정체의 제조방법 | |
| WO2017006791A1 (ja) | フルオレン骨格を有するアルコールの結晶およびその製造方法 | |
| TWI658034B (zh) | 具有茀骨架之醇之結晶及其製造方法 | |
| TW202112735A (zh) | 2,2’-雙(乙氧基羰基甲氧基)-1,1’-聯萘的結晶體 | |
| TWI787515B (zh) | 2,2'-雙(羧甲氧基)-1,1'-聯萘的結晶體 | |
| JP6830304B2 (ja) | フルオレン骨格を有するジヒドロキシ化合物の精製方法 | |
| TWI740943B (zh) | 具有茀骨架之醇化合物的製造方法 | |
| TW202104154A (zh) | 聯萘羧酸類的製造方法 | |
| JP6905281B2 (ja) | (±)−2,2’−ビス−(2−ヒドロキシエトキシ)−1,1’−ビナフチル結晶及び調製方法 | |
| JP6351074B2 (ja) | ビナフタレン誘導体の非晶質体およびその製造方法 | |
| WO2022215460A1 (ja) | 2,2'-ビス(2-ヒドロキシエトキシ)-1,1'-ビナフタレンの製造方法 | |
| JP7315386B2 (ja) | ビナフタレン骨格を有する化合物およびその結晶多形体、ならびにその製造方法 | |
| WO2023176687A1 (ja) | ビフェナントレン化合物又はそのアルカリ金属塩 | |
| JP2024056627A (ja) | 9,9-ビス(6-ヒドロキシ-2-ナフチル)フルオレンの結晶およびその製造方法 | |
| WO2020004207A1 (ja) | 9,9-ビス(4-ヒドロキシフェニル)-2,3-ベンゾフルオレンの結晶体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15762092 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20167027866 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 15762092 Country of ref document: EP Kind code of ref document: A1 |