WO2013129115A1 - 炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 - Google Patents
炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 Download PDFInfo
- Publication number
- WO2013129115A1 WO2013129115A1 PCT/JP2013/053444 JP2013053444W WO2013129115A1 WO 2013129115 A1 WO2013129115 A1 WO 2013129115A1 JP 2013053444 W JP2013053444 W JP 2013053444W WO 2013129115 A1 WO2013129115 A1 WO 2013129115A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- treatment agent
- modified silicone
- fiber
- carbon fiber
- weight
- Prior art date
Links
Classifications
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
- D01F9/12—Carbon filaments; Apparatus specially adapted for the manufacture thereof
- D01F9/14—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments
- D01F9/20—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products
- D01F9/24—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/244—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing sulfur or phosphorus
- D06M13/282—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing sulfur or phosphorus with compounds containing phosphorus
- D06M13/292—Mono-, di- or triesters of phosphoric or phosphorous acids; Salts thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/49—Phosphorus-containing compounds
- C08K5/51—Phosphorus bound to oxygen
- C08K5/52—Phosphorus bound to oxygen only
- C08K5/521—Esters of phosphoric acids, e.g. of H3PO4
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/54—Silicon-containing compounds
- C08K5/541—Silicon-containing compounds containing oxygen
- C08K5/5415—Silicon-containing compounds containing oxygen containing at least one Si—O bond
- C08K5/5419—Silicon-containing compounds containing oxygen containing at least one Si—O bond containing at least one Si—C bond
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F6/00—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof
- D01F6/28—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolymers obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D01F6/38—Monocomponent artificial filaments or the like of synthetic polymers; Manufacture thereof from copolymers obtained by reactions only involving carbon-to-carbon unsaturated bonds comprising unsaturated nitriles as the major constituent
-
- D—TEXTILES; PAPER
- D01—NATURAL OR MAN-MADE THREADS OR FIBRES; SPINNING
- D01F—CHEMICAL FEATURES IN THE MANUFACTURE OF ARTIFICIAL FILAMENTS, THREADS, FIBRES, BRISTLES OR RIBBONS; APPARATUS SPECIALLY ADAPTED FOR THE MANUFACTURE OF CARBON FILAMENTS
- D01F9/00—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments
- D01F9/08—Artificial filaments or the like of other substances; Manufacture thereof; Apparatus specially adapted for the manufacture of carbon filaments of inorganic material
- D01F9/12—Carbon filaments; Apparatus specially adapted for the manufacture thereof
- D01F9/14—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments
- D01F9/20—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products
- D01F9/21—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds
- D01F9/22—Carbon filaments; Apparatus specially adapted for the manufacture thereof by decomposition of organic filaments from polyaddition, polycondensation or polymerisation products from macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds from polyacrylonitriles
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/643—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds containing silicon in the main chain
Definitions
- the present invention relates to an acrylic fiber treatment agent for carbon fiber production (hereinafter sometimes referred to as a precursor treatment agent) used when producing an acrylic fiber for carbon fiber production (hereinafter sometimes referred to as a precursor),
- a precursor treatment agent used when producing an acrylic fiber for carbon fiber production
- the present invention relates to an acrylic fiber for producing carbon fiber that has been treated with a treatment agent, and a method for producing carbon fiber using the treatment agent.
- Carbon fibers are widely used for aerospace applications, sports applications, general industrial applications and the like as reinforcing fibers for composite materials with plastics called matrix resins, utilizing their excellent mechanical properties.
- a precursor production process may be referred to as a yarn production process.
- This precursor is converted to flame-resistant fibers in an oxidizing atmosphere at 200 to 300 ° C. (this process may be referred to as a flame-resistant treatment process hereinafter), followed by carbonization in an inert atmosphere at 300 to 2000 ° C.
- This process may hereinafter be referred to as a carbonization treatment process
- the flameproofing treatment process and the carbonization treatment process may be collectively referred to as a firing process).
- the precursor is manufactured through a drawing process that is drawn at a high magnification even when compared with a normal acrylic fiber. At that time, the fibers tend to stick together and are not uniformly stretched at a high magnification, resulting in a non-uniform precursor.
- the carbon fiber obtained by firing such a precursor has a problem that sufficient strength cannot be obtained. Further, when the precursor is fired, there is a problem that the single fibers are fused with each other, and the quality and quality of the obtained carbon fibers are deteriorated.
- Many techniques for applying an amino-modified silicone-based treating agent to a precursor have been proposed (see Patent Documents 1 and 2) and are widely used industrially.
- a commonly used amino-modified silicone treatment agent is an amino-modified silicone oil that is made into an aqueous emulsion. When it is not a self-emulsifying type amino-modified silicone, a water-based emulsion is formed using a surfactant.
- aqueous emulsion In order to apply this aqueous emulsion to the precursor fiber, it is necessary to remove moisture after application, and it is necessary to pass through a drying step. Thereafter, heat drawing is further performed in a drawing step, and a precursor fiber drawn at a high magnification is obtained. Due to its excellent thermal crosslinkability, amino-modified silicone can be easily cross-linked even with a heat roller in the drying process and stretching process, and roller dirt (hereinafter sometimes referred to as gum-up) is likely to accumulate, resulting in yarn breakage of the precursor. In addition to causing fluff and fluffing, there is a problem that the operability is lowered due to the cleaning.
- Patent Documents 3 and 4 propose a treatment agent composition in which an antioxidant is added in order to suppress gum-up.
- Japanese Unexamined Patent Publication No. 60-181322 Japanese Unexamined Patent Publication No. 2001-172879 Japanese Unexamined Patent Publication No. 2-91224 Japanese Unexamined Patent Publication No. 11-012853
- the crosslinking reaction of the silicone type component in the drying step and the stretching step can be suppressed.
- the antioxidant accelerates the decomposition of the silicone-based component, the heat resistance of the silicone-based treatment agent decreases. For this reason, there has been a problem that in the firing step, carbon fiber fusion cannot be suppressed, and carbon fibers having sufficient strength cannot be obtained.
- the temporal stability of the treatment agent emulsion solution is poor, and there is a problem that it is difficult to uniformly apply to the precursor fiber bundle stably for a long time.
- the object of the present invention is to suppress the gum-up in the precursor production process (yarn production process) by using the precursor solution with excellent solution stability and being used when producing the precursor fiber.
- an acrylic fiber treatment agent for producing carbon fiber capable of suppressing the fusion of carbon fibers in the firing step, an acrylic fiber for producing carbon fiber produced by applying the treatment agent, and the treatment agent were used. It is providing the manufacturing method of carbon fiber.
- the present inventors have found that the above-mentioned problems are obtained as long as the treatment agent contains a modified silicone having a modified group containing a nitrogen atom and a specific acidic phosphate ester having an acidic hydroxyl group. Found to solve.
- the acrylic fiber treating agent for producing carbon fiber of the present invention essentially contains a modified silicone having a modifying group containing a nitrogen atom and an acidic phosphate ester represented by the following general formula (1).
- R 1 represents an alkyl group or alkenyl group having 6 to 22 carbon atoms
- A represents an alkylene group having 2 to 4 carbon atoms
- AO represents an oxyalkylene group
- n represents an oxyalkylene group. Represents the number of moles of the group added and is an integer of 0 to 20.
- the weight ratio of the modified silicone to the acidic phosphate is preferably 99.6 / 0.4 to 90/10.
- the acid value of the acidic phosphate is preferably 15 to 500 mgKOH / g.
- the nitrogen atom content in the modified silicone is preferably 0.35 to 3.2% by weight.
- the modified silicone is preferably an amino-modified silicone.
- the weight ratio of the modified silicone to the non-volatile content of the treatment agent is preferably 50 to 95% by weight.
- the treatment agent of the present invention further contains a nonionic surfactant, and the weight ratio of the surfactant to the nonvolatile content of the treatment agent is preferably 1 to 40% by weight.
- the treatment agent of the present invention is preferably an emulsion dispersed in water.
- the acrylic fiber for producing carbon fiber of the present invention is produced by attaching the above-mentioned treatment agent to the raw acrylic fiber of the acrylic fiber for producing carbon fiber.
- the carbon fiber production method of the present invention was produced by a yarn production step of producing an acrylic fiber for carbon fiber production by attaching the treatment agent to the raw acrylic fiber of the acrylic fiber for carbon fiber production, and the yarn production step.
- the acrylic fiber treatment agent for producing carbon fiber of the present invention is excellent in solution stability. Moreover, the process agent is made to adhere to the raw material acrylic fiber of an acrylic fiber for carbon fiber manufacture, and the gum-up derived from a processing agent can be suppressed, and it is excellent in yarn-manufacturability. Moreover, since the heat resistance of the treatment agent is excellent, it is possible to suppress the fusion of the carbon fibers in the firing step such as the flameproofing treatment step and the carbonization treatment step, and to improve the strength of the carbon fibers. it can. Moreover, in the manufacturing method of the carbon fiber of this invention, since this processing agent is made to adhere, high strength carbon fiber can be manufactured.
- the acrylic fiber treatment agent for carbon fiber production (precursor treatment agent) of the present invention is a step of producing an acrylic fiber for carbon fiber production (precursor), and is first applied to the precursor raw acrylic fiber before the drawing step.
- a treatment agent intended for the above and essentially contains a modified silicone having a modifying group containing a nitrogen atom and a specific acidic phosphate ester. This will be described in detail below.
- the precursor oil agent of the present invention essentially contains a modified silicone having a modifying group containing a nitrogen atom (hereinafter sometimes referred to as a modified silicone (A)).
- the modified silicone (A) is not particularly limited as long as it is a modified group containing a nitrogen atom.
- the modifying group containing a nitrogen atom include a modifying group containing an amino bond or an imino bond (ie, an amino group), a modifying group containing an amide bond (ie, an amide group), and the like.
- a modified group having a plurality of different bonds may be used.
- the modifying group containing a nitrogen atom may be bonded to the side chain of silicone as the main chain, may be bonded to the terminal, or may be bonded to both.
- modified silicone having a modified group containing a nitrogen atom examples include amino-modified silicone, amino polyether-modified silicone, amide-modified silicone, amide-polyether-modified silicone, and one kind of modified silicone may be used. A plurality of modified silicones may be used in combination.
- the nitrogen atom content in the modified silicone (A) is preferably 0.35 to 3.2% by weight, more preferably 0.37 to 2.2% by weight, and 0.40 to 1.3% by weight. Further preferred. When the content of nitrogen atoms is lower than 0.35% by weight, the solution stability of the emulsion may deteriorate when aqueous emulsification is performed.
- amino-modified silicones are preferred because they are excellent in solution stability of emulsions when water-based emulsified, and are excellent in effect when used in combination with acidic phosphates.
- the structure of the amino-modified silicone is not particularly limited. That is, the amino group that is a modifying group may be bonded to the side chain of silicone that is the main chain, may be bonded to the terminal, or may be bonded to both.
- the amino group may be a monoamine type or a polyamine type, and both may be present in one molecule.
- the amino group (NH 2 ) content in the amino-modified silicone (hereinafter referred to as “amino weight%”) is preferably 0.4 to 3.7% by weight, more preferably 0.42 to 2.5% by weight, More preferably, it is 0.46 to 1.5% by weight.
- amino weight% is lower than 0.4 weight%, the solution stability of the emulsion may be deteriorated when emulsified with water.
- content is higher than 3.7% by weight, the tackiness of the amino-modified silicone becomes high due to thermal crosslinking, which causes gum-up. Therefore, even if acidic phosphate ester is used together in a specific ratio, there is a possibility that gum-up cannot be suppressed.
- the viscosity of the amino-modified silicone at 25 ° C. is not particularly limited. However, if the viscosity is too low, the treatment agent is likely to be scattered, and the solution stability of the emulsion is deteriorated when water-based emulsification is performed. It becomes impossible to apply uniformly. As a result, fiber fusion may not be prevented. On the other hand, if the viscosity is too high, gum-up due to adhesiveness may be a problem. From the standpoint of preventing these problems, the viscosity at 25 ° C. of an amino-modified silicone is preferably 100 ⁇ 15,000mm 2 / s, more preferably 500 ⁇ 10,000mm 2 / s, 1,000 ⁇ 5,000mm 2 / s is more preferable.
- the amino-modified silicone can be represented by the following chemical formula (2), for example.
- R 2 represents an alkyl group or aryl group having 1 to 20 carbon atoms.
- R 2 is preferably an alkyl group having 1 to 10 carbon atoms or an aryl group, more preferably an alkyl group having 1 to 5 carbon atoms, and further preferably a methyl group.
- R ⁇ 2 > in Formula (2) may be the same, and may differ.
- R 3 is a group represented by the following chemical formula (3).
- R 4 is a group represented by R 2 , R 3 or —OR 10 , preferably R 2 .
- R ⁇ 4 > in Formula (2) may be the same, and may differ.
- R 10 is a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, preferably a hydrogen atom or an alkyl group having 1 to 4 carbon atoms, and more preferably a hydrogen atom or a methyl group.
- p is a number of 10 to 10,000, preferably 50 to 5000, and more preferably 100 to 2000.
- q is a number of 0.1 to 1000, preferably 0.5 to 500, and more preferably 1 to 100.
- R 5 and R 7 are each independently an alkylene group having 1 to 6 carbon atoms, preferably an alkylene group having 1 to 3 carbon atoms.
- R 6 , R 8 and R 9 are each independently a hydrogen atom, an alkyl group having 1 to 10 carbon atoms or an aryl group, preferably a hydrogen atom or an alkyl group having 1 to 5 carbon atoms, Preferably it is a hydrogen atom.
- r is a number from 0 to 6, preferably from 0 to 3, and more preferably from 0 to 1.
- the precursor treating agent of the present invention essentially contains an acidic phosphate ester represented by the above general formula (1) (hereinafter sometimes referred to as acidic phosphate ester (B)).
- the acidic phosphate ester (B) used in the present invention refers to an unneutralized phosphate ester that does not form a salt and has an acidic hydroxyl group in the molecule.
- a specific acidic phosphate ester and a modified silicone having a modifying group containing a nitrogen atom it is possible to suppress gum-up caused by the treating agent in the yarn making process.
- carbon fiber fusion can be suppressed in firing processes such as a flameproofing process and a carbonization process.
- the treatment agent is excellent in solution stability. 1 type (s) or 2 or more types may be used for acidic phosphate ester (B).
- R 1 is an alkyl group or an alkenyl group having 6 to 22 carbon atoms.
- R 1 is an aromatic hydrocarbon group and when R 1 has more than 22 carbon atoms, the disadvantage is that the precursor is converted into a carbon fiber graphite structure by tarting the acidic phosphate ester in the firing step. Thus, the strength of the carbon fiber is reduced.
- R 1 preferably has 8 to 20 carbon atoms, more preferably 10 to 18 carbon atoms, and still more preferably 10 to 16 carbon atoms.
- R 1 is preferably an alkyl group.
- the carbon number of R 1 may be distributed, and R 1 may be linear or branched.
- A represents an alkylene group having 2 to 4 carbon atoms
- AO represents an oxyalkylene group. That is, it represents an oxyethylene group, an oxypropylene group or an oxybutylene group.
- the oxyalkylene group an oxyethylene group and an oxypropylene group are preferable, and an oxyethylene group is more preferable.
- N which is the number of repeating oxyalkylene groups, is an integer of 0 to 20, preferably 2 to 18, more preferably 5 to 15, and still more preferably 8 to 12.
- the oxyalkylene group AO constituting the polyoxyalkylene group (AO) n may be the same or different.
- the acid value of the acidic phosphate ester (B) is preferably 15 to 500 mgKOH / g, more preferably 20 to 350 mgKOH / g, and further preferably 50 to 200 mgKOH / g.
- the acid value is less than 15 mgKOH / g, it is sometimes impossible to suppress the carbon fiber fusion in the yarn-making process and the baking process.
- the acid value is more than 500 mgKOH / g, the solution stability of the emulsion when water-based emulsified may be deteriorated.
- the acid value as used in the field of this invention means the mg number of potassium hydroxide required in order to neutralize the acidic hydroxyl group which exists in 1 g of acidic phosphoric acid ester.
- the acidic phosphoric acid ester (B) represented by the general formula (1) is a combination of a and b in the formula, and the acidic phosphoric acid monoester (B1) represented by the following general formula (4) and the following general formula It can be distinguished from the acidic phosphoric acid diester (B2) represented by (5).
- acidic phosphate ester (B) examples include hexyl phosphate ester, octyl phosphate ester, decyl phosphate ester, lauryl phosphate ester, tridecyl phosphate ester, myristyl phosphate ester, cetyl phosphate ester, stearyl phosphate ester, behenyl phosphate Acid ester, oleyl phosphate, 2-ethylhexyl phosphate, isoheptyl phosphate, isooctyl phosphate, isononyl phosphate, isodecyl phosphate, isoundecyl phosphate, isolauryl phosphate, isotridecyl Phosphate ester, isomyristyl phosphate ester, isocetyl phosphate ester, isostearyl phosphate ester, and their polyoxyethylene, polyoxypropylene, polyoxy There
- the acidic phosphate ester (B) can be produced by a known method.
- an inorganic phosphoric acid such as anhydrous phosphoric acid P 2 O 5
- a compound having an alcoholic hydroxyl group in the molecule such as an alcohol or a polyoxyalkylene-added alkyl ether (hereinafter sometimes simply referred to as a raw material alcohol)
- a raw material alcohol a compound having an alcoholic hydroxyl group in the molecule
- an alcohol or a polyoxyalkylene-added alkyl ether hereinafter sometimes simply referred to as a raw material alcohol
- an arbitrary It is obtained by reacting at a molar ratio of
- the molar ratio of P 2 O 5 to 1 mol of a compound such as a raw material alcohol is preferably 0.1 to 0.8, more preferably 0.15 to 0.6, and further preferably 0.2 to 0.4.
- the molar ratio exceeds 0.8, the solution stability of the emulsion when water-based emulsified may deteriorate.
- the molar ratio is less than 0.1, it is sometimes impossible to suppress the carbon fiber fusion in the yarn-making process and the baking process.
- a mixture of acidic phosphoric acid monoester (B1) and acidic phosphoric acid diester (B2) can be obtained.
- the ratio of the acidic phosphoric acid monoester (B1) and acidic phosphoric acid diester (B2) obtained can be adjusted by adjusting the molar ratio of P 2 O 5 to 1 mol of the raw material alcohol.
- the acidic phosphoric acid ester (B) may be an acidic phosphoric acid monoester (B1) or an acidic phosphoric acid diester (B2) alone, but the acidic phosphoric acid monoester (B1) and the acidic phosphoric acid diester (B2). And mixtures thereof are preferred.
- the molar ratio (B1 / B2) of the acidic phosphoric acid monoester (B1) to the acidic phosphoric acid diester (B2) is 5/95 to 80/20 from the viewpoint of improving the solution stability of the emulsion when emulsified with water. It is preferably 10/90 to 70/30, more preferably 15/85 to 50/50.
- these molar ratios (B1 / B2) in the mixture of acidic phosphoric acid monoester (B1) and acidic phosphoric acid diester (B2) can be confirmed by a known NMR method.
- the precursor treating agent of the present invention essentially contains the modified silicone (A) and the acidic phosphate ester (B). In order to exhibit the effects of the present invention more than just using these components together, they should be used in a specific ratio such that the amount of the acidic phosphate ester (B) is small with respect to the modified silicone (A). Is preferred. From these points, the weight ratio of the modified silicone (A) to the acidic phosphate (B) (modified silicone / acid phosphate) is preferably 99.6 / 0.4 to 90/10, and 99.3. /0.7 to 92/8 is more preferable, 99/1 to 95/5 is more preferable, and 99/1 to 97/3 is particularly preferable.
- the weight ratio exceeds 99.6 / 0.4, it is sometimes impossible to suppress the carbon fiber fusion in the yarn-making process and the baking process.
- the weight ratio is less than 90/10, the solution stability of the emulsion upon aqueous emulsification may be deteriorated.
- disassembly of a silicone type component is accelerated
- the weight ratio of the modified silicone (A) in the nonvolatile content of the treatment agent is preferably 50 to 95% by weight, more preferably 55 to 90% by weight, further preferably 60 to 90% by weight, and 70 to 90% by weight. Is particularly preferred. When the ratio is less than 50% by weight, the effect of preventing fusion in the firing process is inferior, and it may be difficult to obtain high-strength carbon fibers. On the other hand, when the ratio exceeds 95% by weight, aqueous emulsification becomes difficult and a stable solution may be difficult to obtain.
- the non-volatile content means an absolutely dry component when the treatment agent is heat treated at 105 ° C. to remove the solvent and the like and reach a constant weight.
- the weight ratio of the acidic phosphate ester (B) in the non-volatile content of the treatment agent is preferably 0.3 to 7% by weight, more preferably 0.4 to 6% by weight, and 0.5 to 5% by weight. More preferred is 0.7 to 4% by weight.
- the ratio is less than 0.3% by weight, it is sometimes impossible to suppress the carbon fiber fusion in the yarn-making process and the baking process.
- it exceeds 7% by weight the solution stability of the emulsion upon aqueous emulsification may deteriorate.
- stimulated the heat resistance of a processing agent may fall.
- the precursor treating agent of the present invention preferably further contains a surfactant (however, the acidic phosphate ester (B) is not included).
- the surfactant is used as an emulsifier, and makes it possible to emulsify and disperse the modified silicone and the like in water.
- the surfactant is not particularly limited, and a known one can be appropriately selected from nonionic surfactants, anionic surfactants, cationic surfactants and amphoteric surfactants. Surfactant may use together 1 type (s) or 2 or more types.
- nonionic surfactant examples include polyoxyalkylene linear alkyl ethers such as polyoxyethylene hexyl ether, polyoxyethylene octyl ether, polyoxyethylene decyl ether, polyoxyethylene lauryl ether, and polyoxyethylene cetyl ether; Polyoxyalkylene branched primary alkyl ethers such as polyoxyethylene 2-ethylhexyl ether, polyoxyethylene isocetyl ether, polyoxyethylene isostearyl ether; polyoxyethylene 1-hexyl hexyl ether, polyoxyethylene 1-octyl hexyl ether , Polyoxyethylene 1-hexyloctyl ether, polyoxyethylene 1-pentyl heptyl ether, polyoxyethylene 1-heptyl pentyl Polyoxyalkylene branched secondary alkyl ethers such as ether; polyoxyalkylene alkenyl ethers such as polyoxyethylene oleyl ether; polyoxys such as
- polyoxyalkylene alkylaryl phenyl ether Of polyoxyalkylene alkylaryl phenyl ether; polyoxyethylene monolaur , Polyoxyethylene monooleate, polyoxyethylene monostearate, polyoxyethylene monomyristate, polyoxyethylene dilaurate, polyoxyethylene dioleate, polyoxyethylene dimyristate, polyoxyethylene distearate, etc.
- polyoxyalkylene branched primary alkyl ethers polyoxyalkylene branched secondary alkyl ethers, polyoxyalkylene alkenyl ethers, polyoxyalkylene ethers, Oxyalkylene alkylphenyl ethers, polyoxyalkylene fatty acid esters, oxyethylene-oxypropylene block copolymers, and terminal alkyl etherified products of oxyethylene-oxypropylene block copolymers are preferred, and further, tarred on the fiber in the firing step.
- Oxyethylene-oxypropylene blocks or random copolymers, and terminal alkyl ethers of oxyethylene-oxypropylene block copolymers are more preferred because they are less likely to damage the fiber. .
- anionic surfactant examples include fatty acids (salts) such as oleic acid, palmitic acid, sodium oleate, potassium palmitate, triethanolamine oleate; hydroxyacetic acid, potassium potassium hydroxyacetate, lactic acid, lactic acid Hydroxyl group-containing carboxylic acid (salt) such as potassium salt; polyoxyalkylene alkyl ether acetic acid (salt) such as polyoxyethylene tridecyl ether acetic acid (sodium salt); and many carboxyl groups such as potassium trimellitic acid and potassium pyromellitic acid Salt of substituted aromatic compound; alkylbenzene sulfonic acid (salt) such as dodecylbenzene sulfonic acid (sodium salt); polyoxyalkylene alkyl ether sulfonic acid (salt) such as polyoxyethylene 2-ethylhexyl ether sulfonic acid (potassium salt) Stearoylmethyl taurine (sodium), la
- cationic surfactant examples include quaternary ammonium salts such as lauryltrimethylammonium chloride and oleylmethylethylammonium ethosulphate; (polyoxyethylene) laurylaminoether lactate, stearylaminoether lactate, (polyoxyethylene) ) (Polyoxyalkylene) alkylamino ether salts such as lauryl amino ether trimethyl phosphate salt;
- amphoteric surfactants examples include 2-undecyl-N, N- (hydroxyethylcarboxymethyl) -2-imidazoline sodium and 2-cocoyl-2-imidazolinium hydroxide-1-carboxyethyloxy disodium salt.
- Imidazoline-based amphoteric surfactants 2-heptadecyl-N-carboxymethyl-N-hydroxyethylimidazolium betaine, lauryldimethylaminoacetic acid betaine, alkylbetaines, amide betaines, sulfobetaines, and other betaine-based amphoteric surfactants; N- Examples include amino acid type amphoteric surfactants such as lauryl glycine, N-lauryl ⁇ -alanine, N-stearyl ⁇ -alanine.
- ionic surfactants may change over time after emulsification, and may affect the crosslinkability of silicone.
- nonionic surfactants are preferred because they are excellent in stability over time, have little effect on silicone crosslinkability, and are excellent in emulsifying power of silicone.
- the weight ratio of the surfactant in the nonvolatile content of the treatment agent is preferably 1 to 40% by weight, more preferably 5 to 30% by weight, and further preferably 8 to 25% by weight.
- the weight ratio is less than 1% by weight, good emulsification stability may be difficult to obtain.
- this weight ratio exceeds 40 weight%, the heat resistance of a processing agent falls and it may be unable to suppress the fusion
- the precursor treating agent of the present invention may contain a silicone component other than the modified silicone having a modifying group containing a nitrogen atom within a range not inhibiting the effect of the present invention.
- a silicone component other than the modified silicone having a modifying group containing a nitrogen atom within a range not inhibiting the effect of the present invention.
- the precursor treating agent of the present invention may contain components other than the above-described components as long as the effects of the present invention are not impaired.
- Other components include phenolic, amine-based, sulfur-based, phosphorus-based and quinone-based antioxidants; sulfates and sulfonates of higher alcohols and higher alcohol ethers, phosphoric acids of higher alcohols and higher alcohol ethers
- Antistatic agents such as ester salts, quaternary ammonium salt type cationic surfactants, amine salt type cationic surfactants; smootheners such as alkyl esters of higher alcohols, higher alcohol ethers and waxes; antibacterial agents; antiseptics Agents; rust preventives; and hygroscopic agents.
- the precursor treating agent of the present invention does not substantially contain an antioxidant.
- the antioxidant is contained, the decomposition of the modified silicone having a modifying group containing a nitrogen atom is promoted in the firing step, so that the heat resistance of the precursor treatment agent may be lowered. If it becomes so, the fusion
- “Substantially not contained” means that the weight ratio of the antioxidant is 1.0% by weight or less with respect to the modified silicone having a modifying group containing a nitrogen atom. The weight ratio is more preferably 0.5% by weight or less, further preferably 0.1% by weight or less, and particularly preferably 0% by weight.
- the precursor treatment agent of the present invention preferably does not contain a phosphate ester salt obtained by neutralizing the acidic phosphate ester (B) of the present invention.
- a phosphate ester salt obtained by neutralizing the acidic phosphate ester (B) of the present invention.
- the phosphoric acid ester salt is contained, not only the solution stability of the emulsion upon aqueous emulsification may be deteriorated, but also the strength of the carbon fiber may be reduced.
- the phosphate ester salt include alkali metal salts (Na, K, etc.), alkaline earth metal salts (Ca, Mg, Ba, etc.), ammonium salts, quaternary ammonium of the acidic phosphate ester (B) of the present invention. Examples thereof include salts and amine salts.
- the precursor treatment agent may be composed of the above-mentioned components consisting only of non-volatile components, but from the viewpoint of uniform adhesion to the fiber and safety in the work environment, it contains a surfactant as an emulsifier and is contained in water. A state in which the emulsion is emulsified or dispersed to form an emulsion dispersed in water is preferable.
- the precursor processing agent of this invention contains water, there is no limitation in particular about the weight ratio of the water which occupies for the whole precursor processing agent, and the weight ratio of a non volatile matter.
- the weight ratio of water in the entire precursor treatment agent is preferably 0.1 to 99.9% by weight, more preferably 10 to 99.5% by weight, and particularly preferably 50 to 99% by weight.
- the weight ratio (concentration) of the non-volatile component in the entire precursor treatment agent is preferably 0.01 to 99.9% by weight, more preferably 0.5 to 90% by weight, and particularly preferably 1 to 50% by weight.
- the precursor treating agent of the present invention can be produced by mixing the components described above.
- the method for emulsifying and dispersing the components described above is not particularly limited, and a known method can be employed.
- each component constituting the precursor treatment agent is put into warm water under stirring and emulsified and dispersed, and each component constituting the precursor treatment agent is mixed, a homogenizer, a homomixer,
- the method include phase inversion emulsification by gradually adding water while applying a mechanical shearing force using a ball mill or the like.
- components other than acidic phosphate ester (B) are emulsified and dispersed by the above-described method, and the necessary amount of acidic phosphate ester (B) is mixed with the obtained emulsion to obtain a treatment agent. It can also be prepared.
- a precursor and carbon fiber can be produced using the precursor treating agent of the present invention.
- the precursor and the carbon fiber manufacturing method using the precursor treating agent of the present invention are not particularly limited, examples thereof include the following manufacturing methods.
- the method for producing carbon fiber of the present invention includes a yarn making process, a flameproofing process, and a carbonization process.
- the precursor of the present invention is obtained by this spinning process.
- the yarn making process is a process of manufacturing a precursor by attaching the above-mentioned acrylic fiber treatment agent for carbon fiber production (precursor treatment agent) to the raw acrylic fiber of the acrylic fiber for carbon fiber production (precursor).
- the adhesion treatment process is a process of adhering a precursor treatment agent after spinning precursor acrylic fiber. That is, the precursor treatment agent is adhered to the precursor raw acrylic fiber in the adhesion treatment step.
- the precursor raw acrylic fiber is stretched immediately after spinning, and the high-strength stretching after the adhesion treatment step is particularly called a “stretching step”.
- the stretching process may be a wet heat stretching method using high temperature steam or a dry heat stretching method using a hot roller.
- the precursor is composed of acrylic fibers mainly composed of polyacrylonitrile obtained by copolymerizing at least 95 mol% or more of acrylonitrile and 5 mol% or less of the flame resistance promoting component.
- the flame resistance promoting component a vinyl group-containing compound having copolymerizability with acrylonitrile can be suitably used.
- the fineness of the single fiber of the precursor is not particularly limited, but is preferably 0.1 to 2.0 dTex from the balance between performance and production cost.
- the number of single fibers constituting the precursor fiber bundle is not particularly limited, but is preferably 1,000 to 96,000 from the balance between performance and production cost.
- the precursor treatment agent may be attached to the precursor raw acrylic fiber at any stage of the yarn-making process, but is preferably attached once before the drawing process. It may be attached at any stage before the stretching process, for example, immediately after spinning. Further, it may be reattached at any stage after the stretching process, for example, it may be reattached immediately after the stretching process, it may be reattached at the winding stage, or it may be reattached immediately before the flameproofing process. You may let them.
- the adhesion method when the precursor treatment agent consists only of non-volatile components, it may be adhered using a roller or the like as straight oil, or the precursor treatment agent is emulsified or dispersed in a solvent such as water or an organic solvent. In the case of the emulsion thus formed, it may be attached by dipping, spraying or the like.
- the rate of applying the precursor treatment agent obtains the effect of preventing fiber-to-fiber sticking and the effect of preventing fusion, and prevents the carbon fiber quality from being deteriorated by the tar product of the treatment agent in the carbonization treatment process.
- it is preferably 0.1 to 2% by weight, more preferably 0.3 to 1.5% by weight, based on the weight of the precursor.
- the application rate of the precursor treatment agent exceeds 2% by weight, the precursor treatment agent covers more than necessary between the single fibers, so that the supply of oxygen to the fibers is hindered in the flameproofing treatment step, and the resulting carbon fiber The strength of the may decrease.
- the provision rate of a precursor processing agent here is defined with the percentage of the non volatile matter weight to which the precursor processing agent adhered with respect to the precursor weight.
- the flameproofing treatment step is a step of converting the precursor with the precursor treating agent into flameproofing fibers in an oxidizing atmosphere at 200 to 300 ° C.
- the oxidizing atmosphere is usually an air atmosphere.
- the temperature of the oxidizing atmosphere is preferably 230 to 280 ° C.
- the acrylic fiber after the adhesion treatment is applied for 20 to 100 minutes (preferably 30 minutes) while applying a tension of a stretch ratio of 0.90 to 1.10 (preferably 0.95 to 1.05). Heat treatment is performed for ⁇ 60 minutes.
- a flameproof fiber having a flameproof structure is produced through intramolecular cyclization and oxygen addition to the ring.
- the carbonization treatment step is a step of carbonizing the flameproof fiber in an inert atmosphere of 300 to 2000 ° C.
- a tension of 0.95 to 1.15 is applied to the flame resistant fiber in a firing furnace having a temperature gradient from 300 ° C. to 800 ° C. in an inert atmosphere such as nitrogen or argon.
- first carbonization treatment step it is preferable to carry out a pre-carbonization treatment step (first carbonization treatment step) by applying heat treatment for several minutes while applying.
- a tension ratio of 0.95 to 1.05 is applied to the first carbonization treatment step in an inert atmosphere such as nitrogen or argon.
- the maximum temperature is preferably set to 1000 ° C. or higher (preferably 1000 to 2000 ° C.) while applying a temperature gradient. This maximum temperature is appropriately selected and determined according to the required characteristics (tensile strength, elastic modulus, etc.) of the desired carbon fiber.
- a graphitization treatment step can be performed subsequent to the carbonization treatment step.
- the graphitization treatment step is usually performed at a temperature of 2000 to 3000 ° C. while applying tension to the fiber obtained in the carbonization treatment step in an inert atmosphere such as nitrogen or argon.
- the carbon fiber thus obtained can be subjected to a surface treatment for increasing the adhesive strength with the matrix resin when made into a composite material, depending on the purpose.
- a surface treatment method gas phase or liquid phase treatment can be adopted, and from the viewpoint of productivity, liquid phase treatment with an electrolytic solution of acid, alkali or the like is preferable.
- various sizing agents having excellent compatibility with the matrix resin can be added to improve the processability and handleability of the carbon fiber.
- ⁇ Application rate of treatment agent> The precursor after application of the treatment agent was alkali-melted with potassium hydroxide / sodium butyrate, dissolved in water, and adjusted to pH 1 with hydrochloric acid. This was colored by adding sodium sulfite and ammonium molybdate, and colorimetric determination (wavelength 815 m ⁇ ) of silicomolybdenum blue was performed to determine the silicon content. Using the silicon content obtained here and the value of the silicon content in the treatment agent obtained in advance by the same method, the application rate (wt%) of the precursor treatment agent was calculated.
- the acid value of the mixture p-1 was 28.8 mgKOH / g, and the molar equivalent of phosphoric anhydride relative to 1 mol of polyoxyethylene alkyl ether was 0.252.
- the theoretical molecular weight in all the production examples is a formula weight determined based on a chemical formula.
- the weight ratio of polyoxyethylene alkyl phosphate ester P-1 in mixture p-1 was 35.3 wt% by anion exchange chromatography. Therefore, the acid value of the polyoxyethylene alkyl phosphate ester P-1 is 81.6 mgKOH / g (28.8 / 0.353).
- the molar ratio of phosphoric acid monoester to phosphoric acid diester was 59.4 / 40.6.
- the acid value of the mixture p-2 was 30.0 mgKOH / g, and the molar equivalent of phosphoric anhydride relative to 1 mol of the polyoxyethylene alkyl ether was 0.215.
- the weight proportion of the polyoxyethylene alkyl phosphate ester P-2 in the mixture p-2 was 30.1 wt% by anion exchange chromatography. Therefore, the acid value of the polyoxyethylene alkyl phosphate ester is 99.7 mgKOH / g (30.0 / 0.301).
- the molar ratio of phosphoric acid monoester and phosphoric acid diester was 60.5 / 39.5.
- the acid value of the mixture p-3 was 50.4 mgKOH / g, and the molar equivalent of phosphoric anhydride relative to 1 mol of the polyoxyethylene alkyl ether was 0.333.
- the weight ratio of polyoxyethylene alkyl phosphate ester P-3 in mixture p-3 was 46.7 wt% from anion exchange chromatography. Therefore, the acid value of the polyoxyethylene alkyl phosphate ester is 108.0 mgKOH / g (50.4 / 0.467).
- the molar ratio of phosphoric acid monoester and phosphoric acid diester was 65.2 / 34.8.
- the acid value of the mixture p-4 was 63.6 mgKOH / g, and the molar equivalent of phosphoric anhydride relative to 1 mol of polyoxyethylene alkyl ether was 0.380.
- the weight ratio of polyoxyethylene alkyl phosphate ester P-4 in mixture p-4 was 53.2 wt% by anion exchange chromatography. Therefore, the acid value of the polyoxyethylene alkyl phosphate ester is 119.5 mgKOH / g (63.6 / 0.532).
- the molar ratio of phosphoric acid monoester to phosphoric acid diester was 59.7 / 40.3.
- the acid value of the mixture p-5 was 74.4 mgKOH / g, and the molar equivalent of phosphoric anhydride relative to 1 mol of the polyoxyethylene alkyl ether was 0.456.
- the weight ratio of polyoxyethylene alkyl phosphate ester P-5 in mixture p-5 was 63.9 wt% by anion exchange chromatography. Therefore, the acid value of the polyoxyethylene alkyl phosphate ester is 116.4 mgKOH / g (74.4 / 0.639).
- the molar ratio of phosphoric acid monoester to phosphoric acid diester was 58.8 / 41.2.
- the acid value of the mixture p-6 was 42.0 mgKOH / g, and the molar equivalent of phosphoric anhydride to 1 mol of polyoxyethylene nonylphenol ether was 0.315.
- the weight ratio of polyoxyethylene nonylphenol phosphate P-6 in mixture p-6 was 44.1 wt% by anion exchange chromatography. Therefore, the acid value of polyoxyethylene nonylphenol phosphate is 95.2 mgKOH / g (42.0 / 0.441).
- the molar ratio of phosphoric acid monoester to phosphoric acid diester was 60.2 / 39.8.
- Modified silicone S-1 Diamine type amino modified silicone (25 ° C. viscosity: 1,300 mm 2 / s, amino weight%: 0.8 weight%, nitrogen atom content: 0.7 weight%)
- Modified silicone S-2 Monoamine type amino modified silicone (25 ° C. viscosity: 1,700 mm 2 / s, amino weight%: 0.4 weight%, nitrogen atom content: 0.35 weight%)
- Modified silicone S-3 Diamine type amino-modified silicone (25 ° C. viscosity: 1,300 mm 2 / s, amino weight%: 2.7 weight%, nitrogen atom content: 2.4 weight%)
- the non-volatile concentration of the treatment agent was 3.0% by weight.
- This treating agent emulsion was attached to a precursor raw acrylic fiber obtained by copolymerizing 97 mol% acrylonitrile and 3 mol% itaconic acid so that the application rate was 1.0%, and a stretching step (steam stretching, A precursor was produced through a draw ratio of 2.1 times (single fiber fineness 0.8 dtex, 24,000 filaments).
- the precursor was flameproofed in a flameproofing furnace at 250 ° C. for 60 minutes and then baked in a carbonization furnace having a temperature gradient of 300 to 1400 ° C. in a nitrogen atmosphere to convert into carbon fibers. Table 1 shows the evaluation results of the characteristic values.
- Example 1 a precursor and carbon fiber after the treatment agent was adhered were obtained in the same manner as in Example 1 except that the treatment agent emulsion was prepared so as to have a nonvolatile composition of treatment agents shown in Tables 1 to 4. The evaluation results of each characteristic value are shown in Tables 1 to 4.
- MP-4 (produced by Daihachi Chemical Industry Co., Ltd.): An acidic phosphate ester antioxidant in which R 1 is an alkyl group having 4 carbon atoms, n is 0, a is 1, and b is 2 in the general formula (1) : Tetrakis [methylene-3- (3 ′, 5′-di-t-butyl-4′-hydroxyphenyl) propionate] methane
- the acrylic fiber treatment agent for producing carbon fibers of the present invention is a treatment agent used when producing acrylic fibers for producing carbon fibers, and is useful for producing high-quality carbon fibers.
- the acrylic fiber for producing carbon fiber of the present invention is treated with the treatment agent of the present invention and is useful for producing high-quality carbon fiber.
- High quality carbon fibers can be obtained by the carbon fiber manufacturing method of the present invention.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
- Inorganic Fibers (AREA)
Abstract
Description
炭素繊維を製造する方法としては、まずプレカーサーを製造する(このプレカーサーの製造工程を製糸工程と称することがある)。このプレカーサーを200~300℃の酸化性雰囲気中で耐炎化繊維に転換し(この工程を以下、耐炎化処理工程と称することがある)、続いて300~2000℃の不活性雰囲気中で炭素化する(この工程を以下、炭素化処理工程と称することがある)方法が一般的である(以下、耐炎化処理工程と炭素化処理工程をあわせて、焼成工程と称することがある)。このプレカーサーの製造には通常のアクリル繊維と比較しても高倍率に延伸される延伸工程を経る。その際、繊維同士の膠着が起こり易く、均一に高倍率延伸が行われない為に、不均一なプレカーサーとなる。この様なプレカーサーを焼成して得られる炭素繊維は十分な強度が得られないという問題がある。また、プレカーサーの焼成時には、単繊維同士の融着が発生し、得られた炭素繊維の品質、品位を低下させるという問題がある。
一般的に利用されているアミノ変性シリコーン系処理剤は、アミノ変性シリコーンオイルを水系エマルジョン化したものである。自己乳化型のアミノ変性シリコーンでない場合は、界面活性剤を用いて水系エマルジョン化が行われる。この水系エマルジョンをプレカーサー繊維に付与するため、付与後に水分を除去する必要があり、乾燥工程を通過させる必要がある。その後、さらに延伸工程で熱延伸が行われ、高倍率に延伸されたプレカーサー繊維が得られる。アミノ変性シリコーンは、その優れた熱架橋性のために、乾燥工程や延伸工程における熱ローラーでも架橋が促進され、ローラー汚れ(以後ガムアップと称することがある)が蓄積し易く、プレカーサーの糸切れや毛羽発生の原因となるばかりでなく、その清掃の為に操業性が低下するという問題が起こり易い。
かかる従来の技術背景に鑑み、本発明の目的は、処理剤の溶液安定性に優れ、プレカーサー繊維を製造する際に使用することで、プレカーサーの製造工程(製糸工程)におけるガムアップを抑制することができ、かつ焼成工程における炭素繊維の融着を抑制することができる炭素繊維製造用アクリル繊維処理剤、該処理剤を付与させて製糸した炭素繊維製造用アクリル繊維、及び該処理剤を用いた炭素繊維の製造方法を提供することにある。

前記酸性リン酸エステルの酸価は15~500mgKOH/gであることが好ましい。
処理剤の不揮発分に占める前記変性シリコーンの重量割合は50~95重量%であることが好ましい。
本発明の処理剤は、水中に分散したエマルジョンとなっていることが好ましい。
本発明のプレカーサー油剤は、窒素原子を含む変性基を持つ変性シリコーン(以下変性シリコーン(A)ということがある)を必須に含有する。変性シリコーン(A)は窒素原子を含む変性基であれば変性基の種類は特に限定されない。窒素原子を含む変性基としては、アミノ結合やイミノ結合を含有する変性基(即ち、アミノ基)や、アミド結合を含有する変性基(即ち、アミド基)などが挙げられ、アミノ結合とアミド結合など異なる結合が複数存在する変性基でもよい。窒素原子を含む変性基は、主鎖であるシリコーンの側鎖と結合していてもよいし、末端と結合していてもよいし、また両方と結合していてもよい。また、分子中にポリオキシアルキレン基(例えば、ポリオキシエチレン基、ポリオキシプロピレン基、ポリオキシブチレン基等)を有していてもよい。
また、変性シリコーン(A)における窒素原子の含有量は、0.35~3.2重量%が好ましく、0.37~2.2重量%がより好ましく、0.40~1.3重量%がさらに好ましい。窒素原子の含有量が0.35重量%より低い場合、水系乳化した際にエマルジョンの溶液安定性が悪くなることがある。一方、窒素原子の含有量が3.2重量%より高い場合、熱架橋により変性シリコーン(A)の粘着性が高くなり、ガムアップの原因となる。そのため、酸性リン酸エステルを併用しても、ガムアップを抑制できないおそれがある。
変性シリコーン(A)がアミノ変性シリコーンである場合、そのアミノ変性シリコーンの構造は特に限定されるものではない。即ち、変性基であるアミノ基は、主鎖であるシリコーンの側鎖と結合していてもよいし、末端と結合していてもよいし、また両方と結合していてもよい。また、そのアミノ基は、モノアミン型であってもポリアミン型であってもよく、1分子中に両者が併存していてもよい。


本発明のプレカーサー処理剤は、上記一般式(1)で示される酸性リン酸エステル(以下、酸性リン酸エステル(B)ということがある)を必須に含有する。本発明で用いる酸性リン酸エステル(B)は、塩を形成していない、未中和のリン酸エステルをいい、分子内に酸性水酸基を有するものである。特定の酸性リン酸エステルと窒素原子を含む変性基を持つ変性シリコーンとを併用することにより、製糸工程における処理剤由来のガムアップを抑制できる。また、耐炎化処理工程、炭素化処理工程などの焼成工程において、炭素繊維の融着を抑制することができる。さらに、処理剤は、溶液安定性に優れる。酸性リン酸エステル(B)は1種又は2種以上を用いてもよい。
Aは炭素数2~4のアルキレン基を示し、AOとしてはオキシアルキレン基を示す。つまり、オキシエチレン基、オキシプロピレン基又はオキシブチレン基を示す。オキシアルキレン基としては、オキシエチレン基、オキシプロピレン基が好ましく、オキシエチレン基がさらに好ましい。オキシアルキレン基の繰り返し数であるnは0~20の整数であり、2~18が好ましく、5~15がより好ましく、8~12がさらに好ましい。nが20超の場合、水系乳化した際のエマルジョンの溶液安定性が悪くなることがある。ポリオキシアルキレン基(AO)nを構成するオキシルキレン基AOとしては、同一でも異なっていてもよい。
aとbは、各々1又は2であり、a+b=3を満足する整数である。


原料アルコールなどの化合物1モルに対するP2O5のモル比は0.1~0.8が好ましく、0.15~0.6がより好ましく、0.2~0.4がさらに好ましい。該モル比が0.8を超えると、水系乳化した際のエマルジョンの溶液安定性が悪くなることがある。一方、該モル比が0.1未満では、製糸工程におけるガムアップ及び焼成工程の炭素繊維の融着を抑制することができないことがある。
本発明のプレカーサー処理剤は、上記の変性シリコーン(A)と酸性リン酸エステル(B)とを必須に含有するものである。本発明の効果をより発揮させるためには、これら成分を単に併用するだけではなく、変性シリコーン(A)に対して酸性リン酸エステル(B)が少量になるような特定の割合で併用することが好ましい。このような点から、変性シリコーン(A)と酸性リン酸エステル(B)の重量割合(変性シリコーン/酸性リン酸エステル)は、99.6/0.4~90/10が好ましく、99.3/0.7~92/8がより好ましく、99/1~95/5がさらに好ましく、99/1~97/3が特に好ましい。該重量割合が99.6/0.4超の場合、製糸工程におけるガムアップ及び焼成工程の炭素繊維の融着を抑制することができないことがある。一方、該重量割合が90/10未満の場合、水系乳化した際のエマルジョンの溶液安定性が悪くなることがある。また、シリコーン系成分の分解が促進されるため、処理剤の耐熱性が低下し、焼成工程の炭素繊維の融着を抑制することができないことがある。
本発明のプレカーサー処理剤が水を含む場合、プレカーサー処理剤全体に占める水の重量割合、不揮発分の重量割合については、特に限定はない。例えば、本発明のプレカーサー処理剤を輸送する際の輸送コストや、エマルジョン粘度に因るところの取扱い性等を考慮して適宜決定すればよい。プレカーサー処理剤全体に占める水の重量割合は、0.1~99.9重量%が好ましく、10~99.5重量%がさらに好ましく、50~99重量%が特に好ましい。プレカーサー処理剤全体に占める不揮発分の重量割合(濃度)は、0.01~99.9重量%が好ましく、0.5~90重量%がさらに好ましく、1~50重量%が特に好ましい。
また、酸性リン酸エステル(B)以外の成分を上記の方法により乳化、分散させて、得られた乳化物に対して必要量の酸性リン酸エステル(B)を混合することにより、処理剤を調製することもできる。
本発明の炭素繊維の製造方法は、製糸工程と耐炎化処理工程と炭素化処理工程とを含む。本発明のプレカーサーは、この製糸工程で得られるものである。
製糸工程は、炭素繊維製造用アクリル繊維(プレカーサー)の原料アクリル繊維に上記炭素繊維製造用アクリル繊維処理剤(プレカーサー処理剤)を付着させてプレカーサーを製造する工程であり、付着処理工程と延伸工程とを含む。
付着処理工程は、プレカーサーの原料アクリル繊維を紡糸した後、プレカーサー処理剤を付着させる工程である。つまり、付着処理工程でプレカーサーの原料アクリル繊維にプレカーサー処理剤を付着させる。またこのプレカーサーの原料アクリル繊維は紡糸直後から延伸されるが、付着処理工程後の高倍率延伸を特に「延伸工程」と呼ぶ。延伸工程は高温水蒸気をもちいた湿熱延伸法でもよいし、熱ローラーをもちいた乾熱延伸法でもよい。
処理剤付与後のプレカーサーを水酸化カリウム/ナトリウムブチラートでアルカリ溶融した後、水に溶解して塩酸でpH1に調整した。これを亜硫酸ナトリウムとモリブデン酸アンモニウムを加えて発色させ、ケイモリブデンブルーの比色定量(波長815mμ)を行い、ケイ素の含有量を求めた。ここで求めたケイ素含有量と予め同法で求めた処理剤中のケイ素含有量の値を用いて、プレカーサー処理剤の付与率(wt%)を算出した。
不揮発分濃度が3.0重量%である各処理剤エマルジョンを50℃に調節された恒温槽で保管し、溶液の外観を目視で確認し、下記の評価基準で溶液安定性を判定した。
◎ :60日間分離無し。
○ :30日間分離無し、60日以内には分離。
△ :7日間分離無し、30日以内に分離。
× :7日間以内に分離。
××:乳化当日に分離、または乳化できない。
直径φ60mmのアルミカップ上に不揮発分が1gとなるよう各処理剤エマルジョンを採取し、温風乾燥機にて105℃×3時間処理して水分を除去し、試料を精秤(W1)した。得られた試料をギヤオーブンにて各温度条件(160℃×180min、250℃×60min)で熱処理し、熱処理後の試料を精秤(W2)した。下記式にて減量率を算出した。
(W1-W2)/W1×100=減量率(wt%)
直径φ60mmのアルミカップ上に不揮発分が1gとなるよう各処理剤エマルジョンを採取し、温風乾燥機にて105℃×3時間処理して水分を除去し、試料を精秤(W3)した。得られた試料をギヤオーブンにて各温度条件(160℃×180min、250℃×60min)で熱処理し、熱処理後の試料をクロロフォルムに溶解し、可溶部分と不可溶部分に分離する。不可溶部分からクロロフォルムを室温で3時間、80℃×20分処理してクロロフォルムを除去した試料を精秤(W4)した。下記式にて架橋率を算出した。
W4/W3×100=架橋率(wt%)
プレカーサー50kgに処理剤を付与した後の乾燥ローラーの汚染度合い(ガムアップ)を下記の評価基準で判定した。
◎ :ガムアップによるローラー汚染が無く、製糸操業性問題無し。
○ :ガムアップによるローラー汚染が少なく、製糸操業性問題無し。
△ :ガムアップによるローラー汚染がややあるが、製糸操業性問題無し。
× :ガムアップによるローラー汚染があり、やや製糸操業性に劣る。
××:ガムアップによるローラー汚染が著しく、製糸時に単糸取られ、捲き付きあり。
炭素繊維から無作為に20カ所を選び、そこから長さ10mmの短繊維を切り出し、その融着状態を観察し、下記の評価基準で判定した。
◎:融着無し
○:ほぼ融着無し
△:融着少ない
×:融着多い
JIS-R-7601に規定されているエポキシ樹脂含浸ストランド法に準じ測定し、測定回数10回の平均値を炭素繊維強度(GPa)とした。
〔製造例1〕
窒素気流下の反応容器中に、ポリオキシエチレンが12モル付加された炭素数が11~15のアルキルエーテル(理論分子量728、炭素数13として)976部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)24部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンアルキルエーテルとポリオキシエチレンアルキルリン酸エステルP-1とを含む混合物p-1を得た。混合物p-1の酸価は28.8mgKOH/gであり、ポリオキシエチレンアルキルエーテル1モルに対する無水リン酸のモル当量は0.252であった。なお、製造例全てにおける理論分子量は、化学式に基づいて求めた式量である。
混合物p-1中のポリオキシエチレンアルキルリン酸エステルP-1の重量割合はアニオン交換クロマトグラフィーより35.3wt%であった。よって、ポリオキシエチレンアルキルリン酸エステルP-1の酸価は81.6mgKOH/g(28.8/0.353)である。また、リン酸モノエステルとリン酸ジエステルのモル比は59.4/40.6であった。
窒素気流下の反応容器中に、ポリオキシエチレンが9モル付加された炭素数が11~15のアルキルエーテル(理論分子量596、炭素数13として)975部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)25部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンアルキルエーテルとポリオキシエチレンアルキルリン酸エステルP-2とを含む混合物p-2を得た。混合物p-2の酸価は30.0mgKOH/gであり、ポリオキシエチレンアルキルエーテル1モルに対する無水リン酸のモル当量は0.215であった。
混合物p-2中のポリオキシエチレンアルキルリン酸エステルP-2の重量割合はアニオン交換クロマトグラフィーより30.1wt%であった。よって、ポリオキシエチレンアルキルリン酸エステルの酸価は99.7mgKOH/g(30.0/0.301)である。また、リン酸モノエステルとリン酸ジエステルのモル比は60.5/39.5であった。
窒素気流下の反応容器中に、ポリオキシエチレンが9モル付加された炭素数が12のアルキルエーテル(理論分子量540)958部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)42部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンアルキルエーテルとポリオキシエチレンアルキルリン酸エステルP-3とを含む混合物p-3を得た。混合物p-3の酸価は50.4mgKOH/gであり、ポリオキシエチレンアルキルエーテル1モルに対する無水リン酸のモル当量は0.333であった。
混合物p-3中のポリオキシエチレンアルキルリン酸エステルP-3の重量割合はアニオン交換クロマトグラフィーより46.7wt%であった。よって、ポリオキシエチレンアルキルリン酸エステルの酸価は108.0mgKOH/g(50.4/0.467)である。また、リン酸モノエステルとリン酸ジエステルのモル比は65.2/34.8であった。
窒素気流下の反応容器中に、ポリオキシエチレンが8モル付加された炭素数が8のアルキルエーテル(理論分子量482)947部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)53部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンアルキルエーテルとポリオキシエチレンアルキルリン酸エステルP-4を含む混合物p-4とを得た。混合物p-4の酸価は63.6mgKOH/gであり、ポリオキシエチレンアルキルエーテル1モルに対する無水リン酸のモル当量は0.380であった。
混合物p-4中のポリオキシエチレンアルキルリン酸エステルP-4の重量割合はアニオン交換クロマトグラフィーより53.2wt%であった。よって、ポリオキシエチレンアルキルリン酸エステルの酸価は119.5mgKOH/g(63.6/0.532)である。また、リン酸モノエステルとリン酸ジエステルのモル比は59.7/40.3であった。
窒素気流下の反応容器中に、ポリオキシエチレンが5モル付加された炭素数が18のアルキルエーテル(理論分子量490)938部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)62部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンアルキルエーテルとポリオキシエチレンアルキルリン酸エステルP-5とを含む混合物p-5を得た。混合物p-5の酸価は74.4mgKOH/gであり、ポリオキシエチレンアルキルエーテル1モルに対する無水リン酸のモル当量は0.456であった。
混合物p-5中のポリオキシエチレンアルキルリン酸エステルP-5の重量割合はアニオン交換クロマトグラフィーより63.9wt%であった。よって、ポリオキシエチレンアルキルリン酸エステルの酸価は116.4mgKOH/g(74.4/0.639)である。また、リン酸モノエステルとリン酸ジエステルのモル比は58.8/41.2であった。
〔製造例6〕
窒素気流下の反応容器中に、ポリオキシエチレンが9モル付加されたノニルフェノールエーテル(理論分子量616)965部を仕込み攪拌下約65℃にした。次に無水リン酸P2O5(理論分子量142)35部を攪拌下で添加し、約80℃で2時間エステル化反応させ、未反応ポリオキシエチレンノニルフェノールエーテルとポリオキシエチレンノニルフェノールリン酸エステルP-6を含む混合物p-6を得た。混合物p-6の酸価は42.0mgKOH/gであり、ポリオキシエチレンノニルフェノールエーテル1モルに対する無水リン酸のモル当量は0.315であった。
混合物p-6中のポリオキシエチレンノニルフェノールリン酸エステルP-6の重量割合はアニオン交換クロマトグラフィーより44.1wt%であった。よって、ポリオキシエチレンノニルフェノールリン酸エステルの酸価は95.2mgKOH/g(42.0/0.441)である。また、リン酸モノエステルとリン酸ジエステルのモル比は60.2/39.8であった。
〔製造例7〕
製造例1で得られた混合物p-1970部に攪拌下、水酸化カリウム30部を徐々に添加し中和反応を行い、中和物であるリン酸エステル塩P-1aを含む混合物p-1aを得た。
製造例1で得られた混合物p-1945部に攪拌下、ジエタノールアミン55部を徐々に添加し中和反応を行い、中和物であるリン酸エステル塩P-1bを含む混合物p-1bを得た。
製造例6で得られた混合物p-6957部に攪拌下、水酸化カリウム43部を徐々に添加し中和反応を行い、中和物であるリン酸エステル塩P-6aを含む混合物p-6aを得た。
変性シリコーン S-1:ジアミン型アミノ変性シリコーン(25℃粘度:1,300mm2/s、アミノ重量%:0.8重量%、窒素原子の含有量:0.7重量%)
変性シリコーン S-2:モノアミン型アミノ変性シリコーン(25℃粘度:1,700mm2/s、アミノ重量%:0.4重量%、窒素原子の含有量:0.35重量%)
変性シリコーン S-3:ジアミン型アミノ変性シリコーン(25℃粘度:1,300mm2/s、アミノ重量%:2.7重量%、窒素原子の含有量:2.4重量%)
表1に示す処理剤の不揮発分組成になるように、変性シリコーンS-1、混合物p-1、界面活性剤E-1(ポリオキシエチレンが9モル付加された炭素数が12~14のアルキルエーテル)、界面活性剤E-2(ポリオキシエチレンが5モル付加された炭素数が12~14のアルキルエーテル)及び水を混合して水系乳化し、処理剤の不揮発分に占める変性シリコーン(A)の重量割合が83.3重量%、酸性リン酸エステル(B)の重量割合が1.1重量%、界面活性剤の重量割合が15.6重量%である処理剤エマルジョン(プレカーサー処理剤)を調製した。なお、処理剤の不揮発分濃度は3.0重量%とした。
この処理剤エマルジョンを97モル%のアクリロニトリルと3モル%のイタコン酸を共重合させて得られるプレカーサーの原料アクリル繊維に、付与率1.0%となるように付着し、延伸工程(スチーム延伸、延伸倍率2.1倍)を経てプレカーサーを作製した(単繊維繊度0.8dtex,24,000フィラメント)。このプレカーサーを250℃の耐炎化炉にて60分間耐炎化処理し次いで窒素雰囲気下300~1400℃の温度勾配を有する炭素化炉で焼成して炭素繊維に転換した。各特性値の評価結果を表1に示す。
実施例1において、表1~4に示す処理剤の不揮発分組成になるように処理剤エマルションを調製した以外は実施例1と同様にして、処理剤付着後のプレカーサーおよび炭素繊維を得た。各特性値の評価結果を表1~4に示す。




酸化防止剤:テトラキス[メチレン-3-(3‘,5’-ジ-t-ブチル-4‘-ヒドロキシフェニル)プロピオネート]メタン
酸化防止剤を用いた比較例7、8においては、250℃×60minでの減量率が大きい。つまり、耐炎化処理工程におけるシリコーン成分の分解を引き起こすことを意味する。そのため、これらの比較例では、炭素繊維の融着防止性が悪い。また、溶液安定性も悪く、炭素繊維強度も低くなっている。
リン酸エステル塩を用いた比較例2、3、5においては、溶液安定性が悪く、炭素繊維強度も低くなっている。酸性リン酸エステル(B)以外の酸性リン酸エステルを用いた比較例4、6においては、溶液安定性が悪かったり、炭素繊維強度が低下したりする。
一方、実施例においては、いずれの評価項目に於いてもこれら比較例よりも優れ、またより高い炭素繊維強度の炭素繊維を得ることができた。
Claims (10)
- 窒素原子を含む変性基を持つ変性シリコーンと、下記一般式(1)で示される酸性リン酸エステルとを必須に含有する、炭素繊維製造用アクリル繊維処理剤。
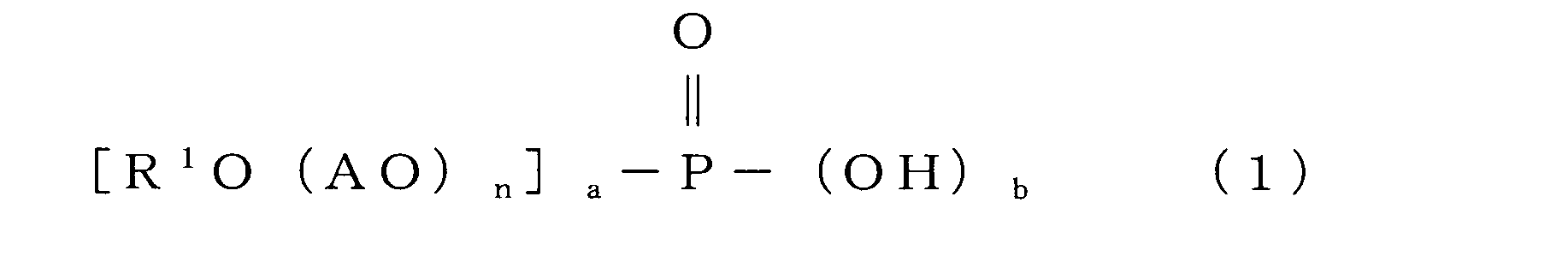
- 前記変性シリコーンと前記酸性リン酸エステルとの重量割合(変性シリコーン/酸性リン酸エステル)が99.6/0.4~90/10である、請求項1に記載の処理剤。
- 前記酸性リン酸エステルの酸価が15~500mgKOH/gである、請求項1又は2に記載の処理剤。
- 前記変性シリコーンにおける窒素原子の含有量が0.35~3.2重量%である、請求項1~3のいずれかに記載の処理剤。
- 前記変性シリコーンがアミノ変性シリコーンである、請求項1~4のいずれかに記載の処理剤。
- 処理剤の不揮発分に占める前記変性シリコーンの重量割合が50~95重量%である、請求項1~5のいずれかに記載の処理剤。
- さらに非イオン性界面活性剤を含有し、処理剤の不揮発分に占める該界面活性剤の重量割合が1~40重量%である、請求項1~6のいずれかに記載の処理剤。
- 水中に分散したエマルジョンとなっている、請求項1~7のいずれかに記載の処理剤。
- 炭素繊維製造用アクリル繊維の原料アクリル繊維に、請求項1~7のいずれかに記載の処理剤を付着させて製糸した、炭素繊維製造用アクリル繊維。
- 炭素繊維製造用アクリル繊維の原料アクリル繊維に請求項1~8のいずれかに記載の処理剤を付着させて、炭素繊維製造用アクリル繊維を製造する製糸工程と、その製糸工程で製造された炭素繊維製造用アクリル繊維を200~300℃の酸化性雰囲気中で耐炎化繊維に転換する耐炎化処理工程と、前記耐炎化繊維をさらに300~2000℃の不活性雰囲気中で炭化させる炭素化処理工程とを含む、炭素繊維の製造方法。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13755204.8A EP2821544B1 (en) | 2012-03-02 | 2013-02-14 | Acrylic-fiber finish for carbon-fiber production, acrylic fiber for carbon-fiber production, and carbon-fiber production method |
| US14/375,346 US9200384B2 (en) | 2012-03-02 | 2013-02-14 | Acrylic-fiber finish for carbon-fiber production, acrylic fiber for carbon-fiber production, and carbon-fiber production method |
| CN201380012043.5A CN104160085B (zh) | 2012-03-02 | 2013-02-14 | 碳纤维制造用丙烯酸纤维处理剂、碳纤维制造用丙烯酸纤维及碳纤维的制造方法 |
| JP2013521687A JP5309280B1 (ja) | 2012-03-02 | 2013-02-14 | 炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 |
| KR1020147027846A KR101930262B1 (ko) | 2012-03-02 | 2013-02-14 | 탄소섬유 제조용 아크릴섬유 처리제, 탄소섬유 제조용 아크릴섬유 및 탄소섬유의 제조방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-046549 | 2012-03-02 | ||
| JP2012046549 | 2012-03-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013129115A1 true WO2013129115A1 (ja) | 2013-09-06 |
Family
ID=49082317
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/053444 WO2013129115A1 (ja) | 2012-03-02 | 2013-02-14 | 炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9200384B2 (ja) |
| EP (1) | EP2821544B1 (ja) |
| JP (1) | JP5309280B1 (ja) |
| KR (1) | KR101930262B1 (ja) |
| CN (1) | CN104160085B (ja) |
| WO (1) | WO2013129115A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6951813B1 (ja) * | 2021-04-05 | 2021-10-20 | 竹本油脂株式会社 | 合成繊維用処理剤及び合成繊維 |
| JP6984930B1 (ja) * | 2021-06-23 | 2021-12-22 | 竹本油脂株式会社 | 炭素繊維前駆体用処理剤及び炭素繊維前駆体 |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106319970B (zh) * | 2015-06-30 | 2020-06-19 | 松本油脂制药株式会社 | 合成纤维用处理剂及其用途 |
| JP6397601B2 (ja) * | 2016-06-30 | 2018-09-26 | 松本油脂製薬株式会社 | 繊維処理剤及びその利用 |
| KR102405528B1 (ko) * | 2017-03-09 | 2022-06-03 | 마쓰모토유시세이야쿠 가부시키가이샤 | 아크릴 섬유 처리제 및 그 용도 |
| US11466400B2 (en) | 2018-06-19 | 2022-10-11 | Hexcel Corporation | Finish composition |
| CN110195272B (zh) * | 2019-05-21 | 2021-06-08 | 湖南东映碳材料科技有限公司 | 一种中间相沥青纤维上油的方法 |
| JP6632016B1 (ja) * | 2019-07-05 | 2020-01-15 | 竹本油脂株式会社 | 炭素繊維前駆体用処理剤、及び炭素繊維前駆体 |
| JP6798734B1 (ja) * | 2020-02-12 | 2020-12-09 | 竹本油脂株式会社 | 炭素繊維前駆体用処理剤、炭素繊維前駆体、及び耐炎化繊維の製造方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5224136B2 (ja) * | 1975-04-04 | 1977-06-29 | ||
| JPS60181322A (ja) | 1984-02-22 | 1985-09-17 | Mitsubishi Rayon Co Ltd | 炭素繊維の製造方法 |
| JPH0291224A (ja) | 1988-09-27 | 1990-03-30 | Toray Ind Inc | 炭素繊維用原糸の製法 |
| JPH1112853A (ja) | 1997-06-20 | 1999-01-19 | Shin Etsu Chem Co Ltd | 炭素繊維前駆体用油剤 |
| JP2001172879A (ja) | 1999-10-08 | 2001-06-26 | Sanyo Chem Ind Ltd | 炭素繊維製造工程用油剤 |
| JP4616934B1 (ja) | 2009-06-04 | 2011-01-19 | 松本油脂製薬株式会社 | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6047382B2 (ja) * | 1982-05-26 | 1985-10-21 | 東レ株式会社 | 炭素繊維製造用原糸油剤 |
| US4938832A (en) * | 1989-05-30 | 1990-07-03 | Hercules Incorporated | Cardable hydrophobic polypropylene fiber, material and method for preparation thereof |
| JP2003201346A (ja) | 2001-12-28 | 2003-07-18 | Mitsubishi Rayon Co Ltd | シリコン系油剤及び炭素繊維前駆体アクリル繊維並びに炭素繊維の製造法 |
| KR101324045B1 (ko) | 2005-12-09 | 2013-11-01 | 마쓰모토유시세이야쿠 가부시키가이샤 | 탄소섬유 제조용 아크릴섬유 유제 및 그것을 사용한탄소섬유의 제조방법 |
| CN101849063B (zh) * | 2007-11-07 | 2012-10-10 | 三菱丽阳株式会社 | 碳纤维前体丙烯腈纤维用油剂组合物、碳纤维前体丙烯腈纤维束及其制造方法 |
| JP5437649B2 (ja) * | 2009-01-30 | 2014-03-12 | 松本油脂製薬株式会社 | 炭素繊維製造用アクリル繊維油剤およびそれを用いた炭素繊維の製造方法 |
| JP5592676B2 (ja) | 2010-03-11 | 2014-09-17 | 松本油脂製薬株式会社 | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 |
| CN101858037B (zh) * | 2010-06-18 | 2011-11-23 | 济南大学 | 一种乳液型碳纤维上浆剂及其制备方法和应用 |
-
2013
- 2013-02-14 US US14/375,346 patent/US9200384B2/en active Active
- 2013-02-14 JP JP2013521687A patent/JP5309280B1/ja active Active
- 2013-02-14 KR KR1020147027846A patent/KR101930262B1/ko active IP Right Grant
- 2013-02-14 EP EP13755204.8A patent/EP2821544B1/en active Active
- 2013-02-14 WO PCT/JP2013/053444 patent/WO2013129115A1/ja active Application Filing
- 2013-02-14 CN CN201380012043.5A patent/CN104160085B/zh active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5224136B2 (ja) * | 1975-04-04 | 1977-06-29 | ||
| JPS60181322A (ja) | 1984-02-22 | 1985-09-17 | Mitsubishi Rayon Co Ltd | 炭素繊維の製造方法 |
| JPH0291224A (ja) | 1988-09-27 | 1990-03-30 | Toray Ind Inc | 炭素繊維用原糸の製法 |
| JPH1112853A (ja) | 1997-06-20 | 1999-01-19 | Shin Etsu Chem Co Ltd | 炭素繊維前駆体用油剤 |
| JP2001172879A (ja) | 1999-10-08 | 2001-06-26 | Sanyo Chem Ind Ltd | 炭素繊維製造工程用油剤 |
| JP4616934B1 (ja) | 2009-06-04 | 2011-01-19 | 松本油脂製薬株式会社 | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2821544A4 |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6951813B1 (ja) * | 2021-04-05 | 2021-10-20 | 竹本油脂株式会社 | 合成繊維用処理剤及び合成繊維 |
| WO2022215571A1 (ja) * | 2021-04-05 | 2022-10-13 | 竹本油脂株式会社 | 合成繊維用処理剤及び合成繊維 |
| JP2022159712A (ja) * | 2021-04-05 | 2022-10-18 | 竹本油脂株式会社 | 合成繊維用処理剤及び合成繊維 |
| JP6984930B1 (ja) * | 2021-06-23 | 2021-12-22 | 竹本油脂株式会社 | 炭素繊維前駆体用処理剤及び炭素繊維前駆体 |
| JP2023003046A (ja) * | 2021-06-23 | 2023-01-11 | 竹本油脂株式会社 | 炭素繊維前駆体用処理剤及び炭素繊維前駆体 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20150044125A1 (en) | 2015-02-12 |
| CN104160085B (zh) | 2016-04-06 |
| EP2821544A4 (en) | 2015-10-21 |
| KR20140131997A (ko) | 2014-11-14 |
| EP2821544B1 (en) | 2018-05-16 |
| US9200384B2 (en) | 2015-12-01 |
| KR101930262B1 (ko) | 2018-12-18 |
| JP5309280B1 (ja) | 2013-10-09 |
| JPWO2013129115A1 (ja) | 2015-07-30 |
| CN104160085A (zh) | 2014-11-19 |
| EP2821544A1 (en) | 2015-01-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5309280B1 (ja) | 炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 | |
| JP4616934B1 (ja) | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 | |
| KR101324045B1 (ko) | 탄소섬유 제조용 아크릴섬유 유제 및 그것을 사용한탄소섬유의 제조방법 | |
| WO2017169632A1 (ja) | アクリル繊維処理剤及びその用途 | |
| JP5914780B1 (ja) | アクリル繊維処理剤及びその用途 | |
| KR102405528B1 (ko) | 아크릴 섬유 처리제 및 그 용도 | |
| JP6397601B2 (ja) | 繊維処理剤及びその利用 | |
| JP5528649B1 (ja) | 炭素繊維製造用アクリル繊維処理剤及びその用途 | |
| JP6190673B2 (ja) | 炭素繊維製造用アクリル繊維処理剤及びその用途 | |
| JP5277005B2 (ja) | 炭素繊維製造用アクリル繊維油剤およびそれを用いた炭素繊維の製造方法 | |
| JP2012102429A (ja) | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 | |
| JP5592676B2 (ja) | 炭素繊維製造用アクリル繊維油剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 | |
| JP2018021263A (ja) | アクリル繊維処理剤及びその用途 | |
| JP2013159868A (ja) | 炭素繊維製造用アクリル繊維処理剤、炭素繊維製造用アクリル繊維および炭素繊維の製造方法 | |
| JP6204211B2 (ja) | アクリル繊維処理剤及びその用途 | |
| JP4217748B6 (ja) | 炭素繊維製造用アクリル繊維油剤およびそれを用いた炭素繊維の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201380012043.5 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2013521687 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13755204 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14375346 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013755204 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20147027846 Country of ref document: KR Kind code of ref document: A |