WO2013115010A1 - 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ - Google Patents
分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ Download PDFInfo
- Publication number
- WO2013115010A1 WO2013115010A1 PCT/JP2013/051186 JP2013051186W WO2013115010A1 WO 2013115010 A1 WO2013115010 A1 WO 2013115010A1 JP 2013051186 W JP2013051186 W JP 2013051186W WO 2013115010 A1 WO2013115010 A1 WO 2013115010A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- conjugated diene
- branched conjugated
- rubber composition
- weight
- copolymer
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F236/04—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F236/06—Butadiene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B60—VEHICLES IN GENERAL
- B60C—VEHICLE TYRES; TYRE INFLATION; TYRE CHANGING; CONNECTING VALVES TO INFLATABLE ELASTIC BODIES IN GENERAL; DEVICES OR ARRANGEMENTS RELATED TO TYRES
- B60C1/00—Tyres characterised by the chemical composition or the physical arrangement or mixture of the composition
- B60C1/0016—Compositions of the tread
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F236/04—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F236/10—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated with vinyl-aromatic monomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/22—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having three or more carbon-to-carbon double bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L9/00—Compositions of homopolymers or copolymers of conjugated diene hydrocarbons
- C08L9/06—Copolymers with styrene
Definitions
- the present invention relates to a branched conjugated diene copolymer, a rubber composition comprising the copolymer, and a pneumatic tire produced using the rubber composition.
- High performance tires such as racing tires used in races are mainly required to have high levels of wear resistance and grip performance.
- a high molecular weight polymer for example, having a molecular weight of 250,000 or more, 500,000 or more, 1,000,000 or more
- carbon black as a filler
- blending to is known.
- rubbers having a high glass transition temperature (Tg) for example, those having a Tg of -20 ° C. or more
- carbon black having a large surface area for example, nitrogen adsorption specific surface area
- Myrcene is a naturally occurring organic compound which is a kind of olefin belonging to monoterpenes. Myrcene has two isomers, ⁇ -myrcene (2-methyl-6-methyleneocta-1,7-diene) and ⁇ -myrcene (7-methyl-3-methyleneocta-1,6-diene) Exists. Patent Document 1 discloses a myrcene polymer.
- Farnesene is one of isoprenoid compounds chemically synthesized by oligomerization of isoprene and dehydration reaction of nerolidol, and is mainly used as a fragrance or its raw material (Patent Document 2).
- the present invention relates to a novel branched conjugated diene copolymer useful for improving processability as a rubber component of a rubber composition for a tire, a rubber composition for a tire comprising the branched conjugated diene copolymer, particularly, A rubber composition for a tire, which improves both the abrasion resistance and the grip performance to high levels and exhibits excellent properties in processability, and a pneumatic tire manufactured using the rubber composition for a tire It is intended to be provided.
- the present invention relates to the general formula (1) (Wherein, R 1 represents an aliphatic hydrocarbon having 6 to 11 carbon atoms). And a branched conjugated diene compound (1) represented by the general formula (2) (Wherein, R 2 and R 3 are the same or different and each represents a hydrogen atom, an aliphatic hydrocarbon group having 1 to 3 carbon atoms, or a halogen atom).
- a conjugated diene compound represented by the general formula (3) (Wherein R 4 represents a hydrogen atom, an aliphatic hydrocarbon group having 1 to 3 carbon atoms, an alicyclic hydrocarbon group having 3 to 8 carbon atoms, or an aromatic hydrocarbon group having 6 to 10 carbon atoms) .
- a branched conjugated diene copolymer obtained by copolymerizing with a vinyl compound represented by Copolymerization ratio (l) of branched conjugated diene compound (1) is 1 to 99% by weight
- copolymerization ratio (m) of conjugated diene compound (2) is less than 99% by weight
- copolymerization ratio of vinyl compound (3) It relates to a branched conjugated diene copolymer in which (n) is less than 99% by weight.
- the copolymerization ratio (l) of the branched conjugated diene compound (1) is less than 2.5 to 75 wt%
- the copolymerization ratio (m) of the conjugated diene compound (2) is 72.5 wt% or less
- the vinyl compound (3) The copolymerization ratio (n) of (a) is preferably 25 to less than 97.5% by weight.
- the present invention has low Mooney viscosity ML 1 + 4 (130 ° C.) in comparison with a copolymer having the same weight average molecular weight in which branched conjugated diene compound (1) is replaced with conjugated diene compound (2).
- the present invention relates to the above-mentioned branched conjugated diene copolymer for improving processability, which is one of the above.
- the branched conjugated diene compound (1) is preferably myrcene and / or farnesene.
- the conjugated diene compound (2) is preferably 1,3-butadiene and / or isoprene.
- the vinyl compound (3) is preferably one or more selected from the group consisting of styrene, ⁇ -methylstyrene, ⁇ -vinylnaphthalene and ⁇ -vinylnaphthalene.
- the present invention also relates to a rubber composition comprising the branched conjugated diene copolymer.
- the present invention relates to a pneumatic tire produced using the above rubber composition.
- a novel branched conjugated diene copolymer useful for improving processability can be provided as a rubber component for a tire, and the use of the branched conjugated diene copolymer makes it possible to prevent abrasion. Both the wear resistance and the grip performance can be improved to high levels, and a tire rubber composition excellent in processability can be provided.
- Such a rubber composition for a tire according to the present invention is particularly useful as a rubber composition for a tire tread and as a rubber composition for tires for racing tires used in a race.
- a polymer having a large weight average molecular weight (Mw) (Mw is, for example, 250,000 or more, 500,000 or more, or 1,000,000 or more
- Mw is, for example, 250,000 or more, 500,000 or more, or 1,000,000 or more
- the branched conjugated diene compound (1) is blended as a component of the polymer
- the blending of the branched conjugated diene compound (1) may also have a feature of having little influence on the glass transition temperature (Tg) of the polymer.
- the branched conjugated diene copolymer of the present invention refers to a copolymer obtained by copolymerizing the branched conjugated diene compound (1), the conjugated diene compound (2) and the vinyl compound (3).
- the weight average molecular weight (Mw) of the branched conjugated diene copolymer of the present invention is not particularly limited as long as it is 3,000 or more, but preferably 250,000 or more, more preferably 500,000 or more, and still more preferably 1,000,000 or more. is there. If the Mw is less than 3,000, the liquid polymer tends to be a highly fluid liquid polymer, and if the Mw is less than 250,000, the hardness of the rubber composition tends to decrease and the processability does not deteriorate. On the other hand, Mw is not particularly limited as long as it is 3,000,000 or less, preferably 2,000,000 or less. If the Mw exceeds 3,000,000, it tends to be a solid which does not have rubber elasticity.
- the number average molecular weight (Mn) of the branched conjugated diene copolymer is preferably 3,000 or more, more preferably 250,000 or more. If it is less than 3000, the hardness of the rubber composition is increased and the processability tends not to be deteriorated. On the other hand, Mn is preferably 3,000,000 or less, more preferably 2,000,000 or less. If Mn exceeds 3,000,000, it tends to be a solid which does not have rubber elasticity.
- the ratio of Mw to the number average molecular weight (Mn), that is, the preferable range of Mw / Mn is 20.0 or less, more preferably 10.0 or less.
- Mw / Mn is more than 20.0, there is a tendency that the problem of deterioration in processability due to the decrease in hardness of the rubber composition does not occur.
- Mw / Mn there is no restriction
- the glass transition temperature (Tg) of the branched conjugated diene copolymer is usually in the range of -110 ° C to 110 ° C.
- Tg glass transition temperature
- the Tg of the branched conjugated diene copolymer contained in a relatively large amount tends to increase as the content of styrene is increased.
- the branched conjugated diene copolymer containing a large amount of high cis-butadiene and the branched conjugated diene copolymer containing a large amount of styrene contain a small amount of the branched conjugated diene compound (1) Although copolymerization only improves the processability, in most cases the Tg does not change much depending on the composition of the branched conjugated diene compound (1).
- the Mooney viscosity ML 1 + 4 (130 ° C.) of the branched conjugated diene copolymer is a copolymer of the same molecular weight obtained by replacing the branched conjugated diene compound (1) constituting the copolymer with the conjugated diene compound (2) Since the effect of the present invention to improve processability can be exhibited as long as it is low in comparison with the polymer, it is not particularly limited, but in general, it is preferably 25 or more, more preferably 30 or more. is there. If the Mooney viscosity is less than 25, it tends to be fluid.
- the Mooney viscosity is preferably 160 or less, more preferably 150 or less, still more preferably 100 or less, and still more preferably 60 or less.
- the Mooney viscosity is more than 160, many softening agents and processing aids tend to be required when processing.
- the copolymerization ratio of the branched conjugated diene compound (1), which is a monomer, the conjugated diene compound (2) and the vinyl compound (3) in the branched conjugated diene copolymer will be described.
- the copolymerization ratio (l) of the branched conjugated diene compound (1) is not particularly limited as long as it is 1 to 99% by weight, but the lower limit is preferably 2.5% by weight or more, and 5% by weight or more More preferable. If it is less than 1%, the effect of the branched conjugated diene compound (1) combination to improve processability tends not to be sufficiently obtained.
- the upper limit value is preferably less than 75% by weight, more preferably less than 60% by weight, still more preferably less than 50% by weight, and still more preferably less than 15% by weight. If it exceeds 99% by weight, it tends to be a fluid polymer, and the effect on the processability of the branched conjugated diene compound (1) tends to be sufficiently exhibited if it is blended at 15% by weight.
- the branched conjugated diene copolymer of the present invention includes both the conjugated diene compound (2) and the vinyl compound (3).
- the lower limit value of the preferable range of the copolymerization ratio (m) of the conjugated diene compound (2) is 1% by weight or more, more preferably 50% by weight or more. If m is less than 1% by weight, it tends to be a flowable polymer.
- the upper limit is less than 99% by weight, more preferably less than 80% by weight, still more preferably less than 72.5% by weight, still more preferably less than 55% by weight. If m is 99% by weight or more, the effect of copolymerizing the branched conjugated diene compound (1) tends to be small to improve processability.
- the lower limit of the preferable range of the copolymerization ratio (n) of the vinyl compound (3) is 10% by weight or more, more preferably 25% by weight or more, and still more preferably 40% by weight or more. If n is less than 10% by weight, the hardness of the rubber is not so high that the processability becomes a problem, and the effect of copolymerizing the branched conjugated diene compound (1) tends to be small for the processability improvement, In addition, when n is 25% by weight or more, the problem arises that the processability of the copolymer is deteriorated while contributing to the improvement of the grip performance of the rubber, so the branched conjugated diene compound (1) It is preferable because the effect of improving the processability by blending it tends to be prominent, and the tendency is more strong when n is 40% by weight or more.
- the upper limit is less than 99% by weight, preferably less than 97.5% by weight, more preferably less than 95% by weight, still more preferably less than 80% by weight, more preferably less than 60% by weight. If n is 99% by weight or more, the copolymer does not become rubbery and resinous, and the effect of copolymerizing the branched conjugated diene compound (1) tends to be small.
- the polymerization ratio l, m, n of the branched conjugated diene compound (1), the conjugated diene compound (2) and the vinyl compound (3) is 100% by weight in total. From this, if any one lower limit value is selected from the above-mentioned preferable range, the possible range of the upper limit value is naturally determined for the other two. In addition, if any two lower limit values are selected from the above preferable range, the upper limit value is naturally determined for the remaining one. Similarly, as for the polymerization ratios l, m and n, as long as any one upper limit is selected from the above preferable range, the possible range of the lower limit is naturally determined for the other two. In addition, if any two upper limit values are selected from the above preferable range, the lower limit value is naturally determined for the remaining one.
- branched conjugated diene compound (1) examples of the aliphatic hydrocarbon group having 6 to 11 carbon atoms include those having a normal structure such as hexyl group, heptyl group, octyl group, nonyl group, decyl group and undecyl group, These isomers and / or unsaturated ones, and their derivatives (for example, halides and hydroxylated compounds etc.) can be mentioned. Among them, 4-methyl-3-pentenyl group, 4,8-dimethyl-nona-3,7-dienyl group and the like, and derivatives thereof are particularly preferable.
- branched conjugated diene compound (1) examples include myrcene, farnesene and the like.
- myrcene includes both ⁇ -myrcene (2-methyl-6-methyleneocta-1,7-diene) and ⁇ -myrcene, and among them, ⁇ having the following structure -Myrcene (7-methyl-3-methyleneocta-1,6-diene) is preferred.
- farnesene includes any isomer such as ⁇ -farnesene ((3E, 7E) -3,7,11-trimethyl-1,3,6,10-dodecatetraene) and ⁇ -farnesene.
- (E) - ⁇ -farnesene (7,11-dimethyl-3-methylene-1,6,10-dodecatriene) having the following structure is preferable.
- branched conjugated diene compound (1) one or more may be used.
- examples of the aliphatic hydrocarbon group having 1 to 3 carbon atoms include a methyl group, an ethyl group, an n-propyl group, an isopropyl group and the like, among which a methyl group is preferable.
- a halogen atom a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom are mentioned, Among these, a chlorine atom is preferable.
- Each of R 2 and R 3 in the conjugated diene compound (2) is independently preferably a hydrogen atom, a methyl group, an ethyl group, an n-propyl group or an isopropyl group, and more preferably a hydrogen atom or a methyl group.
- the conjugated diene compound (2) for example, 1,3-butadiene, isoprene, 2,3-dimethyl-1,3-butadiene and the like are preferable, and among these, 1,3-butadiene, isoprene and the like are preferable. .
- conjugated diene compound (2) one or more kinds can be used.
- examples of the aliphatic hydrocarbon group having 1 to 3 carbon atoms include a methyl group, an ethyl group, an n-propyl group, an isopropyl group and the like, among which a methyl group is preferable.
- Examples of the alicyclic hydrocarbon group having 3 to 8 carbon atoms include cyclopropyl group, cyclobutyl group, cyclopentyl group, cyclohexyl group, cycloheptyl group, cyclooctyl group, cyclopropenyl group, cyclobutenyl group, cyclopentenyl group, A cyclohexenyl group, a cycloheptenyl group, a cyclooctenyl group etc. are mentioned, Among these, a cyclopropyl group and a cyclobutyl group are preferable.
- Examples of the aromatic hydrocarbon group having 6 to 10 carbon atoms include phenyl group, benzyl group, phenethyl group, tolyl group, xylyl group and naphthyl group.
- the substitution position of the methyl group on the benzene ring in the tolyl group includes any position of ortho-, meta- or para-, and the substitution position of the methyl group in the xylyl group is any of arbitrary substitution positions. Is also included.
- a phenyl group, a tolyl group and a naphthyl group are preferable.
- Specific examples of the vinyl compound (3) are preferably styrene, ⁇ -methylstyrene, ⁇ -vinylnaphthalene and ⁇ -vinylnaphthalene.
- vinyl compound (3) 1 type, or 2 or more types of things can be used.
- the branched conjugated diene copolymer of the present invention can be obtained by copolymerizing the branched conjugated diene compound (1), the conjugated diene compound (2) and the vinyl compound (3).
- Such copolymerization is not particularly limited in the order of copolymerization.
- all the monomers may be randomly copolymerized at one time, or a specific monomer (for example, branched conjugated diene compound (1) monomer) may be preliminarily After copolymerizing only the conjugated diene compound (2) monomer only, the vinyl compound (3) monomer only, or any two kinds of monomers selected from these, etc., and then adding the remaining monomers and copolymerizing Alternatively, it is possible to perform block copolymerization by previously copolymerizing each specific monomer.
- a specific monomer for example, branched conjugated diene compound (1) monomer
- Such copolymerization can be carried out by any conventional method, and can be carried out by, for example, anionic polymerization reaction, coordination polymerization reaction and the like.
- the polymerization method is not particularly limited, and any of solution polymerization method, emulsion polymerization method, gas phase polymerization method and bulk polymerization method can be used, but among these, solution polymerization method is preferable.
- the polymerization type may be either batch type or continuous type.
- the anionic polymerization can be carried out in a suitable solvent in the presence of an anionic polymerization initiator.
- an anionic polymerization initiator any conventional one can be suitably used, and as such an anionic polymerization initiator, for example, a compound represented by the general formula RLix (wherein R represents one or more carbon atoms) And an organolithium compound having an aliphatic, aromatic or alicyclic group, and x is an integer of 1 to 20).
- Suitable organolithium compounds include methyllithium, ethyllithium, n-butyllithium, sec-butyllithium, tert-butyllithium, phenyllithium and naphthyllithium.
- Preferred organolithium compounds are n-butyllithium and sec-butyllithium.
- An anionic polymerization initiator can be used individually or in mixture of 2 or more types.
- the amount of the polymerization initiator to be used in the anionic polymerization is not particularly limited. Is more preferred. If the amount of the polymerization initiator used is less than 0.05 mmol, the copolymer tends to be resin-like instead of rubbery, and if it is more than 35 mmol, the copolymer is soft and branched conjugated diene for processability. The effect of copolymerizing the compound (1) tends to be small.
- a solvent used for anion polymerization as long as it does not inactivate an anion polymerization initiator and does not stop a polymerization reaction, all can be used suitably, and any of a polar solvent or a nonpolar solvent can be used. It can be used.
- polar solvents include ether solvents such as tetrahydrofuran
- nonpolar solvents include, for example, chain hydrocarbons such as hexane, heptane, octane and pentane, cyclic hydrocarbons such as cyclohexane, benzene, toluene And aromatic hydrocarbons such as xylene. These solvents may be used alone or in combination of two or more.
- the anionic polymerization is preferably further carried out in the presence of a polar compound.
- polar compounds include dimethyl ether, diethyl ether, ethyl methyl ether, ethyl propyl ether, tetrahydrofuran, dioxane, diphenyl ether, tripropylamine, tributylamine, trimethylamine, triethylamine, N, N, N ', N'-tetramethylethylenediamine (TMEDA) and the like.
- TEDA trimethylamine
- the polar compounds can be used alone or in combination of two or more. This polar compound is useful for reducing the content of 1,2-structure with respect to control of the microstructure of the butadiene moiety.
- the amount of the polar compound used varies depending on the type of the polar compound and the polymerization conditions, but is preferably 0.1 or more as a molar ratio with the anionic polymerization initiator (polar compound / anionic polymerization initiator). If the molar ratio to the anionic polymerization initiator (polar compound / anionic polymerization initiator) is less than 0.1, the effect of the polar substance on controlling the microstructure tends to be insufficient.
- the reaction temperature in the anionic polymerization is not particularly limited as long as the reaction proceeds suitably, but it is preferably -10 ° C to 100 ° C, and more preferably 25 ° C to 70 ° C.
- the reaction time varies depending on the preparation amount, reaction temperature and other conditions, but usually, for example, about 3 hours is sufficient.
- the anionic polymerization can be terminated by the addition of reaction terminators commonly used in this field.
- a reaction terminator for example, polar solvents having active protons such as alcohols such as methanol, ethanol and isopropanol or acetic acid, and mixtures thereof, or polar solvents thereof and nonpolar solvents such as hexane and cyclohexane A mixed solution is mentioned.
- the addition amount of the reaction terminator is usually sufficient in the same molar amount or about 2 times the molar amount with respect to the anionic polymerization initiator.
- the branched conjugated diene copolymer is prepared by removing the solvent from the polymerization solution by a conventional method, or pouring the polymerization solution into one or more times the amount of alcohol to obtain a branched conjugated diene copolymer. It can be easily isolated by precipitation.
- the coordination polymerization can be carried out by using a coordination polymerization initiator in place of the anionic polymerization initiator in the above-mentioned anionic polymerization.
- a coordination polymerization initiator any conventional one can be suitably used, and as such a coordination polymerization initiator, for example, transition metal-containing compounds such as lanthanoid compounds, titanium compounds, cobalt compounds, nickel compounds and the like
- the catalyst which is a compound is mentioned.
- an aluminum compound or a boron compound can be further used as a cocatalyst.
- the lanthanoid compound is not particularly limited as long as it contains any of the elements with atomic numbers 57 to 71 (lanthanoid), and among these lanthanoids, neodymium is particularly preferable.
- lanthanoid compounds include carboxylates of these elements, ⁇ -diketone complexes, alkoxides, phosphates or phosphites, and halides. Among these, carboxylates, alkoxides and ⁇ -diketone complexes are preferred in terms of ease of handling.
- titanium compound for example, a cyclopentadienyl group, an indenyl group, a substituted cyclopentadienyl group or a substituted indenyl group containing one, and a substituent selected from a halogen, an alkoxysil group and an alkyl group is selected.
- titanium-containing compounds are preferred, but from the viewpoint of catalytic performance, compounds having one alkoxysilyl group are preferred.
- cobalt compounds include cobalt halides, carboxylates, ⁇ -diketone complexes, organic base complexes, organic phosphine complexes and the like.
- nickel compounds include nickel halides, carboxylates, ⁇ -diketone complexes, organic base complexes and the like.
- the catalyst used as a coordination polymerization initiator can be used individually or in combination of 2 or more types.
- the use amount of the catalyst as a polymerization initiator at the time of performing the coordination polymerization is not particularly limited.
- the preferable use amount is the same as the use amount of the catalyst in the case of the anionic polymerization.
- Examples of the aluminum compound used as a cocatalyst include organic aluminoxanes, halogenated organic aluminum compounds, organic aluminum compounds, hydrogenated organic aluminum compounds and the like.
- organic aluminoxanes for example, alkylaluminoxanes (methylaluminoxane, ethylaluminoxane, propylaluminoxane, butylaluminoxane, isobutylaluminoxane, octylaluminoxane, etc.)
- a halogenated organoaluminum compound for example, a halogenated alkyl Aluminum compounds (dimethylaluminium chloride, diethylaluminium chloride, methylaluminium dichloride, ethylaluminium dichloride), and as an organic aluminum compound, for example, an alkylaluminium compound (trimethylaluminium, triethylaluminium, triisopropylaluminium, triisobutyla
- boron compound for example, a compound containing an anionic species such as tetraphenyl borate, tetrakis (pentafluorophenyl) borate, (3,5-bistrifluoromethylphenyl) borate and the like can be mentioned. These cocatalysts may also be used alone or in combination of two or more.
- reaction time and reaction temperature are also the same as those described for the anionic polymerization. Termination of the polymerization reaction and isolation of the branched conjugated diene copolymer can also be carried out in the same manner as in the case of anionic polymerization.
- the weight average molecular weight (Mw) of the branched conjugated diene copolymer can be controlled by a conventional method, for example, controlled by adjusting the amount of the branched conjugated diene compound (1) and other monomers charged during polymerization. be able to.
- the Mw can be increased by increasing the total monomer / anion polymerization catalyst ratio, and the Mw can be decreased by decreasing the ratio.
- the Tg of the branched conjugated diene copolymer can be controlled by a conventional method, for example, controlled by adjusting the feed ratio of the conjugated diene compound (2) to the vinyl compound (3) charged during polymerization. it can.
- the Tg can be increased by increasing the feed ratio of the vinyl compound (3), and the Tg can be decreased by decreasing the feed ratio of the vinyl compound (3).
- the Mooney viscosity of the branched conjugated diene copolymer can be controlled by a conventional method, for example, by controlling the amount of the branched conjugated diene compound (1) monomer charged at the time of polymerization. For example, the Mooney viscosity increases as the charged amount of the branched conjugated diene compound (1) decreases, and the Mooney viscosity decreases as the charged amount of the branched conjugated diene compound (1) increases.
- the branched conjugated diene copolymer of the present invention thus obtained can be made into a rubber composition for tires by appropriately blending other components usually used in the field of rubber industry.
- the compounding amount of the branched conjugated diene copolymer in the rubber component is about 3% by weight or more, preferably about 5% by weight or more, more preferably 10% by weight or more Preferably it is 30 weight% or more. If the blending amount of the branched conjugated diene copolymer is less than 3% by weight, the effect of blending the branched conjugated diene compound (1) copolymerization with the processability tends to be small. On the other hand, the upper limit value of the compounding amount of the branched conjugated diene copolymer is not particularly limited, and may be 100% by weight.
- branched conjugated diene copolymer for example, natural rubber (NR), isoprene rubber (IR), butadiene rubber (BR), styrene butadiene rubber (SBR), styrene isoprene And diene rubbers such as styrene isoprene butadiene rubber (SIBR), ethylene propylene diene rubber (EPDM), chloroprene rubber (CR), acrylonitrile butadiene rubber (NBR), and butyl rubber (IIR).
- NR natural rubber
- IR isoprene rubber
- BR butadiene rubber
- SBR styrene butadiene rubber
- EPDM ethylene propylene diene rubber
- CR chloroprene rubber
- NBR acrylonitrile butadiene rubber
- IIR butyl rubber
- NR is preferable to use NR, BR, and SBR because the grip performance and the abrasion resistance can be obtained in a well-balanced manner in combination with the branched conjugated diene copolymer, and it is more preferable to use NR.
- the NR is not particularly limited, and those common in tire manufacture can be used, and examples thereof include SIR20, RSS # 3, and TSR20.
- fillers As the filler, mention may be made of fillers commonly used in this field such as carbon black and silica.
- N 2 SA nitrogen adsorption specific surface area
- N 2 SA of carbon black is about 270 m 2 / g or less, preferably about 260 m 2 / g or less. If the carbon black N 2 SA is greater than 270, the carbon black dispersion tends to be poor.
- the compounding amount of carbon black is about 1% by weight or more, preferably about 3% by weight or more, with respect to 100 parts by weight of the rubber component. If the amount of carbon black is less than 1% by weight, the abrasion resistance tends to decrease. On the other hand, the blending amount of carbon black is about 200% by weight or less, more preferably 150 or less. When the amount of carbon black exceeds 200% by weight, the processability tends to be deteriorated.
- silica examples include silica (anhydrous silicic acid) prepared by a dry method, silica (hydrous silicic acid) prepared by a wet method, and the like. Among them, silica prepared by a wet method is preferable because the number of silanol groups on the surface is large and the number of reaction points with the silane coupling agent is large.
- the N 2 SA of silica is about 50 m 2 / g or more, preferably about 80 m 2 / g or more. If N 2 SA is less than 50, the reinforcing effect is small and the abrasion resistance tends to be lowered.
- the N 2 SA of silica is about 300 m 2 / g or less, preferably about 250 m 2 / g or less.
- N 2 SA is larger than 300 m 2 / g, the dispersion tends to decrease and the processability tends to decrease.
- the compounding amount of silica is about 1 part by weight or more, preferably about 10 parts by weight or more with respect to 100 parts by weight of the rubber component. If the amount of silica is less than 1 part by weight, abrasion resistance tends to be insufficient. On the other hand, the compounding amount of silica is about 150 parts by weight or less, and more preferably 100 parts by weight or less. When the amount of the silica is more than 150 parts by weight, the dispersibility of the silica tends to deteriorate and the processability tends to deteriorate.
- the rubber composition preferably contains a silane coupling agent.
- silane coupling agent conventionally known silane coupling agents can be used.
- silane coupling agent bis (3-triethoxysilylpropyl) tetrasulfide, bis (2-triethoxysilylethyl) tetrasulfide, bis (4- (4) Triethoxysilylbutyl) tetrasulfide, bis (3-trimethoxysilylpropyl) tetrasulfide, bis (2-trimethoxysilylethyl) tetrasulfide, bis (4-trimethoxysilylbutyl) tetrasulfide, bis (3-triethoxy) Silylpropyl) trisulfide, bis (2-triethoxysilylethyl) trisulfide, bis (4-triethoxysilylbutyl) trisulfide, bis (3-trimethoxysilylpropyl
- silane coupling agents may be used alone or in combination of two or more.
- bis (3-triethoxysilylpropyl) tetrasulfide and bis (3-triethoxysilylpropyl) disulfide are preferably contained in view of good processability.
- the blending amount thereof is preferably 1 part by weight or more, and more preferably 2 parts by weight or more with respect to 100 parts by weight of silica. If the content of the silane coupling agent is less than 1 part by weight, effects such as improvement of dispersibility tend not to be sufficiently obtained. In addition, the content of the silane coupling agent is preferably 20 parts by weight or less, and more preferably 15 parts by weight or less. When the content of the silane coupling agent exceeds 20 parts by weight, a sufficient coupling effect can not be obtained, and the reinforcing property tends to be lowered.
- the rubber composition of the present invention may contain, in addition to the components described above, compounding agents conventionally used in the rubber industry, such as other reinforcing fillers, anti-aging agents, vulcanizing agents such as oils, waxes and sulfur, and vulcanizing agents.
- compounding agents conventionally used in the rubber industry such as other reinforcing fillers, anti-aging agents, vulcanizing agents such as oils, waxes and sulfur, and vulcanizing agents.
- a vulcanization accelerator, a vulcanization acceleration auxiliary and the like can be appropriately blended.
- the rubber composition of the present invention thus obtained can be used as various members of a tire, but since both the wear resistance and the grip performance can be improved to high levels, in particular, the tire tread can be used. In particular, it can be suitably used as a tire tread of a high performance tire such as a competition tire.
- the rubber composition of the present invention is used in the manufacture of a tire and can be made into a tire by a conventional method. That is, a mixture in which the above-mentioned components are appropriately compounded as necessary is kneaded, extruded at the unvulcanized stage according to the shape of each component of the tire, and molded by a usual method on a tire molding machine Thus, an unvulcanized tire is formed.
- the unvulcanized tire is heated and pressurized in a vulcanizer to obtain a tire, which can be filled with air to form a pneumatic tire.
- Mw and Mn are measured using a gel permeation chromatograph (GPC) and converted from standard polystyrene.
- the glass transition temperature (Tg) is measured by differential scanning calorimetry (DSC).
- Mooney viscosity is measured according to JIS K6300.
- the term “1 to 99% by weight” simply includes the values at both ends.
- NR Natural rubber (TSR 20) Copolymer: one synthesized according to the description of the present invention
- Carbon black Show black N220 (nitrogen adsorption specific surface area (N2SA): 125 m 2 / g) manufactured by Cabot Japan Ltd.
- Silica Ultrasil VN3 manufactured by Texsa (nitrogen adsorption specific surface area (N 2 SA): 175 m 2 / g)
- Silane coupling agent Si69 (bis (3-triethoxysilylpropyl) tetrasulfide) manufactured by Tegusa Corporation
- Anti-aging agent Noclac 6C (N-1,3-dimethylbutyl-N'-phenyl-p-phenylenediamine) manufactured by Ouchi Emerging Chemical Industry Co., Ltd.
- Stearic acid stearic acid oil manufactured by NOF Corporation: mineral oil PW-380 manufactured by Idemitsu Kosan Co., Ltd.
- Zinc oxide Zinc oxide No.
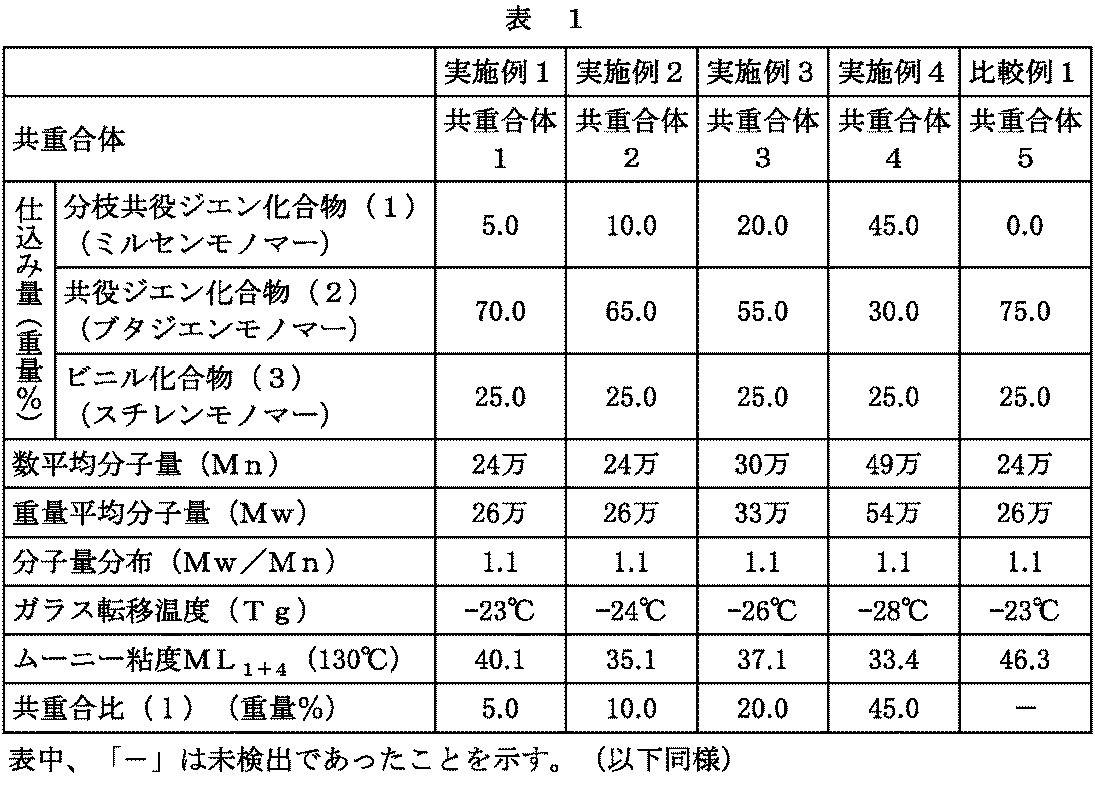
- Example 1 (Synthesis of Copolymer 1) In a dry, nitrogen-substituted 3 L pressure-resistant stainless steel container, 2000 ml of hexane, 10 g of myrcene, 140 g of butadiene, 50 g of styrene and 0.22 mmol of TMEDA are added, and further 1.17 mmol of n-butyllithium (n-BuLi) is added, 50 The polymerization reaction was carried out for 3 hours at ° C. After 3 hours, 1.15 ml of 1 M isopropanol / hexane solution was added dropwise to complete the reaction.
- the obtained polymerization solution was blow-dried to remove the solvent, and then dried under a reduced pressure condition of internal pressure 0.1 kPa or less / temperature 50 ° C. until constant weight was reached, to obtain 200 g (dry weight) of copolymer 1.
- the polymerization conversion (percent of "dry weight / charge") was approximately 100%.
- (2) (Production of Unvulcanized Rubber Composition 1) According to the composition described in Table 2, the copolymer 1 obtained above and various chemicals for producing the above rubber composition (excluding insoluble sulfur and vulcanization accelerator) are mixed for 5 minutes at 150 ° C. with a Banbury mixer It was refined and a kneaded product was obtained.
- Example 2 (1) (Synthesis of Copolymer 2) The same treatment as in Example 1 (1) was carried out except that 20 g of myrcene and 130 g of butadiene were used, to obtain 200 g of a copolymer 2. The polymerization conversion was about 100%.
- Example 3 (Synthesis of Copolymer 3) The same treatment as in Example 1 (1) was carried out except that 40 g of myrcene and 110 g of butadiene were used, to obtain 200 g of a copolymer 3. The polymerization conversion was about 100%.
- (2) (Production of Unvulcanized Rubber Composition 3) An unvulcanized rubber composition 3 was obtained in the same manner as in Example 1 (2) except that the copolymer 3 was used instead of the copolymer 1.
- (3) (Production of Vulcanized Rubber Composition 3) The unvulcanized rubber composition obtained in the above (2) was treated in the same manner as in Example 1 (3) to obtain a vulcanized rubber composition 3.
- Example 4 (Synthesis of copolymer 4) The same treatment as in Example 1 (1) was carried out except that 90 g of myrcene and 60 g of butadiene were used, to obtain 200 g of a copolymer 4. The polymerization conversion was about 100%.
- (2) (Production of Unvulcanized Rubber Composition 4) An unvulcanized rubber composition 4 was obtained in the same manner as in Example 1 (2) except that the copolymer 4 was used instead of the copolymer 1.
- (3) (Production of Vulcanized Rubber Composition 4) The unvulcanized rubber composition obtained in the above (2) was treated in the same manner as in Example 1 (3) to obtain a vulcanized rubber composition 4.
- Comparative Example 1 (1) Synthesis of Copolymer 5 The same treatment as in Example 1 (1) was carried out except that 0 g of myrcene and 150 g of butadiene were used, to obtain 200 g of a copolymer 5. The polymerization conversion was about 100%.
- (3) Production of Vulcanized Rubber Composition 5) The unvulcanized rubber composition obtained in the above (2) was treated in the same manner as in Example 1 (3) to obtain a vulcanized rubber composition 5.
- Tg glass transition temperature
- the copolymerization ratio (l) (% by weight) was measured by a conventional method using pyrolysis gas chromatography (PGC). That is, a calibration curve is prepared for the purified branched conjugated diene compound (1), and the branched conjugated diene in the copolymer is obtained from the area ratio of the thermal decomposition product derived from the branched conjugated diene compound (1) obtained by PGC The weight% of the compound (1) was calculated.
- PGC pyrolysis gas chromatography
- a system comprising a gas chromatograph mass spectrometer spectrometer GCMS-QP 5050A manufactured by Shimadzu Corp. and a pyrolysis apparatus JHP-330 manufactured by Japan Analysis Industry Co., Ltd. was used.
- the copolymer 1 or 2 of the present invention has a Mooney viscosity ML 1+ compared to the copolymer 5 of the same Mw (260,000) in which myrcene is replaced by the conjugated diene compound (2) 4 (130 ° C) is low and processability is excellent. Moreover, the copolymer 3 or 4 is excellent in processability in spite of having a large Mw compared with the copolymer 5.
- Test pieces of a predetermined size were prepared from the unvulcanized rubber composition, and were heated by preheating for 1 minute using a Mooney viscosity tester according to JIS K 6300 “Test method of unvulcanized rubber”. The large rotor was rotated under the temperature condition of 130 ° C., and the Mooney viscosity ML 1 + 10 (130 ° C.) after 10 minutes was measured. The smaller the Mooney viscosity index, the better the processability.
- a tire for a cart (tire size: 11 ⁇ 1.10-5) is formed by molding an unvulcanized rubber sheet into a tread shape, bonding it to other tire members, and press curing it for 12 minutes under conditions of 170 ° C. )created.
- the cart tire was mounted on a cart, and a two-kilometer-round test course was run for eight weeks to conduct an actual vehicle running test.
- the tire grip performance of Comparative Example 1 was set to 100 points, and the test driver performed a sensory evaluation at a full 200 points.
- the initial grip performance indicates the grip performance on the first to fourth rounds, and the second half grip performance indicates the grip on the fifth to eighth rounds. The larger the value, the better the grip characteristics.
- Example 1 As shown in Table 2, in Comparative Example 1, the Mooney viscosity is high and the processability is poor, but in Examples 1 to 4 in which myrcene is blended, the Mooney viscosity is low and the processability is improved in all. Moreover, compared with Comparative Example 1, Examples 1 to 4 are also excellent in tensile strength, elongation at break, grip performance, and abrasion resistance, and the workability is improved while maintaining the strength or performance as a rubber. Has been achieved.
- the obtained polymerization solution was blown and dried to remove the solvent, and then dried under a reduced pressure condition of an internal pressure of 0.1 kPa or less / temperature of 50 ° C. until constant weight was reached, to obtain 200 g of a copolymer 6.
- the polymerization conversion (percent of "dry weight / charge") was approximately 100%.
- (2) (Production of Unvulcanized Rubber Composition 6) According to the composition described in Table 4, the copolymer 6 obtained above and various chemicals for producing the above rubber composition (excluding insoluble sulfur and vulcanization accelerator) are mixed for 5 minutes at 150 ° C. with a Banbury mixer It was refined and a kneaded product was obtained.
- Example 6 (Synthesis of copolymer 7) The same treatment as in Example 5 (1) was carried out except that 20 g of farnesene and 130 g of butadiene were used, to obtain 200 g of a copolymer 7. The polymerization conversion was about 100%.
- (2) (Production of Unvulcanized Rubber Composition 7) An unvulcanized rubber composition 7 was obtained in the same manner as in Example 5 (2) except that the copolymer 7 was used instead of the copolymer 6.
- (3) (Production of Vulcanized Rubber Composition 7) The unvulcanized rubber composition 7 obtained in the above (2) was treated in the same manner as in Example 5 (3) to obtain a vulcanized rubber composition 7.
- Example 7 (Synthesis of copolymer 8) The same processing as in Example 5 (1) was carried out except that 40 g of farnesene and 110 g of butadiene were used, to obtain 200 g of a copolymer 8. The polymerization conversion was about 100%.
- (2) (Production of Unvulcanized Rubber Composition 8) An unvulcanized rubber composition 8 was obtained in the same manner as in Example 5 (2) except that the copolymer 8 was used instead of the copolymer 6.
- (3) (Production of Vulcanized Rubber Composition 8) The unvulcanized rubber composition 8 obtained in the above (2) was treated in the same manner as in Example 5 (3) to obtain a vulcanized rubber composition 8.
- Example 8 (Synthesis of copolymer 9) The same treatment as in Example 5 (1) was conducted except that 90 g of farnesene and 60 g of butadiene were used, to obtain 200 g of a copolymer 9. The polymerization conversion was about 100%.
- (2) (Production of Unvulcanized Rubber Composition 9) An unvulcanized rubber composition 9 was obtained in the same manner as in Example 5 (2) except that the copolymer 9 was used instead of the copolymer 6.
- (3) (Production of Vulcanized Rubber Composition 9) The unvulcanized rubber composition 9 obtained in the above (2) was treated in the same manner as in Example 5 (3) to obtain a vulcanized rubber composition 9.
- Comparative example 2 (1) (Synthesis of copolymer 10) The same treatment as in Example 5 (1) was carried out except that 0 g of farnesene and 150 g of butadiene were used, to obtain 200 g of a copolymer 10. The polymerization conversion was about 100%.
- Tg glass transition temperature
- the copolymerization ratio (l) (% by weight) was measured by a conventional method using pyrolysis gas chromatography (PGC). That is, a calibration curve is prepared for the purified branched conjugated diene compound (1), and the branched conjugated diene in the copolymer is obtained from the area ratio of the thermal decomposition product derived from the branched conjugated diene compound (1) obtained by PGC The weight% of the compound (1) was calculated.
- PGC pyrolysis gas chromatography
- a system comprising a gas chromatograph mass spectrometer spectrometer GCMS-QP 5050A manufactured by Shimadzu Corp. and a pyrolysis apparatus JHP-330 manufactured by Japan Analysis Industry Co., Ltd. was used.
- the copolymers 6 to 9 of the present invention have Mw of 420,000 to 620,000, they have Mw of 260,000 consisting only of the conjugated diene compound (2) and the vinyl compound (3).
- the Mooney viscosity ML 1 + 4 (130 ° C.) is lower than that of the copolymer 10 of Therefore, it is clear that the copolymer of the present invention is lower in Mooney viscosity ML 1 + 4 (130 ° C.) than the copolymer of the same Mw in which farnesene is replaced by the conjugated diene compound (2). And from this, it can be said that it is excellent in processability.
- Test pieces of a predetermined size were prepared from the unvulcanized rubber composition, and were heated by preheating for 1 minute using a Mooney viscosity tester according to JIS K 6300 “Test method of unvulcanized rubber”. The large rotor was rotated under the temperature condition of 130 ° C., and the Mooney viscosity ML 1 + 10 (130 ° C.) after 10 minutes was measured. The smaller the Mooney viscosity index, the better the processability.
- a tire for a cart (tire size: 11 ⁇ 1.10-5) is formed by molding an unvulcanized rubber sheet into a tread shape, bonding it to other tire members, and press curing it for 12 minutes under conditions of 170 ° C. )created.
- the cart tire was mounted on a cart, and a two-kilometer-round test course was run for eight weeks to conduct an actual vehicle running test.
- the tire grip performance of Comparative Example 2 was set to 100 points, and the test driver performed a sensory evaluation at a full 200 points.
- the initial grip performance indicates the grip performance on the first to fourth rounds, and the second half grip performance indicates the grip on the fifth to eighth rounds. The larger the value, the better the grip characteristics.
- a novel branched conjugated diene copolymer useful for improving processability as a rubber component of a rubber composition for a tire a rubber composition for a tire comprising the branched conjugated diene copolymer,
- a rubber composition for a tire that improves both the abrasion resistance and the grip performance to high levels and exhibits excellent properties in processability, and pneumatically produced using the rubber composition for a tire A tire can be provided.
Abstract
Description

で示される分枝共役ジエン化合物(1)と、一般式(2)

で示される共役ジエン化合物と、一般式(3)

で示されるビニル化合物とを共重合して得られる分枝共役ジエン共重合体であって、
分枝共役ジエン化合物(1)の共重合比(l)が1~99重量%、共役ジエン化合物(2)の共重合比(m)が99重量%未満、ビニル化合物(3)の共重合比(n)が99重量%未満である分枝共役ジエン共重合体に関する。
本発明の分枝共役ジエン共重合体とは、分枝共役ジエン化合物(1)と、共役ジエン化合物(2)と、ビニル化合物(3)とを共重合して得られる共重合体をいう。
分枝共役ジエン化合物(1)において、炭素数6~11の脂肪族炭化水素基としては、例えば、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ウンデシル基等のノルマル構造のもの、それらの異性体かつ/または不飽和体、並びに、それらの誘導体(例えば、ハロゲン化物および水酸基化物等)が挙げられる。そのうち、特に、4-メチル-3-ペンテニル基、4,8-ジメチル-ノナ-3,7-ジエニル基等、および、それらの誘導体が好ましい。


共役ジエン化合物(2)において、炭素数1~3の脂肪族炭化水素基としてはメチル基、エチル基、n-プロピル基、イソプロピル基等が挙げられ、このうちメチル基が好ましい。ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられ、このうち、塩素原子が好ましい。
ビニル化合物(3)において、炭素数1~3の脂肪族炭化水素基としてはメチル基、エチル基、n-プロピル基、イソプロピル基等が挙げられ、このうちメチル基が好ましい。炭素数3~8の脂環式炭化水素基としては、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロへキシル基、シクロへプチル基、シクロオクチル基、シクロプロペニル基、シクロブテニル基、シクロペンテニル基、シクロへキセニル基、シクロへプテニル基、シクロオクテニル基等が挙げられ、このうちシクロプロピル基、シクロブチル基が好ましい。炭素数6~10の芳香属炭化水素基としては、フェニル基、ベンジル基、フェネチル基、トリル(tolyl)基、キシリル(xylyl)基、ナフチル基などが挙げられる。但し、トリル基におけるベンゼン環上のメチル基の置換位置はオルト-、メタ-もしくはパラ-のいずれの位置も含むものであり、キシリル基におけるメチル基の置換位置も、任意の置換位置のいずれをも含むものである。これらのうち、フェニル基、トリル(tolyl)基、ナフチル基が好ましい。ビニル化合物(3)の具体例としては、スチレン、α-メチルスチレン、α-ビニルナフタレン、β-ビニルナフタレンが好ましい。
本発明の分枝共役ジエン共重合体は、分枝共役ジエン化合物(1)と、共役ジエン化合物(2)と、ビニル化合物(3)とを、共重合させて得ることができる。
該アニオン重合は、アニオン重合開始剤の存在下、適当な溶媒中で実施することができる。アニオン重合開始剤としては、慣用のものをいずれも好適に使用することができ、そのようなアニオン重合開始剤としては、例えば、一般式RLix(但し、Rは1個またはそれ以上の炭素原子を含む脂肪族、芳香族または脂環式基であり、xは1~20の整数である。)を有する有機リチウム化合物があげられる。適当な有機リチウム化合物としては、メチルリチウム、エチルリチウム、n-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム、フェニルリチウムおよびナフチルリチウムが挙げられる。好ましい有機リチウム化合物はn-ブチルリチウムおよびsec-ブチルリチウムである。アニオン重合開始剤は、単独でまたは2種以上を混合して用いることができる。アニオン重合を行う際の重合開始剤の使用量は特に限定はないが、例えば、重合に供する全モノマー100g当り、約0.05~35mmol用いるのが好ましく、約0.05~0.2mmol用いるのがより好ましい。重合開始剤の使用量が0.05mmol未満では共重合体がゴム状とならず樹脂状となる傾向があり、35mmolより多い場合には、共重合体が軟らかく加工性に対して分枝共役ジエン化合物(1)を共重合させることによる効果が小さくなる傾向がある。
配位重合は、上記アニオン重合におけるアニオン重合開始剤に代えて、配位重合開始剤を用いることにより、実施することができる。配位重合開始剤としては、慣用のものをいずれも好適に用いることができ、そのような配位重合開始剤としては、例えば、ランタノイド化合物、チタン化合物、コバルト化合物、ニッケル化合物等の遷移金属含有化合物である触媒が挙げられる。また、所望により、さらにアルミニウム化合物、ホウ素化合物を助触媒として使用することができる。
ガラス転移温度(Tg)は、示差走査熱量計(DSC)により測定される。
ムーニー粘度は、JIS K6300に準じて測定される。
単に、「1~99重量%」というときは、両端の値を含むものである。
ヘキサン:関東化学(株)製のノルマルヘキサン(特級)
イソプロパノール:関東化学(株)製のイソプロパノール(特級)
TMEDA:関東化学(株)製のテトラメチルエチレンジアミン(試薬)
ブタジエン:高千穂化学工業(株)製の1,3-ブタジエン
スチレン:和光純薬(株)のスチレン(試薬)
ミルセン:和光純薬(株)のβ-ミルセン(試薬)
ファルネセン:日本テルペン化学(株)の(E)-β-ファルネセン(試薬)
NR:天然ゴム(TSR 20)
共重合体:本明細書の記載に従い合成したもの
カーボンブラック:キャボットジャパン(株)製のショウブラックN220(チッ素吸着比表面積(N2SA):125m2/g)
シリカ:テグッサ社製のウルトラシルVN3(チッ素吸着比表面積(N2SA):175m2/g)
シランカップリング剤:テグッサ社製のSi69(ビス(3-トリエトキシシリルプロピル)テトラスルフィド)
老化防止剤:大内新興化学工業(株)製のノクラック6C(N-1,3-ジメチルブチル-N’-フェニル-p-フェニレンジアミン)
ステアリン酸:日油(株)製のステアリン酸
オイル:出光興産(株)製のミネラルオイルPW-380
酸化亜鉛:三井金属鉱業(株)製の亜鉛華1号
ワックス:大内新興化学工業(株)製のサンノックワックスN
硫黄:鶴見化学(株)製の粉末硫黄
加硫促進剤1:大内新興化学工業(株)製のノクセラーCZ(N-シクロヘキシル-2-ベンゾチアゾリルスルフェンアミド)
加硫促進剤2:大内新興化学工業(株)製のノクセラーD(N,N’-ジフェニルグアニジン)
実施例1
(1)(共重合体1の合成)
乾燥し窒素置換した3Lの耐圧ステンレス容器に、ヘキサン 2000ml、ミルセン 10g、ブタジエン 140g、スチレン 50gとともにTMEDA 0.22mmolを加え、更にn-ブチルリチウム(n-BuLi)1.17mmolを加えた後、50℃で3時間重合反応を行った。3時間後、1Mイソプロパノール/ヘキサン溶液を1.15ml滴下し、反応を終了させた。得られた重合溶液を送風乾燥して溶媒を除去したのち内圧0.1kPa以下/温度50℃の減圧条件にて恒量に達するまで乾燥して、共重合体1を200g(乾燥重量)得た。重合転化率(「乾燥重量/仕込量」の百分率)はほぼ100%であった。
(2)(未加硫ゴム組成物1の製造)
表2記載の配合に従い、上記で得た共重合体1と、上記ゴム組成物製造用の各種薬品(不溶性硫黄および加硫促進剤を除く)を、バンバリーミキサーにて、150℃で5分間混錬りし、混練り物を得た。得られた混錬物に、硫黄ならびに加硫促進剤を添加して、オープンロールを用いて、170℃で12分間混錬りし、未加硫ゴム組成物1を得た。
(3)(加硫ゴム組成物1の製造)
上記(2)で得た未加硫ゴム組成物を、170℃で20分間プレス加硫し、加硫ゴム組成物1を得た。
(1)(共重合体2の合成)
ミルセンを20g、ブタジエンを130gとした以外は、実施例1(1)と同様に処理して、共重合体2を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物2の製造)
共重合体1に代えて共重合体2を使用した以外は、実施例1(2)と同様に処理して、未加硫ゴム組成物2を得た。
(3)(加硫ゴム組成物2の製造)
上記(2)で得た未加硫ゴム組成物を、実施例1(3)と同様に処理して、加硫ゴム組成物2を得た。
(1)(共重合体3の合成)
ミルセンを40g、ブタジエンを110gとした以外は、実施例1(1)と同様に処理して、共重合体3を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物3の製造)
共重合体1に代えて共重合体3を使用した以外は、実施例1(2)と同様に処理して、未加硫ゴム組成物3を得た。
(3)(加硫ゴム組成物3の製造)
上記(2)で得た未加硫ゴム組成物を、実施例1(3)と同様に処理して、加硫ゴム組成物3を得た。
(1)(共重合体4の合成)
ミルセンを90g、ブタジエンを60gとした以外は、実施例1(1)と同様に処理して、共重合体4を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物4の製造)
共重合体1に代えて共重合体4を使用した以外は、実施例1(2)と同様に処理して、未加硫ゴム組成物4を得た。
(3)(加硫ゴム組成物4の製造)
上記(2)で得た未加硫ゴム組成物を、実施例1(3)と同様に処理して、加硫ゴム組成物4を得た。
(1)(共重合体5の合成)
ミルセンを0g、ブタジエンを150gとした以外は、実施例1(1)と同様に処理して、共重合体5を200gを得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物5の製造)
共重合体1に代えて共重合体5を使用した以外は、実施例1(2)と同様に処理して、未加硫ゴム組成物5を得た。
(3)(加硫ゴム組成物5の製造)
上記(2)で得た未加硫ゴム組成物を、実施例1(3)と同様に処理して、加硫ゴム組成物5を得た。
上記で得た共重合体1~5について、重量平均分子量Mw、数平均分子量Mn、ガラス転移温度Tg、ムーニー粘度および分枝共役ジエン化合物(1)の共重合比(l)を、以下方法に従い測定した。結果を、表1に示す。
Mw、Mnは、東ソー(株)製GPC-8000シリーズの装置、検知器として示差屈折計を用いて測定し、標準ポリスチレンにより校正した。
各共重合体について、示差走査熱量計(DSC)を用い、昇温速度10℃/分にて開始温度-150℃から最終温度150℃までを測定しTgを算出した。
各共重合体について、JIS K 6300「未加硫ゴムの試験方法」に準じて、ムーニー粘度試験機を用いて、1分間の予熱によって熱せられた130℃の温度条件にて、大ローターを回転させ、4分間経過した時点でのムーニー粘度ML1+4(130℃)を測定した。なお、ムーニー粘度が小さいほど、加工性に優れることを示している。
該共重合比(l)(重量%)は、熱分解ガスクロマトグラフィー(PGC)による定法によって測定した。すなわち、精製した分枝共役ジエン化合物(1)についての検量線を作製し、PGCによって得られる分枝共役ジエン化合物(1)由来の熱分解物の面積比から共重合体中の分枝共役ジエン化合物(1)の重量%を算出した。熱分解クロマトグラフィーは(株)島津製作所製のガスクロマトグラフ質量分析計GCMS-QP5050Aと日本分析工業(株)製の熱分解装置JHP-330から構成されるシステムを使用した。
上記で得た未加硫ゴム組成物1~5、および、加硫ゴム組成物1~5を用いて、下記の試験を行った。結果を表2に示す。
前記未加硫ゴム組成物から所定のサイズの試験片を作成し、JIS K 6300「未加硫ゴムの試験方法」に準じて、ムーニー粘度試験機を用いて、1分間の予熱によって熱せられた130℃の温度条件にて、大ローターを回転させ、10分間経過した時点でのムーニー粘度ML1+10(130℃)を測定した。なお、ムーニー粘度指数が小さいほど、加工性に優れることを示している。
前記加硫ゴム組成物からからなる3号ダンベル型試験片を用いて、JIS K 6251「加硫ゴムおよび熱可塑性ゴム-引張特性の求め方」に準じて引張試験を実施し、破断時の引張強さTB(MPa)および伸びEB(%)を測定した。TBが大きいほどゴム強度が優れることを示し、同様にEBが大きいほどゴム強度が優れることを示す。
未加硫ゴムシートをトレッド形状に成形して、他のタイヤ部材と貼りあわせ、170℃の条件下で12分間プレス加硫することにより、カート用タイヤ(タイヤサイズ:11×1.10-5)を作成した。該カート用タイヤをカートに装着し、1周2kmのテストコースを8週走行して、実車走行試験を行った。比較例1のタイヤグリップ性能を100点とし、200点満点でテストドライバーが官能評価した。なお、初期グリップ性能は1~4周目のグリップ性能、後半グリップ性能は5~8周目のグリップ性能を示す。値が大きいほど、グリップ特性が優れていることを示す。
上記実車走行試験後のタイヤについて、比較例1のタイヤの摩耗外観を100点とし、各配合の摩耗外観を、200点満点で相対評価した。値が大きいほど耐摩耗性が優れていることを示す。

実施例5
(1)(共重合体6の合成)
乾燥し窒素置換した3Lの耐圧ステンレス容器に、ヘキサン 2000ml、ファルネセン10g、ブタジエン 140g、スチレン 50gとともにTMEDA 0.22mmolを加え、更にn-ブチルリチウム(n-BuLi)1.17mmolを加えた後、50℃で3時間重合反応を行った。3時間後、1Mイソプロパノール/ヘキサン溶液を1.15ml滴下し、反応を終了させた。得られた重合溶液を送風乾燥して溶媒を除去したのち内圧0.1kPa以下/温度50℃の減圧条件にて恒量に達するまで乾燥して、共重合体6を200g得た。重合転化率(「乾燥重量/仕込量」の百分率)はほぼ100%であった。
(2)(未加硫ゴム組成物6の製造)
表4記載の配合に従い、上記で得た共重合体6と、上記ゴム組成物製造用の各種薬品(不溶性硫黄および加硫促進剤を除く)を、バンバリーミキサーにて、150℃で5分間混錬りし、混練り物を得た。得られた混錬物に、硫黄ならびに加硫促進剤を添加して、オープンロールを用いて、170℃で12分間混錬りし、未加硫ゴム組成物6を得た。
(3)(加硫ゴム組成物6の製造)
上記(2)で得た未加硫ゴム組成物6を、170℃で20分間プレス加硫し、加硫ゴム組成物6を得た。
(1)(共重合体7の合成)
ファルネセンを20g、ブタジエンを130gとした以外は、実施例5(1)と同様に処理して、共重合体7を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物7の製造)
共重合体6に代えて共重合体7を使用した以外は、実施例5(2)と同様に処理して、未加硫ゴム組成物7を得た。
(3)(加硫ゴム組成物7の製造)
上記(2)で得た未加硫ゴム組成物7を、実施例5(3)と同様に処理して、加硫ゴム組成物7を得た。
(1)(共重合体8の合成)
ファルネセンを40g、ブタジエンを110gとした以外は、実施例5(1)と同様に処理して、共重合体8を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物8の製造)
共重合体6に代えて共重合体8を使用した以外は、実施例5(2)と同様に処理して、未加硫ゴム組成物8を得た。
(3)(加硫ゴム組成物8の製造)
上記(2)で得た未加硫ゴム組成物8を、実施例5(3)と同様に処理して、加硫ゴム組成物8を得た。
(1)(共重合体9の合成)
ファルネセンを90g、ブタジエンを60gとした以外は、実施例5(1)と同様に処理して、共重合体9を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物9の製造)
共重合体6に代えて共重合体9を使用した以外は、実施例5(2)と同様に処理して、未加硫ゴム組成物9を得た。
(3)(加硫ゴム組成物9の製造)
上記(2)で得た未加硫ゴム組成物9を、実施例5(3)と同様に処理して、加硫ゴム組成物9を得た。
(1)(共重合体10の合成)
ファルネセンを0g、ブタジエンを150gとした以外は、実施例5(1)と同様に処理して、共重合体10を200g得た。重合転化率はほぼ100%であった。
(2)(未加硫ゴム組成物10の製造)
共重合体6に代えて共重合体10を使用した以外は、実施例5(2)と同様に処理して、未加硫ゴム組成物10を得た。
(3)(加硫ゴム組成物10の製造)
上記(2)で得た未加硫ゴム組成物10を、実施例5(3)と同様に処理して、加硫ゴム組成物10を得た。
上記で得た共重合体6~10について、重量平均分子量Mw、数平均分子量Mn、ガラス転移温度Tg、ムーニー粘度および分枝共役ジエン化合物(1)の共重合比(l)を、以下方法に従い測定した。
Mw、Mnは、東ソー(株)製GPC-8000シリーズの装置、検知器として示差屈折計を用いて測定し、標準ポリスチレンにより校正した。結果を、表3に示す。
各共重合体について、示差走査熱量計(DSC)を用い、昇温速度10℃/分にて開始温度-150℃から最終温度150℃までを測定しTgを算出した。
各共重合体について、JIS K 6300「未加硫ゴムの試験方法」に準じて、ムーニー粘度試験機を用いて、1分間の予熱によって熱せられた130℃の温度条件にて、大ローターを回転させ、4分間経過した時点でのムーニー粘度ML1+4(130℃)を測定した。なお、ムーニー粘度が小さいほど、加工性に優れることを示している。結果を、表3に示す。
該共重合比(l)(重量%)は、熱分解ガスクロマトグラフィー(PGC)による定法によって測定した。すなわち、精製した分枝共役ジエン化合物(1)についての検量線を作製し、PGCによって得られる分枝共役ジエン化合物(1)由来の熱分解物の面積比から共重合体中の分枝共役ジエン化合物(1)の重量%を算出した。熱分解クロマトグラフィーは(株)島津製作所製のガスクロマトグラフ質量分析計GCMS-QP5050Aと日本分析工業(株)製の熱分解装置JHP-330から構成されるシステムを使用した。

上記で得た未加硫ゴム組成物6~10、および、加硫ゴム組成物6~10を用いて、下記の試験を行った。結果を表4に示す。
前記未加硫ゴム組成物から所定のサイズの試験片を作成し、JIS K 6300「未加硫ゴムの試験方法」に準じて、ムーニー粘度試験機を用いて、1分間の予熱によって熱せられた130℃の温度条件にて、大ローターを回転させ、10分間経過した時点でのムーニー粘度ML1+10(130℃)を測定した。なお、ムーニー粘度指数が小さいほど、加工性に優れることを示している。
前記加硫ゴム組成物からからなる3号ダンベル型試験片を用いて、JIS K 6251「加硫ゴムおよび熱可塑性ゴム-引張特性の求め方」に準じて引張試験を実施し、破断時の引張強さTB(MPa)および伸びEB(%)を測定した。TBが大きいほどゴム強度が優れることを示し、同様にEBが大きいほどゴム強度が優れることを示す。
未加硫ゴムシートをトレッド形状に成形して、他のタイヤ部材と貼りあわせ、170℃の条件下で12分間プレス加硫することにより、カート用タイヤ(タイヤサイズ:11×1.10-5)を作成した。該カート用タイヤをカートに装着し、1周2kmのテストコースを8週走行して、実車走行試験を行った。比較例2のタイヤグリップ性能を100点とし、200点満点でテストドライバーが官能評価した。なお、初期グリップ性能は1~4周目のグリップ性能、後半グリップ性能は5~8周目のグリップ性能を示す。値が大きいほど、グリップ特性が優れていることを示す。
上記実車走行試験後のタイヤについて、比較例2のタイヤの摩耗外観を100点とし、各配合の摩耗外観を、200点満点で相対評価した。値が大きいほど耐摩耗性が優れていることを示す。

Claims (8)
- 一般式(1)

で示される分枝共役ジエン化合物と、一般式(2)

で示される共役ジエン化合物と、一般式(3)

で示されるビニル化合物とを共重合して得られる分枝共役ジエン共重合体であって、
分枝共役ジエン化合物(1)の共重合比(l)が1~99重量%、共役ジエン化合物(2)の共重合比(m)が99重量%未満、ビニル化合物(3)の共重合比(n)が99重量%未満である分枝共役ジエン共重合体。 - 分枝共役ジエン化合物(1)の共重合比(l)が2.5~75重量%未満、共役ジエン化合物(2)の共重合比(m)が72.5重量%以下、ビニル化合物(3)の共重合比(n)が25~97.5重量%未満である請求項1記載の分枝共役ジエン共重合体。
- 分枝共役ジエン化合物(1)を共役ジエン化合物(2)で置き換えた同一の重量平均分子量の共重合体との比較において、ムーニー粘度ML1+4(130℃)が低いものである、加工性改善用の、請求項1または2記載の分枝共役ジエン共重合体。
- 分枝共役ジエン化合物(1)が、ミルセンおよび/またはファルネセンである請求項1~3のいずれか1項に記載の分枝共役ジエン共重合体。
- 共役ジエン化合物(2)が、1,3-ブタジエンおよび/またはイソプレンである請求項1~4のいずれか1項に記載の分枝共役ジエン共重合体。
- ビニル化合物(3)がスチレン、α-メチルスチレン、α-ビニルナフタレンおよびβ-ビニルナフタレンからなる群から選択される1種または2種以上である請求項1~5のいずれか1項に記載の分枝共役ジエン共重合体。
- 請求項1~6のいずれか1項に記載の分枝共役ジエン共重合体を含んでなるゴム組成物。
- 請求項7記載のゴム組成物を用いて作製した空気入りタイヤ。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/368,407 US9328184B2 (en) | 2012-02-01 | 2013-01-22 | Branched conjugated diene copolymer, rubber composition and pneumatic tire |
| JP2013556326A JP6173923B2 (ja) | 2012-02-01 | 2013-01-22 | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
| EP13743537.6A EP2810964B1 (en) | 2012-02-01 | 2013-01-22 | Branched conjugated diene copolymer, rubber composition and pneumatic tire |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-020365 | 2012-02-01 | ||
| JP2012020365 | 2012-02-01 | ||
| JP2012-217554 | 2012-09-28 | ||
| JP2012217554 | 2012-09-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013115010A1 true WO2013115010A1 (ja) | 2013-08-08 |
Family
ID=48905052
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/051186 WO2013115010A1 (ja) | 2012-02-01 | 2013-01-22 | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9328184B2 (ja) |
| EP (1) | EP2810964B1 (ja) |
| JP (3) | JP6173923B2 (ja) |
| WO (1) | WO2013115010A1 (ja) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014088544A (ja) * | 2012-10-04 | 2014-05-15 | Sumitomo Rubber Ind Ltd | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
| JP5555813B2 (ja) * | 2012-04-04 | 2014-07-23 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JP5555814B2 (ja) * | 2012-04-04 | 2014-07-23 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JP5617040B2 (ja) * | 2012-02-24 | 2014-10-29 | 株式会社クラレ | ゴム組成物及びタイヤ |
| JP2014218631A (ja) * | 2013-05-10 | 2014-11-20 | 住友ゴム工業株式会社 | ゴム組成物及び空気入りタイヤ |
| WO2015045450A1 (ja) * | 2013-09-30 | 2015-04-02 | 株式会社クラレ | 樹脂組成物、これを硬化させた硬化物及びこの樹脂組成物を含有する光学用粘着剤 |
| JPWO2013132905A1 (ja) * | 2012-03-06 | 2015-07-30 | 住友ゴム工業株式会社 | 水添分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
| US9334394B1 (en) | 2015-06-03 | 2016-05-10 | Fina Technology, Inc. | Farnesene resins, rubber compositions, and tire compositions |
| WO2017002651A1 (ja) * | 2015-06-30 | 2017-01-05 | 株式会社クラレ | ファルネセン重合体及びその製造方法 |
| JP2017014374A (ja) * | 2015-06-30 | 2017-01-19 | 株式会社クラレ | ゴム組成物、及びタイヤ |
| JP2017014373A (ja) * | 2015-06-30 | 2017-01-19 | 株式会社クラレ | ゴム組成物、及びタイヤ |
| JP2017088769A (ja) * | 2015-11-12 | 2017-05-25 | 住友ゴム工業株式会社 | ゴム組成物及び該ゴム組成物を用いて作製した空気入りタイヤ |
| WO2017158740A1 (ja) * | 2016-03-15 | 2017-09-21 | 株式会社島津製作所 | 共役ジエン化合物と芳香族ビニル化合物の共重合体の分析方法 |
| JP2017200983A (ja) * | 2016-05-06 | 2017-11-09 | 株式会社ブリヂストン | 共役ジエン系共重合体及びその製造方法、ゴム組成物、架橋ゴム組成物、並びにタイヤ |
| US9822245B2 (en) | 2013-03-27 | 2017-11-21 | Sumitomo Rubber Industries, Ltd. | Studless winter tire |
| JP2017214489A (ja) * | 2016-05-31 | 2017-12-07 | 株式会社クラレ | ゴム組成物及びタイヤ |
| US9862816B2 (en) | 2013-09-10 | 2018-01-09 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JP2018048349A (ja) * | 2017-12-14 | 2018-03-29 | 株式会社クラレ | ゴム組成物及びタイヤ |
| WO2018101361A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| WO2018101360A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| WO2018101362A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| WO2019189572A1 (ja) | 2018-03-29 | 2019-10-03 | 株式会社クラレ | ブロック共重合体及びその製造方法、該ブロック共重合体を用いたゴム組成物及びタイヤ |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9434804B2 (en) | 2012-02-02 | 2016-09-06 | Sumitomo Rubber Industries, Ltd. | Branched conjugated diene copolymer, rubber composition and pneumatic tire |
| GB201905379D0 (en) * | 2019-04-16 | 2019-05-29 | Univ Durham | Method of epoxidation |
| CN110256631A (zh) * | 2019-05-06 | 2019-09-20 | 北京化工大学 | 含β-月桂烯的三元无规共聚物及其制备方法和应用 |
| WO2021079564A1 (ja) | 2019-10-23 | 2021-04-29 | 住友ゴム工業株式会社 | タイヤ |
| EP4146737A1 (en) * | 2020-05-08 | 2023-03-15 | Pirelli Tyre S.p.A. | High-performance tyre |
| WO2023000098A1 (en) * | 2021-07-21 | 2023-01-26 | ARLANXEO Canada Inc. | Branched polyisobutylene compounds |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63179908A (ja) | 1987-01-22 | 1988-07-23 | Japan Synthetic Rubber Co Ltd | ミルセン重合体およびその製造方法 |
| JPH05125225A (ja) * | 1991-11-06 | 1993-05-21 | Asahi Chem Ind Co Ltd | 水添ブタジエン系共重合体組成物 |
| JPH05125108A (ja) * | 1991-11-05 | 1993-05-21 | Asahi Chem Ind Co Ltd | 水添ブタジエン系共重合体 |
| WO2005085306A1 (ja) * | 2004-03-04 | 2005-09-15 | Riken | アイソタクチック3,4−イソプレン系重合体 |
| JP2008156516A (ja) | 2006-12-25 | 2008-07-10 | Lion Corp | 香料粒子及び洗剤組成物 |
| WO2010027463A1 (en) * | 2008-09-04 | 2010-03-11 | Amyris Biotechnologies, Inc. | Adhesive compositions comprising a polyfarnesene |
| WO2010027464A1 (en) * | 2008-09-04 | 2010-03-11 | Amyris Biotechnologies, Inc. | Farnesene interpolymers |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3018812A (en) * | 1958-08-25 | 1962-01-30 | Us Rubber Co | Adhering vinylpyridine-butadiene rubbers to other rubbers |
| US4553578A (en) * | 1981-07-13 | 1985-11-19 | Gencorp Inc. | Star-shaped polymers for improved tire treads |
| US7655739B1 (en) * | 2009-06-26 | 2010-02-02 | Amyris Biotechnologies, Inc. | Adhesive compositions comprising a polyfarnesene |
| TW201120213A (en) * | 2009-06-17 | 2011-06-16 | Danisco Us Inc | Polymerization of isoprene from renewable resources |
| CA2837545C (en) * | 2011-09-30 | 2014-06-10 | Amyris, Inc. | Rubber composition and tire |
| CN104364280B (zh) | 2012-04-04 | 2017-03-01 | 株式会社可乐丽 | 共聚物、使用了其的橡胶组合物和轮胎 |
| KR102047639B1 (ko) | 2012-04-04 | 2019-11-21 | 주식회사 쿠라레 | 공중합체, 그것을 사용한 고무 조성물 및 타이어 |
| RU2631318C2 (ru) | 2012-04-04 | 2017-09-21 | Курарей Ко., Лтд. | Сополимер, резиновая композиция с его использованием и шина |
| JP5952788B2 (ja) * | 2012-10-04 | 2016-07-13 | 住友ゴム工業株式会社 | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
-
2013
- 2013-01-22 WO PCT/JP2013/051186 patent/WO2013115010A1/ja active Application Filing
- 2013-01-22 EP EP13743537.6A patent/EP2810964B1/en active Active
- 2013-01-22 JP JP2013556326A patent/JP6173923B2/ja active Active
- 2013-01-22 US US14/368,407 patent/US9328184B2/en active Active
-
2016
- 2016-06-03 JP JP2016111912A patent/JP6106316B2/ja active Active
-
2017
- 2017-03-02 JP JP2017039131A patent/JP6230736B2/ja active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS63179908A (ja) | 1987-01-22 | 1988-07-23 | Japan Synthetic Rubber Co Ltd | ミルセン重合体およびその製造方法 |
| JPH05125108A (ja) * | 1991-11-05 | 1993-05-21 | Asahi Chem Ind Co Ltd | 水添ブタジエン系共重合体 |
| JPH05125225A (ja) * | 1991-11-06 | 1993-05-21 | Asahi Chem Ind Co Ltd | 水添ブタジエン系共重合体組成物 |
| WO2005085306A1 (ja) * | 2004-03-04 | 2005-09-15 | Riken | アイソタクチック3,4−イソプレン系重合体 |
| JP2008156516A (ja) | 2006-12-25 | 2008-07-10 | Lion Corp | 香料粒子及び洗剤組成物 |
| WO2010027463A1 (en) * | 2008-09-04 | 2010-03-11 | Amyris Biotechnologies, Inc. | Adhesive compositions comprising a polyfarnesene |
| WO2010027464A1 (en) * | 2008-09-04 | 2010-03-11 | Amyris Biotechnologies, Inc. | Farnesene interpolymers |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2810964A1 |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5617040B2 (ja) * | 2012-02-24 | 2014-10-29 | 株式会社クラレ | ゴム組成物及びタイヤ |
| JPWO2013132905A1 (ja) * | 2012-03-06 | 2015-07-30 | 住友ゴム工業株式会社 | 水添分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
| US10040877B2 (en) | 2012-03-06 | 2018-08-07 | Sumitomo Rubber Industries, Ltd. | Hydrogenated branched conjugated diene copolymer, rubber composition and pneumatic tire |
| US10759885B2 (en) | 2012-03-06 | 2020-09-01 | Sumitomo Rubber Industries, Ltd. | Hydrogenated branched conjugated diene copolymer, rubber composition and pneumatic tire |
| JP5555814B2 (ja) * | 2012-04-04 | 2014-07-23 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JP5555813B2 (ja) * | 2012-04-04 | 2014-07-23 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JPWO2013151069A1 (ja) * | 2012-04-04 | 2015-12-17 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JPWO2013151068A1 (ja) * | 2012-04-04 | 2015-12-17 | 株式会社クラレ | 共重合体、それを用いたゴム組成物及びタイヤ |
| JP2014088544A (ja) * | 2012-10-04 | 2014-05-15 | Sumitomo Rubber Ind Ltd | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ |
| US9822245B2 (en) | 2013-03-27 | 2017-11-21 | Sumitomo Rubber Industries, Ltd. | Studless winter tire |
| JP2014218631A (ja) * | 2013-05-10 | 2014-11-20 | 住友ゴム工業株式会社 | ゴム組成物及び空気入りタイヤ |
| US9862816B2 (en) | 2013-09-10 | 2018-01-09 | Sumitomo Rubber Industries, Ltd. | Pneumatic tire |
| JPWO2015045450A1 (ja) * | 2013-09-30 | 2017-03-09 | 株式会社クラレ | 樹脂組成物、これを硬化させた硬化物及びこの樹脂組成物を含有する光学用粘着剤 |
| WO2015045450A1 (ja) * | 2013-09-30 | 2015-04-02 | 株式会社クラレ | 樹脂組成物、これを硬化させた硬化物及びこの樹脂組成物を含有する光学用粘着剤 |
| US9334394B1 (en) | 2015-06-03 | 2016-05-10 | Fina Technology, Inc. | Farnesene resins, rubber compositions, and tire compositions |
| JP2017014373A (ja) * | 2015-06-30 | 2017-01-19 | 株式会社クラレ | ゴム組成物、及びタイヤ |
| JP2017014374A (ja) * | 2015-06-30 | 2017-01-19 | 株式会社クラレ | ゴム組成物、及びタイヤ |
| JP2020143297A (ja) * | 2015-06-30 | 2020-09-10 | 株式会社クラレ | ファルネセン重合体 |
| JPWO2017002651A1 (ja) * | 2015-06-30 | 2018-04-12 | 株式会社クラレ | ファルネセン重合体及びその製造方法 |
| WO2017002651A1 (ja) * | 2015-06-30 | 2017-01-05 | 株式会社クラレ | ファルネセン重合体及びその製造方法 |
| JP2017088769A (ja) * | 2015-11-12 | 2017-05-25 | 住友ゴム工業株式会社 | ゴム組成物及び該ゴム組成物を用いて作製した空気入りタイヤ |
| WO2017158740A1 (ja) * | 2016-03-15 | 2017-09-21 | 株式会社島津製作所 | 共役ジエン化合物と芳香族ビニル化合物の共重合体の分析方法 |
| CN108885195B (zh) * | 2016-03-15 | 2021-09-24 | 株式会社岛津制作所 | 共轭二烯化合物与芳香族乙烯基化合物的共聚物的分析方法 |
| JPWO2017158740A1 (ja) * | 2016-03-15 | 2018-11-29 | 株式会社島津製作所 | 共役ジエン化合物と芳香族ビニル化合物の共重合体の分析方法 |
| CN108885195A (zh) * | 2016-03-15 | 2018-11-23 | 株式会社岛津制作所 | 共轭二烯化合物与芳香族乙烯基化合物的共聚物的分析方法 |
| JP2017200983A (ja) * | 2016-05-06 | 2017-11-09 | 株式会社ブリヂストン | 共役ジエン系共重合体及びその製造方法、ゴム組成物、架橋ゴム組成物、並びにタイヤ |
| JP2017214489A (ja) * | 2016-05-31 | 2017-12-07 | 株式会社クラレ | ゴム組成物及びタイヤ |
| WO2018101362A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| WO2018101360A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| WO2018101361A1 (ja) * | 2016-12-01 | 2018-06-07 | 日本ゼオン株式会社 | ゴム組成物および空気入りタイヤ |
| JP2018048349A (ja) * | 2017-12-14 | 2018-03-29 | 株式会社クラレ | ゴム組成物及びタイヤ |
| WO2019189572A1 (ja) | 2018-03-29 | 2019-10-03 | 株式会社クラレ | ブロック共重合体及びその製造方法、該ブロック共重合体を用いたゴム組成物及びタイヤ |
| KR20200138736A (ko) | 2018-03-29 | 2020-12-10 | 주식회사 쿠라레 | 블록 공중합체 및 그 제조 방법, 그 블록 공중합체를 사용한 고무 조성물 및 타이어 |
| JPWO2019189572A1 (ja) * | 2018-03-29 | 2021-04-22 | 株式会社クラレ | ブロック共重合体及びその製造方法、該ブロック共重合体を用いたゴム組成物及びタイヤ |
| JP7291123B2 (ja) | 2018-03-29 | 2023-06-14 | 株式会社クラレ | ブロック共重合体及びその製造方法、該ブロック共重合体を用いたゴム組成物及びタイヤ |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140357824A1 (en) | 2014-12-04 |
| JP6173923B2 (ja) | 2017-08-02 |
| EP2810964A4 (en) | 2014-12-17 |
| EP2810964B1 (en) | 2016-11-16 |
| JPWO2013115010A1 (ja) | 2015-05-11 |
| JP2017106033A (ja) | 2017-06-15 |
| US9328184B2 (en) | 2016-05-03 |
| JP2016164280A (ja) | 2016-09-08 |
| JP6106316B2 (ja) | 2017-03-29 |
| EP2810964A1 (en) | 2014-12-10 |
| JP6230736B2 (ja) | 2017-11-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6230736B2 (ja) | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ | |
| JP6381733B2 (ja) | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ | |
| US10759885B2 (en) | Hydrogenated branched conjugated diene copolymer, rubber composition and pneumatic tire | |
| JP5952788B2 (ja) | 分枝共役ジエン共重合体、ゴム組成物および空気入りタイヤ | |
| JP6159574B2 (ja) | 分枝共役ジエン共重合体および水添分枝共役ジエン共重合体、ゴム組成物、並びに空気入りタイヤ | |
| JP2019099656A (ja) | タイヤ用ゴム組成物 | |
| JP5886782B2 (ja) | スタッドレスタイヤ用ゴム組成物及びスタッドレスタイヤ | |
| JP6164927B2 (ja) | 空気入りタイヤ | |
| JP2016003266A (ja) | スタッドレスタイヤ用ゴム組成物及びスタッドレスタイヤ | |
| CN114058151A (zh) | 共轭二烯系聚合物组合物 | |
| JP2014240470A (ja) | フェランドレン共重合体、タイヤ用ゴム組成物および空気入りタイヤ |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13743537 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14368407 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2013556326 Country of ref document: JP Kind code of ref document: A |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013743537 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013743537 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |