WO2012094820A1 - 蒽吡啶酮磺酸化合物及其制备方法和用途 - Google Patents
蒽吡啶酮磺酸化合物及其制备方法和用途 Download PDFInfo
- Publication number
- WO2012094820A1 WO2012094820A1 PCT/CN2011/070263 CN2011070263W WO2012094820A1 WO 2012094820 A1 WO2012094820 A1 WO 2012094820A1 CN 2011070263 W CN2011070263 W CN 2011070263W WO 2012094820 A1 WO2012094820 A1 WO 2012094820A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- salt
- reaction
- dye
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
- C09D11/30—Inkjet printing inks
- C09D11/32—Inkjet printing inks characterised by colouring agents
- C09D11/328—Inkjet printing inks characterised by colouring agents characterised by dyes
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B5/00—Dyes with an anthracene nucleus condensed with one or more heterocyclic rings with or without carbocyclic rings
- C09B5/02—Dyes with an anthracene nucleus condensed with one or more heterocyclic rings with or without carbocyclic rings the heterocyclic ring being only condensed in peri position
- C09B5/14—Benz-azabenzanthrones (anthrapyridones)
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B62/00—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves
- C09B62/02—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group directly attached to a heterocyclic ring
- C09B62/04—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group directly attached to a heterocyclic ring to a triazine ring
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B62/00—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves
- C09B62/44—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group not directly attached to a heterocyclic ring
- C09B62/465—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group not directly attached to a heterocyclic ring the reactive group being an acryloyl group, a quaternised or non-quaternised aminoalkyl carbonyl group or a (—N)n—CO—A—O—X or (—N)n—CO—A—Hal group, wherein A is an alkylene or alkylidene group, X is hydrogen or an acyl radical of an organic or inorganic acid, Hal is a halogen atom, and n is 0 or 1
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B62/00—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves
- C09B62/44—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group not directly attached to a heterocyclic ring
- C09B62/503—Reactive dyes, i.e. dyes which form covalent bonds with the substrates or which polymerise with themselves with the reactive group not directly attached to a heterocyclic ring the reactive group being an esterified or non-esterified hydroxyalkyl sulfonyl or mercaptoalkyl sulfonyl group, a quaternised or non-quaternised aminoalkyl sulfonyl group, a heterylmercapto alkyl sulfonyl group, a vinyl sulfonyl or a substituted vinyl sulfonyl group, or a thiophene-dioxide group
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D11/00—Inks
- C09D11/02—Printing inks
- C09D11/03—Printing inks characterised by features other than the chemical nature of the binder
- C09D11/037—Printing inks characterised by features other than the chemical nature of the binder characterised by the pigment
Definitions
- the present invention relates to a novel class of anthrapyridone sulfonic acid compounds, and processes for their preparation and use.
- it relates to an anthrapyridone sulfonic acid compound having a sulfonic acid substituent and a magenta inkjet printing ink. Background technique
- an inkjet printing method is one of its typical methods.
- a variety of ink ejection methods have been developed so far, and droplets of ink are formed and adsorbed on various recording materials (such as paper, film, fabric, etc.) to achieve recording. Since the ink jet printing head does not come into contact with the recording material, it has the characteristics of being silent and quiet, and is also easy to realize miniaturization, high speed, and colorization. As a result, it has developed rapidly in recent years.
- inks are prepared by dissolving a water-soluble dye in an aqueous medium, adding a water-soluble organic solvent that prevents the nib from being blocked by the ink, and making a pen or a brush ink.
- ink-jet inks are required to have a high-density printed image, a nozzle that does not block the nozzle, good drying property on the recording material, less penetration, and storage stability.
- images formed by inkjet inks must have water resistance, light resistance, moisture resistance, ozone resistance, solubility, and the fastness of these properties.
- the surface of the substrate can adsorb porous silica, cationic polymer, alumina sol or special ceramics, and the dye can be coated on the paper surface together with these organic or inorganic particles and PVA resin.
- For light resistance Among the four primary colors of yellow, magenta, cyan, and black, the reddish light resistance is the weakest, which seriously affects the image quality. Therefore, the improvement of the light resistance of the magenta dye becomes an important issue.
- the dye in the recording material is required to have a penetration resistance. If there is a phenomenon of dye penetration, especially in the case where the photo coloring requirement is high, the image quality is remarkably lowered. However, it is difficult to achieve improvement in light resistance, moisture resistance, ozone resistance, and solubility of an inkjet image with respect to improvement in water resistance.
- ozone gas is a main substance that promotes oxidation and fading of ink-jet printed images. Therefore, improvement of ozone-resistant gas properties is also an important issue in improving light resistance.
- magenta dyes used in inkjet inks are: xanthene-type rhodamine dyes and azo-type dyes coupled by H acid.
- rhodamine dyes are excellent in color and vividness, but resistant to Light is very poor.
- H-acid azo dye although the color and water resistance are good, the light resistance, the ozone resistance, and the vividness are insufficient, especially compared with the cyan dye represented by copper phthalocyanine and the azo yellow dye. , light resistance is still insufficient.
- magenta dye which is excellent in vividness and light resistance
- an anthrapyridone type dye has been obtained, and the molecular skeleton has no sulfonic acid group on the anthracene ring, indicating that it has the advantages of vividness, light resistance, and ozone resistance.
- Fujifilm's specials lj JP2007138124A, JP2007224119A, CN101370882A, WO2009044094A2, US2010080908A1, GB2464188A
- Canon's specials lj US2002041318A1, US2002036681A1, JP2002069349A, JP2006199922A
- the dyes disclosed in these patents have not met all the requirements of hue, sharpness, light fastness, water resistance, ozone resistance, and solubility and solution stability. Although some of these dyes have improved light fastness and ozone resistance, the solubility of the dye and its long-term stability in inkjet inks are insufficient. In particular, the long-term stability of the dye in the ink is related to the solubility of the dye, especially the solubility of the dye in water. In many cases, it is not ideal, and M has to be changed to Li to limit the solubility to a limited extent. Summary of the invention
- An object of the present invention is to provide a magenta dye compound (an anthracene sulfonate sulfonic acid compound) which not only has improved light resistance, ozone resistance, water resistance, but also excellent water solubility, and inkjet ink. Long-term stability in the middle.
- an anthrapyridone sulfonic acid compound represented by the formula (I) or a salt of the formula (I) in a free acid form In order to solve the above problems, the present inventors have found that the above-mentioned problem can be solved by an anthrapyridone sulfonic acid compound represented by the formula (I) or a salt of the formula (I) in a free acid form.
- a first aspect of the invention relates to a compound of the formula (I) or a salt thereof, the salt having the formula (11),
- R 2 , R 3 , and R 4 are the same or different H, Cw 8 alkyl, cyclohexyl, CH 2 CH 2 OH, CH(CH 3 )CH 2 OH or benzyl;
- R 5 is CL 18 alkyl, phenyl, tolyl, benzyl, CF 3 , or (C 6 H 5 . m XC0 2 M;) m , wherein m is 0-3, (C 6 H 5-m ) (C0 2 M) m is a benzene ring having m C0 2 M substituents, and the substituent C0 2 M may be located at any position on the benzene ring;
- a 2 in the formula L is the same as the same, and 2 in the formula L is the same or different.
- the organic ammonium salt N+R R2R3R4 is selected from the group consisting of monoethanolamine salt, diethanolamine salt, triethanolamine salt, monoisopropanolamine salt, diisopropanolamine salt, or triiso Propanolamine salt.
- the M is selected from the group consisting of Li + , Na + , or H 4 + .
- R 7 is H, Cw 8 alkyl, cyclohexyl, CH 2 CH 2 OH, CH(CH 3 )CH 2 OH, benzyl, CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 C0 2 M, CH 2 CH 2 CH 2 C0 2 M, CH 2 CH 2 CH 2 CH 2 C0 2 M, or CH 2 CH 2 CH 2 CH 2 CH 2 C0 2 M; wherein CC 6 H 5 _ m )CS0 3 M) m is a benzene ring having m S 0 3 M substituents, and the substituent S0 3 M may be located at any position on the benzene ring; (C 1 () H 9 _ m )(C0 2 M) m is a naphthalene ring having m C0 2 M substituents, (C 10 H 9 ⁇ n )(SO 3 M) m is a substituent
- a second aspect of the invention relates to a compound of the formula (III), wherein A 2 , R 6 , M and n have the same meanings as defined in the formulae (I) - (II):
- a third aspect of the invention relates to a process for the preparation of a compound of the formula (I) or a salt of the formula ( ⁇ ), comprising the steps of:
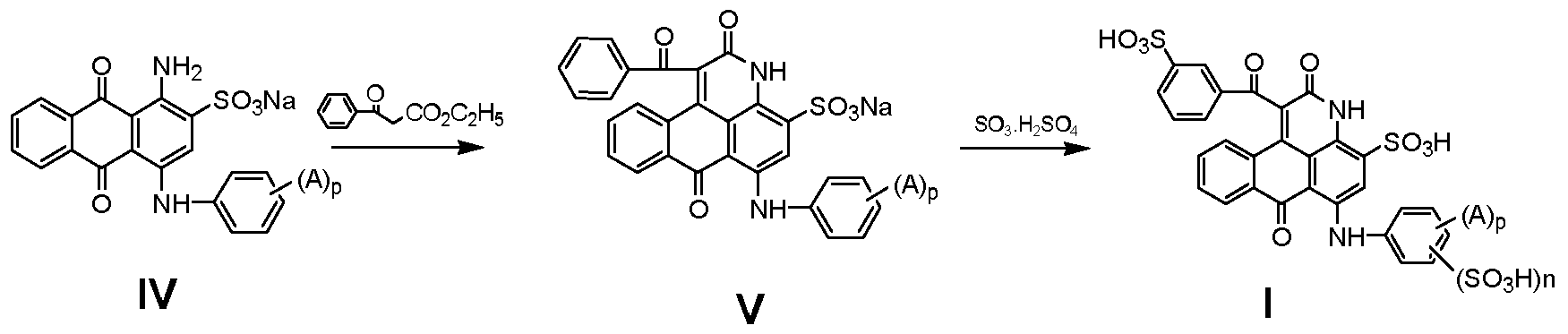
- Cyclization step The compound of the formula (IV) is used as a raw material in an organic solvent at 100 ° C to 250 °.
- the compound of the formula (IV) is cyclically reacted with ethyl benzoylacetate at a temperature for 2 to 10 hours to form a compound of the formula (V) (reaction end point determination: using liquid chromatography or thin layer chromatography, as a raw material The characteristic blue peak disappears, indicating the end of the reaction), wherein the organic solvent is an organic solvent having a boiling point of 100 ° C - 300 ° C, which can dissolve or partially dissolve the reaction starting material (IV);
- step (1) Sulfonation step: using sulphuric acid S0 3 'H 2 S0 4 or chlorosulfonic acid containing 5-30% S0 3 , obtained in step (1) at a temperature of 10 ° C - 100 ° C
- the compound of the formula (V) is subjected to a sulfonation reaction for 1-4 hours to form a compound of the formula (I).
- the liquid chromatography control is still used, and the reverse phase ion pair method is used to judge whether the reaction is finished according to the retention time of the peak of the raw material and the sulfonated product.
- the organic solvent described in the step (1) is selected from the group consisting of: xylene, diethylbenzene, trimethylbenzene, chlorobenzene, dichlorobenzene, nitrobenzene, DMSO, DMF, or mixture.
- the method further comprises the step of: adding a low boiling point to the intermediate product (V) to the reaction system. It is a low boiling organic solvent of 30 ° C to 150 ° C to promote the precipitation of the intermediate product (V).
- the low boiling organic solvent is selected from the group consisting of: methanol, ethanol, and C Alcohol, isopropanol, acetone, acetonitrile, petroleum ether, cyclohexane, or a mixture thereof.
- an inorganic salt selected from ammonium chloride, sodium chloride or lithium chloride is used for salting out to form a compound of the formula (11).
- a fourth aspect of the invention relates to a process for the preparation of the compound of the formula (III), comprising the steps of: (1) using a blue sulfonate compound of the formula (VI) as a starting material, according to the procedure of claim 6. (1) - (2), sequentially obtaining a compound of the formula (VII) - (VIII);
- a fifth aspect of the invention relates to an ink known comprising: the general formula (I) according to the invention a compound thereof or a salt thereof or a compound of the formula (in) (the ink is preferably a printing ink, a coating ink, or an inkjet ink, and the inkjet ink is preferably a water-based or solvent-based or aqueous solvent-based spray. Ink ink).
- a sixth aspect of the invention relates to an inkjet aqueous ink composition
- an inkjet aqueous ink composition comprising: 1 to 20% by weight of the above-mentioned compound of the present invention or a salt thereof, 5 to 50% by weight of a water-miscible organic solvent, and 30 to 94 % by weight water, based on the total weight of the composition;
- the water-miscible organic solvent is selected from one or more of the following: ethanol, propanol, isopropanol, ethylene glycol, diethylene glycol, triethylene glycol, glycerin, ethylene glycol Butyl ether, diethylene glycol monobutyl ether, triethylene glycol monobutyl ether, propylene glycol, butanediol, pentanediol, hexanediol, diglycerin, 2-pyrrolidone, and N -methyl-2-pyrrolidone.
- a seventh aspect of the invention relates to a coating material (preferably an outdoor coating material) comprising the above-mentioned compound of the above formula (1), (II) or (III).
- An eighth aspect of the invention relates to a lacquer (preferably an outdoor lacquer) comprising the above formula of the invention
- a ninth aspect of the invention relates to a toner for laser printing comprising: the above compound of the above formula (1), (11), or (III).
- a tenth aspect of the invention relates to a marker comprising: a compound of the above formula (1), (11), or (III) of the invention.
- An eleventh aspect of the invention relates to the use of a compound of the above formula (1), (II) or (III) according to the invention, which is used as a colorant in the following materials: inks, paints, lacquers, laser-printed toners, Or a marker.
- a twelfth aspect of the invention relates to the use of a compound of the above formula (1), (II) or (III) of the invention for use as a colorant for: paper, fabric (preferably selected from the group consisting of: woven fabrics, Knitted or nonwoven fabric), glass, ceramic, or polymer (preferably selected from: rubber, plastic or fiber).
- the compound of the formula (I) - (III) has such a structural feature that a sulfonic acid group is introduced on the core of the substituted 4-aminopyridinone to increase the water solubility;
- the preparation method of the invention adopts the blue dye derivative of the existing commercial bromine as the basic raw material for synthesis, and the synthesis is convenient and the cost is low.
- the existing patented technology requires a higher reaction rate of the sulfonic acid-free compound as the starting material, and requires more reaction steps.
- the sulfonic acid group is an electron-absorbing water-soluble group, its introduction can not only reduce the electron cloud density of the molecule, improve the ability of the compound to resist photo-oxidation and ozone oxidation, but also simultaneously improve the solubility of the dye and enhance the dye. Long-term stability in ink.
- the preparation method of the present invention uses an industrially mass-produced dye having a bromine-based precursor, which can be shortened Process, reduce costs.
- the dye compound of the present invention has high solubility in water, and has color and vividness suitable for inkjet printing, and an image printed by the inkjet ink prepared from the dye compound is excellent in light fastness, moisture resistance, and ozone fastness. It is also possible to obtain a high vividness hue on the ink jet recording material.
- the anthrapyridone sulfonic acid compound represented by the formula (1) of the present invention is characterized in that it exhibits vividness and extremely high color tone on an ink jet recording paper, and is excellent in water solubility, and in the production process of the ink composition,
- the filter has good filterability.
- the ink composition using the compound does not precipitate crystals, has no physical change, and has no color change after long-term storage, and has good storage stability, whereby the color tone of the color tone of the photograph can be faithfully and long-lasted, even if printing
- a special paper (film) for photo quality coated with inorganic fine particles
- its fastness to light resistance, ozone resistance, moisture resistance, etc. is also very good, and it has long-term stable image preservation. .
- the present invention relates to a class of anthrapyridone sulfonic acid compounds of the formula (I) in the form of a free acid form, wherein a sulfonic acid group is introduced on the core of the substituted 4-aminopyridinone:
- (A) p and P (S0 3 H) n may be at different positions of the benzene ring.
- n is 0-2 and p is 0-3.
- n is preferably 1-2, more preferably n is 2, and p is preferably 0-2.
- the free sulfonic acid group of the compound of the formula (I) is usually in the form of a cation (M) salt of a sulfonic acid having a structural formula (11).
- M is preferably Li + , Na + , K + , ⁇ 4 + , or an organic ammonium salt
- R 2 , R 3 and R 4 are the same or different H, d. 18 alkyl, cyclohexyl, CH 2 CH 2 OH, CH(CH 3 )CH 2 OH, or benzyl.
- the organic ammonium salt N+R R2R3R4 is preferably a monoethanolamine salt, a diethanolamine salt, a triethanolamine salt, a monoisopropanolamine salt, a diisopropanolamine salt, or a triisopropanolamine salt;
- M is preferably Li + , Na + , K + , ⁇ 4 + or the like, and more preferably Li + , Na + , H 4 + .
- the group containing 0, S, and N preferably contains a phenol or a mercapto group, and more preferably contains a mercapto group. Specific examples thereof include and are not limited to: OH, OR 7 , OS0 3 M, 0(C 6 H 5-m )(C0 2 M) m , 0(C6H 5-m )(S03M) m , 0(CioH 9 -m )(C0 2 M) m , 0(CioH 9-m )(S0 3 M) m , H 2 N(R 6 ) 2 R 6 R7 N(R6)(C 6 H 5-m )(C0 2 M) m , N(R6)(C 6 H 5-m )(S0 3 M) m , N(R6)(C 10 H 9-m )(CO 2 M) m , N(R6)(CioH 9- m )(S0 3 M) m , SH, SR 7
- OH, H 2 , NRsRj, SH, and SR 7 are used . More preferred are OH, SR 7 .
- (C 6 H 5 . M ; C0 2 M) m is a benzene ring having m C0 2 M substituents
- (C 6 H 5 . M ; S0 3 M) m is m having S 0 3 M substituents
- the phenyl ring, the substituent C0 2 M or S0 3 M may be located at any position on the phenyl ring, such as an ortho, meta or para position.
- (doHcj.n CCChM is a naphthalene ring having m C0 2 M substituents
- (doH ⁇ XSC M is a naphthalene ring having m S0 3 M substituents
- the substituent C0 2 M or S0 3 M may be located in naphthalene Any position on the ring.
- R 5 is d- 18 alkyl, phenyl, tolyl (o-, m-, or p-tolyl), benzyl, CF 3 , or (C 6 H 5-m )(C0 2 M) m ; Preference is given to Cw 2 alkyl, phenyl, CF 3 .
- R 7 is H, C 1-18 alkyl, cyclohexyl, CH 2 CH 2 OH, CH(CH 3 )CH 2 OH benzyl,
- CH 2 CH 2 S0 3 M CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 C0 2 M, CH 2 CH 2 CH 2 C0 2 M, CH 2 CH 2 CH 2 CH 2 C0 2 M, or CH 2 CH 2 CH 2 CH 2 C0 2 M.
- R7 is preferably H, CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 CH 2 S0 3 M, CH 2 CH 2 C0 2 M , CH 2 CH 2 CH 2 C0 2 M, CH 2 CH 2 CH 2 CH 2 C0 2 M , or CH 2 CH 2 CH 2 CH 2 CH 2 C0 2 M, more preferably H, CH 2 CH 2 S0 3 M, CH 2 CH 2 CH 2 S0 3 M , CH 2 CH 2 CH 2 CH 2 S0 3 M.
- the ring-forming step of the compound of the formula (IV) is carried out by reacting with ethyl benzoylacetate in an organic solvent having a boiling point of from 100 ° C to 300 ° C at a temperature of from 100 ° C to 250 ° C.
- the ethanol and water are removed by heating under reflux or by heating to accelerate the reaction, and after 2 to 10 hours, a compound of the formula (V) is formed.
- the resulting by-product water and ethanol were separated from the reflux condenser using a water separator to promote completion of the reaction.
- the endpoint of the ring formation reaction can be judged by conventional methods in the industry, such as liquid chromatography or thin layer chromatography. In the case of judgment by liquid chromatography, when the characteristic blue peak of the starting material (IV) disappeared, it was indicated that the reaction was completed.
- the molar ratio of the compound of the (IV) to the ethyl benzoylacetate is not particularly limited. It may be 1 : 1-100, preferably 1 : 1-50, more preferably 1 : 1-25, still more preferably 1: 2-15, still more preferably 1: 2-10, still more preferably 1: 2-5.
- One of the reaction raw materials ethyl benzoylacetate
- ethyl benzoylacetate can also be used as a reaction solvent as it is. In this case, the amount of ethyl benzoylacetate is large.
- the organic solvent in the ring-forming reaction needs to dissolve or partially dissolve the starting material (IV) to accelerate the reaction.
- the by-product water and ethanol can be evaporated out of the reaction system during the reaction.
- the organic solvent has a boiling point of from 100 to 300 ° C, preferably from 140 to 250 ° C, more preferably from 140 to 200 ° C.
- the organic solvent includes, but is not limited to, toluene, various isomeric xylenes and isomeric mixtures thereof, various isomeric toluenes and isomeric mixtures thereof, various isomeric diethylbenzenes and their Isomer mixture, various isomeric triethylbenzene and its isomeric mixture, petroleum ether, ethylene glycol dimethyl ether, ethylene glycol diethyl ether, ethylene glycol dipropyl ether, ethylene glycol dibutyl ether, 1,2-propylene glycol dimethyl ether, 1,2-propylene glycol diethyl ether, 1,2-propylene glycol dipropyl ether, 1,2-propylene glycol dibutyl ether, diethylene glycol dimethyl ether, diethylene glycol diethyl ether, Diethylene glycol
- the organic solvent is: xylene, diethylbenzene, trimethylbenzene, chlorobenzene, dichlorobenzene, trichlorobenzene, Nitrobenzene, DMSO, DMF, 2-pyrrolidone, MP, sulfolane, or a mixed solvent thereof.
- the organic solvent is most preferably: a mixture of xylene isomers, o-dichlorobenzene, a mixed solvent of xylene and DMSO, a mixed solvent of o-dichlorobenzene and DMSO.
- the ring-forming reaction temperature is from 100 to 250 ° C, preferably from 100 to 200 ° C, more preferably from 130 to 190 ° C.
- the reaction temperature can also be raised or regulated under pressure or vacuum, and a pressure of 0.5 to 5 atm can be employed.
- the ring formation reaction time is preferably 2-8 hours, more preferably 2-5 hours, still more preferably 2-4 hours.
- the reaction system is cooled to 0-50 ° C, preferably to 0-30 ° C, and the solid intermediate product (V) is precipitated from the liquid reaction system, and filtered to obtain a solid intermediate product (V). ).
- a low-boiling organic solvent having a low solubility in the intermediate product (V) and having a boiling point of from 30 ° C to 150 ° C to promote complete precipitation of the intermediate product (V).
- the low boiling organic solvent includes and is not limited to: methanol, ethanol, propanol, isopropanol, acetone, methyl ethyl ketone, diethyl ether, tetrahydrofuran, dioxane, dichloromethane, chloroform, carbon tetrachloride, cyclohexane. , petroleum ether, ethyl acetate, methyl acetate, butyl acetate, isobutyl acetate, sec-butyl acetate, ethyl formate, propyl formate, butyl formate, isobutyl formate, sec-butyl formate, or mixture.
- the low boiling organic solvent is preferably methanol, ethanol, propanol, isopropanol, acetone, acetonitrile, petroleum ether, cyclohexane, or a mixed solvent thereof. More preferably: methanol, ethanol, propanol, isopropanol, or a mixture thereof.
- a base may also be added to promote the progress of the reaction.
- the base includes but is not limited to: sodium carbonate, sodium hydrogencarbonate, potassium carbonate, potassium hydrogencarbonate, lithium carbonate, lithium hydrogencarbonate, ammonium carbonate, ammonium hydrogencarbonate, sodium phosphate, disodium hydrogen phosphate, potassium phosphate, hydrogen phosphate.
- the base is preferably sodium carbonate or sodium hydrogencarbonate.
- the base is added in an amount such that the molar ratio of the compound of the formula (IV) to the base is 1: 0.1-20, preferably 1: 0.5-10, more preferably 1: 0.5-5, more preferably 1: 0.5-2.5.
- the sulfonation step of the intermediate product (V) comprises: using a fuming sulfuric acid (S(VH 2 S0 4 ) or chlorosulfonic acid containing 5-30% S0 3 , at a temperature of 10 ° C to 100 ° C for the compound (V) Sulfonation is carried out for 1-4 hours to form a compound of the formula (1).
- a fuming sulfuric acid S(VH 2 S0 4 ) or chlorosulfonic acid containing 5-30% S0 3
- the sulfonation of the intermediate (V) is carried out using fuming sulfuric acid or chlorosulfonic acid with stirring.
- the content of sulfur trioxide in the fuming sulfuric acid is 5-30%, preferably 5-15%, more preferably 6-13%, and most preferably 7-12%.
- the weight ratio of the dried intermediate (V) to fuming sulfuric acid in the present invention is 1:5-50, preferably 1:20, more preferably 1:15, still more preferably 1:10.
- the temperature for sulfonation with fuming sulfuric acid is preferably from 10 to 100 ° C, more preferably from 40 to 90 ° C.
- the ratio of the amount of the intermediate (V) to the chlorosulfonic acid is not particularly limited, but preferably: the molar ratio of the intermediate (V) to the chlorosulfonic acid after drying is 1:3. -50, preferably 1:
- the sulfonation reaction temperature using chlorosulfonic acid is preferably 20-100 ° C, more preferably 10-80 ° C, still more preferably 20-60 ° C.
- the reaction time is preferably 2-4 hours, more preferably 3-4 hours, and is completed.
- the endpoint of the sulfonation reaction can also be judged by conventional methods in various industries, such as liquid chromatography or thin layer chromatography to control the endpoint.
- the reverse phase ion pair method is used, and the end of the reaction is judged based on the retention time of the peak of the raw material and the sulfonated product.
- the sulfonated product is poured into ice water with stirring to control the temperature below 40 °C. Then salting out or salt conversion is carried out.
- the salting out or salt conversion of the compound of the formula (I) can be carried out in a conventional manner in the industry.
- the obtained compound of the formula (I) can be salted out using an inorganic salt to form a salt of the formula (II).
- the inorganic salt is preferably and not limited to: ammonium chloride, sodium chloride, lithium chloride or the like, or a mixture thereof.
- salting out of the sulfonated product which is poured into ice water is carried out by adding sodium chloride or ammonium chloride for a plurality of salting out to obtain a salt of the formula (?).
- a wet cake of sodium salt can be obtained.
- hydrochloric acid is added to adjust the pH to 1 to 2, and the crystals are obtained by filtration to obtain a compound of the formula I or II in the form of a free acid (or a portion directly as a sodium salt).
- the wet cake of the free acid is stirred with water, and neutralized by, for example, potassium hydroxide, lithium hydroxide, ammonia water, organic amine, etc., and then salted out by adding the corresponding salt to obtain the corresponding potassium salt and lithium salt.
- ammonium salts organic ammonium salts.
- lithium, sodium and ammonium salts are particularly preferred.
- the method for synthesizing the compound includes the following steps:
- the compound of the formula (VII) and (VIII) is obtained in the order of the ring-forming and sulfonation steps of the above-mentioned compound of the formula (I), starting from the blue sulfonic acid compound (VI).
- the preparation is carried out starting from the blue dye compound (VI), and after the above cyclization and sulfonation steps, the intermediate compound VII and the ring are sequentially obtained. Then, using the magenta dye compound ring as a starting material, it is hydrolyzed and reacted with cyanuric chloride. ! with! ! ? The reaction gave Compound III.
- the compound (VIII) is preferably hydrolyzed in an acid of ⁇ ⁇ ⁇ 4, and the acid includes, but is not limited to, sulfuric acid, hydrochloric acid, sulfonic acid, phosphoric acid, acetic acid and the like.
- the acid includes, but is not limited to, sulfuric acid, hydrochloric acid, sulfonic acid, phosphoric acid, acetic acid and the like.
- sulfuric acid, sulfonic acid, and hydrochloric acid More preferred is sulfuric acid, dilute sulfuric acid, or sulfonic acid.
- the hydrolysis temperature is from 30 ° C to 100 ° C, preferably from 40 to 90 ° C, more preferably from 50 to 80 ° C, still more preferably from 60 to 70 ° C, most preferably from 60 ° C to 65 ° C.
- the product is subjected to salting out to give the M salt form of the amino compound ring-NH 2 .
- the salting out can be carried out by a conventional method known in the art, a conventional salt containing a M cation, for example, salting out using sodium chloride.
- the definition of M is as described above.
- the M salt form of the compound ring-NH 2 is reacted with cyanuric chloride to obtain a dichloro compound (cyclo-Cl 2 ).
- the ratio of the amounts of the two reaction raw materials is not particularly limited, and it is preferably in a ratio of approximately 1:1. The ratio of molecular ratios is reacted.
- cyanuric chloride is 2,4,6-trichloro-S-triazine.
- the pH is preferably from 3 to 8
- the temperature is preferably from 0 to 20 ° C, more preferably from 0 to 10 ° C.
- the reaction time is preferably 2 to 8 hours. More preferably, it is 3-7 hours.
- the reaction raw material HA preferably contains a phenol or a mercapto group, and more preferably contains a mercapto group.
- the HAi containing a phenol is preferably a phenol having a carboxyl group substituent, for example, hydroxybenzoic acid, hydroxydicarboxylic acid, hydroxybenzenesulfonic acid or the like, wherein the substituent on the benzene ring may be in the ortho, meta or para position.
- HA containing a mercapto group examples include, but are not limited to, mercaptoethanol, 3-mercaptopropanesulfonic acid and the like.
- the pH is preferably from 4 to 9
- the reaction temperature is preferably from 10 ° C to 50 ° C, more preferably from 20 ° C to 50 ° C, still more preferably from 30 to 50 ° C.
- the reaction time is usually from 10 minutes to 5 hours, preferably from 30 minutes to 3 hours.
- the compound (VIII-AiCl) thus obtained is further reacted with HA 2 (same as or different from 11), preferably under the reaction conditions of: pH 4-10, more preferably 5-9, and reaction temperature preferably 50 ° C-100 °C, more preferably 60 ° C - 90 ° C.
- the reaction time is usually from 10 minutes to 5 hours, preferably from 30 minutes to 3 hours, to obtain a dye of the structural formula III.
- the blue compound of the formula (IV) and the formula (VI) used as a raw material is usually composed of bromine (i.e., 1-amino-4-bromo-9,10-fluorene- 2-sulfonic acid) is prepared by heating a copper salt with a corresponding aromatic amine in the presence of sodium carbonate in water or an organic solvent.
- bromine i.e., 1-amino-4-bromo-9,10-fluorene- 2-sulfonic acid
- Ai SCH 2 CH 2 CH 2 S0 3 M
- a 2 HC 6 H 3 -2,5-(S0 3 M) 2 H
- Ai OC 6 H 3 -3,5-(C0 2 M) 2
- a 2 N(CH 2 CH 2 OH) 2
- Ai N(CH 2 CH 2 OH) 2
- a 2 N(CH 2 CH 2 OH) 2
- Ai p-OC 6 S0 3 M
- a 2 SCH 2 CH 2 CH 2 S0 3 M
- Ai N(CH 2 CH 2 OH) 2
- a 2 N(CH 2 CH 2 OH) 2
- Ai OC 6 H 3 -3,5-(C0 2 M) 2
- a 2 N(CH 2 CH 2 OH) 2 H
- Ai OC 6 H 3 -3,5-(C0 2 M) 2
- a 2 SCH 2 CH 2 CH 2 S0 3 MH
- Ai OC 6 H 3 -3,5-(C0 2 M) 2
- a 2 OC 6 H 3 -3,5-(C0 2 M) 2
- Ai N(CH 2 CH 2 OH) 2
- a 2 N(CH 2 CH 2 OH) 2 CH 3
- Ai SCH 2 CH 2 CH 2 S0 3 M
- a 2 . SCH 2 CH 2 CH 2 S0 3 M
- the obtained compound of the formula (VIII-H 2 ) may also be not reacted with cyanuric chloride, and the acid chloride or anhydride is at 0-80 ° C.
- the acylation reaction takes place. It is also possible to obtain, by such a reaction, a compound of the formula (I) wherein the group A is NReCORs or NR6S0 2 R 5 .
- the acid chloride or acid anhydride is used as an acylating agent, including but not limited to: d- 18 alkyl acid chloride, d- 18 alkylsulfonyl chloride, benzoyl chloride, benzenesulfonyl chloride, p-toluenesulfonyl chloride, chloroacetyl chloride, dichloroethane Acid chloride, trichloroacetyl Chlorine, C 2 -6 anhydride, succinic anhydride, cis-succinic anhydride, glutaric anhydride, trifluoroacetic anhydride, phthalic anhydride, and the like.
- the dye compounds of the above formulas (1) and (II) preferably have an inorganic salt content of less than 1% by weight.
- the dye can be desalted using a general method such as a high pressure reverse osmosis membrane.
- the ink composition of the present invention is prepared by dissolving a compound represented by the general formulae (1) and (II) in water or an aqueous solvent (water containing the following water-soluble organic solvent) to produce an ink composition.
- the dye of the present invention is usually used in the ink in an amount of from 0.1 to 20% by weight, preferably from 1 to 20% by weight, more preferably from 1 to 15% by weight, still more preferably from 2 to 10% by weight.
- the ink composition of the present invention further contains 0 to 50% by weight of a water-soluble or water-miscible organic solvent, preferably 5 to 50% by weight; and 0 to 5% by weight of an ink control agent.
- the rest is water. Based on the total weight of the above components in the ink composition.
- water-soluble or water-miscible organic solvent used in the present invention are: methanol, ethanol, n-propanol, isopropanol, n-butanol, isobutanol, sec-butanol, tert-butanol, etc.
- a C1 to C4 alkanol an amide of a carboxylic acid such as hydrazine, hydrazine-dimethylformamide or hydrazine, hydrazine-dimethylacetamide; a lactam such as 2-pyrrolidone or hydrazine-methyl-2-pyrrolidone; a cyclic nitrogen-containing solvent such as dimethylmorpholin-2-one or 1,3-dimethylhexahydropyrimidin-2-one; acetone, methyl ethyl ketone, 2-methyl-2-hydroxypenta- a ketone such as a ketone; a cyclic ether such as tetrahydrofuran or dioxane; ethylene glycol, 1,2- or 1,3-propanediol, 1,2- or 1,4-butanediol, 1,6-hexane alcohol.
- Monomers, oligomers or poly Asians having (C2 to C6) alkylene units such as diethylene glycol, triethylene glycol, tetraethylene glycol, dipropylene glycol, thiodiol, polyethylene glycol, polypropylene glycol Alkanediol or thioglycol; polyhydric alcohol (triol) such as glycerin, hexane-1,2,6-triol; ethylene glycol monomethyl ether or ethylene glycol monoethyl ether, diethylene glycol monomethyl ether Diethylene glycol monobutyl Ether; C1 to C4 alkyl ether of polyhydric alcohol such as diethylene glycol monoethyl ether, triethylene glycol monomethyl ether, triethylene glycol monoethyl ether; ⁇ -butyrolactone or dimethyl sulfoxide.
- These water-soluble organic solvents may be used singly or in combination.
- 2-pyrrolidone fluorene-methyl-2-pyrrolidone, mono-, di- or triethylene glycol, dipropylene glycol; and more preferably 2-pyrrolidone, fluorene-methyl-2-pyrrolidone, or diethylene glycol.
- the ink control agent are: antiseptic and antifungal agent, ⁇ adjusting agent, chelating agent, rust preventive agent, water-soluble ultraviolet absorbing agent, water-soluble polymer compound, dye dissolving agent, surfactant, and the like.
- Antiseptic and antifungal agents include, for example, organic sulfurs, organic nitrogen sulfurs, organic halogens, haloallylsulfones, iodopropynes, anthracene-haloalkylsulfides, nitriles, pyridines, 8-hydroxyl groups.
- organohalogen compound examples include sodium pentachlorophenol; and the pyridine oxide compound may, for example, be 2-pyridylthiol-1-oxide; and the inorganic salt compound may, for example, be anhydrous sodium acetate; isothiazoline
- the compound may, for example, be: 1,2-benzisothiazolin-3-one, 2-n-octyl-4-isothiazolin-3-one, 5 -chloro-2-methyl-4-isothiazoline- 3-keto, 5-chloro-2-methyl-4-isothiazolin-3-one magnesium chloride, 5-chloro-2-methyl-4-isothiazolin-3-one calcium chloride, 2-methyl -4-Isothiazolin-3-one calcium chloride and the like.
- Other antiseptic and antifungal agents include, for example, sodium sorbate and sodium benzoate.
- the ⁇ adjusting agent is any substance that controls the ink ⁇ to a range of 7.0 to 11.0.
- examples thereof include an alkanolamine such as diethanolamine or triethanolamine; an alkali metal hydroxide such as lithium hydroxide, sodium hydroxide or potassium hydroxide; ammonium hydroxide or ammonia; or lithium carbonate, sodium carbonate or potassium carbonate.
- An alkali metal carbonate is preferred.
- chelating agent examples include sodium edetate, sodium nitrotriacetate, sodium hydroxyethylethylenediaminetriacetate, sodium diethylenetriaminepentaacetate, sodium urate sodium diacetate and the like.
- rust inhibitor examples include, for example, acidic sulfite, sodium thiosulfate, ammonium thioglycolate, diisopropylammonium nitrite, pentaerythritol tetranitrate, dicyclohexylammonium nitrite, and the like.
- water-soluble ultraviolet absorber examples include, for example, a sulfonated benzophenone or a sulfonated benzotriazole.
- water-soluble high-component compound examples include polyvinyl alcohol, cellulose derivatives, polyamines, polyamines and the like.
- dye solubilizing agent examples include urea, ⁇ -caprolactam, diethyl carbonate and the like.
- the surfactant examples include an anionic surfactant, an amphoteric surfactant, a cationic surfactant, a nonionic surfactant, and the like.
- the anionic surfactant include, for example, an alkylsulfocarboxylate, an ⁇ -olefinsulfonate, a polyoxyethylene alkyl ether acetate, a phosphonyl amino acid, and a salt thereof.
- ⁇ -acylmethyltaurate citronellyl soap, castor oil sulfate, lauryl sulfate, alkylphenol phosphate, alkyl phosphate, alkyl allyl sulfonate, Diethyl sulfosuccinate, diethyl Hexyl sulfosuccinic acid, dioctyl sulfosuccinate, and the like.
- the cationic surfactant include, for example, a 2-vinylpyridine derivative, a poly-4-vinylpyridine derivative, and the like.
- amphoteric surfactants include, for example, lauryl dimethylaminoacetic acid betaine, 2-alkyl-N-carboxymethyl-N-hydroxyethyl imidazoline gun betaine, and coconut oil fatty amide propyl di Methylaminoacetic acid betaine, other imidazoline derivatives of polyoctyl polyaminoethylglycine, and the like.
- nonionic surfactant include, for example, polyoxyethylene nonylphenyl ether, polyoxyethylene octylphenyl ether, polyoxyethylene lauryl phenyl ether, polyoxyethylene octylphenyl group.
- Ethers such as ether, polyoxyethylene oleyl ether, polyoxyethylene lauryl ether, polyoxyethylene alkyl ether; polyoxyethylene oleic acid, polyoxyethylene oleate, polyoxyethylene di-hard Fatty acid ester, sorbitan laurate, sorbitan monostearate, sorbitan monooleate, sorbitan sesquioleate, polyoxyethylene monooleate, Esters such as polyoxyethylene stearate; 2,4,7,9-tetramethyl-5-decyne-4,7-diol, 3,6-dimethyl-4-octyne-3 An alkynediol such as 6-diol or 3,5-dimethyl-1-hexyne-diol (for example, Surfynol 104, 82, 465, Olfine STG, etc. manufactured by Nisshin Chemical Co., Ltd.). These ink modulating agents can be used singly or in combination.
- the ink composition of the present invention is obtained by dissolving the dye compounds represented by the general formulae (1) and (II) in water or the above aqueous solvent (water containing a water-soluble organic solvent) or a water-miscible organic solvent. It is produced by dissolving it together with the above-mentioned ink control agent etc. as needed.
- the order of dissolution of each component is not particularly limited.
- the dye may be dissolved in water or the above aqueous solvent (water containing a water-soluble organic solvent), and an ink control agent may be added thereto to dissolve the dye, or an aqueous solvent or an ink preparation agent may be added after the dye is dissolved in water to dissolve the dye. It can also be different from this order. Further, an aqueous composition or an ink preparation agent may be added to a solution obtained by subjecting the reaction liquid containing the dye or the solution containing the dye to a desalting treatment to produce an ink composition.
- the water to be used is preferably ion-exchanged water or deionized water having less impurities such as distilled water. Then use a filter or the like to perform fine filtration and remove inclusions.
- the pore size of the filter for precision filtration is usually from 1 ⁇ m to 0.01 ⁇ m, preferably from 0.8 ⁇ m to 0.2 ⁇ m.
- the magenta ink composition of the water-soluble anthrapyridone compound of the present invention is suitable for use in stamping, copying, marking, taking, drawing, stamping or printing, particularly for inkjet printing.
- the advantage is that the resulting image has excellent resistance to water, sunlight, ozone and friction, and can also be used for color matching, especially for black.
- the above-mentioned dye compounds represented by the general formulae (1) and (II) can be used as a coloring agent, and can be used for coloring a plurality of substrates such as paper, fiber or cloth (cellulose, nylon, wool, etc.), leather, color filter. A substrate or the like, but is not limited thereto.
- the coloring method may be, for example, a method such as a dip dyeing method, a printing method, a screen printing method, or the like, or an ink jet printing method, and an ink jet printing method is preferred.
- Examples of the recording substrate to which the inkjet printing method of the present invention is applicable include sheets for information transmission such as paper and film, fibers, and leather.
- sheets for information transfer surface treatment is usually required, and an ink absorbing layer is provided in these substrates.
- the ink absorbing layer is a solution of a polymer such as a cation.
- the stain is applied to the substrate, and the coating further contains porous silica, alumina sol or special ceramics, and the white inorganic substance is coated with a hydrophilic polymer such as polyvinyl alcohol or polyvinylpyrrolidone.
- a hydrophilic polymer such as polyvinyl alcohol or polyvinylpyrrolidone.
- Sheets coated with these ink adsorbing layers are generally referred to as inkjet paper (film) or glossy paper (film), such as professional glossy paper, top gloss paper, mat gloss paper (made by Canon), photo paper. Gloss, mat paper, ultra-fine special gloss film (made by Epson), high-quality gloss paper, high-quality gloss film, light paper (made by HP), etc.
- plain paper can of course be used.
- an image is printed on a substrate coated with a porous white inorganic substance, and the discoloration caused by ozone is increased.
- the aqueous magenta ink composition of the present invention is excellent in gas resistance, such a base is The printing of the material can have a special effect.
- porous white inorganic substance examples include: calcium carbonate, kaolin, talc, clay, diatomaceous earth, synthetic amorphous silica, aluminum silicate, magnesium silicate, calcium silicate.
- ink jet printing in addition to the usual yellow and cyan ink compositions, there are also green ink compositions, orange ink compositions, blue (or violet) ink compositions, and magenta ink compositions.
- the dye compound of the present invention can be formulated into a finished red ink composition. These different color compositions may also be used in combination, or may be formulated into a black ink composition or the like as necessary.
- the ink compositions of the respective colors are injected into respective ink cartridges and used in a predetermined position of the ink jet printer. Examples of the ink jet printer include a piezoelectric printer or a foaming printer that generates bubbles by heating.
- the water-based magenta ink composition of the present invention is a vivid magenta color, and particularly has a high vivid color tone in ink-jet glossy paper, and the recorded image has high fastness and high safety to the human body.
- the ink composition of the present invention does not precipitate or separate during storage. Moreover, when the ink of the present invention is used in ink jet printing, the head is not blocked. The ink of the present invention does not undergo physical property changes even if it is used by a continuous ink jet printer for a long period of time or intermittent use.
- the sodium salt of the intermediate B1-2 was obtained in the same manner as in the step 1 of Example 1. Then in the second step of the sulfonation reaction, 10% SO 3 H 2 SO 4 was replaced by 12% S0 3 H 2 S0 4 , the reaction temperature was raised to 85-90 ° C, and the same method as in step 2 of Example 1 was carried out.
- the salt gave 2600 parts of a solution containing 150 parts of a dye (sodium salt, B3 being its free sulfonic acid form). The maximum absorption wavelength of the dye B3 in water was 527 nm.
- the starting material B1-1 was changed to B2-1 to obtain a sodium salt dye of cerium 4 ( ⁇ 4 is its free sulfonic acid form).
- the dye has a maximum absorption wavelength of 528 nm in water.
- the starting material B1-1 is replaced by B5-1, and the sodium salt of the intermediate formula B5-2 is obtained by ring formation, and the sodium salt dye of the formula B5 and B6 is obtained by sulfonation (B5 and B6 are free sulfonic acids thereof). a mixture of forms) (weight ratio of 2:1).
- the maximum absorption wavelength of dye B5 in water is 536 nm.
- the dye of the intermediate product C1-1 has a maximum absorption of 546 nm in water, mass spectrometry (EI-MS) m/z (-): 521.1 ([MH]- dye intermediate C1-1 (free sulfonic acid form)
- the molecular mass number M was 522.1.
- the dye has a maximum absorption wavelength in water of 541 nm.
- Example 5 In the reaction of the step (1) of Example 3, 210 parts of o-dichlorobenzene was replaced with xylene, and the ring-forming condensation reaction was carried out at a temperature of 140 to 145 ° C for 8 hours, and other conditions were the same as in Example 3 to obtain 130.
- a dye of pale reddish purple crystal C1-1 sodium salt, C1-1 is its free sulfonic acid form). The dye has a maximum absorption of 546 nm in water, mass spectrometry (EI-MS) m/z(-): 521.1 ([ ⁇ - ⁇ The dye (in the form of free sulfonic acid) has the most accurate molecular mass number 522.1.
- EI-MS mass spectrometry
- Example 3 According to the similar process of Example 3, starting from the corresponding different raw materials, a sodium salt dye of the formula C2, C3, C4 or the like can be obtained, and the general structure is as shown in the following formula c in the form of a free sulfonic acid, and the specific structure is shown in Table 6. Show.
- Example 7 According to a similar procedure of Example 6, starting from the corresponding different starting materials, a sodium salt dye of the formula C6, C7, C8 or the like can be obtained, and the general structure is represented by the following formula c in the form of a free sulfonic acid, and the specific structure is shown in Table 7. .
- the wet cake is dissolved in 600 parts of water, 90 parts of sodium chloride is added, and then stirred for 2 hours, and the crystal of the red dye is obtained by filtration, and dried to obtain a dye of the crystal C5-H 2 of the red dye (sodium salt, Formula C5-NH 2 is 93 parts in its free sulfonic acid form.
- the dye has a maximum absorption wavelength of 545 nm in water. Mass spectrum (EI-MS) m / z (-): 258.1 ([M-3H] 3 "/ 3) 387.5 ([M-2H] 2" / 2) 776.0 ([MH] -1).
- aqueous solution of % NaOH is maintained at a pH of 2.7 to 3 and is carried out at 25 to 30 ° C for 3 hours to obtain a sodium salt of the intermediate product C5-C1 2 of a condensation reaction (formula C5-C1 2 is its free acidic form) ) the reaction solution.
- the dye has a maximum absorption wavelength of 520 nm in water.
- a reaction liquid containing the formula C5-C1 2 was obtained from Step 1-3 of Example 8.
- the temperature was raised to 27-30 ° C for 1 hour; the temperature was raised to 40-45 ° C for 1 hour, and the pH was maintained at 9.0 ⁇ 0.3. Further, 1.2 parts of diethanolamine (A 2 in the formula III) was added, and the pH was maintained at 8.7 to 9.3 with a 25% aqueous sodium hydroxide solution, and reacted at a temperature of 87 to 93 ° C for 1 hour. After the completion of the reaction, water was added to adjust the amount of the liquid to about 350 parts, and then the insoluble matter was removed by filtration.
- the ink composition shown in Table 8 below was prepared and filtered through a 0.45 mi membrane filter to obtain an aqueous magenta ink composition of the present invention. Further, ion-exchanged water or triethanolamine is added so that the pH of the ink composition is 8 to 10 and the total amount is 100 parts by weight.
- the sulfonate-free fluorenone control dyes Dyel and Dye2 the commercial dye CI active red 180 hydrolyzed derivative (abbreviated as active red 180), and the CI direct red 227 were respectively prepared into comparative ink compositions. .
- An inkjet printer (Epson Model 270 manufactured by Epson Co., Ltd.), a glossy photo paper (manufactured by Epson Co., Ltd.), and inkjet printing were carried out using the ink composition prepared above.
- the glossy paper made by Canon and the glossy paper made by Epson are used in XG-P (Zhe Rui, China), under the conditions of humidity 60% RH and temperature 24 °C.
- the illuminance of 0.36 W/m 2 was irradiated for 50 hours, and the color difference ( ⁇ ) before and after the test was measured.
- the color difference ( ⁇ ) is the value of each L*, a*, b* before and after the measurement test by the above-mentioned color measurement system (Schlab), and the difference between the values of L*, a*, and b* before and after the test is as follows Formula:
- the printed image was placed in an ozone concentration of 40 ppm, a humidity of 60% RH, and a temperature of 24 ° C for 6 hours using an ozone weathering instrument (manufactured by Surrey).
- the color difference ( ⁇ ) before and after the test was measured in the same manner as in the above (1), and evaluated in three levels according to the following criteria:
- thermo-hygrostat manufactured by Suray Co., Ltd.
- 50 ° C and 90% RH 50 ° C and 90% RH for 168 hours
- anthrapyridone sulfonic acid dye of the present invention has extremely excellent solubility and stability as a dye in an inkjet ink, and an image printed by the inkjet ink composition thereof has excellent light resistance, ozone resistance and moisture resistance. Sex. Industrial applicability
- the anthrapyridone sulfonic acid compound of the formula (1) of the present invention has high solubility in water, is stable in water, and has color and vividness suitable for inkjet printing, and is a magenta ink composition containing the compound.
- the printed image using the ink is excellent in light resistance, moisture resistance, and ozone fastness, and thus such a compound is a magenta dye suitable for inkjet printing.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inks, Pencil-Leads, Or Crayons (AREA)
- Plural Heterocyclic Compounds (AREA)
- Paints Or Removers (AREA)
- Ink Jet Recording Methods And Recording Media Thereof (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Description














































Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180003906.3A CN102834467B (zh) | 2011-01-14 | 2011-01-14 | 蒽吡啶酮磺酸化合物及其制备方法和用途 |
| PCT/CN2011/070263 WO2012094820A1 (zh) | 2011-01-14 | 2011-01-14 | 蒽吡啶酮磺酸化合物及其制备方法和用途 |
| JP2013548717A JP6031044B2 (ja) | 2011-01-14 | 2011-01-14 | アントラピリドンスルホン酸化合物およびその調製法 |
| EP11855880.8A EP2546309B1 (en) | 2011-01-14 | 2011-01-14 | Anthrapyridone sulphonic acid compounds, preparation and use thereof |
| US13/583,282 US8968453B2 (en) | 2011-01-14 | 2011-01-14 | Anthrapyridone sulfonic acid compounds and their preparation methods and applications |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2011/070263 WO2012094820A1 (zh) | 2011-01-14 | 2011-01-14 | 蒽吡啶酮磺酸化合物及其制备方法和用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012094820A1 true WO2012094820A1 (zh) | 2012-07-19 |
Family
ID=46506761
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2011/070263 WO2012094820A1 (zh) | 2011-01-14 | 2011-01-14 | 蒽吡啶酮磺酸化合物及其制备方法和用途 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8968453B2 (zh) |
| EP (1) | EP2546309B1 (zh) |
| JP (1) | JP6031044B2 (zh) |
| CN (1) | CN102834467B (zh) |
| WO (1) | WO2012094820A1 (zh) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102634223B (zh) * | 2012-03-20 | 2016-06-29 | 大连理工大学 | 品红染料及其制备方法和用途 |
| CN105368091B (zh) * | 2015-11-20 | 2020-07-17 | 浙江龙盛集团股份有限公司 | 一种蒽吡啶酮活性染料化合物及其制备方法和应用 |
Citations (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0927747A1 (en) | 1996-09-11 | 1999-07-07 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compounds, water-base ink composition, and articles colored therewith |
| JP2000109464A (ja) | 1998-03-25 | 2000-04-18 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| JP2000191660A (ja) | 1998-10-22 | 2000-07-11 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| US6183549B1 (en) | 1998-08-28 | 2001-02-06 | Avecia Limited | Ink compositions containing monoazo dyes |
| GB2353533A (en) | 1999-07-10 | 2001-02-28 | Avecia Ltd | Ink jet dyes based on two aliphatically linked [7-(ortho-carboxyarylazo)-8-hydroxydisulphonaphth-2-ylamino]-(dia/tria)zinyl units |
| JP2002069349A (ja) | 2000-06-12 | 2002-03-08 | Canon Inc | インクセット、インクジェット記録方法、記録ユニット、インクカートリッジ及びインクジェット記録装置 |
| US20020036681A1 (en) | 2000-06-12 | 2002-03-28 | Kumiko Mafune | Ink set, recording method, recording unit, ink cartridge and recording apparatus |
| US20020041318A1 (en) | 2000-06-23 | 2002-04-11 | Koichi Osumi | Ink, ink set, ink-jet recording process, ink cartridge, recording unit and ink-jet recording apparatus |
| US6471760B1 (en) | 1998-03-25 | 2002-10-29 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compounds, water-based magenta ink composition, and method of ink-jet recording |
| JP2002332419A (ja) | 2001-05-09 | 2002-11-22 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| US6648952B1 (en) | 1999-09-03 | 2003-11-18 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compound, aqueous magenta ink compositions and ink-jet recording method |
| US20040134383A1 (en) | 2001-05-09 | 2004-07-15 | Hiroyuki Matsumoto | Anthrapyridone compound, water-based magenta ink composition, and method of ink-jet recording |
| US20050057607A1 (en) | 2003-08-11 | 2005-03-17 | Canon Kabushiki Kaisha | Image-forming method, image-forming apparatus, ink set, and ink |
| US20050115459A1 (en) | 2003-09-30 | 2005-06-02 | Seiko Epson Corporation | Magenta ink composition |
| US20050115458A1 (en) | 2003-09-30 | 2005-06-02 | Seiko Epson Corporation | Ink set |
| EP1626070A1 (en) | 2003-05-22 | 2006-02-15 | Nippon Kayaku Kabushiki Kaisha | Novel anthrapyridone compound, aqueous magenta ink composition and inkjet recording method |
| JP2006199922A (ja) | 2004-12-24 | 2006-08-03 | Canon Inc | インクジェット用インクセット、インクジェット記録方法、インクカートリッジ、記録ユニット及びインクジェット記録装置 |
| US20060219131A1 (en) | 2003-05-22 | 2006-10-05 | Hiroyuki Matsumoto | Anthrapyridone compound, aqueous magenta ink composition and inkjet recording method |
| JP2007138124A (ja) | 2005-10-20 | 2007-06-07 | Fujifilm Corp | インクセット、インクカートリッジ、インクジェットプリンター、インクジェット記録方法、記録物及び褪色改良方法 |
| JP2007224119A (ja) | 2006-02-22 | 2007-09-06 | Fujifilm Corp | インク組成物、インクジェット記録用インク、インクセットおよびインクジェット記録方法 |
| US20070242100A1 (en) | 2006-03-09 | 2007-10-18 | Canon Kabushiki Kaisha | Image forming method, image forming apparatus, and ink jet recording apparatus |
| US20070263055A1 (en) | 2006-05-12 | 2007-11-15 | Seiko Epson Corporation | Magenta ink composition, ink set, ink cartridge, inkjet recording method and recorded product |
| US20080257209A1 (en) | 2006-09-29 | 2008-10-23 | Seiko Epson Corporation. | Inkset, ink cartridge, inkjet recording method, and recorded matter |
| CN101298526A (zh) | 2007-05-01 | 2008-11-05 | 佳能株式会社 | 喷墨墨、喷墨记录方法、墨盒、记录单元和喷墨记录设备 |
| CN101370882A (zh) | 2004-07-13 | 2009-02-18 | 富士胶片株式会社 | 油墨组合物和喷墨记录方法 |
| WO2009044094A2 (en) | 2007-09-29 | 2009-04-09 | Fujifilm Imaging Colorants Limited | Magenta dyes and inks for use in ink-jet printing |
| WO2009116243A1 (ja) | 2008-03-18 | 2009-09-24 | 日本化薬株式会社 | 水溶性アントラピリドン化合物又はその塩、インク組成物及び着色体 |
| CN101547976A (zh) | 2006-12-01 | 2009-09-30 | 日本化药株式会社 | 蒽吡啶酮化合物、其盐、含有该化合物的洋红油墨组合物及着色体 |
| US20100015410A1 (en) | 2006-11-29 | 2010-01-21 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compound, salt thereof, magenta ink composition and colored product |
| US20100075047A1 (en) | 2008-09-24 | 2010-03-25 | Ilford Imaging Switzerland Gmbh | Anthrapyridone dyes and their preparation and use |
| US20100080908A1 (en) | 2008-09-26 | 2010-04-01 | Fujifilm Corporation | Ink set and method for forming image |
| GB2464188A (en) | 2008-10-11 | 2010-04-14 | Fujifilm Imaging Colorants Ltd | Dyes and inks suitable for use in ink jet printing |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2962497A (en) * | 1960-11-29 | Xsoih | ||
| US3632611A (en) * | 1967-10-09 | 1972-01-04 | Sumitomo Chemical Co | Anthraquinone dyes containing a reactive carbamylmethyl group |
| US4740581A (en) * | 1987-02-24 | 1988-04-26 | Eastman Kodak Company | Condensation copolymers containing copolymerized isoquinoline derivative colorants and products therefrom |
| TWI235156B (en) * | 1998-10-22 | 2005-07-01 | Nippon Kayaku Kk | Novel anthrapyridone compound, aqueous magenta ink composition and ink-jet recording method |
| JP2000256857A (ja) * | 1999-03-05 | 2000-09-19 | Sony Corp | ガスマニフォールドの脱着用治具 |
| US6511169B1 (en) * | 2001-08-01 | 2003-01-28 | Eastman Kodak Company | Ink jet printing method |
| JP4374917B2 (ja) * | 2003-06-12 | 2009-12-02 | コニカミノルタホールディングス株式会社 | インクジェット記録液 |
| US7622580B2 (en) * | 2004-08-13 | 2009-11-24 | Xerox Corporation | Colorant compounds |
| KR101431849B1 (ko) * | 2006-08-01 | 2014-08-26 | 오세-테크놀로지스 베파우 | 핫멜트 잉크 |
| WO2008056699A1 (fr) * | 2006-11-09 | 2008-05-15 | Nippon Kayaku Kabushiki Kaisha | Composé d'anthrapyridone, sel de celui-ci, composition d'encre magenta et corps coloré |
| JP2008202011A (ja) * | 2007-02-22 | 2008-09-04 | Nippon Kayaku Co Ltd | アントラピリドン化合物又はその塩、そのアントラピリドン化合物を含有するバイオレットインク組成物及び着色体 |
| US7618484B2 (en) * | 2007-05-01 | 2009-11-17 | Canon Kabushiki Kaisha | Ink jet ink, ink jet recording method, ink cartridge, recording unit and ink jet recording apparatus |
| CN101918502B (zh) * | 2007-10-24 | 2013-07-31 | 惠普发展公司,有限责任合伙企业 | 用于喷墨成像的品红色油墨和油墨组 |
| US7871464B2 (en) * | 2007-11-06 | 2011-01-18 | Nippon Kayaku Kabushiki | Anthrapyridone compound or salt thereof, magenta ink composition and colored product |
| JP5337716B2 (ja) * | 2008-01-25 | 2013-11-06 | 日本化薬株式会社 | アントラピリドン化合物又はその塩、そのアントラピリドン化合物を含有するマゼンタインク組成物及び着色体 |
| US20110195238A1 (en) * | 2008-10-22 | 2011-08-11 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone coloring matter, salt thereof, ink composition and colored body |
| EP2682433B1 (en) * | 2011-09-01 | 2015-06-03 | Dalian University Of Technology | Carbonylpropylsulfonyl anthracene pyridone sulfonic acid compound and preparation method and use thereof |
-
2011
- 2011-01-14 JP JP2013548717A patent/JP6031044B2/ja active Active
- 2011-01-14 WO PCT/CN2011/070263 patent/WO2012094820A1/zh active Application Filing
- 2011-01-14 US US13/583,282 patent/US8968453B2/en active Active
- 2011-01-14 CN CN201180003906.3A patent/CN102834467B/zh active Active
- 2011-01-14 EP EP11855880.8A patent/EP2546309B1/en not_active Not-in-force
Patent Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0927747A1 (en) | 1996-09-11 | 1999-07-07 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compounds, water-base ink composition, and articles colored therewith |
| JP2000109464A (ja) | 1998-03-25 | 2000-04-18 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| US6471760B1 (en) | 1998-03-25 | 2002-10-29 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compounds, water-based magenta ink composition, and method of ink-jet recording |
| US6183549B1 (en) | 1998-08-28 | 2001-02-06 | Avecia Limited | Ink compositions containing monoazo dyes |
| JP2000191660A (ja) | 1998-10-22 | 2000-07-11 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| GB2353533A (en) | 1999-07-10 | 2001-02-28 | Avecia Ltd | Ink jet dyes based on two aliphatically linked [7-(ortho-carboxyarylazo)-8-hydroxydisulphonaphth-2-ylamino]-(dia/tria)zinyl units |
| US6648952B1 (en) | 1999-09-03 | 2003-11-18 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compound, aqueous magenta ink compositions and ink-jet recording method |
| JP2002069349A (ja) | 2000-06-12 | 2002-03-08 | Canon Inc | インクセット、インクジェット記録方法、記録ユニット、インクカートリッジ及びインクジェット記録装置 |
| US20020036681A1 (en) | 2000-06-12 | 2002-03-28 | Kumiko Mafune | Ink set, recording method, recording unit, ink cartridge and recording apparatus |
| US20020041318A1 (en) | 2000-06-23 | 2002-04-11 | Koichi Osumi | Ink, ink set, ink-jet recording process, ink cartridge, recording unit and ink-jet recording apparatus |
| JP2002332419A (ja) | 2001-05-09 | 2002-11-22 | Nippon Kayaku Co Ltd | 新規アントラピリドン化合物、水性マゼンタインク組成物及びインクジェット記録方法 |
| US20040134383A1 (en) | 2001-05-09 | 2004-07-15 | Hiroyuki Matsumoto | Anthrapyridone compound, water-based magenta ink composition, and method of ink-jet recording |
| CN1791643A (zh) * | 2003-05-22 | 2006-06-21 | 日本化药株式会社 | 新颖蒽并吡啶酮化合物、水性洋红油墨组合物及喷墨记录方法 |
| EP1626070A1 (en) | 2003-05-22 | 2006-02-15 | Nippon Kayaku Kabushiki Kaisha | Novel anthrapyridone compound, aqueous magenta ink composition and inkjet recording method |
| US20060219131A1 (en) | 2003-05-22 | 2006-10-05 | Hiroyuki Matsumoto | Anthrapyridone compound, aqueous magenta ink composition and inkjet recording method |
| US20050057607A1 (en) | 2003-08-11 | 2005-03-17 | Canon Kabushiki Kaisha | Image-forming method, image-forming apparatus, ink set, and ink |
| US20050115458A1 (en) | 2003-09-30 | 2005-06-02 | Seiko Epson Corporation | Ink set |
| US20050115459A1 (en) | 2003-09-30 | 2005-06-02 | Seiko Epson Corporation | Magenta ink composition |
| CN101370882A (zh) | 2004-07-13 | 2009-02-18 | 富士胶片株式会社 | 油墨组合物和喷墨记录方法 |
| JP2006199922A (ja) | 2004-12-24 | 2006-08-03 | Canon Inc | インクジェット用インクセット、インクジェット記録方法、インクカートリッジ、記録ユニット及びインクジェット記録装置 |
| JP2007138124A (ja) | 2005-10-20 | 2007-06-07 | Fujifilm Corp | インクセット、インクカートリッジ、インクジェットプリンター、インクジェット記録方法、記録物及び褪色改良方法 |
| JP2007224119A (ja) | 2006-02-22 | 2007-09-06 | Fujifilm Corp | インク組成物、インクジェット記録用インク、インクセットおよびインクジェット記録方法 |
| US20070242100A1 (en) | 2006-03-09 | 2007-10-18 | Canon Kabushiki Kaisha | Image forming method, image forming apparatus, and ink jet recording apparatus |
| US20070263055A1 (en) | 2006-05-12 | 2007-11-15 | Seiko Epson Corporation | Magenta ink composition, ink set, ink cartridge, inkjet recording method and recorded product |
| US20080257209A1 (en) | 2006-09-29 | 2008-10-23 | Seiko Epson Corporation. | Inkset, ink cartridge, inkjet recording method, and recorded matter |
| US20100015410A1 (en) | 2006-11-29 | 2010-01-21 | Nippon Kayaku Kabushiki Kaisha | Anthrapyridone compound, salt thereof, magenta ink composition and colored product |
| CN101547976A (zh) | 2006-12-01 | 2009-09-30 | 日本化药株式会社 | 蒽吡啶酮化合物、其盐、含有该化合物的洋红油墨组合物及着色体 |
| CN101298526A (zh) | 2007-05-01 | 2008-11-05 | 佳能株式会社 | 喷墨墨、喷墨记录方法、墨盒、记录单元和喷墨记录设备 |
| WO2009044094A2 (en) | 2007-09-29 | 2009-04-09 | Fujifilm Imaging Colorants Limited | Magenta dyes and inks for use in ink-jet printing |
| WO2009116243A1 (ja) | 2008-03-18 | 2009-09-24 | 日本化薬株式会社 | 水溶性アントラピリドン化合物又はその塩、インク組成物及び着色体 |
| US20100075047A1 (en) | 2008-09-24 | 2010-03-25 | Ilford Imaging Switzerland Gmbh | Anthrapyridone dyes and their preparation and use |
| CN101684201A (zh) * | 2008-09-24 | 2010-03-31 | 依福德成像瑞士有限公司 | 蒽吡啶酮染料及其制备和用途 |
| US20100080908A1 (en) | 2008-09-26 | 2010-04-01 | Fujifilm Corporation | Ink set and method for forming image |
| GB2464188A (en) | 2008-10-11 | 2010-04-14 | Fujifilm Imaging Colorants Ltd | Dyes and inks suitable for use in ink jet printing |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2546309A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP6031044B2 (ja) | 2016-11-24 |
| EP2546309A1 (en) | 2013-01-16 |
| US20130327248A1 (en) | 2013-12-12 |
| CN102834467A (zh) | 2012-12-19 |
| JP2014508821A (ja) | 2014-04-10 |
| CN102834467B (zh) | 2014-08-27 |
| EP2546309B1 (en) | 2016-06-01 |
| US8968453B2 (en) | 2015-03-03 |
| EP2546309A4 (en) | 2013-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100580937B1 (ko) | 신규 안트라피리돈 화합물, 수성 마젠타 잉크 조성물 및잉크젯 기록 방법 | |
| TWI488921B (zh) | A water-soluble azo compound or a salt thereof, an ink composition and a coloring matter | |
| TWI500705B (zh) | Azo compounds, ink compositions, recording methods and colorants | |
| TWI504687B (zh) | Ink composition, ink jet recording method and chromosome | |
| TWI477562B (zh) | Ink composition, ink jet recording method and chromosome | |
| JP6673687B2 (ja) | インク組成物、インクジェット記録方法及び着色体 | |
| US8734580B2 (en) | Carbonyl propyl sulfuryl anthrapyridone sulfonic acid compounds and their preparation methods and applications | |
| WO2012094820A1 (zh) | 蒽吡啶酮磺酸化合物及其制备方法和用途 | |
| JP2016098274A (ja) | 水溶性アゾ化合物又はその塩、インク組成物及び着色体 | |
| US9011589B2 (en) | Magenta dyes and their preparation methods and applications | |
| TW201437291A (zh) | 偶氮化合物、墨水組成物、記錄方法及著色體 | |
| JP2019077739A (ja) | アゾ化合物又はその塩、及びインク | |
| JP2019167490A (ja) | インク組成物、インクジェット記録方法及び着色体 | |
| JP2018076488A (ja) | アゾ化合物又はその塩、及びインク | |
| JP2018127592A (ja) | アゾ化合物又はその塩、インク組成物及び着色体 | |
| JP4888949B2 (ja) | アゾ色素またはその塩、該アゾ色素またはその塩を含有する水性オレンジインク組成物 | |
| JP2016204436A (ja) | 水溶性アゾ化合物又はその塩、インク組成物及び着色体 | |
| WO2013075286A1 (zh) | 双核蒽吡啶酮磺酸化合物或其盐、其制备方法及应用 | |
| WO2013075287A1 (zh) | 含柔性链的双核蒽吡啶酮磺酸化合物或其盐、其制备方法及应用 | |
| JP2018070792A (ja) | アゾ化合物又はその塩、及びインク。 | |
| JP2018070793A (ja) | アゾ化合物又はその塩、及びインク | |
| JP2018070791A (ja) | アゾ化合物又はその塩、及びインク | |
| JP2018127583A (ja) | 水溶性アゾ化合物又はその塩、インク組成物及び着色体 | |
| JP2018127585A (ja) | アゾ化合物又はその塩、インク組成物及び着色体 | |
| JP2018127591A (ja) | アゾ化合物又はその塩、インク組成物及び着色体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180003906.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11855880 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011855880 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2013548717 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13583282 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |