WO2012070526A1 - ポリ乳酸系フィルム又はシート - Google Patents
ポリ乳酸系フィルム又はシート Download PDFInfo
- Publication number
- WO2012070526A1 WO2012070526A1 PCT/JP2011/076794 JP2011076794W WO2012070526A1 WO 2012070526 A1 WO2012070526 A1 WO 2012070526A1 JP 2011076794 W JP2011076794 W JP 2011076794W WO 2012070526 A1 WO2012070526 A1 WO 2012070526A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- film
- polylactic acid
- sheet
- weight
- meth
- Prior art date
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/04—Polyesters derived from hydroxycarboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G63/00—Macromolecular compounds obtained by reactions forming a carboxylic ester link in the main chain of the macromolecule
- C08G63/02—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds
- C08G63/06—Polyesters derived from hydroxycarboxylic acids or from polycarboxylic acids and polyhydroxy compounds derived from hydroxycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/26—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L27/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers
- C08L27/02—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L27/12—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment containing fluorine atoms
- C08L27/18—Homopolymers or copolymers or tetrafluoroethene
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J7/00—Adhesives in the form of films or foils
- C09J7/20—Adhesives in the form of films or foils characterised by their carriers
- C09J7/22—Plastics; Metallised plastics
- C09J7/25—Plastics; Metallised plastics based on macromolecular compounds obtained otherwise than by reactions involving only carbon-to-carbon unsaturated bonds
- C09J7/255—Polyesters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/04—Polyesters derived from hydroxy carboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2301/00—Additional features of adhesives in the form of films or foils
- C09J2301/30—Additional features of adhesives in the form of films or foils characterized by the chemical, physicochemical or physical properties of the adhesive or the carrier
- C09J2301/302—Additional features of adhesives in the form of films or foils characterized by the chemical, physicochemical or physical properties of the adhesive or the carrier the adhesive being pressure-sensitive, i.e. tacky at temperatures inferior to 30°C
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2427/00—Presence of halogenated polymer
- C09J2427/006—Presence of halogenated polymer in the substrate
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2451/00—Presence of graft polymer
- C09J2451/006—Presence of graft polymer in the substrate
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09J—ADHESIVES; NON-MECHANICAL ASPECTS OF ADHESIVE PROCESSES IN GENERAL; ADHESIVE PROCESSES NOT PROVIDED FOR ELSEWHERE; USE OF MATERIALS AS ADHESIVES
- C09J2467/00—Presence of polyester
- C09J2467/006—Presence of polyester in the substrate
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/28—Web or sheet containing structurally defined element or component and having an adhesive outermost layer
Definitions
- the present invention relates to a polylactic acid film or sheet, and more particularly to a polylactic acid film or sheet that is excellent in heat resistance and that does not break or tear during manufacturing or processing.
- the present invention also relates to a separator, and more particularly to a separator (release liner) based on a polylactic acid film or sheet that has excellent heat resistance and does not break or tear during production or processing.
- This separator is used for protecting the pressure-sensitive adhesive layer surface such as a pressure-sensitive adhesive tape, a pressure-sensitive adhesive sheet, and a label.
- the present invention relates to a pressure-sensitive adhesive tape or sheet, and more particularly to a pressure-sensitive adhesive tape or sheet based on a polylactic acid film or sheet that has excellent heat resistance and does not break or tear during production or processing.
- the present invention relates to a protective film (including a sheet), more specifically, a protective film based on a polylactic acid film or sheet that is excellent in heat resistance and does not break or tear during production or processing.
- This protective film is a protective film for protecting the surface of an automobile wheel; a protective film for protecting the surface of an optical member or electronic component such as a polarizing plate, a wave plate, a retardation plate, a reflective sheet, etc. used in a liquid crystal display or the like.
- Film It can be used for a protective film for protecting the surface of a metal layer or a metal oxide layer used for a plasma display panel, an electromagnetic shielding material for CRT, or the like. The protective film is peeled off and removed when it is no longer needed.
- Polylactic acid is a biomass polymer derived from plants, and has attracted attention as a resin that replaces petroleum-derived polymers.
- polylactic acid has a slow crystallization rate and is hardly crystallized by ordinary film forming means.
- a film made of a resin composition containing polylactic acid has a problem in that the film shape cannot be maintained due to thermal deformation at 60 ° C. or higher, which is the glass transition temperature of polylactic acid. Therefore, several methods have been proposed in the past in order to improve the heat resistance of the polylactic acid resin film.
- a method in which a resin composition containing polylactic acid is formed into a film by a melt extrusion method and then stretched and crystallized by biaxial stretching to increase the heat resistance of the polylactic acid resin film.
- this method has an internal residual stress at the time of stretching, so that there is a disadvantage that the heat shrinkage becomes very large when the use temperature becomes high. Therefore, the temperature that can actually be used is at most about 100 ° C.
- Patent Document 1 proposes a method for improving heat resistance by blending a high melting point material with polylactic acid. However, in this method, the plant-derived component ratio (biomass degree) decreases.
- Patent Document 2 discloses that a resin composition containing polylactic acid is heated from the crystallization temperature (Tc) + 15 ° C. during the temperature lowering process of the resin composition. Melting temperature in the process (Tm) ⁇ 5 ° C. or forming a resin composition containing polylactic acid after melting and forming a film, and then lowering the crystallization temperature ( Tc) A method for producing a polylactic acid-based resin film or sheet characterized by cooling and solidifying after a step of promoting crystallization at a temperature of ⁇ 10 ° C. is disclosed.
- the polylactic acid-based resin film or sheet thus obtained has improved heat resistance, the film or sheet may be broken or torn at the time of production, processing, or winding into a roll. . Therefore, when such a film or sheet is used as a base material such as a separator (release liner), an adhesive tape, or a protective film, the separator or the like may be broken or torn during manufacturing or processing of the separator.
- a separator release liner
- an adhesive tape or a protective film
- the object of the present invention is that there is no melting or deformation of the film or sheet even at a high temperature exceeding 100 ° C., and when the film or sheet is wound into a roll, etc.
- An object of the present invention is to provide a polylactic acid resin film or sheet that does not break or tear.
- Another object of the present invention is that the base material is not melted or deformed even at a high temperature exceeding 100 ° C., and breakage or tearing occurs when the separator is manufactured, processed, or wound into a roll.
- An object of the present invention is to provide a separator based on a polylactic acid film or sheet that does not occur.
- Another object of the present invention is that there is no melting or deformation of the substrate even at a high temperature exceeding 100 ° C., and it is ruptured when it is wound into a roll or the like during the production or processing of an adhesive tape or sheet.
- An object of the present invention is to provide an adhesive tape or sheet based on a polylactic acid film or sheet that does not tear or tear.
- Another object of the present invention is that the base material is not melted or deformed even at a high temperature exceeding 100 ° C., and it is broken or broken when the protective film is manufactured or processed, or wound into a roll.
- An object of the present invention is to provide a protective film based on a polylactic acid film or sheet that does not tear.
- the inventors of the present invention have made the melting endotherm of the crystallization part of the polylactic acid-based resin film or sheet a specific value or more, and the tear strength is a certain value or more. As a result, the present inventors have found that the above problems can be solved and completed the present invention.
- the melting endotherm ⁇ Hc ′ of the crystallized part during film formation determined in step 1 is 10 J / g or more, and the tear strength (according to JIS P 8116 paper-tear strength test method-Elmendorf tear tester method) Both the direction (MD direction) and the width direction (TD direction) are 2.5 N / mm or more.
- the polylactic acid film or sheet may further contain 0.5 to 15 parts by weight of a fluoropolymer (B) with respect to 100 parts by weight of polylactic acid (A).
- the fluoropolymer (B) may be a tetrafluoroethylene polymer.
- the polylactic acid film or sheet may further contain 0.1 to 15 parts by weight of a crystallization accelerator (C) with respect to 100 parts by weight of polylactic acid (A).
- C crystallization accelerator
- the polylactic acid film or sheet further has an acidic functional group-modified olefin polymer (D) having an acid value of 10 to 70 mgKOH / g and a weight average molecular weight of 10,000 to 80,000 per 100 parts by weight of polylactic acid (A). ) May be contained in an amount of 0.1 to 10 parts by weight.
- D acidic functional group-modified olefin polymer
- the polylactic acid film or sheet may be a film or sheet formed by a melt film formation method, for example, a calendar method.
- the present invention is a separator having a release agent treatment layer on at least one surface of a separator substrate, wherein the separator substrate is composed of the polylactic acid film or sheet. Provide a separator.
- the present invention is a pressure-sensitive adhesive tape or sheet having a pressure-sensitive adhesive layer on at least one surface of a substrate, wherein the substrate is composed of the polylactic acid film or sheet.
- An adhesive tape or sheet is provided.
- the present invention is a protective film having a releasable pressure-sensitive adhesive layer on at least one surface of a base material, wherein the base material is composed of the polylactic acid film or sheet.
- a protective film is provided.
- ⁇ Hc is a calorific value (J / g) accompanying crystallization in the temperature rising process of a film or sheet after film formation, as measured by DSC, and ⁇ Hm is an absorption due to subsequent melting.
- a polylactic acid-based film or sheet, wherein the melting endothermic amount ⁇ Hc ′ of the crystallized portion at the time of film formation determined in step 1 is 10 J / g or more.
- an acidic functional group-modified olefin polymer (D) having an acid value of 10 to 70 mg KOH / g and a weight average molecular weight of 10,000 to 80,000 is added to 0.1 parts by weight of 100 parts by weight of polylactic acid (A).
- a separator comprising a lactic acid film or sheet.
- a protective film comprising a polylactic acid film or sheet.
- a resin film or sheet containing polylactic acid (A), which is a core-shell structure polymer (G) having a graft layer outside the particulate rubber with respect to 100 parts by weight of the polylactic acid (A) 1 to 20 parts by weight, and the following formula (1) ⁇ Hc ′ ⁇ Hm ⁇ Hc (1)
- ⁇ Hc is a calorific value (J / g) accompanying crystallization in the temperature rising process of a film or sheet after film formation, as measured by DSC, and ⁇ Hm is an absorption due to subsequent melting.
- a polylactic acid-based film or sheet, wherein the melting endothermic amount ⁇ Hc ′ of the crystallized portion at the time of film formation determined in (1) is 10 J / g or more.
- an acidic functional group-modified olefin polymer (D) having an acid value of 10 to 70 mgKOH / g and a weight average molecular weight of 10,000 to 80,000 is added to 0.1 part of 100 parts by weight of polylactic acid (A).
- a separator comprising a lactic acid film or sheet.
- a protective film comprising a polylactic acid film or sheet.
- the following formula (1) ⁇ Hc ′ ⁇ Hm ⁇ Hc (1)
- ⁇ Hc is a calorific value (J / g) accompanying crystallization in the temperature rising process of a film or sheet after film formation, as measured by DSC, and ⁇ Hm is an absorption due to subsequent melting.
- a polylactic acid-based film or sheet, wherein the melting endothermic amount ⁇ Hc ′ of the crystallized portion at the time of film formation determined in (1) is 10 J / g or more.
- an acidic functional group-modified olefin polymer (D) having an acid value of 10 to 70 mg KOH / g and a weight average molecular weight of 10,000 to 80,000 is added to 0.1 parts by weight of 100 parts by weight of polylactic acid (A).
- a separator comprising a lactic acid film or sheet.
- a protective film comprising a polylactic acid film or sheet.
- the polylactic acid film or sheet of the present invention does not melt or deform the film or sheet even at a high temperature exceeding 100 ° C., and maintains the inherent rigidity while producing or processing the film or sheet. Or, when the sheet is wound into a roll, etc., it has a characteristic that it does not break or tear even if tension is applied. Therefore, it is possible to use biomass polymers in a wide range of fields, and the industrial significance is extremely large.
- the separator of the present invention does not melt or deform the base material even at a high temperature exceeding 100 ° C., and maintains the original rigidity of the base material, and is wound in a roll shape at the time of manufacturing or processing the separator. In this case, even if tension is applied, it has a characteristic that it does not break or tear.
- the pressure-sensitive adhesive tape or sheet of the present invention does not melt or deform the base material even at a high temperature exceeding 100 ° C., and maintains the original rigidity of the base material while manufacturing or processing the pressure-sensitive adhesive tape or sheet.
- the pressure-sensitive adhesive tape or sheet of the present invention When wound in a roll shape, etc., it has a characteristic that it does not break or tear even when tension is applied.
- the protective film of the present invention has no melting or deformation of the base material even at a high temperature exceeding 100 ° C., while maintaining the original rigidity of the base material, at the time of manufacturing or processing the protective film, a roll shape Even when it is wound around, it has a characteristic that it does not break or tear even if tension is applied.
- the polylactic acid film or sheet of the present invention is a resin film or sheet containing polylactic acid (A). Since lactic acid, which is a raw material monomer for polylactic acid, has an asymmetric carbon atom, there are L and D isomers of optical isomers.
- the polylactic acid (A) used in the present invention is a polymer mainly composed of L-form lactic acid. The smaller the content of lactic acid in the D-form that is mixed as an impurity during production, the higher the crystallinity and the higher melting point of the polymer. Therefore, it is preferable to use the L-form purity as high as possible, and the L-form purity is 95%. It is more preferable to use the above.
- polylactic acid (A) may contain other copolymerization components other than lactic acid.
- Examples of the other copolymer components include ethylene glycol, propylene glycol, 1,3-propanediol, butanediol, pentanediol, neopentylglycol, hexanediol, heptanediol, octanediol, nonanediol, decanediol, Polyol compounds such as, 4-cyclohexanedimethanol, glycerin, pentaerythritol, polyethylene glycol, polypropylene glycol, polytetramethylene glycol, bisphenol A; oxalic acid, malonic acid, glutaric acid, adipic acid, sebacic acid, azelaic acid, dodecanedione Acid, cyclohexanedicarboxylic acid, terephthalic acid, isophthalic acid, phthalic acid, naphthalenedicarboxylic acid, bis
- the weight average molecular weight of the polylactic acid (A) is, for example, 10,000 to 400,000, preferably 50,000 to 300,000, and more preferably 80,000 to 150,000.
- the melt flow rate [JIS K-7210 (Test condition 4)] of polylactic acid (A) at 190 ° C. and a load of 21.2 N is, for example, 0.1 to 50 g / 10 minutes, preferably 0.2 to 20 g. / 10 minutes, more preferably 0.5 to 10 g / 10 minutes, and particularly preferably 1 to 7 g / 10 minutes. If the melt flow rate is too high, the mechanical properties and heat resistance of the film or sheet obtained by film formation may be inferior. On the other hand, if the value of the melt flow rate is too low, the load during film formation may become too high.
- weight average molecular weight refers to that measured by gel permeation chromatography (GPC) (in terms of polystyrene).
- GPC gel permeation chromatography
- the measurement conditions of GPC are as follows. Column: TSKgel SuperHZM-H / HZ2000 / HZ1000 Column size: 4.6 mm I.D. D. ⁇ 150mm Eluent: Chloroform Flow rate: 0.3 ml / min Detector: RI Column temperature: 40 ° C Injection volume: 10 ⁇ l
- the production method of polylactic acid is not particularly limited, but typical production methods include lactide method, direct polymerization method and the like.
- lactide method lactic acid is heated and dehydrated and condensed to give low molecular weight polylactic acid, which is then thermally decomposed under reduced pressure to obtain lactide, which is a cyclic dimer of lactic acid, and this lactide is converted into tin (II) octanoate or the like.
- high molecular weight polylactic acid is obtained by ring-opening polymerization in the presence of a metal salt catalyst.
- the direct polymerization method is a method in which polylactic acid is directly obtained by heating lactic acid in a solvent such as diphenyl ether under reduced pressure and polymerizing it while removing water in order to suppress hydrolysis.
- a commercially available product can be used as polylactic acid (A).
- polylactic acid (A) For example, trade names “Lacia H-400”, “Lacia H-100” (manufactured by Mitsui Chemicals), trade names “Terramac TP-4000”, “Terramac TE-4000” (above, Unitika Ltd.) Manufactured) and the like.
- polylactic acid (A) you may use what was manufactured by the well-known thru
- the content of polylactic acid (A) in the polylactic acid film or sheet of the present invention is usually 60% by weight or more, preferably 70% by weight or more, more preferably 80% by weight or more, from the viewpoint of increasing the degree of biomass. Particularly preferred is 85% by weight or more.
- the upper limit of content of the said polylactic acid (A) is 97 weight%, for example, Preferably it is 95 weight%, More preferably, it is 93 weight%.
- the biomass degree is the ratio of the dry weight of the used biomass to the dry weight of the film or sheet. Biomass is a renewable, organic organic resource that excludes fossil resources.
- ⁇ Hc is the amount of heat generated by crystallization in the temperature raising process in the region where the film or sheet could not be crystallized during film formation, and ⁇ Hm was generated in the crystallization part during film formation and in the temperature raising process. This is the amount of heat absorbed when the crystallization part melts.
- DSC Different Scanning Calorimetry
- calorie crystal exothermic peak area
- calorie melting endothermic peak area
- ⁇ Hc ′ is a value obtained by subtracting ⁇ Hc from ⁇ Hm, and corresponds to the melting endotherm of the crystallized portion generated during film formation. Therefore, ⁇ Hc ′ is an index of the crystallinity of the polylactic acid film or sheet, and thus the heat resistance.
- a polylactic acid film or sheet having ⁇ Hc ′ of 10 J / g or more has excellent heat resistance, and the film or sheet does not melt or deform even at a high temperature exceeding 100 ° C. (for example, a temperature of 120 ° C. or higher). Therefore, processing at a high temperature is possible.
- ⁇ Hc ′ is preferably 12 J / g or more, more preferably 14 J / g or more, and particularly preferably 15 J / g or more.
- the value of ⁇ Hc ′ is preferably as high as possible from the viewpoint of heat resistance. However, if the value of ⁇ Hc ′ is too high, the film or sheet becomes hard and the tear resistance may be lowered. Therefore, ⁇ Hc ′ is preferably 40 J / g or less, more preferably 35 J / g or less, and particularly preferably 30 J / g or less.
- the fluorine-based polymer (B) may be used for the polylactic acid film or sheet.
- the fluorine-based polymer (B) is used as, for example, a melt tension adjusting agent or a crystallization accelerator.
- Examples of the fluorine-based polymer (B) include tetrafluoroethylene-based polymers, polychlorotrifluoroethylene, polyvinylidene fluoride, and polyvinyl fluoride.
- a fluorine-type polymer (B) can be used individually by 1 type or in combination of 2 or more types.
- a tetrafluoroethylene polymer (B ′) can be particularly preferably used.
- the tetrafluoroethylene polymer (B ′) may be a tetrafluoroethylene homopolymer or a copolymer of tetrafluoroethylene and another monomer.
- examples of the tetrafluoroethylene polymer (B ′) include polytetrafluoroethylene, perfluoroalkoxyalkane (copolymer of tetrafluoroethylene and perfluoroalkyl vinyl ether), and perfluoroethylene propene copolymer (tetrafluoroethylene and hexafluoro).
- Copolymer with propylene ethylene-tetrafluoroethylene copolymer, tetrafluoroethylene-perfluorodioxole copolymer and the like.
- polytetrafluoroethylene is preferable.
- the tetrafluoroethylene-based polymer (B ′) can be used alone or in combination of two or more.
- the fluorine-based polymer (B) improves the melt tension of the polylactic acid (A) -containing resin composition, and enables, for example, orientation crystallization in a flow field during the melt film formation process, so that the polylactic acid (A) Promotes crystallization. Further, when the fluoropolymer (B) is blended with the polylactic acid (A) -containing resin composition, the melt tension is improved and the melt viscosity is also increased. It is possible to prevent the occurrence of elongation and peeling failure when the formed resin composition leaves the roll.
- fluorine-based polymers such as tetrafluoroethylene-based polymer (B ′) also have a function as a crystal nucleating agent for polylactic acid (A).
- the temperature of the resin composition immediately after film formation is adjusted to the crystallization temperature.
- crystallization of polylactic acid (A) can be further promoted.
- the crystallization of polylactic acid (A) can be promoted by blending the fluorine-based polymer (B) [particularly tetrafluoroethylene-based polymer (B ′)], thereby increasing ⁇ Hc ′ of the polylactic acid-based film or sheet. be able to.
- the function of the tetrafluoroethylene-based polymer (B ′) as a crystal nucleating agent for the polylactic acid (A) is considered to depend on the crystal structure of the tetrafluoroethylene-based polymer (B ′).
- the plane spacing of the polylactic acid crystal lattice was 4.8 angstroms, whereas that of the tetrafluoroethylene polymer (B ′) was 4.9 angstroms. From this, it is considered that the tetrafluoroethylene-based polymer (B ′) can act as a crystal nucleating agent for the polylactic acid (A) by having an epitaxy action.
- the epitaxy action means that polylactic acid (A) grows on the surface of the tetrafluoroethylene-based polymer (B ′) and aligns with the crystal plane of the crystal surface of the tetrafluoroethylene-based polymer (B ′). This refers to the growth pattern in which lactic acid (A) is arranged.
- the surface spacing of the tetrafluoroethylene-based polymer (B ′) is governed by the crystal form of the tetrafluoroethylene portion even if it is a copolymer of tetrafluoroethylene and other monomers. Is the same. Accordingly, the amount of other monomer components of the copolymer is not particularly limited as long as the crystalline form of polytetrafluoroethylene can be maintained and the physical properties are not greatly changed.
- the tetrafluoroethylene polymer (B The ratio of the other monomer component in ′) is desirably 5% by weight or less.
- the tetrafluoroethylene polymer (B ′) may be obtained by any polymerization method, but is preferably obtained by emulsion polymerization. Since the tetrafluoroethylene polymer obtained by emulsion polymerization is easy to be fiberized, it becomes easier to form a network structure in polylactic acid (A), improving the melt tension of the resin composition containing polylactic acid (A) and melting. It is considered that it effectively acts to promote crystallization of polylactic acid (A) in a flow field during film formation.
- the particles of tetrafluoroethylene-based polymer (B ′) are compatible with polylactic acid (A) such as (meth) acrylic acid ester-based polymer. You may use what modified
- the weight average molecular weight of the fluorine-based polymer (B) is not particularly limited, but is usually 1 million to 10 million, preferably 2 million to 8 million.
- fluorine polymer (B) [tetrafluoroethylene polymer (B ′) etc.]
- a commercially available product may be used.
- commercial products of polytetrafluoroethylene trade names “Fluon CD-014”, “Fluon CD-1”, “Fluon CD-145” manufactured by Asahi Glass Co., Ltd. and the like can be mentioned.
- commercially available products of acrylic-modified polytetrafluoroethylene include the Metabrene A series such as “Madebrene A-3000” and “Methbrene A-3800” manufactured by Mitsubishi Rayon Co., Ltd.
- the content of the fluorine-based polymer (B) in the polylactic acid-based film or sheet [particularly, the content of the tetrafluoroethylene-based polymer (B ′)] is usually 0.5% relative to 100 parts by weight of the polylactic acid (A). From the viewpoint of improving the melt tension and maintaining the degree of biomass, and obtaining a good surface state, it is preferably 0.7 to 10 parts by weight, more preferably 1 to 5 parts by weight.
- the content of the fluoropolymer (B) is less than 0.5 parts by weight, the effect of improving the melt tension is difficult to obtain, and exceeds 15 parts by weight. And the effect according to addition amount is not acquired, and the fall of a biomass degree becomes a problem.
- a resin composition containing polylactic acid (A) is melt-formed by a calendar film forming method or the like.
- Examples include film formation using a film method, (2) film formation of a resin composition in which a polycrystallization is added to polylactic acid (A), and (3) combinations thereof.
- the melt film forming method will be described later.
- crystallization accelerator those other than the fluorine-based polymer [for example, tetrafluoroethylene-based polymer (B ′)] that can be used as the crystallization accelerator among the fluorine-based polymer (B) can also be used.
- a crystallization accelerator [may be referred to as a crystallization accelerator (C)] is not particularly limited as long as an effect of promoting crystallization is recognized, but is a crystal lattice of polylactic acid (A). It is desirable to select a substance having a crystal structure having a face spacing close to the face spacing.
- crystallization accelerator (C) examples include organic substances such as melamine polyphosphate, melamine cyanurate, zinc phenylphosphonate, calcium phenylphosphonate, magnesium phenylphosphonate, inorganic talc, clay. Etc. Of these, zinc phenylphosphonate is preferred because the face spacing is closest to the face spacing of polylactic acid (A), and a good crystallization promoting effect is obtained.
- the crystallization accelerator (C) can be used alone or in combination of two or more.
- Crystallization accelerator (C) Commercially available products can be used as the crystallization accelerator (C).
- a trade name “Eco Promote” manufactured by Nissan Chemical Co., Ltd. may be mentioned.
- the content of the crystallization accelerator (C) in the polylactic acid-based film or sheet is usually 0.1 to 15 parts by weight with respect to 100 parts by weight of the polylactic acid (A). From the viewpoint of maintenance, the amount is preferably 0.3 to 10 parts by weight.
- the content of the crystallization accelerator (C) is less than 0.1 parts by weight, it is difficult to obtain the effect of crystallization promotion.
- the content exceeds 15 parts by weight, the effect according to the amount added cannot be obtained. Decrease in biomass degree becomes a problem.
- the crystallization accelerator (C) When the tetrafluoroethylene polymer (B ′) is used as the fluoropolymer (B) at a ratio of 0.5 to 15 parts by weight with respect to 100 parts by weight of the polylactic acid (A), the crystallization accelerator (C)
- the content of is usually 0.1 to 5 parts by weight with respect to 100 parts by weight of polylactic acid (A), and preferably 0.3 to 3 parts by weight from the viewpoint of better crystallization promoting effect and maintaining the degree of biomass. It is.
- the content of the crystallization accelerator (C) is less than 0.1 parts by weight, the effect of promoting crystallization is difficult to obtain, and if it exceeds 5 parts by weight, the effect according to the amount added is obtained. In addition, a decrease in the degree of biomass becomes a problem.
- the tearing strength of the polylactic acid film or sheet of the present invention is 2.5 N / mm or more in both the flow direction (MD direction) and the width direction (TD direction).
- the polylactic acid-based film or sheet having such tear strength does not break or tear even when there is a step where tension is applied when the film or sheet is produced or when the film or sheet is processed. . Further, even when wound into a roll shape or subjected to a punching process or the like, no breakage or tearing occurs.
- a polylactic acid film or sheet having such a tear strength is used as a substrate such as a separator, an adhesive tape or sheet, and a protective film
- a separator, an adhesive tape or sheet, and a protective film when manufacturing the separator, adhesive tape or sheet, protective film, when the separator, adhesive tape or sheet, protective film, when the separator, adhesive tape or sheet, protective film or the like is processed, even if there is a step where tension is applied, no breakage or tearing occurs. Further, even when these are wound into a roll shape or subjected to processing such as punching, no breakage or tearing occurs.
- the tear strength can be measured using a tear strength tester according to JIS P 8116 paper-tear strength test method-Elmendorf tear tester method.
- a polylactic acid film or sheet having a tear strength of 2.5 N / mm or more in both the flow direction (MD direction) and the width direction (TD direction) is not limited to the production of the film or sheet as described above.
- MD direction flow direction
- TD direction width direction
- no breakage or tearing occurs at the time of roll winding, processing, etc., various processing becomes possible, and the range of use is greatly expanded.
- the tear strength is preferably 2.8 N / mm or more, more preferably 3.2 N / mm or more in both the flow direction (MD direction) and the width direction (TD direction). The higher the tear strength, the better. Although it does not specifically limit as an upper limit of tear strength, For example, 30 N / mm or less may be sufficient, 20 N / mm or less may be sufficient, and it is 15 N / mm or less normally.
- a resin composition in which a tear resistance modifier (E) is blended with polylactic acid (A) is formed into a film. The method of doing is mentioned.
- the tear resistance modifier (E) for example, (a) polyglycerol fatty acid ester and / or polyglycerol condensed hydroxy fatty acid ester [“polyglycerol fatty acid ester and / or polyglycerol condensed hydroxy fatty acid ester (F)” and And (b) a core-shell structure polymer having a graft layer outside the particulate rubber [may be referred to as “core-shell structure polymer (G) having a graft layer outside the particulate rubber”]. ], (C) soft aliphatic polyester [“may be referred to as soft aliphatic polyester (H)”] and the like. These can be used individually by 1 type or in combination of 2 or more types.
- the polyglycerol fatty acid ester is an ester obtained by reacting polyglycerol with a fatty acid.
- the polyglycerin that is a constituent of the polyglycerin fatty acid ester include diglycerin, triglycerin, tetraglycerin, pentaglycerin, hexaglycerin, heptaglycerin, octaglycerin, nonaglycerin, decaglycerin, and dodecaglycerin. These are used individually by 1 type or in mixture of 2 or more types.
- the average degree of polymerization of polyglycerol is preferably 2 to 10.
- fatty acid which is the other component of the polyglycerol fatty acid ester
- a fatty acid having 12 or more carbon atoms is used.
- Specific examples of fatty acids include lauric acid, myristic acid, palmitic acid, stearic acid, oleic acid, linoleic acid, linolenic acid, eicosadienoic acid, arachidonic acid, behenic acid, erucic acid, ricinoleic acid, 12-hydroxystearic acid, hydrogenated Castor oil fatty acid and the like. These are used individually by 1 type or in mixture of 2 or more types.
- Polyglycerin condensed hydroxy fatty acid ester is an ester obtained by reacting polyglycerol with condensed hydroxy fatty acid. What was illustrated as a structural component of the said polyglycerol fatty acid ester as a polyglycerol which is a structural component of polyglycerol condensed hydroxy fatty acid ester is mentioned.
- the condensed hydroxy fatty acid which is the other component of the polyglycerol condensed hydroxy fatty acid ester is a condensate of hydroxy fatty acid.
- the hydroxy fatty acid may be any fatty acid having one or more hydroxyl groups in the molecule, and examples thereof include ricinoleic acid, 12-hydroxystearic acid, hydrogenated castor oil fatty acid and the like.
- the condensation degree of the condensed hydroxy acid is, for example, 3 or more, preferably 3 to 8.
- the condensed hydroxy fatty acid is used alone or in combination of two or more.
- polyglycerol fatty acid ester and the polyglycerol condensed hydroxy fatty acid ester.
- examples of commercially available polyglycerin fatty acid esters include the tyrazole series such as “Tirabazole VR-10” and “Tirabazole VR-2” manufactured by Taiyo Kagaku Co., Ltd.
- examples of the particulate rubber constituting the core include acrylic rubber, butadiene rubber, and silicone / acrylic composite rubber. Can be mentioned.
- examples of the polymer constituting the shell include styrene resins such as polystyrene and acrylic resins such as polymethyl methacrylate.
- the average particle size (aggregate of primary particles) of the core-shell structure polymer is, for example, 50 to 500 ⁇ m, preferably 100 to 250 ⁇ m. When this is blended with polylactic acid (A) and melt-kneaded, primary particles are dispersed.
- the average particle diameter of the primary particles is, for example, 0.1 to 0.6 ⁇ m.
- the core-shell structure polymer examples include paraloid series (particularly, paraloid EXL series) such as trade name “Paraloid EXL2315” manufactured by Rohm and Haas Japan, and trade name “Metabrene” manufactured by Mitsubishi Rayon Co., Ltd. Metabrene S type such as “S-2001”, Metabrene W type such as “Metabrene W-450A”, Metabrene C type such as “Metabrene C-223A”, and Metabrene E type such as “Metabrene E-901”.
- paraloid series particularly, paraloid EXL series
- Paraloid EXL2315 trade name “Paraloid EXL2315” manufactured by Rohm and Haas Japan
- Metalabrene manufactured by Mitsubishi Rayon Co., Ltd.
- Metabrene S type such as “S-2001”
- Metabrene W type such as “Metabrene W-450A”
- Metabrene C type such as “Metabrene C-223A”
- the (c) soft aliphatic polyester includes aliphatic polyester and aliphatic-aromatic copolymer polyester.
- the soft aliphatic polyester (aliphatic polyester, aliphatic-aromatic copolymer polyester) is a polyester obtained from a polyhydric alcohol such as a diol and a polycarboxylic acid such as a dicarboxylic acid, Examples include polyesters in which at least aliphatic diols are used and at least aliphatic dicarboxylic acids are used as dicarboxylic acids; polymers of aliphatic hydroxycarboxylic acids having 4 or more carbon atoms, and the like.
- aliphatic diol examples include ethylene glycol, 1,2-propanediol, 1,3-propanediol, 2,2-dimethyl-1,3-propanediol, 1,2-butanediol, and 1,4-butane.
- Aliphatic diels having 2 to 12 carbon atoms such as diol, neopentyl glycol, 1,5-pentanediol, 1,6-hexanediol, cyclohexanediol, 1,3-cyclohexanedimethanol, 1,4-cyclohexanedimethanol ( And an alicyclic diol).
- aliphatic dicarboxylic acid examples include succinic acid, malonic acid, glutaric acid, adipic acid, pimelic acid, suberic acid, azelaic acid, sebacic acid, dodecanedioic acid, 1,3-cyclohexanedicarboxylic acid, 1,4-cyclohexanedicarboxylic acid
- saturated aliphatic dicarboxylic acids having 2 to 12 carbon atoms including alicyclic dicarboxylic acids
- the ratio of the aliphatic diol to the entire diol component is, for example, 80% by weight or more, preferably 90%. % By weight or more, more preferably 95% by weight or more, and the remainder may be an aromatic diol or the like.
- the proportion of the aliphatic dicarboxylic acid in the entire dicarboxylic acid component is, for example, 20% by weight. As mentioned above, Preferably it is 30 weight% or more, More preferably, it is 50 weight% or more, An aromatic dicarboxylic acid (for example, terephthalic acid etc.) etc. may be sufficient as the remainder.
- aliphatic hydroxycarboxylic acid having 4 or more carbon atoms examples include hydroxycarboxylic acids having 4 to 12 carbon atoms such as hydroxybutyric acid, hydroxyvaleric acid, hydroxypentanoic acid, hydroxyhexanoic acid, hydroxydecanoic acid, and hydroxydodecanoic acid. Is mentioned.
- the weight average molecular weight of the soft aliphatic polyester is, for example, 50,000 to 400,000, preferably 80,000 to 250,000.
- Typical examples of soft aliphatic polyesters include polybutylene succinate, polybutylene succinate adipate, polyethylene succinate, polyethylene succinate adipate, polybutylene adipate terephthalate, polybutylene sebacate terephthalate, polyhydroxyl alkanoate Etc.
- (C) Commercially available products can be used as the soft aliphatic polyester.
- polybutylene succinate trade name “GS Pla AZ91T” manufactured by Mitsubishi Chemical Corporation
- polybutylene succinate adipate trade name “GS Pla AD92W” manufactured by Mitsubishi Chemical Corporation
- polybutylene adipate terephthalate trade name “Eco-Flex” manufactured by BASF Japan may be mentioned.
- the content (total amount) of the tear resistance modifier (E) in the polylactic acid film or sheet is usually 1 with respect to 100 parts by weight of the polylactic acid (A) from the viewpoint of the tear resistance modification effect. Part by weight or more, preferably 1.5 parts by weight or more, more preferably 2 parts by weight or more. If the amount of the tear resistance modifier (E) is too large, the degree of crystallization and the crystallization speed are likely to decrease, the heat resistance is likely to decrease, and the tear resistance modifier (E) is reduced. May bleed out. From such a viewpoint, the content (total amount) of the tear resistance modifier (E) in the polylactic acid film or sheet is preferably 30 parts by weight or less with respect to 100 parts by weight of the polylactic acid (A). More preferably, it is 25 parts by weight or less, more preferably 20 parts by weight or less, and particularly preferably 15 parts by weight or less.
- the tear resistance modifier (E) (a) polyglycerol fatty acid ester and / or polyglycerol condensed hydroxy fatty acid ester (F), or (b) a core-shell structure having a graft layer outside the particulate rubber
- the content [total amount of (a) or total amount of (b)] is usually 1 part by weight or more per 100 parts by weight of polylactic acid (A), respectively. Preferably it is 1.5 weight part or more, More preferably, it is 2 weight part or more.
- the soft aliphatic polyester (H) is used as the tear resistance modifier (E)
- the content thereof is usually based on 100 parts by weight of the polylactic acid (A). 5 parts by weight or more, preferably 10 parts by weight or more.
- it is 30 weight part or less, More preferably, it is 25 weight part or less, More preferably, it is 20 weight part or less.
- seat improves significantly by containing soft aliphatic polyester (H) in said range.
- the tear strength of the polylactic acid film or sheet can be 2.5 N / mm or more in both the flow direction (MD direction) and the width direction (TD direction).
- the polylactic acid film or sheet of the present invention may contain an acidic functional group-modified olefin polymer (D) in addition to the above components.
- Roll lubricity can be imparted by blending the acidic functional group-modified olefin polymer (D) with the polylactic acid (A). For this reason, when the polylactic acid film or sheet of the present invention is melted by a calender film forming machine or the like and passed through the gap between the metal rolls, the film or sheet is easily peeled off from the surface of the metal roll. And it can form into a film smoothly.
- the acidic functional group-modified olefin polymer (D) can be used alone or in combination of two or more.
- Examples of the acidic functional group of the acidic functional group-modified olefin polymer (D) include a carboxyl group or a derivative group thereof.
- a derivative group of a carboxyl group is chemically derived from a carboxyl group, and examples thereof include an acid anhydride group, an ester group, an amide group, an imide group, and a cyano group of carboxylic acid. Among these, a carboxylic anhydride group is preferable.
- the acidic functional group-modified olefin polymer (D) is abbreviated as, for example, an unsaturated compound containing the above “acidic functional group” in an unmodified polyolefin polymer (hereinafter referred to as “acidic functional group-containing unsaturated compound”). In some cases).
- Non-modified polyolefin polymers include high density polyethylene, medium density polyethylene, low density polyethylene, polypropylene, polybutene, poly-4-methylpentene-1, copolymers of ethylene and ⁇ -olefins, propylene and ⁇ -olefins.
- Polyolefins such as copolymers or oligomers thereof; ethylene-propylene rubber, ethylene-propylene-diene copolymer rubber, butyl rubber, butadiene rubber, low crystalline ethylene-propylene copolymer, propylene-butene copolymer, Polyolefin such as ethylene-vinyl ester copolymer, ethylene-methyl (meth) acrylate copolymer, ethylene-ethyl (meth) acrylate copolymer, ethylene-maleic anhydride copolymer, blend of polypropylene and ethylene-propylene rubber system Elastomers; and mixtures of two or more of these.
- polypropylene a copolymer of propylene and ⁇ -olefin, low density polyethylene and oligomers thereof
- a copolymer of polypropylene, propylene and ⁇ -olefin and oligomers thereof is particularly preferred.
- oligomers include those obtained from a corresponding polymer by a molecular weight degradation method by thermal decomposition. Oligomers can also be obtained by polymerization methods.
- Examples of the acidic functional group-containing unsaturated compound include a carboxyl group-containing unsaturated compound and a carboxyl group derivative-containing unsaturated compound.
- Examples of the carboxyl group-containing unsaturated compound include maleic acid, itaconic acid, chloroitaconic acid, chloromaleic acid, citraconic acid, and (meth) acrylic acid.
- carboxyl group derivative-containing unsaturated compound examples include, for example, maleic anhydride, itaconic anhydride, chloroitaconic anhydride, chloromaleic anhydride, citraconic anhydride and other carboxylic anhydride group-containing unsaturated compounds; (Meth) acrylate, glycidyl (meth) acrylate, (meth) acrylic acid ester such as 2-hydroxyethyl (meth) acrylate; (meth) acrylamide, maleimide, (meth) acrylonitrile and the like.
- a carboxyl group-containing unsaturated compound and a carboxylic acid anhydride group-containing unsaturated compound are preferable, an acid anhydride group-containing unsaturated compound is more preferable, and maleic anhydride is particularly preferable.
- the weight average molecular weight of the acidic functional group-modified olefin polymer (D) is 10,000 to 80,000, preferably 15,000 to 70,000, more preferably 20,000 to 60,000. If this weight average molecular weight is less than 10,000, it tends to cause bleed-out after film or sheet molding, and if it exceeds 80000, it may separate from polylactic acid (A) during roll kneading.
- bleed-out refers to a phenomenon in which a low molecular weight component appears on the film or sheet surface over time after the film or sheet is formed.
- the acidic functional group in the acidic functional group-modified olefin polymer (D) may be bonded to any position of the olefin polymer, and the modification ratio is not particularly limited, but the acidic functional group-modified olefin polymer (D).
- the acid value of is usually 10 to 70 mgKOH / g, preferably 20 to 60 mgKOH / g. If the acid value is less than 10 mgKOH / g, the effect of improving roll lubricity cannot be obtained, and if it exceeds 70 mgKOH / g, it tends to cause plate-out to the roll.
- the plate-out to the roll means that when the resin composition is melt-formed using a metal roll, the components blended in the resin composition or the oxidized, decomposed, combined, or deteriorated product is a metal. Attaching or depositing on the surface of a roll.
- the “acid value” means a value measured according to the neutralization titration method of JIS K0070-1992.
- the acidic functional group-modified olefin polymer (D) can be obtained by reacting an unmodified polyolefin polymer with an acidic functional group-containing unsaturated compound in the presence of an organic peroxide.
- an organic peroxide those generally used as an initiator in radical polymerization can be used.
- Such a reaction can be performed by either a solution method or a melting method.
- an acidic functional group-modified olefin polymer (D) is obtained by dissolving a mixture of an unmodified polyolefin polymer and an acidic functional group-containing unsaturated compound in an organic solvent together with an organic peroxide and heating. Can do.
- the reaction temperature is preferably about 110 to 170 ° C.
- an acidic functional group-modified olefin polymer (D) is obtained by mixing a mixture of an unmodified polyolefin polymer and an acidic functional group-containing unsaturated compound with an organic peroxide, melting and reacting.
- Melt mixing can be performed with various mixers such as an extruder, a plastic bender, a kneader, and a Banbury mixer, and the kneading temperature is usually in the temperature range of the melting point of the unmodified polyolefin polymer to 300 ° C.
- the acidic functional group-modified olefin polymer (D) is preferably maleic anhydride-modified polypropylene.
- the acidic functional group-modified olefin polymer (D) a commercially available product can be used.
- trade name “Yumex 1010” maleic anhydride group-modified polypropylene, acid value: 52 mgKOH / g, manufactured by Sanyo Chemical Industries, Ltd.) (Weight average molecular weight: 32000, modification ratio: 10% by weight)
- “Yumex 1001” maleic anhydride group-modified polypropylene, acid value: 26 mg KOH / g, weight average molecular weight: 49000, modification ratio: 5% by weight
- “Yumex 2000 Maleic anhydride group-containing modified polyethylene, acid value: 30 mg KOH / g, weight average molecular weight: 20000, modification ratio: 5% by weight
- the content of the acidic functional group-modified olefin polymer (D) in the polylactic acid film or sheet of the present invention is not particularly limited, and is usually 0.1 to 10 parts by weight with respect to 100 parts by weight of the polylactic acid (A). From the viewpoint of sustaining the roll slip effect without plate-out to the roll and maintaining the degree of biomass, the amount is preferably 0.1 to 5 parts by weight, particularly preferably 0.3 to 3 parts by weight. If the content of the acidic functional group-modified olefin polymer (D) is less than 0.1 parts by weight, the effect of improving roll lubricity is difficult to obtain, and if it exceeds 10 parts by weight, the effect according to the amount added cannot be obtained. In addition, a decrease in biomass degree becomes a problem.
- the polylactic acid-based film or sheet of the present invention may contain various additives as necessary within a range not impairing the effects of the present invention.
- additives include antioxidants, ultraviolet absorbers, plasticizers, stabilizers, mold release agents, antistatic agents, colorants (white pigments, etc.), anti-drip agents, flame retardants, hydrolysis inhibitors, A foaming agent, a filler, etc. are mentioned.
- the polylactic acid film or sheet of the present invention Since the polylactic acid film or sheet of the present invention has a high degree of crystallization, it has excellent solvent resistance.
- the degree of swelling of the polylactic acid film or sheet of the present invention is, for example, 2.5 or less, preferably 2.0 or less, with respect to any solvent of ethyl acetate and toluene.
- the degree of swelling was determined by immersing a film or sheet sample (50 mm ⁇ 50 mm ⁇ 0.05 mm thickness) in a solvent for 15 minutes, removing the solvent on the surface of the taken film or sheet sample with a waste cloth, and immersing the weight after immersion. Can be measured by dividing by previous weight.
- the polylactic acid film or sheet of the present invention can maintain a high level of mechanical properties such as rigidity and elastic properties.
- the initial elastic modulus of the polylactic acid film or sheet of the present invention is usually 1000 MPa or more, preferably 1500 MPa or more in the flow direction (MD direction).
- the upper limit of the initial elastic modulus is generally about 3500 MPa (for example, about 3000 MPa) in the flow direction (MD direction).
- the breaking strength of the polylactic acid film or sheet of the present invention is usually 30 MPa or more, preferably 35 MPa or more in the flow direction (MD direction).
- the upper limit of the breaking strength is generally about 150 MPa (for example, about 120 MPa).
- the elongation of the polylactic acid film or sheet of the present invention is usually 2.5% or more, preferably 3.5% or more in the flow direction (MD direction).
- the upper limit of elongation is generally about 15% (for example, about 12%) in the flow direction (MD direction).
- the elongation of the polylactic acid film or sheet is usually 5% or more in the flow direction (MD direction).
- the upper limit of elongation is generally 150%, preferably 120%, more preferably 100% in the flow direction (MD direction).
- the initial elastic modulus, breaking strength, and elongation were measured using a tensile tester according to the plastic-tensile property test method of JIS K 7161.
- Apparatus Tensile tester (Autograph AG-20kNG, manufactured by Shimadzu Corporation) Sample size: thickness 0.05 mm x width 10 mm x length 100 mm (note that the direction parallel to the length direction is the flow direction (MD) during film formation) Measurement condition: Distance between chucks: 50mm Tensile speed: 300 mm / min
- the thickness of the polylactic acid film or sheet of the present invention is not particularly limited, but is usually 10 to 500 ⁇ m, preferably 20 to 400 ⁇ m, more preferably 30 to 300 ⁇ m.
- the polylactic acid-based film or sheet of the present invention can be widely used in the same applications as commonly used films or sheets.
- the base material of the separator, the protective film used for the adhesive tape or sheet substrate, the adhesive tape or sheet, etc. can be suitably used as a base material or the like.
- a method for producing the polylactic acid film or sheet of the present invention is not particularly limited, but a method of forming a resin composition containing polylactic acid (A) by a melt film formation method is preferable.
- the melt film formation method is employed, the melting endotherm ⁇ Hc ′ of the crystallized portion during film formation can be easily set to 10 J / g or more.
- the polylactic acid film or sheet of the present invention is obtained by uniformly dispersing each component using a continuous melt kneader such as a twin screw extruder or a batch melt kneader such as a pressure kneader, a Banbury mixer, or a roll kneader.
- a polylactic acid (A) -containing resin composition can be prepared, and this can be produced by film formation and cooling and solidification by an extrusion method such as a T-die method or an inflation method, a calendar method, a polishing method, or the like.
- the melt film formation method is preferably a technique in which a molten resin composition is formed into a desired thickness by passing through a gap between two metal rolls, and particularly preferably a calendar method. This is a polishing method.
- the temperature of the resin composition at the time of melt film formation (hereinafter referred to as the resin temperature at the time of melt film formation) is not particularly limited, The temperature is preferably between the crystallization temperature (Tc) + 15 ° C. in the temperature-lowering process of the resin composition and the melting temperature (Tm) ⁇ 5 ° C. in the temperature-raising process. By setting this temperature, the crystallization of polylactic acid (A) is promoted, and the film or sheet of the present invention can easily obtain heat resistance.
- the temperature of the resin composition at the time of calender roll rolling (corresponding to the resin temperature at the time of melt film-forming) is reduced in the temperature-decreasing process of the resin composition. Is set to a temperature between the crystallization temperature (Tc) + 15 ° C. and the melting temperature (Tm) ⁇ 5 ° C. in the heating process. By rolling at a temperature below the melting point in this way, oriented crystallization is promoted. This effect of promoting orientation crystallization is markedly improved when the resin composition contains a tetrafluoroethylene-based polymer (B ′).
- the tetrafluoroethylene-based polymer (B ′) is considered to promote oriented crystallization by fibrillating and networking in the resin composition and a synergistic effect of its effect as a crystal nucleating agent. Therefore, by rolling in the above temperature range, the film or sheet of the present invention can realize good heat resistance due to the orientation crystallization promoting effect as well as the smooth surface state.
- a process for suppressing the temperature condition after melt film formation is provided in order to make the crystallization promoting effect of the tetrafluoroethylene polymer (B ′) more effective. It may be.
- the resin composition that has been dissolved and formed is temporarily maintained at a crystallization temperature (Tc) of + 25 ° C. from a temperature of crystallization temperature (Tc) of ⁇ 25 ° C. in the process of lowering the temperature of the resin composition. And may be cooled and solidified after undergoing a step of promoting crystallization (hereinafter sometimes simply referred to as “crystallization promoting step”).
- the crystallization promotion step refers to the temperature of the resin composition in the process of lowering the temperature of the resin composition from the temperature of crystallization temperature (Tc) ⁇ 25 ° C. to the crystallization temperature (Tc) + 10 ° C.
- This is a step of exposing to a temperature-controlled state, and a step of promoting crystallization of the resin composition while maintaining a smooth surface state after melt film formation.
- the temperature control method is not particularly limited, and examples thereof include a method in which the resin composition that has been melt-formed is brought into direct contact with a roll or belt that can be heated to a predetermined temperature.
- the resin composition formed into a melt into contact with a metal roll having a predetermined surface temperature it is desirable to bring the resin composition formed into a melt into contact with a metal roll having a predetermined surface temperature. Therefore, it is desirable that the polylactic acid (A) -containing resin composition be easily peelable from the metal roll also in this step. From this viewpoint, the addition of the acidic functional group-modified olefin polymer (D) is desirable. preferable.
- the time for the crystallization promoting step is preferably as long as possible, and ultimately depends on the degree of crystallization of the resin composition, so it cannot be specified unconditionally, but usually 2 to 10 seconds, preferably 3 to 8 seconds. It is.
- the crystallization promotion step even if the crystallization temperature (Tc) in the temperature-decreasing process of the resin composition changes due to the addition of another crystal nucleating agent or the like, the measurement is performed in advance with a differential scanning calorimeter (DSC).
- DSC differential scanning calorimeter
- the method for producing the polylactic acid film or sheet of the present invention is preferably a method comprising forming a polylactic acid (A) -containing resin composition by a melt film formation method,
- the temperature of the resin composition (resin temperature at the time of melt film formation) is from the temperature of the crystallization temperature (Tc) + 15 ° C. in the temperature lowering process of the resin composition to the melting temperature (Tm) ⁇ 5 in the temperature rising process.
- the crystallization temperature (Tc) of the resin composition that is at a temperature between 0 ° C. and / or melt-deposited is from the crystallization temperature (Tc) in the process of lowering the temperature of the resin composition to 25 ° C. It is a method characterized by cooling and solidifying after a crystallization promoting step at + 10 ° C.
- the resin composition was cooled and solidified after the crystallization promotion step in the crystallization promotion step. It does not cause extreme heat shrinkage when using the film or sheet. Therefore, the highly crystallized film or sheet of the present invention formed by the above manufacturing method can maintain the shape up to the vicinity of the melting point of polylactic acid, and can be used sufficiently even in applications requiring heat resistance that could not be used so far. It becomes possible. Furthermore, since the inefficient process of heating once again after cooling and solidification becomes unnecessary, the above manufacturing method can be said to be a very useful method in terms of economy and productivity.
- the crystallization can be further improved by performing the stretching treatment.
- the stretching temperature is, for example, 60 to 100 ° C.
- a method for producing a polylactic acid-based film or sheet including the crystallization promotion step a method of continuously performing from the melt film formation step to the crystallization promotion step and the cooling and solidification step shortens the processing time. This is desirable. Examples of such a method include a method using a calendar film forming machine, a polishing film forming machine, and the like.
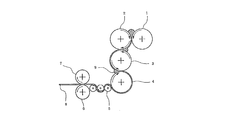
- FIG. 1 shows a schematic diagram of an example of a calendar film forming machine used in such a manufacturing method. In the following, FIG. 1 will be described in detail.
- the resin composition in a molten state is rolled between four calender rolls of the first roll 1, the second roll 2, the third roll 3, and the fourth roll 4, and gradually thinned. It adjusts so that it may become desired thickness when passing between the roll 3 and the 4th roll 4.
- FIG. In the case of calendar film formation the film formation of the resin composition on the first roll 1 to the fourth roll 4 corresponds to the “melt film formation step”.
- the take-off roll 5 that is set from the crystallization temperature (Tc) ⁇ 25 ° C. to the crystallization temperature (Tc) + 10 ° C. in the process of lowering the temperature of the resin composition,
- the roll group which contacts first is shown, is comprised of one or two or more (three in FIG.
- the take-off roll 5 when the take-off roll 5 is composed of a plurality of rolls and the temperature of each roll can be adjusted, the temperature of each roll is preferably the same, but if within the desired temperature range, May be different. The larger the number of take-off rolls 5, the longer the isothermal crystallization time, which is advantageous for promoting crystallization. In the case of calendar film formation, the take-off roll 5 accelerates the crystallization of the resin composition 8 that has been melt-formed, and thus the process of passing the resin composition 8 through the take-off roll 5 is the “crystallization promotion process”. It corresponds to.
- the two cooling rolls 6 and 7 serve to cool and solidify the resin composition 8 by passing the resin composition 8 between them, and to mold the surface thereof into a desired shape. Therefore, one roll (for example, the cooling roll 6) is usually a metal roll, and the roll surface is designed to bring out the surface shape of the resin composition 8, and the other roll (for example, the cooling roll 7). ) Rubber rolls are used.
- the arrow in a figure shows the rotation direction of a roll. 9 is a bank (resin pool).
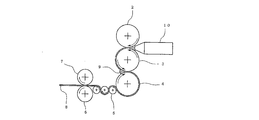
- FIG. 2 shows a schematic diagram of an example of a polishing film forming machine used in such a manufacturing method. In the following, FIG. 2 will be described in detail.
- An extruder tip 10 of an extruder (not shown) is disposed between the heated second roll 2 and third roll 3, and the second roll 3 and third roll 3 are set at a preset extrusion speed.
- the molten resin composition 8 is continuously extruded.
- the extruded resin composition 8 is rolled and thinned between the second roll 2 and the third roll 3, and finally has a desired thickness when it passes between the third roll 3 and the fourth roll 4. It is adjusted to become.
- the film formation of the resin composition 8 on the second roll 2 to the fourth roll 4 corresponds to a “melt film formation process”.
- the resin composition 8 passes through three take-off rolls 5 set to a crystallization temperature (Tc) + 10 ° C.
- the film or sheet of the present invention can be widely used for the same applications as those of generally used films or sheets.
- the separator of the present invention is a separator having a release agent-treated layer on at least one surface of a separator base material, and the separator base material is composed of the polylactic acid film or sheet. .
- the release agent (release agent) used for forming the release agent treatment layer is not particularly limited, and examples thereof include silicone release agents, fluorine release agents, long-chain alkyl release agents, polyolefin release agents, and the like. Can be used.
- the release agents can be used alone or in combination of two or more.
- the release agent is not particularly limited as long as it can form a film on the separator substrate, and the film exhibits an appropriate release property according to the application and does not adversely affect the adhesive.
- the peeling force on the adhesive surface is about 0 to 25 N / 50 mm (preferably about 0.1 to 10 N / 50 mm). That which can form the film which becomes) is preferable.
- silicone release agents are preferable from the viewpoint of release performance. Although it does not specifically limit as a silicone type release agent, The thermosetting addition type silicone type release agent (thermosetting addition type polysiloxane type release agent) is mentioned as the typical thing.
- thermosetting addition-type silicone release agent essentially comprises a polyorganosiloxane containing an alkenyl group as a functional group in the molecule (alkenyl group-containing silicone) and a polyorganosiloxane containing a hydrosilyl group as a functional group in the molecule. Ingredients.
- the polyorganosiloxane having an alkenyl group as a functional group in the molecule a polyorganosiloxane having two or more alkenyl groups in the molecule is preferable.
- the alkenyl group include a vinyl group (ethenyl group), an allyl group (2-propenyl group), a butenyl group, a pentenyl group, and a hexenyl group.
- the alkenyl group is usually bonded to a silicon atom (for example, a terminal silicon atom or a silicon atom inside the main chain) of the polyorganosiloxane forming the main chain or skeleton.
- polyalkylalkylsiloxanes such as polydimethylsiloxane, polydiethylsiloxane, and polymethylethylsiloxane, and polyalkylarylsiloxanes.
- a copolymer in which a plurality of silicon atom-containing monomer components are used [for example, poly (dimethylsiloxane-diethylsiloxane) and the like] and the like. Of these, polydimethylsiloxane is preferred.
- polydimethylsiloxane having a vinyl group, hexenyl group or the like as a functional group is preferably exemplified.
- the polyorganosiloxane crosslinking agent containing a hydrosilyl group as a functional group in the molecule is a polyorgano having a hydrogen atom bonded to a silicon atom (particularly, a silicon atom having a Si—H bond) in the molecule.
- a siloxane, particularly a polyorganosiloxane having two or more silicon atoms having Si—H bonds in the molecule is preferred.
- the silicon atom having the Si—H bond may be either a silicon atom in the main chain or a silicon atom in the side chain, that is, may be contained as a constituent unit of the main chain, or It may be contained as a constituent unit of the side chain.
- the number of silicon atoms in the Si—H bond is not particularly limited as long as it is 2 or more.
- Specific examples of the polyorganosiloxane crosslinking agent containing a hydrosilyl group as a functional group in the molecule are polymethylhydrogensiloxane and poly (dimethylsiloxane-methylhydrogensiloxane).
- a reaction inhibitor may be used in order to impart storage stability at room temperature.
- a thermosetting addition type silicone release agent specifically, for example, 3,5-dimethyl-1-hexyn-3-ol, 3-methyl-1-pentene- Examples include 3-ol, 3-methyl-3-penten-1-yne, and 3,5-dimethyl-3-hexen-1-yne.
- a release control agent or the like may be used as necessary in addition to the above components.
- a release control agent such as MQ resin, a polyorganosiloxane having no alkenyl group or hydrosilyl group (trimethylsiloxy group terminal) Sealed polydimethylsiloxane and the like
- the content of these components in the thermosetting addition-type silicone release agent is not particularly limited, but is preferably 1 to 30% by weight.
- additives may be used for the release agent as necessary.
- examples of the additive used as necessary include a filler, an antistatic agent, an antioxidant, an ultraviolet absorber, a plasticizer, and a colorant (dye, pigment, etc.).
- the release agent-treated layer can be formed by a known or commonly used method. For example, the method etc. which apply
- the coating can be performed by a coater, an extruder, a printing machine or the like generally used for forming a release treatment layer.
- the thickness of the release agent treatment layer can be appropriately selected depending on the application and the like, and is, for example, 0.02 to 1 ⁇ m, preferably 0.05 to 0.7 ⁇ m, and more preferably about 0.1 to 0.5 ⁇ m. .
- the separator of the present invention may have another layer (intermediate layer) between the separator substrate and the release agent treatment layer as necessary.
- the pressure-sensitive adhesive tape or sheet of the present invention is a pressure-sensitive adhesive tape or sheet having a pressure-sensitive adhesive layer on at least one surface of the substrate, and the substrate is composed of the polylactic acid film or sheet.
- the surface of the substrate may have a conventional surface treatment, for example, chromic acid treatment, ozone exposure, flame exposure, high piezoelectric impact exposure, ionization, in order to improve adhesion with an adjacent layer.
- Oxidation treatment or the like by a chemical or physical method such as radiation treatment may be performed.
- the pressure-sensitive adhesive constituting the pressure-sensitive adhesive layer is not particularly limited.
- a rubber-based pressure-sensitive adhesive an acrylic pressure-sensitive adhesive, a vinyl alkyl ether-based pressure-sensitive adhesive, a silicone-based pressure-sensitive adhesive, a polyester-based pressure-sensitive adhesive, and a polyamide-based pressure-sensitive adhesive.
- Known adhesives such as urethane adhesives, styrene-diene block copolymer adhesives, and adhesives with improved creep characteristics in which a hot-melt resin having a melting point of about 200 ° C. or less is blended with these adhesives.
- the pressure-sensitive adhesive may be any known pressure-sensitive adhesive such as a solvent type, an emulsion type, a hot melt type, an energy ray curable pressure sensitive adhesive, and a heat release type.
- the pressure-sensitive adhesive a rubber-based pressure-sensitive adhesive using natural rubber or various synthetic rubbers as a base polymer; an acrylic polymer using one or more (meth) acrylic acid alkyl esters as monomer components
- An acrylic pressure-sensitive adhesive having a base polymer (homopolymer or copolymer) is used.
- an acrylic adhesive having an acrylic polymer as a base polymer is particularly preferably used.
- Examples of the (meth) acrylic acid alkyl ester used as the monomer component of the acrylic polymer include, for example, methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, and (meth) acrylic.
- the acrylic polymer is a unit corresponding to another monomer component copolymerizable with the (meth) acrylic acid alkyl ester, if necessary, for the purpose of modifying cohesive strength, heat resistance, crosslinkability and the like. May be included.
- a monomer component include an aliphatic cyclic skeleton such as cyclopentyl (meth) acrylate, cyclohexyl (meth) acrylate, cyclohexylmethyl (meth) acrylate, and bornyl (meth) acrylate (meta ) Acrylic acid ester; (meth) acrylic acid ester having an aromatic carbon ring such as phenyl (meth) acrylate and benzyl (meth) acrylate; acrylic acid, methacrylic acid, carboxyethyl acrylate, carboxypentyl acrylate, itaconic acid, Carboxylic group-containing monomers such as maleic acid, fumaric acid and crotonic acid; Acid anhydride
- Glycol-based acrylic ester monomers N- (meth) acryloylmorpholine, tetrahydrofurfuryl (meth) acrylate, fluorine (meth) acrylate, silicone (meth) acrylate and other heterocyclic rings, halogen atoms, silicon atoms, etc.
- Monomers hexanediol di (meth) acrylate, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, neopentyl Recall di (meth) acrylate, pentaerythritol di (meth) acrylate, trimethylolpropane tri (meth) acrylate, pentaerythritol tri (meth) acrylate, dipentaerythritol hexa (meth) acrylate, epoxy acrylate, polyester acrylate, urethane acrylate, etc.
- Polyfunctional monomers Olefin monomers such as isoprene, butadiene, and isobutylene
- Vinyl ether monomers such as vinyl ether.
- the acrylic polymer can be produced by a known radical polymerization method such as solution polymerization, bulk polymerization, and emulsion polymerization.
- the acrylic polymer may be any of a random copolymer, a block copolymer, a graft polymer, and the like.
- a commonly used polymerization initiator or chain transfer agent can be used.
- the weight average molecular weight of the base polymer constituting the pressure-sensitive adhesive is, for example, 10,000 to 2,000,000, preferably 300,000 to 1,500,000.
- the weight average molecular weight of the base polymer is too low, although excellent in the followability with the adherend, for example, when peeling by heating, contamination such as adhesive residue tends to occur on the adherend.
- the weight average molecular weight of the base polymer is too high, the followability to the adherend tends to decrease.
- crosslinking agents epoxy crosslinking agents, isocyanate crosslinking agents, melamine crosslinking agents, oxazoline crosslinking agents, aziridine crosslinking agents, metal chelate compounds, etc.
- crosslinking Accelerator crosslinking catalyst
- tackifier for example, rosin derivative resin, polyterpene resin, petroleum resin, oil-soluble phenol resin, etc.
- thickener plasticizer
- filler foaming agent
- an anti-aging agent antioxidant
- an appropriate additive such as an ultraviolet absorber, an antistatic agent, a surfactant, a leveling agent, a colorant, a flame retardant, and a silane coupling agent.
- the formation of the pressure-sensitive adhesive layer can be performed by a known or conventional method. For example, a method of applying the pressure-sensitive adhesive composition onto a base material (if an intermediate layer is present on the base material, the intermediate layer), a method of applying the pressure-sensitive adhesive composition onto an appropriate separator to form a pressure-sensitive adhesive layer Thereafter, a method of transferring (transferring) the pressure-sensitive adhesive layer onto a base material (in the case where an intermediate layer is present on the base material, the intermediate layer) may be mentioned.
- the coating can be performed by a coater, an extruder, a printing machine or the like generally used for forming the pressure-sensitive adhesive layer.
- the thickness of the pressure-sensitive adhesive layer can be appropriately selected depending on the application and the like, and is, for example, about 5 to 3000 ⁇ m, preferably about 10 to 500 ⁇ m.
- the pressure-sensitive adhesive strength of the pressure-sensitive adhesive layer at 25 ° C. can be appropriately selected according to the use (weak adhesion type, strong adhesion type, etc.), for example, 3 0.0 N / 20 mm or more, preferably 5.0 N / 20 mm or more, more preferably 7.0 N / 20 mm or more. In the case of a strong adhesive type, 10.0 N / 20 mm or more is more preferable.
- the pressure-sensitive adhesive tape or sheet of the present invention may have another layer (intermediate layer; for example, an elastic layer, a rigid layer, etc.) between the base material and the pressure-sensitive adhesive layer as necessary.
- a separator for protecting the pressure-sensitive adhesive layer may be provided on the pressure-sensitive adhesive layer until the pressure-sensitive adhesive tape or sheet is used.
- the protective film of the present invention is a protective film having a releasable pressure-sensitive adhesive layer on at least one surface of a base material, and the base material is composed of the polylactic acid film or sheet.
- the surface of the substrate may have a conventional surface treatment, for example, chromic acid treatment, ozone exposure, flame exposure, high piezoelectric impact exposure, ionization, in order to improve adhesion with an adjacent layer.
- Oxidation treatment or the like by a chemical or physical method such as radiation treatment may be performed.
- the pressure-sensitive adhesive constituting the re-peelable pressure-sensitive adhesive layer is not particularly limited.
- a rubber-based pressure-sensitive adhesive an acrylic pressure-sensitive adhesive, a vinyl alkyl ether pressure-sensitive adhesive, a silicone pressure-sensitive adhesive, a polyester pressure-sensitive adhesive, and a polyamide.
- Type adhesives urethane type adhesives, styrene-diene block copolymer type adhesives, known adhesives such as those having improved melting characteristics and blending these adhesives with a hot-melt resin having a melting point of about 200 ° C. or less
- One or more pressure-sensitive adhesives can be used in combination.
- the pressure-sensitive adhesive may be any known removable pressure-sensitive adhesive such as a solvent type, an emulsion type, or a hot melt type.
- a rubber-based pressure-sensitive adhesive having a base polymer of natural rubber or various synthetic rubbers; one or more of (meth) acrylic acid alkyl esters are used as monomer components.
- An acrylic pressure-sensitive adhesive having an acrylic polymer (homopolymer or copolymer) as a base polymer is used.
- an acrylic adhesive having an acrylic polymer as a base polymer is particularly preferably used.
- Examples of the (meth) acrylic acid alkyl ester used as the monomer component of the acrylic polymer include, for example, methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, and (meth) acrylic.
- the acrylic polymer is a unit corresponding to another monomer component copolymerizable with the (meth) acrylic acid alkyl ester, if necessary, for the purpose of modifying cohesive strength, heat resistance, crosslinkability and the like. May be included.
- a monomer component include an aliphatic cyclic skeleton such as cyclopentyl (meth) acrylate, cyclohexyl (meth) acrylate, cyclohexylmethyl (meth) acrylate, and bornyl (meth) acrylate (meta ) Acrylic acid ester; (meth) acrylic acid ester having an aromatic carbon ring such as phenyl (meth) acrylate and benzyl (meth) acrylate; acrylic acid, methacrylic acid, carboxyethyl acrylate, carboxypentyl acrylate, itaconic acid, Carboxylic group-containing monomers such as maleic acid, fumaric acid and crotonic acid; Acid anhydride
- Glycol-based acrylic ester monomers N- (meth) acryloylmorpholine, tetrahydrofurfuryl (meth) acrylate, fluorine (meth) acrylate, silicone (meth) acrylate and other heterocyclic rings, halogen atoms, silicon atoms, etc.
- Monomers hexanediol di (meth) acrylate, (poly) ethylene glycol di (meth) acrylate, (poly) propylene glycol di (meth) acrylate, neopentyl Recall di (meth) acrylate, pentaerythritol di (meth) acrylate, trimethylolpropane tri (meth) acrylate, pentaerythritol tri (meth) acrylate, dipentaerythritol hexa (meth) acrylate, epoxy acrylate, polyester acrylate, urethane acrylate, etc.
- Polyfunctional monomers Olefin monomers such as isoprene, butadiene, and isobutylene
- Vinyl ether monomers such as vinyl ether.
- the acrylic polymer can be produced by a known radical polymerization method such as solution polymerization, bulk polymerization, and emulsion polymerization.
- the acrylic polymer may be any of a random copolymer, a block copolymer, a graft polymer, and the like.
- a commonly used polymerization initiator or chain transfer agent can be used.
- the weight average molecular weight of the base polymer constituting the pressure-sensitive adhesive is, for example, 10,000 to 5,000,000, preferably 20,000 to 1,000,000. If the weight average molecular weight of the base polymer is too low, the adherence to the adherend is excellent, but contamination such as adhesive residue tends to occur on the adherend after re-peeling. On the other hand, when the weight average molecular weight of the base polymer is too high, contamination due to adhesive residue or the like on the adherend after re-peeling is reduced, but the followability to the adherend tends to be lowered.
- crosslinking agents epoxy crosslinking agents, isocyanate crosslinking agents, melamine crosslinking agents, oxazoline crosslinking agents, aziridine crosslinking agents, metal chelate compounds, etc.
- crosslinking Accelerator crosslinking catalyst
- tackifier for example, rosin derivative resin, polyterpene resin, petroleum resin, oil-soluble phenol resin, etc.
- thickener plasticizer
- filler foaming agent
- an anti-aging agent antioxidant
- an appropriate additive such as an ultraviolet absorber, an antistatic agent, a surfactant, a leveling agent, a colorant, a flame retardant, and a silane coupling agent.
- a crosslinking agent in order to impart removability to the pressure-sensitive adhesive.
- the re-peelable pressure-sensitive adhesive layer can be formed by a known or conventional method. For example, a method of applying the pressure-sensitive adhesive composition onto a base material (if an intermediate layer is present on the base material, the intermediate layer), a method of applying the pressure-sensitive adhesive composition onto an appropriate separator to form a pressure-sensitive adhesive layer Thereafter, a method of transferring (transferring) the pressure-sensitive adhesive layer onto a base material (in the case where an intermediate layer is present on the base material, the intermediate layer) may be mentioned.
- the coating can be performed by a coater, an extruder, a printing machine or the like generally used for forming the pressure-sensitive adhesive layer.
- the thickness of the re-peelable pressure-sensitive adhesive layer can be appropriately selected depending on the application and the like, and is, for example, about 1 to 100 ⁇ m, preferably about 1 to 50 ⁇ m.
- the adhesive strength at 25 ° C. of the releasable pressure-sensitive adhesive layer may be in a range where re-peeling is possible, for example, less than 3 N / 20 mm. .
- the lower limit is, for example, about 0.01 N / 20 mm.
- the protective film of the present invention may have another layer (intermediate layer) between the base material and the releasable pressure-sensitive adhesive layer as necessary. Moreover, a separator for protecting the removable pressure-sensitive adhesive layer may be provided on the removable pressure-sensitive adhesive layer until the protective film is used.
- Example I-1 A resin composition was prepared at the blending ratio shown in Table 1 below, melted and kneaded with a Banbury mixer, and then formed into a calendar film so as to have a thickness of 50 ⁇ m with an inverted L-shaped four calendar (melting process). Membrane process). Next, as shown in FIG. 1, immediately after the melt film formation step, three rolls (take-off rolls) that can be heated to an arbitrary temperature are arranged so that the melt-formed resin composition can pass alternately up and down. This promoted crystallization (crystallization promoting step). Thereafter, the resin composition was solidified by passing through a cooling roll to produce a film.
- the temperature of the resin composition in the melt film formation step was the surface temperature of the roll corresponding to the fourth roll 4 in FIG. Moreover, about the temperature of the resin composition in a crystallization promotion process, the surface temperature of the three take-off rolls 5 of FIG. 1 was made substantially the same, and the temperature was made into the crystallization promotion temperature.
- the film formation rate was 5 m / min, and the substantial crystallization promotion step time (take-off roll passage time) was about 5 seconds.
- Examples I-2 to I-5 Resin compositions were prepared at the blending ratios shown in Table 1 below, and films of Examples I-2 to I-5 were produced by the same operations as in Example I-1.
- Comparative Example I-1 A resin composition was prepared at the blending ratio shown in Table 1 below, and a film of Comparative Example I-1 was produced in the same manner as in Example I-1.
- ⁇ Melting temperature (° C)> The temperature at the top of the endothermic peak that accompanies the melting in the re-heating process of the resin composition after film formation, measured by DSC (differential scanning thermal analyzer), is the melting temperature (Tm; also referred to as crystal melting peak temperature). It was.
- ⁇ Crystallization temperature (°C)> The temperature at the peak top of the exothermic peak accompanying the crystallization of the resin composition after film formation, measured by DSC, during the temperature lowering process from 200 ° C. was defined as the crystallization temperature (Tc; also referred to as crystallization peak temperature). .
- the DSC and measurement conditions used in the measurement of the melting temperature (Tm), the crystallization temperature (Tc), and the melting endotherm ⁇ Hc ′ of the crystallization part during film formation are as follows.
- the measurement apparatus and measurement conditions used are as follows. Equipment: Elmendorf-type tear tester manufactured by Tester Sangyo Co., Ltd. Conditions: Sample size: Thickness of about 0.25 mm (5 layers of 0.05 mm film) x width 76 mm x length 63 mm In addition, it cut out so that the direction parallel to a length direction might turn into the flow direction (henceforth MD direction) at the time of film film-forming, and a direction opposite to a flow direction (henceforth TD direction). Tear strength calculation method: calculated using the following formula (2).
- T (A ⁇ p) / (n ⁇ t ⁇ 1000) (2) T: Tear strength (N / mm) A: Reading of scale (mN) p: Number of test specimens to be used as a reference for the scale of the pendulum (16 in this apparatus) n: number of test pieces torn simultaneously t: average thickness (mm) per test piece
- ⁇ Swelling degree> A film or sheet sample (50 mm ⁇ 50 mm ⁇ thickness 0.05 mm) was immersed in a solvent for 15 minutes, the solvent on the surface of the sample taken out was removed with a waste cloth, and the weight after immersion was measured by dividing by the weight before immersion. .
- the solvent used was two types, toluene and ethyl acetate. Acceptance / rejection determination: 2.0 or less is accepted.
- ⁇ Workability test> A sample cut into a thickness of 0.05 mm, a width of 20 mm, and a length of 100 mm is fixed using the above tensile tester so that the stress is 25 N / mm 2 . Thereafter, a 1 mm cut is given with a razor so that the center part between the chucks is perpendicular to the MD direction. At that time, those that did not break were judged as “ ⁇ ” (good), and those that broke were judged as “x” (bad).
- the measurement apparatus and measurement conditions used are as follows.
- Tensile tester Autograph AG-20kNG, manufactured by Shimadzu Corporation
- Sample size thickness 0.05 mm x width 20 mm x length 100 mm (note that the direction parallel to the length direction is the flow direction (MD) during film formation)
- Measurement condition Distance between chucks: 50mm
- Tensile speed 50 mm / min (stops when 25 N / mm 2 is reached)
- Table 1 shows the evaluation results of Examples I-1 to I-5 and Comparative Example I-1.
- Example II-1 A resin composition was prepared at the blending ratio shown in Table 2 below, melted and kneaded with a Banbury mixer, and then formed into a calendar film so as to have a thickness of 50 ⁇ m with an inverted L-shaped four calendar (melting process). Membrane process). Next, as shown in FIG. 1, immediately after the melt film formation step, three rolls (take-off rolls) that can be heated to an arbitrary temperature are arranged so that the melt-formed resin composition can pass alternately up and down. This promoted crystallization (crystallization promoting step). Thereafter, the resin composition was solidified by passing through a cooling roll to produce a film (base material).
- the temperature of the resin composition in the melt film formation step was the surface temperature of the roll corresponding to the fourth roll 4 in FIG. Moreover, about the temperature of the resin composition in a crystallization promotion process, the surface temperature of the three take-off rolls 5 of FIG. 1 was made substantially the same, and the temperature was made into the crystallization promotion temperature.
- the film formation rate was 5 m / min, and the substantial crystallization promotion step time (take-off roll passage time) was about 5 seconds.
- thermosetting addition type silicone (trade name “KS-847T”), manufactured by Shin-Etsu Chemical Co., Ltd., 30 wt% toluene solution) and platinum catalyst (trade name “PL-50T”, Shin-Etsu Chemical Co., Ltd.) Manufactured, toluene solution) 1 part by weight and 3000 parts by weight of heptane were added, mixed and dissolved to prepare a release agent composition.
- This release agent composition was applied onto the film (separator base material) obtained above and dried (after curing) so as to have a thickness of 0.2 ⁇ m, and dried by heating to obtain a separator.
- Examples II-2 to II-5 Resin compositions were prepared at the blending ratios shown in Table 2 below, and the films (separator base materials) of Examples II-2 to II-5 were prepared in the same manner as in Example II-1, respectively. A separator was obtained in the same manner as II-1.
- Comparative Example II-1 A resin composition was prepared at the blending ratio shown in Table 2 below, and a film (base material) of Comparative Example II-1 was prepared by the same operation as in Example II-1, and the same as in Example II-1. A separator was obtained.
- the separator was evaluated by the following method.
- ⁇ Workability test> A sample cut into a thickness of 0.05 mm, a width of 20 mm, and a length of 100 mm is fixed using the above tensile tester so that the stress is 25 N / mm 2 . Thereafter, a 1 mm cut is given with a razor so that the center part between the chucks is perpendicular to the MD direction. At that time, those that did not break were judged as “ ⁇ ” (good), and those that broke were judged as “x” (bad).
- the measurement apparatus and measurement conditions used are as follows.
- Tensile tester Autograph AG-20kNG, manufactured by Shimadzu Corporation
- Sample size thickness 0.05 mm x width 20 mm x length 100 mm (note that the direction parallel to the length direction is the flow direction (MD) during film formation)
- Measurement condition Distance between chucks: 50mm
- Tensile speed 50 mm / min (stops when 25 N / mm 2 is reached)
- Example III-1 A resin composition was prepared at the blending ratio shown in Table 3 below, melt kneaded with a Banbury mixer, and then formed into a calendar film so as to have a thickness of 50 ⁇ m with an inverted L-shaped four calendar (melting process). Membrane process). Next, as shown in FIG. 1, immediately after the melt film formation step, three rolls (take-off rolls) that can be heated to an arbitrary temperature are arranged so that the melt-formed resin composition can pass alternately up and down. This promoted crystallization (crystallization promoting step). Thereafter, the resin composition was solidified by passing through a cooling roll to produce a film (base material).
- the temperature of the resin composition in the melt film formation step was the surface temperature of the roll corresponding to the fourth roll 4 in FIG. Moreover, about the temperature of the resin composition in a crystallization promotion process, the surface temperature of the three take-off rolls 5 of FIG. 1 was made substantially the same, and the temperature was made into the crystallization promotion temperature.
- the film formation rate was 5 m / min, and the substantial crystallization promotion step time (take-off roll passage time) was about 5 seconds.
- an acrylic polymer (weight average molecular weight: about 400,000) composed of ethyl acrylate / 2-ethylhexyl acrylate (60 parts by weight / 40 parts by weight), 10 parts by weight of a rosin phenol tackifier and toluene
- This pressure-sensitive adhesive composition was applied and dried on the film (base material) obtained above so that the thickness after drying was 50 ⁇ m, to obtain a pressure-sensitive adhesive sheet.
- Examples III-2 to III-5 Resin compositions were prepared at the blending ratios shown in Table 3 below, and the films (base materials) of Examples III-2 to III-5 were prepared in the same manner as in Example III-1. Further, Example III A pressure-sensitive adhesive sheet was obtained in the same manner as -1.
- Comparative Example III-1 A resin composition was prepared at the blending ratio shown in Table 3 below, and a film (base material) of Comparative Example III-1 was prepared by the same operation as in Example III-1, and further the same as in Example III-1. Thus, an adhesive sheet was obtained.
- the adhesive sheet was evaluated by the following method.
- ⁇ Workability test> A sample cut into a thickness of 0.05 mm, a width of 20 mm, and a length of 100 mm is fixed using a tensile tester so that the stress is 25 N / mm 2 . Thereafter, a 1 mm cut is given with a razor so that the center part between the chucks is perpendicular to the MD direction. At that time, those that did not break were judged as “ ⁇ ” (good), and those that broke were judged as “x” (bad).
- the measurement apparatus and measurement conditions used are as follows.
- Tensile tester Autograph AG-20kNG, manufactured by Shimadzu Corporation
- Sample size thickness 0.05 mm x width 20 mm x length 100 mm (note that the direction parallel to the length direction is the flow direction (MD) during film formation)
- Measurement condition Distance between chucks: 50mm
- Tensile speed 50 mm / min (stops when 25 N / mm 2 is reached)
- Table 3 shows the evaluation results of Examples III-1 to III-5 and Comparative Example III-1.
- Example IV-1 A resin composition was prepared at a blending ratio shown in Table 4 below, melted and kneaded with a Banbury mixer, and then a calendar film was formed with a reverse L-shaped four-calendar so as to have a thickness of 50 ⁇ m (melting process). Membrane process). Next, as shown in FIG. 1, immediately after the melt film formation step, three rolls (take-off rolls) that can be heated to an arbitrary temperature are arranged so that the melt-formed resin composition can pass alternately up and down. This promoted crystallization (crystallization promoting step). Thereafter, the resin composition was solidified by passing through a cooling roll to produce a film (base material).
- the temperature of the resin composition in the melt film formation step was the surface temperature of the roll corresponding to the fourth roll 4 in FIG. Moreover, about the temperature of the resin composition in a crystallization promotion process, the surface temperature of the three take-off rolls 5 of FIG. 1 was made substantially the same, and the temperature was made into the crystallization promotion temperature.
- the film formation rate was 5 m / min, and the substantial crystallization promotion step time (take-off roll passage time) was about 5 seconds.
- an acrylic polymer was added to a 20 wt% toluene solution of an acrylic polymer (weight average molecular weight: 400,000) consisting of 2-ethylhexyl acrylate / N-acryloylmorpholine / acrylic acid (70 parts by weight / 30 parts by weight / 3 parts). 2 parts by weight of an epoxy-based crosslinking agent (trade name “Tetrad C”, manufactured by Mitsubishi Gas Chemical Co., Ltd.) and 2 parts of an isocyanate-based crosslinking agent (trade name “Coronate L”, manufactured by Nippon Polyurethane Co., Ltd.) per 100 parts by weight of polymer An adhesive composition was prepared by blending parts by weight. This pressure-sensitive adhesive composition was applied and dried on the film (base material) obtained above so that the thickness after drying was 5 ⁇ m to obtain a protective film.
- an epoxy-based crosslinking agent trade name “Tetrad C”, manufactured by Mitsubishi Gas Chemical Co., Ltd.
- an isocyanate-based crosslinking agent
- Examples IV-2 to IV-5 Resin compositions were prepared at the blending ratios shown in Table 4 below, and the films (base materials) of Examples IV-2 to IV-5 were prepared in the same manner as in Example IV-1. Further, Example IV A protective film was obtained in the same manner as -1.
- Comparative Example IV-1 A resin composition was prepared at a blending ratio shown in Table 4 below, and a film (base material) of Comparative Example IV-1 was prepared in the same manner as in Example IV-1. Further, in the same manner as in Example IV-1. A protective film was obtained.
- the protective film was evaluated by the following method.
- ⁇ Workability test> A sample cut into a thickness of 0.05 mm, a width of 20 mm, and a length of 100 mm is fixed using the above tensile tester so that the stress is 25 N / mm 2 . Thereafter, a 1 mm cut is given with a razor so that the center part between the chucks is perpendicular to the MD direction. At that time, those that did not break were judged as “ ⁇ ” (good), and those that broke were judged as “x” (bad).
- the measurement apparatus and measurement conditions used are as follows.
- Tensile tester Autograph AG-20kNG, manufactured by Shimadzu Corporation
- Sample size thickness 0.05 mm x width 20 mm x length 100 mm (note that the direction parallel to the length direction is the flow direction (MD) during film formation)
- Measurement condition Distance between chucks: 50mm
- Tensile speed 50 mm / min (stops when 25 N / mm 2 is reached)
- Table 4 shows the evaluation results of Examples IV-1 to IV-5 and Comparative Example IV-1.
- the polylactic acid film or sheet of the present invention there is no melting or deformation of the film or sheet even at a high temperature exceeding 100 ° C., and the film or sheet is manufactured or processed while maintaining the inherent rigidity.
- the film or sheet When the film or sheet is wound into a roll, etc., it has a characteristic that it does not break or tear even if tension is applied. Therefore, it can be used, for example, as a base material for pressure-sensitive adhesive tapes or sheets, a base material for separators used for pressure-sensitive adhesive tapes or sheets, a base material for protective films, and the like.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Manufacturing & Machinery (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Materials Engineering (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Adhesive Tapes (AREA)
- Laminated Bodies (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上であり、且つ引裂き強度(JIS P 8116 紙-引裂強さ試験方法-エルメンドルフ形引裂試験機法に準拠)が、流れ方向(MD方向)、幅方向(TD方向)ともに、2.5N/mm以上であることを特徴とするポリ乳酸系フィルム又はシートである。
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上であることを特徴とするポリ乳酸系フィルム又はシート。
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上であることを特徴とするポリ乳酸系フィルム又はシート。
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上であることを特徴とするポリ乳酸系フィルム又はシート。
本発明のポリ乳酸系フィルム又はシートは、ポリ乳酸(A)を含む樹脂フィルム又はシートである。ポリ乳酸の原料モノマーである乳酸は、不斉炭素原子を有するため、光学異性体のL体とD体が存在する。本発明で使用するポリ乳酸(A)は、L体の乳酸を主成分とした重合体である。製造時に不純物として混入するD体の乳酸の含有量が少ないものほど、高結晶性で高融点の重合体となるため、できるだけL体純度の高いものを用いるのが好ましく、L体純度が95%以上のものを用いるのがより好ましい。また、ポリ乳酸(A)は、乳酸以外の他の共重合成分を含んでいてもよい。
カラム:TSKgel SuperHZM-H/HZ2000/HZ1000
カラムサイズ:4.6mmI.D.×150mm
溶離液:クロロホルム
流量:0.3ml/min
検出器:RI
カラム温度:40℃
注入量:10μl
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上である。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅10mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:300mm/min
本発明のポリ乳酸系フィルム又はシートを製造する方法としては特に限定されるものではないが、ポリ乳酸(A)を含む樹脂組成物を溶融成膜法により成膜する方法が好ましい。溶融成膜法を採用すると、成膜時結晶化部の融解吸熱量ΔHc′を10J/g以上にすることが容易に可能となる。
図1に、かかる製造方法に用いられるカレンダー成膜機の1例の模式図を示す。以下に、図1を詳細に説明する。
図2に、かかる製造方法に用いられるポリッシング成膜機の1例の模式図を示す。以下に、図2を詳細に説明する。
本発明のセパレータは、セパレータ基材の少なくとも一方の面に剥離剤処理層を有するセパレータであって、該セパレータ基材が、前記のポリ乳酸系フィルム又はシートで構成されていることを特徴とする。
本発明の粘着テープ又はシートは、基材の少なくとも一方の面に粘着剤層を有する粘着テープ又はシートであって、該基材が、前記のポリ乳酸系フィルム又はシートで構成されていることを特徴とする。
本発明の保護フィルムは、基材の少なくとも一方の面に再剥離性粘着剤層を有する保護フィルムであって、該基材が、前記のポリ乳酸系フィルム又はシートで構成されていることを特徴とする。
A1:商品名「テラマックTP-4000」(ユニチカ社製)
<フッ素系ポリマー(B)>
B1:アクリル変性ポリテトラフルオロエチレン(商品名「メタブレンA-3000」、三菱レイヨン社製)
<結晶化促進剤(C)>
C1:フェニルホスホン酸亜鉛(商品名「エコプロモート」、日産化学工業社製)
<酸性官能基変性オレフィン系ポリマー(D)>
D1:無水マレイン酸基含有変性ポリプロピレン(重量平均分子量32000、酸価52mgKOH/g、商品名「ユーメックス1010」、三洋化成工業社製)
<耐引裂性改質剤(E)>
E1:ポリグリセリン脂肪酸エステル(数平均分子量Mn1000、商品名「チラバゾールVR-10」、太陽化学社製)
E2:ポリグリセリン脂肪酸エステル(数平均分子量Mn1700、商品名「チラバゾールVR-2」、太陽化学社製)
E3:コアシェル構造重合体(アクリルゴム/ポリメチルメタクリレート-スチレン コアシェル構造重合体、商品名「パラロイドEXL2315」、ローム・アンド・ハース・ジャパン社製)
E4:軟質脂肪族系ポリエステル(ポリブチレンアジペートテレフタレート、商品名「エコフレックス」、BASF・ジャパン社製)
下記表1に示す配合割合で樹脂組成物を調製し、バンバリーミキサーにて溶融混練を行った後、逆L型4本カレンダーにて厚さ50μmになるようにカレンダー成膜を行った(溶融成膜工程)。次に、図1のように溶融成膜工程の直後に、任意の温度に加熱可能なロール(テイクオフロール)を3本配し、溶融成膜された樹脂組成物が上下交互に通過できるようにすることで結晶化を促進させた(結晶化促進工程)。その後に冷却ロールを通過することで樹脂組成物を固化し、フィルムを作製した。溶融成膜工程における樹脂組成物の温度(溶融成膜時の樹脂温度)は、図1における第4ロール4に相当するロールの表面温度とした。また、結晶化促進工程における樹脂組成物の温度については、図1の3本のテイクオフロール5の表面温度を略同一とし、その温度を結晶化促進温度とした。成膜速度は5m/minとし、実質的な結晶化促進工程の時間(テイクオフロール通過時間)は約5秒間であった。
下記表1に示す配合割合で樹脂組成物を調製し、実施例I-1と同様の操作により実施例I-2~I-5のフィルムをそれぞれ作製した。
下記表1に示す配合割合で樹脂組成物を調製し、実施例I-1と同様の操作により比較例I-1のフィルムを作製した。
DSC(示差走査熱分析装置)にて測定した、成膜後の樹脂組成物の再昇温過程での融解に伴う吸熱ピークのトップ時の温度を融解温度(Tm;結晶融解ピーク温度ともいう)とした。
DSCにて測定した、成膜後の樹脂組成物の200℃からの降温過程での結晶化に伴う発熱ピークのピークトップ時の温度を結晶化温度(Tc;結晶化ピーク温度ともいう)とした。
(1)ロールへのプレートアウト:ロール表面の汚れを目視により評価し、ロール表面の汚れがない状態を「○」(なし)、ロール表面の汚れがある状態を「×」(あり)と判定した。
(2)剥離性:図1の第4ロール4からの溶融成膜された樹脂組成物の剥離性により評価し、テイクオフロール5で引き取り可能である状態を「○」(良好)、テイクオフロール5で引き取り不可である状態を「×」(不良)と判定した。
(3)フィルム面状態:得られたフィルム面を目視で観察し、フィルム表面に粗さがなく平滑である状態を「○」(良好)、バンクマーク(樹脂の流れムラによる凹凸)やサメ肌、ピンホールがある状態を「×」(不良)と判定した。
DSCにて測定した、成膜後のフィルムサンプルの昇温過程での結晶化に伴う発熱ピークの熱量ΔHc(J/g)と、その後の融解に伴う熱量ΔHm(J/g)から、以下の式(1)を用いて算出した。
ΔHc′=ΔHm-ΔHc (1)
装置:エスアイアイ・ナノテクノロジー社製 DSC6220
条件:
測定温度域:20℃→200℃→0℃→200℃(すなわち、まず20℃から200℃への昇温過程での測定に続けて、200℃から0℃への降温過程での測定を行い、最後に0℃から200℃への再昇温過程での測定を行った)
昇温/降温速度:2℃/min
測定雰囲気:窒素雰囲気下(200ml/min)
なお、再昇温過程で、結晶化に伴う発熱ピークが無かったことから、2℃/minの降温速度で結晶化可能領域が100%結晶化するものと判断し、成膜時結晶化部の融解吸熱量の算出式の妥当性を確認した。
JIS P 8116の紙-引裂強さ試験方法-エルメンドルフ形引裂試験機法に準じ測定した。使用した測定装置及び測定条件は、以下の通りである。
装置:テスター産業株式会社製 エルメンドルフ形引裂試験機
条件:試料サイズ:厚さ0.25mm程度(0.05mmのフィルムを5枚重ね)×幅76mm×長さ63mm
なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(以下、MD方向という。)および流れ方向と反対方向(以下、TD方向という)となるように切り出した。
引裂強度算出方法:以下の式(2)を用い算出した。
T=(A×p)/(n×t×1000) (2)
T:引裂強度(N/mm)
A:目盛りの読み値(mN)
p:振子の目盛の基準となる試験片の重ね枚数(この装置では16)
n:同時に引き裂かれる試験片の枚数
t:試験片1枚あたりの平均厚さ(mm)
フィルム又はシートサンプル(50mm×50mm×厚さ0.05mm)を溶剤に15分間浸漬し、取り出したサンプルの表面の溶剤をウエスで取り除き、浸漬後の重量を浸漬前の重量で割ることによって測定した。
なお、使用した溶剤は、トルエンと酢酸エチルの2種類である。
合否判定:2.0以下を合格とする。
JIS K 7161のプラスチック-引張特性の試験方法に準じて測定した。
使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅10mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:300mm/min
厚さ0.05mm×幅200mm×長さ300mmに切り出したサンプルの両端をクリップで挟み、100℃のオーブン中に1分間投入する。その後、取り出したときに元の形状を保てたものを「○」(良好)、元の形状が保てなかったものを「×」(不良)と判定した。
厚さ0.05mm×幅20mm×長さ100mmに切り出したサンプルを、上記の引張試験機を用い、応力が25N/mm2となるように固定する。その後、チャック間の中心部をMD方向に直角になるように、剃刀にて1mmの切れ込みを与える。その時に、破断しなかったものを「○」(良好)、破断したものを「×」(不良)と判定した。使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅20mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:50mm/min(25N/mm2となったときに停止)

下記表2に示す配合割合で樹脂組成物を調製し、バンバリーミキサーにて溶融混練を行った後、逆L型4本カレンダーにて厚さ50μmになるようにカレンダー成膜を行った(溶融成膜工程)。次に、図1のように溶融成膜工程の直後に、任意の温度に加熱可能なロール(テイクオフロール)を3本配し、溶融成膜された樹脂組成物が上下交互に通過できるようにすることで結晶化を促進させた(結晶化促進工程)。その後に冷却ロールを通過することで樹脂組成物を固化し、フィルム(基材)を作製した。溶融成膜工程における樹脂組成物の温度(溶融成膜時の樹脂温度)は、図1における第4ロール4に相当するロールの表面温度とした。また、結晶化促進工程における樹脂組成物の温度については、図1の3本のテイクオフロール5の表面温度を略同一とし、その温度を結晶化促進温度とした。成膜速度は5m/minとし、実質的な結晶化促進工程の時間(テイクオフロール通過時間)は約5秒間であった。
下記表2に示す配合割合で樹脂組成物を調製し、実施例II-1と同様の操作により実施例II-2~II-5のフィルム(セパレータ基材)をそれぞれ作製し、さらに、実施例II-1と同様にしてセパレータを得た。
下記表2に示す配合割合で樹脂組成物を調製し、実施例II-1と同様の操作により比較例II-1のフィルム(基材)を作製し、さらに、実施例II-1と同様にしてセパレータを得た。
幅200mm×長さ300mmに切り出したセパレータサンプルの両端をクリップで挟み、100℃のオーブン中に1分間投入する。その後、取り出したときに元の形状を保てたものを「○」(良好)、元の形状が保てなかったものを「×」(不良)と判定した。
厚さ0.05mm×幅20mm×長さ100mmに切り出したサンプルを、上記の引張試験機を用い、応力が25N/mm2となるように固定する。その後、チャック間の中心部をMD方向に直角になるように、剃刀にて1mmの切れ込みを与える。その時に、破断しなかったものを「○」(良好)、破断したものを「×」(不良)と判定した。使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅20mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:50mm/min(25N/mm2となったときに停止)

下記表3に示す配合割合で樹脂組成物を調製し、バンバリーミキサーにて溶融混練を行った後、逆L型4本カレンダーにて厚さ50μmになるようにカレンダー成膜を行った(溶融成膜工程)。次に、図1のように溶融成膜工程の直後に、任意の温度に加熱可能なロール(テイクオフロール)を3本配し、溶融成膜された樹脂組成物が上下交互に通過できるようにすることで結晶化を促進させた(結晶化促進工程)。その後に冷却ロールを通過することで樹脂組成物を固化し、フィルム(基材)を作製した。溶融成膜工程における樹脂組成物の温度(溶融成膜時の樹脂温度)は、図1における第4ロール4に相当するロールの表面温度とした。また、結晶化促進工程における樹脂組成物の温度については、図1の3本のテイクオフロール5の表面温度を略同一とし、その温度を結晶化促進温度とした。成膜速度は5m/minとし、実質的な結晶化促進工程の時間(テイクオフロール通過時間)は約5秒間であった。
下記表3に示す配合割合で樹脂組成物を調製し、実施例III-1と同様の操作により実施例III-2~III-5のフィルム(基材)をそれぞれ作製し、さらに、実施例III-1と同様にして粘着シートを得た。
下記表3に示す配合割合で樹脂組成物を調製し、実施例III-1と同様の操作により比較例III-1のフィルム(基材)を作製し、さらに、実施例III-1と同様にして粘着シートを得た。
幅200mm×長さ300mmに切り出した粘着シートサンプルの両端をクリップで挟み、100℃のオーブン中に1分間投入する。その後、取り出したときに元の形状を保てたものを「○」(良好)、元の形状が保てなかったものを「×」(不良)と判定した。
厚さ0.05mm×幅20mm×長さ100mmに切り出したサンプルを、引張試験機を用い、応力が25N/mm2となるように固定する。その後、チャック間の中心部をMD方向に直角になるように、剃刀にて1mmの切れ込みを与える。その時に、破断しなかったものを「○」(良好)、破断したものを「×」(不良)と判定した。使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅20mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:50mm/min(25N/mm2となったときに停止)

下記表4に示す配合割合で樹脂組成物を調製し、バンバリーミキサーにて溶融混練を行った後、逆L型4本カレンダーにて厚さ50μmになるようにカレンダー成膜を行った(溶融成膜工程)。次に、図1のように溶融成膜工程の直後に、任意の温度に加熱可能なロール(テイクオフロール)を3本配し、溶融成膜された樹脂組成物が上下交互に通過できるようにすることで結晶化を促進させた(結晶化促進工程)。その後に冷却ロールを通過することで樹脂組成物を固化し、フィルム(基材)を作製した。溶融成膜工程における樹脂組成物の温度(溶融成膜時の樹脂温度)は、図1における第4ロール4に相当するロールの表面温度とした。また、結晶化促進工程における樹脂組成物の温度については、図1の3本のテイクオフロール5の表面温度を略同一とし、その温度を結晶化促進温度とした。成膜速度は5m/minとし、実質的な結晶化促進工程の時間(テイクオフロール通過時間)は約5秒間であった。
下記表4に示す配合割合で樹脂組成物を調製し、実施例IV-1と同様の操作により実施例IV-2~IV-5のフィルム(基材)をそれぞれ作製し、さらに、実施例IV-1と同様にして保護フィルムを得た。
下記表4に示す配合割合で樹脂組成物を調製し、実施例IV-1と同様の操作により比較例IV-1のフィルム(基材)を作製し、さらに、実施例IV-1と同様にして保護フィルムを得た。
幅200mm×長さ300mmに切り出した保護フィルムサンプルの両端をクリップで挟み、100℃のオーブン中に1分間投入する。その後、取り出したときに元の形状を保てたものを「○」(良好)、元の形状が保てなかったものを「×」(不良)と判定した。
厚さ0.05mm×幅20mm×長さ100mmに切り出したサンプルを、上記の引張試験機を用い、応力が25N/mm2となるように固定する。その後、チャック間の中心部をMD方向に直角になるように、剃刀にて1mmの切れ込みを与える。その時に、破断しなかったものを「○」(良好)、破断したものを「×」(不良)と判定した。使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、島津製作所製)
試料サイズ:厚さ0.05mm×幅20mm×長さ100mm(なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)となるように切り出した)
測定条件:
チャック間距離:50mm
引張速度:50mm/min(25N/mm2となったときに停止)

2 第2ロール
3 第3ロール
4 第4ロール
5 テイクオフロール
6 冷却ロール
7 冷却ロール
8 樹脂組成物
9 バンク(樹脂だまり)
10 押出機先端部
Claims (10)
- ポリ乳酸(A)を含む樹脂フィルム又はシートであって、下記式(1)
ΔHc′=ΔHm-ΔHc (1)
[式中、ΔHcは、DSCにて測定される、成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱量(J/g)であり、ΔHmは、その後の融解に伴う吸熱量(J/g)である]
で求められる成膜時結晶化部の融解吸熱量ΔHc′が10J/g以上であり、且つ引裂き強度(JIS P 8116 紙-引裂強さ試験方法-エルメンドルフ形引裂試験機法に準拠)が、流れ方向(MD方向)、幅方向(TD方向)ともに、2.5N/mm以上であることを特徴とするポリ乳酸系フィルム又はシート。 - さらに、ポリ乳酸(A)100重量部に対し、フッ素系ポリマー(B)を0.5~15重量部含む請求項1記載のポリ乳酸系フィルム又はシート。
- フッ素系ポリマー(B)がテトラフルオロエチレン系ポリマーである請求項2記載のポリ乳酸系フィルム又はシート。
- さらに、ポリ乳酸(A)100重量部に対し、結晶化促進剤(C)を0.1~15重量部含む請求項1~3の何れかの項に記載のポリ乳酸系フィルム又はシート。
- さらに、ポリ乳酸(A)100重量部に対し、酸価が10~70mgKOH/g、重量平均分子量が10000~80000である、酸性官能基変性オレフィン系ポリマー(D)を0.1~10重量部含む請求項1~4の何れかの項に記載のポリ乳酸系フィルム又はシート。
- 溶融成膜法により成膜されたフィルム又はシートである請求項1~5の何れかの項に記載のポリ乳酸系フィルム又はシート。
- 溶融成膜法がカレンダー法である請求項6記載のポリ乳酸系フィルム又はシート。
- セパレータ基材の少なくとも一方の面に剥離剤処理層を有するセパレータであって、該セパレータ基材が、請求項1~7の何れかの項に記載のポリ乳酸系フィルム又はシートで構成されていることを特徴とするセパレータ。
- 基材の少なくとも一方の面に粘着剤層を有する粘着テープ又はシートであって、該基材が、請求項1~7の何れかの項に記載のポリ乳酸系フィルム又はシートで構成されていることを特徴とする粘着テープ又はシート。
- 基材の少なくとも一方の面に再剥離性粘着剤層を有する保護フィルムであって、該基材が、請求項1~7の何れかの項に記載のポリ乳酸系フィルム又はシートで構成されていることを特徴とする保護フィルム。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP11843504.9A EP2644643B1 (en) | 2010-11-26 | 2011-11-21 | Polylactic acid-based film or sheet |
| KR1020137013863A KR20130117798A (ko) | 2010-11-26 | 2011-11-21 | 폴리락트산계 필름 또는 시트 |
| CN2011800570438A CN103228713A (zh) | 2010-11-26 | 2011-11-21 | 聚乳酸系薄膜或片 |
| US13/885,407 US20130236723A1 (en) | 2010-11-26 | 2011-11-21 | Polylactic acid-based film or sheet |
Applications Claiming Priority (32)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-264423 | 2010-11-26 | ||
| JP2010264424A JP2012111206A (ja) | 2010-11-26 | 2010-11-26 | 保護フィルム |
| JP2010-264415 | 2010-11-26 | ||
| JP2010264415A JP2012111914A (ja) | 2010-11-26 | 2010-11-26 | ポリ乳酸系フィルム又はシート |
| JP2010-264421 | 2010-11-26 | ||
| JP2010264422A JP2012111918A (ja) | 2010-11-26 | 2010-11-26 | 粘着テープ又はシート |
| JP2010264419A JP2012111204A (ja) | 2010-11-26 | 2010-11-26 | セパレータ |
| JP2010-264417 | 2010-11-26 | ||
| JP2010264423A JP2012111919A (ja) | 2010-11-26 | 2010-11-26 | 粘着テープ又はシート |
| JP2010-264426 | 2010-11-26 | ||
| JP2010264416A JP5913798B2 (ja) | 2010-11-26 | 2010-11-26 | ポリ乳酸系フィルム又はシート |
| JP2010-264420 | 2010-11-26 | ||
| JP2010-264418 | 2010-11-26 | ||
| JP2010-264416 | 2010-11-26 | ||
| JP2010264421A JP2012111917A (ja) | 2010-11-26 | 2010-11-26 | 粘着テープ又はシート |
| JP2010-264424 | 2010-11-26 | ||
| JP2010264420A JP2012111205A (ja) | 2010-11-26 | 2010-11-26 | セパレータ |
| JP2010-264425 | 2010-11-26 | ||
| JP2010264417A JP5731174B2 (ja) | 2010-11-26 | 2010-11-26 | ポリ乳酸系フィルム又はシート |
| JP2010-264422 | 2010-11-26 | ||
| JP2010264425A JP2012111207A (ja) | 2010-11-26 | 2010-11-26 | 保護フィルム |
| JP2010264418A JP2012111203A (ja) | 2010-11-26 | 2010-11-26 | セパレータ |
| JP2010264426A JP2012111208A (ja) | 2010-11-26 | 2010-11-26 | 保護フィルム |
| JP2010-264419 | 2010-11-26 | ||
| JP2010265580A JP2012116888A (ja) | 2010-11-29 | 2010-11-29 | 粘着テープ又はシート |
| JP2010265579A JP2012116012A (ja) | 2010-11-29 | 2010-11-29 | セパレータ |
| JP2010-265581 | 2010-11-29 | ||
| JP2010-265578 | 2010-11-29 | ||
| JP2010265578A JP2012116887A (ja) | 2010-11-29 | 2010-11-29 | ポリ乳酸系フィルム又はシート |
| JP2010-265579 | 2010-11-29 | ||
| JP2010-265580 | 2010-11-29 | ||
| JP2010265581A JP2012116889A (ja) | 2010-11-29 | 2010-11-29 | 保護フィルム |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012070526A1 true WO2012070526A1 (ja) | 2012-05-31 |
Family
ID=46145868
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/076794 WO2012070526A1 (ja) | 2010-11-26 | 2011-11-21 | ポリ乳酸系フィルム又はシート |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20130236723A1 (ja) |
| EP (1) | EP2644643B1 (ja) |
| KR (1) | KR20130117798A (ja) |
| WO (1) | WO2012070526A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013118408A1 (ja) * | 2012-02-10 | 2013-08-15 | 日東電工株式会社 | 保護フィルム |
| JP2013163762A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | ポリ乳酸系フィルム又はシート、及び、粘着テープ又はシート |
| JP2013163764A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | 保護フィルム |
| JP2013163763A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | セパレータ |
| CN103396659A (zh) * | 2013-06-25 | 2013-11-20 | 浙江大学 | 一种加快聚乳酸树脂结晶的方法 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130276701A1 (en) * | 2010-12-27 | 2013-10-24 | Sharp Kabushiki Kaisha | Deposition device, and collection device |
| CN104044251A (zh) * | 2014-05-29 | 2014-09-17 | 六安市金赛特橡塑制品有限公司 | 一种汽车转向机缓冲板的制造工艺 |
| WO2016065455A1 (en) * | 2014-10-27 | 2016-05-06 | Cini Frank Albert | Biodegradable film having an adhesive and methods of use thereof |
| DE102014223470A1 (de) * | 2014-11-18 | 2016-05-19 | Tesa Se | Flexible, thermisch stabile und gleichzeitig transparente biobasierte Folie basierend auf Polymilchsäure, eine Formulierung zur Herstellung der Folie sowie ihre Verwendung |
| CN107109041B (zh) | 2014-12-22 | 2020-10-16 | 3M创新有限公司 | 包含聚乳酸聚合物、聚乙酸乙烯酯聚合物和增塑剂的组合物和膜 |
| WO2017200756A1 (en) | 2016-05-20 | 2017-11-23 | 3M Innovative Properties Company | Oriented polylactic acid polymer based film |
| CN109312147B (zh) | 2016-06-21 | 2021-04-16 | 3M创新有限公司 | 包括半结晶聚乳酸基膜的图形制品 |
| EP3491063B1 (en) * | 2016-07-28 | 2021-12-01 | PTT Public Company Limited | A bioplastic composition comprising biomass as a component and a production process |
| KR101950139B1 (ko) * | 2018-06-25 | 2019-02-19 | 김대영 | 다중 탈부착 기능을 구비한 opp 테이프, 그리고 이의 제조 방법 |
| KR20210086743A (ko) | 2019-12-30 | 2021-07-09 | (주) 에이치피엠글로벌 | 통기성이 우수한 식품 포장 필름용 생분해성 수지 조성물, 이로부터 제조되는 필름 및 그 제조방법 |
| KR102576520B1 (ko) * | 2021-12-22 | 2023-09-12 | 한국전자통신연구원 | 전자파 차폐용 박막 유연 소재의 대형화 이음 시공 방법 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11116788A (ja) | 1997-10-09 | 1999-04-27 | Mitsui Chem Inc | ポリ乳酸系樹脂組成物 |
| JP2003292642A (ja) * | 2002-01-30 | 2003-10-15 | Asahi Kasei Corp | 生分解性フィルム |
| JP2007130893A (ja) * | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2009013271A (ja) * | 2007-07-04 | 2009-01-22 | Lonseal Corp | ポリ乳酸系シートまたはフィルム、およびその製造方法。 |
| JP2009138085A (ja) * | 2007-12-06 | 2009-06-25 | Toray Ind Inc | ポリ乳酸系フィルム |
| JP2009173715A (ja) * | 2008-01-22 | 2009-08-06 | Lonseal Corp | ポリ乳酸系シートまたはフィルム、およびその製造方法 |
| JP2010106272A (ja) | 2008-10-02 | 2010-05-13 | Nitto Denko Corp | ポリ乳酸系フィルムまたはシート |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3344613B2 (ja) * | 1994-12-16 | 2002-11-11 | 王子製紙株式会社 | 粘着シート |
| US5849401A (en) * | 1995-09-28 | 1998-12-15 | Cargill, Incorporated | Compostable multilayer structures, methods for manufacture, and articles prepared therefrom |
| JP5305590B2 (ja) * | 2004-06-16 | 2013-10-02 | ユニチカ株式会社 | ポリ乳酸含有樹脂組成物及びそれより得られる成形体 |
| JP5311828B2 (ja) * | 2005-01-12 | 2013-10-09 | ビーエーエスエフ ソシエタス・ヨーロピア | 生物分解性ポリエステル混合物 |
| TWI464210B (zh) * | 2005-07-08 | 2014-12-11 | Toray Industries | 樹脂組成物及其成形品 |
| US7754826B1 (en) * | 2005-11-15 | 2010-07-13 | Clemson University | Copolymers from lactide |
| CN101336167B (zh) * | 2005-11-30 | 2012-07-18 | 东丽株式会社 | 聚乳酸类树脂叠层片及其成型体 |
| JP4773870B2 (ja) * | 2006-04-27 | 2011-09-14 | 積水化成品工業株式会社 | ポリ乳酸系樹脂発泡成形体の製造方法 |
| US7683117B2 (en) * | 2007-02-02 | 2010-03-23 | Fuji Xerox Co., Ltd. | Resin composition, resin mold and method for producing the same |
| JP5154834B2 (ja) * | 2007-05-07 | 2013-02-27 | デクセリアルズ株式会社 | 異方導電性接着剤フィルム及び異方導電性接着剤フィルムの製造方法 |
| US8512852B2 (en) * | 2009-05-22 | 2013-08-20 | Toyo Boseki Kabushiki Kaisha | Polylactic acid resin composition and film |
| US8901275B2 (en) * | 2009-06-17 | 2014-12-02 | Toray Industries, Inc. | Method for producing crystallized polyester |
| CN102361936A (zh) * | 2010-01-08 | 2012-02-22 | 日东电工株式会社 | 阻燃性聚乳酸系薄膜或薄片及其制造方法 |
| CN102361918B (zh) * | 2010-01-28 | 2015-08-12 | 日东电工株式会社 | 阻燃性聚乳酸系薄膜或薄片及其制造方法 |
| CN102361935B (zh) * | 2010-03-30 | 2015-10-14 | 日东电工株式会社 | 聚乳酸系薄膜或薄片及其制造方法 |
| US8729165B2 (en) * | 2010-03-30 | 2014-05-20 | Nitto Denko Corporation | Flame-retardant poly lactic acid-containing film or sheet, and method for manufacturing thereof |
-
2011
- 2011-11-21 KR KR1020137013863A patent/KR20130117798A/ko not_active Application Discontinuation
- 2011-11-21 EP EP11843504.9A patent/EP2644643B1/en active Active
- 2011-11-21 US US13/885,407 patent/US20130236723A1/en not_active Abandoned
- 2011-11-21 WO PCT/JP2011/076794 patent/WO2012070526A1/ja active Application Filing
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11116788A (ja) | 1997-10-09 | 1999-04-27 | Mitsui Chem Inc | ポリ乳酸系樹脂組成物 |
| JP2003292642A (ja) * | 2002-01-30 | 2003-10-15 | Asahi Kasei Corp | 生分解性フィルム |
| JP2007130893A (ja) * | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2009013271A (ja) * | 2007-07-04 | 2009-01-22 | Lonseal Corp | ポリ乳酸系シートまたはフィルム、およびその製造方法。 |
| JP2009138085A (ja) * | 2007-12-06 | 2009-06-25 | Toray Ind Inc | ポリ乳酸系フィルム |
| JP2009173715A (ja) * | 2008-01-22 | 2009-08-06 | Lonseal Corp | ポリ乳酸系シートまたはフィルム、およびその製造方法 |
| JP2010106272A (ja) | 2008-10-02 | 2010-05-13 | Nitto Denko Corp | ポリ乳酸系フィルムまたはシート |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013118408A1 (ja) * | 2012-02-10 | 2013-08-15 | 日東電工株式会社 | 保護フィルム |
| WO2013118406A1 (ja) * | 2012-02-10 | 2013-08-15 | 日東電工株式会社 | ポリ乳酸系フィルム又はシート、及び、粘着テープ又はシート |
| WO2013118407A1 (ja) * | 2012-02-10 | 2013-08-15 | 日東電工株式会社 | セパレータ |
| JP2013163762A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | ポリ乳酸系フィルム又はシート、及び、粘着テープ又はシート |
| JP2013163764A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | 保護フィルム |
| JP2013163763A (ja) * | 2012-02-10 | 2013-08-22 | Nitto Denko Corp | セパレータ |
| US10118999B2 (en) | 2012-02-10 | 2018-11-06 | Nitto Denko Corporation | Polylactic acid film or sheet, and pressure-sensitive adhesive tape or sheet |
| CN103396659A (zh) * | 2013-06-25 | 2013-11-20 | 浙江大学 | 一种加快聚乳酸树脂结晶的方法 |
| CN103396659B (zh) * | 2013-06-25 | 2015-10-28 | 浙江大学 | 一种加快聚乳酸树脂结晶的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2644643A4 (en) | 2014-04-09 |
| US20130236723A1 (en) | 2013-09-12 |
| KR20130117798A (ko) | 2013-10-28 |
| EP2644643B1 (en) | 2019-01-30 |
| EP2644643A1 (en) | 2013-10-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2012070526A1 (ja) | ポリ乳酸系フィルム又はシート | |
| WO2013118408A1 (ja) | 保護フィルム | |
| WO2010038833A1 (ja) | ポリ乳酸系フィルムまたはシート | |
| JP5883306B2 (ja) | 剥離ライナー | |
| JP5775831B2 (ja) | ポリ乳酸系フィルム又はシート、及び、粘着テープ又はシート | |
| WO2011093302A1 (ja) | 難燃性ポリ乳酸系フィルム又はシート、及びその製造方法 | |
| JP5883307B2 (ja) | 保護フィルム | |
| CN103228713A (zh) | 聚乳酸系薄膜或片 | |
| JP2012111207A (ja) | 保護フィルム | |
| JP2012111206A (ja) | 保護フィルム | |
| JP2007211068A (ja) | 粘着テープ及びその製造方法 | |
| JP2012116889A (ja) | 保護フィルム | |
| JP2012111917A (ja) | 粘着テープ又はシート | |
| JP2012116888A (ja) | 粘着テープ又はシート | |
| JP2012111208A (ja) | 保護フィルム | |
| JP2012111919A (ja) | 粘着テープ又はシート | |
| JP2012111918A (ja) | 粘着テープ又はシート | |
| JP5913798B2 (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012111914A (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012116012A (ja) | セパレータ | |
| JP2012111205A (ja) | セパレータ | |
| JP5731174B2 (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012111203A (ja) | セパレータ | |
| JP2012111204A (ja) | セパレータ | |
| JP2012116887A (ja) | ポリ乳酸系フィルム又はシート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11843504 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13885407 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20137013863 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011843504 Country of ref document: EP |