WO2011049157A1 - ペプチド翻訳合成におけるrapidディスプレイ法 - Google Patents
ペプチド翻訳合成におけるrapidディスプレイ法 Download PDFInfo
- Publication number
- WO2011049157A1 WO2011049157A1 PCT/JP2010/068549 JP2010068549W WO2011049157A1 WO 2011049157 A1 WO2011049157 A1 WO 2011049157A1 JP 2010068549 W JP2010068549 W JP 2010068549W WO 2011049157 A1 WO2011049157 A1 WO 2011049157A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- peptide
- linker
- mrna
- sequence
- translation
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 176
- 238000000034 method Methods 0.000 title claims abstract description 86
- 230000015572 biosynthetic process Effects 0.000 title claims description 23
- 238000003786 synthesis reaction Methods 0.000 title description 17
- 238000013519 translation Methods 0.000 claims abstract description 129
- 150000001413 amino acids Chemical class 0.000 claims abstract description 60
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims abstract description 44
- 239000003054 catalyst Substances 0.000 claims abstract description 16
- 241000023308 Acca Species 0.000 claims abstract description 9
- 102100033047 G-protein coupled receptor 3 Human genes 0.000 claims abstract description 9
- 101000963424 Homo sapiens Acetyl-CoA carboxylase 1 Proteins 0.000 claims abstract description 9
- 101000871088 Homo sapiens G-protein coupled receptor 3 Proteins 0.000 claims abstract description 9
- 230000014616 translation Effects 0.000 claims description 144
- 238000006243 chemical reaction Methods 0.000 claims description 84
- 108090000623 proteins and genes Proteins 0.000 claims description 47
- 108020004414 DNA Proteins 0.000 claims description 40
- 102000004169 proteins and genes Human genes 0.000 claims description 39
- 230000027455 binding Effects 0.000 claims description 27
- 238000009739 binding Methods 0.000 claims description 27
- 238000013518 transcription Methods 0.000 claims description 24
- 230000035897 transcription Effects 0.000 claims description 24
- 108091034117 Oligonucleotide Proteins 0.000 claims description 19
- 108010079855 Peptide Aptamers Proteins 0.000 claims description 19
- 238000000338 in vitro Methods 0.000 claims description 18
- 150000001261 hydroxy acids Chemical class 0.000 claims description 17
- 238000001243 protein synthesis Methods 0.000 claims description 17
- 239000013076 target substance Substances 0.000 claims description 16
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 12
- 230000006229 amino acid addition Effects 0.000 claims description 12
- 229920001223 polyethylene glycol Polymers 0.000 claims description 10
- 239000002202 Polyethylene glycol Substances 0.000 claims description 9
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 239000000126 substance Substances 0.000 claims description 8
- 102000053602 DNA Human genes 0.000 claims description 7
- 150000002148 esters Chemical class 0.000 claims description 7
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 6
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 6
- 229960005305 adenosine Drugs 0.000 claims description 6
- 230000002194 synthesizing effect Effects 0.000 claims description 5
- 238000006276 transfer reaction Methods 0.000 claims description 4
- 230000032050 esterification Effects 0.000 claims description 2
- 238000005886 esterification reaction Methods 0.000 claims description 2
- 102000039446 nucleic acids Human genes 0.000 abstract description 6
- 108020004707 nucleic acids Proteins 0.000 abstract description 6
- 150000007523 nucleic acids Chemical class 0.000 abstract description 6
- 108020004999 messenger RNA Proteins 0.000 abstract description 5
- 229940024606 amino acid Drugs 0.000 description 64
- 235000001014 amino acid Nutrition 0.000 description 57
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 46
- 239000000243 solution Substances 0.000 description 35
- 235000018102 proteins Nutrition 0.000 description 28
- 239000002299 complementary DNA Substances 0.000 description 27
- 239000000047 product Substances 0.000 description 25
- 108020004566 Transfer RNA Proteins 0.000 description 23
- 229950010131 puromycin Drugs 0.000 description 23
- 238000002824 mRNA display Methods 0.000 description 16
- 102000004196 processed proteins & peptides Human genes 0.000 description 16
- 239000000758 substrate Substances 0.000 description 16
- 239000000562 conjugate Substances 0.000 description 12
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 11
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 11
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 11
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 11
- 239000007795 chemical reaction product Substances 0.000 description 11
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 10
- 239000011324 bead Substances 0.000 description 10
- 238000009396 hybridization Methods 0.000 description 10
- 241000588724 Escherichia coli Species 0.000 description 9
- 125000003275 alpha amino acid group Chemical group 0.000 description 9
- 238000002474 experimental method Methods 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 210000004899 c-terminal region Anatomy 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 238000010839 reverse transcription Methods 0.000 description 8
- 108020004705 Codon Proteins 0.000 description 7
- 230000009918 complex formation Effects 0.000 description 7
- 238000005755 formation reaction Methods 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- 239000000203 mixture Substances 0.000 description 6
- -1 polyethylene Polymers 0.000 description 6
- 238000011084 recovery Methods 0.000 description 6
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical group N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 5
- 108010067902 Peptide Library Proteins 0.000 description 5
- 229920002352 Peptidyl-tRNA Polymers 0.000 description 5
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 5
- 108020005038 Terminator Codon Proteins 0.000 description 5
- 230000003321 amplification Effects 0.000 description 5
- 230000000977 initiatory effect Effects 0.000 description 5
- 229910001629 magnesium chloride Inorganic materials 0.000 description 5
- 238000013507 mapping Methods 0.000 description 5
- 238000003199 nucleic acid amplification method Methods 0.000 description 5
- 229960005190 phenylalanine Drugs 0.000 description 5
- 210000003705 ribosome Anatomy 0.000 description 5
- 239000001632 sodium acetate Substances 0.000 description 5
- 235000017281 sodium acetate Nutrition 0.000 description 5
- 125000006850 spacer group Chemical group 0.000 description 5
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 4
- 150000008574 D-amino acids Chemical class 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 4
- 102000005877 Peptide Initiation Factors Human genes 0.000 description 4
- 108010044843 Peptide Initiation Factors Proteins 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 4
- 101710137500 T7 RNA polymerase Proteins 0.000 description 4
- 229960003767 alanine Drugs 0.000 description 4
- 238000001962 electrophoresis Methods 0.000 description 4
- 239000002773 nucleotide Substances 0.000 description 4
- 125000003729 nucleotide group Chemical group 0.000 description 4
- 238000003753 real-time PCR Methods 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102000052866 Amino Acyl-tRNA Synthetases Human genes 0.000 description 3
- 108700028939 Amino Acyl-tRNA Synthetases Proteins 0.000 description 3
- 108090000994 Catalytic RNA Proteins 0.000 description 3
- 102000053642 Catalytic RNA Human genes 0.000 description 3
- 108010069514 Cyclic Peptides Proteins 0.000 description 3
- 102000001189 Cyclic Peptides Human genes 0.000 description 3
- QIVBCDIJIAJPQS-SECBINFHSA-N D-tryptophane Chemical compound C1=CC=C2C(C[C@@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-SECBINFHSA-N 0.000 description 3
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 3
- 102100034343 Integrase Human genes 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 3
- 108091081024 Start codon Proteins 0.000 description 3
- 108010090804 Streptavidin Proteins 0.000 description 3
- 108010006785 Taq Polymerase Proteins 0.000 description 3
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 3
- 238000005917 acylation reaction Methods 0.000 description 3
- 125000000266 alpha-aminoacyl group Chemical group 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 235000020958 biotin Nutrition 0.000 description 3
- 229960002685 biotin Drugs 0.000 description 3
- 239000011616 biotin Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 230000002068 genetic effect Effects 0.000 description 3
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 239000011591 potassium Substances 0.000 description 3
- 230000008929 regeneration Effects 0.000 description 3
- 238000011069 regeneration method Methods 0.000 description 3
- 108091092562 ribozyme Proteins 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 230000014621 translational initiation Effects 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- PGTJUXHMJYBSBW-LLVKDONJSA-N (2r)-2-[(2-chloroacetyl)amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound C1=CC=C2C(C[C@H](C(=O)O)NC(=O)CCl)=CNC2=C1 PGTJUXHMJYBSBW-LLVKDONJSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 102000002281 Adenylate kinase Human genes 0.000 description 2
- 108020000543 Adenylate kinase Proteins 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 102000004420 Creatine Kinase Human genes 0.000 description 2
- 108010042126 Creatine kinase Proteins 0.000 description 2
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 2
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 2
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 150000008575 L-amino acids Chemical class 0.000 description 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- DRBBFCLWYRJSJZ-UHFFFAOYSA-N N-phosphocreatine Chemical compound OC(=O)CN(C)C(=N)NP(O)(O)=O DRBBFCLWYRJSJZ-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 2
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 2
- 108091093037 Peptide nucleic acid Proteins 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 102000009609 Pyrophosphatases Human genes 0.000 description 2
- 108010009413 Pyrophosphatases Proteins 0.000 description 2
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 2
- 108050001681 Signal peptidase complex catalytic subunit SEC11A Proteins 0.000 description 2
- 102100036268 Signal peptidase complex catalytic subunit SEC11A Human genes 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 2
- 229920001222 biopolymer Polymers 0.000 description 2
- 102000043871 biotin binding protein Human genes 0.000 description 2
- 108700021042 biotin binding protein Proteins 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000012790 confirmation Methods 0.000 description 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 239000001177 diphosphate Substances 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- IIRDTKBZINWQAW-UHFFFAOYSA-N hexaethylene glycol Chemical group OCCOCCOCCOCCOCCOCCO IIRDTKBZINWQAW-UHFFFAOYSA-N 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 229930182817 methionine Natural products 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 238000005580 one pot reaction Methods 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- 239000001103 potassium chloride Substances 0.000 description 2
- 235000011164 potassium chloride Nutrition 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000002702 ribosome display Methods 0.000 description 2
- 238000007363 ring formation reaction Methods 0.000 description 2
- 239000012488 sample solution Substances 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- ATHGHQPFGPMSJY-UHFFFAOYSA-N spermidine Chemical compound NCCCCNCCCN ATHGHQPFGPMSJY-UHFFFAOYSA-N 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000001308 synthesis method Methods 0.000 description 2
- 239000003656 tris buffered saline Substances 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- GKQXPTHQTXCXEV-UHFFFAOYSA-N (4-chlorophenyl)methanethiol Chemical compound SCC1=CC=C(Cl)C=C1 GKQXPTHQTXCXEV-UHFFFAOYSA-N 0.000 description 1
- ITVKOVQVBCQQGY-UHFFFAOYSA-N 1-chloro-4-[(4-chlorophenyl)methylsulfanylmethyl]benzene Chemical compound C1=CC(Cl)=CC=C1CSCC1=CC=C(Cl)C=C1 ITVKOVQVBCQQGY-UHFFFAOYSA-N 0.000 description 1
- AUFGTPPARQZWDO-YUZLPWPTSA-N 10-formyltetrahydrofolate Chemical compound C1NC=2NC(N)=NC(=O)C=2NC1CN(C=O)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 AUFGTPPARQZWDO-YUZLPWPTSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- QDGAVODICPCDMU-UHFFFAOYSA-N 2-amino-3-[3-[bis(2-chloroethyl)amino]phenyl]propanoic acid Chemical compound OC(=O)C(N)CC1=CC=CC(N(CCCl)CCCl)=C1 QDGAVODICPCDMU-UHFFFAOYSA-N 0.000 description 1
- RYSMHWILUNYBFW-GRIPGOBMSA-N 3'-amino-3'-deoxy-N(6),N(6)-dimethyladenosine Chemical group C1=NC=2C(N(C)C)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](N)[C@H]1O RYSMHWILUNYBFW-GRIPGOBMSA-N 0.000 description 1
- 125000004042 4-aminobutyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])N([H])[H] 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical compound NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 108020005098 Anticodon Proteins 0.000 description 1
- 101000640990 Arabidopsis thaliana Tryptophan-tRNA ligase, chloroplastic/mitochondrial Proteins 0.000 description 1
- 101000787278 Arabidopsis thaliana Valine-tRNA ligase, chloroplastic/mitochondrial 2 Proteins 0.000 description 1
- 101000787296 Arabidopsis thaliana Valine-tRNA ligase, mitochondrial 1 Proteins 0.000 description 1
- 108010014885 Arginine-tRNA ligase Proteins 0.000 description 1
- 101710132120 Asparagine-tRNA ligase Proteins 0.000 description 1
- 102100023245 Asparagine-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 101710160288 Asparagine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 101710090387 Asparagine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 108010065272 Aspartate-tRNA ligase Proteins 0.000 description 1
- 102100026198 Aspartate-tRNA ligase, mitochondrial Human genes 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 101100268670 Caenorhabditis elegans acc-3 gene Proteins 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 101710152440 Cysteine-tRNA ligase Proteins 0.000 description 1
- 102100030115 Cysteine-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 101710185308 Cysteine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 108010002156 Depsipeptides Proteins 0.000 description 1
- 101000787280 Dictyostelium discoideum Probable valine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 102100021309 Elongation factor Ts, mitochondrial Human genes 0.000 description 1
- 102100033238 Elongation factor Tu, mitochondrial Human genes 0.000 description 1
- 101710202200 Endolysin A Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010015514 Glutamate-tRNA ligase Proteins 0.000 description 1
- 102100024977 Glutamine-tRNA ligase Human genes 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- 108010051724 Glycine-tRNA Ligase Proteins 0.000 description 1
- 102100036589 Glycine-tRNA ligase Human genes 0.000 description 1
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 1
- 101710177011 Histidine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 102100029015 Histidine-tRNA ligase, mitochondrial Human genes 0.000 description 1
- 101000611183 Homo sapiens Tumor necrosis factor Proteins 0.000 description 1
- 102100036015 Isoleucine-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 101710176147 Isoleucine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 108010092041 Lysine-tRNA Ligase Proteins 0.000 description 1
- 102100035529 Lysine-tRNA ligase Human genes 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 1
- 108010003060 Methionine-tRNA ligase Proteins 0.000 description 1
- 102100037206 Methionine-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 102000008109 Mixed Function Oxygenases Human genes 0.000 description 1
- 108010074633 Mixed Function Oxygenases Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108010049977 Peptide Elongation Factor Tu Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 102100036134 Probable arginine-tRNA ligase, mitochondrial Human genes 0.000 description 1
- 101710086402 Probable asparagine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 101710108281 Probable asparagine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 101710121315 Probable cysteine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 101710096715 Probable histidine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 101710149031 Probable isoleucine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 102100028531 Probable proline-tRNA ligase, mitochondrial Human genes 0.000 description 1
- 101710164123 Probable proline-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 101710146427 Probable tyrosine-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 102100026126 Proline-tRNA ligase Human genes 0.000 description 1
- 101710115782 Proline-tRNA ligase Proteins 0.000 description 1
- 101710140381 Proline-tRNA ligase, cytoplasmic Proteins 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- ODHCTXKNWHHXJC-GSVOUGTGSA-N Pyroglutamic acid Natural products OC(=O)[C@H]1CCC(=O)N1 ODHCTXKNWHHXJC-GSVOUGTGSA-N 0.000 description 1
- 108091008103 RNA aptamers Proteins 0.000 description 1
- 101710086015 RNA ligase Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 101800001707 Spacer peptide Proteins 0.000 description 1
- 239000006180 TBST buffer Substances 0.000 description 1
- 108010029287 Threonine-tRNA ligase Proteins 0.000 description 1
- 102100028196 Threonine-tRNA ligase 2, cytoplasmic Human genes 0.000 description 1
- 241000209140 Triticum Species 0.000 description 1
- 235000021307 Triticum Nutrition 0.000 description 1
- 102100034300 Tryptophan-tRNA ligase, cytoplasmic Human genes 0.000 description 1
- 102100025336 Tyrosine-tRNA ligase, mitochondrial Human genes 0.000 description 1
- 101710107268 Tyrosine-tRNA ligase, mitochondrial Proteins 0.000 description 1
- 102100025607 Valine-tRNA ligase Human genes 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 230000006287 biotinylation Effects 0.000 description 1
- 238000007413 biotinylation Methods 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical group OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- PHGUWORRLGKHCK-JTQLQIEISA-N cyanomethyl (2s)-2-amino-3-phenylpropanoate Chemical compound N#CCOC(=O)[C@@H](N)CC1=CC=CC=C1 PHGUWORRLGKHCK-JTQLQIEISA-N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000005370 electroosmosis Methods 0.000 description 1
- 108010063460 elongation factor T Proteins 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 108010051239 glutaminyl-tRNA synthetase Proteins 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 102000057041 human TNF Human genes 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 108010057757 methionyl-tRNA formyltransferase Proteins 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000000863 peptide conjugate Substances 0.000 description 1
- 125000001151 peptidyl group Chemical group 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 229920001281 polyalkylene Polymers 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920002704 polyhistidine Polymers 0.000 description 1
- 108010005636 polypeptide C Proteins 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- GUUBJKMBDULZTE-UHFFFAOYSA-M potassium;2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid;hydroxide Chemical compound [OH-].[K+].OCCN1CCN(CCS(O)(=O)=O)CC1 GUUBJKMBDULZTE-UHFFFAOYSA-M 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 230000004850 protein–protein interaction Effects 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 210000001995 reticulocyte Anatomy 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000010532 solid phase synthesis reaction Methods 0.000 description 1
- 229940063673 spermidine Drugs 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- PIEPQKCYPFFYMG-UHFFFAOYSA-N tris acetate Chemical compound CC(O)=O.OCC(N)(CO)CO PIEPQKCYPFFYMG-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1062—Isolating an individual clone by screening libraries mRNA-Display, e.g. polypeptide and encoding template are connected covalently
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/04—Libraries containing only organic compounds
- C40B40/06—Libraries containing nucleotides or polynucleotides, or derivatives thereof
- C40B40/08—Libraries containing RNA or DNA which encodes proteins, e.g. gene libraries
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B50/00—Methods of creating libraries, e.g. combinatorial synthesis
- C40B50/06—Biochemical methods, e.g. using enzymes or whole viable microorganisms
Definitions
- the present invention relates to a novel method used for producing a linked body of cDNA or mRNA and its translation product peptide or protein in genotype-phenotype association technology (display system).
- this method the RAPID display method.
- This method is suitable for screening a peptide aptamer that can be a drug candidate from a special peptide library constructed by using the previously reported flexizyme system (or RAPID system: Random Peptide Discovery system).
- the genotype and phenotype mapping technology that was born as a tool for evolutionary molecular engineering is also called the display method.
- the mRNA display method ("In vitro virus", Nemoto N, et al. FEBS Lett. 414 , 405-408 ( 1997), International Publication WO 98/16636; or “RNA-peptide fusions”, Roberts, RW & Szostak, JW, Proc. Natl. Acad. Sci. USA., 94 , 12297-12302 (1997), International Publication WO 98/31700. ), STABLE method (noncovalent DNA display), microbead droplet method, covalent DNA display method, phage display method, ribosome display method and the like are known.
- the display method when a functional peptide or protein molecule is selected from a library, the corresponding gene is linked so that the sequence can be easily read, and the genetic information of a polypeptide having a specific function It is useful when selecting.
- the mRNA display method is a technology that uses a cell-free translation system (in ⁇ vitro protein synthesis system) to unify genotype and phenotype by covalently binding mRNA as a genotype and peptide molecule as a phenotype.
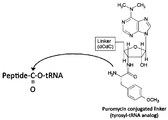
- a method of linking a synthesized peptide molecule and mRNA encoding the same through puromycin, which is an analog of the 3 ′ terminal portion of tyrosyl-tRNA has been employed.
- puromycin is bound to the 3 ′ end of mRNA via an appropriate linker, and this is introduced into a cell-free translation system to synthesize a peptide from mRNA.
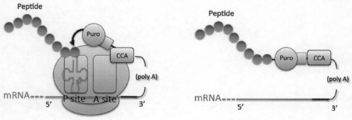
- As a substrate for the reaction it is linked to the C-terminus of the growing peptide chain, and a peptide molecule as a translation product is linked to mRNA via puromycin (FIG. 1A).
- the linker is inserted between mRNA and puromycin mainly in order to efficiently incorporate puromycin into the A site of ribosome.
- a characteristic of puromycin is that, unlike the 3 'end of aminoacyl-tRNA, an adenosine-like moiety and an amino acid (tyrosine) -like moiety form an amide bond instead of an ester bond (FIG. 1B). Therefore, the conjugate of puromycin and peptide linked on the ribosome is not easily hydrolyzed and is stable.
- a puromycin-conjugated linker was prepared by synthesizing a linear polymer spacer from puromycin to the 5 ′ end, and then the linker and the 3 ′ end of mRNA were bound. And a method of binding puromycin to the conjugate after binding a spacer to the 3 ′ end of the mRNA.
- the end of the linear polymer that is a spacer is typically a phosphate group or nucleotide, and the bond between the 3 ′ end of mRNA and the 5 ′ end of the linker is a phosphate group.
- a covalent bond is formed by a reaction using RNA ligase or DNA ligase, or a general organic chemical reaction.
- Patent No. 3683282 Publication WO98 / 16636
- Patent No. 3683902 Japanese Patent No. 3692542 (International Publication WO98 / 31700)
- mRNA having puromycin at the 3 'end is used as a template, and this is added to a cell-free translation system using an extract of wheat germ or rabbit reticulocyte to perform translation into a peptide. Therefore, it is necessary to carry out transcription from DNA to mRNA and ligation reaction between mRNA and puromycin in advance outside the translation system.
- a reconstituted cell-free translation system is a system in which components that are not related to translation are removed by subdividing the cell-free translation system mainly using E. coli extract and reconstituting each factor. Compared with a cell-free translation system using a conventional cell extract, contamination with inhibitors such as nucleases and proteases can be easily prevented. By adding factors necessary for the transcription reaction, transcription from DNA can be performed simultaneously with translation.
- mRNA can be synthesized by transcription from the template DNA in the same system as the translation reaction. Furthermore, if the complex formation of mRNA and linker molecule can be performed in the same system, unlike the conventional mRNA display method, one-pot (from within the same reaction vessel) from the transcription of cDNA to the production of mRNA-peptide conjugate. ) Can be prepared. It is a first object of the present invention to provide a display method that takes advantage of such a reconfigurable cell-free translation system capable of controlling reaction system components.
- the second object of the present invention is to enable the construction of a library.
- the mRNA display method of the present invention is modified to use a linker molecule newly developed by the present inventors in place of the puromycin-binding linker, and the reconstituted cell-free translation system is optimized.
- transcription, translation, formation of a complex between the linker and mRNA, and subsequent binding of the peptide to the linker can be completed in one translation system.
- the main features of the RAPID display method compared with the conventional mRNA display method are as follows.
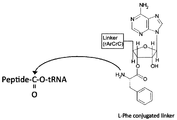
- (A) The 3 ′ end of the linker molecule is not puromycin but has a structure in which an amino acid is linked to an adenosine (aminoacylation) via an ester.
- (A) The aminoacylation reaction is performed with an artificial RNA catalyst (ribozyme).
- D) Linker and mRNA are bound not by ligation but by complex formation by hybridization in the translation system.
- special peptides can be presented as phenotypes.
- An acylation reaction in which a non-protein amino acid or hydroxy acid, which is a structural unit of a special peptide, is supported on tRNA is also performed using an artificial RNA catalyst (ribozyme).
- the outline of the present invention is as follows. (1) In a reconstituted in vitro protein synthesis system, a linker used for preparing a conjugate in which mRNA and a peptide that is a translation product thereof are bound via a linker, A single-stranded structure region having a side chain base paired with a base on the 3 ′ end side of mRNA at one end of the linker; A peptide acceptor region containing a group capable of binding to a translation product by a peptide transfer reaction at the other end of the linker; The peptide acceptor region has a structure in which an amino acid is ester-linked to an oligo RNA consisting of the base sequence ACCA.
- the artificial RNA catalyst used in the aminoacylation reaction is one of the following RNA sequences: GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (SEQ ID NO: 3) GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (SEQ ID NO: 4) GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (SEQ ID NO: 5) GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (SEQ ID NO: 19)
- the linker according to any one of (1) to (3) above, which has a chemical structure consisting of: (5) A method for producing a [mRNA]-[linker]-[peptide] conjugate in which mRNA and a peptide which is a translation product thereof are bonded via any one of the linkers (1) to (4).
- the step of preparing the linker according to any one of (1) to (4) above Synthesizing mRNA having a sequence that can hybridize to the base sequence of the single-stranded structure region of the linker downstream of the sequence encoding the peptide;
- the method comprising the steps of bringing a linker into contact with mRNA in a reconstituted in vitro protein synthesis reaction solution and translating the peptide from mRNA.
- (6) A method for producing a [mRNA]-[linker]-[peptide] conjugate in which mRNA and a peptide which is a translation product thereof are bound via any one of the linkers (1) to (4).
- the step of preparing the linker according to any one of (1) to (4) above, A step of synthesizing DNA serving as a template for mRNA having a sequence that can hybridize to the base sequence of the single-stranded structure region of the linker downstream of the sequence encoding the peptide;
- the method comprising the steps of performing transcription from DNA to mRNA, translation into a peptide, and formation of a complex between the linker and mRNA by providing the linker and DNA in a reconstituted in vitro protein synthesis reaction solution.
- tRNA By including tRNA charged with a non-protein amino acid or hydroxy acid in a reconstituted in vitro protein synthesis reaction solution, the peptide as a translation product becomes a special peptide.
- a peptide as a translation product becomes a special peptide by including tRNA charged with a non-protein amino acid or a hydroxy acid in a reconstituted in vitro protein synthesis reaction solution. (9) or (10 ) Method.
- a method for producing the linker according to (2) above A chimeric oligonucleotide in which a single-stranded structural region and an oligo RNA consisting of the sequence ACCA are linked with polyethylene glycol is synthesized, By attaching an amino acid to an adenosine at the 3 ′ end of the chimeric oligonucleotide by esterification with an artificial RNA catalyst, The method, wherein a linker in which a single chain structure region and a peptide acceptor region are linked with polyethylene glycol is prepared.
- the artificial RNA catalyst is any of the following RNA sequences: GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (SEQ ID NO: 3) GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (SEQ ID NO: 4) GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (SEQ ID NO: 5) GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (SEQ ID NO: 19)
- the method according to (12) which has a chemical structure consisting of:
- linker molecule that can bind to mRNA in a reconstituted cell-free translation reaction solution
- FIG. 1A schematically shows how a peptide molecule as a translation product is linked to mRNA via a puromycin-conjugated linker in an mRNA display method.
- FIG. 1B shows the structure of the peptide acceptor part of the linker (Puromycin conjugated linker).
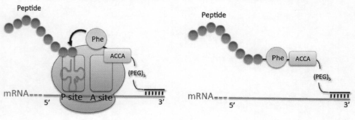
- FIG. 2A schematically shows how peptide molecules as translation products are linked to mRNA via a RAPID linker (L-PhePconjugated linker is exemplified) in the RAPID display method of the present invention.
- FIG. 2B shows the structure of the peptide acceptor portion of the linker (L-Phe conjugated linker).
- the linker molecule (an21-ACCA) is aminoacylated by a reaction catalyzed by flexizyme.
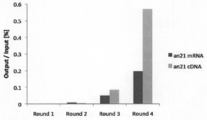
- Example 1 The recovery rate of the complex of a peptide aptamer (TNF- ⁇ -DW) and mRNA when mRNA is added to the reaction solution is shown.
- Example 1 The recovery rate of the complex of peptide aptamer (TNF- ⁇ -DW) and mRNA when cDNA is added to the reaction solution is shown.
- Example 1 It is the result of confirming that the peptide-mRNA complex displaying TNF- ⁇ -DW was recovered more efficiently than the complex of the control (EMP1SS) that did not bind to the target.
- EMP1SS complex of the control
- the linker is linked to the 3 ′ end of the mRNA at one end and the C terminal of the peptide at the other end, as in the known mRNA display method. And the translation product peptide.
- the structures at both ends of the linker are different from the known mRNA display method.
- the linker used in the RAPID display method of the present invention may be referred to as “RAPID linker”.
- the part that binds to the C-terminal of the peptide, which is one end of the linker is described.
- this part is also referred to as “peptide acceptor” or simply “acceptor”. That is, the peptide acceptor means a molecule having a structure capable of binding to a growing peptide (peptidyl tRNA) by a peptide transfer reaction in a ribosome.
- the peptide acceptor may refer to a moiety located at the end of the linker, or may refer to the entire structure including the linker.
- the peptide acceptor in the known mRNA display method is puromycin located at the end of the linker or a puromycin-binding linker that is the entire structure including the linker.
- the RAPID linker of the present invention is characterized by the structure of the peptide acceptor and its production method.
- a linker having an ACCA sequence consisting of 4 ribonucleotides on the 3 ′ end side is synthesized, and then an arbitrary amino acid is bound to the 3 ′ end adenosine, whereby a peptide acceptor is attached to the linker.
- the amino acid bound to the linker end receives the peptide C-terminus of the peptidyl tRNA and binds to the peptide.
- a structure in which an amino acid is ester-linked to the RNA sequence ACCA is referred to as a “peptide acceptor region”.
- Puromycin a peptide acceptor in the known mRNA display method, has an aminonucleoside structure in which an adenosine-like ribose and an amino acid are amide-bonded.
- the RAPID display method of the present invention has a structure in which an amino acid is ester-linked to 3'-O of ribose. That is, the peptide acceptor in the RAPID display of the present invention has a nucleoside structure similar to that of a natural aminoacyl tRNA. As an example of such a peptide acceptor, refer to FIG.
- the peptide acceptor exhibits uptake efficiency equal to or higher than that of puromycin by using a structure that is more natural.
- the formation of the bond between the peptide acceptor and the peptide C-terminus is similar to the normal peptide transfer reaction that takes place in the ribosome, and the peptidyl incorporated into the A-site is linked to the ester bond at the C-terminal of the peptide attached to the peptidyl tRNA at the P-site. It appears to be caused by the proximity of the acceptor amino group. Therefore, the covalent bond formed between the peptide chain and the C-terminus is typically an amide bond, as in the mRNA display method.
- the RAPID display of the present invention has a D-amino acid or ⁇ (beta) -amino acid that is different from a natural (non-protein) amino acid by using an artificial RNA catalyst (flexizyme) described later for linker synthesis. It is also possible to use one.
- the 5 ′ end side of the linker and the 3 ′ end side of the mRNA molecule form a complex by hybridization based on base pair formation. Accordingly, the 5 ′ end side of the linker takes a single-stranded structure having a nucleobase in the side chain.
- This part of the RAPID linker is referred to herein as a “single-stranded structure region”.
- Specific examples of the single-stranded structure having a nucleobase in the side chain include single-stranded DNA, single-stranded RNA, and single-stranded PNA (peptide nucleic acid).
- the complex formed needs to be stably retained during peptide selection.
- 5′-CTCCCGCCCCCCGTCC-3 ′ SEQ ID NO: 1 used in the Examples is not limited in any way. 5'-CCCGCCTCCCGCCCCCCGTCC-3 '(SEQ ID NO: 2) And a single-stranded DNA consisting of 13 to 21 nucleotides.
- the parts other than both ends of the linker are designed to have a simple linear structure that is flexible, hydrophilic, and has few side chains as a whole, similar to the structure of the linker used in the known mRNA display method. Is done. Accordingly, for example, oligonucleotides such as single-stranded or double-stranded DNA and RNA, polyalkylenes such as polyethylene, polyalkylene glycols such as polyethylene glycol, linear substances such as polystyrene and polysaccharides, or combinations thereof are appropriately used. It can be selected and used.
- the length of the linker is preferably 100 mm or more, and more preferably about 100 to 1000 mm.
- Non-limiting specific examples of the linker that can be used in the present invention include a single-stranded DNA having a sequence with a high GC content in a single-stranded structure region, an RNA having an ACCA sequence at the 3 ′ end, and DNA DNA / RNA chimeric oligonucleotides in which RNA and RNA are linked with polyethylene glycol (PEG linker) are raised.
- PEG linker DNA DNA / RNA chimeric oligonucleotides in which RNA and RNA are linked with polyethylene glycol (PEG linker) are raised.
- PEG linker polyethylene glycol
- a typical example is [DNA]-[Spacer18] n -rArCrCrA (wherein Spacer18 is hexaethylene glycol and n is an integer of 4 to 8) synthesized in Example 1.
- Flexizyme is also known by names such as dinitrobenzyl flexizyme (dFx), enhanced flexizyme (eFx), and aminoflexizyme (aFx).
- Flexizyme is composed of a weakly activated amino acid as a substrate, a carbonyl group that is the reactive site of the amino acid, an aromatic ring that is the amino acid side chain or leaving group, and the ACC-3 ′ sequence present at the 3 ′ end of the linker. Recognizes the moiety and has a catalytic ability to aminoacylate to adenosine at the 3 ′ end. This is why the oligo RNA structure consisting of the sequence of ACCA-3 ′ is essential at the end of the RAPID linker.
- the aminoacylation reaction with flexizyme proceeds only by placing an amino acid substrate and a linker molecule having a binding partner oligo RNA moiety on ice for about 2 hours in the presence of flexizyme.
- a flexizyme having a sequence represented by the following formula is preferably used.
- Prototype Flexizyme Fx [GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU-3 ', 45nt] (SEQ ID NO: 3)
- Enhanced Flexizyme eFx [5'-GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU-3 ', 45nt])
- SEQ ID NO: 4 Dinitrobenzyl flexizyme dFx [5'-GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU-3 ', 46nt] (SEQ ID NO: 5)
- Aminoflexizyme aFx [5'-GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU-3 ', 47nt]) (SEQ ID NO: 19)
- aminoacylation can be catalyzed using 4-chlorobenzylthiol as a leaving group and an amino acid having a side chain other than an aromatic ring as a side chain as a substrate.
- 4-chlorobenzylthiol as a leaving group
- an amino acid having a side chain other than an aromatic ring as a side chain as a substrate For details, see the above-mentioned literature. Accordingly, by reacting an amino acid having such a structure with a linker in the presence of flexizyme, a molecule in which the amino acid is ester-bonded to the hydroxyl group at the 3 ′ position on the ribose ring in the adenosine at the linker 3 ′ end is obtained. be able to.
- amino acid substrates those having an arbitrary structure can be bound, and not only amino acids used in natural translation, but also non-protein amino acids such as D-amino acids or ⁇ (beta) -amino acids (ie, natural amino acids) Other than the L-amino acids normally found in proteins present in Furthermore, a hydroxycarboxylic acid can be bound to the linker 3 ′ end instead of an amino acid, and incorporation into a ribosome as a peptide acceptor is also possible.
- a linker is added to a cell-free translation system (also referred to as an “in vitro protein synthesis system”) together with cDNA or mRNA and reacted for a certain period of time. Complex formation, followed by ligation of the peptide and linker.
- a conventional reconstituted cell-free translation system can be appropriately modified and used. If a DNA-dependent RNA polymerase (preferably T7 RNA polymerase) is added, transcription from DNA is also translated and translated. Can be done in the same system.
- a DNA-dependent RNA polymerase preferably T7 RNA polymerase
- reaction vessel one-pot.
- A Reaction to transcribe DNA into mRNA
- a Reaction in which the 3 ′ end of mRNA forms a complex with the single-stranded structure region of the linker end
- c Reaction to translate mRNA into peptide
- D Reaction in which the C-terminus of the translated peptide is amide-bonded to the peptide acceptor region at the other end of the linker
- e Reaction in which the [peptide]-[linker]-[mRNA] complex is dissociated from the ribosome.
- a reconstituted cell-free translation system requires purified ribosome, translation initiation factor, translation elongation factor, mRNA, aminoacyl-tRNA, and ATP or GTP as a substrate (M. H. Schreier, B. Erni and T. Staehelin (1977) “Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors.” Journal of Molecular Biology, Vol. 116, No. 4, 727-53. H. Trachsel, B. Emi, M. M. . Schreier and T. Staehelin (1977) “Initiation of mammalian protein synthesis. II. The assembly of the initiation complex with purified initiation factors.” Journal of Molecular Biology, Vol.
- aminoacyl-tRNA can be replaced by adding tRNA, aminoacyl-tRNA synthetase and its substrate in the same reaction solution.
- translation termination factors in order to increase the efficiency and fidelity of translation reactions, translation termination factors, ribosome regeneration factors, creatine kinase, myokinase, nucleotide diphosphate kinase, pyrophosphatase, etc.
- proteins and enzymes and their substrates PC Jelenc and CG Kurland (1979) “Nucleoside triphosphate regeneration decreases the frequency of translation errors” Proceedings of the Natural Academy Science of the United States of meric No. 7, 3174-3178).
- cDNA and T7 RNA polymerase and its substrate can be added instead of mRNA.
- DNA or RNA having a necessary sequence is provided to a cell-free translation system composed of components optimized for the purpose.
- a sequence is required for the cDNA to be transcribed into mRNA and translation of the mRNA is started in the synthesis system to be used.
- the entire length of the part encoding the amino acid sequence of the peptide needs to be translated to the end, and further, a spacer sequence composed of a peptide for linking the C-terminal side of the translated amino acid is linked, Immediately after the stop codon comes.
- a peptide sequence Cys- (Gly-Ser-) x3
- an amber codon stop codon
- the 3 ′ end of the mRNA has a structure that can hybridize with the single-stranded structural region of the linker to form a double-stranded region, so the downstream of the coding region (immediately after the stop codon) is single-stranded. It is necessary to have a sequence complementary to the sequence of the structural region.
- the sequence of this portion (double-strand forming portion) is referred to as a linker hybridization sequence in the present specification.
- the cDNA or mRNA desirably contains the following sequences.
- TAATACGACTCACTATA for the T7 promoter (SEQ ID NO: 6)
- the SD sequence gene is included. This is similar to normal protein synthesis.
- GGGTTAACTTTAA GAAGGA GATATACAT SEQ ID NO: 7: A modified upstream sequence of gene 10 protein of T7 phage. Underlined part is SD array.
- the release factor corresponding to the amber codon, peptidyl-tRNA hydroxylase (PTH), which is an enzyme that degrades peptidyl-tRNA, and the like are excluded.
- the reconstituted cell-free translation system was charged with the desired non-protein amino acid (or hydroxy acid) in advance (that is, the activated amino acid was bound) to the system given only limited natural amino acids. It is also possible to add acylated tRNA. Based on the genetic information of mRNA, a peptide containing a non-protein amino acid (or hydroxy acid) can be obtained by making the codon of a natural amino acid not added correspond to the anti-codon of tRNA acylated with a non-protein amino acid (or hydroxy acid). Translational synthesis on the ribosome is possible.
- acylated tRNA charged with a non-protein amino acid (or hydroxy acid) to a reconstituted cell-free translation system that does not contain a natural amino acid
- a special peptide that does not contain any natural amino acid can be translated and synthesized. Is possible.
- Acylated tRNA having a non-protein amino acid (or hydroxy acid) can be prepared using the above-mentioned artificial RNA catalyst “flexizyme” having an aminoacyl tRNA synthesis catalytic ability.
- the amino acid recognition site does not contain a substituent at the ⁇ -position, it is not limited to L amino acids, but is also a hydroxy acid ( ⁇ -position is a hydroxyl group), N-methylamino acid ( ⁇ -position is an N-methylamino acid), Acylamino acids ( ⁇ -position is N-acylamino group), D-amino acids and the like can also be used as substrates.
- ⁇ -position is a hydroxyl group
- N-methylamino acid ⁇ -position is an N-methylamino acid
- Acylamino acids ⁇ -position is N-acylamino group
- D-amino acids and the like can also be used as substrates.
- the present applicants have used a peptide synthesis system (concept including both kits and synthesis methods) using tRNA that is acylated with non-protein amino acids and hydroxy acids using “flexizyme” as a core technology.
- the integrated technology for translation synthesis, modification, and screening is called the RAPID system (Random Peptide Integrated Discovery system).
- RAPID system Random Peptide Integrated Discovery system
- various specialized peptides can be translated and synthesized as in vitro translation products based on template mRNAs of appropriate sequences.
- the special peptide includes any translation product that can be synthesized by the RAPID system, and in addition to 20 kinds of natural amino acids, ⁇ ( Consists of the various substrates described above, including beta) -amino acids, ⁇ (gamma) -amino acids and ⁇ (delta) -amino acids, D-amino acids, and derivatives with amino and carboxyl groups substituted on the amino acid skeleton It is a polymer as an element. Furthermore, the special peptide may have a structure different from a normal amide bond as a main chain skeleton.
- depsipeptides composed of amino acids and hydroxy acids, polyesters in which hydroxy acids are continuously condensed, peptides in which the amide bond nitrogen is methylated by the introduction of N-methylamino acids, various acyl groups (acetyl) at the N-terminus Peptides having a group, pyroglutamic acid, fatty acid, etc.) are also included in the special peptides.
- a cyclic peptide N-methyl peptide is obtained by cyclization of an acyclic peptide consisting of an amino acid sequence having a pair of functional groups capable of bond formation reaction between each other under appropriate reaction conditions.
- a cyclic N-methyl peptide can also be synthesized with the RAPID system. Depending on the type of functional group, it may cyclize under the conditions of cell-free translation system. As shown in the examples below, a peptide sequence with chloroacetyl groups and cysteine at both ends can be translated and synthesized. An example is a cyclic peptide obtained by cyclization with a thioether bond.
- RAPID display method of the present invention is suitable for use in order to associate a special peptide synthesized by the RAPID system with its genotype mRNA.
- the special peptide that is the translation product is mRNA via the linker. It is presented in conjunction with.
- the linker-mRNA binding that occurs in the cell-free translation system will be described.
- the RAPID display method of the present invention is characterized in that a complex can be formed in a translation system by hybridization of a linker and mRNA.
- mRNA is subjected to a cell-free translation system together with a RAPID linker, the hybridization between mRNA and linker, the synthesis of peptide molecules, and the linkage between the peptide C-terminus and the linker will be described.
- the binding between mRNA and the linker is based on the fact that the base sequence of the single-stranded structure region of the linker and the complementary base sequence at the 3 'end of the mRNA form a double strand by hydrogen bonding. Since this bond is for associating the mRNA with the translated peptide molecule, the [mRNA]-[linker]-[peptide] complex formed during translation on the ribosome is It also needs to be kept stable during the selection of translation products.
- the ribosome By subjecting the mRNA to a cell-free translation system together with the RAPID linker, the ribosome is placed on the mRNA, the translation reaction starts, the peptide chain is elongated, and the C-terminus of the terminated peptide chain is linked to the linker [rACCA- It is cleaved from tRNA by binding to an amino acid in the peptide acceptor region consisting of [amino acid].
- linker [rACCA- It is cleaved from tRNA by binding to an amino acid in the peptide acceptor region consisting of [amino acid].
- the hybridization between mRNA and RAPID linker begins with the initiation of a peptide chain elongation reaction.
- the present inventors believe that this can occur in the previous stage.
- the transcription and translation reaction from cDNA is performed first, and then a linker is added to the translation reaction solution after removing the termination factor and PTH. Complex formation occurs correctly.
- a covalent bond between the linker terminal [rACCA-amino acid] and the peptide C terminal is considered to occur when this part accidentally enters the ribosome A site at the end of translation.
- this [rACCA-amino acid] substrate is linked to the mRNA on the ribosome by the linker, and the local concentration is very high. It happens with high efficiency.
- a DNA population is first prepared, an RNA population is obtained as an in vitro transcription product, and a peptide population is obtained as an in vitro translation product. From this peptide population, one having a desired function or property is selected by some screening system. For example, when it is desired to obtain peptide molecules that bind to a specific protein, the peptide population can be poured into a column on which the target protein is immobilized, and a mixture of peptide molecules bound to the column can be recovered. At this time, since the mRNA of the template is added to each peptide molecule like a tag, it was returned to DNA from the collected peptide-mRNA complex population with reverse transcriptase and amplified by PCR.
- a similar selection experiment is performed again.
- a reverse transcription reaction can be performed before selection for the purpose of making the nucleic acid portion into a double strand. By repeating this operation, clones having a desired phenotype are enriched in the population as the generation progresses.
- mapping molecules active species
- nucleic acid portion of the selected mapping molecule The peptide aptamer gene that binds to the target substance can be cloned by repeating the step of preparing a nucleic acid library by PCR.
- the step of selecting the mapping molecule bound to the target substance is that the complex consisting of [RNA (or DNA / RNA hybrid)]-[linker]-[peptide] is bound to the target substance and another complex is prepared by an appropriate method. Separation from the body can be performed by identifying peptides having the desired binding properties.
- the target substance may be protein, nucleic acid, carbohydrate, lipid, or any other compound.
- a complex which is an active species that binds to a target substance, from other complexes, it is convenient that the target substance is modified so that it can be recovered by binding to a solid phase.
- the target substance is modified with biotin and recovered using specific binding to the immobilized biotin-binding protein.
- biotin binding protein avidin, streptavidin, etc.
- Biotin combinations such specific binding includes maltose binding protein / maltose, polyhistidine peptide / metal ions (nickel, cobalt, etc.), glutathione- S-transferase / glutathione, antibody / antigen (epitope) and the like can be used, but are not limited thereto.
- linker As a linker, a DNA / RNA chimeric oligonucleotide in which DNA and RNA were linked with polyethylene glycol (5 units of Spacer18) was used. Various linkers were purchased from BEX (Tokyo). In the following sequences, * A and * C correspond to RNA, and SPC18 corresponds to Spacer18 (hexaethylene glycol).
- an21-ACCA 5'-CCCGCCTCCCGCCCCCCGTCC- [SPC18] 5 -A * -C * -C * -A * -3 ' [Aminoacylation of linker]
- L-phenylalanine or ⁇ -L-alanine was bound to the 3 ′ end of the an21-ACCA linker molecule via an ester bond.
- the reaction product was confirmed by acrylamide electrophoresis analysis of the purified reaction product solution under acidic conditions.
- the band derived from the linker molecule is aminoacylated, the mobility decreases. Therefore, aminoacylation efficiency can be determined by comparing the intensity of the band derived from the unreacted product and the band derived from the reaction product.
- Each cDNA obtained by linking these oligo DNAs is composed of a T7 promoter sequence, a ribosome binding sequence, an initiation codon, a peptide aptamer sequence, a spacer peptide sequence (CGSGSGS), an amber codon, and a linker hybridization sequence.
- TNF-a_D-Trp.R66 GCCGCTGCCGCTGCCGCAATGCTTCAGATACAGACAATGCAGACGTTGCATATGTATATCTCCTTC EMP1SS.F63: GAAGGAGATATACATATGGCAGCAGGTGGTACCTATTCTTCTCATTTTGGTCCGCTGACCTGG EMP1SS.R63: GCCGCTGCCGCTGCCGCATGCTGCACCACCTTGCGGCTTAGAAACCCAGGTCAGCGGACCAAA T7g10M.F48: TAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG CGS3an13.R39: TTTCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA CGS3an21.R44: CCCGCCTCCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA PCR ligation was performed according to the following procedure.
- PCR reaction solution (10 mM Tris-HCl (pH 9.0), 50 mM potassium chloride, 2.5 mM magnesium chloride) containing 250 nM T7g10M.F48 and TNF-a_D-Trp.R66 (or a combination of EMP1SS.F63 and EMP1SS.R63) , 250 ⁇ M dNTPs, 0.2% Triton X-100 (Traton X-100, Nacalai Tesque) and Taq polymerase) 20 uL was prepared, and a thermal cycler (TC-3000, Techne) was used at 94 ° C for 1 min, ⁇ 50 The reaction was carried out at 5 ° C. for 30 cycles, 72 ° C. for 30 sec.
- mRNA was synthesized by transcription reaction using T7 RNA polymerase based on the cDNA (TNF- ⁇ -DW (an21)) prepared by the above method, and the resulting reaction product was extracted with phenol / chloroform and chloroform according to conventional methods. Further purification was performed by 2-propanol precipitation. The purified mRNA concentration was determined from UV absorption at 260 nm and diluted to 10 ⁇ M. [Transcription / Translation, Hybridization of linker, Linkage with peptide acceptor] Translation from mRNA and complex formation with a linker molecule or transcription from cDNA, translation and complex formation with a linker molecule were performed using a reconstituted cell-free translation system.
- the reconstituted cell-free translation system used in this example includes the following biopolymers: 70S ribosome (1.2uM), translation initiation factor (IF1 (0.7uM), IF2 (0.4uM), IF3 (1.5uM)), Elongation factor (EF-G (0.26 uM), EF-Tu / EF-Ts complex (10 uM)), translation termination factor (RF2 (0.25 uM), RF3 (0.17 uM)) methionyl tRNA transformylase (MTF (0.6 uM)), ribosome regeneration factor (RRF (0.5uM)), aminoacyl tRNA synthetase (AlaRS (0.73uM), ArgRS (0.03uM), AsnRS (0.38uM), CysRS (0.02uM), GlnRS (0.06uM), GluRS (0.23uM), GlyRS (0.09uM), HisRS (0.02uM), IleRS (0.4uM), LeuRS (0.04
- Ribosomes are purified from Escherichia coli in the logarithmic growth phase, and various proteins other than ribosomes are recombinant proteins that have been cloned, expressed and purified based on E. coli genes, unless otherwise specified.
- the following are contained as components other than a biopolymer: 50 mM HEPES-KOH (pH 7.6), 2 mM NTPs, 20 mM creatine phosphate, 100 mM potassium acetate, 2 mM spermidine, 1 mM dithiothreitol, 6 mM magnesium acetate, 0.1 mM 10-formyl-5, 6, 7, 8-tetrahydrofolic acid .
- mRNA ⁇ ⁇ ⁇ 1.5 ⁇ M or cDNA 0.15 M aminoacyl initiation tRNA carrying N-chloroacetyl-D-tryptophan (ClAc-D-Trp) as an acyl group (prepared by the method disclosed in JP 2008-125396) Then, 2.5 ⁇ L of a transcription / translation reaction solution containing 19 kinds of amino acids (each 5 ⁇ mM) constituting the natural protein excluding methionine was prepared.
- a peptide-mRNA complex was prepared according to the following procedure. First, 4 mM sodium acetate pH 5.0 and mRNA were mixed at 1: 3, heated on a thermoblock at 95 C for 1 minute, and then allowed to stand at room temperature for 5 minutes. A 25- ⁇ M linker solution dissolved in 1 mM sodium acetate was mixed and allowed to stand for 10 minutes to form an mRNA-linker complex. To this, other components of the translation system were added, and incubated at 37 ° C.
- a constant temperature bath (NT-202D, Hatsukashin Rika), followed by 12 minutes at room temperature, and 0.1 ⁇ M EDTA (ethylenediamine 4) adjusted to pH 7.5.
- Acetic acid, for molecular biology experiments, Nacalai Tesque were added to a final concentration of 20 mM, and further incubated in a constant temperature bath at 37 DEG C for 30 minutes.
- a peptide-mRNA complex was prepared by the following procedure. First, a solution containing a substance other than the linker was prepared, and this was reacted at 37 ° C. for 30 minutes in a constant temperature water bath to carry out transcription and translation reactions. To this, 0.25 ⁇ L of a 25 ⁇ M linker solution was added, and further incubated at 37 ° C. for 30 minutes and then at room temperature for 12 minutes.
- RNA-DNA hybrid chain was formed by performing a reverse transcription reaction. Specifically, the following operations were performed.
- the above translation reaction product contains 12 mM Tris-HCl (pH 8.3), 5 ⁇ M reverse transcription primer (CGS3an13.R21), 0.5 mM dNTPs, 18 mM Mg (OAc) 2 , 10 mM KOH (each concentration is the final concentration) ) was added.
- 1 unit of M-MLV Reverse Transcriptase RNaseH Minus, Point Mutant, Promega was added and reacted at 42 ° C. for 10 minutes in a constant temperature bath. Thereafter, EDTA and hydrochloric acid were added so that the final concentrations were 10 mM and 18 mM. ⁇ selection ⁇ Whether or not the peptide-mRNA complex molecule thus prepared was recovered by the interaction between the displayed peptide and the target protein was confirmed by the method shown below.
- TNF- ⁇ Human tumor necrosis factor alpha
- Biotin-modified TNF- ⁇ protein was added to a final concentration of 250 nM to 6 ⁇ L of cell-free translation reaction product solution containing peptide-mRNA complex molecules, and this solution was subjected to low adsorption treatment 0.6 mL
- the tube was transferred to an Eppendorf tube (platinum (ultra-low adsorption) tube, BM equipment), and the mixture was gently agitated on a rotator (RT-30 mini, Taitec) at 4 ° C for 1 hr for binding.
- 3 ⁇ L of a suspension of streptavidin-immobilized magnetic beads (Dynabeads registered trademark Streptavidin M-280, Invitrogen) was added, and the mixture was further stirred for 10 minutes.
- TBS Tris Buffered Saline, 50 mM Tris-
- Tween20 polyoxyethylene sorbitan monolaurate, Nacalai Tesque
- PCR (taq-) buffer (10 mM Tris-HCl (pH 9.0), 50 mM potassium chloride, 2.5 mM magnesium chloride, 250 ⁇ M dNTPs, 0.25 ⁇ M T7g10m.F48, 0.25 ⁇ M CGS3an13.R21, 0.2% 25 ⁇ L of Triton X-100) was added, and the supernatant was recovered after heating at 95 ° C. for 5 minutes on a thermoblock, whereby DNA after reverse transcription was recovered from the magnetic beads.
- PCR (taq-) buffer (10 mM Tris-HCl (pH 9.0), 50 mM potassium chloride, 2.5 mM magnesium chloride, 250 ⁇ M dNTPs, 0.25 ⁇ M T7g10m.F48, 0.25 ⁇ M CGS3an13.R21, 0.2% 25 ⁇ L of Triton X-100
- the real-time PCR measuring instrument uses LightCycler (registered trademark) 1.5 (Roche Applied Science), Taq polymerase, SYBR (registered trademark) Green I (100,000 dilution, Invitrogen), and the above-mentioned PCR (taq-) buffer.
- the reaction solution to which the sample solution was added was used for measurement.
- the soluble TNF- ⁇ which is a recombinant expressed by E. coli, was used as the TNF- ⁇ protein used in this example.
- Recombinant TNF- ⁇ is a fusion of the amino acid sequence of amino acids 77 to 233 of wild-type TNF- ⁇ , the AviTag sequence (GLNDIFEAQKIEWHE) and His ⁇ 6 tag sequence at the N-terminal side.
- Co-expression of this protein and biotin ligase (BirA) using AviTag as a substrate in E. coli results in biotinylation modification of the lysine side chain of AviTag, so TNF- ⁇ used in this example is strept It can be easily separated by avidin-immobilized beads.
- TNF- ⁇ -DW used as a peptide aptamer has a chloroacetyl group of N-chloroacetyl-D-tryptophan (ClAc-D-Trp), a non-protein amino acid, and a thiol group in the side chain of cysteine in the peptide sequence.
- Peptide XQRLHCLYLKH (X: Ac-D-Trp) cyclized by reacting to form thioether.
- a cDNA encoding a sequence in which the spacer amino acid sequence CGSGSGS was fused to the C-terminal side of this peptide was prepared by the method described above, and a peptide and mRNA complex molecule was prepared by a cell-free transcription / translation system.
- the recovery rate of the peptide-mRNA complex when this peptide aptamer and target protein combination was used was as shown in FIG. 4 and FIG.
- the recovery rates of an21- Phe and an21- ⁇ -Ala in which L-phenylalanine was bound to the 3 ′ end via an ester were 1.30% and 0.25%, respectively, when mRNA was added to the translation reaction solution (FIG. 4). In the case of adding (Fig. 5), it was 0.65% and 0.38%.
- the recovery rate of an21-ACCA linkers with no modified 3 'end is several tens of times lower than those (0.015% for mRNA, 0.007% for cDNA), L-phenylalanine, ⁇ It was shown that -L-alanine was efficiently recovered as a peptide-mRNA complex molecule by linking it to the C-terminal side of the peptide synthesized by translation via a covalent bond.
- the presented peptide and the mRNA encoding the sequence are of a single type, so the peptide synthesized by translation from mRNA.
- another linker molecule may be bound as an acceptor, or the linker molecule and mRNA molecule forming a double strand may be exchanged with another linker molecule or mRNA during the operation. Cannot be excluded. Therefore, the following experiment was performed to confirm that the displayed peptide and mRNA of the recovered peptide-mRNA-linker complex molecule were correctly associated.
- mRNA or cDNA encoding a peptide sequence that does not bind to the target protein (EMP1SS: XAAGGTYSSHFGPLTWVSKPQGGAA, X is Ac-D-Trp like TNF- ⁇ -DW) was performed as a control for TNF- ⁇ -DW.
- EMP1SS XAAGGTYSSHFGPLTWVSKPQGGAA, X is Ac-D-Trp like TNF- ⁇ -DW
- a mixture prepared by the method shown in Example 1 and mixed with mRNA or cDNA encoding TNF- ⁇ -DW at a molar ratio of 100: 1 was prepared. Based on this, the same experiment as described above was performed, and the recovered DNA was amplified by PCR.
- reaction product solution 2.5 ⁇ L was separated by electrophoresis using a gel consisting of TAE (40 mM Tris acetate, 1 mM mMEDTA) and 3%% agarose (low electroosmosis, Nacalai Tesque), stained with ethidium bromide and transilluminator An electrophoretic image was obtained using The result is shown in FIG.
- a band derived from mRNA encoding TNF- ⁇ -DW is confirmed at a position of about 90 bp, and a band derived from mRNA encoding EMP1SS as a control is observed at a position of about 150 bp.
- an an21-ACCA linker not containing a peptide acceptor was used, only the band derived from the mRNA of EMP1SS was confirmed, reflecting the molar ratio when two kinds of cDNA were first mixed.
- an21-Phe a band derived from TNF- ⁇ -DW mRNA was confirmed, and the fluorescence intensity was the same as or higher than that of EMP1SS.
- the peptide-mRNA complex presenting TNF- ⁇ -DW binding to the target protein was recovered more efficiently than the peptide-mRNA complex presenting EMP1SS not binding to the target.
- the same results can be obtained both when the mRNA is added to the translation reaction solution to form a peptide-mRNA complex and when the cDNA is added to form a peptide-mRNA complex. It was.
- TNF- ⁇ -DW spiked library random peptide library
- a peptide aptamer library displaying a random sequence of 8-12 amino acids was prepared.
- This library was prepared as mRNA encoding a random sequence.
- the structure of this library is a randomized portion of the TNF- ⁇ -DW or EMP1SS display sequence described above, and the rest of the library has the same structure, but the linker hybridizes.
- the length of the region forming this strand is 13 bases.
- TNF- ⁇ -DW spiked library For this random peptide library, mRNA encoding TNF- ⁇ -DW (TNF- ⁇ -DW (an13): the length of the double-stranded formation part with the linker is 13 bases) in a molar ratio of 1,000,000: 1 A spiked library was prepared so as to be mixed. [Round 1] Using this spiked library mRNA and an21-Phe linker, translation and formation of a peptide-mRNA complex were performed according to the procedure shown in Examples 1 and 2 above (Round 1).
- peptide-mRNA complex that binds to magnetic beads by mixing with streptavidin solid-phased magnetic beads not bound to any peptide-mRNA complex solution as negative selection at 4 ° C for 10 minutes was excluded.
- TNF- ⁇ was added to this solution to a final concentration of 250 nM and mixed at 4 ° C. for 1 hour.
- Streptavidin-immobilized magnetic beads were added to this, and bound at 4 ° C. for 10 minutes, and then washed 4 times with TBST.
- reverse transcription reaction solution (5 units / ⁇ L M-MLV Reverse Transcriptase (Promega), 2 ⁇ M CGS3an21.R44, 0.5 mM dNTP, 10 mM Tris-HCl (pH 8.3), 15 mM chlorination Suspended in 10 ⁇ L of potassium, 0.6 mM magnesium chloride, 2 mM dithiothreitol), heated at 42 C for 1 ⁇ hr to perform reverse transcription reaction.
- peptide aptamers can be selected from peptide libraries consisting of random sequences by this method.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Crystallography & Structural Chemistry (AREA)
- Bioinformatics & Computational Biology (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
Abstract
遺伝子型と表現型の対応付け技術(ディスプレイ法)において、核酸とその翻訳産物であるペプチドとの連結体を再構成型の無細胞翻訳系内で作製する際に好適なリンカーとして、一方の端に、mRNAの3'末端側の塩基と対合する側鎖塩基を有する一本鎖構造領域を含み、もう一方の端に、塩基配列ACCAからなるオリゴRNAにアミノ酸がエステル結合した構造からなるペプチドアクセプター領域を含み、当該エステル結合が人工RNA触媒を用いて形成されたことを特徴とするリンカー、及びそのようなリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を使用するディスプレイ法を提供する。
Description
本発明は、遺伝子型と表現型の対応付け技術(ディスプレイ系)において、cDNAまたはmRNAとその翻訳産物であるペプチドまたはタンパク質との連結体を作製する際に用いられる新規な方法に関する。本発明者らは、この方法をRAPIDディスプレイ法と命名した。この方法は、先に報告されたフレキシザイムシステム(あるいは、RAPIDシステム:Random Peptide Integrated Discovery system)を用いて構築される特殊ペプチドライブラリーからの、薬剤候補となり得るペプチドアプタマーのスクリーニングに好適である。
進化分子工学のツールとして誕生した遺伝子型と表現型の対応付け技術は、ディスプレイ法とも称され、mRNAディスプレイ法 ("In vitro virus"、Nemoto N, et al. FEBS Lett. 414, 405-408 (1997)、国際公開WO98/16636;又は"RNA-peptide fusions"、Roberts, R. W. & Szostak, J. W. , Proc. Natl. Acad. Sci. USA., 94, 12297-12302 (1997)、国際公開WO98/31700)、STABLE法(非共有結合DNAディスプレイ)、マイクロビーズドロップレット法、共有結合DNAディスプレイ法、ファージディスプレイ法、リボソーム・ディスプレイ法などが知られている。ディスプレイ法では、ライブラリから機能のあるペプチドやタンパク質の分子を選択した際に、それに対応する遺伝子が連結しているのでその配列を容易に読み取ることができ、特定の機能を有するポリペプチドの遺伝情報を選択する際に有用である。
mRNAディスプレイ法は無細胞翻訳系(in vitroタンパク質合成系)を用いて、遺伝子型としてのmRNAと表現型としてのペプチド分子を共有結合させることにより、遺伝子型と表現型を一体化する技術であり、現在、チロシルtRNA3’末端部分のアナログであるピューロマイシンを介して、合成されたペプチド分子とこれをコードするmRNAとを連結する方法がとられている。
mRNAディスプレイ法においては、ピューロマイシンを適当なリンカーを介してmRNAの3’末端に結合させておき、これを無細胞翻訳系に導入してmRNAからペプチドを合成すると、ピューロマイシンがリボソームにおけるペプチド転移反応の基質として伸長中のペプチド鎖のC末端に連結し、翻訳産物であるペプチド分子がピューロマイシンを介してmRNAと連結する(図1A)。リンカーは、主として、ピューロマイシンをリボソームのAサイトに効率良く取り込ませるために、mRNAとピューロマイシンの間に挿入される。ピューロマイシンの特徴として、アミノアシルtRNAの3’末端とは異なり、アデノシン様部分とアミノ酸(チロシン)様部分がエステル結合ではなくアミド結合を形成していることが挙げられる(図1B)。そのため、リボソーム上で連結したピューロマイシンとペプチドとの結合体は、加水分解されにくく安定である。
mRNAディスプレイ法においては、ピューロマイシンを介してmRNAと翻訳産物を連結するために、無細胞翻訳系の外で、予め、ピューロマイシンをmRNAの3’末端に連結しておくことが必要である。この連結方法として、まずピューロマイシンから5’末端側へ直鎖高分子からなるスペーサーを合成したピューロマイシン結合リンカー(Puromycin conjugated linker)を作成してから、当該リンカーとmRNAの3’末端を結合する方法と、mRNAの3’末端側にスペーサーを結合してから、当該結合体にピューロマイシンを結合する方法がある。いずれの方法でも、スペーサーである直鎖高分子の端は、典型的には、リン酸基またはヌクレオチドであり、mRNAの3’末端とリンカーの5’末端側との結合は、リン酸基を介した共有結合となっている。この共有結合は、RNAリガーゼまたはDNAリガーゼを用いた反応、または一般的な有機化学反応によって形成される。
Nemoto N, et al. FEBS Lett. 414, 405-408 (1997)
Roberts, R. W. & Szostak, J. W. , Proc. Natl. Acad. Sci. USA., 94, 12297-12302 (1997)
公知のmRNAディスプレイ法においては、3’末端にピューロマイシンを持つmRNAを鋳型として、これを小麦胚芽やウサギ網状赤血球の抽出液を使用する無細胞翻訳系に加えてペプチドへの翻訳を行う。従って、DNAからmRNAへの転写、及びmRNAとピューロマイシンとの連結反応を、予め翻訳系外で行っておく必要がある。
近年、大腸菌のリボソームを用いる系で、翻訳に必要な因子をそれぞれ精製して混ぜ合わせた、再構成型の無細胞翻訳系が開発された(H. F. Kung, B. Redfield, B. V. Treadwell, B. Eskin, C. Spears and H. Weissbach (1977) “DNA-directed in vitrosynthesis of beta-galactosidase. Studies with purified factors” The Journal of Biological Chemistry Vol. 252, No. 19, 6889-6894 ; M. C. Gonza, C. Cunningham and R. M. Green (1985) “Isolation and point of action of a factor from Escherichia coli required to reconstruct translation” Proceeding of National Academy of Sciences of the United States of America Vol. 82, 1648-1652 ; M. Y. Pavlov and M. Ehrenberg (1996) “Rate of translation of natural mRNAs in an optimized in Vitro system” Archives of Biochemistry and Biophysics Vol. 328, No. 1, 9-16 ; Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda (2001) “Cell-free translation reconstituted with purified components” Nature Biotechnology Vol. 19, No. 8, 751-755;H. Ohashi, Y. Shimizu, B. W. Ying, and T. Ueda (2007) “Efficient protein selection based on ribosome display system with purified components” Biochemical and Biophysical Research Communications Vol. 352, No. 1, 270-276)。再構成型の無細胞翻訳系とは、主に大腸菌抽出液を使用する無細胞翻訳系を細分化し、各因子を再構成することにより、翻訳と関係の無い成分が除かれたシステムであり、従来の細胞抽出液を使用する無細胞翻訳系よりもヌクレアーゼやプロテアーゼなどの阻害物質の混入を容易に防ぐことができる。転写反応に必要な因子を加えることにより、DNAからの転写を翻訳と同時に行うこともできる。
このような転写カップル型の無細胞翻訳系を用いれば、翻訳反応と同一の系内で、鋳型DNAからの転写によるmRNAの合成を行うことができる。さらに、mRNAとリンカー分子との複合体形成もその同じ系内で行うことができれば、従来のmRNAディスプレイ法とは異なり、cDNAの転写からmRNA-ペプチド連結体の作製までをワンポット(同一反応容器内)で調製することが可能になる。そのような、反応系成分の制御が可能な再構成型の無細胞翻訳系の利点を生かしたディスプレイ法を提供することが、本発明の第一の課題である。
さらに、このようなディスプレイ法を、先に報告された、アシル化tRNA合成触媒能を有するリボザイム(フレキシザイム)を用いた特殊ペプチド合成技術と組み合わせることで、特殊ペプチドが提示されたmRNAまたはcDNAのライブラリーの構築を可能とすることが、本発明の第二の課題である。
本発明のRAPIDディスプレイ法では、mRNAディスプレイ法を改変して、ピューロマイシン結合リンカーの代わりに本発明者らが今回新たに開発したリンカー分子を利用し、さらに再構成型の無細胞翻訳系を最適化することにより、転写、翻訳、およびリンカーとmRNAとの複合体形成、それに続くペプチドとリンカーとの結合までを、一つの翻訳系内で完結させることが可能となった。
従来のmRNAディスプレイ法と比較した、RAPIDディスプレイ法の主な特徴は以下の通りである。
(ア)リンカー分子の3’末端はピューロマイシンではなく、アミノ酸がエステルを介してアデノシンに結合(アミノアシル化)した構造となっている。
(イ)アミノアシル化反応は、人工RNA触媒(リボザイム)により行う。
(ウ)再構成型の無細胞翻訳系を利用する。
(エ)リンカーとmRNAとの結合は、ライゲーションではなく、翻訳系内でのハイブリダイゼーションによる複合体形成で行う。
(オ)転写、翻訳およびリンカーとの複合体形成を単一の翻訳反応容器中で行うことができる。
(ア)リンカー分子の3’末端はピューロマイシンではなく、アミノ酸がエステルを介してアデノシンに結合(アミノアシル化)した構造となっている。
(イ)アミノアシル化反応は、人工RNA触媒(リボザイム)により行う。
(ウ)再構成型の無細胞翻訳系を利用する。
(エ)リンカーとmRNAとの結合は、ライゲーションではなく、翻訳系内でのハイブリダイゼーションによる複合体形成で行う。
(オ)転写、翻訳およびリンカーとの複合体形成を単一の翻訳反応容器中で行うことができる。
さらに、同じ翻訳系において、特殊ペプチドの翻訳合成技術を適用することにより、表現型として特殊ペプチドを提示することもできる。特殊ペプチドの構成単位である、非タンパク質性のアミノ酸やヒドロキシ酸をtRNAに担持させるアシル化反応も、人工RNA触媒(リボザイム)を使用して行う。
本発明の概略は以下の通りである。
(1)再構成型のin vitroタンパク質合成系において、mRNAと、その翻訳産物であるペプチドとが、リンカーを介して結合した連結体を作製するために使用されるリンカーであって、
リンカーの一方の端に、mRNAの3'末端側の塩基と対合する側鎖塩基を有する一本鎖構造領域を含み、
リンカーのもう一方の端に、ペプチド転移反応によって翻訳産物と結合可能な基を含むペプチドアクセプター領域を含み、
ペプチドアクセプター領域は、塩基配列ACCAからなるオリゴRNAにアミノ酸がエステル結合した構造からなり、
当該エステル結合が、人工RNA触媒を用いるアミノアシル化反応により形成された
ことを特徴とするリンカー。
(2)一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されている、前記(1)のリンカー。
(3)一本鎖構造領域が一本鎖DNAからなる、前記(1)または(2)のリンカー。
(4)アミノアシル化反応に使用される人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、前記(1)~(3)のいずれかのリンカー。
(5)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAとを接触させるとともに、mRNAからペプチドの翻訳を行う工程
を含む、前記方法。
(6)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAの鋳型となるDNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中にリンカーとDNAを供することにより、DNAからmRNAへの転写およびペプチドへの翻訳ならびにリンカーとmRNAとの複合体形成を行う工程
を含む、前記方法。
(7)再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、(5)または(6)に記載の方法。
(8)前記(7)に記載の方法で作製された、〔mRNA〕-〔リンカー〕-〔特殊ペプチド〕連結体からなるライブラリー。
(9)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体ライブラリーから、標的物質に結合するペプチドアプタマーを選択する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ランダムペプチド配列をコードする配列の下流に有するmRNAライブラリーを作製する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAライブラリーとを接触させるとともに、ペプチドの翻訳を行うことにより、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを作製する工程
標的物質と、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを接触させる工程
標的物質に結合したペプチドを提示する連結体を選択する工程
を含む、前記方法。
(10)標的物質がビオチン化修飾されている、(9)に記載の方法。
(11)再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、(9)または(10)に記載の方法。
(12)上記(2)に記載のリンカーを作製する方法であって、
一本鎖構造領域と、配列ACCAからなるオリゴRNAとの間を、ポリエチレングリコールで連結したキメラオリゴヌクレオチドを合成し、
キメラオリゴヌクレオチドの3’末端のアデノシンに、人工RNA触媒を用いる反応により、アミノ酸をエステル結合させることにより、
一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されたリンカーを作製する、前記方法。
(13)人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、(12)に記載の方法。
(1)再構成型のin vitroタンパク質合成系において、mRNAと、その翻訳産物であるペプチドとが、リンカーを介して結合した連結体を作製するために使用されるリンカーであって、
リンカーの一方の端に、mRNAの3'末端側の塩基と対合する側鎖塩基を有する一本鎖構造領域を含み、
リンカーのもう一方の端に、ペプチド転移反応によって翻訳産物と結合可能な基を含むペプチドアクセプター領域を含み、
ペプチドアクセプター領域は、塩基配列ACCAからなるオリゴRNAにアミノ酸がエステル結合した構造からなり、
当該エステル結合が、人工RNA触媒を用いるアミノアシル化反応により形成された
ことを特徴とするリンカー。
(2)一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されている、前記(1)のリンカー。
(3)一本鎖構造領域が一本鎖DNAからなる、前記(1)または(2)のリンカー。
(4)アミノアシル化反応に使用される人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、前記(1)~(3)のいずれかのリンカー。
(5)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAとを接触させるとともに、mRNAからペプチドの翻訳を行う工程
を含む、前記方法。
(6)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAの鋳型となるDNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中にリンカーとDNAを供することにより、DNAからmRNAへの転写およびペプチドへの翻訳ならびにリンカーとmRNAとの複合体形成を行う工程
を含む、前記方法。
(7)再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、(5)または(6)に記載の方法。
(8)前記(7)に記載の方法で作製された、〔mRNA〕-〔リンカー〕-〔特殊ペプチド〕連結体からなるライブラリー。
(9)mRNAと、その翻訳産物であるペプチドが、前記(1)~(4)のいずれかのリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体ライブラリーから、標的物質に結合するペプチドアプタマーを選択する方法であって、
前記(1)~(4)のいずれかのリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ランダムペプチド配列をコードする配列の下流に有するmRNAライブラリーを作製する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAライブラリーとを接触させるとともに、ペプチドの翻訳を行うことにより、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを作製する工程
標的物質と、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを接触させる工程
標的物質に結合したペプチドを提示する連結体を選択する工程
を含む、前記方法。
(10)標的物質がビオチン化修飾されている、(9)に記載の方法。
(11)再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、(9)または(10)に記載の方法。
(12)上記(2)に記載のリンカーを作製する方法であって、
一本鎖構造領域と、配列ACCAからなるオリゴRNAとの間を、ポリエチレングリコールで連結したキメラオリゴヌクレオチドを合成し、
キメラオリゴヌクレオチドの3’末端のアデノシンに、人工RNA触媒を用いる反応により、アミノ酸をエステル結合させることにより、
一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されたリンカーを作製する、前記方法。
(13)人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、(12)に記載の方法。
再構成型の無細胞翻訳反応液中でmRNAと結合可能なリンカー分子を利用することで、単一の反応容器中にリンカーとcDNAまたはmRNAを加えて翻訳反応を行うだけで、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製することができる。
さらに、同じ再構成型の無細胞翻訳反応液中に、非タンパク質性のアミノ酸やヒドロキシ酸を担持させたtRNAを導入することにより、鋳型である核酸分子の配列情報から翻訳により合成された特殊ペプチドを提示した、〔mRNA〕-〔リンカー〕-〔特殊ペプチド〕連結体を得ることができる。
このように、RAPIDディスプレイ法を、特殊ペプチドの翻訳合成技術と組み合わせることにより、生体内での安定性や標的タンパク質への結合力の向上が期待される人工的なペプチドアプタマーの遺伝子ライブラリーを簡便に構築することが可能となる。
1.リンカー
リンカーは、本発明のRAPIDディスプレイ法においても公知のmRNAディスプレイ法と同様に、一方の端でmRNAの3’末端側と、もう一方の端でペプチドのC末端側と結合することにより、mRNAとその翻訳産物であるペプチドを連結する。
リンカーは、本発明のRAPIDディスプレイ法においても公知のmRNAディスプレイ法と同様に、一方の端でmRNAの3’末端側と、もう一方の端でペプチドのC末端側と結合することにより、mRNAとその翻訳産物であるペプチドを連結する。
しかしながら、本発明のRAPIDディスプレイ法では、リンカーの両端の構造が公知のmRNAディスプレイ法と相違する。本明細書では、本発明のRAPIDディスプレイ法で使用されるリンカーのことを、「RAPIDリンカー」と称することもある。
まず、リンカーの一方の端である、ペプチドのC末端と結合する部分について説明する。本明細書においては、この部分を「ペプチドアクセプター(peptidyl acceptor)」あるいは、単に「アクセプター」とも称する。すなわち、ペプチドアクセプターとは、リボソームにおけるペプチド転移反応によって伸長中のペプチド(ペプチジルtRNA)と結合できる構造を持った分子を意味する。ペプチドアクセプターは、リンカーの末端に位置する部分のことを指す場合もあるし、リンカーを含んだ全体の構造を指す場合もある。例えば、公知のmRNAディスプレイ法におけるペプチドアクセプターは、リンカーの末端に位置するピューロマイシン、またはリンカーを含んだ全体の構造であるピューロマイシン結合リンカーである。
本発明のRAPIDリンカーでは、ペプチドアクセプターの構造とその作製方法に特徴がある。
RAPIDディスプレイ法では、3’末端側に4残基のリボヌクレオチドからなるACCA配列を有するリンカーを合成してから、その3’末端のアデノシンに任意のアミノ酸を結合させることにより、リンカーにペプチドアクセプターとしての構造を付与する。リボソーム上のペプチド伸長反応において、リンカー末端に結合したアミノ酸が、ペプチジルtRNAのペプチドC末端を受け取ってペプチドと結合する。本明細書において、RNA配列ACCAにアミノ酸がエステル結合した構造を「ペプチドアクセプター領域」と称する。
公知のmRNAディスプレイ法におけるペプチドアクセプターであるピューロマイシンは、アデノシン様部分のリボースとアミノ酸とがアミド結合したアミノヌクレオシド構造を有する。これに対して、本発明のRAPIDディスプレイ法では、リボースの3’-Oにアミノ酸がエステル結合した構造となっている。つまり、本発明のRAPIDディスプレイにおけるペプチドアクセプターは、天然のアミノアシルtRNAと同様のヌクレオシド構造を有する。このようなペプチドアクセプターの一例として、L-フェニルアラニンが結合したリンカーの構造について、図2B(L-Phe conjugated linker)を参照し、図1B(Puromycin conjugated linker)と比較されたい。本発明では、ペプチドアクセプターが、より天然に近い構造を用いることでピューロマイシンと同等あるいはそれ以上の取り込み効率を示す。
ペプチドアクセプターとペプチドC末端との結合の形成はリボソーム内で起こる通常のペプチド転移反応と同様に、P部位のペプチジルtRNAに付いたペプチドのC末端のエステル結合に、A部位に取り込まれたペプチジルアクセプターのアミノ基が近接することにより起こると思われる。従って、当該ペプチド鎖C末端との間で形成される共有結合は、mRNAディスプレイ法の場合と同様に、典型的にはアミド結合である。なお、本発明のRAPIDディスプレイでは、リンカーの合成に後述する人工RNA触媒(フレキシザイム)を用いることで天然型(非タンパク質性)のアミノ酸とは異なるD-アミノ酸あるいはβ(beta)-アミノ酸を有するものを用いることも可能である。
次に、リンカーのもう一方の端におけるmRNAとの結合について説明する。
本発明のRAPIDディスプレイ法においては、リンカーの5’末端側とmRNA分子の3’末端側とが、塩基対形成に基くハイブリダイゼーションにより複合体を形成する。従って、リンカーの5’末端側は核酸塩基を側鎖に持つ一本鎖構造を取る。RAPIDリンカーにおける、この部分を、本明細書では「一本鎖構造領域」と称する。核酸塩基を側鎖に持つ一本鎖構造の具体的な例は、一本鎖DNA、一本鎖RNA、一本鎖PNA(ペプチド核酸)などである。形成された複合体は、ペプチドのセレクションの間も安定的に保持される必要がある。リンカーの一本鎖構造領域の塩基配列と、mRNA分子の3’末端側配列との、相補性が高いほど二本鎖形成の効率が高くなり、安定性も高くなる。安定性は、GC含量、反応溶液の塩濃度、反応温度にも依存する。特に、高いGC含量を有することが望ましく、具体的には80%以上、好ましくは85%以上のGC含量を有することが望ましい。そのようなリンカーの5’末端構造の具体例を挙げて説明するならば、決して限定されるものではないが、実施例で使用された
5'-CTCCCGCCCCCCGTCC-3’(配列番号1)
5'-CCCGCCTCCCGCCCCCCGTCC-3’(配列番号2)
といった塩基配列を有する、13~21ヌクレオチドからなる一本鎖DNAが挙げられる。
5'-CTCCCGCCCCCCGTCC-3’(配列番号1)
5'-CCCGCCTCCCGCCCCCCGTCC-3’(配列番号2)
といった塩基配列を有する、13~21ヌクレオチドからなる一本鎖DNAが挙げられる。
リンカーの両端以外の部分は、公知のmRNAディスプレイ法で使用されるリンカーの構造と同様に、全体として、柔軟性があり、親水性で、側鎖の少ない単純な直鎖構造を有するように設計される。従って、例えば、一本鎖又は二本鎖DNAやRNA等のオリゴヌクレオチド、ポリエチレン等のポリアルキレン、ポリエチレングリコール等のポリアルキレングリコール、ポリスチレン、多糖類等の直鎖状物質又はこれらの組合わせを適宜選択して用いることができる。リンカーの長さは、100Å以上あることが好ましく、100~1000Å程度であることがさらに好ましい。
本発明で使用可能なリンカーの非限定的な具体例としては、一本鎖構造領域がGC含量の高い配列を有する一本鎖DNAであり、3’末端がACCA配列からなるRNAであり、DNAとRNAの間がポリエチレングリコール(PEGリンカー)で連結されたDNA/RNAキメラオリゴヌクレオチドが上げられる。例えば、実施例1で合成された、〔DNA〕-〔Spacer18〕n-rArCrCrA(但し、Spacer18はヘキサエチレングリコールであり、nは4~8の整数である)が典型的な例として挙げられる。
2.リンカーのアミノ酸修飾
上述の通り、リンカーにペプチドアクセプターとしての構造を付与するためには、リンカー分子末端のオリゴRNA(ACCA配列)部分に、アミノ酸を結合させる必要がある。このアミノ酸の結合を、人工RNA触媒を用いて行うことが、本発明の特徴である。
上述の通り、リンカーにペプチドアクセプターとしての構造を付与するためには、リンカー分子末端のオリゴRNA(ACCA配列)部分に、アミノ酸を結合させる必要がある。このアミノ酸の結合を、人工RNA触媒を用いて行うことが、本発明の特徴である。
固相合成法などの通常の化学合成を用いて合成したオリゴRNAの3'-O部位にエステルを介してアミノ酸を結合させることは理論上可能ではあるが、生産物は反応性が高く安定性を欠くため、これを合成反応後の後処理を行った後に無細胞翻訳系に供することは事実上不可能である。本発明においては、この問題点を、アミノアシルtRNA合成酵素として開発された人工RNA触媒である「フレキシザイム」を用いたオリゴRNAのアミノアシル化反応により解決している。この方法では、アミノアシル化反応は穏和な条件で行うことができ、容易な後処理を行うだけで翻訳系に導入して用いることができる。詳細は以下の文献を参照されたい。
H. Murakami, H. Saito, and H. Suga, (2003), Chemistry & Biology, Vol. 10, 655-662;
H. Murakami, D. Kourouklis, and H. Suga, (2003), Chemistry & Biology, Vol. 10, 1077-1084;
H. Murakami, A. Ohta, H. Ashigai, H. Suga (2006) Nature Methods 3, 357-359;
N. Niwa, Y. Yamagishi, H. Murakami, H. Suga (2009) Bioorganic & Medicinal Chemistry Letters 19, 3892-3894; および
特開2008-125396またはWO2007/066627。
H. Murakami, H. Saito, and H. Suga, (2003), Chemistry & Biology, Vol. 10, 655-662;
H. Murakami, D. Kourouklis, and H. Suga, (2003), Chemistry & Biology, Vol. 10, 1077-1084;
H. Murakami, A. Ohta, H. Ashigai, H. Suga (2006) Nature Methods 3, 357-359;
N. Niwa, Y. Yamagishi, H. Murakami, H. Suga (2009) Bioorganic & Medicinal Chemistry Letters 19, 3892-3894; および
特開2008-125396またはWO2007/066627。
フレキシザイムは、ジニトロベンジルフレキシザイム(dFx)、エンハンスドフレキシザイム(eFx)、アミノフレキシザイム (aFx) 等の呼称でも知られる。フレキシザイムは、弱く活性化されたアミノ酸を基質として、アミノ酸の反応点であるカルボニル基、およびアミノ酸側鎖あるいは脱離基である芳香環、並びにリンカーの3’末端に存在するACC-3'配列部分を認識して、3’末端のアデノシンにアミノアシル化する触媒能を有する。RAPIDリンカーの端にACCA-3'の配列からなるオリゴRNA構造が必須なのはこのためである。フレキシザイムによるアミノアシル化反応は、アミノ酸基質と、結合相手であるオリゴRNA部分を有するリンカー分子を、フレキシザイム存在下、2時間程度氷上に置くだけで進行する。
本発明においては、以下の式で示される配列を有するフレキシザイムが好適に使用される。
原型のフレキシザイム Fx
[GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU-3’, 45nt] (配列番号3)
エンハンスドフレキシザイム eFx
[5'-GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU-3’,45nt]) (配列番号4)
ジニトロベンジルフレキシザイム dFx
[5'-GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU-3’,46nt] (配列番号5)
アミノフレキシザイム aFx
[5'-GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU-3’,47nt]) (配列番号19)
フレキシザイム Fxは、脱離基としてシアノメチル基を、側鎖として芳香環をもつアミノ酸(例えば、フェニルアラニン、チロシンなどのシアノメチルエステル)、フレキシザイム eFx, dFx, aFxはフレキシザイム Fxで利用可能な脱離基とアミノ酸に加え、脱離基として4-クロロベンジルチオールを、側鎖としては芳香環以外の側鎖をもつアミノ酸を基質としたアミノアシル化を触媒することができる。詳細は、前述の文献を参照されたい。したがって、このような構造を持つアミノ酸とリンカーをフレキシザイムの存在下で反応させることにより、リンカー3’末端のアデノシン内のリボース環上の3’位の水酸基に、アミノ酸がエステル結合した分子を得ることができる。アミノ酸基質としては、任意の構造を持つものを結合させることができ、天然の翻訳で使用されるアミノ酸のみならず、D-アミノ酸あるいはβ(beta)-アミノ酸などの非タンパク質性アミノ酸(すなわち、天然に存在するタンパク質中に通常見出されるL-アミノ酸以外のもの)も利用できる。さらに、アミノ酸の代わりにヒドロキシカルボン酸をリンカー3’末端に結合させることもでき、ペプチドアクセプターとしてリボソームへの取り込みも可能である。
原型のフレキシザイム Fx
[GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU-3’, 45nt] (配列番号3)
エンハンスドフレキシザイム eFx
[5'-GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU-3’,45nt]) (配列番号4)
ジニトロベンジルフレキシザイム dFx
[5'-GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU-3’,46nt] (配列番号5)
アミノフレキシザイム aFx
[5'-GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU-3’,47nt]) (配列番号19)
フレキシザイム Fxは、脱離基としてシアノメチル基を、側鎖として芳香環をもつアミノ酸(例えば、フェニルアラニン、チロシンなどのシアノメチルエステル)、フレキシザイム eFx, dFx, aFxはフレキシザイム Fxで利用可能な脱離基とアミノ酸に加え、脱離基として4-クロロベンジルチオールを、側鎖としては芳香環以外の側鎖をもつアミノ酸を基質としたアミノアシル化を触媒することができる。詳細は、前述の文献を参照されたい。したがって、このような構造を持つアミノ酸とリンカーをフレキシザイムの存在下で反応させることにより、リンカー3’末端のアデノシン内のリボース環上の3’位の水酸基に、アミノ酸がエステル結合した分子を得ることができる。アミノ酸基質としては、任意の構造を持つものを結合させることができ、天然の翻訳で使用されるアミノ酸のみならず、D-アミノ酸あるいはβ(beta)-アミノ酸などの非タンパク質性アミノ酸(すなわち、天然に存在するタンパク質中に通常見出されるL-アミノ酸以外のもの)も利用できる。さらに、アミノ酸の代わりにヒドロキシカルボン酸をリンカー3’末端に結合させることもでき、ペプチドアクセプターとしてリボソームへの取り込みも可能である。
3.翻訳系内での反応
RAPIDディスプレイ法では、リンカーをcDNAあるいはmRNAと共に無細胞翻訳系(「in vitroタンパク質合成系」ともいう)に加えて一定時間反応させることで、mRNAからの翻訳およびリンカー分子との複合体形成、それに続くペプチドとリンカーとの連結を行うことができる。
RAPIDディスプレイ法では、リンカーをcDNAあるいはmRNAと共に無細胞翻訳系(「in vitroタンパク質合成系」ともいう)に加えて一定時間反応させることで、mRNAからの翻訳およびリンカー分子との複合体形成、それに続くペプチドとリンカーとの連結を行うことができる。
無細胞翻訳系としては、慣用の再構成型の無細胞翻訳系を適宜改変して利用でき、DNA依存性RNAポリメラーゼ(好ましくはT7RNAポリメラーゼ)を加えた系であればDNAからの転写も翻訳と同じ系で行うことができる。
転写・翻訳型の合成系であれば、以下の(ア)から(エ)の反応を一つの反応容器(one-pot)内で行うことが可能である。
(ア)DNAをmRNAに転写する反応
(イ)mRNAの3’末端側が、リンカー末端の一本鎖構造領域とハイブリダイゼーションにより複合体を形成する反応
(ウ)mRNAからペプチドに翻訳する反応
(エ)翻訳が完了したペプチドのC末端がリンカーのもう一方の末端のペプチドアクセプター領域にアミド結合する反応
(オ)〔ペプチド〕-〔リンカー〕-〔mRNA〕複合体が、リボソームから解離する反応。
(ア)DNAをmRNAに転写する反応
(イ)mRNAの3’末端側が、リンカー末端の一本鎖構造領域とハイブリダイゼーションにより複合体を形成する反応
(ウ)mRNAからペプチドに翻訳する反応
(エ)翻訳が完了したペプチドのC末端がリンカーのもう一方の末端のペプチドアクセプター領域にアミド結合する反応
(オ)〔ペプチド〕-〔リンカー〕-〔mRNA〕複合体が、リボソームから解離する反応。
あるいは、予め調製されたmRNAを、リンカーと一緒に翻訳系に加える場合は、(イ)から始まる一連の反応を再構成型の無細胞翻訳系内で行う。
再構成型の無細胞翻訳系には精製したリボソーム、翻訳開始因子、翻訳伸長因子、mRNA、アミノアシルtRNA、基質としてATPやGTP等が必要である(M. H. Schreier, B. Erni and T. Staehelin (1977) “Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors.” Journal of Molecular Biology, Vol. 116, No. 4, 727-53. H. Trachsel, B. Emi, M. H. Schreier and T. Staehelin (1977)“Initiation of mammalian protein synthesis. II. The assembly of the initiation complex with purified initiation factors.” Journal of Molecular Biology, Vol. 116, No. 4, 755-67.)。これらのうち、アミノアシルtRNAは同一反応液中にtRNAとアミノアシル-tRNA合成酵素およびその基質を加えることで代替が可能である。また、一般の無細胞翻訳系において行われているように、翻訳反応の効率や忠実性を上げるため、翻訳終止因子、リボソーム再生因子、クレアチンキナーゼ、ミオキナーゼ、ヌクレオチド二リン酸キナーゼ、ピロフォスファターゼなどのタンパク質や酵素およびその基質を添加することも可能である(P.C. Jelenc and C.G. Kurland (1979) “Nucleoside triphosphate regeneration decreases the frequency of translation errors” Proceedings of the Natural Academy Science of the United States of America Vol. 76, No. 7, 3174-3178)。また、翻訳と同時に転写反応を行う場合は、mRNAの代わりにcDNAおよびT7 RNAポリメラーゼとその基質を添加することができる。
本発明においては、目的に合わせて最適化された構成因子からなる無細胞翻訳系に、必要な配列を有するDNAまたはRNAが供される。DNAまたはRNAの配列については、使用する合成系で、cDNAがmRNAに転写され、当該mRNAの翻訳が開始されるための配列が必要である。ペプチドのアミノ酸配列をコードする部分の全長が、最後まで翻訳される必要があり、さらに、翻訳されたアミノ酸のC末端側にも柔軟性を持たせるためのペプチドからなるスペーサー配列が連結し、その直後に停止コドンが来る。例えば、ペプチド配列(Cys-(Gly-Ser-)x3)と、その直後にアンバーコドン(停止コドン)がコードされる。また、mRNAの3’末端側は、リンカーの一本鎖構造領域とハイブリダイズして二本鎖を形成できるような構造を有するので、コード領域の下流(停止コドンの直後)は、一本鎖構造領域の配列に相補的な配列を有する必要がある。この部分(二本鎖形成部)の配列を、本明細書中ではリンカーハイブリダイゼーション配列と称する。
cDNAまたはmRNAは、具体的には、以下のような配列を含むことが望ましい。
(1)cDNAにおいて、使用するRNAポリメラーゼに対応したプロモーター配列。
T7プロモーターの場合、TAATACGACTCACTATA (配列番号6)
(2)開始コドン上流の適切な配列をコードする配列。
無細胞翻訳系で大腸菌由来のリボソームを使用する場合、SD配列遺伝子を含む。これは通常のタンパク質合成と同様である。例えば、GGGTTAACTTTAA GAAGGAGATATACAT (配列番号7): T7ファージのgene 10 proteinの上流配列を改変したもの。下線部はSD配列。
T7プロモーターの場合、TAATACGACTCACTATA (配列番号6)
(2)開始コドン上流の適切な配列をコードする配列。
無細胞翻訳系で大腸菌由来のリボソームを使用する場合、SD配列遺伝子を含む。これは通常のタンパク質合成と同様である。例えば、GGGTTAACTTTAA GAAGGAGATATACAT (配列番号7): T7ファージのgene 10 proteinの上流配列を改変したもの。下線部はSD配列。
(3)開始コドン(ATG)から始まる、変異遺伝子ライブラリーのORF〔ペプチドアプタマーのC末端側にスペーサーが融合したアミノ酸配列をコードする配列〕を構成する配列。
ここで使用される停止コドンにより、無細胞翻訳系から対応するrelease factor(解離因子)を除く必要がある。TAG(アンバーコドン)を用いた場合はRF1を、TGA(オパールコドン)を用いた場合はRF2を、TAA(オーカーコドン)を用いた場合はRF1とRF2の両方を除いて無細胞翻訳反応液を調製する。
(4)二本鎖形成部をコードする配列(リンカーハイブリダイゼーション配列)。
本発明で使用される再構成型の無細胞翻訳反応液としては、構成因子を目的に合わせて改変して利用することが好ましい。例えば、実施例においては、アンバーコドンに対応するrelease factor、ペプチジル-tRNAを分解する酵素であるペプチジル-tRNAヒドロキシダーゼ(PTH)などを除いている。
また、再構成無細胞翻訳系に、限られた天然アミノ酸だけを与えた系に、予め所望の非タンパク質性アミノ酸(又はヒドロキシ酸)でチャージされた(つまり、活性化されたアミノ酸が結合した)アシル化tRNAを添加することも可能である。添加しない天然アミノ酸のコドンを、非タンパク質性アミノ酸(又はヒドロキシ酸)でアシル化されたtRNAのアンチコドンに対応させることにより、非タンパク質性アミノ酸(又はヒドロキシ酸)を含むペプチドをmRNAの遺伝情報に基づき、リボソーム上で翻訳合成することが可能である。あるいは、天然アミノ酸を含まない再構成無細胞翻訳系に、非タンパク質性アミノ酸(又はヒドロキシ酸)でチャージされたアシル化tRNAを加えることにより、天然アミノ酸を全く含まない特殊ペプチドを翻訳合成することも可能である。
非タンパク質性アミノ酸(又はヒドロキシ酸)を有するアシル化tRNAは、アミノアシルtRNA合成触媒能を有する上述の人工RNA触媒「フレキシザイム」を用いて調製することができる。上述したように、これらの人工RNA触媒は、任意の側鎖を持つアミノ酸をチャージさせることができ、また、tRNAの3'末端に存在する共通配列である5'-RCC-3'配列部分(R = A or G)のみを認識し、tRNAの3'末端にアシル化する機能を有するので、アンチコドンの異なるどのようなtRNAにも対応することが可能である。さらに、アミノ酸の認識部位にα位の置換基が含まれていないため、Lアミノ酸に限らず、ヒドロキシ酸(α位が水酸基)、N-メチルアミノ酸(α位がN-メチルアミノ酸)、N-アシルアミノ酸(α位がN-アシルアミノ基)、D-アミノ酸なども基質とすることができる。詳細については、上述のフレキシザイムに関する文献に加え、Y. Goto, H. Suga (2009) "Translation initiation with initiator tRNA charged with exotic peptides" Journal of the American Chemical Society, Vol. 131, No. 14, 5040-5041、WO2008/059823「N末端に非天然骨格をもつポリペプチドの翻訳合成とその応用」、Goto et al., ACS Chem. Biol., 2008, 3, 120-129、WO2008/117833「環状ペプチド化合物の合成方法」などに記載されている。
本出願人らは、「フレキシザイム」を用いて非タンパク質性アミノ酸やヒドロキシ酸をアシル化したtRNAを用いた特殊ペプチドの合成系(キットまたは合成方法の両方を含む概念)をコア技術としてペプチドの翻訳合成・修飾・スクリーニングを行う総合技術をRAPIDシステム(Random Peptide Integrated Discovery system)と呼んでいる。RAPIDシステムでは、適当な配列の鋳型mRNAに基くin vitro翻訳産物として、様々な特殊ペプチドを翻訳合成することができる。これまでの説明から自明であるが、本明細書において、特殊ペプチドとは、RAPIDシステムで合成可能なあらゆる翻訳産物を含み、20種類の天然アミノ酸以外に、様々な側鎖を有するアミノ酸、β(beta)-アミノ酸、γ(gamma)-アミノ酸及びδ(delta)-アミノ酸、D-アミノ酸、アミノ酸骨格上のアミノ基やカルボキシル基が置換された構造を有する誘導体を含む、上述した様々な基質を構成要素とする重合体である。さらに、特殊ペプチドは、主鎖骨格として通常のアミド結合とは異なる構造も有し得るものである。例えば、アミノ酸とヒドロキシ酸から構成されるデプシペプチド、ヒドロキシ酸が連続して縮合したポリエステル、N-メチルアミノ酸の導入によりアミド結合の窒素がメチル化されたペプチド、N-末端に様々なアシル基(アセチル基、ピログルタミン酸、脂肪酸など)を有するペプチドも、特殊ペプチドに含まれる。また、両端に互いの間で結合形成反応が可能な一組の官能基を配置したアミノ酸配列からなる非環状ペプチドを適切な反応条件下で環化させて得られる環状ペプチド(N-メチルペプチドが導入された場合は、環状N-メチルペプチド)もRAPIDシステムで合成可能である。一組の官能基の種類によっては無細胞翻訳系の条件下において環化することがあり、後述の実施例のように、両端にそれぞれクロロアセチル基とシステインを配置したペプチド配列を翻訳合成して得られるチオエーテル結合で環化した環状ペプチドはその一例である。
RAPIDシステムでは、リボソームによる翻訳で特殊ペプチドを合成するため、鋳型であるmRNAを代えるだけで様々な構造を有するペプチドを合成できる。ランダム配列を含むmRNA(または対応するDNA)を用いて翻訳合成を行えば、ランダムペプチドライブラリーを容易に構築することができる。本発明のRAPIDディスプレイ法は、RAPIDシステムにより合成される特殊ペプチドを、その遺伝子型であるmRNAと対応付けるために使用するのに、好適である。特殊ペプチドの合成用に最適化された無細胞翻訳系に、鋳型cDNAあるいはmRNAと共に、本発明のRAPIDリンカーを加えて、一定時間反応させることで、翻訳産物である特殊ペプチドがリンカーを介してmRNAと連結して提示される。
次に、無細胞翻訳系内で起こる、リンカーとmRNAの結合について説明する。従来技術の項で述べたように、公知のmRNAディスプレイ法では、翻訳反応の前段階に、リンカーとmRNAの連結(ライゲーション)を翻訳系外で行っておくことが必要であった。これに対し、本発明のRAPIDディスプレイ法ではリンカーとmRNAのハイブリダイゼーションによる複合体形成を翻訳系内で行うことができることが特徴である。
図2を参照しながら、mRNAをRAPIDリンカーと共に無細胞翻訳系に供することにより、mRNAとリンカーとのハイブリダイゼーションおよびペプチド分子の合成、そしてペプチドC末端とリンカーとの連結が起こる過程について説明する。
上述したように、mRNAとリンカーとの結合は、リンカーの一本鎖構造領域の塩基配列とmRNAの3’末端の相補的な塩基配列が水素結合で二本鎖を形成することによる。この結合は、mRNAと翻訳されたペプチド分子とを対応付けるためのものであるから、リボソーム上で翻訳が行われている間に形成された〔mRNA〕-〔リンカー〕-〔ペプチド〕複合体が、翻訳産物であるペプチドのセレクションの間も安定に維持される必要がある。
mRNAをRAPIDリンカーと共に無細胞翻訳系に供することにより、リボソームがmRNA上に配置されて翻訳反応が開始し、ペプチド鎖が伸長し、そして、終結したペプチド鎖のC末端が、リンカーの〔rACCA-アミノ酸〕からなるペプチドアクセプター領域のアミノ酸と結合してtRNAから切り離される。本発明の概念を限定する意図ではなく、あくまでも説明のためであるが、mRNAをRAPIDリンカーと同時に無細胞翻訳系に供する場合、mRNAとRAPIDリンカーとのハイブリダイゼーションは、ペプチド鎖の伸長反応開始よりも前の段階で起こり得ると本発明者らは考えている。あるいは、cDNAからの転写および翻訳反応を先に行ってから、終結因子およびPTHを除いた翻訳反応液中にリンカーを後から添加することによっても、リボソーム上のmRNAとRAPIDリンカーとのハイブリダイゼーションによる複合体形成は正しく起こる。そして、リンカー末端の〔rACCA-アミノ酸〕とペプチドC末端との共有結合は、翻訳終了時に偶然にこの部分がリボソームAサイトに入った場合に起こると考えられる。翻訳終了時には既にmRNAとリンカーとのハイブリダイゼーションが起こっているので、この〔rACCA-アミノ酸〕基質はリンカーでリボソーム上のmRNAとつながっており局所的な濃度が非常に高くなっているため、反応は高い効率で起こる。
このようにしてリボソーム上で形成された〔mRNA〕-〔リンカー〕-〔ペプチド〕複合体が、ペプチド鎖がリボソームから解離した後も安定に維持されていることは、後述の実施例においても裏付けられている。
4.ペプチドアプタマーの選択
進化分子工学では、所望の機能や性質を持つタンパク質やペプチドを創製することを目的として、可能性のある遺伝子を大規模に準備し、その中から狙った表現型を有するクローンを選択する。
進化分子工学では、所望の機能や性質を持つタンパク質やペプチドを創製することを目的として、可能性のある遺伝子を大規模に準備し、その中から狙った表現型を有するクローンを選択する。
基本的には、最初にDNA集団を調製し、in vitro転写産物としてRNA集団を得て、in vitro翻訳産物としてペプチド集団を得る。このペプチド集団から、所望の機能や性質を持つものを何らかのスクリーニング系で選択することになる。例えば、特定のタンパク質に結合するペプチド分子を得たい場合は、標的タンパク質を固相化したカラムにペプチド集団を流し込み、カラムに結合したペプチド分子の混合物を回収することができる。このとき、各ペプチド分子には、その鋳型であるmRNAがタグのように付加されているので、回収したペプチド-mRNA複合体の集団から逆転写酵素でDNAに戻し、PCRで増幅して狙った表現型を有するクローンが多く含まれるバイアスのかかったライブラリーを得た後に、再度同じような選択実験を行う。あるいは、RNAアプタマーを回収してしまう可能性を回避するため、核酸部分を2本鎖にする目的で、選択前に逆転写反応を行うことも可能である。この操作を繰返すことで、世代の経過とともに所望の表現型を有するクローンが集団中で濃縮されていく。
ペプチドアプタマーを同定する場合、対応付け分子のライブラリーと標的物質とを混合し、標的物質に結合したペプチドを提示する対応付け分子(活性種)を選択し、選択された対応付け分子の核酸部分からPCRにより核酸ライブラリーを調製する工程を繰り返すことで、標的物質に結合するペプチドアプタマーの遺伝子をクローニングできる。標的物質に結合した対応付け分子を選択する工程は、〔RNA(あるいはDNA/RNAハイブリッド)〕-〔リンカー〕-〔ペプチド〕からなる複合体を標的物質と結合させ、適当な方法で他の複合体から分離して、目的の結合特性を有するペプチドを同定することにより行うことができる。
標的物質としては、タンパク質、核酸、糖質、脂質、その他どのような化合物でもよい。標的物質と結合する活性種である複合体を、他の複合体から分離するために、標的物質に、固相への結合により回収可能な修飾を施しておくと便利である。例えば、後述の実施例では、標的物質をビオチン修飾しておき、固相化されたビオチン結合タンパク質への特異的な結合を利用して回収している。このような特異的な結合としては、ビオチン結合タンパク質(アビジン、ストレプトアビジンなど)/ビオチンの組み合わせの他にも、マルトース結合タンパク質/マルトース、ポリヒスチジンペプチド/金属イオン(ニッケル、コバルトなど)、グルタチオン-S-トランスフェラーゼ/グルタチオン、抗体/抗原(エピトープ)などが、利用可能であるが、これらに限定されるものでは勿論ない。
進化分子工学を用いることで、遺伝子ライブラリーとして、A、T、G、Cの4つの塩基がランダムに結合したDNA配列から、天然には全く存在しないアミノ酸配列を有するペプチドを得ることが原理的に可能である。また、翻訳系に非タンパク質性アミノ酸(またはヒドロキシ酸)でアシル化されたtRNAを導入することにより、in vitro翻訳産物として非タンパク質性アミノ酸(またはヒドロキシ酸)が導入された特殊ペプチドの翻訳による合成も可能である。特殊ペプチドとmRNA(あるいはcDNA)の複合体の集団から、所望の結合特性を有するペプチドが提示された活性種を選択し、対応付けされた遺伝子部分を増幅、再び翻訳するという作業を繰返すことで、特殊ペプチドのライブラリーを効率的に得ることができる。
これまでの説明および後述の実施例の内容に関し、分子生物学的手法の詳細は、例えば、Sambrook, Molecular Cloning: A Laboratory Manual, 3rd edition, Cold Spring Harbor Laboratory Press, 2001; Golemis, Protein-Protein Interactions: A Molecular Cloning Manual, 2nd edition, Cold Spring Laboratory Press, 2005などを参照されたい。
以下、本発明を実施例によって具体的に説明する。なお、これらの実施例は、本発明を説明するためのものであって、本発明の範囲を限定するものではない。
〔リンカーの合成〕
リンカーとしてDNAとRNA間をポリエチレングリコール(5ユニットのSpacer18)で連結したDNA/RNAキメラオリゴヌクレオチドを使用した。種々リンカーはBEX社(東京)より購入した。以下に示す配列の*Aおよび*CはRNA、SPC18はSpacer18(ヘキサエチレングリコール)に相当する。
リンカーとしてDNAとRNA間をポリエチレングリコール(5ユニットのSpacer18)で連結したDNA/RNAキメラオリゴヌクレオチドを使用した。種々リンカーはBEX社(東京)より購入した。以下に示す配列の*Aおよび*CはRNA、SPC18はSpacer18(ヘキサエチレングリコール)に相当する。
an21-ACCA: 5'-CCCGCCTCCCGCCCCCCGTCC-[SPC18] 5-A*-C*-C*-A*-3'
〔リンカーのアミノアシル化〕
フレキシザイムが触媒する反応により、an21-ACCAリンカー分子の3’末端側にエステル結合を介してL-フェニルアラニンもしくはβ-L-アラニンを結合させた。反応産物は精製した反応産物溶液を酸性条件下でのアクリルアミド電気泳動分析により確認した。リンカー分子に由来するバンドがアミノアシル化されると移動度が少なくなる。そのため、未反応物由来のバンドと反応産物由来のバンドの強度を比較することでアミノアシル化効率を決定することができる。
〔リンカーのアミノアシル化〕
フレキシザイムが触媒する反応により、an21-ACCAリンカー分子の3’末端側にエステル結合を介してL-フェニルアラニンもしくはβ-L-アラニンを結合させた。反応産物は精製した反応産物溶液を酸性条件下でのアクリルアミド電気泳動分析により確認した。リンカー分子に由来するバンドがアミノアシル化されると移動度が少なくなる。そのため、未反応物由来のバンドと反応産物由来のバンドの強度を比較することでアミノアシル化効率を決定することができる。
アシル化反応は、L-フェニルアラニンを結合させる場合は、0.1 M HEPES-カリウムバッファー(pH 7.5)、600 mM 塩化マグネシウム中で、20 μMのフレキシザイムeFx 5 μL、20 μMのan21-ACCAリンカー、および基質(L- フェニルアラニンシアノメチルエステル)を20 %ジメチルスルホキシドに加えて、氷上で2時間反応させた。詳細な手順としては、まず、純水に溶解した40 μMのリンカー分子を0.2 M HEPES-カリウムバッファー(pH 7.5)およびフレキシザイムeFx(200 μM, 0.5 μL)を加え、サーモブロック(ND-MD1, 日伸理化)上で95 ℃で2分間加熱し、室温で5分間静置した。その後、氷上で塩化マグネシウム(3 M, 1 μL) および基質(ジメチルスルホキシド中25 mM, 1 μL)を加えることによりリンカー分子のアシル化反応を開始し、氷上で2時間静置した。β-L-アラニンを結合させる場合は、同様の条件にて、pHを8.0とし、基質としてβ-L-アラニンp-クロロベンジルチオエーテルを用いて行った。反応は0.3 M 酢酸ナトリウム(pH 5.0)を40 μL加えることにより停止した。反応産物をエタノール沈殿し、70% エタノールで洗浄したペレットを10 μLの1 mM酢酸ナトリウムに溶解した。反応後の溶液を酸性条件下、20 %の変性ポリアクリルアミド電気泳動(50 mM 酢酸ナトリウム(pH 5.0)、6 M 尿素)で分離し、泳動後のゲルをSYBR Green II (Invitrogen、SYBRはMolecular Probes Inc.の登録商標)を用いた蛍光染色により解析した。
結果は図3で示したようになり、対照となる未反応のリンカー分子よりも、反応産物のリンカー分子の移動度が小さいことが確認され、リンカー分子がフレキシザイムの触媒する反応によりアミノアシル化されていることが確認された。
〔cDNAの合成〕
ペプチド-mRNA複合体を形成するために用いるcDNAは、合成オリゴヌクレオチドをPCRにより連結することにより調製した。以下に配列を示す合成DNAはオペロン社(東京)より購入した。これらのオリゴDNAを連結することにより得られる各々のcDNAは、T7プロモーター配列、リボソーム結合配列、開始コドン、ペプチドアプタマー配列、スペーサーペプチド配列(CGSGSGS)、アンバーコドンおよびリンカーハイブリダイゼーション配列より構成される。
TNF-a_D-Trp.R66:
GCCGCTGCCGCTGCCGCAATGCTTCAGATACAGACAATGCAGACGTTGCATATGTATATCTCCTTC
EMP1SS.F63: GAAGGAGATATACATATGGCAGCAGGTGGTACCTATTCTTCTCATTTTGGTCCGCTGACCTGG
EMP1SS.R63: GCCGCTGCCGCTGCCGCATGCTGCACCACCTTGCGGCTTAGAAACCCAGGTCAGCGGACCAAA
T7g10M.F48: TAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG
CGS3an13.R39: TTTCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA
CGS3an21.R44: CCCGCCTCCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA
PCRによる連結は以下の手順にて行った。
〔cDNAの合成〕
ペプチド-mRNA複合体を形成するために用いるcDNAは、合成オリゴヌクレオチドをPCRにより連結することにより調製した。以下に配列を示す合成DNAはオペロン社(東京)より購入した。これらのオリゴDNAを連結することにより得られる各々のcDNAは、T7プロモーター配列、リボソーム結合配列、開始コドン、ペプチドアプタマー配列、スペーサーペプチド配列(CGSGSGS)、アンバーコドンおよびリンカーハイブリダイゼーション配列より構成される。
TNF-a_D-Trp.R66:
GCCGCTGCCGCTGCCGCAATGCTTCAGATACAGACAATGCAGACGTTGCATATGTATATCTCCTTC
EMP1SS.F63: GAAGGAGATATACATATGGCAGCAGGTGGTACCTATTCTTCTCATTTTGGTCCGCTGACCTGG
EMP1SS.R63: GCCGCTGCCGCTGCCGCATGCTGCACCACCTTGCGGCTTAGAAACCCAGGTCAGCGGACCAAA
T7g10M.F48: TAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATG
CGS3an13.R39: TTTCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA
CGS3an21.R44: CCCGCCTCCCGCCCCCCGTCCTAGCTGCCGCTGCCGCTGCCGCA
PCRによる連結は以下の手順にて行った。
T7g10M.F48およびTNF-a_D-Trp.R66(もしくはEMP1SS.F63とEMP1SS.R63の組み合わせ)を250 nM含むPCR反応液(10 mM Tris-HCl (pH 9.0), 50 mM 塩化カリウム, 2.5 mM 塩化マグネシウム, 250 μM dNTPs, 0.2 % Triton X-100(トリトン X-100、ナカライテスク)およびTaq ポリメラーゼ) 20 uLを調製し、サーマルサイクラー(TC-3000, Techne社)を用いて94 ℃ 1 min, {50 ℃ 30 sec, 72 ℃ 30 sec}5サイクルによる反応を行った。次に各々500 nM のプライマー、T7g10M.F48およびGCSan21.R44(もしくはGCSan13.R39)を含むPCR反応液100 μLを調製し、これに先のPCR反応後液1 μLを加え、温度サイクル{94 ℃ 40 sec, 50 ℃ 40 sec, 72 ℃ 40 sec}10サイクルで反応を行った。反応産物は常法に従いフェノール・クロロホルム抽出、クロロホルム抽出、さらにエタノール沈殿により精製を行った。
〔mRNAの調製〕
mRNAは前述の方法で作成したcDNA(TNF-α-DW(an21))を元にT7 RNA ポリメラーゼによる転写反応によりRNAを合成し、得られた反応産物は常法に従いフェノール・クロロホルム抽出、クロロホルム抽出、さらに2-プロパノール沈殿により精製を行った。精製後のmRNAの濃度を260 nmにおけるUVの吸収から求め、10 μMになるように希釈した。
〔転写・翻訳、リンカーのハイブリダイゼーション、ペプチドアクセプターとの連結〕
mRNAからの翻訳およびリンカー分子との複合体形成もしくはcDNAからの転写、翻訳およびリンカー分子との複合体形成は再構成型の無細胞翻訳系を用いて行った。
〔mRNAの調製〕
mRNAは前述の方法で作成したcDNA(TNF-α-DW(an21))を元にT7 RNA ポリメラーゼによる転写反応によりRNAを合成し、得られた反応産物は常法に従いフェノール・クロロホルム抽出、クロロホルム抽出、さらに2-プロパノール沈殿により精製を行った。精製後のmRNAの濃度を260 nmにおけるUVの吸収から求め、10 μMになるように希釈した。
〔転写・翻訳、リンカーのハイブリダイゼーション、ペプチドアクセプターとの連結〕
mRNAからの翻訳およびリンカー分子との複合体形成もしくはcDNAからの転写、翻訳およびリンカー分子との複合体形成は再構成型の無細胞翻訳系を用いて行った。
本実施例で使用した再構成型の無細胞翻訳系には以下の生体高分子が含まれる:
70Sリボソーム(1.2uM)、翻訳開始因子(IF1(0.7uM), IF2(0.4uM), IF3(1.5uM))、
伸長因子(EF-G(0.26uM), EF-Tu/EF-Ts複合体(10uM))、翻訳終止因子(RF2(0.25uM), RF3(0.17uM))メチオニルtRNAトランスフォルミラーゼ(MTF(0.6uM))、リボソーム再生因子(RRF(0.5uM))、アミノアシルtRNA合成酵素(AlaRS(0.73uM), ArgRS(0.03uM), AsnRS(0.38uM), CysRS(0.02uM), GlnRS(0.06uM), GluRS(0.23uM), GlyRS(0.09uM), HisRS(0.02uM), IleRS(0.4uM), LeuRS(0.04uM), MetRS(0.03uM), PheRS(0.68uM), ProRS(0.16uM), SerRS(0.04uM), ThrRS(0.09uM), TrpRS(0.03uM), ValRS(0.02uM), AspRS(0.13uM), LysRS(0.11uM), TyrRS(0.02uM))、クレアチンキナーゼ(ロシュ社製CK(4ug/mL)、ミオキナーゼ(ロシュ社製、MK(3ug/mL))、ピロフォスファターゼ(PPa(0.1uM))、
ヌクレオチド二リン酸キナーゼ(NDK(0.1uM))、T7 RNAポリメラーゼ(T7ファージ遺伝子由来の組み換え体、0.1uM)、大腸菌tRNA(ロシュ社製、1.5mg/mL)。リボソームは対数増殖期の大腸菌から精製したもの、リボソーム以外の各種タンパク質は、特に指示がある場合を除き、大腸菌の遺伝子をクローニングし、それを元に発現、精製した組み換えタンパク質である。
70Sリボソーム(1.2uM)、翻訳開始因子(IF1(0.7uM), IF2(0.4uM), IF3(1.5uM))、
伸長因子(EF-G(0.26uM), EF-Tu/EF-Ts複合体(10uM))、翻訳終止因子(RF2(0.25uM), RF3(0.17uM))メチオニルtRNAトランスフォルミラーゼ(MTF(0.6uM))、リボソーム再生因子(RRF(0.5uM))、アミノアシルtRNA合成酵素(AlaRS(0.73uM), ArgRS(0.03uM), AsnRS(0.38uM), CysRS(0.02uM), GlnRS(0.06uM), GluRS(0.23uM), GlyRS(0.09uM), HisRS(0.02uM), IleRS(0.4uM), LeuRS(0.04uM), MetRS(0.03uM), PheRS(0.68uM), ProRS(0.16uM), SerRS(0.04uM), ThrRS(0.09uM), TrpRS(0.03uM), ValRS(0.02uM), AspRS(0.13uM), LysRS(0.11uM), TyrRS(0.02uM))、クレアチンキナーゼ(ロシュ社製CK(4ug/mL)、ミオキナーゼ(ロシュ社製、MK(3ug/mL))、ピロフォスファターゼ(PPa(0.1uM))、
ヌクレオチド二リン酸キナーゼ(NDK(0.1uM))、T7 RNAポリメラーゼ(T7ファージ遺伝子由来の組み換え体、0.1uM)、大腸菌tRNA(ロシュ社製、1.5mg/mL)。リボソームは対数増殖期の大腸菌から精製したもの、リボソーム以外の各種タンパク質は、特に指示がある場合を除き、大腸菌の遺伝子をクローニングし、それを元に発現、精製した組み換えタンパク質である。
また、生体高分子以外の成分として、以下の物が含まれる:
50mM HEPES-KOH (pH7.6), 2mM NTPs, 20mM リン酸クレアチン, 100mM 酢酸カリウム, 2mM スペルミジン, 1mM ジチオスレイトール, 6mM 酢酸マグネシウム, 0.1mM 10-ホルミル-5, 6, 7, 8-テトラヒドロ葉酸。
50mM HEPES-KOH (pH7.6), 2mM NTPs, 20mM リン酸クレアチン, 100mM 酢酸カリウム, 2mM スペルミジン, 1mM ジチオスレイトール, 6mM 酢酸マグネシウム, 0.1mM 10-ホルミル-5, 6, 7, 8-テトラヒドロ葉酸。
これらに加えて、mRNA 1.5 μMもしくはcDNA 0.15 μM、アシル基としてN-クロロアセチル-D-トリプトファン(ClAc-D-Trp)を担持したアミノアシルイニシエーションtRNA(特開2008-125396で開示の方法により調製)、メチオニンを除く天然タンパク質を構成する19種類のアミノ酸(各々5 mM)を含む転写・翻訳反応溶液2.5 μLを調製した。
あらかじめ転写反応を行いmRNAを調製し、これを元に翻訳およびペプチドとリンカーとの結合を行う場合は、以下の手順によりペプチド-mRNA複合体を調製した。まず、4 mM酢酸ナトリウム pH 5.0とmRNAを1:3で混合し、これをサーモブロック上で95 ℃で1分間加熱したのちに室温下で5分静置した。1 mM酢酸ナトリウムに溶解した25 μM のリンカー溶液を混合し10分間静置することでmRNA-リンカーの複合体を形成させた。これに翻訳系の他のコンポーネントを加え、恒温水槽(NT-202D, 日伸理化)中で37 ℃、30分間、続いて室温で12分インキュベートし、pH 7.5に調製した0.1 M EDTA(エチレンジアミン4酢酸、分子生物学実験用、ナカライテスク)を終濃度が20 mMになるように添加し、さらに恒温水槽中で37 ℃で30分間インキュベートした。
反応溶液中にcDNAを加え、この反応溶液中で転写、翻訳およびリンカーとの結合を行う場合は、以下の手順によりペプチド-mRNA複合体を調製した。まず始めにリンカー以外の物を含む溶液を調製し、これを恒温水槽中で37 ℃で30分間反応させることにより、転写および翻訳反応を行った。これに25 μMのリンカー溶液0.25 μLを加え、さらに37 ℃で30分、続いて室温で12分インキュベートした。その後、pH 7.5に調製した0.1 M EDTA(エチレンジアミン4酢酸、分子生物学実験用、ナカライテスク)を終濃度が20 mMになるように添加し、さらに恒温水槽中で37 ℃ で30分間インキュベートした。
〔逆転写〕
ペプチド-mRNA複合体のmRNA部分の安定性を向上させるため、逆転写反応を行うことにより、RNA-DNAハイブリッド鎖を形成させた。具体的には以下の操作を行った。前述の翻訳反応産物に、12 mM Tris-HCl (pH 8.3), 5 μM 逆転写プライマー(CGS3an13.R21), 0.5 mM dNTPs, 18 mM Mg(OAc)2, 10 mM KOH(各々の濃度は終濃度)を添加した。これにM-MLV Reverse Transcriptase (RNaseH Minus, Point Mutant, Promega) 1 unitを添加し恒温水槽中で42 ℃で10分間反応させた。その後、終濃度が10 mMおよび18 mMになるようにEDTAと塩酸を添加した。
〔セレクション〕
このようにして作成したペプチド-mRNA複合体分子が、提示ペプチドとターゲットとなるタンパク質との相互作用により回収されるかどうかを以下に示す方法により確認した。
〔逆転写〕
ペプチド-mRNA複合体のmRNA部分の安定性を向上させるため、逆転写反応を行うことにより、RNA-DNAハイブリッド鎖を形成させた。具体的には以下の操作を行った。前述の翻訳反応産物に、12 mM Tris-HCl (pH 8.3), 5 μM 逆転写プライマー(CGS3an13.R21), 0.5 mM dNTPs, 18 mM Mg(OAc)2, 10 mM KOH(各々の濃度は終濃度)を添加した。これにM-MLV Reverse Transcriptase (RNaseH Minus, Point Mutant, Promega) 1 unitを添加し恒温水槽中で42 ℃で10分間反応させた。その後、終濃度が10 mMおよび18 mMになるようにEDTAと塩酸を添加した。
〔セレクション〕
このようにして作成したペプチド-mRNA複合体分子が、提示ペプチドとターゲットとなるタンパク質との相互作用により回収されるかどうかを以下に示す方法により確認した。
ターゲットとなるタンパク質として、ヒトの腫瘍壊死因子アルファ(以下、TNF-α)を選択した。
ペプチド-mRNA複合体分子を含む無細胞翻訳反応産物液6 μLに対し、ビオチン修飾されたTNF-αタンパク質を終濃度250 nMになるように添加し、この溶液を低吸着処理を施した0.6 mLエッペンドルフチューブ(プラチナ(超低吸着)チューブ、ビーエム機器)に移し、ローテーター(RT-30 mini、タイテック)の上で4 ℃、1 hr 穏やかに攪拌し結合させた。この溶液に対して、ストレプトアビジンを固相化した磁性ビーズ(Dynabeads 登録商標Streptavidin M-280, Invitrogen社)の懸濁液3 μLを加え、さらに10分攪拌した。その後、遠心分離およびマグネットホルダーを使用して磁性ビーズを分離し上清を除き、再度0.05 % のTween20(ポリオキシエチレンソルビタンモノラウレート、ナカライテスク)を含むTBS (Tris Buffered Saline, 50 mM Tris-HCl(トリスヒドロキシメチルアミノメタン、ナカライテスク) pH 7.5, 150 mM 塩化ナトリウム) 50 μL に懸濁した。この洗浄操作を4回繰り返した。
続いて磁性ビーズにPCR(taq-)バッファー(10 mM Tris-HCl (pH 9.0), 50 mM 塩化カリウム, 2.5 mM 塩化マグネシウム, 250 μM dNTPs, 0.25 μM T7g10m.F48, 0.25 μM CGS3an13.R21, 0.2 % Triton X-100)を25 μL加え、サーモブロック上で95 ℃で5分間加熱後に上清を回収することにより、磁性ビーズより逆転写後のDNAを回収した。
〔リアルタイムPCRによる定量〕
ターゲットタンパク質添加前、および磁性ビーズより回収後のDNAの量をリアルタイムPCR法により定量した。リアルタイムPCR測定装置はLightCycler(登録商標)1.5 (Roche Applied Science社)を使用し、前述のPCR(taq-)バッファーにTaqポリメラーゼ、SYBR(登録商標)Green I (100,000希釈、Invitrogen社)、および測定サンプル溶液を添加した反応液を測定に供した。
TNF-αを標的としたモデル実験
リンカーの機能を確認するために用いたターゲットとなるタンパク質およびペプチドアプタマーとして、TNF-αおよびTNF-αに結合するペプチドアプタマーであるTNF-α-DWを選択した。
〔リアルタイムPCRによる定量〕
ターゲットタンパク質添加前、および磁性ビーズより回収後のDNAの量をリアルタイムPCR法により定量した。リアルタイムPCR測定装置はLightCycler(登録商標)1.5 (Roche Applied Science社)を使用し、前述のPCR(taq-)バッファーにTaqポリメラーゼ、SYBR(登録商標)Green I (100,000希釈、Invitrogen社)、および測定サンプル溶液を添加した反応液を測定に供した。
TNF-αを標的としたモデル実験
リンカーの機能を確認するために用いたターゲットとなるタンパク質およびペプチドアプタマーとして、TNF-αおよびTNF-αに結合するペプチドアプタマーであるTNF-α-DWを選択した。
本実施例で用いたTNF-αタンパク質として大腸菌により発現した組換え体である可溶型TNF-αを使用した。組換えTNF-αは野生型TNF-αの77番目から233番目のアミノ酸配列の部分、そのN末端側にAviTag配列(GLNDIFEAQKIEWHE)およびHis×6 tag配列を融合したものである。このタンパク質とAviTagを基質とするビオチンリガーゼ(BirA)を大腸菌内で共発現させることによりAviTagのリジン残基の側鎖がビオチン化修飾されるため、本実施例で用いているTNF-αはストレプトアビジン固相化ビーズにより容易に分離することができる。
ペプチドアプタマーとして用いたTNF-α-DWは、非タンパク質性アミノ酸であるN-クロロアセチル-D-トリプトファン(ClAc-D-Trp)のクロロアセチル基とペプチド配列中のシステインの側鎖のチオール基が反応し、チオエーテルを形成することにより環化したペプチドXQRLHCLYLKH(X: Ac-D-Trp)である。このペプチドのC末端側にスペーサーアミノ酸配列CGSGSGSを融合した配列をコードするcDNAを前述の方法にて作成し、無細胞転写・翻訳系にてペプチドとmRNA複合体分子を作成した。
このペプチドアプタマーと標的タンパク質の組み合わせを用いた場合のペプチド-mRNA複合体の回収率は図4および図5に示すとおりであった。L-フェニルアラニンを3’末端にエステルを介して結合させたan21- Phe、an21-β-Alaの回収率は翻訳反応液にmRNAを添加した場合(図4)は各々1.30 %, 0.25 %、cDNAを添加した場合(図5)は0.65 %, 0.38 %であった。一方、3’末端が修飾されていないan21-ACCAリンカーでの回収率はそれらのものと比較して数十倍程度低く(mRNAの場合0.015%, cDNAの場合0.007 %)、L-フェニルアラニン、β-L-アラニンが翻訳合成されたペプチドのC末端側に共有結合を介して連結することにより、ペプチド-mRNA複合体分子として効率よく回収されることが示された。
提示ペプチドとmRNAとの対応付けの確認
前述までの実験に於いては、提示しているペプチドと、その配列をコードしているmRNAは単一の種類であるため、mRNAから翻訳合成されたペプチドに対して、別のリンカー分子がアクセプターとして結合している可能性や、2本鎖を形成しているリンカー分子とmRNA分子が操作の途中で別のリンカー分子やmRNAと交換している可能性を排除できない。そのため、回収されるペプチド-mRNA-リンカー複合分子の提示ペプチドとmRNAとの対応付けが正確に行われていることを確認するために以下に示す実験を行った。
前述までの実験に於いては、提示しているペプチドと、その配列をコードしているmRNAは単一の種類であるため、mRNAから翻訳合成されたペプチドに対して、別のリンカー分子がアクセプターとして結合している可能性や、2本鎖を形成しているリンカー分子とmRNA分子が操作の途中で別のリンカー分子やmRNAと交換している可能性を排除できない。そのため、回収されるペプチド-mRNA-リンカー複合分子の提示ペプチドとmRNAとの対応付けが正確に行われていることを確認するために以下に示す実験を行った。
まず、TNF-α-DWの対照としてターゲットとするタンパク質に結合しないペプチド配列(EMP1SS:XAAGGTYSSHFGPLTWVSKPQGGAA、XはTNF-α-DWと同様にAc-D-Trpである)をコードするmRNAもしくはcDNAを実施例1に示す方法で調製し、これとTNF-α-DWをコードするmRNAもしくはcDNAをモル比100:1で混合したものを用意した。これを元に前述と同様の実験を行い、回収したDNAをPCRにて増幅した。その際の条件として、PCR(taq-)バッファーにTaqポリメラーゼおよび測定サンプル溶液を添加した反応液20 μLを用意し、サーマルサイクラーを用いて、温度サイクル{94 ℃ 40 sec, 61 ℃ 40 sec, 72 ℃ 40 sec}(61 ℃から72 ℃への昇温は0.5 ℃/sec)を30サイクル行い反応させた。反応産物液2.5 μLをTAE(40 mM Tris酢酸、1 mM EDTA)および3 % アガロース(低電気浸透度、ナカライテスク)からなるゲルを用いた電気泳動により分離し、エチジウムブロミドによる染色およびトランスイルミネーターを用いて泳動像を得た。その結果を図6に示す。
TNF-α-DWをコードするmRNAに由来するバンドは約90 bpの位置に確認され、対照とするEMP1SSをコードするmRNAに由来するバンドは約150 bpの位置に観察される。ペプチドアクセプターを含まないan21-ACCAリンカーを用いた場合は、はじめに2種類のcDNAを混合した際のモル比を反映しEMP1SSのmRNAに由来するバンドのみが確認された。それに対して、an21-Pheにおいて、TNF-α-DWのmRNAに由来するバンドが確認され、その蛍光強度はEMP1SSと同程度もしくはそれ以上であった。したがって、ターゲットタンパク質に結合するTNF-α-DWを提示したペプチド-mRNA複合体が、ターゲットに結合しないEMP1SSを提示したペプチド-mRNA複合体よりも効率よく回収されていることが確認された。このことは、ペプチド-mRNA-リンカー複合分子の提示ペプチドとmRNAとの対応付けが実験の途中で変化しないことを示している。また、この結果は翻訳反応液中にmRNAを添加してペプチド-mRNA複合体を形成させた場合、およびcDNAを添加してペプチド-mRNA複合体を形成させた場合ともに同様の結果が得えられた。
TNF-α-DWスパイクドライブラリーのセレクション
〔ランダムペプチドライブラリー〕
本手法によりランダムペプチド配列をコードするmRNAライブラリー中に含まれる低コピー数のペプチドアプタマーをコードするmRNAもしくはcDNAを、複数回のセレクションを繰り返すことにより選別することが可能かどうかを検討した。
〔ランダムペプチドライブラリー〕
本手法によりランダムペプチド配列をコードするmRNAライブラリー中に含まれる低コピー数のペプチドアプタマーをコードするmRNAもしくはcDNAを、複数回のセレクションを繰り返すことにより選別することが可能かどうかを検討した。
始めに、8-12アミノ酸のランダム配列を提示するペプチドアプタマーライブラリを調製した。このライブラリーはランダム配列をコードするmRNAとして調製したものである。このライブラリーの構造は前述のTNF-α-DWやEMP1SSの提示配列の部分がランダム化された物であり、それ以外の部分は同様の構造を有しているが、リンカーがハイブリダイズし2本鎖を形成する領域の長さは13塩基となっている。
〔TNF-α-DWスパイクドライブラリーの調製〕
このランダムペプチドライブラリーに対して、TNF-α-DWをコードするmRNA(TNF-α-DW(an13): リンカーとの2本鎖形成部の長さが13塩基)をモル比で1,000,000 : 1となるように混合したスパイクドライブラリーを用意した。
〔ラウンド1〕
このスパイクドライブラリーmRNAおよびan21-Pheリンカーを用いて、前述の実施例1および2に示した手順に従い翻訳とペプチド?mRNA複合体の形成を行った(ラウンド1)。
〔TNF-α-DWスパイクドライブラリーの調製〕
このランダムペプチドライブラリーに対して、TNF-α-DWをコードするmRNA(TNF-α-DW(an13): リンカーとの2本鎖形成部の長さが13塩基)をモル比で1,000,000 : 1となるように混合したスパイクドライブラリーを用意した。
〔ラウンド1〕
このスパイクドライブラリーmRNAおよびan21-Pheリンカーを用いて、前述の実施例1および2に示した手順に従い翻訳とペプチド?mRNA複合体の形成を行った(ラウンド1)。
セレクションの際には、ネガティブセレクションとしてペプチド-mRNA複合体溶液を何も結合させていないストレプトアビジン固相化磁性ビーズと4 ℃,10分間混合することにより、磁性ビーズに結合するペプチド-mRNA複合体を除いた。この溶液にTNF-αを終濃度250 nMになるように添加し、4 ℃ で1時間混和した。これにストレプトアビジン固相化磁性ビーズを加え、4 ℃, 10 分で結合させた後にTBSTで4回洗浄した。
次に、洗浄後の磁性ビーズに逆転写反応液(5 units / μL M-MLV Reverse Transcriptase (Promega), 2 μM CGS3an21.R44, 0.5 mM dNTP, 10 mM Tris-HCl (pH 8.3), 15 mM 塩化カリウム, 0.6 mM 塩化マグネシウム, 2 mM ジチオスレイトール)10 μLに懸濁し、42 ℃で1 hr加温し逆転写反応を行った。続いて磁性ビーズ懸濁液にPCR(taq-)バッファー(プライマーとして0.25 μM T7g10m.F48および 0.25 μM CGS3an21.R44を含む)を15 μL加え、これをサーモブロック上で95 ℃で5分間加熱後に上清を回収することにより、磁性ビーズより逆転写後のDNAを回収した。
翻訳直後のmRNAおよび回収したDNAの一部を取り、そのコピー数を実施例1と同様にリアルタイムPCRを用いて定量した。また、実施例2と同様にPCRによる増幅を行い、PCRによる増幅の際には、反応産物同士の再アニーリングを避けるため、増幅反応が飽和する前で終了した。
〔ラウンド2、3および4〕
ラウンド2以降に於いては、前ラウンドのPCR産物から合成したmRNAを翻訳反応に用いた場合と、前ラウンドのPCR産物をcDNAとして転写・翻訳反応を同時に行った場合とを比較した。実験の手順は実施例1および2と同様に行った。その際のネガティブセレクションの操作を複数回行った。具体的にはラウンド毎に1回ずつネガティブセレクションの回数を増やしている。ラウンド2以降もPCRは増幅反応が飽和する前に増幅を終了した。
〔選択された配列の確認〕
ラウンド2の操作を複数回繰り返した結果、ラウンド4において結合率の大幅な増大を認めた。ラウンド毎の結合率の変化を図7に示した。PCR産物をpGEM-T easy Vector System I (Promega社)を用い常法に従いクローニングし、各々5クローンをDNAシーケンシングにより解析した。その結果、得られたクローンは全てTNF-α-DW(an21)と同一の配列であった。
〔ラウンド2、3および4〕
ラウンド2以降に於いては、前ラウンドのPCR産物から合成したmRNAを翻訳反応に用いた場合と、前ラウンドのPCR産物をcDNAとして転写・翻訳反応を同時に行った場合とを比較した。実験の手順は実施例1および2と同様に行った。その際のネガティブセレクションの操作を複数回行った。具体的にはラウンド毎に1回ずつネガティブセレクションの回数を増やしている。ラウンド2以降もPCRは増幅反応が飽和する前に増幅を終了した。
〔選択された配列の確認〕
ラウンド2の操作を複数回繰り返した結果、ラウンド4において結合率の大幅な増大を認めた。ラウンド毎の結合率の変化を図7に示した。PCR産物をpGEM-T easy Vector System I (Promega社)を用い常法に従いクローニングし、各々5クローンをDNAシーケンシングにより解析した。その結果、得られたクローンは全てTNF-α-DW(an21)と同一の配列であった。
本手法によってランダム配列からなるペプチドライブラリーよりペプチドアプタマーを選別しうることが示された。
配列番号1 合成オリゴヌクレオチド(synthetic oligonucleotide)
配列番号2 合成オリゴヌクレオチド
配列番号3 フレキシザイムFx (Flexizyme Fx)
配列番号4 フレキシザイムeFX (Flexizyme eFx)
配列番号5 フレキシザイムdFx (Flexizyme dFx)
配列番号6 合成オリゴヌクレオチド
配列番号7 合成オリゴヌクレオチド
配列番号8 合成オリゴヌクレオチド TNF-a_D-Trp.R66
配列番号9 合成オリゴヌクレオチド EMP1SS.F63
配列番号10 合成オリゴヌクレオチド EMP1SS.R63
配列番号11 合成オリゴヌクレオチド T7g10M.F48
配列番号12 合成オリゴヌクレオチド CGS3an13.R39
配列番号13 合成オリゴヌクレオチド CGS3an21.R44
配列番号14 合成オリゴヌクレオチド CGSan13.R21
配列番号15 TNF-alpha-DW(an21)
配列番号16 TNF-alpha-DW(an13)
配列番号17 EMP1SS (an21)
配列番号18 TNF-alpha
配列番号19 フレキシザイムaFx (Flexizyme aFx)
(注)配列番号15~17のDNAでは、メチオニンのコドンが非タンパク質性アミノ酸(ClAc-D-Trp)に割り当てられている(遺伝暗号のリプログラミング)。
配列番号2 合成オリゴヌクレオチド
配列番号3 フレキシザイムFx (Flexizyme Fx)
配列番号4 フレキシザイムeFX (Flexizyme eFx)
配列番号5 フレキシザイムdFx (Flexizyme dFx)
配列番号6 合成オリゴヌクレオチド
配列番号7 合成オリゴヌクレオチド
配列番号8 合成オリゴヌクレオチド TNF-a_D-Trp.R66
配列番号9 合成オリゴヌクレオチド EMP1SS.F63
配列番号10 合成オリゴヌクレオチド EMP1SS.R63
配列番号11 合成オリゴヌクレオチド T7g10M.F48
配列番号12 合成オリゴヌクレオチド CGS3an13.R39
配列番号13 合成オリゴヌクレオチド CGS3an21.R44
配列番号14 合成オリゴヌクレオチド CGSan13.R21
配列番号15 TNF-alpha-DW(an21)
配列番号16 TNF-alpha-DW(an13)
配列番号17 EMP1SS (an21)
配列番号18 TNF-alpha
配列番号19 フレキシザイムaFx (Flexizyme aFx)
(注)配列番号15~17のDNAでは、メチオニンのコドンが非タンパク質性アミノ酸(ClAc-D-Trp)に割り当てられている(遺伝暗号のリプログラミング)。
Claims (13)
- 再構成型のin vitroタンパク質合成系において、mRNAと、その翻訳産物であるペプチドとが、リンカーを介して結合した連結体を作製するために使用されるリンカーであって、
リンカーの一方の端に、mRNAの3'末端側の塩基と対合する側鎖塩基を有する一本鎖構造領域を含み、
リンカーのもう一方の端に、ペプチド転移反応によって翻訳産物と結合可能な基を含むペプチドアクセプター領域を含み、
ペプチドアクセプター領域は、塩基配列ACCAからなるオリゴRNAにアミノ酸がエステル結合した構造からなり、
当該エステル結合が、人工RNA触媒を用いるアミノアシル化反応により形成された
ことを特徴とするリンカー。 - 一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されている、請求項1に記載のリンカー。
- 一本鎖構造領域が一本鎖DNAからなる、請求項1または2に記載のリンカー。
- アミノアシル化反応に使用される人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、請求項1~3のいずれかに記載のリンカー。 - mRNAと、その翻訳産物であるペプチドが、請求項1~4のいずれかに記載のリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
請求項1~4のいずれかに記載のリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAとを接触させるとともに、mRNAからペプチドの翻訳を行う工程
を含む、前記方法。 - mRNAと、その翻訳産物であるペプチドが、請求項1~4のいずれかに記載のリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体を作製する方法であって、
請求項1~4のいずれかに記載のリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ペプチドをコードする配列の下流に有するmRNAの鋳型となるDNAを合成する工程、
再構成型のin vitroタンパク質合成反応液中にリンカーとDNAを供することにより、DNAからmRNAへの転写およびペプチドへの翻訳ならびにリンカーとmRNAとの複合体形成を行う工程
を含む、前記方法。 - 再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、請求項5または6に記載の方法。
- 請求項7に記載の方法で作製された、〔mRNA〕-〔リンカー〕-〔特殊ペプチド〕連結体からなるライブラリー。
- mRNAと、その翻訳産物であるペプチドが、請求項1~4のいずれかに記載のリンカーを介して結合した〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体ライブラリーから、標的物質に結合するペプチドアプタマーを選択する方法であって、
請求項1~4のいずれかに記載のリンカーを調製する工程、
リンカーの一本鎖構造領域の塩基配列にハイブリダイズできる配列を、ランダムペプチド配列をコードする配列の下流に有するmRNAライブラリーを作製する工程、
再構成型のin vitroタンパク質合成反応液中で、リンカーとmRNAライブラリーとを接触させるとともに、ペプチドの翻訳を行うことにより、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを作製する工程
標的物質と、〔mRNA〕-〔リンカー〕-〔ペプチド〕連結体のライブラリーを接触させる工程
標的物質に結合したペプチドを提示する連結体を選択する工程
を含む、前記方法。 - 標的物質がビオチン化修飾されている、請求項9に記載の方法。
- 再構成型のin vitroタンパク質合成反応液中に、非タンパク質性アミノ酸またはヒドロキシ酸でチャージされたtRNAを含むことにより、翻訳産物であるペプチドが特殊ペプチドとなる、請求項9または10に記載の方法。
- 請求項2に記載のリンカーを作製する方法であって、
一本鎖構造領域と、配列ACCAからなるオリゴRNAとの間を、ポリエチレングリコールで連結したキメラオリゴヌクレオチドを合成し、
キメラオリゴヌクレオチドの3’末端のアデノシンに、人工RNA触媒を用いる反応により、アミノ酸をエステル結合させることにより、
一本鎖構造領域とペプチドアクセプター領域の間がポリエチレングリコールで連結されたリンカーを作製する、前記方法。 - 人工RNA触媒が、以下のいずれかのRNA配列
GGAUCGAAAGAUUUCCGCAGGCCCGAAAGGGUAUUGGCGUUAGGU (配列番号3)
GGAUCGAAAGAUUUCCGCGGCCCCGAAAGGGGAUUAGCGUUAGGU (配列番号4)
GGAUCGAAAGAUUUCCGCAUCCCCGAAAGGGUACAUGGCGUUAGGU (配列番号5)
GGAUCGAAAGAUUUCCGCACCCCCGAAAGGGGUAAGUGGCGUUAGGU (配列番号19)
からなる化学構造を有する、請求項12に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10825006.9A EP2492344B1 (en) | 2009-10-22 | 2010-10-21 | Rapid display method in translational synthesis of peptide |
| JP2011537297A JP5174971B2 (ja) | 2009-10-22 | 2010-10-21 | ペプチド翻訳合成におけるrapidディスプレイ法 |
| US13/502,487 US20120208720A1 (en) | 2009-10-22 | 2010-10-21 | Rapid display method in translational synthesis of peptide |
| US16/807,435 US11970694B2 (en) | 2009-10-22 | 2020-03-03 | Rapid display method in translational synthesis of peptide |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009243240 | 2009-10-22 | ||
| JP2009-243240 | 2011-04-28 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/502,487 A-371-Of-International US20120208720A1 (en) | 2009-10-22 | 2010-10-21 | Rapid display method in translational synthesis of peptide |
| US16/807,435 Continuation US11970694B2 (en) | 2009-10-22 | 2020-03-03 | Rapid display method in translational synthesis of peptide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011049157A1 true WO2011049157A1 (ja) | 2011-04-28 |
Family
ID=43900380
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/068549 WO2011049157A1 (ja) | 2009-10-22 | 2010-10-21 | ペプチド翻訳合成におけるrapidディスプレイ法 |
Country Status (4)
| Country | Link |
|---|---|
| US (2) | US20120208720A1 (ja) |
| EP (1) | EP2492344B1 (ja) |
| JP (2) | JP5174971B2 (ja) |
| WO (1) | WO2011049157A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013183707A1 (ja) | 2012-06-06 | 2013-12-12 | 国立大学法人東京大学 | pH依存的に標的分子に結合するペプチドのスクリーニング方法 |
| WO2014034922A1 (ja) * | 2012-09-03 | 2014-03-06 | 国立大学法人東京大学 | 血管内皮細胞増殖因子受容体阻害ペプチド |
| WO2014119600A1 (ja) * | 2013-01-30 | 2014-08-07 | ペプチドリーム株式会社 | Flexible Display法 |
| WO2015120058A2 (en) | 2014-02-05 | 2015-08-13 | Molecular Templates, Inc. | Methods of screening, selecting, and identifying cytotoxic recombinant polypeptides based on an interim diminution of ribotoxicity |
| US9409952B2 (en) | 2011-12-28 | 2016-08-09 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| WO2017195901A1 (ja) | 2016-05-13 | 2017-11-16 | 国立大学法人 東京大学 | 肝分泌型代謝制御因子阻害作用による肥満関連疾患治療剤 |
| WO2018168999A1 (ja) | 2017-03-17 | 2018-09-20 | 国立研究開発法人理化学研究所 | Rna分子とペプチドとの複合体の製造方法、及びその利用 |
| WO2019026920A1 (ja) | 2017-07-31 | 2019-02-07 | 国立大学法人東京大学 | 環状ペプチドをタンパク質構造に提示させる超汎用法 |
| WO2019077887A1 (ja) * | 2017-10-16 | 2019-04-25 | 国立大学法人東京大学 | D-アミノ酸及びβ-アミノ酸の取り込みを増強するtRNAのD及びTアームの改変 |
| WO2020027224A1 (ja) | 2018-07-31 | 2020-02-06 | 国立大学法人東京大学 | 新たな結合特異性を抗体に付与する超汎用法 |
| US10815489B2 (en) | 2015-03-13 | 2020-10-27 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-tRNA synthetase and use thereof |
| US11492369B2 (en) | 2017-12-15 | 2022-11-08 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide, and method for processing bases |
| WO2022256380A1 (en) | 2021-06-03 | 2022-12-08 | Genentech, Inc. | Cyclic peptide antibiotics |
| US11542299B2 (en) | 2017-06-09 | 2023-01-03 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| US11732002B2 (en) | 2018-11-30 | 2023-08-22 | Chugai Seiyaku Kabushiki Kaisha | Deprotection method and resin removal method in solid-phase reaction for peptide compound or amide compound, and method for producing peptide compound |
| US12071396B2 (en) | 2019-03-15 | 2024-08-27 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing aromatic amino acid derivative |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9428845B1 (en) | 2010-12-28 | 2016-08-30 | Warp Drive Bio, Inc. | Identifying new therapeutic agents |
| US20170175108A1 (en) * | 2014-05-14 | 2017-06-22 | Evorx Technologies, Inc. | Barcoded peptides |
| EP4289950A3 (en) | 2015-01-09 | 2024-01-24 | Revolution Medicines, Inc. | Macrocyclic compounds that participate in cooperative binding and medical uses thereof |
| TW201629069A (zh) | 2015-01-09 | 2016-08-16 | 霍普驅動生物科技股份有限公司 | 參與協同結合之化合物及其用途 |
| AU2016329064B2 (en) | 2015-10-01 | 2023-10-19 | Warp Drive Bio, Inc. | Methods and reagents for analyzing protein-protein interfaces |
| EP3442599B1 (en) | 2016-04-12 | 2022-03-30 | Warp Drive Bio, Inc. | Compositions and methods for the production of compounds |
| WO2017189409A1 (en) * | 2016-04-25 | 2017-11-02 | Evorx Technologies, Inc. | Beta-catenin barcoded peptides |
| US11479797B2 (en) | 2016-10-28 | 2022-10-25 | Ginkgo Bioworks, Inc. | Compositions and methods for the production of compounds |
| US11814621B2 (en) | 2018-06-01 | 2023-11-14 | Northwestern University | Expanding the chemical substrates for genetic code reprogramming |
| WO2021091967A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| CA3159561A1 (en) | 2019-11-04 | 2021-05-14 | Revolution Medicines, Inc. | Ras inhibitors |
| JP2022553859A (ja) | 2019-11-04 | 2022-12-26 | レボリューション メディシンズ インコーポレイテッド | Ras阻害剤 |
| CA3194067A1 (en) | 2020-09-15 | 2022-03-24 | Revolution Medicines, Inc. | Ras inhibitors |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| WO1998031700A1 (en) | 1997-01-21 | 1998-07-23 | The General Hospital Corporation | Selection of proteins using rna-protein fusions |
| JP2004097213A (ja) * | 2002-07-19 | 2004-04-02 | Mitsubishi Chemicals Corp | 核酸および/またはタンパク質の選択方法 |
| JP3683902B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| WO2007066627A1 (ja) | 2005-12-06 | 2007-06-14 | The University Of Tokyo | 多目的アシル化触媒とその用途 |
| WO2008059823A1 (fr) | 2006-11-17 | 2008-05-22 | The University Of Tokyo | Traduction et synthèse de polypeptides ayant une structure non naturelle au niveau de l'extrémité n et leur application |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6261804B1 (en) * | 1997-01-21 | 2001-07-17 | The General Hospital Corporation | Selection of proteins using RNA-protein fusions |
| US8207093B2 (en) * | 1997-01-21 | 2012-06-26 | The General Hospital Corporation | Selection of proteins using RNA-protein fusions |
| JP4963142B2 (ja) * | 2000-12-14 | 2012-06-27 | 学校法人慶應義塾 | 遺伝子型と表現型の対応付け分子とその構成要素、および対応付け分子の製造方法と利用方法 |
| WO2006041194A1 (ja) * | 2004-10-15 | 2006-04-20 | Japan Science And Technology Agency | mRNA-ピューロマイシン-タンパク質連結体作製用リンカー |
| US20090156416A1 (en) * | 2005-10-12 | 2009-06-18 | Weihong Tan | Hybrid Molecular Probe |
-
2010
- 2010-10-21 WO PCT/JP2010/068549 patent/WO2011049157A1/ja active Application Filing
- 2010-10-21 US US13/502,487 patent/US20120208720A1/en not_active Abandoned
- 2010-10-21 EP EP10825006.9A patent/EP2492344B1/en active Active
- 2010-10-21 JP JP2011537297A patent/JP5174971B2/ja active Active
-
2012
- 2012-11-16 JP JP2012252252A patent/JP5837478B2/ja active Active
-
2020
- 2020-03-03 US US16/807,435 patent/US11970694B2/en active Active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| JP3683282B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| JP3683902B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| WO1998031700A1 (en) | 1997-01-21 | 1998-07-23 | The General Hospital Corporation | Selection of proteins using rna-protein fusions |
| JP3692542B2 (ja) | 1997-01-21 | 2005-09-07 | ザ ジェネラル ホスピタル コーポレーション | Rna−蛋白質の融合体を用いた蛋白質の選抜 |
| JP2004097213A (ja) * | 2002-07-19 | 2004-04-02 | Mitsubishi Chemicals Corp | 核酸および/またはタンパク質の選択方法 |
| WO2007066627A1 (ja) | 2005-12-06 | 2007-06-14 | The University Of Tokyo | 多目的アシル化触媒とその用途 |
| WO2008059823A1 (fr) | 2006-11-17 | 2008-05-22 | The University Of Tokyo | Traduction et synthèse de polypeptides ayant une structure non naturelle au niveau de l'extrémité n et leur application |
| JP2008125396A (ja) | 2006-11-17 | 2008-06-05 | Univ Of Tokyo | N末端に非天然骨格をもつポリペプチドの翻訳合成とその応用 |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
Non-Patent Citations (27)
| Title |
|---|
| GOLEMIS: "Protein-Protein Interactions: A Molecular Cloning Manual", 2005, COLD SPRING LABORATORY PRESS |
| GOTO ET AL., ACS CHEM. BIOL., vol. 3, 2008, pages 120 - 129 |
| H. F. KUNG; B. REDFIELD; B. V. TREADWELL; B. ESKIN; C. SPEARS; H. WEISSBACH: "DNA- directed in vitro synthesis of beta-galactosidase. Studies with purified factors", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 252, no. 19, 1977, pages 6889 - 6894 |
| H. MURAKAMI; A. OHTA; H. ASHIGAI; H. SUGA, NATURE METHODS, vol. 3, 2006, pages 357 - 359 |
| H. MURAKAMI; D. KOUROUKLIS; H. SUGA, CHEMISTRY & BIOLOGY, vol. 10, 2003, pages 1077 - 1084 |
| H. MURAKAMI; H. SAITO; H. SUGA, CHEMISTRY & BIOLOGY, vol. 10, 2003, pages 655 - 662 |
| H. OHASHI; Y. SHIMIZU; B. W. YING; T. UEDA: "Efficient protein selection based on ribosome display system with purified components", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 352, no. 1, 2007, pages 270 - 276, XP005727956, DOI: doi:10.1016/j.bbrc.2006.11.017 |
| H. TRACHSEL; B. EMI; M. H. SCHREIER; T. STAEHELIN: "Initiation of mammalian protein synthesis. II. The assembly of the initiation complex with purified initiation factors", JOURNAL OF MOLECULAR BIOLOGY, vol. 116, no. 4, 1977, pages 755 - 67, XP024020909, DOI: doi:10.1016/0022-2836(77)90269-8 |
| M. C. GONZA; C. CUNNINGHAM; R. M. GREEN: "Isolation and point of action of a factor from Escherichia coli required to reconstruct translation", PROCEEDING OF NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 82, 1985, pages 1648 - 1652, XP055124549 |
| M. H. SCHREIER; B. ERNI; T. STAEHELIN: "Initiation of mammalian protein synthesis. I. Purification and characterization of seven initiation factors", JOURNAL OF MOLECULAR BIOLOGY, vol. 116, no. 4, 1977, pages 727 - 53, XP024020908, DOI: doi:10.1016/0022-2836(77)90268-6 |
| M. Y. PAVLOV; M. EHRENBERG: "Rate of translation of natural mRNAs in an optimized in vitro system", ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS, vol. 328, no. 1, 1996, pages 9 - 16, XP001066398, DOI: doi:10.1006/abbi.1996.0136 |
| MURAKAMI, HIROSHI ET AL.: "Flexizyme as a versatile tRNA acylation catalyst and the application for translation.", NUCLEIC ACIDS SYMP. SER., no. 50, 2006, pages 35 - 36, XP008154709 * |
| N. NIWA; Y. YAMAGISHI; H. MURAKAMI; H. SUGA, BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 19, 2009, pages 3892 - 3894 |
| NEMOTO N. ET AL., FEBS LETT., vol. 414, 1997, pages 405 - 408 |
| NEMOTO, NAOTO ET AL.: "In vitro virus: bonding of mRNA bearing puromycin at the 3'-terminal end to the C-terminal end of its encoded protein on the ribosome in vitro.", FEBS LETT., vol. 414, 1997, pages 405 - 408, XP008154713 * |
| OHUCHI, MASAKI ET AL.: "The flexizyme system: a highly flexible tRNA aminoacylation tool for the translation apparatus.", CURR. OPIN. CHEM. BIOL., vol. 11, 2007, pages 537 - 542, XP008154711 * |
| P.C. JELENC; C.G. KURLAND: "Nucleoside triphosphate regeneration decreases the frequency of translation errors", PROCEEDINGS OF THE NATURAL ACADEMY SCIENCE OF THE UNITED STATES OF AMERICA, vol. 76, no. 7, 1979, pages 3174 - 3178 |
| RAMASWAMY, KRISHNA ET AL.: "Designer ribozymes: programming the tRNA specificity into flexizyme.", J. AM. CHEM. SOC., vol. 126, 2004, pages 11454 - 11455, XP008154708 * |
| ROBERTS, R. W.; SZOSTAK, J. W., PROC. NATL. ACAD. SCI. USA., vol. 94, 1997, pages 12297 - 12302 |
| ROBERTS, RICHARD W ET AL.: "RNA-peptide fusions for the in vitro selection of peptides and proteins.", PROC. NATL. ACAD. SCI. USA, vol. 94, 1997, pages 12297 - 12302, XP008154707 * |
| SAMBROOK: "Molecular Cloning: A Laboratory Manual", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| See also references of EP2492344A4 |
| TABUCHI, ICHIRO ET AL.: "An Efficient Ligation Method in the Making of an in vitro Virus for in vitro Protein Evolution.", BIOL. PROCED., vol. 4, no. 1, 2002, pages 49 - 54, XP008154710 * |
| UENO, SHINGO ET AL.: "An mRNA-protein fusion at N-terminus for evolutionary protein engineering.", INT. J. BIOL. SCI., vol. 3, no. 6, 2007, pages 365 - 374, XP008154706 * |
| Y. GOTO; H. SUGA: "Translation initiation with initiator tRNA charged with exotic peptides", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 131, no. 14, 2009, pages 5040 - 5041, XP002564833, DOI: doi:10.1021/ja900597d |
| Y. SHIMIZU; A. INOUE; Y. TOMARI; T. SUZUKI; T. YOKOGAWA; K. NISHIKAWA; T. UEDA: "Cell-free translation reconstituted with purified components", NATURE BIOTECHNOLOGY, vol. 19, no. 8, 2001, pages 751 - 755, XP002677144, DOI: doi:10.1038/90802 |
| YAMAGUCHI, JUNICHI ET AL.: "cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions.", NUCLEIC. ACIDS RES., vol. 37, no. 16, September 2009 (2009-09-01), pages 1 - 13, XP008154704 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9409952B2 (en) | 2011-12-28 | 2016-08-09 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| US11891457B2 (en) | 2011-12-28 | 2024-02-06 | Chugai Seiyaku Kabushiki Kaisha | Peptide-compound cyclization method |
| WO2013183707A1 (ja) | 2012-06-06 | 2013-12-12 | 国立大学法人東京大学 | pH依存的に標的分子に結合するペプチドのスクリーニング方法 |
| WO2014034922A1 (ja) * | 2012-09-03 | 2014-03-06 | 国立大学法人東京大学 | 血管内皮細胞増殖因子受容体阻害ペプチド |
| JPWO2014034922A1 (ja) * | 2012-09-03 | 2016-08-08 | 国立大学法人 東京大学 | 血管内皮細胞増殖因子受容体阻害ペプチド |
| US9815867B2 (en) | 2012-09-03 | 2017-11-14 | The University Of Tokyo | Peptide for inhibiting vascular endothelial growth factor receptor |
| WO2014119600A1 (ja) * | 2013-01-30 | 2014-08-07 | ペプチドリーム株式会社 | Flexible Display法 |
| JPWO2014119600A1 (ja) * | 2013-01-30 | 2017-01-26 | ペプチドリーム株式会社 | FlexibleDisplay法 |
| WO2015120058A2 (en) | 2014-02-05 | 2015-08-13 | Molecular Templates, Inc. | Methods of screening, selecting, and identifying cytotoxic recombinant polypeptides based on an interim diminution of ribotoxicity |
| US10815489B2 (en) | 2015-03-13 | 2020-10-27 | Chugai Seiyaku Kabushiki Kaisha | Modified aminoacyl-tRNA synthetase and use thereof |
| WO2017195901A1 (ja) | 2016-05-13 | 2017-11-16 | 国立大学法人 東京大学 | 肝分泌型代謝制御因子阻害作用による肥満関連疾患治療剤 |
| US11286481B2 (en) | 2017-03-17 | 2022-03-29 | Cubicstars, Inc. | Method for producing complex of RNA molecule and peptide, and utilization thereof |
| WO2018168999A1 (ja) | 2017-03-17 | 2018-09-20 | 国立研究開発法人理化学研究所 | Rna分子とペプチドとの複合体の製造方法、及びその利用 |
| US11542299B2 (en) | 2017-06-09 | 2023-01-03 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| US11787836B2 (en) | 2017-06-09 | 2023-10-17 | Chugai Seiyaku Kabushiki Kaisha | Method for synthesizing peptide containing N-substituted amino acid |
| WO2019026920A1 (ja) | 2017-07-31 | 2019-02-07 | 国立大学法人東京大学 | 環状ペプチドをタンパク質構造に提示させる超汎用法 |
| WO2019077887A1 (ja) * | 2017-10-16 | 2019-04-25 | 国立大学法人東京大学 | D-アミノ酸及びβ-アミノ酸の取り込みを増強するtRNAのD及びTアームの改変 |
| JPWO2019077887A1 (ja) * | 2017-10-16 | 2021-01-14 | 国立大学法人 東京大学 | D−アミノ酸及びβ−アミノ酸の取り込みを増強するtRNAのD及びTアームの改変 |
| JP7079018B6 (ja) | 2017-10-16 | 2024-01-31 | 国立大学法人 東京大学 | D-アミノ酸及びβ-アミノ酸の取り込みを増強するtRNAのD及びTアームの改変 |
| JP7079018B2 (ja) | 2017-10-16 | 2022-06-01 | 国立大学法人 東京大学 | D-アミノ酸及びβ-アミノ酸の取り込みを増強するtRNAのD及びTアームの改変 |
| US11946042B2 (en) | 2017-10-16 | 2024-04-02 | The University Of Tokyo | Modification of D- and T-arms of tRNA enhancing D-amino acid and β-amino acid uptake |
| US11492369B2 (en) | 2017-12-15 | 2022-11-08 | Chugai Seiyaku Kabushiki Kaisha | Method for producing peptide, and method for processing bases |
| WO2020027224A1 (ja) | 2018-07-31 | 2020-02-06 | 国立大学法人東京大学 | 新たな結合特異性を抗体に付与する超汎用法 |
| US11732002B2 (en) | 2018-11-30 | 2023-08-22 | Chugai Seiyaku Kabushiki Kaisha | Deprotection method and resin removal method in solid-phase reaction for peptide compound or amide compound, and method for producing peptide compound |
| US12071396B2 (en) | 2019-03-15 | 2024-08-27 | Chugai Seiyaku Kabushiki Kaisha | Method for preparing aromatic amino acid derivative |
| WO2022256380A1 (en) | 2021-06-03 | 2022-12-08 | Genentech, Inc. | Cyclic peptide antibiotics |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013046637A (ja) | 2013-03-07 |
| JP5174971B2 (ja) | 2013-04-03 |
| EP2492344A1 (en) | 2012-08-29 |
| JP5837478B2 (ja) | 2015-12-24 |
| JPWO2011049157A1 (ja) | 2013-03-14 |
| EP2492344A4 (en) | 2013-08-21 |
| US20200199579A1 (en) | 2020-06-25 |
| EP2492344A9 (en) | 2013-02-20 |
| EP2492344B1 (en) | 2016-04-06 |
| US11970694B2 (en) | 2024-04-30 |
| US20120208720A1 (en) | 2012-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5837478B2 (ja) | ペプチド翻訳合成におけるrapidディスプレイ法 | |
| JP6206943B2 (ja) | ペプチドライブラリーの製造方法、ペプチドライブラリー、及びスクリーニング方法 | |
| CN110637086B (zh) | Rna分子与肽的复合体的制造方法及其利用 | |
| JP2020176121A (ja) | Dnaコードライブラリーをタグ付けするための方法 | |
| KR101789216B1 (ko) | Dna―코딩된 라이브러리의 생성 및 스크리닝 방법 | |
| CA2544153C (en) | Method for the synthesis of a bifunctional complex | |
| WO2016154675A1 (en) | Platform for non-natural amino acid incorporation into proteins | |
| WO2012074129A1 (ja) | 安定化された二次構造を有するペプチド、及びペプチドライブラリー、それらの製造方法 | |
| WO2012033154A1 (ja) | N-メチルアミノ酸およびその他の特殊アミノ酸を含む特殊ペプチド化合物ライブラリーの翻訳構築と活性種探索法 | |
| KR20020033743A (ko) | 펩티드 수용체 리게이션 방법 | |
| JP6057185B2 (ja) | 核酸リンカー | |
| EP2952582A1 (en) | Flexible display method | |
| US6440695B1 (en) | Method for producing diverse libraries of encoded polypeptides | |
| JP2008253176A (ja) | 高親和性分子取得のためのリンカー | |
| JP5759678B2 (ja) | 核酸リンカー | |
| JP2004097213A (ja) | 核酸および/またはタンパク質の選択方法 | |
| EP3262185B1 (en) | Dna display and methods thereof | |
| JPWO2005024018A1 (ja) | 核酸構築物およびその製造方法 | |
| JP4747292B2 (ja) | 翻訳テンプレートおよびそのライブラリー、それらから合成される蛋白質および蛋白質のライブラリー、ならびにそれらを構成する要素、ならびにそれらの製造法および利用方法 | |
| 上野真吾 et al. | Development of mRNA-protein fusion at N-terminus for evolutionary protein engineering |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10825006 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010825006 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011537297 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13502487 Country of ref document: US |