WO2010143540A1 - ヒドラジド化合物及びその製造方法、並びにそれを用いた硬化剤、樹脂組成物及び硬化体 - Google Patents
ヒドラジド化合物及びその製造方法、並びにそれを用いた硬化剤、樹脂組成物及び硬化体 Download PDFInfo
- Publication number
- WO2010143540A1 WO2010143540A1 PCT/JP2010/059034 JP2010059034W WO2010143540A1 WO 2010143540 A1 WO2010143540 A1 WO 2010143540A1 JP 2010059034 W JP2010059034 W JP 2010059034W WO 2010143540 A1 WO2010143540 A1 WO 2010143540A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrazide compound
- hydrazide
- crystalline
- resin
- resin composition
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 329
- 239000011342 resin composition Substances 0.000 title claims abstract description 78
- 238000004519 manufacturing process Methods 0.000 title claims description 19
- 238000000034 method Methods 0.000 title abstract description 19
- 239000000203 mixture Substances 0.000 claims abstract description 85
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 72
- 229910052751 metal Inorganic materials 0.000 claims abstract description 48
- 239000002184 metal Substances 0.000 claims abstract description 42
- 229920005989 resin Polymers 0.000 claims abstract description 40
- 239000011347 resin Substances 0.000 claims abstract description 40
- 239000003822 epoxy resin Substances 0.000 claims abstract description 27
- 229920000647 polyepoxide Polymers 0.000 claims abstract description 27
- 238000001816 cooling Methods 0.000 claims abstract description 21
- 238000010438 heat treatment Methods 0.000 claims abstract description 5
- 238000002441 X-ray diffraction Methods 0.000 claims description 33
- 238000001228 spectrum Methods 0.000 claims description 32
- 239000002245 particle Substances 0.000 claims description 31
- 239000002253 acid Substances 0.000 claims description 30
- 238000002844 melting Methods 0.000 claims description 21
- 230000008018 melting Effects 0.000 claims description 21
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 10
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 8
- 229910052782 aluminium Inorganic materials 0.000 claims description 8
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 8
- 125000003647 acryloyl group Chemical group O=C([*])C([H])=C([H])[H] 0.000 claims description 7
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 claims description 7
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims description 5
- 229910052797 bismuth Inorganic materials 0.000 claims description 5
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 claims description 5
- 229910052742 iron Inorganic materials 0.000 claims description 5
- 238000010298 pulverizing process Methods 0.000 claims description 5
- 229910052726 zirconium Inorganic materials 0.000 claims description 5
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 claims description 4
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims description 4
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 claims description 4
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 4
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 4
- 229910052787 antimony Inorganic materials 0.000 claims description 4
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 claims description 4
- 229910052788 barium Inorganic materials 0.000 claims description 4
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 claims description 4
- 229910052796 boron Inorganic materials 0.000 claims description 4
- 229910017052 cobalt Inorganic materials 0.000 claims description 4
- 239000010941 cobalt Substances 0.000 claims description 4
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 4
- 230000000536 complexating effect Effects 0.000 claims description 4
- 229910052802 copper Inorganic materials 0.000 claims description 4
- 239000010949 copper Substances 0.000 claims description 4
- 229940042795 hydrazides for tuberculosis treatment Drugs 0.000 claims description 4
- 229910052749 magnesium Inorganic materials 0.000 claims description 4
- 239000011777 magnesium Substances 0.000 claims description 4
- 229910052750 molybdenum Inorganic materials 0.000 claims description 4
- 239000011733 molybdenum Substances 0.000 claims description 4
- 229910052759 nickel Inorganic materials 0.000 claims description 4
- 229910052718 tin Inorganic materials 0.000 claims description 4
- 229910052719 titanium Inorganic materials 0.000 claims description 4
- 239000010936 titanium Substances 0.000 claims description 4
- 229910052725 zinc Inorganic materials 0.000 claims description 4
- 239000011701 zinc Substances 0.000 claims description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 3
- 229910052689 Holmium Inorganic materials 0.000 claims description 3
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 3
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 claims description 3
- 229910052792 caesium Inorganic materials 0.000 claims description 3
- TVFDJXOCXUVLDH-UHFFFAOYSA-N caesium atom Chemical compound [Cs] TVFDJXOCXUVLDH-UHFFFAOYSA-N 0.000 claims description 3
- 229910052791 calcium Inorganic materials 0.000 claims description 3
- 239000011575 calcium Substances 0.000 claims description 3
- 229910052732 germanium Inorganic materials 0.000 claims description 3
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 claims description 3
- 229910052737 gold Inorganic materials 0.000 claims description 3
- 239000010931 gold Substances 0.000 claims description 3
- KJZYNXUDTRRSPN-UHFFFAOYSA-N holmium atom Chemical compound [Ho] KJZYNXUDTRRSPN-UHFFFAOYSA-N 0.000 claims description 3
- 229910052738 indium Inorganic materials 0.000 claims description 3
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 claims description 3
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 claims description 3
- 229910052710 silicon Inorganic materials 0.000 claims description 3
- 239000010703 silicon Substances 0.000 claims description 3
- 229910052709 silver Inorganic materials 0.000 claims description 3
- 239000004332 silver Substances 0.000 claims description 3
- 229910052727 yttrium Inorganic materials 0.000 claims description 3
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 claims description 3
- 238000001723 curing Methods 0.000 abstract description 76
- 230000000694 effects Effects 0.000 abstract description 7
- 238000013007 heat curing Methods 0.000 abstract description 3
- 239000000463 material Substances 0.000 abstract description 2
- 230000000052 comparative effect Effects 0.000 description 64
- 239000007788 liquid Substances 0.000 description 35
- 229920001187 thermosetting polymer Polymers 0.000 description 31
- 238000001938 differential scanning calorimetry curve Methods 0.000 description 28
- 230000004927 fusion Effects 0.000 description 25
- ZWLIYXJBOIDXLL-UHFFFAOYSA-N decanedihydrazide Chemical compound NNC(=O)CCCCCCCCC(=O)NN ZWLIYXJBOIDXLL-UHFFFAOYSA-N 0.000 description 17
- -1 acryl group Chemical group 0.000 description 16
- GRGBENNNGZARRZ-UHFFFAOYSA-N dodecanedihydrazide Chemical compound NNC(=O)CCCCCCCCCCC(=O)NN GRGBENNNGZARRZ-UHFFFAOYSA-N 0.000 description 16
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 15
- 239000002994 raw material Substances 0.000 description 15
- 239000011521 glass Substances 0.000 description 13
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 12
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 12
- 239000005297 pyrex Substances 0.000 description 12
- 239000004973 liquid crystal related substance Substances 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 8
- 229910002012 Aerosil® Inorganic materials 0.000 description 8
- 238000005259 measurement Methods 0.000 description 8
- 239000004925 Acrylic resin Substances 0.000 description 7
- 229920000178 Acrylic resin Polymers 0.000 description 7
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 7
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 7
- 239000007789 gas Substances 0.000 description 7
- 239000004593 Epoxy Substances 0.000 description 5
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 5
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 5
- 230000009257 reactivity Effects 0.000 description 5
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 4
- 150000008065 acid anhydrides Chemical class 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- 125000002947 alkylene group Chemical group 0.000 description 4
- 125000003118 aryl group Chemical group 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 238000011109 contamination Methods 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 125000003700 epoxy group Chemical group 0.000 description 4
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 4
- 229920000768 polyamine Polymers 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 239000004135 Bone phosphate Substances 0.000 description 3
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 3
- 125000000732 arylene group Chemical group 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 229920003986 novolac Polymers 0.000 description 3
- 150000001451 organic peroxides Chemical class 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- IVORCBKUUYGUOL-UHFFFAOYSA-N 1-ethynyl-2,4-dimethoxybenzene Chemical compound COC1=CC=C(C#C)C(OC)=C1 IVORCBKUUYGUOL-UHFFFAOYSA-N 0.000 description 2
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 2
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 2
- QEVGZEDELICMKH-UHFFFAOYSA-N Diglycolic acid Chemical compound OC(=O)COCC(O)=O QEVGZEDELICMKH-UHFFFAOYSA-N 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- IBVAQQYNSHJXBV-UHFFFAOYSA-N adipic acid dihydrazide Chemical compound NNC(=O)CCCCC(=O)NN IBVAQQYNSHJXBV-UHFFFAOYSA-N 0.000 description 2
- UTTHLMXOSUFZCQ-UHFFFAOYSA-N benzene-1,3-dicarbohydrazide Chemical compound NNC(=O)C1=CC=CC(C(=O)NN)=C1 UTTHLMXOSUFZCQ-UHFFFAOYSA-N 0.000 description 2
- 229910000416 bismuth oxide Inorganic materials 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 230000009918 complex formation Effects 0.000 description 2
- 229930003836 cresol Natural products 0.000 description 2
- TYIXMATWDRGMPF-UHFFFAOYSA-N dibismuth;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Bi+3].[Bi+3] TYIXMATWDRGMPF-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000010828 elution Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 2
- 238000007561 laser diffraction method Methods 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- 238000000016 photochemical curing Methods 0.000 description 2
- 239000003504 photosensitizing agent Substances 0.000 description 2
- 238000000790 scattering method Methods 0.000 description 2
- 238000004904 shortening Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000011787 zinc oxide Substances 0.000 description 2
- ZNGSVRYVWHOWLX-KHFUBBAMSA-N (1r,2s)-2-(methylamino)-1-phenylpropan-1-ol;hydrate Chemical compound O.CN[C@@H](C)[C@H](O)C1=CC=CC=C1.CN[C@@H](C)[C@H](O)C1=CC=CC=C1 ZNGSVRYVWHOWLX-KHFUBBAMSA-N 0.000 description 1
- MDJZGXRFYKPSIM-JCYAYHJZSA-N (2r,3r)-2,3-dihydroxybutanedihydrazide Chemical compound NNC(=O)[C@H](O)[C@@H](O)C(=O)NN MDJZGXRFYKPSIM-JCYAYHJZSA-N 0.000 description 1
- LTVUCOSIZFEASK-MPXCPUAZSA-N (3ar,4s,7r,7as)-3a-methyl-3a,4,7,7a-tetrahydro-4,7-methano-2-benzofuran-1,3-dione Chemical compound C([C@H]1C=C2)[C@H]2[C@H]2[C@]1(C)C(=O)OC2=O LTVUCOSIZFEASK-MPXCPUAZSA-N 0.000 description 1
- MUTGBJKUEZFXGO-OLQVQODUSA-N (3as,7ar)-3a,4,5,6,7,7a-hexahydro-2-benzofuran-1,3-dione Chemical compound C1CCC[C@@H]2C(=O)OC(=O)[C@@H]21 MUTGBJKUEZFXGO-OLQVQODUSA-N 0.000 description 1
- KMOUUZVZFBCRAM-OLQVQODUSA-N (3as,7ar)-3a,4,7,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C=CC[C@@H]2C(=O)OC(=O)[C@@H]21 KMOUUZVZFBCRAM-OLQVQODUSA-N 0.000 description 1
- SNVRDQORMVVQBI-OWOJBTEDSA-N (e)-but-2-enedihydrazide Chemical compound NNC(=O)\C=C\C(=O)NN SNVRDQORMVVQBI-OWOJBTEDSA-N 0.000 description 1
- SNVRDQORMVVQBI-UPHRSURJSA-N (z)-but-2-enedihydrazide Chemical compound NNC(=O)\C=C/C(=O)NN SNVRDQORMVVQBI-UPHRSURJSA-N 0.000 description 1
- WZCQRUWWHSTZEM-UHFFFAOYSA-N 1,3-phenylenediamine Chemical compound NC1=CC=CC(N)=C1 WZCQRUWWHSTZEM-UHFFFAOYSA-N 0.000 description 1
- ROFVGYAMRSGUSQ-UHFFFAOYSA-N 1-(2-bromoethyl)piperazine;hydrobromide Chemical compound Br.BrCCN1CCNCC1 ROFVGYAMRSGUSQ-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- UYINXWLEUKYMEC-UHFFFAOYSA-N 2-(2-hydrazinyl-2-oxoethoxy)acetohydrazide;4-[1-(4-hydroxyphenyl)ethyl]phenol Chemical compound NNC(=O)COCC(=O)NN.C=1C=C(O)C=CC=1C(C)C1=CC=C(O)C=C1 UYINXWLEUKYMEC-UHFFFAOYSA-N 0.000 description 1
- XSXYESVZDBAKKT-UHFFFAOYSA-N 2-hydroxybenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1O XSXYESVZDBAKKT-UHFFFAOYSA-N 0.000 description 1
- LGWROMGRXCZCLA-UHFFFAOYSA-N 2-hydroxybutanedihydrazide Chemical compound NNC(=O)CC(O)C(=O)NN LGWROMGRXCZCLA-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- GWZMWHWAWHPNHN-UHFFFAOYSA-N 2-hydroxypropyl prop-2-enoate Chemical compound CC(O)COC(=O)C=C GWZMWHWAWHPNHN-UHFFFAOYSA-N 0.000 description 1
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 1
- SXIFAEWFOJETOA-UHFFFAOYSA-N 4-hydroxy-butyl Chemical group [CH2]CCCO SXIFAEWFOJETOA-UHFFFAOYSA-N 0.000 description 1
- ZMZGIVVRBMFZSG-UHFFFAOYSA-N 4-hydroxybenzohydrazide Chemical compound NNC(=O)C1=CC=C(O)C=C1 ZMZGIVVRBMFZSG-UHFFFAOYSA-N 0.000 description 1
- ALEBYBVYXQTORU-UHFFFAOYSA-N 6-hydrazinyl-6-oxohexanoic acid Chemical compound NNC(=O)CCCCC(O)=O ALEBYBVYXQTORU-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 229930185605 Bisphenol Natural products 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- SNUKVQJEARLLTK-UHFFFAOYSA-N C(COCC(=O)NN)(=O)NN.C=1(O)C(O)=CC=CC1 Chemical compound C(COCC(=O)NN)(=O)NN.C=1(O)C(O)=CC=CC1 SNUKVQJEARLLTK-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229910021503 Cobalt(II) hydroxide Inorganic materials 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical class C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 239000006087 Silane Coupling Agent Substances 0.000 description 1
- 229910006404 SnO 2 Inorganic materials 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- 229920001807 Urea-formaldehyde Polymers 0.000 description 1
- 239000006096 absorbing agent Substances 0.000 description 1
- OFLXLNCGODUUOT-UHFFFAOYSA-N acetohydrazide Chemical compound C\C(O)=N\N OFLXLNCGODUUOT-UHFFFAOYSA-N 0.000 description 1
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 125000004653 anthracenylene group Chemical group 0.000 description 1
- 230000003712 anti-aging effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 239000002216 antistatic agent Substances 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- GXGMLHLXTNVBOQ-UHFFFAOYSA-N benzene-1,3-diol;2-(2-hydrazinyl-2-oxoethoxy)acetohydrazide Chemical compound OC1=CC=CC(O)=C1.NNC(=O)COCC(=O)NN GXGMLHLXTNVBOQ-UHFFFAOYSA-N 0.000 description 1
- ALHNLFMSAXZKRC-UHFFFAOYSA-N benzene-1,4-dicarbohydrazide Chemical compound NNC(=O)C1=CC=C(C(=O)NN)C=C1 ALHNLFMSAXZKRC-UHFFFAOYSA-N 0.000 description 1
- VQXDIDTUGQJUFE-UHFFFAOYSA-N benzene-1,4-diol;2-(2-hydrazinyl-2-oxoethoxy)acetohydrazide Chemical compound OC1=CC=C(O)C=C1.NNC(=O)COCC(=O)NN VQXDIDTUGQJUFE-UHFFFAOYSA-N 0.000 description 1
- VJRITMATACIYAF-UHFFFAOYSA-N benzenesulfonohydrazide Chemical compound NNS(=O)(=O)C1=CC=CC=C1 VJRITMATACIYAF-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- HCOMFAYPHBFMKU-UHFFFAOYSA-N butanedihydrazide Chemical compound NNC(=O)CCC(=O)NN HCOMFAYPHBFMKU-UHFFFAOYSA-N 0.000 description 1
- 125000006226 butoxyethyl group Chemical group 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- XEVRDFDBXJMZFG-UHFFFAOYSA-N carbonyl dihydrazine Chemical compound NNC(=O)NN XEVRDFDBXJMZFG-UHFFFAOYSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- ASKVAEGIVYSGNY-UHFFFAOYSA-L cobalt(ii) hydroxide Chemical compound [OH-].[OH-].[Co+2] ASKVAEGIVYSGNY-UHFFFAOYSA-L 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- HUYREUUJFLHEDE-UHFFFAOYSA-N cyclohexanecarbohydrazide Chemical compound NNC(=O)C1CCCCC1 HUYREUUJFLHEDE-UHFFFAOYSA-N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- KBLWLMPSVYBVDK-UHFFFAOYSA-N cyclohexyl prop-2-enoate Chemical compound C=CC(=O)OC1CCCCC1 KBLWLMPSVYBVDK-UHFFFAOYSA-N 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000012933 diacyl peroxide Substances 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- ZZTCPWRAHWXWCH-UHFFFAOYSA-N diphenylmethanediamine Chemical compound C=1C=CC=CC=1C(N)(N)C1=CC=CC=C1 ZZTCPWRAHWXWCH-UHFFFAOYSA-N 0.000 description 1
- YTQHSQQSLTYMSL-UHFFFAOYSA-N dodecanohydrazide Chemical compound CCCCCCCCCCCC(=O)NN YTQHSQQSLTYMSL-UHFFFAOYSA-N 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- SWRGUMCEJHQWEE-UHFFFAOYSA-N ethanedihydrazide Chemical compound NNC(=O)C(=O)NN SWRGUMCEJHQWEE-UHFFFAOYSA-N 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- ANSXAPJVJOKRDJ-UHFFFAOYSA-N furo[3,4-f][2]benzofuran-1,3,5,7-tetrone Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1C(=O)OC2=O ANSXAPJVJOKRDJ-UHFFFAOYSA-N 0.000 description 1
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 description 1
- 239000004845 glycidylamine epoxy resin Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- OXAGUPFRAIIDLT-UHFFFAOYSA-N heptanedihydrazide Chemical compound NNC(=O)CCCCCC(=O)NN OXAGUPFRAIIDLT-UHFFFAOYSA-N 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- PJPRFQPMDKJMRW-UHFFFAOYSA-N hexadecanedihydrazide Chemical compound NNC(=O)CCCCCCCCCCCCCCC(=O)NN PJPRFQPMDKJMRW-UHFFFAOYSA-N 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 150000003949 imides Chemical class 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000004611 light stabiliser Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 125000005439 maleimidyl group Chemical class C1(C=CC(N1*)=O)=O 0.000 description 1
- 229910000000 metal hydroxide Chemical class 0.000 description 1
- 150000004692 metal hydroxides Chemical class 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- BFDHFSHZJLFAMC-UHFFFAOYSA-L nickel(ii) hydroxide Chemical compound [OH-].[OH-].[Ni+2] BFDHFSHZJLFAMC-UHFFFAOYSA-L 0.000 description 1
- ZWLFGLCGZUVIEA-UHFFFAOYSA-N nonanedihydrazide Chemical compound NNC(=O)CCCCCCCC(=O)NN ZWLFGLCGZUVIEA-UHFFFAOYSA-N 0.000 description 1
- HATIEXJZXOLRAO-UHFFFAOYSA-N octanedihydrazide Chemical compound NNC(=O)CCCCCCC(=O)NN HATIEXJZXOLRAO-UHFFFAOYSA-N 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- MMCOUVMKNAHQOY-UHFFFAOYSA-L oxido carbonate Chemical compound [O-]OC([O-])=O MMCOUVMKNAHQOY-UHFFFAOYSA-L 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- PJBQYCIDGYKEMN-UHFFFAOYSA-N pentanehydrazide Chemical compound CCCCC(=O)NN PJBQYCIDGYKEMN-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005562 phenanthrylene group Chemical group 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 239000004848 polyfunctional curative Substances 0.000 description 1
- 125000006225 propoxyethyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000001686 rotational spectrum Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 239000000565 sealant Substances 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000003566 sealing material Substances 0.000 description 1
- 229920002050 silicone resin Polymers 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 125000003011 styrenyl group Chemical class [H]\C(*)=C(/[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 238000012719 thermal polymerization Methods 0.000 description 1
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 1
- 229910001887 tin oxide Inorganic materials 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
Images












































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/24—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C243/00—Compounds containing chains of nitrogen atoms singly-bound to each other, e.g. hydrazines, triazanes
- C07C243/24—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids
- C07C243/38—Hydrazines having nitrogen atoms of hydrazine groups acylated by carboxylic acids with acylating carboxyl groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F15/00—Compounds containing elements of Groups 8, 9, 10 or 18 of the Periodic Table
- C07F15/06—Cobalt compounds
- C07F15/065—Cobalt compounds without a metal-carbon linkage
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F3/00—Compounds containing elements of Groups 2 or 12 of the Periodic Table
- C07F3/003—Compounds containing elements of Groups 2 or 12 of the Periodic Table without C-Metal linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/06—Aluminium compounds
- C07F5/069—Aluminium compounds without C-aluminium linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/003—Compounds containing elements of Groups 4 or 14 of the Periodic Table without C-Metal linkages
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/40—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the curing agents used
- C08G59/4007—Curing agents not provided for by the groups C08G59/42 - C08G59/66
- C08G59/4014—Nitrogen containing compounds
- C08G59/4035—Hydrazines; Hydrazides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/16—Nitrogen-containing compounds
- C08K5/22—Compounds containing nitrogen bound to another nitrogen atom
- C08K5/24—Derivatives of hydrazine
- C08K5/25—Carboxylic acid hydrazides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
Definitions
- thermosetting agent for an acrylic resin
- organic peroxides are used as the thermosetting agent, and these peroxides are liquids, so that elution into the liquid crystal easily occurs and contamination occurs. There is a problem that it is likely to cause a bleed phenomenon.
- the organic peroxide is inhibited from being cured by oxygen, there is a problem in that it becomes poorly cured in the air atmosphere.
- the organic peroxide-based thermosetting agent for example, ketone peroxide, peroxyketal, hydroperoxide, dialkyl peroxide, diacyl peroxide, peroxyester, peroxycarbonate and the like are used.
- thermosetting agent for example, by adding a dibasic acid dihydrazide compound (adipic acid hydrazide; ADH) having an average particle size of 1 to 5 ⁇ m as a thermosetting agent to the photothermosetting resin composition, the curing agent is uniformly matrixed during heat curing.
- ADH dibasic acid dihydrazide
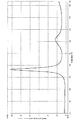
- 1 shows an X-ray diffraction spectrum of a hydrazide compound of the present invention (Example 6: F3-2h).
- 1 shows an X-ray diffraction spectrum of a hydrazide compound of the present invention (Example 7: G3-2h).
- 2 shows an X-ray diffraction spectrum of a hydrazide compound of the present invention (Example 8: hX3-2h).
- 2 shows an X-ray diffraction spectrum of a hydrazide compound of the present invention (Example 9: hY3-2h).
- 1 shows an X-ray diffraction spectrum of a hydrazide compound of the present invention (Example 10: hZ3-2h).
- one type of crystalline hydrazide compound may be used alone, or two or more types of crystalline hydrazide compounds may be used in any combination.
- one kind of monobasic acid hydrazide compound may be used alone, or two or more kinds of monobasic acid hydrazide compounds may be used in combination.
- a dibasic acid hydrazide compound may be used alone, a monobasic acid hydrazide compound and a dibasic acid hydrazide compound may be used in combination, or two or more dibasic acid hydrazide compounds may be used in combination.
- the content of one crystalline hydrazide compound is preferably 1 to 99% by mass, more preferably, based on the total amount of two or more crystalline hydrazide compounds. Is 10 to 90% by mass, more preferably 30 to 70% by mass.
- monobasic acid hydrazide represented by the general formula (1) examples include acetohydrazide, propionic acid hydrazide, pentanoic acid hydrazide, lauric acid hydrazide, cyclohexanecarbohydrazide, salicylic acid hydrazide, p-hydroxybenzoic acid hydrazide, naphthoic acid. And acid hydrazide.
- Specific examples of the other monobasic acid hydrazide include benzenesulfonohydrazide.
- dibasic acid hydrazide represented by the general formula (2) include oxalic acid dihydrazide, malonic acid dihydrazide, succinic acid dihydrazide, adipic acid dihydrazide, pimelic acid dihydrazide, suberic acid dihydrazide, azelaic acid dihydrazide, and sebacine.
- Examples include acid dihydrazide, dodencandioic acid dihydrazide, hexadecanedioic acid dihydrazide, isophthalic acid dihydrazide, and terephthalic acid dihydrazide.
- polyfunctional hydrazide examples include polyacrylic hydrazide.
- the content of the metal element is preferably 0.1 to 20.0 mass%, more preferably 1.0 to 10.0 mass%, based on the total amount of the crystalline hydrazide compound and the metal element. If the content of the metal element is 0.1 to 20.0% by mass based on the total of the crystalline hydrazide compound and the metal element, it is estimated that the metal element and the crystalline hydrazide compound form a good complex.
- the type, number and ratio of the crystalline hydrazide compound and the type, number and ratio of the elements capable of forming a complex depend on various conditions such as the type of resin composition to be cured, application, required curing time, and curing temperature. It can be determined accordingly.
- the average particle size of the hydrazide compound is not particularly limited, but when the hydrazide compound of the present invention is used as a curing agent for a resin, the average particle size is preferably 0.5 to 20.0 ⁇ m, and more The particle shape is preferably 1.0 to 10.0 ⁇ m. When the average particle size of the hydrazide compound is less than 0.5 ⁇ m, the storage stability may be lowered, and the viscosity of the resin composition containing the hydrazide compound as a thermosetting agent is not preferable.
- epoxy resin known ones can be used, for example, bisphenol A type epoxy resin, bisphenol F type epoxy resin, bisphenol AD type epoxy resin, phenol novolac type epoxy resin, cresol novolac type epoxy resin, cycloaliphatic.
- epoxy resins include epoxy resins, glycidyl ester resins, glycidyl amine epoxy resins, heterocyclic epoxy resins, and urethane-modified epoxy resins.
- Epoxy (meth) acrylic acid obtained by modifying a resin having a glycidyl group with (meth) acrylic acid may be used.
- (Meth) acrylic acid alkyl esters such as butyl; (meth) acrylic acid phenyl esters, (meth) acrylic acid aryl esters such as benzyl (meth) acrylate; methoxyethyl (meth) acrylate, ethoxyethyl (meth) acrylate, Alkoxyalkyl (meth) acrylates such as propoxyethyl (meth) acrylate and butoxyethyl (meth) acrylate; 2-hydroxyethyl (meth) acrylate, 3-hydroxypropyl (meth) acrylate, (meth) Such as 2-hydroxypropyl acrylate and 4-hydroxybutyl (meth) acrylate Rokishiru group-containing (meth) acrylic acid esters; (meth) (meth) acrylates of alicyclic alcohols such as cyclohexyl acrylate, and the like.
- (meth) acrylic acid means both acrylic acid and methacrylic acid.
- the temperature for the isothermal treatment is not particularly limited, but is preferably 100 to 280 ° C., more preferably 130 to 250 ° C. If the temperature for the isothermal treatment is too high (for example, a temperature exceeding 280 ° C.), complex formation proceeds rapidly and becomes difficult to control. On the other hand, if the temperature for the isothermal treatment is too low (for example, a temperature of less than 100 ° C.), the isothermal treatment time becomes long and the production efficiency is lowered, which is not preferable.
- the longer the isothermal treatment time the closer to the wavelength of 3300 cm ⁇ 1 .
- the peak derived from NH stretching becomes smaller.
- the peak derived from CH stretching in the wavelength range of 2800 to 3000 cm ⁇ 1 hardly changes.
- the mixture was transferred to a Pyrex (registered trademark) glass tray preheated to 200 ° C. and set in an oven at 200 ° C.
- the molten mixture was cooled to room temperature in an oven at a cooling rate of about 1.0 ° C./min to obtain a solidified body.
- the mixture was transferred to a Pyrex (registered trademark) glass tray preheated to 200 ° C. and set in an oven at 200 ° C.
- the molten mixture was cooled in an oven to 200 to 180 ° C. at a cooling rate of about 1.0 ° C./min, and crystals were grown at 180 ° C. for 5 hours. Furthermore, it cooled at room temperature with the cooling rate of 1.0 degree-C / min, and the solidified body was obtained.
- the solidified product that has been completely cooled to room temperature is roughly pulverized with a cutter mill (made by Orient), and finally pulverized with a high-pressure pulverizer (trade name: NanoJet Mizer, made by Aisin Nanotechnology), with an average particle size of 2 A 3 ⁇ m hydrazide compound (hereinafter, the hydrazide compound of Example 4 is referred to as “C3-2h-5Hold”) was produced.
- Example 9 500 parts by weight of sebacic acid dihydrazide (SDH, manufactured by Otsuka Chemical Co., Ltd.), 500 parts by weight of dodecanedioic acid dihydrazide (DDH, manufactured by Otsuka Chemical Co., Ltd.), and 50 parts by weight of nickel hydroxide (manufactured by Tanaka Chemical Laboratory) Content of 2.2% by mass) was put in a separable flask of 5000 ml and heated at 200 ° C. to obtain a mixture in which two crystalline hydrazide compounds were dissolved so as to be almost in a liquid state. The mixture was subjected to a constant temperature treatment at 200 ° C. for 2 hours.
- SDH sebacic acid dihydrazide
- DDH dodecanedioic acid dihydrazide
- nickel hydroxide manufactured by Tanaka Chemical Laboratory
- Example 10 500 parts by weight of sebacic acid dihydrazide (SDH, manufactured by Otsuka Chemical Co., Ltd.), 500 parts by weight of dodecanedioic acid dihydrazide (DDH, manufactured by Otsuka Chemical Co., Ltd.), and zirconium hydroxide (zirconium hydroxide 999-D, manufactured by Shin Nippon Metal Co., Ltd.)
- SDH sebacic acid dihydrazide
- DDH dodecanedioic acid dihydrazide
- zirconium hydroxide 999-D manufactured by Shin Nippon Metal Co., Ltd.
- Example 13 500 parts by weight of sebacic acid dihydrazide (SDH, manufactured by Otsuka Chemical Co., Ltd.), 500 parts by weight of dodecanedioic acid dihydrazide (DDH, manufactured by Otsuka Chemical Co., Ltd.), and 50 parts by weight of aluminum oxide (AEROXIDE AluC, manufactured by Nippon Aerosil Co., Ltd.) was added to a separable flask of 5000 ml, and heated to 200 ° C. It was confirmed that the two crystalline hydrazide compounds were completely melted, and a molten mixture was obtained.
- SDH sebacic acid dihydrazide
- DDH dodecanedioic acid dihydrazide
- AEROXIDE AluC aluminum oxide
- Example 15 Using the hydrazide compound (T1-2h) of Example 1 as a thermosetting agent, 15 parts by weight of this hydrazide compound and 100 parts by weight of bisphenol A type epoxy acrylic resin (KR-850CRP, manufactured by KSM) are blended, A resin composition was produced.
- KR-850CRP bisphenol A type epoxy acrylic resin
- Example 16 A resin composition was produced in the same manner as in Example 15 except that the hydrazide compound (T1-5h) of Example 2 was used as a thermosetting agent.
- the resin composition of Example 15 started to be cured at around 105 ° C.
- the resin composition of Example 16 had been cured from around 90 ° C.
- the resin composition of Comparative Example 5 has been cured from around 150 ° C. It was confirmed that the resin compositions of Examples 15 and 16 were reduced in the curing temperature by 30 ° C. or more as compared with the resin composition of Comparative Example 5, and the reactivity was improved.
- the hydrazide compound of the present invention can form a suitable curing agent depending on the type of metal element that forms a complex with the hydrazide compound, the isothermal treatment time, the isothermal treatment temperature, and the like. .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Epoxy Resins (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Adhesives Or Adhesive Processes (AREA)
- Liquid Crystal (AREA)
Abstract
Description
光熱硬化型の樹脂組成物としては、一般的に、光硬化成分として(メタ)アクリル基を有する樹脂、加熱硬化成分としてエポキシ基を有する樹脂を任意の割合で配合した組成物が用いられている。
さらに近年、電子部品等は、配線等のピッチがより狭く、より精細化されたことによって、配線やレイアウト等によっては、紫外線等の光が照射されにくい遮光部が増える傾向にあり、光が照射されにくい遮光部であっても、硬化不足にならない硬化剤が求められている。
これらの硬化剤中、アミン化合物は、低温硬化性を有するために、速硬化性が要求される分野で多用されている。しかし、アミン化合物は、耐湿性や電気特性に劣り、ポットライフが短いという問題がある。酸無水物は、透明性が高いという利点があるものの、耐湿性にやや劣るという問題がある。
ここで、アミン化合物としては、例えばジエチルトリアミン、トリエチレンテトラミン等の脂肪酸ポリアミン、脂肪族ポリアミンにエポキシ樹脂、アクリロニトリル、酸化エチレン等を付加した変性ポリアミン、メタフェニレンジアミン、ジアミノジフェニルメタン、ジアミノジフェニルスルホン等の芳香族ポリアミン等が使用されている。
また、酸無水物としては、無水フタル酸、テトラヒドロ無水フタル酸、ヘキサヒドロ無水フタル酸、無水メチルナジック酸、無水ピロメリット酸等が使用されている。ヒドラジド化合物としては、アジピン酸ジヒドラジド、ドデカン二酸ジヒドラジド、セバシン酸ジヒドラジド等の二塩基酸ヒドラジド等が使用されている。
[1]分子内に少なくとも1個のヒドラジド基を有する結晶性ヒドラジド化合物と、該結晶性ヒドラジド化合物と錯体形成可能な金属元素とを含むヒドラジド化合物。
[2]CuKα線(波長1.541Å)に対するX線回折スペクトルにおいて、ブラッグ角度2θ(誤差2θ±0.2°)の5.5~7.5°の範囲にピークを有する、前記[1]に記載のヒドラジド化合物。
[3]融点が60~240℃である、前記[1]又は[2]に記載のヒドラジド化合物。
[4]金属元素が、アルミニウム、チタン、スズ、ジルコニウム、亜鉛、鉄、マグネシウム、コバルト、ニッケル、ビスマス、モリブデン、銅、アンチモン、バリウム、ホウ素、マンガン、インジウム、セシウム、ホルミウム、イットリウム、シリコン、カルシウム、銀、ゲルマニウム、及び金よりなる群から選択される少なくとも1種である、前記[1]~[3]のいずれか1つに記載のヒドラジド化合物。
[5]金属元素の含有量が、結晶性ヒドラジド化合物と金属元素との合計に対して、0.1~20.0質量%である、前記[1]~[4]のいずれか1つに記載のヒドラジド化合物。
[6]2種以上の結晶性ヒドラジド化合物を含む、前記[1]~[5]のいずれか1つに記載のヒドラジド化合物。
[7]二塩基酸ヒドラジド化合物を含む、前記[1]~[6]のいずれか1つに記載のヒドラジド化合物。
[8]2種以上の結晶性ヒドラジド化合物が、全て二塩基酸ヒドラジドである、前記[6]に記載のヒドラジド化合物。
[9]2種以上の結晶性ヒドラジド化合物の合計に対して、1種の結晶性ヒドラジド化合物の含有量が1~99質量%である、前記[6]又は[8]のいずれか1つに記載のヒドラジド化合物。
[10]平均粒子径が0.5~20.0μmである、前記[1]~[9]のいずれか1つに記載のヒドラジド化合物。
[11]前記[1]~[10]に記載のヒドラジド化合物を含む、樹脂用硬化剤。
[12]前記[11]に記載の硬化剤と、分子内に少なくとも1個の不飽和結合を有する樹脂及び/又はエポキシ樹脂とを含む、樹脂組成物。
[13]不飽和結合を有する樹脂が、分子内に少なくとも1個の(メタ)アクリロイル基を有する樹脂である、前記[12]に記載の樹脂組成物。
[14]前記[12]又は[13]に記載の樹脂組成物を硬化させてなる、硬化体。
[15]分子内に少なくとも1個のヒドラジド基を有する結晶性ヒドラジド化合物と、この結晶性ヒドラジド化合物と錯形成可能な金属元素とを加熱し、混合物を得る工程と、混合物を恒温処理する工程と、恒温処理後に混合物を冷却して固化体を得る工程とを含む、ヒドラジド化合物の製造方法。
[16]固化体を、平均粒子径が0.5~20.0μmの粒子状に粉砕する工程を含む、前記[15]に記載の製造方法。
また、本発明のヒドラジド化合物は、原料の結晶性ヒドラジド化合物よりも、融点が低くなる傾向があり、融解熱も小さくなるので、比較的低い温度で不飽和結合(例えば(メタ)アクリル基)やエポキシ基等と反応し、粘度低下や被着体の汚染を引き起こすことなく、均一に硬化し、硬化時間を短縮することができる。このように、本発明のヒドラジド化合物は、硬化温度の低温化及び硬化時間の短縮が可能であり、不飽和結合を有する樹脂及び/又はエポキシ樹脂等の硬化剤として好適に用いることができる。本発明のヒドラジド化合物を硬化剤として用いた樹脂組成物は、ポットライフ的にも安定しており、生産性が良好である。
結晶性ヒドラジド化合物の任意の組み合わせとしては、例えば1種の一塩基酸ヒドラジド化合物を単独で使用してもよく、2種以上の一塩基酸ヒドラジド化合を組み合わせて使用してもよく、1種の二塩基酸ヒドラジド化合物を単独で使用してもよく、一塩基酸ヒドラジド化合物と二塩基酸ヒドラジド化合物とを組み合わせて使用してもよく、2種以上の二塩基酸ヒドラジド化合物を組み合わせて使用してもよく、一塩基酸ヒドラジド化合物と、二塩基酸ヒドラジド化合物と、三塩基酸ヒドラジド化合物を組み合わせて使用してもよい。中でも、2種以上の結晶性ヒドラジド化合物を組み合わせて使用することによって融点が低くなることから、単独の場合と比較し恒温処理の温度調節が行いやすい。


で表される二塩基酸ジヒドラジドが挙げられる。
金属元素の含有量が、結晶性ヒドラジド化合物と金属元素との合計に対して、0.1~20.0質量%であると、金属元素と結晶性ヒドラジド化合物が良好な錯体を形成すると推測される。
[X線回折スペクトル]
本発明のヒドラジド化合物は、結晶性ヒドラジド化合物が配位子となって、金属イオンと配位結合し、少なくとも一部の結晶性ヒドラジド化合物と金属元素が錯体を形成していると推測される。
結晶性ヒドラジド化合物と金属元素が厳密に錯体を形成しているかどうかは明らかではないが、本発明のヒドラジド化合物と、原料の1種類の結晶性ヒドラジド化合物を比較すると、CuKα線に対するX線回折スペクトルに対して、ブラッグ角度2θ(誤差2θ±0.2°)の10°以下の最大強度ピークが現われる範囲が異なる。原料の1種類の結晶性ヒドラジド化合物は、CuKα線に対するX線回折スペクトルに対して、ブラッグ角度2θ(誤差2θ±0.2°)の4.0~5.0の範囲にピークが現われる。
一方、本発明のヒドラジド化合物は、CuKα線に対するX線回折スペクトルに対して、ブラッグ角度2θ(誤差2θ±0.2°)の5.5~7.5°の範囲にピークが現われる。本発明のヒドラジド化合物は、原料の1種類の結晶性ヒドラジド化合物と比較して、ブラッグ角2θの10°以下の最大強度ピークが格子間距離の短くなる方向にシフトしている。このことから、少なくとも一部の結晶性ヒドラジド化合物と金属元素は錯体を形成していると推測される。
X線回折スペクトルは、例えばX線回折装置(製品名:XRD-6100、島津製作所社製)を用いて測定を行う。
なお、金属元素を含んでいない結晶性ヒドラジド化合物も、2種類以上の結晶性ヒドラジド化合物を含むものは、1種類の結晶性ヒドラジド化合物のピークと比べて、ブラッグ角度2θ(誤差2θ±0.2°)の10°以下の最大強度ピークがブラッグ角度の5.5°の方向にシフトする傾向がある。
本発明のヒドラジド化合物は、好ましくは60~260℃、より好ましくは60~230℃、さらに好ましくは60~220℃の範囲に融点を有する。
ヒドラジド化合物の融点は、例えば示差走査熱量測定装置(Pyris6DSC、パーキンエルマー社製、以下、「DSC」と称する。)を用いて、融解による吸熱の最大ピーク温度を融点として求める。
さらに本発明のヒドラジド化合物は、金属元素を含んでいない原料の結晶性ヒドラジド化合物と比較して、融解熱が15%以上、小さくなる。
本発明のヒドラジド化合物は、融解熱が原料の結晶性ヒドラジド化合物よりも小さくなるため、このヒドラジド化合物を不飽和結合を有する樹脂(例えば(メタ)アクリル樹脂)やエポキシ樹脂等の熱硬化剤として用いた場合に、硬化温度を低温化し、硬化時間の短縮化することができる。
なお、融解熱は、融点を測定した装置と同様の装置を用いて、融解による吸熱ピークのピーク面積を融解熱(J/g)として求める。
本発明のヒドラジド化合物は、粉砕した粒子状の形態を有することが好ましい。例えば、ヒドラジド化合物を粉砕して、粒子状の形態にすることによって、このヒドラジド化合物を熱硬化剤として用いた場合に、樹脂との反応性をより高めて、硬化時間をより短縮することが可能である。
ヒドラジド化合物の平均粒子径が0.5μm未満では、保存安定性が低下する可能性があり、ヒドラジド化合物を熱硬化剤として含む樹脂組成物の粘度が高くなるので好ましくない。また、平均粒子径が20.0μmを超えると、反応性が低くなり、熱硬化剤として用いた場合に、不均一な硬化状態となるので、硬化体の耐熱性、耐湿性が低下する場合があり好ましくない。
なお、平均粒子径は、レーザー回折・散乱法による粒度分布測定装置によって求めることができ、平均粒子径は、レーザー回折・散乱法による粒度分布測定における質量平均値D50(D50は累積質量が50%になるときの粒子径、すなわちメジアン径)として測定した値である。
本発明のヒドラジド化合物は、少なくとも一部の結晶性ヒドラジド化合物と金属元素が錯体を形成していると推測されるため、不飽和結合を有する樹脂に対して高い活性を有する。したがって、本発明のヒドラジド化合物は、不飽和結合を有する樹脂に対して優先的に反応する。また、本発明のヒドラジド化合物は、原料の結晶性ヒドラジド化合物と比較して、融点が低下する傾向にあり、融解熱が小さくなるため、比較的低い温度で不飽和結合を有する樹脂、エポキシ樹脂とも反応し、粘度低下、被着体への汚染、ブリード現象等を引き起こすことなく、短い硬化時間で均一に硬化させることができる。このように本発明のヒドラジド化合物は、不飽和結合を有する樹脂、エポキシ樹脂等の熱硬化剤として好適に用いることができ、硬化温度の低温化及び硬化時間の短縮化が可能であり、液安定性も高いため、ポットライフ的に安定であり、生産性が高い。
本発明のヒドラジド化合物を樹脂用硬化剤として用いる場合は、1種又は2種以上を混合して用いてもよい。
樹脂組成物は、本発明のヒドラジド化合物を含む硬化剤と、分子内に少なくとも1個の不飽和結合を有する樹脂及び/又はエポキシ樹脂を含むものであり、例えば、分子内に少なくとも1個の不飽和結合を有する樹脂又はエポキシ樹脂の1種を単独で用いてもよく、複数種を併用してもよく、不飽和結合含有エポキシ樹脂であってもよい。
アクリル酸誘導体としては、具体的には、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル、(メタ)アクリル酸n-ブチル、(メタ)アクリル酸i-ブチル、(メタ)アクリル酸t-ブチル等の(メタ)アクリル酸アルキルエステル;(メタ)アクリル酸フェニル、(メタ)アクリル酸ベンジル等の(メタ)アクリル酸アリールエステル;(メタ)アクリル酸メトキシエチル、(メタ)アクリル酸エトキシエチル、(メタ)アクリル酸プロポキシエチル、(メタ)アクリル酸ブトキシエチル等の(メタ)アクリル酸アルコキシアルキル;(メタ)アクリル酸-2-ヒドロキシエチル、(メタ)アクリル酸-3-ヒドロキシプロピル、(メタ)アクリル酸-2-ヒドロキシプロピル、(メタ)アクリル酸-4-ヒドロキシブチル等のヒドロキシル基含有(メタ)アクリル酸エステル;(メタ)アクリル酸シクロヘキシルのような脂環式アルコールの(メタ)アクリル酸エステル等が挙げられる。ここで(メタ)アクリル酸とは、アクリル酸とメタクリル酸の両方を意味する。
液安定性の測定方法としては、例えばRE-105U型粘度計(東機産業社製)を用いて、この粘度計に3°×R7.7コーンロータを取り付け、測定対象となる樹脂組成物0.1mlを該粘度計にセットし、2.5rpmにて測定を行い、測定対象を25℃で1週間静置した後、再び粘度を測定して、1週間静置前後の粘度の変化率を算出する方法が挙げられる。
本発明の樹脂組成物を硬化させた硬化体は、硬化温度の低温化、硬化時間の短縮化、及びポットライフ的に安定であるので、生産性が良好である。
本発明のヒドラジド化合物の製造方法は、分子内に少なくとも1個のヒドラジド基を有する結晶性ヒドラジド化合物と、この結晶性ヒドラジド化合物と錯形成可能な金属元素とを加熱し、混合物を得る工程と、混合物を恒温処理する工程と、混合物を冷却して固化体を得る工程とを含む。
結晶性ヒドラジド化合物を加熱する温度は、特に限定されないが、結晶性ヒドラジド化合物がほぼ液体の状態となる温度であることが好ましい。結晶性ヒドラジド化合物がほぼ液体の状態となる温度としては、結晶性ヒドラジド化合物の融点付近の温度、例えば結晶性ヒドラジド化合物の融点よりも10℃程度低い温度から該融点よりも10℃程度高い温度であることが好ましい。
恒温処理する温度が高すぎると(例えば280℃を超える温度)、錯体形成が急激に進み、制御し難くなる。一方、恒温処理する温度が低すぎると(例えば100℃未満の温度)、恒温処理時間が長くなり、生産効率が低くなってしまうため好ましくない。
恒温処理する時間は、恒温処理する温度によって異なり、温度が高ければ処理時間は短くなり、温度が低ければ処理時間が長くなるので、硬化剤として使用する際の使用目的等に合わせて最適な処理時間を選択することが可能である。
粉砕する方法としては、固化体は、例えば高圧粉砕機を使用して粉砕することが好ましい。高圧粉砕機としては、例えばクロスジェットミル(栗源鉄工所社製)、カウンタージェットミル(ホソカワミクロン社製)、ナノジェットマイザー(アイシンナノテクノロジーズ社製)等が挙げられる。
〔実施例1〕
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化チタン(AEROXIDE P25、日本アエロジル社製)20重量部(チタンの含有量が1.2質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、目開き500μmふるいにかけて通過したものを、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.8μmのヒドラジド化合物(以下、実施例1のヒドラジド化合物を「T1-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化チタン(AEROXIDE P25、日本アエロジル社製)20重量部(チタンの含有量が1.2質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で5時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径3.4μmのヒドラジド化合物(以下、実施例2のヒドラジド化合物を「T1-5h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化アルミニウム(AEROXIDE AluC、日本アエロジル社製)50重量部(アルミニウムの含有量が2.5質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.5μmのヒドラジド化合物(以下、実施例3のヒドラジド化合物を「C3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化アルミニウム(AEROXIDE AluC、日本アエロジル社製)50重量部(アルミニウムの含有量が2.5質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、200~180℃まで1.0℃/分程度の冷却速度で冷却し、180℃で5時間、結晶を成長させた。さらに、室温まで1.0℃/分の冷却速度で冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.3μmのヒドラジド化合物(以下、実施例4のヒドラジド化合物を「C3-2h-5Hold」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化亜鉛(ZnO、シーアイ化成社製)50重量部(亜鉛の含有量が3.8質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径4.3μmのヒドラジド化合物(以下、実施例5のヒドラジド化合物を「E3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化スズ(SnO2、シーアイ化成社製)50重量部(スズの含有量が3.7質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径3.5μmのヒドラジド化合物(以下、実施例6のヒドラジド化合物を「F3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化ビスマス(酸化ビスマスA、新日本金属社製)50重量部(ビスマスの含有量が4.3質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.6μmのヒドラジド化合物(以下、実施例7のヒドラジド化合物を「G3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、水酸化コバルト(田中化学研究所社製)50重量部(コバルトの含有量が2.0質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径3.3μmのヒドラジド化合物(以下、実施例8のヒドラジド化合物を「hX3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、水酸化ニッケル(田中化学研究所社製)50重量部(ニッケルの含有量が2.2質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.6μmのヒドラジド化合物(以下、実施例9のヒドラジド化合物を「hY3-2h」と称する。)を製造した。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、水酸化ジルコニウム(水酸化ジルコニウム999-D、新日本金属社製)50重量部(ジルコニウムの含有量が2.6質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径2.9μmのヒドラジド化合物(以下、実施例10のヒドラジド化合物を「hZ3-2h」と称する。)を製造した。
〔実施例11〕
セバシン酸ジヒドラジド(SDH、大塚化学社製)1000重量部と、酸化アルミニウム(AEROXIDE AluC、日本アエロジル社製)50重量部(アルミニウムの含有量が2.5質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、2種の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径3.4μmのヒドラジド化合物(以下、実施例11のヒドラジド化合物を「S-C3-2h」と称する。)を製造した。
〔実施例12〕
セバシン酸ジヒドラジド(SDH、大塚化学社製)300重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)300重量部と、イソフタル酸ジヒドラジド(IDH、大塚化学社製)300重量部と、酸化アルミニウム(AEROXIDE AluC、日本アエロジル社製)45重量部(アルミニウムの含有量が2.5質量%)を5000mlのセパラブルフラスコに入れ、200℃で加熱し、3種類の結晶性ヒドラジド化合物をほぼ液体の状態となるように溶解させた混合物を得た。この混合物を200℃で2時間恒温処理を行った。
その後、混合物を200℃に予備加熱したパイレックス(登録商標)製のガラストレーに移し、200℃のオーブンにセットした。そして、溶融混合物を、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
室温まで完全に冷却した固化体をカッターミル(オリエント社製)で粗粉砕し、最終的に高圧粉砕機(商品名:ナノジェットマイザー、アイシンナノテクノロジース社製)で粉砕し、平均粒子径3.0μmのヒドラジド化合物(以下、実施例12のヒドラジド化合物を「DSI-111C3-2h」と称する。)を製造した。
市販のセバシン酸ジヒドラジド(SDH、大塚化学社製)を比較例1とした。
市販のドデカン二酸ジヒドラジド(DDH、大塚化学社製)を比較例2とした。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部とを混合したもの(以下、「SD55」と称する。)を比較例3とした。
比較例3の混合物を200℃まで加熱し、混合物を液状に融解した後、実施例4と同様にして、比較例4のヒドラジド化合物を得た。
X線回折装置(製品名:XRD-6100、島津製作所社製)を用いて、X線出力電圧:40.0KV、X線出力電流:40.0mA、スキャンステップ幅:0.02度で測定した。
示差走査熱量測定装置(Pyris6DSC、パーキンエルマー社製)を用いて、測定試料であるヒドラジド化合物をアルミ製の容器に封入して、融解による吸熱の最大ピーク温度を融点として求めた。また、吸熱のピーク面積を融解熱として求めた。
実施例1~12のヒドラジド化合物を熱硬化剤として用いて、このヒドラジド化合物15重量部と、ビスフェノールA型エポキシアクリル樹脂(KR-850CRP、KSM社製)100重量部とを配合し、樹脂組成物を製造した。なお、ビスフェノールA型エポキシアクリル樹脂は、エポキシ基1当量に対して、アクリロイル基が1当量であった。この樹脂組成物が25℃となるように恒温処理し、RE-105U型粘度計(東機産業社製)に3°×R7.7コーンロータを取り付け、対象となる樹脂組成物0.15mlをコーンロータ内にセットし、2.5rpmで25℃の樹脂組成物の粘度を測定した。測定対象とした樹脂組成物を25℃で1週間静置し、1週間後の該樹脂組成物の粘度を上記方法で測定し、初期の粘度から1週間後の粘度の変化率(%/week)を算出した。測定レンジオーバーした場合は、測定不可とした。なお、測定レンジオーバーの基準は、1,200,000mPa・sである。

なお、図10においてブラッグ角度2θ(誤差2θ±0.2°)の2°付近に現われているピークは散乱光であり、X線回折由来のピークではない。
また、実施例1~12のヒドラジド化合物は、比較例1、2の原料の1種類の結晶性ヒドラジド化合物と比較して、ブラッグ角度2θ(誤差2θ±0.2°)の5.5~7.5°の範囲に現われるピークの強度が小さくなっており、全体的に非晶質の割合が多くなっていると推測される。
さらに、実施例1~12のヒドラジド化合物は、ブラッグ角度2θ(誤差2θ±0.2°)の20.0~25.0°の範囲に現われるピークが、比較例1、2の原料の1種類の結晶性ヒドラジド化合物のピークと比較して、ブロードになっている。この結果から、実施例1~12のヒドラジド化合物は、少なくとも一部が錯体を形成すると共に、非晶状態と結晶状態が混在している部分結晶化合物であると考えられる。
なお、図15に示すように、比較例4の2種類の結晶性ヒドラジド化合物は、ブラッグ角度2θ(誤差2θ±0.2°)の10°以下の最大強度ピークが、5.5°の方向にシフトしていたが、金属元素を含んでいないので、ヒドラジド化合物と金属元素の錯体形成に由来するピークではない。
この結果から、実施例1~12のヒドラジド化合物を熱硬化剤として用いた場合は、硬化温度の低温化及び硬化時間の短縮化が可能である。
また、実施例1~12のヒドラジド化合物は、恒温処理の条件(時間、温度等)を変化させることにより、液安定性の数値も変化させることが可能である。なお、比較例4の融解熱(J/g)は、同一の恒温処理を行った実施例4よりも液安定性が向上していなかった。
ヒドラジド化合物を熱硬化剤として用いる場合は、硬化させる樹脂等によって、恒温処理の条件を変化させることにより、好適な熱硬化剤として使用し得るヒドラジド化合物を形成することが可能である。ヒドラジド化合物を熱硬化剤として用いる場合は、液安定性が、好ましくは80%/week以下、より好ましくは50%/week以下、さらに好ましくは30%/weekとなるように、ヒドラジド化合物を形成する。
〔実施例13〕
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化アルミニウム(AEROXIDE AluC、日本アエロジル社製)50重量部(アルミニウムの含有量が2.5質量%)を5000mlのセパラブルフラスコに入れ、200℃に加熱した。2種の結晶性ヒドラジド化合物が完全に溶融したのを確認し、溶融混合物を得た。この溶融混合物を200℃で0~3時間恒温処理を行い、0、1、2、3時間ごとにサンプリングし、自然冷却を行い、ヒドラジド化合物を得た。各ヒドラジド化合物を、それぞれ、実施例13-1(C3-b12-0h、図31)実施例13-2(C3-b12-1h、図32)、実施例13-3(C3-b12-2h、図33)、実施例13-4(C3-b12-3h、図34)と称する。
セバシン酸ジヒドラジド(SDH、大塚化学社製)500重量部と、ドデカン二酸ジヒドラジド(DDH、大塚化学社製)500重量部と、酸化チタン(AEROXIDE AluC、日本アエロジル社製)20重量部(チタンの含有量が1.2質量%)を5000mlのセパラブルフラスコに入れ、200℃に加熱した。2種の結晶性ヒドラジド化合物が完全に溶融したのを確認し、溶融混合物を得た。この溶融混合物を200℃で0~5時間恒温処理を行い、0、1、2、4、5時間ごとにサンプリングし、自然冷却を行い、ヒドラジド化合物を得た。各ヒドラジド化合物を、それぞれ、「T1-b5S-0h」、「T1-b5S-1h」、「T1-b5S-2h」、「T1-b5S-4h」、「T1-b5S-5h」と称する。
Spectrum One(パーキンエルマー社製)を用いて、FT-IRを測定した。
1H-NMRはJNM-ECA600(日本電子社製)を用いて、サンプル50μlをアセトン-d6(重水素化率99.9%以上)500μlに溶かしたものを25℃で測定した。
また、恒温処理の時間が長くなるほど、ブラッグ角度2θ(誤差2θ±0.2°)の23.0~24.0°の範囲に現われるピーク強度が減少した。
図36に示すように、ヒドラジド化合物の製造時に発生したガスの1H-NMRのチャートは、2.9592ppmにピークが現れる。このピークは、水(H2O))を表すピークである。
図35及び図36に示す結果からも、実施例のヒドラジド化合物は、少なくとも一部の原料の結晶性ヒドラジド化合物と金属元素とが錯体を形成していると推測できる。
〔実施例15〕
実施例1のヒドラジド化合物(T1-2h)を熱硬化剤として用いて、このヒドラジド化合物15重量部と、ビスフェノールA型エポキシアクリル樹脂(KR-850CRP、KSM社製)100重量部とを配合し、樹脂組成物を製造した。
実施例2のヒドラジド化合物(T1-5h)を熱硬化剤として用いたこと以外は、実施例15と同様にして、樹脂組成物を製造した。
比較例3の2種の結晶性ヒドラジド化合物から得られたヒドラジド化合物(SD55)を硬化剤として用いたこと以外は、実施例15と同様にして、樹脂組成物を製造した。
実施例1のヒドラジド化合物(T1-2h)を熱硬化剤として用いて、このヒドラジド化合物30重量部と、ビスフェノールA型エポキシ樹脂(850S、DIC社製)100重量部とを配合し、樹脂組成物を製造した。
実施例2のヒドラジド化合物(T1-5h)を熱硬化剤として用いたこと以外は、実施例17と同様にして、樹脂組成物を製造した。
比較例3の2種の結晶性ヒドラジド化合物から得られたヒドラジド化合物(SD55)を硬化剤として用いたこと以外は、実施例17と同様にして、樹脂組成物を製造した。
実施例1のヒドラジド化合物(T1-2h)を熱硬化剤として用いて、このヒドラジド化合物15重量部と、ビスフェノールA型エポキシ樹脂(850S、DIC社製)をアクリル酸で100%変性したエポキシアクリル樹脂100重量部とを配合し、樹脂組成物を製造した。
実施例2のヒドラジド化合物(T1-5h)を熱硬化剤として用いたこと以外は、実施例19と同様にして、樹脂組成物を製造した。
比較例3の2種の結晶性ヒドラジド化合物から得られたヒドラジド化合物(SD55)を硬化剤として用いたこと以外は、実施例19と同様にして、樹脂組成物を製造した。
実施例15、16の樹脂組成物は、硬化温度が、比較例5の樹脂組成物と比較して30℃以上の低温化し、反応性が向上していることが確認できた。
実施例17、18の樹脂組成物は、硬化温度が、比較例6の樹脂組成物と比較して30℃以上の低温化し、反応性が向上していることが確認できた。
実施例19、20の樹脂組成物は、硬化温度が、比較例7の樹脂組成物と比較して30℃以上の低温化し、反応性が向上していることが確認できた。
比較例3の混合物を200℃まで加熱し、混合物を液状に融解した後、恒温処理することなく、オーブン中で、1.0℃/分程度の冷却速度で室温まで冷却して、固化体を得た。
この固化体を実施例1と同様にして、比較例7のヒドラジド化合物を得た。

一方、比較例のヒドラジド化合物は、恒温処理時間を長くしても、実施例のヒドラジド化合物ほど、融解熱の低下率が大きくならなかった。
即ち、実施例1~4のヒドラジド化合物は、比較例3(原料)のヒドラジド化合物と比べて、融解熱の低下率が57.3~3~83.5%と大きくなった。なお、比較例4、8についても、恒温処理時間が長くなるほど、融解熱、融解熱の低下率が小さくなる傾向が見られたが、同一の恒温処理時間の実施例4と比較例4を比べると、実施例4の融解熱の低下率は83.5%であるのに対して、比較例4の融解熱の低下率は57.9%と小さかった。
同種の金属元素を用いた実施例1及び2、実施例3及び4の液体安定性を比較すると、実施例1に対して、実施例2は液体安定性が92.8%向上した。また、実施例3に対して、実施例4は液体安定性が98.3%向上した。一方、比較例4、8を比較すると、恒温処理時間の長い比較例4に対して、恒温処理時間の短い比較例8の方が却って、液体安定性が低下しており、実施例とは逆の傾向が見られた。
この結果から、本発明のヒドラジド化合物は、ヒドラジド化合物と錯体を形成する金属元素の種類、恒温処理時間、恒温処理温度等によって、好適な硬化剤を形成することが可能であることが確認できた。
Claims (16)
- 分子内に少なくとも1個のヒドラジド基を有する結晶性ヒドラジド化合物と、該結晶性ヒドラジド化合物と錯体形成可能な金属元素とを含むことを特徴とするヒドラジド化合物。
- CuKα線(波長1.541Å)に対するX線回折スペクトルにおいて、ブラッグ角度2θ(誤差2θ±0.2°)の5.5~7.5°の範囲にピークを有する、請求項1に記載のヒドラジド化合物。
- 融点が60~240℃である、請求項1又は2に記載のヒドラジド化合物。
- 金属元素がアルミニウム、チタン、スズ、ジルコニウム、亜鉛、鉄、マグネシウム、コバルト、ニッケル、ビスマス、モリブデン、銅、アンチモン、バリウム、ホウ素、マンガン、インジウム、セシウム、ホルミウム、イットリウム、シリコン、カルシウム、銀、ゲルマニウム、及び金よりなる群から選択される少なくとも1種である、請求項1~3のいずれか1項に記載のヒドラジド化合物。
- 金属元素の含有量が、結晶性ヒドラジド化合物と金属元素との合計に対して、0.1~20.0質量%である、請求項1~4のいずれか1項に記載のヒドラジド化合物。
- 2種以上の結晶性ヒドラジド化合物を含む、請求項1~5のいずれか1項に記載のヒドラジド化合物。
- 二塩基酸ヒドラジド化合物を含む、請求項1~6のいずれか1項に記載のヒドラジド化合物。
- 2種以上の結晶性ヒドラジド化合物が、全て二塩基酸ヒドラジドである、請求項6に記載のヒドラジド化合物。
- 2種以上の結晶性ヒドラジド化合物の合計に対して、1種の結晶性ヒドラジド化合物の含有量が1~99質量%である、請求項6又は8に記載のヒドラジド化合物。
- 平均粒子径が0.5~20.0μmである、請求項1~9のいずれか1項に記載のヒドラジド化合物。
- 請求項1~10に記載のヒドラジド化合物を含む、樹脂用硬化剤。
- 請求項11に記載の硬化剤と、分子内に少なくとも1個の不飽和結合を有する樹脂及び/又はエポキシ樹脂とを含む、樹脂組成物。
- 不飽和結合を有する樹脂が、分子内に少なくとも1個の(メタ)アクリロイル基を有する樹脂である、請求項12に記載の樹脂組成物。
- 請求項12又は13に記載の樹脂組成物を硬化させてなる、硬化体。
- 分子内に少なくとも1個のヒドラジド基を有する結晶性ヒドラジド化合物と、この結晶性ヒドラジド化合物と錯形成可能な金属元素とを加熱し、混合物を得る工程と、混合物を恒温処理する工程と、恒温処理後に混合物を冷却して固化体を得る工程とを含むことを特徴とするヒドラジド化合物の製造方法。
- 固化体を、平均粒子径が0.5~20.0μmの粒子状に粉砕する工程を含む、請求項15に記載の製造方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020127000573A KR101712687B1 (ko) | 2009-06-10 | 2010-05-27 | 히드라지드 화합물 및 그 제조 방법, 및 그것을 이용한 경화제, 수지 조성물 및 경화체 |
| CN201080025073.6A CN102459155B (zh) | 2009-06-10 | 2010-05-27 | 酰肼化合物及其制造方法、以及使用它的固化剂、树脂组合物及固化体 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009139569A JP4961458B2 (ja) | 2009-06-10 | 2009-06-10 | ヒドラジド化合物及びその製造方法、並びにそれを用いた硬化剤、樹脂組成物及び硬化体 |
| JP2009-139569 | 2009-06-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010143540A1 true WO2010143540A1 (ja) | 2010-12-16 |
Family
ID=43308795
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/059034 WO2010143540A1 (ja) | 2009-06-10 | 2010-05-27 | ヒドラジド化合物及びその製造方法、並びにそれを用いた硬化剤、樹脂組成物及び硬化体 |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP4961458B2 (ja) |
| KR (1) | KR101712687B1 (ja) |
| CN (1) | CN102459155B (ja) |
| TW (1) | TWI482750B (ja) |
| WO (1) | WO2010143540A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012077720A1 (ja) * | 2010-12-09 | 2012-06-14 | 協立化学産業株式会社 | 光重合開始剤に適した化合物、光重合開始剤及び光硬化性樹脂組成物 |
| JP2013067569A (ja) * | 2011-09-21 | 2013-04-18 | Nippon Kayaku Co Ltd | 新規ヒドラジド化合物及びそれを用いた樹脂組成物 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6290599B2 (ja) * | 2012-11-13 | 2018-03-07 | 積水化学工業株式会社 | 液晶表示素子用シール剤、上下導通材料、液晶表示素子、及び、ヒドラジド系熱硬化剤 |
| CN103193959B (zh) * | 2013-04-18 | 2014-07-30 | 艾达索高新材料无锡有限公司 | 可降解酰肼类潜伏型环氧树脂固化剂及其应用 |
| CN105734177B (zh) | 2016-03-11 | 2020-08-11 | 广西叶茂食品有限责任公司 | 一种全营养纯净粉糖和液体糖浆的工业化生产工艺及设备 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4730154U (ja) * | 1971-05-04 | 1972-12-05 | ||
| JPH05222296A (ja) * | 1992-02-14 | 1993-08-31 | Mitsubishi Yuka Badische Co Ltd | 水性架橋性樹脂組成物 |
| JPH09104687A (ja) * | 1995-10-06 | 1997-04-22 | Daicel Chem Ind Ltd | トリス(カルボヒドラジド−o,n)マグネシウム(ii)硝酸塩錯体とその製造方法及びガス発生剤組成物 |
| WO1998029425A1 (fr) * | 1996-12-26 | 1998-07-09 | Otsuka Kagaku Kabushiki Kaisha | Complexe constitue par manganese et semicarbazide et generateur de gaz pour airbag |
| JPH11152288A (ja) * | 1997-11-19 | 1999-06-08 | Daicel Chem Ind Ltd | ジ(カルボヒドラジド−o,n)カルシウム(ii)硝酸塩一水和物及びその脱水錯体とそれらの製造方法及びガス発生剤組成物 |
| JP2007054328A (ja) * | 2005-08-25 | 2007-03-08 | Sinanen Zeomic Co Ltd | 消臭剤および消臭性樹脂組成物 |
| JP2007223289A (ja) * | 2006-02-27 | 2007-09-06 | Mitsubishi Chemicals Corp | 光学記録媒体の記録層形成用色素、及びそれを用いた光学記録媒体、その光学記録媒体の記録方法 |
| WO2007114184A1 (ja) * | 2006-03-29 | 2007-10-11 | Sekisui Chemical Co., Ltd. | 液晶滴下工法用シール剤、上下導通材料及び液晶表示素子 |
| JP2007297492A (ja) * | 2006-04-28 | 2007-11-15 | Kansai Paint Co Ltd | 硬化型樹脂組成物 |
| WO2007138870A1 (ja) * | 2006-05-26 | 2007-12-06 | Nippon Kayaku Kabushiki Kaisha | 液晶シール剤およびそれを用いた液晶表示セル |
| WO2008016122A1 (fr) * | 2006-08-04 | 2008-02-07 | Mitsui Chemicals, Inc. | Matière d'étanchéité pour cristaux liquides, procédé pour la production d'écrans à cristaux liquides avec celle-ci et écrans à cristaux liquides |
| WO2008018337A1 (fr) * | 2006-08-08 | 2008-02-14 | Mitsubishi Chemical Corporation | Composé complexe chélate d'hydrazide, support d'enregistrement optique utilisant le composé et procédé d'enregistrement associé |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3030499B2 (ja) * | 1997-02-19 | 2000-04-10 | 大塚化学株式会社 | エポキシ樹脂用硬化剤 |
| JP2006058466A (ja) | 2004-08-18 | 2006-03-02 | Sekisui Chem Co Ltd | 液晶表示素子用光硬化性樹脂組成物、液晶表示素子用シール剤、上下導通材料及び液晶表示素子 |
| JP4974344B2 (ja) * | 2006-06-19 | 2012-07-11 | 日本化薬株式会社 | 液晶シール剤及びそれを用いた液晶表示セル |
| CN101356241A (zh) * | 2006-08-08 | 2009-01-28 | 三菱化学株式会社 | 酰肼螯形配位化合物和使用了该化合物的光学记录介质及其记录方法 |
-
2009
- 2009-06-10 JP JP2009139569A patent/JP4961458B2/ja active Active
-
2010
- 2010-05-27 CN CN201080025073.6A patent/CN102459155B/zh active Active
- 2010-05-27 WO PCT/JP2010/059034 patent/WO2010143540A1/ja active Application Filing
- 2010-05-27 KR KR1020127000573A patent/KR101712687B1/ko active IP Right Grant
- 2010-06-07 TW TW099118377A patent/TWI482750B/zh active
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4730154U (ja) * | 1971-05-04 | 1972-12-05 | ||
| JPH05222296A (ja) * | 1992-02-14 | 1993-08-31 | Mitsubishi Yuka Badische Co Ltd | 水性架橋性樹脂組成物 |
| JPH09104687A (ja) * | 1995-10-06 | 1997-04-22 | Daicel Chem Ind Ltd | トリス(カルボヒドラジド−o,n)マグネシウム(ii)硝酸塩錯体とその製造方法及びガス発生剤組成物 |
| WO1998029425A1 (fr) * | 1996-12-26 | 1998-07-09 | Otsuka Kagaku Kabushiki Kaisha | Complexe constitue par manganese et semicarbazide et generateur de gaz pour airbag |
| JPH11152288A (ja) * | 1997-11-19 | 1999-06-08 | Daicel Chem Ind Ltd | ジ(カルボヒドラジド−o,n)カルシウム(ii)硝酸塩一水和物及びその脱水錯体とそれらの製造方法及びガス発生剤組成物 |
| JP2007054328A (ja) * | 2005-08-25 | 2007-03-08 | Sinanen Zeomic Co Ltd | 消臭剤および消臭性樹脂組成物 |
| JP2007223289A (ja) * | 2006-02-27 | 2007-09-06 | Mitsubishi Chemicals Corp | 光学記録媒体の記録層形成用色素、及びそれを用いた光学記録媒体、その光学記録媒体の記録方法 |
| WO2007114184A1 (ja) * | 2006-03-29 | 2007-10-11 | Sekisui Chemical Co., Ltd. | 液晶滴下工法用シール剤、上下導通材料及び液晶表示素子 |
| JP2007297492A (ja) * | 2006-04-28 | 2007-11-15 | Kansai Paint Co Ltd | 硬化型樹脂組成物 |
| WO2007138870A1 (ja) * | 2006-05-26 | 2007-12-06 | Nippon Kayaku Kabushiki Kaisha | 液晶シール剤およびそれを用いた液晶表示セル |
| WO2008016122A1 (fr) * | 2006-08-04 | 2008-02-07 | Mitsui Chemicals, Inc. | Matière d'étanchéité pour cristaux liquides, procédé pour la production d'écrans à cristaux liquides avec celle-ci et écrans à cristaux liquides |
| WO2008018337A1 (fr) * | 2006-08-08 | 2008-02-14 | Mitsubishi Chemical Corporation | Composé complexe chélate d'hydrazide, support d'enregistrement optique utilisant le composé et procédé d'enregistrement associé |
Non-Patent Citations (4)
| Title |
|---|
| CHARLES E. C. ET AL.: "Synthesis of Oligomeric Tin Polyamines and Polyhydrazides", J. MACROMOL. SCI., CHEMISTRY, vol. 7, no. 6, 1973, pages 1349 - 1357 * |
| M. ARSHAD ET AL.: "Cobalt(II) Complexes of Adipyl Dihydrazide", REVUE ROUMAINE DE CHIMIE, vol. 15, no. 12, 1970, pages 1857 - 1862 * |
| M. ARSHAD ET AL.: "Nickel(II) Complexes of Adipyl Dihydrazide", PAK. J. SCI. IND. RES., vol. 12, no. 4, 1970, pages 339 - 341 * |
| R. C. AGGARWAL ET AL.: "Synthesis and Structural Studies of Simple and Mixed Acid Dihydrazide Complexes of Oxovanadium(IV)", TRANSITION METAL CHEMISTRY, vol. 1, no. 4, 1976, pages 161 - 166 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012077720A1 (ja) * | 2010-12-09 | 2012-06-14 | 協立化学産業株式会社 | 光重合開始剤に適した化合物、光重合開始剤及び光硬化性樹脂組成物 |
| JP2013067569A (ja) * | 2011-09-21 | 2013-04-18 | Nippon Kayaku Co Ltd | 新規ヒドラジド化合物及びそれを用いた樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102459155B (zh) | 2015-03-25 |
| CN102459155A (zh) | 2012-05-16 |
| JP4961458B2 (ja) | 2012-06-27 |
| KR20120032522A (ko) | 2012-04-05 |
| KR101712687B1 (ko) | 2017-03-06 |
| TW201113241A (en) | 2011-04-16 |
| JP2010285370A (ja) | 2010-12-24 |
| TWI482750B (zh) | 2015-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6699145B2 (ja) | 光および熱硬化性樹脂組成物 | |
| JP7070552B2 (ja) | 硬化性組成物および構造物 | |
| WO2010143540A1 (ja) | ヒドラジド化合物及びその製造方法、並びにそれを用いた硬化剤、樹脂組成物及び硬化体 | |
| WO2013005471A1 (ja) | 樹脂組成物 | |
| JP6873489B2 (ja) | 樹脂組成物、接着剤、封止材、ダム剤、および半導体装置 | |
| WO2018047597A1 (ja) | 熱硬化型導電性接着剤 | |
| WO2016143777A1 (ja) | 樹脂組成物、接着剤、および封止剤 | |
| JP7297338B2 (ja) | 硬化性樹脂組成物及びその製造方法 | |
| TW200831541A (en) | Sealant for liquid crystal display device and liquid crystal display device | |
| JP2016023256A (ja) | 熱硬化性樹脂組成物、半導体装置及び電気・電子部品 | |
| JP6048717B2 (ja) | 滴下シール剤組成物 | |
| JP2009191231A (ja) | 電子部品接合用接着剤 | |
| TWI543992B (zh) | A liquid crystal sealant and a liquid crystal display unit using the liquid crystal sealant | |
| JP2020504774A (ja) | チオキサントン誘導体光開始剤 | |
| JP5478061B2 (ja) | ヒドラジド系混晶 | |
| JP2014196437A (ja) | 樹脂組成物 | |
| JP2010053330A (ja) | 硬化性組成物、液晶シール剤及び液晶表示素子 | |
| JP6973768B2 (ja) | インクジェット用樹脂組成物、電子部品、電子部品の製造方法 | |
| JP7217565B1 (ja) | 樹脂組成物、接着剤、封止材、硬化物、半導体装置及び電子部品 | |
| JP5445165B2 (ja) | 硬化剤、硬化性樹脂組成物及び半導体用接着剤 | |
| WO2024090259A1 (ja) | 樹脂組成物、接着剤、封止材、硬化物、半導体装置及び電子部品 | |
| JP2004211007A (ja) | 硬化促進剤、エポキシ樹脂組成物および半導体装置 | |
| JP2024034973A (ja) | 硬化性樹脂組成物、硬化物及び物品 | |
| JP2021088672A (ja) | 電子部品用接着剤 | |
| JP2014065712A (ja) | ヒドラジド系混晶 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080025073.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10786071 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20127000573 Country of ref document: KR Kind code of ref document: A |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10786071 Country of ref document: EP Kind code of ref document: A1 |