WO2009082881A1 - Dérivés de tétrahydroimidazo[1,5-a]pyrazine, leurs procédés de préparation et leurs utilisations médicales - Google Patents
Dérivés de tétrahydroimidazo[1,5-a]pyrazine, leurs procédés de préparation et leurs utilisations médicales Download PDFInfo
- Publication number
- WO2009082881A1 WO2009082881A1 PCT/CN2008/001936 CN2008001936W WO2009082881A1 WO 2009082881 A1 WO2009082881 A1 WO 2009082881A1 CN 2008001936 W CN2008001936 W CN 2008001936W WO 2009082881 A1 WO2009082881 A1 WO 2009082881A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- trifluoromethyl
- pyrazine
- acid
- amino
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
Definitions
- Tetrahydroimidazolium fl,5-aj pyridinium derivative preparation method thereof and application thereof in medicine
- the present invention relates to a novel tetrahydroimidazo[1,5-a]pyrazine derivative of the formula (I), a process for the preparation thereof, and a pharmaceutical composition containing the same, and as a therapeutic agent It is used as a dipeptidyl peptidase IV inhibitor.
- Diabetes is a multi-pathogenic metabolic disease characterized by chronic hyperglycemia accompanied by disorders of sugar, fat and protein metabolism caused by defects in insulin secretion and/or function. Diabetes is a very old disease. It is caused by the absolute or relative lack of insulin in the human body. The blood glucose level is increased, and then the sugar is discharged from the urine, and there are polydipsia, polyuria, polyphagia, weight loss, and head. Halo, fatigue and other symptoms.
- Type I diabetics ie, insulin-dependent diabetes mellitus (IDDM) patients produce little or no insulin.
- Insulin is a hormone used in the body to regulate glucose utilization.
- People with type 2 diabetes ie, insulin-independent diabetes mellitus (NIDDM) have the same or higher plasma insulin levels than non-diabetic patients.
- NIDDM insulin-independent diabetes mellitus
- Glucose and lipid metabolism in tissue cells such as muscle, liver, and 25 adipose tissue are stimulating. Even if the plasma insulin level is improved, the patient's significant resistance to insulin cannot be overcome.
- Insulin resistance is mainly due to a decrease in the number of insulin receptors, as well as insulin receptor defects, which have not been understood so far.
- the resistance to insulin responsiveness causes insulin to fail to activate glucose uptake, oxidation, and storage in muscle tissue, and is ineffective in inhibiting lipolysis of adipose tissue and production and secretion of hepatic glucose.
- DPPIV Dipeptidyl peptidase-IV
- a serine protease that cleaves N-terminal dipeptidase in a peptide chain containing a proline residue at the secondary end, although DPPIV has no physiological effects on mammals. It is fully confirmed, but it plays an important role in neuroenzyme metabolism, T-cell activation, cancer cell metastasis into the endothelium and HIV virus entry into lymphoid cells (W098/19998).
- DPPIV can prevent the secretion of glucagon-like peptide (GLP)-1, in particular, it can cleave the N-terminal group-propadipeptide enzyme of GLJP-1 from the active form of GLP- 1(7-36) NH 2 degraded to inactive GLP-1 (9-36) NH 2 (Endocdnology, 1999, 140: 5356 ⁇ 5363). Due to physiological conditions, the half-life of intact GLP-1 in circulating blood is very short, and the inactive metabolites after DPPIV degrades GLP-1 can bind to GLP-1 receptor antagonistic activity GLP-1, thereby shortening the physiology of GLP-1. reaction.
- DPPIV inhibitors can completely protect endogenous and even exogenous GLP-1 from being inactivated by DPPIV, greatly increasing the physiological activity of GLP-1 (5 to 10 times), due to GLP-1 secretion of pancreatic insulin. It is an important stimulator and can directly affect the distribution of glucose. DPPIV inhibitors play a very good role in the treatment of non-insulin-dependent diabetes mellitus (NIDDM) (US6110949).
- NIDDM non-insulin-dependent diabetes mellitus
- DPP-IV inhibitors have been disclosed (US5462928, US5543396, WO9515309, WO2003004498, WO2003082817, WO2004032836, WO2004085661), wherein the DPPIV inhibitor MK-0431 produced by Merck Company shows good DPPIV inhibitory activity and selectivity, and Listed in 2006.
- An object of the present invention is to provide a compound having a therapeutic or palliative drug which inhibits DPPIV activity and which can be used for diabetes or the like. Summary of the invention
- R 1 is selected from a hydrogen atom, an alkyl group, a trifluoromethyl group, a cycloalkyl group, an aryl group or a heteroaryl group, wherein the alkyl group, heterocycloalkyl group, aryl group, heteroaryl group are optionally further selected by one or more Substituted by a substituent of a halogen, a cyano group, an aryl group, a hydroxyl group or an amino group, preferably a trifluoromethyl group;
- R 2 is selected from the group consisting of hydroxyl, amino, alkyl, alkoxy, cyclodecyl, heterocycloalkyl, aryl, heteroaryl or -NR 4 R 5 , wherein alkyl, decyloxy, cycloalkyl, hetero a cycloalkyl, aryl or heteroaryl group optionally further one or more Substituted from halogen, amino, cyano, hydroxy, alkyl, cycloalkyl, alkoxy, aryl, heteroaryl, -NR 4 R 5 , -OC(0)OR ⁇ carboxylic acid or carboxylic acid ester Substituted by
- R 3 is selected from a hydrogen atom or an alkyl group
- R 4 and R 5 are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocyclic fluorenyl group, an aryl group or a heteroaryl group, wherein an alkyl group, a cycloalkyl group, a heterocyclic fluorenyl group, an aryl group or a heteroaryl group.
- R 6 is selected from halogen, cyano, hydroxy, decyl or decyloxy, wherein alkyl or alkoxy is unsubstituted or substituted with one or more halogens;
- R 7 is a sulfhydryl group
- R 8 is an alkyl group or a cycloalkyl group.
- the pharmaceutically acceptable salt described in the present invention is a salt of the compound of the present invention and an acid selected from the group consisting of malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, Acetic acid or trifluoroacetic acid.
- Typical compounds of the invention include, but are not limited to:
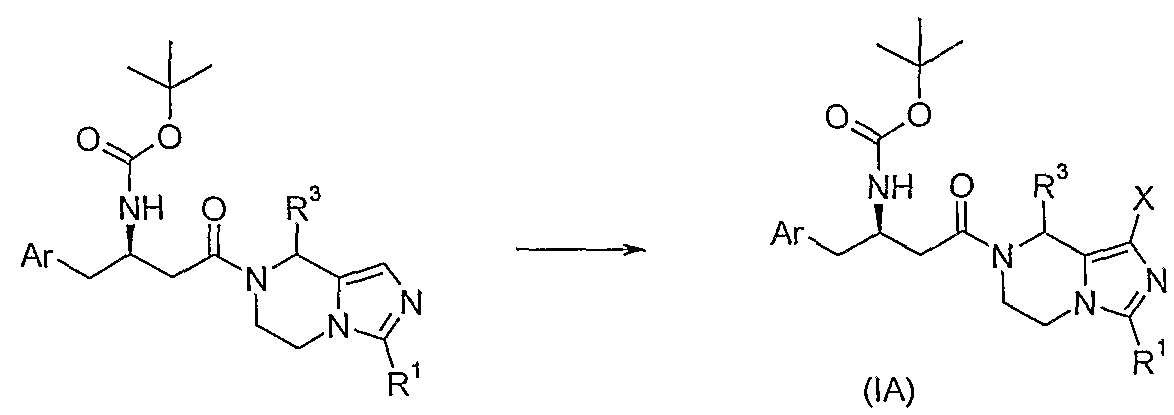
- the present invention includes a compound represented by the following formula (IA) which is an intermediate for the synthesis of the compound of the formula (I):
- Ar is a phenyl group, the phenyl group of which is unsubstituted or further substituted by 1 to 5 R 6 ;
- R 1 is selected from a hydrogen atom, an alkyl group, a trifluoromethyl group, a cycloalkyl group, an aryl group or a heteroaryl group, wherein the alkyl group, heterocycloalkyl group, aryl group, heteroaryl group is optionally further selected by one or more Substituted by a substituent of a halogen, a cyano group, an aryl group, a hydroxyl group or a chloro group, preferably a trifluoromethyl group;
- R 3 is selected from a hydrogen atom or an alkyl group
- R 6 is selected from halogen, cyano, hydroxy, alkyl or alkoxy, wherein the fluorenyl or alkoxy group is unsubstituted or is optionally further substituted by one or more halogens;
- X is a halogen.
- the present invention includes a compound represented by the following formula (IB) which is an intermediate for the synthesis of the compound of the formula (I):
- R 1 is selected from a hydrogen atom, a fluorenyl group, a trifluoromethyl group, a cycloalkyl group, an aryl group or a heteroaryl group, wherein the alkyl group, heterocycloalkyl group, aryl group, heteroaryl group are optionally further selected by one or more Substituted by a substituent of a halogen, a cyano group, an aryl group, a hydroxyl group or an amino group, preferably a trifluoromethyl group;
- R 2 is selected from the group consisting of hydroxyl, amino, alkyl, alkoxy, cycloalkyl, heterocycloalkyl, aryl, heteroaryl or -NR 4 R 5 , wherein alkyl, decyloxy, cycloalkyl, hetero
- the cycloalkyl, aryl or heteroaryl group is further optionally further selected from one or more selected from the group consisting of halogen, amino, cyano, hydroxy, alkyl, cycloalkyl, alkoxy, aryl, heteroaryl, -NR 4 Substituted with a substituent of R 5 , —OC(0)OR 8 , a carboxylic acid or a carboxylic acid ester;
- R 3 is selected from a hydrogen atom or an alkyl group
- R 4 and R 5 are each independently selected from a hydrogen atom, an alkyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group or a heteroaryl group, wherein an alkyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group or a heteroaryl group.
- R 4 and R 5 together form a 4 to 8 membered heterocyclic group, wherein the 4 to 8 membered heterocyclic ring contains one or more N, 0, S atoms, and the 4 to 8 membered heterocyclic ring is optionally further One or more selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, heterocycloalkyl, heteroaryl, carbonyl, hydroxyalkyl, -S0 2 R 7 , -NR 4 R 5 , -C(0)NR 4 R 5 , -C(0)R 7 , substituted with a substituent of a carboxylic acid or a carboxylic acid ester; R 7 is an alkyl group;
- R 8 is selected from an alkyl group or a cycloalkyl group.
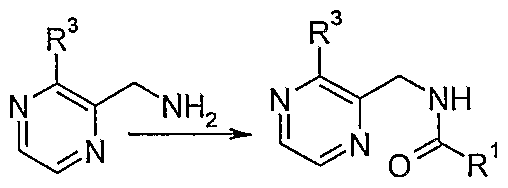
- the anhydride is added dropwise to the starting pyrazine 2-methylamine under an ice bath, and then reacted at room temperature to form an amide product;
- the imidazo[1,5-a]pyrazine ring is reduced in hydrogen in a solvent of palladium on carbon to form an R 1 , R 3 substituted tetrahydroimidazo[1,5-a]pyrazine product;
- R 1 , R 3 substituted imidazo[1,5-a]pyrazine product is dissolved in a dichloromethane solvent, and in the condensing agent bis(2-oxo-3-oxazolidinyl)phosphoryl chloride and Condensation reaction with carboxylic acid under the action of triethylamine;
- R 1 -substituted imidazo[1,5-a]pyrazine is hydrogenated and reduced in an ethanol solvent at room temperature, and then reacted with di-tert-butyl dicarbonate in an ethanol solvent to protect the amino group to obtain an amino-protected R 1 substituted tetrahydroimidazo[1,5-a]pyridinium;
- the obtained halogenated product is reacted in a methanol solvent with an octacarbonyl dicobalt and chloroacetate in a carbon monoxide atmosphere to obtain an ester-substituted tetrahydroimidazo[1,5-a]pyrazine;
- the obtained carboxylic acid compound is reacted with a haloalkyl group in a dry ice-acetone bath to obtain an alkyl-substituted acid;
- the acid of the alkyl substituted product can be further esterified to give a ketone-substituted tetrahydroimidazo[1,5-a]pyrazine;
- the alkyl-substituted acid can be used in a dichloromethane solvent in the condensation reagent bis(2-oxo-3-oxazolidine Reaction with N-methoxymethylamine under the action of hypophosphoryl chloride;
- the product obtained by the condensation reaction is reacted with a Grignard reagent in a tetrahydrofuran solvent to obtain a ketone-substituted tetrahydroimidazo[1,5-a]pyrazine;
- keto-substituted tetrahydroimidazo[1,5-a]pyrazine is deprotected under acidic conditions to give the intermediate (IB).
- another aspect of the present invention is a process for the preparation of the compound (I) of the formula, which comprises:
- the intermediate ( ⁇ ) is reacted with chloroacetate in a carbon monoxide atmosphere under an action of an oil bath under an action of octacarbonyl octacobalt in an oil bath, and then hydrolyzed to a carboxylic acid at room temperature under basic conditions;
- Another aspect of the invention is a process for the preparation of a compound of formula (I), which comprises the following steps -
- the intermediate (IB) is condensed with a carboxylic acid under the conditions of a condensing reagent bis(2-oxo-3-oxazolidinyl)phosphinic chloride, and the obtained product is further subjected to removal of an amino protecting group under acidic conditions to give a general formula.
- Compound (1) Further, a process for producing the compound of the above formula (I), which further comprises an acid addition product salt of the compound of the formula (I).
- the salt is a salt of the above compound with an acid selected from the group consisting of phosphoric acid, malic acid, lactic acid, maleic acid, hydrochloric acid, methanesulfonic acid, sulfuric acid, phosphoric acid, citric acid, tartaric acid, acetic acid or trifluoroacetic acid.
- the preferred acid is hydrochloric acid.
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier or excipient.
- the present invention relates to the use of a compound of the present invention or a pharmaceutically acceptable salt thereof for the preparation of a medicament for the treatment of diabetes mellitus, hyperglycemia, obesity or insulin resistance.
- One aspect of the invention is a method for inhibiting the catalytic activity of dipeptidyl peptidase IV, characterized in that the dipeptidyl peptidase IV is contacted with a compound or salt according to any one of the formula (I).
- Another aspect of the invention is the use of a compound, salt or pharmaceutical composition according to any one of the formula (I) for the treatment of a disease such as type II diabetes, hyperglycemia, obesity or insulin resistance.
- a disease such as type II diabetes, hyperglycemia, obesity or insulin resistance.
- Alkyl means a saturated aliphatic hydrocarbon group including straight chain and branched chain groups of 1 to 20 carbon atoms. Preference is given to alkyl groups having 1 to 10 carbon atoms, such as methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl, tert-butyl, pentyl and the like. More preferred are lower alkyl groups having 1 to 4 carbon atoms, such as methyl, ethyl, propyl, 2-propyl, n-butyl, isobutyl or t-butyl groups and the like.
- the alkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, heterocycle. Alkyl, heteroaryl, carbonyl, hydroxydecyl, -S0 2 R 7 , -NR 4 R 5 , -C(0)NR 4 R 5 , -C(0)R 7 , -OC(0)OR 8 , a carboxylic acid or a carboxylic acid ester.
- Cycloalkyl means a 3 to 8 membered all carbon monocyclic, all carbon 5/6 or 6/6 fused or polycyclic fused ring (“fused" ring system means each in the system The rings share an adjacent pair of carbon atoms) groups with other rings in the system, wherein one or more of the rings may contain one or more double bonds, but none of the rings have a fully conjugated pi-electron system.
- cycloalkyl group examples include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclopentene group, a cyclohexanyl group, a cyclohexadiene group, an adamantane, a cycloheptane, a cycloheptatriene or the like.
- the cycloalkyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, heterocyclic.
- Aryl means a group having at least one aromatic ring structure, that is, an aromatic ring having a conjugated ⁇ -electron system, including a carbocyclic aryl group, a heteroaryl group, and a biaryl group.
- the alkynyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of 3 ⁇ 4, hydroxy, amino, alkoxy, alkyl, cyano, aryl, hetero Cycloalkyl, heteroaryl, carbonyl, hydroxydecyl, -S0 2 R 7 , -NR 4 R 5 , -C(0)NR 4 R 5 , -C(0)R 7 , carboxylic acid or carboxylate .
- Heteroaryl means an aryl group having from 1 to 3 heteroatoms as ring atoms, the remaining ring atoms being carbon, and heteroatoms including oxygen, sulfur and nitrogen.
- the ring may be a 5- or 6-membered ring.
- the heterocyclic aryl group include a furyl group, a thienyl group, a pyridyl group, a pyrrole, an N-alkylpyrrolyl group, a pyrimidinyl group, a pyrazinyl group, an imidazolyl group and the like.
- the heteroaryl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, hetero Cyclodecyl, heteroaryl, carbonyl, hydroxyalkyl, -S0 2 R 7 , -NR 4 R 5 , -C(0)NR 4 R 5 , -C(0)R 7 , carboxylic acid or carboxylate .
- Heterocyclic fluorenyl means a monocyclic or fused ring radical having from 5 to 9 ring atoms in the ring wherein one or two ring atoms are selected from nitrogen, oxygen or S(0)n (where n is an integer) From 0 to 2), the remaining ring atoms are carbon. These rings may also have one or more double bonds. However, these rings do not have a fully conjugated ⁇ -electron system.
- Unsubstituted heterocyclic fluorenyl includes, but is not limited to, pyrrolidinyl, piperidino, piperazino, morpholinyl, thiomorpholinyl, homopiperazine, etc.
- heterocycloalkyl may be substituted or Unsubstituted.
- the alkynyl group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, heterocycle.
- Haldroxy means an -OH group.
- Alkoxy means -0-(alkyl) and -0-(unsubstituted cycloalkyl). Representative examples include, but are not limited to, methoxy, ethoxy, propoxy, butoxy, cyclopropoxy, cyclobutoxy, cyclopentyloxy, cyclohexyloxy, and the like.
- the alkoxy group may be substituted or unsubstituted, and when substituted, the substituent is preferably one or more, independently selected from the group consisting of halogen, hydroxy, amino, alkoxy, alkyl, cyano, aryl, Heterocycloalkyl, heteroaryl, carbonyl, hydroxyalkyl, -S0 2 R 7 , -NR 4 R 5 , -C(0)NR 4 R 5 , -C(0)R 7 , carboxylic acid or carboxylic acid ester.
- Halogen means fluoro, chloro, bromo or iodo, preferably fluoro or chloro.
- Trifluoromethyl means -CF 3 .
- Amino means -N3 ⁇ 4.
- Hydroalkyl means an alkyl group substituted by a hydroxy group.
- “Pharmaceutical composition” means a mixture of one or more of the compounds described herein, or a physiologically/pharmaceutically acceptable salt or prodrug thereof, with other chemical components, such as a physiological/pharmaceutically acceptable carrier. And excipients.
- the purpose of the pharmaceutical composition is to facilitate the administration of the compound to the organism.
- the preparation method of the compound (I) of the general formula of the present invention comprises the following steps:
- the raw pyrazine 2-methylamine is added dropwise with an acid anhydride in an ice bath, and then reacted at room temperature to form an amide product; after the amide product is mixed with phosphorus oxychloride at room temperature, phosphorus pentoxide is added, and the mixture is heated and refluxed.
- An imidazo[1,5-a]pyrazine ring is formed; the imidazo[1,5-a]pyrazine ring is reduced in hydrogen in a solvent of palladium on carbon to form an R 1 -substituted tetrahydroimidazole.
- [1,5-a]pyrazine product the R 1 -substituted tetrahydroimidazo[1,5-a]pyrazine product in dichloromethane solvent and in the condensation reagent bis(2-oxo-3- Condensation reaction with carboxylic acid under the action of oxazolidinylphosphoryl chloride and triethylamine; the obtained condensation product is reacted with halogenated succinimide in an anhydrous ethanol solvent at room temperature to form a compound of formula (IA);
- the compound of the formula (IA) is reacted with chloroacetate in a carbon monoxide atmosphere under an action of an oil bath in an oil bath under an action of dicotid octacarbonyl, and then hydrolyzed to a carboxylic acid under acidic conditions at room temperature;
- An acid compound condensation reaction with an amine or with an alcohol at room temperature, or with a 1-halocarbonate followed by an acid The amino protecting group is removed under the neutral
- the preparation method of the compound (I) of the formula of the present invention comprises the following steps -
- [1,5- a]pyrazine the obtained amino-protected R 1 -substituted tetrahydroimidazo[1,5-a]pyrazine is reacted with a halogenated succinimide at room temperature to obtain a halogenated group.
- the obtained halogenated product is reacted in a solvent of methanol with octacarbonyl hexacarbonyl and chloroacetate under a carbon monoxide atmosphere to obtain an ester-substituted tetrahydroimidazo[1,5-a]pyrazine.
- the resulting ester-substituted tetrahydroimidazo[l,5-a]pyrazine is hydrolyzed under basic conditions Acid
- the obtained carboxylic acid compound is reacted with a haloalkyl group in a dry ice-acetone bath to obtain an alkyl-substituted product; the carboxyl group in the alkyl-substituted product can be further esterified to obtain an intermediate (IB); It can be reacted with N-methoxymethylamine in a dichloromethane solvent under the action of a condensation reagent bis(2-oxo-3-oxazolidinyl)phosphoryl chloride; the product obtained by the condensation reaction and the Grignard reagent Reaction in tetrahydrofuran solvent to give a ketone-substituted tetrahydroimidazo[1,5-a]pyrazine; removing the amino-protecting group under acidic conditions with a ketone-substituted tetrahydroimidazo[1,5-a]pyrazine To obtain a compound of the formula (IB); the intermediate compound (IB) is con
- the compound of the formula (I) is purified and reacted in an acid solution of methanol, dichloromethane or ethyl acetate to obtain an acid addition product salt.
- the structure of the example compounds was determined by nuclear magnetic resonance (H NMR) or mass spectrometry (MS).
- the iHNMR shift ( ⁇ ) is given in parts per million (ppm). !
- the HNMR was measured by a Bruker AVANCE-400 nuclear magnetic instrument.
- the solvent was deuterated methanol (CD 3 OD), deuterated chloroform (CDC1 3 ), and the internal standard of hexamethyl dimethyl sulfoxide (DMSO-d6) was silane (TMS), chemical shifts are 10- 6 (ppm) is given as a unit;
- MS FINNIGAN LCQAd (ESI) mass spectrometer manufactured by Therm, model: Finnigan LCQ advantage MAX;
- the IC 50 value was determined using a NovoStar plate reader (BMG, Germany);
- the nitrogen atmosphere means that the reaction flask is connected to a nitrogen balloon of about 1 L volume.
- the hydrogen atmosphere means that the reaction flask is connected to a hydrogen balloon of about 1 L volume.
- 2,2,2-Trifluoro-N-pyrazine-2-methyl-carboxamide li (21.0 g, 100 mmol) was added to the reaction flask at room temperature, 100 mL of phosphorus oxychloride was added, and stirred at room temperature 30 After a minute, phosphorus pentoxide (17.8 g, 125 mmol) was added, and the reaction was heated under reflux for 5 hours. The reaction was traced by thin layer chromatography until the starting material disappeared, the solvent phosphorus trichloride was removed, and the reaction system was quenched with deionized water. 20% sodium hydroxide solution was adjusted to pH 5-6 in an ice-bath, and extracted with ethyl acetate (250 mL ⁇ 4). Purification gave the title product 3-trifluoromethyl-imidazo[1,5-a]pyrazine lj (12.0 g, yellow solid).
- 3-trifluoromethyl-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine 3-trifluoromethyl-imidazo[1,5-a]pyrazine lj (12.0 g, 64.2 mmol) was dissolved in 150 mL of absolute ethanol with stirring, and 500 mg of 10% palladium on carbon was added and stirred under a hydrogen atmosphere overnight.
- Morpholine (19 mg, 0.218 mmol) and bis(2-oxo-3-oxazolidinyl) Phosphonoyl chloride (53.5 mg, 0.218 mmol) was dissolved in 5 mL of dichloromethane with stirring.
- Triethylamine (0.1 mL, 0.65 mmol) was added and allowed to react overnight at room temperature. The reaction was traced by thin layer chromatography until the starting material disappeared.
- reaction was traced by thin layer chromatography until the starting material disappeared.
- the reaction mixture was concentrated, and then purified mjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj
- reaction was traced by thin layer chromatography until the material disappeared.
- the reaction mixture was purified to silica gel column chromatography toiel [1,5-a]pyrazin-7-yl)-3-oxo-1-(2,4,5-trifluoro-benzyl)-propyl]-carbamic acid tert-butyl ester 8a (150 mg, White solid), Yield: 98.6 %.
- Triethylamine (0.25 mL, 1.62 mmol) was added and allowed to react at room temperature overnight, and the reaction was traced by thin layer chromatography until the starting material disappeared.
- the reaction mixture was concentrated under reduced vacuolululululululululululululu 8H-imidazo[1,5-a]pyrazin-7-yl)-3-oxo-1-(2,4,5-trifluoro-benzyl)-propyl]-carbamic acid tert-butyl ester 9a (120 mg, white solid), Yield: 77%.
- Triethylamine (0.36 g, 3.6 mmol) was added at room temperature, and the reaction mixture was stirred at room temperature for 2 hr. (cyclopropylcarbamoyl)-3-trifluoromethyl-5,6-dihydro-8H-imidazo[1,5-a]pyrazine-7-yl]-3-oxo-1-( 2,4,5-Trifluoro-benzyl)-propyl]-carbamic acid tert-butyl ester 15a (0.1 g, oily liquid), yield: 45 %.
- Triethylamine (0.25 mL, 1.62 mmol)
- bis(2-oxo-3-oxazolidinyl)phosphinic acid chloride (0.138 g, 0.54 mmol) was added and allowed to react at room temperature for 20 hours.
- the reaction mixture was chromatographed until the material was evaporated, and the residue was evaporated.
- reaction mixture was concentrated, and then purified mjjjjjjjjj ,6-Dihydro-8H-imidazo[1,5-a]pyrazin-7-yl]-3-oxo small (2,4,5-trifluorobenzyl)-propyl]-carbamic acid Butyl ester 29a (0.1 g, white solid).
- [1,5-a]pyrazine-1-ethyl 1-(1-ethoxyoxy)carboxylate 34a (0.26 g, 0.39 mmol) and 5 mL of ethyl acetate were added to the reaction flask, and then 3 mL of 6.5 N was added.
- [1,5-a]pyrazine-1-carboxylic acid tert-butyl ester 36a (0.094 g, 0.15 mmol) and 10 mL of ethyl acetate were added to the reaction flask, and 3 mL of 5.5 N hydrogen chloride was added to the reaction flask under ice bath. After the dropwise addition, the ice bath was removed, and the mixture was stirred at room temperature for 3 hours. The reaction mixture was evaporated to dryness, and the mixture was evaporated to dryness.
- the reaction tube was taken out from the oil bath, and after it was cooled to room temperature, 40 mL of water was added thereto, and the mixture was extracted with ethyl acetate (25 mL ⁇ 3).
- the product in the aqueous phase was detected by thin layer chromatography, and the organic phase was collected and washed with water (20 mL> ⁇ 2), the organic phase was combined, dried with EtOAcjHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH 4,5-trifluorophenyl)butyryl)-3-(trifluoromethyl)-5,6,7,8-tetrahydroimidazo[1,5-a]pyrazine-1-carboxylic acid (isopropyl Oxyformyloxy)ethyl ester 37a (0.29 g, pale yellow solid), yield: mp.
- the reaction tube was taken out from the oil bath, and after the reaction mixture was cooled to room temperature, 40 mL of water was added thereto, and the mixture was extracted with ethyl acetate (30 mL ⁇ 3), and the organic phase was combined, washed with 30 mL of water, and then washed with 30 mL of saturated brine, and the organic phase was combined. Drying over anhydrous magnesium sulfate, EtOAc (EtOAc m.
- the inhibition rate or semi-inhibitory concentration of each compound, IC 5Q (the concentration of the compound measured when the enzyme activity is inhibited to 50%), is determined by a fixed amount of the enzyme mixed substrate and different concentrations of the test compound.
- Tris buffer Prepare 100 mL of 2 mM Tris buffer, dissolve 0.0242 g Tris in approximately 90 mL of deionized water, adjust pH to 8.00 with HC1 and NaOH, and finally add deionized water to 100 mL.
- DPPIV enzyme CalBiochem Catalog no. 317630, dissolved in Tris buffer to 2 mM.
- DPPIV—GloTM substrate Promega Catalog no. G8350
- DPPIV -GloTM Buffer Promega Catalog no.G8350
- the ratio of mixed substrate and DPPIV-Glo. is 1:49. Mix the vibrations to mix thoroughly. Before use at room temperature Let stand for 30-60 minutes.
- the compound of the present invention has a half-inhibitory concentration IC 5C value ranging from 0.005 ⁇ M to 0.216 ⁇ M, and most of the compounds of the present invention have good inhibitory activity against DPPIV compared with the IC 5 o value of 0.023 ⁇ M of MK-0431. .
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Diabetes (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description









































































Claims





















Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2008342461A AU2008342461B2 (en) | 2007-12-26 | 2008-11-27 | Tetrahydro-imidazo(1,5-a)pyrazine derivatives, preparation methods and medical uses thereof |
| KR1020107014988A KR101685103B1 (ko) | 2007-12-26 | 2008-11-27 | 테트라하이드로-이미다조[1,5-a]피라진 유도체, 이의 제조 방법 및 의학적 용도 |
| UAA201008000A UA101002C2 (ru) | 2007-12-26 | 2008-11-27 | ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗО[1,5-а]ПИРАЗИНА, СПОСОБ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ |
| JP2010539997A JP5481008B2 (ja) | 2007-12-26 | 2008-11-27 | テトラヒドロ・イミダゾ[1,5−α]ピラジン誘導体、その調製プロセスおよび医薬的使用 |
| CA2706735A CA2706735C (en) | 2007-12-26 | 2008-11-27 | Tetrahydro-imidazo[1,5-.alpha.]pyrazine derivatives, preparation process and medicinal use thereof |
| US12/743,795 US8207161B2 (en) | 2007-12-26 | 2008-11-27 | Tetrahydro-imidazo[1,5-α]pyrazine derivatives, preparation process and medicinal use thereof |
| MX2010006438A MX2010006438A (es) | 2007-12-26 | 2008-11-27 | Derivados de tetrahidro-imidazo [1,5-a] pirazina proceso de preparacion y su uso medicinal. |
| BRPI0821440-9A BRPI0821440A2 (pt) | 2007-12-26 | 2008-11-27 | Derivados de tetra-hidro-imidazo[1,5-a] pirazina, processo de preparação e uso medicinal dos mesmos |
| RU2010125703/04A RU2483070C2 (ru) | 2007-12-26 | 2008-11-27 | ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗО[1,5-a]ПИРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ ИХ В МЕДИЦИНЕ |
| EP08868957.5A EP2230241B1 (en) | 2007-12-26 | 2008-11-27 | Tetrahydro-imidazo[1,5-a]pyrazine derivatives, preparation methods and medical uses thereof |
| CN2008800097616A CN101641361B (zh) | 2007-12-26 | 2008-11-27 | 四氢咪唑并[1,5-a]吡嗪类衍生物,其制备方法及其在医药上的应用 |
| ES08868957.5T ES2529435T3 (es) | 2007-12-26 | 2008-11-27 | Derivados de tetrahidro-imidazo[1,5-A]pirazina, procedimientos de preparación y usos médicos de los mismos |
| HK10102993.1A HK1134817A1 (en) | 2007-12-26 | 2010-03-23 | Tetrahydro-imidazo[1,5-a]pyrazine derivatives, preparation methods and medical uses thereof |
| ZA2010/04154A ZA201004154B (en) | 2007-12-26 | 2010-06-10 | Tetrahydro-imidazo[1,5-a]pyrazine derivatives,preparation methods and medical uses thereof |
| US13/463,532 US8513411B2 (en) | 2007-12-26 | 2012-05-03 | Tetrahydro-imidazo[1,5-α] pyrazine derivatives, preparation process and medicinal use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNA2007103023359A CN101468988A (zh) | 2007-12-26 | 2007-12-26 | 哌嗪类衍生物,其制备方法及其在医药上的应用 |
| CN200710302335.9 | 2007-12-26 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/743,795 A-371-Of-International US8207161B2 (en) | 2007-12-26 | 2008-11-27 | Tetrahydro-imidazo[1,5-α]pyrazine derivatives, preparation process and medicinal use thereof |
| US13/463,532 Division US8513411B2 (en) | 2007-12-26 | 2012-05-03 | Tetrahydro-imidazo[1,5-α] pyrazine derivatives, preparation process and medicinal use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009082881A1 true WO2009082881A1 (fr) | 2009-07-09 |
Family
ID=40823770
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2008/001936 WO2009082881A1 (fr) | 2007-12-26 | 2008-11-27 | Dérivés de tétrahydroimidazo[1,5-a]pyrazine, leurs procédés de préparation et leurs utilisations médicales |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8207161B2 (zh) |
| EP (1) | EP2230241B1 (zh) |
| JP (1) | JP5481008B2 (zh) |
| KR (1) | KR101685103B1 (zh) |
| CN (2) | CN101468988A (zh) |
| AU (1) | AU2008342461B2 (zh) |
| BR (1) | BRPI0821440A2 (zh) |
| CA (1) | CA2706735C (zh) |
| ES (1) | ES2529435T3 (zh) |
| HK (1) | HK1134817A1 (zh) |
| MX (1) | MX2010006438A (zh) |
| PT (1) | PT2230241E (zh) |
| RU (1) | RU2483070C2 (zh) |
| UA (1) | UA101002C2 (zh) |
| WO (1) | WO2009082881A1 (zh) |
| ZA (1) | ZA201004154B (zh) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008157751A2 (en) | 2007-06-21 | 2008-12-24 | Cara Therapeutics, Inc. | Substituted imidazoheterocycles |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012019427A1 (zh) | 2010-08-09 | 2012-02-16 | 上海恒瑞医药有限公司 | 酞嗪酮类衍生物、其制备方法及其在医药上的应用 |
| US20120122875A1 (en) * | 2009-05-27 | 2012-05-17 | Jiangsu Hengrui Medicine Co., Ltd. | Salts of Methyl (R)-7-[3-Amino-4-(2,4,5-Trifluoro-Phenyl)-Butyryl]-3-Trifluoromethyl-5,6,7,8-Tetrahydro-Imidazo[1,5-A]Pyrazine-1-Carboxylate |
| EP2402342A4 (en) * | 2010-03-08 | 2012-09-19 | Jiangsu Hengrui Medicine Co | PHARMACEUTICAL COMPOSITION FOR TREATING TYPE 2 DIABETES |
| EP2551259A1 (en) * | 2010-03-25 | 2013-01-30 | Zhejiang Jiuzhou Pharmaceutical Co., Ltd. | Sitagliptin intermediate compounds, preparation methods and uses thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8518946B2 (en) | 2009-03-05 | 2013-08-27 | Jiangsu Hengrui Medicine Co., Ltd. | Salts of (R)-7-[3-amino-4-(2,4,5-trifluorophenyl)butanoyl]-3-trifluoromethyl-5,6,7,8-tetrahydroimidazo[1,5-a] pyrazine-1-carboxylic acid, preparation method and medical use thereof |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| TWI462925B (zh) * | 2010-09-27 | 2014-12-01 | Jiangsu Hengrui Medicine Co | 治療2型糖尿病的藥物組合物 |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| CN114478536A (zh) * | 2020-10-28 | 2022-05-13 | 上海森辉医药有限公司 | 四氢吡嗪稠环衍生物的制备方法 |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101961336B (zh) * | 2009-07-21 | 2012-05-30 | 江苏恒瑞医药股份有限公司 | 治疗哺乳动物包括人2型糖尿病的药物组合物 |
| CN102471294B (zh) * | 2010-04-12 | 2013-11-20 | 上海源力生物技术有限公司 | 手性环状β-氨基芳基丁酸衍生物、其制备方法及通过其制备手性β-氨基芳基丁酸衍生物的方法 |
| TWI494313B (zh) * | 2010-12-29 | 2015-08-01 | Jiangsu Hengrui Medicine Co | 治療包括人類之哺乳動物中第2型糖尿病的醫藥組成物 |
| WO2012088682A1 (en) * | 2010-12-29 | 2012-07-05 | Shanghai Fochon Pharmaceutical Co Ltd. | 2-(3-aminopiperidin-1-yl)-[1,2,4]triazolo[1,5-c]pyrimidine-5,7(3h,6h)-dione derivates as dipeptidyl peptidase iv(dpp-iv) inhibitors |
| SI2736909T1 (sl) | 2011-07-27 | 2017-08-31 | Farma Grs, D.O.O. | Proces za pripravo sitagliptina in njegovih farmacevtsko sprejemljivih soli |
| CN103087067A (zh) * | 2012-08-02 | 2013-05-08 | 盛世泰科生物医药技术(苏州)有限公司 | 作为二肽基酶抑制剂的化合物及其组合物,以及它们的用途 |
| CN103724352B (zh) * | 2012-10-16 | 2017-11-28 | 江苏盛迪医药有限公司 | 一种dpp‑iv抑制剂的中间体、其制备方法和通过其制备dpp‑iv抑制剂的方法 |
| TWI500613B (zh) | 2012-10-17 | 2015-09-21 | Cadila Healthcare Ltd | 新穎之雜環化合物 |
| JP6412563B2 (ja) * | 2013-10-30 | 2018-10-24 | シャンハイ ヘンルイ ファーマスーティカル カンパニー リミテッドShanghai Hengrui Pharmaceutical Co., Ltd. | ピラゾロピリミドンまたはピロロトリアゾン誘導体、その製造方法、およびそれらの医薬適用 |
| CN103664962A (zh) * | 2013-12-20 | 2014-03-26 | 南京华威医药科技开发有限公司 | 哌嗪类衍生物 |
| EP3087076B1 (en) * | 2013-12-23 | 2017-09-27 | Merck Patent GmbH | Imidazopyrazinone derivatives |
| CN103910734B (zh) * | 2014-03-28 | 2016-01-13 | 南京华威医药科技开发有限公司 | 一种具有哌嗪结构的dpp-4抑制剂 |
| KR101709127B1 (ko) * | 2015-06-16 | 2017-02-22 | 경동제약 주식회사 | Dpp-iv 억제제의 제조를 위한 신규 중간체, 이의 제조방법 및 이를 이용한 dpp-iv 억제제의 제조방법 |
| CN108658990B (zh) * | 2017-03-31 | 2021-03-23 | 南京科技职业学院 | 一类新型咪唑并[1,5-a]吡嗪类布鲁顿激酶抑制剂 |
| AU2018307761A1 (en) * | 2017-07-24 | 2020-02-20 | Isocure Biosciences Inc. | Inhibitors of mutant isocitrate dehydrogenases and compositions and methods thereof |
| EP3773587A4 (en) | 2018-03-29 | 2021-09-29 | Board of Regents, The University of Texas System | IMIDAZOPIPERAZINE INHIBITORS OF TRANSCRIPTION ACTIVATION PROTEINS |
| US10899769B2 (en) | 2018-04-06 | 2021-01-26 | Board Of Regents, The University Of Texas System | Imidazopiperazinone inhibitors of transcription activating proteins |
| CN108383845B (zh) * | 2018-04-26 | 2020-11-06 | 盛世泰科生物医药技术(苏州)有限公司 | 盛格列汀盐的晶型和无定形及其制备方法和用途 |
| CN113620957B (zh) * | 2020-05-08 | 2022-04-19 | 盛世泰科生物医药技术(苏州)有限公司 | 一种胺甲基吡嗪类化合物的制备方法 |
| CN113773323B (zh) * | 2020-06-10 | 2023-05-12 | 江苏恒瑞医药股份有限公司 | 3r-氨基取代丁酰胺衍生物的制备方法 |
| WO2024015889A2 (en) * | 2022-07-14 | 2024-01-18 | The Scripps Research Institute | Small molecule regulators of alveolar type 2 cell proliferation for the treatment of pulmonary diseases |
| CN115583935A (zh) * | 2022-11-03 | 2023-01-10 | 浙江工业大学 | 一种4-(2,4,5-三氟苯基)-3-氧丁酸酯的制备方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995015309A1 (en) | 1993-12-03 | 1995-06-08 | Ferring B.V. | Dp-iv-serine protease inhibitors |
| US5462928A (en) | 1990-04-14 | 1995-10-31 | New England Medical Center Hospitals, Inc. | Inhibitors of dipeptidyl-aminopeptidase type IV |
| US5543396A (en) | 1994-04-28 | 1996-08-06 | Georgia Tech Research Corp. | Proline phosphonate derivatives |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| US6110949A (en) | 1999-06-24 | 2000-08-29 | Novartis Ag | N-(substituted glycyl)-4-cyanothiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| WO2003004498A1 (en) | 2001-07-06 | 2003-01-16 | Merck & Co., Inc. | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2003082817A2 (en) | 2002-03-25 | 2003-10-09 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2004032836A2 (en) | 2002-10-07 | 2004-04-22 | Merck & Co., Inc. | Antidiabetic beta-amino heterocylcic dipeptidyl peptidase inhibitors |
| WO2004085661A2 (en) | 2003-03-24 | 2004-10-07 | Merck & Co., Inc | Process to chiral beta-amino acid derivatives |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4504924B2 (ja) * | 2002-12-20 | 2010-07-14 | メルク・シャープ・エンド・ドーム・コーポレイション | 糖尿病の治療および予防のためのジペプチジルペプチダーゼ阻害薬としての3−アミノ−4−フェニルブタン酸誘導体 |
| ATE545647T1 (de) * | 2006-12-22 | 2012-03-15 | Actelion Pharmaceuticals Ltd | 5,6,7,8-tetrahydro-imidazoä1,5-aüpyrazinderivat |
| NZ582760A (en) * | 2007-06-21 | 2011-12-22 | Cara Therapeutics Inc | Substituted imidazoheterocycles |
| CN101417999A (zh) * | 2007-10-25 | 2009-04-29 | 上海恒瑞医药有限公司 | 哌嗪类衍生物,其制备方法及其在医药上的应用 |
| CN101524082A (zh) | 2009-04-28 | 2009-09-09 | 北京燕化永乐农药有限公司 | 吡唑醚菌酯与咪鲜胺复配农药 |
-
2007
- 2007-12-26 CN CNA2007103023359A patent/CN101468988A/zh active Pending
-
2008
- 2008-11-27 WO PCT/CN2008/001936 patent/WO2009082881A1/zh active Application Filing
- 2008-11-27 US US12/743,795 patent/US8207161B2/en not_active Expired - Fee Related
- 2008-11-27 CN CN2008800097616A patent/CN101641361B/zh active Active
- 2008-11-27 UA UAA201008000A patent/UA101002C2/ru unknown
- 2008-11-27 MX MX2010006438A patent/MX2010006438A/es active IP Right Grant
- 2008-11-27 PT PT88689575T patent/PT2230241E/pt unknown
- 2008-11-27 BR BRPI0821440-9A patent/BRPI0821440A2/pt not_active IP Right Cessation
- 2008-11-27 ES ES08868957.5T patent/ES2529435T3/es active Active
- 2008-11-27 CA CA2706735A patent/CA2706735C/en not_active Expired - Fee Related
- 2008-11-27 EP EP08868957.5A patent/EP2230241B1/en not_active Not-in-force
- 2008-11-27 AU AU2008342461A patent/AU2008342461B2/en not_active Ceased
- 2008-11-27 KR KR1020107014988A patent/KR101685103B1/ko active IP Right Grant
- 2008-11-27 JP JP2010539997A patent/JP5481008B2/ja not_active Expired - Fee Related
- 2008-11-27 RU RU2010125703/04A patent/RU2483070C2/ru not_active IP Right Cessation
-
2010
- 2010-03-23 HK HK10102993.1A patent/HK1134817A1/xx unknown
- 2010-06-10 ZA ZA2010/04154A patent/ZA201004154B/en unknown
-
2012
- 2012-05-03 US US13/463,532 patent/US8513411B2/en active Active
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5462928A (en) | 1990-04-14 | 1995-10-31 | New England Medical Center Hospitals, Inc. | Inhibitors of dipeptidyl-aminopeptidase type IV |
| WO1995015309A1 (en) | 1993-12-03 | 1995-06-08 | Ferring B.V. | Dp-iv-serine protease inhibitors |
| US5543396A (en) | 1994-04-28 | 1996-08-06 | Georgia Tech Research Corp. | Proline phosphonate derivatives |
| WO1998019998A2 (en) | 1996-11-07 | 1998-05-14 | Novartis Ag | N-substituted 2-cyanopyrrolidines |
| US6110949A (en) | 1999-06-24 | 2000-08-29 | Novartis Ag | N-(substituted glycyl)-4-cyanothiazolidines, pharmaceutical compositions containing them and their use in inhibiting dipeptidyl peptidase-IV |
| WO2003004498A1 (en) | 2001-07-06 | 2003-01-16 | Merck & Co., Inc. | Beta-amino tetrahydroimidazo (1, 2-a) pyrazines and tetrahydrotrioazolo (4, 3-a) pyrazines as dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| CN1524082A (zh) * | 2001-07-06 | 2004-08-25 | 作为治疗或预防糖尿病的二肽基肽酶抑制剂的β-氨基四氢咪唑并(1,2-A)吡嗪和四氢三唑并(4,3-A)吡嗪 | |
| WO2003082817A2 (en) | 2002-03-25 | 2003-10-09 | Merck & Co., Inc. | Beta-amino heterocyclic dipeptidyl peptidase inhibitors for the treatment or prevention of diabetes |
| WO2004032836A2 (en) | 2002-10-07 | 2004-04-22 | Merck & Co., Inc. | Antidiabetic beta-amino heterocylcic dipeptidyl peptidase inhibitors |
| WO2004085661A2 (en) | 2003-03-24 | 2004-10-07 | Merck & Co., Inc | Process to chiral beta-amino acid derivatives |
Non-Patent Citations (6)
| Title |
|---|
| ENDOCRINOLOGY, vol. 140, 1999, pages 5356 - 5363 |
| J. ORGANOMET. CHEM, 1985, pages 293 |
| JOURNAL OF ORGANIC CHEMISTRY, vol. 71, no. 3, 2006, pages 1220 - 1225 |
| JOURNAL OF ORGANOMETALLIC CHEMISTRY, vol. 285, no. 1-3, 1985, pages 293 - 303 |
| See also references of EP2230241A4 * |
| TETRAHEDRON ASYMMETRY, vol. 17, no. 2, 2006, pages 205 - 209 |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2170350A2 (en) * | 2007-06-21 | 2010-04-07 | Cara Therapeutics, Inc. | Substituted imidazoheterocycles |
| JP2010530891A (ja) * | 2007-06-21 | 2010-09-16 | カラ セラピューティクス インコーポレイテッド | 置換イミダゾ複素環 |
| EP2170350A4 (en) * | 2007-06-21 | 2011-09-28 | Cara Therapeutics Inc | SUBSTITUTED IMIDAZOHETEROCYCLES |
| WO2008157751A2 (en) | 2007-06-21 | 2008-12-24 | Cara Therapeutics, Inc. | Substituted imidazoheterocycles |
| KR101731602B1 (ko) | 2009-03-05 | 2017-04-28 | 지앙수 헨그루이 메디슨 컴퍼니 리미티드 | 테트라하이드로―이미다조[1,5―a]피라진 유도체 염, 이의 제조방법 및 용도 |
| US8518946B2 (en) | 2009-03-05 | 2013-08-27 | Jiangsu Hengrui Medicine Co., Ltd. | Salts of (R)-7-[3-amino-4-(2,4,5-trifluorophenyl)butanoyl]-3-trifluoromethyl-5,6,7,8-tetrahydroimidazo[1,5-a] pyrazine-1-carboxylic acid, preparation method and medical use thereof |
| EP2436684A4 (en) * | 2009-05-27 | 2012-12-19 | Jiangsu Hengrui Medicine Co | SALTS OF METHYL (R) -7- [3-AMINO-4- (2,4,5-TRIFLUOR-PHENYL) -BUTYRYL] -3-TRIFLUOROMETHYL-5,6,7,8-TETRAHYDRO-IMIDAZO- [1, 5-A] pYRAZINE-1-carboxylate |
| US20120122875A1 (en) * | 2009-05-27 | 2012-05-17 | Jiangsu Hengrui Medicine Co., Ltd. | Salts of Methyl (R)-7-[3-Amino-4-(2,4,5-Trifluoro-Phenyl)-Butyryl]-3-Trifluoromethyl-5,6,7,8-Tetrahydro-Imidazo[1,5-A]Pyrazine-1-Carboxylate |
| US8618104B2 (en) * | 2009-05-27 | 2013-12-31 | Jiangsu Hengrui Medicine Co., Ltd. | Salts of methyl (R)-7-[3-amino-4-(2,4,5-trifluoro-phenyl)-butyryl]-3-trifluoromethyl-5,6,7,8-tetrahydro-imidazo[1,5-A]pyrazine-1-carboxylate |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| KR101686265B1 (ko) | 2010-03-08 | 2016-12-13 | 지앙수 헨그루이 메디슨 컴퍼니 리미티드 | 타입 2 당뇨병 치료용 약학적 조성물 |
| EP2402342A4 (en) * | 2010-03-08 | 2012-09-19 | Jiangsu Hengrui Medicine Co | PHARMACEUTICAL COMPOSITION FOR TREATING TYPE 2 DIABETES |
| JP2013521223A (ja) * | 2010-03-08 | 2013-06-10 | ジエンス ヘンルイ メデイシンカンパニー リミテッド | 2型糖尿病治療用の医薬組成物 |
| KR20130006567A (ko) * | 2010-03-08 | 2013-01-17 | 지앙수 헨그루이 메디슨 컴퍼니 리미티드 | 타입 2 당뇨병 치료용 약학적 조성물 |
| EP2551259A4 (en) * | 2010-03-25 | 2014-04-16 | Zhejiang Jiuzhou Pharm Co Ltd | SITAGLIPTIN INTERMEDIATE COMPOUNDS, PREPARATION METHODS AND USES THEREOF |
| EP2551259A1 (en) * | 2010-03-25 | 2013-01-30 | Zhejiang Jiuzhou Pharmaceutical Co., Ltd. | Sitagliptin intermediate compounds, preparation methods and uses thereof |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012019427A1 (zh) | 2010-08-09 | 2012-02-16 | 上海恒瑞医药有限公司 | 酞嗪酮类衍生物、其制备方法及其在医药上的应用 |
| US9273052B2 (en) | 2010-08-09 | 2016-03-01 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | Phthalazinone ketone derivative, preparation method thereof, and pharmaceutical use thereof |
| US9566277B2 (en) | 2010-08-09 | 2017-02-14 | Jiangsu Hansoh Pharmaceutical Co., Ltd. | Methods of using phthalazinone ketone derivatives |
| TWI462925B (zh) * | 2010-09-27 | 2014-12-01 | Jiangsu Hengrui Medicine Co | 治療2型糖尿病的藥物組合物 |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
| CN114478536A (zh) * | 2020-10-28 | 2022-05-13 | 上海森辉医药有限公司 | 四氢吡嗪稠环衍生物的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2706735A1 (en) | 2009-07-09 |
| PT2230241E (pt) | 2015-02-10 |
| UA101002C2 (ru) | 2013-02-25 |
| CA2706735C (en) | 2016-04-12 |
| US20120220766A1 (en) | 2012-08-30 |
| AU2008342461B2 (en) | 2013-09-05 |
| KR101685103B1 (ko) | 2016-12-09 |
| CN101641361B (zh) | 2012-02-08 |
| MX2010006438A (es) | 2010-10-13 |
| EP2230241B1 (en) | 2014-11-26 |
| AU2008342461A2 (en) | 2010-09-09 |
| US8207161B2 (en) | 2012-06-26 |
| CN101468988A (zh) | 2009-07-01 |
| RU2010125703A (ru) | 2012-02-10 |
| CN101641361A (zh) | 2010-02-03 |
| EP2230241A4 (en) | 2011-06-22 |
| BRPI0821440A2 (pt) | 2015-06-16 |
| EP2230241A1 (en) | 2010-09-22 |
| ES2529435T3 (es) | 2015-02-20 |
| US8513411B2 (en) | 2013-08-20 |
| AU2008342461A1 (en) | 2009-07-09 |
| ZA201004154B (en) | 2011-08-31 |
| JP5481008B2 (ja) | 2014-04-23 |
| JP2011508735A (ja) | 2011-03-17 |
| US20100273786A1 (en) | 2010-10-28 |
| HK1134817A1 (en) | 2010-05-14 |
| RU2483070C2 (ru) | 2013-05-27 |
| KR20110002003A (ko) | 2011-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009082881A1 (fr) | Dérivés de tétrahydroimidazo[1,5-a]pyrazine, leurs procédés de préparation et leurs utilisations médicales | |
| CN109563099B (zh) | 一种化合物的晶型、其制备和用途 | |
| CA2529400C (en) | Phosphoric acid salt of a dipeptidyl peptidase-iv inhibitor | |
| CN103415515B (zh) | 作为jak抑制剂的环丁基取代的吡咯并吡啶和吡咯并嘧啶衍生物 | |
| AU2010220722B2 (en) | Salts of tetrahydroimidazo [1,5-a] pyrazine derivatives, preparation methods and pharmaceutical use thereof | |
| AU2010271270A1 (en) | Substituted pyrazolo[1,5-a]pyrimidine compounds as Trk kinase inhibitors | |
| AU2004268024A1 (en) | Novel crystalline forms of a phosphoric acid salt of a dipeptidyl peptidase-IV inhibitor | |
| WO2009065298A1 (fr) | Dérivés de la pipérazine, leur procédé de préparation et leur utilisation pharmaceutique | |
| CA2301520A1 (en) | Pyrazinone thrombin inhibitors | |
| JP7168149B2 (ja) | Fgfr阻害剤としてのピラジン-2(1h)-オン系化合物 | |
| CN112752757B (zh) | 作为rho-激酶抑制剂的酪氨酸酰胺衍生物 | |
| JP2023533849A (ja) | Alk5阻害剤としてのピリダジニルアミノ誘導体 | |
| TWI716107B (zh) | 6-氟-2-甲基苯并[d]噻唑-5-基化合物 | |
| CN113754635A (zh) | 稠环类化合物及其制备方法和用途 | |
| TWI439271B (zh) | 四氫咪唑並〔1,5-a〕吡類衍生物,其製備方法及其在醫藥上的應用 | |
| RU2824362C1 (ru) | Ингибиторы альдостеронсинтазы (cyp11b2) человека | |
| TW201211043A (en) | Tetrahydro-imidazo[1,5-a]pyrazine derivatives salts, preparation process and pharmaceutical use thereof | |
| MXPA00006906A (en) | Piperazino derivatives as neurokinin antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880009761.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08868957 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12743795 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010050856 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2706735 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3311/CHENP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2010/006438 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010539997 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2008342461 Country of ref document: AU Date of ref document: 20081127 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008868957 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20107014988 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: A201008000 Country of ref document: UA Ref document number: 2010125703 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0821440 Country of ref document: BR Kind code of ref document: A2 Effective date: 20100629 |