WO2009081872A1 - 6-ハロゲノ-3-アリールピリジン誘導体の製造方法 - Google Patents
6-ハロゲノ-3-アリールピリジン誘導体の製造方法 Download PDFInfo
- Publication number
- WO2009081872A1 WO2009081872A1 PCT/JP2008/073221 JP2008073221W WO2009081872A1 WO 2009081872 A1 WO2009081872 A1 WO 2009081872A1 JP 2008073221 W JP2008073221 W JP 2008073221W WO 2009081872 A1 WO2009081872 A1 WO 2009081872A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- bis
- derivative
- substituent
- halogeno
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to a method for producing a 6-halogeno-3-arylpyridine derivative, particularly a 6-halogeno-3,2'-bipyridine derivative.
- the 6-halogeno-3-arylpyridine derivative obtained by the present invention is useful as an organic synthesis intermediate.
- 6-halogeno-3,2'-bipyridine derivatives are useful as intermediates for therapeutic agents for neurological diseases (see WO 2001/96308).
- a method for synthesizing a pyridine derivative is known (J. Molecular Structure 2000, 553, p. 61), but a 5-halogenopyridine derivative having a functional group at the 2-position and magnesium.
- An object of the present invention is to provide an industrially advantageous method for producing a 6-halogeno-3-arylpyridine derivative that does not require cryogenic conditions, is a short process, and has no by-product of difficult-to-separate isomers. There is.
- X 1 and X 2 each independently represent a halogen atom
- R 1 , R 2 , and R 3 each independently represent a hydrogen atom, an optionally substituted alkyl group, a substituted group Represents a cycloalkyl group which may have a group or an aryl group which may have a substituent
- R 1 and R 2 have a substituent together with the carbon atom to which they are bonded.
- the product obtained in the first step is subjected to the following general formula (II) in the presence of a palladium compound:
- X 3 represents a halogen atom
- Y 1 represents a methine group or a nitrogen atom
- R 4 represents a hydrogen atom, an alkyl group which may have a substituent, or a cyclo which may have a substituent.
- n represents an integer of 0 to 5
- the 6-halogeno-3-arylpyridine derivative (III) is represented by the following general formula (IV):
- M represents an element of Group 1 or Group 2 of the periodic table
- R 5 represents an alkyl group or a cycloalkyl group
- M is an element of Group 1
- m is 1.
- alkyl alcohol salt (IV) is an alkyl alcohol salt represented by the following formula (hereinafter also referred to as “alkyl alcohol salt (IV)”):
- a 6-alkoxy-3-arylpyridine derivative comprising a third step to obtain a 6-alkoxy-3-arylpyridine derivative represented by formula (hereinafter also referred to as “6-alkoxy-3-arylpyridine derivative (V)”) ( A manufacturing method of V) is provided.
- the 6-halogeno-3-arylpyridine derivative (III) can be produced industrially advantageously.
- One embodiment of the present invention is a method for producing a 6-halogeno-3-arylpyridine derivative (III).
- a first step in which a 2,5-dihalogenopyridine derivative (I) is reacted with a magnesium agent; and the product obtained in the first step is reacted with a halogeno compound in the presence of a palladium compound. It is characterized in that it comprises a second step of reacting with an aryl derivative (II) to obtain a 6-halogeno-3-arylpyridine derivative (III). Therefore, there are no particular restrictions on specific forms other than these.
- a preferred embodiment of the manufacturing method of the present embodiment will be described in detail.
- the alkyl group represented by each of R 1 , R 2 , R 3 , R 4 , and R 5 , and the alkyl group represented by the alkoxy group represented by R 4 may be either linear or branched. Those having 1 to 12 carbon atoms are preferred, and those having 1 to 4 carbon atoms are more preferred.
- Examples of the alkyl group include a methyl group, an ethyl group, a propyl group, an isopropyl group, an n-butyl group, an isobutyl group, a sec-butyl group, a tert-butyl group, a hexyl group, an octyl group, and a dodecyl group.
- the cycloalkyl group represented by each of R 1 to R 5 and the cycloalkyl group represented by R 4 preferably has 3 to 12 carbon atoms, and has 5 to 7 carbon atoms. Is more preferable.
- Examples of the cycloalkyl group include a cyclopentyl group and a cyclohexyl group.
- R 1 and R 2 may be bonded to each other together with the carbon atom of the pyridine ring to which R 1 and R 2 are attached respectively to form a ring structure. It is preferable that R 1 and R 2 may be formed together with the carbon atom to which they are bonded, and the ring has 4 to 10 carbon atoms. Examples of such a ring include a benzene ring and a naphthalene ring.
- the above alkyl group, cycloalkyl group, and ring structure may have a substituent.
- substituents include a phenyl group, a tolyl group, a methoxyphenyl group, a nitrophenyl group, a naphthyl group, an anthracenyl group, a pyridyl group, a furyl group, and a thienyl group having 4 to 15 carbon atoms,
- An aryl group which may optionally contain a hetero atom such as a nitrogen atom, oxygen atom or sulfur atom in the ring structure; an alkenyl group such as a vinyl group or 1-methylvinyl group; a methoxy group, an ethoxy group, a propoxy group or an isopropoxy group
- Examples of the alkoxy group which may have a substituent represented by R 4 include a methoxy group, an ethoxy group, a propoxy group, an isopropoxy group, an n-butoxy group, an isobutoxy group, a sec-butoxy group, and a tert-butoxy group. Hexyloxy group, octyloxy group, allyloxy group, benzyloxy group and the like.
- Examples of the cycloalkoxy group optionally having a substituent represented by R 4 include a cyclopentyloxy group and a cyclohexyloxy group.
- the aryl group represented by each of R 1 , R 2 , R 3 , and R 4 , and the aryl group represented by the aryloxy group represented by R 4 can be any ring structure of heteroatoms such as nitrogen atoms, oxygen atoms, and sulfur atoms.
- the number of carbon atoms is preferably 4-15.
- Examples of the aryl group include a phenyl group, a naphthyl group, an anthracenyl group, a pyridyl group, a furyl group, and a thienyl group.
- the above aryl group may have a substituent.
- substituents include linear groups such as methyl group, ethyl group, propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, hexyl group, octyl group and dodecyl group.
- a linear or branched alkoxy group having 1 to 12 carbon atoms such
- aryloxy group represented by R 4 include a phenoxy group, a nitrophenoxy group, a naphthyloxy group, a pyridyloxy group, a furyloxy group, and a thienyloxy group.
- examples of the Group 1 element of the periodic table represented by M include a sodium atom, a lithium atom, and a potassium atom.
- examples of the Group 2 element in the periodic table of elements represented by M include a magnesium atom and a calcium atom.
- the production method of the present invention comprises a first step of reacting a 2,5-dihalogenopyridine derivative (I) with a magnesium agent, and the product of the first step in the presence of a palladium compound in the halogenoaryl derivative (II). And a second step of obtaining a 6-halogeno-3-arylpyridine derivative (III).
- a first step of reacting a 2,5-dihalogenopyridine derivative (I) with a magnesium agent and the product of the first step in the presence of a palladium compound in the halogenoaryl derivative (II).
- a second step of obtaining a 6-halogeno-3-arylpyridine derivative (III).
- the route for obtaining the 2,5-dihalogenopyridine derivative (I), which is the reaction raw material in the first step, is not particularly limited.
- a product purchased from the commercially available product can be used for the reaction.
- the 2,5-dihalogenopyridine derivative (I) itself prepared from industrially available raw materials may be used in the first step.
- a method for preparing the 2,5-dihalogenopyridine derivative (I) by itself for example, a method of brominating the 5-position of 2-methoxypyridine and then substituting the methoxy group at the 2-position with chlorine can be mentioned ( Tetrahedron Letters, 1998, 39, 2059).
- a compound used in preparing an organic magnesium halide compound from an organic halogen compound in ordinary organic chemistry can be used.
- examples thereof include Grignard reagents such as magnesium metal; magnesium anthracene complex; ethylmagnesium bromide, isopropylmagnesium bromide, isopropylmagnesium chloride, and t-butylmagnesium chloride.
- Grignard reagents are preferable from the viewpoint of reactivity.
- the amount of the magnesium agent used is preferably in the range of 0.1 to 10 molar equivalents relative to the 2,5-dihalogenopyridine derivative (I), more preferably in the range of 0.5 to 3 equivalents. preferable.
- the reaction is preferably performed in the presence of a solvent.
- the solvent is not particularly limited as long as it does not adversely influence the reaction.
- the solvent include aliphatic hydrocarbons such as hexane, heptane, and octane; aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, and mesitylene; tetrahydrofuran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, 1, And ethers such as 2-dimethoxyethane, 1,4-dioxane, and diethylene glycol dimethyl ether.
- ethers are preferably used as a solvent, and tetrahydrofuran is particularly preferably used from the viewpoint of reaction operation.
- a solvent may be used independently and may be used in combination of 2 or more type.
- the reaction in the first step is preferably performed in the range of ⁇ 20 to 100 ° C., more preferably in the range of 0 to 40 ° C.
- the reaction time in the first step is not particularly limited, but is usually in the range of 0.5 to 24 hours.
- the reaction product of the first step may be isolated and used in the next step, or may be used in the next step as the reaction mixture, but is used in the next step as the reaction mixture. Is preferred.
- the reaction mixture is allowed to stand, and the reaction product precipitated as a solid is collected by filtration; the solvent is distilled off from the reaction mixture and concentrated. It can be used as a separation method.
- organomagnesium compound (VI) an organomagnesium compound represented by the following general formula (VI) (hereinafter referred to as “organomagnesium compound (VI)”). It is estimated that the And in the 2nd process mentioned later, it is understood that the coupling reaction of organomagnesium compound (VI) and halogenoaryl derivative (II) is advancing. However, the scope of rights of the present invention is not restricted by these mechanisms.
- X 3 represents a halogen atom
- X 1 , R 1 , R 2 , and R 3 are as defined above.
- the route for obtaining the halogenoaryl derivative (II) that is the reaction raw material in the second step is not particularly limited.
- a product purchased from the commercially available product can be used for the reaction.
- the halogenoaryl derivative (II) prepared by itself from industrially available raw materials may be used in the second step.
- a method for preparing the halogenoaryl derivative (II) by itself for example, a method of substituting the amino group of 2-aminopyridine with bromine can be mentioned (see Organic Synthesis, Collective Volume 3, page 136, 1955).
- the amount of the halogenoaryl derivative (II) used is preferably in the range of 0.1 to 10 moles relative to 1 mole of the 2,5-dihalogenopyridine derivative (I) used in the first step. A range of ⁇ 2 molar equivalents is more preferred.
- the reaction in the second step is preferably performed in the range of ⁇ 20 to 100 ° C., more preferably in the range of 0 to 40 ° C.
- the palladium compound used in the second step is not particularly limited as long as it is generally used in a carbon-carbon bond generation reaction.
- Examples of the palladium compound include tetrakis (triphenylphosphine) palladium (0), bis [1,2-bis (diphenylphosphino) ethane] palladium (0), bis [1,3-bis (diphenylphosphino) propane.
- dichlorobis [methylenebis (diphenyl) Phosphine)] dipalladium, [1,2-bis (diphenylphosphino) ethane] dichloropalladium (II), [1,3-bis (diphenylphosphino) propa ] Dichlorodichloro (II), [1,4-bis (diphenylphosphino) butane] dichloropalladium (II), [1,5-bis (diphenylphosphino) pentane] dichloropalladium (II), [1,6 More preferred are palladium complex compounds having a bidentate phosphorus ligand such as -bis (diphenylphosphino) hexane] dichloropalladium (II) and [1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium (II).
- the palladium compound used in the second step may be a commercially available product or a product produced in the reaction system.
- a phosphorus ligand capable of coordinating with the palladium compound may be used.
- the phosphorus ligand used is, for example, triphenylphosphine, tri-o-tolylphosphine, tri-m-tolylphosphine, tri-p-tolylphosphine, tris (2,6-dimethoxyphenyl) phosphine, tris [2 -(Diphenylphosphino) ethyl] phosphine, bis (2-methoxyphenyl) phenylphosphine, 2- (di-t-butylphosphino) biphenyl, 2- (dicyclohexylphosphino) biphenyl, 2- (diphenylphosphino)- 2 '-(N, N-dimethylamino) biphenyl, tri-t-butylphosphine, bis (diphenylphosphino) methane, 1,2-bis (dip
- the amount of the palladium compound used is preferably in the range of 0.0001 to 2 equivalents, more preferably in the range of 0.001 to 0.1 equivalents with respect to the halogenoaryl derivative (II).
- the halogenoaryl derivative (II) and the palladium compound are added to the reaction mixture containing the reaction product of the first step, or the reaction of the first step is carried out in a solution of the halogenoaryl derivative (II) and the palladium compound.
- the halogenoaryl derivative (II) may be diluted with the above reaction solvent.
- the dilution concentration is not particularly limited, but is preferably in the range of 1 to 80% by weight of the halogenoaryl derivative (II), and more preferably in the range of 5 to 50% by weight.
- the addition rate is not particularly limited, but is preferably a rate that can be controlled to a temperature at which a preferable reaction result can be exhibited.
- the reaction time in the second step is not particularly limited, but is usually in the range of 0.5 to 24 hours.
- the 6-halogeno-3-arylpyridine derivative (III) thus produced can be isolated and purified by a method used for usual isolation and purification of organic compounds.
- the reaction mixture is neutralized with hydrochloric acid or the like, extracted with an organic solvent such as toluene, and then subjected to treatments such as distillation, silica gel column chromatography, recrystallization, etc., thereby producing the target 6-halogeno-
- the 3-arylpyridine derivative (III) can be purified.
- Another embodiment of the present invention is a method for producing a 6-alkoxy-3-arylpyridine derivative (V).
- This production method includes a third step in which a 6-halogeno-3-arylpyridine derivative (III) is reacted with a salt (IV) of an alkyl alcohol to obtain a 6-alkoxy-3-arylpyridine derivative (V). Characterized by points. Therefore, there are no particular restrictions on specific forms other than these.
- a preferred embodiment of the manufacturing method of the present embodiment will be described in detail.
- the 6-halogeno-3-arylpyridine derivative (III) is reacted with an alkyl alcohol salt (IV). This gives a 6-alkoxy-3-arylpyridine derivative (V).
- the route for obtaining the 6-halogeno-3-arylpyridine derivative (III), which is a reaction raw material in the third step, is not particularly limited. For example, it may be manufactured through the first step and the second step described above, may be manufactured by other general methods, or may be purchased commercially. May be.
- the amount of the alkyl alcohol salt (IV) used is preferably in the range of 0.1 to 10 moles, preferably 0.5 to 3 mole equivalents per mole of 6-halogeno-3-arylpyridine derivative (III). More preferably, it is the range.
- the reaction is preferably performed in the presence of a solvent.
- the solvent is not particularly limited as long as it does not adversely influence the reaction.
- the solvent include aliphatic hydrocarbons such as hexane, heptane, and octane; aromatic hydrocarbons such as benzene, toluene, xylene, ethylbenzene, and mesitylene; tetrahydrofuran, diethyl ether, diisopropyl ether, tert-butyl methyl ether, 1, And ethers such as 2-dimethoxyethane, 1,4-dioxane, and diethylene glycol dimethyl ether.
- aromatic hydrocarbons are preferably used, and toluene is particularly preferably used from the viewpoint of reaction operation.
- a solvent may be used independently and may be used in combination of 2 or more type.
- the reaction in the third step is preferably performed in the range of 0 to 200 ° C, more preferably in the range of 60 to 100 ° C.
- the reaction time in the third step is not particularly limited, but is usually in the range of 0.5 to 24 hours.
- the 6-alkoxy-3-arylpyridine derivative (V) produced in this manner can be isolated and purified by a method used for usual isolation and purification of organic compounds.
- the reaction mixture is neutralized with hydrochloric acid or the like, extracted with an organic solvent such as toluene, and then subjected to treatment such as distillation, silica gel column chromatography, recrystallization, etc., thereby producing the target 6-alkoxy-
- the 3-arylpyridine derivative (V) can be purified.
- Example 1 Synthesis of 6-chloro-3-phenylpyridine 30.3 g of a 19.5 wt% tetrahydrofuran solution of isopropylmagnesium chloride as a magnesium agent (57.5 mmol as isopropylmagnesium chloride) was placed in a flask with an internal volume of 300 mL purged with nitrogen. After charging and cooling to 10 ° C., a solution of 2,5-dihalogenopyridine derivative (I), 5-bromo-2-chloropyridine (9.6 g, 50.0 mmol) in tetrahydrofuran (20 g) was added in an amount of 10-20.
- 2,5-dihalogenopyridine derivative (I) 2,5-dihalogenopyridine derivative (I)
- 5-bromo-2-chloropyridine 9.6 g, 50.0 mmol
- the solution was added dropwise over a period of 1.0 hour in the range of ° C. After completion of the dropwise addition, the mixture was stirred for 1.5 hours in the range of 10 to 20 ° C., and then the reaction mixture was a halogenoaryl derivative (II) bromobenzene (7.9 g, 50.0 mmol) and a palladium compound [1 , 1′-bis (diphenylphosphino) ferrocene] dichloropalladium (II) (0.20 g, 0.25 mmol) in tetrahydrofuran (20 g) was added dropwise over a period of 2.0 hours in the range of 10 to 20 ° C.
- a halogenoaryl derivative (II) bromobenzene 7.9 g, 50.0 mmol
- a palladium compound [1 , 1′-bis (diphenylphosphino) ferrocene] dichloropalladium (II) (0.20 g
- Example 2 Synthesis of 6-chloro-3-phenylpyridine The same procedure as in Example 1 was performed except that chlorobenzene (5.6 g, 50.0 mmol) was used instead of bromobenzene in Example 1, and 6-chloro- 3-Phenylpyridine was obtained with a yield of 40%.
- 6-chloro-3-pyridylboronic acid (6.9 g, 44.0 mmol) was added to 2-bromopyridine (7.0 g, 44.0 mmol), palladium acetate (0.5 g, 2.2 mmol). ), Triphenylphosphine (2.5 g, 9.5 mmol), potassium carbonate (37.0 g, 267.0 mmol), 1,2-dimethoxyethane (60 g), water (80 g), and after stirring, Heated to reflux for 6.0 hours. After cooling the reaction solution, ethyl acetate (50 g) was added and stirred for 1.0 hour, and then allowed to stand to separate the organic layer.
Abstract
Description
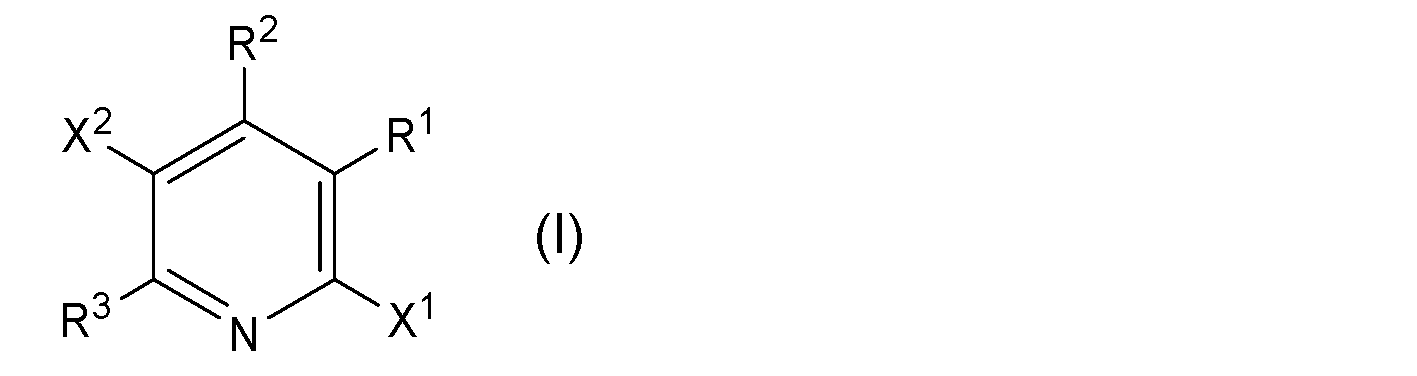
で示される2,5-ジハロゲノピリジン誘導体(以下、「2,5-ジハロゲノピリジン誘導体(I)」とも称する)を、マグネシウム化剤と反応させる第一工程;および、
前記第一工程で得られた生成物を、パラジウム化合物の存在下で、下記一般式(II):
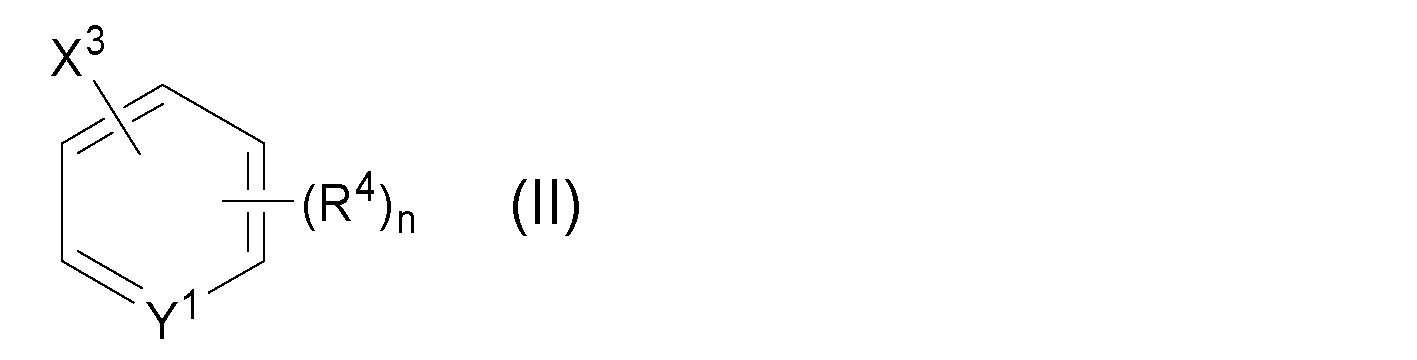
で示されるハロゲノアリール誘導体(以下、「ハロゲノアリール誘導体(II)」とも称する)と反応させて、下記一般式(III):

で示される6-ハロゲノ-3-アリールピリジン誘導体(以下、「6-ハロゲノ-3-アリールピリジン誘導体(III)」とも称する)を得る第二工程;
を含む、6-ハロゲノ-3-アリールピリジン誘導体(III)の製造方法が提供される。
で示されるアルキルアルコールの塩(以下、「アルキルアルコールの塩(IV)」とも称する)と反応させて、下記一般式(V):

で示される6-アルコキシ-3-アリールピリジン誘導体(以下、「6-アルコキシ-3-アリールピリジン誘導体(V)」とも称する)を得る第三工程を含む、6-アルコキシ-3-アリールピリジン誘導体(V)の製造方法が提供される。
本発明の一形態は、6-ハロゲノ-3-アリールピリジン誘導体(III)の製造方法である。この製造方法は、2,5-ジハロゲノピリジン誘導体(I)を、マグネシウム化剤と反応させる第一工程;および、当該第一工程で得られた生成物を、パラジウム化合物の存在下で、ハロゲノアリール誘導体(II)と反応させて、6-ハロゲノ-3-アリールピリジン誘導体(III)を得る第二工程を含む点に特徴を有する。よって、これら以外の具体的な形態について特に制限はない。以下、本形態の製造方法の好ましい実施形態について、詳細に説明する。
第一工程では、2,5-ジハロゲノピリジン誘導体(I)をマグネシウム化剤と反応させる。

第二工程では、上述した第一工程の生成物(有機マグネシウム化合物(VI)と推定される)をパラジウム化合物の存在下で、ハロゲノアリール誘導体(II)と反応させる。これにより、6-ハロゲノ-3-アリールピリジン誘導体(III)を得る。
本発明の他の形態は、6-アルコキシ-3-アリールピリジン誘導体(V)の製造方法である。この製造方法は、6-ハロゲノ-3-アリールピリジン誘導体(III)を、アルキルアルコールの塩(IV)と反応させて、6-アルコキシ-3-アリールピリジン誘導体(V)を得る第三工程を含む点に特徴を有する。よって、これら以外の具体的な形態について特に制限はない。以下、本形態の製造方法の好ましい実施形態について、詳細に説明する。
第三工程では、6-ハロゲノ-3-アリールピリジン誘導体(III)を、アルキルアルコールの塩(IV)と反応させる。これにより、6-アルコキシ-3-アリールピリジン誘導体(V)を得る。なお、第三工程における反応原料である6-ハロゲノ-3-アリールピリジン誘導体(III)の入手経路は特に制限されない。例えば、上述した第一工程および第二工程を経て製造されたものであってもよいし、他の一般的な手法により自ら製造したものであってもよいし、市販品を購入したものであってもよい。
6-クロロ-3-フェニルピリジンの合成
窒素置換した内容積300mLのフラスコに、マグネシウム化剤であるイソプロピルマグネシウムクロリドの19.5重量%テトラヒドロフラン溶液30.3g(イソプロピルマグネシウムクロリドとして57.5ミリモル)を仕込み、10℃に冷却した後、2,5-ジハロゲノピリジン誘導体(I)である5-ブロモ-2-クロロピリジン(9.6g、50.0ミリモル)のテトラヒドロフラン(20g)溶液を10~20℃の範囲で1.0時間かけて滴下した。滴下終了後、10~20℃の範囲で1.5時間攪拌した後、反応混合物を、ハロゲノアリール誘導体(II)であるブロモベンゼン(7.9g、50.0ミリモル)およびパラジウム化合物である[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(0.20g、0.25mmol)のテトラヒドロフラン(20g)溶液に、10~20℃の範囲で2.0時間かけて滴下した。滴下終了後、10~20℃の範囲で4.0時間攪拌した後、飽和塩化アンモニア水溶液(50g)を加えた。反応混合物にトルエン(50g)を加え1.0時間攪拌した後、静置して有機層を分離した。有機層を飽和食塩水(10g)で洗浄した後、減圧下で濃縮して粗6-クロロ-3-フェニルピリジン6.5g(純度82%、収率60%)を得た。得られた粗6-クロロ-3-フェニルピリジン6.5gを、シリカゲルカラムクロマトグラフィー(200g)にて精製し、6-ハロゲノ-3-アリールピリジン誘導体(III)である6-クロロ-3-フェニルピリジン4.5g(収率42%)を得た。
[実施例2]
6-クロロ-3-フェニルピリジンの合成
実施例1においてブロモベンゼンに代えてクロロベンゼン(5.6g、50.0ミリモル)を使用した点以外は実施例1と同様の操作を行ない、6-クロロ-3-フェニルピリジンを収率40%で得た。
6-クロロ-3,2’-ビピリジンの合成
窒素置換した内容積300mLのフラスコに、マグネシウム化剤であるイソプロピルマグネシウムクロリドのテトラヒドロフラン溶液(19.5重量%、30.3g、57.5ミリモル)を仕込み、10℃に冷却した後、2,5-ジハロゲノピリジン誘導体(I)である5-ブロモ-2-クロロピリジン(9.6g、50.0ミリモル)のテトラヒドロフラン(20g)溶液を10~20℃の範囲で1.0時間かけて滴下した。滴下終了後、10~20℃の範囲で1.5時間攪拌した後、反応混合物を、ハロゲノアリール誘導体(II)である2-ブロモピリジン(7.9g、50.0ミリモル)およびパラジウム化合物である[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(0.20g、0.25mmol)のテトラヒドロフラン(20g)溶液に、10~20℃の範囲で2.0時間かけて滴下した。滴下終了後、10~20℃の範囲で4.0時間攪拌した後、飽和塩化アンモニア水溶液(50g)を加えて反応を停止させた。反応混合物にトルエン(50g)を加え1.0時間攪拌した後、静置して有機層を分離した。有機層を飽和食塩水(10g)で洗浄した後、減圧下で濃縮して粗6-クロロ-3,2’-ビピリジン10.6g(純度81%、収率90%)を得た。得られた粗6-クロロ-3,2’-ビピリジン10.6gを、減圧下で単蒸留(0.9mmHg、124℃)して、6-ハロゲノ-3-アリールピリジン誘導体(III)である6-クロロ-3,2’-ビピリジン8.0g(収率68%)を得た。
[実施例4~9]
ハロゲノアリール誘導体(II)、パラジウム化合物として下記の表1に記載の化合物を使用した点以外は実施例3と同様に操作を行なった。生成物および収率を併せて表1に示す。

6-メトキシ-3,2’-ビピリジンの合成
内容積200mLのフラスコに、6-ハロゲノ-3-アリールピリジン誘導体(III)である6-クロロ-3,2’-ビピリジン(6.0g、31.5ミリモル)のトルエン(10g)溶液を仕込み、アルキルアルコールの塩(IV)であるナトリウムメトキシドのメタノール溶液(32.0重量%、8.0g、47.4ミリモル)を10~20℃の範囲で0.5時間かけて滴下した。滴下終了後、70~75℃の範囲で3.0時間攪拌した後、10~20℃の範囲で水(30g)を加えて反応を停止させた。反応混合物にトルエン(20g)を加え1.0時間攪拌した後、静置して有機層を分離した。水層をトルエン(30g+30g)で2回抽出し、抽出液と上記の有機層を合わせた後、減圧下で濃縮して粗6-メトキシ-3,2’-ビピリジン6.2g(純度91%、収率95%)を得た。得られた粗6-メトキシ-3,2’-ビピリジン6.2gを、減圧下で単蒸留(1.0mmHg、104℃)して、6-アルコキシ-3-アリールピリジン誘導体(V)である6-メトキシ-3,2’-ビピリジン5.4g(収率83%)を得た。
[比較例1]
ニッケル化合物存在下での6-クロロ-3,2’-ビピリジンの合成
窒素置換した内容積300mLのフラスコに、イソプロピルマグネシウムクロリドのテトラヒドロフラン溶液(19.5重量%、30.3g、57.5ミリモル)を仕込み、10℃に冷却した後、5-ブロモ-2-クロロピリジン(9.6g、50.0ミリモル)のテトラヒドロフラン(20g)溶液を10~20℃の範囲で1.0時間かけて滴下した。滴下終了後、10~20℃の範囲で1.5時間攪拌した後、反応混合物を、2-ブロモピリジン(7.9g、50.0ミリモル)および[1,1’-ビス(ジフェニルホスホノプロパン)]ニッケルジクロリド(0.14g、0.25ミリモル)のテトラヒドロフラン(20g)溶液に、10~20℃の範囲で2.0時間かけて滴下した。滴下終了後、10~20℃の範囲で4.0時間攪拌したが、6-クロロ-3,2’-ビピリジンは得られなかった。
鈴木カップリングによる6-クロロ-3,2’-ビピリジンの合成(-78℃)
窒素置換したフラスコに5-ブロモ-2-クロロピリジン(9.6g、50.0ミリモル)のテトラヒドロフラン(80g)溶液を仕込み、-78℃に冷却した後、n-ブチルリチウムのn-ヘキサン溶液(15重量%、24.6g、57.5ミリモル)を-78℃にて滴下した。次いで、-78℃にて、トリメトキシボラン(5.7g、55.0ミリモル)を滴下した。滴下終了から30分後、冷却バスをはずし、室温で終夜攪拌した。その後、氷冷下10重量%塩酸(50g)を加えて、1.5時間攪拌した後、40重量%水酸化ナトリウム水溶液(15g)を加えて中和した。反応混合物に酢酸エチル(50g)を加えて1.0時間攪拌した後、静置して有機層を分離した。有機層を無水硫酸マグネシウムで乾燥後、減圧下濃縮して6-クロロ-3-ピリジルボロン酸(6.9g、44.0ミリモル)を得た。
鈴木カップリングによる6-クロロ-3,2’-ビピリジンの合成(10℃)
窒素置換したフラスコに5-ブロモ-2-クロロピリジン(9.6g、50.0mmol)のテトラヒドロフラン(80g)溶液を仕込み、10℃に冷却した後、n-ブチルリチウムのn-ヘキサン溶液(15重量%、24.6g、57.5ミリモル)を10℃にて滴下した。次いで、10℃にて、トリメトキシボラン(5.7g、55.0ミリモル)を滴下した。滴下終了から30分後、冷却バスをはずし、室温で終夜攪拌したが、6-クロロ-3-ピリジルボロン酸は得られなかった。
6-メトキシ-3,2’-ビピリジンの合成
窒素置換した内容積300mLのフラスコにイソプロピルマグネシウムクロリドのテトラヒドロフラン溶液(19.5重量%、30.3g、57.5ミリモル)を仕込み、10℃に冷却した後、5-ブロモ-2-メトキシピリジン(9.4g、50.0ミリモル)のテトラヒドロフラン(20g)溶液を10~20℃の範囲で1.0時間かけて滴下した。滴下終了後、10~20℃の範囲で1.5時間攪拌した後、反応混合物を、2-ブロモピリジン(7.9g、50.0ミリモル)および[1,1’-ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)(0.14g、0.25ミリモル)のテトラヒドロフラン(20g)溶液に、10~20℃の範囲で2.0時間かけて滴下した。滴下終了後、10~20℃の範囲で4.0時間攪拌したが、6-メトキシ-3,2’-ビピリジンは得られなかった。
Claims (3)
- 下記一般式(I):

で示される2,5-ジハロゲノピリジン誘導体を、マグネシウム化剤と反応させる第一工程;および、
前記第一工程で得られた生成物を、パラジウム化合物の存在下で、下記一般式(II):

で示されるハロゲノアリール誘導体と反応させて、下記一般式(III):
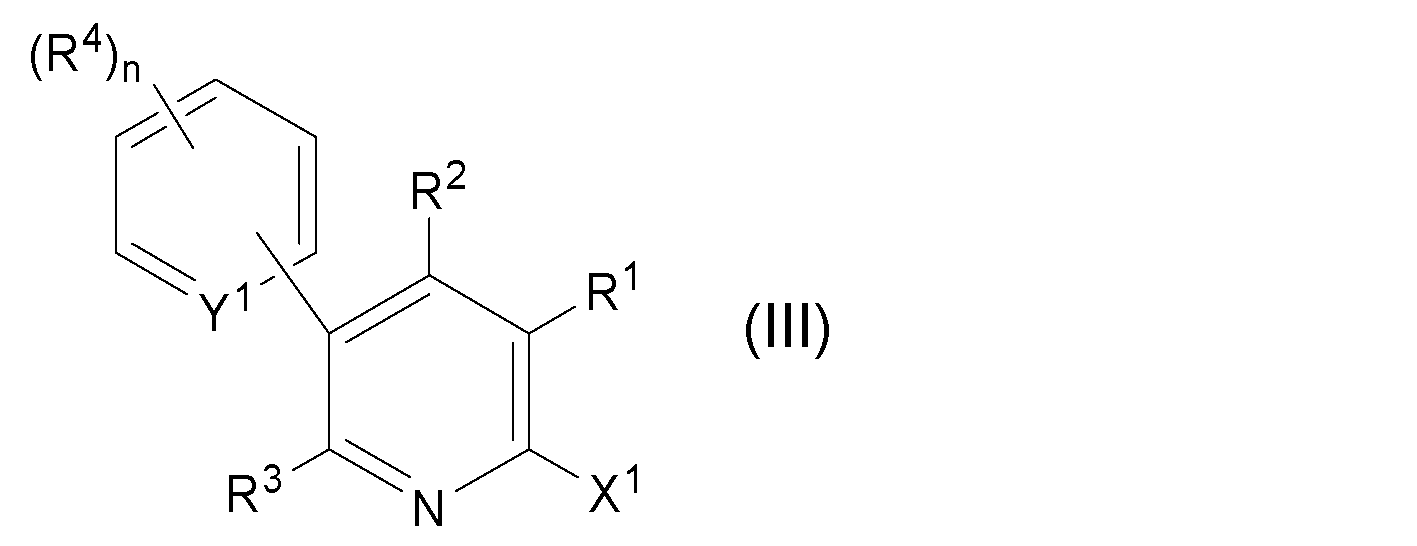
で示される6-ハロゲノ-3-アリールピリジン誘導体を得る第二工程;
を含む、6-ハロゲノ-3-アリールピリジン誘導体の製造方法。 - Y1が窒素原子を表す、請求項1に記載の製造方法。
- 請求項1に記載の一般式(III)で示される6-ハロゲノ-3-アリールピリジン誘導体を、下記一般式(IV):

で示されるアルキルアルコールの塩と反応させて、一般式(V):

で示される6-アルコキシ-3-アリールピリジン誘導体を得る第三工程を含む、6-アルコキシ-3-アリールピリジン誘導体の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP08863865A EP2223912A4 (en) | 2007-12-21 | 2008-12-19 | PROCESS FOR THE PREPARATION OF A 6-HALOGENO-3-ARYLPYRIDINE DERIVATIVE |
| US12/809,868 US8742127B2 (en) | 2007-12-21 | 2008-12-19 | Production method of 6-halogeno-3-arylpyridine derivative |
| JP2009547083A JP5536458B2 (ja) | 2007-12-21 | 2008-12-19 | 6−ハロゲノ−3−アリールピリジン誘導体の製造方法 |
| CN200880118758.8A CN101883757B (zh) | 2007-12-21 | 2008-12-19 | 6-卤代-3-芳基吡啶衍生物的制备方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007-330516 | 2007-12-21 | ||
| JP2007330516 | 2007-12-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009081872A1 true WO2009081872A1 (ja) | 2009-07-02 |
Family
ID=40801173
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2008/073221 WO2009081872A1 (ja) | 2007-12-21 | 2008-12-19 | 6-ハロゲノ-3-アリールピリジン誘導体の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8742127B2 (ja) |
| EP (1) | EP2223912A4 (ja) |
| JP (1) | JP5536458B2 (ja) |
| CN (1) | CN101883757B (ja) |
| WO (1) | WO2009081872A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013095752A (ja) * | 2011-10-27 | 2013-05-20 | Fis Fabbrica Italiana Sintetici Spa | ペランパネルの中間体である2−アルコキシ−5−(ピリジン−2−イル)ピリジンの調製方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106243018B (zh) * | 2016-07-26 | 2018-09-21 | 陕西师范大学 | 一种多氟苯基吡啶类化合物的合成方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000355581A (ja) | 1999-06-14 | 2000-12-26 | Sankio Chemical Co Ltd | 2−ピリジルピリジン誘導体の製造方法 |
| WO2001096308A1 (fr) | 2000-06-12 | 2001-12-20 | Eisai Co., Ltd. | Composes 1,2-dihydropyridine, leur procede de preparation et leur utilisation |
| JP2004051592A (ja) * | 2002-07-23 | 2004-02-19 | Kuraray Co Ltd | 5−(2’−ピリジル)−2−ピリドン誘導体の製造方法 |
| JP2007291092A (ja) * | 2006-03-30 | 2007-11-08 | Chisso Corp | 新規ビピリジン誘導体、およびこれを含む有機電界発光素子 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1442096A (en) * | 1974-05-22 | 1976-07-07 | Ici Ltd | Process for the manufacture of 2/2 and 4/4-bipyridyls |
| US4463008A (en) * | 1982-11-10 | 1984-07-31 | Sterling Drug Inc. | 2-Alkoxy-5-(pyridinyl)pyridines and cardiotonic use thereof |
| US4910312A (en) * | 1985-11-08 | 1990-03-20 | Warner-Lambert Company | Various N-substituted 3-piperidine carboxylic acids or N-substituted 3-pyridinecarboxylic acids and derivatives thereof |
| JPH0930989A (ja) * | 1995-05-15 | 1997-02-04 | Chisso Corp | ビアリール誘導体の製造方法 |
| GB0129260D0 (en) * | 2001-12-06 | 2002-01-23 | Eisai London Res Lab Ltd | Pharmaceutical compositions and their uses |
| JP2007119379A (ja) * | 2005-10-26 | 2007-05-17 | Tosoh Corp | ジハロゲン化ビフェニル類の製造方法 |
-
2008
- 2008-12-19 US US12/809,868 patent/US8742127B2/en not_active Expired - Fee Related
- 2008-12-19 JP JP2009547083A patent/JP5536458B2/ja not_active Expired - Fee Related
- 2008-12-19 WO PCT/JP2008/073221 patent/WO2009081872A1/ja active Application Filing
- 2008-12-19 EP EP08863865A patent/EP2223912A4/en not_active Withdrawn
- 2008-12-19 CN CN200880118758.8A patent/CN101883757B/zh not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000355581A (ja) | 1999-06-14 | 2000-12-26 | Sankio Chemical Co Ltd | 2−ピリジルピリジン誘導体の製造方法 |
| WO2001096308A1 (fr) | 2000-06-12 | 2001-12-20 | Eisai Co., Ltd. | Composes 1,2-dihydropyridine, leur procede de preparation et leur utilisation |
| JP2004051592A (ja) * | 2002-07-23 | 2004-02-19 | Kuraray Co Ltd | 5−(2’−ピリジル)−2−ピリドン誘導体の製造方法 |
| JP2007291092A (ja) * | 2006-03-30 | 2007-11-08 | Chisso Corp | 新規ビピリジン誘導体、およびこれを含む有機電界発光素子 |
Non-Patent Citations (12)
| Title |
|---|
| BONNET, V. ET AL.: "Cross-coupling between 3-pyridylmagnesium chlorides and heteroaromatic halides", SYNLETT, 2002, pages 1008 - 1010, XP008136762 * |
| CHEM. PHARM. BULL., vol. 36, 1988, pages 2244 |
| GRAVE T. B. ET AL.: "Attempts to Prepare 1-methyl-2- methoxypiperidine. The Hydrogenation of Certain Pyridine Derivatives", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 46, no. 6, 1924, pages 1460 - 1470, XP008136812 * |
| HETEROCYCLES, vol. 26, 1987, pages 2711 |
| J. MOLECULAR STRUCTURE, vol. 553, 2000, pages 61 |
| J. ORG. CHEM., vol. 67, 2002, pages 7541 |
| J. ORGANOMETALLIC CHEM., vol. 118, 1976, pages 349 |
| MURMANN, R. K. ET AL.: "The Stability of Silver(I) Complexes of Some 3- and 4-Substituted Pyridines", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 77, no. 13, 1955, pages 3484 - 3486, XP008136763 * |
| ORGANIC SYNTHESIS, COLLECTIVE, vol. 3, 1955, pages 136 |
| PARRY, P. R. ET AL.: "Functionalized Pyridylboronic Acids and Their Suzuki Cross- Coupling Reactions to Yield Novel Heteroarylpyridines", JOURNAL OF ORGANIC CHEMISTRY, vol. 67, no. 21, 2002, pages 7541 - 7543, XP008136759 * |
| See also references of EP2223912A4 |
| TETRAHEDRON LETTERS, vol. 39, 1998, pages 2059 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013095752A (ja) * | 2011-10-27 | 2013-05-20 | Fis Fabbrica Italiana Sintetici Spa | ペランパネルの中間体である2−アルコキシ−5−(ピリジン−2−イル)ピリジンの調製方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US8742127B2 (en) | 2014-06-03 |
| US20100280253A1 (en) | 2010-11-04 |
| CN101883757A (zh) | 2010-11-10 |
| JP5536458B2 (ja) | 2014-07-02 |
| EP2223912A1 (en) | 2010-09-01 |
| JPWO2009081872A1 (ja) | 2011-05-06 |
| CN101883757B (zh) | 2013-06-12 |
| EP2223912A4 (en) | 2012-04-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8586742B2 (en) | Process for preparing amines from alcohols and ammonia | |
| JP5138386B2 (ja) | 置換ビフェニル類の製造方法 | |
| US20090005597A1 (en) | Process for Preparing Substituted Biphenyls | |
| JP5536458B2 (ja) | 6−ハロゲノ−3−アリールピリジン誘導体の製造方法 | |
| CN117550960A (zh) | 用于制备1-(3,5-二氯-4-氟-苯基)-2,2,2-三氟-乙酮的方法 | |
| JP2019147829A (ja) | 5−(トリフルオロメチル)ピリミジン誘導体及びその製造方法 | |
| US10544177B2 (en) | Chiral dihydrobenzooxaphosphole ligands and synthesis thereof | |
| Schull et al. | Synthesis of symmetrical triarylphosphines from aryl fluorides and red phosphorus: scope and limitations | |
| JP2002193845A (ja) | 単官能性、二官能性又は多官能性のビアリールの製造法 | |
| JP5848201B2 (ja) | アリールジクロロホスフィンの製造方法 | |
| JP5261140B2 (ja) | ビニル芳香族化合物の製造方法 | |
| US10889599B2 (en) | 1,1-diborylalkyl-1-metal compounds, preparation method thereof, and their applications toward synthesis of 1,1-diboronate ester compounds | |
| US6096894A (en) | Production method of 2-(p-alkylphenyl)pyridine compound | |
| JP2004352724A (ja) | フェニルアルキレンカルボン酸誘導体の製造方法 | |
| JP4759722B2 (ja) | 置換基を有する芳香族カルボン酸エステルの製造方法 | |
| KR20090085173A (ko) | 신규한 알킬아릴셀레나이드 유도체 및 이의 제조방법 | |
| US6953871B2 (en) | Processes for producing poly-ethynyl-substituted aromatic compound | |
| JP4509327B2 (ja) | N,n−ジ置換−4−アミノクロトン酸エステルの製造方法 | |
| EP3286161B1 (en) | Methods for preparing bridged bi-aromatic ligands | |
| JP4505876B2 (ja) | 4−ハロベンゾフラン誘導体及びその製造方法 | |
| JP2019189569A (ja) | ホスフィン化合物、触媒組成物および芳香族アミン化合物の製造方法 | |
| JP2690038B2 (ja) | ベンゾ−1,4−ジシラシクロヘキセン誘導体及びその製造方法 | |
| TW201945324A (zh) | 製備2,6-二烷基苯基乙酸之方法 | |
| JP2012106964A (ja) | 2‐シクロプロピル‐6‐ハロゲノメチル‐4‐トリフルオロメチルピリジン誘導体及びその製造方法 | |
| JP2003300991A (ja) | ブタジエニルホスホン酸環状エステル及びその製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880118758.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08863865 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009547083 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2151/KOLNP/2010 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008863865 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12809868 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |