2, 4, 5-三取代噻唑类化合物、 其制备方法、
药物组合物及其制药用途 技术领域
本发明涉及作为磷脂转移蛋白 (Phosphol ipid transfer protein (PLTP) )抑制剂和 /或胆固醇酯转移蛋白(Choles teryl es ter transfer protein (CETP) )抑制剂的 2, 4, 5-三取代塞唑类 化合物, 以及它们的制备方法和在医药领域的用途, 特别是用于 制备治疗和预防与血浆 PLTP活性升高相关和 /或与血浆 CETP活性 升高相关的各种疾病的药物的用途,所述疾病包括动脉粥样硬化、 心血管疾病和外周血管疾病等。 背景技术
动脉粥样硬化导致的疾病是发达国家第一位的死亡原因。 在 我国, 随着社会经济的发展和人口的老龄化, 心脑血管病发病率 与死亡率近年来也显著增加。 动脉粥样硬化病因病理复杂, 目前 尚未完全阐明, 但已知与下列因素有密切关系: 血脂异常、 高血 压、 糖尿病、 肥胖、 吸烟等。 在诸多因素中对动脉粥样硬化形成 最重要的诱导因素是血脂异常。 血脂异常主要表现为 LDL胆固醇 水平升高以及 HDL胆固醇水平下降。
磷脂转移蛋白(PLTP)以前称作脂肪转移蛋白 Π, 是存在于血 浆中的糖蛋白, 能在磷脂嚢泡和高密度脂蛋白(high-dens i ty l ipoprotein (HDL) )间以及主要的脂蛋白间介导磷脂的净转移和 交换。 PLTP含 476个氨基酸残基, 分布在所有组织中, 在胎盘、 胰腺、 脂肪组织和肺中高表达, 在肝脏、 肾和心脏中低表达, 生 物合成主要在肝脏和脂肪组织中完成。 血浆中存在两种 PLTP, —
种高活性形式(与 apo E结合),一种低活性形式(与 apo A-I结合), 高活性形式在血浆中占 46% ( Tol, A. V. 2002; Janis M. T. , Siggins S., Tahvanainen E. , et al. J. Lipid. Res. 2004; 45 (12): 2303-2309)。 PLTP对脂蛋白的代谢有非常重要的作用。
PLTP在脂蛋白代谢中的主要作用有三个: 其一是磷脂转移活 性, 即从脂解中的乳糜微粒、 极低密度脂蛋白(very low-density lipoprotein (VLDL) ) 和 ^氐 密 度 脂 蛋 白 (low— density lipoprotein (LDL) )的表面残粒向 HDL转移磷脂, 使 HDL颗粒增 大; 其二是重塑 HDL, 即调节 HDL颗粒大小和亚类组成, 介导两 个 HDL3颗粒的融合, 产生大颗粒 HDL2和 pre- β- HDL, 而且磷脂转 移是重塑 HDL的先决条件, pre- β- HDL在胆固醇逆向转运过程中 是胆固醇的有效接受体; 其三是调节肝细胞分泌载脂蛋白 B (apoB) , 增加血液中 VLDL的含量, PLTP缺乏导致 VLDL分泌的 减少。 另外参与维生素 E从脂蛋白向细胞膜的转移, 活性减小时 可使含 apo B脂蛋白(VLDL和 LDL)中的 VE含量增加,脂蛋白的抗 氧化作用增强; PLTP还有转移脂多糖的活性, 增强机体对炎症的 反应 (Huuskonen J. , Olkkonen V. M. , Jauhiainen Μ. , Ehnholm C. Atherosclerosis 2001, 155, 269—281; Albers J. J., Cheung M. C. Current Opinion In Llpidology 15, 255-260;
Huuskonen J., Olkkonen V. M., Ehnholm C. et al. Biochemistry 2000, 39, 16092-16098)。
PLTP和 CETP同属于脂肪转运 /脂多糖结合蛋白家族, 该蛋白 家族有四个成员,其它成员为杀菌渗透性增强蛋白(bactericidal permeability increasing protein (BPI) )和月旨多糖结合蛋白 (lipopolysaccharide binding protein (LBP))。 CETP以前称作 脂肪转移蛋白 I,介导胆固醇酯等中性脂肪从 HDL向 LDL的转移,
结果使 HDL颗粒减小。发现人遗传性缺失 CETP造成 HDL水平显著 升高, LDL水平中等程度下降, 引发了 CETP抑制剂的研究和开发 (Brown M. L. , Inazu A., Hesler C. B. et al. Nature 1989, 342, 448-451)。 临床研究表明 CETP抑制剂可以升高 HDL, 用于 动 脉 粥 样 硬 化 的 预 防 和 治 疗 , CETP 抑 制 剂 Torcetrapib (Brousseau M. E. , Schaefer E. J. , Wolfe M. L. et al. N. Engl. J. Med. 2004, 350, 1505-1515)和 JTT— 705 (de Grooth G. J. , Kuivenhoven J. A. , Stalenhoef A. F. et al. Circulation 2002, 105, 2159-2165)分别处于三期和二期临床 研究中 , Torcetrapib 在三期临床研究中与阿托伐他汀 (Atorvastatin)联合用于抗动脉粥样硬化和血脂异常治疗。
在这四个成员中,只有人的 BPI报导了 X射线衍射晶体结构。 BPI 的晶体结构显示其为回飞棒形状, 由两个亚单位组成, 呈现 假对称的结构。 在回飞棒的两个凹陷面分别有两个非极性口袋, 每个口袋结合一个的卵磷脂分子,主要与磷脂的酰基链相互作用, 推测在生理过程中 BPI与脂多糖的酰基链相结合。 由 BPI的结构 可以推测该脂肪转运蛋白家族脂肪转运的机理, 两个非极性口袋 是其主要的功能结构(Beamer L. J. , Carroll S. F. , Eisenberg D. Science 1997, 276, 1861—1864)。 Himskonen等人以 BPI 为 模板蛋白同源模建了 PLTP的结构,氨基酸定位突变研究表明 PLTP 的 N端口袋对磷脂转移活性至关重要, C端口袋主要功能是与 HDL 结合 (Huuskonen J. , Wohlfahrt G. , Jauhiainen M. et al. J. lipid Res. 1999, 40, 1123-1130; Pons in G. , Qu S. J. , Fan H. Z. et al. Biochemistry 2003, 42, 4444-4451) 0
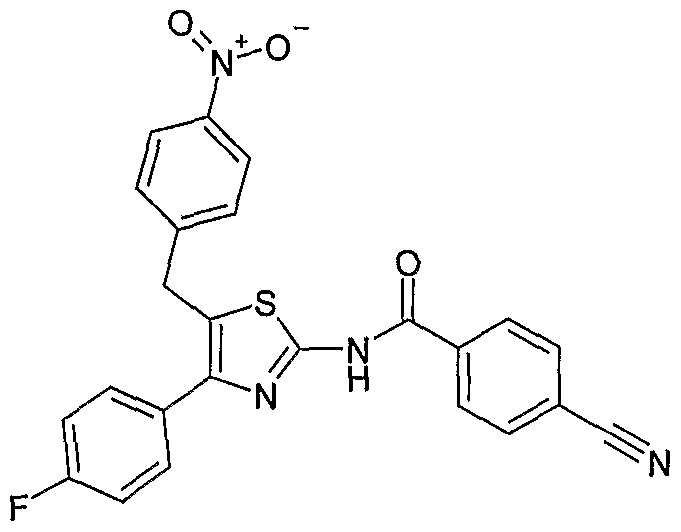
流行病学研究结果表明,冠状动脉疾病患者血浆中 PLTP的含 量显著高于控制组(25.5对 22· 4 pmol/ yL/h; P<0.0001) 0 血浆
中 PLTP 的含量最高的五分之一人群比含量最低的五分之一人群 患冠状动脉疾病的几率高出 1.9倍。 多变量回归分析表明: 在校 正了年龄、 血浆脂肪、 吸烟、 糖尿病、 高血压等因素后, PLTP活 性是冠心病预测的独立影响因子。 血浆中磷脂转移蛋白水平的升 高对冠状动脉疾病是一个危险因素(Schlitt A., Bickel C. , Thumraa P. et al. Arterioscler Thromb. Vase. Biol. 2003, 23 (10), 1857-62.)。 血浆 PLTP活性在 I期和 I I期糖尿病、 肥 胖症和胰岛素耐受症患者中是升高的(Tol, A. V. 2002)。
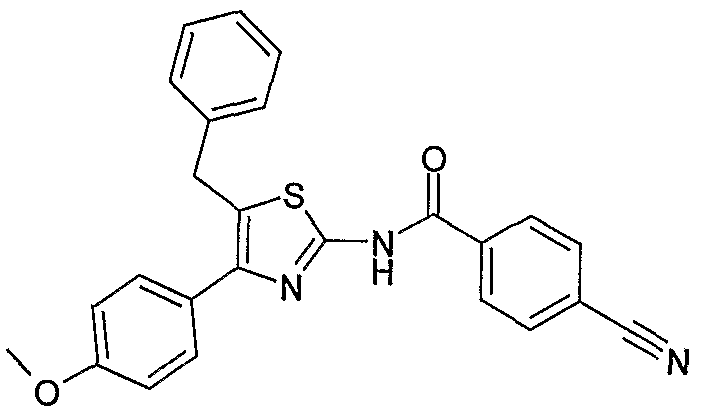
过量表达人 PLTP 的转基因小鼠血浆 PLTP 活性水平升高 2.5- 4.5 倍, 导致血浆 HDL 胆固醇水平与野生型动物相比降低 30 - 40%, 同时增强了 pro-p-HDL 的形成能力(Van Haperen R. , van To 1 A. Verraeulen P. et al. Arterioscler Thromb Vase Biol. 2000, 20, 1082-1088)。 在 LDL受体缺失的杂合子小鼠体内过量 表达 PLTP, 导致 PLTP活性提高 1.3 ~ 2倍, 剂量依赖性地增加动 脉粥样硬化损伤 (Yang X. P. , Yan D., Qiao C. et al. Arterioscler Thromb. Vase. Biol. 2003, 23, 1601—1607)。
PLTP基因敲除小鼠肝脏中载脂蛋白 B分泌减少, 导致 VLDL 分泌减少, 脂蛋白抗氧化能力增强, 改善了 HDL的抗炎作用, 使 其受到动脉粥样硬化的损伤显著减少(Jiang X. C., Zhou H. W. Curr. Opin. Lipidol. 2006, 17, 302-308; Jiang X. C. , Qin S., Qiao C. et al. Nat. Med. 2001, 7, 847—852·)。 给 PLTP 缺失小鼠喂养高脂肪食物没有引起血浆脂蛋白异常, 但是阻碍了 所有主要血浆磷脂从 VLDL向 HDL的转移, 显著减小 HDL的水平 (Jiang, X. C.; Bruce, C.; Mar, J. et al. J. Clin. Invest. 1999, 103, 907-914. ) 0 PLTP过量表达的小鼠肝脏分泌 VLDL增 长 48%,从反方面证明了 PLTP对 VLDL分泌的关键作用(Lie J., de
Crom R., van G. T. et a l. J. Lipid Res. 2002, 43 (11) , 1875 - 80)。 PLTP缺乏状态增强血浆脂蛋白抗氧化能力的原因是增 加了维生素 E的积累, 增加了维生素 E在致动脉粥样硬化的脂蛋 白(LDL和 VLDL)中的生物利用度(J iang X. C., Tal l A. R. , Qin S. et al. J. Biol. Chem. 2002, 277 (35) , 31850—56)。 Schl i t t 等人报导缺失 PLTP的小鼠机体抗炎能力增强 (Schl i t t A. , Liu J., Yan D. et a l. Biochim Biophys Acta. 2005, 1733 (2-3) , 187-91)。
综上所述, 降低血浆 PLTP活性具有抗动脉粥样硬化作用, 其 作用机理至少有以下三个: 其一减少肝脏载脂蛋白 B的分泌, 使 VLDL分泌水平下降; 其二增强 LDL的抗氧化作用, 减轻由于 LDL 被氧化而导致的动脉粥样硬化形成; 其三增强机体抗炎能力, 减 轻由于炎症诱发的动脉粥样硬化损伤。 所以, PLTP抑制剂是预防 和治疗动脉粥样硬化的新药物靶标。
PLTP和 CETP同为脂肪转移 /脂多糖结合蛋白家族成员,研究 表明两者在生理功能上没有交叉, 降低 PLTP活性可以减少 VLDL 的分泌, 而降低 CETP活性可以升高 HDL胆固醇水平,这两方面对 动脉粥样硬化预防和治疗都是有利的, 所以 J iang等人提出 PLTP 和 CETP 汉重抑制剂是一个有希望的动脉粥样硬化治疗靶标 (J iang X. C. , Ta l l A. R. PCT Int. Appl. WO 2002068450 ; J iang X. C. , Zhou H. W. Curr. Opin. Lipidol. 2006, 17, 302-308)。 N- {2- [2- (2, 2-二甲基 -丙酰胺) -苯基二硫基] -苯 基} -2, 2-二甲基-丙酰胺是目前文献中报导的唯 个 PLTP抑制 剂, 并且是 PLTP和 CETP双重抑制剂,对两者抑制的 Ki值分别为 15μΜ和 13μΜ (Ψ0 2002068450) 0
研究表明, PLTP选择性抑制剂、 PLTP和 CETP双重抑制剂以
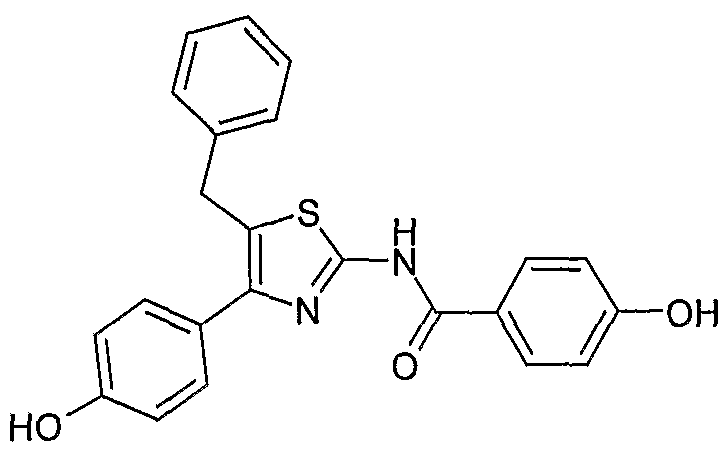
及 CETP 选择性抑制剂均可用于治疗和预防哺乳动物(包括人)患 有的与血浆 PLTP活性升高相关的各种疾病: 如动脉粥样硬化、外 周血管疾病、 血脂异常症、 高脂血症、 高胆固醇血症、 高甘油三 酯血症、 家族性高胆固醇血症、 心血管疾病、 心绞痛、 局部缺血、 心脏缺血、 中风、 心肌梗塞、 再灌注损伤、 高血压、 糖尿病血管 并发症、 肥胖症或内毒素性血症等。
术语 "PLTP选择性抑制剂" 指一个化合物能抑制 PLTP活性, 而对 CETP活性没有抑制作用。
术语" PLTP和 CETP默重抑制剂"指一个化合物既能抑制 PLTP 活性, 又能抑制 CETP活性。
术语 "CETP选择性抑制剂" 指一个化合物能抑制 CETP活性, 而对 PLTP活性没有抑制作用。 发明内容
本发明的目的是寻找并开发磷脂转移蛋白抑制剂和 /或胆固 醇酯转移蛋白抑制剂,通过抑制磷脂转移蛋白活性和 /或抑制胆固 醇酯转移蛋白活性, 调节相关脂蛋白的代谢, 治疗和预防各种与 血浆磷脂转移蛋白活性升高和 /或血浆胆固醇酯转移蛋白活性升 高相关的疾病。
本发明人经过研究发现, 具有下面通式 I的化合物具有抑制 磷脂转移蛋白和 /或抑制胆固醇酯转移蛋白的活性,从而可以降低 肝脏 VLDL的分泌和 /或升高 HDL胆固醇的水平, 因此可以用于治 疗和预防与磷脂转移蛋白活性升高和 /或胆固醇酯转移蛋白活性 升高相关的各种疾病, 从而完成了本发明。
因此, 本发明的一个方面涉及通式 I的化合物, 或其所有可 能的异构体、 前药、 可药用盐、 溶剂合物或水合物:
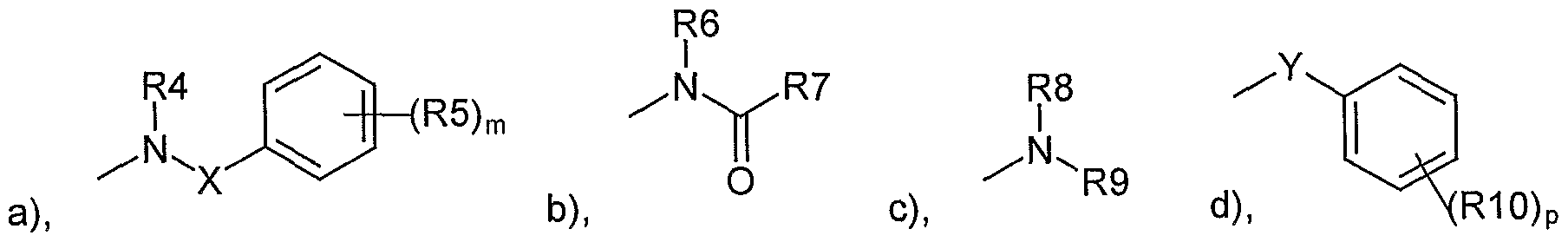
Rl为苯基 -d-C6烷基、 d-C^烷基、 CfC。环烷基 - d-C6烷基或 C3-C1Q环烷基, 其中的苯基未被取代或被一个、 两个或三个独立选 自卤素、 硝基、 羟基、 氨基、 氰基、 d- C6烷基、 d- 烷氧基和 d-6卤代烷基的取代基取代;并且其中的烷基和环烷基可以任选被 羟基、 0- (C -C4) -烷基、氧代、氨基、 NH- (C -C4) -烷基或 N- [ (C -C6)― 烷基 ] 2所取代, 或任选被- 0-、 - S -、 -NH -、 -C00-、 - C0NH-所间隔; 各个 R2独立地为氢、 羟基、 卤素、 氧基、 d- C6烷基、 6 ¾代烷基、 氨基、 硝基、 氰基或烷基酰基;
R 3选自具有以下结构的基团:
其中,
R4为氢、 d- C6坑基、 烷基酰基、 苯甲酰基或苯磺酰基; 其中 苯曱酰基和苯磺酰基中的苯基任选被一个或两个独立选自羟基、 卤素、 硝基、 氨基、 氰基、 - 坑基、 (^- 烷氧基和 d-6卤代烷 基的取代基所取代;
各个 R5独立地选自氢、 羟基、 d- 06浣基、 d- 烷氧基、 CH 卤代烷基、 氨基、 卤素、 硝基、 氰基和烷基酰基;
R6为氢、 d-C6烷基、 烷基酰基、 苯曱酰基或苯磺酰基; 其中 苯甲酰基和苯磺酰基中的苯基任选被一个或两个独立选自羟基、 卤素、 硝基、 氨基、 氰基、 C广 C6烷基、 (广^烷氧基和 ^ 代烷 基的取代基所取代;
R7为 d- C6烷基、 苯基- d-C6烷基, 其中的苯基任选被一个、 两个或三个羟基、 d- C6 ^基、 - 烷氧基、 d— 6卤代烷基、 氨基、 卤素、 硝基、 氰基、 烷基酰基所取代; 并且其中的烷基可以被羟 基、 O- O^-CJ -烷基、氧代、氨基、 NH- (d- C4) -烷基或 - [ (C -C6) - 烷基 ] 2取代, 或任选被 -0-、 - S -、 -NH -、 -C00-、 -C0NH-所间隔;
R8和 R9各自独立选自氢; 任选被羟基、 氨基、 羧基、 d-C6 烷基、 d- C6烷氧基和氧代所取代的并任选被- 0-、 - S-和- NH -、 -C00 -、 - C0NH-所间隔的 d- C22烷基; 苯基 - d-C6烷基; 其中的苯 基任选被一个、 两个或三个羟基、 d- C6烷基、 (广^烷氧基、 d-6 卤代烷基、 氨基、 素、 硝基、 氰基或烷基酰基所取代; 或者
R8和 R9与其所连接的氮原子共同形成一个五元或六元杂环, 该杂环可以含有另外一个或两个独立选自 N、0和 S的杂原子并且 可以与苯环形成稠合杂环, 所述杂环例如为吗啉、 哌嗪、 哌啶、 吡咯、 咪唑烷、 噻唑烷、 噁唑烷、 吲哚或二氢吲哚, 所述杂环可 以任选被羟基、 C广 C6烷基、 氧基、 d-6 i¾代烷基、 氨基、 卤素、 硝基或氰基所取代;
各个 R10独立选自氢、 羟基、 C「C6烷基、 (^- 烷氧基、 d_6 卤代烷基、 氨基、 !¾素、 硝基、 氰基和烷基酰基;
X为 - (c=o) -或 - (so2) -;
Y为 d-d。亚烷基、 d-C1Q亚烯基或 d- 。亚炔基;所述亚烷基、 亚烯基和亚炔基任选被羟基、氨基、氧代、羧基、 d- c6烷基、 c -c6 烷氧基所取代或任选被- 0-、 - S -、 -NH -、 -C00-、 - C0NH-所间隔;
n为 0、 1、 2、 3或 4;
m为 0、 1、 2、 3或 4;
p为 0、 1、 2、 3或 4。 本发明的另一个方面涉及通式 I的化合物的制备方法。
本发明的再一个方面涉及包含本发明通式 I化合物或其所有 可能异构体、 前药、 可药用盐、 溶剂合物或水合物以及至少一种 可药用的载体的药物组合物。
本发明进一步的一个方面涉及包含本发明通式 I化合物用于 制备药物的用途, 所述药物用于治疗和 /或预防哺乳动物的与血浆 PLTP活性升高相关和 /或与血浆 CETP活性升高相关的疾病, 所述 疾病包括但不局限于:动脉粥样硬化、外周血管疾病、血脂异常症、 高脂血症、 高胆固醇血症、高甘油三酯血症、家族性高胆固醇血症、 心血管疾病、 心绞痛、 局部缺血、 心脏缺血、 中风、 心肌梗塞、 再 灌注损伤、 高血压、糖尿病血管并发症、肥胖症或内毒素性血症等。
本文使用的术语 "(V- C6烷基" , 不管是其自身还是作为其它 更大基团如 d-C6烷氧基的一部分,均指直链或支链的含有 1-6个 碳原子的原子团, 包括但并不局限于甲基、 乙基、 正丙基、 异丙 基、 正丁基、 仲丁基、 异丁基和叔丁基等。
本文使用的术语 "d- C6 -烷氧基" 是指 "d- C广烷基 - 0-" , 烷氧基的例子包括但不限于甲氧基、 乙氧基、 异丙氧基等。
本文使用的术语 "d- 代烷基 "是指其中 1个或多个氢被卤 素原子取代的 d-C6烷基,这里特别需要提到的是三氟曱基 ( -CF3 )。
本文使用的术语 素( 代),,是指氟(氟代)、氯(氯代)、 溴 (溴代) 或碘(碘代) ,,。
本文使用的术语"芳基,,指 5-14 元的取代的或非取代的芳环
系统, 或可能包含稠合的双环或三环的芳环系统, 包括但是并不 局限于苯基和萘基。
下列化合物被本发明的通式 I所包括:
其中 Rl-R10、 X、 Y、 n、 m、 p具有如上所述的定义。 在本发明的一个优选的实施方式中, 所述的化合物为具有通式
la的化合物:
其中,
Rl为苯基 -d- C6烷基、 d-C22烷基或 C3- 。环烷基 - d- C6烷基, 其中的苯基未被取代或被一个、 两个或三个独立选自卤素、硝基、 羟基、 氨基、 氰基、 (广 坑基、 d- C6烷氧基和 d— 6 ¾代烷基的取 代基所取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或 氨基所取代, 或任选被 -0-、 -S -、 -NH -、 -C00-、 -C0NH-所间隔; 各个 R2独立地为氢、 羟基、 卤素、 (广^烷氧基、 d- 烷基、 6 |g代烷基、 氨基、 硝基、 氰基或烷基酰基;
R4为氢、 苯甲酰基和苯磺酰基; 其中的苯甲酰基和苯磺酰基 中的苯基任选被一个或两个独立选自羟基、 素、 硝基、 氨基、 氰基、 d-Cs烷基、 Cr 烷氧基和 6卤代烷基的取代基所取代; 各个 R5独立地选自氢、 羟基、 d-C^烷氧基、 d— 6 代烷基、 氨基、 ¾素、 硝基、 氰基和烷基酰基;
X为-(00) -;
n为 0、 1、 2或 3 ;
m为 0、 1、 2、 3或 4。 在本发明的另一个优选的实施方式中, 所述的化合物为具有通 式 lb的化合物:
其中,
Rl为苯基 - d- C6烷基、 d-C22烷基或( Ci。环烷基 -d- C6烷基, 其中的苯基未被取代或被一个、 两个或三个独立选自 iS素、硝基、 羟基、 氨基、 氰基、 C6烷基、 d-C6烷氧基和(^ 1¾代烷基的取 代基所取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或 氨基所取代, 或任选被 -0-、 -S -、 -NH -、 - C00-、 -C0NH-所间隔; 各个 R2独立地为氢、 羟基、 ¾素、 d- ( 6垸氧基、 d- C6烷基、 (^6 1¾代烷基、 氨基、 硝基、 氰基或烷基酰基;
R6为氢或 d-C6烷基;
!^为 ^- C
6烷基、 苯基 -d- C
6烷基, 其中苯基任选被一个、 两 个或三个羟基、 d- C
6烷基、 C广 (:
6烷氧基、 d—
6卤代烷基、 氨基、 卤素、 硝基、 氰基或烷基酰基所取代; 并且其中的烷基可以被羟 基、 0- (d- C
4) -烷基、氧代、氨基、 NH- (d- C
4) -烷基或 N- [ (C -C
6) - 烷基 ]
2取代, 或任选被- 0-、 +、 - NH -、 - C00-、 -C0NH-所间隔; n为 0、 1、 2或 3。 在本发明的另一个优选的实施方式中, 所述的化合物为具有通 式 Ic的化合物:
其中,
Rl为苯基 - d-C6烷基、 d- C22烷基或 C3- 。环烷基- d- C6烷基, 其中的苯基未被取代或被一个、 两个或三个独立选自 |¾素、硝基、 羟基、 氨基、 氰基、 (^炕基、 CH ^烷氧基和 Cw i¾代烷基的取 代基所取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或 氨基所取代, 或任选被- 0-、 -S -、 - NH -、 -C00-> -C0NH-所间隔; 各个 R2独立地为氢、 羟基、 卤素、 -(^烷氧基、 C广 C6烷基、 6 ¾代烷基、 氨基、 硝基、 氰基或烷基酰基;
R8和 R9各自独立选自氢, 任选被羟基、 氨基、 羧基、 d-C6 烷基、 d- C6烷氧基或氧代所取代的并任选被- 0-、 - S-和 -NH -、 - C00 -、 - C0NH-所间隔的 d-C22烷基; 或者
R8和 R9与其所连接的氮原子共同形成一个五元或六元杂环, 该杂环可以含有另外一个或两个独立选自 N、 0和 S的杂原子并且 可以与苯环形成稠合杂环, 所述杂环例如为吗啉、 哌嗪、 哌啶、 吡咯、 咪唑烷、 噻唑烷、 噁唑烷、 吲哚、 二氢吲哚, 所述杂环可 以任选被羟基、 d-C6烷基、 d-C6烷氧基、 d— 6 ¾代烷基、 氨基、 卤素、 硝基或氰基所取代;
n为 0、 1、 2或 3。 在本发明的另一个优选的实施方式中, 所述的化合物为具有通 式 Id的化合物:
其中,
Rl为苯基 -d- C6烷基、 d- C22烷基或 C3- 。环烷基 -d- C6烷基, 其中的苯基未被取代或被一个、 两个或三个独立选自卣素、硝基、 羟基、 氨基、 氰基、 -( 6烷基、 (^-(^烷氧基和( 6 代烷基的取 代基所取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或 氨基所取代, 或任选被 - 0-、 - S -、 -NH -、 -C00-、 -C0NH-所间隔; 各个 R2独立地为氢、 羟基、 |¾素、 d- ( 6烷氧基、 (^- 烷基、 6卤代烷基、 氨基、 硝基、 氰基或烷基酰基;
各个 R10独立地选自氢、 羟基、 d-Cs ^基、 d-C6烷氧基、 d-6 卤代烷基、 氨基、 1¾素、 硝基、 氰基和烷基酰基;
Y为 d- 。亚烷基、 d-Ci。亚烯基、 d-C。亚炔基; 所述亚烷基、 亚烯基和亚炔基任选被羟基、氨基、氧代、羧基、 d-C6坑基、 c -c6 烷氧基所取代或任选被- 0-、 - S -、 - NH -、 -C00-、 - C0NH-所间隔; n为 0、 1、 2或 3;
p为 0、 1、 2或 3。 在本发明的一个更优选的实施方式中, 所述的化合物为具有通 式 Iaa的化合物:
其中,
Rl为苯基 - d- C6烷基、 d-C22烷基或 CrCi。环烷基 -d-C6烷基, 其中的苯基未被取代或被一个或两个独立选自卤素、硝基、羟基、 氨基、 氰基、 C广 C6烷基、 d-C6烷氧基和 d— 6 ¾代烷基的取代基所 取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或氨基所 取代, 或任选被- 0-、 -S -、 -NH -、 - C00-、 - C0NH-所间隔;
各个 R2独立地为氢、 羟基、 卤素、 d- C6烷氧基、 d-C6烷基、 (^-6 |¾代烷基、 氨基、 硝基、 氰基或烷基酰基;
各个 R5独立地选自氢、 羟基、 d-C6烷基、 (^- 烷氧基、 d-6 卤代烷基、 氨基、 !¾素、 硝基、 氰基和烷基酰基;
n为 0、 1或 2 ;
m为 0、 1或 2。 在本发明的另一个更优选的实施方式中, 所述的化合物为具有 通式 Iba的化合物:
R1为苯基 -d-C6烷基、 C广 C22烷基或 C3- 。环烷基- d- C6烷基,
其中的苯基未被取代或被一个或两个独立选自卤素、硝基、羟基、 氨基、 氰基、 d- 烷基、 -(^烷氧基和 代烷基的取代基所 取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代或氨基所 取代, 或任选被 -0-、 - S -、 -NH -、 - C00-、 -C0NH-所间隔;
各个 R2独立地为氢、 羟基、 卤素、 - 烷氧基、 C广 烷基、 卤代烷基、 氨基、 硝基、 氰基或烷基酰基;
各个 R5独立地选自氢、 羟基、 d- (^坑基、 (^- 烷氧基、 d— 6 卤代烷基、 氨基、 1¾素、 硝基、 氰基和烷基酰基;
Z为 d- 。亚烷基;所述亚烷基任选被羟基、氨基、羧基、 d-C6 烷基或 d-C6烷氧基所取代或任选被- 0-、 -S -、 - NH -、 - C00-、 -C0NH- 所间隔;
n为 0、 1或 2 ;
m为 0、 1或 2。 在本发明的另一个更优选的实施方式中, 所述的化合物为具有 通式 Ic的化合物, 其中
R1为苯基 - d-C6烷基、 d- C22烷基或 C3- 。环烷基 - d-C6烷基, 其中的苯基未被取代或被一个或两个独立选自 素、硝基、羟基、 氨基、 氰基、 d-C6烷基、 d- ( 6烷氧基和 d-6 i¾代烷基的取代基所 取代; 并且其中的烷基和环烷基可以任选被羟基、 氧代、 氨基所 取代, 或任选被 -0-、 -S -、 -NH -、 - C00-、 -C0NH-所间隔;
各个 R2独立地为氢、 羟基、 卤素、 C广 ( 6烷氧基、 d-C6烷基、 d-6鹵代烷基、 氨基、 硝基、 氰基或烷基酰基;
R8和 R9各自独立选自氢; 任选被羟基、 氨基、 羧基、 d- 烷基、 d-C6烷氧基或氧代所取代的并任选被 -0-、 - S-和 -NH -、 -C00 -、 - C0NH-所间隔的 d- C22烷基; 或者
R8和 R9与其所连接的氮原子共同形成一个五元或六元杂环, 该杂环可以含有另外一个或两个独立选自 N、 0和 S的杂原子并且 可以与苯环形成稠合杂环, 所述杂环例如为吗啉、 哌嗪、 哌啶、 吡咯、 咪唑烷、 噻唑烷、 噁唑烷、 吲哚、 二氢吲哚, 所述杂环可 以任选被羟基、 d- C6烷基、 d- (^烷氧基、 (^-6 1¾代烷基、 氨基、 卤素、 硝基或氰基所取代;
n为 0、 1或 2。 在本发明的另一个更优选的实施方式中, 所述的化合物为具有 通式 Ida的化合物:
其中,
R1为苯基 -d- C6烷基、 d- C22烷基或 。环烷基 - d- C6烷基, 其中的苯基未被取代或被一个或两个独立选自 素、硝基、羟基、 氨基、 氰基、 CrC6烷基、 d- ( 6烷氧基和 d-6卤代烷基的取代基所 取代; 并且其中的垸基和环烷基可以任选被羟基、 氧代、 氨基所 取代, 或任选被 -0-、 -S -、 -NH -、 - C00 -、 -C0NH-所间隔;
各个 R2独立地为氢、 羟基、 ¾素、 d- Cs烷氧基、 d- C6烷基、 -6 1¾代烷基、 氨基、 硝基、 氰基或烷基酰基;
各个 R10独立地选自氢、 羟基、 d-Cs烷基、 d-C6烷氧基、 6 卤代烷基、 氨基、 ή素、 硝基、 氰基和烷基酰基;
丫为 d- (^亚烷基、 c广 。亚烯基或 d- 。亚炔基, 所述亚烷基、 亚烯基和亚炔基任选被羟基、氨基、氧代、羧基、 d- C6烷基或 d-C6
烷氧基所取代, 或任选被- 0-、 - S -、 -NH -、 - C00-、 - C0NH-所间 隔;
n为 0、 1或 2;
p为 0、 1或 2。 更进一步优选的式 la化合物选自:
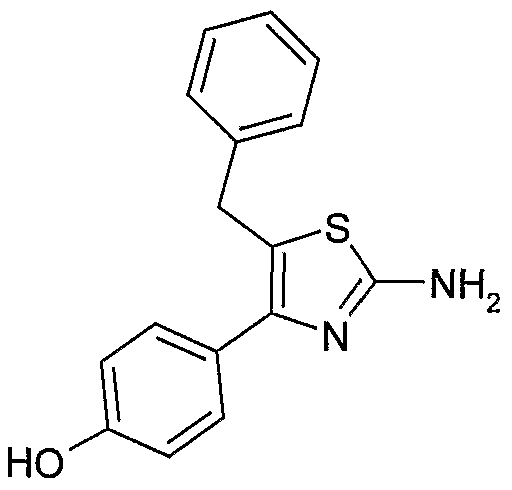
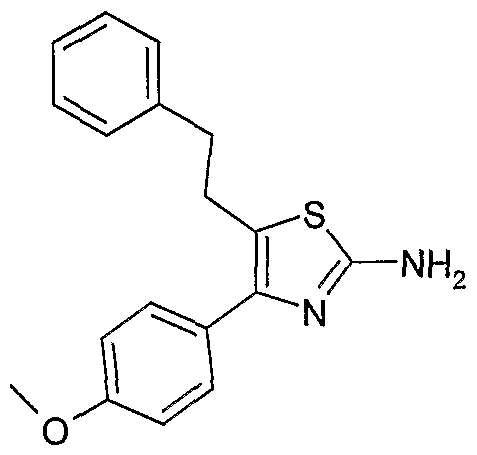
(1) 5-苄基- 4- (4-甲氧基-苯基) -噻唑- 2-基胺;
(2) 5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基胺;
(3) N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -苯曱酰胺;
(4) N- [5-苄基- 4- (4- ψ氧基-苯基) -噻唑 -2-基] -4-氟-苯甲酰 胺;
(5) N- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -4-氯-苯甲酰 胺;
(6) 4- (4-甲氧基-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺;
(7) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -4-氟-苯曱酰 胺;
(8) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -4-氯-苯甲酰 胺;
(9) 4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺;
(10) 4-氟 [4- (4-甲氧基-苯基) -5 -(4-硝基-苄基) -噻唑 -2- 基] -苯甲酰胺;
(11) 4-氟 -N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基] - 苯甲酰胺;
(12) N- [5-苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 4-二甲氧 基 -苯甲酰胺;
(13) N- [5 -苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -4 -氰基-苯
甲酰胺;
(14) 4-氰基- N- [4- (4-甲氧基-苯基) -5- (4-硝基-苄基) -噻唑 -2-基] -苯甲酰胺;
(15) 4-氰基 [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2 - 基] -苯甲酰胺;
(16) N- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 5-二氟- 苯甲酰胺;
(17) 3, 5-二氟 -N - [4- (4-甲氧基-苯基) -5- (4-硝基-苄基) -噻 唑- 2-基] -苯甲酰胺;
(18) 3, 5-二氟 -N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2- 基] -苯甲酰胺;
(19) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -3, 4-二羟基- 苯甲酰胺;
(20) N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基] -3, 4- 二甲氧基-苯甲酰胺;
(21) 5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2-基胺;
(22) 3, 5-二氟- N- [5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2 -基] -苯甲酰胺;
(23) 4-氟- N- [5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2- 基] -苯甲酰胺;
(24) N- [5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2- 基] -N- (3, 4-二甲氧基-苯甲酰胺基) -3, 4-二曱氧基-苯甲酰胺;
(25) 4-氰基 -N- [5- (4-氟-苄基) -4- (4-曱氧基-苯基) -噻唑 -2- 基] -苯甲酰胺;
(26) 5- (4-硝基-苄基) -4-苯基-噻唑- 2-基胺;
(27) N- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -4-曱氧基-
苯甲酰胺;
(28) 3,4-二甲氧基 - [4- (4-甲氧基-苯基)-5-(4-硝基-苄 基) -噻唑 -2-基] -苯甲酰胺;
(29) 3,4-二甲氧基-1^- [5- (4-硝基-苄基)-4-苯基-噻唑-2- 基] -苯 ψ酰胺;
(30) N- [5- (4-氟-苄基) -4- (4-甲氧基 -苯基)-噻唑 -2- 基] -3, 4-二甲氧基-苯甲酰胺;
(31) N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基] -3, 4- 二羟基 -苯甲酰胺;
(32) 3, 4-二羟基 -N- [5- (4-硝基-苄基) -4-苯基-噻唑- 2-基] - 苯曱酰胺;
(33) N- [5- (4-氟-苄基) -4- (4-羟基-苯基) -噻唑 -2-基] -3, 4- 二羟基 -苯曱酰胺;
(34) 3, 4-二羟基 -N- [4- (4-羟基-苯基) -5- (4-硝基-苄基) -噻 唑 -2-基] -苯甲酰胺;
(35) N- [5-苄基 1-4- (4-甲氧基-苯基) -噻唑 -2-基] -2, 4-二甲 氧基 -苯甲酰胺;
(36) N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -2-甲氧基- 苯甲酰胺;
(37) N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4, 5-三甲 氧基 -苯甲酰胺;
(38) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -2, 4-二羟基- 苯甲酰胺;
(39) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -2 -羟基-苯甲 酰胺;
(40) N- [5 -苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -4-甲氧基-苯
甲酰胺;
(41) N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -3, 4, 5-三羟基 -苯甲酰胺;
(42) N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3-甲氧基- 苯甲酰胺;
(43) N- [5 -苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 5-二甲氧 基 -苯甲酰胺;
(44) N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -3-羟基-苯甲 酰胺;
(45) N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 5-二羟基- 苯甲酰胺;
(46) N- [5-苄基- 4- (4 -羟基-苯基) -噻唑 -2-基] -4-羟基-苯甲 酰胺;
(47) 5-环己基甲基 -4- (4-甲氧基-苯基) -噻唑 -2-基胺;
(48) N- [5-环己基曱基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4- 二曱氧基-苯甲酰胺;
(49) N- [5-环己基甲基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 4 -二 羟基 -苯曱酰胺;
(50) N- [5-环己基曱基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 4, 5- 三曱氧基-苯甲酰胺;
(51) N- [5-环己基甲基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 4, 5- 三羟基 -苯甲酰胺;
(52) 4- (4-甲氧基-苯基) -5-甲基-噻唑- 2-基胺;
(53) 3, 4, 5-三甲氧基 -N- [4- (4-甲氧基-苯基) -5-曱基-噻唑 -2 -基] -苯甲酰胺;
(54) 3, 4-二甲氧基 -N- [4- (4-甲氧基-苯基) -5-甲基-噻唑- 2-
基] -苯甲酰胺;
(55) 3,4, 5-三羟基 - N- [4- (4-羟基-苯基)-5-甲基-噻唑-2- 基] -苯甲酰胺;
(56) 3, 4-二羟基 - N- [4- (4-羟基-苯基) -5-曱基-噻唑 -2-基] - 苯甲酰胺;
(57) 4- (4-曱氧基-苯基) -5-苯乙基 -噻唑 -2-基胺;
(58) 3, 4-二甲氧基 -N- [4- (4-甲氧基-苯基) -5-苯基乙基-噻唑 -2 -基] -苯曱酰胺;
(59) 3, 4, 5-三甲氧基 -N- [4- (4-曱氧基-苯基) -5-苯基乙基-噻 唑- 2 -基] -苯曱酰胺;
(60) 3, 4-二羟基 - N- [4- (4-羟基-苯基) -5-苯基乙基 -噻唑 -2- 基] -苯曱酰胺;
(61) 3,4,5-三羟基 - [4- (4-羟基-苯基)-5-苯基乙基-噻唑 - 2 -基] -苯甲酰胺;
(62) 4- (4-甲氧基-苯基) -5-正丁基 -噻唑 -2-基胺;
(63) N- [5-正丁基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 4-二甲 氧基 -苯甲酰胺;
(64) N- [5-正丁基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 4, 5 -三 曱氧基 -苯甲酰胺;
(65) N- [5-正丁基 -4- (4-羟基-苯基) -噻唑 -2-基] -3, 4-二羟基 -苯甲酰胺;
(66) N- [5-正丁基 -4- (4-羟基-苯基) -噻唑 -2-基] -3, 4, 5-三羟 基 -苯曱酰胺; 和
(67) N- [5-苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基] 对甲苯磺 酰基-对甲苯磺酰胺;
或其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。
更进一步优选的式 lb化合物选自:
(68) N- [5-苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3- (3, 4 -二 甲氧基-苯基)-丙酰胺; 和
(69) N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -3- (3, 4-二羟 基-苯基)-丙酰胺;
或其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。 更进一步优选的式 Ic化合物选自:
(70) 4- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2 -基] -吗啉;
(71) 1- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -哌嗪;
(72) 1- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -哌嗪三盐酸 盐;
(73) 2- [ [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] - (2-羟基- 乙基) -氨基 ] _乙醇;
(74) 4- (5-苄基 -2-吗啉- 4-基-噻唑- 4-基) -苯酚;
(75) 4- (5-苄基- 2-哌嗪- 4-基-噻唑 -4-基) -苯酚; 和
(76) 4- {5-苄基 -2- [双-(2-羟基-乙基) -氨基] -噻唑 - 4-基} -苯 酚;
或其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。 更进一步优选的式 Id化合物选自:
(77) 5-苄基 -2- [2- (3, 4-二甲氧基-苯基) -乙基〗 -4- (4-甲氧基 -苯基 X唑'' 和
(78) 4- {2- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -乙基 } -苯 一 1,2-二醇;
或其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。 本发明还涉及如通式 I所示化合物的合适的可药用的盐、 溶 剂合物或水合物, 其中可药用的盐包括但不局限于通式 I化合物 与无机酸如盐酸、 硫酸、 磷酸、 亚磷酸、 氢溴酸和硝酸所成的盐 以及与各种有机酸, 如马来酸、 苹果酸、 延胡索酸、 琥珀酸、 酒 石酸、 柠檬酸、 乙酸、 乳酸、 甲磺酸、 对甲苯磺酸、 椋榈酸等形 成的盐。
本发明中的一些化合物可能用水或各种有机溶剂结晶或重结 晶, 在这种情况下, 可能形成各种溶剂合物。 本发明包括那些化 学计量的溶剂合物, 包括水合物, 也包括在用低压升华干燥法制 备时形成的包含可变量水的化合物。
本发明还涉及通式 I化合物的各种异构体。 本发明中的部分 化合物可能以光学异构体或互变异构体的形式存在, 本发明包括 其所有可能的存在形式, 特别是纯异构体的形式。 不同的异构体 形式可以通过各种常规手段与其它形式的异构体分离或拆分开, 或者某种异构体可通过各种常规的合成方法或立体专一或不对称 合成的方法得到。 既然通式 I化合物是以药用为目的, 可以理解 它们最好以其纯的形式提供, 例如至少 60%纯度、 优选至少 75%、 更优选至少 85%、 最优选至少 98%纯度(°/。是指重量百分比) 。 不 纯化合物的制备方法可用于药用组合物中更纯的形式。 这些不够 纯的产物中至少含有 1%、 优选至少 5%、 更优选至少 10%的如通式 I所示的化合物或其可药用衍生物。
另一方面, 本发明涉及制备通式 I化合物的合成方法。 通式 I 的化合物可以已知的或购商可得的化合物为原料, 经过人工合 成方法制备。如果原料不能购得, 则本发明提供它们的制备方法,
或可以通过文献艮道的方法制备。
具体地说, 本发明提供了制备通式 I化合物或其可药用盐、 溶剂合物或水合物的方法, 下面分别以亚类结构描述。
通式 la化合物的制备方法如下:
(i) 将通式 II 的取代乙酸与过量二氯亚砜在室温至回流温 度下反应,制备得到通式 in的酰氯,通式 II的取代乙酸由商购 得到或由文献方法制备,
(ii) 在路易斯酸催化剂如 A1C13的存在下, 在傅克酰基化常 用溶剂如二氯甲烷、 1,2-二氯乙烷、 石油醚、 硝基苯等中, 在 -20 °C - 150°C的反应温度下, 将通式 III的酰氯与通式 IV的芳烃反 应, 得到通式 V的酮,
其中, Rl、 R2和 n具有如上所述的定义;
(iii) 在路易斯酸催化剂如 A1C1
3的存在下, 在惰性溶剂如 氯仿或四氯化碳中, 在 0°C ~ 50°C的反应温度下, 将通式 V的酮 与溴素反应, 得到通式 VI的 α-溴代酮,
其中, Rl、 R2和 n具有如上所述的定义;
( i V) 在缚酸剂无水乙酸钠的存在下,在低级醇类溶剂如乙醇 中, 在回流温度下, 将通式 VI的 α-溴代酮与硫脲反应得到通式 I I的 2-氨基噻唑,
(V) 在羰基活化剂如 N,N-二甲氨基-吡啶和缚酸剂如三乙胺 的存在下, 在惰性溶剂如四氢呋喃、 乙酸乙酯或二氯曱烷等中, 将通式 VII的 2-氨基噻唑与通式 VII I (其中 A为 Cl、 Br、 I)的羧 酸酰! ¾ (X=- CO-)或磺酰 ή (X=-S02-)反应得到通式 IX的化合物, 化合物 VI I I由相应的羧酸或磺酸与过量 S0C12反应制备,
其中 Rl、 R2、 R5、 X、 n和 m具有如上所述的定义;
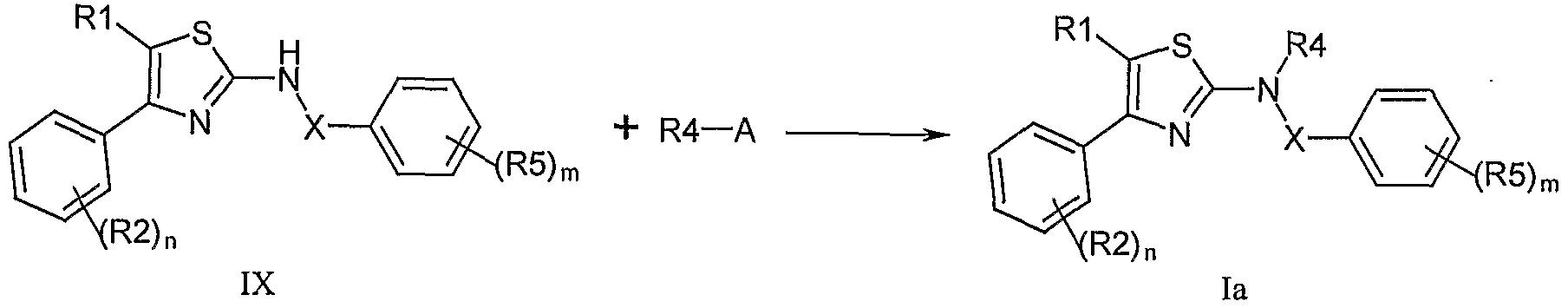
(vi) 在步驟(v)中当化合物 VI I I 过量时可以分离得到相应 的二酰基或二磺酰基取代的化合物,也可以以化合物 IX为原料制 备,在惰性溶剂如四氢呋喃中,将化合物 IX用氢化钠或丁基锂等 强碱处理,然后与卤代化合物或羧酸酰卤 R4-A (其中 A为 Cl、 Br、 I) 反应得到通式 la的化合物,
其中 Rl、 R2、 R4、 R5、 X、 n和 m具有如上所述的定义;
(vi i) 对通式 la 化合物中的取代基可以按照本技术领域已 知的方法进行官能团转化, 例如对于苯环上连接有甲氧基的化合 物, 可以在二氯甲烷中, 在 -78 ~ 50°C的温度范围内, 将其用三 溴化硼处理, 去除甲基得到相应羟基的化合物, 对于有多个苯环 上带有甲氧基或苯环上带有多个曱氧基的化合物相应增加三溴化 硼的摩尔数即可。 通式 lb化合物的制备方法如下:
(i) 在羰基活化剂如 Ν, Ν-二曱氨基-吡啶和縛酸剂如三乙胺 的存在下, 在惰性溶剂如四氢呋喃、 乙酸乙酯或二氯甲烷等中, 将通式 VI I的 2-氨基噻唑与通式 X的羧酸酰卤(其中 Α为 Cl、Br、 I)反应得到通式 XI 的化合物, 化合物 X 由相应的羧酸与过量
S0C 12反应制备
其中, Rl、 R2、 R7和 n具有如上所述的定义;
(i i) 在步骤(i)中当化合物 X过量时可以分离得到相应的二 酰基或二磺酰基取代的化合物。 也可以以化合物 XI为原料制备, 在惰性溶剂如四氢呋喃中,将化合物 XI用氢化钠或丁基锂等强碱 处理, 然后与卤代化合物或羧酸酰卤 XI I (其中 A为 Cl、 Br、 I) 反应得到通式 lb的化合物,
其中 Rl、 R2、 R6、 R7和 n具有如上所述的定义;
(i i i) 对通式 lb化合物中的取代基可以按照本技术领域已 知的方法进行官能团转化, 例如对于苯环上连接有甲氧基的化合 物, 可以在二氯甲烷中, 在 -78 ~ 50Ό的温度范围内, 将其用三 溴化硼处理, 去除甲基得到相应羟基的化合物, 对于有多个苯环
上带有甲氧基或苯环上带有多个曱氧基的化合物相应增加三溴化 硼的摩尔数即可。
通式 Ic化合物的制备方法如下:
(i) 在有机溶剂如乙腈中, 在 0 ~ 10°C的温度范围内, 将 通式 VII的 2-氨基噻唑用亚硝酸异戊酯和氯化铜处理, 得到通式 XIII的化合物,
其中 Rl、 R2和 n具有如上所述的定义;
(ii) 在有机溶剂如 N,N-二甲基甲酰胺(DMF)中, 在 50 ~ 150°C的温度范围内, 将通式 XIII的化合物用通式 XIV的胺或含 氮杂环、 氢氧化锂一水合物和碘化钾处理得到通式 Ic的化合物,
其中 Rl、 R2、 R8、 R9和 n具有如上所述的定义;
(Hi) 对通式 Ic化合物中的取代基可以按照本技术领域已 知的方法进行官能团转化, 例如对于苯环上连接有曱氧基的化合
物, 可以在二氯曱烷中, 在 -78 ~ 50°C的温度范围内, 将其用三 溴化硼处理, 去除甲基得到相应羟基的化合物, 对于有多个苯环 上带有甲氧基或苯环上带有多个甲氧基的化合物相应增加三溴化 硼的摩尔数即可。 通式 Id化合物的制备方法如下:
(i) 在室温至回流温度下, 将通式 XV的羧酸与过量 S0C12 反应, 得到通式 XVI的酰氯; 或者在縛酸剂如三乙胺的存在下, 在惰性溶剂如四氢呋喃中,在 -10 ~ 50°C的温度范围内,将通式 XV的羧酸与等摩尔的氯甲酸异丁酯反应得到通式 XVI I的混合酸 酐, 通式 XV的羧酸由商购得到或者按照本领域技术人员已知的 方法制备,
其中 R10、 Y和 p具有如上所述的定义;
(i i) 在 -10 - 50°C的温度范围内, 将通式 XVI的酰氯或通 式 XVI I的混合酸酐,与氨水或者氨在有机溶剂如四氢呋喃、曱醇、 乙醇等中的溶液反应得到通式 XVI I I的酰胺,
(R10)
XVII 其中 R10、 Y和 p具有如上所述的定义;
(iii) 在惰性溶剂如四氢呋喃中,将通式 XVIII的酰胺与 P2S5 反应得到通式 XIX的硫代酰胺,
其中 R10、 Y和 p具有如上所述的定义;
( i V ) 在缚酸剂无水乙酸钠的存在下,在低级醇类溶剂如乙醇 中, 在回流温度下, 将通式 XIX的硫代酰胺与通式 VI的 α-溴代 酮反应得到通式 Id的噻唑,
其中 Rl、 R2、 RI O, Y、 n和!)具有如上所述的定义;
(V) 对通式 Id 化合物中的取代基可以按照本技术领域已知 的方法进行官能团转化,例如对于苯环上连接有曱氧基的化合物, 可以在二氯甲烷中,在- 78 ~ 50°C的温度范围内,将其用三溴化 硼处理, 去除甲基得到相应羟基的化合物, 对于有多个苯环上带 有甲氧基或苯环上带有多个曱氧基的化合物相应增加三溴化硼的 摩尔数即可。 通式 I化合物可以用常规方法单个合成, 亦可用组合化学的 混-分方法或平行合成的方法以库 (每个库中至少含两个, 或 5-1000个, 最好是 10-100个化合物) 为单位合成, 即可以在液 相中合成也可以用固相合成方法。
关于制备通式 I化合物更详尽的资料见实施例。
另一方面, 本发明涉及通式 I的化合物、 或其所有可能的异 构体、 前药、 可药用盐、 溶剂合物或水合物用于生产药物的用途, 所述药物用于治疗和 /或预防哺乳动物的与血浆 PLTP活性升高相 关和 /或与血浆 CETP活性升高相关的疾病。 所述疾病包括但不局 限于如下疾病: 动脉粥样硬化、 外周血管疾病、 血脂异常症、 高 脂血症、 高胆固醇血症、 高甘油三酯血症、 家族性高胆固醇血症、 心血管疾病、 心绞痛、 局部缺血、 心脏缺血、 中风、 心肌梗塞、 再灌注损伤、 高血压、 糖尿病血管并发症、 肥胖症或内毒素性血 症等。
本发明还涉及治疗哺乳动物的与血浆 PLTP活性升高相关和 /
或与血浆 CETP活性升高相关的疾病的方法,该方法包括用合适的 方式对需要治疗的哺乳动物给予有效剂量的通式 I的化合物、 或 其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。 本 发明还公开了对需要治疗的哺乳动物的给药方式、 通式 I化合物 或其合适的可药用盐或水合物的有效剂量。
另一方面, 本发明涉及通式 I的化合物、 或其所有可能的异 构体、 前药、 可药用盐、 溶剂合物或水合物用于生产可抑制哺乳 动物血浆 PLTP活性和 /或血浆 CETP活性的药物的用途。
本发明还涉及抑制哺乳动物血浆 PLTP 活性和 /或血浆 CETP 活性的方法, 此方法包括对需要抑制血浆 PLTP 活性和 /或血浆 CETP 活性的哺乳动物用合适的方式给予有效剂量的通式 I 化合 物、 其所有可能的异构体、 前药、 可药用盐、 溶剂合物或水合物。
另一方面, 本发明的通式 I的化合物或其可药用的盐可以单 独使用, 或与可药用的载体或赋形剂一起以药物组合物的形式使 用, 当以药物组合物的形式使用时, 通常将有效剂量的本发明通 式 I化合物或其可药用盐或水合物以及一种或多种可药用载体或 稀释剂结合制成适当的施用形式或剂量形式, 这一程序包括通过 合适的方式将组分混合、 粒化、 压缩或溶解。 因此, 本发明提供 了药物组合物, 它包含通式 I的化合物、 其所有可能的异构体、 前药、可药用盐、溶剂合物或水合物以及至少一种可药用的载体。
本发明化合物的药物组合物,可以以下方面的任意方式施与: 口服、 喷雾吸入、 直肠给药、 鼻腔给药、 阴道给药、 局部给药、 非肠道给药如皮下、 静脉、 肌内、 腹膜内、 鞘内、 心室内、 胸骨 内或颅内注射或输入, 或借助一种外植的储器用药, 其中优选口 服、 肌注、 腹膜内或静脉内用药方式。
本发明化合物或含有它的药物组合物可以单位剂量形式给
药。 给药剂型可以是液体剂型、 固体剂型。 液体剂型可以是真溶 液类、 胶体类、 微粒剂型、 乳剂剂型、 混悬剂型。 其他剂型例如 片剂、 胶嚢、 滴丸、 气雾剂、 丸剂、 粉剂、 溶液剂、 混悬剂、 乳 剂、 颗粒剂、 栓剂、 冻干粉针剂、 包合物、 埋植剂、 贴剂、 擦剂 等。
本发明的药物组合物中还可以含有常用的载体, 这里所述可 药用载体包括但不局限于: 离子交换剂, 氧化铝, 硬脂酸铝, 卵 磷脂, 血清蛋白如人血清蛋白, 緩沖物质如磷酸盐, 甘油, 山梨 酸, 山梨酸钾, 饱和植物脂肪酸的部分甘油酯混合物, 水, 盐或 电解质, 如硫酸鱼精蛋白, 磷酸氢二钠, 磷酸氢钾, 氯化钠, 辞 盐, 胶态氧化硅, 三硅酸镁, 聚乙烯吡咯烷酮, 纤维素物质, 聚 乙二醇, 羧甲基纤维素钠, 聚丙烯酸酯, 蜂蜡, 羊毛酯等。 载体 在药物组合物中的含量可以是 1重量%-98重量%, 通常大约占到 80 重量%。 为方便起见, 局部麻醉剂, 防腐剂, 緩沖剂等可直接 溶于载体中。
口服片剂和胶嚢可以含有赋形剂如粘合剂, 如糖浆, 阿拉伯 胶, 山梨醇, 黄蓍胶, 或聚乙烯吡咯烷酮, 填充剂, 如乳糖, 蔗 糖, 玉米淀粉, 磷酸钙, 山梨醇, 氨基乙酸, 润滑剂, 如硬脂酸 镁, 滑石, 聚乙二醇, 硅土, 崩解剂, 如马铃薯淀粉, 或可接受 的增润剂, 如月桂醇钠硫酸盐。 片剂可以用制药学上公知的方法 包衣。
口服液可以制成水和油的悬浮液, 溶液, 乳浊液, 糖浆或酏 剂, 也可以制成干品, 用前补充水或其它合适的媒质。 这种液体 制剂可以包含常规的添加剂, 如悬浮剂, 山梨醇, 纤维素甲醚, 葡萄糖糖浆, 凝胶, 羟乙基纤维素, 羧甲基纤维素, 硬脂酸铝凝 胶, 氢化的食用油脂, 乳化剂, 如卵磷脂, 山梨聚醣单油酸盐,
阿拉伯树胶; 或非水载体(可能包含可食用油) , 如杏仁油, 油 脂如甘油, 乙二醇, 或乙醇; 防腐剂, 如对羟基苯甲酸曱酯或丙 酯, 山梨酸。 如需要可添加调味剂或着色剂。
栓剂可包含常规的栓剂基质, 如可可黄油或其它甘油酯。 对胃外投药,液态剂型通常由化合物和一种消毒的载体制成。 载体首选水。 依照所选载体和药物浓度的不同, 化合物既可溶于 载体中也可制成悬浮溶液, 在制成注射用溶液时先将化合物溶于 水中, 过滤消毒后装入封口瓶或安瓿中。
当皮肤局部施用时, 本发明化合物可以制成适当的软膏, 洗 剂, 或霜剂的形式, 其中活性成分悬浮或溶解于一种或多种的载 体中。 其中软膏制剂可以使用的载体包括但不局限于: 矿物油, 液体凡士林, 白凡士林, 丙二醇, 聚氧化乙烯, 聚氧化丙烯, 乳 化蜡和水; 洗剂和霜剂可使用的载体包括但不限于: 矿物油, 脱 水山梨糖醇单硬脂酸酯, 吐温 60, 十六烷酯蜡, 十六碳烯芳醇, 2 -辛基十二烷醇, 苄醇和水。
依据给药方式的不同, 组分中可以含有重量比 0. 1%, 或更合 适的重量比 10-60%的活性组分。 但組分中包含单位剂量时, 每个 单位最好包含 50- 500亳克活性成分。依据给药途径和给药频率的 不同, 用于成人的适宜治疗剂量为每天 100-3000 亳克, 如每天 1500 亳克。 这一剂量对应于 1. 5 - 50 毫克 /公斤 /天, 合适的剂 量是 5-20亳克 /公斤 /天。
必须认识到, 通式 I化合物的最佳给药剂量和间隔是由化合 物性质和诸如给药的形式、 途径和部位以及所治疗的特定哺乳动 物等外部条件决定的,而这一最佳给药剂量可用常规的技术确定。 同时也必须认识到, 最佳的疗程, 即通式 I化合物在额定的时间 内每日的剂量, 可用本领域内公知的方法确定。
具体实施方式
下面的具体实施例是本发明的优选实施方案, 其不应理解为 对本发明构成任何限制。
化合物的熔点由 RY- 1 熔点仪测定, 温度计未较正。 盾谱由 Micromas s ZabSpec 高分辨率质谱仪(分辨率 1000 )测定。 ^-NMR 由 JNM-ECA- 400超导 11仪测定, 工作频率1 H-NMR 400MHz。
实施例 1 5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基胺的制备
步骤 1 3-苯基丙酰氯的制备
在 100 mL茄形烧瓶中加入 20. 0 g (133. 2 讓 o l) 3 -苯基 丙酸和 24. 5 g (205. 9 irnnol)亚硫酰氯, 安装带有氯化钙干燥管和 氢氧化钠酸性气体吸收装置的回流冷凝管,磁力搅拌回流 1小时, 减压蒸馏除去过量的亚硫酰氯, 得到 3-苯基丙酰氯粗品, 为浅黄 色油状物, 未经纯化直接用于下步反应。
步骤 2 1- (4-甲氧基 -苯基)-3-苯基 -丙烷 -1-酮的制备 在配有机械搅拌器和温度计的 500 raL 三颈瓶中加入步骤 1 制备的理论量为 22. 4 g (133. 2 ramol)的 3-苯基丙酰氯溶于 100 mL 二氯曱烷中的溶液、 14. 4 g (205. 9 mmo l)苯甲醚和 150 mL二氯甲 烷, 用水盐浴冷却至- 5 °C以下, 保持内温在- 5 ~ -10 °C分批加入 17. 8 g (133, 2 腿 ol)无水 A1C13 , 加完后在该温度下搅拌 1小时, 室温下搅拌 1小时, 将反应液在搅拌下倾入 100 g水、 100 g水 和 40 mL浓盐酸的混合物中, 分出有机层, 水层用二氯甲烷萃取
2次 X2QmL, 合并有机层, 用饱和盐水洗涤、 饱和碳酸氢钠水溶 液洗涤, 再用饱和盐水洗涤至中性, 无水硫酸镁干燥, 过滤并减 压浓缩得到粗品, 用石油醚和乙酸乙酯(2:1)重结晶, 得到产物 23.5 g,母液浓缩后再次重结晶得到产物 3.3 g,合计收率 83.7 %, rap: 96-97 °C0 'H-NMR (CDC13, 400 MHz) δ 3.05 (2H, t, J=8.16Hz), 3.25(2H, t, J-8.16Hz), 3.86 (3H, s, 0CH3) , 6.92 (2H, d, J=9. OOHz, ArH), 7.18 - 7.32 (5H, m, ArH), 7.94 (2H, d, J=9. OOHz, ArH); EI -MS m/e (%) : 240.1 (M+, 37), 135 (100)。
步驟 3 2-涣 -1- (4-甲氧基 -苯基)-3-苯基-丙烷- 1-酮的制 备
在装配有温度计的 500 mL三颈瓶中加入 23.0 g(95.7 ramol) 步骤 2 制备的 1-(4-甲氧基 -苯基)- 3-苯基-丙烷 -1-酮、 0.30g(2.25 mmol)无水 A1C13和 150 mL氯仿, 搅拌溶解, 用冰水 浴控制保持温度在 5 ~ 10 滴加溶于 30 raL氯仿中的溴素, 控制 滴加使溴素的颜色很快退去,加完后在室温搅拌 30分钟,转入分 液漏斗中, 水洗涤、 饱和碳酸氢钠水溶液洗涤, 再次水洗涤, 无 水硫酸钠干燥, 过滤并减压浓缩得到粗品, 经硅胶柱层析, 石油 醚 /乙酸乙酯(13 : 1)洗涤脱得到产物 28.7 g, 无色液体, 用无 水乙醇重结晶得到白色固体, 收率 93.9%, mp: 57-58 。C。
^- MR (CDC13, 400 MHz) S: 3.34 (1H, dd, J=14.28, 7. OOHz), 3.66 (1H, dd, J=14.28, 7.00Hz), 3.13 (3H, s, 0CH3), 5.29 (1H, t, J=7.32Hz, CHBr) , 6.91 (2H, d, J=9.00Hz, ArH), 7.18 ~ 7.32 (5H, m, ArH), 7.94 (2H, d, J=9.00Hz, ArH); ESI -MS ra/e(°/o) : 320.8 (M+1, 100), 239.2 (M-Br, 18)。
步骤 4 5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基胺的制备 在 500 mL茄形烧瓶中加入 25.00 g (78.32 mmol)步骤 3制备
的 2-溴- 1- (4-甲氧基 -苯基)-3-苯基 -丙烷 -1-酮和 150raL无水乙 醇, 搅拌下加入 5.96 g(78.32 mmol)硫脲和 6.42 g(78.32 mmol) 无水乙酸钠, 室温下搅拌 1小时, 回流 2小时, 冷却至室温, 析 出固体, 过滤, 用水洗涤, 干燥得到产物 18.01 g, TLC显示单一 斑点,未进一步纯化,收率 77.6°/o,mp: 177-1780C。 1H -醒(DMS( 6 400 MHz) δ: 3.76 (3Η, s, 0CH3), 4.04 (2Η, s, CH2), 6.82 (2H, s, NH2), 6.93 (2H, d, J=9.00Hz, ArH) , 7.18 ~ 7.35 (5H, m, ArH) 7.46 (2H, d, J=9.00Hz, ArH); EI -MS ra/e(%): 296.1 (M+, 100), 219.0(19); HREI-MS Calcd. for C17H16N2OS: 296.0983, found: 296.0978; Anal. Calcd. for C„H16N2OS: C 68.89, H 5.44, N 9.45; found: C 69.08, H 5.32, N 9.57. 实施例 2 5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基胺的制备
在 50 mL茄形烧瓶中加入 0.50 g(l.69 mmol)实施例 1制备 的 5 -苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基胺和 15 mL二氯甲烷, 室温下滴加 2.9mL(5.06 mmol) 1.76 M的 BBr3二氯甲烷溶液, 室 温搅拌 2小时, 倾入 20 mL饱和碳酸氢钠水溶液中, 析出固体, 过滤、 水洗涤并干燥得到粗品, 经硅胶柱层析, 石油醚 /乙酸乙酯 (3 : 2)洗涤脱得到产物 0.46 g, 白色固体, 收率 95.8%, rap: 170-172 °C。 1H-NMR (DMS0- 6, 400 MHz) δ: 4.02 (2H, s, CH2), 6.74 - 6.76 (4H, m, 2ArH+NH2) , 7.15 - 7.40(7H, m, ArH),
9.49(1H, s, OH); EI -MS m/e (%): 282.0 (M+, 100) , 205.0 (37) ; HREI-MS Calcd. for C16H14N2OS: 282.0827, found: 282.0824。 实施例 3 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -苯 甲酰胺的制备
在 100 mL三颈瓶中加入 0.50 g(l.69 mmol)实施例 1制备的 5 -苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基胺、 1.42 g(10.12 匪 ol) 三乙胺和 40 mL 乙酸乙酯, 滴加溶于 10 mL 乙酸乙酯中的 0.48 g(3.38 ramol)苯甲酰氯, 室温下磁力搅拌反应过夜, 抽滤, 乙酸 乙酯洗涤, 滤液用 1 N氢氧化钠水溶液 7洗涤, 水洗涤至中性, 无水硫酸钠干燥, 过滤并减压浓缩得到粗品, 经硅胶柱层析, 石 油醚 /乙酸乙酯(5 : 1)洗涤脱得到产物 0.39 g, 白色固体, 收率 57.4%, mp: 146-147 °C。 !H-NMR (CDC13, 400 MHz) δ: 3.88 (3H, s, 0CH3) , 4.20 (2H, s, CH2), 7.01 (2H, d, J=8.72Hz, ArH),
7.31 (2H, d, J=8.72Hz, ArH), 7.42 ~ 7· 65 (7H, m, 6ArH+C0NH) ,
8.09 (2H, d, J=7.28Hz, ArH), 8.17 (2H, d, J=7.28Hz, ArH) ; EI -MS ra/e(%): 400.0 (M
+, 94), 104.9 (100); HREI-MS Calcd. for C
24H
20N
2O
2S: 400.1245, found: 400.1235。 实施例 4 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -4- 氟-苯甲酰胺的制备
在 50 mL茄形烧瓶中加入 1.00 g(3.37 mmol)实施例 1制备 的 5 -苄基 -4-(4 -曱氧基-苯基)-噻唑 -2 -基胺、 0.68 g (6· 75讓 ol) 三乙胺、 30 mg(0.25 讓 ol)DMAP和 25 mL THF, 室温下滴加溶于 5 mL THF中的 0.53 g (3.37 麵 ol)对氟苯甲酰氯, 室温下搅拌 2 小时, 升温至 35~40 °C反应 10小时, 过滤, THF洗潦, 母液减 压浓缩得到粗品浅棕色油状物, 用 50mL乙酸乙酯和 10mL 1NHC1 分层, 分出有机层, 饱和碳酸氢钠水溶液洗涤, 饱和盐水洗涤, 无水硫酸钠干燥, 过滤浓缩得到粗品, 以乙酸乙酯 /石油醚(1: 2) 重结晶得到产物,白色晶体 1.10 g,收率 78.0%, mp: 164-165 °C。
'H-NMR (CDC 13, 400 MHz) δ: 3.77 (3H, s, 0CH3), 4.22 (2H, s, CH2),
6.76 (2H, d, J=7.84Hz, ArH) , 6.94 (2H, t, 3JF„=3J„„=8.40Hz, ArH)
7.24 - 7.36 (7H, m, ArH), 7.71 (2H, dd,
3J
HH=7.56,
4J
F„=5.32Hz, ArH), 11.62 (1H, br, CONH); EI -MS m/e ( ): 418.2 (M
+, 100) , 123.0 (86); HREI-MS Calcd. for C
24H
19FN
20
2S: 418.1151, found: 418.1138; Anal. Calcd. for C
24H
19FN
20
2S: C 68.88, H 4.58, N 6.69; found: C 68.77, H 4.48, N 6.84. 实施例 5 N- [5 -苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] - 4- 氯-苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4- (4-甲氧基 -苯基)-噻唑 -2-基胺和对氯苯甲酰氯为原料, 得到 粗品后用丙酮重结晶得到产品, 为白色晶体, 收率 69.4%, mp: 182-183 °C。
400 MHz) δ: 3.78 (3H, s, 0CH
3) , 4.21 (2H, s, CH
2), 6.75 (2H, d, J-7.56Hz, ArH), 7.22 - 7.35 (9H ra, ArH), 7.60 (2H, d, J=7.56Hz, ArH), 11.70(1H, br, CONH); EI -MS ra/e(%): 434.2 (M+, 100) , 123.0 (83); HREI-MS Calcd. for C
24H
19C1N
20
2S: 434.0856, found: 434.0859; Anal. Calcd. for C
24H
19C1N
20
2S: C 66.28, H 4· 40, N 6.44; found: C 65.91, H 4.22, N 6.50。 实施例 6 4- (4-甲氧基-苯基) -5- (4-硝基-苄基) -噻唑 -2- 基胺的制备
步骤 1 3- (4-硝基-苯基) -丙酸的制备
在具温度计的 1000 mL三颈瓶中加入 50.0 g(332.9 mmol) 3- 苯基丙酸、 225 mL冰醋酸和 125 mL浓硫酸, 滴加由 25.7 mL 65%
的硝酸(366.2 mmol)和 35 mL浓硫酸预先混合并冷却至 5 °C左右 的混酸,冰水浴保持内温 18 ~ 25 °C,保温 20 25 °C搅拌 5小时, 将反应液倾入 500 g水中, 抽滤析出的固体, 水洗涤至中性, 干 燥后用乙醇重结晶得到产物 35.8 g, 白色固体, 收率 55.1%, 166-167 。C。 一 NMR(CDC13, 400 MHz) δ: 2.62 (2Η, t, J=7.56Hz),
2.96 (2H, t, J=7.56Hz), 7.53 (2H, d, J=8.68Hz, ArH), 8.15 (2H, d, J=8.68Hz, ArH), 12.22 (1H, s, C00H); ESI- MS ra/e(%): 218.0(M+Na, 27), 213.4(M+NH3, 3), 196.1 (M+l, 100)。
步骤 2 3- (4-硝基-苯基)-丙酰氯的制备
釆用与实施例 1 步骤 1 类似的方法制备, 将 3- (4-硝基-苯 基) -丙酸转化为 3- (4-硝基-苯基) -丙酰氯, 其为白色固体, 未经 纯化直接用于下步反应。
步骤 3 1- (4-甲氧基-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮 的制备
采用与实施例 1 步骤 2 类似的方法制备, 以步骤 2 制备的 3-(4-硝基-苯基) -丙酰氯和苯曱醚为原料, 无水 A1C13为催化剂, 得到产物为白色固体,收率 81.3%,mp: 123-125 °C。 - NMR (CDC13, 400 MHz) δ: 3.18 (2H, t, J=7.00Hz), 3.30 (2H, t, J=7.00Hz),
3.87 (3H, s, 0CH3) , 6.93 (2H, d, J=8.96Hz, ArH), 7.42 (2H, d, J=8.60Hz, ArH), 7.93 (2H, d, J=8.96Hz, ArH), 8.15 (2H, d, J=8.60Hz, ArH); EI -MS ra/e( ): 285.0 (M+, 62), 134 (100) 0 步骤 4 2-溴 -1- (4-曱氧基-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮的制备
采用与实施例 1 步骤 3类似的方法制备, 以步骤 3 制备的 1- (4-曱氧基-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮和溴素为原料, 无水 A1C13为催化剂, 得到产物为白色晶体, 收率 96.1%, mp:
120-122 V。 'H-MR (CDC13, 400 MHz) δ: 3.46 (1H, dd, J=14.28, 7.28Hz), 3.74 (1H, dd, J=14.28, 7.28Hz), 3.88 (3H, s, OCH3) , 5.27(1H, t, J=7.28Hz, CHBr) , 6.94 (2H, d, J=8.96Hz, ArH),
7.46 (2H, d, J=8.60Hz, ArH), 7.96 (2H, d, J=8.96Hz, ArH),
8.16 (2H, d, J=8.60Hz, ArH); ESI— MS: 366.1 (M+2, 93), 364.2 (M, 100)。
步骤 5 4- (4-甲氧基-苯基) -5- (4-硝基-苄基) -噻唑 - 2 -基 胺的制备
在 50 mL茄形烧瓶中加入 0.45 g(1.24 醒 ol)步骤 4制备的 2-溴- 1- (4-甲氧基 -苯基)-3- (4-硝基-苯基) -丙烷 -1-酮和 15 mL 无水乙醇,搅拌下加入 94 mg(l.24 mmol)疏脲和 101 mg(l.24 ramol) 无水乙酸钠, 回流 3小时, 减压蒸馏除去溶剂, 以 20 mL乙酸乙 酯和 20 mL 5% NaHC0
3水溶液分层, 分出有机层, 以饱和盐水洗 涤至中性, 无水硫酸钠干燥, 过滤浓缩得粗品, 乙酸乙酯重结晶 得到产物 0.34 g , 收率 79.1%, mp: 209-210 °C (Dec.:)。 ^-NMR (DMS0-i
6, 400 MHz) δ: 3.34 (3H, s, 0CH
3) , 4.20 (2H, s, CH
2), 6.93(2H, s, NH
2) , 6.95 (2H, d, J=8.72Hz, ArH), 7.43- 7.46 (4H, m, ArH), 8.19 (2H, d, J=8.72Hz, ArH); EI-MS m/e (%) : 341.2 (M
+, 100), 219.1 (14); HREI-MS Calcd. for C
17H
15N
30
3S: 341.0834, found: 341.0834; Anal. Calcd. for C
17H
15N
30
3S: C 59.81, H 4.43, N 12.31; found: C 59.76, H 4.36, N 12.05. 实施例 7 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -4-氟- 苯甲酰胺的制备
在 100 mL三颈瓶中加入 0.60 g(l.43 mraol)实施例 4制备的 N- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -4-氟-苯甲酰胺和 15 mL二氯曱烷, 冰盐浴控制温度在- 10 °C以下, 滴加 1.7 mL浓 度为 1.763 mol/L的 BBr3二氯甲烷溶液, 加完后在该温度下搅拌 1小时, 室温搅拌 1小时, 加入 2 g冰块, 减压蒸馏除去二氯甲 烷, 加入 20 mL乙酸乙酯, 分出有机层, 饱和盐水洗, 无水硫酸 钠干燥, 过滤减压浓缩得到粗品, 用乙酸乙酯 /石油醚重结晶得到 产物 0.39 g, 白色固体, 收率 67.2%, mp: 205-206 °C。
^-NMR m 0-d6, 400 MHz) δ: 4.22 (2H, s, CH2), 6.84 (2H, d, J=8.68Hz, ArH), 7.23 - 7.39 (7H, m, ArH), 7.47 (2H, d, J=8.68Hz, ArH), 8.15 (2H, dd, 3JHH=9.00, 4JFH=5.64Hz, ArH), 9.62 (1H, s, OH), 12.66 (1H, s, CONH); EI -MS m/e(%): 404.1 (M+, 100), 123.0 (70); HREI-MS Calcd. for C23H17FN202S: 404.0995, found: 404.0988。 实施例 8 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] - 4-氯- 苯曱酰胺的制备
釆用与实施例 7类似的方法制备, 将实施例 5制备的 N- [5-
苄基— 4- (4-甲氧基-苯基) -噻唑 - 2-基] -4-氯-苯甲酰胺转化为 N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -4-氯-苯甲酰胺, 为白 色固体, 收率 65.1%, mp: 109-110°C。 'H-NMR (DMS0-i6, 00 MHz) δ: 4.20 (2H, s, CH2), 6.83 (2H, d, J=8.68Hz, ArH), 7.26 ~ 7.36 (7H, m, ArH), 7.42 (2H, d, J=7.56Hz, ArH), 7.88 (2H, d, J=8.68Hz, ArH) , CONH and OH disappear; EI -MS m/e (%): 420.1 (M+, 100), 123.0 (94); HREI- S Calcd. for C23H17C1N202S: 420.0699, found: 420.0703。 实施例 9 4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺 的制备
步骤 1 1- (4-氟-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮的制 备
采用与实施例 1步骤 2类似的方法制备, 以实施例 6步骤 2 制备的 3- (4-硝基-苯基) -丙酰氯和氟苯为原料, 无水 A1C13为催 化剂, 得到产物为白色固体, 收率 61.2%, mp: 122-124 °C。 'H-NMR (CDC 13, 400 MHz) δ: 3.19 (2H, t, J=7.28Hz), 3.32 (2H, t, J=7.28Hz), 7.14 (2H, t, 3JFH=3JHH-8.40Hz, ArH), 7.42 (2H, d, J=8.40Hz, ArH), 7.99 (2H, dd, 3JHH=7.84, 4JFH=5.60Hz, ArH), 8.16 (2H, d, J=8.40Hz, ArH); EI -MS m/e(%): 273.1 (M+, 55), 123.0 (100)。
步骤 2 2-溴- 1- (4-氟-苯基) -3- (4-硝基-苯基) -丙烷 -1 -酮 的制备
采用与实施例 1 步骤 3类似的方法制备, 以步驟 1 制备的 1- (4-氟-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮和溴素为原料,无水 A1C13为催化剂,得到产物为浅黄色晶体,收率 95.3%,mp: 126-128 X:。 —NMlUCDCls, 400 MHz) δ: 3· 47 (1Η, dd, J=14.28, 7· 28Ηζ), 3.74 (1Η, dd, J=14.28, 7.28Ηζ), 5· 25 (1Η, t, J=7.28Hz, CHBr) ,
7.15 (2Η, t,
40HZ, ArH) , 7.46 (2Η, d, J-8.權 ζ, ArH)
8.03 (2Η, dd, 3J„„=8.72, 4JF„=5.36Hz, ArH), 8.17 (2H, d, J=8.40Hz, ArH); ESI-MS (negative ion): 352.2 (M-1, 54), 350.2 (M-3, 52), 256.2 (100).
步驟 3 4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺的 制备
釆用与实施例 6步骤 5类似的方法制备, 以步骤 1制备的 2- 溴- 1- (4-氟-苯基) -3- (4-硝基-苯基) -丙烷 -1-酮、 硫脲和无水乙 酸钠为原料, 得到粗品后用乙酸乙酯和丙酮混合溶剂重结晶得到 产物,为黄色固体,收率 71.4%, mp: 217-218 °C 0 'H-NMR (DMS0-i6, 400 MHz) δ: 4.22 (2H, s, CH2), 6.99 (2H, s, NH2), 7.21 (2H, t,
7 (2H, d, J=8.96Hz, ArH), 8.03 (2H,
ArH), 8.19 (2H, d, J=8.68Hz, ArH); EI -MS m/e(%): 329.1 (M+, 100), 207.0 (21); HREI-MS Calcd. for C
16H
12FN
30
2S: 329.0634, found: 329.0627; Anal. Calcd. for Ci
6H
12FN
30
2S: C 58.35, H 3.67, N 12.76; found: C 58.61, H 3.52, N 12.61。 实施例 10 4-氟 -N- [4- (4-甲氧基-苯基) -5- (4-硝基-苄
基) -噻唑 -2-基] -苯曱酰胺的制备
釆用与实施例 4类似的方法制备, 以实施例 6制备的 4- (4- 甲氧基-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺和对氟苯甲酰氯为 原料, 于 35-40 °C反应 48小时, 后处理得到粗品, 经硅胶柱层 析纯化, 石油醚和乙酸乙酯(4: 1)洗脱得到产物, 为黄色固体, 收 率 81.3%, mp: 188-189 X:
0 ^-NMR (CDC1
3, 400 MHz) δ: 3.82 (3H, s, 0CH
3) , 4.33 (2H, s, CH
2) , 6.87 (2H, d, J=8.68Hz, ArH), 7.10 (2H, t,
ArH) 7.39 (2H, d, J=8.68Hz, ArH), 7.89 (2H, dd,
3J
HH=8.68,
4J
F„=5.02Hz, ArH), 8.19 (2H, d, J=8.68Hz, ArH); EI -MS ra/e(%): 463.1 (M
+, 60), 123.0 (100); HREI-MS Calcd. for C
24H
18FN
30
4S: 463.1002, found: 463.1000; Anal. Calcd. for C
24H
18FN
30
4S: C 62.19, H 3.91, N 9.07; found: C 62.21, H 3.82, N 9.02。 实施例 11 4-氟- N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻 唑 -2-基] -苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 9制备的 4-(4- 氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺和对氟苯曱酰氯为原 料, 于 35-40 Ό反应 24小时, 后处理得到粗品, 经硅胶柱层析 纯化, 石油醚和乙酸乙酯(4: 1)洗脱得到产物, 为浅黄色固体, 收 率 69.6%, mp: 120-122 。 ^-NMR (CDC1
3, 400 MHz) δ: 4.33 (2Η,
4JFH=5.32HZ, ArH), 8.20 (2H, d, J=8.68Hz, ArH); EI -MS ra/e(%): 451.0(M+, 32), 123.0 (100); HREI-MS Calcd. for C23H15F2N303S: 451.0802, found: 451.0818; Anal. Calcd. for C23H15F2N303S: C 61.19, H 3.35, N 9.31; found: C 61.16, H 3.08, N 9.35. 实施例 12 N- [5-苄基- 4- (4-甲氧基-苯基)-噻唑- 2- 基] -3, 4-二甲氧基-苯甲酰胺的制备
步骤 1 3, 4-二甲氧基-苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 3, 4-二曱氧基- 苯甲酸转化为 3, 4-二曱氧基-苯曱酰氯, 其为白色固体, 未经纯 化直接用于下步反应。
步骤 2 N-〖5 -苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4- 二甲氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基
-4-(4-曱氧基-苯基) -噻唑 -2-基胺和步骤 1制备的 3, 4-二曱氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶 柱层析纯化, 石油醚和乙酸乙酯(10: 1)洗脱得到产物, 为白色固 体, 收率 46. 5% , mp: 179-180 °C o 'H-NMR (CDC13, 400 MHz) δ: 3. 79 (3H, s, 0CH3) , 3. 83 (3H, s, 0CH3), 3. 89 (3H, s, 0CH3) , 4. 22 (2H, s, CH2), 6. 73 (1H, d, J=8. 40Hz, ArH) , 6. 78 (2H, d, J=8. 40Hz, ArH) , 7. 26 ~ 7. 35 (9H, m, ArH), 11. 03 (1H, br, CONH); EI-MS m/e (%) : 460. 1 (M+, 99) , 164. 9 (100) ; HREI-MS Ca lcd. for C26H24N204S: 460. 1457, found: 460. 1463; Anal. Calcd. for C26H24N204S: C 67. 81, H 5, 25, N 6. 08; found: C 67. 62, H 5. 20, N 6. 50. 实施例 13 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -4- 氰基-苯甲酰胺的制备
步骤 1 对氰基苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将对氰基苯甲酸转 化为对氰基苯甲酰氯, 其为白色固体, 未经纯化直接用于下步反 应。
步骤 2 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -4-氰 基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4-(4-甲氧基 -苯基)-噻唑 -2-基胺和步骤 1制备的对氰基苯甲酰
氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层析 純化, 石油醚和乙酸乙酯(20: 1~ 10: 1)梯度洗脱得到产物, 为白 色固体, 收率 39.5%, mp: 194- 196°C。 】H -醒(CDC13, 400 MHz) δ: 3.77 (3H, s, 0CH3) , 4.21 (2H, s, CH2) , 6.69 (2H, d, J=8.68Hz, ArH), 7.23 - 7.32 (5H, ra, ArH), 7.36 (2H, d, J=8.68Hz, ArH), 7.48(2H, d, J=8.16Hz, ArH), 7.72 (2H, d, J=8.16Hz, ArH); EI-MS ra/e(°/o): 425.2 (M+, 100), 130.0 (40); HREI-MS Calcd. for C25H19N302S: 425.1198, found: 425.1191; Anal. Calcd. for C25H19N302S: C 70.57, H 4.50, N 9.88; found: C 70.50, H 4.39, N 9.99。 实施例 14 4-氰基 -N- [4- (4-甲氧基-苯基) -5- (4-硝基-苄 基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 6制备的 4- (4- 甲氧基 -苯基)-5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 13 的步 骤 1制备的对氰基苯曱酰氯为原料, 于室温下反应过夜, 后处理 得到粗品,经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(20: 1 ~ 10: 1) 梯度洗脱得到产物, 为黄色固体, 收率 26.8%, rap: 190-192X。 ^-NMR (CDCI3, 400 MHz) δ: 3.80(3H, s, 0CH3) , 4.32 (2H, s, CH2) ,
6.75 (2H, d, J=8.72Hz, ArH), 7.25 (2H, d, J=8.68Hz, ArH),
7.37 (2H, d, J=8.68Hz, ArH), 7.59 (2H, d, J=8.40Hz, ArH), 7.82 (2H, d, J=8.40Hz, ArH), 8.19 (2H, d, J=8.72Hz, ArH);
EI-MS m/e(%): 470.1 (M+, 100), 130.0 (76); HREI-MS Calcd. for C25H18N404S: 470.1049, found: 470.1050; Anal. Calcd. for C25H18N404S: C 63.82, H 3.86, N 11.91; found: C 64.16, H 3.68, N 12.19. 实施例 15 4-氰基 -N- [4- (4-氟-苯基) -5- (4-硝基-苄基) - 噻唑- 2-基]-苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 9制备的 4-(4- 氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 13 的步驟 1 制备的对氰基苯甲酰氯为原料, 于室温下反应过夜, 后处理得到 粗品, 以乙酸乙酯和石油醚混合溶剂重结晶得到产物, 为类白色 固体, 收率 62.9%, mp: 244-245°C。 aH-NMR (CDC13, 400 MHz) δ: 4.33 (2H, s, CH2), 7.07 (2H, t, 3JFH-3JHH-8.44Hz, ArH) , 7.38 (2H, d, J=8.40Hz, ArH), 7.43(2H, dd, 3JHH=8.72, 4JF„=5.32Hz, ArH),
7.75 (2H, d, J=7.84Hz, ArH), 7.98 (2H, d, J=8.12Hz, ArH),
8.20 (2H, d, J=8.40Hz, ArH); EI-MS m/e (%): 458.3 (M+, 79), 129.9 (100); HREI-MS Calcd. for C24H15f 03S: 458.0849, found: 458.0850; Anal. Calcd. for C24H15FN403S: C 62.88, H 3.30, N 12.22; found: C 62.52, H 2.96, N 12.04。 实施例 16 N-[5-苄基- 4-(4-甲氧基 -苯基)-噻唑 -2-
基] -3, 5-二氟-苯曱酰胺的制备
步骤 1 3, 5-二氟苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 3, 5-二氟-苯甲 酸转化为 3, 5-二氟苯甲酰氯, 为无色液体, 未经純化直接用于下 步反应。
步骤 1 N- [5-苄基 - 4-(4-甲氧基-苯基) -噻唑 -2-基] -3, 5- 二氟-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 - 4- (4-甲氧基-苯基) -瘘唑 -2-基胺和步骤 1制备的 3, 5-二氟苯甲 酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层 析纯化, 二氯甲烷和乙酸乙酯(20: 1 ~ 10: 1)梯度洗脱得到产物, 为白色固体,收率 59.1%, mp: 154- 155Ό。 - NMR(CDC13, 400 MHz) δ: 3.77 (3Η, s, 0CH3), 4.23 (2H, s, CH2) , 6.72 (2H, d, J=8.44Hz, ArH), 6.82 (1H, t, 3JFH=8.40Hz, ArH) , 7.17 ~ 7.37 (9H, m, ArH), 11.97 (1H, br, CONH); EI -MS m/e (%): 436. l (M+, 100), 141.0(36); HREI-MS Calcd. for C24H18F2N202S: 436.1057, found: 436.1053; Anal. Calcd. for C24H18F2N202S: C 66.04, H 4.16, N 6.42; found: C 66.02, H 4.08, N 6.44。 实施例 17 3, 5-二氟 -N- [4- (4-甲氧基-苯基) -5- (4-硝基- 苄基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 6制备的 4- (4 - 甲氧基 -苯基)-5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 16步骤 1 制备的 3, 5-二氟苯甲酰氯为原料, 于室温下反应过夜, 后处理得 到粗品, 经硅胶柱层析纯化, 二氯甲烷和乙酸乙酯(20: 1~ 10: 1) 梯度洗脱得到产物, 为白色固体, 收率 66.7%, mp: 189 - 190°C。
^-NMR (CDC13, 400 MHz) δ: 3.76 (3Η, s, 0CH3) , 4.34 (2H, s, CH2),
6.72 (2H, d, J=8.68Hz, ArH), 6.85 (1H, t, 3JF„=8.40Hz, ArH) ,
7.18 (2H, d, 3JFH =5.32Hz, ArH), 7.25 (2H, d, J=8.68Hz, ArH),
7.40 (2H, d, J=8.40Hz, ArH), 8.21 (2H, d, J=8.40Hz, ArH), 12.05 (1H, br, CONH); EI -MS m/e (%): 481.1 (M+, 100), 141.0(63); HREI-MS Calcd. for C24H17F2N304S: 481.0908, found: 481.0902; Anal. Calcd. for C24HnF2N30,S: C 59.87, H 3.56, N
8.73; found: C 59.98, H 3.80, N 8.67。 实施例 18 3, 5-二氟 -N- [4-(4-氟-苯基) - 5- (4-硝基-苄 基) -噻唑 -2-基]-苯甲酰胺的制备
釆用与实施例 4类似的方法制备, 以实施例 9制备的 4- (4- 氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 16步骤 1制备 的 3, 5-二氟苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗 品, 经硅胶柱层析纯化, 二氯甲烷和乙酸乙酯(20: 1~ 10: 1)梯度 洗脱得到产物, 为白色固体, 收率 58.0%, mp: 216-218 °C。
'H-NMR (DMS0-i¾, 400 MHz) δ: 4.44 (2H, s, CH
2) , 7.30 (2H, t,
ArH), 7.30 (2H, d, 3JF„=6.16Hz, ArH), 8.20(2H, d, J=8.68Hz, ArH), 12.95 (1H, br, CONH); EI- S ra/e(%): 469.1 (M+, 77), 141.1(100); HREI-MS Calcd. for C23H"F3N303S: 469.0708, found: 469.0710; Anal. Calcd. for C23H"F3N303S: C 58.85, H 3.01, N 8.95; found: C 58.42, H 2.64, N 8.80。 实施例 19 N-〖5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 4- 二羟基 -苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 12制备的 N-[5- 苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4-二甲氧基-苯甲酰胺 和三溴化硼为原料,粗品用柱层析純化,用二氯甲烷和甲醇(20: 1 ) 洗脱得到产物, 浅黄色固体, 收率 38.9%, mp: 240-241 °C (Dec) 0 ^-NMR (DMS0-i/6, 400 MHz) δ: 4.13(2H, s, CH2), 6.79 - 6.86 (3H, m, ArH), 7.21 - 7.36 (5H, m, ArH), 7.44 - 7.56 (4H, m, ArH),
9.33 (1H, br, OH), 9.61 (1H, br, OH), 9.79 (1H, br, OH), 12.26 (1H, br, CONH); EI -MS ra/e (%): 418.3 (M+, 39), 282.4 (100); HREI-MS Calcd. for C23H18N204S: 418.0987, found: 418.0976。 实施例 20 N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2- 基] -3, 4-二甲氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 9制备的 4- (4- 氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 12步骤 1制备 的 3, 4-二甲氧基 -苯曱酰氯为原料, 于室温下反应过夜, 后处理 得到粗品,经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(20: 1 ~ 10: 1) 梯度洗脱得到产物, 为白色固体, 收率 24.0%, mp: 128- 129°C。
'H-NMR (DMSO-i/6, 400 MHz) δ: 3.91 (3H, s, 0CH3), 3.95 (3H, s, 0CH3) , 4.31 (2H, s, CH2), 6.90(1H, d, J=9.00Hz, ArH), 7.08 (2H t, J=8.68Hz, ArH), 7.38 (2H, d, J=8.68Hz, ArH), 7.25 - 7.47(4H, m, ArH), 8.19 (2H, d, J-8.68Hz, ArH), 10.04 (1H, br, CONH); EI -MS ra/e(%): 493.2 (M+, 17), 165.3 (100); HREI-MS Calcd. for C25H2。FN305S: 493.1108, found: 493.1105。 实施例 21 5- (4-氟-苄基) -4- (4-曱氧基-苯基) -噻唑 -2 -基 胺的制备
步驟 1 3- (4-氟-苯基) -丙酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 3- (4-氟-苯基) - 丙酸转化为 3- (4-氟苯基)-丙酰氯, 其为无色液体, 未经纯化直 接用于下步反应。
步骤 2 3- (4-氟-苯基) -1- (4-甲氧基-苯基) -丙烷 -1-酮的 制备
采用与实施例 1 步驟 2 类似的方法制备, 以步骤 1 制备的 3-(4-氟-苯基) -丙酰氯和苯甲醚为原料, 无水 A1C 1
3为催化剂, 得到产物为白色固体, 收率 100%, mp: 80-81 °C。 ^-NMR (CDC 1
3, 400 MHz) δ: 3. 05 (2H, t, J=7. 84Hz, ArH) , 3. 23 (2H, t, J=7. 84Hz ArH) , 3. 87 (3H, s, 0CH
3), 6. 92 (2H, d, J=8. 68Hz, ArH) , 6. 97 (2H t,
60Hz ArH) , 7. 93 (2H, d, J=8. 68Hz, ArH) ; BI-MS ra/e (%): 258. 0 (M+, 27), 135. 0 (100)
0 步骤 3 2-溴 -3- (4-氟-苯基) -1- (4-甲氧基-苯基) -丙烷 -1- 酮的制备
采用与实施例 1 步驟 3类似的方法制备, 以步骤 2 制备的 3- (4-氟-苯基) -1- (4-甲氧基-苯基) -丙烷 -1-酮和溴素为原料,无 水 A1C13为催化剂,得到产物为白色晶体, 收率 95. 0%, rap: 76-77 °C。 ^-NMI CDC 400 MHz) δ: 3. 31 (1H, dd, J=14. 28, 7. 00Hz) , 3. 66 (1H, dd, J=14. 28, 7. 28Hz) , 3. 87 (3H, s, 0CH3) , 5. 23 (1H,
t, J=7.56Hz, CHBr) , 6.93 (2H, d, J=8.96Hz, ArH), 6.96(2H? T, 3JFH=3Jhh=8.72Hz, ArH), 7.23(2H, dd, 3JHH=8.68 Hz, 4JFH=5.60Hz, ArH), 7.95 (2H, d, J=8.96Hz, ArH); ESI-MS: 339.2 (M+2, 89), 337.0 (M, 70), 257.2 (100).
步骤 4 5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2-基胺 的制备
采用与实施例 6步骤 5类似的方法制备, 以步骤 3制备的 2- 溴 -3- (4-氟-苯基) -1- (4-曱氧基-苯基) -丙烷 -1-酮、 硫脲和无水 乙酸钠为原料, 回流反应 2小时, 放置过夜, 过滤, 水洗, 干燥 后得到产品, TLC显示单一斑点, 未进行进一步纯化, 得到产物, 为白色固体, 收率 81.9%, mp: 199-201 °C。 'H-NMR (DMSO-^, 400 MHz) δ: 3.76 (3H, s, 0CH3) , 4.03 (2H, s, CH2) , 6.84 (2H, s, NH2),
6.94 (2H, d, J=8.96Hz, ArH), 7.13(2H, t, W 8.96Hz, ArH),
7.23 (2H, dd, 3JHH=8.72, H=5.64Hz, ArH), 7.46 (2H, d, J=8.96Hz, ArH); EI-MS m/e (%) : 314.2 (M+, 100), 109.1 (12); HREI-MS Calcd. for C17H15FN2OS: 314.0889, found: 314.0890; Anal. Calcd. for C17H15FN2OS: C 64.95, H 4.81, N 8.91; found: C 65.10, H 5.24, N 8.98。 实施例 22 3, 5-二氟 -N- [5- (4-氟-苄基) -4 -(4-甲氧基-苯 基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 21制备的 5-(4-
氟-苄基) -4- (4-甲氧基-苯基) -噬唑 -2-基胺和对氟苯甲酰氯为原 料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层析纯化, 石油醚和乙酸乙酯(2: 1)重结晶得到产物, 为白色固体, 收率 75. 4%, rap: 175-176 X:。 ^-NMR (CDC13, 400 MHz) δ: 3. 76 (3H, s, 0CH3) , 4. 20 (2H, s, CH2), 6. 73 (2H, d, J=8. 68Hz, ArH) ,
6. 83 (1H, t, 3JF„=8. 96HZ, ArH) , 6. 83 (2H, t, 3JP„=8. 72Hz, ArH) ,
7. 15 - 7. 22 (4H, m, ArH) , 7. 30 (2H, d, J=8. 68Hz, ArH) ; EI -MS m/e (%): 454. 1 (M+, 100) , 140. 9 (48) ; HREI-MS Ca lcd. for C24H17F具 02S: 454. 0963, found: 454. 0966; Ana l. Ca lcd. for C24H17F3N202S: C 63. 43, H 3. 77, N 6. 16; found: C 63. 70, H 3. 70, N 6. 26。 实施例 23 4-氟 -N- [5- (4-氟-苄基) -4- (4-曱氧基-苯基) - 噻唑- 2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 21制备的 5- (4- 氟-苄基) -4- (4-甲氧基-苯基)-噻唑- 2-基胺和实施例 16 步骤 1 制备的 3, 5-二氟苯甲酰氯为原料, 于室温下反应过夜, 后处理得 到粗品, 经硅胶柱层析纯化, 二氯曱烷和乙酸乙酯(10: 1)洗脱得 到产物,为白色固体,收率 44. 5%,mp: 184-185 °C
0 'H-NMR (CDC1
3, 400 MHz) δ: 3. 80 (3H, s, 0CH
3) , 4. 19 (2H, s, CH
2) , 6. 80 (2H, d, J-8. 96Hz, ArH) , 6. 98 - 7. 04 (4H, ra, ArH), 7. 20 (2H, dd,
3J
HH=8. 68,
4J
F„=5. 32HZ, ArH) , 7. 35 (2H, d, J=8. 96Hz, ArH),
7.77 (2H, dd,
3J
HH-8.68,
ArH); EI -MS ra/e(%): 436.0 (M
+, 100), 122.9 (87); HREI-MS Calcd. for C
24H
18F
2N
20
2S: 436.1057, found: 436.1056; Anal. Calcd. for C
24H
18F
2N
20
2S: C 66.04, H 4.16, N 6.42; found: C 66.08, H 4.02, N 6.53。 实施例 24 N- [5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2 -基] -N-(3, 4-二甲氧基-苯甲酰胺基) -3, 4-二甲氧基-苯曱酰胺 的制备
采用与实施例 4类似的方法制备, 以实施例 21制备的 5-(4- 氟-苄基) -4-(4-曱氧基-苯基) -噻唑 -2-基胺和实施例 12 步驟 1 制备的 3, 4-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后 处理得到粗品, 经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(20: 1 ~ 10: 1)梯度洗脱得到产物, 为白色固体, 收率 34.2%, mp: 136-137 °C。 'ti-MR (CDC1
3, 400 MHz) δ: 3.70(3H, s, 0CH
3), 3.80 (3H, s, OCH3) , 3.87 (3H, s, OCH3) , 3.89 (6H, s, 2xOCH
3), 3.95 (2H, s, CH
2), 6.75 (IH, d, J=8.72Hz, ArH), 6.88 (3H, d, J=8.68Hz, ArH) 7.01 (2H, t,
4JF„=5.32HZ, ArH), 7.24 (2H, d, J=8.68Hz, ArH), 7.41 - 7.49 (3H m, ArH), 7.52 (IH, d, J=8.68Hz, ArH); EI -MS m/e(°/o): 642.2 (M+, 7), 165.1 (100); HREI-MS Calcd. for C35H31FN207S: 642.1836, found: 642.1867。
实施例 25 4-氰基- N- [5- (4-氟-苄基) -4- (4-甲氧基-苯 基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 13制备的 5- (4- 氟-苄基)- 4- (4-甲氧基-苯基)-噻唑 -2-基胺和实施例 12 步骤 1 制备的对氰基苯甲酰氯为原料, 于室温下反应过夜, 后处理得到 粗品, 用石油醚和乙酸乙酯重结晶得到产物, 为白色固体, 收率 85.9%, mp: 191-192 °C。 ^-NMR (CDC1
3, 400 MHz) δ: 3.79 (3H, s, 0CH
3) , 4.18 (2H, s, CH
2), 6.73 (2H, d, J=8.96Hz, ArH), 7.03 (2H, t,
4JFH=5.60HZ, ArH), 7.24 (2H, d, J=8.96Hz, ArH), 7.53 (2H, d, J=8.40Hz, ArH), 7.76 (2H, d, J=8.40Hz, ArH), 11.74 (1H, br, CONH);; EI -MS ra/e(%): 443.1 (M+, 100), 130.0 (48); HREI-MS Calcd. for C25H18FN302S: 443.1104, found: 443.1108; Anal. Calcd. for C25H18FN302S: C 67.71, H 4.09, N 9.47; found: C 67.89 H 3.97, N 9.42。 实施例 26 5- (4-硝基-苄基) -4-苯基-噻唑 -2-基胺的制备
步驟 1 3- (4-硝基-苯基) -1-苯基-丙烷- 1-酮的制备
采用与实施例 1步骤 1类似的方法制备, 以实施例 6步驟 2 制备的 3- (4-硝基-苯基) -丙酰氯和苯为原料, 无水 A1C13为催化 剂, 得到产物为白色固体, 收率 70.3%, rap: 100-102 °C。 ^-NMR (CDC13, 400 MHz) δ: 3.19 (2H, t, J=7.28Hz), 3.36 (2H, t, J=7.28Hz), 7.42 - 7.60 (5H, m, ArH), 7.96 (2H, d, J=7.56Hz, ArH), 8.16 (2H, d, J=7.56Hz, ArH); ESI-MS: 278.2 (M+Na, 100), 273.3 (M+NH3+H, 49), 256.3 (M+1, 47).
步骤 2 2-溴- 3- (4-硝基-苯基) -1-苯基-丙烷- 1-酮的制备 采用与实施例 1 步骤 3 类似的方法制备, 以步骤 1 制备的 3-(4-硝基-苯基) -1-苯基-丙烷- 1-酮和溴素为原料, 无水 A1C13 为催化剂, 得到产物为白色固体, 收率 90.4%, mp: 97-98 °C。 !H-NMR (CDCI3, 400 MHz) δ: 3.48 (1H, dd, J=14.28, 7.28Hz), 3.75(1H, dd, J=14.28, 7.28Hz), 5.31 (1H, t, J=7.28Hz, CHBr) , 7.46~ 7.51 (4H, m, ArH), 7.61 (1H, t, J=8.00Hz, ArH), 7.98 (2H d, J=8.68Hz, ArH), 8.17 (2H, d, J=8.68Hz, ArH); FAB-MS: 336. KM+2, 90), 334.1 (M, 90), 254.2 (58), 238.2 (36), 105.1 (100).
步骤 3 5- (4-硝基-苄基) -4-苯基-噻唑- 2-基胺的制备 釆用与实施例 6步骤 5类似的方法制备, 以步骤 1制备的 1- 溴 -3- (4-硝基-苯基) -1-苯基-丙烷- 1-酮为原料, 回流 2小时, 室
温放置过夜, 析出固体, 过滤、 水洗并干燥后, 用无水乙醇重结 晶得到产物, 收率 79. 5%, rap: 215-216 °C。 H-NMR (DMS0-i/6, 400 MHz) δ: 4. 23 (2H, s, CH2) , 6. 97 (2H, s, NH2) , 7. 30 (1H, t, J=7. 32Hz, ArH), 7. 38 (2H, t, J=7. 28Hz, ArH), 7. 47 (2H, d, J=8. 68Hz, ArH) , 7. 51 (2H, d, J=7. 56Hz, ArH), 8. 19 (2H, d, J=8. 68Hz, ArH) ; EI-MS m/e ( ) : 311. 1 (M+, 100) , 189. 0 (14) ; HREI-MS Ca lcd. for C16H13N302S: 311. 0728, found: 311. 0721; Ana l. Ca l cd. for C16H13N302S: C 61. 72, H 4. 21, N 13. 50; found: C 62. 05, H 4. 13, N 13. 07。 实施例 27 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -4- 甲氧基 -苯曱酰胺的制备
步骤 1 对甲氧基苯曱酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将甲氧基苯甲酸转 化为对甲氧基苯甲酰氯, 其为无色液体, 未经纯化直接用于下步 反应。
步骤 2 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2 -基] -4-甲 氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基胺和步骤 1制备的对甲氧基苯甲 酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层 析纯化, 二氯甲烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱得到产物,
为白色固体, 收率 58.7%, mp: 58-59 °C。 'H-NMR (CDC13, 400 MHz) δ: 3.73 (3H, s, 0CH3), 3.85 (3H, s, 0CH3), 4.22(2H, s, CH2), 6.89 (4H, t, J=8.44Hz, ArH), 7.22 - 7.35 (5H, ra, ArH), 7.45 (2H t, J=8.68Hz, ArH), 7.82 (2H, t, J=8.68Hz, ArH), 10.27(1H, br, CONH); EI -MS m/e(%): 430.1 (M+, 89), 135.0 (100); HREI-MS Calcd. for C25H22N203S: 430.1351, found: 430.1361。 实施例 28 3, 4-二甲氧基 - N- [4- (4-甲氧基-苯基) -5- (4-硝 基-苄基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 6制备的 4- (4- 曱氧基 -苯基)-5- (4-硝基-苄基) -噻唑 -2-基胺和实施例 12步驟 1 制备的 3, 4-二甲氧基-苯甲酰氯为原料, 于室温下反应过夜, 后 处理得到粗品,经硅胶柱层析純化,二氯甲烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱得到产物, 为白色固体, 收率 60.8%, mp: 106-108 °C。 ^-NMIUCDC 400 MHz) δ: 3.83 (3H, s, 0CH3) , 3.92 (3H, s, 0CH3) , 3.97 (3H, s, OCH3) , 4.33 (2H, s, CH2) , 6.85 - 6.89 (3H, m, ArH), 7.37 ~ 7.45 (6H, m, ArH), 8.18 (2H, t, J=8.68Hz, ArH) 10.48 (1H, br, CONH, exchangable) ; EI -MS m/e (%) : 505.0 (M+, 54), 165.0 (100); HREI-MS Calcd. for C26H23N306S: 505.1308, found: 505.1315; Anal. Calcd. f or C26H23N306S: C 61.77, H 4.59, N 8.31; found: C 61.90, H 4.70, N 8.05。
实施例 29 3, 4-二甲氧基 -N- [5- (4-硝基-苄基) -4-苯基-噻 唑- 2-基] -苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 26制备的 5- (4- 硝基-苄基) -4-苯基-噻唑 -2-基胺和实施例 12步驟 1制备的 3, 4- 二甲氧基-苯甲酰氯为原料,于室温下反应过夜,后处理得到粗品, 经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱 得到产物,为白色固体,收率 64. 7%, mp: 105-106 "C o 'H- MR (CDC13 400 MHz) δ: 3. 90 (3H, s, 0CH3) , 3. 96 (3H, s, 0CH3) , 4. 35 (2H, s, CH2), 6. 85 (1H, t, J=8. 68Hz, ArH) , 7. 30 - 7. 48 (9H, ra, ArH) , 8. 18 (2H, t, J=8. 68Hz, ArH) , 10. 21 (1H, br, CONH); EI -MS m/e (%): 475. 0 (M+, 20), 165. 0 (100) ; HREI-MS Calcd. for C25H21N3O5S: 475. 1202, found: 475. 1210。 实施例 30 N- [5- (4-氟-苄基)- 4- (4-甲氧基-苯基) -噻唑 - 2-基] - 3, 4-二甲氧基-苯甲酰胺的制备
釆用与实施例 4类似的方法制备, 以实施例 21制备的 5- (4- 氟-苄基) -4- (4-甲氧基-苯基) -噻唑- 2-基胺和实施例 12 步驟 1 制备的 3, 4-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后 处理得到粗品,经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱得到产物, 为白色固体, 收率 69. 1%, rap: 140-142 °C。 -NMl CDC , 400 MHz) δ: 3. 80 (3H, s, 0CH
3), 3. 84 (3H, s, OCHs) , 3. 91 (3H, s, 0CH
3) , 4. 18 (2H, s, CH
2) , 6. 79 (1H, d, J=8. 68Hz, ArH), 6. 82 (2H, d, J=8. 68Hz, ArH), 7. 00 (2H, t,
32Hz, ArH) , 7. 35 (4H, d, J=9. 28Hz, ArH), 11. 00 (1H, br, CONH, exchangable) ; EI -MS m/e (%): 478. 2 (M+, 88) , 165. 0 (100) ; HREI-MS Calcd. for C
26H
23FN
20
4S: 478. 1363, found: 478. 1361; Ana l. Calcd. for C
26H
23FN
20
4S: C 65. 26, H 4. 84, N 5. 85; found: C 65. 11, H 4. 68, N 5. 96。 实施例 31 N-〖4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2- 基] - 3, 4-二羟基-苯甲酰胺的制备
采用与实施例 7 类似的方法制备, 以实施例 20 制备的 N- [4- (4-氟-苯基) -5- (4-硝基-苄基) -噻唑 -2-基] -3, 4-二甲氧基 -苯甲酰胺和三溴化硼为原料,粗品用丙酮重结晶得到产物, 浅黄 色固体,收率 63. 3%, mp: 276-278 "C (Dec)。 -NMR (DMS0- , 400
MHz) δ: 4.4K2H, s, CH
2) , 6.81 (1H, d, J=8.12Hz, ArH), 7.29 (2H, t,
ArH), 7.47 ~ 7.54 (4H, m, ArH), 7.67 (2H, dd,
3J
HH=8.68Hz, J
F„=5.60Hz, ArH), 8.20 (2H, d, J=8.40Hz, ArH), 9.32 (1H, br, OH), 9.81 (1H, br, OH), 12.38 (1H, br, CONH); EI -MS m/e(%): 465.0 (M
+, 28), 329.0 (100); HREI-MS Calcd. for C
23H
16FN
30
5S: 465.0795, found: 465.0798。 实施例 32 3, 4-二羟基 - N- [5- (4-硝基-苄基) -4-苯基 -噻唑 - 2-基]-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 29制备的 3, 4- 二曱氧基 -N- [5- (4-硝基-苄基) -4-苯基-噻唑- 2-基] -苯甲酰胺和 三溴化硼为原料, 粗品用丙酮和乙酸乙酯重结晶得到产物, 白色 固体, 收率 62.8%, rap: 244°C (Dec.:)。 ^-NMR (DMS0- , 00 MHz) δ: 4.42(2H, s, CH2), 6.81 (1H, d, J=8.40Hz, ArH), 7.38 (1H, t, J=7.60Hz, ArH), 7.44 - 7.55 (6H, m, ArH), 7.64 (2H, d, J=8.44Hz, ArH), 8.20 (2H, d, J=8.68Hz, ArH), 9.32 (1H, br, OH), 9.81 (1H, br, OH), 12.38 (1H, br, CONH); EI -MS m/e(%): 447.1 (M+, 16), 311.1 (100); HREI-MS Calcd. for C23H17N305S: 447.0889, found: 447.0887。
实施例 33 N- [5- (4-氟-苄基) -4- (4-羟基-苯基) -噻唑- 2- 基] -3, 4-二羟基-苯甲酰胺的制备
采用与实施例 7 类似的方法制备, 以实施例 30 制备的 N- [5- (4-氟-苄基) -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4-二甲氧 基-苯甲酰胺和三溴化硼为原料,粗品用丙酮重结晶得到产物, 白 色固体, 收率 64. 4% , mp: 154-155。C。 ^-NMR (DMS0-i/
6, 400 MHz) δ: 4. 19 (2H, s, CH
2) , 6. 80 - 6. 86 (3H, ra, ArH) , 7. 15 (2H, t,
64Hz, ArH) , 7. 43 - 7. 56 (4H, m, ArH) , 12. 25 (1H, br, CONH); EI -MS ra/e (%): 436. 1 (NT, 24) , 300. 1 (100) ; H EI-MS Ca l cd. for C
23H
17FN
20
4S: 436. 0893, f ound: 436. 0900。 实施例 34 3, 4-二羟基 - N- [4- (4-羟基-苯基) -5- (4-硝基- 苄基) -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 28制备的 3, 4 - 二甲氧基- N- [4- (4-曱氧基-苯基) -5- (4-硝基-苄基) -噻唑 -2- 基] -苯曱酰胺和三溴化硼为原料, 粗品用丙酮重结晶得到产物,
白色固体,收率 66.7%,mp: 195-196 °C。 -NMR (DMS0- 400 MHz) δ: 4.33 (2H, s, CH2), 6.74 - 6.81 (3H, m, ArH), 7.37 ~ 7.50 (6H, ra, ArH), 8.16 (2H, d, J=8.80 Hz, ArH), 9.30 (IH, br, OH), 9.59 (IH, br, OH), 9.77 (IH, br, OH), 12.30(1H, br, CONH); EI-MS m/e(%): 463.1 (M+, 1), 327.1 (100); HREI-MS Calcd. for C23H17N306S: 463.0838, found: 463.0822。 实施例 35 N-[5-苄基 l-4-(4-甲氧基-苯基)-噻唑 -2- 基] -2, 4-二甲氧基-苯曱酰胺的制备
步骤 1 2, 4-二甲氧基-苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 2,4-二甲氧基- 苯甲酸转化为 2, 4-二甲氧基-苯甲酰氯, 其为白色固体, 未经纯 化直接用于下步反应。
步驟 2 N-[5-苄基 1-4- (4-甲氧基-苯基)-噻唑- 2-基] -2,4- 二甲氧基-苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 - 4- (4-甲氧基-苯基) -噻唑 -2-基胺和步骤 1制备的 2, 4-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶 柱层析纯化,二氯甲烷和乙酸乙酯(100: 0~ 10: 1)梯度洗脱得到产 物,为白色固体,收率 71.0%, mp: 196-197 0Co 'H-NMR (CDC13, 400 MHz) δ: 3.85 (3H, s, 0CH3) , 3.88 (3H, s, 0CH3) , 4.08 (3H, s, 0CH3), 4.20 (2H, s, CH2) , 6.54 (IH, d, J=2.24Hz, ArH), 6.65 (IH
dd, J=8.96Hz, 2.24Hz, ArH), 6.97 (2H, d, J=8.96Hz, ArH), 7.22 - 7.34 (5H, m, ArH), 7.56 (2H, d, J=8.96Hz, ArH), 8.22 (IE d, J=8.68Hz, ArH), 11.01 (1H, br, CONH); EI -MS m/e ( ): 460. l(M+, 58), 165.0 (100); HREI-MS Calcd. for C26H24N204S: 460.1457, found: 460.1454; Anal. Calcd. for C26H24N204S: C 67.81, H 5.25, N 6.08; found: C 68.04, H 5.16, N 6.19。 实施例 36 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -2- 曱氧基-苯甲酰胺的制备
步骤 1 邻曱氧基苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将邻曱氧基苯甲酸 转化为邻甲氧基苯甲酰氯, 其为无色液体, 未经纯化直接用于下 步反应。
步骤 2 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -2-甲 氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基胺和步骤 1制备的邻甲氧基苯甲 酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层 析纯化, 二氯甲烷和乙酸乙酯(100: 0~ 10: 1)梯度洗脱得到产物, 为白色固体,收率 65· 6%, mp: 147-148°C0 'H-NMRCCDCls, 400 MHz) δ: 3.84 (3H, s, 0CH3) , 4.09 (3H, s, 0CH3) , 4.21 (2H, s, CH2), 6.97 (2H, d, J=8.68Hz, ArH), 7.05 (1H, d, J=8.12Hz, ArH),
7. 14 (1H, t, J=8. 12Hz, ArH) , 7. 22 - 7. 32 (5H, m, ArH) , 7. 53 - 7. 56 (3H, m, ArH) , 8. 28 (1H, dd, J=7. 84Hz, 1. 68Hz, ArH) , 11. 05 (1H, br, CONH); EI -MS m/e (%) : 430. 2 (M+, 89), 135. 1 (100) ; HREI-MS Ca lcd. for C25H22N203S: 430. 1351, found: 430. 1357。 实施例 37 N- [5-苄基 -4- (4-甲氧基-苯基)-噻唑- 2- 基] - 3, 4, 5-三甲氧基-苯甲酰胺的制备
步驟 1 3, 4, 5-三甲氧基-苯甲酰氯的制备
釆用与实施例 1步骤 1类似的方法制备,将 3, 4, 5-三甲氧基 -苯甲酸转化为 3, 4, 5-三甲氧基 -苯曱酰氯, 其为白色固体, 未经 纯化直接用于下步反应。
步骤 2 N- [5-苄基 -4- (4-甲氧基 -苯基)-噻唑- 2- 基] - 3 , 4, 5 -三曱氧基-苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4-(4-甲氧基 -苯基)-噻唑 -2-基胺和步骤 1制备的 3, 4, 5-三甲氧 基 -苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅 胶柱层析纯化,二氯曱烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱得到 产物, 为白色固体, 收率 61. 7%, mp: 157-158 °C。 'H-NMR (CDC13, 400 MHz) δ: 3. 75 (6H, s, 2xOCH3) , 3. 77 (3H, s, 0CH3) , 3. 87 (3H, s , 0CH3) , 4. 21 (2H, s, CH2), 6. 75 (2H, d, J=8. 96Hz, ArH) , 6. 92 (2H, s, ArH) , 7. 23 ~ 7. 35 (7H, m, ArH) , 11. 72 (1H, br,
C謹); EI -MS m/e (%): 490.3 (M+, 82), 195.1 (100); HREI-MS Calcd. for C27H26N205S: 490.1562, found: 490.1560。 实施例 38 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -2, 4- 二羟基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 35制备的 N-[5- 苄基 L-4- (4- ψ氧基-苯基) -噻唑 -2-基] -2, 4 -二曱氧基-苯曱酰 胺和三溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收率 67.1%, mp: 217-219 。C。 !H-NMR (400Hz, Acetone- d6) δ: 4.40 (2H, s, CH2), 6.53 (IH, d, J=2.24Hz, ArH), 6.58 (IH, dd, J=8.96Hz, 2.24Hz, ArH), 7.04 (2H, d, J=8.68Hz, ArH), 7.32- 7.40(5H, m, ArH), 7.65 (2H, d, J=8.68Hz, ArH), 8.29 (IH, d, J=8.72Hz, ArH); EI-MS ra/e (%) : 418.2 (M+, 33), 282.1 (100); HREI-MS Calcd. for C23H18N204S: 418.0987, found: 418.0970。 实施例 39 N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基〗 -2-羟 基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 36制备的 N-[5-
苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -2-甲氧基-苯甲酰胺和三 溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收率 64.4%, mp: 279-280 。C。 'H-NMR (DMSO-^, 400 MHz) δ: 4·19(2Η, s, CH2), 6.85 (2H, d, J=8.40Hz, ArH), 6.94 - 7.02 (2H, m, ArH), 7.22 - 7.28 (3H, m, ArH), 7.34 (2H, t, J=7.84Hz, ArH), 7.42- 7.48 (3H, ra, ArH), 7.96 (1H, d, J=7.84Hz, ArH), 9.70(1H, br, OH), 12.15 (1H, br, CONH); EI -MS ra/e (%): 402.2 (M+, 63), 282.1 (100); HREI-MS Calcd. for C23H18N203S: 402.1038, found: 402.1037; Anal. Calcd. for C23H18N203S: C 68.64, H 4.51, N 6.96; found: C 68.45, H 4.38, N 6, 80。 实施例 40 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -4 -甲 氧基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 27制备的 N-[5- 苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -4-甲氧基-苯曱酰胺和三 溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收率 41.3%, mp: 124-125 °C。 - R (DMS0-cT6, 400 MHz) δ: 3.84 (3H, s, CH30) , 4.21(2H, s, CH2), 6.84 (2H, t, J-8.68Hz, ArH), 7.06 (2H, t, J=8.68Hz, ArH), 7.23 ~ 7.26 (3H, m, ArH), 7.32- 7.36 (2H, m, ArH) 7.47 (2H, t, J=8.68Hz, ArH), 8.08 (2H, t, J=8.96Hz, ArH), 12.45(1H, br, CONH); EI -MS m/e( ): 416.2 (M+ 62), 282.1 (4), 135.1 (100); HREI-MS Calcd. for C24H20N2O3S:
416. 1195, found: 416. 1194。 实施例 41 N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2- 基] - 3, 4, 5-三羟基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 37制备的 N- [5- 苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4, 5-三甲氧基-苯曱酰 胺和三溴化硼为原料,粗品用丙酮重结晶得到产物, 浅黄色固体, 收率 61. 3%, rap: 245-246 t;。 - NMR (DMS0- , 400 MHz) δ: 4. 19 (2Η, s, CH2), 6. 83 (2H, d, J=8. 68Hz, ArH) , 7. 06 (2H, s, ArH), 7. 22 - 7. 35 (5H, ra, ArH) , 7. 46 (2H, d, J=8. 68Hz, ArH) , 12. 15 (1H, br, CONH); EI-MS m/e (%) : 434. l (M+, 6), 282. 1 (100) ; HREI-MS Calcd. for C23H18N205S: 434. 0936, found: 434. 0936。 实施例 42 N- [5-苄基- 4- (4-曱氧基-苯基) -噻唑 -2-基] - 3- 曱氧基 -苯甲酰胺的制备
步骤 1 间曱氧基苯甲酰氯的制备
采用与实施例 1步驟 1类似的方法制备, 将间甲氧基苯曱酸 转化为间曱氧基苯甲酰氯, 其为无色液体, 未经纯化直接用于下
步骤 2 N- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -3 -甲 氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4-(4-甲氧基-苯基) -噻唑 -2-基胺和步骤 1制备的间曱氧基苯甲 酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶柱层 析纯化, 二氯甲烷和乙酸乙酯(100: 0 ~ 10: 1)梯度洗脱得到产物, 为白色固体, 收率 56. 8%, mp: 71-72 °C。 'H-NMR (CDC13, 400 MHz) δ: 3. 80 (3H, s, 0CH3), 3. 81 (3H, s, 0CH3) , 4. 23 (2H, s, CH2),
6. 85 (2H, d, J=8. 80Hz, ArH), 7. 05 (1H, d, J=2. 30Hz, ArH) ,
7. 20 - 7. 42 (10H, m, ArH) ; EI -MS ra/e (%) : 430. 1 (M+, 100), 135. 0 (85) ; HREI-MS Ca lcd. for C25H22N203S: 430. 1351, found: 430. 1352。 实施例 43 N- [5-苄基- 4- (4 -甲氧基-苯基) -噻唑 - 2- 基] -3, 5-二甲氧基-苯甲酰胺的制备
步骤 1 3, 5-二曱氧基-苯甲酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 3, 5-二甲氧基- 苯甲酸转化为 3, 5-二曱氧基-苯曱酰氯, 其为白色固体, 未经纯 化直接用于下步反应。
步骤 2 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 5- 二曱氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 - 4-(4 -甲氧基-苯基)-噻唑 -2-基胺和步骤 1制备的 3, 5-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗品, 经硅胶 柱层析纯化,二氯曱烷和乙酸乙酯(100: 0~ 10: 1)梯度洗脱得到产 物,为白色固体,收率 65.7%, nip: 152-153 °C。 -腿(CDC13, 400 MHz) δ: 3.78 (6Η, s, 2x0CH3), 3.80 (3H, s, 0CH3), 4.22 (2H, s, CH2), 6.55 (1H, t, J=2.12Hz, ArH), 6.83 (2H, d, J=8.68Hz, ArH) 6.90 (2H, t, J=2.12Hz, ArH), 7.25 - 7.33 (5H, m, ArH), 7.39 (2H d, J-8.68Hz, ArH), 10.75 (1H, br, CONH) ; EI -MS m/e (%): 460.2 (M+, 100), 165.0 (84); HREI-MS Calcd. for C26H24N204S: 460.1457, found: 460.1464。 实施例 44 N-〖5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -3 -羟 基-苯曱酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 42制备的 N- [5- 苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3-曱氧基-苯甲酰胺和三 溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收率 65.0%, mp: 233-234 °C。 -NMR (DMS0-i6, 400 MHz) δ: 4.23 (2H, s, CH2), 6.83 (2H, d, J=7.28Hz, ArH), 6.98 - 7.02 (1H, ra, ArH), 7.22 - 7.35 (6H, m, ArH), 7.40 (1H, s, ArH), 7.46 (2H, d, J=7.28Hz, ArH), 7.52 (1H, d, J=7.56Hz, ArH), 9.50 (1H, br, OH), 12.50 (1H, br, CONH); EI -MS m/e (%): 402. l(M+, 83),
121.0(100); HREI-MS Calcd. for C23H18N203S: 402.1038, found: 402.1042。 实施例 45 N- [5-苄基 -4- (4-羟基-苯基) -噻唑 -2-基] -3, 5- 二羟基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 43制备的 N-[5- 苄基- 4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 5-二甲氧基-苯甲酰胺 和三溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收 率 48.2%,mp: 262-263 "C H-NMR (DMS0-i
6, 400 MHz) δ: 4.21 (2H, s, CH
2), 6.44 (IH, t, J=2.12Hz, ArH), 6.83 (2H, d, J=8.40Hz, ArH), 6.88 (2H, d, J=2.24Hz, ArH), 7.22 - 7.26 (3H, m, ArH), 7.32 - 7.35 (2H, m, ArH), 7.46 (2H, d, J=8.68Hz, ArH), 9.52 (2H br, OH), 12.39 (IH, br, CONH); EI -MS m/e (%) : 418.1(M+, 82), 282.1 (66), 137.0 (100); HREI-MS Calcd. for C
23H
18N
20
4S: 418.0987, found: 418.0981。 实施例 46 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] -4-羟 基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 27制备的 N-[5- 苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -4-甲氧基-苯甲酰胺和三 溴化硼为原料, 粗品用丙酮重结晶得到产物, 白色固体, 收率 67.9%, mp: 254-255 °C。 H-NMR (DMSO- , 400 MHz) δ: 4.20(2H, s, CH2), 6.84 (4H, t, J=7.80Hz, ArH), 7.20 ~ 7.28 (3H, m, ArH) , 7.33 (2H, t, J=8.08Hz, ArH), 7.47 (2H, d, J=8.64Hz, ArH), 7.97 (2H, d, J=8.60Hz, ArH), 9.61(1H, s, OH), 10.29 (1H, s, OH), 12.34 (1H, s, CONH); EI -MS ra/e ( ): 402.0 (M+, 82), 282.1 (74), 121.0 (100); HREI-MS Calcd. for C23H18N203S: 402.1038, found: 402.1027。 实施例 47 5-环己基甲基- 4- (4-甲氧基-苯基) -噻唑 -2 -基 胺的制备
步骤 1 3-环己基丙酰氯的制备
采用与实施例 1步驟 1类似的方法制备, 将 3-环己基丙酸转 化为 3-环己基丙酰氯, 其为无色液体, 未经纯化直接用于下步反 应。
歩骤 2 3-环己基- 1- (4-甲氧基-苯基) -丙烷 -1-酮的制备 在 250 mL三颈瓶中加入理论量 5· 00 g(28.74 mmol)的步骤 1 制备的 3-环己基丙酰氯、 3.11 g(28.74 讓 ol)苯甲醚和 150 mL CH2C12, 水盐浴至- 5 °C, 搅拌下分批加入 3.83 g(28.74 mmol) 无水 A1C13,加完后室温搅拌 2小时,倾入(150 g冰 + 5 mL浓 HC1) 的混合物中, 分出有机层, 減压浓缩至约 10 mL, 水相用乙酸乙 酯萃取 2次 x50 mL, 有机相合并, 饱和碳酸氢钠饱和盐水洗涤, 无水 Na2S04干燥, 过滤浓缩得到浅棕色固体, 以石油醚重结晶得 到产物, 白色固体, 5.60 g, 收率 79.1%。 mp: 64-65 °C。
!H-NMR (CDC13, 400 MHz) δ: 0.85 - 1.00 (2Η, ra, CH2), 1· 10 ~ 1.40 (4Η, m, 2xCH2), 1.61 - 1.1.82 (7H, ra) , 2.92 (2H, t, J=7.56Hz, C0CH2), 3.87 (3H, s, 0CH3) , 6.23 (2H, d, J=8.68Hz, ArH), 7.94 (2H, d, J=8.68Hz, ArH); ESI-MS ra/e(%): 269.2 (M+Na: 100), 247.2 (M+1, 71) 。
步骤 3 2-溴 -3-环己基- 1- (4-曱氧基-苯基) -丙烷 -1-酮的 制备
采用与实施例 1步骤 3类似的方法制备, 以步骤 2制备的 3- 环己基- 1- (4-甲氧基-苯基) -丙烷 -1-酮和溴素为原料,无水 A1C13 为催化剂,得到产物为无色油状物,收率 96.6%。^- NMR(CDC13, 400 MHz) δ: 0.85 - 1.30 (5Η, ra) , 1.45 ~ 1.85 (6H, m), 2.04 (2H, t, J=8.08Hz, CH2CHBr) , 3.89 (3H, s, 0CH3), 5.24 (1H, t, J=7.48Hz, CHBr) , 6.96 (2H, d, J=9.16Hz, ArH), 8.02 (2H, d, J=8.68Hz, ArH); ESI-MS ra/e(%): 346.9 (M+Na, 100), 327.3 (M+1, 71) 。
步骤 4 5-环己基甲基 -4- (4-甲氧基-苯基)-噻唑 -2-基胺的 制备
采用与实施例 6步骤 5类似的方法制备, 以步骤 3制备的 2-
溴 -3-环己基 -1- (4-甲氧基-苯基) -丙烷 -1-酮、硫脲和无水乙酸钠 为原料, 回流 3h, 后处理得到粗品后用减压硅胶柱层析纯化, 石 油醚和乙酸乙酯 3: 1(V:V)洗脱得到产物, 白色固体, 收率 79.8%, mp: 118-119 °C。 ^-NMR (CDC13, 400 MHz) δ: 0.80 - 0.92 (2H, m), 1.05 - 1.25 (3H, m) , 1.40 ~ 1.50 (1H, m, CH), 1.60 ~ 1.80(5H, in), 2.59 (2H, d, J=7.04 Hz, CHCH2Ar) , 3.83 (3H, s, 0CH3) , 5.55 (2H, br, NH2) , 6.93 (2H, d, J=6.72Hz, ArH ), 7.44 (2H, d, J=6.72Hz, ArH ); EI -MS m/e(%): 302.2 (M+, 59), 219.1 (100), 177.1 (44); HREI-MS Calcd. for C17H22N2OS: 302.1453, found: 302.1450; Anal. Calcd. for C17H22N20S: C 67.51, H 7.33, N 9.26; found: C 67.41, H 7.47, N 9.24。 实施例 48 N- [5-环己基甲基 -4-(4-甲氧基-苯基) -噻唑 -2- 基] - 3, 4-二曱氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 47制备的 5 -环 己基甲基 -4-(4-曱氧基-苯基)-噻唑 -2-基胺和实施例 12 步骤 1 制备的 3, 4-二曱氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后 处理得到粗品, 经硅胶柱层析纯化, 二氯曱烷和乙酸乙酯(20:1) 梯度洗脱得到产物, 为白色固体, 收率 67.5%, mp: 171-172 °C。
JH-NMR (CDCI3, 400 MHz) δ: 0.82 ~ 0· 98 (2H, m), 1.08 - 1.28 (3H, m), 1.58 - 1.88 (6H, in), 2.74 (2H, d, J=7.28 Hz, CHCH2Ar), 3.82 (3H, s, OCH3) , 3.89 (3H, s, 0CH3) , 3.94 (3H, s, 0CH3) ,
6.82-6.89 (3H, m, ArH), 7.35~7.47(4H, m, ArH), 11.00 (IH, br, CONH); EI-MS ra/e (%): 466.2 (M+, 46), 383.2 (29), 165.1 (100); HREI- S Calcd. for C26H3。N204S: 466.1926, found: 466.1920; Anal. Calcd. f or C26H3。N204S: C 66.93, H 6.48, N 6.00; found: C 66.88, H 6.68, N 6.02。 实施例 49 N- [5-环己基甲基 -4- (4-羟基-苯基) -噻唑 - 2- 基] -3, 4-二羟基-苯甲酰胺的制备
釆用与实施例 7类似的方法制备, 以实施例 48制备的 N-[5- 环己基曱基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -3, 4-二甲氧基-苯 甲酰胺和三溴化硼为原料, 粗品用乙酸乙酯重结晶纯化, 产物为 白色固体,收率 60.5°/。,mp: 133-134 "C H-NMR (DMS0-i6, 400 MHz) δ: 0.75 - 0.90 (2H, m), 0.95 ~ 1.18 (3H, m) , 1.15-1.17 (6H, m) : 2.65 (2H, d, J=7.0 Hz), 6.75 (IH, d, J=8.0 Hz, ArH), 6.76 (2H, d, J=8.4 Hz, ArH), 7.32 (2H, d, J=8.4 Hz, ArH), 7.41 (IH, d, J=2.4 Hz, ArH), 7.46 (IH, dd, J=8.4, 2.4 Hz, ArH), 9.52 (IH, br, OH), 12.15 (IH, br, CONH); EI-MS m/e (%) : 424.1 (M+, 21), 288.1 (36), 205.1 (100); HREI-MS Calcd. for C23H24N204S: 424.1457, found: 424.1458。 实施例 50 N- [5-环己基甲基 -4- (4-羟基-苯基) -噻唑 -2- 基]- 3, 4, 5-三甲氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 47制备的 5 -环 己基甲基 -4- (4-甲氧基-苯基)-噻唑- 2-基胺和实施例 37 步骤 1 制备的 3, 4, 5-三甲氧基 -苯甲酰氯为原料, 于室温下反应过夜, 后处理得到粗品,经硅胶柱层析纯化,二氯甲烷和乙酸乙酯(20: 1) 梯度洗脱得到产物, 为白色固体, 收率 60.1%, mp: 193- 194°C。 ^-NMR (CDC13, 400 MHz) δ: 0.85 - 0.98 (2H, m), 1.05 - 1.28 (3H, m), 1· 58 ~ 1.88(6H, m), 2.75 (2H, d, J=7.28 Hz, CHCH2Ar) , 3.74 (3H, s, OCH3) , 3.77(3H, s, 0CH3) , 3.79 (3H, s, 0CH3) , 3.97 (3H, s, 0CH3), 6.77 (2H, d, J=8.44 Hz, ArH), 6.98(2H, s, ArH), 7.29 (2H, d, J=8.44 Hz, ArH); EI -MS m/e(%): 496.1 (M+, 64), 413.1 (48), 195.1 (100); HREI-MS Calcd. for C27H32N205S: 496.2032, found: 496.2040; Anal. Calcd. for C27H32N205S: C 65.30, H 6.49, N 5.64; found: C 65.11, H 6.55, N 5.70。 实施例 51 N- [5-环己基甲基- 4- (4-羟基-苯基) -噻唑 -2- 基] -3, 4, 5-三羟基-苯曱酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 50制备的 N- [5-
环己基甲基- 4- (4-羟基-苯基) -噻唑 -2-基] -3, 4, 5-三曱氧基-苯 甲酰胺和三溴化硼为原料, 粗品用丙酮重结晶纯化, 产物为白色 固体, 收率 55.8%, mp: 238-239 °C:。 -NMR (DMS0-i/
6, 400 MHz) δ: 0.80 - 0.90 (2H, in), 1.00~ 1.23(3H, m), 1.45 - 1.75 (6H, m) 2.70 (2H, d, J=7.00 Hz, CHCH
2Ar) , 6.84 (2H, d, J=8.68 Hz, ArH) 7.09 (2H, s, ArH), 7.38 (2H, d, J=8.68 Hz, ArH), 9.17 (4H, br, 4x0H), 12.20 (1H, br, CONH); EI -MS ra/e (%): 440.0 (M
+, 6), 288.1 (38), 205.1 (100), 163.0 (28); HREI-MS Calcd. for C
23H
24N
20
5S: 440.1406, found: 440.1406。 实施例 52 4- (4-曱氧基 -苯基)-5-甲基 -噻唑 -2-基胺的制 备
步骤 1 丙酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将丙酸转化为丙酰 氯, 其为无色液体, 未经纯化直接用于下步反应。
步骤 2 1- (4-甲氧基-苯基) -丙烷 -1-酮的制备
采用与实施例 47步骤 2类似的方法制备,以步骤 1制备的丙 酰氯和苯甲醚为原料, 以无水 A1C13为催化剂在二氯甲烷中反应 制备, -5 。C ~ -10 °C下搅拌 30分钟, 室温下搅拌 1 小时, 得 到粗品后经减压硅胶柱层析纯化, 石油醚和乙酸乙酯 25: 1 (V:V) 洗脱得到产物,收率 71.8%,为无色液体。 ^-NMR (CDC13, 400 MHz) δ: 1.22(3H, t, J=7.00Hz, CH2CH3) , 2.96 (2H, q, J=7.00Hz, CH2CH3) , 3.87(3H, s, 0CH3) , 6.93 (2H, d, J=8.72Hz, ArH),
7.95 (2H, d, J=8.72Hz, ArH); ESI-MS: 187.2(M+Na, 20),
165.2(M+1, 100).
步骤 3 2-溴 -l-(4-甲氧基 -苯基)-丙烷- 1-酮的制备
采用与实施例 1 步驟 3类似的方法制备, 以步驟 2 制备的
1- (4-甲氧基 -苯基)-丙垸- 1-酮和溴素为原料, 无水 A1C13为催化 剂,得到产物为白色固体,收率 90.7%,mp: 69-70 "C.^-NMR (CDC13, 400 MHz) δ: 1.89 (3H, d, J=6.44Hz, CHBrCH3) , 3.88 (3H, s, 0CH3) ,
5.27 (1H, q, J=6.44Hz, CHBrCH3) , 6.96 (2H, d, J=8.72Hz, ArH), 8.02 (2H, d, J=8.72Hz, ArH); ESI-MS m/e(%): 242.0 (M+2, 11), 241.0(M, 11), 135.0 (100)。
步骤 4 4— (4—甲氧基—苯基)_5—甲基—噻唑 -2-基胺的制备 采用与实施例 6步骤 5类似的方法制备, 以步骤 3制备的
2-溴- 1- (4-甲氧基-苯基)-丙烷- 1-酮、硫脲和无水乙酸钠为原料, 回流 4h, 后处理得到粗品后用无水乙醇重结晶得到产物, 白色固 体, 收率 66.8%, rap: 139-140 °C。 'H-NMR (CDC13, 00 MHz) δ: 2.37 (3H, s, ArCH3), 3.84 (3H, s, 0CH3) , 5.09 (2H, br, NH2) ,
6.93 (2H, d, J=8.68Hz, ArH), 7.50 (2H, d, J=8.68Hz, ArH); EI -MS m/e( ): 220.1 (M+, 100), 205.1 (18), 163.1 (31); HREI-MS Calcd. for C
nH
12N
2OS: 220.0670, found: 220.0672; Anal. Calcd. for C
nH
12N
20S: C 59.98, H 5.49, N 12.72; found: C 59.90, H 5.52, N 12.55。 实施例 53 3, 4, 5-三甲氧基 -N- [4- (4-甲氧基-苯基) -5-曱 基 -噻唑 -2 -基] -苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 52制备的 4- (4 - 甲氧基 -苯基)-5-曱基-噻唑- 2-基胺和实施例 37 步骤 1 制备的 3, 4, 5-三甲氧基-苯甲酰氯为原料, 于室温下反应过夜,后处理得 到粗品, 经硅胶柱层析纯化, 二氯甲烷和乙酸乙酯(15: 1)梯度洗 脱得到产物, 为白色固体, 收率 63. 0%, rap: 105-106 °C。 ^-NMR (CDC1
3, 400 MHz) δ: 2. 51 (3H, s, ArCH
3) , 3. 78 (3H, s, OCH3) , 3. 79 (3H, s, OCH3) , 3. 80 (3H, s, 0CH
3) , 3. 89 (3H, s, 0CH
3) 6. 78 (2H, d, J=8. 44 Hz, ArH) , 6. 98 (2H, s, ArH) , 7. 35 (2H, d, J=8. 44 Hz, ArH); EI -MS m/e (%) : 414. 1 (M+, 70) , 195. 1 (100) ; HREI-MS Ca lcd. for C
21H
22N
20
5S: 414. 1249, found: 414. 1258。 实施例 54 3, 4-二曱氧基 -N- [4- (4-甲氧基-苯基) -5-甲基- 噻唑- 2-基] -苯甲酰胺的制备
釆用与实施例 4类似的方法制备, 以实施例 52制备的 4- (4- 曱氧基 -苯基)-5-曱基-噻唑- 2-基胺和实施例 12 步骤 1 制备的 3, 4-二甲氧基 -苯曱酰氯为原料, 于室温下反应过夜,后处理得到 粗品, 经硅胶柱层析純化, 二氯甲烷和乙酸乙酯(20: 1)梯度洗脱 得到产物,为白色固体,收率 63. 5%, mp: 174-175 "C H-NMR (CDC1
3 400 MHz) δ: 2. 51 (3Η, s, ArCH
3) , 3. 83 (3Η, s, 0CH
3) , 3. 92 (3H, s, OCH3) , 3. 95 (3H, s, OCH3) , 6. 82 - 6. 92 (3H, m, ArH) , 7. 42 -
7.56 (4H, m, ArH), 10.83 (1H, br, CONH); EI-MS m/e(%): 384.0 (M
+, 66), 165.0 (100); HREI-MS Calcd. for C
2。H
2。N
20
4S: 384.1144, found: 384.1143; Anal. Calcd. for C
2。H
2。N
20
4S: C 62.48, H 5.24, N 7.29; found: C 62.30, H 5.19, N 7.30。 实施例 55 3, 4, 5-三羟基 - N- [4- (4-羟基-苯基) -5-甲基-噻 唑 -2-基] -苯甲酰胺的制备
采用与实施例 7类似的方法制备,以实施例 53制备的 3,4, 5- 三甲氧基- N- [4- (4-甲氧基-苯基) -5-甲基-噻唑-
2_基] -苯甲酰胺 和三溴化硼为原料, 粗品用丙酮重结晶纯化, 产物为白色固体, 收率 60.5%, mp: 262-263 X:。 ^-NMR (DMSO- , 400 MHz) δ: 2.36 (3Η, s, CH
3), 6.76 (2H, d, J=8.80 Hz, ArH), 7.02 (2H, s, ArH), 7.41(2H, d, J=8.80 Hz, ArH), 9.10 (2H, br, OH), 9.50(1H br, OH), 12.06 (1H, br, CONH); EI-MS m/e (%) : 358.1 (M
+, 25), 206.2 (100), 153.1 (20); HREI-MS Calcd. for C
17H
14N
20
5S: 358.0623, found: 358.0621。 实施例 56 3, 4-二羟基 -N- [4- (4-羟基-苯基) -5-甲基 -噻唑 - 2-基] -苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 54制备的 3,4-
二甲氧基- N- [4- (4-曱氧基-苯基) -5-甲基 -噻唑 -2 -基] -苯甲酰胺 和三溴化硼为原料, 粗品用丙酮重结晶纯化, 产物为白色固体, 收率 61.2°/», mp: 285 °C (Dec. ) 0 :H-NMR (CD30D, 400 MHz) δ: 2.46 (3H s, ArCH3), 6.88 - 6.98 (3H? m, ArH) , 7.40- 7.51 (4H, m, ArH) ; EI -MS m/e(%): 342.1 (M+, 29), 206.2 (100), 137.1 (36); HREI-MS Calcd. for C17H14N204S: 342.0674, found: 342.0670。 实施例 57 4- (4-甲氧基-苯基) -5-苯乙基-噻唑 -2-基胺的 制备
步骤 1 4-苯基丁酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将 4-苯基丁酸转化 为 4-苯基丁酰氯, 其为无色液体, 未经纯化直接用于下步反应。
步骤 2 1- (4-曱氧基 -苯基)-4-苯基-丁烷 -1-酮的制备 采用与 5.3.1步骤 2类似的方法制备, 以步骤 1制备的 4 -苯 基丁酰氯和苯甲醚为原料, 以无水 A1C13为催化剂在二氯甲烷中 反应制备, - 10 °C ~ -15 °C下搅拌 30分钟, 室温下搅拌 3小时, 得到粗品后经减压硅胶柱层析纯化, 石油醚和乙酸乙酯 20: 1 ~ 10: 1 (V: V)梯度洗脱得到产物, 白色晶体, 收率 57.5%, mp: 60-61 °C。 NMR(CDC13, 400 MHz) δ: 2.07 (2Η, quintuple, J=7.56Hz), 2.72 (2H, t, J=7.56Hz), 2.93 (2H, t, J=7.56Hz) , 3.86 (3H, s, 0CH3) , 6.91 (2H, d, J=7.00Hz, ArH), 7.16 - 7.21 (5H, m, ArH), 7.91 (2H, d, J=7.00Hz, ArH); FAB-MS: 255.2 (M+1, 100),
150.1 (27), 135.1 (22), 91.1 (7).
步驟 3 2-溴- 1- (4-甲氧基-苯基) -4-苯基-丁烷 -1-酮的制 备
采用与实施例 1 步驟 3类似的方法制备, 以步骤 2 制备的 1-(4-甲氧基 -苯基 )-4-苯基-丁烷- 1-酮和溴素为原料,无水 A1C13 为催化剂,粗品用石油醚和乙酸乙酯(2:1)混合溶剂重结晶,得到 产物为白色固体, 收率 89.4°/。, mp: 65-66 °C。 !H-NMR (CDC13, 400 MHz) δ: 2.38 - 2.52 (2H, m, CH2) , 2.75 - 2.90 (2H, ra, CH2) , 3.88 (3H, s, 0CH3) , 5.03 (1H, t, J=8.00Hz, CHBr) , 6.93 (2H, d, J=9.2Hz, ArH), 7.18 ~ 7.32 (5H, m, ArH), 7.91 (2H, d, J=9.20Hz, ArH); ESI-MS m/e(%): 334.0 (M+2, 1), 332.0 (M, 1), 229.9 (53), 227.9 (53), 135.0 (100) 0
步骤 4 4- (4-甲氧基-苯基) -5-苯乙基 -噻唑 -2-基胺(4- (4- 曱氧基 -苯基)-5-苯乙基 -噻唑 -2-基胺)的制备
采用与实施例 6步骤 5类似的方法制备, 以步骤 3制备的 1 - 溴 -1- (4-曱氧基-苯基) -4-苯基 -丁烷 -1-酮、硫脲和无水乙酸钠为 原料, 回流 3h, 后处理得到粗品后用无水乙醇重结晶得到产物, 白色固体,收率 85.9%,, mp: 143-144 Ό。 -腿(CDC13, 400 MHz) δ: 2.90 (2Η, t, J=7.60Hz, CH2), 3.05 (2H, t, J=7.60Hz, CH2), 3.82 (3H, s, 0CH3) , 4.99 (2H, br, NH2), 6.89 (2H, d, J=8.40Hz, ArH), 7.12-7.30 (5H, m, ArH), 7.35 (2H, d, J=8.40Hz, ArH); EI-MS m/e(%): 310.1 (M+, 23), 219.1 (100), 177.1 (39); HREI-MS Calcd. for C18H18N2OS: 310.1140, found: 310.1140; Anal. Calcd. for C18H1SN20S: C 69.65, H 5.84, N 9.02; found: C 69.74, H 5.86, N 9.03。
实施例 58 3,4-二 氧基- N- [4- (4-甲氧基 -苯基)-5-苯基 乙基-噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 57制备的 4- (4- 甲氧基 -苯基)-5-苯乙基 -噻唑 -2-基胺和实施例 12步骤 1制备的 3, 4-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜,后处理得到 粗品, 用石油醚和乙酸乙酯 2: 1 (V: V)重结晶得到产物, 为白色固 体,收率 76. 4%,mp: 74-75 。C。 -腿(CDC13, 400 MHz) δ: 3. 02 (2Η t, J=7. 60Hz, CH2) , 3. 19 (2H, t, J=7. 60Hz, CH2) , 3. 83 (3H, s, 0CH3) , 3. 94 (3H, s, OCH3) , 3. 96 (3H, s, 0CH3), 9. 85 - 7. 95 (3H, m, ArH), 7. 15 ~ 7. 37 (7H, m, ArH), 7. 51 (2H, s, ArH) , 10. 48 (1H br, CONH); EI -MS ra/e (%): 474. 1 (M+, 21) , 383. 0 (58) , 165. 0 (100) ; HREI-MS Ca lcd. for C27H26N204S: 474. 1613, found: 474. 1613。 实施例 59 3, 4, 5-三甲氧基 -N- [4- (4-曱氧基-苯基) -5 -苯 基乙基 -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 57制备的 4- (4-
甲氧基 -苯基)-5-苯乙基 -噻唑 -2-基胺和实施例 37步骤 1制备的 3, 4, 5-三甲氧基-苯曱酰氯为原料, 于室温下反应过夜,后处理得 到粗品泡沫状物, 用石油醚和乙酸乙酯 2: 1(V:V)重结晶得到产 物, 为白色固体, 收率 67.2%, rap: 78-79 °C。 -NMR (CDC13, 400 MHz) δ: 3.03 (2H, t, J=7.60Hz, CH2) , 3.19 (2H, t, J=7.60Hz, CH2), 3.80 (6H, s, 2xOCH3), 3.81 (3H, s, 0CH3) , 3.91 (3H, s, 0CH3) , 6.78 (2H, d, J=8.40Hz, ArH), 7.02 (2H, s, ArH) , 7.15- 7.32 (7H, ra, ArH), 11.54 (1H, br, CONH); EI -MS m/e (%): 504.2 (M+, 24), 413.2 (53), 195.1 (100); HREI-MS Calcd. for C28H28N205S: 504.1719, found: 504.1716。 实施例 60 3, 4-二羟基 - N- [4- (4-羟基-苯基) -5-苯基乙基- 噻唑 -2-基]-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 58制备的 3,4- 二甲氧基- N- [4- (4-甲氧基-苯基) -5-苯基乙基-噻唑- 2-基] -苯甲 酰胺和三溴化硼为原料, 粗品用乙酸乙酯重结晶纯化, 产物为白 色固体, 收率 53.8%, mp: 208-209 °C。 -腿(DMS0 - , 400 MHz) δ: 2.94 (2Η, t, J=7.60Hz, CH2), 3.11 (2H, t, J=7.60Hz, CH2) , 6.8K3H, d, J=8.40Hz, ArH), 7· 17 ~ 7.31 (5H, m, ArH), 7.35 (2H t, J=7.60Hz, CH2), 7.47 - 7.55 (2H, ra, ArH), 12.23(1H, br, CONH); EI -MS m/e 00: 432.1 (M+, 4), 205.0 (100), 163.0 (28); HREI-MS Calcd. for C24H2。N204S: 432.1144, found: 432.1140。
实施例 61 3, 4, 5-三羟基 -N- [4- (4-羟基-苯基) -5-苯基乙 基 -噻唑 -2-基] -苯甲酰胺的制备
采用与实施例 7类似的方法制备,以实施例 59制备的 3,4,5- 三甲氧基 -N- [4- (4-甲氧基-苯基) -5-苯基乙基 -噻唑 -2-基] -苯曱 酰胺和三溴化硼为原料, 粗品用丙酮重结晶纯化, 产物为白色固 体, 收率 43.8%, mp: 310 °C (Dec. ) D -NMR (DMSO- , 400 MHz) δ: 2.94 (2H, t, J=7.60Hz, CH2) , 3.13(2H, t, J=7.60Hz, CH2) , 6.84 (2H, d, J=8.40Hz, ArH), 7.07 (2H, s, ArH), 7.15-7.30(5H m, ArH), 7.33 (2H, d, J=8.40Hz, ArH); EI -MS m/e(%): 448.2 (M+, 2), 205.0 (100), 163.0 (29); HREI-MS Calcd. for C24H2。N205S: 448.1093, found: 448.1097。 实施例 62 4- (4-曱氧基-苯基) -5-正丁基 -噻唑 -2-基胺的 制备
步骤 1 正己酰氯的制备
采用与实施例 1步骤 1类似的方法制备, 将正己酸转化为正
己酰氯, 其为无色液体, 未经純化直接用于下步反应。
步骤 2 1- (4-曱氧基 -苯基)-己垸 -1-酮的制备
采用与实施例 52步骤 1类似的方法制备,以步骤 1制备的正 己酰氯和苯甲醚为原料, 以无水 A1C13为催化剂在二氯甲烷中反 应制备, -10 °C - -15 °C下搅拌 30分钟, 室温下搅拌 3小时, 得到粗品后经减压硅胶柱层析纯化,石油醚和乙酸乙酯 20: 1洗脱 得到产物, 蜡状固体, 收率 66.2%, mp: 39-40 °C。 ^-NMR (CDC13, 400 MHz) δ: 0.91 (3H, t, J=7.20Hz, CH3) , 1.30~ 1.42(4H, m, CH2CH2) , 1.73(2H, t, J=7.20Hz, COCH2CH2) , 2.91 (2H, t, J=7.20Hz, COCH2CH2) , 3.87 (3H, s, 0CH3), 6.93 (2H, d, J=8.80Hz; ArH), 7.96 (2H, d, J=8.80Hz, ArH) ; ESI-MS m/e(%): 206.1 (M, 5), 163.0 (6), 150.0 (58), 135.0 (100) 0
步骤 3 2-溴- 1- (4-甲氧基-苯基) -己烷 -1-酮的制备
采用与实施例 1 步骤 3 类似的方法制备, 以步骤 2 制备的 1-(4-曱氧基-苯基) -己烷 -1-酮和溴素为原料, 无水 A1C13为催化 剂,粗品经减压硅胶柱层析纯化,石油醚和乙酸乙酯 20: 1洗脱得 到产物, 为白色晶体, 收率 93.4%, mp: 54-55 °C。 -NMR (CDC13, 400 MHz) δ: 0.92 (3H, t, J=7.2Hz, CH3) , 1.30 - 1.60 (4H, m, CH2CH2), 2.05 - 2.25 (2H, m, CH2CHBr) , 3.89 (3H, s, 0CH3) , 5.11 (1H, t, J=7.2Hz, CHBr) , 6.96 (2H, d, J=8.80Hz, ArH), 8.01 (2H, d, J=8.80Hz, ArH) ; ESI-MS ra/e(%): 286.1 (M+2, 2), 284.1 (M, 2), 230.0 (3), 228.0 (3), 205.1 (5), 135.1 (100)。
步骤 4 5-正丁基 1-4- (4-甲氧基-苯基)-噻唑- 2-基胺的制 备
采用与实施例 6步驟 5类似的方法制备, 以步骤 3制备的 2- 溴- 1 - (4 -甲氧基-苯基) -己烷 - 1 -酮、 硫脲和无水乙酸钠为原料,
回流 3h,后处理得到粗品后用石油醚和乙酸乙酯 1: 1(V: V)重结晶 得到产物,白色固体,收率 57.2%,mp: 118-119 °C。 ^- MR (CDC13, 400 M, Hz) δ: 0.90 (3H, t, J=7.2Hz, CH3) , 1.36 (2H, sextuple, J=7.20Hz, CH2), 1.59 (2H, quintuple, J=7.20Hz, CH2), 2.74 (2H, t, J=7.2Hz, CH3), 3.83 (3H, s, 0CH3) , 4.93 (2H, br, 肌),
6.93 (2H, d, J=8.80Hz, ArH) , 7.45(2H, d, J=8.80Hz, ArH); EI -MS m/e(%): 262.0 (M+, 55), 219.0 (100), 177.0 (36); HREI-MS Calcd. for C"H18N2OS: 262.1140, found: 262.1140; Anal. Calcd. for C14H18N2OS: C 64.09, H 6.92, N 10.68; found: C 64.19, H
7.02, N 10.63。 实施例 63 N- [5-正丁基 -4- (4-曱氧基-苯基) -噻唑 -2- 基]- 3 , 4 -二甲氧基-苯甲酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 62制备的 4-(4- 甲氧基 -苯基)-5-正丁基 -噻唑 -2-基胺和实施例 12步驟 1制备的 3, 4-二甲氧基 -苯甲酰氯为原料, 于室温下反应过夜,后处理得到 粗品, 用石油醚和乙酸乙酯 2: 1(V:V)重结晶得到产物, 为白色固 体, 收率 62.8% mp: 150-151 °C。 ^-NMR (CDC13, 400 MHz) δ:
0.93 (3H, t, J=7.2Hz, CH3) , 1.41 (2H, sextuple, J=7.20Hz, CH2)
1.72 (2H, quintuple, J=7.20Hz, CH2), 2.88 (2H, t, J=7.2Hz, CH3), 3.80 (3H, s, 0CH3), 3.83 (3H, s, 0CH3), 3.94 (3H, s, 0CH3), 6.72 - 6.82 (3H, m, ArH), 7.25 ~ 7.40 (4H, m, ArH), 11.16 (1H,
br, CONH); EI— MS m/e (%): 426.1 (M+, 50), 383.1 (7), 165.1 (100); HREI-MS Calcd. for C23H26N204S: 426.1613, found: 426.1613; Anal. Calcd. for C23H26N20,S: C 64.77, H 6.14, N 6.57; found: C 64.56, H 6.23, N 6.61。 实施例 64 N- [5-正丁基 -4- (4-甲氧基 -苯基)-噻唑- 2- 基]- 3, 4, 5-三曱氧基-苯曱酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 62制备的 4-(4- 甲氧基 -苯基)-5-正丁基 -噻唑 -2-基胺和实施例 37步骤 1制备的 3, 4, 5-三甲氧基 -苯甲酰氯为原料, 于室温下反应过夜,后处理得 到粗品玻璃状物, 用石油醚和乙酸乙酯 2:1(V:V)重结晶得到产 物,为白色固体,收率 51.8°/。, mp: 197-198 °C 0 ^-NMR (CDC13, 400 MHz) δ: 0.93 (3H, t, J=7.2Hz, CH3) , 1.43 (2H, sextuple, J=7.20Hz, CH2), 1.72 (2H, quintuple, J=7.20Hz, CH2) , 2.88 (2H, t, J=7.2Hz, CH3), 3.75 (6H, s, 2x0CH3), 3.78 (3H, s, 0CH3) , 3.89 (3H, s, 0CH3) , 6.74 (2H, d, J=8.80Hz, ArH), 6.91 (2H, s, ArH), 7.28(2H, d, J=8.80Hz, ArH), 11.96 (1H, br, CONH); EI -MS m/e(°/o) : 456. l(M+, 49), 413.1(7), 195.1 (100); HREI-MS Calcd. for C24H28N205S: 456.1719, found: 456.1718; Anal. Calcd. for C24H28N205S: C 63.14, H 6.18, N 6.14; found: C 63.24, H 6.20, N 6.25。 实施例 65 N- [5-正丁基 -4- (4-羟基-苯基) -噻唑 -2-
- 3, 4-二羟基-苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 63制备的 N- [5- 正丁基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 4-二甲氧基-苯甲酰 胺和三溴化硼为原料,粗品用丙酮重结晶纯化,产物为白色固体, 收率 43.6%, mp: 189-190 °C。 - R (DMS(W6, 400 MHz) δ:
0.88 (3H, t, J=7.2Hz, CH3) , 1.35 (2H, sextuple, J=7.20Hz, CH2) ;
1.62 (2H, quintuple, J=7.20Hz, CH2), 2.82 (2H, t, J=7.2Hz, CH3), 6.07 (3H, br, OH), 7.65 ~ 7.85 (3H, ra, ArH), 7.32 - 7.52 (4H, m, ArH), 12.12 (1H, br, CONH); EI -MS m/e (%): 384.1 (M+, 37), 248.2 (83), 205.1 (100); HREI-MS Calcd. for C2。H2。N204S: 384.1144, found: 384.1146。 实施例 66 N- [5-正丁基 -4- (4-羟基-苯基) -噻唑 -2- 基]- 3, 4, 5-三羟基 -苯甲酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 64制备的 N-[5- 正丁基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -3, 4, 5-三甲氧基 -苯甲 酰胺和三溴化硼为原料, 粗品用丙酮重结晶纯化, 产物为白色固 体, 收率 47.5%, mp: 119-120 °C。 ^-NMR (DMS0-i6, 400 MHz) δ:
0.88 (3H, t, J=7.2Hz, CH3) , 1.35 (2H, sextuple, J=7.20Hz, CH2) ,
1.63 (2H, quintuple, J=7.20Hz, CH2), 2.82 (2H, t, J=7.2Hz, CH3), 6.83 (2H, d, J=8.80Hz, ArH) , 7.08 (2H, s, ArH) , 7.40 (2H, d, J=8.80Hz, ArH), 8.80 (4H, br, OH), 12.12 (1H, br, CONH); EI -MS ra/e ( ): 400.1 (M+, 14), 248.2 (55), 205.1 (100), 163.1 (38); HREI-MS Calcd. for C2。H2。N205S: 400.1093, found: 400.1091。 实施例 67 N- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -N- 对曱苯磺酰基-对曱苯磺酰胺的制备
在 100 mL茄形烧瓶中加入 1.00 g(3.37 mraol)实施例 1制备 的 5 -苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基胺、 0.68 g (6.75 mmol) 三乙胺和 25 mL THF, 室温下滴加溶于 5 mL THF中的 1.28 g(6.74 mraol)对甲苯磺酰氯, 室温搅拌 24小时, 过滤, THF洗, 母液减 压浓缩得到粗品, 丙酮重结晶得到产物 1.04 g, 类白色固体, 收 率 51.0%,mp: 182-183 "C e'H-NMR (DMSO-fl^, 400 MHz) δ: 2.45 (6H, s, 2xPhCH3), 3.84 (3H, s, 0CH3) , 4.26(2H, s, CH2) , 6.92 (2H, d, J=8.68Hz, ArH), 7.18 - 7.36 (9H, ra, ArH), 7.46(2H, d, J=8.68Hz, ArH), 7.92 (4H, d, J=8.68Hz, ArH); EI -MS m/e(%): 604.2 (M+, 19), 450.2 (44), 294.2 (78), 91.1 (100); HREI-MS Calcd. for C31H28N205S3: 604.1160, found: 604.1165; Anal. Calcd. for C31H28N205S3: C 61.57, H 4.67, N 4.63; found: C 61.71
H 4.49, N 4.82。 实施例 68 N- [5-苄基 -4- (4-甲氧基-苯基)-噻唑 -2- 基] - 3- ( 3, 4-二甲氧基-苯基) -丙酰胺的制备
步骤 1 3- ( 3, 4 -二甲氧基-苯基) -丙酰氯的制备
釆用与实施例 1步骤 1类似的方法制备, 将 3-(3,4-二甲氧 基-苯基)-丙酸转化为 3- (3, 4-二曱氧基-苯基) -丙酰氯, 其为白 色固体, 未经纯化直接用于下步反应。
步驟 2 N- [5-苄基- 4-(4-曱氧基-苯基)-噻唑- 2- 基] -3- (3, 4-二甲氧基-苯基) -丙酰胺的制备
采用与实施例 4类似的方法制备, 以实施例 1制备的 5-苄基 -4-(4-曱氧基 -苯基)-噻唑- 2-基胺和步驟 1制备的 3-(3,4-二甲 氧基-苯基) -丙酰氯为原料, 于 35- 40 °C反应 10小时, 后处理得 到粗品, 经硅胶柱层析纯化,石油醚和乙酸乙酯(4: 1)洗脱得到产 物, 为白色固体, 收率 65.0%, mp: 142- 143°C。 !H-NMR (CDC13, 400 MHz) δ: 2.12 (2H, t, J=7.72Hz), 2.74 (2H, t, J=7.72Hz), 3.80 (6H, s, 2xOCH3), 3.83 (3H, s, 0CH3) , 4.18 (2H, s, CH2) , 6.51(1H, dd, J-8.16, 1.96Hz, ArH) , 6.56 (1H, d, J=l.96Hz, ArH), 6.69 (1H, d, J=8.16Hz, ArH), 6.83 (2H, d, J=8.68Hz, ArH), 7.21-7.33 (5H, m, ArH), 7.42 (2H, d, J=8.68Hz, ArH), 11.05 (1H, br, CONH); EI -MS m/e (%): 488.1 (M+, 66), 296.0 (100); HREI-MS Calcd. for C28H28N204S: 488.1770, found:
488.1764; Anal. Calcd. for C2SH28N20,S: C 68.83, H 5.78, N 5.73; found: C 68.69, H 5.67, N 6.09. 实施例 69 N- [5-苄基- 4- (4-羟基-苯基) -噻唑 - 2- 基] -3- (3, 4-二羟基-苯基) -丙酰胺的制备
采用与实施例 7类似的方法制备, 以实施例 68制备的 N-[5- 苄基— 4- (4-甲氧基-苯基) -噻唑 -2-基] -3- (3, 4-二甲氧基-苯基) - 丙酰胺和三溴化硼为原料, 粗品用乙酸乙酯重结晶得到产物, 白 色固体, 收率 60.3%, mp: 94-95 °C。 !H-NMR (DMSO- 400 MHz) δ: 2.6K2H, t, J=7.00Hz, CH2) , 2.68 (2H, t, J=7.00Hz, CH2) , 4.17 (2H, s, CH2), 6.43 (1H, d, J-7.84Hz, ArH), 6.59 (2H, t, J=7.84Hz, CH2), 6.80 (2H, d, J=7.84Hz, ArH), 7.18-7.24 (3H, ra, ArH), 7.32 (2H, t, J=7.28Hz, CH2) , 7.41(2H, d, J=7.84Hz, ArH), 8.65 (1H, s, OH), 8.74 (1H, s, OH), 9.59 (1H, s, OH), 12.06 (1H, s, CONH); EI -MS m/e(%): 446.1 (M+, 18), 282.1 (100) 205.1 (17); HREI-MS Calcd. for C25H22N204S: 446.1300, found: 446.1299。 实施例 70 4- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -吗 啉的制备
步骤 1 5-苄基 -2-氯- 4- (4-甲氧基-苯基) -噻唑的制备 在 100 mL三颈瓶中加入 1.00 g (3.37 mmol) 实施例 1制备 的 5-苄基- 4- (4-甲氧基-笨基) -噻唑 -2-基胺和 15 mL乙睛, 搅拌 溶解, 冰浴冷却至 Q °C, 加入 0.69 g (4.04 mmol)氯化铜一水合 物, 滴加溶于 5 mL乙睛中的 0.59 g (5.06 mmol)亚硝酸异戊酯, 加完后在该温度搅拌 lh, 在室温下搅拌 lh, 旋转蒸发除去乙睛, 残余物中加入 10 mL乙酸乙酯, 就悬浮液经短硅胶柱过滤, 乙酸 乙酯洗脱, 浓缩滤液, 经减压硅胶柱层析纯化, 以石油醚和乙酸 乙酯 20: 1 ~ 10: 1 (V:V)梯度洗脱, 得到产物, 白色固体 0.55 g, 收率 51.9%, rap: 78—79 °C。 ^-NMR (CDC13, 400 MHz) δ: 3.84 (3H, s, 0CH3) , 4.21 (2H, s, CH2), 6.95 (2H, d, J=8.96Hz, ArH) , 7.18-7.38 (5H, m, ArH), 7.54 (2H, d, J=8.96Hz, ArH); ESI— MS m/e(%): 315.9(M+1, 100)。
步骤 2 4- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -吗啉 的制备
在 25 mL茄型瓶中加入 1.00 g (3.17讓。 1)步骤 1制备的 5- 苄基- 2-氯- 4-(4-甲氧基 -苯基)-噻唑、 0.27 g (6.34 mmol) LiOH'H20、 20 mg KI、 0.55 g (6.34 mmol)吗啉、 5 mL DMF和 0· 5 mL水,将上述混合物搅拌均勾,加热至 100'C搅拌 20h,加入 20 mL 水, 用乙酸乙酯萃取 3次 xlO mL, 合并有机相, 饱和盐水洗, 无 水硫酸钠干燥, 过滤浓缩得到粘稠粗品, 经减压硅胶柱层析纯化, 石油醚和乙酸乙酯 4: 1 ~ 3: 1 (V: V)洗脱,得到产物,白色固体 0.73
g,收率 62.9%,κ : 88-89 Ό。 !H-NMR (CDC13, 400 MHz) δ: 3.47 (4H m, 2xCH2), 3.80 (4H, t, J=5.2Hz, 2xCH2), 3.82 (3H, s, 0CH3) , 4.12 (2H, s, CH2), 6.92 (2H, d, J=8.40Hz, ArH) , 7.18-7.32 (5H m, ArH), 7.53 (2H, d, J=8.40Hz, ArH); EI-MS m/e(%): 366.1 (M+, 100), 309.1 (23),; 固 I - MS Calcd. for C21H22N202S: 366.1402, found: 366.1404; Anal. Calcd. for C21H22N202S: C 68.83, H 6.05, N 7.64; found: C 68.73, H 6.26, N 7.83。 实施例 71 1- [5-苄基 -4- (4-曱氧基-苯基) -噻唑 -2-基] -哌 嗪的制备
在 25 mL茄型瓶中加入 1.30 g (4.12 mraol) 实施例 70步骤 1制备的 5-苄基- 2-氯- 4- (4-曱氧基-苯基) -噻唑、 0.35 g (8.24 ramol) LiOH'H20、 20 rag KI、 1.77 g (20.6 mmol) p底嗪、 5 mL DMF 和 0.5 mL水, 将上述混合物搅拌均勾, 加热至 ΙΟΟΌ搅拌 12h, 加入 20 mL水, 用乙酸乙酯萃取 3次 xlO raL, 合并有机相, 饱和 盐水洗, 无水硫酸钠干燥, 过滤浓缩得到粘稠粗品, 经减压硅胶 柱层析纯化, 二氯甲烷和曱醇 10: 1 ~ 5: 1 (V:V)洗脱, 得到产物, 粘稠油状物 1.05 g, 收率 69.5%。 ^-NMR (CDC13, 400 MHz) δ:
2.99 (4Η, t, J=5.2Hz, 2xCH2), 3.46 (4H, t, J=5.2Hz, 2xCH2),
3.82 (3H, s, 0CH3) , 4.12 (2H, s, CH2) , 6.90 (2H, d, J=8.80Hz, ArH), 7.18-7.35 (5H, m, ArH), 7.53(2H, d, J=8.80Hz, ArH); EI-MS m/e (%): 365.0 (M+, 86), 323.0 (28), 309.0 (100),
296.0 (49); HREI-MS Calcd. for C21H23N3OS: 365.1562, found: 365.1564。 实施例 72 1- [5-苄基- 4- (4-甲氧基-苯基) -噻唑 -2-基] -哌 嗪三盐酸盐的制备
将 0.4 g (1.09 麵 ol)实施例 71制备的 1- [5-苄基 -4- (4-曱 氧基-苯基) -噻唑 -2-基]-哌嗪溶解在 20 mL乙酸乙酯中, 冰水浴 冷却, 搅拌下加入 2 Hiol/L的氯化氢乙酸乙酯溶液, 生成黄色固 体, 抽滤, 乙酸乙酯洗得到产物, 黄色固体 3.4 g, 收率 77.3%, mp: 111-112 °C。 ^-NMR (DMS0-if
6, 400 MHz) δ: 3.19 (4H, m, 2xCH
2), 3.62 (4H, m, 2xCH
2), 3.79 (3H, s, 0CH
3) , 4.12 (2H, s, CH
2), 5.58 (2H, br, exchangeable, 2xNH), 6.98 (2H, d, J=8.80Hz, ArH), 7.18 ~ 7.32 (5H, m, ArH) , 7.52 (2H, d, J=8.80Hz, ArH), 9.34 (2H, br, exchangeable, 2 NH); EI - MS m/e (%): 365.0 (M-3, 81), 323.0 (26), 309.0 (100), 295.9 (51); HREI-MS Calcd. for C
21H
23N
3OS: 365.1562, found: 365.1564。 实施例 73 2- [[5-苄基 -4-(4-甲氧基-苯基) -噻唑 -2- 基]一 (2-羟基-乙基) -氨基] -乙醇的制备
在 25 mL茄型瓶中加入 1.30 g (4.12 mmol) 实施例 70步骤 1制备的 5-苄基- 2-氯- 4- (4-甲氧基 -苯基)-噻唑、 0.35 g (8.24 mmol) LiOH'H20、 20 mg KI、 0.87 g (8.24 imnol) p束嗪、 5 mL DMF 和 0.5 mL水, 将上述混合物搅拌均勾, 加热至 10(TC搅拌 12h, 加入 20 mL水, 用乙酸乙酯萃取 3次 xlO mL, 合并有机相, 饱和 盐水洗, 无水硫酸钠干燥, 过滤浓缩得到粗品类白色固体, 经减 压硅胶柱层析纯化, 二氯甲烷和曱醇 10: 1(V: V)洗脱, 得到产物, 粘稠油状物, 石油醚乙酸乙酯重结晶得到白色固体 0.98 g, 收率 62.0%, mp: 119-120 °C。 一 NMR (CDC13, 400 MHz) δ: 3.62 (4Η, t, J=5.2Hz, 2xCH2), 3.81 (3H, s, OCH) , 3.88 (4H, t, J=5.2Hz, 2xCH2), 4.09 (2H, s, CH2) , 6.90 (2H, d, J=8.80Hz, ArH), 7.18- 7.35 (5H, m, ArH), 7.48(2H, d, J=8.80Hz, ArH); EI-MS m/e (%): 384.1 (M+, 80), 353.1 (64), 309.0 (100), 295.1 (15); HREI-MS Calcd. for C21H24N203S: 384.1508, found: 384.1504; Anal. Calcd. for C21H24N203S: C 65.60, H 6.29, N 7.29; found: C 65.42, H 6.56, N 7.34。 实施例 74 4- (5-苄基- 2-吗啉- 4-基-噻唑- 4-基) -苯酚的制 备
在 50 mL茄型瓶中加入 0.60 g(l.64 mmol)实施例 70制备的 4- [5-苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基] -吗啉和 20 mL二氯甲 烷, 冰水浴冷却, 滴加 2.4 mL (4.92 ramol) 2.034 mol/L三溴化 硼的二氯甲烷溶液, 冰水浴下搅拌 1小时, 室温下搅拌 3小时, 加入 2 g 水分解复合物, 减压浓缩除去二氯甲烷, 用 100 mL 乙 酸乙酯溶解, 饱和盐水洗, 无水硫酸钠干燥, 浓缩, 减压硅胶柱 层析,二氯甲烷和甲醇 10: 1-5:1洗脱,得到浅黄色粘稠液体 0.26 g, 收率 44.8%。 - NMR(DMS0 - , 400 MHz) δ: 3.50 (2Η, m, CH2) , 3.63 (2H, m, CH2), 3.97 (2H, s, CH2) , 4.27 (4H, s, CH2) , 5.18 (1H m), 6.97 (2H, d, J=8.40Hz, ArH), 7.15 - 7.40 (7H, m, ArH) , 10.14 (1H, s, OH); EI-MS m/e (%) : 352.0 (M+, 37), 322.0 (100), 240.0 (48); HREI-MS Calcd. for C2。H2。N202S: 352.1245, found: 352.1243。 实施例 75 4- (5-苄基 -2-哌嗪- 4-基-噻唑- 4-基) -苯酚的制
采用与实施例 74类似的方法制备,用实施例 71制备的 1- [5 -
苄基 -4- (4-ψ氧基-苯基) -噻唑 -2-基] -哌嗪和三溴化硼二氯甲烷 溶液为原料, 得到粗品后以丙酮重结晶得到产物, 白色固体, 收 率 49.2%,mp: 222-223 H-NMR (DMS0-i¾, 00 MHz) δ: 2·71(2Η, s, CH2), 3.20 (2H, m, CH2), 3.40(1H, br, NH) , 4.01 (2H, s, CH2), 6.70 (2H, d, J=8. OOHz, ArH), 7.05 - 7.25 (2H, m, ArH), 7.31 (2H d, J=8. OOHz, ArH), 9.48 (IH, s, OH); EI -MS m/e(%): 351.1(M+, 77), 309.0 (29), 295.0 (100), 282.0 (42); HREI-MS Calcd. for C2oH2iN3OS: 351.1405, found: 351.1406。 实施例 76 4- {5-苄基 -2- [双- (2-羟基-乙基) -氨基] -噻唑 - 4 -基 } -苯酚的制备
采用与实施例 74 类似的方法制备, 用实施例 73 制备的 2- [ [5 -苄基 -4- (4-甲氧基-苯基) -噻唑 -2-基]一 (2-羟基-乙基) -氨 基] -乙醇和三溴化硼二氯甲烷溶液为原料, 得到粗品后以丙酮重 结晶得到产物, 白色固体, 收率 54.7°/。, mp: 167-168 °C。
^-NMR (DMSO-i6, 400 MHz) δ: 3.46(2H, t, J=6.40Hz, CH2) , 3.59(2H, t, J=6.40Hz, CH2) , 4.04 (2H, s, CH2), 4.88 (2H, t, J=5.20Hz, 2xOH), 6.78 (2H, d, J=8.80Hz, ArH), 7.15-7.28 (5H m, ArH), 7.36 (2H, d, J=8.80Hz, ArH), 9.54 (IH, br, OH); EI -MS m/e (%): 370.1 (M+, 75), 339.1 (74), 309.0 (17), 295.0 (100); HREI-MS Calcd. for C2。H22N203S: 370.1351, found: 370.1349; Anal. Calcd. for C20H22N2O3S: C 64.84, H 5.99, N 7.56; found:
C 64. 86, H 5. 89, N 7. 61 实施例 77 5-苄基- 2- [2- (3, 4-二甲氧基-苯基) -乙 基] -4- (4-甲氧基-苯基) -噻唑的制备
步驟 1 3- ( 3 , 4-二甲氧基-苯基) -丙酸叔丁氧甲酸混酐的制 备
在 50 mL三颈瓶中加入 0· 50 g (2. 38 mmo l) 3-(3, 4-二甲氧 基-苯基) -丙酸、 5 mL THF和 0. 5 g三乙胺,水水浴冷却至 0 ~ 5 "C , 滴加溶于 3 raL THF中的氯甲酸异丁酯溶液, 加完后室温搅拌 lh, 抽滤除去三乙胺的盐酸盐, THF 洗, 得到标题的混酐溶液, 直接 用于下步反应中。
步骤 2 3 -(3, 4-二曱氧基-苯基) -丙酰胺的制备
在 50 raL三颈瓶中加入 5 raL 25%的氨水, 冰水浴冷却至 0 ~ 5 "C , 滴加步骤 1的混酐溶液, 加完后室温搅拌 2h, 减压浓缩, 残 余物用水和乙酸乙酯各 10 mL分层,水相用乙酸乙酯洗 2次 x5 mL, 合并有机相, 依次用 1 mol /L HC1、 饱和盐水、 饱和 NaHC03及饱 和盐水洗涤,无水 Na2S04干燥,过滤并浓缩得到 290 mg白色固体, 收率 58. 0%,mp: 122-123 "C H-NMR (CDC13, 400 MHz) δ: 2. 52 (2H, t, J=7. 84Hz, CH2), 2. 93 (2H, t, J=7. 84Hz, CH2) , 3. 86 (3H, s, CH30) , 3. 87 (3H, s, CH3O) , 5. 40 (2H, s, NH2) , 6. 72 - 6. 82 (3H, m, ArH) ; ESI-MS (m/e, %): 232. 2 (M+Na, 61) , 227. 5 (M+NH3+H, 16), 210. 2 (M+H, 100)。
步骤 3 3- (3, 4-二甲氧基-苯基) -硫代丙酰胺的制备 在 25 raL茄型瓶中加入 0.15 g (0.717 mmol)步驟 2制备的 3 -(3, 4-二甲氧基-苯基)-丙酰胺、 0.16 g(0.717腿 ol) P2S5和 10 mL THF, 安装具 CaC12干燥管的回流冷凝管, 磁力搅拌回流 lh, 减压浓缩, 以乙酸乙酯和饱和 NaHCOr溶液各 15 mL分层, 水层用 乙酸乙酯萃取 2次 xlOmL, 合并有机相, 用饱和盐水洗涤 2次 xl5 mL, 无水 Na2S04干燥, 过滤并浓缩得到固体粗品, 减压硅胶柱层 析, 以石油醚、 乙酸乙酯 10:1(V : V)洗脱得到产物, 白色固体 0.11 g, 收率 68.8%, mp: 140-141 。C。 ^- MR (CDC13, 600 MHz) δ: 2.93 (2H, t, J=7.20Hz, CH2), 3.07 (2H, t, J=7.84Hz, CH2), 3.86 (3H, s, CH30) , 3.87 (3H, s, CH30) , 6.62 (1H, br), 6.75- 6.82 (3H, ra, ArH) , 7.35 (1H, br); ESI-MS (m/e, %): 248.3 (M+Na, 100), 226.3 (M+H, 64)。
步驟 4 5-苄基- 2- [2- (3, 4-二甲氧基-苯基) -乙基] -4- (4- 曱氧基-苯基) -噻唑的制备
在 50 mL茄型瓶中加入 0.12 g(0.533 mmol) 步驟 3制备的 3-(3, 4-二甲氧基-苯基) -硫代丙酰胺、 0.17 g(0.533 mmol) 实 施例 1 的步骤 3制备的 2-溴- 1- (4-曱氧基 -苯基)-3-苯基 -丙烷 -1 -酮、 0.04 g (0.533讓 ol)无水乙酸钠以及 20 mL无水乙醇, 回 流 4h, TLC显示反应完全, 减压浓缩, 以水和乙酸乙酯各 20 mL 分层, 乙酸乙酯层用饱和盐水洗 2次 xlO mL, 无水 ^2304干燥, 过滤并浓缩得到固体粗品,以石油醚和乙酸乙酯重结晶得到产物, 粘稠液体 0.17 g, 收率 70.8%。 :H-NMR (CDCI3, 400 MHz) δ: 3.06 (2Η, t, J=8.40Hz, CH2) , 3.25 (2H, t, J=8.40Hz, CH2),
3.83 (3H, s, CH3O) , 3.84 (3H, s, CH30), 3.86 (3H, s, CH30) ,
4.20 (2H, s, CH2), 6.70 - 6.80 (3H, m, ArH), 6.95 (2H, d,
J-8.80Hz, ArH), 7.15 - 7.35 (5Η, m, ArH), 7.54 (2H, d, J=8.80Hz, ArH); EI -MS ra/e (%): 445.1 (M+, 100), 151.1 (93); 腦 I—MS Calcd. for C27H具 S: 445.1712, found: 445.1716。 实施例 78 4- {2- [5-苄基- 4- (4-羟基-苯基) -噻唑 -2-基] - 乙基 } -苯- 1,2-二醇的制备
采用与实施例 7类似的方法制备,以实施例 77步骤 4制备的 5-苄基- 2- [2- (3, 4-二甲氧基-苯基) -乙基] -4- (4-甲氧基-苯基) - 噻唑和三溴化硼为原料,粗品用丙酮重结晶得到产物, 白色固体, 收率 52.2%, mp: 177-178 。C。 'H-NMR (DMS0-i6, 400 MHz) δ: 2.82 (2H, t, J=8.16Hz, ArH), 3.11 (2H, t, J=8.16Hz, ArH), 4.20 (2H, s, CH2), 6.48 (1H, d, J=8.12Hz, ArH), 6.58 - 6.64 (2H m, ArH), 6.82 (2H, d, J=8.40Hz, ArH), 7.14-7.26 (3H, m, ArH)
7.32 (2H, t, J=7.28Hz, ArH), 7.44 (2H, d, J=8.40Hz, ArH),
8.69 (1H, s, OH), 8.75(1H, s, OH), 9.61 (1H, s, OH); EI -MS m/e(%): 403.2 (M+, 67), 281.2 (100); HREI-MS Calcd. for C24H21N03S: 403.1242, found: 403.1238。 本发明涉及的化合物的 PLTP抑制活性和 CETP抑制活性可用 如下方法检测:
实施例 79 PLTP抑制活性测定方法
将 30 μΐ磷脂脂质体(约 1, OOOcpm/μΙ)、 30 μΐ HDL (6.0 g 蛋
白 / μ1)、 337μ1 TSE和 3 μΐ血浆混合, 在 37 。C水浴中温孵 1 小时。然后加入 300 μΐ沉降溶液(50ml溶液配制: 5. 36 ral 5N NaCl, 9. 075ml 2M MnCl2和 46mg 肝磷脂, 加水至 50 ml) , 在室温下将 该溶液静置 10分钟,然后高速旋转 10分钟(为去除磷脂脂质体), 取 500 μΐ上清液计数。 PLTP活性(cpra/h/ 3 μΐ) = 样品 cpra - 背 景 cpm。 实施例 80 CETP抑制活性测定方法
将含有(3 μΐ供体、 9 μ1接受体 (250 g蛋白)、 3 μΐ的 CETP 溶液和 85 μΐ分析緩冲液、 10 mM Tr is、 0. 15 M NaCl和 2 rnM EDTA, pH 7. 4)的混合物在 37 °C温孵 0. 5小时。 在带有 460 nm发射光和 530 nm 激发光波长的 Victor 3 默重照度计 /荧光计数器(Perkin Elmer)上测定荧光强度。
PLTP抑制活性和 CETP抑制活性测定结果见表 1。
PLTP抑制活性和 CETP抑制活性测定结果
PLTP抑制活性 CETP抑制活性 实施
化合物浓 抑制率 IC5。 化合物浓 抑制率 IC5„ 例号
度(μ Μ) (%) (μΜ) 度(μ Μ) (%) (μΜ)
8 150 0 一 500 80. 4 一
19 250 79. 8 70 500 19. 4 一
21 250 11. 5 一 500 52. 8 一
31 250 100 75 500 0 一
32 250 100 75 500 0 一
33 250 73. 2 100 500 9. 2 一
34 250 67. 6 100 500 30. 4 一
38 150 11. 5 一 500 88. 0 一
40 150 0 一 500 71. 6 一
41 150 89. 4 15 500 44. 1 一