WO2007100101A1 - アスパラギン酸アミノトランスフェラーゼ遺伝子およびl-ホスフィノスリシンの製造方法 - Google Patents
アスパラギン酸アミノトランスフェラーゼ遺伝子およびl-ホスフィノスリシンの製造方法 Download PDFInfo
- Publication number
- WO2007100101A1 WO2007100101A1 PCT/JP2007/054079 JP2007054079W WO2007100101A1 WO 2007100101 A1 WO2007100101 A1 WO 2007100101A1 JP 2007054079 W JP2007054079 W JP 2007054079W WO 2007100101 A1 WO2007100101 A1 WO 2007100101A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- activity
- polynucleotide
- protein
- salt
- seq
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1096—Transferases (2.) transferring nitrogenous groups (2.6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P9/00—Preparation of organic compounds containing a metal or atom other than H, N, C, O, S or halogen
Definitions
- the present invention relates to aspartate aminotransferase and its gene, and a method for producing L-phosphinothricin.
- the present invention also relates to a recombinant vector for expressing an aminotransferase gene and a host into which the vector has been introduced.
- L-phosphinothricin is a constituent of bialaphos
- Ru substance der have herbicidal activity (Tachibana, K, J. pesticide Sci , 11:.. 27-31, 1986) 0 inventors
- OMPB 2-oxobutyric acid
- OMPB 2-oxobutyric acid
- the oxacin acetic acid produced by deamino of aspartate is easily decarboxylated at the normal reaction temperature (for example, 37 ° C) and becomes pyruvic acid, thus leaving the reaction system without affecting the yield. .
- OMPB reduced glutamic acid
- HMPB glutamic acid: aspartic acid
- this technology has a conversion rate to L-phosphinothricin of up to 86%, which is less than the industrial target yield and uses an enzyme from pigs that is difficult to procure at low cost. It can be said that this technology is practically difficult to put into practical use.
- an aminotransferase having aspartic acid as a direct amino group donor is disclosed (Japanese Patent Publication No. 2003-528572).
- this technology uses only OMPB and aspartic acid as substrates, and has the advantage of not using glutamic acid at all.
- the maximum conversion rate is about 75%, which is not industrially sufficient. It can be said that it is a result.
- the present inventors have isolated a novel gene encoding aspartate aminotransferase (hereinafter sometimes referred to as "AAT”) from Streptomvces hvgroscopicus, and using the expression vector containing the gene, Streptomvces hvgroscopicus As a result of comparing the AAT activity of the obtained transformant with the AAT activity of the parent strain, it was found to be about 47 times (Example 3).
- AAT aspartate aminotransferase
- An object of the present invention is to provide an AAT gene and an AAT protein.
- Another object of the present invention is to provide a promoter capable of highly expressing a target gene.
- the present invention further aims to provide a recombinant vector for introducing the AAT gene into the host and a host into which the AAT gene has been introduced.
- Another object of the present invention is to provide a method for producing L-phosphinothricin with a high conversion rate.
- polynucleotide selected from the following (i), (ii), (iii) and (iv):
- a polynucleotide comprising a nucleotide sequence having at least 90% identity with the polynucleotide having the nucleotide sequence ability represented by SEQ ID NO: 1, and encoding a protein having AAT activity.
- (V) a protein consisting of the amino acid sequence represented by SEQ ID NO: 2; (vi) a protein having an amino acid sequence ability represented by SEQ ID NO: 2, comprising an amino acid sequence in which one or more amino acids are deleted, substituted, inserted or added, and having AAT activity;
- a protein comprising an amino acid sequence having at least 90% identity with the amino acid sequence represented by SEQ ID NO: 2 and having AAT activity.
- the present invention also provides a polynucleotide encoding the protein according to the present invention.
- This polynucleotide and the above-mentioned polynucleotides (i), (ii), (iii) and (iv) whose forces are also selected may be collectively referred to as “AAT gene according to the present invention”.
- promoter in which the following), (x), (xi) and (xii) forces are also selected:
- xii a polynucleotide having a promoter activity and comprising a nucleotide sequence having at least 95% identity to the polynucleotide having the nucleotide sequence ability represented by SEQ ID NO: 3.
- the present invention there is provided a recombinant vector (hereinafter sometimes referred to as "the recombinant vector of the first aspect according to the present invention") comprising the AAT gene according to the present invention.
- the host of the first aspect according to the present invention there is provided a host transformed with the recombinant vector of the first aspect according to the present invention (hereinafter sometimes referred to as "the host of the first aspect according to the present invention").
- a recombinant comprising the AAT gene according to the present invention and a polynucleotide encoding a protein having PAT activity (hereinafter sometimes referred to as "PAT gene”).
- a vector hereinafter sometimes referred to as “the recombinant vector of the second aspect according to the present invention”.
- host of the second aspect of the present invention there is provided a host transformed with the recombinant vector of the second aspect of the present invention (hereinafter sometimes referred to as "host of the second aspect of the present invention").
- FIG. 1 is a diagram showing a restriction enzyme map of about 5.8 Kbp containing AAT gene.
- FIG. 2 shows the construction of expression vector pLG04.
- FIG. 3 shows the construction of expression vector pSGl1.
- FIG. 4 shows the construction of plasmid pATSGOl.
- FIG. 5 shows the construction of an expression vector pAHSG7201.
- the Streptomvces hveroscopicus SF1293 NP-50 strain used in the present invention was patented by the National Institute of Advanced Industrial Science and Technology (NTT 305) on May 20, 1987.
- the StreDtomvces hvgroscopicus SF1293 strain used in the present invention is a patent biological deposit center, National Institute of Advanced Industrial Science and Technology (MOL 305-8566, Tsukuba Sakaihigashi 1-chome, Ibaraki Prefecture, Japan, dated May 19, 1982. 1 Deposited in the middle 6).
- the accession number is FER
- Actinomycetes (Streptomvces lividans) transformed with the plasmid OMSB515 became a patent biological deposit center at the National Institute of Advanced Industrial Science and Technology ( ⁇ 305 — 8566, Tsukuba, Ibaraki, Japan 1 Deposited at No. 1 Chome 1 Center No. 6).
- the accession number is FERM BP-2496.
- Streptomvces lividans transformed with the plasmid pAHSG7201 is a patent biological deposit center (National Institute of Advanced Industrial Science and Technology) on February 1, 2006. Deposited at Tsukuba Sakai Higashi 1-chome, 1-chome, 1-Chuo 6th, Ibaraki, Japan. The accession number is FERM BP-10496.
- the origin of the AAT gene according to the present invention is not particularly limited as long as the protein encoded thereby has AAT activity, but is preferably derived from actinomycetes, more preferably from Streptomvces hveroscopicus.
- nucleotides are deleted, substituted, inserted or added in a polynucleotide
- amino acids are deleted or substituted in an amino acid sequence
- ⁇ inserted or added amino acid sequence '' means that the amino acid sequence has been altered by a well-known method such as site-directed mutagenesis, or by substitution of a plurality of nucleotides or amino acids that can occur naturally. To do.
- the number of nucleotide and amino acid modifications is one or more (eg, one to several, or 1, 2, 3, or 4).
- modified base sequences include one or more (eg, one! / Several or 1, 2, 3, or 4) affecting the AAT activity.
- examples include the nucleotide sequence set forth in SEQ ID NO: 1 having a mutation that is not given.
- modified amino acid sequences include one or more (eg, one, several, or one, two, three, or four) mutations that do not affect AAT activity.
- Sequence number No . The amino acid sequence described in 2 can be mentioned.
- AAT activity refers to a reaction in which AAT transfers the amino group of L-aspartic acid to a-ketoglutaric acid to produce L-glutamic acid and oxaloacetic acid (or the reverse reaction). Defined as ability. More specifically, a substrate for measuring AAT activity (150 mM aspartic acid, 50 mM a-ketoglutaric acid, 0. ImM pyridoxalphosphoric acid, 100 mM Tris-HCl buffer; pH 8.5) and AAT are mixed at 37 ° C for 20 minutes.
- Specific activity (U / mg) can be measured using ⁇ -globulin as a standard for AAT activity (U) using the amount of protein (eg, protein assembly kit (Bio-Rad)). ).
- Examples of “mutation without affecting activity” include conservative substitutions.
- conservative substitution means that one or more amino acid residues are replaced with another chemically similar amino acid residue so as not to substantially alter the activity of the protein.
- a certain hydrophobic residue is substituted with another hydrophobic residue
- a certain polar residue is substituted with another polar residue having the same charge, and the like.
- Functionally similar amino acids that can make such substitutions are known in the art for each amino acid. Specific examples include non-polar (hydrophobic) amino acids such as allan, noline, isoleucine, leucine, proline, tryptophan, phenylalanin, and methionine.
- Examples of polar (neutral) amino acids include glycine, serine, threonine, thiocin, glutamine, asparagine, and cysteine.
- Examples of positively charged (basic) amino acids include arginine, histidine, and lysine.
- Examples of negatively charged (acidic) amino acids include aspartic acid and glutamic acid.
- stringent conditions means that the washing operation of the membrane after hybridization is performed in a low salt concentration solution at a high temperature.
- .5 XS SC concentration l X SSC: 15 mM trisodium citrate, 150 mM sodium chloride
- 60 ° C for 15 minutes 60 ° C for 15 minutes, preferably 0.5 X SSC concentration, 0.1% SDS solution at 60 ° C , 15 minutes cleaning condition.
- the hybridization can be performed according to a known method.
- it can be performed according to the method described in the attached instruction manual.
- identity with respect to a base sequence or an amino acid sequence is used to mean the degree of coincidence of bases or amino acid residues constituting each sequence between compared sequences.
- Any numerical value of “identity” shown in the present specification may be a numerical value calculated using a homology search program known to those skilled in the art. For example, FASTA, BLAST, etc. It can be easily calculated by using the parameter of (setting).
- the polynucleotide encoding the amino acid sequence is easily determined, and various polynucleotides encoding the amino acid sequence represented by SEQ ID NO: 2 are determined. Nucleotides can be selected.
- the polynucleotide encoding the amino acid sequence represented by SEQ ID NO: 2 is a DNA sequence encoding the same amino acid in addition to part or all of the DNA sequence represented by SEQ ID NO: 1. It also means a sequence having a degenerate codon as a DNA sequence. Furthermore, RNA sequences corresponding to these are also included.
- Preferred examples of the polynucleotide encoding the amino acid sequence represented by SEQ ID NO: 2 include a polynucleotide having the nucleotide sequence ability represented by SEQ ID NO: 1.
- Example 3 AAT activity was examined for a host transformed with an expression vector containing the BamHI to XhoI fragment in the full-length sequence of the AAT gene isolated from Streptomvces hvgroscopicus. It was twice. This was significantly higher than the activity in the host transformed with the expression vector containing the PstI to XhoI fragment. From this, it was confirmed that the sequence from the transcription start point to the upstream Bam HI site in the promoter region of the AAT gene acts as a strong promoter. Upstream area 5 'When upstream force is cut off, the expression of the structural gene controlled by the upstream region is usually decreased, but the promoter according to the present invention surprisingly has a strong promoter activity.
- the nucleotide sequence of the promoter according to the present invention includes the ability to include the nucleotide sequence set forth in SEQ ID NO: 3. This sequence includes not only this sequence but also a modified sequence having the same level of promoter activity.
- promoter activity is determined by, for example, preparing an AAT gene expression vector as described in Example 3, expressing it in a host, and measuring AAT activity. Can be evaluated. If AAT activity is observed, it is said to have promoter activity. Preferably, if the AAT activity is enhanced by 2 times or more, more preferably 20 times or more, most preferably 40 times or more of the parent strain, the promoter is It has an activity.
- the promoter according to the present invention can be operably linked to a target gene so that the target gene can be highly expressed.
- Examples of the target gene that can be highly expressed by the promoter according to the present invention include genes derived from actinomycetes. Moreover, actinomycetes are mentioned as a host which can express a target gene more highly.
- Streptomvces hvgroscopicus Streptomvces albus. Strept omvces coelicolor. Streptomvces griseus. Streptomvces lividans. onvillei, Saccharopolvspora hirsuta, Kitasatosporia phosalacinea. Micromonospora arbonacae, Streptosporangium pseudovulgare force S, and more preferred is Streptomvce s hvgroscopicus.
- the promoter according to the present invention can be operably linked to a gene derived from actinomycetes (preferably Streptomvces hvgroscopicus) (preferably an AAT gene).
- actinomycetes preferably Streptomvces hvgroscopicus
- AAT gene preferably an AAT gene
- the promoter according to the present invention is also operable to a gene derived from actinomycetes (preferably Streptomyces hvgroscopicus.) (Preferably an AAT gene).
- actinomycetes preferably Streptomyces hvgroscopicus.
- AAT gene preferably an AAT gene.
- Recombinant vectors ligated to each other can be introduced into actinomycetes (preferably Streptomvces hveroscopicus).
- the origin of the PAT gene used in the recombinant vector according to the present invention is not particularly limited as long as the protein encoded thereby has PAT activity, but is preferably derived from actinomycetes, more preferably Streptomvces hygroscopicus. It comes from.
- PAT activity refers to the reaction in which PAT transfers the amino group of L-glutamic acid to OMPB to produce L-phosphinothricin and a-ketoglutaric acid (or the reverse reaction). Defined as the ability to mediate. More specifically, a PAT activity measurement substrate (150 mM glutamic acid, 50 mM OMPB, 0. ImM pyridoxal phosphate, lOOmM Tris—HC1 buffer; pH 8.5) was mixed with PAT and incubated at 37 ° C. for 20 minutes.
- Specific activity (U / mg) can be measured by using PAT activity (U) as a standard using the amount of protein (for example, protein assembly kit (manufactured by Bio-Rad)) as a standard. ).
- a protein selected from the following (v,), (vi,), (vii,) and (viii,) can be used:
- SEQ ID NO: 5 is a protein having an amino acid sequence in which one or more amino acids are deleted, substituted, inserted or added, and having PAT activity;
- ( ⁇ ′) a protein encoding the amino acid sequence represented by SEQ ID NO: 5 and a protein encoded by a polynucleotide that hybridizes under stringent conditions and has PAT activity;
- ( ⁇ ′) A protein having an amino acid sequence having at least 90% identity with the amino acid sequence represented by SEQ ID NO: 5 and having PAT activity.
- the recombinant vector according to the present invention is prepared according to the conventional method of gene recombination technology described in, for example, Sambrook, J. et al., Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, New York (1989). can do.
- the thread replacement vector according to the present invention has a control sequence necessary for expression of each gene, such as a promoter, a transcription initiation signal, a ribosome binding site, a translation termination signal, a transcription termination signal, and the like.
- Transcriptional control signals, translational control signals, etc. may be operably linked to the target gene.
- the recombinant vector according to the present invention may also contain a selection marker for selecting a transformant, and a selection marker can be appropriately selected according to the host to be used.
- Selectable markers include drug resistance genes and genes that complement auxotrophy, and preferred examples include ampicillin resistance genes, kanamycin resistance genes, tetracycline resistance genes, etc. when the host is a bacterium.
- trybutophane biosynthesis gene TRP1
- UAA3 uracil biosynthesis gene
- LEU2 leucine biosynthesis gene
- actinomycetes hygromycin resistance gene, bialaphos resistance
- examples thereof include a gene, a bleomycin resistance gene, an aureobasidin resistance gene, a thiostrepton resistance gene, and the like.
- the recombinant vector according to the present invention can be appropriately selected from viruses, plasmids, cosmid vectors, etc., taking into account the type of host used.
- the host cell is Escherichia coli, it is a phage-type butteriophage, PBR322, pUC-type vector, Bacillus subtilis, PUB110-type, pPL603-type, pC194-type vector, and yeast, pYC-type, pYE-type
- actinomycetes include PU702, pU680, and pAK114 vectors.
- the recombinant vector of the first aspect according to the present invention may preferably further comprise a promoter according to the present invention.
- the promoter according to the present invention can be operably linked to the AAT gene according to the present invention.
- This recombinant vector is advantageous in that the AAT gene can be highly expressed in a host (particularly actinomycetes).
- the recombinant vector of the second aspect according to the present invention may preferably further comprise a promoter according to the present invention.
- the promoter according to the present invention can be operably linked to the AAT gene according to the present invention.
- This recombinant vector is advantageous in that it can highly express the AAT gene and the PAT gene in the host (especially actinomycetes).
- the recombinant vector according to the second aspect of the present invention may further include a promoter that induces expression of the PAT gene, and the promoter can be operably linked to the PAT gene.
- the promoter of the PAT gene can be appropriately selected by those skilled in the art.
- the AT-II gene promoter in the plasmid pMSB515 described in JP-A-2-195889 can be used. .
- the linking to the promoter according to the present invention is performed, for example, by inserting a translation region of a gene encoding the target protein (target gene) in the forward direction downstream of the promoter according to a conventional method. Can do.
- the target gene can be linked to a foreign gene encoding the translation region of another protein and expressed as a fusion protein.
- the AAT gene according to the present invention is used.
- the PAT gene may be linked in the reverse direction or in the forward direction, but can be preferably linked in the reverse direction from the viewpoint of enhancing AAT activity.
- a target gene other than the AAT gene according to the present invention can be linked.
- the host to be transformed may be appropriately selected from actinomycetes, coliforms, Bacillus subtilis, yeasts, filamentous fungi, and other microorganisms depending on the type of recombinant vector used. Streptomvces hygroscopicus, Streptomvces albus, Str eptomvces coelicolor, Streptomvces griseus.Streptomvces lividans. Nirsuta. Kitasatosporia phosalacinea. Micromonospora carbonacae.
- Streptosporangium pseudovukare preferably ⁇ Streptomvces hvgroscoDicus
- Examples include the SF 1293 NP-50 strain deposited or the SF1293 strain deposited at the Patent Organism Depositary, National Institute of Advanced Industrial Science and Technology under the deposit number of FERM BP-130.
- Methods for introducing a recombinant vector into a host are well known, and the most efficient method is selected depending on the type of host and vector.
- transformation actinomycetes transmission by conjugation with E. coli, infection with actinomycetes phage, introduction into host protoplasts, etc. can be performed.
- genetic indicators such as antibiotic resistance, bock formation, melanin biosynthesis, etc. possessed by the vector used can be used.
- a medium and culture conditions usually used in the art can be used according to a conventional method.
- glucose, other saccharides, starch, and the like can be used as usual components, for example, as a carbon source.
- meat extract and peptone, and various extracts such as corn, wheat, soybean, and microorganism can be used.
- inorganic salts such as sodium, potassium, calcium, magnesium, cobalt, chlorine, phosphoric acid, sulfuric acid and various vitamins can be added.
- a commercially available antifoaming agent can be added to suppress firing during the culture.
- Culture conditions can be performed by flask culture using a rotary shaker, aeration and agitation culture using a jar fermenter device or tank equipment, and the like.
- the pH of the medium, the culture temperature, and the number of culture days can be appropriately determined according to the transformant.
- Preference of the transformant of the first aspect according to the present invention U, for example, phenotype by the recombinant vector of the first aspect operably linked to the AAT gene according to the present invention and the promoter according to the present invention.
- Examples include converted actinomycetes (particularly Streptomvces hvgroscopicus). This transformant is advantageous in that it exhibits an activity that exceeds the AAT activity of the parent strain actinomycetes.
- a more preferred example of the transformant of the first aspect according to the present invention is prepared from a transformant deposited at the National Institute of Advanced Industrial Science and Technology under the accession number FERM BP-10495. StreDtomvces hvgroscopicu S transformed with the expression vector OSG11, and most preferably, the National Institute of Advanced Industrial Science and Technology under the accession number of FERM BP-1368 transformed with the expression vector pSGll And the SF12 93 NP-50 strain deposited in Japan.
- This transformant is extremely advantageous in that it exhibits a very high AAT activity compared to the parent strain.
- the transformant of the second aspect according to the present invention is preferred.
- the transformant of the second aspect according to the present invention is operably linked to the AAT gene according to the present invention and the promoter according to the present invention.
- Examples include converted actinomycetes (particularly Streptomvces hvgroscopicus). This transformant is advantageous in that it exhibits an activity exceeding both the AAT activity and the PAT activity of the actinomycete of the parent strain.
- an AAT gene according to the present invention and a promoter according to the present invention are operably linked, and the AAT gene and the PAT gene according to the present invention are linked.
- This transformant is advantageous in that it exhibits activity exceeding the AAT activity and PAT activity of the actinomycete of the parent strain.
- a transformant deposited at the National Institute of Advanced Industrial Science and Technology under the accession number of FERM BP-1 0496 is prepared.
- StreDtomvces hvgro scoDicus transformed with the expression vector OAHSG7201 is bald, most preferably deposited with the National Institute of Advanced Industrial Science and Technology under the accession number of FERM BP-1368 transformed with the expression vector PAHSG7201. SF1293 NP-50 strain.
- This transformant is extremely advantageous in that it exhibits very high AAT activity and PAT activity compared to the parent strain.
- Actinomycetes used for the production method according to the present invention exhibit AAT activity and PAT activity, and are involved in the production of L phosphinothricin. Specifically, this actinomycete is involved in the progress of the following enzyme reaction in the presence of OMPB, L-aspartic acid, and L-glutamic acid.
- the “conversion rate” means the ratio of OMPB as a substrate converted to L-phosphinothricin and can be calculated by the following formula.
- the actinomycetes among actinomycetes that exhibit AAT activity and PAT activity and are capable of producing L-phosphinothricin, the actinomycetes have AAT activity that exceeds the AAT activity of the parent strain.
- a bacterium for the production of L-phosphinothricin, it is possible to produce L-phosphinothricin with high efficiency that has not been achieved so far.
- L-phosphinothricin can be produced with high efficiency even when glutamic acid, which is a raw material for producing L phosphinothricin, is reduced. The viewpoint power is extremely advantageous.
- Example 4 when L-phosphinothricin was produced using a transformant having AAT activity exceeding that of the parent strain, the conversion rate was 97%. Considering that the conversion rate of the parent strain is 93.2% and the conversion rate of the strain that highly expressed only the PAT gene is 92.6%, L-phosphinothria The conversion rate of thin is considered to improve.
- Example 4 when a transformant having an AAT activity exceeding the AAT activity of the parent strain was used to produce L-phosphinothricin from a substrate with reduced glutamic acid, conversion rate was 90.1%. Considering that the conversion rate of the parent strain is 76.2%, and that the conversion rate of the strain expressing only the PAT gene is 82.7%, AAT activity can be obtained even when glutamate-reducing substrate is used. It is considered that the conversion rate of L phosphinothricin is improved by the enhancement of.
- the production method of the first embodiment according to the present invention is an actinomycete that exhibits AAT activity and PAT activity, and is capable of producing phosphinothricin, wherein the activity exceeds the AAT activity of the parent strain. It is characterized by using the actinomycetes shown.
- the AAT activity of the actinomycete used exceeds the activity of the parent strain, preferably 2 times or more, more preferably 10 times or more, particularly preferably. 20 times or more, most preferably 40 times or more.
- Actinomycetes that can be used in the production method of the first aspect according to the present invention include actinomycetes that exhibit an activity that exceeds the AAT activity of the parent strain.
- a single expression vector containing an AAT gene Actinomycetes obtained by conversion and actinomycetes with enhanced AAT activity bred by mutation treatment can be used.
- actinomycetes having AAT activity enhanced by 20 times or more than the parent strain can be used.
- the host according to the first aspect of the present invention (actinomycetes), more preferably FERM BP- StreDtomvces hvgroscopicus s transformed with the expression vector OSG11, most preferably the expression vector pSGl l SF1293 NP-50 strain deposited with the National Institute of Advanced Industrial Science and Technology under the accession number of FERM BP-1368 transformed with
- L-phosphinothricin represented by the formula (I) or a salt thereof comprising the step of contacting 2-hydroxymethylphosphier 2-oxobutyric acid or a salt thereof with the host (actinomycetes) according to the first aspect of the present invention.
- a method for producing, wherein the recombinant vector introduced into the host further comprises a promoter according to the present invention, and the AAT gene according to the present invention and the promoter according to the present invention are operably linked. Is mentioned.
- the production method of the first embodiment according to the present invention is particularly preferred as the embodiment of 4-hydroxymethylphosphier 2-oxobutyric acid represented by the formula ( ⁇ ) or a salt thereof according to the first embodiment of the present invention.
- a method for producing L-phosphinothricin represented by the formula (I) or a salt thereof comprising a step of contacting with a host (actinomycetes), wherein the recombinant vector introduced into the host is in accordance with the present invention.
- a promoter further comprising an AAT gene according to the present invention operably linked to a promoter according to the present invention and whose host is derived from StreDtomv ces hveroscopicus
- L phosphinothricin can be produced with extremely high efficiency which has not been achieved so far.
- L phosphinothricin can be produced with high efficiency even if glutamic acid, which is a raw material for producing L phosphinothricin, is reduced. It is.
- Example 6 the form having activity exceeding the parent strain's AAT activity and PAT activity.
- the conversion rate was 95.5%.
- the conversion rate of the parent strain is 76.2%, and the conversion rate of the strain that expressed only the PAT gene is 82.7%, even when glutamic acid reduction substrate is used, It is considered that the conversion rate of L-phosphinothricin is improved by enhancing the PAT activity in addition to the AAT activity.
- the production method of the second aspect according to the present invention is an actinomycete that exhibits AAT activity and PAT activity and is capable of producing phosphinothricin, wherein the parent strain has AAT activity and PAT activity. It is characterized by using actinomycetes exhibiting an activity exceeding.
- the AAT activity of the actinomycetes used may be greater than that of the parent strain, but is preferably about 2 times or more, more preferably about 20 times or more, and still more preferably. Is about 40 times or more, particularly preferably about 100 times or more, and most preferably about 170 times or more.
- the actinomycete used may have a PAT activity that exceeds the activity of the parent strain, but is preferably about 2 times or more, more preferably about 10 times or more, Most preferably, it is about 16 times or more.
- Example 6 an extremely high production efficiency of L-phosphinothricin was achieved when the enhanced AAT activity was relatively stronger than the enhanced PAT activity.
- a-ketoglutaric acid which is thought to cause product inhibition, is quickly converted to glutamic acid by relatively enhanced AAT. Therefore, in the production method of the second aspect according to the present invention, it is preferred that the actinomycetes used have V and AAT activities that are relatively stronger than PAT activities.
- Actinomycetes that can be used in the production method according to the second aspect of the present invention include actinomycetes that exhibit an activity that exceeds the AAT activity and PAT activity of the parent strain.
- Actinomycetes obtained by transformation with an expression vector, and actinomycetes with enhanced AAT and PAT activities bred by mutation treatment can be used.
- actinomycetes having an AAT activity enhanced by 40 times or more than the parent strain and a PAT activity enhanced by 10 times or more can be used.
- the host according to the second aspect of the present invention ( Actinomycetes), more preferably i ⁇ mi ces hvgroscopicus transformed with the expression vector PAHSG7201 prepared from transformants deposited with the National Institute of Advanced Industrial Science and Technology under the accession number FERM BP-10496 Most preferably, the SF1293 NP-50 strain deposited with the National Institute of Advanced Industrial Science and Technology under the accession number of FERM BP-1368 transformed with the expression vector pAHSG7201 can be used.
- L-phosphinothricin represented by the formula (I) or a salt thereof comprising a step of contacting 2-hydroxymethylphosphier 2-oxobutyric acid or a salt thereof with the host (actinomycetes) according to the second aspect of the present invention.
- a method for producing, wherein the recombinant vector introduced into the host further comprises a promoter according to the present invention, and the AAT gene according to the present invention and the promoter according to the present invention are operably linked. Is mentioned.
- the production method of the second embodiment according to the present invention is particularly preferred as an embodiment, in which 4-hydroxymethylphosphier 2-oxobutyric acid represented by the formula ( ⁇ ) or a salt thereof is added according to the present invention.
- a method for producing L-phosphinothricin represented by the formula (I) or a salt thereof comprising the step of contacting with a host (actinomycetes) of the second embodiment, wherein the recombinant vector is introduced into the host. Further comprising a promoter according to the present invention, wherein the AAT gene according to the present invention and the promoter according to the present invention are operably linked and the host is derived from StreDtomces hygroscopicus.
- the most preferable aspect of the production method of the second aspect according to the present invention is that 4-hydroxymethylphosphier 2-oxobutyric acid represented by the formula ( ⁇ ) or a salt thereof is used according to the second aspect of the present invention.
- a method for producing L-phosphinothricin represented by the formula (I) or a salt thereof comprising a step of contacting with a host (actinomycetes), wherein the recombinant vector introduced into the host is a promoter according to the present invention.
- the AAT gene according to the present invention and the promoter according to the present invention are operably linked, the host is derived from StreDtomvces h vgroscoDicus, and the AAT gene and the PAT gene are linked in the opposite direction. There is a method.
- the production method according to the present invention can be carried out as described in Japanese Patent No. 2638541.
- a mixed solution of 3% OMPB or a salt thereof and 0.75% to 3% glutamic acid or a salt thereof and 3% aspartic acid or a salt thereof is neutralized with sodium hydroxide, pH 7.0-9.5, preferably about pH 8.5, temperature 30-45. C, preferably at 37 ° C for 0.5 to 5 days by allowing actinomycetes with enhanced AAT activity or actinomycetes with enhanced AAT and PAT activities to act . It is also possible to add a small amount of pyridoxalphosphoric acid or a salt thereof during the conversion reaction.
- the present invention it is possible to purify L-phosphinothricin from the obtained mixed solution containing L-phosphinothricin through a purification process.
- a purification method in this case, it is desirable to use a precipitation method using force ion exchange chromatography solvent (for example, methanol), which can be applied to all methods usually used in the field. Since the conversion liquid obtained by the present invention can be easily purified by these purification methods, even if a trace amount of alanine is present in the conversion liquid, there is no particular problem.
- the compound of formula (I), the compound of formula ( ⁇ ), glutamic acid, and aspartic acid are each It can exist as a salt.
- Examples of the salt of the compound of the formula (I) include salts with inorganic bases such as sodium salt, potassium salt, and ammonium salt, as well as salts with organic bases, inorganic acids, and organic acids. Is preferably a sodium salt.
- the salt of the compound of the formula (ii) includes a salt with an inorganic base such as a sodium salt, potassium salt, and ammonium salt, and a salt with an organic base, preferably a sodium salt. is there.
- Examples of the salt of glutamic acid include salts with inorganic bases such as sodium salt, potassium salt and ammonium salt, as well as salts with organic bases, inorganic acids and organic acids, preferably sodium salts. It is.
- Aspartic acid salts include salts with inorganic bases such as sodium salts, potassium salts and ammonium salts, as well as salts with organic bases, inorganic acids and organic acids.
- inorganic bases such as sodium salts, potassium salts and ammonium salts
- organic bases such as sodium salts, potassium salts and ammonium salts
- organic acids such as sodium salts, potassium salts and ammonium salts
- Sodium salt such as sodium salts, potassium salts and ammonium salts.
- Example 1 Cloning of AAT gene derived from StreDtomvces hvgroscopicus
- Genomic DNA was prepared by treating the lyophilized cells by the method described in Example B2 of WO00Z24879.
- AAT from two actinomycetes (StreDtomvces giniae and StreDtomvces coelicolor) obtained from a well-known database (DDBJ homepage httD: //www.ddbi.nig.ac.ip/Welcome-ih tml public database)
- Amino acid sequences were compared to search for regions with homology.
- two homologous amino acid sequences GEPDFP TP and KTYAMTGWRVG
- two types of synthetic oligonucleotide primers corresponding to this part were prepared.
- PCR was carried out using the above-mentioned Streptomvces hvgroscopicus genomic DNA as a cage.
- TaKaRa LA Taq with GC buffer (Takara Bio) was used, and after heat denaturation at 94 ° C for 1 minute, 98 ° C for 30 seconds, 60 ° C for 30 seconds, 72 ° C The cycle of 1 minute and 15 seconds was repeated 30 times, and then incubated at 72 ° C for another 10 minutes. After completion of the reaction, the sample was subjected to 0.8% agarose electrophoresis and analyzed. As a result, amplification of a specific band of about 0.6 kbp was confirmed.
- DNA was purified by the Wizard SV Gel and PCR Clean-up System (Promega) and TOPO TA Subcloning was performed using a cloning kit (Invitrogen) and E. coli competent cells JM109 (Takarabio).
- the obtained plasmid was designated as pSH-AAT.
- pSH-AAT As a result of analyzing the base sequence of the insert fragment of plasmid pSH-AAT, a homologous DNA sequence encoding AAT derived from Streptomvces virriniae was confirmed (the base sequence analyzer is an ABI PRISM310 manufactured by Applied Systems).
- Example l After digesting the genomic DNA obtained in a) with Sau3AI, 0.8% agarose It was subjected to gel electrophoresis, and a 9-23 kbp DNA fragment was recovered from the gel. The recovered fragment was treated with phenol and purified by ethanol precipitation, and then partially digested genomic DNA fragment and EMBL3 using EMBL3ZBamHI Vector Kit (Stratagene) and DNA Ligation Kit ver. 2 (Takarabio). Vectors were ligated. The obtained ligation mixture was knocked for Maxplax Packaging Extract (Epicenter) and then infected with E. coli XL1-Blue MRA strain. Using the 2.0 ⁇ 10 5 phage library obtained by this method, cloning of the target gene was performed.
- Example l The plasmid pSH-AAT obtained in c) was digested with EcoRI and subjected to 0.8% agarose gel electrophoresis, and a DNA fragment of about 0.6 kbp was recovered from the gel. The recovered DNA fragments are purified using Wizard SV Gel and PCR Clean-up System (Promega) and DIG High Prime DNA Labeling and Detection Starter Kit I (Roche Diagnostics). The probe was labeled with DIG.
- Example l (2) a the phage library obtained in Example l (2) a was copied to Hybond-N + membrane (Amersham Biosciences), denatured with 0.5N sodium hydroxide, 5 The DNA was fixed by washing with X SSC (l X SSc: 15 mM trisodium citrate, 150 mM sodium chloride) and drying. As described in the kit above, after 1 hour of prehybridization (42 ° C), a heat-denatured labeled probe was added and hybridization was performed for 16 hours (42 ° C). .
- phage particles were recovered 16 hours later, and Grossberger's method (Grossberger, D., Nucleic Acids. Res., 15: 6737, 1987) After treatment with rotinase K and phenol, phage DNA was prepared by ethanol precipitation.
- Example 1 (1) The genomic DNA obtained in a) and the phage DNA obtained in Example 1 (2) c) were each digested with a plurality of restriction enzymes and then subjected to 0.8% agarose gel electrophoresis. did. After copying DNA to Hybond—N + membrane by the method of Southen (Southern, EM, J. Mol. Biol, 98: 503-517, 1975), ECF Random -Prime Label ling and Detection System (manufactured by Amersham Biosciences) ) was used for hybridization. By the method described in this system, after 30 minutes of prehybridization (60 ° C), a heat-denatured labeled probe was added, and hybridization was performed for 16 hours at 60 ° C.
- the probe used was about 0.6 kbp DNA fragment derived from pSH-AAT obtained in Example l (2) b) and labeled with fluorescein by the method described in this system.
- label washing follow the method described in this system. First, 1 X SSC containing 0.1% SDS, 60. After washing with C for 15 minutes, it was washed with 0.5 X SSC containing 0.1% SDS at 60 ° C for 15 minutes. Furthermore, according to the method described in this system, after reacting with an alkaline phosphatase-labeled anti-fluorescein antibody, a substrate (ECF sub strate) attached to this system is added, and the resulting fluorescence band is converted into a molecular imager FX (Biotechnology).
- the phage DNA obtained in Example 1 (2) c) was digested with the restriction enzyme BamHI, then subjected to 0.8% agarose gel electrophoresis, and a DNA fragment of about 4.3 kbp was recovered from the gel and purified. .
- the obtained DNA fragment was ligated to pUC118 BamHI-BAP-treated DNA (manufactured by Takara Bio Inc.), and the resulting plasmid was designated as pi 18G-411B.
- phage DNA was treated with the restriction enzyme Pstl, and a DNA fragment of about 2.7 kbp was recovered from the gel and then ligated to pUC118 Pstl-BAP-treated DNA.
- the resulting plasmid was designated as p118G-411P.
- the obtained plasmid pi18G-41 IP was digested with the restriction enzyme Pstl, and then subjected to 0.8% agarose gel electrophoresis, and a DNA fragment of about 2.7 kbp was recovered from the gel and purified.
- plasmid pi18G-411B was digested with restriction enzyme Pstl, and then subjected to 0.8% agarose gel electrophoresis, and a DNA fragment of about 6.2 kbp was recovered from the gel and purified.
- the obtained DNA fragment was dephosphorylated by alkaline phosphatase (BAP; manufactured by Takarabio Co., Ltd.) treatment, and then ligated with the about 2.7 kbp DNA fragment derived from pi18G-411P obtained earlier.
- the obtained plasmid was designated as PAAT3-5.
- the obtained plasmid pAAT3-5 was digested with restriction enzymes HindIII and Xhol, and then subjected to 0.8% agarose gel electrophoresis, and a DNA fragment of about 2.9 kbp was recovered from the gel and purified.
- a DNA fragment of about 2.9 kbp was recovered from the gel and purified.
- HindIII and Sail restriction enzymes HindIII and Sail
- Figure 1 shows a restriction enzyme map of approximately 5.8 Kbp containing the AAT gene from Streptomvces hvgroscodcus.
- Example 2 Analysis of salt-and-mouth arrangement of AAT gene derived from StreDtomvces hvgroscopicus
- the sequencing reaction was performed using BigDye Terminator V3.1 Cycle Sequencing Kit (manufactured by Applied Biosystems).
- plasmid pHX-01 was used as the vertical DNA, and a synthetic oligonucleotide having the following sequence was used as the primer.
- the sequencing reaction was performed by repeating 30 cycles of 0 second, 50 ° C '5 seconds, 60 ° C' 4 minutes.
- Example 2 (1) Using the sequence determined in b), a public database (DDBJ home page http: //www.ddbi.nig.ac.ip/Welcome-i.html) ) A homology search with the above sequence was performed.
- the search program uses blastx (DNA sequence X amino acid sequence DB) and squeezed under the condition of filter OFF, Streptomvces ce licolor AAT gene 87%, Streptomvces virginiae AAT gene 88% The same cases were shown.
- Example 3 High expression of AAT gene derived from StreDtomvces hvgroscopicus
- the expression vector PLG04 was constructed as shown in FIG.
- the expression vector pSGll was constructed as shown in FIG.
- the plasmid 3 ⁇ 1 ⁇ 03 obtained in Example 3 (1) &) was cleaved with the restriction enzyme 13 ⁇ 411 (1111, EcoRI) and subjected to 0.8% agarose gel electrophoresis, followed by about 4.9 kbp of DNA.
- the fragment was recovered from the gel and the DNA was purified, and then the plasmid pHXOl obtained in Example 1 (2) e) was used.
- a DNA fragment of about 2.9 kbp was recovered from the gel and purified.
- Streptomvces lividans was performed according to Thompson's method (Thompson, CJ, J. BactrioL, 151: 668-677, 1982). After mixing Strepto mvces lividans protoplast, ligation mixture, T medium, add 20% polyethylene glycol 1000, introduce DNA, and incubate at 30 ° C on R2YE agar plate (20 ml medium Z plate) did.
- a pre-treatment step of adding lysozyme to the buffer PI attached to the kit to a final concentration of lOmgZml and preincubating the bacterial cells with this solution for 30 minutes at room temperature. was carried out.
- the DNA was recovered from 20 mU of the culture and finally dissolved in 50 / zl TE buffer.
- a plurality of transformants were treated, and the obtained plasmid DNA was cleaved with restriction enzymes HindIII and EcoRI, and then subjected to 0.8% agarose electrophoresis to confirm the cleavage pattern. Plasmid DNA exhibiting a band of about 2.9 kbp and about 4.9 kbp was selected as the target DNA, and this was used as the expression vector pLGO4 (Fig. 2).
- Example 1 The plasmid pHXOl obtained in e) was cleaved with the restriction enzyme BamHI, and after electrophoresis, an approximately 1.5 kbp DNA fragment was recovered from the gel and purified. This DNA fragment was ligated to pUCl 18 BamHI-BAP-treated DNA (manufactured by Takara Bio Inc.), and the resulting plasmid was designated as pi 18-G1 (FIG. 3).
- This plasmid pl l8-G1 was cleaved with restriction enzymes HindIII and EcoRI, and after electrophoresis, about 1.
- Example 3 (1) b A 5 kbp DNA fragment was recovered from the gel and purified.
- the plug obtained in Example 3 (1) a) was used.
- rasmid PSYTL03 was cleaved with restriction enzymes HindIII and EcoRI, and after electrophoresis, a DNA fragment of about 4.9 kbp was recovered from the gel and purified.
- These two kinds of DNA fragments were ligated using the DNA Ligation Kit ver. 2, and the resulting ligation mixture was transformed into the actinomycetes StreDtomvces Ife according to the method of Example 3 (1) b).
- the obtained transformant was cultured with shaking in 80 ml YEME medium containing 10 ⁇ gZml of thiostrepton at 30 ° C for 2 days.
- DNA was prepared.
- the obtained plasmid DNA was cleaved with restriction enzymes HindIII and EcoRI and subjected to 0.8% agarose electrophoresis to confirm the cleavage pattern.
- a plasmid DNA having bands of about 1.5 kbp and about 4.9 kbp was selected as a target DNA and used as an expression vector pSG11 (FIG. 3).
- StreDtomvces hvgroscopicus (SF1293 NP—50 strain, FERM BP—1368) strain stock (freeze-dried cells) 10 ml of SI medium (2% starch, 1% polypeptone, 0.3% monolet extratat, 0.05% K ⁇ ; ⁇ ⁇ 7.0) and shake at 28 ° C for 40 hours
- the obtained culture fluid force was also collected by centrifugation, washed with 0.5M sucrose, and then washed with 0.5M sucrose, P3 medium containing final concentration of 2.5mgZml lysozyme and final concentration of 1.25mgZml acropeptidase ( 70 mM NaCl, 0.5 M sucrose, 5 mM MgCl
- the TH medium used for transformation was prepared by the following method. First, a trace element solution was prepared by the Okanishi method (Okanishi, M., J. Gen. Microbiol, 80, 389-400, 1974). Next, sucrose 26.7g, K SO 0.0375g about 60m After dissolving in 1 and adding 0.3 ml of Trace element solution, it was diluted to 77.5 ml with water to make 3 / 2TH medium. In 7.75 ml of this 3 / 2TH medium, add 2M CaCl 0
- the RME medium for protoplast regeneration was prepared by the following method. First, proline 2g, gnore course 10g, yeast extratate 2g, casamino acid 2g, sucrose 174g, KC1 15g, K SO 0.25g, DEXTRAN SULFATE 0. 05g, LAB DEMC
- Olg was dissolved in water, adjusted to pH 7.2 with 10% NaOH, made up to 500 ml with water, and this was used as 2 XRME medium.
- 2 XRME medium 500ml, 0.5% KH PO 10ml, 1M
- the protoplast solution 1001 obtained in Example 3 (2) a) was added and mixed gently.
- RME soft-flager medium approximatelyx. 2mlZ plate.
- Example 3 Culture fluid obtained in a) The bacterial cells were collected by centrifugation, washed with 0.9% saline, and then frozen at 80 ° C. Frozen bacterial cells in 20 ml buffer A2 (20 mM phosphate buffer, 0. ImM pyridoxal phosphate, 5 mM 2 mercaptoethanol,
- the cells were dissolved and suspended in ImM phenol methylsulfur fluoride (pH 7.0), and the cells were disrupted by ultrasonic waves. The crushing fluid force was also removed by centrifuging and the supernatant was used as a crude enzyme solution. Using the obtained crude enzyme solution, Tanaka et al. (Tanaka, T., Agric. Biol. Chem.
- the pSGl1 transformant had a markedly higher activity of about 5.4 times that of the pLG04 transformant (Table 1).
- Example 3 (2) b)-Inoculate the strain stock (lyophilized cells) of the transformant with the expression vector pSGl 1 obtained in 2 in 10 ml of SPY medium containing 10 ⁇ g Zml of tiostrepton. And cultured with shaking at 28 ° C for 2 days. 2 ml of the obtained culture broth was added to 30 ml of P-101 medium containing 10 g / ml thiostrepton (7% glucose, 4.4% soyton, 0.327% KH PO, 0.085% Na HPO, 1 15% TES, 0.001% CoCl6 ⁇ 0; pH 6.0)
- Example 3 Culture fluid obtained in a) The bacterial cells were collected by centrifugation, washed with 0.9% saline, and then frozen at ⁇ 80 ° C. The frozen cells were dissolved and suspended in 20 ml of buffer A2, and the cells were disrupted by ultrasonic waves. Crushing fluid force Cell fragments were removed by centrifugation, and the supernatant was used as a crude enzyme solution. Using the obtained crude enzyme solution, AAT and PAT activities were measured.
- the PAT activity was measured according to the Schulz method (Schulz, A., Appl. Environ. Microbiol, 56, 1-6, 1990). Substrate for PAT activity measurement (150 mM glutamic acid, 50 mM
- OMPB 0. ImM pyridoxal phosphate, lOOmM Tris—HC1 buffer; pH 8.5
- crude enzyme solution was mixed and incubated at 37 ° C for 20 minutes, then the reaction was stopped by heating in a boiling water bath for 5 minutes. .
- the sample was filtered through a 0.45 m filter and subjected to HPLC for amino acid analysis (model LC—VP, manufactured by Shimadzu Corp.), and the concentration of L-phosphinothricin produced was measured.
- Enzyme power was defined as 1 U (unit) as the ability to produce l / z mol of L-phosphinothricin per minute.
- the protein concentration in the crude enzyme solution was measured using a protein assembly kit (Bio-Rad) with ⁇ -globulin as a standard, and the specific activity (U / mg) of the crude enzyme solution was determined.
- AAT activity was measured by the following method.
- Substrate for measuring AAT activity 150 mM aspartic acid, 50 mM a-ketoglutaric acid, 0. ImM pyridoxal phosphate, lOOmM
- Tris-HCl buffer (pH 8.5) and the crude enzyme solution were mixed and incubated at 37 ° C for 20 minutes, and then the reaction was stopped by heating in a boiling water bath for 5 minutes. After stopping the reaction, the sample was filtered through a 0.45 m filter and subjected to HPLC for amino acid analysis (model LC—VP, manufactured by Shimadzu Corporation) to measure the concentration of the produced glutamic acid. Enzyme power was defined as 1 U (unit) as the ability to produce 1 ⁇ mol of glutamic acid per minute. The protein concentration in the crude enzyme solution was measured with the above protein assay kit, and the specific activity (U / mg) of the crude enzyme solution was determined.
- the strain stock of the transformant obtained by pSGl 1 obtained in Example 3 was inoculated into 30 ml of SPY medium containing 10 ⁇ g Zml of thiostrepton and shaken at 28 ° C for 2 days. did. 2 ml of the obtained culture solution was transplanted to 40 ml of SPY medium containing 10 ⁇ g Zml of thiostrepton and cultured with shaking at 28 ° C. for 2 days. 20 ml of the obtained culture broth was transplanted into 400 ml of SBK medium (2% starch, 3% defatted soybean meal, 0.05% KH PO; pH 7.0) containing 10 ⁇ g Zml of thiostrepton, and 28 ° C. Culturing with shaking for 2 days. Obtained
- the parent strain (without plasmid) and the transformant by plasmid PMSB515 were also cultured in the same process (however, the parent strain was cultured in a medium without thiostrepton), and the conversion reaction was performed in the same manner.
- the reaction was started by mixing 440 ml of the culture solution obtained in Example 4 (1) and 1760 ml of the substrate solution A or B obtained in a) or b) of Example 4 (2), respectively.
- the conversion conditions were a temperature of 37 ° C and a pH of 8.5 (adjusted with sodium hydroxide), and the reaction was carried out with stirring for 3 days (about 70 hours).
- the sample was filtered through a 0.45 m filter and subjected to HPLC for amino acid analysis (model LC—VP, manufactured by Shimadzu Corporation), and the concentration of the produced L-phosphinothricin was measured.
- the amount of the conversion solution at the end of the conversion was also accurately measured, and the amount of L-phosphinothricin produced from these products was determined. Furthermore, the conversion ratio to L-phosphinothricin in each culture solution was calculated by determining the molar ratio of the OMPB used and the produced L-phosphinothricin.
- Plasmid pATSGOl was constructed as shown in FIG.
- the expression vector pAHSG 7201 was constructed as shown in FIG.
- the gene of AT-II (see JP-A-2-195889), which is a kind of PAT, was used.
- the gene of Haccho-11 which is a kind of PAT
- Approx. 4.7 kbp DNA fragment is recovered from the gel Purified.
- pUC18 manufactured by Tacarano
- the pUC18 Sacl fragment was treated with alkaline phosphatase (BAP; manufactured by Takarabio Co., Ltd.), and then ligated with an approximately 4.7 kbp Sacl fragment derived from pMSB515.
- BAP alkaline phosphatase
- the obtained plasmid was designated as pAT ⁇ 515-03 (FIG. 4).
- the plasmid pHX-01 obtained in Example 1 (2) e) was cleaved with the restriction enzyme BamHI and subjected to 0.8% agarose gel electrophoresis, and then a DNA fragment of about 1.5 kbp was recovered from the gel. DNA was purified.
- the plasmid ⁇ 515-03 obtained in Example 5 (1) a) was digested with the restriction enzyme BamHI and subjected to 0.8% agarose gel electrophoresis, and then a DNA fragment of about 7.4 kbp was extracted from the gel. Collected and purified DNA.
- the BamHI fragment of ⁇ 515-03 was treated with alkaline phosphatase (BAP), and then ligated with an about 1.5 kbp BamHI fragment derived from pHX-01.
- BAP alkaline phosphatase
- the resulting plasmid was designated as pATSGOl (Fig. 4).
- the plasmid 3 ⁇ 1 ⁇ 03 obtained in Example 3 (1) &) was cleaved with the restriction enzyme 13 ⁇ 411 (1111, EcoRI) and subjected to 0.8% agarose gel electrophoresis, followed by about 4.9 kbp of DNA. The fragment was recovered from the gel, and the DNA was purified.
- the plasmid pATSG01 obtained in Example 5 (1) b) was similarly cleaved with the restriction enzymes HindIII and EcoRI. A 6 kbp DNA fragment was recovered from the gel and purified. These two types of DNA fragments were ligated using DNA Ligation Kit ver.
- Example 3 (l) b) The obtained transformant was cultured with shaking in 80 ml YEME medium containing 10 ⁇ g, ml of thiostrepton at 30 ° C. for 2 days, and then the plasmid DNA was also prepared with the culture fluid power.
- the obtained plasmid DNA was digested with restriction enzymes Hindlll and EcoRI, and subjected to 0.8% agarose electrophoresis to confirm the cleavage pattern. Plasmid DNA showing bands of about 4.9 kbp and about 5.6 kbp was selected as the target DNA and used as the expression vector PAHSG7201 (Fig. 5).
- Example 5 PAT activity and AAT activity were measured using a crude enzyme solution derived from a transformant obtained from the expression vector pAHSG7201 obtained in a). The enzyme activity was measured in accordance with the method of Example 3 (4) b) 1-2 (as a control, the results of Example 3 (4) b) 1-2 were also shown.
- Example 6 L-phosphinothricin produced in cows with a co-expressing strain of AAT gene and PAT gene
- the transformant with the expression vector pAHSG 7201 obtained in Example 5 (2) was cultured in the same steps as in Example 4 (1). Incubation was carried out with P-2 medium at 28 ° C for 4 days under aeration and agitation, and L-phosphinothricin was produced using the obtained culture solution.
- Example 44 Oml of the culture solution of the transformant obtained from the expression vector pAHSG7201 obtained in Example 6 (1) and 1760 ml of the substrate solution A or B prepared according to the method of Example 4 (2) were mixed at 37 ° C, pH 8. Reacted at 5 for 3 days (about 70 hours). The obtained conversion solution was analyzed in the same manner as in Example 4 (2), and the amount of L-phosphinothricin produced was determined (as a control, Example 4 (2) The results of c) are also shown).
- the transformant ie, AAT'PAT co-expressing strain
- the expression vector pAHSG7201 showed a significantly higher conversion rate than other strains (Table 5 ).
- Example 7 Identification of N-terminal amino acid ⁇ R sequence of highly expressed protein in a co-expression strain of AAT gene and PAT gene
- the N-terminal amino acid sequence of the protein highly expressed in the transformant by the expression vector PAHSG7201 was performed according to the method described in Example A2 of Japanese Patent No. 3593134. First, the crude enzyme solution of a transformant obtained from the expression vector PAHSG7201 obtained in Example 5 (2) c) was used as an SDS-PAGE mini
Abstract
本発明は、新規アスパラギン酸アミノトランスフェラーゼ(AAT)遺伝子および高効率のL-ホスフィノスリシンの製造法を提供することを目的とする。本発明によれば、配列番号:1のDNA配列またはその改変配列からなるポリヌクレオチドおよびAAT活性が増強された放線菌を利用したL-ホスフィノスリシンの製造法が提供される。
Description
明 細 書
ァスパラギン酸アミノトランスフェラーゼ遺伝子および L—ホスフィノスリシ ンの製造方法
発明の分野
[0001] 本発明は、ァスパラギン酸アミノトランスフェラーゼおよびその遺伝子、並びに Lーホ スフイノスリシンの製造方法に関する。本発明はまた、アミノトランスフェラーゼ遺伝子 を発現させるための組換えベクターゃ該ベクターが導入された宿主に関する。
背景技術
[0002] L—ホスフィノスリシンは、ビアラホスの構成成分であり、除草活性を有する物質であ る(Tachibana,K., J. pesticide Sci., 11:27-31, 1986) 0本発明者らはこれまでに、 4一 ( ヒドロキシメチルホスフィエル) 2—ォキソ 酪酸(以下「OMPB」 t 、うことがある)を アミノ基供与体の存在下で、微生物の有するアミノトランスフェラーゼの作用を利用す ることにより L—ホスフィノスリシンに変換する技術を構築し (特許第 2638541号公報 )、また、 L—ホスフィノスリシンアミノトランスフェラーゼ遺伝子の利用により、使用する 微生物の菌体量 (培養液量)を削減する技術にっ 、ても確立して 、る(特開平 2 - 19 5889号公報)。
[0003] このような微生物変換 (酵素変換)による L ホスフィノスリシンの製造法においては 、 OMPBと等量 (または等モル量)のグルタミン酸とァスパラギン酸を使用することが 一般的である。これは、グルタミン酸のみを使用した場合、反応は平衡に達し、理論 変換率 50%で終了してしまうが、グルタミン酸とァスパラギン酸を併用した場合、ダル タミン酸の脱ァミノによって生じた aーケトグルタル酸にァスパラギン酸のアミノ基が転 移し、反応が連続的に進行し、高い変換率が得られるからである。この場合、ァスパ ラギン酸の脱ァミノによって生じたォキザ口酢酸は通常の反応温度 (例えば 37°C)で 容易に脱炭酸され、ピルビン酸となり反応系外に出るため、収率に影響を与えない。
[0004] 一方、 L—ホスフィノスリシンを除草剤として製造、販売する場合において、その製 造費用は著しく安価なことが期待される。具体的には、原料 OMPB力 L ホスフィ ノスリシンへの変換率 (少なくとも 90%以上)を高くすることに加え、アミノ基供与体と
なるグルタミン酸ゃァスパラギン酸使用量を削減することも重大な課題となる。この課 題を解決するための一手段として、特許 3018261号公報の実施例 4において、大腸 菌 K— 12由来の L -ホスフィノスリシンアミノトランスフェラーゼとブタ由来の GOT (ァ スパラギン酸アミノトランスフェラーゼ)を混合して用いることにより、グルタミン酸を低 減した基質(OMPB (HMPB):グルタミン酸:ァスパラギン酸 = 1 : 0. 2 : 1)において も、 L—ホスフィノスリシンが合成されることが開示されている。しかしこの技術は、 L— ホスフィノスリシンへの変換率が最高でも 86%であり、工業的な目標収率に満たない 上、安価な大量調達が難しいブタ由来の酵素を使用しているため、実質的に実用化 が難しい技術であるといえる。別の一手段として、ァスパラギン酸を直接のアミノ基供 与体とするアミノトランスフヱラーゼが開示されている(特表 2003— 528572号公報) 。し力しこの技術は、基質として OMPBとァスパラギン酸のみを使用するため、グルタ ミン酸を全く使用しないというメリットを有するものの、最高の変換率は約 75%であり、 工業的には不十分な結果であるといえる。
[0005] したがって現在、微生物(または酵素)による L—ホスフィノスリシンの製造法におい ては、原料 OMPBから L—ホスフィノスリシンへの変換率をより高くすることに加えて、 標準的な基質(すなわち、 OMPB:グルタミン酸:ァスパラギン酸 = 1 : 1 : 1)からダル タミン酸を削減することが重要な課題であり、その解決が切望されている。
発明の概要
[0006] 本発明者らは、 Streptomvces hvgroscopicusより、ァスパラギン酸アミノトランスフェラ ーゼ (以下「AAT」と呼ぶことがある)をコードする新規の遺伝子を単離し、該遺伝子 を含む発現ベクターで Streptomvces hvgroscopicusを形質転換し、得られた形質転 換体の AAT活性を親株の AAT活性と比較した結果、約 47倍であることを見出した( 実施例 3)。該 AAT活性増強株を用いて L—ホスフィノスリシンを製造したところ、 97 %と、比較対照株を用いた場合に比べ高い変換率を示し、更に、グルタミン酸を低減 した基質(OMPB :グルタミン酸:ァスパラギン酸 = 1 : 0. 25 : 1)においても 90. 1%と いう高い変換率を示した(実施例 4)。また、 L—ホスフィノスリシンアミノトランスフェラ ーゼ(以下「PAT」と呼ぶことがある)の一糠である AT— II AATを StreDtomvces hve msSQEk において共発現させることにより、 PAT活性と AAT活性が共に増強され
た菌株を作出し、得られた形質転換体の AAT活性および PAT活性を親株と比較し た結果、 PAT活性は約 16倍、 AAT活性は約 170倍であることを見出した(実施例 5 ) o該 AAT'PAT活性増強株を用いて L—ホスフィノスリシンを製造したところ、比較 対照株を用いた場合に比べ、 95. 3%という高い変換率を示し、更に、グルタミン酸 を低減した基質(OMPB:グルタミン酸:ァスパラギン酸 = 1 : 0. 25 : 1)にお!/ヽても 95 . 5%という極めて高い変換率を示した(実施例 6)。本発明はこれらの知見に基づくも のである。
[0007] 本発明は、 AAT遺伝子および AATタンパク質を提供することをその目的とする。
[0008] 本発明はまた、 目的遺伝子を高発現させることができるプロモーターを提供すること を目的とする。
[0009] 本発明は更に、 AAT遺伝子を宿主に導入するための組換えベクターおよび AAT 遺伝子が導入された宿主を提供することを目的とする。
[0010] 本発明は更に、 L—ホスフィノスリシンを高変換率で製造する方法を提供することを 目的とする。
[0011] 本発明によれば、以下の (i)、(ii)、(iii)および (iv)から選択される、ポリヌクレオチ ドが提供される:
(i)配列番号: 1で表される塩基配列力 なるポリヌクレオチド;
(ii)配列番号: 1で表される塩基配列からなるポリヌクレオチドにおいて、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつ A AT活性を有するタンパク質をコードするポリヌクレオチド;
(iii)配列番号: 1で表される塩基配列とストリンジェントな条件でノ、イブリダィズし、 つ AAT活性を有するタンパク質をコードするポリヌクレオチド;および
(iv)配列番号: 1で表される塩基配列力もなるポリヌクレオチドと少なくとも 90%の同 一性を有する塩基配列を含んでなり、かつ AAT活性を有するタンパク質をコードす るポリヌクレオチド。
[0012] 本発明によれば、以下の (V)、 (vi)、 (vii)および (viii)力 選択されるタンパク質( 以下「本発明によるタンパク質」と呼ぶことがある)が提供される:
(V)配列番号: 2で表されるアミノ酸配列からなるタンパク質;
(vi)配列番号: 2で表されるアミノ酸配列力もなるタンパク質にお 、て、 1もしくは複数 個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列からなり、かつ AAT 活性を有するタンパク質;
(vii)配列番号: 2で表されるアミノ酸配列をコードするポリヌクレオチドとストリンジェン トな条件でノヽイブリダィズするポリヌクレオチドによりコードされ、かつ AAT活性を有 するタンパク質;および
(viii)配列番号: 2で表されるアミノ酸配列と少なくとも 90%の同一性を有するアミノ酸 配列からなり、かつ AAT活性を有するタンパク質。
[0013] 本発明によればまた、本発明によるタンパク質をコードするポリヌクレオチドが提供 される。(このポリヌクレオチドと前述の (i)、(ii)、(iii)および (iv)力も選択されるポリヌ クレオチドを併せて、「本発明による AAT遺伝子」と呼ぶことがある。 )
本発明によれば、以下の )、(x)、 (xi)および (xii)力も選択される、ポリヌクレオ チド (以下「本発明によるプロモーター」と呼ぶことがある)が提供される:
(ix)配列番号: 3で表される塩基配列からなるポリヌクレオチド;
(X)配列番号: 3で表される塩基配列力 なるポリヌクレオチドにおいて、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつプ 口モーター活性を有するポリヌクレオチド;
(xi)配列番号: 3で表される塩基配列とストリンジェントな条件でノ、イブリダィズし、 つプロモーター活性を有するポリヌクレオチド;および
(xii)配列番号: 3で表される塩基配列力もなるポリヌクレオチドと少なくとも 95%の同 一性を有する塩基配列を含んでなり、かつプロモーター活性を有するポリヌクレオチ ド、。
[0014] 本発明によれば、本発明による AAT遺伝子を含んでなる、組換えベクター(以下「 本発明による第一の態様の組換えベクター」と呼ぶことがある)が提供される。
[0015] 本発明によれば、本発明による第一の態様の組換えベクターにより形質転換された 宿主 (以下「本発明による第一の態様の宿主」と呼ぶことがある)が提供される。
[0016] 本発明によれば、本発明による AAT遺伝子と PAT活性を有するタンパク質をコー ドするポリヌクレオチド(以下「PAT遺伝子」と呼ぶことがある)とを含んでなる、組換え
ベクター(以下「本発明による第二の態様の組換えベクター」と呼ぶことがある)が提 供される。
[0017] 本発明によれば、本発明による第二の態様の組換えベクターにより形質転換された 宿主 (以下「本発明による第二の態様の宿主」と呼ぶことがある)が提供される。
[0018] 本発明によれば、式 (I) :
[化 1]

で示される L—ホスフィノスリシンまたはその塩を製造する方法であって、式 (Π): [化 2]
0
CH3-P-CH2CH9CCOOH
OH 0
で示される 4 ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、ダル タミン酸またはその塩およびァスパラギン酸またはその塩の存在下で、 AAT活性お よび PAT活性を示し、かつ L ホスフィノスリシンを産生することができる放線菌と接 触させる工程を含んでなり、使用される放線菌が親株の AAT活性を超える活性を示 すことを特徴とする方法 (以下「本発明による第一の態様の方法」と呼ぶことがある) が提供される。
[0019] 本発明によれば、式 (I) :
[化 3]
— P— CH2CH2CHCOOH
OH
で示される L—ホスフィノスリシンまたはその塩を製造する方法であって、式 (Π): [化 4]

で示される 4 ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、ダル タミン酸またはその塩およびァスパラギン酸またはその塩の存在下で、 ΑΑΤ活性お よび PAT活性を示し、かつ L ホスフィノスリシンを産生することができる放線菌と接 触させる工程を含んでなり、使用される放線菌が親株の AAT活性を超える活性を示 し、かつ、親株の PAT活性を超える活性を示すことを特徴とする方法 (以下「本発明 による第二の態様の方法」と呼ぶことがある)が提供される。
図面の簡単な説明
[0020] [図 1]AAT遺伝子を含む約 5. 8Kbpの制限酵素地図を示した図である。
[図 2]発現ベクター pLG04の構築を示した図である。
[図 3]発現ベクター pSGl 1の構築を示した図である。
[図 4]プラスミド pATSGOlの構築を示した図である。
[図 5]発現ベクター pAHSG7201の構築を示した図である。
発明の具体的な説明
[0021] [微生物の寄託]
本発明で使用される Streptomvces hveroscopicus SF1293 NP— 50株は、 1987 年 5月 20日付で独立行政法人産業技術総合研究所特許生物寄託センター ( τ 305
— 8566日本国茨城県つくば巿東 1丁目 1番地 1中央第 6)に寄託された。受託番号 は、 FERM BP— 1368である。
[0022] 本発明で使用される StreDtomvces hvgroscopicus SF1293株は、 1982年 5月 19 日付で独立行政法人産業技術総合研究所特許生物寄託センター(干 305— 8566 日本国茨城県つくば巿東 1丁目 1番地 1中央第 6)に寄託された。受託番号は、 FER
M BP— 130である。
[0023] プラスミド pSGl 1で形質転換された放線菌 (Streptomvces lividans) (実施例 3 (1) ) は、 2006年 2月 1日付で独立行政法人産業技術総合研究所特許生物寄託センター (〒 305— 8566日本国茨城県つくば巿東 1丁目 1番地 1中央第 6)に寄託された。受 託番号は、 FERM BP— 10495である。
[0024] プラスミド OMSB515で形質転換された放線菌(Streptomvces lividans)は、 1989 年 6月 30日付で独立行政法人産業技術総合研究所特許生物寄託センター ( τ 305 — 8566日本国茨城県つくば巿東 1丁目 1番地 1中央第 6)に寄託された。受託番号 は、 FERM BP— 2496である。
[0025] プラスミド pAHSG7201で形質転換された放線菌 (Streptomvces lividans) (実施例 5 (1) )は、 2006年 2月 1日付で独立行政法人産業技術総合研究所特許生物寄託 センター(〒 305 - 8566日本国茨城県つくば巿東 1丁目 1番地 1中央第 6)に寄託さ れた。受託番号は、 FERM BP— 10496である。
[0026] [AAT遺伝子および AATタンパク質]
本発明による AAT遺伝子は、それによりコードされるタンパク質が AAT活性を有す る限り、その由来は特に限定されないが、好ましくは放線菌由来であり、より好ましく は、 Streptomvces hveroscopicus由来である
[0027] 本願明細書において、「ポリヌクレオチドにおいて、 1もしくは複数個のヌクレオチド が欠失、置換、挿入もしくは付加された」および「アミノ酸配列において、 1もしくは複 数個のアミノ酸が欠失、置換、挿入もしくは付加されたアミノ酸配列」とは、部位特異 的突然変異誘発法等の周知の方法により、または天然に生じ得る程度の複数個のヌ クレオチドあるいはアミノ酸の置換等により改変がなされたことを意味する。ヌクレオチ ドおよびアミノ酸の改変の個数は、 1個または複数個(例えば、 1個ないし数個あるい は 1、 2、 3、または 4個)である。
[0028] 改変された塩基配列の例としては、 1個または複数個(例えば、 1個な!/ヽし数個ある いは 1、 2、 3、または 4個)の、 AAT活性に影響を与えない変異を有する配列番号: 1 に記載の塩基配列が挙げられる。
[0029] 改変されたアミノ酸配列の例としては、 1個または複数個(例えば、 1個な 、し数個 あるいは 1、 2、 3、または 4個)の、 AAT活性に影響を与えない変異を有する配列番
号: 2に記載のアミノ酸配列が挙げられる。
[0030] ここで「AAT活性」とは、 AATが L—ァスパラギン酸のアミノ基を a—ケトグルタル 酸に転移し、 L—グルタミン酸とォキザ口酢酸を生じさせる反応 (またはその逆反応) を触媒する能力として定義される。より具体的には、 AAT活性測定用基質(150mM ァスパラギン酸、 50mM aーケトグルタル酸、 0. ImM ピリドキサルリン酸、 100 mM Tris-HCl buffer ;pH8. 5)と AATを混合し、 37°C、 20分間インキュベート した後、反応を停止し、生成したグルタミン酸の量をアミノ酸分析用 HPLC (model LC—VP、島津製作所社製)にて分析することにより求められる酵素力(1分間に 1 μ molのグルタミン酸を生成する能力を 1U (ユニット)とする)として定義される。
[0031] 本発明にお ヽて、微生物の産生する AAT活性を評価する場合、比活性 (U/mg) を指標として評価することが望ま U、。
[0032] 比活性 (U/mg)は、 AAT活性 (U)をタンパク質量 (例えば、プロテインアツセィキ ット (バイオラッド社製) )を用いて γ —グロブリンをスタンダードとして測定することが できる)で除することにより求められる。
[0033] また、「活性に影響を与えな 、変異」の例としては、保存的置換が挙げられる。ここ で、「保存的置換」とは、タンパク質の活性を実質的に改変しないように 1若しくは複 数個のアミノ酸残基を、別の化学的に類似したアミノ酸残基で置き換えることを意味 する。例えば、ある疎水性残基を別の疎水性残基によって置換する場合、ある極性 残基を同じ電荷を有する別の極性残基によって置換する場合などが挙げられる。こ のような置換を行うことができる機能的に類似のアミノ酸は、アミノ酸毎に当該技術分 野において公知である。具体例を挙げると、非極性 (疎水性)アミノ酸としては、ァラ- ン、ノ リン、イソロイシン、ロイシン、プロリン、トリプトファン、フエ二ルァラニン、メチォ ニンなどが挙げられる。極性(中性)アミノ酸としては、グリシン、セリン、スレオニン、チ 口シン、グルタミン、ァスパラギン、システィンなどが挙げられる。陽電荷をもつ (塩基 性)アミノ酸としては、アルギニン、ヒスチジン、リジンなどが挙げられる。また、負電荷 をもつ(酸性)アミノ酸としては、ァスパラギン酸、グルタミン酸などが挙げられる。
[0034] 本願明細書にぉ 、て「ストリンジェントな条件」とは、ハイブリダィゼーシヨン後のメン プレンの洗浄操作を、高温下低塩濃度溶液中で行うことを意味し、例えば、 0. 5 X S
SC濃度(l X SSC : 15mMクェン酸 3ナトリウム、 150mM塩ィ匕ナトリウム)、 60°C、 15 分間の洗浄条件、好ましくは 0. 5 X SSC濃度、 0. 1%SDS溶液中で 60°C、 15分間 の洗浄条件、を意味する。
[0035] ノ、イブリダィゼーシヨンは、公知の方法に従って行うことができる。また、市販のライ ブラリーを使用する場合、添付の使用説明書に記載の方法に従って行うことができる
[0036] 本願明細書において、塩基配列またはアミノ酸配列についての「同一性」とは、比 較される配列間において、各々の配列を構成する塩基またはアミノ酸残基の一致の 程度の意味で用いられる。本明細書において示した「同一性」の数値はいずれも、当 業者に公知の相同性検索プログラムを用いて算出される数値であればよぐ例えば F ASTA、 BLAST等にぉ 、てデフォルト(初期設定)のパラメータを用いることにより、 容易に算出することができる。
[0037] 本発明において、配列番号: 2で表されるアミノ酸配列が与えられれば、それをコー ドするポリヌクレオチドは容易に決まり、配列番号: 2で表されるアミノ酸配列をコード する種々のポリヌクレオチドを選択することができる。
[0038] 従って、配列番号: 2で表されるアミノ酸配列をコードするポリヌクレオチドとは、配列 番号: 1で表される DNA配列の一部または全部に加え、同一のアミノ酸をコードする DNA配列であって縮重関係にあるコドンを DNA配列として有する配列をも意味する ものである。さらに、これらに対応する RNA配列も含まれる。
[0039] 配列番号: 2で表されるアミノ酸配列をコードするポリヌクレオチドの好まし 、例とし ては、配列番号: 1で表される塩基配列力 なるポリヌクレオチドが挙げられる。
[0040] [プロモーター配列]
実施例3によれば、 Streptomvces hvgroscopicusより単離した AAT遣伝子の全長配 列の内、 BamHI〜XhoI断片を含む発現ベクターにより形質転換された宿主につい て AAT活性を調べたところ、親株の 47倍であった。これは、 PstI〜XhoI断片を含む 発現ベクターにより形質転換された宿主における活性と比べ顕著に高活性であった 。このことから、 AAT遺伝子のプロモーター領域の内、転写開始点から上流の Bam HIサイトまでの配列が強力なプロモーターとして働くことが認められた。上流領域を 5
'上流力 切りつめた場合には、その上流領域により制御される構造遺伝子の発現 が低下するのが通常であるが、本発明によるプロモーターは、意外にも、強力なプロ モーター活性を有する。
[0041] 本発明によるプロモーターの塩基配列としては、配列番号: 3に記載される塩基配 列が挙げられる力 この配列のみならず、プロモーター活性を同等程度有するその 改変配列も含まれる。
[0042] ここで「プロモーター活性」を有するか否かは、例えば、実施例 3に記載のように AA T遺伝子の発現ベクターを作成し、宿主にて発現させ、 AAT活性を測定することによ り評価することができる。 AAT活性が認められれば、プロモーター活性を有するとい えるが、好ましくは親株の 2倍以上、より好ましくは 20倍以上、最も好ましくは 40倍以 上の AAT活性の増強が認められた場合に「プロモーター活性を有する」と評価する ことができる。
[0043] 本発明によるプロモーターは、 目的遺伝子に作動可能に連結することにより、 目的 遺伝子を高発現させることができる。本発明によるプロモーターによってより高発現さ せることができる目的遺伝子としては、放線菌由来の遺伝子が挙げられる。また、 目 的遺伝子をより高発現させることができる宿主としては、放線菌が挙げられる。
[0044] ここで「放線菌」としては、 Streptomvces hvgroscopicus. Streptomvces albus. Strept omvces coelicolor. Streptomvces griseus. Streptomvces lividans. Streptomvces virgi niae、 Streptomvces viridochromogenes、 Streptomvces pilosus. Streptoverticillium ci nnamoneum. Streptomvces morookaensis、 Nocardia mediterranei ゝ Nocardopsis dass onvillei、 Saccharopolvspora hirsuta、 Kitasatosporia phosalacinea. Micromonospora c arbonacae, Streptosporangium pseudovulgare力 S挙げられ、より好ましくま Streptomvce s hvgroscopicusである。
[0045] 本発明の好ましい態様によれば、本発明によるプロモーターを、放線菌 (好ましくは Streptomvces hvgroscopicus)由来の遺伝子(好ましくは AAT遺伝子)に作動可能に 連結することができる。
[0046] 本発明の好ま 、態様によればまた、本発明によるプロモーターを、放線菌 (好まし くは Streptomyces hvgroscopicus.)由来の遺伝子(好ましくは AAT遺伝子)に作動可
能に連結してなる組換えベクターを放線菌 (好ましくは Streptomvces hveroscopicus) に導入することができる。
[0047] [PAT遺伝子]
本発明による組換えベクターに使用される PAT遺伝子は、それによりコードされる タンパク質が PAT活性を有する限り、その由来は特に限定されないが、好ましくは放 線菌由来であり、より好ましくは、 Streptomvces hygroscopicus由来でめる。
[0048] ここで、「PAT活性」とは、 PATが L—グルタミン酸のアミノ基を OMPBに転移し、 L —ホスフィノスリシンと a—ケトグルタル酸を生じさせる反応 (またはその逆反応)を触 媒する能力として定義される。より具体的には、 PAT活性測定用基質(150mM グ ルタミン酸、 50mM OMPB, 0. ImM ピリドキサルリン酸、 lOOmM Tris— HC1 buffer ; pH8. 5)と PATを混合し、 37°C、 20分間インキュベートした後、反応を停 止し、生成した L—ホスフィノスリシンの量をアミノ酸分析用 HPLC (model LC— VP 、島津製作所社製)にて分析することにより求められる酵素力(1分間に 1 μ molの L —ホスフィノスリシンを生成する能力を 1U (ユニット)とする)として定義される。
[0049] 本発明にお ヽて、微生物の産生する PAT活性を評価する場合、比活性 (U/mg) を指標として評価することが望ま U、。
[0050] 比活性 (U/mg)は、 PAT活性 (U)をタンパク質量 (例えば、プロテインアツセィキ ット (バイオラッド社製) )を用いて γ —グロブリンをスタンダードとして測定することが できる)で除することにより求められる。
[0051] PAT活性を有するタンパク質をコードするポリヌクレオチドとしては、以下の(i' )、 (i ί' )、 (ίϋ' )および (vi' )力も選択されるものを使用することができる。
[0052] (!' )配列番号: 4で表される塩基配列力 なるポリヌクレオチド;
(π' )配列番号: 4で表される塩基配列からなるポリヌクレオチドにお 、て、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつ Ρ AT活性を有するタンパク質をコードするポリヌクレオチド;
(iii' )配列番号: 4で表される塩基配列とストリンジヱントな条件でノ、イブリダィズし、か つ PAT活性を有するタンパク質をコードするポリヌクレオチド;および
(vi' )配列番号: 4で表される塩基配列力もなるポリヌクレオチドと少なくとも 90%の同
一性を有する塩基配列を含んでなり、かつ PAT活性を有するタンパク質をコードする ポリヌクレオチド。
[0053] PAT活性を有するタンパク質としては、以下の (v,)、 (vi,)、 (vii,)および (viii,)か ら選択されるものを使用することができる:
(ν' )配列番号: 5で表されるアミノ酸配列からなるタンパク質;
(νί' )配列番号: 5で表されるアミノ酸配列にお 、て、 1もしくは複数個のアミノ酸が欠 失、置換、挿入もしくは付加されたアミノ酸配列からなり、かつ PAT活性を有するタン パク質;
(νη' )配列番号: 5で表されるアミノ酸配列をコードするポリヌクレオチドとストリンジェ ントな条件でノヽイブリダィズするポリヌクレオチドによりコードされ、かつ PAT活性を有 するタンパク質;および
(νίϋ' )配列番号: 5で表されるアミノ酸配列と少なくとも 90%の同一性を有するァミノ 酸配列からなり、かつ PAT活性を有するタンパク質。
[0054] [組換えベクター]
本発明による組換えベクターは、例えば、 Sambrook, J. et al., Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, New York (1989)等に己載される 遺伝子組換え技術の慣行法に従って作製することができる。
[0055] 本発明による糸且換えベクターにお!/、ては、各遺伝子の発現に必要な制御配列、例 えば、プロモーター、転写開始信号、リボソーム結合部位、翻訳停止シグナル、転写 終結信号等の転写調節信号、翻訳調節信号等が、 目的遺伝子に作動可能に連結さ れていてもよい。
[0056] 本発明による組換えベクターはまた、形質転換体を選抜するための選択マーカー を含んでいてもよぐ使用する宿主に応じて適宜選択マーカーを選択することができ る。選択マーカーとしては薬剤耐性遺伝子、栄養要求性を相補する遺伝子などが挙 げられ、好ましい例としては、宿主が細菌の場合は、アンピシリン耐性遺伝子、カナマ イシン耐性遺伝子、テトラサイクリン耐性遺伝子等が、宿主が酵母の場合は、トリブト ファン生合成遺伝子 (TRP1)、ゥラシル生合成遺伝子 (URA3)、ロイシン生合成遺伝 子 (LEU2)等が、放線菌の場合は、ハイグロマイシン耐性遺伝子、ビアラホス耐'性遺
伝子、ブレオマイシン耐性遺伝子、オーレォバシジン耐性遺伝子、チォストレプトン 耐性遺伝子等が、それぞれ挙げられる。
[0057] 本発明による組換えベクターは、使用する宿主の種類を勘案しながら、ウィルス、プ ラスミド、コスミドベクター等力も適宜選択することができる。例えば、宿主細胞が大腸 菌の場合はえファージ系のバタテリオファージ、 PBR322系、 pUC系のベクター、枯 草菌の場合は PUB110系、 pPL603系、 pC194系ベクター、酵母の場合は pYC系 、 pYE系ベクター、放線菌の場合は PU702系、 pU680系、 pAK114系ベクターが 挙げられる。
[0058] 本発明による第一の態様の組換えベクターは、好ましくは、本発明によるプロモー ターを更に含んでいてもよい。この場合、本発明によるプロモーターは、本発明によ る AAT遺伝子に作動可能に連結することができる。この組換えベクターは宿主 (特に 放線菌)にお 、て AAT遺伝子を高発現させることができる点で有利である。
[0059] 本発明による第二の態様の組換えベクターは、好ましくは、本発明によるプロモー ターを更に含んでいてもよい。この場合、本発明によるプロモーターは、本発明によ る AAT遺伝子に作動可能に連結することができる。この組換えベクターは宿主 (特に 放線菌)にお 、て AAT遺伝子および PAT遺伝子を高発現させることができる点で有 利である。
[0060] 本発明による第二の態様の組換えベクターはまた、 PAT遺伝子の発現を誘導する プロモーターを更に含んでいてもよぐそのプロモーターは PAT遺伝子に作動可能 に連結することができる。
[0061] PAT遺伝子のプロモーターについては、当業者であれば適宜選択することができ 、好ましくは、特開平 2— 195889号公報に記載のプラスミド pMSB515中の AT— II 遺伝子プロモーターを使用することができる。
[0062] 本発明によるプロモーターへの連結は、例えば、常法に従い、 目的タンパク質をコ ードする遺伝子(目的遺伝子)の翻訳領域をプロモーターの下流に順方向に挿入す ること〖こよって行うことができる。この場合、 目的遺伝子を他のタンパク質の翻訳領域 をコードする外来遺伝子と連結させて融合タンパク質として発現させることもできる。
[0063] 本発明による第二の態様の組換えベクターにおいては、本発明による AAT遺伝子
と PAT遺伝子とは逆方向に連結されていても、順方向に連結されていてもよいが、 A AT活性を増強する観点から、好ましくは、逆方向に連結することができる。
[0064] 本発明によるプロモーターには、本発明による AAT遺伝子以外の目的遺伝子を連 結することができる。
[0065] [形質転換体]
形質転換される宿主は、使用される組換えベクターの種類に応じて、放線菌、大腸 菌、枯草菌、酵母、糸状菌、その他の微生物の中から適宜選択されてよい。宿主とし て使用でさる ί¾ 菌とし は、 Streptomvces hygroscopicus, Streptomvces albus, Str eptomvces coelicolor、 Streptomvces griseus. Streptomvces lividans. Streptomvces vi rginiae、 Streptomvces vindochromogenes、 Streptomvces pilosus. Streptoverticnlium cinnamoneum. Streptomvces morookaensis、 Nocardia mediterranei ゝ Nocardopsis da ssonvilleu Saccharopolvspora nirsuta. Kitasatosporia phosalacinea. Micromonospora carbonacae. Streptosporangium pseudovukare ) げられ、好ましく【ま Streptomvces hvgroscoDicusであり、より好ましくは、 FERM BP— 1368の受託番号のもと独立行 政法人産業技術総合研究所特許生物寄託センターに寄託された SF 1293 NP- 5 0株または FERM BP— 130の受託番号のもと独立行政法人産業技術総合研究所 特許生物寄託センターに寄託された SF1293株が挙げられる。
[0066] 組換えベクターを宿主に導入する方法は周知であり、宿主やベクターの種類によつ て最も効率のよい方法が選択される。放線菌を形質転換する場合には、大腸菌との 接合による伝達、放線菌ファージによる感染、宿主菌のプロトプラストへの導入等が 実施できる。形質転換によって得られた組換え体の選別〖こは、用いるベクターの保 有する遺伝的指標、例えば、抗生物質耐性、ボック形成、メラニン生合成等が利用で きる。
[0067] 形質転換された宿主の培養は、常法に従って、当該分野で通常用いられる培地、 培養条件を使用することができる。
[0068] 培地については、慣用の成分、例えば炭素源としては、グルコースおよびその他の 糖類、並びに澱粉等が使用できる。また、窒素源としては、肉エキスおよびペプトン、 並びにコーン、小麦、大豆、微生物などの各種抽出物が使用できる。その他必要に
応じ、ナトリウム、カリウム、カルシウム、マグネシウム、コバルト、塩素、リン酸、硫酸な どの無機塩類や各種ビタミンを添加することもできる。培養中の発砲を抑えるため、 市販の消泡剤を添加することもできる。
[0069] 培地の殺菌方法については、蒸気殺菌やフィルター殺菌などの方法を使用するこ とがでさる。
[0070] 培養条件については、ロータリーシェーカーを用いたフラスコ培養や、ジャーファー メンター装置やタンク設備を用いた通気攪拌培養などにより行うことができる。培地の pH、培養温度、培養日数は、形質転換体に応じて適宜決定することができる。
[0071] 本発明による第一の態様の形質転換体の好ま U、例としては、本発明による AAT 遺伝子と本発明によるプロモーターとが作動可能に連結された第一の態様の組換え ベクターにより形質転換された放線菌(特に、 Streptomvces hvgroscopicus)が挙げら れる。この形質転換体は、親株の放線菌の AAT活性を超える活性を示す点で有利 である。
[0072] 本発明による第一の態様の形質転換体のより好ましい例としては、 FERM BP—1 0495の受託番号のもと独立行政法人産業技術総合研究所に寄託された形質転換 体から調製される発現ベクター OSG11で形皙転椽された StreDtomvces hvgroscopicu Sが挙げられ、最も好ましくは、該発現ベクター pSGl lで形質転換された FERM B P— 1368の受託番号のもと独立行政法人産業技術総合研究所に寄託された SF12 93 NP— 50株が挙げられる。この形質転換体は、親株と比較して、非常に高い AA T活性を示す点で極めて有利である。
[0073] 本発明による第二の態様の形質転換体の好ま U、例としては、本発明による AAT 遺伝子と本発明によるプロモーターとが作動可能に連結された第二の態様の組換え ベクターにより形質転換された放線菌(特に、 Streptomvces hvgroscopicus)が挙げら れる。この形質転換体は、親株の放線菌の AAT活性および PAT活性のいずれをも 超える活性を示す点で有利である。
[0074] 本発明による第二の態様の形質転換体のより好ましい例としては、本発明による A AT遺伝子と本発明によるプロモーターとが作動可能に連結され、かつ本発明による AAT遺伝子と PAT遺伝子とが逆方向に連結された第二の態様の組換えベクターに
より形質転換された放線菌(特に、 Streptomvces hveroscopicus)が挙げられる。この 形質転換体は、親株の放線菌の AAT活性および PAT活性を超える活性を示す点 で有利である。
[0075] 本発明による第二の態様の形質転換体のより好ましい例としては、 FERM BP—1 0496の受託番号のもと独立行政法人産業技術総合研究所に寄託された形質転換 体力 調製される発現ベクター OAHSG7201で形質転換された StreDtomvces hvgro scoDicusが举げられ、最も好ましくは、該発現ベクター PAHSG7201で形質転換され た FERM BP— 1368の受託番号のもと独立行政法人産業技術総合研究所に寄託 された SF1293 NP— 50株が挙げられる。この形質転換体は、親株と比較して、非 常に高い AAT活性および PAT活性を示す点で極めて有利である。
[0076] [L ホスフィノスリシンの製造方法] 本発明による製造方法にぉ ヽて使用する放線菌は、 AAT活性および PAT活性を 示し、 L ホスフィノスリシンの製造に関与する。具体的には、この放線菌は、 OMPB 、 Lーァスパラギン酸、 L グルタミン酸の存在下で、以下の酵素反応の進行に関与 する。
[0077] (1) PATが L—グルタミン酸と OMPBに作用することにより、 L—グルタミン酸のアミノ 基が OMPBに転移し、 L ホスフィノスリシンと α ケトグルタル酸が生じる。
[0078] (2) ΑΑΤが Lーァスパラギン酸と aーケトグルタル酸に作用することにより、 Lーァス パラギン酸のアミノ基が α—ケトグルタル酸に転移し、 L—グルタミン酸とォキザ口酢 酸を生じる。
[0079] (3) (2)で生じた L グルタミン酸が(1)において再び利用され、反応が継続する。
[0080] 栾椽率の定義
本発明による製造法において、「変換率」とは、基質としての OMPBのうち、 L ホ スフイノスリシンに変換された割合を意味し、下記の式により算出することができる。
[数 1] 変換率 (%) = (生成した L一ホスフィノスリシンのモル数 I'm o 1 ) ) X 1 0 0
(使用した OM P Bのモル数 (m o 1 ))
[0081] 第一の ¾の製诰方法
本発明による第一の態様の製造方法によれば、 AAT活性および PAT活性を示し 、かつ L ホスフィノスリシンを産生することができる放線菌のうち、親株の AAT活性 を超える AAT活性を有する放線菌を L ホスフィノスリシンの製造に用いることにより 、これまでに達成できな力つた高効率で L—ホスフィノスリシンを製造することができる 。特に、本発明による第一の態様の製造方法では、 L ホスフィノスリシンの製造原 料であるグルタミン酸を削減してもなお、 L—ホスフィノスリシンを高効率で製造できる ことから、コスト削減の観点力 極めて有利である。
[0082] すなわち、実施例 4によれば、親株の AAT活性を超える AAT活性を有する形質転 換体を使用して L ホスフィノスリシンを製造した場合、変換率は 97%であった。親 株の変換率が 93. 2%であり、また PAT遺伝子のみを高発現させた株の変換率が 9 2. 6%であることを考慮すると、 AAT活性の増強により、 L—ホスフィノスリシンの変 換率が向上すると考えられる。
[0083] 実施例 4によればまた、親株の AAT活性を超える AAT活性を有する形質転換体 を使用して、グルタミン酸を減量した基質カゝら L -ホスフィノスリシンを製造した場合、 変換率は 90. 1%であった。親株の変換率が 76. 2%であり、また PAT遺伝子のみ を高発現させた株の変換率が 82. 7%であることを考慮すると、グルタミン酸減量基 質を使用した場合にも、 AAT活性の増強により、 L ホスフィノスリシンの変換率が向 上すると考えられる。
[0084] これまで、 L ホスフィノスリシンの製造においては、 L—ホスフィノスリシンへの変換 工程に直接作用する PATに着目して、放線菌の PAT活性を増強する試みがなされ たが、満足できる高効率での製造は達成できな力つた。また、仮に AAT活性を増強 しても PAT活性が律速になり、結局、 L ホスフィノスリシンの高効率での製造は達 成できな 、と考えられてきた。
[0085] 以下の理論に拘束される訳ではないが、 L—ホスフィノスリシンを産生する放線菌に おいて AAT活性が増強されると、 L ホスフィノスリシンとともに生じた α ケトグルタ ル酸が増強された ΑΑΤにより速やかに L—グルタミン酸に戻されるため、 a ケトグ ルタル酸による生成物阻害が力からなくなり、反応が円滑に進行するようになったと
考えられる。
[0086] 本発明による第一の態様の製造方法は、 AAT活性および PAT活性を示し、かっし ホスフィノスリシンを産生することができる放線菌であって、親株の AAT活性を超え る活性を示す放線菌を使用することを特徴とする。本発明による第一の態様の製造 法では、使用される放線菌の AAT活性が親株の活性を超えるものであればよ ヽが、 好ましくは 2倍以上、より好ましくは 10倍以上、特に好ましくは 20倍以上、最も好まし くは 40倍以上である。
[0087] ここで、「使用される放線菌が親株の AAT活性を超える活性を示す」か否かは、使 用される放線菌が産生する AATの比活性を測定し、親株の比活性と比較すること〖こ より確認することができる。
[0088] 本発明による第一の態様の製造方法で使用できる放線菌としては、親株の AAT活 性を超える活性を示す放線菌であればよぐ例えば、 AAT遺伝子を含む発現べクタ 一で形質転換して得られた放線菌や、変異処理 (ニトロソグァ-ジン処理、 UV処理 等)により育種された AAT活性が増強された放線菌株を使用することができる。好ま しくは、 AAT活性が親株よりも 20倍以上増強された放線菌を用いることができ、具体 的には、本発明による第一の態様の宿主 (放線菌)、より好ましくは、 FERM BP- 1 0495の受託番号のもと独立行政法人産業技術総合研究所に寄託された形質転換 体から調製される発現ベクター OSG11で形皙転椽された StreDtomvces hvgroscopicu s,最も好ましくは、該発現ベクター pSGl lで形質転換された FERM BP— 1368の 受託番号のもと独立行政法人産業技術総合研究所に寄託された SF1293 NP- 5 0株、を使用することができる。
[0089] 本発明による第一の態様の製造方法にぉ 、て使用される基質にっ 、ては、式 (II) の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはその塩 = 1 : 0. 25〜1 : 1とすることができる力 経済的な観点からは、好ましくは、約 1 :約 0. 25 :約 1である。
[0090] 本発明による第一の態様の製造法の好ま U、態様としては、式 (Π)で示される 4 ヒドロキシメチルホスフィエル 2—ォキソ 酪酸またはその塩を、本発明による第一 の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L—ホスフ
ィノスリシンまたはその塩を製造する方法が挙げられる。
[0091] 本発明による第一の態様の製造法のより好ましい態様としては、式 (Π)で示される 4
-ヒドロキシメチルホスフィエル 2—ォキソ 酪酸またはその塩を、本発明による第 一の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L ホス フィノスリシンまたはその塩を製造する方法であって、その宿主に導入された組換え ベクターが本発明によるプロモーターを更に含んでなり、かつ本発明による AAT遺 伝子と本発明によるプロモーターとが作動可能に連結されている方法が挙げられる。
[0092] 本発明による第一の態様の製造法の特に好ま 、態様としては、式 (Π)で示される 4 -ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、本発明による 第一の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L—ホ スフイノスリシンまたはその塩を製造する方法であって、その宿主に導入された組換 えベクターが本発明によるプロモーターを更に含んでなり、本発明による AAT遺伝 子と本発明によるプロモーターとが作動可能に連結され、かつその宿主が StreDtomv ces hveroscopicus由来である方 fe力李げりれる
[0093] 第二の の ¾告 法
本発明による第二の態様の製造方法によれば、 AAT活性および PAT活性を示し 、かつ L ホスフィノスリシンを産生することができる放線菌のうち、親株の AAT活性 および PAT活性を超える活性を有する放線菌を L ホスフィノスリシンの製造に用い ることにより、これまでに達成できなかった極めて高 、効率で L ホスフィノスリシンを 製造することができる。特に、本発明による第二の態様の製造方法では、 L ホスフィ ノスリシンの製造原料であるグルタミン酸を削減してもなお、 L ホスフィノスリシンを 高効率で製造できることから、コスト削減の観点力 極めて有利である。
[0094] すなわち、実施例 6によれば、親株の AAT活性および PAT活性を超える活性を有 する形質転換体を使用して L ホスフィノスリシンを製造した場合、変換率は 95. 3% であった。親株の変換率が 93. 2%であり、また PAT遺伝子のみを高発現させた株 の変換率が 92. 6%であることを考慮すると、 AAT活性にカ卩えて PAT活性を増強す ることにより、 L ホスフィノスリシンの変換率が更に向上すると考えられる。
[0095] 実施例 6によればまた、親株の AAT活性および PAT活性を超える活性を有する形
質転換体を使用して、グルタミン酸を減量した基質力も L -ホスフィノスリシンを製造 した場合、変換率は 95. 5%であった。親株の変換率が 76. 2%であり、また PAT遺 伝子のみを高発現させた株の変換率が 82. 7%であることを考慮すると、グルタミン 酸減量基質を使用した場合にも、 AAT活性に加えて PAT活性を増強することにより 、 L—ホスフィノスリシンの変換率が向上すると考えられる。
[0096] 前述のように、放線菌の PAT活性のみを増強しても L—ホスフィノスリシンの変換率 を向上させることができなかった。また、 L ホスフィノスリシンの製造において AAT 活性のみならず PAT活性を増強することは報告されていな力つた。
[0097] 本発明による第二の態様の製造方法は、 AAT活性および PAT活性を示し、かっし ホスフィノスリシンを産生することができる放線菌であって、親株の AAT活性およ び PAT活性を超える活性を示す放線菌を使用することを特徴とする。本発明による 第二の態様の製造法では、使用される放線菌の AAT活性が親株の活性を超えるも のであればよいが、好ましくは約 2倍以上、より好ましくは約 20倍以上、さらに好ましく は約 40倍以上、特に好ましくは約 100倍以上、最も好ましくは約 170倍以上である。 本発明による第二の態様の製造法ではまた、使用される放線菌の PAT活性が親株 の活性を超えるものであればよいが、好ましくは約 2倍以上、より好ましくは約 10倍以 上、最も好ましくは約 16倍以上である。
[0098] 実施例 6によれば、増強された AAT活性力 増強された PAT活性よりも相対的に 強い場合に、 L—ホスフィノスリシンの極めて高い製造効率が達成された。以下の理 論に拘束される訳ではないが、生成物阻害を起こすと考えられる aーケトグルタル酸 が相対的に増強された AATにより速やかにグルタミン酸に変換されるためであると考 えられる。従って、本発明による第二の態様の製造方法では、使用する放線菌にお V、て、 AAT活性の方が PAT活性よりも相対的に強 、ことが好ま 、。
[0099] ここで、「使用される放線菌が親株の AAT活性を超える活性を示す」か否かは、使 用される放線菌が産生する AATの比活性を測定し、親株の比活性と比較すること〖こ より確認することができる。また、「使用される放線菌が親株の PAT活性を超える活性 を示す」か否かは、使用される放線菌が産生する PATの比活性を測定し、親株の比 活性と比較することにより確認することができる。
[0100] 本発明による第二の態様の製造方法で使用できる放線菌としては、親株の AAT活 性および PAT活性を超える活性を示す放線菌であればよぐ例えば、 AAT遺伝子と PAT遺伝子を含む発現ベクターで形質転換して得られた放線菌や、変異処理 (-ト ロソグァ-ジン処理、 UV処理等)により育種された AAT活性および PAT活性が増 強された放線菌株を使用することができる。好ましくは、 AAT活性が親株よりも 40倍 以上増強されるとともに、 PAT活性が 10倍以上増強された放線菌を用いることがで き、具体的には、本発明による第二の態様の宿主 (放線菌)、より好ましくは、 FERM BP— 10496の受託番号のもと独立行政法人産業技術総合研究所に寄託された 形質転換体から調製される発現ベクター PAHSG7201で形質転換された i^mi ces hvgroscopicus.最も好ましくは、該発現ベクター pAHSG7201で形質転換され た FERM BP— 1368の受託番号のもと独立行政法人産業技術総合研究所に寄託 された SF1293 NP— 50株、を使用することができる。
[0101] 本発明による第二の態様の製造方法にぉ 、て使用される基質にっ 、ては、式 (II) の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはその塩 = 1 : 0. 25〜1 : 1とすることができる力 経済的な観点からは、好ましくは、約 1 :約 0. 25 :約 1である。
[0102] 本発明による第二の態様の製造法の好ましい態様としては、式 (Π)で示される 4 ヒドロキシメチルホスフィエル 2—ォキソ 酪酸またはその塩を、本発明による第二 の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L—ホスフ ィノスリシンまたはその塩を製造する方法が挙げられる。
[0103] 本発明による第二の態様の製造法のより好ましい態様としては、式 (Π)で示される 4
-ヒドロキシメチルホスフィエル 2—ォキソ 酪酸またはその塩を、本発明による第 二の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L ホス フィノスリシンまたはその塩を製造する方法であって、その宿主に導入された組換え ベクターが本発明によるプロモーターを更に含んでなり、かつ本発明による AAT遺 伝子と本発明によるプロモーターとが作動可能に連結されている方法が挙げられる。
[0104] 本発明による第二の態様の製造法の特に好ま 、態様としては、式 (Π)で示される 4 -ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、本発明による
第二の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L ホ スフイノスリシンまたはその塩を製造する方法であって、その宿主に導入された組換 えベクターが本発明によるプロモーターを更に含んでなり、本発明による AAT遺伝 子と本発明によるプロモーターとが作動可能に連結され、かつその宿主が StreDtomv ces hygroscopicus由来である方 fe力孕げりれる。
[0105] 本発明による第二の態様の製造法の最も好ま 、態様としては、式 (Π)で示される 4 -ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、本発明による 第二の態様の宿主 (放線菌)と接触させる工程を含んでなる、式 (I)で示される L ホ スフイノスリシンまたはその塩を製造する方法であって、その宿主に導入された組換 えベクターが本発明によるプロモーターを更に含んでなり、本発明による AAT遺伝 子と本発明によるプロモーターとが作動可能に連結され、その宿主が StreDtomvces h vgroscoDicus由来であり、かつ、 AAT遺伝子と PAT遺伝子とが逆方向に連結されて いる方法が挙げられる。
[0106] 観告 ィ Φ
本発明による製造方法は、特許第 2638541号公報の記載に従って行うことができ る。例えば、 3%OMPBまたはその塩と 0. 75%〜3%グルタミン酸またはその塩と 3 %ァスパラギン酸またはその塩との混合液を基質として、これを水酸ィ匕ナトリウムによ り中和し、 pH7. 0〜9. 5、好ましくは、 pH約 8. 5、温度 30〜45。C、好ましくは、 37 °Cの条件で、 0. 5〜5日間、 AAT活性が増強された放線菌あるいは AAT活性およ び PAT活性が増強された放線菌を作用させることにより行うことができる。また、変換 反応時に、微量のピリドキサルリン酸またはその塩を添加することも可能である。
[0107] 更に本発明においては、得られた L—ホスフィノスリシンを含む混合液より、精製ェ 程を経て、 L—ホスフィノスリシンを純ィ匕することが可能である。この場合の精製方法 は、当該分野で通常用いられている方法は全て適用可能である力 イオン交換クロ マトグラフィーゃ溶媒 (例えばメタノールなど)による沈澱法を使用することが望ま ヽ 。本発明で得られる変換液はこれらの精製法により容易に純ィ匕可能であるため、変 換液に微量のァラニンが存在しても特に問題とはならない。
[0108] 式 (I)の化合物、式 (Π)の化合物、グルタミン酸、およびァスパラギン酸はそれぞれ
塩として存在することができる。
[0109] 式 (I)の化合物の塩としては、ナトリウム塩、カリウム塩、アンモ-ゥム塩等の無機塩 基との塩の他、有機塩基、無機酸、有機酸との塩が挙げられるが、好ましくはナトリウ ム塩である。
[0110] 式 (Π)の化合物の塩としては、ナトリウム塩、カリウム塩、アンモ-ゥム塩等の無機塩 基との塩の他、有機塩基との塩が挙げられる力 好ましくはナトリウム塩である。
[0111] グルタミン酸の塩としては、ナトリウム塩、カリウム塩、アンモ-ゥム塩等の無機塩基 との塩の他、有機塩基、無機酸、有機酸との塩が挙げられるが、好ましくはナトリウム 塩である。
[0112] ァスパラギン酸の塩としては、ナトリウム塩、カリウム塩、アンモ-ゥム塩等の無機塩 基との塩の他、有機塩基、無機酸、有機酸との塩が挙げられるが、好ましくはナトリウ ム塩である。
実施例
[0113] 以下に、本発明を実施例により具体的に説明するが、本発明はこれに限定されるも のではない。
[0114] 実施例 1: StreDtomvces hvgroscopicus由来の AAT遣伝子のクローニング
(1) PCR法による長鎖プローブの作製
a; Streptomvces hygroscopicusのゲノム DN Aの! ^製
StreDtomvces hveroscopicus (SF1293 NP— 50株、 FERM BP— 1368)を SP Y培地(2%でんぷん、 1%ポリペプトン、 0. 5%イーストエキストラタト、 0. 05%KH P
2
O : pH7. 0)に植菌し、 28°Cで 24時間振とう培養した。得られた培養液力も遠心分
4
離により菌体を回収し、凍結乾燥した。この凍結乾燥菌体を WO00Z24879号公報 の実施例 B2記載の方法により処理することにより、ゲノム DNAを調製した。
[0115] b) PCR法による AAT遺伝子部分断片の増幅
公知のァータベース (DDBJホームページ httD://www.ddbi.nig.ac.ip/Welcome-i.h tmlで利用可能な公共データベース)より得られる 2種類の放線菌 (StreDtomvces giniaeおよび StreDtomvces coelicolor)由来の AATのアミノ酸配列を比較し、ホモロジ 一のある領域を検索した。その結果、相同性のある 2ケ所のアミノ酸配列(GEPDFP
TPおよび KTYAMTGWRVG)を特定し、この部分に対応する 2種類の合成オリゴ ヌクレオチドプライマーを作製した。
SV-GOT-N :
5, - GGCGAGCCCGACTTCCCGACCCCG - 3 ' (配列番号: 6)
5,— CCCACGCGCCAGCCGGTCATGGCGTACGTCTT— 3,(配列番号: 7
)
[0116] このプライマーを使用して、上述の Streptomvces hvgroscopicusのゲノム DNAを鎳 型に PCRを実施した。 PCRには、 TaKaRa LA Taq with GC buffer (タカラ バイオ社製)を使用し、 94°C ' l分間の熱変性の後、 98°C ' 30秒間、 60°C ' 30秒間、 72°C ' 1分 15秒間のサイクルを 30回繰り返した後、 72°Cで更に 10分間インキュベー トした。反応終了後、サンプルを 0. 8%ァガロース電気泳動に供し解析したところ、約 0. 6kbpの特異的なバンドの増幅が確認された。
[0117] c)増幅した遺伝子断片のサブクローユング
実施例 1 (1) b)で得られた約 0. 6kbpの遺伝子バンドをゲルから回収した後、 Wiza rd SV Gel and PCR Clean-up System (プロメガ社製)により DN Aを精製 し、 TOPO TAクロー-ングキット(インビトロジェン社製)および E. coli competen t cells JM109 (タカラバィォ社製)を使用してサブクローンした。得られたプラスミド を pSH-AATとした。プラスミド pSH—AATの挿入断片の塩基配列を解析したとこ ろ、 Streptomvces virriniae由来の AATをコードする DNA配列 の相同件が確認 れた (塩基配列の解析装置は、アプライドバイォシステム社製の ABI PRISM310 Genetic Analyzerを使用した。シーケンス反応は、同社の DNA sequencing K it であ dRhodamme Terminator Cycle Sequencing Ready Reaction 用いた。解析用のプライマーは、キット添付の M13プライマーを使用した)。よって、 本揷入断片を Streptomvces hveroscopicus由来の AAT遣伝子の部分断片であると 判断し、以後の実験のプローブとして用いた。
[0118] (2) AAT遺伝子のクローユング
a)ゲノム DNAのライブラリーの構築
実施例 l (l) a)で得られたゲノムDNAをSau3AIで消化した後、 0. 8%ァガロース
ゲル電気泳動に供し、 9〜23kbpの DNA断片をゲルから回収した。回収断片をフエ ノール処理し、エタノール沈殿により精製した後、 EMBL3ZBamHI Vector K it (ストラタジーン社製)、および DNA Ligation Kit ver. 2 (タカラバィォ社製)を 用いて、部分消化したゲノム DNA断片と EMBL3ベクターを連結した。得られた li gation mixtureを Maxplax Packaging Extract ェピセンター社製) 用 ヽて ノ ッケージングした後、大腸菌 XL1— Blue MRA株に感染させた。この方法により 得られた 2. 0 X 105個のファージライブラリーを用いて、 目的遺伝子のクローユングを 行った。
[0119] b)ライブラリーのスクリーニング
実施例 l (l) c)で得られたプラスミド pSH— AATを EcoRIで消化した後、 0. 8%ァ ガロースゲル電気泳動に供し、約 0. 6kbpの DNA断片をゲルから回収した。回収し た DNA断片を Wizard SV Gel and PCR Clean-up System (プロメガ社製 )を用いて精製し、 DIG High Prime DNA Labeling and Detection Start er Kit I (ロシュ'ダイァグノスティックス社製)を用いてプローブを DIG標識した。
[0120] 次に、実施例 l (2) a)で得られたファージラィブラリーをHybond—N+メンブラン( アマシャムバイオサイエンス社製)に写し取り、 0. 5N水酸ィ匕ナトリウムで変性後、 5 X SSC (l X SSc : 15mMクェン酸 3ナトリウム、 150mM塩化ナトリウム)で洗浄し、乾燥 させて DNAを固定した。前述キットの記載に従って、 1時間のプレハイブリダィゼー シヨン (42°C)の後、熱変性した標識ィ匕プローブを添加し、 16時間ハイブリダィゼーシ ヨン (42°C)を行った。ラベルの洗浄についてもキット記載の方法に従い、まず、 0. 1 %SDSを含む 2 X SSCで、室温で 5分間 X 2回洗浄した後、 0. 1%SDSを含む 0. 5 X SSCで、 65°Cで 15分間 X 2回洗浄した。更に、キット記載の方法に従い、アルカリ ホスファターゼ標識抗 DIG抗体を反応させた後、キット添付の基質 (NBTZBCIP) を添加した。結果、青色を呈する 2個の陽性クローンが得られた。
[0121] c)ファージ DNAの調製
陽性のファージを、前述 λ EMBL3ZBamHI Vector Kit記載の方法に従い大 腸菌 XL1— Blue MRA株に感染させた後、 16時間後にファージ粒子を回収し、 Gr ossbergerの方法(Grossberger, D., Nucleic Acids. Res., 15:6737, 1987)に従い、プ
ロティナーゼ Kおよびフエノール処理後、エタノール沈殿によりファージ DNAを調製 した。
[0122] d)サザンハイブリダィゼーシヨン
実施例 1 (1) a)で得られたゲノム DNAおよび、実施例 1 (2) c)で得られたファージ DNAを複数の制限酵素でそれぞれ消化した後、 0. 8%ァガロースゲル電気泳動に 供した。 DNAを Southenの方法(Southern, E. M., J. Mol. Biol, 98:503-517,1975) により、 Hybond— N+メンブランに写し取った後、 ECF Random -Prime Label ling and Detection System (アマシャムバイオサイエンス社製)を用いてハイブ リダィゼーシヨンを実施した。本システム記載の方法により、 30分間のプレハイブリダ ィゼーシヨン (60°C)の後、熱変性した標識ィ匕プローブを添加し、 16時間ノ、イブリダイ ゼーシヨン(60°C)を行った。プローブは、実施例 l (2) b)で得られた pSH— AAT由 来の約 0. 6kbpの DNA断片を、本システム記載の方法によりフルォレセイン標識し て用いた。ラベルの洗浄についても本システム記載の方法に従い、まず、 0. 1%SD Sを含む 1 X SSCで、 60。Cで 15分間洗浄した後、 0. 1%SDSを含む 0. 5 X SSCで 、 60°Cで 15分間洗浄した。更に、本システム記載の方法に従い、アルカリホスファタ ーゼ標識抗フルォレセイン抗体を反応させた後、本システム添付の基質 (ECF sub strate)を添加し、得られた蛍光バンドをモレキュラーイメージヤー FX (バイオ 'ラッド ラボラトリーズ社製)で検出した。その結果、ゲノム DNAおよびファージ DNAを Ba mHIで処理した場合に約 4. 3kbpの共通のバンド力 Pstlで処理した場合に約 2. 7 kbpの共通のバンド力 BamHIと Xholで処理した場合に約 1. 5kbpの共通のバンド がそれぞれ得られることが明らカゝとなった。
[0123] e)目的遺伝子のサブクローユング
まず、実施例 1 (2) c)で得られたファージ DNAを制限酵素 BamHIで消化した後、 0. 8%ァガロースゲル電気泳動に供し、約 4. 3kbpの DNA断片をゲルから回収、精 製した。得られた DNA断片を、 pUC118 BamHI— BAP処理 DNA (タカラバイオ 社製)に連結し、得られたプラスミドを pi 18G— 411Bとした。同様にファージ DNAを 制限酵素 Pstlで処理し、約 2. 7kbpの DNA断片をゲルから回収した後、 pUC118 Pstl - BAP処理 DNAに連結し、得られたプラスミドを p 118G— 411 Pとした。
[0124] 得られたプラスミド pi 18G— 41 IPを制限酵素 Pstlで消化した後、 0. 8%ァガロー スゲル電気泳動に供し、約 2. 7kbpの DNA断片をゲルから回収、精製した。次に、 プラスミド pi 18G— 411Bを制限酵素 Pstlで消化した後、 0. 8%ァガロースゲル電 気泳動に供し、約 6. 2kbpの DNA断片をゲルから回収、精製した。得られた DNA 断片をアルカリホスファターゼ (BAP;タカラバィォ社製)処理により脱リン酸ィ匕した後 、先に得られた pi 18G— 411P由来の約 2. 7kbpの DNA断片と連結した。得られた プラスミドを PAAT3 - 5とした。
[0125] 得られたプラスミド pAAT3— 5を制限酵素 HindIII、 Xholで消化した後、 0. 8%ァ ガロースゲル電気泳動に供し、約 2. 9kbpの DNA断片をゲルから回収、精製した。 次に、プラスミド pUC19 (タカラバィォ社製)を制限酵素 HindIII、 Sailで消化した後 、約 2. 7kbpの DNA断片をゲルから回収、精製し、先に得られたプラスミド pAAT3 5由来の約 2. 9kbpの Hindlll—Xhol断片と連結した。得られたプラスミドを pHX —01とした。
[0126] Streptomvces hvgroscodcus由来の AAT遣伝子を含む約 5. 8Kbpの制限酵素地 図を図 1に示す。
[0127] 実施例 2 : StreDtomvces hvgroscopicus由来の AAT遣伝子の塩某配列解析
(1)プラスミド pHX— 01の塩基配列の解析
a)シーケンス反応
シーケンス反応は、 BigDye Terminator V3. 1 Cycle Sequencing Kit (ァ プライドバイオシステム社製)を用いて実施した。反応には、铸型 DNAとしてプラスミ ド pHX— 01を、プライマーとしては下記の配列を有する合成オリゴヌクレオチドを使 用した。
Ml 3— 20 : 5, - CGACGTTGTAAAACGACGGCCAGT- 3 ' (配列番号: 8)
M13 -R : 5,— GGAAACAGCTATG ACCATGATTAC - 3 ' (配列番号: 9) [0128] 付属のマ-ユアルに従い反応液を調製後、サーマルサイクラ一に使用して 96°C ' l
0秒間、 50°C ' 5秒間、 60°C '4分間のサイクルを 30回繰り返すことによりシーケンス 反応を実施した。
[0129] b)シーケンス解析
実施例 2 (1) a)で得られた反応液から、余剰の蛍光色素およびプライマーをエタ一 ノール沈殿法により除去した後、サンプルを 3730x1 DNA Analyzer (アプライドバ ィォシステム社製)に供した。得られた塩基配列データを基に新規のプライマーを作 製し、シーケンス反応 ·解析を繰り返すことにより、全長を両方向でカバーする配列デ ータを取得した。最後に Sequencher (ジーンコード社製)を使用して得られた配列 データのアセンブルを実施し、塩基配列を確定した (配列番号: 10)。
[0130] (2)公共のデータベースを利用した相同性解析
実施例 2 (1) b)で決定した配列を用いて、公共のデータベース(DDBJホームべ一 ジ http://www.ddbi.nig.ac.ip/Welcome-i.htmlで利用可能な公共データベース)上 の配列との相同性検索を実施した。検索プログラムとして blastx (DNA配列 Xァミノ 酸配列 DB)を使用し、フィルター OFFの条件で枪索を行った結菓、 Streptomvces ce licolorの AAT遣伝子 87 %、 Streptomvces virginiaeの AAT遣伝子 88%の同一件 をそれぞれ示した。
[0131] 実施例 3 : StreDtomvces hvgroscopicus由来の AAT遣伝子の高発現
(1)発現ベクター pLG04、 pSGl lの構築
発現ベクター PLG04は図 2に示すようにして構築した。また、発現ベクター pSGl l は図 3に示すように構築した。
[0132] a)プラスミド pSYTL03の構築
Katz, E., J. Gen. Microbiol, 129, 2703-2714, 1983に記載のプラスミド p!J702を制 限酵素 pstI、 Saclで切断し、 0. 8%ァガロースゲル電気泳動に供した後、約 4. 9kb pの DNA断片をゲルから回収、精製した。次に、 pUC 19 (タカラバィォ社製)も同様 に Pstl、 Saclで切断し、 0. 8%ァガロースゲル電気泳動に供した後、 DNA断片をゲ ルから回収、精製した。得られた 2種類の DNA断片を DNA Ligation Kit ver. 2 (タカラバイオ社製)を用いて連結し、プラスミド PSYTL03を構築した(図 2)。
[0133] b)発現ベクター pLG04の構築
まず、実施例 3 (1) &)で得られたプラスミド 3丫1^03を制限酵素1¾11(1111、 EcoRI で切断し、 0. 8%ァガロースゲル電気泳動に供した後、約 4. 9kbpの DNA断片をゲ ルから回収し、 DNAを精製した。次に、実施例 1 (2) e)で得られたプラスミド pHXOl
も同様に制限酵素 HindIII、 EcoRIで切断し、電気泳動後、約 2. 9kbpの DNA断片 をゲルから回収し、精製した。これら 2種類の DNA断片を DNA Ligation Kit ve r. 2 (タカラバィォ社製)を用いて連結し、得られた ligation mixtureを放線菌^ ^ tomvces lividansに形 換した。 Streptomvces lividansの は、 Thompson の方法(Thompson, C. J., J. BactrioL, 151:668-677, 1982)に従い実施した。 Strepto mvces lividansプロトプラスト、 ligation mixture, T培地を混合した後、 20%ポリエ チレングリコール 1000を添カ卩して DNAを導入し、 R2YE agarプレート(20ml培地 Zプレート)上で 30°C、ー晚培養した。その後、 100 gZmlのチォストレプトン 2ml をプレート一枚に重層した (チォストレプトン終濃度 10 gZml)。 30°Cで更に数日 間培養し、形成したコロニーを形質転換体とした。得られた形質転換体を 10 g,m 1のチォストレプトンを含む 80ml YEME培地で 30°C、 2日間、振とう培養した後、培 養液からプラスミド DNAを調製した。 Streptomvces lividans形皙転椽体からのプラスミ ド DNAの調製は、 QIAfilter Plasmid Midi Kit (キアゲン社製)を使用し、キット 添付の説明書に従い実施した。ただし、菌体の溶菌を促進させるため、キット添付の buffer PIに終濃度 lOmgZmlになるようリゾチームを添カ卩し、本溶液で菌体を室 温で 30分、プレインキュペートするという前処理工程を追加して実施した。培養液 20 mUり DNAを回収し、最終的に 50 /z lの TE bufferに溶解した。複数の形質転換 体を処理し、得られたプラスミド DNAを制限酵素 HindIII、 EcoRIで切断後、 0. 8% ァガロース電気泳動に供し、切断パターンを確認した。約 2. 9kbpと約 4. 9kbpのバ ンドを呈するプラスミド DNAを目的の DNAとして選択し、これを発現ベクター pLGO 4とした(図 2)。
[0134] c)発現ベクター pSGl lの構築
実施例 1 (2) e)で得られたプラスミド pHXOlを制限酵素 BamHIで切断し、電気泳 動後、約 1. 5kbpの DNA断片をゲルから回収し、精製した。この DNA断片を pUCl 18 BamHI— BAP処理 DNA (タカラバイオ社製)に連結し、得られたプラスミドを pi 18— G1とした(図 3)。
[0135] このプラスミ pl l8— G1を制限酵素 HindIII、 EcoRIで切断し、電気泳動後、約 1.
5kbpの DNA断片をゲルから回収し、精製した。また、実施例 3 (1) a)で得られたプ
ラスミド PSYTL03も同様に制限酵素 HindIII、 EcoRIで切断し、電気泳動後、約 4. 9kbpの DNA断片をゲルから回収し、精製した。これら 2種類の DNA断片を DNA Ligation Kit ver. 2を用いて連結し、得られた ligation mixtureを実施例 3 (1) b)の方法に従 ヽ放線菌 StreDtomvces I feに形質転換した。得られた形質転換体 を 10 μ gZmlのチォストレプトンを含む 80mlYEME培地で 30°C、 2日間、振とう培 養した後、培養液力も実施例 3 (1) b)の方法に従い、プラスミド DNAを調製した。得 られたプラスミド DNAを制限酵素 HindIII、 EcoRIで切断後、 0. 8%ァガロース電気 泳動に供し、切断パターンを確認した。約 1. 5kbpと約 4. 9kbpのバンドを呈するプ ラスミド DNAを目的の DNAとして選択し、これを発現ベクター pSG 11とした(図 3)。
[0136] (2)発現ベクター pLG04、 pSGl lによる StreDtomvces hvgroscopicusの形質転換 a)プロトプラストの調製
StreDtomvces hvgroscopicus (SF1293 NP— 50株、 FERM BP— 1368)の菌株 ストック(凍結乾燥菌体)を 10mlの SI培地(2%でんぷん、 1%ポリペプトン、 0. 3% モノレトエキストラタト、 0. 05%K ΗΡΟ ; ρΗ7. 0)に植菌し、 28°Cで 40時間、振とう
2 4
培養した。得られた培養液のうち 2mlを、 5%グリシンを含む 80mlの S2培地(1%グ ルコース、 0. 4%ペプトン、 0. 4%イーストエキス卜ラタ卜、 0. 05%MgSO · 7Η 0、 0
4 2
. 2%KH PO、 0. 4%K HP04 ;pH7. 0)に移植し、 28°Cで 18時間、振とう培養し
2 4 2
た。得られた培養液力も遠心分離により菌体を集め、 0. 5M シュクロースで洗浄し た後、終濃度 2. 5mgZmlのリゾチームと終濃度 1. 25mgZmlのァクロモぺプチダ ーゼを含む P3培地(70mM NaCl、0. 5M シュクロース、 5mM MgCl · 6Η 0、
2 2
5mM CaCl - 2H 0、 25mM TES buffer ;pH7. 2)に懸濁し、 30°Cで一時間
2 2
振とうし、溶菌させた。生じたプロトプラストを P3培地で洗浄した後、約 500 1の P3 培地に懸濁し、これをプロトプラスト溶液とした。
[0137] b)形質転換
b)— 1 培地の調製
形質転換の際に使用する TH培地は以下の方法で調製した。まず、 Okanishiの方 法(Okanishi, M., J. Gen. Microbiol, 80, 389—400, 1974)【こ従!ヽ、 Trace element solutionを調製した。次に、シユークロース 26. 7g、 K SO 0. 0375gを約 60m
1に溶解し、 Trace element solution 0. 3mlをカ卩えた後、水で 77. 5mlにメスァ ップし、これを 3/2TH培地とした。この 3/2TH培地 7. 75mlに、 2M CaCl 0
2
. 75mlと 0. 5M Tris— maleic acid buffer (pH8. 0) 1. 5mlを加え、これを TH 培地とした。
[0138] プロトプラスト再生用の RME培地は以下の方法で調製した。まず、プロリン 2g、グ ノレコース 10g、イーストエキストラタト 2g、カザミノ酸 2g、シユークロース 174g、 KC1 15g、K SO 0. 25g、 DEXTRAN SULFATE 0. 05g, LAB DEMC
2 4
O lgを水に溶解し、 10% NaOHで pH7. 2に調整し、水で 500mlにメスアップし 、これを 2 XRME培地とした。 2 XRME培地 500ml、 0. 5%KH PO 10ml, 1M
2 4
CaC12 50ml、 3%コーンスティープリカ一(NaOHで pH7. 0に調整) 100ml、 5% ァスパラギン酸ナトリウム(NaOHで pH7. 0に調整) 10ml、 3. 03%ァガー 330ml を別々に殺菌後、混合しこれを RME培地 (ァガー終濃度 1%)とした。重層用の R MEソフトァガー培地は、 RME培地のァガー終濃度を 0. 5%に変更したものを用い た。
[0139] b) - 2 形質転換体の作出
実施例 3 (1) b)で得られたベクター pLG04および実施例 3 (1) c)で得られたベクタ 一 pSGl lの DNA溶液各 25 μ 1、 TE buffer 25 μ 1に実施例 3 (2) b) - 1で得られ た TH培地 100 μ 1をカロえよく混合した後、実施例 3 (2) a)で得られたプロトプラスト溶 液 100 1を加え、緩やかに混合した。この DNAZプロトプラスト混合液に PEG sol ution(3g ポリエチレングリコール 1000、 6ml TH培地) 375 1をカ卩え、約 1分間 混合した後、適当量(10 μ 1-100 μ 1)を RME培地(約 20mlZプレート)にプレーテ イングし、さらに RMEソフトァガー培地を重層した (約 2mlZプレート)。 28°Cで約 2日 間培養した後、 100 gZmlのチォストレプトン溶液 2mlをプレートに添カ卩し塗布した 。さらに 28°Cで 7〜10日間培養し、得られたコロニーを形質転換体とした。
[0140] (3)発現ベクター pLG04および pSGl 1による形質転換体の評価
a)形質転換体の培養
実施例 3 (2) b) 2で得られたコロニーを 10 μ gZmlのチォストレプトンを含む 10 mlの SPY培地に植菌し、 28°Cで 2日間振とう培養した。得られた培養液のうち 2mlを
10 μ g/mlのチォストレプトンを含む 30mlの SPY培地に植菌し、 28°Cで 2日間、更 に振とう培養した。
[0141] b)形質転換体の評価
実施例 3 (3) a)で得られた培養液力 遠心分離により菌体を回収し、 0. 9%食塩水 で洗浄した後、菌体を 80°Cで凍結した。凍結菌体を 20mlの buffer A2 (20mM リン酸バッファー、 0. ImM ピリドキサルリン酸、 5mM 2 メルカプトエタノール、
ImM フエ-ルメチルスルホ -ルフルオリド; pH7. 0)に溶解、懸濁し、菌体を超音 波により破砕した。破砕液力も遠心分離により細胞断片を除去し、上清を粗酵素液と した。得られた粗酵素液を用いて、 Tanakaらの方法(Tanaka, T., Agric. Biol. Chem.
, 54:625-631, 1990)に従い、 37°C、 ρΗ7. 5における ΑΑΤ活性を測定した。
[0142] その結果、 pLG04形質転換体の ΑΑΤ活性に対して pSGl l形質転換体の ΑΑΤ 活性が約 5. 4倍と顕著に高活性であったことから (表 1)、 pSGl l形質転換体を AA
T高発現株として以降の実験に用いた。
[表 1]
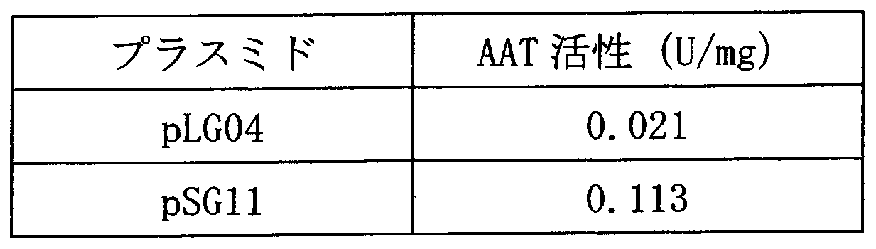
[0143] (4)発現ベクター pSGl 1による形質転換体の評価
a)形質転換体の培養
実施例 3 (2) b) - 2で得られた発現ベクター pSGl 1による形質転換体の菌株ストツ ク(凍結乾燥菌体)を 10 μ gZmlのチォストレプトンを含む 10mlの SPY培地に植菌 し、 28°Cで 2日間振とう培養した。得られた培養液のうち 2mlを 10 g/mlのチォスト レプトンを含む 30mlの P— 101培地(7%グルコース、 4. 4%ソィトン、 0. 327%KH PO、 0. 085%Na HPO、 1. 15% TES、 0. 0001%CoCl · 6Η 0 ;pH6. 0)
2 4 2 4 2 2
に移植し、 28°Cで 4日間振とう培養した。対照として親株 (プラスミドなし)および特開 平 2— 195889号公報記載のプラスミド pMSB515による形質転換体も同様の工程 で培養した (ただし、親株はチォストレプトンを含まな 、培地で培養した)。
[0144] b)酵素活性の測定
b)— 1 粗酵素液の調製
実施例 3 (4) a)で得られた培養液力 遠心分離により菌体を回収し、 0. 9%食塩水 で洗浄した後、菌体を—80°Cで凍結した。凍結菌体を 20mlの buffer A2に溶解、 懸濁し、菌体を超音波により破砕した。破砕液力 遠心分離により細胞断片を除去し 、上清を粗酵素液とした。得られた粗酵素液を用いて、 AATおよび PAT活性を測定 した。
[0145] b) - 2 PAT活性および AAT活性の測定
PAT活性の測定は、 Schulzの方法(Schulz, A., Appl. Environ. Microbiol, 56, 1- 6, 1990)に従って実施した。 PAT活性測定用基質(150mM グルタミン酸、 50mM
OMPB、0. ImM ピリドキサルリン酸、 lOOmM Tris— HC1 buffer ; pH8. 5)と 粗酵素液を混合し、 37°C、 20分間インキュベートした後、沸騰水浴中で 5分間加熱 することにより反応を停止した。反応停止後、サンプルを 0. 45 mのフィルタ一によ りろ過し、アミノ酸分析用 HPLC (model LC— VP、島津製作所社製)に供し、生成 した L—ホスフィノスリシン濃度を測定した。酵素力は、 1分間に l /z molの Lーホスフ ィノスリシンを生成する能力を 1U (ユニット)として定義した。粗酵素液中のタンパク質 濃度をプロテインアツセィキット (バイオラッド社製)を用いて γ —グロブリンをスタンダ ードとして測定し、粗酵素液の比活性 (U/mg)を求めた。
[0146] AAT活性の測定は、以下の方法で実施した。 AAT活性測定用基質(150mM ァ スパラギン酸、 50mM aーケトグルタル酸、 0. ImM ピリドキサルリン酸、 lOOmM
Tris -HCl buffer ; pH8. 5)と粗酵素液を混合し、 37°C、 20分間インキュベート した後、沸騰水浴中で 5分間加熱することにより反応を停止した。反応停止後、サン プルを 0. 45 mのフィルターによりろ過し、アミノ酸分析用 HPLC (model LC— V P、島津製作所社製)に供し、生成したグルタミン酸濃度を測定した。酵素力は、 1分 間に 1 μ molのグルタミン酸を生成する能力を 1U (ユニット)として定義した。粗酵素 液中のタンパク質濃度を上述のプロテインアツセィキットで測定し、粗酵素液の比活 '性 (U/mg)を求めた。
[0147] その結果、 pSG l lの組換え体の PAT活性は親株とほとんど変わらな力つた力 A AT活性は親株の約 47倍であった (表 2)。
[表 2]

[0148] ¾施例 4 : AAT遣伝早の高 現株による L-ホスフィノスリシンの牛産
(1)発現ベクター pSGl 1による形質転換体の培養
実施例 3で得られた pSGl 1による形質転換体の菌株ストック (凍結乾燥菌体)を 10 μ gZmlのチォストレプトンを含む 30mlの SPY培地に植菌し、 28°Cで 2日間振とう 培養した。得られた培養液のうち 2mlを 10 μ gZmlのチォストレプトンを含む 40mlの SPY培地に移植し、 28°Cで 2日間振とう培養した。得られた培養液のうち 20mlを 10 μ gZmlのチォストレプトンを含む 400mlの SBK培地(2%でんぷん、 3%脱脂大豆 かす、 0. 05%KH PO ; pH7. 0)に移植し、 28°Cで 2日間振とう培養した。得られた
2 4
培養液のうち 5mlを 4Lの SBK培地に移植し、 28°Cで 2日間、通気攪拌培養した。得 られた培養液のうち 400mlを 4Lの P2培地(3%グルコース、 3%脱脂大豆かす、 1% グルテンミール、 0. 001%CoCl · 6Η 0、 0. 01%MgSO · 7Η 0、 0. 013%CaCl
2 2 4 2
•2H 0 ;pH7. 0)に移植し、 28°Cで 4日間通気攪拌培養した。得られた培養液を用
2 2
いて、下記の条件で微生物変換を実施し、 L ホスフィノスリシンの生産を行った。尚
、対照として親株 (プラスミドなし)およびプラスミド PMSB515による形質転換体も同 様の工程で培養し (ただし、親株はチォストレプトンを含まない培地で培養した)、同 様に変換反応を行った。
[0149] (2)微生物変換による L ホスフィノスリシンの生産
a)基質溶液 Aの調製
まず、 OMPB、ァスパラギン酸、グルタミン酸を各 66g測りとり、約 1Lの水に懸濁し た。次に、 25%水酸ィ匕ナトリウム水溶液で pH8. 5に調製し、水で 1760mlにメスアツ プし、これを基質溶液 Aとした。
[0150] b)基質溶液 Bの調製
まず、 OMPB、ァスパラギン酸を各 66g、グルタミン酸を各 16. 5g測りとり、約 1Lの 水に懸濁した。次に、 25%水酸ィ匕ナトリウム水溶液で pH8. 5に調製し、水で 1760m
1にメスアップし、これを基質溶液 Bとした。
[0151] c)微生物変換
実施例 4 (1)で得られた培養液 440mlと実施例 4 (2)の a)または b)でそれぞれ得ら れた基質溶液 Aまたは B1760mlを混合し反応を開始した。変換条件は、温度 37°C 、 pH8. 5 (水酸化ナトリウムで調整)とし、攪拌しながら 3日間 (約 70時間)反応を実 施した。変換終了後、サンプルを 0. 45 mのフィルターによりろ過し、アミノ酸分析 用 HPLC (model LC— VP、島津製作所社製)に供し、生成された L—ホスフィノス リシンの濃度を測定した。また、変換終了時の変換液量も正確に測定し、それらの積 カゝら生産された L -ホスフィノスリシン量を求めた。さらに使用した OMPBと生産され た L—ホスフィノスリシンのモル比を求めることにより、各培養液における L—ホスフイノ スリシンへの変換率を算出した。
[0152] その結果、 AAT高発現株は、親株や PMSB515による形質転換体に比べ、グルタ ミン酸濃度を低減した基質溶液 Bにお 、ても、顕著に高!ヽ変換率を示した (表 3)。
[表 3]

[0153] 実施例 5 :AAT遣伝子と PAT遣伝子の共発現
(1)発現ベクター pAHSG7201の構築
プラスミド pATSGOlは図 4に示すようにして構築した。また、発現ベクター pAHSG 7201は図 5に示すようにして構築した。
[0154] a)プラスミド ρΑΤΠ515— 03の構築
PAT遺伝子は、 PATの一種である AT— II (特開平 2— 195889号公報参照)の遺 伝子を使用した。まず、特開平 2— 195889号公報記載のプラスミド pMSB515より、 PATの一種である八丁ー11の遺伝子を制限酵素3&01 (351;1)で切断し、 0. 8%ァガロ ースゲル電気泳動に供した後、約 4. 7kbpの DNA断片をゲルから回収し、 DNAを
精製した。次に、 pUC18 (タカラノィォ社製)も同様に Saclで切断し、 0. 8%ァガロ ースゲル電気泳動に供した後、 DNA断片をゲルから回収し、 DNAを精製した。 pU C18の Sacl断片をアルカリホスファターゼ(BAP ;タカラバィォ社製)により処理した 後、 pMSB515由来の約 4. 7kbpの Sacl断片と連結した。得られたプラスミドを pAT Π515— 03とした(図 4)。
[0155] b)プラスミド pATSGOlの構築
まず、実施例 1 (2) e)で得られたプラスミド pHX— 01を制限酵素 BamHIで切断し、 0. 8%ァガロースゲル電気泳動に供した後、約 1. 5kbpの DNA断片をゲルから回 収し、 DNAを精製した。次に、実施例 5 (1) a)で得られたプラスミド ρΑΤΠ515— 03 を制限酵素 BamHIで切断し、 0. 8%ァガロースゲル電気泳動に供した後、約 7. 4k bpの DNA断片をゲルから回収し、 DNAを精製した。 ρΑΤΠ515— 03の BamHI断 片をアルカリホスファターゼ(BAP)により処理した後、 pHX— 01由来の約 1. 5kbp の BamHI断片と連結した。得られたプラスミドを pATSGOlとした(図 4)。
[0156] c)発現ベクター pAHSG7201の構築
まず、実施例 3 (1) &)で得られたプラスミド 3丫1^03を制限酵素1¾11(1111、 EcoRI で切断し、 0. 8%ァガロースゲル電気泳動に供した後、約 4. 9kbpの DNA断片をゲ ルから回収し、 DNAを精製した。次に、実施例 5 (1) b)で得られたプラスミド pATSG 01も同様に制限酵素 HindIII、 EcoRIで切断し、電気泳動後、約 5. 6kbpの DNA 断片をゲルから回収し、精製した。これら 2種類の DNA断片を DNA Ligation Kit ver. 2を用いて連結し、得られた ligation mixtureを実施例 3 (l) b)と同様の方 法で放線菌 StreDtomvces I teに形質転換した。得られた形質転換体を 10 μ g, mlのチォストレプトンを含む 80ml YEME培地で 30°C、 2日間、振とう培養した後、 培養液力もプラスミド DNAを調製した。得られたプラスミド DNAを制限酵素 Hindlll 、 EcoRIで切断後. 0. 8%ァガロース電気泳動に供し、切断パターンを確認した。約 4. 9kbpと約 5. 6kbpのバンドを呈するプラスミド DNAを目的の DNAとして選択し、 これを発現ベクター PAHSG7201とした(図 5)。
[0157] (2)発現ベクター OAHSG7201による StreDtomvces hvgroscopicusの形質転換と开 質転換体の評価
a)発現ベクター pAHSG7201による形質転換の作出と形質転換体の培養 実施例 5 (1) c)で得られた発現ベクター pAHSG7201により、 Streptomvces hveros coEicus (SF1293 NP— 50株、 FERM BP— 1368)を形質転換した。実施例 3 (2 )の方法に従い形質転換を実施し、得られた形質転換体を培養した。実施例 3 (4) a) と同様の工程で培養を行い、 10 g/mlのチォストレプトンを含む P— 101培地で 2 8°C、 4日間振とう培養した培養液を得た。得られた培養液より菌体を回収し、実施例 3 (4) b)— 1の方法に従い粗酵素液を調製した。
[0158] b)発現ベクター pAHSG7201による形質転換体の評価
実施例 5 (2) a)で得られた発現ベクター pAHSG7201による形質転換体由来の粗 酵素液を用いて、 PAT活性および AAT活性を測定した。酵素活性の測定は、実施 例 3 (4) b)一 2の方法に従った (対照として、実施例 3 (4) b)一 2の結果を併記した)。
[0159] その結果、 PAHSG7201による形質転換体の PAT活性は親株の約 16倍、 AAT 活性は親株の約 170倍であった (表 4)。
[表 4]

[0160] 実施例 6: AAT遣伝子と PAT遣伝子の共発現株による L -ホスフィノスリシンの牛産
( 1 )発現ベクター pAHSG7201による形質転換体の培養
実施例 5 (2)で得られた発現ベクター pAHSG 7201による形質転換体を、実施例 4 (1)と同様の工程で培養した。 P— 2培地で 28°C、 4日の通気攪拌間培養を実施し、 得られた培養液を用いて L -ホスフィノスリシンの生産を行った。
[0161] (2)発現ベクター pAHSG7201による形質転換体の培養
実施例 6 (1)で得られた発現ベクター pAHSG7201による形質転換体の培養液 44 Omlと実施例 4 (2)の方法に従って調製した基質溶液 Aまたは B1760mlを混合し、 3 7°C、 pH8. 5で 3日間 (約 70時間)反応した。得られた変換液を実施例 4 (2)と同様 の方法で分析し、 L—ホスフィノスリシンの生産量を求めた (対照として、実施例 4 (2)
c)の結果を併記した)。
その結果、グルタミン酸を低減した基質溶液 Bにおいても、発現ベクター pAHSG7 201による形質転換体 (すなわち、 AAT'PAT共発現株)は他の株に比べ、顕著に 高い変換率を示した (表 5)。
[表 5]

実施例 7 :AAT遣伝子と PAT遣伝子の共発現株における、高発現タンパク質の N末 端アミノ酸 §R列の同定
発現ベクター PAHSG7201による形質転換体にぉ 、て高発現したタンパク質の N 末端アミノ酸配列を決定するため、特許第 3593134号公報の実施例 A2記載の方 法に従い、 N末端アミノ酸のシーケンスを実施した。まず、実施例 5 (2) c)で得られた 発現ベクター PAHSG7201による形質転換体の粗酵素液を、 SDS— PAGE mini
12% Gel (テフコネ土製)を用いて電気泳動した後、マルチフォー II電気泳動装置( アマシャムバイオサイエンス社製)によりタンパク質を PVDF膜 (ミリポア社製)に写し 取った。次にこの PVDF膜をコマジ一ブリリアントブルー R— 250 (ナカライテスタ社製 )で染色し、脱色した後、水で洗浄し風乾した。ここから 50kDaと 43kDaのタンパク質 がブロットされた部分を切り出し、それぞれをプロテインシーケンサー Model492 (パ 一キンエルマ一社製)に供し、下記のように N末端アミノ酸配列を決定した。
43kDaタンパク質:
Ser— Ala Ala Thr Pro— Ser— Ala Ser (配列番号:11)
50kDaタンパク質:
Thr - Glu - Leu— Ser— Gly— Ala - Pro - Ala (配列番号: 12)
43kDaタンパク質の N末端アミノ酸配列は配列番号: 2の第 2番目〜第 9番目と一 致した。従って、 43kDaタンパク質は、 AATタンパク質であることが明らかとなった。 また、 50kDaタンパク質の N末端アミノ酸配列は配列番号: 5の第 2番目〜第 9番目と 一致した。従って、 50kDaタンパク質は、 PATタンパク質であることが明らかとなった
Claims
請求の範囲
[1] 以下の (i)、 (ii)、 (iii)および (iv)力も選択される、ポリヌクレオチド:
(i)配列番号: 1で表される塩基配列力 なるポリヌクレオチド;
(ii)配列番号: 1で表される塩基配列からなるポリヌクレオチドにおいて、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつァ スパラギン酸アミノトランスフェラーゼ (AAT)活性を有するタンパク質をコードするポリ ヌクレ才チド;
(iii)配列番号: 1で表される塩基配列とストリンジェントな条件でノ、イブリダィズし、 つ AAT活性を有するタンパク質をコードするポリヌクレオチド;および
(iv)配列番号: 1で表される塩基配列力もなるポリヌクレオチドと少なくとも 90%の同 一性を有する塩基配列を含んでなり、かつ AAT活性を有するタンパク質をコードす るポリヌクレオチド。
[2] 以下の (V)、 (vi)、 (vii)および (viii)力 選択されるタンパク質:
(V)配列番号: 2で表されるアミノ酸配列からなるタンパク質;
(vi)配列番号: 2で表されるアミノ酸配列において、 1もしくは複数個のアミノ酸が欠失 、置換、挿入もしくは付加されたアミノ酸配列からなり、かつァスパラギン酸アミノトラン スフエラーゼ (AAT)活性を有するタンパク質;
(vii)配列番号: 2で表されるアミノ酸配列をコードするポリヌクレオチドとストリンジェン トな条件でノヽイブリダィズするポリヌクレオチドによりコードされ、かつ AAT活性を有 するタンパク質;および
(viii)配列番号: 2で表されるアミノ酸配列と少なくとも 90%の同一性を有するアミノ酸 配列からなり、かつ AAT活性を有するタンパク質。
[3] 請求項 2に記載のタンパク質をコードするポリヌクレオチド。
[4] 以下の )、(x)、 (xi)および (xii)力も選択される、ポリヌクレオチド:
(ix)配列番号: 3で表される塩基配列からなるポリヌクレオチド;
(X)配列番号: 3で表される塩基配列力 なるポリヌクレオチドにおいて、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつプ 口モーター活性を有するポリヌクレオチド;
(xi)配列番号: 3で表される塩基配列とストリンジェントな条件でノ、イブリダィズし、か つプロモーター活性を有するポリヌクレオチド;および
(xii)配列番号: 3で表される塩基配列力もなるポリヌクレオチドと少なくとも 95%の同 一性を有する塩基配列を含んでなり、かつプロモーター活性を有するポリヌクレオチ ド、。
[5] 請求項 1または 3に記載のポリヌクレオチドを含んでなる、組換えベクター。
[6] 請求項 4に記載のポリヌクレオチドを更に含んでなり、該ポリヌクレオチドが請求項 1 または 3に記載のポリヌクレオチドに作動可能に連結された、請求項 5に記載の組換 えベクター。
[7] FERM BP— 10495の受託番号のもと独立行政法人産業技術総合研究所特許 生物寄託センターに寄託された放線菌力 調製される発現ベクター pSGl lである、 請求項 5または 6に記載の組換えベクター。
[8] 請求項 5〜7のいずれか一項に記載の組換えベクターにより形質転換された宿主。
[9] 放線菌である、請求項 8に記載の宿主。
[10」 放線菌カ SStreptomvces hvgroscopicusである、 青求項 9に記載の宿王。
[11] FERM BP— 1368の受託番号のもと独立行政法人産業技術総合研究所特許生 物寄託センターに寄託された SF1293 NP— 50株または FERM BP— 130の受 託番号のもと独立行政法人産業技術総合研究所特許生物寄託センターに寄託され た SF1293株が形質転換されてなる、請求項 8〜10のいずれか一項に記載の宿主
[12] 請求項 1または 3に記載のポリヌクレオチドと L—ホスフィノスリシンアミノトランスフエ ラーゼ活性を有するタンパク質をコードするポリヌクレオチドとを含んでなる、組換え ベクター
[13] 請求項 4に記載のポリヌクレオチドを更に含んでなり、該ポリヌクレオチド力 請求項 1または 3に記載のポリヌクレオチドに作動可能に連結された、請求項 12に記載の組 換えベクター。
[14] L—ホスフィノスリシンアミノトランスフェラーゼ (PAT)活性を有するタンパク質をコー ドするポリヌクレオチドが、以下の )、 (ii,)、 (iii' )および (iv' )力も選択される、請
求項 12または 13に記載の組換えベクター:
(ί' )配列番号: 4で表される塩基配列力 なるポリヌクレオチド;
(π' )配列番号: 4で表される塩基配列からなるポリヌクレオチドにお 、て、 1もしくは複 数個のヌクレオチドが欠失、置換、挿入もしくは付加された塩基配列力もなり、かつ Ρ
AT活性を有するタンパク質をコードするポリヌクレオチド;
(iii' )配列番号: 4で表される塩基配列とストリンジヱントな条件でノ、イブリダィズし、か つ PAT活性を有するタンパク質をコードするポリヌクレオチド;および
(ίν' )配列番号: 4で表される塩基配列力もなるポリヌクレオチドと少なくとも 90%の同 一性を有する塩基配列を含んでなり、かつ PAT活性を有するタンパク質をコードする ポリヌクレオチド。
[15] L—ホスフィノスリシンアミノトランスフェラーゼ (PAT)活性を有するタンパク質力 以 下の(v' )、 (vi' )、 (νϋ' )および (viii' )力も選択される、請求項 12または 13に記載 の組換えベクター:
(ν' )配列番号: 5で表されるアミノ酸配列からなるタンパク質;
(νί' )配列番号: 5で表されるアミノ酸配列にお 、て、 1もしくは複数個のアミノ酸が欠 失、置換、挿入もしくは付加されたアミノ酸配列からなり、かつ PAT活性を有するタン パク質;
(νη' )配列番号: 5で表されるアミノ酸配列をコードするポリヌクレオチドとストリンジェ ントな条件でノヽイブリダィズするポリヌクレオチドによりコードされ、かつ PAT活性を有 するタンパク質;および
(νίϋ' )配列番号: 5で表されるアミノ酸配列と少なくとも 90%の同一性を有するァミノ 酸配列からなり、かつ PAT活性を有するタンパク質。
[16] 請求項 1または 3に記載のポリヌクレオチドと L—ホスフィノスリシンアミノトランスフエ ラーゼ活性を有するタンパク質をコードするポリヌクレオチドとが逆方向に連結されて
V、る、請求項 12〜 15の 、ずれか一項に記載の組換えベクター。
[17] 請求項 1または 3に記載のポリヌクレオチドと L—ホスフィノスリシンアミノトランスフエ ラーゼ活性を有するタンパク質をコードするポリヌクレオチドとが正方向に連結されて
V、る、請求項 12〜 15の 、ずれか一項に記載の組換えベクター。
[18] FERM BP— 10496の受託番号のもと独立行政法人産業技術総合研究所特許 生物寄託センターに寄託された放線菌カも調製される発現ベクター PAHSG7201 である、請求項 12〜16のいずれか一項に記載の組換えベクター。
[19] 請求項 12〜 18のいずれか一項に記載の組換えベクターにより形質転換された、 宿主。
[20] 放線菌である、請求項 19に記載の宿主。
[21] 放線菌:^ Streptomvces hvgroscopicusである、 青求項 20に,己载の ί百王。
[22] FERM BP— 1368の受託番号のもと独立行政法人産業技術総合研究所特許生 物寄託センターに寄託された SF1293 NP— 50株または FERM BP— 130の受 託番号のもと独立行政法人産業技術総合研究所特許生物寄託センターに寄託され た SF1293株が形質転換されてなる、請求項 19〜21のいずれか一項に記載の宿主 式 (I) :
[化 1]
0
CH3-P-CH2CH2CHCOOH
OH 丽 2
で示される L—ホスフィノスリシンまたはその塩を製造する方法であって、式 (Π): [化 2]
0
CH3-P-CH2CH9CCOOH
0H 0
で示される 4 ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、ダル タミン酸またはその塩およびァスパラギン酸またはその塩の存在下で、ァスパラギン 酸アミノトランスフェラーゼ活性および L ホスフィノスリシンアミノトランスフェラーゼ活 性を示し、かつ L—ホスフィノスリシンを産生することができる放線菌と接触させる工程
を含んでなり、使用される放線菌が親株のァスパラギン酸アミノトランスフェラーゼ活 性を超える活性を示すことを特徴とする方法。
[24] 使用される放線菌が、請求項 8〜: L 1のいずれか一項に記載の宿主である、請求項 23に記載の方法。
[25] 式 (Π)の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはそ の塩 = 1 : 0. 25〜1 : 1である、請求項 23または 24に記載の方法。
[26] 式 (Π)の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはそ の塩 = 1 : 0. 25 : 1である、請求項 25に記載の方法。
[27] 式 (I) :
[化 3]
0
CH3-P-CH2CH2CHCOOH
OH 丽 2
で示される L—ホスフィノスリシンまたはその塩を製造する方法であって、式 (Π): [化 4]
0
CH3-P-CH2CH CCOOH
OH 0
で示される 4 ヒドロキシメチルホスフィエル 2 ォキソ 酪酸またはその塩を、ダル タミン酸またはその塩およびァスパラギン酸またはその塩の存在下で、ァスパラギン 酸アミノトランスフェラーゼ活性および L ホスフィノスリシンアミノトランスフェラーゼ活 性を示し、かつ L—ホスフィノスリシンを産生することができる放線菌と接触させる工程 を含んでなり、使用される放線菌が親株のァスパラギン酸アミノトランスフェラーゼ活 性を超える活性を示し、かつ、親株の L ホスフィノスリシンアミノトランスフェラーゼ活 性を超える活性を示すことを特徴とする方法。
使用される放線菌が、請求項 19〜22のいずれか一項に記載の宿主である、請求
項 27に記載の方法。
式 (Π)の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはそ の塩 =1:0.25〜1:1である、請求項 27または 28に記載の方法。
式 (Π)の化合物またはその塩:グルタミン酸またはその塩:ァスパラギン酸またはそ の塩 =1:0.25:1である、請求項 29に記載の方法。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2007800075186A CN101395271B (zh) | 2006-03-02 | 2007-03-02 | 天冬氨酸转氨酶基因和l-膦丝菌素的制备方法 |
| JP2008502874A JP5335413B2 (ja) | 2006-03-02 | 2007-03-02 | アスパラギン酸アミノトランスフェラーゼ遺伝子およびl−ホスフィノスリシンの製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006-056726 | 2006-03-02 | ||
| JP2006056726 | 2006-03-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007100101A1 true WO2007100101A1 (ja) | 2007-09-07 |
Family
ID=38459196
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2007/054079 WO2007100101A1 (ja) | 2006-03-02 | 2007-03-02 | アスパラギン酸アミノトランスフェラーゼ遺伝子およびl-ホスフィノスリシンの製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP5335413B2 (ja) |
| CN (1) | CN101395271B (ja) |
| WO (1) | WO2007100101A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018108797A1 (de) | 2016-12-15 | 2018-06-21 | Bayer Cropscience Aktiengesellschaft | Verfahren zur herstellung von l-glufosinat oder dessen salzen unter verwendung von ephedrin |
| WO2018108794A1 (de) | 2016-12-15 | 2018-06-21 | Bayer Cropscience Aktiengesellschaft | Verfahren zur herstellung von d-glufosinat oder dessen salzen unter verwendung von ephedrin |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6427485A (en) * | 1986-06-09 | 1989-01-30 | Meiji Seika Kaisha | Production of l-2-amino-4-(hydroxymethylphosphinyl)-butyric acid |
| JPH02195889A (ja) * | 1988-07-04 | 1990-08-02 | Meiji Seika Kaisha Ltd | 新規なトランスアミナーゼをコードする遺伝子 |
| JPH04248987A (ja) * | 1990-09-27 | 1992-09-04 | Hoechst Ag | 共役酵素反応によるl−ホスフィノトリシンの製造方法 |
| JPH07250694A (ja) * | 1986-06-09 | 1995-10-03 | Meiji Seika Kaisha Ltd | L−2−アミノ−4−(ヒドロキシメチルホスフィニル)−酪酸の製法 |
| JP2003528572A (ja) * | 1999-04-30 | 2003-09-30 | バイエル クロップサイエンス ゲーエムベーハー | アスパルテートでの酵素トランスアミノ化によるl−ホスフィノトリシンの製造方法 |
-
2007
- 2007-03-02 JP JP2008502874A patent/JP5335413B2/ja not_active Expired - Fee Related
- 2007-03-02 CN CN2007800075186A patent/CN101395271B/zh not_active Expired - Fee Related
- 2007-03-02 WO PCT/JP2007/054079 patent/WO2007100101A1/ja active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6427485A (en) * | 1986-06-09 | 1989-01-30 | Meiji Seika Kaisha | Production of l-2-amino-4-(hydroxymethylphosphinyl)-butyric acid |
| JPH07250694A (ja) * | 1986-06-09 | 1995-10-03 | Meiji Seika Kaisha Ltd | L−2−アミノ−4−(ヒドロキシメチルホスフィニル)−酪酸の製法 |
| JPH02195889A (ja) * | 1988-07-04 | 1990-08-02 | Meiji Seika Kaisha Ltd | 新規なトランスアミナーゼをコードする遺伝子 |
| JPH04248987A (ja) * | 1990-09-27 | 1992-09-04 | Hoechst Ag | 共役酵素反応によるl−ホスフィノトリシンの製造方法 |
| JP2003528572A (ja) * | 1999-04-30 | 2003-09-30 | バイエル クロップサイエンス ゲーエムベーハー | アスパルテートでの酵素トランスアミノ化によるl−ホスフィノトリシンの製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| BENTLEY S.D. ET AL.: "Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2)", NATURE, vol. 417, no. 6885, 2002, pages 141 - 147, XP002233530 * |
| KATAYAMA M. ET AL.: "Gene organization in the ada-rplL region of Streptomyces virginiae", GENE, vol. 171, no. 1, 1996, pages 135 - 136, XP004042784 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018108797A1 (de) | 2016-12-15 | 2018-06-21 | Bayer Cropscience Aktiengesellschaft | Verfahren zur herstellung von l-glufosinat oder dessen salzen unter verwendung von ephedrin |
| WO2018108794A1 (de) | 2016-12-15 | 2018-06-21 | Bayer Cropscience Aktiengesellschaft | Verfahren zur herstellung von d-glufosinat oder dessen salzen unter verwendung von ephedrin |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2007100101A1 (ja) | 2009-07-23 |
| CN101395271A (zh) | 2009-03-25 |
| CN101395271B (zh) | 2013-02-27 |
| JP5335413B2 (ja) | 2013-11-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101528918B (zh) | 生产3-羟基丙酸的β-丙氨酸/α-酮戊二酸氨基转移酶 | |
| Kook et al. | Enhancement of γ-amminobutyric acid production by Lactobacillus sakei B2–16 expressing glutamate decarboxylase from Lactobacillus plantarum ATCC 14917 | |
| JPH04507346A (ja) | アルカリ性タンパク質分解酵素およびその製造方法 | |
| CN106906236B (zh) | 唾液酸酶基因重组表达载体及其构建方法,唾液酸酶及其制备方法 | |
| WO2018201160A1 (en) | Compositions and methods for enzyme immobilization | |
| NZ579599A (en) | Transglutaminase having disulfide bond introduced therein | |
| Park et al. | Enhancement of γ-aminobutyric acid production in Chungkukjang by applying a Bacillus subtilis strain expressing glutamate decarboxylase from Lactobacillus brevis | |
| JP2013000099A (ja) | ポリ乳酸分解酵素及びそれを産生する微生物 | |
| CN104073506B (zh) | 编码具有d‑丝氨酸合成活性的酶的dna、该酶的制备方法及d‑丝氨酸制备方法 | |
| US5482843A (en) | Enzyme of use in chitosan hydrolysis | |
| WO2006062189A1 (ja) | ニトリルヒドラターゼを発現する形質転換体 | |
| CN106459962A (zh) | 改良型腈水合酶 | |
| US20080241895A1 (en) | N-Acetyl-(R,S)-beta-Amino Acid Acylase Gene | |
| CA2800283C (en) | A novel 40 kda lipase from tetrasphaera | |
| WO2007100101A1 (ja) | アスパラギン酸アミノトランスフェラーゼ遺伝子およびl-ホスフィノスリシンの製造方法 | |
| EP0897005A1 (en) | Polyester synthase and a gene coding for the same | |
| CN113785069A (zh) | 用于生产3-羟基己二酸、α-氢化己二烯二酸和/或己二酸的基因修饰微生物以及该化学品的制造方法 | |
| WO2001018179A1 (fr) | Synthases de depsipeptides cycliques, genes correspondants et systeme de production de masse de ces depsipeptides cycliques | |
| JP5283154B2 (ja) | トリプシン様酵素 | |
| WO2004108942A1 (ja) | 新規なニトリルヒドラターゼ | |
| EP0579907A1 (en) | Novel nitrile hydratase, the gene encoding it and its use for producing amides from nitriles | |
| CN114651066A (zh) | 具有4-氨基苯甲酸羟化活性的多肽及其用途 | |
| CN107208041B (zh) | 用于生产喹啉酸的重组微生物和使用其生产喹啉酸的方法 | |
| JP4679785B2 (ja) | 新規な(r)−2−ヒドロキシ−3−フェニルプロピオン酸(d−フェニル乳酸)脱水素酵素およびそれをコードする遺伝子 | |
| JP2006055131A (ja) | 新規なd−アミノアシラーゼおよびその遺伝子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008502874 Country of ref document: JP Ref document number: 200780007518.6 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07737700 Country of ref document: EP Kind code of ref document: A1 |