明 細 書
チアゾール誘導体
技術分野
[0001] 本発明は、 11 β -HSD1阻害剤として有用な新規チアゾール誘導体もしくはその医 薬上許容される塩又はその溶媒和物に関する。
背景技術
[0002] 11 β -HSD1はコルチゾン力もコルチゾールへ変換する酵素であり、肝臓、内臓脂肪 などで発現しており、細胞内のコルチゾール濃度を各臓器レベルで増幅するファクタ 一として機能していると考えられている。また、 Il j8 - HSD1は局所的な作用を担って おり、肝臓では糖新生を担い、内臓脂肪の蓄積に関係していることが示唆されるため 、本酵素の活性を阻害することで肝臓においては糖新生の抑制による血糖降下作用
、内臓脂肪においては脂肪蓄積の抑制という効果が期待される。
11 β - HSD1の阻害剤としては、 Barf Τらの報告(非特許文献 1)、 WO01/090090〜W
001/090094 (特許文献 1〜5)、 WO03/043999 (特許文献 6)などに記載されている チアゾール誘導体が開示されて 、るが、本発明が示すチアゾール誘導体は知られて いない。
[0003] 非特許文献 1: J Med Chem. 2002 :3813-5
特許文献 1:国際公開第 01/090090号パンフレット
特許文献 2 :国際公開第 01/090091号パンフレット
特許文献 3:国際公開第 01/090092号パンフレット
特許文献 4:国際公開第 01/090093号パンフレット
特許文献 5:国際公開第 01/090094号パンフレット
特許文献 6:国際公開第 03/043999号パンフレット
発明の開示
発明が解決しょうとする課題
[0004] 公知の 11 β -HSD1阻害剤は阻害活性が十分とは言えず医薬品として満足できるも のではない。このため 11 jS -HSDl阻害作用による治療効果を有し医薬品として満足
できる化合物の開発が望まれている。
課題を解決するための手段
[0005] 本発明者らは、上記目的を達成すべく鋭意検討を重ねた結果、ある種のチアゾー ル誘導体が優れた 11 -HSDl阻害活性を有することを見出し、本発明を完成するに 至った。
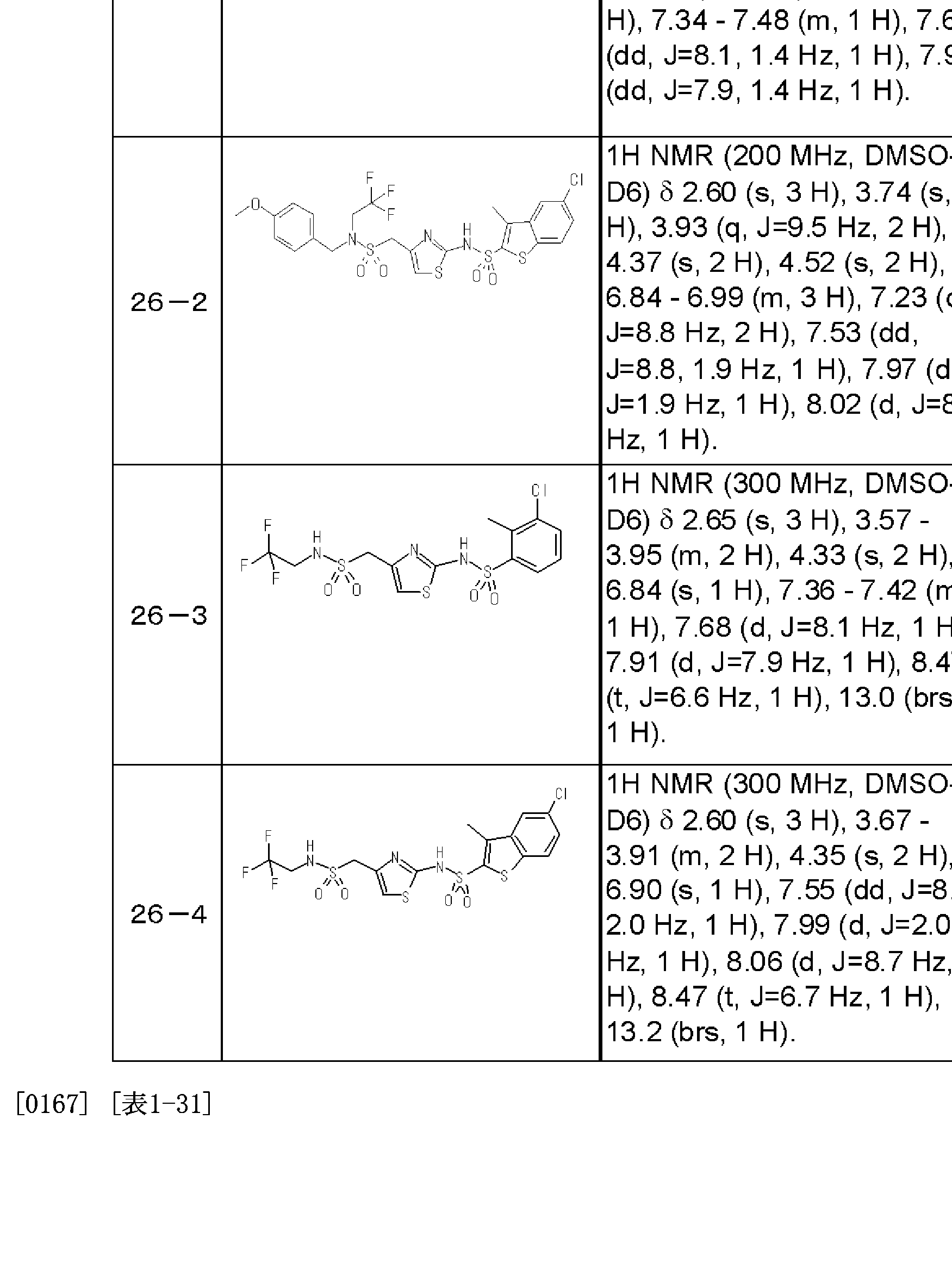
すなわち、本発明は、下記式( I )
[0006] [化 1]
[0007] [式中、 R1は、
式- C(R5)(R6)- S(0)n- R7、
式- C(R51)(R61)- C(R52)(R62)- S(0)n- R71、
式- C(R53)(R63)- C(R54)(R64)- C(R55)(R65)-S(0)n-R72
(式中、 R
5、 R
51、 R
52、 R
53、 R
54、 R
55、 R
6、 R
61、
R
63、 R
64及び R
65は、同一又は異なって 、水素原子;又は置換されてもよい C のアルキル基を表し、
1-6
n=0の場合、
もよい C のシクロアルキル基を表し、
3-6
n= l又は 2の場合、
R R71、及び R72は、水素原子;置換されてもょ 、C のアルキル基;置換されてもよ ヽ
1-6
C のシクロアルキル基;式- NR8R9{式中、 R8及び R9は、同一又は異なって、水素原
3-6
子;置換されてもよい c のアルキル基;置換されてもよい C のシクロアルキル基;置
1-6 3-6
換されてもょ 、ァリールアルキル基;置換されてもょ 、ァリール基;置換されてもよ ヽ ヘテロァリール基;又は式- COR1Q (式中、 R1Qは、置換されてもよい C のアルキル基;
1-6
置換されてもょ 、C のシクロアルキル基;置換されてもよ!/、ァリール基;又は置換さ
3-6
れてもよいへテロァリール基を表す。)を表す。 } ;置換されてもよい 3〜6員環の脂環 式へテロ環基;置換されてもょ 、ァリール基;又は置換されてもよ 、ヘテロァリール基
を表す。 )、
C のシクロアルキル基
3-8
(ここで C のシクロアルキル基は 1つ以上の置換基で置換されていてもよぐその置
3-8
換基とは、ハロゲン原子;置換されてもよい C のアルキル基; -CN;式- NRDRE;式- 0
1-6
てもよい C のアルキル基;式- COR11 ;式- CONR12R13;又は式- S(〇) R14を表し、 RE、 R
1-6 2
F、 RH、 Rj、 RK及び R1!ま、同一又は異なって、水素原子;又は置換されてもよい C の
1-6 アルキル基を表し、 RGは、水素原子;水酸基;置換されてもよ 、C のアルキル基;又
1-6
は置換されてもよい C のアルコキシ基を表す。(式中、 R11は、水素原子;水酸基;置
1-6
換されてもよい C のアルキル基;又は置換されてもよい C のアルコキシ基を表し、 R
1-6 1-6
12及び R13は、同一又は異なって、水素原子;又は置換されてもよい C のアルキル基
1-6
を表し、 R"は、置換されてもょ 、C のアルキル基;又は置換されてもょ 、ァリール基
1-6
を表す。)}を表す。)、
又は酸素原子;硫黄原子;式- S(O)-;式- S(O) -;式- N(RA)-;式- N(Re)S(0) -;又は式
2 2
-S(O) N(Re)_から選ばれる 1〜3個のグループを環に含む 3〜8員環の脂環式へテロ
2
環基
{式中、 RAは、水素原子;置換されてもよい C のアルキル基;式- COR111 ;式- CONR12
1-6
131 ;又は式- S(O) R141 (式中、 Rmは、水素原子;水酸基;置換されてもよい C のァ
2 1-6 ルキル基;又は置換されてもよい C のアルコキシ基を表し、 R121及び R131は、同一又
1-6
は異なって、水素原子;又は置換されてもょ 、C のアルキル基を表し、 R141は、置換
1-6
されてもよい C のアルキル基;又は置換されてもよいァリール基を表す。)を表し、 Rc
1-6
は、水素原子;置換されてもよい C のアルキル基;又は置換されてもよいァリールァ
1-6
ルキル基を表す。(ここで 3〜8員環の脂環式へテロ環基は 1つ以上の置換基で置換 されていてもよぐその置換基とは、ハロゲン原子;ォキソ基;置換されてもよい C の
1-6 アルキル基; -CN ;式- NRD'IT;式- ORF ';式- COR";式- CONRH R1' ;又は式- S(〇) NRK
2
'RL'{式中、 'は、水素原子;置換されてもよい C のアルキル基;式- COR112;式- CO
1-6
NR122R132;又は式- S(O) R142を表し、 RE'、 RF'、 RH'、 Rj'、 RK'及び RL 'は、同一又は異なつ
2
て、水素原子;又は置換されてもょ 、C のアルキル基を表し、 RG'は、水素原子;水
酸基;置換されてもよい C のアルキル基;又は置換されてもよい C のアルコキシ基
1-6 1-6
を表す。(式中、 R112は、水素原子;水酸基;置換されてもよい C のアルキル基;又は
1-6
置換されてもよい C のアルコキシ基を表し、 R122及び R132は、同一又は異なって、水
1-6
素原子;又は置換されてもょ 、C のアルキル基を表し、 R"2は、置換されてもょ 、C
1-6 1- のアルキル基;又は置換されてもよいァリール基を表す。 ) }を表す。 ) }を表し、
6
R2は、水素原子、ハロゲン原子又は置換されてもよい C のアルキル基を表し、
1-6
R3は、水素原子、置換されてもよい C のアルキル基、置換されてもよい C のァルケ
1-6 2-6
-ル基又は置換されてもよい C のアルキ-ル基を表し、
2-6
R4は、ァリール基、ヘテロァリール基、ァリールァルケ-ル基又はへテロアリールアル ケ-ル基
にこでァリール基、ヘテロァリール基、ァリールァルケ-ル基又はへテロアリールァ ルケ二ル基は 1つ以上の置換基で置換されていてもよぐその置換基とは、ハロゲン 原子;シァノ基;ニトロ基;
式- X1- X2- X3
(式中、 X1は、単結合;酸素原子;硫黄原子;式- S(O)-;式- SO -;式- C(O)-;式- NX4-
2
;式- C〇- NX41-;式- NX42- C〇-;式- SO - NX43-;式- NX44- SO -;式- C〇-〇-;又は式-
2 2
〇- CO-を表し、 X2は、単結合;又は c のアルキレン基を表し、 x3、 x4、 x41、 x42、 X43
1-6
及び X44は、同一又は異なって、水素原子; C のアルキル基; C のシクロアルキル
1-6 3-6
基; C のアルケニル基; C のアルキニル基;ァリール基;ヘテロァリール基;又は 3
2-6 2-6
〜8員環の脂環式へテロ環基を表す。ただし、 X2、 X3、 X4、 X41、 X42、 X43及び X44は、下 記置換基 Y群より選ばれる 1〜3個の置換基を有していてもよい。 );
式- X1- X5- X6
(式中、 X1は、前記と同義であり、 X5は、 C のアルキレン基を表し、 X6は、式- ox7;式
1-6
-NX71X81;式— CO— NX72X82;式— NX73— CO— X83;式— SO NX74X84;式— NX75— SO X85;式
2 2
— CO— 0— X76;又 ίま式— 0— CO— X77を表し、 x7、 x71、 x72、 x73、 x74、 x75、 x76、 x77、 X8、 X81 、 x82、 x83、 X84及び X85は、同一又は異なって、水素原子; C のアルキル基;又はフエ
1-6
-ル基を表す。ただし、 x5、 x7、 x71、 x72、 x73、 x74、 x75、 x76、 x77、 x8、 x81、 x82、 x83、 X84 及び X85は、下記置換基 Y群より選ばれる 1〜3個の置換基を有していてもよい。 );
又は隣接する 2個の置換基が一緒になつて式- χ χ χ χ1-
(式中、 X1及び X5は、前記と同義であり、 X9は、単結合;又は C のアルキレン基を表
1-4
す。)で表される一部飽和された 5〜 10員環構造を表し、
置換基 Y群とは、ハロゲン原子;シァノ基;ニトロ基;水酸基;ァセチル基;メチル基;トリ フルォロメチル基;又はメトキシ基カゝらなる群を表す。 }を表す。 ]で表されるチアゾー ル誘導体又はその薬学的に許容される塩もしくはその溶媒和物を提供する。
発明の効果
[0008] 本発明により、優れた 11 β -HSD1阻害活性を示すチアゾール誘導体、その薬学的 に許容される塩、又はその溶媒和物を提供することができた。
発明を実施するための最良の形態
[0009] 本発明の他の態様によると、本発明は、上記式( I )において
[式中、 R1が、
式- C(R5)(R6)- S(0)n-R7 (式中、 R5及び R6が、同一又は異なって、水素原子;又は置換 されてもよい C のアルキル基であり、 R7力 水素原子;置換されてもよい C のアルキ
1-6 1-6 ル基;置換されてもよい C のシクロアルキル基;式- NR 9{式中、 R8及び R9力 同一
3-6
又は異なって、水素原子;置換されてもょ 、C のアルキル基;置換されてもよ 、C
1-6 3-6 のシクロアルキル基;置換されてもょ 、ァリールアルキル基;置換されてもょ 、ァリー ル基;置換されてもよいへテロアリール基;又は式- COR1Q (式中、 R1Qが、置換されても よい C のアルキル基;置換されてもよい C のシクロアルキル基;置換されてもよいァ
1-6 3-6
リール基;又は置換されてもよいへテロアリール基である。)である。 } ;置換されてもよ
V、3〜6員環の脂環式へテロ環基;置換されてもょ 、ァリール基;又は置換されてもよ いへテロァリール基であり、 n= l又は 2である。)、
C のシクロアルキル基(ここで C のシクロアルキル基は 1つ以上の置換基で置換さ
3-8 3-8
れていてもよぐその置換基とは、ハロゲン原子;置換されてもよい C のアルキル基;
1-6
- CN ;式- NRDRE;式- ORF;式- CORG;式- CONRHR1;又は式- S(O) NRKRL{式中、 が
2
、水素原子;置換されてもょ 、C のアルキル基;式- COR11;式- CONR12R13;又は式-
1-6
S(O) R14であり、 R R RH、 R Rk及び^が、同一又は異なって、水素原子;又は置
2
換されてもよい C のアルキル基であり、 RG力 水素原子;水酸基;置換されてもよい
C のアルキル基;又は置換されてもよい C のアルコキシ基である。(式中、 R11が、
1-6 1-6
水素原子;水酸基;置換されてもよい C のアルキル基;又は置換されてもよい C の
1-6 1-6 アルコキシ基であり、 R12及び R13が、同一又は異なって、水素原子;又は置換されても よい C のアルキル基であり、 R14が、置換されてもよい C のアルキル基;又は置換さ
1-6 1-6
れてもよぃァリール基である。 ) }である。 )、
又は酸素原子;硫黄原子;式- S(O)- ;式- S(O) -;式- N(RA)- ;式- N(Re)S(0) -;又は式
2 2
-S(O) N(Re)_から選ばれる 1〜3個のグループを環に含む 3〜8員環の脂環式へテロ
2
環基
{式中、 RAが、水素原子;置換されてもよい C のアルキル基;式- COR111 ;式- CONR12
1-6
131 ;又は式- S(O) R141 (式中、 Rmが、水素原子;水酸基;置換されてもよい C のァ
2 1-6 ルキル基;又は置換されてもよい C のアルコキシ基をであり、 R 及び R131は、同一
1-6
又は異なって、水素原子;又は置換されてもよい C のアルキル基であり、 R141は、置
1-6
換されてもよい C のアルキル基;又は置換されてもよいァリール基である。)であり、
1-6
Reが、水素原子;置換されてもよい C のアルキル基;又は置換されてもよいァリール
1-6
アルキル基である。(ここで 3〜8員環の脂環式へテロ環基は 1つ以上の置換基で置 換されていてもよぐその置換基とは、ハロゲン原子;ォキソ基;置換されてもよい C
1-6 のアルキル基; -CN ;式- NRD RE' ;式- OR';式- CORG' ;式- CONIT R1' ;又は式- S(〇) N
2
RK'RL'{式中、 RD 'が、水素原子;置換されてもよい C のアルキル基;式- COR112 ;式- C
ONR
mR
132 ;又は式- S(O) R
K'及び R
L'が、同一又は異なつ
て、水素原子;又は置換されてもよい C のアルキル基であり、 RG'が、水素原子;水酸
1-6
基;置換されてもよい c のアルキル基;又は置換されてもよい C のアルコキシ基で
1-6 1-6
ある。(式中、 R112が、水素原子;水酸基;置換されてもよい C のアルキル基;又は置
1-6
換されてもよい C のアルコキシ基であり、 R122及び R132が、同一又は異なって、水素
1-6
原子;又は置換されてもよい C のアルキル基であり、 R142力 置換されてもよい C の
1-6 1-6 アルキル基;又は置換されてもよいァリール基である。 ) }である。 ) }であり、
R2が、水素原子又は置換されてもよい C のアルキル基であり、
1-6
R3力 水素原子、置換されてもよい C のアルキル基、置換されてもよい C のァルケ
1-6 2-6
-ル基又は置換されてもよい C のアルキ-ル基であり、
R4が、ァリール基、ヘテロァリール基、ァリールァルケ-ル基又はへテロアリールアル ケ-ル基にこでァリール基、ヘテロァリール基、ァリールァルケ-ル基又はへテロア リールアルケニル基は 1つ以上の置換基で置換されていてもよぐその置換基とは、 ハロゲン原子;シァノ基;ニトロ基;
式- χ χ^χ^式中、 X1が、単結合;酸素原子;硫黄原子;式- s(o)-;式- so -;式- C(
2
〇)-;式- NX4-;式- CO- NX41-;式- NX42- CO-;式- SO -NX43-;式- NX44- SO -;式- CO
2 2
-〇-;又は式 -〇- C〇-であり、 X2が、単結合;又は c のアルキレン基であり、 x3、 X4、
1-6
χ41、χ42、χ43及び X44が、同一又は異なって、水素原子; C のアルキル基; C のシク
1-6 3-6 口アルキル基; C のアルケニル基; C のアルキニル基;ァリール基;ヘテロァリール
2-6 2-6
基;又は 3〜8員環の脂環式へテロ環基である。ただし、 X2、 X3、 X4、 X41、 X42、 X43及び
X"は、下記置換基 Υ群より選ばれる 1〜3個の置換基を有していてもよい。 ); 式- χ χ^χ^式中、 X1は、前記と同義であり、 X5が、 C のアルキレン基であり、 X6が
1-6
、式- 0Χ7;式- ΝΧ71Χ81;式- CO- ΝΧ72Χ82;式- NX73- CO- X83;式- SO - NX74X84;式- NX75
2
-so - x85;式- CO- o- x76;又は式- o- co- x77であり、 x7、 x71、 x72、 x73、 x74、 x75、 x76、
2
x77、 x x81、 x82、 x83、 x84及び x85が、同一又は異なって、水素原子; C のアルキル
1-6
基;又 ίまフエ-ノレ基である。†† χ5、 χ7、 χ71、 χ72、 χ73、 χ74、 χ75、 χ76、 χ77、 χ8、 χ81、
X82、 X83、 X84及び X85は、下記置換基 Y群より選ばれる 1〜3個の置換基を有していて ちょい。)
;又は隣接する 2個の置換基が一緒になつて式- χ χ χ χ1- (式中、 X1及び X5は、 前記と同義であり、 X9が、単結合;又は C のアルキレン基である。)である一部飽和
1-4
された 5〜 10員環構造であり、
置換基 Y群とは、ハロゲン原子;シァノ基;ニトロ基;水酸基;ァセチル基;メチル基;トリ フルォロメチル基;又はメトキシ基カゝらなる群を表す。 }である。 ]である請求項 1記載 のチアゾール誘導体もしくはその薬学的に許容される塩又はその溶媒和物を提供す る。
本発明の他の態様によると、本発明は、式(1)において、 R1が、 C のシクロアルキル
3-8
基である前記チアゾール誘導体もしくはその薬学的に許容される塩又はその溶媒和 物を提供する。
本発明の他の態様によると、本発明は、式(1)において、 R1が、酸素原子;硫黄原子 ;式- S(O)- ;式- S(O) -;式- N(RA)- ;式- N(RE)S(0) -;又は式- S(O) N(RE)-から選ばれ
2 2 2 る 1〜3個のグループを環に含む 3〜8員環の脂環式へテロ環基である前記チアゾー ル誘導体もしくはその薬学的に許容される塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、好ましくは R1が、酸素原子; 硫黄原子;式- S(O)- ;式- S(O) -;式- N(RA)- ;式- N(RE)S(0) -;又は式- S(O) N(RE)-か
2 2 2 ら選ばれる 1〜3個のグループを環に含む 3〜8員環の脂環式へテロ環基で且つ環 内に少なくとも 1つの酸素原子を有する前記チアゾール誘導体もしくはその薬学的に 許容される塩又はその溶媒和物を提供することであり、より好ましくは R1が、テトラヒド 口ピラン又は 1, 3—ジォキサンである前記チアゾール誘導体もしくはその薬学的に許 容される塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、 R1が、
式- C(R5)(R6)- S(0)n- R7、
式- C(R51)(R61)- C(R52)(R62)- S(0)n- R71、
式- C(R53)(R63)- C(R54)(R64)- C(R55)(R65)-S(0)n-R72
(式中、 R5、 R51、 R52、 R53、 R54、 R55、 R6、 R61、 R62、 R63、 R64及び R65が、同一又は異なって 、水素原子;又は置換されてもよい C のアルキル基であり、
1-6
n=0の場合、
てもよい C のシクロアルキル基であり、
3-6
n= l又は 2の場合、
R R71、及び R72は、力 水素原子;置換されてもよ 、C のアルキル基;置換されても
1-6
よ!/、C のシクロアルキル基;式- NR8R9
3-6
{式中、 R8及び R9が、同一又は異なって、水素原子;置換されてもよい C のアルキル
1-6 基;置換されてもよ!/、C のシクロアルキル基;置換されてもよ!/、ァリールアルキル基;
3-6
置換されてもょ 、ァリール基;置換されてもょ 、ヘテロァリール基;又は式— CORW (式中、 R1Qが、置換されてもよい C のアルキル基;置換されてもよい C のシクロアル
1-6 3-6
キル基;置換されてもょ 、ァリール基;又は置換されてもょ 、ヘテロァリール基である
。)である。 }
;置換されてもょ 、3〜6員環の脂環式へテロ環基;置換されてもょ 、ァリール基;又 は置換されてもょ 、ヘテロァリール基である。 )である請求項 1記載のチアゾール誘導 体もしくはその薬学的に許容される塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、好ましくは R1が、式- C(R5)(R 6)-S(0)n-R7であり、且つ R7が、式- NR 9{式中、 R8及び R9は、同一又は異なって、水 素原子;置換されてもよい C のアルキル基;置換されてもよい C のシクロアルキル
1-6 3-6
基;置換されてもよ 、ァリールアルキル基;置換されてもよ 、ァリール基;置換されても よいへテロアリール基;又は式- COR10 (式中、 R10は、置換されてもよい C のアルキル
1-6 基;置換されてもょ 、C のシクロアルキル基;置換されてもょ 、ァリール基;又は置換
3-6
されてもよいへテロアリール基を表す。)を表す。 }であり、 n= 2である前記チアゾー ル誘導体もしくはその薬学的に許容される塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、好ましくは R1が、式- C(R5)(R 6)-S(0)n-R7であり、且つ R7が、置換されてもよい C のアルキル基;置換されてもよい
1-6
C のシクロアルキル基;置換されてもょ 、3〜6員環の脂環式へテロ環基;置換され
3-6
てもよ 、ァリール基;又は置換されてもょ 、ヘテロァリール基であり、 n= 2である前記 チアゾール誘導体もしくはその薬学的に許容される塩又はその溶媒和物を提供する 本発明の他の態様によると、本発明は、式(1)において、より好ましくは R1力 tert-ブ チルスルホニルメチル基である前記チアゾール誘導体もしくはその薬学的に許容さ れる塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、好ましくは R4が、置換されて もよ 、フエ-ル基または置換されてもょ 、ベンゾチォフエ-ル基である前記チアゾー ル誘導体もしくはその薬学的に許容される塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、式(1)において、より好ましくは R2が水素原 子であり、 R3が水素原子である前記チアゾール誘導体もしくはその薬学的に許容さ れる塩又はその溶媒和物を提供する。
本発明の他の態様によると、本発明は、上記いずれかのチアゾール誘導体又はその
薬学的に許容される塩又はそれらの溶媒和物を有効成分として含有する医薬を提供 する。
本発明の他の態様によると、本発明は、 11 jS -HSDlを阻害することで改善しうる疾 患又は状態を予防または治療するための前記医薬を提供する。
本発明の他の態様によると、本発明は、 11 jS -HSDlを阻害することで改善しうる疾 患又は状態が糖尿病、メタボリックシンドローム、肥満症、高血圧症もしくは動脈硬化 症である前記医薬を提供する。
本発明をさらに詳細に説明するが、例示されたものに特に限定されない。 ハロゲン原子とは、フッ素原子、塩素原子、臭素原子又はヨウ素原子を示す。
C のアルキル基としては、例えばメチル基、ェチル基、プロピル基、イソプロピル基
1-6
、ブチル基、イソブチル基、 sec-ブチル基、 tert-ブチル基、ペンチル基、イソペンチ ル基、 1-ェチルプロピル基、へキシル基が挙げられる。
C のアルキル基としては、例えばブチル基、イソブチル基、 sec-ブチル基、 tert-ブ
3-6
チル基、ペンチル基、イソペンチル基、 1-ェチルプロピル基、へキシル基が挙げられ る。
[0010] C のシクロアルキル基としては、例えばシクロプロピル基、シクロブチル基、シクロ
3-6
ペンチル基、シクロへキシル基が挙げられる。
[0011] C のシクロアルキル基としては、例えばシクロプロピル基、シクロブチル基、シクロ
3-8
ペンチル基、シクロへキシル基、シクロへプチル基、シクロォクチル基、シクロペンテ -ル基、シクロへキセニル基が挙げられる。
ァリール基としては、例えばフエニル基、 1-ナフチル基、 2-ナフチル基が挙げられる。 ァリールアルキル基としては、例えばべンジル基、フエネチル基、 1-ナフチルメチル 基、 2-ナフチルメチル基が挙げられる。
[0012] ヘテロァリール基としては、例えばピロリル基、フリル基、チェニル基、ォキサゾリル 基、イソォキサゾリル基、イミダゾリル基、チアゾリル基、イソチアゾリル基、ピラゾリル 基、トリァゾリル基、テトラゾリル基、 1,3,4-ォキサジァゾリル基、 1,2,4-ォキサジァゾリ ル基、 1,2,4-チアジアゾリル基、ピリジル基、ビラジニル基、ピリミジニル基、ピリダジ- ル基、 1,2,4-トリアジ-ル基、 1,2,3-トリアジ-ル基、 1,3,5-トリアジ-ル基、ベンズォキ
サゾリル基、ベンズイソキサゾリル基、ベンゾチアゾリル基、ベンズイソチアゾリル基、 ベンズイミダゾリル基、ベンゾトリァゾリル基、ベンゾチアジアゾリル基、ベンゾフラザ二 ル基、ベンゾビラ-ル基、チアナフテニル基、イソチアナフテュル基、ベンゾフラニル 基、イソべンゾフラニル基、ベンゾチェ二ル基、イソインドリル基、インドリル基、インダ ゾリル基、イソキノリル基、キノリル基、フタラジニル基、キノキサリニル基、キナゾリ二 ル基、シンノリ-ル基、 2, 1,3-ベンズォキサジァゾリル基、ベンゾキサジ-ル基、タマリ ル基、ナフチリジニル基、プリニル基、プテリジニル基、チェノフラニル基、イミダゾチ ァゾリル基、イミダゾピリジ-ル基、ピロ口ピリジ-ル基、ピロ口ピリミジ -ル基、ピリドピリ ミジニル基が挙げられる。
3〜6員環の脂環式へテロ環基としては、例えばァゼチジニル基、ピロリジ -ル基、 ピベリジ-ル基、テトラヒドロフラニル基、ジヒドロフラ -ル基、テトラヒドロビラニル基、 ジヒドロビラ-ル基、テトラヒドロチォフエ-ル基、テトラヒドロチォビラ-ル基、ジヒドロ チォビラ-ル基、テトラヒドロピリジ-ル基、ジヒドロピリジ-ル基、モルホリニル基、チ オモルホリニル基、ピペラジニル基、チアゾリジ-ル基、ジォキサ-ル基、イミダゾリ- ル基、チアゾリニル基、イソチアゾリジ-ル基、チアジナ -ル基、ジォキソラニル基が 挙げられる。
酸素原子;硫黄原子;式- S(O)-;式- S(O) -;式- N(RA)-;式- N(Re)S(0) -;又は式- S(
2 2
0) N(Re)-から選ばれる 1〜3個のグループ {式中、 RAは、水素原子;置換されてもよ
2
いじ のアルキル基;式- COR111 ;式- CONR121Rm ;又は式- S(O) R1" (式中、 Rmは、
1-6 2
水素原子;水酸基;置換されてもよい C のアルキル基;又は置換されてもよい C の
1-6 1-6 アルコキシ基を表し、 R121及び R131は、同一又は異なって、水素原子;又は置換されて もよい C のアルキル基を表し、 R141は、置換されてもよい C のアルキル基;又は置
1-6 1-6
換されてもょ 、ァリール基を表す。 )を表し、 Reは、水素原子;置換されてもょ 、C の
1-6 アルキル基;又は置換されてもよいァリールアルキル基を表す。 }を環に含む 3〜8員 環の脂環式へテロ環基としては、例えばァゼチジニル基、ピロリジニル基、ピペリジニ ル基、テトラヒドロフラ-ル基、ジヒドロフラ -ル基、テトラヒドロビラ-ル基、ジヒドロビラ ニル基、テトラヒドロチオフ ニル基、テトラヒドロチォピラニル基、ジヒドロチォビラ二 ル基、テトラヒドロピリジニル基、ジヒドロピリジニル基、モルホリニル基、チオモルホリ
-ル基、ピペラジニル基、チアゾリジニル基、ジォキサニル基、イミダゾリ-ル基、チア ゾリニル基、イソチアゾリジ-ル基、チアジナ -ル基、ジァゼパ-ル基、ジォキソラ- ル基、 1, 3 プロパンスルタミル基、 1, 4 ブタンスルタミル基が挙げられる。
[0014] C のアルコキシ基としては、例えばメトキシ基、エトキシ基、プロポキシ基、イソプロ
1- 6
ポキシ基、ブトキシ基、イソブトキシ基、 sec-ブトキシ基、 tert-ブトキシ基、ペントキシ 基、イソペントキシ基、 1-ェチルプロポキシ基、へキシルォキシ基が挙げられる。
[0015] C のァルケ-ル基としては、例えばビュル基、ァリル基、 1-プロべ-ル基、 2-プロ
2- 6
ぺニル基、イソプロぺニル基、 2-ブテニル基、 3-ブテニル基、イソブテニル基、 4-ぺ ンテュル基、 5-へキセ-ル基が挙げられる。
ァリールァルケ-ル基とは、ァリール基と C のァルケ-ル基が結合した基であり、例
2-6
えばスチリル基、ナフチルビニル基が挙げられる。
ヘテロァリールァルケ-ル基とは、ヘテロァリール基と C のァルケ-ル基が結合した
2-6
基であり、例えばフラ-ルビ-ル基、チェ-ルビニル基、ピリジルビニル基が挙げられ る。
[0016] C のアルキニル基としては、例えばェチニル基、 1-プロピニル基、 2-プロピニル基
2-6
、 3-ブチュル基、 4-ペンチ-ル基、 5_へキシュル基が挙げられる。
[0017] C のアルキレン基としては、例えばメチレン基、 1,1-エチレン基、 1,2-エチレン基、
1-4
1,1-プロピレン基、 1,3-プロピレン基、テトラメチレン基が挙げられる。
[0018] C のアルキレン基とは、前記定義「C のアルキル基」からさらに任意の水素原子
1-6 1-6
を 1個除いて誘導される二価の基を意味し、例えばメチレン基、 1,1-エチレン基、 1,2- エチレン基、 1,1-プロピレン基、 1,3-プロピレン基、テトラメチレン基、ペンタメチレン 基、へキサメチレン基が挙げられる。
[0019] 置換されてもよい C のアルキル基、置換されてもよい C のアルキル基、置換され
1-6 3-6
てもよい C のシクロアルキル基、置換されてもよい 3〜6員環の脂環式へテロ環基、
3-6
置換されてもよい c のアルコキシ基、置換されてもよい C のアルケニル基又は置
1-6 2-6
換されてもよい C のアルキ-ル基の置換基とは、ハロゲン原子、シァノ基、水酸基、
2-6
カルボキシル基、力ルバモイル基、トリメチルシリル基、メトキシ基、エトキシ基、ヒドロ キシエトキシ基、ォキソ基など力もなる群より選ばれる 1個以上を示す。
[0020] 置換されてもょ 、ァリール基、置換されてもょ 、ヘテロァリール基の置換基とは、ハ ロゲン原子、シァノ基、水酸基、カルボキシル基、力ルバモイル基、トリメチルシリル基 、メトキシ基、エトキシ基、ニトロ基、メチル基、ェチル基、 2-ヒドロキシ- 1,1-ジメチルェ チルァミノカルボニル基、 4,4-ジメチル -4,5-ジヒドロ- 1,3-ォキサゾリル基などからなる 群より選ばれる 1個以上を示す。
置換されてもよいァリールアルキル基の置換基とは、ハロゲン原子、シァノ基、水酸 基、カルボキシル基、力ルバモイル基、トリメチルシリル基、メトキシ基、エトキシ基、二 トロ基、メチル基、ェチル基などカゝらなる群より選ばれる 1個以上を示す。
[0021] 式- Xi-X2-X3で表される置換基を例示すると、メチル基、ェチル基、プロピル基、ィ ソプロピル基、ブチル基、イソブチル基、 sec-ブチル基、 tert-ブチル基、ペンチル基 、イソペンチル基、 1-ェチルプロピル基、へキシル基、 2-ヒドロキシェチル基、 2-フル ォロェチル基、メトキシェチル基、ジフルォロメチル基、トリフルォロメチル基、トリフル ォロェチル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロへキシ ル基、シクロペンテ-ル基、シクロへキセニル基、 4_ヒドロキシシクロへキシル基、シク 口ペンチルメチル基、シクロへキシルメチル基、ビュル基、ァリル基、 1-プロべ-ル基 、 2-プロぺニル基、イソプロぺニル基、 2-ブテニル基、 3-ブテニル基、イソブテニル基 、 4-ペンテ-ル基、 5-へキセ-ル基ェチュル基、 1-プロピ-ル基、 2-プロピ-ル基、 3 -ブチュル基、 4-ペンチ-ル基、 5-へキシュル基、フエ-ル基、 1-ナフチル基、 2-ナ フチル基、 4-ヒドロシフェ-ル基、 4-クロ口フエ-ル基、 2-シァノフエ-ル基、 4-ァセチ ルフエ-ル基、 4-ニトロフエ-ル基、ベンジル基、フエネチル基、 4-メトキシベンジル 基、ピロリル基、 1-メチルピロリル基、フリル基、 2-ニトロフリル基、チェ-ル基、 3-トリ フルォロチェニル基、ォキサゾリル基、イソォキサゾリル基、イミダゾリル基、チアゾリ ル基、イソチアゾリル基、ピラゾリル基、トリァゾリル基、テトラゾリル基、 1,3,4-ォキサジ ァゾリル基、 1,2,4-ォキサジァゾリル基、 1,2,4-チアジアゾリル基、ピリジル基、ピラジ -ル基、ピリミジ -ル基、ピリダジ -ル基、 1,2,4-トリアジ-ル基、 1,2,3-トリアジ-ル基 、 1,3,5-トリアジ-ル基、ベンズォキサゾリル基、ベンズイソキサゾリル基、ベンゾチア ゾリル基、ベンズイソチアゾリル基、ベンズイミダゾリル基、ベンゾトリァゾリル基、ベン ゾチアジアゾリル基、ベンゾフラザニル基、ベンゾピラニル基、チアナフテュル基、ィ
ソチアナフテニル基、ベンゾフラ-ル基、イソべンゾフラ-ル基、ベンゾチェ-ル基、 イソインドリル基、インドリル基、インダゾリル基、イソキノリル基、キノリル基、フタラジュ ル基、キノキサリニル基、キナゾリ-ル基、シンノリ-ル基、 2, 1,3-ベンズォキサジァゾ リル基、ベンゾキサジ-ル基、クマリル基、ナフチリジ-ル基、フタラジュル基、プリ- ル基、プテリジニル基、チェノフラ-ル基、イミダゾチアゾリル基、イミダゾピリジ-ル基 、ピロ口ピリジ-ル基、ピロ口ピリミジニル基、ピリドピリミジニル基、ピリジルメチル基、 イミダゾリルメチル基、水酸基、メトキシ基、エトキシ基、プロポキシ基、イソプロポキシ 基、ブトキシ基、イソブトキシ基、 sec-ブトキシ基、 tert-ブトキシ基、ペントキシ基、イソ ペントキシ基、 1-ェチルプロポキシ基、へキシルォキシ基、シクロペンチルォキシ基、 シクロへキシルォキシ基、プロべ-ルォキシ基、プロパルギルォキシ基、フエ-ノキシ 基、 4-クロ口フエ-ノキシ基、チェ-ルォキシ基、ジフルォロメチルォキシ基、トリフル ォロメチルォキシ基、トリフルォロェチルォキシ基、チオール基、メチルチオ基、ェチ ルチオ基、プロピルチオ基、イソプロピルチオ基、プチルチオ基、イソプチルチオ基、 sec-ブチルチオ基、 tert-ブチルチオ基、ペンチルチオ基、イソペンチルチオ基、 1- ェチルプロピルチオ基、へキシルチオ基、シクロペンチルチオ基、シクロへキシルチ ォ基、プロべ-ルチオ基、プロパルギルチオ基、フヱ-ルチオ基、 4-クロロフヱ-ルチ ォ基、チェ-ルチオ基、メチルスルフィエル基、ェチルスルフィ-ル基、プロピルスル フィエル基、イソプロピルスルフィエル基、ブチルスルフィ-ル基、イソブチルスルフィ -ル基、 sec-ブチルスルフィ-ル基、 tert-ブチルスルフィ-ル基、ペンチルスルフィ ニル基、イソペンチルスルフィエル基、 1-ェチルプロピルスルフィエル基、へキシルス ルフィ-ル基、シクロペンチルスルフィ-ル基、シクロへキシルスルフィ-ル基、プロぺ ニノレスノレフィ-ノレ基、プロノ レギノレスノレフィ-ノレ基、フエ-ノレスノレフィ-ノレ基、 4—クロ口 フエ-ノレスノレフィ-ノレ基、チェ-ノレスノレフィ-ノレ基、メチノレスノレホニノレ基、ェチノレスノレ ホ-ル基、プロピルスルホ-ル基、イソプロピルスルホ-ル基、ブチルスルホ -ル基、 イソブチルスルホ -ル基、 sec-ブチルスルホ -ル基、 tert-ブチルスルホ -ル基、ペン チルスルホ-ル基、イソペンチルスルホ -ル基、 1-ェチルプロピルスルホ-ル基、へ キシルスルホ -ル基、シクロペンチルスルホ -ル基、シクロへキシルスルホ -ル基、プ ロぺニノレスノレホニノレ基、プロノ ノレギノレスノレホニノレ基、フエニノレスノレホニノレ基、 4—クロ口
フエ-ルスルホ-ル基、チェ-ルスルホ-ル基、ァセチル基、プロピオ-ル基、ブタノ ィル基、ペンタノィル基、へキサノィル基、シクロペンタノィル基、シクロへキサノィル 基、ベンゾィル基、 4-クロ口ベンゾィル基、チェ-ルカルポ-ル基、メチルァミノ基、ェ チルァミノ基、プロピルアミノ基、イソプロピルアミノ基、ブチルァミノ基、イソブチルアミ ノ基、 sec-ブチルァミノ基、 tert-ブチルァミノ基、ペンチルァミノ基、イソペンチルァミノ 基、 1-ェチルプロピルアミノ基、へキシルァミノ基、シクロペンチルァミノ基、シクロへ キシルァミノ基、プロべ-ルァミノ基、プロパルギルアミノ基、フエ-ルァミノ基、 4-クロ 口フエ-ルァミノ基、チェ-ルァミノ基、ジメチルァミノ基、ジェチルァミノ基、ジプロピ ルァミノ基、ェチルメチルァミノ基、 1-ピロリジ -ル基、 1-ピベリジ-ル基、 4-モルホリ -ル基、 4-メチルビペラジン- 1-ィル基、メチルァミノカルボ-ル基、ェチルァミノカル ボニル基、プロピルアミノカルボ-ル基、イソプロピルアミノカルボ-ル基、ブチルアミ ノカルボ-ル基、イソブチルァミノカルボ-ル基、 sec-ブチルァミノカルボ-ル基、 tert -ブチルァミノカルボ-ル基、ペンチルァミノカルボ-ル基、イソペンチルァミノカルボ -ル基、 1-ェチルプロピルアミノカルボ-ル基、へキシルァミノカルボ-ル基、シクロ ペンチルァミノカルボ-ル基、シクロへキシルァミノカルボ-ル基、プロべ-ルァミノ力 ルポ-ル基、プロパルギルアミノカルボ-ル基、フエ-ルァミノカルボ-ル基、 4-クロ口 フエ-ルァミノカルボ-ル基、チェ-ルァミノカルボ-ル基、ジメチルァミノカルボ-ル 基、ジェチルァミノカルボ-ル基、ジプロピルアミノカルボ-ル基、ェチルメチルァミノ カルボ-ル基、力ルバモイル基、 1-ピロリジ-ルカルボ-ル基、 1-ピベリジ-ルカルボ ニル基、 4-モルホリ -ルカルボ-ル基、 4-メチルピペラジン- 1-ィルカルボ-ル基、ァ セチルァミノ基、プロピオ-ルァミノ基、ブタノィルァミノ基、ペンタノィルァミノ基、へキ サノィルァミノ基、シクロペンタノィルァミノ基、シクロへキサノィルァミノ基、ベンゾィル アミノ基、 4-クロ口べンゾィルァミノ基、チェ-ルカルポ-ルァミノ基、 1-ピロリジ-ルカ ルボニルァミノ基、 1-ピベリジ-ルカルポ-ルァミノ基、 4-モルホリニルカルボ-ルアミ ノ基、 4-メチルビペラジン- 1-ィルカルボ-ルァミノ基、メチルアミノスルホ -ル基、ェ チルアミノスルホ -ル基、プロピルアミノスルホ -ル基、イソプロピルアミノスルホ -ル 基、ブチルアミノスルホ -ル基、イソブチルアミノスルホ -ル基、 sec-ブチルアミノスル ホ-ル基、 tert-ブチルアミノスルホ -ル基、ペンチルアミノスルホ-ル基、イソペンチ
ルアミノスルホ -ル基、 1-ェチルプロピルアミノスルホ -ル基、へキシルアミノスルホ- ル基、シクロペンチルアミノスルホ-ル基、シクロへキシルアミノスルホ -ル基、プロべ -ルアミノスルホ -ル基、プロパルギルアミノスルホ -ル基、フエ-ルアミノスルホ -ル 基、 4-クロロフヱ-ルアミノスルホ-ル基、チェ-ルアミノスルホ -ル基、スルファモイ ル基、ジメチルアミノスルホ -ル基、ジェチルアミノスルホ -ル基、ジプロピルアミノス ルホ-ル基、ェチルメチルアミノスルホ -ル基、 1-ピロリジ-ルスルホ -ル基、 1-ピぺ リジ-ルスルホ -ル基、 4-モルホリ-ルスルホ -ル基、 4-メチルピペラジン- 1-ィルス ルホ-ル基、メチルスルホ -ルァミノ基、ェチルスルホ -ルァミノ基、プロピルスルホ二 ルァミノ基、イソプロピルスルホ -ルァミノ基、ブチルスルホ -ルァミノ基、イソブチルス ルホ -ルァミノ基、 sec-ブチルスルホ -ルァミノ基、 tert-ブチルスルホ -ルァミノ基、 ペンチルスルホ -ルァミノ基、イソペンチルスルホ -ルァミノ基、 1-ェチルプロピルス ルホ -ルァミノ基、へキシルスルホ -ルァミノ基、シクロペンチルスルホ -ルァミノ基、 シクロへキシルスルホ -ルァミノ基、プロべ-ルスルホ -ルァミノ基、プロパルギルス ルホ -ルァミノ基、フエ-ルスルホ -ルァミノ基、 4-クロ口フエ-ルスルホ -ルァミノ基、 チェ-ルスルホ -ルァミノ基、 1-ピロリジ-ルスルホ -ルァミノ基、 1-ピベリジ-ルスル ホ-ルァミノ基、 4-モルホリ-ルスルホ -ルァミノ基、 4-メチルピペラジン- 1-ィルスル ホ-ルァミノ基、メトキシカルボ-ル基、エトキシカルボ-ル基、プロポキシカルボニル 基、イソプロポキシカルボ-ル基、ブトキシカルボ-ル基、イソブトキシカルボ-ル基、 sec-ブトキシカルボ-ル基、 tert-ブトキシカルボ-ル基、ペントキシカルボ-ル基、ィ ソペントキシカルボ-ル基、 1-ェチルプロポキシカルボ-ル基、へキシルォキシカル ボ-ル基、シクロペンチルォキシカルボ-ル基、シクロへキシルォキシカルボ-ル基 、プロぺ-ルォキシカルボ-ル基、プロパルギルォキシカルボ-ル基、フエ-ノキシ力 ルボニル基、 4-クロ口フエ-ルォキシカルボ-ル基、チェ-ルォキシカルボ-ル基、 ジフルォロメチルォキシカルボ-ル基、トリフルォロメチルォキシカルボ-ル基、トリフ ルォロェチルォキシカルボ-ル基、ァセチルォキシ基、プロピオ-ルォキシ基、ブタ ノィルォキシ基、ペンタノィルォキシ基、へキサノィルォキシ基、シクロペンタノィルォ キシ基、シクロへキサノィルォキシ基、ベンゾィルォキシ基、 4-クロ口べンゾィルォキ シ基、チェ-ルカルポ-ルォキシ基が挙げられる。
式- χ χ χ6で表される置換基を例示すると、メトキシメチル基、メトキシェチル基、 エトキシメチル基、エトキシェチル基、フエノキシメチル基、アミノメチル基、アミノエチ ル基、メチルァミノメチル基、メチルアミノエチル基、ジメチルァミノメチル基、ジメチル アミノエチル基、フエ-ルァミノメチル基、力ルバモイルメチル基、力ルバモイルェチ ル基、メチルァミノカルボ-ルメチル基、メチルァミノカルボ-ルェチル基、ジメチルァ ミノカルボ-ルメチル基、ジメチルァミノカルボ-ルェチル基、フエ-ルァミノカルボ- ルメチル基、ァセチルァミノメチル基、ァセチルアミノエチル基、スルファモイルメチル 基、スルファモイルェチル基、メチルアミノスルホ -ルメチル基、メチルアミノスルホ- ルェチル基、フエ-ルアミノスルホ -ルメチル基、メタンスルホ -ルァミノメチル基、メタ ンスルホ-ルアミノエチル基、フエ-ルスルホ -ルァミノメチル基、メトキシカルボ-ル メチル基、エトキシカルボ-ルメチル基、エトキシカルボ-ルェチル基、ァセチルォキ シメチル基、ァセチルォキシェチル基、ベンゾィルォキシェチル基、メトキシェチルォ キシ基、エトキシェチルォキシ基、フエノキシェチルォキシ基、メトキシェチルチオ基 、エトキシェチルチオ基、フエノキシェチルチオ基、メトキシェチルォキシ基、エトキシ ェチルォキシ基、アミノメチルォキシ基、アミノエチルォキシ基、メチルアミノエチルォ キシ基、ジメチルアミノエチルォキシ基、力ルバモイルメチルォキシ基、力ルバモイル ェチルォキシ基、メチルァミノカルボ-ルメチルォキシ基、メチルァミノカルボ-ルェ チルォキシ基、ジメチルァミノカルボ-ルメチルォキシ基、ジメチルァミノカルボ-ル ェチルォキシ基、フエ-ルァミノカルボ-ルメチルォキシ基、ァセチルアミノエチルォ キシ基、スルファモイルメチルォキシ基、スルファモイルェチルォキシ基、メチルァミノ スルホ-ルメチルォキシ基、メチルアミノスルホ -ルェチルォキシ基、フエ-ルアミノス ルホ-ルメチルォキシ基、メタンスルホ-ルアミノエチルォキシ基、エトキシカルボ- ルェチルォキシ基、ァセチルォキシェチルォキシ基、ベンゾィルォキシェチルォキシ 基、メトキシェチルチオ基、エトキシェチルチオ基、アミノメチルチオ基、アミノエチル チォ基、メチルアミノエチルチオ基、ジメチルアミノエチルチオ基、力ルバモイルメチ ルチオ基、力ルバモイルェチルチオ基、メチルァミノカルボ-ルメチルチオ基、メチル ァミノカルボ-ルェチルチオ基、ジメチルァミノカルボ-ルメチルチオ基、ジメチルアミ ノカルボ-ルェチルチオ基、フエ-ルァミノカルボ-ルメチルチオ基、ァセチルァミノ
ェチルチオ基、スルファモイルメチルチオ基、スルファモイルェチルチオ基、メチルァ ミノスルホ-ルメチルチオ基、メチルアミノスルホ-ルェチルチオ基、フエ-ルアミノス ルホ-ルメチルチオ基、メタンスルホ-ルアミノエチルチオ基、エトキシカルボ-ルェ チルチオ基、ァセチルォキシェチルチオ基、ベンゾィルォキシェチルチオ基、メトキ シェチルスルホ -ル基、アミノメチルスルホ -ル基、ジメチルアミノエチルスルホ-ル 基、力ルバモイルメチルスルホ -ル基、力ルバモイルェチルスルホ -ル基、メチルアミ ノカルボ-ルェチルスルホ -ル基、ジメチルァミノカルボ-ルメチルスルホ -ル基、フ ェ-ルァミノカルボ-ルメチルスルホ -ル基、ァセチルアミノエチルスルホ-ル基、ス ルファモイルメチルスルホ -ル基、メチルアミノスルホ-ルェチルスルホ -ル基、フエ ニノレアミノスノレホニノレメチノレスノレホニノレ基、メタンスノレホニノレアミノエチノレスノレホニノレ 基、エトキシカルボ-ルェチルスルホ -ル基、ァセチルォキシェチルスルホ -ル基、 ベンゾィルォキシェチルスルホ -ル基、メトキシェチルカルボ-ル基、アミノメチルカ ルボニル基、ジメチルアミノエチルカルボ-ル基、力ルバモイルメチルカルボ-ル基、 力ルバモイルェチルカルボ-ル基、メチルァミノカルボ-ルェチルカルボ-ル基、ジ メチルァミノカルボ-ルメチルカルボ-ル基、フエ-ルァミノカルボ-ルメチルカルボ ニル基、ァセチルアミノエチルカルボ-ル基、スルファモイルメチルカルボ-ル基、メ チルアミノスルホ -ルェチルカルボ-ル基、フヱ -ルアミノスルホ -ルメチルカルボ- ル基、メタンスルホ-ルアミノエチルカルボ-ル基、エトキシカルボ-ルェチルカルボ ニル基、ァセチルォキシェチルカルボ-ル基、ベンゾィルォキシェチルカルボ-ル 基、メトキシェチルァミノ基、ジメチルアミノエチルァミノ基、力ルバモイルメチルァミノ 基、力ルバモイルェチルァミノ基、メチルァミノカルボ-ルェチルァミノ基、ジメチルァ ミノカルボ-ルメチルァミノ基、フエ-ルァミノカルボ-ルメチルァミノ基、ァセチルアミ ノエチルァミノ基、スルファモイルメチルァミノ基、メチルアミノスルホ -ルェチルァミノ 基、フエ-ルアミノスルホ -ルメチルァミノ基、メタンスルホ-ルアミノエチルァミノ基、 エトキシカルボ-ルェチルァミノ基、ァセチルォキシェチルァミノ基、ベンゾィルォキ シェチルァミノ基、メトキシェチルァミノカルボ-ル基、力ルバモイルメチルァミノカル ボ-ル基、メトキシェチルカルボ-ルァミノ基、力ルバモイルメチルカルボ-ルァミノ 基、メトキシェチルアミノスルホ -ル基、力ルバモイルメチルアミノスルホ -ル基、メトキ
シェチルスルホ -ルァミノ基、力ルバモイルメチルスルホ -ルァミノ基、メトキシェチル ォキシカルボ-ル基、力ルバモイルメチルォキシカルボ-ル基、メトキシェチルカル ボニルォキシ基、力ルバモイルメチルカルボ-ルォキシ基が挙げられる。
[0023] 式- Χ^Χ^Χ^Χ1-で表される一部飽和された 5〜: L0員環構造を有する R4としては、 例えばインダン、 1,2,3,4-テトラヒドロナフタレン、 6,7,8,9-テトラヒドロ- 5H-ベンゾ [7]ァ ヌレン、 2,3-ジヒドロ- 1-ベンゾフラン、 1,3-ベンゾジォキソール、 2,3-ジヒドロ- 1,4-ベ ンゾジォキサン、 3,4-ジヒドロ- 2H- 1,5-ベンゾジォキセピン、 3,4-ジヒドロ- 2H- 1,4-ベ ンゾキサジン、 1,2,3,4-テトラヒドロキノキサリン、 2,3-ジヒドロ- 1,4-ベンゾキサンチン、 3 ,4-ジヒドロ- 2H- 1,2-ベンゾチアジン 1,1-ジォキシドが挙げられる。
[0024] 本発明のチアゾール誘導体は、その薬学的に許容される塩又はその溶媒和物であ つても良い。以下、本発明のチアゾール誘導体、その薬学的に許容される塩及びそ の溶媒和物を含めて、「本発明の化合物」ともいう。
[0025] 本明細書において、薬学的に許容される塩とは、例えば、塩酸塩、臭化水素酸塩、 ヨウ化水素酸塩、リン酸塩、硫酸塩、硝酸塩のような鉱酸塩;メタンスルホン酸塩、ェ タンスルホン酸塩、ベンゼンスルホン酸塩、 p—トルエンスルホン酸塩のようなスルホ ン酸塩;シユウ酸塩、酒石酸塩、クェン酸塩、マレイン酸塩、コハク酸塩、酢酸塩、安 息香酸塩、マンデル酸塩、ァスコルビン酸塩、乳酸塩、ダルコン酸塩、リンゴ酸塩のよ うな有機酸塩等の酸付加塩、グリシン塩、リジン塩、アルギニン塩、オル-チン塩、グ ルタミン酸塩、ァスパラギン酸塩のようなアミノ酸塩、あるいはリチウム塩、ナトリウム塩 、カリウム塩、カルシウム塩、マグネシウム塩のような無機塩又はアンモ-ゥム塩、トリ ェチルァミン塩、ジイソプロピルアミン塩、シクロへキシルァミン塩のような有機塩基と の塩であり得、好適には塩酸塩、臭化水素酸塩、リン酸塩、硫酸塩、メタンスルホン 酸塩、 P-トルエンスルホン酸塩、シユウ酸塩、酒石酸塩、クェン酸塩、酢酸塩、乳酸 塩、グルタミン酸塩、ァスパラギン酸塩、ナトリウム塩、カリウム塩、アンモ-ゥム塩又は トリェチルァミン塩があげられ、好ましくはナトリウム塩、塩酸塩又は硫酸塩であり、より 好ましくは塩酸塩である。なお、本明細書において、本発明の化合物には、生体内 において代謝されて本発明の本発明の化合物に変換される化合物、いわゆるプロド ラッグち含まれる。
[0026] 本明細書において、溶媒和物としては、水和物など薬学的に許容される溶媒和物 があげられる。本発明のチアゾール誘導体は、大気にさらされ、又は再結晶すること Π.
などにより、水分を吸収し、吸着水がつく場合や、水和物となる場合がある。本発明 のチアゾール誘導体の薬学的に許容される溶媒和物は、そのような水和物をも含む
[0027] 本発明のチアゾール誘導体は、不斉中心を持つことがあり、その場合種々の光学 異性体又は配置のものが存在する。したがって、本発明の化合物は、(+ )もしくは( ―)の別々の光学活性体として、又はラセミ体もしくは(士)混合物として存在し得る。 また、不斉中心を 2個以上持つ化合物の場合には、さらにそれぞれの光学異性によ るジァステレオマーも存在する。本発明のチアゾール誘導体は、これらすベての型を 、任意の割合で含む。たとえば、ジァステレオマーは当業者によく知られた方法、たと えば分別結晶法等によって分離することができ、また、光学活性体はこの目的のため によく知られた有機化学的手法によって得ることができる。また、本発明のチアゾール 誘導体は、シス体、トランス体などの異性体が存在することがある。本発明のチアゾー ル誘導体は、それらの異性体及びそれらの異性体を任意の割合で含んだものも含む
[0028] 以下、本化合物に係る化合物の製造方法を詳細に説明するが、例示されたものに 特に限定されない。また、反応にしょうする溶媒においても、各反応を阻害しないもの であればよぐ特に下記の記載に限定されない。
[反応式 1]
(III) (IV)
(式中、
R
2および!?
3はそれぞれ前記と同意義である。 X
aは塩素原子、臭素原子、 ヨウ素原子、メタンスルホ -ルォキシ基またはトシルォキシ基を示す。 )
この工程は、化合物(Π)と化合物(III)を縮合してァミノチアゾール誘導体 (IV)を得 る工程である。この反応に使用する溶媒としては、エタノール、メタノール、 Ν,Ν-ジメ
チルホルムアミド、クロ口ホルム等が挙げられ、反応は 0〜100°Cで行うことが出来る。 この際、生成する塩酸、または臭化水素酸を捕捉するために塩基を添加してもよい。 添加する塩基としてトリェチルァミン、ジイソプロピルェチルァミン等のアミン類または 炭酸カリウムや炭酸水素ナトリウム等の無機塩基等が挙げられる。また得られた化合 物 (IV)を酸との塩として得た場合は、酢酸ェチル等の有機溶媒に溶解し、無機塩基 の水溶液で洗浄を行うことにより、フリーのァミンとして単離することも出来る。
[反応式 2]
(V) (VI)
(式中、
R
2および!?
3はそれぞれ前記と同意義である。 )
この工程は、化合物 (V)のァミノ基に R3を導入し化合物 (VI)を得る工程である。 例えば、脱離基で置換された R3、即ち R3— Xa (Xaは前記と同意義である)を用い化合 物 (V)と反応を行う場合は、反応に使用する溶媒としてはジクロロメタン、クロ口ホルム 、 Ν,Ν-ジメチルホルムアミド、エタノール等が挙げられ、反応は 0〜100°Cで行うことが 出来る。この際、反応は適当な塩基を用いて行うことが出来、塩基としてとしてトリェチ ルァミン、ジイソプロピルェチルァミン等のアミン類または炭酸カリウムや炭酸水素ナト リウム等の無機塩基等が挙げられる。
[0031] また例えば、別の例としては、アルデヒド誘導体と還元剤を用いる還元的アミノ化反 応を挙げることが出来る。還元剤にはトリァセトキシ水素化ホウ素ナトリウムや水素化 シァノホウ素ナトリウム、水素化ホウ素ナトリウム等が挙げられ、必要に応じて氷酢酸 やトシル酸等の酸を添加することが出来る。この反応に使用する溶媒としてはジクロ口 メタンやクロ口ホルム、テトラヒドロフラン、ジォキサン、 1,2-ジクロロエタン等が挙げら れ、反応は- 20〜100°Cで行うことが出来る。
[反応式 3]
[0032] [化 4]
[0033]
[0034]
[0035]
(式中、
R
2および R
4は前記と同意義である。 x
bはァミノ基の保護基を示す。 ) この工程は、化合物 (vm)の保護基 x
bを脱保護して化合物 (IX)を得る工程である。 例えば、 x
bが 4-メトキシベンジル基等の酸で脱保護される基の場合は、塩酸、硫酸、 トリフルォロ酢酸、 P-トルエンスルホン酸、メタンスルホン酸等の酸を用い、脱保護す
ることができる。この際、有機溶媒又は水で希釈又は溶解して行うことが出来、反応 温度は- 50°Cから 50°Cで行うことができる。有機溶媒としては、例えばエタノール、メ タノール、テトラヒドロフラン、 Ν,Ν-ジメチルホルムアミド、ジクロロメタン、クロ口ホルム、 1,2-ジクロロェタン等が挙げられる。
[反応式 5]
[0036] [化 6]
R " R 3 a
R' 4
S O 》 S o
R2 (X) R- (xi)
(式中、
R
4および X
aは前記と同意義である。 R
4aは、式— X
1— X
2— X
3また は式 X
1— X
5— X
6で表される基のうち X
1力 酸素原子、硫黄原子または式 NX
4— で表されるものを示す。 X
1、 x
2、 x
3、 X
4、 X
5および X
6は、前記と同意義である。 ) この工程は、脱離基を置換基に有する化合物 (X)を用い置換反応を行って R
4を導 入した化合物 (XI)を得る工程である。
[0037] 例えば、 Xaがフッ素の場合、求核置換反応を用いてァミン、ァ-リン、アルコール及 びチオールを導入することが出来る。また例えば、 2-メチルスルホ -ルエタノールな どを用いて同様の置換反応を行うと生成物としてハロゲン原子が水酸基に変換され た化合物 (XI)を得ることができる。これら反応に使用する溶媒としてジメチルスルホキ シド、 Ν,Ν-ジメチルホルムアミド、テトラヒドロフラン、トルエン等が挙げられ、反応は 0 〜200°Cで行うことが出来る。またこの反応は適当な塩基を添加して反応を行うこと が出来、塩基として tert-ブトキシカリウム、 n-ブチルリチウム、リチウムジイソプロピル アミド、リチウムへキサメチルジシラジド等の有機塩基や水素化ナトリウム、炭酸力リウ ム等の無機塩基が挙げられる。また必要に応じて金属触媒を添加することも出来る。 金属触媒としてはトリス (ジベンジリデンアセトン)ジパラジウムや酢酸パラジウム等が 挙げられる。
[0038] また例えば、金属触媒と有機金属化合物を用いたクロスカップリング法により、ビア リール等の炭素 炭素結合を構築することも出来る。この反応に使用する溶媒として テトラヒドロフラン、トルエン、 1,2-ジメトキシェタン等が举げられ、反応温度は 0〜200
°Cで行うことが出来る。金属触媒としてはテトラキス(トリフエニルホスフィン)パラジウム ゃトリス(ジベンジリデンアセトン)ジパラジウムやビス(ァセチルァセトナト)ニッケル等 が挙げ Vられる。有機金属化合物にはグリニャール試薬、有機アルミニウム化合物、ァ リールボロン酸 oが挙げられる。またこの反応は適当な塩基を添加して反応を行うこと が出来、塩基としてはトリエチルァミン、ジイソプロピルアミン等のアミンゃ水酸ィ匕ナトリ ゥム、炭酸カリウム、炭酸ナトリウム等の無機塩基が挙げられる。
[反応式 6]
[0039] [化 7]
[0040]
(式中、 R
2、 R
3および R
4は前記と同意義である。 R
4bは、式— X
1— X
2— X
3または式 ー^ー
5—
6で表される基のぅち^カ 酸素原子で表されるものを示す。 X
1、 X
2、 X
3 、 X
4、 X
5および X
6は、前記と同意義である。 )
この工程は、水酸基を置換基に有する化合物 (XII)を用いて化合物 (XIII)を得るェ 程である。
[0041] 例えば、ハロゲン化アルキルを用いた反応で、水酸基のアルキル化を行うことがで きる。この反応は適当な塩基を添加して反応を行うことが出来、塩基として tert-ブトキ シカリウム、 n-ブチルリチウム、リチウムジイソプロピルアミド、リチウムへキサメチルジ シラジド等の有機塩基や水素化ナトリウム、炭酸カリウム等の無機塩基が挙げられる。 これら反応に使用する溶媒としてジメチルスルホキシド、 Ν,Ν-ジメチルホルムアミド、 テトラヒドロフラン、トルエン等が挙げられ、反応は 0〜200°Cで行うことが出来る。
[反応式 7]
[0042] [化 8]
R ,X1 -X5-NX7Xb R 3 .Χ1 -Χ5-ΝΗΧ'
R4
Ν、
R
o R
-S 〇
2
R (XIV) 2
R (XV)
(式中、
X
1、 x
5および x
7は前記と同意義である。 x
bは、ァミノ基の保護
基を表す。 )
この工程は、 A環置換基に保護されたアミノ基を有する化合物 (XIV)を用いて脱保 護を行 、、ァミン誘導体 (XV)を得る工程である。
この脱保護については PROTECTIVE GROUPS IN ORGANIC SYNTHESIS, THE ODORA W. GREENE and PETER G. M WUTS著に記載の種々の方法を用いること が出来る。例えば X^¾ert-ブトキシカルボニル基、トリチル基、 トロベンゼンスル フエニル基等の酸で脱保護される基の場合は、塩酸、硫酸、トリフルォロ酢酸、 P-トル エンスルホン酸、メタンスルホン酸等の酸を用い、脱保護することができる。この際、 有機溶媒又は水で希釈又は溶解して行うことができ、反応は- 50°C力も 50°Cで行うこ とができる。有機溶媒としては、例えばエタノール、メタノール、テトラヒドロフラン、 Ν,Ν -ジメチルホルムアミド、ジクロロメタン、クロ口ホルム、 1,2-ジクロロェタン等があげられ る。更に例えば、 Xbがべンジルォキシカルボニル基等の加水素分解反応により脱保 護される基の場合は、パラジウム等の金属触媒を用いた加水素分解反応により脱保 護することができる。溶媒としては、エタノール、メタノール、テトラヒドロフラン、酢酸ェ チル等の反応に関与しない溶媒を用いることができる。反応は 0〜100°Cで行うこと ができる。また、この反応に水素ガスを用いることもできるし、ぎ酸一ぎ酸アンモ-ゥム を例とする試薬の組み合わせで行うこともできる。更に例えば、 xbが塩基で脱保護さ れるフルォレニルォキシカルボ-ル基等の保護基の場合は、ジェチルァミン、ピペリ ジン、アンモニア、水酸化ナトリウム、炭酸カリウム等の塩基を用いて脱保護すること ができる。これらの塩基は、単独で、あるいは溶媒に希釈又は懸濁してして用いること ができる。この際、溶媒としては水、エタノール、メタノール、テトラヒドロフラン、 Ν,Ν- ジメチルホルムアミド、ジクロロメタン、クロ口ホルム、 1,2-ジクロロエタン等を用いること ができる。反応温度は 0〜: L00°Cで行うことができる。更に例えば、 Xbがァリルォキシ カルボニル基等の金属触媒により脱保護される基の場合は、テトラキス(トリフエニル ホスフィン)パラジウム等を触媒又は試薬として用いることにより脱保護することができ る。この際、ジクロロメタン、クロ口ホルム、テトラヒドロフラン等の反応に関与しない溶 媒中で行うことができる。反応は 0〜100°Cで行うことができる。
[反応式 8]
[0043] [化 9]
(式中、 R R R3および R4は前記と同意義である。 Xeは、水素原子または炭素数 1か ら 5のアルキル基を表す。)
この工程は、化合物(XVI)を用いて還元反応を行!ヽ、ヒドロキシメチル体 (XVII)を 得る工程である。
[0044] この反応は、適当な還元法を用いた条件下で反応を行うことができる。用いる還元 法としては、例えば水素化リチウムアルミニウムを用いる方法があげられる。用いる溶 媒としては、テトラヒドロフラン、ジォキサン等の反応に関与しない溶媒を用いることが できる。反応は- 20〜100°Cで行うことができる。
[反応式 9]
[0045] [化 10]
[0046] (式中、 、 R2、 R3および R4は前記と同意義である。 XDは、 C のアルキル基を表す。 )
1-6
この工程は、化合物 (xvm)を用いて加水分解反応を行!ヽ、カルボン酸 (XIX)を得る 工程である。
[0047] この反応は、水酸化ナトリウム、水酸ィ匕カリウム、水酸化リチウム等を用いて行うこと ができる。用いる溶媒としては、水、メタノール、エタノール、テトラヒドロフラン、ジォキ サン等を用いることができる。反応は- 20〜100°Cで行うことができる。
[反応式 10]
[0048] [化 11]
-CONXW
(式中、
R
4、 X
7および X
8は前記と同意義である。 )
この工程は、カルボン酸 (XIX)を用いてァミンとの縮合反応を行い、アミド体 (XX)得 る工程である。
[0049] 例えば、脱水縮合剤を用いてアミド化を行うことができる。脱水縮合剤には例えば、 1-ェチル -3- (3-ジメチルァミノプロピル)カルボジイミド ·塩酸塩、ジシクロへキシルカ ルボジイミド、ジフエ-ルホスホリルアジド、カルボ-ルジイミダゾール等があげられ、 必要に応じて 1-ヒドロキシベンゾトリァゾール、ヒドロキシスクシンイミド等の活性化剤 を用いることができる。反応溶媒としては、ジクロロメタン、クロ口ホルム、 1,2-ジクロロ ェタン、 Ν,Ν-ジメチルホルムアミド、テトラヒドロフラン、ジォキサン、トルエン、酢酸ェ チル等があげられる。この際、塩基を用いて反応を行うことができ、塩基の例としては 、トリエチルァミン、ジイソプロピルェチルァミン等のアミン類、 2-ェチルへキサン酸ナ トリウム、 2-ェチルへキサン酸カリウム等の有機酸塩、炭酸カリウム等の無機塩基があ げられる。反応は- 50〜50°Cで行うことができる。
[0050] また、例えば、カルボン酸とクロル炭酸エステル等力 得られる混合酸無水物を用 いてアミド化することができる。これらの反応の溶媒としては、テトラヒドロフラン、ジォ キサン、ジクロロメタン、クロ口ホルム、 Ν,Ν-ジメチルホルムアミド、トルエン、酢酸ェチ ル等の反応に関与しない溶媒があげられる。この際、塩基を用いて行うことができ、 塩基の例としては、トリェチルァミン、ジイソプロピルェチルァミン等のアミン類、 2-ェ チルへキサン酸ナトリウム、 2-ェチルへキサン酸カリウム等の有機酸塩、炭酸カリウム 等の無機塩基があげられる。これらの反応は、 - 50〜50°Cで行うことができる。
[0051] また、例えばアミド化反応は、ァシルク口リドゃァシルプロミド等のァシルハライドに 変換した後に、ァミンとの縮合を行うことができる。ァシルノヽライドへの変換は、チォ- ルクロリド、ォキシ塩化リン、五塩化リン、塩ィ匕オギザリル等を用い、ジクロロメタン、ク ロロホルム、 1,2-ジクロロェタン、テトラヒドロフラン、ジォキサン、トルエン、酢酸ェチル 等の反応に関与しない溶媒中で行うことができる。この反応は、 - 50〜100°Cで行うこ とができる。ァミンとァシルハライドとの縮合は、ジクロロメタン、クロ口ホルム、 1,2-ジク ロロ工タン、テトラヒドロフラン、ジォキサン、トルエン、酢酸ェチル等の反応に関与し ない溶媒中で行うことができる。この際、塩基を用いて行うことができ、塩基の例として
は、トリェチルァミン、ジイソプロピルェチルァミン等のアミン類、 2-ェチルへキサン酸 ナトリウム、 2-ェチルへキサン酸カリウム等の有機酸塩、炭酸カリウム等の無機塩基が あげられる。これらの反応は、 - 50〜100°Cで行うことができる。
[0052] またアミド化反応の別の例として、例えば、 1-ベンゾトリアゾリルエステルゃスクシン ィミジルエステル等の活性エステルを用いて行うことができる。反応溶媒としては、ジ クロロメタン、クロロホノレム、 1,2—ジクロロェタン、 Ν,Ν—ジメチノレホノレムアミド、テトラヒドロ フラン、ジォキサン、トルエン、酢酸ェチル等があげられる。反応は- 50〜50°Cで行う ことができる。
[反応式 11]
[0053] [化 12]
O O R2
R1-C-CH2-R2 " R^C-CH ^
(XXI) (I I)
(式中、
R
2および x
aは前記と同意義である。 )
この工程は、化合物 (XXI)に xaを導入して化合物(Π)を得る工程である。 この工程には、例えば臭素を用いた臭素化反応を用いることができる。用いる溶媒と しては、メタノール、クロ口ホルム、テトラヒドロフラン、ジォキサン等の溶媒を用いること ができる。反応は- 20〜100°Cで行うことができる。
[反応式 12]
[0054] [化 13]
R'— S-CH -S-C- O R6 O R6
(XXI I) (XXI I I)
(式中、 R2、 R5、 R6、 R7および XAは前記と同意義である。 )
この工程は、化合物 (ΧΧΠ)を化合物 (ΧΧΙΠ)に変換する工程である。
例えば、化合物(XXII)に n-ブチルリチウムなどの塩基を作用させ、次いで R2XaCHC 0 Me、 R2XaCHCO Et、 R2XaCHCOClなどを作用させることにより化合物(XXIII)を得
2 2
ることができる。溶媒としては、テトラヒドロフラン、トルエン、ジェチルエーテル、ジォ
キサン、クロ口ホルム等を用いることができる。反応は- 80〜50°Cで行うことができる。
[反応式 13]
[0055] [化 14]
O / ° Me \ O
R1-C-0-Xc R1_C_N'w I *- R1-C-CH,
(XXIV) \ (XXV) ノ (XXVI)
(式中、 R1および Xeは前記と同意義である。 )
この工程は、化合物 (XXIV)を化合物 (XXVI)に変換する工程である。
例えば、メチルリチウムやメチルマグネシウムブロミド /Ν,Ο-ジメチルヒドロキシルァミン 等を用いて、直接ィ匕合物 (XXIV)力 化合物 (XXVI)を得ることができる。用いる溶媒 としては、ジクロロメタン、クロロホノレム、 1,2-ジクロロエタン、テトラヒドロフラン、ジォキ サン、トルエンなどがあげられ、反応は- 78〜50°Cで行うことができる。
[0056] また例えば、化合物 (XXIV)を化合物 (XXV)に変換した後に、化合物 (XXVI)を合 成することができる。化合物 (XXV)は、化合物 (XXIV)を水酸化ナトリウムや水酸化力 リウム、水酸化リチウム等を用いて加水分解しカルボン酸とした後に、縮合剤と Ν,Ο- ジメチルヒドロキシルァミンを用いて縮合を行うことで得ることができる。加水分解は、 溶媒として水、メタノール、エタノール、テトラヒドロフラン、ジォキサン等を用いること ができ、反応は- 20〜100°Cで行うことができる。縮合は、反応式 10に記載した方法と 同様にして行うことができる。また、メチルマグネシウムブロミド /Ν,Ο-ジメチルヒドロキ シルァミン等を用いて、直接化合物 (XXIV)を化合物 (XXV)に変換することができる 。用いる溶媒としては、ジクロロメタン、クロ口ホルム、 1,2-ジクロロェタン、テトラヒドロフ ラン、ジォキサン、トルエン等が挙げられ、反応は- 78〜50°Cで行うことができる。
[反応式 14]
(XXVii) (XXVIil) (XXIX)
(式中、 Xdは前記 R4の置換基として定義した置換基と同意義である。 )
この工程は、化合物 (χχνπ)を化合物 (xxvm)を経由して化合物 (XXIX)に変換す る工程である。
はじめに化合物(XXVII)をィ匕合物(XXVIII)に変換するには、例えば、 NaNO - HC1を
2 用いることができる。溶媒としては水を用いることができ、反応は一 20〜20°Cで行うこ とがでさる。
上記方法で得られたィ匕合物 (XXVIII)をィ匕合物 (XXIX)に変換するには、例えば、 SO と CuClを用いることができる。反応は 20〜50°Cで行うことができる。
2 2
[反応式 15]
(XXX) (XXXI)
(式中、 xeは前記の置換基 Υ群力も選ばれる置換基と同意義である。 )
この工程は、化合物 (XXX)をィ匕合物 (XXXI)に変換する工程である。例えば、クロ口 硫酸を用い、無溶媒又は反応に関与しないジクロロメタン、クロ口ホルム、 1, 2—ジク ロロエタン、テトラヒドロフラン、ジォキサン、トルエンなどを用いて行うことができる。反 応は 20〜100°Cで行うことができる。
[反応式 16]
( XXII) (xxxm)
(式中、 xfは置換されてもよい C のアルキル基を表す。 xdは前記と同意義である。 )
1-6
この工程は、化合物 (XXXII)をィ匕合物 (XXXIII)に変換する工程である。例えば、クロ 口硫酸を用い、無溶媒又は反応に関与しないジクロロメタン、クロ口ホルム、 1, 2—ジ クロ口エタン、テトラヒドロフラン、ジォキサン、トルエンなどを用いて行うことができる。 反応は— 20〜100°Cで行うことができる。
[反応式 17]
[0060] [化 18]
(式中、 Xgは CH又は窒素原子を表す。 Xdは前記と同意義である。 )
この工程は、化合物 (XXXIV)をィ匕合物 (XXXV)に変換する工程である。例えば、 n- ブチルリチウムを作用させた後にスルフリルクロリド用いて合成することができる。溶媒 としては、テトラヒドロフラン、 n キサン、ジクロロメタン、クロロホノレム、 1, 2—ジクロ 口エタン、ジォキサン、トルエンなどを用いて行うことができる。反応は— 78 20°Cで 行うことができる。
[0061] 本発明の化合物は、 11 β - HSD1活性阻害作用を有し、 11 β - HSD1の関与する疾 患、例えば、肝臓における糖新生抑制、又は内臓脂肪の蓄積抑制に有効に使用で きる。すなわち、本発明の化合物は、 11 18 - HSD1の阻害剤;肝臓における糖新生抑 制、又は内臓脂肪の蓄積抑制などの医薬として利用できる。本発明の化合物は、単 独又は薬学的あるいは薬剤学的に許容される担体又は希釈剤と共に投与することが できる。本発明の化合物を 11 18 - HSD1阻害剤などとして使用する場合は、本発明の 化合物をそのまま経口投与、又は非経口投与してもよい。また、本発明の化合物を 有効成分として含む剤として経口投与、又は非経口投与してもよい。非経口投与とし ては、注射による静脈内投与があげられる。
[0062] 上記の剤を経口投与する場合は、希釈剤、賦形剤、崩壊剤、結合剤、滑沢剤、抗 酸化剤、コーティング剤、界面活性剤、可塑剤、着色剤、矯味矯臭剤などを混合して 、本発明の化合物を有効成分として含む顆粒剤、カプセル剤、錠剤、薬用ドロップ、 トローチ、硬質キャンディ、粉末剤、噴霧剤、などの製剤として投与されてもよい。また 、適宜に甘味付け、又は香味付けを行っても良い。上記の剤を非経口投与する場合 は、本発明の化合物を有効成分として含む注射剤、点滴剤、点眼剤、クリーム、膏薬 、坐薬、ゼリー、ジエル、ペースト、ローション、軟膏、水性懸濁液などの製剤として投
与されてもよい。製剤化する際には、通常の製剤化の方法を使用できる。
[0063] 本発明の化合物は経口投与又は非経口投与でき、例えば 1回につき lmg〜1000m g、好ましくは 10mg〜200mg投与でき、例えば 1日当り 1回〜 3回投与すればよい。本 発明の化合物の投与量は、患者の年齢、体重及び症状によって適宜調整することが できる。
[0064] 本発明の化合物の 11 β - HSD1活性阻害を評価するには、例えば、実施例に記載 した方法など、公知の手法に従って行なうことができる。
[0065] 以下に、参考例、実施例及び試験例を示して本発明を具体的に説明するが、例示 されたものに特に限定されない。
[0066] 参考例 1 4- (テトラヒドロ- 2Η-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンの合成
(1) 1- (テトラヒドロ- 2Η-ピラン- 4-ィル)エタノンの合成
窒素雰囲気下、 4-テトラヒドロピランカルボン酸メチルエステル(21. 44g)と Ν,Ο-ジ メチルヒドロキシルァミン塩酸塩(18. 77g)のテトラヒドロフラン(250ml)溶液に、 1 5°Cで 3Mメチルマグネシウムブロミドのジェチルエーテル溶液(197ml)を 1時間かけ て滴下した後、室温で 2時間攪拌した。反応液を氷冷した 1M塩酸水溶液にあけ、ク ロロホルムで抽出し、飽和食塩水で洗浄した。有機層を無水硫酸ナトリウムで乾燥後 、乾燥剤を濾去して溶媒を減圧留去し、黄色油状物質として表題ィ匕合物 (21. 98g) を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.61— 1.86 (m, 4 H), 2.16 (s, 3 H), 2.4 6 - 2.63 (m, 1 H), 3.35 - 3.49 (m, 2 H), 3.93 - 4.08 (m, 2 H).
(2) 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンの合成
1- (テトラヒドロ- 2H-ピラン- 4-ィル)エタノン(21. 98g)のメタノール(360ml)溶液に 臭素(7. 26ml)をカ卩ぇ 65°Cで 30分攪拌した後、室温に戻し、チォゥレア(11. 27g) を加え一晩攪拌した。反応終了後クロ口ホルムと飽和炭酸水素ナトリウム水溶液を加 え、室温で 2時間攪拌した後、クロ口ホルムで抽出し飽和食塩水で洗浄した。有機層 を無水硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。得られた残 渣にへキサンをカ卩ぇ析出した固体をろ取した。この固体をメタノールで再結晶して、 薄黄色粉末として表題ィ匕合物(14. 13g)を得た。
1H NMR (300 MHz, DMSO- D6) δ 1.35 - 1.69 (m, 2 H), 1.68 - 1.87 (m, 2 H), 2.47 - 2.72 (m, 1 H), 3.30 - 3.44 (m, 2 H), 3.77 - 3.94 (m, 2 H), 6.11 (s, 1 H), 6.81 (br s, 2 H).
参考例 2 4-シクロへキシル -1,3-チアゾール -2-アミン臭化水素酸塩の合成
1-シクロへキシルエタノン(2. 00g)のメタノール溶液(20ml)に臭素(812 1)をカロ えて室温で 5分攪拌した後、チォゥレア(1. 02g)を加え、 1晚攪拌した。不溶物を濾 去した後、濾液を減圧下濃縮し、薄黄色固体として表題ィ匕合物(2. 73g)を得た。 1H NMR (300 MHz, DMSO- D6) δ 1.32 - 1.42 (10 Η), 2.44 - 2.60 (1 Η), 6.49 (s, 1 H).
参考例 3 3 フルォロ 2 メチルベンゼンスルホ-ルクロリドの合成
(1)亜硫酸ガスの 30% (w/w)酢酸溶液の調製
3頸フラスコに亜硫酸ナトリウム(100g)を入れ、メカ-カルスターラーで激しく攪拌し ながら、硫酸 (45ml)を滴下漏斗で、ゆっくり滴下した。発生する亜硫酸ガスをテフ口 ンチューブを通して水浴につけた 2頸フラスコ中の酢酸(28g)に吹き込んだ。 2頸フ ラスコの内容物の重量が 11. 78g増えたところで硫酸の滴下を終了した。
(2) 3 -フルォロ 2—メチルベンゼンスルホ-ルクロリドの合成
3 フルオロー 2—メチルァ-リン(1. 06g)に氷冷下で濃塩酸(2. Oml)をカ卩えた。 懸濁液に亜硝酸ナトリウム(507mg)の水溶液 (2. Oml)を内温を 7〜5°Cに保ちな がらゆっくりと約 1時間かけて滴下した。反応液を氷冷下で 1時間攪拌した後、塩ィ匕マ グネシゥム 6水和物(813mg)を加え、 10分間攪拌した後、濾過をした。濾液を参考 例 3—1で調製した亜硫酸ガスの 30% (w/w)酢酸溶液(11. Og)中に塩化第二銅(3 82mg)を含む懸濁液に、氷冷下で少しずつ加えた。そのまま室温で 2時間攪拌した 後、反応液に氷(17g)を加えて攪拌し、ジェチルエーテル(100ml)で抽出した。有 機層を飽和食塩水で洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を濾去 し、溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶 媒 へキサン:クロ口ホルム = 20 : 1〜15 : 1〜12 : 1〜10 : 1〜5 : 1)で精製し、無色油 状物質として表題化合物(789mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.70 (d, J=2.5 Hz, 3 H), 7.37 - 7.45 (m,
2 H), 7.87 - 7.94 (m, 1 H).
参考例 3の方法と同様にして、 3 クロロー 2 メチルベンゼンスルホユルク口リド、 3 ブロモ 2 メチルベンゼンスルホニルクロリド、 3 クロロー 2—メトキシベンゼンス ルホ-ルクロリド、 2 クロ口一 3—メチルベンゼンスルホ-ルクロリド、 2—メチル 3 トリフルォロメチルベンゼンスルホ-ルクロリド、 4ーブロモー 2, 6 ジメチルベンゼ ンスルホ-ルクロリド、 4 クロロー 2, 6 ジメチルベンゼンスルホ-ルクロリド、 3 フ ェ-ノレベンゼンスノレホニノレクロリド、 3— (4—クロ口フエ-ノレ)ベンゼンスノレホニノレクロリ ドなどを合成した。
参考例 4 5 クロ口一 3 イソプロピル 1 ベンゾチォフェン一 2 スルホ-ルクロ リドの合成
5 クロ口一 3—イソプロピル一 1—ベンゾチォフェン(552mg)のクロ口ホルム溶液(1 Oml)に、氷冷下、クロ口硫酸(0. 52ml)のクロ口ホルム(3ml)溶液を滴下し、室温で 5時間攪拌した。反応液を氷水にあけ、クロ口ホルムにて抽出した。有機層を水、飽和 食塩水にて洗浄し、無水硫酸ナトリウムにて乾燥後、乾燥剤を濾去して溶媒を減圧 留去し、褐色粉末として表題ィ匕合物 (469mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.57 (d, J=7.2 Hz, 6 H), 4.17 - 4.36 (m, 1 H), 7.53 (dd, J=8.8, 2.0 Hz, 1 H), 7.82 (d, J=8.8 Hz, 1 H), 8.15 (d, J=2.0 Hz, 1 H) 参考例 4の方法と同様にして、 4一(4 ブロモフエ-ル)ベンゼンスルホユルク口リド、 4一(4 シァノフエ-ル)ベンゼンスルホ-ルクロリド、 4一(4 トリフルォロメチルフエ -ル)ベンゼンスルホ-ルクロリド、 4— (3—クロ口フエ-ル)ベンゼンスルホユルクロリ ド、 7—メトキシカルボ-ルー 3—メチルー 1 ベンゾチォフェン 2—スルホ-ルクロ リド、 5—メトキシ 3—メチルー 1 ベンゾチォフェン 2 スルホユルク口リド、 5—メ トキシカルボ-ル 3 メチル 1 ベンゾチォフェン 2 スルホ-ルクロリド、 7 クロロー 3—メチルー 1 ベンゾチォフェンー2—スルホニルクロリド、 3—メチルー 7— トリフルォロメチルー 1 ベンゾチォフェン 2—スルホ-ルクロリド、 3—メチルー 1 ベンゾチォフェン 2—スルホ-ルクロリド、 3—メチルー 5 トリフルォロメチルー 1 ベンゾチォフェン 2—スルホユルク口リド、などを合成した。
参考例 5 4 クロロー 3—メチルー 1 ベンゾチォフェンー2—スルホ-ルクロリドの 合成
4 クロ口一 3 メチル - 1 -ベンゾチォフェン(810mg)のテトラヒドロフラン溶液(9m 1)を—40°Cに冷却し、 n—ブチルリチウム(2. 71Mのへキサン溶液、 1. 80ml)を 5分 間かけて滴下し、 0°Cに昇温して 10分間攪拌した。これを、 40°Cに冷却したスルフ リルクロリド(1. 79g)のへキサン溶液(9ml)へ滴下し、 0°Cにて 1. 5時間攪拌した。 反応液を飽和炭酸水素ナトリウム水溶液にあけ、酢酸ェチルにて抽出した。有機層 を飽和食塩水にて洗浄し、無水硫酸マグネシウムにて乾燥後、乾燥剤を濾去して溶 媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィ (展開溶媒 n— へキサン:酢酸ェチル = 20: 1〜: LO: 1)で精製し、黄色粉末として表題化合物 (493 mg)を得た。
1H NMR (300 MHz, CHLOROFORM- D) δ 3.15 (s, 3 Η), 7.43 - 7.53 (m, 2 H), 7. 75 - 7.80 (m, 1 H).
参考例 5の方法と同様にして、チエノ [2,3-C]ピリジン 2 スルホユルク口リド、チエノ [3, 2-B]ピリジン一 2 スルホユルク口リド、 5 -シァノ 3 メチルー 1 ベンゾチオフ ェン 2—スルホ-ルクロリド、などを合成した。
参考例 6 4 ブロモー 2, 6 ジメチルベンゼンスルホ-ルクロリドの合成
4ーブロモー 2, 6 ジメチルベンゼンスルホン酸の合成
参考例 3と同様の方法で 4ーブロモー 2, 6 ジメチルァ-リン(1. 69g)から薄緑色 粉末として表題化合物( 1. 15g)を得た。
1H NMR (300 MHz, CHLOROFORM- D) δ 2.18 - 2.20 (m, 3 H), 2.38 - 2.40 (m, 3 H), 7.27 - 7.30 (m, 2 H).
(2) 4-ブロモ 2, 6 ジメチルベンゼンスルホ-ルクロリドの合成
4ーブロモー 2, 6 ジメチルベンゼンスルホン酸(1. 04g)のクロ口ホルム懸濁液(15 ml)にクロ口硫酸(1. 09ml)をカ卩え、室温で 1晚攪拌した。反応液に氷を加え、しばら く攪拌した後、ジェチルエーテルで抽出した。有機層を飽和食塩水で洗浄した後、 無水硫酸マグネシウムで乾燥した。乾燥剤を濾去して、溶媒を減圧留去して得られ た残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 へキサン:クロ口ホルム = 10 :
1〜5: 1)で精製し、薄茶色粘性油状物質として表題ィ匕合物 (257mg)を得た。
1H NMR (300 MHz, CHLOROFORM- D) δ 2.73 - 2.77 (m, 6 H), 7.40 - 7.43 (m,
2 H).
以下に、スルホンアミド誘導体合成にぉ 、て実施例で用いた 5種の製造方法の概 要を記載する。
製造方法 A ピリジンと 4-ジメチルァミノピリジンを塩基として用いるスルホ-ルイ匕反応 4-置換- 1 ,3-チアゾール -2-アミンをクロ口ホルムに溶解又は懸濁し、ピリジン( 1〜4 モル当量)と 4-ジメチルァミノピリジン( 1モル当量)を加え、室温又は氷冷でスルホ二 ルクロリド(1〜4モル当量)をカ卩える。室温〜 50°Cで、 3時間〜 3日間反応を行い目 的物を得る。
製造方法 B トリェチルァミンと 4-ジメチルァミノピリジンを塩基として用いるスルホ- ル化反応
4-置換- 1,3-チアゾール -2-アミンをクロ口ホルムに溶解又は懸濁し、トリェチルアミ ン(1〜4モル当量)と 4-ジメチルァミノピリジン(1〜: L 5モル当量)を加え、室温又は 氷冷でスルホニルクロリド(1〜4モル当量)をカ卩える。室温〜還流で、 1日〜 3日間反 応を行い目的物を得る。
製造方法 C ピリジンを塩基として用いマイクロウエーブ反応装置を使用するスルホ- ル化反応
4-置換- 1 ,3-チアゾール -2-アミンをクロ口ホルムに溶解又は懸濁し、ピリジン( 1〜4 モル当量)とスルホ-ルクロリド(1〜4モル当量)を加える。マイクロウエーブ反応装置 (パーソナルケミストリ一社製)を用いて、 100〜150°Cに加熱して、 15分間〜 1. 5時 間反応を行 ヽ目的物を得る。
製造方法 D ピリジンを塩基として用いるスルホ二ルイ匕反応
4-置換- 1 ,3-チアゾール -2-アミンをクロ口ホルムに溶解又は懸濁し、ピリジン( 1〜4 モル当量)を加え、室温又は氷冷でスルホユルクロリド(1〜4モル当量)をカ卩える。室 温〜 80°Cで、 3時間〜 8時間反応を行い目的物を得る。
製造方法 E ジイソプロピルァミンと 4-ジメチルァミノピリジンを塩基として用いるスル ホ-ル化反応
4-置換- 1,3-チアゾール -2-アミンをクロ口ホルムに溶解又は懸濁し、ジイソプロピル ァミン( 1〜4モル当量)と 4-ジメチルァミノピリジン( 1モル当量)をカ卩え、室温又は氷 冷でスルホユルクロリド(1〜4モル当量)をカ卩える。室温で、 1日〜 3日間反応を行い 目的物を得る。
実施例 1 スルホンアミド誘導体( 1 1)〜( 1 38)の合成
(1) 4-クロ口- 2-フルォロ- N- [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2- ィル]ベンゼンスルホンアミド(1— 1)の合成 (製造方法 A)
4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミン(198mg)をクロ口ホル ム(5ml)に溶解し、 4-ジメチノレアミノピリジン(131mg)、ピリジン(346 μ 1)を加えた 後、氷冷下で 4-クロ口- 2-フルォロベンゼンスルホユルクロリド(490mg)をカ卩え、その 後、室温に戻して 2日間攪拌した。反応液にクロ口ホルムをカ卩ぇ希釈してから 1M塩酸 水溶液、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、乾燥 剤を濾去して溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー( 展開溶媒 n-へキサン:酢酸ェチル = 3 : 1)で精製し、淡桃色粉末の表題化合物 (2 17mg)を得た。 NMRデータは、表 1に記載した。
(2) 2,3,4-トリクロ口- N- [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル] ベンゼンスルホンアミド( 1 2)の合成 (製造方法 B)
4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミン(21 lmg)をクロ口ホル ム(5ml)に溶解し、 4-ジメチノレアミノピリジン(140mg)、トリエチノレアミン(321 1)を 加えた後、氷冷下で 2,3,4-トリクロ口ベンゼンスルホユルクロリド(644mg)をカ卩え、そ の後、室温に戻して 3日間攪拌した。反応液にクロ口ホルムを加え希釈してから 1M 塩酸水溶液、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、 乾燥剤を濾去して溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフ ィー (展開溶媒 n-へキサン:酢酸ェチル = 5 : 1)で精製し、無色粉末の表題化合物 (271mg)を得た。 NMRデータは、表 1に記載した。
(3) 4-クロ口- N- [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル]ベンゼ ンスルホンアミド( 1 3)の合成 (製造方法 C)
4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミン(184mg)をクロ口ホル
ム(2. 5ml)に溶解し、ピリジン(323 μ 1)、 4-クロ口ベンゼンスルホユルクロリド(844 mg)を加えた後、マイクロウエーブ合成装置を用いて 100°Cで 45分攪拌した。反応 液にクロ口ホルムを加え希釈してから 1M塩酸水溶液、飽和食塩水で順次洗浄した。 有機層を無水硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、得られ た残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 n-へキサン:酢酸ェチル = 3 : 1)で精製し、無色粉末の表題ィ匕合物(178mg)を得た。 NMRデータは、表 1に記載 した。
(4)化合物(1 4)〜(1 38)の合成
参考例 1で得た 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ァミンと対応 するスルホユルク口リドを用い、製造方法 A〜Cの何れかを用いて表の化合物を合成 した。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 2 化合物(2— 1)及び(2— 2)の合成
(1) 4-メチルテトラヒドロ- 2H-ピラン- 4-カルボン酸メチルエステルの合成
4-テトラヒドロピランカルボン酸メチルエステル(7. 09g)のテトラヒドロフラン(150ml )溶液に 60°Cで 1. 6Mリチウムへキサメチルジシラジドのテトラヒドロフラン溶液(3 2ml)を滴下し、 15分攪拌した。次にヨウ化メチル (6. 1ml)を加え徐々に昇温して室 温で 2時間攪拌した。再度、反応溶液を 60°Cに冷却し 1. 6Mリチウムへキサメチ ルジシラジドのテトラヒドロフラン溶液(15. 3ml)をカ卩えてからヨウ化メチル(1. 5ml)を 加え室温で 1時間攪拌した。反応液に水を加え、クロ口ホルムで抽出し、飽和塩化ァ ンモ -ゥム水溶液、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥 後、乾燥剤を濾去して溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグ ラフィー(展開溶媒 n-へキサン:酢酸ェチル = 30: 1)で精製し褐色油状物質として 表題化合物(2. 18g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.23 (s, 3 H), 1.43 - 1.56 (m, 2 H), 2.0 2 - 2.14 (m, 2 H), 3.41 - 3.53 (m, 2 H), 3.72 (s, 3 H), 3.74 - 3.85 (m, 2 H).
(2) l-(4-メチルテトラヒドロ- 2H-ピラン- 4-ィル)エタノンの合成
参考例 1 (1)と同様の方法で 4-メチルテトラヒドロ- 2H-ピラン- 4-カルボン酸メチルェ ステル(2. 18g)と 3Mメチルマグネシウムブロミドのジェチルエーテル溶液(38ml)か
ら無色油状物質として表題化合物(1. 45g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.18 (s, 3 H), 1.44 - 1.57 (m, 2 H), 1.9 6 - 2.08 (m, 2 H), 2.15 (s, 3 H), 3.45 - 3.57 (m, 2 H), 3.69 - 3.81 (m, 2 H).
(3) 4-(4-メチルテトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンの合成 参考例 1 (2)と同様の方法で 1-(4-メチルテトラヒドロ- 2H-ピラン- 4-ィル)エタノン(1
. 36g)と臭素(0. 54ml)、チォゥレア(727mg)から薄黄色粉末として表題ィ匕合物(8 60mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.16 (s, 3 H), 1.40 - 1.54 (m, 2 H), 1.93 - 2.05 (m, 2 H), 3.40 - 3.52 (m, 2 H), 3.55 - 3.66 (m, 2 H), 6.16 (s, 1 H), 6.81 (brs, 2 H).
(4)化合物(2— 1)及び(2— 2)の合成
4- (4-メチルテトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ァミンと、 3-クロ口- 2 -メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-ス ルホニルクロリドを用い、製造方法 Aに従い表題化合物を得た。得られた化合物の構 造及び NMRデータを表 1に示す。
実施例 3 化合物(3— 1)〜(3— 10)の合成
(1)ェチル 4-ァセチルテトラヒドロ- 2H-ピラン- 4-カルボキシレートの合成
ェチル 3-ォキソブタノエート(255 μ 1)のアセトン溶液(2. 00ml)に炭酸カリウム(6 91mg)と 2-ブロモェチルエーテル (410 1)をカ卩えて、 8時間加熱還流した。セライト を用いて反応液を濾過し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラム クロマトグラフィー(展開溶媒 n キサン:酢酸ェチル = 20 : 1 15: 1 10: 1)で 精製して、無色油状物質として表題化合物(21 lmg)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.28 (t, J=7.1 Hz, 3 H), 1.96 - 2.06 (m , 2 H), 2.09 - 2.23 (m, 2 H), 2.17 (s, 3 H), 3.51 - 3.67 (m, 2 H), 3.70 - 3.84 (m, 2 H), 4.24 (q, J=7.1 Hz, 2 H).
(2)ェチル 4- (2-ァミノ- 1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボキ シレート臭化水素酸塩の合成
ェチル 4-ァセチルテトラヒドロ- 2H-ピラン- 4-カルボキシレート(21 lmg)のエタノー ル溶液(2. 00ml)に臭素(51 μ 1)をカ卩ぇ 65°Cで 30分攪拌した後、室温に戻し、チ
ォゥレア(75mg)を加え 2日間攪拌した。溶媒を減圧下留去して得られた残渣をシリ 力ゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム〜クロ口ホルム:メタノール = 20: 1)で精製して、無色固体として表題ィ匕合物(134mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.14 (t, J=7.1 Hz, 3 H), 1.88 - 1.99 (m, 2 H), 2. 12 - 2.22 (m, 2 H), 3.40 - 3.50 (m, 2 H), 3.62 - 3.72 (m, 2 H), 4.10 (q, J=7.1 Hz, 2 H), 6.44 (s, 1 H).
(3)化合物(3— 1)及び(3— 2)の合成
ェチル 4- (2-ァミノ- 1 ,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボキシ レート臭化水素酸塩と、 3-クロ口- 2-メチルベンゼンスルホユルクロリド又は 5-クロ口- 3 -メチル- 1-ベンゾチォフェン- 2-スルホユルク口リドを用い製造方法 Aに従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0069] (4) 4- (2- {[(3-クロ口- 2-メチルフエ-ル)スルホ -ル]アミノ}- 1,3-チアゾール -4-ィル) テトラヒドロ- 2H-ピラン- 4_カルボン酸(3— 3)の合成
実施例 3 (3)で得られたェチル 4- (2-{ [ (3-クロ口- 2-メチルフエ-ル)スルホニル]ァ ミノ }-1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボキシレート(3— 1) (1 . OOg)のテトラヒドロフラン エタノール 3 : 1混合溶液 (40ml)に氷冷下で 5M水酸化 ナトリウム水溶液(1. 35ml)を加え、室温に戻して 1時間攪拌した。更に 5M水酸ィ匕 ナトリウム水溶液(1. 35ml)を加え一晩攪拌した。反応液を減圧下濃縮して得られた 溶液に、氷冷下で 6M塩酸水溶液(2. 50ml)をカ卩え、固体を析出させた。水を加え て攪拌した後、吸引濾取して、水と IPEで洗浄し、無色粉末として表題ィ匕合物(932 mg)を得た。 NMRデータは、表 1に記載した。
(5) 4- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホ -ル]アミノ}- 1,3-チア ゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボン酸(3—4)の合成
実施例 3 (4)と同様の方法でェチル 4-(2-{[(5-クロ口- 3-メチル - 1-ベンゾチェン- 2 -ィル)スルホニル]アミノ} -1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボ キシレート(1. 50g)から無色粉末として表題ィ匕合物(1. 38g)を得た。 NMRデータは 、 ¾:上に ti載し 7こ。
[0070] (6) 4- (2- {[(3-クロ口- 2-メチルフエ-ル)スルホ -ル]アミノ}- 1,3-チアゾール -4-ィル)
テトラヒドロ- 2H-ピラン- 4-カルボキサミド(3— 5)の合成
実施例 3 (4)で得られた 4-(2-{ [(3-クロ口- 2-メチルフエ-ル)スルホ -ル]アミノ} -1,3 -チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボン酸(250mg)の N, N -ジメ チルホルムアミド溶液(2. 50ml)に 1-ヒドロキシベンゾトリアゾール 1水和物(119mg) 、 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩酸塩( 150mg)及び 28% アンモニア水(160 μ 1)を加え、室温で 2日間攪拌した。氷冷下で反応液に 5%炭酸 水素ナトリウム水溶液と飽和食塩水の 1: 1混合液(10ml)を加え、クロ口ホルム (60m I X 2)で抽出した。有機層を無水硫酸マグネシウムで乾燥後、乾燥剤を濾去し、溶媒 を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口 ホルム:メタノール = 30: 1)で精製し、無色粉末として表題化合物(55mg)を得た。 N MRデータは、表 1に記載した。
(7) 4- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホ -ル]アミノ}- 1,3-チア ゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボキサミド(3— 6)の合成
実施例 3 (6)と同様の方法で 4-(2-{ [(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)ス ルホ -ル]アミノ}-1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボン酸(250 mg)と 28%アンモニア水(140 μ 1)力も無色粉末として表題ィ匕合物(102mg)を得た 。 NMRデータは、表 1に記載した。
(8) 4- (2- {[(3-クロ口- 2-メチルフエ-ル)スルホ -ル]アミノ}- 1,3-チアゾール -4-ィル) -N-ェチルテトラヒドロ- 2H-ピラン- 4-カルボキサミド(3— 7)の合成
実施例 3 (6)と同様の方法で 4- (2- {[(3-クロ口- 2-メチルフエ-ル)スルホ -ル]ァミノ) -1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボン酸(250mg)とェチルァ ミン 70%水溶液 (62 μ 1)カゝら無色粉末として表題ィ匕合物(122mg)を得た。 NMRデ ータは、表 1に記載した。
(9) 4- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホ -ル]アミノ}- 1,3-チア ゾール -4-ィル) -N-ェチルテトラヒドロ- 2H-ピラン- 4-カルボキサミ(3— 8)の合成 実施例 3 (6)と同様の方法で 4-(2-{ [(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)ス ルホ -ル]アミノ}-1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボン酸(250 mg)とェチルァミン 70%水溶液 (62 μ 1)力も無色粉末として表題ィ匕合物(14 lmg)を
得た。 NMRデータは、表 1に記載した。
[0072] (10) 3-クロ口- N- {4- [4- (ヒドロキシメチル)テトラヒドロ- 2H-ピラン- 4-ィル] -1,3-チアゾ ール -2-ィル }-2-メチルベンゼンスルホンアミド(3— 9)の合成
実施例 3 (3)で得られたェチル 4- (2-{ [ (3-クロ口- 2-メチルフエ-ル)スルホニル]ァ ミノ } 1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボキシレート(220mg)の テトラヒドロフラン溶液(7. 9ml)に氷冷下で水素化リチウムアルミニウム(37mg)をカロ え、氷冷下で 1時間攪拌した後、室温に戻して 1. 5時間攪拌した。氷冷下で反応液 に飽和塩ィ匕アンモ-ゥム水溶液(60ml)をカ卩え、クロ口ホルム(100ml)で抽出した。 有機層を飽和食塩水と水の 1: 1混合液 (60ml X 2)、飽和食塩水(60ml)で順次洗 浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を濾去し、溶媒を減圧留去して 得られた残渣をクロ口ホルムとテトラヒドロフランの 20 : 1混合液で洗浄した後、プレパ ラティブ TLC (展開溶媒 クロ口ホルム:メタノール = 10 : 1)で精製し、無色粉末として 表題ィ匕合物(59mg)を得た。 NMRデータは、表 1に記載した。
(11) 5-クロ口- N- {4- [4- (ヒドロキシメチル)テトラヒドロ- 2H-ピラン- 4-ィル] -1,3-チアゾ ール -2-ィル }-3-メチル -1-ベンゾチォフェン- 2-スルホンアミド(3— 10)の合成 実施例 3 (10)と同様の方法でェチル 4- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホニル]アミノ} -1,3-チアゾール -4-ィル)テトラヒドロ- 2H-ピラン- 4-カルボ キシレート(215mg)力も無色粉末として表題ィ匕合物(129mg)を得た。 NMRデータ は、表上に じ載し 7こ。
[0073] 実施例 4 化合物(4 1A)及び (4 1B)の合成
(1) N-メチル -4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ァミン及び 4- (4 -メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -N-メチル -1,3-チアゾール -2-ァミンの合成
1- (テトラヒドロ- 2H-ピラン- 4-ィル)エタノン(1. 83g)のメタノール(30ml)溶液に臭 素(0. 81ml)を加え 65°Cで 30分攪拌した後、室温に戻し、メチルチオゥレア(1. 29g )を加え 50°Cで 3時間攪拌した。反応終了後クロ口ホルムと飽和炭酸水素ナトリウム 水溶液を加えた後、クロ口ホルムで抽出し飽和食塩水で洗浄した。有機層を無水硫 酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、得られた残渣をシリカ ゲルカラムクロマトグラフィー(展開溶媒 n-へキサン:酢酸ェチル = 10 : 1)で精製し
薄黄色粉末として N-メチル -4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2- ァミンと 4- (4-メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -N-メチル -1,3-チアゾール -2-ァ ミンの混合物(474mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.44— 1.67 (m, 2 H), 1.72 - 2.04 (m, 6 H), 2.55 - 2.72 (m, 1 H), 2.73 - 2.82 (m, 6 H), 2.94 (s, 3 H), 3.34 - 3.48 (m, 2 H), 3.49 - 3 .72 (m, 4 H), 3.77 - 3.97 (m, 2 H), 6.18 (s, 1 H), 6.51 (s, 1 H), 7.28 - 7.38 (m, 1 H) , 7.39 - 7.52 (m, 1 H).
(2) 3-クロ口- N,2-ジメチル- N- [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル]ベンゼンスルホンアミド(4 - 1 A)及び 3-クロ口- N- [4- (4-メトキシテトラヒドロ -2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル] -N,2-ジメチルベンゼンスルホンアミ ド (4—IB)の合成
製造方法 Cを用いて N-メチル -4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール-
2-ァミン、 4- (4-メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -N-メチル -1 ,3-チアゾール -2- ァミンの混合物(58mg)と 3-クロ口- 2-メチルベンゼンスルホユルクロリド(263mg)から
3-クロ口- N,2-ジメチル- N- [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィ ル]ベンゼンスルホンアミド(l lmg)、 3-クロ口- N- [4- (4-メトキシテトラヒドロ- 2H-ピラン -4-ィル) -1,3-チアゾール -2-ィル] -N,2-ジメチルベンゼンスルホンアミド(26mg)を得 た。 NMRデータは、表 1に記載した。
実施例 5 化合物(5— 1)及び(5— 2)の合成
(1) N-メトキシ- N-メチルテトラヒドロフラン- 3-カルボキサミドの合成
テトラヒドロ- 3-フロイックアシッド(10. 33g)と Ν,Ο-ジメチルヒドロキシルァミン塩酸 塩(10. 73g)、トヒドロキシベンゾトリアゾール(13. 63g)、トリェチルァミン(15. 3ml )のァセトニトリル(100ml)溶液に、氷冷下 1-ェチル -3-(3-ジメチルァミノプロピル)力 ルボジイミド塩酸塩(21. 09g)を加え 1時間攪拌した後、室温で一晩攪拌した。反応 終了後、水を加え、クロ口ホルムで抽出し、飽和炭酸水素ナトリウム水溶液、 1M塩酸 水溶液、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、乾燥 剤を濾去して溶媒を減圧留去し、無色油状物質として表題化合物(13. Og)を得た。 1H NMR (300 MHz, CHLOROFORM— D) δ 2.01 - 2.16 (m, 1 H), 2.15 - 2.31 (m, 1
H), 3.21 (s, 3 H), 3.31 - 3.52 (m, 1 H), 3.66 - 3.73 (m, 3 H), 3.75 - 3.98 (m, 3 H), 4.00 - 4.14 (m, 1 H).
(2) 1- (テトラヒドロフラン- 3-ィル)エタノンの合成
窒素雰囲気下、 N-メトキシ -N-メチルテトラヒドロフラン- 3-カルボキサミド(13. Og) のテトラヒドロフラン(200ml)溶液に氷冷下、 3Mメチルマグネシウムブロミドのジェチ ルエーテル溶液(54. 4ml)を滴下し、同温で 1時間攪拌した。反応終了後、反応液 を氷水(200ml)にあけ、クロ口ホルムで抽出し、飽和食塩水で洗浄した。有機層を無 水硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、薄褐色油状物質 として表題ィ匕合物(6. 57g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.03 - 2.18 (m, 2 H), 2.21 (s, 3 H), 3.1 4 - 3.28 (m, 1 H), 3.72 - 3.99 (m, 4 H).
(3) 4- (テトラヒドロフラン- 3-ィル) -1,3-チアゾール -2-ァミンの合成
参考例 1 (2)と同様の方法で 1- (テトラヒドロフラン- 3-ィル)エタノン (6. 50g)と臭素( 1. 90ml)、チォゥレア(3. 12g)から黄色粉末として表題ィ匕合物(3. 67g)を得た。 1H NMR (300 MHz, DMSO— D6) δ 1.88 - 2.18 (m, 2 H), 3.13 - 3.29 (m, 1 H), 3.56
(t, J=7.9 Hz, 1 H), 3.66 - 3.85 (m, 2 H), 3.91 (t, J=7.9 Hz, 1 H), 6.23 (d, J=0.8 Hz,
1 H), 6.87 (brs, 2 H).
(4)化合物(5— 1)及び(5— 2)の合成
実施例 5 (3)で得られた 4- (テトラヒドロフラン- 3-ィル) -1,3-チアゾール -2-ァミンと、 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチオフ ェン -2-スルホユルク口リドを用い製造方法 Aに従 、表表題ィ匕合物を得た。得られた 化合物の構造及び NMRデータを表 1に示す。
[0075] 実施例 6 化合物(6— 1)の合成
参考例 2で得られた 4-シクロへキシル -1,3-チアゾール -2-アミン臭化水素酸塩と、 5 -クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い製造方法 Bに従!ヽ 表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0076] 実施例 7 化合物(7— 1)の合成
(1) 1-(4-ヒドロキシシクロへキシル)エタノンの合成
窒素雰囲気下、 4 ヒドロキシシクロへキサンカルボン酸ェチルエステル(シス ·トラ ンス混合物、 1. 72g)と Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(1. 27g)のテトラヒド 口フラン(50ml)溶液に、一 30〜一 25°Cで 3Mメチルマグネシウムブロミドのジェチル エーテル溶液(27. 7ml)を 10分間かけて滴下した後、室温で 3時間攪拌した。反応 液を氷冷した希塩酸水溶液にあけ、クロ口ホルムで抽出した。有機層を無水硫酸マグ ネシゥムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、淡黄色油状物質として表 題化合物(1. 44g)を得た。
(2) 4-(2-ァミノ- 1,3-チアゾール -4-ィル) -シクロへキサノールの合成
参考例 1 (2)と同様の方法で 1-(4-ヒドロキシシクロへキシル)エタノン(1. 43g)と臭 素(0. 41ml)、チォゥレア(761mg)から薄黄色粉末として表題ィ匕合物 (480mg)を得 た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.14 - 2.10 (m, 9 H), 2.15 (s, 3 H), 2.2 4 - 2.42 (m, 1 H), 3.52 - 3.66 (m, 0.25 H), 3.91 - 4.01 (m, 0.75 H).
(3) 5-クロ口- N- [4- (4-ヒドロキシシクロへキシル )-1,3-チアゾール -2-ィル] -3-メチル- 1-ベンゾチォフェン- 2-スルホンアミド(7— 1)の合成
4- (2-ァミノ- 1,3-チアゾール -4-ィル) -シクロへキサノール(197mg)のクロ口ホルム 懸濁液(5ml)に、氷冷下トリエチルァミン(0. 31ml)とトリメチルシリルクロリド(0. 14 ml)を加え、室温で 5時間攪拌した。トリェチルァミン (0. 28ml)とトリメチルシリルクロ リド (0. 12ml)を追加し、更に室温で 1時間攪拌した。反応液に、 4-ジメチルアミノビ リジン(122mg)と 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホユルクロリド(562 mg)をカ卩ぇ室温で一晩攪拌した。反応液にメタノール(1ml)を加え室温で 1時間攪 拌し、 6M塩酸(2ml)を加え更に 1時間攪拌した。溶媒を減圧留去し、得られた残渣 をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール: 28%アン モ-ァ水 = 100 : 3 : 0. 3〜: L00 : 7 : 0. 7)で精製し、無色粉末として表題化合物(99 mg)を得た。 NMRデータは表 1に記載した。
実施例 8 化合物(8— 1)〜(8— 3)の合成
( 1) tert-ブチル 3- { [メトキシ (メチル)ァミノ]カルボ-ル }ピペリジン- 1-カルボキシレ ートの合成
(±) - 1- (tert-ブトキシカルボ-ル)ピぺリジン- 3-カルボン酸(8. 44g)の N, N—ジ メチルホルムアミド溶液(85ml)に Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(4. 31g)と 1-ヒドロキシベンゾトリアゾール 1水和物(6.47g)を加え、水冷下でトリェチルァミン(6 . 67ml)を滴下した。続いて 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩 酸塩(9. 17g)を加え、室温で 2日間攪拌した。氷冷下で反応液に飽和食塩水と水の 1 : 1混合液(200ml)をカ卩え、クロ口ホルム(500ml)で抽出した。有機層を 1M塩酸 水溶液(100ml)、 5%炭酸水素ナトリウム水溶液(100ml)、水(100ml)、飽和食塩 水(100ml)で順次洗浄した。有機層を無水硫酸マグネシウムで乾燥後、乾燥剤を濾 去し、溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (展開 溶媒 クロ口ホルム:メタノール =80 : 1)で精製し、無色油状物質として表題化合物( 9.49g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.46 (s, 9 H), 1.57 - 1.77 (m, 3 H), 1.8 5 - 1.99 (m, 1 H), 2.63 - 2.76 (m, 1 H), 2.78 - 2.93 (m, 2 H), 3.19 (s, 3 H), 3.73 (s, 3 H), 4.02 - 4.21 (m, 2 H).
(2) tert-ブチル 3-ァセチルビペリジン- 1-カルボキシレートの合成
tert-ブチル 3-{ [メトキシ (メチル)ァミノ]カルボ-ル}ピペリジン- 1-カルボキシレート (7. 42g)のテトラヒドロフラン溶液(130ml)に氷冷下で 3Mメチルマグネシウムブロミ ドのジェチルエーテル溶液(18. 2ml)を 5分かけて滴下し、氷冷下で 1.5時間攪拌し た
氷冷下で反応液に飽和塩ィ匕アンモ-ゥム水溶液 (300ml)を加え、クロ口ホルム (50 0ml)で抽出した。有機層を飽和食塩水と水の 1: 1混合液 (300ml)、飽和食塩水(3 00ml)で順次洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を濾去して、 溶媒を減圧留去し、薄黄色油状物質として表題化合物 (6. 02g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.41 - 1.79 (m, 3 H), 1.46 (s, 9 H), 1.9 1 - 2.08 (m, 1 H), 2.19 (s, 3 H), 2.39 - 2.58 (m, 1 H), 2.68 - 3.02 (m, 2 H), 3.83 - 4.00 (m, 1 H), 4.02 - 4.20 (m, 1 H).
(3) 4-ピぺリジン- 3-ィル- 1,3-チアゾール -2-ァミンの合成
参考例 1 (2)と同様の方法で tert-ブチル 3-ァセチルビペリジン- 1-カルボキシレート
(1. OOg)、臭素(225 /z l)及びチォゥレア(268mg)から得られた粗生成物をシリカ ゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 5 : 1 : 0.1)で精製し、薄茶色固体として表題ィ匕合物(580mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.47 - 2.01 (m, 4 H), 2.74 - 2.90 (m, 3 H), 3.19 - 3.42 (m, 3 H), 6.27 (s, 1 H), 6.91 (brs, 2 H).
(4) tert-ブチル 3- (2-ァミノ- 1,3-チアゾール -4-ィル)ピぺリジン- 1-カルボキシレート の合成
4-ピぺリジン- 3-ィル- 1,3-チアゾール -2-ァミン(3. 27g)にテトラヒドロフラン(29ml )と水(11ml)をカ卩えて懸濁させた後、氷冷下で 5M水酸ィ匕ナトリウム水溶液(7. 14m 1)及びジ- tert-ブチルジカルボネート(4. 10ml)のテトラヒドロフラン溶液(4. 00ml) を加え、氷冷下で 1時間攪拌した。氷冷下で反応液に水(50ml)を加え、酢酸ェチル (100ml)で抽出した。有機層を水(50ml)、飽和食塩水(50ml)で順次洗浄した後、 有機層を無水硫酸マグネシウムで乾燥した。乾燥剤を濾去し、溶媒を減圧留去して 得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノー ル: 28%アンモニア水 = 30: 1: 0.1)で精製し、無色粉末として表題化合物(2. 94g) を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.32 - 1.42 (10 Η), 1.50 - 1.58 (m, 1 H), 1.59 - 1.70 (m, 1 H), 1.86 - 1.98 (m, 1 H), 2.37 - 2.49 (m, 1 H), 2.62 - 2.80 (m, 2 H), 3. 82 - 3.93 (m, 1 H), 3.96 - 4.15 (m, 1 H), 6.19 (s, 1 H), 6.84 (brs, 2 H).
(5)化合物(8— 1)の合成
実施例 8 (4)で得られた tert-ブチル 3- (2-ァミノ- 1,3-チアゾール -4-ィル)ピベリジ ン- 1-カルボキシレートと、 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロ リドを用い製造方法 Bに従 、表題ィ匕合物を得た。得られた化合物の構造及び NMRデ 一タを表 1に示す。
(6) 5-クロ口- 3-メチル -N- (4-ピぺリジン- 3-ィル- 1 ,3-チアゾール -2-ィル) -1-ベンゾ チォフェン- 2-スルホンアミド塩酸塩(8— 2)の合成
実施例 8 (5)で得た tert-ブチル 3- (2-{ [5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル ]スルホ -ルァミノ }- 1,3-チアゾール -4-ィル)ピぺリジン- 1-カルボキシレート(438m
g)に 4M塩酸—ジォキサン (8. OOml)を加え、室温で 2日間攪拌した。析出した固体 を吸引濾取して、淡橙色粉末として表題ィ匕合物(254mg)を得た。 NMRデータは表 1 に己載し 7こ。
[0079] (7) 5-クロ口- 3-メチル - Ν-{4-[1- (メチルスルホ -ル)ピぺリジン- 3-ィル] -1,3-チアゾ ール -2-ィル }-1-ベンゾチォフェン- 2-スルホンアミド(8— 3)の合成
実施例 8 (6)で得た 5-クロ口- 3-メチル -N- (4-ピぺリジン- 3-ィル- 1,3-チアゾール -2 -ィル )-1-ベンゾチォフェン- 2-スルホンアミド塩酸塩(130mg)の Ν,Ν-ジメチルホル ムアミド溶液(1. 30ml)に氷冷下でトリェチルァミン(78 μ 1)とメタンスルホユルクロリ ド(23 μ 1)を滴下して氷冷下で 30分間攪拌した。反応液に 5%硫酸水素カリウム水 溶液と飽和食塩水の 1: 1混合液 (5. 00ml)を滴下してクロ口ホルム( 15ml)で抽出し た。有機層を飽和食塩水(20ml)で洗浄した後、無水硫酸マグネシウムで乾燥した。 乾燥剤を濾去し、溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフ ィー(展開溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 30 : 1 : 0. 1)で精製し 、無色粉末として表題ィ匕合物(lOOmg)を得た。 NMRデータは表 1に記載した。
[0080] 実施例 9 化合物(9 1)〜(9 4)の合成
( 1) tert-ブチル 4- { [メトキシ (メチル)ァミノ]カルボ-ル }ピペリジン- 1-カルボキシレ ートの合成
実施例 8 (1)と同様の方法で l-(tert-ブトキシカルボ-ル)ピぺリジン- 4-カルボン酸 (2. OOg)と Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(1. l lg)から無色油状物質とし て表題化合物(2. 31g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.46 (s, 9 H), 1.58 - 1.78 (m, 4 H), 2.7 0 - 2.88 (m, 3 H), 3.19 (s, 3 H), 3.71 (s, 3 H), 4.15 - 4.25 (m, 2 H).
(2) tert-ブチル 4-ァセチルビペリジン- 1-カルボキシレートの合成
実施例 8 (2)と同様の方法で tert-ブチル 4-{ [メトキシ (メチル)ァミノ]カルボ二ル}ピ ペリジン- 1-カルボキシレート(2. 25g)と 3Mメチルマグネシウムブロミドのジェチルェ 一テル溶液(5. 50ml)力ゝら薄黄色油状物質として表題ィ匕合物(1. 48g)を得た。 1H NMR (300 MHz, CHLOROFORM— D) δ 1.46 (s, 9 H), 1.48 - 1.61 (m, 2 H), 1.7 8 - 1.89 (m, 2 H), 2.17 (s, 3 H), 2.39 - 2.52 (m, 1 H), 2.70 - 2.87 (m, 2 H), 4.00 -
4.20 (m, 2 H).
(3) 4-ピぺリジン- 4-ィル -1,3-チアゾール -2-ァミン 2臭化水素酸塩の合成
参考例 2と同様の方法で tert-ブチル 4-ァセチルビペリジン- 1-カルボキシレート(6 44mg)と臭素( 145 1)及びチォゥレア( 172mg)力も薄黄色粉末として表題ィ匕合物 (715mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.56 - 1.82 (m, 2 H), 2.00 - 2.20 (m, 2 H), 2.78 - 3.12 (m, 3 H), 3.28 ? 3.46 (m, 2 H), 6.56 (s, 1 H), 8.20 - 9.20 (m, 3 H).
(4) tert-ブチル 4- (2-ァミノ- 1,3-チアゾール -4-ィル)ピぺリジン- 1-カルボキシレート の合成
実施例 8 (4)と同様の方法で 4-ピぺリジン- 4-ィル -1,3-チアゾール -2-ァミン 2臭化 水素酸塩(700mg)とジ -tert-ブチルジカルボネート(443mg)力も薄黄色ァモルファ スとして表題ィ匕合物 (418mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.29 - 1.47 (11 Η), 1.76 - 1.88 (m, 2 H), 2.51 - 2.60 (m, 1 H), 2.62-2.88(m, 2 H), 3.88 - 4.05 (m, 2 H), 6.12 (s, 1 H), 6.81 (brs, 2 H).
(5)化合物(9 1)の合成
実施例 9 (4)で得られた tert-ブチル 4- (2-ァミノ- 1,3-チアゾール -4-ィル)ピベリジ ン- 1-カルボキシレートと、 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロ リドを用い製造方法 Bに従 、表題ィ匕合物を得た。得られた化合物の構造及び NMRデ 一タを表 1に示す。
[0081] (6) 5-クロ口- 3-メチル -N- (4-ピぺリジン- 4-ィル- 1,3-チアゾール -2-ィル) -1-ベンゾ チォフ ン- 2-スルホンアミド塩酸塩(9 2)の合成
実施例 9 (5)で得た tert-ブチル 4- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィ ル)スルホ -ル]アミノ}-1, 3-チアゾール -4-ィル)ピぺリジン- 1-カルボキシレート(17 lmg)を氷冷下で 4M塩酸 酢酸ェチル(3. 00ml)に加え、室温に戻してー晚攪拌 した。析出した固体を吸引濾取して、酢酸ェチルで洗浄し薄茶色粉末として表題ィ匕 合物(135mg)を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0082] (7) N- [4- (1-ァセチルビペリジン- 4-ィル) -1,3-チアゾール -2-ィル] -5-クロ口- 3-メチ
ル- 1-ベンゾチォフェン- 2-スルホンアミド(9— 3)の合成
実施例 9 (6)で得た 5-クロ口- 3-メチル -N- (4-ピぺリジン- 4-ィル- 1,3-チアゾール -2 -ィル )-1-ベンゾチォフェン- 2-スルホンアミド塩酸塩(lOOmg)の Ν,Ν-ジメチルホル ムアミド溶液(1. OOml)に氷冷下でジイソプロピルェチルァミン(75 μ 1)とァセチルク ロリド(15 1)を滴下して氷冷下で 30分間攪拌した。更にジイソプロピルェチルァミン (75 μ 1)とァセチルクロリド(15 1)を加えて氷冷下で 30分間攪拌後、反応液に 5% 硫酸水素カリウム水溶液と飽和食塩水の 1: 1混合液(5. OOml)を滴下してクロ口ホル ム(15ml)で抽出した。有機層を飽和食塩水(20ml)で洗浄した後、無水硫酸マグネ シゥムで乾燥した。乾燥剤を濾去し、溶媒を減圧留去して得られた残渣をシリカゲル カラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 20 : 1 : 0. 1)で精製し、無色粉末として表題化合物 (88mg)を得た。得られた化合物の 構造及び NMRデータを表 1に示す。
[0083] (8) 5-クロ口- 3-メチル -N- {4- [1- (メチルスルホ -ル)ピぺリジン- 4-ィル] -1,3-チアゾ ール -2-ィル }-1-ベンゾチォフェン- 2-スルホンアミド(9 4)の合成
実施例 9 (7)と同様の方法で 5-クロ口- 3-メチル -N- (4-ピぺリジン- 4-ィル -1,3-チア ゾール -2-ィル) -1-ベンゾチォフェン- 2-スルホンアミド塩酸塩(lOOmg)とメタンスル ホニルクロリド(17 1)力 無色アモルファスとして表題ィ匕合物(63mg)を得た。得ら れた化合物の構造及び NMRデータを表 1に示す。
[0084] 実施例 10 化合物(10— 1)及び(10— 2)の合成
( 1 ) tert-ブチル(3- { [メトキシ (メチル)ァミノ]カルボ-ル }シクロへキシル)カルバメー トの合成
実施例 8 (1)と同様の方法で 3- [ (tert-ブトキシカルボニル)ァミノ)]シクロへキサン カルボン酸(7. 23g)と Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(3. 48g)から無色泡 状物質として表題ィ匕合物 (8. 23g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 0.99 - 1.17 (m, 1 H), 1.33 - 2.11 (m, 7 H), 1.41 - 1.49 (m, 9 H), 2.71 - 3.01 (m, 1 H), 3.17 (s, 3 H), 3.40 - 3.61 (m, 1 H), 3.69 (s, 3 H), 4.39 - 4.72 (m, 1 H).
(2) tert-ブチル(3-ァセチルシクロへキシル)力ルバメートの合成
実施例 8 (2)と同様の方法で tert-ブチル (3-{ [メトキシ (メチル)ァミノ]カルボ二ル} シクロへキシル)力ルバメート(2. OOg)と 3Mメチルマグネシウムブロミドのジェチルェ 一テル溶液 (4. 66ml)カゝら無色固体として表題ィ匕合物(1. 54g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 0.94 - 1.68 (m, 5 H), 1.39 - 1.48 (m, 9 H), 1.67 - 2.03 (m, 3 H), 2.12 - 2.18 (m, 3 H), 2.37 - 2.66 (m, 1 H), 3.37 - 3.89 ( m, 1 H), 4.32 - 4.65 (m, 1 H).
(3) 4- (3-アミノシクロへキシル) -1,3-チアゾール -2-ァミンの合成
参考例 1 (2)と同様の方法で tert-ブチル(3-ァセチルシクロへキシル)力ルバメート (700mg)と臭素(149 μ 1)及びチォゥレア(177mg)力も得られた粗生成物をシリカ ゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 5 : 1 : 0.1)で精製し、薄茶色油状物質として表題化合物 (43 lmg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.13 - 2.22 (m, 8 H), 2.79 - 3.19 (m, 2 H), 6.09 - 6.18 (m, 1 H), 6.79 - 6.89 (m, 2 H), 7.79 - 7.98 (m, 2 H).
(4) 2- (トリメチルシリル)ェチル [3- (2-ァミノ- 1,3-チアゾール -4-ィル)シクロへキシ ル]力ルバメートの合成
4- (3-アミノシクロへキシル)-1,3-チアゾール -2-ァミン(40 lmg)の水懸濁液(2. 0 Oml)〖こトリエチノレアミン (425 μ 1)のジォキサン溶液(2. 00ml)を滴下した後、 1- [ (2 -トリメチルシリル)エトキシカルボ-ルォキシ]ピロリジン- 2 , 5-ジオン( 580mg)をカロえ 、室温で 2日間攪拌した。反応液に水(10ml)を加えて、クロ口ホルム (20ml)で抽出 した。有機層を飽和食塩水と水の 1: 1混合液(10ml X 3)、飽和食塩水(10ml)で順 次洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を濾去し、溶媒を減圧留 去して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メ タノール =80 : 1)で精製し、薄黄色アモルファスとして表題ィ匕合物(394mg)を得た。 1H NMR (300 MHz, DMSO— D6) δ —0.05 - 0.06 (m, 9 H), 0.85 - 2.05 (m, 12 H), 3. 95 - 4.09 (m, 2 H), 6.03 - 6.10 (m, 1 H), 6.77 (s, 2 H), 6.89 - 7.09 (m, 1 H).
(5) 2- (トリメチルシリル)ェチル [3- (2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル) スルホ -ル]アミノ}- 1,3-チアゾール -4-ィル)シクロへキシル]力ルバメート(10— 1)の 合成
実施例 10 (4)で得られた 2- (トリメチルシリル)ェチル [3- (2-ァミノ- 1,3-チアゾール -4-ィル)シクロへキシル]力ルバメート(379mg)と 5-クロ口- 3-メチル -1-ベンゾチオフ ェン -2-スルホユルクロリド(624mg)から製造方法 Bの方法を用いて薄茶色ァモルフ ァスとして表題ィ匕合物(285mg)を得た。得られた化合物の構造及び NMRデータを 表 1に示す。
[0085] (6) N- [4- (3-アミノシクロへキシル )-1,3-チアゾール -2-ィル] -5-クロ口- 3-メチル -1- ベンゾチォフェン- 2-スルホンアミド(10— 2)の合成
実施例 10 (5)で得られた 2- (トリメチルシリル)ェチル [3- (2- {[(5-クロ口- 3-メチル -1- ベンゾチェン- 2-ィル)スルホ -ル]アミノ} -1,3-チアゾール -4-ィル)シクロへキシル]力 ルバメート(275mg)にトリフルォロ酢酸(1. 5ml)をカ卩え、室温で 30分間攪拌した。 反応液を減圧下濃縮して、得られた残渣をシリカゲルカラムクロマトグラフィー(展開 溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 10 : 1 : 0.1 8 : 1 : 0. 1)で精製 し、薄茶色粉末として表題化合物(113mg)を得た。得られた化合物の構造及び NM Rデータを表 1に示す。
[0086] 実施例 11 化合物(11 1)〜(: L 1 4)の合成
(1) 3-クロ口- N- (4-メトキシベンジル)プロパン- 1-スルホンアミドの合成
4-メトキシベンジルァミン(3. 49g)のテトラヒドロフラン-クロ口ホルム(1: 3)混合溶 液(100ml)〖こ、氷冷下 3-クロ口プロパンスルホユルクロリド(1. 50g)を滴下し、室温 にて 7時間攪拌した。反応液を飽和塩ィ匕アンモ-ゥム水溶液(150ml)にあけ、クロ口 ホルム(100ml X 3)にて抽出した。有機層を飽和食塩水(100ml)にて洗浄し、無水 硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。得られた残渣 をシリカゲルカラムクロマトグラフィー(展開溶媒 n キサン:酢酸ェチル = 3 : 1 1: 1)で精製し無色粉末として表題ィ匕合物 (2. 28g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 2.14 - 2.32 (m, 2 H), 3.03 - 3.14 (m, 2 H), 3.57 - 3.67 (m, 2 H), 3.81 (s, 3 H), 4.21 - 4.30 (m, 2 H), 4.37 - 4.50 (m, 1 H), 6.83 - 6.95 (m, 2 H), 7.21 - 7.32 (m, 2 H).
(2) 2-(4-メトキシベンジル)イソチアゾリジン 1,1-ジォキシドの合成
3-クロ口- N- (4-メトキシベンジル)プロパン- 1-スルホンアミド(3. 18g)のテトラヒドロ
フラン溶液(114ml)に、 0°C下、水素化ナトリウム(60%オイル懸濁、 504mg)をカロえ 、攪拌し、その後 5時間加熱還流した。反応液に飽和塩ィ匕アンモ-ゥム水溶液(150 ml)を加え、酢酸ェチル(150ml)にて抽出した。有機層を飽和食塩水(100ml)にて 洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。 得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 n—へキサン:酢酸ェ チル = 2: 1〜2: 3)で精製し無色油状物質として表題ィ匕合物(2. 64g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 2.19 - 2.38 (m, 2 H), 3.03 - 3.25 (m, 4 H), 3.81 (s, 3 H), 4.12 (s, 2 H), 6.82 - 6.93 (m, 2 H), 7.22 - 7.32 (m, 2 H).
(3) 4-[2-(4-メトキシベンジル) - 1 , 1-ジォキシドイソチアゾリジン- 5-ィル] - 1 ,3-チアゾ ール- 2-ァミンの合成
2-(4-メトキシベンジノレ)イソチアゾリジン 1,1-ジォキシド(2. 64g)のテトラヒドロフラ ン(36ml)溶液を— 60°Cに冷却し、 n—ブチルリチウム(2. 59Mのへキサン溶液、 5. 07ml)を滴下し、—60°Cで 30分間、 40〜一 20°Cで 45分間攪拌した。再び—60 °Cに冷却し、クロ口酢酸ェチル(1. 28ml)を加えて 40〜一 20°Cで 1. 5時間攪拌 した。酢酸(751 /z l)およびチォゥレア(916mg)を加え、室温で 14時間攪拌した。反 応液にメタノール(36ml)をカ卩え、 60°Cで更に 3時間攪拌した。反応液を水にあけ酢 酸ェチルで抽出し、有機層を飽和食塩水にて洗浄し、無水硫酸マグネシウムで乾燥 後、乾燥剤を濾去して溶媒を減圧留去した。得られた残渣にエタノールとイソプロピ ルエーテルの 1: 1混合溶媒を加え、析出した粉末を濾取して淡黄色粉末として表題 化合物(1. 55g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 2.45 - 2.79 (m, 2 H), 3.01 - 3.29 (m, 2 H), 3.81 (s, 3 H), 4.20 (s, 2 H), 4.33 (t, J=8.6 Hz, 1 H), 5.04 (brs, 2 H), 6.60 (s, 1 H), 6.83 - 6.92 (m, 2 H), 7.23 - 7.33 (m, 2 H).
(4)化合物(11 1)及び(11 2)の合成
実施例 11 (3)で得られた 4-[2- (4-メトキシベンジル) -1,1-ジォキシドイソチアゾリジ ン -5-ィル] -1 ,3-チアゾール -2-ァミンと、 3-クロ口- 2-メチルベンゼンスルホユルクロリ ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い製造方法 Bに表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0087] (5) 3-クロ口- N- [4- (1,1-ジォキシドイソチアゾリジン- 5-ィル) -1,3-チアゾール -2-ィル ]-2-メチルベンゼンスルホンアミド(11— 3)の合成
3-クロ口- N- {4-[2-(4-メトキシベンジル) - 1,1-ジォキシドイソチアゾリジン- 5-ィル] -1 ,3-チアゾール -2-ィル } -2-メチルベンゼンスルホンアミド(250mg)にトリフルォロ酢 酸(2. 5ml)とァ-ソール (0. 25ml)をカ卩えて、室温で 19時間攪拌した。溶媒を減圧 留去し、残渣にイソプロピルエーテルを力卩ぇ不溶物を濾取した。これにテトラヒドロフラ ンとイソプロピルエーテルの 1: 1混合溶媒を加えて攪拌後、粉末を濾取し淡褐色粉 末として表題ィ匕合物(133mg)を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(6)化合物(11 4)の合成
化合物(11— 2)を用い、実施例 11 (5)と同様の方法で表題ィ匕合物を得た。得られ た化合物の構造及び NMRデータを表 1に示す。
[0088] 実施例 12 化合物(12— 1)〜(12— 4)の合成
(1) 2-(4-メトキシベンジル) -1,2-チアジナン 1,1-ジォキシドの合成
1,4-ブタンスルタム(2. 16g)の Ν,Ν-ジメチルホルムアミド(100ml)溶液に氷冷下、 水素化ナトリウム(60%オイル懸濁、 700mg)、 4-メトキシベンジルクロリド(3. 5ml)を 加え、室温で 2時間攪拌した。氷冷下で水を加え、酢酸ェチルで抽出した後、有機層 を水、飽和食塩水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、乾燥剤を 濾去して溶媒を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー (展 開溶媒 n-へキサン:酢酸ェチル = 20 : 1)で精製し無色粉末として表題ィ匕合物 (4. 16g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.53— 1.65 (m, 2 H), 2.12 - 2.26 (m, 2 H), 3.02 - 3.13 (m, 2 H), 3.15 - 3.24 (m, 2 H), 3.81 (s, 3 H), 4.24 (s, 2 H), 6.83 - 6.95 (m, 2 H), 7.21 - 7.35 (m, 2 H).
(2) 4-[2-(4-メトキシベンジル) - 1 , 1-ジォキシド- 1 ,2-チアジナン- 6-ィル] - 1 ,3-チアゾ ール- 2-ァミンの合成
実施例 11 (3)と同様の方法で 2-(4-メトキシベンジル) -1,2-チアジナン 1,1-ジォキ シド (4. 16g)から無色粉末として表題ィ匕合物 (4. 38g)を得た。
1H NMR (300 MHz, DMSO- D6) δ 1.48 - 1.62 (m, 1 H), 1.76 - 1.98 (m, 1 H), 2.07 - 2.21 (m, 1 H), 2.24 - 2.43 (m, 1 H), 2.96 - 3.10 (m, 1 H), 3.26 - 3.46 (m, 1 H), 3.75 (s, 3 H), 4.17 - 4.38 (m, 3 H), 6.56 (s, 1 H), 6.89 - 7.04 (m, 4 H), 7.21 - 7.33 (m, 2 H).
(3)化合物(12— 1)及び(12— 2)の合成
実施例 12 (2)で得られた 4-[2- (4-メトキシベンジル) - 1 , 1-ジォキシド -1 ,2-チアジナ ン -6-ィル] -1 ,3-チアゾール -2-ァミンと、 3-クロ口- 2-メチルベンゼンスルホユルクロリ ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い製造方法 Bに表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(4)化合物(12— 3)及び(12— 4)の合成
実施例 12 (3)で得られた化合物を用い、実施例 11 (5)と同様の方法で表題ィ匕合 物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 13 化合物(13— 1)の合成
(1) N,N-ジメチル- 2-ォキソプロパン- 1-スルホンアミドの合成
— 60°Cに冷却した N, N—ジメチルメタンスルホンアミド(500mg)のテトラヒドロフラ ン溶液(15ml)に n—ブチルリチウム(2. 59Mのへキサン溶液、 1. 88ml)を加え、 3 0分間攪拌した後、酢酸ェチルを加え、 0°Cにて 2時間攪拌した。反応溶液に飽和塩 化アンモ-ゥム水溶液(60ml)をカ卩え、酢酸ェチル(60ml X 3)で抽出した。集めた 有機層を無水硫酸マグネシウムで乾燥し、乾燥剤をろ過し、溶媒を減圧留去した。黄 色油状物として表題ィ匕合物(600mg)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 2.45 (s, 3 H), 2.91 (s, 6 H), 3.99 (s, 2 H).
(2) 4-ァセチル- N, N-ジメチルテトラヒドロ- 2H-ピラン- 4-スルホンアミドの合成 Ν,Ν-ジメチル- 2-ォキソプロパン- 1-スルホンアミド(1. 25g)のアセトン溶液(10ml) に炭酸カリウム(2. 62g)及び 2-ブロモェチルエーテル(1. 3ml)を順次加え、 15時 間加熱還流した。セライトを用いて反応液をろ過し、溶媒を減圧留去した。得られた 残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 へキサン:酢酸ェチル = 2 : 1) で精製し、無色固体として表題ィ匕合物(530mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.17 - 2.37 (m, 2 H), 2.39 - 2.49 (m, 2 H), 2.50 (s, 3 H), 2.89 (s, 6 H), 3.11 - 3.36 (m, 2 H), 3.78 - 4.09 (m, 2 H).
(3) 4- (2-ァミノ- 1, 3-チアゾール -4-ィル) -N, N-ジメチルテトラヒドロ- 2H-ピラン- 4-ス ルホンアミドの合成
4-ァセチル- N, N-ジメチルテトラヒドロ- 2H-ピラン- 4-スルホンアミド(550mg)のメタ ノール溶液(15ml)に臭素(0. 24ml)を加え、 8時間加熱還流した。溶媒を減圧留 去して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 へキサン:酢酸 ェチル = 3 : 2)で精製し、無色粉末を得た。この無色粉末をメタノールとテトラヒドロフ ランの 1: 1混合溶媒(10ml)に溶解し、チォゥレア(195mg)を加え、室温で 5日間攪 拌した。溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー (展開 溶媒 クロ口ホルム:メタノール: 28%アンモニア水 = 20 : 1 : 0. 1)で精製し、無色粉 末として表題化合物(240mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 2.02 - 2.26 (m, 2 H), 2.32 - 2.49 (m, 2 H), 2.60 (s, 6 H), 3.09 - 3.25 (m, 2 H), 3.67 - 3.91 (m, 2 H), 6.79 (s, 1 H), 7.07 (brs, 2 H).
(4)化合物(13— 1)の合成
実施例 13 (3)で得られた4-(2-ァミノ-1, 3-チアゾール -4-ィル) -N, N-ジメチルテト ラヒドロ- 2H-ピラン- 4-スルホンアミドと、 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-ス ルホニルクロリドを用い製造方法 Aに従 、表題ィ匕合物を得た。得られた化合物の構 造及び NMRデータを表 1に示す。
実施例 14 化合物(14 1)及び(14 2)の合成
(1) N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-カルボキサミドの合成
4-ォキソチアンから Helvetica Chimica Acta 80, 1997, 1528- 1545.の方法に従って 合成したテトラヒドロ- 2H-チォピラン- 4-カルボン酸 (4. 53g)を用い、実施例 5 (1)と 同様の方法で、 Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(3. 63g)、 1-ヒドロキシベン ゾトリアゾール(4. 19g)、トリエチルァミン(5. 18ml)、 1-ェチル -3- (3-ジメチルァミノ プロピル)カルポジイミド塩酸塩(7. 13g)から、無色油状物質として表題ィ匕合物(5. 8 6g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.78 - 2.13 (m, 4 H), 2.59 - 2.84 (m, 5
H), 3.17 (s, 3 H), 3.70 (s, 3 H).
(2) N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-カルボキサミド 1-ォキシドの 合成
N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-カルボキサミド(3. 70g)のメタノ ール溶液(196ml)に、 0°C下、メタ過ヨウ素酸ナトリウム(5. 02g)の水溶液(23ml) を滴下し、室温にて 3時間攪拌した。反応液を飽和食塩水(150ml)にあけ、クロロホ ルム(150ml)にて抽出した。有機層を飽和食塩水(100ml)にて洗浄し、無水硫酸 マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、無色油状物質として 表題化合物(3. 94g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.78 - 1.99 (m, 2 H), 2.27 - 2.41 (m, 1 .4 H), 2.53 - 2.64 (m, 1.2 H), 2.65 - 2.78 (m, 1.4 H), 2.79 - 2.99 (m, 1 H), 3.07 - 3.15 (m, 0.6 H,) 3.19 (s, 2.1 H), 3.20 (s, 0.9 H), 3.27 - 3.39 (m, 1.4 H), 3.72 (s, 0.9 H), 3.73 (s, 2.1 H).
(3) 1-(1-ォキシドテトラヒドロ- 2H-チォピラン- 4-ィル)エタノンの合成
実施例 5 (2)と同様の方法で N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-力 ルボキサミド 1-ォキシド(3. 94g)と 3Mメチルマグネシウムブロミドのジェチルエーテ ル溶液(12. 8ml)から、淡黄色油状物質として表題ィ匕合物(2. 85g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.81 - 2.02 (m, 2 H), 2.20 (s, 0.9 H), 2 .21 (s, 2.1 H), 2.38 - 2.60 (m, 3 H), 2.62 - 2.80 (m, 2 H), 3.02 - 3.14 (m, 2 H).
(4) 4- (テトラヒドロ- 2H-チォピラン- 4-ィル) -1 ,3-チアゾール -2-ァミンの合成
1-(1-ォキシドテトラヒドロ- 2H-チォピラン- 4-ィル)エタノン(2. 85g)のメタノール溶 液(180ml)に臭素(1. 01ml)を力卩ぇ徐々に加熱し、 80°Cにて 30分間攪拌した。室 温まで戻し、チォゥレア(1. 35g)を加え室温にて 3日間攪拌した。反応溶媒を減圧 留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム: メタノール= 20 : 1〜4 : 1)で精製した。これをクロ口ホルム(50ml)〖こ溶力し、 1M水 酸ィ匕ナトリウム水溶液(30ml)、飽和食塩水(30ml)にて順次洗浄し、無水硫酸マグ ネシゥムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。再び得られた残渣をシリ 力ゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール = 30: 1〜20: 1)
で精製し淡褐色粉末として表題化合物(1. Olg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.64— 1.85 (m, 2 H), 2.26 - 2.37 (m, 2 H), 2.48 - 2.62 (m, 1 H), 2.62 - 2.86 (m, 4 H), 4.87 (brs, 2 H), 6.09 (d, J=0.9 Hz, 1 H).
(5)化合物(14 1)及び(14 2)の合成
実施例 14 (4)で得られた 4- (テトラヒドロ- 2H-チォピラン- 4-ィル) -1,3-チアゾール- 2-ァミンと、 3-クロ口- 2-メチルベンゼンスルホユルクロリド又は 5-クロ口- 3-メチル -1- ベンゾチォフェン- 2-スルホユルク口リドを用い製造方法 Bに従 、表題ィ匕合物を得た。 得られた化合物の構造及び NMRデータを表 1に示す。
[0091] 実施例 15 化合物(15— 1)及び(15— 2)の合成
(1) 3-クロ口- 2-メチル -N- [4- (1-ォキシドテトラヒドロ- 2H-チォピラン- 4-ィル) -1,3-チ ァゾール -2-ィル]ベンゼンスルホンアミド(15— 1)の合成
3-クロ口- 2メチル -N- [4- (テトラヒドロ- 2H-チォピラン- 4-ィル) -1,3-チアゾール -2-ィ ル]ベンゼンスルホンアミド(148mg)のメタノール溶液(4ml)に、 0°C下、メタ過ヨウ素 酸ナトリウム(90mg)の水溶液 (0. 4ml)を滴下し、室温にて 3. 5時間攪拌した。反応 液を飽和食塩水(30ml)にあけ、クロ口ホルム (40ml)にて抽出した。有機層を飽和 食塩水(20ml)にて洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶 媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (展開溶媒 ク ロロホルム:メタノール = 20: 1〜: L0: 1)で精製し無色粉末として表題ィ匕合物(44mg) を得た。 NMRデータは、表 1に記載した。
(2)化合物(15— 2)の合成
化合物(14— 2)を用い、実施例 15 (1)と同様の方法にて表題ィ匕合物を得た。得ら れた化合物の構造及び NMRデータを表 1に示す。
[0092] 実施例 16 化合物(16— 1)の合成
(1) N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-カルボキサミド 1,1-ジォキシ ドの合成
N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-カルボキサミド( 1. 84g)のクロ口 ホルム溶液(97ml)に、 0°C下、 3-クロ口過安息香酸 (純度 65%以上、 5. 87g)を加
え、室温にて 4. 5時間攪拌した。反応液を飽和チォ硫酸ナトリウム水溶液(200ml) にあけ、クロ口ホルム(50ml)にて抽出した。有機層を 1M水酸ィ匕ナトリウム水溶液(1 OOml)、飽和食塩水(70ml)にて洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤 を濾去して溶媒を減圧留去し、無色粉末として表題化合物(1. 79g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.20 - 2.43 (m, 4 H), 2.91 - 3.04 (m, 3 H), 3.20 (s, 3 H), 3.28 - 3.40 (m, 2 H), 3.73 (s, 3 H).
(2) 1-(1,1-ジォキシドテトラヒドロ- 2H-チォピラン- 4-ィル)エタノンの合成
実施例 5 (2)と同様の方法で N-メトキシ- N-メチルテトラヒドロ- 2H-チォピラン- 4-力 ルボキサミド 1,1-ジォキシド(0. 89g)と 3Mメチルマグネシウムブロミドのジェチルェ 一テル溶液(2. 68ml)から、無色粉末として表題化合物(0. 70g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.23 (s, 3 H), 2.27 - 2.38 (m, 4 H), 2.5 7 - 2.69 (m, 1 H), 2.92 - 3.04 (m, 2 H), 3.10 - 3.22 (m, 2 H).
(3) 4- (1,1-ジォキシドテトラヒドロ- 2H-チォピラン- 4-ィル) -1,3-チアゾール -2-ァミン 臭化水素酸塩の合成
1-(1,1-ジォキシドテトラヒドロ- 2H-チォピラン- 4-ィノレ)エタノン(0. 70g)のメタノー ル溶液 (40ml)に臭素(0. 23ml)を加え徐々〖こ加熱し、 80°Cにて 30分間攪拌した。 室温まで戻し、チォゥレア(302mg)をカ卩ぇ室温にて 7時間攪拌した。メタノール (40 ml)と水(8ml)をカ卩え、室温にて 15時間攪拌し、 50°Cにて 10時間攪拌した。反応溶 媒を減圧留去し、得られた残渣をテトラヒドロフランで洗浄して無色粉末として表題ィ匕 合物(992mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.93 - 2.10 (m, 2 H), 2.17 - 2.28 (m, 2 H), 2.88 - 3.00 (m, 1 H), 3.08 - 3.19 (m, 2 H), 3.22 - 3.36 (m, 2 H), 6.63 (d, J=0.6 Hz, 1 H ), 8.92 (brs, 2 H).
(4)化合物(16— 1)の合成
実施例 16 (3)で得られた 4-(1,1-ジォキシドテトラヒドロ- 2H-チォピラン- 4-ィル) -1,3 -チアゾール -2-アミン臭化水素酸塩と、 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-ス ルホニルクロリドを用い製造方法 Bに従 、表題ィ匕合物を得た。得られた化合物の構造 及び NMRデータを表 1に示す。
[0093] 実施例 17 化合物(17— 1)の合成
(1) 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミン及び 4- (4-メトキシテ トラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンの合成
1- (テトラヒドロ- 2H-ピラン- 4-ィル)エタノン(6. 57g)のメタノール(100ml)溶液に臭 素(2. 9ml)をカ卩ぇ 65°Cで 30分攪拌した後、室温に戻し、チォゥレア(3. 90g)を加え 一晩攪拌した。反応終了後クロ口ホルムと飽和炭酸水素ナトリウム水溶液を加え、室 温で 10分攪拌した後、クロ口ホルムで抽出し飽和食塩水で洗浄した。有機層を無水 硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。得られた残留物に へキサンを加え攪拌し、固体を濾取して 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チア ゾール -2-ァミンと少量の 4-(4-メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾー ル- 2-ァミンの混合物(5. 32g)を得た。
(2) 5-クロ口- N- [4- (4-メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィ ル] -3-メチル -1-ベンゾチォフェン- 2-スルホンアミド体(17— 1)の合成
製造方法 Bに従い 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンと 4- (4-メトキシテトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミンの混合物(1. 00 5g)と 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホニルクロリド(3. 07g)のカップ リングを行った。プレパラティブ TLC (展開溶媒 クロ口ホルム:メタノール: 28%アンモ ユア水 = 20 : 1 : 0. 1)で精製し、薄褐色粉末として表題ィ匕合物(17mg)を得た。 得られた化合物の構造及び NMRデータを表 1に示す。
[0094] 実施例 18 化合物(18— 1)〜(18— 84)の合成
参考例 1 (2)で得た 4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ァミン(3 0 mol)のクロ口ホルム(700 1)溶液に、 4ージメチルァミノピリジン(30 mol)、ピ リジン(120 μ mol)をカ卩えた後、それぞれ対応するスルホユルクロリド(120 μ mol)を 加え、 50°Cで一晩攪拌した。反応液に酢酸ェチルとテトラヒドロフランの 1 : 1混合溶 媒を加え、 1M塩酸水溶液で洗浄し、有機層を減圧で濃縮した。得られた残渣を TL Cプレート (メルク社製シリカゲル 60F254)を用いて精製し、目的物を得た。構造式 及び質量分析データを表 2に記載した。
[0095] 実施例 19 化合物(19 1)〜(19 11)の合成
(1) N- (4-メトキシベンジル)メタンスルホンアミドの合成
- 50°Cに冷却した 4-メトキシベンジルァミン(10. 24g)のクロ口ホルム( 150ml)溶 液にトリェチルァミン(10. 24ml)及びメタンスルホユルクロリド(5. 78ml)を順次加え 、室温で 1時間攪拌した。反応溶液をクロ口ホルム(100ml)で希釈し、水、 1M塩酸 水溶液、飽和食塩水で順次洗浄した。その有機層を無水硫酸マグネシウムで乾燥し 、乾燥剤を濾去して溶媒を減圧留去した。無色粉末として、表題化合物(14. 85g) を得た。
1H NMR (200 MHz, DMSO-D6) δ 2.80 (s, 3 H), 3.74 (s, 3 H), 4.07 (d, J=6.6 Hz, 2 H), 6.90 (d, J=8.8 Hz, 2 H), 7.25 (d, J=8.8 Hz, 2 H), 7.38 - 7.56 (m, 1 H).
(2) N- (4-メトキシベンジル) -N-メチルメタンスルホンアミドの合成
0°Cに冷却した水素化ナトリウム(60%オイル懸濁、 1. 76g)の N, N—ジメチルホ ルムアミド(35ml)溶液に、 N- (4-メトキシベンジル)メタンスルホンアミド(8. 61g)をカロ え、 30分間攪拌した。次にヨウ化メチル(3. 23ml)を加え、同温度にて 1時間攪拌し た。反応溶液を酢酸ェチル(120ml)で希釈し、飽和食塩水、 1M塩酸水溶液、飽和 食塩水で順次洗浄した。その有機層を無水硫酸マグネシウムで乾燥し、乾燥剤を濾 去して溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (展開 溶媒 へキサン:酢酸ェチル = 1 : 1)で精製し、無色固体として表題ィ匕合物(7. 6g) を得た。
1H NMR (300 MHz, DMSO-D6) δ 2.62 (s, 3 H), 2.91 (s, 3 H), 3.75 (s, 3 H), 4.15 (s, 2 H), 6.94 (d, J=8.7 Hz, 2 H), 7.25 (d, J=8.7 Hz, 2 H).
(3) 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-メチルメタンス ルホンアミドの合成
- 70°Cに冷却した N- (4-メトキシベンジル) -N-メチルメタンスルホンアミド(2. 49g) のテトラヒドロフラン(35ml)溶液に n—ブチルリチウム(2. 59Mのへキサン溶液、 5. 3 4ml)を加え、 30分間攪拌した後、クロ口酢酸ェチル(1. 27ml)を加え、— 20°Cにて 1時間攪拌した。次に酢酸 (0. 75ml)及びチォゥレア(910mg)を順次カ卩え、室温に て一晩攪拌した。反応溶液を酢酸ェチル(150ml)で希釈し、水、炭酸水素ナトリウム 水溶液、飽和食塩水で順次洗浄した。集めた有機層を無水硫酸マグネシウムで乾燥
し、乾燥剤をろ過し、溶媒を減圧留去した。得られた残渣をエタノールとジイソプロピ ルエーテルの 1 : 1混合溶媒で洗净し、黄色粉末として表題化合物(1. 61g)を得た。 1H NMR (300 MHz, DMSO-D6) δ 2.59 (s, 3 H), 3.74 (s, 3 H), 4.09 (s, 2 H), 4.30 (s, 2 H), 6.59 (s, 1 H), 6.92 (d, J=8.7 Hz, 2 H), 7.03 (brs, 1 H), 7.21 (d, J=8.7 Hz, 2 H).
(4) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-メチルメタンスルホンアミド トリフルォロ 酢酸塩の合成
1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-メチルメタンスル ホンアミド(1. 4g)をトリフルォロ酢酸(15ml)に溶解し、ァ-ソール(1. 5ml)をカロえ、 室温で一晩攪拌した。反応溶液を減圧乾固して得られた残渣にジイソプロピルエー テルを加え、不溶物を濾取し淡黄色粉末として表題ィ匕合物(1. 4g)を得た。
1H NMR (200 MHz, DMSO-D6) δ 2.61 (d, J=4.4 Hz, 3 H), 4.29 (s, 2 H), 6.75 (s, 1 H), 6.98 - 7.24 (m, 1 H).
(5) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-メチルメタンスルホンアミドの合成
1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N-メチルメタンスルホンアミドトリフルォロ酢 酸塩(1. 4g)を水 (40ml)に溶解し、ゆっくりと炭酸水素ナトリウム(380mg)をカ卩えた 。室温で 30分間攪拌した後、塩ィ匕ナトリウムを加え、さらに 30分間攪拌を続けた。反 応溶液を酢酸ェチル(100ml X 3)で抽出し、その有機層を無水硫酸マグネシウムで 乾燥し、乾燥剤を濾去して溶媒を減圧留去した。得られた残渣にジイソプロピルエー テルを加え、不溶物を濾取し淡黄色粉末として表題化合物(624mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 2.58 (d, J=4.9 Hz, 3 H), 4.14 (s, 2 H), 6.51 (s, 1 H), 6.83 (q, J=4. 9Hz, 1 H), 6.96 (brs, 2 H).
(6)化合物(19 1)及び(19 2)の合成
実施例 19 (3)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-メチルメタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリド又は 5 -クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホユルク口リドを用い、製造方法 Bに従 Vヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(7)化合物(19 3)及び(19 4)の合成
I '(H Z <ZH 6'8=f 'Ρ) ΐ6·9 '(Η ΐ SS'9 '(Η Ζ <s) LZ'f '(Η Ζ <s) 9Vf '(Η S 's) ε ' (Η Ζ 'ΖΗ ΓΖ=Γ 90·ε '(Η S 'ΖΗ ΓΖ=Γ 98 9 (9。- oswa 'ζ οοε) Η匪 Ηΐ
^ べ ベ^ ( べ:^ - )- Ν- ^エ- 翁^; (£)6i m ベ^ ( /;^ベ:^^^ ― ) Ν— ^エ— Ν—( / — /— 、 ^— ε' ΐ— — S)— ΐ (ζ)
•(Η Ζ 'ΖΗ
S"8=f 'Ρ) SZ'L '(Η Ζ 'ΖΗ S"8=f 'Ρ) Ζ6'9 '(Η Ζ 's) fZ'f '(Η £ fL'£ '(Η Ζ 'ΖΗ Ζ' L=
[ π·ε '(Η ε £βτ '(Η ε 'ΖΗ
'ζ οζ) Η匪 HI
¾^^0)、 ^ べ ベ^ ( べ:^ - - Ν- ^エ- Ν (I)
¾^^ (0"[—02)〜("[ 02)呦^^ [9600] 。 · コ 挲 ¾ ^^ Ν、 呦 ^ p i^
ベ ベ^ ^ - Ν -( / - /— 、 ^- ε'ΐ- - - ΐ (S) 6ΐ ¾ϊ第
¾ίί^ )(ΐ "[— 6ΐ)〜(9 6ΐ)呦^ Ι 6)
。 ·η»¾コ 挲 «^-^H N (
srao61 ) ^m^
/ 一ェ ΰ。 ^^ m^ ^m^m
nf(¾難 (
Su¾o
^Q) (SuiooS)、 ^ べ ベ^ ^ — N—( / — /— 、 ^— ε'ΐ— — S)— ΐ
(α¾^¾) ^Ο) (9-61)、 べ ベ ベ
- ( - s - /— 、 ^- ε'ΐ- { ^ [ - ( ^ ^ )])- - Ν(8)
。 · コ 挲 ¾ ^ WN、 呦 ^ i^ 。:
6098T0/S00Zdf/X3d 89 Z99TS0/900Z OAV
.01 (brs, 2 H), 7.25 (d, J=8.9 Hz, 2 H).
(3) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-ェチルメタンスルホンアミドの合成
1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-ェチル -N- (4-メトキシベンジル)メタンスル ホンアミドから、実施例 19 (4)及び(5)の方法に従い表題化合物を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.05 (t, J=7.2 Hz, 3 H), 2.79 - 3.05 (m, 2 H), 4. 12 (s, 2 H), 6.50 (s, 1 H), 6.84 - 7.07 (m, 3 H).
(4) 3-クロ口- N- [4- ({ [ェチル (4-メトキシベンジル)ァミノ]スルホ-ル }メチル )-1,3-チ ァゾール -2-ィル] -2-メチルベンゼンスルホンアミド(20— 1)の合成(製造方法 E)
1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-ェチル -N- (4-メトキシベンジル)メタンスル ホンアミド(221mg)をクロ口ホルム(5ml)に溶解し、 4-ジメチルァミノピリジン(79mg )、ジイソプロピルェチルァミン(171 μ 1)をカ卩えた後、氷冷下で 3-クロ口- 2-メチルベ ンゼンスルホユルクロリド(221mg)をカロえ、その後、室温に戻して 5時間攪拌した。さ らに 3-クロ口- 2-メチルベンゼンスルホユルクロリド(293mg)、 4-ジメチルァミノピリジ ン(159mg)を加え室温で一晩攪拌した。反応液に 10%硫酸水素カリウム水溶液を 加え酢酸ェチルで抽出した後、抽出液を 10%硫酸水素カリウム水溶液、飽和食塩 水で順次洗浄した。有機層を無水硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶媒 を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口 ホルム:メタノール =40: 1)で精製し、薄褐色泡状の表題化合物(244mg)を得た。 N MRデータは、表 1に記載した。
(5)化合物(20— 2)の合成
実施例 20 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-ェチルメタンスルホンアミドと 2-クロ口- 4-フルォロベンゼンスルホ-ルクロリドを用 い、製造方法 Bに従い表題ィ匕合物を得た。得られた化合物の構造及び NMRデータを 表 1に示す。
(6)化合物(20— 3)及び(20— 4)の合成
化合物(20— 1)及び(20— 2)を用い、実施例 19 (4)と同様の方法で従!、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(7)化合物(20— 5)〜(20— 10)の合成
実施例 20 (3)で得た l-(2-ァミノ- 1,3-チアゾール -4-ィル) -N-ェチルメタンスルホン アミドと対応するスルホニルクロリドを用い、製造方法 A、 B又は Dに従い表題ィ匕合物を 得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0098] 実施例 21 化合物(21— 1)〜(21— 4)の合成
(1) N-イソプロピル- N- (4-メトキシベンジル)メタンスルホンアミドの合成
実施例 12 (1)と同様の方法で N-イソプロピルメタンスルホンアミド(2. 84g)と p-メト キシベンジルクロリド(3. 31ml)力も薄黄色固体として表題ィ匕合物 (4. 36g)を得た。 1H NMR (200 MHz, CHLOROFORM— D) δ 1.18 (d, J=7.0 Hz, 6 H), 2.76 (s, 3 H), 3.80 (s, 3 H), 4.04 - 4.20 (m, 1 H), 4.30 (s, 2 H), 6.82 - 6.90 (m, 2 H), 7.28 - 7.36 (m, 2 H).
(2) 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル- N-イソプロピル- N- (4-メトキシベンジル)メ タンスルホンアミドの合成
実施例 19 (3)と同様の方法で N-イソプロピル- N- (4-メトキシベンジル)メタンスルホ ンアミド(4. 23g)、 n—ブチルリチウム(2. 59Mのへキサン溶液、 7. 62ml)、クロ口酢 酸ェチル(1. 93ml)およびチォゥレア(1. 38g)から、黄色粉末として表題化合物 (4 . 27g)を得た。
1H NMR (200 MHz, DMSO-D6) δ 1.00 (d, J=7.0 Hz, 6 H), 3.73 (s, 3 H), 3.78 - 3. 94 (m, 1 H), 4.15 (brs, 2 H), 4.20 (brs, 2 H), 6.53 (s, 1 H), 6.83 - 6.93 (m, 2 H), 6. 99 (brs, 2 H), 7.23 - 7.34 (m, 2 H).
(3)化合物(21— 1)及び(21— 2)の合成
実施例 21 (2)で得た 1- (2-ァミノ- 1,3-チアゾール -4-ィル- N-イソプロピル- N- (4-メ トキシベンジル)メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリド 又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方法 Bに従 、表題ィ匕合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0099] (4)化合物(21— 3)及び(21— 4)の合成
化合物( 21— 1 )及び( 21— 2)を用い、実施例 19 (4)と同様の方法で従!、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0100] 実施例 22 化合物(22— 1)及び(22— 2)の合成
(1) N- (4-メトキシベンジル) -N-プロピルメタンスルホンアミドの合成
0°Cに冷却した水素化ナトリウム(60%オイル懸濁、 420mg)の Ν,Ν-ジメチルホル ムアミド(20ml)溶液に、 Ν-プロピルメタンスルホンアミド(1. 32g)を加え、 30分間攪 拌した。次に 4-メトキシベンジルクロリド(1. 36ml)を加え、室温にて 1時間攪拌した。 反応溶液を酢酸ェチル(120ml)で希釈し、飽和食塩水と水の 1: 1混合液、飽和食 塩水で順次洗浄した。その有機層を無水硫酸マグネシウムで乾燥し、乾燥剤を濾去 して溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶 媒 へキサン:酢酸ェチル =4 : 1)で精製し、黄色油状物質として表題ィ匕合物(2. 34 g)を得た。
1H NMR (200 MHz, DMSO— D6) δ 0.72 (t, J=7.5 Hz, 3 H), 1.25 - 1.57 (m, 2 H), 2. 91 (s, 3 H), 2.95 - 3.10 (m, 2 H), 3.74 (s, 3 H), 4.24 (s, 2 H), 6.92 (d, J=8.8 Hz, 2 H), 7.28 (d, J=8.8 Hz, 2 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-n-プロピルメタ ンスルホンアミドの合成
実施例 19 (3)と同様の方法で N- (4-メトキシベンジル) -N-プロピルメタンスルホンァ ミド(2. 34g)から、淡黄色固体として表題ィ匕合物(3. 05g)を得た。
1H NMR (300 MHz, DMSO— D6) δ 0.63 (t, J=7.4 Hz, 3 H), 1.10 - 1.36 (m, 2 H), 2. 83 - 3.10 (m, 2 H), 3.74 (s, 3 H), 4.15 (s, 2 H), 4.27 (s, 2 H), 6.55 (s, 1 H), 6.90 (d , J=8.7 Hz, 2 H), 7.01 (brs, 2 H), 7.26 (d, J=8.7 Hz, 2 H).
(3)化合物(22— 1)の合成
実施例 22 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-n-プロピルメタンスルホンアミドと 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スル ホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られた化合物の構造 及び NMRデータを表 1に示す。
[0101] (4)化合物(22— 2)の合成
化合物(22— 1)を用い、実施例 19 (4)と同様の方法で従い表題ィ匕合物を得た。得 られた化合物の構造及び NMRデータを表 1に示す。
[0102] 実施例 23 化合物(23— 1)〜(23— 4)の合成
(1) N-シクロへキシル -N- (4-メトキシベンジル)メタンスルホンアミドの合成 実施例 22 (1)と同様の方法で N-シクロへキシルメタンスルホンアミド(1. 30g)と 4- メトキシベンジルクロリド(1. 1ml)から、無色粉末として表題化合物(2. 15g)を得た
1H NMR (300 MHz, DMSO— D6) δ 0.85 - 1.05 (m, 1 H), 1.11 - 1.44 (m, 4 H), 1.46 - 1.57 (m, 1 H), 1.58 - 1.75 (m, 4 H), 2.93 (s, 3 H), 3.44 - 3.62 (m, 1 H), 3.74 (s, 3 H), 4.27 (s, 2 H), 6.89 (d, J=8.7 Hz, 2 H), 7.30 (d, J=8.7 Hz, 2 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N-シクロへキシル -N- (4-メトキシベンジル) メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N-シクロへキシル -N- (4-メトキシベンジル)メタンスル ホンアミド(2. 15g)、クロ口酢酸ェチル(0. 84ml)とチォゥレア(602mg)から、無色 粉末として表題化合物(1. 59g)を得た。
1H NMR (200 MHz, DMSO— D6) δ 0.64 - 0.98 (m, 1 H), 1.01 - 1.29 (m, 4 H), 1.34 - 1.53 (m, 1 H), 1.56 - 1.79 (m, 4 H), 3.18 - 3.60 (m, 1 H), 3.73 (s, 3 H), 4.16 (s, 2 H), 4.21 (s, 2 H), 6.54 (s, 1 H), 6.88 (d, J=8.5 Hz, 2 H), 6.98 (brs, 2 H), 7.27 (d, J=8.5 Hz, 2 H).
(3)化合物(23— 1)及び(23— 2)の合成
実施例 23 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N-シクロへキシル -N- (4 -メトキシベンジル)メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリ ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方 法 Bに従 ヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す
[0103] (4)化合物(23— 3)及び(23— 4)の合成
化合物(23— 1)及び(23— 2)を用い、実施例 19 (4)と同様の方法で従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0104] 実施例 24 化合物(24— 1)〜(24— 4)の合成
(1) N- (4-メトキシベンジル) -N- (2-メトキシェチル)メタンスルホンアミドの合成 実施例 22 (1)と同様の方法で N- (2-メトキシェチル)メタンスルホンアミド(850mg)と
Ζ·ε '(Η Ζ 's) 86 '(Η S 's) £S'Z '(Η 6 's) S8 9 (9。- 'ζ OOS) Η匪 Ηΐ
。 频
¾ίί^ ^^ ベ ベ^ ( べ:^ 、^ Η - - Ν- ( ΰ 1 ^ - Ν (I)
。
Λ(—^Λ( : ^ ^Λ(^-Ζ- ^-Ζ 、 べ ベ^ ( ^エ ^ H - - N- ( べ:^ 、^ H — — N—( / — /— 、 ^— ε' ΐ— — — ¾ (Z) Z M
-^ (z→z) ii (i→z) (ε)
•(H Z 'ZH S"8=f 'P) WL '
(H Z Ί WL '(H Z <ZH S'8=f 'Ρ) ΐ6·9 '(Η ΐ 's) W9 '(H Z <s) ZZ'f '(H Z <s) 6Vf '(
H ε 's) L-£ '(H 'ω) zz'z - ers '(Η ε ere g oa-os^a 'z oz) 匪 HI
(3go ·ΐ)、 べ
^^^( ^^/ ^4^-2)-Ν-( / ^>-/ ^4^)-Ν ¾^ ) ^^ (ε) 6ΐίϋ¾ϊ第
¾^^0)、 ^ べ ベ^ (
^エ 、^ Η — — Ν— ( べ:^^^ Η — — Ν—( / — /— 、 ^— ε' ΐ— — s)— ΐ (s)
•(Η Ζ
'ΖΗ 6Γ8=ί" 'Ρ) 62"Ζ '(Η Ζ 'ΖΗ 8·8=ί" 'Ρ) 88·9 '(Η Ζ <s) 0 · '(Η Ζ ΐ8·ε '(Η f 'ω) f ex - esx '(Η ε 's) sex '(Η ε ΐ6 s (a - Ο ΟΗΟΊΗ 'ζ oz) Η匪 HI
6098T0/S00Zdf/X3d 89 Z99TS0/900Z OAV
(s, 3 H), 4.34 (s, 2 H), 6.92 (d, J=8.8 Hz, 2 H), 7.31 (d, J=8.86 Hz, 2 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (2,2-ジメチルプロピル)- N- (4-メトキシべ ンジル)メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N- (2,2-ジメチルプロピル)- N- (4-メトキシベンジル)メ タンスルホンアミド(2. 59g)、クロ口酢酸ェチル(1. 06ml)とチォゥレア(76 lmg)力 ら、無色粉末として表題ィ匕合物(2. 30g)を得た。
1H NMR (200 MHz, DMSO-D6) δ 0.73 (s, 9 H), 2.95 (s, 2 H), 3.73 (s, 3 H), 4.25 (brs, 4 H), 6.53 (s, 1 H), 6.90 (d, J=8.8 Hz, 2 H), 7.01 (brs, 2 H), 7.28 (d, J=8.8 Hz , 2 H).
(3)化合物(25— 1)及び(25— 2)の合成
実施例 25 (2)で得た 1-(2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (2,2-ジメチルプロピ ル)- N- (4-メトキシベンジル)メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホ -ルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い 、製造方法 Bに従い表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0107] (4)化合物(25— 3)及び(25— 4)の合成
化合物(25— 1)及び(25— 2)を用い、実施例 19 (4)と同様の方法で表題ィ匕合物 を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0108] 実施例 26 化合物(26— 1)〜(26— 4)の合成
(1) N- (4-メトキシベンジル) -N-(2-トリフルォロェチル)メタンスルホンアミドの合成 実施例 22 (1)と同様の方法で N-(2-トリフルォロェチル)メタンスルホンアミド(1. 15 g)と 4-メトキシベンジルクロリド (0. 97ml)から、無色粉末として表題化合物(1. 62g )を得た。
1H NMR (200 MHz, DMSO-D6) δ 3.04 (s, 3 H), 3.75 (s, 3 H), 3.86 - 4.05 (m, 2 H ), 4.41 (s, 2 H), 6.93 (d, J=8.8 Hz, 2 H), 7.29 (d, J=8.8 Hz, 2 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N- (2-トリフルォロ ェチル)メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N- (4-メトキシベンジル) -N-(2-トリフルォロェチル)メ
タンスルホンアミド(1. 62g)、クロ口酢酸ェチル(0. 64ml)とチォゥレア(457mg)力 ら、黄色粉末として表題化合物(720mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 3.74 (s, 3 H), 3.82 (q, J=9.5 Hz, 2 H), 4.34 (s, 2 H), 4.43 (s, 2 H), 6.58 (s, 1 H), 6.91 (d, J=8.8 Hz, 2 H), 7.07 (brs, 2 H), 7.27 (d, J=8.8 Hz, 2 H).
(3)化合物(26— 1)及び(26— 2)の合成
実施例 26 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-(2-トリフルォロェチル)メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホ -ルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い 、製造方法 Bに従い表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0109] (4)化合物(26— 3)及び(26— 4)の合成
化合物(26— 1)及び(26— 2)を用い、実施例 19 (4)と同様の方法に従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0110] 実施例 27 化合物(27— 1)〜(27— 4)の合成
(1) N- (4-メトキシベンジル) -N- {2- [(4-メトキシベンジル)ォキシ]ェチル }メタンスルホ ンアミドの合成
実施例 22 (1)と同様の方法で N-(2-ヒドロキシェチル)メタンスルホンアミド(920mg )と 4-メトキシベンジルクロリド(1. 79ml)から、無色粉末として表題化合物(1. 98g) を得た。
1H NMR (200 MHz, DMSO— D6) δ 2.93 (s, 3 H), 3.17 - 3.32 (m, 2 H), 3.36 - 3.50 (m, 2 H), 3.74 (s, 6 H), 4.29 (s, 2 H), 4.34 (s, 2 H), 6.76 - 7.02 (m, 4 H), 7.12 - 7.3 1 (m, 4 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N- {2- [(4-メトキシ ベンジル)ォキシ]ェチル }メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N- (4-メトキシベンジル) - N-{2-[(4-メトキシベンジル) ォキシ]ェチル }メタンスルホンアミド(1. 98g)、クロ口酢酸ェチル(0. 61ml)とチォゥ レア (436mg)から、黄色油状物質として表題化合物(1. 45g)を得た。
1H NMR (200 MHz, DMSO— D6) δ 3.09 - 3.38 (m, 4 H), 3.73 (s, 6 H), 4.18 (s, 2 H ), 4.27 (s, 2 H), 4.31 (s, 2 H), 6.51 (s, 1 H), 6.80 - 6.94 (m, 4 H), 7.02 (brs, 2 H), 7 .16 (d, J=8.8 Hz, 2 H), 7.22 (d, J=8.8 Hz, 2 H).
(3)化合物(27— 1)及び(27— 2)の合成
実施例 27 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N- {2- [(4-メトキシベンジル)ォキシ]ェチル }メタンスルホンアミドと、 3-クロ口- 2-メチ ルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ ユルク口リドを用い、製造方法 Dに従い表題化合物を得た。得られた化合物の構造及 び NMRデータを表 1に示す。
[0111] (4)化合物(27— 3)及び(27— 4)の合成
化合物(27— 1)及び(27— 2)を用い、実施例 19 (4)と同様の方法に従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0112] 実施例 28 化合物(28— 1)〜(28— 4)の合成
(1) N-(2-フルォロェチル) -N- (4-メトキシベンジル)メタンスルホンアミドの合成 実施例 22 (1)と同様の方法で N-(2-フルォロェチル)メタンスルホンアミド(1. 21g) と 4-メトキシベンジルクロリド(1. 28ml)から、無色粉末として表題化合物(2. 08g)を 得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 2.91 (s, 3 H), 3.34 - 3.48 (m, 1 H), 3.5 1 - 3.60 (m, 1 H), 4.31 - 4.41 (m, 1 H), 4.44 (s, 2 H), 4.56 - 4.72 (m, 1 H), 6.89 (d , J=8.8 Hz, 2 H), 7.29 (d, J=8.8 Hz, 2 H).
(2) 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (2-フルォロェチル) -N- (4-メトキシベン ジル)メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N-(2-フルォロェチル) -N- (4-メトキシベンジル)メタン スルホンアミド(2. 08g)、クロ口酢酸ェチル(0. 94ml)とチォゥレア(670mg)から、 無色粉末として表題化合物(2. 28g)を得た。
1H NMR (300 MHz, DMSO— D6) δ 3.20 - 3.44 (m, 2 H), 3.74 (s, 3 H), 4.12 - 4.27 (m, 3 H), 4.31 - 4.47 (m, 3 H), 6.57 (s, 1 H), 6.92 (d, J=8.7 Hz, 2 H), 7.03 (brs, 2 H), 7.26 (d, J=8.7 Hz, 2 H).
( ^ (—^Λ( : ^ ^Λ(^-Ζ- ^-Ζ 、 べ ベ^ ( べ:^ ^ -
ΐ— — — ΐ (Z)
•(Η Ζ 'ΖΗ 8"8=f 'Ρ) ZZ'L '(Η Ζ
) WL '(Η Ζ 'ΖΗ 8·8=ί" 'Ρ) 68·9 '(Η ΐ 9S.9 '(Η Ζ <s) 9Z'f '(Η Ζ <s) fVf '(Η S
S '(Η ΐ 'ω) £q'Z - ££τ '(Η f 'ω) 9Ζ - ZVO 9 (9。- OSW I 'z 002) 匪 Ηΐ
Ww、 W(
3ui0Z9) ^; (I
ra 6 Ό) ^エ邈 3 0 'S)、 べ
(S)
•(Η Ζ 'ΖΗ 8'
8=1" 'Ρ) WL '(Η Ζ 'ΖΗ 8·8=ί" 'Ρ) 88·9 '(Η Ζ <s) SZ'f '(Η S ΐ8·ε '(Η S ΜΖ '(Η ΐ 'ω) VZ - SVZ '(Η 'ω) Ζ6 - 69 9 (a- HOHOHOlHD '^Η 002) Η Ν Ηΐ
°-Μ ^Ο) ^^ ^ Μ^^ ^^ -^)- -Λ^Ά ^ ^-Η (I)
6S)〜("[一 6S)呦 6Sfi ¾?第 [WIO]
^m ^m^ u^ (p)6mm ヽ ¾r¾ (s sz) (x sz)
-^ ( -2Z) i (ε - 82) ( ) [επο]
。
-( ^エ ΰ / - S)- N -( / - /— 、 ^- ε'ΐ- - - ΐ (Z)
-^ (Z-2Z) m (I-8S) (ε)
6098T0/S00Zdf/X3d ZL Z99TS0/900Z OAV
ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方 法 Bに従 ヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す
[0115] (4)化合物(29— 3)及び(29— 4)の合成
化合物(29— 1)及び(29— 2)を用い、実施例 19 (4)と同様の方法に従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0116] 実施例 30 化合物(30— 1)〜(30— 4)の合成
(1) Ν,Ν-ビス (4-メトキシベンジル)メタンスルホンアミドの合成
実施例 22 (1)と同様の方法で Ν- (4-メトキシベンジル)メタンスルホンアミド(7. 10g) と 4-メトキシベンジルクロリド (4. 94ml)から、無色粉末として表題ィ匕合物(11. lg)を 得た。
1H NMR (200 MHz, DMSO-D6) δ 2.88 (s, 3 H), 3.74 (s, 6 H), 4.18 (s, 4 H), 6.89 (d, J=8.4 Hz, 4 H), 7.19 (d, J=8.4 Hz, 4 H).
(2) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -N,N-ビス (4-メトキシベンジル)メタンスルホ ンアミドの合成
実施例 19 (3)と同様の方法で Ν,Ν-ビス (4-メトキシベンジル)メタンスルホンアミド(5 . OOg)、クロ口酢酸ェチル(1. 74ml)とチォゥレア(1. 24g)から、無色粉末として表 題化合物(5. 48g)を得た。
1H NMR (300 MHz, DMSO-D6) δ 3.71 (s, 6 H), 4.14 (s, 4 H), 4.25 (s, 2 H), 6.43 (s, 1 H), 6.82 (d, J=8.8 Hz, 4 H), 7.03 (brs, 2 H), 7.10 (d, J=8.8 Hz, 4 H).
(3)化合物(30— 1)及び(30— 2)の合成
実施例 30 (2)で得た 1- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -Ν,Ν-ビス (4-メトキシベン ジル)メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリド又は 5-クロ 口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方法 Βに従!、表 題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0117] (4)化合物(30— 3)及び(30— 4)の合成
化合物(30— 1)及び(30— 2)を用い、実施例 19 (4)と同様の方法に従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0118] 実施例 31 化合物(31— 1)〜(31— 4)の合成
(1) N- (4-メトキシベンジル) -N-メチルプロパン- 2-スルホンアミドの合成
実施例 22 (1)と同様の方法で N-メチルプロパン- 2-スルホンアミド(1. 61g)と 4—メ トキシベンジルクロリド(1. 59ml)から、無色粉末として表題化合物(1. 61g)を得た。 IH NMR (200 MHz, CHLOROFORM— D) δ 1.37 (s, 3 H), 1.40 (s, 3 H), 2.77 (s, 3 H), 3.13 - 3.44 (m, 1 H), 3.81 (s, 3 H), 4.32 (s, 2 H), 6.88 (d, J=8.8 Hz, 2 H), 7.27 (d, J=8.8 Hz, 2 H).
(2) 2- (2-ァミノ- 1,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-メチルプロパン -2-スルホンアミドの合成
実施例 19 (3)と同様の方法で N- (4-メトキシベンジル) -N-メチルプロパン- 2-スルホ ンアミド(1. 61g)、クロ口酢酸ェチル(1. 23ml)とチォゥレア(999mg)から、黄色粉 末として表題化合物(260mg)を得た。
IH NMR (300 MHz, CHLOROFORM— D) δ 1.81 (s, 6 H), 2.56 (s, 3 H), 3.79 (s, 3 H), 4.06 (brs, 2 H), 5.04 (brs, 2 H), 6.64 (s, 1 H), 6.84 (d, J=8.7 Hz, 2 H), 7.23 (d, J=8.7 Hz, 2 H).
(3)化合物(31— 1)及び(31— 2)の合成
実施例 31 (2)で得た 2- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -N- (4-メトキシベンジル) -N-メチルプロパン- 2-スルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリ ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方 法 Bに従 ヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す
[0119] (4)化合物(31— 3)及び(31— 4)の合成
化合物(31— 1)及び(31— 2)を用い、実施例 19 (4)と同様の方法で従!、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0120] 実施例 32 化合物(32— 1)〜(32— 4)の合成
(1) N- (4-メトキシベンジル) -N-メチル -1- [2- (メチルァミノ)- 1 ,3-チアゾール -4-ィル] メタンスルホンアミドの合成
実施例 19 (3)と同様の方法で N- (4-メトキシベンジル) N-メチルメタンスルホンアミド
(5. 03g)、クロ口酢酸ェチル(2. 55ml)とメチルチオゥレア(2. 16g)から、黄色粉末 として表題ィ匕合物(6. 12g)を得た。
1H NMR (300 MHz, DMSO-D6) δ 2.60 (s, 3 H), 2.84 (d, J=4.8 Hz, 3 H), 3.74 (s, 3 H), 4.11 (s, 2 H), 4.35 (s, 2 H), 6.65 (s, 1 H), 6.92 (d, J=8.8 Hz, 2 H), 7.21 (d, J= 8.8 Hz, 2 H), 7.47 - 7.67 (m, 1 H).
(2)化合物(32— 1)及び(32— 2)の合成
実施例 32 (1)で得た N- (4-メトキシベンジル) -N-メチル -ト [2- (メチルァミノ)- 1,3-チ ァゾール -4-ィル]メタンスルホンアミドと、 3-クロ口- 2-メチルベンゼンスルホユルクロリ ド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、製造方 法 B又は Cに従 、表題ィ匕合物を得た。得られた化合物の構造及び NMRデータを表 1 に示す。
[0121] (3)化合物(32— 3)及び(32— 4)の合成
化合物(32— 1)及び(32— 2)を用い、実施例 19 (4)と同様の方法に従 、表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0122] 実施例 33 化合物(33— 1)の合成
( 1) 4- { [(4-メチルビペラジン- 1-ィル)スルホ -ル]メチル } -1 ,3-チアゾール -2-ァミン の合成
実施例 19 (3)と同様の方法で 1-メチル -4- (メチルスルホニル)ピぺラジン(2. 55g) 、 n—ブチルリチウム(2. 59Mのへキサン溶液、 6. 63ml)、クロ口酢酸ェチル(1. 68 ml)およびチォゥレア(1. 20g)から、薄茶色粉末として表題化合物(3. 07g)を得た
1H NMR (200 MHz, DMSO-D6) δ 2.17 (s, 3 H), 2.23 - 2.36 (m, 4 H), 3.06 - 3.19 (m, 4 H), 4.21 (s, 2 H), 6.55 (s, 1 H), 7.00 (brs, 2 H).
(2)化合物(33— 1)の合成
実施例 33 (1)で得た 4-{[(4-メチルビペラジン- 1-ィル)スルホ -ル]メチル }-1,3-チ ァゾール- 2-ァミンと 3-クロ口- 2-メチルベンゼンスルホユルク口リドを用い、製造方法 B に従 ヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0123] 実施例 34 化合物(34— 1)及び(34— 2)の合成
(1) 2- (2-ァミノ- 1,3-チアゾール -4-ィル) -Ν,Ν-ジメチルプロパン- 2-スルホンアミドの 合成
実施例 19 (3)と同様の方法で Ν,Ν-ジメチルプロパン- 2-スルホンアミド(1. 35g)、 クロ口酢酸ェチル(1. 05ml)とチォゥレア(750mg)から、淡褐色粉末として表題ィ匕 合物(190mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 1.61 (s, 6 H), 2.64 (s, 6 H), 6.65 (s, 1 H), 6.99 (brs, 2 H).
(2)化合物(34— 1)及び(34— 2)の合成
実施例 34 (1)で得た 2- (2-ァミノ- 1 ,3-チアゾール -4-ィル) -Ν,Ν-ジメチルプロパン- 2-スルホンアミドと 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチ ル -1-ベンゾチォフェン- 2-スルホユルク口リドを用い、製造方法 Αに従 ヽ表題化合物 を得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 35 化合物(35— 1)の合成
(1) 1- (メチルスルホ -ル) -1H-インドールの合成
インドール(5. OOg)のテトラヒドロフラン溶液(33ml)に一 60°C下、 n—ブチルリチ ゥム(1. 58Mのへキサン溶液、 28. 4ml)を 10分間かけて滴下した。 0°Cまで昇温し
50分間攪拌した後、 60°Cへ冷却した。 60°Cにてメタンスルホユルクロリド(3. 6
3ml)を滴下し、徐々に室温まで昇温し 22時間攪拌した。反応液を飽和炭酸水素ナ トリウム水溶液(100ml)にあけ、クロ口ホルム(100ml)にて抽出した。有機層を飽和 食塩水(70ml)にて洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶 媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (展開溶媒 n —へキサン:酢酸ェチル = 10 : :L〜1 : Dで精製し無色油状物質として表題ィ匕合物
. 20g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 3.10 (s, 3 H), 6.70 - 6.74 (m, 1 H), 7.2 5 - 7.42 (m, 2 H), 7.45 (d, J=3.7 Hz, 1 H), 7.61 - 7.67 (m, 1 H), 7.92 (dd, J=8.2, 0. 9 Hz, 1 H).
(2) 4-[(lH-インドール- 1-ィルスルホ -ル)メチル ]-1,3-チアゾール -2-ァミンの合成 実施例 19 (3)と同様の方法で 1- (メチルスルホ -ル) -1H-インドール(1. 20g)、 n—
ブチルリチウム(2. 59Mのへキサン溶液、 2. 85ml)、クロ口酢酸ェチル(0. 72ml) およびチォゥレア(515mg)から、無色粉末として表題化合物(240mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 4.70 (s, 2 H), 6.19 (s, 1 H), 6.75 (dd, J=3.7, 0.9
Hz, 1 H), 6.93 (brs, 2 H), 7.22 - 7.34 (m, 2 H), 7.36 (d, J=3.7 Hz, 1 H), 7.62 - 7.6 6 (m, 1 H), 7.70 - 7.75 (m, 1 H).
(3)化合物(35— 1)の合成
実施例 35 (2)で得た 4-[(1Η-インドール- 1-ィルスルホ -ル)メチル ]-1,3-チアゾー ル- 2-ァミンと 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホ-ルクロリドを用い、 製造方法 Bに従 ヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1 に示す。
実施例 36 化合物(36— 1)〜(36— 3)の合成
( 1 ) 3-クロロ- 2-メチル -N- { 4- [( { [5- (トリフルォロメチル)ピリジン- 2-ィル]ァミノ }スルホ -ル)メチル ]-1,3-チアゾール -2-ィル }ベンゼンスルホンアミド(36— 1)の合成 実施例 30 (4)で得た N- {4- [(アミノスルホ -ル)メチル ]-1,3-チアゾール -2-ィル }-3- クロ口- 2-メチルベンゼンスルホンアミド(150mg)と 2-クロ口- 5- (トリフルォロメチル)ピ リジン(86mg)のジメチルスルホキシド溶液(1. 5ml)に炭酸カリウム(120mg)をカロえ 、マイクロウェーブ反応装置を用いて 140°Cにて 15分間、さらに 180°Cにて 15分間 反応を行なった。固形物をろ過して除去し、溶媒を減圧留去した。酢酸ェチル (30m 1)を加え、飽和炭酸水素ナトリウム水溶液(20ml)、飽和食塩水(20ml)にて洗浄し た。有機層を無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し 、得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノ ール = 30: 1〜: LO: 1)で精製し淡褐色粉末として表題化合物(55mg)を得た。 NMR データは表 1に記載した。
(2) 3-クロ口- 2-メチル -N- [4- ({[(4-メチルフエ-ル)ァミノ]スルホ-ル }メチル )-1,3-チ ァゾール _2_ィル]ベンゼンスルホンアミド(36— 2)の合成
実施例 30 (4)で得た N- {4- [(アミノスルホ -ル)メチル ]-1,3-チアゾール -2-ィル }-3- クロ口- 2-メチルベンゼンスルホンアミド(150mg)と 4-メチルフエ-ルボロン酸(64mg )のクロ口ホルム溶液(8ml)にトリェチルァミン、 4 Aモレキュラーシーブス、酢酸銅(Π
)を加え、室温にて 4時間酸素を吹き込んだ。反応液をセライトろ過し、水(15ml)を 加えてクロ口ホルム(20ml)にて抽出した。有機層を飽和食塩水(15ml)にて洗浄し 、無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去し、得られた 残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 クロ口ホルム:メタノール = 20: 1〜5: 1、 n-へキサン:酢酸ェチル = 2 : 1〜1: 1)で精製し無色粉末として表題ィ匕合 物(5mg)を得た。 NMRデータは表 1に記載した。
(3) 3-クロ口- 2-メチル -Ν-{4-[(1Η-ピロール- 1-ィルスルホ -ル)メチル ]-1,3-チアゾ ール -2-ィル }ベンゼンスルホンアミド(36— 3)の合成
実施例 30 (4)で得た N- {4- [(アミノスルホ -ル)メチル ]-1,3-チアゾール -2-ィル }-3- クロ口- 2-メチルベンゼンスルホンアミド(150mg)と 2,5-ジメトキシテトラヒドロフラン(6 2mg)を酢酸 (4ml)へ溶解し、 4時間加熱還流した。反応液を氷水(10ml)にあけ、 1 M水酸ィ匕ナトリウム水溶液(3ml)にて中和し、酢酸ェチル(20ml)にて抽出した有機 層を飽和食塩水(15ml)にて洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤を濾 去して溶媒を減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (展開 溶媒 !1-へキサン:酢酸ェチル= 1 : 1〜2 : 3、又はクロ口ホルム:メタノール = 10 : 1) にて精製を繰り返し行い、無色粉末として表題ィ匕合物(22mg)を得た。 NMRデータ は表 1に己載した。
実施例 37 化合物(37— 1)及び(37— 2)の合成
( 1) 4- [(フエ-ルスルホ -ル)メチル ]-1 ,3-チアゾール -2-ァミンの合成
実施例 19 (3)と同様の方法でメチルフエ-ルスルホン(2. 50g)、 n—ブチルリチウ ム(2. 59Mのへキサン溶液、 7. 40ml)、クロ口酢酸ェチル(1. 88ml)およびチォゥ レア(1. 34g)から、褐色粉末として表題化合物(1. 10g)を得た。
1H NMR (300 MHz, DMSO— D6) δ 4.45 (s, 2 H), 6.31 (s, 1 H), 6.92 (brs, 2 H), 7.5 6 - 7.64 (m, 2 H), 7.68 - 7.80 (m, 3 H).
(2)化合物(37— 1)及び(37— 2)の合成
実施例 37 (1)で得た 4- [(フエ-ルスルホ -ル)メチル ]-1,3-チアゾール -2-ァミンと、 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチオフ ェン -2-スルホユルク口リドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られたィ匕
- s - /— 、 ^- ε' ΐ- [ ^ ( / - / - ベ f )]- (z) emW
•(Η ΐ 'ω) 8·8 - 6Γ8 '(Η ΐ 'ω) 3Γ8 - 90·8 '(Η ΐ 'ω) 96"Ζ - 68"Ζ '(Η ΐ 'ω) 8ΓΖ - ΐ
Ζ·Ζ '(Η Ζ 's-iq) ΐ6·9 '(Η ΐ 's) Γ9 '(Η Ζ 's) 09·, 9 (9。- 'ζ 00S) Η匪 Ηΐ
。 (Su¾68)呦 挲ェつ; ^ ¾^(¾2 ·ΐ)
•^iQTfi (V^SL -\)^^ ^^WM^O ' L、继缀ベ ^ O)W6S ·Ζ)マ ^
ベ ^ - s - /— 、 ^- ε' ΐ- [ ^ ( - / - s -ベ;^ ri )] - (z)
•(H ΐ 'ω) 8Γ8 - ZL'S '(Η ΐ 'ω) ΐ·8 - 80·8 '(Η ΐ 'ω) εθ·8 - f
6"Ζ' (Η ΐ 'ω) WL― WL '(Η S 's) ^Ζ'Ζ 9 (α - Ο ΟΗΟΊΗ 'ζ 00S) Η匪 Ηΐ
iszio
ベ ^ - s - /— 、 ^- ε'ΐ- [ ^ ( - -ェ s)]- (ΐ) 8ε ¾ϊ第
•(H ΐ 'ΖΗ VI '0"S=f 'ΡΡ) 90·8 '(Η ΐ '^Η ·ΐ '8"S=f 'ΡΡ) S9"Z '(Η ΐ '^Η 8·ε '0"S=f 'ΡΡ) S
Z'L '(Η Ζ 's-iq) 96·9 '(Η ΐ 8S"9 '(Η Ζ 's) Z ' 9 (9。- OSWd 'z 00S) 匪 HI
°-Μ (?9ζ -z) ^m ^^w^ ^ ¾^(§6s
'z)マ 4· ベ ^ - s - /— 、 ^- ε'ΐ- [ ^ ( - -ェ s)]-
8Sfi ¾?第 [Ζ2Ϊ0] 。 · コ 挲 ¾ ^^ Ν、 ¾ ^癬 Ο)呦^
6098T0/S00Zdf/X3d 6Z Z99TS0/900Z OAV
ミンと 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾ チォフェン- 2-スルホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得ら れた化合物の構造及び NMRデータを表 1に示す。
[0129] 実施例 40 化合物(40— 1)及び (40— 41)の合成
( 1) tert-ブチルメチルスルホンの合成
実施例 16 (1)と同様の方法で tert-ブチルメチルスルフイド(3. 00g)、 3-クロ口過安 息香酸(19. 9g)から、無色粉末として表題化合物(3. 78g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.43 (s, 9 H), 2.82 (s, 3 H).
(2) 4-[(tert-ブチルスルホ -ル)メチル ]- 1 ,3-チアゾール - 2-ァミンの合成
実施例 19 (3)と同様の方法で tert-ブチルメチルスルホン(3. 78g)、 n—ブチルリチ ゥム(2. 59Mのへキサン溶液、 12. 9ml)、クロ口酢酸ェチル(3. 26ml)およびチォ ゥレア(2. 33g)から、淡褐色粉末として表題化合物(3. 99g)を得た。
1H NMR (300 MHz, DMSO-D6) δ 1.31 (s, 9 H), 4.24 (s, 2 H), 6.56 (s, 1 H), 6.99 (brs, 2 H).
(3)化合物(40— 1)及び (40— 41)の合成
実施例 40 (2)で得た 4-[(tert-ブチルスルホ -ル)メチル ]- 1 ,3-チアゾール - 2-ァミン と、 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチ ォフェン- 2-スルホユルク口リドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られ た化合物の構造及び NMRデータを表 1に示す。
[0130] 実施例 41 化合物 (41 1)及び (41 2)の合成
(1) 4,4-ジメチル- 2- [5- (メチルスルホ-ル)- 2-チェ-ル]- 4,5-ジヒドロ- 1 ,3-ォキサゾ ールの合成
5- (メチルスルホ -ル)チォフェン- 2-カルボン酸(1. 19g)のクロ口ホルム溶液(96m 1)に塩ィ匕チォニル (0. 63ml)を加え、 1時間加熱還流した。テトラヒドロフラン(10ml) と塩化チォ -ル(1. 26ml)を加え、 1時間加熱還流した。 Ν,Ν-ジメチルホルムアミド を 1滴加え、さらに 7時間加熱還流した。溶媒を減圧留去し、クロ口ホルム(23ml)を カロえトリェチルァミン(0. 97ml)、 2-ァミノ- 2-メチル -1-プロパノール(617mg)をカロえ 、室温にて 21時間攪拌した。反応液に飽和塩ィ匕アンモ-ゥム水溶液(50ml)をカロえ
、クロ口ホルム (50ml)にて抽出した。有機層を水(50ml)、飽和食塩水(50ml)にて 洗浄し、無水硫酸マグネシウムで乾燥後、乾燥剤を濾去して溶媒を減圧留去した。 得られた残渣にクロ口ホルム(96ml)をカ卩え、 0°C下、塩化チォ -ル(1. 68ml)を滴 下し、室温にて 3. 5時間攪拌した。反応液を減圧留去し、クロ口ホルム(100ml)、飽 和炭酸水素ナトリウム水溶液(20ml)、水(20ml)および 2M水酸ィ匕ナトリウム水溶液 (20ml)を加え、 30分間攪拌した。クロ口ホルム(50ml)にて抽出し、飽和食塩水(50 ml)にて洗浄して、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去した。得られた 残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 n-へキサン:酢酸ェチル = 2 : 1〜1: 3)で精製し無色粉末として表題ィ匕合物(940mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.39 (s, 6 H), 3.19 (s, 3 H), 4.14 (s, 2 H), 7.57 (d, J=3.8 Hz, 1 H), 7.65 (d, J=3.8 Hz, 1 H).
(2) 4- ({[5- (4,4-ジメチル- 4,5-ジヒドロ- 1,3-ォキサゾール -2-ィル) -2-チェ-ル]スル ホ-ル }メチル )-1,3-チアゾール -2-ァミンの合成
実施例 19 (3)と同様の方法で 4,4-ジメチル -2-[5- (メチルスルホ-ル)- 2-チェ-ル] -4,5-ジヒドロ- 1,3-ォキサゾール(940mg)、 n ブチルリチウム(2. 59Mのへキサン 溶液、 1. 68ml)、クロ口酢酸ェチル(0. 42ml)およびチォゥレア(303mg)から、無 色粉末として表題化合物(348mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.29 (s, 6 H), 4.16 (s, 2 H), 4.61 (s, 2 H), 6.44 (s, 1 H) 6.97 (brs, 2 H), 7.58 - 7.63 (m, 2 H).
(3)化合物 (41 1)の合成
実施例 41 (2)で得た 4-({[5- (4,4-ジメチル- 4,5-ジヒドロ- 1,3-ォキサゾール -2-ィル )-2-チェ-ル]スルホ-ル }メチル )-1,3-チアゾール -2-ァミンと 5-クロ口- 3-メチル -1- ベンゾチォフェン- 2-スルホユルク口リドを用い、製造方法 Bに従 、表題化合物を得た 。得られた化合物の構造及び NMRデータを表 1に示す。
(4) 5- {[(2- {[(5-クロ口- 3-メチル -1-ベンゾチェン- 2-ィル)スルホ -ル]アミノ}- 1,3-チ ァゾール -4-ィル)メチル]スルホ-ル }-N-(2-ヒドロキシ- 1,1-ジメチルェチル)チォフエ ン- 2-カルボキサミド (41 2)の合成
5-クロ口- N- [4- ({[5- (4,4-ジメチル- 4,5-ジヒドロ- 1,3-ォキサゾール -2-ィル) -2-チ
ェニル]スルホ二ル}メチル )-1,3-チアゾール -2-ィル] -3-メチル -1-ベンゾチォフェン -2-スルホンアミド(140mg)の 4. 5M塩酸水溶液(11ml)を 4時間加熱還流した。反 応液をろ過して水で洗浄した。得られた固体を乾燥し、無色粉末として表題化合物( 132mg)を得た。 NMRデータは表 1に記載した。
[0132] 実施例 42 化合物 (42— 1)の合成
(1) 4,4-ジメチル -2-[2- (メチルスルホ -ル)フエ-ル]- 4,5-ジヒドロ- 1 ,3-ォキサゾール の合成
実施例 41 (1)と同様の方法で 2-メチルスルホ -ルベンゾイツクアシッド(1. 25g)、 塩化チォ -ル(0. 68ml)、 2-ァミノ- 2-メチル -1-プロパノール(667mg)および塩化 チォニル(1. 82ml)から、無色粉末として表題ィ匕合物(1. 10g)を得た。
IH NMR (300 MHz, CHLOROFORM— D) δ 1.43 (s, 6 H), 3.37 (s, 3 H), 4.19 (s, 2 H), 7.58 - 7.75 (m, 3 H), 8.09 - 8.16 (m, 1 H).
(2) 4- ({[2- (4,4-ジメチル- 4,5-ジヒドロ- 1,3-ォキサゾール -2-ィル)フエ-ル]スルホ- ル}メチル )-1,3-チアゾール -2-ァミンの合成
実施例 19 (3)と同様の方法で 4,4-ジメチル -2-[2- (メチルスルホ -ル)フエ-ル] -4,5 -ジヒドロ- 1,3-ォキサゾール(1. 10g)、 n—ブチルリチウム(2. 59Mのへキサン溶液 、 2. 01ml)、クロ口酢酸ェチル(0. 51ml)およびチォゥレア(363mg)から、褐色油 状物質として表題ィ匕合物(130mg)を得た。
IH NMR (300 MHz, DMSO-D6) δ 1.35 (s, 6 H), 4.14 (s, 2 H), 4.81 (s, 2 H), 6.36 (s, 1 H), 6.93 (brs, 2 H), 7.64 - 7.82 (m, 4 H).
(3)化合物 (42— 1)の合成
実施例 42 (2)で得た 4-({ [2- (4,4-ジメチル- 4,5-ジヒドロ- 1 ,3-ォキサゾール -2-ィル )フエ-ル]スルホ-ル }メチル )-1,3-チアゾール -2-ァミンと 5-クロ口- 3-メチル -1-ベン ゾチォフェン- 2-スルホユルク口リドを用い、製造方法 Bに従い表題ィ匕合物を得た。得 られた化合物の構造及び NMRデータを表 1に示す。
[0133] 実施例 43 化合物 (43— 1)の合成
(1) 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -Ν,Ν-ジェチルメタンスルホンアミドの合成 実施例 19 (3)と同様の方法で Ν,Ν-ジェチルメタンスルホンアミド(1. 053g)、クロ口
ァセチルクロリド (0. 895g)とチォゥレア (0. 580g)から、淡褐色粉末として表題化合 物(0. 115g)を得た。
1H NMR (200 MHz, DMSO— D6) δ 1.06 (t, J=7.0 Hz, 6 H), 3.09 (q, J=7.0 Hz, 4 H) , 4.17 (s, 2 H), 6.52 (s, 1 H), 6.96 (brs, 2 H).
(2)化合物 (43— 1)の合成
実施例 43 (1)で得た 1- (2-ァミノ- 1,3-チアゾール -4-ィル) -Ν,Ν-ジェチルメタンス ルホンアミドと 3-クロ口- 2-メチルベンゼンスルホ-ルクロリドを用い、製造方法 Εに従 Vヽ表題化合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
[0134] 実施例 44 化合物(44 1)及び (44 2)の合成
(1) 4- [(ピロリジン- 1-ィルスルホ -ル)メチル ]-1,3-チアゾール -2-ァミンの合成 実施例 19 (3)と同様の方法でト (メチルスルホニル)ピロリジン(1. 00g)、 η—ブチ ルリチウム(2. 59Μのへキサン溶液、 2. 60ml)、クロ口酢酸ェチル(0. 79ml)および チォゥレア(0. 62g)から、無色粉末として表題ィ匕合物(0. 37g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.82 - 1.93 (m, 4 H), 3.21 - 3.31 (m, 4 H), 4.23 (s, 2 H), 5.02 (brs, 2 H), 6.61 (s, 1 H).
(2)化合物(44 1)及び (44 2)の合成
実施例 44 (1)で得た 4- [(ピロリジン- 1-ィルスルホ -ル)メチル ]-1 ,3-チアゾール -2- ァミンと、 3-クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベ ンゾチォフェン- 2-スルホユルク口リドを用い、製造方法 Aに従 ヽ表題化合物を得た。 得られた化合物の構造及び NMRデータを表 1に示す。
[0135] 実施例 45 化合物(45— 1)〜(45— 83)の合成
実施例 19 (5)で得た 1-(2-ァミノ- 1,3-チアゾール -4-ィル) -N-メチルメタンスルホン アミド(30 μ mol)のクロ口ホルム(700 μ 1)溶液に、 4 ジメチルァミノピリジン(30 μ mol)、ピリジン(120 μ mol)をカ卩えた後、それぞれ対応するスルホユルクロリド(120 mol)を加え、 50°Cでー晚攪拌した。反応液に酢酸ェチルとテトラヒドロフランの 1 : 1混合溶媒を加え、 1M塩酸水溶液で洗浄し、有機層を減圧で濃縮した。得られた残 渣を TLCプレート (メルク社製シリカゲル 60F254)を用いて精製し、 目的物を得た。 構造式及び質量分析データを表 2に記載した。
実施例 46 化合物(46— 1)〜(46— 88)の合成
実施例 20 (3)で得た 1-(2-ァミノ- 1,3-チアゾール -4-ィル) -N-ェチルメタンスルホン アミド(30 μ mol)のクロ口ホルム(700 μ 1)溶液に、 4-ジメチルァミノピリジン(30 μ m ol)、ピリジン(120 /z mol)をカ卩えた後、それぞれ対応するスルホユルクロリド(120 mol)を加え、 50°Cでー晚攪拌した。反応液に酢酸ェチルとテトラヒドロフランの 1 : 1 混合溶媒を加え、 1M塩酸水溶液で洗浄し、有機層を減圧で濃縮した。得られた残 渣を TLCプレート (メルク社製シリカゲル 60F254)を用いて精製し、 目的物を得た。 構造式及び質量分析データを表 2に記載した。
実施例 47 化合物(47— 1)〜(47— 4)の合成
(1) 3-メチル -2- ({ [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ィル]ァミノ }スルホ -ル) -1-ベンゾチォフェン- 7-カルボン酸(47—1)の合成
化合物番号 1 13、即ちメチル 3-メチル -2-({ [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ィル]アミノ}スルホ -ル) -1-ベンゾチォフェン- 7-カルボキシレート( 362mg)のメタノール (4ml)懸濁液に 5M水酸化ナトリウム水溶液(0. 4ml)を加え、 80°Cで 6時間攪拌した。反応液を減圧下濃縮して得られた溶液に、 6M塩酸水溶液 (0. 4ml)を加えると固体が析出した。水を加えて攪拌した後、吸引濾取して、無色 粉末として表題ィ匕合物(33 lmg)を得た。 NMRデータは、表 1に記載した。
(2) 3-メチル -2- ({[4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル]ァミノ }スルホ -ル) -1-ベンゾチォフェン- 7-カルボキサミド(47— 2)の合成
実施例 47 (1)で得られた 3-メチル -2- ({ [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チア ゾール -2-ィル]アミノ}スルホ -ル) -1-ベンゾチォフェン- 7-カルボン酸(47— 1) (28 lmg)の N, N—ジメチルホルムアミド溶液(2ml)に 1-ヒドロキシベンゾトリアゾール 1 水和物(98mg)、 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩酸塩(147 mg)及び 28%アンモニア水(58 μ 1)を加え、室温でー晚攪拌した。反応液に水(10 ml)をカ卩えて析出した結晶を濾取し、固体を得た。これをエタノール(5ml)に懸濁し、 濾取し、うすいピンク色の固体として表題ィ匕合物(192mg)を得た。 NMRデータは、 表上に己載し 7こ。
(3) 3-メチル -2- ({[4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル]ァミノ
}スルホ -ル) -ベンゾチォフェン- 5-カルボン酸(47— 3)の合成 実施例 47 (1)と同様の方法で、化合物番号(1— 14)メチル 3-メチル -2-({[4- (テ トラヒドロ- 2H-ピラン- 4-ィル) -1,3-チアゾール -2-ィル]アミノ}スルホニル) -1-ベンゾ チォフェン- 5-カルボキシレート(1. 04g)と 5M水酸化ナトリウム水溶液(1. 15ml)か ら、無色粉末として表題ィ匕合物(962mg)を得た。得られた化合物の構造及び NMR データを表 1に示す。
(4) 3-メチル -2- ({ [4- (テトラヒドロ- 2H-ピラン- 4-ィル) -1 ,3-チアゾール -2-ィル]ァミノ
}スルホ -ル) -ベンゾチォフェン- 5-カルボキサミド(47— 4)の合成
実施例 47 (2)と同様の方法で、化合物(47— 3) (300mg)と 1-ヒドロキシベンゾトリア ゾール 1水和物(126mg)、 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩 酸塩(197mg)及び 28%アンモニア水(210 1)から、無色粉末として表題ィ匕合物(
62mg)を得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 48 化合物 (48— 1)及び (48— 2)の合成
(1)ェチル 3 ヒドロキシー2 (ヒドロキシメチル) 2 メチルプロパノエートの合成 2, 2 ビス(ヒドロキシメチル) プロピオン酸(5. 00g)のエタノール溶液(110ml)に 硫酸 (4. 57ml)を滴下して 5時間還流した。室温に戻した後、反応液に 5M水酸ィ匕 ナトリウム水溶液を滴下して弱酸性とした。溶媒を減圧留去した後に、残渣にクロロホ ルムを加えて水層を分液して除き、有機層をセライトを通じて濾過した。濾液を減圧 下濃縮し、無色油状物質として表題化合物 (3. 22g)を得た。
1H NMR (300 MHz, CHLOROFORM- D) δ 1.06 (s, 3 Η), 1.30 (t, J=7.2 Hz, 3 H), 2.91 (brs, 2 H), 3.72 (d, J=11.3 Hz, 2 H), 3.91 (d, J=11.3 Hz, 2 H), 4.23 (q, J=7.2 H z, 2 H).
(2)ェチル 5—メチルー 1, 3 ジォキサン 5 カルボキシレートの合成
ェチル 3 ヒドロキシ一 2— (ヒドロキシメチル) 2—メチルプロパノエート(200mg)と 2, 6—ルチジン(325 1)をジメトキシメタン(9. 7ml)に溶解し、氷冷下でトリメチル シリルトリフルォロメタンスルホネート(915 1)を滴下した。氷冷下で 1. 5時間攪拌後 、反応液に飽和炭酸水素ナトリウム水溶液(20ml)を滴下して中和した。発泡が止ん だ後、酢酸ェチル (40ml)で抽出し、有機層を 10%硫酸水素カリウム水溶液(20ml
X 2)、飽和炭酸水素ナトリウム水溶液 (20ml)、飽和食塩水(50ml)で順次洗浄した 後、有機層を無水硫酸マグネシウムで乾燥した。乾燥剤を濾去し、溶媒を減圧留去 して得られた残渣をシリカゲルカラムクロマトグラフィー(展開溶媒 へキサン:酢酸ェ チル =4: 1)で精製し、無色油状物質として表題ィ匕合物(115mg)を得た。
IH NMR (300 MHz, CHLOROFORM— D) δ 1.15 (s, 3 H), 1.29 (t, J=7.2 Hz, 3 H), 3 .56 (d, J=11.5 Hz, 2 H), 4.17 - 4.32 (m, 4 H), 4.75 (d, J=6.1 Hz, 1 H), 4.88 (d, J=6. 1 Hz, 1 H).
( 3) 1—( 5 メチル—1, 3 ジォキサン 5 ィル)エタノンの合成
参考例 1一(1)と同様の方法でェチル 5—メチルー 1, 3 ジォキサン 5 カルボキ シレート(3. 00g)、 Ν,Ο-ジメチルヒドロキシルァミン塩酸塩(2. 18g)、 3Mメチルマグ ネシゥムブロマイドのジェチルエーテル溶液 (23ml)から無色油状物質として表題ィ匕 合物(1. 61g)を得た。
IH NMR (300 MHz, CHLOROFORM— D) δ 0.99 (s, 3 H), 2.29 (s, 3 H), 3.54 (d, J=l 1.7 Hz, 2 H), 4.32 (d, J=11.7 Hz, 2 H), 4.72 (d, J=6.1 Hz, 1 H), 4.92 (d, J=6.1 Hz, I H).
(4) 4— (5—メチル—1, 3—ジォキサン—5—ィル)—丄, 3—チアゾール—2—ァミン 臭化水素酸塩の合成
参考例 1一( 2)と同様の方法で 1一( 5—メチルー 1 , 3 ジォキサン 5 ィル)ェタノ ン(79 lmg)、臭素(281 1)、チォゥレア(334mg)から薄黄色粘性油状物質として 表題化合物(747mg)を得た。
IH NMR (300 MHz, DMSO-D6) δ 1.13 (s, 3 H), 3.71 (d, J=11.7 Hz, 2 H), 4.06 (d, J=11.7 Hz, 2 H), 4.75 (d, J=6.0 Hz, 1 H), 4.94 (d, J=6.0 Hz, 1 H), 6.69 (s, 1 H), 8. 68 - 9.17 (m, 3 H).
(5) 5 クロ口一 3—メチル N— [4— (5—メチル 1, 3 ジォキサン一 5—ィル) 1, 3 チアゾーノレ - 2 ィル 1 ベンゾチォフェン一 2 スノレホンアミド(48— 1) の合成
実施例 48 (4)で得られた 4— (5—メチル—1, 3 ジォキサン— 5—ィル)—1, 3— チアゾールー 2 アミン臭化水素酸塩(383mg)と 5-クロ口- 3-メチルベンゾ [B]チオフ
ェン -2-スルホ-ルクロライド(766mg)から Method Bの方法を用いて無色粉末として 表題化合物(89mg)を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(6) 4— (5—メチル—1, 3 ジォキサン— 5—ィル)—1, 3 チアゾール—2 ァミン の合成
1 (5—メチルー 1, 3 ジォキサンー5 ィル)エタノン(800mg)のメタノール溶液( 8ml)に炭酸水素ナトリウム(933mg)をカ卩えて懸濁させ、臭素(284 μ 1)を滴下した 後、 1時間還流した。続いて反応液にチォゥレア(338mg)を加え、室温で 1晚攪拌し た。溶媒を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー (展開 溶媒 展開溶媒 クロ口ホルム:メタノール: 28%アンモニア水溶液 = 30 : 1 : 0.1〜25 : 1 : 0.1〜20: 1 : 0.1)で精製し、得られた粗結晶を IPEで洗浄した。薄黄色粉末とし て表題化合物(205mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 1.25 (s, 3 H), 3.66 (d, J=11.0 Hz, 2 H), 3.96 (d, J=11.0 Hz, 2 H), 4.66 (d, J=6.0 Hz, 1 H), 4.87 (d, J=6.0 Hz, 1 H), 6.27 (s, 1 H), 6. 89 (s, 2 H).
(7) 3 クロ口一 2—メチル N [4— (5—メチル 1, 3 ジォキサン一 5—ィル) 1, 3 チアゾール 2 ィル]ベンゼンスルホンアミド(48— 2)の合成
実施例口で得られた 4— (5—メチル 1, 3 ジォキサン一 5—ィル) 1, 3 チアゾ 一ルー 2 ァミン(205mg)と 3-クロ口- 2-メチルベンゼン- 1-スルホ-ルクロライド(46 lmg)力も製造方法 Aの方法を用いて無色粉末として表題ィ匕合物(339mg)を得た。 得られた化合物の構造及び NMRデータを表 1に示す。
実施例 49 5 クロ口一 N— (4— {4— [ (2 ヒドロキシエトキシ)メチノレ]テトラヒドロー 2 H—ピラン 4 ィル } 1 , 3 チアゾール 2 ィル) 3 メチル 1 ベンゾチ オフヱン 2—スルホンアミド(49— 1)の合成
実施例 3 (10)で得られた 5-クロ口- N- {4- [4- (ヒドロキシメチル)テトラヒドロ- 2H-ピラン -4-ィル] -1,3-チアゾール -2-ィル }-3-メチル -1-ベンゾチォフェン- 2-スルホンアミド( 200mg)とピリジン(141 μ 1)のクロ口ホルム溶液(2ml)に氷冷下でメタンスルホ-ル クロリド(933mg)を加えて、懸濁させたまま、氷冷下で 30分間攪拌した後、室温に戻 して 20分間攪拌した。懸濁液にトリェチルァミン(243 1)を加えて、氷冷下で 30分
間攪拌した後、室温に戻して 3時間攪拌した。氷冷下で 10%硫酸水素カリウム水溶 液(10ml)をカ卩え、クロ口ホルム(20ml)で抽出した。有機層を 10%硫酸水素カリウム 水溶液、飽和食塩水で順次洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤 を濾去し、溶媒を減圧留去した。得られた残渣にエチレングリコール(10ml)とフツイ匕 カリウム(203mg)を加え、 100°Cで 12時間攪拌した。室温に戻して 1晚攪拌した後、 110°Cに昇温して 1時間攪拌した。フッ化カリウム(203mg)をカ卩え、 100°Cで 5時間 、 120°Cで 5時間攪拌した。室温に戻して 3日間攪拌した後、更に 120°Cで 12時間 攪拌した。反応液に水(20ml)を加え、クロ口ホルム(50ml)で抽出した。有機層を水 で 3回、飽和食塩水で順次洗浄した後、無水硫酸マグネシウムで乾燥した。乾燥剤を 濾去し、溶媒を減圧留去して得られた残渣をプレパラティブ TLC (展開溶媒 クロロホ ルム:メタノール = 30 : 1 3回展開)で精製し、無色粉末として表題ィ匕合物(58mg)を 得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 50 化合物(50— 1)の合成
(1)ジェチル 1,3-ジォキサン- 5,5-ジカルボキシレートの合成
パラホルムアルデヒド(4. 44g)のトルエン(600ml)溶液にジェチルビス(ヒドロキシメ チル)マロネート(10. 0g)、 p -トルエンスルホン酸(96mg)をカ卩え、 90°Cで 7時間攪 拌した後、 P-トルエンスルホン酸 (400mg)を追加し、 90°Cで 2時間攪拌した。さら〖こ p -トルエンスルホン酸 (400mg)を追力!]、室温でー晚攪拌を行った後、 Dean- Stark装 置を取り付け 130°Cで 40分反応を行った。反応液をろ過後、溶媒を減圧留去し、得ら れた残渣をシリカゲルクロマトグラフィー(展開溶媒 n-へキサン:酢酸ェチル = 10 : 1 )で精製し無色として油状物質として表題ィ匕合物(10. 41g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.28 (t, J=7.3 Hz, 6 H), 3.87 - 4.46 (m , 8 H), 4.83 (s, 2 H).
(2) N-メトキシ- N-メチル -1,3-ジォキサン- 5-カルボキサミドの合成
水酸化カリウム(6. 76g)のエタノール(200ml)溶液にジェチル 1,3-ジォキサン- 5,5 -ジカルボキシレート(6. 41g)をカ卩ぇ 1時間加熱還流を行った後、溶媒を減圧留去し 、クロ口ホルムを加えた。氷冷下、濃塩酸を加え溶媒を減圧留去した。得られた残渣 に氷冷下トリエチルァミン(100ml)をカ卩え、 1時間加熱還流を行った。室温に冷却後
、溶媒を減圧留去した。ここへテトラヒドロフラン(100ml)、 Ν,Ο-ジメチルヒドロキシル ァミン塩酸塩(3. 32g)、 1-ヒドロキシベンゾトリアゾール(4. 29g)、トリェチルァミン(4 . 7ml)をカ卩え、氷冷下 1-ェチル -3-(3-ジメチルァミノプロピル)カルボジイミド塩酸塩( 6. 52g)をカ卩ぇ室温で一晩攪拌した。反応終了、クロ口ホルムで希釈し、飽和食塩水 :水 =4 : 1で洗浄した。有機層を無水硫酸ナトリウムで乾燥後、乾燥剤を濾去して溶 媒を減圧留去し、得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 n-へキサ ン:酢酸ェチル = 5: 1)で精製し無色油状物質として表題ィ匕合物( 1. 78g)を得た。 1H NMR (300 MHz, CHLOROFORM— D) δ 3.16 (s, 3 H), 3.33 - 3.49 (m, 1 H), 3.7 5 (s, 3 H), 3.87 (t, J=11.3 Hz, 2 H), 4.21 (dd, J=11.3, 4.4 Hz, 2 H), 4.69 (d, J=6.1 Hz, 1 H), 5.03 (d, J=6.1 Hz, 1 H).
(3) 1-(1, 3-ジォキサン- 5-ィル)エタノンの合成
実施例 5 (2)と同様の方法で N-メトキシ -N-メチル -1,3-ジォキサン- 5-カルボキサミド (1. 78g)と 1Mメチルマグネシウムブロミドのテトラヒドロフラン溶液(20ml)から淡黄 色油状物質として表題ィ匕合物(630mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 2.22 (s, 3 H), 2.71 - 2.95 (m, 1 H), 3.9 5 (m, 2 H), 4.17 (dd, J=11.9, 4.1 Hz, 2 H), 4.71 (d, J=6.2 Hz, 1 H), 4.93 (d, J=6.2 Hz, 1 H).
(4) 4- (1 ,3-ジォキサン- 5-ィル) -1 ,3-チアゾール -2-ァミンの合成
参考例 1 (2)と同様の方法で 1-(1,3-ジォキサン- 5-ィル)エタノン(630mg)、臭素(2 20 μ 1)及びチォゥレア(368mg)力 得られた粗生成物を NHシリカゲルカラムクロマ トグラフィー (展開溶媒 n-へキサン:酢酸ェチル = 2 : 1)で精製し、黄色粉末として 表題化合物(362mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 2.89 - 3.08 (m, 1 H), 3.62 - 3.80 (m, 2 H), 4.09 (dd, J=10.8, 4.4 Hz, 2 H), 4.61 (d, J=6.1 Hz, 1 H), 4.94 (d, J=6.1 Hz, 1 H), 6.27 (s, 1 H), 6.91 (brs, 2 H).
(5) 3-クロ口- N- [4- (1,3-ジォキサン- 5-ィル) -1,3-チアゾール -2-ィル] -2-メチルベン ゼンスルホンアミド(50—1)の合成
実施例 50 (4)で得られた 4-(1, 3-ジォキサン- 5-ィル) -1,3-チアゾール -2-ァミンと、 3-
クロ口- 2-メチルベンゼンスルホユルク口リドを用い製造方法 Aに従 、表題ィ匕合物を得 た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 51 化合物(51— 1)〜(51— 5)の合成
(1) 4- [(メチルスルホ -ル)メチル ]-1 ,3-チアゾール -2-ァミンの合成
実施例 19 (3)と同様の方法でジメチルスルホン(3. OOg)、n—ブチルリチウム(2. 59 Mのへキサン溶液、 12. 3ml)、クロ口酢酸ェチル(3. 40ml)およびチォゥレア(2. 4 3g)から、無色粉末として表題化合物(1. 48g)を得た。
IH NMR (300 MHz, DMSO-D6) δ 3.01 (s, 3 H), 4.26 (s, 2 H), 6.58 (s, 1 H), 7.04 (brs, 2 H).
(2)化合物(51— 1)及び(51— 2)の合成
実施例 51 (1)で得た 4- [(メチルスルホ -ル)メチル ]-1,3-チアゾール -2-ァミンと、 3-ク ロロ- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン -2-スルホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られた化合 物の構造及び NMRデータを表 1に示す。
(3) 1, 1-ジメチルプロピルメチルスルホンの合成
実施例 16 (1)と同様の方法で 1,1-ジメチルプロピルメチルスルフイド(980mg)、 3- クロ口過安息香酸(5. 50g)から、無色油状物質として表題ィ匕合物(858mg)を得た。 IH NMR (300 MHz, CHLOROFORM— D) δ 1.02 (t, J=7.5 Hz, 3 H), 1.36 (s, 6 H), 1.83 (q, J=7.5 Hz, 2 H), 2.80 (brs, 3 H).
(4) 4- { [(1 , 1-ジメチルプロピル)スルホ -ル]メチル } -1 ,3-チアゾール -2-ァミン 実施例 19 (3)と同様の方法で 1,1-ジメチルプロピルメチルスルホン(858mg)、 n— ブチルリチウム(2. 71Mのへキサン溶液、 2. 53ml)、クロ口酢酸ェチル(0. 67ml) およびチォゥレア (440mg)から、淡褐色粉末として表題化合物(596mg)を得た。 IH NMR (300 MHz, DMSO— D6) δ 0.93 (t, J=7.5 Hz, 3 H), 1.28 (s, 6 H), 1.74 (q, J =7.5 Hz, 2 H), 2.65 (s, 3 H), 4.35 (s, 2 H), 6.86 (s, 1 H), 7.39 (t, J=8.0 Hz, 1 H), 7. 68 (dd, J=8.0, 1.1 Hz, 1 H), 7.90 (dd, J=8.0, 1.1 Hz, 1 H), 13.05 (brs, 1 H).
(5) (メチルスルホ -ル)シクロへキサンの合成
実施例 16 (1)と同様の方法でシクロへキシルメチルスルフイド(2. 5g)、 3-クロ口過
安息香酸(12. 70g)から、無色油状物質として表題化合物(2. 96g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.14—1.40 (m, 2 H), 1.44 - 1.60 (m, 3 H), 1.69 - 1.80 (m, 1 H), 1.91 - 2.01 (m, 2 H), 2.17 - 2.27 (m, 2 H), 2.76 - 2.93 ( m, 1 H), 2.82 (s, 3 H).
(6) 4- [(シクロへキシルスルホ -ル)メチル] -1,3-チアゾール -2-ァミン
実施例 19 (3)と同様の方法で (メチルスルホ -ル)シクロへキサン(1. 48g)、 n—ブ チルリチウム(2. 71Mのへキサン溶液、 3. 37ml)、クロ口酢酸ェチル(0. 972ml)お よびチォゥレア(729mg)から、淡黄色粉末として表題化合物 (400mg)を得た。 1H NMR (300 MHz, DMSO— D6) δ 1.11 - 1.45 (m, 5 H), 1.59 - 1.69 (m, 1 H), 1.76 - 1.86 (m, 2 H), 2.05 - 2.14 (m, 2 H), 3.05 - 3.19 (m, 1 H), 4.21 (s, 2 H), 6.57 (s, 1 H), 7.01 (brs, 2 H).
(7) 4- [(イソプロピルスルホ -ル)メチル] -1,3-チアゾール -2-ァミン
実施例 19 (3)と同様の方法でイソプロピルメチルスルホン(1. 50g)、 n—ブチルリ チウム(2. 71Mのへキサン溶液、 4. 53ml)、クロ口酢酸ェチル(1. 31ml)およびチ ォゥレア(981mg)から、無色粉末として表題化合物(1. 41g)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.26 (d, J=6.8 Hz, 6 H), 3.31 - 3.43 (m, 1 H), 4 .24 (s, 2 H), 6.58 (s, 1 H), 7.03 (brs, 2 H).
(8)化合物(51— 3)〜(51— 5)の合成
実施例 51 (4) (6) (7)で得た 4- {[(1,1-ジメチルプロピル)スルホ -ル]メチル }- 1,3-チ ァゾール- 2-ァミン、 4- [(シクロへキシルスルホ -ル)メチル] -1,3-チアゾール -2-アミ ン、 4- [(イソプロピルスルホ -ル)メチル] -1,3-チアゾール -2-ァミンのそれぞれと、 3- クロ口- 2-メチルベンゼンスルホユルク口リドを用い、製造方法 Bに従 、表題化合物を 得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 52 化合物(52— 1)〜(52— 3)の合成
(1) 1- (tert-ブチルチオ)- 3-クロ口アセトンの合成
ジクロロアセトン(5. 9 lg)のクロ口ホルム溶液(133ml)に 0°C下、 tert-ブチルメル力 プタン(3. OOg)トリェチルァミン(6. 95ml)の混合物を滴下し、 0°Cにて 2時間攪拌し た。反応液にクロ口ホルム(100ml)を加え、 0. 1N塩酸(200ml)、水(200ml)、飽
和食塩水(150ml)にて洗浄した。有機層を無水硫酸マグネシウムにて乾燥後、乾燥 剤を濾去して溶媒を減圧留去した。得られた残渣をシリカゲルクロマトグラフィー (展 開溶媒 n—へキサン:酢酸ェチル = 30 : 1〜20: 1)にて精製し、黄色油状物質とし て表題化合物( 1. 60g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.33 (s, 9 H), 3.50 (s, 2 H), 4.36 (s, 2 H).
(2) 4- [(tert-ブチルチオ)メチル ]-1,3-チアゾール -2-ァミンの合成
実施例 52 (1)で得た 1- (tert-ブチルチオ)- 3-クロ口アセトン(800mg)の 1,4-ジォ キサン溶液(9ml)にチォゥレア(370mg)をカ卩え、室温にてー晚攪拌した。反応液を 減圧留去し、得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 クロ口ホルム:メ タノール:アンモニア水 = 100 : 2 : 0. 2〜: L00 : 7 : 0. 7)にて精製し、淡黄色粉末とし て表題化合物 (450mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 1.28 (s, 9 H), 3.56 (s, 2 H), 6.32 (s, 1 H), 6.86 (brs, 2 H).
(3)化合物(52— 1)及び(52— 3)の合成
実施例 52 (2)で得た 4- [(tert-ブチルチオ)メチル ]-1,3-チアゾール -2-ァミンと、 3-ク ロロ- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン -2-スルホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られた化合 物の構造及び NMRデータを表 1に示す。
(4)化合物(52— 2)の合成
実施例 52 (3)で得たィ匕合物(52— 1) (120mg)のメタノール溶液(3ml)に 0°C下、 メタ過要素酸ナトリウム(79mg)水溶液 (0. 4ml)を加え、室温にて 5時間攪拌した。 反応液に飽和食塩水(15ml)をカ卩え、クロ口ホルム(30ml)にて抽出した。有機層を 飽和食塩水(15ml)にて洗浄し、無水硫酸マグネシウムにて乾燥後、乾燥剤を濾去 して溶媒を減圧留去した。得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 ク ロロホルム:メタノール = 50: 1〜20: 1)にて精製し、無色粉末として表題化合物(10 Omg)を得た。得られた化合物の構造及び NMRデータを表 1に示す。
実施例 53 化合物(53— 1)〜(53— 3)の合成
(1) 4-(tert-ブチルチオ)ブタン- 2-オンの合成
tert-ブチルチオール(4. 51g)とメチルビ-ルケトン(3. 85g)のエーテル溶液(10 Oml)にトリェチルァミン(7. 7ml)を加え、 21時間加熱還流した。反応液を減圧留去 し、得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 n—へキサン:酢酸ェチ ル= 20 : 1〜6 : 1)にて精製し、黄色油状物質として表題ィ匕合物(1. 70g)を得た。 1H NMR (300 MHz, CHLOROFORM— D) δ 1.33 (s, 9 H), 2.17 (s, 3 H), 2.64 - 2.8 0 (m, 4 H).
(2) 4-(tert-ブチルスルホ -ル)ブタン- 2-オンの合成
実施例 16 (1)と同様の方法で、実施例 53 (1)で得た 4-(tert -プチルチオ)ブタン- 2 -オン (425mg)、 3-クロ口過安息香酸(1. 41g)から、無色粉末として表題ィ匕合物(3 20mg)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.43 (s, 9 H), 2.26 (s, 3 H), 3.00 - 3.0 8 (m, 2 H), 3.18 - 3.26 (m, 2 H).
(3) 4-[2-(tert-ブチルスルホ -ル)ェチル ]- 1 ,3-チアゾール - 2-ァミンの合成 実施例 53 (2)で得た 4-(tert-ブチルスルホ -ル)ブタン- 2-オン(320mg)のメタノー ル溶液(17ml)にブロミン(94 1)を加え、徐々に昇温し 90°Cにて 20分間攪拌した。 室温まで戻し、チォゥレア(127mg)をカ卩えて、室温にて一晩攪拌した。反応液を減 圧留去し、シリカゲルカラムクロマトグラフィ(展開溶媒 クロ口ホルム:メタノール:アン モ-ァ水 = 100 : 3 : 0. 3〜: L00 : 7 : 0. 7)にて精製し、淡黄色粉末として表題化合物 (48mg)を得た。
1H NMR (600 MHz, DMSO-D6) δ 1.29 (s, 9 H), 2.75 - 2.80 (m, 2 H), 3.21 - 3.25 (m, 2 H), 6.31 (s, 1 H), 6.89 (brs, 2 H).
(4)化合物(53— 1)の合成
実施例 53 (3)で得た 4-[2-(tert-ブチルスルホ -ル)ェチル ]- 1 ,3-チアゾール - 2-アミ ンと、 3-クロ口- 2-メチルベンゼンスルホニルクロリドを用い、製造方法 Bに従い表題ィ匕 合物を得た。得られた化合物の構造及び NMRデータを表 1に示す。
(5) 4- (イソプロピルチォ)ブタン- 2-オンの合成
2-プロパンチオール(3. 81g)とメチルビ-ルケトン(3. 85g)のテトラヒドロフラン溶
液(75ml)にトリェチルァミン (0. 7ml)を加え、 3時間加熱還流した。反応液を減圧 留去し、得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 n—へキサン:酢酸 ェチル = 20: 1〜6: 1)にて精製し、淡黄色油状物質として表題化合物(2. 88g)を 得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.27 (d, J=6.6 Hz, 6 H), 2.17 (s, 3 H), 2.69 - 2.79 (m, 4 H), 2.85 - 3.01 (m, 1 H).
(6) 4- (イソプロピルスルホ -ル)ブタン- 2-オンの合成
実施例 16 (1)と同様の方法で、実施例 53 (5)で得た 4- (イソプロピルチォ)ブタン- 2 -オン(2. 88g)、3-クロ口過安息香酸(10. 5g)から、無色油状物質として表題ィ匕合 物(500mg)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.42 (d, J=7.0 Hz, 6 H), 2.26 (s, 3 H), 2.97 - 3.28 (m, 5 H).
(7) 4- [2- (イソプロピルスルホ -ル)ェチル ]-1 ,3-チアゾール -2-ァミンの合成 実施例 53 (3)と同様の方法で、実施例 53 (6)で得た 4- (イソプロピルスルホニル)ブ タン- 2-オン(500mg)、ブロミン(159 /z l)、チォゥレア(214mg)から、淡黄色粉末と して表題化合物( 112mg)を得た。
1H NMR (300 MHz, DMSO— D6) δ 1.24 (d, J=6.8 Hz, 6 H), 2.76 - 2.86 (m, 2 H), 3 .18 - 3.33 (m, 3 H), 6.31 (s, 1 H), 6.90 (brs, 2 H).
(8)化合物(53— 2)及び(53— 3)の合成
実施例 53 (7)で得た 4-[2- (イソプロピルスルホ -ル)ェチル ]-1 ,3-チアゾール -2-アミ ンと、 3-クロ口- 2-メチルベンゼンスルホユルクロリド又は 5-クロ口- 3-メチル -1-ベンゾ チォフェン- 2-スルホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得ら れた化合物の構造及び NMRデータを表 1に示す。
実施例 54 化合物(54— 1)〜(54— 4)の合成
(1) 3- (tert-ブチルスルホ-ル)- 1-クロ口- 1-フルォロアセトンの合成
実施例 40 (1)で得られた tert-ブチルメチルスルホン( 1. 55g)のテトラヒドロフラン(4
Oml)溶液にアルゴン雰囲気下、—70°Cで n—ブチルリチウム(2. 71Mのへキサン溶 液、 5. 00ml)を加え、 15分間攪拌した後、クロ口フルォロ酢酸ェチル(1. 4ml)をカロ
え、— 20°Cにて 2時間攪拌した。溶媒を減圧留去し、得られた残渣をシリカゲルクロ マトグラフィー(展開溶媒 n-へキサン:酢酸ェチル = 30 : 1)で精製し無色粉末として 表題化合物(1. 70g)を得た。
1H NMR (300 MHz, CHLOROFORM— D) δ 1.47 (s, 9 H), 4.31 - 4.37 (m, 2 H), 6.6 4 (d, J=50.0 Hz, 1 H).
(2) 4-[(tert-ブチルスルホ -ル)メチル ]-5-フルォロ- 1 ,3-チアゾール -2-ァミンの合成
3- (tert-ブチルスルホ-ル)- 1-クロ口- 1-フルォロアセトン(1. 51g)の N, N—ジメチ ノレホノレムアミド(25ml)溶液にチォゥレア(548mg)を加え、 30°Cにて 2日間攪拌した 。溶媒を減圧留去し、得られた残渣をシリカゲルクロマトグラフィー(展開溶媒 n-へ キサン:酢酸ェチル =4: 1、クロ口ホルム:メタノール: 28%アンモニア水 = 50: 1: 0.1 =)で精製した後、メタノールで再結晶を行い、無色粉末として表題ィ匕合物(249mg) を得た。
1H NMR (200 MHz, DMSO-D6) δ 1.34 (s, 9 H), 4.22 (d, J=1.8 Hz, 2 H), 6.93 (brs , 2 H).
( 3) N- { 4-[(tert-ブチルスルホ -ル)メチル ]- 5-フルォロ - 1 , 3-チアゾール -2-ィル } -3 -クロ口- 2-メチルベンゼンスルホンアミド(54—1)及び N-{4-[(tert-ブチルスルホ- ル)メチル ]-5-フルォロ- 1,3-チアゾール -2-ィル }-5-クロ口- 3-メチル -1-ベンゾチオフ ヱン -2-スルホンアミド(54— 2)の合成
4- [(tert-ブチルスルホ -ル)メチル ]-5-フルォロ- 1 ,3-チアゾール -2-ァミンと、 3-クロ 口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2-スルホニルクロリドを用い、製造方法 Bに従い表題ィ匕合物を得た。得られた化合物 の構造及び NMRデータを表 1に示す。
(4) 3- (tert-ブチルスルホ-ル)- 1,1-ジクロロアセトンの合成
実施例 54 (1)と同様の方法で tert-ブチルメチルスルホン(1. 40g)と n—ブチルリチ ゥム(2. 71Mのへキサン溶液、 4. 60ml)、ジクロ口酢酸ェチル(1. 77g)から無色粉 末として表題化合物( 1. 26g)を得た。
1H NMR (200 MHz, CHLOROFORM— D) δ 1.47 (s, 9 H), 4.41 (s, 2 H), 6.43 (s, 1 H).
(5) 4-[(tert-ブチルスルホ -ル)メチル ]-5-クロ口- 1 ,3-チアゾール -2-ァミン塩酸塩の 合成
3- (tert-ブチルスルホ-ル)- 1 , 1-ジクロロアセトン(770mg)のジォキサン( 15ml)溶 液にチォゥレア(261mg)を加え、 40°Cで 2日間攪拌した。反応液をろ過し、得られ た結晶をジォキサン、へキサンで順次洗浄、乾燥を行い、無色粉末として表題化合 物(750mg)を得た。
1H NMR (300 MHz, DMSO-D6) δ 1.36 (s, 9 H), 4.25 (s, 2 H).
(6) N-{4-[(tert-ブチルスルホ -ル)メチル ]- 5-クロ口- 1,3-チアゾール -2-ィル }-3-ク ロロ- 2-メチルベンゼンスルホンアミド(54— 3)及び N-{4-[(tert-ブチルスルホ -ル)メ チル] -5-クロ口- 1,3-チアゾール -2-ィル }- 5-クロ口- 3-メチル -1-ベンゾチォフェン- 2- スルホンアミド(54— 4)の合成
4- [(tert-ブチルスルホ -ル)メチル ]-5-クロ口- 1 ,3-チアゾール -2-ァミン塩酸塩と、 3- クロ口- 2-メチルベンゼンスルホニルクロリド又は 5-クロ口- 3-メチル -1-ベンゾチォフエ ン -2-スルホニルクロリドを用い製造方法 Bに従 、表題ィ匕合物を得た。得られた化合 物の構造及び NMRデータを表 1に示す。
表 1
[表 1-1]
/v:/ O 6098SS00ifcl£ SS0900ZAV Z6
κ〔〕ΐ∞εΐοι
6098T0/S00Zdf/X3d 66 Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 001· Z99TS0/900Z OAV
Sffi0142T
6098T0/S00Zdf/X3d εοΐ· Z99TS0/900Z OAV
/v:/ O 6098SS00ifcl£ SS0900ZAV0 _■
/v: O 6098SS00ifcl£looiAV
< < <
(9α〇の _Λ_α Η_Λ-〇oc一 ¾d_≥N H -- ) ε9ί (ェ ε)339S· -
993エ 3) ε602(Η Ζ -. - (Hs)ε寸 ε(Η)E s - -- -- as(Hs) 99ε寸- ^ "·
)卜 ε卜 (H H oz「E=- "..- 卜 vspp)∞9卜(HN= - - - 卜卜卜「PP)S6卜(HN = - - - -
(H ίq)寸卜 3L(Hs」 -. -
(s:〇の _Λ_αH_A- ooe) ¾d_A-N H -N- _
§) 3919ν(Η ε)-S - 3エ 3) ε66卜 ί(Η Ζ - - (HS) ζςεζ寸 ε(Η)E S- --· "- X 0Z卜 sPP)(HN= - - - - (Η Η οζ「Ρ) 66卜(Η= - - -
9L(Η Η「) 908= -.
(s:〇の _Λ_αH_A- ooe) ¾d_A-N HN- _
S〇6V(H εXsN - -
S)寸 LZ HS) SOZω -
99ε01 ε(Ηε)寸 9Ζ(Η7S-. - - - τX |卜「) 14寸(H寸)/N= -. - e ^卜卜 (H)£卜9 (H s-一一 .一
CM 36卜「pp)89卜(H)E = -. - - -
316卜「pp)卜(HHN = -.- - - (Ηq)s3_エH.s」N ■ -. -
(9α〇の _Λ_α Η_Λ-〇oc一 ¾d_≥N H -- 〇6ν(Η εXs 9N - -
S) 9SZ寸 LZ HS) εοζω- -
3 εθτε Ιε) 093エ-·
τX卜r)o_寸(H寸)zNb = - - Ϊpp) 99卜エ) 089(Hs -. -. - 66卜 (HX 0ド 8「N=.- - q) 8〇 είェ Hド 8「s」= - (H .
〔 u〔〕οΐΐ90ι
〔〕〔
〔¾014
〔〕〔〕
S〔〕ffi0152T^
6098T0/S00Zdf/X3d S Z99TS0/900Z OAV
S〔〕ffl0154T^
6098T0/S00Zdf/X3d 9 Z99TS0/900Z OAV
S〔〕015812I
6098T0/S00Zdf/X3d 8 Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 6 Z99TS0/900Z OAV
1-25]
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d ZZY Z99TS0/900Z OAV
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 1731- Z99TS0/900Z OAV
[OS- ΐ挲] [9910]
6098T0/S00Zdf/X3d 931- Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 931- Z99TS0/900Z OAV
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d οεΐ· Z99TS0/900Z OAV
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d εεΐ· Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 9εΐ· Z99TS0/900Z OAV
1-42]
〔¾〔〕εϊ6§
[0182] [表 1-46]
訂正された用紙 (規則 1)
/v:/ O 6098SS00ifcl£ SS0900ZAV
〔〔〕寸ΐε∞ΐο卜 ι
-i [ o]
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
6098T0/S00Zdf/X3d Z99TS0/900Z OAV
1-50]
1-54]
IH NMR (300 MHz, DMSO- D6) 61.27 (d =6.8 Hz, 6 H), 2.64(s, 3 H), 3.27-3.38 (m, 1 H), 4.38 (s, 2
-5 / 'S s 00 1 H), 6.89 (s, 1 H),
7.39 (t like, =7.9 Hz, 1 H), B 7.68 (dd, =8.2, 1.1 Hz, 1 H), ''。 7.91 (dd =7.9, 1.1 Hz, 1 H),
13.05 (bra, 1 H).
IH NMR (300 MHz, DMSO- D6)51.25 (s, 9H),2.64 (s, 3 H), 3.62 (s, 2 H), 6.66 (s, 1 H),-1
7.35 - 7.42 (m, 1 H), 7.67 (dd, B
=8.0, 1.3 Hz, 1 H),7.89 (dd, =7.8, 1.3 Hz, 1 H), 12.91 (brs, IH).
IH NMR (300 MHz, DMSO- D6)61.20 (s, 9H),2.65 (s, 3 H), 3.53 (d, =13.6 Hz, 1 H), 3.89 (d, = 13.6 Hz, 1 H), 6.79 (s, 1 H), 7.39 (t like =7.9 Hz,-2
1 H), 7.68 (dd =8.1, 1.2 Hz, 1 H), 7.91 (dd, =7.8, 1.2 Hz, 1 H) 13.07 (brs, 1 H).
CI IH NMR (600 MHz, DMSO- D6)61.21 (s, 9H),2.56 (s, 3 H), 3.60 (s, 2 H), 6.69 (s, 1 H),-3 v o o 7.51 (dd, =8.7, 2.1 Hz, 1 H), B
7.95 (d =2.1 Hz, 1 H), 8.02 (d, =8.7Hz, 1 H), 13.08 (brs, 1 H). 55]
CI
D6) 51.28 - 1.39 (m, 9 H), 2.58
CJl寸
(s, 3 H), 4.31 (s, 2H), 7.52 (dd, N、 s J=8.6, 1.9 Hz, 1 H), 7.96 (d, B
J=1.9Hz, 1 H), 8.03 (d, J=8.6 Hz, 1 H).
1HNMR(300 MHz, DMSO- D6)51.37 (s, 9H),2.65 (s, 3 H), 4.34 (s, 2 H), 7.41 (t, J=7.9
54一 3
7 o o CI Hz, 1 H), 7.71 (dd, J=8.1, 1.3 B
Hz, 1 H), 7.90 (dd, J=7.9, 1.3 Hz, 1 H).
1HNMR(300 MHz, DMSO-d6) δ 1.34 (s, 9 H), 2.58(s, 3 H), 4.29 (s, 2 H), 7.52(dd, J=8.6, 2.0 Hz, 1 H), 7.95 (d, J=2.0 Hz, B
1 H), 8.03 (d, J=8.6 Hz, 1 H).
表 2
[表 2-1]
6098T0/S00Zdf/X3d 9V Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 991- Z99TS0/900Z OAV
/v: O 6098SS00ifcl£looiAV
OS 寸
O in CO
O ォ O O 寸
LL 2s」gH^O
8广1
0ιεο2Ν2Η9〇.
08_.|
〇
6098T0/S00Zdf/X3d 19 V Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 891- Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 691- Z99TS0/900Z OAV
6098T0/S00Zdf/X3d 091· Z99TS0/900Z OAV
6098T0/S00Zdf/X3d I.9I- Z99TS0/900Z OAV
[π-z [soso]
6098T0/S00Zdf/X3d 391- Z99TS0/900Z OAV
/v:/ OS00ifcl£ SS0900ZAV ε9 _■
S〔M02131I
/v:/ OS00ifcl£ SS0900ZAV s _■
^fi^90s
/v:/ O 6098SS00ifcl£ SS0900ZAV 99 _■
寸 ォ 寸 CM CM CM
O O O
寸 CO O CO ォ CO ォ O O O O O CO
寸 O CO O CO 寸 寸 in 寸 ォ 寸 O ォ 〇 ォ 〇
〇 O O
〇 O CO
O O CO
O z O z O z z O O CO CO
CM m 〇 in l_L in l_L
o CM CM ェ X ェ X X
O ェ si ェ si ェ
CM CM
〇 〇 〇 〇 〇
〇 〇 ϋ ェ F
二 O
f ° ェ o
0); C Co
ェ z:、。 、
o ェ o。
00 G 〇 CO 寸
CM CM CM CM CM
1 1 1 I 1 1 1 1
1 1 1 1 1 1 1 1
LD ιο LD to to to to to 寸 寸 寸 寸 寸 寸 寸 寸
/v:/ O 6098SS00ifcl£ SS0900ZAV Z9 _■
〔 s§9ΐ2-
/v:/ O 6098SS00ifcl£ SS0900ZAV 89 _■
¾U〔〕I6§I
/v: O 6098SS00ifcl£looiAV
6098T0/S00Zdf/X3d III Z99TS0/900Z OAV
2- 21]
/v:/ OS00ifcl£ SS0900ZAV ε- _■ O 寸 寸 1 9寸· 0L 6寸寸 CS90寸 N2H9O L SS905J92H8〇
aL寸sgo寸 N9HO
/v:/ OS00ifcl£ SS0900ZAV寸 _■
ss〔〕^ΐ203l
/v:/ O 6098SS00ifcl£ SS0900ZAV _■
ss〔〕^9ΐ20l
/v:/ O 6098SS00ifcl£ SS0900ZAV 9Z _■
〔〕ssISsl
/v:/ O 6098SS00ifcl£ SS0900ZAV LL _■
〔 u922-
S〔¾0212271I
/v:/ O 6098SS00ifcl£ SS0900ZAV 6Z _■
/v:/ O 6098SS00ifcl£900ZAV
〔 s§6SI
/v:/ O 6098SS00ifcl£ SS0900ZAV _ώ_·
〔¾〕 s§οει
OAV
s§
/v:/ OS00ifcl£ SS0900ZAV ε8_·
寸
0990171
9^寸の寸 O寸Η_〇17■-1
LHSLO
ε99017ι
99のの^ 0寸 Ν0工^〇 17171 99 917寸の^ ο寸 Ν寸 ieno1
_ (〔D)干 IA
9991701
(〔D)—干 IA
/v:/ OS00ifcl£ SS0900ZAV 1¾_·
〔 s§εε2ι
表 2- 34]
//:/ OsooaTI>d sso900AV 98 !■
〔 s§sssl
試験例 [11 β HSD1阻害試験]
試験化合物の評価は以下のように行った。
30mM TrisHCl(pH7.4)/lmM EDTA緩衝液中に 200nM NADPHを加えた反応液に酵 素源であるヒト肝臓ミクロソーム(Tissue Transformation Technologies社)を 10 μ g/ml として添加し、さらに試験化合物をカ卩えた。その後、終濃度 100nMとなるように基質で あるコルチゾン溶液をカ卩ぇ反応を開始させた。 37°Cで 80分間インキュベーションした 後、非特異的な阻害剤である 18 βグリチルレチン酸を終濃度 100 Μとして加えること で反応を停止させた。生成したコルチゾール量を HTRF (登録商標)(Homogeneous T ime- Resolved Fluorescence)法による検出キット(日本シエーリング株式会社)を用い て定量した。本系はユーロピウムで標識された抗コルチゾール抗体と XL665が標識さ れたコルチゾールの間で生じる蛍光共鳴エネルギー移動を検出する系であり、未標 識のコルチゾールを加えると競合反応により、結合のシグナルが減弱する。この時、 キット付属の濃度既知のコルチゾールにより標準曲線を作製し、反応によって生成す るコルチゾール量を評価した。酵素を含まな 、ゥエルのコルチゾール生成量をバック グラウンド、化合物を含まな 、ゥエルのコルチゾール生成量を 100%の酵素活性として 、それぞれの化合物について 50 M力 公比 3の希釈系列につき評価し、 IC50値を 算出した。比較対照として化合物 A; 2-(2-{ [(3-クロ口- 2-メチルフエニル)スルホニル] アミノ}- 1,3-チアゾール -4-ィル) -Ν,Ν-ジェチルァセタミド(Journal of Medicinal Che
mistry 2002, 45, 3813-3815.に記載の化合物)を用いた。結果を表 3に示す。 表 3
[0228] [表 3]
[0229] 本発明により、優れた 11 β -HSD1阻害活性を有する化合物の提供が可能となり、 本発明化合物は、 11 18 -HSD1阻害作用として十分な治療効果を有する医薬品の 有効成分として利用することができる。