RU2576036C2 - Модуляторы активности нес1 и способы для них - Google Patents
Модуляторы активности нес1 и способы для них Download PDFInfo
- Publication number
- RU2576036C2 RU2576036C2 RU2012144022/04A RU2012144022A RU2576036C2 RU 2576036 C2 RU2576036 C2 RU 2576036C2 RU 2012144022/04 A RU2012144022/04 A RU 2012144022/04A RU 2012144022 A RU2012144022 A RU 2012144022A RU 2576036 C2 RU2576036 C2 RU 2576036C2
- Authority
- RU
- Russia
- Prior art keywords
- mmol
- nmr
- mhz
- dimethylphenyl
- ethanone
- Prior art date
Links
- 230000000694 effects Effects 0.000 title abstract description 18
- 238000000034 method Methods 0.000 title description 13
- 150000001875 compounds Chemical class 0.000 claims abstract description 170
- 239000000203 mixture Substances 0.000 claims abstract description 80
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 27
- 125000003118 aryl group Chemical group 0.000 claims abstract description 19
- 229910052736 halogen Chemical group 0.000 claims abstract description 17
- 125000003545 alkoxy group Chemical group 0.000 claims abstract description 15
- 150000002367 halogens Chemical group 0.000 claims abstract description 15
- 125000004076 pyridyl group Chemical group 0.000 claims abstract description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 13
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 9
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 9
- 239000001257 hydrogen Substances 0.000 claims abstract description 8
- 201000010099 disease Diseases 0.000 claims abstract description 7
- 239000003937 drug carrier Substances 0.000 claims abstract description 6
- 230000002018 overexpression Effects 0.000 claims abstract description 6
- 230000004064 dysfunction Effects 0.000 claims abstract description 5
- 125000001188 haloalkyl group Chemical group 0.000 claims abstract description 4
- 239000003814 drug Substances 0.000 claims description 15
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 7
- 229910005965 SO 2 Inorganic materials 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims 2
- 238000004519 manufacturing process Methods 0.000 claims 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 abstract description 6
- 239000003795 chemical substances by application Substances 0.000 abstract description 5
- 239000000126 substance Substances 0.000 abstract description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 abstract 2
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 237
- 238000005481 NMR spectroscopy Methods 0.000 description 204
- 239000000243 solution Substances 0.000 description 188
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 153
- 230000002829 reductive effect Effects 0.000 description 124
- 235000019439 ethyl acetate Nutrition 0.000 description 100
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 99
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 99
- 239000007787 solid Substances 0.000 description 99
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 86
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 75
- 239000011541 reaction mixture Substances 0.000 description 72
- 239000002244 precipitate Substances 0.000 description 69
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 66
- 229920006395 saturated elastomer Polymers 0.000 description 57
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 56
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Natural products NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 43
- 239000012044 organic layer Substances 0.000 description 40
- 238000010992 reflux Methods 0.000 description 40
- -1 C 1 -C 6 alkyl Chemical class 0.000 description 39
- 239000003921 oil Substances 0.000 description 33
- 235000019198 oils Nutrition 0.000 description 33
- 210000004027 cell Anatomy 0.000 description 31
- 239000011734 sodium Substances 0.000 description 31
- 239000012267 brine Substances 0.000 description 28
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 28
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 238000010898 silica gel chromatography Methods 0.000 description 23
- 239000003480 eluent Substances 0.000 description 22
- 238000000746 purification Methods 0.000 description 22
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- SFLXUZPXEWWQNH-UHFFFAOYSA-K tetrabutylazanium;tribromide Chemical compound [Br-].[Br-].[Br-].CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC.CCCC[N+](CCCC)(CCCC)CCCC SFLXUZPXEWWQNH-UHFFFAOYSA-K 0.000 description 19
- 239000000706 filtrate Substances 0.000 description 17
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 15
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 14
- 239000000741 silica gel Substances 0.000 description 14
- 229910002027 silica gel Inorganic materials 0.000 description 14
- 238000003818 flash chromatography Methods 0.000 description 13
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 12
- 206010028980 Neoplasm Diseases 0.000 description 12
- 239000000651 prodrug Substances 0.000 description 12
- 229940002612 prodrug Drugs 0.000 description 12
- YTPYRGOFBLEEBD-UHFFFAOYSA-N 1-(4-chloro-2,6-dimethylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(Cl)C=C1C YTPYRGOFBLEEBD-UHFFFAOYSA-N 0.000 description 11
- 125000000753 cycloalkyl group Chemical group 0.000 description 11
- 229940079593 drug Drugs 0.000 description 11
- 125000001072 heteroaryl group Chemical group 0.000 description 11
- 125000001424 substituent group Chemical group 0.000 description 11
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 10
- 201000011510 cancer Diseases 0.000 description 10
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 10
- 239000012065 filter cake Substances 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- 239000003112 inhibitor Substances 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 238000005160 1H NMR spectroscopy Methods 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 0 Cc1cc(Sc2ccc(*)cc2)cc(C)c1-c1c[s]c(NC(c2ccncc2)=O)n1 Chemical compound Cc1cc(Sc2ccc(*)cc2)cc(C)c1-c1c[s]c(NC(c2ccncc2)=O)n1 0.000 description 8
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 8
- 125000000304 alkynyl group Chemical group 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 230000003993 interaction Effects 0.000 description 8
- 230000000394 mitotic effect Effects 0.000 description 8
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 7
- VYFXXRTZMRYFPX-UHFFFAOYSA-N 2-chloro-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(Cl)N=CC=2)=N1 VYFXXRTZMRYFPX-UHFFFAOYSA-N 0.000 description 7
- 125000003342 alkenyl group Chemical group 0.000 description 7
- SACNIGZYDTUHKB-UHFFFAOYSA-N ditert-butyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C(C)(C)C)C(C)(C)C SACNIGZYDTUHKB-UHFFFAOYSA-N 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- IRGVMVPBEQQZEQ-UHFFFAOYSA-N 1-(4-hydroxy-2,6-dimethylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(O)C=C1C IRGVMVPBEQQZEQ-UHFFFAOYSA-N 0.000 description 6
- HTGCEJVPFAWJDU-UHFFFAOYSA-N 2-bromo-1-[4-(4-methoxyphenyl)sulfanyl-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1SC1=CC(C)=C(C(=O)CBr)C(C)=C1 HTGCEJVPFAWJDU-UHFFFAOYSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- 229910021589 Copper(I) bromide Inorganic materials 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 6
- BERDEBHAJNAUOM-UHFFFAOYSA-N copper(i) oxide Chemical compound [Cu]O[Cu] BERDEBHAJNAUOM-UHFFFAOYSA-N 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- RAIPHJJURHTUIC-UHFFFAOYSA-N 1,3-thiazol-2-amine Chemical class NC1=NC=CS1 RAIPHJJURHTUIC-UHFFFAOYSA-N 0.000 description 5
- QZKJDCAHBYUYPU-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-phenoxyphenyl)ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(OC=2C=CC=CC=2)=C1 QZKJDCAHBYUYPU-UHFFFAOYSA-N 0.000 description 5
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 230000001028 anti-proliverative effect Effects 0.000 description 5
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 230000011278 mitosis Effects 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 150000003254 radicals Chemical class 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 125000003107 substituted aryl group Chemical group 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 125000000335 thiazolyl group Chemical group 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- YZVNPIUPAKOREH-UHFFFAOYSA-N 1-(2,6-difluoro-4-methoxyphenyl)ethanone Chemical compound COC1=CC(F)=C(C(C)=O)C(F)=C1 YZVNPIUPAKOREH-UHFFFAOYSA-N 0.000 description 4
- APXHVISGTUKTFI-UHFFFAOYSA-N 1-(2-chloro-4-iodo-6-methylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(I)C=C1Cl APXHVISGTUKTFI-UHFFFAOYSA-N 0.000 description 4
- LSIHTXYDLIUKOL-UHFFFAOYSA-N 1-[4-(2-methoxyethoxy)-2,6-dimethylphenyl]ethanone Chemical compound COCCOC1=CC(C)=C(C(C)=O)C(C)=C1 LSIHTXYDLIUKOL-UHFFFAOYSA-N 0.000 description 4
- VAJGXSNETQJYTM-UHFFFAOYSA-N 1-[4-(3,5-dimethylphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1=CC(C)=CC(C)=C1 VAJGXSNETQJYTM-UHFFFAOYSA-N 0.000 description 4
- WHDSGKPISWHUCX-UHFFFAOYSA-N 2-bromo-1-(2,5-difluoro-4-methoxyphenyl)ethanone Chemical compound COC1=CC(F)=C(C(=O)CBr)C=C1F WHDSGKPISWHUCX-UHFFFAOYSA-N 0.000 description 4
- SJNUHZJDIRUMMI-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-methylsulfanylphenyl)ethanone Chemical compound CSC1=CC(C)=C(C(=O)CBr)C(C)=C1 SJNUHZJDIRUMMI-UHFFFAOYSA-N 0.000 description 4
- XQJAHBHCLXUGEP-UHFFFAOYSA-N 2-bromo-1-(4-methoxyphenyl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C=C1 XQJAHBHCLXUGEP-UHFFFAOYSA-N 0.000 description 4
- MEJFKOWBFAVCDJ-UHFFFAOYSA-N 2-bromo-1-[4-(2-methoxyethoxy)-2,6-dimethylphenyl]ethanone Chemical compound COCCOC1=CC(C)=C(C(=O)CBr)C(C)=C1 MEJFKOWBFAVCDJ-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- 108091000080 Phosphotransferase Proteins 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 4
- 125000004429 atom Chemical group 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 230000015556 catabolic process Effects 0.000 description 4
- 125000004093 cyano group Chemical group *C#N 0.000 description 4
- 125000000392 cycloalkenyl group Chemical group 0.000 description 4
- 238000006731 degradation reaction Methods 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 4
- 102000020233 phosphotransferase Human genes 0.000 description 4
- 150000003839 salts Chemical class 0.000 description 4
- 230000002195 synergetic effect Effects 0.000 description 4
- 210000004881 tumor cell Anatomy 0.000 description 4
- XKFBNVYIXNVCGB-UHFFFAOYSA-N (4-acetyl-3,5-dimethylphenyl) trifluoromethanesulfonate Chemical compound CC(=O)C1=C(C)C=C(OS(=O)(=O)C(F)(F)F)C=C1C XKFBNVYIXNVCGB-UHFFFAOYSA-N 0.000 description 3
- VQEFHKQDLVDMSL-UHFFFAOYSA-N 1-(2,6-dimethyl-4-phenoxyphenyl)ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1=CC=CC=C1 VQEFHKQDLVDMSL-UHFFFAOYSA-N 0.000 description 3
- ZLORLLCWRIKOPO-UHFFFAOYSA-N 1-(2,6-dimethyl-4-phenylphenyl)ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1C1=CC=CC=C1 ZLORLLCWRIKOPO-UHFFFAOYSA-N 0.000 description 3
- MXRIBGPJQHOIFC-UHFFFAOYSA-N 1-(2,6-dimethyl-4-phenylsulfanylphenyl)ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1SC1=CC=CC=C1 MXRIBGPJQHOIFC-UHFFFAOYSA-N 0.000 description 3
- GMGFZYBLCTZAMJ-UHFFFAOYSA-N 1-(4-amino-2-chloro-6-methylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(N)C=C1Cl GMGFZYBLCTZAMJ-UHFFFAOYSA-N 0.000 description 3
- VAXFXZQOIHAZDK-UHFFFAOYSA-N 1-(4-anilino-2,6-dimethylphenyl)ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1NC1=CC=CC=C1 VAXFXZQOIHAZDK-UHFFFAOYSA-N 0.000 description 3
- SVSPKMWEASLYQX-UHFFFAOYSA-N 1-(4-cyclopentyloxy-2,6-dimethylphenyl)ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1CCCC1 SVSPKMWEASLYQX-UHFFFAOYSA-N 0.000 description 3
- CWKKTZPPDCROOA-UHFFFAOYSA-N 1-[2,6-dimethyl-4-(4-methylphenyl)sulfanylphenyl]ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1SC1=CC=C(C)C=C1 CWKKTZPPDCROOA-UHFFFAOYSA-N 0.000 description 3
- YTZDTZNMHMBALQ-UHFFFAOYSA-N 1-[2,6-dimethyl-4-[4-(trifluoromethyl)phenoxy]phenyl]ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1=CC=C(C(F)(F)F)C=C1 YTZDTZNMHMBALQ-UHFFFAOYSA-N 0.000 description 3
- JOAQKYJTXQOBHN-UHFFFAOYSA-N 1-[2-chloro-4-(4-methoxyphenoxy)-6-methylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1OC1=CC(C)=C(C(C)=O)C(Cl)=C1 JOAQKYJTXQOBHN-UHFFFAOYSA-N 0.000 description 3
- AIFVMJJGWNBKAC-UHFFFAOYSA-N 1-[4-(1,3-benzodioxol-5-yloxy)-2,6-dimethylphenyl]-2-bromoethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(OC=2C=C3OCOC3=CC=2)=C1 AIFVMJJGWNBKAC-UHFFFAOYSA-N 0.000 description 3
- KGGHOZLQLWWHPU-UHFFFAOYSA-N 1-[4-(1,3-benzodioxol-5-yloxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1=CC=C(OCO2)C2=C1 KGGHOZLQLWWHPU-UHFFFAOYSA-N 0.000 description 3
- KYCQOVKXHPLKIT-UHFFFAOYSA-N 1-[4-(2,3-dihydroxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC(=O)C1=C(C)C=C(OCC(O)CO)C=C1C KYCQOVKXHPLKIT-UHFFFAOYSA-N 0.000 description 3
- XGQZPRSFAFSWTD-UHFFFAOYSA-N 1-[4-(2-hydroxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC(O)COC1=CC(C)=C(C(C)=O)C(C)=C1 XGQZPRSFAFSWTD-UHFFFAOYSA-N 0.000 description 3
- POYCUQFZQOZNPG-UHFFFAOYSA-N 1-[4-(3-methoxyphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound COC1=CC=CC(OC=2C=C(C)C(C(C)=O)=C(C)C=2)=C1 POYCUQFZQOZNPG-UHFFFAOYSA-N 0.000 description 3
- WAMLSOMYSRIIHN-UHFFFAOYSA-N 1-[4-(3-methoxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound COCCCOC1=CC(C)=C(C(C)=O)C(C)=C1 WAMLSOMYSRIIHN-UHFFFAOYSA-N 0.000 description 3
- FVPUIEAXHIWQHD-UHFFFAOYSA-N 1-[4-(4-ethylphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(CC)=CC=C1OC1=CC(C)=C(C(C)=O)C(C)=C1 FVPUIEAXHIWQHD-UHFFFAOYSA-N 0.000 description 3
- MZDDCRHTCBJZRZ-UHFFFAOYSA-N 1-[4-(4-fluorophenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=C(C)C(C(=O)C)=C(C)C=C1OC1=CC=C(F)C=C1 MZDDCRHTCBJZRZ-UHFFFAOYSA-N 0.000 description 3
- VUKNIWBQUFTZCY-UHFFFAOYSA-N 1-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1OC1=CC(C)=C(C(C)=O)C(C)=C1 VUKNIWBQUFTZCY-UHFFFAOYSA-N 0.000 description 3
- ZGZXAZZGHURLBB-UHFFFAOYSA-N 1-[4-(4-methoxyphenyl)sulfanyl-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1SC1=CC(C)=C(C(C)=O)C(C)=C1 ZGZXAZZGHURLBB-UHFFFAOYSA-N 0.000 description 3
- MWODTVPKPLLOOE-UHFFFAOYSA-N 2-bromo-1-(2,4,6-trimethoxyphenyl)ethanone Chemical compound COC1=CC(OC)=C(C(=O)CBr)C(OC)=C1 MWODTVPKPLLOOE-UHFFFAOYSA-N 0.000 description 3
- HRAZXKYOYNRVMU-UHFFFAOYSA-N 2-bromo-1-(2,4,6-trimethylphenyl)ethanone Chemical compound CC1=CC(C)=C(C(=O)CBr)C(C)=C1 HRAZXKYOYNRVMU-UHFFFAOYSA-N 0.000 description 3
- VYSHUFGSJJRAHL-UHFFFAOYSA-N 2-bromo-1-(2,4,6-trimethylpyridin-3-yl)ethanone;hydrobromide Chemical compound Br.CC1=CC(C)=C(C(=O)CBr)C(C)=N1 VYSHUFGSJJRAHL-UHFFFAOYSA-N 0.000 description 3
- PKVBZABQCCQHLD-UHFFFAOYSA-N 2-bromo-1-(2,4-dimethoxyphenyl)ethanone Chemical compound COC1=CC=C(C(=O)CBr)C(OC)=C1 PKVBZABQCCQHLD-UHFFFAOYSA-N 0.000 description 3
- RPOZZXXRUXYHMP-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-methylsulfonylphenyl)ethanone Chemical compound CC1=CC(S(C)(=O)=O)=CC(C)=C1C(=O)CBr RPOZZXXRUXYHMP-UHFFFAOYSA-N 0.000 description 3
- HHNMDCSHPZGHCX-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-phenylphenyl)ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(C=2C=CC=CC=2)=C1 HHNMDCSHPZGHCX-UHFFFAOYSA-N 0.000 description 3
- CODJLDJJHDWGFW-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-phenylsulfanylphenyl)ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(SC=2C=CC=CC=2)=C1 CODJLDJJHDWGFW-UHFFFAOYSA-N 0.000 description 3
- XSGHTUSQPRWHOG-UHFFFAOYSA-N 2-bromo-1-(2,6-dimethyl-4-propan-2-yloxyphenyl)ethanone Chemical compound CC(C)OC1=CC(C)=C(C(=O)CBr)C(C)=C1 XSGHTUSQPRWHOG-UHFFFAOYSA-N 0.000 description 3
- OLYLHIPGAFRNAE-UHFFFAOYSA-N 2-bromo-1-(3,5-difluoro-4-methoxyphenyl)ethanone Chemical compound COC1=C(F)C=C(C(=O)CBr)C=C1F OLYLHIPGAFRNAE-UHFFFAOYSA-N 0.000 description 3
- IEJRKMYWSPVQAD-UHFFFAOYSA-N 2-bromo-1-(4-chloro-2,6-dimethylphenyl)ethanone Chemical compound CC1=CC(Cl)=CC(C)=C1C(=O)CBr IEJRKMYWSPVQAD-UHFFFAOYSA-N 0.000 description 3
- JTBXGJZZLYFGOW-UHFFFAOYSA-N 2-bromo-1-(4-cyclopentyloxy-2,6-dimethylphenyl)ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(OC2CCCC2)=C1 JTBXGJZZLYFGOW-UHFFFAOYSA-N 0.000 description 3
- VKADWUQIQOXJPI-UHFFFAOYSA-N 2-bromo-1-(4-ethoxy-2,6-dimethylphenyl)ethanone Chemical compound CCOC1=CC(C)=C(C(=O)CBr)C(C)=C1 VKADWUQIQOXJPI-UHFFFAOYSA-N 0.000 description 3
- HXHUHILIRBPSHX-UHFFFAOYSA-N 2-bromo-1-(4-methoxy-2,6-dimethylphenyl)ethanone Chemical compound COC1=CC(C)=C(C(=O)CBr)C(C)=C1 HXHUHILIRBPSHX-UHFFFAOYSA-N 0.000 description 3
- QNCDPGOJVGDTAN-UHFFFAOYSA-N 2-bromo-1-(4-methoxyphenyl)propan-1-one Chemical compound COC1=CC=C(C(=O)C(C)Br)C=C1 QNCDPGOJVGDTAN-UHFFFAOYSA-N 0.000 description 3
- CLHDNXPEVAZXTH-UHFFFAOYSA-N 2-bromo-1-[2,4,6-tri(propan-2-yl)phenyl]ethanone Chemical compound CC(C)C1=CC(C(C)C)=C(C(=O)CBr)C(C(C)C)=C1 CLHDNXPEVAZXTH-UHFFFAOYSA-N 0.000 description 3
- NPXPPXBBQFDRTN-UHFFFAOYSA-N 2-bromo-1-[2,6-dimethyl-4-(4-methylphenyl)sulfanylphenyl]ethanone Chemical compound C1=CC(C)=CC=C1SC1=CC(C)=C(C(=O)CBr)C(C)=C1 NPXPPXBBQFDRTN-UHFFFAOYSA-N 0.000 description 3
- BBBHPYQINQFMTN-UHFFFAOYSA-N 2-bromo-1-[2,6-dimethyl-4-[4-(trifluoromethyl)phenoxy]phenyl]ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(OC=2C=CC(=CC=2)C(F)(F)F)=C1 BBBHPYQINQFMTN-UHFFFAOYSA-N 0.000 description 3
- SVGMKTUTXUGNPL-UHFFFAOYSA-N 2-bromo-1-[4-(2,3-dihydroxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC1=CC(OCC(O)CO)=CC(C)=C1C(=O)CBr SVGMKTUTXUGNPL-UHFFFAOYSA-N 0.000 description 3
- KDKSUZXNKIHYII-UHFFFAOYSA-N 2-bromo-1-[4-(3,5-dimethylphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC1=CC(C)=CC(OC=2C=C(C)C(C(=O)CBr)=C(C)C=2)=C1 KDKSUZXNKIHYII-UHFFFAOYSA-N 0.000 description 3
- PTVNRYUOEHXDJM-UHFFFAOYSA-N 2-bromo-1-[4-(3-methoxyphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound COC1=CC=CC(OC=2C=C(C)C(C(=O)CBr)=C(C)C=2)=C1 PTVNRYUOEHXDJM-UHFFFAOYSA-N 0.000 description 3
- KTLBZWQSXNRMKA-UHFFFAOYSA-N 2-bromo-1-[4-(3-methoxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound COCCCOC1=CC(C)=C(C(=O)CBr)C(C)=C1 KTLBZWQSXNRMKA-UHFFFAOYSA-N 0.000 description 3
- IBQSJJAOUSYGRS-UHFFFAOYSA-N 2-bromo-1-[4-(4-bromoanilino)-2,6-dimethylphenyl]ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(NC=2C=CC(Br)=CC=2)=C1 IBQSJJAOUSYGRS-UHFFFAOYSA-N 0.000 description 3
- DCQWSTHJWRCOGV-UHFFFAOYSA-N 2-bromo-1-[4-(4-ethylphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(CC)=CC=C1OC1=CC(C)=C(C(=O)CBr)C(C)=C1 DCQWSTHJWRCOGV-UHFFFAOYSA-N 0.000 description 3
- ZNRYPXSEYMVXGZ-UHFFFAOYSA-N 2-bromo-1-[4-(4-fluorophenoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC1=C(C(=O)CBr)C(C)=CC(OC=2C=CC(F)=CC=2)=C1 ZNRYPXSEYMVXGZ-UHFFFAOYSA-N 0.000 description 3
- KBSZJKBCDPDKKY-UHFFFAOYSA-N 2-bromo-1-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1OC1=CC(C)=C(C(=O)CBr)C(C)=C1 KBSZJKBCDPDKKY-UHFFFAOYSA-N 0.000 description 3
- WPRSIMQIWSNSRP-UHFFFAOYSA-N 2-bromo-1-[4-(4-methoxyphenyl)sulfinyl-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1S(=O)C1=CC(C)=C(C(=O)CBr)C(C)=C1 WPRSIMQIWSNSRP-UHFFFAOYSA-N 0.000 description 3
- UEKOPPFIISZDJE-UHFFFAOYSA-N 2-bromo-1-[4-(4-methoxyphenyl)sulfonyl-2,6-dimethylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)C1=CC(C)=C(C(=O)CBr)C(C)=C1 UEKOPPFIISZDJE-UHFFFAOYSA-N 0.000 description 3
- XSVXZERBVBHQFO-UHFFFAOYSA-N 2-chloro-1-(2,4,6-trifluorophenyl)ethanone Chemical compound FC1=CC(F)=C(C(=O)CCl)C(F)=C1 XSVXZERBVBHQFO-UHFFFAOYSA-N 0.000 description 3
- TUAMRELNJMMDMT-UHFFFAOYSA-N 3,5-xylenol Chemical compound CC1=CC(C)=CC(O)=C1 TUAMRELNJMMDMT-UHFFFAOYSA-N 0.000 description 3
- VEGXYZYAGLCXMZ-UHFFFAOYSA-N 3-[4-(2-amino-1,3-thiazol-4-yl)-3,5-dimethylphenoxy]propane-1,2-diol Chemical compound CC1=CC(OCC(O)CO)=CC(C)=C1C1=CSC(N)=N1 VEGXYZYAGLCXMZ-UHFFFAOYSA-N 0.000 description 3
- XUZCJDBXXYJXDD-UHFFFAOYSA-N 3-chloro-5-methylaniline Chemical compound CC1=CC(N)=CC(Cl)=C1 XUZCJDBXXYJXDD-UHFFFAOYSA-N 0.000 description 3
- NTPLXRHDUXRPNE-UHFFFAOYSA-N 4-methoxyacetophenone Chemical compound COC1=CC=C(C(C)=O)C=C1 NTPLXRHDUXRPNE-UHFFFAOYSA-N 0.000 description 3
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 3
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 3
- 239000012346 acetyl chloride Substances 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000000973 chemotherapeutic effect Effects 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000004992 fission Effects 0.000 description 3
- 125000002541 furyl group Chemical group 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 125000000842 isoxazolyl group Chemical group 0.000 description 3
- 238000000021 kinase assay Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- PKZPIAGUTDNQJG-UHFFFAOYSA-N n-(3-chloro-5-methylphenyl)acetamide Chemical compound CC(=O)NC1=CC(C)=CC(Cl)=C1 PKZPIAGUTDNQJG-UHFFFAOYSA-N 0.000 description 3
- GLLJNFSFUOENEH-UHFFFAOYSA-N n-(4-acetyl-3-chloro-5-methylphenyl)acetamide Chemical compound CC(=O)NC1=CC(C)=C(C(C)=O)C(Cl)=C1 GLLJNFSFUOENEH-UHFFFAOYSA-N 0.000 description 3
- YYJCDWHFYWRYHA-UHFFFAOYSA-N n-[4-(2-bromoacetyl)-3,5-dimethylphenyl]acetamide Chemical compound CC(=O)NC1=CC(C)=C(C(=O)CBr)C(C)=C1 YYJCDWHFYWRYHA-UHFFFAOYSA-N 0.000 description 3
- VDXVLFMULWHCLT-UHFFFAOYSA-N n-[4-(4-iodo-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(I)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 VDXVLFMULWHCLT-UHFFFAOYSA-N 0.000 description 3
- RKGPQNZLZMBNAM-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]-2-nitropyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(N=CC=2)[N+]([O-])=O)=N1 RKGPQNZLZMBNAM-UHFFFAOYSA-N 0.000 description 3
- NBNNDUZYMXBCOX-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 NBNNDUZYMXBCOX-UHFFFAOYSA-N 0.000 description 3
- RAWUQDVOCPYSLY-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenyl)sulfinyl-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1S(=O)C(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 RAWUQDVOCPYSLY-UHFFFAOYSA-N 0.000 description 3
- YGBMCLDVRUGXOV-UHFFFAOYSA-N n-[6-[6-chloro-5-[(4-fluorophenyl)sulfonylamino]pyridin-3-yl]-1,3-benzothiazol-2-yl]acetamide Chemical compound C1=C2SC(NC(=O)C)=NC2=CC=C1C(C=1)=CN=C(Cl)C=1NS(=O)(=O)C1=CC=C(F)C=C1 YGBMCLDVRUGXOV-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 description 3
- 239000001301 oxygen Chemical group 0.000 description 3
- 239000002574 poison Substances 0.000 description 3
- 231100000614 poison Toxicity 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 125000003373 pyrazinyl group Chemical group 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 125000005308 thiazepinyl group Chemical group S1N=C(C=CC=C1)* 0.000 description 3
- 125000001544 thienyl group Chemical group 0.000 description 3
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- OTGQPYSISUUHAF-UHFFFAOYSA-N 1,3-difluoro-5-methoxybenzene Chemical compound COC1=CC(F)=CC(F)=C1 OTGQPYSISUUHAF-UHFFFAOYSA-N 0.000 description 2
- KPZWHZSIXZXDMW-UHFFFAOYSA-N 1-(2,4,6-trimethoxyphenyl)ethanone Chemical compound COC1=CC(OC)=C(C(C)=O)C(OC)=C1 KPZWHZSIXZXDMW-UHFFFAOYSA-N 0.000 description 2
- XWCIICLTKWRWCI-UHFFFAOYSA-N 1-(2,4,6-trimethylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(C)C=C1C XWCIICLTKWRWCI-UHFFFAOYSA-N 0.000 description 2
- GFVYFAYTYSTBOM-UHFFFAOYSA-N 1-(4-amino-2,6-dimethylphenyl)ethanone Chemical compound CC(=O)C1=C(C)C=C(N)C=C1C GFVYFAYTYSTBOM-UHFFFAOYSA-N 0.000 description 2
- ADQZDYLKQLQHSA-UHFFFAOYSA-N 1-[2,6-dimethyl-4-(2-methylpropoxy)phenyl]ethanone Chemical compound CC(C)COC1=CC(C)=C(C(C)=O)C(C)=C1 ADQZDYLKQLQHSA-UHFFFAOYSA-N 0.000 description 2
- SBPNNIVZIWRVQA-UHFFFAOYSA-N 1-[4-(2-amino-1,3-thiazol-4-yl)-3,5-dimethylphenoxy]propan-2-ol Chemical compound CC1=CC(OCC(O)C)=CC(C)=C1C1=CSC(N)=N1 SBPNNIVZIWRVQA-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- KKVYYGGCHJGEFJ-UHFFFAOYSA-N 1-n-(4-chlorophenyl)-6-methyl-5-n-[3-(7h-purin-6-yl)pyridin-2-yl]isoquinoline-1,5-diamine Chemical compound N=1C=CC2=C(NC=3C(=CC=CN=3)C=3C=4N=CNC=4N=CN=3)C(C)=CC=C2C=1NC1=CC=C(Cl)C=C1 KKVYYGGCHJGEFJ-UHFFFAOYSA-N 0.000 description 2
- IHPYMWDTONKSCO-UHFFFAOYSA-N 2,2'-piperazine-1,4-diylbisethanesulfonic acid Chemical compound OS(=O)(=O)CCN1CCN(CCS(O)(=O)=O)CC1 IHPYMWDTONKSCO-UHFFFAOYSA-N 0.000 description 2
- FHBPWYJQINJCGN-UHFFFAOYSA-N 2-(dimethylamino)-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=NC(N(C)C)=CC(C(=O)NC=2SC=C(N=2)C=2C(=CC(C)=CC=2C)C)=C1 FHBPWYJQINJCGN-UHFFFAOYSA-N 0.000 description 2
- FTBXWKAVSDXIHD-UHFFFAOYSA-N 2-amino-n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(N)N=CC=2)=N1 FTBXWKAVSDXIHD-UHFFFAOYSA-N 0.000 description 2
- GTAZURIDICHFSM-UHFFFAOYSA-N 2-bromo-1-[2,6-dimethyl-4-(2-methylpropoxy)phenyl]ethanone Chemical compound CC(C)COC1=CC(C)=C(C(=O)CBr)C(C)=C1 GTAZURIDICHFSM-UHFFFAOYSA-N 0.000 description 2
- FCFQWSHPZHMSQH-UHFFFAOYSA-N 2-bromo-1-[4-(2-hydroxypropoxy)-2,6-dimethylphenyl]ethanone Chemical compound CC(O)COC1=CC(C)=C(C(=O)CBr)C(C)=C1 FCFQWSHPZHMSQH-UHFFFAOYSA-N 0.000 description 2
- JHMVCCGRLYATDZ-UHFFFAOYSA-N 2-methoxy-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=NC(OC)=CC(C(=O)NC=2SC=C(N=2)C=2C(=CC(C)=CC=2C)C)=C1 JHMVCCGRLYATDZ-UHFFFAOYSA-N 0.000 description 2
- IGSQKSNZUQAONK-UHFFFAOYSA-N 2-morpholin-4-yl-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(N=CC=2)N2CCOCC2)=N1 IGSQKSNZUQAONK-UHFFFAOYSA-N 0.000 description 2
- UAJLKHOUBSFFFU-UHFFFAOYSA-N 2-piperidin-1-yl-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(N=CC=2)N2CCCCC2)=N1 UAJLKHOUBSFFFU-UHFFFAOYSA-N 0.000 description 2
- ASHGTJPOSUFTGB-UHFFFAOYSA-N 3-methoxyphenol Chemical compound COC1=CC=CC(O)=C1 ASHGTJPOSUFTGB-UHFFFAOYSA-N 0.000 description 2
- XRBNIVRCMULMJU-UHFFFAOYSA-N 4-(2,4,6-trifluorophenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C(=CC(F)=CC=2F)F)=C1 XRBNIVRCMULMJU-UHFFFAOYSA-N 0.000 description 2
- CAQIXUVMLQYMIY-UHFFFAOYSA-N 4-(2,4,6-trimethoxyphenyl)-1,3-thiazol-2-amine Chemical compound COC1=CC(OC)=CC(OC)=C1C1=CSC(N)=N1 CAQIXUVMLQYMIY-UHFFFAOYSA-N 0.000 description 2
- QKYORNXSXAOYPV-UHFFFAOYSA-N 4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(N)=N1 QKYORNXSXAOYPV-UHFFFAOYSA-N 0.000 description 2
- SNIBQFOEOVBOKU-UHFFFAOYSA-N 4-(2,4,6-trimethylpyridin-3-yl)-1,3-thiazol-2-amine Chemical compound CC1=NC(C)=CC(C)=C1C1=CSC(N)=N1 SNIBQFOEOVBOKU-UHFFFAOYSA-N 0.000 description 2
- NRGCDEWBJMQJBX-UHFFFAOYSA-N 4-(2,4-dimethoxyphenyl)-1,3-thiazol-2-amine Chemical compound COC1=CC(OC)=CC=C1C1=CSC(N)=N1 NRGCDEWBJMQJBX-UHFFFAOYSA-N 0.000 description 2
- IYRQSNZTCNLWQW-UHFFFAOYSA-N 4-(2,6-difluoro-4-methoxyphenyl)-1,3-thiazol-2-amine Chemical compound FC1=CC(OC)=CC(F)=C1C1=CSC(N)=N1 IYRQSNZTCNLWQW-UHFFFAOYSA-N 0.000 description 2
- QSHGCGUBJXMUTP-UHFFFAOYSA-N 4-(2,6-dimethyl-4-methylsulfanylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(SC)=CC(C)=C1C1=CSC(N)=N1 QSHGCGUBJXMUTP-UHFFFAOYSA-N 0.000 description 2
- RIPJPHUMZHYGOO-UHFFFAOYSA-N 4-(2,6-dimethyl-4-methylsulfonylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(S(C)(=O)=O)=CC(C)=C1C1=CSC(N)=N1 RIPJPHUMZHYGOO-UHFFFAOYSA-N 0.000 description 2
- OHDUBYZWTLQFTH-UHFFFAOYSA-N 4-(2,6-dimethyl-4-phenoxyphenyl)-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1OC1=CC=CC=C1 OHDUBYZWTLQFTH-UHFFFAOYSA-N 0.000 description 2
- ZMTQNZQXINDNBN-UHFFFAOYSA-N 4-(2,6-dimethyl-4-phenylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(C=2C=CC=CC=2)=CC(C)=C1C1=CSC(N)=N1 ZMTQNZQXINDNBN-UHFFFAOYSA-N 0.000 description 2
- ACNCOVBXCPTXFE-UHFFFAOYSA-N 4-(2,6-dimethyl-4-phenylsulfanylphenyl)-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1SC1=CC=CC=C1 ACNCOVBXCPTXFE-UHFFFAOYSA-N 0.000 description 2
- HHZNCVUGOCERGA-UHFFFAOYSA-N 4-(2,6-dimethyl-4-propan-2-yloxyphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(OC(C)C)=CC(C)=C1C1=CSC(N)=N1 HHZNCVUGOCERGA-UHFFFAOYSA-N 0.000 description 2
- WQWKOXCCOAAIMU-UHFFFAOYSA-N 4-(3,5-difluoro-4-methoxyphenyl)-1,3-thiazol-2-amine Chemical compound C1=C(F)C(OC)=C(F)C=C1C1=CSC(N)=N1 WQWKOXCCOAAIMU-UHFFFAOYSA-N 0.000 description 2
- FCGIKUJDENXWFE-UHFFFAOYSA-N 4-(4-chloro-2,6-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(Cl)=CC(C)=C1C1=CSC(N)=N1 FCGIKUJDENXWFE-UHFFFAOYSA-N 0.000 description 2
- JAYCCFWDZPBASH-UHFFFAOYSA-N 4-(4-cyclopentyloxy-2,6-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1OC1CCCC1 JAYCCFWDZPBASH-UHFFFAOYSA-N 0.000 description 2
- LLAAWXTWSLDVFO-UHFFFAOYSA-N 4-(4-ethoxy-2,6-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(OCC)=CC(C)=C1C1=CSC(N)=N1 LLAAWXTWSLDVFO-UHFFFAOYSA-N 0.000 description 2
- QGJAPSOSAWMPKU-UHFFFAOYSA-N 4-(4-methoxy-2,6-dimethylphenyl)-1,3-thiazol-2-amine Chemical compound CC1=CC(OC)=CC(C)=C1C1=CSC(N)=N1 QGJAPSOSAWMPKU-UHFFFAOYSA-N 0.000 description 2
- YPVVEXKDPBRGIK-UHFFFAOYSA-N 4-(4-methoxyphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1C1=CSC(N)=N1 YPVVEXKDPBRGIK-UHFFFAOYSA-N 0.000 description 2
- WUTXFLYFZOMYSF-UHFFFAOYSA-N 4-(4-methoxyphenyl)-5-methyl-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1C1=C(C)SC(N)=N1 WUTXFLYFZOMYSF-UHFFFAOYSA-N 0.000 description 2
- ARLHWYFAPHJCJT-UHFFFAOYSA-N 4-(4-methylphenyl)-1,3-thiazol-2-amine Chemical compound C1=CC(C)=CC=C1C1=CSC(N)=N1 ARLHWYFAPHJCJT-UHFFFAOYSA-N 0.000 description 2
- CPHWMBZBSNHOOJ-UHFFFAOYSA-N 4-[2,4,6-tri(propan-2-yl)phenyl]-1,3-thiazol-2-amine Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CSC(N)=N1 CPHWMBZBSNHOOJ-UHFFFAOYSA-N 0.000 description 2
- QTUJQDKKTJBKAU-UHFFFAOYSA-N 4-[2,6-dimethyl-4-(2-methylpropoxy)phenyl]-1,3-thiazol-2-amine Chemical compound CC1=CC(OCC(C)C)=CC(C)=C1C1=CSC(N)=N1 QTUJQDKKTJBKAU-UHFFFAOYSA-N 0.000 description 2
- MPAAEQDZDYZCHL-UHFFFAOYSA-N 4-[2,6-dimethyl-4-(4-methylphenyl)sulfanylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(C)=CC=C1SC(C=C1C)=CC(C)=C1C1=CSC(N)=N1 MPAAEQDZDYZCHL-UHFFFAOYSA-N 0.000 description 2
- IYCBAISLCRKJJX-UHFFFAOYSA-N 4-[2,6-dimethyl-4-[4-(trifluoromethyl)phenoxy]phenyl]-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1OC1=CC=C(C(F)(F)F)C=C1 IYCBAISLCRKJJX-UHFFFAOYSA-N 0.000 description 2
- ZOXMXVXTBGFLAV-UHFFFAOYSA-N 4-[2-chloro-4-(4-methoxyphenoxy)-6-methylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1OC(C=C1Cl)=CC(C)=C1C1=CSC(N)=N1 ZOXMXVXTBGFLAV-UHFFFAOYSA-N 0.000 description 2
- HLAPUIPSNBUWTG-UHFFFAOYSA-N 4-[4-(1,3-benzodioxol-5-yloxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound CC1=CC(OC=2C=C3OCOC3=CC=2)=CC(C)=C1C1=CSC(N)=N1 HLAPUIPSNBUWTG-UHFFFAOYSA-N 0.000 description 2
- ZGFXSZLRNCTIMY-UHFFFAOYSA-N 4-[4-(2-methoxyethoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound CC1=CC(OCCOC)=CC(C)=C1C1=CSC(N)=N1 ZGFXSZLRNCTIMY-UHFFFAOYSA-N 0.000 description 2
- UCXBUSRPRMIBOD-UHFFFAOYSA-N 4-[4-(3,5-dimethylphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound CC1=CC(C)=CC(OC=2C=C(C)C(C=3N=C(N)SC=3)=C(C)C=2)=C1 UCXBUSRPRMIBOD-UHFFFAOYSA-N 0.000 description 2
- UTPGIFCACYHMFD-UHFFFAOYSA-N 4-[4-(3-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound COC1=CC=CC(OC=2C=C(C)C(C=3N=C(N)SC=3)=C(C)C=2)=C1 UTPGIFCACYHMFD-UHFFFAOYSA-N 0.000 description 2
- FKCYCGVPZMHGMN-UHFFFAOYSA-N 4-[4-(3-methoxypropoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound CC1=CC(OCCCOC)=CC(C)=C1C1=CSC(N)=N1 FKCYCGVPZMHGMN-UHFFFAOYSA-N 0.000 description 2
- LNPSHYXEFMYYBI-UHFFFAOYSA-N 4-[4-(4-ethylphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(CC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(N)=N1 LNPSHYXEFMYYBI-UHFFFAOYSA-N 0.000 description 2
- ATLRDTJFWFUUSF-UHFFFAOYSA-N 4-[4-(4-fluorophenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1OC1=CC=C(F)C=C1 ATLRDTJFWFUUSF-UHFFFAOYSA-N 0.000 description 2
- PFQDGRRBAYHHEU-UHFFFAOYSA-N 4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(N)=N1 PFQDGRRBAYHHEU-UHFFFAOYSA-N 0.000 description 2
- IDNBCYXKQRWKRN-UHFFFAOYSA-N 4-[4-(4-methoxyphenyl)sulfanyl-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1SC(C=C1C)=CC(C)=C1C1=CSC(N)=N1 IDNBCYXKQRWKRN-UHFFFAOYSA-N 0.000 description 2
- VGDVALVRMLXYFD-UHFFFAOYSA-N 4-[4-(4-methoxyphenyl)sulfonyl-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)C(C=C1C)=CC(C)=C1C1=CSC(N)=N1 VGDVALVRMLXYFD-UHFFFAOYSA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- HXDOZKJGKXYMEW-UHFFFAOYSA-N 4-ethylphenol Chemical compound CCC1=CC=C(O)C=C1 HXDOZKJGKXYMEW-UHFFFAOYSA-N 0.000 description 2
- LARKPJHOVACKEM-UHFFFAOYSA-N 5-methyl-4-(4-methylphenyl)-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC(C)=CC=2)=C1C LARKPJHOVACKEM-UHFFFAOYSA-N 0.000 description 2
- HTXQOROHFFYFMC-UHFFFAOYSA-N 5-methyl-4-phenyl-1,3-thiazol-2-amine Chemical compound S1C(N)=NC(C=2C=CC=CC=2)=C1C HTXQOROHFFYFMC-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- 101100381978 Mus musculus Braf gene Proteins 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical class C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 125000004423 acyloxy group Chemical group 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000031709 bromination Effects 0.000 description 2
- 238000005893 bromination reaction Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 2
- 125000002843 carboxylic acid group Chemical group 0.000 description 2
- 150000001768 cations Chemical class 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000005770 chromosome separation Effects 0.000 description 2
- 238000004140 cleaning Methods 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 125000005805 dimethoxy phenyl group Chemical group 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 238000003119 immunoblot Methods 0.000 description 2
- 238000001114 immunoprecipitation Methods 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000003701 inert diluent Substances 0.000 description 2
- VFQXVTODMYMSMJ-UHFFFAOYSA-N isonicotinamide Chemical compound NC(=O)C1=CC=NC=C1 VFQXVTODMYMSMJ-UHFFFAOYSA-N 0.000 description 2
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 2
- 210000002415 kinetochore Anatomy 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 238000012417 linear regression Methods 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- UHOVQNZJYSORNB-UHFFFAOYSA-N monobenzene Natural products C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 2
- FGXOJZDWXAQMCS-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-propan-2-yloxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OC(C)C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 FGXOJZDWXAQMCS-UHFFFAOYSA-N 0.000 description 2
- YVNLUFJWLIXABT-UHFFFAOYSA-N n-[4-(2-amino-1,3-thiazol-4-yl)-3,5-dimethylphenyl]acetamide Chemical compound CC1=CC(NC(=O)C)=CC(C)=C1C1=CSC(N)=N1 YVNLUFJWLIXABT-UHFFFAOYSA-N 0.000 description 2
- LBMUXKTXBJCYOL-UHFFFAOYSA-N n-[4-(3,4,5-trimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound COC1=C(OC)C(OC)=CC(C=2N=C(NC(=O)C=3N=CC=CC=3)SC=2)=C1 LBMUXKTXBJCYOL-UHFFFAOYSA-N 0.000 description 2
- BCHBEMYVGPFERY-UHFFFAOYSA-N n-[4-(3,4,5-trimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound COC1=C(OC)C(OC)=CC(C=2N=C(NC(=O)C=3C=NC=CC=3)SC=2)=C1 BCHBEMYVGPFERY-UHFFFAOYSA-N 0.000 description 2
- RNWZBBLJRVHMCN-UHFFFAOYSA-N n-[4-(4-anilino-3-bromo-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound BrC=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1NC1=CC=CC=C1 RNWZBBLJRVHMCN-UHFFFAOYSA-N 0.000 description 2
- KQMQBMQVQCKRRX-UHFFFAOYSA-N n-[4-(4-benzyl-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1CC1=CC=CC=C1 KQMQBMQVQCKRRX-UHFFFAOYSA-N 0.000 description 2
- XRIDEZVVCZKPPS-UHFFFAOYSA-N n-[4-(4-methoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OC)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 XRIDEZVVCZKPPS-UHFFFAOYSA-N 0.000 description 2
- ILXXCNQLKQLLNW-UHFFFAOYSA-N n-[4-[4-[(4-methoxyphenyl)methyl]-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1CC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 ILXXCNQLKQLLNW-UHFFFAOYSA-N 0.000 description 2
- WYQWKVROZXZBPI-UHFFFAOYSA-N n-[5-methyl-4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound N=1C(C=2C=CC(C)=CC=2)=C(C)SC=1NC(=O)C1=CC=CC=N1 WYQWKVROZXZBPI-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 238000011580 nude mouse model Methods 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000008389 polyethoxylated castor oil Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 125000002755 pyrazolinyl group Chemical group 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 230000008054 signal transmission Effects 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 239000011593 sulfur Chemical group 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- IOGXOCVLYRDXLW-UHFFFAOYSA-N tert-butyl nitrite Chemical compound CC(C)(C)ON=O IOGXOCVLYRDXLW-UHFFFAOYSA-N 0.000 description 2
- 239000012414 tert-butyl nitrite Substances 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 150000003568 thioethers Chemical class 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- GCTFTMWXZFLTRR-GFCCVEGCSA-N (2r)-2-amino-n-[3-(difluoromethoxy)-4-(1,3-oxazol-5-yl)phenyl]-4-methylpentanamide Chemical compound FC(F)OC1=CC(NC(=O)[C@H](N)CC(C)C)=CC=C1C1=CN=CO1 GCTFTMWXZFLTRR-GFCCVEGCSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- FNHHVPPSBFQMEL-KQHDFZBMSA-N (3S)-5-N-[(1S,5R)-3-hydroxy-6-bicyclo[3.1.0]hexanyl]-7-N,3-dimethyl-3-phenyl-2H-1-benzofuran-5,7-dicarboxamide Chemical compound CNC(=O)c1cc(cc2c1OC[C@@]2(C)c1ccccc1)C(=O)NC1[C@H]2CC(O)C[C@@H]12 FNHHVPPSBFQMEL-KQHDFZBMSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- JXUKFFRPLNTYIV-UHFFFAOYSA-N 1,3,5-trifluorobenzene Chemical compound FC1=CC(F)=CC(F)=C1 JXUKFFRPLNTYIV-UHFFFAOYSA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- DJMOXMNDXFFONV-UHFFFAOYSA-N 1,3-dimethyl-7-[2-(n-methylanilino)ethyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCN(C)C1=CC=CC=C1 DJMOXMNDXFFONV-UHFFFAOYSA-N 0.000 description 1
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- RKNKTACHKMTWTD-UHFFFAOYSA-N 1-(2,4,6-trimethylpyridin-3-yl)ethanone Chemical compound CC(=O)C1=C(C)C=C(C)N=C1C RKNKTACHKMTWTD-UHFFFAOYSA-N 0.000 description 1
- ISGSVXDEWCDSII-UHFFFAOYSA-N 1-(2,6-dimethyl-4-propan-2-yloxyphenyl)ethanone Chemical compound CC(C)OC1=CC(C)=C(C(C)=O)C(C)=C1 ISGSVXDEWCDSII-UHFFFAOYSA-N 0.000 description 1
- OUJZNFJFMAKGMS-UHFFFAOYSA-N 1-(3,5-difluoro-4-methoxyphenyl)ethanone Chemical compound COC1=C(F)C=C(C(C)=O)C=C1F OUJZNFJFMAKGMS-UHFFFAOYSA-N 0.000 description 1
- UJPUGSXNZQJEPM-UHFFFAOYSA-N 1-(4-ethoxy-2,6-dimethylphenyl)ethanone Chemical compound CCOC1=CC(C)=C(C(C)=O)C(C)=C1 UJPUGSXNZQJEPM-UHFFFAOYSA-N 0.000 description 1
- LOQOGGQUEVRUSS-UHFFFAOYSA-N 1-(4-methoxy-2,6-dimethylphenyl)ethanone Chemical compound COC1=CC(C)=C(C(C)=O)C(C)=C1 LOQOGGQUEVRUSS-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- SGRDDUKVLKWIBZ-UHFFFAOYSA-N 1-[2,4,6-tri(propan-2-yl)phenyl]ethanone Chemical compound CC(C)C1=CC(C(C)C)=C(C(C)=O)C(C(C)C)=C1 SGRDDUKVLKWIBZ-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- CKKYIPUTRBBCIH-UHFFFAOYSA-N 1-[4-(4-methoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]-3-pyridin-4-ylurea Chemical compound CC1=CC(OC)=CC(C)=C1C1=CSC(NC(=O)NC=2C=CN=CC=2)=N1 CKKYIPUTRBBCIH-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- XTIGGAHUZJWQMD-UHFFFAOYSA-N 1-chloro-2-methoxyethane Chemical compound COCCCl XTIGGAHUZJWQMD-UHFFFAOYSA-N 0.000 description 1
- BQLHMMQUVJCTAN-UHFFFAOYSA-N 1-chloro-3-methoxypropane Chemical compound COCCCCl BQLHMMQUVJCTAN-UHFFFAOYSA-N 0.000 description 1
- IYHHGLULQDOFGC-UHFFFAOYSA-N 1-chloro-3-methyl-5-nitrobenzene Chemical compound CC1=CC(Cl)=CC([N+]([O-])=O)=C1 IYHHGLULQDOFGC-UHFFFAOYSA-N 0.000 description 1
- WKMGUUGGDUDBNN-UHFFFAOYSA-N 1-oxido-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridin-1-ium-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C[N+]([O-])=CC=2)=N1 WKMGUUGGDUDBNN-UHFFFAOYSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- VQTDPCRSXHFMOL-UHFFFAOYSA-N 2,4-Dimethoxyacetophenone Chemical compound COC1=CC=C(C(C)=O)C(OC)=C1 VQTDPCRSXHFMOL-UHFFFAOYSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- VCUXVXLUOHDHKK-UHFFFAOYSA-N 2-(2-aminopyrimidin-4-yl)-4-(2-chloro-4-methoxyphenyl)-1,3-thiazole-5-carboxamide Chemical compound ClC1=CC(OC)=CC=C1C1=C(C(N)=O)SC(C=2N=C(N)N=CC=2)=N1 VCUXVXLUOHDHKK-UHFFFAOYSA-N 0.000 description 1
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 1
- USIOVANNNPLJNG-UHFFFAOYSA-N 2-(4-methylpiperazin-1-yl)-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1CN(C)CCN1C1=CC(C(=O)NC=2SC=C(N=2)C=2C(=CC(C)=CC=2C)C)=CC=N1 USIOVANNNPLJNG-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- KRVGXFREOJHJAX-UHFFFAOYSA-N 2-bromo-1-(4-methylphenyl)ethanone Chemical compound CC1=CC=C(C(=O)CBr)C=C1 KRVGXFREOJHJAX-UHFFFAOYSA-N 0.000 description 1
- OZLUPIIIHOOPNQ-UHFFFAOYSA-N 2-bromo-1-(4-methylphenyl)propan-1-one Chemical compound CC(Br)C(=O)C1=CC=C(C)C=C1 OZLUPIIIHOOPNQ-UHFFFAOYSA-N 0.000 description 1
- NZDYTUSOXDBHOM-UHFFFAOYSA-N 2-bromo-1-[2-chloro-4-(4-methoxyphenoxy)-6-methylphenyl]ethanone Chemical compound C1=CC(OC)=CC=C1OC1=CC(C)=C(C(=O)CBr)C(Cl)=C1 NZDYTUSOXDBHOM-UHFFFAOYSA-N 0.000 description 1
- WPDWOCRJBPXJFM-UHFFFAOYSA-N 2-bromo-1-phenylpropan-1-one Chemical compound CC(Br)C(=O)C1=CC=CC=C1 WPDWOCRJBPXJFM-UHFFFAOYSA-N 0.000 description 1
- WRWQRWFDSRHPLD-UHFFFAOYSA-N 2-chloro-6-methoxy-n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound ClC1=NC(OC)=CC(C(=O)NC=2SC=C(N=2)C=2C(=CC(C)=CC=2C)C)=C1 WRWQRWFDSRHPLD-UHFFFAOYSA-N 0.000 description 1
- JZGFZITYBNEDHQ-UHFFFAOYSA-N 2-fluoro-n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(F)N=CC=2)=N1 JZGFZITYBNEDHQ-UHFFFAOYSA-N 0.000 description 1
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 1
- 125000004204 2-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C(OC([H])([H])[H])C([H])=C1[H] 0.000 description 1
- BUOYTFVLNZIELF-UHFFFAOYSA-N 2-phenyl-1h-indole-4,6-dicarboximidamide Chemical compound N1C2=CC(C(=N)N)=CC(C(N)=N)=C2C=C1C1=CC=CC=C1 BUOYTFVLNZIELF-UHFFFAOYSA-N 0.000 description 1
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical compound C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- WFOVEDJTASPCIR-UHFFFAOYSA-N 3-[(4-methyl-5-pyridin-4-yl-1,2,4-triazol-3-yl)methylamino]-n-[[2-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound N=1N=C(C=2C=CN=CC=2)N(C)C=1CNC(C=1)=CC=CC=1C(=O)NCC1=CC=CC=C1C(F)(F)F WFOVEDJTASPCIR-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- SSZWWUDQMAHNAQ-UHFFFAOYSA-N 3-chloropropane-1,2-diol Chemical compound OCC(O)CCl SSZWWUDQMAHNAQ-UHFFFAOYSA-N 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- ZAKUXPKAPWYKQK-UHFFFAOYSA-N 3h-oxathiadiazole Chemical compound N1SOC=N1 ZAKUXPKAPWYKQK-UHFFFAOYSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- RGEORFDNAPDIIW-UHFFFAOYSA-N 4-[4-(4-bromoanilino)-2,6-dimethylphenyl]-1,3-thiazol-2-amine Chemical compound C=1C(C)=C(C=2N=C(N)SC=2)C(C)=CC=1NC1=CC=C(Br)C=C1 RGEORFDNAPDIIW-UHFFFAOYSA-N 0.000 description 1
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 1
- MPMKMQHJHDHPBE-RUZDIDTESA-N 4-[[(2r)-1-(1-benzothiophene-3-carbonyl)-2-methylazetidine-2-carbonyl]-[(3-chlorophenyl)methyl]amino]butanoic acid Chemical compound O=C([C@@]1(N(CC1)C(=O)C=1C2=CC=CC=C2SC=1)C)N(CCCC(O)=O)CC1=CC=CC(Cl)=C1 MPMKMQHJHDHPBE-RUZDIDTESA-N 0.000 description 1
- GSDQYSSLIKJJOG-UHFFFAOYSA-N 4-chloro-2-(3-chloroanilino)benzoic acid Chemical compound OC(=O)C1=CC=C(Cl)C=C1NC1=CC=CC(Cl)=C1 GSDQYSSLIKJJOG-UHFFFAOYSA-N 0.000 description 1
- HVQRIDUXDNXJPF-UHFFFAOYSA-N 4-cyano-n-(5-methyl-4-phenyl-1,3-thiazol-2-yl)benzamide Chemical compound N=1C(C=2C=CC=CC=2)=C(C)SC=1NC(=O)C1=CC=C(C#N)C=C1 HVQRIDUXDNXJPF-UHFFFAOYSA-N 0.000 description 1
- LJFWMSZYWOKTMT-UHFFFAOYSA-N 4-cyano-n-[4-(4-methoxyphenyl)-5-methyl-1,3-thiazol-2-yl]benzamide Chemical compound C1=CC(OC)=CC=C1C1=C(C)SC(NC(=O)C=2C=CC(=CC=2)C#N)=N1 LJFWMSZYWOKTMT-UHFFFAOYSA-N 0.000 description 1
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- NIFAOMSJMGEFTQ-UHFFFAOYSA-N 4-methoxybenzenethiol Chemical compound COC1=CC=C(S)C=C1 NIFAOMSJMGEFTQ-UHFFFAOYSA-N 0.000 description 1
- WLHCBQAPPJAULW-UHFFFAOYSA-N 4-methylbenzenethiol Chemical compound CC1=CC=C(S)C=C1 WLHCBQAPPJAULW-UHFFFAOYSA-N 0.000 description 1
- 125000001826 4H-pyranyl group Chemical group O1C(=CCC=C1)* 0.000 description 1
- VKLKXFOZNHEBSW-UHFFFAOYSA-N 5-[[3-[(4-morpholin-4-ylbenzoyl)amino]phenyl]methoxy]pyridine-3-carboxamide Chemical compound O1CCN(CC1)C1=CC=C(C(=O)NC=2C=C(COC=3C=NC=C(C(=O)N)C=3)C=CC=2)C=C1 VKLKXFOZNHEBSW-UHFFFAOYSA-N 0.000 description 1
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 1
- IRBAWVGZNJIROV-SFHVURJKSA-N 9-(2-cyclopropylethynyl)-2-[[(2s)-1,4-dioxan-2-yl]methoxy]-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=C2C3=CC=C(C#CC4CC4)C=C3CCN2C(=O)N=C1OC[C@@H]1COCCO1 IRBAWVGZNJIROV-SFHVURJKSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 241001120493 Arene Species 0.000 description 1
- IYHHRZBKXXKDDY-UHFFFAOYSA-N BI-605906 Chemical compound N=1C=2SC(C(N)=O)=C(N)C=2C(C(F)(F)CC)=CC=1N1CCC(S(C)(=O)=O)CC1 IYHHRZBKXXKDDY-UHFFFAOYSA-N 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 1
- ISMDILRWKSYCOD-GNKBHMEESA-N C(C1=CC=CC=C1)[C@@H]1NC(OCCCCCCCCCCCNC([C@@H](NC(C[C@@H]1O)=O)C(C)C)=O)=O Chemical compound C(C1=CC=CC=C1)[C@@H]1NC(OCCCCCCCCCCCNC([C@@H](NC(C[C@@H]1O)=O)C(C)C)=O)=O ISMDILRWKSYCOD-GNKBHMEESA-N 0.000 description 1
- WBOMLGWJPONAPF-UHFFFAOYSA-N CC(C)OC1=CC(C)C(c2c[s]c(NC(c3cc(F)ncc3)=O)n2)C(C)=C1 Chemical compound CC(C)OC1=CC(C)C(c2c[s]c(NC(c3cc(F)ncc3)=O)n2)C(C)=C1 WBOMLGWJPONAPF-UHFFFAOYSA-N 0.000 description 1
- GTGOGGSEIBVREH-UHFFFAOYSA-N CC(c(c(C)c1)c(C)cc1[O](C)=C)=O Chemical compound CC(c(c(C)c1)c(C)cc1[O](C)=C)=O GTGOGGSEIBVREH-UHFFFAOYSA-N 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- ZIJSGBFUAXAZBU-UHFFFAOYSA-N CCCCP(CCCC)c1ccccc1-c(c(C)cc(I)c1)c1-c1ccccc1 Chemical compound CCCCP(CCCC)c1ccccc1-c(c(C)cc(I)c1)c1-c1ccccc1 ZIJSGBFUAXAZBU-UHFFFAOYSA-N 0.000 description 1
- UHNRLQRZRNKOKU-UHFFFAOYSA-N CCN(CC1=NC2=C(N1)C1=CC=C(C=C1N=C2N)C1=NNC=C1)C(C)=O Chemical compound CCN(CC1=NC2=C(N1)C1=CC=C(C=C1N=C2N)C1=NNC=C1)C(C)=O UHNRLQRZRNKOKU-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- 101150012716 CDK1 gene Proteins 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- OMDMIOJTJAQBIY-UHFFFAOYSA-N Cc1c(-c2ccccc2)nc(NC(Cc(cccc2)c2OC)=O)[s]1 Chemical compound Cc1c(-c2ccccc2)nc(NC(Cc(cccc2)c2OC)=O)[s]1 OMDMIOJTJAQBIY-UHFFFAOYSA-N 0.000 description 1
- FIOHGIGGHSQKHU-UHFFFAOYSA-N Cc1cc(Nc(cc2)ccc2OC)cc(Cl)c1C(CBr)=O Chemical compound Cc1cc(Nc(cc2)ccc2OC)cc(Cl)c1C(CBr)=O FIOHGIGGHSQKHU-UHFFFAOYSA-N 0.000 description 1
- BCKGBXLJRXSHQF-UHFFFAOYSA-N Cc1cc(OCC(COC)O)cc(C)c1C(CBr)=O Chemical compound Cc1cc(OCC(COC)O)cc(C)c1C(CBr)=O BCKGBXLJRXSHQF-UHFFFAOYSA-N 0.000 description 1
- ZPWGZOKDTJNKRT-UHFFFAOYSA-N Cc1cc(Sc(cc2)ccc2OC)cc(C)c1-c1c[s]c(NC(c2cc(F)ncc2)=O)n1 Chemical compound Cc1cc(Sc(cc2)ccc2OC)cc(C)c1-c1c[s]c(NC(c2cc(F)ncc2)=O)n1 ZPWGZOKDTJNKRT-UHFFFAOYSA-N 0.000 description 1
- ZEOWTGPWHLSLOG-UHFFFAOYSA-N Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F Chemical compound Cc1ccc(cc1-c1ccc2c(n[nH]c2c1)-c1cnn(c1)C1CC1)C(=O)Nc1cccc(c1)C(F)(F)F ZEOWTGPWHLSLOG-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- VGCXGMAHQTYDJK-UHFFFAOYSA-N Chloroacetyl chloride Chemical compound ClCC(Cl)=O VGCXGMAHQTYDJK-UHFFFAOYSA-N 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 1
- 102000002427 Cyclin B Human genes 0.000 description 1
- 108010068150 Cyclin B Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 230000004543 DNA replication Effects 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- UIOFUWFRIANQPC-JKIFEVAISA-N Floxacillin Chemical compound N([C@@H]1C(N2[C@H](C(C)(C)S[C@@H]21)C(O)=O)=O)C(=O)C1=C(C)ON=C1C1=C(F)C=CC=C1Cl UIOFUWFRIANQPC-JKIFEVAISA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 101150072624 Haus8 gene Proteins 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001112162 Homo sapiens Kinetochore protein NDC80 homolog Proteins 0.000 description 1
- 101000777293 Homo sapiens Serine/threonine-protein kinase Chk1 Proteins 0.000 description 1
- 101000777277 Homo sapiens Serine/threonine-protein kinase Chk2 Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 102100023890 Kinetochore protein NDC80 homolog Human genes 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-L L-tartrate(2-) Chemical compound [O-]C(=O)[C@H](O)[C@@H](O)C([O-])=O FEWJPZIEWOKRBE-JCYAYHJZSA-L 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 101001026869 Mus musculus F-box/LRR-repeat protein 3 Proteins 0.000 description 1
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 1
- FFDGPVCHZBVARC-UHFFFAOYSA-N N,N-dimethylglycine Chemical class CN(C)CC(O)=O FFDGPVCHZBVARC-UHFFFAOYSA-N 0.000 description 1
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 1
- POFVJRKJJBFPII-UHFFFAOYSA-N N-cyclopentyl-5-[2-[[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]amino]-5-fluoropyrimidin-4-yl]-4-methyl-1,3-thiazol-2-amine Chemical compound C1(CCCC1)NC=1SC(=C(N=1)C)C1=NC(=NC=C1F)NC1=NC=C(C=C1)CN1CCN(CC1)CC POFVJRKJJBFPII-UHFFFAOYSA-N 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 101150033450 NUF2 gene Proteins 0.000 description 1
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 1
- KGCZWFOCVNAFFI-UHFFFAOYSA-N Nc1nc([AlH2])c[s]1 Chemical compound Nc1nc([AlH2])c[s]1 KGCZWFOCVNAFFI-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 108010051742 Platelet-Derived Growth Factor beta Receptor Proteins 0.000 description 1
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 235000004443 Ricinus communis Nutrition 0.000 description 1
- 101150094320 SPC25 gene Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 102100031081 Serine/threonine-protein kinase Chk1 Human genes 0.000 description 1
- 102100031075 Serine/threonine-protein kinase Chk2 Human genes 0.000 description 1
- LUSZGTFNYDARNI-UHFFFAOYSA-N Sesamol Natural products OC1=CC=C2OCOC2=C1 LUSZGTFNYDARNI-UHFFFAOYSA-N 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 1
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 102100039102 ZW10 interactor Human genes 0.000 description 1
- 101710177447 ZW10 interactor Proteins 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- VQXNKXDENJHSPI-UHFFFAOYSA-M [Br-].COC1=CC=C(C[Zn+])C=C1 Chemical compound [Br-].COC1=CC=C(C[Zn+])C=C1 VQXNKXDENJHSPI-UHFFFAOYSA-M 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- WREOTYWODABZMH-DTZQCDIJSA-N [[(2r,3s,4r,5r)-3,4-dihydroxy-5-[2-oxo-4-(2-phenylethoxyamino)pyrimidin-1-yl]oxolan-2-yl]methoxy-hydroxyphosphoryl] phosphono hydrogen phosphate Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O[C@H]1N(C=C\1)C(=O)NC/1=N\OCCC1=CC=CC=C1 WREOTYWODABZMH-DTZQCDIJSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- VJHCJDRQFCCTHL-UHFFFAOYSA-N acetic acid 2,3,4,5,6-pentahydroxyhexanal Chemical compound CC(O)=O.OCC(O)C(O)C(O)C(O)C=O VJHCJDRQFCCTHL-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000012382 advanced drug delivery Methods 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229950003476 aminothiazole Drugs 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 229940124599 anti-inflammatory drug Drugs 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- 239000005441 aurora Substances 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 description 1
- 239000007844 bleaching agent Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 230000024321 chromosome segregation Effects 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 238000000749 co-immunoprecipitation Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126086 compound 21 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125961 compound 24 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940127204 compound 29 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 229940126545 compound 53 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 229940125900 compound 59 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- CNXMDTWQWLGCPE-UHFFFAOYSA-N ditert-butyl-(2-phenylphenyl)phosphane Chemical group CC(C)(C)P(C(C)(C)C)C1=CC=CC=C1C1=CC=CC=C1 CNXMDTWQWLGCPE-UHFFFAOYSA-N 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000005553 drilling Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- GWNFQAKCJYEJEW-UHFFFAOYSA-N ethyl 3-[8-[[4-methyl-5-[(3-methyl-4-oxophthalazin-1-yl)methyl]-1,2,4-triazol-3-yl]sulfanyl]octanoylamino]benzoate Chemical compound CCOC(=O)C1=CC(NC(=O)CCCCCCCSC2=NN=C(CC3=NN(C)C(=O)C4=CC=CC=C34)N2C)=CC=C1 GWNFQAKCJYEJEW-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical compound [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 125000004447 heteroarylalkenyl group Chemical group 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005553 heteroaryloxy group Chemical group 0.000 description 1
- 125000005368 heteroarylthio group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 210000005119 human aortic smooth muscle cell Anatomy 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 238000010191 image analysis Methods 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- SNHMUERNLJLMHN-UHFFFAOYSA-N iodobenzene Chemical compound IC1=CC=CC=C1 SNHMUERNLJLMHN-UHFFFAOYSA-N 0.000 description 1
- 125000003010 ionic group Chemical group 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 201000010982 kidney cancer Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 239000012160 loading buffer Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 208000026037 malignant tumor of neck Diseases 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000031864 metaphase Effects 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- ZBELDPMWYXDLNY-UHFFFAOYSA-N methyl 9-(4-bromo-2-fluoroanilino)-[1,3]thiazolo[5,4-f]quinazoline-2-carboximidate Chemical compound C12=C3SC(C(=N)OC)=NC3=CC=C2N=CN=C1NC1=CC=C(Br)C=C1F ZBELDPMWYXDLNY-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 1
- YMZYXRXAOBGMQY-UHFFFAOYSA-N n-(4-acetyl-3,5-dimethylphenyl)acetamide Chemical compound CC(=O)NC1=CC(C)=C(C(C)=O)C(C)=C1 YMZYXRXAOBGMQY-UHFFFAOYSA-N 0.000 description 1
- KMKNTPXMXHHINF-UHFFFAOYSA-N n-(5-methyl-4-phenyl-1,3-thiazol-2-yl)pyridine-2-carboxamide Chemical compound N=1C(C=2C=CC=CC=2)=C(C)SC=1NC(=O)C1=CC=CC=N1 KMKNTPXMXHHINF-UHFFFAOYSA-N 0.000 description 1
- SRNJCTRFTUVKGW-UHFFFAOYSA-N n-(5-methyl-4-phenyl-1,3-thiazol-2-yl)pyridine-3-carboxamide Chemical compound N=1C(C=2C=CC=CC=2)=C(C)SC=1NC(=O)C1=CC=CN=C1 SRNJCTRFTUVKGW-UHFFFAOYSA-N 0.000 description 1
- VPLLUDJRXDCJOT-UHFFFAOYSA-N n-(5-methyl-4-phenyl-1,3-thiazol-2-yl)pyridine-4-carboxamide Chemical compound N=1C(C=2C=CC=CC=2)=C(C)SC=1NC(=O)C1=CC=NC=C1 VPLLUDJRXDCJOT-UHFFFAOYSA-N 0.000 description 1
- KRJQZMLWLUSKFN-UHFFFAOYSA-N n-[3,5-dimethyl-4-[2-(pyridin-4-ylcarbamoylamino)-1,3-thiazol-4-yl]phenyl]acetamide Chemical compound CC1=CC(NC(=O)C)=CC(C)=C1C1=CSC(NC(=O)NC=2C=CN=CC=2)=N1 KRJQZMLWLUSKFN-UHFFFAOYSA-N 0.000 description 1
- IFYDWYVPVAMGRO-UHFFFAOYSA-N n-[3-(dimethylamino)propyl]tetradecanamide Chemical compound CCCCCCCCCCCCCC(=O)NCCCN(C)C IFYDWYVPVAMGRO-UHFFFAOYSA-N 0.000 description 1
- HELXQPRCBAJGCY-UHFFFAOYSA-N n-[4-(2,4,6-trifluorophenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound FC1=CC(F)=CC(F)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 HELXQPRCBAJGCY-UHFFFAOYSA-N 0.000 description 1
- NXYSXNOVVUSONI-UHFFFAOYSA-N n-[4-(2,4,6-trimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound COC1=CC(OC)=CC(OC)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 NXYSXNOVVUSONI-UHFFFAOYSA-N 0.000 description 1
- HFRQICACJOUHNY-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]-1,3-thiazole-5-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2SC=NC=2)=N1 HFRQICACJOUHNY-UHFFFAOYSA-N 0.000 description 1
- SJVWQAPVYDABTO-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]-1h-indazole-5-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C3C=NNC3=CC=2)=N1 SJVWQAPVYDABTO-UHFFFAOYSA-N 0.000 description 1
- AJLVOWQOQWLZOZ-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]-1h-indazole-6-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C3NN=CC3=CC=2)=N1 AJLVOWQOQWLZOZ-UHFFFAOYSA-N 0.000 description 1
- XBOKYHYNVHCSQW-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]-2h-benzotriazole-5-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C3N=NNC3=CC=2)=N1 XBOKYHYNVHCSQW-UHFFFAOYSA-N 0.000 description 1
- HUZNECREVCYXED-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridazine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=NN=CC=2)=N1 HUZNECREVCYXED-UHFFFAOYSA-N 0.000 description 1
- RFUKJHFXBGDHNJ-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 RFUKJHFXBGDHNJ-UHFFFAOYSA-N 0.000 description 1
- NQCJVQQYSANGLV-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]pyrimidine-4-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C=2N=CN=CC=2)=N1 NQCJVQQYSANGLV-UHFFFAOYSA-N 0.000 description 1
- RKNKMACWCKMUQV-UHFFFAOYSA-N n-[4-(2,4,6-trimethylphenyl)-1,3-thiazol-2-yl]thiophene-3-carboxamide Chemical compound CC1=CC(C)=CC(C)=C1C1=CSC(NC(=O)C2=CSC=C2)=N1 RKNKMACWCKMUQV-UHFFFAOYSA-N 0.000 description 1
- BYUXMTPYIVDVBC-UHFFFAOYSA-N n-[4-(2,4,6-trimethylpyridin-3-yl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound CC1=NC(C)=CC(C)=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 BYUXMTPYIVDVBC-UHFFFAOYSA-N 0.000 description 1
- HPEMNVROIYZNAW-UHFFFAOYSA-N n-[4-(2,4,6-trimethylpyridin-3-yl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound CC1=NC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 HPEMNVROIYZNAW-UHFFFAOYSA-N 0.000 description 1
- ARTDELXQZXNTSB-UHFFFAOYSA-N n-[4-(2,4,6-trimethylpyridin-3-yl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=NC(C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 ARTDELXQZXNTSB-UHFFFAOYSA-N 0.000 description 1
- AMFXQEKIBYMRHQ-UHFFFAOYSA-N n-[4-(2,4-dimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound COC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 AMFXQEKIBYMRHQ-UHFFFAOYSA-N 0.000 description 1
- ARYUXVCPBFWHEZ-UHFFFAOYSA-N n-[4-(2,4-dimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound COC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 ARYUXVCPBFWHEZ-UHFFFAOYSA-N 0.000 description 1
- YDKDSJZAQDMTLR-UHFFFAOYSA-N n-[4-(2,4-dimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound COC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 YDKDSJZAQDMTLR-UHFFFAOYSA-N 0.000 description 1
- WOHMSRUPNMRNIZ-UHFFFAOYSA-N n-[4-(2,6-difluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound FC1=CC(OC)=CC(F)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 WOHMSRUPNMRNIZ-UHFFFAOYSA-N 0.000 description 1
- QTIATNOLSVCCKT-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-methylsulfanylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(SC)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 QTIATNOLSVCCKT-UHFFFAOYSA-N 0.000 description 1
- QYVFPUURHBXJJN-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-methylsulfonylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(S(C)(=O)=O)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 QYVFPUURHBXJJN-UHFFFAOYSA-N 0.000 description 1
- XBQOKTQMDBDKED-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-phenoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1OC1=CC=CC=C1 XBQOKTQMDBDKED-UHFFFAOYSA-N 0.000 description 1
- HQMIUYSQALRBDS-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-phenylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C=2C=CC=CC=2)=CC(C)=C1C(N=1)=CSC=1NC(=O)C1=CC=NC=C1 HQMIUYSQALRBDS-UHFFFAOYSA-N 0.000 description 1
- IQDDZEKWZBVUDM-UHFFFAOYSA-N n-[4-(2,6-dimethyl-4-phenylsulfanylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1SC1=CC=CC=C1 IQDDZEKWZBVUDM-UHFFFAOYSA-N 0.000 description 1
- LQUIXOFNXGEXHF-UHFFFAOYSA-N n-[4-(2-fluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]-3-phenylpropanamide Chemical compound FC1=CC(OC)=CC=C1C1=CSC(NC(=O)CCC=2C=CC=CC=2)=N1 LQUIXOFNXGEXHF-UHFFFAOYSA-N 0.000 description 1
- SABFSHVUXZGMAJ-UHFFFAOYSA-N n-[4-(2-fluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound FC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 SABFSHVUXZGMAJ-UHFFFAOYSA-N 0.000 description 1
- IDAKZFULEYLUHZ-UHFFFAOYSA-N n-[4-(2-fluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound FC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 IDAKZFULEYLUHZ-UHFFFAOYSA-N 0.000 description 1
- ZDXZWJIUVAFAMG-UHFFFAOYSA-N n-[4-(2-fluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound FC1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 ZDXZWJIUVAFAMG-UHFFFAOYSA-N 0.000 description 1
- ZBVMWSJQTDXZMN-UHFFFAOYSA-N n-[4-(3,4,5-trimethoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound COC1=C(OC)C(OC)=CC(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)=C1 ZBVMWSJQTDXZMN-UHFFFAOYSA-N 0.000 description 1
- ZBEMOLOVTYSXAH-UHFFFAOYSA-N n-[4-(3,5-difluoro-4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=C(F)C(OC)=C(F)C=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 ZBEMOLOVTYSXAH-UHFFFAOYSA-N 0.000 description 1
- JHKZQBHZJJZZJJ-UHFFFAOYSA-N n-[4-(4-chloro-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(Cl)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 JHKZQBHZJJZZJJ-UHFFFAOYSA-N 0.000 description 1
- URGMRSZTZCFZAN-UHFFFAOYSA-N n-[4-(4-cyclopentyloxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1OC1CCCC1 URGMRSZTZCFZAN-UHFFFAOYSA-N 0.000 description 1
- UYCMSMZGRYCFLT-UHFFFAOYSA-N n-[4-(4-ethoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]-2-fluoropyridine-4-carboxamide Chemical compound CC1=CC(OCC)=CC(C)=C1C1=CSC(NC(=O)C=2C=C(F)N=CC=2)=N1 UYCMSMZGRYCFLT-UHFFFAOYSA-N 0.000 description 1
- QEQIPPIJSXSONT-UHFFFAOYSA-N n-[4-(4-ethoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OCC)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 QEQIPPIJSXSONT-UHFFFAOYSA-N 0.000 description 1
- XNPVEXOHGDYZEK-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]-1,3-thiazole-5-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3SC=NC=3)SC=2)=C1 XNPVEXOHGDYZEK-UHFFFAOYSA-N 0.000 description 1
- SHPDGVUNLATABM-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]furan-3-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C3=COC=C3)SC=2)=C1 SHPDGVUNLATABM-UHFFFAOYSA-N 0.000 description 1
- NSVFXIAASWUSDS-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]pyridazine-4-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3C=NN=CC=3)SC=2)=C1 NSVFXIAASWUSDS-UHFFFAOYSA-N 0.000 description 1
- DPJCVAYPKLXIRE-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3N=CC=CC=3)SC=2)=C1 DPJCVAYPKLXIRE-UHFFFAOYSA-N 0.000 description 1
- YZOOIOMXRGZPCN-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3C=NC=CC=3)SC=2)=C1 YZOOIOMXRGZPCN-UHFFFAOYSA-N 0.000 description 1
- FVRJHRBXHLHVNT-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)=C1 FVRJHRBXHLHVNT-UHFFFAOYSA-N 0.000 description 1
- VFWQPAOOXUNACW-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]pyrimidine-4-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C=3N=CN=CC=3)SC=2)=C1 VFWQPAOOXUNACW-UHFFFAOYSA-N 0.000 description 1
- ZNGRGWIJBSIPAQ-UHFFFAOYSA-N n-[4-(4-hydroxy-3-methoxyphenyl)-1,3-thiazol-2-yl]thiophene-3-carboxamide Chemical compound C1=C(O)C(OC)=CC(C=2N=C(NC(=O)C3=CSC=C3)SC=2)=C1 ZNGRGWIJBSIPAQ-UHFFFAOYSA-N 0.000 description 1
- AGYCNBRKXDDKTO-UHFFFAOYSA-N n-[4-(4-hydroxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound C1=CC(O)=CC=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 AGYCNBRKXDDKTO-UHFFFAOYSA-N 0.000 description 1
- NUDUZDKLFVYUIU-UHFFFAOYSA-N n-[4-(4-hydroxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound C1=CC(O)=CC=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 NUDUZDKLFVYUIU-UHFFFAOYSA-N 0.000 description 1
- XOJGJGZPKRVMKE-UHFFFAOYSA-N n-[4-(4-hydroxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(O)=CC=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 XOJGJGZPKRVMKE-UHFFFAOYSA-N 0.000 description 1
- YASJCGYXCWIACW-UHFFFAOYSA-N n-[4-(4-hydroxyphenyl)-1,3-thiazol-2-yl]pyrimidine-4-carboxamide Chemical compound C1=CC(O)=CC=C1C1=CSC(NC(=O)C=2N=CN=CC=2)=N1 YASJCGYXCWIACW-UHFFFAOYSA-N 0.000 description 1
- UIJSBSLZBLHCJC-UHFFFAOYSA-N n-[4-(4-methoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]-1,3-thiazole-5-carboxamide Chemical compound CC1=CC(OC)=CC(C)=C1C1=CSC(NC(=O)C=2SC=NC=2)=N1 UIJSBSLZBLHCJC-UHFFFAOYSA-N 0.000 description 1
- JANCDTSYDMCUTJ-UHFFFAOYSA-N n-[4-(4-methoxy-2,6-dimethylphenyl)-1,3-thiazol-2-yl]-1-oxidopyridin-1-ium-4-carboxamide Chemical compound CC1=CC(OC)=CC(C)=C1C1=CSC(NC(=O)C=2C=C[N+]([O-])=CC=2)=N1 JANCDTSYDMCUTJ-UHFFFAOYSA-N 0.000 description 1
- VLQYAOPRYCLQFB-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 VLQYAOPRYCLQFB-UHFFFAOYSA-N 0.000 description 1
- YCNJJIPTBKGWCL-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 YCNJJIPTBKGWCL-UHFFFAOYSA-N 0.000 description 1
- OLJPHMZCLHQBTN-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 OLJPHMZCLHQBTN-UHFFFAOYSA-N 0.000 description 1
- IFCSODHNQRFTOY-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-5-methyl-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=C(C)SC(NC(=O)C=2N=CC=CC=2)=N1 IFCSODHNQRFTOY-UHFFFAOYSA-N 0.000 description 1
- XXPCXCKNZDYZOI-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-5-methyl-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=C(C)SC(NC(=O)C=2C=NC=CC=2)=N1 XXPCXCKNZDYZOI-UHFFFAOYSA-N 0.000 description 1
- FROZTBAKRFXALP-UHFFFAOYSA-N n-[4-(4-methoxyphenyl)-5-methyl-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1C1=C(C)SC(NC(=O)C=2C=CN=CC=2)=N1 FROZTBAKRFXALP-UHFFFAOYSA-N 0.000 description 1
- DAIASWMZQYEEEC-UHFFFAOYSA-N n-[4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-2-carboxamide Chemical compound C1=CC(C)=CC=C1C1=CSC(NC(=O)C=2N=CC=CC=2)=N1 DAIASWMZQYEEEC-UHFFFAOYSA-N 0.000 description 1
- VKDYUSUSRNWMMK-UHFFFAOYSA-N n-[4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound C1=CC(C)=CC=C1C1=CSC(NC(=O)C=2C=NC=CC=2)=N1 VKDYUSUSRNWMMK-UHFFFAOYSA-N 0.000 description 1
- PWDRCRZKQHYTSF-UHFFFAOYSA-N n-[4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(C)=CC=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 PWDRCRZKQHYTSF-UHFFFAOYSA-N 0.000 description 1
- IFOGPYRPRNNYNX-UHFFFAOYSA-N n-[4-[2,4,6-tri(propan-2-yl)phenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 IFOGPYRPRNNYNX-UHFFFAOYSA-N 0.000 description 1
- YIXVNJIOWHNFMV-UHFFFAOYSA-N n-[4-[2,6-dimethyl-4-(2-methylpropoxy)phenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OCC(C)C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 YIXVNJIOWHNFMV-UHFFFAOYSA-N 0.000 description 1
- PTGQNPNZEHGRNH-UHFFFAOYSA-N n-[4-[2,6-dimethyl-4-(4-methylphenoxy)phenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(C)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 PTGQNPNZEHGRNH-UHFFFAOYSA-N 0.000 description 1
- DKELVNSHEABYFO-UHFFFAOYSA-N n-[4-[2,6-dimethyl-4-(4-methylphenyl)sulfanylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(C)=CC=C1SC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 DKELVNSHEABYFO-UHFFFAOYSA-N 0.000 description 1
- WSHZTCRPQKJVTG-UHFFFAOYSA-N n-[4-[4-(1,3-benzodioxol-5-yloxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OC=2C=C3OCOC3=CC=2)=CC(C)=C1C(N=1)=CSC=1NC(=O)C1=CC=NC=C1 WSHZTCRPQKJVTG-UHFFFAOYSA-N 0.000 description 1
- PEQHIFUZHRJYJO-UHFFFAOYSA-N n-[4-[4-(2,3-dihydroxypropoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OCC(O)CO)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 PEQHIFUZHRJYJO-UHFFFAOYSA-N 0.000 description 1
- QRMBNAKRAWQWEF-UHFFFAOYSA-N n-[4-[4-(2-methoxyethoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OCCOC)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 QRMBNAKRAWQWEF-UHFFFAOYSA-N 0.000 description 1
- PAKQVEFAIXGQRH-UHFFFAOYSA-N n-[4-[4-(2-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound COC1=CC=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 PAKQVEFAIXGQRH-UHFFFAOYSA-N 0.000 description 1
- JEITUTCWOZUUCM-UHFFFAOYSA-N n-[4-[4-(3,5-dimethylphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(C)=CC(OC=2C=C(C)C(C=3N=C(NC(=O)C=4C=CN=CC=4)SC=3)=C(C)C=2)=C1 JEITUTCWOZUUCM-UHFFFAOYSA-N 0.000 description 1
- KUUJPJWJXWVKAN-UHFFFAOYSA-N n-[4-[4-(3-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound COC1=CC=CC(OC=2C=C(C)C(C=3N=C(NC(=O)C=4C=CN=CC=4)SC=3)=C(C)C=2)=C1 KUUJPJWJXWVKAN-UHFFFAOYSA-N 0.000 description 1
- LUXYBMINOUBUHK-UHFFFAOYSA-N n-[4-[4-(3-methoxypropoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound CC1=CC(OCCCOC)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 LUXYBMINOUBUHK-UHFFFAOYSA-N 0.000 description 1
- LBIBQNISKGDKRN-UHFFFAOYSA-N n-[4-[4-(4-bromoanilino)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1NC1=CC=C(Br)C=C1 LBIBQNISKGDKRN-UHFFFAOYSA-N 0.000 description 1
- XRCPLNCSHVCFOS-UHFFFAOYSA-N n-[4-[4-(4-ethylphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(CC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 XRCPLNCSHVCFOS-UHFFFAOYSA-N 0.000 description 1
- GKDUQUYBQGBCIK-UHFFFAOYSA-N n-[4-[4-(4-fluorophenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C=1C(C)=C(C=2N=C(NC(=O)C=3C=CN=CC=3)SC=2)C(C)=CC=1OC1=CC=C(F)C=C1 GKDUQUYBQGBCIK-UHFFFAOYSA-N 0.000 description 1
- FIMSGPVUOARWQY-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]-1-oxidopyridin-1-ium-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=C[N+]([O-])=CC=2)=N1 FIMSGPVUOARWQY-UHFFFAOYSA-N 0.000 description 1
- IVWIIXKDRITPBE-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenoxy)-2,6-dimethylphenyl]-1,3-thiazol-2-yl]-3-methylpyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1OC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C(=CN=CC=2)C)=N1 IVWIIXKDRITPBE-UHFFFAOYSA-N 0.000 description 1
- DXOHHAQAPLLAMF-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenyl)sulfanyl-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1SC(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 DXOHHAQAPLLAMF-UHFFFAOYSA-N 0.000 description 1
- RRDSDIQVXRIAQD-UHFFFAOYSA-N n-[4-[4-(4-methoxyphenyl)sulfonyl-2,6-dimethylphenyl]-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)C(C=C1C)=CC(C)=C1C1=CSC(NC(=O)C=2C=CN=CC=2)=N1 RRDSDIQVXRIAQD-UHFFFAOYSA-N 0.000 description 1
- MBGWUDAJDVNMKD-UHFFFAOYSA-N n-[5-methyl-4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-3-carboxamide Chemical compound N=1C(C=2C=CC(C)=CC=2)=C(C)SC=1NC(=O)C1=CC=CN=C1 MBGWUDAJDVNMKD-UHFFFAOYSA-N 0.000 description 1
- MQKFNCKVAXIASY-UHFFFAOYSA-N n-[5-methyl-4-(4-methylphenyl)-1,3-thiazol-2-yl]pyridine-4-carboxamide Chemical compound N=1C(C=2C=CC(C)=CC=2)=C(C)SC=1NC(=O)C1=CC=NC=C1 MQKFNCKVAXIASY-UHFFFAOYSA-N 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 229920002113 octoxynol Polymers 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- ZJVAWPKTWVFKHG-UHFFFAOYSA-N p-Methoxypropiophenone Chemical compound CCC(=O)C1=CC=C(OC)C=C1 ZJVAWPKTWVFKHG-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229940124583 pain medication Drugs 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- QULYNCCPRWKEMF-UHFFFAOYSA-N parachlorobenzotrifluoride Chemical compound FC(F)(F)C1=CC=C(Cl)C=C1 QULYNCCPRWKEMF-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 125000003884 phenylalkyl group Chemical group 0.000 description 1
- 150000008105 phosphatidylcholines Chemical class 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 125000000394 phosphonato group Chemical group [O-]P([O-])(*)=O 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 229960004063 propylene glycol Drugs 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 210000000512 proximal kidney tubule Anatomy 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Chemical class COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- KSAVQLQVUXSOCR-UHFFFAOYSA-M sodium lauroyl sarcosinate Chemical compound [Na+].CCCCCCCCCCCC(=O)N(C)CC([O-])=O KSAVQLQVUXSOCR-UHFFFAOYSA-M 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 230000020347 spindle assembly Effects 0.000 description 1
- 206010041823 squamous cell carcinoma Diseases 0.000 description 1
- 208000017572 squamous cell neoplasm Diseases 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000005307 thiatriazolyl group Chemical group S1N=NN=C1* 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 125000002813 thiocarbonyl group Chemical group *C(*)=S 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 230000004565 tumor cell growth Effects 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- HOFQVRTUGATRFI-XQKSVPLYSA-N vinblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 HOFQVRTUGATRFI-XQKSVPLYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 238000012447 xenograft mouse model Methods 0.000 description 1
- 238000001086 yeast two-hybrid system Methods 0.000 description 1
- WRSWIWOVJBYZAW-UHFFFAOYSA-M zinc;methanidylbenzene;bromide Chemical compound Br[Zn+].[CH2-]C1=CC=CC=C1 WRSWIWOVJBYZAW-UHFFFAOYSA-M 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Изобретение относится к соединению, имеющему структуру формулы I, где R1 представляет собой алкокси, ORa, SRa, -S(O)Ra, или -S(O)2Ra; где Ra представляет собой алкил или арил, необязательно замещенный алкилом, галогеналкилом, алкокси и/или галогеном; R2, R3 представляют собой C1-C6алкил; R4 представляет собой водород или C1-C6алкил; R5 представляет собой пиридинил, необязательно замещенный галогеном. Изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение формулы I в концентрации, эффективной для нарушения связывания Hec1/Nek2 у пациента, когда композицию вводят пациенту. Соединения I предназначены для применения при получении лекарственного средства для лечения заболевания, ассоциированного с дисфункцией и/или сверхэкспрессией Hec1, где соединение присутствует в лекарственном средстве в количестве, эффективном для нарушения связывания Nek2/Hec1. Технический результат - соединения, предназначенные для нарушения связывания Nek2/Hec1. 4 н. и 5 з.п. ф-лы, 10 ил., 6 табл.
Description
По данной заявке испрашивается приоритет по находящейся на совместном рассмотрении временной заявке авторов настоящего изобретения с серийным номером 61/314798, которая была подана 17 марта 2010 года.
Область изобретения
Областью изобретения являются различные соединения, композиции и способы, связанные с модулированием активности HEC1, особенно в отношении ингибирования размножения опухолевых клеток.
Уровень техники
Хотя механизмы, ассоциированные с митотической регуляцией, концептуально являются привлекательной мишенью в попытках уменьшить рост опухолевых клеток, соединения с высокой удельной активностью и селективностью и желаемым фармакологическим профилем являются труднодостижимыми. Например, на аппарат веретена деления можно осуществлять нацеливание с помощью действующих на веретено деления ядов (например, таксаны, алкалоиды барвинка и т.д.) с относительно высокой активностью, однако многие действующие на веретено деления яды являются неприемлемыми для фармацевтического вмешательства, поскольку такие яды часто являются неспецифическими.
Для увеличения специфичности лечения можно выбирать компоненты для регуляции веретена деления и кинетохора или регуляции митотической системы контроля, для которых было показано, что они являются функционально ассоциированными со злокачественной опухолью. Например, Hec1 является важным компонентом передачи сигнала в системе контроля веретена деления, который в высокой степени экспрессируется при злокачественной опухоли и помогает обеспечить правильное расхождение хромосом в процессе деления клеток. Hec1 взаимодействует с различными другими компонентами кинетохора, включая Nuf2, Spc 24, Spc25 и Zwint-1, а также с митотическими киназами Nek2 и Aurora B. Сверхэкспрессия Hec1 является частой среди большого множества злокачественных опухолей и злокачественных клеточных линий, и часто она может служить в качестве прогностического маркера при первичном раке молочной железы и других злокачественных опухолях. Исходя из кажущейся важности Hec1 при росте опухолевых клеток, используют РНКi для снижения экспрессии Hec1, и это оказалось достаточно перспективным, по меньшей мере в моделях на животных. Однако доставка миРНК in vivo с высокой специфичностью в опухоль часто является проблематичной.
Позднее были разработаны различные низкомолекулярные ингибиторы, которые препятствуют взаимодействию Nek2/Hec1. Поскольку Nek2 является регуляторным компонентом Hec1 при митозе, ожидалось, что устранение функции Hec1/Nek2 приведет к неправильному расхождению хромосом и гибели клеток. Описано несколько перспективных соединений (см. J. Med. Chem., 2009, 52 (6), pp 1757-1767, Cancer Res. 2008 Oct 15; 68(20):8393-9), которые имели значительную активность уничтожения клеток и были прямо нацелены на каскад Hec1/Nek2. Этот и все другие посторонние материалы, обсуждаемые в настоящем описании, включены в качестве ссылки в полном объеме. Когда определение или использование термина во включенной ссылке не соответствует или противоречит определению этого термина, предоставленному в настоящем описании, применяется определение этого термина, предоставленное в настоящем описании, и определение этого термина в ссылке не применяется. Однако, хотя наблюдаемая активность была по меньшей мере в некоторых случаях перспективной, тем не менее, оставались проблемы, ассоциированные с растворимостью, токсичностью и относительно высокими полумаксимальными ингибиторными концентрациями.
Таким образом, все еще остается потребность в усовершенствованных соединениях, композициях и способах для ингибирования Hec1, особенно в отношении применения таких соединений для лечения злокачественной опухоли.
Сущность изобретения
Объектом изобретения являются различные соединения, композиции и способы для ингибирования Hec1. Более конкретно, предусматриваемые соединения включают соединения формулы I

где R1, R2, R3, R4 и R5 описаны дополнительно ниже. Кроме того, особенно предпочтительные соединения имеют структуру формул II и III (где соответствующие радикалы также более подробно описаны ниже).

В одном аспекте объекта изобретения рассматриваемые соединения представляют собой ингибиторы Hec1, и/или они могут быть охарактеризованы как нарушающие взаимодействие Hec1/Nek2. Следовательно, соединения, представленные в настоящем описании, особенно пригодны для применения в качестве лекарственных средств, которые нарушают каскад митоза. Таким образом и с другой точки зрения, главным образом, рассматриваемые композиции включают фармацевтические композиции, которые содержат одно или несколько из рассматриваемых соединений в концентрации, эффективной для нарушения связывания Hec1/Nek2 у пациента, когда композицию вводят пациенту.
Таким образом, в другом аспекте объекта изобретения предусмотрен способ нарушения взаимодействия Nek2/Hec1, и он включает стадию контактирования комплекса Nek2/Hec1 с одним или несколькими соединениями, представленными в настоящем описании, в количестве, которое является эффективным для нарушения связывания Nek2/Hec1. Хотя, главным образом, предусматриваются все пути контактирования, как правило, предпочтительно, чтобы стадию контактирования комплекса Nek2/Hec1 проводили in vivo у млекопитающего, и чтобы стадию контактирования также можно было проводить в комбинации со средством, которое препятствует образованию или деградации микротрубочек.
Различные задачи, признаки, аспекты и преимущества объекта изобретения станут более понятными из представленного ниже подробного описания предпочтительных вариантов осуществления, а также из прилагаемых чертежей, на которых подобные цифры соответствуют подобным компонентам.
Краткое описание чертежей
На фиг.1A и 1B представлены таблицы, иллюстрирующие цитотоксический эффект выбранных соединений на опухолевые клетки (1A) и нормальные клетки (1B).
На фиг.2A-2D представлены фотографии вестерн-блотов, на которых представлено нарушение взаимодействия Hec1/Nek2 (2A, 2B), деградация Nek2 (2C) и нестабильность Nek2 (2D), вызванная выбранными соединениями.
На фиг.3 представлена таблица, иллюстрирующая процент митотических клеток, поврежденных рассматриваемыми соединениями.
На фиг.4 представлена таблица, иллюстрирующая высокую специфичность рассматриваемых соединений в отношении протеинкиназ.
На фиг.5A и 5B представлены графики, на которых изображен эффект выбранных соединений in vivo на объем опухоли у мышей nude.
Подробное описание
Рассматриваемые соединения
Авторы изобретения открыли, что можно получить определенные соединения формулы I, и что они имеют преимущественные свойства в качестве структур, которые препятствуют Hec1. Особенно предпочтительные соединения включают соединения формулы I

В особенно предпочтительных аспектах R1 представляет собой водород, алкил, алкенил, алкинил, алкокси, арил, галоген, нитро, циано, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, ORa, SRa, NRaRb, -S(O)2Ra, -S(O)2NRaRb, -C(O)Ra, -C(O)NRaRb, -NRaC(O)Rb, -NRaS(O)2Rb, -N=CRaRb или -NRaC(O)NHRb; Ra и Rb независимо представляют собой водород, алкил, алкенил, алкинил, арил, арилокси, алкокси, гидрокси, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил или гетероциклоалкенил, или Ra и Rb, вместе с атомом азота, к которому они присоединены, представляют собой гетероарил, гетероциклоалкил или гетероциклоалкенил; R2, R3 и R4 независимо представляют собой водород, C1-C6алкил, галоген или ORa; и R5 представляет собой алкил, фенилалкил, гетероарилалкил, фенилалкенил, гетероарилалкенил, фенил, гетероарил, гетероциклоалкил или гетероциклоалкенил; где каждый из R1, R2, R3, R4, R5, Ra и Rb независимо необязательно является замещенным. Менее предпочтительные соединения включают соединения, где (I) R1 и R2 представляют собой метил и где R3 представляет собой водород, R5 не является тиазолилом, N-метилимидазолилом, пиразинилом, пиридинилом, морфолинилом, фенилом или диметоксифенилом; где (II) R1, R2 и R3 представляют собой метил, R5 не является тиазолилом, N-метилимидазолилом, пиразинилом, пиридинилом, морфолинилом, фенилом, метоксифенилом, дигидроксифенилом, гидроксиметоксифенилом, трифторметилфенилом или диметоксифенилом; и где (III) R1 и R2 представляют собой метил и где R3 представляет собой гидроксил или метокси, R5 не является фенилом.
Особенно предпочтительно, чтобы R1 представлял собой алкокси, SRa, ORa или -S(O)2Ra, чтобы Ra представлял собой алкил или необязательно замещенный арил, чтобы R2, R3 и R4 независимо представляли собой водород или C1-C6алкил, и чтобы R5 представлял собой необязательно замещенный гетероарил. Еще более предпочтительными соединениями среди них являются соединения, где R1 представляет собой алкокси, SRa, ORa или -S(O)2Ra, где Ra представляет собой алкил или необязательно замещенный арил, где R2 и R3 представляют собой C1-C6алкил, и где R5 представляет собой необязательно замещенный (например, галогенированный) пиридинил. Наиболее предпочтительно, R1 представляет собой ORa, где Ra представляет собой необязательно замещенный арил, R2 и R3 представляют собой C1-C6алкил, и R5 представляет собой необязательно замещенный пиридинил.
Следовательно, с другой точки зрения, также являются предпочтительными соединения, которые имеют структуру согласно формуле II

где X1 и X2 независимо представляют собой H, алкил, алкенил, алкинил, галоген, нитро, циано, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, ORa, NRaRb, -S(O)2Ra, -S(O)2NRaRb, -C(O)Ra, -C(O)NRaRb, -NRaC(O)Rb, -NRaS(O)2Rb, -N=CRaRb или -NRaC(O)NHRb; Y представляет собой CH2, CHRa, CRaRb, O, NH, NRa, S, SO или SO2; R1, R2 и R3 независимо представляют собой H, алкил, алкокси или галоген; n равно 0, 1 или 2; и в которых A представляет собой необязательно замещенный арил или необязательно замещенный гетероарил, и наиболее предпочтительно соединение, представленное ниже

где каждый из X1 и X2 независимо необязательно является замещенным, и где Rc и Rd независимо представляют собой Ra. Среди таких соединений, кроме того, предпочтительно, чтобы Y представлял собой O, S или SO2, и/или чтобы A представлял собой необязательно замещенный пиридинил. Наиболее часто X1 и X2 в таких соединениях независимо представляют собой H, алкил и алкокси, и n равно 0 или 1. Что касается остальных радикалов, применимы те же принципы, которые предоставлены для формулы I.
Следующие предпочтительные соединения имеют структуру формулы III

где X1, X2 и X3 независимо представляют собой H, алкил, алкенил, алкинил, галоген, нитро, циано, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, ORa, NRaRb, -S(O)2Ra, -S(O)2NRaRb, -C(O)Ra, -C(O)NRaRb, -NRaC(O)Rb, -NRaS(O)2Rb, -N=CRaRb или -NRaC(O)NHRb; Y представляет собой CH2, CHRa, CRaRb, O, NH, NRa, S, SO или SO2; R1, R2 и R3 независимо представляют собой H, алкил, алкокси или галоген; n представляет собой 0, 1 или 2; где каждый из X1 и X2 независимо необязательно является замещенным; где Rc и Rd независимо представляют собой Ra, и в которых A и Het независимо и предпочтительно представляют собой ароматический и необязательно замещенный арил или гетероарил. Среди других пригодных выборов, обычно предпочтительно, чтобы

и чтобы

Что касается остальных радикалов, применяются те же принципы, как и для формулы I. Особенно предпочтительные соединения формулы II включают соединения, в которых A представляет собой необязательно замещенный пиридинил, и/или в которых Y представляет собой O, S или SO2.
Ввиду вышесказанного и дополнительных экспериментальных данных (см. ниже), особенно предпочтительные соединения имеют структуру, как показано ниже


Термин "алкил", как используют в рамках изобретения, относится к углеводородному радикалу, который может быть прямым, циклическим или разветвленным. Термин "алкенил" относится к алкилу, имеющему по меньшей мере одну двойную связь. Когда присутствует более одной двойной связи, предусматривается, что двойные связи могут быть сопряженными или несопряженными. Термин "алкинил", как используют в рамках изобретения, относится к алкилу, имеющему по меньшей мере одну тройную связь. Предусматриваемые алкинилы, кроме того, могут включать другую тройную связь или двойную связь, которая может быть сопряженной или может не быть сопряженной с первой тройной связью. Термин "алкокси", как используют в рамках изобретения, относится к O-алкильной группе, где "алкил" определяют, как указано выше.
"Циклоалкил", как используют в рамках изобретения, относится к неароматическому одновалентному моноциклическому или полициклическому радикалу, имеющему от 3 до 14 атомов углерода, каждый из которых может быть насыщенным или ненасыщенным, и может быть незамещенным или замещенным одним или несколькими пригодными заместителями, как определено в настоящем описании, и с которым может быть конденсирована одна или несколько арильных групп, гетероарильных групп, циклоалкильных групп или гетероциклоалкильных групп, которые сами по себе могут быть незамещенными или замещены одним или несколькими заместителями. Примеры циклоалкильных групп включают циклопропил, циклогептил, циклооктил, циклодецил, циклобутил, адамантил, норпинанил, декалинил, норборнил, циклогексил и циклопентил.
"Гетероциклоалкил", как используют в рамках изобретения, относится к неароматическому одновалентному моноциклическому или полициклическому радикалу, имеющему 1-5 гетероатомов, выбранных из азота, кислорода и серы, и он может быть незамещенным или замещен одним или несколькими пригодными заместителями, как определено в настоящем описании, и с которым может быть конденсирована одна или несколько арильных групп, гетероарильных групп, циклоалкильных групп или гетероциклоалкильных групп, которые сами по себе могут быть незамещенными или замещены одним или несколькими заместителями. Примеры гетероциклоалкильных групп включают оксиранил, пирролидинил, пиперидил, тетрагидропиран и морфолинил.
"Арил" (Ar), как используют в рамках изобретения, относится к ароматическому моноциклическому или полициклическому радикалу, содержащему, как правило, от 5 до 18 членов углеродного кольца, который может быть незамещенным или замещен одним или несколькими пригодными заместителями, как определено в настоящем описании, и с которым может быть конденсирована одна или несколько циклоалкильных групп, гетероциклоалкильных групп или гетероарильных групп, которые сами по себе могут быть незамещенными или замещены одним или несколькими пригодными заместителями. Таким образом, термин "арильная группа" включает бензильную группу (Bzl). Примеры включают фенил, бифенил, 1,2,3,4-тетрагидронафтил, нафтил, антрил и фенантрил.
"Гетероарил", как используют в рамках изобретения, относится к ароматическому моноциклическому или полициклическому радикалу, содержащему, главным образом, от 4 до 18 членов кольца, включая 1-5 гетероатомов, выбранных из азота, кислорода и серы, который может быть незамещенным или замещен одним или несколькими пригодными заместителями, как определено ниже, и с которым может быть конденсирована одна или несколько циклоалкильных групп, гетероциклоалкильных групп или арильных групп, которые сами по себе могут быть незамещенными или замещены одним или несколькими пригодными заместителями. Примеры включают тиенил, фуранил, тиазолил, триазолил, имидазолил, изоксазолил, оксадиазолил, тетразолил, пиридил, пирролил, тиадиазолил, оксадиазолил, оксатиадиазолил, тиатриазолил, пиримидинил, изохинолинил, хинолинил, нафтиридинил, фталимидил, бензимидазолил и бензоксазолил.
Термин "гетероцикл" или "гетероциклический", как используют в рамках изобретения, относится к ароматическим и неароматическим гетероциклическим группам, как правило, с 4-10 атомами, образующими кольцо, и содержащим один или несколько гетероатомов (как правило, O, S или N). Неароматические гетероциклические группы включают группы, имеющие только 4 атома в их кольцевой системе, однако ароматические гетероциклические группы, как правило, имеют по меньшей мере 5 атомов в их кольцевой системе. Примеры неароматических гетероциклических групп включают пирролидинил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, пиперидино, морфолино, тиоморфолино, тиоксанил, пиперазинил, азетидинил, оксетанил, тиетанил, гомопиперидинил, оксепанил, тиепанил, оксазепинил, диазепинил, тиазепинил, 1,2,3,6-тетрагидропиридинил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, дитианил, дитиоланил, дигидропиранил, дигидротиенил, дигидрофуранил, пиразолидинил, имидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3H-индолил и хинолизинил.
Примерами ароматических гетероциклических групп являются пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, изобензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, хиназолинил, бензотиазолил, бензоксазолил, хиноксалинил, нафтиридинил и фуропиридинил. Рассматриваемые 4-10-членные гетероциклы могут быть C-связанными или N-связанными (когда это целесообразно). Например, группа, образованная из пиррола, может представлять собой пиррол-1-ил (N-связанный) или пиррол-3-ил (C-связанный).
Далее, как используют в рамках изобретения, термин "замещенный", как используют в рамках изобретения, относится к замене или модификации атома (радикала) или химической группы (например, NH2 или OH) в молекуле функциональной группой с получением замещенной молекулы, и, в частности, рассматриваемые функциональные группы включают нуклеофильные группы (например, -NH2, -OH, -SH, -NC и т.д.), электрофильные группы (например, C(O)OR, C(O)OH и т.д.), полярные группы (например, -OH), неполярные группы (например, арил, алкил, алкенил, алкинил и т.д.), ионные группы (например, -NH3 +), и галогены (например, -F, -Cl), и все их химически допустимые комбинации. Например, когда молекула представляет собой алкил, замещенный радикал представляет собой водородный радикал, и функциональная группа представляет собой гидроксильную группу, атом H замещен OH-группой с образованием замещенного алкила. В другом примере, когда молекула представляет собой аминокислоту, модифицированная группа представляет собой аминогруппу и функциональная группа представляет собой алкильную группу, аминогруппа является алкилированной с образованием N-замещенной аминокислоты.
Например, пригодные заместители включают галоген (хлор, йод, бром или фтор); C1-6алкил; C1-6алкенил; C1-6алкинил, гидроксил, C1-6алкоксил; амино; нитро; тиол; простой тиоэфир; имин; циано; амидо; фосфонато; фосфин; карбоксил; карбонил; аминокарбонил; тиокарбонил; сульфонил; сульфонамин; сульфонамид; кетон; альдегид; сложный эфир; кислород (=O); галогеналкил (например, трифторметил); карбоциклический циклоалкил, который может быть моноциклическим или конденсированным или неконденсированным полициклическим радикалом (например, циклопропил, циклобутил, циклопентил или циклогексил), или гетероциклоалкил, который может быть моноциклическим или конденсированным или неконденсированным полициклическим радикалом (например, пирролидинил, пиперидинил, пиперазинил, морфолинил или тиазинил); карбоциклическим или гетероциклическим, моноциклическим или конденсированным или неконденсированным полициклическим арилом (например, фенил, нафтил, пирролил, индолил, фуранил, тиофенил, имидазолил, оксазолил, изоксазолил, тиазолил, триазолил, тетразолил, пиразолил, пиридинил, хинолинил, изохинолинил, акридинил, пиразинил, пиридазинил, пиримидинил, бензимидазолил, бензотиофенил или бензофуранил); амино (первичный, вторичный или третичный); нитро; тиол; простой тиоэфир, O-низший алкил; O-арил, арил; арил-низший алкил; CO2CH3; CONH2; OCH2CONH2; NH2; SO2NH2; OCHF2; CF3; OCF3; и т.д. Кроме того, следует отметить, что все заместители, предусматриваемые в рамках изобретения, кроме того, могут быть необязательно замещены одним или несколькими заместителями, указанными выше. Особенно предпочтительные заместители включают гидроксильные группы, галогены, оксогруппы, алкильные группы (и особенно низший алкил), ацильные группы, сульфонильные группы, меркаптогруппы, алкилтиогруппы, алкилоксильные группы, циклоалкильные группы, гетероциклоалкильные группы, арильные группы, гетероарильные группы, карбоксильные группы, аминогруппы, алкиламиногруппы, диалкиламиногруппы, карбамоильные группы, арилоксильные группы, гетероарилоксильные группы, арилтиогруппы, гетероарилтиогруппы.
Более того, должно быть понятно, что соединения согласно объекту изобретения содержат один или несколько асимметричных центров, и, таким образом, могут существовать в различных энантиомерных формах, и все энантиомерные формы рассматриваемых соединений конкретно предусмотрены в рамках изобретения. Аналогично, когда рассматриваемые соединения проявляют оптическую активность и/или имеют стереоизомеры, все случаи оптической активности и/или изомерные формы предусмотрены в рамках изобретения. Когда двойные связи отличают Z-форму от E-формы (или цис- от транс-), предусматриваются оба изомера. Более того, следует отметить, что соединения согласно объекту изобретения также могут быть изотопно меченными. Примерами пригодных изотопов являются 2H, 3H, 13C, 14C, 15N, 18O, 17O, 18F или 36Cl. Определенные изотопно меченные соединения согласно объекту изобретения, например, соединения, в которые включен 14C или 3H, могут быть пригодны в анализах распределения в тканях лекарственного средства и/или субстрата. С другой стороны, замена нерадиоактивными изотопами (например, 2H или 13C) может обеспечить определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличенное время полужизни in vivo или сниженные потребности в дозировках и, таким образом, они могут быть предпочтительными в некоторых случаях.
Рассматриваемые соединения можно получать в качестве фармацевтически приемлемой соли(ей), которая, главным образом, включает соли кислотных или основных групп, которые могут присутствовать в рассматриваемых соединениях. Например, когда рассматриваемые соединения являются основными, следует отметить, что такие соединения могут образовывать различные соли с различными неорганическими и органическими кислотами. Пригодные кислоты обеспечивают фармакологически приемлемые анионы, включая хлоридный, бромидный, йодидный, нитратный, сульфатный, бисульфатный, фосфатный, кислый фосфатный, изоникотинатный, ацетатный, лактатный, салицилатный, цитратный, кислый цитратный, тартратный, пантотенатный, битартратный, аскорбатный, сукцинатный, малеатный, гентисинатный, фумаратный, глюконатный, глюкаронатный, сахаратный, формиатный, бензоатный, глутаматный, метансульфонатный, этансульфонатный, бензолсульфонатный, п-толуолсульфонатный и памоатный [1,1'-метилен-бис-(2-гидрокси-3-нафтоатный)] анионы. Аналогично, когда рассматриваемые соединения являются кислотными, следует отметить, что такие соединения могут образовывать основные соли с различными фармакологически приемлемыми катионами, и особенно пригодные катионы включают ионы щелочных металлов или щелочноземельных металлов (например, катионы натрия и калия).
В следующих предусматриваемых аспектах соединения, представленные в настоящем описании, можно получать в качестве пролекарств, и все известные формы и типы пролекарств считаются пригодными для применения в настоящем описании при условии, что такое пролекарство увеличивает концентрацию лекарственного средства (или метаболита пролекарства) в органе-мишени, клетке-мишени и/или Hec1. Например, когда рассматриваемые соединения имеют свободную амино, амидо, гидрокси, тио или карбоновую группу, предусматривается, что такие группы можно использовать для ковалентного и поддающегося высвобождению связывания группы, которая конвертирует лекарственное средство в пролекарство. Таким образом, пролекарства, в частности, включают пролекарства, в которых рассматриваемые соединения образуют сложноэфирную, амидную или дисульфидную связь с другой расщепляемой группой. Такие группы могут способствовать специфической доставке в орган или клетку лекарственного средства и, таким образом, в частности, включают лиганды рецепторов и их аналоги, фрагменты антител или другие высокоаффинные лиганды (Kd<106 М).
Например, карбоксильная группа может быть образована из амида или алкильного сложного эфира, которые могут включать простую эфирную группу, аминогруппу и/или группу карбоновой кислоты. Свободные гидроксильные группы могут быть образованы с использованием гемисукцинатов, фосфатных сложных эфиров, диметиламиноацетатов и фосфорилоксиметилоксикарбонилов, как описано в D. Fleisher, R. Bong, B. H. Stewart, Advanced Drug Delivery 40 Reviews (1996) 19, 115. Также включены карбаматные пролекарства гидроксильной и аминогрупп, а также карбонатные пролекарства и сульфатные сложные эфиры гидроксильных групп. Также предусматривается преобразование гидроксильных групп в простые (ацилокси)метил- и (ацилокси)этилэфиры, где ацильная группа может представлять собой алкильный сложный эфир (необязательно замещенный), или где ацильная группа представляет собой сложный эфир аминокислоты (пролекарства этого типа описаны в R. P. Robinson et al., J. Medicinal Chemistry (1996) 39:p.10).
В следующих предусматриваемых аспектах, следует понимать, что соединения согласно объекту изобретения также могут быть активны в качестве метаболита (пролекарства или непролекарственной формы) и что все такие метаболиты конкретно предусматриваются в рамках изобретения. Например, пригодные метаболиты включают гидроксилированные формы, окисленные формы, глюкуронидированные формы, сульфатированные формы и т.д. Более того, также следует отметить, что метаболиты могут быть более активными, чем первоначально вводимая форма.
Рассматриваемые композиции и составы
Исходя из активности соединений в качестве модуляторов Hec1, авторы изобретения предполагают, что соединения и композиции согласно объекту изобретения можно использовать для профилактики и/или лечения различных заболеваний, ассоциированных с дисфункцией и/или сверхэкспрессией Hec1, и, в действительности, всех заболеваний, которые положительно отвечают на введение рассматриваемых соединений. Термин "дисфункция Hec1", как используют в рамках изобретения, относится к любому нарушению Hec1, особенно он относится к его ассоциации с функцией Nek2 и передачей сигнала системы контроля веретена деления. Такие нарушения могут быть следствием одного или нескольких из мутации (например, увеличение или снижение аффинности в отношении партнера по связыванию), временной или постоянной сверхэкспрессии (например, активированной ненадлежащим или мутантным промотором), необратимого или более прочного связывания активатора, ненадлежащей активации нефизиологической молекулой, и т.д. Следовательно, особенно предусматриваемые заболевания включают неопластические заболевания, и особенно злокачественные неопластические заболевания (например, рак молочной железы, плоскоклеточный рак, рак мочевого пузыря, рак желудка, рак поджелудочной железы, рак головы, рак шеи, рак пищевода, рак предстательной железы, рак ободочной и прямой кишки, рак легкого, рак почки, гинекологический рак или рак щитовидной железы). Незлокачественные неопластические заболевания включают гиперплазию кожи (например, псориаз) или предстательной железы (например, доброкачественная гипертрофия предстательной железы (BPH).
Таким образом, автор изобретения также предусматривает многочисленные фармацевтические композиции, которые включают соединения, представленные в настоящем описании, и, главным образом, предусматривает, что соединения согласно объекту изобретения могут быть изготовлены в виде фармацевтических композиций, которые содержат терапевтически эффективное количество рассматриваемых соединений (или их фармацевтически приемлемой соли, гидрата или пролекарства) и фармацевтически приемлемый носитель.
Активность, токсичность и другие фармакологические и фармакодинамические параметры можно устанавливать для соединений, представленных в настоящем описании, с использованием многочисленных известных протоколов. Аналогично, цитотоксичность можно устанавливать с помощью анализа MTS в различных клеточных линиях, в то время как мониторинг нарушения взаимодействия Hec1-Nek2 можно проводить с помощью коиммунопреципитации или дрожжевой двухгибридной системы. Анализ клеточного цикла можно проводить путем мониторинга различных популяций различных стадий (например, суб-G1, G0/G1, S, и т.д.), и количественное определение нарушения ориентации хромосом в метафазе можно проводить с использованием иммунофлуоресцентных способов, хорошо известных в данной области. Активность in vivo можно устанавливать с использованием различных моделей на животных и особенно моделей с ксенотрансплантатами. Иллюстративные результаты предоставлены в прилагаемой таблице и нормализованных данных. Следовательно, авторы изобретения предусматривают фармацевтическую композицию, которая включает фармацевтически приемлемый носитель и рассматриваемые в настоящем описании соединения, где соединения присутствуют в концентрации, эффективной для нарушения связывания Hec1/Nek2 у пациента, когда композицию вводят пациенту. Авторы изобретения также открыли, что многочисленные соединения согласно объекту изобретения являются биодоступными при пероральном введении и их можно выявить в сыворотке в течение длительных периодов после перорального введения или внутривенного (в/в) введения (см. ниже).
Наиболее предпочтительно, рассматриваемые соединения включают в состав с одним или несколькими нетоксичными фармацевтически приемлемыми носителями, предпочтительно их включают в состав для перорального введения в твердой или жидкой форме или для парентеральной инъекции. Таким образом, следует понимать, что фармацевтические композиции согласно объекту изобретения можно вводить человеку и другим животным с использованием различных путей, в том числе перорально, ректально, парентерально, внутрибрюшинно, вагинально или местно.
Например, пригодные фармацевтические композиции для инъекции предпочтительно содержат фармацевтически приемлемые стерильные водные или неводные растворы, дисперсии, эмульсии или суспензии, а также стерильные порошки для восстановления в стерильные инъецируемые растворы или дисперсии перед применением. Примеры пригодных водных и неводных носителей, разбавителей, растворителей или наполнителей включают воду, этанол, полиолы (например, глицерин, пропиленгликоль, полиэтиленгликоль и т.д.), и их пригодные смеси, масла и инъецируемые органические сложные эфиры (например, этилолеат). Рассматриваемые композиции также могут содержать различные неактивные ингредиенты, в том числе консерванты, смачивающие вещества, эмульгаторы и/или диспергирующие средства. Стерильность можно обеспечивать путем включения антибактериальных и/или противогрибковых средств (например, парабен, фенолсорбиновая кислота, хлорбутанол и т.д.). Когда это целесообразно, могут быть включены осмотически активные средства (например, сахара, хлорид натрия и т.д.).
Альтернативно рассматриваемые композиции могут быть изготовлены в виде твердых дозированных форм для перорального введения, и, таким образом, они могут представлять собой капсулы, таблетки, пилюли, порошки и гранулы. В предпочтительных твердых дозированных формах рассматриваемое соединение смешивают по меньшей мере с одним из фармацевтически приемлемого эксципиента или носителя (например, цитрат натрия или дикальцийфосфат), наполнителя или разбавителя (например, крахмал, лактоза, сахароза, глюкоза, маннит или кремниевая кислота), связующего вещества (например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и т.д.), увлажнителя (например, глицерин), дезинтегрирующего вещества (например, агар-агар, карбонат кальция, картофельный или маниоковый крахмал, альгиновая кислота, определенные силикаты или карбонат натрия), замедлителя растворения (например, парафин), ускорителя всасывания (например, четвертичное соединение аммония), смачивающих веществ (например, цетиловый спирт и глицерин моностеарат), и абсорбентов (например, каолин или бентонитовая глина), и смазывающих веществ (например, тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия).
Твердые композиции сходного типа также можно использовать в качестве наполнителей в мягких и твердых заполненных желатиновых капсулах с использованием таких эксципиентов, как лактоза или молочный сахар, а также высокомолекулярные полиэтиленгликоли и т.д. Твердые дозированные формы в виде таблеток, драже, капсул, пилюль и гранул можно получать с покрытиями и оболочками, такими как желудочно-резистентные покрытия и другие покрытия, хорошо известные в области изготовления фармацевтических составов. Рассматриваемые композиции, кроме того, можно изготавливать так, чтобы они высвобождали активный ингредиент(ы) только, или предпочтительно, в определенной части кишечного тракта, необязательно, замедленным образом. Примеры композиций для окружения, которые можно использовать, включают полимерные вещества и воски. Рассматриваемые соединения также могут быть в микроинкапсулированной форме, в соответствующим случае, с одним или несколькими из упомянутых выше эксципиентов.
Жидкие дозированные формы для перорального введения включают фармацевтически приемлемые эмульсии, растворы, суспензии, сиропы и эликсиры. В дополнение к активным соединениям, жидкие дозированные формы могут содержать инертные разбавители, обычно используемые в данной области (например, воду или другой растворитель, солюбилизирующие агенты), эмульгаторы (например, этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, диметилформамид), масла (и, в частности, хлопковое, арахисовое, кукурузное, зародышевое, оливковое, касторовое и кунжутное масла), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот и сорбитана, и их смеси. Помимо инертных разбавителей, пероральные композиции также могут включать адъюванты, такие как смачивающие вещества, эмульгирующие и суспендирующие вещества, подсластители, вкусовые добавки и отдушки.
Соединения согласно объекту изобретения также можно вводить в форме липосом, которые могут быть моноламеллярными, олиголамеллярными или полиламеллярными. Рассматриваемые композиции в форме липосом, кроме того, могут содержать стабилизаторы, консерванты, эксципиенты и т.д. Предпочтительные липиды для липосомального состава включают фосфолипиды и фосфатидилхолины (лецитины), как природные, так и синтетические. Способы получения липосом известны в данной области. См., например, Prescott, Ed., Methods in Cell Biology, Volume XIV, Academic Press, New York, N.Y. (1976), p. 33 et seq.
Истинные уровни дозирования рассматриваемых соединений в фармацевтических композициях согласно объекту изобретения можно варьировать, так чтобы получить количество рассматриваемого соединения(ий), которое является эффективным для достижения желаемого терапевтического ответа для конкретного пациента, композиции и способа введения. Таким образом, выбранный уровень дозирования зависит от различных факторов, включая активность конкретного соединения, путь введения, тяжесть подвергаемого лечению состояния, и состояние и предшествующий медицинский анамнез у пациента, подвергаемого лечению. Однако в пределах квалификации специалиста в данной области находится начало дозирования соединения на уровнях ниже требуемого для достижения желаемого терапевтического эффекта и постепенное увеличение дозировки, пока не будет достигнут желаемый эффект. Как правило, пациенту-млекопитающему перорально вводят дозы на уровне от приблизительно 0,01 мг до приблизительно 500 мг, более предпочтительно от приблизительно 0,5 мг до приблизительно 50 мг рассматриваемого соединения на килограмм массы тела в сутки. Если желательно, эффективную суточную дозу можно разделять на множество доз для целей введения, например, на от двух до четырех отдельных доз в сутки. Таким образом, рассматриваемые составы, главным образом, включают составы, пригодные для перорального введения, парентерального введения, для введения в качестве крема или в качестве глазных капель или другого жидкого местного состава.
Более того, предварительные данные показали, что различные ингибиторы Hec1 проявляли синергический эффект с выбранными химиотерапевтическими ингибиторами. Среди других химиотерапевтических ингибиторов, соединения, включающие таксол, винкристин и винбластин, продемонстрировали синергический эффект, и также ожидается, что они имеют синергический эффект в отношении ингибиторов образования или полимеризации тубулина, а также ингибиторов претубулина. Таким образом, пригодные химиотерапевтические ингибиторы, главным образом, включают одно или несколько лекарственных средств, которые препятствуют образованию или деградации микротрубочек. Таким образом, любые лекарственные средства, которые воздействуют на деление клеток, и любые антиметаболиты считаются пригодными в комбинации с ингибиторами Hec1, предусматриваемыми в рамках изобретения. Напротив, было показано, что антрациклины (например, доксорубицин) имеют только самое большее аддитивный эффект и не имеют синергического эффекта с ингибиторами Hec1.
Далее, следует понимать, что рассматриваемые фармацевтические композиции также могут включать дополнительные фармацевтически активные соединения, и, главным образом, предусматриваемые дополнительные фармацевтически активные соединения включают антинеопластические средства, которые могут действовать на репликацию ДНК, клеточный цикл, метаболизм клеток, ангиогенез, или индуцировать апоптоз. Кроме того, пригодные активные вещества включают иммунологически активные вещества (например, противовоспалительные средства, иммунодепрессанты, стероиды, интерфероны (альфа, бета или гамма) и их фрагменты, и молекулы, которые селективно увеличивают или подавляют экспрессию Th1- и/или Th2-цитокинов). Другие пригодные активные вещества включают антибактериальные и противовирусные средства, лекарственные средства, которые стимулируют или модифицируют метаболизм, неврологически активные лекарственные средства и/или обезболивающие лекарственные средства. Безусловно, следует понимать, что дополнительные фармацевтически активные соединения могут быть включены в ту же фармацевтическую композицию, или их можно вводить отдельно, и специалист в данной области легко определит расписание и путь пригодного совместного введения дополнительных фармацевтических активных соединений.
Примеры/эксперименты
Иллюстративный синтез 4-арил-2-амидотиазолов
Рассматриваемые соединения 4-арил-2-амидотиазола можно получать многочисленными способами синтеза, и представленный ниже способ предоставлен только для иллюстративного руководства. Хотя приведенную ниже схему можно использовать для получения большинства соединений, представленных в настоящем описании, для других соединений могут требоваться незначительные модификации общей схемы, которые будут очевидны квалифицированному специалисту.

Схема выше иллюстрирует способ синтеза 4-арил-2-аминотиазола Е. Ароматические соединения структуры A, включая замещенный бензол, пиридин или другое гетероциклическое соединение (5-, 6- или 7-членное) подвергают реакции с ацетилхлоридом в присутствии AlCl3 с получением ацетилированных аренов B. Бромирование B дает α-Br-ацетилированные арены C, которым позволяют реагировать с тиомочевиной с получением аминотиазолов D с арильным заместителем в положении C-4. Затем полученные таким образом аминотиазолы подвергают реакции с различными кислотами с образованием конечных 4-арил-2-амидотиазолов E.
Ацетилирование Ar1:

Ацетилирование Ar1 можно проводить с использованием различных реагентов, как представлено на указанной выше схеме.
Бромирование ацетил-Ar1:

Пригодные бромирующие агенты включают Br2, HBr, NBS, TBABr3, CuBr2 и т.д. в различных растворителях, включая простой эфир, THF, галогенированные углеводороды, сложный эфир и т.д.
Амидирование аминотиазолов:

Пригодные агенты реакции сочетания включают CDI, EDC, CDC и т.д.

X представляет собой, как правило, Cl или Br; основание представляет собой, как правило, Et3N, Me3N, DIPEA, K2CO3, Na2CO3, DMAP и т.д. Альтернативно 4-арил-2-амидотиазолы можно получать следующим образом:

Альтернативно реакцию сочетания можно проводить следующим образом:

К раствору 4-арилтиазол-2-амина (1,0 экв.) в CH2Cl2 добавляли триэтиламин (3,0 экв.) или DMAP (3,0 экв.), а затем арилоксихлорид (1,5 экв.) или арилоксихлорида гидрохлорид (1,5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении и добавляли горячую воду. Полученный осадок фильтровали и сушили в вакууме с получением соответствующих 4-арил-2-амидотиазолов. Для конкретных примеров синтеза см. ниже.
Синтез иллюстративных аминотиазолов и родственных промежуточных соединений

2-Бром-1-мезитилэтанон. К раствору 1-мезитилэтанона (1,02 г, 6,27 ммоль) в EtOAc (50 мл) добавляли бромид меди(II) (CuBr2, 2,85 г, 12,8 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 90 мин. Раствору позволяли остыть и полученные твердые вещества отфильтровывали и промывали EtOAc. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-бром-1-мезитилэтанона (1,67 г) в виде желтого масла: 1H ЯМР (500 МГц, CDCl3) δ 6,87 (2H, с), 4,27 (2H, с), 2,31 (3H, с), 2,22 (6H, с).

4-Мезитилтиазол-2-амин. 2-бром-1-мезитилэтанон (2,43 г, 10,1 ммоль) и тиомочевину (0,810 г, 10,6 ммоль) растворяли в 95% этаноле (20 мл). Реакционную смесь нагревали с обратным холодильником в течение 2,0 ч. Раствор концентрировали при пониженном давлении и осадок перекристаллизовывали из 2-пропанола с получением желаемого 4-мезитилтиазол-2-амина (2,36 г) в виде белого твердого вещества: 1H ЯМР (500 МГц, CD3OD) δ 7,00 (2H, с), 6,67 (1H, с), 2,31 (3H, с), 2,19 (6H, с).

4-(п-Толил)тиазол-2-амин. Смесь 2-бром-1-(п-толил)этанона (5,00 г, 23,5 ммоль) и тиомочевины (1,97 г, 25,9 ммоль) в 95% EtOH (33,5 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (1,0 мл). Полученный осадок фильтровали и промывали горячей водой. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(п-толил)тиазол-2-амина (4,40 г) в виде белого твердого вещества с выходом 99%: 1H ЯМР (500 МГц, CDCl3) δ 7,66 (д, J=8,0 Гц, 2H), 7,18 (д, J=7,5 Гц, 2H), 6,66 (с, 1H), 5,25 (уш.с, 2H), 2,36 (с, 6H).

5-Метил-4-(п-толил)тиазол-2-амин. Смесь 2-бром-1-(п-толил)пропан-1-она (6,88 г, 30,3 ммоль) и тиомочевины (2,54 г, 33,4 ммоль) в 95% EtOH (43 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 5-метил-4-(п-толил)тиазол-2-амина (6,10 г) в виде белого твердого вещества с выходом 99%: 1H ЯМР (500 МГц, CDCl3) δ 7,40 (д, J=8,0 Гц, 2H), 7,23 (д, J=8,0 Гц, 2H), 3,18 (уш.с, 2H), 2,37 (с, 3H), 2,35 (с, 3H).

2-Бром-1-(4-метокси-2,6-диметилфенил)этанон. К раствору 1-(4-метокси-2,6-диметилфенил)этанона (5,7 г, 32 ммоль) в ацетонитриле (64 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 15,4 г, 32,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 80 мин. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(4-метокси-2,6-диметилфенил)этанона (9,14 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-метокси-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-метокси-2,6-диметилфенил)этанона (8,65 г, 33,6 ммоль) и тиомочевины (2,56 г, 33,6 ммоль) в 95% EtOH (48 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (50 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-метокси-2,6-диметилфенил)тиазол-2-амина (5,9 г) в виде белого твердого вещества с выходом 66%: 1H ЯМР (500 МГц, CDCl3) δ 6,61 (с, 2H), 6,27 (с, 1H), 4,91 (уш.с, 2H), 3,79 (с, 3H), 2,15 (с, 6H).

2-Бром-1-(2,4,6-триметилпиридин-3-ил)этанона гидробромид. К раствору 1-(2,4,6-триметилпиридин-3-ил)этанона (5,0 г, 30,6 ммоль) в 33% HBr в растворе уксусной кислоты (10,2 мл) капельно добавляли бром (1,57 мл, 30,6 ммоль) в уксусной кислоте (10,2 мл). Реакционную смесь перемешивали при 70°С в течение 2,0 ч. Раствор охлаждали до комнатной температуры и промывали простым эфиром. Осадок сушили при пониженном давлении с получением 2-бром-1-(2,4,6-триметилпиридин-3-ил)этанона гидробромида, который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,4,6-Триметилпиридин-3-ил)тиазол-2-амин. Смесь 2-бром-1-(2,4,6-триметилпиридин-3-ил)этанона гидробромида (9,00 г, 27,9 ммоль) и тиомочевины (2,12 г, 27,9 ммоль) в 95% EtOH (39,8 мл) нагревали с обратным холодильником в течение 120 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,4,6-триметилпиридин-3-ил)тиазол-2-амина (3,80 г) в виде желтого твердого вещества с выходом 62%: 1H ЯМР (500 МГц, CDCl3) δ 6,87 (с, 1H), 6,31 (с, 1H), 5,07 (уш.с, 2H), 2,49 (с, 3H), 2,38 (с, 3H), 2,14 (с, 3H).

2-Бром-1-(4-этокси-2,6-диметилфенил)этанон. К раствору 1-(4-этокси-2,6-диметилфенил)этанона (4,00 г, 20,8 ммоль) в ацетонитриле (41,6 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 10,0 г, 20,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-этокси-2,6-диметилфенил)этанона (6,40 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-Этокси-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-этокси-2,6-диметилфенил)этанона (6,35 г, 23,4 ммоль) и тиомочевины (1,78 г, 23,4 ммоль) в 95% EtOH (33,5 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-этокси-2,6-диметилфенил)тиазол-2-амина (4,18 г) в виде белого твердого вещества с выходом 72%: 1H ЯМР (500 МГц, ДМСО-d6) δ 6,84 (с, 2H), 6,60 (с, 2H), 6,27 (с, 1H), 3,99 (кв, J=6,5 Гц, 2H), 2,06 (с, 6H), 1,31 (т, J=6,95 Гц, 3H).

4-Ацетил-3,5-диметилфенилтрифторметансульфонат. Раствор 1-(4-гидрокси-2,6-диметилфенил)этанона (3,30 г, 20,1 ммоль), триэтиламина (4,07 г, 40,2 ммоль) в безводном CH2Cl2 (20,1 мл) охлаждали до 0°С, а затем капельно добавляли трифторметансульфоновый ангидрид (4,0 мл, 24 ммоль). После завершения добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1,0 ч. В раствор добавляли воду и экстрагировали этилацетатом (60 мл). Органический слой отделяли, сушили над MgSO4(s), концентрировали при пониженном давлении. Осадок очищали флэш-хроматографией на силикагеле с получением 4-ацетил-3,5-диметилфенилтрифторметансульфоната (5,0 г) в виде желтого масла с выходом 85%.

1-(3,5-Диметил-[1,1'-бифенил]-4-ил)этанон. К раствору 4-ацетил-3,5-диметилфенилтрифторметансульфоната (1,00 г, 3,38 ммоль), KF (0,65 г, 11 ммоль) и фенилбороновой кислоты (0,49 г, 4,0 ммоль) в THF (4,0 мл) добавляли трициклогексилфосфин (11,4 мг, 0,04 ммоль) и Pd(OAc)2 (7,6 мг, 0,03 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 5,0 ч в атмосфере N2. Реакционную смесь фильтровали через небольшой слой целита, и осадок на фильтре промывали этилацетатом (40 мл). Фильтрат концентрировали при пониженном давлении и осадок очищали флэш-хроматографией на силикагеле с получением 1-(3,5-диметил-[1,1'-бифенил]-4-ил)этанона (0,68 г) с выходом 90%: 1H ЯМР (500 МГц, CDCl3) δ 7,56 (д, J=8,0 Гц, 2H), 7,44 (т, J=7,0 Гц, 2H), 7,35 (м, 1H), 7,25 (с, 2H), 2,52 (с, 3H), 2,33 (с, 6H).

2-Бром-1-(3,5-диметил-[1,1'-бифенил]-4-ил)этанон. К раствору 1-(3,5-диметил-[1,1'-бифенил]-4-ил)этанона (1,89 г, 8,43 ммоль) в ацетонитриле (16,9 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 4,07 г, 8,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(3,5-диметил-[1,1'-бифенил]-4-ил)этанона (3,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(3,5-Диметил-[1,1'-бифенил]-4-ил)тиазол-2-амин. Смесь 2-бром-1-(3,5-диметил-[1,1'-бифенил]-4-ил)этанона (2,56 г, 8,44 ммоль) и тиомочевины (0,64 г, 8,44 ммоль) в 95% EtOH (12,1 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (1,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (10 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(3,5-диметил-[1,1'-бифенил]-4-ил)тиазол-2-амина (0,66 г) в виде желтого твердого вещества с выходом 28%: 1H ЯМР (500 МГц, CDCl3) δ 7,60 (д, J=1 Гц, 2H), 7,43 (т, J=7,5 Гц, 1H), 7,32 (м, 1H), 7,25 (с, 2H), 6,34 (с, 1H), 5,03 (уш.с, 2H), 2,24 (с, 6H).

1-(4-Хлор-2,6-диметилфенил)этанон. Безводный хлорид меди(II) (98,9 г, 0,74 моль) смешивали с трет-бутилнитритом (94,8 г, 0,83 моль) в ацетонитриле (1,02 л). Раствор охлаждали до 0°С и медленно добавляли 1-(4-амино-2,6-диметилфенил)этанон (100 г, 0,61 моль) в течение 5,0 мин. После завершения добавления реакционную смесь нагревали до комнатной температуры и выливали в водный раствор хлористоводородной кислоты (20%, 1,0 л). Раствор экстрагировали EtOAc (800 мл) и органический слой отделяли, промывали H2O (1,0 л), сушили над MgSO4(s) и концентрировали при пониженном давлении. Жидкость подвергали перегонке с получением 1-(4-хлор-2,6-диметилфенил)этанона (85,0 г) в виде желтого масла с выходом 76%: 1H ЯМР (500 МГц, CDCl3) δ 7,02 (с, 2H), 2,45 (с, 3H), 2,22 (с, 6H).

2-Бром-1-(4-хлор-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (5,0 г, 27 ммоль) в ацетонитриле (54,8 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 13,2 г, 27,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(4-хлор-2,6-диметилфенил)этанона (7,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-Хлор-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-хлор-2,6-диметилфенил)этанона (6,54 г, 25,0 ммоль) и тиомочевины (1,90 г, 25,0 ммоль) в 95% EtOH (35,7 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл), а затем насыщенный водный Na2CO3 (4,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-хлор-2,6-диметилфенил)тиазол-2-амина (4,30 г) в виде белого твердого вещества с выходом 72%: 1H ЯМР (500 МГц, ДМСО-d6) δ 7,16 (с, 2H), 6,43 (с, 1H), 2,10 (с, 6H).

N-(4-(2-Бромацетил)-3,5-диметилфенил)ацетамид. К раствору N-(4-ацетил-3,5-диметилфенил)ацетамида (5,00 г, 24,4 ммоль) в ацетонитриле (48,7 мл) добавляли тетрабутиламмонийтрибромид (ΤΒΑΒr3, 11,7 г, 24,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением N-(4-(2-бромацетил)-3,5-диметилфенил)ацетамида (7,00 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

N-(4-(2-Аминотиазол-4-ил)-3,5-диметилфенил)ацетамид. Смесь N-(4-(2-бромацетил)-3,5-диметилфенил)ацетамида (7,34 г, 25,8 ммоль) и тиомочевины (1,97 г, 25,9 ммоль) в 95% EtOH (36,9 мл) нагревали с обратным холодильником в течение 120 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (50 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением N-(4-(2-аминотиазол-4-ил)-3,5-диметилфенил)ацетамида (5,83 г) в виде желтого твердого вещества с выходом 86%: 1H ЯМР (500 МГц, ДМСО-d6) δ 9,80 (с, 1H), 7,26 (с, 2H), 6,90 (уш.с, 2H), 6,30 (с, 1H), 2,06 (с, 6H), 2,02 (с, 3H).

2-Бром-1-(2,4,6-триизопропилфенил)этанон. К раствору 1-(2,4,6-триизопропилфенил)этанона (10,0 г, 65,3 ммоль) в ацетонитриле (81 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 19,6 г, 40,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3,0 ч. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(2,4,6-триизопропилфенил)этанона (13,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,4,6-Триизопропилфенил)тиазол-2-амин. Смесь 2-бром-1-(2,4,6-триизопропилфенил)этанона (13,9 г, 42,7 ммоль) и тиомочевины (3,24 г, 42,6 ммоль) в 95% EtOH (60,9 мл) нагревали с обратным холодильником в течение ночи. Раствор концентрировали и добавляли воду (100 мл), насыщенный водный Na2CO3 (10 мл) и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении, после чего очищали колоночной хроматографией на силикагеле (33% EtOAc в гексане в качестве элюента) с получением 4-(2,4,6-триизопропилфенил)тиазол-2-амина (3,28 г) в виде белого твердого вещества с выходом 25%: 1H ЯМР (500 МГц, CDCl3) δ 7,03 (с, 2H), 6,22 (с, 1H), 4,75 (уш.с, 2H), 2,89 (м, 1H), 2,68 (м, 2H), 1,27-1,14 (м, 18H).

1-(2,6-Диметил-4-феноксифенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (4,50 г, 24,6 ммоль), K3PO4 (10,5 г, 49,3 ммоль) и фенола (2,78 г, 29,5 ммоль) в толуоле (49,3 мл) добавляли 2-(ди-трет-бутилфосфино)бифенил (221 мг, 0,74 ммоль) и Pd(OAc)2 (233 мг, 1,04 ммоль). Реакционную смесь нагревали при 100°С в течение 2,0 ч в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(2,6-диметил-4-феноксифенил)этанона в виде желтого масла с выходом 68%: 1H ЯМР (500 МГц, CDCl3) δ 7,35 (т, J=8,0 Гц, 2H), 7,12 (т, J=7,5 Гц, 1H), 7,00 (д, J=7,5 Гц, 2H), 6,65 (с, 2H), 2,48 (с, 3H), 2,22 (с, 6H).

2-Бром-1-(2,6-диметил-4-феноксифенил)этанон. К раствору 1-(2,6-диметил-4-феноксифенил)этанона (3,60 г, 15,0 ммоль) в ацетонитриле (30 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 7,95 г, 15,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду, и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(2,6-диметил-4-феноксифенил)этанона (4,8 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,6-Диметил-4-феноксифенил)тиазол-2-амин. Смесь 2-бром-1-(2,6-диметил-4-феноксифенил)этанона (5,18 г, 16,2 ммоль) и тиомочевины (1,24 г, 16,3 ммоль) в 95% EtOH (23,2 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,6-диметил-4-феноксифенил)тиазол-2-амина (2,84 г) в виде желтого твердого вещества с выходом 59%: 1H ЯМР (500 МГц, CDCl3) δ 7,33 (т, J=7,5 Гц, 2H), 7,26 (т, J=7,5 Гц, 1H), 7,10 (д, J=7,3, 2H), 6,72 (с, 2H), 6,30 (с, 1H), 5,18 (уш.с, 2H), 2,14 (с, 6H).

2-Бром-1-(4-изопропокси-2,6-диметилфенил)этанон. К раствору 1-(4-изопропокси-2,6-диметилфенил)этанона (4,3 г, 20,9 ммоль) в ацетонитриле (41,7 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 11,1 г, 22,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-изопропокси-2,6-диметилфенил)этанона (5,9 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-Изопропокси-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-изопропокси-2,6-диметилфенил)этанона (5,18 г, 18,2 ммоль) и тиомочевины (1,38 г, 18,2 ммоль) в 95% EtOH (26 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-изопропокси-2,6-диметилфенил)тиазол-2-амина (3,44 г) в виде желтого твердого вещества с выходом 72,2%: 1H ЯМР (500 МГц, CDCl3) δ 6,60 (с, 2H), 6,26 (с, 1H), 4,97 (уш.с, 2H), 4,54 (м, 1H), 2,13 (с, 6H), 1,32 (д, J=6,1 Гц, 6H).

2-Бром-1-(4-(циклопентилокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(циклопентилокси)-2,6-диметилфенил)этанона (4,60 г, 1,98 ммоль) в ацетонитриле (39,6 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 10,5 г, 21,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду, и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-(циклопентилокси)-2,6-диметилфенил)этанона (6,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(Циклопентилокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(циклопентилокси)-2,6-диметилфенил)этанона (6,16 г, 19,8 ммоль) и тиомочевины (1,51 г, 19,8 ммоль) в 95% EtOH (28,3 мл) нагревали с обратным холодильником в течение 90 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(циклопентилокси)-2,6-диметилфенил)тиазол-2-амина (4,2 г) в виде белого твердого вещества с выходом 73,7%: 1H ЯМР (500 МГц, CDCl3) δ 6,58 (с, 2H), 6,24 (с, 1H), 4,75 (м, 1H), 2,13 (с, 6H), 1,88-1,78 (м, 6H), 1,62-1,59 (м, 2H).

1-(4-(4-Метоксифенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (10,0 г, 54,8 ммоль), K3PO4 (23,2 г, 110 ммоль), 4-метоксифенола (8,16 г, 65,7 ммоль) в толуоле (78,2 мл), добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (349 мг, 0,82 ммоль), Pd(OAc)2 (259 мг, 1,15 ммоль). Реакционную смесь нагревали при 100°С в течение 5,0 ч в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок перекристаллизовывали в MeOH с получением 1-(4-(4-метоксифенокси)-2,6-диметилфенил)этанона (11,1 г) в виде белого твердого вещества с выходом 75,0%: 1H ЯМР (500 МГц, CDCl3) δ 6,96 (м, 2H), 6,88 (м, 2H), 6,57 (с, 2H), 3,81 (с, 3H), 2,46 (с, 3H), 2,20 (с, 6H).

2-Бром-1-(4-(4-метоксифенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(4-метоксифенокси)-2,6-диметилфенил)этанона (3,80 г, 14,1 ммоль) в ацетонитриле (28,1 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 7,46 г, 15,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду, и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-(4-метоксифенокси)-2,6-диметилфенил)этанона (5,25 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(4-метоксифенокси)-2,6-диметилфенил)этанона (4,90 г, 14,0 ммоль) и тиомочевины (1,07 г, 14,1 ммоль) в 95% EtOH (20,0 мл) нагревали с обратным холодильником в течение 100 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(4-метоксифенокси)-2,6-диметилфенил)тиазол-2-амина (3,10 г) в виде желтого твердого вещества с выходом 68%: 1H ЯМР (500 МГц, CDCl3) δ 6,98 (м, 2H), 6,88 (м, 2H), 6,64 (с, 2H), 6,27 (с, 1H), 5,40 (уш.с, 2H), 3,81 (с, 3H), 2,13 (с, 6H).

1-(4-(4-Фторфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (4,50 г, 24,6 ммоль), K3PO4 (10,5 г, 49,3 ммоль), 4-фторфенола (3,31 г, 29,5 ммоль) в толуоле (49,3 мл), добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (314 мг, 0,74 ммоль), Pd(OAc)2 (233 мг, 1,04 ммоль). Реакционную смесь нагревали при 100°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (100 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(4-(4-фторфенокси)-2,6-диметилфенил)этанона (4,40 г) в виде желтого масла с выходом 68%: 1H ЯМР (500 МГц, CDCl3) δ 7,03 (м, 2H), 6,98 (м, 2H), 6,60 (с, 2H), 2,47 (с, 3H), 2,22 (с, 6H).

2-Бром-1-(4-(4-фторфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(4-фторфенокси)-2,6-диметилфенил)этанона (4,40 г, 17,0 ммоль) в ацетонитриле (34,1 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 9,04 г, 18,8 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду, и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(4-(4-фторфенокси)-2,6-диметилфенил)этанона (5,8 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(4-Фторфенокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(4-фторфенокси)-2,6-диметилфенил)этанона (5,74 г, 17,0 ммоль) и тиомочевины (1,30 г, 17,1 ммоль) в 95% EtOH (24,3 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(4-фторфенокси)-2,6-диметилфенил)тиазол-2-амина (4,50 г) в виде желтого твердого вещества с выходом 84%: 1H ЯМР (500 МГц, CDCl3) δ 7,05-6,97 (м, 4H), 6,66 (с, 2H), 6,28 (с, 1H), 5,95 (уш.с, 2H), 2,14 (с, 6H).

2-Бром-1-(4-изобутокси-2,6-диметилфенил)этанон. К раствору 1-(4-изобутокси-2,6-диметилфенил)этанона (4,3 г, 19,5 ммоль) в ацетонитриле (39 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 9,41 г, 19,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 1-(4-изобутокси-2,6-диметилфенил)этанона (6,1 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-Изобутокси-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-изобутокси-2,6-диметилфенил)этанона (5,84 г, 19,5 ммоль) и тиомочевины (1,49 г, 19,6 ммоль) в 95% EtOH (28 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-изобутокси-2,6-диметилфенил)тиазол-2-амина (4,4 г) в виде белого твердого вещества с выходом 82%: 1H ЯМР (500 МГц, CDCl3) δ 6,61 (с, 2H), 6,24 (с, 1H), 3,70 (д, J=6,5 Гц, 2H), 2,15 (с, 6H), 2,07 (м, 1H), 1,01 (д, J=6,7 Гц, 6H).

1-(4-(Бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (5,0 г, 27,4 ммоль), K3PO4 (11,6 г, 54,7 ммоль), сезамола (4,54 г, 32,9 ммоль) в толуоле (54,8 мл), добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (349 мг, 0,82 ммоль), Pd(OAc)2 (259 мг, 1,15 ммоль). Реакционную смесь нагревали при 100°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок перекристаллизовывали в MeOH с получением 1-(4-(бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)этанона (4,80 г) в виде белого твердого вещества с выходом 62%: 1H ЯМР (500 МГц, CDCl3) δ 6,77 (д, J=8,5 Гц, 1H), 6,59 (с, 2H), 6,56 (с, 1H), 6,48 (м, 1H), 5,98 (с, 2H), 2,46 (с, 3H), 2,21 (с, 6H).

1-(4-(Бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)-2-бромэтанон. К раствору 1-(4-(бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)этанона (4,80 г, 16,9 ммоль) в ацетонитриле (33,8 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 8,14 г, 16,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 1-(4-(бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)-2-бромэтанона (6,70 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(Бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 1-(4-(бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)-2-бромэтанона (6,13 г, 16,9 ммоль) и тиомочевины (1,29 г, 16,9 ммоль) в 95% EtOH (24,1 мл) нагревали с обратным холодильником в течение 90 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)тиазол-2-амина (5,50 г) в виде желтого твердого вещества с выходом 96%: 1H ЯМР (500 МГц, CDCl3) δ 6,75 (д, J=8,5 Гц, 1H), 6,66 (с, 2H), 6,58 (м, 1H), 6,49 (м, 1H), 6,28 (с, 1H), 5,98 (с, 2H), 5,05 (уш.с, 2H), 2,13 (с, 6H).

1-(4-(3,5-Диметилфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (5,0 г, 27,4 ммоль), K3PO4 (11,6 г, 54,7 ммоль), 3,5-диметилфенола (4,01 г, 32,8 ммоль) в толуоле (54,8 мл), добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (349 мг, 0,82 ммоль), Pd(OAc)2 (259 мг, 1,15 ммоль). Реакционную смесь нагревали при 100°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(4-(3,5-диметилфенокси)-2,6-диметилфенил)этанона (6,3 г) в виде желтого твердого вещества с выходом 86%: 1H ЯМР (500 МГц, CDCl3) δ 6,76 (с, 1H), 6,63 (с, 2H), 6,62 (с, 2H), 2,48 (с, 3H), 2,29 (с, 6H), 2,22 (с, 6H).

2-Бром-1-(4-(3,5-диметилфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(3,5-диметилфенокси)-2,6-диметилфенил)этанона (6,30 г, 23,5 ммоль) в ацетонитриле (47,0 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 11,9 г, 24,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду, и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-(3,5-диметилфенокси)-2,6-диметилфенил)этанона (8,3 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(3,5-Диметилфенокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(3,5-диметилфенокси)-2,6-диметилфенил)этанона (8,15 г, 23,5 ммоль) и тиомочевины (1,79 г, 23,5 ммоль) в 95% EtOH (33,5 мл) нагревали с обратным холодильником в течение 120 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(3,5-диметилфенокси)-2,6-диметилфенил)тиазол-2-амина (4,50 г) в виде желтого твердого вещества с выходом 59%: 1H ЯМР (500 МГц, CDCl3) δ 6,76 (с, 1H), 6,68 (с, 2H), 6,64 (с, 2H), 6,26 (с, 1H), 2,29 (с, 6H), 2,16 (с, 6H).

1-(4-(3-Метоксифенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (5,00 г, 27,4 ммоль), K3PO4 (11,6 г, 54,7 ммоль), 3-метоксифенола (4,08 г, 32,9 ммоль) в толуоле (54,8 мл) добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (349 мг, 0,82 ммоль), Pd(OAc)2 (259 мг, 1,15 ммоль). Реакционную смесь нагревали при 100°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(4-(3-метоксифенокси)-2,6-диметилфенил)этанона (5,4 г) в виде желтого масла с выходом 73%: 1H ЯМР (500 МГц, CDCl3) δ 7,24 (м, 1H), 6,68-6,66 (м, 3H), 6,57-6,56 (м, 2H), 3,79 (с, 3H), 2,48 (с, 3H), 2,22 (с, 6H).

2-Бром-1-(4-(3-метоксифенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(3-метоксифенокси)-2,6-диметилфенил)этанона (5,40 г, 20,0 ммоль) в ацетонитриле (40,0 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 10,1 г, 21,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(4-(3-метоксифенокси)-2,6-диметилфенил)этанона (7,00 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(3-Метоксифенокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(3-метоксифенокси)-2,6-диметилфенил)этанона (6,98 г, 20,0 ммоль) и тиомочевины (1,52 г, 20,0 ммоль) в 95% EtOH (28,5 мл) нагревали с обратным холодильником в течение 5,0 ч. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (1,0 мл), и экстрагировали этилацетатом (100 мл). Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 4-(4-(3-метоксифенокси)-2,6-диметилфенил)тиазол-2-амина (4,30 г) в виде темно-коричневого масла, которое использовали непосредственно на следующей стадии без дальнейшей очистки.

1-(2,6-Диметил-4-(4-(трифторметил)фенокси)фенил)этанон. К раствору 1-хлор-4-(трифторметил)бензола (6,60 г, 36,6 ммоль), K3PO4 (12,9 г, 60,9 ммоль), 1-(4-гидрокси-2,6-диметилфенил)этанона (5,00 г, 30,5 ммоль) в толуоле (60,9 мл) добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (388 мг, 0,91 ммоль) и Pd(OAc)2 (288 мг, 1,28 ммоль). Реакционную смесь нагревали при 100°С в течение 120 мин в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)этанона (1,8 г) в виде желтого масла с выходом 19%: 1H ЯМР (500 МГц, CDCl3) δ 7,58 (д, J=8,5 Гц, 2H), 7,04 (д, J=8,5 Гц, 2H), 6,70 (с, 2H), 2,50 (с, 3H), 2,25 (с, 6H).

2-Бром-1-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)этанон. К раствору 1-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)этанона ( 1,80 г, 5,84 ммоль) в ацетонитриле (11,7 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 2,82 г, 5,84 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s), и концентрировали при пониженном давлении с получением 2-бром-1-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)этанона (2,16 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,6-Диметил-4-(4-(трифторметил)фенокси)фенил)тиазол-2-амин. Смесь 2-бром-1-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)этанона (2,20 г, 5,68 ммоль) и тиомочевины (0,43 г, 5,68 ммоль) в 95% EtOH (8,1 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (1,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,6-диметил-4-(4-(трифторметил)фенокси)фенил)тиазол-2-амина (1,30 г) в виде желтого твердого вещества с выходом 63%: 1H ЯМР (500 МГц, CDCl3) δ 7,56 (д, J=8,5 Гц, 2H), 7,05 (д, J=8,5 Гц, 2H), 6,76 (с, 2H), 6,32 (с, 1H), 5,03 (с, 2H), 2,17 (с, 6H).

1-(4-(4-Этилфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-хлор-2,6-диметилфенил)этанона (5,0 г, 27,4 ммоль), K3PO4 (11,6 г, 54,7 ммоль), 4-этилфенола (4,01 г, 32,8 ммоль) в толуоле (54,8 мл) добавляли 2-ди-трет-бутилфосфино-2',4',6'-триизопропилбифенил (349 мг, 0,82 ммоль) и Pd(OAc)2 (259 мг, 1,15 ммоль). Реакционную смесь нагревали при 100°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(4-(4-этилфенокси)-2,6-диметилфенил)этанона (6,0 г) в виде желтого масла с выходом 82%: 1H ЯМР (500 МГц, CDCl3) δ 7,17 (д, J=8,5 Гц, 2H), 6,93 (д, J=8,5 Гц, 2H), 6,63 (с, 2H), 2,64 (кв, J=7,5 Гц, 2H), 2,47 (с, 3H), 2,21 (с, 6H), 1,25 (т, J=7,5 Гц, 3H).

2-Бром-1-(4-(4-этилфенокси)-2,6-диметилфенил)этанон. К раствору 1-(4-(4-этилфенокси)-2,6-диметилфенил)этанона (6,00 г, 22,4 ммоль) в ацетонитриле (44,7 мл) добавляли тетрабутиламмонийтрибромид (ΤΒΑΒr3, 10,8 г, 22,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(4-(4-этилфенокси)-2,6-диметилфенил)этанона (8,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(4-Этилфенокси)-2,6-диметилфенил)тиазол-2-амин. Смесь 2-бром-1-(4-(4-этилфенокси)-2,6-диметилфенил)этанона (7,70 г, 22,2 ммоль) и тиомочевины (1,69 г, 22,2 ммоль) в 95% EtOH (31,7 мл) нагревали с обратным холодильником в течение 180 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-(4-этилфенокси)-2,6-диметилфенил)тиазол-2-амина (6,30 г) в виде желтого твердого вещества с выходом 88%: 1H ЯМР (500 МГц, CDCl3) δ 7,18 (д, J=7,5 Гц, 2H), 6,95 (д, J=8,5, 2H), 6,71 (с, 2H), 6,29 (с, 1H), 5,45 (уш.с, 2H), 2,64 (кв, J=7,5 Гц, 2H), 2,14 (с, 6H), 1,25 (т, J=8,0 Гц, 3H).

2-Бром-1-(3,5-дифтор-4-метоксифенил)этанон. К раствору CH3CN (56 мл), содержащему 1-(3,5-дифтор-4-метоксифенил)этанон (5,0 г, 26,9 ммоль, 1,0 экв.), добавляли TBABr3 (12,95 г, 26,9 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (2,0% EtOAc в гексане в качестве элюента) с получением 2-бром-1-(3,5-дифтор-4-метоксифенил)этанона (5,05 г, 19,0 ммоль) в виде белого твердого вещества с выходом 71%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,76-7,81 (м, 2H), 4,91 (с, 2H), 4,07 (с, 3H).

4-(3,5-Дифтор-4-метоксифенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(3,5-дифтор-4-метоксифенил)этанон (2,0 г, 7,5 ммоль, 1,0 экв.) и тиомочевину (0,57 г, 7,5 ммоль, 1,0 экв.) в EtOH (20,0 мл) нагревали с обратным холодильником в течение 3,0 ч. Осадок подщелачивали насыщенным водным NaHCO3 (20 мл) и экстрагировали EtOAc (3×30 мл). Органический слой отделяли, сушили над MgSO4(s) и концентрировали при пониженном давлении. Полученные твердые вещества промывали гексаном с получением 4-(3,5-дифтор-4-метоксифенил)тиазол-2-амина (1,54 г, 6,4 ммоль) в виде белого твердого вещества с выходом 84%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,49-7,54 (м, 2H), 7,12-7,14 (м, 3H), 3,92 (с, 3H); ESI-MS: m/z 243,0 (M+H)+.

1-(2,6-Дифтор-4-метоксифенил)этанон. Смесь хлорида алюминия (10,0 г, 69,4 ммоль, 5,0 экв.) и ацетилхлорида (2,0 мл, 28 ммоль, 2,0 экв.) в CH2Cl2 (50,0 мл) перемешивали при 0°С в течение 30 мин. К реакционной смеси медленно добавляли 1,3-дифтор-5-метоксибензол (2,0 г, 13,9 ммоль, 1,0 экв.) в CH2Cl2 (10,0 мл), и полученный раствор перемешивали при комнатной температуре в течение дополнительных 2,0 ч. Раствор подщелачивали насыщенным водным NaHCO3 (20 мл) до pH 8-9. Органический слой отделяли, сушили над MgSO4(s), и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (15% EtOAc в гексане в качестве элюента) с получением 1-(2,6-дифтор-4-метоксифенил)этанона (1,5 г, 8,1 ммоль) в виде желтого масла с выходом 58%: 1H ЯМР (CDCl3, 500 МГц) δ 6,46-6,48 (м, 2H), 3,83 (с, 3H), 2,56 (с, 3H); ESI-MS: m/z 187,0 (M+H)+.

2-Бром-1-(2,5-дифтор-4-метоксифенил)этанон. К раствору CH3CN (20 мл), содержащему 1-(2,6-дифтор-4-метоксифенил)этанон (1,5 г, 8,1 ммоль, 1,0 экв.) добавляли TBABr3 (3,88 г, 8,1 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (2,0% EtOAc в гексане в качестве элюента) с получением 2-бром-1-(2,5-дифтор-4-метоксифенил)этанона (5,05 г, 19,1 ммоль) в виде желтого масла с выходом 84%: 1H ЯМР (CDCl3, 500 МГц) δ 6,50-6,52 (м, 2H), 4,34 (с, 2H), 3,85 (с, 3H).

4-(2,6-Дифтор-4-метоксифенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(2,5-дифтор-4-метоксифенил)этанон (1,5 г, 5,7 ммоль, 1,0 экв.) и тиомочевину (430,8 мг, 5,7 ммоль, 1,0 экв.) в EtOH (15,0 мл), нагревали с обратным холодильником в течение 6,0 ч. Осадок подщелачивали насыщенным водным NaHCO3 (20 мл) и экстрагировали EtOAc (3×30 мл). Органический слой отделяли, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 4-(2,6-дифтор-4-метоксифенил)тиазол-2-амина (928,6 мг, 3,8 ммоль) в виде белого твердого вещества с выходом 68%: 1H ЯМР (CDCl3, 500 МГц) δ 6,68 (с, 1H), 6,50-6,52 (м, 2H), 5,07 (уш.с, 2H), 3,81 (с, 3H); ESI-MS: m/z 243,7 (M+H)+.

1-(4-(2-Гидроксипропокси)-2,6-диметилфенил)этанон. В стеклянную емкость высокого давления помещали 1-(4-гидрокси-2,6-диметилфенил)этанон (500 мг, 3,1 ммоль, 1,0 экв.) и 2-метилоксиран (0,22 мл, 3,1 ммоль, 1,0 экв.) в 50% водном растворе NaOH (5,0 мл) и перемешивали при 140°С в течение 4,0 ч. Смесь разбавляли H2O (20 мл) и экстрагировали EtOAc. Органический слой собирали, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30% EtOAc в гексане в качестве элюента) с получением 1-(4-(2-гидроксипропокси)-2,6-диметилфенил)этанона (445,9 мг, 2,1 ммоль) в виде желтого масла с выходом 66%: 1H ЯМР (CDCl3, 500 МГц) δ 6,57 (с, 2H), 4,10-4,20 (уш.с, 1H), 3,90-3,93 (м, 2H), 3,75-3,79 (м, 1H), 2,45 (с, 3H), 2,23 (с, 6H), 1,25-1,28 (м, 3H); ESI-MS: m/z 223,4 (M+H)+.

2-Бром-1-(4-(2-гидроксипропокси)-2,6-диметилфенил)этанон. К раствору CH3CN (6,0 мл), содержащему 1-(4-(2-гидроксипропокси)-2,6-диметилфенил)этанон (445,9 мг, 2,0 ммоль, 1,0 экв.), добавляли TBABr3 (967,3 мг, 2,0 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель концентрировали при пониженном давлении, и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. 2-бром-1-(4-(2-гидроксипропокси)-2,6-диметилфенил)этанон (547,8 мг, 1,8 ммоль) получали в качестве коричневого масла с выходом 91%: 1H ЯМР (CDCl3, 500 МГц) δ 6,60 (с, 2H), 4,25 (с, 2H), 4,10-4,20 (уш.с, 1H), 3,91-3,94 (м, 2H), 3,79-3,80 (с, 1H), 2,24 (с, 6H), 1,27-1,29 (м, 3H).

1-(4-(2-Аминотиазол-4-ил)-3,5-диметилфенокси)пропан-2-ол. Реакционную смесь, содержащую 2-бром-1-(2,5-дифтор-4-метоксифенил)этанон (547,8 мг, 1,8 ммоль, 1,0 экв.) и тиомочевину (138,5 мг, 1,8 ммоль, 1,0 экв.) в EtOH (3,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (30 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30% EtOAc в гексане в качестве элюента) с получением 1-(4-(2-аминотиазол-4-ил)-3,5-диметилфенокси)пропан-2-ола (332,5 мг, 1,2 ммоль) в виде желтого масла с выходом 66%: 1H ЯМР (CDCl3, 500 МГц) δ 6,62 (с, 2H), 6,26 (с, 1H), 4,95 (уш.с, 2H), 4,10-4,20 (уш.с, 1H), 3,91-3,94 (м, 2H), 3,75-3,79 (м, 1H), 2,15 (с, 6H), 1,26-1,28 (м, 3H); ESI-MS: m/z 279,7 (M+H)+.

1-(4-(2,3-Дигидроксипропокси)-2,6-диметилфенил)этанон. В стеклянную емкость высокого давления помещали 1-(4-гидрокси-2,6-диметилфенил)этанон (2,00 г, 12,2 ммоль, 1,0 экв.) и 3-хлорпропан-1,2-диол (1,02 мл, 12,2 ммоль, 1,0 экв.) в 50% водном растворе NaOH (20,0 мл) и нагревали при 140°С в течение 16 ч. Смесь разбавляли H2O (20 мл) и экстрагировали EtOAc. Органический слой собирали, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30% EtOAc в гексане в качестве элюиента) с получением 1-(4-(2,3-дигидроксипропокси)-2,6-диметилфенил)этанона (1,66 г, 7,0 ммоль) в виде желтого масла с выходом 57%: 1H ЯМР (CDCl3, 500 МГц) δ 6,57 (с, 2H), 4,10-4,11 (м, 1H), 4,08-4,09 (м, 2H), 4,01-4,02 (м, 1H), 3,74-3,75 (м, 1H), 2,58-2,59 (уш.с, 1H), 2,45 (с, 3H), 2,23 (с, 6H), 2,05-2,10 (уш.с, 1H); ESI-MS: m/z 239,9 (M+H)+.

2-Бром-1-(4-(2,3-дигидроксипропокси)-2,6-диметилфенил)этанон. К раствору CH3CN (10,0 мл), содержащему 1-(4-(2,3-дигидроксипропокси)-2,6-диметилфенил)этанон (1,0 г, 4,2 ммоль, 1,0 экв.), добавляли TBABr3 (2,04 г, 4,2 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. 2-бром-1-(4-(2,3-дигидроксипропокси)-2,6-диметилфенил)этанон (741,9 мг, 2,3 ммоль) получали в виде желтого твердого вещества с выходом 56%: 1H ЯМР (CDCl3, 500 МГц) δ 6,60 (с, 2H), 4,25 (с, 2H), 4,10-4,11 (м, 1H), 4,03-4,04 (м, 2H), 3,82-3,85 (м, 1H), 3,75-3,76 (м, 1H), 2,24 (с, 6H).

3-(4-(2-Аминотиазол-4-ил)-3,5-диметилфенокси)пропан-1,2-диол. Реакционную смесь, содержащую 2-бром-1-(4-(2,3-дигидроксипропокси)-2,6-диметилфенил)этанон (741,9 мг, 2,3 ммоль, 1,0 экв.) и тиомочевину (178,1 мг, 2,3 ммоль, 1,0 экв.), в EtOH (10,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении, и осадок перерастворяли в EtOAc (30 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30,0% EtOAc в гексане в качестве элюента) с получением 3-(4-(2-аминотиазол-4-ил)-3,5-диметилфенокси)пропан-1,2-диола (694,1 мг, 2,4 ммоль) в виде желтого твердого вещества с выходом >99%: 1H ЯМР (CDCl3, 500 МГц) δ 6,76 (с, 2H), 5,31-5,32 (м, 1H), 3,99-4,00 (м, 1H), 3,79-3,87 (м, 1H), 3,78-3,79 (м, 1H), 3,43-3,44 (м, 2H), 3,37 (уш.с, 2H), 2,15 (с, 6H); ESI-MS: m/z 295,6 (M+H)+.

1-(4-(2-Метоксиэтокси)-2,6-диметилфенил)этанон. В стеклянную емкость высокого давления помещали 1-(4-гидрокси-2,6-диметилфенил)этанон (500 мг, 3,1 ммоль, 1,0 экв.) и 1-хлор-2-метоксиэтан (0,28 мл, 3,1 ммоль, 1,0 экв.) в 50% водном растворе NaOH (5,0 мл) и нагревали при 140°С в течение 16 ч. Осадок разбавляли H2O (20 мл) и экстрагировали EtOAc (3×30 мл). Органический слой отделяли, сушили MgSO4 и концентрировали при пониженном давлении с получением 1-(4-(2-метоксиэтокси)-2,6-диметилфенил)этанона (430,9 мг, 1,9 ммоль) в виде желтого масла с выходом 64%: 1H ЯМР (CDCl3, 500 МГц) δ 6,58 (с, 2H), 4,09-4,10 (м, 2H), 3,72-3,74 (м, 2H), 3,44 (с, 3H), 2,45 (с, 3H), 2,23 (с, 6H); ESI-MS: m/z 223,6 (M+H)+.

2-Бром-1-(4-(2-метоксиэтокси)-2,6-диметилфенил)этанон. К раствору CH3CN (6,0 мл), содержащему 1-(4-(2-метоксиэтокси)-2,6-диметилфенил)этанон (400 мг, 1,8 ммоль, 1,0 экв.), добавляли TBABr3 (867,7 мг, 1,8 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении, и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4(s) и концентрировали при пониженном давлении. 2-бром-1-(4-(2-метоксиэтокси)-2,6-диметилфенил)этанон (322,9 мг, 1,1 ммоль) получали в виде желтого твердого вещества с выходом 60%: 1H ЯМР (CDCl3, 500 МГц) δ 6,61 (с, 2H), 4,25 (с, 2H), 4,09-4,11 (м, 2H), 3,73-3,75 (м, 2H), 3,45 (с, 3H), 2,24 (с, 6H).

4-(4-(2-Метоксиэтокси)-2,6-диметилфенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(4-(2-метоксиэтокси)-2,6-диметилфенил)этанон (322,9 мг, 1,1 ммоль, 1,0 экв.) и тиомочевину (81,61 мг, 1,1 ммоль, 1,0 экв.) в EtOH (3,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (20 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30% EtOAc в гексане в качестве элюента) с получением 4-(4-(2-метоксиэтокси)-2,6-диметилфенил)тиазол-2-амина (281,0 мг, 1,0 ммоль) в виде желтого твердого вещества с выходом 94%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 6,76 (с, 2H), 5,31-5,33 (м, 1H), 4,09-4,11 (м, 2H), 3,64-3,65 (м, 2H), 3,30 (с, 3H), 2,12 (с, 6H); ESI-MS: m/z 279,7 (M+H)+.

1-(4-(3-Метоксипропокси)-2,6-диметилфенил)этанон. В стеклянную емкость высокого давления помещали 1-(4-гидрокси-2,6-диметилфенил)этанон (800 мг, 4,9 ммоль, 1,0 экв.) и 1-хлор-3-метоксипропан (528,97 мг, 4,9 ммоль, 1,0 экв.) в 50% водном растворе NaOH (10,0 мл) и перемешивали при 140°С в течение 16 ч. Осадок разбавляли H2O (20 мл) и экстрагировали EtOAc (3×30 мл). Органический слой отделяли, сушили над MgSO4(s) и концентрировали при пониженном давлении с получением 1-(4-(3-метоксипропокси)-2,6-диметилфенил)этанона (987,8 мг, 4,2 ммоль) в виде желтого масла с выходом 86%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 6,56 (с, 2H), 4,02-4,04 (м, 2H), 3,53-3,55 (м, 2H), 3,36 (с, 3H), 2,45 (с, 3H), 2,23 (с, 6H), 2,02-2,04 (м, 3H); ESI-MS: m/z 237,7 (M+H)+.

2-Бром-1-(4-(3-метоксипропокси)-2,6-диметилфенил)этанон. К раствору CH3CN (15,0 мл), содержащему 1-(4-(3-метоксипропокси)-2,6-диметилфенил)этанон (987,8 мг, 4,2 ммоль, 1,0 экв.), добавляли TBABr3 (2,02 г, 4,2 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. 2-бром-1-(4-(3-метоксипропокси)-2,6-диметилфенил)этанон (1,23 г, 3,9 ммоль) получали в виде желтого масла с выходом 93%: 1H ЯМР (CDCl3, 500 МГц) δ 6,58 (с, 2H), 4,24-4,35 (м, 2H), 4,03-4,05 (м, 2H), 3,53-3,55 (м, 2H), 3,35 (с, 3H), 2,24 (с, 6H), 2,01-2,06 (м, 2H).

4-(4-(3-Метоксипропокси)-2,6-диметилфенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(4-(3-метоксипропокси)-2,6-диметилфенил)этанон (500,0 мг, 1,6 ммоль, 1,0 экв.) и тиомочевину (126,8 мг, 1,6 ммоль, 1,0 экв.) в EtOH (10,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (30% EtOAc в гексане в качестве элюента) с получением 4-(4-(3-метоксипропокси)-2,6-диметилфенил)тиазол-2-амина (328,9 мг, 1,1 ммоль) в виде желтого твердого вещества с выходом 71%: 1H ЯМР (CDCl3, 500 МГц) δ 9,35 (уш.с, 1H), 9,00 (уш.с, 1H), 6,64 (с, 2H), 6,22 (с, 1H), 4,04-4,05 (м, 2H), 3,54-3,56 (м, 2H), 3,37 (с, 3H), 2,19 (с, 6H), 2,03-2,06 (м, 2H); ESI-MS m/z 293,8 (M+H)+.

1-(2,6-Диметил-4-(фенилтио)фенил)этанон. Смесь 1-(4-йод-2,6-диметилфенил)этанона (1,5 г, 5,5 ммоль, 1,0 экв.), бензолтиола (0,60 мл, 8,2 ммоль, 1,5 экв.), оксида меди(I) (39,2 мг, 0,3 ммоль, 0,05 экв.) и гидроксида калия (614,1 мг, 11,0 ммоль, 2,0 экв.) в DMF (4,4 мл) и H2O (1,1 мл) нагревали с обратным холодильником в течение 20 ч. Смесь гасили H2O (10 мл) и экстрагировали простым эфиром (2×20 мл). Органический слой собирали, сушили над MgSO4(s), и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 1-(2,6-диметил-4-(фенилтио)фенил)этанона (931 мг, 3,6 ммоль) в виде желтого масла с выходом 66%: 1H ЯМР (CD3OD, 500 МГц) δ 7,34-7,35 (м, 5H), 6,97 (с, 2H), 2,46 (с, 3H), 2,17 (с, 6H); ESI-MS: m/z 257,0 (M+H)+.

2-Бром-1-(2,6-диметил-4-(фенилтио)фенил)этанон. К раствору CH3CN (15,0 мл), содержащему 1-(2,6-диметил-4-(фенилтио)фенил)этанон (816,3 мг, 3,2 ммоль, 1,0 экв.), добавляли TBABr3 (1,54 г, 3,2 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 2-бром-1-(2,6-диметил-4-(фенилтио)фенил)этанона (591,7 мг, 1,6 ммоль) в виде желтого масла с выходом 55%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,36-7,42 (м, 5H), 7,01 (с, 2H), 4,75 (с, 2H), 2,13 (с, 6H).

4-(2,6-Диметил-4-(фенилтио)фенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(2,6-диметил-4-(фенилтио)фенил)этанон (591,7 мг, 1,8 ммоль, 1,0 экв.) и тиомочевину (134,3 мг, 1,8 ммоль, 1,0 экв.) в EtOH (15,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (5,0% EtOAc в гексане в качестве элюента) с получением 4-(2,6-диметил-4-(фенилтио)фенил)тиазол-2-амина (483,7 мг, 1,6 ммоль) в виде желтого твердого вещества с выходом 88%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,33-7,38 (м, 2H), 7,29-7,33 (м, 3H), 7,06 (уш.с, 2H), 6,89 (с, 2H), 6,38 (с, 1H), 2,07 (с, 6H); ESI-MS: m/z 313,8 (M+H)+.

1-(2,6-Диметил-4-(п-толилтио)фенил)этанон. Смесь 1-(4-йод-2,6-диметилфенил)этанона (1,5 г, 5,5 ммоль, 1,0 экв.), 4-метилбензолтиола (1,02 г, 8,2 ммоль, 1,5 экв.), оксида меди(I) (39,2 мг, 0,3 ммоль, 0,05 экв.) и гидроксида калия (614,1 мг, 11,0 ммоль, 2,0 экв.) в DMF (4,4 мл) и H2O (1,1 мл) нагревали с обратным холодильником в течение 20 ч. Смесь гасили H2O (10 мл) и экстрагировали простым эфиром (2×20 мл). Органический слой собирали, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 1-(2,6-диметил-4-(п-толилтио)фенил)этанона (1,16 г, 4,3 ммоль) в виде желтого масла с выходом 79%: 1H ЯМР (CD3OD, 500 МГц) δ 7,29 (д, J=8,0 Гц, 2H), 7,20 (д, J=8,0 Гц, 2H), 6,88 (с, 2H), 2,45 (с, 3H), 2,35 (с, 3H), 2,15 (с, 6H); ESI-MS: m/z 271,8 (M+H)+.

2-Бром-1-(2,6-диметил-4-(п-толилтио)фенил)этанон. К раствору CH3CN (20,0 мл), содержащему 1-(2,6-диметил-4-(п-толилтио)фенил)этанон (1,0 г, 3,7 ммоль, 1,0 экв.), добавляли TBABr3 (1,79 г, 3,7 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 2-бром-1-(2,6-диметил-4-(п-толилтио)фенил)этанона (394,8 мг, 1,1 ммоль) в виде желтого масла с выходом 31%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,33 (д, J=8,0 Гц, 2H), 7,25 (д, J=8,0 Гц, 2H), 6,92 (с, 2H), 4,76 (с, 2H), 2,32 (с, 3H), 2,11 (с, 6H).
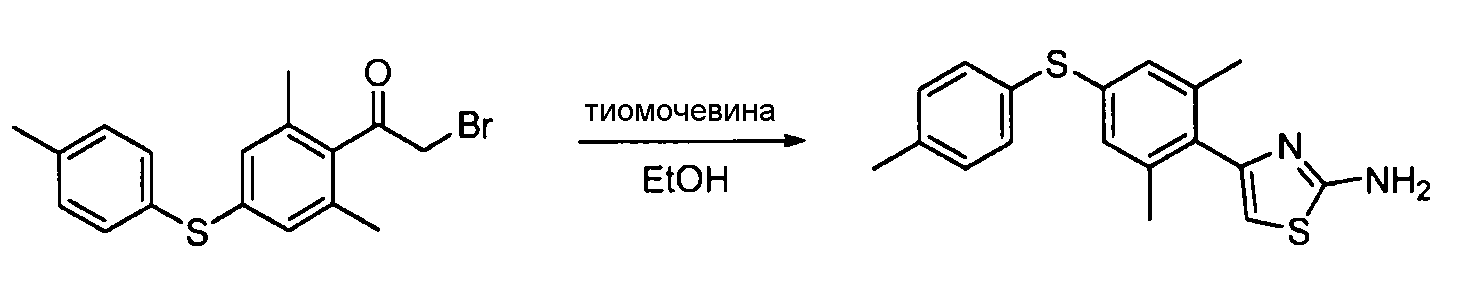
4-(2,6-Диметил-4-(п-толилтио)фенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(2,6-диметил-4-(п-толилтио)фенил)этанон (394,8 мг, 1,1 ммоль, 1,0 экв.) и тиомочевину (86,04 мг, 1,1 ммоль, 1,0 экв.) в EtOH (10,0 мл) нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (5,0% EtOAc в гексане в качестве элюента) с получением 4-(2,6-диметил-4-(п-толилтио)фенил)тиазол-2-амина (371,9 мг, 1,1 ммоль) в виде желтого твердого вещества с выходом >99%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,27 (д, J=8,0 Гц, 2H), 7,21 (д, J=8,0 Гц, 2H), 6,97 (с, 2H), 6,87 (с, 2H), 6,36 (с, 1H), 2,30 (с, 3H), 2,05 (с, 6H); ESI-MS: m/z 327,0 (M+H)+.

1-(4-(4-Метоксифенилтио)-2,6-диметилфенил)этанон. Смесь 1-(4-йод-2,6-диметилфенил)этанона (1,5 г, 5,5 ммоль, 1,0 экв.), 4-метоксибензолтиола (1,01 мл, 8,2 ммоль, 1,5 экв.), оксида меди(I) (39,2 мг, 0,3 ммоль, 0,05 экв.) и гидроксида калия (614,1 мг, 11,0 ммоль, 2,0 экв.) в DMF (4,4 мл) и H2O (1,1 мл) нагревали с обратным холодильником в течение 20 ч. Смесь гасили H2O (10 мл) и экстрагировали простым эфиром (2×20 мл). Органический слой собирали, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанона (1,41 г, 4,9 ммоль) в виде желтого масла с выходом 90%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,39 (д, J=8,5 Гц, 2H), 6,96 (д, J=8,5 Гц, 2H), 6,79 (с, 2H), 3,82 (с, 3H), 2,43 (с, 3H), 2,13 (с, 6H); ESI-MS: m/z 287,6 (M+H)+.

2-Бром-1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанон. К раствору CH3CN (20,0 мл), содержащему 1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанон (1,0 г, 3,5 ммоль, 1,0 экв.), добавляли ΤΒΑΒr3 (1,684 г, 3,5 ммоль, 1,0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (3,0% EtOAc в гексане в качестве элюента) с получением 2-бром-1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанона (1,06 г, 2,9 ммоль) в виде желтого масла с выходом 83%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,44 (д, J=8,7 Гц, 2H), 7,03 (д, J=8,7 Гц, 2H), 6,83 (с, 2H), 4,71 (с, 2H), 3,80 (с, 3H), 2,10 (с, 6H).

4-(4-(4-Метоксифенилтио)-2,6-диметилфенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанон (1,06 г, 2,9 ммоль, 1,0 экв.) и тиомочевину (221,5 мг, 2,9 ммоль, 1,0 экв.) в EtOH (20,0 мл), нагревали с обратным холодильником в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (5,0% EtOAc в гексане в качестве элюента) с получением 4-(4-(4-метоксифенилтио)-2,6-диметилфенил)тиазол-2-амина (890,9 мг, 2,6 ммоль) в виде желтого твердого вещества с выходом 90%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,40 (д, J=8,7 Гц, 2H), 7,00 (д, J=8,7 Гц, 2H), 6,86-6,87 (м, 4H), 6,33 (с, 1H), 3,78 (с, 3H), 2,03 (с, 6H); ESI-MS m/z 343,9 (M+H)+.

2-Бром-1-(4-(4-метоксифенилсульфонил)-2,6-диметилфенил)этанон. Смесь 2-бром-1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанона (1,0 г, 2,7 ммоль, 1,0 экв.) и м-хлорпероксибензойной кислоты (1,69 г, 6,8 ммоль, 2,5 экв.) в дихлорметане (10,0 мл) перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 2-бром-1-(4-(4-метоксифенилсульфонил)-2,6-диметилфенил)этанона (1,09 г, 2,7 ммоль) в виде белого твердого вещества с выходом >99%: 1H ЯМР (CDCl3, 500 МГц) δ 7,85-7,90 (м, 2H), 7,57-7,60 (м, 2H), 6,97-6,90 (м, 2H), 4,21 (с, 2H), 3,85 (с, 3H), 2,30 (с, 6H).

4-(4-(4-Метоксифенилсульфонил)-2,6-диметилфенил)тиазол-2-амин. Реакционную смесь, содержащую 2-бром-1-(4-(4-метоксифенилсульфонил)-2,6-диметилфенил)этанон (1,33 г, 3,4 ммоль, 1,0 экв.) и тиомочевину (254,8 мг, 3,4 ммоль, 1,0 экв.) в EtOH (5,0 мл) нагревали с обратным холодильником в течение 1,0 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Полученные твердые вещества промывали гексаном с получением 4-(4-(4-метоксифенилсульфонил)-2,6-диметилфенил)тиазол-2-амина (839,2 мг, 2,2 ммоль) в виде желтого твердого вещества с выходом 65%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,89 (д, J=8,9 Гц, 2H), 7,61 (с, 2H), 7,13 (д, J=8,9 Гц, 2H), 6,95 (уш.с, 2H), 6,43 (с, 1H), 3,83 (с, 3H), 2,16 (с, 6H); ESI-MS: m/z 375,6 (M+H)+.

2-Бром-1-(4-(4-метоксифенилсульфинил)-2,6-диметилфенил)этанон. Смесь 2-бром-1-(4-(4-метоксифенилтио)-2,6-диметилфенил)этанона (500,0 мг, 1,3 ммоль, 1,0 экв.), уксусного ангидрида (0,14 мл, 1,5 ммоль, 1,1 экв.), 30% пероксида водорода (55,86 мг, 1,6 ммоль, 1,2 экв.) и силикагеля (273,75 мг, 230-400 меш) в дихлорметане (10,0 мл) перемешивали при комнатной температуре в течение 16 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении с получением 2-бром-1-(4-(4-метоксифенилсульфинил)-2,6-диметилфенил)этанона (235,4 мг, 0,6 ммоль) в виде светло-желтого масла с выходом 48%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,65 (д, J=8,8 Гц, 2H), 7,41 (с, 2H), 7,09 (д, J=8,8 Гц, 2H), 4,78 (с, 2H), 3,79 (с, 3H), 2,21 (с, 6H).

N-(4-(4-(4-Метоксифенилсульфинил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид. Реакционную смесь, содержащую 2-бром-1-(4-(4-метоксифенилсульфинил)-2,6-диметилфенил)этанон (235,4 мг, 0,6 ммоль, 1,0 экв.) и тиомочевину (47,0 мг, 0,60 ммоль, 1,0 экв.) в EtOH (5,0 мл), нагревали с обратным холодильником в течение 1,0 ч. Раствор концентрировали при пониженном давлении и осадок перерастворяли в EtOAc (50 мл). Раствор промывали насыщенным водным NaHCO3 (30 мл), сушили над MgSO4 и концентрировали при пониженном давлении. Полученные твердые вещества промывали гексаном с получением N-(4-(4-(4-метоксифенилсульфинил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамида (236,7 мг, 0,70 ммоль) в виде желтого твердого вещества с выходом >99%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 7,64 (д, J=8,9 Гц, 2H), 7,34 (с, 2H), 7,09 (д, J=8,9 Гц, 2H), 6,90 (уш.с, 2H), 6,39 (с, 1H), 3,79 (с, 3H), 2,13 (с, 6H); ESI-MS m/z 359,0 (M+H)+.

5-Метил-4-фенилтиазол-2-амин. Смесь 2-бром-1-фенилпропан-1-она (3,00 г, 19,5 ммоль) и тиомочевины (1,56 г, 20,5 ммоль) в 95% EtOH (30 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 5-метил-4-фенилтиазол-2-амина (4,07 г) в виде желтого твердого вещества с выходом 77%: 1H ЯМР (500 МГц, ДМСО-d6) δ 8,85 (с, 2H), 7,54-7,49 (м, 5H), 2,28 (с, 3H).

2-Бром-1-(4-метоксифенил)пропан-1-он. К раствору 1-(4-метоксифенил)пропан-1-она (5,01 г, 30,2 моль) в EtOAc (120 мл) добавляли бромид меди(II) (CuBr2, 13,6 г, 6,8 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 90 мин. Раствору позволяли остыть и полученные твердые вещества отфильтровывали и промывали EtOAc. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-бром-1-(4-метоксифенил)пропан-1-она (10,4 г) в виде желтого масла: 1H ЯМР (500 МГц, CDCl3) δ 8,02 (2H, м), 6,96 (2H, м), 5,28-5,25 (м, 1H), 3,89 (с, 3H), 1,89 (д, 3H).

4-(4-Метоксифенил)-5-метилтиазол-2-амин. Смесь 2-бром-1-(4-метоксифенил)пропан-1-она (10,4 г, 36,1 ммоль) и тиомочевины (2,76 г, 36,2 ммоль) в 95% EtOH (70 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-метоксифенил)-5-метилтиазол-2-амина (6,16 г) в виде желтого твердого вещества с выходом 78%: 1H ЯМР (500 МГц, ДМСО-d6) δ 8,90 (с, 2H), 7,46-7,44 (м, 2H), 7,09-7,07 (м, 2H), 3,81 (с, 3H), 2,47 (с, 3H).

2-Бром-1-(2,4,6-триметоксифенил)этанон. К раствору 1-(2,4,6-триметоксифенил)этанона (5,0 г, 23,3 ммоль) в EtOAc (100 мл) добавляли бромид меди(II) (CuBr2, 10,4 г, 46,7 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 90 мин. Раствору позволяли остыть и полученные твердые вещества отфильтровывали и промывали EtOAc. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-бром-1-(2,4,6-триметоксифенил)этанона (2,70 г) в виде желтого масла: 1H-ЯМР (500 МГц, CDCl3) δ 6,11 (м, 2H), 4,36 (м, 2H), 3,86 (с, 3H), 3,82 (с, 6H).

4-(2,4,6-Триметоксифенил)тиазол-2-амин. Смесь 2-бром-1-(2,4,6-триметоксифенил)этанона (2,49 г, 8,6 ммоль) и тиомочевины (0,67 г, 8,7 ммоль) в 95% EtOH (16 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,4,6-триметоксифенил)тиазол-2-амина (1,75 г) в виде желтого твердого вещества с выходом >99%: 1H ЯМР (500 МГц, ДМСО-d6) δ 9,00 (с, 2H), 6,78 (с, 1H), 6,36 (с, 2H), 3,84 (с, 3H), 3,79 (с, 6H).

2-Бром-1-(4-метоксифенил)этанон. К раствору 1-(4-метоксифенил)этанона (15,2 г, 0,10 моль) в EtOAc (250 мл) добавляли бромид меди(II) (CuBr2, 45,1 г, 0,20 моль). Реакционную смесь нагревали с обратным холодильником в течение 90 мин. Раствору позволяли остыть и полученные твердые вещества отфильтровывали и промывали EtOAc. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-бром-1-(4-метоксифенил)этанона (15,8 г) в виде желтого масла: 1H ЯМР (500 МГц, CDCl3) δ 7,98 (м, 2H), 6,97 (м, 2H), 4,41 (с, 3H), 3,89 (с, 6H).

4-(4-Метоксифенил)тиазол-2-амин. Смесь 2-бром-1-(4-метоксифенил)этанона (5,00 г, 21,8 ммоль) и тиомочевины (1,72 г, 22,6 ммоль) в 95% EtOH (40 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(4-метоксифенил)тиазол-2-амина (5,24 г) в виде желтого твердого вещества с выходом >99%: 1H ЯМР (500 МГц, ДМСО-d6) δ 7,72 (д, 2H), 6,99 (с, 2H), 6,92-6,91 (м, 2H), 6,82 (с, 1H), 3,76 (с, 3H).

2-Бром-1-(2,4-диметоксифенил)этанон. К раствору 1-(2,4-диметоксифенил)этанона (10,0 г, 54,4 ммоль) в EtOAc (220 мл) добавляли бромид меди(II) (CuBr2, 24,3 г, 0,11 моль). Реакционную смесь нагревали с обратным холодильником в течение 90 мин. Раствору позволяли остыть и полученные твердые вещества отфильтровывали и промывали EtOAc. Фильтрат концентрировали при пониженном давлении с получением неочищенного 2-бром-1-(2,4-диметоксифенил)этанона (14,5 г) в виде желтого масла: 1H ЯМР (500 МГц, CDCl3) δ 7,91 (м, 2H), 6,52 (м, 2H), 4,57 (с, 3H), 3,98 (с, 3H), 3,85 (с, 3H).

4-(2,4-Диметоксифенил)тиазол-2-амин. Смесь 2-бром-1-(2,4-диметоксифенил)этанона (14,5 г, 55,8 ммоль) и тиомочевины (4,32 г, 56,7 ммоль) в 95% EtOH (110 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле. Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,4-диметоксифенил)тиазол-2-амина (10,9 г) в виде желтого твердого вещества с выходом 62%: 1H ЯМР (500 МГц, ДМСО-d6) δ 8,60 (с, 2H), 7,53 (с, 1H), 6,97 (с, 1H), 6,69 (с, 1H), 6,67-6,63 (м, 1H), 3,86 (с, 3H), 3,80 (с, 3H).

2-Хлор-1-(2,4,6-трифторфенил)этанон. К механически перемешиваемому раствору 1,3,5-трифторбензола (6,0 мл, 58 ммоль) в дихлорэтане (14,0 мл) осторожно постепенно добавляли AlCl3 (15,5 г, 116 ммоль) в течение 15 мин. Наблюдали энергичное бурление и выделение газообразного HCl. Смесь осторожно нагревали с обратным холодильником и капельно добавляли хлорацетилхлорид (5,5 мл, 69 ммоль) в течение 45 мин. Реакционную смесь нагревали с обратным холодильником в течение дополнительных 6,0 ч. Раствор охлаждали, осторожно выливали в смесь лед/вода (200 мл) и водный раствор экстрагировали простым эфиром (3×50 мл). Объединенные слои простого эфира промывали 10% водной HCl (2×30 мл), 1,0н водным NaOH (3×30 мл) и рассолом (25 мл). Раствор сушили над MgSO4 и концентрировали при пониженном давлении с получением 2-хлор-1-(2,4,6-трифторфенил)этанона (5,28 г) в виде желтого твердого вещества с выходом 51%: 1H ЯМР (500 МГц, CDCl3) δ 6,79-6,76 (м, 2H), 4,50 (с, 2H).

4-(2,4,6-Трифторфенил)тиазол-2-амин. Смесь 2-хлор-1-(2,4,6-трифторфенил)этанона (9,04 г, 43,5 ммоль) и тиомочевины (3,51 г, 46,1 ммоль) в 95% EtOH (50 мл) нагревали с обратным холодильником в течение ночи. Раствор концентрировали и смешивали с водой (100 мл) и насыщенным водным Na2CO3 (5,0 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,4,6-трифторфенил)тиазол-2-амина (9,71 г) в виде розовато-белого твердого вещества с выходом 97%: 1H ЯМР (500 МГц, ДМСО-d6) δ 7,26-7,22 (м, 2H), 7,09 (с, 2H), 6,77 (с, 1H).

1-(2,6-Диметил-4-(фениламино)фенил)этанон. К раствору 1-(4-амино-2,6-диметилфенил)этанона (3,26 г, 20,0 ммоль), K3PO4 (9,2 г, 40 ммоль) и 1-йодбензола (4,08 г, 20,0 ммоль) в DMF (35,0 мл) добавляли CuI (761,8 мг, 40 ммоль). Реакционную смесь нагревали при 110°С в течение ночи в атмосфере N2. Раствор охлаждали до комнатной температуры и фильтровали через небольшой слой целита. Осадок на фильтре промывали этилацетатом (50 мл) и объединенный фильтрат концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(2,6-диметил-4-(фениламино)фенил)этанона в виде красно-коричневого сиропа: 1H ЯМР (500 МГц, CDCl3) δ 7,30 (д, J=8,2 Гц, 2H), 7,10 (д, J=7,7 Гц, 2H), 6,99 (д, J=4,2 Гц, 1H), 6,71 (с, 1H), 2,47 (с, 3H) , 2,18 (с, 6H); ESI-MS: m/z 239,5 (M+H)+.

1-(4-(4-Бромфениламино)-2,6-диметилфенил)-2-бромэтанон. К раствору 1-(2,6-диметил-4-(фениламино)фенил)этанона (2,10 г, 8,78 ммоль) в ацетонитриле (50 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 4,24 г, 8,78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 60 мин. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 1-(4-(4-бромфениламино)-2,6-диметилфенил)-2-бромэтанона (2,01 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(4-(4-Бромфениламино)-2,6-диметилфенил)тиазол-2-амин. Раствор 1-(4-(4-бромфениламино)-2,6-диметилфенил)-2-бромэтанона (1,6 г, 4,0 ммоль) и тиомочевины (0,79 г, 7,2 ммоль) в ацетонитриле (30 мл) нагревали с обратным холодильником в течение 90 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (1,0 мл) и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением продукта (1,1 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

3-Хлор-5-метилфениламин. В раствор этанола (75 мл), содержащий 1-хлор-3-метил-5-нитробензол (5,0 г, 29 ммоль), добавляли SnCl2·2H2O (32,8 г, 146 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 3,0 ч. Раствор концентрировали в вакууме и осадок перерастворяли в водном NaOH, фильтровали и экстрагировали EtOAc. Органический слой собирали, промывали рассолом, сушили над MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле с получением 3-хлор-5-метилфениламина (4,0 г) в виде светло-желтого твердого вещества с выходом 97%: 1H ЯМР (500 МГц, CDCl3) δ 6,56 (с, 1H), 6,48 (с, 1H), 6,36 (с, 1H), 3,66 (уш.с, 2H), 2,23 (с, 3H); ESI-MS: m/z 141,7 (M+H)+.

N-(3-Хлор-5-метилфенил)ацетамид. Уксусный ангидрид (6,7 мл) и 3-хлор-5-метилфениламин (5,0 г, 35 ммоль) смешивали и выдерживали в течение 2,0 ч. Реакционную смесь охлаждали до комнатной температуры с получением N-(3-хлор-5-метилфенил)ацетамида (5,1 г) в виде светло-желтого твердого вещества с выходом 79%: 1H ЯМР (500 МГц, CDCl3) δ 7,38 (с, 1H), 7,19 (с, 1H), 7,12 (с, 1H), 6,91 (с, 1H), 2,31 (с, 3H), 2,16 (с, 3H).
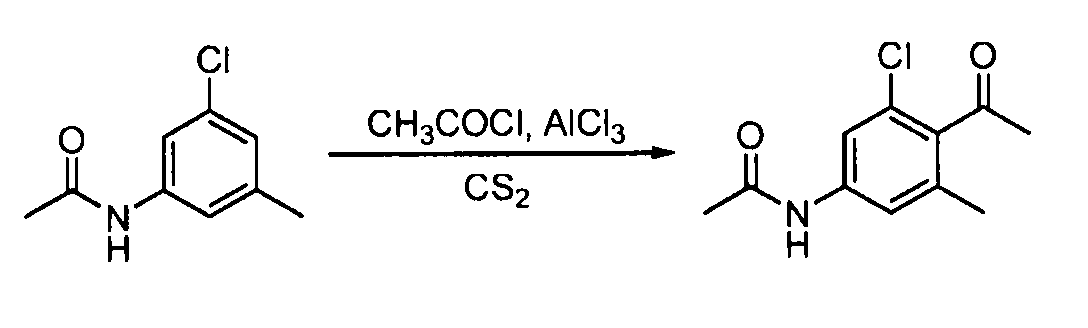
N-(4-Ацетил-3-хлор-5-метилфенил)ацетамид. В сухой раствор CS2 (30 мл), содержащий N-(3-хлор-5-метилфенил)ацетамид (5,0 г, 27 ммоль) и ацетилхлорид (2,9 мл, 40,8 ммоль), медленно добавляли хлорид алюминия (9,1 г, 68 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 30 мин, охлаждали до комнатной температуры и выдерживали в течение 4,0 ч. CS2 удаляли и оставшийся сироп выливали в ледяную HCl. Полученные твердые вещества собирали, перерастворяли в EtOH и обесцвечивали с помощью угля. Раствор фильтровали и фильтрат концентрировали в вакууме с получением N-(4-ацетил-3-хлор-5-метилфенил)ацетамида (5,2 г) в виде светло-желтого твердого вещества с выходом 85%: 1H ЯМР (500 МГц, CDCl3) δ 7,48 (с, 1H), 7,26 (с, 1H), 7,21 (с, 1H), 2,52 (с, 3H), 2,24 (с, 3H), 2,18 (с, 3H).

1-(4-Амино-2-хлор-6-метилфенил)этанон. Раствор этанола (4,0 мл), содержащий N-(4-ацетил-3-хлор-5-метилфенил)ацетамид (0,53 г, 2,3 ммоль) и концентрированную хлористоводородную кислоту (1,6 мл), нагревали с обратным холодильником в течение 15 ч. В раствор добавляли 10% водный NaOH и полученные твердые вещества собирали с получением 1-(4-амино-2-хлор-6-метилфенил)этанона (0,37 г) в виде светло-желтого твердого вещества с выходом 88%: 1H ЯМР (500 МГц, CDCl3) δ 6,46 (д, J=1,77 Гц, 1H), 6,34 (с, 1H), 3,85 (уш.с, 2H), 2,49 (с, 3H), 2,14 (с, 3H): ESI-MS: m/z 183,4 (M+H)+.

1-(2-Хлор-4-йод-6-метилфенил)этанон. К раствору CH3CN (20 мл), содержащему KI (2,5 г, 15 ммоль) и трет-бутилнитрит (2,00 мл, 16,9 ммоль), добавляли 1-(4-амино-2-хлор-6-метилфенил)этанон (2,3 г, 12,5 ммоль) в CH3CN (13 мл) при -10°С. Реакционную смесь нагревали до комнатной температуры и выливали в водный раствор HCl (20%, 23 мл). Раствор экстрагировали EtOAc (20 мл) и органический слой отделяли, промывали H2O (23 мл), сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-(2-хлор-4-йод-6-метилфенил)этанона (1,28 г) в виде желтого масла с выходом 35%: 1H ЯМР (500 МГц, CDCl3) δ 7,58 (с, 1H), 7,49 (с, 1H), 2,51 (с, 3H), 2,21 (с, 3H).

1-[2-Хлор-4-(4-метоксифенокси)-6-метилфенил]этанон. К раствору 1-(2-хлор-4-йод-6-метилфенил)этанона (1,1 г, 3,7 ммоль), K3PO4 (1,6 г, 7,4 ммоль) и 4-метоксифенола (0,55 г, 4,44 ммоль) в DMF (55 мл) добавляли бромид тетрабутиламмония (0,12 г, 0,37 ммоль) и йодид меди(I) (70 мг, 0,37 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 22 ч. Раствор экстрагировали EtOAc (10 мл) и органический слой отделяли, промывали H2O (11 мл), сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением 1-[2-хлор-4-(4-метоксифенокси)-6-метилфенил]этанона в виде желтого масла с выходом 19%: 1H ЯМР (500 МГц, CDCl3) δ 6,97 (м, 2H), 6,90 (м, 2H), 6,73 (д, J=2,19 Гц, 1H), 6,67 (д, J=1,99 Гц, 1H), 3,81 (с, 3H), 2,52 (с, 3H), 2,20 (с, 3H).

2-Бром-1-[2-хлор-4-(4-метоксифенокси)-6-метилфенил]этанон. К раствору 1-[2-хлор-4-(4-метоксифенокси)-6-метилфенил]этанона (0,20 г, 0,69 ммоль) в ацетонитриле (6,0 мл) добавляли ΤΒΑΒr3 (0,33 г, 0,69 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(2,6-диметил-4-феноксифенил)этанона, который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-[2-Хлор-4-(4-метоксифенокси)-6-метилфенил]тиазол-2-иламин. Смесь 2-бром-1-(2,6-диметил-4-феноксифенил)этанона и тиомочевины (63 мг, 0,83 ммоль) в 95% EtOH (3,0 мл) нагревали с обратным холодильником в течение 60 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный NaHCO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-[2-хлор-4-(4-метоксифенокси)-6-метилфенил]тиазол-2-иламина (0,10 г) в виде желтого твердого вещества с выходом 42%: 1H ЯМР (500 МГц, CDCl3) δ 6,98 (м, 2H), 6,90 (м, 2H), 6,83 (д, J=2,4 Гц, 1H), 6,73 (д, J=2,3 Гц, 1H), 6,41 (с, 1H), 4,97 (уш.с, 2H), 3,81 (с, 3H), 2,16 (с, 3H).

2-Бром-1-(2,6-диметил-4-(метилтио)фенил)этанон. К раствору 1-(4-(циклопентилокси)-2,6-диметилфенил)этанона (3,30 г, 17,0 ммоль) в ацетонитриле (34,0 мл) добавляли тетрабутиламмонийтрибромид (TBABr3, 8,19 г, 17,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали при пониженном давлении, добавляли воду и экстрагировали этилацетатом. Органический слой промывали рассолом, сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(2,6-диметил-4-(метилтио)фенил)этанона (5,2 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,6-Диметил-4-(метилтио)фенил)тиазол-2-амин. Смесь 2-бром-1-(2,6-диметил-4-(метилтио)фенил)этанона (4,64 г, 17,0 ммоль) и тиомочевины (1,29 г, 17,0 ммоль) в 95% EtOH (24,3 мл) нагревали с обратным холодильником в течение 120 мин. Раствор концентрировали и добавляли воду (50 мл) и насыщенный водный Na2CO3 (4,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (30 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,6-диметил-4-(метилтио)фенил)тиазол-2-амина (1,9 г) в виде светло-желтого твердого вещества с выходом 45%: 1H ЯМР (500 МГц, CDCl3) δ 6,97 (с, 2H), 6,26 (с, 1H), 2,47 (с, 3H), 2,15 (с, 6H).

2-Бром-1-(2,6-диметил-4-(метилсульфонил)фенил)этанон. К раствору 2-бром-1-(2,6-диметил-4-(метилтио)фенил)этанона (4,92 г, 0,653 моль) в CH2Cl2 (36 мл) при 0°C добавляли mCPBA (70%, 11,1 г, 1,63 моль). Смесь перемешивали при комнатной температуре в течение 7,0 ч. Раствор фильтровали и к фильтрату добавляли насыщенный водный NaHCO3 (50 мл). Органический слой сушили над безводным MgSO4(s) и концентрировали при пониженном давлении с получением 2-бром-1-(2,6-диметил-4-(метилсульфонил)фенил)этанона (7,6 г), который использовали непосредственно на следующей стадии без дальнейшей очистки.

4-(2,6-Диметил-4-(метилсульфонил)фенил)тиазол-2-амин. Смесь 2-бром-1-(2,6-диметил-4-(метилсульфонил)фенил)этанона (7,60 г, 24,9 ммоль) и тиомочевины (1,90 г, 25,0 ммоль) в 95% EtOH (35,6 мл) нагревали с обратным холодильником в течение 90 мин. Раствор концентрировали и добавляли воду (100 мл) и насыщенный водный Na2CO3 (5,0 мл). Полученный осадок фильтровали и перекристаллизовывали в толуоле (20 мл). Твердые вещества отфильтровывали и сушили в вакууме с получением 4-(2,6-диметил-4-(метилсульфонил)фенил)тиазол-2-амина (3,28 г) в виде желтого твердого вещества с выходом 47%: 1H ЯМР (500 МГц, CDCl3) δ 7,64 (с, 2H), 6,34 (с, 1H), 5,1 (м, 1H), 3,04 (с, 3H), 2,26 (с, 6H).

2-Амино-N-(4-(4-(4-метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид. Смесь N-(4-(4-(4-метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)-2-нитроизоникотинамида (0,20 г, 0,40 ммоль) и Pd/C (0,15 г, 10% масс./масс.) в этаноле (10 мл) перемешивали в атмосфере H2 в течение ночи. Реакционную смесь фильтровали через диатомовую землю и концентрировали при пониженном давлении с получением 2-амино-N-(4-(4-(4-метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамида (0,11 г) в виде желтого твердого вещества с выходом 59%: 1H-ЯМР (500 МГц, ДМСО-d6) δ 7,88-7,89 (м, 1H), 7,10-7,11 (м, 2H), 6,95-6,97 (м, 2H), 6,62 (с, 1H), 5,76 (с, 1H), 3,29 (с, 3H), 2,03 (с, 6H); ESI-MS: m/z 446,6 [M+H]+.

N-(4-Мезитилтиазол-2-ил)-2-морфолиноизоникотинамид. Смесь 2-хлор-N-(4-мезитилтиазол-2-ил)изоникотинамида (500,0 мг, 1,4 ммоль, 1,0 экв.) и морфолина (1,5 мл, 16,8 ммоль, 12 экв.) в метилпирролидоне (15,0 мл) перемешивали при 150°С в течение 16 ч. Смесь выливали в ледяную H2O (20,0 мл) и полученное твердое вещество фильтровали с получением N-(4-мезитилтиазол-2-ил)-2-морфолиноизоникотинамида (358,6 мг, 0,90 ммоль) в виде желтого твердого вещества с выходом 63%: 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,30 (д, J=5,1 Гц, 2H), 7,50 (с, 1H), 7,22 (д, J=5,1 Гц, 2H), 7,10 (с, 1H), 6,92 (с, 2H), 3,70-3,73 (м, 4H), 3,53-3,55 (м, 4H), 2,26 (с, 3H), 2,05 (с, 6H); ESI-MS: m/z 409,3 (M+H)+.

N-(4-Мезитилтиазол-2-ил)-2-(4-метилпиперазин-1-ил)изоникотинамид. Смесь 2-хлор-N-(4-мезитилтиазол-2-ил)изоникотинамида (300,0 мг, 0,8 ммоль, 1,0 экв.) и 1-метилпиперазина (1,12 мл, 10,1 ммоль, 12 экв.) в метилпирролидоне (9,0 мл) перемешивали при 150°С в течение 16 ч. Смесь выливали в ледяную H2O (15,0 мл) и полученные твердые вещества фильтровали с получением N-(4-мезитилтиазол-2-ил)-2-(4-метилпиперазин-1-ил)изоникотинамида (95,6 мг, 0,20 ммоль) в виде желтого твердого вещества с выходом 27%: 1H ЯМР (CDCl3, 500 МГц) δ 8,27 (д, J=5,1 Гц, 1H), 7,12 (с, 1H), 6,83-6,86 (м, 3H), 6,78 (с, 1H), 3,63-3,65 (м, 4H), 2,35 (с, 3H), 2,27 (с, 3H), 2,04 (с, 6H); ESI-MS: m/z 422,1 (M+H)+.

N-(4-Мезитилтиазол-2-ил)-2-(пиперидин-1-ил)изоникотинамид. Смесь 2-хлор-N-(4-мезитилтиазол-2-ил)изоникотинамида (200 мг, 0,60 ммоль, 1,0 экв.) и пиперидина (0,70 мл, 6,7 ммоль, 12 экв.) в метилпирролидоне (6,0 мл) перемешивали при 150°С в течение 16 ч. Смесь выливали в ледяную H2O (10,0 мл) и полученные твердые вещества фильтровали. Твердые вещества очищали колоночной хроматографией на силикагеле (15% EtOAc в гексане в качестве элюента) с получением N-(4-мезитилтиазол-2-ил)-2-(пиперидин-1-ил)изоникотинамида (87,2 мг, 0,20 ммоль) в виде желтого твердого вещества с выходом 38%: 1H ЯМР (CDCl3, 500 МГц) δ 8,29 (д, J=5,1 Гц, 1H), 7,15 (с, 1H), 6,83-6,90 (м, 3H), 6,79 (с, 1H), 3,61-3,63 (м, 4H), 2,31 (с, 3H), 2,08 (с, 6H), 1,57-1,67 (м, 6H); ESI-MS: m/z 407,2 (M+H)+.

2-(Диметиламино)-N-(4-мезитилтиазол-2-ил)изоникотинамид. Смесь 2-хлор-N-(4-мезитилтиазол-2-ил)изоникотинамида (200 мг, 0,60 ммоль, 1,0 экв.), карбоната цезия (2,73 г, 0,6 ммоль, 15 экв.) и 2,0 M диметиламина в THF (3,4 мл, 6,7 ммоль, 12 экв.) в DMF (6,0 мл) нагревали с обратным холодильником в течение 16 ч. Смесь выливали в ледяную H2O (10,0 мл) и экстрагировали EtOAc. Органический слой собирали, сушили над MgSO4(s) и концентрировали при пониженном давлении. Осадок очищали колоночной хроматографией на силикагеле (15% EtOAc в гексане в качестве элюента) с получением 2-(диметиламино)-N-(4-мезитилтиазол-2-ил)изоникотинамида (5,5 мг, 0,10 ммоль) в виде желтого твердого вещества с выходом 3,0%: 1H ЯМР (CDCl3, 500 МГц) δ 8,32 (д, J=5,1 Гц, 1H), 7,02 (с, 1H), 6,92 (с, 2H), 6,85 (д, J=5,1 Гц, 1H), 6,80 (с, 1H), 3,16 (с, 6H), 2,31 (с, 3H), 2,09 (с, 6H); ESI-MS: m/z 367,1 (M+H)+.

N-(4-(4-Бензил-2,6-диметилфенил)тиазол-2-ил)изоникотинамид. Раствор бромида бензилцинка(II) (4,0 мл, 2,0 ммоль) в THF добавляли к дегазированному раствору N-(4-(4-йод-2,6-диметилфенил)тиазол-2-ил)изоникотинамида (435 мг, 1,0 ммоль) и тетракистрифенилфосфинпалладия (57,8 мг, 0,10 ммоль) в THF (5,0 мл). Реакционную смесь нагревали с обратным холодильником в течение 16 ч в атмосфере N2, а затем выливали в насыщенный водный NaHCO3. Смесь экстрагировали этилацетатом, промывали рассолом, сушили MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением N-(4-(4-бензил-2,6-диметилфенил)тиазол-2-ил)изоникотинамида: 1H ЯМР (500 МГц, CDCl3) δ 8,70 (д, J=4,9 Гц, 2H), 7,67 (д, J=4,9 Гц, 2H), 7,33 (д, J=8,6 Гц, 2H), 7,10-7,26 (м, 3H), 6,80 (с, 1H), 6,24 (с, 1H), 3,86 (с, 2H), 2,04 (с, 6H); ESI-MS: m/z 399,9 (M+H)+.

N-(4-(4-(4-Метоксибензил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид. Раствор бромида 4-метоксибензилцинка(II) (4,0 мл, 2,0 ммоль) в THF добавляли к дегазированному раствору N-(4-(4-йод-2,6-диметилфенил)тиазол-2-ил)изоникотинамида (435 мг, 1,0 ммоль) и тетракистрифенилфосфинпалладия (57,8 мг, 0,10 ммоль) в THF (5,0 мл). Реакционную смесь нагревали с обратным холодильником в течение 16 ч в атмосфере N2, затем выливали в насыщенный водный NaHCO3. Смесь экстрагировали этилацетатом, промывали рассолом, сушили MgSO4 и концентрировали при пониженном давлении. Осадок очищали колоночной флэш-хроматографией на силикагеле с получением N-(4-(4-(4-метоксибензил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамида: 1H ЯМР (500 МГц, CDCl3) δ 8,69 (д, J=5,2 Гц, 2H), 7,66 (д, J=4,9 Гц, 2H), 7,11 (д, J=8,4 Гц, 2H), 6,86 (д, 2H), 6,80 (с, 1H), 6,75 (с, 2H), 3,80 (с, 2H), 3,78 (с, 2H), 1,98 (с, 6H); ESI-MS: m/z 399,9 (M+H)+.
Иллюстративные соединения и физико-химические данные

4-Циано-N-(мезитилтиазол-2-ил)бензамид
Соединение 4
Выход: 67%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,23 (д, 2Н), 8,02 (д, 2Н), 7,09 (с, 1Н), 6,92 (с, 2Н), 2,26 (с, 3Н), 2,05 (с, 6Н); ESI-MS: m/z 348,0 (M+H)+.

N-(4-Мезитилтиазол-2-ил)пиримидин-4-карбоксамид
Соединение 5
Выход: 62%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,41 (с, 1Н), 9,15 (д, 1Н), 8,14 (д, 1H), 7,17 (с, 1Н), 6,89 (с, 2Н), 2,25 (с, 3Н), 2,03 (с, 6Н); ESI-MS: m/z 325,1 (M+H)+ .

N-(4-п-толилтиазол-2-ил)изоникотинамид
Соединение 6
Выход: 8,6%; 1H ЯМР (500 МГц, CDCl3) δ 8,70 (д, J=5,5 Гц, 2Н), 7,65-7,63 (м, 2Н), 7,60 (д, J=8,0 Гц, 2Н), 7,17 (с, 1Н), 7,14 (д, J=7,5 Гц, 2Н), 2,34 (с, 6Н); ESI-MS: m/z 295,9 (M+H)+.

4-Циано-N-(4-п-толилтиазол-2-ил)бензамид
Соединение 7
Выход: 63%; 1H ЯМР (500 МГц, CDCl3) δ 7,89 (д, J=8,5 Гц, 2Н), 7,62 (д, J=8,5 Гц, 2Н), 7,55 (д, J=8,5 Гц, 2Н), 7,17 (с, 1Н), 7,12 (д, J=8,0 Гц, 2Н), 2,34 (с, 6Н); ESI-MS: m/z 317,9 (M-H)-.

N-(4-Мезитилтиазол-2-ил)пиридазин-4-карбоксамид
Соединение 8
Выход: 62%; 1Н ЯМР (500 МГц, ДМСО-d6) δ 9,72 (с, 1Н), 9,50 (д, 1Н), 8,21 (м, 1Н), 7,13 (с, 1Н), 6,94 (с, 2H), 2,27 (с, 3H), 2,06 (с, 6Н); ESI-MS: m/z 324,5 (M+H)+.

N-(4-Мезитилтиазол-2-ил)тиазол-5-карбоксамид
Соединение 9
Выход: 40%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,36 (с, 1Н), 8,82 (уш.с, 1Н), 7,06 (с, 1Н), 6,93 (с, 2Н), 2,26 (с, 3H), 2,05 (с, 6Н): ESI-MS: m/z 329,3 (M+Н)+.

N-(4-(4-Метоксифенил)-5-метилтиазол-2-ил)никотинамид
Соединение 14
Выход: 74%; 1H ЯМР (500 МГц, CDCl3) δ 9,09 (д, 1Н), 8,73-8,72 (м, 1Н), 8,15-8,14 (м, 1H), 7,42-7,41 (м, 2H), 7,35 (м, 1Н), 6,87-6,85 (м, 2Н), 3,82 (с, 3H), 2,51 (с, 6H); ESI-MS: m/z 325,3 [M+H]+.

N-(4-(2,4-Диметоксифенил)тиазол-2-ил)никотинамид
Соединение 15
Выход: 87%; 1H ЯМР (500 МГц, CDCl3) δ 9,30 (с, 1H), 8,82-8,81 (м, 1H), 8,39-8,36 (м, 1Н), 7,80-7,79 (м, 1Н), 7,48-7,46 (м, 1Н), 7,43-7,39 (м, 1Н), 6,58-6,55 (м, 2Н), 3,92 (с, 3Н), 3,86 (с, 3Н); ESI-MS: m/z 341,4 [M+H]+.

N-(4-(4-Метоксифенил)-5-метилтиазол-2-ил)пиколинамид
Соединение 16
Выход: >99%; 1H ЯМР (500 МГц, CDCl3) δ 10,55 (с, 1Н), 8,65-8,64 (м, 1Н), 8,30-8,29 (м, 1H), 7,93 (м, 1Н), 7,60-7,58 (м, 2Н), 7,54-7,53 (м, 1Н), 6,99-6,98 (м, 2Н), 3,86 (с, 3Н), 2,54 (с, 3Н); ESI-MS: m/z 325,6 [M+H]+.

N-(4-(4-Метоксифенил)тиазол-2-ил)пиколинамид
Соединение 17
Выход: >99%; 1H ЯМР (500 МГц, ДМСО-d6) δ 11,98 (с, 1Н), 8,78-8,77 (м, 1Н), 8,19-8,17 (м, 1Н), 8,11-8,09 (м, 1Н), 7,89-7,87 (м, 2Н), 7,74-7,71 (м, 1Н), 7,58 (м, 1Н), 7,00-6,99 (м, 2Н), 3,79 (с, 3Н), 2,54 (с, 3Н); ESI-MS: m/z 310,0 [M-Н]-.

N-(4-(2,4-Диметоксифенил)тиазол-2-ил)пиколинамид
Соединение 18
Выход: 89%; 1H ЯМР (500 МГц, ДМСО-d6) δ 11,9 (с, 1Н), 8,79-8,78 (м, 1Н), 8,20-8,19 (м, 1H), 8,12-8,07 (м, 1Н), 7,75-7,74 (м, 2Н), 7,61 (с, 1Н), 6,68-6,63 (м, 2H), 3,92 (с, 3H), 3,82 (с, 3H); ESI MS: m/z 340,3 [M-H]-.

N-(5-Метил-4-фенилтиазол-2-ил)пиколинамид
Соединение 19
Выход: 90%: 1H ЯМР (500 МГц, ДМСО-d6) δ 11,9 (с, 1Н), 8,76-8,77 (м, 1Н), 8,18-8,19 (м, 1Н), 8,10 (м, 1H), 7,69-7,73 (м, 3Н), 7,45-7,48 (м, 1Н), 7,36-7,38 (м, 1Н), 2,50 (с, 3Н); ESI-MS: m/z 295,4 [M+H]+.

N-(4-(3,4,5-Триметоксифенил)тиазол-2-ил)никотинамид
Соединение 20
Выход: 78%: 1H ЯМР (500 МГц, ДМСО-d6) δ 9,24 (д, 1Н), 8,79 (т, 1Н), 8,44 (д, 1Н), 7,75 (с, 1Н), 7,58-7,60 (м, 1Н), 7,26 (с, 2Н), 3,85 (с, 6Н), 3,69 (с, 3Н); ESI-MS: m/z 372,5 [M+H]+.

N-(4-(2-Фтор-4-метоксифенил)тиазол-2-ил)никотинамид
Соединение 21
Выход: 81%: 1H ЯМР (500 МГц, ДМСО-d6) δ 9,23 (д, 1Н), 8,80 (т, 1Н), 8,44 (д, 1Н), 8,00-8,03 (м, 1H), 7,58-7,60 (м, 1H), 7,46 (д, 1Н), 6,90-6,98 (м, 2H), 3,82 (с, 3Н); ESI-MS: m/z 330,0 [M+H]+.

N-(4-(2-Фтор-4-метоксифенил)тиазол-2-ил)-3-(4-метоксифенил)пропанамид
Соединение 22
Выход: 53%: 1H ЯМР (500 МГц, CDCl3) δ 11,01 (с, 1Н), 7,82-7,86 (м, 1Н), 7,27 (д, 1Н), 6,63-6,84 (м, 6Н), 3,80 (с, 3Н), 3,76 (с, 3Н), 2,77 (т, 2Н), 2,29 (т, 3Н); ESI-MS: m/z 387,0 [M+H]+.

N-(4-(2-Фтор-4-метоксифенил)тиазол-2-ил)-3-фенилпропанамид
Соединение 23
Выход: 45%: 1H ЯМР (500 МГц, CDCl3) δ 10,87 (с, 1Н), 7,83-7,87 (м, 1Н), 7,15-7,27 (м, 5Н), 7,95 (д, 2Н), 6,62-6,73 (м, 2Н), 3,80 (с, 3Н), 2,86 (т, 2Н), 2,36 (т, 2Н); ESI-MS: m/z 356,0 [M+H]+.

N-(4-(2-Фтор-4-метоксифенил)тиазол-2-ил)пиколинамид
Соединение 24
Выход: 77%: 1H ЯМР (500 МГц, ДМСО-d6) δ 12,04 (с, 1H), 8,79 (д, 2H), 8,02-8,21 (м, 3Н), 7,74 (т, 1Н), 7,49 (д, 2Н), 6,90-6,97 (м, 2Н), 3,82 (с, 3Н); ESI-MS: m/z 330,0 [M+H]+.

N-(4-(3,4,5-Триметоксифенил)тиазол-2-ил)пиколинамид
Соединение 25
Выход: 75%: 1H ЯМР (500 МГц, ДМСО-d6) δ 12,04 (с, 1Н), 8,78 (с, 1Н), 8,18 (д, 1Н), 8,11 (т, 1Н), 7,78-7,82 (м, 2Н), 7,28 (с, 2Н), 3,86 (с, 6Н), 3,69 (с, 3Н); ESI-MS: m/z 372,0 [M+H]+.

N-(4-(2-Фтор-4-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 26
Выход: 84%: 1H ЯМР (500 МГц, ДМСО-d6) δ 8,82 (д, 2Н), 7,99-8,03 (м, 3Н), 7,48 (д, 1Н), 6,91-6,98 (м, 2Н), 3,83 (с, 3Н); ESI-MS: m/z 330,0 [M+H]+.

N-(4-(3,4,5-Триметоксифенил)тиазол-2-ил)изоникотинамид
Соединение 27
Выход: 82%: 1H ЯМР (500 МГц, ДМСО-d6) δ 8,79 (д, 2H), 8,00-8,01 (м, 2H), 7,71 (с, 1Н), 7,26 (с, 2Н), 3,85 (с, 6Н), 3,70 (с, 3H); ESI-MS: m/z 372,0 [M+H]+.

N-(4-п-толилтиазол-2-ил)пиколинамид
Соединение 28
Выход: 6,7%; 1H ЯМР (500 МГц, CDCl3) δ 10,96 (уш.с, 1Н), 9,11 (уш.с, 1Н), 8,75 (уш.с, 1Н), 8,14 (д, J=8,0 Гц, 1H), 7,60 (д, J=7,5 Гц, 2Н), 7,34 (с, 1Н), 7,15 (с, 1Н), 7,12 (д, J=7,5 Гц, 2Н), 2,33 (с, 6Н); ESI-MS: m/z 293,7 (M-H)-.

N-(4-п-толилтиазол-2-ил)никотинамид
Соединение 29
Выход: 83%; 1H ЯМР (500 МГц, CDCl3) δ 11,24 (с, 1Н), 8,68 (д, J=4,5 Гц, 1Н), 8,31 (д, J=8,0 Гц, 1Н), 7,95 (м, 1H), 7,78 (д, J=8,0 Гц, 2Н), 7,54 (м, 1Н), 7,24 (м, 2H), 7,17 (с, 1Н), 2,39 (с, 6Н); ESI-MS: m/z 295,6 (M+H)+.

4-Циано-N-(5-метил-4-п-толилтиазол-2-ил)бензамид
Соединение 30
Выход: 36%; 1H ЯМР (500 МГц, CDCl3) δ 7,72 (д, J=8,5 Гц, 2Н), 7,49 (д, J=8,5 Гц, 2Н), 7,20 (д, J=8,0 Гц, 2Н), 6,99 (д, J=8,0 Гц, 2Н), 2,51 (с, 3Н), 2,27 (с, 3Н); ESI-MS: m/z 332,0 (M-H)-.

N-(5-Метил-4-п-толилтиазол-2-ил)никотинамид
Соединение 31
Выход: 56%; 1H ЯМР (500 МГц, CDCl3) δ 8,99 (с, 1Н), 8,63 (д, J=5,0 Гц, 1Н), 8,01 (д, J=7,9 Гц, 1Н), 7,29-7,21 (м, 3Н), 7,03 (д, J=7,8 Гц, 2Н), 2,49 (с, 3Н), 2,29 (с, 3Н); ESI-MS: m/z 310,3 (M+H)+.

N-(5-Метил-4-п-толилтиазол-2-ил)пиколинамид
Соединение 32
Выход: 79%; 1H ЯМР (500 МГц, CDCl3) δ 11,11 (с, 1Н), 8,64 (д, J=4,5 Гц, 1H), 8,29 (д, J=7,5 Гц, 1H), 7,93 (т, J=8,0 Гц, 1Н), 7,55-7,51 (м, 3Н), 7,25 (д, J=7,5 Гц, 1H), 2,54 (с, 3Н), 2,40 (с, 3Н); ESI-MS: m/z 309,0 (M-H)-.

N-(4-Мезитилтиазол-2-ил)тиофен-3-карбоксамид
Соединение 33
Выход: 37%; 1Н ЯМР (500 МГц, DMSO-d6) δ 12,59 (с, 1Н), 8,60 (с, 1Н), 7,69-7,76 (м, 2H), 7,04 (с, 1Н), 6,93 (с, 2Н), 2,27 (с, 3H), 2,06 (с, 6H); ESI-MS: m/z 327,1 (M-H)-.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 34
Выход: 54%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,89 (д, 2Н), 8,00 (д, 2Н), 7,57 (д, 1H), 7,44-7,46 (м, 1Н), 7,26 (д, 1Н), 7,11 (с, 3Н), 3,81 (с, 3Н); ESI-MS: m/z 327,9 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)никотинамид
Соединение 35
Выход: 44%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,25 (с, 1Н), 8,91 (с, 1Н), 8,45-8,48 (м, 1Н), 7,65-7,67 (м, 1H), 7,57 (д, 1H), 7,44-7,46 (м, 1Н), 7,26 (д, 1Н), 7,10 (с, 2H), 3,81 (с, 3Н); ESI-MS: m/z 328,0 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)пиколинамид
Соединение 36
Выход: 37%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,82 (д, 1Н), 8,23 (д, 1Н), 8,08 (д, 1Н), 7,74-7,75 (м, 1Н), 7,57 (д, 1Н), 7,44-7,46 (м, 1H), 7,23 (д, 1Н), 7,10 (с, 2Н), 3,81 (с, 3Н); ESI-MS: m/z 328,1 (M+H)+.

N-(4-(4-Метоксифенил)тиазол-2-ил)никотинамид
Соединение 37
Выход: 94%; 1H ЯМР (500 МГц, ДМСО-d6) δ 12,98 (с, 1Н), 9,23 (м, 1Н), 8,80 (м, 1Н), 8,46-8,43 (м, 1H), 7,90-7,88 (м, 2H), 7,61-7,56 (м, 1H), 7,02-7,00 (м, 2Н), 3,80 (с, 3Н); ESI-MS: m/z 310,0 [M-H]-.

4-Циано-N-(5-метил-4-фенилтиазол-2-ил)бензамид
Соединение 38
Выход: 99%; 1H ЯМР (500 МГц, ДМСО-d6) δ 7,82-7,80 (м, 2Н), 7,60-7,58 (м, 2Н), 7,41-7,40 (м, 2H), 7,30-7,29 (м, 2Н), 7,22-7,19 (м, 1Н), 2,54 (с, 3Н); ESI-MS: m/z 320,0 [M+H]+.

4-Циано-N-(4-(4-метоксифенил)-5-метилтиазол-2-ил)бензамид
Соединение 39
Выход: 66%; 1H ЯМР (500 МГц, CD3OD) δ 8,18-8,17 (м, 2Н), 7,92-7,90 (м, 2Н), 7,60-7,58 (м, 2H), 7,01-7,00 (м, 2H), 3,84 (с, 3Н), 2,50 (c, 2Н); ESI-MS m/z 349,5 [M+H]+.

N-(4-(2,4-Диметоксифенил)тиазол-2-ил)-2-(2-метоксифенил)ацетамид
Соединение 40
Выход: 85%; 1H ЯМР (500 МГц, ДМСО-d6) δ 12,3 (с, 1Н), 7,99-7,97 (м, 1Н), 7,64 (с, 1Н), 7,24 (м, 1Н), 6,91 (м, 2Н), 6,90 (м, 1Н), 6,66-6,61 (м, 2Н), 3,89 (с, 3Н), 3,80 (с, 3Н), 3,75-3,74 (м, 5Н); ESI-MS: m/z 385,1 [M+H]+.

2-(2-Метоксифенил)-N-(5-метил-4-фенилтиазол-2-ил)ацетамид
Соединение 41
Выход: 76%; 1H ЯМР (500 МГц, CDCl3) δ 8,90 (с, 1Н), 7,56-7,55 (м, 2Н), 7,43-7,40 (м, 2Н), 7,34-7,33 (м, 1Н), 7,26-7,22 (м, 1Н), 3,77 (с, 3Н), 3,47 (с, 2Н), 2,49 (с, 3Н); ESI-MS: m/z 339,2 [M+H]+.

N-(4-(4-Метокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 42
Выход: 69%; 1H ЯМР (500 МГц, CDCl3) δ 8,67 (д, J=5,5 Гц, 2Н), 7,55 (д, J=6,0 Гц, 2Н), 6,77 (с, 1Н), 6,32 (с, 2Н), 3,73 (с, 3Н), 1,91 (с, 6Н); ESI-MS: m/z 340,0 (M+H)+.

N-(5-Метил-4-п-толилтиазол-2-ил)изоникотинамид
Соединение 43
Выход: 54%; 1H ЯМР (500 МГц, CDCl3) δ 8,57 (д, J=5,0 Гц, 2Н), 7,46 (д, J=5,5 Гц, 2Н), 7,25 (д, J=4,5 Гц, 2Н), 7,02 (д, J=7,5 Гц, 2Н), 2,51 (с, 3Н), 2,28 (с, 3Н); ESI-MS: m/z 309,9 (M+H)+.

N-(4-(2,4,6-Триметилпиридин-3-ил)тиазол-2-ил)изоникотинамид
Соединение 44
Выход: 51%; 1H ЯМР (500 МГц, CDCl3) δ 8,79 (д, J=5,5 Гц, 2H), 7,70 (д, J=5,5 Гц, 2Н), 6,86 (с, 1Н), 6,77 (с, 1Н), 2,45 (с, 3Н), 2,23 (с, 3Н), 2,03 (с, 3Н); ESI-MS: m/z 324,5 (M+H)+.

N-(4-(2,4,6-Триметилпиридин-3-ил)тиазол-2-ил)пиколинамид
Соединение 45
Выход: 18%; 1H ЯМР (500 МГц, CDCl3) δ 11,22 (с, 1Н), 8,65 (д, J=4,5 Гц, 1Н), 8,31 (д, J=7,5 Гц, 1Н), 7,96 (т, J=7,5 Гц, 1Н), 7,54 (м, 1Н), 6,92 (с, 1Н), 6,85 (с, 1Н), 2,52 (с, 3Н), 2,37 (с, 3Н), 2,133 (с, 3Н); ESI-MS: m/z 325,0 (M+H)+.

N-(4-(2,4,6-Триметилпиридин-3-ил)тиазол-2-ил)никотинамид
Соединение 46
Выход: 18%; 1H ЯМР (500 МГц, CDCl3) δ 9,11 (с, 1Н), 8,77 (с, 1Н), 8,17 (д, J=7,5 Гц, 1Н), 7,42 (м, 1Н), 6,85 (с, 1Н), 6,78 (с, 1Н), 2,45 (с, 3Н), 2,27 (с, 3Н), 2,02 (с, 3Н); ESI-MS: m/z 325,1 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)пиридазин-4-карбоксамид
Соединение 47
Выход: 54%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,75 (с, 1Н), 9,61 (с, 1H), 8,28-8,30 (м, 1Н), 7,58 (д, 1Н), 7,46-7,48 (м, 1Н), 7,29 (д, 1Н), 7,12 (с, 3Н), 3,82 (с, 3Н); ESI-MS: m/z 329,4 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)пиримидин-4-карбоксамид
Соединение 48
Выход: 44%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,49 (д, 1H), 9,19 (д, 1Н), 8,22-8,23 (м, 1Н), 7,58 (д, 1Н), 7,45-7,47 (м, 1Н), 7,27 (д, 1H), 7,12 (с, 3Н), 3,82 (с, 3Н); ESI-MS: m/z 328,9 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)тиофен-3-карбоксамид
Соединение 49
Выход: 37%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,58 (д, 1Н), 7,73-7,75 (м, 1Н), 7,60 (д, 1Н), 7,55 (д, 1Н), 7,42-7,44 (м, 1Н), 7,18 (д, 1Н), 7,09 (м, 3Н); ESI-MS: m/z 333,0 (M+H)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)тиазол-5-карбоксамид
Соединение 50
Выход: 37%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 8,75 (с, 1Н), 7,56 (с, 1Н), 7,43-7,45 (м, 1Н), 7,24 (д, 1Н), 7,11 (м, 3Н), 3,81 (с, 3Н); ESI-MS: m/z 333,9 (M+Н)+.

N-(4-(4-Гидрокси-3-метоксифенил)тиазол-2-ил)фуран-3-карбоксамид
Соединение 51
Выход: 32%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,63 (с, 1Н), 7,91 (с, 1Н), 7,54 (с, 1Н), 7,41-7,43 (м, 1Н), 7,08-7,18 (м, 4Н), 6,93 (с, 1Н), 3,80 (с, 3Н); ESI-MS: m/z 316,9 (M+H)+.

N-(4-(4-Этокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 52
Выход: 88%; 1H ЯМР (500 МГц, CDCl3) δ 8,70 (д, J=6,0 Гц, 2Н), 7,58 (д, J=6,0 Гц, 2H), 6,78 (с, 1Н), 6,37 (с, 2Н), 3,95 (кв, J=7,0 Гц, 2Н), 1,94 (с, 6Н), 1,41 (т, J=7,0 Гц, 3Н); ESI-MS: m/z 353,6 (M+H)+.

N-(4-(3,5-Диметилбифенил-4-ил)тиазол-2-ил)изоникотинамид
Соединение 53
Выход: 78%; 1Н ЯМР (500 МГц, CDCl3) δ 8,58 (м, 2H), 7,53-7,44 (м, 5H), 7,37 (м, 1Н), 7,03 (с, 2Н), 6,86 (с, 1Н), 2,02 (с, 6Н); ESI-MS: m/z 385,7 (M+H)+.

N-(4-(4-Хлор-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 55
Выход: 89%; 1H ЯМР (500 МГц, CDCl3) δ 8,76 (д, J=6,0 Гц, 2Н), 7,57 (м, 2Н), 6,81 (м, 3Н), 1,92 (с, 6H); ESI-MS: m/z 343,8 (M+H)+.

4-Циано-N-(4-(4-(гидроксифенил)тиазол-2-ил)бензамид
Соединение 56
Выход: 38%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,28 (д, 2Н), 8,09 (д, 2Н), 7,88 (д, 2Н), 7,31 (д, 2Н), 7,04-7,08 (м, 3Н); ESI-MS: m/z 322,0 (M+H)+.

N-(4-(4-Гидроксифенил)тиазол-2-ил)изоникотинамид
Соединение 57
Выход: 75%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,89 (д, 2Н), 8,01-8,06 (м, 2Н), 7,89 (д, 2Н), 7,32 (д, 2Н), 7,05-7,09 (м, 3Н); ESI-MS: m/z 297,6 (M+H)+.

N-(4-(4-Гидроксифенил)тиазол-2-ил)пиримидин-4-карбоксамид
Соединение 58
Выход: 48%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,49 (с, 1Н), 9,18 (д, 1Н), 8,23 (д, 1Н), 7,86-7,91 (м, 2H), 7,33 (д, 2Н), 7,06-7,09 (м, 3Н); ESI-MS: m/z 192,5 (M-106, аминотиазол).

N-(4-(4-Гидроксифенил)тиазол-2-ил)пиколинамид
Соединение 59
Выход: 49%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,82 (д, 1Н), 8,24 (д, 1Н), 8,07-8,10 (м, 1Н), 7,88 (д, 2Н), 7,73-7,75 (м, 1Н), 7,30 (д, 2Н), 7,05-7,08 (м, 3Н); ESI-MS: m/z 297,7 (M+H)+.

N-(4-(4-Гидроксифенил)тиазол-2-ил)никотинамид
Соединение 60
Выход: 49%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,26 (д, 1Н), 8,91 (д, 1Н), 8,47 (д, 1Н), 7,89 (д, 2Н), 7,65-7,67 (м, 1Н), 7,32 (д, 2Н), 7,05-7,08 (м, 3Н); ESI-MS: m/z 297,6 (M+H)+.

4-Циано-N-(4-(4-гидрокси-2-метилфенил)тиазол-2-ил)бензамид
Соединение 61
Выход: 48%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,24-8,30 (м, 2Н), 8,04-8,11 (м, 2Н), 7,63 (д, 1Н), 7,00-7,20 (м, 4Н), 6,67 (с, 1Н); ESI-MS: m/z 335,7 (M+H)+.

N-(4-(3,5-Дифтор-4-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 62
Выход: 67%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 13,10 (с, 1Н), 8,82 (д, J=5,6 Гц, 2Н), 8,00 (д, J=5,6 Гц, 2Н), 7,88 (с, 1Н), 7,70-7,72 (м, 2Н), 3,96 (с, 3Н); ESI-MS m/z 348,0 (M+Н)+.

1-(4-(4-Метокси-2,6-диметилфенил)тиазол-2-ил)-3-(пиридин-4-ил)мочевина
Соединение 67
Выход: 40%; 1H ЯМР (500 МГц, ДМСО-d6) δ 10,49 (уш.с, 1Н), 8,54 (д, J=6,5 Гц, 2Н), 7,94 (с, 2Н), 6,88 (с, 1Н), 6,74 (с, 2Н), 3,76 (с, 3Н), 2,10 (с, 6Н); ESI-MS: m/z 354,8 (M+H)+.

N-(3,5-Диметил-4-(2-(3-пиридин-4-илуреидо)тиазол-4-ил)фенил)ацетамид
Соединение 68
Выход: 73%; 1H ЯМР (500 МГц, ДМСО-d6) δ 9,87 (с, 1H), 9,40 (с, 1Н), 8,39 (д, J=6,0 Гц, 2Н), 7,49 (д, J=6,5 Гц, 2Н), 7,32 (с, 2Н), 6,92 (с, 1Н), 2,06 (с, 6Н), 2,04 (с, 3Н); ESI-MS: m/z 381,8 (M+H)+.

N-(4-(2,4,6-Триизопропилфенил)тиазол-2-ил)изоникотинамид
Соединение 73
Выход: 62%; 1H ЯМР (500 МГц, CDCl3) δ 8,83 (д, J=5,5 Гц, 2H), 7,83 (д, J=6,0 Гц, 2Н), 7,06 (с, 2Н), 6,82 (с, 1H), 2,94 (м, 1Н), 2,64 (м, 2Н), 1,29 (д, J=7,0 Гц, 6Н), 1,13 (д, J=7,0 Гц, 12Н); ESI-MS: m/z 407,9 (M+H)+.

N-(4-(4-Ацетамидо-2,6-диметилбифенил)тиазол-2-ил)изоникотинамид
Соединение 74
Выход: 39%; 1H ЯМР (500 МГц, ДМСО-d6) δ 13,03 (уш.с, 1H), 9,86 (с, 1Н), 8,80 (д, J=5,5 Гц, 2H), 7,99 (д, J=5,5 Гц, 2Н), 7,34 (с, 2Н), 7,13 (с, 1Н), 2,06 (с, 6H); ESI-MS: m/z 385,7 (M+H)+.

3-Фтор-N-(4-мезитилтиазол-2-ил)изоникотинамид
Соединение 77
Выход: 23%; 1H ЯМР (500 МГц, CDCl3) δ 11,32 (уш.с, 1Н), 8,57 (м, 2Н), 7,80 (т, J=5,5 Гц, 1Н), 6,80 (с, 1H), 6,71 (c, 2Н), 2,23 (с, 3Н), 1,96 (с, 6Н); ESI-MS: m/z 341,9 (M+H)+.

N-(4-(2,6-Дифтор-4-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 78
Выход: 49%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 13,13 (с, 1Н), 8,81 (д, J=5,9 Гц, 2Н), 8,00 (д, J=5,9 Гц, 2Н), 7,46 (с, 1Н), 6,86-6,88 (м, 2Н), 3,83 (с, 3Н); ESI-MS: m/z 348,7 (M+H)+.

N-(4-(2,6-Диметил-4-феноксифенил)тиазол-2-ил)изоникотинамид
Соединение 79
Выход: 30%; 1H ЯМР (500 МГц, CDCl3) δ 8,81 (д, J=5,5 Гц, 2Н), 7,99 (д, J=5,5 Гц, 2Н), 7,41 (т, J=8,0 Гц, 2Н), 7,19 (с, 1Н), 7,15 (т, J=7,5 Гц, 1H), 7,04 (д, J=8,2 Гц, 2Н), 6,78 (с, 2Н) 2,07 (с, 6Н); ESI-MS: m/z 401,8 (M+H)+.

N-(4-(4-Изопропокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 82
Выход: 80%; 1Н ЯМР (500 МГц, CDCl3) δ 8,67 (д, J=6,0 Гц, 2Н), 7,55 (д, J=6,0 Гц, 2H), 6,77 (с, 1Н), 6,30 (с, 2Н), 4,43 (м, 1Н), 1,89 (с, 6Н), 1,31 (д, J=6,0 Гц, 6H); ESI-MS: m/z 368,1 (M+H)+.

N-(4-(4-(Циклопентилокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 85
Выход: 62%; 1H ЯМР (500 МГц, CDCl3) δ 8,71 (д, J=6,0 Гц, 2Н), 7,60 (д, J=6,0 Гц, 2Н), 6,78 (с, 1Н), 6,37 (с, 2H), 4,68 (м, 1Н), 1,95 (с, 6Н), 1,80-1,92 (м, 6Н), 0,85 (м, 2Н); ESI-MS: m/z 394,1 (M+H)+.

N-(4-(4-(Гидроксипропокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 94
Выход: 4,0%; 1H ЯМР (CDCl3, 500 МГц) δ 8,80 (д, J=5,9 Гц, 2Н), 7,69 (д, J=5,9 Гц, 2Н), 6,81 (с, 1Н), 6,55 (с, 2Н), 4,19-4,25 (уш.с, 1Н), 4,10-4,15 (м, 1Н), 3,91-3,98 (м, 1Н), 3,78-3,82 (м, 1Н), 2,05 (с, 6Н), 1,28 (с, 3Н); ESI-MS: m/z 384,7 (M+H)+.
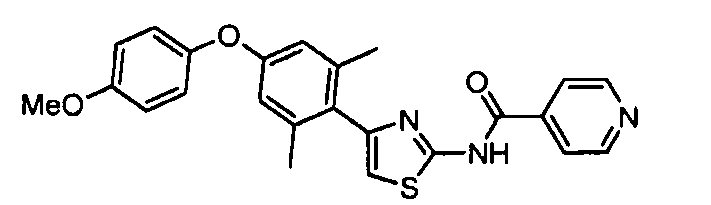
N-(4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 95
Выход: 95%; 1H ЯМР (500 МГц, CDCl3) δ 8,73 (м, 2Н), 7,62 (м, 2Н), 6,90-6,96 (м, 4Н), 6,80 (с, 1Н), 6,45 (с, 2H), 3,83 (с, 3Н), 1,92 (с, 6H); ESI-MS: m/z 431,7 (M+H)+.

N-(4-(4-(4-Фторфенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 96
Выход: 17%; 1H ЯМР (500 МГц, CDCl3) δ 8,72 (д, J=5,5 Гц, 2Н), 7,60 (д, J=5,5 Гц, 2Н), 7,04 (м, 2Н), 6,94 (м, 2Н), 6,81 (с, 1Н), 6,43 (с, 2Н), 1,92 (с, 6Н); ESI-MS: m/z 420,2 (M+H)+.

N-(4-(4-(2,3-Дигидроксипропокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 97
Выход: 12%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,78 (д, J=4,5 Гц, 2Н), 7,98 (д, J=4,5 Гц, 2Н), 7,15 (с, 1Н), 6,69 (с, 2Н), 4,95-4,96 (м, 1Н), 4,68-4,69 (м, 1Н), 3,97-3,98 (м, 1Н), 3,84-3,85 (м, 1Н), 3,78-3,79 (м, 1Н), 2,06 (с, 6Н); ESI-MS: m/z 400,7 (M+H)+.

N-(4-(4-Изобутокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 100
Выход: 99%; 1H ЯМР (500 МГц, CDCl3) δ 8,68 (д, J=6,0 Гц, 2Н), 7,56 (д, J=6,0 Гц, 2H), 6,77 (с, 1H), 6,33 (с, 2Н), 3,62 (д, J=6,5 Гц, 2H), 2,08 (м, 1Н), 1,91 (с, 6Н), 1,05 (д, J=6,7 Гц, 6H); ESI-MS: m/z 381,1 (M+H)+.

N-(4-(4-(Бензо[d][1,3]диоксол-5-илокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 101
Выход: 92%; 1H ЯМР (500 МГц, CDCl3) δ 8,73 (м, 2Н), 7,62 (м, 2Н), 6,81 (с, 1Н), 6,78 (д, J=8,5 Гц, 1Н), 6,43-6,52 (м, 4Н), 6,00 (с, 2Н), 1,93 (с, 6Н); ESI-MS: m/z 445,9 (M+H)+.

N-(4-(4-(3,5-Диметилфенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 102
Выход: 94%; 1H ЯМР (500 МГц, CDCl3) δ 8,81 (д, J=5,1 Гц, 2H), 7,78 (д, J=5,6 Гц, 2Н), 6,84 (с, 1Н), 6,78 (с, 1Н), 6,64 (с, 2Н), 6,61 (с, 2Н), 2,31 (с, 6Н), 2,02 (с, 6Н); ESI-MS: m/z 429,8 (M+H)+.

N-(4-(4-(3-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 107
Выход: 51%; 1H ЯМР (500 МГц, CDCl3) δ 8,75 (м, 2Н), 7,62 (м, 2Н), 7,25 (м, 1Н), 6,82 (с, 1Н), 6,69 (м, 1Н), 6,56 (д, 1Н), 6,51 (м, 2H), 6,47 (с, 2Н), 3,81 (с, 3Н), 1,94 (с, 6Н); ESI-MS: m/z 431,6 (M+H)+.

N-(4-(2,6-Диметил-4-(4-трифторметил)фенокси)фенил)тиазол-2-ил)изоникотинамид
Соединение 108
Выход: 87%; 1H ЯМР (500 МГц, CDCl3) δ 8,76 (м, 2Н), 7,64 (м, 2Н), 7,58 (д, J=8,5 Гц, 2Н), 7,01 (д, J=8,5 Гц, 2Н), 6,86 (с, 1Н), 6,57 (с, 2Н), 1,98 (с, 6Н); ESI-MS: m/z 469,7 (M+H)+.

N-(4-(4-(2-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 111
Выход: 85%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,80-8,81 (м, 2H), 7,98-7,99 (м, 2Н), 7,16-7,21 (м, 3H), 6,99-7,06 (м, 2H), 6,59 (с, 2Н), 3,76 (с, 3Н), 2,03 (с, 6Н); ESI-MS: m/z 431,5 [M+H]+.

N-(4-(2,6-Диметил-4-(п-толилокси)фенил)тиазол-2-ил)изоникотинамид
Соединение 112
Выход: 89%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,80-8,81 (м, 2Н), 7,98-7,99 (м, 2Н), 7,18-7,22 (м, 3Н), 6,94-6,95 (м, 2Н), 6,73 (с, 2H), 2,30 (с, 3Н), 2,06 (с, 6Н): ESI-MS: m/z 414,9 [M-H]-.

N-(4-(4-(4-Этилфенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 113
Выход: 91%; 1H ЯМР (500 МГц, CDCl3) δ 8,78 (д, J=6,0 Гц, 2Н), 8,67 (м, 2Н), 7,18 (д, J=8,0 Гц, 2Н), 6,93 (д, J=8,5 Гц, 2Н), 6,83 (с, 1Н), 6,56 (с, 2Н), 2,65 (м, 2Н), 1,98 (с, 6H), 1,26 (т, J=7,5 Гц, 3Н); ESI-MS: m/z 429,6 (M+H)+.

N-(4-(4-Йод-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 114
Выход: 61%; 1H ЯМР (500 МГц, CDCl3) δ 8,76 (м, 2H), 7,52 (м, 2H), 7,11 (с, 2H), 6,80 (с, 1Н), 1,84 (с, 6Н); ESI-MS: m/z 435,6 (M+H)+.

N-(4-(2,6-Диметил-4-(фенилтио)фенил)тиазол-2-ил)изоникотинамид
Соединение 115
Выход: 63%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,77 (д, J=5,4 Гц, 2H), 7,98 (д, J=5,4 Гц, 2Н), 7,38-7,40 (м, 2Н), 7,31-7,35 (м, 3Н), 7,11 (с, 3Н), 2,07 (с, 6Н); ESI-MS: m/z 418,8 (M+H)+.

N-(4-(2,6-Диметил-4-(п-толилтио)фенил)тиазол-2-ил)изоникотинамид
Соединение 116
Выход: 84%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,78 (д, J=5,2 Гц, 2Н), 7,98 (д, J=5,2 Гц, 2Н), 7,30 (д, J=8,0 Гц, 2Н), 7,23 (д, J=8,0 Гц, 2Н), 7,10 (с, 1H), 7,02 (с, 2Н), 2,36 (с, 3Н), 2,04 (с, 6Н); ESI-MS: m/z 432,5 (M+H)+.

N-(4-(4-(4-Метоксифенилтио)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 117
Выход: 64%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,79 (д, J=5,0 Гц, 2H), 7,98 (д, J=5,0 Гц, 2Н), 7,43 (д, J=8,4 Гц, 2Н), 7,12 (с, 1Н), 7,02 (д, J=8,4 Гц, 2Н), 6,92 (с, 2Н), 3,79 (с, 3Н), 2,02 (с, 6H); ESI-MS: m/z 448,1 (M+H)+.

N-(4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)-3-метилизоникотинамид
Соединение 121
Выход: 62%; 1H ЯМР (500 МГц, ДМСО-d6) δ 12,8 (с, 1Н), 8,54-8,58 (м, 2Н), 7,55-7,56 (м, 1Н), 7,14 (с, 1Н), 6,96-7,03 (м, 4Н), 6,67 (с, 2Н), 3,76 (с, 3Н), 2,40 (с, 3Н), 2,05 (с, 6Н); ESI-MS: m/z 445,7 [M+H]+.

N-(4-(4-(4-Бромфениламино)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 122
Выход: 60%; 1H ЯМР (500 МГц, CDCl3) δ 8,79 (д, J=4,5 Гц, 2Н), 7,85 (д, J=4,5 Гц, 2Н), 7,39 (д, J=8,6 Гц, 2Н), 6,97 (д, J=8,6 Гц, 2Н), 6,83 (с, 1Н), 2,05 (с, 6Н); ESI-MS: m/z 479,2 (M+H)+.

N-(4-(3-Бром-2,6-диметил-4-(фениламино)фенил)тиазол-2-ил)изоникотинамид
Соединение 123
Выход: 58%; 1H ЯМР (500 МГц, CDCl3) δ 8,87 (д, J=4,5 Гц, 2Н), 8,02 (д, J=4,5 Гц, 2Н), 7,47 (д, J=8,6 Гц, 2Н), 7,08 (д, J=8,6 Гц, 2Н), 6,89 (с, 1Н), 6,85 (с, 1Н), 6,24 (с, 1Н), 2,04 (с, 6Н); ESI-MS: m/z 479,3 (M+H)+.

N-(4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)-2-нитроизоникотинамид
Соединение 124
Выход: 94%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,82 (с, 1Н), 8,71-8,72 (м, 1Н), 8,39-8,40 (м, 1Н), 7,01-7,03 (м, 2Н), 6,96-6,99 (м, 2Н), 6,64-6,67 (м, 3Н), 3,75 (с, 3Н), 2,03 (с, 6Н); ESI-MS: m/z 476,8 [M+H]+.

N-(4-(2,6-Диметил-4-(метилтио)фенил)тиазол-2-ил)изоникотинамид
Соединение 125
Выход: 94%: 1H ЯМР (500 МГц, CDCl3) δ 8,66-8,68 (м, 2Н), 7,49-7,50 (м, 2Н), 6,77 (с, 1Н), 6,57 (с, 2Н), 2,42 (с, 3Н), 1,87 (с, 6Н); ESI-MS: m/z 355,6 (M+H)+.

N-(4-(2,6-Диметил-4-(метилсульфонил)фенил)тиазол-2-ил)изоникотинамид
Соединение 127
Выход: 39%; 1H ЯМР (500 МГц, CDCl3) δ 8,80-8,81 (м, 2Н), 7,98-8,00 (м, 2Н), 7,70 (с, 2Н), 7,30 (с, 1Н), 3,30 (с, 1Н), 2,20 (с, 6Н); ESI-MS: m/z 387,6 (M+H)+.

N-(4-(4-(4-Метоксифенилсульфонил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 129
Выход: 61%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,79 (с, 2Н), 7,97 (д, J=6,0 Гц, 2Н), 7,91 (д, J=8,9 Гц, 2Н), 7,69 (с, 2Н), 7,27 (с, 1Н), 7,15 (д, J=8,9 Гц, 2Н), 3,84 (с, 3Н), 2,16 (с, 6H); ESI-MS: m/z 480,6 (M+H)+.

N-(4-(4-(4-Метоксифенилсульфинил)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 130
Выход: 43%; 1Н ЯМР (ДМСО-d6, 500 МГц) δ 13,1 (уш.с, 1Н), 8,80 (д, J=6,0 Гц, 2Н), 7,97 (д, J=6,0 Гц, 2Н), 7,66 (д, J=8,8 Гц, 2Н), 7,42 (с, 2Н), 7,23 (с, 1Н), 7,10 (д, J=8,8 Гц, 2Н), 3,80 (с, 3Н), 2,13 (с, 6H); ESI-MS: m/z 464,7 (M+H)+.

N-(4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 131
Выход: 24%; 1H ЯМР (500 МГц, CDCl3) δ 8,80 (с, 2H), 7,70 (д, J=5,1 Гц, 2Н), 6,97 (м, 2Н), 6,92 (м, 3Н), 6,70 (д, J=2,4 Гц, 1Н), 6,61 (д, J=2,3, 1Н), 3,83 (с, 3Н), 2,02 (с, 3H); ESI-MS: m/z 452,4 (M+H)+.

Общая методика II для синтеза 4-арил-2-амидотиазолов. К суспензии арилкарбоновой кислоты (1,5 экв.) в дихлорметане добавляли 1,1'-карбонилдиимидазол (CDI, 3,0 экв.). После перемешивания при комнатной температуре в течение 2,0 ч в раствор добавляли 4-арилтиазол-2-амин (q.0 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Раствор концентрировали и осадок перерастворяли в дихлорметане. Раствор промывали рассолом, сушили над MgSO4 и концентрировали при пониженном давлении с получением соответствующих 4-арил-2-амидотиазолов.

N-(4-Мезитилтиазол-2-ил)изоникотинамид
Соединение 1
Выход: 77%; 1H ЯМР (500 МГц, CD3OD) δ 8,75-8,76 (м, 2Н), 7,96-7,99 (м, 2Н), 6,90-6,92 (м, 3Н), 2,29 (с, 3Н), 2,08 (с, 6Н); ESI-MS: m/z 324,0 [M+H]+.

N-(5-Метил-4-фенилтиазол-2-ил)изоникотинамид
Соединение 2
Выход: 77%; 1H ЯМР (500 МГц, CDCl3) δ 11,7 (с, 1Н), 8,61-8,62 (м, 2Н), 7,51-7,53 (м, 2Н), 7,41-7,43 (м, 2Н), 7,26-7,30 (м, 2Н), 7,20-7,22 (м, 1Н), 2,54 (с, 3Н); ESI-MS: m/z 295,3 [M+H]+.

N-(5-Метил-4-фенилтиазол-2-ил)никотинамид
Соединение 3
Выход: 15%; 1H ЯМР (500 МГц, CDCl3) δ 11,7 (с, 1Н), 9,03 (с, 1Н), 8,68-8,69 (м, 1Н), 8,06-8,08 (м, 1Н), 7,45-7,47 (м, 2Н), 7,22-7,31 (м, 4Н), 2,54 (с, 3Н); ESI-MS: m/z 295,9 [M+H]+.

N-(4-(4-Метоксифенил)-5-метилтиазол-2-ил)изоникотинамид
Соединение 10
Выход: 12%; 1H ЯМР (500 МГц, ДМСО-d6) δ 12,9 (с, 1Н), 8,80-8,81 (м, 2Н), 7,99-8,00 (м, 2Н), 7,61-7,63 (м, 2Н), 7,02-7,04 (м, 2Н), 3,80 (с, 3Н), 2,49 (с, 3Н); ESI-MS: m/z 326,0 [M+H]+.

N-(4-(2,4,6-Триметоксифенил)тиазол-2-ил)изоникотинамид
Соединение 11
Выход: 12%; 1H ЯМР (500 МГц,, ДМСО-d6) δ 13,0 (с, 1H), 8,78 (с, 2Н), 7,98-8,00 (м, 3Н), 6,98 (с, 1Н), 6,29 (с, 2Н), 3,82 (с, 3Н), 3,68 (с, 3Н); ESI-MS: m/z 369,9 [M-H]-.
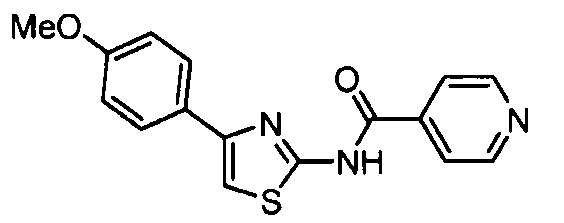
N-(4-(4-Метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 12
Выход: 50%; 1H ЯМР (500 МГц, CDCl3) δ 11,7 (с, 1Н), 8,61-8,62 (м, 2Н), 7,51-7,53 (м, 2H), 7,41-7,43 (м, 2Н), 7,26-7,30 (м, 2H), 7,20-7,22 (м, 1H), 2,54 (с, 3H); ESI-MS: m/z 310,1 [M-H]-.

N-(4-(2,4-Диметоксифенил)тиазол-2-ил)изоникотинамид
Соединение 13
Выход: 10%; 1H ЯМР (500 МГц, ДМСО-d6) δ 12,97 (с, 1Н), 8,81-8,82 (м, 2Н), 8,00-8,07 (м, 3H), 7,59 (с, 1Н), 6,64-6,68 (м, 2Н), 3,92 (с, 3Н), 3,81 (с, 3Н); ESI-MS: m/z 340,3 [M-H]-.

2-Хлор-N-(4-(4-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 54
Выход: 95%; 1H ЯМР (500 МГц, CDCl3) δ 8,33-8,34 (м, 1Н), 7,47-7,54 (м, 4Н), 7,11 (с, 1Н), 6,79-6,80 (м, 2Н), 3,81 (с, 3Н); ESI-MS: m/z 345,7 [M+Н]+.

2-Хлор-N-(4-(3-фтор-4-метоксифенил)тиазол-2-ил)изоникотинамид
Соединение 63
Выход: 83%; 1H ЯМР (500 МГц, CDCl3) δ 8,52-8,53 (м, 1Н), 7,69-7,70 (м, 1H), 7,59-7,60 (м, 1Н), 7,28-7,47 (м, 2H), 7,16 (с, 1Н), 6,93-6,97 (м, 1Н), 3,93 (с, 3Н); ESI-MS: m/z 363,7 [M+H]+.

2-Хлор-N-(4-мезитилтиазол-2-ил)изоникотинамид
Соединение 64
Выход: 87%; 1H ЯМР (500 МГц, CDCl3) δ 8,49-8,50 (м, 1Н), 7,74 (м, 1Н), 7,62 (м, 1Н), 6,83 (с, 1Н), 6,72 (м, 2H), 2,26 (с, 3Н), 1,97 (с, 6Н): ESI-MS: m/z 357,7 [M+H]+.

2-Хлор-N-(4-(4-метокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 65
Выход: 63%; 1H ЯМР (500 МГц, CDCl3) δ 8,60-8,61 (м, 1Н), 7,91-7,96 (м, 2Н), 6,87 (с, 1H), 6,58 (м, 2H), 3,81 (с, 3Н), 2,11 (с, 6Н); ESI-MS: m/z 373,9 [M+H]+.

2-Хлор-N-(4-(4-этокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 66
Выход: 95%; 1H ЯМР (500 МГц, CDCl3) δ 8,53-8,54 (м, 1Н), 7,74-7,84 (м, 2Н), 6,83 (с, 1Н), 6,48 (м, 2Н), 3,98-4,02 (м, 2Н), 2,01 (с, 6Н), 1,41-1,44 (м, 3Н); ESI-MS: m/z 387,9 [M+H]+.

2-Фтор-N-(4-(4-метокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 69
Выход: 68%; 1H ЯМР (500 МГц, CDCl3) δ 8,36-8,38 (м, 1Н), 7,72 (с, 1Н), 7,42 (с, 1Н), 6,84 (с, 1Н), 6,48 (с, 2Н), 3,78 (с, 3Н), 2,03 (с, 6Н); ESI-MS: m/z 357,5 [M+H]+.

N-(4-(4-Этокси-2,6-диметилфенил)тиазол-2-ил)-2-фторизоникотинамид
Соединение 70
Выход: 94%; 1H ЯМР (500 МГц, CDCl3) δ 8,39-8,40 (м, 1H), 7,78 (с, 1Н), 7,48 (с, 1Н), 6,85 (с, 1Н), 6,50 (с, 2Н), 3,98-4,01 (кв, 2Н), 2,05 (с, 6Н), 1,42-1,44 (т, 3Н); ESI-MS: m/z 371,8 [M+H]+.

4-(4-Мезитилтиазол-2-илкарбамоил)пиридина 1-оксид
Соединение 71
Выход: 75%; 1Н ЯМР (500 МГц, CDCl3) δ 8,15-8,16 (д, 2H), 7,78-7,79 (д, 2H), 6,83 (с, 1Н), 6,80 (с, 2Н), 2,23 (с, 3Н), 2,01 (с, 6Н); ESI-MS: m/z 340,1 [M+H]+.

4-(4-(4-Метокси-2,6-диметилфенил)тиазол-2-илкарбамоил)пиридина 1-оксид
Соединение 72
Выход: 43%; 1H ЯМР (500 МГц, CDCl3) δ 8,23 (с, 2Н), 8,04 (с, 2Н), 6,85 (с, 1Н), 6,60 (с, 2Н), 3,80 (с, 3Н), 2,12 (с, 6Н); ESI-MS: m/z 355,8 [M+H]+.

N-(4-(4-Метокси-2,6-диметилфенил)тиазол-2-ил)тиазол-5-карбоксамид
Соединение 75
Выход: 18%; 1H ЯМР (500 МГц, CDCl3) δ 9,01 (с, 1Н), 8,66 (с, 1Н), 6,82 (с, 1Н), 6,59 (с, 2H), 3,81 (с, 3Н), 2,13 (с, 6Н); ESI-MS: m/z 345,6 [M+H]+.

N-(4-(2,4,6-Трифторфенил)тиазол-2-ил)изоникотинамид
Соединение 76
Выход: 46%; 1H ЯМР (500 МГц, CDCl3) δ 8,78-8,79 (м, 2Н), 7,73-7,74 (м, 2Н), 7,26-7,28 (м, 1Н), 6,76-6,79 (м, 1H), 6,67-6,70 (м, 2H); ESI-MS: m/z 335,5 [M+H]+.

3-хлор-N-(4-Мезитилтиазол-2-ил)изоникотинамид
Соединение 80
Выход: 21%; 1H ЯМР (500 МГц, CDCl3) δ 8,68 (с, 1Н), 8,59-8,60 (м, 1Н), 7,54-7,55 (м, 1H), 6,81-6,83 (м, 2Н), 2,30 (с, 3Н), 2,01 (с, 6Н); ESI-MS: m/z 357,8 [M+H]+.

2-хлор-N-(4-Мезитилтиазол-2-ил)изоникотинамид
Соединение 81
Выход: 42%; 1H ЯМР (500 МГц, ДМСО-d6) δ 8,63-8,64 (м, 1Н), 8,11 (с, 1Н), 7,98-7,99 (м, 1Н), 7,12 (с, 1Н), 6,93 (м, 2Н), 2,50 (с, 3Н), 2,05 (с, 6Н); ESI-MS: m/z 357,9 [M+H]+.

N-(4-Мезитилтиазол-2-ил)-2-метоксиизоникотинамид
Соединение 86
Выход: 40%; 1H ЯМР (500 МГц, CDCl3) δ 8,38-8,39 (м, 1Н), 7,55-7,56 (м, 1Н), 7,41 (с, 1Н), 6,91-6,93 (м, 2Н), 6,86 (с, 1Н), 5,30 (с, 1Н), 4,00 (с, 3Н), 2,32 (с, 3Н), 2,12 (с, 6Н); ESI-MS: m/z 355,0 [M+H]+.

2-Хлор-N-(4-мезитилтиазол-2-ил)-6-метоксиизоникотинамид
Соединение 87
Выход: 63%; 1H ЯМР (500 МГц, CDCl3) δ 7,31 (с, 1Н), 7,06 (с, 1Н), 6,81 (с, 1Н), 6,76 (с, 2Н), 4,00 (с, 3Н), 2,32 (с, 3Н), 1,98 (с, 6Н); ESI-MS: m/z 387,9 [M+H]+.

2,6-Дихлор-N-(4-мезитилтиазол-2-ил)изоникотинамид
Соединение 88
Выход: 70%; 1H ЯМР (500 МГц, CDCl3) δ 7,61 (с, 2Н), 6,80 (с, 1Н), 6,73 (с, 2Н), 2,29 (с, 3Н), 1,94 (с, 6Н); ESI-MS: m/z 392,0 [M+H]+.

2-Ацетамидо-N-(4-мезитилтиазол-2-ил)изоникотинамид
Соединение 89
Выход: 61%; 1H-ЯМР (500 МГц, CDCl3) δ 8,81 (с, 1Н), 8,37 (с, 1H), 7,80-7,77 (м, 1Н), 6,91 (с, 2Н), 6,80 (с, 1Н), 2,30 (с, 3Н), 2,26 (с, 3Н), 2,11 (с, 6Н); ESI-MS: m/z 381,2 [M+H]+.

2,6-Дифтор-N-(4-мезитилтиазол-2-ил)изоникотинамид
Соединение 90
Выход: 67%; 1H-ЯМР (500 МГц, CDCl3) δ 7,11 (с, 2Н), 6,81 (с, 1Н), 6,68 (с, 1Н), 2,30 (с, 3Н), 1,91 (с, 6Н); ESI-MS: m/z 360,0 [M+H]+.

N-(4-Мезитилтиазол-2-ил)-2-(пиридин-4-ил)ацетамид
Соединение 93
Выход: 65%; 1Н-ЯМР (500 МГц, ДМСО-d6) δ 8,52-8,53 (м, 2Н), 7,35-7,36 (м, 2H), 6,99 (с, 1Н), 6,91 (с, 1Н), 3,84 (с, 1Н), 2,25 (с, 3Н), 2,02 (с, 6Н); ESI-MS: m/z 338,1 [M+H]+.

2-Фтор-N-(4-(4-(4-метоксифенокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 98
Выход: 70%; 1Н-ЯМР (500 МГц, CDCl3) δ 8,38-8,40 (м, 1Н), 7,66-7,67 (м, 2Н), 7,43 (с, 1Н), 6,98-7,00 (м, 2Н), 6,91-6,93 (м, 2Н), 6,84 (с, 1Н), 6,54 (с, 1H), 3,83 (с, 3H), 2,0 (с, 6Н); ESI-MS: m/z 450,0 [M+H]+.

2-Фтор-N-(4-(4-изопропокси-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 99
Выход: 83%; 1H-ЯМР (500 МГц, CDCl3) δ 8,40 (м, 1H), 7,78 (с, 1H), 7,50 (с, 1H), 6,84 (с, 1Н), 6,53 (с, 2Н), 4,52-4,56 (м, 1Н), 2,06 (с, 6H), 1,33 (с, 6Н); ESI-MS: m/z 385,8 [M+H]+.

(Е)-N-(4-Мезитилтиазол-2-ил)-3-(пиридин-3-ил)акриламид
Соединение 103
Выход: 34%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 12,48 (уш.с, 1Н), 8,82-8,83 (м, 1Н), 8,60-8,61 (м, 1Н), 8,04-8,05 (м, 1Н), 7,76-7,79 (м, 1Н), 7,49-7,51 (м, 1Н), 7,00-7,03 (м, 2Н), 6,92 (с, 2Н), 2,26 (с, 3Н), 2,05 (с, 6Н); ESI-MS: m/z 350,7 (M+H)+.

N-(4-Мезитилтиазол-2-ил)-1Н-индазол-6-карбоксамид
Соединение 104
Выход: 25%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 13,20 (уш.с, 1Н), 8,36 (с, 1Н), 8,19 (с, 1Н), 7,87-7,88 (м, 1Н), 7,72-7,83 (м, 1Н), 7,00 (с, 1Н), 6,93 (с, 2Н), 2,27 (с, 3Н), 2,07 (с, 6Н); ESI-MS: m/z 363,9 (M+H)+.

N-(4-Мезитилтиазол-2-ил)-1Н-индазол-5-карбоксамид
Соединение 105
Выход: 38%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 13,20 (уш.с, 1Н), 8,66 (с, 1Н), 8,26 (с, 1Н), 8,08 (д, J=8,4 Гц, 1Н), 7,65 (д, J=8,4 Гц, 1Н), 7,02 (с, 1Н), 6,93 (с, 2Н), 2,36 (с, 3Н), 2,07 (с, 6Н); ESI-MS: m/z 363,9 (M+H)+.

N-(4-Мезитилтиазол-2-ил)-1Н-бензо[d][1,2,3]триазол-5-карбоксамид
Соединение 106
Выход: 41%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,78 (с, 1H), 8,11 (д, J=8,4 Гц, 1Н), 7,96 (д, J=8,4 Гц, 1Н), 7,07 (с, 1Н), 6,93 (с, 2Н), 2,27 (с, 3Н), 2,07 (с, 6Н); ESI-MS: m/z 364,9 (M+H)+.

N-(4-(4-(2-Метоксиэтокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 109
Выход: 19%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,79-8,80 (м, 12H), 7,98-7,99 (м, 2Н), 7,08 (с, 1Н), 6,70 (с, 2H), 4,08-4,10 (м, 2Н), 3,65-3,66 (м, 2Н), 3,31 (с, 3Н), 2,06 (с, 6Н); ESI-MS: m/z 384,6 (M+H)+.

N-(4-(4-(3-Метоксипропокси)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 110
Выход: 58%; 1H ЯМР (ДМСО-d6, 500 МГц) δ 8,78-8,79 (м, 2Н), 7,98-7,99 (м, 2Н), 7,05 (с, 1Н), 6,69 (с, 2Н), 4,00-4,02 (м, 2Н), 3,46-3,48 (м, 2Н), 3,25 (с, 3Н), 2,06 (с, 6Н), 1,93-1,95 (м, 2Н), ESI-MS: m/z 398,8 (M+H)+.

4-(4-(4-(4-Метоксифенокси)-2,6-диметилфенил)тиазол-2-илкарбамоил)пиридина 1-оксид
Соединение 120
Выход: 69%; 1H ЯМР (500 МГц, CDCl3) δ 8,46-8,48 (м, 1Н), 8,39-8,43 (м, 2Н), 8,32-8,33 (м, 1Н), 7,02-7,05 (м, 2Н), 6,93-6,95 (м, 3Н), 6,70 (с, 2Н), 3,84 (с, 3Н), 2,19 (с, 3Н), 2,16 (с, 3Н); ESI-MS: m/z 447,8 [M+H]+.
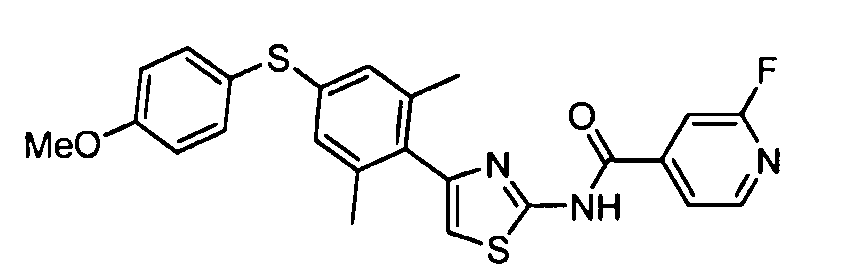
2-Фтор-N-(4-(4-(4-метоксифенилтио)-2,6-диметилфенил)тиазол-2-ил)изоникотинамид
Соединение 126
Выход: 65%; 1Н-ЯМР (500 МГц, ДМСО-d6) δ 13,1 (с, 1Н), 8,46-8,47 (м, 1H), 7,93-7,94 (м, 1Н), 7,78 (с, 1Н), 7,42-7,44 (м, 2Н), 7,19 (с, 1Н), 7,01-7,03 (м, 2Н), 6,91 (с, 2Н), 3,79 (с, 3H), 2,02 (с, 6Н); ESI-MS: m/z 465,4 [M+H]+.
Иллюстративные соединения и ингибиторная активность
В представленной ниже таблице приведены результаты для выбранных соединений, иллюстрирующих антипролиферативную активность в отношении выбранных злокачественных клеток с использованием воздействия на клетки в среде для роста соединений, как указано. Антипролиферативный эффект выражен в качестве величин IC50 в микромолярной конечной концентрации.












Иллюстративные виды биологической активности выбранных соединений
Представленные ниже данные обеспечивают иллюстративные указания в отношении биологической активности определенных соединений in vitro и in vivo. Когда соединения указаны с помощью числа, это число относится к соединениям, приведенным в таблице выше.
Цитотоксичность и антипролиферативная активность: клетки из установленных клеточных линий (например, из клеточных линий, таких как MDA-MB-231, MDA-MB-468, Hela и K562) культивировали в 10% FBS (Hylcone) в среде DMEM (Sigma, D5523). Клетки выращивали при 37°C в увлажненной атмосфере с 5% CO2 и 95% воздуха. Клетки высевали в 96-луночные планшеты для культивирования тканей.
Обработку соединением начинали после инкубации клеток в течение ночи (T0). Соединение приготавливали в концентрации от 10 мкМ до 4,6 нМ в 3× разведении из восьми точек. Соединение добавляли в лунки планшета в трех экземплярах, а затем планшеты инкубировали в течение 96 часов. Также включали ДМСО (разбавитель соединений) и добавляли его в контрольные лунки планшета. Затем определяли жизнеспособность клеток с помощью анализа с MTS с использованием нерадиоактивной системы для анализа пролиферации клеток CellTiter 96® AQueous (Promega). Для считывания оптической плотности использовали устройство для считывания планшетов (Molecular Devices, Vmax), и результаты использовали для построения кривых концентрация-ответ. Все данные соответствуют результатам экспериментов в трех экземплярах и представляют собой среднее значение трех отдельных определений с отклонением менее ±20%. Результаты анализировали с использованием программного обеспечения для вычисления линейной регрессии (GraphPad Prism 5; GraphPad Software Inc.).
Величины IC50 относятся к концентрации, которая вызывает 50% ингибирование роста. Величины GI50 (активность ингибирования роста) определяли, чтобы сделать акцент на коррекции по числу клеток в нулевой момент времени; таким образом, % ингибирование тестируемого лекарственного средства представляло собой: [1-(T-T0)/(C-T0)]×100; и эти величины использовали для нанесения на график концентрация-ответ, а затем анализировали с помощью программного обеспечения, вычисляющего линейную регрессию (GraphPad Prism 5).
Как можно видеть из фиг.1A, выбранные соединения имели значительный цитотоксический и антипролиферативный эффект на множество клеток солидной опухоли, а также на клетки лейкоза. Напротив, как можно видеть из фиг.1B, те же соединения не проявляли выраженного цитотоксического и антипролиферативного эффекта на несколько нормальных клеточных линий. Здесь, WI-38 представляет собой нормальную клеточную линию фибробластов легких человека, RPTEC представляют собой эпителиальные клетки проксимальных канальцев почек, HuVec представляют собой эндотелиальные клетки пупочной вены человека и HAoSMC представляют собой гладкомышечные клетки аорты человека.
Выбранные соединения нарушают взаимодействие Hec1/Nek2, запускают деградацию Nek2 и повышают нестабильность белка Nek2: клетки ресуспендировали в ледяном буфере для лизиса 250 (50 мМ Tris-HCl, pH 7,4, 250 мМ NaCl, 0,3% Nonidet P-40, 10 мМ NaF, дополненный ингибиторами протеаз), подвергали трем циклам замораживания/размораживания и центрифугировали при 14000 об/мин в течение 2 мин при комнатной температуре. Супернатанты использовали для анализа лизата или иммунопреципитации. Для иммунопреципитации супернатанты инкубировали с антителом против Hec1 mAb1 9G3 или поликлональным антителом против Nek2 в течение 1 ч, затем с гранулами с белком A-Sepharose в течение дополнительного часа. Гранулы собирали и промывали пять раз буфером для лизиса, содержащим гипертонический NaCl, и кипятили в SDS-буфере для нагрузки для иммуноблот-анализа. После иммуноблоттинга на мембрану Immobilon-P (Millipore Corp., Bedford, MA), блоты контактировали с антителами против Hec1 или антителами против Nek2 (Genetex, Irvine, CA). Блоты проявляли с использованием набора для хемилюминесценции ECL (Amersham Biosciences). Дополнительные детали могут быть найдены в литературе (Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J Biol Chem. 2002 Dec 20; 277(51):49408-16. Epub 2002 Oct 16).
На фиг.2A и 2B представлен иллюстративный результат такого эксперимента, где хорошо понятно, что выбранные протестированные соединения значительно нарушали взаимодействие Hec1/Nek2. На фиг.2C представлены типичные результаты снижения уровней Nek2 при инкубации клеток K562 в течение 24 ч с 1 мкМ протестированных соединений, и на фиг.2D представлены результаты, демонстрирующие нестабильность белка Nek2 с течением времени после обработки клеток K562, подвергнутых воздействию выбранных соединений в конечной концентрации 1 мкМ.
Выбранные соединения индуцируют нарушенный митоз: Клетки выращивали на покровных стеклах и осторожно промывали буфером PEMG [80 мМ пиперазин-N,N-бис(2-этансульфоновая кислота) (PIPES), pH 6,8, 5 мМ EGTA, 1 мМ MgCl2 и 4 M глицерин] или фосфатно-солевым буфером (PBS). Затем клетки фиксировали 100% метанолом при -20°C или 4% параформальдегидом в буфере PEMG или PBS и обеспечивали их проницаемость с помощью 0,4% Triton-X 100. Клетки блокировали 5% нормальной сывороткой козы (NGS) в PBS и инкубировали с первичными антителами в PBS с 5% NGS (1-2 ч при комнатной температуре). Вторичные антитела конъюгировали с Alexa 488 или 594 (Invitrogen, Carlsbad, CA). После инкубации с вторичным антителом проводили окрашивание 4,6-диамидино-2-фенилиндолом (DAPI) и из клеток приготавливали препарат на покровных стеклах с помощью реагента против обесцвечивания Prolong gold (Invitrogen). Изображения получали с помощью микроскопа Nikon H550L, оборудованного цифровыми камерами и программным обеспечением для цифровой визуализации SPOT (версия 4, Diagnostic Instruments, Inc). Дальнейший анализ изображений или количественное определение проводили с помощью Image-Pro Plus (MediaCybernetics, Bethesda, MD) или программного обеспечения Adobe Photoshop (Adobe Systems, Mountain View, CA). Дальнейшие детали описаны в литературе (Hec1 contributes to mitotic centrosomal microtubule growth for proper spindle assembly through interaction with Hice1. Mol Biol Cell. 2009 Nov; 20(22):4686-95. Epub 2009 Sep 23).
На фиг.3 представлена таблица, на которой изображен эффект выбранных соединений на митоз. Более конкретно, результаты выражены в качестве процентов неправильной ориентации хромосом в митотических клетках в течение 48 ч. Как можно видеть, все протестированные соединения значительно влияли на митоз в большом количестве клеток.
Выбранные соединения являются высокоселективными ингибиторами киназ: Ингибирование киназной активности тестируемым соединением измеряли путем количественного определения включения [33P] субстрата в присутствии тестируемого соединения. Стандартные анализы киназ начинали с MgATP в присутствии тестируемого соединения (разбавленного до конечной концентрации 4% ДМСО) или контроля в виде ДМСО, останавливали добавлением 3% фосфорной кислоты и проводили сбор в планшет для фильтрования с использованием устройства для сбора с одним фильтром (PerkinElmer, Boston, MA, США) и подсчитывали с использованием TopCount. Для первичного скрининга ингибирования киназной активности каждое тестируемое соединение оценивали в двух концентрациях (10 мМ и 1 мМ) в двух экземплярах. Результаты представляли собой среднее значение измерений в двух экземплярах и их выражали в качестве процентного ингибирования (обработка соединением против контроля в виде ДМСО). Доступные анализы киназ являются следующими: VEGFR2, PDGFR-β, FGFR1, Flt3, c-Met, CHK1, CHK2, Cdk1/циклин B, Aurora A, Aurora B, B-Raf, B-Raf (V600E), C-Raf и mTOR. Концентрация ATP, используемая в большинстве анализов киназ, находится на уровне или ниже Km для ATP для каждого фермента.
Несмотря на существенные эффекты рассматриваемых соединений при очень низкой IC50, ингибиторный профиль был высоко селективным, как можно видеть из таблицы на фиг.4.
Биодоступность: выбранные соединения вводили крысам перорально или путем инъекции в соответствии с хорошо известными методиками. Например, соединение 82 инъецировали в/в в концентрации 2 мг/кг в составе, содержащем 5% ДМСО, 10% Cremophor и 85% WFI. В таблице ниже приведены иллюстративные фармакокинетические данные.

Соединение 82 также вводится перорально в концентрации 20 мг/кг в составе, содержащем 1% метилцеллюлозу. В таблице ниже представлены иллюстративные фармакокинетические данные

Аналогично, данные PK получали для соединений 42 и 95 с в остальном идентичными составами и путями введения. В представленных ниже таблицах показаны иллюстративные результаты.
Соединение 42 в/в и перорально представлено в соответствующих таблицах ниже:


Соединение 95 в/в и перорально представлено в соответствующих таблицах ниже:

Выбранные соединения являются эффективными в модели с ксенотрансплантатом мыши: Методика была адаптирована из предшествующего опубликованного протокола (Small molecule targeting the Hec1Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008 Oct 15;68(20):8393-9). Более конкретно, самок мышей BALB/c nude (nu/nu) (5-8 недель) приобретали от Lasco (Taiwan). Животных поддерживали в определенных условиях без патогенов, и пищу и еду давали без ограничений. Содержание и все процедуры, вовлекающие животных, проводили согласно протоколам, одобренным IACUC в DCB. Для подкожной имплантации клеток MDA-MB-468 и MDA-MB-231, клетки (1×107 в матриксном геле/животном, и 0,5×107/животное, соответственно) инъецировали подкожно в правую подмышечную область. Через 10 суток после имплантации опухоли мышам вводили (в/в, QD/21 курс или п/о, QD/28 курсов в целом) носитель A (5% ДМСО, 10% Cremophor, 85% H2O), или соединения-кандидаты, включенные в состав с носителем A (7,5-150 мг/кг массы тела). Измерение перпендикулярного диаметра каждой опухоли проводили с помощью цифровых толщиномеров и объем опухоли вычисляли с использованием формулы (L×W×W)/2, где L и W соответствуют длине и ширине, соответственно. Массу тела измеряли три раза в неделю. Среднее ингибирование роста опухоли в каждой группе введения сравнивали с контролем в виде носителя и величину ингибирования роста опухоли вычисляли с использованием формулы: [1-(T/C)×100%].
Эффект in vivo рассматриваемых соединений на объем опухоли у мышей nude хорошо понятен из графиков на фиг.5A и 5B. Несмотря на уменьшение размера опухоли, масса тела оставалась постоянной во всех случаях (данные не представлены).
Специалистам в данной области должно быть очевидно, что возможно множество других модификаций, помимо уже описанных, без отклонения от идеи изобретения, описанной в настоящем описании. Таким образом, объект изобретения не должен ограничиваться чем-либо, кроме сущности прилагаемой формулы изобретения. Более того, при интерпретации как описания, так и формулы изобретения, все термины должны быть интерпретированы в наиболее широком значении, согласующимся с контекстом. В частности, термины “содержит” и “содержащий” должны быть интерпретированы как указывающие на элементы, компоненты или стадии неисключающим образом, показывая, что указанные элементы, компоненты или стадии могут быть представлены, или использованы, или комбинированы с другими элементами, компонентами или стадиями, которые прямо не указаны. Когда формула изобретения в описании относится по меньшей мере к одному элементу, выбранному из группы, состоящей из A, B, C…. и N, текст следует интерпретировать как требование только одного элемента из группы, а не A плюс N, или B плюс N, и т.д.
Claims (9)
1. Соединение, имеющее структуру формулы I

R1 представляет собой алкокси, ORa, SRa, -S(O)Ra, или -S(O)2Ra, где
Ra представляет собой алкил или арил, необязательно замещенный алкилом, галогеналкилом, алкокси и/или галогеном;
R2, R3 представляют собой C1-C6алкил;
R4 представляет собой водород или C1-C6алкил;
R5 представляет собой пиридинил, необязательно замещенный галогеном.
R1 представляет собой алкокси, ORa, SRa, -S(O)Ra, или -S(O)2Ra, где
Ra представляет собой алкил или арил, необязательно замещенный алкилом, галогеналкилом, алкокси и/или галогеном;
R2, R3 представляют собой C1-C6алкил;
R4 представляет собой водород или C1-C6алкил;
R5 представляет собой пиридинил, необязательно замещенный галогеном.
2. Соединение по п.1, где Ra представляет собой арил, необязательно замещенный галогеналкилом, алкокси и/или галогеном, R2, R3 представляют собой С1-С6алкил, R5 представляет собой пиридинил, необязательно замещенный галогеном.
3. Соединение по п.1, имеющее структуру формулы II

где X1 и X2 независимо представляют собой Н, галоген или ORa, где Ra представляет собой алкил;
Y представляет собой О, S, SO или SO2;
R1, R2 представляют собой C1-C6алкил;
R3 представляет собой Н;
n равно 0;
 ; и
; и
где Ra, Rb, Rc и Rd в независимо представляют собой водород или галоген.
независимо представляют собой водород или галоген.
где X1 и X2 независимо представляют собой Н, галоген или ORa, где Ra представляет собой алкил;
Y представляет собой О, S, SO или SO2;
R1, R2 представляют собой C1-C6алкил;
R3 представляет собой Н;
n равно 0;
где Ra, Rb, Rc и Rd в
4. Соединение по п.3, где Y представляет собой О или S.
5. Соединение по п.1, имеющее структуру, выбранную из группы, состоящей из



 и
и

6. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и соединение по п.1 в концентрации, эффективной для нарушения связывания Hec1/Nek2 у пациента, когда композицию вводят пациенту.
7. Фармацевтическая композиция по п.6, где соединение представляет собой соединение по п.5.
8. Применение соединения по п.1 для получения лекарственного средства для лечения заболевания, ассоциированного с дисфункцией и/или сверхэкспрессией Hec1, где соединение присутствует в лекарственном средстве в количестве, эффективном для нарушения связывания Nek2/Hec1.
9. Применение соединения по п.5 для получения лекарственного средства для лечения заболевания, ассоциированного с дисфункцией и/или сверхэкспрессией Hec1, где соединение присутствует в лекарственном средстве в количестве, эффективном для нарушения связывания Nek2/Hec1.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US31479810P | 2010-03-17 | 2010-03-17 | |
| US61/314,798 | 2010-03-17 | ||
| PCT/US2011/028532 WO2011115998A2 (en) | 2010-03-17 | 2011-03-15 | Modulators of hec1 activity and methods therefor |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2012144022A RU2012144022A (ru) | 2014-04-27 |
| RU2576036C2 true RU2576036C2 (ru) | 2016-02-27 |
Family
ID=44647719
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2012144022/04A RU2576036C2 (ru) | 2010-03-17 | 2011-03-15 | Модуляторы активности нес1 и способы для них |
Country Status (16)
| Country | Link |
|---|---|
| US (2) | US8946268B2 (ru) |
| EP (1) | EP2547676B1 (ru) |
| JP (2) | JP5825535B2 (ru) |
| KR (1) | KR101609856B1 (ru) |
| CN (2) | CN103038231B (ru) |
| AU (2) | AU2011227398C1 (ru) |
| BR (1) | BR112012023355A2 (ru) |
| CA (1) | CA2793311C (ru) |
| ES (1) | ES2557465T3 (ru) |
| HK (1) | HK1176055A1 (ru) |
| MX (2) | MX2012010664A (ru) |
| MY (1) | MY192693A (ru) |
| NZ (1) | NZ602121A (ru) |
| RU (1) | RU2576036C2 (ru) |
| SG (1) | SG183853A1 (ru) |
| WO (1) | WO2011115998A2 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2773957C2 (ru) * | 2017-11-30 | 2022-06-14 | Юниверсити Оф Цукуба | Модулятор активности |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2011227398C1 (en) * | 2010-03-17 | 2014-11-27 | Taivex Therapeutics Corporation | Modulators of Hec1 activity and methods therefor |
| CN102336720B (zh) * | 2011-03-02 | 2016-01-13 | 华中科技大学 | 2-氨基噻唑衍生物及制备方法和应用 |
| DE102011083271A1 (de) * | 2011-09-23 | 2013-03-28 | Beiersdorf Ag | Aromatische Amidothiazole, deren kosmetische oder dermatologische Verwendung sowie kosmetische oder dermatologische Zubereitungen mit einem Gehalt an solchen Aromatischen Amidothiazolen |
| CN103058949A (zh) * | 2011-10-18 | 2013-04-24 | 华东理工大学 | 做为dhodh抑制剂的噻唑衍生物及其应用 |
| SG11201402143YA (en) | 2011-11-21 | 2014-06-27 | Taivex Therapeutics Corp | Biomarkers for cancers responsive to modulators of hec1 activity |
| US11071736B2 (en) | 2011-11-21 | 2021-07-27 | Taivex Therapeutics Corporation | Modulators of HEC1 activity and methods therefor |
| TWI640519B (zh) * | 2011-11-29 | 2018-11-11 | 泰緯生命科技股份有限公司 | Hec1活性調控因子及其調節方法 |
| ES2681027T3 (es) * | 2012-02-01 | 2018-09-11 | City Of Hope | Inhibidores de la ribonucleótida reductasa y métodos de uso |
| US9422275B2 (en) * | 2013-07-20 | 2016-08-23 | The Regents Of The University Of California | Small molecule modifiers of the HEC1-NEK2 interaction in G2/M |
| US11028061B2 (en) * | 2015-07-27 | 2021-06-08 | Sanford Burnham Prebys Medical Discovery Institute | Modulators of myocyte lipid accumulation and insulin resistance and methods of use thereof |
| EP3746124A4 (en) | 2018-01-30 | 2021-10-27 | Foghorn Therapeutics Inc. | COMPOUNDS AND USES THEREOF |
| CN115023226A (zh) | 2020-01-29 | 2022-09-06 | 福宏治疗公司 | 化合物及其用途 |
| CN115702148B (zh) * | 2020-06-15 | 2024-09-06 | 帝斯曼知识产权资产管理有限公司 | 用于制造烷基酰胺噻唑的方法 |
| CN111925377B (zh) * | 2020-08-31 | 2022-05-31 | 上海应用技术大学 | 对位取代的二氢呋喃香豆素类神经氨酸酶抑制剂及其制备方法和应用 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2059637C1 (ru) * | 1991-06-05 | 1996-05-10 | Елф Санофи | Гетероциклические производные замещенных 2-ациламино-5-тиазолов, способы их получения, производное замещенного 2-аминотиазола, производные 2-амино-4-фенилтиазола |
| RU2125569C1 (ru) * | 1993-02-19 | 1999-01-27 | Елф Санофи | Производные полизамещенных 2-амидо-4-фенилтиазолов, способ их получения, промежуточные соединения синтеза и фармацевтическая композиция на их основе |
| US20060140956A1 (en) * | 2002-10-11 | 2006-06-29 | Wen-Hwa Lee | Method and compounds for inhibiting hec1 ativity for the treatment of proliferative diseases |
| EA200702445A1 (ru) * | 2005-05-09 | 2008-04-28 | Ачиллион Фармасьютикалз, Инк. | Соединения тиазола и способы их применения |
| RU2348630C2 (ru) * | 2003-04-25 | 2009-03-10 | Санофи-Авентис | Производные 2-ациламино-4-фенилтиазола, их получение и их применение в терапии |
Family Cites Families (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5011A (en) * | 1847-03-13 | V boring-machine | ||
| US6014A (en) * | 1849-01-09 | Stop-motion for drawing-frames | ||
| CN100357283C (zh) * | 2002-04-02 | 2007-12-26 | 中国科学院上海药物研究所 | 一类甲硫氨酰氨肽酶抑制剂 |
| EP1519736A2 (en) | 2002-06-12 | 2005-04-06 | Max-Planck-Gesellschaft Zur Förderung Der Wissenschaften E.V. | Use of hec1 antagonists in the treatment of proliferative disorders and cancer |
| KR20060088537A (ko) * | 2003-09-06 | 2006-08-04 | 버텍스 파마슈티칼스 인코포레이티드 | Atp-결합 카세트 수송체의 조절자 |
| FR2872813B1 (fr) | 2004-07-09 | 2007-01-19 | Sanofi Synthelabo | Derives de 2-carbamide-4-phenylthiazole, leur preparation et leur application en therapeutique |
| CN101048158A (zh) * | 2004-08-13 | 2007-10-03 | 健泰科生物技术公司 | 利用atp的酶的噻唑-类抑制剂 |
| JP4612487B2 (ja) * | 2005-06-30 | 2011-01-12 | キョーラク株式会社 | 容器のキャップ |
| WO2007004038A1 (en) * | 2005-07-05 | 2007-01-11 | Pfizer Products Inc. | Aminothiazole derivatives as agonists of the thrombopoietin receptor |
| WO2007008541A2 (en) * | 2005-07-08 | 2007-01-18 | Kalypsys, Inc. | Cellular cholesterol absorption modifiers |
| JP2009503107A (ja) * | 2005-08-04 | 2009-01-29 | アポジー・バイオテクノロジー・コーポレイション | スフィンゴシンキナーゼ阻害剤およびそれらの使用方法 |
| WO2007131071A2 (en) | 2006-05-02 | 2007-11-15 | Washington State University | Mig-7 as a specific anticancer target |
| WO2008124000A2 (en) * | 2007-04-02 | 2008-10-16 | Ligand Pharmaceuticals Incorporated | Thiazole derivatives as androgen receptor modulator compounds |
| JP4986749B2 (ja) * | 2007-07-09 | 2012-07-25 | 富士フイルム株式会社 | 圧力測定用材料 |
| WO2009014674A1 (en) * | 2007-07-23 | 2009-01-29 | Sirtris Pharmaceuticals, Inc. | Heterocyclylamides as gut microsomal triglyceride transport protein inhibitors |
| CN102088973A (zh) * | 2008-05-15 | 2011-06-08 | 杜克大学 | 与热休克转录因子激活化合物及其靶标有关的组合物和方法 |
| US8324385B2 (en) * | 2008-10-30 | 2012-12-04 | Madrigal Pharmaceuticals, Inc. | Diacylglycerol acyltransferase inhibitors |
| EP2309273B1 (en) | 2009-09-16 | 2016-05-18 | ZEILLINGER, Robert | Novel tumor marker determination |
| AU2011227398C1 (en) | 2010-03-17 | 2014-11-27 | Taivex Therapeutics Corporation | Modulators of Hec1 activity and methods therefor |
| SG11201402143YA (en) | 2011-11-21 | 2014-06-27 | Taivex Therapeutics Corp | Biomarkers for cancers responsive to modulators of hec1 activity |
-
2011
- 2011-03-15 AU AU2011227398A patent/AU2011227398C1/en active Active
- 2011-03-15 KR KR1020127027026A patent/KR101609856B1/ko active IP Right Grant
- 2011-03-15 BR BR112012023355A patent/BR112012023355A2/pt not_active Application Discontinuation
- 2011-03-15 MY MYPI2012003984A patent/MY192693A/en unknown
- 2011-03-15 MX MX2012010664A patent/MX2012010664A/es active IP Right Grant
- 2011-03-15 CA CA2793311A patent/CA2793311C/en active Active
- 2011-03-15 CN CN201180024772.3A patent/CN103038231B/zh active Active
- 2011-03-15 NZ NZ602121A patent/NZ602121A/en unknown
- 2011-03-15 JP JP2013500159A patent/JP5825535B2/ja active Active
- 2011-03-15 WO PCT/US2011/028532 patent/WO2011115998A2/en active Application Filing
- 2011-03-15 SG SG2012064762A patent/SG183853A1/en unknown
- 2011-03-15 ES ES11756862.6T patent/ES2557465T3/es active Active
- 2011-03-15 US US13/048,709 patent/US8946268B2/en active Active
- 2011-03-15 CN CN201610159130.9A patent/CN105906617B/zh active Active
- 2011-03-15 EP EP11756862.6A patent/EP2547676B1/en active Active
- 2011-03-15 MX MX2015015934A patent/MX346395B/es unknown
- 2011-03-15 RU RU2012144022/04A patent/RU2576036C2/ru active
-
2013
- 2013-03-12 HK HK13103078.4A patent/HK1176055A1/zh unknown
-
2014
- 2014-08-11 AU AU2014210678A patent/AU2014210678B2/en active Active
- 2014-10-29 US US14/527,087 patent/US9409902B2/en active Active
-
2015
- 2015-10-01 JP JP2015196085A patent/JP6294277B2/ja active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2059637C1 (ru) * | 1991-06-05 | 1996-05-10 | Елф Санофи | Гетероциклические производные замещенных 2-ациламино-5-тиазолов, способы их получения, производное замещенного 2-аминотиазола, производные 2-амино-4-фенилтиазола |
| RU2125569C1 (ru) * | 1993-02-19 | 1999-01-27 | Елф Санофи | Производные полизамещенных 2-амидо-4-фенилтиазолов, способ их получения, промежуточные соединения синтеза и фармацевтическая композиция на их основе |
| US20060140956A1 (en) * | 2002-10-11 | 2006-06-29 | Wen-Hwa Lee | Method and compounds for inhibiting hec1 ativity for the treatment of proliferative diseases |
| RU2348630C2 (ru) * | 2003-04-25 | 2009-03-10 | Санофи-Авентис | Производные 2-ациламино-4-фенилтиазола, их получение и их применение в терапии |
| EA200702445A1 (ru) * | 2005-05-09 | 2008-04-28 | Ачиллион Фармасьютикалз, Инк. | Соединения тиазола и способы их применения |
Non-Patent Citations (3)
| Title |
|---|
| Database accession no.CID 2322088, введено 15.07.2005); Database accession no.CID 1554161, введено 11.07.2005; Database accession no.CID 1185425, введено 10.07.2005. * |
| GUIKAI WU et al.:"Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal", Cancer Research, 2008, vol.68, no.20, pp.8393-8399. * |
| XIAO-LONG QOI et al.:"Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues", JOURNAL OF MEDICINAL CHEMISTRY, 2009, vol.52, no.6, р.1757-1767. * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2773957C2 (ru) * | 2017-11-30 | 2022-06-14 | Юниверсити Оф Цукуба | Модулятор активности |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2576036C2 (ru) | Модуляторы активности нес1 и способы для них | |
| US20220227732A1 (en) | Pyridine carboxamide compounds for inhibiting nav1.8 | |
| ES2761295T3 (es) | Métodos y composiciones para su uso en el tratamiento del cáncer y para reducir los efectos mediados por Wnt en una célula | |
| JP4751567B2 (ja) | 新規アミド誘導体およびその医薬としての用途 | |
| ES2865182T3 (es) | Inhibidor de URAT1 | |
| JPWO2002072145A1 (ja) | Ep1アンタゴニストを有効成分として含有するうつ病の治療剤 | |
| KR20080097456A (ko) | 소화관 궤양의 치료제 또는 예방제 | |
| US11767297B2 (en) | Compounds for inhibiting TNIK and medical uses thereof | |
| KR101983585B1 (ko) | Hec1 활성의 개선된 조절인자 및 이를 위한 방법 | |
| US10532987B2 (en) | Compounds and methods for inducing browning of white adipose tissue | |
| EP3129102A1 (en) | Iodonium analogs as inhibitors of nadph oxidases and other flavin dehydrogenases; formulations thereof; and uses thereof | |
| KR100903974B1 (ko) | 2,4,5-삼중치환-1,3-티아졸 유도체 및 약제학적으로 허용가능한 그의 염, 그의 제조방법 및 그를 유효성분으로함유하는 spc 수용체 활성으로 유발되는 염증관련 질환치료제 | |
| JPS6132287B2 (ru) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| HE9A | Changing address for correspondence with an applicant | ||
| HE9A | Changing address for correspondence with an applicant | ||
| HZ9A | Changing address for correspondence with an applicant |