RU2564032C2 - Производные пептидо-нуклеиновых кислот с хорошей клеточной пенетрацией и сильной аффинностью к нуклеиновой кислоте - Google Patents
Производные пептидо-нуклеиновых кислот с хорошей клеточной пенетрацией и сильной аффинностью к нуклеиновой кислоте Download PDFInfo
- Publication number
- RU2564032C2 RU2564032C2 RU2010135635/04A RU2010135635A RU2564032C2 RU 2564032 C2 RU2564032 C2 RU 2564032C2 RU 2010135635/04 A RU2010135635/04 A RU 2010135635/04A RU 2010135635 A RU2010135635 A RU 2010135635A RU 2564032 C2 RU2564032 C2 RU 2564032C2
- Authority
- RU
- Russia
- Prior art keywords
- group
- substituted
- unsubstituted
- hydrogen atom
- independently selected
- Prior art date
Links
- 108020004707 nucleic acids Proteins 0.000 title claims abstract description 32
- 102000039446 nucleic acids Human genes 0.000 title claims abstract description 32
- 150000007523 nucleic acids Chemical class 0.000 title claims abstract description 32
- 230000035515 penetration Effects 0.000 title claims abstract description 23
- 108091093037 Peptide nucleic acid Proteins 0.000 title claims abstract description 14
- 150000001875 compounds Chemical class 0.000 claims description 65
- 125000003277 amino group Chemical group 0.000 claims description 57
- -1 (9H-fluoren-9-yl) methoxycarbonyl group Chemical group 0.000 claims description 53
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 claims description 52
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 claims description 47
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 41
- 238000000034 method Methods 0.000 claims description 36
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 claims description 34
- 125000000217 alkyl group Chemical group 0.000 claims description 33
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 claims description 32
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 claims description 28
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 27
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 claims description 26
- 229940104302 cytosine Drugs 0.000 claims description 26
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 claims description 25
- 229930024421 Adenine Natural products 0.000 claims description 24
- 229960000643 adenine Drugs 0.000 claims description 24
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 21
- 108090000623 proteins and genes Proteins 0.000 claims description 18
- 150000003839 salts Chemical class 0.000 claims description 18
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 17
- 229940113082 thymine Drugs 0.000 claims description 17
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 claims description 16
- 125000002252 acyl group Chemical group 0.000 claims description 15
- 230000001413 cellular effect Effects 0.000 claims description 15
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 229940035893 uracil Drugs 0.000 claims description 13
- 125000003545 alkoxy group Chemical group 0.000 claims description 11
- 102000004169 proteins and genes Human genes 0.000 claims description 11
- 229910052717 sulfur Inorganic materials 0.000 claims description 10
- 125000004434 sulfur atom Chemical group 0.000 claims description 10
- 125000004104 aryloxy group Chemical group 0.000 claims description 8
- 125000005647 linker group Chemical group 0.000 claims description 8
- 230000001225 therapeutic effect Effects 0.000 claims description 8
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 5
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 5
- 229910052805 deuterium Inorganic materials 0.000 claims description 3
- 125000004431 deuterium atom Chemical group 0.000 claims description 3
- 230000014509 gene expression Effects 0.000 claims description 3
- 238000000338 in vitro Methods 0.000 claims description 3
- 125000001424 substituent group Chemical group 0.000 claims description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims 6
- 239000008194 pharmaceutical composition Substances 0.000 claims 1
- 238000009739 binding Methods 0.000 abstract description 25
- 230000027455 binding Effects 0.000 abstract description 23
- 230000000694 effects Effects 0.000 abstract description 17
- 239000000126 substance Substances 0.000 abstract description 6
- 101001007348 Arachis hypogaea Galactose-binding lectin Proteins 0.000 description 164
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 142
- 239000007787 solid Substances 0.000 description 62
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 59
- 210000004027 cell Anatomy 0.000 description 59
- 238000004949 mass spectrometry Methods 0.000 description 53
- 239000000243 solution Substances 0.000 description 44
- 108020004414 DNA Proteins 0.000 description 39
- 239000000178 monomer Substances 0.000 description 37
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 33
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 31
- 125000001145 hydrido group Chemical group *[H] 0.000 description 31
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 28
- 230000000692 anti-sense effect Effects 0.000 description 28
- 230000002829 reductive effect Effects 0.000 description 28
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 27
- 239000011541 reaction mixture Substances 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 21
- 230000003595 spectral effect Effects 0.000 description 21
- 239000012044 organic layer Substances 0.000 description 19
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 18
- 108091034117 Oligonucleotide Proteins 0.000 description 17
- 125000006633 tert-butoxycarbonylamino group Chemical group 0.000 description 17
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Natural products NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 16
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 16
- 238000006243 chemical reaction Methods 0.000 description 15
- 238000004440 column chromatography Methods 0.000 description 15
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 15
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 239000002253 acid Substances 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 14
- 108020004635 Complementary DNA Proteins 0.000 description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 13
- 238000010804 cDNA synthesis Methods 0.000 description 13
- 239000002299 complementary DNA Substances 0.000 description 13
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 239000004471 Glycine Substances 0.000 description 11
- 125000004494 ethyl ester group Chemical group 0.000 description 11
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 11
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 10
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 10
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 10
- 238000000942 confocal micrograph Methods 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 239000007858 starting material Substances 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 9
- 235000015165 citric acid Nutrition 0.000 description 9
- 238000004128 high performance liquid chromatography Methods 0.000 description 9
- 239000000203 mixture Substances 0.000 description 9
- 230000008859 change Effects 0.000 description 8
- 150000004985 diamines Chemical class 0.000 description 8
- 108010010705 glutarylphenylalanine-4-nitroanilide Proteins 0.000 description 8
- MSSXOMSJDRHRMC-UHFFFAOYSA-N 9H-purine-2,6-diamine Chemical group NC1=NC(N)=C2NC=NC2=N1 MSSXOMSJDRHRMC-UHFFFAOYSA-N 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 7
- 239000007864 aqueous solution Substances 0.000 description 6
- 238000004624 confocal microscopy Methods 0.000 description 6
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 6
- 230000000670 limiting effect Effects 0.000 description 6
- 230000014616 translation Effects 0.000 description 6
- WMZTYIRRBCGARG-UHFFFAOYSA-N 2-azaniumyl-5-(4-nitroanilino)-5-oxopentanoate Chemical compound OC(=O)C(N)CCC(=O)NC1=CC=C([N+]([O-])=O)C=C1 WMZTYIRRBCGARG-UHFFFAOYSA-N 0.000 description 5
- 210000000170 cell membrane Anatomy 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 238000001638 lipofection Methods 0.000 description 5
- 210000004962 mammalian cell Anatomy 0.000 description 5
- 108091070501 miRNA Proteins 0.000 description 5
- 239000002679 microRNA Substances 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 239000003921 oil Substances 0.000 description 5
- 235000019198 oils Nutrition 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 238000001243 protein synthesis Methods 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- GEGLCBTXYBXOJA-UHFFFAOYSA-N 1-methoxyethanol Chemical compound COC(C)O GEGLCBTXYBXOJA-UHFFFAOYSA-N 0.000 description 4
- PIINGYXNCHTJTF-UHFFFAOYSA-N 2-(2-azaniumylethylamino)acetate Chemical compound NCCNCC(O)=O PIINGYXNCHTJTF-UHFFFAOYSA-N 0.000 description 4
- 239000004475 Arginine Substances 0.000 description 4
- 108020004394 Complementary RNA Proteins 0.000 description 4
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 4
- 108010081734 Ribonucleoproteins Proteins 0.000 description 4
- 102000004389 Ribonucleoproteins Human genes 0.000 description 4
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 4
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 235000011054 acetic acid Nutrition 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 230000003915 cell function Effects 0.000 description 4
- 239000003184 complementary RNA Substances 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 101150024228 mdm2 gene Proteins 0.000 description 4
- 238000002844 melting Methods 0.000 description 4
- 230000008018 melting Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- 239000013259 porous coordination polymer Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- XFNJVJPLKCPIBV-UHFFFAOYSA-N trimethylenediamine Chemical compound NCCCN XFNJVJPLKCPIBV-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 238000001262 western blot Methods 0.000 description 4
- IZGFERLHOCMZQF-UHFFFAOYSA-N 2-(10h-phenoxazin-1-yloxy)ethanamine Chemical group O1C2=CC=CC=C2NC2=C1C=CC=C2OCCN IZGFERLHOCMZQF-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 108090000189 Neuropeptides Proteins 0.000 description 3
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000001243 acetic acids Chemical class 0.000 description 3
- 201000011510 cancer Diseases 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 108091060283 mipomersen Proteins 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 210000004940 nucleus Anatomy 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 230000009870 specific binding Effects 0.000 description 3
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- GCTFTMWXZFLTRR-GFCCVEGCSA-N (2r)-2-amino-n-[3-(difluoromethoxy)-4-(1,3-oxazol-5-yl)phenyl]-4-methylpentanamide Chemical compound FC(F)OC1=CC(NC(=O)[C@H](N)CC(C)C)=CC=C1C1=CN=CO1 GCTFTMWXZFLTRR-GFCCVEGCSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 2
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 2
- MHSLDASSAFCCDO-UHFFFAOYSA-N 1-(5-tert-butyl-2-methylpyrazol-3-yl)-3-(4-pyridin-4-yloxyphenyl)urea Chemical compound CN1N=C(C(C)(C)C)C=C1NC(=O)NC(C=C1)=CC=C1OC1=CC=NC=C1 MHSLDASSAFCCDO-UHFFFAOYSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 2
- TZLADKNAQIWWPK-UHFFFAOYSA-N 1-amino-7h-purin-6-one Chemical compound O=C1N(N)C=NC2=C1NC=N2 TZLADKNAQIWWPK-UHFFFAOYSA-N 0.000 description 2
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 2
- ONXCBJOMYNPZNI-UHFFFAOYSA-N 2-bromo-3,7-dihydropurin-6-one Chemical compound N1C(Br)=NC(=O)C2=C1N=CN2 ONXCBJOMYNPZNI-UHFFFAOYSA-N 0.000 description 2
- WIEATFFSFYPBQD-UHFFFAOYSA-N 2-chloro-3,7-dihydropurin-6-one Chemical compound N1C(Cl)=NC(=O)C2=C1N=CN2 WIEATFFSFYPBQD-UHFFFAOYSA-N 0.000 description 2
- HBJGQJWNMZDFKL-UHFFFAOYSA-N 2-chloro-7h-purin-6-amine Chemical compound NC1=NC(Cl)=NC2=C1NC=N2 HBJGQJWNMZDFKL-UHFFFAOYSA-N 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 2
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 2
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 2
- 0 C*CN(C=C(C=C(COCCC*)C1(C2)NC1)C2=N1)C1=O Chemical compound C*CN(C=C(C=C(COCCC*)C1(C2)NC1)C2=N1)C1=O 0.000 description 2
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 2
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 102100034343 Integrase Human genes 0.000 description 2
- 101710203526 Integrase Proteins 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 2
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 2
- 108010015847 Non-Receptor Type 1 Protein Tyrosine Phosphatase Proteins 0.000 description 2
- 101710163270 Nuclease Proteins 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 102000011931 Nucleoproteins Human genes 0.000 description 2
- 108010061100 Nucleoproteins Proteins 0.000 description 2
- 102000000536 PPAR gamma Human genes 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 108010017842 Telomerase Proteins 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 102100033001 Tyrosine-protein phosphatase non-receptor type 1 Human genes 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 2
- 150000000475 acetylene derivatives Chemical group 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 239000000074 antisense oligonucleotide Substances 0.000 description 2
- 238000012230 antisense oligonucleotides Methods 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- ICFSDLPZJDDPHP-UHFFFAOYSA-M benzyl 3-methylimidazol-3-ium-1-carboxylate;trifluoromethanesulfonate Chemical compound [O-]S(=O)(=O)C(F)(F)F.CN1C=C[N+](C(=O)OCC=2C=CC=CC=2)=C1 ICFSDLPZJDDPHP-UHFFFAOYSA-M 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- TZRFSLHOCZEXCC-HIVFKXHNSA-N chembl2219536 Chemical compound N1([C@H]2C[C@@H]([C@H](O2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2CO)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(NC(=O)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@@H]2[C@H]([C@H]([C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C3=NC=NC(N)=C3N=C2)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2[C@H](O)[C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)C=C(C)C(N)=NC1=O TZRFSLHOCZEXCC-HIVFKXHNSA-N 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000011278 co-treatment Methods 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 229940127113 compound 57 Drugs 0.000 description 2
- 229940125900 compound 59 Drugs 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 210000000172 cytosol Anatomy 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 238000004520 electroporation Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical group O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 230000001771 impaired effect Effects 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 238000005040 ion trap Methods 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 2
- 238000000520 microinjection Methods 0.000 description 2
- 229960004778 mipomersen Drugs 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 238000003752 polymerase chain reaction Methods 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 210000003705 ribosome Anatomy 0.000 description 2
- 229920002477 rna polymer Polymers 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- WMSUFWLPZLCIHP-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 9h-fluoren-9-ylmethyl carbonate Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1COC(=O)ON1C(=O)CCC1=O WMSUFWLPZLCIHP-UHFFFAOYSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- FPQMTWQFFRTHQP-LURJTMIESA-N (2s)-2-(2-aminoethylamino)-5-(diaminomethylideneamino)pentanoic acid Chemical group NCCN[C@H](C(O)=O)CCCNC(N)=N FPQMTWQFFRTHQP-LURJTMIESA-N 0.000 description 1
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 1
- FSEJIWDJVPXHJU-ZETCQYMHSA-N (2s)-6-amino-2-(2-aminoethylamino)hexanoic acid Chemical group NCCCC[C@@H](C(O)=O)NCCN FSEJIWDJVPXHJU-ZETCQYMHSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- HSILAFDVJZUQPI-UHFFFAOYSA-N 1,3-benzothiazole-2-sulfonyl chloride Chemical compound C1=CC=C2SC(S(=O)(=O)Cl)=NC2=C1 HSILAFDVJZUQPI-UHFFFAOYSA-N 0.000 description 1
- HDHOUYSWJQKQIV-UHFFFAOYSA-N 1-(10h-phenoxazin-1-yloxy)propan-2-amine Chemical compound O1C2=CC=CC=C2NC2=C1C=CC=C2OCC(N)C HDHOUYSWJQKQIV-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- TZMSYXZUNZXBOL-UHFFFAOYSA-N 10H-phenoxazine Chemical compound C1=CC=C2NC3=CC=CC=C3OC2=C1 TZMSYXZUNZXBOL-UHFFFAOYSA-N 0.000 description 1
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 1
- ZDLSZXWATPDDQS-UHFFFAOYSA-N 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxy]ethyl methanesulfonate Chemical compound CC(C)(C)OC(=O)NCCOCCOS(C)(=O)=O ZDLSZXWATPDDQS-UHFFFAOYSA-N 0.000 description 1
- RZGIMULKYLBTEI-UHFFFAOYSA-N 2-[2-[2-[bis(phenylmethoxycarbonylamino)methylideneamino]ethylamino]-6-(phenylmethoxycarbonylamino)purin-9-yl]acetic acid Chemical compound N1=C(NCCNC(NC(=O)OCC=2C=CC=CC=2)=NC(=O)OCC=2C=CC=CC=2)N=C2N(CC(=O)O)C=NC2=C1NC(=O)OCC1=CC=CC=C1 RZGIMULKYLBTEI-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- BPTVYKQNFQPCGR-UHFFFAOYSA-N 2-[6-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethoxymethyl]-2-oxo-7h-pyrrolo[2,3-d]pyrimidin-3-yl]acetic acid Chemical compound OC(=O)CN1C(=O)N=C2NC(COCCNC(=O)OC(C)(C)C)=CC2=C1 BPTVYKQNFQPCGR-UHFFFAOYSA-N 0.000 description 1
- SMLJSDLXJRGOKW-UHFFFAOYSA-N 2-[9h-fluoren-9-ylmethoxycarbonyl-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethyl]amino]acetic acid Chemical compound C1=CC=C2C(COC(=O)N(CC(O)=O)CCNC(=O)OC(C)(C)C)C3=CC=CC=C3C2=C1 SMLJSDLXJRGOKW-UHFFFAOYSA-N 0.000 description 1
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- ASJSAQIRZKANQN-CRCLSJGQSA-N 2-deoxy-D-ribose Chemical compound OC[C@@H](O)[C@@H](O)CC=O ASJSAQIRZKANQN-CRCLSJGQSA-N 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- BGAJNPLDJJBRHK-UHFFFAOYSA-N 3-[2-[5-(3-chloro-4-propan-2-yloxyphenyl)-1,3,4-thiadiazol-2-yl]-3-methyl-6,7-dihydro-4h-pyrazolo[4,3-c]pyridin-5-yl]propanoic acid Chemical compound C1=C(Cl)C(OC(C)C)=CC=C1C1=NN=C(N2C(=C3CN(CCC(O)=O)CCC3=N2)C)S1 BGAJNPLDJJBRHK-UHFFFAOYSA-N 0.000 description 1
- DQAZPZIYEOGZAF-UHFFFAOYSA-N 4-ethyl-n-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]piperazine-1-carboxamide Chemical compound C1CN(CC)CCN1C(=O)NC(C(=CC1=NC=N2)OC)=CC1=C2NC1=CC=CC(C#C)=C1 DQAZPZIYEOGZAF-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- VKKXEIQIGGPMHT-UHFFFAOYSA-N 7h-purine-2,8-diamine Chemical compound NC1=NC=C2NC(N)=NC2=N1 VKKXEIQIGGPMHT-UHFFFAOYSA-N 0.000 description 1
- XJGFWWJLMVZSIG-UHFFFAOYSA-N 9-aminoacridine Chemical compound C1=CC=C2C(N)=C(C=CC=C3)C3=NC2=C1 XJGFWWJLMVZSIG-UHFFFAOYSA-N 0.000 description 1
- 102000008873 Angiotensin II receptor Human genes 0.000 description 1
- 108050000824 Angiotensin II receptor Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 108020004513 Bacterial RNA Proteins 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- QIKPEQVGXKJLEY-UHFFFAOYSA-N CC(C)(C)OC(NCCCOCC#C)=O Chemical compound CC(C)(C)OC(NCCCOCC#C)=O QIKPEQVGXKJLEY-UHFFFAOYSA-N 0.000 description 1
- POPGBGYTSRQYLK-UHFFFAOYSA-N CC(C)(C)OC(NCCOCC#C)=O Chemical compound CC(C)(C)OC(NCCOCC#C)=O POPGBGYTSRQYLK-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical group NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- 238000008214 LDL Cholesterol Methods 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 108010077850 Nuclear Localization Signals Proteins 0.000 description 1
- WSDRAZIPGVLSNP-UHFFFAOYSA-N O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O Chemical group O.P(=O)(O)(O)O.O.O.P(=O)(O)(O)O WSDRAZIPGVLSNP-UHFFFAOYSA-N 0.000 description 1
- 102100026056 Oligodendrocyte transcription factor 3 Human genes 0.000 description 1
- 101710195927 Oligodendrocyte transcription factor 3 Proteins 0.000 description 1
- 239000012124 Opti-MEM Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 235000019482 Palm oil Nutrition 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- WUGQZFFCHPXWKQ-UHFFFAOYSA-N Propanolamine Chemical compound NCCCO WUGQZFFCHPXWKQ-UHFFFAOYSA-N 0.000 description 1
- 102000004005 Prostaglandin-endoperoxide synthases Human genes 0.000 description 1
- 108090000459 Prostaglandin-endoperoxide synthases Proteins 0.000 description 1
- 239000012083 RIPA buffer Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 206010041660 Splenomegaly Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- NOPMDSPMBLWBER-ZCFIWIBFSA-N [(2R)-1-hydroxy-4,4-dimethylpentan-2-yl] carbamate Chemical compound C(N)(O[C@@H](CO)CC(C)(C)C)=O NOPMDSPMBLWBER-ZCFIWIBFSA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000005904 alkaline hydrolysis reaction Methods 0.000 description 1
- 229960001441 aminoacridine Drugs 0.000 description 1
- WNDSRDGPTXWTJN-UHFFFAOYSA-N aminophosphonous acid morpholine Chemical compound P(O)(O)N.N1CCOCC1 WNDSRDGPTXWTJN-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 230000003178 anti-diabetic effect Effects 0.000 description 1
- 230000003276 anti-hypertensive effect Effects 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000003356 anti-rheumatic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 1
- GXLAQDOOECCSJG-UHFFFAOYSA-N benzyl n-[9-[2-[4-(1,3-benzothiazol-2-ylsulfonyl)-3-oxopiperazin-1-yl]-2-oxoethyl]-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propylamino]purin-6-yl]carbamate Chemical compound C=12N=CN(CC(=O)N3CC(=O)N(CC3)S(=O)(=O)C=3SC4=CC=CC=C4N=3)C2=NC(NCCCNC(=O)OC(C)(C)C)=NC=1NC(=O)OCC1=CC=CC=C1 GXLAQDOOECCSJG-UHFFFAOYSA-N 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- VHRGRCVQAFMJIZ-UHFFFAOYSA-N cadaverine Chemical compound NCCCCCN VHRGRCVQAFMJIZ-UHFFFAOYSA-N 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 230000024203 complement activation Effects 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000001218 confocal laser scanning microscopy Methods 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- AQEFLFZSWDEAIP-UHFFFAOYSA-N di-tert-butyl ether Chemical compound CC(C)(C)OC(C)(C)C AQEFLFZSWDEAIP-UHFFFAOYSA-N 0.000 description 1
- 238000013154 diagnostic monitoring Methods 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000294 dose-dependent toxicity Toxicity 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- RSCSJFUGQHJOJP-UHFFFAOYSA-N ethyl 2-[6-amino-2-[2-[bis(phenylmethoxycarbonylamino)methylideneamino]ethylamino]purin-9-yl]acetate Chemical compound N1=C2N(CC(=O)OCC)C=NC2=C(N)N=C1NCCNC(=NC(=O)OCC=1C=CC=CC=1)NC(=O)OCC1=CC=CC=C1 RSCSJFUGQHJOJP-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 238000002073 fluorescence micrograph Methods 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019256 formaldehyde Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- 235000011167 hydrochloric acid Nutrition 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940030980 inova Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 210000001865 kupffer cell Anatomy 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- FUJCRWPEOMXPAD-UHFFFAOYSA-N lithium oxide Chemical compound [Li+].[Li+].[O-2] FUJCRWPEOMXPAD-UHFFFAOYSA-N 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- SUUIECYTKGRQCI-UHFFFAOYSA-N methyl 2-[2-(9h-fluoren-9-ylmethoxycarbonylamino)ethylamino]acetate Chemical compound C1=CC=C2C(COC(=O)NCCNCC(=O)OC)C3=CC=CC=C3C2=C1 SUUIECYTKGRQCI-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- OSGPYAHSKOGBFY-KMHHXCEHSA-A mipomersen sodium Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].N1([C@H]2C[C@@H]([C@H](O2)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C(C)=C2)=O)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP([O-])(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP([O-])(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP([O-])(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP([O-])(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP([O-])(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP([O-])(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2CO)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(NC(=O)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP([O-])(=O)OC[C@H]2O[C@H](C[C@@H]2SP([O-])(=O)OC[C@H]2O[C@H](C[C@@H]2SP([O-])(=O)OC[C@H]2O[C@H](C[C@@H]2SP([O-])(=O)OC[C@@H]2[C@H]([C@H]([C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)SP([O-])(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP([O-])(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C3=NC=NC(N)=C3N=C2)OCCOC)SP([O-])(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP([O-])(=O)OC[C@H]2[C@H](O)[C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)C=C(C)C(N)=NC1=O OSGPYAHSKOGBFY-KMHHXCEHSA-A 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- RNFZFHUPJYZJBA-UHFFFAOYSA-N n,n-dimethylpurin-4-amine Chemical compound C1=NC=NC2(N(C)C)C1=NC=N2 RNFZFHUPJYZJBA-UHFFFAOYSA-N 0.000 description 1
- KUQWAXPYFIYBBY-UHFFFAOYSA-N n-(5-iodo-2-oxo-1h-pyrimidin-6-yl)benzamide Chemical compound IC1=CNC(=O)N=C1NC(=O)C1=CC=CC=C1 KUQWAXPYFIYBBY-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229940100692 oral suspension Drugs 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 239000002540 palm oil Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 108010043655 penetratin Proteins 0.000 description 1
- MCYTYTUNNNZWOK-LCLOTLQISA-N penetratin Chemical compound C([C@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CCCNC(N)=N)[C@@H](C)CC)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(N)=O)C1=CC=CC=C1 MCYTYTUNNNZWOK-LCLOTLQISA-N 0.000 description 1
- 230000007903 penetration ability Effects 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- AOHJOMMDDJHIJH-UHFFFAOYSA-N propylenediamine Chemical compound CC(N)CN AOHJOMMDDJHIJH-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000009711 regulatory function Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000006296 sulfonyl amino group Chemical group [H]N(*)S(*)(=O)=O 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 108091035539 telomere Proteins 0.000 description 1
- 102000055501 telomere Human genes 0.000 description 1
- 210000003411 telomere Anatomy 0.000 description 1
- WGCGURPHUSJFKY-UHFFFAOYSA-N tert-butyl 2-[2-(9h-fluoren-9-ylmethoxycarbonylamino)ethylamino]acetate Chemical compound C1=CC=C2C(COC(=O)NCCNCC(=O)OC(C)(C)C)C3=CC=CC=C3C2=C1 WGCGURPHUSJFKY-UHFFFAOYSA-N 0.000 description 1
- UQRCWKHNNZOONU-UHFFFAOYSA-N tert-butyl N-[2-[2-(1-amino-6-oxo-7H-purin-2-yl)ethoxy]ethyl]carbamate Chemical compound C(C)(C)(C)OC(=O)NCCOCCC=1N(C(C=2N=CNC=2N=1)=O)N UQRCWKHNNZOONU-UHFFFAOYSA-N 0.000 description 1
- VZSGDNVJJDXYCY-UHFFFAOYSA-N tert-butyl N-[3-(1-amino-6-oxo-7H-purin-2-yl)propyl]carbamate Chemical compound C(C)(C)(C)OC(=O)NCCCC=1N(C(C=2N=CNC2N1)=O)N VZSGDNVJJDXYCY-UHFFFAOYSA-N 0.000 description 1
- ZXNHHLAQFLJNFI-UHFFFAOYSA-N tert-butyl N-[3-(6,8-diamino-7H-purin-2-yl)propyl]carbamate Chemical compound C(C)(C)(C)OC(=O)NCCCC1=NC(=C2NC(=NC2=N1)N)N ZXNHHLAQFLJNFI-UHFFFAOYSA-N 0.000 description 1
- GPTXCAZYUMDUMN-UHFFFAOYSA-N tert-butyl n-(2-hydroxyethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCO GPTXCAZYUMDUMN-UHFFFAOYSA-N 0.000 description 1
- XDJCYKMWJCYQJM-UHFFFAOYSA-N tert-butyl n-(3-hydroxypropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCO XDJCYKMWJCYQJM-UHFFFAOYSA-N 0.000 description 1
- VULKFBHOEKTQSF-UHFFFAOYSA-N tert-butyl n-[2-(2-aminoethoxy)ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCOCCN VULKFBHOEKTQSF-UHFFFAOYSA-N 0.000 description 1
- LPHXDOGUEPNTJE-UHFFFAOYSA-N tert-butyl n-[3-(2-aminoethoxy)propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCOCCN LPHXDOGUEPNTJE-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- USFPINLPPFWTJW-UHFFFAOYSA-N tetraphenylphosphonium Chemical compound C1=CC=CC=C1[P+](C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 USFPINLPPFWTJW-UHFFFAOYSA-N 0.000 description 1
- 238000001269 time-of-flight mass spectrometry Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 108010062760 transportan Proteins 0.000 description 1
- PBKWZFANFUTEPS-CWUSWOHSSA-N transportan Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(N)=O)[C@@H](C)CC)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CN)[C@@H](C)O)C1=CC=C(O)C=C1 PBKWZFANFUTEPS-CWUSWOHSSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/18—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one oxygen and one nitrogen atom, e.g. guanine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/001—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof by chemical synthesis
- C07K14/003—Peptide-nucleic acids (PNAs)
Abstract
Изобретение относится к новому новый классу производных пептидо-нуклеиновых кислот, которые показывают хорошую клеточную пенетрацию и сильную связывающую аффинность к нуклеиновой кислоте. 10 н. и 6 з.п. ф-лы, 10 ил., 2 табл., 73 пр.
Description
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к производным пептидо-нуклеиновых кислот, химически модифицированных для обеспечения хорошей клеточной пенетрации и сильной аффинности к нуклеиновой кислоте.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На Фиг.1 представлены хроматограммы высокоэффективной жидкостной хроматографии ВЭЖХ до и после очистки Олиго 17 с помощью ВЭЖХ с обращенной фазой. Сокращения, используемые в настоящей заявке, определены в таблице на страницах 27-29.
На Фиг.2 представлен масс-спектр MALDI-TOF для очищенной серии Олиго 17.
На Фиг.3 представлены кривые изменения поглощения в зависимости от температуры для Олиго 17 для комплементарной или с нарушенной комплементарностью ДНК.
На Фиг.4(а) и 4(б) представлены изображения конфокальной микроскопии (при 63-кратном увеличении) для 1, 2, 3 и 24 часов после обработки HeLa клеток Олиго 1 и Олиго 2 при 5 мкМ соответственно.
На Фиг.5(а) и 5(б) представлены изображения конфокальной микроскопии (при 63-кратном увеличении) для 0,5 и 1 часа после обработки MCF-7 клеток Олиго 6 и Олиго 7 при 2,5 мкМ соответственно.
На Фиг.6(а) и 6(б) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 6 или 24 часов после обработки HeLa клеток Олиго 1 и Олиго 6 при 1 мкМ соответственно.
На Фиг.7(а) и 7(б) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 24 часов после обработки JAR клеток Олиго 21 и Олиго 28 при 2 мкМ соответственно.
На Фиг.7(в) и 7(г) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 24 часов после обработки А549 клеток Олиго 21 и Олиго 28 при 2 мкМ соответственно.
На Фиг.7(д) и 7(е) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 12 часов после обработки HeLa клеток Олиго 21 и Олиго 28 при 2 мкМ соответственно.
На Фиг.7(ж) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 24 часов после обработки HeLa клеток Одиго 21 при 2 мкМ.
На Фиг.8(а), 8(б) и 8(в) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) для 24 часов после обработки HeLa, A549 и JAR клеток Олиго 22 при 2 мкМ соответственно.
На Фиг.9 представлены результаты вестерн-блоттинга для JAR клеток, обработанных Олиго 9 при 5 мкМ или 10 мкМ, Олиго 10 при 5 мкМ или 10 мкМ, при совместной обработке олигомерами при 5 мкМ или 10 мкМ каждый, и контрольный опыт (без обработки олигомером).
На Фиг.10 представлена типичная структура олигомеров ПНК, описанных в данном изобретении.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Олигонуклеотиды были использованы для различных биологических целей, включая антисмысловое ингибирование экспрессии генов, ПЦР (полимеразную цепную реакцию), диагностический контроль с помощью генных чипов и т.д. Поскольку олигонуклеотиды взаимодействуют в определенной последовательности с нуклеиновыми кислотами, такими как ДНК и РНК, они являются весьма полезными для предсказуемого модулирования биологических процессов с участием ДНК или РНК в клетке. Однако, в отличие от малых молекул лекарственных препаратов, олигонуклеотиды не легко пенетрируют через мембрану клеток млекопитающих и, поэтому, вряд ли влияют на биологические процессы внутри клетки, в случае если они соответствующим образом не модифицированы или не входят в состав, дающий возможность им легко пенетрировать через цитоплазматическую мембрану.
Белки в качестве мишеней лекарственных препаратов. Белки являются посредниками различных клеточных функций. Не удивительно, что большинство в настоящее время продаваемых на рынке лекарственных препаратов показывают терапевтическую активность через модулирующие функции белка(ов). Например, нестероидный противовоспалительный лекарственный препарат - аспирин - ингибирует ферменты, названные циклооксигеназами, в отношении их противовоспалительной активности.
Лосартан связывается и является антагонистом функции трансмембранного рецептора, названного рецептором ангиотензина II, в отношении его противогипертонической активности. Росиглитазон селективно активирует внутриклеточный рецептор, названный рецептором γ, активируемым пролифератором пероксисом (PPARγ), с появлением антидиабетической активности. Этанерцепт является слитым белком, который связывает цитокин, названным фактором некроза опухоли-α (tumor necrosis factor-α, TNF-α), и нейтрализует биологическую активность TNF-α в отношении его противоревматической активности. Герцептин является моноклональным антителом для лечения рака молочной железы путем селективного связывания с erbB2, сверхэкспрессированного в некоторых типах клеток рака молочной железы.
Антисмысловое ингибирование синтеза белка. Белки кодируются ДНК (2-дезоксирибонуклеиновая кислота). В ответ на клеточную стимуляцию ДНК транскрибируется с продуцированием пре-мРНК (незрелая рибонуклеиновая кислота) в ядре. Интроновая(ые) часть(и) пре-мРНК ферментативно сплайсируется, давая мРНК (зрелая рибонуклеиновая кислота), которая затем перемещается к питозольному компоненту. В цитозоле комплекс механизма трансляции, называемый рибосомой, связывает мРНК и осуществляет синтез белка, поскольку он сканирует генетическую информацию, закодированную в мРНК. (Biochemistry vol.41, 4503-4510, 2002; Cancer Res. vol.48, 2659-2668, 1988).
Олигонуклеотид, связанный в определенной последовательности с мРНК или пре-мРНК, называется антисмысловым олигонуклеотидом (antisense oligonucleotide, АО). АО может прочно связывать мРНК и ингибировать синтез белка с помощью рибосомы на мРНК в цитозоле. АО должен находиться внутри клетки, чтобы ингибировать синтез его белка-мишени. АО может прочно связывать пре-мРНК в ядре и влиять на сплайсинг пре-мРНК, продуцируя мРНК с измененной последовательностью и, следовательно, измененным белком.

Неприродные олигонуклеотиды. Олигонуклеотиды ДНК или РНК чувствительны к деградации эндогенными нуклеазами, ограничивающими их терапевтическую полезность. На сегодняшний день были разработаны и интенсивно изучены многие типы неприродных олигонуклеотидов. (Clin. Exp. Pharmacol. Physiol. vol.33, 533-540, 2006). Некоторые из них показывают увеличенную метаболическую стабильность по сравнению с ДНК и РНК. Приведенные выше структуры являются химическими структурами некоторых типичных представителей неприродных олигонуклеотидов. Как и ожидалось, такие олигонуклеотиды связывают комплементарную нуклеиновую кислоту, как это делает ДНК или РНК.
Фосфоротиоатный олигонуклеотид - ФТО (phosphorothioate oligonucleotide, РТО) представляет собой аналог ДНК с одним из фосфатных атомов кислорода в остове, замененного на атом серы в расчете на мономер. Такие небольшие структурные изменения делают ФТО сравнительно устойчивым к деградации нуклеазами. (Ann. Rev. Biochem. Vol.54, 367-402, 1985).
Отражая структурное сходство ФТО и ДНК, оба они плохо пенетрируют через клеточную мембрану в большинстве типов клеток млекопитающих. Однако, для некоторых типов клеток, в большом количестве экспрессирующих переносчик(и) ДНК, ДНК и ФТО показывают хорошую клеточную пенетрацию. Известно, что системно введенные ФТО(ы) быстро распределяются в печени и почках. (Nucleic Acids Res. vol.25, 3290-3296, 1997).
В целях облегчения клеточной пенетрации ФТО in vitro на практике обычно осуществляли липофекцию. Однако, липофекция физически изменяет клеточную мембрану, вызывает цитотоксичность, и, следовательно, не будет идеальной при длительном терапевтическом применении.
За последние 20 лет, антисмысловые ФТО(ы) и варианты ФТО(ов) были клинически оценены для лечения злокачественных новообразований, иммунологических нарушений, болезней обмена веществ и так далее. (Biochemistry vol.41, 4503-4510, 2002; Clin. Exp. Pharmacol. Physiol. vol.33, 533-540, 2006). Многие такие антисмысловые кандидаты лекарственных препаратов не были эффективными отчасти из-за плохой клеточной пенетрации ФТО. Для того, чтобы преодолеть плохую клеточную пенетрацию, для проявления терапевтической активности ФТО необходимо вводить в высоких дозах. Однако, известно, что ФТО(ы) связаны с зависимыми от дозы токсичностями, как например увеличенным временем свертывания крови, активацией комплемента, тубулярной нефропатией, активацией клеток Купфера и иммунной стимуляцией, включая спленомегалию, лимфоидную гиперплазию, мононуклеарную клеточную инфильтрацию. (Clin. Exp. Pharmacol. Physiol. vol.33, 533-540, 2006).
Было найдено, что многие антисмысловые ФТО(ы) показывают соответствующую клиническую активность для заболеваний с существенным вкладом печени или почек. ISIS-301012 (мипомерсен) является аналогом ФТО, который ингибирует синтез ароВ-100, белка, вовлеченного в транспорт LDL холестерина (липопротеид низкой плотности, ЛПНП). Мипомерсен проявлял соответствующую клиническую активность у некоторой группы больных атеросклерозом, скорее всего, в силу его преимущественного распределения в печени. (www.medscape.com/viewarticle/556073: имеющий доступ с 19 февраля 2009). ISIS-113715 является аналогом антисмыслового ФТО, ингибирующего синтез протеинтирозинфосфатазы 1В (РТР1В), и было найдено, что он проявляет терапевтическую активность у больных диабетом II типа. (Curr. Opin. Mol. Ther. vol.6, 331-336, 2004).
В фосфорамидитном морфолиновом олигонуклеотиде - ФМО (phosphoroamidite morpholino oligonucleotide, PMO) остов фосфата и 2-дезоксирибозы ДНК заменяются фосфорамидитом и морфолином, соответственно. (Appl. Microbiol. Biotechnol. vol 71, 575-586, 2006). В то время как остов ДНК отрицательно заряжен, остов ФМО является незаряженным. Таким образом, связывание ФМО и мРНК происходит без электростатического отталкивания между остовами и обычно бывает сильнее, чем между ДНК и мРНК. Поскольку ФМО структурно очень отличается от ДНК, ФМО не будет распознаваться гепатическим(ими) переносчиком(ами), распознающим(и) ДНК. Однако, ФМО с трудом пенетрирует через клеточную мембрану.
Пептидо-нуклеиновая кислота - ПНК (peptide nucleic acid, PNA) представляет собой полипептид с N-(2-аминоэтил)глицином в качестве элементарного звена остова, и была описана Nielsen с сотр.. (Science vol.254, 1497-1500, 1991). Подобно ДНК и РНК, ПНК также селективно связывает комплементарную нуклеиновую кислоту [Nature (London) vol.365, 566-568, 1992]. Аналогично ФМО, остов ПНК не заряжен. Таким образом, связывание между ПНК и РНК обычно бывает сильнее, чем между ДНК и РНК. Поскольку ПНК структурно значительно отличается от ДНК, ПНК не будет распознаваться гепатическим(ими) переносчиком(ами), распознающим(и) ДНК, и покажет(ут) профиль распределения в тканях сильно отличающийся от такового для ДНК или ФТО. Однако, ПНК также плохо пенетрирует через мембрану клетки млекопитающего. (Adv. Drug Delivery Rev. vol.55, 267-280, 2003).
В закрытой нуклеиновой кислоте - ЗНК (locked nucleic acid, LNA) рибозное кольцо остова РНК структурно ограничивает увеличение связывающей аффинности к РНК или ДНК. Таким образом, ЗНК(ы) можно рассматривать как производные ДНК или РНК с высокой аффинностью. (Biochemistry vol.45, 7347-7355, 2006).
Антисмысловые механизмы. Антисмысловой механизм отличается в зависимости от типов АО(ов). РНКаза Н распознает дуплекс мРНК с ДНК, РНК или ФТО, и разрушает часть двойной спирали мРНК. Таким образом, антисмысловая активность ФТО существенно амплифицируется РНКазой Н. В то же время, РНКаза Н не распознает дуплекс мРНК с ФМО, ПНК или ЗНК. Иными словами, ФМО, ПНК и ЗНК должны полагаться исключительно на стерическое блокирование мРНК в отношении их антисмысловой активности. (Biochemistry vol.41, 4501-4510, 2002).
Для олигонуклеотидов с такой же связывающей аффинностью к мРНК, ФТО поэтому будет показывать более сильную антисмысловую активность, чем ФМО, ПНК и ЗНК. Для стерического блока АО(ов), как например ФМО, ПНК и ЗНК, для антисмысловой активности желательна сильная аффинность к мРНК.
Антисмысловая активность ПНК. Связывающая аффинность ПНК к мРНК будет возрастать по мере увеличения до определенного момента длины ПНК. Однако, антисмысловая активность ПНК, как представляется, не всегда увеличивается с длиной ПНК. Были случаи, когда антисмысловая активность ПНК достигала максимальной активности при 12-13-мер и после уменьшалась. (Nucleic acids Res. vol.32, 4893-4902, 2004). С другой стороны, оптимальная антисмысловая активность достигалась с 15-18-мер ПНК(ами) по сравнению с конкретной мРНК, отражая то, что структурная доступность направленного связывающего участка (сайта) мРНК будет иметь важное значение. (Biochemistry vol.40, 53-64, 2001).
Во многих случаях сообщалось, что ПНК(ы) ингибируют синтез белка рибосомой при микромолярном уровне при условиях хорошей клеточной пенетрации. (Science vol.258, 1481-85, 1992; Biochemistry vol.40, 7853-7859, 2001; Nucleic acids Res. vol.32, 4893-4902, 2004). Однако, было найдено, что ПНК(ы), нацеленные на весьма доступное положение мРНК, показывают антисмысловую активность при субмикромолярном уровне. (Neuropeptides vol.38, 316-324, 2004; Biochemistry vol.40, 53-64, 2001) или даже при субнаномолярном уровне (Nucleic Acids Res. vol.36, 4424-4432, 2008) при хороших условиях трансфекции.
Кроме того, нацеливание на весьма доступный центр мРНК, сильная связывающая аффинность ПНК к мРНК будет очень необходима для хорошей антисмысловой активности. В отличие от ДНК, ФТО и ЗНК, остов ПНК не заряжен. ПНК проявляет тенденцию к агрегации и становится менее пригодной для связывания с мРНК, поскольку ее размер увеличивается. Желательно улучшить связывающую аффинность ПНК к мРНК без увеличения длины ПНК. Включение мономеров ПНК с точечным зарядом было бы полезным для предотвращения ПНК от агрегации.
Стратегии клеточной пенетрации для ПНК. ПНК(ы) не легко пенетрируют через клеточную мембрану и, как правило, показывают недостаточную антисмысловую активность, если только должным образом не трансфицировались. В начале, антисмысловая активность ПНК оценивалась с помощью микроинъекции (Science vol.258, 1481-85, 1992) или электропорации (Biochemistry vol.40, 7853-7859, 2001). Микроинъекция и электропорация являются инвазивными и неподходящими для применения в терапевтических целях. Чтобы улучшить клеточную пенетрацию, были разработаны различные стратегии. (Adv. Drug Delivery Rev. vol.55, 267-280, 2003; Curr. Top. Med. Chem. vol.7, 727-737, 2007).
ПНК(ы) были эффективно доставлены в клетку с помощью ковалентной инкорпорации пенетрирующих в клетку пептидов (Neuropeptides vol.38, 316-324, 2004), липофекции с последующим образованием дуплекса с комплементарной ДНК (Biochemistry vol.40, 53-64, 2001), липофекции ПНК(т) с ковалентно присоединенным 9-аминоакридином (Nucleic Acids Res. vol.32, 2695-2706, 2004), липофекции ПНК(т) с ковалентно присоединенными фосфонат-анионами (Nucleic Acids Res. vol.36,4424-4432, 2008), и так далее. Клеточная пенетрация также улучшалась при присоединении к ПНК липофильного остатка, как например адамантана (Bioconjugate Chem. vol.10, 965-972, 1999), или амфифильной группы, как например тетрафенилфосфония. (Nucleic Acids Res. vol.29, 1852-1863, 2001). Тем не менее, такая ковалентная модификация вряд ли увеличивает связывающую аффинность к мРНК, несмотря на заметное улучшение в клеточной пенетрации.
ПНК(ы) с ковалетно присоединеным ПКП. Пенетрирующие в клетку пептиды - ПКП(-ы) (cell penetrating peptides, CPPs) представляют собой полипептиды, показывающие хорошую клеточную пенетрацию, и они имеют множество положительных зарядов от остатков аргинина или лизина. На сегодняшний день описаны многие ПКП(-ы), как например транспортан, пенетратин, клеточный сигнал внутриядерной локализации - КСВЛ (nuclear localization signal, NLS) и Tat. Известно, что ПКП(-ы) эффективно несут ковалентно присоединенный груз в клетку. ПНК(-ы) с ковалентно присоединенным ПКП также показали хорошую клеточную пенетрацию.
Хотя некоторые ПНК(-ы) с ковалентно присоединенным ПКП показывали антисмысловую IC50s около 100 нм (Neuropeptides vol.38, 316-324, 2004), для таких ПНК(-т) более распространенной является микромолярная антисмысловая IC50s.
ПНК(-ы) с ковалентно связанным ПКП состоят из двух частей, гидрофобного домена ПНК и положительно заряженного домена ПКП. Такая ПНК имеет тенденцию к агрегации, она захватывается эндосомами внутри клетки и становится недоступной для антисмыслового ингибирования синтеза белка. (Curr. Top. Med. Chem. vol.7, 727-737, 2007; Nucleic Acids Res. vol.33, 6837-6849, 2005). Кроме того, такой ковалентно присоединенный ПКП вряд ли увеличивает связывающую аффинность ПНК к мРНК.
ПНК(-ы) с хиральным остовом. Предпринимались попытки ввести хиральный заместитель на остов 2-аминоэтил-глицина ПНК (2-aminoethyl-glycme, Aeg). Например, растворимость ПНК в воде значительно улучшилась при включении мономера(ов) ПНК с остовом 2-аминоэтил-лизина вместо Aeg. (Angew. Chem. Int. Ed. Engl. vol.35, 1939-1941, 1996).
При включении остова L-(2-амино-2-метил)этил-глицина вместо Aeg, связывающая аффинность ПНК к ДНК и РНК значительно улучшалась. 10-мер ПНК со всем остовом L-(2-амино-2-метил)этил-глицина вместо 2-аминоэтил-глицина показала увеличение температуры плавления (Т. пл.) на 19°С и 10°С по сравнению с комплементарной ДНК и РНК, соответственно. Такое увеличение, по-видимому, не является все же пропорциональным числу замещений L-(2-амино-2-метил)этил-глицином. (J. Am. Chem. Soc. vol.128, 10258-10267, 2006).
GPNA. Сообщалось, что клеточная пенетрация ПНК значительно улучшалась при включении мономеров ПНК с остовом 2-аминоэтил-аргинина вместо Aeg. (J. Am. Chem. Soc. vol.125, 6878-6879, 2003). Такие ПНК(ы) назывались 'GPNA', так как они на остове имеют остаток гуанидиния.
Сообщалось, что GPNAs с остовом 2-аминоэтил- 
-аргинина имеют более сильную аффинность к ДНК и РНК, чем соответствующие GPNAs с остовом 2-аминоэтил- 
-аргинина. (Chem. Commun. 244-246, 2005). Для 10-мер GPNA с пятью мономерами GPNA с остовом 2-аминоэтил- 
-аргинина имелось увеличение Т. пл. на 7°С против комплементарной ДНК по сравнению с соответствующей немодифированной ПНК. (Bioorg. Med. Chem. Lett. vol.16, 4931-4935, 2006).
Сообщалось, что 16-мер антисмысловая GPNA против человеческого EGFR-TK проявляет противоопухолевую активность при перитонеальном введении бестимусным голым мышам, хотя антисмысловая активность для антисмысловой GPNA in vitro в предшествующем уровни техники не документировалась. (WO 2008/061091).
ПНК(-ы) с модифицированным нуклеиновым основанием. Для улучшения аффинности ПНК к нуклеиновым кислотам, как в случае ДНК, были осуществлены модификации нуклеиновых оснований.
ПНК(-ы) с аденином, замещенным 2,6-диаминопурином, оценивались по их аффинности к комплементарной ДНК или РНК. Установлено, что замещение 2,6-диаминопурином вызывает увеличение Т.пл. на 2,5 ~ 6°С на одно замещение. {Nucleic Acids Res. vol.25, 4639-4643, 1997).

ПНК(-ы) с цитозином, замещенным 9-(2-аминоэтокси)феноксазином, оценивались по их аффинности к комплементарной ДНК или РНК. Единственное замещение 9-(2-аминоэтокси)феноксазином вызывало увеличение Т.пл. на 10,7 ~ 23,7°С, хотя такое увеличение существенно зависело от нуклеотидной последовательности. Нуклеиновое основание 9-(2-аминопророкси)феноксазин также вызывал большое увеличение Т. пл.. В связи с огромным увеличением Т.пл., мономер ПНК с 9-(2-аминоэтокси)феноксазином или 9-(2-аминопропокси)-феноксазином, который заменил цитозин, был назван 'G-clamp' (G-зажим) (Org. Lett. vol.4, 4395-4398, 2002). Однако, о данных по клеточной пенетрации для ПНК(-т) с G-зажимом(ами)) не сообщались.
ПНК(-ы) с цитозином, замещенным 6-{2-(2-аминоэтокси)фенил}-пирролоцитозином или 6-{2,6-ди(2-аминоэтокси)фенил}пирролоцитозином, оценивались по их аффинности к комплементарной ДНК или РНК. Единственное замещение 6-{2-(2-аминоэтокси)фенил}-пирролоцитозином или 6-{2,6-ди(2-аминоэтокси)фенил}пирролоцитозином повышало Т. пл. на 3 ~ 11,5°С. (J. Am. Chem. Soc. vol.130, 12574-12575, 2008). Однако, такие ПНК(-ы) не оценивались в отношении клеточной пенетрации.
Другое применение ПНК(-т). При прочном связывании с микроРНК, ПНК может ингибировать регуляторную функцию микроРНК, приводя к увеличению уровня экспрессии белка(ов), непосредственно регулируемого(ых) микроРНК. (RNA vol.14, 336-346, 2008). При прочном связывании с рибонуклеопротеидом, как например теломеразой, ПНК может модулировать клеточную функцию рибонуклеопротеида. (Bioorg. Med. Chem. Lett. vol.9, 1273-78, 1999). При прочном связывании с определенной частью гена в ядре, ПНК может модулировать уровень транскрипции гена. (Biochemistry vol.46, 7581-89, 2007).
Поскольку ПНК прочно связывает ДНК и РНК и конкурентно распознает одно ошибочное спаривание оснований, ПНК будет пригодна для высокоточного обнаружения однонуклеотидного полиморфизма (single nucleotide polymorphism, SNP). Так как ПНК прочно связывает ДНК и РНК с высокой специфичностью, обеспечиваемой последовательностью, ПНК может найти различные другие терапевтические и диагностические приложения с участием ДНК или РНК. (FASEB vol.14, 1041-1060, 2000).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение предлагает новый класс олигомеров ПНК, представленных формулой I, или ее фармацевтически приемлемой соли:

где:
n - целое число, равное или большее 5;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1, и Tn независимо обозначают гидридогруппу, дейтеридогруппу, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
Х и Y независимо обозначают гидридогруппу, дейтеридогруппу, гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу, замещенную или незамещенную алкилгруппу, замещенную или незамещенную ацилгруппу, замещенную или незамещенную сульфонилгруппу или замещенную или незамещенную арилгруппу;
Z обозначает гидридогруппу, дейтеридогруппу, гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
B1, B2,…, Bn-1, и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и по меньшей мере одна из B1, B2,…, Bn-1 и Bn независимо обозначает неприродное нуклеиновое основание с замещенной или незамещенной аминогруппой, ковалентно связанной с остатком, ответственным за соответствующие свойства спаривания нуклеиновых оснований.
Олигомер ПНК формулы I показывает улучшенную связывающую аффинность к нуклеиновой кислоте и клеточную пенетрацию по сравнению с соответствующим «немодифицированным» олигомером ПНК. Олигомеры ПНК, описанные в изобретении, пригодны к циклу специфического ингибирования или модулирования клеточных и физиологических функций, обусловленных нуклеиновыми кислотами или физиологически активными молекулами с доменом нуклеиновой кислоты, как например рибонуклеопротеинами. Также олигомеры ПНК, описанные в данном изобретении, пригодны для диагностических целей вследствие их специфической связывающей способности к нуклеиновым кислотам.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное изобретение предлагает новый класс олигомеров ПНК, представленных формулой I, или ее фармацевтически приемлемой соли:
где:
n - целое число, равное или большее 5;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1, и Tn независимо обозначают гидридогруппу, дейтеридогруппу, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
Х и Y независимо обозначают гидридогруппу, дейтеридогруппу, гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу, замещенную или незамещенную алкилгруппу, замещенную или незамещенную ацилгруппу, замещенную или незамещенную сульфонилгруппу или замещенную или незамещенную арилгруппу;
Z обозначает гидридогруппу, дейтеридогруппу, гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
B1, B2,…, Bn-1, и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и
по меньшей мере одна из B1, B2,…, Bn-1 и Bn независимо обозначает неприродное нуклеиновое основание с замещенной или незамещенной аминогруппой, ковалентно связанной с остатком, ответственным за соответствующие свойства спаривания нуклеиновых оснований.
Олигомер ПНК, описанный в данном изобретении, показывает улучшенную клеточную пенетрацию и связывание с нуклеиновой кислотой по сравнению с «немодифицированным» олигомером ПНК. В данном изобретении «немодифицированный» олигомер ПНК относится к олигомеру ПНК формулы I, где S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1, и Tn представляют собой гидридогруппу; и B1, B2,…, Bn-1, и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин и цитозин.
Олигомер ПНК, описанный в данном изобретении, легко пенетрирует через мембрану клетки млекопитающего и может повлиять или изменить клеточные функции путем последовательного специфического связывания с нуклеиновой кислотой или нуклеопротеином внутри клетки.
Олигомер ПНК формулы I может эффективно ингибировать синтез рибосомных белков прочным связыванием с мРНК. Олигомер ПНК, описанный в данном изобретении, может прочно связывать пре-мРНК и изменять сплайсинг пре-мРНК в мРНК. Кроме того, олигомер ПНК, описанный в данном изобретении, может прочно связывать микроРНК и ингибировать деградацию мРНК, вызванную микроРНК.
Олигомер ПНК формулы I может предсказуемо связывать домен нуклеиновой кислоты рибонуклеопротеина, например, теломеразу, и модулировать ее физиологическую(ие) функцию(и). Олигомер ПНК, описанный в данном изобретении, может связывать ген и модулировать транскрипцию гена. Олигомер ПНК формулы I может связывать вирусный ген или его транскрипт и ингибировать пролиферацию вируса. Олигомер ПНК, описанный в данном изобретении, может повлиять на клеточные функции, отличающиеся от тех, которые описаны выше, путем последовательного специфического связывания нуклеиновой кислоты или нуклеопротеина внутри клетки млекопитающего. Кроме того, олигомер ПНК, описанный в данном изобретении, может прочно связывать бактериальную РНК, нуклеиновую кислоту или ген и ингибировать бактериальную пролиферацию или изменять профили бактериального биосинтеза.
Олигомер ПНК, описанный в данном изобретении, является весьма чувствительным к ошибочному спариванию оснований при связывании с неотъемлемой частью комплементарной ДНК, и будет пригоден для детектирования с высокой точностью однонуклеотидного полиморфизма (SNP). Олигомеры ПНК, описанные в данном изобретении, прочно связывают их комплементарные ДНК(-ы) с высокой специфичностью, обеспечиваемой последовательностью, и могут быть полезны для определения профиля гена. Олигомер ПНК формулы I может быть полезным для зондирования или обнаружения молекулы, содержащей нуклеиновую кислоту, такой как теломер внутри клетки, если метку хромофора, например флуорофора, вводили соответствующим образом. Олигомеры ПНК, описанные в данном изобретении, могут быть полезными для различных диагностических и аналитических целей, отличных от подробно описанных выше.
Олигомер ПНК, описанный в данном изобретении, обладает хорошей растворимостью в воде по сравнению с соответствующим «немодифицированным» олигомером ПНК, и может применяться в виде водного, солевого или буферного раствора. Олигомер ПНК формулы I может входить в состав с катионным липидом, например липофектамином. Олигомер ПНК, описанный в данном изобретении, может образовать дуплекс с комплементарной ДНК и полученный в результате дуплекс может входить в состав с катионным липидом.
Олигомер ПНК, описанный в данном изобретении, может входить в состав различных лекарственных форм, включая инъекционный состав, назальный спрей, таблетку, гранулы, твердую капсулу, мягкую капсулу, липосомный состав, пероральную суспензию, трансдермальный состав и т.д., но выбор ими не ограничивается.
Олигомер ПНК, описанный в данном изобретении, может вводиться субъекту в терапевтически эффективных дозах, которые будут меняться в зависимости от показаний, способа введения, режима дозирования, состояний субъекта и т.д.
Олигомер ПНК, описанный в данном изобретении, может вводиться субъекту различными способами, включая внутривенную инъекцию, подкожную инъекцию, внутрибрюшинную инъекцию, носовую ингаляцию, пероральное введение, трансдермальное применение и т.д., но выбор ими не ограничивается.
Олигомер ПНК формулы I может вводиться субъекту вместе с фармацевтически приемлемым вспомогательным веществом, включая лимонную кислоту, соляную кислоту, винную кислоту, стеариновую кислоту, полиэтиленгликоль, полипропиленгликоль, этанол, бикарбонат натрия, дистиллированную воду, гиалуроновую кислоту, катионный липид, как например липофектамин, крахмал, желатин, тальк, аскорбиновую кислоту, оливковое масло, пальмовое масло, метилцеллюлозу, окись титана, натрий карбоксиметилцеллюлозу, подсластитель, консервант и т.д., но выбор ими не ограничивается.
Олигомер ПНК, описанный в данном изобретении, в зависимости от наличия основной или кислотной функциональной группы (групп), может применяться в виде, нейтрализованном эквивалентным количеством фармацевтически приемлемой кислоты или основания, включая гидроксид натрия, гидроксид калия, соляную кислоту, метансульфоновую кислоту, лимонную кислоту и т.д., но выбор ими не ограничивается.
Предпочтительные олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 5, но меньшее или равное 30;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1 и Tn представляют собой гидридогруппы;
X и Y независимо выбраны из гидридогруппы, замещенной или незамещенной алкилгруппы, замещенной или незамещенной ацилгруппы, замещенной или незамещенной сульфонилгруппы и замещенной или незамещенной арилгруппы;
Z обозначает гидридогруппу, гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную аминогруппу, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
B1, B2,…, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и,
по меньшей мере одна из B1, B2,…, Bn-1 и Bn независимо выбрана из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV:



где:
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы, гидридогруппы, гидроксилгруппы и замещенной или незамещенной алкилоксигруппы; и,
L1, L2 и L3 представляют собой ковалентный линкер, отвечающий формуле V, соединяющий основную аминогруппу в остаток, ответственный за свойства спаривания нуклеиновых оснований:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу (-СН2-), и Qm непосредственно связана с основной аминогруппой;
Q2, Q3,… и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода (-O-), атома серы (-S-) и замещенной или незамещенной аминогруппы [-N(Н)-группы или -N(заместители-группы]; и
m - целое число, равное или большее 2, но меньшее или равное 15.
Представляющие особенный интерес олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 8, но меньшее или равное 25;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1 и Tn представляют собой гидридогруппу;
Х и Y независимо выбраны из гидридогруппы, замещенной или незамещенной алкилгруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, B2,…, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере две из B1, В2,…, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и гидридогруппы;
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу, и Qm непосредственно связана с основной аминогруппой;
Q2, Q3,… и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода и аминогруппы; и
m - целое число, равное или большее 2, но меньшее или равное 12.
Представляющие большой интерес олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 10, но меньшее или равное 25;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1 и Tn представляют собой гидридогруппу;
X и Y независимо выбраны из гидридогруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу, алкилоксигруппу или замещенную или незамещенную аминогруппу; и
B1, B2,…, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тамин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2,…, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и гидридогруппы;
Q1 и Qm представляют собой метиленгруппу, и Qm непосредственно связана с основной аминогруппой;
Q2, Q3,… и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число, равное или большее 2, но меньшее или равное 10.
Представляющие более повышенный интерес олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 10, но меньшее или равное 20;
S1, S2,…, Sn-1, Sn, T1, Т2,…, Tn-1 и Tn представляют гидридогруппу;
X и Y независимо выбраны из гидридогруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2,…, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, B2,…, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3, и R5 представляют собой гидридогуппу, и R2, R4 и R6 независимо обозначают гидридогруппу или замещенную или незамещенную амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу, и Qm непосредственно связана с основной аминогруппой;
Q2, Q3,… и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число, равное или большее 2, но меньшее или равное 10.
Представляющие наибольший интерес олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 10, но меньшее или равное 20;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1 и Tn представляют собой гидридогруппу;
X и Y независимо выбраны из гидридогруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2,…, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, B2,…, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой гидридогруппу, и R2, R4 и R6 независимо обозначают гидридогруппу или амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу, и Qm непосредственно связана с основной аминогруппой;
Q2, Q3,… и Qm-1 независимо выбраны из метиленгруппы и атома кислорода; и
m - целое число, равное или большее 2, но меньшее или равное 8.
Представляющие большой интерес специфические олигомеры ПНК включают олигомеры ПНК формулы I или ее фармацевтически приемлемую соль,
где:
n - целое число, равное или большее 8, но меньшее или равное 20;
S1, S2,…, Sn-1, Sn, T1, T2,…, Tn-1 и Tn представляют собой гидридогруппу;
Х представляет собой гидридогруппу;
Y обозначает гидридогруппу или замещенную или незамещенную ацилгруппу;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2,…, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2,…, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой гидридогруппу, и R2, R4 и R6 независимо обозначают гидридогруппу или амидинилгруппу;
L1 обозначает -(CH2)2-O-(CH2)2-группу, -СН2-O-(СН2)2-группу или -СН2-O-(СН2)3-группу с правой частью непосредственно связанной с основной аминогруппой; и
L2 и L3 независимо выбраны из -(СН2)2-O-(СН2)2-группы, -(СН2)3-O-(СН2)2-группы, -(СН2)2-O-(СН2)3-группы, -(СН2)2-группы, -(СН2)3-группы, -(СН2)4-группы, -(СН2)5-группы, -(СН2)6-группы, -(СН2)7-группы и -(СН2)8-группы с правой частью непосредственно связанной с основной аминогруппой.
Использованные выше термины и сокращения для олигомеров ПНК данного изобретения иллюстрируются ниже в таблице.
| Термин/Сокращение | Иллюстрация или определение |
| олигомер | Олигонуклеотид |
| гидридогруппа | отдельный атом водорода (-Н) |
| дейтеридогруппа | отдельный атом дейтерия (-D) |
| алкилгруппа | линейная или разветвленная алкилгруппа |
| арилгруппа | ароматическая группа, например фенилгруппа, пиридилгруппа, фурилгруппа, нафтилгруппа и др. |
| метиленгруппа | -(СН2)- |
| ацилгруппа | «-С(О)-», замещенная гидридогруппой, алкилгруппой или арилгруппой |
| сульфонилгруппа | «-S(O)2-», замещенная алкилгруппой или арилгруппой |
| алкилоксигруппа | «R-O-», где R представляет замещенную или незамещенную алкилгруппу |
| атом кислорода | «-O-» |
| атом серы | «-S-» |
| амидинилгруппа |
 |
| цитозин (С) |
 |
| тимин (Т) |
 |
| урацил (U) |
 |
| аденин (А) |
 |
| гуанин (G) |
 |
ОБЩИЕ СИНТЕТИЧЕСКИЕ МЕТОДИКИ
Для характеристики молекул, описанных в данном изобретении, снимались спектры ЯМР на ЯМР-спектрометре Varian Mercury 300 МГц, Bruker Avance 400 МГц или Varian Inova 500 МГц. Для определения молекулярного веса применялся Broker Daltonics UltraFlex MALDI-TOF или Agilent LC/MS Ion Trap System (система ионной ловушки). Олигомеры ПНК анализировались и очищались с помощью ВЭЖХ с C18-обращенной фазой или на Hewlett Packard 1050 HPLC или Shimazu LC-6AD HPLC. Если не указано иначе, для хроматографического разделения малых молекул, полученных в данном изобретении, применялся силикагель. Сообщалось, что точка плавления неоткорректирована.
Производные неприродных нуклеиновых оснований, применяемые для синтеза мономеров ПНК, описанной в данном изобретении, получали в соответствии с одним из методов (методы А, Б и В), приведенным ниже, или с незначительной(ыми) модификацией(ями), если только не указано особо в приведенных примерах синтеза.
Метод А. Производные 6-алкил-пирролоцитозина синтезировались в виде соответствующим образом защищенных производных в соответствии со схемой 1 или с незначительным(и) изменением(ями) ее. Такие производные 6-алкил-пирролоцитозина применялись для синтеза мономеров ПНК, содержащей нуклеиновое основание, представленное формулой II, в качестве эквивалента цитозина.
Сначала соединение а депротонировали с помощью NaH и затем алкилировали этилбромацетатом с получением соединения б. Соединение б подвергали действию палладия, катализирующего связывание с терминальным ацетиленовым производным, которое циклизовалось in situ в продукт в согласно литературе. (Nucleosides Nucleotides & Nucleic Acids vol.22, 1029-1033, 2003).
Схема 1

Метод Б. Производные 2,6-диаминопурина синтезировались в виде соответствующим образом защищенных производных в соответствии со схемой 2 или с незначительным(и) изменением(ями) ее. Такие производные 2,6-диаминопурина применялись для синтеза мономеров ПНК, содержащей нуклеиновое основание, представленное Формулой III, в качестве эквивалента аденина.
Сначала 2-галогенаденин реагировал с диамином при высокой температуре с получением соединения г, которое затем реагировало с Вос2О с получением соединения д. Соединение д депротонировали с помощью NaH и алкилировали этилбромацетатом с получением соединения е. Ароматическую аминогруппу соединения е защищали или Cbz- или Boc- группой с получением соединения ж.
Схема 2

Метод В. N-Алкилированные производные гуанина синтезировались в виде соответствующим образом защищенных производных в соответствии со схемой 3 или с незначительными изменениями ее. Такие производные гуанина применялись для синтеза мономеров ПНК, содержащих нуклеиновое основание, представленное Формулой IV, в качестве эквивалента гуанина
Схема 3

Сначала 2-галогенгипоксантин реагировал с диамином при высокой температуре с получением соединения з, которое затем реагировало с Вос2О с получением соединения и. Соединение и депротонировали с помощью NaH и алкилировали этилбромацетатом с получением соединения к.
Два вида мономеров ПНК синтезировались в соответствии с методом Г или методом Д с получением олигомеров ПНК формулы I. Олигомеры ПНК были получены от компании Panagene, Inc. (www.panagene.com, Daejon, South Korea), используя мономеры ПНК типа п из схемы 4 по запросу CTI Bio. Наоборот, мономеры ПНК типа с из схемы 5 применялись на месте для синтеза олигомеров ПНК в соответствии с методом, описанным ранее или с незначительной(ыми) модификапией(ями) его. (USP 6,133,444).
Метод Г. Мономеры ПНК с модифицированным нуклеиновым основанием получали по схеме 4 или с незначительным(и) изменением(ями) ее в виде соответствующим образом защищенных мономеров для метода синтеза олигомера ПНК, описанного в литературе. (Org. Lett. Том 9, 3291-3293, 2006). В схеме 4, соединение л может соответствовать соединению в схемы 1, соединению ж схемы 2 или соединению к схемы 3, однако, возможно, нет необходимости ограничиваться одним из этих сложноэфирных соединений.
Схема 4

Сначала сложный эфир л подвергали щелочному гидролизу с получением кислоты м, которая затем связывалась с этиловым эфиром N-[2-{N-(2-бензотиазолинил)сульфониламино}этил]-глициновой кислоты с получением соединения н. Соединение н гидролизовалось в мягких условиях с помощью LiOH с получением кислоты о, которая подвергалась циклизации по реакции связывания с EDCI с получением модифицированного мономера ПНК - п. Химическая структура мономера ПНК были принята такой, как на схеме 4 в данном изобретении, учитывая, что согласно литературе таким образом обозначенные мономеры ПНК успешно давали олигомеры ПНК. (Org. Lett. vol.9, 3291-3293, 2006).
Метод Д. Или же, мономеры ПНК с модифицированным нуклеиновым основанием получали в соответствии со схемой 5 или с незначительным(и) изменением(ями) ее в виде соответствующим образом защищенных мономеров для метода синтеза олигомера ПНК, описанного в предшествующем уровне техники. (USP 6,133,444). В схеме 5 соединение л может соответствовать соединению в на схеме 1, соединению ж на схеме 2 или соединению к на схеме 3, однако, возможно нет необходимости ограничиваться одним из этих сложноэфирных соединений.
Схема 5

Сначала кислота м связывалась с этиловым эфиром N-[2-{N-(9Н-флуорен-9-ил)амино}этил]-глициновой кислоты с получением соединения р по реакции связывания с EDCI. Затем соединение р гидролизовалось в мягких условиях с помощью LiOH с получением мономера ПНК с модифицированным нуклеиновым основанием.
Следующие примеры содержат подробное описание способов получения соединений, описанных в данном изобретении. Подробные описания этих примеров приведены только для иллюстрации и не должно истолковываться как ограничивающие данное изобретение. Большинство из этих подробных описаний находятся в пределах объема изобретения и служат примером для вышеописанных ОБЩИХ СИНТЕТИЧЕКСИХ МЕТОДИК, которые являются частью изобретения. Сокращения, используемые в следующих примерах, определены в нижеприведенной таблице.
| Категория | Обозначение (Объем понятия) |
| 1H ЯМР | Протонный магнитный резонанс. В представленных данных по ЯМР применяли следующие широко распространенные сокращения: с - для синглета, д - для дублета, т - для триплета, кв - для квартета, м - для мультиплета, уш. - для уширенного пика, J - для константы взаимодействия, CDCl3 - для дейтерированного хлороформа, DMSOd6 - для гекса-дейтерированного диметилсульфоксида (ДМСО) DMSO и т.д.. |
| МС | Масс-спектроскопия. В представленных данных по масс-спектроскопия (МС) применяли следующие всеми принятые сокращения: MALDI-TOF - для времяпролетной масс-спектрометрии с лазерной десорбцией и ионизацией в присутствии матрицы, ESI - для ионизации электрораспылением, М.в. - для молекулярного веса, (m+1) - для МН+ ионного пика, (m+23) - для MNa+ ионного пика, и т.д.. |
| Растворители | Следующие общепринятые сокращения применялись для растворителей: THF - для тетрагидрофурана, МС - для метиленхлорида, DMF - для диметилформамида, EtOH - для этанола, МеОН - для метанола, DMSO - для диметилсульфоксида, ЕА - для этилацетата и т.д. |
| Реагенты | Для реагентов применялись следующие всеми принятые сокращения: NaH - для гидрида натрия, HCl - для соляной кислоты, EDCI - для 1-этил-3-(3-диметиламинопропил)-карбодиимида гидрохлорида, НОВТ - для 1-гидрокси-бензотриазола, Boc - для трет-бутилоксикарбонила, Boc2O - для Вое ангидрида или ди-трет-бутил-дикарбоната, Cbz - для бензилоксикарбонила, Fmoc - для (9Н-флуорен-9-ил)-метоксикарбонила, Bts - для (бензо[d]тиазол-2-сульфонила), Bts-Cl - для (бензо-[d]тиазол-2-сульфонил)хлорида, TFA - для трифторуксусной кислоты, TEA - для триэтиламина, DIEA - для N,N-диизопропилэтиламина, LiOH - для гидроксида лития, Aeg - для N-(2-аминоэтил)глицина, Fmoc-Aeg-OH - для N-[2-{(9Н-флуорен-9-ил)-метоксикарбонил}амино-этил]глицина, Fmoc-Aeg-OMe - для |
| Категория | Обозначение (Объем понятия) |
| метилового эфира N-[2-(Fmoc-амино)этил]-глициновой кислоты, Fmoc-Aeg-OtBu - для трет-бутилового эфира N-[2-(Fmoc-амино)этил]-глициновой кислоты, Fmoc-Aeg-OSu - для N-янтарного эфира N-[2-(Fmoc-амино)-этил]-глициновой кислоты, HBTU - для O-(бензотриазол-1-ил)-1,1,3,3-тетраметилурания гексафторфосфата, DCC - для 1,3-дицикло-гексилкарбодиимида и т.д. | |
| Другие | Для терминологии применялись следующие общепринятые сокращения: Т. пл. - для точки плавления, °С - для градуса по Цельсию, час - для часов, мин - для минуты, г - для грамма, мг - для миллиграмма, кг - для килограмма, л - для литра, мл - для миллилитра, М - для моль/л, соед. - для соединения, водн. - для водного раствора, RT - для комнатной температуры и т.д. |
Пример 1. Получение 3-{(трет-бутоксикарбонил)амино}-1-пропанола
К 14 г 3-амино-1-пропанола, растворенного в 150 мл THF и 150 мл воды, по каплям в течение 30 минут прибавляли 40,7 г Вос2О, растворенного в 100 мл THF. После того как реакционную смесь перемешивали в течение 24 часов, THF удаляли при пониженном давлении. Полученный водный слой экстрагировали 200 мл ЕА и органический слой промывали 0,5 М водной лимонной кислотой и дистиллированной водой и затем сушили над безводным сульфатом магния. Сульфат магния отфильтровывали и полученный фильтрат концентрировали в вакууме с получением 25 г соединения 1 в виде бесцветной жидкости. 1Н ЯМР (400 МГц; CDCl3): 
4,84 (уш.с, 1Н), 3,66 (т, J=5,6 Гц, 2Н), 3,28 (кв, J=6,0 Гц, 2Н), 3,05 (уш.с, 1Н), 1,66 (м, 2Н), 1,45 (с, 9Н).
Пример 2. Получение этилового эфира {(N-бензоил)-5-йодцитозин-1-ил}уксусной кислоты (2).
К перемешиваемому раствору 8,3 г N-бензоил-5-йодцитозина, растворенного в 60 мл DMF, при 0°С прибавляли 1,06 г 55% NaH в минеральном масле и раствор перемешивали при комнатной температуре в течение 2 часов. После прибавления к реакционной смеси 2,7 мл этилбромацетата, реакционный раствор перемешивали в течение еще 24 часов при комнатной температуре, за которым следовало удаление растворителя при пониженном давлении. Полученный остаток растворяли и нерастворившееся вещество отфильтровывали. Фильтрат промывали два раза насыщенным водным раствором хлорида аммония, сушили над безводным сульфатом магния и концентрировали в вакууме. Полученный остаток очищали с помощью колоночной хроматографии (1:1 гексан/ЕА) с получением 6,5 г соединения 2 (соединение 6 в схеме 1) в виде желтого твердого вещества. Т. пл. 154-5°С. 1H ЯМР (400 МГц; CDCl3): 
13,31 (уш.с, 1Н), 8,37 (д, J=7,2 Гц, 2Н), 7,69 (с, 1Н), 7,55 (т, J=7,4 Гц, 1Н), 7,46 (т, J=7,6 Гц, 2Н), 4,49 (с, 2Н), 4,27 (кв, J=7,2 Гц, 2Н), 1,32 (т, J=7,2 Гц, 3Н).
Пример 3. Получение 3-{3-(трет-бутоксикарбониламино)пропилокси}-1-пропина (3).
К 6,5 г 55% NaH в минеральном масле, диспергированном в 150 мл THF при 0°С, по каплям в течение 15 минут прибавляли 25 г соединения 1 и смесь перемешивали в течение 1 часа. После прибавления по каплям 17,5 мл пропаргилбромида (80% толуольный раствор) в течение 30 минут, реакционную смесь перемешивали при комнатной температуре в течение 20 часов. Реакцию останавливали медленным прибавлением 250 мл воды и THF удаляли при пониженном давлении. Затем полученную водную смесь экстрагировали 250 мл ЕА, которую промывали 3 раза 250 мл воды. Органический слой сушили над безводным сульфатом магния, и сульфат магния отфильтровывали. Полученный фильтрат концентрировали в вакууме и подвергали колоночной хроматографии (5:1 гексан/ЕА) с получением 22,7 г соединения 3 в виде желтой жидкости. 1H ЯМР (400 МГц; DMSOd6): δ 6,78 (т, J=5,2 Гц, 1Н), 4,09 (д, J=2,4 Гц, 2Н), 3,43-3,39 (м, 3Н), 2,95 (кв, J=6,4 Гц, 2Н), 1,60 (м, 2Н), 1,37 (с, 9Н).
Пример 4. Получение 4-{2-(трет-бутоксикарбониламино)этокси}-1-бутина (4)
К 3,8 г 4-(2-азидоэтокси)-1-бутана, растворенного в 17 мл THF, прибавляли 7,2 г трифенилфосфина и 0,7 мл воды и реакционную смесь перемешивали в течение 8 часов, за которым последовало удаление растворителя при пониженном давлении. Затем полученный остаток растворяли в 20 мл ЕА и экстрагировали дважды 10 мл 1 М водной HCl. К водному слою прибавляли водный раствор карбоната натрия с доведением рН до 9~10. К раствору прибавляли 5,96 г Boc2O, растворенного в 15 мл THF, и реакционную смесь перемешивали 12 часов. После удаления в вакууме THF, полученный раствор экстрагировали ЕА. Органический слой промывали 0,5 М водной лимонной кислотой и сушили над безводным сульфатом магния. Органический слой концентрировали и очищали колоночной хроматографией (9:1 гексан/ЕА) с получением 3,4 г соединения 4 в виде желтого масла. 1H ЯМР (400 МГц; CDCl3):
4,95 (с, 1Н), 3,58 (т, J=6,8 Гц, 2Н), 3,53 (т, J=5,0 Гц, 2Н), 3,32 (м, 2Н), 2,46 (м, 2Н), 2,00 (т, J=2,8 Гц, 1Н), 1,45 (с, 9Н).
Пример 5. Получение 3-{2-(трет-бутоксикарбониламино)этокси}-1-пропина (5).
20 г 2-{(трет-бутоксикарбонил)амино}-1-этанола реагировал и его очищали аналогично описанной в примере 3 методики с получением 23,7 г соединения 5 в виде бледно-желтого масла. 1H ЯМР (400 МГц; DMSOd6): δ 6,81 (т, 1Н), 4,11 (д, J=2,4 Гц, 2Н), 3,41 (м, 3Н), 3,07 (кв, J=6,0 Гц, 2Н), 1,38 (с, 9Н).
Пример 6. Получение 3-[N-{3-(трет-бутоксикарбониламино)пропил}-N-(трет-бутокси-карбонил)амино]-1-пропина (6)
К перемешиваемому раствору N-[3-(трет-бутоксикарбониламино)пропил]-N-(2-пропинил)амина в 83 мл THF и 95 мл воды, прибавляли по каплям 42 г Вос2О при комнатной температуре. Реакционную смесь перемешивали в течение 1,5 часов и концентрировали в вакууме. Полученный водный слой экстрагировали ЕА. ЕА слой промывали последовательно 0,5 М водной лимонной кислотой и рассолом, сушили над безводным сульфатом магния, концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (1:1 гексан/ЕА) с получением 19 г соединения 6 в виде желтого масла. 1H ЯМР (400 МГц; CDCl3):
5,26 (уш.с, 0,6Н), 4,74 (уш.с, 0,4Н), 4,07 (уш.с, 1Н), 3,98 (уш.с, 1Н), 3,40 (т, J=6,4 Гц, 2Н), 3,13 (м, 2Н), 2,21 (т, 1Н), 1,73 (м, 2Н), 1,49 (с, 9Н), 1,45 (с, 9Н).
Пример 7. Получение 3-[2-{2,3-бис(бензилоксикарбонил)гуанидино}-этокси]-1-пропина (7)

К перемешиваемому раствору 10,9 г соединения 5 в 110 мл МС, прибавляли 110 мл TFA по каплям в течение 2 часов при 0°С и реакционную смесь перемешивали еще 3 часа. Реакционный раствор концентрировали при пониженном давлении и полученный остаток растворяли в 40 мл МС при 0°С, к которому прибавляли 12,3 мл TEA и затем 8,8 г 1,3-бис(бензилоксикарбонил)-2-(метилтио)псевдомочевины при комнатной температуре. Реакционный раствор перемешивали в течение 4 часов и промывали дважды водой. Органический слой сушили над безводным сульфатом магния, концентрировали в вакууме и подвергали колоночной хроматографии (5:1 гексан/ЕА) с получением 9,8 г соединения 7 в виде белого твердого вещества. 1H ЯМР (400 МГц; DMSOd6):
11,72 (с, 1Н), 8,58 (с, 1Н), 7,40-7,35 (м, 10Н), 5,18 (с, 2Н), 5,12 (с, 2Н), 4,18 (д, 2Н), 3,67-3,66 (м, 4Н), 2,43 (т, 1Н).
Пример 8. Получение 2-{(трет-бутоксикарбонил)амино}-1-(2-пропинил-1-окси)}-(R)-пропана (8).
10,8 г трет-бутил-(R)-1-гидрокси-(пропан-2-ил)карбамата реагировал и его очищали по аналогичной методике, описанной в примере 3. с получением 10,1 г соединения 8 в виде желтого масла. 1H ЯМР (500 МГц; DMSOd6): δ 6,63 (д, 1Н), 4,11 (д, 2Н), 3,60 (м, 1Н), 3,37-3,33 (м, 2Н), 3,26-3,23 (м, 1Н), 1,38 (с, 9Н), 1,05 (д, 3Н).
Пример 9. Получение N-[2-{2-(трет-бутоксикарбонил)аминоэтокси}этил]-N-[2-{(3-бутинил)-1-окси}этил]-N-(трет-бутоксикарбонил)амина (9).
К перемешиваемому раствору 5 г 2-{(3-бутинил)-1-окси}этил метансульфоната и 5,32 г 2-[2-{2-(трет-бутоксикарбонил)амино}этил-1-окси]этиламина в 60 мл ацетонитрила прибавляли по каплям 3,6 г карбоната калия, растворенного в воде при 0°С. Реакционный раствор оставляли медленно нагреваться до комнатной температуры и перемешивали еще 24 часа, а затем концентрировали при пониженном давлении. Полученный остаток растворяли в МС и промывали водой. Органический слой концентрировали и растворяли в 80 мл THF и 80 мл воды, к которым прибавляли 8,4 г Вос2О, растворенного в 50 мл THF. Реакционную смесь перемешивали при комнатной температуре в течение 16 часов, за которым следовало удаление THF в вакууме и экстракция ЕА. Органический слой промывали последовательно 0,5 М водной лимонной кислотой, водой и рассолом. Органический слой сушили над безводным сульфатом натрия, концентрировали и очищали с помощью колоночной хроматографии (гексан→1:4 ЕА/гексан) с получением 2,45 г соединения 9 в виде бледно-желтого масла. 1H ЯМР (400 МГц; CDCl3): δ 5,08 (уш.с, 0,5Н), 4,93 (уш.с, 0,5Н), 3,61-3,46 (м, 12Н), 3,31 (м, 2Н), 2,48 (м, 2Н), 1,99 (т, 1Н), 1,48 (с, 9Н), 1,46 (с, 9Н).
Пример 10. Получение этилового эфира 2-[6-{3-(трет-бутоксикарбонил-амино)пропил-1-окси}-метил-2-оксо-2Н-пирроло[2,3-d]пиримидин-3(7Н)-ил]уксусной кислоты (10).

К перемешиваемому раствору 6,5 г соединения 2 в 120 мл DMF, прибавляли последовательно 580 мг CuI, 4,2 мл TEA, 9,74 г соединения 3 и 1,76 г Pd(PPh3)4. Затем реакционную смесь перемешивали в течение 24 часов при 50°С с экранированием от света и концентрировали при пониженном давлении. Полученный остаток растворяли в 250 мл EtOH и перемешивали при кипении в течение 18 часов. Затем раствор концентрировали в вакууме и подвергали хроматографическому разделению (95:5 EA/EtOH) с получением 2,3 г соединения 10 в виде темно-красного губчатого/твердого вещества. 1H ЯМР (400 МГц; DMSOd6):
11,30 (уш.с, 1Н), 8,37 (с, 1Н), 6,78 (м, 1Н), 6,19 (с, 1Н), 4,70 (с, 2Н), 4,37 (с, 2Н), 4,14 (кв, J=7,2 Гц, 2Н), 3,42 (т, J=6,4 Гц, 2Н), 2,98 (м, 2Н), 1,63 (м, 2Н), 1,36 (с, 9Н), 1,20 (т, J=7,2 Гц, 3Н).
Пример 11. Получение 2-[6-{3-(трет-бутоксикарбониламино)пропил-1-окси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]уксусной кислоты (11).

К 3,3 г соединения 10 прибавляли 15 мл THF, 30 мл воды и затем 760 мг LiOH и затем смесь перемешивали при комнатной температуре в течение 20 минут. После удаления THF при пониженном давлении, полученный водный раствор промывали диэтиловым эфиром. Водный слой подкисляли до рН 3 с помощью 1 М водной HCl и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 2,46 г соединения 11 в виде белого твердого вещества. 1H ЯМР (400 МГц; DMSOd6):
11,05 (с, 1Н), 8,16 (с, 1Н), 6,79 (т, 1Н), 6,12 (с, 1Н), 4,35 (с, 2Н), 4,23 (с, 2Н), 3,41 (т, 2Н), 2,97 (кв, J=6,4 Гц, 2Н), 1,64 (м, 2Н), 1,36 (с, 9Н).
Пример 12. Получение этилового эфира N-[2-{(бензо[d]тиазол-2-сульфонил)амино}-этил]-N-[2-[6-{3-(трет-бутоксикарбониламино)пропил-1-окси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]ацетил]глициновой кислоты (12).

К 4,0 г соединения 11 и 3,6 г этилового эфира N-[2-{(бензо[d]тиазол-2-сульфонил)амино}этил]глициновой кислоты, растворенных в 30 мл DMF, прибавляли при комнатной температуре 2,42 г EDCI и 1,70 г HOBt. Реакционную смесь перемешивали в течение 8 часов. После удаления растворителя в вакууме, полученный остаток растворяли в МС и промывали 1 М водной HCl и затем водой. МС слой концентрировали при пониженном давлении и очищали с помощью колоночной хроматографии (95:5 МС/МеОН) с получением 4,6 г соединения 12 в виде желтого пенистого/твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 11,09 (уш.с, 1Н), 8,74 (с, 0,6Н), 8,58 (с, 0,4Н), 8,27 (м, 1Н), 8,20-8,14 (м, 2Н), 7,66 (м, 2Н), 6,56 (уш.с, 1Н), 6,16 (м, 1Н), 4,91 (с, 1,2Н), 4,73 (с, 0,8Н), 4,38 (с, 2,6Н), 4,17 (м, 0,9Н), 4,07 (м, 2,5Н), 3,67 (м, 1,1Н), 3,49-3,44 (м, 4Н), 3,26 (м, 0,9Н), 3,01 (м, 2Н), 1,66 (м, 2Н), 1,38 (с, 9Н), 1,24 (т, J=7,0 Гц, 1,2Н), 1,17 (т, J=7,0 Гц, 1,8Н).
Пример 13. Получение N-[2-{(бензо[d]тиазол-2-сульфонил)амино}-этил]-N-[2-[6-{3-(трет-бутоксикарбониламино)пропил-1-окси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]ацетил]глицина (13).

4,5 г соединения 12 и 670 мг LiOH диспергировали в 20 мл THF и 20 мл воды и перемешивали при комнатной температуре в течение 20 минут. THF удаляли в вакууме и полученный водный раствор промывали диэтиловым эфиром. Водный слой подкисляли до рН 3 с помощью 1 М водной HCl и экстрагировали ЕА. Слой ЕА сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 4,4 г соединения 13 в виде темно-желтого твердого вещества. 1Н ЯМР (400 МГц; DMSOd6): δ 11,32 (уш.с, 1Н), 8,36 (м, 1Н), 8,28 (м, 1,6Н), 8,22 (с, 0,4Н), 7,73 (м, 2Н), 6,78 (м, 1Н), 6,20 (с, 1Н), 4,94 (с, 1,2Н), 4,84 (с, 0,8Н), 4,52 (с, 0,8Н), 4,37 (с, 2Н), 4,30 (с, 1,2Н), 4,26 (м, 1,2Н), 4,07 (м, 2Н), 3,87 (м, 0,8Н), 3,43 (м, 2Н), 2,99 (м, 2Н), 1,63 (м, 2Н), 1,37 (с, 9Н).
Пример 14. Получение 1-{(бензо[d]тиазол-2-сульфонил)}-2-оксо-4-[6-{3-(трет-бутоксикарбониламино)пропил-1-окси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]ацетил]пиперазина (14).

4,4 г соединения 13 и 1,49 г EDCI в 50 мл DMF перемешивали при комнатной температуре в течение 16 часов. После концентрирования реакционной смеси в вакууме, полученный остаток растворяли в 50 мл МС. Раствор МС промывали последовательно 1 М водной HCl и водой, концентрировали в вакууме и затем очищали с помощью колоночной хроматографии (ацетон) с получением 1,5 г соединения 14 в виде коричневого губчатого/твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 11,25 (уш.с, 1Н), 8,36 (м, 1Н), 8,29 (м, 1Н), 8,25 (с, 0,6Н), 8,19 (0,4Н), 7,72 (м, 2Н), 6,78 (т, J=5,2 Гц, 1Н), 6,18 (с, 1Н), 4,92 (с, 1,2Н), 4,82 (с, 0,8Н), 4,51 (с, 0,8Н), 4,37 (с, 2Н), 4,29 (с, 1,2Н), 4,23 (м, 1,2Н), 4,06 (м, 2Н), 3,87 (м, 0,8Н), 3,41 (т, J=6,4 Гц, 2Н), 2.98 (кв, J=6,8 Гц, 2Н), 1,62 (м, 2Н), 1,36 (с, 9Н). MS/ESI (m+23/MNa+)=682,2 (экспериментальный), М.в.=659,8 (C28H33N7O8S2).
Исходя из ацетиленовых производных 4~9, производные пирролоцитозина 15~20 получали с помощью аналогичной методики, описанной в примере 10. Спектральные и физические данные для соединений 15~20 приводятся ниже в таблице.
Примеры 15~20. Аналитические данные для производных пирроло-цитозина 15~20.
| Соединение | Исходное вещество | X | Спектральные и физические данные |
| 15 | 4 |
 |
1H ЯМР (400 МГц; DMSOd6): δ 11,12 (с, 1Н), 8,27 (с, 1Н), 6,79 (т, J=5,4 Гц, 1Н), 6,00 (с, 1Н),4,68 (с, 2Н), 4,14 (кв, J=7,2 Гц, 2Н), 3,65 (т, J=6,6,2Н), 3,39 (т, J=6,2 Гц, 2Н), 3,08 (м, 2Н),2,78 (т, J=6,6 Гц, 2Н), 1,37 (с, 9Н), 1,20 (т, J=7,2 Гц, 3Н). Бледно-зеленое губчатое/твердое вещество. |
| 16 | 5 |
 |
1H ЯМР (400 МГц; DMSOd6): δ 11,35 (с, 1Н), 8,39 (с, 1Н), 6,87 (т, J=5,2 Гц, 1Н), 6,21 (с, 1Н), 4,70 (с, 2Н), 4,41 (с, 2Н), 4,15 (кв, J=7,2 Гц, 2Н), 3,43 (м, 2Н), 3,12 (м, 2Н), 1,38 (с, 9Н), 1,21 (т, J=7,2 Гц, 3Н). Бледно-желтое губчатое/твердое вещество. |
| Соединение | Исходное вещество | X | Спектральные и физические данные |
| 17 | 6 |
 |
1H ЯМР (400 МГц; DMSOd6) δ 11,20 (уш.с, 0,6Н), 8,86 (уш.с, 0,4Н), 8,57 (с, 0,2Н), 8,35 (с, 0,8Н), 6,83-6,76 (м, 1Н), 6,00 (с, 0,8Н), 5,76 (с, 0,2Н), 4,75 (с, 0,3Н), 4,70 (с, 1,7Н), 4,55 (с, 0,3Н), 4,30 (с, 1,7Н), 4,14 (кв, J=7,2 Гц, 2Н), 3,18 (м, 2Н), 2,90-2,88 (м, 2Н), 1,58 (м, 2Н), 1,40-1,36 (м, 18Н), 1,20 (т, 3Н). Коричневое губчатое/твердое вещество. |
| 18 | 7 |
 |
1H ЯМР (400 МГц; DMSOd6) δ 11,57 (с, 1Н), 11,33 (с, 1Н), 8,50 (м, 1Н), 8,37 (с, 1Н), 7,44-7,31 (м, 10Н), 6,22 (с, 1Н), 5,22 (с, 2Н), 5,03 (с, 2Н), 4,70 (с, 2Н), 4,44 (с, 2Н), 4,14 (кв, 2Н), 3,57-3,53 (м, 4Н), 1,21 (т, 3Н). Бледно-коричневое твердое вещество. |
| 19 | 8 |
 |
1H ЯМР (500 МГц; DMSOd6): δ 11,32 (с, 1Н), 8,38 (с, 1Н), 6,71 (д, 1Н), 6,20 (с, 1Н), 4,70 (с,2Н), 4,41 (м, 2Н), 4,14 (кв, 2Н), 3,65 (м, 1Н), 3,37-3,34 (м, 1Н), 3,26-3,22 (м, 1Н), 1,37 (с, 9Н), 1,20 (т, 3Н), 1,02 (д, 3Н). Бледно-коричневое твердое вещество. |
| 20 | 9 |
 |
1H ЯМР (500 МГц; DMSOd6): δ 11,13 (с, 1Н), 8,25 (с, 1Н), 6,73 (с, 1Н), 5,99 (с, 1Н), 4,68 (с,2Н), 4,12 (кв, 2Н), 3,67 (т, 2Н), 3,48-3,27 (м, 10Н), 3,04 (кв, 2Н), 2,78 (т, 2Н), 1,38 (с, 9Н), 1,36 (с, 1Н), 1,19 (т, 3Н). Коричневое твердое вещество. |
Исходя из производных пирролоцитозина 15, 16, 17 и 20, мономеры ПНК с модифицированным цитозином 21~24 получали аналогично методикам, описанным в примерах 11~14. Спектральные и физические данные для соединений 21~24 приводятся ниже в таблице.
Примеры 21~24. Аналитические данные для цитозиновых мономеров ПНК 21~24.

| Соединение | Исходное вещество | X | Спектральные и физические данные |
| 21 | 15 |
|
1H ЯМР (400 МГц; DMSOd6): δ 11,06 (с, 1Н), 8,36 (м, 1Н), 8,28 (м, 1Н), 8,14 (с, 0,6Н), 8,08 (2,0,4Н), 7,72 (м, 2Н), 6,78 (т, 1Н), 5,98 (с, 1Н), 4,91 (с, 1,2Н), 4,80 (с, 0,8Н), 4,51 (с, 0,8Н), 4,29 (с, 1,2Н), 4,24 (м, 1,2Н), 4,06 (м, 2Н), 3,86 (м, 0,8Н), 3,64 (т, J=6,4 Гц, 2Н), 3,38 (т, J=6,0 Гц, 2Н), 3,07 (м, 2Н), 2,78 (м, 2Н), 1,37 (с, 9Н). MS/ESI (m+1)=660,2 (экспериментальный), М.в.=659,8(C28H33N7O8S2). Коричневое губчатое/твердое вещество. |
| 22 | 16 |
|
1Н ЯМР (400 МГц; DMSOd6): δ 11,31 (с, 1Н), 8,36 (м, 1Н), 8,30-8,27 (м, 1,6Н), 8,22 (с, 0,4Н), 7,73 (м, 2Н), 6,87 (т, J=5,6 Гц, 1Н), 6,20 (м, 1Н), 4,94 (с, 1,2Н), 4,83 (с, 0,8Н), 4,52 (с, 0,7Н), 4,41 (с, 2,1Н), 4,30 (с, 1,1Н), 4,25 (м, 1,2Н), 4,06 (м, 2Н), 3,87 (м, 0,8Н), 3,42 (т, 2Н), 3,12 (м, 2Н), 1,38 (с, 9Н). MS/ESI (m+1)=646,2 (экспериментальный), М.в=645,7 (C27H31N7O8S2). Красное губчатое/твердое вещество. |
| 23 | 17 |
|
1Н ЯМР (400 МГц; DMSOd6): δ 11,16 (уш.с, 1Н), 8,36 (м, 1Н), 8,28 (м, 1Н), 8,21 (с, 0,6Н), 8,15 (с, 0,4Н), 7,73 (м, 2Н), 6,77 (уш.с, 1Н), 6,00 (уш.с, |
| Соединение | Исходное вещество | X | Спектральные и физические данные |
| 1Н), 4,92 (с, 1,2Н), 4,82 (с, 0,8Н), 4,52 (с, 0,9Н), 4,30 (с, 3,1Н), 4,25 (м, 1,2Н), 4,07 (м, 2Н), 3,87 (м, 0,8Н), 3,19 (м, 2Н), 2,89 (м, 2Н), 1,59 (м, 2Н), 1,41-1,36 (м, 18Н); MS/ESI (m+23/MNa+)=781,3 (экспериментальный), М.в=758,9 (C33H42N8O9S2). Красное губчатое/твердое вещество. | |||
| 24 | 20 |
|
1H ЯМР (500 МГц; DMSOd6): δ 11,10 (м, 1Н), 8,35 (м, 1Н), 8,28 (м, 1Н), 8,14 (с, 0,6Н), 8,08 (с, 0,4Н), 7,72 (м, 2Н), 6,76 (м, 1Н), 5,97-5,96 (с, 1Н), 4,90 (с, 1,2Н), 4,80 (с, 0,8Н), 4,51 (с, 0,8Н), 4,29 (с, 1,2Н), 4,25 (т, 1,2Н), 4,08-4,04 (м, 2Н), 3,86 (т, 0,8Н), 3,66 (м, 2Н), 3,47 (м, 2Н), 3,41 (м, 2Н), 3,32-3,30 (м, 4Н), 3,27 (м, 2Н), 3,04 (м, 2Н), 2,77 (м, 2Н), 1,37 (с, 9Н), 1,35 (с, 9Н). MS/ESI(m+23/MNa+)=869,3 (экспериментальный), М.в.=847,0 (C37H50N8O11S2). Желтое твердое вещество. |
Пример 25. Получение 2-{3-(трет-бутоксикарбониламино)пропил}амино-аденина (25).

6,8 г 2-хлораденина, растворенного в 68 мл 1,3-диаминопропана и 68 мл монометоксиэтанола, перемешивали при кипении в течение 24 часов, и реакционную смесь концентрировали в вакууме. Полученный остаток растворяли в 100 мл THF и 100 мл воды, к которым медленно прибавляли 60 г Boc2O, растворенного в 79 мл THF. Реакционную смесь перемешивали при комнатной температуре в течение 6 часов и затем органический растворитель удаляли при пониженном давлении. Полученный водный слой экстрагировали дважды 100 мл ЕА. Органический слой промывали 0,5 М водной лимонной кислотой и рассолом и сушили над безводным сульфатом магния. Органический слой концентрировали при пониженном давлении и подвергали хроматографическому разделению (1:10 МеОН/МС) с получением 4,07 г соединения, защищенного двумя Вос-группами. Данное соединение растворяли в 100 мл МеОН, к которому медленно прибавляли 45 мл водного насыщенного раствора карбоната натрия. Реакционный раствор перемешивали при 50°С в течение 1 часа и затем концентрировали в вакууме. Полученный остаток растворяли в 50 мл МеОН и нерастворенное вещество отфильтровывали. Затем фильтрат концентрировали с получением 3,16 г соединения 25 в виде белого твердого вещества. 1H ЯМР (400 МГц; DMSOd6): 
12,11 (уш.с, 1Н), 7,63 (с, 1Н), 6,78 (т, 1Н), 6,55 (с, 2Н), 6,07 (т, 1Н), 3,20 (кв, 2Н), 2,96 (кв, 2Н), 1,60 (м, 2Н), 1,37 (с, 9Н).
Исходя из 2-хлораденина и соответствующего диамина, производные 2,6-диаминопурина 26~30 получали аналогично методике, описанной в примере 25. Спектральные и физические данные для соединений 26~30 приводятся ниже в таблице.
Пример 26~30. Аналитические данные для производных 2,6-диаминопурина 26~30.

| Соединение | Исходный диамин | L2 | Спектральные и физические данные |
| 26 |
|
-(СН2)2- | 1H ЯМР (400 МГц; DMSOd6): δ 12,20 (уш.с, 1Н), 7,66 (с, 1Н), 6,84 (т, 1Н), 6,62 (с, 2Н), 6,10 (т, 1Н), 3,25 (кв, 2Н), 3,08 (кв, 2Н), 1,36 (с, 9Н). Бледно-желтое твердое вещество. |
| Соединение | Исходный диамин | L2 | Спектральные и физические данные |
| 27 |
|
-(CH2)4- | 1H ЯМР (500 МГц; DMSOd6): δ 12,07 (уш.с, 1Н), 7,63 (с, 1Н), 6,75 (с, 1Н), 6,50 (с, 2Н), 6,02(с, 1Н), 3,18 (кв, 2Н), 2,91 (кв, 2Н), 1,48-1,36 (м,13Н). Желтовато-зеленое твердое вещество. |
| 28 |
 |
-(CH2)5- | 1H ЯМР (400 МГц; DMSOd6): δ 12,14 (уш.с, 1Н), 7,65 (с, 1Н), 6,77 (т, 1Н), 6,55 (с, 2Н), 6,01 (с, 1Н), 3,17 (м, 2Н), 2,89 (кв, 2Н), 1,48 (м, 2Н), 1,41-1,36 (м, 11Н), 1,26 (м, 2Н). Бледно-желтое твердое вещество. |
| 29 |
|
-(CH2)7- | 1Н ЯМР (500 МГц; DMSOd6): δ 12,11 (уш.с, 1Н), 7,64 (с, 1Н), 6,78 (т, J=5,6 Гц, 1Н), 6,56 (с,2Н), 6,04 (т, J=5,5 Гц, 1Н), 3,17 (тд, J=6,3, 6,3Гц, 2Н), 2,88 (тд, J=6,7, 6,7 Гц, 2Н), 1,49-1,47(м, 2Н), 1,36-1,31 (м, 11Н), 1,29-1,22 (м, 6Н). Желтовато-зеленое твердое вещество. |
| 30 |
 |
|
1Н ЯМР (500 МГц; DMSOd6): δ 12,15 (с, 1Н), 7,64 (с, 1Н), 6,84 (т, 1Н), 6,56 (с, 2Н), 6,05 (т, 1Н), 3,48 (т, 2Н), 3,39-3,34 (м, 4Н), 3,07 (кв, 2Н), 1,37 (с, 9Н). Желтое губчатое вещество. |
Пример 31. Получение 2-[2-{2-(трет-бутоксикарбониламино)-2-метил}этил]-амино-1Н-пурин-6(9Н)-она (31).

11 г 2-хлоргипоксантина и 4,96 мл 1,2-диаминопропана (рацемата) диспергировали в 33 мл монометоксиэтанола и перемешивали в течение 24 часов при 130°С. Растворитель удаляли в вакууме и полученный остаток растворяли в 97 мл THF и 97 мл воды, к которым медленно прибавляли 22,8 г Вос20, растворенного в 64 мл THE Реакционную смесь перемешивали при комнатной температуре в течение 6 часов и к раствору прибавляли ЕА. Полученный осадок собирали с помощью фильтрации с получением соединения 31 в виде серого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): 
12,42 (с, 1Н), 10,44 (уш.с, 1Н), 7,61 (с, 1Н), 6,76 (д, 1Н), 6,27 (м, 1Н), 3,67 (м, 1Н), 3,32 (м, 1Н), 3,14 (м, 1Н), 1,36 (с, 9Н), 1,02 (д, 3Н).
Пример 32. Получение этилового эфира 2-[6-амино-2-{3-(трет-бутоксикарбонил-амино)-пропил}амино-9Н-пурин-9-ил]уксусной кислоты (32).

К перемешиваемому раствору 3,16 г соединения 25, растворенного в 100 мл DMF, прибавляли 480 мг 55% NaH в минеральном масле. Реакционный раствор перемешивали в течение 2 часов, после чего медленно прибавляли 1,98 мл этилбромацетата. Двумя часами позже реакционную смесь концентрировали в вакууме и очищали с помощью колоночной хроматографии (1:10 EtOH/EA) с получением 2,92 г аналога диаминопурина 32 в виде бледно-желтого твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 7,67 (с, 1Н), 6,80 (т, 1Н), 6,71 (с, 2Н), 6,28 (т, 1Н), 4,85 (с, 2Н), 4,15 (кв, 2Н), 3,20 (кв, 2Н), 2,94 (кв, 2Н), 1,57 (м, 2Н), 1,37 (с, 9Н), 1,21 (т, 3Н).
Пример 33. Получение этилового эфира 2-[6-(бензилоксикарбонил)амино-2-{3-(трет-бутокси-карбониламино)пропил}амино-9Н-пурин-9-ил]уксусной кислоты (33).

К перемешиваемому раствору 4,68 г соединения 32 в 100 мл DMF, при комнатной температуре прибавляли 13,2 г N-(бензилоксикарбонил)-N'-метил-имидазолий трифлата. Через 12 часов реакционную смесь концентрировали при пониженном давлении и подвергали колоночной хроматографии (5% МеОН в МС) с получением 5,4 г соединения 33 в виде белого твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 10,19 (с, 1Н), 7,92 (с, 1Н), 7,45-7,33 (м, 5Н), 6,88 (т, 1Н), 6,77 (т, 1Н), 5,18 (с, 2Н), 4,94 (с, 2Н), 4,16 (кв, 2Н), 3,25 (кв, 2Н), 2,95 (кв, 2Н), 1,60 (м, 2Н), 1,36 (с, 9Н), 1,21 (т, 3Н).
Пример 34. Получение этилового эфира N-[2-{2-(бензо[d]тиазол)сульфонил}-амино-этил]-N-[2-[6-(бензилоксикарбонил)амино-2-{3-(трет-бутоксикарбонил-амино)-пропил}амино-9Н-пурин-9-ил]ацетил]глициновой кислоты (34).

5,4 г соединения 33 и 950 мг LiOH растворяли в 40 мл THF и 40 мл воды и перемешивали при комнатной температуре в течение 1 часа. THF удаляли в вакууме и полученный водный раствор подкисляли до рН 3 с помощью 1 М водной HCl и затем экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток и 2,92 г этилового эфира 2-N-[2-{(бензо[d]тиазол-2-сульфонил)амино}этил]глициновой кислоты растворяли в 240 мл DMF, к которым при комнатной температуре прибавляли 1,95 г EDCI и 1,38 г HOBt. Реакционную смесь перемешивали в течение 20 часов, концентрировали при пониженном давлении и растворяли в МС. МС раствор промывали 1 М водной HCl, концентрировали в вакууме и затем очищали колоночной хроматографией (5% МеОН/МС) с получением 2,7 г соединения 34 в виде бледно-желтого губчатого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 10,18 (м, 1Н), 8,97 (уш.с, 0,6Н), 8,80 (уш.с, 0,4Н), 8,28 (д, 1Н), 8,18 (м, 1Н), 7,80 (с, 0,6Н), 7,76 (с, 0,4Н), 7,66 (м, 2Н), 7,46-7,32 (м, 5Н), 6,77 (м, 2Н), 5,18 (с, 2Н), 5,10 (с, 1,2Н), 4,89 (с, 0,8Н), 4,45 (с, 0,8Н), 4,17 (кв, 0,8Н), 4,07-4,00 (м, 2,4Н), 3,68 (м, 1,2Н), 3,47 (м, 1,2Н), 3,41 (м, 0,9Н), 3,22 (м, 2,7Н), 2,94 (м, 2Н), 1,59 (м, 2Н), 1,36 (с, 9Н), 1,31-1,12 (м, 3Н).
Пример 35. Получение 1-(бензо[d]тиазол-2-сульфонил)-2-оксо-4-[[6-(бензил-оксикарбонил)амино-2-{3-(трет-бутоксикарбониламино)пропиламино}-9Н-пурин-9-ил]-ацетил]пиперазина (35).

2,7 г соединения 34 и 340 мг LiOH диспергировали в 15 мл THF и 20 мл воды и перемешивали в течение 30 минут при комнатной температуре. THF удаляли при пониженном давлении. Затем полученный водный слой подкисляли до рН 3 с помощью 1 М водной HCl и экстрагировали ЕА. ЕА слой сушили над безводным сульфатом натрия и концентрировали в вакууме с получением 2,48 г неочищенного продукта. Неочищенный продукт и 716 мг EDCI, растворенного в 70 мл DMF, перемешивали при комнатной температуре в течение 20 часов. Растворитель удаляли при пониженном давлении и полученный в результате остаток растворяли в МС и промывали 1 М водной HCl и затем водой. Органический слой концентрировали в вакууме и очищали колоночной хроматографией (ацетон) с получением 1,4 г соединения 35 в виде белого губчатого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 10,16 (с, 1Н), 8,35 (м, 1Н), 8,26 (м, 1Н), 7,81 (с, 0,6Н), 7,77 (с, 0,4Н), 7,72 (м, 2Н), 7,45-7,31 (м, 5Н), 6,78 (м, 2Н), 5,18 (с, 2Н), 5,12 (с, 1,2Н), 5,01 (с, 0,8Н), 4,55 (с, 0,8Н), 4,29-4,27 (м, 2,4Н), 4,09 (м, 2Н), 3,88 (м, 0,8Н), 3,26 (м, 2Н), 2,95 (м, 2Н), 1,61 (м, 2Н), 1,36 (с, 9Н); MS/ESI (m+1)=779,2 (экспериментальный), М.в.=778,9 (C34H38N10O8S2).
Исходя из производных 2,6-диаминопурина 26~30, модифицированные адениновые мономеры ПНК получали аналогично методикам, описанным в примерах 32~35. Спектральные и физические данные для соединений 36~40 приводятся ниже в таблице.
Примеры 36~40. Аналитические данные для адениновых мономеров ПНК 36~40.

| Соединение | Исходное вещество | L2 | Спектральные и физические данные |
| 36 | 26 | -(СН2)2- | 1H ЯМР (400 МГц; DMSOd6): δ 10,17 (с, 1Н), 8,36 (м, 1Н), 8,26 (м, 1Н), 7,82 (с, 0,6Н), 7,78(с, 0,4Н), 7,72 (м, 2Н), 7,45-7,31 (м, 5Н), 6,79 (2Н), 5,18 (с, 2Н), 5,12 (с, 1,2Н), 5,01 (с, 0,8Н), 4,55 (с, 0,8Н), 4,29-4,25 (м, 2,4Н), 4,09 (м, 2Н), 3,87 (м, 0,8Н), 3,29 (м, 2Н), 3,11 (м, 2Н), 1,33(д, 9Н). MS/ESI (m+1)=765,2 (экспериментальный), М.в.=764,8 (C33H36N10O8S2). Белое губчатое вещество. |
| 37 | 27 | -(СН2)4- | 1Н ЯМР (400 МГц; DMSOd6): δ 10,10 (с, 1Н), 8,36 (м, 1Н), 8,26 (м, 1Н), 7,80 (с, 0,6Н), 7,76-7,71 (м, 2,4Н), 7,46-7,31 (м, 5Н), 6,81-6,73 (м, 2Н), 5,18 (с, 2Н), 5,12 (с, 1,2Н), 5,01 (с, 0,8Н), 4,55 (с, 0,8Н), 4,30-4,25 (м, 2,4Н), 4,09 (м, 2Н), 3,88 (м, 0,8Н), 3,26 (м, 2Н), 2,90 (м, 2Н), 1,50-1,36 (м, 13Н); MS/ESI (m+1)=793,3 (экспериментальный), М.в.=792,9 (C35H40N10O8S2). Желтовато-красное губчатое/твердое вещество. |
| 38 | 28 | -(СН2)5- | 1H ЯМР (400 МГц; DMSOd6): δ 10,09 (с, 1Н), 8,35 (м, 1Н), 8,26 (м, 1Н), 7,80 (с, 0,6Н), 7,76 (с, 0,4Н), 7,74-7,72 (м, 2,0Н), 7,46-7,31 (м, 5Н), 6,79-6,72 (м, 2Н), 5,18 (с, 2Н), 5,12 (с, 1,2Н), 5,01 (с, 0,8Н), 4,56 (с, 0,8Н), 4,30-4,27 (м, |
| Соединение | Исходное вещество | L2 | Спектральные и физические данные |
| 2,4Н), 4,09 (м, 2Н), 3,88 (м, 0,8Н), 3,25 (м, 2Н), 2,89 (м, 2Н), 1,49 (м, 2Н), 1,36 (м, 11Н), 1,25 (м, 2Н); MS/ESI (m+1)=807,3 (экспериментальный), М.в.=806,9 (C36H42N10O8S2). Желтое губчатое/твердое вещество. | |||
| 39 | 29 | -(СН2)7- | 1H ЯМР (500 МГц; DMSOd6): δ 10,11 (д, J=3,1 Гц, 1Н), 8,37-8,34 (м, 1Н), 8,28-8,24 (м, 1Н), 7,80 (с, 0,6Н), 7,76 (с, 0,4 Гц), 7,75-7,70 (м, 2Н), 7,75-7,31 (м, 5Н), 6,82-6,74 (м, 2Н),5,18 (с, 2Н), 5,12 (с, 1,2Н), 5,01 (с, 0,8Н), 4,58(с, 0,8Н), 4,29 (м, 1,2Н), 4,27 (кв, J=4,9 Гц, 1Н), 4,06-4,03 (м, 2Н), 3,88 (т, J=5,2 Гц, 1Н), 3,26-3,20 (м, 2Н), 2,88-2,85 (м, 2Н), 1,51-1,45(м, 2Н), 1,39-1,32 (м, 11Н), 1,28-1,15 (м, 6Н). MS/ESI (m+1)=834,8 (экспериментальный), М.в.=835,0 (C38H46N10O8S2). Красновато-желтое губчатое/твердое вещество. |
| 40 | 30 |
|
1H ЯМР (400 МГц; DMSOd6); δ 10,14 (с, 1Н), 8,35 (м, 1Н), 8,26 (м, 1Н), 7,82 (с, 0,6Н), 7,78 (с, 0,4Н), 7,73 (м, 2Н), 7,46-7,31 (м, 5Н), 6,81-6,74 (м, 2Н), 5,18 (с, 2Н), 5,13 (с, 1,2Н), 5,02 (с, 0,8Н), 4,55 (с, 0,8Н), 4,30-4,26 (м, 2,4Н), 4,09 (м, 2Н), 3,88 (м, 0,8Н), 3,50 (м, 2Н), 3,43-3,38 (м, 4Н), 3,07 (м, 2Н), 1,36 (с, 9Н); MS/ESI(m+1)=809,3 (экспериментальный), М.в.=808,9 (C35H40N10O9S2). Бледно-желтое губчатое вещество. |
Пример 41. Получение этилового эфира 2-[2-[2-{2-(трет-бутоксикарбониламино)-2-метил}этил]амино-6-оксо-6,9-дигидро-1Н-пурин-2-ил]уксусной кислоты (41).

К перемешиваемому раствору 4,69 г соединения 31 в 47 мл DMF прибавляли 790 мг 55% NaH в минеральном масле и реакционный раствор перемешивали в течение 2 часов. После медленного прибавления 1,85 мл этилбромацетата, реакционный раствор перемешивали еще в течение 2 часов. Реакционную смесь концентрировали в вакууме и очищали колоночной хроматографией (5:95 МеОН/МС) с получением 5,04 г соединения 41 в виде бледно-желтого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 10,55 (с, 1Н), 7,67 (с, 1Н), 6,74 (д, 1Н), 6,40 (м, 1Н), 4,87 (с, 2Н), 4,17 (кв, 2Н), 3,65 (м, 1Н), 3,28 (м, 1Н), 3,16 (м, 1Н), 1,36 (с, 9Н), 1,21 (т, 3Н), 1,01 (д, 3Н).
Пример 42. Получение 2-{2-(трет-бутоксикарбониламино)этокси}этиламина (42).
К 146 г [2-{2-(трет-бутоксикарбониламино)этокси}этил]метансульфоната, растворенного в 500 мл DMF, прибавляли 134 г азида натрия. Реакционную смесь перемешивали при 70°С в течение 20 часов и затем концентрировали при пониженном давлении. Полученный остаток растворяли в 1200 мл воды и экстрагировали ЕА. Органический слой сушили над безводным сульфатом натрия и концентрировали в вакууме. Полученный в результате остаток растворяли в 2000 мл THF, к которому прибавляли 162 г трифенилфосфина. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, после чего прибавляли 200 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение 18 часов и концентрировали до 500 мл при пониженном давлении. Затем полученный в результате осадок отфильтровывали. Фильтрат далее концентрировали при пониженном давлении для удаления THF и промывали МС. Водный слой концентрировали с получением 86,2 г соединения 42 в виде жидкости. 1H ЯМР (400 МГц; CDCl3): δ 4,96 (уш.с, 1Н), 3,54-3,48 (м, 4Н), 3,34 (кв, 2Н), 2,88 (т, 2Н), 1,48-1,46 (м, 11Н).
Пример 43. Получение 2-[2-{2-(трет-бутоксикарбониламино)-этокси}этил]амино-1Н-пурин-6(9Н)-она (43).

6,3 г соединения 42 и 2,0 г 2-бромгипоксантина диспергировали в 55 мл монометоксиэтанола и 17,5 мл воды. Реакционную смесь перемешивали при кипении в течение 16 часов и растворитель удаляли при пониженном давлении. Затем концентрат перемешивали в 20 мл МС и 10 мл воды в течение 30 минут и полученный в результате осадок собирали фильтрацией с получением 2,1 г соединения 43 в виде бледно-желтого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 12,43 (уш.с, 1Н), 10,45 (уш.с, 1Н), 7,89 (с, 0,2Н), 7,61 (с, 0,8Н), 6,77 (м, 1Н), 6,34 (с, 0,8Н), 6,12 (с, 0,2Н), 3,52 (т, 2Н), 3,41 (м, 4Н), 3,09 (кв, 2Н), 1.36 (с, 9Н).
Пример 44. Получение 2-[2-[3-(трет-бутоксикарбониламино)пропилокси}-этил]]-амино-1Н-пурин-6(9Н)-она (44).

2-{3-(трет-бутоксикарбониламино)пропилокси}этиламин и 2-бромгипо-ксантин реагировали аналогично методике, описанной в примере 43, с получением соединения 44 в виде белого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 12,43 (уш.с, 1Н), 10,45 (уш.с 1Н), 7,61 (м, 1Н), 6,80 (т, 1Н), 6,30 (с, 0,7Н), 6,08 (с, 0,3Н), 3,49 (т, 2Н), 3,41 (т, 4Н), 2,99 (кв, 2Н), 1,61 (м, 2Н), 1,37 (с, 9Н).
Пример 45. Получение 2-{3-(трет-бутоксикарбониламино)пропил}амино-1Н-пурин-6(9Н)-она (45).

Смесь из 10 г хлоргипоксантина и 19,6 мл 1,3-диаминопропана, диспергированную в 40 мл монометоксиэтанола, перемепгавали при 130°С в течение 10 часов. Затем растворитель удаляли при пониженном давлении и полученный в результате остаток растворяли в 150 мл THF и 150 мл воды, к которым медленно прибавляли 19,2 г Boc2O, растворенного в 100 мл THF. Смесь перемешивали при комнатной температуре в течение 6 часов. После прибавления ЕА полученный в результате осадок собирали фильтрацией с получением 6,31 г соединения 45 в виде темно-зеленого твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 11,13 (уш.с, 1Н), 7,64 (с, 1Н), 6,87 (с, 1Н), 6,31 (с, 1Н), 3,23 (кв, 2Н), 2,98 (м, 2Н), 1,62 (м, 2Н), 1,38 (с, 9Н).
Производные гуанина 46~47 получали с помощью соответствующего диамина аналогично методике, описанной в примере 45. Спектральные и физические данные для соединений 46~47 приводятся ниже в таблице.
Примеры 46~47. Аналитические данные для производных гуанина 46~47.

| Соединение | Примененный диамин | n | Спектральные и физические данные |
| 46 | Этилендиамин | 2 | 1H ЯМР (500 МГц; DMSOd6): δ 12,43 (уш.с, 1Н), 10,61 (уш.с, 1Н), 7,62 (с, 1Н), 6,93 (т, 1Н), 6,32 (с, 1Н), 3,29 (кв, 2Н), 3,10 (кв, 2Н), 1,37 (с, 9Н). Серое твердое вещество. |
| 47 | Пентилендиамин | 5 | 1H ЯМР (500 МГц; DMSOd6): δ 12,44 (с,1Н), 10,35 (с, 1Н), 7,60 (с, 1Н), 6,80 (м, |
| Соединение | Примененный диамин | n | Спектральные и физические данныеа |
| 1Н), 6,29 (м, 1Н), 3,21 (м, 2Н), 2,90 (м,2Н), 1,49 (м, 2Н), 1,39-1,35 (м, 11Н), 1,27-1,23 (м, 2Н). Бледно-коричневое твердое вещество. |
Соединения 43-46 превращались в соединения 48~51 с помощью методики аналогичной описанной в примере 32. Спектральные и физические данные для соединений 48~51 приводятся ниже в таблице.
Примеры 48~51. Аналитические данные для производных гуанина 48~51.

| Соединение | Исходное вещество | L3 | Спектральные и физические данные |
| 48 | 43 |
|
1Н ЯМР (500 МГц; DMSOd6): δ 10,67 (с, 1Н), 7,69 (с, 1Н), 6,78 (м, 1Н), 6,15 (т, 1Н), 4,87 (с,2Н), 4,15 (кв, 2Н), 3,51 (м, 2Н), 3,41 (м, 4Н), 3,10(м, 2Н), 1,37 (с, 9Н), 1,20 (т, 3Н). Белое губчатое/твердое вещество. |
| 49 | 44 |
|
1H ЯМР (500 МГц; DMSOd6): δ 10,57 (с, 1Н), 7,69 (с, 1Н), 6,79 (м, 1Н), 6,44 (м, 1Н), 4,87 (с, 2Н), 4,16 (кв, 2Н), 3,48 (т, 2Н), 3,40 (м, 4Н), 2,99 (кв, 2Н), 1,61 (м, 2Н), 1,37 (с, 9Н), 1,21 (т, 3Н). Желтое губчатое/твердое вещество. |
| 50 | 45 |
|
1H ЯМР (500 МГц; DMSOd6): δ 10,64 (с, 1Н), 7,68 (с, 1Н), 6,91 (т, 1Н), 6,47 (с, 1Н), 4,88 (с, 2Н), 4,16 (кв, 2Н), 3,28 (кв, 2Н), 3,08 (кв, 2Н), 1,36 (с, 9Н), 1,21 (т, 3Н). Темно-красное твердое вещество. |
| Соединение | Исходное вещество | Ls | Спектральные и физические данные |
| 51 | 46 |
|
1Р ЯМР (400 МГц; DMSOd6): δ 10,44 (уш. с, 1Н), 7,66 (с, 1Н), 6,77 (м, 1Н), 6,41 (м, 1Н), 4,86 (с, 2Н), 4,16 (кв, 2Н), 3,21 (кв, 2Н), 2,89 (кв, 2Н),1,48 (м, 2Н), 1,41-1,36 (м, 11Н), 1,28-1,19 (м, 5Н). Темно-серое твердое вещество. |
Исходя из производных гуанина 48, 49 и 51, модифицированные гуаниновые мономеры ПНК 52~54 получали аналогично методикам, описанным в примерах 34~35. Спектральные и физические данные для соединений 52~54 приводятся ниже в таблице.
Примеры 52~54. Аналитические данные для гуаниновых мономеров ПНК 52~54.

| Соединение | Исходное вещество | L3 | Спектральные и физические данные |
| 52 | 48 |
|
1H ЯМР (400 МГц; DMSOd6): δ 10,61 (м,1Н), 8,36 (м, 1Н), 8,25 (м, 1Н), 7,76-7,65(м, 3Н), 6,78 (т, 1Н), 6,54 (м, 1Н), 5,07 (с, 1,2Н), 4,96 (с, 0,8Н), 4,54 (с, 0,8Н), 4,30 (с,1,2Н), 4,25 (м, 1,2Н), 4,07 (м, 2Н), 3,88 (м, 0,8Н), 3,49 (м, 2,4Н), 3,40 (м, 3,6Н), 3,09 (м, 2Н), 1,36 (с, 9Н); MS/ESI (m+1)=676,1 (экспериментальный), MW=675,8 (C27H33N9O8S2). Темно-коричневое губчатое/твердое вещество. |
| Соединение | Исходное вещество | L3 | Спектральные и физические данные |
| 53 | 49 |
|
1H ЯМР (400 МГц; DMSOd6): 8 10,69 (с, 1Н), 8,36 (м, 1Н), 8,25 (м, 1Н), 7,73 (м, 2Н), 7,64-7,60 (м, 1Н), 6,80 (т, 1Н), 6,65 (уш.с, 1Н), 5,05 (с, 1,2Н), 4,94 (с, 0,8Н), 4,54 (с, 0,8Н), 4,29 (с, 1,2Н), 4,24 (м, 1,2Н), 4,07 (м, 2Н), 3,87 (м, 0,8Н), 3,46-3,39 (м, 6Н), 2,97 (м, 2Н), 1,60 (м, 2Н), 1,36 (с, 9Н); MS/ESI (m+1)=689,8 (экспериментальный), М.в.=689,8 (C28H35N9O8S2). Желтое губчатое/твердое вещество. |
| 54 | 51 |
|
1H ЯМР (400 МГц; DMSOd6): δ 10,42-10,40 (м, 1Н), 8,37-8,32 (м, 1Н), 8,28-8,25 (м, 1Н), 7,73-7,70 (м, 2Н), 7,58-7,54 (м,1Н), 6,76 (т, 1Н), 6,39-6,38 (м, 1Н), 5,03 (с, 1,2Н), 4,92 (с, 0,8Н), 4,54 (с, 0,8Н), 4,29 (с, 1,2Н), 4,25 (м, 1,2Н), 4,08-4,07 (м, 2Н), 3,87 (м, 0,8Н), 3,18 (м, 2Н), 2,89 (м, 2Н), 1,47 (м, 2Н), 1,40-1,30 (м, 11Н), 1,24 (м, 2Н). MS/ESI (m+23/MNa+)=696,2 (экспериментальный), MW=673,8 (C28H35N9O7S2). Красное губчатое/твердое вещество. |
Пример 55. Получение этилового эфира 2-[6-амино-2-{2-(трет-бутоксикарбонил-амино)этил}-амино-9Н-пурин-9-ил]уксусной кислоты (55).

Соединение 55 получали из соединения 26 с помощью методики, аналогичной описанной в примере 32. Бледно-желтое твердое вещество. 1H ЯМР (400 МГц, DMSOd6): δ 7,70 (с, 1Н), 6,84 (т, 1Н), 6,79 (с, 2Н), 6,30 (т, 1Н), 4,87 (с, 2Н), 4,16 (кв, 2Н), 3,25 (кв, 2Н), 3,08 (кв, 2Н), 1,37 (с, 9Н), 1,22 (т, 3Н).
Пример 56. Получение этилового эфира 2-[6-амино-2-[2-{2,3-бис(бензилокси-карбонил)гуанидино}этил]амино-9Н-пурин-9-ил]уксусной кислоты (56).

К 4,42 г соединения 55, растворенного в 22 мл МС, медленно прибавляли 22 мл TFA при 0°С и раствор перемешивали в течение 2,5 часов. Реакционный раствор концентрировали при пониженном давлении, к которому прибавляли 100 мл диэтилового эфира. Полученный в результате осадок собирали фильтрацией с получением 5,79 г бледно-коричневого твердого промежуточного продукта. 3,9 г промежуточного продукта растворяли в 39 мл МС, к которому медленно прибавляли 6,9 мл TEA при 0°С. Раствор перемешивали в течение 15 мин при комнатной температуре, к которому прибавляли 2,48 г 1,3-бис(бензилоксикарбонил)-2-(метилтио)псевдомочевины. Затем реакционную смесь перемешивали еще 24 часа и промывали 0,5 М водной HCl. Органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением 4,58 г соединения 56 в виде бледно-желтого твердого вещества. 1Н ЯМР (500 МГц; DMSOd6): δ 11,59 (с, 1Н), 8,56 (т, 1Н), 7,69 (с, 1Н), 7,39-7,29 (м, 10Н), 6,75 (с, 2Н), 6,53 (с, 1Н), 5,15 (с, 2Н), 5,02 (с, 2Н), 4,86 (с, 2Н), 4,13 (кв, 2Н), 3,50 (кв, 2Н), 3,37 (м, 2Н), 1,19 (т, 3Н).
Пример 57. Получение этилового эфира 2-[6-(бензилоксикарбониламино)-2-[2-{2,3-бис-(бензилоксикарбонил)гуанидино}этил]амино-9Н-пурин-9-ил]уксусной кислоты (57).

4,54 г соединения 56 и 8,22 г N-(бензилоксикарбонил)-N'-метил-имидазолий трифлата растворяли в 90 мл DMF и перемешивали в течение 29 часов при комнатной температуре. Растворитель удаляли при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией (1:3 гексан/ЕА) с получением 3,06 г соединения 57 в виде белого губчатого/твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 11,60 (с, 1Н), 10,25 (с, 1Н), 8,57 (т, 1Н), 7,95 (с, 1Н), 7,45-7,29 (м, 15Н), 7,14 (т, 1Н), 5,18 (с, 2Н), 5,14 (с, 2Н), 5,02 (с, 2Н), 4,95 (с, 2Н), 4,15 (кв, 2Н), 3,54 (кв, 2Н), 3,42 (кв, 2Н), 1,19 (т, 3Н).
Пример 58. Получение этилового эфира 2-[6-амино-2-{4-(трет-бутоксикарбонил-амино)бутил}-амино-9Н-пурин-9-ил]уксусной кислоты (58).

Соединение 58 получали из соединения 27 в виде красновато-желтого губчатого/твердого вещества по методике, аналогичной описанной в примере 32. 1H ЯМР (500 МГц; DMSOd6): δ 7,67 (с, 1Н), 6,79 (т, 1Н), 6,69 (с, 2Н), 6,30 (м, 1Н), 4,85 (с, 2Н), 4,15 (кв, 2Н), 3,22-3,17 (м, 2Н), 2,93-2,89 (м, 2Н), 1,45 (м, 2Н), 1,40-1,36 (м, 11Н), 1,21 (т, 3Н).
Пример 59. Получение этилового эфира 2-[6-(бензилоксикарбониламино)-2-[4-{2,3-бис-(бензилоксикарбонил)гуанидино}бутил]амино-9Н-пурин-9-ил]уксусной кислоты (59).

Соединение 59 получали из соединения 58 в виде бледно-желтого губчатого/твердого вещества по методикам, аналогичным описанным в примерах 56~57. 1Н ЯМР (500 МГц; DMSOd6): δ 11,49 (с, 1Н), 10,12 (с, 1Н), 8,28 (т, 1Н), 7,91 (с, 1Н), 7,45-7,31 (м, 5Н), 6,95 (т, 1Н), 5,17 (с, 2Н), 4,93 (с, 2Н), 4,16 (кв, 2Н), 3,28 (м, 4Н), 1,51 (м, 4Н), 1,46 (с, 9Н), 1,38 (с, 9Н), 1,21 (т, 3Н).
Пример 60. Получение этилового эфира 2-[6-амино-2-{5-(трет-бутоксикарбонил-амино)-пентил}амино-9Н-пурин-9-ил] уксусной кислоты (60).

Соединение 60 получали из соединения 28 в виде красновато-желтого губчатого/твердого вещества по методике, аналогичной описанной в примере 32. 1H ЯМР (500 МГц; DMSOd6): δ 7,67 (с, 1Н), 6,78 (т, 1Н), 6,69 (с, 2Н), 6,28 (м, 1Н), 4,85 (с, 2Н), 4,15 (кв, 2Н), 3,18 (кв, 2Н), 2,89 (кв, 2Н), 1,47 (м, 2Н), 1,40-1,34 (м, 11Н), 1,25 (м, 2Н), 1,21 (т, 3Н).
Пример 61. Получение этилового эфира 2-[6-{ди-(трет-бутоксикарбонил)}амино-2-[5-{(трет-бутоксикарбонил)амино}пентил]амино-9Н-пурин-9-ил]уксусной кислоты (61).

К 6,98 г соединения 60, растворенного в 100 мл THF, прибавляли 7,95 г Вос2О и 186 мг 4-(N,N-диметиламино)пурина, и раствор перемешивали в течение 10 минут. Затем раствор смешивали с 4,62 мл TEA, перемешивали в течение 30 минут, медленно нагревали до 50°С и затем перемешивали при температуре еще 24 часа. Реакционный раствор концентрировали в вакууме и полученный в результате остаток растворяли в 170 мл ЕА и промывали последовательно 0,5 М водной HCl и водой. Органический слой сушили над безводным сульфатом натрия, концентрировали и подвергали хроматографическому разделению (1:1 гексан/МС→МС) с получением соединения 61 в виде желтого губчатого/твердого вещества. 1Н ЯМР (500 МГц; DMSOd6): δ 8,05 (с, 1Н), 7,23 (т, 1Н), 6,77 (т, 1Н), 5,00 (с, 2Н), 4,19 (кв, 2Н), 3,25 (кв, 2Н), 2,91 (кв, 2Н), 1,53 (м, 2Н), 1,40-1,39 (м, 29Н), 1,28 (м, 2Н), 1,22 (т, 3Н).
Пример 62. Получение этилового эфира 2-[2-[2-{2,3-бис-(бензилоксикарбонил)-гуанидино}этил]амино-6-оксо-6,9-дигидро-1Н-пурин-2-ил]уксусной кислоты (62).

Соединение 50 превращали в соединение 62 в виде белого твердого вещества по методике, аналогичной описанной в примере 57. 1H ЯМР (500 МГц; DMSOd6): δ 11,59 (с, 1Н), 10,68 (с, 1Н), 8,50 (т, 1Н), 7,68 (с, 1Н), 7,42-7,29 (м, 10Н), 6,58 (м, 1Н), 5,13 (с, 2Н), 5,02 (с, 2Н), 4,86 (с, 2Н), 4,12 (кв, 2Н), 3,50 (м, 2Н), 3,46 (м, 2Н), 1,18 (т, 3Н).
Пример 63. Получение 2-[6-(бензилоксикарбониламино)-2-[2-{2,3-бис-(бензил-оксикарбонил)гуанидино)этил]амино-9Н-пурин-9-ил]уксусной кислоты (63).

К 2,57 г соединения 57, растворенного в 7,1 мл THF и 7,1 мл воды, прибавляли 340 мг LiOH при 0°С и раствор перемешивали при комнатной температуре в течение 40 минут. Реакционный раствор подкисляли до рН 5~6 1 н водной HCl при 0°С и полученное в результате твердое вещество собирали фильтрацией с получением 2,33 г соединения 63 в виде белого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 11,59 (с, 1Н), 10,21 (с, 1Н), 8,57 (т, 1Н), 7,93 (с, 1Н), 7,45-7,28 (м, 15Н), 7,12 (т, 1Н), 5,17 (с, 2Н), 5,13 (с, 2Н), 5,02 (с, 2Н), 4,83 (с, 2Н), 3,53 (кв, 2Н), 3,42 (кв, 2Н).
Пример 64. Получение трет-бутилового эфира N-[2-{(9Н-флуорен-9-ил)метокси-карбонил-амино}этил)]-N-[2-{6-(бензилоксикарбониламино)-2-[2-{2,3-бис-(бензилокси-карбонил)-гуанидино}этил]амино-9Н-пурин-9-ил}ацетил]глициновой кислоты (64).

К 1,6 г соединения 63, растворенного в 30 мл DMF, при 0°С прибавляли 660 мг EDCI и 910 мг Fmoc-Aeg-OtBu. Реакционный раствор перемешивали в течение 2 часов при комнатной температуре и затем концентрировали при пониженном давлении. Полученный в результате остаток растворяли в 50 мл МС и промывали 0,5 М водной HCl и органический слой сушили над безводным сульфатом натрия. Затем органический слой концентрировали и подвергали хроматографическому разделению (65:1 МС/МеОН) с получением 500 мг соединения 64 в виде белого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): 
11,59 (с, 0,4Н), 11,58 (с, 0,6Н), 10,21 (с, 1Н), 8,55 (м, 1Н), 7,47-7,28 (м, 20Н), 7,06 (уш.с, 1Н), 5,17-4,89 (м, 8Н), 4,34-4,28 (м, 2,8Н), 4,20 (м, 1Н), 3,95 (с, 1,2Н), 3,52 (м, 3,4Н), 3,43 (м, 2,2Н), 3,34 (м, 1,7Н), 3,12 (м, 0,7Н), 1,43 (с, 3Н), 1,34 (с, 6Н).
Пример 65. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-[2-{6-(бензилоксикарбониламино)-2-[2-{2,3-бис(бензилоксикарбонил)-гуанидино}этил]амино-9Н-пурин-9-ил}ацетил]глицина (65).

К 460 мг соединения 64, растворенного в 3,6 мл МС, медленно прибавляли 3,6 мл TFA при 0°С. Реакционный раствор перемешивали при комнатной температуре в течение 3,5 часов и затем прибавляли 50 мл диэтилового эфира. Полученный осадок собирали фильтрацией с получением 430 мг соединения 65 в виде белого твердого вещества. 1H ЯМР (400 МГц; DMSOd6): δ 11,57 (с, 1Н), 10,77 (уш.с, 1Н), 8,66 (с, 1Н), 8,54 (с, 1Н), 7,87 (м, 2Н), 7,63 (м, 2Н), 7,50-7,28 (м, 21Н), 5,26-4,96 (м, 8Н), 4,34-4,18 (м, 4Н), 4,03 (с, 1Н), 3,52-3,36 (м, 7Н), 3,13 (м, 1Н). MS/ESI (m+1)=1019,4 (экспериментальный), М.в.=1018,0 (C53H51N11O11).
Пример 66. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-[2-{6-(бензилоксикарбониламино)-2-[4-{2,3-бис(бензилоксикарбонил)-гуанидино}-бутил]амино-9Н-пурин-9-ил} ацетил]глицина (66).

Соединение 59 превращали в соединение 66 в виде белого губчатого/твердого вещества по методикам, аналогичным описанным в примерах 63~65. 1H ЯМР (500 МГц; DMSOd6): δ 12,84 (уш.с, 1Н), 11,50 (с, 1Н), 10,14-10,13 (м, 1Н), 8,28 (м, 1Н), 7,88 (м, 2Н), 7,80-7,77 (м, 1Н), 7,68-7,66 (м, 2Н), 7,49 (т, 1Н), 7,45-7,29 (м, 9Н), 6,90 (м, 1Н), 5,17 (с, 2Н), 5,07 (с, 1,2Н), 4,89 (с, 0,8Н), 4,35-4,18 (м, 3Н), 4,00 (с, 1Н), 3,52 (м, 1Н), 3,35-3,25 (м, 6Н), 3,12 (м, 1Н), 1,49 (м, 4Н), 1,44 (д, 9Н), 1,37 (д, 9Н). MS/ESI (m+1)=978,4 (экспериментальный), М.в.=978,1 (C49H59N11O11).
Пример 67. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-[2-[6-[2-{2,3-бис(бензилоксикарбонил)гуанидино}этокси]метил-2-оксо-2Н-пирроло[2,3-d]пиримидин-3(7Н)-ил]ацетил]глипина (67).

Соединение 18 превращали в соединение 67 в виде бледно-желтого твердого вещества по методикам, аналогичным описанным в примерах 63~65. 1H ЯМР (500 МГц; DMSOd6): δ 11,99 (уш., с, 1Н), 11,57 (уш.с, 1Н), 8,56 (м, 1Н), 8,48-8,45 (м, 1Н), 7,89-7,87 (м, 2Н), 7,70-7,65 (м, 2Н), 7,49-7,26 (м, 15Н), 6,36-6,33 (м, 1Н), 5,20 (с, 2Н), 5,03-5,01 (м, 3,3Н), 4,83 (с, 0,7Н), 4,49-4,17 (м, 5,7Н), 4,01 (м, 1,3Н), 3,57-3,11 (м, 8Н); MS/ESI (m+1)=899,7 (экспериментальный), М.в.=898,9 (C47H46N8O11).
Пример 68. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-{2-[2-{2,3-бис-(бензилоксикарбонил)гуанидино}этил]амино-6-оксо-6,9-дигидро-1Н-пурин-2-ил]ацетил}глицина (68).

Соединение 62 превращали в соединение 68 в виде белого губчатого/твердого вещества по методикам, аналогичным описанным в примерах 63~65. 1Н ЯМР (500 МГц; DMSOd6): δ 11,58 (с, 1Н), 10,88 (с, 1Н), 8,51 (м, 1Н), 7,93 (м, 1Н), 7,87 (м, 2Н), 7,64 (м, 2Н), 7,47 (т, 1Н), 7,41-7,26 (м, 14Н), 6,66 (уш.с, 1Н), 5,16-4,89 (м, 8Н), 4,34-4,18 (м, 3,8Н), 4,00 (м, 1,2Н), 3,50-3,35 (м, 7Н), 3,13 (м, 1Н); MS/ESI (m+1)=885,3 (экспериментальный), М.в.=884,9 (C45H44N10O10).
Пример 69. Получение 2-[6-{2-(трет-бутоксикарбониламино)этокси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]уксусной кислоты (69).

Соединение 16 гидролизовали до соединения 69 в виде бледно-коричневого твердого вещества по методике, аналогичной описанной в примере 11. 1H ЯМР (500 МГц; DMSOd6): δ 13,03 (уш.с, 1Н), 11,31 (с, 1Н), 8,37 (с, 1Н), 6,85 (т, 1Н), 6,19 (с, 1Н), 4,63 (с, 2Н), 4,40 (с, 2Н), 3,42 (т, 2Н), 3,11 (кв, 2Н), 1,37 (с, 9Н).
Пример 70. Получение метилового эфира N-[2-{(9Н-флуорен-9-ил)метокси-карбонил-амино}этил)]-N-{2-[6-{2-(трет-бутоксикарбониламино)-этокси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]ацетил}глициновой кислоты (70).

3,6 г соединения 69, 3,6 г Fmoc-Aeg-OMe, 2,5 г EDCI, 1,73 г HOBt и 2,24 мл DIEA растворяли в 70 мл DMF и перемешивали при комнатной температуре в течение 1,5 часа. Реакционный растворитель удаляли при пониженном давлении и полученный в результате остаток растворяли в 100 мл МС и промывали последовательно 1 М водной HCl, дистиллированной водой и рассолом. Органический слой сушили над безводным сульфатом натрия, концентрировали в вакууме и очищали с помощью колоночной хроматографии (100:2 МС/МеОН) с получением 2,5 г соединения 70 в виде желтого губчатого/твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 11,30 (с, 1Н), 8,24 (с, 0,65Н), 8,21 (с, 0,35Н), 7,89-7,87 (м, 2Н), 7,71-7,67 (м, 2Н), 7,48-7,25 (м, 5Н), 6,87 (т, 1Н), 6,17 (с, 0,7Н), 6,15 (с, 0,3Н), 4,93 (с, 1,3Н), 4,74 (с, 0,7Н), 4,40-4,39 (м, 2,7Н), 4,35-4,21 (м, 3Н), 4,08 (с, 1,3Н), 3,73 (с, 0,8Н), 3,62 (с, 2,2Н), 3,51 (т, 1,4Н), 3,43-3,30 (м, 3,6Н), 3,13-3,10 (м, 3Н), 1,37 (с, 9Н).
Пример 71. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-{2-[6-{2-(трет-бутоксикарбониламино)этокси}метил-2-оксо-2Н-пирроло-[2,3-d]пиримидин-3(7Н)-ил]ацетил}глицина (71).

К 5,0 г соединения 70, растворенного в 75 мл 1:1:1 ацетонитрила/ацетона/воды, медленно при 0°С прибавляли 28,5 мл 2,5 н водной LiOH. Реакционный раствор перемешивали в течение 10 минут и нейтрализовали 20%-ной водной лимонной кислотой. После доведения рН раствора до 8 насыщенным водным раствором бикарбоната натрия, к раствору прибавляли 516 мг Fmoc-OSu и раствор перемешивали в течение 2 часов при комнатной температуре. Затем раствор подкисляли до рН 3 с помощью 20%-ной водной лимонной кислоты и перемешивали в течение 90 минут при 0°С. Полученный в результате осадок собирали фильтрацией с получением 4,0 г соединения 71 в виде желтовато-зеленого твердого вещества. 1H ЯМР (500 МГц; DMSOd6): δ 12,02 (уш.с, 1Н), 8,51-8,49 (м, 1Н), 7,89-7,88 (д, 2Н), 7,70-7,50 (м, 2Н), 7,49 (т, 1Н), 7,42-7,28 (м, 4Н), 6,87 (т, 1Н), 6,36 (с, 0,7Н), 6,33 (с, 0,3Н), 5,02 (с, 1,2Н), 4,84 (0,8Н), 4,43-4,42 (м, 2,4Н), 4,34-4,19 (м, 3,2Н), 4,01 (с, 1,4Н), 3,48 (т, 1,2Н), 3,44-3,41 (м, 2,1Н), 3,37-3,29 (м, 2Н), 3,12-3,10 (м, 2,7Н), 1,37 (с, 9Н); MS/ESI (m+1)=689,3 (экспериментальный), М.в.=688,7 (C35H40N6O9).
Пример 72. Получение N-[2-{(9Н-флуорен-9-ил)метоксикарбониламино}-этил)]-N-{2-[5-{(трет-бутоксикарбонил)амино}пентил]амино-6-оксо-6,9-дигидро-1Н-пурин-2-ил]ацетил}глицина (72).

Соединение 51 превращали в соединение 72 в виде белого губчатого/твердого вещества по методикам, аналогичным описанным в примерах 69~71. 1H ЯМР (500 МГц; DMSOd6): δ 13,01 (уш.с, 1Н), 10,52-10,46 (м, 1Н), 7,88 (д, 2Н), 7,65 (д, 2Н), 7,54 (с, 0,5Н), 7,50 (с, 0,5Н), 7,48 (м, 1Н), 7,40 (т, 2Н), 7,31 (м, 2Н), 6,81 (т, 0,5Н), 6,72 (т, 0,5Н), 6,52-6,48 (м, 1Н), 4,98 (с, 1Н), 4,77 (с, 1Н), 4,33 (д, 1Н), 4,23-4,21 (м, 2Н), 4,05 (м, 1Н), 3,96 (с, 1Н), 3,50 (м, 1Н), 3,35 (м, 2Н), 3,21 (м, 2Н), 3,14 (кв, 1Н), 2,88 (м, 2Н), 1,46 (кв, 2Н), 1,39-1,35 (м, 11Н), 1,23 (м, 2Н); MS/ESI (m+1)=717,4 (экспериментальный), М.в.=716,8 (C36H44N8O8)
Пример 73. Получение N-[2-{(9H-флуорен-9-ил)метоксикарбониламино}-этил)]-N-[2-[6-{бис(трет-бутоксикарбонил)амино}-2-{5-(трет-бутоксикарбониламино)-пентил}амино-9Н-пурин-9-ил]ацетил]глицина (73).

Соединение 61 превращали в соединение 73 в виде белого губчатого/твердого вещества по методикам, аналогичным описанным в примерах 69~71. 1H ЯМР (500 МГц; DMSOd6): δ 12,71 (уш.с, 1Н), 7,90-7,87 (м, 3Н), 7,67 (м, 2Н), 7,44-7,39 (м, 3Н), 7,31 (м, 2Н), 7,07 (м, 1Н), 6,69 (м, 1Н), 5,11 (с, 1,2Н), 4,93 (с, 0,8Н), 4,37-4,21 (м, 3,8Н), 4,01 (с, 1,2Н), 3,52 (м, 1Н), 3,36 (м, 2Н), 3,23 (м, 2Н), 3,13 (м, 1Н), 2,88 (м, 2Н), 1,49 (м, 2Н), 1,38-1,35 (м, 27Н), 1,27-1,25 (м, 4Н); MS/ESI (m+1)=916,5 (экспериментальный), М.в.=916,0 (C46H61N9O11).
Получение олигомеров ПНК: Мономеры ПНК - п, которые синтезировались по схеме 4, отправлялись в компанию Panagene, Inc (www.panagene.com, Daejon, South Korea) для приготовления олигомеров ПНК формулы I по методу, описанному в литературе или с помощью их незначигельного(ных) изменения(й). (Org. Lett. vol 9, 3291-3293, 2006). Олигомеры ПНК получали от Panagene, охарактеризованные методом MALDI-TOF и проанализированные высокоэффективной жидкостной хроматографией - ВЭЖХ с C18-обращенной фазой. Олигомеры ПНК, полученные от Panagene, применялись без дополнительной очистки.
Мономеры ПНК - с - схемы 5 применялись для синтеза олигомеров ПНК формулы I по способу, раскрытому в предшествующем уровне техники или путем его незначительного(ных) изменения(й). (USP 6,133,444). Эти олигомеры ПНК очищались ВЭЖХ с C18-обращенной фазой (водный ацетонитрил с 0,1% TFA) и охарактеризовывались методом MALDI-TOF. На Фиг.1 представлены хроматограммы ВЭЖХ до и после очистки Олиго 17 с помощью ВЭЖХ с обращенной фазой. На Фиг.2 представлен масс-спектр MALDI-TOF для очищенной серии Олиго 17. Фиг.1 и 2 представлены только для иллюстративных целей и не должны истолковываться как ограничивающие данное изобретение.
Олигомеры ПНК, синтезированные для данного изобретения, представлены в таблице 1 наряду с данными их молекулярного веса, полученными с помощью MALDI-TOF. Сокращения, использованные в таблице 1, А, Т, G и С относятся к немодифицированному нуклеиновому основанию аденина, тимина, гуанина и цитозина, соответственно. Модифицированные нуклеиновые основания C(mXn), C(mXng), A(mXn), A(m), A(mg) и G(m) таковы, как определено ниже в таблице 1 наряду с Lys, Fam, L(1) и L(2). Данные олигомеры ПНК представлены только для иллюстративных целей и не должны истолковываться как ограничивающие данное изобретение.
| Таблица 1. | |||
| Олигомеры ПНК, описанные в данном изобретении, и их масс-спектрометрические данныеа | |||
| Название | Последовательность (N→С) | М.в. | (m+1)б |
| Олиго 1 | Fam-L(1)L(1)-TGC(103)-TAC(103)-TAC(103)-TG-Lys-NH2 | 4079,0 | 4078,3 |
| Олиго 2 | Fam-L(1)L(1)-TGC-TAC-TAC-TG-Lys-NH2 | 3745,6 | 3745,5 |
| Олиго 3 | TGC(103)-TAC-TAC(103)-TG-Lys-NH2 | 3319,4 | 3318,5 |
| Название | Последовательность (N→С) | М.в. | (m+1)б |
| Олиго 4 | TGC-TAC(103)-TAC-TG-Lys-NH2 | 3208,3 | 3208,3 |
| Олиго 5 | TGC-TAC-TAC-TG-Lys-NH2 | 3097,2 | 3097,8 |
| Олиго 6 | Fam-L(1)L(1)-TC(103)T-CC(103)C-AGC(103)-GTG-C(103)GC-C(103)AT-Lys-NH2 | 6140,1 | 6141,8 |
| Олиго 7 | Fam-L(1)L(1)-TCT-CCC-AGC-GTG-CGC-CAT-Lys-NH2 | 5584,4 | 5583,1 |
| Олиго 8 | TGC(202)-TAC-TAC(202)-TG-Lys-NH2 | 3319,4 | 3318,9 |
| Олиго 9 | GC(202)A-C(202)AT-TTG-C(202)CT-NH2 | 3553,7 | 3552,7 |
| Олиго 10 | GC(102)A-C(102)AT-TTG-C(102)CT-NH2 | 3511,6 | 3511,1 |
| Олиго 11 | GCA-CAT-TTG-CCT-Lys-NH2 | 3348,3 | 3345,8 |
| Олиго 12 | CA(3)T-A(3)GT-A(3)TA-A(3)GT-NH2 | 3580,8 | 3580,9 |
| Олиго 13 | CA(4)T-A(4)GT-A(4)TA-A(4)GT-NH2 | 3636,9 | 3634,9 |
| Олиго 14 | CA(5)T-A(5)GT-A(5)TA-A(5)GT-NH2 | 3693,0 | 3691,5 |
| Олиго 15 | CA(7)T-A(7)GT-A(7)TA-A(7)GT-NH2 | 3805,0 | 3803,4 |
| Олиго 16 | CAT-AGT-ATA-AGT-Lys-NH2 | 3420,3 | 3418,3 |
| Олиго 17 | CA(5)T-A(5)GT-A(5)TA-A(5)GT-Lys-NH2 | 3820,9 | 3819,8 |
| Олиго 18 | CA(202)T-A(202)GT-A(202)TA-A(202)GT-NH2 | 3700,7 | 3701,4 |
| Олиго 19 | L(1)-TAG(203)-CTG(203)-CTG-ATT-Lys-NH2 | 3746,9 | 3748,9 |
| Олиго 20 | TG(5)G-C(102)AA-C(102)TG-A(5)T-Lys-NH2 | 3525,6 | 3523,8 |
| Олиго 21 | Fam-L(2)-TG(5)G-C(102)AA-C(102)TG-A(5)T-Lys-NH2 | 3997,0 | 3996,1 |
| Олиго 22 | Fam-L(2)-TT-C(102)AT-A(5)GT-A(5)TA-AG(5)T-Lys-NH2 | 4806,9 | 4806,7 |
| Олиго 23 | Fam-L(2)L(2)-TC(102)A-GA(5)A-C(102)TT-A(5)T-Lys-NH2 | 4084,2 | 4083,8 |
| Олиго 24 | Fam-L(2)-CA(5)T-A(4g)GT-A(4g)TA(5)-AGT-Lys-NH2 | 4348,5 | 4347,4 |
| Олиго 25 | TT-C(1O2g)AT-A(5)GT-A(5)TA-AG(5)T-Lys-NH2 | 4377,4 | 4375,6 |
| Олиго 26 | GC(1N3)A-C(1N3)AT-TTG-C(1N3)CT-NH2 | 3550,8 | 3550,9 |
| Олиго 27 | CAT-AGT-ATA-AGT-NH2 | 3292,3 | 3292,5 |
| Олиго 28 | Fam-L(2)-TGG-CAA-CTG-AT-Lys-NH2 | 3617,5 | 3616,3 |
| а. Использованные сокращения для мономеров определены ниже. | |||
| б. Экспериментальный ионный пик для МН+, если не указано иначе. |
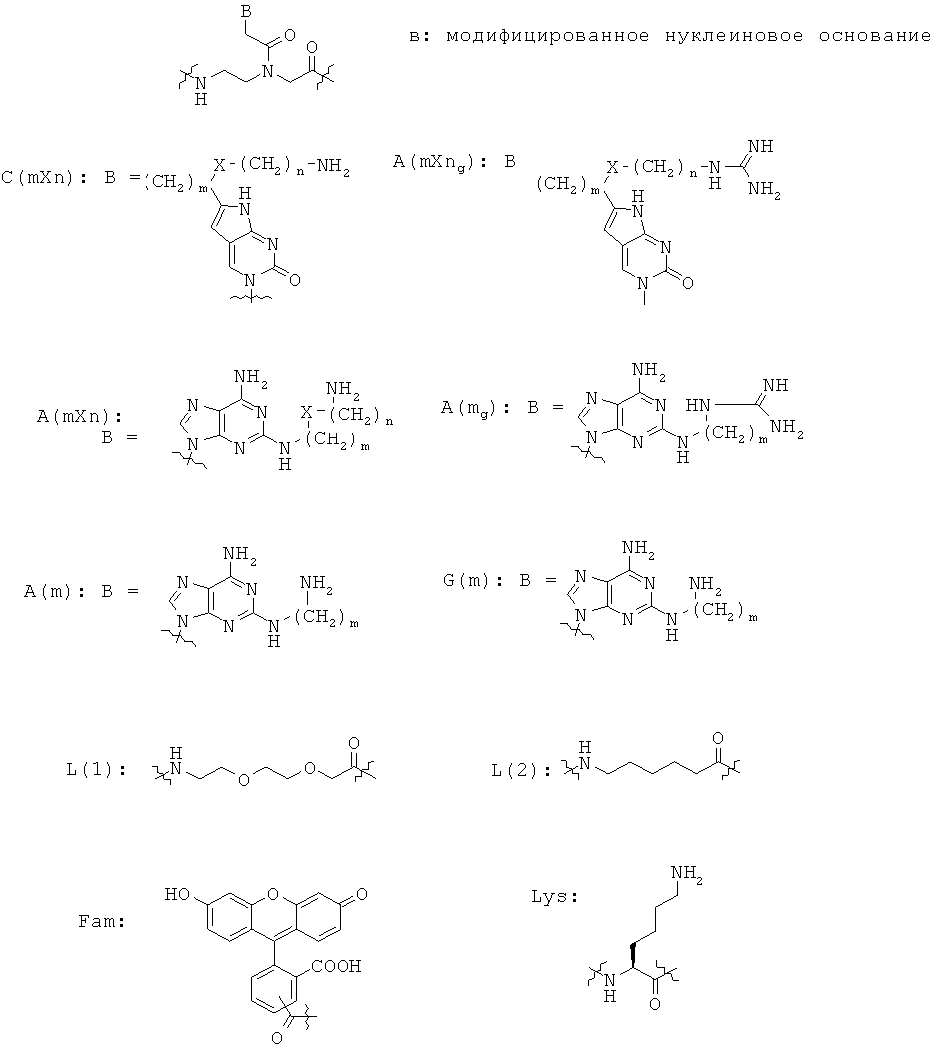
Связывающая аффинность к ДНК. Олигомеры ПНК, описанные в данном изобретении, оценивались в отношении их связывающей аффинности к ДНК путем измерения величины Т. пл. следующим образом.
4 мкМ олигомера ПНК и 4 мкМ ДНК смешивали в водном буферном растворе (рН 7,16, 10 мМ фосфат натрия, 100 мМ NaCl) и выдерживали при 90°С в течение нескольких минут и медленно охлаждали до комнатной температуры. Затем раствор переносили в 4 мл кварцевую кювету и кювету плотно закрывали. Кювету помещали в работающий в ультрафиолетовой/видимой области спектрофотометр Agilent 8453 и регистрировали изменения поглощения при 260 нм при повышении температуры кюветы со скоростью 0,5 или 1,0°С в минуту. Из кривой зависимости «поглощения от времени», температура, показывающая наибольшее увеличение скорости поглощения, считывалась как температура плавления - Т. пл. между ПНК и ДНК. ДНК(ы) для измерения Т.пл. закупались или у фирмы Bioneer, Inc. (www.bioneer.com, Daejon, South Korea) или у Ahram Biosystems (www.ahrambio.com, Seoul, South Korea), и применялись без дополнительной очистки.
На Фиг.3 представлены кривые изменения поглощения с температурой для Олиго 17 по сравнению с комплементарной или с нарушенной комплементарностью ДНК. Для последовательностей ДНК(т) с нарушенной комплементарностью против Олиго 17 см. таблицу 2. На Фиг.3 имеется температура перехода в каждой кривой, которая считывается как величина Т. пл. для кривой.
Величины Т. пл. для олигомеров ПНК, описанных в данном изобретении, представлены в таблице 2. Данные величины Т.пл. представлены только для иллюстративных целей и не должны истолковываться как ограничивающие данное изобретение.
| Таблица 2. | |||
| Величины Т. пл. между ПНК и комплементарной или с нарушенной комплементарностью ДНК. | |||
| Название | Последовательность ДНК (5'→3') | Т. пл, °C | Примечание |
| Олиго5 | CAG-TAG-TAG-CA | 55 | немодифицированный олигомер ПНК |
| Олиго3 | 65 | С(1O3)×2 | |
| Олиго4 | 61 | С(1O3)×1 | |
| Олиго 8 | 68 | С(2O2)×2 | |
| Олиго 10 | AGG-CAA-TTG-TGC | >85 | С(1O2)×3 |
| Олиго 11 | 59 | немодифицированный олигомер ПНК | |
| Олиго 12 | ACT-TAT-ACT-ATG | 60 | А(3)×4 |
| Олиго 13 | 64 | А(4)×4 | |
| Олиго 14 | 69 | А(5)×4 | |
| Олиго 15 | 71 | А(7)×4 | |
| Олиго 18 | 66 | А(2O2)×4 | |
| Олиго 27 | 55 | немодифицированный олигомер ПНК | |
| Олиго 16 | ACT-TAT-ACT-ATG | 56 | немодифицированный олигомер ПНК |
| Олиго 17 | ACT-TAT-ACT-ATG | 72 | комплементарная |
| ACT-TAC-ACT-ATG | 61 | Нарушение комплементарности (Т→С) | |
| ACT-TAA-ACT-ATG | 59 | Нарушение комплементарности (Т→А) | |
| ACT-TAG-ACT-ATG | 58 | Нарушение комплементарности (Т→G) | |
| Олиго 24 | ACT-TAT-ACT-ATG | 70 | А(5)×2 плюс A(4g)×2 |
| Олиго 20 | ATC-AGT-TGC-CA | 84 | комплементарная |
| ATC-ATT-TGC-CA | 62 | Нарушение комплементарности (G→Т) | |
| ATC-AAT-TGC-CA | 65 | Нарушение комплементарности (G→А) |
Замена цитозина производным пирролоцитозина в неприродном нуклеиновом основании, описанном в данном изобретении, значительно повышало аффинность олигомера ПНК к комплементарной ДНК. Например, Олиго 10 с тремя «модифицированными» цитозиновыми «С (1O2)» мономерами показал Т. пл., превышающую 85°С, в то время как соответствующий «немодифицированный» Олиго 11 показали Т. пл., равную 58°С. Другие мономеры с модифицированным цитозином, таким как «С (1O3)» или «С (2O2)» также значительно увеличивали аффинность олигомера ПНК к комплементарной ДНК, как представлено на примере Олиго 3 и Олиго 8.
«Модифицированные» адениновые нуклеиновые основания, описанные в данном изобретении, также значительно увеличивали аффинность олигомера ПНК к комплементарной ДНК. Например, Олиго 15 с четырьмя «модифицированными» адениновыми А(7) мономерами показал Т. пл., равную 71°С, которая существенно выше, чем Т. пл., равная 55°С, наблюдаемая для «немодифицированного» Олиго 27. Другие «модифицированные» адениновые мономеры, как например А(4) и А(5) также заметно увеличивали аффинность к комплементарной ДНК.
Найдено, что «модифицированные» мономеры ПНК, описанные в данном изобретении, весьма чувствительны к ошибочному спариванию оснований. Например, уменьшения Т. пл. на 11~14°С наблюдались с ошибочными спариваниями пар оснований для А(5) мономера в Одиго 17. Ошибочные спаривания пар оснований для С(1O2) мономера в Олиго 20 приводило к уменьшениям Т. пл. на 19~22°С.
Клеточная пенетрация. Для того, чтобы оценить способность к клеточной пенетрации олигомеров ПНК, описанной в данном изобретении, раковые клетки человеческого происхождения обрабатывались олигомерами ПНК, ковалентно связанной с флуоресцеином. Примененный метод представлен в краткой форме, как изложено ниже.
На каждое покровное стекло (стерилизованное в автоклаве), помещенное в каждую лунку 24-луночного планшета, засевали 20000~100000 клеток в зависимости от скорости роста примененной линии клеток, и клетки культивировали при 37°С и 5% СО2 в течение 16-24 часов. Затем среду заменяли 500 мкл свежеприготовленной среды Opti-MEM (с или без 1% FBS), к которой прибавляли аликвоту водного исходного раствора олигомера ПНК ковалентно меченого (связанного с) флуоресцеином. После того, как клетки культивировались в течение предварительно обозначенного интервала, их промывали с помощью PBS (phosphate-buffered saline-забуференный фосфатом физиологический раствор) и фиксировали путем инкубирования в 3,7% или 4% параформальдегиде. Клетки тщательно промывали несколько раз PBS или PBS, содержащим 0,1% твина-20. Затем покровное стекло устанавливали на предметное стекло с помощью капли раствора для закрепления и герметизировали лаком для ногтей для конфокальной флуоресцентной микроскопии. Флуоресцентные изображения получали или на конфокальном микроскопе Zeiss LSM 510 (Германия) при 63-кратном увеличении или на конфокальном микроскопе Nikon C1Si при 40-кратном увеличении.
Изображения клеточной пенетрации на фиг.4~8 представлены только для иллюстрации и не должны истолковываться как ограничивающие данное изобретение.
На фиг.4(а) и 4(б) представлены изображения конфокальной микроскопии (при 63-кратном увеличении) для 1, 2, 3 и 24 часов после того, как клетки HeLa обрабатывались Олиго 1 и Олиго 2 при концентрации 5 мкМ, соответственно (без FBS). В то время как интенсивность флуоресценции не вызывает сомнения и становится интенсивной при 24 часах на фиг.4(а), интенсивность флуоресценции на фиг.4(б) является слабой, указывая, что Олиго 1 пенетрирует в клетки HeLa значительно быстрее, чем «немодифицированный» Олиго 2.
На фиг.5(а) и 5(б) представлены изображения конфокальной микроскопии (при 63-кратном увеличении) через 0,5 и 1 час после того, как MCF-7 клетки были обработаны Олиго 6 и Олиго 7 при концентрации 2,5 мкМ, соответственно (без FBS). В то время как интенсивность флуоресценции не вызывает сомнения и становится интенсивной при 1 часе на фиг.5(а), интенсивность флуоресценции на фиг.5(б) является слабой, указывая, что Олиго 6 пенетрирует в MCF-7 клетки значительно быстрее, чем «немодифицированный» Олиго 7.
На фиг.6(а) и 6(б) представлены фотографии конфокальной микроскопии (при 40-кратном увеличении) через 6 или 24 часа после того, как HeLa клетки были обработаны Олиго 1 и Олиго 6 при концентрации 1 мкМ, соответственно (с 1% FBS). В то время как интенсивность флуоресценции является слабой даже при 24 часах на фиг.6(а), интенсивность флуоресценции на фиг.6(б) не вызывает сомнения и становится интенсивной при 24 часах, означая, что Олиго 6 пенетрирует в HeLa клетки значительно быстрее, чем Олиго 1.
На фиг.7(а) и 7(б) представлены фотографии конфокальной микроскопии (при 40-кратном увеличении) через 24 часа после того, как JAR клетки были обработаны Олиго 21 и Олиго 28 при концентрации 2 мкМ, соответственно (без FBS). В то время как интенсивность флуоресценции является сильной на фиг.7(а), на фиг.7(б) интенсивность флуоресценции незначительна, означая, что Олиго 21 пенетрирует в JAR клетки значительно быстрее, чем «немодифицированный» Олиго 28.
На фиг.7(в) и 7(г) представлены фотографии конфокальной микроскопии (при 40-кратном увеличении) через 24 часа после того, как А549 клетки были обработаны Олиго 21 и Олиго 28 при концентрации 2 мкМ, соответственно (без FBS). В то время как интенсивность флуоресценции является сильной на фиг.7(в), на фиг.7(г) интенсивность флуоресценции незначительна, означая, что Олиго 21 пенетрирует в А549 клетки значительно быстрее, чем «немодифицированный» Олиго 28.
На фиг.7(д) и 7(е) представлены фотографии конфокальной микроскопии (при 40-кратном увеличении) через 12 часов после того, как HeLa клетки были обработаны Олиго 21 и Олиго 28 при концентрации 2 мкМ, соответственно (без FBS). В то время как интенсивность флуоресценции на фиг.7(д) является выраженной, на фиг.7(е) интенсивность флуоресценции незначительна, означая, что Олиго 21 пенетрирует в HeLa клетки значительно быстрее, чем «немодифицированный» Олиго 28.
На фиг.7(ж) представлены фотографии конфокальной микроскопии (при 40-кратном увеличении) через 24 часа после того, как HeLa клетки были обработаны Олиго 21 при концентрации 2 мкМ (без FBS). Учитывая, что клеточная флуоресценция на фиг.7 (ж) значительно сильнее, чем на фиг.7(е), Олиго 21 по-видимому пенетрирует через 24 часа вместо 12 часов.
На фиг.8(а), 8(б) и 8(в) представлены изображения конфокальной микроскопии (при 40-кратном увеличении) через 24 часа после того, как HeLa А549 и JAR клетки были обработаны 2 мкМ Олиго 22. соответственно (без FBS). Все изображения связанны с флуоресценцией внутри клетки, указывая, что Олиго 22 обладает хорошей клеточной пенетрацией в тестируемые клетки.
Антисмысловые примеры. Олиго 9 и Олиго 12 обладают теми же последовательностями оснований, как Т1-12 и Т5-12, соответственно, о которых в литературе сообщалось, что они ингибируют рибосомный синтез mdm2. (Nucleic Acids Res. vol.32, 4893-4902, 2004). Олиго 9 и Олиго 12 оценивались в отношении их способности ингибировать рибосомный синтез mdm2 в JAR клетках следующим образом. Такой антисмысловый пример приводится только для иллюстрации и не должен истолковываться как ограничивающий данное изобретение.
JAR клетки (АТСС каталог # НТВ-144) выращивали в RPMI-1640 среде, дополненной 10% FBS и 1% пенициллина-стрептомицина при 37°С и 5% СО2. Клетки затем высевали в каждую лунку 12-луночного планшета, содержащего 1 мл указанной среды, и обрабатывали аликвотой водного исходного раствора Олиго 9 или Олиго 12 при обозначенной выше концентрации. Затем клетки инкубировали при 37°С и 5% СО2 в течение 15 часов.
Клетки в каждой лунке промывали холодным PBS и обрабатывали 80 мкл буфера RIPA, содержащего 1% коктейль ингибиторов протеаз, и планшет инкубировали при 4°С и медленно встряхивали в течение 15 минут. Содержимое каждой лунки отправляли в микропробирку. Микропробирку выдерживали на льду в течение 10 минут и центрифугировали при 10000 g. Полученный в результате супернатант собирали и подвергали количественному определению белка по анализу Брэдфорда (Bradford) и вестерн-блоттингу. Для электрофореза 20 мкг белка помещали на каждой дорожке геля в аппарате MiniGel, отделяли и переносили на мембраны PVDF (0,45 мкн, Millipore). Первое mdm2 антитело, примененное для вестерн-блоттинга, представляло собой SC-965 (Santa Cruz Biotechnology).
На фиг.9 представлены результаты вестерн-блоттинга для клеток JAR, обработанных Олиго 9 при 5 мкМ или 10 мкМ, Олиго 10 при 5 мкМ или 10 мкМ, совместной обработке олигомерами при концентрации 5 мкМ или 10 мкМ каждый, и контрольный опыт (без обработки олигомером). На фиг.9 обработка Олиго 9 или Олиго 10. или совместная обработка с Олиго 9 и Олиго 10 существенно ингибировала рибосомный синтез mdm2 в клетках JAR как при 5 мкМ, так и при 10 мкМ.
Claims (16)
1. Производное пептидо-нуклеиновой кислоты формулы I или ее фармацевтически приемлемой соли:

где:
n - целое число, равное или большее 5;
S1, S2, …, Sn-1, Sn, T1, T2,…, Tn-1 и Tn независимо обозначают атом водорода, атом дейтерия, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
X и Y независимо обозначают атом водорода, атом дейтерия, замещенную или незамещенную алкилгруппу, замещенную или незамещенную ацилгруппу, замещенную или незамещенную сульфонилгруппу или замещенную или незамещенную арилгруппу;
Z обозначает гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и
по меньшей мере одна из B1, В2, …, Bn-1 и Bn независимо выбрана из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV:

где:
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода; и
L1, L2 и L3 представляют собой ковалентный линкер, отвечающий формуле V, соединяющий основную аминогруппу в остаток, ответственный за свойства спаривания нуклеиновых оснований:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу (-СН2-) и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода (-O-), атома серы (-S-) и замещенной или незамещенной аминогруппы [-N(H)-группы или -N(заместитель)-группы]; и
m - целое число от 2 до 15.
где:
n - целое число, равное или большее 5;
S1, S2, …, Sn-1, Sn, T1, T2,…, Tn-1 и Tn независимо обозначают атом водорода, атом дейтерия, замещенную или незамещенную алкилгруппу или замещенную или незамещенную арилгруппу;
X и Y независимо обозначают атом водорода, атом дейтерия, замещенную или незамещенную алкилгруппу, замещенную или незамещенную ацилгруппу, замещенную или незамещенную сульфонилгруппу или замещенную или незамещенную арилгруппу;
Z обозначает гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и
по меньшей мере одна из B1, В2, …, Bn-1 и Bn независимо выбрана из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV:
где:
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода; и
L1, L2 и L3 представляют собой ковалентный линкер, отвечающий формуле V, соединяющий основную аминогруппу в остаток, ответственный за свойства спаривания нуклеиновых оснований:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу (-СН2-) и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода (-O-), атома серы (-S-) и замещенной или незамещенной аминогруппы [-N(H)-группы или -N(заместитель)-группы]; и
m - целое число от 2 до 15.
2. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 5 до 30;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода, замещенной или незамещенной алкилгруппы, замещенной или незамещенной ацилгруппы, замещенной или незамещенной сульфонилгруппы;
Z обозначает гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и
по меньшей мере одна из B1, В2, …, Bn-1 и Bn независимо выбрана из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV.
где:
n - целое число от 5 до 30;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода, замещенной или незамещенной алкилгруппы, замещенной или незамещенной ацилгруппы, замещенной или незамещенной сульфонилгруппы;
Z обозначает гидроксилгруппу, замещенную или незамещенную алкилоксигруппу, замещенную или незамещенную арилоксигруппу, замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований; и
по меньшей мере одна из B1, В2, …, Bn-1 и Bn независимо выбрана из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV.
3. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 8 до 25;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода, замещенной или незамещенной алкилгруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере две из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода;
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 12.
где:
n - целое число от 8 до 25;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода, замещенной или незамещенной алкилгруппы и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере две из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода;
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 12.
4. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 10 до 25;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу; и
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 10.
где:
n - целое число от 10 до 25;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу; и
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R2, R3, R4, R5 и R6 независимо выбраны из замещенной или незамещенной алкилгруппы и атома водорода;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 10.
5. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 10 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или замещенную или незамещенную амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 10.
где:
n - целое число от 10 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из природных нуклеиновых оснований, включая аденин, тимин, гуанин, цитозин и урацил, и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или замещенную или незамещенную амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы, атома кислорода и аминогруппы; и
m - целое число от 2 до 10.
6. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 10 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина и урацила и ненатуральных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы и атома кислорода; и
m - целое число от 2 до 8.
где:
n - целое число от 10 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X и Y независимо выбраны из атома водорода и замещенной или незамещенной ацилгруппы;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина и урацила и ненатуральных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или амидинилгруппу;
Q1 и Qm представляют собой метиленгруппу и Qm непосредственно связана с основной аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из метиленгруппы и атома кислорода; и
m - целое число от 2 до 8.
7. Производное пептидо-нуклеиновой кислоты по п. 1 или ее фармацевтически приемлемой соли,
где:
n - целое число от 8 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X представляет собой атом водорода;
Y обозначает атом водорода или замещенную или незамещенную ацилгруппу;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или амидинилгруппу;
L1 обозначает -(СН2)2-O-(СН2)2-группу, -СН2-O-(СН2)2-группу или -СН2-O-(СН2)3-группу с правой частью, непосредственно связанной с основной аминогруппой; и
L2 и L3 независимо выбраны из -(СН2)2-O-(СН2)2-группы, -(СН2)3-O-(СН2)2-группы, -(СН2)2-O-(СН2)3-группы, -(СН2)2-группы, -(СН2)3-группы, -(СН2)4-группы, -(СН2)5-группы, -(СН2)6-группы, -(СН2)7-группы и -(СН2)8-группы с правой частью, непосредственно связанной с основной аминогруппой.
где:
n - целое число от 8 до 20;
S1, S2, …, Sn-1, Sn, T1, T2, …, Tn-1 и Tn представляют собой атом водорода;
X представляет собой атом водорода;
Y обозначает атом водорода или замещенную или незамещенную ацилгруппу;
Z обозначает гидроксилгруппу или замещенную или незамещенную аминогруппу;
B1, В2, …, Bn-1 и Bn независимо выбраны из аденина, тимина, гуанина, цитозина и неприродных нуклеиновых оснований;
по меньшей мере три из B1, В2, …, Bn-1 и Bn независимо выбраны из неприродных нуклеиновых оснований, представленных формулой II, формулой III или формулой IV;
R1, R3 и R5 представляют собой атом водорода и R2, R4 и R6 независимо обозначают атом водорода или амидинилгруппу;
L1 обозначает -(СН2)2-O-(СН2)2-группу, -СН2-O-(СН2)2-группу или -СН2-O-(СН2)3-группу с правой частью, непосредственно связанной с основной аминогруппой; и
L2 и L3 независимо выбраны из -(СН2)2-O-(СН2)2-группы, -(СН2)3-O-(СН2)2-группы, -(СН2)2-O-(СН2)3-группы, -(СН2)2-группы, -(СН2)3-группы, -(СН2)4-группы, -(СН2)5-группы, -(СН2)6-группы, -(СН2)7-группы и -(СН2)8-группы с правой частью, непосредственно связанной с основной аминогруппой.
8. Фармацевтическая композиция для обеспечения клеточной пенетрации, содержащая терапевтически эффективное количество производного пептидо-нуклеиновой кислоты по любому из пп. 1-7 или ее фармацевтически приемлемой соли для терапевтической цели.
9. Способ применения производного пептидо-нуклеиновой кислоты по любому из пп. 1-7 или ее фармацевтически приемлемой соли для диагностической цели.
10. Способ применения производного пептидо-нуклеиновой кислоты по любому из пп. 1-7 или ее фармацевтически приемлемой соли для модуляции экспрессии клеточного белка in vitro.
11. Соединение формулы VI:

где:
R7 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L1 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R7 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L1 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
12. Соединение формулы VII:

где:
R8 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы;
Р3 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы;
Р4 выбрана из атома водорода и трет-бутоксикарбонилгруппы; и
L2 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R8 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы;
Р3 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы;
Р4 выбрана из атома водорода и трет-бутоксикарбонилгруппы; и
L2 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
13. Соединение формулы VIII:

где:
R9 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L3 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R9 представляет собой атом водорода, N-сукцинилгруппу или замещенную или незамещенную алкилгруппу;
P1 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы и замещенной или незамещенной арилсульфонилгруппы;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L3 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
14. Соединение формулы IX:

где:
R10 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L1 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R10 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L1 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
15. Соединение формулы X:

где:
R11 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы;
Р3 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы;
Р4 выбрана из атома водорода и трет-бутоксикарбонилгруппы; и
L2 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R11 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы;
Р3 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы;
Р4 выбрана из атома водорода и трет-бутоксикарбонилгруппы; и
L2 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбраны из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
16. Соединение формулы XI:

где:
R12 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L3 представляет собой линкер, отвечающий формуле V:

где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
где:
R12 представляет собой гидроксилгруппу, замещенную или незамещенную алкилоксигруппу или замещенную или незамещенную аминогруппу;
Р2 выбрана из атома водорода, трет-бутоксикарбонилгруппы, (9Н-флуорен-9-ил)метокси-карбонилгруппы, замещенной или незамещенной бензилоксикарбонилгруппы, замещенной алкилоксикарбонилгруппы, замещенной или незамещенной алкилгруппы, амидинилгруппы, 1,3-бис(трет-бутокси-карбонил)амидинилгруппы, 1,3-бис-(бензилоксикарбонил)амидинилгруппы; и
L3 представляет собой линкер, отвечающий формуле V:
где:
Q1 и Qm представляют собой замещенную или незамещенную метиленгруппу и Qm непосредственно связана с аминогруппой;
Q2, Q3, … и Qm-1 независимо выбрана из замещенной или незамещенной метиленгруппы, атома кислорода, атома серы и замещенной или незамещенной аминогруппы; и
m - целое число от 2 до 15.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR20080023658 | 2008-03-14 | ||
| KR10-2008-23658 | 2008-03-14 | ||
| KR20080111459 | 2008-11-11 | ||
| KR10-2008-111459 | 2008-11-11 | ||
| PCT/KR2009/001256 WO2009113828A2 (en) | 2008-03-14 | 2009-03-13 | Peptide nucleic acid derivatives with good cell penetration and strong affinity for nucleic acid |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2010135635A RU2010135635A (ru) | 2012-03-10 |
| RU2564032C2 true RU2564032C2 (ru) | 2015-09-27 |
Family
ID=41065678
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2010135635/04A RU2564032C2 (ru) | 2008-03-14 | 2009-03-13 | Производные пептидо-нуклеиновых кислот с хорошей клеточной пенетрацией и сильной аффинностью к нуклеиновой кислоте |
Country Status (19)
| Country | Link |
|---|---|
| US (4) | US8680253B2 (ru) |
| EP (1) | EP2268607B9 (ru) |
| JP (1) | JP5620282B2 (ru) |
| KR (2) | KR20090098710A (ru) |
| CN (1) | CN102015628B (ru) |
| AU (1) | AU2009224149B2 (ru) |
| BR (1) | BRPI0906104B8 (ru) |
| CA (1) | CA2715844C (ru) |
| DK (1) | DK2268607T5 (ru) |
| ES (1) | ES2533769T3 (ru) |
| HK (1) | HK1155717A1 (ru) |
| IL (4) | IL207617A (ru) |
| MX (1) | MX2010009687A (ru) |
| NZ (1) | NZ587448A (ru) |
| PL (1) | PL2268607T3 (ru) |
| PT (1) | PT2268607E (ru) |
| RU (1) | RU2564032C2 (ru) |
| WO (1) | WO2009113828A2 (ru) |
| ZA (1) | ZA201005960B (ru) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2748834C2 (ru) * | 2016-09-16 | 2021-05-31 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды против scn9a |
| RU2753517C2 (ru) * | 2016-10-11 | 2021-08-17 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды к hif-1-альфа |
| RU2753966C2 (ru) * | 2016-08-08 | 2021-08-24 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды андрогенового рецептора |
| RU2766701C2 (ru) * | 2017-01-06 | 2022-03-15 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды для snap25 |
| RU2786637C2 (ru) * | 2016-12-30 | 2022-12-23 | Олипасс Корпорейшн | Пропуск экзонов с помощью производных пептидо-нуклеиновых кислот |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9234059B2 (en) | 2008-07-16 | 2016-01-12 | Outlast Technologies, LLC | Articles containing functional polymeric phase change materials and methods of manufacturing the same |
| US8221910B2 (en) | 2008-07-16 | 2012-07-17 | Outlast Technologies, LLC | Thermal regulating building materials and other construction components containing polymeric phase change materials |
| MX359743B (es) | 2009-10-16 | 2018-10-08 | Melinta Therapeutics Inc | Compuestos antimicrobianos y métodos para fabricar y utilizar los mismos. |
| CN104628729B (zh) * | 2009-10-16 | 2018-11-06 | 梅琳塔治疗公司 | 抗微生物化合物和其制备和使用方法 |
| CN102725274A (zh) | 2009-10-16 | 2012-10-10 | Rib-X制药公司 | 抗微生物化合物和其制备和使用方法 |
| ES2675798T3 (es) * | 2009-10-16 | 2018-07-12 | Melinta Therapeutics, Inc. | Compuestos antimicrobianos y métodos para fabricar y usar los mismos |
| ES2665988T3 (es) | 2011-04-15 | 2018-04-30 | Melinta Therapeutics, Inc. | Compuestos antimicrobianos y métodos de fabricación y uso de los mismos |
| CN103031337A (zh) * | 2012-09-28 | 2013-04-10 | 北京吉利奥生物科技发展有限公司 | 一种小核酸分子快递技术 |
| MX2016003046A (es) | 2013-09-09 | 2016-09-08 | Melinta Therapeutics Inc | Compuestos antimicrobianos y métodos de fabricación y utilización de los mismos. |
| KR20160070066A (ko) | 2013-09-09 | 2016-06-17 | 멜린타 테라퓨틱스, 인크. | 항균 화합물, 및 이의 제조 방법 및 이용 방법 |
| BR112017019349A2 (pt) | 2015-03-11 | 2018-06-05 | Melinta Therapeutics Inc | compostos antimicrobianos e métodos de fabricação e uso dos mesmos |
| AU2016341208A1 (en) * | 2015-10-20 | 2018-05-10 | Sorrento Therapeutics, Inc. | Intracellular delivery compounds |
| WO2018091544A1 (en) | 2016-11-16 | 2018-05-24 | Biomarin Pharmaceutical, Inc. | Substances for targeting various selected organs or tissues |
| BR112019011692A2 (pt) * | 2016-12-30 | 2019-10-22 | Olipass Corp | salto de exon por derivados de ácido nucleico de peptídeo |
| KR102592876B1 (ko) * | 2017-01-24 | 2023-10-23 | 올리패스 주식회사 | Scn9a 안티센스 진통제 |
| WO2019022434A1 (en) * | 2017-07-24 | 2019-01-31 | Olipass Corporation | ANTISENSE OLIGONUCLEOTIDES OF TYROSINASE |
| KR20190011181A (ko) * | 2017-07-24 | 2019-02-01 | 올리패스 주식회사 | 티로시나아제 안티센스 올리고뉴클레오티드 |
| TWI832851B (zh) * | 2018-05-18 | 2024-02-21 | 韓商奧利通公司 | 基質金屬蛋白酶-1之反義寡核苷酸 |
| KR102304280B1 (ko) | 2018-08-14 | 2021-09-23 | 올리패스 주식회사 | 아세틸코에이카복실라제2 안티센스 올리고뉴클레오티드 |
| CN114502571A (zh) * | 2019-07-18 | 2022-05-13 | 奥利通公司 | 黑素亲和素反义寡核苷酸 |
| KR20210151593A (ko) | 2020-06-05 | 2021-12-14 | 넥스올리고(주) | 신규한 몰포리노 올리고뉴클레오티드 유도체 |
| KR102633299B1 (ko) * | 2023-09-08 | 2024-02-02 | 국방과학연구소 | 세포 투과가 가능한 펩타이드 핵산 올리고머를 유효성분으로 포함하는 한탄바이러스 감염 질환 치료용 약학적 조성물 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992020702A1 (en) * | 1991-05-24 | 1992-11-26 | Ole Buchardt | Peptide nucleic acids |
| US6617422B1 (en) * | 1997-05-23 | 2003-09-09 | Peter Nielsen | Peptide nucleic acid monomers and oligomers |
| US20040265885A1 (en) * | 2000-04-18 | 2004-12-30 | Aventis Pharma Deutschland Gmbh | Polyamide nucleic acid derivatives, and agents, and processes for preparing them |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5539082A (en) | 1993-04-26 | 1996-07-23 | Nielsen; Peter E. | Peptide nucleic acids |
| US6133444A (en) | 1993-12-22 | 2000-10-17 | Perseptive Biosystems, Inc. | Synthons for the synthesis and deprotection of peptide nucleic acids under mild conditions |
| CN1814614A (zh) * | 2005-02-06 | 2006-08-09 | 中国人民解放军军事医学科学院毒物药物研究所 | 核酸、肽核酸衍生物及它们的用途 |
| WO2008061091A2 (en) | 2006-11-13 | 2008-05-22 | Grandis Jennifer R | Antisense guanidinium peptide nucleic acid (gpna) oligonucleotides as antitumor agents |
-
2009
- 2009-03-12 KR KR1020090021040A patent/KR20090098710A/ko not_active Application Discontinuation
- 2009-03-13 ES ES09719041.7T patent/ES2533769T3/es active Active
- 2009-03-13 PL PL09719041T patent/PL2268607T3/pl unknown
- 2009-03-13 WO PCT/KR2009/001256 patent/WO2009113828A2/en active Application Filing
- 2009-03-13 CA CA2715844A patent/CA2715844C/en active Active
- 2009-03-13 EP EP20090719041 patent/EP2268607B9/en active Active
- 2009-03-13 JP JP2010550603A patent/JP5620282B2/ja active Active
- 2009-03-13 US US12/922,322 patent/US8680253B2/en active Active
- 2009-03-13 NZ NZ587448A patent/NZ587448A/en unknown
- 2009-03-13 BR BRPI0906104A patent/BRPI0906104B8/pt active IP Right Grant
- 2009-03-13 RU RU2010135635/04A patent/RU2564032C2/ru active
- 2009-03-13 AU AU2009224149A patent/AU2009224149B2/en active Active
- 2009-03-13 CN CN200980109059.1A patent/CN102015628B/zh active Active
- 2009-03-13 MX MX2010009687A patent/MX2010009687A/es active IP Right Grant
- 2009-03-13 PT PT97190417T patent/PT2268607E/pt unknown
- 2009-03-13 DK DK09719041.7T patent/DK2268607T5/en active
- 2009-03-13 KR KR1020107018868A patent/KR101598423B1/ko active IP Right Grant
-
2010
- 2010-08-15 IL IL207617A patent/IL207617A/en active IP Right Grant
- 2010-08-20 ZA ZA2010/05960A patent/ZA201005960B/en unknown
-
2011
- 2011-09-14 HK HK11109665.2A patent/HK1155717A1/xx unknown
-
2013
- 2013-12-17 US US14/109,327 patent/US8884008B2/en active Active
- 2013-12-18 US US14/133,045 patent/US8859766B2/en active Active
- 2013-12-19 US US14/134,279 patent/US8895734B2/en active Active
-
2014
- 2014-12-30 IL IL236514A patent/IL236514A0/en active IP Right Grant
- 2014-12-30 IL IL236515A patent/IL236515A0/en active IP Right Grant
- 2014-12-30 IL IL236513A patent/IL236513A0/en active IP Right Grant
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1992020702A1 (en) * | 1991-05-24 | 1992-11-26 | Ole Buchardt | Peptide nucleic acids |
| US6617422B1 (en) * | 1997-05-23 | 2003-09-09 | Peter Nielsen | Peptide nucleic acid monomers and oligomers |
| US20040265885A1 (en) * | 2000-04-18 | 2004-12-30 | Aventis Pharma Deutschland Gmbh | Polyamide nucleic acid derivatives, and agents, and processes for preparing them |
Non-Patent Citations (1)
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2753966C2 (ru) * | 2016-08-08 | 2021-08-24 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды андрогенового рецептора |
| RU2748834C2 (ru) * | 2016-09-16 | 2021-05-31 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды против scn9a |
| RU2753517C2 (ru) * | 2016-10-11 | 2021-08-17 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды к hif-1-альфа |
| RU2786637C2 (ru) * | 2016-12-30 | 2022-12-23 | Олипасс Корпорейшн | Пропуск экзонов с помощью производных пептидо-нуклеиновых кислот |
| RU2766701C2 (ru) * | 2017-01-06 | 2022-03-15 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды для snap25 |
| RU2802418C2 (ru) * | 2018-05-18 | 2023-08-28 | Олипасс Корпорейшн | Антисмысловые олигонуклеотиды матриксной металлопротеиназы-1 |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2564032C2 (ru) | Производные пептидо-нуклеиновых кислот с хорошей клеточной пенетрацией и сильной аффинностью к нуклеиновой кислоте | |
| JP2020513036A (ja) | 協同的結合に関与する化合物及びその使用 | |
| US20100317593A1 (en) | 2,3-dihydro-1h-indene compounds | |
| WO2002097134A2 (en) | Modified peptide nucleic acid | |
| JPH07112969A (ja) | 治療および診断用のc分枝を有する核酸結合性オリゴマー類 | |
| KR102515760B1 (ko) | 펩타이드 핵산 단량체와 소중합체 | |
| US20130231480A1 (en) | Sequence specific double-stranded dna/rna binding compounds and uses thereof | |
| WO2013183656A1 (ja) | Gタンパク質共役受容体結合リガンドと核酸分子とのコンジュゲート | |
| CA3066698A1 (en) | Novel compounds activating the nrf2 pathway | |
| CN109608524B (zh) | 兼具dpp-4抑制及glp1r激活活性的多肽衍生物、制备和应用 | |
| EP3819006A1 (en) | Compounds with thymine skeleton for use in medicine | |
| NO317225B1 (no) | Nye overforingsmidler for nukleinsyrer, preparater inneholdende disse samt midlenes anvendelse | |
| JP2003219874A (ja) | Rnaに選択的に結合するペプチド核酸をアンチセンスオリゴヌクレオチドとして用いる方法、及び該ペプチド核酸の製造方法 |