KR20120101411A - 치환된 3-페닐프로피온산 및 그의 용도 - Google Patents
치환된 3-페닐프로피온산 및 그의 용도 Download PDFInfo
- Publication number
- KR20120101411A KR20120101411A KR1020127013589A KR20127013589A KR20120101411A KR 20120101411 A KR20120101411 A KR 20120101411A KR 1020127013589 A KR1020127013589 A KR 1020127013589A KR 20127013589 A KR20127013589 A KR 20127013589A KR 20120101411 A KR20120101411 A KR 20120101411A
- Authority
- KR
- South Korea
- Prior art keywords
- mmol
- methyl
- mixture
- ethyl
- added
- Prior art date
Links
- 0 CCO*(c1c(*)ccc(*)c1*)OCC Chemical compound CCO*(c1c(*)ccc(*)c1*)OCC 0.000 description 10
- HTCQECRVUCYCQI-VQHVLOKHSA-N CC(C)(C)OC(/C=C(\C)/c(cc1[N+]([O-])=O)ccc1Cl)=O Chemical compound CC(C)(C)OC(/C=C(\C)/c(cc1[N+]([O-])=O)ccc1Cl)=O HTCQECRVUCYCQI-VQHVLOKHSA-N 0.000 description 1
- WEXMIQFTKROSLG-UHFFFAOYSA-N CC(C)(C)OC(C(Cc(cc1Br)ccc1Cl)C1CC1)=O Chemical compound CC(C)(C)OC(C(Cc(cc1Br)ccc1Cl)C1CC1)=O WEXMIQFTKROSLG-UHFFFAOYSA-N 0.000 description 1
- QGSHLYWBRRCHDF-UHFFFAOYSA-N CC(C)(C)OC(CCc(cc1N)ccc1C#N)=O Chemical compound CC(C)(C)OC(CCc(cc1N)ccc1C#N)=O QGSHLYWBRRCHDF-UHFFFAOYSA-N 0.000 description 1
- ZLKOACIDIBZPEW-UHFFFAOYSA-N CC(C)(C)OC(CCc(cc1N)ccc1F)=O Chemical compound CC(C)(C)OC(CCc(cc1N)ccc1F)=O ZLKOACIDIBZPEW-UHFFFAOYSA-N 0.000 description 1
- AEALYRMFQRFINN-UHFFFAOYSA-N CCC(C)(Cc(cc1)cc(NCc2ccccc2)c1Cl)C(OC(C)(C)C)=O Chemical compound CCC(C)(Cc(cc1)cc(NCc2ccccc2)c1Cl)C(OC(C)(C)C)=O AEALYRMFQRFINN-UHFFFAOYSA-N 0.000 description 1
- CSEAKZJDYIBAGQ-UHFFFAOYSA-N CCOC(C(C1)C1c(cc1NCc2ccccc2)ccc1F)=O Chemical compound CCOC(C(C1)C1c(cc1NCc2ccccc2)ccc1F)=O CSEAKZJDYIBAGQ-UHFFFAOYSA-N 0.000 description 1
- YXGIFBAKTNIKAY-CGCSKFHYSA-N CCOC(C([C@@H](C)C(F)(F)F)c(cc1)ccc1C(F)=C)=O Chemical compound CCOC(C([C@@H](C)C(F)(F)F)c(cc1)ccc1C(F)=C)=O YXGIFBAKTNIKAY-CGCSKFHYSA-N 0.000 description 1
- SRVTXLPAWBTQSA-UHFFFAOYSA-N CCOC(CC(C)C(F)(F)F)=O Chemical compound CCOC(CC(C)C(F)(F)F)=O SRVTXLPAWBTQSA-UHFFFAOYSA-N 0.000 description 1
- RSICLPQEAGBEJX-UHFFFAOYSA-N CCOC(CC(CCC1)C1(F)F)=O Chemical compound CCOC(CC(CCC1)C1(F)F)=O RSICLPQEAGBEJX-UHFFFAOYSA-N 0.000 description 1
- SRVTXLPAWBTQSA-RXMQYKEDSA-N CCOC(C[C@@H](C)C(F)(F)F)=O Chemical compound CCOC(C[C@@H](C)C(F)(F)F)=O SRVTXLPAWBTQSA-RXMQYKEDSA-N 0.000 description 1
- QODMSEUUCUVSJO-JZKQVHKSSA-N CCOC([C@H](C)Cc(cc1)cc(NC([C@@H]([C@@H](C)C(F)(F)F)c(cc2)ccc2Cl)=O)c1Cl)=O Chemical compound CCOC([C@H](C)Cc(cc1)cc(NC([C@@H]([C@@H](C)C(F)(F)F)c(cc2)ccc2Cl)=O)c1Cl)=O QODMSEUUCUVSJO-JZKQVHKSSA-N 0.000 description 1
- IOILCXWFSBNSBD-MRVPVSSYSA-N CCOC([C@H](C)Cc(cc1N)ccc1F)=O Chemical compound CCOC([C@H](C)Cc(cc1N)ccc1F)=O IOILCXWFSBNSBD-MRVPVSSYSA-N 0.000 description 1
- FPNDYJYPKKELQF-JZKQVHKSSA-N CCc1ccc([C@H]([C@@H](C)C(F)(F)F)C(Nc(cc(C[C@@H](C)C(O)=O)cc2)c2Cl)=O)cc1 Chemical compound CCc1ccc([C@H]([C@@H](C)C(F)(F)F)C(Nc(cc(C[C@@H](C)C(O)=O)cc2)c2Cl)=O)cc1 FPNDYJYPKKELQF-JZKQVHKSSA-N 0.000 description 1
- IXPYWLWJAQXIRT-LKBUQDJMSA-N CCc1ccc([C@H]([C@@H](C)C(F)(F)F)C(Nc(cc([C@H](C)CC(O)=O)cc2)c2Cl)=O)cc1 Chemical compound CCc1ccc([C@H]([C@@H](C)C(F)(F)F)C(Nc(cc([C@H](C)CC(O)=O)cc2)c2Cl)=O)cc1 IXPYWLWJAQXIRT-LKBUQDJMSA-N 0.000 description 1
- LDDGFZFZLWONPJ-XCLFUZPHSA-N C[C@H]([C@H](C(Nc1cc(CCC(O)=O)ccc1F)=O)c1ccc(C=C)cc1)C(F)(F)F Chemical compound C[C@H]([C@H](C(Nc1cc(CCC(O)=O)ccc1F)=O)c1ccc(C=C)cc1)C(F)(F)F LDDGFZFZLWONPJ-XCLFUZPHSA-N 0.000 description 1
- BTDMTOBUCWREFS-JRQSSSKMSA-N C[C@H]([C@H](C(Nc1cc([C@H](CC(OC(C)(C)C)=O)C(F)(F)F)ccc1Cl)=O)c(cc1)ccc1Cl)C(F)(F)F Chemical compound C[C@H]([C@H](C(Nc1cc([C@H](CC(OC(C)(C)C)=O)C(F)(F)F)ccc1Cl)=O)c(cc1)ccc1Cl)C(F)(F)F BTDMTOBUCWREFS-JRQSSSKMSA-N 0.000 description 1
- QADCNGZPRUSTJL-UHFFFAOYSA-N [O-][N+](c1cc(C(C(F)(F)F)=O)ccc1)=O Chemical compound [O-][N+](c1cc(C(C(F)(F)F)=O)ccc1)=O QADCNGZPRUSTJL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/88—Carboxylic acid amides having nitrogen atoms of carboxamide groups bound to an acyclic carbon atom and to a carbon atom of a six-membered aromatic ring wherein at least one ortho-hydrogen atom has been replaced
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/58—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton
- C07C255/60—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and singly-bound nitrogen atoms, not being further bound to other hetero atoms, bound to the carbon skeleton at least one of the singly-bound nitrogen atoms being acylated
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/196—Carboxylic acids, e.g. valproic acid having an amino group the amino group being directly attached to a ring, e.g. anthranilic acid, mefenamic acid, diclofenac, chlorambucil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/655—Azo (—N=N—), diazo (=N2), azoxy (>N—O—N< or N(=O)—N<), azido (—N3) or diazoamino (—N=N—N<) compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/16—Central respiratory analeptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/12—Drugs for disorders of the metabolism for electrolyte homeostasis
- A61P3/14—Drugs for disorders of the metabolism for electrolyte homeostasis for calcium homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/27—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups
- C07C205/35—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups having nitro groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C205/36—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups having nitro groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton to carbon atoms of the same non-condensed six-membered aromatic ring or to carbon atoms of six-membered aromatic rings being part of the same condensed ring system
- C07C205/38—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by etherified hydroxy groups having nitro groups and etherified hydroxy groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton to carbon atoms of the same non-condensed six-membered aromatic ring or to carbon atoms of six-membered aromatic rings being part of the same condensed ring system the oxygen atom of at least one of the etherified hydroxy groups being further bound to a carbon atom of a six-membered aromatic ring, e.g. nitrodiphenyl ethers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/02—Preparation of carboxylic acid amides from carboxylic acids or from esters, anhydrides, or halides thereof by reaction with ammonia or amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/53—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
- C07C233/55—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring having the carbon atom of the carboxamide group bound to a carbon atom of an unsaturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D305/00—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms
- C07D305/02—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D305/04—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D305/06—Heterocyclic compounds containing four-membered rings having one oxygen atom as the only ring hetero atoms not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Abstract
본 발명은 신규 3-페닐프로피온산 유도체, 그의 제조 방법, 질환의 치료 및/또는 예방을 위한 그의 용도, 및 질환의 치료 및/또는 예방, 특히 심혈관 장애의 치료 및/또는 예방을 위한 의약의 제조에 있어서의 그의 용도에 관한 것이다.
Description
본원은 신규 3-페닐프로피온산 유도체, 그의 제조 방법, 질환의 치료 및/또는 예방을 위한 그의 용도, 및 질환의 치료 및/또는 예방, 특히 심혈관 장애의 치료 및/또는 예방을 위한 의약의 제조에 있어서의 그의 용도에 관한 것이다.
시클릭 구아노신 모노포스페이트 (cGMP)는 포유동물 세포에서 가장 중요한 세포 전달 시스템 중 하나이다. 내피로부터 방출되며, 호르몬 및 기계 신호를 전달하는 산화질소 (NO)와 함께, 이것은 NO/cGMP 시스템을 형성한다. 구아닐레이트 시클라제는 구아노신 트리포스페이트 (GTP)로부터의 cGMP의 생합성에 촉매 작용을 한다. 현재까지 개시된 상기 패밀리의 대표물은 구조적 특징 및 리간드의 유형 둘 모두에 따라 하기 2개의 군으로 나뉘어질 수 있다: 나트륨이뇨 펩티드에 의해 자극될 수 있는 미립자 구아닐레이트 시클라제 및 NO에 의해 자극될 수 있는 가용성 구아닐레이트 시클라제. 가용성 구아닐레이트 시클라제는 2개의 서브유닛으로 이루어지며, 조절 부위의 일부인 이종이량체 당 하나의 헴을 함유할 가능성이 매우 높다. 후자는 활성화의 메카니즘에 있어서 중추적으로 중요하다. NO는 헴의 철 원자에 결합할 수 있고, 따라서 효소의 활성을 두드러지게 증가시킬 수 있다. 반면에, 헴-무함유 제제는 NO에 의해 자극될 수 없다. 일산화탄소 (CO)도 또한 헴의 중심 철 원자에 부착될 수 있지만, CO에 의한 자극은 NO에 의한 자극보다 현저히 낮다.
cGMP의 생성 및 그로 인한 포스포디에스테라제, 이온 채널 및 단백질 키나제의 조절을 통해, 구아닐레이트 시클라제는 다양한 생리적 과정, 특히 평활근 세포의 이완 및 증식, 혈소판 응집과 유착 및 뉴런의 신호 전달에서, 및 상기 언급한 과정의 손상에 의해 초래된 장애에서 결정적인 역할을 한다. 병리생리학적 조건 하에서, NO/cGMP 시스템은 억제될 수 있으며, 이는, 예를 들어, 고혈압, 혈소판 활성화, 세포 증식 증가, 내피 기능장애, 아테롬성동맥경화증, 협심증, 심부전, 혈전증, 졸중 및 심근경색을 야기할 수 있다.
NO에 비의존성이고 유기체에서 cGMP 신호전달 경로에 영향을 미치는 것을 목적으로 하는, 이러한 장애를 치료하는 가능한 방법은 고효율적이며 부작용이 거의 예견되지 않으므로 유망한 접근법이다.
NO에 근거한 효과를 갖는 화합물, 예컨대 유기 니트레이트는 현재까지 독점적으로 가용성 구아닐레이트 시클라제의 치료학적 자극을 위해 사용되어 왔다. NO는 생물전환에 의해 생성되고, 헴의 중심 철 원자에 부착함으로써 가용성 구아닐레이트 시클라제를 활성화시킨다. 부작용 이외에, 내성의 발생은 이러한 방식의 치료의 결정적 단점 중 하나이다 (문헌 [O.V. Evgenov et al., Nature Rev. Drug Disc. 5 (2006), 755]).
가용성 구아닐레이트 시클라제를 직접적으로, 즉, NO의 선행 방출없이 자극하는 물질이 최근 몇 년 동안 동정되었다. 인다졸 유도체 YC-1은 첫번째 NO-비의존성 그러나 헴-의존성 sGC 자극물질로 기재되었다 (상기 문헌 [Evgenov et al.]). YC-1을 기반으로, YC-1보다 더 강력하며 포스포디에스테라제 (PDE)의 관련 억제를 나타내지 않는 추가적 물질이 발견되었다. 이것은 피라졸로피리딘 유도체 BAY 41-2272, BAY 41-8543과 BAY 63-2521의 동정으로 이어졌다. 최근에 발표된 구조적으로 상이한 물질 CMF-1571 및 A-350619와 함께, 이들 화합물은 sGC 자극물질의 신규한 클래스를 형성한다 (상기 문헌 [Evgenov et al.]). 이 물질 유형의 공통 특성은 헴-함유 sGC의 선택적 활성화 및 NO-비의존성이다. 또한, NO와 결합된 sGC 자극물질은 니트로실-헴 착체의 안정화를 기반으로 sGC 활성화에 대해 상승작용 효과를 갖는다. sGC에서의 sGC 자극물질의 정확한 결합 부위는 여전히 논의되고 있다. 헴 기가 가용성 구아닐레이트 시클라제에서 제거되는 경우, 효소는 검출가능한 촉매 기저 활성을 여전히 가지고 있으며, 즉, cGMP가 여전히 형성된다. 헴-무함유 효소의 남아있는 촉매 기저 활성은 상기에 언급된 임의의 자극물질에 의해 자극될 수 없다 (상기 문헌 [Evgenov et al.]).
또한, NO- 및 헴-비의존성 sGC 활성화제가, 상기 클래스의 원형으로서의 BAY 58-2667과 함께 동정되었다. 이들 물질의 공통 특성은 NO와 함께 이들이 단지 효소 활성화에 대해 부가적인 효과만을 가지고 있고, 산화 또는 헴-무함유 효소의 활성화가 헴-함유 효소의 활성화보다 두드러지게 높다는 것이다 (문헌 [Evgenov et al., ibid.; J.P. Stasch et al., Br. J. Pharmacol. 136 (2002), 773]; [J.P. Stasch et al., J. Clin. Invest. 116 (2006), 2552]). BAY 58-2667이 산화 헴 기 (이는, 철-히스티딘 결합의 약화의 결과로서, sGC에 단지 약하게 부착되어 있음)를 대체한다는 것이 분광학적 연구를 통해 나타났다. 특징적 sGC 헴 결합 모티프 Tyr-x-Ser-x-Arg가 헴 기의 음으로 하전된 프로피온산의 상호작용에 대해 및 BAY 58-2667의 작용에 대해 모두 절대적으로 필수적이라는 것이 또한 나타났다. 이러한 배경에 대하여, sGC에서의 BAY 58-2667의 결합 부위는 헴 기의 결합 부위와 동일한 것으로 추정된다 (문헌 [J.P. Stasch et al., J. Clin. Invest. 116 (2006), 2552]).
본 발명에 기재된 화합물은 이제 마찬가지로 가용성 구아닐레이트 시클라제의 헴-무함유 형태를 활성화시킬 수 있다. 이것은 또한 이러한 신규 활성화제가 우선 헴-함유 효소에서 NO와의 상승 작용을 갖지 않으며, 둘째로 그의 작용이 가용성 구아닐레이트 시클라제, 1H-1,2,4-옥사디아졸로[4,3-a]퀴녹살린-1-온 (ODQ)의 헴-의존성 억제제에 의해 차단될 수 없을 뿐 아니라, 심지어 이 억제제에 의해 강화된다는 사실로 확인된다 (문헌 [O.V. Evgenov et al., Nature Rev. Drug Disc. 5 (2006), 755]; [J.P. Stasch et al., J. Clin. Invest. 116 (2006), 2552] 참조).
따라서, 본 발명의 목적은 상기 기재된 방식으로 가용성 구아닐레이트 시클라제의 활성화제로서 작용하고, 특히 심혈관 장애의 치료 및 예방에 그 자체로 사용될 수 있는 신규한 화합물을 제공하는 것이었다.
WO 00/64888-A1, EP 1 216 980-A1, EP 1 375 472-A1, EP 1 452 521-A1, US 2005/0187266-A1 및 US 2005/0234066-A1에는 당뇨병, 이상지혈증, 동맥경화증, 비만 및 다른 장애의 치료를 위한 PPAR 효능제로서의 다양한 아릴알칸카르복실산 유도체가 기재되어 있다. EP 1 312 601-A1 및 EP 1 431 267-A1에는, 예를 들어 통증, 비뇨기 장애, 알츠하이머병 및 암의 치료를 위한 PGE2 수용체 길항제로서의 치환된 아릴알칸카르복실산이 개시되어 있다. 또한, 아릴알칸카르복실산은 WO 2005/086661-A2에서 당뇨병 및 이상지혈증의 치료를 위한 GPR40 조절인자로서 청구되며, WO 2004/099170-A2, WO 2006/050097-A1 및 WO 2006/055625-A2에는 당뇨병, 암 및 신경변성 장애의 치료를 위한 PTP-1B 억제제로서의 페닐-치환된 카르복실산이 기재되어 있다. 또한, 비-공유 혼합물의 형태로 신체 내 활성 펩티드 화합물의 제공을 향상시키는 별개의 페닐아세트아미도-치환된 페닐알칸카르복실산이 WO 96/12473-A1 및 WO 96/30036-A1로부터 공지되어 있다. 최근에, 가용성 구아닐레이트 시클라제의 활성화제로서 작용하는 옥소헤테로시클릭 치환된 카르복실산 유도체가 WO 2009/127338-A1에 개시되었다.
본 발명은 하기 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 제공한다.
<화학식 I>

상기 식에서,
R1A는 수소, 플루오린, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸, n-프로필, 시클로프로필 또는 시클로부틸을 나타내고,
R1B는 수소 또는 메틸을 나타내고,
R2A는 수소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸 또는 n-프로필을 나타내고,
R2B는 수소 또는 메틸을 나타내거나, 또는
R1A 및 R2A는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, R1B 및 R2B는 상기 언급된 의미를 가짐)의 시클로프로필 고리를 형성하거나, 또는
(여기서, R1B 및 R2B는 상기 언급된 의미를 가짐)의 시클로프로필 고리를 형성하거나, 또는
R2A 및 R2B는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, n은 1, 2 또는 3의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1, 2 또는 3의 수를 나타냄)의 시클릭 기를 형성하고,
R3은 수소, 플루오린, 메틸 또는 트리플루오로메틸을 나타내고,
R4는 수소, 플루오린, 염소, 시아노, 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5A는 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5B는 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로알킬 고리를 형성하고,
의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6은 수소, 플루오린, 염소, 브로민, 시아노, (C1-C4)-알킬, (C2-C4)-알케닐, 시클로프로필 또는 시클로부틸을 나타내고, 여기서
(C1-C4)-알킬 및 (C2-C4)-알케닐은 플루오린에 의해 3회 이하 치환될 수 있고,
시클로프로필 및 시클로부틸은 플루오린에 의해 2회 이하 치환될 수 있고,
R7은 수소, 플루오린, 염소, 시아노, 메틸, 트리플루오로메틸, 에틸, 메톡시 또는 트리플루오로메톡시를 나타낸다.
본 발명에 따른 화합물은 화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물, 하기 언급되는 화학식들 중 화학식 I에 포함되는 화합물, 및 그의 염, 용매화물 및 염의 용매화물, 및 화학식 I에 포함되고 실시양태 실시예로서 하기 언급되는 화합물, 및 그의 염, 용매화물 및 염의 용매화물이며, 여기서 화학식 I에 포함되고 하기에 언급되는 화합물은 염, 용매화물 및 염의 용매화물이 아니다.
본 발명에 따른 화합물은 그의 구조에 따라 다양한 입체이성질체 형태, 즉 배위 이성질체, 또는 적절한 경우에는 또한 형태 이성질체 (거울상이성질체 및/또는 부분입체이성질체 (회전장애이성질체의 경우의 것들 포함))의 형태로 존재할 수 있다. 따라서, 본 발명은 거울상이성질체 및 부분입체이성질체, 및 이들의 특정 혼합물을 포함한다. 입체이성질체적으로 균일한 성분은 상기 거울상이성질체 및/또는 부분입체이성질체의 혼합물로부터 공지된 방식으로 단리될 수 있고; 바람직하게는 이에 대해 크로마토그래피 방법, 특히 비키랄 또는 키랄 상 상에서의 HPLC 크로마토그래피가 이용된다.
본 발명에 따른 화합물이 호변이성질체 형태로 존재할 수 있는 경우, 본 발명은 모든 호변이성질체 형태를 포함한다.
본 발명의 문맥에서 바람직한 염은 본 발명에 따른 화합물의 생리학적으로 허용되는 염이다. 그 자체가 제약 용도에 적합하지는 않지만, 예를 들어 본 발명에 따른 화합물을 단리 또는 정제하는데 사용될 수 있는 염이 또한 포함된다.
본 발명에 따른 화합물의 생리적으로 허용되는 염에는 또한, 특히 통상의 염기의 염, 예컨대 예로서 및 바람직하게는 알칼리 금속염 (예를 들어, 나트륨 및 칼륨 염), 알칼리 토금속염 (예를 들어, 칼슘 및 마그네슘 염), 및 암모니아 또는 1 내지 16개의 C 원자를 갖는 유기 아민, 예컨대, 예로서 및 바람직하게는 에틸아민, 디에틸아민, 트리에틸아민, 에틸디이소프로필아민, 모노에탄올아민, 디에탄올아민, 트리에탄올아민, 디시클로헥실아민, 디메틸아미노에탄올, 프로카인, 디벤질아민, N-메틸모르폴린, N-메틸피페리딘, 아르기닌, 리신 및 에틸렌디아민에서 유래된 암모늄 염이 포함된다.
본 발명의 문맥에서 용매화물은 용매 분자와의 배위에 의해 고체 또는 액체 상태로 복합체를 형성하는 본 발명에 따른 화합물의 형태로서 정의된다. 수화물은 배위가 물과 함께 일어나는 특정 형태의 용매화물이다. 본 발명의 문맥에서, 수화물이 바람직한 용매화물이다.
또한, 본 발명은 본 발명에 따른 화합물의 전구약물도 포함한다. 여기서, 용어 "전구약물"은 그 자체가 생물학적으로 활성일 수 있거나, 또는 불활성일 수 있지만 체내에서 그의 체류 시간 동안 본 발명에 따른 화합물로 (예를 들어, 대사적으로 또는 가수분해적으로) 전환되는 화합물을 나타낸다.
본 발명은 특히 본 발명에 따른 화학식 I의 카르복실산의 가수분해성 에스테르 유도체를 포함한다. 이들은, 이하 기재될 생물학적 시험의 조건 하에 생리적 매질 중에서, 특히 생체내 효소적 또는 화학적 경로에 의해, 대부분 생물학적으로 활성인 화합물로서 유리 카르복실산으로 가수분해될 수 있는 에스테르를 의미하는 것으로 이해된다. (C1-C4)-알킬 에스테르 (여기서, 알킬기는 직쇄 또는 분지형일 수 있음)가 이러한 에스테르로서 바람직하다. 특히 메틸, 에틸 또는 tert-부틸 에스테르가 바람직하다.
본 발명의 문맥에서, 치환기는 달리 명시되지 않는 한 하기 의미를 갖는다.
본 발명의 문맥에서 (C1-C4)-알킬은 1 내지 4개의 탄소 원자를 갖는 직쇄 또는 분지형 알킬 라디칼을 나타낸다. 예로써 및 바람직하게는 메틸, 에틸, n-프로필, 이소프로필, n-부틸, iso-부틸, sec-부틸 및 tert-부틸을 언급할 수 있다.
본 발명의 문맥에서 (C2-C4)-알케닐 및 (C2-C3)-알케닐은 각각 2 내지 4개, 또는 2 또는 3개의 탄소 원자, 및 이중 결합을 갖는 직쇄 또는 분지형 알케닐 라디칼을 나타낸다. 2 또는 3개의 탄소 원자를 갖는 직쇄 또는 분지형 알케닐 라디칼이 바람직하다. 예로서 및 바람직하게는, 비닐, 알릴, n-프로프-1-엔-1-일, 이소프로페닐, n-부트-1-엔-1-일, n-부트-2-엔-1-일, n-부트-3-엔-1-일, 2-메틸프로프-1-엔-1-일 및 2-메틸프로프-2-엔-1-일을 언급할 수 있다.
본 발명의 문맥에서, 1회 초과로 발생하는 모든 라디칼에 대해 그의 의미는 서로 독립적이다. 본 발명에 따른 화합물에서의 라디칼이 치환되는 경우, 라디칼은 달리 명시되지 않는다면 일치환 또는 다치환될 수 있다. 1개 또는 2개 또는 3개의 별개의 또는 상이한 치환기에 의한 치환이 바람직하다. 1개 또는 2개의 동일하거나 상이한 치환기에 의한 치환이 특히 바람직하다.
특정 실시양태에서, 본 발명은
R1A가 수소, 플루오린, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸 또는 n-프로필을 나타내고,
R1B가 수소 또는 메틸을 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸 또는 n-프로필을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R1A 및 R2A가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, R1B 및 R2B는 상기 의미를 가짐)의 시클로프로필 고리를 형성하거나, 또는
(여기서, R1B 및 R2B는 상기 의미를 가짐)의 시클로프로필 고리를 형성하거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, n은 1, 2 또는 3의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1, 2 또는 3의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소, 플루오린, 메틸 또는 트리플루오로메틸을 나타내고,
R4가 수소, 플루오린, 염소, 시아노, 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5A가 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로알킬 고리를 형성하고,
의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6이 수소, 플루오린, 염소, 브로민, 시아노, (C1-C4)-알킬 또는 (C2-C4)-알케닐을 나타내고, 여기서 (C1-C4)-알킬 및 (C2-C4)-알케닐이 그의 일부에 대해 플루오린에 의해 3회 이하 치환될 수 있고,
R7이 수소, 플루오린, 염소 또는 메틸을 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 문맥에서,
R1A가 수소, 메틸, 트리플루오로메틸, 에틸, n-프로필, 시클로프로필 또는 시클로부틸을 나타내고,
R1B가 수소 또는 메틸을 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소, 플루오린 또는 메틸을 나타내고,
R4가 수소, 플루오린, 염소, 시아노, 메틸 또는 트리플루오로메틸을 나타내고,
R5A가 메틸 또는 에틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로알킬 고리를 형성하고,
의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6이 플루오린, 염소, (C1-C4)-알킬, (C2-C3)-알케닐, 시클로프로필 또는 시클로부틸을 나타내고, 여기서
(C1-C4)-알킬 및 (C2-C3)-알케닐이 플루오린에 의해 3회 이하 치환될 수 있고,
시클로프로필 및 시클로부틸이 플루오린에 의해 2회 이하 치환될 수 있고,
R7이 수소, 플루오린, 염소, 메틸 또는 메톡시를 나타내는 것인
화학식 I의 화합물, 그의 염, 용매화물, 및 그의 염의 용매화물이 바람직하다.
본 발명의 추가의 바람직한 실시양태는
R1A가 수소, 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R1B가 수소 또는 메틸을 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식 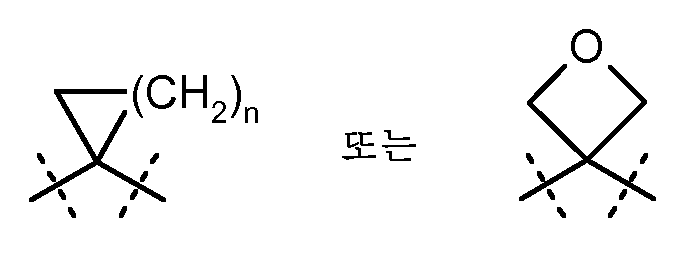 (여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소 또는 플루오린을 나타내고,
R4가 수소, 플루오린, 염소, 시아노, 메틸 또는 트리플루오로메틸을 나타내고,
R5A가 메틸 또는 에틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로알킬 고리를 형성하고,
의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6이 플루오린, 염소, (C1-C4)-알킬 또는 (C2-C3)-알케닐을 나타내고, 여기서 (C1-C4)-알킬 및 (C2-C3)-알케닐이 그의 일부에 대해 플루오린에 의해 3회 이하 치환될 수 있고,
R7이 수소, 플루오린 또는 염소를 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 문맥에서,
R1A가 수소, 메틸 또는 에틸을 나타내고,
R1B가 수소를 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소를 나타내고,
R4가 플루오린, 염소 또는 메틸을 나타내고,
R5A가 메틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로펜틸 고리를 형성하고,
의 디플루오로-치환된 시클로펜틸 고리를 형성하고,
R6이 플루오린, 염소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸, 이소프로필, tert-부틸, 1,1,1-트리플루오로-2-메틸프로판-2-일, 비닐, 1-플루오로비닐, 시클로프로필, 2,2-디플루오로시클로프로필, 시클로부틸 또는 3,3-디플루오로시클로부틸을 나타내고,
R7이 수소, 플루오린, 염소 또는 메틸을 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물이 특히 바람직하다.
본 발명의 추가의 특히 바람직한 실시양태는
R1A가 수소, 메틸 또는 에틸을 나타내고,
R1B가 수소를 나타내고,
R2A가 수소 또는 메틸을 나타내고,
R2B가 수소를 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  (여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소를 나타내고,
R4가 플루오린, 염소 또는 메틸을 나타내고,
R5A가 메틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로펜틸 고리를 형성하고,
의 디플루오로-치환된 시클로펜틸 고리를 형성하고,
R6이 염소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸, 이소프로필, tert-부틸, 1,1,1-트리플루오로-2-메틸프로판-2-일, 비닐 또는 1-플루오로비닐을 나타내고,
R7이 수소 또는 플루오린을 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 특정한 실시양태는
R1A가 수소, 메틸 또는 에틸을 나타내고,
R1B, R2A 및 R2B가 각각 수소를 나타내는 것인
화학식 I의 화합물 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R2A가 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R1A, R1B 및 R2B가 각각 수소를 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R1A 및 R1B가 각각 수소를 나타내고,
R2A 및 R2B가 각각 메틸을 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R1A 및 R1B가 각각 수소를 나타내고,
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 시클로프로필 또는 시클로부틸 고리를 형성하는 것인
의 시클로프로필 또는 시클로부틸 고리를 형성하는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R3이 수소를 나타내고,
R4가 플루오린 또는 염소를 나타내는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R5A가 메틸을 나타내고,
R5B가 트리플루오로메틸을 나타내는 것인
화학식 I의 화합물, 또는 그의 염, 용매화물 또는 그의 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식  의 디플루오로-치환된 시클로펜틸 고리를 형성하는 것인
의 디플루오로-치환된 시클로펜틸 고리를 형성하는 것인
화학식 I의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다.
본 발명의 추가의 특정한 실시양태는 하기 화학식 I-A의 화합물, 및 그의 염, 용매화물 및 염의 용매화물을 포함한다
<화학식 I-A>

상기 식에서,
페닐아세트아미드 기의 * 부호로 표시된 탄소 원자는 제시된 S 배위를 갖고,
라디칼 R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6 및 R7은 각각 상기 주어진 의미를 갖는다.
라디칼의 각각의 조합 또는 바람직한 조합에 구체적으로 나타낸 라디칼의 정의는 원하는 경우, 라디칼에 대해 나타낸 특정 조합과는 독립적으로, 또한 다른 조합의 라디칼 정의로 대체된다.
상기 언급된 2가지 이상의 바람직한 범위의 조합이 매우 특히 바람직하다.
본 발명은 하기 화학식 II의 카르복실산을 축합제의 보조 하에 또는 염기의 존재 하에 상응하는 카르보닐 클로라이드의 중간체를 통해 불활성 용매 중에서 하기 화학식 III의 아민과 커플링시켜 하기 화학식 IV의 카르복스아미드를 수득하고,
<화학식 II>

(상기 식에서, R5A, R5B, R6 및 R7은 상기 주어진 의미를 가짐)
<화학식 III>

(상기 식에서, R1A, R1B, R2A, R2B, R3 및 R4는 상기 주어진 의미를 갖고,
T1은 (C1-C4)-알킬 또는 벤질을 나타냄)
<화학식 IV>

(상기 식에서, R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6, R7 및 T1은 상기 주어진 의미를 가짐)
이어서 에스테르 라디칼 T1을 염기성 또는 산성 가용매분해에 의해 제거하거나, 또는 T1이 벤질을 나타내는 경우, 또한 가수소분해에 의해 제거하여 화학식 I의 카르복실산을 수득하고, 화학식 I의 화합물을, 적절한 경우, 당업자에게 공지된 방법에 의해 그의 거울상이성질체 및/또는 부분입체이성질체로 분리하고/거나, 적절한 경우, 적절한 (i) 용매 및/또는 (ii) 염기와 반응시켜 그의 용매화물, 염 및/또는 염의 용매화물을 수득하는 것을 특징으로 하는,
본 발명에 따른 화학식 I의 화합물의 제조 방법을 추가로 제공한다.
공정 단계 II + III → IV [아미드 커플링]에 대한 불활성 용매는, 예를 들어 에테르, 예컨대 디에틸 에테르, tert-부틸 메틸 에테르, 테트라히드로푸란, 1,4-디옥산, 글리콜 디메틸 에테르 또는 디에틸렌 글리콜 디메틸 에테르, 탄화수소, 예컨대 벤젠, 톨루엔, 크실렌, 헥산, 시클로헥산 또는 미네랄 오일 분별유, 할로겐화 탄화수소, 예컨대 디클로로메탄, 트리클로로메탄, 사염화탄소, 1,2-디클로로에탄, 트리클로로에틸렌 또는 클로로벤젠, 또는 다른 용매, 예컨대 아세톤, 아세토니트릴, 에틸 아세테이트, 피리딘, 디메틸 술폭시드 (DMSO), N,N-디메틸포름아미드 (DMF), N,N'-디메틸프로필렌우레아 (DMPU) 또는 N-메틸피롤리디논 (NMP)이다. 언급된 용매의 혼합물을 이용하는 것이 또한 가능하다. 디클로로메탄, 테트라히드로푸란, 디메틸포름아미드 또는 이들 용매의 혼합물을 사용하는 것이 바람직하다.
상기 커플링 반응에 적합한 축합제는, 예를 들어 카르보디이미드, 예컨대 N,N'-디에틸-, N,N'-디프로필-, N,N'-디이소프로필-, N,N'-디시클로헥실카르보디이미드 (DCC) 또는 N-(3-디메틸아미노이소프로필)-N'-에틸카르보디이미드 히드로클로라이드 (EDC), 포스겐 유도체, 예컨대 N,N'-카르보닐디이미다졸 (CDI), 1,2-옥사졸륨 화합물 예컨대 2-에틸-5-페닐-1,2-옥사졸륨 3-술페이트 또는 2-tert-부틸-5-메틸이속사졸륨 퍼클로레이트, 아실아미노 화합물, 예컨대 2-에톡시-1-에톡시카르보닐-1,2-디히드로퀴놀린, 또는 이소부틸 클로로포르메이트, 1-클로로-2-메틸-1-디메틸아미노-1-프로펜, 프로판포스폰산 무수물, 디에틸 시아노포스포네이트, 비스(2-옥소-3-옥사졸리디닐)포스포릴 클로라이드, 벤조트리아졸-1-일옥시트리스(디메틸아미노)포스포늄 헥사플루오로포스페이트, 벤조트리아졸-1-일옥시트리스(피롤리디노)포스포늄 헥사플루오로포스페이트 (PyBOP), O-(벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트 (TBTU), O-(벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트 (HBTU), 2-(2-옥소-1-(2H)-피리딜)-1,1,3,3-테트라메틸우로늄 테트라플루오로보레이트 (TPTU), O-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트 (HATU) 또는 O-(1H-6-클로로벤조트리아졸-1-일)-1,1,3,3-테트라메틸우로늄 테트라플루오로보레이트 (TCTU)이고, 적절한 경우에 추가의 보조제, 예컨대 1-히드록시벤조트리아졸 (HOBt) 또는 N-히드록시숙신이미드 (HOSu), 및 염기, 알칼리 금속 탄산염, 예를 들어 탄산나트륨 또는 탄산칼륨, 또는 유기 염기 예컨대 트리에틸아민, N-메틸모르폴린, N-메틸피페리딘, N,N-디이소프로필에틸아민, 피리딘 또는 4-N,N-디메틸아미노피리딘과 조합된다. O-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트 (HATU) 또는 O-(벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 테트라플루오로보레이트 (TBTU) (각 경우에 피리딘 또는 N,N-디이소프로필에틸아민과 조합하여), 또는 N-(3-디메틸아미노이소프로필)-N'-에틸카르보디이미드 히드로클로라이드 (EDC) (1-히드록시벤조트리아졸 (HOBt) 및 트리에틸아민과 조합하여), 또는 피리딘과 함께 1-클로로-2-메틸-1-디메틸아미노-1-프로펜을 사용하는 것이 바람직하다.
반응 II + III → IV는 일반적으로 0℃ 내지 +60℃, 바람직하게는 +10℃ 내지 +40℃의 온도 범위에서 수행된다.
화합물 II에 상응하는 카르보닐 클로라이드를 사용하는 경우, 아민 성분 III과의 커플링은 통상적인 유기 보조 염기, 예컨대 트리에틸아민, N-메틸모르폴린, N-메틸피페리딘, N,N-디이소프로필에틸아민, 피리딘, 4-N,N-디메틸아미노피리딘, 1,8-디아자비시클로[5.4.0]운데스-7-엔 (DBU) 또는 1,5-디아자비시클로[4.3.0]논-5-엔 (DBN)의 존재 하에 수행된다. 트리에틸아민 또는 N,N-디이소프로필에틸아민을 사용하는 것이 바람직하다.
아민 III과 카르보닐 클로라이드의 반응은 일반적으로 -20℃ 내지 +60℃, 바람직하게는 -10℃ 내지 +30℃ 범위의 온도에서 수행된다.
카르보닐 클로라이드는 카르복실산 II를 티오닐 클로라이드 또는 옥살릴 클로라이드로 처리함으로써 통상적인 방식으로 제조된다.
공정 단계 IV → I에서의 에스테르 기 T1의 제거는 불활성 용매 중에서 에스테르를 산 또는 염기로 처리하는 것에 의한 통상적인 방법에 의해 수행되며, 후자의 변이체의 경우 처음에 형성된 염을 산으로 처리하여 유리 카르복실산으로 전환시킨다. tert-부틸 에스테르의 경우에, 에스테르 절단은 바람직하게는 산을 사용하여 수행된다. 벤질 에스테르는 바람직하게는 적합한 촉매, 예컨대 예를 들어 활성 탄소 상 팔라듐의 존재 하에 가수소분해 (수소화)에 의해 절단된다.
이러한 반응에 적합한 불활성 용매는 물 또는 에스테르 절단에 통상적인 유기 용매이다. 이들은 바람직하게는 알콜, 예컨대 메탄올, 에탄올, n-프로판올, 이소프로판올, n-부탄올 또는 tert-부탄올, 또는 에테르, 예컨대 디에틸 에테르, 테트라히드로푸란, 디옥산 또는 글리콜 디메틸 에테르 또는 다른 용매, 예컨대 아세톤, 디클로로메탄, 디메틸포름아미드 또는 디메틸 술폭시드를 포함한다. 언급된 용매의 혼합물을 사용하는 것이 또한 가능하다. 염기성 에스테르 가수분해의 경우, 물과, 디옥산, 테트라히드로푸란, 메탄올 및/또는 에탄올의 혼합물을 사용하는 것이 바람직하다. 트리플루오로아세트산과의 반응의 경우, 디클로로메탄을 사용하는 것이 바람직하고, 염화수소와의 반응의 경우, 테트라히드로푸란, 디에틸 에테르, 디옥산 또는 물을 사용하는 것이 바람직하다.
적합한 염기는 통상적인 무기 염기이다. 이들은 특히 알칼리 금속 또는 알칼리 토금속 수산화물, 예컨대 예를 들어 수산화리튬, 수산화나트륨, 수산화칼륨 또는 수산화바륨, 또는 알칼리 금속 또는 알칼리 토금속 탄산염, 예컨대 탄산나트륨, 탄산칼륨 또는 탄산칼슘을 포함한다. 수산화리튬, 수산화나트륨 또는 수산화칼륨이 바라직하다.
에스테르 절단에 적합한 산은 일반적으로 황산, 염화수소산/염산, 브로민화수소산/브롬화수소산, 인산, 아세트산, 트리플루오로아세트산, 톨루엔술폰산, 메탄술폰산 또는 트리플루오로메탄술폰산 또는 그의 혼합물이고, 적절한 경우에 물이 첨가된다. tert-부틸 에스테르의 경우에는 염화수소 또는 트리플루오로아세트산이 적합하고, 메틸 에스테르의 경우에는 염산이 적합하다.
에스테르 절단은 일반적으로 -20℃ 내지 +100℃, 바람직하게는 0℃ 내지 +60℃ 범위의 온도에서 수행된다.
화학식 II의 중간체는, 예를 들어 먼저 하기 화학식 V의 카르복실산 에스테르를 불활성 용매 중에서 염기의 보조 하에 탈양성자화시키고, 이어서 이를 적합한 팔라듐 촉매의 존재 하에 하기 화학식 VI의 페닐 브로마이드를 사용하여 아릴화시킴으로써 하기 화학식 VII의 화합물을 수득하고,
<화학식 V>

(상기 식에서, R5A 및 R5B는 상기 주어진 의미를 갖고,
T2는 (C1-C4)-알킬 또는 벤질을 나타냄)
<화학식 VI>

(상기 식에서, R6 및 R7은 상기 주어진 의미를 가짐)
<화학식 VII>

(상기 식에서, R5A, R5B, R6, R7 및 T2는 상기 주어진 의미를 가짐)
후속으로 에스테르 라디칼 T2를 염기성 또는 산성 가용매분해에 의해 제거하거나, 또는 T2가 벤질을 나타내는 경우, 또한 가수소분해에 의해 제거하여, 카르복실산 II를 수득함으로써 제조될 수 있다.
공정 단계 V + VI → VII의 아릴화 반응은 바람직하게는 +20℃ 내지 +100℃ 범위의 온도에서 톨루엔 또는 톨루엔/테트라히드로푸란 혼합물 중에서 수행된다. 여기서, 에스테르 V의 탈양성자화에 사용된 염기는 바람직하게는 리튬 비스(트리메틸실릴)아미드이다. 적합한 팔라듐 촉매는, 예를 들어 아세트산팔라듐(II) 또는 트리스(디벤질리덴아세톤)디팔라듐 (각 경우에 입체 장애 전자-풍부 포스핀 리간드, 예컨대 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노)비페닐 또는 2-디-tert-부틸포스피노-2'-(N,N-디메틸아미노)비페닐과 조합됨)이다 (예를 들어, 문헌 [W.A. Moradi, S.L. Buchwald, J. Am. Chem. Soc. 123, 7996-8002 (2001)] 참조).
공정 단계 VII → II에서의 에스테르 기 T2의 제거는 에스테르 라디칼 T1에 대해 상기 기재된 것과 유사한 방식으로 수행된다.
대안적으로, 하기 화학식 II-A의 중간체를 또한, 먼저 하기 화학식 VIII의 페닐아세트산 에스테르를 2-시클로펜텐-1-온으로의 염기-유도된 첨가에 의해 하기 화학식 IX의 화합물로 전환시키고,
<화학식 II-A>

(상기 식에서, R6 및 R7은 상기 주어진 의미를 가짐)
<화학식 VIII>

(상기 식에서, R6, R7 및 T2는 상기 주어진 의미를 가짐)
<화학식 IX>

(상기 식에서, R6, R7 및 T2는 상기 주어진 의미를 가짐)
이어서 상기 화합물을 삼플루오린화붕소 촉매작용 하에 1,1'-[(트리플루오로-λ4-술파닐)이미노]비스(2-메톡시에탄)을 사용하여 플루오린화시켜 하기 화학식 VII-A의 화합물을 수득하고,
후속으로 에스테르 기 T2를 다시 제거하여 카르복실산 II-A를 수득함으로써 제조될 수 있다.
<화학식 VII-A>

(상기 식에서, R6, R7 및 T2는 상기 주어진 의미를 가짐)
에스테르 VIII의 탈양성자화에 대한 공정 단계 VIII → IX에서, 아미드 염기, 예컨대 리튬 디이소프로필아미드 또는 리튬 비스(트리메틸실릴)아미드를 사용하는 것이 바람직하다. 변환 IX → VII-A에서의 데옥시플루오린화에 대해, 상기 언급된 1,1'-[(트리플루오로-λ4-술파닐)이미노]비스(2-메톡시에탄) ("데스옥소플루오르") 대신에, 적절한 경우, 다른 공지된 플루오린화제, 예컨대 디에틸아미노황 트리플루오라이드 (DAST) 또는 모르폴리노황 트리플루오라이드 (모르포-DAST)를 사용하는 것이 또한 가능하다 (반응 순서 VIII → IX → VII-A에 대해, 예를 들어 문헌 [T. Mase et al., J. Org. Chem. 66 (20), 6775-6786 (2001)] 참조).
화학식 III의 중간체는, 예를 들어
[A] 하기 화학식 X의 포스포노아세트산 에스테르를 불활성 용매 중에서 염기-유도된 올레핀화 반응으로 하기 화학식 XI의 3-니트로벤조일 화합물과 반응시켜 하기 화학식 XII의 화합물을 수득하고, 이어서 적합한 팔라듐 또는 백금 촉매의 존재 하에 이를 수소화시켜, 하기 화학식 III-A의 3-(3-아미노페닐)프로피온산 에스테르를 수득함으로써 제조될 수 있거나, 또는
<화학식 X>

(상기 식에서, R1A 및 T1은 상기 주어진 의미를 갖고,
R8은 (C1-C4)-알킬을 나타냄)
<화학식 XI>

(상기 식에서, R2A, R3 및 R4는 상기 주어진 의미를 가짐)
<화학식 XII>

(상기 식에서, R1A, R2A, R3, R4 및 T1은 상기 주어진 의미를 가짐)
<화학식 III-A>

(상기 식에서, R1A, R2A, R3, R4 및 T1은 상기 주어진 의미를 가짐)
[B] 하기 화학식 XIII의 아크릴산 에스테르를 불활성 용매 중에서 (i) 로듐(I) 촉매작용 하에 하기 화학식 XIV의 페닐보론산과 반응시키거나, 또는 (ii) 구리(I) 촉매작용 하에 하기 화학식 XV의 페닐마그네슘 시약과 반응시켜 하기 화학식 XVI의 화합물을 수득하고, 후속으로 아미노보호기 PG를 가수소분해에 의해 또는 산화적으로 통상적인 방법에 따라 제거하여, 하기 화학식 III-B의 3-(3-아미노페닐)프로피온산 에스테르를 수득함으로써 제조될 수 있거나, 또는
<화학식 XIII>

(상기 식에서, R1A, R2A, R2B 및 T1은 상기 주어진 의미를 가짐)
<화학식 XIV>

(상기 식에서, R3 및 R4는 상기 주어진 의미를 갖고,
PG는 불활성 아미노보호기로서의 벤질 또는 p-메톡시벤질을 나타냄)
<화학식 XV>

(상기 식에서, R3, R4 및 PG가 상기 주어진 의미를 갖고,
Hal1은 염소 또는 브로민을 나타냄)
<화학식 XVI>

(상기 식에서, R1A, R2A, R2B, R3, R4, PG 및 T1은 상기 주어진 의미를 가짐)
<화학식 III-B>

(상기 식에서, R1A, R2A, R2B, R3, R4 및 T1은 상기 주어진 의미를 가짐)
[C] 하기 화학식 XVII의 아크릴산 에스테르를 불활성 용매 중에서 팔라듐 촉매작용 하에 하기 화학식 XVIII의 3-아미노- 또는 3-니트로페닐 브로마이드와 커플링시켜, 하기 화학식 XIX의 화합물을 수득하고, 이를 적합한 팔라듐 또는 백금 촉매의 존재 하에 수소화시켜, 하기 화학식 III-C의 3-(3-아미노페닐)프로피온산 에스테르를 수득함으로써 제조될 수 있거나, 또는
<화학식 XVII>

(상기 식에서, R1A, R2A 및 T1은 상기 주어진 의미를 가짐)
<화학식 XVIII>

(상기 식에서, R3 및 R4는 상기 주어진 의미를 갖고,
R9는 아미노 또는 니트로를 나타냄)
<화학식 XIX>

(상기 식에서, R1A, R2A, R3, R4, R9 및 T1은 상기 주어진 의미를 가짐)
<화학식 III-C>

(상기 식에서, R1A, R2A, R3, R4 및 T1은 상기 주어진 의미를 가짐)
[D] 하기 화학식 XX의 에스테르를 α-탈양성자화 후에 불활성 용매 중에서 하기 화학식 XXI의 3-브로모벤질 할라이드를 사용하여 알킬화하여, 하기 화학식 XXII의 화합물을 수득하고, 이어서 이를 염기 및 팔라듐 촉매의 존재 하에 벤질아민과 반응시켜 하기 화학식 XXIII의 화합물을 수득하고, 이어서 N-벤질 기를 가수소분해에 의해 제거하여 하기 화학식 III-D의 3-(3-아미노페닐)프로피온산 에스테르를 수득함으로써 제조될 수 있다.
<화학식 XX>

(상기 식에서, R1A, R1B 및 T1은 상기 주어진 의미를 가짐)
<화학식 XXI>

(상기 식에서, R3 및 R4는 상기 주어진 의미를 갖고,
Hal2는 염소, 브로민 또는 아이오딘을 나타냄)
<화학식 XXII>

(상기 식에서, R1A, R1B, R3, R4 및 T1은 상기 주어진 의미를 가짐)
<화학식 XXIII>

(상기 식에서, R1A, R1B, R3, R4 및 T1은 상기 주어진 의미를 가짐)
<화학식 III-D>

(상기 식에서, R1A, R1B, R3, R4 및 T1은 상기 주어진 의미를 가짐)
올레핀화 반응 X + XI → XII에서의 포스폰산 에스테르 X의 탈양성자화에는 특히 비-친핵성 강염기, 예컨대 예를 들어 수소화나트륨 또는 수소화칼륨, 리튬 비스(트리메틸실릴)아미드, 나트륨 비스(트리메틸실릴)아미드 또는 칼륨 비스(트리메틸실릴)아미드 또는 리튬 디이소프로필아미드가 적합하고; 수소화나트륨을 사용하는 것이 바람직하다.
공정 단계 XII → III-A, 또는 XIX → III-C에서의 수소화는 일반적으로 대기압에서 고정 수소 분위기 하에 수행된다. 여기서, 사용된 촉매는 바람직하게는 (지지체로서의) 활성 탄소 상 팔라듐이다. 변환 XVI → III-B, 및 XXIII → III-D에서의 아미노보호기(들)의 제거는 통상적으로 동일한 절차에 따른 가수소분해에 의해 수행되며; XVI에서의 PG가 p-메톡시벤질을 나타내는 경우에 이는 또한 대안적으로, 예를 들어 2,3-디클로로-5,6-디시아노-1,4-벤조퀴논 (DDQ) 또는 질산세륨(IV)암모늄의 보조 하에 산화적으로 수행될 수 있다.
반응 XVII + XVIII → XIX [헤크(Heck) 반응]에 대해 바람직한 팔라듐 촉매는 포스핀 리간드 예컨대 예를 들어 트리페닐- 또는 트리-2-톨릴포스핀과 조합된 아세트산팔라듐(II)이다 (반응 XIII + XIV → XVI에 대해서는, 예를 들어 문헌 [N. Miyaura et al., Organometallics 16, 4229 (1997)] 및 또한 [T. Hayashi, Synlett, Special Issue 2001, 879-887] 참조; 반응 XIII + XV → XVI에 대해서는, 예를 들어 문헌 [P. Knochel et al., Tetrahedron 56, 2727-2731 (2000)], [Angew. Chem. 120, 6907-6911 (2008)] 참조).
마찬가지로, 알킬화 반응 XX + XXI → XXII에서의 에스테르 XX의 α-탈양성자화에는 비-친핵성 강염기, 예컨대 예를 들어 수소화나트륨 또는 수소화칼륨, 리튬 비스(트리메틸실릴)아미드, 나트륨 비스(트리메틸실릴)아미드 또는 칼륨 비스(트리메틸실릴)아미드 또는 리튬 디이소프로필아미드가 특히 적합하고; 여기서, 리튬 디이소프로필아미드를 사용하는 것이 바람직하다.
반응 XXII + 벤질아민 → XXIII [부흐발트-하르트비히 커플링]에 대해, 팔라듐 촉매로서의 트리스(디벤질리덴아세톤)디팔라듐(0)을 포스핀 리간드로서의 (±)-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 및 염기로서의 나트륨 tert-부톡시드 또는 칼륨 tert-부톡시드와 조합하여 사용하는 것이 바람직하다 (예를 들어, 문헌 [J. P. Wolfe and S. L. Buchwald, Organic Syntheses, Coll. Vol. 10, 423 (2004), Vol. 78, 23 (2002)] 참조).
상기 기재된 공정 단계는 대기압 하에, 승압 하에 또는 감압 하에 (예를 들어, 0.5 내지 5 bar 범위에서) 수행될 수 있고; 일반적으로, 각 경우에 이는 대기압 하에 수행된다.
본 발명에 따른 화합물의 상응하는 거울상이성질체 및/또는 부분입체이성질체로의 분리가, 적절한 경우, 편의에 따라, 심지어 화합물 II, III, IV, VII, XVI, XXII 또는 XXIII의 단계에서 수행될 수 있고, 이어서 이를 상기 기재된 방법 순서에 따라 분리된 형태로 추가 반응시킨다. 입체이성질체의 이러한 분리는 당업자에게 공지된 통상적인 방법에 의해 수행될 수 있다. 비키랄 또는 키랄 분리 상 상에서 크로마토그래피 방법을 이용하는 것이 바람직하며; 중간체 또는 최종 생성물로서의 카르복실산의 경우, 분리는 또한 대안적으로 부분입체이성질체 염을 통해 이루어질 수 있다.
화학식 V, VI, VIII, X, XI, XIII, XIV, XV, XVII, XVIII, XX 및 XXI의 화합물은 상업적으로 입수가능하거나, 문헌에 기재되어 있거나, 또는 문헌에 공개된 방법와 유사하게 당업자에게 명백한 방식으로 제조될 수 있다. 출발 물질을 제조하기 위한 다수의 상세 절차 및 참고문헌은 또한 출발 물질 및 중간체의 제조에 대한 섹션 내의 실험 부분에서 찾아볼 수 있다.
본 발명에 따른 화합물의 제조는 하기 반응식에 의해 예시적 방식으로 설명될 수 있다.
<반응식 1>

<반응식 2>

<반응식 3>
<반응식 4>

<반응식 5>

[PMB = p-메톡시벤질; A = CH2 또는 O; R = 메틸 또는 벤질].
<반응식 6>

[Bn = 벤질].
<반응식 7>

<반응식 8>

<반응식 9>

본 발명에 따른 화합물은 유익한 약리 특성을 가지며, 인간 및 동물에서 장애의 예방 및 치료에 사용될 수 있다.
본 발명에 따른 화합물은 가용성 구아닐레이트 시클라제의 강력한 활성화제이다. 이는 혈관이완, 혈소판 응집의 억제 및 혈압의 저하 및 관상동맥 혈류의 증가를 야기한다. 이러한 효과는 가용성 구아닐레이트 시클라제의 직접적 헴-비의존성 활성화 및 세포내 cGMP의 증가에 의해 매개된다.
또한 본 발명에 따른 화합물은, 특히 그의 신체 내 생체이용률 및 반감기와 관련하여, 우수한 약동학 특성을 갖는다.
따라서, 본 발명에 따른 화합물은 심혈관 장애, 예컨대 예를 들어, 높은 혈압 (고혈압) 및 심부전, 안정형 및 불안정형 협심증, 폐 동맥 고혈압 (PAH) 및 다른 형태의 폐고혈압 (PH), 신성 고혈압, 말초 및 심장 혈관 장애 및 또한 부정맥의 치료 및/또는 예방, 혈전색전성 장애 및 허혈, 예컨대 심근경색, 졸중, 일과성 허혈 발작 및 또한 말초 혈류 장애의 치료, 혈전용해 요법, 경피 경혈관 혈관성형술 (PTA), 경피 경관 관상 동맥성형술 (PTCA) 및 우회술 이후의 재협착 방지, 동맥경화증의 치료, 상처 치유의 촉진 및 골다공증, 녹내장 및 위마비의 치료를 위한 의약에 사용될 수 있다.
본 발명의 문맥에서, 용어 심부전은 심부전의 급성 및 만성 둘 모두의 징후, 뿐만 아니라 보다 특정한 또는 관련된 유형의 질환, 예컨대 급성 비대상성 심부전, 우심부전, 좌심부전, 전체 심부전, 허혈성 심근병증, 확장성 심근병증, 비후성 심근병증, 특발성 심근병증, 선천성 심장 결손, 심장 판막 결손, 심장 판막 결손과 관련된 심부전, 승모판 협착, 승모판 폐쇄부전, 대동맥 협착, 대동맥 폐쇄부전, 삼천판 협착, 삼천판 폐쇄부전, 폐동맥 협착, 폐동맥판 폐쇄부전, 복합 심장 판막 결손, 심근 염증 (심근염), 만성 심근염, 급성 심근염, 바이러스성 심근염, 당뇨병 심부전, 알콜성 심근병증, 심장 축적 장애 및 또한 확장기 및 수축기 심부전을 포함한다.
본 발명에 따른 화합물은 추가로, 원발성 및 속발성 레이노 현상, 미세순환 장애, 파행, 이명, 말초 및 자율 신경병증, 당뇨병성 미세혈관병증, 당뇨병성 망막병증, 사지의 당뇨병성 궤양, 크레스트 증후군, 홍반증, 조갑진균증 및 류마티스성 장애의 치료에 사용될 수 있다.
또한, 본 발명에 따른 화합물은, 특히 수술적 개입에서 또는 이식 의약의 분야에서, 기관 또는 조직에 대한 허혈- 및/또는 재관류-관련 손상의 방지를 위하여, 및 또한 인간 또는 동물 기원의 기관, 기관부, 조직 또는 조직부의 관류 및 보존액을 위한 첨가제로서 사용될 수 있다.
또한, 본 발명에 따른 화합물은 신장 질환, 특히 신기능부전 및 신부전의 치료 및/또는 예방에 적합하다. 본 발명의 문맥에서, 용어 신기능부전 및 신부전은 그의 급성 및 만성 둘 모두의 징후, 뿐만 아니라 기본적인 또는 관련된 신장 질환, 예컨대 신장 저관류, 투석중 고혈압, 폐쇄성 요로병증, 사구체병증, 사구체신염, 급성 사구체신염, 사구체경화증, 세뇨관간질 질환, 신병증성 질환, 예컨대 원발성 선천성 신장 질환, 신염, 면역 신장 질환, 예컨대 신장 이식편 거부 및 면역복합체-유발성 신장 질환, 독성 물질에 의해 유발된 신병증, 조영제에 의해 유발된 신병증, 당뇨병성 및 비-당뇨병성 신병증, 신우신염, 신낭종, 신경화증, 고혈압 신경화증, 및 진단학적으로 예를 들어 비정상적으로 감소된 크레아티닌 및/또는 물 배출, 우레아, 질소, 칼륨 및/또는 크레아티닌의 비정상적으로 상승된 혈중 농도, 신장 효소, 예를 들어 글루타밀 신테타제의 변경된 활성, 변경된 요삼투압 또는 뇨량을 특징으로 할 수 있는 신증후군, 상승된 미세알부민뇨증, 거대알부민뇨증, 사구체 및 세동맥에 대한 병변, 세뇨관 확장, 고인산혈증 및/또는 투석의 필요를 포함한다. 본 발명은 또한 신기능부전의 후유증, 예를 들어 고혈압, 폐 부종, 심부전, 요독증, 빈혈, 전해질 장애 (예를 들어, 고칼슘혈증, 저나트륨혈증) 및 골 및 탄수화물 대사 장애의 치료 및/또는 예방을 위한 본 발명에 따른 화합물의 용도를 포함한다.
또한, 본 발명에 따른 화합물은 비뇨생식기계의 장애, 예컨대 예를 들어, 과민성 방광, 배뇨 장애, 하부 요로 증후군 (LUTS), 실금, 양성 전립선 비대증 (BPH), 발기 기능장애 및 여성 성 기능장애의 치료 및/또는 예방에 적합하다.
본 발명에 따른 화합물은 또한 천식성 장애, 만성 폐쇄성 폐 장애 (COPD) 및 호흡 곤란 증후군의 치료에 사용될 수 있다.
본 발명에 기재된 화합물은 또한 NO/cGMP 시스템의 교란을 특징으로 하는 중추신경계 질환을 제어하기 위한 활성 성분을 나타낸다. 이들은 경도 인지 손상, 연령-관련 학습 및 기억 손상, 연령-관련 기억 상실, 혈관성 치매, 두개뇌 외상, 졸중, 졸중 후에 발생하는 치매 (후-졸중 치매), 외상후 두개뇌 외상, 일반적 집중력 손상, 학습 및 기억 문제를 갖는 어린이에서의 집중력 손상, 알츠하이머병, 루이 소체 치매, 전두엽의 변성으로 인한 치매 예컨대 픽 증후군, 파킨슨병, 진행성 핵성 마비, 피질기저핵 변성으로 인한 치매, 근위축성 측삭 경화증 (ALS), 헌팅턴병, 다발성 경화증, 시상 변성, 크로이츠펠트-야콥 치매, HIV 치매, 치매로 인한 정신분열증 또는 코르사코프의 정신병과 같은 상황/질환/증후군과 연관되어 발생하는 것과 같은 인지 손상 후의 지각, 집중력, 학습 또는 기억의 개선에 특히 적합하다. 이들은 또한 불안, 긴장 및 우울증 상태, CNS-관련 성 기능장애 및 수면 장애와 같은 중추신경계 장애의 치료, 및 식품, 자극제 및 중독성 물질 흡입의 병리학적 교란 조절에 적합하다.
본 발명에 따른 화합물은 또한 또한 뇌 혈류를 제어하는데 적합하며, 따라서 편두통의 제어에 효과적인 작용제이다. 이들은 또한 뇌경색 (뇌졸중)의 후유증, 예컨대 졸중, 뇌 허혈 및 두개뇌 외상의 예방 및 조절에서 적합하다. 본 발명에 따른 화합물은 또한 통증 상태의 조절에 사용될 수 있다.
또한, 본 발명에 따른 화합물은 항염증 작용을 가지며, 따라서 패혈증, 다발성 기관 부전, 신장의 염증성 장애, 만성 장 염증, 예컨대 궤양성 결장염 및 크론병, 췌장염, 복막염, 류마티스 장애, 염증성 피부 질환 및 염증성 안질환의 치료 및/또는 예방을 위한 항염증제로서 사용될 수 있다.
본 발명에 따른 화합물은 그의 활성 프로파일로 인하여, 심혈관 장애, 예컨대 심부전, 협심증, 고혈압, 폐고혈압, 허혈, 혈관 장애, 미세순환 장애, 혈전색전성 장애 및 동맥경화증의 치료 및/또는 예방에 특히 적합하다.
본 발명은 또한 장애, 특히 상기 언급된 장애의 치료 및/또는 예방에서의 본 발명에 따른 화합물의 용도에 관한 것이다.
본 발명은 또한 장애, 특히 상기 언급된 장애의 치료 및/또는 예방을 위한 의약의 제조에서의 본 발명에 따른 화합물의 용도에 관한 것이다.
본 발명은 또한 장애, 특히 상기 언급된 장애의 치료 및/또는 예방 방법에서의 본 발명에 따른 화합물의 용도에 관한 것이다.
본 발명은 또한 유효량의 본 발명에 따른 하나 이상의 화합물을 사용하여, 장애, 특히 상기 언급된 장애를 치료 및/또는 예방하는 방법에 관한 것이다.
본 발명에 따른 화합물은 단독으로, 또는 필요한 경우 다른 활성 화합물과 조합되어 사용될 수 있다. 추가로, 본 발명은 하나 이상의 본 발명에 따른 화합물, 및 하나 이상의 추가의 활성 성분을 포함하는, 특히 상기 장애의 치료 및/또는 예방을 위한 의약에 관한 것이다. 바람직하게 언급할 수 있는 적합한 조합 활성 화합물의 예는 다음과 같다:
ㆍ 유기 니트레이트 및 NO 공여자, 예를 들어 나트륨 니트로프루시드, 니트로글리세린, 이소소르비드 모노니트레이트, 이소소르비드 디니트레이트, 몰시도민 또는 SIN-1 및 흡입 NO;
ㆍ 시클릭 구아노신 모노포스페이트 (cGMP)의 분해를 억제하는 화합물, 예를 들어 포스포디에스테라제 (PDE) 1, 2 및/또는 5의 억제제, 특히 PDE 5 억제제, 예컨대 실데나필, 바르데나필 및 타달라필;
ㆍ 구아닐레이트 시클라제의 NO-비의존성이지만 헴-의존성인 자극물질, 예컨대 특히 WO 00/06568, WO 00/06569, WO 02/42301 및 WO 03/095451에 기재된 화합물;
ㆍ 항혈전 작용을 갖는 작용제, 예를 들어 바람직하게는 혈소판 응집 억제제, 항응고제 또는 전섬유소용해 물질의 군으로부터의 작용제;
ㆍ 혈압을 강하시키는 활성 성분, 예를 들어 바람직하게는 칼슘 길항제, 안지오텐신 AII 길항제, ACE 억제제, 엔도텔린 길항제, 레닌 억제제, 알파-수용체 차단제, 베타-수용체 차단제, 미네랄로코르티코이드 수용체 길항제 및 이뇨제의 군으로부터의 활성 성분; 및/또는
ㆍ 지질 대사를 변경시키는 활성 성분, 예를 들어 바람직하게는 갑상선 수용체 효능제, 콜레스테롤 합성 억제제, 예컨대, 예를 들어 바람직하게는, HMG-CoA 리덕타제 억제제 또는 스쿠알렌 합성 억제제, ACAT 억제제, CETP 억제제, MTP 억제제, PPAR-알파, PPAR-감마 및/또는 PPAR-델타 효능제, 콜레스테롤 흡수 억제제, 리파제 억제제, 중합체성 담즙산 흡착제, 담즙산 재흡수 억제제 및 지단백질 (a) 길항제의의 군으로부터의 활성 성분.
항혈전 활성을 갖는 작용제는 바람직하게는 혈소판 응집 억제제, 항응고제 또는 전섬유소용해 물질의 군으로부터의 화합물을 의미한다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 혈소판 응집 억제제, 예컨대, 예를 들어 바람직하게는, 아스피린, 클로피도그렐, 티클로피딘 또는 디피리다몰과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 트롬빈 억제제, 예컨대, 예를 들어 바람직하게는, 크시멜라가트란, 멜라가트란, 비발리루딘 또는 크렉산과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 GPIIb/IIIa 길항제, 예컨대, 예를 들어 바람직하게는, 티로피반 또는 압식시맙과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 인자 Xa 억제제, 예컨대, 예를 들어 바람직하게는, 리바록사반, 아픽사반, 피덱사반, 라작사반, 폰다파리눅스, 이드라파리눅스, DU-176b, PMD-3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 또는 SSR-128428과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 헤파린 또는 저분자량 (LMW) 헤파린 유도체와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 비타민 K 길항제, 예컨대, 예를 들어 바람직하게는, 쿠마린과 조합하여 투여된다.
혈압을 강하시키는 작용제는 바람직하게는 칼슘 길항제, 안지오텐신 AII 길항제, ACE 억제제, 엔도텔린 길항제, 레닌 억제제, 알파-수용체 차단제, 베타-수용체 차단제, 미네랄로코르티코이드 수용체 길항제, 및 이뇨제의 군으로부터의 화합물을 의미한다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 칼슘 길항제, 예컨대, 예를 들어 바람직하게는, 니페디핀, 암로디핀, 베라파밀 또는 딜티아젬과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 알파-1-수용체 차단제, 예컨대, 예를 들어 바람직하게는, 프라조신과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 베타-수용체 차단제, 예컨대, 예를 들어 바람직하게는, 프로프라놀롤, 아테놀롤, 티몰롤, 핀돌롤, 알프레놀롤, 옥스프레놀롤, 펜부톨롤, 부프라놀롤, 메티프라놀롤, 나돌롤, 메핀돌롤, 카라잘롤, 소탈롤, 메토프롤롤, 베탁솔롤, 셀리프롤롤, 비소프롤롤, 카르테올롤, 에스몰롤, 라베탈롤, 카르베딜롤, 아다프롤롤, 란디올롤, 네비볼롤, 에파놀롤 또는 부신돌롤과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 안지오텐신 AII 길항제, 예컨대, 예를 들어 바람직하게는, 로사르탄, 칸데사르탄, 발사르탄, 텔미사르탄 또는 엠부사르탄과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 ACE 억제제, 예컨대, 예를 들어 바람직하게는, 에날라프릴, 카프토프릴, 리시노프릴, 라미프릴, 델라프릴, 포시노프릴, 퀴노프릴, 페린도프릴 또는 트란도프릴과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 엔도텔린 길항제, 예컨대, 예를 들어 바람직하게는, 보센탄, 다루센탄, 암브리센탄 또는 시탁센탄과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 레닌 억제제, 예컨대, 예를 들어 바람직하게는, 알리스키렌, SPP-600 또는 SPP-800과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 미네랄로코르티코이드 수용체 길항제, 예컨대, 예를 들어 바람직하게는, 스피로놀락톤 또는 에플레레논과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 이뇨제, 예컨대, 예를 들어 바람직하게는, 푸로세미드와 조합하여 투여된다.
지질 대사를 변경시키는 작용제는 바람직하게는 CETP 억제제, 갑상선 수용체 효능제, 콜레스테롤 합성 억제제, 예컨대 HMG-CoA 리덕타제 억제제 또는 스쿠알렌 합성 억제제, ACAT 억제제, MTP 억제제, PPAR-알파, PPAR-감마 및/또는 PPAR-델타 효능제, 콜레스테롤 흡수 억제제, 중합체성 담즙산 흡착제, 담즙산 재흡수 억제제, 리파제 억제제 및 지단백질 (a) 길항제의 군으로부터의 화합물을 의미한다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 CETP 억제제, 예컨대, 예를 들어 바람직하게는, 토르세트라피브 (CP-529 414), JJT-705 또는 CETP 백신 (아반트(Avant))과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 갑상선 수용체 효능제, 예컨대, 예를 들어 바람직하게는, D-티록신, 3,5,3'-트리아이오도티로닌 (T3), CGS 23425 또는 악시티롬 (CGS 26214)과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 스타틴 클래스로부터의 HMG-CoA 리덕타제 억제제, 예컨대, 예를 들어 바람직하게는 로바스타틴, 심바스타틴, 프라바스타틴, 플루바스타틴, 아토르바스타틴, 로수바스타틴 또는 피타바스타틴과 조합되어 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 스쿠알렌 합성 억제제, 예컨대, 예를 들어 바람직하게는, BMS-188494 또는 TAK-475와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 ACAT 억제제, 예컨대, 예를 들어 바람직하게는, 아바시미브, 멜린아미드, 팩티미브, 에플루시미브 또는 SMP-797과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 MTP 억제제, 예컨대, 예를 들어 바람직하게는, 임플리타피드, BMS-201038, R-103757 또는 JTT-130과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 PPAR-감마 효능제, 예컨대, 예를 들어 바람직하게는, 피오글리타존 또는 로시글리타존과 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 PPAR-델타 효능제, 예컨대, 예를 들어 바람직하게는, GW 501516 또는 BAY 68-5042와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 콜레스테롤 흡수 억제제, 예컨대, 예를 들어 바람직하게는, 에제티미브, 티퀘시드 또는 파마퀘시드와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 리파제 억제제, 예컨대, 예를 들어 바람직하게는, 오를리스타트와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 중합체성 담즙산 흡착제, 예컨대, 예를 들어 바람직하게는, 콜레스티라민, 콜레스티폴, 콜레솔밤, 콜레스타겔 또는 콜레스티미드와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 담즙산 재흡수 억제제, 예컨대, 예를 들어 바람직하게는, ASBT (= IBAT) 억제제, 예컨대, 예를 들어, AZD-7806, S-8921, AK-105, BARI-1741, SC-435 또는 SC-635와 조합하여 투여된다.
본 발명의 바람직한 실시양태에서, 본 발명에 따른 화합물은 지단백질 (a) 길항제, 예컨대, 예를 들어 바람직하게는, 겜카벤 칼슘 (CI-1027) 또는 니코틴산과 조합하여 투여된다.
본 발명은 또한 하나 이상의 본 발명에 따른 화합물을, 통상적으로 하나 이상의 제약상 적합한 비-독성의 불활성 보조제와 함께 포함하는 의약, 및 상기 목적을 위한 그의 용도에 관한 것이다.
본 발명에 따른 화합물은 전신적으로 및/또는 국소적으로 작용할 수 있다. 이러한 목적을 위해, 이를 적합한 방식으로, 예를 들어, 경구, 비경구, 폐, 비강, 설하, 설측, 협측, 직장, 피부, 경피, 결막, 귀 경로로, 또는 임플란트 또는 스텐트로서 투여할 수 있다.
본 발명에 따른 화합물은 이러한 투여 경로에 적합한 투여 형태로 투여될 수 있다.
경구 투여에 적합한 것은 선행기술에 따라 기능하며 본 발명에 따른 화합물을 신속하게 및/또는 변형된 방식으로 전달하는, 본 발명에 따른 화합물을 결정질 및/또는 무정형 및/또는 용해된 형태로 함유하는 투여 형태, 예를 들어, 정제 (비코팅 또는 코팅 정제, 예컨대 본 발명에 따른 화합물의 방출을 지연 및 제어하는 불용성 또는 용해성 코팅, 또는 장용 코팅을 갖는 정제), 구강 내에서 신속하게 붕해되는 정제, 또는 필름/웨이퍼, 필름/동결건조물, 캡슐 (예를 들어, 경질 또는 연질 젤라틴 캡슐), 당-코팅 정제, 과립, 펠릿, 분말, 에멀젼, 현탁액, 에어로졸 또는 용액이다.
비경구 투여는 흡수 단계 없이 (예를 들어, 정맥내, 동맥내, 심장내, 척수내 또는 요추내), 또는 흡수 단계를 포함하여 (예를 들어, 근육내, 피하, 피내, 경피 또는 복강내) 수행될 수 있다. 비경구 투여에 적합한 투여 형태는 특히 용액, 현탁액, 에멀젼, 동결건조물 또는 멸균 분말 형태의 주사 및 주입을 위한 제제이다.
그밖의 투여 경로에 적합한 것은, 예를 들어, 흡입용 약제 형태 (특히, 분말 흡입기, 네뷸라이저), 점비제, 용액 또는 스프레이; 설측, 설하 또는 협측 투여용 정제, 필름/웨이퍼 또는 캡슐, 좌제, 귀 또는 눈을 위한 제제, 질 캡슐, 수현탁액 (로션, 진탕 혼합물), 친유성 현탁액, 연고, 크림, 경피 치료 시스템 (예를 들어, 패치), 유제, 페이스트, 발포체, 살포용 분말, 임플란트 또는 스텐트이다.
경구 또는 비경구 투여가 바람직하고, 특히 경구 및 정맥내 투여가 바람직하다.
본 발명에 따른 화합물을 언급된 투여 형태로 전환시킬 수 있다. 이는 공지된 방식 그 자체로 불활성, 비독성, 제약상 적합한 부형제와 혼합함으로써 수행될 수 있다. 이들 부형제는 특히 담체 (예를 들어 미세결정질 셀룰로스, 락토스, 만니톨), 용매 (예를 들어 액체 폴리에틸렌 글리콜), 유화제 및 분산제 또는 습윤제 (예를 들어 나트륨 도데실 술페이트, 폴리옥시소르비탄 올레에이트), 결합제 (예를 들어 폴리비닐피롤리돈), 합성 및 천연 중합체 (예를 들어 알부민), 안정화제 (예를 들어 항산화제, 예를 들어 아스코르브산), 착색제 (예를 들어 무기 안료, 예컨대 산화철) 및 향 및/또는 냄새 차폐제를 포함한다.
일반적으로, 비경구 투여의 경우 효과적인 결과를 달성하기 위해 약 0.001 내지 1 mg/체중 kg, 바람직하게는 약 0.01 내지 0.5 mg/체중 kg의 양을 투여하는 것이 유리하며, 경구 투여의 경우, 투여량이 약 0.01 내지 100 mg/체중 kg, 바람직하게는 약 0.01 내지 20 mg/체중 kg, 매우 특히 바람직하게는 0.1 내지 10 mg/체중 kg인 것이 유리한 것으로 입증되었다.
그럼에도 불구하고, 적절한 경우, 특히 체중, 투여 경로, 활성 성분에 대한 개별 반응, 제제의 성질, 및 투여가 수행되는 시간 또는 간격의 함수로서, 언급된 양으로부터 벗어나는 것이 필요할 수 있다. 따라서, 일부 경우에서 상기 언급한 최소량 미만으로 실시하는 것으로 충분할 수 있는 한편, 다른 경우에서는 상기 명시한 상한값을 초과해야 한다. 더 많은 양을 투여하는 경우에는 이것을 하루에 걸쳐서 복수개의 개별 용량으로 나누는 것이 바람직할 수 있다.
하기 예시적인 실시양태는 본 발명을 설명한다. 본 발명은 하기 실시예에 의해 제한되지 않는다.
달리 나타내지 않는 한, 하기 시험 및 실시예에서의 백분율 데이터는 중량%이고, 부는 중량부이다. 액체/액체 용액에 대한 용매 비, 희석 비 및 농도 데이터는 각 경우에서 부피에 기초한다.
A. 실시예
약어 및 두문자어:


GC-MS 및 LC-MS 방법:
방법 1 (GC-MS):
기기: 마이크로매스(Micromass) GCT, GC 6890; 칼럼: 레스텍(Restek) RTX-35, 15 m x 200 μm x 0.33 μm; 일정한 헬륨 유량: 0.88 ml/분; 오븐: 70℃; 주입구: 250℃; 구배: 70℃, 30℃/분 → 310℃는 (3분 동안 유지).
방법 2 (LC-MS):
MS 기기 유형: 마이크로매스 ZQ; HPLC 기기 유형: HP 1100 시리즈; UV DAD; 칼럼: 페노메넥스 제미니(Phenomenex Gemini) 3 μ 30 mm x 3.00 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 90% A → 2.5 분 30% A → 3.0 분 5% A → 4.5 분 5% A; 유량: 0.0 분 1 ml/분 → 2.5 분/3.0 분/4.5 분 2 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 3 (LC-MS):
MS 기기 유형: 마이크로매스 ZQ; HPLC 기기 유형: 워터스 얼라이언스(Waters Alliance) 2795; 칼럼: 페노메넥스 시너지(Synergi) 2.5 μ MAX-RP 100A 머큐리(Mercury) 20 mm x 4 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 90% A → 0.1 분 90% A → 3.0 분 5% A → 4.0 분 5% A → 4.01 분 90% A; 유량: 2 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 4 (LC-MS):
기기: 워터스 UPLC 액퀴티(Acquity)가 구비된 마이크로매스 쿼트로 프리미어(Micromass Quattro Premier); 칼럼: 써모 하이퍼실 골드(Thermo Hypersil GOLD) 1.9 μ 50 mm x 1 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 90% A → 0.1 분 90% A → 1.5 분 10% A → 2.2 분 10% A; 유량: 0.33 ml/분; 오븐:50℃; UV 검출: 210 nm.
방법 5 (LC-MS):
MS 기기 유형: 워터스 마이크로매스 쿼트로 마이크로(Quattro Micro); HPLC 기기 유형: 애질런트(Agilent) 1100 시리즈; 칼럼: 써모 하이퍼실 골드 3 μ 20 mm x 4 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 100% A → 3.0 분 10% A → 4.0 분 10% A → 4.01 분 100% A (유량 2.5 ml/분) → 5.00 분 100% A; 오븐: 50℃; 유량: 2 ml/분; UV 검출: 210 nm.
방법 6 (LC-MS):
기기: 워터스 액퀴티 SQD UPLC 시스템; 칼럼: 워터스 액퀴티 UPLC HSS T3 1.8 μ, 50 mm x 1 mm; 이동상 A: 물 1 l + 99% 농도 포름산 0.25 ml, 이동상 B: 아세토니트릴 1 l + 99% 농도 포름산 0.25 ml; 구배: 0.0 분 90% A → 1.2 분 5% A → 2.0 분 5% A; 유량: 0.40 ml/분; 오븐: 50℃; UV 검출: 210-400 nm.
방법 7 (LC-MS):
MS 기기 유형: 워터스 ZQ; HPLC 기기 유형: 애질런트 1100 시리즈; UV DAD; 칼럼: 써모 하이퍼실 골드 3 μ 20 mm x 4 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 100% A → 3.0 분 10% A → 4.0 분 10% A → 4.1 분 100% A (유량 2.5 ml/분); 오븐: 55℃; 유량: 2 ml/분; UV 검출: 210 nm.
방법 8 (GC-MS):
기기: 마이크로매스 GCT, GC 6890; 칼럼: 레스텍 RTX-35, 15 m x 200 μm x 0.33 μm; 일정한 헬륨 유량: 0.88 ml/분; 오븐: 70℃; 주입구: 250℃; 구배: 70℃, 30℃/분 → 310℃ (12분 동안 유지).
방법 9 (LC-MS):
기기: 워터스 UPLC 액퀴티가 구비된 마이크로매스 쿼트로 프리미어; 칼럼: 써모 하이퍼실 골드 1.9 μ 50 mm x 1 mm; 이동상 A: 물 1 l + 50% 농도 포름산 0.5 ml, 이동상 B: 아세토니트릴 1 l + 50% 농도 포름산 0.5 ml; 구배: 0.0 분 90% A → 0.3 분 90% A → 3.0 분 10% A → 4.8 분 10% A; 유량: 0.33 ml/분; 오븐: 50℃; UV 검출: 210 nm.
방법 10 (GC-MS):
기기: 써모 DFS, 트레이스 GC 울트라(Trace GC Ultra); 칼럼: 레스텍 RTX-35, 15 m x 200 μm x 0.33 μm; 일정한 헬륨 유량: 1.20 ml/분; 오븐: 60℃; 주입구: 220℃; 구배: 60℃, 30℃/분 → 300℃ (3.33분 동안 유지).
출발 물질 및 중간체:
실시예 1A
tert-부틸 3-(3-아미노-2-메틸페닐)프로파노에이트

아르곤 하에, tert-부틸-프로프-2-에노에이트 201 ml (1.39 mol)를 DMF 2 리터 중 1-브로모-2-메틸-3-니트로벤젠 100 g (463 mmol), 트리에틸아민 322 ml (2.31 mol), 트리-2-톨릴포스핀 28.18 g (92.58 mmol) 및 아세트산팔라듐(II) 10.39 g (46.29 mmol)의 용액에 적가하고, 이어서 혼합물을 125℃에서 36 시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 포화 수성 염화암모늄 용액과 함께 교반하고, 유기 상을 분리하였다. 수성 상을 tert-부틸 메틸 에테르로 3회 추출하고, 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하여 건조상태로 만들었다. 수득한 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 석유 에테르/에틸 아세테이트 9:1). 이로써 중간체 tert-부틸-(2E)-3-(2-메틸-3-니트로페닐)프로프-2-에노에이트 89 g (338 mmol, 이론치의 73%)을 무색 고체로서 수득하였다. 상기 고체 88 g (334 mmol)을 에탄올 2 리터에 용해시키고, 탄소 상 팔라듐 (10%) 7 g을 실온에서 첨가하고, 혼합물을 대기압 하에 18 시간 동안 수소화시켰다. 전환이 완료된 후, 반응 용액을 규조토를 통해 여과하고, 수득한 여과물을 감압 하에 농축시켰다. 이로써 표제 화합물 61.3 g (260.5 mmol, 이론치의 78%)을 무색 고체로서 수득하였다.

실시예 2A
에틸 3-(3-아미노-2-메틸페닐)프로파노에이트

아르곤 하에, 에틸 프로프-2-에노에이트 10.844 g (108 mmol)을 DMF 200 ml 중 1-브로모-2-메틸-3-니트로벤젠 7.8 g (36.1 mmol), 트리에틸아민 25 ml (180.5 mmol), 트리-2-톨릴포스핀 2.197 g (7.22 mmol) 및 아세트산팔라듐(II) 810 mg (3.6 mmol)의 용액에 적가하고, 이어서 혼합물을 125℃에서 36 시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 포화 수성 염화암모늄 용액과 함께 교반하고, 유기 상을 분리하였다. 수성 상을 tert-부틸 메틸 에테르로 3회 추출하고, 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하여 건조상태로 만들었다. 수득한 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 석유 에테르/에틸 아세테이트 3:1). 이로써 중간체 에틸 (2E)-3-(2-메틸-3-니트로페닐)프로프-2-에노에이트 6.6 g (27.2 mmol, 함량 97%, 이론치의 75%)을 무색 고체로서 수득하였다. 상기 고체 6.6 g (27.2 mmol, 함량 97%)을 에탄올 200 ml에 용해시키고, 탄소 상 팔라듐 (10%) 500 mg을 실온에서 첨가하고, 혼합물을 대기압 하에 밤새 수소화시켰다. 반응이 완료된 후, 반응 용액을 규조토를 통해 여과하고, 수득한 여과물을 감압 하에 농축시켰다. 이로써 표제 화합물 5.47 g (26.38 mmol, 함량 97%, 이론치의 97%)을 무색 고체로서 수득하였다.
실시예 3A
tert-부틸 (2E)-3-(4-플루오로-3-니트로페닐)아크릴레이트

아르곤 하에, 먼저 수소화나트륨 (미네랄 오일 중 60% 현탁액으로서) 0.65 g (16.3 mmol)을 THF 25 ml에 충전하고, 0℃로 냉각시켰다. 이어서, tert-부틸 디에틸포스포노아세테이트 4.29 g (17 mmol)을 서서히 적가하였다. 30 분 후, 4-플루오로-3-니트로벤즈알데히드 2.5 g (14.8 mmol)을 첨가하였다. 반응 혼합물을 실온에서 3 시간 동안 교반하고, 이어서 물 100 ml에 붓고, 에틸 아세테이트 (각 경우에 100 mL)로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축시켰다. 잔류물을 플래쉬 크로마토그래피에 의해 정제하였다 (실리카 겔, 이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 표제 화합물 3.37 g (이론치의 85%)을 수득하였다.

실시예 4A
tert-부틸 3-(3-아미노-4-플루오로페닐)프로파노에이트

tert-부틸 (2E)-3-(4-플루오로-3-니트로페닐)프로프-2-에노에이트 535 mg (2.00 mmol)을 에탄올 1 ml 및 THF 1 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 21.3 mg을 첨가하였다. 실온에서, 혼합물을 수소의 분위기 하에 대기압에서 밤새 수소화시켰다. 이어서, 반응 혼합물을 규조토를 통한 흡인으로 여과하고, 잔류물을 THF로 세척하고, 여과물을 농축시켰다. 이로써 표제 화합물 479 mg (이론치의 100%)을 수득하였다.

실시예 5A
tert-부틸 (2E)-3-(4-클로로-3-니트로페닐)프로프-2-에노에이트

아르곤 하에, 수소화나트륨 1.19 g (29.64 mmol, 60%)을 톨루엔 25 ml 및 THF 25 ml 중에 현탁시키고, 혼합물을 0℃로 냉각시켰다. 이어서, tert-부틸 (디에톡시포스포릴)아세테이트 7.28 ml (30.99 mmol)를 서서히 적가하고, 혼합물을 0℃에서 30 분 동안 교반하였다. 이어서, 4-클로로-3-니트로벤즈알데히드 5 g (26.94 mmol)을 반응 혼합물에 첨가하고, 후속으로 혼합물을 실온으로 가온하였다. 혼합물을 실온에서 2 시간 동안 교반하고, 이어서 물 50 ml를 첨가하였다. 유기 상을 분리하고, 이어서 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 9:1). 이로써 표제 화합물 6.77 g (23.86 mmol, 이론치의 77%)을 수득하였다.

실시예 6A
tert-부틸-3-(3-아미노-4-클로로페닐)프로파노에이트

실온에서, 탄소 상 팔라듐 (10%) 500 mg을 에탄올 200 ml 및 THF 20 ml 중 tert-부틸 (2E)-3-(4-클로로-3-니트로페닐)프로프-2-에노에이트 6.74 g (23.76 mmol)의 용액에 첨가하고, 혼합물을 대기압 하에 12 시간 동안 수소화시켰다. 반응이 완료된 후 (TLC에 의해 모니터링; 이동상 시클로헥산/에틸 아세테이트 1:1), 반응 용액을 규조토를 통해 여과하고, 여과물을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 4:1 → 2:1). 이로써 표제 화합물 1.40 g (5.47 mmol, 이론치의 23%)을 수득하였다.

실시예 7A
메틸 3-(3-아미노-4-클로로페닐)프로파노에이트

환류 하에, 티오닐 클로라이드 0.86 ml (11.7 mmol)를 메탄올 20 ml 중 tert-부틸 3-(3-아미노-4-클로로페닐)프로파노에이트 1.0 g (3.91 mmol)의 용액에 적가하였다. 혼합물을 환류 하에 1.5 시간 동안 교반하고, 이어서 냉각시킨 후, 디클로로메탄으로 희석하였다. 용액을 물에 첨가하고, 상을 분리한 후, 유기 상을 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 고진공 하에 건조시켰다. 이로써 표적 화합물 745 mg (이론치의 90.3%)을 수득하였다.

실시예 8A
메틸 {1-[3-(디벤질아미노)-4-플루오로페닐]시클로프로필}아세테이트

용액 A의 제조: 아르곤 하에, 염화리튬 688 mg (16.2 mmol)을 THF 50 ml 중에 용해시키고, 이어서, 마그네슘 터닝 789 mg (32.5 mmol) 및 THF 중 디이소부틸알루미늄 히드라이드의 1 M 용액 23 μl (0.023 mmol)를 첨가하였다. 반응 용액을 실온에서 10 분 동안 교반하고, 이어서 -10℃로 냉각시켰다. 이어서, N,N-디벤질-5-브로모-2-플루오로아닐린 (CAS 등록 번호 869529-97-5) 5 g (13.5 mmol)을 첨가하고, 용액을 -10℃에서 약 1 시간 동안 교반하였다.
용액 B의 제조: 아르곤 하에, 염화리튬 110 mg (2.6 mmol) 및 염화구리(I) 128 mg (1.3 mmol)을 실온에서 THF 10 ml 중에 현탁시키고, 이어서 클로로(트리메틸)실란 1.65 ml (12.98 mmol) 및 메틸 시클로프로필리덴 아세테이트 (CAS 등록 번호 110793-87-8) 1.46 g (12.98 mmol)을 첨가하였다. 후속으로, 용액을 실온에서 추가로 1 시간 동안 교반하였다.
상기 수득한 용액 A를 -40℃로 냉각시켰다. 이어서, 용액 B를 서서히 적가하였다. 합한 용액을 -20℃로 서서히 가온하고, 상기 온도에서 1 시간 동안 교반하였다. 이어서, 빙냉 반-포화 염화암모늄 용액 50 ml를 반응 혼합물에 첨가하였다. 상을 분리하고, 이어서 수성 상을 에틸 아세테이트로 3회 더 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표제 화합물 2.1 g (5.2 mmol, 이론치의 39%)을 수득하였다.

실시예 9A
메틸 [1-(3-아미노-4-플루오로페닐)시클로프로필]아세테이트
실온에서, 탄소 상 팔라듐 (10%) 200 mg을 에탄올 100 ml 중 메틸 {1-[3-(디벤질아미노)-4-플루오로페닐]시클로프로필}아세테이트 2.1 g (5.2 mmol)의 용액에 첨가하고, 혼합물을 대기압에서 12 시간 동안 수소화시켰다. 반응이 완료된 후 (TLC에 의해 모니터링; 이동상 시클로헥산/에틸 아세테이트 1:1), 반응 용액을 규조토를 통해 여과하고, 여과물을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표제 화합물 647 mg (2.9 mmol, 이론치의 56%)을 수득하였다.

실시예 10A
5-브로모-2-클로로-N,N-비스(4-메톡시벤질)아닐린

아르곤 하에, 수소화나트륨 5.07 g (126.93 mmol, 60%)을 THF 150 ml 중에 현탁시키고, 혼합물을 0℃로 냉각시켰다. 이어서, THF 10 ml 중에 용해된 5-브로모-2-클로로아닐린 10.70 g (51.81 mmol)을 서서히 적가하고, 혼합물을 0℃에서 30 분 동안 교반하였다. 이어서, 4-메톡시벤질 클로라이드 25 g (124.34 mmol)을 반응 혼합물에 첨가하고, 후속으로 혼합물을 실온으로 가온하였다. 혼합물을 실온에서 2 시간 동안 교반하고, 이어서 빙수 150 ml에 서서히 부었다. 유기 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 크로마토그래피로 정제하였다 [칼럼: 크로마실(Kromasil) Si 6012, 350 mm x 30 mm; 이동상 A: 이소헥산, 이동상 B: 에틸 아세테이트; 구배: 0 분 98% A → 4.65 분 98% A → 13 분 87% A → 13.01 분 98% A → 13.28 분 98% A; 유량: 70 ml/분; 온도: 20℃; UV 검출: 265 nm]. 이로써 표제 화합물 12.37 g (27.69 mmol, 이론치의 57%)을 수득하였다.

실시예 11A
{3-[비스(4-메톡시벤질)아미노]-4-클로로페닐}보론산

-78℃에서 아르곤 하에, 헥산 중 n-부틸리튬의 2.5 M 용액 6.1 ml (15.25 mmol)를 THF/디에틸 에테르 (1:1) 100 ml 중 5-브로모-2-클로로-N,N-비스(4-메톡시벤질)아닐린 5.2 g (11.64 mmol)의 용액에 서서히 적가하였다. 반응 용액을 -78℃에서 60 분 동안 교반하고, 이어서 트리이소프로필 보레이트 4.3 ml (18.62 mmol)를 서서히 첨가하였다. 이어서, 반응 용액을 -78℃에서 추가로 15 분 동안 교반한 후, 실온으로 서서히 가온하고, 상기 온도에서 추가로 3 시간 동안 교반하였다. 이어서, 빙수 150 ml를 계량하여 넣었다. 유기 상을 분리하고, 이어서 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상: 먼저 시클로헥산/에틸 아세테이트 10:1 → 9:1 → 4:1, 이어서 디클로로메탄/메탄올 95:5). 이로써 표제 화합물 2.54 g (6.17 mmol, 이론치의 53%)을 수득하였다.
실시예 12A
벤질 옥세탄-3-일리덴아세테이트

0℃에서 아르곤 하에, 옥세탄-3-온 (CAS 등록 번호 6704-31-0) 3.0 g (41.63 mmol)을 디클로로메탄 50 ml 중에 용해시키고, 이어서 벤질 (트리페닐-λ5-포스파닐리덴)아세테이트 18.8 g (45.79 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온으로 서서히 가온하고, 추가로 15 분 동안 교반하였다. 이어서, 반응 용액을 농축 건조시켰다. 잔류물을 디에틸 에테르 25 ml에 녹이고, 교반하고, 혼합물을 4℃에서 12 시간 동안 정치시켰다. 침전된 트리페닐포스핀 옥시드를 여과하고, 여과물을 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 4:1 → 1:1). 이로써 표제 화합물 4.2 g (20.57 mmol, 이론치의 49%)을 수득하였다.

실시예 13A
벤질 (3-{3-[비스(4-메톡시벤질)아미노]-4-클로로페닐}옥세탄-3-일)아세테이트

실온에서 아르곤 하에, 1.5 M 수성 수산화칼륨 용액 1.6 ml (2.37 mmol), 벤질 옥세탄-3-일리덴아세테이트 272 mg (1.82 mmol) 및 {3-[비스(4-메톡시벤질)아미노]-4-클로로페닐}보론산 750 mg (1.82 mmol)을 디옥산 25 ml 중 (1Z,5Z)-시클로옥타-1,5-디엔/염화로듐(I) 이량체 45 mg (0.09 mmol)의 용액에 연속적으로 첨가하였다. 이어서, 반응 용액을 실온에서 4 시간 동안 교반하였다. 반응이 완료된 후, 용액을 농축 건조시키고, 잔류물을 물 25 ml 및 에틸 아세테이트 25 ml에 녹였다. 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 4:1 → 1:1). 이로써 표제 화합물 669 mg (1.17 mmol, 이론치의 64%)을 수득하였다.
하기 화합물을 합성 실시예 13A와 유사하게 수득하였다.

실시예 15A
벤질 [3-(3-아미노-4-클로로페닐)옥세탄-3-일]아세테이트

실온에서, 2,3-디클로로-5,6-디시아노-1,4-벤조퀴논 (DDQ) 576 mg (2.54 mmol)을 디클로로메탄 30 ml 및 물 6 ml 중 벤질 (3-{3-[비스(4-메톡시벤질)아미노]-4-클로로페닐}옥세탄-3-일)아세테이트 660 mg (1.15 mmol)의 용액에 첨가하고, 혼합물을 2 시간 동안 교반하였다. 반응이 완료된 후 (TLC에 의해 모니터링; 이동상 시클로헥산/에틸 아세테이트 2:1), 포화 중탄산나트륨 용액 25 ml를 반응 용액에 첨가하였다. 상을 분리하고, 이어서 수성 상을 디클로로메탄으로 3회 더 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 5:1). 이로써 표제 화합물 360 mg (0.98 mmol, 함량 90%, 이론치의 85%)을 수득하였다.
하기 화합물을 합성 실시예 15A와 유사하게 수득하였다.

실시예 17A
3-브로모-2-플루오로아닐린

3-브로모-2-플루오로니트로벤젠 2.0 g (9.09 mmol)을 디옥산 10 ml 중에 용해시키고, 염화주석(II) 8.62 g (45.45 mmol)을 실온에서 첨가하였다. 1 N 염산 몇 방울을 첨가한 후, 혼합물을 70℃에서 2 시간 동안 가열하였다. 냉각시킨 후, 반응 혼합물을 감압 하에 농축시키고, 잔류물을 에틸 아세테이트에 녹였다. 용액을 1N 수성 수산화나트륨 용액 (2회), 물 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표적 화합물 997 mg (이론치의 57.7%)을 수득하였다.

실시예 18A
tert-부틸 (2E)-3-(3-아미노-2-플루오로페닐)아크릴레이트

트리에틸아민 5.2 ml (37.1 mmol)를 DMF 8 ml 중 3-브로모-2-플루오로아닐린 1.41 g (7.42 mmol) 및 tert-부틸 아크릴레이트 2.85 g (22.3 mmol)의 용액에 첨가하였다. 플라스크를 배기시키고 아르곤으로 환기시켰고 (3회), 이어서 트리-2-톨릴포스핀 451 mg (1.48 mmol) 및 아세트산팔라듐(II) 166.6 mg (0.74 mmol)을 첨가하였다. 한 번 더, 반응 용기를 2회 배기시키고 아르곤으로 환기시키고, 이어서 혼합물을 약 140℃로 가열하였다. 2 시간 격렬하게 교반한 후, 반응 혼합물을 냉각시키고, 포화 중탄산나트륨 용액에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표적 생성물 1660 mg (이론치의 94.3%)을 수득하였다.

실시예 19A
tert-부틸 3-(3-아미노-2-플루오로페닐)프로파노에이트

탄소 상 팔라듐 (10%)을 에탄올 5 ml 및 THF 3 ml의 혼합물 중 tert-부틸 (2E)-3-(3-아미노-2-플루오로페닐)아크릴레이트 1660 mg (7.0 mmol)의 용액에 첨가하고, 혼합물을 대기압에서 수소의 분위기 하에 밤새 격렬하게 교반하였다. 이어서, 반응 혼합물을 규조토를 통해 여과하고, 여과 잔류물을 에탄올/THF로 반복적으로 세척하였다. 합한 여과물을 감압 하에 농축시키고, 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1 → 10:1). 이로써 표적 생성물 1350 mg (이론치의 80.6%)을 수득하였다.

실시예 20A
에틸 (E/Z)-3-(4-플루오로-3-니트로페닐)-2-메틸프로프-2-에노에이트

수소화나트륨 (미네랄 오일 중 60% 현탁액, 79.36 mmol) 3.17 g을 THF/DMF 혼합물 (2:1) 90 ml 중에 현탁시켰다. 혼합물을 0℃로 냉각시키고, THF/DMF (2:1) 60 ml 중 트리에틸 2-포스포노프로피오네이트 19.76 g (82.96 mmol)의 용액을 적가하였다. 30 분 후, THF/DMF (2:1) 60 ml 중 4-플루오로-3-니트로벤즈알데히드 12.2 g (72.14 mmol)의 용액을 0℃에서 적가하였다. 첨가가 종료된 후, 반응 혼합물을 실온으로 서서히 가온하고, 상기 온도에서 2 시간 동안 교반하였다. 이어서, 반응 혼합물을 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표적 생성물 15.2 g (이론치의 83.2%)을 E/Z 이성질체 혼합물 (E/Z 91:9)로서 수득하였다.

실시예 21A
에틸 (+/-)-3-(3-아미노-4-플루오로페닐)-2-메틸프로파노에이트

탄소 상 팔라듐 (10%)을 에탄올 100 ml 및 THF 100 ml의 혼합물 중 에틸 (E/Z)-3-(4-플루오로-3-니트로페닐)-2-메틸프로프-2-에노에이트 (E/Z 91:9) 15.2 g (60.02 mmol)에 첨가하고, 혼합물을 대기압에서 수소의 분위기 하에 밤새 격렬하게 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에탄올/디클로로메탄으로 세척하고, 합한 여과물을 감압 하에 농축시켰다. 생성물을 고진공 하에 건조시켰다. 이로써 표적 생성물 13.34 g (이론치의 98.7%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩(Daicel Chiralpak) AD-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.15 ml; 온도: 30℃; 이동상: 90% 이소헥산/10% 에탄올; 유량: 15 ml/분; 검출: 220 nm]. 라세미체 7.25 g으로 거울상이성질체 1 (실시예 22A) 3.43 g 및 거울상이성질체 2 (실시예 23A) 3.35 g을 수득하였다.
실시예 22A
에틸 (+)-(2S)-3-(3-아미노-4-플루오로페닐)-2-메틸프로파노에이트

실시예 23A
에틸 (-)-(2R)-3-(3-아미노-4-플루오로페닐)-2-메틸프로파노에이트

실시예 24A
에틸 (E/Z)-3-(4-클로로-3-니트로페닐)-2-메틸프로프-2-에노에이트

수소화나트륨 (미네랄 오일 중 60% 현탁액, 118.56 mmol) 4.74 g을 THF/DMF 혼합물 (1:1) 93 ml 중에 현탁시켰다. 혼합물을 0℃로 냉각시키고, 트리에틸 2-포스포노프로피오네이트 26.6 ml (123.95 mmol)를 적가하였다. 30 분 후, 4-클로로-3-니트로벤즈알데히드 20.0 g (107.78 mmol)을 0℃에서 첨가하였다. 첨가가 종결된 후, 반응 혼합물을 실온으로 서서히 가온하고, 상기 온도에서 추가로 3 시간 동안 교반하였다. 이어서, 반응 혼합물을 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 70:1 → 50:1). 이로써 표적 생성물 26.7 g (이론치의 91.9%)을 E/Z 이성질체 혼합물 (E/Z 91:9)로서 수득하였다.

실시예 25A
에틸 (+/-)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트

에틸 (E/Z)-3-(4-클로로-3-니트로페닐)-2-메틸프로프-2-에노에이트 (E/Z 91:9) 10.0 g (37.08 mmol)을 에틸 아세테이트 25 ml 및 아세트산 25 ml 중에 용해시키고, 탄소 상 팔라듐 (10%)을 첨가하였다. 반응 혼합물을 대기압에서 수소의 분위기 하에 총 6 시간 동안 격렬하게 교반하였다 (2 시간 후에 추가의 아세트산 25 ml 및 추가 분량의 10% 탄소 상 팔라듐을 첨가함). 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에탄올/디클로로메탄으로 세척하였다. 합한 여과물을 포화 중탄산나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 30:1 → 10:1). 이로써 표적 생성물 4.01 g (이론치의 44.7%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.15 ml; 온도: 35℃; 이동상: 50% 이소헥산/50% 이소프로판올; 유량: 15 ml/분; 검출: 220 nm]. 라세미체 10.3 g으로 거울상이성질체 1 (실시예 26A) 4.0 g 및 거울상이성질체 2 (실시예 27A) 3.7 g을 수득하였다.
실시예 26A
에틸 (-)-(2R)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트

실시예 27A
에틸 (+)-(2S)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트

실시예 28A
에틸 (2E/Z)-2-(4-클로로-3-니트로벤질리덴)부타노에이트

수소화나트륨 (미네랄 오일 중 60% 현탁액, 29.64 mmol) 1.19 g을 THF/DMF 혼합물 (1:1) 50 ml 중에 현탁시켰다. 혼합물을 0℃로 냉각시키고, 트리에틸 2-포스포노부티레이트 7.3 ml (30.99 mmol)를 적가하였다. 30 분 후, 4-클로로-3-니트로벤즈알데히드 5.0 g (26.94 mmol)을 -10℃에서 조금씩 첨가하였다. 첨가가 종료된 후, 반응 혼합물을 0℃에서 5 시간 동안 교반하고, 이어서 밤새 실온으로 서서히 가온하였다. 이어서, 반응 혼합물을 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 6:1). 이로써 표적 생성물 7.05 g (이론치의 92.1%)을 E/Z 이성질체 혼합물로서 수득하였다.
실시예 29A
(+/-)-에틸 2-(3-아미노-4-클로로벤질)부타노에이트

에틸 (2E/Z)-2-(4-클로로-3-니트로벤질리덴)부타노에이트 7.05 g (24.84 mmol)을 에틸 아세테이트 35 ml 및 아세트산 35 ml 중에 용해시키고, 탄소 상 팔라듐 (10%)을 첨가하였다. 반응 혼합물을 대기압에서 수소의 분위기 하에 총 6 시간 동안 격렬하게 교반하였다 (4 시간 후에 추가 분량의 10% 탄소 상 팔라듐을 첨가함). 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트/THF로 세척하였다. 합한 여과물을 포화 중탄산나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 30:1 → 10:1). 이로써 표적 생성물 4.12 g (이론치의 64.9%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.43 ml; 온도: 30℃; 이동상: 에탄올; 유량: 15 ml/분; 검출: 220 nm]. 라세미체 3.22 g으로 거울상이성질체 1 (실시예 30A) 1.22 g 및 거울상이성질체 2 (실시예 31A) 1.27 g을 수득하였다.
실시예 30A
(-)-에틸 (2R)-2-(3-아미노-4-클로로벤질)부타노에이트

실시예 31A
(+)-에틸 (2S)-2-(3-아미노-4-클로로벤질)부타노에이트

실시예 32A
tert-부틸 (2E/Z)-3-(4-클로로-3-니트로페닐)부트-2-에노에이트

수소화나트륨 (미네랄 오일 중 60% 현탁액, 71.65 mmol) 2.87 g을 THF 80 ml 중에 현탁시켰다. 혼합물을 0℃로 냉각시키고, tert-부틸 (디에톡시포스포릴)아세테이트 17.6 ml (74.9 mmol)를 적가하였다. 0℃에서 30 분 후, 4-클로로-3-니트로아세토페논 13.0 g (65.1 mmol)을 첨가하였다. 첨가가 종료된 후, 반응 혼합물을 실온으로 서서히 가온하고, 실온에서 추가로 1.5 시간 동안 교반하고, 이어서 혼합물을 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1 → 10:1). 이로써 표적 생성물 17.03 g (이론치의 87.8%)을 E/Z 이성질체 혼합물로서 (E/Z 약 1:1) 수득하였다.
실시예 33A
tert-부틸 (+/-)-3-(3-아미노-4-클로로페닐)부타노에이트

tert-부틸 (2E/Z)-3-(4-클로로-3-니트로페닐)부트-2-에노에이트 (E/Z 약 1:1) 11.5 g (38.62 mmol)을 에틸 아세테이트 60 ml 및 아세트산 60 ml 중에 용해시키고, 탄소 상 팔라듐 (10%)을 첨가하였다. 반응 혼합물을 대기압에서 수소의 분위기 하에 6 시간 동안 격렬하게 교반하였다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하였다. 합한 여과물을 포화 중탄산나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 30:1). 이로써 표적 생성물 3.90 g (이론치의 37.4%)을 수득하였다.
상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.15 ml; 온도: 30℃; 이동상: 90% 이소헥산/10% 에탄올; 유량: 15 ml/분; 검출: 220 nm]. 라세미체 5.0 g 으로 거울상이성질체 1 (실시예 34A) 2.1 g 및 거울상이성질체 2 (실시예 35A) 1.8 g을 수득하였다.
실시예 34A
tert-부틸 (+)-(3S)-3-(3-아미노-4-클로로페닐)부타노에이트

실시예 35A
tert-부틸 (-)-(3R)-3-(3-아미노-4-클로로페닐)부타노에이트

실시예 36A
tert-부틸 (2E/Z)-3-(4-플루오로-3-니트로페닐)부트-2-에노에이트

수소화나트륨 (미네랄 오일 중 60% 현탁액, 120.13 mmol) 4.81 g을 THF 120 ml 및 DMF 120 ml의 혼합물 중에 현탁시켰다. 혼합물을 0℃로 냉각시키고, tert-부틸 (디에톡시포스포릴)아세테이트 29.5 ml (125.59 mmol)를 적가하였다. 30 분 후, 4-플루오로-3-니트로아세토페논 20.0 g (109.21 mmol)을 0℃에서 첨가하였다. 첨가가 종료된 후, 반응 혼합물을 실온으로 서서히 가온하고, 실온에서 추가로 3.5 시간 동안 교반하고, 그 후에 혼합물을 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 표적 생성물 7.24 g (이론치의 23.6%)을 E/Z 이성질체 혼합물로서 (E/Z 약 1.2:1) 수득하였다.
실시예 37A
tert-부틸 (+/-)-3-(3-아미노-4-플루오로페닐)부타노에이트

tert-부틸 (2E/Z)-3-(4-플루오로-3-니트로페닐)부트-2-에노에이트 (E/Z 약 1.2:1) 7.24 g (25.74 mmol)을 에탄올 200 ml 중에 용해시키고, 탄소 상 팔라듐 (10%)을 첨가하였다. 반응 혼합물을 대기압에서 수소의 분위기 하에 밤새 격렬하게 교반하였다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 2회 세척하였다. 합한 여과물을 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표적 생성물 6.02 g (이론치의 92.4%)을 수득하였다.
상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.15 ml; 온도: 35℃; 이동상: 65% 이소헥산/35% 에탄올; 유량: 15 ml/분; 검출: 220 nm]. 라세미체 6.0 g으로 거울상이성질체 1 (실시예 38A) 2.44 g 및 거울상이성질체 2 (실시예 39A) 1.92 g을 수득하였다.
실시예 38A
tert-부틸 (+)-(3S)-3-(3-아미노-4-플루오로페닐)부타노에이트

실시예 39A
tert-부틸 (-)-(3R)-3-(3-아미노-4-플루오로페닐)부타노에이트

실시예 40A
에틸 3-(3-브로모-4-플루오로페닐)아크릴레이트

이산화망가니즈 9.65 g (111 mmol)을 톨루엔 390 ml 중 3-브로모-4-플루오로벤질 알콜 6.5 g (31.7 mmol) 및 에톡시카르보닐메틸렌포스포란 13.25 g (38 mmol)의 용액에 첨가하였다. 반응 혼합물을 환류하에 가열하고, 1 시간 후에 추가의 이산화망가니즈 9.65 g을 첨가하고, 환류 하에 가열을 밤새 계속하였다. 냉각시킨 후, 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켰다. 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 5:1). 이로써 표적 생성물 7.05 g (이론치의 81%)을 E/Z 이성질체 혼합물의 형태로 수득하였다.
실시예 41A
rac-에틸 2-(3-브로모-4-플루오로페닐)-트랜스-시클로프로판카르복실레이트

아르곤 하에, 먼저 수소화나트륨 (파라핀 오일 중 60%) 381 mg (9.52 mmol)을 DMSO 20 ml에 충전하고, 트리메틸술폭소늄 아이오다이드 2.1 g (9.52 mmol)을 실온에서 한번에 첨가하였다. 기체 발생이 중지된 후, DMSO 10 ml 중에 용해된 에틸 3-(3-브로모-4-플루오로페닐)아크릴레이트 2.0 g (7.3 mmol)을 서서히 적가하였다. 반응 혼합물을 50℃에서 밤새 가열하고, 이어서 실온으로 냉각시키고, 추가의 후처리 없이, 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 이소헥산/에틸 아세테이트 100:1). 이로써 표적 생성물 907 mg (이론치의 43%)을 수득하였다.

실시예 42A
rac-에틸 2-[3-(벤질아미노)-4-플루오로페닐]-트랜스-시클로프로판카르복실레이트

아르곤 하에, 나트륨 tert-부톡시드 361.5 mg (3.8 mmol)을 톨루엔 12.9 ml 중에 현탁시키고, (+/-)-트랜스-에틸 2-(3-브로모-4-플루오로페닐)시클로프로판카르복실레이트 900 mg (3.1 mmol), 벤질아민 403 mg (3.8 mmol), 트리스(디벤질리덴아세톤)디팔라듐 28.7 mg (0.03 mmol) 및 rac-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 19.5 mg (0.03 mmol)을 연속적으로 첨가하였다. 혼합물을 110℃에서 4 시간 동안 가열하였다. 이어서, 반응 혼합물을 실온으로 냉각시키고, 에틸 아세테이트 100 ml 및 포화 염화암모늄 용액 50 ml를 첨가하고, 혼합물을 셀라이트를 통해 여과하였다. 유기 상을 분리하고, 포화 염화암모늄 용액 및 포화 염화나트륨 용액 (각 경우에 50 ml)으로 세척하고, 황산마그네슘 상에서 건조시키고, 농축시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다. 이로써 66% 순도의 표적 화합물 262 mg (이론치의 18%)을 수득하였다.
실시예 43A
rac-에틸 2-[3-아미노-4-플루오로페닐]-트랜스-시클로프로판카르복실레이트

(+/-)-에틸 2-[3-(벤질아미노)-4-플루오로페닐]-트랜스-시클로프로판카르복실레이트 262 mg (순도 66%, 0.55 mmol)을 에탄올/THF (1:1) 5 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 26 mg을 첨가하고, 혼합물을 1 bar의 수소압을 이용하여 실온에서 24 시간 동안 수소화시켰다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에탄올로 세척하고, 여과물을 농축시켰다. 상기 방식으로 수득한 조 생성물을 정제용 HPLC에 의해 정제하였다. 이로써 표적 화합물 87 mg (이론치의 69%)을 수득하였다.

실시예 44A
3-아미노-4-플루오로아세토페논

0℃에서, 물 12 ml 중 염화주석 2수화물 11.1 g (89 mmol)의 용액을 12 N 염산 7.8 ml 중 4-플루오로-3-니트로아세토페논 3 g (16.4 mmol)의 용액에 15 분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 환류하에 15 분 동안 가열하고, 후속으로 실온에서 밤새 교반하였다. 이어서, 반응 혼합물을 얼음 상에 붓고, 50% 농도의 수성 수산화나트륨 용액을 사용하여 pH 12로 조정하고, 에틸 아세테이트로 추출하였다. 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 농축시켰다. 이로써 표적 화합물 2.47 g (순도 90%, 이론치의 87%)을 수득하였다.
실시예 45A 및 실시예 46A
에틸 (2E)-3-(3-아미노-4-플루오로페닐)-2-메틸부트-2-에노에이트
및
에틸 (2Z)-3-(3-아미노-4-플루오로페닐)-2-메틸부트-2-에노에이트

0℃에서, 트리에틸 2-포스포노프로피오네이트 6.92 ml (7.68 g, 32.3 mmol)를 THF 24.7 ml 중 수소화나트륨 (파라핀 오일 중 60%; 32.3 mmol) 1.29 g의 현탁액에 서서히 적가하였다. 반응 혼합물을 30 분 동안 교반하고, 이어서 3-아미노-4-플루오로아세토페논 2.47 g (순도 90%, 14.5 mmol)을 첨가하였다. 먼저 반응 혼합물을 실온에서 1 시간 동안 교반하고, 이어서 환류 하에 2 시간 동안 교반한 후, 다시 실온으로 냉각시키고, 밤새 교반하였다. 이어서, 혼합물을 물에 붓고, 에틸 아세테이트 (각 경우에 100 ml)로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축시키고, 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 톨루엔/에틸 아세테이트 5:1). 이로써 분리된 형태의 2E 이성질체 (실시예 45A) 612 mg (이론치의 15%) 및 2Z 이성질체 (실시예 46A) 529 mg (이론치의 13%)을 수득하였다.
2E 이성질체 (실시예 45A):

2Z 이성질체 (실시예 46A):

실시예 47A
rac-트레오-에틸 3-(3-아미노-4-플루오로페닐)-2-메틸부타노에이트
탄소 상 팔라듐 (10%) 4.8 mg을 메탄올 5 ml 중 에틸 (2E)-3-(3-아미노-4-플루오로페닐)-2-메틸부트-2-에노에이트 48 mg (0.2 mmol)의 용액에 첨가하였다. 반응 혼합물을 1 bar의 수소압에서 밤새 수소화시켰다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켰다. 이로써 약 20%의 에리트로 이성질체를 함유하는 표제 화합물 35.8 mg (이론치의 74%)을 수득하였다.
실시예 48A
rac-에리트로-에틸 3-(3-아미노-4-플루오로페닐)-2-메틸부타노에이트

탄소 상 팔라듐 (10%) 3 mg을 메탄올 3.1 ml 중 에틸 (2Z)-3-(3-아미노-4-플루오로페닐)-2-메틸부트-2-에노에이트 30 mg (0.13 mmol)의 용액에 첨가하였다. 반응 혼합물을 1 bar의 수소압에서 밤새 수소화시켰다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켰다. 이로써 약 5%의 트레오 이성질체를 함유하는 표제 화합물 22.5 mg (이론치의 74%)을 수득하였다.
실시예 49A
tert-부틸 (2E)-3-(4-플루오로-3-니트로페닐)아크릴레이트

아르곤 하에, 먼저 수소화나트륨 (파라핀 오일 중 60%; 16.3 mmol) 0.65 g을 THF 25 ml에 충전하고, 혼합물을 0℃로 냉각시켰다. 이어서, tert-부틸 디에틸포스포노아세테이트 4.29 g (17 mmol)을 서서히 적가하였다. 30 분 교반 후, 4-플루오로-3-니트로벤즈알데히드 2.5 g (14.8 mmol)을 첨가하였다. 반응 혼합물을 실온에서 3 시간 동안 교반하고, 이어서 물 100 ml에 붓고, 에틸 아세테이트 (각 경우에 100 ml)로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축시켰다. 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 표적 생성물 3.37 g (이론치의 85%)을 수득하였다.

실시예 50A
tert-부틸 (2E)-3-(4-시아노-3-니트로페닐)아크릴레이트

시안화칼륨 134 mg (2.06 mmol)을 DMF 5.4 ml 중 tert-부틸 (2E)-3-(4-플루오로-3-니트로페닐)아크릴레이트 500 mg (1.87 mmol)의 용액에 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 직접 정제하였다 (이동상 시클로헥산/에틸 아세테이트 혼합물). 이로써 표제 화합물 57 mg (이론치의 11%)을 수득하였다.

실시예 51A
tert-부틸 3-(3-아미노-4-시아노페닐)프로파노에이트

탄소 상 팔라듐 (10%) 4.9 mg을 에탄올 4.4 ml 중 tert-부틸 (2E)-3-(4-시아노-3-니트로페닐)아크릴레이트 48.9 mg (0.18 mmol)의 용액에 첨가하였다. 반응 혼합물을 1 bar의 수소압을 이용하여 실온에서 밤새 수소화시켰다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켰다. 이로써 85% 순도의 표적 화합물 43.5 mg (이론치의 99%)을 수득하였다.
실시예 52A
에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트

실온에서, 티오닐 클로라이드 133 ml (1.82 mmol)를 에탄올 580 ml 중 (3R)-4,4,4-트리플루오로-3-메틸부탄산 (문헌 [A. Gerlach and U. Schulz, Speciality Chemicals Magazine 24 (4), 37-38 (2004)]; CAS 등록 번호 142:179196) 287 g (1.65 mol)을 서서히 첨가하였다. 이어서, 반응 용액을 80℃로 가열하고, 상기 온도에서 2 시간 동안 교반하였다. 이어서, 혼합물을 실온으로 냉각시키고, 물 250 ml를 서서히 첨가하고, 혼합물을 tert-부틸 메틸 에테르 (각 경우에 150 ml)로 3회 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 30℃에서 300 mbar의 감압 하에 제거하였다. 이어서, 조 생성물을 100 mbar 및 65℃의 헤드 온도에서 증류시켰다. 이로써 표제 화합물 225.8 g (113 mol, 이론치의 74%)을 무색 액체로서 수득하였다.

실시예 53A
에틸 4,4,4-트리플루오로-3-메틸-2-(4-메틸페닐)부타노에이트 (부분입체이성질체 혼합물)

아르곤 하에, 먼저 아세트산팔라듐(II) 196.9 mg (0.88 mmol) 및 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노)비페닐 724.8 mg (1.84 mmol)을 무수 톨루엔 50 ml에 충전하였다. 이어서, THF 중 리튬 헥사메틸디실라지드의 1 M 용액 43.8 ml (43.8 mmol)를 서서히 첨가하고, 반응 용액을 실온에서 10 분 동안 교반하였다. 이어서, 반응 용액을 -10℃로 냉각시키고, (+/-)-에틸 4,4,4-트리플루오로-3-메틸부타노에이트 7 g (38.0 mmol)을 서서히 첨가하고, 혼합물을 -10℃에서 10 분 동안 교반하였다. 이어서, 톨루엔 50 ml 중에 용해된 4-브로모톨루엔 5 g (29.2 mmol)을 적가하고, 반응 용액을 먼저 실온으로, 이어서 80℃로 가온하였다. 혼합물을 상기 온도에서 2 시간 동안 교반하고, 이어서 실온으로 냉각시키고, 밤새 교반하였다. 반응이 완료된 후 (TLC에 의해 모니터링; 이동상 시클로헥산/디클로로메탄 2:1), 반응 혼합물을 규조토를 통해 여과하고, 잔류물을 에틸 아세테이트 및 디클로로메탄으로 반복적으로 세척하고, 합한 여과물을 감압 하에 농축시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 석유 에테르/디클로로메탄 4:1 → 3:1). 이로써 표제 화합물 3.91 g (14.3 mmol, 이론치의 48.8%)을 무색 액체로서 수득하였다.

실시예 54A
에틸 (3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트

용액 A의 제조: 아르곤 하에, 톨루엔 중 리튬 헥사메틸디실라지드의 1 M 용액 163.9 ml를 -10℃에서 -20℃로 냉각시키고 (아세톤/드라이아이스로 냉각), 톨루엔 150 ml 중에 용해된 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 20 g (108.6 mmol)을, -10℃의 온도를 초과하지 않도록 주의하면서 서서히 첨가하였다. 이어서, 용액을 -10℃ 이하에서 10 분 동안 교반하였다.
용액 B의 제조: 아르곤 하에, 1-브로모-4-클로로벤젠 27.03 g (141.2 mmol)을 실온에서 톨루엔 100 ml 중에 용해시키고, 아세트산팔라듐(II) 731 mg (3.26 mmol) 및 2'-(디시클로헥실포스파닐)-N,N-디메틸비페닐-2-아민 2.693 g (6.84 mmol)을 첨가하였다. 용액을 실온에서 10 분 동안 교반하였다.
먼저, 냉각 조를 용액 A로부터 제거하였다. 이어서, 용액 B를 여전히 차가운 용액 A에 서서히 적가하였다. 합한 용액을 실온으로 서서히 가온하고, 상기 온도에서 1 시간 동안 교반하였다. 이어서, 반응 용액을 80℃ (내부 온도)로 가온하고, 상기 온도에서 3 시간 동안 교반하였다. 이어서, 반응 용액을 서서히 실온으로 냉각시키고, 추가로 12 시간 동안 교반하였다. 이어서, 반응 혼합물을 규조토를 통해 여과하고, 잔류물을 톨루엔으로 반복적으로 세척하고, 합한 여과물을 감압 하에 농축시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/디클로로메탄 4:1). 이로써 표제 화합물 27.4 g (92.98 mmol, 이론치의 86%)을 황색 오일로서 3:1의 부분입체이성질체 비로 수득하였다.
하기 화합물을 합성 실시예 53A 및 54A와 유사하게 수득하였다.
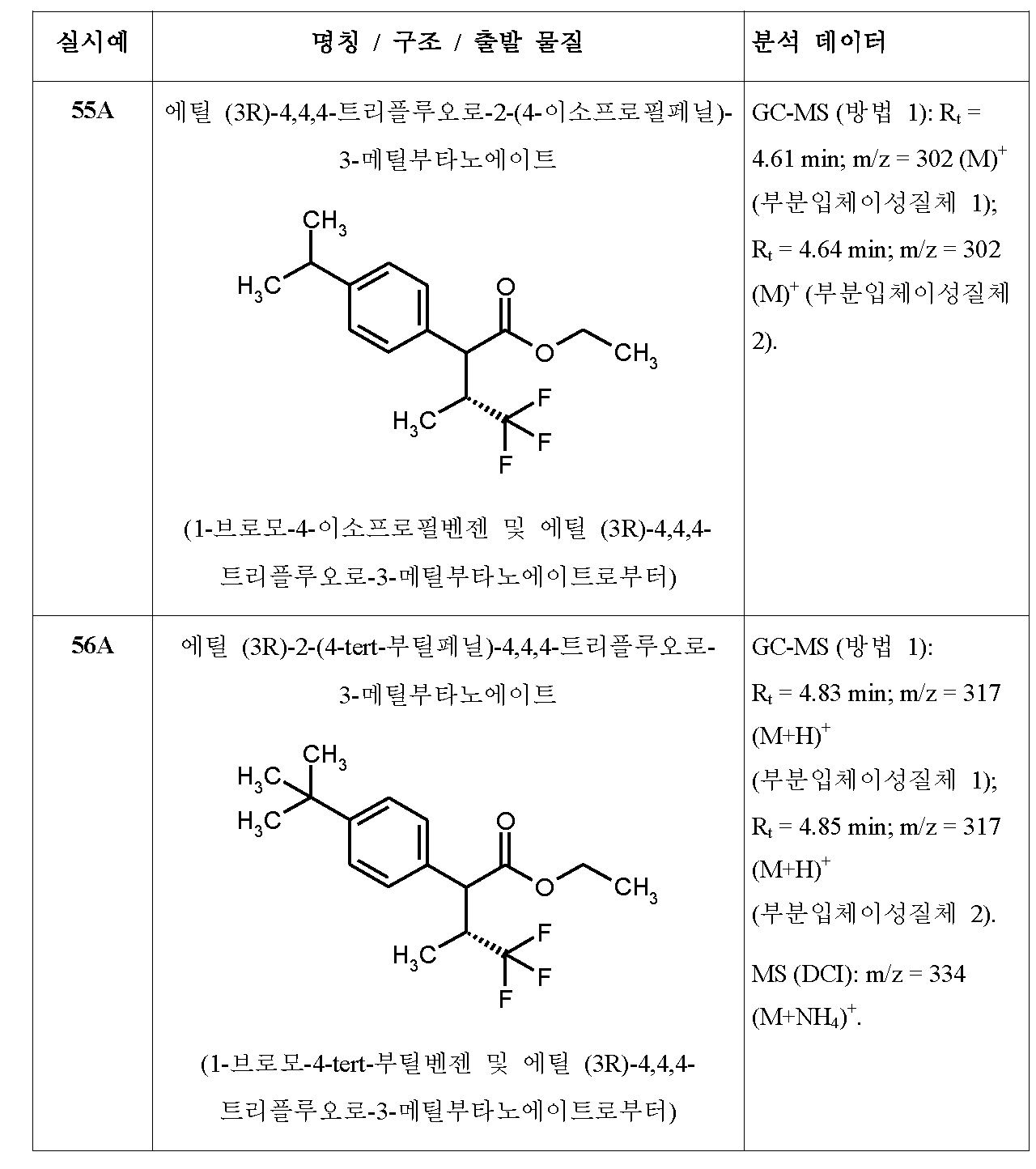

실시예 59A
에틸 2-[4-(브로모메틸)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트

트리클로로메탄 36 ml 중 에틸 4,4,4-트리플루오로-3-메틸-2-(4-메틸페닐)부타노에이트 2.25 g (8.2 mmol), N-브로모숙신이미드 1.53 g (8.6 mmol) 및 2,2'-아조비스-2-메틸프로판니트릴 67 mg (0.41 mmol)을 환류 하에 밤새 교반하였다. 반응이 완료된 후, 숙신이미드를 여과하고, 여과 잔류물을 디클로로메탄으로 세척하고, 여과물을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 40:1). 이로써 황색빛 오일 2.667 g (7.5 mmol, 이론치의 92%)을 수득하였다.
실시예 60A
에틸 4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노에이트

아이오딘화구리(I) 529 mg (2.78 mmol) 및 메틸 2,2-디플루오로-2-(플루오로술포닐) 아세테이트 4 g (20.82 mmol)을 1-메틸피롤리딘-2-온 40 ml 중 에틸 2-[4-(브로모메틸)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트 3.77 g (10.67 mmol)에 첨가하고, 혼합물을 80℃에서 밤새 교반하였다. 반응이 완료된 후, 반응 용액을 빙수 100 ml에 서서히 부었다. 이어서, 수득한 혼합물을 디에틸 에테르로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/디클로로메탄 4:1). 이로써 표제 화합물 1.48 g (4.32 mmol, 이론치의 41%)을 황색빛 오일로서 수득하였다.

실시예 61A
1-브로모-4-(2-브로모-1-플루오로에틸)벤젠

4-브로모스티렌 5.0 g (27.31 mmol)을 디클로로메탄 40 ml 중에 용해시키고, 0℃로 냉각시키고, 트리에틸아민 트리히드로플루오라이드 13.21 g (81.94 mmol)을 첨가하였다. 이어서, N-브로모숙신이미드 5.83 g (32.78 mmol)을 3번으로 나누어 첨가하였다. 혼합물을 실온에서 밤새 교반하였다. 디클로로메탄으로 희석한 후, 반응 혼합물을 빙수에 부었다. 유기 상을 1 N 염산, 물 및 포화 중탄산나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 펜탄). 이로써 표제 화합물 4.14 g (이론치의 53.8%)을 수득하였다.

실시예 62A
1-브로모-4-(1-플루오로비닐)벤젠

칼륨 tert-부톡시드 796 mg (7.09 mmol)을 펜탄 10 ml 중 1-브로모-4-(2-브로모-1-플루오로에틸)벤젠 1.0 g (3.55 mmol)의 0℃로 냉각시킨 용액에 수 회 분량으로 나누어 첨가하였다. 생성된 현탁액을 0℃에서 30 분 동안, 이어서 실온에서 1 시간 동안 교반하였다. 고체를 여과하고, 여과물을 포화 염화암모늄 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 조심스럽게 농축시켰다. 이로써 표제 화합물 0.61 g (이론치의 85.6%)을 수득하였다.

실시예 63A
에틸 (3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

톨루엔 중 리튬 헥사메틸디실라지드 1 M 용액 24.4 ml (24.4 mmol)를 -10℃로 냉각시키고, 무수 톨루엔 15 ml 중 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 3.0 g (16.29 mmol)의 용액을 적가하였다. 혼합물을 10 분 동안 교반하였다. 이어서, -10℃에서, 무수 톨루엔 20 ml 중 1-브로모-4-에틸벤젠 3.92 g (21.18 mmol), 아세트산팔라듐(II) 110 mg (0.49 mmol) 및 2'-디시클로헥실포스피노-2-(N,N-디메틸아미노)비페닐 404 mg (1.03 mmol)의 미리 제조한 용액을 적가하였다. 생성된 반응 혼합물을 먼저 실온에서 1 시간 동안, 이에서 80℃에서 3 시간 동안 교반하였다. 이어서, 혼합물을 감압 하에 농축시키고, 잔류물을 에틸 아세테이트에 녹이고, 물에 첨가하였다. 수성 상을 에틸 아세테이트로 역-추출하고, 합한 유기 상을 포화 염화암모늄 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피한 후 (이동상: 먼저 시클로헥산, 이어서 구배 시클로헥산/에틸 아세테이트 200:1 → 50:1), 표제 화합물 3.051 g (이론치의 64.9%, 부분입체이성질체 비 약 3:1)을 수득하였다.

하기 두 화합물을 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 및 적절한 페닐 브로마이드로부터 유사한 방식으로 제조하였다.
실시예 64A
에틸 (3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노에이트 (부분입체이성질체 혼합물)


실시예 65A
에틸 (3R)-4,4,4-트리플루오로-2-[4-(1-플루오로비닐)페닐]-3-메틸부타노에이트
실시예 66A
메틸 (4-클로로페닐)(3-옥소시클로펜틸)아세테이트

아르곤 하에, 먼저 디이소프로필아민 14.8 ml (105.6 mmol)를 THF 150 ml에 충전하고, 혼합물을 -30℃로 냉각시키고, 헥산 중 n-부틸리튬의 2.5 M 용액 42.3 ml (105.75 mmol)를 서서히 첨가하였다. 이어서, 반응 용액을 -20℃로 가온하고, THF 90 ml 중에 용해된 메틸 (4-클로로페닐)아세테이트 15 g (81.25 mmol)을 서서히 첨가하고, 혼합물을 상기 온도에서 2 시간 동안 교반하였다. 이어서, 반응 용액을 -78℃로 냉각시키고, THF 60 ml 중에 용해된 2-시클로펜텐-1-온 7.2 ml (86.1 mmol)를 서서히 첨가하였다. 첨가가 종료된 후, 용액을 상기 온도에서 1 시간 동안 교반하였다. TLC 확인 후 (이동상 시클로헥산/에틸 아세테이트 9:1), 포화 염화암모늄 용액을 첨가하고, 혼합물을 에틸 아세테이트에 녹였다. 수성 상을 에틸 아세테이트로 2회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 4:1). 이로써 표제 화합물 15.65 g (58.67 mmol, 이론치의 72%)을 황색빛 오일로서 수득하였다.

실시예 67A
메틸 (4-클로로페닐)(3,3-디플루오로시클로펜틸)아세테이트

아르곤 하에, 먼저 톨루엔 200 ml로 희석된, THF 중 1,1'-[(트리플루오로-λ4-술파닐)이미노]비스(2-메톡시에탄) (데속소플루오르(Desoxofluor))의 50% 농도 용액 82.5 ml (82.14 mmol)를 충전하고, 5℃로 냉각시키고, 삼플루오린화붕소/디에틸 에테르 복합체의 1 M 용액 744 μl (5.87 mmol)를 서서히 첨가하였다. 혼합물을 5℃에서 2 시간 동안 교반하였다. 이어서, 톨루엔 200 ml 중에 용해된 메틸 (4-클로로페닐)(3-옥소시클로펜틸)아세테이트 15.65 g (58.67 mmol)을 서서히 첨가하고, 후속으로 혼합물을 55℃로 가온하고, 상기 온도에서 60 시간 동안 교반하였다. 이어서, 반응 혼합물을, 톨루엔 100 ml 및 2 M 수성 수산화나트륨 용액 100 ml로 이루어진 0℃로 냉각된 혼합물에 첨가하였다. 유기 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 7:1). 이로써 표제 화합물 13.24 g (45.86 mmol, 이론치의 78%)을 무색 오일로서 수득하였다.

실시예 68A
(+/-)-에틸 (2,2-디플루오로시클로펜틸)아세테이트

실온에서, (+/-)-에틸 2-옥소시클로펜틸아세테이트 17.0 g (99.88 mmol)을 무수 디클로로메탄 150 ml 중 디에틸아미노황 트리플루오라이드 (DAST) 52.8 ml (399.5 mmol)의 용액에 적가하였다. 혼합물을 환류 하에 밤새 가열하였다. 냉각시킨 후, 추가의 디에틸아미노황 트리플루오라이드 (DAST) 13.2 ml (99.88 mmol)를 첨가하고, 혼합물을 한 번 더 환류 하에 36 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 디클로로메탄으로 희석하고, 포화 중탄산나트륨 용액을 조심스럽게 첨가하고, 이어서 혼합물을 격렬하게 교반하였다. 유기 상을 포화 중탄산나트륨 용액, 1 N 염산 (2회) 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 암갈색 잔류물로부터, 생성물을 실리카 겔 상에서 칼럼 크로마토그래피에 의해 단리하였다 (이동상 펜탄/디클로로메탄 10:1 → 1:1). 이로써 표제 화합물 7.52 g (이론치의 39%)을 수득하였다.

실시예 69A
에틸 (4-클로로페닐)(2,2-디플루오로시클로펜틸)아세테이트 (부분입체이성질체 혼합물)

톨루엔 중 리튬 헥사메틸디실라지드의 1 M 용액 22.6 ml (22.6 mmol)를 -20℃로 냉각시키고, 무수 톨루엔 20 ml 중 (+/-)-에틸 (2,2-디플루오로시클로펜틸)아세테이트 2.90 g (15.09 mmol)의 용액을 적가하였다. 혼합물을 -20℃에서 10 분 동안 교반하였다. 냉각을 제거하고, 이어서 무수 톨루엔 20 ml 중 4-브로모클로로벤젠 3.75 g (19.61 mmol), 아세트산팔라듐(II) 110 mg (0.49 mmol) 및 2'-디시클로헥실포스피노-2-(N,N-디메틸아미노)비페닐 374 mg (0.95 mmol)의 미리 제조한 용액을 적가하였다. 생성된 반응 혼합물을 먼저 실온에서 1 시간 동안, 이어서 90℃에서 2 시간 동안 교반하였다. 냉각시킨 후, 반응 혼합물을 물에 첨가하였다. 수성 상을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 50:1), 표제 화합물 2.70 g (이론치의 59.1%, 부분입체이성질체 비 약 1:4.3)을 수득하였다.

실시예 70A
(+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산

에틸 (3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 5.086 g (17.26 mmol)을 디옥산 68 ml 중에 용해시키고, 1 N 수성 수산화나트륨 용액 34 ml를 첨가하였다. 혼합물을 50℃에서 2 시간 동안 교반하였다. 이어서, 1 N 염산을 사용하여 반응 혼합물을 pH 1로 산성화시키고, 디클로로메탄으로 반복적으로 추출하였다. 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 표적 화합물 3.9 g (14.63 mmol, 이론치의 85%, 83% de)을 수득하였다.

하기 표에 열거된 화합물을 합성 실시예 70A와 유사하게 제조하였다.




실시예 78A
(3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

에틸 (3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (순도 약 88%, 약 9.16 mmol; 부분입체이성질체 혼합물) 3.0 g을 메탄올, THF 및 물 (각 경우에 12.4 ml)의 혼합물 중에 용해시키고, 수산화나트륨 5.49 g (137.35 mmol)을 조금씩 첨가하였다. 반응 혼합물을 40℃에서 9 시간 동안 교반하였다. 냉각시킨 후, 대부분의 휘발성 용매를 감압 하에 제거하고, 잔류물을 물로 희석하였다. 염산을 첨가하여 혼합물을 산성화시키고, 수성 상을 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표제 화합물 2.61 g을 조 생성물로서 수득하였고, 이를 더이상 정제하지 않았다 (부분입체이성질체 비 약 9:1).

유사한 방식으로 (반응 온도: 실온 내지 40℃; 반응 시간: 9 내지 12 시간), 하기 두 카르복실산 유도체를 상응하는 에스테르로부터 제조하였다.
실시예 79A
(3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부탄산 (부분입체이성질체 혼합물)

실시예 80A
(3R)-4,4,4-트리플루오로-2-[4-(1-플루오로비닐)페닐]-3-메틸부탄산 (부분입체이성질체 혼합물)

실시예 81A
(4-클로로페닐)(2,2-디플루오로시클로펜틸)아세트산 (부분입체이성질체 혼합물)

에틸 (4-클로로페닐)(2,2-디플루오로시클로펜틸)아세테이트 (부분입체이성질체 혼합물) 2.70 g (8.92 mmol)을 메탄올 10 ml, THF 10 ml 및 물 5 ml 중에 용해시키고, 50% 농도 수성 수산화나트륨 용액 7.13 g (89.18 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 이어서, 혼합물을 물로 희석하고, 염산으로 산성화시켰다. 수성 상을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표제 화합물 2.39 g (이론치의 97.6%, 부분입체이성질체 비 약 1:1)을 수득하였다.
실시예 82A
(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드

(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 19.5 g (73.13 mmol)을 디클로로메탄 860 ml 중에 용해시키고, DMF 0.5 ml를 첨가하였다. 이어서, -5℃로부터 -10℃까지 (얼음/아세톤 냉각 조)에서, 디클로로메탄 중 옥살릴 클로라이드의 2 M 용액 73 ml (146.26 mmol)를 서서히 적가하고, 혼합물을 상기 온도에서 1 시간 동안 교반하였다. 반응이 완료된 후, 반응 용액을 감압 하에 증발시키고, 수득한 잔류물을 디클로로메탄 200 ml에 녹이고, 이어서 한번 더 농축 건조시켰다. 이로써 표제 화합물 20.1 g (70.5 mmol, 이론치의 96%)을 무색 오일로서 수득하였다. 추가 정제없이, 그리고 추가 분광학적 특성화없이, 상기 방식으로 수득한 생성물을 후속 반응에 사용하였다.
하기 표에 열거된 화합물을 유사한 방식으로 제조하였다.


실시예 89A
tert-부틸-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)프로파노에이트

(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 18 g (70.38 mmol)을 THF 500 ml 중에 용해시키고, N,N-디이소프로필에틸아민 18.4 ml (105.57 mmol)를 첨가하고, 혼합물을 -10℃로 냉각시켰다. 이어서, THF 500 ml 중에 용해된 tert-부틸-3-(3-아미노-4-클로로페닐)프로파노에이트 20.07 g (70.38 mmol)을, 첨가하는 동안 반응 온도가 0℃를 초과하지 않도록 주의하면서 서서히 첨가하였다. 이어서, 혼합물을 추가로 1 시간 동안 교반하였다. 이어서, 물 및 에틸 아세테이트를 반응 용액에 첨가하고, 유기 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시키고, 회전 증발기 상에서 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표제 화합물 30.13 g (59.74 mmol, 이론치의 85%)을 수득하였다.

하기 표에 열거된 화합물을 유사한 방식으로 수득하였다.





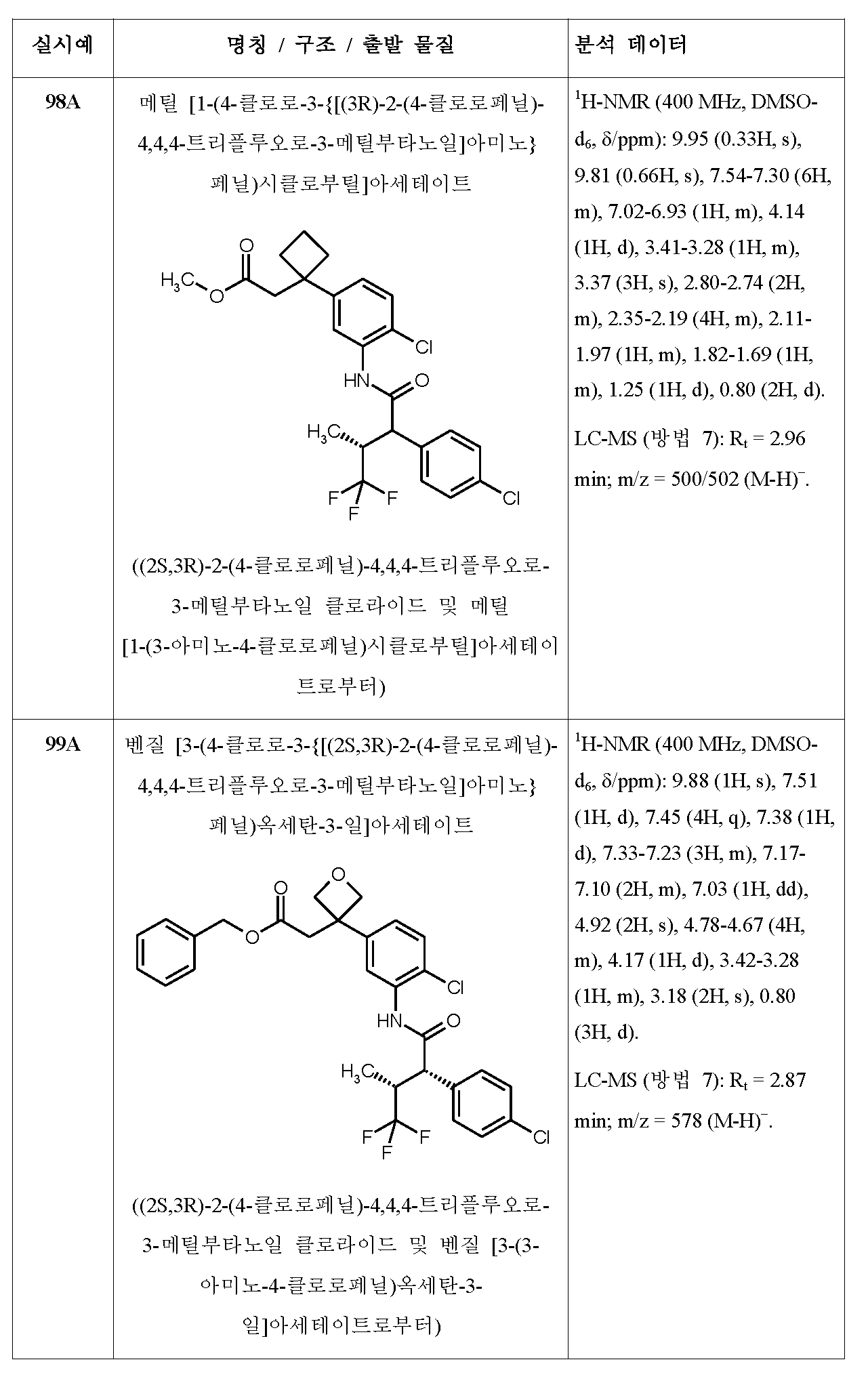
실시예 100A
메틸 [1-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)시클로프로필]아세테이트

DMF 2.4 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 70 mg (0.31 mmol), 메틸 [1-(3-아미노-4-플루오로페닐)시클로프로필]아세테이트 84 mg (0.31 mmol), 2-(1H-7-아자벤조트리아졸-1-일)-1,1,3,3-테트라메틸우로늄 헥사플루오로포스페이트 (HATU) 179 mg (0.47 mmol) 및 피리딘 0.6 ml의 용액을 실온에서 밤새 교반하였다. 반응이 종료된 후, 혼합물을 추가의 후처리 없이 정제용 HPLC에 의해 직접 분리하였다. 이로써 표제 화합물 106 mg (0.22 mmol, 이론치의 72%)을 무색 오일로서 수득하였다.

하기 화합물을 유사한 방식으로 수득하였다.

일반적 절차 1: 4,4,4-트리플루오로-3-메틸-2-페닐부탄산 유도체와 아닐린의 HATU-매개 아미드 커플링
실온에서, HATU (1.0 내지 2.0 당량)를 DMF 및 피리딘의 혼합물 (약 3:1 내지 1.5:1 비율로 혼합) 중 당해 4,4,4-트리플루오로-3-메틸-2-페닐부탄산 유도체 (약 0.8 내지 1.5 당량, 0.15 내지 1.5 mol/l) 및 아닐린 (약 0.8 내지 1.5 당량, 0.15 내지 1.5 mol/l)의 용액에 첨가하였다. 대안적으로, 피리딘 대신에, N,N-디이소프로필에틸아민 (2.0 내지 5.0 당량)을 사용하는 것 또한 가능하다. 생성된 혼합물을 실온에서 60℃의 온도에서 4 시간 내지 48 시간 동안 교반하였다. 적절한 경우, 추가 분량의 아닐린 또는 카르복실산 및 HATU를 24 시간 후에 첨가한다. 반응이 종료된 후, 감압 하에 용매를 제거한 다음 조 생성물을 정제용 RP-HPLC (이동상: 아세토니트릴/물 구배)에 의해, 또는 대안적으로, 반응 혼합물의 수성 후처리 후에, 실리카 겔 상에서의 크로마토그래피 (이동상: 시클로헥산/에틸 아세테이트 또는 디클로로메탄/메탄올 혼합물)에 의해 정제할 수 있다.
하기 실시예를 일반적 절차 1에 따라 제조하였다.








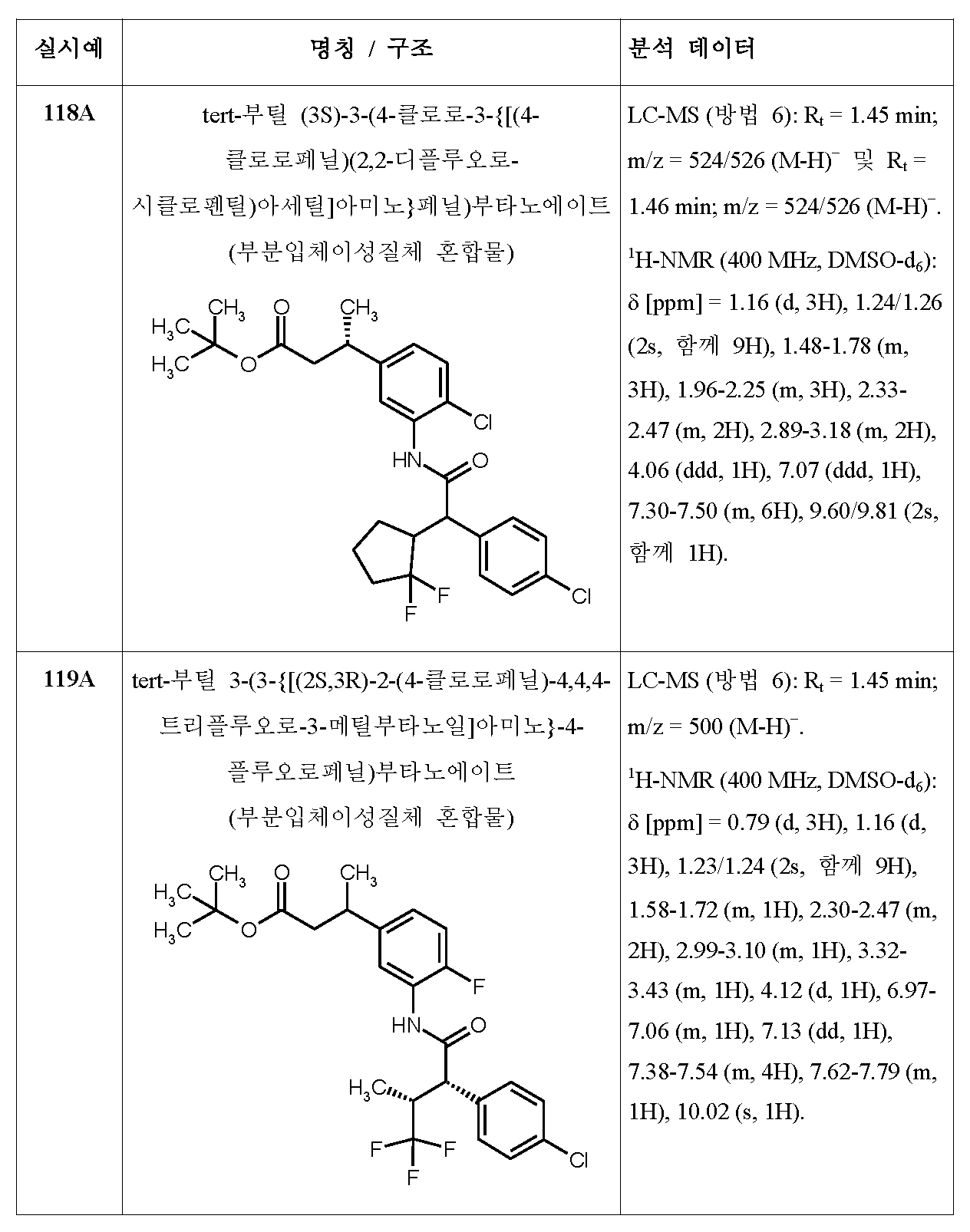


실시예 123A
에틸 (2R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트

에틸 (-)-(2R)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트 500 mg (2.07 mmol) 및 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 607 mg (2.28 mmol)을 DMF 2.0 ml 및 피리딘 1.0 ml의 혼합물 중에 용해시키고, HATU 1022 mg (2.69 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 혼합물을 에틸 아세테이트로 희석하고, 용액을 1 N 염산, 물, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 40:1). 이로써 표적 화합물 998 mg (이론치의 98.4%)을 수득하였다.

실시예 124A
(+)-에틸 (2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트

방법 A:
에틸 (+)-(2S)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트 1.50 g (6.21 mmol) 및 (3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (약 9:1 부분입체이성질체 혼합물로서) 1.82 g (6.83 mmol)을 DMF 6.3 ml 및 피리딘 3.2 ml의 혼합물 중에 용해시키고, HATU 2.83 g (7.45 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 이어서, 혼합물을 에틸 아세테이트로 희석하고, 용액을 포화 염화암모늄 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1 → 40:1). 정제 중에 수득한 혼합 분획 (부 부분입체이성질체 함유)을 실리카 겔 상에서 또 다른 크로마토그래피에 의해 (이동상 시클로헥산/에틸 아세테이트 40:1) 분리하였다. 이로써 표적 화합물 총 2.46 g (이론치의 80.8%)을 수득하였다.
방법 B:
디클로로메탄 30 ml 및 DMF 한 방울을 (3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (약 9:1-부분입체이성질체 혼합물) 7.60 g (28.50 mmol)에 첨가하였다. 옥살릴 클로라이드 4.48 ml (51.3 mmol)를 -10℃로 냉각시킨 상기 용액에 온도가 -5℃를 초과하지 않도록 하면서 적가하였다. 반응 혼합물을 -5℃로부터 0℃까지에서 1 시간 동안 교반하고, 이어서 냉각없이 실온으로 가온하면서 약 30 분 동안 교반하고, 후속으로 감압 하에 농축시켰다. 잔류물을 디클로로메탄에 녹이고, 용액을 한 번 더 감압 하에 농축시켰다. 상기 절차를 한 번 더 반복하고, 이어서 수득한 산 클로라이드를 고진공 하에 가볍게 건조시키고, 추가 정제없이 추가로 직접 반응시켰다.
N,N-디이소프로필에틸아민 6.1 ml (35.04 mmol)를 무수 THF 25 ml 중 에틸 (+)-(2S)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트 6.05 g (25.03 mmol)의 용액에 첨가하였다. 생성된 용액을 -10℃로 냉각시키고, 온도를 0℃ 미만으로 유지하면서 무수 THF 약 10 ml 중 상기 제조한 산 클로라이드 (7.85 g, 27.5 mmol)의 용액을 적가하였다. 이어서, 반응 혼합물을 -10℃로부터 0℃까지에서 1 시간 동안 교반하고, 이어서, 에틸 아세테이트 및 3 방울의 물을 첨가하였다. 10 분 후, 에틸 아세테이트로 추가로 희석한 혼합물을 1 N 염산, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 디이소프로필 에테르 50 ml로 4 시간 동안 연화처리하였다. 여과한 후, 수득한 고체를 디이소프로필 에테르 40 ml로 한 번 더 연화처리하였다. 수득한 고체를 고진공 하에 철저하게 건조시켰다. 이로써 표적 화합물 8.32 g을 수득하였다. 상기 수득한 여과물을 합하고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 50:1), 추가의 생성물 1.75 g을 수득하였다. 상기 방식으로, 총 10.07 g (이론치의 82.1%)의 표제 화합물을 수득하였다.

실시예 125A
에틸 (+)-(2S)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)부타노에이트

디클로로메탄 13 ml 및 DMF 한 방울을 (3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (약 9:1 부분입체이성질체 혼합물로서) 3.3 g (12.38 mmol)에 첨가하였다. 옥살릴 클로라이드 1.94 ml (22.28 mmol)를 -10℃로 냉각시킨 상기 용액에, 온도가 -5℃를 초과하지 않도록 하면서 적가하였다. 이어서, 반응 혼합물을 -5℃로부터 0℃까지에서 1 시간 동안 교반하고, 후속으로 감압 하에 농축시켰다. 잔류물을 디클로로메탄에 녹이고, 용액을 한 번 더 감압 하에 농축시켰다. 상기 절차를 한 번 더 반복하고, 이어서 수득한 산 클로라이드를 고진공 하에 가볍게 건조시키고, 추가 정제없이 추가로 직접 반응시켰다.
N,N-디이소프로필에틸아민 1.2 ml (6.68 mmol)를 무수 THF 4.8 ml 중 (+)-에틸 (2S)-2-(3-아미노-4-클로로벤질)부타노에이트 1.22 g (4.77 mmol)의 용액에 첨가하였다. 생성된 용액을 -10℃로 냉각시키고, 온도를 0℃ 미만으로 유지하면서 무수 THF 2 ml 중 상기 제조한 산 클로라이드 (1.5 g, 5.25 mmol)의 용액을 적가하였다. 이어서, 반응 혼합물을 -10℃로부터 0℃까지에서 1 시간 동안 교반하고, 이어서 에틸 아세테이트 및 3 방울의 물을 첨가하였다. 10 분 후, 에틸 아세테이트로 추가로 희석한 혼합물을 1 N 염산, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 목적 생성물을 실리카 겔 상에서 잔류물의 크로마토그래피 (이동상 시클로헥산/에틸 아세테이트 40:1)에 의해 단리하였다. 이로써 표제 화합물 2.13 g (이론치의 88.5%)을 수득하였다.

하기 화합물을 유사한 절차에 따라 제조하였다.
실시예 126A
에틸 (+)-(2R)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)부타노에이트


실시예 127A
에틸 2-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-trifluoor-3-메틸부타노일]아미노}-4-플루오로페닐)-트랜스-시클로프로판카르복실레이트

HATU 167 mg (0.44 mmol)을 DMF 및 피리딘의 4:1 혼합물 1.5 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 90 mg (0.34 mmol)의 용액에 첨가하였다. 실온에서 30 분 교반 후, rac-에틸 2-[3-아미노-4-플루오로페닐]-트랜스-시클로프로판카르복실레이트 83 mg (0.37 mmol)을 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 에틸 아세테이트 (약 50 ml)로 희석하고, 포화 염화나트륨 용액으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 농축시키고, 잔류물을 정제용 HPLC에 의해 정제하였다. 이로써 표제 화합물 123 mg (이론치의 77%)을 수득하였다.

실시예 128A
에틸 트레오-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸부타노에이트

피리딘 15 μl (0.19 mmol) 및 HATU 75.8 mg (0.2 mmol)을 DMF 0.98 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 35.5 mg (0.13 mmol)의 용액에 첨가하였다. 반응 혼합물을 실온에서 30 분 동안 교반하고, 이어서 rac-트레오-에틸 3-(3-아미노-4-플루오로페닐)-2-메틸부타노에이트 35 mg (0.15 mmol)을 첨가하였다. 반응 혼합물을 밤새 교반하고, 이어서 정제용 HPLC에 의해 직접 정제하였다. 이로써 표제 화합물 34 mg (이론치의 52%)을 수득하였다.
실시예 129A
에틸 에리트로-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸부타노에이트

피리딘 9.5 μl (117 μmol) 및 HATU 47.7 mg (125 μmol)을 DMF 0.62 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 22.3 mg (84 μmol)의 용액에 첨가하였다. 반응 혼합물을 실온에서 30 분 동안 교반하고, 이어서 rac-에리트로-에틸 3-(3-아미노-4-플루오로페닐)-2-메틸부타노에이트 22 mg (92 μmol)을 첨가하였다. 반응 혼합물을 밤새 교반하고, 이어서 정제용 HPLC에 의해 직접 정제하였다. 이로써 표제 화합물 10.7 mg (이론치의 24%)을 수득하였다.
실시예 130A
tert-부틸 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-시아노페닐)프로파노에이트

0℃에서, 디클로로메탄 중 옥살릴 클로라이드의 2 M 용액 155 μl (0.31 mmol) 및 한 방울의 DMF를 디클로로메탄 1.16 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 41.3 mg (0.16 mmol)의 용액에 첨가하였다. 0℃에서 1 시간 교반 후, 혼합물을 농축시키고, 남아 있는 잔류물을 THF 1 ml 중에 용해시키고, N,N-디이소프로필에틸아민 32 μl (0.19 mmol)를 첨가하고, 혼합물을 0℃로 냉각시키고, THF 2 ml 중 tert-부틸 3-(3-아미노-4-시아노페닐)프로파노에이트 42 mg (0.17 mmol)의 용액을 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 이어서, 혼합물을 물 10 ml에 붓고, 에틸 아세테이트로 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 농축시켰다. 잔류물을 정제용 HPLC에 의해 정제하였다. 이로써 표제 화합물 16.5 mg (이론치의 22%)을 수득하였다.

실시예 131A
메틸 (2E)-3-(2-메틸-3-니트로페닐)아크릴레이트

아르곤 하에, 메틸 아크릴레이트 119.5 g (1.39 mol)을 DMF 2.0 리터 중 2-브로모-6-니트로톨루엔 100 g (0.463 mol), 트리에틸아민 323 ml (2.31 mol), 트리-2-톨릴포스핀 28.2 g (92.6 mmol) 및 아세트산팔라듐(II) 10.4 g (46.3 mmol)의 혼합물에 적가하고, 이어서 혼합물을 125℃에서 36 시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 포화 수성 염화암모늄 용액 4 리터와 함께 교반하고, 총 5 리터의 디에틸 에테르로 3회 추출하였다. 합한 유기 상을 물 및 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하여 건조상태로 만들었다. 수득한 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 석유 에테르/에틸 아세테이트 6:1). 생성물을 헵탄으로 연화처리하고, 수득한 고체를 흡인 여과하고, 고진공 하에 건조시켰다. 이로써 표제 화합물 48.7 g (이론치의 46.6%)을 수득하였다.

실시예 132A
메틸 3-(3-아미노-2-메틸페닐)프로파노에이트

메틸 (2E)-3-(2-메틸-3-니트로페닐)아크릴레이트 48.7 g (220.1 mmol)을 메탄올 2.2 리터 중에 용해시키고, 용액을 연속-흐름 수소화 반응기 (탈레스 나노(Thales Nano; 부다페스트)로부터의 "H-큐브 미디(H-Cube midi)")에서 6 내지 10 ml/분의 유량으로 35 내지 40℃의 반응 온도에서 최대 수소압하에 수소화시켰다. 반응이 종료된 후, 생성물-함유 용액을 감압 하에 농축시켰다. 이로써 고체로서의 표적 생성물 40.0 g (이론치의 92.1%)을 수득하였다.

실시예 133A
메틸 3-(3-아미노-4-클로로-2-메틸페닐)프로파노에이트

실온에서, N-클로로숙신이미드 1.38 g (10.3 mmol)을 아세토니트릴 10 ml 중 메틸 3-(3-아미노-2-메틸페닐)프로파노에이트 2.0 g (10.3 mmol)의 용액에 첨가하였다. 반응 혼합물을 30 분 동안 교반하고, 이어서 에틸 아세테이트로 희석하였다. 혼합물을 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 10:1), 표적 생성물 279 mg (이론치의 11.8%)을 수득하였다.

실시예 134A
3-브로모-6-클로로-2-메틸아닐린

실온에서, N-클로로숙신이미드 8.61 g (64.5 mmol)을 아세토니트릴 150 ml 중 3-브로모-2-메틸아닐린 12.0 g (64.5 mmol)의 용액에 첨가하였다. 반응 혼합물을 60℃에서 7 시간 동안 교반하고, 냉각시킨 후, 감압 하에 농축시켰다. 잔류물을 디클로로메탄에 녹이고, 혼합물을 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 연속적으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 40:1 → 20:1), 표적 생성물 3.78 g (이론치의 26.6%)을 수득하였다.
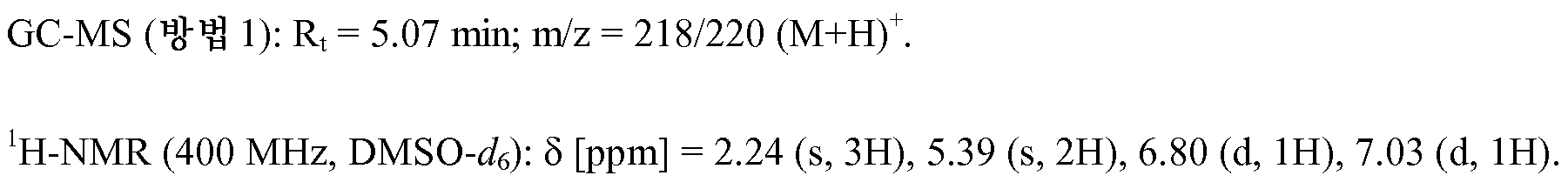
실시예 135A
tert-부틸 (2E)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸아크릴레이트
및
tert-부틸 2-(3-아미노-4-클로로-2-메틸벤질)아크릴레이트

3-브로모-6-클로로-2-메틸아닐린 1.50 g (6.80 mmol), tert-부틸 메타크릴레이트 2.90 g (20.4 mmol) 및 트리에틸아민 4.74 ml (34.0 mmol)를 DMF 10.0 ml 중에 용해시켰다. 반응 용액을 3회 배기시키고, 각 경우에 아르곤으로 다시 환기시켰다. 아세트산팔라듐(II) 152.7 mg (0.68 mmol) 및 트리-2-톨릴포스핀 414.1 mg (1.36 mmol)을 첨가한 후, 혼합물을 한 번 더 2회 배기시키고, 각 경우에 아르곤으로 환기시켰다. 이어서, 반응 혼합물을 150℃에서 2 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 DMF로 세척하였다. 여과물을 감압 하에 농축시키고, 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 100:1). 이로써 두 표제 화합물의 혼합물 1.59 g (비율 약 2:1, 이론치의 83%)을 수득하였다.

실시예 136A
(+/-)-tert-부틸 3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸프로파노에이트

메탄올 5.0 ml 중 tert-부틸 (2E)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸아크릴레이트 및 tert-부틸 2-(3-아미노-4-클로로-2-메틸벤질)아크릴레이트의 혼합물 (실시예 135A) 1.58 g (5.61 mmol)의 용액을 마그네슘 터닝 354 mg (14.6 mmol) 및 몇 알갱이의 아이오딘에 첨가하였다. 혼합물을 실온에서 (처음에는 냉각시키면서) 밤새 교반하였다. 이어서, 1 N 염산 50 ml를 얼음-냉각시키면서 첨가하였다. 이어서, 10% 농도의 수성 수산화나트륨 용액을 첨가하여, 혼합물의 pH를 약 10으로 조정하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 50:1 → 40:1), 표적 생성물 962 mg (이론치의 60.5%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 유량: 20 ml/분; 검출: 220 nm; 주입 부피: 0.28 ml; 온도: 22℃; 이동상: 93% 이소헥산 / 7% 이소프로판올]. 라세미체 962 mg으로 거울상이성질체 1 (실시예 137A) 434 mg 및 거울상이성질체 2 (실시예 138A) 325 mg을 수득하였다.
실시예 137A
(-)-tert-부틸 (2R)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸프로파노에이트

실시예 138A
(+)-tert-부틸 (2S)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸프로파노에이트
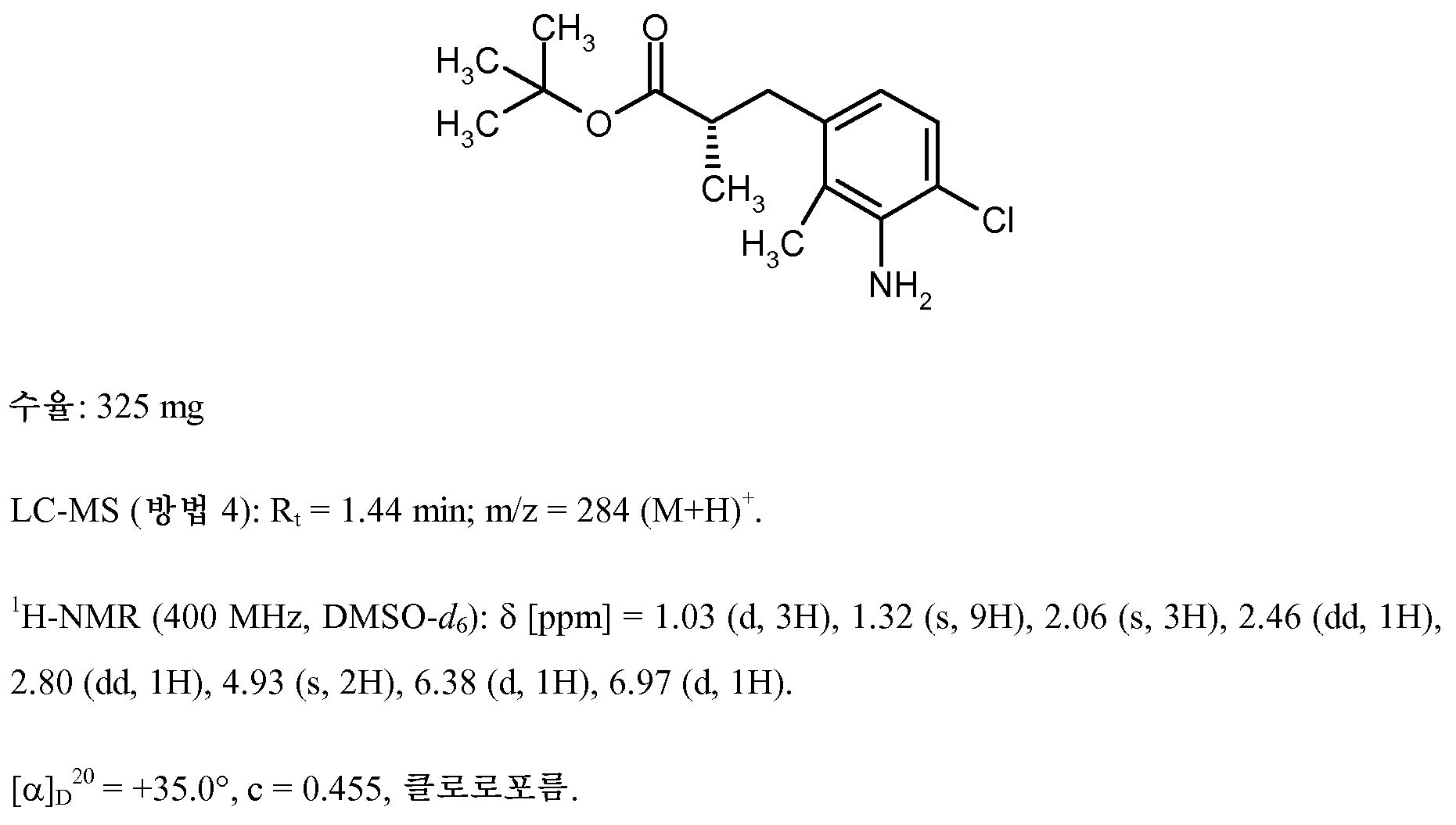
실시예 139A
에틸 4,4,4-트리플루오로-3-(4-플루오로-3-니트로페닐)부트-2-에노에이트

에틸 디에틸포스포노아세테이트 10.9 g (48.5 mmol)을 THF 70 ml 및 DMF 20 ml의 혼합물 중 수소화나트륨 1.86 g (미네랄 오일 중 60%, 46.4 mmol)의 얼음-냉각 현탁액에 서서히 적가하였다. 첨가가 종료된 후, 혼합물을 0℃에서 추가로 30 분 동안 교반하고, 이어서 2,2,2-트리플루오로-1-(4-플루오로-3-니트로페닐)에타논 10.0 g (42.2 mmol)을 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 20:1 → 10:1), 표적 생성물 9.23 g (이론치의 71.2%)을 수득하였다.

실시예 140A
(+/-)-에틸 3-(3-아미노-4-플루오로페닐)-4,4,4-트리플루오로부타노에이트

에틸 4,4,4-트리플루오로-3-(4-플루오로-3-니트로페닐)부트-2-에노에이트 5.0 g (16.3 mmol)을 에탄올 133 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 866 mg을 아르곤 하에 첨가하였다. 실온에서, 반응 혼합물을 수소의 분위기 하에 (대기압) 밤새 격렬하게 교반하였다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하였다. 여과물을 감압 하에 농축시키고, 수득한 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 표적 생성물 3.91 g (이론치의 85.9%)을 수득하였다.

실시예 141A
tert-부틸 2-메틸부타노에이트

2-메틸부티릴 클로라이드 15.0 g (124.4 mmol)을 무수 THF 150 ml 중에 용해시키고, 0℃로 냉각시키고, THF 중 칼륨 tert-부틸레이트의 1 M 용액 114 ml (114 mmol)를 적가하였다. 첨가가 종료된 후, 혼합물을 0℃에서 1 시간 동안, 이어서 실온에서 5 시간 동안 교반하고, 이어서 용매의 약 절반을 감압 하에 제거하였다. 디에틸 에테르를 첨가한 후, 포화 중탄산나트륨 용액을 격렬한 교반하에 적가하였다. 상 분리 후, 수성 상을 디에틸 에테르로 추출하고, 합한 유기 상을 포화 탄산나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 진공 증류에 의해 정제하였다 (19 mm Hg, 40-45℃). 이로써 표적 생성물 총 6.35 g (이론치의 32.3%)을 수득하였다.

실시예 142A
2-브로모-4-(브로모메틸)-1-클로로벤젠

단계 1:
3-브로모-4-클로로벤조산 199.0 g (0.845 mol)을 THF 2.5 리터 중에 용해시키고, 혼합물을 -10℃로 냉각시키고, THF 중 보란의 1 M 용액 1.69 리터 (1.69 mol)를 상기 온도에서 첨가하였다. 반응 혼합물을 밤새 실온으로 가온하고, 이어서 포화 수성 염화암모늄 용액을 첨가하였다. 물을 첨가한 후, 혼합물을 에틸 아세테이트로 2회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 (3-브로모-4-클로로페닐)메탄올 206 g을 조 생성물로서 수득하였고, 이를 후속 단계에 추가 정제없이 사용하였다.
단계 2:
조 (3-브로모-4-클로로페닐)메탄올 260 g (약 1.05 mol)을 디클로로메탄 2.86 리터 중에 용해시키고, 혼합물을 -5℃로 냉각시키고, 삼브로민화인 127.1 g (44.6 ml, 460 mmol)을 서서히 첨가하였다. 첨가가 종료된 후, 교반을 -5℃에서 1 시간 동안 계속하고, 이어서 혼합물을 디클로로메탄 및 물로 희석하였다. 유기 상을 분리하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 2-브로모-4-(브로모메틸)-1-클로로벤젠 280.5 g (이론치의 약 84%)을 조 생성물로서 수득하였다.

실시예 143A
(+/-)-tert-부틸 2-(3-브로모-4-클로로벤질)-2-메틸부타노에이트

아르곤 하에, 디이소프로필아민 5.8 ml (41.6 mmol)를 건조 THF 50 ml 중에 용해시키고, 혼합물을 -30℃로 냉각시켰다. n-부틸리튬 용액 (헥산 중 2.5 M) 16.6 ml (41.6 mmol)를 적가하고, 생성된 혼합물을 0℃로 가온하고, 이어서 -70℃로 냉각시켰다. 반응 온도를 -60℃ 미만으로 유지하면서 THF 20 ml 중 tert-부틸 2-메틸부타노에이트 5.06 g (32.0 mmol)의 용액을 첨가하였다. -60℃에서 4 시간 교반 후, THF 30 ml 중 2-브로모-4-(브로모메틸)-1-클로로벤젠 10.0 g (35.2 mmol)의 용액을 첨가하고, 온도를 한 번 더 -60℃ 미만으로 유지하였다. 이어서, 반응 혼합물을 밤새 서서히 실온으로 가온한 후, 포화 수성 염화암모늄 용액 및 에틸 아세테이트를 첨가하였다. 상 분리 후, 수성 상을 에틸 아세테이트로 2회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 100:1). 이로써 표제 화합물 7.62 g (이론치의 65.9%)을 수득하였다.

실시예 144A
(+/-)-tert-부틸 2-[3-(벤질아미노)-4-클로로벤질]-2-메틸부타노에이트

아르곤 하에, 나트륨 tert-부톡시드 1.59 g (16.6 mmol)을 건조 플라스크 내로 칭량하여 넣고, 무수 톨루엔 34.6 ml를 첨가하였다. (+/-)-tert-부틸-2-(3-브로모-4-클로로벤질)-2-메틸부타노에이트 5.0 g (13.8 mmol), 벤질아민 1.8 ml (16.6 mmol), 트리스(디벤질리덴아세톤)디팔라듐 633 mg (0.69 mmol) 및 (+/-)-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 344 mg (0.55 mmol)을 연속하여 첨가하였다. 이어서, 반응 혼합물을 110℃에서 2.0 시간 동안 교반하였다. 냉각시킨 후, 포화 수성 염화암모늄 용액 및 에틸 아세테이트를 첨가하고, 반응 혼합물을 규조토를 통한 흡인에 의해 여과하였다. 상 분리 후, 유기 상을 포화 염화암모늄 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 80:1). 이로써 여전히 약간 오염된 형태의 표제 화합물 4.44 g (이론치의 약 83%)을 수득하였다.

실시예 145A
(+/-)-tert-부틸 2-(3-아미노-4-클로로벤질)-2-메틸부타노에이트

(+/-)-tert-부틸 2-[3-(벤질아미노)-4-클로로벤질]-2-메틸부타노에이트 2.20 g (약 5.67 mmol)을 에틸 아세테이트 130 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 100 mg을 첨가하였다. 실온에서, 반응 혼합물을 수소의 분위기 하에 대기압에서 밤새 교반하였다. 이어서, 반응 혼합물을 규조토를 통한 흡인에 의해 여과하고, 잔류물을 에틸 아세테이트로 철저하게 세척하고, 합한 여과물을 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1 → 30:1). 이로써 표적 화합물 924 mg (이론치의 54.7%)을 수득하였다.
상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 유량: 20 ml/분; 검출: 220 nm; 주입 부피: 0.30 ml; 온도: 35℃; 이동상: 70% 이소헥산 / 30% 에탄올]. 라세미체 924 mg으로 거울상이성질체 1 (실시예 146A) 405 mg 및 거울상이성질체 2 (실시예 147A) 403 mg을 수득하였다.
실시예 146A
(-)-tert-부틸 2-(3-아미노-4-클로로벤질)-2-메틸부타노에이트 (거울상이성질체 1)

실시예 147A
(+)-tert-부틸-2-(3-아미노-4-클로로벤질)-2-메틸부타노에이트 (거울상이성질체 2)

실시예 148A
2,2,2-트리플루오로-1-(3-니트로페닐)에타논

먼저, 2,2,2-트리플루오로아세토페논 20.0 g (114.9 mmol)을 진한 황산 80 ml에 충전하고, 혼합물을 -10℃로 냉각시켰다. -10℃에서 미리 제조한 진한 황산 20 ml 중 질산 4.8 ml (114.8 mmol)의 용액을 반응 온도가 -5℃를 초과하지 않도록 하면서 상기 혼합물에 적가하였다. 첨가가 종료된 후, 반응 혼합물을 -10℃ 내지 0℃에서 1 시간 동안 교반하고, 이어서 빙수에 조심스럽게 첨가하였다. 50% 농도의 수성 수산화나트륨 용액을 첨가하여 혼합물의 pH를 약 9 내지 10으로 조정하였다. 혼합물을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 먼저 시클로헥산/디클로로메탄 2:1 → 1:1, 최종적으로 순수 디클로로메탄). 이로써 표적 생성물 19.2 g (이론치의 76.2%)을 수득하였다.

실시예 149A
tert-부틸 (2E/Z)-4,4,4-트리플루오로-3-(3-니트로페닐)부트-2-에노에이트

tert-부틸 (디에톡시포스포릴)아세테이트 25.9 ml (110.4 mmol)를 THF 37.2 ml 및 DMF 37.2 ml의 혼합물 중 수소화나트륨 4.41 g (미네랄 오일 중 60%, 110.4 mmol)의 0℃로 냉각시킨 현탁액에 적가하였다. 30 분 후, 2,2,2-트리플루오로-1-(3-니트로페닐)에타논 18.6 g (84.9 mmol)을 첨가하고, 냉각 조를 제거하고, 반응 혼합물을 실온에서 2 시간 동안 교반하였다. 이어서, 반응 혼합물을 물에 첨가하고, 염화나트륨으로 포화시킨 후, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 100:1 → 20:1). 이로써 표적 생성물 18.0 g (이론치의 66.8%)을 E/Z 이성질체 혼합물로서 수득하였다.

실시예 150A
(+/-)-tert-부틸 3-(3-아미노페닐)-4,4,4-트리플루오로부타노에이트

tert-부틸 (2E/Z)-4,4,4-트리플루오로-3-(3-니트로페닐)부트-2-에노에이트 18.0 g (56.7 mmol)을 에탄올 100 ml 중에 용해시키고, 아르곤을 사용하여 산소를 제거하였다. 탄소 상 팔라듐 (10%) 1.21 g을 첨가한 후, 혼합물을 실온에서 수소의 분위기 하에 대기압에서 밤새 격렬하게 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에탄올로 철저하게 세척하고, 여과물을 감압 하에 농축시키고, 수득한 생성물을 고진공 하에 밤새 건조시켰다. 이로써 표적 생성물 13.7 g (이론치의 83.7%)을 수득하였다.

실시예 151A
(+/-)-tert-부틸 3-(3-아미노-4-클로로페닐)-4,4,4-트리플루오로부타노에이트

먼저, (+/-)-tert-부틸-3-(3-아미노페닐)-4,4,4-트리플루오로부타노에이트 13.6 g (47.0 mmol)을 아세토니트릴 100 ml에 충전하고, N-클로로숙신이미드 6.28 g (47.0 mmol)을 실온에서 첨가하였다. 먼저, 반응 혼합물을 60℃에서 12 시간 동안 교반하고, 이어서 실온에서 3 일 동안 정치시켰다. 감압 하에 농축시킨 후, 잔류물을 실리카 겔 상에서 크로마토그래피의 의해 분리하고 (이동상 시클로헥산/에틸 아세테이트 100:1), 목적 표적 생성물을 단리하였다. 이로써 표제 화합물 4.49 g (이론치의 29.5%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 20 mm; 유량: 15 ml/분; 검출: 220 nm; 주입 부피: 0.20 ml; 온도: 35℃; 이동상: 70% 이소헥산 / 30% 이소프로판올]. 라세미체 4.49 g으로 거울상이성질체 1 (실시예 152A) 2.02 g 및 거울상이성질체 2 (실시예 153A) 2.04 g을 수득하였다.
실시예 152A
(-)-tert-부틸 (3R)-3-(3-아미노-4-클로로페닐)-4,4,4-트리플루오로부타노에이트

실시예 153A
(+)-tert-부틸 (3S)-3-(3-아미노-4-클로로페닐)-4,4,4-트리플루오로부타노에이트

실시예 154A
tert-부틸 시클로부틸아세테이트

시클로부틸아세트산 4.0 g (35.0 mmol)을 디클로로메탄 20 ml 중에 용해시키고, DMF 한 방울을 첨가하고, 0℃로 냉각시킨 후에 옥살릴 클로라이드 4.0 ml (45.6 mmol)를 적가하였다. 반응 혼합물을 0℃ 내지 10℃에서 2 시간 동안 교반하고, 이어서 냉각 감압 하에 농축시켰다. 잔류물을 무수 디클로로메탄에 녹이고, 한 번 더 냉각 감압 하에 농축시켰다. 상기 절차를 한 번 더 반복하고, 이어서 수득한 산 클로라이드를 고진공 하에 5 분 동안 가볍게 건조시켰다. 이어서, 잔류물을 무수 THF 20 ml에 녹이고, 0℃로 냉각시키고, THF 중 칼륨 tert-부톡시드의 1 M 용액 28 ml (28 mmol)를 적가하였다. 첨가가 종료된 후, 냉각을 제거하고, 혼합물을 실온에서 1 시간 동안 교반하고, 이어서 물에 첨가하였다. 혼합물을 디클로로메탄으로 3회 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 조 표적 생성물 3.88 g (이론치의 약 65%)을 수득하였다.
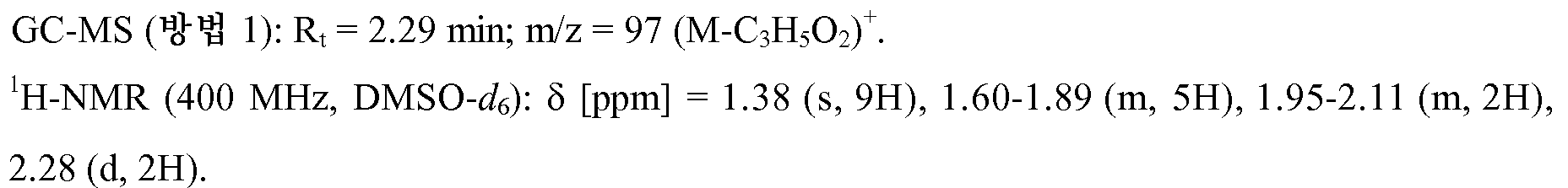
실시예 155A
tert-부틸 시클로프로필아세테이트

시클로프로필아세트산 10.0 g (99.9 mmol)을 디클로로메탄 50 ml 중에 용해시키고, DMF 한 방울을 첨가하고, 0℃로 냉각시킨 후, 옥살릴 클로라이드 9.6 ml (109.9 mmol)를 적가하였다. 반응 혼합물을 0℃ 내지 10℃에서 2 시간 동안 교반하고, 이어서 냉각 감압 하에 농축시켰다. 잔류물을 가볍게 (약 5 분) 고진공 하에 건조시키고, 이어서 무수 THF 20 ml에 녹이고, 0℃로 냉각시키고, THF 중 칼륨 tert-부톡시드의 1 M 용액 89.9 ml (89.9 mmol)를 적가하였다. 첨가가 종료된 후, 냉각을 제거하고, 혼합물을 실온에서 2 시간 동안 교반한 후, 대부분의 THF를 감압 하에 제거하였다 (150 mm Hg 이하, 수조 약 30℃). 디에틸 에테르 및 0.5 N 수성 수산화나트륨 용액을 잔류물에 첨가하였다. 상 분리 후, 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 가볍게 건조시켰다. 이로써 조 표적 생성물 8.38 g (이론치의 약 53%)을 수득하였다.

실시예 156A
(+/-)-tert-부틸 3-(3-브로모-4-클로로페닐)-2-시클로부틸프로파노에이트

아르곤 하에, 디이소프로필아민 2.9 ml (20.8 mmol)를 건조 THF 30 ml 중에 용해시키고, 혼합물을 -20℃로 냉각시켰다. n-부틸리튬 용액 (헥산 중 2.5 M) 8.3 ml (20.8 mmol)를 적가하고, 생성된 혼합물을 -20℃로 30 분 동안 교반하고, 이어서 -78℃로 냉각시켰다. 상기 온도에서, THF 10 ml 중 tert-부틸 시클로부틸아세테이트 2.60 g (약 15.3 mmol, 조 물질)의 용액을 첨가하였다. -78℃에서 4 시간 교반 후, THF 10 ml 중 2-브로모-4-(브로모메틸)-1-클로로벤젠 3.95 g (13.9 mmol)의 용액을 첨가하였다. 반응 혼합물을 밤새 서서히 실온으로 가온하고, 이어서 포화 수성 염화암모늄 용액을 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 남아 있는 고체를 시클로헥산/디클로로메탄 (1:1) 30 ml로 연화처리하고, 여과하였다. 고체를 한 번 더 시클로헥산/디클로로메탄 (1:1) 10 ml로 연화처리하고, 다시 여과하고; 상기 절차를 한 번 더 반복하였다. 모든 여과물을 수집하고, 합하고, 감압 하에 농축시켰다. 이어서, 상기 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 추가로 정제하였다 (이동상 순수 시클로헥산 → 시클로헥산/디클로로메탄 20:1 → 10:1). 이로써 표제 화합물 2.24 g (이론치의 43.2%)을 수득하였다.
하기 실시예를 유사한 방식으로 수득하였다.
실시예 157A
tert-부틸 3-(3-브로모-4-클로로페닐)-2-시클로프로필프로파노에이트

tert-부틸 시클로프로필아세테이트 및 2-브로모-4-(브로모메틸)-1-클로로벤젠으로부터, 표제 화합물 3.13 g을 수득하였다 (이론치의 45%).

실시예 158A
(+/-)-tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2-시클로부틸프로파노에이트

아르곤 하에, 나트륨 tert-부톡시드 848.6 mg (8.83 mmol), 트리스(디벤질리덴아세톤)디팔라듐 337 mg (0.39 mmol) 및 rac-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 183 mg (0.29 mmol)을 건조 플라스크 내로 칭량하여 넣고, 고진공 하에 10 분 동안 유지하고, 이어서 아르곤으로 환기시켰다. 무수 톨루엔 5 ml, 벤질아민 0.96 ml (8.83 mmol), 및 무수 톨루엔 5 ml 중 (+/-)-tert-부틸 3-(3-브로모-4-클로로페닐)-2-시클로부틸프로파노에이트 2.75 g (7.36 mmol)의 용액을 연속하여 첨가하였다. 3회 더, 반응 혼합물을 배기시키고, 각 경우에 아르곤으로 환기시키고, 이어서 혼합물을 110℃에서 3 시간 동안 교반하였다. 냉각시킨 후, 반응 혼합물을 포화 수성 염화암모늄 용액에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기 상을 합하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/디클로로메탄 4:1 → 2:1, 이어서 순수 디클로로메탄). 이로써 표제 화합물 1.95 g (이론치의 65.1%)을 수득하였다.

하기 실시예를 유사한 방식으로 수득하였다.
실시예 159A
tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2-시클로프로필프로파노에이트

tert-부틸 3-(3-브로모-4-클로로페닐)-2-시클로프로필프로파노에이트 및 벤질아민으로부터, 표제 화합물 2.51 g을 수득하였다 (이론치의 74.7%).
실시예 160A
(+/-)-tert-부틸 3-(3-아미노-4-클로로페닐)-2-시클로부틸프로파노에이트

(+/-)-tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2-시클로부틸프로파노에이트 1.85 g (4.63 mmol)을 에틸 아세테이트 10 ml 중에 용해시키고, 아르곤을 사용하여 산소를 제거하고, 탄소 상 팔라듐 (10%) 98 mg (0.093 mmol)을 첨가하였다. 반응 혼합물을 실온에서 수소의 분위기 하에 대기압에서 4 시간 동안 교반하고, 이어서 주말에 걸쳐 정치시켰다. 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 감압 하에 농축시키고, 여과물의 잔류물을 고진공 하에 건조시켰다. 상기 잔류물 (출발 물질 및 표적 생성물의 약 1:1 혼합물)을 한 번 더 에틸 아세테이트 약 10 ml 중에 용해시키고, 아르곤을 사용하여 산소를 제거하고, 한 번 더 탄소 상 팔라듐 (10%) 98 mg (0.093 mmol)을 첨가하였다. 다시, 반응 혼합물을 실온에서 수소의 분위기 하에 대기압에서 4 시간 동안 교반하였다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 실리카 겔 상에서 크로마토그래피 정제하여 (이동상 시클로헥산/에틸 아세테이트 20:1) 표적 생성물 1.12 g (이론치의 78.2%)을 수득하였다.

상기 수득한 라세미체를 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 유량: 20 ml/분; 검출: 230 nm; 주입 부피: 0.80 ml; 온도: 25℃; 이동상: 95% 이소헥산 / 5% 에탄올]. 라세미체 790 mg으로 거울상이성질체 1 (실시예 161A) 318 mg 및 거울상이성질체 2 (실시예 162A) 339 mg을 수득하였다.
실시예 161A
tert-부틸 3-(3-아미노-4-클로로페닐)-2-시클로부틸프로파노에이트 (거울상이성질체 1)

실시예 162A
tert-부틸 3-(3-아미노-4-클로로페닐)-2-시클로부틸프로파노에이트 (거울상이성질체 2)

실시예 163A
(+/-)-tert-부틸 3-(3-아미노-4-클로로페닐)-2-시클로프로필프로파노에이트

(+/-)-tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2-시클로프로필프로파노에이트 2.50 g (4.63 mmol)을 에틸 아세테이트 160 ml 중에 용해시키고, 혼합물을 아르곤을 사용하여 산소를 제거하고, 탄소 상 팔라듐 (10%) 150 mg을 첨가하였다. 반응 혼합물을 실온에서 수소의 분위기 하에 대기압에서 8 시간 동안 교반하였다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 순수 시클로헥산 → 시클로헥산/에틸 아세테이트 20:1). 이로써 표적 생성물 1.41 g (이론치의 73.6%)을 수득하였다.

실시예 164A
메틸 3-(3-아미노-4-클로로페닐)헥스-2-에노에이트
및
메틸 3-(3-아미노-4-클로로페닐)헥스-3-에노에이트

트리에틸아민 33.8 ml (242.2 mmol)를 DMF 100 ml 중 5-브로모-2-클로라닐린 10.0 g (48.4 mmol) 및 메틸 (2E)-헥스-2-에노에이트 8.69 g (67.8 mmol)의 혼합물에 첨가하였다. 혼합물을 3회 배기시키고, 각 경우에 아르곤으로 환기시켰다. 아세트산팔라듐(II) 1.09 g (4.84 mmol) 및 트리-2-톨릴포스핀 2.95 g (9.69 mmol)을 첨가한 후, 혼합물을 2회 더 배기시키고, 각 경우에 아르곤으로 환기시키고, 이어서 반응 혼합물을 150℃에서 4 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 물에 첨가하고, 염화나트륨으로 포화시키고, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 최종적으로 고진공 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 두 표제 화합물의 혼합물 7.70 g (이론치의 62.7%, 약 1.5:1 비율의 α,β-불포화 이성질체)을 수득하였다.

실시예 165A
(+/-)-메틸 3-(3-아미노-4-클로로페닐)헥사노에이트
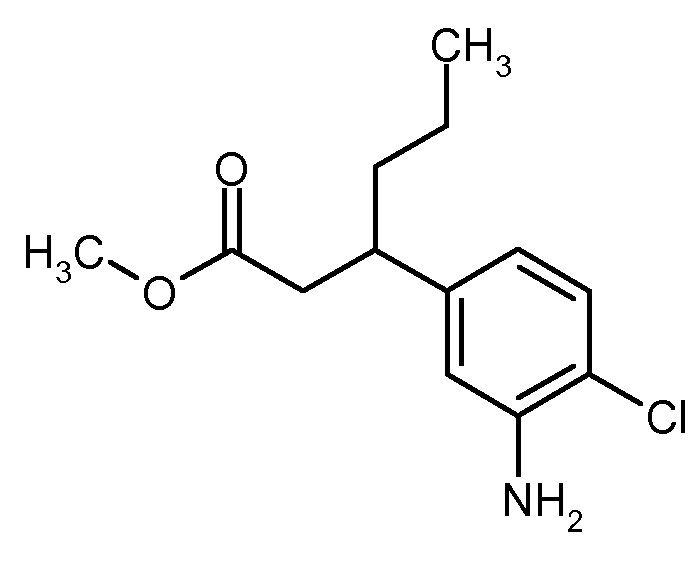
메틸 3-(3-아미노-4-클로로페닐)헥스-2-에노에이트 및 메틸 3-(3-아미노-4-클로로페닐)헥스-3-에노에이트의 혼합물 (약 1.5:1, 실시예 164A) 7.70 g (30.3 mmol)을 에틸 아세테이트 45 ml 중에 용해시키고, 탄소 상 팔라듐 (5%) 646 mg (0.303 mmol)을 첨가하고, 혼합물을 수소의 분위기 하에 대기압 및 실온에서 교반하였다. 10 시간 후, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시켰다. 잔류물을 에틸 아세테이트 약 50 ml에 녹이고, 추가의 탄소 상 팔라듐 (5%) 약 650 mg을 첨가하고, 혼합물을 수소의 분위기 하에 대기압 및 실온에서 교반하였다. 추가로 36 시간 후, 반응 혼합물을 한 번 더 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시켰다. 잔류물을 에틸 아세테이트 800 ml에 녹이고, 한 번 더 탄소 상 팔라듐 (5%) 약 650 mg을 첨가하고, 혼합물을 수소의 분위기 하에 대기압 및 실온에서 24 시간 동안 교반하였다. 다시, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1 → 10:1). 이로써 표적 생성물 및 출발 물질의 혼합물 총 4.79 g을 수득하였다. 상기 혼합물을 에틸 아세테이트 180 ml 중에 용해시키고, 추가의 탄소 상 팔라듐 (10%) 603 mg (0.566 mmol)을 첨가하고, 혼합물을 수소의 분위기 하에 대기압 및 실온에서 밤새 교반하였다. 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표적 생성물 4.45 g (이론치의 약 57%)을 수득하였다.

실시예 166A 및 실시예 167A
메틸 3-(3-아미노-4-클로로페닐)펜트-2-에노에이트
및
메틸 3-(3-아미노-4-클로로페닐)펜트-3-에노에이트

트리에틸아민 16.9 ml (121 mmol)를 DMF 50 ml 중 5-브로모-2-클로라닐린 5.0 g (24.2 mmol) 및 메틸 2-펜테노에이트 5.53 g (48.4 mmol)의 혼합물에 첨가하였다. 혼합물을 3회 배기시키고, 각 경우에 아르곤으로 환기시켰다. 아세트산팔라듐(II) 544 mg (2.42 mmol) 및 트리-2-톨릴포스핀 1.47 g (4.84 mmol)을 첨가한 후, 혼합물을 2회 더 배기시키고, 각 경우에 아르곤으로 환기시키고, 이어서 반응 혼합물을 150℃에서 6 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 실온에서 밤새 정치시키고, 이어서 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기 상을 합하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피하여 (이동상 시클로헥산/에틸 아세테이트 50:1 → 10:1), 두 이성질체 표적 생성물을 분리된 형태로 수득하였다. 이로써 메틸 3-(3-아미노-4-클로로페닐)펜트-2-에노에이트 (이론치의 14.6%) 0.85 g 및 메틸 3-(3-아미노-4-클로로페닐)펜트-3-에노에이트 (이론치의 52.5%) 3.05 g을 수득하였다.
메틸 3-(3-아미노-4-클로로페닐)펜트-2-에노에이트 (실시예 166A):

메틸 3-(3-아미노-4-클로로페닐)펜트-3-에노에이트 (실시예 167A):

실시예 168A
(+/-)-메틸 3-(3-아미노-4-클로로페닐)펜타노에이트

메틸 3-(3-아미노-4-클로로페닐)펜트-3-에노에이트 3.05 g (12.7 mmol) 및 메틸 3-(3-아미노-4-클로로페닐)펜트-2-에노에이트 0.85 g (3.55 mmol)을 함께 에틸 아세테이트 500 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 346 mg (0.325 mmol)을 첨가하고, 혼합물을 실온에서 수소의 분위기 하에 대기압에서 밤새 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시켰다. 잔류물을 고진공 하에 건조시켜 표적 생성물 3.73 g (이론치의 94.8%)을 수득하였다.

실시예 169A
에틸 (3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

톨루엔 중 리튬 헥사메틸디실라지드의 1 M 용액 81.5 ml (81.5 mmol)을 -20℃로 냉각시키고, 무수 톨루엔 50 ml 중 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 10.0 g (50.3 mmol)의 용액을 적가하였다. 혼합물을 10 분 동안 교반하였다. 이어서, -20℃에서, 미리 제조한 무수 톨루엔 50 ml 중 1-브로모-4-클로로-2-플루오로벤젠 14.8 g (70.6 mmol), 아세트산팔라듐(II) 366 mg (1.63 mmol) 및 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노) 비페닐 1.35 g (3.42 mmol)의 용액을 적가하였다. 첨가가 종료된 후, 냉각을 제거하고, 먼저 생성된 반응 혼합물을 실온에서 1 시간 동안 교반하고, 이어서 80℃에서 밤새 교반하였다. 냉각시킨 후, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 톨루엔으로 반복적으로 세척하고, 수득한 여과물을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피한 후 (이동상 시클로헥산/에틸 아세테이트 100:1 → 100:4), 표제 화합물 4.26 g (이론치의 25.1%)을 수득하였다.
하기 실시예를 유사한 방식으로 수득하였다.
실시예 170A
에틸 (3R)-2-(4-클로로-3-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 2.0 g 및 1-브로모-4-클로로-3-플루오로벤젠 2.96 g으로부터, 표적 화합물 2.47 g을 수득하였다.

실시예 171A
에틸 (3R)-2-(4-클로로-2-메틸페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

톨루엔 중 리튬 헥사메틸디실라지드의 1 M 용액 22.5 ml (22.5 mmol)을 -20℃로 냉각시키고, 무수 톨루엔 15 ml 중 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 2.76 g (50.3 mmol)의 용액을 적가하였다. 혼합물을 10 분 동안 교반하였다. 이어서, -20℃에서, 미리 제조한 무수 톨루엔 15 ml 중 2-브로모-5-클로로톨루엔 4.0 g (19.5 mmol), 아세트산팔라듐(II) 101 mg (0.45 mmol) 및 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노)비페닐 371 mg (0.94 mmol)의 용액을 적가하였다. 첨가가 종료된 후, 냉각을 제거하고, 생성된 반응 혼합물을 먼저 실온에서 1 시간 동안, 이어서 100℃에서 밤새 교반하였다. 냉각시킨 후, 혼합물을 셀라이트를 통해 여과하고, 잔류물을 톨루엔으로 반복적으로 세척하고, 수득한 여과물을 감압 하에 농축시켰다. 이로써 표제 화합물 3.10 g을 조 생성물로서 수득하였고, 이를 추가로 직접 반응시켰다.
실시예 172A
에틸 (3R)-2-(4-클로로-3-메틸페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

톨루엔 중 리튬 헥사메틸디실라지드의 1 M 용액 29.2 ml (29.2 mmol)를 -10℃로 냉각시키고, 무수 톨루엔 26 ml 중 에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 4.30 g (23.4 mmol)의 용액을 적가하였다. 혼합물을 10 분 동안 교반하였다. 이어서, -10℃에서, 미리 제조한 무수 톨루엔 26 ml 중 5-브로모-2-클로로톨루엔 5.0 g (19.5 mmol, 80% 순수), 아세트산팔라듐(II) 131 mg (0.58 mmol) 및 2-디시클로헥실포스피노-2'-(N,N-디메틸아미노)비페닐 483 mg (1.23 mmol)의 용액을 적가하였다. 생성된 반응 혼합물을 먼저 실온에서 1 시간 동안, 이어서 80℃에서 4 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 에틸 아세테이트로 희석하고, 포화 수성 중탄산나트륨 용액으로 2회, 포화 염화나트륨 용액으로 1회 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 표제 화합물 7.80 g을 조 생성물로서 수득하였고, 이를 추가로 직접 반응시켰다.
하기 실시예를 유사한 방식으로 수득하였다.
실시예 173A
에틸 (3R)-2-(3,4-디클로로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물)

에틸 (3R)-4,4,4-트리플루오로-3-메틸부타노에이트 3.91 g 및 4-브로모-1,2-디클로로벤젠 5.0 g으로부터, 표적 화합물 7.54 g을 조 생성물로서 수득하였다.

실시예 174A
(3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

에틸 (3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물) 4.26 g (13.6 mmol)을 메탄올 22 ml, THF 22 ml 및 물 11 ml의 혼합물 중에 용해시키고, 50% 농도의 수성 수산화나트륨 용액 10.9 g을 0℃에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 이어서, 대부분의 유기 용매를 감압 하에 제거하였다. 남아 있는 혼합물을 물로 희석하고, 디에틸 에테르로 추출하였다. 상 분리 후, 유기 상을 폐기하고, 반-진한 염산 (pH 약 2)을 사용하여 수성 상을 산성화시키고, 에틸 아세테이트로 반복적으로 추출하였다. 합한 에틸 아세테이트 상을 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 표적 생성물 3.38 g (이론치의 76.7%)을 부분입체이성질체의 혼합물로서 수득하였고, 이를 후속 반응에 추가 정제없이 사용할 수 있었다.

하기 두 카르복실산을 유사한 방식으로 수득하였다.
실시예 175A
(3R)-2-(4-클로로-3-플루오로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

부분입체이성질체 비 약 1:1.

실시예 176A
(3R)-2-(4-클로로-3-메틸페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

부분입체이성질체 비 약 5:1.
실시예 177A
(3R)-2-(4-클로로-2-메틸페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

에틸 (3R)-2-(4-클로로-2-메틸페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물) 3.10 g (조 물질, 약 10.04 mmol)을 메탄올 10 ml, THF 10 ml 및 물 5 ml의 혼합물 중에 용해시키고, 50% 농도의 수성 수산화나트륨 용액 8.03 g을 0℃에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하였다. 이어서, 1 N 염산 (pH 약 2)을 사용하여 혼합물을 산성화시키고, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1 → 4:1). 이로써 표적 생성물 1.46 g (이론치의 51.8%)을 부분입체이성질체의 혼합물 (약 5:1)로서 수득하였다.

실시예 178A
(3R)-2-(3,4-디클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물)

에틸 (3R)-2-(3,4-디클로로페닐)-4,4,4-트리플루오로-3-메틸부타노에이트 (부분입체이성질체 혼합물) 3.77 g (조 물질, 약 11.5 mmol)을 메탄올 14 ml, THF 14 ml 및 물 5 ml의 혼합물 중에 용해시키고, 50% 농도의 수성 수산화나트륨 용액 9.16 g을 0℃에서 첨가하였다. 반응 혼합물을 40℃에서 약 6 시간 동안 교반하였다. 이어서, 1 N 염산 (pH 약 2)을 사용하여 혼합물을 산성화시키고, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 표적 화합물 3.94 g을 조 생성물로서 수득하였고, 이를 후속 반응에 추가 정제없이 사용할 수 있었다.
실시예 179A
(3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 (부분입체이성질체 혼합물)

(3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부탄산 (부분입체이성질체 혼합물) 660 mg (2.32 mmol)을 디클로로메탄 2 ml 중에 용해시켰다. 작은 방울의 DMF를 첨가한 후, 반응 용액을 -5℃에서 0℃로 냉각시키고, 옥살릴 클로라이드 0.4 ml (4.64 mmol)를 적가하였다. 냉각을 제거하고, 기체 발생이 중지될 때까지 반응 혼합물을 실온에서 1 시간 동안 교반하였다. 이어서, 혼합물을 감압 하에 농축시켰다. 잔류물을 무수 디클로로메탄에 2회 녹이고, 각 경우에 감압 하에 재농축시키고, 최종적으로 잔류물을 고진공 하에 건조시켰다. 이로써 표적 생성물 640 mg을 수득하였고, 이를 추가 정제없이 직접 추가로 반응시켰다.
하기 실시예를 일반적 절차 1 (염기로서 피리딘 또는 N,N-디이소프로필에틸아민을 사용한 DMF 중에서의 4,4,4-트리플루오로-3-메틸-2-페닐부탄산 유도체와 아닐린의 HATU-매개 아미드 커플링)에 따라 제조하였다.













실시예 196A
(+)-에틸 (2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트

에틸 (+)-(2S)-3-(3-아미노-4-클로로페닐)-2-메틸프로파노에이트 280.7 mg (1.16 mmol)을 무수 THF 1.5 ml 중에 용해시키고, N,N-디이소프로필에틸아민 0.26 ml (1.48 mmol)를 첨가하고, -10℃로 냉각시킨 후, 무수 THF 0.5 ml 중 (3R)-2-(4-클로로-2-플루오로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 320 mg (조 물질, 약 1.06 mmol)의 계내 제조된 용액을 적가하였다. 첨가가 종료된 후, 반응 혼합물을 -10℃ 내지 0℃에서 30 분 동안 교반하였다. 몇 방울의 물을 첨가한 후, 이어서 혼합물을 디클로로메탄으로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 먼저 정제용 RP-HPLC에 의해 (이동상 메탄올/물), 이어서 실리카 겔 상에서 크로마토그래피에 의해 (이동상 시클로헥산/에틸 아세테이트 40:1) 정제하였다. 이로써 표적 화합물 144 mg (이론치의 26.9%)을 수득하였다.

실시예 197A
메틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-메틸페닐)프로파노에이트

메틸 3-(3-아미노-4-클로로-2-메틸페닐)프로파노에이트 265 mg (1.16 mmol)을 무수 THF 1.5 ml 중에 용해시키고, N,N-디이소프로필에틸아민 0.28 ml (1.63 mmol)을 첨가하고, -10℃로 냉각시킨 후, 무수 THF 0.5 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 398 mg (조 물질, 약 1.40 mmol)의 계내 제조된 용액을 적가하였다. 첨가가 종료된 후, 반응 혼합물을 -10℃에서 실온으로 1 시간에 걸쳐 가온하고, 이어서 에틸 아세테이트로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 RP-HPLC에 의해 정제하였다 (이동상 메탄올/물). 이로써 표적 화합물 485 mg (이론치의 87.5%)을 수득하였다.
실시예 198A
(+)-tert-부틸 (2R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-메틸페닐)-2-메틸프로파노에이트

(-)-tert-부틸 (2R)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸프로파노에이트 200 mg (0.705 mmol)을 무수 THF 1 ml 중에 용해시키고, N,N-디이소프로필에틸아민 0.17 ml (0.987 mmol)를 첨가하고, -10℃로 냉각시킨 후, 무수 THF 0.2 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 241 mg (조 물질, 약 0.846 mmol)의 계내 제조된 용액을 적가하였다. 첨가가 종료된 후, 반응 혼합물을 -10℃에서 실온으로 2 시간에 걸쳐 가온하고, 이어서 물에 첨가하였다. 수성 상을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표적 화합물 282 mg (이론치의 75.2%)을 수득하였다.

하기 실시예를 유사한 방식으로 수득하였다.
실시예 199A
(+)-tert-부틸 (2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-메틸페닐)-2-메틸프로파노에이트

(+)-tert-부틸 (2S)-3-(3-아미노-4-클로로-2-메틸페닐)-2-메틸프로파노에이트 200 mg 및 새로 제조한 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일 클로라이드 241 mg으로부터, 표적 생성물 287 mg을 수득하였다 (이론치의 75.2%).

실시예 200A
(+)-tert-부틸 2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)-2-메틸부타노에이트 (부분입체이성질체 A)

(-)-tert-부틸 2-(3-아미노-4-클로로벤질)-2-메틸부타노에이트 (거울상이성질체 1) 225 mg (0.756 mmol) 및 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 231 mg (0.907 mmol)을 피리딘 0.9 ml 및 DMF 2.7 ml 중에 용해시키고, HATU 345 mg (0.907 mmol)을 실온에서 첨가하였다. 반응 혼합물을 45℃에서 밤새 교반하고, 이어서 추가의 0.5 당량의 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 및 0.6 당량의 HATU를 첨가하였다. 반응 혼합물을 45℃에서 추가로 3 시간 동안 교반하고, 이어서 냉각시킨 후, 에틸 아세테이트로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 RP-HPLC에 의해 (이동상 아세토니트릴/물), 후속으로 실리카 겔 상에서 크로마토그래피에 의해 (이동상 시클로헥산/에틸 아세테이트 40:1) 정제하였다. 이로써 표적 생성물 177 mg (이론치의 35.6%)을 수득하였다.

실시예 201A
메틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥사노에이트 (부분입체이성질체 혼합물)

(+/-)-메틸 3-(3-아미노-4-클로로페닐)헥사노에이트 1.45 g (5.67 mmol) 및 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 1.81 g (6.80 mmol)을 피리딘 5.0 ml 및 DMF 10.0 ml 중에 용해시키고, HATU 2.80 g (7.37 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 에틸 아세테이트로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 먼저 시클로헥산, 이어서 시클로헥산/에틸 아세테이트 50:1). 두 분획에서, 총 2.02 g의 표적 생성물을 수득하였다 (이론치의 70.6%).

실시예 202A
메틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜타노에이트 (부분입체이성질체 혼합물)

(+/-)-메틸 3-(3-아미노-4-클로로페닐)펜타노에이트 500 mg (2.07 mmol) 및 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 668.9 mg (2.48 mmol)을 피리딘 1.7 ml 및 DMF 3.3 ml 중에 용해시키고, HATU 1.02 g (2.69 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 에틸 아세테이트로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 먼저 시클로헥산, 이어서 시클로헥산/에틸 아세테이트 50:1). 이로써 표적 생성물 675 mg (이론치의 66.6%)을 수득하였다.

실시예 203A
(+)-tert-부틸 (3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부타노에이트

(+)-tert-부틸 (3S)-3-(3-아미노-4-클로로페닐)-4,4,4-트리플루오로부타노에이트 2.0 g (6.18 mmol) 및 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 1.98 g (7.41 mmol)을 피리딘 5.0 ml 및 DMF 10.0 ml 중에 용해시키고, HATU 3.05 g (8.03 mmol)을 실온에서 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 추가의 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 1.98 g (7.41 mmol) 및 HATU 3.05 g (8.03 mmol)을 첨가하였다. 반응 혼합물을 40℃에서 추가로 8 시간 동안 교반하고, 이어서 냉각시킨 후, 에틸 아세테이트로 희석하였다. 혼합물을 1 N 염산 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 먼저 시클로헥산, 이어서 시클로헥산/에틸 아세테이트 50:1). 상기 방식으로 수득한 생성물 (2.7 g)을 실리카 겔 상에서 또 다른 크로마토그래피에 의해 다시 재정재하였다 (이동상 시클로헥산/에틸 아세테이트 100:1). 이로써 표적 생성물 1.80 g (이론치의 50.9%)을 수득하였다.

하기 실시예를 유사한 방식으로 제조하였다.
실시예 204A
(+)-tert-부틸 (3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부타노에이트

(-)-tert-부틸 (3R)-3-(3-아미노-4-클로로페닐)-4,4,4-트리플루오로부타노에이트 1.0 g (3.09 mmol) 및 (+)-(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 988 mg (3.71 mmol)으로부터 표적 생성물 1.2 g (이론치의 68%)을 수득하였다.

실시예 205A
tert-부틸 3-(3-아미노-2-메틸페닐)프로파노에이트

아르곤 하에, tert-부틸 프로프-2-에노에이트 201 ml (1.39 mol)을 DMF 2 리터 중 1-브로모-2-메틸-3-니트로벤젠 100 g (463 mmol), 트리에틸아민 322 ml (2.31 mol), 트리-2-톨릴포스핀 28.18 g (92.58 mmol) 및 아세트산팔라듐(II) 10.39 g (46.29 mmol)의 용액에 적가하고, 이어서 혼합물을 125℃에서 36 시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 포화 수성 염화암모늄 용액과 함께 교반하고, 유기 상을 분리하였다. 수성 상을 tert-부틸 메틸 에테르로 3회 추출하고, 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하여 건조상태로 만들었다. 수득한 잔류물을 실리카 겔 상에서 플래쉬 크로마토그래피에 의해 정제하였다 (이동상 석유 에테르/에틸 아세테이트 9:1). 이로써 중간체 tert-부틸 (2E)-3-(2-메틸-3-니트로페닐)프로프-2-에노에이트 89 g (338 mmol, 이론치의 73%)을 무색 고체로서 수득하였다.
상기 고체 88 g (334 mmol)을 에탄올 2 리터 중에 용해시키고, 탄소 상 팔라듐 (10%) 7 g을 실온에서 첨가하고, 혼합물을 대기압 하에 18 시간 동안 수소화시켰다. 반응이 완료된 후, 반응 용액을 규조토를 통해 여과하고, 수득한 여과물을 감압 하에 농축시켰다. 이로써 표제 화합물 61.3 g (260.5 mmol, 이론치의 78%)을 무색 고체로서 수득하였다.

실시예 206A
tert-부틸 3-(3-브로모-4-클로로페닐)-2,2-디메틸프로파노에이트

아르곤 하에, 디이소프로필아민 4.0 ml (28.8 mmol)를 건조 THF 50 ml 중에 용해시키고, 혼합물을 -30℃로 냉각시켰다. n-부틸리튬 용액 (헥산 중 2.5 M) 11.5 ml (28.8 mmol)를 적가하였다. 생성된 혼합물을 0℃로 가온하고, 이어서 -70℃로 냉각시켰다. 이어서, 온도를 -60℃ 미만으로 유지하면서 THF 20 ml 중 tert-부틸-2-메틸프로파노에이트 2.77 g (19.2 mmol)의 용액을 첨가하였다. -60℃에서 4 시간 교반 후, 반응 온도를 한 번 더 -60℃ 미만으로 유지하면서 THF 30 ml 중 2-브로모-4-(브로모메틸)-1-클로로벤젠 6.0 g (21.1 mmol)의 용액을 첨가하였다. 반응 혼합물을 실온으로 서서히 가온하면서 밤새 교반하고, 이어서 포화 수성 염화암모늄 용액 및 에틸 아세테이트를 첨가하였다. 상 분리 후, 수성 상을 에틸 아세테이트로 2회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1 → 4:1). 이로써 표제 화합물 5.6 g (이론치의 84%)을 수득하였다.
실시예 207A
tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2,2-디메틸프로파노에이트

아르곤 하에, 나트륨 tert-부톡시드 1.73 g (17.95 mmol)을 건조 플라스크 내로 칭량하여 넣고, 무수 톨루엔 40 ml를 첨가하였다. tert-부틸 3-(3-브로모-4-클로로페닐)-2,2-디메틸프로파노에이트 5.2 g (14.96 mmol), 벤질아민 1.96 ml (17.95 mmol), 트리스(디벤질리덴아세톤)디팔라듐 685 mg (0.75 mmol) 및 (+/-)-2,2'-비스(디페닐포스피노)-1,1'-비나프틸 373 mg (0.60 mmol)을 연속하여 첨가하였다. 반응 혼합물을 110℃에서 2.0 시간 동안 교반하고, 이어서 실온으로 냉각시키고, 상기 온도에서 밤새 교반하였다. 이어서, 포화 수성 염화암모늄 용액 및 에틸 아세테이트를 첨가하고, 반응 혼합물을 규조토를 통한 흡인에 의해 여과하였다. 상 분리 후, 유기 상을 포화 염화암모늄 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다 (이동상 아세토니트릴/물). 이로써 표제 화합물 2.78 g (이론치의 50%)을 수득하였다.
실시예 208A
tert-부틸 3-(3-아미노-4-클로로페닐)-2,2-디메틸프로파노에이트

tert-부틸 3-[3-(벤질아미노)-4-클로로페닐]-2,2-디메틸프로파노에이트 2.7 g (약 7.22 mmol)을 에틸 아세테이트 150 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 100 ml를 첨가하였다. 반응 혼합물을 실온에서 수소의 분위기 하에 대기압에서 밤새 교반하였다. 이어서, 혼합물을 규조토를 통한 흡인에 의해 여과하고, 잔류물을 에틸 아세테이트로 철저하게 세척하고, 합한 여과물을 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1 → 7:1). 이로써 표적 화합물 1.49 g (이론치의 72.7%)을 수득하였다.

실시예 209A
N,N-디벤질-5-브로모-2-클로로아닐린

아르곤 하에, 수소화나트륨 9.69 g (242.16 mmol, 미네랄 오일 중 60%)을 THF 100 ml 중에 현탁시키고, 혼합물을 0℃로 냉각시켰다. 이어서, THF 50 ml 중에 용해된 5-브로모-2-클로로아닐린 20.0 g (96.86 mmol)을 서서히 적가하고, 혼합물을 0℃에서 30 분 동안 교반하였다. 이어서, THF 150 ml 중에 용해된 벤질 브로마이드 39.76 g (232.47 mmol)을 서서히 반응 혼합물에 첨가하고, 이어서 혼합물을 실온으로 가온하였다. 혼합물을 실온에서 밤새 교반하고, 이어서 빙수 150 ml에 조심스럽게 부었다. 유기 상을 분리하고, 이어서 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 이소프로판올을 수득한 조 생성물에 첨가하고, 형성된 결정을 흡인 여과하고, 40℃에서 고진공 하에 건조시켰다. 이로써 표제 화합물 14 g을 수득하였다. 여과물을 증발시키고, 수득한 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 추가의 표제 화합물 7.57 g (총 수율: 21.57 g, 이론치의 58%)을 수득하였다.
하기 화합물을 실시예 209A와 유사하게 수득하였다.

실시예 211A
[4-클로로-3-(디벤질아미노)페닐]보론산

-78℃에서 아르곤 하에, 헥산 중 n-부틸리튬의 2.5 M 용액 20.2 ml (50.42 mmol)를 THF/디에틸 에테르 (1:1) 350 ml 중 N,N-디벤질-5-브로모-2-클로로아닐린 15 g (38.79 mmol)의 용액에 서서히 적가하였다. 반응 용액을 -78℃에서 60 분 동안 교반하고, 이어서 트리이소프로필 보레이트 14.3 ml (62.1 mmol)를 서서히 첨가하였다. 후속으로, 반응 용액을 -78℃에서 추가로 15 분 동안 교반하고, 이어서 실온으로 서서히 가온하고, 교반을 상기 온도에서 밤새 계속하였다. 이어서, 빙수 150 ml를 계량하여 넣었다. 유기 상을 분리하고, 이어서 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1 → 4:1). 이로써 표제 화합물 9 g (이론치의 66%)을 수득하였다.
실시예 212A
tert-부틸 시클로부틸리덴아세테이트

실온에서 아르곤 하에, 시클로부타논 3.0 g (42.8 mmol)을 디클로로메탄 160 ml 중에 용해시키고, 이어서 tert-부틸 (트리페닐-λ5-포스파닐리덴) 아세테이트 20.95 g (55.64 mmol) 및 벤조산 0.68 g (5.56 mmol)을 첨가하였다. 반응 혼합물을 실온에서 밤새 교반하고, 이어서 농축 건조시켰다. 잔류물을 디에틸 에테르 25 ml로 연화처리하고, 혼합물을 4℃에서 12 시간 동안 보관하였다. 침전된 트리페닐포스판 옥시드를 여과하고, 여과물을 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표제 화합물 9.3 g (이론치의 99%)을 수득하였다.

실시예 213A
tert-부틸 시클로프로필리덴아세테이트

실온에서, THF 중 테트라-n-부틸암모늄 플루오라이드의 1 M 용액 55 ml (55 mmol)를 THF 240 ml 중 [(1-에톡시시클로프로필)옥시](트리메틸)실란 9.65 g (55.34 mmol), tert-부틸 (트리페닐-λ5-포스파닐리덴)아세테이트 25 g (66.41 mmol) 및 벤조산 8.11 g (66.41 mmol)의 용액에 적가하였다. 교반 1 시간 후, 반응 혼합물을 80℃로 가열하고, 상기 온도에서 2 시간 동안 교반하였다. 이어서, 회전 증발기를 이용하여, 용매를 증류시켰다 (200 mbar, 조 온도 40℃). 수득한 잔류물을 디에틸 에테르에 녹이고, 혼합물을 4℃로 냉각시키고, 상기 온도에서 1 시간 동안 정치시켰다. 생성된 침전물 (트리페닐포스판 옥시드)을 여과하였다. 이어서, 회전 증발기를 이용하여, 여과물을 용매로부터 분리해냈다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표제 화합물 3.58 g (이론치의 42%)을 수득하였다.

실시예 214A
에틸 (3,3-디메톡시시클로부틸리덴)아세테이트
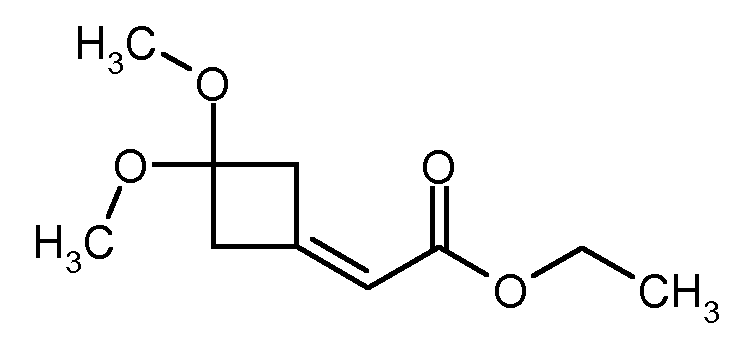
톨루엔 50 ml 중 1,1-디메톡시에텐 3.93 g (44.59 mmol) 및 에틸 부타-2,3-디에노에이트 5 g (44.59 mmol)의 용액을 환류하에 가열하고, 24 시간 동안 교반하였다. 실온으로 냉각시킨 후, 반응 혼합물을 용매로부터 분리해내고, 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표제 화합물 1.9 g (이론치의 21%)을 무색 액체로서 수득하였고, 이를 후속 반응에 추가 특성화 없이 사용하였다.
실시예 215A
tert-부틸 {1-[4-클로로-3-(디벤질아미노)페닐]시클로프로필}아세테이트
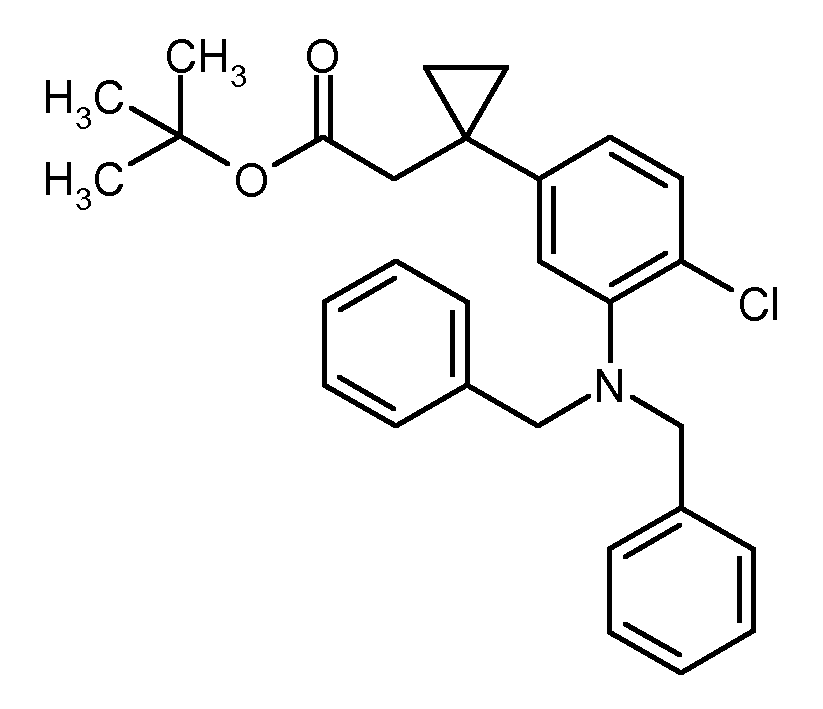
용액 A의 제조: 아르곤 하에, N,N-디벤질-2-클로로-5-요오도아닐린 300 mg (0.69 mmol)을 THF 3 ml 중에 용해시키고, 용액을 -78℃로 냉각시켰다. 이어서, THF 중 이소프로필마그네슘 클로라이드의 2M 용액 0.4 ml (0.80 mmol)를 서서히 적가하였다. 이어서, 반응 용액을 -40℃로 서서히 가온하고, 상기 온도에서 30 분 동안 교반하였다.
용액 B의 제조: 실온에서 아르곤 하에, 염화리튬 6 mg (0.14 mmol) 및 염화구리(I) 13 mg (0.07 mmol)을 THF 12 ml 중에 현탁시키고, 이어서 클로로(트리메틸)실란 84 μl (0.66 mmol) 및 tert-부틸 시클로프로필리덴 아세테이트 102 mg (0.66 mmol)을 첨가하였다. 이어서, 용액을 실온에서 추가로 1 시간 동안 교반하였다.
용액 B를 -40℃로 냉각시키고, 용액 A에 서서히 적가하였다. 이어서, 수득한 반응 혼합물을 -40℃에서 추가로 1 시간 동안 교반하였다. 이어서, 빙냉 반포화 수성 염화암모늄 용액 20 ml를 반응 혼합물에 첨가하였다. 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하고, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 농축 건조시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 20:1). 이로써 표제 화합물 135 mg (이론치의 42%)을 수득하였다.
하기 화합물을 실시예 13A와 유사하게 수득하였다.

실시예 218A
에틸 {1-[4-클로로-3-(디벤질아미노)페닐]-3-옥소시클로부틸}아세테이트

에틸 {1-[4-클로로-3-(디벤질아미노)페닐]-3,3-디메톡시시클로부틸}아세테이트 770 mg (1.52 mmol)을 THF 10 ml 중에 용해시키고, 1 M 염산 2 ml를 첨가하고, 혼합물을 50℃에서 1 시간 동안 교반하였다. 이어서, 반응 용액을 물 10 ml 및 에틸 아세테이트 10 ml로 희석하였다. 상을 분리하고, 이어서 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 회전 증발기를 이용하여 농축 건조시켰다. 이로써 표제 화합물 607 mg (이론치의 87%)을 수득하였다.
실시예 219A
에틸 {1-[4-클로로-3-(디벤질아미노)페닐]-3,3-디플루오로시클로부틸}아세테이트

아르곤 하에, [에틸(트리플루오로-λ4-술파닐)아미노]에탄 0.3 ml (2.27 mmol)를 디클로로메탄 2 ml에 첨가하였다. 반응 용액을 0℃로 냉각시키고, 이어서 디클로로메탄 3 ml 중 에틸 {1-[4-클로로-3-(디벤질아미노)페닐]-3-옥소시클로부틸}아세테이트 175 mg (0.38 mmol)을 서서히 첨가하였다. 이어서, 용액을 실온으로 서서히 가온하고, 상기 온도에서 밤새 교반하였다. 이어서, 반응 혼합물을 빙수 50 ml에 붓고, 유기 상을 분리하였다. 수성 상을 디클로로메탄으로 3회 더 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하고, 수득한 조 생성물을 정제용 HPLC에 의해 정제하였다 (이동상 메탄올/물 8:2). 이로써 표제 화합물 59 mg (이론치의 32%)을 수득하였다.
실시예 220A
tert-부틸 [1-(3-아미노-4-클로로페닐)시클로프로필]아세테이트

tert-부틸 {1-[4-클로로-3-(디벤질아미노)페닐]시클로프로필}아세테이트 135 mg (0.29 mmol)을 에틸 아세테이트 10 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 15 mg을 첨가하고, 혼합물을 실온에서 수소의 분위기 하에 대기압에서 2 시간 동안 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 잔류물을 에틸 아세테이트로 세척하고, 여과물을 농축시켰다. 이로써 표제 화합물 73 mg (이론치의 89%)을 수득하였다.
하기 화합물을 실시예 220A와 유사하게 수득하였다.

실시예 223A
tert-부틸 (2E)-3-(3-아미노-4-시아노페닐)-2-메틸아크릴레이트

아르곤 하에, 디옥산 20 ml 중 2-아미노-4-브로모벤조니트릴 2.0 g (10.15 mmol), tert-부틸 2-메틸아크릴레이트 2.165 g (2.5 ml, 15.23 mmol), 트리스(디벤질리덴아세톤)디팔라듐 93 mg (0.10 mmol), 트리-tert-부틸포스핀 41 mg (0.20 mmol) 및 N,N-디시클로헥실메틸아민 2.4 ml (11.17 mmol)의 혼합물을 120℃로 가열하고, 상기 온도에서 밤새 교반하였다. 반응을 확인하고 (TLC, 이동상 시클로헥산/에틸 아세테이트 9:1), 이어서 추가의 트리스(디벤질리덴아세톤)디팔라듐 10 mg, 트리-tert-부틸포스핀 10 mg 및 tert-부틸 2-메틸아크릴레이트 500 μl를 첨가하고, 혼합물을 120℃에서 추가로 4 시간 동안 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 여과물을 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸아세테이트 9:1 → 4:1). 이로써 표제 화합물 1.375 g (이론치의 52%)을 수득하였다.
실시예 224A
tert-부틸 3-(3-아미노-4-시아노페닐)-2-메틸프로파노에이트
tert-부틸 (2E)-3-(3-아미노-4-시아노페닐)-2-메틸아크릴레이트 1370 mg (5.3 mmol)을 에틸 아세테이트 30 ml 중에 용해시키고, 탄소 상 팔라듐 (10%) 282 mg을 첨가하고, 혼합물을 실온에서 수소의 분위기 하에 대기압에서 3 일 동안 교반하였다. 이어서, 반응 혼합물을 셀라이트를 통해 여과하고, 여과 잔류물을 에틸 아세테이트로 세척하고, 합한 여과물을 농축시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다 (이동상 아세토니트릴/물). 이로써 표제 화합물 870 mg (이론치의 63%)을 수득하였다.
하기 화합물을 실시예 54A와 유사하게 수득하였다.

실시예 226A
에틸 (3R)-2-[4-(2,2-디클로로-3-옥소시클로부틸)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트

에틸 (3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노에이트 3.83 g (13.38 mmol)을 디에틸 에테르 50 ml 중에 용해시키고, 아연-구리 커플 2.67 g (20.74 mmol) 및 1,2-디메톡시에탄 6.5 ml를 연속하여 첨가하였다. 이어서, 트리클로로아세틸 클로라이드 4 ml (36.1 mmol)를 수득한 현탁액에 서서히 적가하였다. 이어서, 반응 용액을 환류 하에 가열하고, 밤새 교반하였다. 디클로로메탄을 첨가한 후, 반응 혼합물을 물 및 포화 염화나트륨 용액으로 연속적으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 감압 하에 농축시켰다. 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상: 시클로헥산/에틸 아세테이트 4:1). 이로써 표제 화합물 4.57 g (이론치의 86%)을 황색빛 오일의 형태로 수득하였고, 이를 후속 반응에 추가 특성화 없이 사용하였다.
실시예 227A
에틸 (3R)-4,4,4-트리플루오로-3-메틸-2-[4-(3-옥소시클로부틸)페닐]부타노에이트

포화 수성 염화암모늄 용액 100 ml를 THF 100 ml 중 에틸 (3R)-2-[4-(2,2-디클로로-3-옥소시클로부틸)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트 4.57 g (11.51 mmol) 및 아연 분말 3.76 g (57.5 mmol)에 첨가하고, 이어서 혼합물을 75℃에서 5 시간 동안 교반하였다. 실온으로 냉각시키고 디클로로메탄을 첨가한 후, 반응 혼합물을 물로 세척하였다. 상 분리 후, 수성 상을 디클로로메탄으로 3회 역추출하였다. 이어서, 합한 유기 상을 황산마그네슘 상에서 건조시키고, 여과하고, 감압 하에 농축시켰다. 이로써 표제 화합물 1.21 g (이론치의 32%)을 수득하였다.

실시예 228A
에틸 (3R)-2-[4-(3,3-디플루오로시클로부틸)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트

아르곤 하에, 톨루엔 20 ml로 희석한 THF 중 1,1'-[(트리플루오로-λ4-술파닐)이미노]비스(2-메톡시에탄) (데속소플루오르)의 50% 농도의 용액 7.3 ml (5.16 mmol)을 먼저 충전하고, 혼합물을 5℃로 냉각시키고, 1 M 삼플루오린화붕소 디에틸 에테르 복합체 용액 47 μl (0.37 mmol)를 서서히 첨가하였다. 혼합물을 5℃에서 2 시간 동안 교반하였다. 이어서, 톨루엔 20 ml 중에 용해된 에틸 (3R)-4,4,4-트리플루오로-3-메틸-2-[4-(3-옥소시클로부틸)페닐]부타노에이트 1.21 g (3.69 mmol)을 반응 용액에 서서히 첨가한 후, 혼합물을 55℃로 가온하고, 상기 온도에서 48 시간 동안 교반하였다. 이어서, 반응 혼합물을 톨루엔 20 ml 및 2 M 수성 수산화나트륨 용액 20 ml로 이루어진 0℃로 냉각시킨 혼합물에 첨가하였다. 유기 상을 분리하고, 수성 상을 에틸 아세테이트로 3회 더 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시켰다. 여과한 후, 용매를 감압 하에 제거하였다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표제 화합물 558 mg (이론치의 43%)을 황색빛 액체로서 수득하였다.

실시예 229A
에틸 (3R)-2-[4-(2,2-디플루오로시클로프로필)페닐]-4,4,4-트리플루오로-3-메틸부타노에이트
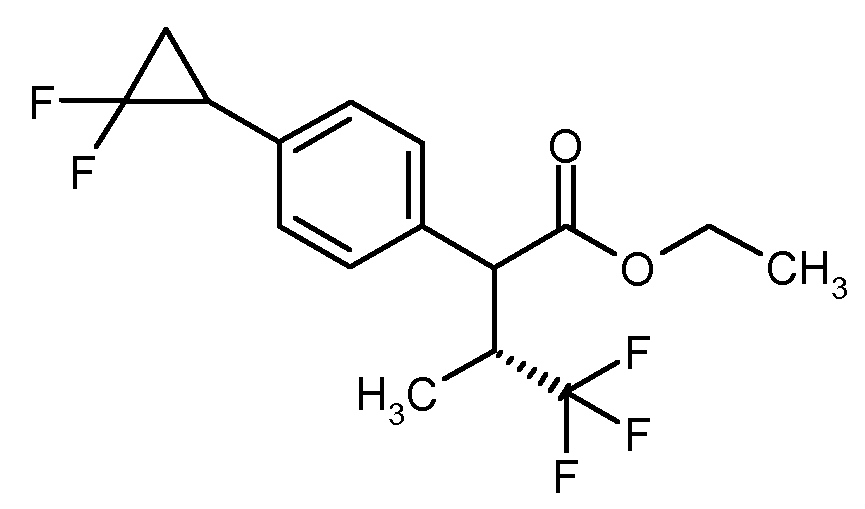
에틸 (3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노에이트 1.58 g (5.52 mmol), 플루오린화나트륨 23 mg (0.55 mmol) 및 2,6-디-tert-부틸 4-메틸페놀 24 mg (0.11 mmol)을 110℃로 가열하고, 5 분 동안 교반하였다. 이어서, 트리메틸실릴 디플루오로(플루오로술포닐)아세테이트 1.9 ml (9.38 mmol)를 서서히 적가하고, 혼합물을 110℃에서 60 분 동안 교반하였다 (주의: 약 30 분 후 기체 발생). 실온으로 냉각시키고 에틸 아세테이트 및 포화 수성 탄산수소나트륨 용액을 첨가한 후, 유기 상을 분리하고, 황산마그네슘 상에서 건조시키고, 여과하고, 농축 건조시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피로 정제하였다 (이동상 시클로헥산/ 디클로로메탄 4:1). 이로써 표제 화합물 1.5 g (이론치의 81%)을 수득하였다.

하기 표에 열거된 화합물을 실시예 70A와 유사하게 제조하였다.


하기 표에 열거된 화합물을 실시예 82A와 유사하게 제조하였다.

실시예 235A
tert-부틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2,2-디메틸프로파노에이트

(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 400 mg (1.50 mmol)을 디클로로메탄 24 ml 중에 용해시키고, 1-클로로-N,N,2-트리메틸프로프-1-엔-1-아민 320 mg (2.40 mmol)을 첨가하고, 혼합물을 실온에서 30 분 동안 교반하였다. 이어서, 피리딘 364 μl (4.5 mmol) 및 tert-부틸 3-(3-아미노-4-클로로페닐)-2,2-디메틸프로파노에이트 510 mg (1.80 mmol)을 첨가하고, 혼합물을 실온에서 2 시간 동안 교반하였다. 이어서, 반응 혼합물을 감압 하에 농축시키고, 수득한 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸아세테이트 20:1). 이로써 표적 화합물 462 mg (이론치의 58%)을 수득하였다.

하기 표에 열거된 화합물을 유사한 방식으로 제조하였다.





하기 표에 열거된 화합물을 실시예 89A와 유사하게 제조하였다.



실시예 247A
tert-부틸 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-메틸페닐)프로파노에이트

DMF 4 ml 중 (2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부탄산 100 mg (0.38 mmol), tert-부틸 3-(3-아미노-2-메틸페닐)프로파노에이트 88 mg (0.38 mmol), 2-(1H-7-아자벤조트리아졸-1-일)-1,1,3,3-테트라메틸우로늄 헥사플루오로포스페이트 (HATU) 213 mg (0.56 mmol) 및 피리딘 1 ml의 혼합물을 실온에서 밤새 교반하였다. 반응이 종료된 후, 반응 혼합물을 추가의 후처리 없이 직접 정제용 HPLC에 의해 (이동상 아세토니트릴/물) 그의 성분으로 분리하였다. 이로써 표제 화합물 151 mg (이론치의 83%)을 무색 오일로서 수득하였다.

하기 화합물을 유사한 방식으로 수득하였다.

실시예 249A
디에틸 [2-(4-클로로페닐)프로판-2-일]말로네이트

아르곤 하에, 디에틸 에테르 2.5 ml 중 1-브로모-4-클로로벤젠 1 g (5.23 mmol)을 디에틸 에테르 5 ml 중 마그네슘 터닝 254 mg (10.45 mmol)에 서서히 첨가하였다. 반응이 시작된 후, 추가의 디에틸 에테르 2.5 ml 중 1-브로모-4-클로로벤젠 1 g (5.23 mmol)을 반응 혼합물에 계량하여 넣었다. 반응 혼합물을 실온에서 30 분 동안 교반하고, 염화구리(I) 103 mg (1.05 mmol)을 첨가하고, 이어서 혼합물을 -10℃로 냉각시켰다. 이어서, 디에틸 프로판-2-일리덴말로네이트 2.09 g (10.45 mmol)을 서서히 적가하였다. 후속으로, 반응 혼합물을 환류하에 가열하고, 상기 온도에서 3 시간 동안 교반하였다. 이어서, 빙냉 1 M 염산 20 ml를 매우 서서히 첨가하였다. 상 분리 후, 수성 상을 디에틸 에테르로 3회 더 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 이어서 농축 건조시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다 (이동상 메탄올/물 70:30). 이로써 표제 화합물 800 mg (이론치의 25%)을 수득하였다.

실시예 250A
에틸 3-(4-클로로페닐)-3-메틸부타노에이트

DMSO 5 ml 중 디에틸 [2-(4-클로로페닐)프로판-2-일]말로네이트 796 mg (2.55 mmol), 염화리튬 216 mg (5.10 mmol) 및 물 46 μl (2.55 mmol)의 용액을 환류하에 가열하고, 상기 온도에서 4 시간 동안 교반하였다. 실온으로 냉각시킨 후, 디에틸 에테르 20 ml 및 물 20 ml를 반응 혼합물에 첨가하였다. 상 분리 후, 유기 상을 물로 3회 더 세척하고, 유기 상을 황산마그네슘 상에서 건조시키고, 이어서 농축 건조시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다 (이동상 메탄올/물 70:30). 이로써 표제 화합물 276 mg (이론치의 45%)을 수득하였다.

실시예 251A
에틸 3-(4-클로로-3-니트로페닐)-3-메틸부타노에이트

에틸 3-(4-클로로페닐)-3-메틸부타노에이트 276 mg (1.45 mmol)을 디클로로메탄 10 ml 중에 용해시키고, 혼합물을 0℃로 냉각시켰다. 이어서, 니트로늄 테트라플루오로보레이트 278 mg (1.38 mmol)을 조금씩 첨가하고, 혼합물을 0℃ 내지 10℃의 온도에서 4 시간 동안 교반하였다. 이어서, 물 10 ml 및 디클로로메탄 10 ml를 첨가하고, 상을 분리하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 농축 건조시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1). 이로써 표제 화합물 223 mg (이론치의 68%)을 수득하였다.

실시예 252A
에틸 3-(3-아미노-4-클로로페닐)-3-메틸부타노에이트

탄소 상 팔라듐 (10%) 40 mg을 에틸 아세테이트 10 ml 중 에틸 3-(4-클로로-3-니트로페닐)-3-메틸부타노에이트 213 mg (0.75 mmol)의 용액에 첨가하였다. 반응 혼합물을 실온에서 1 bar의 수소압을 이용하여 밤새 수소화시켰다. 이어서, 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켰다. 이로써 표적 화합물 166 mg (이론치의 87%)을 황색빛 오일로서 수득하였다.
하기 화합물을 실시예 235A와 유사하게 수득하였다.

예시적 실시양태:
일반적 절차 2: 트리플루오로아세트산을 사용한 tert-부틸 에스테르의 상응하는 카르복실산으로의 절단
0℃ 내지 실온에서, 트리플루오로아세트산 (TFA)을 약 2:1 - 1:2 (v/v)의 디클로로메탄/TFA 비에 도달할 때까지 디클로로메탄 중 당해 tert-부틸 에스테르의 용액 (농도 약 0.1 - 2.0 mol/l; 추가로 임의로 한 방울의 물)에 적가하였다. 혼합물을 실온에서 1-24 시간 동안 교반하고; 필요한 경우, 완전한 전환이 달성될 때까지 혼합물을 40℃로 가온하였다. 이어서, 반응 혼합물을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 (디클로로메탄/에틸 아세테이트 또는 시클로헥산/에틸 아세테이트 혼합물로 용리, 적절한 경우 소량의 아세트산 첨가, 또는 디클로로메탄/메탄올 혼합물 사용), 아세토니트릴 또는 물/아세토니트릴 혼합물로부터의 결정화에 의해, 또는 정제용 RP-HPLC에 의해 (이동상: 아세토니트릴/물 구배) 정제할 수 있었다.
하기 실시예를 일반적 절차 2에 따라 제조하였다.







실시예 12
(+)-3-(4-플루오로-3-{[(2S,3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노일]아미노}페닐)프로판산

tert-부틸 (+)-3-(4-플루오로-3-{[(2S,3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노일]아미노}페닐)프로파노에이트 283 mg (0.590 mmol)을 디옥산 중 염화수소의 4 N 용액 5.9 ml 중에 용해시키고, 혼합물을 실온에서 24 시간 동안 교반하였다. 이어서, 휘발성 성분을 감압 하에 제거하였다. 잔류물을 두 정제용 RP-HPLC에 의해 정제하였다 (이동상: 아세토니트릴/물 구배). 이로써 표제 화합물 48 mg (이론치의 19.2%)을 수득하였다.

실시예 13
(+)-3-(4-클로로-3-{[(2S,3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노일]아미노}페닐)프로판산

tert-부틸 (+)-3-(4-클로로-3-{[(2S,3R)-4,4,4-트리플루오로-3-메틸-2-(4-비닐페닐)부타노일]아미노}페닐)프로파노에이트 249.0 mg (0.502 mmol)을 디옥산 중 염화수소의 4 N 용액 3.8 ml 중에 용해시키고, 혼합물을 실온에서 24 시간 동안 교반하였다. 이어서, 반응 혼합물을 동결시키고 (-78℃), 후속으로 고진공 하에 동결건조시켰다. 잔류물을 정제용 RP-HPLC에 의해 정제하였다 (이동상: 아세토니트릴/물 구배). 이로써 표제 화합물 167.4 mg (이론치의 75.8%)을 수득하였다.

실시예 14
(+)-3-[4-클로로-3-({(2S,3R)-4,4,4-트리플루오로-2-[4-(1-플루오로비닐)페닐]-3-메틸부타노일}아미노)페닐]프로판산

수산화리튬 16.1 mg (0.674 mmol)을 메탄올, THF 및 물 (각 경우에 1.0 ml)의 혼합물 중 (+)-3-[4-클로로-3-({(2S,3R)-4,4,4-트리플루오로-2-[4-(1-플루오로비닐)페닐]-3-메틸부타노일}아미노)페닐]프로파노에이트 212 mg (0.449 mmol)의 0℃로 냉각시킨 용액에 첨가하였다. 이어서, 혼합물을 실온으로 가온하고, 실온에서 3 시간 동안 교반하고, 이어서 물로 희석하고, 1 N 염산 (pH 약 2)을 사용하여 산성화시켰다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기 상을 합하고, 감압 하에 농축시켰다. 조 생성물을 먼저 RP-HPLC에 의해 재정제하였다 (이동상: 아세토니트릴/물 구배). 이어서, 염기성 가수분해 동안 형성된 2R 부분입체이성질체를 키랄 상 상에서 정제용 HPLC에 의해 제거하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.25 ml; 온도: 35℃; 이동상: 90% 이소헥산/10% 에탄올; 유량: 15 ml/분; 검출: 220 nm]. 이로써 표제 화합물 74.0 mg (이론치의 36.0%)을 수득하였다.

실시예 15
(+)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)프로판산

tert-부틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)프로파노에이트 30.13 g (59.74 mmol)을 디클로로메탄 1000 ml 중에 용해시키고, 트리플루오로아세트산 92 ml를 실온에서 첨가하였다. 반응 혼합물을 실온에서 3.5 시간 동안 교반하였다. 이어서, 디클로로메탄 및 물을 첨가하였다. 유기 상을 분리하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 고진공 하에 철저하게 건조시켰다. 이로써 표적 화합물 26.31 g (이론치의 98.3%)을 수득하였다.

실시예 16
(+)-(2R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산

에틸 (2R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트 990 mg (2.02 mmol)을 아세트산 5.7 ml 중에 용해시키고, 진한 염산 2.7 ml를 첨가하였다. 혼합물을 100℃에서 1 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 감압 하에 농축시켰다. 잔류물을 에틸 아세테이트에 녹이고, 몇 방울의 포화 중탄산나트륨 용액을 첨가하면서 물로 반복적으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상: 먼저 디클로로메탄, 이어서 디클로로메탄/에틸 아세테이트 10:1). 이로써 표제 화합물 652 mg (이론치의 69.9%)을 수득하였다.

실시예 17
(+)-(2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산

방법 A:
에틸 (2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트 2.45 g (5.0 mmol), 아세트산 6.0 ml 및 20% 농도의 수성 황산 20 ml의 혼합물을 환류 하에 7 시간 동안 교반하였다. 냉각시킨 후, 반응 혼합물을 물에 첨가하였다. 수성 상을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 잔류물을 한 번 더 에틸 아세테이트에 녹이고, 몇 방울의 포화 중탄산나트륨 용액을 첨가하면서 물로 반복적으로 세척하였다. 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 10:1 → 4:1). 이로써 표제 화합물 1.88 g (이론치의 81.4%)을 수득하였다.

방법 B:
(+)-에틸 (2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로파노에이트 12.99 g (26.49 mmol), 아세트산 60 ml 및 30% 농도의 수성 황산 60 ml의 혼합물을 환류 하에 3 시간 동안 (조 온도 140℃) 교반하였다. 냉각시킨 후, 반응 혼합물을 물에 첨가하였다. 수성 상을 에틸 아세테이트로 3회 추출하고, 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 고진공 하에 밤새 건조시켰다. 상기 방식으로 수득한 조 생성물을 디이소프로필 에테르 90 ml와 함께 먼저 50℃에서 1 시간 동안, 이어서 실온에서 4 시간 동안 교반하였다. 여과한 후, 고체를 고진공 하에 건조시켰다. 이로써 표적 화합물 (분획 1) 7.84 g (이론치의 64%)을 수득하였다. 농축시키고 디이소프로필 에테르 30 ml로 새로이 처리한 후 여과물로부터 추가 분량을 단리하였다. 고진공 하에 건조시켜 약간 오염된 표적 화합물 (분획 2) 1.65 g (이론치의 13.5%)을 수득하였다.

실시예 18
(+)-[1-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-시클로프로필]아세트산

메틸 [1-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)시클로프로필]아세테이트 106 mg (0.23 mmol)을 빙초산 4 ml 및 진한 염산 2 ml 중에 용해시키고, 혼합물을 100℃에서 1 시간 동안 교반하였다. 이어서, 반응 혼합물을 물 10 ml로 희석하고, 후속으로 수성 용액을 에틸 아세테이트 (각 경우에 10 ml)로 3회 추출하였다. 합한 유기 상을 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 수득한 조 생성물을 정제용 RP-HPLC에 의해 정제하였다 (이동상: 아세토니트릴/물 구배). 이로써 표제 화합물 64 mg (0.14 mmol, 이론치의 89%)을 수득하였다.

일반적 절차 3: 염산 또는 황산과 아세트산의 혼합물을 사용한 에틸 또는 메틸 에스테르의 상응하는 카르복실산으로의 절단
아세트산 및 진한 염산의 혼합물, 또는 아세트산 및 10% 농도의 또는 반-진한 황산의 혼합물 중 당해 에틸 또는 메틸 에스테르의 용액을 80℃ 내지 130℃의 온도에서 (적절한 경우, 환류 하에) 30 분 내지 12 시간 동안 교반하였다. 냉각시킨 후, 반응 혼합물을 감압 하에 직접 농축시키거나, 또는 물에 첨가하고, 수성 상을 에틸 아세테이트 또는 디클로로메탄으로 추출하고, 합한 유기 상을 감압 하에 농축시켰다. 조 생성물을 실리카 겔 상에서 크로마토그래피에 의해 (디클로로메탄/에틸 아세테이트 또는 시클로헥산/에틸 아세테이트 혼합물로 용리, 적절한 경우 소량의 아세트산 첨가, 또는 디클로로메탄/메탄올 혼합물 사용), 아세토니트릴 또는 물/아세토니트릴 혼합물로부터의 결정화에 의해, 또는 정제용 RP-HPLC에 의해 (이동상: 아세토니트릴/물 구배) 정제할 수 있었다.
하기 실시예를 일반적 절차 3에 따라 제조하였다.



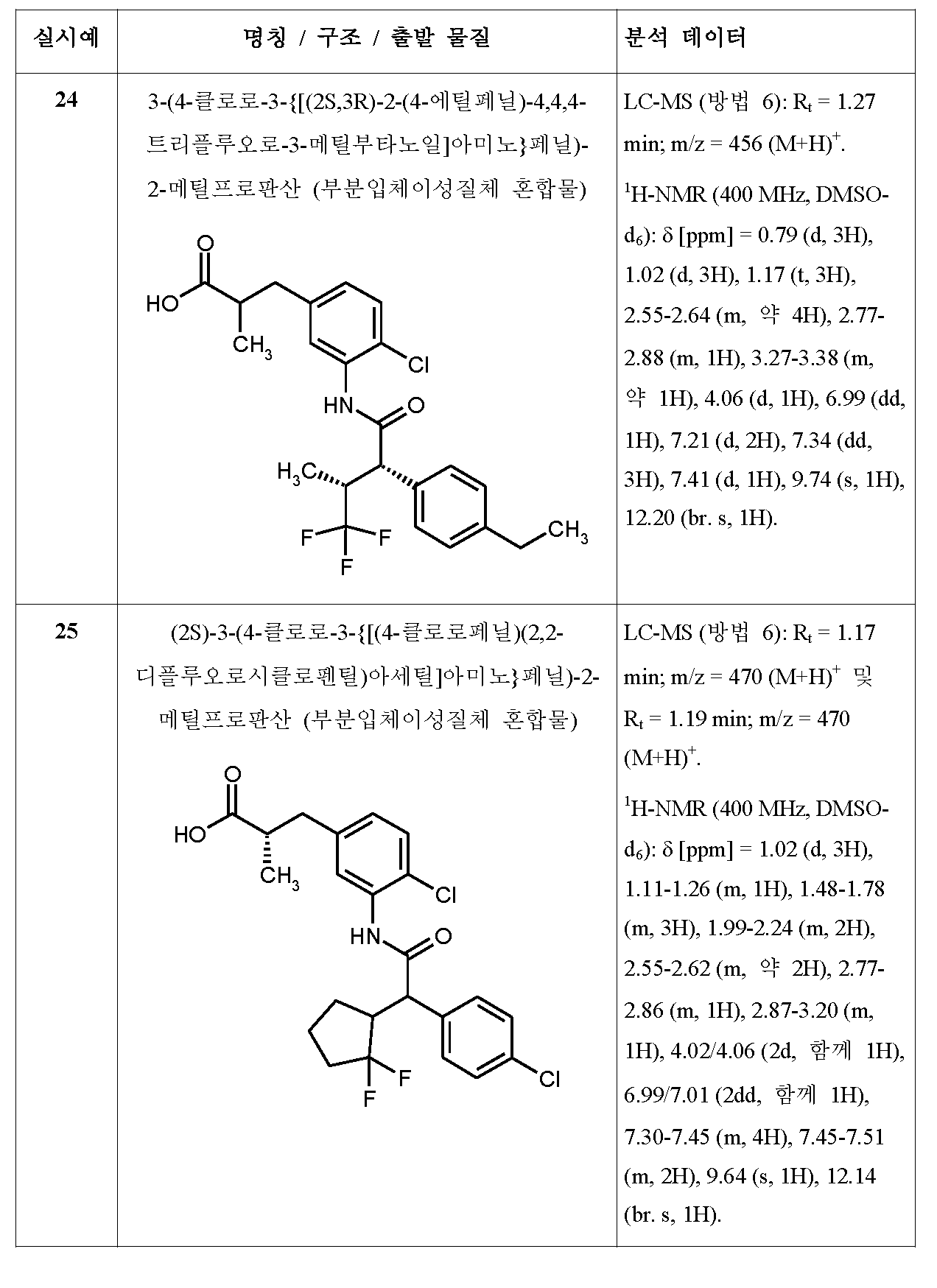

실시예 27
(+)-[3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-옥세탄-3-일]아세트산

탄소 상 팔라듐 (10%) 25 mg을 에틸 아세테이트 15 ml 중 벤질 [3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)옥세탄-3-일]아세테이트 120 mg (0.21 mmol)의 용액에 첨가하였다. 수소의 분위기 하에, 혼합물을 대기압에서 2 시간 동안 수소화시켰다. 이어서, 반응 혼합물을 톤실(Tonsil)을 통해 여과하고, 여과 잔류물을 에틸 아세테이트로 세척하고, 합한 여과물을 회전 증발기 상에서 농축시켰다. 이로써 표제 화합물 98 mg (0.2 mmol, 이론치의 97%)을 수득하였다.

실시예 28
[1-(4-클로로-3-{[(3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)시클로부틸]아세트산

메틸-[1-(4-클로로-3-{[(3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)시클로부틸]아세테이트 38 mg (0.08 mmol)을 디옥산 9.5 ml 중에 용해시키고, 1 N 수성 수산화나트륨 용액 0.15 ml를 첨가하였다. 혼합물을 80℃에서 밤새 교반하였다. 이어서, 1 N 염산을 사용하여 반응 혼합물을 pH 1로 산성화시키고, 에틸 아세테이트로 반복적으로 추출하였다. 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 HPLC에 의해 정제하였다. 이로써 표적 화합물 22 mg (0.05 mmol, 이론치의 60%)을 수득하였다.

실시예 29
(+)-(2R)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)부탄산

아세트산 15.2 ml 및 진한 염산 7.6 ml를 에틸 (+)-(2R)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)부타노에이트 1.96 g (3.89 mmol)에 첨가하였다. 반응 혼합물을 환류 하에 5 시간 동안 (조 온도 140℃) 교반하였다. 냉각시킨 후, 물을 첨가하였다. 혼합물을 디클로로메탄으로 반복적으로 추출하고, 합한 유기 상을 포화 염화나트륨 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 잔류물을 실리카 겔 상에서 크로마토그래피하여 (이동상 시클로헥산/에틸 아세테이트 10:1 → 2:1), 표제 화합물 1.46 g (이론치의 78.6%)을 수득하였다.

하기 화합물을 유사한 절차에 따라 제조하였다.
실시예 30
(+)-(2S)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)부탄산


실시예 31 및 실시예 32
3-[4-클로로-3-({4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}아미노)페닐]프로판산 (거울상이성질체 1 및 2)

라세미 3-[4-클로로-3-({4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}아미노)페닐]프로판산 (실시예 5) 120 mg (0.24 mmol)을 키랄 상 상에서 정제용 HPLC에 의해 거울상이성질체로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 85:15 (v/v); 유량: 15 ml/분; UV 검출: 220 nm; 온도: 35℃].
실시예 31
(+)-3-[4-클로로-3-({(2S,3R)-4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}-아미노)페닐]프로판산 (거울상이성질체 1)

수율: 48 mg
Rt = 5.75 분; 화학적 순도 >99%; >99% ee
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/(에탄올 + 0.2% TFA + 1% 물) 85:15 (v/v); 유량: 1 ml/분; 온도: 35℃; UV 검출: 220 nm].

실시예 32
(-)-3-[4-클로로-3-({(2R,3S)-4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}아미노)페닐]프로판산 (거울상이성질체 2)

수율: 52 mg
Rt = 6.85 분; 화학적 순도 >97.4%; >99% ee
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/(에탄올 + 0.2% TFA + 1% 물) 85:15 (v/v); 유량: 1 ml/분; 온도: 35℃; UV 검출: 220 nm].

실시예 33 내지 36
3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)프로판산
(이성질체 1 내지 4)

3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)프로판산의 부분입체이성질체 혼합물 (실시예 6) 44 mg (0.096 mmol)을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄셀(Daicel Chiralcel) OJ-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 70:30 (v/v); 유량: 15 ml/분; UV 검출: 220 nm; 온도: 35℃]. 이로써 각각 2개의 이성질체의 혼합물로 이루어진 4개의 상이한 분획을 수득하였다. 키랄 상 상에서 정제용 HPLC를 반복하여, 이들 분획을 개별 이성질체로 분리하였다 [분획 1 및 2: 칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/이소프로판올 75:25 (v/v); 유량: 15 ml/분; UV 검출: 220 nm; 온도: 35℃. 분획 3 및 4: 칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 80:20 (v/v); 유량: 15 ml/분; UV 검출: 220 nm; 온도: 35℃].
실시예 33 (이성질체 1):
수율: 8 mg
Rt = 6.49 분; 화학적 순도 >99%
[칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/이소프로판올 75:25 (v/v); 유량: 1 ml/분; UV 검출: 220 nm; 온도: 35℃].
실시예 34 (이성질체 2):
수율: 11 mg
Rt = 9.08 분; 화학적 순도 >98.5%
[칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/이소프로판올 75:25 (v/v); 유량: 1 ml/분; UV 검출: 220 nm; 온도: 35℃].
실시예 35 (이성질체 3):
수율: 12 mg
Rt = 7.19 분; 화학적 순도 >99%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/에탄올 80:20 (v/v); 유량: 1 ml/분; UV 검출: 220 nm; 온도: 30℃].
실시예 36 (이성질체 4):
수율: 9 mg
Rt = 8.58 분; 화학적 순도 >97.5%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4.6 mm; 이동상: 이소헥산/에탄올 80:20 (v/v); 유량: 1 ml/분; UV 검출: 220 nm; 온도: 30℃].
실시예 37
3-(3-{[(3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산 (부분입체이성질체 혼합물)

에틸 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로파노에이트 (부분입체이성질체 혼합물) 300 mg (0.633 mmol)을 메탄올, THF 및 물 (각 경우에 1.0 ml)의 혼합물 중에 용해시키고, 수산화리튬 265.5 mg (6.33 mmol)을 0℃에서 첨가하였다. 혼합물을 먼저 0℃에서 1 시간 동안, 이어서 실온에서 1 시간 동안 교반하였다. 이어서, 용액을 물로 희석하고, 1 N 염산 (pH 약 2)을 사용하여 산성화시켰다. 수성 상을 디에틸 에테르로 3회, 에틸 아세테이트로 1회 추출하였다. 합한 유기 상을 황산나트륨 상에서 건조시키고, 감압 하에 농축시켰다. 이로써 표제 화합물 294 mg (이론치의 99.7%)을 4개의 부분입체이성질체의 혼합물로서 수득하였다.
실시예 38 및 실시예 39
3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(3-{[(3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산 (실시예 37)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.25 ml; 온도: 40℃; 이동상: 90% 이소헥산 / 10% (에탄올 + 0.2% TFA + 1% 물); 유량: 15 ml/분; 검출: 220 nm]. 부분입체이성질체 혼합물 260 mg으로 2개의 추가의 이성질체 이외에, 이성질체 1 (실시예 38) 52 mg 및 이성질체 2 (실시예 39) 54 mg을 수득하였다.
실시예 38 (부분입체이성질체 1):
(+)-(2S)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산

이성질체 1을 정제용 RP-HPLC에 의해 다시 재정제하였다 (이동상 아세토니트릴/물). 이로써 32 mg을 수득하였다.

실시예 39 (부분입체이성질체 2):
(+)-(2R)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산

이성질체 2를 정제용 RP-HPLC에 의해 다시 재정제하였다 (이동상 아세토니트릴/물). 이로써 21 mg을 수득하였다.

실시예 40 및 실시예 41
3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산 (실시예 24)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 키랄 실리카 겔 상, 선택기 폴리(N-메타크릴로일-L-이소류신-3-펜틸아미드) 기반, 430 mm x 40 mm; 주입 부피: 2.0 ml; 온도: 24℃; 이동상: 40% 이소헥산 / 60% 에틸 아세테이트; 유량: 80 ml/분; 검출: 265 nm]. 부분입체이성질체 혼합물 514 mg으로 부분입체이성질체 1 (실시예 40) 178 mg 및 부분입체이성질체 2 (실시예 41) 218 mg을 수득하였다.
실시예 40 (부분입체이성질체 1):
(+)-(2R)-3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산


실시예 41 (부분입체이성질체 2):
(+)-(2S)-3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-2-메틸프로판산


실시예 42 및 실시예 43
3-(3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산 (실시예 23)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.30 ml; 온도: 30℃; 이동상: 92% 이소헥산 / 8% (에탄올 + 0.2% TFA + 1% 물); 유량: 15 ml/분; 검출: 220 nm]. 부분입체이성질체 혼합물 509 mg으로 부분입체이성질체 1 (실시예 42) 209 mg 및 부분입체이성질체 2 (실시예 43) 220 mg을 수득하였다.
실시예 42 (부분입체이성질체 1):
(+)-(2S)-3-(3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산


실시예 43 (부분입체이성질체 2):
(+)-(2R)-3-(3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-2-메틸프로판산


실시예 44 및 실시예 45
3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)부탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)부탄산 (실시예 7)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.20 ml; 온도: 30℃; 이동상: 90% 이소헥산 / 10% (이소프로판올 + 0.2% TFA + 1% 물); 유량: 15 ml/분; 검출: 220 nm]. 부분입체이성질체 혼합물 210 mg으로 부분입체이성질체 1 (실시예 44) 110 mg 및 부분입체이성질체 2 (실시예 45) 99 mg을 수득하였다.
실시예 44 (부분입체이성질체 1):
(+)-(3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-페닐)부탄산


실시예 45 (부분입체이성질체 2):
(+)-(3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-에틸페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-페닐)부탄산


실시예 46 및 실시예 47
3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-클로로페닐)부탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-클로로페닐)부탄산 (실시예 8)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.30 ml; 온도: 30℃; 이동상: 90% 이소헥산 / 10% 이소프로판올; 유량: 15 ml/분; 검출: 220 nm]. 부분입체이성질체 혼합물 250 mg으로 부분입체이성질체 1 (실시예 46) 116 mg 및 부분입체이성질체 2 (실시예 47) 113 mg을 수득하였다.
실시예 46 (부분입체이성질체 1):
(+)-(3S)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-클로로페닐)부탄산


실시예 47 (부분입체이성질체 2):
(+)-(3R)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-클로로페닐)부탄산


실시예 48 및 실시예 49
3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)부탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)부탄산 (실시예 9)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AS-H, 5 μm, 250 mm x 20 mm; 주입 부피: 0.25 ml; 온도: 30℃; 이동상: 85% 이소헥산 / 15% 이소프로판올; 유량: 15 ml/분; 검출: 220 nm]. 부분입체이성질체 혼합물 295 mg으로 부분입체이성질체 1 (실시예 48) 121 mg 및 부분입체이성질체 2 (실시예 49) 111 mg을 수득하였다.
실시예 48 (부분입체이성질체 1):
(+)-(3S)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)부탄산


실시예 49 (부분입체이성질체 2):
(+)-(3R)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)부탄산


일반적 절차 4: 에틸 에스테르의 산성 가수분해
당해 에틸 에스테르를 빙초산 및 진한 염산 (약 10 ml / 기질의 mmol) 7:2 혼합물 중에 용해시키고, 반응이 완료될 때까지 (일반적으로 1 시간 내지 8 시간) 100℃에서 가열하였다. 이어서, 혼합물을 냉각시키고, 물에 붓고, 디클로로메탄으로 반복적으로 추출하였다. 합한 유기 상을 포화 염화나트륨 용액으로 3회 세척하고, 황산마그네슘 상에서 건조시키고, 농축시켰다. 필요한 경우, 잔류물을 플래쉬 크로마토그래피 또는 정제용 HPLC에 의해 정제하였다.
하기 예시적인 화합물을 일반적 절차 4에 따라 제조하였다.

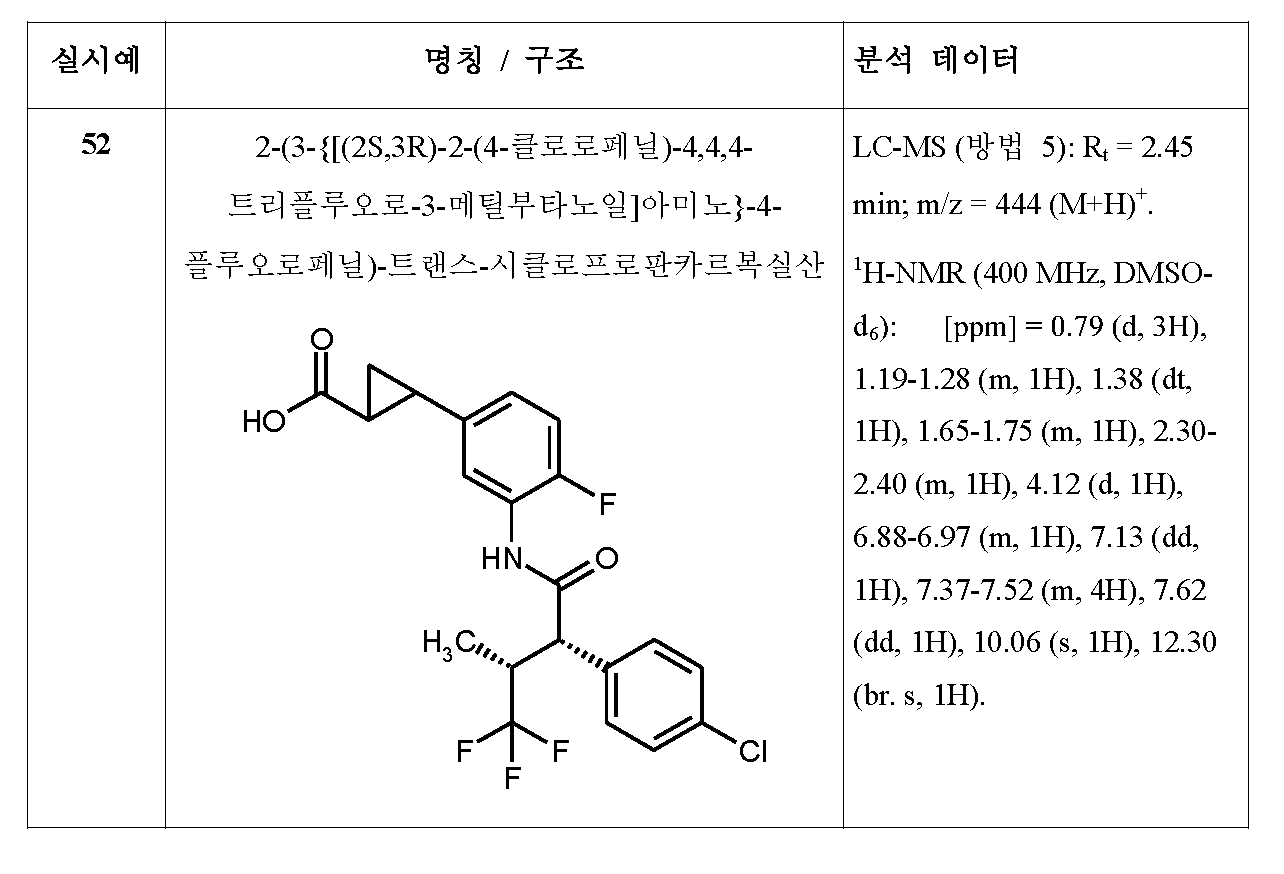
실시예 53 및 실시예 54
2-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-트랜스-시클로프로판카르복실산 (부분입체이성질체 1 및 2)

2-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-트랜스-시클로프로판카르복실산의 부분입체이성질체 혼합물 (실시예 52) 71 mg을 에탄올 2 ml 및 이소헥산 2 ml 중에 용해시키고, 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 200 mm; 주입 부피: 0.25 ml; 온도: 30℃; 이동상: 15% 이소프로판올 / 85% 이소헥산; 유량: 15 ml/분; 검출: 220 nm]. 이로써 부분입체이성질체 1 (실시예 53) 36 mg 및 부분입체이성질체 2 (실시예 54) 37 mg을 수득하였다.
실시예 53 (부분입체이성질체 1):

실시예 54 (부분입체이성질체 2):

실시예 55
3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-시아노페닐)프로판산

tert-부틸 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-시아노페닐)프로파노에이트 16.5 mg (33 μmol)을 디클로로메탄 1.1 ml 중에 용해시키고, 트리플루오로아세트산 275 μl를 첨가하였다. 반응 혼합물을 실온에서 1.5 시간 동안 교반하고,이어서 디클로로메탄 20 ml로 희석하고, 감압 하에 농축시켰다. 잔류물을 고진공 하에 밤새 건조시켰다. 이로써 표제 화합물 14.8 mg (이론치의 97%)을 수득하였다.

실시예 56
(+/-)-3-(3-{[2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-플루오로페닐)프로판산 (부분입체이성질체 1)

tert-부틸 (+/-)-3-(3-{[2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-플루오로페닐)프로판산 (부분입체이성질체 1, 실시예 102A) 270 mg (0.553 mmol)을 디클로로메탄 0.2 ml 중에 용해시키고, 트리플루오로아세트산 0.85 ml을 실온에서 첨가하였다. 반응 혼합물을 실온에서 4 시간 동안 교반하고, 이어서 감압 하에 농축시켰다. 잔류물을 정제용 RP-HPLC에 의해 정제하였다 (아세토니트릴/물 혼합물). 이로써 표적 화합물 188 mg (이론치의 78.7%)을 수득하였다.

하기 화합물을 유사한 방식으로 제조하였다.
실시예 57
(+/-)-3-(3-{[2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-2-플루오로페닐)프로판산 (부분입체이성질체 2)


하기 실시예를 일반적 절차 2 (트리플루오로아세트산을 사용한 tert-부틸 에스테르의 상응하는 카르복실산으로의 절단)에 따라 제조하였다.












실시예 71
(+)-2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)-2-메틸부탄산 (부분입체이성질체 A)

(+)-tert-부틸 2-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}벤질)-2-메틸부타노에이트 (부분입체이성질체 A) 302 mg (0.553 mmol)을 디클로로메탄 2.3 ml 중에 용해시키고, TFA 2 ml를 실온에서 첨가하였다. 30 분 후, 반응 혼합물을 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이어서, 잔류물을 정제용 RP-HPLC에 의해 정제하였다 (이동상 아세토니트릴/물). 이로써 표적 생성물 110.8 mg (이론치의 40.9%)을 수득하였다.

하기 실시예를 일반적 절차 3 (염산 또는 황산과 아세트산의 혼합물을 사용한 메틸 또는 에틸 에스테르의 상응하는 카르복실산으로의 절단)에 따라 제조하였다.






실시예 79 및 실시예 80
(+)-3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-4,4,4-트리플루오로부탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-4-플루오로페닐)-4,4,4-트리플루오로부탄산 (실시예 78)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 유량: 15 ml/분; 검출: 230 nm; 주입 부피: 0.80 ml; 온도: 45℃; 이동상: 92% 이소헥산 / 8% 이소프로판올]. 부분입체이성질체 혼합물 1.95 g으로 부분입체이성질체 1 (실시예 79) 556 mg 및 부분입체이성질체 2 (실시예 80) 730 mg을 수득하였다.
실시예 79 (부분입체이성질체 1):
부분입체이성질체 1을 한 번 더 정제용 RP-HPLC에 의해 재정제하였다 (이동상 메탄올/물). 이로써 418 mg을 수득하였다.

실시예 80 (부분입체이성질체 2):
부분입체이성질체 2를 한 번 더 정제용 RP-HPLC에 의해 재정제하였다 (이동상 메탄올/물). 이로써 352 mg을 수득하였다.

실시예 81
3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥산산 (부분입체이성질체 혼합물)

메틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥사노에이트 (부분입체이성질체 혼합물) 1.50 g (2.97 mmol)을 아세트산 5 ml 중에 용해시키고, 30% 농도의 황산 5 ml를 첨가하고, 혼합물을 환류하에 (조 온도 약 140℃) 가열하였다. 1.5 시간 후, 추가의 아세트산 2.5 ml를 첨가하고, 반응 혼합물을 환류 하에 추가로 2.5 시간 동안 교반하였다. 냉각시킨 후, 혼합물을 실온에서 밤새 정치시키고, 이어서 물에 첨가하고, 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 5% 농도의 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시키고, 잔류물을 고진공 하에 건조시켰다. 이로써 표적 생성물 1.44 g (이론치의 98.8%)을 수득하였다.

실시예 82 및 실시예 83
(+)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥산산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥산산 (실시예 81)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 유량: 20 ml/분; 검출: 230 nm; 주입 부피: 0.60 ml; 온도: 25℃; 이동상: 95% 이소헥산 / 5% 이소프로판올]. 부분입체이성질체 혼합물 59.2 mg으로 부분입체이성질체 1 (실시예 82) 19 mg 및 부분입체이성질체 2 (실시예 83) 17 mg을 수득하였다
실시예 82 (부분입체이성질체 1):
(+)-(3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-페닐)헥산산

보다 많은 양 (1.40 g)의 부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)헥산산 (실시예 81)을 마찬가지로 동일한 정제용 HPLC 방법에 의해 분리하였다. 본 경우에, 수득한 부분입체이성질체 1을 한 번 더 RP-HPLC에 의해 재정제하였다 [칼럼: 선파이어(Sunfire) 250 mm x 20 mm; 이동상: 80% 아세토니트릴 / 5% 수성 TFA (1% 농도) / 15% 물]. 이로써 337 mg의 순수 부분입체이성질체 1을 수득하였다.

실시예 83 (부분입체이성질체 2):
(+)-(3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}-페닐)헥산산

실시예 84 및 실시예 85
(+)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산 (실시예 66)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 유량: 15 ml/분; 검출: 220 nm; 주입 부피: 0.25 ml; 온도: 30℃; 이동상: 93% 이소헥산 / 7% 이소프로판올]. 부분입체이성질체 혼합물 150 mg으로 부분입체이성질체 1 (실시예 84) 70 mg 및 부분입체이성질체 2 (실시예 85) 79 mg을 수득하였다.
실시예 84 (부분입체이성질체 1):
(+)-(3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산

대안적으로, (+)-(3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산을 또한 하기 경로에 의해 제조할 수 있었다.
(+)-tert-부틸 (3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부타노에이트 (실시예 203A) 1.76 g (3.08 mmol)을 디클로로메탄 4.9 ml 중에 용해시키고, TFA 4.7 ml를 실온에서 첨가하였다. 반응 혼합물을 실온에서 2 시간 동안 교반하고, 이어서 감압 하에 농축시켰다. 잔류물을 에틸 아세테이트에 녹이고, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 RP-HPLC에 의해 정제하였다 (이동상 메탄올/물). 이로써 표적 생성물 1.30 g (이론치의 81.9%)을 수득하였다.

실시예 85 (부분입체이성질체 2):
(+)-(3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산

대안적으로, (+)-(3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부탄산을 또한 하기 경로에 의해 제조할 수 있었다.
(+)-tert-부틸 (3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)-4,4,4-트리플루오로부타노에이트 (실시예 204A) 1.17 g (2.04 mmol)을 디클로로메탄 4.9 ml 중에 용해시키고, TFA 3.2 ml를 실온에서 첨가하였다. 반응 혼합물을 실온에서 2 시간 동안 교반하고, 이어서 감압 하에 농축시켰다. 잔류물을 에틸 아세테이트에 녹이고, 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 조 생성물을 정제용 RP-HPLC에 의해 정제하였다 (이동상 메탄올/물). 이로써 표적 생성물 0.76 g (이론치의 72%)을 수득하였다.

실시예 86
3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜탄산 (부분입체이성질체 혼합물)

메틸 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜타노에이트 (부분입체이성질체 혼합물) 650 mg (1.33 mmol)을 아세트산 2 ml 중에 용해시키고, 30% 농도의 황산 1 ml를 첨가하고, 혼합물을 환류하에 (조 온도 약 140℃) 가열하였다. 1.5 시간 후, 반응 혼합물을 냉각시키고, 물에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 합한 유기 상을 포화 중탄산나트륨 용액 및 포화 염화나트륨 용액으로 세척하고, 황산마그네슘 상에서 건조시키고, 감압 하에 농축시켰다. 고진공 하에 건조시킨 후, 잔류물을 실리카 겔 상에서 크로마토그래피에 의해 정제하였다 (이동상 시클로헥산/에틸 아세테이트 50:1). 이로써 표적 생성물 600 mg (이론치의 95%)을 수득하였다.

실시예 87 및 실시예 88
(+)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜탄산 (부분입체이성질체 1 및 2)

부분입체이성질체 3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜탄산 (실시예 86)의 상기 수득한 혼합물을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 유량: 18 ml/분; 검출: 230 nm; 주입 부피: 0.25 ml; 온도: 25℃; 이동상: 95% 이소헥산 / 5% 이소프로판올]. 부분입체이성질체 혼합물 545 mg으로 부분입체이성질체 1 (실시예 87) 140 mg 및 부분입체이성질체 2 (실시예 88) 156 mg을 수득하였다.
실시예 87 (부분입체이성질체 1):
(+)-(3S)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜탄산


실시예 88 (부분입체이성질체 2):
(+)-(3R)-3-(4-클로로-3-{[(2S,3R)-2-(4-클로로페닐)-4,4,4-트리플루오로-3-메틸부타노일]아미노}페닐)펜탄산

하기 실시예를 일반적 절차 2 (트리플루오로아세트산을 사용한 tert-부틸 에스테르의 상응하는 카르복실산으로의 절단)에 따라 제조하였다.

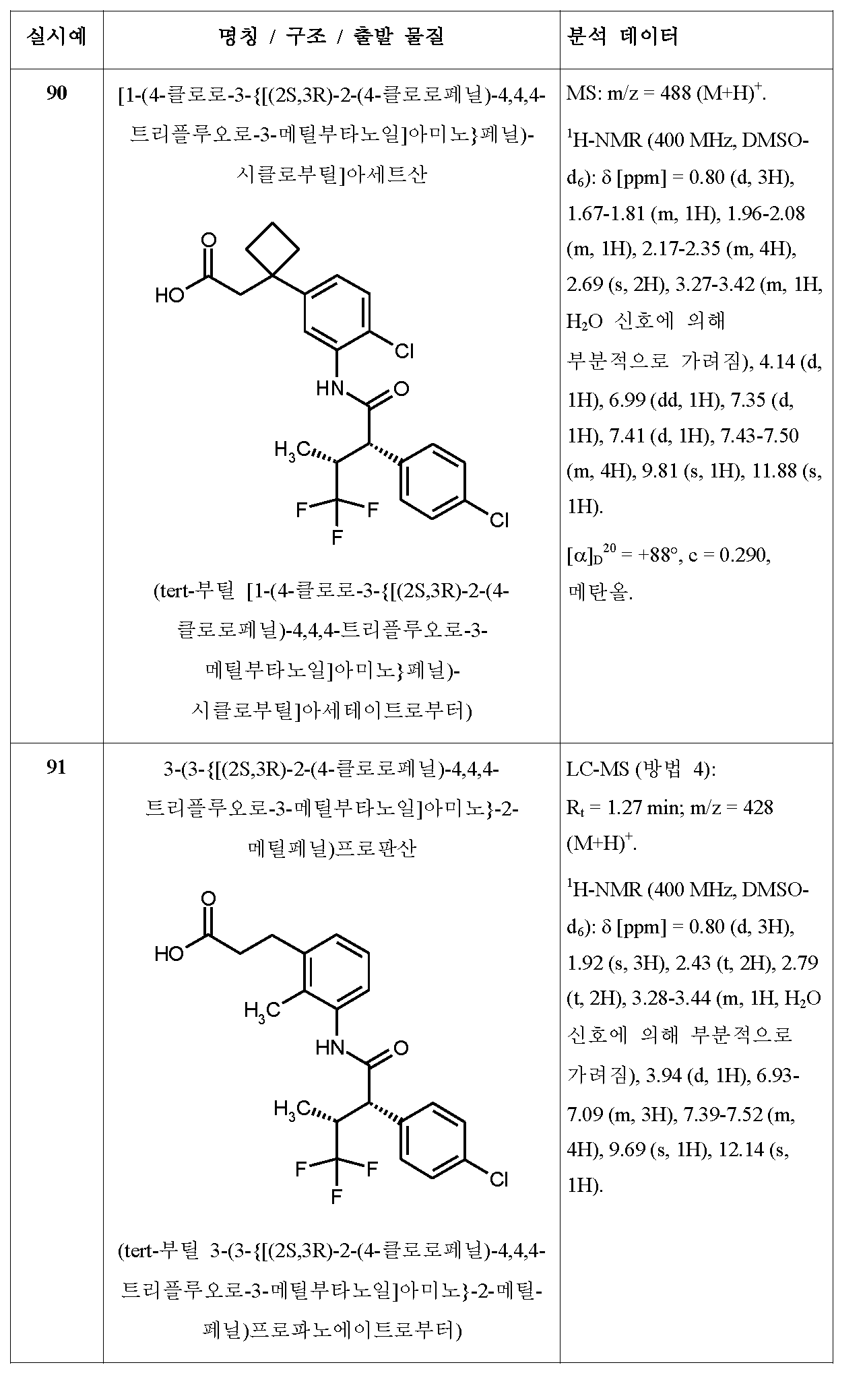




하기 실시예를 일반적 절차 3 (염산 또는 황산과 아세트산의 혼합물을 사용한 메틸 또는 에틸 에스테르의 상응하는 카르복실산으로의 절단)에 따라 제조하였다.
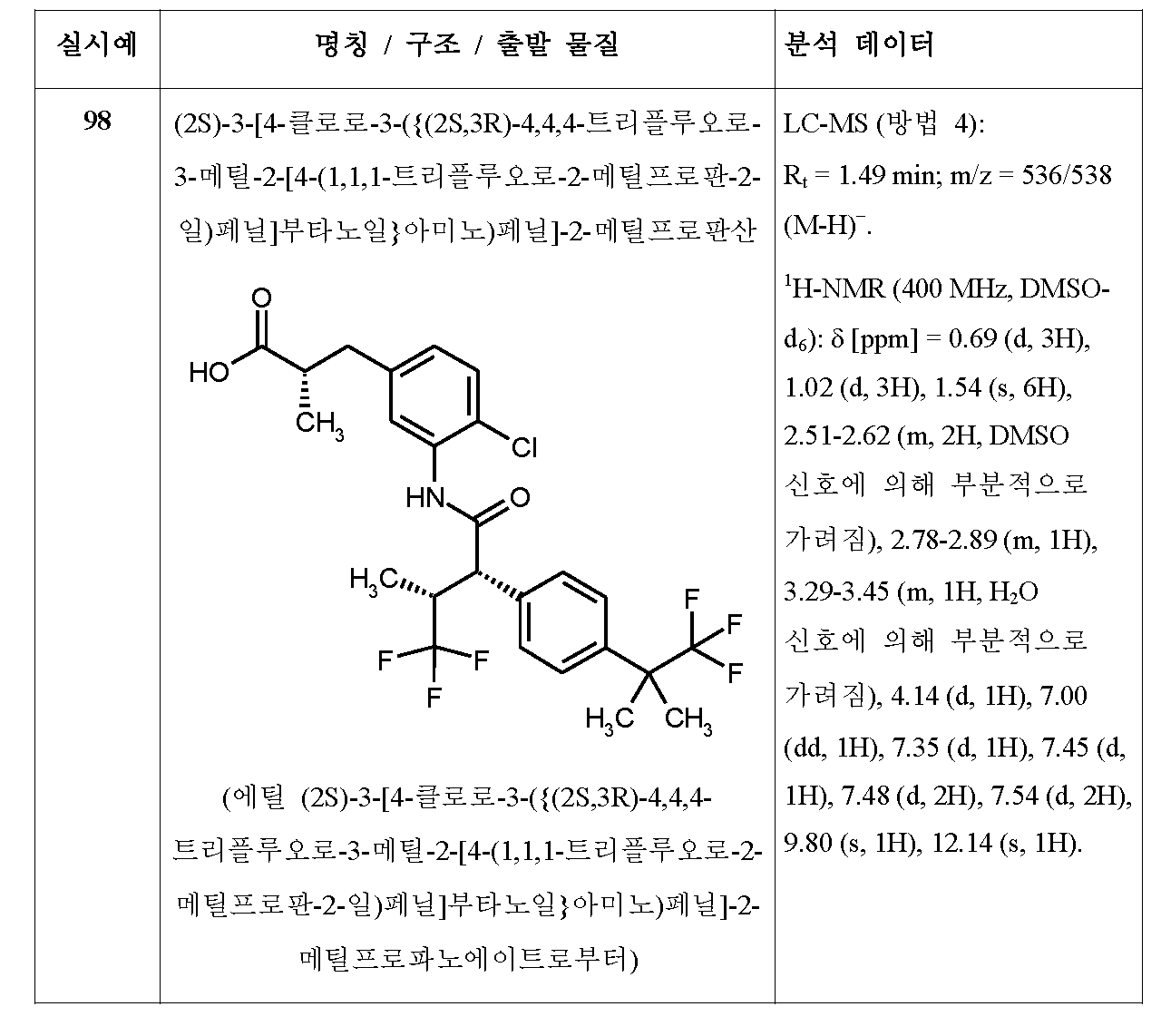




실시예 103 및 실시예 104
(2S)-3-[4-클로로-3-({4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}아미노)페닐]-2-메틸프로판산 (부분입체이성질체 1 및 2)

(2S)-3-[4-클로로-3-({4,4,4-트리플루오로-3-메틸-2-[4-(2,2,2-트리플루오로에틸)페닐]부타노일}아미노)페닐]-2-메틸프로판산의 이성질체 혼합물 (실시예 99) 74 mg (0.15 mmol)을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄팩 AY-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 1:1 (v/v); 유량: 15 ml/분; UV 검출: 220 nm; 온도: 45℃].
실시예 103 (부분입체이성질체 1):
수율: 39 mg
Rt = 3.72 분; 화학적 순도 >98%; >99% de
[칼럼: 다이셀 키랄팩 AY-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/(이소프로판올 + 0.2% TFA + 1% 물) 70:30 (v/v); 유량: 1 ml/분; 온도: 45℃; UV 검출: 220 nm].
실시예 104 (부분입체이성질체 2):
수율: 39 mg
Rt = 6.09 분; 화학적 순도 >98%; >99% de
[칼럼: 다이셀 키랄팩 AY-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/(이소프로판올 + 0.2% TFA + 1% 물) 70:30 (v/v); 유량: 1 ml/분; 온도: 45℃; UV 검출: 220 nm].
실시예 105 내지 108
(2S)-3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)-2-메틸프로판산 (이성질체 1 내지 4)

(2S)-3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)-2-메틸프로판산의 부분입체이성질체 혼합물 (실시예 20) 205 mg (0.44 mmol)을 키랄 상 상에서 정제용 HPLC에 의해 추가로 분리하였다 [칼럼: 다이셀 키랄셀 OJ-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 70:30 (v/v); 유량: 25 ml/분; UV 검출: 230 nm; 온도: 25℃]. 이로써 각각 2개의 이성질체의 혼합물로 이루어진 2개의 상이한 상이한 분획을 수득하였다. 이들 2개의 분획을 추가로 키랄 상 상에서 정제용 HPLC에 의해 개별 이성질체로 분리하였다 [분획 1: 칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/이소프로판올 80:20 (v/v); 유량: 20 ml/분; UV 검출: 230 nm; 온도: 25℃. 분획 2: 칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올 90:10 (v/v); 유량: 20 ml/분; UV 검출: 230 nm; 온도: 25℃].
실시예 105 (이성질체 1):
수율: 30 mg
Rt = 15.70 분; 화학적 순도 >89.8%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/이소프로판올 70:30 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].

실시예 106 (이성질체 2):
수율: 35 mg
Rt = 20.07 분; 화학적 순도 >98.9%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/이소프로판올 70:30 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].

실시예 107 (이성질체 3):
수율: 37 mg
Rt = 14.17 분; 화학적 순도 >95.7%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올 90:10 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].

실시예 108 (이성질체 4):
수율: 29 mg
Rt = 17.77 분; 화학적 순도 >99.5%
[칼럼: 다이셀 키랄팩 AD-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올 90:10 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].

실시예 109 내지 112
(2R)-3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)-2-메틸프로판산 (이성질체 1 내지 4)

(2R)-3-(4-클로로-3-{[(4-클로로페닐)(3,3-디플루오로시클로펜틸)아세틸]아미노}페닐)-2-메틸프로판산의 부분입체이성질체 혼합물 (실시예 21) 160 mg (0.32 mmol)을 정제용 HPLC에 의해 4개의 이성질체로 분리하였다 [칼럼: 다이셀 키랄셀 OJ-H, 5 μm, 250 mm x 20 mm; 이동상: 이소헥산/에탄올/메탄올 90:5:5 (v/v); 유량: 20 ml/분; UV 검출: 230 nm; 온도: 25℃].
실시예 109 (이성질체 1):
수율: 16.7 mg
Rt = 10.49 분; 화학적 순도 >92.8%
[칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올/ 메탄올 90:5:5 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].
실시예 110 (이성질체 2):
수율: 24.4 mg
Rt = 12.26 분; 화학적 순도 >94.7%
[칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올/ 메탄올 90:5:5 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].
실시예 111 (이성질체 3):
수율: 22 mg
Rt = 18.89 분; 화학적 순도 >97.8%
[칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올/ 메탄올 90:5:5 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].
실시예 112 (이성질체 4):
수율: 25 mg
Rt = 28.37 분; 화학적 순도 >97.8%
[칼럼: 다이셀 키랄팩 OJ-H, 5 μm, 250 mm x 4 mm; 이동상: 이소헥산/에탄올/ 메탄올 90:5:5 (v/v); 유량: 1 ml/분; UV 검출: 230 nm; 온도: 25℃].
하기 실시예를 일반적 절차 3에 따라 제조하였다.

B. 약리 활성의 평가
본 발명에 따른 화합물의 약리 효과는 하기 검정에서 확인할 수 있다.
B-1. 시험관내 재조합 가용성 구아닐레이트 시클라제 (sGC)의 자극:
나트륨 니트로프루시드의 존재 및 부재 하에, 그리고 헴-의존성 sGC 억제제 1H-1,2,4-옥사디아졸로-(4,3a)-퀴녹살린-1-온 (ODQ)의 존재 및 부재 하에 본 발명에 따른 화합물에 의한 재조합 가용성 구아닐레이트 시클라제 (sGC)의 자극에 대한 조사를 참고문헌 [M. Hoenicka, E.M. Becker, H. Apeler, T. Sirichoke, H. Schroeder, R. Gerzer and J.-P. Stasch, "Purified soluble guanylyl cyclase expressed in a baculovirus/Sf9 system: Stimulation by YC-1, nitric oxide, and carbon oxide", J. Mol. Med. 77 (1999), 14-23]에 상세히 기재된 방법에 의해 수행하였다. 헴-무함유 구아닐레이트 시클라제는 샘플 완충제에 트윈 20을 첨가함으로써 수득하였다 (최종 농도 0.5%).
시험 물질에 의한 sGC의 활성화를 기저 활성의 x-배 자극으로 보고하였다. 실시예 15에 대한 결과를 하기 표 1A에, 실시예 17에 대한 결과를 하기 표 1B에 나타내었다.
<표 1A>

<표 1B>
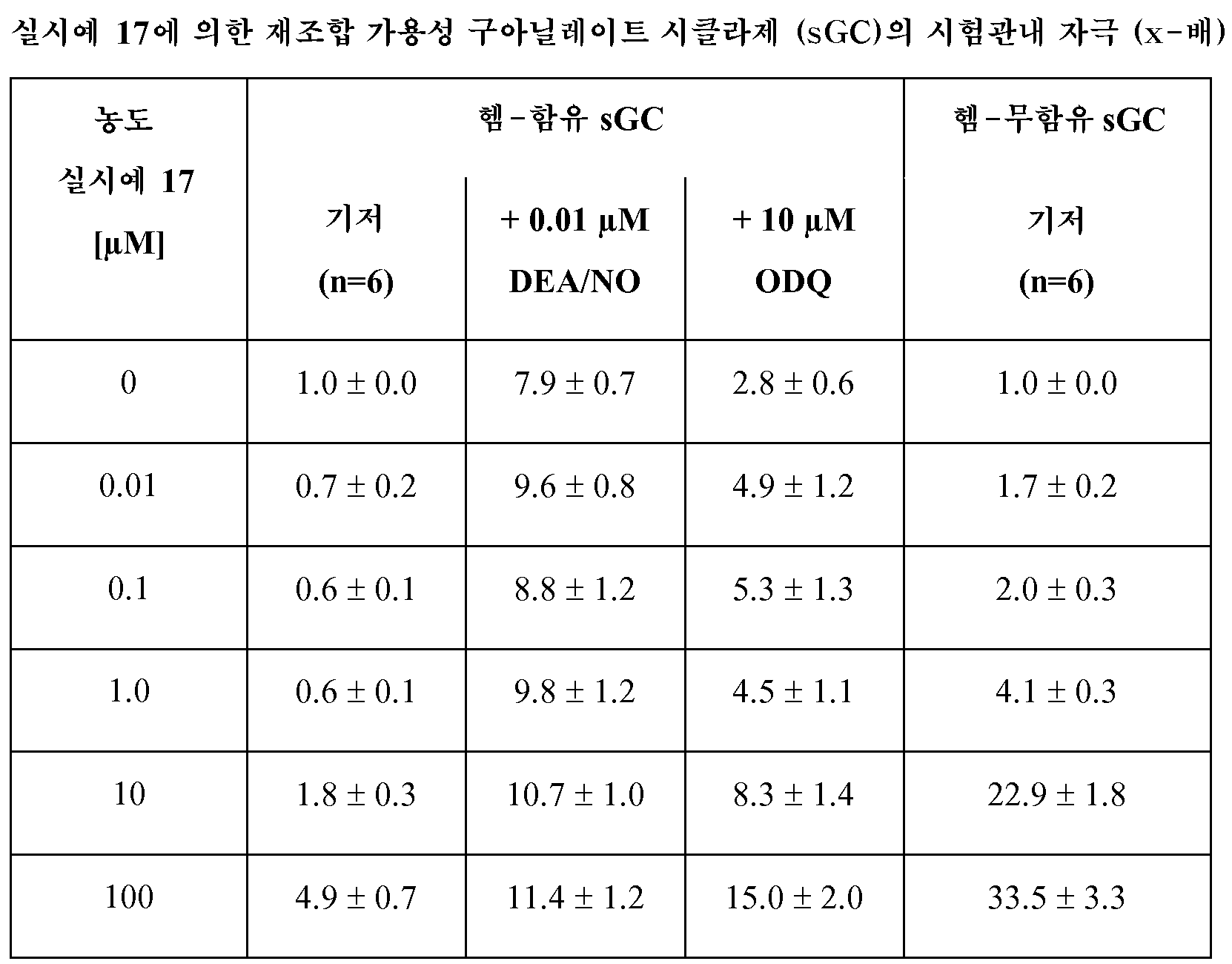
[DEA/NO = 2-(N,N-디에틸아미노)디아제놀레이트 2-옥시드; ODQ = 1H-1,2,4-옥사디아졸로[4,3-a]퀴녹살린-1-온].
헴-함유 및 헴-무함유 효소 둘 모두의 자극이 달성되었다는 것이 표 1A 및 1B로부터 명백하다. 또한, 실시예 15 또는 실시예 17 및 2-(N,N-디에틸아미노)디아제놀레이트 2-옥시드 (DEA/NO), NO 공여자의 조합은 상승작용 효과를 보이지 않았으며, 즉 DEA/NO의 효과가 헴-의존성 메카니즘을 통한 sGC 활성화제 작용으로 예상된 바와 같이 강화되지 않았다. 또한, 본 발명에 따른 sGC 활성화제의 효과는 가용성 구아닐레이트 시클라제 1H-1,2,4-옥사디아졸로[4,3-a]퀴녹살린-1-온 (ODQ)의 헴-의존성 억제제에 의해 차단되지 않고, 사실상 증가했다. 따라서, 표 1A 및 1B의 결과는 가용성 구아닐레이트 시클라제의 활성화제로서의 본 발명에 따른 화합물의 작용의 메카니즘을 확인해준다.
B-2. 재조합 구아닐레이트 시클라제 리포터 세포주에서의 작용
본 발명에 따른 화합물의 세포 작용을 문헌 [F. Wunder et al., Anal. Biochem. 339, 104-112 (2005)]에 기재된 바와 같이, 재조합 구아닐레이트 시클라제 리포터 세포주에서 측정하였다.
본 발명에 따른 화합물에 대한 대표적인 결과를 하기 표 2에 수록하였다.
<표 2>

B-3. sGC 효소 활성의 자극
가용성 구아닐레이트 시클라제 (sGC)는 자극시 GTP를 cGMP 및 피로포스페이트 (PPi)로 전환시켰다. PPi를 하기 기재된 검정에 의해 검출하였다. 검정에서 생성된 신호는 반응 진행에 따라 증가하였고, 주어진 자극 하에 sGC 효소 활성의 척도로서 기능하였다.
검정을 수행하기 위해, 29 μl의 효소 용액 [50 mM TEA, 2 mM MgCl2, 0.1% BSA (분획 V), 0.005% Brij®, pH 7.5 중 0-10 nM 가용성 구아닐레이트 시클라제 (문헌 [Hoenicka et al., J. Mol. Med. 77, 14-23 (1999)]에 따라 제조)]을 먼저 마이크로플레이트에 도입하고, 1 μl의 시험할 물질 (DMSO 중에 연속적으로 희석된 용액으로서)을 첨가하였다. 혼합물을 실온에서 10분 동안 인큐베이션하였다. 이어서, 20 μl의 검출 믹스 [50 mM TEA, 2 mM MgCl2, 0.1% BSA (분획 V), 0.005% Brij®, pH 7.5 중 1.2 nM 반딧불이 루시페라제 (포티누스 피랄리스(Photinus pyralis) 루시페라제, 프로메가(Promega)), 29 μM 데히드로루시페린 (문헌 [Bitler & McElroy, Arch. Biochem. Biophys. 72, 358 (1957)]에 따라 제조), 122 μM 루시페린 (프로메가), 153 μM ATP (시그마) 및 0.4 mM DTT (시그마)]를 첨가하였다. 20 μl의 기질 용액 [50 mM TEA, 2 mM MgCl2, 0.1% BSA (분획 V), 0.005% Brij®, pH 7.5 중 1.25 mM 구아노신 5'-트리포스페이트 (시그마)]을 첨가하여 효소 반응을 시작시키고, 발광측정기에서 계속적으로 측정하였다. 시험할 물질에 의한 자극의 정도를 비자극된 반응의 신호와 비교하여 측정할 수 있었다.
효소 용액에 25 μM 의 1H-1,2,4-옥사디아졸로[4,3-a]퀴녹살린-1-온 (ODQ)을 첨가하고, 후속으로 30분 동안 인큐베이션하여 헴-무함유 구아닐레이트 시클라제의 활성화를 검사하였고, 천연 효소의 자극과 비교하였다.
본 발명에 따른 화합물에 대한 대표적인 결과를 하기 표 3에 수록하였다.
<표 3>

B-4. 의식이 있는 SH 래트에 대한 혈압 및 심박수의 무선 원격 측정
데이터 사이언시스 인터내셔널 디에스아이(Data Sciences International DSI, 미국 소재)로부터 시판되는 원격 측정 시스템을 하기 기재된 의식이 있는 SH 래트에 대한 측정을 위해 사용하였다.
시스템은 하기 3가지의 주요 구성요소로 이루어졌다: (1) 주입형 송신기, (2) 수신기 (이는 다중화기를 통해 하기 데이터 수집 컴퓨터에 연결됨), (3) 데이터 수집 컴퓨터. 원격 측정 시스템은, 의식이 있는 동물의 혈압과 심박수를 그의 일반적 서식지에서 계속적으로 기록하는 것을 가능하게 한다.
조사는 체중이 > 200 g인 암컷 성체 자연발생 고혈압 래트 (SH 래트)에서 수행하였다. 송신기 이식 후에, 실험 동물들을 유형 3 마크롤론(Makrolon) 케이지에 단독으로 수용하였다. 이들이 표준 먹이와 물에 자유롭게 접근하도록 하였다. 실험용 실험실에서의 주/야 리듬을 실내 조명에 의해 6.00am 및 7.00pm에 바꾸었다.
사용된 원격 송신기 (TAM PA-C40, 디에스아이)를 첫번째 실험적 사용의 적어도 14 일 전에 실험용 동물에게 무균 조건 하에 외과적으로 이식하였다. 이런 방식으로 기기장착 동물을, 그 상처가 치유되고 이식물이 안착된 후에 반복적으로 사용할 수 있었다.
이식을 위해, 단식시킨 동물을 펜토바르비탈 (넴부탈(Nembutal), 사노피 (Sanofi), 50 mg/kg i.p.)로 마취시키고, 그의 복부를 넓은 영역에 걸쳐 면도하고, 소독하였다. 백선을 따라 복강을 개복한 후, 시스템의 액체-충전 측정 카테터를 대동맥갈림을 따라 두개 방향으로 하행 대동맥에 삽입하고, 조직 아교제 (베트본드 (VetBonD)™, 쓰리엠(3M))로 고정하였다. 송신기 하우징을 복강내로 복벽 근육에 고정하고, 그 상처의 층별 봉합을 수행하였다. 항생제 (타르도미오셀 콤프(Tardomyocel COMP), 바이엘(Bayer), 1 ml/kg s.c.)를 감염의 예방을 위해 수술 후 투여하였다.
실험의 개요:
연구할 물질을 한 군의 동물 (n = 6)에게 각 경우에 위관 영양법에 의해 경구로 투여하였다. 시험 물질을 적합한 용매 혼합물 중에 용해시키거나, 5 ml/체중 kg의 투여 부피에 적절한 0.5% 농도의 틸로스(Tylose) 중에 현탁시켰다. 용매-처리 군의 동물을 대조군으로서 사용하였다.
원격 측정 장치를 24 마리의 동물에 대해 구성하였다. 각 실험을 실험 번호 하에 기록하였다.
시스템 내에 사는 각각의 기기장착 래트를 별도의 수신 안테나 (1010 리시버(Receiver), 디에스아이)에 배정하였다. 이식된 송신기를, 포함된 전자 개폐기에 의하여 외부적으로 활성화시킬 수 있고, 실험의 전단계에서 송신으로 전환하였다. 방출된 신호를 데이터 수집 시스템 (윈도우즈용 데이터퀘스트(Dataquest)™ A.R.T., 디에스아이)에 의해 온라인으로 검출하고, 적절하게 처리할 수 있었다. 데이터는 각 경우에 이러한 목적을 위해 생성한 파일에 저장하고, 실험 번호를 부여하였다.
표준 절차에서, 각각의 경우에 10-초 기간 동안 다음을 측정하였다: (1) 수축기 혈압 (SBP), (2) 이완기 혈압 (DBP), (3) 평균 동맥압 (MAP) 및 (4) 심박수 (HR).
측정값의 수득은 컴퓨터 제어 하에 5-분 간격으로 반복하였다. 절대값으로서 수득된 원시 데이터를 현재 측정된 대기압을 사용하여 다이어그램에서 보정하고, 개별 데이터로 저장하였다. 추가의 기술적 세부사항은 제조 회사 (디에스아이)로부터의 서류에 제공되어 있다.
시험 물질을 실험 당일의 9.00am에 투여하였다. 투여 후에, 상기 기재된 파라미터를 24시간에 걸쳐 측정하였다. 실험 종료 후, 수집된 개별 데이터를 분석 소프트웨어 (데이터퀘스트™ A.R.T. 분석)를 이용하여 분류하였다. 무효 값(void value)을 물질의 투여 2시간 전의 시간으로 가정하여, 선택된 데이터 세트는 실험 당일 7.00am으로부터 다음날 9.00am까지의 기간을 포함하였다.
평균 (15-분 평균, 30-분 평균)의 측정에 의해 미리 설정가능한 시간에 걸쳐 데이터를 평탄화시키고, 텍스트 파일로서 저장 매체로 이동시켰다. 이런 방식으로 사전분류되고 압축된 측정값을 엑셀 템플렛으로 이동시켜 표로 만들었다.
C. 제약 조성물의 예시적 실시양태
본 발명에 따른 화합물을 하기 방식으로 제약 제제로 전환시킬 수 있었다:
정제:
조성:
100 mg의 본 발명에 따른 화합물, 50 mg의 락토스 (일수화물), 50 mg의 메이즈 전분 (천연), 10 mg의 폴리비닐피롤리돈 (PVP 25) (바스프 (BASF), 독일 루드빅샤펜 소재) 및 2 mg의 스테아르산마그네슘.
정제 중량 212 mg, 직경 8 mm, 곡률 반경 12 mm.
제조:
본 발명에 따른 화합물, 락토스 및 전분의 혼합물을, 물 중 PVP의 5% 농도의 용액 (m/m)을 사용하여 과립화하였다. 과립을 건조시킨 후에, 스테아르산마그네슘과 5분 동안 혼합하였다. 상기 혼합물을 통상의 정제 프레스에서 압축시켰다 (정제의 포맷에 대해서는 상기 참조). 압축을 위한 지침 압축력은 15 kN이었다.
경구 투여할 수 있는 현탁액:
조성:
1000 mg의 본 발명에 따른 화합물, 1000 mg의 에탄올 (96%), 400 mg의 로디겔(Rhodigel)® (미국 펜실베이니아주 소재의 에프엠씨(FMC)로부터의 크산탄 검) 및 99 g의 물.
경구 현탁액 10 ml는 본 발명에 따른 화합물 100 mg의 단일 용량에 상응한다.
제조:
로디겔을 에탄올에 현탁시키고, 본 발명에 따른 화합물을 현탁액에 첨가하였다. 교반하면서 물을 첨가하였다. 로디겔의 팽윤이 완료될 때까지 혼합물을 약 6시간 동안 교반하였다.
경구 투여할 수 있는 용액:
조성:
500 mg의 본 발명에 따른 화합물, 2.5 g의 폴리소르베이트 및 97 g의 폴리에틸렌 글리콜 400. 경구 용액 20 g은 본 발명에 따른 화합물 100 mg의 단일 용량에 상응한다.
제조:
본 발명에 따른 화합물을 폴리에틸렌 글리콜 및 폴리소르베이트의 혼합물중에 교반하면서 현탁시켰다. 본 발명에 따른 화합물이 완전히 용해될 때까지 교반 과정을 지속하였다.
i.v. 용액:
본 발명에 따른 화합물을 생리학상 허용되는 용매 (예를 들어, 등장성 염수, 5% 글루코스 용액 및/또는 30% PEG 400 용액) 중에 포화 용해도 미만의 농도로 용해시켰다. 용액을 여과에 의해 멸균하고, 이를 사용하여 멸균 및 발열원 무함유 주사 용기를 충전하였다.
Claims (11)
- 하기 화학식 I의 화합물, 또는 그의 염, 용매화물 또는 염의 용매화물.
<화학식 I>

상기 식에서,
R1A는 수소, 플루오린, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸, n-프로필, 시클로프로필 또는 시클로부틸을 나타내고,
R1B는 수소 또는 메틸을 나타내고,
R2A는 수소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸 또는 n-프로필을 나타내고,
R2B는 수소 또는 메틸을 나타내거나, 또는
R1A 및 R2A는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식(여기서, R1B 및 R2B는 상기 언급된 의미를 가짐)의 시클로프로필 고리를 형성하거나, 또는
R2A 및 R2B는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식(여기서, n은 1, 2 또는 3의 수를 나타냄)의 시클릭 기를 형성하고,
R3은 수소, 플루오린, 메틸 또는 트리플루오로메틸을 나타내고,
R4는 수소, 플루오린, 염소, 시아노, 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5A는 메틸, 트리플루오로메틸 또는 에틸을 나타내고,
R5B는 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B는 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6은 수소, 플루오린, 염소, 브로민, 시아노, (C1-C4)-알킬, (C2-C4)-알케닐, 시클로프로필 또는 시클로부틸을 나타내고, 여기서
(C1-C4)-알킬 및 (C2-C4)-알케닐은 플루오린에 의해 3회 이하 치환될 수 있고,
시클로프로필 및 시클로부틸은 플루오린에 의해 2회 이하 치환될 수 있고,
R7은 수소, 플루오린, 염소, 시아노, 메틸, 트리플루오로메틸, 에틸, 메톡시 또는 트리플루오로메톡시를 나타낸다. - 제1항에 있어서,
R1A가 수소, 메틸, 트리플루오로메틸, 에틸, n-프로필, 시클로프로필 또는 시클로부틸을 나타내고,
R1B가 수소 또는 메틸을 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소, 플루오린 또는 메틸을 나타내고,
R4가 수소, 플루오린, 염소, 시아노, 메틸 또는 트리플루오로메틸을 나타내고,
R5A가 메틸 또는 에틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식의 디플루오로-치환된 시클로알킬 고리를 형성하고,
R6이 플루오린, 염소, (C1-C4)-알킬, (C2-C3)-알케닐, 시클로프로필 또는 시클로부틸을 나타내고, 여기서
(C1-C4)-알킬 및 (C2-C3)-알케닐이 플루오린에 의해 3회 이하 치환될 수 있고,
시클로프로필 및 시클로부틸이 플루오린에 의해 2회 이하 치환될 수 있고,
R7이 수소, 플루오린, 염소, 메틸 또는 메톡시를 나타내는 것인
화학식 I의 화합물, 또는 그의 염, 용매화물 또는 염의 용매화물. - 제1항 또는 제2항에 있어서,
R1A가 수소, 메틸 또는 에틸을 나타내고,
R1B가 수소를 나타내고,
R2A가 수소, 메틸, 트리플루오로메틸, 에틸 또는 n-프로필을 나타내고,
R2B가 수소 또는 메틸을 나타내거나, 또는
R2A 및 R2B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식(여기서, n은 1 또는 2의 수를 나타냄)의 시클릭 기를 형성하고,
R3이 수소를 나타내고,
R4가 플루오린, 염소 또는 메틸을 나타내고,
R5A가 메틸을 나타내고,
R5B가 트리플루오로메틸을 나타내거나, 또는
R5A 및 R5B가 서로 부착되고, 이들이 부착되어 있는 탄소 원자와 함께 화학식의 디플루오로-치환된 시클로펜틸 고리를 형성하고,
R6이 플루오린, 염소, 메틸, 트리플루오로메틸, 에틸, 1,1-디플루오로에틸, 2,2,2-트리플루오로에틸, 이소프로필, tert-부틸, 1,1,1-트리플루오로-2-메틸프로판-2-일, 비닐, 1-플루오로비닐, 시클로프로필, 2,2-디플루오로시클로프로필, 시클로부틸 또는 3,3-디플루오로시클로부틸을 나타내고,
R7이 수소, 플루오린, 염소 또는 메틸을 나타내는 것인
화학식 I의 화합물, 또는 그의 염, 용매화물 또는 염의 용매화물. - 하기 화학식 II의 카르복실산을 축합제의 보조 하에 또는 염기의 존재 하에 상응하는 카르보닐 클로라이드의 중간체를 통해 불활성 용매 중에서 하기 화학식 III의 아민과 커플링시켜 하기 화학식 IV의 카르복스아미드를 수득하고,
<화학식 II>

(상기 식에서, R5A, R5B, R6 및 R7은 제1항 내지 제3항 중 어느 한 항에 주어진 의미를 가짐)
<화학식 III>

(상기 식에서, R1A, R1B, R2A, R2B, R3 및 R4는 제1항 내지 제3항 중 어느 한 항에 주어진 의미를 갖고,
T1은 (C1-C4)-알킬 또는 벤질을 나타냄)
<화학식 IV>

(상기 식에서, R1A, R1B, R2A, R2B, R3, R4, R5A, R5B, R6, R7 및 T1은 상기 주어진 의미를 가짐)
이어서 에스테르 라디칼 T1을 염기성 또는 산성 가용매분해에 의해 제거하거나, 또는 T1이 벤질을 나타내는 경우, 또한 가수소분해에 의해 제거하여 화학식 I의 카르복실산을 수득하고,
화학식 I의 화합물을, 적절한 경우, 당업자에게 공지된 방법에 의해 그의 거울상이성질체 및/또는 부분입체이성질체로 분리하고/거나, 적절한 경우, 적절한 (i) 용매 및/또는 (ii) 염기와 반응시켜 그의 용매화물, 염 및/또는 염의 용매화물을 수득하는 것을 특징으로 하는,
제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 화학식 I의 화합물의 제조 방법. - 제1항 내지 제3항 중 어느 한 항에 있어서, 질환의 치료 및/또는 예방을 위한 화합물.
- 제1항 내지 제3항 중 어느 한 항에 있어서, 심부전, 협심증, 고혈압, 폐고혈압, 허혈, 혈관 장애, 미세순환 장애, 혈전색전성 장애 및 동맥경화증의 치료 및/또는 예방 방법에 사용하기 위한 화합물.
- 심부전, 협심증, 고혈압, 폐고혈압, 허혈, 혈관 장애, 미세순환 장애, 혈전색전성 장애 및 동맥경화증의 치료 및/또는 예방을 위한 의약의 제조에 있어서의 제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 화합물의 용도.
- 제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 화합물을 하나 이상의 불활성, 비-독성, 제약상 적합한 부형제와 조합하여 포함하는 의약.
- 제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 화합물을 유기 니트레이트, NO 공여자, cGMP-PDE 억제제, 구아닐레이트 시클라제의 자극물질, 항혈전 활성을 갖는 작용제, 혈압 강하제 및 지질 대사를 변경하는 작용제로 이루어진 군으로부터 선택된 하나 이상의 추가의 활성 화합물과 조합하여 포함하는 의약.
- 제8항 또는 제9항에 있어서, 심부전, 협심증, 고혈압, 폐고혈압, 허혈, 혈관 장애, 미세순환 장애, 혈전색전성 장애 및 동맥경화증의 치료 및/또는 예방을 위한 의약.
- 유효량의 제1항 내지 제3항 중 어느 한 항에 정의된 바와 같은 하나 이상의 화합물 또는 제8항 내지 제10항 중 어느 한 항에 정의된 바와 같은 의약의 투여에 의한, 인간 및 동물에서의 심부전, 협심증, 고혈압, 폐고혈압, 허혈, 혈관 장애, 미세순환 장애, 혈전색전성 장애 및 동맥경화증의 치료 및/또는 예방 방법.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102009046115.9 | 2009-10-28 | ||
| DE102009046115A DE102009046115A1 (de) | 2009-10-28 | 2009-10-28 | Substituierte 3-Phenylpropansäuren und ihre Verwendung |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20120101411A true KR20120101411A (ko) | 2012-09-13 |
Family
ID=43232960
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020127013589A KR20120101411A (ko) | 2009-10-28 | 2010-10-21 | 치환된 3-페닐프로피온산 및 그의 용도 |
Country Status (23)
| Country | Link |
|---|---|
| US (2) | US20110130445A1 (ko) |
| EP (1) | EP2493845B1 (ko) |
| JP (1) | JP5801312B2 (ko) |
| KR (1) | KR20120101411A (ko) |
| CN (1) | CN102712577B (ko) |
| AR (1) | AR079015A1 (ko) |
| AU (1) | AU2010311678B2 (ko) |
| BR (1) | BR112012010057A2 (ko) |
| CA (1) | CA2777152C (ko) |
| DE (1) | DE102009046115A1 (ko) |
| DK (1) | DK2493845T3 (ko) |
| ES (1) | ES2446970T3 (ko) |
| HK (1) | HK1177195A1 (ko) |
| HR (1) | HRP20140270T1 (ko) |
| IL (1) | IL218989A (ko) |
| MX (1) | MX2012004951A (ko) |
| PL (1) | PL2493845T3 (ko) |
| PT (1) | PT2493845E (ko) |
| RU (1) | RU2553263C2 (ko) |
| TW (1) | TWI487519B (ko) |
| UY (1) | UY32970A (ko) |
| WO (1) | WO2011051165A1 (ko) |
| ZA (1) | ZA201202465B (ko) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140021008A (ko) * | 2011-04-13 | 2014-02-19 | 바이엘 인텔렉쳐 프로퍼티 게엠베하 | 분지 3-페닐프로피온산 유도체 및 그의 용도 |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102008018675A1 (de) | 2008-04-14 | 2009-10-15 | Bayer Schering Pharma Aktiengesellschaft | Oxo-heterocyclisch substituierte Carbonsäure-Derivate und ihre Verwendung |
| DE102009012314A1 (de) * | 2009-03-09 | 2010-09-16 | Bayer Schering Pharma Aktiengesellschaft | Oxo-heterocyclisch substituierte Alkylcarbonsäuren und ihre Verwendung |
| DE102009046115A1 (de) | 2009-10-28 | 2011-09-08 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 3-Phenylpropansäuren und ihre Verwendung |
| US9284301B2 (en) | 2010-03-25 | 2016-03-15 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| EP2575473B1 (en) | 2010-05-27 | 2016-01-20 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2012058132A1 (en) | 2010-10-28 | 2012-05-03 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| AP2013006953A0 (en) | 2010-12-07 | 2013-06-30 | Bayer Ip Gmbh | Substituted 1-benzylcycloalkylcarboxlic acids and use thereof |
| CA2866210A1 (en) * | 2012-02-28 | 2013-09-06 | Piramal Enterprises Limited | Phenyl alkanoic acid derivatives as gpr agonists |
| DE102012208530A1 (de) | 2012-05-22 | 2013-11-28 | Bayer Pharma AG | Substituierte Piperidinoacetamide und ihre Verwendung |
| TWI594975B (zh) * | 2013-04-24 | 2017-08-11 | 第一三共股份有限公司 | 二羧酸化合物 |
| TW201625601A (zh) | 2014-07-02 | 2016-07-16 | 諾華公司 | 噻吩-2-基-吡啶-2-基-1h-吡唑-4-羧酸衍生物及其作為可溶性鳥苷酸環化酶活化劑之用途 |
| TW201625584A (zh) | 2014-07-02 | 2016-07-16 | 諾華公司 | 茚滿及吲哚啉衍生物及其作為可溶性鳥苷酸環化酶活化劑之用途 |
| TW201625586A (zh) | 2014-07-02 | 2016-07-16 | 諾華公司 | 環己烯-1-基-吡啶-2-基-1h-吡唑-4-羧酸衍生物及其作為可溶性鳥苷酸環化酶活化劑之用途 |
| EP3191441B1 (en) * | 2014-09-09 | 2020-02-19 | Boehringer Ingelheim International Trading (Shanghai) Co. Ltd. | Novel process for preparation of spiro[2.5]octane-5,7-dione and spiro[3.5]nonane-6,8-dione |
| US20170327457A1 (en) | 2014-09-09 | 2017-11-16 | Bristol-Myers Squibb Company | Phenyl-(aza)cycloalkyl carboxylic acid gpr120 modulators |
| CN107074793B (zh) | 2014-09-26 | 2019-08-23 | 第一三共株式会社 | 二羧酸化合物的盐 |
| DK3207023T3 (da) | 2014-10-14 | 2019-12-09 | Syngenta Participations Ag | Fremgangsmåde til fremstilling af 1-(3,5-dichlorphenyl)-2,2,2-trifluorethanon og derivater deraf |
| SG11201704971UA (en) * | 2014-12-18 | 2017-07-28 | Bayer Pharma AG | Substituted pyridyl-cycloalkyl-carboxylic acids, compositions containing them and medical uses thereof |
| BR112017023855A2 (pt) | 2015-05-06 | 2018-07-17 | Bayer Pharma Aktiengesellschaft | o uso de estimulantes de sgc, ativadores de scg, em separado e combinações com inibidores de pde5 para o tratamento de úlceras digitais (du) concomitantes à esclerose múltipla (ssc) |
| EP3325013B2 (de) | 2015-07-23 | 2023-07-19 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und einem angiotensin aii-antagonisten und ihre verwendung |
| EP3525778A1 (de) | 2016-10-11 | 2019-08-21 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc aktivatoren und mineralocorticoid-rezeptor-antagonisten |
| CN106565637A (zh) * | 2016-11-04 | 2017-04-19 | 天津雅奥科技发展有限公司 | 一种3‑氧杂环丁烷乙酸的合成方法 |
| WO2019081456A1 (en) | 2017-10-24 | 2019-05-02 | Bayer Aktiengesellschaft | USE OF SGC ACTIVATORS AND STIMULATORS COMPRISING A BETA2 SUBUNIT |
| CN111433204A (zh) | 2017-12-01 | 2020-07-17 | 拜耳制药股份公司 | 制备用作药物活性物质的(3s)-3-(4-氯-3-{[(2s,3r)-2-(4-氯苯基)-4,4,4-三氟-3-甲基丁酰基]氨基}苯基)-3-环丙基丙酸及其结晶形式的方法 |
| EP3498298A1 (en) | 2017-12-15 | 2019-06-19 | Bayer AG | The use of sgc stimulators and sgc activators alone or in combination with pde5 inhibitors for the treatment of bone disorders including osteogenesis imperfecta (oi) |
| US20210052528A1 (en) | 2018-04-30 | 2021-02-25 | Bayer Aktiengesellschaft | The use of sgc activators and sgc stimulators for the treatment of cognitive impairment |
| CN112384213A (zh) | 2018-05-15 | 2021-02-19 | 拜耳公司 | 用于治疗与神经纤维敏化有关的疾病的1,3-噻唑-2-基取代的苯甲酰胺 |
| US11508483B2 (en) | 2018-05-30 | 2022-11-22 | Adverio Pharma Gmbh | Method of identifying a subgroup of patients suffering from dcSSc which benefits from a treatment with sGC stimulators and sGC activators in a higher degree than a control group |
| US10905667B2 (en) | 2018-07-24 | 2021-02-02 | Bayer Pharma Aktiengesellschaft | Orally administrable modified-release pharmaceutical dosage form |
| CA3126778A1 (en) | 2019-01-17 | 2020-07-23 | Bayer Aktiengesellschaft | Methods to determine whether a subject is suitable of being treated with an agonist of soluble guanylyl cyclase (sgc) |
| WO2020164008A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| WO2022237797A1 (zh) * | 2021-05-14 | 2022-11-17 | 南京明德新药研发有限公司 | 烷基羧酸化合物及其应用 |
| TW202342420A (zh) * | 2022-02-18 | 2023-11-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 羧酸類化合物、其製備方法及其在醫藥上的應用 |
| WO2023237577A1 (en) | 2022-06-09 | 2023-12-14 | Bayer Aktiengesellschaft | Soluble guanylate cyclase activators for use in the treatment of heart failure with preserved ejection fraction in women |
| CN115850102A (zh) * | 2022-12-29 | 2023-03-28 | 浙江工业大学 | 一种奥司他韦的制备方法 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5640710B2 (ko) * | 1973-01-18 | 1981-09-22 | ||
| US5041453A (en) * | 1990-05-30 | 1991-08-20 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Quinolinyl-benzoheterobicyclic derivatives as antagonists of leukotriene D4 |
| DE4301900A1 (de) | 1993-01-25 | 1994-07-28 | Bayer Ag | 2-Oxochinolin-1-yl-methylphenylessigsäurederivate |
| US5643957A (en) | 1993-04-22 | 1997-07-01 | Emisphere Technologies, Inc. | Compounds and compositions for delivering active agents |
| DE4443892A1 (de) * | 1994-12-09 | 1996-06-13 | Bayer Ag | 4-(Chinolin-2-yl-methoxy)-phenyl-essigsäurederivate |
| US5650386A (en) | 1995-03-31 | 1997-07-22 | Emisphere Technologies, Inc. | Compositions for oral delivery of active agents |
| DE19546918A1 (de) * | 1995-12-15 | 1997-06-19 | Bayer Ag | Bicyclische Heterocyclen |
| EP0802192A1 (de) * | 1996-04-17 | 1997-10-22 | Bayer Ag | Heterocyclisch-substituierte Phenylglycinolamide mit antiatherosklerotischer Wirkung und Verfahren zu ihrer Herstellung |
| DE19821483A1 (de) * | 1998-05-14 | 1999-11-18 | Hoechst Marion Roussel De Gmbh | Imidazolidinderivate, ihre Herstellung, ihre Verwendung und sie enthaltende pharmazeutische Präparate |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE19834044A1 (de) * | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| BR0010605A (pt) | 1999-04-28 | 2002-02-13 | Aventis Pharma Gmbh | Derivados ácidos de di-aril como ligantes de receptores ppar |
| BR0010126A (pt) * | 1999-04-28 | 2002-02-26 | Aventis Pharma Gmbh | Derivados de ácido tri-arìlico como ligandos receptores de ppar |
| EP1229010A1 (en) | 1999-10-01 | 2002-08-07 | Japan Energy Corporation | Novel diarylamide derivatives and use thereof as medicines |
| TWI262185B (en) * | 1999-10-01 | 2006-09-21 | Eisai Co Ltd | Carboxylic acid derivatives having anti-hyperglycemia and anti-hyperlipemia action, and pharmaceutical composition containing the derivatives |
| PT1285908E (pt) | 2000-05-29 | 2008-12-04 | Kyorin Seiyaku Kk | Derivados do ácido fenilpropiónico substituídos |
| AU2001278771A1 (en) | 2000-08-22 | 2002-03-04 | Ono Pharmaceutical Co. Ltd. | Carboxylic acid derivatives, process for producing the same and drugs containingthe same as the active ingredient |
| AR031176A1 (es) * | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| US7176204B2 (en) * | 2000-12-05 | 2007-02-13 | Kyorin Pahrmaceutical Co., Ltd. | Substituted carboxylic acid derivatives |
| WO2002053547A1 (fr) * | 2000-12-28 | 2002-07-11 | Takeda Chemical Industries, Ltd. | Derives d'acide alcanoique, procede de production et utilisation correspondants |
| WO2002076959A1 (fr) * | 2001-03-23 | 2002-10-03 | Takeda Chemical Industries, Ltd. | Derive heterocyclique a cinq membres d'acide alcanoique |
| EP1375472A4 (en) | 2001-03-30 | 2008-12-10 | Eisai R&D Man Co Ltd | BENZENE COMPOUND AND SALT THEREOF |
| US20030105097A1 (en) | 2001-05-14 | 2003-06-05 | Pfizer Inc. | Alkylamide compounds |
| JP4529119B2 (ja) * | 2001-08-09 | 2010-08-25 | 小野薬品工業株式会社 | カルボン酸誘導体化合物およびその化合物を有効成分として含有する薬剤 |
| JPWO2003016265A1 (ja) | 2001-08-17 | 2004-12-02 | エーザイ株式会社 | 環状化合物およびpparアゴニスト |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| AU2003262023A1 (en) * | 2002-09-10 | 2004-04-30 | Takeda Pharmaceutical Company Limited | Five-membered heterocyclic compounds |
| US20050187266A1 (en) | 2003-04-15 | 2005-08-25 | Pfizer Inc | Alpha substituted carboxylic acids |
| US20050234066A1 (en) | 2004-04-15 | 2005-10-20 | Agouron Pharmaceuticals, Inc. | Alpha substituted carboxylic acids |
| EP1620422A2 (en) | 2003-04-30 | 2006-02-01 | The Institutes for Pharmaceutical Discovery, LLC | Phenyl substituted carboxylic acids as inhibitors of protein tyrosine phosphatase-1b |
| KR20070004769A (ko) | 2004-02-27 | 2007-01-09 | 암젠 인코포레이션 | 대사 장애의 치료에 사용되는 화합물, 약제학적 조성물 및그 사용방법 |
| US20060100251A1 (en) | 2004-10-28 | 2006-05-11 | The Institutes For Pharmaceutical Discovery, Llc | Substituted phenylalkanoic acids |
| JP2008520683A (ja) | 2004-11-18 | 2008-06-19 | ジ インスチチュート フォー ファーマシューティカル ディスカバリー、エルエルシー | タンパク質チロシンホスファターゼ阻害剤としてのヘテロシクリルビフェニル誘導体 |
| MX2008000779A (es) * | 2005-07-18 | 2008-02-21 | Bayer Healthcare Ag | Uso de activadores y estimuladores de guanilatociclasa solubles para la prevencion o tratamiento de trastornos renales. |
| US20110092554A1 (en) * | 2007-11-19 | 2011-04-21 | Richard Chesworth | 1,3,5 tri-subtituted benzenes for treatment of alzheimer's disease and other disorders |
| DE102008018675A1 (de) | 2008-04-14 | 2009-10-15 | Bayer Schering Pharma Aktiengesellschaft | Oxo-heterocyclisch substituierte Carbonsäure-Derivate und ihre Verwendung |
| DE102009012314A1 (de) * | 2009-03-09 | 2010-09-16 | Bayer Schering Pharma Aktiengesellschaft | Oxo-heterocyclisch substituierte Alkylcarbonsäuren und ihre Verwendung |
| DE102009046115A1 (de) | 2009-10-28 | 2011-09-08 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 3-Phenylpropansäuren und ihre Verwendung |
| AP2013006953A0 (en) | 2010-12-07 | 2013-06-30 | Bayer Ip Gmbh | Substituted 1-benzylcycloalkylcarboxlic acids and use thereof |
| DE102011007272A1 (de) | 2011-04-13 | 2012-10-18 | Bayer Pharma Aktiengesellschaft | Verzweigte 3-Phenylpropionsäure-Derivate und ihre Verwendung |
-
2009
- 2009-10-28 DE DE102009046115A patent/DE102009046115A1/de not_active Withdrawn
-
2010
- 2010-10-21 WO PCT/EP2010/065910 patent/WO2011051165A1/de active Application Filing
- 2010-10-21 CN CN201080059787.9A patent/CN102712577B/zh not_active Expired - Fee Related
- 2010-10-21 DK DK10768933.3T patent/DK2493845T3/en active
- 2010-10-21 CA CA2777152A patent/CA2777152C/en not_active Expired - Fee Related
- 2010-10-21 RU RU2012121577/04A patent/RU2553263C2/ru not_active IP Right Cessation
- 2010-10-21 BR BR112012010057A patent/BR112012010057A2/pt not_active IP Right Cessation
- 2010-10-21 PT PT107689333T patent/PT2493845E/pt unknown
- 2010-10-21 AU AU2010311678A patent/AU2010311678B2/en not_active Ceased
- 2010-10-21 PL PL10768933T patent/PL2493845T3/pl unknown
- 2010-10-21 KR KR1020127013589A patent/KR20120101411A/ko not_active Application Discontinuation
- 2010-10-21 EP EP10768933.3A patent/EP2493845B1/de not_active Not-in-force
- 2010-10-21 MX MX2012004951A patent/MX2012004951A/es active IP Right Grant
- 2010-10-21 ES ES10768933.3T patent/ES2446970T3/es active Active
- 2010-10-21 JP JP2012535750A patent/JP5801312B2/ja not_active Expired - Fee Related
- 2010-10-25 UY UY0001032970A patent/UY32970A/es not_active Application Discontinuation
- 2010-10-26 AR ARP100103934A patent/AR079015A1/es unknown
- 2010-10-27 TW TW099136615A patent/TWI487519B/zh not_active IP Right Cessation
- 2010-10-28 US US12/914,101 patent/US20110130445A1/en not_active Abandoned
-
2012
- 2012-04-02 IL IL218989A patent/IL218989A/en not_active IP Right Cessation
- 2012-04-04 ZA ZA2012/02465A patent/ZA201202465B/en unknown
-
2013
- 2013-04-03 HK HK13104154.9A patent/HK1177195A1/xx not_active IP Right Cessation
- 2013-10-03 US US14/045,630 patent/US9018414B2/en active Active
-
2014
- 2014-03-21 HR HRP20140270AT patent/HRP20140270T1/hr unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20140021008A (ko) * | 2011-04-13 | 2014-02-19 | 바이엘 인텔렉쳐 프로퍼티 게엠베하 | 분지 3-페닐프로피온산 유도체 및 그의 용도 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5801312B2 (ja) | 2015-10-28 |
| AU2010311678B2 (en) | 2015-09-03 |
| JP2013509369A (ja) | 2013-03-14 |
| US9018414B2 (en) | 2015-04-28 |
| ZA201202465B (en) | 2013-08-28 |
| IL218989A (en) | 2015-10-29 |
| EP2493845A1 (de) | 2012-09-05 |
| TW201127373A (en) | 2011-08-16 |
| CA2777152A1 (en) | 2011-05-05 |
| AU2010311678A1 (en) | 2012-05-17 |
| TWI487519B (zh) | 2015-06-11 |
| US20140142069A1 (en) | 2014-05-22 |
| RU2012121577A (ru) | 2013-12-10 |
| CN102712577A (zh) | 2012-10-03 |
| MX2012004951A (es) | 2012-06-13 |
| DK2493845T3 (en) | 2014-03-24 |
| WO2011051165A1 (de) | 2011-05-05 |
| BR112012010057A2 (pt) | 2019-09-24 |
| DE102009046115A1 (de) | 2011-09-08 |
| CN102712577B (zh) | 2014-10-15 |
| PL2493845T3 (pl) | 2014-05-30 |
| PT2493845E (pt) | 2014-02-24 |
| CA2777152C (en) | 2017-09-19 |
| RU2553263C2 (ru) | 2015-06-10 |
| EP2493845B1 (de) | 2013-12-25 |
| AR079015A1 (es) | 2011-12-21 |
| UY32970A (es) | 2011-05-31 |
| US20110130445A1 (en) | 2011-06-02 |
| HRP20140270T1 (hr) | 2014-04-25 |
| HK1177195A1 (en) | 2013-08-16 |
| ES2446970T3 (es) | 2014-03-11 |
| IL218989A0 (en) | 2012-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11377417B2 (en) | Branched 3-phenylpropionic acid derivatives and their use | |
| KR20120101411A (ko) | 치환된 3-페닐프로피온산 및 그의 용도 | |
| JP5989660B2 (ja) | 置換1−ベンジルシクロアルキルカルボン酸およびその使用 | |
| DE102010062544A1 (de) | Substituierte 1-Benzylcycloalkylcarbonsäuren und ihre Verwendung | |
| DE102011006974A1 (de) | Substituierte 1-Benzylcycloalkylcarbonsäuren und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| N231 | Notification of change of applicant | ||
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E601 | Decision to refuse application |