KR101106812B1 - 아릴옥시 및 아릴알킬렌옥시 치환된 이미다조퀴놀린 - Google Patents
아릴옥시 및 아릴알킬렌옥시 치환된 이미다조퀴놀린 Download PDFInfo
- Publication number
- KR101106812B1 KR101106812B1 KR1020067003863A KR20067003863A KR101106812B1 KR 101106812 B1 KR101106812 B1 KR 101106812B1 KR 1020067003863 A KR1020067003863 A KR 1020067003863A KR 20067003863 A KR20067003863 A KR 20067003863A KR 101106812 B1 KR101106812 B1 KR 101106812B1
- Authority
- KR
- South Korea
- Prior art keywords
- group
- alkyl
- imidazo
- independently
- independently selected
- Prior art date
Links
- 0 CC*C1=CN1*(C)C Chemical compound CC*C1=CN1*(C)C 0.000 description 5
- CVQWVTFIXRJTGS-UHFFFAOYSA-N CC(C[n]1c(c(ccc(OCc2ccccc2)c2)c2nc2N)c2nc1)O Chemical compound CC(C[n]1c(c(ccc(OCc2ccccc2)c2)c2nc2N)c2nc1)O CVQWVTFIXRJTGS-UHFFFAOYSA-N 0.000 description 1
- FPUHAVHOAWLLEW-UHFFFAOYSA-N CCC[n]1c(c(ccc(OCc2ccccc2)c2)c2nc2N)c2nc1COCC Chemical compound CCC[n]1c(c(ccc(OCc2ccccc2)c2)c2nc2N)c2nc1COCC FPUHAVHOAWLLEW-UHFFFAOYSA-N 0.000 description 1
- YKIFJBUKHZZZIN-UHFFFAOYSA-N CCOCc1nc2c(N)nc(ccc(OCc3cc(CO)ccc3)c3)c3c2[n]1CC(C)C Chemical compound CCOCc1nc2c(N)nc(ccc(OCc3cc(CO)ccc3)c3)c3c2[n]1CC(C)C YKIFJBUKHZZZIN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Virology (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
본 출원은 6번, 7번, 8번 또는 9번-위치에 아릴옥시 또는 아릴알킬렌옥시 또는 히드록시 치환기를 갖는 이미다조퀴놀린 화합물; 이 화합물을 함유하는 제약 조성물; 중간체; 동물에서 사이토카인을 조절하고, 바이러스성 질환 및 신생물성 질환을 비롯한 질환을 치료하는데 면역조정제로서 이들 화합물을 사용하는 방법을 개시한다.
이미다조퀴놀린 화합물, 사이토카인, 바이러스성 질환, 신생물성 질환
Description
<관련 출원>
본 발명은 2003년 8월 27일 출원된 미국 가출원 제60/498270호 및 2004년 6월 18일 출원된 미국 가출원 제60/581254호를 우선권으로 주장하며, 상기 2개의 문헌은 모두 참고로 본원에 포함된다.
최근, 면역계를 조절하는 화합물을 찾아내기 위한 방대한 노력이 이루어져 왔다. 사이토카인 유도 활성 및 면역조절 활성이 입증되어 다양한 장애의 치료에 유용한 상기 화합물의 예로는 특정 1H-이미다조[4,5-c]퀴놀린-4-아민, 1H-이미다조[4,5-c]피리딘-4-아민, 테트라히드로퀴놀린-4-아민, 나프티리딘-4-아민 및 테트라히드로나프티리딘-4-아민 화합물, 및 특정한 유사 티아졸로 및 옥사졸로 화합물이 있다.
그러나, 면역조절 화합물을 찾아내기 위한 노력의 중대한 진전에도 불구하고, 사이토카인 생합성 또는 기타 메카니즘을 유도하거나 억제함으로써 면역 반응의 측면을 조절할 수 있는 부가적인 화합물에 대한 중대한 과학적 요구 및 의학적 요구가 여전히 존재하는 실정이다.
<발명의 개요>
본 발명에 이르러 사이토카인 생합성을 조절하는데 유용한 신규 부류의 화합물이 발견되었다. 한 측면에서, 본 발명은 그러한 화합물을 제공하며, 이 화합물은 하기 화학식 I의 화합물 및 제약상 허용되는 그의 염이다.

상기 식에서, R, n, R', R" 및 R3은 하기 정의되는 바와 같다.
화학식 I의 화합물 및(또는) 염은, 동물에게 투여시 사이토카인 생합성을 조절하는 능력 (예컨대, 1종 이상의 사이토카인의 생합성 또는 생성을 유도하거나 억제함) 및 달리 면역 반응을 조절하는 능력으로 인해 면역 반응 조절인자 (IRM)로서 유용하다. 이로 인해, 상기 화합물 또는 염은 면역 반응에서의 그러한 변화에 대해 반응성인 바이러스성 질환 및 신생물성 질환과 같은 각종 증상의 치료에 유용하다.
다른 측면에서, 본 발명은 유효량의 본 발명의 화합물 또는 염을 함유하는 제약 조성물, 및 유효량의 1종 이상의 화학식 I 화합물 및(또는) 제약상 허용되는 그의 염을 동물에게 투여함으로써 상기 동물에서 사이토카인 생합성을 조절 (예컨대, 유도 또는 억제)하고, 상기 동물에서 바이러스성 질환을 치료하고, 상기 동물에서 신생물성 질환을 치료하는 방법을 제공한다.
다른 측면에서, 본 발명은 화학식 I의 화합물을 합성하는 방법, 및 이들 화합물의 합성에 유용한 중간체 화합물을 제공한다. 또한, 이러한 특정 중간체 화합물 (예를 들어, 하기 화학식 VII의 화합물)은 상기와 같은 면역 반응 조절인자로서 유용함이 밝혀졌다. 따라서, 본 발명은 유효량의 1종 이상의 상기 화합물 및(또는) 그의 염을 함유하는 제약 조성물, 및 유효량의 1종 이상의 상기 화합물 및(또는) 제약상 허용되는 그의 염을 동물에게 투여함으로써 상기 동물에서 사이토카인 생합성을 유도하고, 상기 동물에서 바이러스성 질환을 치료하고, 상기 동물에서 신생물성 질환을 치료하는 방법을 제공한다.
본원에 사용된 "하나", "한", "그", "하나 이상" 및 "1종 이상"은 상호 교환하여 사용될 수 있다.
용어 "포함하는" 및 그의 변형 형태는 상세한 설명 및 청구의 범위에 그 용어가 나타나는 곳에서 제한된 의미를 갖지 않는다.
상기 발명의 개요는 본 발명에 개시된 각각의 실시양태 또는 본 발명의 모든 실시양태를 설명하려는 의도는 아니다. 하기 상세한 설명은 보다 구체적으로 본 발명의 실시양태를 예시한다. 또한, 실시예의 목록을 통해 본 발명에 대한 길잡이가 제공되며, 실시예는 다양하게 조합되어 이용될 수 있다. 각각의 예에서, 언급된 목록은 대표적인 군으로서만 작용하며, 다른 것을 배제하는 목록으로 해석해서는 안된다.
한 측면에서, 본 발명은 화학식 I의 화합물 또는 제약상 허용되는 그의 염을 제공한다.
<화학식 I>

상기 식에서,
R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;
Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;
Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;
Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;
X는 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;
Y는 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 수소 및 알킬로 이루어진 군으로부터 선택되고;
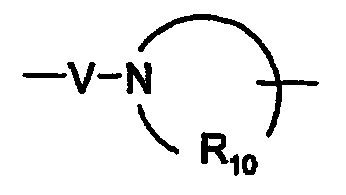
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이고;
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
R' 및 R"은 독립적으로 수소 및 비-간섭 치환기로 이루어진 군으로부터 선택되며;
n은 0 또는 1이다.
한 실시양태에서, 본 발명은 화학식 II의 화합물 또는 제약상 허용되는 그의 염을 제공한다.

상기 식에서,
R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;
Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;
Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;
Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
n은 0 또는 1이고;
R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;
Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아 미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는 각각 독립적으로  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)- W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
다른 실시양태에서, 본 발명은 화학식 III의 화합물 또는 제약상 허용되는 그의 염을 제공한다.

상기 식에서,
R3 -1은 -Z-Ar이고;
Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;
Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아 릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
n은 0 또는 1이고;
R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;
Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는 각각 독립적으로  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택 되며;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
다른 측면에서, 본 발명은 화학식 VII, IX 및 XI의 중간체 화합물을 제공한다.
한 실시양태에서, 본 발명은 하기 화학식 VII의 화합물 또는 제약상 허용되는 그의 염을 제공한다.

상기 식에서,
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
n은 0 또는 1이고;
R1 -1은 -R4 -1, -X'-R4 -1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;
R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알 키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;
X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;
Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시 알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R4 -1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는 각각 독립적으로 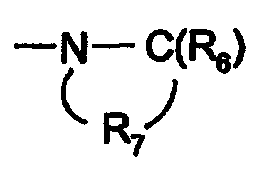 ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택 되고;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이며;
단, R1 -1이 수소 또는 2-메틸프로필인 경우에는 R2가 수소가 아니고; R1 -1이 2-메틸프로페닐 또는 2-히드록시-2-메틸프로필인 경우에는 R2가 메틸, 에톡시메틸 및 히드록시메틸이 아니다.
다른 실시양태에서, 본 발명은 하기 화학식 IX의 화합물 또는 제약상 허용되는 그의 염을 제공한다.

상기 식에서,
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
n은 0 또는 1이고;
R1 -1은 -R4 -1, -X'-R4 -1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;
R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;
X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;
Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아 릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R4 -1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는 각각 독립적으로  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
다른 실시양태에서, 본 발명은 하기 화학식 XI의 화합물 또는 제약상 허용되는 그의 염을 제공한다.

상기 식에서,
R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;
Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;
Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;
Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;
R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택되고;
n은 0 또는 1이고;
R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;
X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;
Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,  ,
,  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;
R5는 각각 독립적으로  ,
,  ,
,  및
및  로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고;
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;
R7은 각각 독립적으로 C2 -7 알킬렌이고;
R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;
R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;
R10은 각각 독립적으로 C3 -8 알킬렌이고;
A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;
Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;
V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;
W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;
a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
본원에 사용된 바와 같이, 용어 "알킬", "알케닐", "알키닐" 및 접두어 "알크-"는 직쇄 및 분지쇄기 양쪽 모두와 시클릭기, 즉 시클로알킬 및 시클로알케닐을 포함한다. 달리 특정하지 않는 한, 이들 기는 1 내지 20개의 탄소 원자를 함유하며, 알케닐기는 2 내지 20개의 탄소 원자를 함유하고, 알키닐기는 2 내지 20개의 탄소 원자를 함유한다. 일부 실시양태에서, 이들 기는 총 10개 이하의 탄소 원자, 8개 이하의 탄소 원자, 6개 이하의 탄소 원자, 또는 4개 이하의 탄소 원자를 갖는다. 시클릭기는 모노시클릭 또는 폴리시클릭일 수 있고, 바람직하게는 3 내지 10개의 고리 탄소 원자를 갖는다. 시클릭기의 예로는, 시클로프로필, 시클로프로필메틸, 시클로펜틸, 시클로헥실, 아다만틸, 및 치환 및 비치환 보르닐, 노르보르닐 및 노르보르네릴이 포함된다.
달리 특정하지 않는 한, "알킬렌", "-알킬렌-", "알케닐렌", "-알케닐렌-", "알키닐렌" 및 "-알키닐렌-"은 상기에 정의한 "알킬", "알케닐" 및 "알키닐"기의 2가 형태이다. 용어 "알킬레닐", "알케닐레닐" 및 "알키닐레닐"은 "알킬렌", "알케닐렌" 및 "알키닐렌"이 각각 치환되는 경우에 사용된다. 예를 들어, 아릴알킬레닐기는 아릴기가 결합된 "알킬렌" 잔기를 포함한다.
용어 "할로알킬"은 퍼플루오르화기를 비롯한, 하나 이상의 할로겐 원자로 치환된 알킬기를 포함한다. 이는 접두어 "할로-"를 포함하는 다른 기에도 적용된다. 적합한 할로알킬기의 예로는, 클로로메틸, 트리플루오로메틸 등이 있다.
본원에 사용된 바와 같이, 용어 "아릴"은 카르보시클릭 방향족 고리 또는 고리계를 포함한다. 아릴기의 예로는, 페닐, 나프틸, 비페닐, 플루오레닐 및 인데닐 이 포함된다.
용어 "헤테로원자"는 O, S 또는 N 원자를 지칭한다.
용어 "헤테로아릴"은 하나 이상의 고리 헤테로원자 (예를 들어, O, S, N)를 함유하는 방향족 고리 또는 고리계를 포함한다. 적합한 헤테로아릴기로는, 푸릴, 티에닐, 피리딜, 퀴놀리닐, 이소퀴놀리닐, 인돌릴, 이소인돌릴, 트리아졸릴, 피롤릴, 테트라졸릴, 이미다졸릴, 피라졸릴, 옥사졸릴, 티아졸릴, 벤조푸라닐, 벤조티오페닐, 카르바졸릴, 벤족사졸릴, 피리미디닐, 벤즈이미다졸릴, 퀴녹살리닐, 벤조티아졸릴, 나프티리디닐, 이속사졸릴, 이소티아졸릴, 퓨리닐, 퀴나졸리닐, 피라지닐, 1-옥시도피리딜, 피리다지닐, 트리아지닐, 테트라지닐, 옥사디아졸릴, 티아디아졸릴 등이 포함된다.
용어 "헤테로시클릴"은 하나 이상의 고리 헤테로원자 (예를 들어, O, S, N)를 함유하는 비-방향족 고리 또는 고리계를 포함하며, 상기에 언급된 헤테로아릴기의 완전 포화 및 부분 불포화 유도체 모두를 포함한다. 헤테로시클릭기의 예로는, 피롤리디닐, 테트라히드로푸라닐, 모르폴리닐, 티오모르폴리닐, 피페리디닐, 피페라지닐, 티아졸리디닐, 이미다졸리디닐, 이소티아졸리디닐, 테트라히드로피라닐, 퀴누클리디닐, 호모피페리디닐, 호모피페라지닐 등이 포함된다.
용어 "아릴렌", "헤테로아릴렌" 및 "헤테로시클릴렌"은 상기에 정의한 "아릴", "헤테로아릴" 및 "헤테로시클릴"기의 2가 형태이다. 용어 "아릴레닐", "헤테로아릴레닐" 및 "헤테로시클릴레닐"은 "아릴렌", "헤테로아릴렌" 및 "헤테로시클릴렌"이 각각 치환되는 경우에 사용된다. 예를 들어, 알킬아릴레닐기는 알킬기가 결 합된 아릴렌 잔기를 포함한다.
기 (또는 치환기 또는 변수)가 본원에 기재된 임의의 화학식에서 하나 이상 존재하는 경우, 각각의 기 (또는 치환기 또는 변수)는 명시되어 있든 없든 독립적으로 선택된다. 예를 들어, 화학식 -N(R8)-C(R6)-N(R8)-에서, R8기는 각각 독립적으로 선택된다. 또다른 예로, R2 및 R3기가 모두 R4기를 함유하는 경우, R4기는 각각 독립적으로 선택된다. 다른 예로, 하나 이상의 Y기가 존재하고 (즉, R2 및 R3이 모두 Y기를 함유하고) Y기가 각각 하나 이상의 R8기를 함유하는 경우, Y기는 각각 독립적으로 선택되고, R8기도 각각 독립적으로 선택된다.
본 발명은, 본원에 기재된 화합물 및 그의 제약상 허용되는 임의의 형태의 그의 염, 예를 들어 부분입체이성질체 및 거울상이성질체 등의 이성질체, 용매화물, 폴리모르프 등을 포함한다. 특히, 화합물이 광학적으로 활성인 경우, 본 발명은 특히 상기 화합물의 거울상이성질체 뿐만 아니라 거울상이성질체의 라세미 혼합물을 각각 포함한다.
당업자에게 이해되는 바와 같이, 본원에 기재된 임의의 화합물에 대하여, 임의의 실시양태에서의 하기 변수들 (예를 들어, R, R', R", R1, R2, R3, n, A, X, Z 등) 각각은 임의의 실시양태에서의 임의의 하나 이상의 다른 변수들과 조합될 수 있다. 형성된 변수들의 조합은 각각 본 발명의 실시양태가 된다.
일부 실시양태에서, R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택된다. 일부 실시양태에서, R3은 -Z-Ar, -Z-Ar'-X-Y-R4 및 -Z-Ar'-Y-R4로 이루어진 군으로부터 선택된다. 일부 실시양태에서, R3 또는 R3 -1은 -Z-Ar이다. 일부 실시양태에서, Ar은 알킬, 알콕시, 니트로, 시아노, 카르복시, 할로겐, 히드록시알킬, 아미노, 알킬아미노, 디알킬아미노, 트리플루오로메틸, 트리플루오로메톡시 및 티에닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않은 페닐 또는 헤테로아릴이다. 특정한 이들 실시양태에서, 헤테로아릴은 벤조티아졸릴, 푸라닐, 이미다졸릴, 인돌릴, 이속사졸릴, 옥사디아졸릴, 피라지닐, 피리디닐, 피롤릴, 티아졸릴 및 티에닐로 이루어진 군으로부터 선택된다. 일부 이들 실시양태에서, Z는 결합, 알킬렌, 또는 -O-가 개재된 알킬렌이다. 특정한 이들 실시양태에서, Z는 C1 -3 알킬렌이다. 특정한 이들 실시양태에서, Z는 결합이다.
일부 실시양태에서, R3 또는 R3 -1은 벤질, 피리딘-3-일메틸, 4-클로로벤질, 4-플루오로벤질 또는 3-피리딘-3-일프로필이다.
일부 실시양태에서, R3은 -Z-Ar'-Y-R4 또는 -Z-Ar'-X-Y-R4이다. 특정한 이들 실시양태에서, X는 C1 -2 알킬렌이고; Y는 -N(R8)-S(O)2-, -S(O)2-, -C(R6)- 또는 -C(R6)-O-이고; R4는 알킬 또는 페닐이다. 특정한 이들 실시양태에서, X는 C1 -2 알킬렌이고; Y는 -NH-S(O)2-, -S(O)2-, -C(O)- 또는 -C(O)-O-이고; R4는 C1 -4 알킬 또는 페닐이다. 일부 이들 실시양태에서, Z는 결합, 알킬렌, 또는 -O-가 개재된 알킬렌이다. 특정한 이들 실시양태에서, Z는 C1 -3 알킬렌이다. 특정한 이들 실시양태에서, Z는 결합이다.
일부 실시양태에서, R3은 -Z-Ar'-Y-R4이다. 특정한 이들 실시양태에서, Y는 -S(O)2- 및 -C(O)O-로 이루어진 군으로부터 선택되고, R4는 C1 -4 알킬, 예를 들어 메틸이다. 특정한 이들 실시양태에서, Ar'는 알킬, 알콕시, 니트로, 시아노, 할로겐, 아미노, 알킬아미노, 디알킬아미노, 트리플루오로메틸 및 트리플루오로메톡시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않은 페닐렌이다.
일부 실시양태에서, R, R' 및 R"는 각각 독립적으로 비-간섭 치환기이다. 특정 실시양태에서, R' 및 R"는 각각 수소 및 비-간섭 치환기로 이루어진 군으로부터 독립적으로 선택된다. 본원에서, "비-간섭"는 화합물의 면역조절인자 활성 (예를 들어, 하나 이상의 사이토카인의 생합성을 유도하거나, 또는 하나 이상의 사이토카인의 생합성을 억제하는 능력)을 손상시키지 않는 것을 의미한다.
일부 실시양태에서, R은 알킬, 알콕시, 히드록시, 할로겐 및 트리플루오로메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, n은 0이다.
일부 실시양태에서, n은 0 또는 1이다.
일부 실시양태에서, R'은 수소 및 비-간섭 치환기로 이루어진 군으로부터 선 택된다.
일부 실시양태에서, R'은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R'은 알킬, 아릴알킬레닐, 아릴옥시알킬레닐, 히드록시알킬, 디히드록시알킬, 알킬술포닐알킬레닐, 헤테로시클릴알킬레닐 (여기서, 헤테로시클릴은 하나 이상의 알킬기로 임의로 치환됨), -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되며; 상기 X는 알킬렌이고, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2-, -N(R8)-C(R6)-N(R8)- 또는  이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는
이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는  이다.
이다.
일부 실시양태에서, R'는 2-히드록시-2-메틸프로필, 2-메틸프로필, 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아미노]부틸, 2-메틸-2-[(메틸술포닐)아미노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R'은 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아 미노]부틸, 2-메틸-2-[(메틸술포닐)아미노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1은 알킬, 아릴알킬레닐, 아릴옥시알킬레닐, 히드록시알킬, 디히드록시알킬, 알킬술포닐알킬레닐, 헤테로시클릴알킬레닐 (여기서, 헤테로시클릴은 하나 이상의알킬기로 임의로 치환됨), -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되며; 상기 X는 알킬렌이고, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2-, -N(R8)-C(R6)-N(R8)- 또는  이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는
이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는  이다.
이다.
일부 실시양태에서, R1은 2-히드록시-2-메틸프로필, 2-메틸프로필, 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아미노]부틸, 2-메틸-2-[(메틸술포닐)아미 노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1은 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아미노부틸, 2-메틸-2-[(메틸술포닐)아미노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1 -1은 -R4 -1, -X'-R4 -1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1 -1은 알킬, 아릴알킬레닐, 히드록시알킬, 디히드록시알킬, 헤테로시클릴알킬레닐 (여기서, 헤테로시클릴은 하나 이상의 알킬기로 임의로 치환됨), -X'-Y'-R4 및 -X'-R5로 이루어진 군으로부터 선택된다. 특정한 이들 실시양태에서, X'는 알킬렌이고, Y'는 -N(R8)-Q-이고, Q는 -C(R6)-, -S(O)2- 또는 -C(R6)-N(R8)-W-이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는  이다.
이다.
일부 실시양태에서, R1 -1은 2-히드록시-2-메틸프로필, 2-메틸프로필, 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아미노]부틸, 2-메틸-2-[(메틸술포닐)아미노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R1 -1은 프로필, 2,3-디히드록시프로필, 4-[(메틸술포닐)아미노]부틸, 2-메틸-2-[(메틸술포닐)아미노]프로필, 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필, 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸, 테트라히드로-2H-피란-4-일메틸 및 (2,2-디메틸-1,3-디옥솔란-4-일)메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R"는 수소 또는 비-간섭 치환기이다.
일부 실시양태에서, R"는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R"는 수소, 알킬, 알콕시알킬레닐 및 -X-N(R8)-C(R6)-N(R8)-R4로 이루어진 군으로부터 선택된다. 특정한 이들 실시양태에서, X는 C1 -4 알킬렌이고, R4는 C1 -4 알킬이다. 일부 이들 실시양태에서, X는 C1 -2 알킬렌이다.
일부 실시양태에서, R"는 수소, 메틸, 에틸, 프로필, 부틸, 에톡시메틸, 메톡시메틸, 2-메톡시에틸 및 메틸아미노카르보닐아미노메틸로 이루어진 군으로부터 선택된다. 일부 이들 실시양태에서, R"는 에틸, 프로필, 에톡시메틸, 2-메톡시에틸 및 메톡시메틸로 이루어진 군으로부터 선택된다. 일부 이들 실시양태에서, R"는 에틸, 프로필, 2-메톡시에틸 및 메톡시메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R2는 수소, 알킬, 알콕시알킬레닐 및 -X-N(R8)-C(R6)-N(R8)-R4로 이루어진 군으로부터 선택된다. 특정한 이들 실시양태에서, X는 C1 -4 알킬렌이고, R4는 C1 -4 알킬이다. 일부 이들 실시양태에서, X는 C1 -2 알킬렌이다.
일부 실시양태에서, R2는 수소, 메틸, 에틸, 프로필, 부틸, 에톡시메틸, 메톡시메틸, 2-메톡시에틸 및 메틸아미노카르보닐아미노메틸로 이루어진 군으로부터 선택된다. 일부 이들 실시양태에서, R2는 에틸, 프로필, 에톡시메틸, 2-메톡시에틸 및 메톡시메틸로 이루어진 군으로부터 선택된다. 일부 이들 실시양태에서, R2는 에틸, 프로필, 2-메톡시에틸 및 메톡시메틸로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R4는 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며; 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴 레닐 및 헤테로시클릴기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있다. 일부 실시양태에서, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이다. 일부 실시양태에서, R4는 알킬 또는 페닐이다. 일부 실시양태에서, R4는 C1 -4 알킬 또는 페닐이다. 일부 실시양태에서, R4는 C1 -4 알킬이다.
일부 실시양태에서, R4 -1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며; 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있 다. 일부 실시양태에서, R4 -1은 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기로 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이다. 일부 실시양태에서, R4 -1은 알킬 또는 페닐이다. 일부 실시양태에서, R4 -1은 C1 -4 알킬 또는 페닐이다. 일부 실시양태에서, R4 -1은 C1 -4 알킬이다.
일부 실시양태에서, R5는



일부 실시양태에서, R6은 =O 및 =S로 이루어진 군으로부터 선택된다. 일부 실시양태에서, R6은 =O이다. 일부 실시양태에서, R6은 =S이다.
일부 실시양태에서, R7은 C2 -7 알킬렌이다. 일부 실시양태에서, R7은 C3 -4 알킬렌이다. 일부 실시양태에서, R7은 프로필렌이다.
일부 실시양태에서, R8은 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택된다. 일부 실시양태에서, R8은 수소 또는 알킬이다. 일부 실시양태에서, R8은 수소이다.
일부 실시양태에서, R9는 수소 및 알킬로 이루어진 군으로부터 선택된다.
일부 실시양태에서, R10은 독립적으로 C3 -8 알킬렌이다.
일부 실시양태에서, Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Ar은 알킬, 알콕시, 니트로, 시아노, 카르복시, 할로겐, 히드록시알킬, 아미노, 알킬아미노, 디알킬아미노, 트리플루오로메틸, 트리플루오로메톡시 및 티에닐로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않은 페닐 또는 헤테로아릴이다. 특정한 이들 실시양태에서, 헤테로아릴은 벤조티아졸릴, 푸라닐, 이미다졸릴, 인돌릴, 이속사졸릴, 옥 사디아졸릴, 피라지닐, 피리디닐, 피롤릴, 티아졸릴 및 티에닐로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Ar은 페닐이다.
일부 실시양태에서, Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Ar'는 알킬, 알콕시, 니트로, 시아노, 할로겐, 아미노, 알킬아미노, 디알킬아미노, 트리플루오로메틸 및 트리플루오로메톡시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기로 치환되거나 치환되지 않은 페닐렌이다.
일부 실시양태에서, A는 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택된다. 일부 실시양태에서, A는 -O-이다.
일부 실시양태에서, Q는 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Q는 -C(R6)-, -S(O)2- 또는 -C(R6)-N(R8)-W-이다. 일부 실시양태에서, Q는 -S(O)2-이다. 일부 실시양태에서, Q는 -C(R6)-N(R8)-W-이다.
일부 실시양태에서, V는 -C(R6)-, -O-C(R6)-, -N-(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택된다.
일부 실시양태에서, W는 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택된다. 일부 실시양태에서, W는 결합이다.
일부 실시양태에서, X는 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며; 상기 알킬렌, 알케닐렌 및 알키닐렌기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나, 하나 이상의 -O-기가 임의로 개재될 수 있다. 일부 실시양태에서, X는 알킬렌이다. 일부 실시양태에서, X는 C1 -4 알킬렌이다. 일부 실시양태에서, X는 C1 -2 알킬렌이다.
일부 실시양태에서, X'는 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며; 상기 알킬렌, 알케닐렌 및 알키닐렌기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌기가 임의로 개재될 수 있다. 일부 실시양태에서, X'는 알킬렌이다. 일부 실시양태에서, X'는 C1 -4 알킬렌이다. 일부 실시양태에서, X'는 C1 -2 알킬렌이다.
일부 실시양태에서, Y는 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,

로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2-, -N(R8)-C(R6)-N(R8)- 또는  이다. 일부 실시양태에서, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2- 또는 -N(R8)-C(R6)-N(R8)-이다. 일부 실시양태에서, Y는 -N(R8)-S(O)2-, -S(O)2-, -C(R6)- 또는 -C(R6)-O-이다. 일부 실시양태에서, Y는 -NH-S(O)2-, -S(O)2-, -C(O)- 또는 -C(O)-O-이다. 일부 실시양태에서, Y는 -S(O)2- 또는 -C(O)O-이다.
이다. 일부 실시양태에서, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2- 또는 -N(R8)-C(R6)-N(R8)-이다. 일부 실시양태에서, Y는 -N(R8)-S(O)2-, -S(O)2-, -C(R6)- 또는 -C(R6)-O-이다. 일부 실시양태에서, Y는 -NH-S(O)2-, -S(O)2-, -C(O)- 또는 -C(O)-O-이다. 일부 실시양태에서, Y는 -S(O)2- 또는 -C(O)O-이다.
일부 실시양태에서, Y'는 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R9)-,

로 이루어진 군으로부터 선택된다. 일부 실시양태에서, Y'는 -N(R8)-C(R6)-, -N(R8)-S(O)2- 또는 -N(R8)-C(R6)-N(R8)-W-이다. 일부 실시양태에서, Y'는 -N(R8)- C(O)-이다. 일부 실시양태에서, Y'는 -N(R8)-S(O)2-이다. 일부 실시양태에서, Y'는 -N(R8)-C(R6)-N(R8)-이다.
일부 실시양태에서, Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌로 이루어진 군으로부터 선택되며; 상기 알킬렌, 알케닐렌 및 알키닐렌에는 -O-가 임의로 개재된다. 일부 실시양태에서, Z는 결합, 알킬렌, 또는 -O-가 개재된 알킬렌이다. 특정 실시양태에서, Z는 C1 -3 알킬렌이다. 특정 실시양태에서, Z는 결합이다.
일부 실시양태에서, a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다. 일부 실시양태에서, a 및 b는 각각 정수 2이다.
화학식 I 내지 III 및 XI의 일부 실시양태에서, R3-O- 또는 R3 -1-O-는 7번 또는 8번 위치에 있다. 일부 실시양태에서 R3-O- 또는 R3 -1-O-은 7번 위치에 있다. 일부 실시양태에서, R3-O- 또는 R3 -1-O-는 8번 위치에 있다.
화학식 VII 및 IX의 일부 실시양태에서, HO-는 7번 또는 8번 위치에 있다. 일부 실시양태에서, HO-는 7번 위치에 있다. 일부 실시양태에서, HO-는 8번 위치에 있다.
일부 실시양태에서, 본 발명의 화합물은 하나 이상의 사이토카인 (예를 들어, IFN-α및(또는) TNF-α)의 생합성을 유도한다. 본 발명의 화합물은, 예를 들어 화학식 I, II, III 및 VII의 화합물 뿐만 아니라 본원에 기재된 그의 임의의 실시양태를 포함한다.
일부 실시양태에서, 화학식 I, II, III의 화합물 또는 본원에 기재된 그의 실시양태는 하나 이상의 사이토카인 (예를 들어, TNF-α)의 생합성을 억제한다.
화합물의 제조
본 발명의 화합물은 반응식 I (식 중, R, R1, R2 및 n은 상기에 정의한 바와 같음)에 따라 제조할 수 있다. 반응식 I의 단계 (1)에서는, 화학식 XV의 벤질옥시아닐린을 2,2-디메틸-1,3-디옥산-4,6-디온 (멜드럼(Meldrum)산) 및 트리에틸 오르토포르메이트로부터 생성된 축합 생성물로 처리하여 화학식 XVI의 이민을 얻는다. 반응은 화학식 XV의 벤질옥시아닐린 용액을 멜드럼산 및 트리에틸 오르토포르메이트의 가열된 혼합물에 첨가하고, 반응물을 승온, 예를 들어 45 ℃에서 가열함으로써 편리하게 수행된다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (2)에서는, 화학식 XVI의 이민을 열분해 및 고리화하여 화학식 XVII의 벤질옥시퀴놀린-4-올을 얻는다. 반응은 200 내지 250 ℃의 온도에서 DOWTHERM A 열 전달 유체와 같은 매질 중에서 편리하게 수행된다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (3)에서는, 화학식 XVII의 벤질옥시퀴놀린-4-올을 통상의 니트로화 조건 하에 니트로화하여 화학식 XVIII의 벤질옥시-3-니트로퀴놀린-4-올을 얻는다. 반응은 질산을 프로피온산과 같은 적합한 용매 중의 화학식 XVII의 벤질옥시퀴놀린-4-올에 첨가하고, 혼합물을 승온, 예를 들어 125 ℃에서 가열함으로써 편리하게 수행된다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (4)에서는, 화학식 XVIII의 벤질옥시-3-니트로퀴놀린-4-올을 통상의 염소화 화학을 이용하여 염소화하여 화학식 XIX의 벤질옥시-4-클로로-3-니트로퀴놀린을 얻는다. 반응은 화학식 XVIII의 벤질옥시-3-니트로퀴놀린-4-올을 N,N-디메틸포름아미드 (DMF)와 같은 적합한 용매 중에서 옥시염화인으로 처리함으로써 편리하게 수행된다. 반응은 승온, 예를 들어 100 ℃에서 수행할 수 있고, 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (5)에서는, 화학식 XIX의 벤질옥시-4-클로로-3-니트로퀴놀린을 화학식 R1-NH2의 아민으로 처리하여 화학식 XX의 벤질옥시-3-니트로퀴놀린-4-아민을 얻는다. 여러가지 화학식 R1-NH2의 아민이 상업적으로 입수가능하며, 다른 것들은 공지된 합성 방법으로 제조할 수 있다. 반응은 화학식 R1-NH2의 아민을 트리에틸아민과 같은 3급 아민의 존재 하에 디클로로메탄 또는 메탄올과 같은 적합한 용매 중의 화학식 XIX의 벤질옥시-4-클로로-3-니트로퀴놀린 용액에 첨가함으로써 편리하게 수행된다. 반응은 주변 온도, 또는 예를 들어 용매의 환류 온도와 같은 승온에서 수행할 수 있다. 반응 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (6)에서는, 화학식 XX의 벤질옥시-3-니트로퀴놀린-4-아민을 환원시켜 화학식 XXI의 벤질옥시퀴놀린-3,4-디아민을 얻는다. 반응은 탄소 상 백금과 같은 불균일 수소화 촉매를 사용하여 수소화함으로써 수행할 수 있다. 수소화는 톨루엔, 메탄올 또는 아세토니트릴과 같은 적합한 용매 중에서 파르(Parr) 장 치에서 편리하게 수행된다. 반응은 주변 온도, 또는 승온, 예를 들어 55 ℃에서 수행할 수 있고, 반응 생성물은 통상의 방법을 이용하여 단리할 수 있다.
별법으로, 단계 (6)에서의 환원은, 붕수소화나트륨 및 염화니켈(II)로부터 동일계 제조된 붕소화니켈을 사용하여 수행할 수 있다. 환원은 적합한 용매 또는 디클로로메탄/메탄올과 같은 용매 혼합물 중의 화학식 XX의 벤질옥시-3-니트로퀴놀린-4-아민 용액을 메탄올 중의 과량의 붕수소화나트륨 및 촉매 염화니켈(II)의 혼합물에 첨가함으로써 편리하게 수행된다. 반응은 주변 온도에서 수행할 수 있다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (7)에서는, 화학식 XXI의 벤질옥시퀴놀린-3,4-디아민을 카르복실산 등가물로 처리하여 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린을 얻는다. 적합한 카르복실산 등가물로는 화학식 R2C(O-알킬)3의 오르토에스테르, 화학식 R2C(O-알킬)2(O-C(O)-알킬)의 1,1-디알콕시알킬 알카노에이트 및 화학식 R2C(O)Cl의 산 염화물이 포함된다. 카르복실산 등가물의 선택은 R2에서의 원하는 치환기에 따라 결정된다. 예를 들어, 트리에틸오르토포르메이트는 R2가 수소인 화합물을 제공하고, 트리메틸 오르토발레레이트는 R2가 부틸기인 화합물을 제공한다. 반응은 카르복실산 등가물을 톨루엔 또는 크실렌과 같은 적합한 용매 중에서 화학식 XXI의 벤질옥시퀴놀린-3,4-디아민에 첨가함으로써 편리하게 수행된다. 임의로는, 촉매 피리딘 히드로클로라이드를 첨가할 수 있다. 반응은 반응 동안 형성된 알콜 또는 물이 제거되기에 충분히 높은 온도에서 수행한다. 편리하게는, 딘-스탁(Dean-Stark) 트랩을 사용하여 휘발성 물질을 수거할 수 있다.
별법으로, 화학식 R2C(O)Cl의 산 염화물을 카르복실산 등가물로서 사용하는 경우에는 단계 (7)을 2 단계로 수행할 수 있다. 단계 (7)의 부분 (i)은 산 염화물을 디클로로메탄 또는 아세토니트릴과 같은 적합한 용매 중의 화학식 XXI의 벤질옥시퀴놀린-3,4-디아민 용액에 첨가하여 아민을 수득함으로써 편리하게 수행된다. 임의로는, 트리에틸아민, 피리딘 또는 4-디메틸아미노피리딘과 같은 3급 아민을 첨가할 수 있다. 반응은 주변 온도 또는 승온에서 수행할 수 있다. 아미드 생성물은 통상의 기술을 이용하여 단리하고 임의로는 정제할 수 있다. 단계 (7)의 부분 (ii)는 부분 (i)에서 제조된 아미드를 가열하여 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린을 얻는 것을 포함한다. 반응은 반응 동안 형성된 물을 제거하기에 충분한 온도에서 톨루엔과 같은 적합한 용매 중에서 편리하게 수행된다. 단계 (7)의 부분 (ii)에서, 이미다조 고리 형성 반응은 트리에틸렌아민 또는 수성 수산화나트륨과 같은 염기의 존재 하에 에탄올 또는 메탄올과 같은 용매 중에서 수행할 수도 있다. 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린은 통상의 방법을 이용하여 단리할 수 있다.
한 실시양태에서, 본 발명은, (1) 5번, 6번, 7번 또는 8번 위치가 하나의 벤질옥시기 및 임의로는 하나의 R기로 치환된 3-아미도-4-아미노퀴놀린 (여기서, 아미도는 -NH-C(O)-R2이고, 아미노는 -NH-R1이고, R, R1 및 R2는 상기에 정의한 바와 같음)을 얻는 단계; (2) 알콜 및 단계 (1)에서 얻어진 화합물을 포함하는 혼합물을 제조하는 단계; 및 (3) 단계 (2)의 혼합물을 염기 (여기서, 염기는 수성 수산화나트륨 또는 트리에틸렌아민임)와 접촉시켜 1번 위치가 R1으로, 2번 위치가 R2로, 5번, 6번, 7번 또는 8번 위치가 하나의 벤질옥시기 및 임의로는 하나의 R기로 치환된 1H-이미다조[4,5-c]퀴놀린을 얻는 단계를 포함하는 방법을 제공한다. 특정 실시양태에서, 알콜은 메탄올, 에탄올 또는 이들의 혼합물이다. 특정 실시양태에서, 염기는 수성 수산화나트륨이다.
반응식 I의 단계 (8)에서는, 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린을 N-옥시드를 형성할 수 있는 통상의 산화제를 사용하여 산화시켜 화학식 XXIII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 얻는다. 반응은 3-클로로퍼옥시벤조산을 디클로로메탄 또는 클로로포름과 같은 용매 중의 화학식 XXII의 화합물 용액에 첨가함으로써 편리하게 수행된다. 반응은 주변 온도에서 수행할 수 있고, 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 I의 단계 (9)에서는, 화학식 XXIII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 아민화하여 화학식 I, II 및 III의 아속인 화학식 XXIV의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다. 단계 (9)는 화학식 XXIII의 N-옥시드를 에스테르로 전환시켜 활성화한 후, 에스테르를 아민화제와 반응시킴으로써 수행할 수 있다. 적합한 활성화제로는, 알킬- 또는 아릴술포닐 클로라이드, 예를 들어 벤젠술포닐 클로라이드, 메탄술포닐 클로라이드 또는 p-톨루엔술포닐 클로라 이드가 포함된다. 적합한 아민화제로는, 예를 들어 수산화암모늄 형태의 암모니아 및 암모늄염, 예컨대 탄산암모늄, 중탄산암모늄 및 인산암모늄이 포함된다. 반응은 수산화암모늄을 디클로로메탄 또는 클로로포름과 같은 적합한 용매 중의 화학식 XXIII의 N-옥시드 용액에 첨가한 후, p-톨루엔술포닐 클로라이드를 첨가함으로써 편리하게 수행된다. 반응은 주변 온도에서 수행할 수 있다. 반응은 화학식 XXIII의 N-옥시드를 단리하지 않고 수산화암모늄 및 p-톨루엔술포닐 클로라이드를 단계 (8)로부터의 반응 혼합물에 첨가함으로써 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
별법으로, 단계 (9)는 화학식 XXIII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 트리클로로아세틸 이소시아네이트와 반응시킨 후, 생성된 중간체를 가수분해하여, 화학식 XXIV의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻음으로써 수행할 수 있다. 반응은 (i) 트리클로로아세틸 이소시아네이트를 디클로로메탄과 같은 용매 중의 화학식 XXIII의 N-옥시드 용액에 첨가하고, 주변 온도에서 교반하여, 단리가능한 아미드 중간체를 얻는 단계를 포함한 2 단계로 편리하게 수행될 수 있다. 단계 (ii)에서는, 메탄올 중의 중간체 용액을 주변 온도에서 나트륨 메톡시드 또는 수산화암모늄과 같은 염기로 처리한다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.

일부 실시양태에서, 반응식 I에 나타낸 화합물은 통상의 합성 방법을 이용하여 추가로 조작할 수 있다. 예를 들어, 화학식 R1-NH2 (식 중, R1은 R4임)의 아민을 히드록시 또는 2급 아미노기로 치환할 수 있고, 이것을 반응식 I의 단계 (6) 이전에 추가로 관능화할 수 있다. 예를 들어, 화학식 XX (식 중, R1은 히드록시 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민을 통상의 염소화제를 사용하여 염소화하고, 이어서 티오알콕시드염과 반응시켜 화학식 XX (식 중, R1은 -X-Y-R4이고, X 및 R4는 상기에 정의한 바와 같고, Y는 -S-임)의 벤질옥시-3-니트로퀴놀린-4-아민을 얻을 수 있다. 염소화 반응은 티오닐 클로라이드를 디클로로메탄과 같은 용매 중의 화학식 XX (식 중, R1은 히드록시 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민 용액에 첨가하고, 반응물을 승온에서 가열함으로써 편리하게 수행된다. 티오에테르기는 나트륨 티오메톡시드와 같은 티오알콕시드염을 DMF와 같은 용매 중의 화학식 XX (식 중, R1은 클로로 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민 용액에 첨가함으로써 편리하게 도입된다. 반응은 주변 온도 또는 승온에서 수행할 수 있다. 이와 같이 도입된 티오에테르기를 반응식 I의 단계 (8)에서 과량의 산화제를 사용하여 술폰기로 산화시켜 화학식 XXIII (식 중, R1은 -X-Y-R4이고, Y는 -S(O)2-임)의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 얻을 수 있다.
화학식 XX (식 중, R1은 아미노 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민은 통상의 방법을 이용하여 반응식 I의 단계 (6) 이전에 관능화할 수도 있다. 예를 들어, 화학식 XX (식 중, Rl은 아미노 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민을 화학식 R4-S(O)2Cl의 술포닐 클로라이드 또는 화학식 (R4-S(O)2)2O의 술폰산 무수물과 반응시켜 화학식 XX (식 중, R1은 -X-Y-R4이고, Y는 -N(R8)-S(O)2-이며, R8은 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 많은 술포닐 클로라이드 및 술폰산 무수물이 상업적으로 입수가능하며, 다른 것들은 공지된 합성 방법을 이용하여 용이하게 제조할 수 있다. 반응은 술포닐 무수물을 디클로로메탄과 같은 적합한 용매 중의 화학식 XX (식 중, R1은 아미노 치환기를 갖는 R4임)의 벤질옥시-3-니트로퀴놀린-4-아민 및 트리에틸아민과 같은 염기 용액에 첨가함으로써 편리하게 수행될 수 있다. 반응은 주변 온도에서 수행할 수 있다. 이어서, 생성물을 반응식 I의 단계 (6) 내지 (9)에 따라 처리할 수 있다.
일부 실시양태에서, Rl의 추가 조작은 반응식 II (식 중, R, R2, R4, R5, X, Q 및 n은 상기에 정의한 바와 같음)에 따라 도입된다. 반응식 II의 단계 (1)에서는, 화학식 XIX의 벤질옥시-4-클로로-3-니트로퀴놀린을 화학식 (CH3)3CO-C(O)-NH-X-NH2의 Boc-보호된 디아민으로 처리하여 화학식 XXV의 벤질옥시-3-니트로퀴놀린-4-아민을 얻는다. 여러가지 화학식 (CH3)3CO-C(O)-NH-X-NH2의 Boc-보호된 디아민이 상업적으로 입수가능하고, 다른 것들은 공지된 합성 방법으로 제조할 수 있다. 반응은 화학식 (CH3)3CO-C(O)-NH-X-NH2의 Boc-보호된 디아민을 트리에틸아민과 같은 3급 아민의 존재 하에 디클로로메탄 또는 물과 같은 적합한 용매 중의 화학식 XIX의 벤질옥시-4-클로로-3-니트로퀴놀린의 냉각된 용액에 첨가함으로써 편리하게 수행된다. 반응은 주변 온도, 또는 예를 들어 용매의 환류 온도와 같은 승온에서 수행할 수 있다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 II의 단계 (2) 내지 (5)에서는, 화학식 XXV의 벤질옥시-3-니트로퀴놀린-4-아민을 먼저 환원시켜 화학식 XXVI의 벤질옥시퀴놀린-3,4-디아민을 얻고, 이것을 카르복실산 등가물과 반응시킴으로써 화학식 XXVII의 벤질옥시-1H-이미다조 [4,5-c]퀴놀린으로 전환시킨다. 이어서, 화학식 XXVII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린을 산화시켜 화학식 XXVIII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 수득하고, 이것을 아민화하여 화학식 I, II 및 III의 아속인 화학식 XXIX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다. 반응식 II의 단계 (2), (3), (4) 및 (5)를 각각 반응식 I의 단계 (6), (7), (8) 및 (9)에 기재된 바와 같이 수행하여, 단계 (5) 후에 형성된 생성물로서 화학식 XXIX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다. 단계 (5)에서, 바람직한 아민화 조건은 화학식 XXVIII의 N-옥시드를 에스테르로 전환시켜 활성화한 후, 에스테르를 아민화제와 반응시키는 것이다. 단계 (5)는 수산화암모늄을 디클로로메탄 또는 디클로로에탄과 같은 적합한 용매 중의 화학식 XXVIII의 N-옥시드 용액에 첨가한 후, p-톨루엔술포닐 클로라이드를 첨가하고, 주변 온도에서 교반함으로써 편리하게 수행된다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.
반응식 II의 단계 (6)에서는, 화학식 XXIX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민의 Boc-보호기를 제거하여 화학식 I, II 및 III의 아속인 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다. 반응은 에탄올 중의 염산 용액을 화학식 XXIX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민에 첨가함으로써 편리하게 수행된다. 반응은 승온, 예를 들어 용매의 환류 온도에서 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법으로 단리할 수 있다.
반응식 II의 단계 (7)에서는, 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 통상의 방법을 이용하여 화학식 XXXI의 벤질옥시-1H-이미다조[4,5- c]퀴놀린-1-일 화합물로 전환시킨다. 예를 들어, 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 화학식 R4C(O)Cl의 산 염화물과 반응시켜 화학식 XXXI (식 중, Q는 -C(O)-임)의 화합물을 얻을 수 있다. 또한, 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 화학식 R4S(O)2Cl의 술포닐 클로라이드 또는 화학식 (R4S(O)2)2O의 술폰산 무수물과 반응시켜 화학식 XXXI (식 중, Q는 -S(O)2-임)의 화합물을 얻을 수 있다. 많은 화학식 R4C(O)Cl의 산 염화물, 화학식 R4S(O)2Cl의 술포닐 클로라이드 및 화학식 (R4S(O)2)2O의 술폰산 무수물이 상업적으로 입수가능하고, 다른 것들은 공지된 합성 방법을 이용하여 용이하게 제조할 수 있다. 반응은 화학식 R1C(O)Cl의 산 염화물, 화학식 R1S(O)2Cl의 술포닐 클로라이드 또는 화학식 (R4S(O)2)2O의 술폰산 무수물을 클로로포름, 디클로로메탄 또는 아세토니트릴과 같은 적합한 용매 중의 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민 및 트리에틸아민과 같은 염기의 냉각된 용액에 첨가함으로써 편리하게 수행될 수 있다. 반응은 주변 온도 또는 주변 온도 미만, 예를 들어 0 ℃에서 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
화학식 XXXI (식 중, Q는 -C(R6)-N(R8)-W-이고, R6은 =O이고, R8은 상기에 정의한 바와 같고, W는 결합임)의 우레아는, 화학식 XXX의 벤질옥시-1H-이미다조 [4,5-c]퀴놀린-4-아민을 화학식 R4N=C=O의 이소시아네이트 또는 화학식 R4N-(R8)-C(O)Cl의 카르바모일 클로라이드와 반응시킴으로써 제조할 수 있다. 많은 화학식 R4N=C=O의 이소시아네이트 및 화학식 R4N-(R8)-C(O)Cl의 카르바모일 클로라이드가 상업적으로 입수가능하고, 다른 것들은 공지된 합성 방법을 이용하여 용이하게 제조할 수 있다. 반응은 이소시아네이트 또는 카르바모일 클로라이드를 디클로로메탄 또는 클로로포름과 같은 적합한 용매 중의 화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민 및 트리에틸아민과 같은 염기의 냉각된 용액에 첨가함으로써 편리하게 수행될 수 있다. 반응은 주변 온도 또는 주변 온도 미만, 예를 들어 0 ℃에서 수행할 수 있다. 별법으로, 화학식 XXX의 화합물을 화학식 R4(CO)N=C=O의 이소시아네이트, 화학식 R4N=C=S의 티오이소시아네이트 또는 화학식 R4S(O)2N=C=O의 술포닐 이소시아네이트로 처리하여 화학식 XXXI (식 중, Q는 -C(R6)-N(R8)-W-이고, R6, R8 및 W는 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
화학식 XXX의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을, 반응식 II의 단계 (7a)에 나타낸 바와 같이, 화학식 XXXIa (식 중, R5는  이고, V는 -N(R8)-C(R6)-이고, a, b, R6, R7, R8 및 A는 상기에 정의한 바와 같음)의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일 화합물 로 전환시킬 수도 있다.
이고, V는 -N(R8)-C(R6)-이고, a, b, R6, R7, R8 및 A는 상기에 정의한 바와 같음)의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일 화합물 로 전환시킬 수도 있다.
화학식 XXX의 화합물을 단계 (7)에 기재된 조건 하에 화학식  의 카르바모일 클로라이드로 처리하여 화학식 XXXIa (식 중, R5는
의 카르바모일 클로라이드로 처리하여 화학식 XXXIa (식 중, R5는  이고, V는 -NH-C(O)-이고, A는 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
이고, V는 -NH-C(O)-이고, A는 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
단계 (7a)에서는, 화학식 XXX의 화합물을 화학식 Cl-R7S(O)2Cl의 클로로알칸술포닐 클로라이드 또는 화학식 Cl-R7C(O)Cl (식 중, R7은 상기에 정의한 바와 같음)의 클로로알카노일 클로라이드와 반응시킬 수도 있다. 반응은 클로로알칸술포닐 클로라이드 또는 클로로알카노일 클로라이드를 주변 온도에서 트리에틸렌아민과 같은 염기의 존재 하에 클로로포름 또는 디클로로메탄과 같은 적합한 용매 중의 아민 용액에 첨가함으로써 편리하게 수행된다. 이어서, 단리가능한 중간체 클로로알칸술폰아미드 또는 클로로알칸아미드를 주변 온도에서 DMF와 같은 적합한 용매 중에서 1,8-디아자비시클로[5.4.0]운데스-7-엔과 같은 염기로 처리하여 고리화를 수행하여 화학식 XXXIa (식 중, R5는  임)의 화합물을 수득할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
임)의 화합물을 수득할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
일부 예에서는 반응식 II의 단계들을 상이한 순서로 수행하는 것이 바람직할 수 있다. 예를 들어, 화학식 XXVII의 화합물을 단계 (6)에 따라 탈보호시킬 수 있고, 생성된 아민을 단계 (7) 또는 (7a)에서와 같이 관능화한 후, 산화 및 아민화 단계 (4) 및 (5)를 각각 수행할 수 있다.

예를 들어, 반응식 I의 단계 (7)에서 사용된 산 염화물이 보호된 히드록시 또는 아미노기 또는 할로겐을 함유하는 경우에는, 반응식 II의 단계 (7) 및 (7a)에 기재된 것과 유사한 합성 변환을 화학식 XXIV의 벤질옥시-1H-이미다조[4,5-c]퀴놀 린의 R2에서 수행할 수도 있다. 이러한 유형의 여러가지 산 염화물, 예를 들어 아세톡시아세틸 클로라이드 및 클로로아세틸 클로라이드가 상업적으로 입수가능하고, 다른 것들, 예를 들어 5-tert-부톡시카르보닐아미노)발레릴 클로라이드는 공지된 합성 방법을 이용하여 제조할 수 있다. 이어서, 이러한 방식으로 도입된 R2 관능기를 아미노기가 나타나도록 조작하여, 이것을 반응식 II의 단계 (7) 및 (7a)에 기재된 방법에 따라 다양한 관능기로 전환시킬 수 있다. 예를 들어, 클로로아세틸 클로라이드를 반응식 I의 단계 (7)에 사용하여 클로로메틸 치환된 벤질옥시-1H-이미다조[4,5-c]퀴놀린을 도입할 수 있고, 이것을 반응식 I의 단계 (8) 및 (9)에 따라 산화 및 아민화할 수 있다. 생성된 클로로메틸 치환된 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 메탄올과 같은 적합한 용매 중의 암모니아로 처리하여 아미노메틸 치환된 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻을 수 있고, 이어서 이것을 반응식 II의 단계 (7) 또는 (7a)에 기재된 방법에 따라 처리하여 다양한 화합물을 얻을 수 있다.
본 발명의 화합물은 반응식 III (식 중, R, R1, R2 및 n은 상기에 정의한 바와 같고, R3은 -Z-Ar, -Z-Ar'-Y-R4 또는 -Z-Ar'-X-Y-R4이고, Z, Ar, Ar', X, Y 및 R4는 상기에 정의한 바와 같음)에 따라 제조할 수 있다. 반응식 III의 단계 (1)에서는, 화학식 XXIV의 벤질옥시-1H-이미다조[4,5-c]퀴놀린-4-아민의 벤질기를 절단하여 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀을 얻는다. 절단은 에탄올과 같은 용매 중에서 탄소 상 팔라듐과 같은 적합한 불균일 촉매를 이용하여 수소첨가분해 조건 하에 파르 장치 상에서 편리하게 수행된다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
반응식 III의 단계 (2)에서는, 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀을 윌리암슨형(Williamson-type) 에테르 합성을 이용하여 화학식 II의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린-4-아민으로 전환시킨다. 반응은 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀을 염기의 존재 하에 화학식 할라이드-Z-Ar, 할라이드-Z-Ar'-Y-R4 또는 할라이드-Z-Ar'-X-Y-R4의 알킬 또는 아릴 할라이드로 처리함으로써 수행한다. 치환된 벤질 브로마이드 및 클로라이드, 치환되거나 치환되지 않은 아릴알킬레닐 브로마이드 및 클로라이드 및 치환된 플루오로벤젠을 비롯한, 많은 상기 화학식의 알킬 또는 아릴 할라이드가 상업적으로 입수가능하다. 다른 상기 화학식의 알킬 또는 아릴 할라이드는 통상의 합성 방법을 이용하여 제조할 수 있다. 반응은 화학식 할라이드-Z-Ar, 할라이드-Z-Ar'-Y-R4 또는 할라이드-Z-Ar'-X-Y-R4의 시약을 탄산세슘 또는 탄산칼륨과 같은 적합한 염기의 존재 하에 DMF와 같은 용매 중의 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀과 조합함으로써 편리하게 수행된다. 임의로는, 촉매 테트라부틸암모늄 브로마이드를 첨가할 수 있다. 반응은 화학식 할라이드-Z-Ar, 할라이드-Z-Ar'-Y-R4 또는 할라이드 -Z-Ar'-X-Y-R4의 시약의 반응성에 따라 주변 온도, 또는 승온, 예를 들어 65 ℃ 또는 85 ℃에서 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.
별법으로, 단계 (2)는 울만(Ullmann) 에테르 합성을 이용하여 수행할 수 있고, 여기서는 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀의 알칼리 금속 아릴옥시드를 구리염의 존재 하에 아릴 할라이드와 반응시켜 화학식 II (식 중, R3은 -Z-Ar 또는 -Z-Ar'-Y-R4이고, Z는 결합임)의 화합물을 얻는다.
별법으로, 반응식 III의 단계 (2)는 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀을 미쯔노부(Mitsunobu) 반응 조건 하에 화학식 HO-Z-Ar의 알콜로 처리함으로써 수행할 수 있다. 상기 화학식의 일부 알콜, 예를 들어 3-피리딜카르비놀 및 3-푸란메탄올이 상업적으로 입수가능하고, 다른 것들은 통상의 합성 방법을 이용하여 제조할 수 있다. 반응은 트리페닐포스핀 및 화학식 HO-Z-Ar의 알콜을 테트라히드로푸란과 같은 적합한 용매 중의 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀 용액에 첨가한 후, 디이소프로필 아조디카르복실레이트를 서서히 첨가함으로써 편리하게 수행된다. 반응은 주변 온도 또는 주변 온도 미만, 예를 들어 0 ℃에서 수행할 수 있다. 생성물은 통상의 방법을 이용하여 단리할 수 있다.

본 발명의 화합물은 반응식 IV (식 중, R, R1, R2 및 n은 상기에 정의한 바 와 같고, R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4이고, Z, Ar, Ar', X, Y 및 R4는 상기에 정의한 바와 같음)에 따라 제조할 수 있다. 반응식 IV의 단계 (1)에서는, 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린의 벤질기를 절단하여 화학식 XXXIII의 1H-이미다조[4,5-c]퀴놀리놀을 얻는다. 반응은 반응식 III의 단계 (1)에 기재된 바와 같이 수행할 수 있거나, 또는 적합한 수소화 촉매의 존재 하에 수소 전달 반응에 의해 수행할 수 있다. 수소 전달 반응은 포름산암모늄을 탄소 상 팔라듐과 같은 촉매의 존재 하에 에탄올과 같은 적합한 용매 중의 화학식 XXII의 벤질옥시-1H-이미다조[4,5-c]퀴놀린 용액에 첨가함으로써 편리하게 수행된다. 반응은 승온, 예를 들어 용매의 환류 온도에서 수행할 수 있다.
반응식 IV의 단계 (2)에서는, 화학식 XXXIII의 1H-이미다조[4,5-c]퀴놀리놀을 화학식 할라이드-Z-Ar, 할라이드-Z-Ar'-Y-R4 또는 할라이드-Z-Ar'-X-Y-R4의 알킬 또는 아릴 할라이드로 처리하여 화학식 XI의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린을 수득한다. 반응은 반응식 III의 단계 (2)에 기재된 바와 같이 수행할 수 있다.
반응식 IV의 단계 (3) 및 (4)에서는, 화학식 XI의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린을 산화시켜 화학식 X의 1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 수득하고, 이것을 아민화하여 화학식 II의 1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다. 단계 (3) 및 (4)는 각각 반응식 I의 단계 (8) 및 (9)에 기재된 바와 같이 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.

반응식 III 또는 IV에서 제조된 화학식 II의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린-4-아민, 또는 반응식 IV에서의 중간체인 화학식 XI의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린의 추가의 합성 조작이 가능하다. 예를 들어, 화학식 II (식 중, R3은 -Z-Ar임)의 화합물 상의 아릴 또는 헤테로아릴기의 니트로 치환기를 통상의 방법을 이용하여 아미노기로 환원시킬 수 있다. 반응은 반응식 I의 단계 (6)에 기재된 방법을 이용하여 수행할 수 있다. 생성된, 화학식 II 또는 XI (식 중, R3은 -Z-Ar임)의 화합물 상의 아릴 또는 헤테로아릴기의 아미노 치환기를 하기와 같이 추가로 조작할 수 있다.
화학식 II 또는 XI (식 중, R3은 -Z-Ar임)의 화합물 상의 아릴 또는 헤테로아릴기의 아미노 치환기를 알데히드와 반응시켜 이민을 얻고, 이것을 통상의 방법을 이용하여 환원시켜 화학식 II 또는 XI (식 중, R3은 -Z-Ar'-N(R8)-H이고, R8은 상기에 정의한 바와 같음)의 화합물 또는 제약상 허용되는 그의 염을 얻을 수 있다. 화학식 II 또는 XI (식 중, R3은 -Z-Ar'-N(R8)-H임)의 화합물을 반응식 II의 단계 (7) 또는 (7a)에 기재된 방법에 따라 처리하여 화학식 II 또는 XI (식 중, R3은 -Z-Ar'-N(R8)-Q-R4 또는 -Z-Ar'-R5이고, Q, R4 및 R8은 상기에 정의한 바와 같고, R5는  이고, V는 -N(R8)-C(R6)-이고, a, b, R6, R7, R8 및 A는 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 화학식 XI (식 중, R3은 -Z-Ar'-N(R8)-Q-R4 또는 -Z-Ar'-R5임)의 화합물을 반응식 IV의 단계 (3) 및 (4)에 기재된 화학을 이용하여 화학식 II의 화합물 또는 제약상 허용되는 그의 염으로 전환시킬 수 있다.
이고, V는 -N(R8)-C(R6)-이고, a, b, R6, R7, R8 및 A는 상기에 정의한 바와 같음)의 화합물을 얻을 수 있다. 화학식 XI (식 중, R3은 -Z-Ar'-N(R8)-Q-R4 또는 -Z-Ar'-R5임)의 화합물을 반응식 IV의 단계 (3) 및 (4)에 기재된 화학을 이용하여 화학식 II의 화합물 또는 제약상 허용되는 그의 염으로 전환시킬 수 있다.
별법으로, 화학식 II (식 중, R, R1, R2 및 n은 상기에 정의한 바와 같고, R3은 -Z-Ar 또는 -Z-Ar'-Y-R4이고, Z는 결합이고, Ar, Ar', Y 및 R4는 상기에 정의한 바와 같음)의 화합물은 반응식 V 및 반응식 VI (식 중, Hal은 할로겐임)에 나타낸 바와 같이 제조할 수 있다. 반응식 V 및 반응식 VI의 단계 (1)은 울만 에테르 합성을 이용하여 수행할 수 있고, 여기서는 화학식 ArOH 또는 HOAr-Y-R4의 아릴 알콜의 알칼리 금속 아릴옥시드를 구리염의 존재 하에 화학식 XXXIV의 할로겐 치환된 1H-이미다조[4,5-c]퀴놀린-4-아민 또는 화학식 XXXV의 할로겐 치환된 1H-이미다조 [4,5-c]퀴놀린과 반응시킨다. 많은 화학식 XXXIV 및 XXXV의 화합물이 공지되어 있다. 예를 들어, 미국 특허 제4,689,338호, 동 제4,929,624호, 동 제5,268,376호, 동 제5,346,905호, 동 제5,389,640호, 동 제5,756,746호, 동 제6,331,539호, 동 제6,451,810호, 동 제6,541,485호, 동 제6,545,016호, 동 제6,660,747호, 동 제6,683,088호, 동 제6,656,938호, 동 제6,664,264호 및 동 제6,664,260호, 유럽 특허 출원 제1 104 764호, 및 일본 특허 출원 제9-255926호를 참조한다. 다른 것들은 공지된 합성 방법을 이용하여 용이하게 제조할 수 있다. 예를 들어, 미국 특허 제4,988,815호, 동 제5,175,296호, 동 제5,367,076호, 동 제5,395,937호 및 동 제5,741,908호를 참조한다. 여러가지 화학식 ArOH 또는 HOAr-Y-R4의 아릴 알콜이 공지되어 있고, 다른 것들은 공지된 합성 방법을 이용하여 제조할 수 있다. 반응식 I에서, 울만 조건 하의 화학식 XXXIV의 할로겐 치환된 1H-이미다조[4,5-c]퀴놀린-4-아민의 반응에 의해 화학식 II의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린-4-아민을 얻는다.

반응식 VI에서는, 화학식 XXXV의 할로겐 치환된 1H-이미다조[4,5-c]퀴놀린의 반응에 의해 화학식 XI의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린을 얻는다. 반 응식 VI의 단계 (2) 및 (3)에서는, 화학식 XI의 에테르 치환된 1H-이미다조[4,5-c]퀴놀린을 산화시켜 화학식 X의 1H-이미다조[4,5-c]퀴놀린-5N-옥시드를 수득하고, 이것을 아민화하여 화학식 II의 1H-이미다조[4,5-c]퀴놀린-4-아민을 수득한다. 반응식 VI의 단계 (2) 및 (3)은 반응식 IV의 단계 (3) 및 (4)에 기재된 바와 같이 수행할 수 있다.

본 발명의 화합물은 반응식 VII (식 중, Ar, R, R1, R2 및 n은 상기에 정의한 바와 같음)에 따라 제조할 수 있다.
반응식 VII의 단계 (1)에서는, 화학식 XXXII의 1H-이미다조[4,5-c]퀴놀리놀을 화학식 Br-(CH2)m-C≡CH의 브롬화물로 알킬화하여 화학식 XXXVI의 1H-이미다조[4,5-c]퀴놀리닐 에테르를 얻는다. 화학식 XXXII의 화합물 및 브롬화물을 탄산세슘의 존재 하에 DMF와 같은 적합한 용매 중에서 합한다. 반응은 실온에서 수행할 수 있다.
반응식 VII의 단계 (2)에서, 화학식 XXXVI의 1H-이미다조[4,5-c]퀴놀린을 소노가시라(Sonogashira) 반응 조건을 이용하여 화학식 할라이드-Ar의 할라이드와 커플링하여 화학식 II의 아속인 화학식 IIa의 1H-이미다조[4,5-c]퀴놀린을 얻는다. 화학식 XXXVI의 화합물을 DMF와 같은 적합한 용매 중에서 요오드화구리(I), 디클로로비스(트리페닐포스핀)팔라듐(II) 및 과량의 트리에틸아민의 존재 하에 할라이드와 배합한다. 반응은 바람직하게는 승온 (60 내지 80 ℃)에서 수행한다.
반응식 VII의 단계 (3)에서, 화학식 IIa의 1H-이미다조[4,5-c]퀴놀린의 알킨 결합을 환원시켜 화학식 II의 아속인 화학식 IIb의 1H-이미다조[4,5-c]퀴놀린을 얻는다. 환원은 탄소 상 팔라듐과 같은 통상의 불균일 수소화 촉매를 사용하여 수소화함으로써 수행할 수 있다. 수소화는 에탄올과 같은 적합한 용매 중에서 파르 장치에서 편리하게 수행된다. 반응은 주변 온도에서 수행할 수 있다. 생성물 또는 제약상 허용되는 그의 염은 통상의 방법을 이용하여 단리할 수 있다.

제약 조성물 및 생물학적 활성
본 발명의 제약 조성물은 치료 유효량의 상기한 바와 같은 본 발명의 화합물을 제약상 허용되는 담체와 함께 함유한다.
용어 "치료 유효량" 또는 "유효량"은 사이토카인 유도, 사이토카인 억제, 면역조절, 항종양 활성 및(또는) 항바이러스 활성과 같은 치료 또는 예방 효과를 유도하기에 충분한 화합물의 양을 의미한다. 본 발명의 제약 조성물에 사용되는 활성 화합물의 정확한 양은 화합물의 물리적 및 화학적 성질, 담체의 성질 및 의도된 투여 요법과 같은 당업자에게 공지된 요인에 따라 달라지지만, 본 발명의 조성물은 약 100 나노그램/킬로그램 (ng/kg) 내지 약 50 밀리그램/킬로그램 (mg/kg), 바람직하게는 약 10 마이크로그램/킬로그램 (㎍/kg) 내지 약 5 mg/kg의 투여량의 화합물을 대상체에게 제공하기에 충분한 활성 성분을 함유하는 것으로 예상된다. 정제, 로젠지, 캡슐, 비경구 제제, 시럽, 크림, 연고, 에어로졸 제제, 경피 패치, 경점막 패치 등과 같은 다양한 제형을 사용할 수 있다.
본 발명의 화합물은 치료 요법에 단일 치료 제제로서 투여할 수 있거나, 또는 본 발명의 화합물을 서로 조합하거나 추가의 면역 반응 조절인자, 항바이러스제, 항생제, 항체, 단백질, 펩티드, 올리고뉴클레오티드 등을 비롯한 다른 활성 제제와 조합하여 투여할 수 있다.
본 발명의 화합물은 하기하는 시험 세트에 따라 수행된 실험에서 특정 사이토카인의 생성을 유도하는 것으로 나타났고, 특정 본 발명의 화합물은 특정 사이토카인의 생성을 억제할 수 있다. 이들 결과는 본 발명의 화합물이 많은 상이한 방식으로 면역 반응을 조절할 수 있는 면역 반응 조절인자로서 유용하여 이들 화합물이 다양한 장애의 치료에 유용하게 됨을 보여준다.
본 발명에 따른 화합물의 투여에 의해 생성이 유도될 수 있는 사이토카인은, 일반적으로 인터페론-α(IFN-α) 및(또는) 종양 괴사 인자-α (TNF-α) 뿐만 아니라 특정 인터루킨 (IL)을 포함한다. 본 발명의 화합물에 의해 생합성이 유도될 수 있는 사이토카인은, IFN-α, TNF-α, IL-1, IL-6, IL-10 및 IL-12 및 다양한 다른 사이토카인을 포함한다. 여러 효과 중에서 특히, 이들 및 다른 사이토카인은 바이러스 생성 및 종양 세포 성장을 억제할 수 있어, 본 발명의 화합물이 바이러스성 질환 및 신생물성 질환의 치료에 유용하게 한다. 따라서, 본 발명은 유효량의 본 발명의 화합물 또는 조성물을 동물에게 투여하는 것을 포함하는, 상기 동물에서 사이토카인 생합성을 유도하는 방법을 제공한다. 사이토카인 생합성의 유도를 위해 본 발명의 화합물 또는 조성물이 투여되는 동물은 하기 질환, 예를 들어 바이러스성 질환 또는 신생물성 질환을 가질 수 있고, 상기 화합물의 투여는 치료적 처치를 제공할 수 있다. 별법으로, 본 발명의 화합물은 동물이 질환에 걸리기 전에 동물에게 투여할 수 있어, 상기 화합물의 투여가 예방적 처치를 제공할 수 있다.
사이토카인의 생성을 유도하는 능력 이외에, 본 발명의 화합물은 선천적 면역 반응의 다른 면에 영향을 줄 수 있다. 예를 들어, 사이토카인 유도에 기인할 수 있는 효과로 자연 살세포 활성을 자극할 수 있다. 또한, 본 발명의 화합물은 대식세포를 활성화시킴으로써 산화질소의 분비 및 추가의 사이토카인의 생성을 자극할 수 있다. 또한, 본 발명의 화합물은 B-림프구의 증식 및 분화를 일으킬 수 있다.
본 발명의 화합물은 또한 후천적 면역 반응에 영향을 줄 수 있다. 예를 들어, 본 발명의 화합물을 투여함에 따라 조력 T 세포 타입 1 (TH1) 사이토카인 IFN-γ의 생성이 간접적으로 유도될 수 있고, 조력 T 세포 타입 2 (TH2) 사이토카인 IL-4, IL-5 및 IL-13의 생성이 억제될 수 있다.
본 발명에 따른 특정 화합물의 투여에 의해 생성이 억제될 수 있는 다른 사이토카인은 종양 괴사 인자-α(TNF-α)를 포함한다. 여러 효과 중에서 특히, TNF-α 생성의 억제가 TNF가 매개되는 동물의 질환의 예방적 또는 치료적 처치를 제공할 수 있어, 본 발명의 화합물이 예를 들어 자가면역 질환의 치료에 유용하게 된다. 따라서, 본 발명은 유효량의 본 발명의 화합물 또는 조성물을 동물에게 투여 하는 것을 포함하는, 상기 동물에서 TNF-α 생합성을 억제하는 방법을 제공한다. TNF-α 생합성의 억제를 위해 본 발명의 화합물 또는 조성물이 투여되는 동물은 하기 질환, 예를 들어 자가면역 질환을 가질 수 있고, 상기 화합물의 투여는 치료적 처치를 제공할 수 있다. 별법으로, 본 발명의 화합물은 동물이 질환에 걸리기 전에 동물에게 투여할 수 있어, 상기 화합물의 투여가 예방적 처치를 제공할 수 있다.
질환의 예방적 처치이든 치료적 처치이든, 또한 선천적 면역에 효과를 주든 또는 후천적 면역에 효과를 주든, 본 발명의 화합물 또는 조성물은 단독으로, 또는 백신 보조제에서와 같이 하나 이상의 활성 성분과 조합하여 투여할 수 있다. 다른 성분과 함께 투여하는 경우, 본 발명의 화합물과 다른 성분(들)은 별도로 투여하거나, 예를 들어 용액 중에서 함께 독립적으로 투여하거나, 또는 예를 들어 콜로이드 현탁액 중에서 (a) 공유 결합 또는 (b) 비-공유 결합 등에 의해 서로 결합되어 함께 투여할 수 있다.
치료에 사용될 수 있는 본원에서 확인된 IRM에 대한 증상은,
(a) 예를 들어, 아데노바이러스, 헤르페스바이러스 (예를 들어, HSV-I, HSV-II, CMV 또는 VZV), 폭스바이러스 (예를 들어, 오르토폭스바이러스, 예컨대 두창 또는 우두, 또는 전염성 연속종), 피코르나바이러스 (예를 들어, 리노바이러스 또는 엔테로바이러스), 오르토믹소바이러스 (예를 들어, 인플루엔자바이러스), 파라믹소바이러스 (예를 들어, 파라인플루엔자바이러스, 멈프스 바이러스, 홍역 바이러스 및 호흡기 세포융합 바이러스 (RSV), 코로나바이러스 (예를 들어, SARS), 파포 바바이러스 (예를 들어, 파필로마바이러스, 예컨대 생식기 사마귀, 심상성 사마귀 또는 발바닥 사마귀를 일으키는 바이러스), 헤파드나바이러스 (예를 들어, 헤파티티스 B 바이러스), 플라비바이러스 (예를 들어, 헤파티티스 C 바이러스 또는 뎅기 바이러스), 또는 레트로바이러스 (예를 들어, 렌티바이러스, 예컨대 HIV)에 의한 감염에 기인하는 질환 등의 바이러스성 질환;
(b) 예를 들어, 속명 에스케리치아(Escherichia), 엔테로박터(Enterobacter), 살모넬라(Salmonella), 스타필로코쿠스(Staphylococcus), 시겔라(Shigella), 리스테리아(Listeria), 에어로박터(Aerobacter), 헬리코박터(Helicobacter), 클레브시엘라(Klebsiella), 프로테우스(Proteus), 슈도모나스(Pseudomonas), 스트렙토코쿠스(Streptococcus), 클라미디아(Chlamydia), 미코플라즈마(Mycoplasma), 뉴모코쿠스(Pneumococcus), 나이세리아(Neisseria), 클로스트리듐(Clostridium), 바실루스(Bacillus), 코리네박테리움(Corynebacterium), 미코박테륨(Mycobacterium), 캄필로박터(Campylobacter), 비브리오(Vibrio), 세라티아(Serratia), 프로비덴시아(Providencia), 크로모박테륨(Chromobacterium), 브루셀라(Brucella), 예르시니아(Yersinia), 헤모필루스(Haemophilus) 또는 보르데텔라(Bordetella)의 박테리아에 의한 감염에 기인하는 질환 등의 세균성 질환;
(c) 기타 감염성 질환, 예를 들어 클라미디아, 진균 질환, 예컨대 칸디다증, 아스페르길루스증, 히스토플라스마증, 효모균성 뇌막염, 또는 기생충병, 예컨대 말라리아, 주폐포자층 폐렴, 리슈마니아증, 크립토스포리디움병, 톡소플라스마증 및 트리파노솜 감염;
(d) 신생물성 질환, 예를 들어 상피내종양, 자궁목형성이상, 광선각화증, 기저세포암, 편평세포암, 신세포 백혈병, 카포시육종, 흑색종, 신세포암, 백혈병, 예컨대 골수성 백혈병, 만성 림프성 백혈병 및 다발성 골수종, 비-호지킨 림프종, 피부 T-세포 림프종, B-세포 림프종, 모발상 세포 백혈병 및 기타 암; 및
(e) TH2-매개된, 아토피 및 자가면역 질환, 예를 들어 아토피 피부염 또는 습진, 호산구 증가증, 천식, 알레르기, 알레르기성 비염, 전신성 홍반성 루푸스, 본태성 혈소판혈증, 다발성 경화증, 옴멘 증후군(Ommen's syndrome), 원반상 루푸스, 원형 탈모증, 켈로이드 형성 및 기타 유형의 흉터형성, 및 만성 상처를 비롯한 상처 치유 향상의 억제를 포함하나, 이들로 제한되는 것은 아니다.
본원에서 확인된 IRM은, 예를 들어 BCG, 콜레라, 흑사병, 장티푸스, A형 간염, B형 간염 및 C형 간염, 인플루엔자 A 및 인플루엔자 B, 파라인플루엔자, 폴리오바이러스, 광견병, 홍역, 유행성 이하선염, 풍진, 황열병, 파상풍, 디프테리아, 헤모필루스 인플루엔자 b, 결핵, 수막구균 및 폐렴구균 백신, 아데노바이러스, HIV, 수두, 거대세포바이러스, 뎅기, 고양이 백혈병, 가금페스트, HSV-1 및 HSV-2, 돼지 콜레라, 일본 뇌염, 호흡기 세포융합 바이러스, 로타바이러스, 파필로마 바이러스, 황열병 및 알츠하이머병과 관련하여 사용하기 위해, 체액 및(또는) 세포 매개 면역 반응을 증진시키는 임의의 물질, 예를 들어 생존 바이러스, 세균 또는 기생충 면역원; 불활성 바이러스, 종양-유도 원충, 균-유도 진균 또는 세균 면역원, 유독소, 독소; 자기-항원; 다당류; 단백질; 당단백질; 펩티드; 세포 백신; DNA 백 신; 재조합 단백질 등과 함께 사용되는 백신 보조제로서도 유용할 수 있다.
IRM은 또한, 손상된 면역 기능을 갖는 개인에게 특히 유용하다. 예를 들어, IRM 화합물은, 예를 들어 이식 환자, 암 환자 및 HIV 환자에서 세포 매개 면역 억제 후에 발생하는 기회 감염 및 종양을 치료하는 데 유용할 수 있다.
따라서, 하나 이상의 상기 질환 또는 질환 유형, 예를 들어 바이러스성 질환 또는 신생물성 질환의 치료가 필요한 동물에게 치료 유효량의 본 발명의 화합물 또는 염 또는 이들의 조합을 투여함으로써, 상기 동물의 상기 질환 또는 질환 유형을 치료할 수 있다. 동물에게 백신 보조제로서 치료 유효량의 본 발명의 화합물 또는 염 또는 이들의 조합을 투여함으로써, 동물을 백신접종할 수 있다.
사이토카인 생합성을 유도하기에 효과적인 화합물의 양은, 예를 들어 단핵세포, 대식세포, 수상세포 및 B-세포 등의 하나 이상의 세포 유형이, 예를 들어 IFN-α, TNF-α,IL-1, IL-6, IL-10 및 IL-12 등의 하나 이상의 사이토카인의 기저 수준 에 비해 증가된 양의 상기 사이토카인의 생성을 일으키기에 충분한 양이다. 정확한 양은 당업계에 공지된 요인에 따라 변하지만, 약 100 ng/kg 내지 약 50 mg/kg, 바람직하게는 약 10 ㎍/kg 내지 약 5 mg/kg의 투여량으로 예상된다. 또한, 본 발명은 동물에게 유효량의 본 발명의 화합물 또는 조성물을 투여하는 것을 포함하는, 동물의 바이러스성 감염을 치료하는 방법 및 동물의 신생물성 질환을 치료하는 방법을 제공한다.
바이러스성 감염을 치료 또는 억제하기에 효과적인 양은, 미처리된 대조군 동물에 비해 바이러스 병소, 바이러스 부하, 바이러스 생성 속도 및 사망률 등의 하나 이상의 바이러스 감염 증상의 감소를 일으키는 양이다. 이러한 치료에 효과적인 정확한 양은 당업계에 공지된 요인에 따라 변하지만, 약 100 ng/kg 내지 약 50 mg/kg, 바람직하게는 약 10 ㎍/kg 내지 약 5 mg/kg의 투여량으로 예상된다. 신생물성 증상을 치료하기에 효과적인 화합물의 양은, 종양 크기 또는 종양 병터 수의 감소를 일으키는 양이다. 역시, 정확한 양은 당업계에 공지된 요인에 따라 변하지만, 약 100 ng/kg 내지 약 50 mg/kg, 바람직하게는 약 10 ㎍/kg 내지 약 5 mg/kg의 투여량으로 예상된다.
특정 실시양태에서는, 동물에게 유효량의 본원에 기재된 화합물 또는 염을 투여하는 것을 포함하는, 동물의 사이토카인 생합성을 유도하는 방법이 제공된다. 또다른 실시양태에서는, 동물에게 치료 유효량의 본원에 기재된 화합물 또는 염을 투여하는 것을 포함하는, 동물의 바이러스성 질환을 치료하는 방법이 제공된다. 또다른 실시양태에서는, 동물에게 치료 유효량의 본원에 기재된 화합물 또는 염을 투여하는 것을 포함하는, 동물의 신생물성 질환을 치료하는 방법이 제공된다.
실시예 1
7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
트리에틸 오르토포르메이트 (92 mL, 0.55 mol) 및 2,2-디메틸-[1,3]-디옥산-4,6-디온 (75.3 g, 0.522 mol) (멜드럼산 (Meldrum's acid))의 혼합물을 55 ℃에서 90분간 가열한 후 45 ℃로 냉각시켰다. 반응 온도를 50 ℃ 미만으로 유지하면서 메탄올 (200 mL) 중 3-벤질옥시아닐린 (100.2 g, 0.5029 mol)의 용액을 반응물에 45분에 걸쳐 서서히 첨가하였다. 이어서 반응물을 1시간 동안 45 ℃로 가열하고 실온으로 냉각시킨 후 하룻밤 동안 교반하였다. 반응 혼합물을 1 ℃로 냉각하고 생성물을 여과하여 단리한 후 여액이 무색이 될 때까지 차가운 에탄올 (~400 mL)로 세척하였다. 황갈색의 분말상 고체로서 5-{[(3-벤질옥시)페닐이미노]메틸}-2,2-디메틸-[1,3]-디옥산-4,6-디온 (170.65 g)을 단리하였다.

B부
5-{[(3-벤질옥시)페닐이미노]메틸}-2,2-디메틸-[1,3]-디옥산-4,6-디온 (170.65 g, 0.483 mol) 및 DOWTHERM A 열 전달 유체 (800 mL)의 혼합물을 100 ℃로 가열한 후 40분에 걸쳐 DOWTHERM A 열 전달 유체 (1.3 L, 210 ℃로 가열함)를 함유하는 플라스크에 서서히 첨가하였다. 첨가 도중, 반응 온도가 207 ℃ 미만이 되지 않도록 하였다. 첨가 후, 반응물을 210 ℃에서 1시간 동안 교반하고 상온으로 냉각시켰다. 형성된 침전물을 여과하여 단리하고, 디에틸에테르 (1.7 L) 및 아세톤 (0.5 L)으로 세척하고 오븐으로 건조시켜 황갈색 분말인 76.5 g의 7-벤질옥시퀴놀린-4-올을 수득하였다.

C부
7-벤질옥시퀴놀린-4-올 (71.47 g, 0.2844 mol) 및 프로피온산 (700 mL)의 혼합물을 격렬한 교반하에 125 ℃로 가열하였다. 반응 온도를 121 내지 125 ℃로 유지하면서 30분에 걸쳐 질산 (16 M, 23.11 mL)을 서서히 첨가하였다. 첨가 후, 반응물을 125 ℃에서 1시간 동안 교반한 후 상온으로 냉각시켰다. 생성된 고체를 여과하여 단리하고, 물로 세척한 후 1.5일간 오븐에서 건조시켜 회색빛이 도는 분말로서 69.13 g의 7-벤질옥시-3-니트로퀴놀린-4-올을 수득하였다.

D부
N,N-디메틸포름아미드 (100 mL) (DM상온에서)를 0 ℃로 냉각하고 포스포러스 옥시클로라이드 (27.5 mL, 0.295 mol)를 적가하였다. 생성된 용액을 25분간 교반한 후 DMF (400 mL) 중 7-벤질옥시-3-니트로퀴놀린-4-올 (72.87 g, 0.2459 mol)의 혼합물에 첨가하였다. 첨가 후, 반응물을 100 ℃에서 5분간 가열하고 상온으로 냉각시킨 후 교반하에 빙수에 부었다. 형성된 황갈색 침전물을 여과하여 단리한 후 디클로로메탄에 용해시켰다. 생성된 용액을 황산마그네슘 상에서 건조시키고 여과 한 후 감압하에 농축시켜 담갈색 고체인 72.9 g의 7-벤질옥시-4-클로로-3-니트로퀴놀린을 수득하였다.

E부
트리에틸아민 (38.6 mL, 0.277 mol)을 디클로로메탄 (1200 mL) 중 7-벤질옥시-4-클로로-3-니트로퀴놀린 (72.9 g, 0.232 mol)의 용액에 첨가하였다. 이어서 이소부틸아민 (25.24 mL, 0.2540 mol)을 첨가하고, 반응 혼합물을 상온에서 18시간 동안 교반하였다. 반응 혼합물을 디클로로메탄으로 희석시키고 물 (2 x) 및 염수로 순차적으로 세척한 후 무수 황산나트륨 상에서 건조시키고 여과한 후 감압하에 농축시켜 갈색 고체인 67.4 g의 (7-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민을 수득하였다.

F부
메탄올 (1.25 L) 중 니켈 (II) 클로라이드 (22.8 g, 0.096 mol)의 용액에 소듐 보로하이드라이드 (29.0 g, 0.767 mol)를 조금씩 나누어 첨가하였다. 메탄올 (300 mL) 및 디클로로메탄 (300 mL) 중 7-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민 (67.4 g, 0.192 mol)의 용액을 생성된 혼합물에 첨가하였다. 침전물 이 존재하였으며 디클로로메탄 (500 mL)을 첨가하여 이를 용해시켰다. (7-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민이 소비될 때까지 추가의 소듐 보로하이드라이드 (~10 g)를 조금씩 첨가하였다. 반응 혼합물을 셀라이트(CELITE) 여과 보조제 층을 통해 여과하고 여과 케이크를 50:50 디클로로메탄:메탄올로 세척하였다. 여액을 감압하에 농축시키고 검은 유성 잔류물을 물 및 디클로로메탄으로 처리하였다. 유기 용액을 물 및 염수로 세척한 후 황산마그네슘 상에서 건조시키고 여과하였다. 여액을 활성탄으로 처리하고 여과한 후 감압하에 농축시켜 유성의 갈색 고체로서 55.4 g의 7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민을 수득하였다.

G부
트리메틸 오르토부티레이트 (29.75 mL, 0.1859 mol)를 3회로 나누어 톨루엔 (795 mL) 중 7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 (54.6 g, 0.170 mol)의 용액에 첨가하였다. 이어서 피리딘 하이드로클로라이드 (1.96 g)를 첨가하고, 반응물을 105 ℃로 가열한 후 4시간 동안 교반하였다. 이어서 추가의 트리메틸 오르토부티레이트 (7 mL, 40 mmol)를 첨가하고, 반응물을 3시간 동안 교반하였다. 반응물을 상온으로 냉각시키고, 감압하에 용매를 제거하였다. 유성 잔류물을 클로 로포름으로 처리하고, 이를 감압하에 제거하여 잔류 톨루엔을 제거한 후 다시 클로로포름 (1.2 L)으로 희석시켰다. 생성된 용액을 5% 중탄산나트륨 수용액, 물 및 염수로 세척하고, 황산마그네슘 상에서 건조시킨 후 여과하고 감압하에 농축시켜 소량의 톨루엔 (0.93 당량)을 함유하는 유성의 갈색 고체로서 60.3 g의 7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린을 수득하였다.

H부
3-클로로퍼옥시벤조산 (순도 60%, 22.9 g, 79.6 mmol)(mCPBA)을 일부씩 디클로로메탄 (1 L) 중 7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린 (27.0 g, 72.3 mmol)의 용액에 첨가하고 반응물을 30분간 교반하였다. 물 (1 L)을 첨가하고, 생성된 혼합물을 30분간 교반하였다. 유기층을 1% 탄산나트륨 수용액 (2 x 200 mL)으로 세척하고 황산마그네슘 상에서 건조하고 여과한 후 감압하에 농축시켰다.
I부
H부에서 얻은 물질을 디클로로메탄 (800 mL)에 용해시키고, 진한 수산화암모늄 (300 mL)을 첨가하였다. p-톨루엔술포닐 클로라이드 (16.6 g, 86.8 mmol)를 조금씩 나누어 생성된 혼합물에 첨가하고 반응물을 30분간 교반한 후 물로 희석하였다. 유기층을 황산마그네슘 상에서 건조시키고 여과한 후 감압하에 농축시켰다. 조 생성물을 아세토니트릴로부터 재결정화하여 깃털 형상의 회백색 결정으로서 21.4 g의 7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 206.2-208.2 ℃.

H부 및 I부를 작은 규모로 수행하여 얻은 물질을 사용하여 상기 특성 데이터를 얻었다.
실시예 2
7-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민의 제조는 실시예 1의 A부 내지 F부에 기재되어 있다. 에톡시아세틸 클로라이드 (12.2 g, 99.2 mmol)의 농축 된 용액을 50:50 톨루엔:피리딘 중 7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 (29 g, 90 mmol)의 용액에 적가하였다. 반응 온도는 40 ℃에 도달하였으며 침전물이 형성되었다. 트리에틸아민 (15-20 g) 및 피리딘을 첨가하여 침전물이 용해되는 것을 도왔다. 반응물을 환류하에 3시간 동안 가열한 후 하룻밤 동안 상온으로 냉각시켰다. 감압하에 용매를 제거하고 흑색 잔류물을 디클로로메탄에 용해시켰다. 생성된 용액을 탄산나트륨 수용액 (150 mL)으로 수 회 세척하고 황산마그네슘 상에서 건조시키고 여과한 후 감압하에 농축시켜 황갈색 고체인 29.2 g의 7-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
B부
7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린 대신 7-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (29.2 g, 78.2 mmol)을 사용하여 실시예 1의 H부 및 I부에 기재된 일반적인 방법에 따랐다. 조 생성물을 아세토니트릴로 연화처리하고 여과하여 단리한 후 68 ℃의 진공 오븐에서 18시간 동안 건조시켜 회백색 고체로서 16.3 g의 7-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 201.0-203.0 ℃.

실시예 3
7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민의 제조는 실시예 1의 A부 내지 F부에 기재되어 있다. 질소 분위기하에, 트리에틸 오르토아세테이트 (4.59 mL, 25.0 mmol)를 크실렌 (130 mL) 중 7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 (8.05 g, 25.0 mmol)의 용액에 첨가하고, 생성된 용액을 하룻밤 동안 환류하에 가열 (160 ℃) 하였다. 딘-스타크 트랩을 사용하여 용매 부피를 70 mL로 감소시켰다. 수일의 기간에 걸쳐 침전물이 형성되었다. 디에틸에테르를 첨가하고, 여 과에 의해 침전물을 단리하고 디에틸에테르로 세척하여 담갈색 분말로서 6.81 g의 7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
B부
mCPBA (순도 65%, 2.31 g, 8.70 mmol)를 클로로포름 (100 mL) 중 7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (3.01 g, 8.71 mmol)의 용액에 2회로 나누어 교반하에 첨가하고, 반응물을 4시간 동안 교반하였다. 박층 크로마토그래피 (TLC)에 의한 분석 결과 반응이 완결되지 않았음을 나타내었으며, 추가의 mCPBA를 첨가하였다. TLC로 측정하여 반응이 완결되었을 때까지 용액을 교반하고, 이어서 포화 중탄산나트륨 수용액으로 세척한 후, 황산마그네슘 상에서 건조하고 여과한 후 감압하에 농축시켰다. 잔류물을 정제하지 않고 사용하였다.
C부
질소 분위기하에, 트리클로로아세틸 이소시아네이트 (1.60 mL, 13.4 mmol)를 디클로로메탄 (100 mL) 중 B부에서 얻은 물질의 용액에 적가하고, 반응물을 1시간 동안 교반하였다. 감압하에 용매를 제거하였다. 잔류물을 메탄올로 희석시키고, 소듐 메톡사이드 용액 (3.06 mL, 13.4 mmol, 메탄올 중 25%)을 서서히 첨가하였다. 반응물을 하룻밤 동안 교반하였으며, 침전물이 형성되었다. 침전물을 여과하여 단리하고, 차가운 헥산으로 세척한 후 (3 x), 아세토니트릴로부터 재결정화하고 2일간 60 ℃에서 건조시켜 백색 고체로서 1.15 g의 7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 171.9 ℃.

실시예 4
7-벤질옥시-2-에틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
실시예 3의 A부에 기재된 일반적인 방법에 따랐다. 트리에틸 오르토아세테이트 대신 트리에틸 오르토프로피오네이트 (7.66 mL, 58.1 mmol)를 크실렌 (200 mL) 중 7-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 (18.68 g, 58.11 mmol)의 용액에 첨가하였다. 반응 말기에 침전물을 3회 수집하여 담갈색 고체로서 7.16 g의 7-벤질옥시-2-에틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린을 수득하였다. mp 127 ℃.

B부
출발 물질로서 7-벤질옥시-2-에틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린을 사용하여 실시예 3의 B부에 기재된 일반적인 방법의 변법을 따랐으며, 반응은 4시간 내에 완결되었다. 반응 생성물을 고진공 하에 하룻밤 동안 건조시켜 발포체상의 오렌지색 고체로서 1.38 g의 7-벤질옥시-2-에틸-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
C부
실시예 3의 C부에 기재된 일반적인 방법을 이용하여 7-벤질옥시-2-에틸-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (1.38 g, 3.67 mmol)을 0.460 g의 7-벤질옥시-2-에틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민으로 전환시켰으며, 이는 백색 고체로서 수득되었다. mp 193.2-193.5 ℃.

실시예 5
4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올

실시예 1에 기재된 바와 같이 제조된 7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 (21.4 g, 55.1 mmol)을 환류하의 에탄올 (2 L)에 용해시키고, 따뜻한 용액에 10% 탄소상 팔라듐 (5.4 g, 5.1 mmol)을 첨가하였다. 반응물을 수소압 (50 psi, 3.4 x 105 Pa) 하에 하룻밤 동안 두었다. 여과에 의해 촉매를 제거하고 고온의 에탄올 (500 mL) 및 메탄올 (400 mL)로 세척하였다. 여액을 감압하에 농축시켜 14.5 g의 회백색 고체를 수득하였다. 고체의 적은 일부를 2-프로판올로부터 재결정화하여 백색 결정으로서 4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올을 수득하였다. mp>265 ℃.

실시예 6
4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올

실시예 2에 기재된 바에 따라 제조된 7-벤질옥시-2-에톡시메틸-1-(2-메틸프 로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (16.3 g, 40.3 mmol)을 환류하의 에탄올 (1.5 L)에 용해시키고, 따뜻한 용액에 10% 탄소상 팔라듐 (4.3 g, 4.0 mmol)을 첨가하였다. 상온으로 냉각되면서 침전물이 형성되었다. 반응 혼합물을 수소압 (50 psi, 3.4 x 105 Pa) 하에 하룻밤 동안 두었다. 여과에 의해 촉매를 제거하고 고온의 에탄올 (500 mL) 및 비등하는 DMF로 세척하였다. 여액을 감압하에 농축시켜 11.5 g의 회백색 고체를 수득하였다. 고체의 적은 일부를 2-프로판올로부터 재결정화하여 백색 결정으로서 4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올을 수득하였다. mp>265 ℃.

실시예 7-20
DMF (25-50 mL) 중 실시예 5에 기재된 바와 같이 제조된 4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올 (1 g, 3 mmol)의 따뜻한 용액을 대략 0 ℃로 냉각시켰다. 고형 탄산세슘 (2 당량)을 첨가하였으며, 반응물은 엷은 황색이 되었다. 하기 표에 기재된 벤질 할라이드 (1.1 당량)를 서서히 첨가하고 반응물을 실온으로 승온시킨 후 고성능 액체 크로마토그래피 (HPLC) 분석에 의해 완결된 것으로 판단될 때까지 하룻밤 동안 교반을 계속하였다. 반응물을 탈이온수 (500-750 mL)에 붓고 수 분간 교반하였다. 형성된 침전물을 여과에 의해 단리하였다. 각각의 실시예에 대하여 생성물의 정제 및 특성화를 표 아래에 기재하였다.


실시예 7
7-(3-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 아세토니트릴로부터 재결정화하고 여과에 의해 단리한 후 적은 부 피의 아세토니트릴로 세척하여 백색 고체인 750 mg의 7-(3-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 200-203 ℃.

실시예 8
7-(4-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 실리카겔 상 컬럼 크로마토그래피 (98:2 클로로포름:메탄올로 용출시킴)로 정제하고, 고온의 아세토니트릴로 연화처리한 후 적은 부피의 아세토니트릴로 세척하고 65 ℃의 진공 오븐에서 2시간 동안 건조시켜 밝은 백색 결정인 1.16 g의 7-(4-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 215-217 ℃.

실시예 9
7-(4-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 실리카겔 상 컬럼 크로마토그래피 (99:1 및 98:2 클로로포름:메탄올로 순차적으로 용출시킴)로 정제하고 아세토니트릴 (36 mL/g)로부터 재결정화한 후 여과에 의해 단리하고 적은 부피의 아세토니트릴로 세척한 후 65 ℃의 진공 오븐에서 2일간 건조시켜 밝은 백색 결정인 1.12 g의 7-(4-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 205-207 ℃.

실시예 10
7-(3,4-디클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 실리카겔 상 컬럼 크로마토그래피 (99.5:0.5로부터 98:2 비율로 변화시킨 클로로포름:메탄올로 용출시킴)로 정제하고 2-프로판올로부터 재결정화한 후 여과하여 단리하고 60 ℃의 진공 오븐에서 하룻밤 동안 건조시켜 백색 고체인 1.11 g의 7-(3,4-디클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 183-184 ℃.

실시예 11
7-(3-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 실시예 10에 기재된 바와 같이 정제하여 백색 결정인 1.00 g의 7-(3-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 182-183 ℃.

실시예 12
1-(2-메틸프로필)-7-(4-니트로벤질옥시)-2-프로필-1H-이미다조[4,5-c]퀴놀린 -4-아민
반응물을 하룻밤 동안 교반한 후 냉각시키고 추가의 4-니트로벤질 브로마이드 및 탄산세슘을 첨가한 것을 제외하면 실시예 7 내지 20에 기재된 일반적인 절차에 따랐다. 반응물을 4시간 동안 교반한 후 탈이온수 (~450 mL)에 부어 갈색의 유액상 침전물을 형성하였다. 침전물을 여과하여 단리하였으며, HPLC로 분석한 결과 여액이 출발 물질을 함유하는 것으로 나타났다. 실시예 10에 기재된 바와 같이 생성물을 정제한 후 70 ℃의 진공 오븐에서 1.5일간 2회 건조하여 담황색 고체인 320 mg의 1-(2-메틸프로필)-7-(4-니트로벤질옥시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다.

실시예 13
7-[4-(tert-부틸)벤질옥시]-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 10에 기재된 바와 같이 생성물을 정제하여 백색 고체인 820 mg의 7-[4-(tert-부틸)벤질옥시]-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4- 아민을 수득하였다. mp 203.5-205.0 ℃.

실시예 14
7-(4-플루오로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 10에 기재된 바와 같이 생성물을 정제하여 백색의 플레이크상 결정인 900 mg의 7-(4-플루오로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 198.0-199.7 ℃.

실시예 15
1-(2-메틸프로필)-2-프로필-7-[4-(트리플루오로메틸)벤질옥시]-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 10에 기재된 바와 같이 생성물을 정제하여 백색의 플레이크상 결정인 1.14 g의 1-(2-메틸프로필)-2-프로필-7-[4-(트리플루오로메틸)벤질옥시]-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 191.8-193.5 ℃.

실시예 16
1-(2-메틸프로필)-7-(3-니트로벤질옥시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 10에 기재된 바와 같이 생성물을 정제하여 담황색 고체인 1.15 mg의 1-(2-메틸프로필)-7-(3-니트로벤질옥시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 175-176 ℃.

실시예 17
7-(2-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
실리카겔 상 컬럼 크로마토그래피 (99.5:0.5로부터 98:2 비율로 변화시킨 클로로포름:메탄올로 용출시킴)로 생성물을 정제하고, 고온의 2-프로판올로 연화처리하고 여과하여 단리한 후, 60 ℃의 진공 오븐에서 하룻밤 동안 건조시켜 백색 분말인 980 mg의 7-(2-메틸벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 227.5-228.5 ℃.

실시예 18
7-(2-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 10에 기재된 바와 같이 생성물을 정제하여 백색 분말인 1.16 g의 7-(2-클로로벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 을 수득하였다. mp 216.0-217.5 ℃.

실시예 19
7-(2-메톡시벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 7 내지 20의 일반적 절차에 따랐다. 2-메톡시벤질 클로라이드 (578 mg, 3.69 mmol)를 적가한 후, 테트라부틸암모늄 브로마이드 (110 mg, 0.34 mmol)를 첨가하였다. 반응물을 하룻밤 동안 교반하였다. 생성물을 실리카겔 상 컬럼 크로마토그래피 (99:1 및 98:2 클로로포름:메탄올로 순차적으로 용출시킴)로 정제하고, 2-프로판올 (28 mL/g)로부터 재결정화한 후 여과하여 단리하고 65 ℃의 진공 오븐에서 2일간 건조시켜 백색 결정인 950 mg의 7-(2-메톡시벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 205.0-206.0 ℃.

실시예 20
7-(4-메톡시벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
2-메톡시벤질 브로마이드 대신 4-메톡시벤질 클로라이드 (551 mg, 3.52 mmol)를 사용하여 실시예 19에 기재된 변법에 따랐다. 반응물을 6시간 동안 교반하고 소량의 4-메톡시벤질 브로마이드를 더 첨가하였다. 정제 후 백색 분말인 750 mg의 7-(4-메톡시벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 186.5-188.0 ℃.

실시예 21
7-(3-아미노벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 16에 기재된 바와 같이 제조된 1-(2-메틸프로필)-7-(3-니트로벤질옥시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 (700 mg, 1.6 mmol)을 아세토니트릴 (35 mL)과 혼합하고, 촉매량의 5% 탄소상 백금을 첨가하였다. 반응물을 수소압 (50 psi, 3.4 x 105 Pa) 하에 3시간 동안 두었다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고 여과 케이크를 고온의 메탄올로 세척하였다. 여액을 감압하에 농축시켜 600 mg의 고체를 수득하였다. 이 고체를 실리카겔 상 컬럼 크로마토그래피 (99.5:0.5로부터 98:2 비율로 변화시킨 클로로포름:메탄올로 용출시킴)로 정제하고 75:25 아세토니트릴:2-프로판올로부터 재결정화한 후 여과하여 단리하고 진공 오븐에서 건조시켜 회백색 결정인 270 mg의 7-(3-아미노벤질옥시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 228.0-230.0 ℃.

실시예 22-32
다음과 같이 변경한 것을 제외하면 실시예 7 내지 20의 일반적인 방법에 따랐다. DMF (50 mL) 중 실시예 6에 기재된 바와 같이 제조된 4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (1 g, 3 mmol)의 따뜻한 용액을 실온으로 냉각한 후 탄산세슘 (2 당량)을 첨가하였다. 하기 표로부터 선택된 할라이드 (1.1 당량)를 사용하였다. 하기 표에 달리 나타내지 않는다면 실시예 10에 기재된 정제 방법을 이용하였다. 각각의 실시예에 대하여, 생성물의 특성화 데이터는 표 아래 포함되어 있다.


실시예 22
2-에톡시메틸-7-(4-메틸벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 22 내지 32에 대해 기재한 일반적인 방법을 이용하였다. 4-메틸벤질 브로마이드 (630 mg, 3.40 mmol)를 첨가한 후, 반응물을 6시간 동안 교반하였다. HPLC 분석으로 측정한 결과 반응이 완결되지 않았으며, 소량의 4-메틸벤질 브로마이드를 더 첨가하였다. 생성물을 실리카겔 상 컬럼 크로마토그래피 (클로로포름에 이어서 99:1로부터 97:3 비율로 변화시킨 클로로포름:메탄올로 용출시킴)로 정제하고 2-프로판올로부터 재결정화한 후 여과하여 단리하고 65 ℃의 진공 오븐에서 2일간 건조시켜 회백색 고체인 1.06 g의 2-에톡시메틸-7-(4-메틸벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 209.0-210.2 ℃.

실시예 23
2-에톡시메틸-7-(3-메틸벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀 린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 22 내지 32에 대해 기재한 일반적인 방법을 이용하였다. 가열하에 4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (310 mg, 0.97 mmol)을 DMF (15 mL)에 용해시켰으나 실온으로 냉각됨에 따라 용액으로부터 침전되었다. DMF (0.5 mL) 중 3-메틸벤질 브로마이드 (197 mg, 1.07 mmol)의 용액에 이어서 탄산세슘 (643 mg, 1.97 mmol)을 첨가하였다. N-메틸피롤리돈 (1 mL)을 반응물에 첨가하고, 반응물을 하룻밤 동안 교반하였다. 조 생성물을 실리카겔 상 컬럼 크로마토그래피로 정제하고 (클로로포름:메탄올로 용출시킴) 아세토니트릴로 연화처리한 후 여과에 의해 단리하여 백색 고체인 175 mg의 2-에톡시메틸-7-(3-메틸벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 207.0-208.0 ℃.

실시예 24
2-에톡시메틸-1-(2-메틸프로필)-7-[4-(트리플루오로메틸)벤질옥시]-1H-이미 다조[4,5-c]퀴놀린-4-아민
회백색 고체로서 생성물 (1.07 g)을 수득하였다. mp 181.3-182.7 ℃.

실시예 25
2-에톡시메틸-7-(3-메톡시벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민
회백색 고체로서 생성물 (710 mg)을 수득하였다. mp 144.0-145.0 ℃.

실시예 26
2-에톡시메틸-1-(2-메틸프로필)-7-[3-(트리플루오로메틸)벤질옥시]-1H-이미다조[4,5-c]퀴놀린-4-아민
회백색 고체로서 생성물 (990 mg)을 수득하였다. mp 151.0-152.0 ℃.

실시예 27
2-에톡시메틸-1-(2-메틸프로필)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 22 내지 32에 대해 기재한 일반적인 방법에 따랐다. 탄산세슘을 첨가한 후 DMF 중 4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올의 용액을 실온으로 냉각시켰다. 생성물인 2-에톡시메틸-1-(2-메틸프로필)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-4-아민 (1.00 g)을 엷은 황색 고체로서 수득하였다. mp 162.5-164.5 ℃.

실시예 28
7-(3,5-디메톡시벤질옥시)-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민
실시예 22 내지 32에 대해 기재한 일반적인 방법의 절반 규모로 반응을 수행하였다. 회백색 고체로서 생성물 (370 mg)을 수득하였다. mp 130.0-131.0 ℃.

실시예 29
2-에톡시메틸-1-(2-메틸프로필)-7-[3-(피롤-1-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 22 내지 32에 대해 기재한 일반적인 방법을 이용하였다. 탈이온수 중 반응 혼합물을 플라스크로부터 경사분리하여 조 생성물을 남기고 이를 물로 세정한 후 건조시켰다. 크로마토그래피로 정제한 후 생성물을 아세토니트릴로부터 재결정화하였다. 생성물을 건조시킨 후, 회백색 고체로서 730 mg의 2-에톡시메틸-1-(2-메틸프로필)-7-[3-(피롤-1-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 160.0-162.0 ℃.

실시예 30
7-(2-클로로벤질옥시)-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민
다음과 같이 변경한 것을 제외하면 실시예 22 내지 32에 대해 기재한 일반적인 방법을 이용하였다. 탄산세슘 (1.55 g, 4.77 mmol)을 첨가한 후 DMF (50 mL) 중 4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (0.750 g, 2.39 mmol)의 용액을 실온으로 냉각시켰다. 생성물인 7-(2-클로로벤질옥시)-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (810 mg)을 회백색 고체로서 수득하였다. mp 211.0-212.0 ℃.

실시예 31
2-에톡시메틸-7-(4-메톡시벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민
2-메톡시벤질 클로라이드 대신 4-메톡시벤질 클로라이드 (548 mg, 3.50 mmol)를 사용하여 실시예 19에 기재된 변법에 따랐다. 생성물 (1.04 g)을 회백색 고체로서 수득하였다. mp 181.0-182.5 ℃.

실시예 32
2-에톡시메틸-7-(2-메톡시벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴 놀린-4-아민
반응물을 이틀밤 동안 교반시킨 것을 제외하면 실시예 19에 기재된 변법에 따랐다. 실시예 10에 기재된 정제 방법을 이용하여 회백색 고체인 820 mg의 2-에톡시메틸-7-(2-메톡시벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 206.0-207.0 ℃.

실시예 33
7-(3-아미노벤질옥시)-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 27에 기재된 바와 같이 제조된 2-에톡시메틸-1-(2-메틸프로필)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-4-아민 (980 mg, 2.2 mmol)을 아세토니트릴 (30 mL)과 혼합하고, 5% 탄소상 백금 (~25 mg)을 첨가하였다. 반응물을 수소압 (50 psi, 3.4 x 105 Pa)하에 하룻밤 동안 두었다. 액체 크로마토그래피/질량 분광법 (LC/MS) 분석 결과 출발 물질이 존재함을 나타내었으며, 5% 탄소상 백금 (~25 mg)을 더 첨가하였다. 반응물을 수소압 (50 psi, 3.4 x 105 Pa) 하에 추가로 3시간 동안 두었다. 다시 LC/MS로 분석한 결과 반응이 완결되지 않은 것으로 나타났으며, 메탄올 (100 mL) 및 5% 탄소상 백금 (200 mg)을 첨가하였다. 반응물을 수소압 (50 psi, 3.4 x 105 Pa) 하에 하룻밤 동안 두었다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고, 여과 케이크를 고온의 메탄올로 세척한 후 여액을 감압하에 농축시켜 고체를 수득하였다. 이 고체를 실리카겔 상 컬럼 크로마토그래피 (45 g, 99:1로부터 95:5 비율로 변화시킨 클로로포름:메탄올로 용출시킴)로 정제하여 두 생성물을 수득하였다. 첫 번째 생성물을 2-프로판올로부터 재결정화하여 회백색 결정인 90 mg의 7-(3-아미노벤질옥시)-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 228.0-230.0 ℃.


실시예 34
2-에톡시메틸-7-(3-에틸아미노벤질옥시)-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 33에서 수득한 두 번째 생성물을 크로마토그래피로 정제하여 황갈색 고체로서 단리하였다. mp 60.0-65.0 ℃.

실시예 35-40
A부
실시예 1의 A부 내지 G부에 기재된 바와 같이 제조된 7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린 (60.3 g, 0.188 mol) 및 10% 탄소상 팔라듐 (10 g)을 에탄올 (500 mL)과 혼합하였다. 이어서 암모늄 포르메이트 (101.53 g, 1.61 mol) 및 에탄올 (500 mL)을 첨가하고 반응 혼합물을 환류하에 2시간 동안 가열하였다. 혼합물을 서서히 상온으로 냉각시키고 하룻밤 동안 교반하였다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고, 여과 케이크를 에탄올 (1 L), 메탄올 (2 L) 및 디클로로메탄 (2 L)로 세척하였다. 취합한 여액을 감압하에 농축시켜 황갈색 고체를 얻은 후 이를 차가운 에탄올로 연화처리하고 여과에 의해 단리하여 황갈색 과립상 고체인 30 g의 1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올을 수득하였다.

B부
1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올 (1 당량) 및 탄산세슘 (1.6 당량)을 DMF 중에서 교반하였다. 하기 표에 나타낸 플루오로벤젠 (1.6 당량)을 한 번에 첨가하고 반응물을 16시간 동안 65 ℃로 가열하였다. 감압하에 용매를 제거하고 잔류물을 물과 디클로로메탄 사이에 분배하였다. 수성층을 디클로로메탄으로 추출하였다. 취합한 유기 분획물을 물 (2 x) 및 염수로 순차적으로 세척하고 황산마그네슘 상에서 건조하고 여과한 후 감압하에 농축시켜 흑색 오일을 수득하였다. 이 오일을 실리카겔 상 컬럼 크로마토그래피 (50:50 에틸 아세테이트:헥산으로 용출시킴)로 정제하여 고체를 수득하였다.
C부
클로로포름 중 B부에서 얻은 물질의 용액에 mCPBA (1.2 당량)를 첨가하였다. 반응물을 1시간 동안 교반한 후 포화 중탄산나트륨 수용액에 붓고 20분간 교반하였다. 수성층을 클로로포름으로 추출하였다. 유기 용액을 취합하여 물 및 염수로 순차적으로 세척하고 황산마그네슘 상에서 건조한 후, 여과하고 감압하에 농축시켰다.
D부
C부로부터의 잔류물을 디클로로메탄에 용해시켰다. 수산화암모늄을 첨가한 후 p-톨루엔술포닐 클로라이드 (1.5 당량)를 첨가하였다. 반응물을 16시간 동안 교반한 후 디클로로메탄으로 희석하였다. 유기층을 수산화암모늄 수용액 (100 mL)으로 세척하였다. 취합한 수성 분획물을 디클로로메탄으로 추출하였다. 유기층을 취합하여 물 및 염수로 순차적으로 세척하고 황산마그네슘 상에서 건조한 후 여과하고 감압하에 농축시켰다. 각각의 실시예에 대하여 생성물의 정제 및 특성화를 표 아래에 기재하였다.

실시예 35
1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
2.00 g의 1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올을 사용 하여 실시예 35 내지 40에 대해 기재한 일반적인 방법의 B부에 따라 황색 고체인 2.33 g의 1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린을 수득하였다. 실시예 35 내지 40에 대해 기재한 일반적인 방법의 C부 및 D부를 다음 절차로 대체하였다. 클로로포름 (17 mL) 중 1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린 (1.00 g, 2.47 mmol)의 용액에 mCPBA (0.853 g, 2.97 mmol)를 한번에 첨가하고 반응물을 30분간 교반하였다. 이어서 수산화암모늄 (17 mL)을 첨가한 후 p-톨루엔술포닐 클로라이드 (0.710 g, 3.70 mmol)를 첨가하였다. 반응물을 16시간 동안 교반한 후 디클로로메탄으로 희석하였다. 수성층을 디클로로메탄으로 추출하였다. 유기 용액을 취합하여 물 및 염수로 순차적으로 세척하고 황산마그네슘 상에서 건조한 후, 여과하고 감압하에 농축시켜 갈색 고체를 수득하였다. 이 고체를 아세토니트릴로부터 재결정화하여 붉은 빛의 갈색 결정질 고체인 0.680 g의 1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 209-211 ℃.

실시예 36
1-(2-메틸프로필)-7-(2-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린- 4-아민
구배 펌프 시스템에 부착된 35 g 실리카겔 카트리지 (RediSep, ISCO, 230-400 메쉬, 직경 13.5 cm x 2.7 cm), 254 nm UV 검출기 및 분획 회수기 (ISCO COMBIFLASH Sg100c 시스템)를 이용하여 플래시 크로마토그래피로 조 생성물을 정제하였다. 컬럼을 디클로로메탄으로 평형화하고 반응 혼합물을 컬럼상에 주입하였다. 혼합물을 100% 디클로로메탄으로부터 2% 메탄올/디클로로메탄으로 이루어지는 선형 구배 프로그램으로 35 mL/분으로 5분간 용출시키고 2% 메탄올/디클로로메탄에서 5분간 유지한 후 화합물이 검출되지 않을 때까지 7% 메탄올/디클로로메탄으로 용출시켰다. 분획물을 TLC로 조사하여 목적 화합물을 함유하는 것들을 취합하여 농축시켰다. 크로마토그래피로 정제한 후 생성물을 아세토니트릴로부터 재결정화하여 황색 침상물인 0.079 g의 1-(2-메틸프로필)-7-(2-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 207-208.5 ℃.

실시예 37
1-(2-메틸프로필)-7-(3-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민
조 생성물을 아세토니트릴로부터 재결정화하여 황색 결정질 고체인 1-(2-메틸프로필)-7-(3-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 198-200 ℃.

실시예 38
2-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-일옥시]벤조니트릴
조 생성물을 에탄올로부터 재결정화하여 황갈색 결정인 2-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-일옥시]벤조니트릴을 수득하였다. mp 230-232 ℃.

실시예 39
4-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-일옥시]벤조니트릴
조 생성물을 에탄올로부터 재결정화하여 황갈색 결정인 4-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-일옥시]벤조니트릴을 수득하였다. mp 223-225 ℃.

실시예 40
메틸 2-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7- 일옥시]벤조에이트
조 생성물을 실리카겔 상 컬럼 크로마토그래피로 정제하고 (98:2로부터 95:5 비율로 변화시킨 디클로로메탄:메탄올로 용출시킴) 아세토니트릴로부터 재결정화하여 황갈색 결정인 메틸 2-[4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-일옥시]벤조에이트를 수득하였다. mp 167-168.5 ℃.

실시예 41
7-(4-아미노페녹시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

20:80 디클로로메탄:메탄올 (30 mL) 중 실시예 35에 기재된 바에 따라 제조된 1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아 민 (0.436 g, 1.04 mmol)의 용액에 니켈(II)클로라이드 (0.044 g, 0.34 mmol)를 첨가하였다. 생성된 용액에 소듐 보로하이드라이드 (0.300 g)를 조금씩 나누어 첨가하고 반응물을 25분간 교반하였다. 반응 중 고체가 형성되었으며 이를 여과하여 제거하고 여액을 감압하에 농축시켰다. 잔류물을 클로로포름에 용해시키고 생성된 용액을 물로 세척하였다. 유기 용액을 AMBERLITE IRA-756 이온 교환 수지와 함께 30분간 교반한 후 수지를 여과하여 제거하였다. 여액을 감압하에 농축시켜 갈색 오일을 얻고, 이를 구배 펌프 시스템에 부착된 10 g 실리카겔 카트리지 (RediSep, ISCO, 230-400 메쉬), 254 nm UV 검출기 및 분획 회수기 (ISCO COMBIFLASH Sg100c 시스템)를 사용하여 플래시 크로마토그래피로 정제하였다. 컬럼을 디클로로메탄 중 1% 메탄올로 평형화하고 반응 혼합물을 컬럼상에 주입하였다. 혼합물을 1% 메탄올/디클로로메탄으로부터 5% 메탄올/디클로로메탄으로 이루어지는 선형 구배 프로그램으로 35 mL/분으로 5분간 용출시키고 5% 메탄올/디클로로메탄에서 5분간 유지하여 목적 화합물을 용출시켰다. 분획물을 TLC로 조사하여 목적 화합물을 함유하는 것들을 취합하여 농축시켰다. 이어서 생성물을 아세토니트릴로부터 재결정화하여 황갈색 결정질 고체인 0.242 g의 7-(4-아미노페녹시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 190-191 ℃.

실시예 42
7-(2-아미노페녹시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

1-(2-메틸프로필)-7-(4-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 대신 실시예 36에 기재된 바와 같이 제조된 1-(2-메틸프로필)-7-(2-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 사용하여 실시예 41에 기재된 일반적인 방법의 변법에 따랐다. 반응이 완결되었을 때 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고, 여과 케이크를 메탄올 및 메탄올:디클로로메탄으로 세척하였다. 조 생성물을 이온 교환 수지로 처리한 후 실리카겔 상 컬럼 크로마토그래피로 정제하였다 (98:2로부터 94:6 비율로 변화시킨 디클로로메탄:메탄올로 용출시킴). 백색의 왁스질 고체를 아세토니트릴로부터 재결정화하여 황갈색 결정인 0.150 g의 7-(2-아미노페녹시)-1-(2-메틸프로필)-2-프로필-1H-이미다조 [4,5-c]퀴놀린-4-아민을 수득하였다. mp 197-199 ℃.

실시예 43
7-(3-에틸아미노페녹시)-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 37에 기재된 바와 같이 제조된 1-(2-메틸프로필)-7-(3-니트로페녹시)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 (0.254 g, 0.606 mmol)을 에탄올 (10 mL) 중에서 교반하고 5% 탄소상 백금 (0.025 g)을 첨가하였다. 반응물을 양압의 수소하에 두고 1시간 동안 교반하였다. 반응 혼합물을 셀라이트 여과 보조제의 층을 통해 여과하고 여과 케이크를 메탄올로 세척하였다. 여액을 감압하에 농축시키고 잔류물을 실리카겔 상 컬럼 크로마토그래피 (98:2로부터 95:5 비율로 변화시킨 디클로로메탄:메탄올로 용출시킴)로 정제하였다. 생성물을 아세토니트릴로부터 재결정화하여 4각의 오렌지색 결정인 0.032 g의 7-(3-에틸아미노페녹시)-1-(2-메틸프 로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 168-174 ℃.

실시예 44
7-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 1에 기재된 일반적인 방법에 따랐다. E부에서 이소부틸아민 대신 프로필아민을 사용하고, G부에서 트리메틸 오르토부티레이트 대신 에톡시아세틸 클로라이드를 사용하였다. 조 생성물을 아세토니트릴로부터 재결정화하여 응집형 백색 고체인 7-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 188-189 ℃.

실시예 45
4-아미노-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-7-올

실시예 44에서 제조한 7-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 (3.9 g, 9.99 mmol)을 에탄올과 혼합하고 에탄올 중 10% 탄소상 팔라듐 (0.390 g)이 충전된 파르 플라스크에 첨가하였다. 플라스크를 수소압 하에 두고 18시간 동안 진탕하였다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고, 여과 케이크를 따뜻한 DMF로 세척하였다. 여액을 감압하에 농축시키고 잔류물을 메탄올로부터 재결정화하여 백색 고체인 2.4 g의 4-아미노-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-7-올을 수득하였다. mp>250 ℃.

실시예 46
7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
디클로로메탄 (250 mL) 중 실시예 1의 A부 내지 D부에서 제조된 7-벤질옥시-4-클로로-3-니트로퀴놀린 (13.45 g, 42.7 mmol)의 용액에 트리에틸아민 (8.93 mL, 64.1 mmol)을 첨가하였다. 10분에 걸쳐 2-페녹시에틸아민 (6.15 mL, 47.0 mmol)을 적가하고 반응 혼합물을 상온에서 3시간 동안 교반하였다. 용액에 증류수 (200 mL)를 첨가하고 수성층을 디클로로메탄 (2 x 200 mL)으로 세척하였다. 취합한 유기 용액을 황산마그네슘 상에서 건조시키고, 여과한 후 감압하에 농축시켜 고체를 수득하였다. 이 고체를 헥산으로 세척하고 여과하여 단리한 후 감압하에 건조하여 황색 고체인 17.14 g의 (7-벤질옥시-3-니트로퀴놀린-4-일)-(2-페녹시에틸)아민을 수득하였다.
B부
가열하에 (7-벤질옥시-3-니트로퀴놀린-4-일)-(2-페녹시에틸)아민 (14.24 g, 34.28 mmol)을 톨루엔 (900 mL)에 용해시키고 5% 탄소상 백금 (6.7 g, 34.28 mmol) 및 톨루엔 (100 mL)이 충전된 파르 용기에 첨가하였다. 용기를 수소압 (35 psi, 2.4 x 105 Pa) 하에 두고 2시간 동안 진탕하였다. 촉매를 여과에 의해 제거하고 헥산 및 디클로로메탄으로 세척하였다. 여액을 감압하에 농축시키고 잔류물을 메탄올로부터 재결정화한 후 여과하여 단리하고 헥산으로 세척하고 고진공하에 건조시켜 고체인 13.82 g의 7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민을 수득하였다.
C부
질소 분위기하에, 톨루엔 (30 mL) 중 7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민 (3.0 g, 7.8 mmol)의 용액에 디에톡시메틸 아세테이트 (3.0 mL, 18 mmol)를 적가하고, 반응물을 환류하에 3시간 동안 가열하였다. 적은 부피의 용매가 남을 때까지 반응 혼합물을 감압하에 농축시켰다. 헥산을 첨가하고 생성된 혼합물을 20분간 냉장고에서 냉각시켰다. 형성된 침전물을 여과하여 단리하고 헥산으로 세척한 후 감압하에 하룻밤 동안 건조시켜 고체인 3.12 g의 7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
D부
실시예 3의 B부에 기재된 일반적인 방법의 변법에 따랐다. 클로로포름 (80 mL) 중 7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린 (2.0 g, 5.1 mmol)의 용액에 30분에 걸쳐 mCPBA (1.5 g, 5.1 mmol, 순도 60%)를 나누어 첨가하고 반응물을 24시간 동안 교반하였다. 후처리를 거쳐 1.59 g의 7-벤질옥시-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
E부
실시예 3의 C부에 기재된 일반적인 방법의 변법을 이용하여 7-벤질옥시-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린 (1.59 g, 3.86 mmol)을 처리하였다. 출발 물질이 용액으로 유지되는 것을 돕기 위해 클로로포름 (10 mL)을 첨가하였다. 트리클로로아세틸 이소시아네이트 (0.6 mL, 5 mmol)와의 반응물을 4.5시간 동안 교반하였다. 소듐 메톡사이드와 반응시킨 후 황갈색 침전물이 형성되었으며, 이를 여과에 의해 단리하고 헥산으로 세척한 후 진공 오븐에서 하룻밤 동안 건조시켰다. 이어서 고체를 물과 함께 하룻밤 동안 교반하고 여과에 의해 단리한 후 진공 오븐에서 건조시켜 황갈색 분말인 0.94 g의 7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 200.3-200.9 ℃.

실시예 47
7-벤질옥시-2-부틸-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
질소 분위기하에, 크실렌 (30 mL) 중 실시예 46의 A부 및 B부에 기재된 바와 같이 제조된 7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민 (3.0 g, 7.8 mmol)의 용액에 트리메틸 오르토발레레이트 (2.7 mL, 16 mmol)를 적가하고, 반응물을 환류하에 24시간 동안 가열하였다. 반응 혼합물을 상온으로 냉각시키고 침전물이 형성되면 이를 여과하여 단리하고 헥산으로 세척한 후 진공 오븐에서 2시간 동안 건조하여 2.96 g의 7-벤질옥시-2-부틸-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
B부
실시예 46의 D부에 기재된 일반적인 방법을 이용하여 7-벤질옥시-2-부틸-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린 (1.0 g, 2.2 mmol)을 1.0 g의 7-벤질옥시-2-부틸-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린으로 전환시켰다.
C부
실시예 3의 C부에 기재된 일반적인 방법의 변법을 이용하여 7-벤질옥시-2-부 틸-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린 (1.0 g, 2.1 mmol)을 처리하였다. TLC에 의한 분석 결과 2시간 후 트리클로로아세틸 이소시아네이트 (1.35 mL, 11.3 mmol)와의 반응은 완결되지 않았으며, 추가의 트리클로로아세틸 이소시아네이트 (1 mL)를 첨가하고 1시간 동안 교반하였다. 소듐 메톡사이드와 반응시킨 후 황갈색 침전물이 형성되었으며, 혼합물을 2일간 교반하였다. 침전물을 여과에 의해 단리하고 에탄올로부터 재결정화하고 여과에 의해 단리하고 헥산으로 세척한 후 진공 오븐에서 하룻밤 동안 건조시켜 백색 결정질 고체인 0.63 g의 7-벤질옥시-2-부틸-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 188.0-189.0 ℃.

실시예 48
7-벤질옥시-2-메톡시메틸-1-페네틸-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
2-페녹시에틸아민 대신 페네틸아민 (9.8 g, 81 mmol)을 사용하여 실시예 46의 A부에 기재된 일반적인 방법에 따랐다. 반응물을 6시간 동안 교반하고 후처리하여 9.9 g의 (7-벤질옥시-3-니트로퀴놀린-4-일)(페네틸)아민을 수득하였다.
B부
실시예 46의 B부에 기재된 일반적인 방법을 이용하여 (7-벤질옥시-3-니트로퀴놀린-4-일)(페네틸)아민 (6.2 g, 16 mmol)을 7-벤질옥시-N4-(페네틸)퀴놀린-3,4-디아민으로 전환시켰다.
C부
질소 분위기하에 디클로로메탄 (150 mL) 중 B부에서 얻은 물질의 용액을 대략 0 ℃로 냉각하고 피리딘 (10 mL)을 첨가하였다. 이어서 디클로로메탄 (40 mL) 중 메톡시아세틸 클로라이드 (1.67 g, 15.4 mmol)의 용액을 적가하고 반응물을 실온으로 승온시킨 후 TLC 분석 결과 출발 물질이 사라진 것으로 나타날 때까지 교반하였다. 감압하에 용매를 제거하고 잔류물을 톨루엔과 혼합한 후 TLC 분석 결과 반응이 완결된 것으로 나타날 때까지 딘-스타크 트랩이 장착된 플라스크 내에서 환 류하에 가열하였다. 감압하에 톨루엔을 제거하고 잔류물을 디클로로메탄 (300 mL) 및 물 (100 mL) 사이에 분배하였다. 유기층을 감압하에 농축시켰다. 조 생성물을 실리카겔 상 컬럼 크로마토그래피로 정제하여 (소량의 트리에틸아민을 함유하는 95:5 에틸 아세테이트:메탄올로 용출시킴) 4.1 g의 7-벤질옥시-2-메톡시메틸-1-페네틸-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
D부
실시예 3의 B부에 기재된 일반적인 방법의 변법에 따라 7-벤질옥시-2-메톡시메틸-1-페네틸-1H-이미다조[4,5-c]퀴놀린 (4.1 g, 9.7 mmol)을 7-벤질옥시-2-메톡시메틸-5-옥시도-1-페네틸-1H-이미다조[4,5-c]퀴놀린으로 전환시켰다. 용매로서 디클로로메탄 (50 mL)을 사용하고 반응을 4시간 후에 종료하였다. 정제하지 않고 물질을 사용하였다.
E부
D부에서 얻은 물질을 사용하여 실시예 3의 C부에 기재된 일반적인 방법에 따랐다. 반응으로부터의 침전물을 여과하여 단리하고 메탄올로 세척한 후 메탄올로부터 재결정화하여 백색 고체인 1.7 g의 7-벤질옥시-2-메톡시메틸-1-페네틸-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 195-197 ℃.

실시예 49
N-{4-[4-아미노-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}-N'-페닐우레아

A부
질소 분위기하에, 디클로로에탄 (30 mL) 중 5-(tert-부톡시카르보닐아미노)발레르산 (1.50 g, 6.91 mmol)의 용액을 -25 ℃로 냉각하였다. 트리에틸아민 (2.40 mL, 17.2 mmol) 및 피발로일 클로라이드 (0.85 mL, 6.91 mmol)를 첨가하고, 반응물을 3시간 동안 교반하였으며, 이때 온도가 -10 ℃로 상승하였다. 실시예 46의 A부 및 B부에 기재된 바와 같이 제조된 7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민 (0.850 g, 2.20 mmol)을 한 번에 첨가한 후 작은 주걱 분량의 4-디메틸아미노피리딘을 첨가하였다. 반응물을 실온으로 승온시키고 하룻밤 동안 교반한 후 환류하에 3시간 동안 가열하였다. 반응 용액을 포화 중탄산나트륨, 물 (2 x) 및 염수로 세척하고 황산나트륨 상에서 건조시킨 후, 여과하고 감압하에 농축시켰다. 조 생성물을 실리카겔 상 컬럼 크로마토그래피로 정제하여 (98:2 및 95:5 클로로포름:메탄올로 순차적으로 용출시킴) 고체를 수득하였다. 이 고체를 디에틸에테르로 처리하고 감압하에 농축시켜 백색 분말인 1.03 g의 tert-부틸 {4-[7-벤질옥 시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트를 수득하였다.
B부
디클로로메탄 (50 mL) 중 tert-부틸 {4-[7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트 (0.89 g, 1.6 mmol)의 용액에 mCPBA (순도 77%, 0.36 g, 1.6 mmol)를 첨가하고 질소 분위기하에 반응물을 2.5시간 동안 교반하였다. 추가의 mCPBA (150 mg)를 첨가하고 반응물을 이틀밤 동안 교반하였다. 용액을 물, 포화 중탄산나트륨 수용액, 물 및 염수로 세척하고 황산나트륨 상에서 건조시키고 감압하에 농축시켜 황갈색 발포체인 0.90 g의 tert-부틸 {4-[7-벤질옥시-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트를 수득하였다.
C부
출발 물질로서 tert-부틸 {4-[7-벤질옥시-5-옥시도-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트 (0.890 g, 1.53 mmol)를 사용하여 실시예 1의 I부에 기재된 일반적인 방법에 따랐다. 후처리를 거쳐 tert-부틸 {4-[4-아미노-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트 (823 mg)를 수득하였으며 이를 정제하지 않고 사용하였다.
D부
tert-부틸 {4-[4-아미노-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트 (823 mg, 1.41 mmol) 및 에탄올의 혼합물에 염산 용 액 (5 mL, 에탄올 중 1.5 M)을 첨가하였다. 반응물을 환류하에 30분간 가열하였으며 균질화되었다. 감압하에 용매를 제거하였다. 잔류물을 메탄올로부터 재결정화하고 여과에 의해 단리한 후 디에틸에테르로 세척하고 감압하에 건조하여 황색 결정인 534 mg의 2-(4-아미노부틸)-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다.
E부
질소 분위기하에, 2-(4-아미노부틸)-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민 (0.40 g, 0.83 mmol) 및 피리딘 (10 mL)의 혼합물을 0 ℃로 냉각하고 페닐 이소시아네이트 (91 μL, 0.84 mmol)를 첨가하였다. 반응물을 1시간 동안 교반하고 추가의 페닐 이소시아네이트 (10 μL)를 첨가하였다. 반응물을 1시간 동안 교반한 후 감압하에 농축시켰다. 잔류물을 실리카겔 상 컬럼 크로마토그래피 (95:5 클로로포름:메탄올로 용출시킴)로 정제하고 톨루엔으로부터 재결정화한 후 진공 오븐에서 건조시켜 백색 결정질 고체인 150 mg의 N-{4-[4-아미노-7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}-N'-페닐우레아를 수득하였다. mp 186.3-186.9 ℃.

실시예 50
N-{4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드

A부
증류수 중 실시예 1의 A부 내지 D부에서 제조된 7-벤질옥시-4-클로로-3-니트로퀴놀린 (36.80 g, 116.9 mmol)의 용액에 tert-부틸 N-(4-아미노부틸)카르바메이트 (22.01 g, 116.9 mmol)를 첨가하였다. 반응물을 1.5시간 동안 80 ℃로 가열하고 실온에서 4시간 동안 교반하였다. 반응은 완결되지 않았으며 트리에틸아민 (16 mL, 115 mmol)을 첨가하였다. 반응물을 실온에서 하룻밤 동안 교반하였다. 침전물이 형성되었으며 이를 여과에 의해 단리하고 헥산으로 세척하여 황색 고체인 39.92 g의 tert-부틸 [4-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트를 수득하였다.
B부
tert-부틸 [4-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트 (39.92 g, 85.57 mmol)를 톨루엔 (1700 mL)에 용해시키고 5% 탄소상 백금 (3.9 g) 및 적은 부피의 톨루엔이 충전된 파르 용기에 첨가하였다. 용기를 수소압 (50 psi, 3.4 x 105 Pa) 하에 두었다. 수소를 3회 교체하고 반응물을 하룻밤 동안 진탕하였다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고 여과 케이크를 에탄올 (700 mL)로 세척하였다. 여액을 감압하에 농축시켜 갈색 고체인 28.62 g의 tert-부틸 {4-[3-아미노-7-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트를 수득하였다.
C부
질소 분위기하에, 디클로로메탄 (1 L) 중 tert-부틸 {4-[3-아미노-7-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 (28.62 g, 65.5 mmol)의 용액을 대략 0 ℃로 냉각시키고 트리에틸아민 (10.0 mL, 72.1 mmol)을 첨가하였다. 메톡시프로피오닐 클로라이드 (8.57 mL, 78.6 mmol)를 적가하고 반응물을 상온에서 2시간 동안 교반하였다. 감압하에 휘발성 물질을 제거하고 잔류물을 에탄올 (840 mL)에 용해시켰다. 트리에틸아민 (33 mL)을 첨가하고 반응물을 환류하에 하룻밤 동안 가열하고 상온으로 냉각시켰다. 감압하에 휘발성 물질을 제거하여 갈색 오일인 30.77 g의 tert-부틸 {4-[7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트를 수득하였으며, 이를 정제하지 않고 사용하였다.
D부
7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 대신 tert-부틸 {4-[7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 (9.08 g, 17.9 mmol)를 사용하여 실시예 3의 B부에 기재된 일반적인 방법 의 변법에 따랐다. 28시간에 걸쳐 3 당량의 mCPBA를 첨가하였다. 후처리를 거쳐 8.07 g의 tert-부틸 {4-[7-벤질옥시-2-(2-메톡시에틸)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트를 수득하였다.
E부
출발 물질로서 tert-부틸 {4-[7-벤질옥시-2-(2-메톡시에틸)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 (8.07 g, 15.5 mmol)를 사용하여 실시예 1의 I부에 기재된 일반적인 방법에 따랐다. 후처리를 거쳐 투명한 갈색 오일로서 tert-부틸 {4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 (8.00 g)를 수득하였으며 정제하지 않고 사용하였다.
F부
에탄올 중 염산 용액 (9.25 mL, 2 M)을 tert-부틸 {4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 (2 g, 4 mmol)에 첨가하고 반응물을 5시간 동안 70 ℃로 가열하였다. 반응물을 상온으로 냉각시키고 용액중으로 질소 가스를 하룻밤 동안 버블링하였다. 감압하에 용매를 제거하고 잔류물을 디에틸에테르로 연화처리하여 끈적이는 고체를 수득하고, 이를 물에 용해시키고 pH가 11이 될 때까지 진한 수산화암모늄으로 처리하였다. 용액을 디클로로메탄으로 수회 추출하고, 추출물을 취합하고 감압하에 농축시켜 암갈색 오일인 1-(4-아미노부틸)-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다.
G부
무수 아세토니트릴 (255 mL) 중 1-(4-아미노부틸)-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-4-아민 (2.55 g, 6.07 mmol)의 용액에 트리에틸아민 (0.89 mL, 6.4 mmol)을 첨가하고 균질해질 때까지 혼합물을 가열하였다. 반응물을 상온으로 냉각시키고 메탄술폰산 무수물 (1.11 g, 6.38 mmol)을 서서히 첨가하였다. 반응물을 4시간 동안 상온에서 교반하고 감압하에 용매를 제거하였다. 잔류물을 에틸 아세테이트에 용해시키고 생성된 용액을 중탄산나트륨 수용액으로 세척하였다. 수용액을 에틸 아세테이트로 1회 세척하고, 취합한 유기 용액을 황산마그네슘 상에서 건조시키고 여과한 후 감압하에 농축시켰다. 조 생성물을 에탄올로부터 재결정화하고 85 ℃의 진공 오븐에서 24시간 동안 건조시켜 갈색 고체인 0.350 g의 N-{4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드를 수득하였다. mp 144.5-147.6 ℃.

실시예 51
N-{4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1- 일]부틸}우레아

출발 물질로서 실시예 50의 A부 내지 D부에 기재된 바와 같이 제조된 tert-부틸 {4-[7-벤질옥시-2-(2-메톡시에틸)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 (17.18 g, 33.00 mmol)를 사용하여 실시예 3의 C부에 기재된 일반적인 방법에 따랐다. 소듐 메톡사이드와 반응시킨 후, 반응 침전물을 여과에 의해 단리하여 황색 분말인 0.267 g의 N-{4-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}우레아를 수득하였다. mp 169.6-170.70 ℃.

실시예 52
N-[2-(4-아미노-2-에톡시메틸-7-하이드록시-1H-이미다조[4,5-c]퀴놀린-1-일) -1,1-디메틸에틸]메탄술폰아미드

A부
실시예 1의 E부에 기재된 일반적인 방법을 이용하여 7-벤질옥시-4-클로로-3-니트로퀴놀린 (14.5 g, 46.0 mmol)을 처리하였다. 이소부틸아민 대신 1,2-디아미노-2-메틸프로판 (5.29 mL, 50.6 mmol)을 사용하였다. 후처리를 거쳐, 조 생성물을 실리카겔 층을 통과시켜 (클로로포름 및 96:4 클로로포름:메탄올로 순차적으로 용출시킴) 황색 고체인 12.4 g의 (2-아미노-2-메틸프로필)-(7-벤질옥시-3-니트로퀴놀린-4-일)아민을 수득하였다.
B부
질소 분위기하에, 디클로로메탄 (400 mL) 중 (2-아미노-2-메틸프로필)-(7-벤질옥시-3-니트로퀴놀린-4-일)아민 (12.4 g, 33.9 mmol)의 용액을 0 ℃로 냉각하였다. 트리에틸아민 (9.43 mL, 67.8 mmol) 및 메탄술폰산 무수물 (5.90 g, 33.9 mmol)을 순차적으로 첨가하고 반응물을 상온에서 2시간 동안 교반하였다. HPLC에 의한 분석 결과 반응은 완결되지 않았으며 메탄술폰산 무수물 (1.4 g, 8.0 mmol)을 더 첨가하였다. 반응물을 90분간 더 교반하고 추가의 메탄술폰산 무수물 (0.7 g, 4 mmol)을 첨가하였다. 반응물을 추가로 3시간 동안 교반하고 포화 중탄산나트륨 수용액 (200 mL)을 첨가하였다. 유기층 내에서 침전물의 형성이 개시되었으며, 이를 분리하고 감압하에 농축시켜 황색 고체를 수득하였다. 고체를 가열하에 물 (200 mL)로 연화처리하고 여과에 의해 단리한 후 물 (3 x 100 mL) 및 디에틸에테르 (3 x 50 mL)로 세척하고 진공하에 하룻밤 동안 건조하여 황색 분말인 14.8 g의 N-[1,1-디메틸-2-(3-니트로-7-벤질옥시퀴놀린-4-일아미노)에틸]메탄술폰아미드를 수득하였다.
C부
N-[1,1-디메틸-2-(3-니트로-7-벤질옥시퀴놀린-4-일아미노)에틸]메탄술폰아미드 (14.8 g, 33.3 mmol)를 아세토니트릴 (300 mL)과 혼합하고 파르 플라스크에 첨가한 후 5% 탄소상 백금 (2 g)을 첨가하였다. 반응물을 질소로 플러싱하고 수소압 (40 psi, 2.8 x 105 Pa) 하에 5.5시간 동안 두고 2시간 후 수소를 교체하였다. TLC 분석 결과 출발 물질의 존재를 나타내었다. 추가의 아세토니트릴 (200 mL) 및 5% 탄소상 백금 (2 g)을 첨가하고 반응물을 하룻밤 동안 수소압 하에 두었다. 반응 혼합물을 셀라이트 여과 보조제 층을 통해 여과하고, 여과 케이크를 아세토니트릴로 세척하였다. 여액을 감압하에 농축시켰다. 톨루엔 및 디클로로메탄을 첨가하고 2회 감압하에 제거하여 발포체인 12.6 g의 N-[2-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]메탄술폰아미드를 수득하였다.
D부
tert-부틸 {4-[3-아미노-7-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 대신 N-[2-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]메탄술폰아미드 (12.6 g, 30.4 mmol), 메톡시프로피오닐 클로라이드 대신 에톡시아세틸 클로라이드 (3.33 mL, 30.4 mmol)를 사용하여 실시예 50의 C부에 기재된 일반적인 방법의 변법에 따랐다. 조 생성물을 디클로로메탄 (300 mL)에 용해시키고 생성된 용액을 물 (2 x 100 mL) 및 염수로 세척하고 황산나트륨 상에서 건조시킨 후 감압하에 농축시켜 갈색 오일을 수득하였다. 이 오일을 실리카겔 상 컬럼 크로마토그래피 (97.5:2.5 클로로포름:메탄올로 용출시킴)로 정제하여 베이지색 발포체인 12.4 g의 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드를 수득하였다.
E부
tert-부틸 {4-[7-벤질옥시-1-(2-페녹시에틸)-1H-이미다조[4,5-c]퀴놀린-2-일]부틸}카르바메이트 대신 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (1.71 g, 3.55 mmol)를 사용하여 실시예 49의 B부에 기재된 일반적인 방법의 변법에 따랐다. 반응은 2.5시간 내에 완결되었으며 담갈색 고체인 1.75 g의 N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드를 수득하였다.
F부
진한 수산화암모늄 (3 내지 4 mL)을 빠른 교반하에 디클로로메탄 (35 mL) 중 N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (1.75 g, 3.51 mmol)의 용액에 첨가하였다. p-톨루엔술 포닐 클로라이드 (670 mg, 3.51 mmol)를 첨가하였다. 반응물을 1시간 동안 교반하였으며 침전물이 형성되었다. 물 (100 mL)을 첨가하고 감압하에 디클로로메탄을 제거하였다. 이어서 빠른 교반하에 디클로로메탄 (5 mL)을 첨가하고 생성된 분말을 여과에 의해 단리하여 90:10 클로로포름:메탄올 (200 mL)에 용해시켰다. 생성된 용액을 감압하에 농축시키고 잔류물을 고온의 프로필 아세테이트 (50 mL)로 연화처리한 후 여과에 의해 단리하고 감압하에 건조하여 황갈색 분말인 1.45 g의 N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드를 수득하였다.
G부
7-벤질옥시-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 대신 N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (1.00 g, 2.01 mmol)를 사용하여 실시예 5에 기재된 일반적인 방법에 따랐다. 조 생성물을 메탄올 (20 mL)로부터 재결정화하였다. 결정을 3회 수집하여 메탄올 및 에틸 아세테이트로 세척하였다. 수집물을 취합하고 실리카겔 상 컬럼 크로마토그래피 (89.1:9.9:1 클로로포름:메탄올:수산화암모늄으로 용출시킴)로 정제하여 백색 분말인 330 mg의 N-[2-(4-아미노-2-에톡시메틸-7-하이드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드를 수득하였다. mp 255-256 ℃.

실시예 53
7-(4-메탄술포닐페녹시)-2-(2-메톡시에틸)-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

실시예 1의 A부 내지 G부에 기재된 일반적인 방법을 이용하여 7-벤질옥시-2-(2-메톡시에틸)-1-프로필-1H-이미다조[4,5-c]퀴놀린을 제조하였다. E부에서 이소부틸아민 대신 프로필아민을 사용하고 G부에서 트리메틸 오르토부티레이트 대신 메톡시프로피오닐 클로라이드를 사용하였다. 실시예 35 내지 40의 A부 내지 D부에 기재된 일반적인 방법에 따라 7-벤질옥시-2-(2-메톡시에틸)-1-프로필-1H-이미다조[4,5-c]퀴놀린을 처리하였다. B부에서, 사용된 플루오로벤젠은 4-플루오로페닐 메틸 술폰이었다. 크로마토그래피로 정제한 후 생성물을 아세토니트릴로부터 재결정화하여 암황갈색 침상물인 0.890 g의 7-(4-메탄술포닐페녹시)-2-(2-메톡시에틸)-1- 프로필-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 210-212 ℃.

실시예 54
1-[4-아미노-7-벤질옥시-2-(하이드록시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

A부
메탄올 (1.84 L) 중 실시예 1의 A부 내지 D부에서 제조한 7-벤질옥시-4-클로로-3-니트로퀴놀린 (230.0 g, 0.731 mmol)의 용액 230.0 g (0.731 mmol)에 트리에틸아민 (81.3 g, 0.803 mol)을 첨가하였다. 이어서 하이드록시이소부틸아민 (71.6 g, 0.803 mol)을 5분에 걸쳐 첨가하였으며 첨가 도중 온도가 30 ℃로부터 39 ℃로 상승하였다. 반응 혼합물을 환류하에 4시간 동안 가열한 후 실온으로 냉각시켰다. 고체 생성물을 여과에 의해 단리하고 에탄올 (1 L)로 세척한 후 55 ℃에서 진공하에 건조시켜 261.2 g의 1-[(7-벤질옥시-3-니트로퀴놀린-4-일)아미노]-2-메틸프로판 -2-올을 수득하였다.
B부
메탄올 (3 L) 중 1-[(7-벤질옥시-3-니트로퀴놀린-4-일)아미노]-2-메틸프로판-2-올 (245.0 g, 0.667 mol)의 용액을 5% 탄소상 백금 (7.35 g)을 함유하는 압력 용기에 첨가하였다. 용기를 55 ℃에서 5시간 동안 수소압 (30 psi, 2.1 x 105 Pa) 하에 두었다. 반응물을 40 ℃까지 냉각시키고 디클로로메탄 (2.5 L) 을 첨가하였다. 이어서 반응 혼합물을 30 내지 40 ℃에서 클라셀(CLARCEL) 여과 보조제를 통해 여과하고 여과 케이크를 메탄올로 세척하였다. 여액을 1.5 L의 부피로 농축하고 5 내지 10 ℃로 냉각한 후 2시간 동안 교반하였다. 고체가 형성되었으며 이를 여과에 의해 단리한 후 소량의 메탄올로 세척하고 50 ℃에서 진공하에 건조시켜 199.4 g의 1-{[3-아미노-7-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올을 수득하였다.
C부
질소 분위기하에, 1-{[3-아미노-7-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올 (45.0 g, 0.133 mol) 및 아세토니트릴 (180 mL)의 혼합물을 40 ℃로 가열하였다. 생성된 회색 현탁액에 아세톡시아세틸 클로라이드 (21.8 g, 0.160 mol)를 15분에 걸쳐 첨가하였으며, 첨가 도중 온도를 55±5 ℃로 유지하였다. 첨가 후, 침전물이 형성되었으며, 반응물을 15분간 55 ℃로 가열하였다. 이어서 반응 혼합물을 대략 0 ℃로 냉각시키고 생성물을 여과에 의해 단리하여 소량의 아세토니트 릴 및 아세톤으로 순차적으로 세척하고 50 ℃에서 진공하에 건조시켜 57.6 g의 2-({7-벤질옥시-4-[(2-하이드록시-2-메틸프로필)아미노]퀴놀린-3-일}아미노)-2-옥소에틸 아세테이트 하이드로클로라이드를 수득하였다.
D부
메탄올 (270 mL) 중 2-({7-벤질옥시-4-[(2-하이드록시-2-메틸프로필)아미노]퀴놀린-3-일}아미노)-2-옥소에틸 아세테이트 하이드로클로라이드 (54.0 g, 0.114 mol)의 혼합물을 55 ℃로 가열하여 용액을 수득하였다. 이어서 환류하에 반응 혼합물을 가열하면서 물 (90 mL) 중 수산화나트륨 (9.2 g, 0.23 mol)의 용액을 5분에 걸쳐 첨가하였다. 첨가 후, 추가의 깔때기를 물 (10 mL)로 세정하고 반응 혼합물을 1시간 동안 환류하에 가열하였다. 이어서 반응 혼합물을 대략 0 ℃로 냉각하고 고체 생성물을 여과에 의해 단리한 후 메탄올, 탈이온수 및 적은 부피의 아세톤으로 세척하고 50 ℃에서 진공하에 건조시켜 회백색 고체인 42.1 g의 1-[7-벤질옥시-2-(하이드록시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올을 수득하였다.
E부
1-[7-벤질옥시-2-(하이드록시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (39.0 g, 0.133 mol), 피리딘 (390 mL) 및 아세트산 무수물 (195 mL)의 혼합물을 염화칼슘 건조관이 장착된 반응 플라스크 내에서 1시간 동안 35±5 ℃로 가열하였다. 반응 혼합물을 얼음 (2.5 kg) 및 탈이온수의 혼합물에 붓고 대략 15분간 교반하였다. 형성된 침전물을 여과에 의해 단리하고 탈이온수 (500 mL)로 세척한 후 50 ℃에서 진공하에 건조시켜 41.8 g의 [7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트를 수득하였다.
F부
불화수소 (48%, 7.5 g, 0.18 mol)를 DMF (910 mL) 및 메탄올 (300 mL) 중 [7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트 (40.0 g, 0.0953 mol)의 용액에 첨가하였다. 이어서 mCPBA (60.6 g, 0.200 mol, 순도 57%)를 한번에 첨가하고 반응물을 상온에서 5.5시간 동안 교반하였다. 이어서 얼음 및 탈이온수 (4 L)의 혼합물을 반응 혼합물에 첨가하고, 생성된 혼합물을 30분간 격렬하게 교반하였다. 고체 생성물을 여과에 의해 단리하고 탈이온수로 세척한 후 50 ℃에서 진공하에 건조시켰다. 이어서 고체를 1시간 동안 디에틸에테르 (500 mL)로 연화처리하고 여과에 의해 단리한 후 디에틸에테르 (400 mL)로 세척하고 40 ℃에서 진공하에 건조시켜 41.7 g의 [7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트를 수득하였다.
G부
디클로로메탄 (650 mL) 중 [7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트 (40.0 g, 0.0918 mol)의 용액을 0 ℃로 냉각시키고 수산화암모늄 (28%, 250 mL)을 첨가하였다. 반응 온도를 5.5 ℃ 미만으로 유지하면서 디클로로메탄 (200 mL) 중 p-톨루엔술포닐 클로라이드 (29.1 g, 0.153 mol)의 용액을 12분에 걸쳐 첨가하였다. 이어서 반응 혼합물을 실 온으로 승온시키고 1시간 동안 교반하였다. 반응 혼합물을 디클로로메탄 (100 mL)으로 희석시키고 유기층을 탈이온수로 세척하고 (2 x 400 mL), 황산마그네슘 상에서 건조시킨 후 여과하였다. 이어서 용액을 1시간 동안 활성탄으로 처리하고 셀라이트 여과 보조제의 층을 통해 여과하고 감압하에 농축시킨 후 60 ℃에서 진공하에 추가로 건조시켜 40.1 g의 [4-아미노-7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트를 수득하였다.
H부
메탄올 (150 mL) 중 [4-아미노-7-벤질옥시-1-(2-하이드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-2-일]메틸 아세테이트 (34.0 g, 0.0782 mol)의 용액을 환류하에 가열하고 수산화나트륨 수용액 (1.7 M, 50 mL)을 5분에 걸쳐 첨가하였다. 백색 침전물이 형성되었으며 반응물을 환류하에 1시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고 약 3시간 동안 교반하였다. 여과에 의해 침전물을 단리하고 탈이온수 및 메탄올로 세척한 후 50 ℃에서 진공하에 건조시켜 조 생성물 24.9 g을 수득하였다. 조 생성물 (6.0 g)을 2-프로판올:아세트산 9:1 (470 mL)의 혼합물로부터 재결정화하고 고온의 용액을 활성탄으로 처리한 후 셀라이트 여과 보조제의 층을 통해 여과하였다. 여과에 의해 촉매를 단리하고 적은 부피의 2-프로판올:아세트산으로 세척한 후 50 ℃에서 진공하에 건조시켰다. 생성물을 수산화나트륨 수용액 (0.15 M, 200 mL)과 3시간 동안 교반하고 여과에 의해 단리한 후 탈이온수로 세척하고 1시간 동안 메탄올 (100 mL)과 교반하고, 메탄올로 세척하고 60 ℃에서 진공하에 건조시켰다. 끝으로, 생성물 (4.5 g)을 DMF로부터 재결정화하여 여과에 의해 단리하고 진공하에 건조시켜 백색 고체로서 1-[4-아미노-7-벤질옥시-2-(하이드록시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올을 수득하였다. mp 284.5-285.5 ℃.

실시예 55
(4-아미노-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올

실시예 54의 A부 및 B부에 기재된 바와 같이 제조된 1-{[3-아미노-7-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올을 실시예 1의 G부의 일반적인 방법에 따라 트리메틸 오르토부티레이트 대신 트리에틸 오르토포르메이트를 사용하여 처리하였다. 생성물인 (7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올을 실시예 1의 H부 및 I부의 일반적인 방법에 따라 처리하여 백색 분말인 (4-아미노-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올을 수득하였다. mp 254-257 ℃.
실시예 56
(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올

실시예 54의 A부 및 B부에 기재된 바와 같이 제조된 1-{[3-아미노-7-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올을 실시예 54의 C부 및 D부에 기재된 일반적인 방법에 따라 C부에서 아세톡시아세틸 클로라이드 대신 에톡시아세틸 클로라이드를 사용하여 처리하였다. 생성물인 (7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올을 실시예 54의 F부 및 G부의 일반적인 방법에 따라 처리하여 백색 분말인 (4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올을 수득하였다. mp 215.1-215.5 ℃.
실시예 57
8-벤질옥시-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
다음과 같이 변경한 것을 제외하면 실시예 1의 A부의 일반적 절차를 이용하였다. 3-벤질옥시아닐린의 용액 대신 메탄올 (150 mL) 중 4-벤질옥시아닐린 (100 g, 0.5 mol)의 용액을 사용하였다. 온도를 57 내지 60 ℃로 유지하면 1시간에 걸쳐 이 용액의 첨가를 수행하였다. 반응 생성물인 5-{[(4-벤질옥시)페닐이미노)]메틸}-2,2-디메틸-[1,3]디옥산-4,6-디온 (136.7 g)을 황색 분말로서 단리하였다.

B부
5-{[(4-벤질옥시)페닐이미노)]메틸}-2,2-디메틸-[1,3]디옥산-4,6-디온 (127.2 g, 0.360 mol) 및 DOWTHERM A 열 전달 유체 (500 mL)의 용액을 100 ℃로 가열한 후 90분에 걸쳐 DOWTHERM A 열 전달 유체 (1 L, 250 ℃로 가열됨)를 함유하는 플라스크에 서서히 첨가하였다. 첨가 도중, 반응 온도는 245 ℃ 미만이 되지 않도록 하였다. 첨가 후 반응물을 250 ℃에서 30분간 교반하고 상온으로 냉각시켰다. 침전물이 형성되었으며, 이를 여과에 의해 단리하고 디에틸에테르 (1 L) 및 아세톤 (250 mL)으로 세척하고 진공하에 2시간 동안 건조시켜 황색 분말인 65.7 g의 6-벤질옥시퀴놀린-4-올을 수득하였다.

C부
7-벤질옥시퀴놀린-4-올 대신 6-벤질옥시퀴놀린-4-올 (65.7 g, 0.261 mol)을 사용하여 실시예 1의 C부에 기재된 일반적인 방법에 따랐다. 반응 침전물을 여과에 의해 단리하고 이를 프로피온산 (600 mL), 이소프로판올 (500 mL) 및 디에틸에테르 (500 mL)로 세척한 후 진공하에 2일간 건조시켜 황갈색 분말인 46.01 g의 6-벤질옥시-3-니트로퀴놀린-4-올을 수득하였다 (5% 6-벤질옥시퀴놀린-4-올 함유).

D부
실시예 1의 D부에 기재된 일반적인 방법을 이용하여 6-벤질옥시-3-니트로퀴놀린-4-올 (13.26 g, 44.7 mmol)을 13.74 g의 6-벤질옥시-4-클로로-3-니트로퀴놀린으로 전환시켰으며 이를 황갈색 고체로서 단리시켰다.

E부
메틸아민 (수중 40% 용액으로서 얻을 수 있음, 21 mL, 0.25 mol)을 증류수 (300 mL) 중 6-벤질옥시-4-클로로-3-니트로퀴놀린 (13.74 g, 43.65 mmol)의 현탁액에 첨가하고, 반응물을 1.5시간 동안 100 ℃에서 교반하였다. 반응물을 상온으로 냉각시키고 3시간 동안 교반하였다. 침전물이 형성되었으며, 이를 여과에 의해 단리하고 증류수로 세척하고 (3 x) 2-프로판올 (44.2 mL/g)로부터 재결정화하였다. 결정을 여과에 의해 단리하고 차가운 헥산 (2 x 100 mL)으로 세척하여 오렌지색 결 정인 11.36 g의 N-(6-벤질옥시-3-니트로퀴놀린-4-일)-N-메틸아민을 수득하였다.
F부
(7-벤질옥시-3-니트로퀴놀린-4-일)-(2-페녹시에틸)아민 대신 N-(6-벤질옥시-3-니트로퀴놀린-4-일)-N-메틸아민 (11.36 g, 36.7 mmol)을 사용하여 실시예 46의 B부에 기재된 일반적인 방법에 따랐다. 암황색 오일로서 6-벤질옥시-N4-메틸퀴놀린-3,4-디아민 (7.91 g)을 수득하였으며 정제하지 않고 사용하였다.
G부
7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민 대신 6-벤질옥시-N4-메틸퀴놀린-3,4-디아민 (7.91 g, 28.3 mmol)을 사용하고 트리메틸 오르토발레레이트 대신 트리에틸 오르토프로피오네이트 (12.69 mL, 56.6 mmol)를 사용하여 실시예 47의 A부에 기재된 일반적인 방법에 따랐다. 반응으로부터의 침전물을 2회 수집하여 황색 고체인 7.56 g의 8-벤질옥시-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린을 수득하였다. mp 168.2-169.0 ℃.
H부
실시예 3의 B부에 기재된 일반적인 방법의 변법에 따랐다. 클로로포름 (75 mL) 중 8-벤질옥시-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린 (1.5 g, 47 mmol)의 용액에 mCPBA (순도 60%, 1.39 g, 47.3 mmol)를 나누어 첨가하고 반응물을 5.5시간 동안 교반하였다. 후처리 도중, 취합한 수성 세척물을 디클로로메탄으로 추출하였으며 용액으로부터 생성물이 침전되었다. 취합한 디클로로메탄 및 클로로포름 용액을 결정이 형성될 때까지 감압하에 농축시키고 하룻밤 동안 방치하였다. 여과에 의해 결정을 단리하였다. 수용액을 클로로포름으로 추출하고, 취합한 추출물을 물 (2 x)로 세척한 후 감압하에 적은 부피로 농축시켰다. 헥산을 첨가하고 생성된 결정을 여과에 의해 단리하였다. 모액을 감압하에 농축시켜 고체를 얻고 이를 2-프로판올로부터 재결정화하였다. 결정의 세 배치를 취합하여 1.30 g의 8-벤질옥시-2-에틸-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
I부
실시예 3의 C부의 일반적인 방법의 변법을 이용하였다. 디클로로메탄 (35 mL) 중 8-벤질옥시-2-에틸-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (1.30 g, 3.90 mmol)의 용액에 클로로포름 (10 mL)을 첨가하여 용해도를 높였다. 트리클로로아세틸 이소시아네이트 (0.633 mL, 5.31 mmol)를 첨가한 후, 반응물을 3.5시간 동안 교반하였다. 두 번째 단계에서, 클로로포름 (10 mL)을 또한 메탄올 (30 mL) 중 현탁액에 첨가하였다. 소듐 메톡사이드와 반응시킨 후, 반응물을 이틀밤 동안 교반하고, 침전물을 여과에 의해 단리하고 헥산으로 세척한 후 메탄올 (278 mL/g)로부터 재결정화하고 여과에 의해 단리한 후 헥산으로 세척하고 진공 오븐에서 2일간 건조시켰다. 모액으로부터의 두 번째 수집된 결정을 첫 번째 수집물과 취합하여 백색 결정질 고체인 0.95 g의 8-벤질옥시-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 238.4-238.9 ℃.

실시예 58
8-벤질옥시-2-(2-메톡시에틸)-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
질소 분위기하에, 디클로로메탄 (300 mL) 중 실시예 57의 A부 내지 F부에 기재된 바와 같이 제조된 6-벤질옥시-N4-메틸퀴놀린-3,4-디아민 (7.88 g, 28.2 mmol)의 용액을 대략 0 ℃로 냉각하고 트리에틸아민 (4.2 mL, 30.3 mmol)을 첨가하였다. 5분에 걸쳐 메톡시프로피오닐 클로라이드 (3.3 mL, 30.6 mmol)를 적가하고 반응물을 상온에서 90분간 교반하였다. 휘발성 물질을 감압하에 제거하고 잔류물을 에탄올 (300 mL) 및 트리에틸아민 (13 mL)에 용해시키고 이틀밤 동안 75 ℃로 가열하였다. 휘발성 물질을 감압하에 제거하고 잔류물을 클로로포름에 용해시켰다. 생성된 용액을 탈이온수 (3 x 200 mL)로 세척하고 감압하에 농축시켰다. 적은 부피의 헥산 및 디클로로메탄을 첨가하고 백색 침전물이 형성되었으며 이를 여과에 의해 단리하고 헥산으로 세척하여 백색 고체인 3.76 g의 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
B부
실시예 3의 B부에 기재된 일반적인 방법을 이용하여 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-1H-이미다조[4,5-c]퀴놀린 (2.0 g, 5.8 mmol)을 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린으로 전환시켰으며, 이를 담오렌지색 고체로서 수득하여 정제하지 않고 사용하였다.
C부
실시예 3의 C부에 기재된 일반적인 방법을 이용하여 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (2.09 g, 5.76 mmol)을 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민으로 전환시켰다. 조 생성물 (1.6 g)을 메틸 아세테이트 (3 L)로부터 재결정화하였다. 용액을 감압하에 600 mL의 부피로 농축시키고 생성된 첫 번째 결정 수집물을 여과에 의해 단리하고 진공 오븐에서 건조시켰다. 모액을 감압하에 300 mL의 부피로 농축시키고 생성된 두 번째 결정 수집물을 여과에 의해 단리하였다. 수집물을 취합하여 회백색 분말인 0.91 g의 8-벤질옥시-2-(2-메톡시에틸)-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 192-194 ℃.

실시예 59
8-벤질옥시-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민

A부
7-벤질옥시-N4-(2-페녹시에틸)퀴놀린-3,4-디아민 대신 실시예 57의 A부 내지 F부에 기재된 바와 같이 제조한 6-벤질옥시-N4-메틸퀴놀린-3,4-디아민 (1.53 g, 5.48 mmol)을 사용하여 실시예 47의 A부에 기재된 일반적인 방법에 따랐다. 반응물을 환류하에 3일간 가열하였다. 반응으로부터의 침전물을 여과에 의해 단리하고 여액을 감압하에 농축시켰다. 잔류물을 실리카겔 상 컬럼 크로마토그래피 (98:2 디클로로메탄:메탄올 및 97:3 디클로로메탄:메탄올로 순차적으로 용출시킴)로 정제하였다. 반응 침전물 및 크로마토그래피로 정제한 생성물을 합쳐 갈색 고체인 0.89 g의 8-벤질옥시-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린을 수득하였다.
B부
실시예 3의 B부에 기재된 일반적인 방법의 변법에 따라 8-벤질옥시-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린 (0.500 g, 1.44 mmol)을 0.50 g의 8-벤질옥시-2-부틸-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린으로 전환시켰다. 반응은 1시간 내에 완결되었으며 생성물을 고진공하에 하룻밤 동안 건조시켰다.
C부
실시예 3의 C부에 기재된 일반적인 방법을 이용하여 8-벤질옥시-2-부틸-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (0.50 g, 1.4 mmol)을 아민화시켰다. 반응으로부터 단리된 고체를 메탄올로부터 재결정화하여 백색 고체인 0.32 g의 8-벤질옥시-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민을 수득하였다. mp 197.9-199.1 ℃.

실시예 60
8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
에탄올을 용매로 사용한 것을 제외하고는, 실시예 46의 파트 B에 기재된 방법을 사용하여 실시예 57의 파트 A 내지 E에 기재된 바와 같이 제조된 N-(6-벤질옥시-3-니트로퀴놀린-4-일)-N-메틸아민 (9.9 g, 32 mmol)을 6-벤질옥시-N4-메틸퀴놀린-3,4-디아민으로 전환시켰다.
파트 B
질소 대기하에, 디클로로메탄 중 파트 A로부터의 물질 및 트리에틸아민 (5.02 g, 49.6 mmol)의 용액을 약 0 ℃로 냉각시키고, 메톡시에톡시아세틸 클로라이드 (6.99 g, 45.8 mmol)를 천천히 첨가하였다. 반응을 실온으로 가온시키고, TLC에 의한 분석이 출발 물질이 없음을 지시할 때까지 교반하였다. 용매를 감압하에 제거하고, 잔류물을 톨루엔과 혼합하고, 딘-스탁 트랩을 사용하여 환류 온도에서 가열하였다. 톨루엔을 감압하에 제거하고, 잔류물을 디클로로메탄 및 물에 분배시켰다. 유기층을 물 (2 x 100 mL) 및 염수 (200 mL)로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (95:5 에틸 아세테이트:메탄올로 용출)에 의해 정제하였다. 반응이 완결되지 않은 것으로 밝혀졌고, 생성물 혼합물을 톨루엔에 용해시켰다. 피리 딘 히드로클로라이드를 첨가하고, TLC에 의한 분석이 반응이 완결되었음을 지시할 때까지 반응을 환류 온도에서 가열하였다. 상기 기재된 바와 같이 후처리 및 정제를 반복하여 고체로서 8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-1H-이미다조[4,5-c]퀴놀린 (m.p. 128-132 ℃) 6.3 g을 얻었다.
파트 C
7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 대신 8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-1H-이미다조[4,5-c]퀴놀린 (5.4 g, 14 mmol)을 사용하는, 실시예 3의 파트 B에 기재된 일반적인 방법의 변법을 따랐다. 디클로로메탄 (100 mL)을 용매로 사용하고, 3시간 후에 TLC에 의한 분석은 반응이 완결되지 않은 것으로 지시하였다. 추가의 mCPBA (0.5 당량)를 첨가하고, 추가의 1시간 후에 반응을 종결시켰다. 후처리 후에, 생성물을 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하여 오일로서 8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 1.5 g을 얻었다.
파트 D
실시예 48의 파트 E에 기재된 실시예 3의 파트 C의 변법을 사용하여 8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (1.4 g, 3.6 mmol)을 전환시켜 8-벤질옥시-2-[2-메톡시(에톡시메틸)]-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민 0.6 g을 회백색 침상 결정체로서 수득하였다 (m.p. 159-162 ℃).

실시예 61 내지 66
파트 A
에탄올 (1.2 L) 중 실시예 59의 파트 A 내지 C에 기재된 바와 같이 제조된 8-벤질옥시-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민 (14.65 g, 42.4 mmol)의 용액을 파르 용기 중 탄소 상 10% 팔라듐 (6.77 g, 63.6 mmol) 및 소량의 에탄올에 첨가하였다. 반응을 수소압 (50 psi, 3.4 x 105 Pa)하에 3.5시간 동안 정치시켰다. 촉매를 여과에 의해 제거하고, 에탈 아세테이트로 세척하였다. 여액을 감압하에 소 부피로 농축하였다. 헥산을 첨가하고, 생성된 혼합물을 냉장기에서 밤새 정치시켰다. 결정이 형성되었고, 이를 여과에 의해 단리하고, 헥산 (500 mL)으로 세척하고, 고 진공 상태하에 3일 동안 건조시켜 백색 고체로서 2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-올 9.40 g (m.p. 219-220.2 ℃)을 얻었다.

파트 B
질소 대기하에, DMF 중 2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-올 (1 당량, 약 1 g)의 0.08 M 용액을 균질해질 때까지 85 ℃에서 가열하였다. 고체 탄산세슘 (2 당량)을 가열하고, 반응을 85 ℃에서 20 내지 40분 동안 교반하였다. 열을 제거하고, DMF (5 내지 10 mL) 중 하기 표에 지시된 알킬 브로마이드 (1.2 당량)의 용액을 첨가하였다. 반응을 85 ℃에서 2 내지 45시간 동안 또는 TLC에 의한 분석이 출발 물질이 소모되었음을 나타낼 때까지 교반하였다. 임의로 메탄올 (1 내지 2 mL)을 첨가하고, 반응 혼합물을 여과하여 고체를 제거하였다. 휘발성 물질을 감압하에 제거하고, 잔류물을 에틸 아세테이트에 용해시켰다. 생성된 용액을 물로 세척하고, 감압하에 농축하여 고체를 얻었다.
파트 C
20분의 기간에 걸쳐, mCPBA (1 당량, 순도 65%)를 클로로포름 중 파트 B (1 당량)로부터의 물질의 0.05 내지 0.1 M 용액에 4부분으로 나누어 첨가하였다. 실시예 61에 대해, 클로로포름 및 DMF의 1.5:1 혼합물을 용매로 사용하였다. 반응을 상온에서 4 내지 28시간 동안 교반하고, TLC에 의한 분석이 출발 물질이 소모되었음을 나타낼 때까지 임의로 추가의 mCPBA를 소량 첨가하였다. 그 후, 용액을 포화 수성 중탄산나트륨으로 세척하고, 감압하에 농축하였다. 실시예 66에 대해, 반응 혼합물을 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하였다. 각 실시예에 대한 합성의 마지막 단계, 정제 및 특징에 대한 기술은 표에 나타냈다.
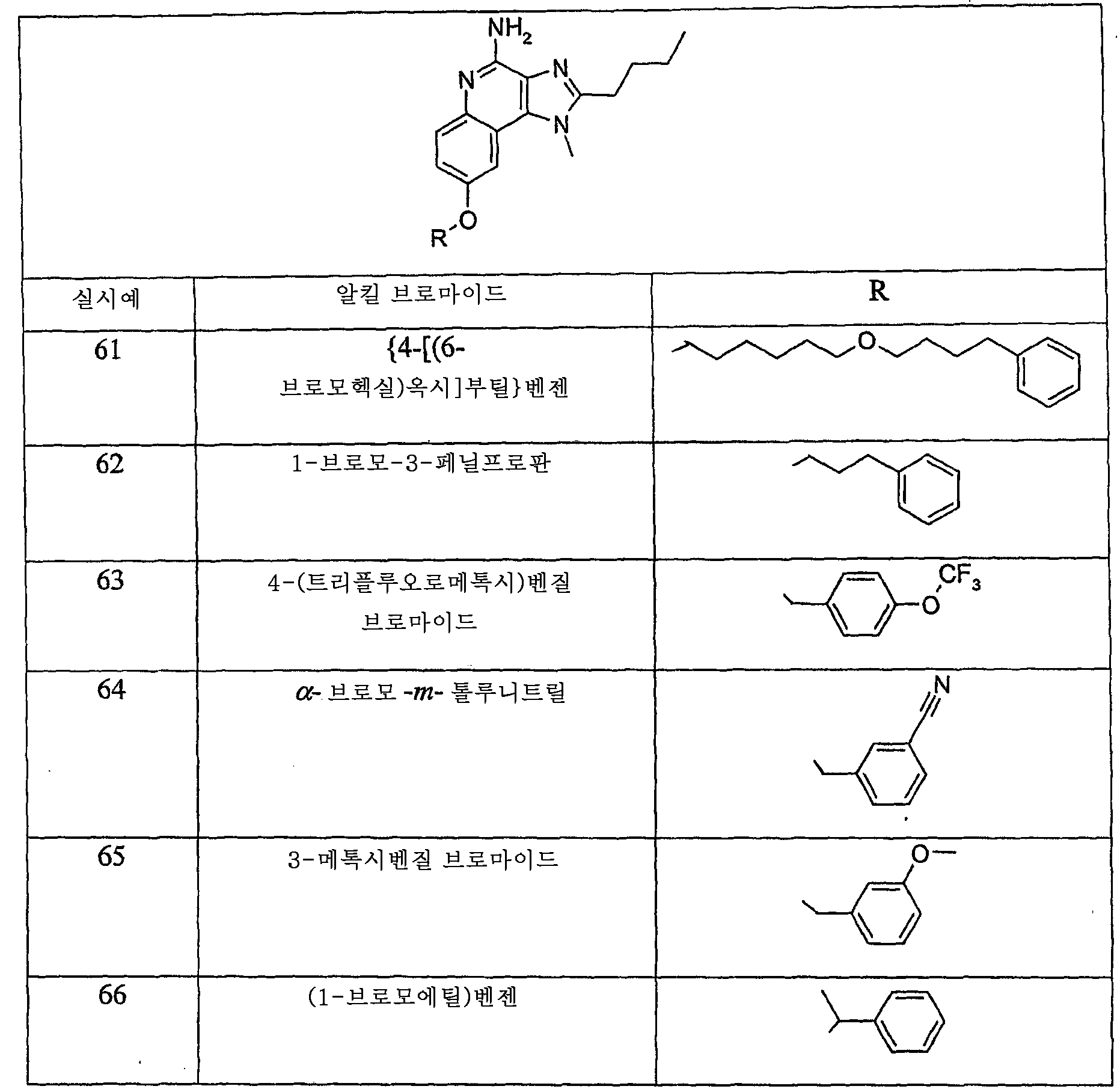
실시예 61, 64 및 66에 대해, 생성물을 정제용 고성능 액체 크로마토그래피 (정제용 HPLC)에 의해 정제하고, UV 트리거(trigger)에 의해 분획물을 수거하였다. 정제용 HPLC 분획물을 마이크로매스 플랫폼 LC/MS를 사용하여 분석하고, 적절한 분획물을 원심분리 증발시켜 목적 화합물의 트리플루오로아세테이트염을 얻었다. 페노메넥스 LUNA C18(2) 컬럼으로 역상 크로마토그레피에 의해 정제용 HPLC 분리를 수행하였다. 이동상은 물 및 아세토니트릴의 구배 혼합물 (물 중 0.05% 트리플루오로아세트산 및 아세토니트릴 중 0.05% 트리플루오로아세트산)이었다. 분리 조건은 하기 표에 나타내었다.

실시예 61
2-부틸-1-메틸-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린-4-아민 트리플루오로아세테이트
질소 대기하에, 트리클로로아세틸 이소시아네이트 (0.9 mL, 7 mmol)를 무수 디클로로메탄 (45 mL) 중 2-부틸-1-메틸-5-옥시도-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린 (2.5 g, 5.0 mmol)의 용액에 적가하였다. 반응을 5시간 동안 교반한 후, 4 방울의 수산화암모늄 (메탄올 중 7 중량%)을 첨가하였다. 반응을 밤새 교반하고, 대부분의 휘발성 물질을 감압하에 제거하였다. 디에틸 에테르를 나머지 용액에 첨가하고, 고체를 침전시켰다. 고체를 상기 기재된 바와 같은 정제용 HPLC에 의해 정제하여 오일을 얻고, 이를 메탄올에 용해시켰다. 생성된 용액을 여과하고, 감압하에 농축하여 백색 왁스로서 2-부틸-1-메틸-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린-4-아민 트리플루오로아세테이트 43.8 mg을 얻었다.

실시예 62
2-부틸-1-메틸-8-(3-페닐프로폭시)-1H-이미다조[4,5-c]퀴놀린-4-아민
출발 물질로서 2-부틸-1-메틸-5-옥시도-8-(3-페닐프로폭시)-1H-이미다조[4,5-c]퀴놀린 (0.360 g, 0.924 mmol)을 사용하는, 실시예 3의 파트 C에 기재된 일반적인 방법의 변법을 따랐다. 나트륨 메톡시드와 반응시킨 후, 반응 생성물을 2가지 수확물로 수거하였다. 제1 수확물을 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하여 밝은 황색 고체로서 2-부틸-1-메틸-8-(3-페닐프로폭시)-1H-이미다조[4,5-c]퀴놀린-4-아민 17.6 mg을 얻었다.

실시예 63
2-부틸-1-메틸-8-{[4-(트리플루오로메톡시)벤질]옥시}-1H-이미다조[4,5-c]퀴놀린-4-아민
2-부틸-1-메틸-5-옥시도-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린 대신 2-부틸-1-메틸-8-{[4-(트리플루오로메톡시)벤질]옥시}-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (0.380 g, 0.853 mmol)을 사용하여 실시예 61에 기재된 일반적인 방법을 따랐다. 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하여 백색 분말로서 2-부틸-1-메틸-8-{[4-( 트리플루오로메톡시)벤질]옥시}-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 219.4-220.2 ℃) 93.9 mg을 얻었다.

실시예 64
3-{[(4-아미노-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조니트릴 트리플루오로아세테이트
질소 대기하에, 수산화암모늄 (10 mL)을 무수 디클로로메탄 (46 mL) 중 3-[(4-아미노-2-부틸-1-메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조니트릴 (0.490 g, 1.26 mmol)의 용액에 첨가하고, 반응을 0 ℃로 냉각시키고, 신속하게 교반하였다. p-톨루엔술포닐 클로라이드 (0.241 g, 1.26 mmol)를 첨가하고, 반응을 천천히 가온시키고, 3일 동안 교반하였다. 침전물이 반응물에 존재하였다. 물을 첨가하고, 혼합물을 4시간 동안 교반하였다. 침전물을 여과에 의해 단리하고, 물로 세척하고, 메탄올로부터 재결정화하고, 진공 오븐에서 밤새 건조시켰다. 고체를 상기 기재된 바와 같이 정제용 HPLC에 의해 더 정제하여 백색 분말로서 3-{[(4-아미노-2-부틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조니트릴 트리플루오로아세테이트 41.8 mg을 얻었다.

실시예 65
2-부틸-8-[(3-메톡시벤질)옥시]-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민
2-부틸-1-메틸-5-옥시도-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린 대신 2-부틸-1-메틸-8-[3-(메톡시벤질)옥시]-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (0.420 g, 1.07 mmol)을 사용하여 실시예 61에 기재된 일반적인 방법을 따랐다. 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하여 백색 고체로서 2-부틸-8-[3-(메톡시벤질)옥시]-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 191.6-192.5 ℃) 34 mg을 얻었다.

실시예 66
2-부틸-1-메틸-8-(1-페닐에톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민 트리플루오로아세테이트
2-부틸-1-메틸-5-옥시도-8-{[6-(4-페닐부톡시)헥실]옥시}-1H-이미다조[4,5-c]퀴놀린 대신 2-부틸-1-메틸-5-옥시도-8-(2-페닐에톡시)-1H-이미다조[4,5-c]퀴놀린 (0.150 g, 0.399 mmol)을 사용하여 실시예 61에 기재된 일반적인 방법을 따랐다. 조 생성물을 상기 기재된 바와 같은 정제용 HPLC에 의해 정제하여 백색 분말로서 2-부틸-1-메틸-8-(1-페닐에톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민 트리플루오로아세테이트 (m.p. 183.4-184.20 ℃) 100 mg을 얻었다.

실시예 67
8-벤질옥시-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
실시예 1의 파트 E에 기재된 일반적인 방법을 사용하여 실시예 57의 파트 A 내지 D에 기재된 바와 같이 제조된 6-벤질옥시-4-클로로-3-니트로퀴놀린 (16.0 g, 50.8 mmol)을 (6-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민 16.7 g으로 전환시켰다. 반응이 30분내에 완결되었고, 생성물을 오렌지색 고체로서 수득하였 다.
파트 B
(7-벤질옥시-3-니트로퀴놀린-4-일)-(2-페녹시에틸)아민 대신 (6-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민 (4.6 g, 14 mmol)을 사용하는, 실시예 46의 파트 B에 기재된 일반적인 방법의 변법을 따랐다. 반응을 수소압하에 4시간 동안 유지시키고, 반응 혼합물을 셀라이트 여과기 보조물을 통해 여과하였다. 여과 케이크를 톨루엔으로 세척한 후, 여액을 100 mL 부피로 농축하였다.
파트 C
트리에틸 오르토포르메이트 (2.55 g, 17.2 mmol) 및 촉매량의 피리딘 히드로클로라이드를 파트 B로부터의 용액에 첨가하였다. 반응을 5시간 동안 환류 온도에서 가열하고, 실온으로 냉각시키고, 밤새 교반하였다. 그 후, 반응 혼합물을 냉각시키고, 침전물이 형성되었다. 침전물을 여과에 의해 단리하고, 헥산으로 세척하여 담황색 고체로서 8-벤질옥시-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (m.p. 129-133 ℃) 3.74 g을 얻었다.
파트 D
7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 대신 8-벤질옥시-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (2.1 g, 6.3 mmol)을 사용하여 실시예 3의 파트 B에 기재된 일반적인 방법을 따랐다. 후처리 후, 백색 고체로서 8-벤질옥시-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린 2.16 g을 수득하였으며, 이를 정제없이 사용하였다.
파트 E
출발 물질로서 8-벤질옥시-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (2.0 g, 5.8 mmol)을 사용하는, 실시예 3의 파트 C에 기재된 일반적인 방법의 변법을 따랐다. 트리클로로아세틸 이소시아네이트와의 반응은 TLC 분석에 의해 증명된 바와 같이 1시간 후에 완결되지 않았다. 추가의 트리클로로아세틸 이소시아네이트 (0.1 당량)를 첨가하고, 반응을 추가의 1시간 동안 교반하였다. 나트륨 메톡시드와 반응시킨 후, 고체 생성물을 여과에 의해 단리시키고, 에탄올로부터 재결정화하였다. 결정체를 진공 오븐에서 45 ℃에서 밤새 건조시켜 백색 고체로서 8-벤질옥시-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 214-216 ℃) 1.6 g을 얻었다.

실시예 68
8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
실시예 67의 파트 A에 기재된 바와 같이 제조된 (6-벤질옥시-3-니트로퀴놀린-4-일)-(2-메틸프로필)아민 (10.6 g, 30.2 mmol)을 실시예 67의 파트 B에 기재된 일반적인 방법의 변법을 사용하여 환원시켰다. 수소화가 5시간 동안 진행되게 하였다. 반응 후, 톨루엔을 감압하에 제거하여 갈색 오일로서 6-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 9.1 g을 얻었다.
파트 B
에톡시아세틸 클로라이드 (3.81 g, 31.1 mmol)를 피리딘 (60 mL) 중 6-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민 (9.1 g, 28.3 mmol)의 냉각된 용액에 첨가하였다. 반응을 상온로 가온시킨 후, 환류 온도에서 3시간 동안 가열하였다. 용매를 감압하에 제거하고, 잔류물을 디클로로메탄 (200 mL)에 용해시켰다. 생성된 용액을 물 (3 x 100 mL)로 세척하고, 감압하에 농축하였다. 농축된 용액을 실리카 겔 (디클로로메탄:메탄올 95:5로 용출)의 층을 통해 통과시키고, 감압하에 농축하여 오일로서 8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 3.6 g을 얻었다.
파트 C
실시예 60의 파트 C에 기재된 일반적인 방법의 변법을 사용하여 8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (3.7 g, 9.4 mmol)을 8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린 2.6 g으로 전환시켰다. 반응은 3시간내에 완결되었고, 생성물은 정제없이 사용하였다.
파트 D
출발 물질로서 8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-5-옥시도-1H-이미다조[4,5-c]퀴놀린 (2.6 g, 6.4 mmol) 및 용매로서 클로로포름을 사용하는, 실시예 3의 파트 C에 기재된 일반적인 방법의 변법을 따랐다. 트리클로로아세틸 이소시아네이트와의 반응은 TLC 분석에 의해 증명된 바와 같이 1시간 후에 완결되지 않았다. 추가의 트리클로로아세틸 이소시아네이트를 첨가하고, 반응을 밤새 교반하였다. 나트륨 메톡시드와 반응시킨 후, 고체 생성물을 여과에 의해 단리하고, 메탄올, 및 디클로로메탄과 물의 혼합물로 세척하였다. 고체를 여과에 의해 단리하고, 2-메톡시에틸 에테르로부터 재결정화하였다. 결정체를 고 진공 상태하에 건조시켜 백색 고체로서 8-벤질옥시-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 183-184 ℃)을 얻었다.

실시예 69
8-벤질옥시-2-메틸-1-[5-(메틸술포닐)펜틸]-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
7-벤질옥시-4-클로로-3-니트로퀴놀린 대신에 6-벤질옥시-4-클로로-3-니트로퀴놀린 (실시예 57의 파트 A 내지 D에 기재된 바와 같이 제조됨) (10.5 g, 34 mmol) 및 2-페녹시에틸아민 대신에 5-아미노-1-펜탄올 (3.5 g, 34 mmol)을 사용하는, 실시예 46의 파트 A에 기재된 일반적인 방법의 변법을 따랐다. 반응을 환류 온도에서 2시간 동안 가열한 후, 상온로 천천히 냉각시키고, 밤새 교반하였다. TLC에 의한 분석은 출발 물질의 존재를 나타내었고, 추가의 5-아미노-1-펜탄올 (0.2 당량)을 첨가하였다. 반응이 TLC에 의해 지시된 바와 같이 완결될 때까지, 반응을 환류 온도에서 가열하였다. 후처리 후에, 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (95:5 디클로로메탄:메탄올로 용출)에 의해 정제하여 황색 고체로서 5-{[6-(벤질옥시)-3-니트로퀴놀린-4-일]아미노}펜탄-1-올 5.95 g을 수득하였다.
파트 B
질소 대기하에, 티오닐 클로라이드 (1.7 mL, 23 mmol)를 5-{[6-(벤질옥시)-3-니트로퀴놀린-4-일]아미노}펜탄-1-올 (5.95 g, 15.6 mmol) 및 무수 디클로로메탄 (78 mL)의 혼합물에 첨가하였다. 반응은 균질화되었고, 1.5 시간 동안 환류 온도에서 가열하자, 황색 침전물이 형성되었다. 휘발성 물질을 감압하에 제거하고, 잔 류물을 묽은 수성 탄산나트륨 (100 mL)과 디클로로메탄 (150 mL)에 분배시켰다. 수성층을 디클로로메탄 (50 mL)으로 세척하고, 합한 유기 용액을 염수 (50 mL)로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하여 황색 오일로서 6-벤질옥시-N-(5-클로로펜틸)-3-니트로퀴놀린-4-아민 6.24 g을 얻었다.
파트 C
질소 대기하에, 고체 나트륨 티오메톡시드 (1.38 g, 18.7 mmol, 순도 95%)를 DMF 중 6-벤질옥시-N-(5-클로로펜틸)-3-니트로퀴놀린-4-아민 (6.24 g, 15.6 mmol)의 용액에 첨가하였다. 반응을 상온에서 30분 동안 교반한 후, 80 ℃에서 1시간 동안 가열하자, 황색 침전물이 형성되었다. 반응 혼합물을 물 (390 mL) 및 디클로로메탄 (150 mL)에 분배시켰다. 수성상을 디클로로메탄 (100 mL)으로 세척하고, 합한 유기 분획물을 포화 수성 중탄산나트륨 (100 mL) 및 염수 (100 mL)로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하여 적색 오일을 얻었다. 핵자기 공명 분광법에 의한 분석은 다량의 출발 물질이 존재함을 나타내었다. 반응 생성물을 DMF (78 mL)에 용해시키고, 나트륨 티오메톡시드로 처리하고, 환류 온도에서 2시간 동안 가열하였다. 상기 기재된 바와 같은 후처리를 반복하고, 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (70:30 에틸 아세테이트:헥산으로 용출)에 의해 정제하여 황색 오일로서 6-벤질옥시-N-[5-(메틸티오)펜틸]-3-니트로퀴놀린-4-아민 5.1 g을 얻었다.
파트 D
(7-벤질옥시-3-니트로-퀴놀린-4-일)-(2-페녹시에틸)아민 대신 6-벤질옥시-N [5-(메틸티오)펜틸]-3-니트로퀴놀린-4-아민 (5.1 g, 12 mmol)을 사용하는, 실시예 46의 파트 B에 기재된 일반적인 방법의 변법을 따랐다. 반응을 수소압 (49 psi, 3.4 x 105 Pa)하에 3시간 동안 유지시켰다. 반응 혼합물을 셀라이트 여과기 보조물의 층을 통해 여과하고, 여과 케이크를 메탄올 (100 mL) 및 클로로포름 (50 mL)으로 세척하였다. 여액을 감압하에 농축하여 황색 오일로서 6-벤질옥시-N4-[5-(메틸티오)펜틸]퀴놀린-3,4-디아민을 얻고, 이를 정제없이 사용하였다.
파트 E
트리메틸 오르토아세테이트 (1.7 mL, 14 mmol)를 톨루엔 (41 mL) 중 파트 D로부터의 물질의 용액에 첨가하였다. 그 후, 피리딘 히드로클로라이드 (0.1 g)를 첨가하고, 반응을 1시간 동안 환류 온도에서 가열하였다. 딘-스탁 트랩을 사용하여 휘발성 물질을 수거하였다. 반응을 상온로 냉각시키고, 용매를 감압하에 제거하였다. 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (95:5 디클로로메탄:메탄올로 용출)에 의해 정제하여 백색 고체로서 8-벤질옥시-2-메틸-1-[5-(메틸티오)펜틸]-1H-이미다조[4,5-c]퀴놀린 5.03 g을 얻었다.
파트 F
13분의 기간에 걸쳐, mCPBA (6.2 g, 27 mmol, 순도 75%)를 클로로포름 (41 mL) 중 8-벤질옥시-2-메틸-1-[5-(메틸티오)펜틸]-1H-이미다조[4,5-c]퀴놀린 (3.3 g, 8.1 mmol)의 용액에 일부분씩 나누어 첨가하였다. 반응을 15분 동안 교반한 후, 침전물이 형성되었다. 추가의 클로로포름 (41 mL)을 첨가하였지만, 침전물이 용 해되지 않았다. 그 후, 수산화암모늄 (40 mL) 및 p-톨루엔술포닐 클로라이드를 혼합물에 첨가하고, 반응을 10분 동안 교반하였다. 수성층을 디클로로메탄 (2 x 50 mL)으로 세척하고, 합한 유기 용액을 감압하에 농축하여 적색의 반-고체를 얻었다. 고체를 실리카 겔 상 컬럼 크로마토그래피 (90:10 디클로로메탄:메탄올로 용출)에 의해 정제하고, 메탄올 (80 mL/g)로부터 재결정화하여 무색의 침상 결정체로서 8-벤질옥시-2-메틸-1-[5-(메틸술포닐)펜틸]-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 215-217 ℃) 1.4 g 을 얻었다.

실시예 70 내지 73
파트 A
디클로로메탄 (200 mL) 중 실시예 57의 파트 A 내지 D에서 제조된 6-벤질옥시-4-클로로-3-니트로퀴놀린 (14.47 g, 46.29 mmol) 및 트리에틸아민 (8.4 mL, 60.2 mmol)의 용액을 0 ℃로 냉각시켰다. tert-부틸 N-(4-아미노부틸)카르바메이트 (8.71 g, 46.3 mmol)를 첨가하고, 반응을 15분 동안 0 ℃에서 교반한 후, 상온로 가온시키고, 5시간 동안 교반하였다. TLC에 의한 분석은 출발 물질의 존재를 지시하였고, 따라서, 추가의 tert-부틸 N-(4-아미노부틸)카르바메이트 (0.5 mL, 2.6 mmol)를 첨가하였다. 반응을 밤새 교반한 후, 물 (2 x 200 mL)로 세척하였다. 염화나트륨을 첨가한 후, 합한 세척액을 클로로포름으로 추출하였다. 합한 유기 용액을 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 조 생성물을 에틸 아세테이트 (100 mL)로부터 재결정화하였다. 결정체를 여과에 의해 단리시키고, 냉각된 헥산으로 세척하여 오렌지색 분말로서 tert-부틸 [4-(6-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트 17.62 g을 얻었다.
파트 B
tert-부틸 [4-(6-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트 (17.62 g, 37.77 mmol)를 가열하면서 톨루엔 (600 mL)에 용해시키고, 탄소 상 5% 백금 (2.20 g)으로 충전된 파르 용기에 첨가하였다. 용기를 수소압 (30 psi, 2.1 x 105 Pa)하에 3시간 동안 정치시켰다. 반응 혼합물을 셀라이트 여과기 보조물의 층을 통해 여과하고, 여액을 감압하에 농축하고, 갈색 고체로서 tert-부틸 {4-[3-아미노-6-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 15.5 g을 얻었다.
파트 C
실시예 70 내지 72에 대해, 다음의 방법을 사용하였다. 질소 대기하에, 하기 표로부터의 시약 (1.5 당량) 및 피리딘 히드로클로라이드 (0.01 내지 0.02 당량)를 톨루엔 (200 mL) 중 tert-부틸 {4-[3-아미노-6-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 (29 내지 36 mmol, 1 당량)의 용액에 첨가하고, 반응을 3 내지 5시간 동안 환류 온도에서 가열하였다. 톨루엔을 감압하에 제거하였다. 잔류물을 소량의 톨루엔에 용해시키고, 톨루엔은 감압하에 제거하였다. 이것을 3회 반 복하였다. 생성된 고체를 고 진공하에 100 ℃에서 건조시켜 하기 표에 지시된 바와 같은 치환기를 갖는 tert-부틸 {4-(8-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트를 얻었다.
실시예 73에 대해, tert-부틸 {4-[3-아미노-7-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 대신 tert-부틸 {4-[3-아미노-6-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 (9.25 g, 21.2 mmol) 및 메톡시프로피오닐 클로라이드 대신 에톡시아세틸 클로라이드 (2.86 g, 23.3 mmol)를 사용하는, 실시예 50의 파트 C에 기재된 일반적인 방법의 변법을 사용하였다. 고리화 반응후에, 용매를 감압하에 제거하고, 잔류물을 클로로포름 (400 mL)에 용해시켰다. 생성된 용액을 물 (2 x 200 mL) 및 염수 (1 x 200 mL)로 세척하고, 감압하에 농축하고, 고 진공하에 건조시켜 갈색 오일을 얻었다. 조 생성물을 실리카 겔 (400 g, 95:5 디클로로메탄:메탄올로 용출) 상 컬럼 크로마토그래피에 의해 정제하였다. 생성된 고체를 디에틸 에테르로 연화처리하고, 여과에 의해 단리시켜 회백색 분말로서 tert-부틸 {4-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}카르바메이트 5.37 g을 얻었다.
파트 D
mCPBA (1 당량, 순도 60%)를 클로로포름 중 파트 C로부터의 물질 (1 당량)의 0.1 M 용액에 첨가하였다. 반응을 질소 대기하에 밤새 교반하였다. 반응 동안 반응이 지연되는 것으로 보일 경우, 추가의 mCPBA를 첨가하였다. 그 후, 반응을 1% 수성 탄산나트륨 (2 내지 3 x) 및 염수 (1 x)로 세척하고, 황산나트륨으로 건조 시키고, 여과하고, 감압하에 농축하였다.
파트 E
과량의 수산화암모늄 및 p-톨루엔술포닐 클로라이드 (1 당량)를 디클로로에탄 중 파트 D로부터의 물질 (1 당량)의 0.5 내지 2 M 용액에 첨가하였다. 반응을 70 ℃에서 1 내지 2시간 동안 가열한 후, 실온으로 냉각시켰다. 유기 용액을 1% 수성 탄산나트륨 (3 x)으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 실시예 70, 71 및 73에 대해, 물질을 정제없이 사용하였다. 실시예 72에 대해, 조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (92.5:7.5 디클로로메탄:메탄올 및 90:10 디클로로메탄:메탄올로 용출)에 의해 정제하였다.
파트 F
실시예 50의 파트 F에 기재된 일반적인 방법의 변법을 따랐다. 에탄올 중 파트 E로부터의 물질 및 염산의 용액을 환류 온도에서 30분 내지 2시간 동안 가열하였다. 반응으로부터 얻은 염을 물에 용해시키고, 수용액을 클로로포름으로 세척하였다. 진한 수산화암모늄을 pH가 염기성이 될 때까지 수용액에 첨가하였다. 목적 생성물을 여과 (실시예 70)에 의해 단리하거나, 클로로포름 (실시예 71 내지 73)으로 추출하였다. 합한 유기 추출물을 염수 또는 1% 수성 탄산나트륨으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하여 하기 표에 도시된 화합물을 얻었다. 각각의 실시예에 대하여, 생성물의 정제 및 특징을 하기 표에 나타내었다.

실시예 70
1-(4-아미노부틸)-8-벤질옥시-2-부틸-1H-이미다조[4,5-c]퀴놀린-4-아민
단리된 고체를 진공 오븐에서 60 ℃에서 밤새 건조시켜 백색 분말로서 1-(4-아미노부틸)-8-벤질옥시-2-부틸-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 161.2-163.6 ℃)을 얻었다.

실시예 71
1-(4-아미노부틸)-8-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 황갈색 고체로서 단리하였다.

실시예 72
1-(4-아미노부틸)-8-벤질옥시-2-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 회백색 분말로서 단리하였다.

실시예 73
1-(4-아미노부틸)-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-아민
생성물을 오렌지색 고체로서 단리하였다.

실시예 74 내지 77
질소 대기하에, 하기 표에 지시된 용매 중 하기 표에 지시된 출발 물질 (1 내지 2 g)의 0.50 내지 0.1 M 용액을 0 ℃로 냉각시켰다. 페닐 이소시아네이트 (1 당량)를 적가하였다. 반응을 15분 동안 0 ℃에서 교반한 후, 상온로 가온시키고, 2시간 동안 또는 밤새 교반하자, 침전물이 형성되었다. 각 화합물의 정제 및 특징 은 하기 표에 제공하였다.

실시예 74
N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아
휘발성 물질을 감압하에 제거하였다. 잔류물을 디클로로메탄에 용해시키고, 헥산으로 희석하였다. 생성된 침전물을 여과에 의해 단리시키고, 이후에 진공 오븐에서 60 ℃에서 밤새 건조시켰다. 생성물을 실리카 겔 (200 g, 85:15 디클로로메탄:메탄올로 용출) 상 컬럼 크로마토그래피에 의해 정제하였다. 정제된 생성물을 진공 오븐에서 60 ℃에서 건조시켜 황색 미세결정체로서 N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아 (m.p. 190-194 ℃) 0.83 g을 얻었다.

실시예 75
N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아 히드로클로라이드
휘발성 물질을 감압하에 제거하고, 1 M 수성 염산을 잔류물에 첨가하였다. 반응을 환류 온도에서 2일 동안 가열하였다. 침전물이 형성되었고, 여과에 의해 단리하였다. 생성물을 메탄올 및 소량의 디클로로메탄으로부터 재결정화하였다. 결정체를 여과에 의해 단리하고, 냉각된 메탄올로 세척하고, 진공 오븐에서 60 ℃에서 건조시켜 황색 결정질 고체로서 mp 250 ℃ 초과의 N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아 히드로클로라이드 0.59 g을 얻었다. 모액을 감압하에 농축한 후 추가의 0.66 g을 얻었다.

실시예 76
N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아
반응 침전물을 여과에 의해 단리하고, 진공 오븐에서 60 ℃에서 밤새 건조시켰다. 고체를 소량의 메탄올 및 디클로로메탄을 함유하는 클로로포름 (250 mL)으로 연화처리하고, 여과에 의해 단리하고, 묽은 수산화암모늄 및 클로로포름과 혼합하였다. 수용액을 클로로포름 (4 x 200 mL)으로 추출하고, 합한 유기 분획물을 감압하에 농축하였다. 잔류물을 메탄올로부터 재결정화한 후, 실리카 겔 (200 mL, 90:10 및 87:13 클로로포름:메탄올로 연속적으로 용출) 상 컬럼 크로마토그래피에 의해 정제하여 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아 (m.p. 202.1-204.5 ℃) 0.74 g을 얻었다.

실시예 77
N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아
반응 침전물을 여과에 의해 단리하고, 진공 오븐에서 60 ℃에서 밤새 건조시켜 회백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}-N'-페닐우레아 (m.p. 186.1-188.5 ℃)를 얻었다.

실시예 78 내지 89
질소 대기하에, 무수 디클로로메탄 중 하기 표에 지시된 출발 물질 (1 내지 2 g, 1 당량) 및 트리에틸아민 (1.1 당량)의 0.50 내지 0.1 M 용액을 0 ℃로 냉각 시켰다. 하기 표에 지시된 시약 (1 당량)을 5 내지 10분의 기간에 걸쳐 적가하였다. 반응을 15분 동안 0 ℃에서 교반한 후, 상온로 가온시키고, 2시간 또는 밤새 교반하였다. 반응을 물 (100 mL) 및 염수 (2 x 100 mL)로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다.
각 화합물의 정제 및 특징을 하기 표에 제공하였다.


실시예 78
N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드
조 생성물을 에틸 아세테이트로부터 재결정화하였다. 결정체를 진공 오븐에서 60 ℃에서 밤새 건조시켜 오렌지색 미세결정체로서 N-{4-[4-아미노-8-(벤질옥 시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드 (m.p. 178.5-181.6 ℃) 1.74 g을 얻었다.

실시예 79
N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드
조 생성물을 소 부피의 디클로로메탄:메탄올에 용해시키고, 헥산을 첨가하였다. 생성된 침전물을 여과에 의해 단리하고, 헥산으로 세척하고, 고 진공하에 70 ℃에서 건조시켰다. 고체를 디클로로에탄으로부터 재결정화하고, 여과에 의해 단리하고, 냉각된 디클로로에탄으로 세척하고, 진공 오븐에서 건조시켰다. 결정체를 소량의 디클로로메탄 및 메탄올에 용해시키고, 감압하에 농축하여 황갈색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드 (m.p. 173.3-174.5 ℃) 1.56 g을 얻었다.

실시예 80
N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤젠술폰아미드
반응이 완결되었을 때, 침전물이 존재하였고, 이를 여과에 의해 단리하였다. 수성 후처리를 수행하지 않았다. 고체를 디클로로에탄으로부터 재결정화하고, 여과에 의해 단리하고, 냉각된 디클로로에탄으로 세척하고, 진공 오븐에서 80 ℃에서 건조시켜 황갈색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤젠술폰아미드 (m.p. 203.9-205.1 ℃) 1.88 g을 얻었다.

실시예 81
N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸} 모르폴린-4-카르복스아미드
반응이 완결되었을 때, 침전물이 존재하였고, 이를 여과에 의해 단리하였다. 여액을 수성 후처리 조건을 겪게하고, 생성된 고체를 디클로로에탄으로부터 재결정화하고, 여과에 의해 단리하고, 고 진공하에 건조시켰다. 반응 침전물을 물로 밤새 연화처리하고, 여과에 의해 단리하고, 물로 세척하고, 2일 동안 진공 오븐에서 80 ℃에서 건조시켜 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-부틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드 (m.p. 177.2-178.6 ℃)를 얻었다.

실시예 82
N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드
반응이 완결되었을 때, 침전물이 존재하였고, 이를 여과에 의해 단리하였다. 침전물을 물로 연화처리하고, 여과에 의해 단리하고, 고 진공 오븐하에 건조시켜 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드 (m.p. 226-230 ℃) 1.55 g을 얻었다.

실시예 83
N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드
조 생성물을 디클로로에탄으로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 디클로로에탄으로 세척하고, 진공 오븐에서 70 ℃에서 건조시켰다. 모액을 감압하에 농축하고, 잔류물을 실리카 겔 (200 mL, 90:10 디클로로메탄:메탄올로 용출) 상 컬럼 크로마토그래피에 의해 정제하였다. 컬럼 크로마토그래피로부터 회수된 고체를 2일 동안 진공 오븐에서 60 ℃에서 건조시켜 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드 (m.p. 177.8-179.5 ℃)를 얻었다.

실시예 84
N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드
조 생성물을 실리카 겔 상 컬럼 크로마토그래피에 의해 정제하고, 생성된 고체를 진공 오븐에서 60 ℃에서 건조시켜 황색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드 (m.p. 150.4-152.8 ℃) 1.21 g을 얻었다.

실시예 85
N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드
클로로포름을 용매로 사용한 것을 제외하고는, 실시예 78 내지 89에 대해 기재된 일반적인 방법을 따랐다. 조 생성물을 디클로로에탄으로부터 재결정화하고, 여과에 의해 단리하고, 냉각된 메탄올로 세척하고, 진공 오븐에서 60 ℃에서 건조시켰다. 생성된 고체를 실리카 겔 (200 mL, 93:7 및 90:10 클로로포름:메탄올로 연속적으로 용출) 상 컬럼 크로마토그래피에 의해 정제하고, 디클로로에탄으로부터 제2 재결정화하여 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}벤즈아미드 (m.p. 244.2-245.2 ℃) 0.50 g을 얻었다.

실시예 86
N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드
클로로포름을 용매로 사용한 것을 제외하고는, 실시예 78 내지 89에 대해 기재된 일반적인 방법을 따랐다. 조 생성물을 실리카 겔 (120 g, 92.5:7.5 디클로로메탄:메탄올로 용출) 상 컬럼 크로마토그래피에 의해 정제하고, 진공 오븐에서 60 ℃에서 건조시켜 회백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드 (m.p. 182.3-184.6 ℃)를 얻었다.

실시예 87
N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드
조 생성물을 디클로로에탄 (100 mL)으로부터 재결정화하고, 여과에 의해 단리하고, 냉각된 디클로로에탄으로 세척하고, 진공 오븐에서 60 ℃에서 건조시켜 백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}모르폴린-4-카르복스아미드 (m.p. 190.1-191.3 ℃) 1.40 g을 얻었다.

실시예 88
N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드
클로로포름을 용매로 사용한 것을 제외하고는, 실시예 78 내지 89에 대해 기재된 일반적인 방법을 따랐다. 반응이 완결되었을 때, 침전물이 존재하였고, 이를 여과에 의해 단리하였다. 생성된 고체를 물로 연화처리하고, 여과에 의해 단리하여 염화수소염을 얻었다. 염을 수산화암모늄과 혼합하고, 생성된 혼합물을 클로로포름 (5x)으로 추출하였다. 합한 추출물을 황산나트륨으로 건조시키고, 여과하고, 농축하여 회백색 고체 0.57 g을 얻었다. 고체를 메탄올로부터 재결정화하고, 여과에 의해 단리하고, 디에틸 에테르로 세척하고, 2일 동안 진공 오븐에서 60 ℃에서 건조시켜 회백색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드를 얻었다.

실시예 89
N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드
아세토니트릴을 용매로 사용한 것을 제외하고는, 실시예 78 내지 89에 대해 기재된 일반적인 방법을 따랐다. 출발 물질을 용해시키기 위하여 가열이 필요하였고, 용액이 가온되었을 때, 트리에틸아민 및 메탄술폰산 무수물을 첨가하였다. 조 생성물을 실리카 겔 (130 g, 92.5:7.5 디클로로메탄:메탄올로 용출) 상 컬럼 크로마토그래피로에 의해 정제하여 백색 분말 1.4 g을 얻었다. 분말을 디클로로에탄으로부터 재결정화하고, 여과에 의해 단리하고, 헥산으로 세척하고, 진공 오븐에서 건조시켜 황색 분말로서 N-{4-[4-아미노-8-(벤질옥시)-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일]부틸}메탄술폰아미드 (m.p. 186-191 ℃) 1.05 g을 얻었다.

실시예 90
4-아미노-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-올

7-벤질옥시-2-프로필-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 대신 실시예 57에 기재된 바와 같이 제조된 8-벤질옥시-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-4-아민 (0.47 g, 1.4 mmol)을 사용하여 실시예 5에 기재된 일반적인 방법을 따랐다. 수소화가 4시간 이내에 완결되었고, 촉매를 여과에 의해 제거한 후, 여액을 감압하에 소 부피로 농축하였다. 헥산을 첨가하고, 침전물이 형성되었 고, 이를 여과에 의해 단리하고, 헥산으로 세척하였다. 고체를 메탄올로부터 재결정화하여 백색 결정질 고체로서 mp 240 ℃ 초과인 4-아미노-2-에틸-1-메틸-1H-이미다조[4,5-c]퀴놀린-8-올 70 mg을 얻었다.

실시예 91 내지 103
에탄올 중 하기 표에 지시된 출발 물질의 0.01 내지 0.03 M 용액을 탄소 상 10% 팔라듐 (0.3 내지 0.7 g)으로 충전된 파르 용기에 첨가하고, 반응을 수소압 (50 psi, 3.4 x 105 Pa)하에 4 내지 24시간 동안 정치시켰다. 반응 혼합물을 셀라이트 여과기 보조물의 층을 통해 여과하고, 여과 케이크를 에탄올로 세척하였다. 여액을 감압하에 농축하였다. 각 혼합물의 정제 및 특징은 하기 표에 제공하였다.


실시예 91
N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아
염화수소염으로 수득된 조 생성물을 수성 수산화암모늄 (pH 11)과 함께 2시간 동안 교반하고, 여과에 의해 단리하고, 물로 세척하였다. 그 후, 고체를 메탄올로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 메탄올로 세척하 고, 진공 오븐에서 60 ℃에서 건조시켜 갈색 고체로서 N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아 0.44 g을 얻었다.

실시예 92
N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아
조 생성물을 실리카 겔 상 컬럼 크로마토그래피 (89:10:1 디클로로메탄:메탄올:수산화암모늄으로 용출)에 의해 정제하여 복숭아색 고체로서 N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아 0.270 g을 얻었다.

실시예 93
N-[4-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아
조 생성물을 메탄올로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 2일 동안 진공 오븐에서 60 ℃에서 건조시켜 미세한 핑크빛-백색 분말로서 N-[4-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]-N'-페닐우레아 (m.p. 218.4-220.8 ℃) 0.72 g을 얻었다.

실시예 94
N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드
조 생성물을 에탄올로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 에탄올로 세척하고, 고 진공하에 건조시켜 황갈색 분말로서 N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (m.p. 223-227 ℃) 0.74 g을 얻었다.

실시예 95
N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드
조 생성물을 에탄올로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 에탄올로 세척하고, 진공 오븐에서 60 ℃에서 건조시켜 백색 분말로서 N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드 (m.p. 188.2-189.5 ℃) 0.54 g을 얻었다.

실시예 96
N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤젠술폰아미드
조 생성물을 메탄올 (31 mL/g)로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 메탄올로 세척하고, 2일 동안 진공 오븐에서 80 ℃에서 건조시켜 미세한 백색 분말로서 N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤젠술폰아미드 (m.p. 219-223 ℃) 0.25 g을 얻었다.

실시예 97
N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드
조 생성물을 메탄올 (29 mL/g)로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 냉각된 메탄올로 세척하고, 며칠 동안 진공 오븐에서 건조시켜 백색 고체로서 N-[4-(4-아미노-2-부틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드 (m.p. 177-182 ℃) 0.21 g을 얻었다.

실시예 98
N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드
조 생성물을 클로로포름으로부터 재결정화하고, 며칠 동안 진공 오븐에서 70 ℃에서 건조시켰다. 고체를 디에틸 에테르로 연화처리한 후, 디클로로메탄과 메탄올의 혼합물에 용해시켰다. 용매를 감압하에 제거하고, 생성된 분말을 며칠 동안 진공 오븐에서 70 ℃에서 건조시켜 회백색 분말로서 N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드를 얻었다.

실시예 99
N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모 르폴린-4-카르복스아미드
조 생성물을 실리카 겔 (100 mL, 84:15:1 디클로로메탄:메탄올:수산화암모늄으로 용출) 상 컬럼 크로마토그래피에 의해 정제하여 회백색 분말로서 N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드 0.280 g을 얻었다.

실시예 100
N-[4-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드
조 생성물을 디에틸 에테르로 연화처리하고, 진공 오븐에서 60 ℃에서 밤새 건조시켰다. 그 후, 고체를 메탄올과 혼합하고, 혼합물을 감압하에 농축하였다. 그 후, 고체를 2일 동안 진공 오븐에서 60 ℃에서 건조시켜 미세한 핑크빛-백색 분말로서 N-[4-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드를 얻었다.

실시예 101
N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드
조 생성물을 메탄올 (10 mL)로부터 재결정화하고, 결정체를 여과에 의해 단리하고, 진공 오븐에서 60 ℃에서 건조시켜 핑크색 결정질 고체로서 N-[4-(4-아미노-8-히드록시-2-메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]벤즈아미드 0.106 g을 얻었다.

실시예 102
N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드
조 생성물을 2-프로판올로부터 재결정화하고, 결정체를 2일 동안 진공 오븐에서 건조시킨 후, 에탄올, 메탄올 및 디클로로메탄의 혼합물에 용해시켰다. 용매를 감압하에 제거하고, 생성된 고체를 디에틸 에테르로 연화처리하고, 진공 오븐에서 건조시켜 회백색 분말로서 N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]모르폴린-4-카르복스아미드를 얻었다.

실시예 103
N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드
조 생성물을 활성 탄소 (0.1 g)의 존재하에 에탄올로부터 재결정화하였다. 결정체를 여과에 의해 단리하고, 냉각된 에탄올로 세척한 후, 메탄올, 디클로로메탄, 및 메탄올의 혼합물에 용해시켰다. 용매를 여과하고, 감압하에 농축하고, 고 진공하에 70 ℃에서 건조시켜 백색 고체로서 N-[4-(4-아미노-2-에틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (m.p. 127.8-130.2 ℃) 0.36 g을 얻었다.

실시예 104
2-(에톡시메틸)-7-[(2-메틸벤질)옥시]-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (750 mg, 2.39 mmol)을 가열하면서 DMF (50 mL)에 용해시켰다. 고체 탄산세슘 (1.55 g, 4.77 mmol)을 첨가하였다. 15분 후에, 2-메틸벤질 클로라이드 (487 mg, 2.63 mmol)를 혼합물에 첨가하였다. 반응을 상온에서 교반한 후, 물 (500 내지 750 mL)에 붓자, 유백색 침전물이 즉시 형성되었다. 혼합물을 1시간 동안 교반한 후, 침전물을 여과에 의해 단리하였다. 고체를 플래시 크로마토그래피 (실리카 겔, 클로로포름 중 0.5 내지 2% 메탄올로 구배 용출)에 의해 정제한 후, 2-프로판올로부터 재결정화하였다. 최종 생성물을 여과에 의해 단리하고, 진공하에 60 ℃에서 밤새 건조시킨 후, 회백색 고체로서 2-(에톡시메틸)-7-[(2-메틸벤질)옥시]-1- (2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 216.0-218.0 ℃) 780 mg을 얻었다.

실시예 105
(4-아미노-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올

파트 A
1-{[3-아미노-6-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올을 실시예 54의 파트 A 및 B에 기재된 일반적인 방법에 따라, 파트 A에서 7-벤질옥시-4-클로로-3-니트로퀴놀린 대신 6-벤질옥시-4-클로로-3-니트로퀴놀린을 사용하여 제조하였다. 에톡시아세틸 클로라이드 (5.37 g, 43.8 mmol)를 아세토니트릴 (230 mL) 중 1-{[3-아미노-6-(벤질옥시)퀴놀린-4-일]아미노}-2-메틸프로판-2-올 (7.45 g, 22.1 mmol)의 용액에 첨가하고, 반응을 밤새 교반하였다. 용매를 감압하에 제거하고, 잔류물을 디클로로메탄에 용해시켰다. 생성된 용액을 수성 중탄산나트륨으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 잔류물을 HORIZON HPFC 시스템 (미국 버지니아주 샬롯데스빌 소재 비오티지 인코포레이션 (Biotage, Inc)으로부터 입수가능한 자동화 모듈 고성능 플래시 정제 생성물) (실리카 겔 카트리지, 디클로로메탄:메탄올 98:2 내지 95:5 비 범위로 용출) 상 컬럼 크로마토그래피에 의해 정제하여 N-{6-벤질옥시-4-[(2-히드록시-2-메틸프로필)아미노]퀴놀린-3-일}-2-에톡시아세트아미드 히드로클로라이드를 얻었다.
파트 B
파트 A로부터의 물질을 압력 용기에서 메탄올 (2 M) 중 암모니아의 용액으로 처리하였다. 반응을 160 ℃에서 6시간 동안 가열한 후, 감압하에 농축하여 백색 고체로서 (8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (m.p. 138-140 ℃)을 얻었다.
파트 C
과아세트산 (묽은 아세트산 중 32 중량% 2.0 mL, 9.5 mmol)을 에틸 아세테이트 (400 mL) 중 (8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (2.5 g, 6.2 mmol)의 용액에 첨가하고, 반응을 2일 동안 상온에서 교반하였다. 휘발성 물질을 감압하에 제거하고, 잔류물을 디클로로메탄에 용해시켰다. 생성된 용액을 수성 중탄산나트륨 및 물로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 조 생성물을 에틸 아세테이트, 헥산 및 2-프로판올의 혼합물로부터 재결정화한 후, HORIZON HPFC 시스템 (실리카 겔 카트리지, 디클로로메탄:메탄올 98:2 내지 90:10 비 범위로 용출) 상 컬럼 크로마토그래피에 의해 정제하여 고체로서 (8-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (m.p. 228-230 ℃) 1.9 g을 얻었다.
파트 D
실시예 54의 파트 G에 기재된 일반적인 방법의 변법을 사용하여 (8-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (1.45 g, 3.44 mmol)을 아민화하였다. 반응을 3시간 동안 교반하고, 수성 후처리한 후, 생성물을 2-프로판올로부터 재결정화하여 백색의 결정질 고체로서 (4-아미노-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (m.p. 181-182 ℃)을 얻었다.
실시예 106
N-[4-(4-아미노-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드

파트 A
tert-부틸 N-(4-아미노부틸)카르바메이트 (28.6 g, 152.0 mmol)를 디클로로메탄 중 실시예 1의 파트 A 내지 D에서 제조된 7-벤질옥시-4-클로로-3-니트로퀴놀린 (40.7 g, 138 mmol) 및 트리에틸아민 (38.5 mL, 276 mmol)의 용액에 1.5 시간의 기간에 걸쳐 적가하였다. 반응을 18시간 동안 교반한 후, 반응 혼합물을 물로 2회 및 포화 수성 염화나트륨으로 1회 세척하였다. 유기물을 무수 황산마그네슘으로 건조시키고, 여과하고, 감압하에 농축하였다. 조 생성물을 2-프로판올로부터 재결정화하여 금빛 갈색 분말로서 tert-부틸 [4-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트 51.4 g을 얻었다.
파트 B
tert-부틸 [4-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)부틸]카르바메이트 (20.36 g, 43.6 mmol)를 톨루엔 (450 mL)에 슬러리화하고, 탄소 상 5% 백금 (4.3 g) 및 소량의 톨루엔으로 충전된 파르 용기에 첨가하였다. 2-프로판올 (50 mL)을 첨가하고, 용기를 수소 (30 psi, 2.1 x 105 Pa)로 충전시켰다. 수소를 3회 교체하였다. 4시간 후에, 촉매를 셀라이트 여과기 보조물의 층을 통해 여과에 의해 제거하고, 여과 케이크를 톨루엔 (0.5 L), 50% 톨루엔/2-프로판올 (0.5 L) 및 2-프로판올 (0.25 L)로 세정하였다. 여액을 감압하에 농축하고, 잔류물을 톨루엔과 혼합하고, 감압하에 농축하여 점성 흑색 오일로서 tert-부틸 [4-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)부틸]카르바메이트 20.7 g을 얻었다.
파트 C
tert-부틸 [4-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)부틸]카르바메이트 (20.7 g, 43.6 mmol)를 톨루엔 (225 mL)에 용해시켰다. 피리딘 히드로클로라이드 (2.03 g) 및 트리메틸 오르토프로피오네이트 (6.2 mL, 43.6 mmol)를 첨가하고, 혼 합물을 환류 온도에서 3.25시간 동안 가열하였다. 반응을 실온으로 냉각시키고, 미세한 침전물이 형성되었다. 침전물을 여과에 의해 단리시키고, 톨루엔으로 세척하였다. 회백색 고체를 2시간 동안 진공하에 건조시켰다. 여액을 증발시키고, 잔류물을 디클로로메탄에 용해시켰다. 생성된 용액을 물 및 염수로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하여 갈색 무정형 고체를 얻었다. 고체를 합하여 tert-부틸 [4-(7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 17.2 g을 얻었다.
파트 D
mCPBA (10.6 g, 33.7 mmol)를 한 부분으로 클로로포름 (270 mL) 중 tert-부틸 [4-(7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 (16.0 g, 33.7 mmol)의 용액에 첨가하였다. 반응을 1.5시간 동안 교반한 후, 수산화암모늄 (270 mL)을 첨가하였다. 혼합물을 15분 동안 교반하고, p-톨루엔술포닐 클로라이드 (6.42 g, 33.7 mmol)를 3부분씩 나누어 첨가하였다. 반응을 18시간 동안 교반하였다. 층을 분리하고, 수성층을 디클로로메탄으로 추출하였다. 합한 유기 분획물을 물 및 염수로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하였다. 잔류물을 아세토니트릴로부터 재결정화하여 회색 분말로서 tert-부틸 [4-(4-아미노-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 9.73 g을 얻었다.
파트 E
진한 염산 (18 mL)을 에탄올 (100 mL) 중 tert-부틸 [4-(4-아미노-7-벤질옥 시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 (9.0 g, 18 mmol)의 용액에 첨가하고, 반응을 15분 동안 교반하였다. 에탄올을 감압하에 제거하고, 잔류물을 에탄올에 2회 용해시키고, 감압하에 농축하였다. 고체 잔류물을 염수 (25 mL) 및 물 (100 mL)과 함께 교반하고, 혼합물의 pH를 50% 수성 수산화나트륨을 첨가하여 14로 조정하였다. 디클로로메탄을 첨가하고, 층을 분리하였다. 수성층을 디클로로메탄 (100 mL)으로 2회 및 클로로포름 (100 mL)으로 2회 추출하였다. 합한 유기 분획물을 황산마그네슘으로 건조시키고, 여과하고, 감압하에 농축하여 회백색 분말로서 1-(4-아미노부틸)-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-4-아민 7.00 g을 얻었다.
파트 F
1-(4-아미노부틸)-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-4-아민 (7.0 g, 18 mmol)을 클로로포름 (180 mL)과 혼합하고, 현탁액을 얼음/물 조로 냉각시켰다. 메탄술포닐 클로라이드 (1.59 mL, 20.5 mmol)를 적가하고, 반응을 18시간 동안 교반하였다. 포화 수성 탄산나트륨을 용액에 첨가하고, 층을 분리하였다. 수성층 중 침전물을 여과에 의해 단리하고, 에탄올 및 디에틸 에테르로 세척하고, 메탄올 및 클로로포름의 혼합물로부터 재결정화하였다. 재결정화 동안, 고온의 용액을 유리 섬유 여과기를 통해 여과하였다. N-[4-(4-아미노-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (2.96 g)를 회백색 고체로서 단리하였다. 유기층을 감압하에 농축하고, 잔류물을 비등 메탄올과 함께 교반하고, 여과에 의해 단리하여 백색 고체로서 N-[4-(4-아미노-7-벤질옥시-2-에틸-1H-이미다 조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (m.p. 213.5-215 ℃) 1.90 g을 얻었다.

실시예 107
N-[4-(4-아미노-2-에틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드

아세토니트릴 (250 mL) 및 메탄올 (50 mL) 중 실시예 106에서 제조된 N-[4-(4-아미노-7-벤질옥시-2-에틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (2.00 g, 4.28 mmol)의 가온 용액을 탄소 상 10% 팔라듐 (1.00 g) 및 소량의 아세토니트릴을 함유하는 파르 용기에 첨가하였다. 용기를 수소압 (25 psi, 1.7 x 105 Pa)하에 정치시키고, 21시간의 기간에 걸쳐 수소를 3회 재충전하였다. 그 후, 진한 염산 (30 mL)을 반응 혼합물에 첨가하고, 셀라이트 여과기 보조물의 층을 통해 여과하였다. 여과 케이크를 물 (200 mL)로 세척하고, 여액을 감압하에 농축하였 다. 에탄올 (2 x 100 mL)을 잔류물에 첨가하고, 감압하에 농축하여 백색 고체를 얻었다. 고체를 물 (30 mL)과 혼합하고, 1 M 수성 수산화나트륨을 첨가하여 pH를 8로 조정하였다. 생성된 백색 과립 침전물을 여과에 의해 단리하고, 물 및 에탄올로 세척하고, 고온 에탄올과 함께 교반하고, 여과에 의해 단리하여 백색 분말로서 N-[4-(4-아미노-2-에틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]메탄술폰아미드 (m.p. 220 ℃) (분해) 0.860 g을 얻었다.

실시예 108
2-에톡시메틸-1-(2-메틸프로필)-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
5-히드록시안트라닐산 (100 g, 0.653 mol) 및 아세트산 무수물 (500 mL, 5.29 mol)의 혼합물을 2시간 동안 환류 온도에서 교반하였다. 용액을 감압하에 농축하고, 잔류물을 감압하에 밤새 건조시켜 회백색 고체로서 6-아세톡시-2-메틸벤조[d][1,3]옥사진-4-온 143 g을 얻었다.
파트 B
빙초산 1.44 L 중 6-아세톡시-2-메틸벤조[d][1,3]옥사진-4-온 (143 g, 0.653 mol)의 용액을 나트륨 아지드 (44.66 g, 0.687 mol)와 함께 밤새 교반하였다. 용액을 감압하에 농축한 후, 톨루엔 (1.2 L)을 첨가하였다. 1시간 동안 교반한 후, 고체가 형성되었다. 고체를 여과에 의해 회수한 후, 감압하에 밤새 건조시켰다. 고체를 물 2 L에 용해시키고, 진한 염산으로 pH를 1로 조정하였다. 3시간 동안 교반한 후, 생성된 고체를 여과하고, 공기 흐름하에 밤새 건조시켜 회백색 고체로서 5-아세톡시-2-(5-메틸테트라졸-1-일)벤조산 164.1 g을 얻었다.
파트 C
탄산세슘 (176 g, 1.13 mol)을 아세톤 1.8 L 중 5-아세톡시-2-(5-메틸테트라졸-1-일)벤조산 (164 g, 0.620 mol)의 용액에 첨가하였다. 혼합물을 격렬하게 혼합하고, 아세톤 100 mL로 희석한 요오도에탄 (116 g, 0.682 mol)을 1시간 기간에 걸쳐 적가하였다. 반응을 상온에서 밤새 교반한 후, 감압하에 농축하였다. 생성된 고체를 디클로로메탄 및 물에 분배시켰다. 수성 분획물을 디클로로메탄으로 추출하고, 합한 유기 분획물을 황산나트륨으로 건조시키고, 여과하고, 농축하여 갈색 오일로서 에틸 5-아세톡시-2-(5-메틸테트라졸-1-일)벤조에이트 166 g을 얻었다.
파트 D
에틸 5-아세톡시-2-(5-메틸테트라졸-1-일)벤조에이트 (164 g, 0.565 mol)를 DMF 500 mL에 용해시키고, 수조에 침지시켰다. 칼륨 에톡시드 (52.3 g, 0.621 mol)를 이 용액에 일부분씩 나누어 첨가하고, 반응 온도는 40 ℃ 미만으로 유지시켰다. 30분 후에, 반응을 물 4 L에 붓고, 염화암모늄을 생성물이 침전될 때까지 용액에 첨가하였다. 1시간 동안 교반한 후, 침전물을 여과하고, 공기 흐름하에 백색 고체로서 에틸 5-히드록시-2-(5-메틸테트라졸-1-일)벤조에이트 98.5 g을 얻었다.
파트 E
탄산칼륨 (110 g, 0.793 mol)을 DMF 500 mL 중 에틸 5-히드록시-2-(5-메틸테트라졸-1-일)벤조에이트 (98.5 g, 0.397 mol)의 용액에 첨가하였다. 30분 동안 격렬하게 교반한 후, 브롬화벤질 (75 g, 0.438)을 첨가하고, 반응을 밤새 교반하였다. 용액을 여과한 후, 감압하에 농축하였다 (50 ℃). 생성된 고체를 디클로로메탄 400 mL에 슬러리화하고, 여과하였다. 여액을 감압하에 농축하여 갈색 오일로서 에틸 5-벤질옥시-2-(5-메틸테트라졸-1-일)벤조에이트 115 g을 얻었다.
파트 F
칼륨 에톡시드 (57.5 g, 0.682 mol)를 DMF 550 mL 중 에틸 5-벤질옥시-2-(5-메틸테트라졸-1-일)벤조에이트 (115.6 g, 0.341 mol)의 용액에 일부분씩 나누어 첨가하였다. 반응은 발열하였고, 온도는 35도까지 상승하였다. 반응 혼합물을 얼음조에서 냉각시키고, 1시간 동안 교반하였다. 그 후, 용액을 염화암모늄 500 g을 함유하는 물 6 L에 부었다. 황색-오렌지색 침전물이 형성되었다. 밤새 교반한 후 , 반응을 여과하였다. 회수된 고체를 진공 오븐에서 50 ℃에서 건조시켜 7-(벤질옥시)테트라졸로[1,5-α]퀴놀린-5-올 100 g을 얻었다.
파트 G
아세트산 500 mL 중 7-(벤질옥시)테트라졸로[1,5-α]퀴놀린-5-올 (99.7 g, 0.341 mol)의 슬러리를 제조하고, 여기에 질산 (70%, 28.2 mL, 0.443 mol)을 첨가하였다. 혼합물을 100 ℃에서 30분 동안 가열하였다. 그 후, 반응을 15 ℃로 냉각시키고, 여과하였다. 고체를 2일 동안 공기 흐름하에 건조시켜 밝은 황갈색 고체로서 7-벤질옥시-4-니트로테트라졸로[1,5-α]퀴놀린-5-올 81 g을 얻었다.
파트 H
DMF (50 mL)를 0 ℃로 냉각시키고, 인 옥시클로라이드 (10.92 g, 71.2 mmol)를 적가하였다. 냉각조를 제거하고, 30분 동안 교반한 후, 생성된 복숭아색 용액을 DMF (15 mL) 중 7-벤질옥시-4-니트로테트라졸로[1,5-α]퀴놀린-5-올 (20 g, 59. 3 mmol)의 슬러리에 적가하였다. 반응을 출발 물질이 소모될 때까지 교반한 후, 얼음 2 L에 부었다. 생성된 황색-갈색 침전물을 여과하고, 물로 세정한 후, 디클로로메탄에 슬러리화하였다. 이 슬러리에 이소부틸아민 (21.6 g, 297 mmol)을 첨가하고, 혼합물을 상온에서 밤새 교반하였다. 그 후, 혼합물을 여과하고, 여액을 황산나트륨으로 건조하고, 감압하에 농축하여 어두운 녹색 고체 23 g을 얻었다. 물질을 클로로포름 중 0 내지 4% 메탄올의 구배로 용출하는 실리카 상 크로마토그래피를 이용하여 정제하였다. 아세토니트릴로부터의 최종 재결정화로 황색-갈색 고체로서 7-(벤질옥시)-N-(2-메틸프로필)-4-니트로테트라졸로[1,5-α]퀴놀린-5-아 민 10 g을 얻었다.
파트 I
7-(벤질옥시)-N-(2-메틸프로필)-4-니트로테트라졸로[1,5-α]퀴놀린-5-아민 (10 g, 25.4 mmol)을 탄소 상 5% 백금 (1 g)과 함께 아세토니트릴 200 mL 중에서 수소 50 psi (3.45 x 105 Pa)하에 진탕시켰다. 5시간 후에, 혼합물을 셀라이트 여과제를 통해 여과하였다. 일부 생성물이 여과 케이크의 상부에 고체로서 단리되었다. 이 고체를 수거하고, 가온 DMF에 용해시키고, 새로운 셀라이트 여과제를 통해 다시 여과하였다. 합한 여액을 냉각시키고, 밤새 정치시켰다. 생성된 침전물을 2가지 수확물로 수거하여 갈색 고체로서 7-(벤질옥시)-N5-(2-메틸프로필)테트라졸로[1,5-α]퀴놀린-4,5-디아민 7.6 g을 얻었다.
파트 J
에톡시아세틸 클로라이드 (2.7 g, 22 mmol)를 피리딘 200 mL 중 7-(벤질옥시)-N5-(2-메틸프로필)테트라졸로[1,5-α]퀴놀린-4,5-디아민 (7.6 g, 21 mmol)의 용액에 적가하였다. 용액을 1시간 동안 교반하고, 추가의 에톡시아세틸 클로라이드를 첨가하였다. 용액을 밤새 환류시킨 후, 용매를 제거하였다. 고체 물질을 클로로포름 300 mL에 용해시키고, 1% 수성 탄산나트륨 150 mL와 함께 교반하였다. pH를 10% 수산화나트륨으로 10으로 조정하였다. 유기 분획물을 1% 탄산나트륨 2 부분 및 염수 용액으로 1회 세척하였다. 그 후, 유기 분획물을 황산나트륨으로 건조시키고 농축하여 황갈색 고체로서 8-(벤질옥시)-5-(에톡시메틸)-6-(2-메틸프로 필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린 8.8 g을 얻었다. 소부분을 에탄올로부터 재결정화하여 분석용 샘플 (m.p. 183.5-184.5 ℃)을 얻었다.
파트 K
8-(벤질옥시)-5-(에톡시메틸)-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린 (8 g, 18.6 mmol)을 에탄올 100 mL 및 아세토니트릴 300 mL 중에서 탄소 상 10% 팔라듐 (1 g)과 함께 수소 50 psi (3.45 x 105 Pa)하에 밤새 진탕하였다. 혼합물을 추가의 촉매와 함께 추가의 날 동안 진탕하였다. 혼합물을 메탄올 500 mL로 희석하고, 가열하여 거의 환류시키고, 셀라이트 여과제를 통해 여과하였다. 냉각시키자, 침전물이 형성되었다. 고체 (4.2 g)를 여과에 의해 회수하였다. 여액을 농축하여 건조상태로 만들고, 메탄올에 다시 슬러리화하고, 여과하여 제2 수확물인 고체 (1 g)를 얻었다. 제2 수확물을 메탄올:아세토니트릴로부터 재결정화하여 백색 고체로서 5-(에톡시메틸)-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린-8-올 650 mg을 얻었다.
파트 L
5-(에톡시메틸)-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린-8-올 (260 mg, 0.76 mmol)을 테트라히드로푸란 (THF) 10 mL, 3-피리딜카르빈 올 (87 mg, 0.80 mmol) 및 트리페닐포스핀 (299 mg, 1.14 mmol) 중에서 교반하였다. 디이소프로필 아조디카르복실레이트 (231 mg, 1.14 mmol)를 혼합물에 적가하였다. 30분 후, 추가의 2 방울의 3-피리딜카르빈올을 첨가하고, 반응을 상온에서 밤새 교반하였다. 박층 크로마토그래피에 의한 분석은 소량의 출발 물질이 여전히 남아있음을 나타내었다. 추가량의 시약을 모든 출발 물질이 소모될 때까지 첨가하였다. 반응을 산성 이온 교환 수지를 통해 여과하고, 수지를 메탄올로 세척한 후, 메탄올 중 1 M 암모니아로 세척하였다. 여액을 감압하에 농축하여 회백색 고체로서 5-(에톡시메틸)-6-(2-메틸프로필)-8-(피리딘-3-일메톡시)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린 280 mg을 얻었다.
파트 M
5-(에톡시메틸)-6-(2-메틸프로필)-8-(피리딘-3-일메톡시)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린 (300 mg, 0.69 mmol) 및 트리페닐포스핀 (1 g, 4.17 mmol)을 밀봉된 바이알에서 합하고, 교반하면서 4시간 동안 150 ℃에서 가열하였다. 반응을 상온로 냉각하고, 클로로포름으로 희석하고, 클로로포름 중 메탄올의 5 내지 10% 구배로 용출하는 실리카 상 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 진공하에 농축하여 갈색 고체를 얻었다. 이 고체를 1시간 동안 3 M 수성 HCl 4 mL 중에서 환류시켰다. 고체 수산화칼륨을 첨가하여 pH를 14로 조정하고, 생성된 침전물을 여과하였다. 고체를 감압하에 건조시켜 회백색 고체로서 2-에톡시메틸-1-(2-메틸프로필)-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 204.0-206.0 ℃)을 얻었다.

실시예 109
3-{[(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조산

파트 A
5-(에톡시메틸)-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린-8-올 (4.0 g, 11.75 mmol)을 DMF 100 mL에 용해시키고, 탄산칼륨 (3.3 g, 23.5 mmol)과 함께 30분 동안 교반하였다. 이 혼합물에 메틸 3-(브로모메틸)벤조에이트 (2.8 g, 12.33 mmol)를 첨가하고, 반응을 밤새 교반하였다. 반응을 물 10 부피에 붓고, 이어서 침전물을 여과하고, 디클로로메탄에 용해시키고, 황산나트륨으로 건조시키고, 여과하고, 농축하여 백색 고체로서 메틸 3-{[(5-에톡시메틸-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1,5-α]퀴놀린-8-일)옥시]메틸}벤조에이트 6.1 g을 얻었다.
파트 B
메틸 3-{[(5-에톡시메틸-6-(2-메틸프로필)-6H-이미다조[4,5-c]테트라졸로[1, 5-α]퀴놀린-8-일)옥시]메틸}벤조에이트 (2.6 g, 5.32 mmol)를 실시예 108, 파트 M에 기재된 일반적인 방법을 사용하여 3-{[(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조산으로 환원시켜 백색 고체로서 나트륨염을 얻었다. 고체를 메탄올에 용해시키고, pH를 8로 조정하였다. 생성된 침전물을 여과하고, 회백색 분말로서 건조시켜 3-{[(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조산 800 mg을 얻었다.

실시예 110
3-{(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸)}벤질 알코올

3-{[(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸}벤조산 (800 mg, 1.78 mmol)을 불활성 조건하에 무수 THF 중에서 교반하고, 수소화리튬알루미늄 (250 mg, 6.58 mmol)을 일부분씩 나누어 첨가하였다. 15분 후에, 물 0.25 mL를 첨가한 후, 15% 수성 수산화나트륨 0.25 mL, 그 후 물 0.75 mL를 첨가하였다. 생성된 혼합물을 농축하고, 클로로포름 중 0 내지 10% 메탄올의 구배로 용출하는 실리카 겔 상 플래시 컬럼 크로마토그래피를 사용하여 정제하였다. 순수한 분획물을 농축하고 아세토니트릴로부터 재결정화하여 핑크색 결정으로서 3-{(4-아미노-2-에톡시메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-8-일)옥시]메틸)}벤질 알코올 (m.p. 164.2-167.2 ℃) 360 mg을 얻었다.

실시예 111
1-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

파트 A
7-벤질옥시-4-클로로-3-니트로퀴놀린 (66.4 g, 0.21 mol)을 디클로로메탄 (1.5 L) 및 트리에틸아민 (59 mL, 0.422 mol)의 혼합물에 용해시켰다. 디클로로메탄 (100 mL) 중 1-아미노-2-메틸-2-프로판올 (22.5 g, 0.25 mol)의 용액을 적가하였다. 반응을 상온에서 3시간 동안 교반한 후, 1% 수성 탄산나트륨 용액으로 켄칭시켰다. 침전물이 형성되었고, 이를 여과하여 황색-갈색 고체로서 조 1-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)-2-메틸프로판-2-올 60 g을 얻었다.
MS(APCI) m/z 368 (M + H)+.
파트 B
아세토니트릴 1.6 L에 파트 A로부터의 생성물 60 g을 첨가하였다. 그 후, 이 슬러리에 탄소 상 5% 백금 촉매 11 g을 첨가하고, 혼합물을 14시간 동안 수소 50 psi (3.4 x 105 Pa)하에 진탕하였다. 그 후, 반응 혼합물을 메탄올 2.5 L에 용해시키고, 셀라이트 여과제를 통해 여과하고, 고온의 메탄올 1 L로 세척하였다. 합한 여액을 감압하에 농축하여 갈색 고체로서 1-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-2-메틸프로판-2-올 53.6 g을 얻었다.
파트 C
1-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-2-메틸-프로판-2-올 (25 g, 74.09 mmol)을 피리딘 500 mL에 첨가하였다. 이 혼합물에 톨루엔 20 mL에 용해된 메톡시프로피오닐 클로라이드 (10 g, 81.5 mmol)의 용액을 적가하였다. 산 염화물을 약 절반 첨가한 후, 반응 혼합물이 완전히 균질화되었다. 산 염화물을 첨가한 지 2시간 후에, 반응 혼합물을 가열하여 환류시키고 밤새 유지시켰다. 용매를 딘-스탁 트랩의 사용을 통해 농축하였다. 상온로 냉각시켜 백색 침전물을 얻고, 이를 여과에 의해 수거하였다. 고체를 1시간 동안 1% 고온 수성 탄산나트륨 (75 내지 80 ℃) 1 L 중에 슬러리화하였다. 그 후, 혼합물을 얼음조에서 냉각시키고, 고체 물질을 여과하고, 물로 세정하고, 건조시켜 백색 고체로서 1-[7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (m.p. 170-171.5 ℃ ) 22.7 g을 얻었다.
파트 D
3-클로로퍼옥시벤조산 (mCPBA, 순도 60%, 11.7 g, 40.7 mmol)을 디클로로메탄 (300 mL) 중 1-[7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (15 g, 37.0 mmol)의 용액에 일부분씩 나누어 첨가하였다. 혼합물을 상온에서 밤새 교반하였다. 소량의 출발 물질이 남아있었고, mCPBA 2 g을 첨가하였다. 반응을 45분 동안 교반한 후, 수성 수산화암모늄 150 mL를 첨가하였다. p-톨루엔술폰산 (8.5 g, 44.4 mmol)을 15분에 걸쳐 일부분씩 나누어 첨가하고, 반응을 1시간 동안 교반하였다. 층을 분리하고, 유기 분획물을 1% 수성 탄산나트륨 용액 100 mL로 2회 세척하였다. 유기 분획물을 무수 황산나트륨으로 건조시키고, 여과하고, 감압하에 농축하여 갈색 고체 18 g을 얻었다. 소량의 고체 (1 g)를 아세토니트릴로부터 재결정화하여 회백색 고체로서 1-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (m.p. 185-186.5 ℃) 0.6 g을 얻었다.
실시예 112
4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올

실시예 5에 기재된 일반적인 방법을 사용하여 1-[4-아미노-7-벤질옥시-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (14.0 g)을 회백색 고체로서 4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올 7 g으로 전환시켰다. 소량의 고체를 메탄올로부터 재결정화하여 분석용 샘플을 제공하였다. mp 258.0-260.0 ℃.
실시예 113
1-[4-아미노-2-(2-메톡시에틸)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

탄산칼륨 (2.1 g, 15.0 mmol)을 DMF (50 mL) 중 4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올 (2.5 g, 7.5 mmol)의 용액에 첨가하였다. 15분 동안 교반한 후, 3-니트로벤질 브로마이드 (1.7 g, 8.3 mmol)를 첨가하였다. 반응을 상온에서 밤새 교반한 후, 물 600 mL에 부었다. 밝은 황색 침전물이 형성되었다. 추가의 탄산칼륨 20 g을 첨가하고, 혼합물을 30분 동안 교반하였다. 침전물을 여과하고, 메탄올 700 mL에 용해시키고, 감압하에 농축하여 황색 고체 3.3 g을 얻었다. 고체를 메탄올로부터 재결정화하여 황색 침상 결정체로서 1-[4-아미노-2-(2-메톡시에틸)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (m.p. 125.0-127.0 ℃) 175 mg을 얻었다.

실시예 114
1-[4-아미노-7-(3-아미노벤질옥시)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

1-[4-아미노-2-(2-메톡시에틸)-7-(3-니트로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (3.1 g, 6.66 mmol)을 가온 에탄올 100 mL 및 탄소 상 5% 백금 1 g과 혼합하였다. 혼합물을 수소압 (50 psi, 3.4 x 105 pa)하에 5시간 동안 진탕하였다. 용액으로부터 물질이 석출되어서 추가의 메탄올 100 mL 및 백금 촉매 0.5 g을 첨가하였다. 수소압하에 밤새 진탕한 후, 혼합물을 셀라이트 여과제를 통해 여과하였다. 여과 케이크를 고온의 메탄올 3 x 75 mL로 세척하고, 여액을 합하고, 감압하에 농축하였다. 오렌지색 발포체가 생성되었고, 이를 실리카 겔을 이용하여 컬럼 크로마토그래피에 의해 정제하였다. 용출액의 극성 성분은 클로로포름:메탄올:2% 수성 수산화암모늄 80:18:2 (CMA)이었고, 용출는 CMA:클로로포름으로 0:100 내지 40:60의 구배로 수행하였다. 생성물을 함유하는 분획물을 합하고, 농축하여 회백색 고체로서 1-[4-아미노-7-(3-아미노벤질옥시)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 1.55 g을 얻었다. 소부분을 아세토니트릴로부터 재결정화하여 분석용 샘플을 얻었다. mp 203.0-206.0 ℃.

실시예 115
1-[4-아미노-2-(2-메톡시에틸)-7-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올 (2.5 g, 7.5 mmol)을 THF 75 mL에 슬러리화하고, 여기에 3-피리딜카르빈올 (350 mg, 3.18 mmol) 및 트리페닐포스핀 (1.59 g, 6.06 mmol)을 첨가하였다. 여기에 디이소프로필 아조디카르복실레이트 (DIAD) 1.2 g (5.93 mmol)을 첨가 하고, 반응 혼합물을 45분 동안 교반하였다. 약 3 당량의 추가량의 각 시약을 첨가하여 반응을 완결시켰다. 그 후, 반응 혼합물을 농축하고, 클로로포름 및 수성 탄산나트륨에 분배시켰다. 유기 분획물을 건조시키고, 용출액으로서 메탄올과 함께 이온 교환 수지를 통해 통과시킨 후, 용출액으로서 메탄올 중 1 M 암모니아와 함께 이온 교환 수지를 통해 통과시켰다. 용매를 농축하고, 잔류물을 클로로포름 중 0 내지 40% CMA의 구배를 이용하여 실리카 겔 상 플래시 컬럼 크로마토그래피에 의해 정제하였다. 목적 분획물을 농축하여 오일을 생성하고, 이를 환류하는 아세토니트릴 20 mL에 용해시켰다. 냉각시키자, 백색 결정이 형성되었고, 이를 여과하고, 건조시켜 1-[4-아미노-2-(2-메톡시에틸)-7-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (m.p. 200.0-202.0 ℃) 0.55 g을 얻었다.

실시예 116
7-(푸란-3-일메톡시)-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
7-벤질옥시-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (16.11 g, 46.6 mmol), 탄소 상 10% 팔라듐 (4.96 g) 및 에탄올 (380 mL)을 조합하였다. 혼합물을 수소 50 psi (3.4 x 105 Pa) 하에 밤새 진탕한 후, 여과하였다. 고체를 에틸 아세테이트로 세척하고, 여액을 농축하였다. 디에틸 에테르를 잔류물에 첨가하고, 녹색 고체가 형성되었다. 고체를 아세토니트릴로부터 재결정화하여 베이지색 고체로서 2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 6.63 g을 얻었다.
파트 B
2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (300 mg, 1.17 mmol), 트리페닐포스핀 (921 mg, 3.51 mmol) 및 3-푸란메탄올 (173 mg, 1.76 mmol)을 무수 THF 20 mL에 슬러리화하였다. 5분 후에 디이소프로필 아조디카르복실레이트 (710 mg, 3.51 mmol)를 혼합물에 적가하였다. 생성된 균질 용액을 6일 동안 교반하였다. HPLC에 의한 반응의 분석은 대부분의 출발 물질이 존재함을 지시하였다. 추가량의 트리페닐포스핀 (150 mg), 3-푸란메탄올 (0.5 mL)을 첨가한 후, 디이소프로필 아조디카르복실레이트 (1 mL)를 첨가하였다. 1시간 후에 반응의 분석은 모든 2-메틸-1-(2-메틸프로필)-1H-이미다조-[4,5-c]퀴놀린-7-올이 소모되었음을 지시하였다. 반응을 메탄올 그 후, 메탄올 중 1 M 암모니아와 함께 산성 이온 교환 수지를 통해 여과하였다. 여액을 농축하여 어두운 오렌지색 오일로서 7-(푸란-3-일메톡시)-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 500 mg을 얻었다.
파트 C
7-(푸란-3-일메톡시)-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린 (500 mg)을 디클로로메탄 30 mL에 용해시키고, 3-클로로퍼옥시벤조산 (순도 60%, 403 mg)을 첨가하였다. 반응을 30분 동안 상온에서 교반한 후, 수성 수산화암모늄 30 mL를 첨가하였다. 10분 후, p-톨루엔술포닐 클로라이드 (268 mg, 1.40 mmol)를 첨가하고, 반응을 추가의 15분 동안 교반하였다. 층을 분리하고, 수성 분획물을 디클로로메탄 (20 mL)으로 2회 세척하였다. 유기 분획물을 합하고, 수성 탄산나트륨 20 mL로 3회 세척하였다. 합한 탄산나트륨 분획물을 클로로포름 (20 mL)으로 4회 추출하였다. 모든 유기 분획물을 합하고, 황산나트륨으로 건조시키고, 농축하여 오렌지색 발포체를 생성하였다. 조 생성물을 클로로포름 중 메탄올의 구배로 용출하는 실리카 겔 상 플래시 컬럼 크로마토그래피를 사용하여 정제하였다. 순수한 분획물을 합하고, 농축하고, 생성된 고체를 아세토니트릴로부터 재결정화하여 백색 분말로서 7-(푸란-3-일메톡시)-2-메틸-1-(2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-4-아민 (m.p. 181.0-183.0 ℃) 102 mg을 얻었다.

실시예 117
3-{[(4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-일)옥시]메틸}벤조니트릴

4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올 (3.5 g, 10.6 mmol), 탄산칼륨 (2.9 g, 21.2 mmol) 및 DMF (100 mL)를 조합하였다. 10분 후에, α-브로모-m-톨루니트릴 (2.25 g, 11.6 mmol)을 첨가하고, 반응을 밤새 교반하였다. 혼합물을 물 1.5 L에 붓고, 백색 침전물이 형성되었다. 물을 경사분리하고, 잔류물을 클로로포름 350 mL에 용해시켰다. 용액을 물 (100 mL)로 2회 세척하였다. 유기 분획물을 건조시키고, 농축하여 담황색 발포체 4.8 g을 얻었다. 실리카 겔 상 플래시 컬럼 크로마토그래피 (클로로포름 중 0 내지 20% CMA의 구배로 용출)에 의해 정제한 후, 아세토니트릴로부터 재결정화하여 담황색 결정으로서 3-{[(4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-일)옥시]메틸}벤조니트릴 (m.p. 195.7-197.2 ℃) 550 mg을 얻었다.

실시예 118
N-{3-[(4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-일옥시)메틸]페닐}메탄술폰아미드

1-[4-아미노-7-(3-아미노벤질옥시)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (850 mg, 1.95 mmol) 및 트리에틸 아민 (600 mg, 5.85 mmol)을 1:1 아세토니트릴:디클로로메탄 100 mL에 용해시켰다. 용액을 얼음조에서 냉각시키고, 메탄술폰산 무수물 (375 mg, 2.15 mmol)을 소부분으로 첨가하였다. 4시간 후에, 제2 등몰량의 메탄술폰산 무수물을 첨가하고, 반응을 밤새 교반하였다. 용매를 감압하에 제거하였다. 생성된 갈색 오일을 디클로로메탄에 용해시키고, 1% 수성 탄산나트륨으로 세척하고, 건조시키고, 감압하에 농축하였다. 생성된 고체를 실리카 겔 상 플래시 컬럼 크로마토그래피 (클로로포름 중 0 내지 20% CMA의 구배로 용출)에 의해 정제한 후, 아세토니트릴로부터 재결정화하여 백색 결정으로서 N-{3-[(4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-일옥시)메틸]페닐}메탄술폰아미드 439 mg을 얻었다.

실시예 119 내지 121
파트 A
(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸 프로판-2-올을 실시예 5의 일반적인 방법을 사용하여 탈벤질화하였다. 반응이 완결되었을 때, 촉매를 폴리테트라플루오로에틸렌 (PTFE) 막을 통해 여과에 의해 제거하고, 여액을 감압하에 농축하여 황갈색 고체를 얻었다. 고체를 1% 수성 탄산나트륨에 슬러리화하였다. 이것으로 점성 슬러리가 형성되었다. 진한 염산을 첨가하여 pH를 1로 감소시켰다. 균질 용액이 형성되었을 때, 1% 수성 탄산나트륨을 첨가하여 pH를 다시 5로 조정하였다. 고체가 침전되었고, 이를 여과에 의해 수거하였다. 고체를 공기 스트림하에 건조시켜 회백색 고체로서 4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올을 얻었다.
MS(APCI) m/z 331 (M + H)+.
파트 B
일반적인 절차에서의 벤질할라이드를 하기 표의 것으로 대체하여 실시예 7 내지 20에 대해 기재된 일반적인 방법을 따랐다. 각각의 실시예에 대하여, 생성물의 정제 및 특징을 하기 표에 기재하였다.

실시예 119
1-[4-아미노-7-(2-벤질옥시에톡시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올
생성물을 물로부터 침전시켰을 때, 물/DMF 용액을 경사분리하고, 잔류물을 클로로포름에 용해시키고, 황산마그네슘으로 건조시켰다. 용액을 여과하고, 클로로포름 중 0 내지 10% 메탄올의 구배를 사용하여 실리카 겔 크로마토그래피 컬럼을 통해 용출하였다. 순수한 분획물을 농축하고, 고체 물질을 빙냉된 아세토니트릴에 슬러리화하였다. 침전물을 여과에 의해 회수하여 백색 고체로서 1-[4-아미노-7-(2-벤질옥시에톡시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (m.p. 131.0-132.0 ℃)을 얻었다.
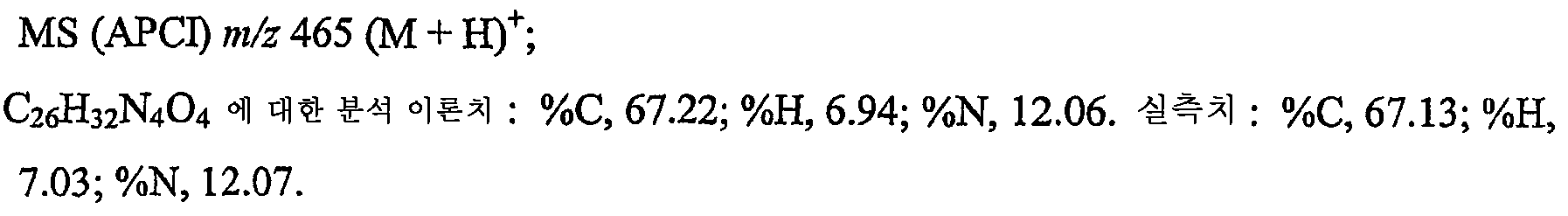
실시예 120
1-{4-아미노-2-에톡시메틸-7-[3-(피롤-1-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올
생성물을 물로부터 침전시켰을 때, 물/DMF 용액을 경사분리하고, 잔류물을 클로로포름에 용해시키고, 황산마그네슘으로 건조시켰다. 용액을 여과하고, 0 내지 10% 클로로포름:메탄올 구배를 사용하여 실리카 겔 크로마토그래피 컬럼을 통해 용출하였다. 순수한 분획물을 농축하고, 고체 물질을 아세토니트릴로부터 재결정화하여 담황색 결정으로서 1-{4-[아미노-2-에톡시메틸-7-[3-(피롤-1-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올 (m.p. 182.5-183.5 ℃)을 얻었다.

실시예 121
1-{4-아미노-7-[2-(벤젠술포닐메틸)벤질옥시]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올
생성물을 물로부터 침전시켰을 때, 물/DMF 용액을 경사분리하고, 잔류물을 클로로포름에 용해시키고, 황산마그네슘으로 건조시켰다. 용액을 여과하고, 클로로포름:메탄올 구배를 사용하여 실리카 겔 크로마토그래피 컬럼을 통해 용출하였다. 순수한 분획물을 농축하고, 황색 오일을 이소프로판올에 용해시키고, 농축하여 황색 발포체로서 1-{4-아미노-7-[2-(벤젠술포닐메틸)벤질옥시]-2-에톡시메틸- 1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올을 얻었다.

실시예 122
1-{4-아미노-2-에톡시메틸-7-[3-(피리딘-3-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올

파트 A
4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (2.35 g, 7.1 mmol), 탄산세슘 (4.6 g, 14.2 mmol) 및 DMF (50 mL)를 배합하였다. 프로파르길 브로마이드 (톨루엔 중 80%, 3.2 g, 21.3 mmol)를 첨가하고, 반응물을 밤새 교반하였다. 반응물을 물 500 mL에 붓고, 고체 탄산칼륨를 첨가하여 혼합물의 염기성을 유지하였다. 침전물이 형성되었다. 반응물의 여과에 이어서 회수된 고체를 공기 건조하여 회백색 분말로서 1-[4-아미노-2-에톡시메틸-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 1.8 g을 제공하였다.
파트 B
1-[4-아미노-2-에톡시메틸-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린 -1-일]-2-메틸프로판-2-올 (1.50 g, 4.07 mmol), 3-요오도피리딘 (920 mg, 4.48 mmol), 무수 트리에틸아민 1.5 mL, PdCl2(PPh3)2 (57 mg, 0.08 mmol), 구리(I) 요오다이드 (30 mg, 0.16 mmol) 및 DMF (50 mL)를 배합하였다. 반응 혼합물을 5일 동안 60℃로 가열하였다. 촉매, 구리 요오다이드 및 요오도피리딘의 추가분을 반응 과정 중에 첨가하였다. 반응물을 물 (500 mL)과 포화 수성 탄산칼륨 (50 mL)의 용액에 부었다. 반응물을 밤새 교반하였으며, 생성 침전물을 여과에 의해 회수하였다. 고체를 디클로로메탄에 용해시키고, 황산마그네슘에서 건조하고, 실리카 겔에서의 플래시 컬럼 크로마토그래피를 사용하여 여과 및 정제하여 (디클로로메탄 중 0-7% 메탄올의 구배로 용출시킴) 오렌지-갈색 반고체로서 1-{4-아미노-2-에톡시메틸-7-[3-(피리딘-3-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올 800 mg를 산출하였다.
파트 C
1-{4-아미노-2-에톡시메틸-7-[3-(피리딘-3-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올 (800 mg, 1.79 mmol)을 가온 에탄올에 용해시켰다. 이것에 탄소상 10% 팔라듐 200 mg을 첨가하고, 혼합물을 수소 50 psi (3.4×105 Pa)하에서 진탕하였다. 24시간 후에, 팔라듐 촉매 200 mg을 추가로 첨가하고, 수소 50 psi하에서 24시간 동안 더 진탕하였다. 촉매를 여과에 의해 제거하고, 여액을 감압 농축하여 고체를 얻었다. 이 물질을 디클로로메탄 중 0-10% 메탄올의 구배로 용출하는 실리카에서의 플래시 컬럼 크로마토그래피를 사용하여 정제하였다. 순수한 분획물들을 합하고 농축하였다. 생성 고체를 아세토니트릴로부터 재결정화화해서 회백색 결정으로서 융점이 160.0-161.0 ℃인 1-{4-아미노-2-에톡시메틸-7-[3-(피리딘-3-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올 160 mg을 제공하였다.

실시예 123
1-{4-아미노-7-[3-(3-아미노페닐)프로폭시]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올

파트 A
1-[4-아미노-2-에톡시메틸-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (0.5 g, 1.35 mmol), 1-요오도-3-니트로피리딘 (565 mg, 2.27 mmol), 무수 트리에틸아민 0.28 mL, PdCl2(PPh3)2 (21 mg, 0.03 mmol), 구리(I) 요오다이드 (12 mg, 0.066 mmol) 및 DMF (15 mL)를 배합하였다. 반응 혼합물을 75℃에서 4시간 동안 가열하였다. 반응물을 포화 수성 NaCl 250 mL에 붓고, 24시간 동안 교반하였다. 생성 갈색 침전물을 디클로로메탄에 용해시키고, 용출액으 로서 디클로로메탄 중 20% 메탄올을 사용하는 실리카 겔의 플러그 (plug)를 통해 여과하였다. 여액을 감압 농축하여 고체로서 1-{4-아미노-2-에톡시메틸-7-[3-(3-니트로페닐)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올을 제공하였다.
파트 B
파트 A로부터의 생성물을 에탄올 (100 mL), 메탄올 (100 mL) 및 탄소상 10% 팔라듐 200 mg과 배합하였다. 혼합물을 수소 50 psi (3.4 ×105 Pa)하에서 밤새 진탕하였다. 추가 팔라듐 촉매를 첨가하고, 반응물을 수소 35 psi (2.4 ×105 Pa)하에서 6시간 동안 추가로 진탕하였다. 촉매를 여과에 의해 제거하고, 여액을 감압 농축하여 고체를 얻었다. 이 물질을 디클로로메탄 중 0-8% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피를 사용하여 정제하였다. 순수 분획물을 합하고, 농축하였다. CMA:디클로로메탄으로 용출하는 실리카겔 상의 플래시 컬럼 크로마토그래피에 의해 회수된 고체를 다시 정제하였다. 아세토니트릴로부터 최종 재결정화하여 회백색 고체로서 융점이 164.0-166.0 ℃인 1-{4-아미노-7-[3-(3-아미노페닐)프로폭시]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일}-2-메틸프로판-2-올 141 mg을 제공하였다.

실시예 124-130
4-아미노-1-(2-메틸프로필)-2-프로필-1H-이미다조[4,5-c]퀴놀린-7-올 대신 N-[2-(4-아미노-2-에톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드로 대체하여 실시예 7-20에 기재된 일반적인 방법을 따랐다. 사용된 할라이드를 하기 표에 나타냈다. 각각의 실시예에 대하여, 생성물의 정제 및 특징이 표 밑에 기재하였다.

실시예 124
N-{2-[4-아미노-2-에톡시메틸-7-(3-메톡시벤질옥시)-lH-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
물로부터 생성물이 침전되어 고체 탄산나트륨 20 g을 침전물/물 혼합물에 첨가하고, 이어서 30분 동안 교반하였다. 침전물을 여과에 의해 회수하고, 고체를 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 생성물을 아세토니트릴로부터 재결정화화하여 백색 고체로서 융점이 221.0-222.0 ℃인 N-{2-[4-아미노-2-에톡시메틸-7-(3-메톡시벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 550 mg를 제공하였다.

실시예 125
N-{2-[4-아미노-7-(2-클로로벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
물로부터 생성물이 침전되어 고체 탄산나트륨 20 g을 침전물/물 혼합물에 첨가하고 이어서 30분 동안 교반하였다. 침전물을 여과에 의해 회수하고, 고체를 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 고온의 메탄올에서 슬러리화된 생성물을 이후 몇 시간 동안 환류하였다. 용액을 농축하여 메탄올의 부피가 보다 작아졌으며, 고체가 침전될 때까지 냉각시켰다. 여과된 고체를 건조하여 백색 고체로서 융점이 253.0-255.0 ℃인 N-{2-[4-아미노-7-(2-클로로벤질옥시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 325 mg을 얻었다.

실시예 126
N-{2-[4-아미노-2-에톡시메틸-7-(4-플루오로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
물로부터 생성물이 침전되어 고체 탄산나트륨 20 g을 침전물/물 혼합물에 첨가하고 이어서 30분 동안 교반하였다. 침전물을 여과하고, 생성물을 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 생성물을 고온의 아세토니트릴에서 슬러리화 하였다. 여과된 고체를 건조하여 백색 고체로서 융점이 251.0-253.0 ℃인 N-{2-[4-아미노-2-에톡시메틸-7-(4-플루오로벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 325 mg을 얻었다.

실시예 127
N-{2-[4-아미노-2-에톡시메틸-7-(3-메틸벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
물로부터 생성물이 침전되어 고체 탄산나트륨 20 g을 침전물/물 혼합물에 첨가하고 이어서 30분 동안 교반하였다. 침전물을 여과에 의해 회수하고, 고체를 1- 5%로 증가하는 메탄올:디클로로메탄의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 생성물을 고온의 아세토니트릴에서 슬러리화 하였다. 여과된 고체를 건조하여 백색 고체로서 융점이 252.0-254.0 ℃인 N-{2-[4-아미노-2-에톡시메틸-7-(3-메틸벤질옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 600 mg을 얻었다.

실시예 128
N-{2-[4-아미노-7-(벤조티아졸-2-일메톡시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
침전물을 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 고체를 아세토니트릴로부터 재결정화하여 백색 고체로서 융점이 258.0-259.0 ℃인 N-{2-[4-아미노-7-(벤즈티아졸-2-일메톡시)-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 300 mg을 얻었다.

실시예 129
N-{2-[4-아미노-2-에톡시메틸-7-(5-트리플루오로메틸푸란-2-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드
침전물을 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔상의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 고체를 아세토니트릴로부터 재결정화하여 백색 고체로서 융점이 186.0-188.0 ℃인 N-{2-[4-아미노-2-에톡시메틸-7-(5-트리플루오로메틸푸란-2-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸-에틸}메탄술폰아미드 200 mg을 얻었다.

실시예 130
에틸 5-[4-아미노-2-에톡시메틸-1-(2-메탄술포닐아미노-2-메틸프로필)-1H- 이미다조[4,5-c]퀴놀린-7-일옥시메틸]푸란-2-카르복실레이트
침전물을 디클로로메탄 중 1-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하고, 고체를 빙냉된 아세토니트릴로부터 슬러리화하였다. 고체를 여과하고 건조하여 백색 고체로서 융점이 122.0-123.0 ℃인 에틸 5-[4-아미노-2-에톡시메틸-1-(2-메탄술포닐아미노-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-일옥시메틸]푸란-2-카르복실레이트 125 mg을 얻었다.

실시예 131
1-(2-메틸프로필)-2-메톡시메틸-8-[3-(피리딘-3-일)프로폭시]-1H-이미다조 [4,5-c]퀴놀린-4-아민

파트 A
6-벤질옥시-N4-(2-메틸프로필)퀴놀린-3,4-디아민을 디클로로메탄 200 mL에 용해시키고, 디클로로메탄 10 mL에 희석된 메톡시아세틸 클로라이드 (1.85 g, 17.1 mmol)를 적가하였다. 1시간 이후에, 용매를 제거하고, 생성 고체를 3:1 메탄올:물 250 mL에 용해시켰다. 수성 탄산칼륨 (6M)을 첨가하고, 반응물을 환류 온도에서 3시간 동안 가열하였다. 용매를 제거하고, 잔류물을 디클로로메탄 200 mL과 물 100 mL 사이에 분배시켰다. 수성 분획물을 디클로로메탄으로 3회 단리 및 추출하였다. 합쳐진 유기 분획물을 건조하고, 여과하고, 감압 농축하였다. 생성 물질을 디클로로메탄 중 0-4% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 농축하여 밝은 갈색 고체로서 8-벤질옥시-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린 4.2 g을 얻었다.
파트 B
3-클로로퍼옥시벤조산 (60% 순도, 3.5 g, 12.3 mmol)을 디클로로메탄 200 mL 중 8-벤질옥시-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린 (4.2 g, 11.19 mmol)의 용액에 나누어 첨가하였다. 반응물을 상온에서 2시간 동안 교반하였으며, 이후 2% 수성 탄산나트륨 100 mL을 첨가하였다. 수성층을 디클로로메탄 100 mL로 2회 추출하였으며, 유기 분획물을 합하고, 건조하고, 농축하여 밝은 갈색 고체로서 8-벤질옥시-1-(2-메틸프로필)-2-메톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린 조 생성물 4.3 g을 얻었다.
파트 C
옥시염화인 (1.78 g, 11.6 mmol)을 DMF 10 mL 중 8-벤질옥시-1-(2-메틸프로필)-2-메톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린의 용액에 적가하였다. 1시간 후에, 반응물을 얼음 700 mL 위에 부었으며, 밤새 교반하였다. 이후, 고체 탄산칼륨을 pH 10이 될 때까지 첨가하였다. 30분 동안 교반한 이후, 혼합물을 여과하였다. 생성 고체를 물로 세척하였으며, 공기 스트림하에서 건조하여 노란 황갈색 고체로서 8-벤질옥시-4-클로로-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린 3.75 g을 얻었다.
파트 D
8-벤질옥시-4-클로로-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린 (2.73 g, 6.65 mmol)을 65 ℃에서 아세트산 중 30% 브롬화수소 25 mL에서 1시간 동안 교반하였다. 반응물을 빙조에서 냉각시키고, 수성 NaOH (50%)를 첨가하여 pH를 7로 조절하였다. 침전물을 여과에 의해 회수하고, 공기 건조하여 갈색 고체로서 4-브로모-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린-8-올 2.75 g을 제공하였다.
파트 E
7N 메탄올성 암모니아 15 mL 중 4-브로모-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린-8-올 (600 mg, 1.64 mmol)을 파르 봄브 장치 (Parr bomb apparatus)에서 9시간 동안 120 ℃로 가열하였다. 36시간 동안 150 ℃로 추가 가열한 이후, 반응물을 냉각하고 농축하여 건조하였다. 4-아미노-1-(2-메틸프로필)-2-메톡시메틸-1H-이미다조[4,5-c]퀴놀린-8-올 조 생성물을 DMF 10 mL에 용해시키고, 탄산세슘 (800 mg, 2.64 mmol)을 첨가하였다. 10분 후에, 프로파르길 브로마이드 (톨루엔 중 80%, 268 mg, 1.80 mmol)를 첨가하고, 반응물을 밤새 교반하였다. 반응물을 이후 물 250 mL에 부었으며, 고체 탄산칼륨을 첨가하여 혼합물 염기성을 유지하였다. 형성된 침전물을 여과하고, 공기 흐름 하에서 건조하여 1-(2-메틸프로필)-2-메톡시메틸-8-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-4-아민 450 mg을 얻었다.
파트 F
DMF 10 mL 중 1-(2-메틸프로필)-2-메톡시메틸-8-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-4-아민 (300 mg, 0.88 mmol), 3-요오도피리딘 (200 mg, 0.98 mmol), 무수 트리에틸아민 (0.3 mL, 2.22 mmol), PdCl2(PPh3)2 (12 mg, 0.01 mmol) 및 구리(Ⅰ) 요오다이드 (6 mg, 0.03 mmol)의 혼합물을 60 ℃로 가열하였다. 4시간 후에, 반응물을 상온으로 냉각시키고 밤새 교반하였다. 반응 혼합물을 물 200 mL 및 포화 탄산칼륨 30 mL에 붓고, 밤새 교반하였다. 생성 블랭크 침전물을 여과하고, 디클로로메탄 중 0-7% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 농축하여 유리와 유사한 고체로서 1-(2-메틸프로필)-2-메톡시메틸-8-[(3-피리딘-3-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-4-아민 160 mg을 얻었다.
파트 G
1-(2-메틸프로필)-2-메톡시메틸-8-[(3-피리딘-3-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-4-아민 (150 mg, 0.36 mmol)을 1:1 에탄올:메탄올의 최소량에 용해시키고, 탄소상 10% 팔라듐 200 mg를 첨가하였다. 혼합물을 수소 50 psi (3.4×105 Pa)하에서 2일 동안 진탕하였다. 여과된 반응물을 농축하여 건조하였다. 고체 물질을 클로로포름 중 0-30% CMA의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고, 농축하고, 아세토니트릴로부터 재결정화하여 회백색 결정으로서 융점이 158.0-159.0 ℃인 1-(2-메틸프로필)-2-메톡시메틸-8-[3-(피리딘-3-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-4-아민 25 mg을 얻었다.

실시예 132
1-[4-아미노-2-에톡시메틸-7-[3-(피라진-2-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

파트 A
1-(4-아미노-2-에톡시메틸-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 (800 mg, 2.17 mmol)을 DMF 40 mL 중 요오도피라진 (492 mg, 2.39 mmol), 무수 트리에틸아민 (0.786 mL, 5.64 mmol), PdCl2(PPh3)2 (30 mg, 0.04 mmol) 및 구리(Ⅰ) 요오다이드 (17 mg, 0.09 mmol)와 배합하였다. 혼합물을 60 ℃로 밤새 가열하였다. 추가 시약을 첨가하였으며, 5일 후에 반응물을 상온으로 냉각시키고 밤새 교반하였다. 이후 매우 우유빛 갈색 침전물이 형성될 때까지 반응 혼합물을 50% 수성 탄산칼륨 400 mL에 부었다. 수용액을 경사분리하고, 고체를 디클로로메탄에 용해시키고, 건조하고, 여과하였다. 여액을 디클로로메탄 중 0-8% 메탄올의 구배로 용출하는 실리카 겔 크로마토그래피를 사용하여 정제하였다. 정제된 분획물을 농축하여 백색 고체로서 1-[4-아미노-2-에톡시메틸-7- [3-(피라진-2-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올을 얻었다.
파트 B
1-[4-아미노-2-에톡시메틸-7-[3-(피라진-2-일)프로프-2-이닐옥시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (400 mg, 0.89 mmol)을 1:1 메탄올:에탄올의 최소량에 용해시키고, 18시간 동안 수소 45 psi (3.1 × 105 Pa)하에서 탄소상 10% 팔라듐 (400 mg)과 진탕하였다. 혼합물을 PTFE 필터를 통해 여과하였으며 여액을 감압 농축하였다. 생성된 암색 오일를 디클로로메탄 중 0-6% 메탄올의 구배로 용출하는 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 농축하였다. 고체를 아세토니트릴로부터 재결정화하여 백색 고체로서 융점이 166.0-167.0 ℃인 1-[4-아미노-2-에톡시메틸-7-[3-(피라진-2-일)프로폭시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 (57 mg)을 제공하였다.
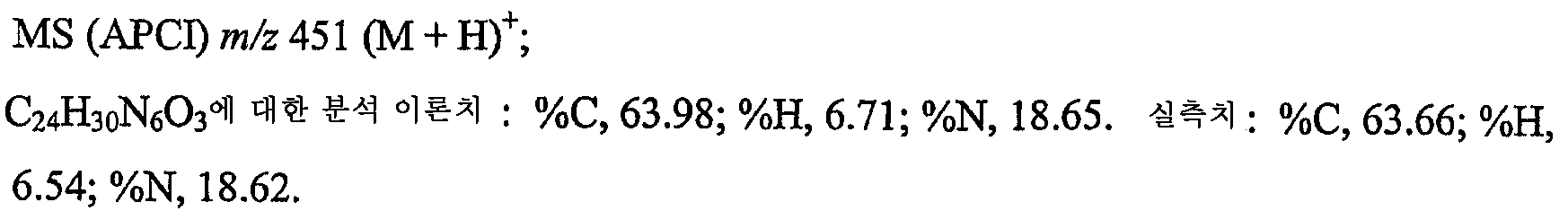
실시예 133
2-에톡시메틸-1-프로필-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
실시예 5에 기재된 방법에 따라서, 실시예 150의 파트 A-F에서 제조된 8-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린을 처리하여 2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-8-올을 제조하였다.
2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-8-올 (680 mg, 2.38 mmol)을 디클로로메탄에 용해하였으며, mCPBA (60%, 750 mg, 2.62 mmol)을 첨가하였다. 반응을 TLC에 의해 모니터링하였으며, 출발 물질의 모두가 소모되었다고 결정되었을 때 2% 수성 탄산나트륨 20 mL을 첨가하였다. 유기층을 2% 탄산나트륨으로 추가로 2회 추출하였다. 수성 분획물을 합하고 진한 염산을 pH 1이 될 때까지 적가하였다. 이후 2% 탄산나트륨을 나누어 첨가하여 pH를 5로 조정하였다. 형성된 침전물을 여과에 의해 수집하여 회백색 분말로서 2-에톡시메틸-5-옥시도-1-프로필-1H-이미다조[4,5-c]퀴놀린-8-올 조 생성물을 얻었다. 고체를 무수 THF 50 mL에서 트리페닐포스핀 (1.2 g, 4.76 mmol) 및 피리딘-3-메탄올 (390 mg, 3.57 mmol)과 배합하였다. 디이소프로필 아조디카르복실레이트 (1.2 g, 5.95 mmol)를 적가하였다. 4시간 이후, 트리페닐포스핀 및 피리딘-3-메탄올의 추가분에 이어서 디이소프로필디카르복실레이트를 첨가하였으며, 반응물을 1시간 동안 교반하였다. 물 (1 mL)을 첨가하고 반응물을 밤새 교반하였다. 반응 혼합물을 감압 농축하였으며, 생성 고체를 디클로로메탄에 용해시키고, 2% 수성 탄산나트륨 50 mL 양으로 3회 세척하였다. 수성 분획물을 합하고, 클로로포름으로 추출하였다. 유기 분획물을 합하고, 황산마그네슘에서 건조하고, 여과하고, 농축하여 2-에톡시메틸-5-옥시도-1-프로필-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린 조 생성물을 얻었다.
파트 B
파트 A로부터의 2-에톡시메틸-5-옥시도-1-프로필-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린 조 생성물을 디클로로메탄 50 mL에 용해시키고, 트리클로로아세틸 이소시아네이트 (540 mg, 2.85 mmol)를 적가하였다. 반응을 이후 박층크로마토그래피에 의해 모니터링하였으며, 출발 물질이 소비되었을 때 진한 수산화암모늄 2 mL를 첨가하였다. 반응물을 30분 동안 교반하고 이후 양이온 교환 수지를 함유하는 컬럼에 통과시켰다. 컬럼을 메탄올로 세척하여 불순물을 제거하고, 이어서 메탄올 중 2M 암모니아로 컬럼으로부터 목적 생성물을 용출시켰다. 휘발성 물질을 감압하에 제거하였다. 생성 잔류물을 디클로로메탄에 용해시키고, HORIZON HPFC 시스템에서 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 디클로로메탄 중 0-8% 메탄올의 구배로 용출하였다. 고체를 아세토니트릴로부터의 재결정화에 의해 더 정제하여 밝은 호박색 결정으로서 융점이 190.0-191.0 ℃인 2-에톡시메틸-1-프로필-8-(피리딘-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-4-아민을 얻었다.
실시예 134
1-[4-아미노-2-(2-메톡시에틸)-7-[2-(피롤-1-일)에톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

탄산세슘 2 당량 및 1-(2-브로모에틸)피롤 1.1 당량을 사용하여, 4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올을 실시예 7-20을 위한 일반 절차에 기재된 바와 같이 알킬화하였다. 반응물을 50℃에서 밤새 가열하였으며 이후 추가로 4시간 동안 80℃로 가열하였다. 반응물을 냉각시키고 염화나트륨 20 g을 함유하는 물에 부었다. 생성 침전물을 디클로로메탄 중 0-8% 메탄올의 구배로 용출하는 컬럼 크로마토그래피를 사용하여 여과하고 정제하였다. 순수한 분획물을 합하고, 농축하고, 아세토니트릴로부터 재결정화하여 회백색 고체로서 융점이 168.0-169.0 ℃인 1-[4-아미노-2-(2-메톡시에틸)-7-[2-(피롤-1-일)에톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 160 mg을 얻었다.

실시예 135
1-(4-아미노-2-에톡시메틸-7-[2-(1H-인돌-3-일)에톡시]-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (500 mg, 1.51 mmol), 탄산세슘 (980 mg, 3.02 mmol), 3-(2-브로모에틸)인돌 (375 mg, 1.66 mmol) 및 DMF (10 mL)를 배합하고, 상온에서 밤새 교반하고, 이어서 80 ℃에서 4시간 동안 가열하였다. 반응 혼합물을 상온으로 냉각시키고, 염화나트륨 20 g을 함유하는 물 200 mL에 부었다. 생성 침전물을 여과하고, 디클로로메탄에 용해시키고, 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 디클로로메탄 중 0-8% 메탄올의 구배로 용출시켜 정제하였다. 순수한 분획물을 합하고 농축하였다. 고체를 고온의 아세토니트릴에서 슬러리화하고, 여과하고, 감압 건조하여 회백색 고체로서 융점이 220.0-222.0 ℃인 1-(4-아미노-2-에톡시메틸-7-[2-(1H-인돌-3-일)에톡시]-1H-이미다조[4,5-c]퀴놀린-1-일)-2-메틸프로판-2-올 305 mg을 산출하였다.

실시예 136
1-[4-아미노-2-(2-메톡시에틸)-7-(5-메틸이속사졸-3-일메톡시)-1H-이미다조 [4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

탄산세슘 2 당량 및 3-(브로모메틸)-5-메틸이속사졸 1.1 당량을 사용하여, 4-아미노-1-(2-히드록시-2-메틸프로필)-2-(2-메톡시에틸)-1H-이미다조[4,5-c]퀴놀린-7-올을 실시예 7-20을 위한 일반 절차에 기재된 바와 같이 알킬화하였다. 실온에서 밤새 교반한 이후, 반응 혼합물을 염화나트륨 20 g을 함유한 물 200 mL에 부었다. 생성 침전물을 여과하고, 디클로로메탄에 용해시켰으며, 디클로로메탄 중 0-8% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 농축하였다. 생성 고체를 냉 아세토니트릴에서 슬러리화하고, 여과하고, 감압 건조하여 회백색 고체로서 융점이 169.0-170.0 ℃인 1-[4-아미노-2-(2-메톡시에틸)-7-(5-메틸이속사졸-3-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2메틸프로판-2-올 315 mg을 산출하였다.

실시예 137
1-[4-아미노-2-에톡시메틸-7-(티아졸-4-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (100 mg, 0.3 mmol), 탄산세슘 (488 mg, 1.5 mmol), 4-(클로로메틸)티아졸·HCl (102 mg, 0.6 mmol), 테트라부틸암모늄 브로마이드 (96 mg, 0.3 mmol), 트리에틸아민 (0.5 ml) 및 DMF (10 mL)를 배합하고 상온에서 밤새 교반하였다. 반응물을 감압 농축하였으며, 디클로로메탄을 잔류물에 첨가하였다. 용해되지 않은 고체를 여과에 의해 제거하였다. 여액을 감압 농축하고, 후속적으로 디클로로메탄 중 0-4%의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 감압 농축하였다. 생성 고체를 아세토니트릴로부터 재결정화하여 백색 분말로서 융점이 190.0-191.0 ℃인 1-[4-아미노-2-에톡시메틸-7-(티아졸-4-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 62 mg을 수득하였다.

실시예 138
1-[4-아미노-2-에톡시메틸-7-(2-메틸티아졸-4-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (100 mg, 0.3 mmol), 탄산세슘 (488 mg, 1.5 mmol), 4-(클로로메틸)-2-메틸티아졸·HCl (110 mg, 0.6 mmol), 테트라부틸암모늄 브로마이드 (96 mg, 0.3 mmol), 트리에틸아민 (0.5 ml) 및 DMF (10 mL)를 배합하고, 상온에서 밤새 교반하였다. 반응물을 감압 농축하고, 잔류물에 디클로로메탄을 첨가하였다. 용해되지 않은 고체를 여과에 의해 제거하였다. 여액을 감압 농축하고, 후속적으로 디클로로메탄 중 0-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 감압 농축하였다. 생성 고체를 아세토니트릴로부터 재결정화하여 백색 분말로서 융점이 200.0-201.0 ℃인 1-[4-아미노-2-에톡시메틸-7-(2-메틸티아졸-4-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 77 mg을 수득하였다.

실시예 139
1-[4-아미노-2-에톡시메틸-7-[2-(티오펜-2-일)티아졸-4-일메톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (100 mg, 0.3 mmol), 탄산세슘 (195 mg, 0.6 mmol), 4-(클로로메틸)-2-(2-티에닐)-1,3-티아졸 (71 mg, 0.33 mmol), 테트라부틸암모늄 브로마이드 (10 mg, 0.03 mmol), 트리에틸아민 (0.5 ml) 및 아세토니트릴 (10 mL)을 배합하고 상온에서 밤새 교반하였다. 반응물을 감압 농축하고, 디클로로메탄을 잔류물에 첨가하였다. 용해되지 않은 고체를 여과하여 제거하였다. 여액을 감압 농축하고 후속적으로 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 디클로로메탄 중 0-5% 메탄올의 구배로 용출시켜 정제하였다. 순수한 분획물을 합하고, 감압 농축하였다. 고체를 아세토니트릴로부터 재결정화하여 회백색 분말로서 융점이 192.0-194.0 ℃인 1-[4-아미노-2-에톡시메틸-7-[2-(티오펜-2-일)티아졸-4-일메톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 82 mg을 수득하였다.

실시예 140
1-[4-아미노-2-에톡시메틸-7-[3-(티오펜-2-일)-[1,2,4]옥사디아졸-5-일메톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸-프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (100 mg, 0.3 mmol), 탄산세슘 (195 mg, 0.6 mmol), 5-(클로로메틸)-3-(2-티에닐)-1,2,4-옥사디아졸 (71 mg, 0.33 mmol), 테트라부틸암모늄 브로마이드 (10 mg, 0.03 mmol), 트리에틸아민 (0.5 ml) 및 아세토니트릴 (10 mL)을 배합하고, 상온에서 밤새 교반하였다. 반응물을 감압 농축하고, 디클로로메탄을 잔류물에 첨가하였다. 용해되지 않은 고체를 여과하여 제거하였다. 여액을 감압 농축하고, 후속적으로 디클로로메탄 중 0-5% 메탄올의 구배로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 순수한 분획물을 합하고 감압 농축하였다. 아세토니트릴로부터 고체를 재결정화하여 백색 고체로서 융점이 173.0-175.0 ℃인 1-[4-아미노-2-에톡시메틸-7-[3-(티오펜-2-일)-[1,2,4]옥사디아졸-5-일메톡시]-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 71 mg을 수득하였다.

실시예 141
1-[4-아미노-2-에톡시메틸-7-(1-메틸-1H-이미다졸-2-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올

4-아미노-2-에톡시메틸-1-(2-히드록시-2-메틸프로필)-1H-이미다조[4,5-c]퀴놀린-7-올 (100 mg, 0.3 mmol), 탄산세슘 (488 mg, 1.5 mmol), 2-클로로메틸-1-메틸-1H-이미다졸·HCl (100 mg, 0.6 mmol), 테트라부틸암모늄 브로마이드 (96 mg, 0.3 mmol), 트리에틸아민 (0.5 ml) 및 DMF (10 mL)를 배합하고, 상온에서 밤새 교반하였다. 반응물을 감압 농축하고, 디클로로메탄을 잔류물에 첨가하였다. 용해되지 않은 고체를 여과에 의해 제거하였다. 여액을 감압 농축하고, 후속적으로 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 디클로로메탄 중 0-5% 메탄올의 구배로 용출시켜 정제하였다. 순수한 분획물을 합하고 감압 농축하였다. 생성 고체를 이소프로판올/디에틸에테르로부터 재결정화하여 회백색 고체로서 융점이 200.0-202.0 ℃인 1-[4-아미노-2-에톡시메틸-7-(1-메틸-1H-이미다졸-2-일메톡시)-1H-이미다조[4,5-c]퀴놀린-1-일]-2-메틸프로판-2-올 40 mg을 산출하였다.

실시예 142
N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드

파트 A
(2-아미노-2-메틸프로필)-(7-벤질옥시-3-니트로퀴놀린-4-일)아민 (5.29 g, 14.44 mmol)을 THF 100 mL 중 교반하였으며, 수산화나트륨 (물 50 mL 중 0.64 g)을 첨가하였다. 이후, THF 50 mL 중 디-tert-부틸 디카르보네이트 3.82 g을 적가하였으며, 반응물을 상온에서 24시간 동안 교반하였다. TLC에 의한 반응의 분석은 출발 물질이 여전히 존재함을 나타내었다. THF 20 mL 중 디-tert-부틸 디카르보네이트 0.55 g을 추가로 첨가하고, 반응물을 추가로 24시간 동안 교반하였다. 감압 하에서 THF를 제거하였으며, 디클로로메탄을 첨가하였다. 유기 분획물을 수성 분획물로부터 분리하였다. 유기 분획물을 이후 순차적으로 물로, 이어서 염수로 세척하고, 황산마그네슘에서 건조하고, 감압 농축하여 황색 발포체로서 tert-부틸 [2-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 7.04 g을 산출하였다.
파트 B
tert-부틸 [2-(7-벤질옥시-3-니트로퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 (6.74 g), 탄소상 5% 백금 (1.02 g) 및 아세토니트릴 (125 mL)을 배합하였다. 혼합물을 수소 50 psi (3.4×105 Pa)하에서 밤새 진탕하였다. 혼합물을 셀 라이트 여과제를 통해 여과하였으며 여액을 감압 농축하여 오렌지색 발포체로서 tert-부틸 [2-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 6.04 g을 얻었다.
파트 C
에톡시아세틸 클로라이드 (1.82 mL, 16.60 mmol)를 무수 디클로로메탄 150 mL 중 tert-부틸 [2-(3-아미노-7-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 (6.04 g, 13.8 mmol) 및 트리에틸아민 (3.86 mL, 27.68 mmol)의 냉각 (0 ℃) 용액에 첨가하였다. 30분 동안 교반한 이후, 냉조를 제거하고, 반응물을 상온에서 24시간 동안 교반하였다. 휘발성 물질을 감압하에서 제거하고, 생성 잔류물을 에탄올에 용해시키고, 이어서 추가로 트리에틸아민 3.86 mL을 용해시켰다. 반응물을 환류상태에서 2.5일 동안 가열하고, 이후 상온으로 냉각하였다. 용매를 감압하에서 제거하고, 디클로로메탄을 첨가하였다. 용액을 수성 나트륨 비카르보네이트로, 이어서 염수로 순차적으로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축시켰다. 잔류물을 실리카 겔에서의 플래시 컬럼 크로마토그래피를 사용하여 디클로로메탄 중 5% 메탄올로 용출시켜 정제하였다. 순수한 분획물은 농축한 한편, 생성물 및 출발 물질 모두가 함유된 분획물은 반응 조건으로 되돌아가서 상기 기재된 바와 같이 재정제하였다. tert-부틸 [2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]카르바메이트 총 5.07 g의 배합 로트를 오렌지색 발포체로서 얻었다.
파트 D
에탄올 (3M, 75 mL) 중 HCl을 tert-부틸 [2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]카르바메이트 (5.07 g, 10.04 mmol)에 첨가하고, 상기 용액을 환류 상태에서 15분 동안 가열하였다. 휘발성 물질을 감압하에서 제거하고, 오렌지색 잔류물을 묽은 수성 염산 및 디틀로로메탄 사이에 분배하였다. 수성 층을 디클로로메탄으로 2회 세척하였으며, 이후 추가 수성 수산화암모늄으로 염기성으로 제조하였다. 이후 수성 분획물을 디클로로메탄으로 3회 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 농축하여 갈색 오일로서 2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 3.77 g을 산출하였다.
파트 E
2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 (1.88 g, 4.65 mmol), 트리에틸아민 (1.3 mL, 9.3 mmol) 및 디클로로메탄 (50 mL)을 배합하고, 0 ℃로 냉각하였다. 아세틸 클로라이드 (0.36 mL, 5.11 mmol)를 첨가하고, 반응물을 30분 동안 교반하였다. 냉조를 제거하고, 반응물을 추가로 3.5시간 동안 교반하였다. 물을 첨가하고, 층을 분리하였다. 순차적으로, 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 황갈색 고체를 제공하였다. 고체를 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하여 (디클로로메탄 중 6-7.5% 메탄올의 구배로 용출시킴) 크림색 고체로서 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 1.71 g을 산출하였다.
파트 F
mCPBA (60% 순도, 1.70 g)을 클로로포름 (100 mL) 중 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 (1.71 g, 3.83 mmol) 용액에 첨가하였다. 반응물을 4시간 동안 교반하고, 이후 추가로 mCPBA 0.57 g을 첨가하였다. 반응물을 2시간 더 교반하고, 이어서 수성 1% 탄산나트륨을 첨가하여 켄칭하였다. 층을 분리하고, 수성 분획물을 클로로포름으로 5회 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 농축하여 오렌지색 발포체로서 N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 1.78 g을 얻었다.
파트 G
N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 (1.77 g, 3.83 mmol)를 디클로로메탄 (100 mL)에 용해시키고, 수성 수산화암모늄 (10 mL), 이어서 파라-톨루엔술포닐 클로라이드 (0.73 g, 3.83 mmol)를 첨가하였다. 5시간 동안 교반한 이후, 유기상 및 수성상을 분리하고, 유기 분획물을 포화 나트륨 비카르보네이트로 2회, 이후 염수로 1회 세척하였다. 이후 유기 분획물을 황산나트륨에서 건조하고, 여과하고, 농축하여 어두운 오렌지색 고체를 얻었다. 상기 고체를 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하고 (디클로로메탄 중 6-7.5% 메탄올의 구배로 용출시킴), 이어서 아세토니트릴로부터 재결정화하여 백색 분말로서 융점이 202.0-205.0 ℃인 N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 1.27 g을 제공하였다.

실시예 143
N-[2-(4-아미노-2-에톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드

N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 (0.72 g, 1.56 mmol)를 가온 에탄올 200 mL에 용해시켰다. 탄소상 10% 팔라듐 (0.33 g)을 첨가하고, 혼합물을 수소 50 psi (3.4×105 Pa)하에서 밤새 진탕하였다. 반응물을 셀라이트 여과제를 통해 여과하였으며, 여액을 감압하에서 제거하였다. 생성 갈색 고체를 디클로로메탄 중 14% 메탄올로 용출하는 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하였다. 목적 생성물을 함유하는 분획물을 합하고, HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 더 정제하였다. 실리카 겔 카트리지를 클로로포름 중 30-50% CMA의 구배가 되는 용출액과 사용하였다. 순수한 분획물을 합하고, 감압 농축하여 회백색 고체로서 융점이 140℃인 N-[2-(4-아미노-2-에톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]아세트아미드 0.23 g을 제공하였다.

실시예 144
N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아

파트 A
2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 (1.88 g, 4.65 mmol) 및 디클로로메탄 (50 mL)을 배합하고, 0℃로 냉각하였다. 이소프로필 이소시아네이트 (0.50 mL, 5.11 mmol)를 첨가하고, 반응물을 30분 동안 교반하였다. 냉조를 제거하고, 반응물을 밤새 교반하였다. 휘발성 물질을 감압하에서 제거하여 갈색 고체를 얻었다. 고체를 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하여 (디클로로메탄 중 6% 메탄올로 용출시킴) 크림색 고체로서 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아 1.96 g을 산출하였다.
파트 B
mCPBA (60% 순도, 1.18 g)를 클로로포름 (50 mL) 중 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아 (1.96 g, 4.0 mmol) 용액에 첨가하였다. 반응물을 3시간 동안 교반하고, 이후 mCPBA 0.53 g을 추가로 첨가하였다. 반응물을 2시간 더 교반하고, 이후 수성 1% 탄산나트륨으로 켄칭하였다. 층을 분리하고, 수성 분획물을 디클로로메탄으로 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 오렌지색 발포체로서 N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아 조 생성물 2.2 g을 산출하였다.
파트 C
N-[2-(7-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N-이소프로필우레아 (2.02 g, 4.0 mmol)를 디클로로메탄 (50 mL)에 용해하고, 수성 수산화암모늄 (10 mL), 이어서 파라-톨루엔술포닐클로라이드 (0.76 g, 4.0 mmol)를 첨가하였다. 4시간 동안 교반한 이후, 유기상 및 수성상을 분리하고, 유기 분획물을 포화 나트륨 비카르보네이트로 2회, 이후 염수로 1회 세척하였다. 이후 유기 분획물을 황산나트륨에서 건조하고, 여과하고, 농축하여 오렌지색 고체 2.03 g을 얻었다. 상기 고체를 아세토니트릴로부터 재결정화하였다. 고체를 1:1 디클로로메탄/메탄올에 용해하여 잔류 아세토니트릴을 생성물로부터 제거하고, 감압하에서 용매를 제거하였다. 크림색 고체로서 융점이 110 ℃인 N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아를 회수하였다.

실시예 145
N-[2-(4-아미노-2-에톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아

N-[2-(4-아미노-7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)- 1,1-디메틸에틸]-N'-이소프로필우레아 (0.94 g, 1.66 mmol)를 가온 에탄올 100 mL에 용해시키고, 이후 상온으로 다시 냉각하였다. 탄소상 10% 팔라듐 (0.35 g)을 첨가하고 혼합물을 수소 50 psi (3.4×105 Pa)하에서 밤새 진탕하였다. 반응물을 셀라이트 여과제를 통해 여과하였으며, 필터 패드를 에탄올 및 메탄올로 순차적으로 세척하였다. 여액을 감압 농축하였다. 생성 회백색 고체를 실리카 겔에서의 플래시 컬럼 크로마토그래피에 의해 정제하여 (90:10:1 디클로로메탄/메탄올/수성 수산화암모늄으로 용출시킴) 회백색 분말로서 융점이 163-168 ℃인 N-[2-(4-아미노-2-에톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]-N'-이소프로필우레아 0.53 g을 제공하였다.

실시예 146
N-[2-(4-아미노-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드

파트 A
7-벤질옥시-4-클로로-3-니트로퀴놀린 대신에 6-벤질옥시-4-클로로-3-니트로퀴놀린을 사용하여 실시예 52의 파트 A에 기재된 방법에 따라 (2-아미노-2-메틸프로필)(6-벤질옥시-3-니트로퀴놀린-4-일)아민을 제조하였다.
THF 400 mL 중 (2-아미노-2-메틸프로필)(6-벤질옥시-3-니트로퀴놀린-4-일)아민 (24.33 g, 66.4 mmol)을 교반하고, 수산화나트륨 (물 100 mL 중 2.92 g)을 첨가하였다. 이후, THF 100 mL 중 디-tert-부틸 디카르보네이트 17.40 g을 적가하였으며, 반응물을 상온에서 밤새 교반하였다. THF를 감압하에서 제거하고, 디클로로메탄 및 물을 첨가하였다. 유기 분획물을 수성 분획물로부터 분리하였다. 유기 분획물을 이후 물, 이어서 염수로 순차적으로 세척하고, 황산나트륨 및 황산마그네슘에서 건조하고, 감압 농축하여 갈색 발포체로서 tert-부틸 [2-(6-벤질옥시-3-니트로퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 조 생성물 31.05 g을 산출하였다.
파트 B
tert-부틸 [2-(6-벤질옥시-3-니트로퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 (13.75 g), 탄소상 5% 백금 (1.03 g) 및 아세토니트릴 (250 mL)을 배합 하였다. 혼합물을 수소 50 psi (3.4×105 Pa)하에서 밤새 진탕하였다. 반응물을 셀라이트 여과제를 통해 여과하였으며, 필터 패드를 아세토니트릴로 세정하였다. 여액을 감압 농축하였다. 잔류 물을 톨루엔으로 공비혼합하여 제거하였다. 모든 휘발성 물질을 감압하에 제거하여 갈색 발포체로서 tert-부틸 [2-(3-아미노-6-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 조 생성물 13.34 g을 제공하였다.
파트 C
에톡시아세틸 클로라이드 (3.5 mL, 32.0 mmol)를 무수 디클로로메탄 약 250 mL 중 tert-부틸 [2-(3-아미노-6-벤질옥시퀴놀린-4-일아미노)-1,1-디메틸에틸]카르바메이트 (12.71 g, 29.1 mmol) 및 트리에틸아민 (8.1 mL, 58.2 mmol)의 용액에 첨가하였다. 2시간 동안 교반한 이후, 휘발성 물질을 감압하에서 제거하고, 생성 갈색 잔류물을 에탄올에, 이어서 추가로 트리에틸아민 8.1 mL에 용해시켰다. 반응물을 환류 상태에서 2.5일 동안 가열하고, 이후 상온으로 냉각시켰다. 용매를 감압하에서 제거하고, 디클로로메탄을 첨가하였다. 용액을 수성 나트륨 비카르보네이트 (2×)로, 이어서 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축하였다. 잔류물을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 0-15% CMA의 구배로 용출시켜 오렌지색 발포체로서 tert-부틸 [2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]카르바메이트 8.4 g을 제공하였다.
파트 D
에탄올 (4.3 M) 중 HCl 용액 50 mL를 에탄올 (50 mL) 중 tert-부틸 [2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]카르바메이트 (8.4 g, 16.6 mmol)의 용액에 첨가하고, 환류 온도에서 1시간 동안 가열하였다. 반응물을 상온으로 냉각시키고, 용액을 통해 질소를 버블링하였다. 용매를 감압하에서 제거하고, 물을 잔류물에 첨가하였다. 용액을 염기성으로 제조하고, 이후 디클로로메탄으로 3회 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 농축하여 갈색 고체로서 2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 6.26 g을 산출하였다.
파트 E
2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 (2.19 g, 5.4 mmol), 트리에틸아민 (1.5 mL, 10.8 mmol) 및 디클로로메탄 (50 mL)을 배합하였다. 메탄술폰산 무수물 (1.04 g, 6.0 mmol)을 첨가하고, 반응물을 밤새 교반하였다. 박층 크로마토그래피에 의한 분석은 반응이 완료되지 않았음을 나타내었다. 메탄술폰산 무수물 0.2 g을 추가로 첨가하고 반응물을 2시간 더 교반하였다. 포화 나트륨 비카르보네이트를 첨가하고 층을 분리하였다. 유기 분획물을 포화 나트륨 비카르보네이트로 2회, 및 이후 염수로 순차적으로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 오렌지색 발포체를 제공하였다. 잔류물을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 0-20% CMA의 구배로 용출시켜 백색 고체로서 N-[2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 2.37 g을 제공하였다.
파트 F
mCPBA (60% 순도, 1.45 g)을 클로로포름 (100 mL) 중 N-[2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (2.37 g, 4.91 mmol)의 용액에 첨가하였다. 반응물을 2시간 동안 교반하고, 수성 2% 탄산나트륨 50 mL을 첨가하여 켄칭하였다. 층을 분리하고, 수성 분획물을 클로로포름으로 2회 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 농축하여 오렌지색 발포체로서 N-[2-(8-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 조 생성물 2.29 g을 얻었다.
파트 G
N-[2-(8-벤질옥시-2-에톡시메틸-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (2.29 g, 4.59 mmol)를 디클로로메탄 (75 mL)에 현탁하고, 트리클로로아세틸 이소시아네이트 (0.71 mL, 5.97 mmol)를 적가하였다. 1시간 동안 교반한 이후, 휘발성 물질을 감압하에서 제거하였다. 생성 오렌지색 잔류물을 메탄올 (75 mL)에 용해시키고, 나트륨 메톡시드 (메탄올 중 21%) 6 mL을 첨가하였다. 반응물을 2시간 동안 추가로 교반하고, 이후 메탄올을 감압하에서 제거하였다. 잔류물을 디클로로메탄에 용해시키고, 물, 이어서 염수로 세척하였다. 유기 분획물을 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 오렌지색 발포 체를 얻었다. 잔류물을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 10-30% CMA의 구배로 용출시켜 백색 분말로서 융점이 90℃인 N-[2-(4-아미노-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 1.88 g을 제공하였다.
실시예 147
N-[2-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드

N-[2-(4-아미노-8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (1.16 g, 2.33 mmol)를 에탄올 50 mL에 용해시키고, 탄소상 10% 팔라듐 (0.46 g)을 첨가하였다. 혼합물을 수소 50 psi (3.4×105 Pa) 하에서 밤새 진탕하였다. 디클로로메탄 (100 mL) 및 메탄올 (100 mL)을 첨가하고, 반응물을 셀라이트 여과제를 통해 여과시켰다. 여과 케이크를 1:1 디클로로메탄:메탄올 200 mL로 세척하였다. 여액을 감압 농축하여 백색 고체를 얻었다. 고체를 최소량의 에탄올로부터 침전시키고, 이후 최소량의 메탄올을 함유하는 디클 로로메탄에 재용해시켰다. 수성 수산화암모늄을 첨가하였다. 2개 상을 진탕에 의해 혼합하고, 이후 분리하였다. 유기 분획물을 버리고, 수성 분획물을 디클로로메탄으로 4회, 이어서 에틸 아세테이트로 2회 추출하였다. 합쳐진 유기 분획물을 농축하였다. 수성 분획물의 부피가 감소되고, 침전물이 형성되었다. 수성 수산화암모늄의 첨가에 의해 수성 분획물을 염기성으로 제조하고, 생성 침전물을 여과에 의해 회수하였다. 고체 침전물을 유기 분획물로부터의 잔류물과 합하여 조 생성물 0.75 g을 얻었다. 이 물질을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 클로로포름 중 30-50% CMA의 구배인 용출액을 사용하는 실리카 겔 카트리지를 사용하여 백색 분말로서 융점이 252-254 ℃인 N-[2-(4-아미노-2-에톡시메틸-8-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 0.57 g을 제공하였다.

실시예 148
N-{2-[4-아미노-8-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드

파트 A
2-(8-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸아민 (2.19 g, 5.4 mmol), 트리에틸아민 (1.5 mL, 10.8 mmol) 및 디클로로메탄 (50 mL)을 배합하였다. 시클로헥산카르보닐 클로라이드 (0.95 mL, 6.0 mmol)를 첨가하고, 반응물을 밤새 교반하였다. 포화 나트륨 비카르보네이트를 첨가하고, 층을 분리하였다. 유기 분획물을 포화 나트륨 비카르보네이트로 2회, 이어서 염수로 순차적으로 세척하고, 황산나트륨으로 건조하고, 여과하고, 감압 농축하여 오렌지색 발포체를 제공하였다. 잔류물을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 0-20% CMA 구배로 용출시켜 황갈색 고체로서 N-{2-[8-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 2.34 g을 제공하였다.
파트 B
mCPBA (60% 순도, 1.34 g)를 클로로포름 (100 mL) 중 N-{2-[8-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 (2.34 g, 4.55 mmol)의 용액에 첨가하였다. 반응물을 2시간 동안 교반하 고, 수성 2% 탄산나트륨 50 mL를 첨가하여 켄칭하였다. 층을 분리하고, 수성 분획물을 클로로포름으로 2회 추출하였다. 합쳐진 유기 분획물을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 농축하여 오렌지색 발포체로서 N-{2-[8-(벤질옥시)-2-(에톡시메틸)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 조 생성물 2.68 g을 얻었다.
파트 C
N-{2-[8-(벤질옥시)-2-(에톡시메틸)-5-옥시도-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 (2.41 g, 4.55 mmol)를 디클로로메탄 (75 mL)에 현탁하고, 트리클로로아세틸 이소시아네이트 (0.70 mL, 5.92 mmol)를 적가하였다. 1시간 동안 교반한 이후, 휘발성 물질을 감압하에서 제거하였다. 생성 오렌지색 잔류물을 메탄올 (75 mL)에 용해시키고, 나트륨 메톡시드 (메탄올 중 21%) 6 mL를 첨가하였다. 반응물을 밤새 교반하고, 이후 메탄올을 감압하에서 제거하였다. 잔류물을 디클로로메탄에 용해하고, 물, 이어서 염수로 세척하였다. 유기 분획물을 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 오렌지색 발포체를 얻었다. 잔류물을 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 10-20% CMA의 구배로 용출시켜 황갈색 고체로서 N-{2-[4-아미노-8-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 1.51 g을 산출하였다. 상기 기재된 조건을 사용한 컬럼 크로마토그래피에 의해 제품을 2차 정제하여 융점이 90 ℃인 분석 샘플을 제공하였다.
실시예 149
N-{2-[4-아미노-8-히드록시-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드

N-{2-[4-아미노-8-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 (1.25 g, 2.36 mmol)를 에탄올 40 mL에 용해시키고, 탄소상 10% 팔라듐 (0.58 g)을 첨가하였다. 혼합물을 수소 50 psi (3.4×105 Pa)하에서 밤새 진탕하였다. 추가로 탄소상 10% 팔라듐 0.21 g을 첨가하고, 반응물을 수소 50 psi (3.4×105 Pa)하에서 3시간 더 진탕하였다. 반응물을 셀라이트 여과제를 통해 여과시키고, 여과 케이크를 에탄올 100 mL로 세척하였다. 여액을 감압 농축하였다. 생성 회백색 고체를 HORIZON HPFC 시스템에서의 컬럼 크로마토그래피에 의해 정제하였다. 실리카 겔 카트리지를 사용하여 클로로포름 중 10-30% CMA의 구배로 용출시켜 백색 분말로서 융점이 231-232 ℃인 N-{2-[4-아미노-8-히드록시-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}시클로헥산카르복스아미드 0.56 g을 제공하였다.

실시예 150
8-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
트리에틸 오르토포르메이트 (170 mL, 1.0 mol) 및 2,2-디메틸-[1,3]-디옥산-4,6-디온 (53.5 g, 0.37 mol)의 혼합물 (멜드럼산)을 55 ℃에서 90분 동안 가열하였다. 메탄올 (150 mL) 중 4-벤질옥시아닐린 (84.8 g, 0.43 mol) 용액을 반응 온도를 57-60 ℃사이로 유지하면서 1시간 동안 반응물에 서서히 첨가하였다. 반응물을 45 ℃로 냉각시키고, 1.5시간 동안 격렬히 교반하고, 실온으로 냉각시기고, 이후 밤새 교반하였다. 반응 혼합물을 1 ℃로 냉각시키고, 생성물을 여과에 의해 단리하였다. 여액이 무색으로 될 때까지 고체를 냉 에탄올 (약 400 mL)로 세척하였다. 황갈색 분말로서 5-{[(4-벤질옥시)페닐이미노]메틸}-2,2-디메틸-[1,3]-디옥산-4,6-디온 (142.4 g, 잔류 용매로 습윤됨)을 단리하였다.
파트 B
5-{[(4-벤질옥시)페닐이미노)]메틸}-2,2-디메틸-[1,3]-디옥산-4,6-디온 (127.2 g) 및 다우섬 에이 (DOWTHERM A, 500 mL) 열 전달 유체의 용액을 100 ℃로 가열하고, 이후 다우섬 에이 열 전달 유체 (1 L, 250 ℃에서 가열됨)를 함유하는 플라스크에 90분 동안 서서히 첨가하였다. 첨가 도중에, 반응 온도를 245 ℃ 미만으로 떨어지게 하지 않았다. 첨가 이후에, 반응물을 250 ℃에서 30분 동안 교반하고, 이후 상온으로 냉각시켰다. 형성된 침전물을 여과에 의해 단리하고, 디에틸 에테르 (1 L) 및 아세톤 (250 mL)으로 세척하고, 진공 하에서 2시간 동안 건조하여 황색 분말로서 6-벤질옥시퀴놀린-4-올 65.7 g을 제공하였다.
파트 C
6-벤질옥시퀴놀린-4-올 (65.7g) 및 프로피온산 (660 mL)의 혼합물을 격렬히 교반하면서 110 ℃로 가열하였다. 반응 온도를 120 ℃ 미만으로 유지하면서 질산 (16 M, 19.2 mL)을 30분 동안 서서히 첨가하였다. 첨가 이후, 반응물을 상온으로 냉각시켰다. 생성 고체를 여과에 의해 단리하고, 프로피온산, 이소프로판올 및 디에틸 에테르로 순차적으로 세척하였다. 물질을 진공 데시케이터에서 2일 동안 건조하여 황갈색 분말로서 6-벤질옥시-3-니트로퀴놀린-4-올 46.0 g을 제공하였다.
상기 분말을 DMF (300 mL)에 현탁하였다. DMF 중 옥시염화인의 예비 형성된 용액 (실시예 1의 파트 D에서 기재된 것과 같이 제조됨)을 반응물에 적가하였다. 첨가 이후에, 반응물을 100 ℃에서 5분 동안 가열하고, 상온으로 냉각시키고, 교반하면서 빙수에 부었다. 형성된 황갈색 침전물을 여과에 의해 단리하고, 디클로로 메탄에 용해하였다. 생성 용액을 황산마그네슘에서 건조하고, 여과하고, 감압 농축하여 황갈색 고체로서 6-벤질옥시-4-클로로-3-니트로퀴놀린 39.1 g을 산출하였다.
고체를 디클로로메탄 (790 mL)에 용해하고, 트리에틸아민 (38.5 mL, 0.28 mol)을 첨가하였다. n-프로필아민 (19.5 mL, 0.24 mol)을 이후 25분의 기간 동안 첨가하였으며, 반응물을 18시간 동안 교반하였다. 반응물을 디클로로메탄 (500 mL)으로 희석하고, 물 및 염수로 순차적으로 세척하고, 황산마그네슘에서 건조하고, 여과하고, 감압 농축하였다. 조 생성물을 2-프로판올로부터 재결정화하여 미세한 황갈색 침상으로서 (6-벤질옥시-3-니트로퀴놀린-4-일)프로필아민 39.1 g을 제공하였다.
파트 D
(6-벤질옥시-3-니트로퀴놀린-4-일)프로필아민 (26.2 g, 77.7 mmol), 탄소상 5% 백금 (5.2 g), 톨루엔 (600 mL) 및 2-프로판올 (75 mL)을 파르 용기에 첨가하였다. 용기를 질소로 퍼징하고, 이후 수소압 (30 psi, 2.1×105 Pa)하에 두고 20분 동안 진탕하였다. 반응 혼합물을 셀라이트 여과 조제의 층을 통해 여과하고, 여과 케이크를 톨루엔 (1 L) 및 2-프로판올 (1 L)로 순차적으로 세척하였다. 오렌지색 여액을 감압 농축하였다. 헵탄을 잔류물에 첨가하고, 후속적으로 감압하에서 제거하였다. 잔류물을 진공하에서 (0.1 torr, 13.3 Pa) 30분 동안 건조하여 약간의 톨루엔을 함유한 갈색 점성 오일로서 6-벤질옥시-N4-프로필퀴놀린-3,4-디아민 24.3 g 을 제공하였다.
파트 E
디클로로메탄 (65 mL) 중 에톡시아세틸 클로라이드 (10.46 g, 85.4 mmol)의 용액을 디클로로메탄 (200 mL) 중 파트 D로부터의 조 생성물 용액에 적가하고, 반응물을 16시간 동안 교반하였다. 침전물이 형성되었다. 고체를 여과에 의해 단리하고, 냉 헥산으로 세척하고, 감압 하에서 30분 동안 건조하여 황갈색 분말로서 N-(6-벤질옥시-4-프로필아미노퀴놀린-3-일)-2-에톡시아세트아미드 히드로클로라이드 25.4 g을 산출하였다.
파트 F
트리에틸아민 (32.9 mL, 0.24 mol)을 에탄올 (250 mL) 중 N-(6-벤질옥시-4-프로필아미노퀴놀린-3-일)-2-에톡시아세트아미드 히드로클로라이드 (25.4 g) 용액에 첨가하고, 반응 혼합물을 환류 상태에서 3시간 동안 가열하였다. 반응 혼합물을 상온으로 냉각시켰다. 반응 혼합물을 감압하에서 제거하고, 잔류물을 클로로포름에 용해시켰다. 용액을 물 및 염수로 순차적으로 세척하고, 황산마그네슘에서 건조하고, 여과하고, 감압 농축하였다. 생성 오일을 아세토니트릴에 용해시키고, 감압 농축하여 갈색 결정질 고체로서 8-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조 [4,5-c]퀴놀린 22.3 g을 산출하였다.
파트 G
8-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린 (2.0 g, 5.3 mmol)을 클로로포름 (20 mL)에 용해하였다. 3-클로로퍼옥시벤조산 (60% 순도, 1.53 g, 5.3 mmol)을 한꺼번에 첨가하고, 혼합물을 25분 동안 교반하였다. 수산화암모늄 (20 mL)을 첨가하고, 2상 혼합물을 10분 동안 교반하였다. p-톨루엔술포닐 클로라이드 (1.0 g, 5.3 mmol)를 한꺼번에 첨가하고, 반응물을 추가로 1시간 동안 교반하였다. 층을 분리하고, 수성 분획물을 디클로로메탄으로 추출하였다. 유기 분획물을 합하고 5% 수성 탄산나트륨, 물 및 포화 수성 염화나트륨으로 연속해서 세척하였다. 유기 분획물을 무수 황산나트륨에서 건조하고, 여과하고, 감압 농축시켰다. 잔류물을 플래시 컬럼 크로마토그래피에 의해 정제하였다. 99:1 내지 93:7 클로로포름:CMA의 구배로 용출시켜 정제를 수행하여 적갈색 고체를 제공하였다. 고체를 아세토니트릴로부터 재결정화하여 적갈색 결정으로서 융점이 152.5-154.0 ℃인 8-벤질옥시-2-에톡시메틸-1-프로필-1H-이미다조[4,5-c]퀴놀린-4-아민 1.1 g을 산출하였다.

실시예 151
7-벤질옥시-1-[4-(1,1-디옥소이소티아졸리딘-2-일)부틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-아민

파트 A
tert-부틸 {4-[3-아미노-7-(벤질옥시)퀴놀린-4-일아미노]부틸}카르바메이트 (28.1 g, 64.3 mmol)를 디클로로메탄 (319 mL)에 용해시켰다. 에톡시아세틸 클로라이드 (7.87 g, 64.5 mmol)를 적하 깔때기를 통해 적가하고, 혼합물을 1시간 동안 교반하였다. 디클로로메탄을 감압하에서 제거하고, 생성 잔류물을 트리에틸아민 (35.84 mL, 0.26 mol) 및 에탄올 (319 mL)의 용액에 용해시켰다. 반응물을 환류 상태에서 4시간 동안 가열하였으며, 이후 밤새 냉각시켰다. 용매를 감압하에서 증발시키고 잔류물을 다클로로메탄에 용해시켰다. 유기 분획물을 물 및 포화 수성 염화나트륨으로 순차적으로 세척하고, 무수 황산나트륨에서 건조하고, 여과하고, 감압 농축하였다. 오일상 잔류물을 아세토니트릴에 용해시켰다. 후속적으로 아세토니트릴을 감압하에서 제거하여 갈색 고체로서 tert-부틸 [4-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 30 g을 제공하였다.
파트 B
tert-부틸 [4-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸]카르바메이트 (21.0 g)를 에탄올 (100 mL) 및 진한 염산 (13.0 mL)에 용해시켰다. 반응물을 환류 상태에서 1시간 동안 가열하고, 이후 상온으로 냉각시켰다. 황갈색 침전물이 형성되었다. 고체를 여과하고, 진공하에서 16시간 동안 건조하여 베이지색 고체로서 4-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)부틸아민 디히드로클로라이드 12.1 g을 수득하였다.
고체를 디클로로메탄 (168 mL) 및 트리에틸아민 (14.0 mL, 0.1mol)의 용액에 용해시켰다. 3-클로로프로판술포닐 클로라이드 (4.58 mL, 37.5 mmol)를 용액에 적가하고, 반응물을 추가로 1.5시간 동안 교반하였다. 반응 혼합물을 5% 수성 탄산나트륨으로 처리하고 분획물을 분리하였다. 유기 분획물을 물 및 포화 수성 염화나트륨으로 순차적으로 세척하고, 무수 황산나트륨에서 건조하고, 여과하고, 감압 건조하여 오일을 수득하였다.
오일을 DMF에 용해시켰다. 1,8-디아자비시클로[5.4.0]운데크-7-엔, DBU, (5.70 mL, 38.0 mmol)를 첨가하고, 반응물을 44시간 동안 교반하였다. 용매를 감압하에서 제거하였다. 잔류물을 디클로로메탄에 용해시키고, 물로 세척하였다 (3×). 유기 분획물을 무수 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 갈색 점성 오일로서 7-벤질옥시-1-[4-(1,1-디옥소이소티아졸리딘-2-일)부틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린 13.1 g을 산출하였다.
파트 C
7-벤질옥시-1-[4-(1,1-디옥소이소티아졸리딘-2-일)부틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린 (2.83 g)을 클로로포름 (30 mL)에 용해시켰다. 3-클로로퍼옥시벤조산 (60% 순도, 2.21 g, 7.7 mmol)을 한꺼번에 첨가하고, 혼합물을 1시간 동안 교반하였다. 수산화암모늄 (30 mL)을 첨가하고, 2상 혼합물을 20분 동안 교반 하였다. p-톨루엔술포닐 클로라이드 (1.07 g, 5.6 mmol)를 한꺼번에 첨가하고, 반응물을 16시간 동안 교반하였다. 백색 침전물이 형성되었다. 혼합물을 디클로로메탄으로 희석하고 (유기 분획물에서 고체가 남음), 층을 분리하였다. 유기 분획물을 5% 수성 나트륨 비카르보네이트로 세척하였다 (고체는 수성 분획물로 이동함). 층을 분리하고, 이어서 수성 분획물로부터 고체를 여과하였다. 고체를 물 및 디에틸 에테르로 순차적으로 세척하고, 아세토니트릴에서 슬러리화하고, 여과하고, 건조하여 회백색 고체로서 융점이 225-227 ℃인 7-벤질옥시-1-[4-(1,1-디옥소이소티아졸리딘-2-일)부틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-아민 1.5 g을 산출하였다.

실시예 152
7-벤질옥시-1-[(2,2-디메틸-[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-아민)

파트 A
트리에틸아민 (31.88 mL, 228.77 mmol, 1.5 eq)을 디클로로메탄 (400 mL) 중 7-벤질옥시-4-클로로-3-니트로퀴놀린 (48.00 g, 152.51 mmol, 1 eq) 용액에 첨가하였다. 반응 혼합물에 2,2-디메틸-1,3-디옥솔란-4-메탄아민 (20.0 g, 152.51 mmol, 1 eq)의 적가 이후에 상온에서 6시간 동안 교반하였다. 조 반응 혼합물을 감압 농축하고, 생성 고체를 물로 연화처리하고, 이후 1시간 동안 교반하였다. 침전물을 여과에 의해 수집하고, 물로 세척하고, 건조하고, 디에틸 에테르 (400 mL)에 현탁하고, 초음파 처리하였으며, 생성 침전물 물질을 여과에 의해 수집하였다. 생성물을 40 ℃에서 12시간 동안 진공하에서 건조하여 황색 고체로서 융점이 154-155 ℃인 (7-벤질옥시-3-니트로-퀴놀린4-일)[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]아민 60.1 g을 수득하였다.

파트 B
물 (450 mL) 중 나트륨 디티오네이트 (85% 순도, 135.07 g, 659.42 mmol) 및 탄산칼륨 (101.27 g, 732.73 mmol)의 용액을 디클로로메탄 (500 mL) 및 물 (50 mL) 중 에틸 비올로겐 디브로마이드 (1.1 g, 2.93 mmol) 및 (7-벤질옥시-3-니트로-퀴놀린4-일)[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]아민 (60.0 g, 146.54 mmol)의 기계적으로 교반된 혼합물에 적가하였다. 반응 혼합물을 상온에서 밤새 교반하고, 이후 물 (600 mL)로 희석시키고, 추가로 10분 동안 교반하였다. 유기상을 분리하고, 수성 층을 디클로로메탄 (400 mL)로 재추출하였다. 합쳐진 유기 층을 물 (800 mL) 및 염수 (800 mL)로 세척하고, 황산나트륨에서 건조하고, 감압 농축하여 갈색 발포체로서 7-벤질옥시-N4-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]퀴놀린-3,4-디아민 55.60 g을 수득하였다.

파트 C
트리에틸아민 (25.53 mL, 183.17 mmol)을 0 ℃에서 디클로로메탄 (500 mL) 중 7-벤질옥시-N4-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]퀴놀린-3,4-디아민 (55.60 g, 146.54 mmol) 용액에 첨가하였다. 반응 혼합물에 에톡시아세틸 클로라이드 (22.45 g, 183.17 mmol)를 적가하고, 이어서 반응 혼합물을 상온에서 4시간 동안 교반하였다. 반응 혼합물을 감압 농축하고, 잔류물을 에탄올 (350 mL) 중 트리에틸아민 (61.26 mL, 439.54 mmol) 혼합물에 첨가하고, 환류 상태로 16시간 동안 가열하였다. 반응 혼합물을 감압 농축하고, 디클로로메탄으로 추출하고 (3×300 mL), 물 (300 mL) 및 염수 (300 mL)로 세척하고, 황산나트륨에서 건조하였다. 조 생성물 물질을 실리카 겔에서의 컬럼 크로마토그래피에 의해 정제하고 (용출액으로 클로로포름:CMA의 95:5 혼합물을 사용함), 감압 농축하여 갈색 고체로서 물질 42.5 g을 얻었다. 상기 물질을 디에틸 에테르로부터 재결정화하여 백색 결정질 고체로서 융점이 110-111 ℃인 7-벤질옥시-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린 37.5 g을 수득하였다.

파트 D
3-클로로퍼옥시벤조산 (mCPBA) (75% 순도, 11.57 g, 50.27 mmol, 1.5 eq)을 디클로로메탄 (200 mL) 중 7-벤질옥시-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린 (15.00 g, 33.52 mmol, 1 eq) 용액에 첨가하고, 반응 혼합물을 2시간 동안 교반하였다. 박층 크로마토그래피에 의한 분석은 반응이 완료되지 않았음을 나타내며, 추가로 mCPBA (1.2 g)를 첨가하고, 반응물을 밤새 교반하였다. 반응 혼합물을 디클로로메탄 (300 mL)으로 희석하고, 4% 수성 탄산나트륨 용액 (2×300 mL) 및 염수 (300 mL)로 순차적으로 세척하고, 감압 농축하여 잔류물을 수득하였다. 이후 진한 수산화암모늄 (75 mL)을 디클로로메탄 (200 mL) 중 잔류물의 혼합물에 첨가하고, 4 ℃로 냉각시켰다. p-톨루엔술포닐 클로라이드 (2.75 g, 14.44 mmol, 1.1 eq)를 반응 혼합물에 나누어서 첨가하고, 상온에서 16시간 동안 교반하였다. 이후 반응 혼합물을 디클로로메탄 (200 mL)으로 희석하고, 4M 수성 탄산나트륨 용액으로 세척하고, 분리하였다. 수성 층을 디클로로메탄 (200 mL)으로 재추출하고, 합쳐진 유기 층을 염수로 세척하고, 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 갈색 고체를 수득하였다. 조 생성물 물질을 디에틸 에테르로부터의 결정화에 의해 정제하여 백색 고체 판으로서 융점이 186-187 ℃인 7-벤질옥시-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-일아민 9.8 g을 제공하였다.

실시예 153
4-아미노-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-7-올

팔라듐 히드록시드 (펄만 촉매 (Pearlman's catalyst)) (탄소상 20% 팔라듐 w/w, 900 mg)를 밀봉 용기에서 아세토니트릴 (300 mL) 및 메탄올 (300 mL) 중 실시예 152에 기재된 것과 같이 제조된 7-벤질옥시-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-4-일아민 (9.00 g, 19.46 mmol, 1 eq) 용액에 첨가하고, 반응 혼합물을 수소압 (30 psi, 2.1×105 Pa)하에서 24시간 동안 두었다. 조 반응 혼합물을 셀라이트 여과 조제의 층을 통해 여과하고, 여액을 감압 농축하고, 아세토니트릴로 연화처리하였다. 생성 결정질 물질을 여과에 의해 수집하고, 아세토니트릴로 세척하여 회백색 고체로서 4-아미노-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-7-올 3.66 g을 수득하였다. 추가 생성물 (0.36 g)을 감압 농축에 의해 초기 연화의 여액으로부터 단리하고, 아세토니트릴로 연화하고, 회백색 분말로 단리된, 융점이 240-242 ℃인 4-아미노-1-[(2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-7-올 4.2 g의 총 수율을 얻기 위하여 여과하였다.

실시예 155-173
하기 표로부터의 알킬화 시약 (0.125 mmol, 1.0 eq)을 DMF (1 mL) 중 4-아미 노-1-[2,2-디메틸[1,3]디옥솔란-4-일)메틸]-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-7-올 (73 mg, 0.2 mmol, 1.6 eq) 및 탄산칼륨 (55 mg, 2 eq)을 함유하는 시험관에 첨가하였다. 시험관을 마개로 막고 상온에서 밤새 진탕하였다. 이후 각각의 반응 혼합물을 여과하고 물 한 방울을 각각의 혼합물에 첨가하였다. 이후 각각의 혼합물을 메탄올 (5 mL)로 희석하고 하기 실시예 174-181에서 사용하기 위하여 각각의 용액의 절반을 제거하였다. 각각의 반응물로부터 남는 용액을 진공 원심분리에 의해 농축시켰다. 화합물을 워터스 프렉션 링스 (Waters Fraction Lynx) 자동 정제 시스템을 사용하는 정제용 HPLC에 의해 정제하였다. 상기 정제용 HPLC 분획물을 마이크로매스 (Micromass) LC/TOF-MS를 사용하여 분석하고, 적합한 분획물을 원심분리 증발하여 목적 화합물의 트리플루오로아세테이트 염을 제공하였다. 컬럼: 조르박스 보너스 알피 (ZORBAX Bonus RP), 21.2×50 밀리미터 (mm), 입도 5 마이크론; 5-95% B로부터의 비선형 구배 용출 (여기서, A는 0.05% 트리플루오로아세트산/물이고 B는 0.05% 트리플루오로아세트산/아세토니트릴임); 질량 선택적 트리거에 의한 분획물 수거. 하기 표는 각각의 시험관에 첨가된 시약, 생성 화합물의 구조 및 단리된 트리플루오로아세테이트 염에 대한 정확한 측정 질량값을 나타낸다.



실시예 174-191
실시예 154-173의 케탈을 가수분해하였다. 1N 수성 염산 (0.50 mL) 및 THF를, 실시예 154-173으로부터의 용액을 함유하는 각각의 시험관에 첨가하였다. 시험관을 마개로 막고 56시간 동안 상온에서 진탕하였다. 이후 각각의 반응 혼합물을 진공 원심분리에 의해 농축하였다. 화합물을 워터스 프렉션 링스 자동 정제 시스템을 사용하여 정제용 HPLC에 의해 정제하였다. 정제용 HPLC 분획물을 마이크로매스 LC/TOF-MS를 사용하여 분석하고, 적합한 분획물을 원심분리 증발하여 목적 화합물의 트리플루오로아세테이트 염을 제공하였다. 컬럼: 조르박스 보너스 알피, 21.2×50 밀리미터 (mm), 입도 5 마이크론; 5-95% B로부터의 비선형 구배 용출 ( 여기서, A는 0.05% 트리플루오로아세트산/물이고 B는 0.05% 트리플루오로아세트산/아세토니트릴임); 질량 선택적 트리거에 의한 분획물 수거. 하기 표는 케탈 출발 물질 제조에 사용된 알킬화제, 생성 화합물의 구조 및 단리된 트리플루오로아세테이트 염에 대한 정확한 측정 질량값을 나타낸다.



실시예 192-225
파트 A
트리에틸아민 (58.9 mL, 422.4 mmol, 1.5 eq) 및 tert-부틸 N-(2-아미노에틸) 카르바메이트 (54.1 g, 337.9 mmol, 1.2 eq)를 DMF (800 mL) 중 7-벤질옥시-4-클로로-3-니트로퀴놀린 (88.63 g, 281.6 mmol) 용액에 순차적으로 첨가하고, 4시간 동안 상온에서 교반하였다. 조 생성물 반응 혼합물을 계속 교반하면서 뜨거운 물에 부어서 선황색 침전물을 수득하였다. 황색 고체를 여과하고, 65 ℃에서 감압하에서 건조하여 tert-부틸 2-[(7-벤질옥시-3-니트로퀴놀린-4-일)아미노]에틸카르바메이트 123.65 g을 수득하였다.
파트 B
tert-부틸 2-[(7-벤질옥시-3-니트로퀴놀린-4-일)아미노]에틸카르바메이트 (40.0 g, 91.22 mmol)를 에틸 아세테이트 (550 mL)에 용해시키고, 탄소상 5% 백금 (10.68 g, 54.73 mmol, 0.03 eq)으로 충전된 파르 수소화 용기 (Parr hydrogenation vessel)로 보냈다. 용기를 질소 기체로 퍼징하고, 수소압 (30 psi, 2.07×105 Pa)하에서 밤새 두었다. 촉매를 셀라이트 여과 조제의 층을 통한 여과에 의해 제거하고, 여과 케이크를 메탄올 및 디클로로메탄으로 세정하였다. 여액을 감압 농축하여 tert-부틸 2-[(3-아미노-7-벤질옥시퀴놀린-4-일)아미노]에틸카르바메이트 35.25 g을 제공하였다.
파트 C
트리에틸아민 (24.0 mL, 172.58 mmol)을 디클로로메탄 (400 mL) 중 tert-부틸 2-[(3-아미노-7-벤질옥시퀴놀린-4-일)아미노]에틸카르바메이트 (35.25 g, 86.29 mmol) 용액에 첨가하고, 상온에서 교반하였다. 클로로아세틸 클로라이드 (6.87 mL, 86.29 mmol)를 상온에서 신속히 첨가하여 튀는 것을 막고 10분 동안 교반하였다. 반응 혼합물을 감압 농축하고, 에탄올 (500 mL)에 용해시키고, 상온에서 2일 동안 교반하였다. 혼합물을 감압 농축하고, 잔류물을 디클로로메탄으로부터 재결정화하여 tert-부틸 2-(7-벤질옥시-2-클로로메틸-1H-이미다조[4,5-c]퀴놀린-1-일)에틸카르바메이트 6.23 g을 수득하였다.
파트 D
3-클로로퍼옥시벤조산 (mCPBA)(77% 순도, 3.53 g, 15.76 mmol, 1.2 eq)을 클로로포름 (60 mL) 중 tert-부틸 2-(7-벤질옥시-2-클로로메틸-1H-이미다조[4,5-c]퀴놀린-1-일)에틸카르바메이트 (6.13 g, 13.13 mmol, 1 eq) 용액에 첨가하고, 반응 혼합물을 1시간 동안 교반하였다. 이후 수산화암모늄 (25 mL, 과량의 eq)을 반응 혼합물에 첨가하고, 5분 동안 교반하였다. p-톨루엔술포닐 클로라이드 (2.75 g, 14.44 mmol, 1.1 eq)를 첨가하고, 반응 혼합물을 밤새 교반하였다. 액체 크로마토그래피 질량 분석기에 의한 분석은 반응이 완료되지 않았음을 나타내었다.
조 생성물을 클로로포름 및 물로 희석하고, 상을 분리하였다. 수성 층을 추가 클로로포름으로 추출하고, 합쳐진 유기 층을 감압 농축하였다. HORIZON HPFC 시스템에서의 크로마토그래피 정제가 잔류물을 산출하며, 메탄올 (60 mL)에 용해시키고, 30분 동안 반응 혼합물을 통해 암모니아 기체를 버블링하여 포화된다. 이후 반응 혼합물을 밤새 상온에서 교반하였다. 조 생성물을 감압 농축하고, 컬럼 크로마토그래피에 의해 정제하여 tert-부틸 2-(4-아미노-2-아미노메틸-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일)에틸카르바메이트 2.32 g를 수득하였다.
파트 E
메틸 이소시아네이트 (322.9 mg, 5.66 mmol, 1.1 eq)를 DMF 중 tert-부틸 2-(4-아미노-2-아미노메틸-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-1-일)에틸카르바메이트 (2.38 g, 5.14 mmol, 1 eq) 용액에 첨가하고, 상온에서 2일 동안 교반하였다. 조 반응 혼합물을 감압 농축하고, 플래시 컬럼 크로마토그래피 (실리카 겔 상, 디클로로메탄 중 0-6% 메탄올)에 의해 정제하고, 감압 농축하여 tert-부틸 2-{4-아미노-7-벤질옥시-2-[(3-메틸우레이도)메틸]-1H-이미다조[4,5-c]퀴놀린-1-일}에틸카르바메이트 2.73 g을 산출하였다.
파트 F
tert-부틸 2-{4-아미노-7-벤질옥시-2-[(3-메틸우레이도)메틸]-1H-이미다조[4,5-c]퀴놀린-1-일}에틸카르바메이트 (2.73 g, 5.25 mmol, 1 eq)를 디클로로메탄 (100 mL)에 용해시켰다. 1,4-디옥산 (100 mL) 중 4 N 염산을 반응 혼합물에 첨가하고, 30분 동안 교반하였다. 반응 혼합물을 감압 농축하고, 잔류물을 메탄올에 용해시켰다. 이후 용액을 디에틸 에테르 (300 mL)에 부어 침전물을 생성하였다. 침전물을 여과에 의해 단리하여 1-{[4-아미노-1-(2-아미노에틸)-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-2-일]메틸}-3-메틸우레아의 히드로클로라이드 염 2.63 g을 산출하였다.
파트 G
하기 표로부터의 시약 (0.14 mmol, 1.1 eq)을 클로로포름 (1 mL) 중 1-{[4-아미노-1-(2-아미노에틸)-7-벤질옥시-1H-이미다조[4,5-c]퀴놀린-2-일]메틸}-3-메틸우레아 (54 mg, 0.13 mmol, 1.0 eq) 및 트리에틸아민 (0.080 mL, 6.0 eq)을 함유하는 시험관에 첨가하였다. 시험관을 마개로 막고 상온에서 밤새 진탕하였다. 물 2방울을 각각의 반응 혼합물에 첨가하고, 이후 혼합물을 진공 원심분리에 의해 농축하였다. 화합물을 워터스 프렉션 링스 자동 정제 시스템을 사용하는 정제용 HPLC에 의해 정제하였다. 정제용 HPLC 분획물을 마이크로매스 LC/TOF-MS를 사용하여 분석하고, 적합한 분획물을 원심분리 증발하여 목적 화합물의 트리플루오로아세테이트 염을 제공하였다. 컬럼: 조르박스 보너스 알피, 21.2×50 밀리미터 (mm), 입도 5 마이크론; 5-95% B로부터의 비선형 구배 용출 (여기서, A는 0.05% 트리플루오로아세트산/물이고 B는 0.05% 트리플루오로아세트산/아세토니트릴임); 질량 선 택적 트리거에 의한 분획물 수거.
하기 표는 각각의 시험관에 첨가된 시약, 생성 화합물의 구조 및 단리된 트리플루오로아세테이트 염에 대한 정확한 측정 질량값을 나타낸다.




실시예 226
N-{2-[4-아미노-7-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드

mCPBA (11.2 g, 39.2 mmol)를 디클로로메탄 (350 mL) 중 N-[2-(7-벤질옥시-2-에톡시메틸-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (실시예 52의 파트 A-D에 기재된 것과 같이 제조됨, 17.2 g, 35.6 mmol)에 첨가하였 다. 용액을 밤새 교반하고, 추가로 mCPBA (약 1-2 g)를 첨가하였다. 20분 후에, 진한 수산화암모늄 (150 mL)을 첨가하였다. 혼합물을 10분 동안 교반하고, p-톨루엔술포닐 클로라이드 (8.20 g, 42.8 mmol)을 나누어 첨가하였다. 혼합물을 상온에서 2일 동안 교반하였으며, 이후 여과하였다. 백색 고체를 얻었으며, 물에 슬러리화 하고, 여과하고, 건조하여 N-{2-[4-아미노-7-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 11.7 g을 산출하였다. 소량의 샘플 500 mg을 정제하여 백색 고체로서 융점이 264-266 ℃인 분석적으로 순수한 N-{2-[4-아미노-7-(벤질옥시)-2-(에톡시메틸)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 300 mg을 산출하였다.

실시예 227
N-{2-[4-아미노-2-(에톡시메틸)-7-(3-티엔-2-일프로폭시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드

파트 A
프로파르길 브로마이드 (1.30 g, 9.19 mmol)를 DMF 중 N-[2-(4-아미노-2-에 톡시메틸-7-히드록시-1H-이미다조[4,5-c]퀴놀린-1-일)-1,1-디메틸에틸]메탄술폰아미드 (2.5 g, 6.13 mmol) 및 탄산세슘 (5.00 g, 12.3 mmol)의 혼합물에 첨가하였다. 혼합물을 밤새 교반하고, 이후 물 (600 mL)에 부었다. 혼합물을 밤새 교반하고, 검상 고체를 여과에 의해 단리하였다. 고체를 디클로로메탄에 용해시키고, 생성 용액을 황산마그네슘에서 건조하고, 여과하고, 감압 농축하였다. 조 생성물을 플래시 크로마토그래피에 의해 정제하여 (실리카겔, 디클로로메탄 중 0-30% 1:5 메탄올/디클로로메탄 용액의 구배로 용출시킴) 회백색 고체로서 N-{2-[4-아미노-2-(에톡시메틸)-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 675 mg을 제공하였다. 소량을 메탄올로부터 재결정화하여 융점이 175.0-176.0 ℃인 회백색 고체를 수득하였다.

파트 B
N-{2-[4-아미노-2-(에톡시메틸)-7-(프로프-2-이닐옥시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 (600 mg, 1.34 mmol), 2-요오도티오펜 (565 mg, 2.69 mmol), 트리에틸아민 (0.50 mL, 3.5 mmol), 디클로로비스(트리페닐포스핀)팔라듐(II) (19 mg, 0.03 mmol), 구리(I) 요오다이드 (10 mg, 0.05 mmol) 및 DMF (10 mL)를 배합하고, 60℃에서 밤새 가열하였다. 상온으로 냉각한 이후, 반응물을 물 (200 mL)에 부어 미세 침전물이 형성되었다. 혼합물을 1시간 동안 교반한 이후, 고체를 여과에 의해 단리하였다. 고체를 디클로로메탄 및 메탄올에 용 해시키고, 실리카 겔의 플러그를 통해 이동시키고, 디클로로메탄 중 20% 메탄올로 세정하였다. 여액을 감압 농축하여 다음 파트에서 추가적인 정제 없이 사용되는 진한 갈색 반 고체로서 N-(2-{4-아미노-2-(에톡시메틸)-7-[(3-티엔-2-일프로프-2-이닐)옥시]-1H-이미다조[4,5-c]퀴놀린-1-일}-1,1-디메틸에틸)메탄술폰아미드 약 900 mg을 산출하였다.
파트 C
파트 B로부터의 물질 (약 1.34 mmol)을 1:1 에탄올/메탄올에 용해시키고 탄소상 10% 팔라듐 (150 mg)을 첨가하였다. 50 psi (3.4×105 Pa)에서 밤새 혼합물을 파르 장치상에서 수소화하였다. 추가로 탄소상 10% 팔라듐 (100 mg)을 조심스럽게 첨가하고, 혼합물을 추가로 4시간 수소화하였다. 혼합물을 셀라이트 여과제를 통해 여과하고 여액을 농축하였다. 조 생성물을 상이한 용매 시스템 (메탄올/디클로로메탄 또는 CMA/클로로포름)으로 여러번 실리카 겔에서의 크로마토그래피에 의해 정제하고, 아세토니트릴 및 부틸 아세테이트로부터 여러번 재결정화하여 몇몇 미량 불순물이 함유된 백색 고체로서 융점이 202.0-203.0 ℃인 N-{2-[4-아미노-2-(에톡시메틸)-7-(3-티엔-2-일프로폭시)-1H-이미다조[4,5-c]퀴놀린-1-일]-1,1-디메틸에틸}메탄술폰아미드 90 mg을 제공하였다.

실시예 228-235
파트 A
수산화암모늄 (1 L)을 메탄올 (500 mL) 중 메틸 테트라히드로-2H-피란-4-카르복실레이트 (20 mL, 150 mmol) 용액에 첨가하고, 반응물을 상온에서 밤새 교반하였다. 추가로 수산화암모늄 (500 mL)을 첨가하고, 반응물을 추가로 4일 동안 교반하였다. 메탄올을 감압하에서 제거하였다. 고체 염화나트륨을 수성 층에 첨가하고, 클로로포름 (3×150 mL)으로 추출하였다. 합쳐진 추출물을 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 백색 고체로서 테트라히드로-2H-피란-4-카르복스아미드 11.4 g을 제공하였다.
파트 B
TFT (441 mL) 중 테트라히드로-2H-피란-4-카르복스아미드 (11.4 g, 88.3 mmol)를 0 ℃로 냉각시켰다. 리튬 알루미늄 수소화물 (10.0 g, 265 mmol)을 10분에 걸쳐 6회 첨가하였다. 첨가 사이에 반응 플라스크를 질소로 퍼징하였다. 반응 혼합물이 더이상 버블링하지 않을 때, 환류에서 6시간 동안 가열하였다. 반응물을 이후 0 ℃로 냉각시키고, 버블링이 중지될 때까지 에틸 아세테이트를 적가하였다. 이후, 버블링이 중지될 때까지 메탄올을 적가하였다. 물 (10 mL), 15% 수성 수산화나트륨 (10 mL) 및 물 (30 mL)을 순차적으로 첨가하였다. 유기 분획물을 경사분리하고, 잔류 회색 고체를 클로로포름으로 세척하였다. 합쳐진 유기 분획물을 황산나트륨에서 건조하고, 여과하고, 감압 농축하여 테트라히드로-2H-피란-4-일메틸아민을 제공하였다.
파트 C
7-벤질옥시-4-클로로-3-니트로퀴놀린을 테트라히드로-2H-피란-4-일메틸아민으로 처리하기 위해 실시예 1의 파트 E에 기재된 방법을 사용하여 (7-벤질옥시-3-니트로퀴놀린-4-일)(테트라히드로-2H-피란-4-일메틸)아민을 수득할 수 있었다. 6-벤질옥시-4-클로로-3-니트로퀴놀린을 테트라히드로-2H-피란-4-일메틸아민으로 처리하기 위해 동일한 방법을 사용하여 (6-벤질옥시-3-니트로퀴놀린-4-일)(테트라히드로-2H 피란-4-일메틸)아민을 제공할 수 있다.
파트 D
실시예 1의 파트 F에 기재된 방법을 사용하여 (7-벤질옥시-3-니트로퀴놀린-4-일)(테트라히드로-2H-피란-4-일메틸)아민 또는 (6-벤질옥시-3-니트로퀴놀린-4-일)(테트라히드로-2H-피란-4-일메틸)아민을 각각 7-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민 또는 6-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민으로 환원할 수 있었다.
파트 E
실시예 228 및 230의 7-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민 (실시예 228) 또는 6-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민 (실시예 230)을 실시예 50의 파트 C에 기재된 방법에 따라 에톡시아세틸 클로라이드로 처리하여 7-벤질옥시-2-에톡시메틸-1-(테트라히드로-2H-피란-4-일메틸)-1H-이미다조[4,5-c]퀴놀린 (실시예 228) 또는 8-벤질옥시-2-에톡시메틸- 1-(테트라히드로-2H-피란-4-일메틸)-1H-이미다조[4,5-c]퀴놀린 (실시예 230)을 수득할 수 있었다.
실시예 229 및 231의 7-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민 (실시예 229) 또는 6-벤질옥시-N4-(테트라히드로-2H-피란-4-일메틸)퀴놀린-3,4-디아민 (실시예 231)을 실시예 1의 파트 G에 기재된 일반적인 방법에 따라 트리에틸 오르토프로피오네이트로 처리하여 7-벤질옥시-2-에틸-1-(테트라히드로-2H-피란-4-일메틸)-1H-이미다조[4,5-c]퀴놀린 (실시예 229) 또는 8-벤질옥시-2-에틸-1-(테트라히드로-2H-피란-4-일메틸)-1H-이미다조[4,5-c]퀴놀린 (실시예 231)을 수득할 수 있었다.
파트 F
실시예 1의 파트 H 및 I에 기재된 방법을 사용하여 파트 E로부터 생성물을 산화 및 아민화함으로써 하기 표에 나타낸 화합물을 수득할 수 있었다. 각 실시예에 대한 출발 물질로 사용될 수 있는 파트 E로부터의 생성물 및 생성물의 구조를 표에 나타냈다.
실시예 228-231

파트 G
실시예 5에서 기재된 방법을 사용하여 실시예 228 내지 231을 환원함으로써 하기 표에 나타낸 화합물을 수득할 수 있었다.

예시 화합물
상기 실시예에 기재된 것들의 일부를 비롯한 특정 예시 화합물은 하기 화학식 IV 또는 V, 및 하기 R1, R2 및 R3 치환체를 가지며, 여기서 표의 각 줄은 각각의 화학식 IV 또는 화학식 V과 부합하여 본 발명의 특정 실시양태를 나타낸다.

| R1 | R2 | R3 |
| 2-히드록시-2-메틸프로필 | 에틸 | 히드록시 |
| 2-히드록시-2-메틸프로필 | 에틸 | 피리딘-3-일메톡시 |
| 2-히드록시-2-메틸프로필 | 에틸 | (4-클로로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 에틸 | 3-피리딘-3-일프로폭시 |
| 2-히드록시-2-메틸프로필 | 프로필 | 히드록시 |
| 2-히드록시-2-메틸프로필 | 프로필 | 피리딘-3-일메톡시 |
| 2-히드록시-2-메틸프로필 | 프로필 | (4-클로로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 2-히드록시-2-메틸프로필 | 메톡시메틸 | 히드록시 |
| 2-히드록시-2-메틸프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 2-히드록시-2-메틸프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 2-히드록시-2-메틸프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 2-히드록시-2-메틸프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 2-히드록시-2-메틸프로필 | 2-메톡시에틸 | 히드록시 |
| 2-히드록시-2-메틸프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 2-히드록시-2-메틸프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 2-히드록시-2-메틸프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 2-메틸프로필 | 에틸 | 히드록시 |
| 2-메틸프로필 | 에틸 | 피리딘-3-일메톡시 |
| 2-메틸프로필 | 에틸 | (4-클로로벤질)옥시 |
| 2-메틸프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 2-메틸프로필 | 에틸 | 3-피리딘-3-일프로필 |
| 2-메틸프로필 | 프로필 | 히드록시 |
| 2-메틸프로필 | 프로필 | 피리딘-3-일메톡시 |
| 2-메틸프로필 | 프로필 | (4-클로로벤질)옥시 |
| 2-메틸프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 2-메틸프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 2-메틸프로필 | 메톡시메틸 | 히드록시 |
| 2-메틸프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 2-메틸프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 2-메틸프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 2-메틸프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 2-메틸프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 2-메틸프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 2-메틸프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 2-메틸프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 2-메틸프로필 | 2-메톡시에틸 | 히드록시 |
| 2-메틸프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 2-메틸프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 2-메틸프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 2-메틸프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 프로필 | 에틸 | 히드록시 |
| 프로필 | 에틸 | 피리딘-3-일메톡시 |
| 프로필 | 에틸 | (4-클로로벤질)옥시 |
| 프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 프로필 | 에틸 | 3-피리딘-3-일프로필 |
| 프로필 | 프로필 | 히드록시 |
| 프로필 | 프로필 | 피리딘-3-일메톡시 |
| 프로필 | 프로필 | (4-클로로벤질)옥시 |
| 프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 프로필 | 메톡시메틸 | 히드록시 |
| 프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 프로필 | 에톡시메틸 | 히드록시 |
| 프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 프로필 | 2-메톡시에틸 | 히드록시 |
| 프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 2,3-디히드록시프로필 | 에틸 | 히드록시 |
| 2,3-디히드록시프로필 | 에틸 | 피리딘-3-일메톡시 |
| 2,3-디히드록시프로필 | 에틸 | (4-클로로벤질)옥시 |
| 2,3-디히드록시프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 2,3-디히드록시프로필 | 에틸 | 3-피리딘-3-일프로필 |
| 2,3-디히드록시프로필 | 프로필 | 히드록시 |
| 2,3-디히드록시프로필 | 프로필 | 피리딘-3-일메톡시 |
| 2,3-디히드록시프로필 | 프로필 | (4-클로로벤질)옥시 |
| 2,3-디히드록시프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 2,3-디히드록시프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 2,3-디히드록시프로필 | 메톡시메틸 | 히드록시 |
| 2,3-디히드록시프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 2,3-디히드록시프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 2,3-디히드록시프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 2,3-디히드록시프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 2,3-디히드록시프로필 | 에톡시메틸 | 히드록시 |
| 2,3-디히드록시프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 2,3-디히드록시프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 2,3-디히드록시프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 2,3-디히드록시프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 2,3-디히드록시프로필 | 2-메톡시에틸 | 히드록시 |
| 2,3-디히드록시프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 2,3-디히드록시프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 2,3-디히드록시프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 2,3-디히드록시프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 4-[(메틸술포닐)아미노]부틸 | 에틸 | 히드록시 |
| 4-[(메틸술포닐)아미노]부틸 | 에틸 | 피리딘-3-일메톡시 |
| 4-[(메틸술포닐)아미노]부틸 | 에틸 | (4-클로로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 에틸 | (4-플루오로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 에틸 | 3-피리딘-3-일프로필 |
| 4-[(메틸술포닐)아미노]부틸 | 프로필 | 히드록시 |
| 4-[(메틸술포닐)아미노]부틸 | 프로필 | 피리딘-3-일메톡시 |
| 4-[(메틸술포닐)아미노]부틸 | 프로필 | (4-클로로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 프로필 | (4-플루오로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 프로필 | 3-피리딘-3-일프로필 |
| 4-[(메틸술포닐)아미노]부틸 | 메톡시메틸 | 히드록시 |
| 4-[(메틸술포닐)아미노]부틸 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 4-[(메틸술포닐)아미노]부틸 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 4-[(메틸술포닐)아미노]부틸 | 에톡시메틸 | 히드록시 |
| 4-[(메틸술포닐)아미노]부틸 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 4-[(메틸술포닐)아미노]부틸 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 4-[(메틸술포닐)아미노]부틸 | 2-메톡시에틸 | 히드록시 |
| 4-[(메틸술포닐)아미노]부틸 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 4-[(메틸술포닐)아미노]부틸 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 4-[(메틸술포닐)아미노]부틸 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에틸 | 히드록시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에틸 | 피리딘-3-일메톡시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에틸 | (4-클로로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에틸 | 3-피리딘-3-일프로필 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 프로필 | 히드록시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 프로필 | 피리딘-3-일메톡시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 프로필 | (4-클로로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 메톡시메틸 | 히드록시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에톡시메틸 | 히드록시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 2-메톡시에틸 | 히드록시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 2-메틸-2-[(메틸술포닐)아미노]프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에틸 | 히드록시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에틸 | 피리딘-3-일메톡시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에틸 | (4-클로로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에틸 | (4-플루오로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에틸 | 3-피리딘-3-일프로필 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 프로필 | 히드록시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 프로필 | 피리딘-3-일메톡시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 프로필 | (4-클로로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 프로필 | (4-플루오로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 프로필 | 3-피리딘-3-일프로필 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 메톡시메틸 | 히드록시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에톡시메틸 | 히드록시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 2-메톡시에틸 | 히드록시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 2-[(시클로헥실카르보닐)아미노]-2-메틸프로필 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에틸 | 히드록시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에틸 | 피리딘-3-일메톡시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에틸 | (4-클로로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에틸 | (4-플루오로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에틸 | 3-피리딘-3-일프로필 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 프로필 | 히드록시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 프로필 | 피리딘-3-일메톡시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 프로필 | (4-클로로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 프로필 | (4-플루오로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 프로필 | 3-피리딘-3-일프로필 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 메톡시메틸 | 히드록시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 메톡시메틸 | 피리딘-3-일메톡시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 메톡시메틸 | (4-클로로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 메톡시메틸 | (4-플루오로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 메톡시메틸 | 3-피리딘-3-일프로필 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에톡시메틸 | 히드록시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에톡시메틸 | 피리딘-3-일메톡시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에톡시메틸 | (4-클로로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에톡시메틸 | (4-플루오로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 에톡시메틸 | 3-피리딘-3-일프로필 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 2-메톡시에틸 | 히드록시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 2-메톡시에틸 | 피리딘-3-일메톡시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 2-메톡시에틸 | (4-클로로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 2-메톡시에틸 | (4-플루오로벤질)옥시 |
| 4-(1,1-디옥시도이소티아졸리딘-2-일)부틸 | 2-메톡시에틸 | 3-피리딘-3-일프로필 |
본 발명의 화합물들은 하기 기재된 방법을 사용한 시험시 사이토카인 생합성을 유도하기 위해 사용되었다.
인간 세포에서의 사이토카인 유도
시험관내 인간 혈액 세포 시스템을 이용하여 사이토카인 유도를 평가하였다. 활성은 문헌 [Testerman et. al., "Cytokine Induction by the Immunomodulators Imiquimod and S-27609", Journal of Leukocyte Biology, 58, 365-372 (September, 1995)]에 기재된 바와 같이 배양 배지로 분비된 인터페론 (α) 및 종양 괴사 인자 (α) (각각 IFN 및 TNF)의 측정치에 기초하였다.
배양을 위한 혈액 세포 제조
건강한 인간 공여자로부터 채취한 전체 혈액을 정맥 천자에 의해 EDTA 진공채혈관 (vacutainer tube)에 수집하였다. HISTOPAQUE-1077을 이용한 밀도 구배 원심분리에 의해 전체 혈액으로부터 말초 혈액 단핵 세포 (PBMC)를 분리하였다. 둘베코 포스페이트 완충 염수 (Dulbecco's Phosphate Buffered Saline; DPBS) 또는 행크 평형 염 용액 (Hank's Balanced Salts Solution; HBSS)로 혈액을 1:1 희석하였다. PBMC 층을 수집하여 DPBS 또는 HBSS로 2회 세척하고, RPMI 완전 배지 중에 4×106개 세포/mL로 재현탁하였다. PBMC 현탁액을, 시험 화합물을 함유하는 동일 부피의 RPMI 완전 배지가 들어있는 48 웰 편평 바닥 멸균 조직 배양 플레이트 (Costar, Cambridge, MA; 또는 Becton Dickinson Labware, Lincoln Park, NJ)에 첨가하였다.
화합물 제조
화합물을 디메틸 술폭시드 (DMSO)에 가용화시켰다. DMSO 농도는 배양 웰에 첨가하기 위해 최종 농도 1%를 초과하지 않아야 한다. 30 내지 0.014 마이크로몰농도 (μM)의 농도 범위에서 화합물을 전반적으로 시험하였다.
인큐베이션
시험 화합물의 용액을 RPMI 완전 배지가 들어있는 첫번째 웰에 60 μM로 첨가하고, 이 웰에서 일련의 3배 희석을 수행하였다. 이어서, PBMC 현탁액을 동일한 부피로 웰에 첨가하여, 시험 화합물의 농도가 원하는 범위 (30 내지 0.014 μM)가 되도록 하였다. PBMC 현탁액의 최종 농도는 2×106개 세포/mL이었다. 플레이트를 멸균 플라스틱 뚜껑으로 덮고, 부드럽게 혼합한 후, 5% 이산화탄소 분위기 중 37 ℃에서 18 내지 24시간 동안 인큐베이션하였다.
분리
인큐베이션 후, 플레이트를 4 ℃에서 1000 rpm (대략 200 x g)으로 10분 동안 원심분리하였다. 무세포 배양 상등액을 멸균 폴리프로필렌 피펫으로 꺼내어, 멸균 폴리프로필렌 튜브로 옮겼다. 분석할 때까지 샘플을 -30 ℃ 내지 -70 ℃에 유지하였다. 샘플을 ELISA에 의해 인터페론 (α)에 대해 분석하고, ELISA 또는 IGEN 분석법에 의해 종양 괴사 인자 (α)에 대해 분석하였다.
ELISA에 의한 인터페론 (α) 및 종양 괴사 인자 (α) 분석
PBL 바이오메디칼 래보라토리즈 (PBL Biomedical Laboratories, New Brunswick, NJ)로부터의 인간 다중-종 키트 (Human Multi-Species kit)를 이용하여 ELISA에 의해 인터페론 (α)의 농도를 측정하였다. 결과는 pg/mL로 표시된다.
바이오소스 인터내셔널 (Biosource International, Camarillo, CA)로부터 입수가능한 ELISA 키트를 이용하여 종양 괴사 인자 (α) (TNF)의 농도를 측정하였다. 별법으로, TNF 농도는 오리겐 M-시리즈 면역분석법 (ORIGEN M-Series Immunoassay) 에 의해 측정하여, IGEN 인터내셔널(IGEN International, Gaithersburg, MD)로부터의 IGEN M-8 분석기 상에서 판독할 수도 있다. 이 면역분석법은 바이오소스 인터내셔널 (Biosource International, Camarillo, CA)로부터의 인간 TNF 포획 및 검출 항체 쌍을 이용한다. 결과는 pg/mL로 표시된다.
본 발명의 특정 화합물은 하기 방법을 이용하여 시험했을 때 TNF-α의 생성을 억제할 수 있다.
마우스 세포에서의 사이토카인 억제
마우스 대식세포 세포주인 Raw 264.7을 이용하여, 리포폴리사카라이드 (LPS)에 의한 자극시 화합물이 종양 괴사 인자-α (TNF-α) 생성을 억제하는 능력을 평가하였다.
단일 농도 분석법:
배양을 위한 혈액 세포 제조
Raw 세포 (ATCC)를 부드럽게 기울여 수거한 후에 이를 계수하였다. 10% 소 태아 혈청 (FBS)이 함유된 RPMI 중에 3×105개 세포/mL이 되도록 세포 현탁액을 제조하였다. 세포 현탁액 (100 ㎕)을 96-웰 편평 바닥 멸균 조직 배양 플레이트 (Becton Dickinson Labware, Lincoln Park, NJ)에 첨가하였다. 세포의 최종 농도는 3×104개 세포/웰이었다. 플레이트를 3시간 동안 인큐베이션하였다. 시험 화합물을 첨가하기 전에, 배지를 3% FBS가 함유된 무색 RPMI 배지로 교체하였다.
화합물 제조
화합물을 디메틸 술폭시드 (DMSO)에 가용화시켰다. DMSO 농도는 배양 웰에 첨가하기 위해 최종 농도 1%를 초과하지 않아야 한다. 5 μM에서 화합물을 시험하였다. LPS (살모넬라 티피무리움 (Salmonella typhimurium)으로부터의 리포폴리사카라이드; Sigma-Aldrich)를 무색 RPMI로 희석하여, 투여량 반응 분석법에 의해 측정된 바와 같은 EC70 농도가 되도록 하였다.
인큐베이션
시험 화합물의 용액 (1 ㎕)을 각 웰에 첨가하였다. 플레이트를 마이크로타이터 플레이트 진탕기에서 1분 동안 혼합한 후, 인큐베이터에 넣었다. 20분 후, LPS의 용액 (1 ㎕, EC70 농도 약 10 ng/ml)을 첨가하고, 플레이트를 진탕기에서 1분 동안 혼합하였다. 플레이트를 5% 이산화탄소 분위기 중 37 ℃에서 18 내지 24시간 동안 인큐베이션하였다.
TNF
-α 분석
상기 인큐베이션 후, 피펫으로 상등액을 제거하였다. 마우스 TNF-α 키트 (Biosource International, Camarillo, CA)를 사용하여 ELLISA에 의해 TNF-α의 농도를 측정하였다. 결과는 pg/mL로 표시된다. LPS만으로 자극시 TNF-α 발현은 100% 반응으로 간주된다.
투여량 반응 분석법:
배양을 위한 혈액 세포 제조
Raw 세포 (ATCC)를 부드럽게 기울여 수거한 후에 이를 계수하였다. 10% FBS가 함유된 RPMI 중에 4×105개 세포/mL이 되도록 세포 현탁액을 제조하였다. 세포 현탁액 (250 ㎕)을 48-웰 편평 바닥 멸균 조직 배양 플레이트 (Costar, Cambridge, MA)에 첨가하였다. 세포의 최종 농도는 1×105개 세포/웰이었다. 플레이트를 3시간 동안 인큐베이션하였다. 시험 화합물을 첨가하기 전에, 배지를 3% FBS가 함유된 무색 RPMI 배지로 교체하였다.
화합물 제조
화합물을 디메틸 술폭시드 (DMSO)에 가용화시켰다. DMSO 농도는 배양 웰에 첨가하기 위해 최종 농도 1%를 초과하지 않아야 한다. 0.03, 0.1, 0.3, 1, 3, 5 및 10 μM에서 화합물을 시험하였다. LPS (살모넬라 티피무리움 (Salmonella typhimurium)으로부터의 리포폴리사카라이드; Sigma-Aldrich)를 무색 RPMI로 희석하여, 투여량 반응 분석법에 의해 측정된 바와 같은 EC70 농도가 되도록 하였다.
인큐베이션
시험 화합물의 용액 (200 ㎕)을 각 웰에 첨가하였다. 플레이트를 마이크로타이터 플레이트 진탕기에서 1분 동안 혼합한 후, 인큐베이터에 넣었다. 20분 후, LPS의 용액 (200 ㎕, EC70 농도 약 10 ng/ml)을 첨가하고, 플레이트를 진탕기에서 1분 동안 혼합하였다. 플레이트를 5% 이산화탄소 분위기 중 37 ℃에서 18 내지 24시간 동안 인큐베이션하였다.
TNF
-α 분석
상기 인큐베이션 후, 피펫으로 상등액을 제거하였다. 마우스 TNF-α 키트 (Biosource International, Camarillo, CA)를 사용하여 ELLISA에 의해 TNF-α의 농도를 측정하였다. 결과는 pg/mL로 표시된다. LPS만으로 자극시 TNF-α 발현은 100% 반응으로 간주된다.
본원에 인용된 특허, 특허 문서 및 공개 공보의 완전한 개시 내용은, 각 문헌이 개별적으로 본원에 포함되어 있는 것과 마찬가지로 그 전문이 본원에 포함된 것으로 한다. 본 발명은 여러가지 실시양태를 언급하여 기재되었다. 상기 예시적 실시양태 및 실시예는 분명한 이해를 위해 제공된 것일 뿐이며, 본 발명이 이들로 제한되는 것은 아니다. 당업자라면, 본 발명의 범주 및 범위로부터 벗어나지 않으면서 기재된 실시양태에 대한 다수의 변화가 가능함을 분명히 알 것이다. 따라서, 본 발명의 범위는 하기 청구의 범위에 의해서만 제한되는 것이다.
Claims (54)
- 화학식 II-7 또는 화학식 II-8의 화합물 또는 제약상 허용되는 그의 염.<화학식 II-7>
 <화학식 II-8>
<화학식 II-8> 상기 식에서,L은 -O-R3이고;R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
상기 식에서,L은 -O-R3이고;R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로,
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다. - 화학식 III-7 또는 화학식 III-8의 화합물 또는 제약상 허용되는 그의 염.<화학식 III-7>
 <화학식 III-8>
<화학식 III-8> 상기 식에서,L은 -O-R3-1이고;R3-1은 -Z-Ar이고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
상기 식에서,L은 -O-R3-1이고;R3-1은 -Z-Ar이고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로,
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다. - 화학식 VII-7 또는 화학식 VII-8의 화합물 또는 제약상 허용되는 그의 염.<화학식 VII-7>
 <화학식 VII-8>
<화학식 VII-8> 상기 식에서,L은 -OH이고;R1-1은 -R4-1, -X'-R4-1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
상기 식에서,L은 -OH이고;R1-1은 -R4-1, -X'-R4-1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R4-1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로,
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R4-1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이며;단, R1-1이 수소 또는 2-메틸프로필인 경우에는 R2가 수소가 아니고; R1-1이 2-메틸프로페닐 또는 2-히드록시-2-메틸프로필인 경우에는 R2가 메틸, 에톡시메틸 및 히드록시메틸이 아니다.
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이며;단, R1-1이 수소 또는 2-메틸프로필인 경우에는 R2가 수소가 아니고; R1-1이 2-메틸프로페닐 또는 2-히드록시-2-메틸프로필인 경우에는 R2가 메틸, 에톡시메틸 및 히드록시메틸이 아니다. - 치료 유효량의 제1항 내지 제3항 중 어느 한 항에 따른 화합물 또는 염을 제약상 허용되는 담체와 함께 포함하는, 바이러스성 질환 또는 신생물성 질환(neoplastic disease)을 치료하는 데 유용한 제약 조성물.
- 바이러스성 질환의 치료가 필요한, 인간을 제외한 동물에게 치료 유효량의 제1항 내지 제3항 중 어느 한 항에 따른 화합물 또는 염을 투여하는 것을 포함하는, 바이러스성 질환의 치료 방법.
- 신생물성 질환의 치료가 필요한, 인간을 제외한 동물에게 치료 유효량의 제1항 내지 제3항 중 어느 한 항에 따른 화합물 또는 염을 투여하는 것을 포함하는, 신생물성 질환의 치료 방법.
- 화학식 IX-7 또는 화학식 IX-8의 화합물 또는 제약상 허용되는 그의 염.<화학식 IX-7>
 <화학식 IX-8>
<화학식 IX-8> 상기 식에서,L은 -OH이고;R1-1은 -R4-1, -X'-R4-1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
상기 식에서,L은 -OH이고;R1-1은 -R4-1, -X'-R4-1, -X'-Y'-R4, -X'-Y'-X-Y-R4 및 -X'-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;X'은 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
Y'은 -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R4-1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로,
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R4-1은 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 헤테로아릴, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다. - 화학식 XI-7 또는 화학식 XI-8의 화합물 또는 제약상 허용되는 그의 염.<화학식 XI-7>
 <화학식 XI-8>
<화학식 XI-8> 상기 식에서,L은 -O-R3이고;R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,,
상기 식에서,L은 -O-R3이고;R3은 -Z-Ar, -Z-Ar'-Y-R4, -Z-Ar'-X-Y-R4, -Z-Ar'-R5 및 -Z-Ar'-X-R5로 이루어진 군으로부터 선택되고;Z는 결합, 알킬렌, 알케닐렌 및 알키닐렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌에는 임의로 -O-가 개재될 수 있고;Ar은 알킬, 알케닐, 알콕시, 메틸렌디옥시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴 및 헤테로아릴로 이루어진 군으로부터 선택되고;Ar'은 알킬, 알케닐, 알콕시, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 히드록시알킬, 메르캅토, 시아노, 카르복시, 포르밀, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 헤테로시클릴알킬레닐, 아미노, 알킬아미노 및 디알킬아미노로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있는 아릴렌 및 헤테로아릴렌으로 이루어진 군으로부터 선택되고;R1은 -R4, -X-R4, -X-Y-R4, -X-Y-X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;R2는 -R4, -X-R4, -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되고;X는 각각 독립적으로 알킬렌, 알케닐렌, 알키닐렌, 아릴렌, 헤테로아릴렌 및 헤테로시클릴렌으로 이루어진 군으로부터 선택되며, 상기 알킬렌, 알케닐렌 및 알키닐렌 기에는 아릴렌, 헤테로아릴렌 또는 헤테로시클릴렌이나 하나 이상의 -O- 기가 임의로 개재될 수 있고;Y는 각각 독립적으로 -S(O)0-2-, -S(O)2-N(R8)-, -C(R6)-, -C(R6)-O-, -O-C(R6)-, -O-C(O)-O-, -N(R8)-Q-, -C(R6)-N(R8)-, -O-C(R6)-N(R8)-, -C(R6)-N(OR9)-,, ,
, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로,
R4는 각각 독립적으로 수소, 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴로 이루어진 군으로부터 선택되며, 상기 알킬, 알케닐, 알키닐, 아릴, 아릴알킬레닐, 아릴옥시알킬레닐, 알킬아릴레닐, 헤테로아릴, 헤테로아릴알킬레닐, 헤테로아릴옥시알킬레닐, 알킬헤테로아릴레닐 및 헤테로시클릴 기는 알킬, 알콕시, 히드록시알킬, 할로알킬, 할로알콕시, 할로겐, 니트로, 히드록시, 메르캅토, 시아노, 아릴, 아릴옥시, 아릴알킬렌옥시, 헤테로아릴, 헤테로아릴옥시, 헤테로아릴알킬렌옥시, 헤테로시클릴, 아미노, 알킬아미노, 디알킬아미노, (디알킬아미노)알킬렌옥시로 이루어진 군으로부터 독립적으로 선택된 하나 이상의 치환기에 의해 치환되거나 치환되지 않을 수 있고, 알킬, 알케닐, 알키닐 및 헤테로시클릴의 경우에는 옥소로 치환되거나 치환되지 않을 수 있고;R5는 각각 독립적으로, ,
, 및
및 로 이루어진 군으로부터 선택되고;
로 이루어진 군으로부터 선택되고; R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다.
R6은 각각 독립적으로 =O 및 =S로 이루어진 군으로부터 선택되고;R7은 각각 독립적으로 C2-7 알킬렌이고;R8은 각각 독립적으로 수소, 알킬, 알콕시알킬레닐 및 아릴알킬레닐로 이루어진 군으로부터 선택되고;R9는 각각 독립적으로 수소 및 알킬로 이루어진 군으로부터 선택되고;R10은 각각 독립적으로 C3-8 알킬렌이고;A는 각각 독립적으로 -O-, -C(O)-, -S(O)0-2-, -CH2- 및 -N(R4)-로 이루어진 군으로부터 선택되고;Q는 각각 독립적으로 결합, -C(R6)-, -C(R6)-C(R6)-, -S(O)2-, -C(R6)-N(R8)-W-, -S(O)2-N(R8)-, -C(R6)-O- 및 -C(R6)-N(OR9)-로 이루어진 군으로부터 선택되고;V는 각각 독립적으로 -C(R6)-, -O-C(R6)-, -N(R8)-C(R6)- 및 -S(O)2-로 이루어진 군으로부터 선택되고;W는 각각 독립적으로 결합, -C(O)- 및 -S(O)2-로 이루어진 군으로부터 선택되며;a 및 b는 독립적으로 1 내지 6의 정수이되, 단 a + b는 7 이하이다. - 제8항에 있어서, R3가 벤질인 화합물 또는 염.
- 제8항 또는 제9항에 있어서, R1이 알킬, 아릴알킬레닐, 아릴옥시알킬레닐, 히드록시알킬, 알킬술포닐알킬레닐, 헤테로시클릴알킬레닐 (여기서, 헤테로시클릴이 하나 이상의 알킬기에 의해 임의로 치환됨), -X-Y-R4 및 -X-R5로 이루어진 군으로부터 선택되며; 상기 X는 알킬렌이고, Y는 -N(R8)-C(O)-, -N(R8)-S(O)2-, -N(R8)-C(O)-N(R8)- 또는이고, R4는 각각 알킬, 알콕시, 할로겐 또는 디알킬아미노로 이루어진 군으로부터 선택된 하나 이상의 치환기에 의해 임의로 치환된 알킬, 아릴, 아릴알킬레닐 또는 헤테로아릴이고, R5는
 ,
, 또는
또는 인 화합물 또는 염.
인 화합물 또는 염.
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US49827003P | 2003-08-27 | 2003-08-27 | |
| US60/498,270 | 2003-08-27 | ||
| US58125404P | 2004-06-18 | 2004-06-18 | |
| US60/581,254 | 2004-06-18 | ||
| PCT/US2004/028021 WO2005020999A1 (en) | 2003-08-27 | 2004-08-27 | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20070026298A KR20070026298A (ko) | 2007-03-08 |
| KR101106812B1 true KR101106812B1 (ko) | 2012-01-19 |
Family
ID=34278585
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020067003863A KR101106812B1 (ko) | 2003-08-27 | 2004-08-27 | 아릴옥시 및 아릴알킬렌옥시 치환된 이미다조퀴놀린 |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US7897597B2 (ko) |
| EP (1) | EP1658076B1 (ko) |
| JP (1) | JP5128815B2 (ko) |
| KR (1) | KR101106812B1 (ko) |
| AR (1) | AR045529A1 (ko) |
| AU (1) | AU2004268625B2 (ko) |
| BR (1) | BRPI0413998A (ko) |
| CA (1) | CA2536136C (ko) |
| ES (1) | ES2406730T3 (ko) |
| IL (1) | IL173728A0 (ko) |
| MX (1) | MXPA06002199A (ko) |
| MY (1) | MY145983A (ko) |
| NZ (1) | NZ545412A (ko) |
| RU (1) | RU2006105101A (ko) |
| WO (1) | WO2005020999A1 (ko) |
Families Citing this family (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6677347B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| US20040214851A1 (en) * | 2003-04-28 | 2004-10-28 | 3M Innovative Properties Company | Compositions and methods for induction of opioid receptors |
| CA2534313C (en) | 2003-08-05 | 2013-03-19 | 3M Innovative Properties Company | Formulations containing an immune response modifier |
| TW200510412A (en) * | 2003-08-12 | 2005-03-16 | 3M Innovative Properties Co | Oxime substituted imidazo-containing compounds |
| CA2536136C (en) | 2003-08-27 | 2012-10-30 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
| JP2007504269A (ja) | 2003-09-05 | 2007-03-01 | スリーエム イノベイティブ プロパティズ カンパニー | Cd5+b細胞リンパ腫の治療方法 |
| JP5043435B2 (ja) | 2003-10-03 | 2012-10-10 | スリーエム イノベイティブ プロパティズ カンパニー | アルコキシ置換イミダゾキノリン |
| US20090075980A1 (en) * | 2003-10-03 | 2009-03-19 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and Analogs Thereof |
| NZ546274A (en) | 2003-10-03 | 2009-12-24 | 3M Innovative Properties Co | Pyrazolopyridines and analags thereof |
| US7544697B2 (en) * | 2003-10-03 | 2009-06-09 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and analogs thereof |
| CN1906193A (zh) | 2003-11-14 | 2007-01-31 | 3M创新有限公司 | 肟取代的咪唑环化合物 |
| AU2004291122A1 (en) | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Hydroxylamine substituted imidazo ring compounds |
| AR046781A1 (es) | 2003-11-25 | 2005-12-21 | 3M Innovative Properties Co | Derivados de imidazoquinolinas. composiciones farmaceuticas. |
| WO2005066170A1 (en) | 2003-12-29 | 2005-07-21 | 3M Innovative Properties Company | Arylalkenyl and arylalkynyl substituted imidazoquinolines |
| WO2005066169A2 (en) | 2003-12-30 | 2005-07-21 | 3M Innovative Properties Company | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinyl sulfonamides |
| WO2005094531A2 (en) | 2004-03-24 | 2005-10-13 | 3M Innovative Properties Company | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| US8017779B2 (en) * | 2004-06-15 | 2011-09-13 | 3M Innovative Properties Company | Nitrogen containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| US7897609B2 (en) * | 2004-06-18 | 2011-03-01 | 3M Innovative Properties Company | Aryl substituted imidazonaphthyridines |
| WO2006009826A1 (en) | 2004-06-18 | 2006-01-26 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines |
| US8541438B2 (en) | 2004-06-18 | 2013-09-24 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| WO2006009832A1 (en) * | 2004-06-18 | 2006-01-26 | 3M Innovative Properties Company | Substituted imidazo ring systems and methods |
| US20090270443A1 (en) * | 2004-09-02 | 2009-10-29 | Doris Stoermer | 1-amino imidazo-containing compounds and methods |
| WO2006063072A2 (en) * | 2004-12-08 | 2006-06-15 | 3M Innovative Properties Company | Immunomodulatory compositions, combinations and methods |
| US8080560B2 (en) | 2004-12-17 | 2011-12-20 | 3M Innovative Properties Company | Immune response modifier formulations containing oleic acid and methods |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| JP2008526765A (ja) | 2004-12-30 | 2008-07-24 | スリーエム イノベイティブ プロパティズ カンパニー | 皮膚転移の処置 |
| WO2006074003A2 (en) | 2004-12-30 | 2006-07-13 | 3M Innovative Properties Company | CHIRAL FUSED [1,2]IMIDAZO[4,5-c] RING COMPOUNDS |
| WO2006084251A2 (en) * | 2005-02-04 | 2006-08-10 | Coley Pharmaceutical Group, Inc. | Aqueous gel formulations containing immune reponse modifiers |
| AU2006338521A1 (en) | 2005-02-09 | 2007-10-11 | Coley Pharmaceutical Group, Inc. | Oxime and hydroxylamine substituted thiazolo(4,5-c) ring compounds and methods |
| AU2006212765B2 (en) | 2005-02-09 | 2012-02-02 | 3M Innovative Properties Company | Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines |
| CA2597446A1 (en) * | 2005-02-11 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Substituted imidazoquinolines and imidazonaphthyridines |
| JP2008530113A (ja) | 2005-02-11 | 2008-08-07 | コーリー ファーマシューティカル グループ,インコーポレイテッド | オキシムおよびヒドロキシラミン置換イミダゾ[4,5−c]環化合物および方法 |
| US8178677B2 (en) * | 2005-02-23 | 2012-05-15 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazoquinolines |
| JP2008531567A (ja) * | 2005-02-23 | 2008-08-14 | コーリー ファーマシューティカル グループ,インコーポレイテッド | ヒドロキシアルキル置換イミダゾキノリン化合物および方法 |
| AU2006216799A1 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Hydroxyalkyl substituted imidazonaphthyridines |
| AU2006216686A1 (en) * | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Method of preferentially inducing the biosynthesis of interferon |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| JP2008539252A (ja) * | 2005-04-25 | 2008-11-13 | スリーエム イノベイティブ プロパティズ カンパニー | 免疫活性化組成物 |
| EA200800782A1 (ru) * | 2005-09-09 | 2008-08-29 | Коли Фармасьютикал Груп, Инк. | ПРОИЗВОДНЫЕ АМИДА И КАРБАМАТА N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗОЛО[4,5-c]ХИНОЛИН-1-IL]-1,1-ДИМЕТИЛЭТИЛ}МЕТАНСУЛЬФОНАМИДА И СПОСОБЫ |
| ZA200803029B (en) | 2005-09-09 | 2009-02-25 | Coley Pharm Group Inc | Amide and carbamate derivatives of alkyl substituted /V-[4-(4-amino-1H-imidazo[4,5-c] quinolin-1-yl)butyl] methane-sulfonamides and methods |
| US8889154B2 (en) | 2005-09-15 | 2014-11-18 | Medicis Pharmaceutical Corporation | Packaging for 1-(2-methylpropyl)-1H-imidazo[4,5-c] quinolin-4-amine-containing formulation |
| JP5247458B2 (ja) | 2005-11-04 | 2013-07-24 | スリーエム・イノベイティブ・プロパティーズ・カンパニー | ヒドロキシ及びアルコキシ置換1h−イミダゾキノリン及び方法 |
| WO2007100634A2 (en) | 2006-02-22 | 2007-09-07 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US8088788B2 (en) | 2006-03-15 | 2012-01-03 | 3M Innovative Properties Company | Substituted fused[1,2] imidazo[4,5-c] ring compounds and methods |
| WO2007106854A2 (en) * | 2006-03-15 | 2007-09-20 | Coley Pharmaceutical Group, Inc. | Hydroxy and alkoxy substituted 1h-imidazonaphthyridines and methods |
| US7906506B2 (en) | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US8178539B2 (en) * | 2006-09-06 | 2012-05-15 | 3M Innovative Properties Company | Substituted 3,4,6,7-tetrahydro-5H-1,2a,4a,8-tetraazacyclopenta[cd]phenalenes and methods |
| US20080149123A1 (en) | 2006-12-22 | 2008-06-26 | Mckay William D | Particulate material dispensing hairbrush with combination bristles |
| HUE033901T2 (en) | 2010-08-17 | 2018-01-29 | 3M Innovative Properties Co | Formulations and formulations for lipidized immune response modifying compounds and related processes |
| DE102010035744A1 (de) * | 2010-08-28 | 2012-03-01 | Merck Patent Gmbh | Imidazolonylchinoline |
| US8728486B2 (en) | 2011-05-18 | 2014-05-20 | University Of Kansas | Toll-like receptor-7 and -8 modulatory 1H imidazoquinoline derived compounds |
| BR112013031039B1 (pt) | 2011-06-03 | 2020-04-28 | 3M Innovative Properties Co | compostos de hidrazino 1h-imidazoquinolina-4-aminas, conjugados feitos destes compostos, composição e composição farmacêutica compreendendo ditos compostos e conjugados, usos dos mesmos e método de fabricação do conjugado |
| JP6460789B2 (ja) | 2011-06-03 | 2019-01-30 | スリーエム イノベイティブ プロパティズ カンパニー | ポリエチレングリコールセグメントを有するヘテロ2官能性リンカー及び該リンカーから調製された免疫反応調節複合体 |
| MX2015008773A (es) | 2013-01-07 | 2015-11-06 | Univ Pennsylvania | Composiciones y metodos para tratar linfoma cutaneo de celulas t. |
| AU2016322813B2 (en) | 2015-09-14 | 2021-04-01 | Pfizer Inc. | Novel imidazo (4,5-c) quinoline and imidazo (4,5-c)(1,5) naphthyridine derivatives as LRRK2 inhibitors |
| WO2017059280A1 (en) | 2015-10-02 | 2017-04-06 | The University Of North Carolina At Chapel Hill | Novel pan-tam inhibitors and mer/axl dual inhibitors |
| KR20180134395A (ko) | 2016-04-19 | 2018-12-18 | 인네이트 튜머 이뮤니티, 인코포레이티드 | Nlrp3 조정제 |
| US10533007B2 (en) | 2016-04-19 | 2020-01-14 | Innate Tumor Immunity, Inc. | NLRP3 modulators |
| CN115317603A (zh) | 2016-07-07 | 2022-11-11 | 小利兰·斯坦福大学托管委员会 | 抗体佐剂缀合物 |
| EP3510036B1 (en) * | 2016-09-07 | 2021-07-21 | GlaxoSmithKline Biologicals SA | Imidazoquinoline derivatives and their use in therapy |
| EP3558971B1 (en) | 2016-12-22 | 2022-02-23 | Global Blood Therapeutics, Inc. | Histone methyltransferase inhibitors |
| TWI674261B (zh) | 2017-02-17 | 2019-10-11 | 美商英能腫瘤免疫股份有限公司 | Nlrp3 調節劑 |
| EP3728255B1 (en) | 2017-12-20 | 2022-01-26 | 3M Innovative Properties Company | Amide substituted imidazo[4,5-c]quinoline compounds with a branched chain linking group for use as an immune response modifier |
| CA3034912A1 (en) | 2018-02-28 | 2019-08-28 | Pfizer Inc. | Il-15 variants and uses thereof |
| EP3796983A2 (en) | 2018-05-23 | 2021-03-31 | Pfizer Inc. | Antibodies specific for gucy2c and uses thereof |
| TWI793325B (zh) | 2018-05-23 | 2023-02-21 | 美商輝瑞大藥廠 | 對cd3具特異性之抗體及其用途 |
| US20220370606A1 (en) | 2018-12-21 | 2022-11-24 | Pfizer Inc. | Combination Treatments Of Cancer Comprising A TLR Agonist |
| WO2020190725A1 (en) | 2019-03-15 | 2020-09-24 | Bolt Biotherapeutics, Inc. | Immunoconjugates targeting her2 |
| IL293926A (en) | 2019-12-17 | 2022-08-01 | Pfizer | Antibodies unique to d47, pd-l1 and their uses |
| TW202216779A (zh) | 2020-07-17 | 2022-05-01 | 美商輝瑞股份有限公司 | 治療性抗體類和彼等之用途 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000076519A1 (en) * | 1999-06-10 | 2000-12-21 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| WO2002046193A2 (en) * | 2000-12-08 | 2002-06-13 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| WO2003050121A1 (en) | 2001-12-06 | 2003-06-19 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
Family Cites Families (331)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3314941A (en) * | 1964-06-23 | 1967-04-18 | American Cyanamid Co | Novel substituted pyridodiazepins |
| DE1645976A1 (de) | 1966-06-18 | 1971-01-21 | Ajinomoto Kk | Verfahren zur Herstellung von Adenosin und 2',3'-O-Isopropylidenadenosin |
| ZA704419B (en) | 1969-07-21 | 1971-04-28 | Ici Australia Ltd | Injectable aqueous solutions of tetramisole |
| US3692907A (en) | 1970-10-27 | 1972-09-19 | Richardson Merrell Inc | Treating viral infections with bis-basic ethers and thioethers of fluorenone and fluorene and pharmaceutical compositons of the same |
| US3917624A (en) | 1972-09-27 | 1975-11-04 | Pfizer | Process for producing 2-amino-nicotinonitrile intermediates |
| US4006237A (en) * | 1973-10-11 | 1977-02-01 | Beecham Group Limited | Tetrahydrocarbostyril derivatives for the prophylaxis of asthma, hayfever and rhinitis |
| US3891660A (en) | 1974-02-07 | 1975-06-24 | Squibb & Sons Inc | Derivatives of 1H-imidazo{8 4,5-c{9 pyridine-7-carboxylic acids and esters |
| US3899508A (en) | 1974-04-12 | 1975-08-12 | Lilly Co Eli | 5-(2-Aminophenyl)pyrazole-3-carboxylic acids and esters thereof |
| DE2423389A1 (de) | 1974-05-14 | 1975-12-04 | Hoechst Ag | Arzneimittel mit psychotroper wirkung und verfahren zu ihrer herstellung |
| US4381344A (en) * | 1980-04-25 | 1983-04-26 | Burroughs Wellcome Co. | Process for producing deoxyribosides using bacterial phosphorylase |
| DE3204126A1 (de) | 1982-02-06 | 1983-08-11 | Bayer Ag, 5090 Leverkusen | Pyrazoloxazine, -thiazine, -chinoline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US4758574A (en) | 1982-05-03 | 1988-07-19 | Eli Lilly And Company | 2-phenylimidazio (4,5-c) pyridines |
| US4563525A (en) * | 1983-05-31 | 1986-01-07 | Ici Americas Inc. | Process for preparing pyrazolopyridine compounds |
| HU190109B (en) | 1983-06-14 | 1986-08-28 | Egyt Gyogyszervegyeszeti Gyar,Hu | Process for preparing imidazo/4,5-c/quinolines |
| CA1271477A (en) * | 1983-11-18 | 1990-07-10 | John F. Gerster | 1h-imidazo[4,5-c]quinolin-4-amines |
| ZA848968B (en) | 1983-11-18 | 1986-06-25 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinolines and 1h-imidazo(4,5-c)quinolin-4-amines |
| IL73534A (en) | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| JPS61112075A (ja) | 1984-11-05 | 1986-05-30 | Shionogi & Co Ltd | チエニルピラゾロキノリン誘導体 |
| US4593821A (en) | 1985-04-25 | 1986-06-10 | Laros Equipment Company, Inc. | Belt separator for blow molding parts |
| US4668686A (en) | 1985-04-25 | 1987-05-26 | Bristol-Myers Company | Imidazoquinoline antithrombrogenic cardiotonic agents |
| US4698346A (en) | 1985-05-08 | 1987-10-06 | Usv Pharmaceutical Corporation | Thiazolo[5,4-h]quinoline compounds useful as anti-allergy agents |
| US4826830A (en) | 1985-07-31 | 1989-05-02 | Jui Han | Topical application of glyciphosphoramide |
| CA1306260C (en) | 1985-10-18 | 1992-08-11 | Shionogi & Co., Ltd. | Condensed imidazopyridine derivatives |
| HU197019B (en) * | 1985-11-12 | 1989-02-28 | Egyt Gyogyszervegyeszeti Gyar | Process for producing thiqzolo (4,5-c) quinoline derivatives and pharmaceuticals comprising same |
| US4837378A (en) | 1986-01-15 | 1989-06-06 | Curatek Pharmaceuticals, Inc. | Topical metronidazole formulations and therapeutic uses thereof |
| US4775674A (en) | 1986-05-23 | 1988-10-04 | Bristol-Myers Company | Imidazoquinolinylether derivatives useful as phosphodiesterase and blood aggregation inhibitors |
| CA1287061C (en) * | 1986-06-27 | 1991-07-30 | Roche Holding Ltd. | Pyridine ethanolamine derivatives |
| US5500228A (en) * | 1987-05-26 | 1996-03-19 | American Cyanamid Company | Phase separation-microencapsulated pharmaceuticals compositions useful for alleviating dental disease |
| US4880779A (en) | 1987-07-31 | 1989-11-14 | Research Corporation Technologies, Inc. | Method of prevention or treatment of AIDS by inhibition of human immunodeficiency virus |
| US4774339A (en) | 1987-08-10 | 1988-09-27 | Molecular Probes, Inc. | Chemically reactive dipyrrometheneboron difluoride dyes |
| US5536743A (en) | 1988-01-15 | 1996-07-16 | Curatek Pharmaceuticals Limited Partnership | Intravaginal treatment of vaginal infections with buffered metronidazole compositions |
| US5225183A (en) | 1988-12-06 | 1993-07-06 | Riker Laboratories, Inc. | Medicinal aerosol formulations |
| US5736553A (en) * | 1988-12-15 | 1998-04-07 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo 4,5-C!quinolin-4-amine |
| US5238944A (en) | 1988-12-15 | 1993-08-24 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo[4,5-c]quinolin-4-amine |
| US5756747A (en) | 1989-02-27 | 1998-05-26 | Riker Laboratories, Inc. | 1H-imidazo 4,5-c!quinolin-4-amines |
| DK0385630T3 (da) | 1989-02-27 | 1997-05-12 | Riker Laboratories Inc | 1H-imidazo(4,5-c)quinolin-4-aminer som antivirale midler |
| US5457183A (en) | 1989-03-06 | 1995-10-10 | Board Of Regents, The University Of Texas System | Hydroxylated texaphyrins |
| US5037986A (en) | 1989-03-23 | 1991-08-06 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo[4,5-c]quinolin-4-amines |
| US4929624A (en) | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| NZ232740A (en) | 1989-04-20 | 1992-06-25 | Riker Laboratories Inc | Solution for parenteral administration comprising a 1h-imidazo(4,5-c) quinolin-4-amine derivative, an acid and a tonicity adjuster |
| US4988815A (en) | 1989-10-26 | 1991-01-29 | Riker Laboratories, Inc. | 3-Amino or 3-nitro quinoline compounds which are intermediates in preparing 1H-imidazo[4,5-c]quinolines |
| US5750134A (en) | 1989-11-03 | 1998-05-12 | Riker Laboratories, Inc. | Bioadhesive composition and patch |
| EP0507863A4 (en) | 1989-12-28 | 1993-07-07 | Virginia Commonwealth University | Sigma receptor ligands and the use thereof |
| US5274113A (en) | 1991-11-01 | 1993-12-28 | Molecular Probes, Inc. | Long wavelength chemically reactive dipyrrometheneboron difluoride dyes and conjugates |
| EP0527818A4 (en) | 1990-04-30 | 1993-09-15 | Isis Pharmaceuticals, Inc. | Oligonucleotide modulation of arachidonic acid metabolism |
| AU653524B2 (en) | 1990-06-08 | 1994-10-06 | Roussel-Uclaf | New imidazole derivatives, their preparation process, the new intermediates obtained, their use as medicaments and the pharmaceutical compositions containing them |
| ES2071340T3 (es) | 1990-10-05 | 1995-06-16 | Minnesota Mining & Mfg | Procedimiento para la preparacion de imidazo(4,5-c)quinolin-4-aminas. |
| MX9203481A (es) | 1990-10-18 | 1992-07-01 | Minnesota Mining & Mfg | Formulaciones. |
| US5248782A (en) | 1990-12-18 | 1993-09-28 | Molecular Probes, Inc. | Long wavelength heteroaryl-substituted dipyrrometheneboron difluoride dyes |
| US5175296A (en) | 1991-03-01 | 1992-12-29 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-c]quinolin-4-amines and processes for their preparation |
| US5389640A (en) * | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5342784A (en) | 1991-04-12 | 1994-08-30 | Mitsubishi Paper Mills Limited | Electrophotographic lithographic printing plate |
| JPH04327587A (ja) | 1991-04-26 | 1992-11-17 | Asahi Chem Ind Co Ltd | 6’−c−アルキル−3−デアザネプラノシンa誘導体、その製造法およびその用途 |
| US5187288A (en) * | 1991-05-22 | 1993-02-16 | Molecular Probes, Inc. | Ethenyl-substituted dipyrrometheneboron difluoride dyes and their synthesis |
| US5268376A (en) | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| TW300219B (ko) | 1991-09-14 | 1997-03-11 | Hoechst Ag | |
| PH31245A (en) | 1991-10-30 | 1998-06-18 | Janssen Pharmaceutica Nv | 1,3-Dihydro-2H-imidazoÄ4,5-BÜ-quinolin-2-one derivatives. |
| US5266575A (en) | 1991-11-06 | 1993-11-30 | Minnesota Mining And Manufacturing Company | 2-ethyl 1H-imidazo[4,5-ciquinolin-4-amines |
| US5378848A (en) * | 1992-02-12 | 1995-01-03 | Shionogi & Co., Ltd. | Condensed imidazopyridine derivatives |
| IL105325A (en) | 1992-04-16 | 1996-11-14 | Minnesota Mining & Mfg | Immunogen/vaccine adjuvant composition |
| DK0641192T3 (da) | 1992-05-18 | 1998-03-02 | Minnesota Mining & Mfg | Anordning til transmucosal lægemiddelafgivelse |
| US5352680A (en) | 1992-07-15 | 1994-10-04 | Regents Of The University Of Minnesota | Delta opioid receptor antagonists to block opioid agonist tolerance and dependence |
| US6608201B2 (en) | 1992-08-28 | 2003-08-19 | 3M Innovative Properties Company | Process for preparing 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5395937A (en) * | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| ES2122261T3 (es) | 1993-03-17 | 1998-12-16 | Minnesota Mining & Mfg | Formulacion de aerosol que contiene un adyuvante de dispersion derivado de un ester, una amida o un mercaptoester. |
| DE4309969A1 (de) | 1993-03-26 | 1994-09-29 | Bayer Ag | Substituierte heteroanellierte Imidazole |
| DE69314318T2 (de) | 1993-04-27 | 1998-04-09 | Agfa Gevaert Nv | Verfahren zum Einfügen von einer Wasserumlöslichen Verbindung in eine hydrophile Schicht |
| JPH09500128A (ja) | 1993-07-15 | 1997-01-07 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | イミダゾ〔4,5−c〕ピリジン−4−アミン |
| US5648516A (en) | 1994-07-20 | 1997-07-15 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| US5352784A (en) | 1993-07-15 | 1994-10-04 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| CA2131680C (en) | 1993-09-17 | 2006-11-07 | Gerhard Stucky | Process for preparing imidazopyridine derivatives |
| US5837809A (en) | 1995-08-11 | 1998-11-17 | Oregon Health Sciences University | Mammalian opioid receptor ligand and uses |
| JPH07163368A (ja) * | 1993-12-15 | 1995-06-27 | Hayashibara Biochem Lab Inc | 組換えdnaとその組換えdnaを含む形質転換体 |
| TW574214B (en) | 1994-06-08 | 2004-02-01 | Pfizer | Corticotropin releasing factor antagonists |
| US6207646B1 (en) * | 1994-07-15 | 2001-03-27 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| CA2194761C (en) * | 1994-07-15 | 2006-12-19 | Arthur M. Krieg | Immunomodulatory oligonucleotides |
| US6239116B1 (en) | 1994-07-15 | 2001-05-29 | University Of Iowa Research Foundation | Immunostimulatory nucleic acid molecules |
| US5612377A (en) * | 1994-08-04 | 1997-03-18 | Minnesota Mining And Manufacturing Company | Method of inhibiting leukotriene biosynthesis |
| US5644063A (en) | 1994-09-08 | 1997-07-01 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-c]pyridin-4-amine intermediates |
| GB9420168D0 (en) | 1994-10-06 | 1994-11-23 | Boots Co Plc | Therapeutic agents |
| US5571819A (en) | 1994-11-22 | 1996-11-05 | Sabb; Annmarie L. | Imidazopyridines as muscarinic agents |
| US5482936A (en) * | 1995-01-12 | 1996-01-09 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-C]quinoline amines |
| US6071949A (en) | 1995-03-14 | 2000-06-06 | The United States Of America As Represented By The Department Of Health And Human Services | Use of lipoxygenase inhibitors as anti-cancer therapeutic and intervention agents |
| US5585612A (en) | 1995-03-20 | 1996-12-17 | Harp Enterprises, Inc. | Method and apparatus for voting |
| FR2732605B1 (fr) | 1995-04-07 | 1997-05-16 | Pasteur Merieux Serums Vacc | Composition destinee a l'induction d'une reponse immunitaire mucosale |
| US5766789A (en) | 1995-09-29 | 1998-06-16 | Energetics Systems Corporation | Electrical energy devices |
| DK0778277T3 (da) | 1995-12-08 | 2003-10-27 | Pfizer | Substituerede heterocycliske derivater som CRF antagonister |
| US5939047A (en) | 1996-04-16 | 1999-08-17 | Jernberg; Gary R. | Local delivery of chemotherapeutic agents for treatment of periodontal disease |
| US5861268A (en) * | 1996-05-23 | 1999-01-19 | Biomide Investment Limited Partnership | Method for induction of tumor cell apoptosis with chemical inhibitors targeted to 12-lipoxygenase |
| DE19621241C2 (de) * | 1996-05-25 | 2000-03-16 | Manfred Kessler | Membranelektrode zur Messung der Glucosekonzentration in Flüssigkeiten |
| US5741908A (en) * | 1996-06-21 | 1998-04-21 | Minnesota Mining And Manufacturing Company | Process for reparing imidazoquinolinamines |
| US5693811A (en) | 1996-06-21 | 1997-12-02 | Minnesota Mining And Manufacturing Company | Process for preparing tetrahdroimidazoquinolinamines |
| ES2232871T3 (es) * | 1996-07-03 | 2005-06-01 | Sumitomo Pharmaceuticals Company, Limited | Nuevos derivados de purina. |
| US6387938B1 (en) | 1996-07-05 | 2002-05-14 | Mochida Pharmaceutical Co., Ltd. | Benzimidazole derivatives |
| US6039969A (en) | 1996-10-25 | 2000-03-21 | 3M Innovative Properties Company | Immune response modifier compounds for treatment of TH2 mediated and related diseases |
| US5939090A (en) | 1996-12-03 | 1999-08-17 | 3M Innovative Properties Company | Gel formulations for topical drug delivery |
| JP4101302B2 (ja) | 1997-01-09 | 2008-06-18 | テルモ株式会社 | 新規アミド誘導体および合成中間体 |
| US6406705B1 (en) | 1997-03-10 | 2002-06-18 | University Of Iowa Research Foundation | Use of nucleic acids containing unmethylated CpG dinucleotide as an adjuvant |
| US6426334B1 (en) | 1997-04-30 | 2002-07-30 | Hybridon, Inc. | Oligonucleotide mediated specific cytokine induction and reduction of tumor growth in a mammal |
| US6113918A (en) | 1997-05-08 | 2000-09-05 | Ribi Immunochem Research, Inc. | Aminoalkyl glucosamine phosphate compounds and their use as adjuvants and immunoeffectors |
| US6303347B1 (en) | 1997-05-08 | 2001-10-16 | Corixa Corporation | Aminoalkyl glucosaminide phosphate compounds and their use as adjuvants and immunoeffectors |
| EP1003531B1 (en) * | 1997-05-20 | 2007-08-22 | Ottawa Health Research Institute | Processes for preparing nucleic acid constructs |
| US6123957A (en) | 1997-07-16 | 2000-09-26 | Jernberg; Gary R. | Delivery of agents and method for regeneration of periodontal tissues |
| US6077349A (en) | 1997-08-20 | 2000-06-20 | Sony Corporation | Method and apparatus for manufacturing disc-shaped recording medium |
| US6309623B1 (en) | 1997-09-29 | 2001-10-30 | Inhale Therapeutic Systems, Inc. | Stabilized preparations for use in metered dose inhalers |
| US6329381B1 (en) | 1997-11-28 | 2001-12-11 | Sumitomo Pharmaceuticals Company, Limited | Heterocyclic compounds |
| US6121323A (en) | 1997-12-03 | 2000-09-19 | 3M Innovative Properties Company | Bishydroxyureas |
| UA67760C2 (uk) | 1997-12-11 | 2004-07-15 | Міннесота Майнінг Енд Мануфакчурінг Компані | Імідазонафтиридин та тетрагідроімідазонафтиридин, фармацевтична композиція, спосіб індукування біосинтезу цитокінів та спосіб лікування вірусної інфекції, проміжні сполуки |
| TW572758B (en) * | 1997-12-22 | 2004-01-21 | Sumitomo Pharma | Type 2 helper T cell-selective immune response inhibitors comprising purine derivatives |
| GB9804377D0 (en) | 1998-03-03 | 1998-04-22 | Speights Limited | Lighting device |
| US6114058A (en) | 1998-05-26 | 2000-09-05 | Siemens Westinghouse Power Corporation | Iron aluminide alloy container for solid oxide fuel cells |
| US6110929A (en) | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| JP2000119271A (ja) * | 1998-08-12 | 2000-04-25 | Hokuriku Seiyaku Co Ltd | 1h―イミダゾピリジン誘導体 |
| US5962636A (en) | 1998-08-12 | 1999-10-05 | Amgen Canada Inc. | Peptides capable of modulating inflammatory heart disease |
| US6518280B2 (en) * | 1998-12-11 | 2003-02-11 | 3M Innovative Properties Company | Imidazonaphthyridines |
| EP1495758A3 (en) | 1999-01-08 | 2005-04-13 | 3M Innovative Properties Company | Formulations and methods for treatment of mucosal associated conditions with an immune response modifier |
| US20020058674A1 (en) | 1999-01-08 | 2002-05-16 | Hedenstrom John C. | Systems and methods for treating a mucosal surface |
| CA2361936C (en) | 1999-01-08 | 2009-06-16 | 3M Innovative Properties Company | Formulations comprising imiquimod or other immune response modifiers for treating mucosal conditions |
| US6486168B1 (en) | 1999-01-08 | 2002-11-26 | 3M Innovative Properties Company | Formulations and methods for treatment of mucosal associated conditions with an immune response modifier |
| US6558951B1 (en) | 1999-02-11 | 2003-05-06 | 3M Innovative Properties Company | Maturation of dendritic cells with immune response modifying compounds |
| US6294271B1 (en) | 1999-02-12 | 2001-09-25 | Shin-Etsu Chemical Co., Ltd. | Flip-chip type semiconductor device sealing material and flip-chip type semiconductor device |
| JP2000247884A (ja) | 1999-03-01 | 2000-09-12 | Sumitomo Pharmaceut Co Ltd | アラキドン酸誘発皮膚疾患治療剤 |
| WO2000061765A2 (en) | 1999-04-12 | 2000-10-19 | Lexicon Genetics Incorporated | Lipoxygenase proteins and polynucleotides encoding the same |
| US6541485B1 (en) | 1999-06-10 | 2003-04-01 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6573273B1 (en) | 1999-06-10 | 2003-06-03 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6756382B2 (en) | 1999-06-10 | 2004-06-29 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6451810B1 (en) | 1999-06-10 | 2002-09-17 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6315985B1 (en) | 1999-06-18 | 2001-11-13 | 3M Innovative Properties Company | C-17/21 OH 20-ketosteroid solution aerosol products with enhanced chemical stability |
| EP2314693A3 (en) | 1999-08-13 | 2012-11-28 | Idera Pharmaceuticals, Inc. | Modulation of oligonucleotide CpG-mediated immune stimulation by positional modification of nucleosides |
| US6432989B1 (en) | 1999-08-27 | 2002-08-13 | Pfizer Inc | Use of CRF antagonists to treat circadian rhythm disorders |
| CO5271670A1 (es) | 1999-10-29 | 2003-04-30 | Pfizer Prod Inc | Antagonistas del factor de liberacion de corticitropina y composiciones relacionadas |
| ATE313318T1 (de) * | 1999-10-29 | 2006-01-15 | Nektar Therapeutics | Trockenpulverzusammensetzungen mit verbesserter dispersität |
| US6376669B1 (en) | 1999-11-05 | 2002-04-23 | 3M Innovative Properties Company | Dye labeled imidazoquinoline compounds |
| US6916925B1 (en) | 1999-11-05 | 2005-07-12 | 3M Innovative Properties Co. | Dye labeled imidazoquinoline compounds |
| US6313156B1 (en) | 1999-12-23 | 2001-11-06 | Icos Corporation | Thiazole compounds as cyclic-AMP-specific phosphodiesterase inhibitors |
| US20040023870A1 (en) * | 2000-01-21 | 2004-02-05 | Douglas Dedera | Methods of therapy and diagnosis using targeting of cells that express toll-like receptor proteins |
| GB0001704D0 (en) * | 2000-01-25 | 2000-03-15 | Glaxo Group Ltd | Protein |
| MXPA02007525A (es) | 2000-02-09 | 2002-12-13 | Hokuriku Pharmaceutical | Derivados 1h-imidazopiridina.. |
| WO2001070663A2 (en) | 2000-03-17 | 2001-09-27 | Corixa Corporation | Novel amphipathic aldehydes and their use as adjuvants and immunoeffectors |
| US20010046968A1 (en) | 2000-03-23 | 2001-11-29 | Zagon Ian S. | Opioid growth factor modulates angiogenesis |
| US6894060B2 (en) * | 2000-03-30 | 2005-05-17 | 3M Innovative Properties Company | Method for the treatment of dermal lesions caused by envenomation |
| HUP0300580A3 (en) | 2000-03-30 | 2006-06-28 | Shionogi & Co | Novel process for producing fused imidazopyridine derivative and novel crystal form |
| US20020137101A1 (en) | 2000-05-19 | 2002-09-26 | Meyers Rachel A. | 46638, a novel human lipoxygenase family member and uses thereof |
| DE10020465A1 (de) | 2000-04-26 | 2001-11-08 | Osram Opto Semiconductors Gmbh | Strahlungsemittierendes Halbleiterbauelement mit Lumineszenzkonversionselement |
| DE10029580C1 (de) * | 2000-06-15 | 2002-01-10 | Ferton Holding Sa | Vorrichtung zum Entfernen von Körpersteinen mit einem intrakorporalen Lithotripter |
| US6387383B1 (en) | 2000-08-03 | 2002-05-14 | Dow Pharmaceutical Sciences | Topical low-viscosity gel composition |
| US6900016B1 (en) | 2000-09-08 | 2005-05-31 | Applera Corporation | Polymorphisms in known genes associated with inflammatory autoimmune disease, methods of detection and uses thereof |
| WO2002022809A2 (en) | 2000-09-15 | 2002-03-21 | Coley Pharmaceutical Gmbh | PROCESS FOR HIGH THROUGHPUT SCREENING OF CpG-BASED IMMUNO-AGONIST/ANTAGONIST |
| US20020055517A1 (en) | 2000-09-15 | 2002-05-09 | 3M Innovative Properties Company | Methods for delaying recurrence of herpes virus symptoms |
| GB0023008D0 (en) | 2000-09-20 | 2000-11-01 | Glaxo Group Ltd | Improvements in vaccination |
| JPWO2002046479A1 (ja) * | 2000-12-07 | 2004-04-08 | 株式会社青山製作所 | 鋼材部品のベイキング処理方法 |
| US6664260B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| US6545016B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Amide substituted imidazopyridines |
| US6667312B2 (en) | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6677348B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Aryl ether substituted imidazoquinolines |
| US6660735B2 (en) | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Urea substituted imidazoquinoline ethers |
| UA74593C2 (en) | 2000-12-08 | 2006-01-16 | 3M Innovative Properties Co | Substituted imidazopyridines |
| US6525064B1 (en) | 2000-12-08 | 2003-02-25 | 3M Innovative Properties Company | Sulfonamido substituted imidazopyridines |
| WO2006091720A2 (en) | 2000-12-08 | 2006-08-31 | 3M Innovative Properties Company | Compositions and methods for targeted delivery of immune response modifiers |
| US6677347B2 (en) | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| US20020107262A1 (en) | 2000-12-08 | 2002-08-08 | 3M Innovative Properties Company | Substituted imidazopyridines |
| US20020110840A1 (en) | 2000-12-08 | 2002-08-15 | 3M Innovative Properties Company | Screening method for identifying compounds that selectively induce interferon alpha |
| US6664265B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6660747B2 (en) | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6545017B1 (en) | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Urea substituted imidazopyridines |
| KR100455713B1 (ko) | 2001-01-29 | 2004-11-06 | 호남석유화학 주식회사 | 올레핀 중합용 다중핵 메탈로센 촉매 및 이를 이용한중합방법 |
| US20020182274A1 (en) * | 2001-03-21 | 2002-12-05 | Kung-Ming Lu | Methods for inhibiting cancer growth, reducing infection and promoting general health with a fermented soy extract |
| CA2444130C (en) | 2001-04-17 | 2010-12-21 | Sumitomo Pharmaceuticals Company, Limited | Adenine derivatives |
| US20020193729A1 (en) | 2001-04-20 | 2002-12-19 | Cormier Michel J.N. | Microprojection array immunization patch and method |
| WO2002085933A1 (en) * | 2001-04-20 | 2002-10-31 | The Institute For Systems Biology | Toll-like receptor 5 ligands and methods of use |
| US6627639B2 (en) | 2001-04-26 | 2003-09-30 | Wyeth | Uses for indoletetrahydropyridine derivatives of 2,3-dihydro-7H-[1,4]dioxino-[2,3-e]indole |
| US7226928B2 (en) | 2001-06-15 | 2007-06-05 | 3M Innovative Properties Company | Methods for the treatment of periodontal disease |
| US6756399B2 (en) | 2001-06-29 | 2004-06-29 | The United States Of America As Represented By The Department Of Health And Human Services | Use of lipoxygenase inhibitors and PPAR ligands as anti-cancer therapeutic and intervention agents |
| CA2453664A1 (en) | 2001-07-16 | 2003-01-30 | Takayuki Kasai | Process for preparation of amidine derivatives |
| JP2005501550A (ja) | 2001-08-30 | 2005-01-20 | スリーエム イノベイティブ プロパティズ カンパニー | 免疫反応調整剤分子を用いた形質細胞様樹状細胞を成熟させる方法 |
| ATE511840T1 (de) | 2001-10-09 | 2011-06-15 | Amgen Inc | Imidazolderivate als entzündungshemmende mittel |
| US20030139364A1 (en) | 2001-10-12 | 2003-07-24 | University Of Iowa Research Foundation | Methods and products for enhancing immune responses using imidazoquinoline compounds |
| DE60230340D1 (de) * | 2001-11-16 | 2009-01-22 | 3M Innovative Properties Co | N-Ä4-(4-Amino-2-ethyl-1H-imidazoÄ4,5-cÜchinolin-1-yl)butylÜmethanesulfonamide, diese enthaltende pharmazeutische Zusammensetzung und deren Verwendung |
| AU2002364897A1 (en) * | 2001-11-17 | 2003-06-10 | Maria Martinez-Colon | Imiquimod therapies |
| CN101033242A (zh) | 2001-11-27 | 2007-09-12 | 安那迪斯药品股份有限公司 | 3-β-呋喃核糖基噻唑并[4,5-d]嘧啶核苷及其应用 |
| NZ532769A (en) | 2001-11-29 | 2005-12-23 | 3M Innovative Properties Co | Pharmaceutical formulations comprising an immune response modifier |
| US6677349B1 (en) | 2001-12-21 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US6775514B2 (en) | 2002-01-11 | 2004-08-10 | Xerox Corporation | Substrate size monitoring system for use in copier/printers |
| US6525028B1 (en) * | 2002-02-04 | 2003-02-25 | Corixa Corporation | Immunoeffector compounds |
| US20050281813A1 (en) | 2002-02-14 | 2005-12-22 | Nuvelo, Inc. | Methods of therapy and diagnosis using targeting of cells that express toll-like receptor proteins |
| ES2541132T3 (es) | 2002-02-22 | 2015-07-16 | Meda Ab | Método para reducir y tratar la inmunosupresión inducida por UV-B |
| ATE491471T1 (de) * | 2002-03-19 | 2011-01-15 | Powderject Res Ltd | Adjuvantien für dns impstoffe basierend auf imidazochinoline |
| US20030185835A1 (en) | 2002-03-19 | 2003-10-02 | Braun Ralph P. | Adjuvant for vaccines |
| EP3006043B1 (en) | 2002-04-04 | 2019-05-29 | Zoetis Belgium S.A. | Immunostimulatory g,u-containing oligoribonucleotides |
| CN101837003B (zh) | 2002-04-30 | 2013-09-04 | 尤尼根公司 | 作为治疗剂的包含无取代b环类黄酮和黄烷混合物的组合物 |
| GB0211649D0 (en) | 2002-05-21 | 2002-07-03 | Novartis Ag | Organic compounds |
| WO2003101949A2 (en) | 2002-05-29 | 2003-12-11 | 3M Innovative Properties Company | Process for imidazo[4,5-c]pyridin-4-amines |
| MXPA04012199A (es) | 2002-06-07 | 2005-02-25 | 3M Innovative Properties Co | Imidazopiridinas sustituidas con eter. |
| CN1315828C (zh) * | 2002-07-23 | 2007-05-16 | 特瓦药厂私人有限公司 | 通过1h-咪唑并[4,5-c]喹啉-4-邻苯二甲酰亚胺类中间体制备1h-咪唑并[4,5-c]喹啉-4-胺类化合物 |
| JP2005537287A (ja) * | 2002-07-26 | 2005-12-08 | テバ ジョジセルジャール レースベニュタールシャシャーグ | 新規1H−イミダゾ[4,5−c]キノリン−4−シアノ及び1H−イミダゾ[4,5−c]キノリン−4−カルボキサミド中間体による1H−イミダゾ[4,5−c]キノリン−4−アミン類の製造 |
| EP1545597B1 (en) | 2002-08-15 | 2010-11-17 | 3M Innovative Properties Company | Immunostimulatory compositions and methods of stimulating an immune response |
| AU2003299082A1 (en) | 2002-09-26 | 2004-04-19 | 3M Innovative Properties Company | 1h-imidazo dimers |
| EP1556053A4 (en) | 2002-10-31 | 2006-04-19 | Amgen Inc | ANTI-INFLAMMATORY AGENTS |
| WO2004043913A2 (en) | 2002-11-08 | 2004-05-27 | Trimeris, Inc. | Hetero-substituted benzimidazole compounds and antiviral uses thereof |
| WO2004053452A2 (en) | 2002-12-11 | 2004-06-24 | 3M Innovative Properties Company | Assays relating to toll-like receptor activity |
| AU2003287324A1 (en) | 2002-12-11 | 2004-06-30 | 3M Innovative Properties Company | Gene expression systems and recombinant cell lines |
| WO2004058759A1 (en) * | 2002-12-20 | 2004-07-15 | 3M Innovative Properties Company | Aryl / hetaryl substituted imidazoquinolines |
| CA2511538C (en) | 2002-12-30 | 2013-11-26 | 3M Innovative Properties Company | Immunostimulatory combinations |
| US7375180B2 (en) | 2003-02-13 | 2008-05-20 | 3M Innovative Properties Company | Methods and compositions related to IRM compounds and Toll-like receptor 8 |
| EP1599726A4 (en) | 2003-02-27 | 2009-07-22 | 3M Innovative Properties Co | SELECTIVE MODULATION OF TLR-MEDIATED BIOLOGICAL ACTIVITY |
| CA2517528A1 (en) | 2003-03-04 | 2004-09-16 | 3M Innovative Properties Company | Prophylactic treatment of uv-induced epidermal neoplasia |
| US7163947B2 (en) * | 2003-03-07 | 2007-01-16 | 3M Innovative Properties Company | 1-Amino 1H-imidazoquinolines |
| EP1605943A4 (en) * | 2003-03-07 | 2008-01-16 | 3M Innovative Properties Co | 1-AMINO-1H-imidazoquinolines |
| US7699057B2 (en) | 2003-03-13 | 2010-04-20 | 3M Innovative Properties Company | Methods for treating skin lesions |
| AU2004220465A1 (en) * | 2003-03-13 | 2004-09-23 | 3M Innovative Properties Company | Method of tattoo removal |
| CA2518282C (en) | 2003-03-13 | 2012-11-06 | 3M Innovative Properties Company | Methods of improving skin quality |
| WO2004087049A2 (en) | 2003-03-25 | 2004-10-14 | 3M Innovative Properties Company | Selective activation of cellular activities mediated through a common toll-like receptor |
| US20040192585A1 (en) | 2003-03-25 | 2004-09-30 | 3M Innovative Properties Company | Treatment for basal cell carcinoma |
| US7893096B2 (en) | 2003-03-28 | 2011-02-22 | Novartis Vaccines And Diagnostics, Inc. | Use of small molecule compounds for immunopotentiation |
| US20040202720A1 (en) | 2003-04-10 | 2004-10-14 | 3M Innovative Properties Company | Delivery of immune response modifier compounds using metal-containing particulate support materials |
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| US20040214851A1 (en) | 2003-04-28 | 2004-10-28 | 3M Innovative Properties Company | Compositions and methods for induction of opioid receptors |
| US7731967B2 (en) | 2003-04-30 | 2010-06-08 | Novartis Vaccines And Diagnostics, Inc. | Compositions for inducing immune responses |
| WO2004110991A2 (en) | 2003-06-06 | 2004-12-23 | 3M Innovative Properties Company | PROCESS FOR IMIDAZO[4,5-c]PYRIDIN-4-AMINES |
| AR044466A1 (es) | 2003-06-06 | 2005-09-14 | 3M Innovative Properties Co | Proceso para la preparacion de imidazo [4,5-c] piridin-4-aminas |
| BRPI0411514A (pt) | 2003-06-20 | 2006-08-01 | Coley Pharm Gmbh | antagonistas de receptor toll-like de molécula pequena |
| MY157827A (en) | 2003-06-27 | 2016-07-29 | 3M Innovative Properties Co | Sulfonamide substituted imidazoquinolines |
| US20050106300A1 (en) | 2003-06-30 | 2005-05-19 | Purdue Research Foundation | Method for producing a material having an increased solubility in alcohol |
| CA2534042A1 (en) * | 2003-07-31 | 2005-02-10 | 3M Innovative Properties Company | Compositions for encapsulation and controlled release |
| CA2534313C (en) | 2003-08-05 | 2013-03-19 | 3M Innovative Properties Company | Formulations containing an immune response modifier |
| TW200510412A (en) * | 2003-08-12 | 2005-03-16 | 3M Innovative Properties Co | Oxime substituted imidazo-containing compounds |
| CA2535338C (en) | 2003-08-14 | 2013-05-28 | 3M Innovative Properties Company | Substituted 1h-imidazo[4,5-c]pyridin-4-amines,1h-imidazo[4,5-c]quinolin -4-amines and 1h-imidazo[4,5-c]naphthyridin-4-amines as immune response modifiers |
| JP2007504145A (ja) * | 2003-08-25 | 2007-03-01 | スリーエム イノベイティブ プロパティズ カンパニー | 免疫刺激性の組み合わせおよび治療 |
| US8961477B2 (en) | 2003-08-25 | 2015-02-24 | 3M Innovative Properties Company | Delivery of immune response modifier compounds |
| CA2536136C (en) | 2003-08-27 | 2012-10-30 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
| JP2007504172A (ja) | 2003-09-02 | 2007-03-01 | スリーエム イノベイティブ プロパティズ カンパニー | 粘膜に関連した症状の処置に関する方法 |
| NZ545536A (en) * | 2003-09-05 | 2010-04-30 | Anadys Pharmaceuticals Inc | TLR7 ligands for the treatment of hepatitis C |
| JP2007504269A (ja) * | 2003-09-05 | 2007-03-01 | スリーエム イノベイティブ プロパティズ カンパニー | Cd5+b細胞リンパ腫の治療方法 |
| GB0321615D0 (en) | 2003-09-15 | 2003-10-15 | Glaxo Group Ltd | Improvements in vaccination |
| EP1664342A4 (en) | 2003-09-17 | 2007-12-26 | 3M Innovative Properties Co | SELECTIVE MODULATION OF TLR GENE EXPRESSION |
| WO2005033049A2 (en) | 2003-10-01 | 2005-04-14 | Taro Pharmaceuticals U.S.A., Inc. | METHOD OF PREPARING 4-AMINO-1H-IMIDAZO(4,5-c)QUINOLINES AND ACID ADDITION SALTS THEREOF |
| US7544697B2 (en) | 2003-10-03 | 2009-06-09 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and analogs thereof |
| JP5043435B2 (ja) * | 2003-10-03 | 2012-10-10 | スリーエム イノベイティブ プロパティズ カンパニー | アルコキシ置換イミダゾキノリン |
| US20090075980A1 (en) * | 2003-10-03 | 2009-03-19 | Coley Pharmaceutical Group, Inc. | Pyrazolopyridines and Analogs Thereof |
| AU2004285575A1 (en) | 2003-10-31 | 2005-05-12 | 3M Innovative Properties Company | Neutrophil activation by immune response modifier compounds |
| US20050239733A1 (en) | 2003-10-31 | 2005-10-27 | Coley Pharmaceutical Gmbh | Sequence requirements for inhibitory oligonucleotides |
| ITMI20032121A1 (it) | 2003-11-04 | 2005-05-05 | Dinamite Dipharma Spa In Forma Abbr Eviata Dipharm | Procedimento per la preparazione di imiquimod e suoi intermedi |
| CN1906193A (zh) * | 2003-11-14 | 2007-01-31 | 3M创新有限公司 | 肟取代的咪唑环化合物 |
| AU2004291122A1 (en) | 2003-11-14 | 2005-06-02 | 3M Innovative Properties Company | Hydroxylamine substituted imidazo ring compounds |
| CU23404A1 (es) | 2003-11-19 | 2009-08-04 | Ct Ingenieria Genetica Biotech | Polisacáridos capsulares de neisseria meningitidis como inmunopotenciadores mucosales y formulaciones resultantes |
| CN1902200A (zh) | 2003-11-21 | 2007-01-24 | 诺瓦提斯公司 | 用做蛋白激酶抑制剂的1h-咪唑并喹啉类衍生物 |
| AR046845A1 (es) | 2003-11-21 | 2005-12-28 | Novartis Ag | Derivados de 1h-imidazo[4,5-c]quinolina para tratamiento de enfermedades dependientes de las proteino-quinasas |
| EP1686992A4 (en) | 2003-11-25 | 2009-11-04 | 3M Innovative Properties Co | HYDROXYLAMINE, AND IMIDAZOQUINOLEINS, AND IMIDAZOPYRIDINES AND IMIDAZONAPHTYRIDINE SUBSTITUTED WITH OXIME |
| AR046781A1 (es) * | 2003-11-25 | 2005-12-21 | 3M Innovative Properties Co | Derivados de imidazoquinolinas. composiciones farmaceuticas. |
| US20050226878A1 (en) | 2003-12-02 | 2005-10-13 | 3M Innovative Properties Company | Therapeutic combinations and methods including IRM compounds |
| EP1689361A4 (en) | 2003-12-02 | 2009-06-17 | 3M Innovative Properties Co | THERAPEUTIC COMBINATIONS AND PROCESSES WITH IRM COMPOUNDS |
| WO2005076783A2 (en) | 2003-12-04 | 2005-08-25 | 3M Innovative Properties Company | Sulfone substituted imidazo ring ethers |
| WO2005066170A1 (en) * | 2003-12-29 | 2005-07-21 | 3M Innovative Properties Company | Arylalkenyl and arylalkynyl substituted imidazoquinolines |
| CA2552101A1 (en) | 2003-12-29 | 2005-07-21 | 3M Innovative Properties Company | Piperazine, [1,4]diazepane, [1,4]diazocane, and [1,5]diazocane fused imidazo ring compounds |
| WO2005066169A2 (en) * | 2003-12-30 | 2005-07-21 | 3M Innovative Properties Company | Imidazoquinolinyl, imidazopyridinyl, and imidazonaphthyridinyl sulfonamides |
| US20050239735A1 (en) | 2003-12-30 | 2005-10-27 | 3M Innovative Properties Company | Enhancement of immune responses |
| WO2005065678A1 (en) | 2003-12-30 | 2005-07-21 | 3M Innovative Properties Company | Immunomodulatory combinations |
| AU2005222995B2 (en) | 2004-03-15 | 2010-08-26 | 3M Innovative Properties Company | Immune response modifier formulations and methods |
| WO2005094531A2 (en) | 2004-03-24 | 2005-10-13 | 3M Innovative Properties Company | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines |
| EP1735010A4 (en) | 2004-04-09 | 2008-08-27 | 3M Innovative Properties Co | METHODS, COMPOSITIONS AND PREPARATIONS FOR ADMINISTRATION OF IMMUNE RESPONSE MODIFIERS (MRI) |
| JP2008505857A (ja) * | 2004-04-28 | 2008-02-28 | スリーエム イノベイティブ プロパティズ カンパニー | 粘膜ワクチン接種のための組成物および方法 |
| US20050267145A1 (en) | 2004-05-28 | 2005-12-01 | Merrill Bryon A | Treatment for lung cancer |
| US20080015184A1 (en) * | 2004-06-14 | 2008-01-17 | 3M Innovative Properties Company | Urea Substituted Imidazopyridines, Imidazoquinolines, and Imidazonaphthyridines |
| US8017779B2 (en) | 2004-06-15 | 2011-09-13 | 3M Innovative Properties Company | Nitrogen containing heterocyclyl substituted imidazoquinolines and imidazonaphthyridines |
| WO2006009826A1 (en) | 2004-06-18 | 2006-01-26 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| EP1765348B1 (en) * | 2004-06-18 | 2016-08-03 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| US7897609B2 (en) | 2004-06-18 | 2011-03-01 | 3M Innovative Properties Company | Aryl substituted imidazonaphthyridines |
| WO2006009832A1 (en) | 2004-06-18 | 2006-01-26 | 3M Innovative Properties Company | Substituted imidazo ring systems and methods |
| WO2006093514A2 (en) | 2004-06-18 | 2006-09-08 | 3M Innovative Properties Company | Aryl and arylalkylenyl substituted thiazoloquinolines and thiazolonaphthyridines |
| EP1799256A4 (en) * | 2004-08-27 | 2009-10-21 | 3M Innovative Properties Co | METHOD FOR PROVIDING AN IMMUNE RESPONSE AGAINST HIV |
| US20060045886A1 (en) * | 2004-08-27 | 2006-03-02 | Kedl Ross M | HIV immunostimulatory compositions |
| US20090270443A1 (en) | 2004-09-02 | 2009-10-29 | Doris Stoermer | 1-amino imidazo-containing compounds and methods |
| AU2005282726B2 (en) | 2004-09-02 | 2011-06-02 | 3M Innovative Properties Company | 1-alkoxy 1H-imidazo ring systems and methods |
| AU2005282523A1 (en) * | 2004-09-02 | 2006-03-16 | 3M Innovative Properties Company | 2-amino 1H imidazo ring systems and methods |
| US20080193468A1 (en) | 2004-09-08 | 2008-08-14 | Children's Medical Center Corporation | Method for Stimulating the Immune Response of Newborns |
| US20080213308A1 (en) | 2004-09-14 | 2008-09-04 | Nicholas Valiante | Imidazoquinoline Compounds |
| JP2008515928A (ja) | 2004-10-08 | 2008-05-15 | スリーエム イノベイティブ プロパティズ カンパニー | Dnaワクチンのためのアジュバント |
| US20100113565A1 (en) | 2004-12-08 | 2010-05-06 | Gorden Keith B | Immunostimulatory combinations and methods |
| WO2006063072A2 (en) | 2004-12-08 | 2006-06-15 | 3M Innovative Properties Company | Immunomodulatory compositions, combinations and methods |
| US8080560B2 (en) * | 2004-12-17 | 2011-12-20 | 3M Innovative Properties Company | Immune response modifier formulations containing oleic acid and methods |
| US8436176B2 (en) | 2004-12-30 | 2013-05-07 | Medicis Pharmaceutical Corporation | Process for preparing 2-methyl-1-(2-methylpropyl)-1H-imidazo[4,5-c][1,5]naphthyridin-4-amine |
| EP1830880A4 (en) | 2004-12-30 | 2008-03-26 | Coley Pharm Group Inc | MULTI-CHANNEL ADMINISTRATION OF IMMUNE RESPONSE MODIFIER COMPOUNDS |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| CA2592897A1 (en) | 2004-12-30 | 2006-07-13 | Takeda Pharmaceutical Company Limited | 1-(2-methylpropyl)-1h-imidazo[4,5-c][1,5]naphthyridin-4-amine ethanesulfonate and 1-(2-methylpropyl)-1h-imidazo[4,5-c][1,5]naphthyridin-4-amine methanesulfonate |
| WO2006074003A2 (en) | 2004-12-30 | 2006-07-13 | 3M Innovative Properties Company | CHIRAL FUSED [1,2]IMIDAZO[4,5-c] RING COMPOUNDS |
| JP2008526765A (ja) * | 2004-12-30 | 2008-07-24 | スリーエム イノベイティブ プロパティズ カンパニー | 皮膚転移の処置 |
| WO2006083400A2 (en) | 2005-02-02 | 2006-08-10 | Mocon, Inc. | Instrument and method for detecting and reporting the size of leaks in hermetically sealed packaging |
| WO2006084251A2 (en) | 2005-02-04 | 2006-08-10 | Coley Pharmaceutical Group, Inc. | Aqueous gel formulations containing immune reponse modifiers |
| AU2006212765B2 (en) | 2005-02-09 | 2012-02-02 | 3M Innovative Properties Company | Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines |
| AU2006338521A1 (en) | 2005-02-09 | 2007-10-11 | Coley Pharmaceutical Group, Inc. | Oxime and hydroxylamine substituted thiazolo(4,5-c) ring compounds and methods |
| AU2006213745A1 (en) * | 2005-02-11 | 2006-08-17 | Coley Pharmaceutical Group, Inc. | Substituted fused [1,2]imidazo[4,5-c] ring compounds and methods |
| CA2597446A1 (en) | 2005-02-11 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Substituted imidazoquinolines and imidazonaphthyridines |
| JP2008530113A (ja) | 2005-02-11 | 2008-08-07 | コーリー ファーマシューティカル グループ,インコーポレイテッド | オキシムおよびヒドロキシラミン置換イミダゾ[4,5−c]環化合物および方法 |
| AU2006216799A1 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Hydroxyalkyl substituted imidazonaphthyridines |
| JP2008531567A (ja) | 2005-02-23 | 2008-08-14 | コーリー ファーマシューティカル グループ,インコーポレイテッド | ヒドロキシアルキル置換イミダゾキノリン化合物および方法 |
| AU2006216686A1 (en) * | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Method of preferentially inducing the biosynthesis of interferon |
| US8178677B2 (en) * | 2005-02-23 | 2012-05-15 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazoquinolines |
| MX2007011112A (es) | 2005-03-14 | 2007-11-07 | Graceway Pharmaceuticals Llc | Metodo para tratar queratosis actinica. |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| CA2602853A1 (en) | 2005-04-01 | 2006-11-16 | Coley Pharmaceutical Group, Inc. | Ring closing and related methods and intermediates |
| WO2006107771A2 (en) | 2005-04-01 | 2006-10-12 | Coley Pharmaceutical Group, Inc. | PYRAZOLO[3,4-c]QUINOLINES, PYRAZOLO[3,4-c]NAPHTHYRIDINES, ANALOGS THEREOF, AND METHODS |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| JP2008539252A (ja) | 2005-04-25 | 2008-11-13 | スリーエム イノベイティブ プロパティズ カンパニー | 免疫活性化組成物 |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| RS20080128A (en) | 2005-09-02 | 2009-05-06 | Pfizer Inc., | Hydroxy substituted 1h-imidazopyridines and methods |
| ZA200803029B (en) | 2005-09-09 | 2009-02-25 | Coley Pharm Group Inc | Amide and carbamate derivatives of alkyl substituted /V-[4-(4-amino-1H-imidazo[4,5-c] quinolin-1-yl)butyl] methane-sulfonamides and methods |
| EA200800782A1 (ru) | 2005-09-09 | 2008-08-29 | Коли Фармасьютикал Груп, Инк. | ПРОИЗВОДНЫЕ АМИДА И КАРБАМАТА N-{2-[4-АМИНО-2-(ЭТОКСИМЕТИЛ)-1Н-ИМИДАЗОЛО[4,5-c]ХИНОЛИН-1-IL]-1,1-ДИМЕТИЛЭТИЛ}МЕТАНСУЛЬФОНАМИДА И СПОСОБЫ |
| US8889154B2 (en) | 2005-09-15 | 2014-11-18 | Medicis Pharmaceutical Corporation | Packaging for 1-(2-methylpropyl)-1H-imidazo[4,5-c] quinolin-4-amine-containing formulation |
| AU2006292119A1 (en) | 2005-09-23 | 2007-03-29 | 3M Innovative Properties Company | Method for 1H-imidazo[4,5-c]pyridines and analogs thereof |
| JP5247458B2 (ja) | 2005-11-04 | 2013-07-24 | スリーエム・イノベイティブ・プロパティーズ・カンパニー | ヒドロキシ及びアルコキシ置換1h−イミダゾキノリン及び方法 |
| WO2007062043A1 (en) | 2005-11-23 | 2007-05-31 | Coley Pharmaceutical Group Inc. | Method of activating murine toll-like receptor 8 |
| MX2008007864A (es) | 2005-12-16 | 2009-03-04 | Coley Pharm Group Inc | Imidazoquinolinas, imidazonaftiridinas, e imidazopiridinas sustituidas, composiciones y metodos. |
| EP1968582A4 (en) | 2005-12-28 | 2011-02-16 | 3M Innovative Properties Co | TREATMENT OF LYMPHOMA T CUTANE |
| WO2007079169A2 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc. | Treatment for acute myeloid leukemia |
| WO2007079146A1 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc | Treatment for non-hodgkin's lymphoma |
| WO2007079171A2 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc. | Treatment for hodgkin's lymphoma |
| WO2007079086A1 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc. | Pyrazoloalkyl substituted imidazo ring compounds and methods |
| WO2007079202A2 (en) | 2005-12-28 | 2007-07-12 | Coley Pharmaceutical Group, Inc. | Treatment for acute lymhoblastic leukemia |
| US20090306388A1 (en) | 2006-02-10 | 2009-12-10 | Pfizer Inc. | Method for substituted ih-imidazo[4,5-c] pyridines |
| WO2007100634A2 (en) * | 2006-02-22 | 2007-09-07 | 3M Innovative Properties Company | Immune response modifier conjugates |
| WO2007106854A2 (en) | 2006-03-15 | 2007-09-20 | Coley Pharmaceutical Group, Inc. | Hydroxy and alkoxy substituted 1h-imidazonaphthyridines and methods |
| US8088788B2 (en) | 2006-03-15 | 2012-01-03 | 3M Innovative Properties Company | Substituted fused[1,2] imidazo[4,5-c] ring compounds and methods |
| WO2007143526A2 (en) | 2006-06-05 | 2007-12-13 | Coley Pharmaceutical Group, Inc. | Substituted tetrahydroimidazonaphthyridines and methods |
| US20100028381A1 (en) * | 2006-06-19 | 2010-02-04 | 3M Innovative Properties Company | Formulation for delivery of immune response modifiers |
| US7906506B2 (en) * | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US8124096B2 (en) * | 2006-07-31 | 2012-02-28 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| US8178539B2 (en) | 2006-09-06 | 2012-05-15 | 3M Innovative Properties Company | Substituted 3,4,6,7-tetrahydro-5H-1,2a,4a,8-tetraazacyclopenta[cd]phenalenes and methods |
| WO2008036312A1 (en) | 2006-09-19 | 2008-03-27 | Coley Pharmaceutical Group, Inc. | Fungicidal methods using immune response modifier compounds |
| WO2008045543A1 (en) | 2006-10-13 | 2008-04-17 | Coley Pharmaceutical Group, Inc. | Substituted 4h-imidazo [4, 5, 1-ij] [1, 6] naphthyridine-9-amines and their pharmaceutical use |
| US20080149123A1 (en) | 2006-12-22 | 2008-06-26 | Mckay William D | Particulate material dispensing hairbrush with combination bristles |
-
2004
- 2004-08-27 CA CA2536136A patent/CA2536136C/en not_active Expired - Fee Related
- 2004-08-27 AR ARP040103100A patent/AR045529A1/es unknown
- 2004-08-27 AU AU2004268625A patent/AU2004268625B2/en not_active Ceased
- 2004-08-27 ES ES04782492T patent/ES2406730T3/es active Active
- 2004-08-27 US US10/595,103 patent/US7897597B2/en not_active Expired - Fee Related
- 2004-08-27 BR BRPI0413998-4A patent/BRPI0413998A/pt not_active IP Right Cessation
- 2004-08-27 KR KR1020067003863A patent/KR101106812B1/ko not_active IP Right Cessation
- 2004-08-27 EP EP04782492A patent/EP1658076B1/en not_active Not-in-force
- 2004-08-27 JP JP2006524906A patent/JP5128815B2/ja not_active Expired - Fee Related
- 2004-08-27 MX MXPA06002199A patent/MXPA06002199A/es active IP Right Grant
- 2004-08-27 WO PCT/US2004/028021 patent/WO2005020999A1/en active Application Filing
- 2004-08-27 RU RU2006105101/04A patent/RU2006105101A/ru unknown
- 2004-08-27 MY MYPI20043526A patent/MY145983A/en unknown
- 2004-08-27 NZ NZ545412A patent/NZ545412A/en not_active IP Right Cessation
-
2006
- 2006-02-14 IL IL173728A patent/IL173728A0/en unknown
-
2011
- 2011-01-18 US US13/008,453 patent/US8263594B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000076519A1 (en) * | 1999-06-10 | 2000-12-21 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| WO2002046193A2 (en) * | 2000-12-08 | 2002-06-13 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| WO2003050121A1 (en) | 2001-12-06 | 2003-06-19 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2004268625A1 (en) | 2005-03-10 |
| KR20070026298A (ko) | 2007-03-08 |
| EP1658076A1 (en) | 2006-05-24 |
| AR045529A1 (es) | 2005-11-02 |
| AU2004268625B2 (en) | 2011-03-31 |
| MXPA06002199A (es) | 2006-05-22 |
| IL173728A0 (en) | 2006-07-05 |
| BRPI0413998A (pt) | 2006-11-07 |
| US7897597B2 (en) | 2011-03-01 |
| RU2006105101A (ru) | 2007-10-10 |
| ES2406730T3 (es) | 2013-06-07 |
| CA2536136A1 (en) | 2005-03-10 |
| CA2536136C (en) | 2012-10-30 |
| US20110144099A1 (en) | 2011-06-16 |
| WO2005020999A1 (en) | 2005-03-10 |
| MY145983A (en) | 2012-05-31 |
| NZ545412A (en) | 2008-12-24 |
| US20090018122A1 (en) | 2009-01-15 |
| JP5128815B2 (ja) | 2013-01-23 |
| EP1658076A4 (en) | 2009-02-25 |
| EP1658076B1 (en) | 2013-03-06 |
| US8263594B2 (en) | 2012-09-11 |
| JP2007504161A (ja) | 2007-03-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101106812B1 (ko) | 아릴옥시 및 아릴알킬렌옥시 치환된 이미다조퀴놀린 | |
| US7163947B2 (en) | 1-Amino 1H-imidazoquinolines | |
| US7648997B2 (en) | Hydroxylamine substituted imidazoquinolines | |
| US8778963B2 (en) | Hydroxylamine and oxime substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines | |
| KR101154101B1 (ko) | 알콕시 치환된 이미다조퀴놀린 | |
| US7943610B2 (en) | Pyrazolopyridine-1,4-diamines and analogs thereof | |
| US8026366B2 (en) | Aryloxy and arylalkyleneoxy substituted thiazoloquinolines and thiazolonaphthyridines | |
| JP5247458B2 (ja) | ヒドロキシ及びアルコキシ置換1h−イミダゾキノリン及び方法 | |
| US20090270443A1 (en) | 1-amino imidazo-containing compounds and methods | |
| US20040176367A1 (en) | 1-Amino 1H-imidazoquinolines | |
| US20090030030A1 (en) | Arylalkenyl and arylalkynyl substituted imidazoquinolines | |
| US20090023720A1 (en) | 2-Amino 1H-Imidazo Ring Systems and Methods | |
| WO2006028451A1 (en) | 1-amino 1-h-imidazoquinolines | |
| JP2006513212A (ja) | アリール/ヘタリール置換されたイミダゾキノリン | |
| JP2008538203A (ja) | インターフェロンの生合成を優先的に誘導する方法 | |
| US20180222906A1 (en) | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20141230 Year of fee payment: 4 |
|
| FPAY | Annual fee payment |
Payment date: 20151217 Year of fee payment: 5 |
|
| FPAY | Annual fee payment |
Payment date: 20161220 Year of fee payment: 6 |
|
| FPAY | Annual fee payment |
Payment date: 20171219 Year of fee payment: 7 |
|
| LAPS | Lapse due to unpaid annual fee |