KR101083631B1 - C형 간염 바이러스의 마크로사이클릭 억제제 - Google Patents
C형 간염 바이러스의 마크로사이클릭 억제제 Download PDFInfo
- Publication number
- KR101083631B1 KR101083631B1 KR1020087004894A KR20087004894A KR101083631B1 KR 101083631 B1 KR101083631 B1 KR 101083631B1 KR 1020087004894 A KR1020087004894 A KR 1020087004894A KR 20087004894 A KR20087004894 A KR 20087004894A KR 101083631 B1 KR101083631 B1 KR 101083631B1
- Authority
- KR
- South Korea
- Prior art keywords
- alkyl
- compound
- formula
- mmol
- methyl
- Prior art date
Links
- 0 CC[C@@](CC*N(*)C(*(C(*)C1(C)C)C2CC1O*)=O)C(C1)C1(C(*)=O)NC2=O Chemical compound CC[C@@](CC*N(*)C(*(C(*)C1(C)C)C2CC1O*)=O)C(C1)C1(C(*)=O)NC2=O 0.000 description 8
- BQHNWMSZACYBGE-PMDINCESSA-N CC(C)(C)OC([C@](C1)([C@@H]1C=C)NC(C(C1)C(C(OC)=O)=CC1O)=O)=O Chemical compound CC(C)(C)OC([C@](C1)([C@@H]1C=C)NC(C(C1)C(C(OC)=O)=CC1O)=O)=O BQHNWMSZACYBGE-PMDINCESSA-N 0.000 description 1
- UCLGAKSFBCVVOQ-DFAJJQHTSA-N CC(C)Nc1nc(O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C=C\CCCCN3C)C(NS(C4CC4)(=O)=O)=O)=O)C[C@H]2[IH]3=O)c(cccc2)c2n1 Chemical compound CC(C)Nc1nc(O[C@H](C[C@H]2C(N[C@](C3)([C@@H]3/C=C\CCCCN3C)C(NS(C4CC4)(=O)=O)=O)=O)C[C@H]2[IH]3=O)c(cccc2)c2n1 UCLGAKSFBCVVOQ-DFAJJQHTSA-N 0.000 description 1
- RXGIAFQRQYBVIC-YFDKIRSFSA-N CC(C)c1c[s]c(-c2nc(OC(C[C@H]3C(NC(C4)([C@@H]4/C=C\CCCCN4C)C(NS(C5CC5)(=O)=O)=O)=O)CN3C4=O)c(ccc(OC)c3C)c3n2)n1 Chemical compound CC(C)c1c[s]c(-c2nc(OC(C[C@H]3C(NC(C4)([C@@H]4/C=C\CCCCN4C)C(NS(C5CC5)(=O)=O)=O)=O)CN3C4=O)c(ccc(OC)c3C)c3n2)n1 RXGIAFQRQYBVIC-YFDKIRSFSA-N 0.000 description 1
- AOFCGSHJMOLSTI-UHFFFAOYSA-N CC(C)c1c[s]c(C(Nc2c(C)c(OC)ccc2C(N)=O)=O)n1 Chemical compound CC(C)c1c[s]c(C(Nc2c(C)c(OC)ccc2C(N)=O)=O)n1 AOFCGSHJMOLSTI-UHFFFAOYSA-N 0.000 description 1
- OYOFMEDPWSXIKW-UHFFFAOYSA-N CC(C)c1c[s]c(C(Nc2ccccc2C(N)=O)=O)n1 Chemical compound CC(C)c1c[s]c(C(Nc2ccccc2C(N)=O)=O)n1 OYOFMEDPWSXIKW-UHFFFAOYSA-N 0.000 description 1
- RODBMEIPZUUYIM-MZTSMEHPSA-N CCOC(C(C1)([C@@H]1C=C)NC([C@H](C[C@H](C1)Oc2c(ccc(OC)c3C)c3nc(-c3ccccc3)n2)[C@@H]1C(O)=O)=O)=O Chemical compound CCOC(C(C1)([C@@H]1C=C)NC([C@H](C[C@H](C1)Oc2c(ccc(OC)c3C)c3nc(-c3ccccc3)n2)[C@@H]1C(O)=O)=O)=O RODBMEIPZUUYIM-MZTSMEHPSA-N 0.000 description 1
- ZOKFYEHVRJASAY-VOLPKPDMSA-N CCOC([C@@](C1)([C@@H]1/C=C\CCCCN(C)C(N(C1)[C@H]2C[C@H]1Oc1c(ccc(OC)c3C)c3nc(-c3cccc(F)c3)n1)=O)NC2=O)=O Chemical compound CCOC([C@@](C1)([C@@H]1/C=C\CCCCN(C)C(N(C1)[C@H]2C[C@H]1Oc1c(ccc(OC)c3C)c3nc(-c3cccc(F)c3)n1)=O)NC2=O)=O ZOKFYEHVRJASAY-VOLPKPDMSA-N 0.000 description 1
- QYPNVARUOIUHPB-KJWPEZJGSA-N CCOC([C@](C1)(CCCC([C@H](C2)N3C[C@H]2OC(c(cc2)ccc2[N+]([O-])=O)=O)=O)[C@@H]1/C=C\CCCCN(C)C3=O)=O Chemical compound CCOC([C@](C1)(CCCC([C@H](C2)N3C[C@H]2OC(c(cc2)ccc2[N+]([O-])=O)=O)=O)[C@@H]1/C=C\CCCCN(C)C3=O)=O QYPNVARUOIUHPB-KJWPEZJGSA-N 0.000 description 1
- VRQDYQBGGWADRE-DWHMSSSTSA-N CCO[IH]([C@@](C1)([C@@H]1C=C)NC([C@H](C[C@@H](C1)OC(c(cc2)ccc2[N+]([O-])=O)=O)N1C(N(CCCCCC=C)Cc(cc1)ccc1OC)=O)=O)=O Chemical compound CCO[IH]([C@@](C1)([C@@H]1C=C)NC([C@H](C[C@@H](C1)OC(c(cc2)ccc2[N+]([O-])=O)=O)N1C(N(CCCCCC=C)Cc(cc1)ccc1OC)=O)=O)=O VRQDYQBGGWADRE-DWHMSSSTSA-N 0.000 description 1
- YXGIXELWCSNGRU-SOSRDRKYSA-N CN(CCCCCC[C@H](C1)C1(C(O)=O)NC([C@H](C1)[C@H]2C[C@@H]1Oc1c(ccc(OC)c3)c3nc(-c3ccccc3)n1)=O)C2=O Chemical compound CN(CCCCCC[C@H](C1)C1(C(O)=O)NC([C@H](C1)[C@H]2C[C@@H]1Oc1c(ccc(OC)c3)c3nc(-c3ccccc3)n1)=O)C2=O YXGIXELWCSNGRU-SOSRDRKYSA-N 0.000 description 1
- RVKZDHKAALGKSL-CRMQODIRSA-N CN(CCCCCC[C@H](C1)[C@]1(C(O)=O)NC([C@H](C1)N2C[C@@H]1Oc1c(ccc(OC)c3)c3nc(-c3ccccc3)n1)=O)C2=O Chemical compound CN(CCCCCC[C@H](C1)[C@]1(C(O)=O)NC([C@H](C1)N2C[C@@H]1Oc1c(ccc(OC)c3)c3nc(-c3ccccc3)n1)=O)C2=O RVKZDHKAALGKSL-CRMQODIRSA-N 0.000 description 1
- FNXZMHNQECQCNX-UHFFFAOYSA-N COc(cc1)cc(N)c1C(N)=O Chemical compound COc(cc1)cc(N)c1C(N)=O FNXZMHNQECQCNX-UHFFFAOYSA-N 0.000 description 1
- PMIRQABZZUUPJI-UHFFFAOYSA-N Cc(c(O)c(cc1C#N)C(O)=O)c1N Chemical compound Cc(c(O)c(cc1C#N)C(O)=O)c1N PMIRQABZZUUPJI-UHFFFAOYSA-N 0.000 description 1
- RURNIKQBKQSAQB-UHFFFAOYSA-N Cc(c(OC)c1)cc(C(N)=O)c1N Chemical compound Cc(c(OC)c1)cc(C(N)=O)c1N RURNIKQBKQSAQB-UHFFFAOYSA-N 0.000 description 1
- PGRJUTLFMUYRKN-UHFFFAOYSA-N Cc(c(OC)ccc12)c1ncnc2O Chemical compound Cc(c(OC)ccc12)c1ncnc2O PGRJUTLFMUYRKN-UHFFFAOYSA-N 0.000 description 1
- CDZMIQSRVJVDFS-UHFFFAOYSA-N Cc(c(OC)ccc1C#N)c1N Chemical compound Cc(c(OC)ccc1C#N)c1N CDZMIQSRVJVDFS-UHFFFAOYSA-N 0.000 description 1
- ITVRAEZTYQSUIS-UHFFFAOYSA-N Cc(c(OC)ccc1c(O)n2)c1nc2Cl Chemical compound Cc(c(OC)ccc1c(O)n2)c1nc2Cl ITVRAEZTYQSUIS-UHFFFAOYSA-N 0.000 description 1
- VHUAAFCMNYCPLH-ZHQUQFPWSA-N Cc1nc(-c2nc(O[C@H](C[C@H]3C(NC(C4)([C@@H]4/C=C\CCCCN4C)C(NS(C5CC5)(=O)=O)=O)=O)CN3C4=O)c(ccc(OC)c3C)c3n2)ccc1 Chemical compound Cc1nc(-c2nc(O[C@H](C[C@H]3C(NC(C4)([C@@H]4/C=C\CCCCN4C)C(NS(C5CC5)(=O)=O)=O)=O)CN3C4=O)c(ccc(OC)c3C)c3n2)ccc1 VHUAAFCMNYCPLH-ZHQUQFPWSA-N 0.000 description 1
- YWHQIVJPIBHBSV-UHFFFAOYSA-N OC(C(CC(C1)O2)C1C2=O)=O Chemical compound OC(C(CC(C1)O2)C1C2=O)=O YWHQIVJPIBHBSV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/10—Spiro-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D245/00—Heterocyclic compounds containing rings of more than seven members having two nitrogen atoms as the only ring hetero atoms
- C07D245/04—Heterocyclic compounds containing rings of more than seven members having two nitrogen atoms as the only ring hetero atoms condensed with carbocyclic rings or ring systems
Abstract
화학식 (I)의 화합물, 이의 N-옥사이드, 염, 또는 입체이성체(A는 OR1, NHS(=O)pR2를 나타내며; 여기서, R1은 수소, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고; R2는 C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내며; p는 독립적으로 1 또는 2를 나타내고; n은 3, 4, 5 또는 6을 나타내며; -----은 임의의 이중 결합을 나타내고; L은 N 또는 CRz를 나타내며; Rz는 H이거나, 별표 표시된 탄소와 이중 결합을 형성하고; Rq는 H이거나, L이 CRz일 때, Rq는 C1-C6알킬일 수도 있으며; Rr은 C1-C6알킬, C1-C6알콕시, 하이드록시, 할로, 할로C1-C6알킬, 아미노, 모노- 또는 디알킬아미노, 모노- 또는 디알킬아미노카보닐, C1-C6알킬카보닐-아미노, C0-C3알킬렌카보사이클릴 및 C0-C3알킬렌헤테로사이클릴에서 각각 독립적으로 선택된 하나 둘 또는 세개의 치환체로 임의로 치환된 퀴나졸리닐을 나타내고; R5는 수소, C1-C6알킬, C1-C6알콕시C1-C6알킬 또는 C3-C7사이클로알킬을 나타내며; R6는 수소, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, 하이드록시, 브로모, 클로로 또는 플루오로 를 나타낸다)는 플라비바이러스, 이를 테면, HCV 감염의 치료 또는 예방에 유용성을 갖는다.
Description
본 발명은 C형 간염 바이러스(HCV)의 복제에 억제 활성을 지니는 마크로사이클릭 화합물에 관한 것이다. 또한 활성 성분으로 이들 화합물을 함유하는 조성물 뿐 아니라 이들 화합물 및 조성물을 제조하는 방법에 관한 것이다.
C형 간염 바이러스는 전세계적으로 만성 간 질환의 주요 원인이며, 많은 의학 연구의 중심이 되고 있다. HCV는 C형 간염 바이러스 속 중 플라비바이러스 과 바이러스의 구성원이며, 인간 질환, 이를 테면, 뎅기 바이러스 및 황열 바이러스에 관련된 수많은 바이러스를 포함하는 플라비바이러스 속 및 소 바이러스성 설사 바이러스 (BVDV)를 포함하는 동물 페스티바이러스 과와 밀접하게 연관된다. HCV는 약 9,600 염기의 게놈을 갖는 파지티브-센스, 단일-가닥 RNA 바이러스이다. 게놈은 RNA 2차 구조를 결정하는 5' 및 3' 비번역 영역과 약 3,010-3,030 아미노산의 단일 폴리단백질을 암호화하는 중심의 개환 리딩 프레임을 함유한다. 폴리단백질은 숙주와 바이러스 프로테아제에 의해 매개되는, 조정된 일련의 번역과 동시 및 번역후 단백질 내부 분해효소 절단에 의하여 전구 폴리단백질에서 생성되는 10개의 유전자 산물을 암호화한다. 바이러스 구조 단백질은 코어 뉴클레오캡시드 단백질과 두개의 외피 당단백질 E1 및 E2를 포함한다. 비구조(NS) 단백질은 일부 주요한 바이러스 효소 작용(헬리카아제, 폴리머라제, 프로테아제), 뿐 아니라 알려지지 않은 작용의 단백질을 암호화한다. 바이러스 게놈의 복제는 비구조 단백질 5b (NS5B)에 의해 암호화되는 RNA-의존성 RNA 폴리머라제에 의해 매개된다. 폴리머라제 외에, 이작용성 NS3 단백질에서 암호화된 바이러스 헬리카아제 및 프로테아제 작용은 HCV RNA의 복제에 필수적인 것으로 보인다. NS3 세린 프로테아제 외에, HCV는 NS2 영역에서 메탈로프로테아제도 암호화한다.
최초 급성 감염 후에, HCV가 우선적으로 간세포에서 복제되나, 직접적으로 세포 변성되지 않으므로, 감염된 대부분의 개체에서 만성 간염이 발달한다. 특히, 격렬한 T-림프구 반응의 결여와 돌연변이가 되려는 바이러스의 높은 성향은 만성 감염을 높은 비율로 증진시키는 것으로 나타난다. 만성 간염은 간 경화, 말기 간질환 및 HCC(간세포암종)이 되어 간 이식의 주요 원인이 되는 간 섬유증으로 진행될 수 있다.
6개의 주요 HCV 유전자형과 50개 이상의 아형이 존재하며, 이는 지리적으로 다르게 존재한다. HCV 제1 타입은 유럽과 미국에서 우세한 유전자형이다. HCV의 광범위한 유전적인 이질성은 중요한 진단적 및 임상적 의미를 가지며, 이는 아마도, 백신 개발에 어려움과 치료법에 대하여 결여된 반응을 설명할 것이다.
HCV의 전염은 오염된 혈액 및 혈액 산물, 예를 들어 다음의 혈액 수혈 또는 정맥내 약물 사용으로의 접촉을 통하여 발생할 수 있다. 혈액 스크리닝에서 이용되는 진단 테스트를 도입하여 수혈-후 HCV 발병률이 낮아지도록 하였다. 그러나 말기 간 질환으로 천천히 진행된다면, 존재하고 있는 감염은 수십년 동안 심각한 의료 및 경제적인 부담을 지속적으로 나타낼 것이다.
현행 HCV 요법들은 리바비린과 배합한 (페길화) 인터페론-알파(IFN-α)를 기초로 한다. 이러한 배합 요법은 제1 유전자 타입 바이러스에 의해 감염된 환자 중 40% 이상과 제2 및 제3 유전자 타입에 의해 감염된 환자들 중 약 80%에서 지속된 바이러스학적 반응을 나타낸다. 제1 HCV 타입에 대한 제한된 효능 이외에, 배합 요법은 유의적인 부작용이 있으며 많은 환자에서 불충분하게 허용된다. 주요 부작용은 인플루엔자류 증후군, 혈액 장애, 및 신경정신병 증후군을 포함한다. 보다 효과적이고, 간편하며 더욱 허용되는 치료법의 개발이 필요하다.
2종의 펩티도미메틱 HCV 프로테아제 억제제, 즉, WO 00/59929호 공보에 개시된 BILN-2061 및 WO 03/87092호 공보에 개시된 VX-950도 임상시험단계에 들어가 있는 것으로 여겨진다. 많은 유사한 HCV 프로테아제 억제제도 학술 및 특허 문헌에 제안되어 있다. BILN-2061 또는 VX-950의 서방형 투여가 각각의 약물에 대해 내성이 있는 HCV 돌연변이체, 소위 약물 탈출 돌연변이체(drug escape mutant)를 선택하는 것은 이미 명백해져 있다. 이들 약물 탈출 돌연변이체는 HCV 프로테아제 게놈, 특히 D168V, D168A 및/또는 A156S에서 특징적인 돌연변이를 지닌다. 그러므로, 치료 옵션에 따른 결함 환자를 제공하기 위해 상이한 내성 패턴을 지닌 추가의 약물이 필요하게 될 것이고, 복수의 약물과의 배합 요법은 제 1라인 처치(first line treatment)에 대해서도 장래에 표준으로 되기 쉽다.
HIV 약물 및 특히 HIV 프로테아제 억제제의 경험은 부차적으로 적합한 약물 동태학 및 복합제 투약 요법이 신속하게 부적합한 순응 실패를 가져올 것임이 더욱 강조된다. 따라서, 이것은 HIV 요법에 있어서 각각의 약물에 대한 24시간 최저 농도(최소 혈장 농도)가 빈번하게 하루의 대부분에서 IC90 또는 ED90 역치 이하로 떨어지게 되는 것을 의미한다. 적어도 IC50, 더욱 현실적으로는 IC90 또는 ED90의 24시간 최저 레벨이 약물 탈출 돌인변이체의 발달을 늦추는 데 필수적이라고 여겨진다.
이러한 최저 레벨을 얻기 위하여 필요한 약물동태학 및 약물 대사를 얻는 것은 약물 설계에 대한 설득력있는 도전을 제공한다. 다수의 펩타이드 결합을 지닌 종래 기술의 HCV 프로테아제 억제제의 강력한 펩타이드미메틱 성질은 유효한 투여 요법에 대한 약물동태학적 장애를 지닌다.
현재 HCV 치료법의 단점, 이를 테면, 부작용, 제한된 효능, 내성 출현 및 순응 실패를 극복할 수 있는 HCV 억제제가 필요하다.
본 발명은 종래 기술 화합물보다 적어도 하나의 개선된 특성을 보유하는 HCV 복제 억제제에 관한 것이다. 특히, 본 발명의 억제제는 다음 약리 관련 특성, 예를 들어, 효능, 감소된 세포독성, 개선된 약물동태학적 특성, 개선된 내성 프로필, 허용가능한 용량 및 환의 부하 중 하나 이상에서 우수하다.
또한, 본 발명의 화합물은 상대적으로 작은 분자량을 가지며, 상업적으로 입수가능하거나 공지된 합성 과정을 통하여 즉시 입수할 수 있는 출발 물질에서 시작하여 합성하기 쉽다.
본 발명은 화학식 (I)으로 나타낼 수 있는 HCV 복제 억제제 및 이의 N-옥사 이드, 염 및 입체이성체에 관한 것이다:

상기 식에서,
A는 OR1, NHS(=O)pR2를 나타내며;
여기서, R1은 수소, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고;
R2는 C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내며;
p는 독립적으로 1 또는 2를 나타내고;
n은 3, 4, 5 또는 6을 나타내며;
-----은 임의의 이중 결합을 나타내고;
L은 N 또는 CRz를 나타내며;
Rz는 H이거나, 별표 표시된 탄소와 이중 결합을 형성하고;
Rq는 H이거나, L이 CRz일 때, Rq는 C1-C6알킬일 수도 있으며;
Rr은 C1-C6알킬, C1-C6알콕시, 하이드록시, 할로, 할로C1-C6알킬, 아미노, 모노- 또는 디알킬아미노, 모노- 또는 디알킬아미노카보닐, C1-C6알킬카보닐-아미노, C0-C3알킬렌카보사이클릴 및 C0-C3알킬렌헤테로사이클릴에서 각각 독립적으로 선택된 하나 둘 또는 세개의 치환체로 임의로 치환된 퀴나졸리닐을 나타내고;
R5는 수소, C1-C6알킬, C1-C6알콕시C1-C6알킬 또는 C3-C7사이클로알킬을 나타내며;
여기서, 각 C1-C6알킬, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴은 할로, 옥소, 니트릴, 아지도, 니트로, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, NH2C(=O)-, Y-NRaRb, Y-O-Rb, Y-C(=O)Rb, Y-(C=O)NRaRb, Y-NRaC(=O)Rb, Y-NHSOpRb, Y-S(=O)pRb 및 Y-S(=O)pNRaRb, Y-C(=O)ORb, Y-NRaC(=O)ORb로 구성된 군에서 독립적으로 선택된 1개 내지 3개 치환체로 임의로 치환되고;
Y는 독립적으로 결합 또는 C1-C3알킬렌을 나타내며;
Ra는 독립적으로 H, C1-C6알콕시, C1-C3알킬을 나타내거나; 또는
Rb는 독립적으로 H, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴을 나타내거나;
Ra 및 Rb는 그들이 부착되는 질소와 함께 헤테로사이클릴기를 형성하는 데 관여한다.
또한, 본 발명은 화학식 (I)의 화합물, 이의 전구 약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 및 입체이성체의 제조 방법, 이의 중간체 및 화학식 (I)의 화합물의 제조에서 중간체의 용도에 관한 것이다.
본 발명은 의약으로 이용하기 위한 본래 화학식 (I)의 화합물, 이의 전구 약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 및 입체이성체에 관한 것이다. 또한 본 발명은 HCV 감염으로 고통받는 대상에 투여하기 위한 상기 언급된 화합물을 포함하는 약제학적 조성물에 관한 것이다. 약제학적 조성물은 상기 언급된 화합물과 다른 항-HCV 약제의 배합물을 포함할 수 있다.
또한 본 발명은 HCV 복제를 억제하기 위한 의약 제조용 화학식 (I)의 화합물 또는 이의 전구 약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 또는 입체이성체의 용도에 관한 것이다. 또는 본 발명은 화학식 (I)의 화합물 또는 이의 전구약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 또는 입체이성체의 유효량의 투여를 포함하여 온혈 동물에서 HCV 복제를 억제하는 방법에 관한 것이다.
본 발명은 또한 하기 화학식 (It)에 나타낸 화학식 (I)의 화합물 및 이의 N-옥사이드, 염 및 입체이성체에 관한 것이다:

상기 식에서,
Xt는 N, CH를 나타내나, Xt가 이중 결합을 갖는 경우 그것은 C를 나타내며,
Rt1은 -ORt5, -NH-SO2Rt6를 나타내고;
Rt2는 수소이나, Xt가 C 또는 CH인 경우, Rt2는 C1 - 6알킬을 나타낼 수 있으며;
Rt3는 수소, C1 - 6알킬, C1 - 6알콕시C1 - 6알킬, 또는 C3 - 7사이클로알킬을 나타내고;
Rt4는 C1 - 6알킬, C1 - 6알콕시, 하이드록시, 할로, 폴리할로C1 - 6알킬, 폴리할로C1 - 6알콕시, 아미노, 모노- 또는 디C1 - 6알킬아미노, 모노- 또는 디C1 - 6알킬아미노카보닐, C1-6알킬카보닐-아미노, 아릴 및 Het에서 각각 독립적으로 선택된 하나 둘 또는 세개의 치환체로 임의로 치환된 퀴나졸리닐을 나타내고;
n은 3, 4, 5, 또는 6을 나타내며;
여기서, 각각의 파선(-----로 나타냄)은 임의의 이중 결합을 나타내고;
Rt5는 수소; 아릴; Het; C1 - 6알킬로 임의로 치환된 C3 - 7사이클로알킬; 또는 C3-7사이클로알킬, 아릴 또는 Het로 임의로 치환된 C1 - 6알킬을 나타내며;
Rt6는 아릴; Het; C1 - 6알킬로 임의로 치환된 C3 - 7사이클로알킬; 또는 C3 - 7사이클로알킬, 아릴 또는 Het로 임의로 치환된 C1 - 6알킬을 나타내며;
기 또는 기의 일부로서 각각의 아릴은 할로, 하이드록시, 니트로, 시아노, 카복실, C1 - 6알킬, C1 - 6알콕시, C1 - 6알콕시C1 - 6알킬, C1 - 6알킬카보닐, 아미노, 모노- 또는 디C1 - 6알킬아미노, 아지도, 머캅토, 폴리할로C1 - 6알킬, 폴리할로C1 - 6알콕시, 사이클로프로필, 피롤리디닐, 피페리디닐, 피페라지닐, 4-C1 - 6알킬피페라지닐, 4-C1 - 6알킬카보닐피페라지닐 및 모르폴리닐에서 선택된 하나, 둘 또는 세개의 치환체로 임의로 치환된 페닐을 나타내고;
기 또는 기의 부분으로서 각 Het는 질소, 산소 및 황에서 각각 독립적으로 선택된 1 내지 4개 헤테로 원자를 함유하며, 할로, 하이드록시, 니트로, 시아노, 카복실, C1 - 6알킬, C1 - 6알콕시, C1 - 6알콕시C1 - 6알킬, C1 - 6알킬카보닐, 아미노, 모노- 또는 디C1 - 6알킬아미노, 아지도, 머캅토, 폴리할로C1 - 6알킬, 폴리할로C1 - 6알콕시, 사이클로프로필, 피롤리디닐, 피페리디닐, 피페라지닐, 4-C1 - 6알킬-피페라지닐, 4-C1 - 6알킬카보닐-피페라지닐 및 모르폴리닐로 구성된 군으로부터 각각 독립적으로 선택된 하 나, 둘 또는 세개의 치환체로 임의로 치환되는 5 또는 6 원 포화, 부분 불포화 또는 완전 불포화 헤테로사이클 환을 나타낸다.
상기 구문에서 본 발명의 택일적 구체예 중, Rt1은 광범위하게 A에 상응하고, Rt2는 광범위하게 Rq에 상응하고, Rt3는 광범위하게 R5에 상응하며, X는 광범위하게 L에 상응하고, 아릴은 광범위하게 C0-C3알킬렌이 0(예를 들어, 하나의 결합)일 때, C0-C3알킬렌카보사이클릴을 포함하며, Het는 광범위하게 C0-C3알킬렌이 0(예를 들어, 하나의 결합)일 때, C0-C3알킬헤테로사이클릴을 포함한다. 화학식 I을 위해 하기에 나타낸 선택은 화학식 It에서 상응하는 값에 적용될 것이며, 화학식 (I)에 대한 참조는 화학식 (It)의 상응하는 화합물을 포함하는 것으로 해석될 것이다.
달리 언급되는 바가 없으면, 상기에서 그리고 하기에서 이용된 바와 같이 다음 정의가 적용된다.
용어 할로는 플루오로, 클로로, 브로모 및 아이오도를 총칭한다.
기 또는 기의 일부로서 용어 "할로C1 - 6알킬"은 예를 들어, 할로-C1 - 6알콕시 중에서, 모노- 또는 폴리할로 치환 C1 - 6알킬, 특히, 최대 1, 2, 3, 4, 5, 6개 이상의 할로 원자로 치환된 C1 - 6알킬, 예를 들어, 하나 이상의 플루오로 원자를 갖는 메틸 또는 에틸, 예를 들어, 디플루오로메틸, 트리플루오로메틸, 트리플루오로에틸로 정의된다. 바람직한 것은 트리플루오로메틸이다. 또한 포함된 것은 모든 수소 원자가 플루오로 원자로 치환된 C1 - 6알킬 그룹인 퍼플루오로C1 - 6알킬 그룹이고, 예를 들어, 펜타플루오로에틸이다. 폴리할로C1 - 6알킬의 정의내에서, 하나 이상의 할로겐 원자가 알킬기에 부착된 경우, 할로겐 원자는 같거나 또는 다를 수 있다.
본원에서 사용되었듯이, 기 또는 기의 일부로서 "C1 - 4알킬"은 1 ~ 4개의 탄소 원자를 갖는 직쇄 또는 분지쇄 포화 탄화수소 라디칼을 정의하고, 예를 들어 메틸, 에틸, 1-프로필, 2-프로필, 1-부틸, 2-부틸, 2-메틸-1-프로필이며; "C1 - 6알킬"은 C1 - 4알킬 라디칼 및 5 또는 6개의 탄소 원자를 갖는 이의 고급 동족체, 예를 들어, 1-펜틸, 2-펜틸, 3-펜틸, 1-헥실, 2-헥실, 2-메틸-1-부틸, 2-메틸-1-펜틸, 2-에틸-1-부틸, 3-메틸-2-펜틸 등을 포함한다. C1 - 6알킬 중에서 가장 중요한 것은 C1 - 4알킬이다.
기 또는 기의 일부로서, 용어 "C2 - 6알케닐"은 포화 탄소-탄소 결합 및 적어도 하나의 이중 결합을 갖고, 2 ~ 6개의 탄소 원자를 갖는 직쇄 및 분지쇄 탄화수소 라디칼을 정의하고, 예를 들어, 에테닐 (또는 비닐), 1-프로페닐, 2-프로페닐 (또는 알릴), 1-부테닐, 2-부테닐, 3-부테닐, 2-메틸-2-프로페닐, 2-펜테닐, 3-펜테닐, 2-헥세닐, 3-헥세닐, 4-헥세닐, 2-메틸-2-부테닐, 2-메틸-2-펜테닐 등이다. C2 - 6알케닐중에서 가장 중요한 것은 C2 - 4알케닐이다.
기 또는 기의 일부로서, 용어 "C2 - 6알키닐"은 포화 탄소-탄소 결합 및 적어도 하나의 삼중 결합을 갖고, 2 ~ 6개의 탄소 원자를 갖는 직쇄 및 분지쇄 탄화수소 라디칼을 정의하고, 예를 들어, 에티닐, 1-프로피닐, 2-프로피닐, 1-부티닐, 2-부티닐, 3-부티닐, 2-펜티닐, 3-펜티닐, 2-헥시닐, 3-헥시닐 등이다. C2 - 6알키닐 중에서 중요한 것은 C2 - 4알키닐이다.
C3 - 7사이클로알킬은 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실 및 사이클로헵틸을 총칭한다.
C0 - 3알킬렌은 결합 (C0) 또는 1 내지 3개의 탄소 원자를 갖는 2가 직쇄 및 분지쇄 포화 탄화수소 라디칼, 이를 테면, 메틸렌, 에틸렌 1,3-프로판디일, 1,2-프로판디일 등, 특히 메틸렌으로 정의된다.
C1 - 6알콕시는 C1 - 6알킬이 상기 정의된 바와 같은 C1 - 6알킬옥시를 의미한다.
본원에서 앞서 사용되었듯이, 용어 (=O) 또는 옥소는 탄소 원자에 부착될 때 카보닐 부분을, 황 원자에 부착될 때 설폭시드 부분을, 그리고 상기 용어 두개가 황 원자에 부착될 때 설포닐 부분을 형성한다. 환 또는 환 시스템이 옥소기로 치환될 때마다, 옥소가 연결되는 탄소 원자는 포화 탄소이다.
문맥상 달리 제안되지 않는다면, '아미노'는 NH2, NHC1-C6알킬 또는 N(C1-C6-알킬)2를 포함하며, 아미노 정의 내에서, 각 C1-C6알킬은 특히 C1-C3알킬 변형체 또는 포화 사이클릭 아민, 이를 테면, 피롤리디닐, 피페리디닐, 피페라지닐, 4-C1-C6 알킬피페라지닐, 이를 테면, 4-메틸피페라지닐, 4-C1-C6알킬-카보닐피페라지닐 및 모르폴리닐이다.
'아미도'는 C(O)NH2 및 알킬아미도, 이를 테면, C(=O)NHC1-C6알킬, C(=O)N(C1-C6알킬)2 특히, C(=O)NHC1-C3알킬, C(=O)N(C1-C3알킬)2 또는 -NH(C=O)C1-C6알킬, 예를 들어, -NHC(=O)CHC(CH3)3, 이를 테면, -NH(C=O)C1-C3알킬을 포함한다.
본원에 이용된 바와 같은 'C0-C3알킬렌아릴'은 아릴 부분, 이를 테면, 페닐, 나프틸, 또는 C3-C7사이클로알킬(예를 들어, 인다닐)에 융합된 페닐을 포함하는 것을 의미하며, 아릴은 직접적으로 결합되거나(예를 들어, CO), 상기 C1-C3알킬렌에서 정의된 바와 같은 중간체 메틸, 에틸 또는 프로필기를 통하여 결합된다. 달리 언급되지 않는다면, 아릴 및/또는 그의 융합된 사이클로알킬 부분은 할로, 하이드록시, 니트로, 시아노, 카복시, C1-C6알킬, C1-C6알콕시, C1-C6알콕시C1-C6알킬, C1-C6알카노일, 아미노, 아지도, 옥소, 머캅토, 니트로 C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴에서 선택된 1-3개 치환체로 임의로 치환되며, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴 치환체에서 헤테로사이클릭 및 카보사이클릭 부분은 본원에 제공된 바와 같이 치환될 수 있으나, 전형적으로 C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴로 추가로 치환되지 않는 것으로 이해된다. " 아릴"은 예를 들어 C0-C3알킬 결합이 부재인 경우 상응하는 의미를 갖는다.
본원에 이용된 바와 같은 'C0-C3알킬렌C3C7사이클로알킬'은 C3-C7사이클로알킬기, 이를 테면, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실 또는 사이클로헵틸을 포함하는 것을 의미하며, 여기서 사이클로알킬은 직접적으로 결합되거나(예를 들어, CO알킬), 상기 C1-C3알킬렌에서 정의된 바와 같은 중간체 메틸, 에틸, 프로필 또는 이소프로필기를 통하여 결합된다. 사이클로알킬기는 불포화 결합을 함유할 수 있다. 달리 언급되지 않는다면, 사이클로알킬 부분은 할로, 하이드록시, 니트로, 시아노, 카복시, C1-C6알킬, C1-C6알콕시, C1-C6알콕시C1-C6알킬, C1-C6알카노일, 아미노, 아지도, 옥소, 머캅토, 니트로 C0-C3알킬카보사이클릴, C0-C3알킬헤테로사이클릴에서 선택된 1-3개 치환체로 임의로 치환되며, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴에서 헤테로사이클릭 및 카보사이클릭 부분은 본원에 제공된 바와 같이 치환될 수 있으나, 전형적으로 C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴로 추가로 치환되지 않는다.
본원에 이용된 바와 같은 'C0-C3알킬카보사이클릴'은 C0-C3알킬아릴 및 C0-C3알킬C3-C7사이클로알킬을 포함하는 것을 의미한다. 달리 언급되지 않는다면, 아릴 또는 사이클로알킬기는 할로, 하이드록시, 니트로, 시아노, 카복시, C1-C6알킬, C1- C6알콕시, C1-C6알콕시C1-C6알킬, C1-C6알카노일, 아미노, 아지도, 옥소, 머캅토, 니트로, C0-C3알킬카보사이클릴 및/또는 C0-C3알킬헤테로사이클릴에서 선택된 1-3개 치환체로 임의로 치환되며, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴에서 헤테로사이클릭 및 카보사이클릭 부분은 본원에 제공된 바와 같이 치환될 수 있으나, 전형적으로 C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴로 추가로 치환되지 않는다. "카보사이클릴"은 예를 들어, C0-C3알킬 결합이 부재인 경우 상응하는 의미를 갖는다.
본원에 이용된 바와 같은 'C0-C3알킬렌헤테로사이클릴'은 모노사이클릭, 포화 또는 불포화, 헤테로원자-함유 환, 이를 테면, 피페리디닐, 모르폴리닐, 피페라지닐, 피라졸릴, 이미다졸릴, 옥사졸릴, 이속사졸릴, 티아지놀릴, 이소티아지놀릴, 티아졸릴, 옥사디아졸릴, 1,2,3-트리아졸릴, 1,2,4-트리아졸릴, 테트라졸릴, 푸라닐, 티에닐, 피리딜, 피리미딜, 피리다지닐, 또는 페닐 환에 융합된 임의의 기, 이를 테면, 퀴놀리닐, 벤즈이미다졸릴, 벤족사졸릴, 벤즈이속사졸릴, 벤조티아지놀릴, 벤즈이소티아지놀릴, 벤조티아졸릴, 벤족사디아졸릴, 벤조-1,2,3-트리아졸릴, 벤조-1,2,4-트리아졸릴, 벤조테트라졸릴, 벤조푸라닐, 벤조티에닐, 벤조피리딜, 벤조피리미딜, 벤조피리다지닐, 벤조피라졸릴 등을 포함하는 것을 의미하며, 여기서 환은 직접적으로 연결되거나, 예를 들어, (C0), 상기 C1-C3알킬렌에서 정의된 바와 같은 중간체 메틸, 에틸, 프로필 또는 이소프로필기를 통하여 결합된다. 방향족 특 성을 갖는 임의의 불포화 환은 본원에서 헤테로아릴 로 언급될 수 있다. 달리 언급되지 않는다면, 헤테로 환 및/또는 이의 융합된 페닐 부분은 할로, 하이드록시, 니트로, 시아노, 카복시, C1-C6알킬, C1-C6알콕시, C1-C6알콕시C1-C6알킬, C1-C6알카노일, 아미노, 아지도, 옥소, 머캅토, 니트로, C0-C3알킬카보사이클릴, C0-C3알킬헤테로사이클릴에서 선택된 1-3개 치환체로 임의로 치환된다. "헤테로사이클릴" 및 "헤테로아릴"은 예를 들어, C0-C3알킬 결합이 부재인 경우 상응하는 의미를 갖는다.
전형적으로 상기 정의의 관점 내에서 헤테로사이클릴 및 카보사이클릴 부분은 5 또는 특히 6 환 원자를 지닌 모노사이클릭 환이거나, 4, 5, 또는 6 원 환에 융합된 6 원 환을 포함하는 바이사이클릭 환 구조이다.
이러한 전형적인 기는 C3-C8사이클로알킬, 페닐, 벤질, 테트라하이드로나프틸, 인데닐, 인다닐, 헤테로사이클릴, 이를 테면, 아제파닐, 아조카닐, 피롤리디닐, 피페리디닐, 모르폴리닐, 티오모르폴리닐, 피페라지닐, 인돌리닐, 피라닐, 테트라하이드로-피라닐, 테트라하이드로티오피라닐, 티오피라닐, 푸라닐, 테트라하이드로푸라닐, 티에닐, 피롤릴, 옥사졸릴, 이속사졸릴, 티아졸릴, 이미다졸릴, 피리디닐, 피리미디닐, 피라지닐, 피리다지닐, 테트라졸릴, 피라졸릴, 인돌릴, 벤조푸라닐, 벤조티에닐, 벤즈이미다졸릴, 벤즈티아졸릴, 벤족사졸릴, 벤즈이속사졸릴, 퀴놀리닐, 테트라하이드로퀴놀리닐, 이소퀴놀리닐, 테트라하이드로이소퀴놀리닐, 퀴나졸리닐, 테트라하이드로퀴나졸리닐 및 퀴녹살리닐을 포함하여, 이 중 임의의 것은 본원에 정의된 바와 같이 임의로 치환될 수 있다.
포화 헤테로사이클 부분은 라디칼, 이를 테면, 피롤리닐, 피롤리디닐, 피라졸리닐, 피라졸리디닐, 피페리디닐, 모르폴리닐, 티오모르폴리닐, 피라닐, 티오피라닐, 피페라지닐, 인돌리닐, 아제티디닐, 테트라하이드로피라닐, 테트라하이드로티오-피라닐, 테트라하이드로푸라닐, 헥사하이드로피리미디닐, 헥사하이드로피리다지닐, 1,4,5,6-테트라하이드로피리미디닐아민, 디하이드로-옥사졸릴, 1,2-티아지나닐-1,1-디옥사이드, 1,2,6-티아디아지나닐-1,1-디옥사이드, 이소티아졸리디닐-1,1-디옥사이드 및 이미다졸리디닐-2,4-디온을 더욱 포함하는 반면, 불포화 헤테로사이클은 방향족 특성을 지닌 라디칼, 이를 테면, 푸라닐, 티에닐, 피롤릴, 옥사졸릴, 티아졸릴, 이미다졸릴, 피라졸릴, 이속사졸릴, 이소티아졸릴, 옥사디아졸릴, 트리아졸릴, 테트라졸릴, 티아디아졸릴, 피리디닐, 피리다지닐, 피리미디닐, 피라지닐, 인돌리지닐, 인돌릴, 이소인돌릴을 포함한다. 각 경우에, 헤테로사이클은 페닐 환과 축합되어, 바이사이클릭 환 시스템을 형성할 수 있다.
라디칼 Het는 본 명세서 및 청구항에서 정의된 바와 같은 헤테로사이클이다. Het의 예는, 예를 들어, 피롤리디닐, 피페리디닐, 모르폴리닐, 티오모르폴리닐, 피페라지닐, 피롤릴, 피라졸릴, 이미다졸릴, 옥사졸릴, 이속사졸릴, 티아지놀릴(thiazinolyl), 이소티아지놀릴, 티아졸릴, 이소티아졸릴, 옥사디아졸릴, 티아디아졸릴, 트리아졸릴 (1,2,3-트리아졸릴, 1,2,4-트리아졸릴 포함), 테트라졸릴, 푸라닐, 티에닐, 피리딜, 피리미딜, 피리다지닐, 피라졸릴, 트리아지닐 등을 포함한다. Het 라디칼 중에서 중요한 것은 불포화, 특히, 방향족 특성을 갖는 것들이다. 더욱 중요한 것은 하나 또는 두개의 질소를 갖는 Het 라디칼이다.
상기에서 언급된 각각의 Het 라디칼은 화학식 (I), (It) 또는 화학식 (I)의 화합물의 임의의 서브그룹의 정의에서 언급된 치환체의 개수 및 종류로 임의로 치환될 수 있다. 여기에서 및 이후의 단락에서 언급된 일부 Het 라디칼은 1, 2 또는 3개의 하이드록시 치환체로 치환될 수 있다. 이러한 하이드록시 치환 환은 케토기를 가진 그들의 토토머로 발생할 수 있다. 예를 들어, 3-하이드록시피리다진 부분은 이의 토토머 2H-피리다진-3-온으로 발생할 수 있다.
정의에서 사용된 임의의 분자 부분상의 라디칼 위치는 화학적으로 안정하다면 이러한 부분의 어디라도 될 수 있다.
변수의 정의에서 사용된 라디칼은 달리 지시되지 않는 한 모든 가능한 이성체를 포함한다. 예를 들어 피리딜은 2-피리딜, 3-피리딜 및 4-피리딜을 포함하고; 펜틸은 1-펜틸, 2-펜틸 및 3-펜틸을 포함한다.
임의의 구성물 (constituent)에서 임의의 변수가 1회 이상 발생할 때, 각각의 정의는 독립적이다.
이후 사용될 때마다, 용어 "화학식 (I)의 화합물", 또는 "본원의 화합물" 또는 유사한 용어는, 화학식 (I)의 화합물, 이의 전구약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 및 입체화학적 이성체를 포함하는 것을 의미한다. 일 구체예는 화학식 (I)의 화합물 또는 본원에 기술된 화학식 (I)의 화합물의 임의의 서브그룹, 및 이의 N-옥사이드, 염, 가능한 입체이성체를 포함한다. 다른 구체예는 화학식 (I)의 화합물 또는 본원에 기술된 화학식 (I)의 화합물의 임의의 서브그룹 및 이의 염 및 가능한 입체이성체를 포함한다.
화학식 (I)의 화합물은 몇몇 키랄 중심을 가지며, 입체화학적 이성체로 존재한다. 본원에서 사용된 용어 "입체화학적 이성체"는 화학식 (I)의 화합물이 가질 수 있는, 같은 결합 순서에 의하지만, 상호교환될 수 없는 다른 3차원 구조를 가진 동일한 원자 결합으로 구성된 모든 가능한 화합물을 정의한다.
(R) 또는 (S)가 치환체내의 키랄 원자의 절대배치를 지정하는데 사용되는 경우와 관련하여, 지정 (designation)은 전체 화합물을 고려하여 수행되고, 분리한 치환체는 고려의 대상이 아니다.
달리 언급되거나, 또는 지시되지 않는 한, 화합물의 화학적 지정은 상기 화합물이 가질 수 있는 모든 가능한 입체화학적 이성체의 혼합물을 포함한다. 상기 혼합물은 상기 화합물의 기본 분자 구조의 모든 디아스테레오머 및/또는 거울상 이성체를 함유할 수 있다. 본 발명의 화합물의 모든 입체화학적 이성체는 순수한 형태 또는 서로 혼합된 상태 모두, 본 발명의 범위내에 포함되는 것으로 의도된다.
본원에서 언급된 화합물 및 중간체의 순수한 입체이성체는 상기 화합물 또는 중간체의 동일한 기본 분자 구조의 다른 거울상 이성체 또는 디아스테레오머가 실질적으로 없는 이성체로 정의된다. 특히, 용어 "입체이성체적으로 순수"는 적어도 80% (즉, 최소 90%의 하나의 이성체 및 최대 10%의 다른 가능한 이성체)에서 100%(즉, 100%의 하나의 이성체 및 다른 것은 전혀 없음) 이하의 입체이성체적 과량을 갖는 화합물 또는 중간체, 더욱 특히, 90%에서 100% 이하의 입체이성체적 과량을 갖고, 더더욱 특히, 94%에서 100% 이하의 입체이성체적 과량을 갖고, 가장 특히, 97%에서 100% 이하의 입체이성체적 과량을 갖는 화합물 또는 중간체에 관한 것 이다. 용어 "거울상 이성체적으로 순수한" 및 "디아스테레오머적으로 순수한"은 유사한 방식으로 이해되어야 하지만, 중요한 혼합물의 각각의 거울상 이성체적 과량,및 디아스테레오머적 과량을 고려해야 한다.
본 발명의 화합물 및 중간체의 순수 입체이성체는 해당 분야에 알려져 있는 방식을 적용하여 수득될 수 있다. 예를 들어, 거울상 이성체는 그들의 디아스테레오머적 염을 광학적으로 활성인 산 또는 염기로 선택적 결정화함으로써 서로 분리될 수 있다. 이의 예는 타르타르산, 디벤조일타르타르산, 디톨루오일타르타르산 및 캄포설폰산이다. 택일적으로, 거울상 이성체는 키랄 정지상을 사용하는 크로마토그래피 기술로 분리될 수 있다. 상기 순수한 입체화학적 이성체는 또한, 적절한 출발 물질의 상응하는 순수한 입체화학적 이성체로부터 유발될 수 있지만, 단, 반응은 입체특이적으로 발생한다. 바람직하게, 만약 특정한 입체이성체를 원한다면, 상기 화합물은 제조의 입체특이적인 방법으로 합성될 것이다. 이들 방법은 거울상 이성체적으로 순수한 출발 물질을 유리하게 사용할 것이다.
화학식 (I)의 화합물의 디아스테레오머적 라세미화합물은 통상적인 방법으로 개별적으로 수득될 수 있다. 유리하게 사용될 수 있는 적절한 물리적 분리 방법은 예를 들어, 선택성 결정화 및 크로마토그래피, 예를 들어, 컬럼 크로마토그래피이다.
일부의 화학식 (I)의 화합물, 이의 전구약물, N-옥사이드, 염, 용매화물, 4차 아민, 또는 금속 복합물 및 이의 제조에 사용되는 중간체에 대하여, 절대 입체화학 배치는 실험적으로 결정될 수 없다. 해당 분야의 숙련자는 이러한 화합물의 절대배치를 해당 분야에 알려져 있는 방법, 예를 들어, X-선 회절로 결정할 수 있다.
본 발명은 또한, 본 화합물에서 존재하는 원자의 모든 동위원소를 포함하는 것으로 의도된다. 동위원소는 같은 원자 번호를 갖지만, 다른 질량수를 갖는 원자를 포함한다. 일반적인 예로서, 그리고 제한 없이, 수소의 동위원소는 삼중수소 및 중수소를 포함한다. 탄소의 동위원소는 C-13 및 C-14를 포함한다.
본 문헌에 걸쳐 사용된 용어 "전구약물"은 수득한 유도체의 생체내 생체전환 생성물이 화학식 (I)의 화합물에 정의된 바와 같은 활성 약물이 되도록 하는 약리학적으로 허용가능한 유도체, 예를 들어 에스테르, 아미드 및 포스페이트를 의미한다. 일반적인 전구약물을 기술한 Goodman 및 Gilman에 의한 참조 문헌 (The Pharmacological Basis of Therapeutics, 8th ed, McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs", p 13-15)은 본원에 포함된다. 바람직하게, 우수한 수용해성, 증가된 생체이용률을 가진 전구약물은 생체내에서 활성 억제제로 바로 대사화된다. 본 발명의 화합물의 전구약물은 화합물중에 존재하는 작용기를 일상적인 조작으로, 또는 생체내에서 모 화합물이 분리되는 방식으로 변형시킴으로써 제조될 수 있다.
바람직한 것은 생체내에서 가수분해되고, 하이드록시 또는 카복실기를 갖는 화학식 (I)의 화합물로부터 유발되는 약제학적으로 허용가능한 에스테르 전구약물이다. 생체내에서 가수분해되는 에스테르는 인간 또는 동물의 체내에서 가수분해되어 모 산 (parent acid) 또는 알콜을 생성하는 에스테르이다. 카복시에 대하여, 적 절한 약제학적으로 허용가능한 에스테르는 본 발명의 화합물에서 임의의 카복시기에 형성될 수 있는, C1-6알콕시메틸 에스테르, 예를 들어 메톡시메틸, C1-6알카노일옥시메틸 에스테르, 예를 들어 피발로일옥시메틸, 프탈리딜 에스테르, C3-8사이클로알콕시카보닐옥시C1-6알킬 에스테르, 예를 들어 1-사이클로헥실카보닐옥시에틸; 1,3-디옥솔렌-2-오닐메틸 에스테르, 예를 들어 5-메틸-1,3-디옥솔렌-2-오닐메틸; 및 C1-6알콕시카보닐옥시에틸 에스테르, 예를 들어 1-메톡시카보닐옥시에틸을 포함한다.
하이드록시기를 함유한 화학식 (I)의 화합물의 생체내 가수분해되는 에스테르는 무기 에스테르, 예를 들어 포스페이트 에스테르 및 α-아실옥시알킬 에테르 및 에스테르 분해 (breakdown)의 생체내 가수분해가 모 하이드록시기를 제공하는 결과물인 연관된 화합물을 포함한다. α-아실옥시알킬 에테르의 예는 아세톡시메톡시 및 2,2-디메틸프로피오닐옥시-메톡시를 포함한다. 하이드록시에 대한, 일련의 생체내 가수분해되는 에스테르 형성기는 알카노일, 벤조일, 페닐아세틸 및 치환된 벤조일 및 페닐아세틸, 알콕시카보닐 (알킬 카보네이트 에스테르 제공), 디알킬카바모일 및 N-(디알킬아미노에틸)-N-알킬카바모일 (카바메이트 제공), 디알킬아미노아세틸 및 카복시아세틸을 포함한다. 벤조일상의 치환체의 예는 환 질소 원자로부터 메틸렌기를 통해 벤조일 환의 3- 또는 4-위치에 연결된 모르폴리노 및 피페라지노를 포함한다.
치료용으로, 화학식 (I)의 화합물의 염은 상대이온이 약제학적으로 허용가능한 것들이다. 그러나, 약제학적으로 허용가능하지 않은 산 및 염기의 염도 또한, 약제학적으로 허용가능한 화합물의 제조 또는 정제에서 용도를 찾을 수도 있다. 모든 염은 약제학적으로 허용가능하거나 또는 가능하지 않거나, 본 발명의 범위에 포함된다.
상기 언급된 약제학적으로 허용가능한 산 및 염기 부가염은 화학식 (I)의 화합물이 형성할 수 있는 치료적으로 활성인 비독성 산 및 염기 부가염 형태를 포함하는 것을 의미한다. 약제학적으로 허용가능한 산 부가염은 염기 형태를 이러한 적절한 산으로 처리함으로써 편리하게 수득될 수 있다. 적절한 산은, 예를 들어, 무기산, 예를 들어 할로겐화수소산, 예를 들어, 염산 또는 브롬화수소산, 황산, 질산, 인산 등; 또는 유기산, 예를 들어, 아세트산, 프로판산, 하이드록시아세트산, 락트산, 피루브산, 옥살산 (즉, 에탄디오산), 말론산, 숙신산 (즉, 부탄디오산), 말레산, 푸마르산, 말산 (즉, 하이드록시부탄디오산), 타르타르산, 시트르산, 메탄설폰산, 에탄설폰산, 벤젠설폰산, p-톨루엔설폰산, 시클람산, 살리실산, p-아미노살리실산, 파모산 (pamoic acid) 등을 포함한다.
반대로, 상기 염 형태는 적절한 염기로 처리함으로써 유리 염기 형태로 전환될 수 있다.
산성 양자를 함유한 화학식 (I)의 화합물은 또한, 적절한 유기 및 무기 염기로 처리함으로써 그들의 비독성 금속 또는 아민 부가염 형태로 전환될 수 있다. 적절한 염기 염 형태는 예를 들어, 암모늄 염, 알칼리 및 알칼리토금속 염, 예를 들어, 리튬, 나트륨, 포타슘, 마그네슘, 칼슘 염 등, 유기 염기와의 염, 예를 들어, 벤자틴, N-메틸-D-글루카민, 하이드라바민 염, 및 아미노산과의 염, 예를 들어, 아 르기닌, 리신 등을 포함한다.
상기에서 사용된 용어 부가염은 또한, 화학식 (I)의 화합물 및 이의 염이 형성할 수 있는 용매화물을 포함한다. 이러한 용매화물은 예를 들어 수화물, 알콜화물 등이다.
상기에서 사용된 용어 "4차 아민"은 화학식 (I)의 화합물이 화학식 (I)의 화합물의 염기성 질소와 적절한 4차화제, 예를 들어, 임의로 치환된 알킬할라이드, 아릴할라이드 또는 아릴알킬할라이드, 예를 들어, 메틸아이오다이드 또는 벤질아이오다이드 사이의 반응으로 형성될 수 있는 4차 암모늄 염을 정의한다. 우수한 이탈기를 가진 다른 반응물이 또한 사용될 수 있고, 예를 들어, 알킬 트리플루오로메탄설포네이트, 알킬 메탄설포네이트 및 알킬 p-톨루엔설포네이트이다. 4차 아민은 양성으로 하전된 질소를 갖는다. 약제학적으로 허용가능한 상대이온은 클로로, 브로모, 아이오도, 트리플루오로아세테이트 및 아세테이트를 포함한다. 선택한 상대이온은 이온 교환 수지를 사용해 도입될 수 있다.
본 발명의 화합물의 N-옥사이드 형태는 하나 또는 수개의 질소 원자가 소위 N-옥사이드로 산화되는 화학식 (I)의 화합물을 포함하는 것을 의미한다.
화학식 (I)의 화합물이 금속 결합, 킬레이팅, 복합물 형성 특성을 가질 수 있고, 따라서, 금속 복합물 또는 금속 킬레이트로 존재할 수 있음을 알 수 있을 것이다. 이러한 화학식 (I)의 화합물의 금속화된 유도체는 본 발명의 범위내에 포함되는 것으로 의도된다.
일부 화학식 (I)의 화합물은 또한, 이의 토토머로 존재할 수 있다. 이러한 형태는 상기 화학식중에 명백하게 지시되지 않았음에도 본 발명의 범위내에 포함되는 것으로 의도된다.
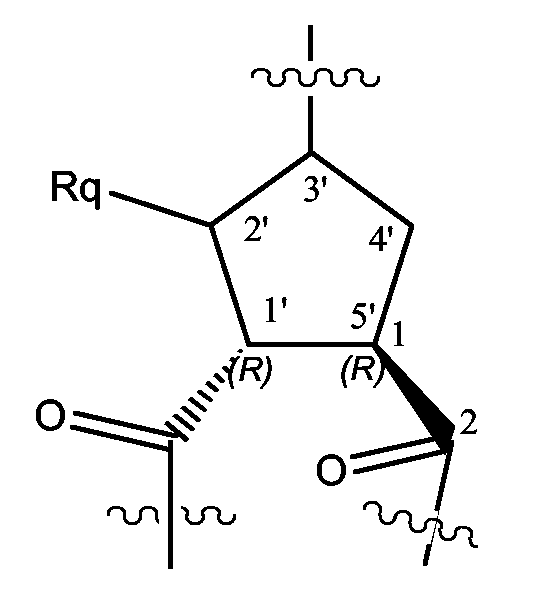
상기 언급되었듯이, 화학식 (I)의 화합물은 수개의 비대칭 중심을 갖는다. 각각의 이들 비대칭 중심을 더욱 효율적으로 나타내기 위해, 하기 구조 화학식에 나타낸 바와 같은 넘버링 시스템이 사용될 것이다.

비대칭 중심은 마크로사이클의 1, 4 및 6번의 위치 및 5원 환의 탄소 원자 3'에, Rq 치환체가 C1 - 6알킬일 때 탄소 원자 2', 그리고, L이 CH일 때 탄소 원자 1'에 존재한다. 각각의 이들 비대칭 중심은 그들의 R 또는 S 배치로 존재할 수 있다.
1번 위치에서의 입체화학은 바람직하게, L-아미노산의 배치의 입체화학, 즉, L-프롤린의 입체화학에 상응한다.
L이 CH일 때, 사이클로펜탄 환의 2 카보닐기는 바람직하게, 하기에 나타낸 바와 같이 트랜스 배치로 있다.
화학식 (I)의 구조는 하기 P1 단편에 나타낸 바와 같은 사이클로프로필기를 포함한다:

상기 식에서,
C7은 7번 위치의 탄소를 나타내고, 4 및 6번의 탄소는 사이클로프로판 환의 비대칭 탄소 원자이다.
본 발명의 화합물의 다른 부분에서 다른 비대칭 중심이 가능하더라도, 이들 두개의 비대칭 중심의 존재는 화합물이 디아스테레오머의 혼합물로 존재할 수 있음을 의미하고, 예를 들어, 7번 위치에서 탄소가 카보닐에 대하여 신 (syn)으로, 또는 아미드에 대하여 신으로 하기 나타낸 바와 같이 배치된 화학식 (I)의 화합물의 디아스테레오머이다.

화학식 (I)의 구조는 프롤린 잔기 (L이 N일 때)를 포함할 수 있다. 바람직한 것은 1 (또는 5')번 위치의 치환체 및 3'번 위치의 치환체 -O-Rr이 트랜스 배치인 화학식 (I)의 화합물이다. 특히 중요한 것은, 1번 위치가 L-프롤린에 상응하는 배치를 갖고, -O-Rr 치환체가 1번 위치에 대하여 트랜스 배치인 화학식 (I)의 화합물이다. 바람직하게, 화학식 (I)의 화합물은 하기의 화학식 (I-a) 및 (I-b)의 구조로 나타낸 바와 같은 입체화학을 갖는다:

본 발명의 일 구체예는 하나 이상의 하기 조건을 적용하는, 화학식 (I) 또는 화학식 (I-a), 또는 화학식 (I)의 화합물의 임의의 서브그룹의 화합물에 관한 것이다:
(a) Rq가 수소이고;
(b) L이 질소이며;
(c) 탄소 원자 7 및 8 사이에 이중 결합이 존재한다.
본 발명의 일 구체예는 하나 이상의 하기 조건을 적용하는, 화학식 (I) 또는 화학식 (I-a), (I-b), 또는 화학식 (I)의 화합물의 임의의 서브그룹의 화합물에 관한 것이다:
(a) Rq가 수소이고;
(b) X가 CH이며;
(c) 탄소 원자 7 및 8 사이에 이중 결합이 존재한다.
본 발명의 일구체예는 하기의 부분 구조를 포함하는 화합물을 포함한다:

화학식 (I)의 화합물의 특별한 서브그룹은 하기 구조식으로 나타내어진다:

화학식 (I-c) 및 (I-d)의 화합물중에서, 각각 화학식 (I-a), 및 (I-b)의 화합물의 입체화학적 배치를 갖는 것이 특히 중요하다.
화학식 (I)의 화합물, 또는 화학식 (I)의 화합물의 임의의 서브그룹중에서 탄소 원자 7 및 8 사이의 이중 결합은 시스 또는 트랜스 배치로 있을 수 있다. 바람직하게 탄소 원자 7 및 8 사이의 이중 결합은 화학식 (I-c) 및 (I-d)에 묘사한 바와 같이 시스 배치로 있다.
탄소 원자 7과 8 사이의 이중 결합은 화학식 (I)의 화합물, 또는 화학식 (I)의 화합물의 임의의 서브그룹중에서 시스 또는 트랜스 배열로 있을 수 있다. 바람직하게, 탄소 원자 7 및 8 사이의 이중 결합은 화학식 (I-c) 및 (I-d)에 나타낸 바와 같이 시스 배열로 있다.
(I-a), (I-b), (I-c) 및 (I-d) 중에서, 적절한 경우, A, L, n, Rr, Rq, R5는 화학식 (I)의 화합물의 정의에서, 또는 본원에서 상술된 화학식 (I)의 화합물의 임의의 서브그룹에서 명기된 바와 같다.
상기 정의된 화학식 (I-a), (I-b), (I-c), (I-d)의 화합물의 서브그룹 및 본원에서 정의된 임의의 다른 서브그룹은 또한, 이러한 화합물의 임의의 전구 약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 및 입체화학적 이성체를 포함하는 것을 의미하는 것으로 이해된다.
n이 2일 때, "n"으로 괄호가 묶인 부분 -CH2-는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹에서 에탄디일에 상응한다. n이 3일 때, "n"으로 괄호가 묶인 부분 -CH2-는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹에서 프로판디일에 상응한다. n이 4일 때, "n"으로 괄호가 묶인 부분 -CH2-는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹에서 부탄디일에 상응한다. n이 5일 때, "n"으로 괄호가 묶인 부분 -CH2-는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹에서 펜탄디일에 상응한다. n이 6일 때, "n"으로 괄호가 묶인 부분 -CH2-는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹에서 헥산디일에 상응한다. 화학식 (I)의 화합물의 특별한 서브그룹은 n이 4 또는 5인 이들 화합물이다.
본 발명의 구체예는
(a) A가 -OR1이거나 (특히, Rl이 C1 - 6알킬, 예를 들어 메틸, 에틸, 또는 tert-부틸이며, 가장 바람직하게는 R1이 수소이다); 또는,
(b) A가 -NHS(=O)2R2인 (특히, R2가 C3-C7사이클로알킬로 임의로 치환된 C1-C6알킬, C1-C6알킬로 임의로 치환된 C3-C7사이클로알킬 또는 아릴이며, 예를 들어, R2는 메틸, 사이클로프로필 또는 페닐이다) 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다. 예를 들어, R2는 1-메틸사이클로프로필일 수 있다.
본 발명의 다른 구체예는
(a) Rq는 수소이고; L은 CH 또는 N이거나;
(b) Rq는 메틸, L은 C이고, 점선은 이중 결합을 나타내는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다.
본 발명의 추가적인 구체예는
(a) R5가 수소이거나;
(c) R5가 C1-C6알킬이거나;
(d) R5가 C1-C6알콕시C1-C6알킬 또는 C3-C7사이클로알킬인 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다.
본 발명의 바람직한 구체예는 R5이 수소, 또는 C1 - 6알킬이고, 더욱 바람직하게는 수소 또는 메틸인 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다.
본 발명의 구체예는 Rr이 퀴나졸린-4-일인 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다. 전형적으로, Rr 퀴나졸린-4-일은 임의로 일, 이, 또는 삼 치환되며, 예를 들어, C1-C6알킬, C1-C6알콕시, 하이드록시, 할로, 트리플루오로메틸, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 아릴, 헤테로아릴 또는 헤테로사이클릴로 치환된다(아릴 헤테로아릴 또는 헤테로사이클릴이 각각 독립적으로 할로, C1-C6알킬, C1-C6알콕시, 폴리할로C1-C6알콕시, 아미노, 모노- 또는 디C1-C6알킬아미노, 사이클로프로필, 피롤리디닐, 피페리디닐, 피페라지닐, N-메틸-피페라지닐 또는 모르폴리닐로 임의로 치환되는 경우).
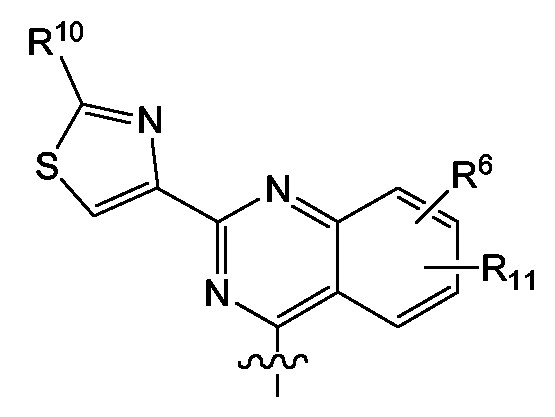
Rr의 퀴나졸린 구체예는 하기의 라디칼 (f-1) 또는 특정 라디칼 (f-1-a)을 포함한다:


상기 식에서,
R9, R6 및 R11은 Rr 또는 Rt4를 위해 언급된 의미를 가지며, 여기서, R9는 특별히 C3-C7사이클로알킬, 아릴 또는 Het이며, 이중 임의의 것은 하나, 둘 또는 세개 의 (특히 하나의) R10으로 임의로 치환되며; 여기서, R10은 수소, C1-C6알킬, C3-C7사이클로알킬, 아릴, Het(바람직하게, C1-C6알킬로 일- 또는 이치환), 피롤리디닐, 피페리디닐, 피페라지닐, 4-메틸-피페라지닐, 티오모르폴리닐 또는 모르폴리닐, 아미노카보닐, 모노 또는 디C1-C6알킬아미노카보닐이며; 여기서, 피페리디닐, 모르폴리닐 또는 티오모르폴리닐은 하나 또는 두 C1-C6알킬 라디칼로 임의로 치환될 수 있거나; R9는 C1-C6알콕시이고;
R6는 수소, 할로겐, C1-C6알킬, 특히 메틸, C3-C7사이클로알킬, 아릴, Het, 할로, 특히 브로모, 클로로 또는 플루오로이며;
R11은 수소 또는 C1-C6알콕시이다.
퀴나졸린을 위한 R9의 바람직한 구체예는 아릴 또는 Het를 포함하며, 특히 여기서 R9는 페닐, 피리딜, 티아졸릴, 옥사졸릴 또는 피라졸릴이며, 둘다는 정의된 바와 같은 하나, 둘 또는 세개의 (특히 하나의) R10으로 임의로 치환된다.
R9의 추가의 바람직한 구체예는 알콕시, 특히 에톡시 및 이소프로폭시이다.
퀴나졸린을 위한 R10의 구체예는 수소, 메틸, 에틸, 이소프로필, tert-부틸, 알콕시, 이를 테면, 메톡시, 할로(디할로, 이를 테면 디플루오로 포함), 피롤리디닐, 피페리디닐, 피페라지닐, 4-C1-C6알킬피페라지닐(예를 들어, 4-메틸피페라지닐), 티오모르폴리닐 또는 모르폴리닐, C1-C6알킬아미노, (C1-C6알킬)2아미노, 아미노카보닐, 모노 또는 디C1-C6알킬아미노카보닐 또는 C3-C7사이클로알킬(특히 사이클로프로필)을 포함한다.
퀴나졸린의 바람직한 R9 구체예는 하나 또는 두개의 R10기, 이를 테면, 수소, 메틸, 에틸, 이소프로필, tert-부틸, 알콕시, 이를 테면, 메톡시, 포화 모노사이클릭 아미노, C1-C6알킬아미노, (C1-C6알킬)2아미노, 또는 C1-C6알킬-아미도 또는 할로(특히 플루오로)로 치환된 페닐을 포함한다(특히 R6가 수소, 메틸 또는 브로모인 경우). 바람직하게, 페닐 치환체는 파라(para) 위치에 있다. 본 구체예에 따른 R9를 위한 특별하게 바람직한 구조는 페닐, p-메톡시페닐 및 p-플루오로페닐이다.
(f-1) 또는 (f-1-a) 하에서 상술된 퀴나졸릴 라디칼에서 R9을 위한 추가의 배열은 하기의 임의의 라디칼을 포함한다:

상기 식에서, R10은 상기 정의된 바와 같거나, 특히 수소, C1-C6알킬(이를 테 면, 메틸, 에틸, 이소프로필, tert-부틸), 피롤리디닐, 피페리디닐, 피페라지닐, 4-C1-C6알킬-피페라지닐, N-메틸피페라지닐, 티오모르폴리닐 또는 모르폴리닐, C1-C6알킬아미노, (C1-C6알킬)2아미노 또는 아미노카보닐, 모노 또는 디C1-C6알킬아미노카보닐이다.
퀴나졸린을 위한 R9는 다음을 포함할 수 있다:

상기 식에서, R10은 하나 또는 두개의 C1-C6알킬기로 임의로 치환된 피페리딘, 모르폴린 또는 피페리딘-1-일, 수소, C1-C6알킬(이를 테면, 메틸, 에틸, 이소프로필, tert-부틸), C1-C6알킬아미노, (C1-C6알킬)2아미노, C1-C6알킬아미도, 모르폴리닐, 티오모르폴리닐이다.
퀴나졸린을 위한 R6의 구체예는 C1-C6알킬, 특히 메틸, 할로(예를 들어, 브로모, 클로로, 플루오로)특히 브로모를 포함한다.
퀴나졸린을 위한 R11의 구체예는 수소, C1-C6알킬옥시(특히 메톡시)를 포함한다.
화학식 (I)의 화합물 또는 화학식 (I)의 임의의 다른 서브그룹의 특정 구체 예는 Rr이 하기와 같은 것이며:

상기 식에서, R10, R10' 및 R11은 상기에서 상술한 바와 같고, 특히 R11은 수소 또는 C1-C6알콕시(예를 들어, 메톡시)이며, R10 및 R10'는 특히 수소, 메톡시 또는 할로, 이를 테면, 플루오로 또는 디플루오로이다. 간편하게, R10 및 R10'가 수소가 아닐 때, 그것은 페닐환의 파라 위치에 있다.
화학식 (I)의 화합물 또는 화학식 (I)의 임의의 다른 서브그룹의 더욱 바람직한 구조는 Rr이 하기와 같은 것이며:

상기 식에서, R10, R10' 및 R11은 상기에서 상술한 바와 같고, 특히 R11은 수소 또는 C1-C6알콕시(예를 들어, 메톡시)이며, R10 및 R10'는 특히 수소, 메톡시 또는 할 로, 이를 테면, 플루오로 또는 디플루오로이다. 간편하게, R10 및 R10'는 페닐환의 파라 위치에 있다.
본 구체예의 특히 바람직한 화합물은 Rr이 화학식 (f-4), (f-5) 또는 (f-6)에 따르는 것이다:

본 발명의 화합물은 하기에서 기술된 바와 실험 부분에서 상술한 바와 같이 일반적으로 제조된다. Rr이 8-메틸 치환된 퀴나졸리닐 유도체인 화학식 (I)의 화합물에 대한 통상의 중간체는 화학식 (II)의 트리-치환된 아닐린이며:

이 아닐린 유도체는 본 발명의 다른 면을 구성한다.
화학식 (I)의 화합물의 제조를 위한 유용한 중간체는 일반식 (III) 및 특히 화학식 (III-a)을 갖는 퀴나졸리닐 유도체이다:


상기 식에서, X는 OH이거나 이탈기, 이를 테면, 할리드, 예컨대, 클로라이드, 브로마이드 또는 아이오다이드 또는 설폰산의 유도체, 이를 테면, 토실레이트, 트리플레이트, 메실레이트 등이고, 바람직하게, X는 OH이다. R6, R9, 및 R11은 화학식(f-1) 및 (f-1-a)의 화합물을 위해 상기에 정의된 바와 같다. 화합물 (III) 및 (IIIa)는 신규한 화합물이며, 본 발명의 다른 면을 구성한다.
퀴나졸리닐 부분을 위해 상기 기재된 다양한 구체예는 또한 화학식 (III) 및 (IIIa)의 화합물에도 적용된다.
화학식 (III) 및 (IIIa)의 화합물을 위한 바람직한 R9 구체예는 하나 또는 두개의 R10기, 이를 테면, 수소, 메틸, 에틸, 이소프로필, tert-부틸, 포화 모노사이클릭 아미노, C1-C6알킬아미노, (C1-C6알킬)2아미노 또는 C1-C6알킬아미도 또는 특히 R6가 수소, 메틸 또는 브로모일 때 할로(특히 플루오로)로 임의로 치환된 페닐 및 피리딜을 포함한다.
화학식 (III)의 화합물의 특정 구체예는 화학식 (III-2) 및 (III-3)에 나타낸 구조를 갖는 것이다:

상기 식에서, X, R10, R10' 및 R11은 상기에서 상술한 바와 같고, 특히 R11은 수소 또는 C1-C6알콕시(예를 들어, 메톡시)이며, R10 및 R10'는 특히 수소, 메톡시 또는 할로, 이를 테면, 플루오로 또는 디플루오로이다. 간편하게, R10 및 R10'는 페닐환의 파라 위치에 있다.
화학식 (III)의 화합물의 더욱 바람직한 구조는 화학식 (III-2-Me) 및 (III-3-Me)에 따른 것이다:

상기 식에서, X, R10, R10' 및 R11은 상기에서 상술한 바와 같고, 특히 R11은 수소 또는 C1-C6알콕시(예를 들어, 메톡시)이며, R10 및 R10'는 특히 수소, 메톡시 또는 할로, 이를 테면, 플루오로 또는 디플루오로이다. 간편하게, R10 및 R10'는 페닐환 의 파라 위치에 있다.
화학식 (III)의 특히 바람직한 화합물은 화학식 (III-4) 또는 (III-5)를 갖는 것이다:

상기 식에서, X는 상기 기술된 바와 같다.
본 발명의 구체예는 Rr이 임의로 메틸, 에틸, 이소프로필, tert-부틸(또는 t.부틸), 메톡시, 트리플루오로메틸, 트리플루오로메톡시, 플루오로, 클로로, 브로모, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 페닐, 메톡시페닐, 시아노페닐, 할로페닐, 피리딜, C1-C4알킬피리딜, 피리미디닐, 모르폴리닐, 피페라지닐, C1-C4알킬피페라지닐, 피롤리디닐, 피라졸릴, C1-C4알킬피라졸릴, 티아졸릴, C1-C4알킬티아졸릴, 사이클로프로필-티아졸릴, 또는 모노- 또는 디C1-C4알킬아미노티아졸릴로 일, 이 또는 삼 치환된 퀴나졸린-4-일인 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다.
본 발명의 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R9는 수소, 할로, C1-C6알킬, C1-C6알콕시, 모노- 또는 C1-C6알킬아미노, 아미노, 아릴, 헤테로아릴 또는 헤테로사이클릴이며, 상기 아릴 또는 헤테로아릴 또는 헤테로사이클릴은 각각 독립적으로 하나 또는 두개의 C1-C6알킬, C1-C6알콕시, 폴리할로C1-C6알콕시, 할로, 아미노, 모노- 또는 디C1-C6알킬아미노로 임의로 치환되며; 각각의 R6 및 R11'는 독립적으로 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로메틸, 아릴, 헤테로아릴 또는 헤테로사이클릴이며; 상기 아릴, 헤테로아릴 또는 헤테로사이클릴은 각각 독립적으로, C1-C6알킬, C1-C6알콕시, 폴리할로C1-C6알콕시, 아미노, 포화 사이클릭 아미노, 모노- 또는 디C1-C6알킬아미노로 임의로 치환된다.
본 발명의 구체예는 R9가 다음으로 구성된 군에서 선택되는 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R10은 각각 독립적으로, 수소, 할로, C1-C6알킬, 아미노, 포화 사이클릭 아미노, 모노- 또는 디-C1-C6알킬아미노이다.
본 발명의 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸, 아릴, 헤테로아릴 또는 헤테로사이클릴이며;
R10은 독립적으로 수소, C1-C6알킬, C1-C6알콕시 또는 할로이다.
본 발명의 다른 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸, 아릴, 또는 Het이며;
R10은 수소, C1-C6알킬, C1-C6알콕시 또는 할로이다.
삭제
삭제
삭제
삭제
본 발명의 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:
삭제
상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸이며; 바람직하게, R4b는 C1-C6알콕시, 가장 바람직하게 메톡시이며; R10은 수소, C1-C6알킬, 아미노, 모노- 또는 디C1-C6알킬아미노, 피롤리디닐, 피페리디닐, 피페라지닐, N-메틸-피페라지닐 또는 모르폴리닐이다.
본 발명의 구체예는 R4가 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

삭제
상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸이며; 바람직하게, R4b는 C1-C6알콕시, 가장 바람직하게 메톡시, 할로 또는 C1 - 3알킬이며;
R10은 수소, C1-C6알킬, 아미노, 모노- 또는 디C1-C6알킬아미노, 피롤리디닐, 피페리디닐, 피페라지닐, N-메틸-피페라지닐 또는 모르폴리닐이다.
본 발명의 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

삭제
상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸이며; 바람직하게, R4b는 C1-C6알콕시, 가장 바람직하게 메톡시, 할로 또는 C1 - 3알킬이며;
R10은 수소, C1-C6알킬, 아미노, 모노- 또는 디C1-C6알킬아미노, 피롤리디닐, 피페리디닐, 피페라지닐, N-메틸-피페라지닐 또는 모르폴리닐이다.
본 발명의 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R6 및 R11은 독립적으로, 수소, C1-C6알킬, C1-C6알콕시, 모노- 또는 디C1-C6알킬아미노, 모노- 또는 디C1-C6알킬아미노카보닐, 하이드록시, 할로, 트리플루오로-메틸이며; 바람직하게, R4b는 C1-C6알콕시, 가장 바람직하게 메톡시, 할로 또는 C1 - 3알킬이며;
R4i는 수소, C1-C6알킬, 아미노, 모노- 또는 디C1 - 6알킬아미노, 피롤리디닐, 피페리디닐, 피페라지닐, N-메틸-피페라지닐 또는 모르폴리닐이다.
본 발명의 바람직한 구체예는 Rr이 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R9는 화학식 (I)의 화합물의 임의의 그룹 또는 서브그룹에서 정의된 바와 같으며;
R6는 수소, 할로 또는 트리플루오로메틸이다.
본 발명의 다른 구체예는 R4가 하기와 같은 화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 임의의 서브그룹이다:

상기 식에서, R6는 수소, 할로 또는 트리플루오로메틸이다.
본 발명의 다른 구체예는 R9가 하기와 같은 것을 포함한다:

상기 식에서, R10은 C1-C3알킬로 임의로 치환된 피페리딘, 수소, 메틸, 에틸, 이소프로필, tert-부틸, C1-C3알킬아미노, (C1-C3알킬)2아미노, (C1-C6알킬)아미도 모르폴린-4-일, 피페리딘-1-일 및 모르폴린이다.
본 발명의 다른 구체예는 Rr이 하기와 같은 것을 포함한다:

상기 식에서, R11은 H 또는 메톡시이다.
마크로사이클 내에 이중 결합을 갖는 화학식 (I-c) 및 (I-d)의 화합물(예를 들어, 탄소 원자 7 및 8 사이에, 하기 화학식 (I-d), (I-e) 및 (I-f)로 나타냄)은 세개의 빌딩 블록(building block) P1, P2 및 P3로 구성된다. 화학적 목적을 위하여, 화학식 (I-d) 및 (I-e) 화합물의 빌딩 블록 P2는 위치 1'에 부착된 카보닐기를 편입한다.
빌딩 블록 P1과 P2, P2와 P3의 연결은 아미드 결합의 형성을 포함한다. 블록 (block) P1 및 P3의 연결은 이중 결합 형성을 포함한다. 화합물 (I-c) 또는 (I-d)를 제조하기 위한 빌딩 블록 P1, P2 및 P3의 연결은 임의의 제공된 순서로 수행될 수 있다. 마지막 단계는 명백하게 마크로사이클이 형성되는 고리화를 포함한다.

바람직한 구체예에서, 화합물 (I-c)는 처음으로 아미드 결합 형성과 그 다음으로 마크로사이클로 고리화에 수반되는 P3 및 P1 사이의 이중 결합 형성에 의해 제조된다.
택일적으로, 화학식 (I-c)의 화합물에서, 우선, 빌딩 블록 P2 및 P1 사이의 아미드 결합이 형성되고, 그 후, P3 빌딩 블록이 커플링되고, 이어서 P3과 P2 사이에 아미드 결합이 폐환 (ring closure)과 동시적으로 형성된다. 또다른 택일적인 합성 방법은 빌딩 블록 P2 및 P3 사이의 아미드 결합 형성 후, 빌딩 블록 P1의 P3 로의 커플링과 P1과 P2 사이의 마지막 아미드 결합이 폐환과 동시적으로 형성된다.
화학식 (I-c)의 화합물에서, 블록 P2와 P3 사이의 아미드 결합 형성이 우레아 모티프의 두개의 다른 위치에서 성취될 수 있다. 첫번째 아미드 결합은 피롤리딘 환의 질소 및 근접한 카보닐(별표로 표시)를 포함한다. 택일적인 두번째 아미드 결합 형성은 별표 표시된 카보닐과 -NHR3 기의 반응을 포함한다. 빌딩 블록 P2 및 P3 사이의 아미드 결합 형성은 둘다 가능하다.
화학식 (I-d)의 화합물은 P1을 P2에, 또는 그 반대로 연결하고, P3 및 P2 빌딩 블록 사이에 두번째 아미드 결합을 마크로사이클로의 고리화와 동시에 형성하여 제조될 수 있다.
택일적으로, 화학식 (I-d)의 화합물에서, 빌딩 블록 P1-P3는 빌딩 블록 P2에 커플링되기 전에 합성될 수 있다. 빌딩 블록 P1-P3는 복분해 반응, 비티그형 반응 (Wittig reaction) 등에 의해 인지될 수 있으며, 이후 빌딩 블록 P2 와 두개의 아미드 결합이 폐환과 동시적으로 형성된다.
개별의 빌딩 블록이 처음으로 제조될 수 있으며, 그 다음, 함께 커플링 되거나, 택일적으로, 빌딩 블록의 전구체가 함께 커플링 되고, 후의 단계에서 목적하는 분자 조성물로 변형될 수 있다.
빌딩 블록 각각에서 작용성은 보호되어 부반응을 피할 수 있다.
아미드 결합의 형성은 펩티드 합성에서 아미노산을 커플링하는 데 이용된 것과 같은 표준 방법을 이용하여 수행될 수 있다. 후자는 한 반응물의 카복실기와 다 른 반응물의 아미노기가 탈수 커플링되어, 아미드 결합 연결을 형성하는 것을 포함한다. 아미드 결합 형성은 커플링제의 존재 하에서 출발 물질을 반응시키거나, 카복실 작용성을 활성 형태, 이를 테면, 활성 에스테르 또는 염화 아실로 전환시켜 수행될 수 있다. 거기에 이용된 커플링 반응 및 시약의 일반적인 설명은 펩티드 화학에서 일반적인 책자, 예를 들어, M. Bodanszky, "Peptide Chemistry", 2nd rev. ed., Springer-Verlag, Berlin, Germany, (1993)에서 찾을 수 있다.
아미드 결합 형성이 있는 커플링 반응의 예는 아자이드법 (azide method), 혼합 탄소(carbonic)-카복실산 무수물 (이소부틸 클로로포르메이트)법, 카보디이미드 (디사이클로헥실카보디이미드, 디이소프로필카보디이미드, 또는 수용해성 카보디이미드, 예를 들어, N-에틸-N'-[(3-디메틸아미노)프로필]카보디이미드)법, 활성 에스테르법 (예: p-니트로페닐 에스테르, N-하이드록시숙신 이미도 에스테르), 우드워드 (Woodward) 시약 K-법, 카보닐디이미다졸 법, 인 시약 또는 산화-환원법을 포함한다. 이들 방법 중 일부는 적절한 촉매를 첨가하여, 예를 들어, 카보디이미드법에서 1-하이드록시벤조트리아졸, 또는 4-DMAP를 첨가함으로써 강화될 수 있다. 추가적인 커플링제는 (벤조트리아졸-1-일옥시)트리스-(디메틸아미노) 포스포늄 헥사플루오로포스페이트 (그 자체로, 또는 1-하이드록시- 벤조트리아졸 또는 4-DMAP의 존재하에서); 또는 2-(1H-벤조트리아졸-1-일)-N,N,N',N'-테트라-메틸우로늄 테트라플루오로보레이트, 또는 O-(7-아자벤조트리아졸-1-일)-N,N,N',N'-테트라메틸우로늄 헥사플루오로포스페이트이다. 이들 커플링 반응은 용액 (액상) 또는 고체상으로 수행될 수 있다.
커플링 반응은 바람직하게, 불활성 용매, 예를 들어 할로겐화된 탄화수소, 예를 들어, 디클로로메탄, 클로로포름, 2극성 비양자성 용매, 예를 들어 아세토니트릴, 디메틸포름아미드, 디메틸아세트아미드, 에테르, 이를테면, 테트라하이드로푸란 중에서 수행된다.
많은 예에서, 커플링 반응은 적절한 염기, 예를 들어, 3차 아민, 예를 들어, 트리에틸아민, 디이소프로필에틸아민 (DIPEA), N-메틸-모르폴린, N-메틸피롤리딘, 또는 4-DMAP의 존재하에서 수행된다. 반응 온도는 0 ℃ 및 50 ℃ 사이의 범위일 수 있고, 반응 시간은 15분 및 24시간 사이일 수 있다.
서로 연결된 빌딩 블록중의 작용기는 원하지 않는 결합의 형성을 피하기 위해 보호될 수 있다. 사용될 수 있는 적절한 보호기는 예를 들어, Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1999) 및 "The Peptides: Analysis, Synthesis, Biology", Vol. 3, Academic Press, New York (1987)에 열거된다.
카복실기는 쪼개져서 카복실산을 제공할 수 있는 에스테르로 보호될 수 있다. 사용될 수 있는 보호기는 하기를 포함한다:
1) 알킬 에스테르, 예를 들어 메틸, 트리메틸실릴 및 tert-부틸;
2) 아르알킬 에스테르, 예를 들어 벤질 및 치환된 벤질; 또는
3) 순한 염기 또는 순한 환원제 (mild reductive)에 의해 쪼개질 수 있는 에스테르, 예를 들어, 트리클로로에틸 및 펜아실 에스테르.
아미노기는 다양한 N-보호기, 예를 들어 하기에 의해 보호될 수 있다:
1) 아실기, 예를 들어 포르밀, 트리플루오로아세틸, 프탈릴 및 p-톨루엔설포닐;
2) 방향족 카바메이트기, 예를 들어 벤질옥시카보닐 (Cbz 또는 Z) 및 치환된 벤질옥시카보닐, 및 9-플루오레닐메틸옥시카보닐 (Fmoc);
3) 지방족 카바메이트기, 예를 들어 tert-부틸옥시카보닐 (Boc), 에톡시카보닐, 디이소프로필메톡시-카보닐 및 알릴옥시카보닐;
4) 사이클릭 알킬 카바메이트기, 예를 들어 사이클로펜틸옥시카보닐 및 아다만틸옥시카보닐;
5) 알킬기, 예를 들어 트리페닐메틸 및 벤질;
6) 트리알킬실릴, 예를 들어 트리메틸실릴; 및
7) 티올 함유 기, 예를 들어 페닐티오카보닐 및 디티아숙시노일.
중요한 아미노 보호기는 Boc 및 Fmoc이다.
바람직하게 α-아미노 보호기는 다음 커플링 단계 이전에 쪼개진다. Boc기가 사용될 때, 선택 방법은 순수하거나, 또는 디클로로메탄중의 트리플루오로아세트산, 또는 디옥산중, 또는 에틸 아세테이트중의 HCl이다. 수득한 암모늄 염은 그 후, 커플링 전에, 또는 염기성 용액, 예를 들어 수성 완충제, 또는 3차 아민과 디클로로메탄 또는 아세토니트릴 또는 디메틸포름아미드중에서 인 시추 (in situ)로 중화된다. Fmoc기가 사용될 때, 선택 시약은 디메틸포름아미드중에 피페리딘 또는 치환된 피페리딘이나, 임의의 2차 아민이 사용될 수 있다. 탈보호는 0 ℃ 및 실온 사이의 온도에서, 보통 약 20-22 ℃에서 수행된다.
합성 과정의 반응, 예를 들어, 빌딩 블록의 커플링 반응에서 간섭할 수 있는 다른 작용기도 역시 보호될 수 있다. 예를 들어 하이드록실기는 Greene, "Protective Groups in Organic Chemistry", John Wiley & Sons, New York (1981)에 열거된 것과 같은 보호기에 의해 보호될 수 있다. 하이드록시 보호기는 치환된 메틸 에스테르, 예를 들어, 메톡시메틸, 벤질옥시메틸, 2-메톡시에톡시메틸, 2-(트리메틸실릴)에톡시메틸, t-부틸 및 다른 저급 알킬 에테르, 이를 테면, 이소프로필, 에틸 및 특히 메틸, 벤질 및 트리페닐메틸; 테트라하이드로피라닐 에테르; 치환된 에틸 에테르, 예를 들어, 2,2,2-트리클로로에틸; 실릴 에테르, 예를 들어, 트리메틸실릴, t-부틸디메틸실릴 및 t-부틸디페닐실릴; 및 하이드록실기와 카복실기를 반응시켜 제조된 에스테르, 이를 테면, 아세테이트, 프로피오네이트, 벤조에이트 등을 포함한다.
추가적인 아미노기는 선택적으로 쪼개질 수 있는 보호기에 의해 보호될 수 있다. 예를 들어, Boc가 α-아미노 보호기로 사용될 때, 하기의 측쇄 보호기가 적절하다: 추가적인 아미노기를 보호하는데 사용될 수 있는 p-톨루엔설포닐 (토실) 부분; 하이드록시기를 보호하는데 사용될 수 있는 벤질 (Bn) 에테르; 및 추가적인 카복실기를 보호하는데 사용될 수 있는 벤질 에스테르. 또는, Fmoc가 α-아미노 보호용으로 선택될 때, 보통 tert-부틸 기반의 보호기가 허용가능하다. 예를 들어, Boc는 추가적인 아미노기에; tert- 부틸 에테르는 하이드록실기에; 그리고, tert-부틸 에스테르는 추가적인 카복실기에 사용될 수 있다.
임의의 보호기는 합성 순서의 임의의 단계에서 제거될 수 있지만, 바람직하 게는 반응 단계에 포함되지 않는 임의의 작용성의 보호기는 마크로사이클의 형성 (build-up)의 완료 후 제거된다. 보호기의 제거는 보호기의 선택에 의해 지시되는, 해당 분야의 숙련자에게 잘 알려져 있는 어떠한 방식으로도 수행될 수 있다.
화합물 (I-c) 및 (I-d)를 위한 빌딩 블록 P1, P2 및 P3는 공지된 중간체로부터 시작하여 제조될 수 있다. 수많은 그러한 합성은 이하에서 더욱 상세하게 기술된다.
P2
빌딩 블록의 합성
P2 빌딩 블록은 -O-Rr로 치환된 사이클로펜텐 부분, 피롤리딘 또는 사이클로펜탄을 함유한다. Rr기는 본 발명에 따른 화합물의 합성의 임의의 통상의 탄계에서 이러한 환 중 임의의 것에 커플링될 수 있다. 하나의 접근법은 먼저 Rr기를 적절한 환에 커플링시키고, 그 다음 다른 목적하는 빌딩 블록, 예를 들어, P1 및 P3를 첨가한 다음, 마크로사이클을 형성시키는 것이다. 또다른 접근법은 Rr 치환체를 갖고 있지 않은 빌딩 블록 P2를 커플링시키고, 마크로사이클 형성 전 또는 후에, Rr 기를 첨가하는 것이다.
P2
치환체의 합성 및 도입
사이클릭 P2 골격에서 목적하는 퀴나졸린기는 다양한 방법에 의하여 합성의 임의의 통상의 단계에서 도입될 수 있다. 반응식 1은 미츠노부 반응에 의하여 P2 치환체의 도입을 예시한다(Mitsunobu, 1981, Synthesis, January, 1-28; Rano et al., Tetrahedron Lett., 1995, 36, 22, 3779-3792; Krchnak et al., Tetrahedron Lett., 1995, 36, 5, 6193-6196; Richter et al., Tetrahedron Lett., 1994, 35, 27, 4705-4706).

반응식 1
트리페닐포스핀 및 활성화제 이를 테면, 디에틸 아조디카복실레이트(DEAD), 디이소프로필 아조디카복실레이트(DIAD) 등의 존재 하에서 적절한 사이클릭 하이드록시 치환된 P2 골격 (1a)를 목적하는 퀴나졸리놀 (1b)로 처리하여 알킬화 화합물 (1c)가 수득된다. 사이클릭 골격(1a)의 하이드록시기는 피리딘과 같은 용매 중 원하는 산의 무수물 또는 할리드로 처리와 같은 적절한 설폰화 조건으로 알콜을 종속시킴으로써 또는 톨루엔과 같은 용매 중 DEAD의 존재 하에 트리페닐 포스핀 및 목적하는 설폰산을 이용하여 택일적으로 임의의 다른 적합한 이탈기, 이를 테면, 설폰산의 유도체, 이를 테면, 토실레이트, 메실레이트 또는 트리플레이트 등으로 전환될 수 있거나, 하이드록시기는 적합한 할로겐화제와 함께 알콜의 처리에 의하여 할리드로 전환될 수 있으며, 예를 들어, 브로마이드는 포스포러스 트리브로마이드 등과 같은 시약을 이용하여 제조될 수 있다. 그 다음, 수득된 이탈기는 목적하는 퀴나졸리놀에 의해 치환되어 알킬화 유도체 (1c)를 제공한다.
반대의 전략이 택일적으로 이용될 수 있으며, 여기서, 하이드록시 화합물 (1a)이 친핵제로서 이용되며, 디메틸포름아미드(DMF)와 같은 용매 중, 염기, 이를 테면, 소듐 하이드라이드 또는 포타슘 t-부톡사이드 등으로 처리하고, 수득된 알콕사이드를 알킬화제 Q-Lg(Lg는 적합한 이탈기, 이를 테면, 할리드, 예를 들어, 클로라이드, 브로마이드 또는 아이오다이드 또는 설폰산 등의 유도체이며, Q는 퀴나졸린 유도체이다)와 반응시키면, 목적하는 치환된 유도체가 수득된다. 프롤린 유도체에 적용되는 예는 E. M. Smith et al. in J. Med. Chem. (1988), 31, 875-885에 기술되어 있다.
퀴나졸린기를 사이클릭 P2 골격에 도입하기 위한 상기 방법을 본 발명에 따른 화합물의 합성의 임의의 통상의 단계에서 수행될 수 있는 것이 명백할 것이다. 예를 들어, R8 치환체는 화합물의 다른 성분이 도입되기 전에 적합한 사이클릭 골격에 도입될 수 있거나, 하이드록시 보호 사이클릭 골격은 합성에서 이용될 수 있고, 퀴나졸린기는 합성의 마지막 단계로서 도입된다.
치환된 퀴나졸린 유도체의 합성의 예는 반응식 2에 나타내었다.

반응식 2
예를 들어, 산을 Vilsmeyer 조건으로 처리하고, Raney-니켈에 대한 촉매적 수소화와 같은 조건을 이용하여 니트로기를 환원시켜, 니트로 치환된 벤조산 유도체 (2a)를 상응하는 벤즈아미드로 전환시키면, 상응하는 아민 (2c)이 수득된다. 수득된 아민은 그 다음 펩티드 커플링 조건 하에서, 이를 테면, HOBt 및 EDAC 또는 본 분야에 공지된 임의의 다른 적합한 커플링 조건 하에서 헤테로사이클릭 카복실산(2d)에 커플링될 수 있다. 이후 폐환과 탈수는 염기, 이를 테면, 탄산수소나트륨으로 처리하여 유효하게 될 수 있으며, 이로 퀴나졸린 유도체 (2f)가 수득된다. 퀴나졸린 유도체 (2f)는 상기 기술된 바와 같은 미츠노부 반응에서 P2 골격의 하이드록시기에 커플링될 수 있거나, 퀴나졸린의 하이드록시기는 퀴나졸린 (2f)을 적절한 할로겐화제, 예를 들어, 포스포릴 클로라이드 등으로 처리함으로써, 적합한 이탈기, 이를 테면, 할리드, 예를 들어, 클로라이드, 브로마이드 또는 아이오다이드에 의해 치환될 수 있다.
8-메틸 퀴나졸린 유도체는 반응식 2A에 나타낸 바와 같은 택일적인 트리-치환된 중간체 산 또는 아미드로부터 취득될 수도 있다.

반응식 2A
적합한 염기, 바람직하게 에톡사이드, 이를 테면, 에탄올 중 소듐 에톡사이드의 존재 하에서 에틸프로피오닐 아세테이트 및 에톡시메틸렌말로니트릴의 축합으로 테트라-치환된 벤조산 유도체 (2Aa)가 수득된다. 염기, 이를 테면, 리튬 하이드록사이드로 처리하여 에틸 에스테르를 가수분해하고, 수득된 산을 가열하여 탈카복실화 단계를 수행한 다음, 트리 치환된 페놀 유도체 (2Ab)가 수득된다. 예를 들어, 염기, 이를 테면, 포타슘 카보네이트 등의 존재 하에 메틸 아이오다이드를 이용하여 하이드록시 작용을 알킬화시키면, 상응하는 알콕시 유도체 (2Ac)가 수득된다. 그 다음, 예를 들어, 수산화 나트륨와 같은 염기의 존재 하에 물 및 에탄올 중 시아노 유도체 용액을 가열하여 시아노기를 가수 분해하여 트리-치환된 아미드 (2Ad)가 상응하는 산 (2Ae)과 함께 수득될 수 있다.
그 다음 반응식 2에 기재된 바와 같은 펩티드 커플링 조건 하에서 아미드 (2Ad)는 목적하는 산과 반응하여, 8-메틸 치환된 퀴나졸리놀을 제공할 수 있으며, 필요에 따라, 추가로 반응하여 상응하는 4-할로 유도체가 될 수 있다.
반응식 2A에서 수득된 산 (2Ae)은 반응식 2B에 나타낸 8-메틸 치환된 취나졸린 유도체의 제조를 위해 이용될 수도 있다.

반응식 2B
예를 들어, 메틸 에스테르와 같은 산 (2Ae)의 산 작용의 보호는 산을 포타슘 카보네이트와 같은 염기의 존재 하에서 메틸 아이오다이드와 처리하는 것 같은 알킬화 조건에 적용하여 유효하게 될 수 있다. 그 다음, 수득된 에스테르 유도체의 아미노 작용은 임의의 통상의 펩티드 커플링 기술, 이를 테면, 트리에틸아민 등과 같은 염기의 존재 하에서 산 클로라이드를 이용하여 목적하는 산과 커플링되어, 아미드 (2Bb)가 제공될 수 있다. 리튬 하이드록사이드와 같은 염기로 처리하고, 수득된 산을 포름아미드의 존재 하에서 가열하여 메틸 에스테르를 가수분해하여 퀴나졸리놀 (2Bc)이 수득된다. 상기 기술된 바와 같이, 퀴나졸리놀은 더욱 반응되어 상응하는 4-할로 유도체가 제공될 수 있다.
일반식 (2d)를 갖는 다양한 카복실산은 반응식 2에서 이용될 수 있다. 이러한 산은 상업적으로 또는 문헌에서와 같이 입수가능하다. Berdikhina et al. Chem. Heterocycl. Compd. (Engl. Transl.) (1991), 427-433에 의한 과정을 따른 2-(치환된)-아미노-카복시-아미노티아졸 유도체의 제조 예를 하기에 나타내었다.

R'는 C1-C6알킬이며; R"는 C1-C6알킬 또는 H이다.
반응식 3
다른 알킬 치환체 R' 및 R"를 지닌 티오우레아(3c)는 디클로로메탄과 같은 용매 중 디이소프로필에틸아민과 같은 염기의 존재 하에서 적절한 아민 (3a)과 tert-부틸이소티오시아네이트의 반응 후, 산성 조건 하에서 tert-부틸기의 제거에 의하여 형성될 수 있다. 택일적으로, 티오우레아(3c)는 아민 (3a)과 티오카보닐디이미다졸, 이후, 메탄올 중 암모니아 포화용액과 반응에 의하여 형성될 수 있다. 그 다음 수득된 티오우레아 유도체 (3c)와 3-브로모피루브산의 축합으로 산 (3d)가 수득된다.
반응식 2에서 아민 2c와의 반응에서 이용되는 4-치환된 티아졸-2-카복신산은 반응식 4에 나타낸 바와 같이 제조될 수 있다.

반응식 4
에틸 티오옥사메이트 (4a)와 목적하는 α-브로모케톤 (4d)의 축합 후, 리튬 하이드록사이드와 같은 염기의 처리에 의한 에스테르 가수분해로 티아졸 카복신살 (4d)가 수득된다. α-브로모케톤 (4d)은 상업적으로 입수가능하거나, 그들은 공지된 과정에 따라 상응하는 케톤의 α-브롬화에 의하여 제조될 수 있다.
P1
빌딩 블록의 합성 및 도입
P1 단편의 제조에 유용한 아미노산은 상업적으로 또는 문헌(예를 들어, WO 00/09543 및 WO00/59929 참조)에서 입수가능하다. 반응식 5는 P1 단편으로 이용될 설폰아미드 유도체의 제조예를 나타낸다.

반응식 5
아미노산을 커플링제, 예를 들어, THF와 같은 용매 중 N,N'-카보닐-디이미다졸 (CDI) 등으로 처리하고, 강염기, 이를 테면, 1,8-디아자비사이클로-[5.4.0]운데크-7-엔 (DBU)의 존재 하에서 목적하는 설폰아미드 (5b)와 반응함으로써, 설폰아미드기는 적합하게 보호된 아미노산 (6a)에 도입될 수 있다. 택일적으로, 아미노산은 디이소프로필 에틸아민과 같은 염기의 존재 하에서 목적하는 설폰아미드 (5b)와 처리된 다음, PyBOP®와 같은 커플링제와 처리되어 설폰아미드기가 도입될 수 있다. 표준 방법과 그 후에 P2 부분 또는 이의 전구체로의 커플링에 의하여 아미노 보호기가 제거된다.
A가 에스테르인 일반식 I에 따른 화합물의 제조를 위한 P1 빌딩 블록은 예를 들어, 에스테르 형성을 위한 표준 조건 하에서 아미노산 (5a)를 적절한 아민 또는 알콜과 반응시켜 제조될 수 있다.
P1 빌딩 블록을 P2 골격의 산 작용에 커플링시키는 일반적인 예를 반응식 7에 나타내었다.

Q는 퀴나졸린 유도체 또는 하이드록시 보호기이며,
A'는 보호된 카복실산 또는 치환된 아미드이다.
반응식 7
아미드 결합 형성을 위한 표준 방법을 이용하여, 이를 테면, 디메틸포름아미드와 같은 용매 중 디이소프로필아민과 같은 염기의 존재 하에서 HATU와 같은 커플링제를 이용하여, 상기에 개시된 바와 같이 제조된 P1 빌딩 블록(7b)을 P2 부분의 산 작용으로의 커플링시켜 아미드 (7c)가 수득된다.
택일적으로, 설폰아미드기는 합성의 후기 단계에, 예를 들어, 마지막 단계에서 도입될 수 있다. 이러한 경우, 반응식 7의 A'는 적절하게 보호된 카복실산, 예를 들어, 메틸 에스테르이며, 예를 들어, 설폰아미드기의 커플링 전에 리튬 하이드록시드 수용액으로 적절하게 탈보호된다.
우레아
연결된 ω-불포화
알킬
사슬의
헤테로사이클릭
P2
골격으로 도입
우레아 작용성을 통하여 P2 골격에 연결된 알킬 사슬은 반응식 10에 나타낸 바와 같이 도입될 수 있다.

Q는 퀴나졸린 유도체 또는 하이드록시 보호기이며;
Rx는 ω-불포화 5-8원 알킬 사슬이고;
A'는 보호된 카복실산 또는 치환된 아미드이다.
반응식 10
하이드라진 유도체 (10a)와 탄산 수소 나트륨과 같은 염기의 존재 하에 포르밀화제, 이를 테면, p-니트로페닐 클로로포르메이트, 카보닐 디이미다졸, 포스겐 등의 반응 후, P2 빌딩 블록을 첨가하여 우레아 유도체 (10c)가 수득된다.
적합하게 반응식 10에서 이용될 알케닐아민은 예를 들어 목적하는 tert-부틸카바메이트의 알킬화에 의하여 제조될 수 있으며, 일반적인 예는 반응식 11에 나타내었다.

반응식 11
목적하는 아민 R5-NH2와 tert-부틸 디카보네이트의 반응으로 boc 보호 아민(11a)가 수득된다. ω-불포화 알킬화제(11b), 이를 테면 알케닐할리드, 예를 들어, 브로마이드 또는 클로라이드로 수득된 카바메이트를 알킬화하고, 표준 조건을 이용하여, 이를 테면, 디클로로메탄과 같은 용매 중 TFA 용액으로 처리하여 boc기를 제거하여, 유리 아민(11c)이 수득된다.
A 또는 Rt1기는 합성의 임의의 단계에, 예를 들어, 고리화 전 또는 후에, 또는 상기 기술된 바와 같은 고리화 및 환원 전 또는 후에, P1 빌딩 블록에 연결될 수 있다. A 또는 Rt1이 -NHSO2R2를 나타내는 화학식 (I)의 화합물(화학식 (I-k-1)로 나타냄)은 두 부분 사이에 아미드 결합을 형성하여 A 또는 Rt1기를 P1에 연결시켜 제조될 수 있다. 유사하게, A 또는 Rt1이 -OR1를 나타내는 화학식 (I)의 화합물, 예를 들어, 화학식 (I-k-2)는 에스테르 결합을 형성시켜, A 또는 Rt1기를 P1에 연결시켜 제조될 수 있다. 일구체예에서, OR1기는 G가 하기의 기를 나타내는 다음 반응식에 개요된 바와 같은 화합물 (I)의 합성 마지막 단계에서 도입된다:

중간체 (2a)는 이하에서 기술된 아미드 결합 형성을 위한 과정 중 임의의 것과 같은 아미드 형성 반응에 의하여 아민 (2b)와 커플링될 수 있다. 특히, (2a)는 용매, 이를 테면, 에테르, 예를 들어, THF, 또는 할로겐화 탄화수소, 예를 들어, 디클로로메탄, 클로로포름, 디클로로에탄 중의 커플링제, 이를 테면, N,N'-카보닐-디이미다졸(CDI), EEDQ, HDQ, EDCI 또는 벤조트리아졸-1-일-옥시-트리스-피롤리디노-포스포늄 헥사플루오로포스페이트(PyBOP®으로 상업적으로 입수가능)와 처리되고, 바람직하게 (2a)와 커플링제의 반응 후, 목적하는 설폰아미드 (2b)와 반응될 수 있다. (2a) 와 (2b)의 반응은 바람직하게, 염기, 예를 들어, 트리알킬아민, 이를 테면, 트리에틸-아민 또는 디이소프로필에틸아민 또는 1,8-디아자비사이클[5.4.0]운데크-7-엔 (DBU)의 존재 하에 수행된다. 중간체 (2a)는 또한 활성 형태, 예를 들어, 일반식 G-CO-Z의 활성 형태로 전환될 수 있으며, 여기서, Z는 할로, 또는 활성 에스테르의 나머지를 나타내고, 예를 들어, Z는 아릴옥시기, 이를 테면, 페녹시, p.니트로페녹시, 펜타플루오로페녹시, 트리클로로페녹시, 펜타클로로페녹시 등이거나; Z는 혼합된 무수물의 나머지일 수 있다. 일구체예에서, G-CO-Z는 산 클로라이드(G-CO-Cl)이거나 혼합된 산 무수물 (G-CO-O-CO-R 또는 G-CO-O-CO-OR, 후자에서 R은 예를 들어, C1 - 4알킬, 이를 테면, 메틸, 에틸, 프로필, 이소프로필, 부틸, t.부틸, 이소부틸 또는 벤질)이다. 활성화 형태 G-CO-Z는 설폰아미드 (2b)와 반응된다.
상기 반응에 기술된 바와 같은 (2a) 중 카복실산의 활성화는 다음 화학식의 아자락톤 중간체로의 내부 고리화 반응을 유발할 수 있다:

상기 식에서, L, Rr, Rq, R5 , n은 상기에서 상술한 바와 같고, 여기서, 입체 중심은 상기에서, 예를 들어, (I-a) 또는 (I-b)에서 상술한 바와 같은 입체화학적 배치를 가질 수 있다. 중간체 (2a-1)는 통상의 방법을 이용하여 반응 혼합물로부터 분리될 수 있고, 그 다음, 분리된 중간체 (2a-1)는 (2b)와 반응하거나, (2a-1)를 함유하는 반응 혼합물은 (2a-1)의 분리 없이 (2b)와 추가로 반응될 수 있다. 일 구체예에서, 커플링제와의 반응이 물과 혼합할 수 없는 용매 중에서 수행되는 경우, (2a-1)를 함유하는 반응 혼합물을 물 또는 약염기 물로 세척하여, 수용성 부 산물이 제거될 수 있다. 이에 따라, 수득된 세척액은 그 다음 추가의 정제 단계 없이 (2b)와 반응될 수 있다. 한편으로, 중간체 (2a-1)의 분리는 분리된 생성물에서 특정한 장점을 제공할 수 있고, 임의의 추가적인 정제 후, (2b)와 반응되어, 더 적은 부생성물과 반응의 더 용이한 워크업 (work-up)을 초래할 수 있다.
중간체 (2a)는 에스테르 형성 반응에 의하여 알콜 (2c)과 커플링될 수 있다. 예를 들어, (2a) 및 (2c)는 예를 들어, 아제오트로피컬 물 제거에 의하여 물리적으로 또는 탈수제를 이용하여 화학적으로 물을 제거하면서 함께 반응시킨다. 중간체 (2a)는 또한 활성 형태 G-CO-Z, 이를 테면 상기 언급된 활성 형태로 전환될 수도 있으며, 이후 알콜 (2c)와 반응된다. 에스테르 형성 반응은 바람직하게, 염기, 이를 테면, 알칼리 금속 카보네이트 또는 탄산수소, 예를 들어, 탄산수소나트륨 또는 칼륨 또는 3차 아민, 이를 테면, 아미드 형성 반응과 관련하여 본원에 언급된 아민, 특히 트리알킬아민, 예를 들어, 트리에틸아민의 존재 하에서 수행된다. 에스테르 형성 반응에서 이용될 수 있는 용매는 에테르, 이를 테면, THF; 할로겐화 탄화수소, 이를 테면, 디클로로메탄, CH2Cl2; 탄화수소, 이를 테면 톨루엔; 극성 비양성자성 용매, 이를 테면, DMF, DMSO, DMA; 유사 용매를 포함한다.
카보사이클릭
P2
유니트를
함유하는 화합물의 합성
예를 들어, 일반식 1에서 L이 CH인 포화 카보사이클릭 P2 골격을 함유하는 화합물로의 전형적인 과정을 반응식 14에 나타내었다.

Rx는 ω-불포화 5-8 원 알킬 사슬이며;
A'는 보호된 카복실산, 치환된 아미드이다.
반응식 14
포화 사이클로알킬 골격(14b)을 예를 들어, Rosenquist et al. in Acta Chem. Scand. 46 (1992) 1127-1129에 개시된 3,4-비스-(메톡시카보닐)사이클로펜타논 (14a)로부터 메탄올과 같은 용매 중 소듐 보로하이드라이드와 같은 환원제로 케토기를 환원시키고, 에스테르를 가수분해하고, 마지막으로 피리딘의 존재 하에서 아세틱 무수물 중 폐환을 수행하여 제조하였다. 그 다음 제공된 비사이클릭산 (14b)은 디메틸 포름아미드와 같은 용매 중 디이소프로필 아민 및 HATU과 같은 통상의 펩티드 커플링 조건을 이용하여, 목적하는 하이드라진 유도체 (14c)의 아민 작용에 커플링되어, (14d)를 제공할 수 있다. 예를 들어 리튬 하이드록사이드로 (14d)의 락톤 개방으로 산 (14e)이 수득되며, 이는 그 다음, 통상의 펩티드 커플링 조건을 이용하여 P1 빌딩 블록의 아미노기 또는 목적하는 P1 단편 (14f)의 전구체에 커플링될 수 있다. 그 다음, 카보사이클의 R8-기의 도입은 예를 들어, 미츠노부 반응으로 적절한 알콜과 상기 개시된 바와 같이 또는 이전에 개시된 임의의 다른 적합한 방법으로 수행될 수 있다.
반응식 15는 포화 P2 골격을 포함하는 화학식 I의 화합물로의 택일적 경로를 나타내며, 여기서 빌딩 블록은 반대 순서로 도입되며, 예를 들어 P1 단편은 하이드라진 부분 전에 도입된다.

Q는 퀴나졸린 유도체이며;
Rx는 ω-불포화 5 내지 8 원 알킬 사슬이고;
A'는 보호된 카복실산, 치환된 아민이다.
반응식 15
예를 들어 tert-부틸 에스테르와 같은 (15a)의 산 그룹의 보호는 디클로로메탄과 같은 용매 중 디메틸아미노피리딘 및 트리에틸아민과 같은 염기의 존재 하에서 디-tert-부틸 디카보네이트로 처리되어, 에스테르 (15b)가 수득된다. 예를 들어, 리튬 하이드록사이드를 이용하여 락톤을 개방시키고, 그 다음, 반응식 12에 나타낸 바와 같이 또는 P1 단편의 아미노기에 의하여 직접적으로 P1 빌딩 블록 (15c) 을 커플링시켜, (15d)가 수득된다. 상기 기술된 바와 같이 R8-기를 도입한 다음, 에스테르를 메틸렌 클로라이드와 같은 용매 중 트리플루오로아세트산 및 트리에틸실란과 같은 산성 조건에 도입하여, 산 보호기가 제거되고, 최종적으로 상기 개시된 바와 같이 펩티드 커플링 조건을 이용하여 하이드라진 부분(15e)을 커플링시켜 하이드라지드 유도체 (15f)가 수득된다.
화학식 I의 화합물의 제조에 유용한 불포화 P2 골격은 반응식 16에 나타낸 바와 같이 제조될 수 있다.

반응식 16
Dolby et al. in J. Org. Chem. 36 (1971) 1277-1285에 의해 개시된 바와 같이 3,4-비스(메톡시카보닐)사이클로펜타논 (15a)의 브롬화-제거 반응 후, 소듐 보로하이드라이드와 같은 환원제로 케토 작용성을 환원시켜, 불포화 하이드록시 화합물 (15b)를 얻었다. 디옥산과 물의 혼합물과 같은 용매 중 예를 들어 리튬 하이드록사이드를 이용한 선택적 에스테르 가수분해로 하이드록시 치환된 모노에스테르 유도체 (15c)가 수득된다.
Rq가 수소가 아닌, 이를 테면, 메틸인 P2 골격은 반응식 17에 나타낸 바와 같이 제조될 수 있다.

반응식 17
피리디늄 클로로크로메이트와 같은 산화제를 이용하여 상업적으로 입수가능한 3-메틸-3-부텐-1-올 (17a)의 산화 후, 아세틸 클로라이드, 브로민 및 메탄올로 처리하여, α-브로모 에스테르 (17c)가 수득된다. 그 다음, 수득된 에스테르 (17c)는 상응하는 tert-부틸 에스테르를 테트라하이드로푸란과 같은 용매 중 리튬 디이소프로필 아미드와 같은 염기와 처리하여 수득된 에놀레이트 (17e)와 반응되어, 알킬화 화합물 (17f)가 수득된다. tert-부틸 에스테르 (17e)는 상응하는 상업적으로 입수가능한 산 (17d)을 디메틸아미노-피리딘과 같은 염기의 존재 하에서 디-tert-부틸 디카보네이트로 처리하여 제조될 수 있다. 상술된 바와 같이 수행된 올레핀 복분해 반응에 의한 (17f)의 고리화로 사이클로펜텐 유도체 (17g)가 제공된다. (17g)의 입체선택성 에폭시화는 야콥센 (Jacobsen) 비대칭 에폭시화 방법을 사용하여 수행되어 에폭시드 (17h)가 수득될 수 있다. 최종적으로, 염기, 특히 DBN (1,5-디아자바이사이클로-[4.3.0]논-5-엔) 첨가로 알콜 (17i)이 수득된다. 임의로, 화합 물 (17i)중에 이중 결합은 예를 들어, 탄소상 팔라듐과 같은 촉매를 사용하는 촉매성 수소화로 환원될 수 있고, 상응하는 포화 화합물을 수득시킨다.
그 다음, 수득된 사이클릭 골격이 상기 개시된 바와 같이 이용되어, 화학식 1의 화합물의 합성을 완료한다. 예는 반응식 18에 나타내었다.

Q는 퀴나졸린 유도체이고;
Rx는 ω-불포화 5 내지 8 원의 알킬 사슬이고;
A'는 보호된 카복실산, 치환된 아미드이다.
반응식 18
P1-빌딩 블록의 아미노기 또는 이의 적합한 전구체 (18b)는 이를 테면, 디이소프로필 페닐아민 등과 같은 염기의 존재 하에서 HATU를 이용하는 표준 아미드 커플링 조건을 이용하여 사이클로펜텐 유도체(18a)의 산에 커플링되고, 예를 들어, 상기 개시된 바와 같은 미츠노부 조건에 의하여 퀴나졸린기가 도입되어 (18d)가 수득될 수 있다. 남아있는 에스테르의 가수분해와 그 후, 목적하는 ω-불포화 아민 (18e)의 아미드 커플링 후, 임의로 P1 부분을 조작하여, 일반식 I에 따른 화합물 (18f)를 함유하는 사이클로펜텐이 수득된다.
마크로고리화
본 발명의 화합물에 존재하는 마크로사이클은 전형적으로 올레핀 복분해 반응(마크로고리화)에 의하여 형성된다. 사이클릭 P2 골격의 퀴나졸린기는 마크로사이클의 형성 전 또는 후에, 이전에 기술된 전략 중 임의의 것에 의하여 도입될 수 있다.
마크로사이클릭 우레아 화합물로의 전형적인 경로는 반응식 19에 나타내었다.

Q는 퀴나졸린 유도체 또는 하이드록시 보호기이며, n은 1,2,3 또는 4이다.
반응식 19
P1 부분으로서 비닐 사이클로프로필 글리신 에틸 아스테르를 이용하여 상기 개시된 바와 같이 제조된 화합물 (19a)은 올레핀 복분해 반응을 수행하여 마크로사이클릭 화합물 (19b)로 전환될 수 있다. Miller, S.J., Blackwell, H.E.; Grubbs, R.H. J. Am. Chem. Soc. 118, (1996), 9606- 9614, Kingsbury, J. S., Harrity, J. P. A., Bonitatebus, P. J., Hoveyda, A. H., J. Am. Chem. Soc. 121, (1999), 791-799 and Huang et al., J. Am. Chem. Soc. 121, (1999), 2674-2678에 기록된 것과 같은 Ru-계 촉매가 복분해 반응을 위해 이용될 수 있다. Mo와 같은 다른 전이 금속을 함유하는 촉매가 본 반응에 이용될 수 있음이 인지될 것이다. 임의로, 이중 결합은 본 분야에 표준 수소화법을 이용하여 환원되어, 상응하는 포화 마크로사이클릭 유도체가 수득된다.
반응식 19에 개시된 마크로고리화는 반응식 20에 예시한 바와 같은 포화 또는 불포화 카보사이클릭 P2 골격을 포함하는 화합물에 적용될 수도 있다.

Q는 퀴나졸린 유도체 또는 하이드록시 보호기이며, n은 1,2,3 또는 4이다.
반응식 20
적합한 염기, 예를 들어, 디이소프로필아민의 존재 하에 표준 펩티드 커플링 조건, 이를 테면 HATU를 이용하여 하이드라진 유도체 (20b)를 반응식 13 또는 14에 나타낸 바와 같이 제조된 P2-P1 빌딩 블록 (21a)에 커플링시켜, 중간체 (20c)가 수득된다. 반응식 18에 개시된 바와 같은 올레핀 복분해 반응에 의한 (20c)의 폐환으로 마크로사이클릭 화합물(2Od)가 수득된다.
상기 개시된 반응식 중 중간체가 작용기(들)를 함유할 때, 이들은 필요에 따라 적합하게 보호되며, 이후 본 분야에 숙련자에게 인지된 방법에 의해 탈보호된다. 광범위한 설명을 위하여, 예를 들어 상기 개시된 Bodanzky 또는 Greene 을 참조한다.
P3
빌딩 블록의 합성
P3 빌딩 블록은 본 분야의 숙련자에게 공지된 방법을 따라 제조될 수 있다. 이러한 방법 중 하나는 하기 반응식 28에 나타내었으며, 모노아실화 아민, 이를 테면, 트리플루오로아세트아미드 또는 Boc-보호 아민을 이용한다.

반응식 28
상기 식에서, R은 t-부톡시, 트리플루오로메틸이며; R5 및 n은 본 발명에서 정의된 바와 같고; LG는 이탈기, 이를 테면, 할로겐이다.
모노아실화 아민 (18a)은 강염기, 이를 테면, 소듐 하이드라이드로 처리되며, 이후 할로C3 - 6알케닐 (28b)과 반응되어 상응하는 보호 아민(28c)이 수득된다. (28c)의 탈보호로 빌딩 블록 P3 또는 (28d)가 수득된다. 탈보호는 작용기 R에 의존할 것이며, 이에 따라, R이 t-부톡시라면, 상응하는 Boc-보호 아민의 탈보호는 산,예를 들어, 트리플루오로아세트 산 처리에 의해 성취될 수 있다. 택일적으로, R이 예를 들어 트리플루오로메틸일 때, R기의 제거는 염기, 예를 들어, 수산화 나트륨으로 성취된다.
반응식 29는 P3 빌딩 블록을 제조하는 또다른 방법을 예시한다.

상기 식에서, X는 수소이며, n은 본 발명에 정의된 바와 같다.
반응식 29
일차 C3 - 6알케닐아민의 Gabriel 합성은 프탈이미드 (29a)를 염기, 이를 테면, 수산화 칼륨 및 할로C3 - 6알케닐 (29b)로 처리하여 수행될 수 있고, 그 후, 중간체 N-알킬 이미드의 가수분해로 일차 C3 - 6알케닐아민 (29c)가 수득된다.
P2-P1 부분에 적절한 P3 빌딩 블록의 커플링은 본원에 설명된 바와 같이 아미드 결합을 형성하여 성취될 수 있을 것이다.
마크로사이클의
형성
마크로사이클의 형성은 올레핀 복분해 (metathesis) 반응을 통해, 적절한 금속 촉매, 예를 들어, Miller, S. J., Blackwell, H.E., Grubbs, R.H. J. Am. Chem. Soc. 118, (1996), 9606-9614; Kingsbury, J. S., Harrity, J. P. A., Bonitatebus, P. J., Hoveyda, A. H., J. Am. Chem. Soc. 121, (1999), 791-799; 및 Huang et al., J. Am. Chem. Soc. 121, (1999), 2674-2678에 의해 보고된 Ru계 촉매; 예를 들어 Hoveyda-Grubbs 촉매의 존재하에서 수행될 수 있다. 공기 중에서 안정한 루테늄 촉매, 예를 들어, 비스(트리사이클로헥실포스핀)-3-페닐-1H-인덴-1-일리덴 루테늄 클로라이드 (Neolyst M1®) 또는 비스(트리사이클로헥실포스핀)-[(페닐티오)메틸렌]루테늄 (IV) 디클로라이드가 대량 생산에 사용될 수 있다. 다른 전이 금속, 예를 들어 Mo을 함유한 다른 촉매가 이 반응에 사용될 수 있다.
복분해 반응은 적절한 용매, 예를 들어 에테르, 예를 들어, THF, 디옥산; 할로겐화 탄화수소, 예를 들어, 디클로로메탄, CHCl3, 1,2-디클로로에탄 등, 탄화수소, 예를 들어, 톨루엔 중에서 수행될 수 있다. 바람직한 구체예에서, 복분해 반응은 톨루엔 중에서 수행된다. 이들 반응은 증가된 온도에서 질소 분위기하에서 수행된다.
임의로 이중 결합은 본 분야에 공지된 표준 수소화법에 의해, 예를 들어, 귀금속 촉매, 이를 테면, Pd 또는 Pt의 존재 하에서 수소와 함께 환원된다.
화학식 (I)의 화합물 또는 화학식 (I)의 화합물의 특정 서브그룹을 제조하는 수많은 특수한 합성 경로는 다음 반응식에, 반응식 30-33에서 더욱 자세하게 개요되었다.

반응식 30
반응식 30에 나타낸 바와 같이 본 발명의 화합물이 화학식 A, B 및 F의 화합물로부터 합성될 수 있다. 락톤 A는 DIPEA와 같은 염기의 존재 하에, 펩티드 커플링제, 이를 테면, HATU 또는 EDCI/HOAt의 존재 하에서, 구조 B의 C3 - 6알케닐아민과 커플링되어, 화학식 C의 화합물이 형성된다. 그 다음 락톤 개방과 DIPEA와 같은 염기의 존재 하에, 펩티드 커플링제, 이를 테면, HATU 또는 EDCI/HOAt의 존재 하에서, 1-(아미노)-2-(비닐)사이클로프로판-카복실산 에틸 에스테르와 커플링으로 화학식 E의 화합물이 수득된다. 화합물 E는 미츠노부형 반응을 이용하여 화학식 F의 퀴나졸린에 커플링될 수 있다. 수득된 디올레핀 G는 올레핀 복분해 촉매, 이를 테면, 호베이다-구럽 촉매 또는 적절한 용매, 이를 테면, 1,2-디클로로에탄, 디클로로메탄 또는 톨루엔 중 비스(트리사이클로헥실-포스핀)[(페닐티오)메틸렌]리테늄 (IV) 디클로라이드, 비스(트리사이클로헥실-포스핀)-3-페닐-1H-인덴-1-일아이덴루테늄 (IV) 디클로라이드 (Neolyst M1®)를 이용하여 폐환되어, 화학식 I의 상응하는 산으로 가수분해될 수 있는 화학식 H의 화합물이 형성될 수 있다. 화학식 I의 산은 펩티드 커플링제, 이를 테면, CDI, EDAC의 존재 하에서 그리고 1,8-디아자비사이클로[5.4.0]운데크-7-엔(DBU) 또는 DMAP와 같은 염기의 존재 하에서 R6SO2NH2와 커플링되어, 화학식 J의 화합물이 수득된다.

반응식 31
반응식 31에서, 화학식 K의 화합물은 염기, 이를 테면 NaH 또는 tBuOK의 존재 하에 클로로퀴나졸린 L과 반응되어, 화학식 M의 화합물을 형성한다. 수득된 산 M은 펩티드 커플링제, 이를 테면, HATU 또는 EDCI/HOAt의 존재 하에서, DIPEA와 같은 염기의 존재 하에 1-(아미노)-2-(비닐)사이클로프로판카복실산 에틸 에스테르 또는 상응하는 토실레이트로 처리되어, 화학식 N의 산물이 수득될 수 있다. 화학식 N의 화합물의 Boc 부분의 탈보호는 메틸렌 클로라이드와 같은 용매 중 산, 이를 테면, TFA로 처리되어 화학식 O의 유리 아민이 수득된다. 그 다음, 화학식 P의 우레아는 염기, 이를 테면 NaHCO3의 존재 하에 포스겐, 또는 포스겐의 등가물 및 화학식 B의 아민으로 처리하여 화학식 O의 화합물로부터 제조될 수 있다. 수득된 디올레핀 P는 올레핀 복분해 촉매, 이를 테면, 호베이다-구럽 촉매 또는 적절한 용매, 이를 테면, 1,2-디클로로에탄, 디클로로메탄 또는 톨루엔 중 비스(트리사이클로헥실-포스핀)[(페닐티오)메틸렌]리테늄 (IV) 디클로라이드, 비스(트리사이클로헥실-포스핀)-3-페닐-1H-인덴-1-일아이덴루테늄 (IV) 디클로라이드 (Neolyst M1®)를 이용하여 폐환되어, 화학식 R의 상응하는 산으로 가수분해될 수 있는 화학식 Q의 화합물이 형성될 수 있다. 화학식 R의 산은 펩티드 커플링제, 이를 테면, CDI, EDAC의 존재 하에서 그리고 1,8-디아자비사이클로[5.4.0]운데크-7-엔(DBU) 또는 DMAP와 같은 염기의 존재 하에서 R6SO2NH2와 커플링되어, 화학식 S의 화합물이 수득된다.
화학식 Q의 화합물의 합성하기 위한 택일적 방법은 하기 반응식 32에 나타내었다.

반응식 32
이에 따라, Boc-하이드록시프롤린은 펩티드 커플링제, 이를 테면, HATU 또는 EDCI/HOAt의 존재 하에서, DIPEA와 같은 염기의 존재 하에 1-(아미노)-2-(비닐)사이클로프로판카복실산 에틸 에스테르로 처리되어, 에스테르(1)가 수득된다. p-니트로벤조일 클로라이드로 유리 하이드록실기의 보호 후, Boc이 제거되어 유리 아민 (3)이 수득된다. 그 다음, 화학식 T의 우레아는 염기, 이를 테면 NaHCO3의 존재 하에 포스겐, 또는 포스겐의 등가물 및 화학식 B의 아민으로 처리하여 (3)으로부터 제조될 수 있다. 수득된 디올레핀 T는 올레핀 복분해 촉매, 이를 테면, 호베이다-구럽 촉매 또는 적절한 용매, 이를 테면, 1,2-디클로로에탄, 디클로로메탄 또는 톨루엔 중 비스(트리사이클로헥실-포스핀)[(페닐티오)메틸렌]리테늄 (IV) 디클로라이드, 비스(트리사이클로헥실-포스핀)-3-페닐-1H-인덴-1-일아이덴루테늄 (IV) 디클로라이드 (Neolyst M1®)를 이용하여 폐환되어, 화학식 U의 화합물이 형성될 수 있으 며, 이는 하이드록시드, 이를 테면 리튬 하이드록시드를 이용하여 탈보호되어, 화학식 V의 상응하는 알콜이 수득될 수 있다. P2 퀴나졸린의 도입은 염기 이를 테면, NaH 또는 tBuOK의 존재 하에서 화학식 V의 화합물과 클로로이소퀴놀린 L로부터 시작하여 화학식 Q의 화합물이 수득될 수 있다.
화학식 Q의 화합물의 합성하기 위한 택일적 방법은 하기 반응식 33에 나타내었다.

반응식 33
이에 따라, 프롤린 유도체(1)는 p-니트로벤조산으로 보호된 다음, Boc이 제거되어 유리 아민 (5)이 수득된다. 그 다음, 화학식 W의 우레아는 염기, 이를 테면 NaHCO3의 존재 하에 포스겐, 또는 포스겐의 등가물 및 화학식 B의 아민으로 처리하여 (5)으로부터 제조될 수 있다. 화학식 W의 화합물은 하이드록시드, 이를 테면 리튬 하이드록시드를 이용하여 탈보호되어, 화학식 X의 상응하는 알콜이 수득될 수 있다. P2 퀴나졸린의 도입은 미츠노부 반응을 이용하여 화학식 X의 화합물 및 하이드록시이소퀴놀린 F로부터 시작될 수 있으며, 화학식 Y의 화합물이 제공된다. 수득된 디올레핀 Y는 적절한 용매, 이를 테면, 1,2-디클로로에탄, 디클로로메탄 또는 톨루엔 중 올레핀 복분해 촉매, 이를 테면, 호베이다-구럽 촉매 등을 이용하여 폐환되어, 화학식 Q의 화합물이 수득될 수 있다.
상기 식 28-33에서(만), 화학식 (I)의 화합물 또는 이의 임의의 서브그룹을 위해 상기에서 정의된 바와 같은 R3는 R5에 상응하고, X는 L에 상응하며, R4a는 R9에 상응하고, R4b 및 R4b'는 R6 및 R11에 상응하며, R5는 R1에 상응하고, R6는 R2에 상응한다.
상기 반응식의 반응은 염기, 이를 테면, 알칼리 금속 카보네이트 또는 하이드록시드, 예를 들어, 소듐, 포타슘 또는 세슘 카보네이트; 또는 유기 염기, 이를 테면, 트리알킬아민, 예를 들어, 트리에틸아민의 존재 하에 적합한 용매 중에서 수행될 수 있다. 본 반응에 적합한 용매는 예를 들어, 에테르, 이를 테면, THF, 디옥산; 할로겐화 탄화수소, 예를 들어, 디클로로메탄, CHCl3, 톨루엔, 극성 비양자성 용매, 이를 테면, DMF, DMSO, DMA 등이다.
화학식 (I)의 화합물은 하기 기술된 것을 포함하는 공지된 작용기 전환 반응에 따라 서로 전환될 수 있다.
화학식 (I)의 화합물을 제조하는 데 이용되는 수많은 중간체는 공지의 화합물이거나, 공지의 화합물의 유사체이며, 이는 숙련자가 즉시 이용할 수 있는 공지 의 방법의 변형을 따라 제조될 수 있다. 수많은 중간체의 제조는 이하에서 더욱 자세하게 기술하였다.
화학식 (I)의 화합물은 3가 질소를 이의 N-옥사이드 형태로 전환하는 공지된 과정에 따라 상응하는 N-옥사이드 형태로 전환될 수 있다. 상기 N-산화 반응은 일반적으로 화학식 (I)의 출발 물질을 적절한 유기 또는 무기 과산화물과 반응시켜 수행될 수 있다. 적절한 무기 과산화물은 예를 들어, 과산화수소, 과산화 알칼리금속 또는 알칼리 토금속, 예를 들어, 과산화 나트륨, 과산화 칼륨을 포함하며; 적절한 유기 과산화물은 과산화산, 예를 들어, 벤젠카보-퍼옥소산 또는 할로 치환된 벤젠카보퍼옥소산, 예를 들어, 3-클로로벤젠-카보퍼옥소산, 퍼옥소알칸산, 예를 들어, 퍼옥소아세트산, 알킬하이드로퍼옥사이드, 예를 들어, tert-부틸 하이드로-퍼옥사이드를 포함할 수 있다. 적합한 용매는 예를 들어, 물, 저급 알코올, 예를 들어, 에탄올 등, 탄화수소, 예를 들어 톨루엔, 케톤, 예를 들어, 2-부탄온, 할로겐화 탄화수소, 예를 들어 디클로로메탄 및 그러한 용매의 혼합물이다.
화학식 (I) 화합물의 순수 입체 이성체는 공지된 과정을 적용하여 수득될 수 있다. 디아스테레오머는 물리적 방법, 이를 테면 선택적 결정화 및 크로마토그래피 기술, 예를 들어, 역류 분배, 액체 크로마토그래피 등에 의하여 분리될 수 있다.
화학식 (I)의 화합물은 공지된 분할 방법에 따라 서로 분리될 수 있는 거울상 이성체의 라세미 혼합물로 수득될 수 있다. 충분하게 염기성이거나 산성인 화학식 (I)의 라세미 화합물은 적합한 키랄 산, 각각 키랄 염기와 반응하여 상응하는 디아스테레오머 염 형태로 전환될 수 있다. 그 다음 상기 디아스테레오머 염 형태 가 예를 들어 선택 또는 분별 결정에 의해 분리되며, 거울상 이성체는 알칼리 또는 산에 의해 그로부터 해방된다. 화학식 (I)의 화합물의 거울상 이성체를 분리하는 선택적인 방법은 액체 크로마토그래피, 특히 키랄 정지상을 이용하는 액체 크로마토그래피를 포함한다. 상기 순수한 입체화학적 이성체는 또한, 적절한 출발 물질의 상응하는 순수한 입체화학적 이성체로부터 유도될 수 있으나, 단, 반응은 입체특이적으로 발생한다. 바람직하게, 만약 특이 입체이성체를 원한다면, 상기 화합물은 입체특이적 제조 방법으로 합성수 있다. 이들 방법은 유리하게 거울상 이성체적으로 순수한 출발 물질을 사용할 것이다.
다른 면에서, 본 발명은 본원에 상술된 바와 같은 화학식 (I)의 화합물 또는 본원에 상술된 바와 같은 화학식 (I)의 화합물의 서브그룹 중 임의의 화합물의 치료적 유효량과 약제학적으로 허용되는 담체를 포함하는 약제학적 조성물에 관한 것이다. 본원에서 치료적 유효량은 감염된 대상 또는 감염될 위험에 있는 대상에서 바이러스 감염 및 특히 HCV 바이러스 감염에 예방적으로 활성이 있거나, 이를 안정하게 하거나, 이를 감소시키기에 충분한 양이다. 또다른 면에서, 본 발명은 본원에 상술된 바와 같은 약제학적 조성물을 제조하는 방법에 관한 것이며, 이는 약제학적으로 허용되는 담체를 본원에 상술된 바와 같은 화학식 (I)의 화합물 또는 본원에 상술된 바와 같은 화학식 (I)의 화합물의 서브그룹 중 임의의 화합물의 치료적 유효량과 혼합하는 것을 포함한다.
이에 따라, 본 발명의 화합물 또는 이의 임의의 서브그룹은 투여 목적을 위하여 다양한 약제학적 형태로 제제화될 수 있다. 적절한 조성물은 보통 전신 투여 약물로 사용되는 것으로 언급된 모든 조성물일 수 있다. 본 발명의 약제학적 조성물을 제조하기 위하여, 활성 성분으로서 특정 화합물의 유효량, 임의로 부가 염 형태 또는 금속 복합물는 약제학적으로 허용되는 담체와 잘 혼합된 혼합물로 결합되고, 이러한 담체는 투여에 바람직한 제조 형태에 의존하여 매우 다양한 형태를 취할 수 있다. 이들 약제학적 조성물은 특히 경구, 직장, 경피 투여 또는 비경구 주사에 적합한 단위 제형이 바람직하다. 예를 들어 경구 제형으로 조성물을 제조하는데 있어서, 임의의 통상 약제 매질은 이를 테면 현탁액, 시럽, 엘릭시르, 에멀젼 및 용액과 같은 경구 액체 제제의 경우 물, 글리콜, 오일, 알코올 등; 또는 분말, 환약, 캡슐, 및 정제의 경우 전분, 슈가, 카올린, 윤활제, 결합제, 붕해제 등과 같은 고체 담체가 사용될 수 있다. 이들의 투여 용이성으로, 정제와 캡슐이 가장 유용한 경구 단위 제형을 대표하며, 이 경우 고체 약제 담체가 분명히 사용된다. 비경구 조성물을 위해, 담체는 다른 성분, 예를 들어 용해도를 돕는 성분이 포함될 수 있지만, 통상 무균수를 적어도 대부분 포함할 것이다. 예를 들어, 담체가 식염수, 글루코스 용액 또는 식염수와 글루코스 용액의 혼합물을 포함하는, 주사가능한 용액이 제조될 수 있다. 적합한 액체 담체, 현탁화제 등이 사용될 수 있는 주사가능한 현탁액이 또한 제조될 수 있다. 사용 직전 액체 제제로 전환되는 고체 제제가 포함된다. 경피 투여에 적합한 조성물에서, 담체는 임의로 침투 증진제 및/또는 적합한 습윤제를 포함하며, 임의로 어떠한 특성의 적합한 첨가제와 소량 비율로 배합되며, 첨가제는 피부 상에 상당한 유해 효과를 유발하지 않는다.
본 발명의 화합물은 경구 흡입 또는 통기를 통해 투여하기 위하여 본 분야에 이용된 방법과 제제의 수단으로 경구 흡입 또는 통기를 통해 투여될 수 있다. 이에 따라, 일반적으로 본 발명의 화합물은 용액, 현탁액 또는 건조 분말의 형태로 폐로 투여될 수 있으며, 용액이 바람직하다. 경구 흡입 또는 통기를 통하여 용액, 현탁액 또는 건조 분말을 전달하기 위해 발달된 임의의 시스템은 본 화합물의 투여에 적합하다.
이에 따라, 본 발명은 또한 화학식 (I)의 화합물 및 약제학적으로 허용되는 담체를 포함하며 경구 흡입 또는 통기에 의해 투여되기 적합한 약제학적 조성물을 제공한다. 바람직하게, 본 발명의 화합물은 분무 또는 연무된 용량으로 용액의 흡입을 통해 투여된다.
상기 언급된 약제학적 조성물은 투여 용이성과 투여량의 균일성을 위해 단위 제형으로 제제화되는 것이 특히 바람직하다. 본원에 이용된 단위 제형은 단위 투여량으로 적합한 물리적 분할 단위를 뜻하며, 각 단위는 필요한 약제학적 담체와 회합되어 목적하는 치료 효과를 나타내도록 계산된 활성 성분의 일정량을 함유한다. 이 단위 제형의 일예는 정제(금이 새겨있거나 코팅된 정제 포함), 캡슐, 환제, 좌제, 분말 패킷, 웨이퍼, 주사 용액 또는 현탁액 등, 및 이의 분할된 다중회분이다.
화학식 (I)의 화합물은 항바이러스성을 나타낸다. 본 발명의 화합물 및 방법을 이용하여 치료할 수 있는 바이러스 감염과 이의 연관된 질환은 HCV 및 다른 병원성 플라비바이러스, 이를 테면 황열, 뎅기열(1-4형), 세인트루이스 뇌염, 일본 뇌염, 머레이계곡 뇌염, 웨스트 나일 바이러스 및 쿤진 바이러스에 의한 감염을 포함한다. HCV와 연관된 질환은 경화증, 말기 간 질환 및 HCC를 유발하는 진행성 간 섬유증, 염증 및 괴사를 포함하며; 다른 병인성 플라비바이러스에서 질환은 황열, 뎅기열, 출혈열 및 뇌염을 포함한다. 더욱이, 본 발명의 수많은 화합물은 HCV의 돌연변이 스트레인(strain)에 대하여 활성이 있다. 또한, 본 발명의 많은 화합물은 바람직한 약물동태학적 프로필을 나타내며, 허용되는 반감기, AUC(곡선 아래 영역) 및 피크치를 포함하는 생체이용률에서 유용한 특성을 가지며, 바람직하지 않은 현상, 이를 테면 불충분하게 빠른 개시 및 조직 잔류가 없다.
화학식 (I)의 화합물의 HCV에 대한 시험관내 항바이러스성을 Lohmann et al.(1999) Science 285:110-113을 기준으로 하고, Krieger et al.(2001) Journal of Virology 75: 4614-4624(본원에 참조로 삽입, 실시예 섹션에 더욱 구체화됨)에 의해 변형된 세포 HCV 레플리콘(replicon) 시스템에서 시험하였다. 이 모델은 HCV에 대한 완전 감염 모델이 아니지만, 현재 이용가능한 자기 HCV RNA 복제의 가장 확고하고 효율적인 모델로서 널리 인정되고 있다. 이 세포 모델에서 항-HCV 활성을 지닌 화합물은 포유 동물에서 HCV 감염의 치료에 추가 개발을 위한 후보군으로서 고려된다. HCV 레플리콘 모델에서 세포독성 또는 세포 증가 억제 효과를 나타내며 그 결과 HCV RNA 또는 연결된 리포터 효소 농도의 감소를 야기하는 화합물들로부터 HCV 작용을 특이하게 간섭하는 화합물들을 구분하는 것은 중요하다고 이해될 것이다. 예를 들어 레사주린과 같은 플루오로게닉 레독스 염료를 이용하여 미토콘드리아 효소의 활성을 기초로 세포의 세포독성을 평가하는 분석이 본 분야에서 알려져 있다. 또한, 세포의 카운터-스크린은 연결된 리포터 유전자 활성, 이를테면 반딧불이 루시퍼라제의 비선택적 억제 평가를 위해 존재한다. 적합한 세포형은 발현이 구 조적으로 활성인 유전자 프로모터에 의존하는 루시퍼라제 리포터 유전자의 안정한 형질감염에 의해 구비될 수 있으며, 이러한 세포는 비선택적 억제제를 제거하기 위해 카운터스크린으로서 사용될 수 있다.
이의 항바이러스성, 특히 항-HCV 특성 때문에, 화학식 (I)의 화합물 또는 이의 임의의 서브그룹, 전구 약물, N-옥사이드, 부가염, 4차 아민, 금속 복합물 및 입체화학적 이성체는 바이러스 감염, 특히 HCV 감염된 개체의 치료와 이러한 감염의 예방에 유용하다. 일반적으로, 본 발명의 화합물은 바이러스, 특히 HCV와 같은 플라비바이러스에 감염된 온혈 동물의 치료에 유용할 수 있다.
더욱이, 본 발명의 화합물 또는 이의 임의의 서브그룹은 의약으로 이용될 수 있다. 상기 의약으로서 용도 또는 치료 방법은 바이러스 감염, 특히 HCV 감염과 연관된 증상을 없애기 위하여 유효량을 바이러스 감염 대상에, 또는 바이러스 감염에 감수성인 대상에 전신 투여하는 것을 포함한다.
또한, 본 발명은 바이러스 감염, 특히 HCV 감염의 치료 또는 예방을 위한 의약의 제조에서 본 화합물 또는 이의 임의의 서브그룹의 용도에 관한 것이다.
게다가, 본 발명은 바이러스에 의해 감염된, 또는 바이러스, 특히 HCV에 의해 감염될 위험에 있는 온혈 동물을 치료하는 방법에 관한 것이며, 상기 방법은 본원에 상술한 바와 같은 화학식 (I)의 화합물 또는 본원에 상술한 바와 같은 화학식 (I) 화합물의 임의의 서브그룹의 화합물의 항-바이러스적 유효량을 투여하는 것을 포함한다.
일반적으로, 하루에 항바이러스에 효율적인 양은 체중 kg당 0.01 mg 내지 500 mg, 더욱 바람직하게 체중 kg 당 0.1 mg 내지 50 mg라고 고려된다. 필요량을 하루 중 적절한 간격에 둘, 셋, 넷 이상의 단위-투여로 투여하는 것이 적절할 수 있다. 상기 단위-투여는 예를 들어, 단위 제형 당 1 내지 1000 mg 및 특히 5 내지 200 mg의 활성 성분을 함유하는 단일 제형으로 제제화될 수 있다.
투여의 정확한 용량과 빈도는 이용되는 화학식 (I)의 특정 화합물, 치료될 특정 증상, 치료될 증상의 중증도, 나이, 체중, 성별, 장애의 범위 및 특정 환자의 일반적 신체 상태, 뿐 아니라 개체가 취하고 있는 다른 의약에 의존하며, 본 분야의 숙련자에게 공지된 것에 따른다. 더욱이, 상기 하루 유효량은 치료되는 대상의 반응 및/또는 본 발명의 화합물을 처방하는 의사의 판단에 따라 더 낮아지거나 더 증가될 수 있다. 이에 따라, 상기 언급된 하루 유효량은 가이드라인일 뿐이다.
또한 공지된 항-HCV 화합물, 이를 테면, 인터페론-α (IFN-α), 페길화 인터페론-α 및/또는 리바비린과 화학식 (I)의 화합물의 배합물이 배합 요법에서 의약으로 이용될 수 있다. 용어 "배합 요법"은 바이러스 감염의 치료에서 특히 HCV 감염의 치료에서, 동시, 분리 또는 연속적 이용을 위하여 배합된 제제로, 필수로 (a) 화학식 (I)의 화합물 및 임의로 (b) 또다른 항-HCV 화합물을 함유하는 산물에 관한 것이다. 이에 따라, HCV 감염을 없애거나 치료하기 위하여, 화학식 (I)의 화합물은 예를 들어 인터페론-α (IFN-α), 페길화 인터페론-α 및/또는 리바비린, 뿐 아니라 HCV 에피토프에 대하여 표적화된 항체, 소간섭 RNA(siRNA), 리보자임, DNAzyme, 안티센스 RNA, 예를 들어 NS3 프로테아제, NS3 헬리카제 및 NS5B 폴리머라제의 소분자 길항제에 기초한 치료제와 배합되어 공동-투여될 수 있다.
이에 따라, 본 발명은 HCV 바이러스에 감염된 포유동물에서 HCV 활성을 억제하는 데 유용한 의약을 제조하기 위한, 상기에서 정의된 바와 같은 화학식 (I)의 화합물 또는 이의 임의의 서브그룹의 용도에 관한 것이며, 여기에서 상기 의약은 배합 요법으로 이용되고, 상기 배합 요법은 바람직하게 화학식 (I)의 화합물 및 (페길화) IFN-α 및/또는 리바비린 및 임의로 항-HIV 화합물을 포함한다. 예를 들어, Cyp3A4에 의하여 빠르게 대사되는 경향이 있는 약물에서, HIV 프로테아제 억제제, 이를 테면 리토나비르의 공동-투여는 더 적은 투여 요법을 허용한다.
실시예
하기 실시예는 본 발명을 설명하려는 것이며, 이에 한정되지 않는다. 빌딩 블록의 제조를 나타내는 예는 본원에 개시된 임의의 적절한 다른 빌딩 블록에 커플링하는 경향이 있으며, 간단하게 성분을 예시된 화학식 (I)의 말단 화합물에 나타내지 않았다.
실시예
1

1-
브로모
-3-
메틸부탄
-2-온 (1)
EtOH (250 ml) 중 3-메틸-2-부타논 (25.8g, 300 mmol)의 얼음처럼 차가운 용액에 브롬(12.9 ml, 250 mmol)을 점적 첨가하고, 혼합물을 얼음 배쓰에서 2 시간 동안 교반하였다. 석유 에테르(600ml)를 첨가하였다. 유기상을 물로 두번 세척하였다. 결합된 수상을 석유 에테르로 2회 추출하였다. 결합된 유기상을 차가운 탄산 나트륨 용액과 염수로 2회 세척하였다. 유기상을 황산 나트륨으로 건조시키고, 감압하에 증발시켰다(실온). 수율: 50%.
실시예
2

에틸 4-
이소프로필티아졸
-2-
카복실레이트
(2)
EtOH 중 에틸 티오옥사메이트 (16.0 g, 120 mmol)의 끓는 용액에 1-브로모-3-메틸-2-부타논을 15분에 걸쳐 점적 첨가하였다. 혼합물을 1.5 시간 동안 환류시켰다. 용액을 300 ml의 얼음 물에 첨가하고, 암모니아 농축액으로 염기성화하였다. 혼합물을 에틸 아세테이트로 2회 추출하였다. 유기상을 염수로 세척하고, 황산 나트륨으로 건조시키고, 감압하에 증발시켰다. 생성물을 크로마토그래피 실리카 겔 상 컬럼으로 정제하고, 헥산 및 20% 에틸 아세테이트로 용리시켰다. 수율: 15.2 g, 67%.
실시예
3

4-
이소프로필티아졸
-2-
카복실산
(3)
THF (100 ml) 및 MeOH (30 ml) 중 에틸 4-이소프로필티아졸-2-카복실레이트 (9.1g, 46mmol) 용액에 수산화 리튬 (1.16 g, 48.5 mmol) 용액을 첨가하고, 혼합물을 실온에서 2일 동안 교반하였다. 혼합물을 2M 염산으로 산성화시키고, 디에틸 에테르로 4회 추출하였다. 유기상을 황산 나트륨으로 건조시키고 감압 하에 증발시켰다. 수율: 7.1g, 90%.
실시예
4

4-
메톡시
-2-니트로-
벤즈아미드
(4)
DCM (150 ml) 중 DMF 몇 방울과 4-메톡시-2-니트로-벤조산 (14.1 g, 71.5 mmol)의 얼음처럼 차가운 현탁액에 염화 옥살릴(19.0 g, 150 mmol)를 점적 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 용매를 증발시키고, 물을 첨가하였다. 생성물을 여과하고 물과 헥산으로 세척하였다. 생성물을 진공 하에 건조시켰다. 수율: 10 g, 71%.
실시예
5

4-
메톡시
-2-아미노-
벤즈아미드
(5)
EtOH (200 ml) 중 4-메톡시-2-니트로-벤즈아미드 (6.9 g, 35.1 mmol)의 현탁액을 2일 동안 실온 및 50 psi에서 Raney-Ni (4.0 g)로 수소화시켰다. 촉매를 여과하고 DMF로 세척하였다. 용매를 감압하에 증발시켰다. 수율: 5.6g, 95%.
실시예
6

4-
이소프로필티아졸
-2-
카복실산
(2-
카바모일
-5-
메톡시
-
페닐
)-아미드 (6)
DMF (150 ml) 중 Hobt-수화물 (6.4 g, 42 mmol), 4-이소프로필-티아졸-2-카복실산 (7.1 g, 42 mmol) 및 4-메톡시-2-아미노벤즈아미드 (5.6 g, 33.7 mmol)의 차가운 용액에 EDAC (8.6 g, 45 mmol) 및 TEA (6.4 ml, 45 mmol)를 첨가하고, 혼합물을 실온에서 밤새 교반하였다. 2.5% 시트르산 수용액(600 ml)을 첨가하고, 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 염수 및 포화 탄산수소나트륨으로 세척하였다. 용액을 황산 나트륨으로 건조시키고, 감압하에 증빌시켰다. 수율: 9.Og, 91%.
실시예
7

2-(4-
이소프로필티아졸
-2-일)-7-
메톡시
-
퀴나졸린
-4-올(7)
EtOH 물 50/50 (300 ml) 중 4-이소프로필-2-카복실산 (2-카바모일-5-메톡시-페닐)-아미드 (9.0 g, 28.2 mmol) 및 탄산 나트륨 (7.5 g, 71 mmol)의 혼합물을 2시간 동안 환류시켰다. 혼합물을 냉각시키고, 시트르산으로 산성화시킨 다음, 에틸 아세테이트로 4회 추출하였다. 유기 상을 황산 나트륨으로 건조시키고, 감압하에 증발시켰다. 생성물을 EtOH로부터 결정화하였다. 수율: 4.8g, 60%.
실시예
8

4-
이소프로필티아졸
-2-
카복실산
(2-
카바모일
-
페닐
)-아미드 (8)
2-아미노벤즈아미드 (2.04 g, 15 mmol)를 실시예 6에 개시된 바와 같은 4-이소프로필티아졸-2- 카복실산 (2.5 g, 14.6 mmol)과 반응시켜, 표제 화합물 (2.4 g, 56%)을 얻었다.
실시예
9

2-(4-
이소프로필티아졸
-2-일)-
퀴나졸린
-4-올(9)
4-이소프로필티아졸-2-카복실산(2-카바모일-페닐)-아미드 (2.4 g, 8.3 mmol)를 실시예 7에서 개시된 과정에 따라 처리하여 표제 화합물 (1.7 g, 77%)을 얻었다.
실시예
10

2-아미노-5-
메톡시
-
벤즈아미드
(10)
Raney-니켈에 대하여 5-메톡시-니트로-벤즈아미드 (3.6 g)를 촉매 수소화하 여, 표제 화합물(2.75 g, 90%)을 얻었다.
실시예
11

7-
메톡시
-2-
페닐
-
퀴나졸린
-4-올 (11)
2-페닐-퀴나졸린 4-올의 제조를 위해 Raid J. Abdel- Jalil, Wolfgang Voelter and Muhammad Saeed in Tetrahedron Letters 45 (2004) 3475-3476에 개시된 과정을 따라 2-아미노-5-메톡시-벤즈아미드를 처리하여 표제 화합물을 수득하였다.
실시예
12

트랜스(3R,4R)-비스(메톡시카보닐)사이클로펜타놀 (12)
0℃에서 소듐 보로하이드라이드 (1.11 g, 0.029 mol)를 메탄올 (300 mL) 중 (1R,2S)-4-옥소-사이클로펜탄1,2-디카복실산 디메틸 에스테르 (4.88 g, 0.0244 mol)의 교반 용액에 첨가하였다. 1시간 후, 90 mL 염수로 반응을 퀘엔칭하고, 농축 시키고, 에틸 아세테이트로 추출하였다. 유기상을 풀링하고, 건조시키고, 여과하고, 농축시켰다. 조 생성물을 플래쉬 컬럼 크로마토그래피(톨루엔/에틸 아세테이트 1:1)로 정제하여, 노란색 오일로서 표제 화합물 (3.73 g, 76%)을 얻었다.
실시예
13
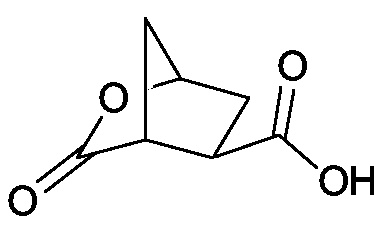
3-옥소-2-옥사-
비사이클로[2.2.1]헵탄
-5-
카복실산
(13)
수산화나트륨 (1M, 74 mL, 0.074 mol)을 실온에서 메탄올 (105 mL) 중 12의 교반 용액 (3.73 g, 0.018 mol)에 첨가하였다. 4 시간 후, 반응 혼합물을 3M HCl로 중화시키고, 증발시키고 톨루엔으로 수회 공동-증발시켰다. 피리딘 (75 mL) 및 Ac2O (53 mL)을 첨가하고, 반응 혼합물을 실온에서 밤새 쉐이킹하였다. 그 다음, 반응물을 톨루엔으로 공동-증발시키고, 플래쉬 컬럼 크로마토그래피(에틸 아세테이트 + 1% 아세트산)로 정제하여, 노란색 오일로서 표제 화합물 (2.51 g, 88%)을 얻었다.
실시예
14

3-옥소-2-옥사-
비사이클로[2.2.1]헵탄
-5-
카복실산
tert
-부틸 에스테르 (14)
DMAP (14 mg, 0.115 mmol) 및 Boc2O (252 mg, 1.44 mmol)를 0℃에서 불활성 아르곤 대기하에 2 mL CH2Cl2 중 13 (180 mg, 1.15 mmol)의 교반 용액에 첨가하였다. 반응은 실온으로 가온시키고, 밤새 교반하였다. 반응 혼합물을 농축시키고, 조 생성물을 플래쉬 컬럼 크로마토그래피 (톨루엔/에틸 아세테이트 구배 15:1, 9:1, 6:1, 4:1, 2:1)로 정제하여 백색 결정으로서 표제 화합물(124 mg, 51%)을 얻었다.

화합물 14를 제조하는 택일적 방법

화합물 13 (13.9 g, 89 mmol)을 디클로로메탄 (200 ml) 중에 용해시킨 다음, 질소 하에서 약 -10℃로 냉각시켰다. 그 다음, 전체 부피가 약 250 ml으로 증가할 때까지 이소부틸렌을 용액을 통하여 버블링시켜, "흐린 액체(clowdy solution)"을 얻었다. BF3 x Et2O (5.6 ml, 44.5 mmol, 0.5 당량)을 첨가하고, 반응 혼합물을 질소 하에서 약 -10℃로 유지하였다. 10분 후, 투명한 용액을 수득하였다. 반응을 TLC(아세트산 몇방울로 산성화된 EtOAc-톨루엔 3:2, 및 헥산-EtOAc 4:1, 염기성 과망간산 용액으로 염색)로 모니터하였다. 70분에 화합물 13의 흔적만이 남았으며, NaHCO3 포화 수용액 (200 ml)을 반응 혼합물에 첨가하고, 그 다음, 10분 동안 격렬하게 교반하였다. 유기층을 NaHCO3 포화 용액 (3 x 200 ml)과 염수(1 x 150 ml)로 세척한 다음, 소듐 설피트로 건조시키고, 여과시킨 다음 몇 방울의 오일로 농축시켰다. 잔류물에 헥산을 첨가함에 따라, 생성물이 결정화되었다. 헥산을 더 첨가하고, 환류 하에 가열하여 생성물이 결정화된 투명한 용액을 얻었다. 여과로 결정을 수집하고, 헥산으로 세척한 다음(실온), 72 시간 동안 자연 건조시켜, 무색의 침상을 얻었다(12.45 g, 58.7 mmol, 처음 수득물로부터 66%).
실시예
15

(1R,2R,4S)-2-((1R,2S)-1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-하이드록시-
사이클로펜탄카복실산
tert
-부틸 에스테르 (15)
화합물 14 (56 mg, 0.264 mmol)를 디옥산/ 물 1:1 (5 mL) 중에 용해시키고, 혼합물을 0℃로 냉각시켰다. 1 M 수산화 리튬 (0.52 mL, 0.520 mmol)을 첨가하고, 혼합물을 0℃에서 45분 동안 교반한 다음, 혼합물을 1M 염산으로 중화시키고, 증발시켰으며, 톨루엔으로 공동증발시켰다. 결정 잔류물을 DMF (5 mL) 중에 용해시키고, (1R,2S)-1-아미노-2-비닐-사이클로프로판 카복실산 에틸 에스테르 하이드로클 로라이드 (60 mg, 0.313 mmol) 및 디이소프로필에틸아민 (DIEA) (138 μL, 0.792 mmol)를 첨가하고, 용액을 0℃로 냉각시켰다. HATU (120 mg, 0.316 mmol)를 첨가하고, 혼합물을 0℃에서 0.5 시간 동안 교반하고, 실온에서 추가로 2시간 동안 교반하였다. 그 다음, 혼합물을 증발시키고, EtOAc로 추출하고, 염수로 세척, 건조, 여과 및 농축시켰다. 플래쉬 컬럼 크로마토그래피(톨루엔/EtOAc 1:1)로 정제하여, 무색 오일로서 표제 화합물 (86 mg, 89 %)을 얻었다. 수득된 오일을 에틸 아세테이트-헥산으로부터 결정화시켰다.
실시예
16

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄카복실산
tert
-부틸 에스테르 (16)
화합물 15 (700 mg, 1.9 mmol), 7- 메톡시-2-페닐-퀴나졸린-4-올 (670 mg, 2.66 mmol) 및 트리페닐 포스핀 (1245 mg, 4.75 mmol)을 THF (50 ml) 중에 용해시키고, 0℃로 냉각시켰다. 디이소프로필 아지도카복실레이트 (960 mg, 4.75 mmol)를 천천히 첨가하고, 슬러리가 실온에 도달되도록 하였다. 12시간 후, 용매를 감압하 에 제거하고, 잔류물을 에테르 중에서 취하고, 여과하였다. 컬럼 크로마토그래피(SiO2; 디클로로메탄 중 1 % 메탄올)로 정제하여 순수한 표제 화합물 (778 mg, 68 %)을 얻었다. MS (M+H)+ 603.
실시예
17

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄카복실산
(17)
화합물 16 (780 mg, 1.29 mmol)을 디클로로메탄 (20 mL) 및 트리에틸실란 (0.4 mL) 중에 용해시켰다. 트리플루오로메탄설폰산을 실온에서 점적 첨가하였다. 그 다음, 혼합물을 실온에서 2 시간 동안 놔두었다. 용매를 제거하여 순수한 표제 산물 (700 mg, 99%)을 얻었다. MS (M+H)+ 546.
실시예
18

1-{[2-
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
)- 사이클로펜탄카보닐]-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (18)
화합물 17 (700 mg, 1.28 mmol), N-메틸-1-헥센 하이드로클로라이드 (291mg, 1.94 mmol), 디이소프로필 에틸아민 (750 mg, 5.8 mmol) 및 HATU (736 mg, 1.94 mmol)를 DMF (30 mL) 중 용해시키고, 혼합물을 실온에서 밤새 교반하였다. 용매를 제거하고, 잔류물을 디클로로메탄과 소듐 비카보네이트 수용액에 분배하였다. 유기상을 수집하고, 조 생성물을 컬럼 크로마토그래피(실리카 겔, 디클로로메탄 중 2 % 메탄올 → 디클로로메탄 중 4 % 메탄올)로 정제하였다. 용매를 증발시켜 순수한 표제 화합물 (700 mg, 85 %)을 얻었다. MS (M+H)+ 641.
실시예
19

17-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
-13-
메틸
-2,14-
디옥소
-3,13-
디아자
- 트리사이클로[
13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (19s)
화합물 18 (700 mg, 1.1 mmol) 및 호베이다-구럽 1차 촉매(55 mg, 0.091 mmol)를 탈기된 무수 1,2-디클로로에탄 (1000 mL) 중에 용해시켰다. 혼합물을 아르곤 대기 하에 밤새 환류 온도로 가열하였다. 용매를 증발시키고, 컬럼 크로마토그래피(실리카 겔; 에테르)로 정제하여 240 mg (40 %) 의 순수 표제 화합물을 얻었다. MS (M+H)+ 613.
실시예
20

17-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
-13-
메틸
-2,14-
디옥소
-3,13-
디아자
- 트리사이클로[
13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(20)
화합물 19 (240 mg, 0.39 mmol)를 40 mL 용매 혼합물(THF 2: 메탄올 1: 메탄올 1) 중에 용해시켰다. 수산화 리튬 수용액 (1.9 mL, 1M)을 첨가하고, 반응 혼합물을 밤새 40℃로 가열하였다. HPLC와 컬럼 크로마토그래피(실리카 겔, 디클로로메탄 중 5 % 메탄올)로 정제하여 표제 화합물 (75 mg, 33 %)을 얻었다. MS (M+H)+ 585.
실시예
21

사이클로프로판설폰산
[17-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
- 2.14-디옥소-3.13-
디아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (21)
THF (7 mL) 중 화합물 20 (75 mg, 0.13 mmol) 및 N,N,-카보닐디이미다졸 (43 mg, 0.26 mmol)을 2 시간 동안 환류 하에 가열하였다. 임의로 형성된 아자락톤을 분리할 수 있다. 그 다음, DBU (29 μl), 및 WO03/053349에 개시된 바와 같이 제조 된 사이클로프로판설폰아미드 (47 mg, 0.39 mmol)를 첨가하고, 혼합물을 60℃에서 밤새 교반하였다. 반응 혼합물을 에틸 아세테이트 (25 mL)로 희석하고, 0.5 M 시트르산으로 세척하였다. HPLC로 정제하여, 30 mg의 순수 표제 화합물을 얻었다. MS (M+H)+ 688.
실시예
22

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
(22)
화합물 15 (850.0 mg, 2.30 mmol), PPh3 (1.60 g, 6 mmol), 및 티아졸 퀴나졸린 7 (820 mg, 2.72 mmol)을 얼음 배쓰에서 THF (30 mL) 중 용해시켰다. 30분 동안 교반시킨 다음, 혼합물을 실온에서 2시간 동안 교반하고, 진공 하에 농축시켰다. 플래쉬 컬럼 크로마토그래피 (실리카, EtOAc-헥산)를 수행하여 미츠노부 산물을 수득하였다. DCM (30 mL) 중 트리에틸실란 (460 mg, 4.00 mmol) 및 이러한 생성물(1.04 g, 1.60 mmol)의 용액에 TFA (30 mL)를 실온에서 점적 첨가하였다. 혼합물을 실온에서 2 시간 동안 교반시키고, 감압 하에 증발시키고, 톨루엔으로 두번 공 동증발시켰다. 플래쉬 컬럼 크로마토그래피 (실리카, 94/6 DCM-MeOH)로 백색 고체로서 표제 화합물(950 mg, 70%)을 얻었다.
실시예
23

1-({2-
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-[2-(4-이소프로필-티아졸-2일)-7-
메톡시
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카보닐
}-아미노)-2-비닐-
사이클로프로판카복실
산 에틸 에스테르 (23)
얼음 배쓰에서 35 mL DMF 중 카복실산 22 (1.6Ommol), N-메틸-5-헥스에닐아민 HCl 염 (360 mg, 2.40 mmol) 및 HATU (920 mg, 2.40 mmol) 용액에 DIEA (1.30 mL, 7.2 mmol)를 첨가하고 30분 동안 교반하였다. 혼합물을 실온에서 3 시간 동안 교반한 다음, 탄산 수소 나트륨 포화 수용액에 첨가하였다. 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 염수로 세척하고, 황산 나트륨으로 건조시키고, 감압하에 증발시켰다. 생성물을 실리카 겔 상 컬럼 크로마토그래피로 분리하고 헥산 에틸 아세테이트로 용리하였다(920 mg, 83%).
실시예
24

17-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2, 14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (24)
디엔 23 (900 mg)을 환류 중 900 mL DCE에 용해시켰다. 이러한 시스템을 성공적으로 비우고 아르곤으로 채웠다 (3x). 호베이다-구럽 2차 촉매 (90 mg)를 첨가하고, 이러한 시스템을 비우고, 아르곤으로 채우는 과정을 2회 실시하였다. 혼합물을 90℃에서 밤새 환류시키고, 농축하고, 플래쉬 컬럼 크로마토그래피(실리카, EtOAc-헥산)로 정제하여 회-갈색 고체로서 표제 화합물을 얻었다 (380 mg, 46%). MS (M+H)+ 662.
실시예
25

1-[(3-옥소-2-옥사-
비사이클로[2.2.1]헵탄
-5-
카보닐
)-아미노]-2-비닐-사이클로프로판
카복실산
에틸 에스테르 (25)
실온에서 DMF (14 mL) 및 DCM (25 mL) 중 13 (857 mg, 5.5 mmol)의 용액에 WO03/099274에 개시된 바와 같이 제조된 1-아미노-2-비닐-사이클로프로판카복실산 에틸 에스테르의 하이드로클로라이드 (1.15 g, 6.0 mmol), HATU (2.29 g, 6.0 mmol) 및 DIPEA (3.82 mL, 22 mmol)를 첨가하였다. 반응을 주위 온도에서 1시간 동안 N2-대기 하에 교반하였다. LC/MS 분석은 완전한 전환을 나타냈으며, 반응 혼합물을 진공 하에 농축시켰다. 잔류물을 DCM (100 mL) 및 0.1 M HCl (수용액) 중에 용해시키고, 상을 분리하였다. 유기상을 NaHCO3 (수용액) 및 염수로 세척하고, 건조 (MgSO4) 및 여과하였다. 진공 하에 용매의 제거로 표제 화합물 (1.6 g, 99%)을 수득하였다. LC/MS >95%, m/z (ESI+)= 294(MH+)
실시예
26

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-
하이드록시
-
사이클로
펜탄
카복실산
디이소프로필에틸아민
염 (26)
20 mL 마이크로파 반응 용기에서 물 (15 mL) 중 25 (800 mg, 2.73 mmol)의 용액에 DIPEA (1.2 mL, 6.8 mmol)와 교반 막대를 첨가하였다. 반응 용기를 밀봉하고, 비혼합성(immiscible) 슬러리를 마이크로파 공간에 삽입하기 전에 격렬하게 쉐이킹하였다. 전-교반을 1시간 한 다음, 반응물을 100℃의 세팅 온도로 40분 동안 조사하였다. 40 ℃로 냉각시킨 다음, 진공 하에 농축시키고, 잔류의 갈색 오일을 MeCN으로 3회 공동증발시켜 임의의 잔류 수분을 제거하였다. DIPEA 염의 형태의 조 표제 화합물을 즉시 다음 단계로 취하였다. LC/MS >95%, m/z (ESI+)= 312(MH+).
실시예
27

1-{[2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-
하이드록시
-
사이클로펜탄카보닐
]-아미노}-2-
이닐
-사이클로프로판
카복실산
에틸 에스테르 (27)
조 화합물 26 (5.5 mol)을 DCM (50 mL) 및 DMF (14 mL) 중 용해시키고, HATU (2.09 g, 5.5 mmol), N-메틸-N-헥스-5-에닐아민 (678 mg, 6.0 mmol) 및 DIPEA (3.08 mL, 17.5 mmol)를 실온에서 첨가하였다. 반응을 주위 온도에서 1시간 동안 교반하였다. LC/MS 분석은 시작 물질의 완전한 전환을 나타냈으며, 반응 혼합물을 진공 하에 농축하였다. 잔류물을 EtOAc (100 mL) 중에 용해하고, 유기상을 0.1 M HCl (수용액), K2CO3 (수용액) 및 염수로 세척시키고, 건조 (MgSO4) 및 여과하였다. 진공 하에 용매를 제거하여, 오일을 수득하였으며, 이를 플래쉬 크로마토그래피(실리카, EtOAc:MeOH)로 정제하여 표제 화합물 (1.65 g, 74%)을 얻었다. TLC(실리카): MeOH:EtOAc 5:95, Rf=0.5; LC/MS >95%, m/z (ESI+)= 407(MH+).
실시예
28

1-{[2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-(2-
페닐
-
퀴나졸린
-4-
일옥시
)-사이클로-펜
탄카보닐
l-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (28)
화합물 27 (0.15 g, 0.37 mmol)을 DMF 중 용해시키고, 용액을 0℃로 냉각하였다. NaH (미네랄 오일 중 60%, 0.04 g, 1.10 mmol)를 한번에 첨가하였다. 0.5 시간 후, 4-클로로-2-페닐퀴나졸린 (Aldrich에서 구입) (0.98 g, 0.41 mmol)를 첨가하고, 0℃에서 0.5 시간 동안 교반한 다음, 반응 혼합물을 실온으로 가온하였다. 실온에서 2 시간 동안 교반시킨 다음, 반응을 시트르산(5%, 수용액)으로 퀘엔칭하고, EtOAc (3 x 20 mL)로 추출하였다. 결합된 유기상을 시트르산(5%, 수용액, 2 x 20 mL), H2O ( 2 x 20 mL)으로 세척하였다. 그 다음, 유기상을 MgSO4로 건조, 여과 및 증발시켰다. DCM/MeOH와 함께 플래쉬 크로마토그래피로 정제하여, 166 mg의 생성물 (9)/가수분해 생성물 (48/52)을 얻었다. 이러한 혼합물을 다음 단계에서 이용하였다.
실시예
29

1-{[2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-(2-
페닐
-
퀴나졸린
-4-
일옥시
)-사이클로-펜
탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
(29)
화합물 28 (0.17 g, 0.27 mmol)을 DMF (2.5 mL) 중 용해시키고, 마이크로파 바이얼로 옮겼다. LiOH (수용액, 2 M, 8 mL)를 첨가하고, 반응을 1시간 동안 130℃에서 마이크로파로 가열하였다. HCl (수용액, 1M)로 반응을 pH 1로 퀘엔칭하고, DCM (3 x 20 mL)으로 추출하였다. 결합된 유기상을 HCl (수용액, 1M, 20 mL) 및 H2O (3 x 30 mL)로 세척하였다. 수상을 DCM (2 x 30 mL)로 백-추출(back-extract)하였다. 유기상을 MgSO4로 건조, 여과 및 증발시켰다. DCM/MeOH와 함께 플래쉬 크로마토그래피로 정제하여, 표제 화합물 (0.08 g, 49%)을 얻었다.
실시예
30

4-(2-
페닐
-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄
-1,2-
디카복실산
1-[(
1사이클로
-프로판설폰카보닐-2-
비닐사이클로프로필
)-아미드] 2-(
헥스
-5-
에닐메틸아미드
) (30)
화합물 29 (0.05 g, 80.70 μmol)를 DMF/DCM (1:3, 1200 μL) 중에 용해시키고, EDAC를 로딩한 바이얼에 옮겼다. 혼합물을 실온에서 10분 동안 인큐베이션하였다. DMAP를 첨가하고, 실온에서 20분동안 인큐베이션하였다. DCM/DMF (1:1, 800 μL) 중 DBU (49.1 mg, 0.32 mmol) 및 WO03/053349에 개시된 바와 같이 제조된 사이클로프로판 설폰아미드의 혼합물 (39.1 mg, 0.32 mmol)을 활성화된 화합물 10에 첨가하였다. 반응 혼합물을 100℃에서 30분 동안 가열하였다. 진공 하에 용매를 증발시킨 후, 잔류물을 DCM 중에 용해시켰다. 유기 상을 HCl (1M, 3 x 20 mL)로 세척하였다. 그 다음, 수상을 DCM (1 x 20 mL)로 백-추출하였다. 결합된 유기상을 HCl (1M, 수용액), 염수 및 물로 세척하였다. 유기상을 MgSO4로 건조하고, 증발시켰다. 진공 하에 건조시켜 표제 화합물 (50 mg, 90%)을 얻었다.
실시예
31

사이클로프로판설폰산[13-
메틸
-2,14-
디옥소
-17-(2-
페닐퀴나졸린
-4-일-
옥시
)3,13-디아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]아미드 (31)
무수 DCE (15 mL) 중 화합물 30 (0.02 g, 25.80 μmol)의 용액을 호베이다 구럽 2차 촉매 (83.1 mg, 5.0 μmol)로 로딩된 건조 마이크로파 바이얼에 첨가하였다. 용액을 질소 가스로 탈기시키고, 150℃에서 10분 동안 마이크로파로 가열하였다. 용매를 증발시킨 다음, 분취용-LC에서 정제를 수행하여 표제 화합물 (3.00 mg, 29%)을 얻었다.
실시예
32

1-{[4-(2-
클로로
-
퀴나졸린
-4-
일옥시
)-2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-사이클로-
펜탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (32)
DMF (1 mL) 중 화합물 27 (0.49 g, 1.21 mmol)을 용해시키고 마그네틱 교반 막대가 구비된 20 mL 마이크로파 반응 용기에 옮기고, LiOH (2 M, 10.5 mL) 수용액을 첨가하였다. 반응 용기를 밀봉하고, 비혼합성(immiscible) 슬러리를 마이크로파 공간에 삽입하기 전에 격렬하게 쉐이킹하였다. 반응물을 130℃로 30분 동안 조사하였다. 반응 혼합물을 40 ℃로 냉각시키고, 투명 용액을 HCl (1 M, 24 mL) 수용액으로 pH2로 산성화시키고, EtOAc (20 mL)로 3회 추출하였다. 풀링된 유기상을 염수로 세척하고, 건조 (MgSO4) 및 여과하였다. 용매를 진공 하에 제거하여, 산 (0.41 g, 90%)을 얻었다. 불순한 산 (410 mg, 1.09 mmol)을 DMF (1.5 mL) 및 DCM (4.5 mL) 중에 용해시킨 다음, EDAC (417 mg, 2.18 mmol)를 실온에서 첨가하였따. 혼합물을 실온에서 교반하면서 인큐베이션하였다. 10분 후, DMAP (133 mg, 1.09 mmol)를 첨가하고, 실온에서 20분 동안 추가로 인큐베이션하였다. 그 다음, DMF (2 mL) 및 DCM (2 mL) 중 DBU (663 mg, 4.36 mmol) 및 WO03/053349에 개시된 바와 같이 제조된 사이클로프로판설폰산 아미드(527 mg, 4.36 mmol)의 전-혼합된 용액을 첨가하고, 100℃에서 30분 동안 마이크로파로 가열하였다. 수득된 빨간색 용액을 진공 하에 농축시키고, EtOAc (20 mL)로 재용해하였다. 유기상을 1 M HCl (수용액) (3x 10 mL) 및 염수 (10 mL)로 세척하고, 건조 (MgSO4) 및 여과시켰다. 용매를 진공 하에 제거하고, 잔류물을 크로마토그래피(실리카, EtOAc:MeOH, 97.5:2.5) 로 정제하여, 설폰아미드 유도체 (0,40 g, 77%)를 얻었다; LC/MS >95%, m/z (ESI+)= 482(MH+).
설폰아미드 유도체 (0.33 g, 0.69 mmol) 를 DMF (9 mL)중에 용해시키고, 용 액을 0℃로 냉각시켰다. NaH (미네랄 오일 중 60%, 0.04 g, 1.10 mmol)를 한번에 첨가하였다. 0.5 시간 후, 2,4-디클로로-퀴나졸린 (0.15 g, 0.75 mmol)를 첨가하고, 0℃에서 1 시간 동안 교반한 다음, 반응을 실온으로 가온하였다. 시트르산(5%, 수용액)을 첨가하여 반응을 퀘엔칭하고, DCM (3 x 20 mL)으로 추출하였다. 결합된 유기상을 시트르산(5%, 수용액, 2 x 20 mL), H2O ( 2 x 20 mL)으로 세척하였다. 그 다음, 유기상을 MgSO4로 건조, 여과 및 증발시켜, 표제 화합물 (0.38 g, 79%)을 얻었다.
실시예
33

4-[2-(4-
메틸
-피페라진-1-일)-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄
-1,2-
디카복
실산 1-[(1-
사이클로프로판설포닐아미노카보닐
-2-비닐-
사이클로프로필
)아미드] 2-(헥스-5-
에닐
-
메틸
-아미드) (33)
화합물 32 (0.03 g, 46.6 μmol)를 1-메틸-피페라진 (0.5 mL)과 함께 마이크로파 바이얼에 로딩하였다. 혼합물을 마이크로파 시스템에서 120℃로 10분 동안 가열하였다. 시트르산(5%, 수용액)을 첨가하여 반응을 pH 5로 퀘엔칭하고, DCM (15 mL x 2)으로 추출하였다. 결합된 유기상을 시트르산(10 mL x 3)으로 세척하였다. 백-추출된 수상을 DCM (20 mL x 2)으로 세척하고, 결합된 유기상을 Na2SO4로 건조, 여과 및 증발시켜 표제 화합물 (27 mg, 82%)을 얻었다.
실시예
34

사이클로프로판설폰산
{13-
메틸
-17-[2-(4-
메틸
-피페라진-1일)
퀴나졸린
-4-
일옥시
]-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-
엔4
-
카보닐
}-아미드(34)
무수 DCE (20 mL) 중 화합물 33 (23.5 mg, 32.2 μmol)의 용액을 각각 호베이다 구럽 2차 촉매 (2.6 mg, 4.2 μmol)가 로딩된 두개의 마이크로파 바이얼에 첨가하였다. 용액을 질소로 탈기시키고, 마이크로파로 150℃에서 10 분 동안 가열하였다. 가열 후 두개의 뱃취(batch)를 합하고, 용매를 증발시켰다. 분취용-LC로 정제하여 표제 화합물 (5.00 mg, 22%)을 얻었다.
실시예
35

4-(2-모르폴린-4-일-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄
-1,2-
디카복실산1
-[(1-사이클로프로판설포닐아미노카보닐-2-비닐-
사이클로프로필
)-아미드]2-(
헥스
-5-
에닐
-메틸-아미드) (35)
화합물 32 (0.03 g, 46.6 μmol)를 모르폴린(0.5 mL)과 함께 마이크로파 바이얼에 로딩하였다. 혼합물을 마이크로파 시스템에서 120℃로 10분 동안 가열하였다. 반응을 pH 5로 퀘엔칭하기 위하여 시트르산 (5%, 수용액)을 첨가하고, DCM (15 mL x 2)으로 추출하였다. 결합된 유기상을 시트르산 (10 mL x 3)으로 세척하였다. DCM (20 mL x 2)으로 수상을 백-추출시키고, 결합된 유기상을 Na2SO4로 건조시키고, 여과 및 증발시켜, 표제 화합물 (17 mg, 52%)을 얻었다.
실시예
36

사이클로프로판설폰산
[13-
메틸
-17-(2-모르폴린-4-일-
퀴나졸린
-4-
일옥시
)-2, 14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (36)
무수 DCE (15 mL) 중 화합물 35 (17 mg, 24.5 μmol)의 용액을 호베이다 구럽 2차 촉매 (3.8 mg, 6.1 μmol)로 로딩된 무수 마이크로파 바이얼에 첨가하였다. 용액을 질소 기체로 탈기시키고, 150℃에서 10분 동안 가열하였다. 용매를 증발시키고, 분취용-LC로 정제하여 표제 화합물 (9.2 mg, 56%)을 얻었다.
실시예
37

1-{[2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-
하이드록시
-
사이클로펜탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
(37)
화합물 27 (493 mg, 1.21 mmol)을 DMF (1 mL) 중에 용해시키고, 마그네틱 교반 막대가 구비된 20 mL 마이크로파 반응 용기로 옮기고, LiOH (2 M, 10.5 mL) 수용액을 첨가하였다. 반응 용기를 밀봉하고, 비혼합성(immiscible) 슬러리를 마이크로파 공간에 삽입하기 전에 격렬하게 쉐이킹하였다. 반응물을 130℃로 30분 동안 조사하였다. 반응 혼합물을 40 ℃로 냉각시키고, 투명 용액을 HCl (1 M, 24 mL) 수용액으로 pH2로 산성화시키고, EtOAc (20 mL)로 3회 추출하였다. 풀링된 유기상을 염수로 세척하고, 건조 (MgSO4) 및 여과하였다. 용매를 진공 하에 제거하여, 표제 화합물 (410 mg, 90 %)을 얻었다. LC/MS >95%, m/z (ESI+)= 379(MH+).
실시예
38

4-
클로로
-2-(4-이소프로필-티아졸-2-일)-
퀴나졸린
(38)
화합물 9 (lOOmg, 0.37 mmol)을 포스포러스 옥시클로라이드 (2 mL)에 첨가하고, 100℃에서 2시간 동안 가열하였다. 그 다음, 반응 혼합물을 격렬하게 교반하면서 얼음에 붓고, NaOH (수용액)로 염기성이 되도록 하였다. 만들어진 슬러리를 에테르(3x20 mL)로 추출하고, 결합된 유기상을 건조 (MgSO4) 및 여과하였다 진공 하에 용매를 제거하여 수율로 표제 화합물을 얻었다. LC/MS >95%, m/z (ESI+)= 290(MH+).
실시예
39

4-
클로로
-2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸린
(39)
화합물 7 (300mg, 1 mmol)을 포스포러스 옥시클로라이드 (2 mL)에 첨가하고, 100℃에서 2시간 동안 가열하였다. 그 다음, 반응 혼합물을 격렬하게 교반하면서 얼음에 붓고, NaOH (수용액)로 염기성이 되도록 하였다. 만들어진 슬러리를 에테르(3x20 mL)로 추출하고, 결합된 유기상을 건조 (MgSO4) 및 여과하였다 진공 하에 용매를 제거하여 정량적 수율로 표제 화합물을 얻었다. LC/MS >95%, m/z (ESI+)= 320(MH+).
실시예
40

1-({2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-[2-(4-이소프로필-티아졸-2-일)-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카보닐
}-아미노)-2-비닐-
사이클로프로판카복실산
(40)
화합물 37 (26 mg, 70 μmol)을 THF (3 mL, 분자체로 건조) 중에 용해시켰다. 이러한 용액에 NaH (오일 중 60 %, 8.2 mg, 210 μmol)를 첨가하고, 반응을 주위 온도에서 10 분 동안 인큐베이션하였다. 그 다음, 반응 혼합물에 화합물 39 (17.6 mg, 61 μmol)를 첨가하고, 주위 온도에서 16 시간 동안 인큐베이션하였다. 그 다음, 반응에 0.1 M HCl (수용액) 및 EtOAc를 첨가하고, 상을 분리하고, 수상을 또다른 부분의 EtOAc로 추출하였다. 풀링한 유기상을 건조(MgSO4), 여과 및 진공 하에 농축시켜, 조 생성물을 수득하였으며, 플래쉬-크로마토그래피 (실리카; DCM:MeOH)로 더욱 정제하여 표제 화합물 (30 mg, 78%)을 얻었다. LC/MS >95%, m/z (ESI+)= 632(MH+).
실시예
41

1-({2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카보닐
}-아미노)-2-비닐-
사이클로프로판카복
실산 (41)
39 대신 퀴나졸린 유도체 38을 이용하여 실시예 40에 기술된 과정에 따라 표제 화합물을 얻었다. LC/MS >95%, m/z (ESI+)= 662(MH+).
실시예
42

4-[2-(4-이소프로필-티아졸-2-일)-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄
-1,2-
디
카복실산 1-[(1-
사이클로프로판설포닐아미노카보닐
-2-비닐-
사이클로프로필
)-아미 드]2-(
헥스
-5-
에닐
-
메틸
-아미드) (42)
화합물 40 (25 mg, 0.0395 mmol)을 DMF:DCM (1:4, 700 μL) 중에 용해시킨 다음, 25℃에서 EDAC (15.2 mg, 0.079 mmol)를 첨가하였다. 혼합물을 10분 동안 인큐베이션한 다음, DMAP (4.8 mg, 0.0395 mmol)를 첨가하고, 추가로 20 분 동안 인큐베이션하였다. DCM:DMF (1:1, 200 μL) 중 DBU (23.8 μL, 0.158 mmol) 및 WO03/053349에 개시된 바와 같이 제조된 사이클로-프로필설폰아미드(19.3 mg, 0.158 mmol)의 전-혼합된 용액을 첨가한 다음, 마이크로파에서 100℃로 30분 동안 가열하였다. 만들어진 빨간색 용액을 진공 하에 농축시켜, 조 생성물을 수득하고, 분취용 LCMS로 더욱 정제하여 화합물 MS-103-156 (19 mg, 65%)를 수득하였다, m/z (ESI+)= 735.28 (MH+).
실시예
43

4-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸린
-4-
일옥시
]-
사이클로펜
탄-1,2-
디카복실산
1-[(1-
사이클로프로판설포닐아미노카보닐
-2-비닐-
사이클로프로필
)-아미드] 2-(
헥스
-5-
에닐
-
메틸
-아미드) (43)
화합물 40 대신 화합물 41을 이용하여 실시예 42에 나타낸 과정에 따라 표제 화합물 (12.3 mg, 36%)을 수득하였다, m/z (ESI+)= 765.28 (MH+).
실시예
44
사이클로프로판설폰산
{17-[2-(4-이소프로필-티아졸-2-일)-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-카
보
닐}-아미드 (44)
화합물 42 (14.9 mg, 0.02 mmol)를 질소 하에서 무수 DCE (8 mL) 중에 용해시킨 다음, 무수 DCE (4mL) 중 용해된 호베이다-구럽 2차 촉매 (3.17 mg, 0.005 mmol)를 첨가하였다. 혼합물을 150℃에서 10 분 동안 마이크로파 가열시킨 다음, 진공 하에 농축시켜 조 생성물을 얻고, 분취용 LCMS로 정제하여 표제 화합물 (9 mg, 64%)을 얻었다, m/z (ESI+)= 707.27 (MH+).
실시예
45

사이클로프로판설폰산
{17-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-
퀴나졸
린-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (45)
화합물 42 대신에 화합물 43(12.3 mg, 0.016 mmol)을 이용하여 실시예 44에 나타낸 과정을 따라 표제 화합물 (4.7 mg, 40%)을 수득하였다, m/z (ESI+)= 737.11 (MH+).
실시예
46

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-
하이드록시
-
피롤리딘
- 1-카
복실
산
tert
-부틸 에스테르 (46)
Boc-보호 프롤린 (4 g, 17.3 mmol), HATU (6.9 g, 18.2 mmol) 및 WO03/099274에 개시된 바와 같이 제조된 1-아미노-2-비닐-사이클로프로판카복실산 에틸 에스테르 (3.5 g, 18.3 mmol)를 DMF (60 ml) 중에 용해시키고, 얼음 배쓰에서 0℃로 냉각시켰다. 디이소프로필에틸 아민 (DIPEA) (6ml)을 첨가하였다. 얼음 배쓰를 제거하고, 혼합물을 주위 온도에서 밤새 놔두었다. 그 다음, 디클로로메탄 (-80 ml)을 첨가하고, 유기상을 탄산 수소 나트륨 수용액, 시트르산, 물, 염수로 세척하고, 황산 나트륨으로 건조시켰다. 플래쉬 크로마토그래피(에테르 → 에테르 중 7% 메탄올)로 정제하여 순수 표제 화합물 (6.13 g, 96%)을 얻었다.
실시예
47

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-(4-니트로-
벤조일옥시
)-
피롤리딘
-1-
카복실산
tert
-부틸 에스테르 (47)
화합물 46 (6.13 g, 16.6 mmol), 4-니트로벤조산 (4.17 g, 25 mmol) 및 PPh3 (6.55 g, 25 mmol)를 THF (130 ml) 중에 용해시켰다. 용액을 ~0℃로 냉각시키고, 디이소프로필 아지도카복실레이트 (5.1 g, 25 mmol)를 천천히 첨가하였다. 그 다음 냉각을 그만 두고, 혼합물을 주위 환경에서 밤새 놔두었다. 탄산 수소 나트륨 수용 액 (60 ml)을 첨가하고, 반응물을 디클로로-메탄으로 추출하였다. 플래쉬 크로마토그래피(펜탄-에테르, 2:1 → 펜탄-에테르, 1:2 → 에테르 중 2 % 메탄올)로 정제하여 순수 표제 화합물 (6.2 g, 72%)을 얻었다.
실시예
48

4-니트로-벤조산 5-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-
피롤리
딘-3-일 에스테르 (48)
화합물 47 (6.2 g, 12 mmol)을 디클로로메탄 중 트리플루오로-메탄설폰산33 %의 얼음처럼 차가운 혼합물 중에 용해시켰다. 그 다음 얼음 배쓰를 제거하고, 혼합물을 실온에서 ~1.5 시간 동안 놔두었다. 용매를 증발시키고, 0.25M 탄산 나트륨을 첨가하고, 혼합물을 디클로로메탄으로 추출하였다. 증발시켜, 노란색을 띄는 분말로서 표제 화합물 (4.8g, 95 %)을 얻었다.
실시예
49

4-니트로-벤조산 5-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-1-(
헥스
-5-
에닐
-
메틸
-
카바모일
)
피롤리딘
-3-일 에스테르 (49)
화합물 48 (4.5 g, 10.8 mmol)을 THF (160 ml) 중 용해시켰다. 탄산 수소 나트륨 1 큰술과 그 후, 포스겐 (11.3 ml, 톨루엔 중 20%)을 첨가하였다. 혼합물을 1시간 동안 격렬하게 교반하였다. 혼합물을 여과하고, 디클로로메탄 (160 ml) 중에 용해하였다. 탄산 수소 나트륨(~1 큰술)을 첨가한 다음, 아민 하이드로클로라이드 (2.9 g, 21.6 mmol)를 첨가하였다. 반응물을 밤새 실온에 놔두었다. 플래쉬 크로마토그래피 (에테르 → 에테르 중 3% 메탄올)로 정제하여 순수 표제 화합물 (5.48 g, 91%)을 얻었다.
실시예
50

13-
메틸
-17-(4-니트로-
벤조일옥시
)-2.14-
디옥소
-3.13.15-
트리아자
-트리사이 클로[13.3.0.0*4.6*]-
옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (50)
화합물 49 (850 mg, 1.53 mmol)를 1.5 l 탈기된 무수 1,2-디클로로에탄 중에 용해시키고, 아르곤 대기 하에 밤새 환류시켰다. 스캐빈져(Scavenger (MP-TMT, P/N 800470, Argonaut technologies, ~1/2 티스푼)를 첨가하고, 혼합물을 2시간 동안 교반하고, 여과 및 감압하에 농축시켰다. 조 생성물을 디클로로메탄/n-헥산으로부터 결정화하여 표제 화합물 (600mg, 74 %)을 얻었다.
실시예
51

17-
하이드록시
-13-
메틸
-2.14-
디옥소
-3.13.15-
트리아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (51)
화합물 50 (200 mg, 0.38 mmol)을 메탄올/THF/ 물, 1:2:1, (20 ml)의 혼합물 중에 용해시키고, 얼음 배쓰에서 냉각시켰다. 수산화 리튬 (1.9 ml, 1M)을 천천히 첨가하였다. 혼합물을 0℃에서 4 시간 동안 교반한 다음, 아세트산 수용액 (20 ml)으로 중화시키고, 디클로로메탄으로 추출하였다. 유기상을 비카보네이트, 물, 염수로 세척하고, 황산 마그네슘으로 건조시켰다. 크로마토그래피(디클로로메탄 중 2 % 메탄올 → 4%)로 정제하여 회색을 띄는 분말로서 표제 화합물을 얻었다 (80%).
실시예
52

17-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
-13-
메틸
-2,14-
디옥소
-3,13,15-
트리
아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(52)
화합물 51 (220 mg, 0.58 mmol), 화합물 11 (220 mg, 0.87 mmol) 및 트리페닐-포스핀 (228 mg, 0.87 mmol)을 무수 THF (20 mL) 중에 현탁시키고, 0℃로 냉각시켰다. 디이소프로필 아지도카복실레이트 (176 mg, 0.87 mmol)를 점적 첨가하였다. 첨가 후 반응 혼합물이 실온에 도달하도록 하고, 밤새 놔두었다. 용매를 제거하고, 탄산 수소 나트륨 수용액을 첨가하고, 혼합물을 디클로로메탄으로 추출하였다. 유기상을 수집하고, 용매를 제거하였다. 수득된 조 생성물을 THF/메탄올/물 (2:1:1)의 10 mL 혼합물 중에 용해시켰다. 수산화 리튬 수용액 (1 mL, 1M)을 첨가하고, 혼합물을 밤새 50℃로 가열하였다. 그 다음, 물 (20 mL)을 첨가하고, 부피를 반으로 감소시켰다. 수산화 리튬 수용액 (1 mL, 1M)을 첨가하고, 수상을 디클로로메탄으로 수회 세척하였다. 그 다음, 수상을 시트르산으로 산성화시키고, 디클로로메탄으로 추출하였다. 용매를 증발시키고, HPLC로 정제하여 순수 표제 화합물 (79 mg, 23%)을 얻었다. M+H+ 586.
실시예
53

사이클로프로판설폰산
[17-(7-
메톡시
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
- 2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (53)
화합물 52 (79 mg, 0.13 mmol) 및 N,N-카보닐디이미다졸 (33 mg, 0.2 mmol)을 니트로 대기 하에 밀봉된 마이크로파 튜브에서 THF (5 mL) 중에 용해시켰다. 혼합물을 10분 동안 100℃로 가열시킨 다음, 냉각시켰다. THF (5 mL) 중 DBU (62 mg, 0.4 mmol) 및 사이클로프로판설폰아미드 (45 mg, 0.4 mmol)의 혼합물을 첨가하였다. 그 다음 100℃에서 60분 동안 가열을 계속하였다. 냉각시킨 다음, 용매를 제거하고, 잔류물을 에틸 아세테이트 중에 용해시켰다. 유기상을 0.5M 시트르산으로 세척하였다. HPLC로 정제하여 순수 표제 화합물 (29 mg, 32 %)을 얻었다. MS (M+H)+ 689.
실시예
54
헵트
-6-
에날
(54)
DCM (17 mL) 중 헵트-6-엔-1-올 (1 mL, 7.44 mmol) 및 N-메틸모르폴린 N-옥사이드 (1.308 g, 11.17 mmol) 용액에 그라운드 분자체(ground molecular sieves) (3.5 g, 4 A)를 첨가하였다. 혼합물을 질소 대기 하에 실온에서 10분 동안 교반하고, 테트라프로필암모늄 퍼루테네이트(TPAP) (131 mg, 0.37 mmol)를 첨가하였다. 추가로 2.5 시간 동안 교반한 다음, 용액을 셀라이트를 통하여 여과하였다. 그 다음, 용매를 조심스럽게 증발시키고, 남아있는 액체를 플래쉬 컬럼 크로마토그래피(DCM)로 정제하여, 오일로서 휘발성 표제 화합물 (620 mg, 74%)을 얻었다.
실시예
55

N'
-
헵트
-6-엔-(E)-
일이덴
-
하이드라진카복실산
tert
-부틸 에스테르 (55)
MeOH (5 mL) 중 54 (68 mg, 0.610 mmol) 및 tert-부틸 카바제이트 (81 mg, 0.613 mmol)의 용액에 그라운드 분자체 (115 mg, 3A)를 첨가하였다. 혼합물을 3 시간 동안 교반하고, 셀라이트를 통해 여과하고 증발시켰다. 잔류물을 무수 THF (3 mL) 및 AcOH (3mL) 중에 용해시켰다. NaBH3CN (95 mg, 1.51 mmol)을 첨가하고, 용액 을 밤새 교반하였다. 반응 혼합물을 NaHCO3 포화 수용액 (6 mL) 및 EtOAc (6 mL)로 희석하였다. 유기상을 염수, NaHCO3 포화 용액, 염수로 세척하고, MgSO4로 건조시키고, 증발시켰다. 시아노보란 첨가물을 MeOH (3 mL) 및 2 M NaOH (1.9 mL)로 처리하여 가수분해하였다. 혼합물을 2시간 동안 교반하고, MeOH을 증발시켰다. H2O (5 mL) 및 DCM (5 mL)을 첨가하고, 수상을 DCM으로 3회 추출하였다. 결합된 유기상을 건조 및 증발시켰다. 플래쉬 컬럼 크로마토그래피(1 % 트리에틸아민과 함께 톨루엔/에틸 아세테이트 9:1 및 1 % 트리에틸아민과 함께 톨루엔/에틸 아세테이트 6:1)로 정제하여 오일로서 표제 화합물 (85 mg, 61 %)을 얻었다.
실시예
56

N-(
tert
-부틸)-N-
이소프로필티오우레아
(56)
CH2Cl2 (200 mL) 중 tert-부틸이소티오시아네이트 (5.0 mL, 39 mmol)의 용액에 이소프로필아민 (4.0 mL, 47 mmol) 및 디이소프로필에틸아민 (DIEA) (6.8 mL, 39 mmol)을 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 반응 혼합물을 EtOAc로 희석시키고, 10 % 시트르산 (2x), NaHCO3 포화용액 (2x), H2O (2x), 및 염수 (1x)로 세척하였다. 유기상을 건조시키고 (MgSO4) 증발시켜, 추가의 정제 없이 이용되는 백색 고체로서 화합물 94 (3.3 g, 52 %)를 얻었다.
실시예
57

N-
이소프로필티오우레아
(57)
화합물 56 (3.3 g, 20 mmol)을 콘(cone)에서 용해시켰다. HCl (45 mL) 및 용액을 40 분 동안 환류시켰다. 혼합물이 실온이 되게 한 다음, 얼음 배쓰에서 냉각시키고, 고체 및 포화 NaHCO3를 이용해 pH 9.5로 염기성화시키고, 생성물을 EtOAc로 추출하였다 (3x). 결합된 유기상을 H2O (2x) 및 염수 (1x)로 세척하고, 건조 (MgSO4) 및 증발시켜 조 표제 화합물 (2.1 g, 90 %)을 수득하였으며, 이는 추가의 정제 없이 이용된다.
실시예
58

2-(
이소프로필아미노
)-1,3-티아졸-4-
카복실산
하이드로브로마이드
(58)
디옥산 (180 mL) 중 화합물 57 (2.1 g, 18 mmol) 및 3-브로모피루브산 (3.0 g, 18 mmol)의 현탁액을 80℃로 가열하였다. 80℃에 가열하면, 혼합물이 투명해지 고, 그 후 즉시 생성물이 백색 고체로 침전하기 시작한다. 2시간 동안 가열한 후, 반응 혼합물을 실온으로 냉각시키고, 침전물을 여과하고, 수집하였다. 순수 표제 화합물 (4.4 g, 94 %)을 수득하였다.
실시예
59

(2S,4R)-2-((1S,2R)1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-
하이드록시
-
피롤리딘
-1-
카복실산
tert
.부틸 에스테르 (59)
디클로로메탄 중 HATU (6 g), 디이소프로필에틸아민 (6.8 mL), (1R,2S)-1-아미노-2-비닐-사이클로프로판카복실산 에틸 에스테르 (1.5 g) 및 BOC-L-하이드록시 프롤린 (1.6 g)의 용액을 1시간 동안 교반하였다. 혼합물을 DCM-NaHCO3 (수용액)로 추출하고, 건조 및 농축시켰다. HPLC 순도 약 90% (M+H)+ 369.1.
실시예
60

(1S,2R)-1-[(2S,4R)-(4-
하이드록시
-
피롤리딘
-2-
카보닐
)-아미노]-2-비닐-사이클로프로판-
카복실산
에틸 에스테르 (60)
화합물 59를 디클로로메탄 및 1% MeOH 중 30% 트리플루오로아세트산에 2시간 동안 유지시키고, 건조될 때까지 농축하였다. 잔류물을 디클로로메탄 중에 재용해하고, 교반하면서 pH 10-11까지 1N NaOH를 첨가하였다. 유기층을 분리하고 농축하여 1.6 g의 표제 화합물을 얻었다. HPLC 순도 약 90% (M+H)+ 269.1.
실시예
61

(
Rac
)-4-
옥소사이클로펜트
-2-엔-1,2-
디카복실산
디메틸 에스테르 (61)
(1R,2S)-4-옥소-사이클로펜탄-1,2-디카복실산 디메틸 에스테르 (4.8 g, 23.8 mmol) 및 CuBr2 (11.9 g, 53.2 mmol)를 무수 THF (70 mL) 중에 용해시키고, 혼합물을 90℃에서 2 시간 동안 환류시켰다. 형성된 CuBr을 여과하고, 유기상을 농축시켰다. CaCO3 (2.7 g, 27.2 mmol) 및 DMF (70 mL)를 첨가하고, 혼합물을 100℃에서 한 시간 동안 놔두었다. 어두운 갈색 혼합물을 얼음(35 g)에 붓고 형성된 침전물을 여과하였다. 수층을 에틸 아세테이트(1 x 30OmL + 3 x 150 mL)로 추출하였다. 유기상을 건조, 여과 및 농축시켰다. 플래쉬 크로마토그래피 (톨루엔/EtOAc 9:1)로 정제하여 노란색 결정으로서 2 (2.1 g, 45 %)를 얻었다.
실시예
62

((1S,4R)&(1R,4S))-4-
하이드록시
-
사이클로펜트
-2-엔-1,2-
디카복실산
디메틸 에스테르 (62)
MeOH (23 mL) 중에 용해된 화합물 61 (3.18 g, 16.1 mmol)의 차가운 용액(-30℃)에 NaBH4 (0.66 g, 17.5 mmol)를 첨가하였다. 9분 후, 과량의 NaBH4를 염수(80 mL)를 첨가하여 파괴하였다. 혼합물을 농축시키고, 에틸 아세테이트 (4 x 80 mL)로 추출하였다. 유기상을 건조시키고, 여과 및 농축시켜 노란색 오일로서 표제 화합물 (3.0 g, 92 %)을 얻었다.
실시예
63

(1S,4R)&(1R,4S)-4-
하이드록시
-
사이클로펜트
-2-엔-1,2-
디카복실산
2-
메틸
에스테르 (63)
디옥산 및 물 (1:1, 110 mL) 중 용해된 62 (3.4 g, 22 mmol)의 얼음처럼 차가운 용액에 LiOH (0.52 g, 22 mmol)를 첨가하였다. 2.5 시간 후, 혼합물을 톨루엔 및 메탄올으로 공동-증발시켰다. 플래쉬 크로마토그래피(톨루엔/에틸 아세테이트 3:1 + 1 % HOAc)로 정제하여, 노란색-백색 결정으로서 표제 화합물 (1.0 g, 27 %)을 얻었다.

실시예
64

((3S,5R)&(3R,5S))-5-((1R,2S)-1-
tert
-
부톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-3-
하이드록시
-
사이클로펜트
-1-
엔카복실산
메틸
에스테르 (64)
59의 제조를 위해 개시된 방법에 따라 화합물 63 (50 mg, 37 mmol)을 (1R, 2S)-1-아미노-2-비닐-사이클로-프로판 카복실산 tert-부틸 에스테르와 반응시켜, 약간 노란색을 띄는 오일로서 표제 화합물을 얻었다.

실시예
65

3-옥소-2-옥사-
비사이클로[2.2.1]헵탄
-5-
카복실산
헥스
-5-
에닐
-
메틸아미드
(65)
아르곤 하, 얼음 배쓰에서 5 mL DMF 중 HATU (2.17 g, 5.7 mmol) 및 N-메틸 헥스-5-에닐아민 하이드로클로라이드 (6.47 mmol)에 11 mL DMF 중 1R,4R,5R-3-옥소-2-옥사-비사이클로[2.2.1]헵탄-5-카복실산 (835.6 mg, 5.35 mmol)을 첨가한 다음, DIEA (2.80 mL, 16 mmol)를 첨가하였다. 40분 동안 교반한 후, 혼합물을 실온에서 5 시간 동안 교반하였다. 용매를 증발시키고, 잔류물을 EtOAc (70 mL) 중에 용해시키고, NaHCO3 포화 용액 (10 mL)으로 세척하였다. 수상을 EtOAc (2 x 25 mL)로 추출하였다. 유기상을 결합시키고, NaCl 포화 용액 (20 mL)으로 세척하고, Na2SO4로 건조 및 증발시켰다. 플래쉬 컬럼 크로마토그래피 (150 g 실리카 겔, 2/1 EtOAc - 석유 에테르 (PE), KMnO4 수용액으로 검출, 4/1 EtOAc 중 Rf 0.55 - PE)로 정제하여 노란색 오일로서 표제 화합물을 얻었다 (1.01 g, 75%).
실시예
66

4-
하이드록시사이클로펜탄
-1,2-
디카복실산
1-[(1-
사이클로프로판설포닐아미노
-
카보닐
-2-
비닐사이클로프로필
)-아미드] 2-(
헥스
-5-
에닐
-
메틸아미드
(66)
얼음 배쓰 중에서 LiOH 용액 (0.15M, 53 mL, 8 mmol)을 락톤 아미드 65 (996 mg, 3.96 mmol)에 첨가하고, 1 시간 동안 교반하였다. 혼합물을 1N HCl을 이용하여 pH 2-3으로 산성화시키고, 증발시키고, 톨루엔으로 수회 공동-증발시킨 다음, 밤새 진공 하에 건조시켰다. (1R,2S)-사이클로프로판설폰산 (1-아미노-2-비닐-사이클로프로판-카보닐)아미드 하이드로클로라이드 (4.21 mmol) 및 HATU (1.78 g, 4.68 mmol)를 첨가하였다. 혼합물을 아르곤 하에 얼음 배쓰에서 냉각시키고, DMF (25 mL) 다음, DIEA (2.0 mL, 11.5 mmol)를 첨가하였다. 30분 동안 교반한 다음, 혼합물을 실온에서 3시간 동안 교반하였다. 용매를 증발시킨 후, 잔류물을 EtOAc (120 mL)에 용해시키고, 0.5 N HCl (20 mL) 및 NaCl 포화 용액 (2 x 20 mL)으로 성공적으로 세척하고, Na2SO4로 건조시켰다. 플래쉬 컬럼 크로마토그래피 (20Og YMC 실리카 겔, CH2Cl2 중 2-4% MeOH)로 백색 고체를 얻었다 (1.25 g, 66%).
실시예
67

사이클로프로판설폰산
(17-
하이드록시
-13-
메틸
-2,14-
디옥소
-3,13-
디아자트리사이클로
-[13.3.0.0*4.6*]
옥타데크
-7-엔-4-
카보닐
)-아미드 (67)
사이클로펜타놀 66 (52.0 mg, 0.108 mmol)을 19 mL 1,2-디클로로-에탄 중에 용해시켰다(이용 전 아르곤으로 버블링). 호베이다-구럽 2차 촉매 (6.62 mg, 10 mole %)를 DCE (2 x 0.5 mL) 중에 용해시키고 첨가하였다. 녹색 용액을 Ar로 1분 동안 버블링시켰다. 분주액 (각각 4mL)을 2 내지 5mL 마이크로파 튜브에 옮겼다. 마지막 튜브는 0.8 mL 용매로 헹구어 첨가하였다. 각각의 튜브를 마이크로파로 가열하였다(5분 동안 실온 내지 160℃). 모든 분주액을 합하고, 용매를 증발시켰다. 플래쉬 컬럼 크로마토그래피 (실리카 겔, CH2Cl2 중 3 → 7% MeOH)로 정제하여 24.39 mg 고체 (Rf 0.28, 두 점으로 10% MeOH - CH2Cl2 중)를 얻었다. 고체를 9.66-mg 샘플과 결합시키고, 2차 크로마토그래피(EtOAc 중 2 → 8% MeOH)를 하여 목적 화합물의 80%를 지닌 크림 고체 (23 mg)를 얻었다 (26% 수율).
실시예
68

N.N-디메틸-
티오우레아
(68)
디메틸아민 (THF 중 2M, 27.5 mL, 55 mmol)을 무수 THF (50 mL) 중 티오카보닐디이미다졸 (10 g, 56.1 mmol)의 교반 용액에 첨가하였다. 첨가로 인하여 반응 혼합물이 투명하게 되었고, 50℃에서 2 시간 동안 교반하였다. 반응 혼합물이 실온이 되도록 한 다음, 실리카 상에서 증발시키고, 플래쉬 크로마토그래피(MeOH: DCM 2:98)로 정제하였다. 용매를 회전 증발로 제거하고, 남아있는 산물을 고진공으로 건조시킨 다음, NH3로 포화된 MeOH (125 mL) 용액에 첨가하였다. TLC가 시작물질의 완전한 소비를 나타내고, LC-MS가 생성물 피크를 나타낼 때까지 반응 혼합물을 60 시간 동안 교반하였다. 회전 증발로 용매를 제거하면서 생성물을 침전시켰다. 남아있는 용매를 디에틸 에테르로 희석시키고, 흰색 결정을 여과 및 건조시켜 1.16 g (20%)를 얻었다. 남아있는 오일을 플래쉬 크로마토그래피(MeOH: DCM 5:95)로 정제하고, 또다른 1.87 g (32%)을 수득하였다.
실시예
69

2-디메틸아미노-티아졸-4-
카복실산
*
HBr
(69)
3-브로모피루브산 (2.94 g, 17.6 mmol)을 무수 THF (60 mL) 중 N,N-디- 메틸-티오우레아 (1.87 g, 17.6 mmol) 교반 용액에 첨가하였다. 반응 혼합물을 실온에서 4시간 동안 교반하였다. 형성된 침전물을 여과하고, 차가운 THF로 세척하고, 고 진공으로 건조시켰다. LC-MS는 생성물 피크를 나타냈다. 백색 고체로서 표제 화합물(2.64 g, 59%)을 수득하였다.
실시예
70

사이클로프로판설폰산
{13-
메틸
-2,14-
디옥소
-17-[2-(5H-
피라졸
-1-일)-
퀴나졸린
-4-
일옥시
]-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
-아미드 (70)
2-피라졸린을 이용하여 실시예 33의 과정과 유사하게 표제 화합물을 합성하였다.
실시예
71

사이클로프로판설폰산
[17-(2-
이소프로필아미노
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (71)
이소프로필아민을 이용하여 실시예 33의 과정과 유사하게 상기 화합물을 합성하였다.
실시예
72

사이클로프로판설폰산
[13-
메틸
-2,14-
디옥소
-17-[2-
피롤리딘
-1-일)-
퀴나졸린
-4-일옥시]-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (72)
피롤리딘을 이용하여 실시예 33의 과정과 유사하게 상기 화합물을 합성하였 다.
실시예
73

사이클로프로판설폰산
{17-[2-(4-
시아노
-
페닐
)-
퀴나졸린
-4-
일옥시
]-13-
메틸
- 2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (73)
4-시아노벤조산을 이용하여 실시예 6 및 7에서와 유사하게 상기 화합물을 합성하였다.
실시예
74

4-아미노-5-
시아노
-2-
하이드록시
-3-
메틸벤조산
에틸 에스테르 (74)
0℃에서 소듐 에톡사이드 용액(1.3 L) (에탄올 (1.3L)에 나트륨 금속 (7.9 g, 0.35 mol)의 첨가로 신선하게 제조)에 에틸프로피오닐 아세테이트 (25 g, 0.17 mol)를 첨가하고, 용액을 실온에서 1시간 동안 교반하였다. 실온에서 상기 용액에 에톡시메틸렌 말로니트릴(21 g, 0.17 mol)을 첨가하고, 반응 혼합물을 80℃에서 2 시간 동안 환류시켰다. 반응 혼합물을 냉각시키고, 1.5 N HCl의 첨가에 의하여 pH=7로 중화시키고, 진공 하에 농축하였다. 수득된 잔류물을 물 (100 mL)로 희석시키고, 여과하였다. 고체를 물로 세척하고, 50℃에서 진공 하에 건조시켜 조 생성물 (27 g)을 얻었다. 불순한 고체를 석유 에테르 중 5% 에틸 아세테이트로 세척하여 순수 표제 화합물 (22.5 g, 59%)을 얻었다.
실시예
75

4-아미노-5-
시아노
-2-
하이드록시
-3-
메틸벤조산
(75)
실온에서 에탄올/물 (1:1, 300 mL) 중 LiOHxH2O (8.4 g, 0.2 mol)의 용액에 화합물 74 (22 g, 0.1 mol)를 첨가하고, 반응 혼합물을 80℃에서 4 시간 동안 환류시켰다. 반응 혼합물을 진공 하에 농축시키고, 수득된 잔류물을 물(100 mL)로 희석하고, 석유 에테르/ 에틸 아세테이트 (1:1, 2x200 mL)로 세척하였다. 수층을 분리하고 1.5N HCl을 이용하여 pH=5로 산성화시키고, 수득된 고체 생성물을 여과하였다. 수층을 에틸 아세테이트 (2x300 mL)로 더욱 추출하고, 건조 및 농축시켜, 생성물을 더 얻었다. 합한 생성물을 석유 에테르 중 5% 에틸 아세테이트로 세척하여 순 수 표제 화합물을 얻었다 (19 g, >95%).
실시예
76

2-아미노-4-
하이드록시
-3-
메틸벤조니트릴
(76)
퀴놀린 (50 mL) 중 화합물 75 (19 g, 0.1 mol)의 혼합물을 2 시간 동안 170℃로 가열하였다(기포 생성이 멈출 때까지). 반응 혼합물을 실온으로 냉각시키고, NaOH 수용액(1M, 500 mL)을 첨가하고, 석유 에테르 (500mL)를 첨가하였다. 반응 혼합물을 15 분 동안 교반하고, 수층을 분리하였다. 수층을 석유 에테르 (2x300 mL)로 더 세척하여 퀴놀린을 완전하게 제거하였다. 1.5N HCl을 이용하여 수층을 pH=5로 산성화시키고, 고체를 여과하고 진공하에 건조시켰다. 수득된 고체를 석유 에테르 중 5% 에틸 아세테이트로 더욱 세척하여 순수 표제 화합물 (12 g, 82%)을 수득하였다.
실시예
77

2-아미노-4-
메톡시
-3-
메틸벤조니트릴
(77)
무수 DMF (200 mL) 중 화합물 76 (12 g, 0.08 mol), K2CO3 (11 g, 0.08 mol)의 혼합물을 실온에서 15 분 동안 교반하였다. 이에 MeI (13.6 g, 0.096 mol)를 첨가하고, 혼합물을 실온에서 4 시간 동안 교반하였다. 반응 혼합물을 물 (800 mL)로 희석시키고, 석유 에테르 중 30% 에틸 아세테이트(3x300 mL)로 추출하였다. 결합된 유기층을 물 및 염수로 세척하고, 건조 및 농축시켜, 조 생성물을 얻었다. 조 생성물을 석유 에테르로 세척하여 순수 표제 화합물을 얻었다 (12 g, 93%).
실시예
78

2-아미노-4-
메톡시
-3-
메틸
-
벤즈아미드
(78-아미드) 및 2-아미노-4-
메톡시
-3-메틸-벤조산 (78-산)
EtOH (150 ml) 중 2-아미노-4-메톡시-3-메틸-벤조니트릴 (9,4 g, 58 mmol) 및 2M 수산화 나트륨 용액 (150 ml)의 혼합물을 8 시간 동안 환류시켰다. 혼합물을 물로 희석시키고, 에틸 아세테이트-THF (9:1)의 혼합물로 3회 추출하였다. 유기상을 물로 세척하고, 황산 나트륨으로 건조시키고, 감압 하에 증발시켰다. 생성물을 디에틸 에테르로부터 결정화하여 표제 아미드를 수득하였다 (5.6 g, 58%).
결합된 수상을 시트르산으로 산성화시키고, 에틸 아세테이트로 3회 추출하 고, 황산 나트륨으로 건조시키고 감압하에 증발시켜 표제 산을 수득하였다 (3.2 g, 30%).
실시예
79

4-
이소프로필티아졸
-2-
카복실산
(6-
카바모일
-3-
메톡시
-2-
메틸
-
페닐
)-아미드 (79)
무수 DMF (80 ml) 중 Hobt-수화물 (2.2 g, 14 mmol), 2-아미노-4-메톡시-3-메틸-벤즈아미드 (2.0 g, 11 mmol) 및 4-이소프로필-티아졸-2-카복실산 (2.4 g, 14 mmol)의 교반 용액에 EDAC (2.88 g, 15 mmol) 및 TEA (2.1 ml, 15 mmol)를 첨가하고, 혼합물을 밤새 교반하였다. 5% 시트르산 수용액을 첨가하고 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 탄화 수소 나트륨 포화 용액(2회) 및 염수로 세척하고, 황산 나트륨으로 건조시키고 감압하에 증발시켜 표제 화합물을 얻었다 (3.1 g).
실시예
80

2-(4-
이소프로필티아졸
-2-일)-7-
메톡시
-8-
메틸퀴나졸린
-4-올(80)
상기 아미드 (79) (3.0 g, 9 mmol)를 EtOH (70 ml) 및 물 (70 ml) 중 탄산 나트륨 (2.4 g, 22.5 mmol)의 혼합물에서 3 시간 동안 환류시켰다. 혼합물을 5% 시트르산으로 산성화시키고, 에틸 아세테이트-THF (4:1)의 혼합물로 3회 추출하였다. 유기상을 황산 나트륨으로 건조시키고, 감압 하에 증발시켰다. 생성물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 3% MeOH을 함유하는 DCM으로 용리하였여 표제 화합물을 얻었다 (1.95 g).
실시예
81

2-아미노-4-
메톡시
-3-
메틸벤조산
메틸
에스테르 (81)
무수 DMF (40 ml) 중 2-아미노-4-메톡시-3-메틸 벤조산 (3.1 g, 17.1 mmol)의 용액에 탄산 칼륨(2.4 g, 17.1 mmol)을 첨가하고, 혼합물을 실온에서 30 분 동 안 교반하였다. 요오드화 메틸 (3.1 g, 22 mmol)을 첨가하고, 혼합물을 실온에서 3 시간 동안 교반하였다. 5% 시트르산 수용액을 첨가하고, 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 물로 세척하고, 황산 나트륨으로 건조하고 감압하에 증발시켰다. 생성물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리하여 표제 화합물을 얻었다 (2.75 g).
실시예
82

2-
벤조일아미노
-4-
메톡시
-3-
메틸벤조산
메틸
에스테르 (82)
무수 DCM (30 ml) 중 TEA (2 ml) 및 상기 에스테르 (81) (1.5 g, 7.68 mmol)의 얼음처럼 차가운 용액에 벤조일 클로라이드 (1.4 g, 10 mmol)를 첨가하고, 혼합물을 실온에서 4 시간 동안 교반하였다. 벤조일 클로라이드 (0.14 g, 1 mmol)를 첨가하고, 혼합물을 실온에서 1시간 동안 더 교반하였다. 5% 시트르산 수용액을 첨가하고 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 탄산 수소 나트륨 포화 수용액과 염수로 세척하고, 황산 나트륨으로 건조하고 감압하에 증발시켰다. 생성물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리하여 표제 화합물을 얻었다 (1.6 g).
실시예
83

7-
메톡시
-8-
메틸
-2-
페닐퀴나졸린
-4-올 (83)
EtOH (6 ml) 중 상기 산(82) (1.5 g, 5 mmol) 및 1m LiOH 용액 (6ml)의 혼합물을 60℃에서 2 시간 동안 교반하였다. 5% 시트르산 수용액을 첨가하고 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 황산 나트륨으로 건조하고 감압하에 증발시켰다. 잔류물을 포름아미드와 함께 150℃에서 5시간 동안 교반하였다. 포름아미드를 감압하에 증류시키고, 생성물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리하여 표제 화합물을 얻었다 (1.2 g).
실시예
84

2-(1-
에톡시카보닐
-2-
비닐사이클로프로필카바모일
)-4-(7-
메톡시
-8-
메틸
-2-
페
닐-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄카복실산
tert
-부틸 에스테르 (84)
실시예 16에 나타낸 바와 같이 퀴나졸리놀 유도체 (83) (480 mg, 1.8 mmol)를 화합물 15 (0.55 mg, 1.5 mmol)에 커플링시켜, 표제 화합물을 수득하였다 (700 mg, 75%) MS (M+H+) 616.
실시예
85

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-(7-
메톡시
-8-
메틸
-2- 페닐-
퀴나졸린
-4-
일옥시
)-
사이클로펜탄카복실산
(85)
화합물 84 (0.68 mg)를 실시예 17에서 기재된 바와 같이 처리하여 표제 화합물을 수득하였다 (620 mg, 100%).
실시예
86

1-{[2-
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4- 일옥시)-
사이클로펜탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (86)
N-메틸-1-헥스에닐아민 (192 mg, 1.7 mmol)을 실시예 18에 나타낸 바와 같이 화합물 85 (615 mg, 1.1 mmol)에 커플링시켜 표제 화합물을 수득하였다 (490 mg, 68%). MS (M+H+) 655.
실시예
87

17-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13- 디아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (87)
화합물 86 (480 mg, 0.73mmol)의 폐환 복분해 반응을 실시예 19에 나타낸 바와 같이 수행하여 표제 화합물을 수득하였다 (290 mg, 46%). MS (M+H+) 627.
실시예
88

17-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13- 디아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(88)
화합물 87 (280 mg, 0.45 mmol)의 에틸 에스테르를 실시예 20에 나타낸 바와 같이 가수 분해하여 표제 화합물을 수득하였다 (210 mg, 78%). MS (M+H+) 599.
실시예
89

사이클로프로판설폰산
[17-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13- 메틸-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (89)
사이클로프로판설폰아미드 (202 mg)를 실시예 21에 나타낸 바와 같이 산 88 (200 mg)에 커플링시켜 표제 화합물 (100 mg,42 %)을 수득하였다. MS (M+H+) 702.
실시예
90

2-(4-
플루오로
-
벤조일아미노
)-4-
메톡시
-3-
메틸
-벤조산
메틸
에스테르 (90)
4-플루오로벤조산 (700 mg, 5 mmol)을 디클로로메탄 (20 ml) 및 피리딘 (2 ml) 중에 용해시켰다. 2-아미노-4-메톡시-3-메틸-벤조산 메틸 에스테르 (81) (878 mg, 4.5 mmol)를 첨가하고, 혼합물을 5 시간 동안 환류시켰다. 물을 첨가하고, 혼합물을 디클로로메탄으로 추출하였다. 유기상을 건조, 여과 및 증발시키고, 수득된 잔류물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 에테르-펜탄 1:1로 용리하여 순수 표제 화합물을 수득하였다 (870 mg, 61 %). MS (M+H+) 318.
실시예
91

2-(4-
플루오로
-
벤조일아미노
)-4-
메톡시
-3-
메틸
-벤조산 (91)
LiOH (1M, 4 mL)를 테트라하이드로푸란 (15 ml), 물 (7.5 ml) 및 메탄올 (7.5 ml) 중 2-(4-플루오로-벤조일아미노)-4-메톡시-3- 메틸-벤조산 메틸 에스테르 (90) (870 mg, 2.7 mmol)에 첨가하였다. 혼합물을 50℃에서 4 시간 동안 가열하였다. 그 다음, 물(30 ml)을 첨가하고, 부피를 반으로 줄였다. 아세트산으로 산성화시킨 다음, 여과하여 순수 표제 화합물을 수득하였다 (830 mg, 100 %). MS (M+H+) 304.
실시예
92

2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-올 (92)
2-(4-플루오로-벤조일아미노)-4-메톡시-3-메틸-벤조산 (91) (830 mg, 2.7 mmol)을 포름아미드 (20 ml) 중 150℃에서 4시간 동안 가열하였다. 과량의 포름아미드를 증류시켜 제거하였다. 물을 첨가하고, 침전된 생성물을 여과하여 순수 표제 화합물을 수득하였다 (642 mg, 83 %). MS (M+H+) 285.
실시예
93

17-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-트리아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(93)
퀴나졸리놀 유도체 (83) (449 mg, 1.7 mmol)를 실시예 52에 나타낸 바와 같이 화합물 51 (400 mg, 1.1 mmol)에 커플링 시킨 다음, 에틸 에스테르를 가수분해시켜 표제 화합물을 수득하였다 (112 mg, 17%). MS (M+H+) 600.
실시예
94

사이클로프로판설폰산
[17-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13- 메틸-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-카보닐]-아미드 (94)
사이클로프로판설폰아미드 (115 mg, 0.95 mmol)를 실시예 53에 개시된 바와 같이 산 93 (112 mg, 0.19 mmol)에 커플링시켜, 표제 화합물을 수득하였다 (25 mg, 19%). MS (M+H+) 703.
실시예
95

17-[2-(4-이소프로필-티아졸-2-일)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-메틸-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-카복실산 (95)
퀴나졸리놀 유도체 (98) (141 mg, 0.5 mmol)를 실시예 52에 나타낸 바와 같이 화합물 51 (170 mg, 0.45 mmol)에 커플링시킨 후, 에틸 에스테르를 가수분해시켜, 표제 화합물을 수득하였다 (125 mg, 45%). MS (M+H+) 618.
실시예
96

사이클로프로판설폰산
{17-[2-(4-이소프로필-티아졸-2-일-(7-
메톡시
-8-
메틸
- 퀴나졸린-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-
트리아자
-트리사이클로[13.3.0.0*4,6*]-옥타데크-7-엔-4-
카보닐
}-아미드 (96)
사이클로프로판설폰아미드 (61 mg, 0.5 mmol)를 실시예 53에 나타낸 바와 같이 산 95 (125 mg, 0.2 mmol)에 커플링시켜 표제 화합물을 수득하였다 (52 mg, 36%). MS (M+H+) 721.
실시예
97

17-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복 실산
(97)
퀴나졸리놀 유도체 (92) (141 mg, 0,5 mmol)를 실시예 52에 나타낸 바와 같이 화합물 51 (170 mg, 0,45 mmol)에 커플링시켜, 표제 화합물의 조 에틸 에스테르를 얻었다. 조 에스테르를 실리카 겔 상 플래쉬 크로마토그래피로 정제하고, 디에틸 에테르 중 5 → 15% MeOH로 용리시켜 수득된 잔류물을 디클로로메탄 중에 용해시키고, 여과하여 남아있는 실리카를 제거하여, 표제 화합물의 에틸 에스테르를 수득하였다 (135 mg, 46%). 그 다음 에틸 에스테르를 실시예 52에 나타낸 바와 같이 가수분해하여, 표제 화합물을 수득하였다 (125 mg, 100%) MS (M+H)+ 618.3.
실시예
98

사이클로프로판설폰산
{17-[2-(4-
플루오로
-
페닐
-(7-
메톡시
-8-
메틸
-
퀴나졸린
- 4-일
옥
시)-13-
메틸
-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (98)
사이클로프로판설폰아미드 (61 mg, 0.5 mmol)를 실시예 53에 나타낸 바와 같이 산 97 (125 mg, 0.2 mmol)에 커플링시켜 표제 화합물을 수득하였다 (52 mg, 36%). MS (M+H+) 721.
실시예
99

4-니트로-벤조산 5-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-1-[
헵
트-6-
에닐
-(4-
메톡시
-벤질)-
카바모일
]-
피롤리딘
-3-일 에스테르 (99)
THF (160 mL) 중 화합물 48 (4.5 g, 10.8 mmol)의 용액에 톨루엔 중 포스겐 (1.93 M, 11.5 mL, 22 mmol) 및 NaHCO3 (1 큰술)를 첨가하였다. 혼합물을 실온에서 1 시간 동안 교반한 다음, 여과 및 증발시켰다. 잔류물을 CH2Cl2 (160 mL) 중에 용해시키고 NaHCO3 (1 큰술) 및 헵트-5-에닐-(p-메톡시벤질)-아민 (4.3 g, 18.5 mmol)를 첨가하였다. 실온에서 밤새 교반한 다음, 반응 혼합물을 여과하고 증발시켜 건조하였다. 실리카 겔 상 플래쉬 컬럼 크로마토그래피 (EtOAc: 톨루엔 25:75 →40:60)로 밝은 갈색 시럽으로서 표제 화합물을 수득하였다 (6.59 g, 90%).
실시예
100

18-
하이드록시
-14-[4-
메톡시
-벤질)-2.15-
디옥소
-3.14.16-
트리아자
-트리사이클로[14.3.0.0*4.6]-
노나데크
-7-엔-4-
카복실산
에틸 에스테르 (100)
화합물 99 (1g, 1.48 mmol)를 1,2-디클로로에탄(2 l) 중에 용해시켰다. 아르곤 스팀을 이용하여 혼합물을 15 분 동안 탈기시켰다. 호베이다-구럽 촉매 (II) (50 mg, 5 mol%)를 첨가하고, 혼합물을 4 시간 동안 환류시켰다. 용매를 증발시키고, 조 에스테르를 테트라하이드로푸란 (100 ml), 메탄올 (50 ml) 및 물 (50 ml) 중에 용해시켰다. 혼합물을 얼음 배쓰 상 0℃로 냉각시켰다. 수산화 리튬 수용액 (20 ml, 1M)을 첨가하고, 혼합물을 0℃에서 교반하였다. 그 다음, 물로 부피를 두배로 증가시키고, 혼합물을 아세트산으로 산성화시켰다. 추출 (디클로로메탄) 후, 플래쉬 크로마토그래피 (에테르 중 메탄올 1 → 5 %)로 순수 표제 화합물을 수득하였다 (450 mg, 61 %). MS (M+H)+ 500.
실시예
101

18-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-14-(4-메톡시-벤질)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-카
복실
산 에틸 에스테르 (101)
퀴나졸리놀 유도체 (92) (125 mg, 0.44 mmol)를 실시예 52에 개시된 바와 같이 화합물 100 (200 mg, 0.4 mmol)에 커플링시켰다. 수득된 조 생성물을 실리카 겔 상 플래쉬 크로마토그래피로 정제하고, 디에틸 에테르 중 1% MeOH로 용리하여 표제 화합물을 수득하였다 (240 mg, 78%). MS (M+H)+ 766.3.
실시예
102

18-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-14-(4-
메톡시
-벤질)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔- 4-카
복실
산 (102)
화합물 101 (240 mg, 0.31 mmol)의 에틸 에스테르를 실시예 20에 나타낸 바와 같이 가수분해하여, 표제 화합물을 수득하였다 (200 mg, 86%). MS (M+H+) 738.
실시예
103

사이클로프로판설폰산
[18-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-일
옥
시]-14-(4-
메톡시
-벤질)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.
0.0*4,6*]노나데크-7-엔-4-
카보닐
]-아미드 (103)
사이클로프로판설폰아미드 (99 mg, 0.8 mmol)를 실시예 21에 나타낸 바와 같이 산 102 (200 mg, 0.27 mmol)에 커플링시켰다. HPLC로 정제하여, 표제 화합물을 수득하였다 (75 mg, 33 %). MS (M-H)- 839.
실시예
104

사이클로프로판설폰산
{18-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-일
옥
시]-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카보닐
}-아미드 (104)
디클로로-메탄-트리플루오로아세트산; 2:1의 혼합물 중 화합물 103 (75 mg, 0.09 mmol)을 2 시간 동안 교반하였다. 증발시키고, HPLC로 정제하여 순수 표제 화합물을 얻었다 (25 mg, 38%). MS (M-H)- 719.0.
실시예
105
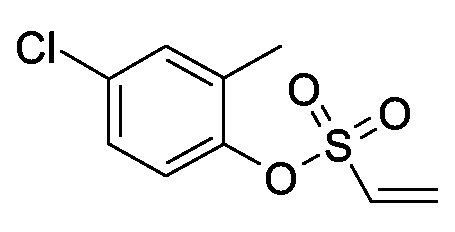
에텐설폰산
4-
클로로
-2-
메틸페닐
에스테르 (105)
아세톤 (10 ml) 중 4-클로로-2-메틸페놀 (24.7g, 173mmol)의 교반 혼합물에 디클로로에탄 (25 ml) 및 물 (45 ml)을 0-5℃에서 점적 첨가하는 동시에, 2-클로로-1-에탄 설포닐 클로라이드 (28.2 g, 173 mmol) 및 25% 수산화 나트륨 용액 (60 g)을 약 1 시간 동안 첨가하였다. 혼합물을 5°에서 1시간 동안 교반하고, 실온에서 1 시간 동안 교반하였다. 물을 첨가하고, 혼합물을 DCM으로 2회 추출하였다. 유기상을 황산 나트륨으로 건조시키고, 여과 및 감압 하에 증발시켰다. 잔류물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리하여, 표제 화합물을 수득하였다 (33.4 g, 83%).
실시예
106

에텐설폰산
4-
클로로
-2-
메틸
-5-
니트로페닐
에스테르 (106)
차가운 농축 설푸릭산 (70 ml) 중에 화합물 105 (33.2 g, 142 mmol)를 용해시키고, 98% 니트릭산 (9.8 g)을 점적 첨가하면서 온도를 10℃ 미만으로 유지하도록 냉각시켰다. 혼합물을 약 5℃에서 한 시간 동안 교반하였다. 혼합물을 얼음물에 첨가하고, 에틸 아세테이트로 3회 추출하였다. 유기산을 염수로 2회 세척하고, 황산 나트륨으로 건조시키고, 감압 하에 증발시켰다. 생성물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리시켰다. 수율 : 30 g = 75%
실시예
107

4-
클로로
-2-
메틸
-5-니트로페놀 (107)
에탄올 물 1/1 (600 ml) 중 탄산 칼륨 (27,6 g, 200 mmol) 및 화합물 106 (27.8g, 100 mmol)의 용액을 1시간 동안 환류시켰다. 시트르산 (5%)을 첨가하고, 혼합물을 DCM으로 3회 추출하였다. 유기상을 황산 나트륨으로 건조시키고, 감압 하에 증발시켜, 표제 화합물을 수득하였다 (19 g, 100%).
실시예
108

1-
클로로
-4-
메톡시
-5-
메틸
-2-니트로벤젠 (108)
DMF (200 ml) 중 4-클로로-2-메틸-5-니트로페놀 (18.8 g, 100 mmol)의 교반 용액에 탄산 칼륨 (13.8 g, 100 mmol)과 요오드화 메틸 (21.3 g, 150 mmol)을 첨가하였다. 혼합물을 실온에서 약 2 시간 동안 교반하였다. 5% 시트르산을 첨가하고, 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 염수로 세척하고, 황산 나트륨으로 건조시키고, 감압 하에 증발시켜 표제 화합물을 수득하였다 (20 G, 100%).
실시예
109

4-
메톡시
-5-
메틸
-2-니트로-
벤조니트릴
(109)
n-메틸-피롤리돈-2 (60 ml) 중 구리 시아나이드 (11.25 g, 125 mmol) 및 화합물 108 (20 g, 100 mmol) 혼합물을 140-150℃에서 20시간 동안 교반하였다. 혼합물을 에틸 아세테이트로 희석하고, 여과시키고, 물로 4회 세척하였다. 유기상을 황산 나트륨으로 건조시키고 감압하에 증발시켰다. 잔류물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리하여 표제 화합물을 수득하였다 (8 g, 41%).
실시예
110

4-
메톡시
-5-
메틸
-2-
니트로벤즈아미드
(110)
4-메톡시-5-메틸-2-니트로-벤조니트릴 (8 g, 40 mmol) 및 물 (50 ml)의 혼합물에 농축된 설푸릭 산(65 ml)을 첨가하고, 혼합물을 100-110℃에서 2.5 시간 동안 교반하였따. 혼합물을 밤새 놔두고, 여과하고, 물로 세척한 다음, 건조시켜 표제 화합물을 수득하였다 (7 g, 83%).
실시예
111

4-
메톡시
-5-
메틸
-2-아미노-
벤즈아미드
(111)
실온, 50 psi에서 EtOH (200 ml) 및 raney-ni (5.0 g) 중 화합물 110 (7.0 g, 33.3 mmol)을 밤새 수소화시켰다. 촉매를 여과하고, 디옥산 및 에탄올로 세척하였다. 진공 하에 용매를 제거하고, 생성물을 실리카 겔 상 컬럼 크로마토그래피로 분리하고, 디클로로메탄 및 3% 메탄올로 용리시켰다. 수율: 3.4 g = 56%.
실시예
112

7-
메톡시
-6-
메틸
-2-
페닐
-
퀴나졸린
-4-올 (112)
무수 DMF (60 ml) 중 화합물 111 (1.8 g, 10 mmol), 벤조산 (1.46 g, 12 mmol) 및 hobt-수화물 (1.87 g, 12 mmol)의 혼합물에 EDAC (2.4 g, 12.5 mmol) 및 TEA (1.75 ml, 12.5 mmol)를 첨가하고, 혼합물을 실온에서 60 시간 동안 교반하였다. 5% 시트르산을 첨가하고, 혼합물을 에틸 아세테이트로 3회 증발시켰다. 유기상을 염수 및 탄산 수소 나트륨 포화 용액으로 세척하였다. 유기상을 황산 나트륨으로 건조시키고, 감압하에 증발시켰다. 잔류물을 100 ml 에탄올-물 1/1 중 탄산 나 트륨 (2.65 g, 25 mmol)과 함께 2 시간 동안 환류시켰다. 5% 시트르산을 첨가하고, 혼합물을 10% THF를 함유하는 에틸 아세테이트로 3회 증발시켰다. 실리카 겔을 첨가하고, 용매를 증발시키고, 생성물을 실리카 겔 상의 컬럼 크로마토그래피로 정제하고, 헥산-에틸 아세테이트로 용리시켰다. 수율 : 1.3 g = 50%
실시예
113

17-(7-
메톡시
-6-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-트리아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(113)
퀴나졸리놀 유도체 (112) (480 mg, 1.8 mmol)를 실시예 16에 나타낸 바와 같이 화합물 15 (550 mg, 1.5 mmol)에 커플링시키고, 실시예 17에 나타낸 바와 같이 boc기를 제거하고, 실시예 18에 나타낸 바와 같이 N-메틸-1-헥스에닐아민을 커플링시키고, 실시예 19에 나타낸 바와 같이 폐환 복분해 반응을 수행하고, 실시예 52에 나타낸 바와 같이 에틸 에스테르를 가수분해시켜 표제 화합물을 수득하였다 (290 mg, 30%). MS (M+H)+ 599.
실시예
114

사이클로프로판설폰산
[17-(7-
메톡시
-6-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)- 13-메틸-2.14-
디옥소
-3.13.15-
트리아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
-7-엔-4-카보닐]-아미드 (114)
사이클로프로판설폰아미드 (202 mg, 1.67 mmol)를 실시예 89에 나타낸 바와 같이 산 113 (200 mg, 0.33 mmol)에 커플링시켜 표제 화합물을 수득하였다 (90 mg, 38%). MS (M+H)+ 702.
실시예
115

4-
메톡시
-3-
메틸
-2-[(5-
메틸
-피리딘-2-
카보닐
)-아미노]-벤조산
메틸
에스테르 (115)
2-아미노-4-메톡시-3-메틸-벤조산 메틸 에스테르 (400 mg, 2 mmol) 및 5-메 틸-피리딘-2-카복실산 (280 mmol, 2 mmol)을 디클로로-메탄 (8 ml) 및 피리딘 (1 ml) 중에 용해시켰다. 포스포러스 옥시클로라이드 (0.37 ml)를 첨가하면서 얼음 배쓰에 냉각시켰다. 혼합물을 0℃에서 1 시간 동안 놔둔 다음, 실온이 되도록 하였다. 수산화 나트륨 수용액 (20 ml, 1M)을 첨가하고, 혼합물을 디클로로메탄으로 추출하였다. 실리카 겔 상 컬럼 크로마토그래피(에테르-펜탄 1:1)로 정제하여 순수 표제 화합물을 수득하였다 (410 mg, 65 %). MS (M-H)+ 315.1
실시예
116

4-
메톡시
-3-
메틸
-2-[(5-
메틸
-피리딘-2-
카보닐
)-아미노]-벤조산 (116)
화합물 115 (620 mg, 1.9 mmol)을 실시예 91에 나타낸 과정에 의하여 가수분해하여 순수 표제 화합물을 수득하였다 (590 mg, 100 %). MS (M-H)+ 301.1.
실시예
117

7-
메톡시
-8-
메틸
-2-(6-
메틸
-피리딘-2-일)-
퀴나졸린
-4-올 (117)
화합물 116을 포름아미드 중에 150℃에서 5-6 시간 동안 가열하였다. 그 다음, 물을 첨가하고, 침전된 생성물을 여과하여 순수 표제 화합물을 수득하였다 (397 mg, 71 %). MS (M-H)+ 282.1
실시예
118

17-[7-
메톡시
-8-
메틸
-2-(6-
메틸
-피리딘-2-일)-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(118)
퀴나졸리놀 유도체 (117) (198 mg, 0.7 mmol)를 실시예 52에 나타낸 바와 같이 화합물 51 (268 mg, 0.7 mmol)에 커플링시킨 다음, 에틸 에스테르를 가수분해하여 표제 화합물을 수득하였다 (50 mg, 10%). MS (M+H)+ 615.3.
실시예
119

사이클로프로판설폰산
{17-[7-
메톡시
-8-
메틸
-2-(6-
메틸
-피리딘-2-일)-
퀴나졸
린-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13,15-트리아자트리사이클로[13.3.0.0*4,6*]옥타데크-7-엔-4-
카보닐
}-아미드 (119)
화합물 118 (50 mg, 0.08 mmol)을 실시예 53에 나타낸 과정에 따라 사이클로프로판설폰산 아미드 (44 mg, 0.36 mmol)와 반응시켜, 표제 화합물을 수득하였다 (13 mg, 22 %). MS (M-H)+ 718.2
실시예
120

17-(7-
메톡시
-6-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-트리아자-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(120)
퀴나졸리놀 유도체 112 (200 mg, 0.53 mmol)를 실시예 52에 나타낸 바와 같이 화합물 51 (268 mg, 0.7 mmol)에 커플링시킨 다음, 에틸 에스테르를 가수분해하여 표제 화합물을 수득하였다 (36 mg, 11%). MS (M-H)+ 600.
실시예
121

사이클로프로판설폰산
[17-(7-
메톡시
-6-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-13-메틸-2.14-
디옥소
-3.13.15-
트리아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
-7-엔-4-카보닐]-아미드 (121)
산 120 (36 mg, 0.06 mmol)을 실시예 53에 나타낸 과정에 따라 사이클로프로판설폰산 아미드와 반응시켜 표제 화합물을 수득하였다 (8 mg, 19 %). MS (M-H)+ 703.
치환된
퀴나졸린
-4-올의 제조를 위한 일반적 방법

무수 THF (60 ml) 중 치환된 2-아미노-벤즈아미드 [A] (1 당량)의 현탁액에 피리딘(2 당량)을 첨가하고, 혼합물을 5℃로 냉각시켰다. 산 클로라이드 [B] (1.25 당량)을 천천히 첨가하고, 혼합물을 실온에서 밤새 교반하였다. 혼합물을 감압하에 증발시킨 다음, 물 중에 현탁시켰다. 화합물을 수 시간 동안 물 중에 놔두고, 여과시키고, 차가운 물과 디에틸 에테르로 세척하였다. 생성물 [C]을 진공 하에 건조시켰다. 수율: 90-100%. 산 클로라이드 [B]가 이용될 때, 니코티닐 클로라이드 하이드로클로라이드 후, 2.5 당량의 피리딘을 이용하였으며, 혼합물을 실온에서 밤새 대신 2-3 일 동안 교반하였다.
형성된 아미드 [C] (1 당량)를 물 및 EtOH 의 1:1 혼합물 중 탄산 나트륨 (2.5 당량)의 현탁액에 첨가하고, 혼합물을 두 시간 동안 환류시켰다. EtOH를 감압 하에 제거하고, 5% 시트르산 용액을 첨가하고, 혼합물을 밤새 놔두었다. 생성물 [D]를 여과로 분리한 다음, 물과 디에틸 에테르로 세척하고 진공 하에 건조시켰다.
실시예
122

7-
메톡시
-8-
메틸
-2-피리딘-3일-
퀴나졸린
-4-올 (122)
벤즈아미드 유도체로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드로서 니코티닐 클로라이드 하이드로클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (2.5g, 92%), [M+H]=268.
실시예
123

7-
메톡시
-8-
메틸
-2-피리딘-4일-
퀴나졸린
-4-올 (123)
벤즈아미드 유도체로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드로서 이소니코티노일 클로라이드 하이드로클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (1.6 g, 60%), [M+H]=268.
실시예
124

7-
메톡시
-8-
메틸
-2-에틸-
퀴나졸린
-4-올 (124)
벤즈아미드 유도체 [A] 로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드 [B] 로서 아세트산 클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (2.2 g, 100%).
실시예
125

7-
메톡시
-8-
메틸
-2-(4-
메톡시페닐
)-
퀴나졸린
-4-올 (125)
벤즈아미드 유도체 [A] 로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드 [B] 로서 4-메톡시벤조산 클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (5.5 g, 92%).
실시예
126

8-
메톡시
-2-
페닐
-
퀴나졸린
-4-올 (126)
벤즈아미드 유도체 [A] 로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드 [B] 로서 벤조일 클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (2.0 g, 80%), [M+H]=253.
실시예
127

2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-올 (127)
벤즈아미드 유도체 [A] 로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드 [B] 로서 3-플루오로-벤조일 클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (2.1 g, 73%), [M+H]=271.
실시예
128

2-(3,5-
디플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-올 (128)
벤즈아미드 유도체 [A] 로서 2-아미노-4-메톡시-3-메틸 벤즈아미드와 산 클로라이드 [B] 로서 3,5-디플루오로-벤조일 클로라이드를 이용하여 상기 나타낸 일반적 과정을 수행하여 표제 화합물을 수득하였다 (2.1 g, 85%), [M+H]=3O3.
실시예
129

7-
메톡시
-8-
메틸
-
퀴나졸린
-4-올 (129)
폐환 반응, 일반적 방법의 단계 [B] 내지 [C]가 EtOH 중에서 보다 DMF 중에서 수행될 때 표제 화합물이 형성된다.
실시예
130

17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
130)
퀴나졸리놀 유도체 (125) (281 mg, 0.949 mmol)를 실시예 52에 나타낸 바와 같이 알콜 51 (300 mg, 0.791 mmol)에 커플링시킨 다음, 에틸 에스테르를 가수분해하여 표제 화합물을 수득하였다 (185 mg, 47%). MS (M+H)=630
실시예
131

사이클로프로판설폰산
{17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-일옥시]-13-
메틸
-2.14-
디옥소
-3.13.15-
트리아자
-트리사이클로[13.3.0.0*4.6*]-
옥타데크
-7-엔-4-
카보닐
}-아미드 (131)
산 (130) (70 mg, 0.111 mmol)을 DCM (2ml) 중에 용해시켰다. EDAC (26 mg, 0.133 mmol)를 첨가하고, 혼합물을 실온에서 밤새 교반하였다. 사이클로프로판설폰산 아미드 (15 mg, 0.122 mmol) 및 DBU (35μl, 0.233 mmol)를 첨가하고, 반응 혼합물을 밤새 실온에서 교반하였다. 5% 시트르산을 반응 혼합물에 첨가하고, 혼합물을 염수로 추출하고, Na2SO4로 건조시키고, 컬럼 크로마토그래피 (DCM/MeOH 20:1)로 정제하여 표제 화합물을 수득하였다 (29 mg, 36%). MS(M+H)=733
실시예
132

1-
메틸
-
사이클로프로판설폰산
{17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나
졸린-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4.6
*]옥
타데
크-7-엔-4-
카보닐
]-아미드 (132)
산 (130) (35 mg, 0.056mmol)을 DCM (2ml) 중에 용해시켰다. EDAC (13 mg, 0.067 mmol)를 첨가하고, 혼합물을 실온에서 밤새 교반하였다. 메틸사이클로프로판설폰산 아미드 (8.2 mg, 0.061 mmol) 및 DBU (18 μl, 0.117 mmol)를 첨가하고, 반응 혼합물을 밤새 실온에서 교반하였다. 5% 시트르산을 반응 혼합물에 첨가하고, 혼합물을 염수로 추출하고, Na2SO4로 건조시키고, HPLC로 정제하여 표제 화합물을 수득하였다 (9 mg, 22%). MS (M+H)= 747.
실시예
133

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[7-
메톡시
-2-(4-
메톡
시-
페닐
)-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
tert
-부틸 에스테르 (133)
알콜 15 (550 mg, 1.5 mmol), 퀴나졸리놀 125 (533 mg, 1.8 mmol) 및 트리페닐 포스핀 (990 mg, 3.75 mmol)을 THF (40 ml) 중에 용해시키고, 0℃로 냉각시켰 다. 디이소프로필 아지도카복실레이트(0.74 ml, 3.75 mmol)를 천천히 첨가하고, 슬러리가 실온이 되도록 하였다. 12 시간 후, 용매를 감압 하에 제거하고, 잔류물을 에테르 중에 취하고, 여과하였다. 컬럼 크로마토그래피로 정제하여 표제 화합물을 수득하였다 (919 mg, 95 %). MS (M+H)+ 646.
실시예
134

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
(134)
화합물 133 (915 mg, 1.417 mmol)을 디클로로메탄 (20 mL) 및 트리에틸 실란 (0.56 mL) 중에 용해시켰다. TFA (20 ml)를 실온에서 점적 첨가하고, 혼합물을 실온에서 3 시간 동안 놔두었다. 용매를 제거하여 표제 화합물을 수득하였다 (737 mg, 88%) MS (M+H)+ 590.
실시예
135

1-({2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
- 퀴나졸린-4-
일옥시
]-
사이클로펜탄카보닐
}-아미노)-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (135)
산 134 (723 mg, 1.227 mmol)를 DMF (25 mL) 중에 용해시켰다. 디이소프로필 에틸아민 (633 mg, 4.91 mmol)을 첨가하고, 반응 혼합물을 얼음 배쓰에 놔두었다. N-메틸-1-헥센 하이드로클로라이드 (266 mg, 1.78 mmol) 및 HATU (676 mg, 1.78 mmol)를 첨가하고, 혼합물을 실온에서 1 시간 동안 교반하였다. 용매를 제거하고, 잔류물을 EtOAc와 소듐 비카보네이트로 분배하였다. 유기상을 수집하고, 조 생성물을 컬럼 크로마토그래피(실리카 겔, 헵탄/EtOAc 80:20→50:50)로 정제하였다. 용매를 증발시켜 표제 화합물을 수득하였다 (585 mg, 70 %). MS (M+H)+ 685.
실시예
136

17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (136)
디엔 135 (585 mg, 0.854 mmol) 및 호베이다-구럽 II차 촉매(50 mg)를 탈기 및 무수 1,2-디클로로에탄 (500 mL) 중에 용해하였다. 혼합물을 아르곤 대기 하에 밤새 환류 온도로 가열하였다. 용매를 증발시키고, 컬럼 크로마토그래피(실리카 ㄱ겔; 헵탄/EtOAc 70:30)로 정제하여, 표제 화합물을 수득하였다 (420 mg, 75 %). MS (M+H)+ 658.
실시예
137

17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(137)
화합물 136 (420 mg, 0.639 mmol)을 96 mL 용매 혼합물 (THF 2: 메탄올 1: 물 1) 중에 용해하였다. 수산화 리튬 수용액 (6.4 mL, 1M)을 첨가하고, 반응 혼합물을 50℃로 밤새 가열하였다. 컬럼 크로마토그래피 (실리카 겔, 디클로로메탄 중 5 % 메탄올)로 정제하여, 표제 화합물을 수득하였다 (230 mg, 57 %). MS (M+H)+ 629.
실시예
138

사이클로프로판설폰산
{17-[7-
메톡시
-2-(4-
메톡시
-
페닐
)-8-
메틸
-
퀴나졸린
-4-일옥시]-13-
메틸
-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (138)
THF (7 mL) 중 산 137 (130 mg, 0.207 mmol) 및 N,N,-카보닐디이미다졸 (43 mg, 0.26 mmol)을 환류 하에 2 시간 동안 가열하였다. 그 다음, DBU (29 μl) 및 WO03/053349에 개시된 바와 같이 제조된 사이클로프로판설폰아미드(28 mg, 0.23 mmol)를 첨가하고, 혼합물을 밤새 60℃에서 교반하였다. 반응 혼합물을 에틸 아세테이트 (25 mL)로 세척하고, 0.5M 시트르산으로 세척하였다. HPLC로 정제하여 30g의 표제 화합물을 수득하였다. MS (M+H)+ 732.
실시예
139

2,4-
디클로로
-7-
메톡시
-8-
메틸퀴나졸린
(139)
트리클로로메틸 클로로포르메이트 (3.60 mL, 29.8 mmol)를 질소 하에서 아세토니트릴 (0.809 g, 19.7 mmol) 중 6-시아노-3-메톡시-2-메틸아닐린 (77) (3.2 g, 19.7 mmol) 용액에 첨가하였다. 수득된 반응 혼합물을 밀봉된 튜브에서 130℃로 가열하였다. 12 시간 후, 반응 혼합물을 실온으로 성공적으로 냉각시키고, 얼음처럼 차가운 물과 EtOAc로 분배하고, 건조 (Na2SO4)시키고, 증발시켰다. 컬럼 크로마토그래피 (EtOAc/CH2Cl2 구배, 1:9 내지 1:1)로 정제하여 오렌지색 고체로서 표제 화합물을 수득하였다 (3.17 g, 85%): m/z = 243 (M+H)+.
실시예
140

2-
클로로
-4-
하이드록시
-7-
메톡시
-8-
메틸퀴나졸린
(140)
물 (40 mL) 중 NaOH (1.58 g, 39.6 mmol) 용액을 THF (20 mL) 중 2,4-디클로로-7-메톡시-8-메틸퀴나졸린 (139) (3.2 g, 13.05 mmol)에 첨가하였다. 만들어진 혼합물을 40℃에서 24시간 동안 가열하였다. 그 다음, 반응 혼합물을 실온으로 냉각시키고, THF를 증발시키고, 추가적으로 물 (30 mL)을 첨가하였다. 침전물을 여과시켰다. 그 다음, 여액의 pH를 AcOH를 이용하여 5로 조정하여, 고체를 수득하였고, 이는 그 다음 여과하고, 물과 이소프로필에테르로 성공적으로 세척하여, 노란색을 띄는 분말로서 표제 화합물을 수득하였다 (2.91 g, 99%): m/z = 225 (M+H)+.
실시예
141

4-
하이드록시
-2-(3-
이소프로필피라졸
-1-일)-7-
메톡시
-8-
메틸퀴나졸린
(141)
2-클로로-4-하이드록시-7-메톡시-8-메틸퀴나졸린 (140) (502 mg, 2.23 mmol) 및 3-이소프로필피라졸 (500 mg, 4.55 mmol)의 혼합물을 155℃에서 10 분 동안 가 열하였다. 그 다음, 반응 혼합물을 실온으로 성공적으로 냉각시키고, CH2Cl2 와 물로 분배하고, 건조 (Na2SO4)시키고, 증발시켰다. 잔류물을 에테르 중에서 분쇄하고, 여과하여 백색 침상으로서 표제 화합물을 수득하였다 (422 mg, 63%): m/z = 299 (M+H)+.
실시예
142

2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-
하이드록시
-
사이클로펜탄카복실산
(142)
LiOH (물 4mL 중 105 mg)를 0℃에서 락톤 아미드 (65)에 첨가하였다. 1 시간 후, 전환이 완료되었다 (HPLC). HCl을 이용하여 혼합물을 pH 2 - 3으로 산성화시키고, EtOAc로 추출하고, 건조(MgSO4), 증발시키고, 톨루엔으로 수회 공동-증발시키고, 밤새 고진공하에서 건조시켜 표제 화합물을 수득하였다 (520 mg, 88%), m/z = 270 (M+H)+.
실시예
143

1-{[2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-
하이드록시
-
사이클로펜탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (143)
1-(아미노)-2-(비닐)사이클로프로판카복실산 에틸 에스테르 하이드로클로라이드 (4.92 g, 31.7 mmol) 및 HATU (12.6 g, 33.2 mmol)를 산 (142) (8.14 g, 30.2 mmol)에 첨가하였다. 혼합물을 아르곤 하에서 얼음 배쓰 중 냉각시킨 다음, DMF (100 mL) 및 DIPEA (12.5 mL, 11.5 mmol)를 첨가하였다. 0℃에서 30분 후, 용액을 실온에서 추가의 3시간 동안 교반하였다. 그 다음, 반응 혼합물을 EtOAc와 물로 분배하고, 0.5 N HCl (20 mL) 및 NaCl 포화 용액 (2 x 20 mL)로 성공적으로 세척하고 건조 (Na2SO4)시켰다. 플래쉬 크로마토그래피 (EtOAc/CH2Cl2/석유 에테르, 1:1:1)로 정제하여 무색의 오일로서 표제 화합물을 수득하였다 (7.41, g 60%), m/z = 407 (M+H)+.
실시예
144

1-({2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-4-[2-(3-이소프로필-
피라졸
-1-일)-7-
메
톡시-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카보닐
}-아미노)-2-비닐-사이클로프로판-
카복실산
에틸 에스테르 (144)
DIAD (280 μL, 1.42 mmol)를 -20℃, 질소 대기 하에서 무수 DMF (35 mL) 중 알콜 (143) (367 mg, 0.90 mmol), 4-하이드록시-2-(3-이소프로필피라졸-1-일)-7-메톡시-8-메틸퀴나졸린 (141) (270 mg, 0.90 mmol) 및 트리페닐-포스핀 (288 mg, 1.42 mmol)용액에 첨가하였다. 2 시간 후, 용액을 실온으로 가온시켰다. 12 시간 후, 반응 혼합물을 얼음처럼 차가운 물과 에테르로 분배하고, 유기층을 건조 (Na2SO4) 및 증발시켰다. 잔류물을 컬럼 크로마토그래피(AcOEt/CH2Cl2 구배, 1:9 내지 10:0)로 정제하여 표제 화합물을 수득하였다 (230 mg, 34 %), m/z = 687 (M+H)+.
실시예
145

17-[2-(3-이소프로필-
피라졸
-1-일)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-메틸-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복
실산 에틸 에스테르 (145)
무수 및 탈기 1,2-디클로로에탄 (230 mL) 중 호베이다-구럽 1차 촉매 (60.8 mg, 0.101 mmol) 및 디엔 용액(144) (230 mg, 0.335 mmol)을 질소 하에서 80℃로 18 시간 동안 가열하였다. 그 다음, 용매를 증발시키고, 잔류물을 실리카 겔 크로마토그래피(에테르)로 정제하여 표적 화합물을 수득하였다, m/z = 659 (M+H)+.
실시예
146

17-[2-(3-
이소프로필피라졸
-1-일)-7-
메톡시
-8-
메틸퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-
디아자트리사이클로[13.3.0.0
4,6
]옥타데크
-7-엔-4-
카복실산
(146)
물 (10 mL) 중 수산화 리튬 수화물 용액(796 mg, 18.6 mmol)을 THF (30 mL) 중 에스테르 (145) (346 mg, 0.526 mmol)의 교반 용액에 첨가하였다. 실온에서 5일 후, 반응 혼합물을 진공 하에 농축하였다. 1N HCl을 이용해 pH를 4로 조정하고, 수득된 용액을 AcOEt로 성공적으로 추출하고, 염수로 세척하고, 건조 (Na2SO4) 및 증발시켰다. 잔류물을 컬럼 크로마토그래피 (CH2Cl2/MeOH, 97.5:2.5)로 정제한 다음, 이소프로필에테르 중에서 분쇄하여 고체로서 표제 화합물을 얻었다, m/z = 631 (M+H)+.
실시예
147

N-[17-[2-(3- 이소프로필피라졸 -1-일)-7- 메톡시 -8- 메틸퀴나졸린 -4- 일옥시 ]- 13-메틸-2,14- 디옥소 -3,13- 디아자트리사이클로[13.3.0.0 4,6 ]옥타데크 -7-엔-4- 카보닐](사이클로- 프로필설폰아미드 (147)
무수 THF (10 mL) 중 카보닐디이미다졸 (29.4 mg, 0.181 mmol) 및 산 (146) (53 mg, 0.084 mmol)의 용액을 질소 하에 2 시간 동안 환류 교반시켰다. 반응 혼합물을 실온으로 냉각시키고, 사이클로프로필설폰아미드 (50.3 mg, 0.415 mmol) 및 DBU (34.1 mg, 0.224 mmol)를 첨가하였다. 이러한 용액을 50℃에서 15 시간 동안 가열하였다. 그 다음, 반응 혼합물을 실온으로 냉각시키고, 감압하에 농축시켰다. 잔류물을 AcOEt와 희석 HCl로 분배하고, 유기층을 염수로 세척하고, 건조 (Na2SO4) 및 증발시켰다. 플래쉬 크로마토그래피(EtOAc/CH2Cl2, 2:8)로 정제하여, 목적 산물을 수득하고, 그 다음 최소의 에탄올에 용해하고, 물로 희석하였다. 침전물을 여과시켜 백색 분말로서 표제 화합물을 수득하였다 (14.8 mg, 24%), m/z = 734 (M+H)+.

실시예 148

2-
에톡시
-4-
하이드록시
-7-
메톡시
-8-
메틸퀴나졸린
(148)
퀴나졸리놀 (140) (530 mg, 2.36 mmol)을 새롭게 제조된 EtONa (20 mL EtOH 중 첨가된 740 mg의 Na)에 조금씩 첨가하였다. 만들어진 용액을 환류 하에 가열하고, 24 시간 후, 반응 혼합물을 실온으로 냉각시키고, 증발시켰다. 잔류물을 물 (10 mL) 중에 재용해시키고, AcOH를 이용하여 만들어진 용액의 pH를 5로 조정하였다. 침전물을 여과하여 수집하고, 얼음처럼 차가운 물로 세척하고 건조시켜 백색 고체로서 표제 화합물 (534 mg, 96.6%)을 얻었다, m/z = 235 (M+H)+.
실시예
149

17-[2-
에톡시
-7-
메톡시
-8-
메틸퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-디
아자트리사이클로[13.3.0.0
4,6
]옥타
데크-7-엔-4-
카복실산
(149)
퀴나졸리놀 (148) 및 알콜 (143)의 반응을 실시예 144-146에 나타낸 과정을 따라 수행하여 표제 화합물을 수득하였다 m/z = 567 (M+H)+.
실시예
150

N-[17-[2-
에톡시
-7-
메톡시
-8-
메틸퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-디
아자트리사이
클로 [13.3.
0
.04,6]
옥타데크
-7-엔-4-
카보닐
](
사이클로프로필설
폰아미드 (150)
산 (149)을 실시예 147에 기재된 과정을 따라 사이클로프로필설폰아미드와 반응시켜, 표제 화합물을 수득하였다, m/z = 670 (M+H)+.
1H NMR (CDCl3):
실시예
151

17-(7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-
트리 아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(126)
퀴나졸리놀 유도체 (126) (460 mg, 2.4 mmol)를 실시예 16에 나타낸 바와 같이 알콜 15 (740 mg, 2 mmol)에 커플링시킨 다음, 실시예 17에 나타낸 바와 같이 boc 기를 제거하고, 실시예 18에 나타낸 바와 같이 N-메틸-1-헥스에닐아민과 커플링시키고, 실시예 19에 나타낸 바와 같이 폐환 복분해 반응을 수행하고, 실시예 52에 나타낸 바와 같이 에틸 에스테르를 가수분해하여 표제 화합물을 수득하였다 (82 mg, 8%), MS (M+H) 523.
실시예
152

사이클로프로판설폰산
[17-(7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
]-아미드 (152)
무수 DCM (2 ml) 중 EDC (40 mg, 0.21 mmol) 및 산 (151) (81 mg, 0.155 mmol)의 용액을 밤새 실온에서 교반하였다. 사이클로프로필설폰아미드 (48mg, 0,4mmol) 및 DBU (76 mg, 0.5 mmol)를 첨가하고, 혼합물을 실온에서 6 시간 동안 교반하였다. 5% 시트르산을 첨가하고, 혼합물을 에틸 아세테이트로 3회 추출하였 다. 유기상을 5% 시트르산과 염수로 세척하고, 황산 나트륨으로 건조시키고, 감압 하에 증발시켰다. 잔류물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 에테르-메탄올로 용리하여, 표제 화합물을 수득하였다 (32 mg, 31%), MS (M+H) 626.
실시예
153

17-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(153)
퀴나졸리놀 유도체 (92) (520 mg, 1.8 mmol)를 실시예 16에 나타낸 바와 같이 알콜 15 (550 mg, 1.5 mmol)에 커플링시킨 다음, 실시예 17에 나타낸 바와 같이 boc 기를 제거하고, 실시예 18에 나타낸 바와 같이 N-메틸-1-헥스에닐아민과 커플링시키고, 실시예 19에 나타낸 바와 같이 폐환 복분해 반응을 수행하고, 실시예 52에 나타낸 바와 같이 에틸 에스테르를 가수분해하여 표제 화합물을 수득하였다 (185 mg, 20%), MS (M+H) 617.
실시예
154

사이클로프로판설폰산
{17-[2-(4-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
- 4-일
옥
시]-13-
메틸
-2.14-
디옥소
-3.13.15-
트리아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (154)
무수 DCM (2 ml) 중 EDC (38 mg, 0.2 mmol) 및 산 (153) (92mg, 0.15 mmol)의 용액을 실온에서 밤새 교반하였다. 사이클로프로필설폰아미드(48 mg, 0.4 mmol) 및 DBU (76 mg, 0.5 mmol)를 첨가하고, 혼합물을 실온으로 6 시간 동안 교반하였다. 5% 시트르산을 첨가하고, 혼합물을 에틸 아세테이트로 3회 추출하였다. 유기상을 5% 시트르산과 염수로 세척하고, 황산 나트륨으로 건조시키고, 감압 하에 증발시켰다. 잔류물을 실리카 겔 상 컬럼 크로마토그래피로 정제하고, 에테르-메탄올로 용리시켜 표제 화합물을 수득하였다 (70 mg, 65%), MS (M+H) 720.
실시예
155

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[2-(3-
플루오로
-
페닐
)-7-메톡시-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
tert
-부틸 에스테르 (155)
PPh3 (787 mg, 3.0 mmol)를 무수 THF (40 mL) 및 무수 DMF 10 mL의 혼합물 중 퀴나졸리놀 127 (430 mg, 1.5 mmol) 및 알콜 15 (550 mg, 1.5 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 실온에서 불활성 대기 (N2)하에 두고, DIAD (591 μL, 3.0 mmol)를 첨가하였다. 반응 혼합물을 18 시간 동안 교반한 다음, 용매를 증발시켰다. 잔류물을 CHCl3 중 용해시키고, 분리 펀넬(funnel)에서 염수로 세척하였다. 유기상을 Na2SO4로 건조시키고, 실리카 상에서 증발시키고, 플래쉬 크로마토그래피(헵탄:에틸 아세테이트 2:1 내지 1:1)로 정제하여 백-베이지색 고체로서 표제 화합물을 수득하였다 (896 mg, 94%), LRMS (M+H) 634.
실시예
156

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[2-(3-
플루오로
-
페닐
)-7-메톡시-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
(156)
화합물 155 (0.896 g, 1.41 mmol)를 DCM (30 mL), TFA (10 mL), 몇 방울의 TES 및 한 방울의 H2O의 혼합물 중에 용해하였다. 반응을 30 분 동안 교반한 다음, 증발로 용매를 제거하였다. 조 잔류물을 CHCl3와 NaHCO3 (포화 수용액)으로 분배하였다. 유기상을 건조시키고 (Na2SO4), 증발시켜 백색 고체로서 화합물을 수득하였다 (0.81 g, 99%). LRMS (M+H) 578.
실시예
157

1-{[4-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-2-(
헥스
- 5-에닐-
메틸
-
카바모일
)-
사이클로펜탄카보닐
]-아미노}-2-비닐-
사이클로프로판카복실산
에틸 에스테르 (157)
화합물 156 (0.81 g, 1.40 mmol) 및 헥스-5-에닐-메틸-아민 하이드로클로라이드 (272 mg, 1.82 mmol)를 무수 DMF (50 mL) 중에 용해하였다. DIEA (975 μL, 5,6 mmol)를 첨가하고, 반응 플라스크를 얼음 배쓰에 두었다. 10 분 후, HATU (559 mg, 1.47 mmol)를 용액에 첨가하였다. 반응 플라스크가 실온이 되도록 하고, 3시간 동안 교반을 계속한 다음, 용매를 증발로 제거하였다. 조 생성물을 CHCl3로 추출하고 NaHCO3 (포화 수용액)으로 세척하였다. 유기상을 건조시키고 (Na2SO4), 실리카 상에서 증발시키고 플래쉬 크로마토그래피(헵탄:에틸 아세테이트 1:1)로 정제하여 표제 화합물을 수득하였다 (0.716 g, 76% ), LRMS (M+H) 673.
실시예
158

17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르 (158)
디엔 157 ( 0.70 g, 1.041 mmol)을 무수 DCE (0.7 L) 중에 용해시켰다. 용액을 불활성 대기 (N2) 하로 가져오고, 촉매(호베이다 구럽 2차 촉매, 70 mg, 0.113 mmol)를 용액에 첨가하였다. 반응 혼합물을 16 시간 동안 환류시키고, 실온으로 냉각시키고, 실리카 상에서 회전 증발로 증발시켰다. 생성물을 플래쉬 크로마토그래피(헵탄:에틸 아세테이트 1:1)로 정제하여 표제 화합물을 수득하였다 (0.466 g, 70%), LRMS (M+H) 645.
실시예
159

17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(159)
에틸 에스테르 158 (460 mg, 0.713 mmol)를 THF: MeOH: H2O (2:1:1, 100 mL) 중에 용해시키고, LiOH (1M) (7.13 mL mg, 7.13 mmol)을 용액에 첨가하였다. 반응을 50℃로 16 시간 동안 가열하였다. 그 다음, THF와 MeOH를 회전 증발로 제거하고, 남아있는 용액을 20 mL 10% 시트르산 (수용액)으로 산성화시켰다.
수상을 CHCl3 (3x50 mL)로 추출하고, 유기상을 염수로 세척하였다. 유기상을 Na2SO4로 건조시키고, 여과 및 회전 증발에 의하여 농축시켰다. 생성물을 백색 고체로서 수득하였다 (0.363 g, 82%), LRMS (M+H) 617.
실시예
160

사이클로프로판설폰산
{17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-일
옥
시]-13-
메틸
-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (160)
무수 THF (12 mL) 중 산 159 (200 mg, 0.324 mmol) 및 CDI (105 mg, 0.649 mmol)의 혼합물을 N2 하에서 환류 하에 2 시간 동안 가열하였다. 반응 혼합물을 50℃로 냉각시키고, 2 ml의 무수 THF 중 사이클로프로필 설폰아미드 (118 mg, 0.973 mmol) 및 DBU (138 μL, 0.908 mmol)의 전-혼합된 용액을 반응 혼합물에 첨가하였다. 반응을 50℃에서 18 시간 동안 교반하였다. 용액을 분리 펀넬에 붓고, 약 20 mL 시트르산 10% (수용액)으로 산성화시켰다. 추가의 염수 (20 mL) 및 EtOAc (40 mL)를 첨가하였다. 혼합물을 EtOAc로 추출하고, 염수로 세척한 다음, Na2SO4로 건조 시키고, 여과하였으며, 용매를 회전 증발로 제거하였다. 조 생성물을 H2O (0.1% TFA) 중 35 에서 60% 아세토니트릴 (0.1 % TFA)으로의 구배를 지닌 Ace-5 C8 컬럼(100x21.2 mm)상에서 HPLC로 8 분에 걸쳐 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (144 mg, 62%), LRMS (M+H) 720.

실시예
161

1-
메틸
-
사이클로프로판설폰산
{17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
- 퀴나졸린-4-
일옥시
l-13-
메틸
-2,14-
디옥소
-3,13-디아자트리사이클로[13.3.0.0*4,6*]옥타데크-7-엔-4-
카보닐
}-아미드 (161)
무수 THF (7mL) 중 산 159 (100 mg, 0.162 mmol) 및 CDI (53 mg, 0.325 mmol)의 혼합물을 N2 하에서 환류 하에 2 시간 동안 가열하였다. 반응 혼합물을 50℃로 냉각시키고, 1 ml의 무수 THF 중 메틸-사이클로프로필 설폰아미드(66 mg, 0.486 mmol) 및 DBU (69 μL, 0.454 mmol)의 전-혼합된 용액을 반응 혼합물에 첨가 하였다. 반응을 50℃에서 18 시간 동안 교반하였다. 용매를 증발시키고, 잔류물을 CHCl3 중에 용해시키고, 시트르산 (10% 수용액)으로 세척하였다. 유기상을 Na2SO4로 건조시키고, 여과하였으며, 용매를 회전 증발로 제거하였다. 조 생성물을 H2O (0.1% TFA) 중 35 에서 60% 아세토니트릴 (0.1 % TFA)으로의 구배를 지닌 Ace-5 C8 컬럼(100x21.2 mm)상에서 HPLC로 8 분에 걸쳐 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (23 mg, 19 %). LRMS (M+H) 734.

실시예
162

프로판-2- 설폰산 {17-[2-(3- 플루오로 - 페닐 )-7- 메톡시 -8- 메틸 - 퀴나졸린 -4- 일 옥시]-13- 메틸 -2.14- 디옥소 -3.13-디아자트리사이클로[13.3.0.0*4.6*]옥타데크-7-엔-4-카보닐}-아미드 (162)
무수 THF (10 mL) 중 산 159 (71 mg, 0.115 mmol) 및 CDI (37 mg, 0.228 mmol)의 혼합물을 N2 하에서 환류 하에 2 시간 동안 가열하였다. 반응 혼합물을 50 ℃로 냉각시키고, 2 ml의 무수 THF 중 메틸-사이클로프로필 설폰아미드(43 mg, 0.349 mmol) 및 DBU (49 μL, 0.322 mmol)의 전-혼합된 용액을 반응 혼합물에 첨가하였다. 반응을 50℃에서 18 시간 동안 교반하였다. 용매를 증발시키고, 잔류물을 CHCl3 중에 용해시키고, 시트르산 (10% 수용액)으로 세척하였다. 유기상을 Na2SO4로 건조시키고, 여과하였으며, 용매를 회전 증발로 제거하였다. 조 생성물을 H2O (0.1% TFA) 중 35 에서 60% 아세토니트릴 (0.1 % TFA)으로의 구배를 지닌 Ace-5 C8 컬럼(100x21.2 mm)상에서 HPLC로 8 분에 걸쳐 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (30 mg, 36 %), LRMS (M+H) 722.

실시예
163

17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
에틸 에스테르
PPh3 (415 mg, 1.58 mmol)를 무수 THF (35 mL) 및 무수 DMF 7 mL 중 알콜 51 (300 mg, 0.79 mmol) 및 퀴나졸리놀 127 (247 mg, 0.87 mmol)의 교반 용액에 첨가하였다. 반응을 실온에서 불활성 대기(N2) 하에 두었다. DIAD (311 μL, 1.58 mmol)를 첨가하였다. 반응 혼합물을 18 시간 동안 교반하였다. 침전물이 플라스크에 형성되었으며, 40 mL 디에틸 에테르를 첨가한 후 백색 고체가 더 침전하였다. 침전물을 여과하고, 디에틸 에테르로 세척하고, 진공 하에 건조시켜 순수 표제 화합물을 수득하였다 (381 mg, 75%). LRMS (M+H) 646.
실시예
164

17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(164)
에틸 에스테르 163을 실시예 159에서와 같이 반응시켰다. 용해도와 느린 반응의 문제 때문에, 반응을 40 시간 동안 수행하였다. LC-MS는 남아있는 시작 물질이 없음을 나타냈으나, 시작 물질의 거의 삼분의 이가 분해되었다. 산성화로 형성 된 침전물을 여과하고, 물로 세척하고, 고진공하에 건조하였다. 생성물의 수율은 중량 및 HPLC에 의해 약 35%로 측정되었다. LRMS (M+H) 618.
실시예
165

사이클로프로판설폰산
{17-[2-(3-
플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-일
옥
시]-13-
메틸
-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드 (165)
산 164를 실시예 160에 나타낸 과정에 따라 반응시켰다. 조 생성물을 5% 아세토니트릴, 암모늄 아세테이트 완충액 5 mM, pH 6.8 중 35 에서 60% 아세토니트릴으로의 구배를 지닌 Ace-5 C8 컬럼(100x21.2 mm)상에서 HPLC로 8 분에 걸쳐 정제하였다. 표제 화합물을 백색 고체로서 수득하였다 (28 mg, 47 %), LRMS (M+H) 721.

실시예
166

2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[2-(3,5-
디플루오로
-페닐)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
tert
-부틸 에스테르(166)
알콜 15 및 퀴나졸리놀 128을 실시예 155에 개시된 동일한 과정을 따라 반응시켜, 트리페닐포스핀 옥사이드로 약간 오염된 백색 고체로서 표제 화합물을 수득하였다 (1.245 g, >100%), LRMS (M+H) 652.
실시예
167
2-(1-
에톡시카보닐
-2-비닐-
사이클로프로필카바모일
)-4-[2-(3,5-
디플루오로
-페닐)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-
사이클로펜탄카복실산
(167)
tert. 부틸 에스테르 166을 실시예 156에서와 같이 반응시켜, 백색 고체(여 전히 약간 POPH3로 오염됨)로서 표제 화합물을 >100% 수율로 수득하였다. LRMS (M+H) 596.
실시예
168
1-{[4-[2-(3,5-
디플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-2-(
헥스
-5-
에닐
-
메틸
-
카바모일
)-
사이클로펜탄카보닐
]-아미노}-2-비닐-사이클로프로판- 카복실산 에틸 에스테르 (168)
산 167을 실시예 157에 개시된 동일한 과정에 따라 헥스-5-에닐-메틸-아민 하이드로클로라이드와 반응시켜, 표제 화합물을 수득하였다 (0.838 g, 81%), LRMS (M+H) 691.
실시예
169

17-[2-(3,5-
디플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나졸린
-4-
일옥시
]-13-
메틸
-2,14-디옥소-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(169)
폐환 복분해 반응을 디엔 168로 실시예 158에 개시된 과정에 따라 수행하여, 약간 오염된 표제 화합물을 수득하였다 (0.509g, 66%), LRMS (M+H) 635.
실시예
170

사이클로프로판설폰산
{17-[2-(3,5-
디플루오로
-
페닐
)-7-
메톡시
-8-
메틸
-
퀴나
졸린-4-
일옥시
]-13-
메틸
-2,14-
디옥소
-3,13-
디아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카보닐
}-아미드.
MV065673
산 169와 사이클로프로판 설폰산 아미드의 반응을 실시예 159에 개시된 과정을 따라 수행한 다음, Ace-5 C8 컬럼(100x21.2 mm) 및 암모늄 아세테이트 완충액 5 mM, pH 6.8, 5% 아세토니트릴, 35 에서 60 %으로의 아세토니트릴을 이용하여 HPLC로 정제하여 백색 고체로서 표제 화합물을 수득하였다 (8 mg, 7%). LRMS (M+H) 738.

실시예
171

14-(4-
메톡시
-벤질)-18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-2,15- 디옥소-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카복실산
에틸 에스테르 (171)
퀴나졸리놀 유도체 83 (352 mg, 0.1.2 mmol)를 실시예 52에 개시된 미츠노부 조건을 이용하여 알콜 100 (600 mg, 1.2 mmol)에 커플링시켰다. 수득된 조 생성물을 실리카 겔 상 플래쉬 크로마토그래피로 정제하고, 디에틸 에테르 중 1% MeOH로 용리시키고, 표제 화합물을 수득하였다 (842 mg, 93%). MS (M+H)+ 748.3
실시예
172

14-(4-
메톡시
-벤질)-18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-2,15- 디옥소-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카복실산
에틸 에스테르 (172)
화합물 171 (842 mg, 1.3 mmol)의 에틸 에스테르를 실시예 20 에서와 같이 가수분해하였다. 4 시간 후, 부피를 반으로 줄인 다음, 물로 부피를 두배가 되게 하였다. 아세트산으로 산성화시킨 다음, 침전된 산물을 여과하여 표제 화합물을 얻었다 MSR-489 (688 mg, 85 %). MS (M+H)+ 720.3
실시예
173

사이클로프로판설폰산
[14-(4-
메톡시
-벤질)-18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴
나졸린-4-
일옥시
)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카보닐
]-아미드 (173)
사이클로프로판설폰아미드 (102 mg, 0.84 mmol)를 실시예 53에 나타낸 바와 같이 산 172 (300 mg, 0.42 mmol)에 커플링시켰다. HPLC로 정제하여 표제 화합물을 수득하였다 (157 mg, 45 %), MS (M-H)+ 823.3.
실시예
174

사이클로프로판설폰산
[18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥시
)-2, 15-디옥소-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카보닐
]-아미드 (174)
화합물 173 (150 mg, 0.18 mmol)을 디클로로메탄-트리플루오로아세트산; 2:1의 혼합물 중에서 30 분 동안 교반하였다. 증발시키고 컬럼 크로마토그래피(에테르 중 5% 메탄올)로 정제하여 표제 화합물을 수득하였다 (81 mg, 62 %). MS (M-H)+ 703
실시예
175

1-
메틸
-
사이클로프로판설폰산
[14-(4-
메톡시
-벤질)-18-(7-
메톡시
-8-
메틸
-2-페닐-
퀴나졸린
-4-
일옥시
)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-
카보닐
]-아미드 (175)
1-메틸-사이클로프로판설폰산 아미드 (218 mg, 1.62 mmol)를 실시예 53에 나타낸 바와 같이 산 172 (388 mg. 0.54 mmol)에 커플링시켰다. 컬럼 크로마토그래피로 정제하여 표제 화합물을 수득하였다 (150 mg, 33%), MS (M-H)+ 837.
실시예
176

1-
메틸
-
사이클로프로판설폰산
[18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥
시)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-카보닐]-아미드 (176)
화합물 175 (150 mg, 0.18 mmol)을 디클로로메탄-트리플루오로아세트산; 2:1의 혼합물 중에서 30 분 동안 교반하였다. 증발시키고, 컬럼 크로마토그래피(에테르 중 5% 메탄올)로 정제하여 표제 화합물을 수득하였다 (74 mg, 57 %), MS (M-H)+ 717.3.
실시예
177

1-
메틸
-
사이클로프로판설폰산
[18-(7-
메톡시
-8-
메틸
-2-
페닐
-
퀴나졸린
-4-
일옥
시)-2,15-
디옥소
-3,14,16-
트리아자
-
트리사이클로[14.3.0.0*4,6*]노나데크
-7-엔-4-카보닐]-아미드.
퀴나졸리놀 유도체 123 (155 mg, 0.58 mmol)을 실시예 52에 나타낸 미츠노부 조건을 이용하여 알콜 51 (200 mg, 0.53 mmol)에 커플링시켰다. 목적 산물이 반응 혼합물 중 침전하며, 여과로 수집하여 순수 표제 화합물을 수득하였다 (152 mg, 45 %), MS (M+H)+ 629.3
실시예
178

17-(7-
메톡시
-8-
메틸
-2-피리딘-4-일-
퀴나졸린
-4-
일옥시
)-13-
메틸
-2,14-
디옥소
-3,13,15-
트리아자
-
트리사이클로[13.3.0.0*4,6*]옥타데크
-7-엔-4-
카복실산
(178)
화합물 177 (152 mg, 0.24 mmol)의 에틸 에스테르를 화합물 20을 위해 기재된 과정에 따라 가수분해하였다. 반응하는 동안 산물은 일부 분해되었다. 크로마토그래피(에테르 + 0.1% 아세트산 중 0 내지 15% 메탄올)로 정제하여 순수 표제 화합물을 수득하였다 (46 %), MS (M+H)+ 601.
실시예
179

사이클로프로판설폰산
[17-(7-
메톡시
-8-
메틸
-2-피리딘-4-일-
퀴나졸린
-4-
일옥
시)-13-
메틸
-2.14-
디옥소
-3.13.15-
트리아자
-
트리사이클로[13.3.0.0*4.6*]옥타데크
- 7-엔-4-
카보닐
]-아미드 (179)
산 178 (67 mg, 0.11 mmol)과 EDAC (26 mg, 0.13 mmol)를 디클로로메탄 (3 ml) 중에 용해하였다. 실온에서 5 시간 동안 교반한 다음, 혼합물을 디클로로메탄 (10 ml)으로 희석시키고, 유기상을 물로 세척하고, 건조(황산 나트륨)시켰다. 그 다음 부피를 2 ml로 줄이고, 사이클로프로필 설폰아미드 (20 mg, 0.17 mmol) 및 DBU (36 mg, 2.3 mmol)를 첨가하였다. 혼합물을 실온에서 밤새 교반한 다음, 5% 시트르산 수용액으로 세척하였다. 크로마토그래피(디클로로메탄 중 0 내지 2% 메탄올)로 정제하여 표제 화합물을 수득하였다 (57 mg, 73 %), MS (M+H)+ 704.
생물학적
실시예
1 : 화학식 (I)의 화합물 활성
레플리콘
어세이
세포 어세이의 HCV RNA 복제 억제 활성에 대해 화학식 (I)의 화합물을 분석하였다. 화학식 (I)의 화합물이 세포 배양에서 작용하는 HCV 레플리콘에 대한 활성을 보유하고 있음이 어세이에서 나타났다. 세포 어세이는 다중-표적 스크리닝 전략에서, Lohmann et al.에 의해 문헌(1999) Science vol. 285 pp. 110-113에 제시되고, 변형법이 Krieger et al.에 의해 문헌 (2001) Journal of Virology 75: 4614-4624에 기재된 이시스트론 발현 작제물을 기준으로 하였다. 본질적으로, 이 방법은 다음과 같다.
어세이에서 안정하게 형질감염된 세포주 Huh-7 luc/neo(이하 Huh-Luc로 지칭 함)를 이용하였다. 이 세포주는 리포터 부분(FfL-루시퍼라제) 및 선별가능한 마커 부분(neoR, 네오마이신 포스포트랜스퍼라제)이 앞에 있는, 뇌심근염 바이러스(EMCV)로부터 내부 리보솜 결합 부위(IRES)에서 번역된 HCV 1b형의 야생형 NS3-NS5B 영역 을 포함하는 이시스트론 발현 작제물을 암호화하는 RNA를 지닌다. 이 작제물은 HCV 1b형으로부터 5' 및 3' NTRs(비번역 영역)과 접하여 있다. G418(neoR)의 존재하에 레플리콘 세포의 연속 배양은 HCV RNA의 복제에 의존한다. 자발적으로 그리고 높은 수준으로 복제하고, 특히 루시퍼라제를 암호화하는 HCV RNA를 발현하는, 안정하게 형질감염된 레플리콘 세포를 항바이러스 화합물을 스크리닝하는데 사용하였다.
다양한 농도로 첨가된 시험 및 대조 화합물의 존재하에 레플리콘 세포를 384 웰 플레이트에서 평판 배양하였다. 3일간 배양한 후, 루시퍼라제 활성을 분석함으로써(표준 루시퍼라제 어세이 기질 및 시약과 Perkin Elmer ViewLuxTm ultraHTS 마이크로플레이트 이미저를 이용함) HCV 복제를 측정하였다. 대조 배양에서 레플리콘 세포는 억제제의 부재하에 높은 루시퍼라제 발현율을 갖는다. 루시퍼라제 활성에 서 화합물의 억제 활성을 Huh-Luc 세포 상에서 모니터하였고, 각 시험 화합물에 대해 투여량-반응 곡선을 작제하였다. 그 후 EC50 값을 계산하였으며, 이 값은 검측된 루시퍼라제 활성 수준, 또는 더 구체적으로, 유전학적으로 연결된 HCV 레플리콘 RNA가 복제하는 능력을 50%까지 감소시키는데 필요한 화합물의 양을 나타낸다.
생물학적
실시예
2 : 화학식 (I)의 화합물 활성
억제
어세이
본 시험관내 어세이의 목적은 본 발명의 화합물에 의한 HCV NS3/4A 프로테아제 복합체의 억제를 측정하는 것이다. 본 어세이로 HCV NS3/4A 단백질 가수 분해 활성을 억제하는 데 본 발명의 화합물이 얼마나 효율적인지 알 수 있다.
전장 C형 간염 NS3 프로테아제 효소의 억제는 Poliakov, 2002 Prot Expression & Purification 25 363 371에 개시된 바와 같이 측정하였다. 간단하게, 데프시펩티드 물질, Ac-DED(Edans)EEAbuψ[COO]ASK(Dabcyl)-NH2 (AnaSpec, San Jose, USA)의 가수 분해를 펩티드 공동 인자 KKGSVVIVGRIVLSGK (Ake Engstrom, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden)의 존재 하에서 분광광도로 측정하였다. [Landro, 1997 #Biochem 36 9340-9348]. 이 효소(1 nM)를 50 mM HEPES, pH 7.5, 10 mM DTT, 40% 글리세롤, 0.1% n-옥틸-D-글루코시드 중에 25 μM NS4A 공동 인자 및 억제제와 함께 30℃에서 10분 동안 인큐베이션하였으며, 반응을 0.5 μM 물질의 첨가에 의해 개시하였다. 억제제를 DMSO 중에 용해시키고, 30초 동안 초음파 처리하고, 보텍싱(vortex)하였다. 측정 사이에 용액을 -20℃에 보관하였다.
어세이 샘플에서 DMSO의 최종 농도를 3.3%로 조정하였다. 공개된 방법에 따라 내부 필터 효과를 위해 가수분해율을 보정하였다. [Liu, 1999 Analytical Biochemistry 267 331-335]. Km (0.15 μM)의 고정 값과 경쟁 억제 모델을 이용한 비-선형 회귀 분석에 의하여 Ki 값을 측정하였다(GraFit, Erithacus Software, Staines, MX, UK). 두 반복값 중 최소치를 모든 측정에서 수행하였다.
다음 표 1에 상기 실시예에 따라 제조된 대표적인 화합물을 열거하였다. 시험된 화합물의 활성도 표 1에 나타내었다. 값 A, B, C, D, E 및 F에 대한 설명은 다음과 같다:
- 값 A는 EC50 > lOμM에 상응하며;
- 값 B는 1OμM 과 1 μM 사이의 EC50에 상응하고;
- 값 C는 0.99μM 과 20OnM 사이의 EC50에 상응하며;
- 값 D는 199nM 과 0.5nM 사이의 EC50에 상응하고;
- 값 E는 Ki > 1 μM에 상응하며;
- 값 F는 1 μM 과 10OnM 사이의 Ki에 상응하고;
- 값 G는 99.9nM 과 5nM 사이의 Ki에 상응하며;
- 값 H는 4.9nM 과 0.1nM 사이의 Ki에 상응한다.

Claims (29)
- 하기 화학식 (I)의 화합물, 그의 염 또는 입체이성체:
 상기 식에서,A는 OR1, NHS(=O)pR2를 나타내며;여기서, R1은 수소, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고;R2는 C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내며;p는 독립적으로 1 또는 2를 나타내고;n은 3, 4, 5 또는 6을 나타내며;-----은 임의의 이중 결합을 나타내고;L은 N 또는 CRz를 나타내며;Rz는 H이거나, 별표 표시된 탄소와 이중 결합을 형성하고;Rq는 H이거나, L이 CRz일 때, Rq는 C1-C6알킬일 수도 있으며;Rr은 C1-C6알킬, C1-C6알콕시, 하이드록시, 할로, 할로C1-C6알킬, 아미노, 모노- 또는 디알킬아미노, 모노- 또는 디알킬아미노카보닐, C1-C6알킬카보닐-아미노, C0-C3알킬렌카보사이클릴 및 C0-C3알킬렌헤테로사이클릴에서 각각 독립적으로 선택된 하나, 둘 또는 세개의 치환체로 임의로 치환된 퀴나졸리닐을 나타내고;R5는 수소, C1-C6알킬, C1-C6알콕시C1-C6알킬 또는 C3-C7사이클로알킬을 나타내며;여기서, 각 C1-C6알킬, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴은 할로, 옥소, 니트릴, 아지도, 니트로, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, NH2C(=O)-, Y-NRaRb, Y-O-Rb, Y-C(=O)Rb, Y-(C=O)NRaRb, Y-NRaC(=O)Rb, Y-NHS(=Op)Rb, Y-S(=O)pRb 및 Y-S(=O)pNRaRb, Y-C(=O)ORb, Y-NRaC(=O)ORb로 구성된 군에서 독립적으로 선택된 1개 내지 3개의 치환체로 임의로 치환되고;Y는 독립적으로 결합 또는 C1-C3알킬렌을 나타내며;Ra는 독립적으로 H, C1-C6알콕시, C1-C3알킬을 나타내거나; 또는Rb는 독립적으로 H, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴을 나타내거나;Ra 및 Rb는 그들이 부착되는 질소와 함께 헤테로사이클릴기를 형성한다.
상기 식에서,A는 OR1, NHS(=O)pR2를 나타내며;여기서, R1은 수소, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고;R2는 C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내며;p는 독립적으로 1 또는 2를 나타내고;n은 3, 4, 5 또는 6을 나타내며;-----은 임의의 이중 결합을 나타내고;L은 N 또는 CRz를 나타내며;Rz는 H이거나, 별표 표시된 탄소와 이중 결합을 형성하고;Rq는 H이거나, L이 CRz일 때, Rq는 C1-C6알킬일 수도 있으며;Rr은 C1-C6알킬, C1-C6알콕시, 하이드록시, 할로, 할로C1-C6알킬, 아미노, 모노- 또는 디알킬아미노, 모노- 또는 디알킬아미노카보닐, C1-C6알킬카보닐-아미노, C0-C3알킬렌카보사이클릴 및 C0-C3알킬렌헤테로사이클릴에서 각각 독립적으로 선택된 하나, 둘 또는 세개의 치환체로 임의로 치환된 퀴나졸리닐을 나타내고;R5는 수소, C1-C6알킬, C1-C6알콕시C1-C6알킬 또는 C3-C7사이클로알킬을 나타내며;여기서, 각 C1-C6알킬, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴은 할로, 옥소, 니트릴, 아지도, 니트로, C1-C6알킬, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, NH2C(=O)-, Y-NRaRb, Y-O-Rb, Y-C(=O)Rb, Y-(C=O)NRaRb, Y-NRaC(=O)Rb, Y-NHS(=Op)Rb, Y-S(=O)pRb 및 Y-S(=O)pNRaRb, Y-C(=O)ORb, Y-NRaC(=O)ORb로 구성된 군에서 독립적으로 선택된 1개 내지 3개의 치환체로 임의로 치환되고;Y는 독립적으로 결합 또는 C1-C3알킬렌을 나타내며;Ra는 독립적으로 H, C1-C6알콕시, C1-C3알킬을 나타내거나; 또는Rb는 독립적으로 H, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴 또는 C0-C3알킬렌헤테로사이클릴을 나타내거나;Ra 및 Rb는 그들이 부착되는 질소와 함께 헤테로사이클릴기를 형성한다. - 제 1항에 있어서, 하기의 부분 구조를 갖는 화합물:

- 제 1항에 있어서, n이 4 또는 5인 화합물.
- 제 3항에 있어서, 사이클로프로필 부분에 근접한 -----은 이중 결합인 화합물.
- 제 1항에 있어서, R5는 수소 또는 메틸을 나타내는 화합물.
- 제 1항에 있어서, A는 -OH 또는 -NHS(=O)2-사이클로프로필을 나타내는 화합물.
- 제 1항에 있어서, A는 NHS(=O)2-C1-C6알킬치환된 사이클로프로필을 나타내는 화합물.
- 제 1항에 있어서, 다음 화학식을 갖는 화합물:
 상기 식에서,n, A, L, Rq 및 R5는 제 1항에서 정의된 바와 같고;R6는 수소, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, 하이드록시, 브로모, 클로로 또는 플루오로를 나타내며;R9는 수소, C1-C6알킬, C1-C6알콕시, NRaRb, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고; 여기서, 상기 R9 카보사이클릴 또는 헤테로사이클릴은 R10으로 임의로 치환되며;R10은 C1-C6알킬, C0-C3알킬렌사이클로알킬, C0-C3알킬렌헤테로사이클릴, C1-C6알콕시, C1-C6알콕시C1-C6알킬, 아미노, 아미도, 아지도, 머캅토, 시아노, 설포닐, (C1-C3알킬)설포닐, 니트로, 하이드록시, 카복시, 머캅토, 할로, 할로C1-C6알킬, 할로C1-C6알킬옥시를 나타내고;Ra 및 Rb는 제 1항에서 정의된 바와 같으며;R11은 수소 또는 C1-C6알콕시를 나타낸다.
상기 식에서,n, A, L, Rq 및 R5는 제 1항에서 정의된 바와 같고;R6는 수소, C1-C6알킬, C1-C6알콕시, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴, 하이드록시, 브로모, 클로로 또는 플루오로를 나타내며;R9는 수소, C1-C6알킬, C1-C6알콕시, NRaRb, C0-C3알킬렌카보사이클릴, C0-C3알킬렌헤테로사이클릴을 나타내고; 여기서, 상기 R9 카보사이클릴 또는 헤테로사이클릴은 R10으로 임의로 치환되며;R10은 C1-C6알킬, C0-C3알킬렌사이클로알킬, C0-C3알킬렌헤테로사이클릴, C1-C6알콕시, C1-C6알콕시C1-C6알킬, 아미노, 아미도, 아지도, 머캅토, 시아노, 설포닐, (C1-C3알킬)설포닐, 니트로, 하이드록시, 카복시, 머캅토, 할로, 할로C1-C6알킬, 할로C1-C6알킬옥시를 나타내고;Ra 및 Rb는 제 1항에서 정의된 바와 같으며;R11은 수소 또는 C1-C6알콕시를 나타낸다. - 제 8항에 있어서, R6는 C1-C3알킬, 클로로, 플루오로, 브로모 또는 수소를 나타내는 화합물;
- 제 8항에 있어서, R11은 수소 또는 메톡시를 나타내는 화합물.
- 제 8항에 있어서, R9은 하나 또는 두개의 R10으로 임의로 치환되는 페닐 또는 헤테로아릴을 나타내며;여기서 R10은 수소, C1-C6알킬, C3-C7사이클로알킬, C3-C7헤테로사이클릴, C1- C6알콕시, 할로, C1-C6알킬로 임의로 일- 또는 이치환된 아미노 또는 C1-C6알킬로 임의로 일- 또는 이치환된 아미도를 나타내는 화합물.
- 제 11항에 있어서, R9은 제 11항에 정의된 바와 같은 R10으로 각각 임의로 치환된 페닐, 피리딜, 티아졸릴, 옥사졸릴 또는 피라졸릴을 나타내는 화합물.
- 제 12항에 있어서, R10은 수소, 플루오로, 디플루오로, 메틸, 에틸, 이소프로필, tert-부틸, C1-C6알콕시, 아미노, 모노- 또는 디C1-C6알킬아미노 피롤리디닐, 피페리닐, 피페라지닐, N-메틸피페라지닐, 모르폴리닐, 또는 모노- 또는 디-C1-C6알킬아미도를 나타내는 화합물.
- 제 13항에 있어서, R10은 수소, 플루오로 또는 메톡시를 나타내는 화합물.
- 제 12항에 있어서, R9은 하기로부터 선택되는 화합물:

- 제 12항에 있어서, R9은 하기로부터 선택되는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 하기 화학식을 갖는 화합물:

- 약제학적으로 허용되는 담체 및 제 1항 내지 제 24항 중 어느 한 항의 화합물을 포함하는, HCV를 포함한 플라비바이러스 감염의 치료 또는 예방용 약제학적 조성물.
- 제 25항에 있어서, 뉴클레오시드 유사 폴리머라제 억제제, 프로테아제 억제제, 리바비린 및 인터페론에서 선택된 추가의 HCV 항바이러스제를 더 포함하는 약제학적 조성물.
- 삭제
- 제 1항 내지 제 24항 중 어느 한 항에 따른 화합물을 포함하는, HCV를 포함한 플라비바이러스 감염의 치료 또는 예방용 약제.
- 삭제
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05107057.1 | 2005-07-29 | ||
| EP05107057 | 2005-07-29 | ||
| EP06113097.7 | 2006-04-25 | ||
| EP06113097 | 2006-04-25 | ||
| PCT/EP2006/064822 WO2007014927A2 (en) | 2005-07-29 | 2006-07-28 | Macrocyclic inhibitors of hepatitis c virus |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20080033475A KR20080033475A (ko) | 2008-04-16 |
| KR101083631B1 true KR101083631B1 (ko) | 2011-11-16 |
Family
ID=37708968
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020087004894A KR101083631B1 (ko) | 2005-07-29 | 2006-07-28 | C형 간염 바이러스의 마크로사이클릭 억제제 |
Country Status (32)
| Country | Link |
|---|---|
| US (1) | US7659245B2 (ko) |
| EP (1) | EP1912981B1 (ko) |
| JP (1) | JP5230418B2 (ko) |
| KR (1) | KR101083631B1 (ko) |
| AP (1) | AP2585A (ko) |
| AR (1) | AR057702A1 (ko) |
| AT (1) | ATE455775T1 (ko) |
| AU (1) | AU2006274866B2 (ko) |
| BR (1) | BRPI0614153A2 (ko) |
| CA (1) | CA2617103C (ko) |
| CY (1) | CY1110008T1 (ko) |
| DE (1) | DE602006011905D1 (ko) |
| DK (1) | DK1912981T3 (ko) |
| EA (1) | EA014188B1 (ko) |
| ES (1) | ES2339702T3 (ko) |
| GT (1) | GT200600343A (ko) |
| HK (1) | HK1116488A1 (ko) |
| HR (1) | HRP20100209T1 (ko) |
| IL (1) | IL188320A (ko) |
| ME (1) | ME01454B (ko) |
| MY (1) | MY151026A (ko) |
| NO (1) | NO20081075L (ko) |
| NZ (1) | NZ564540A (ko) |
| PE (1) | PE20070343A1 (ko) |
| PL (1) | PL1912981T3 (ko) |
| PT (1) | PT1912981E (ko) |
| RS (1) | RS51227B (ko) |
| SI (1) | SI1912981T1 (ko) |
| SV (1) | SV2008002641A (ko) |
| TW (1) | TWI387597B (ko) |
| UY (1) | UY29704A1 (ko) |
| WO (1) | WO2007014927A2 (ko) |
Families Citing this family (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| DE602005017582D1 (en) | 2004-01-30 | 2009-12-24 | Medivir Ab | Hcv ns-3 serine protease inhibitoren |
| PT1912997E (pt) | 2005-07-29 | 2011-12-19 | Tibotec Pharm Ltd | Inibidores macrocíclicos do vírus da hepatite c |
| JO2768B1 (en) * | 2005-07-29 | 2014-03-15 | تيبوتيك فارماسيوتيكالز ليمتد | Large cyclic inhibitors of hepatitis C virus |
| PE20070211A1 (es) | 2005-07-29 | 2007-05-12 | Medivir Ab | Compuestos macrociclicos como inhibidores del virus de hepatitis c |
| EP1913015B1 (en) | 2005-07-29 | 2013-12-11 | Janssen R&D Ireland | Macrocyclic inhibitors of hepatitis c virus |
| DK1999129T3 (da) | 2005-10-11 | 2011-02-07 | Intermune Inc | Forbindelser og fremgangsmåder til inhibering af replikationen af hepatitis C-virus |
| US7741281B2 (en) | 2005-11-03 | 2010-06-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| KR20090024834A (ko) | 2006-07-05 | 2009-03-09 | 인터뮨, 인크. | C형 간염 바이러스 복제의 신규 억제제 |
| EP2049474B1 (en) | 2006-07-11 | 2015-11-04 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2008008502A1 (en) | 2006-07-13 | 2008-01-17 | Achillion Pharmaceuticals, Inc. | 4-amino-4-oxobutanoyl peptides as inhibitors of viral replication |
| US8343477B2 (en) | 2006-11-01 | 2013-01-01 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| US7772180B2 (en) | 2006-11-09 | 2010-08-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8003604B2 (en) | 2006-11-16 | 2011-08-23 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7888464B2 (en) | 2006-11-16 | 2011-02-15 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7763584B2 (en) | 2006-11-16 | 2010-07-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CA2661338C (en) * | 2006-11-17 | 2015-05-12 | Tibotec Pharmaceuticals Ltd. | Macrocyclic inhibitors of hepatitis c virus |
| TWI455936B (zh) * | 2007-02-01 | 2014-10-11 | Tibotec Pharm Ltd | 用於製備hcv之大環蛋白酶抑制劑之方法及中間物 |
| RU2481340C2 (ru) * | 2007-02-08 | 2013-05-10 | Тиботек Фармасьютикалз Лтд. | Пиримидин-замещенные макроциклические ингибиторы hcv |
| ITMI20071616A1 (it) | 2007-08-03 | 2009-02-04 | Cosmo Spa | Processo enzimatico per l'ottenimento di 17-alfa monoesteri del cortexolone e/o suoi 9,11-deidroderivati. |
| CA2700383A1 (en) * | 2007-09-24 | 2009-04-02 | Achillion Pharmaceuticals, Inc. | Urea-containing peptides as inhibitors of viral replication |
| US8383583B2 (en) | 2007-10-26 | 2013-02-26 | Enanta Pharmaceuticals, Inc. | Macrocyclic, pyridazinone-containing hepatitis C serine protease inhibitors |
| US8426360B2 (en) * | 2007-11-13 | 2013-04-23 | Enanta Pharmaceuticals, Inc. | Carbocyclic oxime hepatitis C virus serine protease inhibitors |
| US8263549B2 (en) | 2007-11-29 | 2012-09-11 | Enanta Pharmaceuticals, Inc. | C5-substituted, proline-derived, macrocyclic hepatitis C serine protease inhibitors |
| US8202996B2 (en) | 2007-12-21 | 2012-06-19 | Bristol-Myers Squibb Company | Crystalline forms of N-(tert-butoxycarbonyl)-3-methyl-L-valyl-(4R)-4-((7-chloro-4-methoxy-1-isoquinolinyl)oxy)-N- ((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-L-prolinamide |
| WO2009084695A1 (ja) | 2007-12-28 | 2009-07-09 | Carna Biosciences Inc. | 2-アミノキナゾリン誘導体 |
| US8372802B2 (en) | 2008-03-20 | 2013-02-12 | Enanta Pharmaceuticals, Inc. | Fluorinated macrocyclic compounds as hepatitis C virus inhibitors |
| US8163921B2 (en) | 2008-04-16 | 2012-04-24 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US7964560B2 (en) | 2008-05-29 | 2011-06-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8044023B2 (en) | 2008-05-29 | 2011-10-25 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8207341B2 (en) | 2008-09-04 | 2012-06-26 | Bristol-Myers Squibb Company | Process or synthesizing substituted isoquinolines |
| UY32099A (es) | 2008-09-11 | 2010-04-30 | Enanta Pharm Inc | Inhibidores macrocíclicos de serina proteasas de hepatitis c |
| US8044087B2 (en) | 2008-09-29 | 2011-10-25 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8563505B2 (en) | 2008-09-29 | 2013-10-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| MX2011006244A (es) | 2008-12-10 | 2011-06-27 | Achillion Pharmaceuticals Inc | Nuevos peptidos de 4-amino-4-oxobutanoilo como inhibidores de replica viral. |
| US8283310B2 (en) | 2008-12-15 | 2012-10-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP5711672B2 (ja) * | 2009-02-27 | 2015-05-07 | ジヤンセン・フアーマシユーチカルズ・インコーポレーテツドJanssen Pharmaceuticals,Inc. | Hcvの大環状阻害剤の無定形塩 |
| US8377962B2 (en) | 2009-04-08 | 2013-02-19 | Idenix Pharmaceuticals, Inc. | Macrocyclic serine protease inhibitors |
| CA2762347A1 (en) * | 2009-05-29 | 2010-12-02 | Syngenta Participations Ag | Substituted quinazolines as fungicides |
| TW201117812A (en) | 2009-08-05 | 2011-06-01 | Idenix Pharmaceuticals Inc | Macrocyclic serine protease inhibitors |
| BR112013016480A2 (pt) | 2010-12-30 | 2016-09-20 | Abbvie Inc | macrocíclo da fenantridina inibadores da protease da serina da hepatite c |
| JP2014502620A (ja) | 2010-12-30 | 2014-02-03 | エナンタ ファーマシューティカルズ インコーポレイテッド | 大環状c型肝炎セリンプロテアーゼ阻害剤 |
| US9353100B2 (en) | 2011-02-10 | 2016-05-31 | Idenix Pharmaceuticals Llc | Macrocyclic serine protease inhibitors, pharmaceutical compositions thereof, and their use for treating HCV infections |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10201584B1 (en) | 2011-05-17 | 2019-02-12 | Abbvie Inc. | Compositions and methods for treating HCV |
| US8691757B2 (en) | 2011-06-15 | 2014-04-08 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| MX340033B (es) | 2011-12-28 | 2016-06-21 | Janssen Sciences Ireland Uc | Derivados heterobiciclicos como inhibidores del virus de la hepatitis c. |
| IN2015DN02588A (ko) * | 2012-10-15 | 2015-09-11 | Albemarle Corp | |
| JP6154474B2 (ja) | 2012-10-19 | 2017-06-28 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | C型肝炎ウイルス阻害剤 |
| WO2014070964A1 (en) | 2012-11-02 | 2014-05-08 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2014071007A1 (en) | 2012-11-02 | 2014-05-08 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9006423B2 (en) | 2013-03-15 | 2015-04-14 | Achillion Pharmaceuticals Inc. | Process for making a 4-amino-4-oxobutanoyl peptide cyclic analogue, an inhibitor of viral replication, and intermediates thereof |
| US9085607B2 (en) | 2013-03-15 | 2015-07-21 | Achillion Pharmaceuticals, Inc. | ACH-0142684 sodium salt polymorph, composition including the same, and method of manufacture thereof |
| EP3089757A1 (en) | 2014-01-03 | 2016-11-09 | AbbVie Inc. | Solid antiviral dosage forms |
| EP3108879A1 (en) | 2015-06-25 | 2016-12-28 | Cassiopea S.p.A. | High concentration formulation |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004037855A1 (en) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis c virus |
| WO2005028501A1 (en) | 2003-09-22 | 2005-03-31 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis c virus |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| UA74546C2 (en) * | 1999-04-06 | 2006-01-16 | Boehringer Ingelheim Ca Ltd | Macrocyclic peptides having activity relative to hepatitis c virus, a pharmaceutical composition and use of the pharmaceutical composition |
| US6867185B2 (en) * | 2001-12-20 | 2005-03-15 | Bristol-Myers Squibb Company | Inhibitors of hepatitis C virus |
| US7273885B2 (en) | 2002-04-11 | 2007-09-25 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly HCV NS3-NS4A protease |
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| JP2007524576A (ja) * | 2003-02-07 | 2007-08-30 | エナンタ ファーマシューティカルズ インコーポレイテッド | 大環状のc型肝炎セリンプロテアーゼ阻害剤 |
| US20040180815A1 (en) * | 2003-03-07 | 2004-09-16 | Suanne Nakajima | Pyridazinonyl macrocyclic hepatitis C serine protease inhibitors |
| ES2386161T3 (es) * | 2003-04-16 | 2012-08-10 | Bristol-Myers Squibb Company | Proceso para separar una mezcla de enantiómeros de éster alquílico usando una enzima |
| US7125845B2 (en) * | 2003-07-03 | 2006-10-24 | Enanta Pharmaceuticals, Inc. | Aza-peptide macrocyclic hepatitis C serine protease inhibitors |
| DE602005017582D1 (en) * | 2004-01-30 | 2009-12-24 | Medivir Ab | Hcv ns-3 serine protease inhibitoren |
| PE20070211A1 (es) * | 2005-07-29 | 2007-05-12 | Medivir Ab | Compuestos macrociclicos como inhibidores del virus de hepatitis c |
| US7700552B2 (en) * | 2005-07-29 | 2010-04-20 | Medivir Ab | Macrocyclic inhibitors of hepatitis C virus |
| AR055105A1 (es) * | 2005-07-29 | 2007-08-08 | Tibotec Pharm Ltd | Inhibidores macrociclicos del virus de la hepatitis c |
| CN101273030B (zh) * | 2005-07-29 | 2012-07-18 | 泰博特克药品有限公司 | 丙型肝炎病毒的大环抑制剂 |
-
2006
- 2006-07-26 PE PE2006000903A patent/PE20070343A1/es not_active Application Discontinuation
- 2006-07-28 EA EA200800482A patent/EA014188B1/ru not_active IP Right Cessation
- 2006-07-28 MY MYPI20063670 patent/MY151026A/en unknown
- 2006-07-28 AP AP2008004306A patent/AP2585A/xx active
- 2006-07-28 KR KR1020087004894A patent/KR101083631B1/ko not_active IP Right Cessation
- 2006-07-28 AT AT06764268T patent/ATE455775T1/de active
- 2006-07-28 GT GT200600343A patent/GT200600343A/es unknown
- 2006-07-28 AU AU2006274866A patent/AU2006274866B2/en not_active Ceased
- 2006-07-28 DK DK06764268.6T patent/DK1912981T3/da active
- 2006-07-28 PT PT06764268T patent/PT1912981E/pt unknown
- 2006-07-28 CA CA2617103A patent/CA2617103C/en not_active Expired - Fee Related
- 2006-07-28 RS RSP-2010/0183A patent/RS51227B/sr unknown
- 2006-07-28 ME MEP-2012-119A patent/ME01454B/me unknown
- 2006-07-28 SI SI200630627T patent/SI1912981T1/sl unknown
- 2006-07-28 BR BRPI0614153-6A patent/BRPI0614153A2/pt not_active Application Discontinuation
- 2006-07-28 UY UY29704A patent/UY29704A1/es unknown
- 2006-07-28 TW TW095127585A patent/TWI387597B/zh not_active IP Right Cessation
- 2006-07-28 EP EP06764268A patent/EP1912981B1/en active Active
- 2006-07-28 NZ NZ564540A patent/NZ564540A/en not_active IP Right Cessation
- 2006-07-28 ES ES06764268T patent/ES2339702T3/es active Active
- 2006-07-28 JP JP2008523383A patent/JP5230418B2/ja not_active Expired - Fee Related
- 2006-07-28 AR ARP060103303A patent/AR057702A1/es not_active Application Discontinuation
- 2006-07-28 WO PCT/EP2006/064822 patent/WO2007014927A2/en active Application Filing
- 2006-07-28 PL PL06764268T patent/PL1912981T3/pl unknown
- 2006-07-28 US US11/995,900 patent/US7659245B2/en not_active Expired - Fee Related
- 2006-07-28 DE DE602006011905T patent/DE602006011905D1/de active Active
- 2006-07-29 SV SV2006002641A patent/SV2008002641A/es unknown
-
2007
- 2007-12-20 IL IL188320A patent/IL188320A/en not_active IP Right Cessation
-
2008
- 2008-02-29 NO NO20081075A patent/NO20081075L/no not_active Application Discontinuation
- 2008-06-20 HK HK08106903.5A patent/HK1116488A1/xx not_active IP Right Cessation
-
2010
- 2010-04-14 HR HR20100209T patent/HRP20100209T1/hr unknown
- 2010-04-20 CY CY20101100355T patent/CY1110008T1/el unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004037855A1 (en) | 2002-10-25 | 2004-05-06 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis c virus |
| WO2005028501A1 (en) | 2003-09-22 | 2005-03-31 | Boehringer Ingelheim International Gmbh | Macrocyclic peptides active against the hepatitis c virus |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101083631B1 (ko) | C형 간염 바이러스의 마크로사이클릭 억제제 | |
| RU2419619C2 (ru) | Макроциклические ингибиторы вируса гепатита с | |
| KR101381176B1 (ko) | C형 간염 바이러스의 마크로사이클릭 억제제 | |
| KR101059419B1 (ko) | C형 간염 바이러스의 마크로사이클릭 억제제 | |
| JP6153495B2 (ja) | C型肝炎ウイルスの大環状阻害剤 | |
| KR20090110314A (ko) | 피리미딘 치환된 마크로사이클릭 hcv 억제제 | |
| JP2009502884A (ja) | C型肝炎ウイルスの大環状阻害剤 | |
| JP2010518049A (ja) | Hcv阻害大環状フェニルカルバメート | |
| MX2008001393A (en) | Macrocyclic inhibitors of hepatitis c virus |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E902 | Notification of reason for refusal | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| FPAY | Annual fee payment |
Payment date: 20141022 Year of fee payment: 4 |
|
| FPAY | Annual fee payment |
Payment date: 20151016 Year of fee payment: 5 |
|
| FPAY | Annual fee payment |
Payment date: 20161019 Year of fee payment: 6 |
|
| FPAY | Annual fee payment |
Payment date: 20171018 Year of fee payment: 7 |
|
| LAPS | Lapse due to unpaid annual fee |