JP7585359B2 - 反応性末端基を有するポリベンズイミダゾールオリゴマー - Google Patents
反応性末端基を有するポリベンズイミダゾールオリゴマー Download PDFInfo
- Publication number
- JP7585359B2 JP7585359B2 JP2023000307A JP2023000307A JP7585359B2 JP 7585359 B2 JP7585359 B2 JP 7585359B2 JP 2023000307 A JP2023000307 A JP 2023000307A JP 2023000307 A JP2023000307 A JP 2023000307A JP 7585359 B2 JP7585359 B2 JP 7585359B2
- Authority
- JP
- Japan
- Prior art keywords
- oligomer
- polybenzimidazole
- agents
- hours
- pbi
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000004693 Polybenzimidazole Substances 0.000 title claims description 76
- 229920002480 polybenzimidazole Polymers 0.000 title claims description 76
- 239000011347 resin Substances 0.000 claims description 25
- 229920005989 resin Polymers 0.000 claims description 25
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 claims description 24
- 239000003795 chemical substances by application Substances 0.000 claims description 15
- 238000000034 method Methods 0.000 claims description 14
- KSSJBGNOJJETTC-UHFFFAOYSA-N COC1=C(C=CC=C1)N(C1=CC=2C3(C4=CC(=CC=C4C=2C=C1)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)C1=CC(=CC=C1C=1C=CC(=CC=13)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)C1=CC=C(C=C1)OC Chemical compound COC1=C(C=CC=C1)N(C1=CC=2C3(C4=CC(=CC=C4C=2C=C1)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)C1=CC(=CC=C1C=1C=CC(=CC=13)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)N(C1=CC=C(C=C1)OC)C1=C(C=CC=C1)OC)C1=CC=C(C=C1)OC KSSJBGNOJJETTC-UHFFFAOYSA-N 0.000 claims description 13
- 239000003063 flame retardant Substances 0.000 claims description 12
- 239000000654 additive Substances 0.000 claims description 8
- 238000006243 chemical reaction Methods 0.000 claims description 8
- 239000002904 solvent Substances 0.000 claims description 7
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 claims description 6
- 239000003963 antioxidant agent Substances 0.000 claims description 6
- 239000002216 antistatic agent Substances 0.000 claims description 6
- 239000003086 colorant Substances 0.000 claims description 6
- 239000000945 filler Substances 0.000 claims description 6
- 230000007062 hydrolysis Effects 0.000 claims description 6
- 238000006460 hydrolysis reaction Methods 0.000 claims description 6
- 239000003112 inhibitor Substances 0.000 claims description 6
- 239000003607 modifier Substances 0.000 claims description 6
- 239000000049 pigment Substances 0.000 claims description 6
- 239000004014 plasticizer Substances 0.000 claims description 6
- 239000012744 reinforcing agent Substances 0.000 claims description 6
- 125000000304 alkynyl group Chemical group 0.000 claims description 5
- 239000006227 byproduct Substances 0.000 claims description 4
- 239000007795 chemical reaction product Substances 0.000 claims description 3
- XFEGRFIENDJTCK-UHFFFAOYSA-N 2-phenyl-2,3-dihydroindene-1,1-dicarboxylic acid Chemical compound C1C2=CC=CC=C2C(C(=O)O)(C(O)=O)C1C1=CC=CC=C1 XFEGRFIENDJTCK-UHFFFAOYSA-N 0.000 claims description 2
- 230000001376 precipitating effect Effects 0.000 claims description 2
- CPFKVVLZVUUQGV-UHFFFAOYSA-N 4-[2-(3,4-diaminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]benzene-1,2-diamine Chemical group C1=C(N)C(N)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(N)C(N)=C1 CPFKVVLZVUUQGV-UHFFFAOYSA-N 0.000 claims 2
- PHQYMDAUTAXXFZ-UHFFFAOYSA-N 4-[2-(4-carboxyphenyl)-1,1,1,3,3,3-hexafluoropropan-2-yl]benzoic acid Chemical group C1=CC(C(=O)O)=CC=C1C(C(F)(F)F)(C(F)(F)F)C1=CC=C(C(O)=O)C=C1 PHQYMDAUTAXXFZ-UHFFFAOYSA-N 0.000 claims 2
- 125000001142 dicarboxylic acid group Chemical group 0.000 claims 1
- 239000006082 mold release agent Substances 0.000 claims 1
- 238000007493 shaping process Methods 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 33
- 239000008188 pellet Substances 0.000 description 27
- 230000015572 biosynthetic process Effects 0.000 description 22
- 238000003786 synthesis reaction Methods 0.000 description 22
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 22
- 239000000843 powder Substances 0.000 description 21
- 229910052757 nitrogen Inorganic materials 0.000 description 17
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 17
- 125000003118 aryl group Chemical group 0.000 description 13
- 150000003839 salts Chemical class 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- -1 aldehyde bisulfite Chemical class 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 10
- 238000009835 boiling Methods 0.000 description 9
- 238000006116 polymerization reaction Methods 0.000 description 9
- 239000002244 precipitate Substances 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 229920000137 polyphosphoric acid Polymers 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 238000000465 moulding Methods 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- PHPIMLZTBYCDSX-UHFFFAOYSA-N 3-ethynylbenzoic acid Chemical compound OC(=O)C1=CC=CC(C#C)=C1 PHPIMLZTBYCDSX-UHFFFAOYSA-N 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 125000000217 alkyl group Chemical group 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 description 5
- ZCVOVYKBJCUUKC-UHFFFAOYSA-M sodium (3-ethynylphenyl)-hydroxymethanesulfonate Chemical compound C(#C)C=1C=C(C=CC=1)C(S(=O)(=O)[O-])O.[Na+] ZCVOVYKBJCUUKC-UHFFFAOYSA-M 0.000 description 5
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 4
- 239000000908 ammonium hydroxide Substances 0.000 description 4
- 235000010290 biphenyl Nutrition 0.000 description 4
- 239000004305 biphenyl Substances 0.000 description 4
- 238000004132 cross linking Methods 0.000 description 4
- PTGTZZGUTNGGEV-UHFFFAOYSA-L disodium;hydroxy-[3-[hydroxy(sulfonato)methyl]phenyl]methanesulfonate Chemical compound [Na+].[Na+].[O-]S(=O)(=O)C(O)C1=CC=CC(C(O)S([O-])(=O)=O)=C1 PTGTZZGUTNGGEV-UHFFFAOYSA-L 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 238000005259 measurement Methods 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- SJXHLZCPDZPBPW-UHFFFAOYSA-N 4-ethynylbenzoic acid Chemical compound OC(=O)C1=CC=C(C#C)C=C1 SJXHLZCPDZPBPW-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 239000006057 Non-nutritive feed additive Substances 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- KKEYFWRCBNTPAC-UHFFFAOYSA-N Terephthalic acid Chemical compound OC(=O)C1=CC=C(C(O)=O)C=C1 KKEYFWRCBNTPAC-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 150000005690 diesters Chemical class 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- QQVIHTHCMHWDBS-UHFFFAOYSA-N isophthalic acid Chemical compound OC(=O)C1=CC=CC(C(O)=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-N 0.000 description 2
- 238000003754 machining Methods 0.000 description 2
- 238000000691 measurement method Methods 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 2
- XKZGIJICHCVXFV-UHFFFAOYSA-N 2-ethylhexyl diphenyl phosphite Chemical compound C=1C=CC=CC=1OP(OCC(CC)CCCC)OC1=CC=CC=C1 XKZGIJICHCVXFV-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- HSTOKWSFWGCZMH-UHFFFAOYSA-N 3,3'-diaminobenzidine Chemical group C1=C(N)C(N)=CC=C1C1=CC=C(N)C(N)=C1 HSTOKWSFWGCZMH-UHFFFAOYSA-N 0.000 description 1
- HEMGYNNCNNODNX-UHFFFAOYSA-N 3,4-diaminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1N HEMGYNNCNNODNX-UHFFFAOYSA-N 0.000 description 1
- DPZVOQSREQBFML-UHFFFAOYSA-N 3h-pyrrolo[3,4-c]pyridine Chemical group C1=NC=C2CN=CC2=C1 DPZVOQSREQBFML-UHFFFAOYSA-N 0.000 description 1
- JKETWUADWJKEKN-UHFFFAOYSA-N 4-(3,4-diaminophenyl)sulfonylbenzene-1,2-diamine Chemical compound C1=C(N)C(N)=CC=C1S(=O)(=O)C1=CC=C(N)C(N)=C1 JKETWUADWJKEKN-UHFFFAOYSA-N 0.000 description 1
- WVDRSXGPQWNUBN-UHFFFAOYSA-N 4-(4-carboxyphenoxy)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1OC1=CC=C(C(O)=O)C=C1 WVDRSXGPQWNUBN-UHFFFAOYSA-N 0.000 description 1
- NEQFBGHQPUXOFH-UHFFFAOYSA-N 4-(4-carboxyphenyl)benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C1=CC=C(C(O)=O)C=C1 NEQFBGHQPUXOFH-UHFFFAOYSA-N 0.000 description 1
- ILPWTQGYOZFLBN-UHFFFAOYSA-N 4-[(3,4-diaminophenyl)methyl]benzene-1,2-diamine Chemical compound C1=C(N)C(N)=CC=C1CC1=CC=C(N)C(N)=C1 ILPWTQGYOZFLBN-UHFFFAOYSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000002947 alkylene group Chemical group 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- ANUAIBBBDSEVKN-UHFFFAOYSA-N benzene-1,2,4,5-tetramine Chemical compound NC1=CC(N)=C(N)C=C1N ANUAIBBBDSEVKN-UHFFFAOYSA-N 0.000 description 1
- LOOOVFMUCRDVNN-UHFFFAOYSA-N bicyclo[4.2.0]octa-1(6),2,4-triene-4-carboxylic acid Chemical compound OC(=O)C1=CC=C2CCC2=C1 LOOOVFMUCRDVNN-UHFFFAOYSA-N 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 150000001923 cyclic compounds Chemical class 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 229910003460 diamond Inorganic materials 0.000 description 1
- 239000010432 diamond Substances 0.000 description 1
- 150000001991 dicarboxylic acids Chemical class 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- GWZCCUDJHOGOSO-UHFFFAOYSA-N diphenic acid Chemical compound OC(=O)C1=CC=CC=C1C1=CC=CC=C1C(O)=O GWZCCUDJHOGOSO-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- UIMPSNPKHVULAQ-UHFFFAOYSA-N naphthalene-1,2,5,6-tetramine Chemical compound NC1=C(N)C=CC2=C(N)C(N)=CC=C21 UIMPSNPKHVULAQ-UHFFFAOYSA-N 0.000 description 1
- ABMFBCRYHDZLRD-UHFFFAOYSA-N naphthalene-1,4-dicarboxylic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=C(C(O)=O)C2=C1 ABMFBCRYHDZLRD-UHFFFAOYSA-N 0.000 description 1
- DKYADULMEONPQX-UHFFFAOYSA-N naphthalene-2,3,6,7-tetramine Chemical compound NC1=C(N)C=C2C=C(N)C(N)=CC2=C1 DKYADULMEONPQX-UHFFFAOYSA-N 0.000 description 1
- RXOHFPCZGPKIRD-UHFFFAOYSA-N naphthalene-2,6-dicarboxylic acid Chemical compound C1=C(C(O)=O)C=CC2=CC(C(=O)O)=CC=C21 RXOHFPCZGPKIRD-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 238000010943 off-gassing Methods 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- ZHEDAEGYRDOQER-UHFFFAOYSA-M sodium (4-ethynylphenyl)-hydroxymethanesulfonate Chemical compound C(#C)C1=CC=C(C=C1)C(S(=O)(=O)[O-])O.[Na+] ZHEDAEGYRDOQER-UHFFFAOYSA-M 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/18—Benzimidazoles; Hydrogenated benzimidazoles with aryl radicals directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/18—Polybenzimidazoles
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08L79/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
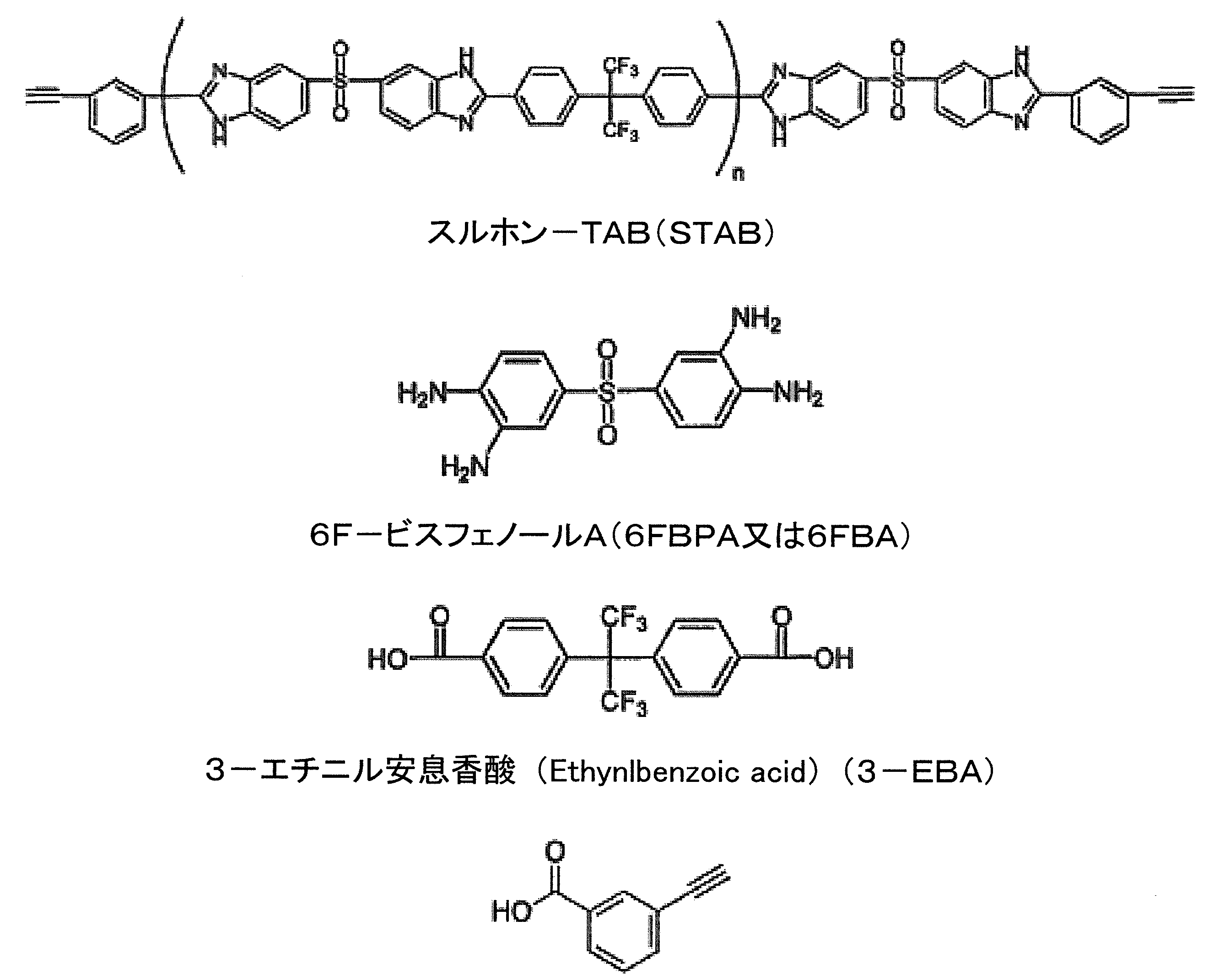
。次の一般化された式は、PBIオリゴマーを形成する際に起こる縮合反応を示す。
テトラアミン + ジカルボン酸成分 + 反応性末端基部分 → PBIオリゴマー。

ルである。


る。イソフタル酸、テレフタル酸、4,4’-ビフェニルジカルボン酸、1,4-ナフタレン-ジカルボン酸、ジフェニル酸(2,2’-ビフェニルジカルボン酸)、フェニルインダンジカルボン酸、1,6-ナフタレンジカルボン(napthalenedicarboxylic)酸、2,6-ナフタレンジカルボン酸、4,4’-ジフェニルエーテルジカルボン酸、4,4’-ジフェニルスルホンジカルボン酸、4,4’-ジフェニルチオエーテルジカルボン酸。ジカルボン酸成分は次の組み合わせのうちの1つであることができる。(1)少なくとも1つの遊離ジカルボン酸とジカルボン酸の少なくとも1つのジフェニルエステル、(2)少なくとも1つの遊離ジカルボン酸とジカルボン酸の少なくとも1つのジアルキルエステル、(3)ジカルボン酸の少なくとも1つのジフェニルエステルとジカルボン酸の少なくとも1つのジアルキルエステル、及び(4)ジカルボン酸の少なくとも1つのジアルキルエステル。各組み合わせの化合物のジカルボキシル部分は、同じであっても異なっていてもよく、組み合わせ(2)、(3)及び(4)のアルキルエステルのアルキル基は、一般に1~5個の炭素原子を含み、最も好ましくはメチルである。ジカルボン酸成分は、1モル当たりに対して約1モルの全ジカルボン酸成分の比率で用いることができ、または芳香族テトラアミンを用いることができる。しかし、特定の重合系における反応物の最適な比率は、当技術分野の当業者によって容易に決定することができる。

(3-エチニルフェニル)(ヒドロキシ)メタンスルホン酸ナトリウム(3EHMS):





合成実施例(Synthesis Example)(「合成実施例(Svn. Ex.)」)1 目標分子量が3
,000Daのエチニル末端m-PBIオリゴマーの合成
目標分子量が3,000Daのエチニル末端m-PBIオリゴマー(DP=9)を次のように合成した。TAB(10mmol、2.1427g)、IBA(8.86mmol、3.034g)、3EHMS(2.27mmol、0.532g)、及びDMAc(又はN,N’-ジメチルアセトアミド)(28mL)を、メカニカルスターラー、冷却器、窒素入口及びディーン・スタークトラップを備えた150mL容の三口丸底フラスコに充填した。この混合物を160~165℃のオイルバス中で48時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、30分間攪拌した。沈殿物を濾過し、次いで沸騰したDl水(500ml)中で4時間撹拌した。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.24dL/g。



分子量が5,000Daのエチニル末端m-PBIオリゴマー(DP=15)を次のように合成した。TAB(10mmol、2.1427g)、IBA(9.35mmol、3.2g)、3EHMS(1.31mmol、0.306g)、及びDMAc(28mL)を、メカニカルスターラー、冷却器、窒素入口及びディーン・スタークトラップを備えた150mL容の三口丸底フラスコに充填した。この混合物を160~165℃のオイルバス中で48時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、30分間攪拌した。沈殿物を濾過し、次いで沸騰したDl水(500ml)中で4時間撹拌した。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.37dL/g。

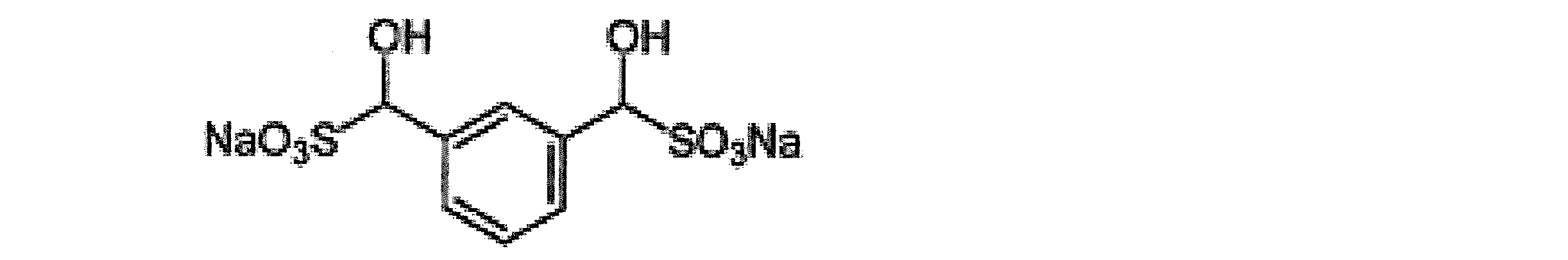

分子量が7,000Daのエチニル末端m-PBIオリゴマー(DP=22)を次のように合成した。TAB(10mmol、2.1427g)、IBA(9.54mmol、3.265g)、3EHMS(0.92mmol、0.215g)、及びDMAc(28ml)を、メカニカルスターラー、冷却器、窒素入口及びディーン・スタークトラップを備えた150mL容の三口丸底フラスコに充填した。この混合物を160~165℃のオイルバス中で48時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、30分間攪拌した。沈殿物を濾過し、次いで沸騰したDl水(500ml)中で4時間撹拌した。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.44dL/g。2回目の実験を同一条件下で行い、同様の粉末を生成した。IV(H2SO4、於23℃)=0.46dL/g。



目標分子量が10,000Daのエチニル末端m-PBIオリゴマー(DP=31)を次のように合成した。TAB(10mmol、2.1427g)、IBA(9.97mmol、3.314g)、3EHMS(0.63mmol、0.1485g)、及びDMAc(28mL)を、メカニカルスターラー、冷却器、窒素入口及びディーン・スタークトラップを備えた150mL容の三口丸底フラスコに充填した。この混合物を160~165℃のオイルバス中で48時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、30分間攪拌した。沈殿物を濾過し、次いで沸騰したDl水(500ml)中で4時間撹拌した。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.51dL/g。



目標分子量が1,200Daのエチニル末端m-PBIオリゴマー(DP=2)を次のように合成した。STAB(10mmol、2.7833g)、IPA(5.8mmol、0.964g)、3EBA(6.6mmol、0.97g)、及びPPA、又はポリリン酸(60g)を、メカニカルスターラー、窒素入口及び窒素出口を備えた150mL容の三口円筒形ケトルフラスコに充填した。この混合物をオイルバス中で、50℃で1時間、120℃で6時間、170℃で10時間、190℃で2時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、ブレンダーで15分間混ぜて、微粉末とした。沈殿物を濾過し、次いでDl水(500ml)中で2時間撹拌した。この溶液に水酸化アンモニウムを加えて、溶液を中和した。中和した溶液を濾過し、固形粉末を沸
騰した水(500ml)中でさらに2時間撹拌し、生成した塩を溶かした。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.20dL/g。


目標分子量が5,000Daのエチニル末端m-PBIオリゴマー(DP=13)を次のように合成した。STAB(10mmol、2,7833g)、IPA(9.2mmol、1.53g)、3EBA(1.5mmol、0.223g)、及びPPA(57g)を、メカニカルスターラー、窒素入口及び窒素出口を備えた150mL容の三口円筒形ケトルフラスコに充填した。この混合物をオイルバス中で、50℃で1時間、120℃で6時間、170℃で10時間、190℃で2時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、ブレンダーで15分間混ぜて、微粉末とした。沈殿物を濾過し、次いでDl水(500ml)中で2時間撹拌した。この溶液に水酸化アンモ
ニウムを加えて、溶液を中和した。中和した溶液を濾過し、固形粉末を沸騰した水(500ml)中でさらに2時間撹拌し、生成した塩を溶かした。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.31dL/g。



目標分子量が2,000DaのSTAB-6FBPAエチニル末端オリゴマー(DP=3)を次のように合成した。STAB(5mmol、1.391g)、6FBA(3.3mmol、1.3g)、3EBA(2.77mmol、0.4g)、及びPPA(103g)を、メカニカルスターラー、窒素入口及び窒素出口を備えた150mL容の三口円筒形ケトルフラスコに充填した。この混合物をオイルバス中で、50℃で1時間、120℃で6時間、170℃で10時間、190℃で2時間撹拌して重合を完了させた。得られた
溶液をDl水(500ml)に沈殿させ、ブレンダーで15分間混ぜて、微粉末とした。沈殿物を濾過し、次いでDl水(500ml)中で2時間撹拌した。この溶液に水酸化アンモニウムを加えて、溶液を中和した。中和した溶液を濾過し、固形粉末を沸騰した水(500ml)中でさらに2時間撹拌し、生成した塩を溶かした。最後のステップをさらに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.39dL/g。

目標分子量が4,000DaのSTAB-6FBPAエチニル末端オリゴマー(DP=6)を次のように合成した。STAB(5mmol、1.391g)、6FBA(4.2mmol、1.65g)、3EBA(1.5mmol、0.215g)、及びPPA(108g)を、メカニカルスターラー、窒素入口及び窒素出口を備えた150mL容の三口円筒形ケトルフラスコに充填した。この混合物をで、50℃で1時間、120℃で6時間、170℃で10時間、190℃で2時間撹拌して重合を完了させた。得られた溶液をDl水(500ml)に沈殿させ、ブレンダーで15分間混ぜて、微粉末とした。沈殿物を濾過し、次いでDl水(500ml)中で2時間撹拌した。この溶オイルバス中液に水酸化アンモニウムを加えて、溶液を中和した。中和した溶液を濾過し、固形粉末を沸騰した水(500ml)中でさらに2時間撹拌し、生成した塩を溶かした。最後のステップをさ
らに1回繰り返し、残留塩をすべて溶かした。濾過してすぐに、固体粉末を減圧下、真空オーブン中で120℃にて12時間乾燥させた。IV(H2SO4、於23℃)=0.46dL/g。
直径0.5および0.78インチの大きさの試料のペレットを、さまざまな条件下(温度135~190℃、圧力25~120kpsi[170~830キロパスカル])で、ハイドロリック圧縮機を使用して調製した。ペレット調製物を調べると、色が淡い茶色から濃い茶色に及ぶ生成物が生じていた。エチニル末端m-PBI末端オリゴマーの温度がおよそ150~160℃、圧力が25~60kpsi[170~415キロパスカル]でのペレットの調製により、光沢のある滑らかな表面をもつ均質で均一な融合試料(fused sample)が得られた。同じ結果が、温度135~160℃、圧力25~30kpsi[170~207.5キロパスカル]でのSTABをベースとするオリゴマーについても観察された。ペレットを作製してすぐに、窒素ガス環境下で300~500℃の範囲の温度で硬化させた。硬化前、ペレットはすべて室温で濃硫酸に溶け、24時間未満で溶解した。硬化後、試料は低い溶解度を示すか、濃硫酸および沸騰DMAcに完全に不溶であった。
ペレットの調製に記載のエチニル末端m-PBIオリゴマーの加工条件の例を表1に示す。


機械的な実施例(Mechanical Example)1 目標分子量が7,000Daのメタ-エチニル末端m-PBIオリゴマーの機械的特性
目標分子量が7,000Daのメタ-エチニルm-PBIオリゴマーを、合成実施例3
に従って作製した。その粉末をイヌの骨の形をした金型に充填し、160℃で3時間、10トンの圧力を用いて加熱プレスした。成形したイヌの骨を金型から取り出し、続いて窒素オーブン中で450℃にて1時間硬化させた。イヌの骨の厚さは1.86mmであった。機械的試験機を使用してこの材料を試験し、破断応力63.5MPa、破断伸び2.1%、初期モジュラス3437MPaの特性を示した。
目標分子量が7,000Daのパラ-エチニルm-PBIオリゴマーを合成実施例3に従って作製した。その粉末をイヌの骨の形をした金型に充填し、160℃で3時間、10トンの圧力を用いて加熱プレスした。成形したイヌの骨を金型から取り出し、続いて窒素オーブン中で450℃にて1時間硬化させた。イヌの骨の厚さは2.04mmであった。機械的試験機を使用してこの材料を試験し、破断応力36.5MPa、破断伸び1.4%、初期モジュラス2823MPaの特性を示した。
目標分子量が10,000Daのパラ-エチニルm-PBIオリゴマーを合成実施例4に従って作製した。その粉末をイヌの骨の形をした金型に充填し、160℃で3時間、10トンの圧力を用いて加熱プレスした。成形したイヌの骨を金型から取り出し、続いて窒素オーブン中で450℃にて1時間硬化させた。イヌの骨の厚さは2.25mmであった。機械的試験機を使用してこの材料を試験し、破断応力77.0MPa、破断伸び3.4%、初期モジュラス2511MPaの特性を示した。
目標分子量が15,000Daのパラ-エチニルm-PBIオリゴマーを合成実施例4に従って、次の量の単量体を使用して分子量を調整することによって作製した。10.7135gのTAB、16.7535gのIBA、及び0.4906gの4EHMS。その粉末をイヌの骨の形をした金型に充填し、160℃で3時間、10トンの圧力を用いて加熱プレスした。成形したイヌの骨を金型から取り出し、続いて窒素オーブン中で450℃にて1時間硬化させた。イヌの骨の厚さは2.54mmであった。機械的試験機を使用してこの材料を試験し、破断応力43.4MPa、破断伸び1.9%、初期モジュラス2221MPaの特性を示した。
硬化実施例1 Mn=5,000Daのペレット実施例2に記載のTABをベースとするオリゴマーから作製したペレットを350℃で2時間硬化させた。試料を硫酸中に24時間置いたが、試料の大部分は溶けずに残った。
24時間置いたが、試料の大部分は溶けずに残った。
CELAZOLE U-60製品は多段階過程で製造される。U-60の原料は、100メッシュのスクリーンを通過するポリベンズイミダゾール樹脂(高分子量、非末端基官能化)である。不活性環境で160℃に4時間加熱することによって樹脂を完全に乾燥させる。次いで、本質的に水を含まない乾燥樹脂を成形ツール中に置き、そこで最終的に非常に高い熱(>420℃で>8時間)に供する。
0.050gのポリベンズイミダゾールを25ml容のメスフラスコに加える。0.2g/dLの最終濃度で、フラスコを濃硫酸で満たす。ポリベンズイミダゾールがすべて溶けるまで、フラスコを機械的シェーカー上で振盪する。ポリベンズイミダゾール溶液を0.45pm PTFEシリンジフィルターを通して濾過し、200pmウベローデ粘度計に加える。その粘度計を23.0℃の水浴に置き、30分間平衡化させる。連続する3回が0.1秒以内になるまで測定を記録する。これら3回の平均値を使用して、次式を用いて固有粘度を計算する。

t(秒):溶液フロー時間
t0(秒):溶媒フロー時間(96%硫酸)
C(g/dL):溶液濃度
機械的特性はASTM D638に従って測定した。
Claims (5)
- 少なくとも2つのアルキニル末端基を有するポリベンズイミダゾールオリゴマーを含み、前記オリゴマーが、0.015~0.60dL/gのIV範囲又は1000~15,000ダルトン(Da)の分子量を有し、
前記ポリベンズイミダゾールオリゴマーが、テトラアミン、ジカルボン酸成分、及び前記アルキニル末端基に至る反応性末端基部分の反応生成物であり、
前記ジカルボン酸成分は、2,2-ビス(4-カルボキシフェニル)ヘキサフルオロプロパンであり、前記テトラアミンは2,2-ビス(3,4-ジアミノフェニル)ヘキサフルオロプロパンである、ポリベンズイミダゾール樹脂。 - フィラー、難燃剤、難燃助剤、可塑剤、酸化防止剤、離型剤、耐光剤、耐候剤、着色剤、顔料、改質剤、帯電防止剤、加水分解阻害剤、及び補強剤からなる群より選択される1つ又は複数の添加剤をさらに含む、請求項1に記載のポリベンズイミダゾール樹脂。
- 少なくとも2つの反応性アルキニル末端基をもつポリベンズイミダゾールオリゴマーを製造する方法であって、
2,2-ビス(3,4-ジアミノフェニル)ヘキサフルオロプロパン、ジカルボン酸成分及び反応性末端基部分を溶媒中で室温より高い温度で一定時間反応させるステップ、
反応後に前記溶媒から0.015~0.60dL/gのIV範囲又は1000~15,000ダルトン(Da)の分子量を有する前記オリゴマーを沈殿させるステップ、並びに
前記オリゴマーから反応副生成物をすべて除去するステップ
を含み、
前記ジカルボン酸成分はフェニルインダンジカルボン酸及び2,2-ビス(4-カルボキシフェニル)ヘキサフルオロプロパンの少なくとも一方である、方法。 - 成形ポリベンズイミダゾール物品を製造する方法であって、
請求項1に記載されるポリベンズイミダゾールオリゴマーを成形するステップ、
成形されたポリベンズイミダゾールオリゴマーを、前記末端基を反応させるのに十分な温度で硬化させるステップ、及び
前記成形ポリベンズイミダゾール物品を得るステップ
を含む、方法。 - 前記少なくとも2つの反応性アルキニル末端基をもつポリベンズイミダゾールオリゴマーが、フィラー、難燃剤、難燃助剤、可塑剤、酸化防止剤、離型剤、耐光剤、耐候剤、着色剤、顔料、改質剤、帯電防止剤、加水分解阻害剤、及び補強剤からなる群より選択される1つ又は複数の添加剤と混合される、請求項4に記載の方法。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862685315P | 2018-06-15 | 2018-06-15 | |
| US62/685,315 | 2018-06-15 | ||
| US16/432,994 | 2019-06-06 | ||
| US16/432,994 US10934395B2 (en) | 2018-06-15 | 2019-06-06 | Polybenzimidazole oligomers with reactive end groups |
| PCT/US2019/035926 WO2019241046A1 (en) | 2018-06-15 | 2019-06-07 | Polybenzimidazole oligomers with reactive end groups |
| JP2021507516A JP2021529875A (ja) | 2018-06-15 | 2019-06-07 | 反応性末端基を有するポリベンズイミダゾールオリゴマー |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021507516A Division JP2021529875A (ja) | 2018-06-15 | 2019-06-07 | 反応性末端基を有するポリベンズイミダゾールオリゴマー |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023026598A JP2023026598A (ja) | 2023-02-24 |
| JP7585359B2 true JP7585359B2 (ja) | 2024-11-18 |
Family
ID=68839181
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021507516A Pending JP2021529875A (ja) | 2018-06-15 | 2019-06-07 | 反応性末端基を有するポリベンズイミダゾールオリゴマー |
| JP2023000307A Active JP7585359B2 (ja) | 2018-06-15 | 2023-01-04 | 反応性末端基を有するポリベンズイミダゾールオリゴマー |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021507516A Pending JP2021529875A (ja) | 2018-06-15 | 2019-06-07 | 反応性末端基を有するポリベンズイミダゾールオリゴマー |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10934395B2 (ja) |
| EP (1) | EP3807340A4 (ja) |
| JP (2) | JP2021529875A (ja) |
| KR (1) | KR102503565B1 (ja) |
| CN (1) | CN112543781B (ja) |
| WO (1) | WO2019241046A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4397999A (en) | 1979-08-21 | 1983-08-09 | Siemens Aktiengesellschaft | Polyimidazole and polyimidazopyrrolone precursor stages and the preparation thereof |
| JP2009108036A (ja) | 2007-09-28 | 2009-05-21 | Fujifilm Corp | 新規アセチレン化合物、その製造方法、それを構成単位として含むポリマー、該化合物及び/又は該ポリマーを含む組成物、該組成物を硬化させてなる硬化物 |
| JP2010006760A (ja) | 2008-06-27 | 2010-01-14 | Fujifilm Corp | 新規アセチレン化合物縮合物、重縮合物、それらを含む組成物、及びその硬化物 |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3969430A (en) * | 1974-11-18 | 1976-07-13 | Celanese Corporation | Processability of intractable polymers |
| US4672104A (en) * | 1984-08-31 | 1987-06-09 | Celanese Corporation | Two stage polybenzimidazole process and product |
| US4912176A (en) | 1987-09-03 | 1990-03-27 | Hoechst Celanese Corporation | Sintered polybenzimidazole/polyaryleneketone articles and method of manufacture |
| US5089592A (en) * | 1988-06-09 | 1992-02-18 | The Dow Chemical Company | Biscyclobutarene monomer comprising two cyclobutarene moieties bridged by a divalent radical comprising at least one benzothiazole or benzimidazole linkage |
| US5089568A (en) | 1988-10-11 | 1992-02-18 | The Dow Chemical Company | Process of making thermoplastic copolymers containing polybenzoxazole, polybenzothiazole and polybenzimidazole moieties |
| DE3912922A1 (de) * | 1989-04-20 | 1990-10-25 | Merck Patent Gmbh | Verzweigte, dipolar orientierte polymermaterialien |
| JPH0693117A (ja) * | 1992-09-11 | 1994-04-05 | Kanegafuchi Chem Ind Co Ltd | 耐熱積層材料及びその製造方法 |
| US7259230B2 (en) * | 2004-06-07 | 2007-08-21 | Battelle Energy Alliance, Llc | Polybenzimidazole compounds, polymeric media, and methods of post-polymerization modifications |
| WO2006104974A1 (en) * | 2005-03-28 | 2006-10-05 | E.I. Du Pont De Nemours And Company | Process for the production of polyareneazole polymer |
| US20090004508A1 (en) | 2007-06-14 | 2009-01-01 | Daicel Chemical Industries, Ltd. | Thin-film materials, thin films and producing method thereof |
| EP2481119A1 (en) | 2009-09-24 | 2012-08-01 | EWE-Forschungszentrum Für Energietechnologie E.V. | Proton exchange membrane comprising polymer blends for use in high temperature proton exchange membrane fuel cells |
| US20110189581A1 (en) | 2010-02-04 | 2011-08-04 | Samsung Electronics Co., Ltd. | Compound, cross-linked material thereof, double cross-linked polymer thereof, and electrolyte membrane, electrode for fuel cell and fuel cell including same |
| US20110189484A1 (en) * | 2010-02-04 | 2011-08-04 | Hopkins Jr John B | Porous polybenzimidazole resin and method of making same |
| US9598541B2 (en) * | 2013-06-04 | 2017-03-21 | Pbi Performance Products, Inc. | Method of making polybenzimidazole |
-
2019
- 2019-06-06 US US16/432,994 patent/US10934395B2/en active Active
- 2019-06-07 EP EP19819777.4A patent/EP3807340A4/en active Pending
- 2019-06-07 WO PCT/US2019/035926 patent/WO2019241046A1/en not_active Ceased
- 2019-06-07 JP JP2021507516A patent/JP2021529875A/ja active Pending
- 2019-06-07 CN CN201980051341.2A patent/CN112543781B/zh active Active
- 2019-06-07 KR KR1020217001466A patent/KR102503565B1/ko active Active
-
2023
- 2023-01-04 JP JP2023000307A patent/JP7585359B2/ja active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4397999A (en) | 1979-08-21 | 1983-08-09 | Siemens Aktiengesellschaft | Polyimidazole and polyimidazopyrrolone precursor stages and the preparation thereof |
| JP2009108036A (ja) | 2007-09-28 | 2009-05-21 | Fujifilm Corp | 新規アセチレン化合物、その製造方法、それを構成単位として含むポリマー、該化合物及び/又は該ポリマーを含む組成物、該組成物を硬化させてなる硬化物 |
| JP2010006760A (ja) | 2008-06-27 | 2010-01-14 | Fujifilm Corp | 新規アセチレン化合物縮合物、重縮合物、それらを含む組成物、及びその硬化物 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2019241046A1 (en) | 2019-12-19 |
| JP2023026598A (ja) | 2023-02-24 |
| KR20210020144A (ko) | 2021-02-23 |
| US10934395B2 (en) | 2021-03-02 |
| EP3807340A4 (en) | 2022-04-20 |
| EP3807340A1 (en) | 2021-04-21 |
| US20190382531A1 (en) | 2019-12-19 |
| CN112543781A (zh) | 2021-03-23 |
| CN112543781B (zh) | 2024-02-20 |
| KR102503565B1 (ko) | 2023-02-24 |
| JP2021529875A (ja) | 2021-11-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0388803A2 (en) | Thermoplastic compositions containing polybenzoxazole; polybenzothiazole and polybenzimidazole moieties and process for making shaped articles therefrom | |
| KR20150135737A (ko) | 공중합 폴리아미드 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| KR101771781B1 (ko) | 폴리아미드에스테르 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| JP7585359B2 (ja) | 反応性末端基を有するポリベンズイミダゾールオリゴマー | |
| EP0415276A1 (en) | Branched polybenzazole polymers and method of preparation | |
| JPH0443928B2 (ja) | ||
| US6974857B1 (en) | End-capped quinoxaline-containing hyperbranched aromatic poly(ether-ketones) | |
| JPS6339616B2 (ja) | ||
| JP2008505209A (ja) | 固相重合工程が改良された2段階溶融重合法によるポリベンゾイミダゾールの製造方法。 | |
| JP2008505209A5 (ja) | ||
| JPS6322833A (ja) | 2段階ポリベンズイミダゾ−ル法の改良 | |
| US7790831B1 (en) | Hyperbranched polybenzazoles via an A3+ B2 polymerization | |
| JPS62252427A (ja) | 熱可塑的に加工できる芳香族ポリアミドおよび−ポリアミドイミドの製造方法 | |
| KR101557535B1 (ko) | 공중합 폴리아미드 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| KR101557543B1 (ko) | 폴리아미드 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| KR101987540B1 (ko) | 공중합 폴리아미드 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| KR20160017197A (ko) | 공중합 폴리아미드 수지, 이의 제조방법 및 이를 포함하는 성형품 | |
| JP7291518B2 (ja) | アスパラギン酸-乳酸共重合体の製造方法 | |
| JPH04502168A (ja) | 複素環式ポリマーの改良合成方法 | |
| KR102263527B1 (ko) | 카프로락탐을 기재로 하는 반-방향족 코폴리아미드 | |
| US5140092A (en) | Rigid-rod benzimidazole pendant benzobisazo polymer | |
| JPH04114062A (ja) | 芳香族ポリチアゾールの分子複合材の製造方法 | |
| JP2009500509A5 (ja) | ||
| Mallakpour et al. | Rapid formation of optically active and organosoluble polyamides containing L-alaninephthalimide side chain via microwave irradiation | |
| JPH04202528A (ja) | 含フッ素ポリベンツオキサゾール及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20230127 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230127 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20231107 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20231121 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240219 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240409 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240705 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20241008 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20241106 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7585359 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |