JP5741641B2 - 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 - Google Patents
感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 Download PDFInfo
- Publication number
- JP5741641B2 JP5741641B2 JP2013148919A JP2013148919A JP5741641B2 JP 5741641 B2 JP5741641 B2 JP 5741641B2 JP 2013148919 A JP2013148919 A JP 2013148919A JP 2013148919 A JP2013148919 A JP 2013148919A JP 5741641 B2 JP5741641 B2 JP 5741641B2
- Authority
- JP
- Japan
- Prior art keywords
- photosensitive resin
- film
- acid
- resin composition
- pattern
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- MCACTZQMAQDCBI-UHFFFAOYSA-N Cc(cc1Cc(cc(C)cc2COC)c2O)cc(COC)c1O Chemical compound Cc(cc1Cc(cc(C)cc2COC)c2O)cc(COC)c1O MCACTZQMAQDCBI-UHFFFAOYSA-N 0.000 description 1
- YXFVVABEGXRONW-UHFFFAOYSA-N Cc1ccccc1 Chemical compound Cc1ccccc1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 1
- BGQWVIOOEFRPOV-UHFFFAOYSA-N OCc1cc(C(C(F)(F)F)(C(F)(F)F)c(cc2CO)cc(CO)c2O)cc(CO)c1O Chemical compound OCc1cc(C(C(F)(F)F)(C(F)(F)F)c(cc2CO)cc(CO)c2O)cc(CO)c1O BGQWVIOOEFRPOV-UHFFFAOYSA-N 0.000 description 1
- MWPXMEJTTYFHGR-UHFFFAOYSA-N OCc1cc(Cc(cc2CO)cc(CO)c2O)cc(CO)c1O Chemical compound OCc1cc(Cc(cc2CO)cc(CO)c2O)cc(CO)c1O MWPXMEJTTYFHGR-UHFFFAOYSA-N 0.000 description 1
Images




Landscapes
- Materials For Photolithography (AREA)
- Polyamides (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
Description
さらに、本発明は、良好な形状と特性のパターンを有し、さらには低温プロセスで硬化可能な感光性樹脂組成物を用いることにより、デバイスへのダメージが避けられ、信頼性の高い電子部品を歩留まり良く提供するものである。

まず、本発明による低温硬化用の感光性樹脂組成物について説明する。本発明による感光性樹脂組成物は、(a)一般式(I):
((a)成分:ポリベンゾオキサゾール前駆体およびポリベンゾオキサゾール前駆体の共重合体)
ポリベンゾオキサゾール前駆体およびポリベンゾオキサゾール前駆体の共重合体は、通常、アルカリ水溶液で現像するので、アルカリ水溶液可溶性であることが好ましい。本発明においては、例えば、前記一般式(I)、(III)以外のポリベンゾオキサゾール前駆体ではないポリアミドの構造、ポリベンゾオキサゾールの構造、ポリイミドやポリイミド前駆体(ポリアミド酸やポリアミド酸エステル)の構造を、前記一般式(I)、(III)のポリオキサゾール前駆体およびポリベンゾオキサゾール前駆体の共重合体の構造と共に有していても良い。
例えば、下記一般式(V)、(VI)で表されるポリイミド前駆体であることが好ましい。
ここで、式中のjとlのモル分率は、j=80〜100モル%、l=20〜0モル%であることがより好ましい。
ここで、式中のjとkとlのモル分率は、j+k=80〜100モル%、l=20〜0モル%であることがより好ましい。
ここで、式中のjとkのモル分率は、j=5〜85モル%、k=15〜95モル%であることがパターン性、機械特性、耐熱性、耐薬品性の点でより好ましい。
炭素数1〜30の脂肪族鎖状構造を含む基は、一般式(I)、(III)において、UとしてもVとして存在していても良い。脂肪族鎖状構造を含む基として、下記一般式(UV1)または(UV2)に示す構造を含む基であると、280℃の以下での脱水閉環率が高い点で望ましい。さらに炭素数7〜30の脂肪族直鎖構造であると、弾性率が低くかつ破断伸びが高く、より好ましい。
一般式(I)、(III)においてVで表される2価の有機基とは、一般に、ジアミンと反応してポリアミド構造を形成する、ジカルボン酸の残基である。
本発明において、Vとしては炭素数1〜30の脂肪族鎖状構造を含む基は、一般式(I)、(III)において、UとしてもVとしても存在していても良い。脂肪族鎖状構造を含む基としては、下記一般式(UV1)または(UV2)に示す構造を有すると、耐熱性、紫外及び可視光量域での高い透明性を有し、280℃の以下での脱水閉環率が高い点で優れる。さらに炭素数7〜30の脂肪族直鎖構造であると、そのポリマーはN−メチル−2−ピロリドン以外にもγ−ブチロラクトンやプロピレングリコールモノメチルエーテルアセテートといった溶剤にも易溶となり、保存安定性も高い。さらに弾性率が低くかつ破断伸びが高く、より好ましい。
本発明の感光性樹脂組成物は、上記ポリベンゾオキサゾール前駆体とともに(b)感光剤を含む。この感光剤とは、光に反応して、その組成物から形成された膜の現像液に対する機能を有するものである。本発明で(b)成分として用いられる感光剤に特に制限はないが、光により酸又はラジカルを発生するものであることが好ましい。
本発明に使用される(c)成分(溶剤)としては、γ−ブチロラクトン、乳酸エチル、プロピレングリコールモノメチルエーテルアセテート、酢酸ベンジル、n−ブチルアセテート、エトキシエチルプロピオネート、3−メチルメトキシプロピオネート、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、ヘキサメチルホスホリルアミド、テトラメチレンスルホン、シクロヘキサノン、シクロペンタノン、ジエチルケトン、ジイソブチルケトン、メチルアミルケトン等が挙げられる。
本発明において、さらに、(d)成分として熱酸発生剤(熱潜在酸発生剤)を使用することができる。熱酸発生剤を使用すると、ポリベンゾオキサゾール前駆体のフェノール性水酸基含有ポリアミド構造が脱水反応を起こして環化する際の触媒として効率的に働くので好ましい。また、本発明の約280℃の以下での脱水閉環率が高い特定の樹脂に、この熱酸発生剤を併用することにより、脱水環化反応をさらに低温化できるので、低温での硬化でも硬化後の膜の物性が、高温で硬化したものと遜色ない性能が得られる。
これらが好ましいのは、分解開始温度が150〜250℃の範囲にあり、280℃以下でのポリベンゾオキサゾール前駆体の環化脱水反応に際して効率的に分解するためである。これに対してトリフェニルスルホニウム塩は、本発明の熱酸発生剤としては望ましくない。トリフェニルスルホニウム塩は熱安定性が高く、一般に分解温度が300℃を超えているため、280℃以下でのポリベンゾオキサゾール前駆体の環化脱水反応に際しては分解が起きず、環化脱水の触媒としては十分に働かないと考えられるためである。
本発明による感光性樹脂組成物において、上記(a)〜(d)成分に加えて、(1)溶解促進剤、(2)溶解阻害剤、(3)密着性付与剤、(4)界面活性剤又はレベリング剤などの成分を配合しても良い。
本発明においては、さらに、(a)成分であるベース樹脂のアルカリ水溶液に対する溶解性を促進させる溶解促進剤、例えばフェノール性水酸基を有する化合物を含有させることができる。フェノール性水酸基を有する化合物は、加えることでアルカリ水溶液を用いて現像する際に露光部の溶解速度が増加し感度が上がり、また、パターン形成後の膜の硬化時に、膜の溶融を防ぐことができる。
本発明においては、さらに、(a)成分であるベース樹脂のアルカリ水溶液に対する溶解性を阻害する化合物である溶解阻害剤を含有させることができる。具体的には、ジフェニルヨードニウムニトラート、ビス(p−tert−ブチルフェニル)ヨードニウムニトラート、ジフェニルヨードニウムブロマイド、ジフェニルヨードニウムクロリド、ジフェニルヨードニウムヨーダイト等である。
本発明の感光性樹脂組成物は、硬化膜の基板との接着性を高めるために、有機シラン化合物、アルミキレート化合物等の密着性付与剤を含むことができる。
また、本発明の感光性樹脂組成物は、塗布性、例えばストリエーション(膜厚のムラ)を防いだり、現像性を向上させたりするために、適当な界面活性剤又はレベリング剤を添加することができる。
次に、本発明によるパターン硬化膜の製造方法について説明する。本発明のパターン硬化膜の製造方法は、上述した感光性樹脂組成物を支持基板上に塗布し乾燥して感光性樹脂膜を形成する感光性樹脂膜形成工程、前記感光性樹脂膜を所定のパターンに露光する露光工程、前記露光後の感光性樹脂膜を現像してパターン樹脂膜を得る現像工程、及び前記パターン樹脂膜を加熱処理してパターン硬化膜を得る加熱処理工程を経て、ポリベンゾオキサゾールのパターン硬化膜を得ることができる。以下、各工程について説明する。
まず、この工程では、ガラス基板、半導体、金属酸化物絶縁体(例えばTiO2、SiO2等)、窒化ケイ素などの支持基板上に、前記本発明の感光性樹脂組成物をスピンナーなどを用いて回転塗布後、ホットプレート、オーブンなどを用いて乾燥する。これにより、感光性樹脂組成物の被膜である感光性樹脂膜が得られる。
次に、露光工程では、支持基板上で被膜となった感光性樹脂膜に、マスクを介して紫外線、可視光線、放射線などの活性光線を照射することにより露光を行う。
現像工程では、活性光線が露光した感光性樹脂膜の露光部を現像液で除去することによりパターン硬化膜が得られる。現像液としては、例えば、水酸化ナトリウム,水酸化カリウム,ケイ酸ナトリウム,アンモニア,エチルアミン,ジエチルアミン,トリエチルアミン,トリエタノールアミン,テトラメチルアンモニウムヒドロキシドなどのアルカリ水溶液が好ましいものとして挙げられる。これらの水溶液の塩基濃度は、0.1〜10重量%とされることが好ましい。さらに上記現像液にアルコール類や界面活性剤を添加して使用することもできる。これらはそれぞれ、現像液100重量部に対して、好ましくは0.01〜10重量部、より好ましくは0.1〜5重量部の範囲で配合することができる。
次いで、加熱処理工程では、現像後得られたパターン樹脂膜を加熱処理することにより、オキサゾール環や他の官能基を有する耐熱性のポリオキサゾールのパターン硬化膜を形成することができる。加熱処理工程における加熱温度は、280℃以下、望ましくは120〜280℃、より望ましくは、160〜250℃である。
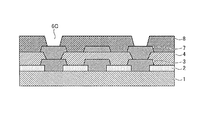
次に、本発明によるパターンの製造方法の一例として、半導体装置の製造工程を図面に基づいて説明する。図1〜図5は、多層配線構造を有する半導体装置の製造工程を説明する概略断面図であり、第1の工程から第5の工程へと一連の工程を表している。
なお、上記例において、層間絶縁膜を本発明の感光性樹脂組成物を用いて形成することも可能である。
次に、本発明による電子部品について説明する。本発明による電子部品は、上述した感光性樹脂組成物を用いて上記パターン硬化膜の製造方法によって形成されるパターン硬化膜を有する。ここで、電子部品としては、半導体装置や多層配線板、各種電子デバイス等を含む。特に、耐熱性の低いMRAM(磁気抵抗メモリ:Magnet Resistive Random Access Memory)が好ましいものとして挙げられる。
攪拌機、温度計を備えた0.2リットルのフラスコ中に、N−メチルピロリドン60gを仕込み、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン13.92gを添加し、攪拌溶解した。続いて、温度を0〜5℃に保ちながら、マロン酸ジクロリド5.64gを10分間で滴下した後、60分間攪拌を続けた。得られた溶液を3リットルの水に投入し、析出物を回収し、これを、純水で3回洗浄した後、減圧してポリヒドロキシアミド(ポリベンゾオキサゾール前駆体)を得た(以下、ポリマーIとする)。ポリマーIのGPC法標準ポリスチレン換算により求めた重量平均分子量は11,500、分散度は1.7であった。
合成例1で使用したマロン酸ジクロリドをスクシン酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーIIとする)の標準ポリスチレン換算により求めた重量平均分子量は20,400、分散度は1.8であった。
合成例1で使用したマロン酸ジクロリドをグルタル酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーIIIとする)の標準ポリスチレン換算により求めた重量平均分子量は12,800、分散度は1.7であった。
合成例1で使用したマロン酸ジクロリドをアジピン酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーIVとする)の標準ポリスチレン換算により求めた重量平均分子量は29,300、分散度は1.9であった。
合成例1で使用したマロン酸ジクロリドをスベリン酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーVとする)の標準ポリスチレン換算により求めた重量平均分子量は36,900、分散度は2.0であった。
合成例1で使用したマロン酸ジクロリドをセバシン酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーVIとする)の標準ポリスチレン換算により求めた重量平均分子量は33,100、分散度は2.0であった。
合成例1で使用したマロン酸ジクロリドをドデカン二酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーVIIとする)の標準ポリスチレン換算により求めた重量平均分子量は31,600、分散度は2.0であった。
合成例1で使用したマロン酸ジクロリドをジメチルマロン酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーVIIIとする)の標準ポリスチレン換算により求めた重量平均分子量は22,200、分散度は1.8であった。
合成例1で使用したマロン酸ジクロリドをヘキサフルオログルタル酸ジクロリドに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーVIVとする)の標準ポリスチレン換算により求めた重量平均分子量は10,700、分散度は1.6であった。
合成例1で使用したマロン酸ジクロリドをグルタル酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXとする)の標準ポリスチレン換算により求めた重量平均分子量は16,400、分散度は1.6であった。
合成例1で使用したマロン酸ジクロリドをスベリン酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXIとする)の標準ポリスチレン換算により求めた重量平均分子量は33,400、分散度は2.0であった。
合成例1で使用したマロン酸ジクロリドをセバシン酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXIIとする)の標準ポリスチレン換算により求めた重量平均分子量は28,800、分散度は1.9であった。
合成例1で使用したマロン酸ジクロリドをドデカン二酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXIIIとする)の標準ポリスチレン換算により求めた重量平均分子量は24,300、分散度は1.9であった。
合成例1で使用したマロン酸ジクロリドをジメチルマロン酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は、合成例1と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXIVとする)の標準ポリスチレン換算により求めた重量平均分子量は29,100、分散度は1.9であった。
合成例3で使用したグルタル酸ジクロリドの50mol%を4,4'−ジフェニルエーテルジカルボン酸ジクロリドに置き換えた以外は合成例3と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXVとする)の標準ポリスチレン換算により求めた重量平均分子量は34,800、分散度は2.0であった。
合成例8で使用したジメチルマロン酸ジクロリドの50mol%を4,4'−ジフェニルエーテルジカルボン酸ジクロリドに置き換えた以外は合成例8と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXVIとする)の標準ポリスチレン換算により求めた重量平均分子量は36,200、分散度は2.0であった。
合成例6で使用したセバシン酸ジクロリドの40mol%を4,4'−ジフェニルエーテルジカルボン酸ジクロリドに置き換えた以外は合成例6と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXVIIとする)の標準ポリスチレン換算により求めた重量平均分子量は45,100、分散度は2.0であった。
合成例7で使用したドデカン二酸ジクロリドの40mol%を4,4'−ジフェニルエーテルジカルボン酸ジクロリドに置き換えた以外は合成例7と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXVIIIとする)の標準ポリスチレン換算により求めた重量平均分子量は41,800、分散度は2.0であった。
合成例8で使用したジメチル酸ジクロリドの50mol%を4,4'−ジフェニルエーテルジカルボン酸ジクロリドに、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)イソプロピリデンに置き換えた以外は合成例8と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXVIVとする)の標準ポリスチレン換算により求めた重量平均分子量は36,200、分散度は2.0であった。
攪拌機、温度計を備えた0.5リットルのフラスコ中に、4,4'−ジフェニルエーテルジカルボン酸15.48g、N−メチルピロリドン90gを仕込み、フラスコを5℃に冷却した後、塩化チオニル12.64gを滴下し、30分間反応させて、4,4'−ジフェニルエーテルジカルボン酸クロリドの溶液を得た。次いで、攪拌機、温度計を備えた0.5リットルのフラスコ中に、N−メチルピロリドン87.5gを仕込み、2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパン18.31gを添加し、攪拌溶解した後、ピリジン8.53gを添加し、温度を0〜5℃に保ちながら、4,4'−ジフェニルエーテルジカルボン酸クロリドの溶液を30分間で滴下した後、30分間攪拌を続けた。溶液を3リットルの水に投入し、析出物を回収、純水で3回洗浄した後、減圧してポリヒドロキシアミド(以下、ポリマーXXとする)を得た。標準ポリスチレン換算により求めた重量平均分子量は17,900、分散度は1.5であった。
2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)ヘキサフルオロプロパンを2,2'−ビス(3−アミノ−4−ヒドロキシフェニル)スルホンに置き換えた以外は合成例20と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXXIとする)の標準ポリスチレン換算により求めた重量平均分子量は13,700、分散度は1.5であった。
合成例20で使用した4,4'−ジフェニルエーテルジカルボン酸をイソフタル酸に置き換えた以外は合成例20と同様の条件にて合成を行った。得られたポリヒドロキシアミド(以下、ポリマーXXIIとする)の標準ポリスチレン換算により求めた重量平均分子量は21,600、分散度は1.6であった。
測定装置;検出器 株式会社日立製作所社製L4000 UV
ポンプ:株式会社日立製作所社製L6000
株式会社島津製作所社製C−R4A Chromatopac
測定条件:カラム Gelpack GL−S300MDT−5 x2本
溶離液:THF/DMF=1/1 (容積比)
LiBr(0.03mol/l)、H3PO4(0.06mol/l)
流速:1.0ml/min、検出器:UV270nm
ポリマー0.5mgに対して溶媒[THF/DMF=1/1(容積比)]1mlの溶液を用いて測定した。
前記合成例1〜22で得られたポリベンゾオキサゾール前駆体10.0gをN−メチルピロリドン15gに溶解して得た樹脂溶液をシリコンウエハ上にスピンコートして、120℃で3分間乾燥し、膜厚12μmの塗膜(A)を得た。この塗膜を光洋サーモシステム社製イナートガスオーブン中、窒素雰囲気下、150℃で30分加熱した後、さらに200℃で1時間又は350℃で1時間加熱して硬化膜(200℃で加熱した硬化膜(B)、350℃で加熱した硬化膜(C))を得た。これらの塗膜(A)及び硬化膜(B)、(C)の赤外吸収スペクトルを測定し、1540cm-1付近のC−N伸縮振動に起因するピークの吸光度を求めた。赤外吸収スペクトルの測定は、測定装置として「FTIR‐8300」(商品名、株式会社島津製作所製)を使用した。塗膜(A)の脱水閉環率を0%、硬化膜(C)の脱水閉環率を100%として、次の式から硬化膜(B)の脱水閉環率を算出した。
前記合成例1〜22で得られたポリベンゾオキサゾール前駆体およびポリイミド前駆体10.0gをN−メチルピロリドン15gに溶解して得た樹脂溶液をシリコンウエハ上にスピンコートしてホットプレート上120℃で3分間乾燥し、膜厚10μmの塗膜を得た。この塗膜の透過率を紫外可視分光光度計(日立製作所社製、商品名「U−3310」)により10μm膜厚における波長365nm(i線)の透過率を測定した。
(感光特性評価)
前記(a)成分であるポリベンゾオキサゾール前駆体100重量部に対し、(b)、(c)成分及びその他の添加成分を表2に示した所定量にて配合し、感光性樹脂組成物の溶液を得た。
2 保護膜
3 第1導体層
4 層間絶縁膜層
5 感光樹脂層
6A、6B、6C 窓
7 第2導体層
8 表面保護膜層
Claims (10)
- (a)一般式(IV):
(b)感光剤と、
(c)溶剤と、
を含有してなることを特徴とする感光性樹脂組成物。 - 前記(b)成分が、光により酸又はラジカルを発生する感光剤であることを特徴とする請求項1に記載の感光性樹脂組成物。
- さらに(d)成分として加熱により酸を発生する熱酸発生剤を含むことを特徴とする請求項1又は2に記載の感光性樹脂組成物。
- さらに(e)成分として溶解阻害剤を含むことを特徴とする請求項1〜3のいずれか1項に記載の感光性樹脂組成物。
- 請求項1〜4のいずれか1項に記載の感光性樹脂組成物を用い、その組成分である(a)ポリベンゾオキサゾール前駆体を脱水閉環して形成されたことを特徴とするポリベンゾオキサゾール膜。
- 請求項1〜4のいずれか1項に記載の感光性樹脂組成物を支持基板上に塗布し乾燥して感光性樹脂膜を形成する感光性樹脂膜形成工程と、前記感光性樹脂膜を所定のパターンに露光する露光工程と、前記露光後の感光性樹脂膜をアルカリ水溶液を用いて現像してパターン樹脂膜を得る現像工程と、前記パターン樹脂膜を加熱処理してパターン硬化膜を得る加熱処理工程とを含むことを特徴とするパターン硬化膜の製造方法。
- 前記加熱処理工程が、前記パターン樹脂膜にマイクロ波をその周波数を変化させながらパルス状に照射するものであることを特徴とする請求項6に記載のパターン硬化膜の製造方法。
- 前記加熱処理工程における加熱処理温度が280℃以下であることを特徴とする請求項6又は7に記載のパターン硬化膜の製造方法。
- 請求項6〜8のいずれか1項に記載のパターン硬化膜の製造方法により得られたパターン硬化膜を、層間絶縁膜層及び/又は表面保護膜層として有することを特徴とする電子部品。
- 磁気抵抗メモリであることを特徴とする請求項9に記載の電子部品。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013148919A JP5741641B2 (ja) | 2013-07-17 | 2013-07-17 | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013148919A JP5741641B2 (ja) | 2013-07-17 | 2013-07-17 | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007062341A Division JP5386781B2 (ja) | 2007-03-12 | 2007-03-12 | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013250566A JP2013250566A (ja) | 2013-12-12 |
| JP5741641B2 true JP5741641B2 (ja) | 2015-07-01 |
Family
ID=49849256
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013148919A Active JP5741641B2 (ja) | 2013-07-17 | 2013-07-17 | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5741641B2 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114746736A (zh) * | 2019-12-26 | 2022-07-12 | 住友电气工业株式会社 | 热固性树脂的固化度的评价方法 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001220443A (ja) * | 2000-02-08 | 2001-08-14 | Sumitomo Bakelite Co Ltd | ポリベンゾオキサゾール前駆体樹脂及びそれを用いた感光性樹脂組成物 |
| JP2004118123A (ja) * | 2002-09-27 | 2004-04-15 | Sumitomo Bakelite Co Ltd | ポジ型感光性プラスチック光導波路用材料及び光導波路 |
| JP2005165185A (ja) * | 2003-12-05 | 2005-06-23 | Hitachi Chemical Dupont Microsystems Ltd | 耐熱感光性樹脂組成物 |
| JP4639956B2 (ja) * | 2005-05-18 | 2011-02-23 | 日立化成デュポンマイクロシステムズ株式会社 | 感光性樹脂組成物、パターンの製造方法及び電子部品 |
-
2013
- 2013-07-17 JP JP2013148919A patent/JP5741641B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013250566A (ja) | 2013-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5434588B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP5386781B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP5169446B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたポリベンゾオキサゾール膜、パターン硬化膜の製造方法及び電子部品 | |
| JP5115635B2 (ja) | ポジ型感光性樹脂組成物、パターンの製造方法及び電子部品 | |
| JP4639956B2 (ja) | 感光性樹脂組成物、パターンの製造方法及び電子部品 | |
| JP4682764B2 (ja) | ポジ型感光性樹脂組成物、パターン形成方法及び電子部品 | |
| JP5920345B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP5577688B2 (ja) | ポジ型感光性樹脂組成物、それを用いた硬化膜及び電子部品 | |
| JP2009134282A (ja) | ポジ型感光性樹脂組成物、パタ−ンの製造方法及び電子部品 | |
| JP2011053458A (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP5387750B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP2007213032A (ja) | ポジ型感光性樹脂組成物、パターン形成方法及び電子部品 | |
| JP2010128011A (ja) | ポジ型感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP2009175356A (ja) | 低温硬化用のポジ型感光性樹脂組成物、パターン硬化膜の製造方法及び電子部品 | |
| JP2013256603A (ja) | 樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP5636680B2 (ja) | ポジ型感光性樹脂組成物、パターン硬化膜の製造方法及び電子部品 | |
| JP5625549B2 (ja) | 感光性重合体組成物、パターンの製造方法及び電子部品 | |
| JP5741641B2 (ja) | 感光性樹脂組成物、該樹脂組成物を用いたパターン硬化膜の製造方法及び電子部品 | |
| JP2011128359A (ja) | ポジ型感光性樹脂組成物、それを用いた硬化膜及び電子部品 | |
| JP5110145B2 (ja) | ポジ型感光性樹脂組成物、パターン形成方法及び電子部品 | |
| JP5029385B2 (ja) | ポジ型感光性樹脂前駆体組成物、パターン硬化膜の製造方法及び電子部品 | |
| JP2009175358A (ja) | ポジ型感光性樹脂前駆体組成物、パターン硬化膜の製造方法及び電子部品 | |
| JP2007322935A (ja) | 感光性樹脂組成物、パターンの製造方法及び電子部品 | |
| JP4529566B2 (ja) | マイクロ波硬化用ポジ型感光性樹脂組成物を用いたパターンの製造方法 | |
| JP5110144B2 (ja) | ポジ型感光性樹脂組成物、パターン形成方法及び電子部品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20140509 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140603 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140729 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20141125 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150115 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150331 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150413 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5741641 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |