JP5708306B2 - エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 - Google Patents
エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 Download PDFInfo
- Publication number
- JP5708306B2 JP5708306B2 JP2011145746A JP2011145746A JP5708306B2 JP 5708306 B2 JP5708306 B2 JP 5708306B2 JP 2011145746 A JP2011145746 A JP 2011145746A JP 2011145746 A JP2011145746 A JP 2011145746A JP 5708306 B2 JP5708306 B2 JP 5708306B2
- Authority
- JP
- Japan
- Prior art keywords
- epoxy resin
- resin composition
- resin
- phenol
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003822 epoxy resin Substances 0.000 title claims description 164
- 229920000647 polyepoxide Polymers 0.000 title claims description 164
- 239000011342 resin composition Substances 0.000 title claims description 61
- 239000004065 semiconductor Substances 0.000 title claims description 20
- 239000003566 sealing material Substances 0.000 title claims description 11
- -1 β-naphthol compound Chemical class 0.000 claims description 50
- 239000003795 chemical substances by application Substances 0.000 claims description 24
- 125000004432 carbon atom Chemical group C* 0.000 claims description 20
- JWAZRIHNYRIHIV-UHFFFAOYSA-N beta-hydroxynaphthyl Natural products C1=CC=CC2=CC(O)=CC=C21 JWAZRIHNYRIHIV-UHFFFAOYSA-N 0.000 claims description 18
- 125000003545 alkoxy group Chemical group 0.000 claims description 16
- 239000003960 organic solvent Substances 0.000 claims description 16
- 239000000463 material Substances 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 11
- 125000003055 glycidyl group Chemical group C(C1CO1)* 0.000 claims description 11
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 10
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 claims description 9
- 239000011256 inorganic filler Substances 0.000 claims description 9
- 229910003475 inorganic filler Inorganic materials 0.000 claims description 9
- 239000011889 copper foil Substances 0.000 claims description 5
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 claims description 4
- 230000003014 reinforcing effect Effects 0.000 claims description 4
- 229920001187 thermosetting polymer Polymers 0.000 claims description 4
- 238000006266 etherification reaction Methods 0.000 claims description 3
- 238000006068 polycondensation reaction Methods 0.000 claims 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 43
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 39
- 229920005989 resin Polymers 0.000 description 35
- 239000011347 resin Substances 0.000 description 35
- 239000003063 flame retardant Substances 0.000 description 32
- 229920003986 novolac Polymers 0.000 description 32
- 238000000034 method Methods 0.000 description 29
- 239000000047 product Substances 0.000 description 25
- RNFJDJUURJAICM-UHFFFAOYSA-N 2,2,4,4,6,6-hexaphenoxy-1,3,5-triaza-2$l^{5},4$l^{5},6$l^{5}-triphosphacyclohexa-1,3,5-triene Chemical compound N=1P(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP(OC=2C=CC=CC=2)(OC=2C=CC=CC=2)=NP=1(OC=1C=CC=CC=1)OC1=CC=CC=C1 RNFJDJUURJAICM-UHFFFAOYSA-N 0.000 description 23
- 239000000203 mixture Substances 0.000 description 22
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 21
- 239000005011 phenolic resin Substances 0.000 description 20
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 17
- 238000006243 chemical reaction Methods 0.000 description 17
- 150000001875 compounds Chemical class 0.000 description 17
- 238000005259 measurement Methods 0.000 description 17
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 15
- 229910052751 metal Inorganic materials 0.000 description 14
- 239000002184 metal Substances 0.000 description 14
- 229910052736 halogen Inorganic materials 0.000 description 12
- 150000002367 halogens Chemical class 0.000 description 12
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 12
- 150000002989 phenols Chemical class 0.000 description 12
- 239000000155 melt Substances 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 10
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 10
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 10
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 239000003054 catalyst Substances 0.000 description 9
- 239000000945 filler Substances 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 8
- 125000003118 aryl group Chemical group 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 8
- 150000003839 salts Chemical class 0.000 description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical group OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 7
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 7
- 239000011521 glass Substances 0.000 description 7
- JDSHMPZPIAZGSV-UHFFFAOYSA-N melamine Chemical compound NC1=NC(N)=NC(N)=N1 JDSHMPZPIAZGSV-UHFFFAOYSA-N 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 239000002904 solvent Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 229920000877 Melamine resin Polymers 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 6
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 6
- 239000003513 alkali Substances 0.000 description 6
- 239000004305 biphenyl Substances 0.000 description 6
- 235000010290 biphenyl Nutrition 0.000 description 6
- 239000011248 coating agent Substances 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- 238000000465 moulding Methods 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- 238000005406 washing Methods 0.000 description 6
- 239000004593 Epoxy Substances 0.000 description 5
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 5
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 5
- 239000000654 additive Substances 0.000 description 5
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 5
- 239000000347 magnesium hydroxide Substances 0.000 description 5
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 5
- 238000002156 mixing Methods 0.000 description 5
- 229920001296 polysiloxane Polymers 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical compound C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 4
- QTWJRLJHJPIABL-UHFFFAOYSA-N 2-methylphenol;3-methylphenol;4-methylphenol Chemical compound CC1=CC=C(O)C=C1.CC1=CC=CC(O)=C1.CC1=CC=CC=C1O QTWJRLJHJPIABL-UHFFFAOYSA-N 0.000 description 4
- GSKNLOOGBYYDHV-UHFFFAOYSA-N 2-methylphenol;naphthalen-1-ol Chemical compound CC1=CC=CC=C1O.C1=CC=C2C(O)=CC=CC2=C1 GSKNLOOGBYYDHV-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 4
- 239000004793 Polystyrene Substances 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 4
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 4
- IISBACLAFKSPIT-UHFFFAOYSA-N bisphenol A Chemical compound C=1C=C(O)C=CC=1C(C)(C)C1=CC=C(O)C=C1 IISBACLAFKSPIT-UHFFFAOYSA-N 0.000 description 4
- 238000013329 compounding Methods 0.000 description 4
- 229930003836 cresol Natural products 0.000 description 4
- 150000007973 cyanuric acids Chemical class 0.000 description 4
- 239000005350 fused silica glass Substances 0.000 description 4
- 150000002484 inorganic compounds Chemical class 0.000 description 4
- 239000012796 inorganic flame retardant Substances 0.000 description 4
- 229910010272 inorganic material Inorganic materials 0.000 description 4
- VSWALKINGSNVAR-UHFFFAOYSA-N naphthalen-1-ol;phenol Chemical compound OC1=CC=CC=C1.C1=CC=C2C(O)=CC=CC2=C1 VSWALKINGSNVAR-UHFFFAOYSA-N 0.000 description 4
- 238000006386 neutralization reaction Methods 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 235000013824 polyphenols Nutrition 0.000 description 4
- 229920002223 polystyrene Polymers 0.000 description 4
- 238000010926 purge Methods 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 3
- GZVHEAJQGPRDLQ-UHFFFAOYSA-N 6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 GZVHEAJQGPRDLQ-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 239000006087 Silane Coupling Agent Substances 0.000 description 3
- 239000000853 adhesive Substances 0.000 description 3
- 230000001070 adhesive effect Effects 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000004744 fabric Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 229910000000 metal hydroxide Inorganic materials 0.000 description 3
- 150000004692 metal hydroxides Chemical class 0.000 description 3
- 150000004780 naphthols Chemical class 0.000 description 3
- 125000001624 naphthyl group Chemical group 0.000 description 3
- 150000002903 organophosphorus compounds Chemical class 0.000 description 3
- 239000003973 paint Substances 0.000 description 3
- 229910052698 phosphorus Inorganic materials 0.000 description 3
- 239000011574 phosphorus Substances 0.000 description 3
- 150000003018 phosphorus compounds Chemical class 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 3
- 238000001228 spectrum Methods 0.000 description 3
- BPSIOYPQMFLKFR-UHFFFAOYSA-N trimethoxy-[3-(oxiran-2-ylmethoxy)propyl]silane Chemical compound CO[Si](OC)(OC)CCCOCC1CO1 BPSIOYPQMFLKFR-UHFFFAOYSA-N 0.000 description 3
- UGZADUVQMDAIAO-UHFFFAOYSA-L zinc hydroxide Chemical compound [OH-].[OH-].[Zn+2] UGZADUVQMDAIAO-UHFFFAOYSA-L 0.000 description 3
- 229910021511 zinc hydroxide Inorganic materials 0.000 description 3
- 229940007718 zinc hydroxide Drugs 0.000 description 3
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- WBODDOZXDKQEFS-UHFFFAOYSA-N 1,2,3,4-tetramethyl-5-phenylbenzene Chemical group CC1=C(C)C(C)=CC(C=2C=CC=CC=2)=C1C WBODDOZXDKQEFS-UHFFFAOYSA-N 0.000 description 2
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 2
- NKTOLZVEWDHZMU-UHFFFAOYSA-N 2,5-xylenol Chemical compound CC1=CC=C(C)C(O)=C1 NKTOLZVEWDHZMU-UHFFFAOYSA-N 0.000 description 2
- GJYCVCVHRSWLNY-UHFFFAOYSA-N 2-butylphenol Chemical compound CCCCC1=CC=CC=C1O GJYCVCVHRSWLNY-UHFFFAOYSA-N 0.000 description 2
- KXGFMDJXCMQABM-UHFFFAOYSA-N 2-methoxy-6-methylphenol Chemical class [CH]OC1=CC=CC([CH])=C1O KXGFMDJXCMQABM-UHFFFAOYSA-N 0.000 description 2
- 0 CCc(c1ccccc1cc1)c1O* Chemical compound CCc(c1ccccc1cc1)c1O* 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- NJYZCEFQAIUHSD-UHFFFAOYSA-N acetoguanamine Chemical compound CC1=NC(N)=NC(N)=N1 NJYZCEFQAIUHSD-UHFFFAOYSA-N 0.000 description 2
- XFSBVAOIAHNAPC-WSORPINJSA-N acetylbenzoylaconine Chemical compound O([C@H]1[C@]2(O)C[C@H]3C45[C@@H]6[C@@H]([C@@]([C@H]31)(OC(C)=O)[C@@H](O)[C@@H]2OC)[C@H](OC)C4[C@]([C@@H](C[C@H]5OC)O)(COC)CN6CC)C(=O)C1=CC=CC=C1 XFSBVAOIAHNAPC-WSORPINJSA-N 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- ZRIUUUJAJJNDSS-UHFFFAOYSA-N ammonium phosphates Chemical compound [NH4+].[NH4+].[NH4+].[O-]P([O-])([O-])=O ZRIUUUJAJJNDSS-UHFFFAOYSA-N 0.000 description 2
- 239000004760 aramid Substances 0.000 description 2
- 229920003235 aromatic polyamide Polymers 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 229910000416 bismuth oxide Inorganic materials 0.000 description 2
- PXKLMJQFEQBVLD-UHFFFAOYSA-N bisphenol F Chemical compound C1=CC(O)=CC=C1CC1=CC=C(O)C=C1 PXKLMJQFEQBVLD-UHFFFAOYSA-N 0.000 description 2
- 150000001639 boron compounds Chemical class 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 239000004203 carnauba wax Substances 0.000 description 2
- 235000013869 carnauba wax Nutrition 0.000 description 2
- 238000005266 casting Methods 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 238000002485 combustion reaction Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- GDVKFRBCXAPAQJ-UHFFFAOYSA-A dialuminum;hexamagnesium;carbonate;hexadecahydroxide Chemical compound [OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[OH-].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Mg+2].[Al+3].[Al+3].[O-]C([O-])=O GDVKFRBCXAPAQJ-UHFFFAOYSA-A 0.000 description 2
- PPQREHKVAOVYBT-UHFFFAOYSA-H dialuminum;tricarbonate Chemical compound [Al+3].[Al+3].[O-]C([O-])=O.[O-]C([O-])=O.[O-]C([O-])=O PPQREHKVAOVYBT-UHFFFAOYSA-H 0.000 description 2
- TYIXMATWDRGMPF-UHFFFAOYSA-N dibismuth;oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Bi+3].[Bi+3] TYIXMATWDRGMPF-UHFFFAOYSA-N 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 239000008393 encapsulating agent Substances 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000011049 filling Methods 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 2
- 229910001701 hydrotalcite Inorganic materials 0.000 description 2
- 229960001545 hydrotalcite Drugs 0.000 description 2
- AMWRITDGCCNYAT-UHFFFAOYSA-L hydroxy(oxo)manganese;manganese Chemical compound [Mn].O[Mn]=O.O[Mn]=O AMWRITDGCCNYAT-UHFFFAOYSA-L 0.000 description 2
- 239000011810 insulating material Substances 0.000 description 2
- 239000011229 interlayer Substances 0.000 description 2
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 2
- 239000001095 magnesium carbonate Substances 0.000 description 2
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000012778 molding material Substances 0.000 description 2
- 239000005078 molybdenum compound Substances 0.000 description 2
- 150000002752 molybdenum compounds Chemical class 0.000 description 2
- JKQOBWVOAYFWKG-UHFFFAOYSA-N molybdenum trioxide Chemical compound O=[Mo](=O)=O JKQOBWVOAYFWKG-UHFFFAOYSA-N 0.000 description 2
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 2
- IWDCLRJOBJJRNH-UHFFFAOYSA-N p-cresol Chemical compound CC1=CC=C(O)C=C1 IWDCLRJOBJJRNH-UHFFFAOYSA-N 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 239000003444 phase transfer catalyst Substances 0.000 description 2
- 229920001568 phenolic resin Polymers 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- 239000000049 pigment Substances 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 239000002798 polar solvent Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 2
- 229960001755 resorcinol Drugs 0.000 description 2
- 238000007788 roughening Methods 0.000 description 2
- 239000001488 sodium phosphate Substances 0.000 description 2
- 229910000162 sodium phosphate Inorganic materials 0.000 description 2
- 238000004381 surface treatment Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- LLZRNZOLAXHGLL-UHFFFAOYSA-J titanic acid Chemical compound O[Ti](O)(O)O LLZRNZOLAXHGLL-UHFFFAOYSA-J 0.000 description 2
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 2
- 238000001721 transfer moulding Methods 0.000 description 2
- 150000003918 triazines Chemical class 0.000 description 2
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- XAEWLETZEZXLHR-UHFFFAOYSA-N zinc;dioxido(dioxo)molybdenum Chemical compound [Zn+2].[O-][Mo]([O-])(=O)=O XAEWLETZEZXLHR-UHFFFAOYSA-N 0.000 description 2
- 229910001928 zirconium oxide Inorganic materials 0.000 description 2
- LTVUCOSIZFEASK-MPXCPUAZSA-N (3ar,4s,7r,7as)-3a-methyl-3a,4,7,7a-tetrahydro-4,7-methano-2-benzofuran-1,3-dione Chemical compound C([C@H]1C=C2)[C@H]2[C@H]2[C@]1(C)C(=O)OC2=O LTVUCOSIZFEASK-MPXCPUAZSA-N 0.000 description 1
- MUTGBJKUEZFXGO-OLQVQODUSA-N (3as,7ar)-3a,4,5,6,7,7a-hexahydro-2-benzofuran-1,3-dione Chemical compound C1CCC[C@@H]2C(=O)OC(=O)[C@@H]21 MUTGBJKUEZFXGO-OLQVQODUSA-N 0.000 description 1
- KMOUUZVZFBCRAM-OLQVQODUSA-N (3as,7ar)-3a,4,7,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C=CC[C@@H]2C(=O)OC(=O)[C@@H]21 KMOUUZVZFBCRAM-OLQVQODUSA-N 0.000 description 1
- KGSFMPRFQVLGTJ-UHFFFAOYSA-N 1,1,2-triphenylethylbenzene Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)CC1=CC=CC=C1 KGSFMPRFQVLGTJ-UHFFFAOYSA-N 0.000 description 1
- VDFVNEFVBPFDSB-UHFFFAOYSA-N 1,3-dioxane Chemical compound C1COCOC1 VDFVNEFVBPFDSB-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- IVORCBKUUYGUOL-UHFFFAOYSA-N 1-ethynyl-2,4-dimethoxybenzene Chemical compound COC1=CC=C(C#C)C(OC)=C1 IVORCBKUUYGUOL-UHFFFAOYSA-N 0.000 description 1
- HECLRDQVFMWTQS-RGOKHQFPSA-N 1755-01-7 Chemical compound C1[C@H]2[C@@H]3CC=C[C@@H]3[C@@H]1C=C2 HECLRDQVFMWTQS-RGOKHQFPSA-N 0.000 description 1
- VILCJCGEZXAXTO-UHFFFAOYSA-N 2,2,2-tetramine Chemical compound NCCNCCNCCN VILCJCGEZXAXTO-UHFFFAOYSA-N 0.000 description 1
- QLVPICNVQBBOQP-UHFFFAOYSA-N 2-(4,6-diamino-1,3,5-triazin-2-yl)guanidine Chemical compound NC(N)=NC1=NC(N)=NC(N)=N1 QLVPICNVQBBOQP-UHFFFAOYSA-N 0.000 description 1
- VVHFXJOCUKBZFS-UHFFFAOYSA-N 2-(chloromethyl)-2-methyloxirane Chemical compound ClCC1(C)CO1 VVHFXJOCUKBZFS-UHFFFAOYSA-N 0.000 description 1
- SFRDXVJWXWOTEW-UHFFFAOYSA-N 2-(hydroxymethyl)propane-1,3-diol Chemical compound OCC(CO)CO SFRDXVJWXWOTEW-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- YZEZMSPGIPTEBA-UHFFFAOYSA-N 2-n-(4,6-diamino-1,3,5-triazin-2-yl)-1,3,5-triazine-2,4,6-triamine Chemical compound NC1=NC(N)=NC(NC=2N=C(N)N=C(N)N=2)=N1 YZEZMSPGIPTEBA-UHFFFAOYSA-N 0.000 description 1
- RNLHGQLZWXBQNY-UHFFFAOYSA-N 3-(aminomethyl)-3,5,5-trimethylcyclohexan-1-amine Chemical compound CC1(C)CC(N)CC(C)(CN)C1 RNLHGQLZWXBQNY-UHFFFAOYSA-N 0.000 description 1
- ZUHMEUFBTDOKPX-UHFFFAOYSA-N 6-[2-(4,6-diamino-1,3,5-triazin-2-yl)ethyl]-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(CCC=2N=C(N)N=C(N)N=2)=N1 ZUHMEUFBTDOKPX-UHFFFAOYSA-N 0.000 description 1
- MWSKJDNQKGCKPA-UHFFFAOYSA-N 6-methyl-3a,4,5,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1CC(C)=CC2C(=O)OC(=O)C12 MWSKJDNQKGCKPA-UHFFFAOYSA-N 0.000 description 1
- HDXGUOZQUVDYMC-UHFFFAOYSA-N 6h-benzo[c][2,1]benzoxaphosphinine Chemical compound C1=CC=C2OPC3=CC=CC=C3C2=C1 HDXGUOZQUVDYMC-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 1
- 239000004254 Ammonium phosphate Substances 0.000 description 1
- 239000004114 Ammonium polyphosphate Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- QPLDLSVMHZLSFG-UHFFFAOYSA-N Copper oxide Chemical compound [Cu]=O QPLDLSVMHZLSFG-UHFFFAOYSA-N 0.000 description 1
- 239000005751 Copper oxide Substances 0.000 description 1
- 244000241257 Cucumis melo Species 0.000 description 1
- 235000015510 Cucumis melo subsp melo Nutrition 0.000 description 1
- MQJKPEGWNLWLTK-UHFFFAOYSA-N Dapsone Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=C1 MQJKPEGWNLWLTK-UHFFFAOYSA-N 0.000 description 1
- 239000005696 Diammonium phosphate Substances 0.000 description 1
- RPNUMPOLZDHAAY-UHFFFAOYSA-N Diethylenetriamine Chemical compound NCCNCCN RPNUMPOLZDHAAY-UHFFFAOYSA-N 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 244000290594 Ficus sycomorus Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- IGFHQQFPSIBGKE-UHFFFAOYSA-N Nonylphenol Natural products CCCCCCCCCC1=CC=C(O)C=C1 IGFHQQFPSIBGKE-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 240000002834 Paulownia tomentosa Species 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 229920000388 Polyphosphate Polymers 0.000 description 1
- 235000001537 Ribes X gardonianum Nutrition 0.000 description 1
- 235000001535 Ribes X utile Nutrition 0.000 description 1
- 235000016919 Ribes petraeum Nutrition 0.000 description 1
- 244000281247 Ribes rubrum Species 0.000 description 1
- 235000002355 Ribes spicatum Nutrition 0.000 description 1
- 208000034189 Sclerosis Diseases 0.000 description 1
- 229910052581 Si3N4 Inorganic materials 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- WGLPBDUCMAPZCE-UHFFFAOYSA-N Trioxochromium Chemical compound O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 1
- 229910021536 Zeolite Inorganic materials 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- FMRLDPWIRHBCCC-UHFFFAOYSA-L Zinc carbonate Chemical compound [Zn+2].[O-]C([O-])=O FMRLDPWIRHBCCC-UHFFFAOYSA-L 0.000 description 1
- FJJCIZWZNKZHII-UHFFFAOYSA-N [4,6-bis(cyanoamino)-1,3,5-triazin-2-yl]cyanamide Chemical compound N#CNC1=NC(NC#N)=NC(NC#N)=N1 FJJCIZWZNKZHII-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 238000007259 addition reaction Methods 0.000 description 1
- 239000012790 adhesive layer Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 229940118662 aluminum carbonate Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- LFVGISIMTYGQHF-UHFFFAOYSA-N ammonium dihydrogen phosphate Chemical compound [NH4+].OP(O)([O-])=O LFVGISIMTYGQHF-UHFFFAOYSA-N 0.000 description 1
- 229910000387 ammonium dihydrogen phosphate Inorganic materials 0.000 description 1
- 229910000148 ammonium phosphate Inorganic materials 0.000 description 1
- 235000019289 ammonium phosphates Nutrition 0.000 description 1
- 235000019826 ammonium polyphosphate Nutrition 0.000 description 1
- 229920001276 ammonium polyphosphate Polymers 0.000 description 1
- 229940058905 antimony compound for treatment of leishmaniasis and trypanosomiasis Drugs 0.000 description 1
- 150000001463 antimony compounds Chemical class 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 238000010533 azeotropic distillation Methods 0.000 description 1
- QBLDFAIABQKINO-UHFFFAOYSA-N barium borate Chemical compound [Ba+2].[O-]B=O.[O-]B=O QBLDFAIABQKINO-UHFFFAOYSA-N 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- AYJRCSIUFZENHW-DEQYMQKBSA-L barium(2+);oxomethanediolate Chemical compound [Ba+2].[O-][14C]([O-])=O AYJRCSIUFZENHW-DEQYMQKBSA-L 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 229940049676 bismuth hydroxide Drugs 0.000 description 1
- TZSXPYWRDWEXHG-UHFFFAOYSA-K bismuth;trihydroxide Chemical compound [OH-].[OH-].[OH-].[Bi+3] TZSXPYWRDWEXHG-UHFFFAOYSA-K 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 229910021538 borax Inorganic materials 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 230000000711 cancerogenic effect Effects 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 231100000315 carcinogenic Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 238000010538 cationic polymerization reaction Methods 0.000 description 1
- 239000007809 chemical reaction catalyst Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 229910000423 chromium oxide Inorganic materials 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 229910021446 cobalt carbonate Inorganic materials 0.000 description 1
- 229910000428 cobalt oxide Inorganic materials 0.000 description 1
- ZOTKGJBKKKVBJZ-UHFFFAOYSA-L cobalt(2+);carbonate Chemical compound [Co+2].[O-]C([O-])=O ZOTKGJBKKKVBJZ-UHFFFAOYSA-L 0.000 description 1
- IVMYJDGYRUAWML-UHFFFAOYSA-N cobalt(ii) oxide Chemical compound [Co]=O IVMYJDGYRUAWML-UHFFFAOYSA-N 0.000 description 1
- 239000004020 conductor Substances 0.000 description 1
- 229910000431 copper oxide Inorganic materials 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 229910002026 crystalline silica Inorganic materials 0.000 description 1
- 238000007766 curtain coating Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- MNNHAPBLZZVQHP-UHFFFAOYSA-N diammonium hydrogen phosphate Chemical compound [NH4+].[NH4+].OP([O-])([O-])=O MNNHAPBLZZVQHP-UHFFFAOYSA-N 0.000 description 1
- 229910000388 diammonium phosphate Inorganic materials 0.000 description 1
- 235000019838 diammonium phosphate Nutrition 0.000 description 1
- QGBSISYHAICWAH-UHFFFAOYSA-N dicyandiamide Chemical compound NC(N)=NC#N QGBSISYHAICWAH-UHFFFAOYSA-N 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 150000002013 dioxins Chemical class 0.000 description 1
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 description 1
- ZZTCPWRAHWXWCH-UHFFFAOYSA-N diphenylmethanediamine Chemical compound C=1C=CC=CC=1C(N)(N)C1=CC=CC=C1 ZZTCPWRAHWXWCH-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000010459 dolomite Substances 0.000 description 1
- 229910000514 dolomite Inorganic materials 0.000 description 1
- 238000005553 drilling Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 238000007772 electroless plating Methods 0.000 description 1
- 238000009713 electroplating Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- GKIPXFAANLTWBM-UHFFFAOYSA-N epibromohydrin Chemical compound BrCC1CO1 GKIPXFAANLTWBM-UHFFFAOYSA-N 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- 125000003700 epoxy group Chemical group 0.000 description 1
- WLPKFQRBARNCNR-UHFFFAOYSA-N ethene 1,3,5-triazine-2,4,6-triamine Chemical compound C=C.NC1=NC(N)=NC(N)=N1.NC1=NC(N)=NC(N)=N1 WLPKFQRBARNCNR-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 1
- RAQDACVRFCEPDA-UHFFFAOYSA-L ferrous carbonate Chemical compound [Fe+2].[O-]C([O-])=O RAQDACVRFCEPDA-UHFFFAOYSA-L 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- SLGWESQGEUXWJQ-UHFFFAOYSA-N formaldehyde;phenol Chemical compound O=C.OC1=CC=CC=C1 SLGWESQGEUXWJQ-UHFFFAOYSA-N 0.000 description 1
- ANSXAPJVJOKRDJ-UHFFFAOYSA-N furo[3,4-f][2]benzofuran-1,3,5,7-tetrone Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1C(=O)OC2=O ANSXAPJVJOKRDJ-UHFFFAOYSA-N 0.000 description 1
- 150000002357 guanidines Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- RXPAJWPEYBDXOG-UHFFFAOYSA-N hydron;methyl 4-methoxypyridine-2-carboxylate;chloride Chemical compound Cl.COC(=O)C1=CC(OC)=CC=N1 RXPAJWPEYBDXOG-UHFFFAOYSA-N 0.000 description 1
- 150000002460 imidazoles Chemical class 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 238000001746 injection moulding Methods 0.000 description 1
- 239000012774 insulation material Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- QDLAGTHXVHQKRE-UHFFFAOYSA-N lichenxanthone Natural products COC1=CC(O)=C2C(=O)C3=C(C)C=C(OC)C=C3OC2=C1 QDLAGTHXVHQKRE-UHFFFAOYSA-N 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 1
- 235000021388 linseed oil Nutrition 0.000 description 1
- 239000000944 linseed oil Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 150000007974 melamines Chemical class 0.000 description 1
- YSRVJVDFHZYRPA-UHFFFAOYSA-N melem Chemical compound NC1=NC(N23)=NC(N)=NC2=NC(N)=NC3=N1 YSRVJVDFHZYRPA-UHFFFAOYSA-N 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- VYKXQOYUCMREIS-UHFFFAOYSA-N methylhexahydrophthalic anhydride Chemical compound C1CCCC2C(=O)OC(=O)C21C VYKXQOYUCMREIS-UHFFFAOYSA-N 0.000 description 1
- 238000001471 micro-filtration Methods 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 239000006082 mold release agent Substances 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- 229910000476 molybdenum oxide Inorganic materials 0.000 description 1
- 235000019837 monoammonium phosphate Nutrition 0.000 description 1
- 239000006012 monoammonium phosphate Substances 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229910000480 nickel oxide Inorganic materials 0.000 description 1
- QGLKJKCYBOYXKC-UHFFFAOYSA-N nonaoxidotritungsten Chemical compound O=[W]1(=O)O[W](=O)(=O)O[W](=O)(=O)O1 QGLKJKCYBOYXKC-UHFFFAOYSA-N 0.000 description 1
- 239000004745 nonwoven fabric Substances 0.000 description 1
- SNQQPOLDUKLAAF-UHFFFAOYSA-N nonylphenol Chemical compound CCCCCCCCCC1=CC=CC=C1O SNQQPOLDUKLAAF-UHFFFAOYSA-N 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical group 0.000 description 1
- 125000001477 organic nitrogen group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- PQQKPALAQIIWST-UHFFFAOYSA-N oxomolybdenum Chemical compound [Mo]=O PQQKPALAQIIWST-UHFFFAOYSA-N 0.000 description 1
- GNRSAWUEBMWBQH-UHFFFAOYSA-N oxonickel Chemical compound [Ni]=O GNRSAWUEBMWBQH-UHFFFAOYSA-N 0.000 description 1
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 150000002990 phenothiazines Chemical class 0.000 description 1
- ACVYVLVWPXVTIT-UHFFFAOYSA-N phosphinic acid Chemical class O[PH2]=O ACVYVLVWPXVTIT-UHFFFAOYSA-N 0.000 description 1
- 150000003009 phosphonic acids Chemical class 0.000 description 1
- VBQCHPIMZGQLAZ-UHFFFAOYSA-N phosphorane Chemical class [PH5] VBQCHPIMZGQLAZ-UHFFFAOYSA-N 0.000 description 1
- 229920002120 photoresistant polymer Polymers 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229920006122 polyamide resin Polymers 0.000 description 1
- 239000002685 polymerization catalyst Substances 0.000 description 1
- 150000008442 polyphenolic compounds Chemical class 0.000 description 1
- 239000001205 polyphosphate Substances 0.000 description 1
- 235000011176 polyphosphates Nutrition 0.000 description 1
- 238000011417 postcuring Methods 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 238000010125 resin casting Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 238000007650 screen-printing Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- HQVNEWCFYHHQES-UHFFFAOYSA-N silicon nitride Chemical compound N12[Si]34N5[Si]62N3[Si]51N64 HQVNEWCFYHHQES-UHFFFAOYSA-N 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 229920002050 silicone resin Polymers 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 239000004945 silicone rubber Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000004328 sodium tetraborate Substances 0.000 description 1
- 235000010339 sodium tetraborate Nutrition 0.000 description 1
- 229910000679 solder Inorganic materials 0.000 description 1
- 238000005507 spraying Methods 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- KCNSDMPZCKLTQP-UHFFFAOYSA-N tetraphenylen-1-ol Chemical compound C12=CC=CC=C2C2=CC=CC=C2C2=CC=CC=C2C2=C1C=CC=C2O KCNSDMPZCKLTQP-UHFFFAOYSA-N 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000011135 tin Substances 0.000 description 1
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 1
- 229910001887 tin oxide Inorganic materials 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- FOZHTJJTSSSURD-UHFFFAOYSA-J titanium(4+);dicarbonate Chemical compound [Ti+4].[O-]C([O-])=O.[O-]C([O-])=O FOZHTJJTSSSURD-UHFFFAOYSA-J 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- AAAQKTZKLRYKHR-UHFFFAOYSA-N triphenylmethane Chemical compound C1=CC=CC=C1C(C=1C=CC=CC=1)C1=CC=CC=C1 AAAQKTZKLRYKHR-UHFFFAOYSA-N 0.000 description 1
- BIKXLKXABVUSMH-UHFFFAOYSA-N trizinc;diborate Chemical compound [Zn+2].[Zn+2].[Zn+2].[O-]B([O-])[O-].[O-]B([O-])[O-] BIKXLKXABVUSMH-UHFFFAOYSA-N 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- 229910001930 tungsten oxide Inorganic materials 0.000 description 1
- 239000002966 varnish Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
- 150000003739 xylenols Chemical class 0.000 description 1
- 239000010457 zeolite Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 239000011667 zinc carbonate Substances 0.000 description 1
- 229910000010 zinc carbonate Inorganic materials 0.000 description 1
- 235000004416 zinc carbonate Nutrition 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- BNEMLSQAJOPTGK-UHFFFAOYSA-N zinc;dioxido(oxo)tin Chemical compound [Zn+2].[O-][Sn]([O-])=O BNEMLSQAJOPTGK-UHFFFAOYSA-N 0.000 description 1
- PZRXQXJGIQEYOG-UHFFFAOYSA-N zinc;oxido(oxo)borane Chemical compound [Zn+2].[O-]B=O.[O-]B=O PZRXQXJGIQEYOG-UHFFFAOYSA-N 0.000 description 1
Images





Landscapes
- Epoxy Resins (AREA)
- Structures Or Materials For Encapsulating Or Coating Semiconductor Devices Or Solid State Devices (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description

(式中、R1、R2、R3、及びR4はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)で表されることを特徴とするエポキシ樹脂に関する。
(式中、R1、R2、R3、及びR4はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)
で表されるエポキシ樹脂(x2)、及び、下記構造式(3)
(式中、R1、及びR2はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)
で表されるエポキシ樹脂(x3)を含有することを特徴とするエポキシ樹脂に関する。
本発明のエポキシ樹脂は、下記構造式(1)
(式中、R1、R2、R3、及びR4はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)で表される分子構造を有することを特徴としている。
(式中、R1、R2、R3、及びR4はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)
で表されるエポキシ樹脂(以下、これを「エポキシ樹脂(x2)」と略記する。)、及び構造式(3)
(式中、R1、及びR2はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表す。)
で表されるエポキシ樹脂(以下、これを「エポキシ樹脂(x3)」と略記する。)を必須成分とするエポキシ樹脂混合物(以下、このエポキシ樹脂混合物を「エポキシ樹脂(α)」と略記する。)として用いることが、硬化物における耐熱性が良好となる点から好ましい。
で表されるものが挙げられる。構造式(4)中のR1、R2、R3、及びR4はそれぞれ独立して水素原子、炭素原子数1〜4のアルキル基、炭素原子数1〜4のアルコキシ基を示し、Grはグリシジル基を表し、nは繰り返し単位であり、0以上の整数である。また、Yは水酸基、又は下記構造式(5)
で表される構造部位を表す。なお、構造式(5)中のR1、R2、及びGrは構造式(4)と同義である。
<GPC測定条件>
測定装置 :東ソー株式会社製「HLC−8220 GPC」、
カラム:東ソー株式会社製ガードカラム「HXL−L」
+東ソー株式会社製「TSK−GEL G2000HXL」
+東ソー株式会社製「TSK−GEL G2000HXL」
+東ソー株式会社製「TSK−GEL G3000HXL」
+東ソー株式会社製「TSK−GEL G4000HXL」
検出器: RI(示差屈折径)
データ処理:東ソー株式会社製「GPC−8020モデルIIバージョン4.10」
測定条件: カラム温度 40℃
展開溶媒 テトラヒドロフラン
流速 1.0ml/分
標準 : 前記「GPC−8020モデルIIバージョン4.10」の測定マニュアルに準拠して、分子量が既知の下記の単分散ポリスチレンを用いた。
(使用ポリスチレン)
東ソー株式会社製「A−500」
東ソー株式会社製「A−1000」
東ソー株式会社製「A−2500」
東ソー株式会社製「A−5000」
東ソー株式会社製「F−1」
東ソー株式会社製「F−2」
東ソー株式会社製「F−4」
東ソー株式会社製「F−10」
東ソー株式会社製「F−20」
東ソー株式会社製「F−40」
東ソー株式会社製「F−80」
東ソー株式会社製「F−128」
試料 : 樹脂固形分換算で1.0質量%のテトラヒドロフラン溶液をマイクロフィルターでろ過したもの(50μl)。
本発明における有機溶剤の使用量は、原料成分であるフェノール化合物及びβ−ナフトール化合物、更に、他のノボラック樹脂を併用する場合には、原料となるフェノール化合物及びβ−ナフトール化合物の総質量100質量部あたり、5〜80質量部の範囲であることが、前記エポキシ樹脂(x1)のエポキシ樹脂中の存在割合を所定範囲に調整し易い点から好ましい。
ナフトールアラルキル型エポキシ樹脂、ナフトール−フェノール共縮ノボラック型エポキシ樹脂、ナフトール−クレゾール共縮ノボラック型エポキシ樹脂、芳香族炭化水素ホルムアルデヒド樹脂変性フェノール樹脂型エポキシ樹脂、ビフェニルノボラック型エポキシ樹脂等が挙げられる。これらのなかでもフェノールアラルキル型エポキシ樹脂、ビフェニルノボラック型エポキシ樹脂や、ナフタレン骨格を含有するナフトールノボラック型エポキシ樹脂、ナフトールアラルキル型エポキシ樹脂、ナフトール−フェノール共縮ノボラック型エポキシ樹脂、ナフトール−クレゾール共縮ノボラック型エポキシ樹脂や、結晶性のビフェニル型エポキシ樹脂、テトラメチルビフェニル型エポキシ樹脂、キサンテン型エポキシ樹脂や、アルコキシ基含有芳香環変性ノボラック型エポキシ樹脂(ホルムアルデヒドでグリシジル基含有芳香環及びアルコキシ基含有芳香環が連結された化合物)等が耐熱性に優れる硬化物が得られる点から特に好ましい。
測定装置 :東ソー株式会社製「HLC−8220 GPC」、
カラム:東ソー株式会社製ガードカラム「HXL−L」
+東ソー株式会社製「TSK−GEL G2000HXL」
+東ソー株式会社製「TSK−GEL G2000HXL」
+東ソー株式会社製「TSK−GEL G3000HXL」
+東ソー株式会社製「TSK−GEL G4000HXL」
検出器: RI(示差屈折径)
データ処理:東ソー株式会社製「GPC−8020モデルIIバージョン4.10」
測定条件: カラム温度 40℃
展開溶媒 テトラヒドロフラン
流速 1.0ml/分
標準 : 前記「GPC−8020モデルIIバージョン4.10」の測定マニュアルに準拠して、分子量が既知の下記の単分散ポリスチレンを用いた。
(使用ポリスチレン)
東ソー株式会社製「A−500」
東ソー株式会社製「A−1000」
東ソー株式会社製「A−2500」
東ソー株式会社製「A−5000」
東ソー株式会社製「F−1」
東ソー株式会社製「F−2」
東ソー株式会社製「F−4」
東ソー株式会社製「F−10」
東ソー株式会社製「F−20」
東ソー株式会社製「F−40」
東ソー株式会社製「F−80」
東ソー株式会社製「F−128」
試料 : 樹脂固形分換算で1.0質量%のテトラヒドロフラン溶液をマイクロフィルターでろ過したもの(50μl)。
3)13C−NMR:測定条件は以下の通り。
装置:日本電子(株)製 AL−400
測定モード:SGNNE(NOE消去の1H完全デカップリング法)
溶媒 :ジメチルスルホキシド
パルス角度:45℃パルス
試料濃度 :30wt%
積算回数 :10000回
4)MS :日本電子株式会社製 JMS−T100GC
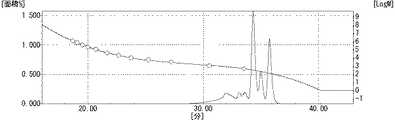
温度計、滴下ロート、冷却管、分留管、撹拌器を取り付けたフラスコに、β−ナフトール216部(1.5モル)、オルソクレゾール97部(0.90モル)、イソプロピルアルコール133部、37%ホルマリン水溶液138部(1.70モル)、49%水酸化ナトリウム49部(0.60モル)を仕込み、室温から75℃まで攪拌しながら昇温し、75℃で3時間撹拌した。反応終了後、第1リン酸ソーダ72質量部を添加して中和した後、メチルイソブチルケトン500部加え、水250質量部で3回洗浄を繰り返した後に、加熱減圧下乾燥してフェノール樹脂(A−1)310質量部得た。得られたフェノール樹脂(A−1)の水酸基当量は139グラム/当量であり、150℃における溶融粘度が0.7dPa・sであった。得られたフェノール樹脂(A−1)のGPCチャートを図1に示す。また、GPCチャートから下記構造式(x’1)で表される化合物の含有率は29.3%、下記構造式(x’2)で表される化合物の含有率は36.7%、下記構造式(x’3)で表される化合物の含有率は29.3%、その他高分子量体(x’4)の含有率は4.7%であった。
次いで、温度計、冷却管、撹拌器を取り付けたフラスコに窒素ガスパージを施しながら上記反応で得られたフェノール樹脂(A−1)139質量部(水酸基1.0当量)、エピクロルヒドリン463質量部(5.0モル)、n−ブタノール53質量部を仕込み攪拌しながら溶解させた。50℃に昇温した後に、20%水酸化ナトリウム水溶液220質量部(1.10モル)を3時間要して添加し、その後更に50℃で1時間反応させた。反応終了後、攪拌を停止し、下層に溜まった水層を除去し、攪拌を再開し150℃減圧下で未反応エピクロルヒドリンを留去した。それで得られた粗エポキシ樹脂にメチルイソブチルケトン300質量部とn−ブタノール50質量部とを加え溶解した。更にこの溶液に10質量%水酸化ナトリウム水溶液15質量部を添加して80℃で2時間反応させた後に洗浄液のpHが中性となるまで水100質量部で水洗を3回繰り返した。次いで共沸によって系内を脱水し、精密濾過を経た後に、溶媒を減圧下で留去して目的のエポキシ樹脂(A−2)186質量部を得た。得られたエポキシ樹脂(A−2)のエポキシ当量は229グラム/当量であり、150℃における溶融粘度が0.5dPa・sであった。エポキシ樹脂(A−2)のGPCチャートを図2に、13C−NMRスペクトルを図3に、MSスペクトルを図4に示す。また、GPCチャートから下記構造式(x1)で表される化合物の含有率は18.4%、下記構造式(x2)で表される化合物の含有率は28.9%、下記構造式(x3)で表される化合物の含有率は29.3%、その他高分子量体(x4)の含有率は23.4%であった。
オルソクレゾールを81部(0.75モル)、イソプロピルアルコール125部、37%ホルマリン水溶液128部(1.58モル)、49%水酸化ナトリウム46部(0.56モル)に変更した以外は、実施例1と同様にしてフェノール樹脂(A−3)305質量部得た。得られたフェノール樹脂(A−3)の水酸基当量は141グラム/当量であり、150℃における溶融粘度は1.0dPa・sであった。フェノール樹脂(A−3)のGPCチャートを図5に示す。また、GPCチャートから前記構造式(x’1)で表される化合物の含有率は19.0%、前記構造式(x’2)で表される化合物の含有率は47.5%、前記構造式(x’3)で表される化合物の含有率は27.8%、その他高分子量体(x’4)の含有率は8.2%であった。
次いで、温度計、冷却管、撹拌器を取り付けたフラスコに窒素ガスパージを施しながら上記反応で得られたフェノール樹脂(A−4)141質量部(水酸基1.0当量)を実施例1と同様にして、目的のエポキシ樹脂(A−4)190質量部を得た。得られたエポキシ樹脂(A−4)のエポキシ当量は228グラム/当量であり、150℃における溶融粘度が0.7dPa・sであった。エポキシ樹脂(A−4)のGPCチャートを図6に示す。また、GPCチャートから前記構造式(x1)で表される化合物の含有率は12.5%、前記構造式(x2)で表される化合物の含有率は36.0%、前記構造式(x3)で表される化合物の含有率は25.9%、その他高分子量体(x4)の含有率は25.6%であった。
エポキシ樹脂として上記A−2及びA−4、比較用のエポキシ樹脂として特許文献1、実施例1に記載されているものと同一の構造を有するビフェニルアラルキル型エポキシ樹脂(日本化薬株式会社製「NC−3000」、エポキシ当量:274g/eq)、硬化剤(B)としてフェノールアラルキル樹脂(三井化学株式会社製「XLC−3L」水酸基当量:176g/eq)を用い、硬化促進剤としてトリフェニルホスフィン(TPP)、無機充填材として球状シリカ(株式会社マイクロン製「S−COL」)、シランカップリング剤としてγ−グリシドキシトリエトキシキシシラン(信越化学工業株式会社製「KBM−403」)、カルナバワックス(株式会社セラリカ野田製「PEARL WAX No.1−P」)、カーボンブラックを用いて表1に示した組成で配合し、2本ロールを用いて85℃の温度で5分間溶融混練して目的の組成物を得た。硬化物の物性は、上記組成物を用いて、評価用サンプルを下記の方法で作成し、難燃性、硬化性(ゲルタイム)、耐熱性を下記の方法で測定し結果を表1に示した。
硬化性樹脂組成物0.15gを150℃に加熱したキュアプレート(THERMO ELECTRIC社製)上に載せ、ストップウォッチで計時を開始する。棒の先端にて試料を均一に攪拌し、糸状に試料が切れてプレートに残るようになった時、ストップウォッチを止める。この試料が切れてプレートに残るようになるまでの時間をゲルタイムとした。
粘弾性測定装置(レオメトリック社製固体粘弾性測定装置RSAII、二重カレンチレバー法;周波数1Hz、昇温速度3℃/min)を用いて測定した。
幅12.7mm、長さ127mm、厚み1.6mmの評価用サンプルを、トランスファー成形機を用い175℃の温度で90秒成形した後、175℃の温度で5時間後硬化して作成した。作成した試験片を用いUL−94試験法に準拠し、厚さ1.6mmの試験片5本を用いて、燃焼試験を行った。
*1:1回の接炎における最大燃焼時間(秒)
*2:試験片5本の合計燃焼時間(秒)
表1中の略号は以下の通りである。
NC−3000:下記構造式で表されるビフェニルアラルキル型エポキシ樹脂(日本化薬株式会社製「NC−3000」、エポキシ当量:274g/eq)
XLC−3L:フェノールアラルキル樹脂(三井化学株式会社製「XLC−3L」水酸基当量:176g/eq)
TPP:トリフェニルホスフィン
シリカ S−COL:球状シリカ(株式会社マイクロン製「S−COL」)
KBM−403:γ−グリシドキシトリエトキシキシシラン(信越化学工業株式会社製シランカップリング剤「KBM−403」)
PEARL WAX No.1−P:カルナバワックス(株式会社セラリカ野田製「PEARL WAX No.1−P」
Claims (6)
- β−ナフトール化合物、フェノール化合物、及びホルムアルデヒドの重縮合体をポリグリシジルエーテル化したエポキシ樹脂(α)であって、該エポキシ樹脂(α)中に、下記構造式(1)
で表されるエポキシ樹脂(x1)、下記構造式(2)
で表されるエポキシ樹脂(x2)、及び、下記構造式(3)
で表されるエポキシ樹脂(x3)を含有することを特徴とするエポキシ樹脂。 - エポキシ樹脂(A)と硬化剤(B)とを必須成分とする熱硬化性樹脂組成物であって、前記エポキシ樹脂(A)として、請求項1記載のエポキシ樹脂を用いることを特徴とする硬化性樹脂組成物。
- 前記エポキシ樹脂(A)及び硬化剤(B)に加え、さらに、無機充填剤を含有する請求項2記載の硬化性樹脂組成物。
- 請求項2又は3記載の硬化性樹脂組成物を硬化させてなる硬化物。
- 請求項3記載の硬化性樹脂組成物からなる半導体封止材料。
- 請求項2記載のエポキシ樹脂(A)及び硬化剤(B)に加え、更に、有機溶剤を配合してワニス化した樹脂組成物を、補強基材に含浸し銅箔を重ねて加熱圧着させることにより得られたプリント配線基板。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011145746A JP5708306B2 (ja) | 2011-06-30 | 2011-06-30 | エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011145746A JP5708306B2 (ja) | 2011-06-30 | 2011-06-30 | エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013010903A JP2013010903A (ja) | 2013-01-17 |
| JP5708306B2 true JP5708306B2 (ja) | 2015-04-30 |
Family
ID=47685053
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2011145746A Active JP5708306B2 (ja) | 2011-06-30 | 2011-06-30 | エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5708306B2 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6198038B2 (ja) * | 2013-03-27 | 2017-09-20 | Dic株式会社 | 硬化性組成物、硬化物、及びプリント配線基板 |
| JP6277595B2 (ja) * | 2013-03-28 | 2018-02-14 | Dic株式会社 | 硬化性組成物、硬化物、及びプリント配線基板 |
| JP6277594B2 (ja) * | 2013-03-28 | 2018-02-14 | Dic株式会社 | 硬化性組成物、硬化物、及びプリント配線基板 |
| JP6125967B2 (ja) | 2013-09-30 | 2017-05-10 | 明和化成株式会社 | エポキシ樹脂組成物、封止材、その硬化物、及びフェノール樹脂 |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH03124723A (ja) * | 1989-10-09 | 1991-05-28 | Sumitomo Metal Ind Ltd | 新規エポキシ樹脂とその製法 |
| JP3074013B2 (ja) * | 1989-11-20 | 2000-08-07 | 日本化薬株式会社 | エポキシ樹脂組成物及びその硬化物 |
| JPH03203922A (ja) * | 1989-12-28 | 1991-09-05 | Sumitomo Metal Ind Ltd | 新規エポキシ樹脂の製造方法 |
| JPH03212419A (ja) * | 1990-01-18 | 1991-09-18 | Sumitomo Metal Ind Ltd | 液状レゾールの製造方法 |
| JP2963260B2 (ja) * | 1991-12-13 | 1999-10-18 | 住友ベークライト株式会社 | エポキシ樹脂組成物 |
| JP3982659B2 (ja) * | 1997-07-18 | 2007-09-26 | 日本化薬株式会社 | ナフトール樹脂、エポキシ樹脂、エポキシ樹脂組成物及びその硬化物 |
| JP4349683B2 (ja) * | 1998-04-28 | 2009-10-21 | 三井化学株式会社 | エポキシ樹脂組成物およびその用途 |
| JP2000026572A (ja) * | 1998-07-07 | 2000-01-25 | Nippon Kayaku Co Ltd | イミド骨格を含有するエポキシ樹脂及びこれを含有する硬化性エポキシ樹脂組成物 |
| JP2000273144A (ja) * | 1999-03-25 | 2000-10-03 | Nippon Kayaku Co Ltd | エポキシ樹脂の製造方法 |
| JP2002167416A (ja) * | 2000-11-30 | 2002-06-11 | Hitachi Chem Co Ltd | フェノール樹脂、これを用いた樹脂組成物及び封止用樹脂成形材料並びに電子部品装置 |
| JP4487625B2 (ja) * | 2004-05-07 | 2010-06-23 | 宇部興産株式会社 | フェノールノボラック樹脂の製造方法 |
-
2011
- 2011-06-30 JP JP2011145746A patent/JP5708306B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013010903A (ja) | 2013-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5071602B2 (ja) | エポキシ化合物、硬化性組成物、及びその硬化物 | |
| JP5293911B1 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP2006274236A (ja) | エポキシ樹脂組成物、その硬化物、半導体封止材料、新規フェノール樹脂、および新規エポキシ樹脂 | |
| JP5136729B2 (ja) | 硬化性樹脂組成物、その硬化物、フェノール樹脂、エポキシ樹脂、及び半導体封止材料 | |
| JP5561571B1 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP5380763B2 (ja) | エポキシ樹脂組成物、その硬化物、半導体封止材料、新規フェノール樹脂、新規エポキシ樹脂、新規フェノール樹脂の製造方法、および新規エポキシ樹脂の製造方法 | |
| JP5708306B2 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、半導体封止材料、及びプリント配線基板 | |
| JP5246481B2 (ja) | 硬化性樹脂組成物、その硬化物、新規エポキシ樹脂、及びその製造方法 | |
| JP5532368B1 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP6083169B2 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP2012077120A (ja) | 硬化性樹脂組成物、その硬化物、フェノール樹脂、エポキシ樹脂、及び半導体封止材 | |
| JP2012201732A (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP2014005338A (ja) | 硬化性組成物、硬化物、及びプリント配線基板 | |
| JP6257020B2 (ja) | フェニルフェノール−ナフトール樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP2013087212A (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP5958104B2 (ja) | 硬化性組成物、硬化物、及びプリント配線基板 | |
| JP6155587B2 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP6032476B2 (ja) | クレゾール−ナフトール樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP6065215B2 (ja) | エポキシ樹脂、硬化性樹脂組成物、その硬化物及び半導体封止材料 | |
| JP5035604B2 (ja) | エポキシ樹脂組成物、その硬化物、および新規エポキシ樹脂 | |
| JP2014058633A (ja) | ビフェノール−ナフトール樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP6048035B2 (ja) | クレゾール−ナフトール樹脂、硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP6002987B2 (ja) | 硬化性樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP2013023560A (ja) | エポキシ樹脂組成物、その硬化物、及びプリント配線基板 | |
| JP5024604B2 (ja) | エポキシ樹脂組成物、その硬化物、新規エポキシ樹脂及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20131125 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140314 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140401 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140523 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20141209 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150114 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20150203 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150216 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5708306 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |