JP5598125B2 - 逆フォトクロミック材料 - Google Patents
逆フォトクロミック材料 Download PDFInfo
- Publication number
- JP5598125B2 JP5598125B2 JP2010157036A JP2010157036A JP5598125B2 JP 5598125 B2 JP5598125 B2 JP 5598125B2 JP 2010157036 A JP2010157036 A JP 2010157036A JP 2010157036 A JP2010157036 A JP 2010157036A JP 5598125 B2 JP5598125 B2 JP 5598125B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- benzyl
- diformylbiphenyl
- photochromic material
- substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images





Landscapes
- Plural Heterocyclic Compounds (AREA)
Description
本発明は逆フォトクロミック材料に関し、特に新規なビイミダゾール化合物からなる逆フォトクロミック材料に関する。
フォトクロミック材料は、光を照射することで色(可視光の透過率)を可逆的に変化させる機能(調光機能)を有し、まぶしさを防ぐためのメガネや、光スイッチ、または表示・非表示の切り替え能を有するインクなどの表示材料として利用される。また、光ディスクなどの記録材料やホログラムとしての応用が研究されている。
フォトクロミック材料による色の変化は光照射による材料の可逆的な化学変化によって発現される。代表的なフォトクロミック材料としては、スピロピラン系化合物、スピロオキサジン系化合物、ナフトピラン系化合物、フルギド系化合物およびジアリールエテン系化合物などが知られている。
フォトクロミック材料は大きく分けて、無色から光照射による構造変化に伴い着色する正のフォトクロミズムと呼ばれる現象を示すものと、逆に、光照射により着色体から無色体へと変化する負のフォトクロミズムと呼ばれる現象を示すもの(逆フォトクロミック材料)とがある。
これらのうち逆フォトクロミック材料については、スピロベンゾピラン誘導体(特許文献1)やジメチルジヒドロピレン誘導体(特許文献2)、ジアリールエテン誘導体(特許文献3)など数例の材料が報告されている。
ところで、調光機能にはその用途に適した色や発色濃度や発色速度などの特性が求められる。そのため、様々な種類の誘導体や新しい分子骨格を有する化合物の開発が必要である。
しかし、逆フォトクロミック材料については、正のフォトクロミック材料に比べて、いまだ種類が少ないというのが現状である。
従って、新たな構造を有する逆フォトクロミック材料が求められていた。
本発明の目的は、初期状態として着色しており、可視光領域に感受性があり、光照射により可逆的な構造変化(色変化)を呈する新しいタイプの逆フォトクロミック材料を提供することにある。
本発明者らは、上記課題を解決するため鋭意研究を重ねた結果、高速フォトクロミック材料であるシクロファン連結型ヘキサアリールビイミダゾールの連結部位であるシクロファンをビフェニル型に改良する過程で、全く新しい逆フォトクロミック分子を見出した。具体的には、本発明者らは、ビイミダゾールを基本骨格とし、一般式(1)のR4およびR5に嵩高い置換基を導入することで負のフォトクロミズムを示す新しい化合物を見出した。この分子は安定状態では着色体であり、可視光を照射することで消色体へと異性化する逆フォトクロミック特性を示した。

(式中R4及びR5はそれぞれ独立してハロゲン原子又はアルキル基を示し、R1〜R3及びR6〜R8はそれぞれ独立して水素原子、ハロゲン原子、アルキル基、フルオロアルキル基、ヒドロキシル基、アルコキシル基、アミノ基、アルキルアミノ基、アルキルカルボニル基、ニトロ基、シアノ基又はアリール基を示す。Ar1〜Ar4はそれぞれ独立して置換又は無置換のアリール基を示す。R4は、R3と共に、縮合した置換又は無置換のアリール環を形成してもよく、R5は、R6と共に、縮合した置換又は無置換のアリール環を形成してもよい。)
即ち本発明は、上記一般式(1)で表されるビイミダゾール化合物からなることを特徴とする逆フォトクロミック材料である。
本発明が提供する逆フォトクロミック材料は、可視光照射により容易に着色体から消色体へと色調が変化する。この性質はフォトクロミック分子が利用されているあらゆる用途への適用が考えられるものである。具体的な用途としては、光スイッチ、印刷用材料、記録材料、ホログラム材料などがある。しかも、本発明の逆フォトクロミック材料は従来の逆フォトクロミズムを示す分子と全く構造が異なるので、逆フォトクロミズムを利用したデバイス開発に新しい選択肢を提供することができる。
本発明の逆フォトクロミック材料は、一般式(1)で表されるビイミダゾール化合物のR4、R5の両部位に立体的に嵩高い置換基を導入することでスピロ環構造を持った構造を得るものである。
R4、R5の両部位の嵩高い置換基としては、ハロゲン原子又はアルキル基が挙げられる。上記置換基は、アルキル基のうちの最も小さいメチル基であってもよい。即ち、逆フォトクロミック材料は、下記一般式(2)で表される。
あるいは、R4は、R3と共に、縮合した置換又は無置換のアリール環を形成し、且つ、R5は、R6と共に、縮合した置換又は無置換のアリール環を形成するものであってもよい。ここで、アリール環がベンゼン環であることが好ましい。この場合の逆フォトクロミック材料は、下記一般式(3)で表される。
(式中R1〜R2及びR7〜R14はそれぞれ独立して水素原子、ハロゲン原子、アルキル基、フルオロアルキル基、ヒドロキシル基、アルコキシル基、アミノ基、アルキルアミノ基、アルキルカルボニル基、ニトロ基、シアノ基又はアリール基を示す。Ar1〜Ar4はそれぞれ独立して置換又は無置換のアリール基を示す。)
本発明の逆フォトクロミック材料は、例えば下記一般式(4)で表される2,2’−ジホルミルビフェニル誘導体と下記一般式(5)及び(6)で表されるジアリールエタンジオン誘導体とを窒素化合物の存在下に反応させてイミダゾール環を含む中間体を得た後にその中間体を酸化反応させることで得られる。ここで、一般式(4)で表される2,2’−ジホルミルビフェニル誘導体に代えて、2,2’−ジホルミル−1,1’−ビナフタレン誘導体を用いることもできる。また2,2’−ジホルミルビフェニル誘導体には、2,2’−ジホルミルビフェニルは含まれない。2,2’−ジホルミルビフェニルが含まれると、本発明の逆フォトクロミック材料が得られなくなるためである。尚、本発明の逆フォトクロミック材料は、二つのホルミル基に異なるジアリールエタンジオン誘導体を反応させることによっても得られる。本発明の効果は、本発明の構造体分子を得られるのであればどのような合成ルートをとっていてもよく、合成ルートによって何ら限定されるものではない。
上記一般式(2)で表される本発明の逆フォトクロミック分子の合成原料に使用する2,2’−ジホルミルビフェニル誘導体は、6,6’位に立体的に大きな置換基を有することが好ましい。このような2,2’−ジホルミルビフェニル誘導体としては、例えば6,6’ジメチル−2,2’−ジホルミルビフェニル、6,6’ジエチル−2,2’−ジホルミルビフェニル、6,6’ジメチル−2,2’−ジ−nプロピルビフェニル、6,6’ジメチル−2,2’−ジイソプロピルビフェニル、6,6’ジメチル−2,2’−ジ−nブチルビフェニル、6,6’ジメチル−2,2’−ジイソブチルビフェニル、6,6’ジメチル−2,2’−ジ−tertブチルビフェニル、6,6’ジトリフルオロメチル−2,2’−ジホルミルビフェニル、4,4’ジヒドロキシ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジメトキシ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジアセトキシ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジアミノ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ビス(ジメチルアミノ)−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジフルオロ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジクロロ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジブロモ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジヨード−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジニトロ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジシアノ−6,6’−ジメチル−2,2’−ジホルミルビフェニル、4,4’ジメトキシカルボニル−6,6’−ジメチル−2,2’−ジホルミルビフェニル、3,3’,6,6’−テトラメチル−2,2’−ジホルミルビフェニル、4,4’,6,6’−テトラメチル−2,2’−ジホルミルビフェニル、5,5’,6,6’−テトラメチル−2,2’−ジホルミルビフェニル、6,6’−ジクロロ−2,2’−ジホルミルビフェニル、6,6’−ジブロモ−2,2’−ジホルミルビフェニル、6,6’−ジヨード−2,2’−ジホルミルビフェニル、4,4’,6,6’−テトラクロロ−2,2’−ジホルミルビフェニル、4,4’,6,6’−テトラブロモ−2,2’−ジホルミルビフェニル、4,4’−ジフルオロ−6,6’−ジクロロ−2,2’−ジホルミルビフェニル、4,4’−ジフルオロ−6,6’−ジブロモ−2,2’−ジホルミルビフェニル、4,4’−ジフルオロ−6,6’−ジヨード−2,2’−ジホルミルビフェニル、3,3’,4,4’,5,5’−ヘキサフルオロ−6,6’
−ジブロモ−2,2’−ジホルミルビフェニル、5,5’ジメトキシ−4,4’,6,6’−テトラメチル−2,2’−ジホルミルビフェニル、などが例示できるが、これらに限定されるものではない。
−ジブロモ−2,2’−ジホルミルビフェニル、5,5’ジメトキシ−4,4’,6,6’−テトラメチル−2,2’−ジホルミルビフェニル、などが例示できるが、これらに限定されるものではない。
また、2,2’−ジホルミル−1,1’−ビナフタレン誘導体としては、2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジブロモ−2,2’−ジホルミル−1,1’−ビナフタレン、4,4’−ジブロモ−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジブロモ−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジクロロ−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジヨード−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジメトキシメチル−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジフェニル−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジメトキシカルボニル−2,2’−ジホルミル−1,1’−ビナフタレン、4,4’−ジシアノ−2,2’−ジホルミル−1,1’−ビナフタレン、7,7’−ジヒドロキシ−2,2’−ジホルミル−1,1’−ビナフタレン、7,7’−ジメトキシ−2,2’−ジホルミル−1,1’−ビナフタレン、7,7’−ビス(メタクロロベンジロキシ)−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジブロモ−3,3’−ジメトキシ−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジメトキシカルボニル−4,4’−ジヒドロキシ−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’,6,6’−テトラフェニル−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジメチル−7,7’−ジヒドロキシ−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジメチル−7,7’−ビス(メタクロロベンジロキシ)−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’,4,4’−テトラヒドロキシ−2,2’−ジホルミル−1,1’−ビナフタレン、3,3’−ジメトキシ−4,4’−ジアミノ−2,2’−ジホルミル−1,1’−ビナフタレン、6,6’−ジメチル−7,7’−ジヒドロキシ−2,2’−ジホルミル−1,1’−ビナフタレン、6−メトキシカルボニルエチル−2,2’−ジホルミル−1,1’−ビナフタレンなどが例示できるが、これらに限定されるものではない。
本発明の逆フォトクロミック材料の原料に使用するジアリールエタンジオン誘導体は、例えば2−フルオロベンジル、3−フルオロベンジル、4−フルオロベンジル、2−クロロベンジル、3−クロロベンジル、4−クロロベンジル、2−ブロモベンジル、3−ブロモベンジル、4−ブロモベンジル、2−ヨードベンジル、3−ヨードベンジル、4−ヨードベンジル、2−ヒドロキシベンジル、3−ヒドロキシベンジル、4−ヒドロキシベンジル、2−メトキシベンジル、3−メトキシベンジル、4−メトキシベンジル、2−エトキシベンジル、3−エトキシベンジル、4−エトキシベンジル、2−フェノキシベンジル、3−フェノキシベンジル、4−フェノキシベンジル、2−アセトキシベンジル、3−アセトキシベンジル、4−アセトキシベンジル、2−メチルベンジル、3−メチルシベンジル、4−メチルベンジル、2−カルボキシベンジル、3−カルボキシベンジル、4−カルボキシベンジル、2−(メチルカルボキシ)ベンジル、3−(メチルカルボキシ)ベンジル、4−(メチルカルボキシ)ベンジル、2−(フェニルカルボキシ)ベンジル、3−(フェニルカルボキシ)ベンジル、4−(フェニルカルボキシ)ベンジル、2−ニトロベンジル、3−ニトロベンジル、4−ニトロベンジル、2−シアノベンジル、3−シアノベンジル、4−シアノベンジル、2−メチルチオベンジル、3−メチルチオベンジル、4−メチルチオベンジル、2−フェニルチオベンジル、3−フェニルチオベンジル、4−フェニルチオベンジル、2−フェニルアセチレニルベンジル、3−フェニルアセチレニルベンジル、4−フェニルアセチレニルベンジル、2−スチリルベンジル、3−スチリルベンジル、4−スチリルベンジル、2−フェニルメチルベンジル、3−フェニルメチルベンジル、4−フェニルメチルベンジル、2−アミノベンジル、3−アミノベンジル、4−アミノベンジル、2−ジメチルアミノベンジル、3−ジメチルアミノベンジル、4−ジメチルアミノベンジル、2−ブロモメチルベンジル、3−ブロモメチルベンジル、4−ブロモメチルベンジル、2−メトキシメチルベンジル、3−メトキシメチルベンジル、4−メトキシメチルベンジル、2−(N−メチルアミノカルボニル)ベンジル、3−(N−メチルアミノカルボニル)ベンジル、4−(N−メチルアミノカルボニル)ベンジル、2−(N−フェニルアミノカルボニル)ベンジル、3−(N−フェニルアミノカルボニル)ベンジル、4−(N−フェニルアミノカルボニル)ベンジル、2−トリフルオロメチルベンジル、3−トリフルオロメチルベンジル、4−トリフルオロメチルベンジル、3,4−メチレンジオキシベンジル、3,4−エチレンジオキシベンジル、3,4−エチレンジチオベンジル、2,4−ジヒドロキシベンジル、2,4−ジメチルベンジル、2,3−ジメチルベンジル、3,4−ジメチルベンジル、3,4−ジメトキシベンジル、2,4−ジメトキシベンジル、2,4−ジニトロベンジル、2−メチル−3−ニトロベンジル、2−メトキシ−3−ニトロベンジル、2−クロロ−4−メトキシベンジル、3−クロロ−4−アミノベンジル、2,3−ジクロロベンジル、3,4−ジクロロベンジル、2−ジフルオロメチル−3−メチルベンジル、3−フルオロ−4−ブロモベンジル、3,5−ジメトキシベンジル、3,5−ジフルオロベンジル、3,4,5−トリメトキシベンジル、4−(2−オキソ−フェニルアセチル)ナフタレン−1,8−ジカルボキシリックアシッドアンハイドライド、1−(9−オキソフルオレン−2−イル)−2−フェニルエタンジオン、1−(4−ニトロフェニル)−3−[4−(2−オキソ−2−フェニルアセチル)フェニル]ウレア、エチル−4−メチル−2−{[4−(2−オキソ−2−フェニルアセチル)フェニル]カルボニルアミノ}−1,3−チアゾール−5−カルボキシレート、N−(2−メトキシ−5−メチルフェニル)[4−(2−オキソ−2−フェニルアセチル)フェニル]カルボキシアミド、N−(2−メトキシエチル)[4−(2−オキソ−2−フェニルアセチル)フェニル]カルボキシアミド、4−クロロ−2−メチルフェニル−4−(2−オキソ−2−フェニルアセチル)ベンゾエイト、1−フェニル−2−(2−ナフチル)エタンジオン、1−フェニル−2−(1−ナフチル)エタンジオン、1−フェニル−2−[4−(4−ニトロフェノキシ)フェニル]エタンジオン、カルバモイルメチル−4−(2−オキソ−2−フェニルアセチル)ベンゾエイト、1−アセナフテン−5−イル−2−フェニルエタンジオン、2,3,4,5,6−ペンタクロロベンジル、ベンジル、2,2’−ジフルオロベンジル、3,3’−ジフルオロベンジル、4,4’−ジフルオロベンジル、2,2’−ジクロロベンジル、3,3’−ジクロロベンジル、4,4’−ジクロロベンジル、2,2’−ジブロモベンジル、3,3’−ジブロモベンジル、4,4’−ジブロモベンジル、2,2’−ジヨードベンジル、3,3’−ジヨードベンジル、4,4’−ジヨードベンジル、2,2’−ジヒドロキシベンジル、3,3’−ジヒドロキシベンジル、4,4’−ジヒドロキシベンジル、2,2’−ジメトキシベンジル、3,3’−ジメトキシベンジル、4,4’−ジメトキシベンジル、2,2’−ジエトキシベンジル、3,3’−ジエトキシベンジル、4,4’−ジエトキシベンジル、2,2’−ジフェノキシベンジル、3,3’−ジフェノキシベンジル、4,4’−ジフェノキシベンジル、2,2’−ジアセトキシベンジル、3,3’−ジアセトキシベンジル、4,4’−ジアセトキシベンジル、2,2’−ジメチルベンジル、3,3’−ジメチルシベンジル、4,4’−ジメチルベンジル、2,2’−ジカルボキシベンジル、3,3’−ジカルボキシベンジル、4,4’−ジカルボキシベンジル、2,2’−ビス(メチルカルボキシ)ベンジル、3,3’−ビス(メチルカルボキシ)ベンジル、4,4’−ビス(メチルカルボキシ)ベンジル、2,2’−ビス(フェニルカルボキシ)ベンジル、3,3’−ビス(フェニルカルボキシ)ベンジル、4,4’−ビス(フェニルカルボキシ)ベンジル、2,2’−ジニトロベンジル、3,3’−ジニトロベンジル、4,4’−ジニトロベンジル、2,2’−ジシアノベンジル、3,3’−ジシアノベンジル、4,4’−ジシアノベンジル、2,2’−ジメチルチオベンジル、3,3’−ジメチルチオベンジル、4,4’−ジメチルチオベンジル、2,2’−ジフェニルチオベンジル、3,3’−ジフェニルチオベンジル、4,4’−ジフェニルチオベンジル、2,2’−ジフェニルアセチレニルベンジル、3,3’−ジフェニルアセチレニルベンジル、4,4’−ジフェニルアセチレニルベンジル、2,2’−ジスチリルベンジル、3,3’−ジスチリルベンジル、4,4’−ジスチリルベンジル、2,2’−ジフェニルメチルベンジル、3,3’−ジフェニルメチルベンジル、4,4’−ジフェニルメチルベンジル、2,2’−ジアミノベンジル、3,3’−ジアミノベンジル、4,4’−ジアミノベンジル、2,2’−ビス(ジメチルアミノ)ベンジル、3,3’−ビス(ジメチルアミノ)ベンジル、4,4’−ビス(ジメチルアミノ)ベンジル、2,2’−ジブロモメチルベンジル、3,3’−ジブロモメチルベンジル、4,4’−ジブロモメチルベンジル、2,2’−ジメトキシメチルベンジル、3,3’−ジメトキシメチルベンジル、4,4’−ジメトキシメチルベンジル、2,2’−ビス(N−メチルアミノカルボニル)ベンジル、3,3’−ビス(N−メチルアミノカルボニル)ベンジル、4,4’−ビス(N−メチルアミノカルボニル)ベンジル、2,2’−ビス(N−フェニルアミノカルボニル)ベンジル、3,3’−ビス(N−フェニルアミノカルボニル)ベンジル、4,4’−ビス(N−フェニルアミノカルボニル)ベンジル、2,2’−ビス(トリフルオロメチル)ベンジル、3,3’−ビス(トリフルオロメチル)ベンジル、4,4’−ビス(トリフルオロメチル)ベンジル、3,4,3’,4’−ジメチレンジオキシベンジル、3,4,3’,4’−ジエチレンジオキシベンジル、3,4,3’,4’−ジエチレンジチオベンジル、2,2’,4,4’−テトラメチルベンジル、3,3’,4,4’−テトラメトキシベンジル、2,2’,4,4’−テトラメトキシベンジル、2,2’,4,4’−テトラニトロベンジル、2,2’−ジメチル−3,3’−ジニトロベンジル、2,2’−ジメトキシ−3,3’−ジニトロベンジル、2,2’−ジクロロ−4,4’−ジメトキシベンジル、3,3’−ジクロロ−4,4’−ジアミノベンジル、2,2’,3,3’−テトラクロロベンジル、3,3’,4,4’−テトラクロロベンジル、2,2’−ビス(ジフルオロメチル)−3,3’−ジメチルベンジル、3,3’−ジフルオロ−4,4’−ジブロモベンジル、3,3’,5,5’−テトラメトキシベンジル、3,3’,5,5’−テトラフルオロベンジル、3,3’,4,4’,5,5’−ヘキサメトキシベンジル、4,4’−ビス(3−メチル−3−ヒドロキシ−1−ブチニル)ベンジル、4,4’−ジフェニルベンジル、4,4’−ビス(N−モルフォリニル)ベンジル、1,2−ビス(2,3,6,7−テトラヒドロ−1H,5H−ピリド[3,2,1−ij]キノリン−9−イル)エタンジオン、3,3’−ジニトロ−4,4’−ジクロロベンジル、3,3’−ジニトロ−4,4’−ジメトキシベンジル、3,3’−ジニトロ−4,4’−ジブロモベンジル、4−メチル−4’−クロロベンジル、2−クロロ−3’,4’−ジメトキシベンジル、2,4’−ジブロモベンジル、2,3,4,5,6−ペンタクロロ−2’,3’,4’,5’,6’
ペンタフルオロベンジルなどが挙げられるが、これらに限定されるものではない。
ペンタフルオロベンジルなどが挙げられるが、これらに限定されるものではない。
一般式(4)で表される2,2’−ジホルミルビフェニル誘導体と一般式(5)及び(6)で表されるジアリールエタンジオン誘導体とを反応させるための温度は、触媒の有無及び使用する触媒によって異なるため一概には言えないが、例えば触媒を使用せず溶媒として酢酸を使用する場合、反応温度は、通常80〜120℃であり、好ましくは100〜120℃である。また触媒として、例えばZrCl4やヨウ素を使用した場合、反応温度は、触媒の存在下、20℃〜75℃とすることができる。
反応時間も、触媒の有無及び使用する触媒によって異なるため一概には言えないが、例えば触媒を使用せず溶媒として酢酸を使用する場合、反応温度は4〜32時間であり、好ましくは12〜24時間である。また、触媒として例えばZrCl4やヨウ素を使用した場合は、反応時間は、触媒存在下、1〜10時間とすることができる。
上記窒素化合物としては、例えば酢酸アンモニウム及びアンモニアなどが挙げられるが、中でも、加熱時に揮発しにくいという理由から、酢酸アンモニウムが好ましい。
上記反応の溶媒としては、原料を溶解できるものであれば特に制限なく使用可能である。このような溶媒としては、例えば酢酸、アセトニトリルのような極性溶媒が好ましい。
中間体の酸化反応は、例えば中間体を溶媒中に溶解させて溶液を調製し、この溶液に酸化剤を加えることによって行うことができる。
溶媒は、中間体を溶解できるものであれば特に制限なく使用可能である。このような溶媒としては、例えばベンゼン、塩化メチレンなどを用いることができる。
酸化剤は、中間体を酸化して逆フォトクロミック材料を得ることができるものであれば特に制限なく使用可能である。このような酸化剤としては、例えばフェリシアン化カリウム、酸化鉛が挙げられるが、中でも、フェリシアン化カリウムが好ましい。これは、フェリシアン化カリウムの反応性がより高いためである。
尚、上記溶液には、酸化剤のほか、さらにアルカリを添加する。このようなアルカリとしては、例えば水酸化カリウムなどを用いることができる。
酸化反応は、不活性ガス雰囲気下、遮光条件下で行うことが好ましい。不活性ガス雰囲気とするのは、酸素との反応抑制のためである。不活性ガスとしては、例えば窒素を用いることが可能である。遮光条件下で酸化反応を行うことが好ましいのは、得られる逆フォトクロミック材料が光によって着色体から無色体に変化することを防止するためである。
以下、本発明について実施例により具体的に説明するが、本発明は下記実施例に限定されるものではない。
原料として、6,6’ジメチル−2,2’−ジホルミルビフェニル、2,2’−ジホルミル−1,1’−ビナフタレン、2,2’−ジホルミルビフェニルは合成したものを用い、ベンジル、4,4’−ジメトキシベンジル、4,4’−ビス(ジメチルアミノ)ベンジル、4,4’−ジブロモベンジル、酢酸アンモニウム、酢酸、フェリシアン化カリウム、水酸化カリウムは市販の試薬(東京化成工業株式会社製)を用いた。
評価方法として、フォトクロミック分子は、2.0×10−4Mに調整したベンゼン溶液にUVスポット光源L8333(浜松ホトニクス製)による紫外可視光を励起光として照射し、可視光領域で最も透過率変化の大きな波長の透過率変化を観測する紫外可視吸収スペクトル分析によって発色体の及び消色体の透過率測定を行った。
<合成例1>
6,6’ジメチル−2,2’−ジホルミルビフェニル100mgと4,4’−ジメトキシベンジル270mgと酢酸アンモニウム960mgと酢酸4.0mlを混合し、110℃のオイルバスで16時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(I)を273mg得た。NMRの分析によって中間体(I)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.92(2H s)、8.38(2H d)、7.53(2H t)、7.46(4H d)、7,40(2H d)、6.94(4H d)、6.80(4H d)、6.75(4H d)3.78(12H s)、1.99(6H s)
6,6’ジメチル−2,2’−ジホルミルビフェニル100mgと4,4’−ジメトキシベンジル270mgと酢酸アンモニウム960mgと酢酸4.0mlを混合し、110℃のオイルバスで16時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(I)を273mg得た。NMRの分析によって中間体(I)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.92(2H s)、8.38(2H d)、7.53(2H t)、7.46(4H d)、7,40(2H d)、6.94(4H d)、6.80(4H d)、6.75(4H d)3.78(12H s)、1.99(6H s)
上記の化合物120mgをベンゼン25mlに溶解させ、フェリシアン化カリウム4.1gと水酸化カリウム1.8gを30mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して再結晶を行い、逆フォトクロミック分子[1]を含む混合物を102mg、逆フォトクロミック分子[1]として24mg得た。NMRの分析によって逆フォトクロミック分子[1]の生成を確認した。尚、NMRの分析は混合物について行ったものであり、逆フォトクロミック分子[1]単品のNMRの分析結果は得られていないが、メトキシ基とメチル基の特徴的なピークと逆フォトクロミック特性を示すことから混合物中に下記構造式で表される逆フォトクロミック分子[1]が含まれていることは明らかと考えられる。
<合成例2>
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ジメトキシベンジル183mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで14時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(II)を158mg得た。NMRの分析によって中間体(II)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.78(2H d)、8.45(2H s)、8.19(2H d)、8.02(2H d)7.55-7.51(2H m)、7.42(4H d)、7.32-7.30(4H m)、6.78(4H d)6.69-6.64(8H m)、3.77(12H s)
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ジメトキシベンジル183mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで14時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(II)を158mg得た。NMRの分析によって中間体(II)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.78(2H d)、8.45(2H s)、8.19(2H d)、8.02(2H d)7.55-7.51(2H m)、7.42(4H d)、7.32-7.30(4H m)、6.78(4H d)6.69-6.64(8H m)、3.77(12H s)
上記の化合物84mgをベンゼン25mlに溶解させ、フェリシアン化カリウム2.5gと水酸化カリウム1.1gを20mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して再結晶を行い、逆フォトクロミック分子[2]を79mg得た。NMRの分析とX線結晶構造解析によって逆フォトクロミック分子[2]の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.24(1H d)、7.92(1H d)、7.90(1H d)、7.77(1H d)、7.50(2H d)、7.47-7.42(5H m)、7.30-7.20(4H m)、7.09-7.05(1H m)、6.93(1H d)、6.89(1H d)、6.85(2H d)、6.80(2H d)6.73(2H d)、6.54(2H d)、6.44(2H d)、3.82(3H s)、3.81(3H s)、3.73(3H s)、3.73(3H s)
<合成例3>
2,2’−ジホルミル−1,1’−ビナフタレン100mgとベンジル149mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで14時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(III)を162mg得た。NMRの分析によって中間体(III)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.76(2H d)、8.69(2H s)、8.20(2H d)、8.04(2H d)7.56-7.51(2H m)、7.51-7.49(4H m)、7.33-7.31(4H m)、7.24-7.14(12H m)6.75-6.73(4H m)
2,2’−ジホルミル−1,1’−ビナフタレン100mgとベンジル149mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで14時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(III)を162mg得た。NMRの分析によって中間体(III)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.76(2H d)、8.69(2H s)、8.20(2H d)、8.04(2H d)7.56-7.51(2H m)、7.51-7.49(4H m)、7.33-7.31(4H m)、7.24-7.14(12H m)6.75-6.73(4H m)
上記の化合物87mgをベンゼン25mlに溶解させ、フェリシアン化カリウム2.9gと水酸化カリウム1.3gを20mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して再結晶を行い、逆フォトクロミック分子[3]を75mg得た。NMRの分析によって逆フォトクロミック分子[3]の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.26(1H d)、7.93(2H d)、7.80(1H d)、7.57(2H d)、7.47(2H d)、7.47(2H d)、7.44(3H d)、7.40(1H d)、7.35-7.31(4H m)、7.30-7.21(3H m)、7.19-7.15(3H m)、7.13-7.07(2H m)、7.01(2H t)、6.98(1H d)、6.94(1H d)、6.55(2H d)
<合成例4>
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ビス(ジメチルアミノ)ベンジル210mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで18時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(IV)を114mg得た。NMRの分析によって中間体(IV)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.84(2H br.s)、8.28(2H br.s)、8.18(2H d)、8.00(2H d)7.51-7.48(2H m)、7.43(4H br.s)、7.30-7.28(4H m)、6.63(8H br.s)6.49(4H br.s)、2.91(24H s)
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ビス(ジメチルアミノ)ベンジル210mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで18時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(IV)を114mg得た。NMRの分析によって中間体(IV)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.84(2H br.s)、8.28(2H br.s)、8.18(2H d)、8.00(2H d)7.51-7.48(2H m)、7.43(4H br.s)、7.30-7.28(4H m)、6.63(8H br.s)6.49(4H br.s)、2.91(24H s)
上記の化合物80mgをベンゼン25mlに溶解させ、フェリシアン化カリウム2.3gと水酸化カリウム1.0gを20mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して固体を析出させた。析出した固体をエタノールに溶解させて再結晶を行い、逆フォトクロミック分子[4]を59mg得た。NMRの分析によって逆フォトクロミック分子[4]の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.23(1H d)、7.91(1H d)、7.86(1H d)、7.74(1H d)、7.52-7.50(3H m)、7.47(4H d)、7.23(1H t)、7.19-7.17(3H m)、7.02-6.98(1H m)、6.89(1H d)、6.76(1H d)、6.62(2H d)、6.59-6.56(4H m)6.40(2H d)、6.33(2H d)、3.00(6H s)、2.99(6H s)、2.87(6H s)、2.86(6H s)
<合成例5>
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ジブロモベンジル261mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで18時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(V)を249mg得た。NMRの分析によって中間体(V)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.82(2H s)、8.62(2H d)、8.19(2H d)、8.04(2H d)7.84-7.82(4H m)、7.69-7.67(4H m)、7.56(2H t)、7.35-7.29(8H m)、6.61(4H d)
2,2’−ジホルミル−1,1’−ビナフタレン100mgと4,4’−ジブロモベンジル261mgと酢酸アンモニウム750mgと酢酸4.0mlを混合し、110℃のオイルバスで18時間加熱攪拌を行い反応させた後に28%アンモニア水8.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(V)を249mg得た。NMRの分析によって中間体(V)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.82(2H s)、8.62(2H d)、8.19(2H d)、8.04(2H d)7.84-7.82(4H m)、7.69-7.67(4H m)、7.56(2H t)、7.35-7.29(8H m)、6.61(4H d)
上記の化合物80mgをベンゼン10mlに溶解させ、フェリシアン化カリウム2.0gと水酸化カリウム0.90gを15mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して固体を析出させた。析出した固体をエタノールに溶解させて再結晶を行い、逆フォトクロミック分子[5]を22mg得た。NMRの分析によって逆フォトクロミック分子[5]の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.21(1H d)、7.95(2H d)、7.83(1H d)、7.49(2H d)、7.43(2H d)、7.40-7.29(12H m)、7.23(2H d)、7.16(2H d)、7.08(1H d)、6.96(1H d)6.36(2H d)
<合成例6>
2,2’−ジホルミルビフェニル50mgと4,4’−ジメトキシベンジル142mgと酢酸アンモニウム550mgと酢酸3.0mlを混合し、110℃のオイルバスで24時間加熱攪拌を行い反応させた後に28%アンモニア水6.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(VI)を136mg得た。NMRの分析によって中間体(VI)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ9.37(2H s)、8.31(2H d)、7.56(2H t)、7.48-7.38(8H m)、7,29(2H d)、7.00(2H br.s)、6.77(8H d)3.78(12H s)
2,2’−ジホルミルビフェニル50mgと4,4’−ジメトキシベンジル142mgと酢酸アンモニウム550mgと酢酸3.0mlを混合し、110℃のオイルバスで24時間加熱攪拌を行い反応させた後に28%アンモニア水6.0mlを加えて固体を析出させながら中和して、固体を水洗浄後にろ過して真空乾燥機で乾燥した。乾燥した固体をシリカゲルカラムで分離精製した後に溶媒を濃縮して中間体(VI)を136mg得た。NMRの分析によって中間体(VI)の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ9.37(2H s)、8.31(2H d)、7.56(2H t)、7.48-7.38(8H m)、7,29(2H d)、7.00(2H br.s)、6.77(8H d)3.78(12H s)
上記の化合物89mgをベンゼン35mlに溶解させ、フェリシアン化カリウム3.2gと水酸化カリウム1.4gを25mlの水に溶解させた溶液を窒素下遮光条件で加えて、室温で2時間撹拌して反応させた後に水層を分離してベンゼンで抽出し、溶媒を濃縮して固体を析出させた。析出した固体をエタノールに溶解させて再結晶を行ったがフォトクロミズムを示さない化合物[6]を86mg得た。NMRの分析及びマススペクトルによって化合物[6]の生成を確認した。尚、NMRの分析結果は下記の通りである。1H‐NMR(500MHz CDCl3)δ8.06(2H d)、7.45(2H t)、7.42(8H d)、7,19(2H t)、6.99(2H d)、6.79(8H d)3.80(12H s)
FD-MS m/z=708(M+)
FD-MS m/z=708(M+)
<実施例1>
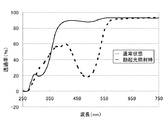
合成例1で合成した逆フォトクロミック分子[1]を含む混合物を用いて1.0×10−3Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図1に示す。尚、図1において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図1に示すように、発色体の最大吸収波長496nmの透過率は60%だったものから93%まで増加した。
合成例1で合成した逆フォトクロミック分子[1]を含む混合物を用いて1.0×10−3Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図1に示す。尚、図1において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図1に示すように、発色体の最大吸収波長496nmの透過率は60%だったものから93%まで増加した。
<実施例2>
合成例2で合成した逆フォトクロミック分子[2]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図2に示す。尚、図2において、実線は励起光を照射している状態を示し、破線は、光を照射しない通常の状態を示す。表1及び図2に示すように、発色体の最大吸収波長493nmの透過率は17%だったものから88%まで増加した。
合成例2で合成した逆フォトクロミック分子[2]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図2に示す。尚、図2において、実線は励起光を照射している状態を示し、破線は、光を照射しない通常の状態を示す。表1及び図2に示すように、発色体の最大吸収波長493nmの透過率は17%だったものから88%まで増加した。
<実施例3>
合成例3で合成した逆フォトクロミック分子[3]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図3に示す。尚、図3において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図3に示すように、発色体の最大吸収波長475nmの透過率は16%だったものから73%まで増加した。
合成例3で合成した逆フォトクロミック分子[3]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図3に示す。尚、図3において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図3に示すように、発色体の最大吸収波長475nmの透過率は16%だったものから73%まで増加した。
<実施例4>
合成例4で合成した逆フォトクロミック分子[4]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図4に示す。尚、図4において、実線は、励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図4に示すように、発色体の最大吸収波長544nmの透過率は23%だったものから74%まで増加した。
合成例4で合成した逆フォトクロミック分子[4]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図4に示す。尚、図4において、実線は、励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図4に示すように、発色体の最大吸収波長544nmの透過率は23%だったものから74%まで増加した。
<実施例5>
合成例5で合成した逆フォトクロミック分子[5]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図5に示す。尚、図5において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図5に示すように、発色体の最大吸収波長486nmの透過率は39%だったものから74%まで増加した。
合成例5で合成した逆フォトクロミック分子[5]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、可視光領域で最も透過率変化の大きな波長の透過率を観測する紫外可視吸収スペクトル分析によって発色体と消色体の測定をした。結果を表1及び図5に示す。尚、図5において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図5に示すように、発色体の最大吸収波長486nmの透過率は39%だったものから74%まで増加した。
<比較例1>
合成例6で合成した化合物[6]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、500nmの透過率を観測する紫外可視吸収スペクトル分析によって励起光を照射しない状態の透過率と励起光を照射している状態の透過率を測定した。結果を表1及び図6に示す。尚、図6において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図6に示すように、透過率は93%から変化しなかった。
合成例6で合成した化合物[6]を用いて2.0×10−4Mのベンゼン溶液を調製した。この溶液を四面石英セルに入れ励起光を照射し、500nmの透過率を観測する紫外可視吸収スペクトル分析によって励起光を照射しない状態の透過率と励起光を照射している状態の透過率を測定した。結果を表1及び図6に示す。尚、図6において、実線は励起光を照射している状態を示し、破線は、通常の状態(励起光を照射していない状態)を示す。表1及び図6に示すように、透過率は93%から変化しなかった。
表1に示す結果より、一般式(1)で表されるビイミダゾール化合物のR4およびR5に立体的に嵩高い置換基を導入することで、逆フォトクロミズムが発現する分子が得られた。
本発明の逆フォトクロミック材料は赤色から紫色を呈しており、可視光に感受性を持ちその光を吸収して透過率を大きく上げる逆フォトクロミック特性を示す。この特性を利用して、これまで応用が提案されてきた光スイッチ、印刷用材料、記録材料などの分野で利用できる。例えば、光メモリ素子のマスク層材料として用いれば、再生信号を劣化させることなく高密度記録されたビットから良好な再生信号を得ることが可能となる。しかも、本発明の分子は従来の逆フォトクロミズムを示す分子と全く構造が異なるので、逆フォトクロミズムを利用したデバイス開発に新しい選択肢を提供する。
Claims (3)
- 下記一般式(1)で表されるビイミダゾール化合物からなることを特徴とする逆フォトクロミック材料。
- 前記一般式(1)中、R4及びR5がメチル基である請求項1に記載の逆フォトクロミック材料であって、下記一般式(2)で表されることを特徴とする逆フォトクロミック材料。
- 前記一般式(1)中、R4は、R3と共に、縮合した置換又は無置換のベンゼン環を形成し、R5は、R6と共に、縮合した置換又は無置換のベンゼン環を形成する請求項1に記載の逆フォトクロミック材料であって、下記一般式(3)で表されることを特徴とする逆フォトクロミック材料。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010157036A JP5598125B2 (ja) | 2010-07-09 | 2010-07-09 | 逆フォトクロミック材料 |
| EP11803688.8A EP2592130B1 (en) | 2010-07-09 | 2011-07-08 | Photochromic material |
| US13/807,461 US8748629B2 (en) | 2010-07-09 | 2011-07-08 | Photochromic material |
| CN201180021083.7A CN102906215B (zh) | 2010-07-09 | 2011-07-08 | 光致变色材料 |
| PCT/JP2011/065702 WO2012005354A1 (ja) | 2010-07-09 | 2011-07-08 | フォトクロミック材料 |
| KR1020127025504A KR20130091243A (ko) | 2010-07-09 | 2011-07-08 | 포토크로믹 재료 |
| AU2011274914A AU2011274914B2 (en) | 2010-07-09 | 2011-07-08 | Photochromic material |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010157036A JP5598125B2 (ja) | 2010-07-09 | 2010-07-09 | 逆フォトクロミック材料 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2012017442A JP2012017442A (ja) | 2012-01-26 |
| JP2012017442A5 JP2012017442A5 (ja) | 2014-05-29 |
| JP5598125B2 true JP5598125B2 (ja) | 2014-10-01 |
Family
ID=45602920
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010157036A Expired - Fee Related JP5598125B2 (ja) | 2010-07-09 | 2010-07-09 | 逆フォトクロミック材料 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5598125B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN106478843B (zh) * | 2016-09-21 | 2017-12-26 | 同济大学 | 一种含噻吩环的双肟酯类光引发剂及其制备方法和应用 |
| CN110234642A (zh) | 2017-01-31 | 2019-09-13 | 富士胶片和光纯药株式会社 | 逆光致变色化合物 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2009321034A1 (en) * | 2008-11-28 | 2011-07-14 | Jiro Abe | Novel crosslinked hexaaryl bisimidazole compound and derivative thereof, method for producing the compound and precursor compound to be used in the production method |
-
2010
- 2010-07-09 JP JP2010157036A patent/JP5598125B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012017442A (ja) | 2012-01-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2558334C (en) | Reimageable paper | |
| US6475590B1 (en) | Aminium salt or diimonium salt compounds and use thereof | |
| JP2013136603A (ja) | 水溶性テトラゾリウム塩 | |
| JP5598125B2 (ja) | 逆フォトクロミック材料 | |
| JP5920082B2 (ja) | カリックス[4]アレーン骨格を有する化合物 | |
| JPH0745509B2 (ja) | ニッケル錯体 | |
| WO2012005354A1 (ja) | フォトクロミック材料 | |
| JP5640868B2 (ja) | フォトクロミック材料 | |
| JPS6014243A (ja) | カラ−写真記録材料 | |
| JP5747735B2 (ja) | フォトクロミック材料 | |
| US4882428A (en) | Azaporphyrin compounds | |
| JPH04178646A (ja) | ピラゾロアゾールアゾメチン色素 | |
| JP2013180996A (ja) | フォトクロミック化合物 | |
| EP0325742B1 (en) | Phthalocyanine compound and optical recording material using it | |
| JP5928004B2 (ja) | フォトクロミック化合物 | |
| JPS6330617B2 (ja) | ||
| JP3200649B2 (ja) | 遷移金属スピロベンゾピラン錯体、その製造方法及び該錯体からなるフォトクロミック材料 | |
| EP0095324B1 (en) | Photographic recording material employing a nondiffusible magenta dye-releasing compound or precursor thereof | |
| Yokoyama | Creation of Molecularly Integrated Multi-responsive Photochromic Systems | |
| WO2015147126A1 (ja) | ペンタアリールビイミダゾール化合物および該化合物の製造方法 | |
| JPH08245627A (ja) | 架橋スピロピラン化合物 | |
| JPH0262280A (ja) | 光情報記録媒体 | |
| JPH0262281A (ja) | 光情報記録媒体 | |
| JPH0439383A (ja) | フォトクロミック材料 | |
| Robinson | A study of hydrogen transfer photochromism |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130123 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140415 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140715 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140728 |
|
| LAPS | Cancellation because of no payment of annual fees |