JP5113045B2 - 高イソタクチックポリ(プロピレンオキシド)または高イソタクチックポリ(ブチレンオキシド)を直接製造するためのイソタクチック特異的触媒 - Google Patents
高イソタクチックポリ(プロピレンオキシド)または高イソタクチックポリ(ブチレンオキシド)を直接製造するためのイソタクチック特異的触媒 Download PDFInfo
- Publication number
- JP5113045B2 JP5113045B2 JP2008518292A JP2008518292A JP5113045B2 JP 5113045 B2 JP5113045 B2 JP 5113045B2 JP 2008518292 A JP2008518292 A JP 2008518292A JP 2008518292 A JP2008518292 A JP 2008518292A JP 5113045 B2 JP5113045 B2 JP 5113045B2
- Authority
- JP
- Japan
- Prior art keywords
- salph
- isotactic
- poly
- oxide
- catalyst
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/04—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers only
- C08G65/06—Cyclic ethers having no atoms other than carbon and hydrogen outside the ring
- C08G65/08—Saturated oxiranes
- C08G65/10—Saturated oxiranes characterised by the catalysts used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/04—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers only
- C08G65/06—Cyclic ethers having no atoms other than carbon and hydrogen outside the ring
- C08G65/08—Saturated oxiranes
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Polyethers (AREA)
Description
(関連出願の相互参照)
本出願は、仮特許出願番号第60/692,237号(出願日:2005年6月21日)の利益を享受し、ここに参照することによって、その全体が本出願の一部を構成するものとする。
Yoshino,N.ら、Macromol.Chem..189,1903−1913(1988)
本発明の目的は、rac−プロピレンオキシド由来の高イソタクチックPPOおよびrac−1−ブチレンオキシド由来の高イソタクチックポリ(ブチレンオキシド)を直接的に(即ち、分画なしに)与え、低PDIを有する高Mnを得ることを可能とする触媒を提供することにある。
次に、rac−プロピレンオキシド重合とrac−1−ブチレンオキシド重合に有用なイソタクチック特異的な触媒を対象とする、本発明の第一の実施形態に言及する。触媒もまた、C2〜C10アルキレンオキシドとCO2の共重合体であって環式アルキレンカーボネート副生成物が10%未満のものを、米国仮特許出願番号60/616,630または米国特許出願番号11/244,231(ここに参照することによって、その全体が本明細書の一部を構成するものとする)に記載された条件下で調製するのに有用である。
〔ここで開始配位子は、好ましくは、ハロゲン(例えば、Cl、Br、I)、C1〜C20アミド、シアノ、アジド、C1〜C20アルキルカルボキシレート、例えばモノ−、ジ−およびトリカルボキシレート、並びにヒドロキシル置換されたもの、C1〜C20アリールカルボキシレート、例えばアダマンチルカルボキシレート、C1〜C20アルコキシドおよびフェノキシドおよびOHからなる群から選択される〕
からなる群から選択される。
本発明を、以下の実施例によって説明する。
(salph)Coの調製
N,N’−ビス−(3,5−ジ−tert−ブチルサリシリデン)−1,2−フェニレンジアミノコバルト(1)。
テフロンコートした撹拌バー、N,N’−ビス−(3,5−ジ−tert−ブチルサリシリデン)−1,2−フェニレンジアミン(3.0g、5.5mmol)およびコバルト(II)アセテート四水和物(0.98g、3.9mmol)が充填されたフラスコに、窒素下で、脱気エタノール(150mL)を添加した。フラスコを80℃に20分間加熱し、その後、22℃に冷却した。空気中で真空濾過によって溶媒を除去し、固体をメタノールで洗浄した。暗赤色粉末を、塩化メチレン(30mL)中に溶解し、ヘキサン(700mL)で層状とし、次いで0℃に冷却することによって、これを再結晶化した。24時間後、暗赤色結晶1が得られ、これを真空濾過によって単離した(2.3g、98%収率)。IR(KBr、cm−1):2960、2873、1575、1523、1465−1359、1260。結晶データ(固体状態構造、以下に示す):三斜晶系、a=9.2149(3)Å、b=12.9485(5)Å、c=14.3796(6)Å、α=107.645(2)°、β=93.624(2)°、γ=95.851(2)°、V=1618.5(1)Å3、空間群P−1;Z=2、(C36H46CoN2O2)についての式量597.68g/molおよび密度(計算値)=1.226mg/m3;R1=0.0326およびRw2=0.0945(I>2σ(I))。
(salph)CoOAcの調製
(N,N’−ビス−(3,5−ジ−tert−ブチルサリシリデン)−1,2−フェニレンジアミノコバルトアセテート(2)。
テフロン撹拌バーが充填された200mLのビーカーに、塩化メチレン(10mL)中に溶解した1(1.00g、1.67mmol)、および酢酸(0.100mL、1.75mmol)を添加し、溶媒を蒸発させながら溶液を空気中で撹拌し、鮮赤色粉末を産出した。粉末2をペンタンで洗浄し、真空で12時間乾燥した(1.1g、97%収率)。IR(KBr、cm−1):2967、2876、1613、1580、1524、1490−1361、1252。
(salph)CoBzOAcを製造するためには、等モル量のベンジル酢酸を酢酸に代えて使用する。
(salph)CoNpOAcを製造するためには、等モル量のナフチル(naphthl)酢酸を酢酸に代えて使用する。
(salph)CoOMeを製造するためには、過剰量のメタノールを酢酸に代えて使用し、メタノール中への溶解を塩化メチレン中への溶解に代えて実施する。
(メトキシsalph)CoOAcの調製
化合物3を下記に表す。
3の調製:
テフロン撹拌バーが装備された100mLの丸底フラスコ中で、3,5−ジ−tert−ブチル−2−ヒドロキシベンズアルデヒド(1.47g、6.27mmol)を、THF(10mL)に溶解した4−メトキシ−1,2−フェニレンジアミン(0.433g、3.14mmol)に添加した。エタノール(40mL)を添加し、溶液を24°で撹拌した。その後、還流冷却器をフラスコに装着し、これを95℃へ18時間加熱した。反応物を24℃に冷却した。溶液を20mLに濃縮すると、固形物が沈殿した。固形物を濾過および冷エタノールで洗浄し、黄色/オレンジ色の固体を得た(0.424g、23.7%)。
4の調製:
3(0.400g、0.701mmol)およびコバルトアセテート四水和物(0.124g、0.498mmol)を、50mLの丸底フラスコ中で混ぜ合わせた。N2下で、脱気エタノールを添加し、24℃で2時間撹拌した。混合物をN2下で濾過し、H2O(15mL)、次いでMeOH(15mL)で洗浄した。固形物を回収し、真空下で数時間乾燥して、暗赤色/茶色粉末(0.289g、95.4%)を得た。
5の調製:
4(0.2g、0.4mmol)をN2下で乾式CH2Cl2(30mL)中に溶解し、5分間撹拌した後、酢酸(0.05mL、0.9mmol)を添加した。溶液をさらに5分間撹拌した後、フラスコを大気に開放した。溶媒を蒸発させながら溶液を18時間大気中で撹拌した。固形物を真空下で18時間乾燥した後、ペンタンで洗浄し、真空下で乾燥して、暗赤色粉末(0.2g、80%)を得た。
イソタクチックPPOを製造するためのrac−POの重合
(salph)CoOAc(2)を用いてrac−POを重合するために、以下の手順を利用した。
rac−POの重合のための代表的な手順。ドライボックス内で、シュレンク管に2(9.4mg、0.014mmol)およびテフロン撹拌バーを充填した後、シールして、ドライボックスから取り出した。トルエン(6.6mL)をN2下で添加し、溶液を0℃とした。rac−PO(0.50mL、7.2mmol)を気密シリンジ経由で添加し、0℃で2時間撹拌した。NMR解析のため反応混合物からアリコートを取り出した後、1NのHCl(5.0mL)でクエンチした。未反応POを真空で除去し、沈降ポリマーを溶解するために塩化メチレン(20mL)を添加した。有機層を分離し、回転蒸発によって22℃で溶媒を除去した(370mg、89%)。この段階で、ポリマーは触媒残渣を含有する。熱アセトン(5.0mL)中で溶解することによってポリマーを精製した後、得られた溶液を25℃のアセトン(150mL)に滴加した。ポリマー溶液を0℃に3時間冷却した。白色沈殿物を濾過し、真空で恒量(360mg、86%)になるまで乾燥した。
結果および条件を下記表1に示す。
(salph)CoOAc、(salph)CoBzOAc、(salph)CoNpOAc、(salph)CoOMeおよび(メトキシsalph)CoOAcを用いたrac−POのイソ特異的重合
Salphがジ−tertブチルを含み、開始配位子を変化させた触媒によるrac−POの重合
使用した触媒は構造式:
下記表3に示す反応条件および収率結果で、実験ランを行った。重合はニートまたはトルエン中ランのいずれかであった。いかなる場合でも、mm−triadは99%より大きかった。
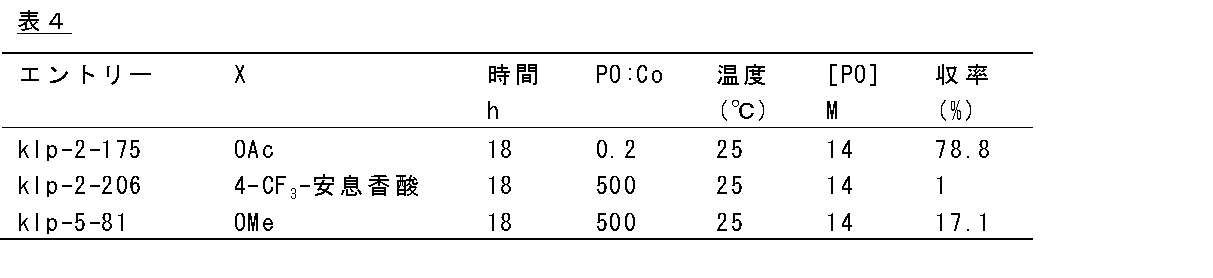
メトキシsalphがジ-tertブチルを含み、開始配位子を変化させた触媒によるrac−POの重合
触媒は構造式:
下記表4に示す反応条件および収率結果で、実験ランを行った。全ての反応をニートで行い、mm−triadは99%より大きかった。
Salphが3−tert−ブチル−5−イソプロピルを含み、XがOMeである触媒によるrac−POの重合
触媒は構造式:
を有する。
[PO]:[Co]を500:1とし、[PO]をトルエン中で7Mとし、他の下記表5に示す条件および結果で、実験ランを行った。
サリシリジンのアルキル置換基を変化させ、XはOAcである、rac−POの重合
触媒は構造式:
重合条件および収率結果を下記表6に示す。
(salph)CoOAcである2を用いたrac−1−ブチレンオキシドの重合
rac−1−ブチレンオキシド(BO)の最適化重合。ドライボックス内で、バイアルに2(7.5mg、0.011mmol)、テフロン撹拌バーおよびトルエン(0.65mL)を充填した。混合物を撹拌しながら、rac−BO(0.50mL、5.8mmol)をバイアルに添加した。直ちにテフロンで裏打ちした蓋でバイアルをシールし、ドライボックスから取り出した。反応物を25℃で4時間撹拌した後、1NのHClでクエンチした。塩化メチレン(20mL)を溶液に添加し、有機層を分離した。回転蒸発によって22℃で溶媒を除去した。残存するコバルトを除去するため、ポリマーをトルエン(40mL)中に溶解し、活性化アルミナを用いて撹拌した後、真空濾過によって濾過を行った。回転蒸発によって22℃で溶媒を濾液から除去した後、真空で乾燥し、淡黄色ゲル(110mg、26%)を得た。1H−NMR(CDCl3、500MHz):δ0.94(ブロード、3H)、1.51−1.58(ブロード、2H)、3.33(ブロード、1H)、3.55(ブロード、2H)。13C{1H}NMR(CDCl3、125MHz):δ9.92、25.07、72.62、81.11。GPCデータ:Mn=185,000g/mol、Mw/Mn=2.08。
(salph)CoOAcを用いた1−ヘキセンオキシドの重合
1−ヘキセンオキシド(HO)の重合。ドライボックス中で、2(6.6mg、0.012mmol)をテフロン撹拌バーが充填されたバイアルに添加した。撹拌しながら、HO(0.50mL、4.1mmol)をバイアルに添加した。直ちにテフロンで裏打ちした蓋でバイアルをシールし、ドライボックスから取り出した。反応物を25℃で18時間撹拌し、次いで得られた重合混合物のアリコートを1H−NMR分析のためにバイアルから取り出した(3.0%)。
未分画イソタクチックPPOと非イソタクチックPPOの混合物の製造
(N,N’−ビス−(5−tert−ブチル−3−イソプロピルサリシリデン)−1,2−フェニレンジアミノ)コバルトアセテート(6)の調製。ドライボックス内で、シュレンク管にAlCl3(1.5g、11.2mmol)およびテフロン被覆撹拌バーを充填した。N2下で、2−イソプロピルフェノール(15.1mL、112mmol)、次いで2−クロロ−2−メチルプロパン(14.5mL、134mmol)を添加した。得られた混合物を25℃で18時間撹拌した。氷水とジエチルエーテルを添加した。エーテル層をH2Oで2度、次いで塩水で1度洗浄した。有機層をMgSO4上で乾燥した。粗生成物をカラムクロマトグラフィ(20%EtOAc/ヘキサン)によって精製して、黄色油を得た(19g、87%)。精製フェノール(12g、62mmol)を、乾燥トルエンおよび2,6−ルチジン(12mL、100mmol)中に溶解した。溶液を0℃に冷却し、SnCl4をゆっくり添加した。反応物を15分間撹拌した後、パラホルムアルデヒドを添加した。乾燥管をシュレンク管に装着し、次いで、これを100℃に16時間加熱した。反応物を25℃に冷却した後、1NのHClを添加し、得られた懸濁液をセリット上で濾過した。有機層をH2Oで洗浄し、MgSO4上で乾燥した。濃度によって、粘性の黄色/茶色油(10g、74%)が生成した。油をMeOH(75mL)中に溶解し、1,2−ジアミノベンゼン(2.4g、23mmol)を添加した。溶液を4時間還流し、次いで、25℃に冷却した。黄色固形物を壊し、これを濾過、MeOHで洗浄して、真空下で乾燥した(4.0g、34%)。得られた配位子(1.5g、2.9mmol)をトルエン(15mL)中に溶解し、コバルトアセテート四水和物(.73g、2.9mmol)をMeOH中に溶解した。トルエン溶液をMeOH溶液に添加し、溶液を25℃で1時間撹拌した。赤色固形物を濾過、MeOHで洗浄、および真空下で乾燥した(1.4g、83%)。固形物をCH2Cl2中に溶解し、酢酸(0.10mL、2.4mmol)を添加した。全溶媒が蒸発するまで溶液を大気中で撹拌した。固形物をペンタンで洗浄し、真空下で乾燥した(1.4g、93%)。
ドライボックス内で、6(9.0mg、.014mmol)およびテフロン撹拌バーをバイアルに充填した。トルエン(0.50mL)を添加し、懸濁液を3分間撹拌した。次に、rac−PO(0.50mL、7.1mmol)を添加して、テフロンで裏打ちした蓋でバイアルを素早くシールした。重合物を25℃で20時間撹拌した。重合物を1NのHClでクエンチし、残存するPOを真空下で除去した。ポリマー残渣をCH2Cl2中に溶解し、HClを夜通し反応させた。溶液をH2Oで洗浄し、Na2SO4上で乾燥し、次いで、真空下で濃縮した(.063g、15%)。得られたポリマーは13C{1H}NMR分光法によれば84%のm−dyadを有し、したがって、およそ20%のアタクチックPPOと80%のイソタクチックPPOの混合物として作用する。
本発明の上述記載は、特定の操作可能で好適な態様を述べるために提示したものである。本発明はそのように限定すべきことを意図するものではなく、その変形および変更は当業者にとって自明であって、その全ては本発明の精神および範囲に含まれる。
本明細書の当初の開示は、少なくとも下記の態様を包含する。
〔1〕(Salphまたはメトキシsalph)Co(開始配位子)。
〔2〕〔1〕に記載の化合物であって、開始配位子は、該化合物と溶媒および/またはエポキシドとを混合すると、化合物が部分的に溶解して隣接コバルト中心を含有する非溶解錯体を与えるものである、化合物。
〔3〕凝集またはインサイチュ配位子修飾によってキラル環境で存在する、〔1〕に記載の化合物。
〔4〕開始配位子は、ハロゲン、C 1 〜C 20 アミド、シアノ、アジド、C 1 〜C 20 カルボキシレート、C 1 〜C 20 アリールカルボキシレート、C 1 〜C 20 アルコキシド、フェノキシド、およびヒドロキシドからなる群から選択される、〔1〕に記載の化合物。
〔5〕salphは、N,N’−ビス(3,5−ジ−tert−ブチルサリシリジン)−ベンゼンジアミンである、〔4〕に記載の化合物。
〔6〕(salph)CoOAcである、〔5〕に記載の化合物。
〔7〕(salph)CoBzOAcである、〔5〕に記載の化合物。
〔8〕(salph)CoNpOAcである、〔5〕に記載の化合物。
〔9〕(salph)CoOMeである、〔5〕に記載の化合物。
〔10〕(メトキシsalph)CoOAcである、〔5〕に記載の化合物。
〔11〕(salph)Coまたは(メトキシsalph)Coを塩化メチレン中に溶解し、H(開始配位子)を添加し、および溶液の塩化メチレンを大気中に蒸発させることによって製造された、(Salphまたはメトキシsalph)Co(開始配位子)。
〔12〕rac−POをイソ特異的触媒の存在下で単独重合する工程を含んでなる、純粋イソタクチックPPOの製造方法。
〔13〕イソ特異的触媒は、(salphまたはメトキシsalph)Co(開始配位子)であって、ここで、salphはN,N’−ビス(3,5−ジ−tert−ブチルサリシリジン)−ベンゼンジアミンである、〔12〕に記載の方法。
〔14〕rac−1−ブチレンオキシドをイソ特異的触媒の存在下で重合する工程を含んでなる、イソタクチックポリ(ブチレンオキシド)の製造方法。
〔15〕イソ特異的触媒は、(salphまたはメトキシsalph)Co(開始配位子)であって、ここで、salphはN,N’−ビス(3,5−ジ−tert−ブチルサリシリジン)−ベンゼンジアミンである、〔14〕に記載の方法。
〔16〕81%より大きいm−dyad含量を有し、かつ、重合触媒残渣を含有し、および/またはアタクチックPPOの痕跡を含有せず、および/またはアタクチックイソタクチックPPO混合物の分画を示す残渣を含有しない、イソタクチックPPO。
〔17〕m−dyad含量は90%より大きい、〔16〕に記載のイソタクチックPPO。
〔18〕m−dyad含量は少なくとも99%である、〔16〕に記載のイソタクチックPPO。
〔19〕81%より大きいm−dyad含量および150,000g/molより大きいM n を有する、イソタクチックPPO。
〔20〕200,000g/molより大きいM n を有する、〔19〕に記載のイソタクチックPPO。
〔21〕81%より大きいm−dyad含量および2.0より小さいPDIを有する、イソタクチックPPO。
〔22〕PDIは1.75より小さい、〔21〕に記載のイソタクチックPPO。
〔23〕PDIは1.50より小さい、〔22〕に記載のイソタクチックPPO。
〔24〕非イソタクチックPPOと未分画イソタクチックPPOとの混合物を与える工程を含んでなる、未分画イソタクチックPPOの性質を変性する方法。
〔25〕コバルトを3+酸化状態で含有する金属錯体を含有するシッフ塩基である触媒の存在下でエポキシドを重合する工程を含んでなる、ポリエポキシドの製造方法。
Claims (12)
- C3〜C 6 アルキルエポキシドをイソ特異的触媒の存在下で単独重合する工程を含んでなる、81%より大きいm−dyad含量を有するイソタクチックポリ(C3〜C 6 アルキレンオキシド)の製造方法であって、
該イソ特異的触媒は
a)salph−Co−(開始配位子);
b)メトキシsalph−Co−(開始配位子);および
c)(a)と(b)の組み合わせ
からなる群から選択され、
ここで、salphは、N,N’−ビス(3,5−ジ−C 1 〜C 4 −アルキルサリシリジン)−1,2−ベンゼンジアミンであり、
該開始配位子は、
a)ハロゲン化物;
b)シアノ;
c)アジド;
d)ヒドロキシド;
e)C 1 〜C 20 アミド;
f)C 1 〜C 20 カルボキシレート;
g)C 2 〜C 20 ジカルボキシレート;
h)C 4 〜C 20 トリカルボキシレート;
i)C 6 〜C 20 アリールカルボキシレート;
j)C 1 〜C 20 アルコキシド;
k)フェノキシド;および
l)(a)ないし(k)の任意の2以上の組み合わせ;
からなる群から選択され、開始配位子(f)〜(h)のいずれかが所望によりヒドロキシル基によって置換されていてよい、
製造方法。 - rac−POをN,N’−ビス(3,5−ジ−tert−ブチルサリシリジン)−ベンゼンジアミン−Co−開始配位子の存在下で単独重合する工程を含んでなる、81%より大きいm−dyad含量を有するイソタクチックポリプロピレンオキシドの製造方法であって、
該開始配位子は、
a)ハロゲン化物;
b)シアノ;
c)アジド;
d)ヒドロキシド;
e)C 1 〜C 20 アミド;
f)C 1 〜C 20 カルボキシレート;
g)C 2 〜C 20 ジカルボキシレート;
h)C 4 〜C 20 トリカルボキシレート;
i)C 6 〜C 20 アリールカルボキシレート;
j)C 1 〜C 20 アルコキシド;
k)フェノキシド;および
l)(a)ないし(k)の任意の2以上の組み合わせ;
からなる群から選択され、開始配位子(f)〜(h)のいずれかが所望によりヒドロキシル基によって置換されていてよい、
製造方法。 - 分画を要さずに前記m−dyad含量を達成する、請求項1に記載の方法。
- salphは、式:
a)メチル;
b)エチル;
c)イソプロピル;および
d)tert−ブチル
からなる群から選択される。〕
を有する、請求項1に記載の方法。 - メトキシsalphは、式:
a)メチル;
b)エチル;
c)イソプロピル;および
d)tert−ブチル
からなる群から選択される。〕
を有する、請求項1に記載の方法。 - 開始配位子は、
a)−Br;
b)−Cl;
c)−OH;
d)−OMe;
e)−OCOMe;
f)−OCOBn;
g)
h)
i)
j)
k)
l)
m)
からなる群から選択される、請求項1に記載の方法。 - C3〜C 6 アルキルエポキシドはプロピレンオキシドである、請求項1に記載の方法。
- C3〜C 6 アルキルエポキシドはブチレンオキシドである、請求項1に記載の方法。
- 生成したポリ(C3〜C 6 アルキレンオキシド)は少なくとも150,000g/molの数平均分子量を有する、請求項1に記載の方法。
- ポリ(C3〜C 6 アルキレンオキシド)のm−dyad含量は少なくとも90%である、請求項1に記載の方法。
- ポリ(C3〜C 6 アルキレンオキシド)のm−dyad含量は少なくとも99%である、請求項1に記載の方法。
- C3〜C 6 アルキルエポキシドを含むモノマーを、構造:
a)メチル;
b)エチル;
c)イソプロピル;および
d)tert−ブチル
からなる群から選択され、
R3は−Hまたは−OMeであり;および
Xは
a)ハロゲン化物;
b)シアノ;
c)アジド;
d)ヒドロキシド;
e)C 1 〜C20アミド;
f)C1〜C20カルボキシレート;
g)C2〜C20ジカルボキシレート;
h)C4〜C20トリカルボキシレート;
i)C6〜C20アリールカルボキシレート;
j)C1〜C20アルコキシド;
k)フェノキシド;および
l)(a)ないし(k)の任意の2以上の組み合わせ;
からなる群から選択され、開始配位子(f)〜(h)のいずれかが所望によりヒドロキシル基によって置換されていてよい。〕
を有する触媒と接触させる工程を含んでなる、イソタクチックポリアルキレンオキシドの製造方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US69223705P | 2005-06-21 | 2005-06-21 | |
| US60/692,237 | 2005-06-21 | ||
| US11/454,945 US7399822B2 (en) | 2005-06-21 | 2006-06-19 | Isotactic specific catalyst for direct production of highly isotactic poly (propylene oxide) or highly isotactic poly (butylene oxide) |
| US11/454,945 | 2006-06-19 | ||
| PCT/US2006/023888 WO2007002023A2 (en) | 2005-06-21 | 2006-06-20 | Isotactic specific catalyst for direct production of highly isotactic poly (propylene oxide) or highly isotactic poly (butylene oxide) |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2008546785A JP2008546785A (ja) | 2008-12-25 |
| JP2008546785A5 JP2008546785A5 (ja) | 2009-08-06 |
| JP5113045B2 true JP5113045B2 (ja) | 2013-01-09 |
Family
ID=37568467
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008518292A Expired - Fee Related JP5113045B2 (ja) | 2005-06-21 | 2006-06-20 | 高イソタクチックポリ(プロピレンオキシド)または高イソタクチックポリ(ブチレンオキシド)を直接製造するためのイソタクチック特異的触媒 |
Country Status (5)
| Country | Link |
|---|---|
| US (3) | US7399822B2 (ja) |
| EP (1) | EP1901845A4 (ja) |
| JP (1) | JP5113045B2 (ja) |
| CA (1) | CA2612547C (ja) |
| WO (1) | WO2007002023A2 (ja) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8093351B2 (en) * | 2006-08-24 | 2012-01-10 | Cornell Research Foundation, Inc. | Copolymerization of propylene oxide and carbon dioxide and homopolymerization of propylene oxide |
| JP5349806B2 (ja) * | 2007-01-30 | 2013-11-20 | 三洋化成工業株式会社 | ウレタン樹脂組成物 |
| EP2195305A2 (en) | 2007-08-17 | 2010-06-16 | Cornell Research Foundation, Inc. | Isoselective polymerization of epoxides |
| JP5334424B2 (ja) * | 2008-02-15 | 2013-11-06 | 住友化学株式会社 | 安定化ポリ(アルキレンオキシド)の製造方法 |
| US8633123B2 (en) | 2008-08-22 | 2014-01-21 | Novomer, Inc. | Catalysts and methods for polymer synthesis |
| CA2736482C (en) * | 2008-09-08 | 2018-01-02 | Novomer, Inc. | Polycarbonate polyol compositions and methods |
| CN101412809B (zh) * | 2008-11-28 | 2011-04-27 | 大连理工大学 | 用于合成聚碳酸酯的单活性点催化剂 |
| WO2010075232A1 (en) | 2008-12-23 | 2010-07-01 | Novomer, Inc. | Tunable polymer compositions |
| US8389660B1 (en) | 2009-06-10 | 2013-03-05 | The Florida State University Research Foundation | Polyolefins having reduced crystallinity |
| JP2011057966A (ja) * | 2009-08-12 | 2011-03-24 | Sumitomo Chemical Co Ltd | 高分子量のアイソタクチックポリ(アルキレンオキシド)及びそれを製造する方法 |
| KR101805648B1 (ko) | 2010-09-14 | 2017-12-14 | 사우디 아람코 테크놀로지스 컴퍼니 | 중합체 합성용의 촉매 및 방법 |
| ES2947327T3 (es) * | 2010-11-23 | 2023-08-04 | Saudi Aramco Tech Co | Composiciones de poli(poliol de carbonato) |
| WO2012166889A2 (en) | 2011-05-31 | 2012-12-06 | Cornell University | Polyethers, methods of making same, and uses thereof |
| JP6110379B2 (ja) | 2011-07-18 | 2017-04-05 | ノボマー, インコーポレイテッド | 金属錯体 |
| CN112979938A (zh) | 2011-07-25 | 2021-06-18 | 沙特阿美技术公司 | 用于聚氨酯的脂族聚碳酸酯 |
| CN103842082B (zh) | 2011-08-08 | 2017-07-18 | 诺沃梅尔公司 | 聚合物合成的催化剂和方法 |
| KR101945168B1 (ko) | 2011-11-04 | 2019-02-07 | 사우디 아람코 테크놀로지스 컴퍼니 | 이산화탄소와 에폭사이드의 공중합용의 금속 착체 |
| WO2014031811A1 (en) * | 2012-08-24 | 2014-02-27 | Novomer, Inc. | Metal complexes |
| WO2014074706A1 (en) | 2012-11-07 | 2014-05-15 | Novomer, Inc. | High strength polyurethane foam compositions and methods |
| US20140275433A1 (en) * | 2013-03-14 | 2014-09-18 | Exxonmobil Research And Engineering Company | Amination of polymers terminated with aldehyde group and their functionalized derivatives for fouling mitigation in hydrocarbon refining processes |
| CN109054011B (zh) * | 2018-07-16 | 2021-01-08 | 中国科学院长春应用化学研究所 | 一种席夫碱钴化合物、其制备方法及聚碳酸酯的制备方法 |
| WO2020028606A1 (en) | 2018-08-02 | 2020-02-06 | Saudi Aramco Technologies Company | Sustainable polymer compositions and methods |
| US12195576B2 (en) | 2021-06-23 | 2025-01-14 | Saudi Aramco Technologies Company | Polyol compositions and methods |
| EP4626445A1 (en) * | 2022-12-02 | 2025-10-08 | University of Maryland, Baltimore | Gallium-salophen antimicrobial compounds and methods of use thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2706181A (en) | 1952-06-05 | 1955-04-12 | Dow Chemical Co | Catalysts for the polymerization of olefin oxides |
| US4962281A (en) * | 1985-12-13 | 1990-10-09 | The Dow Chemical Company | Synthesis of stereoregular poly(propylene oxide) |
| US6262278B1 (en) * | 1995-03-14 | 2001-07-17 | President And Fellows Of Harvard College | Stereoselective ring opening reactions |
| JP3962587B2 (ja) * | 2000-05-24 | 2007-08-22 | アールエステック カンパニー リミテッド | 新規キラルサレン触媒及びこれを使用してラセミエポキシドからキラル化合物を製造する方法 |
| JP2004315588A (ja) * | 2003-04-11 | 2004-11-11 | Sanyo Chem Ind Ltd | ヘテロ環状化合物の開環重合用触媒 |
-
2006
- 2006-06-19 US US11/454,945 patent/US7399822B2/en not_active Expired - Fee Related
- 2006-06-20 WO PCT/US2006/023888 patent/WO2007002023A2/en not_active Ceased
- 2006-06-20 EP EP06785143.6A patent/EP1901845A4/en not_active Withdrawn
- 2006-06-20 CA CA2612547A patent/CA2612547C/en not_active Expired - Fee Related
- 2006-06-20 JP JP2008518292A patent/JP5113045B2/ja not_active Expired - Fee Related
-
2008
- 2008-06-12 US US12/155,962 patent/US8026317B2/en not_active Expired - Fee Related
-
2011
- 2011-08-30 US US13/221,631 patent/US20110313128A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007002023A3 (en) | 2007-11-29 |
| US20110313128A1 (en) | 2011-12-22 |
| CA2612547C (en) | 2015-03-17 |
| JP2008546785A (ja) | 2008-12-25 |
| US20060293501A1 (en) | 2006-12-28 |
| EP1901845A4 (en) | 2014-11-19 |
| WO2007002023A2 (en) | 2007-01-04 |
| US7399822B2 (en) | 2008-07-15 |
| EP1901845A2 (en) | 2008-03-26 |
| CA2612547A1 (en) | 2007-01-04 |
| US20080262164A1 (en) | 2008-10-23 |
| US8026317B2 (en) | 2011-09-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5113045B2 (ja) | 高イソタクチックポリ(プロピレンオキシド)または高イソタクチックポリ(ブチレンオキシド)を直接製造するためのイソタクチック特異的触媒 | |
| Cohen et al. | Copolymerization of cyclohexene oxide and carbon dioxide using (salen) Co (III) complexes: synthesis and characterization of syndiotactic poly (cyclohexene carbonate) | |
| CN106536045B (zh) | 催化剂 | |
| CN111393630B (zh) | 一种聚合物多元醇及其制备方法 | |
| CN104903333B (zh) | 一种液态锡(ii)醇盐的制备方法 | |
| JP4590284B2 (ja) | ポリカーボネートの製造方法 | |
| JP3414029B2 (ja) | 単分散重合体およびそれらの製造方法 | |
| KR101217954B1 (ko) | 극성 시클릭 단량체의 개환 중합을 위한 비스(나프톡시)피리딘 및 비스(나프톡시)티오펜 리간드 기재의 3 족 포스트-메탈로센 착물 | |
| Tang et al. | Highly active catalysts for the ring-opening polymerization of ethylene oxide and propylene oxide based on products of alkylaluminum compounds with bulky tetraphenol ligands | |
| Liao et al. | Alcoholysis of Methyl Aluminium Biphenoxides: Excellent Initiators for the Ring Opening Polymerisation of ε‐Caprolactone | |
| CN114478635A (zh) | 一种铬化合物、其制备方法与多嵌段聚酯材料的制备方法 | |
| CN113024787A (zh) | 一种利用[OSSO]型配合物催化ε-己内酯开环聚合的方法 | |
| Magliozzi et al. | Enantioselective crystallization of diglycerol dicarbonate: impact of the microstructure on polyhydroxyurethane properties | |
| JPS6164721A (ja) | ブロツクコポリマ−の製造方法 | |
| JP4431790B2 (ja) | レゾルシノールノボラック誘導体 | |
| CN119930674B (zh) | 一种钒联吡啶络合物及其制备方法和应用 | |
| Kosolsumollamas | Synthesis and Properties of Fluorine-Containing poly alpha-esters | |
| JP5256498B2 (ja) | 高分子配位子、アルミニウム錯体及びポリラクチドの製造方法 | |
| JP3043779B2 (ja) | エチレン重合体及びその製造方法 | |
| Dunn | Synthesis Of Poly (Hydroxyalkanoates): Routes To Poly (3-Hydroxybutyrate) And Poly (3-Hydroxypropionate) From The Carbonylation And Ring-Opening Polymerization Of Epoxides | |
| JP2010174241A (ja) | ポリエーテルの製造方法 | |
| KR20240031388A (ko) | 향상된 촉매적 카보닐화 활성을 위한 입체적으로 변형된 시프 염기 리간드 | |
| JPH04288334A (ja) | ポリシランの製造方法 | |
| JP2006283016A (ja) | 3位に置換基を有するオキセタン誘導体の重合方法および重合物 | |
| US20200002472A1 (en) | Sustainable process for preparing polyesters having high glass transition temperature |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090617 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090617 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120201 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120228 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120525 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120601 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120625 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120702 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120828 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120918 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121011 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151019 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |