JP4928075B2 - 有機薄膜表面物性の変換方法 - Google Patents
有機薄膜表面物性の変換方法 Download PDFInfo
- Publication number
- JP4928075B2 JP4928075B2 JP2004372988A JP2004372988A JP4928075B2 JP 4928075 B2 JP4928075 B2 JP 4928075B2 JP 2004372988 A JP2004372988 A JP 2004372988A JP 2004372988 A JP2004372988 A JP 2004372988A JP 4928075 B2 JP4928075 B2 JP 4928075B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- thin film
- organic thin
- metal
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Description
例えば、
(a)非特許文献3には、クロロメチルフェニルトリメトキシシランから形成された自己組織化単分子膜に193nm及び248nmの紫外線を照射すると脱ハロゲン化水素反応してアルデヒドが形成するため、これを利用してパターンを作製できる旨が記載されている。
(b)特許文献1には、メルカプト基を有する自己組織化単分子膜に対し、193nmで露光してスルホン化することでパターン化できる旨が記載されている。
特許文献2には、ジアゾナフトキノン基を有する自己組織化単分子膜に対し、360nmの紫外線を照射して選択的に表面をインデンカルボン酸基に変化させることでパターン化できる旨が記載されている。
(d)非特許文献5では、ベンゾフェノン骨格を有する自己組織化単分子膜に対し、340nm以上の波長の光を照射させてラジカルを発生させることにより、ポリマーを選択的にグラフト重合させることでパターン化を行う旨が記載されている。
(f)特許文献4では、オルトニトロベンジル基をスルホニル基の光脱保護基として用いた自己組織化単分子膜を設計し、親水−撥水パターニングを行う旨が記載されている。
(h)また特許文献6では、ベンジルスルホニル基を有する有機薄膜に対し、254nmの紫外線を照射することで選択的に表面をスルフィン酸基に変化させてパターン化を行う旨が記載されている。
本発明の変換方法においては、前記感光性化合物として、式(I)
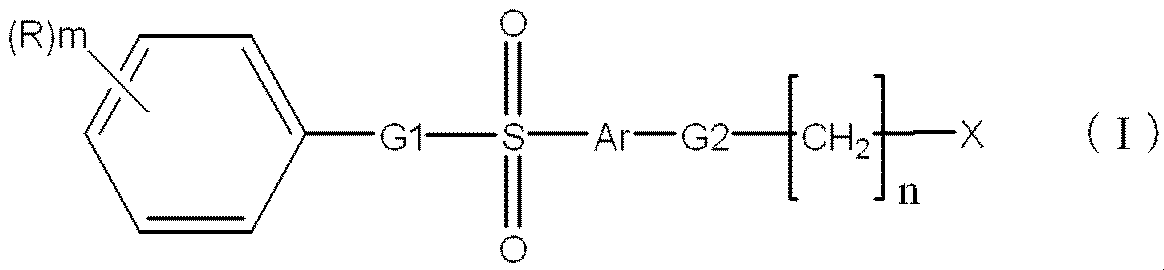
Rは、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数1〜20のアルコキシ基、置換基を有していてもよいアリール基、置換基を有していてもよい炭素数2〜20のアルコキシカルボニル基、またはハロゲン原子を表す。
nは1〜30の整数を表し、mは0〜5の整数を表す。mが2以上のとき、Rは同一でも相異なっていてもよく、2つのRが結合して環を形成していてもよい。
G1は、単結合又は炭素数1〜3の2価の飽和又は不飽和の炭化水素基を表す。
Arは置換基を有していてもよい2価の芳香族基を表す。
G2は、酸素原子、硫黄原子又はNr(rは、水素原子又は炭素数1〜4のアルキル基を表す。)で表される基を表す。〕
で表される化合物を用いることが好ましい。
本発明によれば、電子部品や超微細構造を有する構造体等のパターン形成を効率よく行うことができる。
本発明の有機薄膜の表面物性の変換方法は、基板上に形成された、光照射により脱離する保護基を有する感光性化合物を含む有機薄膜に、(A)有機溶媒を介して光照射する、又は(B)前記有機薄膜を50〜250℃に加熱しながら光照射することを特徴とする。
本発明に用いる有機薄膜は、光照射により脱離する保護基を有する感光性化合物を含む有機薄膜である。
前記式(I)中、式(I−a)
本発明において、官能基とは、基体表面、好ましくは金属又は金属酸化物表面と相互作用できる基を意味する。またヘテロ原子とは、周期律表(長周期型)の第13〜17族の炭素原子以外の原子をいい、例えば、ホウ素原子、アルミニウム原子、珪素原子、窒素原子、りん原子、酸素原子、硫黄原子、フッ素原子、塩素原子等が挙げられる。
炭素数1〜10のアシルチオ基としては、アセチルチオ基、プロピオニルチオ基、ベンゾイルチオ基等が挙げられ、炭素数1〜4のアルキルジスルフィド基としては、メチルジスルフィド基等が挙げられる。
また、置換基を有していてもよいホスホノ基としては、−P(=O)(OH)2、−P(=O)(OCH3)2、−P(=O)(OC2H5)2、−P(=O)(OC3H7)2、−P(=O)(OC4H9)2、−P(=O)(OPh)2等が挙げられる。
また、ハロゲン原子としては、フッ素原子、塩素原子、臭素原子、ヨウ素原子が挙げられる。
mが2以上のとき、2つのRは結合して環を形成してもよい。形成される環としては、例えば、ナフタレン環、アントラセン環、ベンゾシクロブテン環、インダン環等の縮合環が挙げられる。
G1は、単結合又は炭素数が1〜3である2価の飽和又は不飽和の炭化水素基を表す。
G1の具体例としては、単結合、メチレン基、エチレン基、プロピレン基、ビニレン基等が挙げられる。
2価の芳香族基としては、フェニレン基、ビフェニレン基、トリフェニレン基、ナフチレン基等が挙げられる。これらの中でも、パラフェニレン基、パラビフェニレン基、パラトリフェニレン基又はナフチレン基が好ましい。
前記2価の芳香族基の置換基としては、フッ素原子、塩素原子等のハロゲン原子;メチル基等のアルキル基;メトキシ基等のアルコキシ基等が挙げられる。
G2の具体例としては、酸素原子(−O−)、硫黄原子(−S−)、−NH−、−N(CH3)−、−N(C2H5)−、−N(n−C3H7)−、−N(i−C3H7)−等が挙げられる。これらの中でも、酸素原子(−O−)が好ましい。
前記式(I)で表される化合物は、例えば以下のようにして製造することができる。
先ず、芳香族化合物(2)に対し、1〜2当量の塩基存在下、該当する置換基Rを有する化合物(3)を反応させて、スルフィド化合物(4)を得る。次に、得られたスルフィド化合物(4)を、適当な酸化剤によって酸化して、対応するスルホン体(5)を得る。次いで、得られたスルホン体(5)と、末端がオレフィンであるアルキルハライド又はアルコールとを、塩基又は縮合剤の存在下に反応させることにより、オレフィン体(6)を得る。 さらに、得られたオレフィン体(6)を、適当な触媒存在下にヒドロシランと反応させて、Xがシリル基(X=SiQ3)で表される化合物(1−1)を得ることができる。
用いる酸化剤としては、過酸化水素、過酢酸、m−クロロ過安息香酸等の過酸化物類が挙げられる。
用いる縮合剤としては、アゾジカルボン酸ジエチルエステル等のアゾジカルボン酸エステル類と、トリフェニルホスフィン等のホスフィン類の組合せが挙げられる。
また、ヒドロシランの反応に用いる触媒としては、白金、パラジウム、ニッケル、ルテニウム、ロジウム等の遷移金属又はそのハロゲン化物、及びその有機金属錯体が挙げられる。
Xがホスホノ基(X=P(=O)(OQ)2)である化合物(1−2)は、化合物(6)に亜リン酸ジエステル(HO−P(OQ)2)を適当な触媒存在下に反応させ、次いで、加水分解することによって得ることができる。亜リン酸ジエステルの反応に用いる触媒としては、白金、パラジウム、ニッケル、ルテニウム、ロジウム等の遷移金属又はそのハロゲン化物、及びその有機金属錯体が挙げられる。
また、Xがホスホノ基である化合物(1−2)は、後述する化合物(7)と亜リン酸トリエステル(P(OQ)3)を反応させ、加水分解させる方法によっても製造することができる。
Xがメルカプト基(X=SH)、炭素数1〜4のアルキルチオ基(X=SQ)またはアミノ基(X=NH2)である化合物(1−3)は、化合物(5)に、α,ω−ジハロアルカンを反応させて化合部(7)を得、得られた化合物(7)の末端ハロゲンを、硫黄又は窒素原子と求核置換反応させることにより得ることができる。
Xがジスルフィド基(X=SS−)である化合物(1−3)は、Xがメルカプト基である化合物を酸化的に二量化させることにより得ることができる。
前記式(I)において、nが0である化合物(1)は、4,4’−ジヒドロキシジフェニルスルホンの一方の水酸基のみをアルキル化し、残りの水酸基を、末端がオレフィンもしくはハロゲン原子のアルキル化剤を反応させて得た化合物に、上記製造法1〜4のXを導入する方法を適用することで、製造することができる。
前記式(I)において、G2がSである化合物(1)は、前記化合物(5)を公知の方法により硫黄化し、その後、前記製造法1〜5と同様にして得ることができる。
有機薄膜形成用組成物は、光照射により脱離する保護基を有する感光性化合物を、有機溶媒中、金属、金属酸化物、金属塩、金属水酸化物、金属アルコキシド類、キレート化又は配位化された金属化合物、金属アルコキシド類を水で処理して得られる加水分解生成物、及び酸から選ばれる少なくとも1種、並びに水で処理することにより調製することができる。
前記金属酸化物としては、特に制限されないが、好ましい具体例として、酸化パラジウム(IV)、酸化白金(IV)が挙げられる。
これらの金属アルコキシド類は1種単独で、あるいは2種以上を組み合わせて用いることができる。
水の使用量は、前記金属アルコキシド類に対し2倍当量以上用いる場合は、2.0〜8倍当量が好ましく、3〜5倍当量がより好ましい。前記金属アルコキシド類に対し2倍当量未満用いる場合は、0.5〜2.0倍当量が好ましい。
金属アルコキシド類に対して0.5から1.0倍当量未満の水を反応させる場合、その反応温度は特に制限されず、通常−100〜+100℃、好ましくは、−20℃から、用いる有機溶媒又は加水分解によって遊離するアルコールの沸点の範囲である。
塩基としては、例えば、トリエタノールアミン、トリエチルアミン、1,8−ジアザビシクロ[5.4.0]−7−ウンデセン、アンモニア、ジメチルホルムアミド、ホスフィン等が挙げられる。
具体的には、有機溶媒中で凝集せずに安定に分散している性質を有する分散質となっている。この場合、分散質とは、分散系中に分散している微細粒子のことをいい、具体的には、コロイド粒子等を例示することができる。
シラノール縮合触媒としては、カルボン酸金属塩、カルボン酸エステル金属塩、カルボン酸金属塩ポリマー、カルボン酸金属塩キレート、チタン酸エステル、及びチタン酸エステルキレート等が挙げられる。具体例としては、酢酸第一スズ、ジブチルスズジラウレート、ジブチルスズジオクテート、ジブチルスズジアセテート、ジオクチルスズジラウレート、ジオクチルスズジオクテート、ジオクチルスズジアセテート、ジオクタン酸第一スズ、ナフテン酸鉛、ナフテン酸コバルト、2−エチルヘキセン酸鉄、ジオクチルスズビスオクチルチオグリコール酸エステル塩、ジオクチルスズマレイン酸エステル塩、ジブチルスズマレイン酸塩ポリマー、ジメチルスズメルカプトプロピオン酸塩ポリマー、ジブチルスズビスアセチルアセテート、ジオクチルスズビスアセチルラウレート、チタンテトラエトキサイド、チタンテトラブトキサイド、チタンテトライソプロポキサイド、チタンビス(アセチルアセトニル)ジプロポキシサイド等が挙げられる。
また、この場合においては、均一な有機薄膜形成用溶液を得るために、超音波処理を施すことも好ましい。
基材の形状は特に限定されないが、フィルム状、シート状又は板状が好ましい。
ここで、自己組織化単分子膜とは、基板表面上に自発的に形成される化学結合を介して機能分子が配列・固定されてなる単分子膜をいい、単分子相当膜厚の有機薄膜とは、単分子膜とほとんど大差ない膜厚を有する有機薄膜をいう。
このようにして光照射することにより、有機薄膜を単に光照射する場合と比して、より効率よく、かつ確実に有機薄膜の表面物性を変換させることができる。
有機溶媒を介して光照射する方法としては、有機薄膜表面上に有機溶媒の層を設け、この有機用溶媒の層の上部から光照射する方法であれば特に制限されない。例えば、有機溶媒中に前記有機薄膜が形成された基板を浸漬し、該有機溶媒の液面上部から前記有機薄膜を光照射する方法が挙げられる。この方法によれば、光照射により脱離した保護基が、前記有機溶媒に速やかに溶解・除去されると考えられるため、光照射による脱離反応の効率が向上し、より顕著な表面物性の変化を得ることができる。
パターン形成に用いる場合は、フォトマスクと有機薄膜の間を有機溶媒で満たして光照射行うこともできる。
有機薄膜を加熱しながら光照射する方法としては、所定温度に加熱した有機薄膜に光照射するものであれば、特に制限されない。具体的には、前記有機薄膜が形成された基板をホットプレート上に載置し、所定温度に加熱しながら該有機薄膜に光照射する方法や、高い放射熱を発生する光源のもとで光照射する方法等が挙げられる。これらの方法によれば、光照射により脱離した保護基が、速やかに加熱除去されると考えられるため、光照射による脱離反応の効率が向上し、より顕著な表面物性の変化を得ることができる。
(1)有機薄膜形成用溶液の調製
下記式(II)
ソーダライムガラス基板を用意し、それぞれを洗剤とともに超音波洗浄した後、イオン交換水、エタノールで順次洗浄後、60℃で乾燥し、オゾン発生装置中でさらに洗浄した。
前記式(II)で表される化合物に代えて、下記式(III)
なお、以下の実験は、各実施例及び比較例毎にそれぞれ3枚の有機薄膜(SAM−1、2)付ガラス基板を用いて行った。
製造例1で作製した有機薄膜(SAM−1)付ガラス基板の有機薄膜表面に、マイクロシリンジからテトラデカン2μlを滴下し、60秒後に、接触角測定器(エルマ(株)社製、360S型)を用いて有機薄膜表面の接触角をそれぞれ測定した。SAM−1のテトラデカンに対する接触角は、50〜54°の値を示した。
製造例2で作製した有機薄膜(SAM−2)付ガラス基板の有機薄膜表面に、マイクロシリンジから水2μlを滴下し、60秒後に、接触角測定器(エルマ(株)社製、360S型)を用いて有機薄膜表面の接触角をそれぞれ測定した。SAM−2の水に対する接触角は98〜100°の値を示した。
参考例4:参考例2と同様
比較例4:比較例1と同様
比較例5:比較例2と同様
Claims (4)
- 基板上に形成された、光照射により脱離する保護基を有する感光性化合物を、有機溶媒中、金属、金属酸化物、金属塩、金属水酸化物、金属アルコキシド類、キレート化又は配位化された金属化合物、金属アルコキシド類を水で処理して得られる加水分解生成物、及び酸から選ばれる少なくとも1種、並びに水で処理することによって得られる有機薄膜形成用組成物を用いて形成されたものであって、自己組織化単分子膜又は単分子相当膜厚の有機薄膜に、該有機薄膜を50〜200℃に加熱しながら光照射することを特徴とする有機薄膜の表面物性の変換方法。
- 前記感光性化合物として、式(I)
Rは、置換基を有していてもよい炭素数1〜20のアルキル基、置換基を有していてもよい炭素数1〜20のアルコキシ基、置換基を有していてもよいアリール基、置換基を有していてもよい炭素数1〜20のアルコキシカルボニル基またはハロゲン原子を表す。
nは1〜30の整数を表し、mは0〜5の整数を表す。mが2以上のとき、Rは同一でも相異なっていてもよく、2つのRが結合して環を形成していてもよい。
G1は、単結合又は炭素数1〜3の2価の飽和又は不飽和の炭化水素基を表す。
Arは置換基を有していてもよい2価の芳香族基を表す。
G2は、酸素原子、硫黄原子又はNr(rは、水素原子又は炭素数1〜4のアルキル基を表す。)で表される基を表す。〕
で表される化合物を用いることを特徴とする請求項1に記載の有機薄膜の表面物性の変換方法。 - 前記有機薄膜が、光照射により脱離する保護基を有する感光性化合物を、有機溶媒中、金属、金属酸化物、金属塩、金属水酸化物、金属アルコキシド類、キレート化又は配位化された金属化合物、金属アルコキシド類を水で処理して得られる加水分解生成物、及び酸から選ばれる少なくとも1種、並びに水で処理することによって得られる有機薄膜形成用組成物を、ディップ法により、基板の表面に接触させることにより形成されたものであることを特徴とする請求項1又は2に記載の有機薄膜の表面物性の変換方法。
- 光照射されることにより、前記有機薄膜の照射部位のみの感光性化合物の保護基が脱離して、前記有機薄膜の照射部位表面が撥液性から親液性へ変化することを特徴とする請求項1〜3のいずれかに記載の有機薄膜の表面物性の変換方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004372988A JP4928075B2 (ja) | 2004-12-24 | 2004-12-24 | 有機薄膜表面物性の変換方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004372988A JP4928075B2 (ja) | 2004-12-24 | 2004-12-24 | 有機薄膜表面物性の変換方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006178274A JP2006178274A (ja) | 2006-07-06 |
| JP4928075B2 true JP4928075B2 (ja) | 2012-05-09 |
Family
ID=36732436
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004372988A Expired - Fee Related JP4928075B2 (ja) | 2004-12-24 | 2004-12-24 | 有機薄膜表面物性の変換方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4928075B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI618985B (zh) * | 2011-08-10 | 2018-03-21 | 日產化學工業股份有限公司 | 具有碸構造之含矽阻劑底層膜形成組成物 |
| JP6562220B2 (ja) | 2014-06-17 | 2019-08-21 | 日産化学株式会社 | フェニル基含有クロモファーを有するシリコン含有レジスト下層膜形成組成物 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5754317A (en) * | 1980-09-19 | 1982-03-31 | Hitachi Ltd | Method and device for forming pattern |
| US5959098A (en) * | 1996-04-17 | 1999-09-28 | Affymetrix, Inc. | Substrate preparation process |
| AU2747999A (en) * | 1998-03-26 | 1999-10-18 | Nikon Corporation | Projection exposure method and system |
| JP4201173B2 (ja) * | 2001-11-20 | 2008-12-24 | 大日本印刷株式会社 | パターン形成体の製造方法 |
| SG121829A1 (en) * | 2002-11-29 | 2006-05-26 | Asml Netherlands Bv | Lithographic apparatus and device manufacturing method |
| WO2004067540A1 (ja) * | 2003-01-31 | 2004-08-12 | Nippon Soda Co., Ltd. | 光変換性有機薄膜を形成する新規化合物および有機薄膜形成体 |
-
2004
- 2004-12-24 JP JP2004372988A patent/JP4928075B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006178274A (ja) | 2006-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2011052443A1 (ja) | 保護膜形成用薬液 | |
| JP4997765B2 (ja) | 含フッ素化合物、撥水性組成物および薄膜 | |
| JP4718463B2 (ja) | パーフルオロポリエーテルアミド連結ホスホネート、ホスフェートおよびそれらの誘導体 | |
| JP4602994B2 (ja) | 剥離層を有する成形用金型又は電鋳用母型及びそれらの製造方法 | |
| JP4746032B2 (ja) | チタン酸化物粒子の分散液、チタン酸化物薄膜、有機機能膜形成用溶液、有機機能膜形成基体及びその製造方法 | |
| JP4995467B2 (ja) | フッ素系薄膜基材の製造方法 | |
| JP4020247B2 (ja) | 高分子グラフト基板製造方法 | |
| JP2010094583A (ja) | 有機薄膜の製造方法 | |
| JP2003321479A (ja) | 光分解性シランカップリング剤 | |
| WO2012002146A1 (ja) | 保護膜形成用薬液及びウェハ表面の洗浄方法 | |
| Konishi et al. | Substituent effects at the benzyl position and aromatic ring of silane-coupling agents containing 2-nitrobenzyl esters on photosensitivity and hydrophobic surface of a self-assembled monolayer (SAM) | |
| JP2006122748A (ja) | 有機薄膜形成方法及び有機薄膜形成用溶液 | |
| JP4928075B2 (ja) | 有機薄膜表面物性の変換方法 | |
| JP4906305B2 (ja) | 光感応性新規化合物及び有機薄膜形成体 | |
| JP4757474B2 (ja) | 有機薄膜形成方法 | |
| GB2415959A (en) | Fabrication of self-assembled dendron monolayers | |
| JP4286832B2 (ja) | 光変換性有機薄膜を形成する新規化合物および有機薄膜形成体 | |
| WO2006013886A1 (ja) | 光感応性新規化合物及び有機薄膜形成体 | |
| JP2000247799A (ja) | 高密度有機分子薄膜の製造方法 | |
| JP4904026B2 (ja) | 光反応性有機薄膜を形成する新規化合物及び有機薄膜形成体 | |
| JP5347359B2 (ja) | o−ニトロベンジル基含有シラザン化合物及び用途 | |
| US7687145B2 (en) | Compounds containing a thiol protected group bonded to a protecting group via a polarized bond, and structure comprising the compounds on its surface | |
| JPWO2014006885A1 (ja) | 有機ケイ素化合物、それを用いた薄膜形成用組成物および有機薄膜 | |
| JP2007206552A (ja) | 光処理基材の製造方法 | |
| JP2008254977A (ja) | 有機薄膜の製造方法及びパターン形成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070718 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100406 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100607 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20100914 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101213 |
|
| A911 | Transfer of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20101220 |
|
| A912 | Removal of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20110210 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20111114 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120210 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150217 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |