JP4780549B2 - キナーゼ阻害活性を有するピロロピラジン誘導体およびその生物学的用途 - Google Patents
キナーゼ阻害活性を有するピロロピラジン誘導体およびその生物学的用途 Download PDFInfo
- Publication number
- JP4780549B2 JP4780549B2 JP2004528508A JP2004528508A JP4780549B2 JP 4780549 B2 JP4780549 B2 JP 4780549B2 JP 2004528508 A JP2004528508 A JP 2004528508A JP 2004528508 A JP2004528508 A JP 2004528508A JP 4780549 B2 JP4780549 B2 JP 4780549B2
- Authority
- JP
- Japan
- Prior art keywords
- alkyl
- alkoxy
- hal
- pharmaceutical composition
- alk
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 108091000080 Phosphotransferase Proteins 0.000 title description 20
- 102000020233 phosphotransferase Human genes 0.000 title description 20
- LJWZSXDLNMOUIP-UHFFFAOYSA-N N1C=CN=C2N=CC=C21 Chemical class N1C=CN=C2N=CC=C21 LJWZSXDLNMOUIP-UHFFFAOYSA-N 0.000 title description 5
- 230000002401 inhibitory effect Effects 0.000 title description 4
- 125000000217 alkyl group Chemical group 0.000 claims description 205
- 229910052739 hydrogen Inorganic materials 0.000 claims description 84
- 125000003545 alkoxy group Chemical group 0.000 claims description 65
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 claims description 60
- 102000002254 Glycogen Synthase Kinase 3 Human genes 0.000 claims description 60
- 239000008194 pharmaceutical composition Substances 0.000 claims description 48
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 claims description 47
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 claims description 47
- 108010025454 Cyclin-Dependent Kinase 5 Proteins 0.000 claims description 44
- 102100026805 Cyclin-dependent-like kinase 5 Human genes 0.000 claims description 44
- 108010068150 Cyclin B Proteins 0.000 claims description 36
- 102000002427 Cyclin B Human genes 0.000 claims description 36
- -1 -R7 is H Chemical group 0.000 claims description 20
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 14
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 9
- 239000004480 active ingredient Substances 0.000 claims description 9
- 238000000034 method Methods 0.000 claims description 8
- 150000001875 compounds Chemical class 0.000 claims description 7
- 230000004770 neurodegeneration Effects 0.000 claims description 7
- 125000001424 substituent group Chemical group 0.000 claims description 7
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 6
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 6
- 239000003814 drug Substances 0.000 claims description 5
- 239000000203 mixture Substances 0.000 claims description 5
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 5
- 208000024827 Alzheimer disease Diseases 0.000 claims description 4
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 4
- 229910052801 chlorine Inorganic materials 0.000 claims description 4
- 125000002541 furyl group Chemical group 0.000 claims description 4
- 239000000126 substance Substances 0.000 claims description 4
- 230000001028 anti-proliverative effect Effects 0.000 claims description 3
- 229910052794 bromium Inorganic materials 0.000 claims description 3
- 229910052731 fluorine Inorganic materials 0.000 claims description 3
- 229910052740 iodine Inorganic materials 0.000 claims description 3
- 125000001624 naphthyl group Chemical group 0.000 claims description 3
- 125000004076 pyridyl group Chemical group 0.000 claims description 3
- 230000002194 synthesizing effect Effects 0.000 claims description 3
- 125000001544 thienyl group Chemical group 0.000 claims description 3
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 claims description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 2
- 102000001267 GSK3 Human genes 0.000 claims description 2
- 108060006662 GSK3 Proteins 0.000 claims description 2
- 208000018737 Parkinson disease Diseases 0.000 claims description 2
- 239000003937 drug carrier Substances 0.000 claims description 2
- 229910052736 halogen Inorganic materials 0.000 claims description 2
- 150000002367 halogens Chemical class 0.000 claims description 2
- 239000007924 injection Substances 0.000 claims description 2
- 238000002347 injection Methods 0.000 claims description 2
- NFVJNJQRWPQVOA-UHFFFAOYSA-N n-[2-chloro-5-(trifluoromethyl)phenyl]-2-[3-(4-ethyl-5-ethylsulfanyl-1,2,4-triazol-3-yl)piperidin-1-yl]acetamide Chemical compound CCN1C(SCC)=NN=C1C1CN(CC(=O)NC=2C(=CC=C(C=2)C(F)(F)F)Cl)CCC1 NFVJNJQRWPQVOA-UHFFFAOYSA-N 0.000 claims description 2
- WMHRXFNTQPIYDT-UHFFFAOYSA-N pyrrolo[2,3-b]pyrazine Chemical class C1=C[N]C2=NC=CC2=N1 WMHRXFNTQPIYDT-UHFFFAOYSA-N 0.000 claims 13
- BHHMPZQRVWVAAR-UHFFFAOYSA-N 7-bromo-8-methylpyrido[2,3-b]pyrazine Chemical class C1=CN=C2C(C)=C(Br)C=NC2=N1 BHHMPZQRVWVAAR-UHFFFAOYSA-N 0.000 claims 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims 1
- 125000003107 substituted aryl group Chemical group 0.000 claims 1
- 230000000699 topical effect Effects 0.000 claims 1
- 229910052757 nitrogen Inorganic materials 0.000 description 50
- 229910052799 carbon Inorganic materials 0.000 description 49
- 238000000921 elemental analysis Methods 0.000 description 46
- 238000002844 melting Methods 0.000 description 42
- 230000008018 melting Effects 0.000 description 42
- 210000004027 cell Anatomy 0.000 description 37
- 108091007914 CDKs Proteins 0.000 description 19
- 239000003112 inhibitor Substances 0.000 description 17
- 238000000354 decomposition reaction Methods 0.000 description 15
- 102000015792 Cyclin-Dependent Kinase 2 Human genes 0.000 description 13
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 13
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 239000000460 chlorine Substances 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 10
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 10
- 229950006344 nocodazole Drugs 0.000 description 10
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 9
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- 230000022131 cell cycle Effects 0.000 description 7
- 230000005764 inhibitory process Effects 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 6
- 206010028980 Neoplasm Diseases 0.000 description 6
- 239000002246 antineoplastic agent Substances 0.000 description 6
- 201000011510 cancer Diseases 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 5
- 102000019058 Glycogen Synthase Kinase 3 beta Human genes 0.000 description 5
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 5
- 108010049611 glycogen synthase kinase 3 alpha Proteins 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 235000018102 proteins Nutrition 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 5
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 4
- DHCLVCXQIBBOPH-UHFFFAOYSA-N Glycerol 2-phosphate Chemical compound OCC(CO)OP(O)(O)=O DHCLVCXQIBBOPH-UHFFFAOYSA-N 0.000 description 4
- 108010033040 Histones Proteins 0.000 description 4
- 102000006947 Histones Human genes 0.000 description 4
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 4
- 102000001253 Protein Kinase Human genes 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000013078 crystal Substances 0.000 description 4
- 230000017858 demethylation Effects 0.000 description 4
- 238000010520 demethylation reaction Methods 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 4
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 108060006633 protein kinase Proteins 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 150000003212 purines Chemical class 0.000 description 4
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- PRIGRJPRGZCFAS-UHFFFAOYSA-N 6-phenyl[5h]pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(CCCC)=C1C1=CC=C(O)C=C1 PRIGRJPRGZCFAS-UHFFFAOYSA-N 0.000 description 3
- WVMANZPBOBRWCB-UHFFFAOYSA-N 7-butyl-6-(4-methoxyphenyl)-5H-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(CCCC)=C1C1=CC=C(OC)C=C1 WVMANZPBOBRWCB-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 102000004877 Insulin Human genes 0.000 description 3
- 108090001061 Insulin Proteins 0.000 description 3
- ZKHQWZAMYRWXGA-KNYAHOBESA-N [[(2r,3s,4r,5r)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] dihydroxyphosphoryl hydrogen phosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)O[32P](O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KNYAHOBESA-N 0.000 description 3
- 238000001042 affinity chromatography Methods 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 238000002050 diffraction method Methods 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 239000012737 fresh medium Substances 0.000 description 3
- QPCBNXNDVYOBIP-WHFBIAKZSA-N hymenialdisine Chemical compound NC1=NC(=O)C([C@@H]2[C@@H]3C=C(Br)N=C3C(=O)NCC2)=N1 QPCBNXNDVYOBIP-WHFBIAKZSA-N 0.000 description 3
- ATBAETXFFCOZOY-UHFFFAOYSA-N hymenialdisine Natural products N1C(N)=NC(=O)C1=C1C(C=C(Br)N2)=C2C(=O)NCC1 ATBAETXFFCOZOY-UHFFFAOYSA-N 0.000 description 3
- 229940125396 insulin Drugs 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000001360 synchronised effect Effects 0.000 description 3
- PMYDPQQPEAYXKD-UHFFFAOYSA-N 3-hydroxy-n-naphthalen-2-ylnaphthalene-2-carboxamide Chemical compound C1=CC=CC2=CC(NC(=O)C3=CC4=CC=CC=C4C=C3O)=CC=C21 PMYDPQQPEAYXKD-UHFFFAOYSA-N 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- XZKIHKMTEMTJQX-UHFFFAOYSA-N 4-Nitrophenyl Phosphate Chemical compound OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XZKIHKMTEMTJQX-UHFFFAOYSA-N 0.000 description 2
- CXRXLHKIJAVGGC-UHFFFAOYSA-N 6-thiophen-3-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound S1C=CC(C=2NC3=NC=CN=C3C=2)=C1 CXRXLHKIJAVGGC-UHFFFAOYSA-N 0.000 description 2
- 229920000936 Agarose Polymers 0.000 description 2
- 108010039627 Aprotinin Proteins 0.000 description 2
- 102000006311 Cyclin D1 Human genes 0.000 description 2
- 108010058546 Cyclin D1 Proteins 0.000 description 2
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 2
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 2
- 241000701022 Cytomegalovirus Species 0.000 description 2
- 206010059866 Drug resistance Diseases 0.000 description 2
- 108010024636 Glutathione Proteins 0.000 description 2
- 235000010469 Glycine max Nutrition 0.000 description 2
- 244000068988 Glycine max Species 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 101000715854 Homo sapiens Cyclin-dependent kinase 2 Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 2
- 229930182555 Penicillin Natural products 0.000 description 2
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 229940123924 Protein kinase C inhibitor Drugs 0.000 description 2
- 239000005700 Putrescine Substances 0.000 description 2
- 230000018199 S phase Effects 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- 101710162629 Trypsin inhibitor Proteins 0.000 description 2
- 229940122618 Trypsin inhibitor Drugs 0.000 description 2
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000001093 anti-cancer Effects 0.000 description 2
- 230000002555 anti-neurodegenerative effect Effects 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- 239000003096 antiparasitic agent Substances 0.000 description 2
- 229940125687 antiparasitic agent Drugs 0.000 description 2
- 239000003443 antiviral agent Substances 0.000 description 2
- 229960004405 aprotinin Drugs 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- QVQLCTNNEUAWMS-UHFFFAOYSA-N barium oxide Chemical compound [Ba]=O QVQLCTNNEUAWMS-UHFFFAOYSA-N 0.000 description 2
- PXXJHWLDUBFPOL-UHFFFAOYSA-N benzamidine Chemical compound NC(=N)C1=CC=CC=C1 PXXJHWLDUBFPOL-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000018486 cell cycle phase Effects 0.000 description 2
- BYYOTMYLPPUWCF-UHFFFAOYSA-N chembl1207227 Chemical compound C1=CC=C2C(=O)C(C=3C4=CC(=CC=C4NC=3O)S(O)(=O)=O)=NC2=C1 BYYOTMYLPPUWCF-UHFFFAOYSA-N 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012894 fetal calf serum Substances 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 229960003180 glutathione Drugs 0.000 description 2
- 239000011539 homogenization buffer Substances 0.000 description 2
- 102000054634 human CDK2 Human genes 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 230000003463 hyperproliferative effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 2
- 108010052968 leupeptin Proteins 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- GTVPOLSIJWJJNY-UHFFFAOYSA-N olomoucine Chemical compound N1=C(NCCO)N=C2N(C)C=NC2=C1NCC1=CC=CC=C1 GTVPOLSIJWJJNY-UHFFFAOYSA-N 0.000 description 2
- 229940049954 penicillin Drugs 0.000 description 2
- CMPQUABWPXYYSH-UHFFFAOYSA-N phenyl phosphate Chemical compound OP(O)(=O)OC1=CC=CC=C1 CMPQUABWPXYYSH-UHFFFAOYSA-N 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 229940080469 phosphocellulose Drugs 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 238000003825 pressing Methods 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 239000000186 progesterone Substances 0.000 description 2
- 229960003387 progesterone Drugs 0.000 description 2
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 2
- 239000003881 protein kinase C inhibitor Substances 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000022983 regulation of cell cycle Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 2
- BTIHMVBBUGXLCJ-OAHLLOKOSA-N seliciclib Chemical compound C=12N=CN(C(C)C)C2=NC(N[C@@H](CO)CC)=NC=1NCC1=CC=CC=C1 BTIHMVBBUGXLCJ-OAHLLOKOSA-N 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229960001866 silicon dioxide Drugs 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- 235000018716 sodium selenate Nutrition 0.000 description 2
- 239000011655 sodium selenate Substances 0.000 description 2
- 229960001881 sodium selenate Drugs 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 2
- 239000002753 trypsin inhibitor Substances 0.000 description 2
- 241001529453 unidentified herpesvirus Species 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- PNTBUPYLDXGORE-UHFFFAOYSA-N 2-(5H-pyrrolo[2,3-b]pyrazin-6-yl)phenol hydrobromide Chemical compound Br.OC1=CC=CC=C1C1=CC2=NC=CN=C2N1 PNTBUPYLDXGORE-UHFFFAOYSA-N 0.000 description 1
- NQVIIUBWMBHLOZ-UHFFFAOYSA-N 2-[2-hydroxyethyl-[6-[(4-methoxyphenyl)methylamino]-9-propan-2-ylpurin-2-yl]amino]ethanol Chemical compound C1=CC(OC)=CC=C1CNC1=NC(N(CCO)CCO)=NC2=C1N=CN2C(C)C NQVIIUBWMBHLOZ-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 1
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 1
- WBJWXIQDBDZMAW-UHFFFAOYSA-N 2-hydroxynaphthalene-1-carbonyl chloride Chemical compound C1=CC=CC2=C(C(Cl)=O)C(O)=CC=C21 WBJWXIQDBDZMAW-UHFFFAOYSA-N 0.000 description 1
- KLLLJCACIRKBDT-UHFFFAOYSA-N 2-phenyl-1H-indole Chemical compound N1C2=CC=CC=C2C=C1C1=CC=CC=C1 KLLLJCACIRKBDT-UHFFFAOYSA-N 0.000 description 1
- FTZIQBGFCYJWKA-UHFFFAOYSA-N 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Chemical compound S1C(C)=C(C)N=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 FTZIQBGFCYJWKA-UHFFFAOYSA-N 0.000 description 1
- SNWWLPJZLSCQPU-UHFFFAOYSA-N 3-(5h-pyrrolo[2,3-b]pyrazin-6-yl)phenol;hydrobromide Chemical compound Br.OC1=CC=CC(C=2NC3=NC=CN=C3C=2)=C1 SNWWLPJZLSCQPU-UHFFFAOYSA-N 0.000 description 1
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 1
- IYNDTACKOAXKBJ-UHFFFAOYSA-N 3-[[4-[2-(3-chloroanilino)-4-pyrimidinyl]-2-pyridinyl]amino]-1-propanol Chemical compound C1=NC(NCCCO)=CC(C=2N=C(NC=3C=C(Cl)C=CC=3)N=CC=2)=C1 IYNDTACKOAXKBJ-UHFFFAOYSA-N 0.000 description 1
- WOSGPBFDGVRZKT-UHFFFAOYSA-N 3-methyl-6-phenyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC(C)=CN=C2C=C1C1=CC=CC=C1 WOSGPBFDGVRZKT-UHFFFAOYSA-N 0.000 description 1
- KJPRLNWUNMBNBZ-UHFFFAOYSA-N 3-phenylprop-2-enal Chemical class O=CC=CC1=CC=CC=C1 KJPRLNWUNMBNBZ-UHFFFAOYSA-N 0.000 description 1
- ADSFHUBLYMXMJY-UHFFFAOYSA-N 4-(2-cyclopropyl-7-methyl-5H-pyrrolo[2,3-b]pyrazin-6-yl)phenol hydrobromide Chemical compound Br.N1=C2C(C)=C(C=3C=CC(O)=CC=3)NC2=NC=C1C1CC1 ADSFHUBLYMXMJY-UHFFFAOYSA-N 0.000 description 1
- MJZMGRJISCKSIH-UHFFFAOYSA-N 4-(5H-pyrrolo[2,3-b]pyrazin-6-yl)phenol hydrobromide Chemical compound Br.C1=CC(O)=CC=C1C1=CC2=NC=CN=C2N1 MJZMGRJISCKSIH-UHFFFAOYSA-N 0.000 description 1
- LJVIXSSNTIXTNB-UHFFFAOYSA-N 4-(5h-pyrrolo[2,3-b]pyrazin-6-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1=CC2=NC=CN=C2N1 LJVIXSSNTIXTNB-UHFFFAOYSA-N 0.000 description 1
- XOAUHNBBFAVVQD-UHFFFAOYSA-N 4-(7-methyl-5H-pyrrolo[2,3-b]pyrazin-6-yl)phenol hydrobromide Chemical compound Br.N1C2=NC=CN=C2C(C)=C1C1=CC=C(O)C=C1 XOAUHNBBFAVVQD-UHFFFAOYSA-N 0.000 description 1
- XBXQRCJKDCEQBS-UHFFFAOYSA-N 4-[(6-aminopyrimidin-3-ium-4-yl)amino]benzenesulfonamide;chloride Chemical compound Cl.C1=NC(N)=CC(NC=2C=CC(=CC=2)S(N)(=O)=O)=N1 XBXQRCJKDCEQBS-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- VOGDKGBOBCBLFY-UHFFFAOYSA-N 5H-pyrrolo[2,3-b]pyrazine hydrobromide Chemical compound Br.N1=C2C(=NC=C1)NC=C2 VOGDKGBOBCBLFY-UHFFFAOYSA-N 0.000 description 1
- HFTVJMFWJUFBNO-UHFFFAOYSA-N 5h-pyrrolo[2,3-b]pyrazine Chemical class C1=CN=C2NC=CC2=N1 HFTVJMFWJUFBNO-UHFFFAOYSA-N 0.000 description 1
- GPBWBXKELCXCMJ-UHFFFAOYSA-N 6-(2-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound COC1=CC=CC=C1C1=CC2=NC=CN=C2N1 GPBWBXKELCXCMJ-UHFFFAOYSA-N 0.000 description 1
- GDVOQQJJTAVRRG-UHFFFAOYSA-N 6-(3,4,5-trimethoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound COC1=C(OC)C(OC)=CC(C=2NC3=NC=CN=C3C=2)=C1 GDVOQQJJTAVRRG-UHFFFAOYSA-N 0.000 description 1
- NOQBRRDCHJWUNA-UHFFFAOYSA-N 6-(3,4-dimethoxyphenyl)-7-methyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=C(OC)C(OC)=CC=C1C1=C(C)C2=NC=CN=C2N1 NOQBRRDCHJWUNA-UHFFFAOYSA-N 0.000 description 1
- ZUJJUWNSTXNVII-UHFFFAOYSA-N 6-(3,5-dichlorophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound ClC1=CC(Cl)=CC(C=2NC3=NC=CN=C3C=2)=C1 ZUJJUWNSTXNVII-UHFFFAOYSA-N 0.000 description 1
- YOIQNVSHTXFKAL-UHFFFAOYSA-N 6-(3,5-dimethoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound COC1=CC(OC)=CC(C=2NC3=NC=CN=C3C=2)=C1 YOIQNVSHTXFKAL-UHFFFAOYSA-N 0.000 description 1
- MFDLPDIVXMMWSI-UHFFFAOYSA-N 6-(3-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound COC1=CC=CC(C=2NC3=NC=CN=C3C=2)=C1 MFDLPDIVXMMWSI-UHFFFAOYSA-N 0.000 description 1
- ZIPGRNBNZKOCNS-UHFFFAOYSA-N 6-(4-bromophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(Br)=CC=C1C1=CC2=NC=CN=C2N1 ZIPGRNBNZKOCNS-UHFFFAOYSA-N 0.000 description 1
- VFKHBJJSGUYSDC-UHFFFAOYSA-N 6-(4-chlorophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(Cl)=CC=C1C1=CC2=NC=CN=C2N1 VFKHBJJSGUYSDC-UHFFFAOYSA-N 0.000 description 1
- VFPIRSDEIXZQJN-UHFFFAOYSA-N 6-(4-chlorophenyl)-7-(cyclohexylmethyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(Cl)=CC=C1C1=C(CC2CCCCC2)C2=NC=CN=C2N1 VFPIRSDEIXZQJN-UHFFFAOYSA-N 0.000 description 1
- SZRBJCSOUDDITM-UHFFFAOYSA-N 6-(4-chlorophenyl)-7-methyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(C)=C1C1=CC=C(Cl)C=C1 SZRBJCSOUDDITM-UHFFFAOYSA-N 0.000 description 1
- GFJIABMYYUGNEC-UHFFFAOYSA-N 6-(4-chlorophenyl)-7-propan-2-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(C(C)C)=C1C1=CC=C(Cl)C=C1 GFJIABMYYUGNEC-UHFFFAOYSA-N 0.000 description 1
- OGRORAFGBNLUSP-UHFFFAOYSA-N 6-(4-fluorophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(F)=CC=C1C1=CC2=NC=CN=C2N1 OGRORAFGBNLUSP-UHFFFAOYSA-N 0.000 description 1
- XQYJBGKMIPFFJS-UHFFFAOYSA-N 6-(4-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=CC2=NC=CN=C2N1 XQYJBGKMIPFFJS-UHFFFAOYSA-N 0.000 description 1
- ZUHJPCPWABPZDY-UHFFFAOYSA-N 6-(4-methoxyphenyl)-7-methyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(C)C2=NC=CN=C2N1 ZUHJPCPWABPZDY-UHFFFAOYSA-N 0.000 description 1
- RYNPAHUWYURCCW-UHFFFAOYSA-N 6-(4-methoxyphenyl)-7-prop-2-enyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(CC=C)C2=NC=CN=C2N1 RYNPAHUWYURCCW-UHFFFAOYSA-N 0.000 description 1
- CAFYKPIUWNVPMG-UHFFFAOYSA-N 6-(4-methoxyphenyl)-7-propan-2-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(C(C)C)C2=NC=CN=C2N1 CAFYKPIUWNVPMG-UHFFFAOYSA-N 0.000 description 1
- WMFVCLKZYKMDNK-UHFFFAOYSA-N 6-(4-methoxyphenyl)-7-propyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(CCC)=C1C1=CC=C(OC)C=C1 WMFVCLKZYKMDNK-UHFFFAOYSA-N 0.000 description 1
- NUXPRKBWNZAEBF-UHFFFAOYSA-N 6-(4-methylphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(C)=CC=C1C1=CC2=NC=CN=C2N1 NUXPRKBWNZAEBF-UHFFFAOYSA-N 0.000 description 1
- PGQDMRRDTMOOLS-UHFFFAOYSA-N 6-(furan-2-yl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=COC(C=2NC3=NC=CN=C3C=2)=C1 PGQDMRRDTMOOLS-UHFFFAOYSA-N 0.000 description 1
- YWLJQPJXGXMBHN-UHFFFAOYSA-N 6-[1-(4-chlorophenyl)cyclopropyl]-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(Cl)=CC=C1C1(C=2NC3=NC=CN=C3C=2)CC1 YWLJQPJXGXMBHN-UHFFFAOYSA-N 0.000 description 1
- QYVQNAFKQYTJEL-UHFFFAOYSA-N 6-[4-(1,3-dioxol-2-yl)phenyl]-5h-pyrrolo[2,3-b]pyrazine Chemical compound O1C=COC1C1=CC=C(C=2NC3=NC=CN=C3C=2)C=C1 QYVQNAFKQYTJEL-UHFFFAOYSA-N 0.000 description 1
- VDTPNYRFOHKFRY-UHFFFAOYSA-N 6-[4-(trifluoromethyl)phenyl]-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(C(F)(F)F)=CC=C1C1=CC2=NC=CN=C2N1 VDTPNYRFOHKFRY-UHFFFAOYSA-N 0.000 description 1
- DGWXOLHKVGDQLN-UHFFFAOYSA-N 6-cyclohexylmethyloxy-5-nitroso-pyrimidine-2,4-diamine Chemical compound NC1=NC(N)=C(N=O)C(OCC2CCCCC2)=N1 DGWXOLHKVGDQLN-UHFFFAOYSA-N 0.000 description 1
- GOOPKIMTLVKCFF-UHFFFAOYSA-N 6-naphthalen-1-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC=C2C(C3=CC4=NC=CN=C4N3)=CC=CC2=C1 GOOPKIMTLVKCFF-UHFFFAOYSA-N 0.000 description 1
- IXQOVNQMUSNLCC-UHFFFAOYSA-N 6-pyridin-2-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C=C1C1=CC=CC=N1 IXQOVNQMUSNLCC-UHFFFAOYSA-N 0.000 description 1
- KNOUXIMJDQGMDM-UHFFFAOYSA-N 6-thiophen-2-yl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CSC(C=2NC3=NC=CN=C3C=2)=C1 KNOUXIMJDQGMDM-UHFFFAOYSA-N 0.000 description 1
- DGEBIHRCXYJULM-UHFFFAOYSA-N 7-(3-chloropropyl)-6-(4-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(CCCCl)C2=NC=CN=C2N1 DGEBIHRCXYJULM-UHFFFAOYSA-N 0.000 description 1
- KSMRAMWQJUGFSL-UHFFFAOYSA-N 7-(cyclohexylmethyl)-6-(4-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(CC2CCCCC2)C2=NC=CN=C2N1 KSMRAMWQJUGFSL-UHFFFAOYSA-N 0.000 description 1
- JREMEPDATDYPDA-UHFFFAOYSA-N 7-(cyclopropylmethyl)-6-(4-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(OC)=CC=C1C1=C(CC2CC2)C2=NC=CN=C2N1 JREMEPDATDYPDA-UHFFFAOYSA-N 0.000 description 1
- RIFUFLYTGPSIHT-UHFFFAOYSA-N 7-benzyl-6-(4-chlorophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound C1=CC(Cl)=CC=C1C1=C(CC=2C=CC=CC=2)C2=NC=CN=C2N1 RIFUFLYTGPSIHT-UHFFFAOYSA-N 0.000 description 1
- CBALVKLWBKZXIX-UHFFFAOYSA-N 7-benzyl-6-phenyl-5h-pyrrolo[2,3-b]pyrazine Chemical compound C=1C=CC=CC=1CC(C1=NC=CN=C1N1)=C1C1=CC=CC=C1 CBALVKLWBKZXIX-UHFFFAOYSA-N 0.000 description 1
- GKGBCENYMVXALT-UHFFFAOYSA-N 7-butyl-6-(4-chlorophenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(CCCC)=C1C1=CC=C(Cl)C=C1 GKGBCENYMVXALT-UHFFFAOYSA-N 0.000 description 1
- OCQOWMWCDNLMCO-UHFFFAOYSA-N 7-heptyl-6-(4-methoxyphenyl)-5h-pyrrolo[2,3-b]pyrazine Chemical compound N1C2=NC=CN=C2C(CCCCCCC)=C1C1=CC=C(OC)C=C1 OCQOWMWCDNLMCO-UHFFFAOYSA-N 0.000 description 1
- PETCVZZPKYJZAU-UHFFFAOYSA-O 8-(3-bicyclo[2.2.1]heptanyl)-2-[4-[4-(3-hydroxypropyl)piperidin-1-ium-1-yl]anilino]pyrido[2,3-d]pyrimidin-7-one Chemical compound C1CC(CCCO)CC[NH+]1C(C=C1)=CC=C1NC1=NC=C(C=CC(=O)N2C3C4CCC(C4)C3)C2=N1 PETCVZZPKYJZAU-UHFFFAOYSA-O 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- 241000258957 Asteroidea Species 0.000 description 1
- 108060000903 Beta-catenin Proteins 0.000 description 1
- 102000015735 Beta-catenin Human genes 0.000 description 1
- UOGOFLOYGDAWKE-DXNYSGJVSA-N C/C=N\c(cc(-c1c[s]cc1)[nH]1)c1N=C Chemical compound C/C=N\c(cc(-c1c[s]cc1)[nH]1)c1N=C UOGOFLOYGDAWKE-DXNYSGJVSA-N 0.000 description 1
- 101150012716 CDK1 gene Proteins 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 102000008122 Casein Kinase I Human genes 0.000 description 1
- 108010049812 Casein Kinase I Proteins 0.000 description 1
- 102000011632 Caseins Human genes 0.000 description 1
- 108010076119 Caseins Proteins 0.000 description 1
- 101150053721 Cdk5 gene Proteins 0.000 description 1
- 102000003909 Cyclin E Human genes 0.000 description 1
- 108090000257 Cyclin E Proteins 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 101100059559 Emericella nidulans (strain FGSC A4 / ATCC 38163 / CBS 112.46 / NRRL 194 / M139) nimX gene Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 101001105683 Homo sapiens Pre-mRNA-processing-splicing factor 8 Proteins 0.000 description 1
- 101000772173 Homo sapiens Tubby-related protein 1 Proteins 0.000 description 1
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 1
- 101000610557 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp31 Proteins 0.000 description 1
- 101001104102 Homo sapiens X-linked retinitis pigmentosa GTPase regulator Proteins 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 229930182816 L-glutamine Natural products 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 230000027311 M phase Effects 0.000 description 1
- 241000257473 Marthasterias glacialis Species 0.000 description 1
- 238000007476 Maximum Likelihood Methods 0.000 description 1
- 102000047918 Myelin Basic Human genes 0.000 description 1
- 101710107068 Myelin basic protein Proteins 0.000 description 1
- ZYWAASADIMEXLI-UHFFFAOYSA-N NC1=NC=CC=N1.N1=CN=CC2=C1C=CC=N2 Chemical compound NC1=NC=CC=N1.N1=CN=CC2=C1C=CC=N2 ZYWAASADIMEXLI-UHFFFAOYSA-N 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 229920002562 Polyethylene Glycol 3350 Polymers 0.000 description 1
- 102100021231 Pre-mRNA-processing-splicing factor 8 Human genes 0.000 description 1
- 101000862778 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 40S ribosomal protein S3 Proteins 0.000 description 1
- 101000677914 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 40S ribosomal protein S5 Proteins 0.000 description 1
- 101000643078 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 40S ribosomal protein S9-A Proteins 0.000 description 1
- 101000729607 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 40S ribosomal protein S9-B Proteins 0.000 description 1
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 1
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 1
- 101001109965 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L7-A Proteins 0.000 description 1
- 101001109960 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L7-B Proteins 0.000 description 1
- 229940124639 Selective inhibitor Drugs 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- XOKJUSAYZUAMGJ-UHFFFAOYSA-N Toyocamycin Natural products C1=C(C#N)C=2C(N)=NC=NC=2N1C1OC(CO)C(O)C1O XOKJUSAYZUAMGJ-UHFFFAOYSA-N 0.000 description 1
- 102100029293 Tubby-related protein 1 Human genes 0.000 description 1
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 1
- 102100040118 U4/U6 small nuclear ribonucleoprotein Prp31 Human genes 0.000 description 1
- 230000004156 Wnt signaling pathway Effects 0.000 description 1
- 102100040092 X-linked retinitis pigmentosa GTPase regulator Human genes 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- 101100273808 Xenopus laevis cdk1-b gene Proteins 0.000 description 1
- XKWAORSEBHYNSB-UHFFFAOYSA-N [4-(7-methyl-5h-pyrrolo[2,3-b]pyrazin-6-yl)phenyl] n-(dimethylamino)sulfamate Chemical compound C1=CC(OS(=O)(=O)NN(C)C)=CC=C1C1=C(C)C2=NC=CN=C2N1 XKWAORSEBHYNSB-UHFFFAOYSA-N 0.000 description 1
- XJGBORQTNMLWPF-UHFFFAOYSA-N [Li]Cc1cnccn1 Chemical compound [Li]Cc1cnccn1 XJGBORQTNMLWPF-UHFFFAOYSA-N 0.000 description 1
- OLBVUFHMDRJKTK-UHFFFAOYSA-N [N].[O] Chemical compound [N].[O] OLBVUFHMDRJKTK-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- FRTNIYVUDIHXPG-UHFFFAOYSA-N acetic acid;ethane-1,2-diamine Chemical compound CC(O)=O.CC(O)=O.CC(O)=O.CC(O)=O.NCCN FRTNIYVUDIHXPG-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- PYHXGXCGESYPCW-UHFFFAOYSA-N alpha-phenylbenzeneacetic acid Natural products C=1C=CC=CC=1C(C(=O)O)C1=CC=CC=C1 PYHXGXCGESYPCW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- BIIVYFLTOXDAOV-YVEFUNNKSA-N alvocidib Chemical compound O[C@@H]1CN(C)CC[C@@H]1C1=C(O)C=C(O)C2=C1OC(C=1C(=CC=CC=1)Cl)=CC2=O BIIVYFLTOXDAOV-YVEFUNNKSA-N 0.000 description 1
- 229950010817 alvocidib Drugs 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 230000002927 anti-mitotic effect Effects 0.000 description 1
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000000376 autoradiography Methods 0.000 description 1
- 230000003376 axonal effect Effects 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 101150073031 cdk2 gene Proteins 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000005138 cryopreservation Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000002435 cytoreductive effect Effects 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- FHHZOYXKOICLGH-UHFFFAOYSA-N dichloromethane;ethanol Chemical compound CCO.ClCCl FHHZOYXKOICLGH-UHFFFAOYSA-N 0.000 description 1
- AASUFOVSZUIILF-UHFFFAOYSA-N diphenylmethanone;sodium Chemical compound [Na].C=1C=CC=CC=1C(=O)C1=CC=CC=C1 AASUFOVSZUIILF-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 230000028023 exocytosis Effects 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 239000004009 herbicide Substances 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000006951 hyperphosphorylation Effects 0.000 description 1
- TVZISJTYELEYPI-UHFFFAOYSA-N hypodiphosphoric acid Chemical compound OP(O)(=O)P(O)(O)=O TVZISJTYELEYPI-UHFFFAOYSA-N 0.000 description 1
- XSTBCICLDQSQIQ-UHFFFAOYSA-N indeno[2,1-c]pyrazole Chemical class C1=CC=C2C3=CN=NC3=CC2=C1 XSTBCICLDQSQIQ-UHFFFAOYSA-N 0.000 description 1
- HBDSHCUSXQATPO-BGBJRWHRSA-N indirubin-3'-monoxime Chemical compound O=C/1NC2=CC=CC=C2C\1=C\1/C(=N/O)/C2=CC=CC=C2N/1 HBDSHCUSXQATPO-BGBJRWHRSA-N 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 125000005439 maleimidyl group Chemical class C1(C=CC(N1*)=O)=O 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 238000004452 microanalysis Methods 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000004001 molecular interaction Effects 0.000 description 1
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 1
- MCWHFEZTUZXKFF-UHFFFAOYSA-N n,n-dimethyl-4-(5h-pyrrolo[2,3-b]pyrazin-6-yl)aniline Chemical compound C1=CC(N(C)C)=CC=C1C1=CC2=NC=CN=C2N1 MCWHFEZTUZXKFF-UHFFFAOYSA-N 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000007135 neurotoxicity Effects 0.000 description 1
- 231100000228 neurotoxicity Toxicity 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 210000000287 oocyte Anatomy 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 150000005623 oxindoles Chemical class 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 238000000059 patterning Methods 0.000 description 1
- 108010091212 pepstatin Proteins 0.000 description 1
- 229950000964 pepstatin Drugs 0.000 description 1
- FAXGPCHRFPCXOO-LXTPJMTPSA-N pepstatin A Chemical compound OC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)C[C@H](O)[C@H](CC(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)CC(C)C FAXGPCHRFPCXOO-LXTPJMTPSA-N 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000031877 prophase Effects 0.000 description 1
- 238000012342 propidium iodide staining Methods 0.000 description 1
- 239000003909 protein kinase inhibitor Substances 0.000 description 1
- 230000009822 protein phosphorylation Effects 0.000 description 1
- 244000000040 protozoan parasite Species 0.000 description 1
- ZLIBICFPKPWGIZ-UHFFFAOYSA-N pyrimethanil Chemical compound CC1=CC(C)=NC(NC=2C=CC=CC=2)=N1 ZLIBICFPKPWGIZ-UHFFFAOYSA-N 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical class N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000006965 reversible inhibition Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- XOKJUSAYZUAMGJ-WOUKDFQISA-N toyocamycin Chemical compound C1=C(C#N)C=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O XOKJUSAYZUAMGJ-WOUKDFQISA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 229910000166 zirconium phosphate Inorganic materials 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Tropical Medicine & Parasitology (AREA)
- Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
「Cyclin-dependent kinases inhibitors as potential anticancer,anti-neurodegenerative,anti-viral and anti-parasitic agent」 Drug Resistance Update 2000,3,83-88 Meijer,L. 「Cyclin-dependent kinase inhibitors for treating cancer.」Med.Res.Rev.2001,21,487~498 Toogood,P.L. 「ATP-site directed inhibitors of cyckin-dependent kinases.」Curr.Med.Chem.1999,6,859~876 Gray,N;Detivaud,L;Doerig,C.;Meijer,L. 「Inhibitors of cyclin-dependent kinases as anti-cancer therapeutics.」Curr.Med.Chem.2000,7,1213~1245. Fischer,P.M.;Lane,D.P. 「Cyclin-dependent kinase inhibitors:novel anticancer agents.」Expert Opin.Investig.Drugs 2000,9,1849~1870.Pestell,R.;Mani,S.;Wange,C.;Wu,K.;Francis,R. 「Targeting hyperproliferative disorders with cyclin dependent kinase inhibitors.」Exp.Opin.Ther.Patents 2000,10,1~16. Rosiana,G.R.;Chang,Y.T. 「Cyclin-dependent kinase inhibitors:useful targets in cell cycle regulation.」J.Med.Chem.2000,43,1~18. Sielecki,T.M.;Boylan,J.F.;Benfield,P.A.;Trainor,G.L. 「Cyclin-dependent kinase and protein kinase C inhibitors: a novel class of antineoplastic agents in clinical dependent.」Cancer J.2000,6,192~212. Kaubisch,A.;Schwartz G.K.

−R2およびR3は、同一または異なって、H、C1−C6アルキルを示し、該アルキルは、直鎖または分枝鎖アルキルであり、置換されていてもよく、
−R6は、置換されていてもよい芳香環Arまたはシクロアルキルであり、該シクロアルキルは、アリール基によって置換されていてもよく、このアリール基もまた置換されていてもよく、
−R7は、H、C1−C6アルキル、(alk.)n−hal.、CH2−CH=CH2、CH2−シクロアルキル、CH2−Arであり、
−Zは、HまたはCH3である。
− 6位のフェニル基は、1、2または3つのR置換基によって置換され、該Rは、
− H、OH、アルキル、−O−アルキル、hal.、−NH2、−N(H,アルキル)、−N(アルキル)2、−O−SO2−NH2、−O−SO2−N(H,アルキル)、−O−SO2−N(アルキル)2、−COOH、−COO−アルキル、−CONH2、−CON(H,アルキル)およびCON(アルキル)2
を含む群内で選択され、
− R7は、H、アルキル、(alk.)nhal.、−CH2−CH=CH2、(alk.)n−シクロアルキルまたはalk.−Arであり、そして、
− Zは、HまたはCH3である。
R=H、OH、アルコキシ、hal.またはアルキルであり、かつ、R7=Hである化学式(II)の誘導体または、R=アルコキシであり、かつ、R7=アルキル、(alk.)n−hal.またはCH2−CH=CH2である誘導体、または、
R=O−SO2−N−(アルキル)2、好適には、hal.またはOHであり、かつ、R7=アルキルであり、かつ、n=1〜3であり、かつ、Z=Hである誘導体に対応する。
−フェニル基は無置換であり、かつ、R7はHであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはhal.であり、かつ、R7=Hであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシであり、かつ、R7=アルキルであるか、または、
−RaおよびRd=Hであり、かつ、RbおよびRc=アルコキシであり、かつ、R7=アルキルであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシであり、かつ、R7=アルキルであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはhal.であり、かつ、R7=アルキル、(alk.)n−hal.またはCH2−CH=CH2であるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=OHであり、かつ、R7=アルキルである
化合物(a〜eは、フェニル基上のRの位置に対応する)に対応する。
R=p−アルコキシ、p−およびm−ジアルコキシ、hal.、p−O−SO2−N(アルキル)2またはp−OHであり、かつ、R7=Hまたはアルキルである。
R1およびR3は、上記定義の通りであり、Alkylは、芳香族ニトリル類R6CNを有するC1〜C6アルキルであり、ここで、R6は、上記定義の通りである。
融点は、Electrothermal 9200装置によりオープンキャピラリーチューブにおいて測定され、無修正とした。IRスペクトルは、ATI Mattson genesis series FTIRによりKBrにおいて得られた。1H NMRは、Varian EM 360 A分光計(60 MHz)により記録され、ケミカルシフト(ppm)は、TMSに関して記録された。シグナルは、以下のように表される。:bs(ブロード・シングレット:broad singlet)、s(シングレット:singlet)、d(ダブレット:doublet)、dd(ダブレット・オフ・ダブレット:doublet of doublets)、t(トリプレット:triplet)、m(マルチプレット:multiplet)。マススペクトルは、LKB 209(70eVでのEI)により決定された。元素微量分析は、元素記号によって示され、その結果は、他の記載がなければ理論値の±0.4%の範囲内であった。それらは、Perkin Elmer 240装置により行われた。
ジイソプロピルアミン(2.23g;0.022モル)(THF(50mL)中)を0℃に冷却し、ブチルリチウム(0.022モル)を滴下して加えた。0℃で30分攪拌した後、溶液を−40℃に冷却してからアルキルピラジン誘導体(0.02モル)(THF(20mL)中)を加えた。30分後、ニトリル誘導体R6−CN(0.01モル)(THF(20mL)中)を加え、溶液を−40℃で30分、さらに、20℃で1時間〜20時間攪拌し、その後、10%のNH4Cl水溶液で加水分解した。有機層をNa2SO4で乾燥し、減圧下に濃縮した。粗生成物をシリカゲルによるクロマトグラフィー精製に付し、塩化メチレン、次いで、酢酸エチルで溶離した。必要な場合には、生成物を、エタノールまたは塩化メチレン−エタノール混合物から結晶化した。
6−(2−フリル)[5H]ピロロ[2,3−b]ピラジン(1、RP19):融点 232.6℃;IR 3157、3143、3102cm−1;1H NMR δ(60MHz,DMSO−d6) 6.50−6.70(m,1H)、6.80(s,1H)、7.05(d,1H,J=3Hz)、7.80(bs,1H)、8.20および8.35(2d,それぞれ1H,J=3Hz)、12.45(bs,1H)。元素分析(C10H7N3O)C,H,N。
まず、痕跡量の50%の次リン酸により臭化水素酸を再蒸留した。:48%臭化水素酸の各100gに対して1g。メトキシ化合物(0.003モル)を臭化水素酸(20ml)とともに加熱した。前留水(aqueous forerun)を除去した後、温度は126℃に達する。脱メチル化に要する時間は、3〜10時間で変動する。過剰の臭化水素酸を減圧下に除去し、粗生成物をエタノールから結晶化した。
(生化学試薬)
オルトバナジン酸ナトリウム、EGTA(ethylene glycol bis(2-aminoethylether)tetraacetic acid)、EDTA(ethylendiaminetetraacetic acid)、Mops(3-(N-morpholino)propanesulfonic acid)、β−グリセロリン酸塩、フェニルリン酸塩、フッ化ナトリウム、ジチオトレイトール(dithiothreitol:DTT)、グルタチオン−アガロース、グルタチオン、ウシ血清アルブミン(bovine serum albumin:BSA)、ニトロフェニルリン酸塩、ロイペプチン、アプロチニン、ペプスタチン、ダイズトリプシンインヒビター、ベンズアミジン、ヒストンH1(タイプIII−S)が、Sigma Chemicalsから得られた。[γ−32P]−ATP(PB 168)がAmershamから得られた。GS−1ペプチドは、配列番号1 YRRAAVPPSPSLSRHSSPHQSPEDEEEを有する。
均質化緩衝液:60mMのβ−グリセロリン酸塩、15mMのp−ニトロフェニルリン酸塩、25mMのMops(pH7.2)、15mMのEGTA、15mMのMgCl2、1mMのDTT、1mMのバナジン酸ナトリウム、1mMのNaF、1mMのフェニルリン酸塩、10μgのロイペプチン/ml、10μgのアプロチニン/ml、10μgのダイズトリプシンインヒビター/mlおよび100μMのベンズアミジン。
キナーゼ活性は、30℃で、最終的なATP濃度が15μMとなるように、(別の記載がなければ)緩衝液AまたはCにおいてアッセイされた。ブランク値が差し引かれ、10分のインキュベートの間に取り込まれたリン酸塩のpモルとして活性が計算された。活性は、通常、最大活性、すなわち、阻害剤がないときの%で表現される。コントロールは、ジメチルスルホキシドの適切な希釈液で行われた。2,3の場合には、基質のリン酸化は、SDS−PAGE(sodium dodecyl sulfate-polyacrylamide gel electrophoresis)後のオートラジオグラフィーによって評価された。
(CDK1、CDK5およびGSK−3についてのアロイシン類の阻害効果)
複数のキナーゼは、増加する濃度のアロイシン類の存在下に、上記のようにアッセイされた。IC50は、投与量−応答曲線から計算され、表1に与えられ、以降、μMで示す。
アロイシン作用の機構を調べるために、反応速度論実験を、ATPレベルおよびアロイシンA濃度の双方を変更することによって行った。アロイシンAの異なる濃度におけるCDK1/サイクリンB(A)、CDK5/p25(B)およびGSK−3β(C)のキナーゼ活性のアッセイからの速度論データの両逆数プロット(double reciprocal plot)が図2に与えられる。酵素活性は、実験項に記載されるようにアッセイされた。反応混合物におけるATP濃度は、0.1から0.25mMに(CDK1およびCDK5)、または、0.015から0.15mMに(GSK−3β)に変動した。ヒストンH1(A、B)およびGS−1(C)の濃度は、0.7mg/mlおよび6.7μMの一定にそれぞれ維持された。
(ヒトCDK2の圧搾、精製および結晶化)
ヒトCDK2は、Sf9昆虫細胞における組み換えバキュロウイルスから圧搾し、そして、精製することにより得られる。モノマーのリン酸化されていないCDK2結晶は、参照(11)にすでに記載されるように成長される。
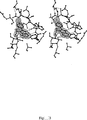
CDK2−アロイシンBのデータセットを、(1×母液溶液(50mM酢酸アンモニウム、10%のPEG3350、15mMのNaCl、100mMのHEPES(pH7.4)+5%DMSO)中の1mMのアロイシンBに60時間浸漬されたモノマーのCDK2結晶から収集した。結晶が一時的に低温予備保護剤(20%グリセロールを含むように調整された母液)に移された後、100KでElettra Light SourcrによるX−RAY DIFFRACTIONの光線によりデータを収集した。画像を、MOSFLMパッケージを用いて積算し(参照(12))、続いて、SCALAを用いて反射光をスケール調整および併合した(参照(13))。続いて、データ整復および構造細分を、CCP4スート(suite)のプログラムを通じて実行した。
ここまでで最も活性が強いアロイシンであるアロイシンAが、26の高度に精製されたキナーゼに関する選択性について試験された。キナーゼの活性は、適切な基質(例えば、ヒストンH1、カゼイン、ミエリン塩基性タンパク質およびペプチド)を用いて、15μMのATPとともに、アロイシンAの増加する濃度の存在中でアッセイされた。IC50値は、投与量−応答曲線から評価され、表3に表されている。
(試薬)
ペニシリン、ストレプトマイシン、ノコダゾール、インシュリン、トランスフェリン、プロゲステロン、プトレシン、セレン酸ナトリウム、臭化3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウム(MTT)、RNAse A、ヨウ化プロピジウムがSigmaから購入された
(細胞培養)
クローンヒトNT2テラテロカルシノーマ(teraterocarcinoma)細胞を、Stratagen(La Jolla,CA)から取得し、ダルベッコ改変イーグル液体培地(Dulbecco’s Modified Eagle Medium):5%FCS(fetal calf serum:牛胎児血清)が補足され、37℃でペニシリン(20UI/ml)およびストレプトマイシン(20μg/ml)を含む2mMのL−グルタミン(BIO WHITTAKER)含有F−12培地(Nutrient Mixture F-12)において、空気中に5%のCO2を含む湿気のある雰囲気で成長させた。
NT2細胞のhNT細胞への分化は、Soulieらによって改変されたPleasureらの方法(参照(17))にしたがって誘導された。簡単には、2回目の再塗布の後、細胞は、処理前5日間、分裂抑制剤(1μMのサイトシン・アラビノシッド(cytosine arabinoside)、10μMのフルオロデオキシウリジンおよび10μMのウリジン)と、塩とホルモン(25μg インシュリン/ml、100μg トランスフェリン/ml、20nMのプロゲステロン、60μMのプトレッシンおよび30nMのセレン酸ナトリウム)の混合物とを加えた血清除去培地において培養された。
指数関数的な増殖細胞をアロイシンA(ジメチルスルホキシドに溶解された保存液)存在下に24時間インキュベートした。細胞のノコダゾール処理を0.04μgノコダゾール/ml(培養液)の濃度で24時間行った。ノコダゾール処理の後、新しい培養液で細胞を2回洗浄し、アロイシンA存在下または非存在下に24時間培養した。血清廃除を実施するために、血清がない培地中に40時間細胞を維持した。血清廃除の後、細胞を2回洗浄し、アロイシンAの存在下または非存在下に新しい血清含有培地中で40時間培養した。
NT2細胞およびhNTヒトニューロンについてのアロイシンAの毒性を定量するために、NTTホルマザンに対するNTTの細胞減少の阻害を、Sailleらにしたがって評価した。アロイシンAの暴露の後、0.5mg・MTT/ml(新しい培地)の存在下に37℃1時間細胞をインキュベートした。ホルマザン生成物をDMSOに溶解し、562nmでの吸収の測定によって定量した。
細胞をトリプシン処理し、遠心分離により収集し、冷却70%エタノールで少なくとも4時間凝集させた。凝集された細胞をPBS(phosphate buffered saline:リン酸緩衝生理食塩水)で洗浄し、10μg RNAse/mlの存在下にインキュベートし、25μg/mlのヨウ化プロピジウムにより37℃で1時間染色した。次いで、FACSort流動細胞計測器(Becton Dickinson)を用いて染色された細胞を細胞周期分布について分析した。multiCYCLEにより細胞周期分析を実施した(参照(18))。
1. Meijer, L. Cyclin-dependent kinases inhibitors as potential anticancer,anti-neurodegenerative,anti-viral and anti-parasitic agent. Drug Resistance Update 2000,3,83-88.
2. Toogood, P.L. Cyclin-dependent kinase inhibitors for treating cancer. Med.Res.Rev.2001,21,487~498.
3. Gray,N.; Detivaud,L.; Doerig,C.; Meijer,L. ATP-site directed inhibitors of cyckin-dependent kinases.Curr. Med.Chem.1999,6,859~876.
4. Fischer, P.M.; Lane, D.P. Inhibitors of cyclin-dependent kinases as anti-cancer therapeutics. Curr.Med.Chem.2000,7,1213~1245.
5. Pestell,R.; Mani,S.; Wange,C.; Wu,K.; Francis,R. Cyclin-dependent kinase inhibitors:novel anticancer agents. Expert Opin.Investig.Drugs 2000,9,1849~1870.
6. Rosiana,G.R.; Chang,Y.T. Targeting hyperproliferative disorders with cyclin dependent kinase inhibitors. Exp.Opin.Ther.Patents 2000,10,1~16.
7. Sielecki,T.M.; Boylan,J.F.; Benfield,P.A.; Trainor,G.L. Cyclin-dependent kinase inhibitors:useful targets in cell cycle regulation. J.Med.Chem.2000,43,1~18.
8. Kaubisch,A.; Schwartz G.K. Cyclin-dependent kinase and protein kinase C inhibitors: a novel class of antineoplastic agents in clinical dependent.Cancer J.2000,6,192~212.
9. Meijer,L.; Borgne,A.; Mulner,O.; Chong,J.P.J.; Blow,J.J.; Inagaki,M.; Delcros,J.G.; Moulinoux,J.P. Biochemical and cellular effects of roscovitine,a potent and selective inhibitor of the cyclin-dependent kinases cdc2,cdk2 and cdk5. Eur.J.Biochem.1997,243,527-536.
10. Meijer,L.; Thunissen,A.M.W.H.; White,A.; Garnier,M.; Nikolic,M.; Tsai,L.H.; Walter,J.; Cleveley,K.E.; Salinas,P.C.; Wu,Y.Z.; Biernat,J.; Mandelkow,E.M.; Kim,S.-H.; Pettit,G.R. Inhibition of cyclin-dependent kinases,GSK-3β and casein kinase 1 by hymenialdisine,a marine sponge constituent. Chem. & Biol.2000,,7-51-63.
11. Lawrie,A.M.; Noble,N.E.; Tunnah,P.; Brown,N.R.; Johnson,L.N.; Endicott,J.A. Protein kinase inhibition by staurosporine revealed in details of the molecular interaction with CDK2. Nature Structural Biol.1997,4,796-800.
12. Leslie,A.G.W. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography.1992,vol.26.
13. CCP4. The CCP4(Collaborative Computational Project Number 4)suite:Programs for protein crystallography.Acta Cryst.D,1994,50,760-763.
14. Navaza,J.AMoRe: An automated package for molecular replacement.Acta.Cryst.,1994,A50,157-163.
15. Murshudov,G.N.; Vagin,A.A.; and Dodson,E.J.Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst.D,1997,53,240-255.
16. Jones,T.A.,Zou,J.Y.,Cowan,S.W.,Kjeldgaard,M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst.A,1991,47,110-119.
17. Pleasure,S.J.; Page,C.; Lee,V.M.Y. Pure,post-mitotic polarized human neurons derived from NT2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J.Neurosci.1992,12,1802-1815.
18. Damiens,E.; Baratte,B.; Marie,D.; Eisenbrand,G.; and Meijer,L. Anti-mitotic properties of indirubin-3’-monoxime,a CDK/GSK-3 inhibitor: induction of endoreplication following prophase arrest. Oncogene,2001,20,3786-3797.
Claims (60)
- ピロロ[2,3b]−ピラジン誘導体を含む、神経変性疾患を治療または予防するための医薬組成物であって、該誘導体は、一般式(I):
− R2およびR3は、同一または異なって、HまたはC1−C6アルキルを示し、前記アルキルは、直鎖または分枝鎖アルキルであり、置換されてもよく、
− R6は、置換されてもよい芳香環Arまたはシクロアルキルであり、前記シクロアルキルは、置換されてもよいアリール基によって置換されてもよく、
− R7は、H、C1−C6アルキル、(alk.)n−hal.、CH2−CH=CH2、CH2−シクロアルキルまたはCH2−Arであり、ここで、「alk.」はC1−C6アルキレン基であり、nは1〜6であり、
− ZはHまたはCH3である)
により示される、医薬組成物。 - Arはフェニル、ナフチル、フリル、チエニル、ピリジル、シクロプロピルフェニルまたはフェニルジオキソリルである、請求項1に記載の医薬組成物。
- シクロアルキル基はC3−C6シクロアルキルである、請求項1に記載の医薬組成物。
- 置換基は、1種以上のハロゲン(F、Cl、Br、I、CF3)、OH、NH2、N(H,アルキル)、N(アルキル)2、O−アルキル、COOH、COO−アルキル、CONH2、CON(H,アルキル)、CON(アルキル)2、NHCONH2、NHCON(H,アルキル)、NHCON(アルキル)2、N(アルキル)CONH2、N(アルキル)CON(H,アルキル)、N(アルキル)CON(アルキル)2、アルコキシ、CN、O−SO2−NH2、O−SO2−N(H,アルキル)、−O−SO2−N(アルキル)2、SHおよびS−アルキルからなる群から選択される、請求項1〜3のいずれか1つに記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよび/またはCDK5/p25および/またはGSK−3に対してIC50≦10μMを有し、化学式(II):
−6位のフェニル基は、
−H、−OH、アルキル、−Oアルキル、hal.、−NH2、−N(H,アルキル)、−N(アルキル)2、−O−SO2−NH2、−SO2−N(H,アルキル)、−O−SO2−N(アルキル)2、−COOH、−COO−アルキル、CONH2、−CON(H,アルキル)および−CON(アルキル)2
からなる群から選択される1、2または3つの置換基Rによって置換され、
−R7は、H、アルキル、(alk.)nhal.、−CH2−CH=CH2、(alk.)n−シクロアルキルまたはalk.−Arであり、
−ZはHまたはCH3である)
により示される、請求項1〜4のいずれか1つに記載の医薬組成物。 - 式(II)の誘導体において、
R=H、OH、アルコキシ、hal.またはアルキルであり、かつ、R7=Hであるか、または、
R=アルコキシであり、かつ、R7=アルキル、(alk.)n−hal.またはCH2−CH=CH2であるか、または、
R=O−SO2−N(アルキル)2、hal.またはOHであり、R7=アルキルであり、n=1〜3であり、かつ、Z=Hである、
請求項5に記載の医薬組成物。 - 前記誘導体は、CDK1/サイクリンB、CDK5/p25およびGSK−3に対してIC50値≦5μMを有する、請求項5に記載の医薬組成物。
- 式(II)の誘導体において、R=H、p−アルコキシ、p−およびm−アルコキシ、p−OH、p−hal.、p−アルキルまたはp−O−SO2−N(アルキル)2であり、かつ、R7はアルキル、(alk.)n−hal.、CH2−CH=CH2またはHであり、ZはHであり、かつ、n=1〜3である、請求項7に記載の医薬組成物。
- −フェニル基は、無置換であり、かつ、R7はHであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはhal.であり、かつ、R7=Hであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシであり、かつ、R7=アルキルであるか、または、
−RaおよびRd=Hであり、かつ、RbおよびRc=アルコキシであり、かつ、R7=アルキルであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシであり、かつ、R7=アルキルであるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはhal.であり、かつ、R7=アルキル、(alk.)n−hal.またはCH2−CH=CH2であるか、または、
−Ra、RbおよびRd=Hであり、かつ、Rc=OHであり、かつ、R7=アルキルである(a−eはフェニル基上のRの位置に対応する)、請求項8に記載の医薬組成物。 - 前記誘導体は、CDK1/サイクリンB、CDK5/p25およびGSK−3に対してIC50≦1μMを有する、請求項5に記載の医薬組成物。
- 式(II)の誘導体において、Rがp−アルコキシ、p−O−SO2−N−(アルキル)2またはp−OHであり、かつ、R7がアルキルである、請求項10に記載の医薬組成物。
- Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、O−SO2−N(アルキル)2またはOHであり、かつ、R7=アルキルである化合物を含む、請求項11に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンB、CDK5/p25およびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項5に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよびCDK5/p25またはGSK−3に対して、または、CDK5/p25およびGSK−3に対してIC50値≦10μMを有する、請求項1〜4のいずれか1つに記載の医薬組成物。
- 前記誘導体は、CDK5/p25およびGSK−3に対してIC50≦10μMを有する、請求項14に記載の医薬組成物。
- R=H、OH、アルコキシ、hal.、アルキルまたはO−SO2−N(アルキル)2であり、かつ、R7=H、アルキル、(alk.)n−hal.またはCH2−CH=CH2である、請求項15に記載の医薬組成物。
- 前記誘導体は、CDK5/p25およびGSK−3に対してIC50値≦5μMを有し、RはH、p−アルコキシ、OH、hal.またはO−SO2−N−(アルキル)2であり、かつ、R7はH、アルキル、(alk.)nhal.またはCH2−CH=CH2である、請求項15に記載の医薬組成物。
- 前記誘導体において、Ra、Rb、Rc、RdおよびR7=Hであるか、または、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、hal.、(alk.)n−hal.またはOHであり、かつ、R7=Hであるか、または、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OSO2−N(アルキル)2、hal.またはOHであり、かつ、R7=アルキルであるか、または、RaおよびRd=Hであり、かつ、RbおよびRc=アルコキシであり、かつ、R7=アルキルである、請求項17に記載の医薬組成物。
- 前記誘導体は、CDK5/p25およびGSK−3に対してIC50値≦1μMを有し、R=p−アルコキシ、p−およびm−ジアルコキシ、hal.、p−O−SO2−N(アルキル)2またはp−OHであり、かつ、R7=Hまたはアルキルである、請求項15に記載の医薬組成物。
- 前記誘導体において、Ra、Rb、Rd=Hであり、かつ、Rc=アルコキシであり、かつ、R7=アルキルであるか、または、RaおよびRd=Hであり、かつ、RbおよびRc=アルコキシであり、かつ、R7=アルキルであるか、または、Ra、RbおよびRd=Hであり、かつ、Rc=O−SO2−N(アルキル)2またはOHであり、かつ、R7=アルキルである、請求項19に記載の医薬組成物。
- 前記誘導体は、CDK5/p25およびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項15に記載の医薬組成物。
- 前記誘導体は、CDK1およびGSK3に対してIC50≦10μMを有し、R=H、OH、アルコキシ、hal.、アルキル、CNまたはO−SO2−N(アルキル)2であり、かつ、R7=H、アルキル、(alk.)n−hal.、CH2−CH=CH2、alk.−シクロアルキルまたはalk.−アリールである、請求項14に記載の医薬組成物。
- 前記誘導体は、CDK1およびGSK−3に対してIC50≦5μMを有し、R=H、p−アルコキシ、p−およびm−アルコキシ、p−OH、p−hal.、p−O−SO2−N(アルキル)2またはp−CNであり、かつ、R7=H、アルキル、(alk.)n−hal.、CH2−CH=CH2、(alk.)n−シクロアルキルまたは(alk.)n−アリールである、請求項22に記載の医薬組成物。
- 前記誘導体は、Ra、RbおよびRd=H、Rc=アルコキシ、OH、hal.、アルキル、CN、およびR7=H、または、Ra、Rb、Rd=H、Rc=アルコキシおよびR7=アルキル、(alk.)n−hal.またはCH2−CH=CH2、または、RaおよびRd=H、RbおよびRc=アルコキシ、およびR7=アルキル、または、Ra、RbおよびRc=H、Rd=O−SO2−N−(アルキル)2、およびR7=アルキル、または、Ra、RbおよびRd=H、Rc=hal.および、R7=(alk.)n−アリールを有する、請求項23に記載の医薬組成物。
- 前記誘導体は、CDK1およびGSK−3に対してIC50値≦1μMを有し、R=p−アルコキシ、p−O−SO2−N(アルキル)2、p−hal.、Hまたはp−OHであり、かつ、R7=アルキル、(alk.)n−hal.、CH2−CH=CH2、(alk.)n−シクロアルキルまたは(alk.)n−アリールである、請求項22に記載の医薬組成物。
- Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OH、O−SO2−N(アルキル)2またはhal.であり、かつ、R7=アルキル、CH2−CH=CH2またはCH2−シクロアルキルである、請求項25に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50値≦0.5μMを有し、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項22に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよびCDK5/p25に対してIC50≦10μMを有し、R=H、OH、アルコキシ、hal.、アルキルまたはO−SO2−N(アルキル)2であり、かつ、R7=H、アルキル、(alk.)n−hal.またはCH2−CH=CH2である、請求項14に記載の医薬組成物。
- CDK1/サイクリンBおよびGSK−3に対してIC50≦5μMを有し、RはH、o−アルコキシ、p−アルコキシ、m−およびp−アルコキシ、p−OH、p−hal.、p−O−SO2−N(アルキル)2であり、かつ、R7はH、アルキル、(alk.)n−hal.またはCH2−CH=CH2である、請求項28に記載の医薬組成物。
- Ra、Rb、Rc、RdおよびR7=Hであるか、または、Ra=OHであり、かつ、Rb、Rc、RdおよびR7=Hであるか、または、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはhal.であり、かつ、R7=H、(alk.)n−hal.、CH2−CH=CH2またはアルキルであるか、または、RaおよびRd=Hであり、かつ、RbおよびRc=アルコキシであり、かつ、R7=Hであるか、または、Ra、RbおよびRd=Hであり、かつ、Rc=O−SO2−N−(アルキル)2またはhal.であり、かつ、R7=アルキルである、請求項29に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50≦1μMを有し、R=p−アルコキシ、p−O−SO2−N(アルキル)2、p−hal.またはp−OHであり、かつ、R7=アルキルである、請求項28に記載の医薬組成物。
- Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシ、OHまたはO−SO2−N(アルキル)2であり、かつ、R7=アルキルである、請求項31に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、かつ、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項28に記載の医薬組成物。
- 前記誘導体は、CDK1/サイクリンB、CDK5およびGSK−3に対してIC50≦10μMを有し、式(III):
- 前記誘導体は、式(IV):
- R2およびR3および/またはZおよび/またはR7はHではない、請求項1〜35のいずれか1つに記載の医薬組成物。
- アルツハイマー病またはパーキンソン病を治療または予防するための、請求項1〜36のいずれか1つに記載の医薬組成物。
- ピロロ[2,3b]−ピラジン誘導体であって、CDK1/サイクリンBおよび/またはCDK5/p25および/またはGSK−3に対してIC50≦1μMを有し、式(I):
R6は、置換フェニル基または置換されてもよいナフチル、フリル、チエニル、ピリジル、シクロプロピル、フェニルジオキソリルであり、
ここで、式(II):
−H、アルキル、−O−アルキル、−NH2、−N(H,アルキル)、―N(アルキル)2、−O−SO2−NH2、−O−SO2−N(H,アルキル)、−O−SO2−N(アルキル)2、−COOH、−COO−アルキル、CONH2、−CON(H,アルキル)、−CON(アルキル)2
からなる群において選択される1、2または3つの置換基Rによって置換され、
− R7は、H、アルキル、(alk.)nhal.、−CH2−CH=CH2、(alk.)n−シクロアルキル、alk.−Arであり、
−ZはHまたはCH3である)
により示され、ただし、
−R6がフェニル基である場合、R2、R3、R7およびZはHではなく、
−R3、R7およびZがHであり、R6がフェニル基である場合、R2はメチルではなく、
−R3およびZがHであり、R7がメチルであり、R6がフェニル基である場合、R2はHではなく、
−R6がフリル基である場合、R2、R3、R7およびZはHではなく、および
−R6が、アルキル、−O−アルキル、−N(H,アルキル)、−N(アルキル) 2 および−NH 2 からなる群において選択される1、2または3つの置換基Rによって置換されたフェニル基である場合、R2、R3、R7およびZはHではない、
ピロロ[2,3b]−ピラジン誘導体。 - CDK1/サイクリンB、CFK5/p25およびGSK−3に対してIC50≦1μMを有する、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- 式(II)の誘導体に対応し、Rは、p−アルコキシ、p−O−SO2−N−(アルキル)2、p−OHであり、R7はアルキルである、請求項39に記載のピロロ[2,3b]−ピラジン誘導体。
- Ra、RbおよびRd=H、Rc=アルコキシ、O−SO2−N(アルキル)2またはOHであり、かつ、R7=アルキルである化合物に対応する、請求項40に記載のピロロ[2,3b]−ピラジン誘導体。
- CDK1/サイクリンB、CDK5/p25およびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- CDK5/p25およびGSK−3に対してIC50値≦1μMを有し、R=p−アルコキシ、p−およびm−ジアルコキシ、hal.、p−O−SO2−N(アルキル)2、p−OHであり、かつ、R7=Hまたはアルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- Ra、Rb、Rd=H、Rc=アルコキシおよびR7=アルキル、またはRaおよびRd=H、RbおよびRc=アルコキシおよびR7=アルキル、または、Ra、RbおよびRd=H、Rc=O−SO2−N(アルキル)2またはOHおよびR7=アルキルである、請求項43に記載のピロロ[2,3b]−ピラジン誘導体。
- CDK5/p25およびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、Rc=アルコキシまたはOHであり、かつR7=アルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- CDK1およびGSK−3に対してIC50値≦1μMを有し、R=p−アルコキシ、p−O−SO2−N(アルキル)2、p−hal.、H、p−OHであり、R7=アルキルまたは(alk.)n−hal、CH2−CH=CH2、(alk.)n−シクロアルキル、(alk.)n−アリールである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- Ra、RbおよびRd=Hであり、Rc=アルコキシ、OH、O−SO2−N(アルキル)2、hal.であり、R7=アルキル、CH2−CH=CH2、CH2−シクロアルキルである、請求項46に記載のピロロ[2,3b]−ピラジン誘導体。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50値≦0.5μMを有し、Ra、RbおよびRd=Hであり、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50≦1μMを有し、R=p−アルコキシ、p−O−SO2−N(アルキル)2、p−hal.、p−OHであり、かつ、R7=アルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- Ra、RbおよびRd=Hであり、Rc=アルコキシ、OHまたはO−SO2−N(アルキル)2であり、かつ、R7=アルキルである、請求項49に記載のピロロ[2,3b]−ピラジン誘導体。
- 前記誘導体は、CDK1/サイクリンBおよびGSK−3に対してIC50≦0.5μMを有し、Ra、RbおよびRd=Hであり、Rc=アルコキシまたはOHであり、かつ、R7=アルキルである、請求項38に記載のピロロ[2,3b]−ピラジン誘導体。
- 式(III):
- 式(IV):
- 化学式(V):
R2およびR3は、同一または異なって、HまたはC1−C6アルキルを示し、前記アルキルは、直鎖または分枝鎖アルキルであり、置換されてもよく、Alkylは、芳香族ニトリル類であるR6CNを有するC1−C6アルキルであり、ここで、R6は請求項38のように定義される)
のアルキルピラジン類を反応させる工程を包含する、請求項38〜53のいずれか1つに記載された化学式Iのピロロ[2,3b]−ピラジン誘導体を合成する方法。 - 有効成分として請求項38〜53のいずれかの少なくとも一つの誘導体の有効量を、医薬的に許容される担体と組み合わせて含む医薬組成物。
- 神経変性疾患を治療または予防するための請求項55の医薬組成物。
- 抗増殖疾患を治療または予防するための請求項55に記載の医薬組成物。
- 経口により、局所に、または、注射(静脈内、皮下、腹腔、または直腸)によって投与される、請求項56または57に記載の医薬組成物。
- 投与量単位当たり100〜1000mgの有効成分を含む経口ルートによる投与のための請求項58の医薬組成物。
- 注射可能な形式の下、投与量単位当たり100〜1000mgの有効成分を含む、請求項58の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20020292019 EP1388541A1 (en) | 2002-08-09 | 2002-08-09 | Pyrrolopyrazines as kinase inhibitors |
| EP02292019.3 | 2002-08-09 | ||
| PCT/EP2003/009515 WO2004016614A2 (en) | 2002-08-09 | 2003-08-08 | Pyrrolopyrazines as kinase inhibitors |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2006502137A JP2006502137A (ja) | 2006-01-19 |
| JP2006502137A5 JP2006502137A5 (ja) | 2006-08-03 |
| JP4780549B2 true JP4780549B2 (ja) | 2011-09-28 |
Family
ID=30129256
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004528508A Expired - Fee Related JP4780549B2 (ja) | 2002-08-09 | 2003-08-08 | キナーゼ阻害活性を有するピロロピラジン誘導体およびその生物学的用途 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8106050B2 (ja) |
| EP (2) | EP1388541A1 (ja) |
| JP (1) | JP4780549B2 (ja) |
| AU (1) | AU2003271566A1 (ja) |
| CA (1) | CA2495060C (ja) |
| WO (1) | WO2004016614A2 (ja) |
Families Citing this family (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1388541A1 (en) | 2002-08-09 | 2004-02-11 | Centre National De La Recherche Scientifique (Cnrs) | Pyrrolopyrazines as kinase inhibitors |
| FR2876377B1 (fr) * | 2004-10-11 | 2007-03-16 | Univ Claude Bernard Lyon | Nouveaux derives de 9h-pyrido[2,3-b]indole, leur procede de preparation, ainsi que les compositions pharmaceutiques contenant de tels composes |
| FR2876582B1 (fr) * | 2004-10-15 | 2007-01-05 | Centre Nat Rech Scient Cnrse | Utilisation de derives de pyrrolo-pyrazines pour la fabrication de medicaments pour le traitement de la mucoviscidose et de maladies liees a un defaut d'adressage des proteines dans les cellules |
| FR2899107B1 (fr) * | 2006-03-30 | 2008-06-13 | Neurokin Entpr Unipersonnelle | Utilisation de la (s)-roscovitine pour la fabrication d'un medicament |
| DK2012822T3 (da) * | 2006-04-28 | 2010-05-25 | Univ Pennsylvania | Modificeret adenovirushexonprotein og anvendelser deraf |
| EP2057164A1 (en) * | 2006-08-07 | 2009-05-13 | Incyte Corporation | Triazolotriazines as kinase inhibitors |
| CL2007002617A1 (es) | 2006-09-11 | 2008-05-16 | Sanofi Aventis | Compuestos derivados de pirrolo[2,3-b]pirazin-6-ilo; composicion farmaceutica que comprende a dichos compuestos; y su uso para tratar inflamacion de las articulaciones, artritis reumatoide, tumores, linfoma de las celulas del manto. |
| WO2008063888A2 (en) | 2006-11-22 | 2008-05-29 | Plexxikon, Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| FR2918061B1 (fr) | 2007-06-28 | 2010-10-22 | Sanofi Aventis | Derives de 6-cycloamino-3-(pyridin-4-yl)imidazo°1,2-b!- pyridazine,leur preparation et leur application en therapeutique. |
| CN102016011B (zh) | 2008-03-04 | 2013-12-11 | 宾夕法尼亚大学托管会 | 猿猴腺病毒sadv-36、-42.1、-42.2和-44及其应用 |
| WO2009152087A1 (en) * | 2008-06-10 | 2009-12-17 | Plexxikon, Inc. | Bicyclic heteroaryl compounds and methods for kinase modulation, and indications therefor |
| SG173178A1 (en) | 2009-04-03 | 2011-09-29 | Hoffmann La Roche | Propane- i-sulfonic acid {3- [5- (4 -chloro-phenyl) -1h-pyrrolo [2, 3-b] pyridine-3-carbonyl] -2, 4-difluoro-pheny l } -amide compositions and uses thereof |
| JP2013510166A (ja) | 2009-11-06 | 2013-03-21 | プレキシコン インコーポレーテッド | キナーゼ調節のための化合物、方法およびその適用 |
| WO2012088266A2 (en) | 2010-12-22 | 2012-06-28 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of fgfr3 |
| RS58455B1 (sr) | 2011-02-07 | 2019-04-30 | Plexxikon Inc | Jedinjenja i postupci za modulaciju kinaze, i indikacije za njih |
| US20140303386A1 (en) * | 2011-08-25 | 2014-10-09 | Rutgers, The State University Of New Jersey | Compositions and methods for enhancing lipid production in microalgae via induction of cell cycle arrest |
| WO2013173702A2 (en) | 2012-05-18 | 2013-11-21 | The Trustees Of The University Of Pennsylvania | Subfamily e simian adenoviruses a1302, a1320, a1331 and a1337 and uses thereof |
| US9150570B2 (en) | 2012-05-31 | 2015-10-06 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| WO2014007951A2 (en) | 2012-06-13 | 2014-01-09 | Incyte Corporation | Substituted tricyclic compounds as fgfr inhibitors |
| WO2014026125A1 (en) | 2012-08-10 | 2014-02-13 | Incyte Corporation | Pyrazine derivatives as fgfr inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| DK2986610T5 (en) | 2013-04-19 | 2018-12-10 | Incyte Holdings Corp | BICYCLIC HETEROCYCLES AS FGFR INHIBITORS |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| MA41551A (fr) | 2015-02-20 | 2017-12-26 | Incyte Corp | Hétérocycles bicycliques utilisés en tant qu'inhibiteurs de fgfr4 |
| NZ773116A (en) | 2015-02-20 | 2024-05-31 | Incyte Holdings Corp | Bicyclic heterocycles as fgfr inhibitors |
| WO2016190847A1 (en) * | 2015-05-26 | 2016-12-01 | Calitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| AR111960A1 (es) | 2017-05-26 | 2019-09-04 | Incyte Corp | Formas cristalinas de un inhibidor de fgfr y procesos para su preparación |
| BR112020022373A2 (pt) | 2018-05-04 | 2021-02-02 | Incyte Corporation | sais de um inibidor de fgfr |
| SG11202010636VA (en) | 2018-05-04 | 2020-11-27 | Incyte Corp | Solid forms of an fgfr inhibitor and processes for preparing the same |
| JP2022508648A (ja) | 2018-10-05 | 2022-01-19 | アンナプルナ バイオ インコーポレイテッド | Apj受容体活性に関連する状態を処置するための化合物および組成物 |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| WO2021007269A1 (en) | 2019-07-09 | 2021-01-14 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| WO2021067374A1 (en) | 2019-10-01 | 2021-04-08 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| BR112022007163A2 (pt) | 2019-10-14 | 2022-08-23 | Incyte Corp | Heterociclos bicíclicos como inibidores de fgfr |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| JP2023505258A (ja) | 2019-12-04 | 2023-02-08 | インサイト・コーポレイション | Fgfr阻害剤としての三環式複素環 |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2022221170A1 (en) | 2021-04-12 | 2022-10-20 | Incyte Corporation | Combination therapy comprising an fgfr inhibitor and a nectin-4 targeting agent |
| EP4352059A1 (en) | 2021-06-09 | 2024-04-17 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001506980A (ja) * | 1996-11-19 | 2001-05-29 | アムジエン・インコーポレーテツド | アリールおよびヘテロアリール置換縮合ピロール抗炎症剤 |
| WO2001047922A2 (en) * | 1999-12-24 | 2001-07-05 | Aventis Pharma Limited | Azaindoles |
| JP2001520227A (ja) * | 1997-10-20 | 2001-10-30 | エフ.ホフマン−ラ ロシュ アーゲー | 二環式キナーゼ阻害剤 |
| WO2001096336A2 (en) * | 2000-06-14 | 2001-12-20 | Warner-Lambert Company | 6,5-fused bicyclic heterocycles |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5286860A (en) * | 1992-11-12 | 1994-02-15 | Neurogen Corporation | Certain aryl substituted pyrrolopyrazines; a new class of GABA brain receptor ligands |
| EP0766684A4 (en) * | 1994-06-09 | 1997-07-16 | Smithkline Beecham Corp | ENDOTHELINE RECEPTOR ANTAGONISTS |
| RS50087B (sr) * | 1998-06-19 | 2009-01-22 | Pfizer Products Inc., | Pirolo (2,3-d) pirimidin jedinjenja |
| PA8474101A1 (es) * | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | Compuestos de pirrolo [2,3-d] pirimidina |
| ES2253930T3 (es) * | 1998-09-18 | 2006-06-01 | ABBOTT GMBH & CO. KG | 4-aminopirrolopirimidinas como inhibidores de quinasa. |
| RU2331640C2 (ru) * | 1999-05-21 | 2008-08-20 | Бристол-Маерс Сквибб Ко. | Пирролтриазиновые ингибиторы киназ |
| PT1235830E (pt) * | 1999-12-10 | 2004-04-30 | Pfizer Prod Inc | Compostos de pirrolo¬2,3-d|pirimidina como inibidores das proteina cinases |
| AU782775B2 (en) | 2000-02-05 | 2005-08-25 | Vertex Pharmaceuticals Incorporated | Pyrazole compositions useful as inhibitors of ERK |
| SI1294724T1 (sl) * | 2000-06-26 | 2006-08-31 | Pfizer Prod Inc | Pirolo(2,3-d)primidinske spojine kot imunosupresivna sredstva |
| CN100537571C (zh) | 2002-04-19 | 2009-09-09 | 细胞基因组公司 | 咪唑并[1,2-а]吡嗪-8-基胺,其制备方法及使用方法 |
| EP1388541A1 (en) | 2002-08-09 | 2004-02-11 | Centre National De La Recherche Scientifique (Cnrs) | Pyrrolopyrazines as kinase inhibitors |
| KR20050033659A (ko) | 2002-09-04 | 2005-04-12 | 쉐링 코포레이션 | 사이클린 의존성 키나제 억제제로서의 피라졸로[1,5-a]피리미딘 화합물 |
| RU2005114010A (ru) | 2002-10-09 | 2006-01-20 | Сайос Инк. (Us) | Производные азаиндола в качестве ингибиторов киназы р38 |
| FR2876582B1 (fr) | 2004-10-15 | 2007-01-05 | Centre Nat Rech Scient Cnrse | Utilisation de derives de pyrrolo-pyrazines pour la fabrication de medicaments pour le traitement de la mucoviscidose et de maladies liees a un defaut d'adressage des proteines dans les cellules |
-
2002
- 2002-08-09 EP EP20020292019 patent/EP1388541A1/en not_active Withdrawn
-
2003
- 2003-08-08 CA CA2495060A patent/CA2495060C/en not_active Expired - Fee Related
- 2003-08-08 EP EP03753362A patent/EP1527077A2/en not_active Withdrawn
- 2003-08-08 WO PCT/EP2003/009515 patent/WO2004016614A2/en active Application Filing
- 2003-08-08 US US10/524,044 patent/US8106050B2/en not_active Expired - Fee Related
- 2003-08-08 JP JP2004528508A patent/JP4780549B2/ja not_active Expired - Fee Related
- 2003-08-08 AU AU2003271566A patent/AU2003271566A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001506980A (ja) * | 1996-11-19 | 2001-05-29 | アムジエン・インコーポレーテツド | アリールおよびヘテロアリール置換縮合ピロール抗炎症剤 |
| JP2001520227A (ja) * | 1997-10-20 | 2001-10-30 | エフ.ホフマン−ラ ロシュ アーゲー | 二環式キナーゼ阻害剤 |
| WO2001047922A2 (en) * | 1999-12-24 | 2001-07-05 | Aventis Pharma Limited | Azaindoles |
| WO2001096336A2 (en) * | 2000-06-14 | 2001-12-20 | Warner-Lambert Company | 6,5-fused bicyclic heterocycles |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2004016614A2 (en) | 2004-02-26 |
| CA2495060A1 (en) | 2004-02-26 |
| US20080161312A1 (en) | 2008-07-03 |
| US8106050B2 (en) | 2012-01-31 |
| JP2006502137A (ja) | 2006-01-19 |
| EP1388541A1 (en) | 2004-02-11 |
| EP1527077A2 (en) | 2005-05-04 |
| AU2003271566A1 (en) | 2004-03-03 |
| WO2004016614A3 (en) | 2004-05-06 |
| CA2495060C (en) | 2012-01-03 |
| AU2003271566A8 (en) | 2004-03-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4780549B2 (ja) | キナーゼ阻害活性を有するピロロピラジン誘導体およびその生物学的用途 | |
| JP5542196B2 (ja) | 1−複素環−1,5−ジヒドロ−ピラゾロ[3,4−d]ピリミジン−4−オン誘導体及びpde9aモジュレーターとしてのそれらの使用 | |
| Mettey et al. | Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects | |
| ES2234300T3 (es) | 3-(3-cloro-4-hidroxifenilamino)-4-(2-nitrofenil)-1h-pirrol-2,5-diona como inhibidor de glucogeno cinasa-3 (gsk-3)sintetasa. | |
| RU2753520C2 (ru) | Производные n-(замещенный фенил)-сульфонамида в качестве ингибиторов киназы | |
| US9328120B2 (en) | 6-cycloalkyl-pyrazolopyrimidinones for the treatment of CNS disorders | |
| EP1641805B1 (en) | Thiazolo-, oxazalo and imidazolo-quinazoline compounds capable of inhibiting protein kinases | |
| EP2675807B1 (en) | 6-cyclobutyl-1, 5-dihydro-pyrazolo [3, 4-d] pyrimidin-4-one derivatives and their use as pde9a inhibitors | |
| ES2879645T3 (es) | Inhibidor de PDE1 | |
| EA038635B1 (ru) | 2-замещенные соединения хиназолина, содержащие замещенную гетероциклическую группу, и способы их применения | |
| Horiuchi et al. | Discovery of novel thieno [2, 3-d] pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: Synthesis, biological evaluation and structure–activity relationships. Part 2 | |
| JP2005526765A (ja) | Cdk−阻害性2−ヘテロアリール−ピリミジン類、それらの生成及び医薬としての使用 | |
| KR20070009546A (ko) | 단백질 키나제 의존성 질환의 치료에 사용하기 위한피라졸로[1,5-a]피리미딘-7-일-아민 유도체 | |
| JP2009512693A (ja) | カゼインキナーゼii(ck2)モジュレーターとしてのピラゾロ−ピリミジン類 | |
| JP2004516297A (ja) | サイクリン依存性キナーゼ(cdk)及びグリコーゲンシンターゼキナーゼ−3(gsk−3)の阻害剤 | |
| JP2009520740A (ja) | スルホキシイミン置換ピリミジン、それらの調製及び医薬としての使用 | |
| FR2959510A1 (fr) | Derives de pyrido[3,2-d]pyrimidine, leurs procedes de preparation et leurs utilisations therapeutiques | |
| EP2935272B1 (en) | Pyrazole substituted imidazopyrazines as casein kinase 1 d/e inhibitors | |
| Dou et al. | Rational modification, synthesis and biological evaluation of 3, 4-dihydroquinoxalin-2 (1H)-one derivatives as potent and selective c-Jun N-terminal kinase 3 (JNK3) inhibitors | |
| EA024295B1 (ru) | N-[4-(1H-ПИРАЗОЛО[3,4-b]ПИРАЗИН-6-ИЛ)ФЕНИЛ]СУЛЬФОНАМИДЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ | |
| JP2007504159A (ja) | プロテインキナーゼ阻害剤としての化合物および組成物 | |
| JP2010522226A (ja) | 酵素pde7及び/又はpde4の二重阻害化合物、薬剤組成物および利用法 | |
| JP2019510822A (ja) | Cdk阻害剤としてのヘテロ環置換ピリジノピリミジノン誘導体およびその用途 | |
| EP3077383B1 (en) | Sulfoximine substituted quinazolines for pharmaceutical compositions | |
| Demange et al. | Synthesis and evaluation of new potent inhibitors of CK1 and CDK5, two kinases involved in Alzheimer’s disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060614 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060614 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100119 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100414 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100421 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100512 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100519 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100618 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100625 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100716 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20100907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110107 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110330 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20110412 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110531 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110627 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140715 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |