JP4635644B2 - シクロヘキサノンオキシムの製造方法 - Google Patents
シクロヘキサノンオキシムの製造方法 Download PDFInfo
- Publication number
- JP4635644B2 JP4635644B2 JP2005052876A JP2005052876A JP4635644B2 JP 4635644 B2 JP4635644 B2 JP 4635644B2 JP 2005052876 A JP2005052876 A JP 2005052876A JP 2005052876 A JP2005052876 A JP 2005052876A JP 4635644 B2 JP4635644 B2 JP 4635644B2
- Authority
- JP
- Japan
- Prior art keywords
- cyclohexanone
- water
- cyclohexanone oxime
- ammonia
- organic solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/04—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/04—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes
- C07C249/08—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes by reaction of hydroxylamines with carbonyl compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C249/00—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C249/04—Preparation of compounds containing nitrogen atoms doubly-bound to a carbon skeleton of oximes
- C07C249/14—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C251/00—Compounds containing nitrogen atoms doubly-bound to a carbon skeleton
- C07C251/32—Oximes
- C07C251/34—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C251/44—Oximes with oxygen atoms of oxyimino groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with the carbon atom of at least one of the oxyimino groups being part of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
(1)反応工程:チタノシリケート触媒の存在下に、シクロヘキサノン、過酸化水素及びアンモニアを反応させて、シクロヘキサノンオキシム、水、未反応のアンモニア及び未反応のシクロヘキサノンを含む反応液を得る工程、
(2)第一蒸留工程:工程(1)で得られた反応液を蒸留して、アンモニアを留出させ、シクロヘキサノンオキシム、水及びシクロヘキサノンを含む缶出液を得る工程、
(3)抽出工程:工程(2)で得られた缶出液を有機溶媒と混合した後、有機層と水層とに分離する工程、
(4)洗浄工程:工程(3)で得られた有機層を水と混合した後、有機層と水層とに分離する工程、
(5)第二蒸留工程:工程(4)で得られた有機層を蒸留して、有機溶媒及び水を留出させ、シクロヘキサノンオキシム及びシクロヘキサノンを含む缶出液を得る工程、
(6)第三蒸留工程:工程(5)で得られた缶出液を蒸留して、シクロヘキサノンを留出させ、シクロヘキサノンオキシムを含む缶出液を得る工程、
から構成され、工程(4)で使用される水及び工程(5)に付される工程(4)からの有機層の少なくとも一方に、ホウ素又はリンの酸化物、オキソ酸、オキソ酸塩、オキソ酸エステル及びオキソ酸アミドから選ばれる化合物を添加することを特徴とするシクロヘキサノンオキシムの製造方法を提供するものである。
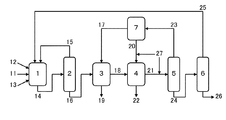
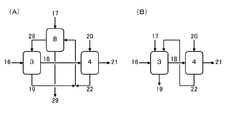
図1に示される連続式製造プロセスに、図2(A)に示される回収操作を適用して、以下の各工程及び熱安定性試験を実施した。
攪拌槽型の反応器1に、チタノシリケート触媒が分散した反応液を滞留させ、この中に、シクロヘキサノン11、60%過酸化水素水12、アンモニア13、及び15%含水t−ブチルアルコールを供給しながら、フィルターを介して反応液14を抜き出すことにより、シクロヘキサノン/過酸化水素/アンモニア/水/t−ブチルアルコール=1/1.1/1.8/4.29/4.0の供給モル比で、圧力0.25MPa(ゲージ圧)の条件下に、温度85℃で反応を行った。反応で副生する窒素ガスは系外に除去し、その際、同伴するアンモニアガスは水に吸収させて回収した。得られた反応液14中の成分濃度は、シクロヘキサノンオキシム20.43%、水22.13%、アンモニア2.32%、シクロヘキサノン0.10%、及びt−ブチルアルコール52.32%であった。
反応器1から抜き出した反応液14を上記アンモニア吸収水と共に、第一蒸留塔2に導入し、50kPa(ゲージ圧)にて温度83℃で蒸留を行い、留分15として、アンモニア及び含水t−ブチルアルコールを回収し、缶出液16として、シクロヘキサノンオキシム47.97%、水51.20%、及びシクロヘキサノン0.317%からなる混合物を得た。留分15は、反応器1に供給するアンモニアの一部及び15%含水t−ブチルアルコールとして使用した。
第一蒸留塔2からの缶出液16と、該缶出液16中に含まれるシクロヘキサノンオキシムと同重量のトルエン28とを、抽出器3に導入し、約72℃にて攪拌した後、トルエン層18と水層19に分離した。
抽出器3からのトルエン層18と、該トルエン層18中に含まれるシクロヘキサノンオキシムと同重量の水20とを、洗浄器4に導入し、約72℃にて攪拌した後、トルエン層21と水層22に分離した。
抽出器3からの水層19と洗浄器4からの水層22とを混合し、この混合水層とトルエン17を回収器8に導入し、約72℃にて攪拌した後、トルエン層28と水層29とに分離した。トルエン層28は、抽出器3に導入するトルエン28として使用した。
洗浄器4からのトルエン層21に、該トルエン層21中に含まれるシクロヘキサノンオキシムに対し3モルppmのオルトリン酸を少量の水に溶かして添加して、第二蒸留塔5に導入し、圧力30kPaの減圧下に温度107℃で蒸留を行い、留分23として、トルエン及び水の混合物を回収し、缶出液24として、シクロヘキサノン1.077%を含む純度98.10%の粗製シクロヘキサノンオキシムを得た。留分23を分液器7に導入し、約35℃にてトルエン層17と水層20とに分離した。トルエン層17は、回収器8に導入するトルエン17として使用し、水層20は、洗浄器4に導入する水20として使用した。
第二蒸留塔5からの缶出液24を第三蒸留塔6に導入し、圧力4kPaの減圧下に温度120℃で蒸留を行い、留分25として、シクロヘキサノンを回収し、缶出液26として、シクロヘキサノン0.04%を含む純度99.78%の精製シクロヘキサノンオキシムを得た。留分25は、反応器1に供給するシクロヘキサノンの一部として使用した。
缶出液26として得られた精製シクロヘキサノンオキシムを、圧力15torr(2kPa)の減圧下に温度120℃で、留出が見られなくなるまで減圧蒸留したところ、残留したタール分の量は、蒸留仕込量に対し0.01%であった(加熱処理前のタール量)。また、同シクロヘキサノンオキシムを、窒素気流下に200℃で5時間加熱処理した後、上記と同様に減圧蒸留したところ、残留したタール分の量は蒸留仕込量に対し0.12%であった(加熱処理後のタール量)。加熱処理によるタールの増加量は0.11%であった。
第二蒸留工程(5)において、第二蒸留塔5に導入される洗浄器4からのトルエン層にオルトリン酸を添加しなかった以外は、実施例1と同様の操作を行い、第三蒸留塔5の缶出液26として、シクロヘキサノン0.01%を含む純度99.23%の精製シクロヘキサノンオキシムを得た。得られたシクロヘキサノンオキシムについて、実施例1と同様に熱安定性試験を行ったところ、加熱処理前のタール量は0.02%であり、加熱処理後のタール量は1.80%であり、加熱処理によるタールの増加量は1.78%であった。
2……第一蒸留塔、
3……抽出器、
4……洗浄器、
5……第二蒸留塔、
6……第三蒸留塔、
7……分液器、
8……回収器、
11……シクロヘキサノン、
12……過酸化水素、
13……アンモニア、
17……有機溶媒、
20……水、
24……粗製シクロヘキサノンオキシム、
26……精製シクロヘキサノンオキシム。
27……ホウ素又はリンの化合物
Claims (6)
- 下記工程(1)〜(6)、
(1)反応工程:チタノシリケート触媒の存在下に、シクロヘキサノン、過酸化水素及びアンモニアを反応させて、シクロヘキサノンオキシム、水、未反応のアンモニア及び未反応のシクロヘキサノンを含む反応液を得る工程、
(2)第一蒸留工程:工程(1)で得られた反応液を蒸留して、アンモニアを留出させ、シクロヘキサノンオキシム、水及びシクロヘキサノンを含む缶出液を得る工程、
(3)抽出工程:工程(2)で得られた缶出液を有機溶媒と混合した後、有機層と水層とに分離する工程、
(4)洗浄工程:工程(3)で得られた有機層を水と混合した後、有機層と水層とに分離する工程、
(5)第二蒸留工程:工程(4)で得られた有機層を蒸留して、有機溶媒及び水を留出させ、シクロヘキサノンオキシム及びシクロヘキサノンを含む缶出液を得る工程、
(6)第三蒸留工程:工程(5)で得られた缶出液を蒸留して、シクロヘキサノンを留出させ、シクロヘキサノンオキシムを含む缶出液を得る工程、
から構成され、工程(4)で使用される水及び工程(5)に付される工程(4)からの有機層の少なくとも一方に、オルトリン酸、メタリン酸、ホスホン酸、ホスフィン酸、及びこれらの縮合酸から選ばれる化合物を添加することを特徴とするシクロヘキサノンオキシムの製造方法。 - 工程(3)で使用される有機溶媒が、炭化水素系溶媒、エーテル系溶媒及びエステル系溶媒から選ばれる請求項1に記載の方法。
- 工程(2)で留出させたアンモニアを工程(1)にリサイクルする請求項1又は2に記載の方法。
- 工程(5)で留出させた有機溶媒を工程(3)にリサイクルする請求項1〜3のいずれかに記載の方法。
- 工程(5)で留出させた水を工程(4)にリサイクルする請求項1〜4のいずれかに記載の方法。
- 工程(6)で留出させたシクロヘキサノンを工程(1)にリサイクルする請求項1〜5のいずれかに記載の方法。
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005052876A JP4635644B2 (ja) | 2005-02-28 | 2005-02-28 | シクロヘキサノンオキシムの製造方法 |
| KR1020060018212A KR20060095476A (ko) | 2005-02-28 | 2006-02-24 | 시클로헥사논 옥심의 제조 방법 |
| EP06003801A EP1700846B1 (en) | 2005-02-28 | 2006-02-24 | Process for producing cyclohexanone oxime |
| SG200601242A SG125242A1 (en) | 2005-02-28 | 2006-02-24 | Process for producing cyclohexanone oxime |
| CN2006100095505A CN1827592B (zh) | 2005-02-28 | 2006-02-24 | 制备环己酮肟的方法 |
| TW095106464A TWI363747B (en) | 2005-02-28 | 2006-02-24 | Process for producing cyclohexanone oxime |
| US11/360,631 US7449600B2 (en) | 2005-02-28 | 2006-02-24 | Process for producing cyclohexanone oxime |
| DE602006001531T DE602006001531D1 (de) | 2005-02-28 | 2006-02-24 | Verfahren zur Herstellung von Cyclohexanon-oxim |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005052876A JP4635644B2 (ja) | 2005-02-28 | 2005-02-28 | シクロヘキサノンオキシムの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006232774A JP2006232774A (ja) | 2006-09-07 |
| JP4635644B2 true JP4635644B2 (ja) | 2011-02-23 |
Family
ID=36591358
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005052876A Expired - Fee Related JP4635644B2 (ja) | 2005-02-28 | 2005-02-28 | シクロヘキサノンオキシムの製造方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7449600B2 (ja) |
| EP (1) | EP1700846B1 (ja) |
| JP (1) | JP4635644B2 (ja) |
| KR (1) | KR20060095476A (ja) |
| CN (1) | CN1827592B (ja) |
| DE (1) | DE602006001531D1 (ja) |
| SG (1) | SG125242A1 (ja) |
| TW (1) | TWI363747B (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5029162B2 (ja) * | 2007-06-18 | 2012-09-19 | 住友化学株式会社 | シクロヘキサノンの製造方法 |
| CN101781232B (zh) * | 2010-01-29 | 2012-09-05 | 河北瑞通美邦工程有限公司 | 一种环己酮肟制备工艺 |
| TWI480100B (zh) * | 2011-04-27 | 2015-04-11 | China Petrochemical Dev Corp Taipei Taiwan | Titanium-silicon molecular sieve and its preparation method and method for producing cyclohexanone oxime using the molecular sieve |
| CN103282344B (zh) * | 2011-07-08 | 2015-02-18 | 宁波欧迅化学新材料技术有限公司 | 一种酮肟化合物的制备方法和烃氧基胺盐酸盐的制备方法 |
| WO2013061752A1 (ja) * | 2011-10-28 | 2013-05-02 | 住友化学株式会社 | 廃水処理方法 |
| TW201350463A (zh) * | 2012-05-04 | 2013-12-16 | Dsm Ip Assets Bv | 純化由肟之合成部所獲得之有機產物溶液的方法 |
| CN103382164B (zh) * | 2012-05-04 | 2017-08-11 | Cap Iii 有限公司 | 一种纯化从环己酮肟合成区获得的有机物溶液的方法 |
| CN103193672B (zh) * | 2013-03-11 | 2014-09-17 | 浙江圣安化工有限公司 | 一种肟的蒸馏方法 |
| CN105439898B (zh) * | 2014-08-26 | 2017-10-13 | 湖北三宁化工股份有限公司 | 一种甲苯肟溶液洗涤装置及方法 |
| CN105837468B (zh) * | 2015-01-15 | 2018-09-28 | 湖北金湘宁化工科技有限公司 | 一种环己酮肟的制备方法 |
| CN110423206B (zh) * | 2019-07-17 | 2022-07-08 | 天津大学 | 从氨肟化反应产物中分离环己酮肟、环己酮与甲苯的方法 |
| CN113105358A (zh) * | 2021-04-15 | 2021-07-13 | 河南资环检测科技有限公司 | 一种臭氧水氧化环己酮制备环己酮肟的方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB977812A (en) * | 1961-11-17 | 1964-12-16 | Basf Ag | Separation of cycloalkanone oximes |
| GB1056124A (en) * | 1963-07-24 | 1967-01-25 | Ici Ltd | Preparation of oximes |
| JPS541295B1 (ja) * | 1967-04-06 | 1979-01-23 | ||
| IT1127311B (it) * | 1979-12-21 | 1986-05-21 | Anic Spa | Materiale sintetico,cristallino,poroso costituito da ossidi di silicio e titanio,metodo per la sua preparazione e suoi usi |
| IT1214622B (it) | 1985-07-10 | 1990-01-18 | Montedipe Spa | Processo catalitico per laproduzione di cicloesanonossima. |
| JPH0610181B2 (ja) * | 1986-11-14 | 1994-02-09 | モンテデイペ・ソチエタ・ペル・アツイオニ | オキシム製造のための接触方法 |
| IT1244680B (it) | 1991-01-23 | 1994-08-08 | Montedipe Srl | Processo a piu' stadi per l'ammossimazione in fase liquida dei composti carbonilici |
| IT1255745B (it) * | 1992-04-01 | 1995-11-15 | Enichem Anic Srl | Processo in due stadi per la produzione in fase liquida di ossime |
| ITMI20011361A1 (it) * | 2001-06-28 | 2002-12-28 | Enichem Spa | Metodo di purificazione della cicloesanossima |
| TWI341830B (en) * | 2003-11-28 | 2011-05-11 | Sumitomo Chemical Co | Stabilization method of cycloalkanone oxime |
-
2005
- 2005-02-28 JP JP2005052876A patent/JP4635644B2/ja not_active Expired - Fee Related
-
2006
- 2006-02-24 TW TW095106464A patent/TWI363747B/zh not_active IP Right Cessation
- 2006-02-24 CN CN2006100095505A patent/CN1827592B/zh not_active Expired - Fee Related
- 2006-02-24 KR KR1020060018212A patent/KR20060095476A/ko not_active Ceased
- 2006-02-24 US US11/360,631 patent/US7449600B2/en not_active Expired - Fee Related
- 2006-02-24 SG SG200601242A patent/SG125242A1/en unknown
- 2006-02-24 EP EP06003801A patent/EP1700846B1/en not_active Expired - Lifetime
- 2006-02-24 DE DE602006001531T patent/DE602006001531D1/de not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| KR20060095476A (ko) | 2006-08-31 |
| US7449600B2 (en) | 2008-11-11 |
| CN1827592A (zh) | 2006-09-06 |
| CN1827592B (zh) | 2011-06-15 |
| EP1700846A1 (en) | 2006-09-13 |
| US20060205939A1 (en) | 2006-09-14 |
| TW200639142A (en) | 2006-11-16 |
| TWI363747B (en) | 2012-05-11 |
| JP2006232774A (ja) | 2006-09-07 |
| DE602006001531D1 (de) | 2008-08-07 |
| EP1700846B1 (en) | 2008-06-25 |
| SG125242A1 (en) | 2006-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4635644B2 (ja) | シクロヘキサノンオキシムの製造方法 | |
| KR20160029021A (ko) | 2,3,6-트리메틸페놀의 산화에 의한 2,3,5-트리메틸 벤조퀴논의 제조 방법 | |
| US8183406B2 (en) | Process for recovering valued compounds from a stream derived from purification of methyl methacrylate | |
| WO2016148200A1 (ja) | ε-カプロラクタムの製造方法 | |
| CN1793114B (zh) | 环己酮肟的制备方法 | |
| US5074967A (en) | Separation of methoxyisopropylamine from methoxyisopropylamine-water azeotrope | |
| JP4595804B2 (ja) | シクロヘキサノンオキシムの製造方法 | |
| JP3803771B2 (ja) | エチルアミン類の製造方法 | |
| JP7359141B2 (ja) | ヘキサフルオロ-1,3-ブタジエンの製造方法 | |
| KR102218342B1 (ko) | Hpo 추출 구역으로부터 배출된 무기 공정 액체의 스팀 스트리핑 및 응축 열의 이용 | |
| JP6004884B2 (ja) | ε−カプロラクタムの製造方法 | |
| WO2013164371A1 (en) | A process for purifying organic product solution obtained from oxime synthesis section | |
| CN102648049B (zh) | 制备脱过氧化催化剂的方法 | |
| JP2003160561A (ja) | 2,2,6,6−テトラメチル−4−ピペリドンの製造法 | |
| JPH09100258A (ja) | エチルアミン類の製造方法 | |
| JPH05301858A (ja) | カプロラクタムの製法 | |
| JP2007210980A (ja) | 酢酸エチル/メチルエチルケトン混合系からの酢酸エチルの回収方法 | |
| WO1999008998A1 (en) | Process for producing formamide | |
| JP4576834B2 (ja) | シクロアルカノンオキシムの処理方法 | |
| JPS63139176A (ja) | 1,1′−パ−オキシジシクロヘキシルアミンの製造方法 | |
| JP2002068718A (ja) | フリーヒドロキシルアミン水溶液の高収率製造法 | |
| JPH07258239A (ja) | インデンオキサイドの精製方法 | |
| JPH11124359A (ja) | ホルムアミドの製造法 | |
| JP2015196658A (ja) | ヒドロキシルアミンを含有する有機溶媒の蒸留方法 | |
| JP2005272386A (ja) | 高純度カプロラクタムの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080131 |
|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080514 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090303 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100126 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100727 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100922 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20101026 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101108 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |