JP4564098B2 - 徐放性組成物およびその製造法 - Google Patents
徐放性組成物およびその製造法 Download PDFInfo
- Publication number
- JP4564098B2 JP4564098B2 JP2009524632A JP2009524632A JP4564098B2 JP 4564098 B2 JP4564098 B2 JP 4564098B2 JP 2009524632 A JP2009524632 A JP 2009524632A JP 2009524632 A JP2009524632 A JP 2009524632A JP 4564098 B2 JP4564098 B2 JP 4564098B2
- Authority
- JP
- Japan
- Prior art keywords
- sustained
- solution
- solvent
- release composition
- composition according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images








Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5089—Processes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
- A61K47/6925—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a microcapsule, nanocapsule, microbubble or nanobubble
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
- A61K9/5031—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/28—Antiandrogens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
- A61P5/32—Antioestrogens
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/095—Oxytocins; Vasopressins; Related peptides
Description
さらに、非特許文献1には、酢酸リュープロライドとポリ乳酸からなるマイクロスフィア型の徐放性製剤が記載され、該製剤の製法として、リュープロライドのメタノール溶液とポリ乳酸のジクロロメタン溶液を混合し、ポリビニルアルコールの水溶液中で分散させた後に有機溶媒を除去することによりマイクロスフィア型の徐放性製剤を製造する方法が記載されている。これは180〜240日にわたって薬物を放出する特性を有するが、初期バースト後の投与初期から1または2ヶ月における薬物放出が少なく、典型的な三相性の薬物放出を示すものである
また、本願発明は、上記の長期にわたる安定した薬物の放出により、薬物の血中濃度を長期間安定させる、徐放性製剤の提供をも目的とする。
さらに、本願発明は、製剤中における生理活性物質の含有量を高含量化することにより、徐放性製剤の単位容積あたりの生理活性物質含量を高めることができ、有効成分の単位用量あたりに必要な徐放性製剤全体の容積または重量を低減させた徐放性製剤を提供することを目的とする。
また、本願発明は、上記の薬物高含量化製剤により、投与時の疼痛や投与後の硬結など、単位容積の大きな製剤を投与することが原因と考えられる患者の身体的負担を軽減した徐放性製剤を提供することをも目的とする。
あわせて、本願発明は、長期にわたる安定した持続放出と薬物の高含量化の双方を同時に達成した上記製剤により、投与時の身体的負担と投与回数の減少による通院負担の軽減という相反する目的を同時に達成するものである。
本発明者らは、これらの知見に基づいて、さらに研究を重ねた結果、本発明を完成するに至った。
(1)乳酸重合体またはその塩からなるマイクロカプセル中に水溶性の生理活性ペプチドからなる生理活性物質が実質的に均一に分散した徐放性組成物であって、該生理活性物質をマイクロカプセル全体に対して15〜35(重量/重量)%含み、該乳酸重合体の重量平均分子量(Mw)が約11,000〜約27,000である徐放性組成物;
(2)乳酸重合体の重量平均分子量(Mw)が、
(i)約11,600〜約20,000、および
(ii)約19,000〜約27,000、
のいずれかから選択されるものである、上記(1)記載の徐放性組成物;
(3)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、徐放性組成物からのインビボにおける生理活性物質の放出が、約60日〜130日にわたって有効血中薬物濃度を維持できることを特徴とする徐放性組成物;
(4)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、徐放性組成物からのインビボにおける生理活性物質の放出が、約120日〜400日にわたって有効血中薬物濃度を維持できることを特徴とする徐放性組成物;
(5)生理活性物質がLH−RH誘導体である上記(1)記載の徐放性組成物;
(6)生理活性物質が式:
5−oxo−Pro−His−Trp−Ser−Tyr−Y−Leu−Arg−Pro−Z [式中、YはDLeu、DAla、DTrp、DSer(tBu)、D2NalまたはDHis(ImBzl)を示し、ZはNH−C2H5またはGly−NH2を示す。]で表されるペプチドまたはその塩である上記(1)記載の徐放性組成物;
(7)生理活性物質が式:
5−oxo−Pro−His−Trp−Ser−Tyr−DLeu−Leu−Arg−Pro−NH−C2H5で表されるペプチドまたはその酢酸塩である上記(1)記載の徐放性組成物;
(8)マイクロカプセル全体に対して、含まれる生理活性物質の含量が17〜26(重量/重量)%であることを特徴とする上記(1)記載の徐放性組成物;
(9)乳酸重合体またはその塩を揮発性の水非混和性の第一溶媒に溶解して第一溶液を調製し、
水溶性の生理活性ペプチドからなる生理活性物質を水混和性の第二溶媒に溶解して第二溶液を調製し、
得られた第一溶液と第二溶液を混合して乳酸重合体またはその塩および生理活性物質が均質に溶解した第三溶液を調製し、
得られた第三溶液を乳化剤の水溶液からなる第四溶液に分散してO/Wエマルションを調製し、
生成したマイクロカプセルから第一溶媒と第二溶媒を除去することにより得られるものである上記(1)記載の徐放性組成物;
(10)上記第一溶液を調製する際における乳酸重合体またはその塩を溶解する溶媒として、水混和性の第三溶媒を第一溶媒にさらに加えた混合溶媒を用いることを特徴とする上記(9)記載の徐放性組成物;
(11)マイクロカプセルから第一溶媒と第二溶媒を除去する工程において、乳化工程の管理温度を約15〜約35℃に調節することを特徴とする上記(9)記載の徐放性組成物;
(12)乳化工程の温度管理が、O/Wエマルションの温度を約15〜35℃に調節することにより行われること特徴とする上記(11)記載の徐放性組成物;
(13)O/Wエマルションを調製する際の第三溶液と第四溶液の各温度が約15〜約35℃である上記(9)記載の徐放性組成物;
(14)マイクロカプセルから第一溶媒と第二溶媒を除去する工程において、水中乾燥法により行うことを特徴とする上記(9)記載の徐放性組成物;
(15)第一溶媒がジクロロメタンである上記(9)記載の徐放性組成物;
(16)第二溶媒および/または第三溶媒が低級アルコールである上記(9)記載の徐放性組成物;
(17)低級アルコールがメタノール、エタノールまたはプロパノールである上記(16)記載の徐放性組成物;
(18)第三溶液中の水非混和性溶媒と水混和性溶媒の容量比率が35:65〜55:45であることを特徴とする上記(9)記載の徐放性組成物;
(19)第一溶液中のポリマー濃度が約33〜45重量%であることを特徴とする上記(9)記載の徐放性組成物;
(20)第三溶液を製造する際における生理活性物質のローディング量が17〜50重量%であることを特徴とする上記(9)記載の徐放性組成物;
(21)マイクロカプセル全体に対して含まれる生理活性物質の含量が17〜26(重量/重量)%であることを特徴とする上記(9)記載の徐放性組成物;
(22)第三溶液を製造する際における生理活性物質のローディング量が19〜38重量%であることを特徴とする上記(21)記載の徐放性組成物;
(23)第三溶液を製造する際における生理活性物質のローディング量が20〜23重量%であることを特徴とする上記(21)記載の徐放性組成物;
(24)徐放性組成物がさらに脂肪酸を含有することを特徴とする上記(1)記載の徐放性組成物;
(25)脂肪酸がステアリン酸、安息香酸、ヒドロキシナフトエ酸およびパモ酸から選択される少なくとも1種である上記(24)記載の徐放性組成物;
(26)マイクロカプセル全体に対する脂肪酸の比率が約0.01〜約50重量%であることを特徴とする上記(24)記載の徐放性組成物;
(27)添加する脂肪酸の量が、水溶性の生理活性ペプチドまたはその塩の1モルに対して0.1〜10モルであることを特徴とする上記(24)記載の徐放性組成物;
(28)分散媒に易分散性であることを特徴とする上記(1)記載の徐放性組成物;
(29)分散媒に分散後24時間以上安定であることを特徴とする上記(28)記載の徐放性組成物;
(30)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、重量平均分子量(Mw)の数平均分子量(Mn)に対する比が1.9よりも大きいことを特徴とする徐放性組成物;
(31)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、重量平均分子量(Mw)の数平均分子量(Mn)に対する比が1.5よりも大きいことを特徴とする徐放性組成物;
(32)乳酸重合体がポリ乳酸またはポリラクチドであることを特徴とする上記(1)記載の徐放性組成物;
(33)乳酸重合体がポリDL−乳酸またはポリDL−ラクチドであることを特徴とする上記(1)記載の徐放性組成物;
(34)乳酸重合体が乳酸−グリコール酸重合体であることを特徴とする上記(1)記載の徐放性組成物;
(35)乳酸−グリコール酸重合体の、乳酸/グリコール酸の組成比が60/40〜99.9/0.1であることを特徴とする上記(34)記載の徐放性組成物;
(36)乳酸重合体が、分子量5000以下の重合体含有量が約5.0重量%以下のものである上記(1)記載の徐放性組成物;
(37)乳酸重合体が、分子量3000以下の重合体含有量が約1.5重量%以下のものである上記(1)記載の徐放性組成物;
(38)乳酸重合体が、分子量1000以下の重合体含有量が約0.1重量%以下のものである上記(1)記載の徐放性組成物;
(39)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、乳酸重合体の重量平均分子量(Mw)が12,000〜19,000である徐放性組成物;
(40)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、乳酸重合体の重量平均分子量(Mw)が13,000〜18,000である徐放性組成物;
(41)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、乳酸重合体の重量平均分子量(Mw)が19,500〜26,500である徐放性組成物;
(42)生理活性物質をマイクロカプセル全体に対して15〜35重量%含むマイクロカプセル型の徐放性組成物の製造方法であって、
(i)乳酸重合体またはその塩を揮発性の水非混和性の第一溶媒に溶解して第一溶液を調製し、
(ii)水溶性の生理活性ペプチドからなる生理活性物質を水混和性の第二溶媒に溶解して第二溶液を調製し、
(iii)得られた第一溶液と第二溶液を混合して乳酸重合体またはその塩および生理活性物質が均質に溶解した第三溶液を調製し、
(iv)得られた第三溶液を界面活性剤の水溶液からなる第四溶液に分散してO/Wエマルションを調製し、
(v)約15〜約35℃の管理温度において、マイクロカプセルから上記第一溶媒と第二溶媒を水中乾燥法により除去する、
工程を含むことを特徴とする製造方法;
(43)乳酸重合体の重量平均分子量(Mw)が、約11,600〜約20,000である、上記(42)記載の製造方法;
(44)乳酸重合体の重量平均分子量(Mw)が、約19,000〜約27,000である、上記(42)記載の製造方法;
(45)上記(i)の工程において、乳酸重合体またはその塩を溶解する溶媒として、水混和性の第三溶媒を第一溶媒にさらに加えた混合溶媒を用いることを特徴とする上記(42)記載の製造方法;
(46)O/Wエマルションを調製する際の第三溶液と第四溶液の各温度を約15〜約35℃に調節することを特徴とする上記(42)記載の製造方法;
(47)さらに脂肪酸またはその塩を第一溶液および/または第二溶液、あるいは第三溶液に添加することを特徴とする上記(42)記載の徐放性組成物の製造方法;
(48)脂肪酸またはその塩を第二溶液に溶解させることを特徴とする上記(42)記載の徐放性組成物の製造方法;
(49)第三溶液を製造する際における生理活性物質のローディング量が17〜50重量%であることを特徴とする上記(42)記載の徐放性組成物の製造方法;
(50)マイクロカプセル全体に対して含まれる生理活性物質の含量が17〜26(重量/重量)%であることを特徴とする上記(42)記載の徐放性組成物の製造方法;
(51)第三溶液を製造する際における生理活性物質のローディング量が19〜38重量%であることを特徴とする上記(50)記載の徐放性組成物の製造方法;
(52)第三溶液を製造する際における生理活性物質のローディング量が20〜23重量%であることを特徴とする上記(50)記載の徐放性組成物の製造方法;
(53)徐放性組成物からの生理活性物質のインビボ放出により、約60日〜130日にわたって有効薬物血中濃度を維持できることを特徴とする上記(42)記載の徐放性組成物の製造方法;
(54)徐放性組成物からの生理活性物質のインビボ放出により、約120日〜400日にわたって有効薬物血中濃度を維持できることを特徴とする上記(42)記載の徐放性組成物の製造方法;
(55)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、投与後24時間以内の活性成分の最高血中濃度の投与後24時間から1ヶ月以内の活性成分の平均血中濃度に対する比が、2〜50であることを特徴とする徐放性組成物;
(56)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、投与後24時間以内の活性成分の最高血中濃度の投与後1ヶ月から3ヶ月以内の活性成分の平均血中濃度に対する比が、20〜350であることを特徴とする徐放性組成物;
(57)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、血中濃度から計算される、投与後24時間以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの3%〜30%であることを特徴とする徐放性組成物;
(58)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、血中濃度から計算される、投与後24時間から1ヶ月以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの40%〜80%であり、優れた徐放特性を有することを特徴とする徐放性組成物;
(59)乳酸重合体の重量平均分子量(Mw)が(i)約11,600〜約20,000である上記(2)記載の徐放性組成物において、投与後1ヶ月から3ヶ月以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの10%〜35%であり、優れた徐放特性を有することを特徴とする徐放性組成物;
(60)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、投与後24時間以内の活性成分の最高血中濃度の投与後24時間から1ヶ月以内の活性成分の平均血中濃度に対する比が、10〜90であることを特徴とする徐放性組成物;
(61)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、投与後24時間以内の活性成分の最高血中濃度の投与後1ヶ月から6ヶ月以内の活性成分の平均血中濃度に対する比が、20〜500であることを特徴とする徐放性組成物;
(62)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、血中濃度から計算される、投与後24時間以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの1%〜20%であることを特徴とする徐放性組成物;
(63)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、血中濃度から計算される、投与後24時間から1ヶ月以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの10%〜50%であり、優れた徐放特性を有することを特徴とする徐放性組成物;
(64)乳酸重合体の重量平均分子量(Mw)が(ii)約19,000〜約27,000である上記(2)記載の徐放性組成物において、投与後1ヶ月から6ヶ月以内の活性成分の血中濃度−時間曲線下面積(AUC)が全体のAUCの40%〜90%であり、優れた徐放特性を有することを特徴とする徐放性組成物;
(65)上記(1)記載の徐放性組成物を含有してなる医薬組成物;
(66)上記(1)記載の徐放性組成物を含有してなる前立腺癌、前立腺肥大症、子宮内膜症、子宮筋腫、子宮線維腫、思春期早発症、月経困難症もしくは乳癌の予防、治療剤または避妊剤;
(67)上記(1)記載の徐放性組成物を含有してなる閉経前乳癌術後再発予防剤;
(68)哺乳動物に対して、上記(1)記載の徐放性組成物の有効量を投与することを特徴とする前立腺癌、前立腺肥大症、子宮内膜症、子宮筋腫、子宮線維腫、思春期早発症、月経困難症もしくは乳癌の予防、治療方法または避妊方法;
(69)哺乳動物に対して、上記(1)記載の徐放性組成物の有効量を投与することを特徴とする閉経前乳癌術後再発予防方法;
(70)徐放性組成物が、上記(42)〜(54)記載のいずれかの方法で製造されることを特徴とする上記(68)または(69)記載の方法;
(71)前立腺癌、前立腺肥大症、子宮内膜症、子宮筋腫、子宮線維腫、思春期早発症、月経困難症もしくは乳癌の予防、治療剤または避妊剤を製造するための上記(1)記載の徐放性組成物の使用;および
(72)閉経前乳癌術後再発予防剤を製造するための上記(1)記載の徐放性組成物の使用を提供する。
該生理活性ペプチドとしては、例えば、黄体形成ホルモン放出ホルモン(LH−RH)、インスリン、ソマトスタチン、成長ホルモン、成長ホルモン放出ホルモン(GH−RH)、プロラクチン、エリスロポイエチン、副腎皮質ホルモン、メラノサイト刺激ホルモン、甲状腺ホルモン放出ホルモン、甲状腺刺激ホルモン、黄体形成ホルモン、卵胞刺激ホルモン、バソプレシン、オキシトシン、カルシトニン、ガストリン、セクレチン、パンクレオザイミン、コレシストキニン、アンジオテンシン、ヒト胎盤ラクトーゲン、ヒト絨毛性ゴナドトロピン、エンケファリン、エンドルフィン、キョウトルフィン、タフトシン、サイモポイエチン、サイモシン、サイモチムリン、胸腺液性因子、血中胸腺因子、腫瘍壊死因子、コロニー誘導因子、モチリン、デイノルフィン、ボンベシン、ニューロテンシン、セルレイン、ブラジキニン、心房性ナトリウム排泄増加因子、神経成長因子、細胞増殖因子、神経栄養因子、エンドセリン拮抗作用を有するペプチド類などおよびその誘導体、さらにはこれらのフラグメントまたはフラグメントの誘導体などが挙げられる。
本発明で用いられる生理活性物質はそれ自身であっても、薬理学的に許容される塩であってもよい。
このような塩としては、該生理活性物質がアミノ基等の塩基性基を有する場合、無機酸(無機の遊離酸とも称する)(例、炭酸、重炭酸、塩酸、硫酸、硝酸、ホウ酸等)、有機酸(有機の遊離酸とも称する)(例、コハク酸、酢酸、プロピオン酸、トリフルオロ酢酸等)などとの塩が挙げられる。
生理活性物質がカルボキシル基等の酸性基を有する場合、塩としては、無機塩基(無機の遊離塩基とも称する)(例、ナトリウム、カリウム等のアルカリ金属、カルシウム、マグネシウム等のアルカリ土類金属など)や有機塩基(有機の遊離塩基とも称する)(例、トリエチルアミン等の有機アミン類、アルギニン等の塩基性アミノ酸類等)などとの塩が挙げられる。また、生理活性ペプチドは金属錯体化合物(例、銅錯体、亜鉛錯体等)を形成していてもよい。
LH−RH誘導体またはその塩の具体例としては、例えば、トリートメント ウイズ GnRH アナログ:コントラバーシス アンド パースペクテイブ(Treatment with GnRH analogs: Controversies and perspectives)[パルテノン バブリッシング グループ(株)(The Parthenon Publishing Group Ltd.)発行1996年]、特表平3−503165号公報、特開平3−101695号、同7−97334号および同8−259460号公報などに記載されているペプチド類が挙げられる。
X-D2Nal-D4ClPhe-D3Pal-Ser-A-B-Leu-C-Pro-DAlaNH2
〔式中、XはN(4H2-furoyl)GlyまたはNAcを、AはNMeTyr、Tyr、Aph(Atz)、NMeAph(Atz)から選ばれる残基を、BはDLys(Nic)、DCit、DLys(AzaglyNic)、DLys(AzaglyFur)、DhArg(Et2)、DAph(Atz)およびDhCi から選ばれる残基を、CはLys(Nisp)、ArgまたはhArg(Et2)をそれぞれ示す〕で表わされる生理活性ペプチドまたはその塩などが用いられる。
LH−RHアゴニストとしては、例えば、一般式〔II〕
5-oxo-Pro-His-Trp-Ser-Tyr-Y-Leu-Arg-Pro-Z
〔式中、YはDLeu、DAla、DTrp、DSer(tBu)、D2NalおよびDHis(ImBzl)から選ばれる残基を、ZはNH-C2H5またはGly-NH2をそれぞれ示す〕で表わされる生理活性ペプチドまたはその塩などが用いられる。特に、YがDLeuで、ZがNH-C2H5であるペプチド(即ち、5-oxo-Pro-His-Trp-Ser-Tyr-DLeu-Leu-Arg-Pro-NH-C2H5で表されるペプチドA;リュープロレリン)またはその塩(例、酢酸塩)が好適である。
これらのペプチドは、前記文献あるいは公報記載の方法あるいはこれに準じる方法で製造することができる。
略 号 : 名称
N(4H2-furoyl)Gly: N-テトラヒドロフロイルグリシン残基
NAc : N-アセチル基
D2Nal : D-3-(2-ナフチル)アラニン残基
D4ClPhe : D-3-(4-クロロ)フェニルアラニン残基
D3Pal : D-3-(3-ピリジル)アラニン残基
NMeTyr : N-メチルチロシン残基
Aph(Atz) : N-[5'-(3'-アミノ-1'H-1',2',4'-トリアゾリル)]フェニル
アラニン残基
NMeAph(Atz) : N-メチル-[5'-(3'-アミノ-1'H-1',2',4'-トリアゾリル)]
フェニルアラニン残基
DLys(Nic) : D-(e-N-ニコチノイル)リシン残基
Dcit : D-シトルリン残基
DLys(AzaglyNic) : D-(アザグリシルニコチノイル)リシン残基
DLys(AzaglyFur) : D-(アザグリシルフラニル)リシン残基
DhArg(Et2) : D-(N,N'-ジエチル)ホモアルギニン残基
DAph(Atz) : D-N-[5'-(3'-アミノ-1'H-1',2',4'-トリアゾリル)]
フェニルアラニン残基
DhCi : D-ホモシトルリン残基
Lys(Nisp) : (e-N-イソプロピル)リシン残基
hArg(Et2) : (N,N'-ジエチル)ホモアルギニン残基
その他アミノ酸に関し、略号で表示する場合、IUPAC-IUBコミッション・オン・バイオケミカル・ノーメンクレーチュアー(Commission on Biochemical Nomenclature) (ヨーロピアン・ジャーナル・オブ・バイオケミストリー(European Journal of Biochemistry)第138巻、9〜37頁(1984年))による略号または該当分野における慣用略号に基づくものとし、また、アミノ酸に関して光学異性体がありうる場合は、特に明示しなければL体を示すものとする。
さらに、本発明の徐放性製剤で使用される乳酸重合体の重量平均分子量は、通常約11,000〜約27,000であり、約11,600〜約20,000または約19,000〜約27,000が好ましい。特に、製剤からのインビボにおける生理活性物質の放出が約60日〜130日にわたって有効薬物血中濃度を維持できる徐放性製剤においては、約11,600〜約20,000が好ましく、さらに好ましくは約12,000〜約19,000であり、より好ましくは約13,000〜約18,000である。一方、製剤からのインビボにおける生理活性物質の放出が約120日〜400日にわたって有効薬物血中濃度を維持できる徐放性製剤においては、約19,000〜約27,000が好ましく、さらに好ましくは約19,500〜約26,500であり、より好ましくは約20,000〜約26,000である。
乳酸重合体の重量平均分子量(Mw)が約11,600〜約20,000である場合には、重量平均分子量(Mw)の数平均分子量(Mn)に対する比が1.9よりも大きいものが好ましく、乳酸重合体の重量平均分子量(Mw)が約19,000〜約27,000である場合には、重量平均分子量(Mw)の数平均分子量(Mn)に対する比が1.5よりも大きいものが好ましい。ここで、重量平均分子量は(Mw)および数平均分子量(Mn)はゲルパーミエーションクロマトグラフィー(GPC)により測定することができる。
また、製剤からのインビボにおける生理活性物質の放出が約120日〜400日にわたって有効濃度の薬物血中濃度を維持できる徐放性製剤において使用される乳酸重合体は、例えば、通常分子量5000以下の重合体含有量が約5重量%以下、好ましくは分子量5000以下の重合体含有量が約5重量%以下であり且つ分子量3000以下の重合体含有量が約1.5重量%以下、更に好ましくは分子量5000以下の重合体含有量が約5重量%以下、分子量3000以下の重合体含有量が約1.5重量%以下であり且つ分子量1000以下の重合体含有量が約0.1重量%以下のものである。
公知の重合方法としては、例えば、乳酸及び要すればグリコール酸とを縮合重合させる方法、例えばラクチドを、要すればグリコリドと共に、例えばジエチル亜鉛、トリエチルアルミニウム、オクチル酸スズ等のルイス酸又は金属塩等の触媒を用いて開環重合させる方法、前記方法に更にカルボキシル基が保護されたヒドロキシカルボン酸誘導体を存在させてラクチドを開環重合させる方法(例えば国際公開WO00/35990等)、その他ラクチドに加熱下で触媒を添加して開環重合させる方法(例えばJ. Med. Chem, 16, 897(1973)等)、例えばラクチドとグリコリドとを共重合させる方法等が挙げられる。
重合形態としては、ラクチド等を溶融させて重合反応に付すバルク重合、ラクチド等を適当な溶媒に溶解して重合反応に付す溶液重合が挙げられるが、中でも溶液重合によって得られる重合体を本発明の乳酸重合体の原料として使用することが工業生産上好ましい。
溶液重合においてラクチドを溶解する溶媒としては、例えばベンゼン,トルエン,キシレン等の芳香族炭化水素類、デカリン、ジメチルホルムアミド等が挙げられる。
高分子量の乳酸重合体を溶解する溶媒としては、乳酸重合体の10重量倍以下の量で該重合体を溶解し得るものであればよく、具体的には、例えばクロロホルム,ジクロロメタン等のハロゲン化炭化水素、例えばトルエン,o−キシレン,m−キシレン,p−キシレン等の芳香族炭化水素、例えばテトラヒドロフラン等の環状エーテル、アセトン、N,N−ジメチルホルムアミド等が挙げられる。尚、高分子量の乳酸重合体の重合時に、高分子量の乳酸重合体の加水分解で使用できる溶媒を用いた場合には、重合した高分子量の乳酸重合体を単離せず、重合および加水分解の操作を連続して行うことができる。
添加する水の量は、高分子量乳酸重合体に対して通常0.001〜1倍重量、好ましくは0.01〜0.1倍重量である。
必要に応じて添加する酸としては、例えば塩酸,硫酸,硝酸等の無機酸、例えば乳酸,酢酸,トリフルオロ酢酸等の有機酸等が挙げられ、好ましくは乳酸が挙げられる。
添加する酸の量は、高分子量乳酸重合体に対して通常0〜10倍重量、好ましくは0.1〜1倍重量である
加水分解反応温度は、通常0〜150℃、好ましくは20〜80℃である。
加水分解反応時間は、高分子量の乳酸重合体の重量平均分子量および反応温度によっても異なり、通常10分〜100時間、好ましくは1〜20時間である。
加水分解処理の終了時期は、加水分解生成物の重量平均分子量に基づいて判断する。即ち、加水分解処理中に適宜サンプリングを行い、サンプル中の加水分解生成物の重量平均分子量をゲルパーミエーションクロマトグラフィー(GPC)により測定し、当該分子量が目標とする数値範囲になっていることが確認できたら加水分解処理を停止させる。
加水分解生成物含有溶液の好ましい態様としては、例えばクロロホルム,ジクロロメタン等のハロゲン化炭化水素、例えばトルエン,o−キシレン,m−キシレン,p−キシレン等の芳香族炭化水素、例えばテトラヒドロフラン等の環状エーテル、アセトン、N,N−ジメチルホルムアミド等の高分子量乳酸重合体を溶解する溶媒に、重量平均分子量15000〜50000、好ましくは15000〜30000、より好ましくは17000〜26000、特に好ましくは17500〜25500の乳酸重合体が約10〜50wt%溶解している溶液が挙げられる。本発明の徐放性製剤にヒドロキシナフトエ酸が含まれない場合は、重量平均分子量15000〜50000、好ましくは15000〜40000の乳酸重合体が約10〜50wt%溶解している溶液等が挙げられる。
加水分解生成物含有溶液中に含有される目的の乳酸重合体を析出させ得る溶媒としては、例えばメタノール,エタノール等のアルコール類、例えばイソプロピルエーテル等の鎖状エーテル類、例えばヘキサン等の脂肪族炭化水素、水等が挙げられる。
この様な各溶媒の種類と使用量の組み合わせの好ましい具体例としては、例えば溶質の1〜5倍重量のジクロロメタンを溶媒として用いられている加水分解生成物含有溶液に、溶解度を低下させる溶媒としてイソプロピルエーテルを、該ジクロロメタンに対して2〜10倍重量使用する態様等が挙げられる。
目的の乳酸重合体溶質を析出させ得る溶媒を加水分解生成物含有溶液に接触させる際の、溶媒の温度は、通常−20〜60℃、好ましくは0〜40℃であり、加水分解生成物含有溶液の温度は通常0〜40℃、好ましくは10〜30℃である。
溶媒と加水分解生成物含有溶液とを接触させる方法としては、加水分解生成物含有溶液を溶媒中に一度に加える方法、加水分解生成物含有溶液を溶媒中に滴下する方法、溶媒を加水分解生成物含有溶液中に一度に加える方法、或いは溶媒を加水分解生成物含有溶液中に滴下する方法等が挙げられる。
上記のようにして得られた本発明の乳酸重合体は、末端カルボキシル基量が徐放性製剤用基材として好ましい範囲にあるため、徐放性製剤用基材として好ましいものである。
添加する脂肪酸の量としては、脂肪酸の種類、水溶性の生理活性ペプチドの種類およびその添加量、持続性放出の期間などによって異なるが、水溶性の生理活性ペプチドまたはその塩の1モルに対して、好ましくは、0.1〜10モル、0.2〜5モル、さらに好ましくは0.25〜2モル、特に好ましくは0.5〜1.5モルである。
また、マイクロカプセル全体に対する脂肪酸の重量比としては、約0.01〜約50重量%であり、好ましくは約0.1〜約25重量%、さらに好ましくは約2〜10重量%である。
本発明の組成物における生理活性物質の重量比は、生理活性物質の種類、所望の薬理効果および効果の持続期間などによって異なるが、生理活性物質またはその塩と乳酸重合体またはその塩を含有する徐放性組成物において、その和に対して、生理活性ペプチドまたはその塩が、約0.001〜約50重量%、好ましくは約0.02〜約40重量%、より好ましくは約0.1〜約30重量%、最も好ましくは約14〜約24重量%であり、非ペプチド性生理活性物質またはその塩の場合、約0.01〜約80重量%、好ましくは約0.1〜約50重量%である。
以下の製造工程中、必要に応じて、薬物保持剤(例えば、ゼラチン、サリチル酸など)を自体公知の方法により添加してもよい。
本方法においては、まず本発明の乳酸重合体(以下、本発明の生体内分解性ポリマーと記載する場合がある。)またはその塩を揮発性で水非混和性の第一溶媒に溶解して第一溶液を調製する。上記の第一溶媒として使用する溶媒は、沸点が100℃以下であることが好ましい。
該第一溶媒としては、例えば、ハロゲン化炭化水素(例、ジクロロメタン、クロロホルム、ジクロロエタン、トリクロロエタン、四塩化炭素等)、エーテル類(例、エチルエーテル、イソプロピルエーテル等)、脂肪酸エステル(例、酢酸エチル、酢酸ブチル等)、芳香族炭化水素(例、ベンゼン、トルエン、キシレン等)が用いられる。なかでもハロゲン化炭化水素が好ましく、特にジクロロメタンが好適である。また、これらは適宜の割合で混合して用いてもよい。
該第二溶媒としては、例えば、低級アルコール(例えば、メタノール、エタノール、プロパノールなど)、アセトニトリル、アセトン、テトラヒドロフランなどが用いられる。なかでも低級アルコールが好ましく、特にメタノールまたはエタノールが好適である。また、これらは適宜の割合で混合して用いてもよい。
この際、生理活性物質の添加量は、生理活性物質をマイクロカプセル全体に対して15〜35(重量/重量)%含むようにする(マイクロカプセル中の生理活性物質の含量)。そのため、生理活性物質などの薬物のローディング量としては、約17〜約50重量%、好ましくは約18〜約43重量、より好ましくは約19〜約38重量%、さらに好ましくは約19〜約25重量%、最も好ましくは約20〜約23重量%である。ここで、ローディング量とは、マイクロカプセルを構成する各成分の、製造時における添加量の合計に対し、生理活性物質の添加量の割合を計算したものである。一方、生理活性物質などの薬物のトラップ率としては、約75重量%以上、好ましくは80重量%以上、より好ましくは82重量%以上、さらに好ましくは約85%以上、最も好ましくは約89%以上である。ここで、トラップ率とは、生理活性物質の添加量に対してマイクロカプセル中に取り込まれる薬物の割合を計算したものである。
上記の水相中に含まれる乳化剤は、一般に安定なO/Wエマルションを形成できるものであればいずれでもよい。具体的には、例えば、アニオン性界面活性剤(オレイン酸ナトリウム、ステアリン酸ナトリウム、ラウリル硫酸ナトリウムなど)、非イオン性界面活性剤(ポリオキシエチレンソルビタン脂肪酸エステル〔ツイーン(Tween)80、ツイーン(Tween)60、アトラスパウダー社〕、ポリオキシエチレンヒマシ油誘導体〔HCO-60、HCO-50、日光ケミカルズ〕など)、ポリビニルピロリドン、ポリビニルアルコール、カルボキシメチルセルロース、レシチン、ゼラチン、ヒアルロン酸が用いられる。これらの中の1種類か、いくつかを組み合わせて使用してもよい。使用の際の濃度は、好ましくは約0.01〜10重量%の範囲で、さらに好ましくは約0.05〜約5重量%の範囲で用いられる。
該浸透圧調節剤としては、例えば、多価アルコール類、一価アルコール類、単糖類、二糖類、オリゴ糖およびアミノ酸類またはそれらの誘導体などがあげられる。
上記の多価アルコール類としては、例えば、グリセリン等の三価アルコール類、アラビトール,キシリトール,アドニトール等の五価アルコール類、マンニトール,ソルビトール,ズルシトール等の六価アルコール類が用いられる。なかでも、六価アルコール類が好ましく、特にマンニトールが好適である。
上記の一価アルコール類としては、例えば、メタノール、エタノール、イソプロピルアルコールなどがあげられ、このうちエタノールが好ましい。
上記の単糖類としては、例えば、アラビノース,キシロース,リボース,2ーデオキシリボース等の五炭糖類、ブドウ糖,果糖,ガラクトース,マンノース,ソルボース,ラムノース,フコース等の六炭糖類が用いられ、このうち六炭糖類が好ましい。
上記のオリゴ糖としては、例えば、マルトトリオース,ラフィノース糖等の三糖類、スタキオース等の四糖類が用いられ、このうち三糖類が好ましい。
上記のアミノ酸類としては、L−体のものであればいずれも用いることができ、例えば、グリシン、ロイシン、アルギニンなどがあげられる。このうちL−アルギニンが好ましい。
これらの浸透圧調節剤は単独で使用しても、混合して使用してもよい。
これらの浸透圧調節剤は、外水相の浸透圧が生理食塩水の浸透圧の約1/50〜約5倍、好ましくは約1/25〜約3倍となる濃度で用いられる。浸透圧調節剤としてマンニトールを用いた場合、0.5%〜1.5%の濃度が好ましい。
本発明の徐放性製剤においては、患者への投与後1ヶ月以降におけるメンテナンス部の血中薬物濃度を理想化し、有効成分である水溶性の生理活性ペプチドを安定的に持続放出させるため、第一溶媒と第二溶媒を除去する際における乳化工程の管理温度を調節することができる。
第一溶媒と第二溶媒を除去する際における乳化工程の管理温度としては、例えば、約5〜約50℃に調節することができる。さらに、約15〜約35℃に調節することが望ましく、特に約15〜約30℃に調節することが望ましい。この場合における温度管理の方法としては、乳化工程の全工程において管理温度に設定された環境に置く方法のほか、上記のO/Wエマルションを上記温度に調節する方法、上記温度にそれぞれ調整した第三溶液と第四溶液を混合してエマルションを調製する方法がある。
このようにして得られたマイクロカプセルは遠心分離または濾過して分取した後、マイクロカプセルの表面に付着している遊離の生理活性物質、乳化剤などを蒸留水で数回繰り返し洗浄し、再び蒸留水などに分散して凍結乾燥する。
マンニトール等の凝集防止剤の添加量は、マイクロカプセル全体に対して、通常0〜約24重量%である。
また、凍結乾燥後、必要であれば、減圧下マイクロカプセル同士が融着しない条件下で加温してマイクロカプセル中の水分および有機溶媒の除去を行ってもよい。好ましくは、毎分10〜20℃の昇温速度の条件下で示差走査熱量計で求めた生体内分解性ポリマーの中間点ガラス転移温度付近あるいは若干高い温度で加温する。より好ましくは生体内分解性ポリマーの中間点ガラス転移温度付近あるいはこれより約30℃高い温度範囲内で加温する。とりわけ,生体内分解性ポリマーとして乳酸-グリコール酸重合体を用いる場合には好ましくはその中間点ガラス転移温度付近から中間点ガラス転移温度より10℃高い温度範囲,さらに好ましくは、中間点ガラス転移温度付近から中間点ガラス転移温度より5℃高い温度範囲で加温する。
加温時間はマイクロカプセルの量などによって異なるものの、一般的にはマイクロカプセル自体が所定の温度に達した後、約12時間〜約168時間、好ましくは約24時間〜約120時間、特に好ましくは約48時間〜約96時間である。
加温方法は、マイクロカプセルの集合が均一に加温できる方法であれば特に限定されない。
該加温乾燥方法としては、例えば、恒温槽、流動槽、移動槽またはキルン中で加温乾燥する方法、マイクロ波で加温乾燥する方法などが用いられる。なかでも恒温槽中で加温乾燥する方法が好ましい。
本方法においては、まず本発明の乳酸重合体またはその塩を揮発性で水非混和性の第一溶媒に溶解して第一溶液を調製する。上記の第一溶媒として使用する溶媒は、沸点が100℃以下であることが好ましい。
該第一溶媒としては、例えば、ハロゲン化炭化水素(例、ジクロロメタン、クロロホルム、ジクロロエタン、トリクロロエタン、四塩化炭素等)、エーテル類(例、エチルエーテル、イソプロピルエーテル等)、脂肪酸エステル(例、酢酸エチル、酢酸ブチル等)、芳香族炭化水素(例、ベンゼン、トルエン、キシレン等)などが用いられる。なかでもハロゲン化炭化水素が好ましく、特にジクロロメタンが好適である。また、これらは適宜の割合で混合して用いてもよい。
該第二溶媒としては、例えば、低級アルコール(例えば、メタノール、エタノール、プロパノールなど)、アセトニトリル、アセトン、テトラヒドロフランなどが用いられる。なかでも低級アルコールが好ましく、特にメタノールまたはエタノールが好適である。また、これらは適宜の割合で混合して用いてもよい。
この際、生理活性物質の添加量は、生理活性物質をマイクロカプセル全体に対して15〜35(重量/重量)%含むようにする(マイクロカプセル中の生理活性物質の含量)。そのため、生理活性物質などの薬物のローディング量としては、約17〜約50重量%、好ましくは約18〜約43重量、より好ましくは約19〜約38重量%、さらに好ましくは約19〜約25重量%、最も好ましくは約20〜約22重量%である。ここで、ローディング量とは、マイクロカプセルを構成する各成分の、製造時における添加量の合計に対し、生理活性物質の添加量の割合を計算したものである。一方、生理活性物質などの薬物のトラップ率としては、約75重量%以上、好ましくは80重量%以上、より好ましくは82重量%以上、さらに好ましくは約85%以上、最も好ましくは約89%以上である。ここで、トラップ率とは、生理活性物質の添加量に対してマイクロカプセル中に取り込まれる薬物の割合を計算したものである。
上記の水相中に含まれる乳化剤は、上記の脂肪酸を含まない徐放性製剤で例示した乳化剤と同様のものを用いることができる。
また、上記の水相中には浸透圧調節剤を加えてもよい。該浸透圧調節剤としては、水溶液とした場合に浸透圧を示すものであればよく、上記の脂肪酸を含まない徐放性製剤で例示した浸透圧調節剤と同様のものを用いることができる。
第一溶媒と第二溶媒を除去する際における乳化工程の管理温度としては、例えば、約5〜約50℃に調節することができる。さらに、約15〜約35℃に調節することが望ましい。この場合における温度管理の方法としては、乳化工程の全工程において管理された環境に置く方法のほか、上記のO/Wエマルションを上記温度に調節する方法、上記温度にそれぞれ調整した第三溶液と第四溶液を混合してエマルションを構成する方法がある。
このようにして得られたマイクロカプセルは遠心分離または濾過して分取した後、マイクロカプセルの表面に付着している遊離の生理活性物質、乳化剤などを蒸留水で数回繰り返し洗浄し、再び蒸留水などに分散して凍結乾燥する。
マンニトール等の凝集防止剤の添加量は、マイクロカプセル全体に対して、通常0〜約24重量%である。
また、凍結乾燥後、必要であれば、減圧下マイクロカプセル同士が融着しない条件下で加温してマイクロカプセル中の水分および有機溶媒の除去を行ってもよい。好ましくは、毎分10〜20℃の昇温速度の条件下で示差走査熱量計で求めた生体内分解性ポリマーの中間点ガラス転移温度付近あるいは若干高い温度で加温する。より好ましくは生体内分解性ポリマーの中間点ガラス転移温度付近あるいはこれより約30℃高い温度範囲内で加温する。とりわけ,生体内分解性ポリマーとして乳酸-グリコール酸重合体を用いる場合には好ましくはその中間点ガラス転移温度付近から中間点ガラス転移温度より10℃高い温度範囲,さらに好ましくは、中間点ガラス転移温度付近から中間点ガラス転移温度より5℃高い温度範囲で加温する。
加温時間はマイクロカプセルの量などによって異なるものの、一般的にはマイクロカプセル自体が所定の温度に達した後、約12時間〜約168時間、好ましくは約24時間〜約120時間、特に好ましくは約48時間〜約96時間である。
加温方法は、マイクロカプセルの集合が均一に加温できる方法であれば特に限定されない。
該加温乾燥方法としては、例えば、恒温槽、流動槽、移動槽またはキルン中で加温乾燥する方法、マイクロ波で加温乾燥する方法などが用いられる。なかでも恒温槽中で加温乾燥する方法が好ましい。
本発明の徐放性組成物は、そのまままたはこれらを原料物質として種々の剤形に製剤化し、筋肉内、皮下、臓器などへの注射剤または埋め込み剤、鼻腔、直腸、子宮などへの経粘膜剤、経口剤(例、カプセル剤(例、硬カプセル剤、軟カプセル剤等)、顆粒剤、散剤等の固形製剤、シロップ剤、乳剤、懸濁剤等の液剤等)などとして投与することができる。
例えば、本発明の徐放性組成物を注射剤とするには、これらを分散剤(例、ツイーン(Tween)80,HCO−60等の界面活性剤、ヒアルロン酸ナトリウム,カルボキシメチルセルロース,アルギン酸ナトリウム等の多糖類など)、保存剤(例、メチルパラベン、プロピルパラベンなど)、等張化剤(例、塩化ナトリウム,マンニトール,ソルビトール,ブドウ糖,プロリンなど)等と共に水性懸濁剤とするか、ゴマ油、コーン油などの植物油と共に分散して油性懸濁剤として実際に使用できる徐放性注射剤とすることができる。
本発明の徐放性組成物を無菌製剤にするには、製造全工程を無菌にする方法、ガンマ線で滅菌する方法、防腐剤を添加する方法等が挙げられるが、特に限定されない。
本発明の徐放性組成物は、低毒性であるので、哺乳動物(例、ヒト、牛、豚、犬、ネコ、マウス、ラット、ウサギ等)に対して安全な医薬などとして用いることができる。
本発明の徐放性組成物の投与量は、主薬である生理活性物質の種類と含量、剤形、生理活性物質放出の持続時間、対象疾病、対象動物などによって種々異なるが、生理活性物質の有効量であればよい。主薬である生理活性物質の1回当たりの投与量としては、例えば、徐放性製剤が6カ月製剤である場合、好ましくは、成人1人当たり約0.01mg〜10mg/kg体重の範囲,さらに好ましくは約0.05mg〜5mg/kg体重の範囲から適宜選ぶことができる。
1回当たりの徐放性組成物の投与量は、成人1人当たり好ましくは、約0.05mg〜50mg/kg体重の範囲、さらに好ましくは約0.1mg〜30mg/kg体重の範囲から適宜選ぶことができる。
投与回数は、数週間に1回、1か月に1回、または数か月(例、3ヵ月、4ヵ月、6ヵ月など)に1回等、主薬である生理活性物質の種類と含量、剤形、生理活性物質放出の持続時間、対象疾病、対象動物などによって適宜選ぶことができる。
すなわち、本願発明の徐放性製剤を投与するとき、例えばラットなどの試験動物に適用した場合、投与後24時間以内の活性成分の最高血中濃度と投与後24時間から1ヶ月以内の活性成分の平均血中濃度の比は2〜90であり、投与後24時間以内の活性成分の最高血中濃度と投与後1ヶ月から該製剤が予定する持続放出期間内の活性成分の平均血中濃度の比は20〜500である。また、血中濃度から計算される投与後24時間以内の活性成分の血中濃度−時間曲線下面積(以下、AUCという。)は全体のAUCの1%〜30%であり、同じく血中濃度から計算される投与後24時間から1ヶ月以内の活性成分のAUCは全体のAUCの10%〜80%であり、投与後1ヶ月から該製剤が予定する持続放出期間内の活性成分のAUCが全体のAUCの10%〜90%である。
特に、重量平均分子量(Mw)が約11,600〜約20,000である乳酸重合体を使用した本願発明の徐放性製剤を患者に投与した場合、徐放性組成物からのインビボにおける生理活性物質の放出を、約60日〜130日にわたって有効濃度の薬物血中濃度で維持できる。この場合、例えばラットなどの試験動物に適用したとき、投与後24時間以内の活性成分の最高血中濃度と投与後24時間から1ヶ月以内の活性成分の平均血中濃度の比は2〜50であり、投与後24時間以内の活性成分の最高血中濃度と投与後1ヶ月から3ヶ月以内の活性成分の平均血中濃度の比は20〜350である。また、血中濃度から計算される投与後24時間以内の活性成分のAUCは全体のAUCの3%〜30%であり、同じく血中濃度から計算される投与後24時間から1ヶ月以内の活性成分のAUCは全体のAUCの40%〜80%であり、投与後1ヶ月から3ヶ月以内の活性成分のAUCが全体のAUCの10%〜35%である。
また、重量平均分子量(Mw)が約19,000〜約27,000である乳酸重合体を使用した本願発明の徐放性製剤を患者に投与した場合、徐放性組成物からのインビボにおける生理活性物質の放出を、約120日〜400日にわたって有効濃度の薬物血中濃度で維持できる。この場合、例えばラットなどの試験動物に適用したとき、投与後24時間以内の活性成分の最高血中濃度と投与後24時間から1ヶ月以内の活性成分の平均血中濃度の比は10〜90であり、投与後24時間以内の活性成分の最高血中濃度と投与後1ヶ月から6ヶ月以内の活性成分の平均血中濃度の比は20〜500である。また、血中濃度から計算される投与後24時間以内の活性成分のAUCは全体のAUCの1%〜20%であり、同じく血中濃度から計算される投与後24時間から1ヶ月以内の活性成分のAUCは全体のAUCの10%〜50%であり、投与後1ヶ月から6ヶ月以内の活性成分のAUCは全体のAUCの40%〜90%である。
したがって、哺乳動物に本願治療・予防剤を有効量投与することにより、ホルモン依存性疾患、特に性ホルモン依存性癌(例、前立腺癌、子宮癌、乳癌、下垂体腫瘍など)、前立腺肥大症、子宮内膜症、子宮筋腫、思春期早発症、月経困難症、無月経症、月経前症候群、多房性卵巣症候群等の性ホルモン依存性の疾患の予防・治療または避妊をすることができ、さらに閉経前乳癌術後再発予防することができる。
以下の実施例・参考例における重量平均分子量及び各重合体含有量は、単分散ポリスチレンを基準物質としてゲルパーミエーションクロマトグラフィー(GPC)で測定したポリスチレン換算の重量平均分子量及びそれらから算出した各重合体含有量である。また、測定は全て高速GPC装置(東ソー(株)製;HLC−8120GPC)で行い、カラムはSuperH4000×2及びSuperH2000(何れも東ソー(株)製)を使用し、移動相としてテトラヒドロフランを流速0.6mL/minで使用した。尚、検出方法は示差屈折率によるものである。
血中薬物濃度の測定方法としては以下の方法がある。酢酸リュープロレリンの場合、例えば、試料血清中の酢酸リュープロレリンと125I標識酢酸リュープロレリンをウサギ抗酢酸リュープロレリン血清と競合反応させる。生成した結合物に第二抗体としてヤギ抗ウサギγグロブリン血清溶液及び、正常ウサギ血清溶液を加えて反応させ遠心分離後、沈殿物の放射活性を測定する。同時に作製した検量線から試料血清中の酢酸リュープロレリン濃度を求める。
なお、図面において、「酢酸リュープロレリン」を「TAP−144」という場合がある。
DL−乳酸重合体(重量平均分子量14,300)3.83gをジクロロメタン6.4gで溶解した溶液を、酢酸リュープロレリン凍乾末0.96gにメタノール4.56g加え約50℃で加温溶解し室温(25℃)とした溶液に加えて分散混合して油相(O相)を調製した。このときの薬物のローディング量は20%である。次いでこのO相を約15℃に冷却後、予め約15℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液0.8リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(日立CR5DL)を用いてマイクロカプセルを沈降させて捕集した(CR5DL;日立;回転数約2,500rpm)。捕集されたマイクロカプセルは蒸留水に再分散し、再度同様に、遠心分離操作によりマイクロカプセルを沈降させて捕集後、少量の水で再分散しマンニトール0.507gとともにナス型フラスコへ回収凍結後、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してリュープロレリン含有のマイクロカプセルとマンニトールの混合粉末(以下「マイクロカプセル末」という。)を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.3%であり、収率は約63%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.0%であることが分かった。ここでいう「マイクロカプセル中の酢酸リュープロレリン含量」とは、各原料(酢酸リュープロレリン、乳酸重合体及びマンニトール)の仕込み重量の合計と収率を乗じて計算される値(以下、収量)に「マイクロカプセル末中の酢酸リュープロレリン含量」を乗じたものを、該収量からマンニトールの量を減じた値で除して計算される割合のことを示し、以下の計算式で示される。
[マイクロカプセル中の酢酸リュープロレリン含量(%)]=[[各原料の仕込み重量の合計(g)]×[収率(%)]×[マイクロカプセル末中の酢酸リュープロレリン含量(%)]]/[[各原料の仕込み重量の合計(g)]×[収率(%)]−[マンニトールの量(g)]]
ここで[各原料の仕込み重量の合計(g)]×[収率(%)]=[収量(g)] とする。
また、この数値は生理活性物質である酢酸リュープロレリンのマイクロカプセル全体に対する含量に相当する(以下同じ)。
油相(O相)調製後の温度及び0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液の温度を約20℃とし、これ以外については、実施例1と同様の方法でマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.1%であり、収率は約64%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は17.8%であることが分かった。
油相(O相)調製後の温度及び0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液の温度を約25℃とし、これ以外については、実施例1と同様の方法でマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は14.3%であり、収率は約64%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は16.8%であることが分かった。
油相(O相)調製後の温度及び0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液の温度を約30℃とし、これ以外については、実施例1と同様の方法でマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は13.4%であり、収率は約67%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は15.6%であることが分かった。
DL−乳酸重合体(重量平均分子量14,100)119.5gをジクロロメタン200.0gで溶解した溶液を約30℃に温調したものを、酢酸リュープロレリン30.0gにメタノール142.5g加え約40℃で加温溶解し室温(25℃)とした溶液に加えて分散混合して油相(O相)を調製した。このときの薬物のローディング量は20%である。次いでこのO相を約15℃に冷却後、予め約15℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600ml/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール21.1gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は約14%であり、収率は約55%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.1%であることが分かった。
DL−乳酸重合体(重量平均分子量14,100)119.5gをジクロロメタン200gで溶解した溶液を約30℃に温調したものを、酢酸リュープロレリン30.0gにメタノール142.5g加え約40℃で加温溶解し室温(25℃)とした溶液に加えて分散混合して油相(O相)を調製した。このときの薬物のローディング量は20%である。次いでこのO相を約20℃に冷却後、予め約20℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600ml/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、95μmの標準篩を用いて篩過した後、マンニトール17.2gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は16.0%であり、回収率は約76%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.5%であることが分かった。
酢酸リュープロレリン0.87gに蒸留水1gを加え加温溶解した。これにDL−乳酸重合体(重量平均分子量13,900)7.65gをジクロロメタン12.8gに溶解した溶液を加え、手で軽く分散させた後ポリトロン(キネマティカ社製)で約30秒間1次乳化しW/Oエマルションを作製した(回転数約1,000rpm)。このときの薬物のローディング量は10%である。次いでこのW/Oエマルションを約15℃に冷却後、予め約15℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1.6リットル中に注入し、ホモミキサー(特殊機化製)を用いて2次乳化しW/O/W乳化物とした(タービン回転数約7,000rpm)。このW/O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(日立 CR5DL)を用いてマイクロカプセルを沈降させて捕集した(回転数約2,500rpm)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、マンニトール0.9gを添加し凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は7.7%であり、収率は約62%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は9.1%であることが分かった。
DL−乳酸重合体(重量平均分子量13,900)3.83gをジクロロメタン6.4gで溶解した溶液を、酢酸リュープロレリン凍結乾燥末1.64gにメタノール7.79gを加え約50℃で加温溶解し室温(25℃)とした溶液に加えて分散混合して油相(O相)を調製した。このときの薬物のローディング量は30%である。次いでこのO相を、予め約15℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液0.8リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(日立CR5DL)を用いてマイクロカプセルを沈降させて捕集した(回転数約2,500rpm)。捕集されたマイクロカプセルは蒸留水に再分散し、再度同様に、遠心分離操作によりマイクロカプセルを沈降させて捕集後、少量の水で再分散しマンニトール0.578gとともにナス型フラスコへ回収凍結後、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は16.8%であり、収率は約74%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.3%であることが分かった。
DL−乳酸重合体(重量平均分子量14,300)3.83gをジクロロメタン6.4gで溶解した溶液を、酢酸リュープロレリン凍結乾燥末2.06gにメタノール9.80gを加え約50℃で加温溶解し室温(25℃)とした溶液に加えて分散混合して油相(O相)を調製した。このときの薬物のローディング量は35%である。次いでこのO相を、予め約15℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液0.8リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(日立CR5DL)を用いてマイクロカプセルを沈降させて捕集した(回転数約2,500rpm)。捕集されたマイクロカプセルは蒸留水に再分散し、再度同様に、遠心分離操作によりマイクロカプセルを沈降させて捕集後、少量の水で再分散しマンニトール0.623gとともにナス型フラスコへ回収凍結後、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.0%であり、収率は約73%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は17.3%であることが分かった。
実施例1で調製したマイクロカプセル末29mg、比較例1で調製したマイクロカプセル末34mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして4.5mgの投与量)して、血清中の酢酸リュープロレリン濃度を測定した。投与後24時間以内と13週までの血中濃度推移を図1に示す。また、投与後24時間以内の最高血中濃度(Cmax)及び血中濃度−時間曲線下面積(AUC)と、投与後24時間から1ヶ月まで(オンセット部)のAUCを計算した結果を表1に示す。図1及び表1から分かるように、比較例1に対して実施例1は投与後24時間以内のCmax及びAUCが低くなり、オンセット部の血中濃度及びAUCが高くなった。すなわち、O/W法で製することにより、投与後24時間以内の過剰な薬物放出を抑制し、オンセット部の血中濃度推移を大幅に改善することができた。
実施例1、比較例2、及び比較例3で調製したマイクロカプセル末について、それぞれにおける酢酸リュープロレリンのトラップ率を計算した。計算した結果を表2に示す。ここでいう「酢酸リュープロレリンのトラップ率」とは、「マイクロカプセル中の酢酸リュープロレリン含量」を「酢酸リュープロレリンのローディング量」で除して計算される割合のことを示す。表2から明らかなように、該ローディング量が30%以上の比較例ではトラップ率が大きく減少していた。
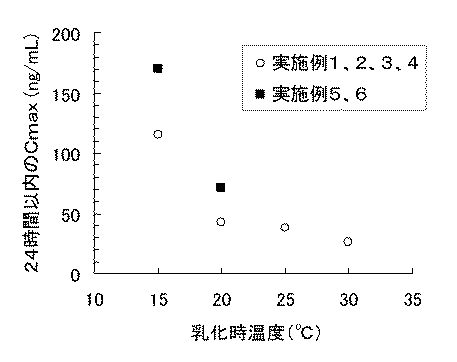
実施例1で調製したマイクロカプセル末29mg、実施例2で調製したマイクロカプセル末30mg、実施例3で調製したマイクロカプセル末32mg、及び実施例4で調製したマイクロカプセル末34mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして4.5mgの投与量)して、血清中の酢酸リュープロレリン濃度を測定した。投与後24時間以内と13週までの血中濃度推移を図2に示す。また、乳化温度と投与後24時間以内のCmaxの関係を調べた結果を図3に、乳化温度と投与後24時間以内のAUCの関係を調べた結果を図4に、及び乳化温度とメンテナンス部(投与後4週以降)のAUCの関係を調べた結果を図5に示す。図2、図3及び図4から分かるように、投与後24時間以内のCmax及びAUCは乳化温度に依存して低下した。すなわち、乳化温度を上げることにより、投与後初期の過剰な薬物の放出を抑制することができた。さらに図2及び図5から分かるように、メンテナンス部の血中濃度レベル及びAUCは乳化温度に依存して増大した。すなわち、乳化温度を上げることにより、メンテナンス部の血中濃度推移を改善することができた。
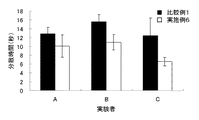
実施例5で調製したマイクロカプセル末32mg、実施例6で調製したマイクロカプセル末28mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして4.5mgの投与量)して、血清中の酢酸リュープロレリン濃度を測定した。投与後13週までの血中濃度推移を図6に示す。また、乳化温度と投与後24時間以内のCmaxの関係を調べた結果を図3に、乳化温度と投与後24時間以内のAUCの関係を調べた結果を図4に、及び乳化温度とメンテナンス部(投与後4週以降)のAUCの関係を調べた結果を図5に示す。図3、図4及び図6から分かるように、投与後24時間以内のCmax及びAUCは乳化温度に依存して低下した。すなわち、乳化温度を上げることにより、投与後初期の過剰な薬物放出をする。さらに図5及び図6から分かるように、メンテナンス部の血中濃度レベル及びAUCは乳化温度に依存して増大した。すなわち、乳化温度を上げることにより、メンテナンス部の血中濃度推移を改善することがでる。
実施例6及び比較例1それぞれについて、マイクロカプセル末(酢酸リュープロレリンとして45mg)と分散媒(容量として1mL)とを混合し、手で軽く振盪攪拌して均一に分散させた。混合を始めてから均一に分散するまでの時間をそれぞれ計測した。結果を図7に示す。実施例6のものは、比較例1のものよりも短時間で分散した。
酢酸リュープロレリン2.4gにメタノール11.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,700)9.6gをジクロロメタン16.8gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は20%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液2リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール1.27gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.3%であり、収率は約65%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は17.9%であることが分かった。ここでいう「マイクロカプセル中の酢酸リュープロレリン含量」とは、各原料(酢酸リュープロレリン、乳酸重合体及びマンニトール)の仕込み重量の合計と収率を乗じて計算される値(以下、収量)に「マイクロカプセル末中の酢酸リュープロレリン含量」を乗じたものを、該収量からマンニトールの量を減じた値で除して計算される割合のことを示し、生理活性物質である酢酸リュープロレリンのマイクロカプセル全体に対する含量に相当する(以下同じ)。
酢酸リュープロレリン1.2gにメタノール5.7gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量26,100)4.8gをジクロロメタン8.4gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は19%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は14.7%であり、収率は約54%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は17.9%であることが分かった。
酢酸リュープロレリン1.35gにメタノール6.41gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,700)4.65gをジクロロメタン8.14gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は16.8%であり、収率は約55%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は20.3%であることが分かった。
酢酸リュープロレリン1.14g及びステアリン酸0.269gにメタノール5.7gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,700)4.53gをジクロロメタン7.93gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は19%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.4%であり、収率は約57%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.5%であることが分かった。ここでいう「マイクロカプセル中の酢酸リュープロレリン含量」とは、各原料(酢酸リュープロレリン、乳酸重合体、ステアリン酸、及びマンニトール)の仕込み重量の合計と収率を乗じて計算される値(以下、収量)に「マイクロカプセル末中の酢酸リュープロレリン含量」を乗じたものを、該収量からマンニトールの量を減じた値で除して計算される割合のことを示し、生理活性物質である酢酸リュープロレリンのマイクロカプセル全体に対する含量に相当する(以下同じ)。
酢酸リュープロレリン1.14g及びステアリン酸0.269gにメタノール5.7gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量26,100)4.53gをジクロロメタン7.93gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は19%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は14.7%であり、収率は約56%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は17.8%であることが分かった。
酢酸リュープロレリン1.29g及びステアリン酸0.3025gにメタノール6.413gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,700)4.347gをジクロロメタン7.608gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は21.7%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は17.1%であり、収率は約58%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は20.5%であることが分かった。
酢酸リュープロレリン1.29g及びステアリン酸0.3025gにメタノール6.413gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量26,100)4.347gをジクロロメタン7.608gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は21.7%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.2%であり、収率は約59%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.2%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸0.079g(酢酸リュープロレリンの0.25倍モル)にメタノール6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.57gをジクロロメタン8.0gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は18.1%であり、収率は約68%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は21.1%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸0.157g(酢酸リュープロレリンの0.5倍モル)にメタノール6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.5gをジクロロメタン7.9gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は17.5%であり、収率は約56%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は21.1%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸0.315g(酢酸リュープロレリンの1倍モル)にメタノール6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.34gをジクロロメタン7.6gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は16.7%であり、収率は約52%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は20.5%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸0.471g(酢酸リュープロレリンの1.5倍モル)にメタノールの6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.19gをジクロロメタン7.34gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は17.1%であり、収率は約73%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.7%であることが分かった。
酢酸リュープロレリン1.23g及びステアリン酸0.287g(酢酸リュープロレリンの1倍モル)にメタノール5.8gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.49gをジクロロメタン7.9gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は20.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.8%であり、収率は約66%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.5%であることが分かった。
酢酸リュープロレリン1.29g及びステアリン酸0.300g(酢酸リュープロレリンの1倍モル)にメタノール6.1gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.42gをジクロロメタン7.7gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は21.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は15.9%であり、収率は約56%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.2%であることが分かった。
酢酸リュープロレリン1.41g及びステアリン酸0.328g(酢酸リュープロレリンの1倍モル)にメタノール6.7gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.27gをジクロロメタン7.5gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は23.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は17.0%であり、収率は約71%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.6%であることが分かった。
酢酸リュープロレリン1.47g及びステアリン酸0.342g(酢酸リュープロレリンの1倍モル)にメタノール7.0gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)4.20gをジクロロメタン7.4gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は24.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は16.3%であり、収率は約77%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.6%であることが分かった。
酢酸リュープロレリン33.75g及びステアリン酸7.56g(酢酸リュープロレリンの1倍モル)にメタノール160.33gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)108.68gをジクロロメタン190.2gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール17.2gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は18.2%であり、収率は約80%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は20.9%であることが分かった。
酢酸リュープロレリン30.0g及びステアリン酸6.725g(酢酸リュープロレリンの1倍モル)にメタノール142.5gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)113.28gをジクロロメタン198.23gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は20.0%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール17.2gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は16.6%であり、収率は約80%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.0%であることが分かった。
酢酸リュープロレリン22.68g及びステアリン酸5.08g(酢酸リュープロレリンの1倍モル)にメタノール107.7gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量26,100)80.24gをジクロロメタン140.4gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は21.0%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液18リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール13.3gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は16.5%であり、収率は約73%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は19.4%であることが分かった。
酢酸リュープロレリン31.5gにメタノール130.08gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量26,300)111.44g及びステアリン酸7.06g(酢酸リュープロレリンの1倍モル)をジクロロメタン195.0g及びメタノール19.50gの混液で溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は21.0%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール17.2gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は17.5%であり、収率は約78%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は20.2%であることが分かった。
酢酸リュープロレリン33.75gにメタノール141.31gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量22,100)108.68g及びステアリン酸7.56g(酢酸リュープロレリンの1倍モル)をジクロロメタン190.2g及びメタノール19.02gの混液で溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール17.2gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は18.4%であり、収率は約77%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は21.2%であることが分かった。
酢酸リュープロレリン30gにメタノール122.68gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量22,100)113.28g及びステアリン酸6.725g(酢酸リュープロレリンの1倍モル)をジクロロメタン198.2g及びメタノール19.82gの混液で溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は20%である。該O相を約30℃に温調後、予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液25リットル中に注入し、HOMOMIC LINE FLOW(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm、循環ポンプ回転数約2000rpm)。このO/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、次いで遠心機(H−600S,国産遠心器製)を用いて連続的にマイクロカプセルを沈降させて捕集した(回転数約2,000rpm、流量約600mL/min)。捕集されたマイクロカプセルは少量の蒸留水に再分散し、90μmの標準篩を用いて篩過した後、マンニトール19.9gを添加し、凍結乾燥機(DFM-05A-S,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。得られたマイクロカプセル末の酢酸リュープロレリン含量は15.4%であり、収率は約66%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は18.7%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸0.907g(酢酸リュープロレリンの3倍モル)にメタノール6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)3.74gをジクロロメタン6.5gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は9.6%であり、収率は約44%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は12.2%であることが分かった。
酢酸リュープロレリン1.35g及びステアリン酸1.513g(酢酸リュープロレリンの5倍モル)にメタノール6.4gを加え約40℃で加温溶解した後、これを30℃に調整した。この溶液に、DL−乳酸重合体(重量平均分子量21,800)3.14gをジクロロメタン5.5gで溶解した溶液を加え、攪拌混合して均一な油相(O相)を調製した。このときの薬物のローディング量は22.5%である。該O相を予め約18℃に温調しておいた0.1%(w/w)ポリビニルアルコール(EG−40、日本合成化学製)水溶液1リットル中に注入し、ホモミキサー(特殊機化製)を用いて乳化しO/W乳化物とした(タービン回転数約7,000rpm)。該O/W乳化物を約3時間水中乾燥し、75μmの標準篩を用いて篩過し、通過したものについて遠心機(日立 CR5DL)を用いて遠心分離し、マイクロカプセルを沈降させ、これを捕集した(回転数約3,000rpm)。該マイクロカプセルに蒸留水を加えて洗浄した後、再度遠心分離してマイクロカプセルを沈降させた。上澄み除去後、これにマンニトール0.635gを加えて少量の蒸留水で分散させ、これをナス型フラスコに回収した。これを凍結し、凍結乾燥機(DF−01H,ULVAC製)で凍結乾燥してマイクロカプセル末を得た。
得られたマイクロカプセル末の酢酸リュープロレリン含量は5.3%であり、収率は約53%であった。これより、マイクロカプセル中の酢酸リュープロレリン含量は6.5%であることが分かった。
比較例4で調製したマイクロカプセル末59mg、及び実施例7で調製したマイクロカプセル末59mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして9mgの投与量)して、血清中の酢酸リュープロレリン濃度を測定した。血中濃度推移を図8に示す。投与後1週から10週において、実施例7の血中濃度レベルは比較例2のそれを大きく上回った。すなわち、ステアリン酸を添加することで、薬物の放出速度を改善し、オンセット部(投与後24時間から1ヶ月まで)及びメンテナンス部(投与後1ヶ月以降)血中濃度レベルを改善することが可能となった。
比較例5で調製したマイクロカプセル末61mg、及び実施例8及び実施例10で調製したマイクロカプセル末61mg及び59mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして9mgの投与量)して、血清中の濃度を測定した。血中濃度推移を図9に示す。投与後4週までにおいて、実施例8及び実施例10の血中濃度レベルは比較例3のそれを大きく上回った。また実施例10は、実施例8で観察される投与後2日目における凹みにおいて高値なレベルのまま維持され、より良好な血中濃度パターンを示した。すなわち、ステアリン酸の添加量及び薬物のローディング量を制御することで、オンセット部(投与後24時間から1ヶ月まで)放出速度を制御し理想的な血中濃度パターンを達成することが可能となった。
実施例15、実施例16、実施例13、実施例17及び実施例18で調製したマイクロカプセル末について、それぞれにおける酢酸リュープロレリンのトラップ率を計算した。計算した結果を表3に示す。ここでいう「酢酸リュープロレリンのトラップ率」とは、「マイクロカプセル中の酢酸リュープロレリン含量」を「酢酸リュープロレリンのローディング量」で除して計算される割合のことを示す。表3から明らかなように、該ローディング量が23.5%を超えてからはトラップ率が若干減少する傾向を示した。
比較例4、実施例11、実施例12、実施例13、実施例14、実施例25及び実施例26で調製したマイクロカプセル末について、それぞれにおける酢酸リュープロレリンのトラップ率を計算した。計算した結果を表4に示す。酢酸リュープロレリンに対し添加するステアリン酸のモル比が1.5を超えてからはトラップ率が減少する傾向を示した。
比較例4で調製したマイクロカプセル末59mg、比較例6で調製したマイクロカプセル末54mg、実施例19で調製したマイクロカプセル末50mg、実施例20で調製したマイクロカプセル末54mg及び実施例21で調製したマイクロカプセル末55mgをそれぞれ分散媒約0.4mLで懸濁しラットに皮下注射(いずれも酢酸リュープロレリンとして9mgの投与量)して、血清中の酢酸リュープロレリン濃度を測定した。投与後24時間以内の最高血中濃度(Cmax)及びAUCと、投与後24時間から1ヶ月まで(オンセット部)のAUC、及び投与後6ヶ月の血清中濃度の結果を表5に示す。表5から分かるように、薬物のローディング量が約20〜22.5%でステアリン酸を含まない比較例4及び比較例6の製剤に対して、薬物のローディング量が約20〜22.5%でステアリン酸を含む実施例19、実施例20及び実施例21の製剤はオンセット部の血中濃度及びAUCが高くなった。一方、投与後6ヶ月においても血中から薬物が検出され、長期にわたり薬物が放出されていることも確認できた。すなわち、ステアリン酸を含有させることで、オンセット部の血中濃度推移を大幅に改善することができた。
すなわち、投与後における該生理活性ペプチドの初期過剰放出の抑制とオンセット部における安定した薬物放出を達成すると共に、投与後約60日〜400日という長期にわたって有効濃度の薬物血中濃度で生理活性物質を安定的に持続放出することができる。
また、製剤中における生理活性物質の含有量が従来製剤より高含量化されているため、有効成分の単位用量あたりに必要な徐放性製剤全体の容積および重量を低減させることができる。これにより、投与時の疼痛や投与後の硬結など、単位容量の大きな製剤を投与することが原因と考えられる患者における身体的負担を軽減することができる。
Claims (55)
- 乳酸重合体またはその塩からなるマイクロカプセル中に水溶性の生理活性ペプチドからなる生理活性物質が実質的に均一に分散した徐放性組成物であって、該生理活性物質をマイクロカプセル全体に対して15〜35(重量/重量)%含み、
該生理活性物質が式:
5−oxo−Pro−His−Trp−Ser−Tyr−Y−Leu−Arg−Pro−Z [式中、YはDLeu、DAla、DTrp、DSer(tBu)、D2NalまたはDHis(ImBzl)を示し、ZはNH−C 2 H 5 またはGly−NH 2 を示す。]で表されるペプチドまたはその塩であり、
該乳酸重合体の重量平均分子量(Mw)が約19,000〜約27,000であり、さらに該ペプチドまたはその塩の1モルに対して0.25〜2モルのステアリン酸を含有する徐放性組成物。 - 徐放性組成物からのインビボにおける生理活性物質の放出が、約120日〜400日にわたって有効血中薬物濃度を維持できることを特徴とする請求項1記載の徐放性組成物。
- 生理活性物質が式:
5−oxo−Pro−His−Trp−Ser−Tyr−DLeu−Leu−Arg−Pro−NH−C2H5で表されるペプチドまたはその酢酸塩である請求項1記載の徐放性組成物。 - 生理活性物質が酢酸リュープロレリンである請求項1記載の徐放性組成物。
- 生理活性物質の含量が、マイクロカプセル全体に対して、17〜26(重量/重量)%である請求項1記載の徐放性組成物。
- 生理活性物質の含量が、マイクロカプセル全体に対して、18〜22(重量/重量)%である請求項1記載の徐放性組成物。
- ステアリン酸の含量が、ペプチドまたはその塩の1モルに対して0.5〜1.5モルである請求項1記載の徐放性組成物。
- 乳酸重合体またはその塩を揮発性の水非混和性の第一溶媒に溶解して第一溶液を調製し、
水溶性の生理活性ペプチドからなる生理活性物質を水混和性の第二溶媒に溶解して第二溶液を調製し、
得られた第一溶液と第二溶液を混合して乳酸重合体またはその塩および生理活性物質が均質に溶解した第三溶液を調製し、
得られた第三溶液を乳化剤の水溶液からなる第四溶液に分散してO/Wエマルションを調製し、
生成したマイクロカプセルから第一溶媒と第二溶媒を除去することにより得られるものである請求項1記載の徐放性組成物。 - 第一溶液を調製する際における乳酸重合体またはその塩を溶解する溶媒として、水混和性の第三溶媒を第一溶媒にさらに加えた混合溶媒を用いることを特徴とする請求項8記載の徐放性組成物。
- マイクロカプセルから第一溶媒と第二溶媒を除去する工程において、乳化工程の管理温度が約15〜約35℃である請求項8記載の徐放性組成物。
- 乳化工程の温度管理が、O/Wエマルションの温度を約15〜35℃に調節することにより行われること特徴とする請求項10記載の徐放性組成物。
- O/Wエマルションを調製する際の第三溶液と第四溶液の各温度が約15〜約35℃である請求項8記載の徐放性組成物。
- マイクロカプセルから第一溶媒と第二溶媒を除去する工程を、水中乾燥法により行うことを特徴とする請求項8記載の徐放性組成物。
- 第一溶媒がジクロロメタンである請求項8記載の徐放性組成物。
- 第二溶媒が低級アルコールである請求項8記載の徐放性組成物。
- 低級アルコールがメタノール、エタノールまたはプロパノールである請求項15記載の徐放性組成物。
- 第三溶液中の水非混和性溶媒と水混和性溶媒の容量比率が35:65〜55:45であることを特徴とする請求項8記載の徐放性組成物。
- 第一溶液中の乳酸重合体またはその塩の濃度が約33〜45重量%であることを特徴とする請求項8記載の徐放性組成物。
- 第三溶液を製造する際における生理活性物質のローディング量が17〜50重量%であることを特徴とする請求項8記載の徐放性組成物。
- 第三溶液を製造する際における生理活性物質のローディング量が19〜38重量%であることを特徴とする請求項8記載の徐放性組成物。
- 第三溶液を製造する際における生理活性物質のローディング量が20〜23重量%であることを特徴とする請求項8記載の徐放性組成物。
- 分散媒に易分散性であることを特徴とする請求項1記載の徐放性組成物。
- 分散媒に分散後24時間以上安定であることを特徴とする請求項22記載の徐放性組成物。
- 乳酸重合体の重量平均分子量(Mw)の数平均分子量(Mn)に対する比が1.5よりも大きいことを特徴とする請求項1記載の徐放性組成物。
- 乳酸重合体がポリ乳酸またはポリラクチドであることを特徴とする請求項1記載の徐放性組成物。
- 乳酸重合体がポリDL−乳酸またはポリDL−ラクチドであることを特徴とする請求項1記載の徐放性組成物。
- 乳酸重合体が乳酸−グリコール酸重合体であることを特徴とする請求項1記載の徐放性組成物。
- 乳酸−グリコール酸重合体の、乳酸/グリコール酸の組成比が60/40〜99.9/0.1であることを特徴とする請求項27記載の徐放性組成物。
- 乳酸重合体が、分子量5000以下の重合体含有量が約5.0重量%以下のものである請求項1記載の徐放性組成物。
- 乳酸重合体が、分子量3000以下の重合体含有量が約1.5重量%以下のものである請求項1記載の徐放性組成物。
- 乳酸重合体が、分子量1000以下の重合体含有量が約0.1重量%以下のものである請求項1記載の徐放性組成物。
- 乳酸重合体の重量平均分子量(Mw)が19,500〜26,500である請求項1記載の徐放性組成物。
- (i)乳酸重合体またはその塩を揮発性の水非混和性の第一溶媒に溶解して第一溶液を調製し、
(ii)水溶性の生理活性ペプチドからなる生理活性物質を水混和性の第二溶媒に溶解して第二溶液を調製し、
(iii)得られた第一溶液と第二溶液を混合して乳酸重合体またはその塩および生理活性物質が均質に溶解した第三溶液を調製し、
(iv)得られた第三溶液を界面活性剤の水溶液からなる第四溶液に分散してO/Wエマルションを調製し、
(v)生成したマイクロカプセルから第一溶媒と第二溶媒を除去する工程を含むことを特徴とする請求項1記載の徐放性組成物の製造方法。 - (i)の工程において、乳酸重合体またはその塩を溶解する溶媒として、水混和性の第三溶媒を第一溶媒にさらに加えた混合溶媒を用いることを特徴とする請求項33記載の製造方法。
- マイクロカプセルから第一溶媒と第二溶媒を除去する工程において、乳化工程の管理温度が約15〜約35℃である請求項33記載の製造方法。
- 乳化工程の温度管理が、O/Wエマルションの温度を約15〜35℃に調節することにより行われること特徴とする請求項35記載の製造方法。
- O/Wエマルションを調製する際の第三溶液と第四溶液の各温度を約15〜約35℃に調節することを特徴とする請求項33記載の製造方法。
- マイクロカプセルから第一溶媒と第二溶媒を除去する工程を、水中乾燥法により行うことを特徴とする請求項33記載の製造方法。
- 第一溶媒がジクロロメタンである請求項33記載の製造方法。
- 第二溶媒が低級アルコールである請求項33記載の製造方法。
- 低級アルコールがメタノール、エタノールまたはプロパノールである請求項40記載の製造方法。
- 第三溶液中の水非混和性溶媒と水混和性溶媒の容量比率が35:65〜55:45であることを特徴とする請求項33記載の製造方法。
- 第一溶液中の乳酸重合体またはその塩の濃度が約33〜45重量%であることを特徴とする請求項33記載の製造方法。
- ステアリン酸を第一溶液および/または第二溶液、あるいは第三溶液に添加することを特徴とする請求項33記載の製造方法。
- ステアリン酸を第二溶液に溶解させることを特徴とする請求項33記載の製造方法。
- ステアリン酸を第一溶液に添加することを特徴とする請求項33記載の製造方法。
- 第三溶液を製造する際における生理活性物質のローディング量が17〜50重量%であることを特徴とする請求項33記載の製造方法。
- 第三溶液を製造する際における生理活性物質のローディング量が19〜38重量%であることを特徴とする請求項33記載の徐放性組成物の製造方法。
- 第三溶液を製造する際における生理活性物質のローディング量が20〜23重量%であることを特徴とする請求項33記載の徐放性組成物の製造方法。
- 6ヶ月製剤である請求項1記載の徐放性組成物。
- 請求項1記載の徐放性組成物を含有してなる医薬組成物。
- 請求項1記載の徐放性組成物を含有してなる前立腺癌、前立腺肥大症、子宮内膜症、子宮筋腫、子宮線維腫、思春期早発症、月経困難症もしくは乳癌の予防、治療剤または避妊剤。
- 請求項1記載の徐放性組成物を含有してなる閉経前乳癌術後再発予防剤。
- 前立腺癌、前立腺肥大症、子宮内膜症、子宮筋腫、子宮線維腫、思春期早発症、月経困難症もしくは乳癌の予防、治療剤または避妊剤を製造するための請求項1記載の徐放性組成物の使用。
- 閉経前乳癌術後再発予防剤を製造するための請求項1記載の徐放性組成物の使用。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US87536406P | 2006-12-18 | 2006-12-18 | |
| US91740107P | 2007-05-11 | 2007-05-11 | |
| PCT/JP2007/074617 WO2008075762A1 (en) | 2006-12-18 | 2007-12-17 | Sustained-release composition and method for producing the same |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010130277A Division JP5258844B2 (ja) | 2006-12-18 | 2010-06-07 | 徐放性組成物およびその製造法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010513225A JP2010513225A (ja) | 2010-04-30 |
| JP2010513225A5 JP2010513225A5 (ja) | 2010-07-22 |
| JP4564098B2 true JP4564098B2 (ja) | 2010-10-20 |
Family
ID=39015849
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009524632A Active JP4564098B2 (ja) | 2006-12-18 | 2007-12-17 | 徐放性組成物およびその製造法 |
| JP2010130277A Active JP5258844B2 (ja) | 2006-12-18 | 2010-06-07 | 徐放性組成物およびその製造法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010130277A Active JP5258844B2 (ja) | 2006-12-18 | 2010-06-07 | 徐放性組成物およびその製造法 |
Country Status (27)
| Country | Link |
|---|---|
| US (3) | US8921326B2 (ja) |
| EP (1) | EP2094246B1 (ja) |
| JP (2) | JP4564098B2 (ja) |
| KR (1) | KR101522035B1 (ja) |
| CN (1) | CN101563068B (ja) |
| AR (1) | AR064381A1 (ja) |
| AU (1) | AU2007335406B2 (ja) |
| BR (1) | BRPI0720582B1 (ja) |
| CA (1) | CA2671670C (ja) |
| CL (1) | CL2007003658A1 (ja) |
| CO (1) | CO6180498A2 (ja) |
| CR (1) | CR10867A (ja) |
| DO (1) | DOP2009000144A (ja) |
| EA (1) | EA016176B1 (ja) |
| EC (1) | ECSP099439A (ja) |
| ES (1) | ES2791698T3 (ja) |
| GE (1) | GEP20125597B (ja) |
| IL (1) | IL198845A (ja) |
| MA (1) | MA31006B1 (ja) |
| MX (1) | MX2009006653A (ja) |
| MY (1) | MY148370A (ja) |
| NO (1) | NO347209B1 (ja) |
| NZ (1) | NZ577281A (ja) |
| PE (1) | PE20081842A1 (ja) |
| TN (1) | TN2009000197A1 (ja) |
| TW (1) | TWI481424B (ja) |
| WO (1) | WO2008075762A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2094246B1 (en) | 2006-12-18 | 2020-04-01 | Takeda Pharmaceutical Company Limited | Sustained-release composition and method for producing the same |
| WO2011078394A2 (en) * | 2009-12-22 | 2011-06-30 | Takeda Pharmaceutical Company Limited | Sustained-release formulation |
| UY33465A (es) * | 2010-06-25 | 2012-01-31 | Takeda Pharmaceutical | Formulacion de liberacion sostenida |
| US20150087624A1 (en) * | 2012-04-24 | 2015-03-26 | Osaka University | Method for producing an aqueous dispersion of drug nanoparticles and use thereof |
| KR101558083B1 (ko) * | 2014-04-07 | 2015-10-07 | 에스케이케미칼주식회사 | 약물함유 고분자미립구 제조방법 |
| WO2015158823A1 (de) | 2014-04-16 | 2015-10-22 | Veyx-Pharma Gmbh | Veterinärpharmazeutische zusammensetzung und deren verwendung |
| WO2016020901A1 (en) | 2014-08-07 | 2016-02-11 | Acerta Pharma B.V. | Methods of treating cancers, immune and autoimmune diseases, and inflammatory diseases based on btk occupancy and btk resynthesis rate |
| MX2020003681A (es) * | 2017-09-26 | 2020-08-03 | Nanomi B V | Metodo para preparar microparticulas por tecnica de doble emulsion. |
| KR101936040B1 (ko) * | 2018-04-23 | 2019-01-08 | 주식회사 씨트리 | 안정화된 단상 혼합액을 이용하는 생분해성 미립구의 제조방법 |
| BR112022000413A2 (pt) * | 2019-07-12 | 2022-03-03 | G2Gbio Inc | Formulação de longa duração contendo rivastigmina e método para preparar a mesma |
Family Cites Families (72)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3297033A (en) * | 1963-10-31 | 1967-01-10 | American Cyanamid Co | Surgical sutures |
| US3565869A (en) * | 1968-12-23 | 1971-02-23 | American Cyanamid Co | Extrudable and stretchable polyglycolic acid and process for preparing same |
| US3773919A (en) * | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US3755558A (en) * | 1971-02-23 | 1973-08-28 | Du Pont | Polylactide drug mixtures for topical application atelet aggregation |
| US3839297A (en) * | 1971-11-22 | 1974-10-01 | Ethicon Inc | Use of stannous octoate catalyst in the manufacture of l(-)lactide-glycolide copolymer sutures |
| US3912692A (en) * | 1973-05-03 | 1975-10-14 | American Cyanamid Co | Process for polymerizing a substantially pure glycolide composition |
| US3890283A (en) * | 1973-06-04 | 1975-06-17 | American Cyanamid Co | Process for post-polymerizing polyglycolic acid |
| US4258063A (en) * | 1978-06-23 | 1981-03-24 | Henkel Corporation | Self-emulsifying cosmetic base |
| US4249531A (en) * | 1979-07-05 | 1981-02-10 | Alza Corporation | Bioerodible system for delivering drug manufactured from poly(carboxylic acid) |
| US4273920A (en) * | 1979-09-12 | 1981-06-16 | Eli Lilly And Company | Polymerization process and product |
| PH19942A (en) | 1980-11-18 | 1986-08-14 | Sintex Inc | Microencapsulation of water soluble polypeptides |
| IE52535B1 (en) | 1981-02-16 | 1987-12-09 | Ici Plc | Continuous release pharmaceutical compositions |
| US5366734A (en) * | 1981-02-16 | 1994-11-22 | Zeneca Limited | Continuous release pharmaceutical compositions |
| US4479911A (en) * | 1982-01-28 | 1984-10-30 | Sandoz, Inc. | Process for preparation of microspheres and modification of release rate of core material |
| US4605730A (en) * | 1982-10-01 | 1986-08-12 | Ethicon, Inc. | Surgical articles of copolymers of glycolide and ε-caprolactone and methods of producing the same |
| US4539981A (en) * | 1982-11-08 | 1985-09-10 | Johnson & Johnson Products, Inc. | Absorbable bone fixation device |
| FR2537980B1 (fr) * | 1982-12-17 | 1986-12-19 | Sandoz Sa | Derives d'acides hydroxycarboxyliques oligomeres, leur preparation et leur utilisation |
| CH656884A5 (de) | 1983-08-26 | 1986-07-31 | Sandoz Ag | Polyolester, deren herstellung und verwendung. |
| JPS60100516A (ja) | 1983-11-04 | 1985-06-04 | Takeda Chem Ind Ltd | 徐放型マイクロカプセルの製造法 |
| CA1256638A (en) * | 1984-07-06 | 1989-06-27 | Motoaki Tanaka | Polymer and its production |
| JPH0678425B2 (ja) | 1984-07-06 | 1994-10-05 | 和光純薬工業株式会社 | 重合体の新規製造法 |
| DE3678308D1 (de) | 1985-02-07 | 1991-05-02 | Takeda Chemical Industries Ltd | Verfahren zur herstellung von mikrokapseln. |
| JP2551756B2 (ja) * | 1985-05-07 | 1996-11-06 | 武田薬品工業株式会社 | ポリオキシカルボン酸エステルおよびその製造法 |
| MY107937A (en) * | 1990-02-13 | 1996-06-29 | Takeda Chemical Industries Ltd | Prolonged release microcapsules. |
| JP2653255B2 (ja) | 1990-02-13 | 1997-09-17 | 武田薬品工業株式会社 | 長期徐放型マイクロカプセル |
| FR2658432B1 (fr) | 1990-02-22 | 1994-07-01 | Medgenix Group Sa | Microspheres pour la liberation controlee des substances hydrosolubles et procede de preparation. |
| EP0452111B1 (en) * | 1990-04-13 | 1998-07-15 | Takeda Chemical Industries, Ltd. | Biodegradable high-molecular polymers, production and use thereof |
| JP3116311B2 (ja) * | 1990-06-13 | 2000-12-11 | エーザイ株式会社 | マイクロスフィアの製法 |
| FR2693905B1 (fr) * | 1992-07-27 | 1994-09-02 | Rhone Merieux | Procédé de préparation de microsphères pour la libération prolongée de l'hormone LHRH et ses analogues, microsphères et formulations obtenues. |
| DE69316101T2 (de) * | 1992-08-07 | 1998-10-22 | Takeda Chemical Industries Ltd | Herstellung von Mikrokapseln, die wasserlösliche Arzneimittel enthalten |
| JP3277342B2 (ja) * | 1992-09-02 | 2002-04-22 | 武田薬品工業株式会社 | 徐放性マイクロカプセルの製造法 |
| TW333456B (en) * | 1992-12-07 | 1998-06-11 | Takeda Pharm Ind Co Ltd | A pharmaceutical composition of sustained-release preparation the invention relates to a pharmaceutical composition of sustained-release preparation which comprises a physiologically active peptide. |
| DE4342092B4 (de) | 1993-12-09 | 2007-01-11 | Zentaris Gmbh | Langwirkende Injektionssuspension und Verfahren zur Herstellung |
| CA2143044C (en) * | 1994-02-21 | 2005-04-12 | Yasutaka Igari | Matrix for sustained-release preparation |
| JP3490171B2 (ja) | 1994-02-21 | 2004-01-26 | 武田薬品工業株式会社 | 生体内分解性ポリマーの末端カルボキシル基におけるエステル |
| JPH07273447A (ja) | 1994-03-29 | 1995-10-20 | Sumitomo Kinzoku Ceramics:Kk | セラミック回路基板及びその製造方法 |
| US5763513A (en) * | 1994-05-19 | 1998-06-09 | Mitsui Toatsu Chemicals, Inc. | L-lactic acid polymer composition, molded product and film |
| FR2736508B1 (fr) | 1995-07-13 | 1997-09-19 | Francais Prod Ind Cfpi | Nouvelle forme solide de stockage et de commercialisation pour compositions phytosanitaires et moyens pour sa preparation |
| TW448055B (en) * | 1995-09-04 | 2001-08-01 | Takeda Chemical Industries Ltd | Method of production of sustained-release preparation |
| CA2192782C (en) * | 1995-12-15 | 2008-10-14 | Nobuyuki Takechi | Production of microspheres |
| CA2192773C (en) * | 1995-12-15 | 2008-09-23 | Hiroaki Okada | Production of sustained-release preparation for injection |
| CA2219698C (en) * | 1996-10-31 | 2007-09-04 | Takeda Chemical Industries, Ltd. | Sustained-release preparation |
| JPH10273447A (ja) | 1997-01-29 | 1998-10-13 | Takeda Chem Ind Ltd | 徐放性マイクロスフィア、その製造法および用途 |
| WO1998032423A1 (en) | 1997-01-29 | 1998-07-30 | Takeda Chemical Industries, Ltd. | Sustained-release microspheres, their production and use |
| US5945126A (en) * | 1997-02-13 | 1999-08-31 | Oakwood Laboratories L.L.C. | Continuous microsphere process |
| GB9718986D0 (en) | 1997-09-09 | 1997-11-12 | Danbiosyst Uk | Controlled release microsphere delivery system |
| US5989463A (en) | 1997-09-24 | 1999-11-23 | Alkermes Controlled Therapeutics, Inc. | Methods for fabricating polymer-based controlled release devices |
| IL136951A0 (en) * | 1998-01-16 | 2001-06-14 | Takeda Chemical Industries Ltd | Sustained-release composition, method of its production and use thereof |
| JPH11269094A (ja) | 1998-01-16 | 1999-10-05 | Takeda Chem Ind Ltd | 徐放性組成物、その製造法および用途 |
| AU2745199A (en) | 1998-03-04 | 1999-09-20 | Takeda Chemical Industries Ltd. | Sustained-release preparation for aii antagonist, production and use thereof |
| US6114495A (en) * | 1998-04-01 | 2000-09-05 | Cargill Incorporated | Lactic acid residue containing polymer composition and product having improved stability, and method for preparation and use thereof |
| US6270802B1 (en) * | 1998-10-28 | 2001-08-07 | Oakwood Laboratories L.L.C. | Method and apparatus for formulating microspheres and microcapsules |
| US6565874B1 (en) * | 1998-10-28 | 2003-05-20 | Atrix Laboratories | Polymeric delivery formulations of leuprolide with improved efficacy |
| AU1797800A (en) * | 1998-12-15 | 2000-07-03 | Takeda Chemical Industries Ltd. | Process for producing polymer |
| WO2001005380A1 (fr) | 1999-07-15 | 2001-01-25 | Takeda Chemical Industries, Ltd. | Compositions a liberation lente, techniques de production et methodes d'utilisation |
| AU6510400A (en) | 1999-08-04 | 2001-03-05 | Oakwood Laboratories L.L.C. | Slow release microspheres |
| WO2002012369A1 (fr) | 2000-08-07 | 2002-02-14 | Wako Pure Chemical Industries, Ltd. | Polymere d'acide lactique et son procede de preparation |
| US6824822B2 (en) * | 2001-08-31 | 2004-11-30 | Alkermes Controlled Therapeutics Inc. Ii | Residual solvent extraction method and microparticles produced thereby |
| WO2002043766A1 (fr) | 2000-11-29 | 2002-06-06 | Takeda Chemical Industries, Ltd. | Compositions medicinales et leur procede de preparation |
| AU2002221139A1 (en) | 2000-12-15 | 2002-06-24 | Takeda Chemical Industries Ltd. | Medicinal compositions of nonpeptidyl gonadotropin-releasing hormone agonist or antagonist, process for producing the same and use thereof |
| TWI225416B (en) * | 2001-06-29 | 2004-12-21 | Takeda Chemical Industries Ltd | Sustained-release composition and process for producing the same |
| TW200526267A (en) * | 2001-06-29 | 2005-08-16 | Takeda Chemical Industries Ltd | Controlled release composition and method of producing the same |
| CA2471521C (en) * | 2001-12-26 | 2010-11-02 | Takeda Chemical Industries, Ltd. | Novel microsphere and method for production thereof |
| CA2490351C (en) * | 2002-06-25 | 2011-11-01 | Takeda Pharmaceutical Company Limited | Process for producing sustained-release composition |
| US6803708B2 (en) * | 2002-08-22 | 2004-10-12 | Cdream Display Corporation | Barrier metal layer for a carbon nanotube flat panel display |
| EP1660039B1 (en) | 2003-07-18 | 2016-09-28 | Oakwood Laboratories L.L.C. | Prevention of molecular weight reduction of the polymer, impurity formation and gelling in polymer compositions |
| AU2004259209A1 (en) | 2003-07-23 | 2005-02-03 | Pr Pharmaceuticals, Inc. | Controlled release compositions |
| JP2007537137A (ja) * | 2003-09-24 | 2007-12-20 | モンタナ ステート ユニバーシティ | ノロウィルスモノクローナル抗体及びペプチド |
| TW200613012A (en) * | 2004-07-02 | 2006-05-01 | Takeda Pharmaceuticals Co | Sustained-release composition, process for producing the same and use of the same |
| EP3173072A1 (en) | 2004-10-01 | 2017-05-31 | Ramscor, Inc. | Conveniently implantable sustained release drug compositions |
| WO2006053175A2 (en) | 2004-11-10 | 2006-05-18 | Qlt Usa Inc. | A stabilized polymeric delivery system |
| EP2094246B1 (en) | 2006-12-18 | 2020-04-01 | Takeda Pharmaceutical Company Limited | Sustained-release composition and method for producing the same |
-
2007
- 2007-12-17 EP EP07859925.5A patent/EP2094246B1/en active Active
- 2007-12-17 ES ES07859925T patent/ES2791698T3/es active Active
- 2007-12-17 CN CN2007800469578A patent/CN101563068B/zh active Active
- 2007-12-17 BR BRPI0720582-1A patent/BRPI0720582B1/pt active IP Right Grant
- 2007-12-17 US US12/312,751 patent/US8921326B2/en active Active
- 2007-12-17 GE GEAP200711311A patent/GEP20125597B/en unknown
- 2007-12-17 AR ARP070105656A patent/AR064381A1/es not_active Application Discontinuation
- 2007-12-17 NZ NZ577281A patent/NZ577281A/en unknown
- 2007-12-17 JP JP2009524632A patent/JP4564098B2/ja active Active
- 2007-12-17 KR KR1020097011993A patent/KR101522035B1/ko active IP Right Review Request
- 2007-12-17 CL CL200703658A patent/CL2007003658A1/es unknown
- 2007-12-17 MX MX2009006653A patent/MX2009006653A/es active IP Right Grant
- 2007-12-17 NO NO20092662A patent/NO347209B1/no unknown
- 2007-12-17 CA CA2671670A patent/CA2671670C/en not_active Expired - Fee Related
- 2007-12-17 AU AU2007335406A patent/AU2007335406B2/en active Active
- 2007-12-17 MY MYPI20092544A patent/MY148370A/en unknown
- 2007-12-17 EA EA200970600A patent/EA016176B1/ru not_active IP Right Cessation
- 2007-12-17 TW TW096148177A patent/TWI481424B/zh active
- 2007-12-17 WO PCT/JP2007/074617 patent/WO2008075762A1/en active Application Filing
-
2008
- 2008-01-02 PE PE2008000035A patent/PE20081842A1/es active IP Right Grant
-
2009
- 2009-05-20 IL IL198845A patent/IL198845A/en active IP Right Grant
- 2009-05-20 TN TNP2009000197A patent/TN2009000197A1/fr unknown
- 2009-06-12 CR CR10867A patent/CR10867A/es unknown
- 2009-06-17 EC EC2009009439A patent/ECSP099439A/es unknown
- 2009-06-17 MA MA32010A patent/MA31006B1/fr unknown
- 2009-06-17 DO DO2009000144A patent/DOP2009000144A/es unknown
- 2009-06-18 CO CO09063558A patent/CO6180498A2/es not_active Application Discontinuation
-
2010
- 2010-06-07 JP JP2010130277A patent/JP5258844B2/ja active Active
-
2014
- 2014-11-19 US US14/547,467 patent/US9617303B2/en active Active
-
2017
- 2017-02-28 US US15/444,712 patent/US9713595B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5258844B2 (ja) | 徐放性組成物およびその製造法 | |
| JP5931147B2 (ja) | 徐放性組成物およびその製造法 | |
| JP4819173B2 (ja) | 徐放性組成物およびその製造法 | |
| JP5622760B2 (ja) | 乳酸重合体及びその製造方法 | |
| JPH10273447A (ja) | 徐放性マイクロスフィア、その製造法および用途 | |
| JP3902518B2 (ja) | 徐放性組成物用乳酸−グリコール酸重合体の製造法 | |
| JP5188670B2 (ja) | 徐放性組成物およびその製造法 | |
| JP2004155792A (ja) | 徐放性組成物およびその製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100607 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100607 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20100607 |
|
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20100630 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100706 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100729 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130806 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4564098 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130806 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130806 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R153 | Grant of patent term extension |
Free format text: JAPANESE INTERMEDIATE CODE: R153 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |