JP4548864B2 - 直接発酵手段によるd―アミノ酸の調製 - Google Patents
直接発酵手段によるd―アミノ酸の調製 Download PDFInfo
- Publication number
- JP4548864B2 JP4548864B2 JP51665098A JP51665098A JP4548864B2 JP 4548864 B2 JP4548864 B2 JP 4548864B2 JP 51665098 A JP51665098 A JP 51665098A JP 51665098 A JP51665098 A JP 51665098A JP 4548864 B2 JP4548864 B2 JP 4548864B2
- Authority
- JP
- Japan
- Prior art keywords
- gene
- cell
- amino
- amino acid
- phenylalanine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 150000008574 D-amino acids Chemical class 0.000 title claims abstract description 63
- 238000000855 fermentation Methods 0.000 title abstract description 25
- 230000004151 fermentation Effects 0.000 title abstract description 25
- 238000002360 preparation method Methods 0.000 title abstract description 6
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 101
- 238000000034 method Methods 0.000 claims abstract description 58
- COLNVLDHVKWLRT-MRVPVSSYSA-N D-phenylalanine Chemical compound OC(=O)[C@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-MRVPVSSYSA-N 0.000 claims abstract description 32
- 229930182832 D-phenylalanine Natural products 0.000 claims abstract description 32
- 238000004519 manufacturing process Methods 0.000 claims abstract description 32
- BTNMPGBKDVTSJY-UHFFFAOYSA-N keto-phenylpyruvic acid Chemical compound OC(=O)C(=O)CC1=CC=CC=C1 BTNMPGBKDVTSJY-UHFFFAOYSA-N 0.000 claims abstract description 16
- 239000006143 cell culture medium Substances 0.000 claims abstract description 12
- 238000012258 culturing Methods 0.000 claims abstract description 6
- 108700026220 vif Genes Proteins 0.000 claims abstract 2
- 239000013612 plasmid Substances 0.000 claims description 71
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 claims description 43
- 239000000758 substrate Substances 0.000 claims description 39
- 229940024606 amino acid Drugs 0.000 claims description 35
- 235000001014 amino acid Nutrition 0.000 claims description 35
- 241000193386 Lysinibacillus sphaericus Species 0.000 claims description 25
- 229960005190 phenylalanine Drugs 0.000 claims description 25
- 241000588724 Escherichia coli Species 0.000 claims description 24
- 150000008575 L-amino acids Chemical class 0.000 claims description 23
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 22
- 239000000203 mixture Substances 0.000 claims description 21
- 239000002609 medium Substances 0.000 claims description 16
- 108090001066 Racemases and epimerases Proteins 0.000 claims description 13
- 108010041525 Alanine racemase Proteins 0.000 claims description 11
- 101150047092 dadA gene Proteins 0.000 claims description 11
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 claims description 7
- 235000008729 phenylalanine Nutrition 0.000 claims description 7
- 206010064571 Gene mutation Diseases 0.000 claims description 6
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 claims description 6
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 6
- 101150018055 aroH gene Proteins 0.000 claims description 6
- 229960000310 isoleucine Drugs 0.000 claims description 6
- 235000014705 isoleucine Nutrition 0.000 claims description 6
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 6
- 101150023849 pheA gene Proteins 0.000 claims description 6
- 241001087672 Cosenzaea myxofaciens Species 0.000 claims description 5
- 241000588770 Proteus mirabilis Species 0.000 claims description 5
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 claims description 5
- 238000004113 cell culture Methods 0.000 claims description 5
- 244000063299 Bacillus subtilis Species 0.000 claims description 4
- 235000014469 Bacillus subtilis Nutrition 0.000 claims description 4
- 241000193385 Geobacillus stearothermophilus Species 0.000 claims description 4
- 108700016167 Glutamate racemases Proteins 0.000 claims description 4
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 claims description 4
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 claims description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 claims description 4
- 241000589516 Pseudomonas Species 0.000 claims description 4
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 claims description 4
- 230000008569 process Effects 0.000 claims description 4
- 229960004441 tyrosine Drugs 0.000 claims description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 claims description 4
- 235000002374 tyrosine Nutrition 0.000 claims description 4
- 101000798396 Bacillus licheniformis Phenylalanine racemase [ATP hydrolyzing] Proteins 0.000 claims description 3
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 claims description 3
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 3
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 3
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 3
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 3
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 claims description 3
- 229960003136 leucine Drugs 0.000 claims description 3
- 235000005772 leucine Nutrition 0.000 claims description 3
- 108090000554 phenylalanine racemase (ATP-hydrolyzing) Proteins 0.000 claims description 3
- 229960004295 valine Drugs 0.000 claims description 3
- 235000014393 valine Nutrition 0.000 claims description 3
- 239000004474 valine Substances 0.000 claims description 3
- 241000193830 Bacillus <bacterium> Species 0.000 claims description 2
- 239000002253 acid Substances 0.000 claims description 2
- 241000186216 Corynebacterium Species 0.000 claims 6
- 241000186146 Brevibacterium Species 0.000 claims 3
- 241000588748 Klebsiella Species 0.000 claims 3
- 241000192041 Micrococcus Species 0.000 claims 3
- 241000607142 Salmonella Species 0.000 claims 3
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 claims 2
- 239000004475 Arginine Substances 0.000 claims 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 claims 2
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 claims 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 claims 2
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 claims 2
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 claims 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 claims 2
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 claims 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 claims 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 claims 2
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 claims 2
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 claims 2
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 claims 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims 2
- 239000004472 Lysine Substances 0.000 claims 2
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 claims 2
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 claims 2
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 claims 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 claims 2
- 229960003121 arginine Drugs 0.000 claims 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims 2
- 229960001230 asparagine Drugs 0.000 claims 2
- 235000009582 asparagine Nutrition 0.000 claims 2
- 229960002173 citrulline Drugs 0.000 claims 2
- 235000013477 citrulline Nutrition 0.000 claims 2
- 229960002433 cysteine Drugs 0.000 claims 2
- 235000018417 cysteine Nutrition 0.000 claims 2
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims 2
- 229960002743 glutamine Drugs 0.000 claims 2
- 229960002885 histidine Drugs 0.000 claims 2
- 235000014304 histidine Nutrition 0.000 claims 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 claims 2
- 229960004502 levodopa Drugs 0.000 claims 2
- 235000018977 lysine Nutrition 0.000 claims 2
- 229960003646 lysine Drugs 0.000 claims 2
- 229930182817 methionine Natural products 0.000 claims 2
- 229960004452 methionine Drugs 0.000 claims 2
- 229960003104 ornithine Drugs 0.000 claims 2
- 229960001153 serine Drugs 0.000 claims 2
- 229960004799 tryptophan Drugs 0.000 claims 2
- 239000001903 2-oxo-3-phenylpropanoic acid Substances 0.000 claims 1
- DEDGUGJNLNLJSR-UHFFFAOYSA-N alpha-hydroxycinnamic acid Natural products OC(=O)C(O)=CC1=CC=CC=C1 DEDGUGJNLNLJSR-UHFFFAOYSA-N 0.000 claims 1
- 239000000463 material Substances 0.000 abstract description 5
- 210000004027 cell Anatomy 0.000 description 92
- 108020004414 DNA Proteins 0.000 description 49
- 239000012634 fragment Substances 0.000 description 48
- QNAYBMKLOCPYGJ-UWTATZPHSA-N D-alanine Chemical compound C[C@@H](N)C(O)=O QNAYBMKLOCPYGJ-UWTATZPHSA-N 0.000 description 37
- QNAYBMKLOCPYGJ-UHFFFAOYSA-N D-alpha-Ala Natural products CC([NH3+])C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-N 0.000 description 37
- 150000001413 amino acids Chemical class 0.000 description 30
- 150000004715 keto acids Chemical class 0.000 description 30
- 108020004707 nucleic acids Proteins 0.000 description 30
- 102000039446 nucleic acids Human genes 0.000 description 30
- 150000007523 nucleic acids Chemical class 0.000 description 30
- 108090000790 Enzymes Proteins 0.000 description 29
- 102000004190 Enzymes Human genes 0.000 description 27
- 239000002243 precursor Substances 0.000 description 21
- 229960003767 alanine Drugs 0.000 description 20
- 238000010276 construction Methods 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 238000003556 assay Methods 0.000 description 14
- 238000010586 diagram Methods 0.000 description 13
- 238000006243 chemical reaction Methods 0.000 description 11
- 238000005891 transamination reaction Methods 0.000 description 11
- 210000000349 chromosome Anatomy 0.000 description 10
- 101150049467 dadX gene Proteins 0.000 description 10
- 101150012893 dat gene Proteins 0.000 description 9
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 9
- 230000001580 bacterial effect Effects 0.000 description 8
- 238000003776 cleavage reaction Methods 0.000 description 8
- 101150099953 ilvE gene Proteins 0.000 description 8
- 230000007017 scission Effects 0.000 description 8
- 239000013598 vector Substances 0.000 description 8
- 229930195713 D-glutamate Natural products 0.000 description 7
- WHUUTDBJXJRKMK-GSVOUGTGSA-N D-glutamic acid Chemical compound OC(=O)[C@H](N)CCC(O)=O WHUUTDBJXJRKMK-GSVOUGTGSA-N 0.000 description 7
- 101100492609 Talaromyces wortmannii astC gene Proteins 0.000 description 7
- 101150116772 aatA gene Proteins 0.000 description 7
- 101150005925 aspC gene Proteins 0.000 description 7
- 101150100742 dapL gene Proteins 0.000 description 7
- 238000001962 electrophoresis Methods 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 238000002955 isolation Methods 0.000 description 7
- 239000013615 primer Substances 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 101100055397 Wigglesworthia glossinidia brevipalpis alr gene Proteins 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 239000013611 chromosomal DNA Substances 0.000 description 6
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 6
- 101150028338 tyrB gene Proteins 0.000 description 6
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 5
- 229920000936 Agarose Polymers 0.000 description 5
- 239000003155 DNA primer Substances 0.000 description 5
- 101500006448 Mycobacterium bovis (strain ATCC BAA-935 / AF2122/97) Endonuclease PI-MboI Proteins 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- 230000003287 optical effect Effects 0.000 description 5
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 108091034117 Oligonucleotide Proteins 0.000 description 4
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 4
- 230000003321 amplification Effects 0.000 description 4
- 210000002421 cell wall Anatomy 0.000 description 4
- 229960005091 chloramphenicol Drugs 0.000 description 4
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical group ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 239000008103 glucose Substances 0.000 description 4
- 229930027917 kanamycin Natural products 0.000 description 4
- 229960000318 kanamycin Drugs 0.000 description 4
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 4
- 229930182823 kanamycin A Natural products 0.000 description 4
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 4
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 4
- 238000003199 nucleic acid amplification method Methods 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- RWQNBRDOKXIBIV-UHFFFAOYSA-N thymine Chemical compound CC1=CNC(=O)NC1=O RWQNBRDOKXIBIV-UHFFFAOYSA-N 0.000 description 4
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 241001302584 Escherichia coli str. K-12 substr. W3110 Species 0.000 description 3
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 3
- 102000003855 L-lactate dehydrogenase Human genes 0.000 description 3
- 108700023483 L-lactate dehydrogenases Proteins 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 3
- 238000012181 QIAquick gel extraction kit Methods 0.000 description 3
- 108090000340 Transaminases Proteins 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 229960000723 ampicillin Drugs 0.000 description 3
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 238000006481 deamination reaction Methods 0.000 description 3
- 230000029087 digestion Effects 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 229910000160 potassium phosphate Inorganic materials 0.000 description 3
- 235000011009 potassium phosphates Nutrition 0.000 description 3
- 238000003498 protein array Methods 0.000 description 3
- 230000006340 racemization Effects 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- PJWIPEXIFFQAQZ-PUFIMZNGSA-N 7-phospho-2-dehydro-3-deoxy-D-arabino-heptonic acid Chemical compound OP(=O)(O)OC[C@@H](O)[C@@H](O)[C@H](O)CC(=O)C(O)=O PJWIPEXIFFQAQZ-PUFIMZNGSA-N 0.000 description 2
- 108700028369 Alleles Proteins 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 101000644386 Brevibacillus parabrevis Phenylalanine racemase [ATP-hydrolyzing] Proteins 0.000 description 2
- 101000906861 Chondromyces crocatus ATP-dependent tyrosine adenylase Proteins 0.000 description 2
- NGHMDNPXVRFFGS-IUYQGCFVSA-N D-erythrose 4-phosphate Chemical compound O=C[C@H](O)[C@H](O)COP(O)(O)=O NGHMDNPXVRFFGS-IUYQGCFVSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 229930195714 L-glutamate Natural products 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- 101100109871 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) aro-8 gene Proteins 0.000 description 2
- DTBNBXWJWCWCIK-UHFFFAOYSA-N Phosphoenolpyruvic acid Natural products OC(=O)C(=C)OP(O)(O)=O DTBNBXWJWCWCIK-UHFFFAOYSA-N 0.000 description 2
- 241000588769 Proteus <enterobacteria> Species 0.000 description 2
- 102000004879 Racemases and epimerases Human genes 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 102000003929 Transaminases Human genes 0.000 description 2
- 150000001294 alanine derivatives Chemical class 0.000 description 2
- 101150070828 alr gene Proteins 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000006037 cell lysis Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 230000002427 irreversible effect Effects 0.000 description 2
- 101150011498 lad gene Proteins 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 238000005192 partition Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 238000000638 solvent extraction Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 229940113082 thymine Drugs 0.000 description 2
- 238000010361 transduction Methods 0.000 description 2
- 230000026683 transduction Effects 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- JVTAAEKCZFNVCJ-UWTATZPHSA-M (R)-lactate Chemical compound C[C@@H](O)C([O-])=O JVTAAEKCZFNVCJ-UWTATZPHSA-M 0.000 description 1
- OSBLTNPMIGYQGY-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;2-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetic acid;boric acid Chemical compound OB(O)O.OCC(N)(CO)CO.OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O OSBLTNPMIGYQGY-UHFFFAOYSA-N 0.000 description 1
- QDGAVODICPCDMU-UHFFFAOYSA-N 2-amino-3-[3-[bis(2-chloroethyl)amino]phenyl]propanoic acid Chemical compound OC(=O)C(N)CC1=CC=CC(N(CCCl)CCCl)=C1 QDGAVODICPCDMU-UHFFFAOYSA-N 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- 108010080376 3-Deoxy-7-Phosphoheptulonate Synthase Proteins 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical group C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- 108020000946 Bacterial DNA Proteins 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- MNQZXJOMYWMBOU-VKHMYHEASA-N D-glyceraldehyde Chemical compound OC[C@@H](O)C=O MNQZXJOMYWMBOU-VKHMYHEASA-N 0.000 description 1
- 108010067770 Endopeptidase K Proteins 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- 241000701959 Escherichia virus Lambda Species 0.000 description 1
- 241000702191 Escherichia virus P1 Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000006137 Luria-Bertani broth Substances 0.000 description 1
- 102000006746 NADH Dehydrogenase Human genes 0.000 description 1
- 108010086428 NADH Dehydrogenase Proteins 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 238000009004 PCR Kit Methods 0.000 description 1
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 1
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 1
- 108010013639 Peptidoglycan Proteins 0.000 description 1
- FSXRLASFHBWESK-HOTGVXAUSA-N Phe-Tyr Chemical compound C([C@H](N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)C1=CC=CC=C1 FSXRLASFHBWESK-HOTGVXAUSA-N 0.000 description 1
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 1
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 239000008051 TBE buffer Substances 0.000 description 1
- 108010006785 Taq Polymerase Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 150000004716 alpha keto acids Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- -1 aromatic amino acid Chemical class 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 230000008840 asymmetric binding Effects 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000027455 binding Effects 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 1
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 101150056924 dadA1 gene Proteins 0.000 description 1
- 230000009615 deamination Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- FSXRLASFHBWESK-UHFFFAOYSA-N dipeptide phenylalanyl-tyrosine Natural products C=1C=C(O)C=CC=1CC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FSXRLASFHBWESK-UHFFFAOYSA-N 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 150000002306 glutamic acid derivatives Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- FEMOMIGRRWSMCU-UHFFFAOYSA-N ninhydrin Chemical compound C1=CC=C2C(=O)C(O)(O)C(=O)C2=C1 FEMOMIGRRWSMCU-UHFFFAOYSA-N 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 238000007833 oxidative deamination reaction Methods 0.000 description 1
- 238000010979 pH adjustment Methods 0.000 description 1
- 230000005501 phase interface Effects 0.000 description 1
- 229930029653 phosphoenolpyruvate Natural products 0.000 description 1
- 230000004260 plant-type cell wall biogenesis Effects 0.000 description 1
- 230000027086 plasmid maintenance Effects 0.000 description 1
- 230000004683 plasmid partitioning Effects 0.000 description 1
- 239000013600 plasmid vector Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000007100 recyclization reaction Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 238000005204 segregation Methods 0.000 description 1
- UQDJGEHQDNVPGU-UHFFFAOYSA-N serine phosphoethanolamine Chemical compound [NH3+]CCOP([O-])(=O)OCC([NH3+])C([O-])=O UQDJGEHQDNVPGU-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 230000010415 tropism Effects 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/90—Isomerases (5.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1096—Transferases (2.) transferring nitrogenous groups (2.6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/78—Hydrolases (3) acting on carbon to nitrogen bonds other than peptide bonds (3.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P13/00—Preparation of nitrogen-containing organic compounds
- C12P13/04—Alpha- or beta- amino acids
- C12P13/22—Tryptophan; Tyrosine; Phenylalanine; 3,4-Dihydroxyphenylalanine
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Wood Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
Description
発明の分野
本発明は、D−アミノ酸を製造するための材料および方法に関する。特に、本発明は、組換え宿主細胞を用いる天然および非天然の両方のD−アミノ酸の調製に関する。具体的には、本発明は、エナンチオマー的に純粋なD−アミノ酸を製造するために組換え細胞を用いる発酵プロセスに関する。
発明の背景
グリシン、トレオニンおよびイソロイシンを除いて、共通の天然産アミノ酸の各々は、偏光面を回転させる方向により左旋性または右旋性と呼ばれる2つの光学異性体の1つとして存在する。不斉炭素を有さないグリシンは光学異性体を有さない。各々2つの不斉炭素を有するトレオニンおよびイソロイシンは、各々4つの光学異性体を有する。アラニンおよびグルタミンのような一部のアミノ酸は右旋性であり、正(右回り)回転を発生させる。フェニルアラニンやトリプトファンのような他のものは、左旋性であり、負(左回り)回転を発生させる。すなわち、アミノ酸は、単離時に光学異性を反映するように1−またはd−アミノ酸と呼ぶことができる。所定のアミノ酸により生じる特定の回転は、温度およびpHにより変化する。
従来法により、アミノ酸のα−炭素の回りの立体配置がグリセルアルデヒドのD立体異性体(エナンチオマー)に相当するかL立体異性体に相当するかに基づき、アミノ酸は(前記dまたはlの表示に対して)DまたはLとも呼ばれるが、これは独断的な基準である。一部のアミノ酸は中性pHで水溶液中に入れたときに右旋性であることにも拘わらず、その基準によれば大部分の天然産アミノ酸はLアミノ酸となる。アミノ酸に作用する大部分の酵素は、アミノ酸のL−型のみを認識する非対称結合領域を有する。従って、大部分の天然産タンパクはL−アミノ酸を含む。
しかしながら、D−アミノ酸が生成され細胞により利用される例外がある。これらの中で主用なものは、特定の微生物によるD−グルタメートおよびD−アラニンの生成である。D−グルタメートおよびD−アラニンは、主に細菌細胞中で生成され、ムレインの合成において利用される。D−グルタメートおよびD−アラニンの不存在下では、不完全な細菌細胞壁が形成され、細胞溶解につながる。大部分の細菌は、直接合成ではなく、アミノ酸特異的ラセミ体によって対応するL−アミノ酸を転化させることによりD−アミノ酸を生成する。例えば、多くの細菌細胞は、L−アラニンとD−アラニンとの間の双方向転化を触媒してL−アラニンとD−アラニンとのラセミ(50:50)混合物を発生させるアラニンラセミ体を有する。同様に、グルタメートラセミ化酵素はD−グルタメートとL−グルタメートとのラセミ混合物を生成し、前者は細胞壁中に取りこまれ、後者は特にタンパクの生成に利用される。これらの2つの酵素の特異性は、いずれか一方が欠けると不完全な細胞壁形成により細胞が溶解することにより示される。
Bacillus属の一員のような特定の細菌は、D−アミノトランスフェラーゼとして知られている酵素の状態でD−アミノ酸を形成するためにラセミ化酵素の代替物を有する。そのような酵素は、種々のD−アミノ酸および対応するα−ケト酸のアミノ交換を可逆的に触媒する。PCT公開WO91/05870においてマンニング(Manning)は、アミノ転移酵素(アミノトランスフェラーゼ:aminotransferase)で触媒することによりD−アラニンおよびD−グルタメートを微生物的に合成する方法を報告している。マンニングは、2頁において、Bacillus sphaericus D−アミノ転移酵素の使用を報告しているが、該公報は、実際には、痕跡量以上のD−アミノ酸の合成を効果的に触媒することができないD−アミノ転移酵素の好熱性種のクローニング、単離および使用のみを報告している。さらに、マンニングは、エナンチオマー的に純粋なD−アミノ酸を触媒するBacillus sphaericus D−アミノ転移酵素または他の任意のD−アミノ転移酵素を単離または使用するいかなる手段も報告していない。
マンニングのBacillus sphaericus D−アミノ転移酵素への言及は誤りであるという証拠が、マンニングの公報の2頁に見られ、ここでマンニングはD−アミノ転移酵素DNAをプラスミドpICT113上にクローニングしたと述べている。StoddardらのJ.Mol.Biol.,第196巻:441頁および442頁(1987年)に報告されているように、プラスミドpICT113は、Bacillus sphaericus属ではなくD−アミノ転移酵素の好熱性種を担持する。この事実の重要性は、好熱性種がD−フェニルアラニンの多量の生成を効果的に触媒することができず、従って、D−フェニルアラニン酸の生成のための組換え法において役に立たないということである。
本出願の前において、Bacillus sphaericus D−アミノ転移酵素の唯一の報告はTransaminases,Christenら(編)、464頁(1985年)において見られる部分的C−末端配列である。しかしながら、本発明から明らかであるように、その部分的配列は誤りであり、Bacillus sphaericus D−アミノ転移酵素の単離において有用ではない。従って、組換え手段または他の手段によるD−アミノ酸の製造においてBacillus sphaericus D−アミノ転移酵素を報告している先行文献はない。他のD−アミノ転移酵素は単離されているが、Bacillus sphaericus種により生成されるものとは違って、D−フェニルアラニンがこれらの酵素にとって比較的乏しい基質である。Tanizawaら,J.Biol.Chem.,第264巻:2445および2449頁(1989年)。
本発明は、エナンチオマー的に純粋な天然および非天然D−アミノ酸を生成する組換え材料および方法を提供する。
発明の概要
本発明は、天然および非天然D−アミノ酸の生成のための材料および方法に関する。特に、本発明は、組換え宿主細胞を用いてD−アミノ酸を生成するための発酵法に関する。
特に、本発明は、
(a)D−アミノ転移酵素遺伝子およびL−アミノ脱アミノ酵素(デアミナーゼ:deaminase)遺伝子を細胞中に組み込む工程、
(b)細胞培地中で細胞を培養する工程、および
(c)細胞培地からD−アミノ酸を単離する工程
を含んでなる、細胞中でD−アミノ酸を生成する方法に関する。
本発明は、また、
(a)D−アミノ転移酵素遺伝子、L−アミノ脱アミノ酵素遺伝子および、フェニルピルベートの生成を増加させるための手段を細胞中に組み込む工程、
(b)細胞培地中で細胞を培養する工程、および
(c)細胞培地からD−フェニルアラニンを単離する工程
を含んでなる、細胞中でD−フェニルアラニンを生成する方法にも関する。
本発明の方法は、さらに、D−アミノ脱アミノ酵素遺伝子が非機能的になるようにD−アミノ脱アミノ酵素遺伝子突然変異種を細胞中に導入する工程を含んでよい。
本発明は、また、エネンチオマー的に純粋なD−アミノ酸の生成において用いるための組換え細胞の調製にも関する。
【図面の簡単な説明】
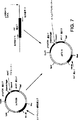
図1は、D−アミノ酸を生成するための本発明の方法を説明する一般的概略図である。
図2は、本発明の方法を用いるD−フェニルアラニンの生成を説明するスキームである。図2において、以下の略語を用いる:E4Pはエリスロース−4−ホスフェート、PEPはホスホエノールピルベート、およびDAHPは3−デオキシ−D−アラビノヘプツロソネート−7−ホスフェートである。
図3は、プラスミドpIF1002の構築を示す概略図である。
図4は、プラスミドpIF1003の構築を示す概略図である。
図5は、プラスミドpIF318の構築を示す概略図である。
図6は、プラスミドpJN326の構築を示す概略図である。
図7は、プラスミドpIF319の構築を示す概略図である。
図8は、プラスミドpIF320の構築を示す概略図である。
図9は、プラスミドpIF321の構築を示す概略図である。
図10は、プラスミドpIF333の構築を示す概略図である。
図11は、プラスミドpALR18の構築を示す概略図である。
図12は、プラスミドpPT362の構築を示す概略図である。
図13は、プラスミドpPT363の構造を示す概略図である。
図面の簡単な説明
本発明は、D−アミノ酸を生成するための材料および方法に関する。本発明の一般的方法を図1に示す。本発明は、細菌細胞中に、D−アミノ転移酵素遺伝子(dat)およびL−アミノ脱アミノ酵素遺伝子(lad)が導入される方法に関する。D−アミノ転移酵素遺伝子産物、すなわち、D−アミノ転移酵素(Dat)が、D−アミノ酸基質とケト酸前駆体との間のアミノ基転移反応を触媒する。アミノ基転移反応において、ケト酸前駆体をその対応するD−アミノ酸に転化し、D−アミノ酸基質をそのケト酸型に転化する。すなわち、D−アミノ酸基質は、アミノ基転移反応においてアミノ供与体として機能する。
L−アミノ転移酵素遺伝子産物、すなわち、L−アミノ転移酵素(Lat)は細胞中に天然に存在する。D−アミノ転移酵素遺伝子産物は、細胞中において基質としてのケト酸前駆体に関してL−アミノ転移酵素遺伝子産物と競合する。L−アミノ転移酵素は、ケト酸前駆体の状態のL−アミノ酸を形成するようなL−アミノ酸基質とケト酸前駆体との間のアミノ基転移反応を触媒する。しかしながら、L−アミノ脱アミノ酵素遺伝子を細胞中に導入されると、その遺伝子産物は、細胞中に存在する任意のL−アミノ酸のその対応するケト酸状態への脱アミノ反応を触媒する。L−アミノ酸の脱アミノ反応によりに形成されるケト酸は、D−アミノ転移酵素遺伝子により、基質として用いられるさらなるケト酸前駆体を提供する。D−アミノ転移酵素によりケト酸前駆体をその対応するD−アミノ酸型に転化することは、D−アミノ酸産物を脱アミノ化するD−アミノ脱アミノ酵素を生成するように細胞中に存在するD−アミノ脱アミノ酵素遺伝子が存在しないので、不可逆的である。
本発明の好ましい態様において、D−アミノ転移酵素およびL−アミノ転移酵素に利用できる望ましい基質を過剰に生成するために、アミノ酸基質およびケト酸前駆体の生成のための酵素をコードする遺伝子を細胞中に組み込むこともできる。組み込まれる遺伝子は、ラセミ化酵素遺伝子または、アミノ酸基質またはケト酸前駆体の生合成に関与する律速酵素をコードする遺伝子であることができる。あるいは、アミノ酸基質及び/又はケト酸前駆体を、D−アミノ酸の生成中に細胞のための培地の一部として提供することができる。基質としてL−アミノ酸またはラセミ体アミノ酸を含む細胞培地の場合、細胞培地の一部として添加されたL−アミノ酸をD−アミノ酸に転化するためのラセミ化酵素を過剰に製造するように、ラセミ化酵素遺伝子が好ましく細胞中に導入される。さらに、L−アミノ脱アミノ酵素遺伝子産物の存在は、細胞中に存在するL−アミノ酸を脱アミノ化してD−アミノ転移酵素が基質として用いるための対応するケト酸前駆体を生成させる。
本発明の方法で用いるのに好適な細胞は、限定はされないが、以下の細菌細胞を含む:Bacillus subtilis、Bacillus sphaericus、Bacillus stearothermophilus、Pseudomonas、Klebsiella、Salmonella、Brevibacterium、Micrococccus,CorynebacteriumおよびEscherichia coli。本発明のもう一つの好ましい態様において、細胞はEscherichia coliである。
本発明のもう一つの好ましい態様において、Bacillus stearothermophilus細胞の使用は、穏やかな好熱性であり、それにより、反応速度が速い高温においてD−アミノ酸の調製を行うことができるというさらなる利点を有する。従って、D−アミノ酸を調製するための製造時間が短縮される。
一つの好ましい態様において、Proteus myxofaciensからのL−アミノ脱アミノ酵素遺伝子、およびBacillus sphaericusからのD−アミノ転移酵素遺伝子が細胞に導入される。これらの遺伝子の両方が、以下の表1に示すような非常に広い基質範囲を有する酵素をコードする。基質は、天然と非天然の両方のD−およびL−アミノ酸を含む。さらに、これらの酵素の基質範囲は、標準的突然変異手段を用いてそれぞれの遺伝子を突然変異させることにより増える。

本発明の一つの好ましい態様において、好ましい宿主細胞はEscherichia coli菌株pIF3である。Escherichia coli菌株pIF3は、American Type Culture Collection,12301 Parklawn Drive,Rockville,Maryland 20852、U.S.A.から得ることのできるRY347菌株(ACTT受託番号69766)である。pIF3菌株は、Millerら著,ここで参考として取り入れる、A Short Course in Bacterial Genetics,Cold Spring Harbor Laboratory Press(1992年)に記載のように、バクテリオファージP1での形質導入によりL−アミノ転移酵素遺伝子typB+およびilvEの野生型複製が細胞中の染色体に導入されている点において、RY347と異なる。typB+およびilvE遺伝子は、ケト酸前駆体をその対応するL−アミノ酸型に転化するL−アミノ転移酵素をコードする。
野生型アミノ転移酵素遺伝子tyrB+およびilvEをpIF3細胞に再度導入することは、RY347よりも細胞成長が向上するというさらなる利益を有するが、これは、おそらくL−アミノ転移酵素遺伝子産物のなんらかの定義されていないさらなる機能によるものである。特に、好ましいL−アミノ転移酵素遺伝子は、限定はされないが、aspC、tyrBおよびilvEを含む。
本発明のD−アミノ酸の生成において用いられる細胞の染色体は、Millerら著,ここで参考として取り入れる、A Short Course in Bacterial Genetics,Cold Spring Harbor Laboratory Press(1992年)に記載のような標準的技術を用いて突然変異させることができる。一つの特別の態様において、dadA遺伝子が非機能的になるようにEscherichia coli細胞にdadA遺伝子突然変異種が導入される。Escherichia coli細胞は、遺伝子dadAおよびdadXを含むdadオペロンを有する。dadX遺伝子は、D型とL型の間でのアミノ酸のラセミ化に関与するアラニン性ラセミ化酵素をコードする。dadA遺伝子は、ある範囲のD−アミノ酸の酸化的脱アミノ反応を行うD−アミノ脱アミノ酵素をコードする。dadオペロンはD−アラニンの存在下に誘発され、D−アミノ脱アミノ酵素およびD−アラニンラセミ化酵素を生成する。DadXおよびDadA酵素は、D−アラニンの取り込みおよびピルベートへの異化に関与する膜複合体を形成する。DadA酵素は、D−フェニルアラニンのような他のD−アミノ酸を脱アミノ化することもできる。従って、D−アミノ酸の過剰生成に関与するEscherichia coli細胞において、DadA酵素の生成を防止するようにdadA遺伝子を突然変異させることが有利である。
さらに、L−アミノ転移酵素遺伝子aspC、ilvE、tyrBまたはD−アミノ脱アミノ酵素dadA遺伝子中に突然変異を含むEscheria coli菌株を、Coli Genetic Stock Center(エール大学、New Haven、CT)から得ることができる。例えば、L−アミノ転移酵素遺伝子aspC、ilvE、およびtyrB中に突然変異を有するEscherichia coli菌株DG30、DG31、DG34およびDG、およびD−アミノ脱アミノ酵素dadA遺伝子中に突然変異を有するEscherichia coli菌株EB105を、Coli Genetic Stock Centerから得ることができる。
標的遺伝子の生体外発生フラグメントを宿主細胞染色体に運び込む温度感受性組換えプラスミドを用いて部位特異的に、欠失を含む突然変異種を細胞の染色体に導入することができる。例えば、プラスミドと宿主細胞との間の組換えを認識するためにプラスミド複製制御領域の温度感受性性質を用いることができるプラスミドpHSG415が米国特許第5,354,672号に開示されている。プラスミド上の標的遺伝子の欠失複製を、pHSG415を用いて細胞の染色体上の同じ遺伝子の野生型複製に交換することができる。その後の宿主細胞からのプラスミドの損失により、標的遺伝子において細胞が突然変異される。従って、pHSG415は、宿主細胞染色体を突然変異させる、または突然変異された宿主細胞染色体中に野生型遺伝子を再導入して戻す効果的手段を提供する。
本発明の一つの好ましい態様において、細胞中にD−フェニルアラニンを生成する方法は、細胞中にD−アミノ転移酵素遺伝子およびL−アミノ脱アミノ酵素遺伝子を組み込むことを含む。D−アミノ転移酵素遺伝子産物は、D−アラニン基質とケト酸前駆体、フェニルピルベートの間のアミノ基転移反応を触媒して、D−フェニルアラニンおよびピルベートが生成される。基質D−アラニンおよびフェニルピルベートは、通常、細胞中に存在し、前者は細胞壁中への組み込まれるものであり、後者は、L−フェニルアラニン生合成の反応経路の最後の前駆体である。さらに、天然に存在するL−アミノ転移酵素遺伝子産物が、L−フェニルアラニンとピルベートを生成するようなL−アラニンとフェニルピルベートとの間のアミノ基転移反応を触媒する。しかしながら、細胞中にL−アミノ脱アミノ酵素遺伝子を導入すると、合成されたL−フェニルアラニンの大部分を脱アミンしてフェニルピルベートに戻すL−アミノ脱アミノ酵素が生成され、一方、存在するL−フェニルアラニンの残りはタンパクの生成に用いられる。脱アミノ反応の結果として生成されるフェニルピルベートは、基質としてD−アミノ転移酵素により利用されてさらにD−フェニルアラニンを生成することができる。細胞中でのD−フェニルアラニンの生成は、D−フェニルアラニンを脱アミノするようなD−アミノ脱アミノ酵素遺伝子が細胞中に存在しないので、不可逆である。
本発明の方法を用いるD−アミノ酸の製造において、アミノ基転移反応においてアミノ供与体として用いるためのD−アミノ酸基質を高水準で有することが望ましい。例えば、D−フェニルアラニンの調製において、細胞へのD−アラニンの添加により、アミノ基転移反応のためのD−アラニン基質の高水準が保証される。
本発明の好ましい態様において、アラニンのラセミ混合物が、発酵中に、細胞培養の一部として細胞に添加される。さらに、アラニンラセミ化酵素をコードする細胞質アラニンラセミ化酵素遺伝子(alr)が細胞中に導入される。アラニンラセミ化酵素が、細胞中において、50/50のD−、L−アラニン平衡を維持する。D−アミノ転移酵素の作用により細胞中でD−アラニンが消費されるので、アラニンラセミ化酵素はL−アラニンをD−アラニンに転化させる。このようにして、細胞壁中に導入される少量は別として、アミノ基転移反応においてアミノ供与体として用いるためのD−アラニン基質として、D−、L−アラニン混合物の全てがD−アミノ転移酵素遺伝子に利用される。一つの好ましい態様において、細胞中に組み込まれたalr遺伝子は、Salmonella typhimuriumからクローニングされる。
D−アミノ酸の生成中に細胞培養に添加することができる他の適当なアミノ供与体は、L−アラニン、L−グルタメート、L−フェニルアラニン、L−アスパルテート、または前記L−アミノ酸のラセミ混合物である。好ましくは、存在するアミノ供与体に依存して、グルタメートラセミ化酵素、アスパルテートラセミ化酵素またはフェニルアラニンラセミ化酵素のようなラセミ化酵素遺伝子も細胞中に組み込まれる。従って、D−アミノ転移酵素は、アミノ基転移反応に用いるのに有用なD−アミノ供与体基質を多量に有する。
細胞中でのD−フェニルアラニンの生成を増加させるために、フェニルピルベートを生成する律速酵素をコードする遺伝子を導入することにより、細胞中でケト酸前駆体、すなわちフェニルピルベートの量を増加させることができる。細胞性芳香族アミノ酸生合成経路からのフェニルピルペート生成が、二つの律速酵素、PheAおよびAroHにより制御される。PheAおよびAroHをコードする遺伝子を細胞中に導入すると、フェニルピルベートが過剰に生成される。従って、フェニルピルベートの量の増加は、D−フェニルピルベートに転化するための、D−アミノ転移酵素遺伝子産物のより多くの基質を提供する。
細胞中でのケト酸前駆体の量は、対応するL−アミノ酸を細胞に添加することにより増加させることもできる。L−アミノ酸の添加の場合、L−アミノ脱アミノ酵素がL−アミノ酸を脱アミノして対応するケト酸前駆体を形成させる。次に、ケト酸前駆体を、D−アミノ転移酵素が対応するD−アミノ酸に転化する基質として用いることができる。
本発明は、外因性D−アミノ転移酵素遺伝子および外因性L−アミノ脱アミノ酵素遺伝子を含む組換え細胞にも関する。本発明の組換え細胞は、さらに、細胞中にD−アミノ脱アミノ酵素遺伝子突然変異を含むことができ、それによりD−アミノ脱アミノ酵素遺伝子が非機能的になる。本発明の組換え細胞は、さらに、外因性アラニンラセミ化酵素遺伝子、外因性aroH遺伝子および外因性pheA遺伝子を含むことができる。外因性D−アミノ転移酵素遺伝子は、Bacillus sphaericus D−アミノ転移酵素遺伝子であってよく、外因性L−アミノ脱アミノ酵素遺伝子はProteus myxofaciens L−アミノ脱アミノ酵素遺伝子またはProteus mirabilis L−アミノ脱アミノ酵素遺伝子であってよく、外因性ラセミ化酵素遺伝子は、Salmonella typhimuriumラセミ化酵素遺伝子であってよい。
本発明の組換え細胞の培養は、エナンチオマー的に純粋なD−アミノ酸を生成するために用いられる。開示された方法を用いて生成されるL−アミノ酸に対するD−アミノ酸のエナンチオマー過剰率(ee)は、存在するD−アミノ酸の量からL−アミノ酸の量を引き、D−とL−の両方のアミノ酸の合計量で割り、100をかけることにより決められる。好ましい態様において、D−フェニルアラニンは実質的に純粋な状態で高収率で得られる。D−フェニルアラニンを生成する方法は図2に示す。
本発明の組換え細胞の培養を用い、D−アミノ転移酵素遺伝子産物のためのフェニルピルベート基質およびD−アラニンのさらなる原料としてD−、L−アラニンおよびL−フェニルアラニンを添加すると、D−フェニルアラニン13.66g/lおよびL−フェニルアラニン0.47g/lが得られ、エナンチオマー過剰率は94%であった。発酵プロセス中にD−、L−アラニンのみを培養に添加すると、D−フェニルアラニン4.15g/lが得られL−フェニルアラニンは得られず、エナンチオマー過剰率は100%であった。対照的に、発酵プロセス中にD−、L−アラニンまたはL−フェニルアラニンを細胞培養に添加しない場合、D−フェニルアラニン1.12g/lおよびL−フェニルアラニン0.47g/lが得られ、エナンチオマー過剰率は41%であった。
本発明の方法により生成したD−アミノ酸は、当業者に良く知られている手順を用いて単離することができる。例えば、開示された方法を用いて調製されたD−アミノ酸を単離する一つの方法を以下に示す。発酵完了時に、発酵ブロスを細胞から傾瀉する。D−アミノ酸産物の濃度を上げるためにブロスの体積を減らすことができる。ブロスの減少は、典型的には、減圧下にブロスを30℃〜100℃の温度に加熱することにより行われる。次に、D−アミノ酸を、ブロスのpHをアミノ酸産物の等電点から±1℃の範囲に調節することにより沈澱させる。pH調整中、D−アミノ酸産物は沈澱する。沈澱に続いて、D−アミノ酸を、濾過、遠心分離または傾瀉を含む標準的方法によりブロスから分離する。単離したD−アミノ酸産物を次に洗浄し、乾燥する。
Escherichia coli中において、アミノ酸アラニン、アスパラギン酸、グルタミン酸、フェニルアラニン、チロシン、バリン、ロイシンおよびイソロイシンが、それらのケト酸前駆体から直接合成される。発酵中に組換え細胞にL−アミノ酸またはラセミ混合物を添加することに加えて、特定のケト酸のための律速酵素を生成する遺伝子を導入することにより、所望のアミノ酸のケト酸前駆体を過剰に生成することができる。
以下の実施例は、本発明を実施する態様をより具体的にかつ詳細に説明するものである。それらは単なる例示であり、出発材料及び/又はプロセスパラメーターに小さな変化および修正を加え得ることが理解される。そのような変化がプロセスを実質的に変えない程度まで、以下の請求の範囲に記載の本発明の精神および範囲に入るものと考えられる。
実施例1
D−アミノ転移酵素DNAの単離
Bacillus sphaericusの培養物を、D−アミノ転移酵素DNAの源としてアメリカン・タイプ・カルチャー・コレクション(ATCC)(ATCC受託番号10208号)から得た。培養物を、未補足LB培地上にすじを付け、そして37℃で一晩増殖させた。染色体DNAを製造するために、単一コロニーを、使用して、1Lフラスコで50mLルリアブロスに接種し、それを300rpm、37℃で一晩振盪させた。その後、細胞を5分間、10,000Gで遠心分離することによって回収し、0.85%生理食塩水で洗浄し、そして5分間再度、10,000Gで遠心分離した。生じたペレットを、5mlの10mMグルコース、25mMトリスHCl、pH8.0、および10mMエチレンジアミンテトラ酢酸(EDTA)に再懸濁させた。アリコート量の50μlRNaseAを添加し、そして溶液を、穏やかに混合した。続いて、10mlの0.4%ドデシル硫酸ナトリウム(SDS)および100μg/mlプロテアーゼKを混合溶液に添加し、それをその後、透明になるまで37℃でインキュベートした。その後、酢酸ナトリウム(pH5.2)を、300mMの最終濃度まで添加した。穏やかなフェノール抽出を、相の界面に見ることができる白色沈澱がなくなるまで水相にほとんど等しい量のフェノールを用いて行った。その後、水相を取出し、そして染色体のDNAを2.5容積のエタノールを用いて沈澱させた。DNAペレットを、取出し、300mM酢酸ナトリウム(pH5.2)に再溶解させた。エタノール沈澱を行い、そしてDNAペレットを取出し、乾燥させ、2ml蒸留水に溶解させた。DNA濃度は、150μg/mlであると測定した。上に記述される方法に加えて、細菌のDNAの単離のための標準的方法が知られ、そして例えば、分子生物学における最新のプロトコール(Current Protocols in Molecular Biology)、2.4.1−2.4.5(Ausubelら編、1994年)で報告されており、ここに参照して組込まれる。
その後、上に記載されるとおりに得られた染色体のDNAを、MboIで部分的に消化させた。2−10kbの範囲でフラグメントを生じる理想的な消化を、13μgの染色体のDNAを用い、そして2.5MboI(ニューイングランド・バイオラボズ(New England Biolabs)マサチューセッツ州ビバリー(Beverly,MA))で40分間消化させて得た。上で示されるとおりおよそ13μgの染色体のDNAを、バイオラボズ(Biolabs)のMboI緩衝液中、37℃で100μlの総量で2.5UのMboIを用いて部分的に消化させた。17μlのサンプルを、5、10、20、30、40分に取り、そして15μlのサンプルを、50分に取った。氷上に載せたサンプルに存在するあらゆる制限酵素を破壊するため、全サンプルを65℃に加熱た。5μlアリコート量の各サンプルを、ここで参照して組込まれるSambrookら(編集)、分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)(コールド・スプリング・ハーバー・ラボラトリーズ・プレス(Cold Spring Harbor Laboratory Press):6.3−6.32(1989年)で記述されるとおりのTBE緩衝液を使用して、0.8%アガロースゲル上で電気泳動させた。電気泳動のデータから、40分で取られたサンプルは、2−10kbサイズ範囲で大部分のDNAを含有すること、およびそれが、D−アミノ転移酵素の発現のためのプラスミドpIF306でライブラリーを構築するのに使用されるそれらのフラグメントであることが決定された。
プラスミドpIF306を、pBR322(ニューイングランド・バイオラボズ(New England Biolabs)マサチューセッツ州ビバリー(Beverly,MA))から誘導した。pIF306を構築するために、修飾pheAプロモーターを、pBR322の単一のHindIIIおよびSphI部位の間に挿入した。HindIII〜SphI挿入部内に、単一のBamHIおよびBglII部位が存在する。修飾pheAプロモーターを、その配列が以下の:
その各々がMboIによって製造されたものに適合しうる末端を生じるBamHIとBglIIで、pIF306を完全に消化することによって、ベクターDNAを製造した。消化を、2時間、0.5μgのプラスミドDNAおよび2単位の各酵素を用いて、20μlの総量で、37℃で行った。4.25kbおよび1.25kbのフラグメントを生成し、1%アガロースTBEゲル上での電気泳動によって分離した。所望の4.25kbのフラグメントを、ゲルから切り取り、そしてゲル抽出キット(キアゲン社(Qiagen Inc.)、カリフォルニア州チャッツワース(Chatsworth))を用いて回収した。その後、そのフラグメントを、DNAの末端を脱リン酸化して、再環化を防ぐために、37℃で1時間、製造業者の指示にしたがってバイオラボズの緩衝液番号2番中の1単位の酵素と一緒に、20μlの容積で子牛腸のフォスファターゼ(ニューイングランド・バイオラボズ(New England Biolabs)マサチューセッツ州ビバリー(Beverly,MA))で処理した。その後、酵素を含まないDNAフラグメントを単離するために、混合物をPCR精製キット(キアゲン)で処理した。
pIF306ベクターフラグメントを、およそ20ngのベクターフラグメントを、残りのおよそ12μlの40分の部分的消化物と合わせることによって、ATCC第10208号の染色体のDNAの40分の部分的消化物(上を参照)から得たフラグメントにライゲートした。ライゲーションは、製造業者の指示にしたがってタカラ・ライゲーションキット(タカラ・バイオケミカルズ,パンベラ・コーポレーション(Takara Biochemicals,PanVera Corporation)、ウィスコンシン州マディソン(Madison,WI))を用いて達成された。ライゲーションを、17℃で2時間行い、その時に、DNAを、PCR精製キット(キアゲン)を用いて50μlの最終容量で回収した。生じたプラスミドを、25μF電気容量で2.5kvに設定したバイオ−ラッド・ジーンパルサーTMおよび200オームの抵抗に設定したバイオ−ラッドのパルス・コントローラーを使用して電気泳動にかけることによって、Escherichia coli、XL1−Bluc(ストラタゲン(Stratagene)、カリフォルニア州ラホーラ(La Jolla,CA))に導入した。
形質転換体を、50μg/mlアンピシリンで補足したLB培地の上に載せた。およそ20,000の形質転換体を、生成しそして貯蔵した。その後、プラスミドDNAを、ここで参照して組込まれる分子クローニング:実験室マニアル(Molecular Cloning:A Laboratory Manual)(Sambrookら編、2版、1989年)で報告されたとおり単離した。生じるプラスミドDNAを、25μF電気容量で2.5kvに設定したバイオ−ラッド・ジーン・パルサーTMおよび200オーム抵抗に設定したバイオ−ラッドのパルス・コントローラーを用いた電気泳動によって、Escherichia coli株WM335に組込んだ。株WM335を、オランダ国(The Netherlands)、ステイト・ユニバーシティー・オブ・ユトレヒト(State University of Utrecht)、デパートメント・オブ・モレキュラー・セル・バイオロジー(Department of Molecular Cell Biology)のファバゲン・コレクション(Phabagen Collection)から得ることができ、そしてLugtenbergら、J.Bacteriol.114巻:499−506頁(1973年)で報告され、ここに参照して組込まれた。細胞を、0.2cmのギャップでバイオラッド・ジーン・パルサーTMのキューベットでパルスにかけた。形質転換されるEscherichia coli細胞を、600nmで0.7の光学密度まで育成(50ml培養物)した。その後、細胞を、5分間10,000Gで遠心分離することによって回収し、そして30mlの脱イオン化蒸留水で洗浄した。細胞を、再回転させ、そして200μl脱イオン化蒸留水で再懸濁させ、そして40μlの細胞を、10μlの回収ライゲーションミックスと混合し、そして電気穿刺キューベット上に載せた。単一パルスを、そのキューベットにかけ、500μlSOC培地(GIBCO/BRL、Gaithersburg,MD)を添加し、そして細胞懸濁液と混合した。その後、キューベットの内容物を、20mlのpvc管に移し、そして37℃で30分間インキュベートした。その後、細胞を、適切な培地に載せ、そして以下に示すとおり選択した。DNAを微生物に形質転換/トランスフェクションさせる多数の培地が知られており、そして本発明による方法で使用できる。例えば、Changら(編集)、電気泳動および電気融合についての指針(Guide to Electroporation and Electrofusion)(アカデミック・プレス(Academic Press)、1992年)参照。
形質転換体を、50μg/mlチミンおよび60μg/mlアンピシリンを補足されるが、D−グルタメートを欠く、LB培地に載せた。D−グルタメートを作ることのできる形質転換体のもののみが、その培地で生存できる。文献中の報告により、Bacillus sphaericusが、グルタメートのラセミ体を欠くと考えられるので、そのような細胞の全ては、Bacillus sphaericusのdat遺伝子を担持する形質転換体であることが必要とされる。しかし、2つの異なるクラスの形質転換体は、上で記述された手段によって単離し、一方は、dat遺伝子を担持し、そして他方は、グルタメートのラセミ体を担持した。ラセミ体含有クローンを、pIF1001と表し、そしてdat含有クローンをpIF1002として表した。図3は、pIF1002の構築を示す概要図である。
各々の場合に、クローンを、制限エンドヌクレアーゼ消化によってマッピングし、そして遺伝子を、配列した。コードしたタンパク質のdat遺伝子の配列および推定アミノ酸配列は、配列番号:2番および3番に示される。dat遺伝子が、他の公知dat遺伝子配列のみと高度の配列相同性を示すことが分かった。Tanizawaら、J.Biol.Chem.、264巻:2450−2454頁(1989年)参照。しかし、pIF1002中のBacillus sphaericusのdat遺伝子によってコードされたD−アミノ転移酵素のC−末端アミノ酸配列は、C−末端配列のみが公表されているBacillus sphaericusのD−アミノ転移酵素の他の公表された報告のみのものと一致しなかった。Christenら(編集)、464頁(1995年)のTransaminasesで報告されたその配列は、Val−Ile−(Phe−Tyr)−Leu−Ala−Leu(配列番号:4)であった。反対に、本発明に提供されるとおりの修正C−末端配列は、Leu−Pro−Ile−Ser−Ile−Asn−Ala(配列番号:5)である。Bacillus sphaericusのD−アミノ転移酵素コード遺伝子を単離するために、Christenで報告される配列を使用することが、試みられたが、結果が得られなかった。
その後、両方のクローンを、dat遺伝子の存在についての生物学的アッセイにかけた。そのアッセイは、Methods in Enzymology、113巻:108−113頁(19)で報告され、ここで参照して組込まれた。簡便には、WM335細胞中のpIF1001またはpIF1002の培養を、50μl/mlのチミンおよび200μl/mlアンピシリンで補足された50mlのLB培地に設定した。培養物を、37℃で振盪インキュベーター内の500mlフラスコで一晩育成した。5分間、10,000Gで遠心分離することによって、細胞を収穫し、そしてpH8.5で50mMリン酸カリウムで洗浄した。細胞を、再度遠心し、そしてpH8.5で1mlの50mMリン酸カリウムに取った。その後、細胞を、1000 lbs/インチ2でフレンチ・プレッシャー・セル(French Pressure Cell)を用いて溶解させ、そして溶解物を、30分間、マイクロフュージ内で14,000Cで遠心分離し、その時、上清を、マイクロピペットで抽出した。生じた細胞抽出物を、ここで参照して組込まれた、Methods in Enzymology、113巻:108−113頁(19)で報告されたとおり乳酸デヒドロゲナーゼ結合アッセイを用いて分析にかけた。アッセイ混合物は、0.3Mリン酸カリウム、pH8.5、25mMのD−アラニン、25mMα−ケトグルタレート、0.1mM NADH、70μg/ml乳酸デヒドロゲナーゼおよび50μlの細胞抽出物を含有した。反応は、25℃で、1mlキューベット中のNADHおよび乳酸デヒドロゲナーゼを、他の成分に添加することによって開始させた。反応は、NADHの酸化の証拠として338nmでの吸収での変化を生じた。非特異的酸化について修正するために、細胞抽出物を欠くアッセイ混合物を使用する対照アッセイを行った。追加の対照として、D−アラニンを欠くアッセイ混合物を用いてアッセイを行った。未形質転換WM335細胞の抽出物および対照は、吸収で基本的に同一の変化を生じた、一方で、pTF1002を担持するWM335細胞は、対照より30倍多く過剰で吸収における変化を示した。dat含有クローンは、高いコピー数のプラスミドpIF306での過剰発現の結果であるBacillus sphaericusの抽出物より約100倍大きい活性のレベルを示した。プラスミドpIF1001は、対照のものと同一の活性を示した。
実施例2
プラスミドpIF1003の構築
プラスミドpIF1003は、プラスミドpLG338(Stokerら、Gene 18巻:355−341頁(1982年))の分配(Par)遺伝子座を担持するpIF1002の誘導体である。プラスミドpLG338の分配遺伝子座(Stokerら、Gene 18巻:355−341頁(1982年))。分配遺伝子座は、細胞分裂の間にプラスミド分配を制御し、そうすることで、プラスミドベクターで分離安定性が増加することに寄与する。それはプラスミド維持において抗生物質の選択の必要性を減少または排除させる上で有用である。分配遺伝子座は、オリゴヌクレオチドプライマー:
その後、生じる992bpフラグメントを、制限酵素SphIおよびBspEI(ニューイングランド・バイオラボズ(New England Biolabs)、マサチューセッツ州ビバリー(Beverly,MA))で消化させ、そして生じた965bpのSphIからBspEIのフラグメントを、キアクイック・ゲル抽出キット(QIAquick gel extraction kit)(キアゲン)を用いて単離し、続いて1%アガロースTBEゲルで電気泳動を行った。その後、このフラグメントを、pIF1002のBspEI開裂および部分的SphI開裂によって生じた5.8kBのDNAフラグメントにライゲートして、pIF1003を生じた。図4は、pIF1003の構築物を示す概要図である。
実施例3
プラスミドpIF321の構築
宿主細胞でD−フェニルアラニンの生成を可能にするベクターを構築するために、dat遺伝子を、PCRを用いてpIF1002から単離した。dat−コーディング領域の増幅を、0.2mlの商標マイクロアンプ(MicorAmpTM)反応試験管(パーキン−エルマー(Perkin−Elmer)、コネチカット州ノーウォーク(Norwalk,CT))中の商標アンプリタック(AmplitaqTM)PCRキット(パーキン−エルマー(Perkin−Elmer)、コネチカット州ノーウォーク(Norwalk,CT))を用いて達成し、それに100ngのpIF1002DNA(1μl)、10ナノモル/mlの濃度でプラスミド:
生じたおよそ914bpPCR産物を、BglIIおよびNcoIで消化させ、そしてその後産物を、製造業者の指示に従ってライゲーション・キット(Ligation Kit)(タカラ・バイオケミカルズ(Takara Biochemicals))を用いてpIF306の4.5kb BamHIからNcoIのフラグメントにライゲートさせた。生じるプラスミドは、pIF318と示された。pIF318の構築物は、図5に示される。
pIF319プラスミドは、ここに参照して組込まれた共同所有の米国特許第5,354,672号に開示されたpLG338プラスミドに基づき、カナマイシン耐性遺伝子を担持する強力な宿主株Escherichia coli HW857と争うことを避けるために、カナマイシン耐性マーカーをクロラムフェニコール耐性マーカーに置換えられた。プラスミドpIF319は、ここに参照して組込まれる共同所有の米国特許第5,120,837号に開示されるとおりのpheA34遺伝子、およびpLC338中の特徴的なEcoRIおよびSalI部位の間の合成オペロンでのaroH遺伝子を含む。pheA34対立遺伝子は、酵素のフェニルアラニン媒介フィードバック阻害を実質的に減少させるpheAコーディング配列中での改変を含む。それは、アテニュエーター配列を欠き、そして結合遺伝子の発現を増大させるpheAプロモーター領域の解除向性をも含む。pheA34およびproHの存在は、Escherichia coli W3110、およびあらゆるEscherichia coli、K12株でのフェニルピルベートへの経路を効果的に解除する。プラスミドpIF319も、pJN307で特徴的なBamHIおよびSalI部位の間にEscherichia coliのaroH遺伝子を導入し、続いてBamHI部位にEscherichia coliのaspCプロモーターを導入することによって、米国特許第5,120,837号に開示されるpJN307から誘導できる。aroH遺伝子を、プライマー
pIF320プラスミドを構築するために、pIF318プラスミドを、dadX遺伝子を挿入するためのBamHIおよびSphIで開裂した。上に示されたMB1810プライマーは、そのプライマーにNcoI部位を重ね合せるBamHI部位(GGATCC)を含む。それは、dadXを導入して、datおよびdadXを含む合成オペロンを形成するのに使用されるBamHI部位(および下流SphI部位)である。dadX遺伝子配列は、ジーンバンク・データベース(Genbank database)、参照コードECODADAXから得た。その配列から、PCRプライマー
その後、pIF321と示されるさらなるプラスミドを構築した。プラスミドpIF321は、HindIIIおよびSphIでpIF320を開裂し、そしてdatおよびdadX遺伝子を担持する2.1kbのフラグメントを単離することによって生成され、その後、それはpIF319の同様の開裂によって産生された9.2kbフラグメントにライゲートされた。pIF321の構築物は、図9に示される。pIF321プラスミドは、HindIII−から−SphIのフラグメント(HindIII−プロモーター−dat−dadX−SphI)で単離され、そしてEscherichia coliのトリプトファン−依存性DAHPシンターゼをコードするaroH遺伝子と一緒に、上述のpheA34対立遺伝子を含むpIF319にライゲートされたpIF320のdatおよびdadX遺伝子を含んだ。
実施例4
プラスミドpIF333の構築
プラスミドpIF333を生成するために、プラスミドpIF321を、最初に、酵素SphIおよびSalIを用いて開裂して、6.9kBおよび4.5kBのフラグメントを得た。6.9kBのフラグメントは、キアクイック(QIAquick)のゲル抽出キット(キアゲン)を用いて単離し、続いて1%アガロースTBEゲルで電気泳動にかけた。その後、このフラグメントを、pBR322(ニューイングランド・バイオラボズ(New England Biolabs)マサチューセッツ州ビバリー(Beverly,MA))のSphIおよびSalI開裂から生成された89bpのフラグメントにライゲートし、そして同様に2%アガロースTBEゲルから単離した。生じたプラスミドは、pIF333である。pIF333の構築物は、図10に示される。
実施例5
pALR18の構築
アラニン・ラセミ体をコードするalr遺伝子を、ATCCから得たSalmonella typhimurium株ATCC受託番号第19585号から単離した。alr遺伝子を、オリゴヌクレオチドプライマー:
1098bpのPCR産物を、BamHIおよびSphIで開裂し、キアクイック(QIAquick)のゲル抽出キット(キアゲン)を用いて単離し、続いて1%アガロースTBEゲルで電気泳動にかけた1082BamHIからSphIのフラグメントを生じた。その後、このフラグメントを、pIF333の5.7kBのフラグメントにライゲートして、pALR18を生成した。pALR18の構築物は、図11に示される。
実施例6
L−アミノデアミナーゼ遺伝子の単離およびpPT363プラスミドの構築
L−アミノデアミナーゼ遺伝子(lad)を、2分の延長時間および以下のオリゴヌクレオチド:
フラグメントを、酵素SphIおよびSalIによって開裂し、そして同様の開裂から産生されたpALR18の6.84kbフラグメントにライゲートした。生じたプラスミドは、pPT362と名づけられた。pPT362の構築物は、図12に示される。
プラスミドpPT363を、pPT362およびプラスミドpIF321から生成した。pPT362およびpIF321の両方は、XhoIおよびApoIで開裂した。pPT362の4.67kBフラグメントおよびpIF321の7.49kBフラグメントを単離し、そしてライゲートしてpPT363を生成した。pPT363の構築物は、図13に示される。
実施例7
株IF3の構築
Escherichia coli株pIF3は、RY347(ATCC受託番号第69766号)から誘導された。RY347は、ここに参照して組込まれる、Millerら、「細菌遺伝学での短いコース」(A Short Course in Bacterial Genetics)で記述されるとおりの標準P1形質導入方法論を用いてtyrB+に形質導入した。tyrB+形質導入体を選択することは、チロシンの栄養要求性突然変異の損失であり、同様にその株は、イソロイシンの栄養要求性突然変異の損失のために選択するilvE+に形質導入された。生じた単離物は、pIF3と示された。
実施例8
外部アミノ供与体の添加なしのD−フェニルアラニンの製造の発酵プロセス
株IF3を、プラスミドpPT363およびpIF1003で形質転換させた。形質転換IF3株を、1Lの以下の育成培地:
その株を、800−900クレット単位に育成し、そして発酵槽に接種するのに使用した。発酵槽は、バイオラフィット78−100(セント・ゲルメイン−エン・ライ、フランス国(St Germain−en Laye,France))20Lであった。以下は、発酵槽が操作される条件である。
実施例9
アミノ供与体として供給されたD−、L−アラニンの添加を伴うD−フェニルアラニンの産生のための発酵
実施例9の発酵プロセスは、以下の局面以外は実施例8の発酵プロセスと同じであった。総グルコース供給は、48時間で1976gであった。酵母抽出物を、2g/lで使用した。発酵培地は、D−、L−アラニン供給を含み、それにより総量1400mlの167g/lのD−、L−アラニンが、発酵の最初から12時間を初めとして1.9ml/分の速度で供給された。発酵により、産生されるべき4.15g/lのD−フェニルアラニンおよび0g/lのL−フェニルアラニンを生じた。
実施例10
アミノ供与体としてD−、L−アラニンの、そしてケト酸前駆体としてL−フェニルアラニンの添加を伴うD−フェニルアラニンの産生のためのプロセス
実施例10の発酵プロセスは、以下の局面以外は、実施例8と同じであった。発酵に使用される育成培地は、以下の表に列記される。
実施例11
プラスミドpPT361の構築
プラスミドpPT361を、以下のとおりpIF306から誘導した。pIF306を、酵素BamHIおよびSphIで開裂した。3.9kbフラグメントを、単離し、そして以下のオリゴヌクレオチドプライマー:
生じたベクターは、pIF307と名づけられた。プラスミドpIF307を、酵素EcoRIおよびPstIで開裂し、そして4.1kBフラグメントを単離した。これを、pLG338から得たカナマイシン耐性遺伝子を含有する、同様に開裂および精製された982bpのDNAフラグメントにライゲートした。これは、以下のオリゴヌクレオチドプライマー:
生じる開裂プラスミドは、pIF312と名づけられた。プラスミドpIF12を、EcoRIおよびBamHIで開裂し、そしてファージ・ラムダ・C1857遺伝子にライゲートし、それは、同様に開裂され、続いてテンプレートとしてラムダ・ZapIIベクター(ストラジェン(Stragene)、カリフォルニア州ラホーラ(La Jolla,CA))、および以下のオリゴヌクレオチドプライマー:
生じたプラスミドをpPT353と名づけた。その後、このプラスミドを、PstIおよびEagIで開裂し、そして3.17kbのフラグメントを単離した。これを、pIF1003の同様の開裂によって生成された同様に開裂した2.5kbフラグメントにライゲートした。生じたベクターを、単離された4.7kbフラグメントと名づけた。これを、以下のオリゴヌクレオチドリンカー
生じたプラスミドは、pPOT2と名づけられた。このプラスミドを、XhoIおよびPstIで開裂し、そして3.9kbフラグメントを単離した。これを、テンプレートとしてpIF319プラスミドDNAを、そして以下のオリゴヌクレオチドプライマーを用いたPCRによって単離されたクロラムフェニコール耐性遺伝子を含むフラグメントにライゲートした。
実施例12
Ladアミノ酸基質の測定
表1に列記された各々のアミノ酸基質を、以下の薄層クロマトグラフィー(TLC)Ladアッセイを用いたLad酵素に適切な基質であることが測定された。使用される全ての化合物は、ミズーリー州セントルイス(St.Louis,MO)のシグマ・ケミカル社(Sigma Chemical Company)から得た。
アッセイミックスは、表1に列記される10mg/mlのアミノ酸基質の内の1つ、および7.5のpHを示す100mMトリスHClを含む。アッセイミックス(2ml)を、Lad遺伝子を含有するプラスミドpPT361を含む株W3110から得た100mgの細胞ペレットに添加した。
細胞を、1L振盪フラスコ内で、37℃で200mlのLB培地(ディフコ(Difco)、ミシガン州デトロイト(Detroit,Michigan))の一晩培養から製造した。細胞を、100mMトリスHCl(pH7.5)で一度洗浄し、そして遠心分離でペレットにした。37℃で16時間、0.005mlの反応ミックスをシリカTLCプレート番号60番F−254(イーエム・サイエンス(EM Science)、オハイオ州シンシナティー(Cincinnati,OH))上にスポットを付けて、反応を行った。
以下の溶媒:水(40%);メタノール(40%);およびアセトニトリル(20%)を用いて、クロマトグラフィーを行った。TLCプレートを、空気乾燥し、そしてエタノール中の2%ニンヒドリンを噴霧し、そしてその後10分間焼いた。
表1に列記されたアミノ酸の各々をそれらの対応のケト酸に転化することを、同時クロマトグラフィーの公知水準に対するアミノ酸誘導スポットの不在によって測定した。表1に列記されたアミノ酸基質の各々が、Lad酵素に適切な基質であることが分かった。
実施例13
Datケト酸基質の測定
Dat酵素を、以下の条件下の結合酵素アッセイで表1に列記した各ケト酸基質で分析した。使用される化合物の全てを、ミズーリー州セントルイス(St.Louis,MO)のシグマ・ケミカル社(Sigma Chemical Company)から得た。
アッセイミックスは、500u/mlのDat;30mM D−アラニン;30mMケト酸基質;0.2mM NADH;および100mMトリスHClを含んだ。アッセイ混合物のpHは、8.3であった。0.85mlのアッセイミックス、0.05mlのD−ラクテート、および0.5−1.0のO.D.650でプラスミドpIF1003を含む0.1mlのW3110細胞(ATCC第27325号)を含む1mlの溶液を用いて、アッセイを行った。
細胞を、1L振盪フラスコ内で、37℃で200mlのLB培地(ディフコ(Difco)、ミシガン州デトロイト(Detroit,Michigan))の一晩培養から製造した。細胞を、100mMトリスHCl(pH7.5)で一度洗浄し、そして遠心分離し、そして水に取った。表1中のケト酸基質の各々の反応を、37℃でΔA340を測定することによって観察した。表1中のケト酸基質アッセイの各々は、Dat酵素に適切な基質であることが分かった。
【配列表】
配列番号:1
配列の長さ:95
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:1424
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:cDNA
配列の特徴
特徴を表す記号:CDS
存在位置:427..1275
配列
配列の長さ:283
配列の型:アミノ酸
トポロジー:直鎖状
配列の種類:タンパク質
配列
配列の長さ:7
配列の型:アミノ酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:タンパク質
配列
配列の長さ:7
配列の型:アミノ酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:タンパク質
配列
配列の長さ:25
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:40
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:30
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:39
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:37塩基対
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:36
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:34
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:29
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:28
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33塩基対
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:47
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:38
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:42
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:38
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:34
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:27
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:74
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:74
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:36
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:36
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:33
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
配列の長さ:41
配列の型:核酸
鎖の数:一本鎖
トポロジー:直鎖状
配列の種類:genomic DNA
配列
Claims (37)
- (a)D−アミノ基転移酵素遺伝子およびL−アミノ脱アミノ酵素遺伝子を細胞中に組み込む工程、
(b)L−アミノ酸またはそのラセミ混合物を含む細胞培地中で細胞を培養する工程、および
(c)細胞培地からD−アミノ酸を単離する工程
を含んでなる、細胞中でD−アミノ酸を生成する方法であって、
細胞が、Bacillus subtilis、Bacillus sphaericus、Bacillus stearothermophilus、Pseudomonas、Klebsiella、Salmonella、Brevibacterium、Micrococccus、CorynebacteriumおよびEscherichia coliからなる群より選択される前記方法。 - D−アミノ脱アミノ酵素遺伝子が非機能的になるように細胞中にD−アミノ脱アミノ酵素遺伝子突然変異を導入する工程をさらに含む請求項1に記載の方法。
- 細胞が、Escherichia coliである請求項1に記載の方法。
- dadA遺伝子が非機能的になるようにEscherichia coli細胞にdadA遺伝子突然変異を導入する工程をさらに含む請求項3に記載の方法。
- D−アミノ基転移酵素遺伝子がBacillus sphacricus D−アミノ基転移酵素遺伝子である請求項1に記載の方法。
- L−アミノ脱アミノ酵素遺伝子がProteus myxofaciens L−アミノ脱アミノ酵素遺伝子またはProteus mirabilis L−アミノ脱アミノ酵素遺伝子である請求項1に記載の方法。
- 細胞中にラセミ化酵素遺伝子を組み込む工程をさらに含む請求項1に記載の方法。
- ラセミ化酵素遺伝子が、アラニンラセミ化酵素、グルタメートラセミ化酵素、アスパルテートラセミ化酵素およびフェニルアラニンラセミ化酵素からなる群より選択される請求項7に記載の方法。
- ラセミ化酵素遺伝子が、アラニンラセミ化酵素である請求項8に記載の方法。
- D−アミノ酸が天然または非天然D−アミノ酸である請求項1に記載の方法。
- 天然または非天然D−アミノ酸が、イソロイシン、ロイシン、トリプトファン、チロシン、バリン、アルギニン、アスパラギン、グルタミン、メチオニン、オルニチン、セリン、ノルロイシン、ノルバリン、フェニルアラニン、ジヒドロキシフェニルアラニン、シトルリン、システイン、ヒスチジンおよびリシンからなる群より選択される請求項10に記載の方法。
- 天然D−アミノ酸がフェニルアラニンである請求項11に記載の方法。
- L−アミノ酸が、L−アラニン、L−グルタメート、L−フェニルアラニン、L−アルパルテートおよび前記L−アミノ酸のラセミ混合物からなる群から選択される請求項1に記載の方法。
- L−アミノ酸がアセパルテートである請求項13に記載の方法。
- L−アミノ酸が、イソロイシン、ロイシン、トリプトファン、チロシン、バリン、アルギニン、アスパラギン、グルタミン、メチオニン、オルニチン、セリン、ノルロイシン、ノルバリン、フェニルアラニン、ジヒドロキシフェニルアラニン、シトルリン、システイン、ヒスチジンおよびリシンからなる群より選択される請求項1に記載の方法。
- D−アミノ酸のエナンチオマー過剰率が94%より高い収量でD−アミノ酸を生成することを特徴とする請求項1の細胞の培養を用いてD−アミノ酸を調製する方法。
- エナンチオマー的に純粋なD−アミノ酸を調製する請求項16に記載の方法。
- D−アミノ基転移酵素遺伝子およびL−アミノ脱アミノ酵素遺伝子がプラスミドを用いて細胞中に組み込まれる請求項1に記載の方法。
- (a)D−アミノ基転移酵素遺伝子、L−アミノ脱アミノ酵素遺伝子および、フェニルピルビン酸の生成を増加させるためのpheA遺伝子、aroH遺伝子またはそれらの混合物を細胞中に組み込む工程、
(b)L−アミノ酸またはそのラセミ混合物を含む細胞培地中で細胞を培養する工程、および
(c)細胞培地からD−フェニルアラニンを単離する工程
を含んでなる、細胞中でD−フェニルアラニンを生成する方法であって、
細胞が、Bacillus subtilis、Bacillus sphaericus、Bacillus stearothermohilus、Pseudomonas、Klebsiella、Salmonella、Brevibacterium、Micrococccus,CorynebacteriumおよびEscherichia coliからなる群より選択される前記方法。 - D−アミノ脱アミノ酵素遺伝子が非機能的になるように細胞中にD−アミノ脱アミノ酵素遺伝子突然変異を導入する工程をさらに含む請求項19に記載の方法。
- 細胞が、Escherichia coliである請求項19に記載の方法。
- dadA遺伝子が非機能的になるようにEscherichia coli細胞にdadA遺伝子突然変異を導入する工程をさらに含む請求項21に記載の方法。
- D−アミノ基転移酵素遺伝子がBacillus sphaericus D−アミノ転移酵素遺伝子である請求項19に記載の方法。
- L−アミノ脱アミノ酵素遺伝子がProteus myxofaciens L−アミノ脱アミノ酵素遺伝子またはProteus mirabilis L−アミノ脱アミノ酵素遺伝子である請求項19に記載の方法。
- 細胞中にラセミ化酵素遺伝子を組み込む工程をさらに含む請求項19に記載の方法。
- ラセミ化酵素遺伝子が、アラニンラセミ化酵素、グルタメートラセミ化酵素、アスパルテートラセミ化酵素およびフェニルアラニンラセミ化酵素からなる群より選択される請求項25に記載の方法。
- ラセミ化酵素遺伝子が、アラニンラセミ化酵素である請求項26に記載の方法。
- L−アミノ酸が、L−アラニン、L−グルタメート、L−フェニルアラニン、L−アルパルテートおよび前記L−アミノ酸のラセミ混合物からなる群から選択される請求項19に記載の方法。
- L−アミノ酸がアセパルテートである請求項28に記載の方法。
- 培地が基質としてL−フェニルアラニンを含む請求項19に記載の方法。
- D−フェニルアラニンのエナンチオマー過剰率が94%より高い収量でD−フェニルアラニンを生成することを特徴とする請求項19の細胞の培養を用いてD−フェニルアラニン酸を調製する方法。
- エナンチオマー的に純粋なD−フェニルアラニンを調製する請求項30に記載の方法。
- D−アミノ基転移酵素遺伝子およびL−アミノ脱アミノ酵素遺伝子がプラスミドを用いて細胞中に組み込まれる請求項31に記載の方法。
- Bacillus sphaericus D−アミノ基転移酵素遺伝子およびProteus myxofaciens L−アミノ脱アミノ酵素遺伝子またはProteus mirabilis L−アミノ脱アミノ酵素遺伝子を含んでなる組換え細胞であって、
細胞が、Bacillus subtilis、Bacillus sphaericus、Bacillus stearothermophilus、Pseudomonas、Klebsiella、Salmonella、Brevibacterium、Micrococccus,CorynebacteriumおよびEscherichia coliからなる群より選択される前記方法である前記組換え細胞。 - D−アミノ脱アミノ酵素遺伝子が非機能的になるように細胞中にD−アミノ脱アミノ酵素遺伝子突然変異を更に含む請求項34に記載の組換え細胞。
- Salmonella typhimurium ラセミ化酵素遺伝子をさらに含む請求項34に記載の組換え細胞。
- Salmonella typhimurium遺伝子がアラニンラセミ化酵素である請求項36に記載の組換え細胞。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/723,896 US5728555A (en) | 1996-09-30 | 1996-09-30 | Preparation of d-amino acids by direct fermentative means |
| US08/723,896 | 1996-09-30 | ||
| PCT/US1997/017133 WO1998014602A2 (en) | 1996-09-30 | 1997-09-25 | Preparation of d-amino acids by direct fermentative means |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2001505048A JP2001505048A (ja) | 2001-04-17 |
| JP2001505048A5 JP2001505048A5 (ja) | 2005-02-10 |
| JP4548864B2 true JP4548864B2 (ja) | 2010-09-22 |
Family
ID=24908159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP51665098A Expired - Fee Related JP4548864B2 (ja) | 1996-09-30 | 1997-09-25 | 直接発酵手段によるd―アミノ酸の調製 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US5728555A (ja) |
| EP (1) | EP0937156B1 (ja) |
| JP (1) | JP4548864B2 (ja) |
| KR (1) | KR20000048776A (ja) |
| AT (1) | ATE234933T1 (ja) |
| AU (1) | AU728884B2 (ja) |
| CA (1) | CA2267205A1 (ja) |
| DE (1) | DE69720026T2 (ja) |
| WO (1) | WO1998014602A2 (ja) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6358714B1 (en) * | 1995-04-03 | 2002-03-19 | The Nutrasweet Company | Materials and methods for the production of D-phenylalanine |
| EP0859060A3 (en) * | 1997-02-14 | 2000-07-26 | DAICEL CHEMICAL INDUSTRIES, Ltd. | Method of producing D-amino acid and amine |
| WO2000023609A1 (en) * | 1998-10-19 | 2000-04-27 | Nsc Technologies Llc | Transaminase biotransformation process employing glutamic acid |
| US6365380B2 (en) * | 2000-02-23 | 2002-04-02 | Pcbu Services, Inc. | Method for stereoselectively inverting a chiral center of a chemical compound using an enzyme and a metal catalyst |
| AU2001285833A1 (en) * | 2000-07-24 | 2002-02-05 | Degussa A.G. | Process for the fermentative preparation of l-glutamic acid using coryneform bacteria |
| US8048665B2 (en) * | 2000-07-24 | 2011-11-01 | Evonik Degussa Gmbh | Nucleotide sequences encoding alanine racemase from coryneform |
| DE10046934A1 (de) * | 2000-09-21 | 2002-04-18 | Consortium Elektrochem Ind | Verfahren zur fermentativen Herstellung von nicht-proteinogenen L-Aminosäuren |
| AU2002212690A1 (en) | 2000-11-01 | 2002-05-15 | Kyowa Hakko Kogyo Co. Ltd. | Process for producing d-serine |
| US7297800B2 (en) * | 2001-12-27 | 2007-11-20 | Ajinomoto Co., Inc. | Process of producing glutamate derivatives |
| US7572607B2 (en) * | 2002-04-23 | 2009-08-11 | Cargill, Incorporated | Polypeptides and biosynthetic pathways for the production of monatin and its precursors |
| US8372989B2 (en) * | 2002-04-23 | 2013-02-12 | Cargill, Incorporated | Polypeptides and biosynthetic pathways for the production of monatin and its precursors |
| CN103525847A (zh) | 2002-08-26 | 2014-01-22 | 味之素株式会社 | 新型醛缩酶和取代α-酮酸的制备方法 |
| DE10251184A1 (de) * | 2002-11-04 | 2004-05-13 | Degussa Ag | Mutanten zur Herstellung von D-Aminosäuren |
| KR100723826B1 (ko) | 2002-12-09 | 2007-06-04 | 아지노모토 가부시키가이샤 | 변이형 d-아미노트랜스퍼라제 및 이를 사용하는 광학활성글루탐산 유도체의 제조방법 |
| US20050112260A1 (en) * | 2003-08-01 | 2005-05-26 | Cargill, Inc. | Monatin tabletop sweetener compositions and methods of making same |
| KR101191542B1 (ko) * | 2003-08-25 | 2012-10-15 | 카아길, 인코포레이팃드 | 모나틴을 포함하는 음료 조성물 및 이의 제조 방법 |
| AU2004286207B2 (en) * | 2003-10-21 | 2011-02-24 | Cargill, Incorporated | Production of monatin and monatin precursors |
| US7180370B2 (en) * | 2004-09-01 | 2007-02-20 | Micron Technology, Inc. | CMOS amplifiers with frequency compensating capacitors |
| US8158389B2 (en) * | 2005-04-20 | 2012-04-17 | Cargill, Incorporated | Products and methods for in vivo secretion of monatin |
| WO2006113897A2 (en) * | 2005-04-20 | 2006-10-26 | Cargill, Incorporated | Products and methods for in vivo secretion of monatin |
| CN101365787B (zh) | 2005-04-26 | 2014-03-05 | 嘉吉有限公司 | 用于生产莫纳甜的立体异构体和其前体的多肽和生物合成途径 |
| CA2643416C (en) * | 2006-03-07 | 2016-07-26 | Cargill, Incorporated | Aldolases, nucleic acids encoding them and methods for making and using them |
| US20090198072A1 (en) * | 2006-05-24 | 2009-08-06 | Cargill, Incorporated | Methods and systems for increasing production of equilibrium reactions |
| JP2009538145A (ja) * | 2006-05-24 | 2009-11-05 | カーギル・インコーポレイテッド | 平衡反応の収量を増加させるための方法およびシステム |
| US8133717B2 (en) | 2006-12-22 | 2012-03-13 | Richmond Chemical Corporation | Stereoinversion of amino acids in a single reactor |
| WO2008108998A2 (en) * | 2007-03-02 | 2008-09-12 | Richmond Chemical Corporation | Method to increase the yield and improve purification of products from transaminase reactions |
| KR20120098235A (ko) * | 2011-02-28 | 2012-09-05 | 이화여자대학교 산학협력단 | 비천연 아미노산 생산 균주 및 이를 이용한 비천연 아미노산의 제조 방법 |
| CN104744278A (zh) * | 2015-03-09 | 2015-07-01 | 黄冈威尔曼生物科技有限责任公司 | 一种新型l-缬氨酸的制备工艺 |
| CN108384818A (zh) * | 2018-05-18 | 2018-08-10 | 南京大学 | 一种酶法转化制备d-组氨酸的方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0785718B2 (ja) * | 1986-03-07 | 1995-09-20 | ダイセル化学工業株式会社 | D−アミノ酸の製造方法 |
| US4753883A (en) * | 1986-05-07 | 1988-06-28 | Biotechnica International, Inc. | Enzyme deregulation |
| JPS63112978A (ja) * | 1986-10-31 | 1988-05-18 | Daicel Chem Ind Ltd | エシエリチア・コリ |
| JPH01285193A (ja) * | 1988-05-12 | 1989-11-16 | Daicel Chem Ind Ltd | D−アスパラギン酸の製造方法 |
| US5120837A (en) * | 1989-09-20 | 1992-06-09 | The Nutrasweet Company | Dna encoding phe a feedback inhibition resistant enzyme analogues |
| WO1991005870A1 (en) * | 1989-10-17 | 1991-05-02 | The Rockefeller University | Enzymatic production of d-amino acids |
| US5354672A (en) * | 1992-11-24 | 1994-10-11 | Ian Fotheringham | Materials and methods for hypersecretion of amino acids |
| JP3151075B2 (ja) * | 1992-12-22 | 2001-04-03 | 協和醗酵工業株式会社 | アラニンの製造法 |
-
1996
- 1996-09-30 US US08/723,896 patent/US5728555A/en not_active Expired - Lifetime
-
1997
- 1997-09-25 KR KR1019990702764A patent/KR20000048776A/ko not_active Application Discontinuation
- 1997-09-25 AT AT97943546T patent/ATE234933T1/de not_active IP Right Cessation
- 1997-09-25 EP EP97943546A patent/EP0937156B1/en not_active Expired - Lifetime
- 1997-09-25 WO PCT/US1997/017133 patent/WO1998014602A2/en active IP Right Grant
- 1997-09-25 DE DE69720026T patent/DE69720026T2/de not_active Expired - Lifetime
- 1997-09-25 JP JP51665098A patent/JP4548864B2/ja not_active Expired - Fee Related
- 1997-09-25 AU AU44995/97A patent/AU728884B2/en not_active Ceased
- 1997-09-25 CA CA002267205A patent/CA2267205A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| CA2267205A1 (en) | 1998-04-09 |
| AU728884B2 (en) | 2001-01-18 |
| ATE234933T1 (de) | 2003-04-15 |
| WO1998014602A3 (en) | 1998-07-09 |
| WO1998014602A2 (en) | 1998-04-09 |
| DE69720026T2 (de) | 2004-01-15 |
| US5728555A (en) | 1998-03-17 |
| DE69720026D1 (de) | 2003-04-24 |
| KR20000048776A (ko) | 2000-07-25 |
| EP0937156A2 (en) | 1999-08-25 |
| JP2001505048A (ja) | 2001-04-17 |
| AU4499597A (en) | 1998-04-24 |
| EP0937156B1 (en) | 2003-03-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4548864B2 (ja) | 直接発酵手段によるd―アミノ酸の調製 | |
| JP2817400B2 (ja) | 物質の製造法 | |
| US5618716A (en) | Materials and methods for biosynthesis of serine and serine-related products | |
| KR19990022521A (ko) | L-리신의 제조 방법 | |
| JP4471402B2 (ja) | 非調節スレオニンデヒドラターゼを用いて組換え微生物によるl−イソロイシンの生成方法 | |
| JP2001136991A (ja) | 発酵法によるl−アミノ酸の製造法 | |
| JP2022185040A (ja) | 新規l-トリプトファン排出タンパク質変異体及びそれを用いてl-トリプトファンを生産する方法 | |
| JP2004049237A (ja) | 微生物株、プラスミド、微生物株の製造方法及びホスホグリセレート−ファミリーのアミノ酸の製造方法 | |
| PH12014501556B1 (en) | Microorganism able to produce l-amino acid, and method for producing l-amino acid by using same | |
| EP0983372B1 (en) | Improved transaminase biotransformation process | |
| JP4695855B2 (ja) | L−セリンによる阻害の減少を示す3−ホスホグリセレートデヒドロゲナーゼの変異体およびそれをコードする遺伝子、ならびにその使用 | |
| JP4821606B2 (ja) | 効率的にL−グルタミン酸を生産するためのプロモーター変異による細菌のsucAB発現の最適化 | |
| JP2001231584A (ja) | 変異型ilvH遺伝子及びL−バリンの製造法 | |
| US6358714B1 (en) | Materials and methods for the production of D-phenylalanine | |
| US5856148A (en) | Materials and methods for biosynthesis of serine and serine-related products | |
| US7585959B2 (en) | Feedback resistant acetohydroxy acid synthethase mutants | |
| JP2011507485A (ja) | (2s,3r,4s)−4−ヒドロキシ−l−イソロイシンの製造方法 | |
| US20230407351A1 (en) | Recombinant host cells to produce anthranilic acid | |
| US20050089974A1 (en) | Fermentative production of d-hydroxyphenylglycine and d-phenylglycine | |
| JP4162383B2 (ja) | ホモグルタミン酸の生産に関与する遺伝子およびその使用 | |
| JP2004024259A (ja) | L−ロイシンの製造方法 | |
| JP2023503218A (ja) | アセトヒドロキシ酸シンターゼ新規変異体及びこれを含む微生物 | |
| RU2268941C2 (ru) | Способ производства l-аминокислот посредством ферментации | |
| JP2003189863A (ja) | アスパラギン酸アミド並びにその誘導体の製造方法 | |
| MXPA01013445A (es) | Regulacion de la asimilacion de carbono. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20040426 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20040426 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20070814 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071113 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080108 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080404 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080519 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090929 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20091222 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100113 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100208 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100608 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100706 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130716 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |