JP4365221B2 - 親水性ポリマー−マルチカルボキシルオリゴペプチドと薬剤分子との結合生成物、医薬組成物およびその薬学上の使用 - Google Patents
親水性ポリマー−マルチカルボキシルオリゴペプチドと薬剤分子との結合生成物、医薬組成物およびその薬学上の使用 Download PDFInfo
- Publication number
- JP4365221B2 JP4365221B2 JP2003573050A JP2003573050A JP4365221B2 JP 4365221 B2 JP4365221 B2 JP 4365221B2 JP 2003573050 A JP2003573050 A JP 2003573050A JP 2003573050 A JP2003573050 A JP 2003573050A JP 4365221 B2 JP4365221 B2 JP 4365221B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- binding product
- product according
- hydrophilic polymer
- binding
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images

Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Preparation (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Polyamides (AREA)
- Polyesters Or Polycarbonates (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Other Resins Obtained By Reactions Not Involving Carbon-To-Carbon Unsaturated Bonds (AREA)
Description

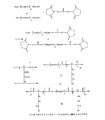
Pは、水溶性ポリマーであり、
mは、2〜12の整数であり、
jは、1〜6の整数であり、
Riは、H、C1-12のアルキル基、置換アリール基、アラルキル基、ヘテロアルキル基および置換アルキル基からなる群から選択される基であり、
Xは、架橋基であり、
Zは、OおよびNHから選択される架橋基であり、
TAは、薬剤分子である。
Rは、H又はC1-12のアルキル基であり、
nは、重合度を表す整数である。
mは、重合度を表す2〜12の整数であり、
jは、1〜6の整数であり、
Riは、H、C1-12のアルキル基、置換アリール基、アラルキル基、ヘテロアルキル基および置換アルキル基からなる群から選択される基である。
a:アミノ化
親水性ポリマーがアミノ化されると、ヒドロキシル基が、反応性により優れるアミノ基になる。前記ポリマーがカルボキシル酸基を含む分子と反応して結合生成物を産出する場合に特に重要である。
b:カルボキシル化
親水性ポリマーのカルボキシル化は、その反応性を向上し、アミノ基又はカルボキシル基を含む分子に結合することを可能とする。
c:塩化アシル、ヒドラジン、マレイミド、二硫化ピリジン等による修飾といった他の方法も、すべて同様に、適宜採用できる。
Pは、水溶性ポリマーであり、ポリエチレングリコール、ポリプロピレングリコール、ポリビニルアルコール、ポリアクリルモルホリン又はこれらの共重合体であってもよく、好ましくは、ポリエチレングリコールおよびその共重合体であり、
mは、2〜12の整数であり、
jは、1〜6の整数であり、
Riは、H、C1-12のアルキル基、置換アリール基、アラルキル基、ヘテロアルキル基および置換アルキル基からなる群から選択される基であり、
Xは、架橋基であり、好ましくは、CO、OCO、NHCO、(CH2)i、(CH2)iOCO、(CH2)iNHCOおよび(CH2)iCOであって、iは、1〜10の整数であり、
Zは、OおよびNHから選択される架橋基であり、
TAは、薬剤分子である。
本発明の結合生成物およびその調製方法について下記実施例を参照してさらに説明する。但し、これらの実施例はいかなる場合であっても本発明の範囲を制限するものではない。本発明の範囲は、請求の範囲にのみ制限される。
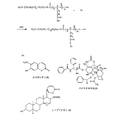
図1は、メトキシポリエチレングリコール-グルタミン酸オリゴペプチドの合成方法を示す。10 gのメトキシポリエチレングリコール(methoxypolyethylene glycol、分子量:5000)と2 gのN,N'-ジスクシンイミジルカーボネート(N, N'-disuccinimidyl carbonate)とを100 mlのアセトニトリルに溶解し、さらに、0.5 mlの乾燥ピリジンを加えた。反応混合物を窒素保護下、室温、オーバーナイトで攪拌した。過剰の溶媒を回転蒸発により除去し、その残留物を真空下で乾燥した。得られた固形物に20 mlの乾燥ジクロロメタンを加えた後、その混合物を濾過して未溶解物を除去した。その有機層を酢酸ナトリウムバッファー溶液(0.1 M, pH 5.5)で一度洗浄し、無水硫酸ナトリウムで乾燥し、濃縮した。その生成物をエーテルに移し、濾過し、真空下で乾燥した。収率:9.0 g (90%)。NMR (DMSO): 3.5 (br m, Hs of PEG), 3.24(3H, s), 4.45(2H, t), 2.82(4H, s)。
図1は、ポリエチレングリコール-ジグルタミン酸オリゴペプチドの合成方法を示す。30 gのポリエチレングリコール(分子量:35,000)と2 gのN,N'-ジスクシンイミジルカーボネートとを200 mlのアセトニトリルに溶解し、さらに、0.5 mlの乾燥ピリジンを加えた。反応混合物を窒素ガス保護下、室温、オーバーナイトで攪拌した。過剰の溶媒を回転蒸発により除去し、その残留物を真空下で乾燥した。得られた固形物に50 mlの乾燥ジクロロメタンを加えた後、その混合物を濾過して未溶解物を除去した。その有機層を酢酸ナトリウムバッファー溶液(0.1 M, pH 5.5)で一度洗浄し、無水硫酸ナトリウムで乾燥し、濃縮した。その生成物をエーテルに移し、濾過し、真空下で乾燥した。収率:27.2 g (90%)。NMR (DMSO): 3.5 (br m, Hs of PEG,), 4.45 (4H, t), 2.82 (8H, s)。
図2は、メトキシポリエチレングリコール-グルタミン酸オリゴペプチドとパクリタキセル(paclitaxel)との結合生成物(conjugate)の合成方法を示す。実施例1で調製された1.25 gのメトキシポリエチレングリコールジグルタミン酸ジペプチド、0.7 gのパクリタキセルおよび0.1 gの4-ジメチルアミノピリジン(4-dimethylamino pyridine:DMAP)を15 mlの乾燥ジクロロメタンに溶解し、そこへ、0.2 gのジシクロヘキシルカルボジイミド(dicyclohexylcarbodiimide:DCC)を加えた。その反応混合物を窒素ガス保護下、室温、オーバーナイトで攪拌した。過剰な溶媒は回転蒸発により除去し、その残留物を8 mlの1,4-ジオキサンに溶解した。この混合物を濾過し沈殿物を除去し、その溶液を濃縮した。30 mlのイソプロピルアルコールをその残留物に加え、濾過し、真空下で乾燥した。収率:1.6 g (80%)。M.p.:59〜62℃。
実施例2で調製された4.0 gのポリエチレングリコールジグルタミン酸ジペプチド、0.4 gのパクリタキセルおよび0.08 gの4-ジメチルアミノピリジン(DMAP)を20 mlの乾燥ジクロロメタンに溶解した。その後、そこへ、0.15 gのジシクロヘキシルカルボジイミド(DCC)を加えた。その反応混合物を窒素ガス保護下、室温、オーバーナイトで攪拌し、過剰な溶媒は回転蒸発により除去した。その残留物を10 mlの1,4-ジオキサンに溶解し、濾過して沈殿物を除去し、その母液を濃縮した。その残留物を50 mlのイソプロピルアルコールに加え、濾過した。その生成物を真空下で乾燥した。収率:3.7 g (85%)。M.p.:61〜64℃。
図3は、メトキシポリエチレングリコール-グルタミン酸ペプチドとカンプトテシン(camptothecin)との結合生成物の合成を示す。0.7 gのカンプトテシンと0.5 gのN-tert-ブチオキシルカルボキシルグリシン(N-tert-butyoxylcarboxylglycine:BOC-gly)とを10 mlの乾燥ジクロロメタンに溶解した。さらに、0.62 gのジシクロヘキシルカルボジイミド(DCC)と0.36 gの4-ジメチルアミノピリジン(DMAP)を加えた。その反応混合物を室温、オーバーナイトで攪拌した。反応中に形成された固形物を濾過して除去し、母液を減圧下で濃縮した。その混合物を50 mlのエーテルに加え濾過した。その沈殿物を回収し、真空下で乾燥した。
図2は、メトキシポリエチレングリコール-グルタミン酸オリゴペプチドとシノブファギン(cinobufagin)との結合生成物の合成を示す。実施例1で調製した1.0 gのメトキシポリエチレングリコールグルタミン酸ジペプチドを10 mlのジクロロメタンに溶解した。そこへ、60 mgのシノブファギン、32 mgの4-ジメチルアミノピリジン(DMAP)および40 mgのジシクロヘキシルカルボジイミド(DCC)を加えた。その反応混合物を窒素ガス保護下、室温、オーバーナイトで攪拌した。過剰な溶媒を回転蒸発により除去した。その残留物を20 mlの1,4-ジオキサンに溶解して濾過し、その母液を濃縮した。その残留物を100 mlのイソプロピルアルコールに加えて濾過した。得られた固形生成物を真空下で乾燥した。収率:0.8 g (60%)。M.p.:58〜80℃。
図3は、メトキシポリエチレングリコール-グルタミン酸オリゴペプチドとグリシレチン酸(glycyrrhetinic acid)との結合生成物の合成を示す。実施例1で調製した1.0 gのメトキシポリエチレングリコールグルタミン酸ジペプチドを10 mlのジクロロメタンに溶解した。そこへ、0.2 mlの塩化チオニルを滴下して加えた。反応混合液を2時間攪拌した。溶媒および低沸点の不純物を、減圧下の蒸留により除去した。10 mlのジクロロメタンに溶解した70 mgのグリシレチン酸溶液を攪拌しながら加えた。その後、60 mgの4-ジメチルアミノピリジン(DMAP)を加えた。その反応混合物を窒素ガス保護下、室温で12時間攪拌した。その溶媒を真空下で濃縮した。その残留物を20 mlのイソプロピルアルコールに加えて濾過した。その沈殿物を回収し、ジエチルエーテルで洗浄し、吸気乾燥し、さらに、真空下で乾燥した。その生成物は、イオン交換クロマトグラフィーで精製できる。収率:0.8 g (60%)。M.p.:60〜62℃。
図2は、メトキシポリエチレングリコール-グルタミン酸オリゴペプチドとスコポレチン(scopoletin)との結合生成物の合成を示す。実施例1で調製した5 gのメトキシポリエチレングリコールグルタミン酸ジペプチドを50 mlのジクロロメタンに溶解した。その後、0.70 gのスコポレチン、0.1 gの4-ジメチルアミノピリジン(DMAP)および0.82 gのジシクロヘキシルカルボジイミド(DCC)を加えた。その反応混合物を窒素ガス保護下、室温で12時間攪拌した。その溶媒を真空下で濃縮した。その残留物を20 mlの1,4-ジオキサンに加え、濾過した。その沈殿物を回収し、エーテルで洗浄し、吸気乾燥した。その母液を減圧下で蒸発させた。100 mlのイソプロピルアルコールをその残留物に加えた。沈殿物を回収し、ジエチルエーテルで洗浄し、真空下で乾燥した。その沈殿物を集結し、真空下で乾燥した。収率:4 g (80%)。M.p.:58〜61℃。
成分:実施例3で調製した結合生成物 ・・・2 g
0.9%食塩水 ・・・100 mlまで
実施例3で調製された結合生成物を0.9%の食塩水に溶解し、100 mlの静脈注射用溶液を得た。これを、0.2 μm膜で濾過し、無菌包装した。
Claims (17)
- 結合生成物であって、親水性ポリマー−マルチカルボキシルオリゴペプチドと薬剤分子との結合生成物であり、下記式で表される結合生成物。
Pは、ポリエチレングリコール、ポリプロピレングリコール、ポリビニルアルコール、ポリアクリルモルホリンおよびこれらの共重合体からなる群から選択される親水性ポリマーであり、
mは、2〜12の整数であり、
jは、1〜6の整数であり、
Riは、H、C1-12のアルキル基、置換アリール基、アラルキル基、ヘテロアルキル基および置換アルキル基からなる群から選択される基であり、
Xは、CO、OCO、NHCO、(CH2)i、(CH2)iOCO、(CH2)iNHCO又は(CH2)iCOであって、iが、1〜10の整数である架橋基であり、
Zは、OおよびNHから選択される架橋基であり、
TAは、薬剤分子である。 - 請求項1に記載の結合生成物であって、前記親水性ポリマーが、ポリエチレングリコールである結合生成物。
- 請求項1又は2に記載の結合生成物であって、前記ポリエチレングリコールの分子量が、300〜60,000である結合生成物。
- 請求項1から3のいずれかに記載の結合生成物であって、前記親水性ポリマーの遊離ヒドロキシル基が、C1-12のアルコキシル基、シクロアルコキシル基又はアロキシル基により置換されうる結合生成物。
- 請求項1から3のいずれかに記載の結合生成物であって、前記親水性ポリマーの遊離ヒドロキシル基が、下記式のものにより置換されうる結合生成物。
x、m、j、Ri、ZおよびTAは、請求項1における定義と同じである。 - 請求項1から5のいずれかに記載の結合生成物であって、前記親水性ポリマーが標的分子を保持し、前記結合生成物の標的を定めたデリバリーがなされ得る結合生成物。
- 請求項6に記載の結合生成物であって、前記標的分子が、抗体である結合生成物。
- 請求項1から7のいずれかに記載の結合生成物であって、前記薬剤部分TAが、アミノ酸、タンパク質、酵素、ヌクレオシド、糖類、有機酸、グルコシド、フラボノイド、キノン、テルペノイド、フェニルプロパノイドフェノール、ステロイドおよびこれらのグルコシドならびにアルカロイドからなる群から選択されるいずれか1つである結合生成物。
- 請求項8に記載の結合生成物であって、前記薬剤部分TAが、天然医薬の有効成分である結合生成物。
- 請求項9に記載の結合生成物であって、前記天然有効成分が、シノブファギン、グリシレチン酸又はスコポレチンである結合生成物。
- 請求項8に記載の結合生成物であって、前記薬剤部分TAが、抗腫瘍剤である結合生成物。
- 請求項11に記載の結合生成物であって、前記抗腫瘍剤が、パクリタキセル、カンプトテシン、ヒドロキシルカンプトテシン、エトポシドおよびこれらの誘導体からなる群から選択される抗腫瘍剤である結合生成物。
- 結合生成物であって、メトキシポリエチレングリコール−グルタミン酸オリゴペプチドと薬剤分子との結合生成物であり、下記式で表される結合生成物。
nは、10〜1200の整数であり、
mは、2〜12の整数であり、
Xは、CO、OCO、NHCO、(CH2)i、(CH2)iOCO、(CH2)iNHCO又は(CH2)iCOであって、iが、1〜10の整数である架橋基であり、
Zは、OおよびNHから選択される架橋基であり、
PTは、パクリタキセル、カンプトテシン、シノブファギン、グリシレチン酸、スコポレチンおよびこれらの誘導体からなる群から選択される薬剤である。 - 組成物であって、請求項1から13のいずれかに記載の結合生成物、および、薬学的に許容されるキャリヤ又は賦形剤を含む組成物。
- 請求項14に記載の組成物であって、さらに、他の治療上の有効成分を含む組成物。
- 請求項14に記載の組成物であって、タブレット、坐剤、ピル、ソフトおよびハードゼラチンカプセル剤、粉剤、液剤、懸濁剤、又は、エアロゾル剤の形態に形成されうる組成物。
- 製薬における、請求項1から13のいずれかに記載の結合生成物の使用。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CNB021066914A CN100475269C (zh) | 2002-03-05 | 2002-03-05 | 亲水性聚合物-谷氨酸寡肽与药物分子的结合物、包含该结合物的组合物及用途 |
| PCT/CN2003/000164 WO2003074586A1 (fr) | 2002-03-05 | 2003-03-05 | Compose d'oligopeptide de polycarboxyle/de polymere hydrophile et medicaments, composition medicale renfermant le compose susmentionne et utilisation dudit compose dans des medicaments |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005519122A JP2005519122A (ja) | 2005-06-30 |
| JP2005519122A5 JP2005519122A5 (ja) | 2005-12-22 |
| JP4365221B2 true JP4365221B2 (ja) | 2009-11-18 |
Family
ID=27768504
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003573050A Expired - Lifetime JP4365221B2 (ja) | 2002-03-05 | 2003-03-05 | 親水性ポリマー−マルチカルボキシルオリゴペプチドと薬剤分子との結合生成物、医薬組成物およびその薬学上の使用 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7498035B2 (ja) |
| EP (1) | EP1489125B1 (ja) |
| JP (1) | JP4365221B2 (ja) |
| CN (2) | CN100475269C (ja) |
| AT (1) | ATE456379T1 (ja) |
| AU (1) | AU2003221208A1 (ja) |
| DE (1) | DE60331135D1 (ja) |
| ES (1) | ES2340374T3 (ja) |
| WO (1) | WO2003074586A1 (ja) |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003221291A1 (en) | 2002-03-13 | 2003-09-22 | Beijing Jiankai Technology Co., Ltd. | Hydrophilic polymer derivate with y type branch and preparation method of it medical composite comprising above compound |
| CN1613868A (zh) * | 2003-11-07 | 2005-05-11 | 北京键凯科技有限公司 | 亲水性聚合物-黄杨木提取物的结合物及其药物组合物 |
| CN100374443C (zh) * | 2004-03-29 | 2008-03-12 | 北京键凯科技有限公司 | 亲水性聚合物-雷公藤提取物的结合物及其药物组合物 |
| CN100475837C (zh) * | 2004-06-11 | 2009-04-08 | 北京键凯科技有限公司 | 多叉分支的聚乙二醇-氨基酸寡肽及其活性衍生物和药物结合物 |
| PT1792927E (pt) * | 2004-09-22 | 2013-05-15 | Nippon Kayaku Kk | Novo copolímero em bloco, preparação de micelas e agente anticancerígeno contendo o mesmo como ingrediente activo |
| JP5249016B2 (ja) | 2006-03-28 | 2013-07-31 | 日本化薬株式会社 | タキサン類の高分子結合体 |
| US8940332B2 (en) | 2006-05-18 | 2015-01-27 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight conjugate of podophyllotoxins |
| BRPI0716812A2 (pt) * | 2006-09-15 | 2013-11-05 | Enzon Pharmaceuticals Inc | Pro drogas polimericas direcionadas contendo ligantes multifuncionais |
| WO2008041610A1 (fr) * | 2006-10-03 | 2008-04-10 | Nippon Kayaku Kabushiki Kaisha | Mélange d'un dérivé de résorcinol avec un polymère |
| CN101168594B (zh) * | 2006-10-24 | 2011-07-27 | 北京键凯科技有限公司 | 寡肽为骨架的聚乙二醇活性衍生物、其制备方法及与药物分子的结合物 |
| JP5503872B2 (ja) * | 2006-11-06 | 2014-05-28 | 日本化薬株式会社 | 核酸系代謝拮抗剤の高分子誘導体 |
| EP2090607B1 (en) * | 2006-11-08 | 2015-05-20 | Nippon Kayaku Kabushiki Kaisha | Polymeric derivative of nucleic acid metabolic antagonist |
| USRE46190E1 (en) * | 2007-09-28 | 2016-11-01 | Nippon Kayaku Kabushiki Kaisha | High-molecular weight conjugate of steroids |
| KR101589582B1 (ko) | 2008-03-18 | 2016-01-28 | 니폰 가야꾸 가부시끼가이샤 | 생리활성물질의 고분자량 결합체 |
| WO2009136572A1 (ja) | 2008-05-08 | 2009-11-12 | 日本化薬株式会社 | 葉酸若しくは葉酸誘導体の高分子結合体 |
| JP2011162569A (ja) * | 2008-05-23 | 2011-08-25 | Nano Career Kk | カンプトテシン高分子誘導体及びその用途 |
| ITRM20080551A1 (it) * | 2008-10-15 | 2010-04-16 | Univ Catania | Derivati anfifilici del poliossietilenglicole (peg), procedimento di preparazione e loro usi nella preparazione di sistemi farmaceutici. |
| EP2431403B1 (en) * | 2009-05-15 | 2016-09-28 | Nipponkayaku Kabushikikaisha | Polymer conjugate of bioactive substance having hydroxy group |
| JP2013234207A (ja) * | 2010-08-31 | 2013-11-21 | Jsr Corp | 新規重合体及び新規n−カルボキシアミノ酸無水物、ならびにそれらの製造方法 |
| EP2641605B1 (en) | 2010-11-17 | 2018-03-07 | Nippon Kayaku Kabushiki Kaisha | Polymer derivative of cytidine metabolism antagonist |
| EP2754682B1 (en) | 2011-09-11 | 2017-06-07 | Nippon Kayaku Kabushiki Kaisha | Method for manufacturing block copolymer |
| CN103083680B (zh) * | 2011-11-07 | 2014-12-24 | 北京键凯科技有限公司 | 聚乙二醇-氨基酸寡肽-依诺替康药物结合物及其药物组合物 |
| WO2014093343A2 (en) * | 2012-12-10 | 2014-06-19 | Massachusetts Institute Of Technology | Multistage nanoparticle drug delivery system for the treatment of solid tumors |
| WO2014114262A1 (zh) * | 2013-01-28 | 2014-07-31 | 天津键凯科技有限公司 | 水溶性聚合物-氨基酸寡肽-药物结合物及其制备方法和用途 |
| US9700633B2 (en) | 2013-01-28 | 2017-07-11 | Jenkem Technology Co., Ltd., Tianjin Branch | Conjugates of water soluble polymer-amino acid oligopeptide-drug, preparation method and use thereof |
| WO2015081821A1 (zh) * | 2013-12-02 | 2015-06-11 | 北京键凯科技有限公司 | 聚乙二醇-多爪寡肽键合的雷帕霉素衍生物 |
| CN107033305B (zh) * | 2017-05-20 | 2019-03-01 | 西南大学 | 一种还原性响应的两亲性蠕虫状单分子前药的制备方法 |
| CN114948850B (zh) * | 2022-05-26 | 2024-02-27 | 中国药科大学 | 可载药寡肽及其可溶性分层微针的制备方法和应用 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6096605A (ja) * | 1983-10-31 | 1985-05-30 | Mitsubishi Chem Ind Ltd | 低分子量の水溶性物質の分離方法 |
| DE3730797A1 (de) * | 1987-09-14 | 1989-03-23 | Siegel Rolf | Materialien mit nicht-thrombogener oberflaeche |
| AU6138396A (en) * | 1995-06-26 | 1997-01-30 | Daiichi Pure Chemicals Co., Ltd. | Stable plasmin solution |
| US5672662A (en) * | 1995-07-07 | 1997-09-30 | Shearwater Polymers, Inc. | Poly(ethylene glycol) and related polymers monosubstituted with propionic or butanoic acids and functional derivatives thereof for biotechnical applications |
| EP0932399B1 (en) * | 1996-03-12 | 2006-01-04 | PG-TXL Company, L.P. | Water soluble paclitaxel prodrugs |
| CN1110681C (zh) * | 1996-04-10 | 2003-06-04 | 丰田自动车株式会社 | 有色金属熔化保持炉与有色金属熔液保持炉 |
| US6824766B2 (en) * | 1998-04-17 | 2004-11-30 | Enzon, Inc. | Biodegradable high molecular weight polymeric linkers and their conjugates |
| US7060708B2 (en) * | 1999-03-10 | 2006-06-13 | New River Pharmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| AU2001228742A1 (en) * | 2000-02-04 | 2001-08-14 | Supratek Pharma, Inc. | Ligand for vascular endothelial growth factor receptor |
| CN1376785A (zh) * | 2001-03-22 | 2002-10-30 | 上海博德基因开发有限公司 | 一种多肽——硫氨焦磷酸激酶-26.73和编码这种多肽的多核苷酸 |
| US6489153B1 (en) * | 2001-10-31 | 2002-12-03 | Pe Corporation (Ny) | Isolated human kinase proteins, nucleic acid molecules encoding human kinase proteins, and uses thereof |
| KR101057102B1 (ko) * | 2002-10-31 | 2011-08-16 | 니폰 가야꾸 가부시끼가이샤 | 캄프토테신의 고분자량 유도체 |
-
2002
- 2002-03-05 CN CNB021066914A patent/CN100475269C/zh not_active Expired - Lifetime
-
2003
- 2003-03-05 EP EP03711787A patent/EP1489125B1/en not_active Expired - Lifetime
- 2003-03-05 ES ES03711787T patent/ES2340374T3/es not_active Expired - Lifetime
- 2003-03-05 AU AU2003221208A patent/AU2003221208A1/en not_active Abandoned
- 2003-03-05 WO PCT/CN2003/000164 patent/WO2003074586A1/zh active Application Filing
- 2003-03-05 JP JP2003573050A patent/JP4365221B2/ja not_active Expired - Lifetime
- 2003-03-05 AT AT03711787T patent/ATE456379T1/de not_active IP Right Cessation
- 2003-03-05 US US10/506,524 patent/US7498035B2/en not_active Expired - Lifetime
- 2003-03-05 DE DE60331135T patent/DE60331135D1/de not_active Expired - Lifetime
- 2003-12-31 CN CNB2003801109158A patent/CN100400657C/zh not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| CN100475269C (zh) | 2009-04-08 |
| DE60331135D1 (de) | 2010-03-18 |
| CN100400657C (zh) | 2008-07-09 |
| EP1489125A4 (en) | 2006-09-13 |
| AU2003221208A1 (en) | 2003-09-16 |
| US20050147617A1 (en) | 2005-07-07 |
| ATE456379T1 (de) | 2010-02-15 |
| WO2003074586A1 (fr) | 2003-09-12 |
| JP2005519122A (ja) | 2005-06-30 |
| EP1489125B1 (en) | 2010-01-27 |
| CN1442440A (zh) | 2003-09-17 |
| US7498035B2 (en) | 2009-03-03 |
| EP1489125A1 (en) | 2004-12-22 |
| ES2340374T3 (es) | 2010-06-02 |
| CN1886500A (zh) | 2006-12-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4365221B2 (ja) | 親水性ポリマー−マルチカルボキシルオリゴペプチドと薬剤分子との結合生成物、医薬組成物およびその薬学上の使用 | |
| JP4272537B2 (ja) | Y型分鎖親水性ポリマー誘導体、それらの調製方法、前記誘導体および薬剤分子の結合生成物、ならびに前記結合生成物を含む医薬組成物 | |
| WO2005121181A1 (fr) | Polyethyleneglycol-oligopeptides de type branchement d'arbre multipoint et leurs derives actifs et composes | |
| JP5544357B2 (ja) | 水酸基を有する生理活性物質の高分子結合体 | |
| CN103755949B (zh) | 多臂聚乙二醇衍生物及其与药物的结合物和凝胶 | |
| JP4339399B2 (ja) | 高分子量のポリマーを基剤とするプロドラッグ | |
| WO2010060260A1 (zh) | 新型的多臂聚乙二醇及其制备方法和应用 | |
| JP6790133B2 (ja) | マルチアームポリエチレングリコール及びその活性誘導体 | |
| US11180615B2 (en) | Cationic polyphosphazene compound, polyphosphazenes-drug conjugate compound and method for preparing same | |
| WO2005092898A1 (fr) | Conjugue d'extraits de tripterydium a polymere hydrophile et leurs compositions pharmaceutiques | |
| ES2659778T3 (es) | Conjugado farmacéutico de polietilenglicol-oligopéptido de aminoácidos-irinotecán y composición farmacéutica del mismo | |
| US20050220753A1 (en) | Hydrophilic polymers-flavoids conjugates and pharmaceutical compositions comprising them | |
| US7708978B2 (en) | Targeted hydrophilic polymer, binders with interferon and medical composite comprising above binders | |
| WO2005044837A1 (fr) | Conjugues d'extraits de buxus et de polymere hydrophile et compositions pharmaceutiques correspondantes | |
| CN1756758A (zh) | 新型的斑蝥胺和去甲斑蝥胺衍生物及其在医药中的应用 | |
| CN1556801A (zh) | 亲水性聚合物-黄酮结合物以及包含该结合物的药物组合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050401 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050401 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081127 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090227 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090409 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090703 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090728 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090820 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120828 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4365221 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130828 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |