JP4171069B2 - 非環状ヌクレオシド誘導体 - Google Patents
非環状ヌクレオシド誘導体 Download PDFInfo
- Publication number
- JP4171069B2 JP4171069B2 JP52927997A JP52927997A JP4171069B2 JP 4171069 B2 JP4171069 B2 JP 4171069B2 JP 52927997 A JP52927997 A JP 52927997A JP 52927997 A JP52927997 A JP 52927997A JP 4171069 B2 JP4171069 B2 JP 4171069B2
- Authority
- JP
- Japan
- Prior art keywords
- butyl
- guanine
- valyloxy
- acid
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- -1 Acyclic nucleoside derivatives Chemical class 0.000 title claims description 224
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 claims description 277
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 145
- 150000001875 compounds Chemical class 0.000 claims description 115
- 150000003839 salts Chemical class 0.000 claims description 27
- 229920006395 saturated elastomer Polymers 0.000 claims description 15
- 239000003795 chemical substances by application Substances 0.000 claims description 12
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 11
- ACBSZTQIFTYFGH-IAPPQJPRSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-amino-6-oxo-3h-purin-9-yl)methyl]butyl] octadecanoate Chemical compound N1C(N)=NC(=O)C2=C1N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCCCCCC)C=N2 ACBSZTQIFTYFGH-IAPPQJPRSA-N 0.000 claims description 9
- 208000036142 Viral infection Diseases 0.000 claims description 6
- 239000003085 diluting agent Substances 0.000 claims description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims description 6
- 230000009385 viral infection Effects 0.000 claims description 6
- 230000002265 prevention Effects 0.000 claims description 5
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 claims description 4
- 206010038997 Retroviral infections Diseases 0.000 claims description 4
- YZMDTCUUDPCFHM-OFSOJUDTSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] octadecanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCCCCCC)C=NC2=C1 YZMDTCUUDPCFHM-OFSOJUDTSA-N 0.000 claims description 4
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 4
- 208000031886 HIV Infections Diseases 0.000 claims description 3
- 206010019973 Herpes virus infection Diseases 0.000 claims description 3
- 241000701044 Human gammaherpesvirus 4 Species 0.000 claims description 2
- 241000701027 Human herpesvirus 6 Species 0.000 claims description 2
- 241000713772 Human immunodeficiency virus 1 Species 0.000 claims description 2
- 241000713340 Human immunodeficiency virus 2 Species 0.000 claims description 2
- JWNMJTOKXBWOSA-IAPPQJPRSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-amino-6-oxo-3h-purin-9-yl)butyl] octadecanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCCCCCC)C=N2 JWNMJTOKXBWOSA-IAPPQJPRSA-N 0.000 claims description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims 5
- 241000701085 Human alphaherpesvirus 3 Species 0.000 claims 1
- 241000700584 Simplexvirus Species 0.000 claims 1
- 230000000069 prophylactic effect Effects 0.000 claims 1
- 230000001225 therapeutic effect Effects 0.000 claims 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 104
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 102
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 66
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 62
- 238000006243 chemical reaction Methods 0.000 description 62
- 239000000047 product Substances 0.000 description 59
- 239000000243 solution Substances 0.000 description 59
- SCBFBAWJWLXVHS-ZCFIWIBFSA-N 2-amino-9-[(2r)-4-hydroxy-2-(hydroxymethyl)butyl]-3h-purin-6-one Chemical compound N1C(N)=NC(=O)C2=C1N(C[C@H](CO)CCO)C=N2 SCBFBAWJWLXVHS-ZCFIWIBFSA-N 0.000 description 58
- 239000000203 mixture Substances 0.000 description 58
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 55
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 46
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 46
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 46
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 39
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 38
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 33
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 30
- 238000000034 method Methods 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 30
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 29
- 229940024606 amino acid Drugs 0.000 description 26
- 235000002639 sodium chloride Nutrition 0.000 description 26
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 239000007787 solid Substances 0.000 description 25
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 24
- 235000001014 amino acid Nutrition 0.000 description 24
- 150000002148 esters Chemical class 0.000 description 23
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 23
- 239000000194 fatty acid Substances 0.000 description 22
- 239000002253 acid Substances 0.000 description 20
- 235000019439 ethyl acetate Nutrition 0.000 description 20
- 239000012442 inert solvent Substances 0.000 description 20
- 235000014113 dietary fatty acids Nutrition 0.000 description 19
- 229930195729 fatty acid Natural products 0.000 description 19
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 18
- 230000000052 comparative effect Effects 0.000 description 18
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 17
- 238000002360 preparation method Methods 0.000 description 17
- 238000004519 manufacturing process Methods 0.000 description 16
- 235000019441 ethanol Nutrition 0.000 description 15
- 239000002904 solvent Substances 0.000 description 15
- WTBAHSZERDXKKZ-UHFFFAOYSA-N octadecanoyl chloride Chemical compound CCCCCCCCCCCCCCCCCC(Cl)=O WTBAHSZERDXKKZ-UHFFFAOYSA-N 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 239000012043 crude product Substances 0.000 description 13
- 150000004665 fatty acids Chemical class 0.000 description 13
- 238000004128 high performance liquid chromatography Methods 0.000 description 13
- 229960004295 valine Drugs 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000000706 filtrate Substances 0.000 description 12
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 12
- 239000004367 Lipase Substances 0.000 description 11
- 102000004882 Lipase Human genes 0.000 description 11
- 108090001060 Lipase Proteins 0.000 description 11
- 238000005917 acylation reaction Methods 0.000 description 11
- 150000001413 amino acids Chemical class 0.000 description 11
- 229940040461 lipase Drugs 0.000 description 11
- 235000019421 lipase Nutrition 0.000 description 11
- 125000006239 protecting group Chemical group 0.000 description 11
- 239000004474 valine Substances 0.000 description 11
- 238000009472 formulation Methods 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M sodium bicarbonate Substances [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 239000000460 chlorine Substances 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 230000010933 acylation Effects 0.000 description 8
- 229910052739 hydrogen Inorganic materials 0.000 description 8
- 239000001257 hydrogen Substances 0.000 description 8
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 8
- 239000002777 nucleoside Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- 125000000547 substituted alkyl group Chemical group 0.000 description 8
- SZXBQTSZISFIAO-ZETCQYMHSA-N (2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoic acid Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)OC(C)(C)C SZXBQTSZISFIAO-ZETCQYMHSA-N 0.000 description 7
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 7
- 125000002015 acyclic group Chemical group 0.000 description 7
- 125000002252 acyl group Chemical group 0.000 description 7
- 125000000217 alkyl group Chemical group 0.000 description 7
- 150000008064 anhydrides Chemical class 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 239000003054 catalyst Substances 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 229940002612 prodrug Drugs 0.000 description 7
- 239000000651 prodrug Substances 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 229960004150 aciclovir Drugs 0.000 description 6
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 6
- 239000003443 antiviral agent Substances 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 238000005886 esterification reaction Methods 0.000 description 6
- IRSCQMHQWWYFCW-UHFFFAOYSA-N ganciclovir Chemical compound O=C1NC(N)=NC2=C1N=CN2COC(CO)CO IRSCQMHQWWYFCW-UHFFFAOYSA-N 0.000 description 6
- 229960002963 ganciclovir Drugs 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 229910000027 potassium carbonate Inorganic materials 0.000 description 6
- 235000011181 potassium carbonates Nutrition 0.000 description 6
- 239000000741 silica gel Substances 0.000 description 6
- 229910002027 silica gel Inorganic materials 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 239000007858 starting material Substances 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- CVVBPBHBMYSGCY-UHFFFAOYSA-N 4-chloropurin-2-amine Chemical compound C1=NC(N)=NC2(Cl)N=CN=C21 CVVBPBHBMYSGCY-UHFFFAOYSA-N 0.000 description 5
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 5
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 5
- 125000001931 aliphatic group Chemical group 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 230000032050 esterification Effects 0.000 description 5
- 239000012065 filter cake Substances 0.000 description 5
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 5
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 5
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 239000002002 slurry Substances 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- 210000002700 urine Anatomy 0.000 description 5
- QJCNLJWUIOIMMF-YUMQZZPRSA-N (2s,3s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]pentanoic acid Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)OC(C)(C)C QJCNLJWUIOIMMF-YUMQZZPRSA-N 0.000 description 4
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 4
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 4
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical class OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- YAASAQVLXCSMDE-PSXMRANNSA-N [(2r)-2-[(2-amino-6-oxo-3h-purin-9-yl)methyl]-4-[tert-butyl(diphenyl)silyl]oxybutyl] octadecanoate Chemical compound C([C@@H](COC(=O)CCCCCCCCCCCCCCCCC)CN1C2=C(C(NC(N)=N2)=O)N=C1)CO[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 YAASAQVLXCSMDE-PSXMRANNSA-N 0.000 description 4
- MBPKKIHHVGAEAW-HSZRJFAPSA-N [(2r)-2-[(2-amino-6-oxo-3h-purin-9-yl)methyl]-4-hydroxybutyl] octadecanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@@H](CCO)COC(=O)CCCCCCCCCCCCCCCCC)C=N2 MBPKKIHHVGAEAW-HSZRJFAPSA-N 0.000 description 4
- KABOZVVUDJJHLI-HSZRJFAPSA-N [(3r)-3-[(2-amino-6-oxo-3h-purin-9-yl)methyl]-4-hydroxybutyl] octadecanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@H](CO)CCOC(=O)CCCCCCCCCCCCCCCCC)C=N2 KABOZVVUDJJHLI-HSZRJFAPSA-N 0.000 description 4
- 235000019445 benzyl alcohol Nutrition 0.000 description 4
- 238000010511 deprotection reaction Methods 0.000 description 4
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 4
- 229910052736 halogen Inorganic materials 0.000 description 4
- 150000002367 halogens Chemical class 0.000 description 4
- 238000005984 hydrogenation reaction Methods 0.000 description 4
- 239000013067 intermediate product Substances 0.000 description 4
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 4
- 229950003188 isovaleryl diethylamide Drugs 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 239000012074 organic phase Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 239000000523 sample Substances 0.000 description 4
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 4
- 238000000825 ultraviolet detection Methods 0.000 description 4
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 description 3
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 3
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 208000007514 Herpes zoster Diseases 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 3
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 3
- 235000019483 Peanut oil Nutrition 0.000 description 3
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 3
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 239000003638 chemical reducing agent Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 230000020176 deacylation Effects 0.000 description 3
- 238000005947 deacylation reaction Methods 0.000 description 3
- ZQPPMHVWECSIRJ-MDZDMXLPSA-N elaidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCC(O)=O ZQPPMHVWECSIRJ-MDZDMXLPSA-N 0.000 description 3
- 230000002255 enzymatic effect Effects 0.000 description 3
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 125000002883 imidazolyl group Chemical group 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- 239000000312 peanut oil Substances 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- 125000002987 valine group Chemical group [H]N([H])C([H])(C(*)=O)C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- CANZBRDGRHNSGZ-NSHDSACASA-N (2s)-3-methyl-2-(phenylmethoxycarbonylamino)butanoic acid Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)OCC1=CC=CC=C1 CANZBRDGRHNSGZ-NSHDSACASA-N 0.000 description 2
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- SYTBZMRGLBWNTM-SNVBAGLBSA-N (R)-flurbiprofen Chemical compound FC1=CC([C@H](C(O)=O)C)=CC=C1C1=CC=CC=C1 SYTBZMRGLBWNTM-SNVBAGLBSA-N 0.000 description 2
- MLQBTMWHIOYKKC-MDZDMXLPSA-N (e)-octadec-9-enoyl chloride Chemical compound CCCCCCCC\C=C\CCCCCCCC(Cl)=O MLQBTMWHIOYKKC-MDZDMXLPSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical compound CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- YQTCQNIPQMJNTI-UHFFFAOYSA-N 2,2-dimethylpropan-1-one Chemical group CC(C)(C)[C]=O YQTCQNIPQMJNTI-UHFFFAOYSA-N 0.000 description 2
- ZIIUUSVHCHPIQD-UHFFFAOYSA-N 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound CC1=CC(C)=CC(C)=C1S(=O)(=O)NC1=CC=CC(C(F)(F)F)=C1 ZIIUUSVHCHPIQD-UHFFFAOYSA-N 0.000 description 2
- QVSXOTGFWFGTKX-LJQANCHMSA-N 2-amino-9-[(2r)-4-[tert-butyl(diphenyl)silyl]oxy-2-(hydroxymethyl)butyl]-3h-purin-6-one Chemical compound C([C@@H](CO)CN1C2=C(C(NC(N)=N2)=O)N=C1)CO[Si](C(C)(C)C)(C=1C=CC=CC=1)C1=CC=CC=C1 QVSXOTGFWFGTKX-LJQANCHMSA-N 0.000 description 2
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 2
- HDECRAPHCDXMIJ-UHFFFAOYSA-N 2-methylbenzenesulfonyl chloride Chemical compound CC1=CC=CC=C1S(Cl)(=O)=O HDECRAPHCDXMIJ-UHFFFAOYSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- UNSAJINGUOTTRA-UHFFFAOYSA-N 3-(3-bromophenyl)prop-2-yn-1-ol Chemical compound OCC#CC1=CC=CC(Br)=C1 UNSAJINGUOTTRA-UHFFFAOYSA-N 0.000 description 2
- QGXQKGLWWDUOEY-UHFFFAOYSA-N 3-n-hydroxybicyclo[2.2.1]hept-5-ene-2,3-dicarboxamide Chemical compound C1C2C=CC1C(C(=O)N)C2C(=O)NO QGXQKGLWWDUOEY-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical class OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- 125000000590 4-methylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 2
- YYROPELSRYBVMQ-UHFFFAOYSA-N 4-toluenesulfonyl chloride Chemical compound CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- 241000282567 Macaca fascicularis Species 0.000 description 2
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- JNTOCHDNEULJHD-UHFFFAOYSA-N Penciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(CCC(CO)CO)C=N2 JNTOCHDNEULJHD-UHFFFAOYSA-N 0.000 description 2
- 102000011420 Phospholipase D Human genes 0.000 description 2
- 108090000553 Phospholipase D Proteins 0.000 description 2
- 102000015439 Phospholipases Human genes 0.000 description 2
- 108010064785 Phospholipases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- IULLBJGQDWUJMH-ZJUUUORDSA-N [(2r)-2-[(2-amino-6-oxo-3h-purin-9-yl)methyl]-4-hydroxybutyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@@H](CCO)COC(=O)[C@@H](N)C(C)C)C=N2 IULLBJGQDWUJMH-ZJUUUORDSA-N 0.000 description 2
- ZPIWQKRPFIDOCG-VEEOACQBSA-N [(3r)-3-[(2-amino-6-oxo-3h-purin-9-yl)methyl]-4-[(2s)-3-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]butanoyl]oxybutyl] octadecanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@H](COC(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)C)CCOC(=O)CCCCCCCCCCCCCCCCC)C=N2 ZPIWQKRPFIDOCG-VEEOACQBSA-N 0.000 description 2
- VMXFMYKLBRDSPN-RDBSUJKOSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-amino-6-oxo-3h-purin-9-yl)butyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)NC(=O)C2=C1N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)[C@@H](N)C(C)C)C=N2 VMXFMYKLBRDSPN-RDBSUJKOSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 125000006241 alcohol protecting group Chemical group 0.000 description 2
- 235000008206 alpha-amino acids Nutrition 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- 230000003602 anti-herpes Effects 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- CSKNSYBAZOQPLR-UHFFFAOYSA-N benzenesulfonyl chloride Chemical compound ClS(=O)(=O)C1=CC=CC=C1 CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- JBDSSBMEKXHSJF-UHFFFAOYSA-N cyclopentanecarboxylic acid Chemical compound OC(=O)C1CCCC1 JBDSSBMEKXHSJF-UHFFFAOYSA-N 0.000 description 2
- IPIVAXLHTVNRBS-UHFFFAOYSA-N decanoyl chloride Chemical compound CCCCCCCCCC(Cl)=O IPIVAXLHTVNRBS-UHFFFAOYSA-N 0.000 description 2
- 230000001627 detrimental effect Effects 0.000 description 2
- 150000005690 diesters Chemical class 0.000 description 2
- GGSUCNLOZRCGPQ-UHFFFAOYSA-N diethylaniline Chemical compound CCN(CC)C1=CC=CC=C1 GGSUCNLOZRCGPQ-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- UKMSUNONTOPOIO-UHFFFAOYSA-N docosanoic acid Chemical compound CCCCCCCCCCCCCCCCCCCCCC(O)=O UKMSUNONTOPOIO-UHFFFAOYSA-N 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000003255 drug test Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 238000003304 gavage Methods 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000002140 halogenating effect Effects 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 208000015181 infectious disease Diseases 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 150000002519 isoleucine derivatives Chemical class 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 229940011051 isopropyl acetate Drugs 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical class CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 235000021313 oleic acid Nutrition 0.000 description 2
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 2
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- 125000003170 phenylsulfonyl group Chemical group C1(=CC=CC=C1)S(=O)(=O)* 0.000 description 2
- ZJAOAACCNHFJAH-UHFFFAOYSA-N phosphonoformic acid Chemical compound OC(=O)P(O)(O)=O ZJAOAACCNHFJAH-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- GRJJQCWNZGRKAU-UHFFFAOYSA-N pyridin-1-ium;fluoride Chemical compound F.C1=CC=NC=C1 GRJJQCWNZGRKAU-UHFFFAOYSA-N 0.000 description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000006798 ring closing metathesis reaction Methods 0.000 description 2
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 2
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 150000003871 sulfonates Chemical class 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 150000003461 sulfonyl halides Chemical class 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- MHYGQXWCZAYSLJ-UHFFFAOYSA-N tert-butyl-chloro-diphenylsilane Chemical compound C=1C=CC=CC=1[Si](Cl)(C(C)(C)C)C1=CC=CC=C1 MHYGQXWCZAYSLJ-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N thiocyanic acid Chemical compound SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 2
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical class CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 2
- 238000001291 vacuum drying Methods 0.000 description 2
- 229920001567 vinyl ester resin Polymers 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- KWMLJOLKUYYJFJ-GASJEMHNSA-N (2xi)-D-gluco-heptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C(O)=O KWMLJOLKUYYJFJ-GASJEMHNSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 description 1
- ADFXKUOMJKEIND-UHFFFAOYSA-N 1,3-dicyclohexylurea Chemical compound C1CCCCC1NC(=O)NC1CCCCC1 ADFXKUOMJKEIND-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 1
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 1
- LHJGJYXLEPZJPM-UHFFFAOYSA-N 2,4,5-trichlorophenol Chemical compound OC1=CC(Cl)=C(Cl)C=C1Cl LHJGJYXLEPZJPM-UHFFFAOYSA-N 0.000 description 1
- HKZYWLNVGYRMDG-UHFFFAOYSA-N 2-(2,2-diethoxyethyl)propane-1,3-diol Chemical compound CCOC(OCC)CC(CO)CO HKZYWLNVGYRMDG-UHFFFAOYSA-N 0.000 description 1
- IBAOFQIOOBQLHE-UHFFFAOYSA-N 2-amino-3,9-dihydropurin-9-ium-6-one;chloride Chemical compound Cl.N1C(N)=NC(=O)C2=C1N=CN2 IBAOFQIOOBQLHE-UHFFFAOYSA-N 0.000 description 1
- LILXDMFJXYAKMK-UHFFFAOYSA-N 2-bromo-1,1-diethoxyethane Chemical compound CCOC(CBr)OCC LILXDMFJXYAKMK-UHFFFAOYSA-N 0.000 description 1
- JBMBVWROWJGFMG-UHFFFAOYSA-N 2-chloro-7h-purine Chemical compound ClC1=NC=C2NC=NC2=N1 JBMBVWROWJGFMG-UHFFFAOYSA-N 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical class C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- 125000002672 4-bromobenzoyl group Chemical group BrC1=CC=C(C(=O)*)C=C1 0.000 description 1
- RJWBTWIBUIGANW-UHFFFAOYSA-N 4-chlorobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Cl)C=C1 RJWBTWIBUIGANW-UHFFFAOYSA-N 0.000 description 1
- 125000000242 4-chlorobenzoyl group Chemical group ClC1=CC=C(C(=O)*)C=C1 0.000 description 1
- CNPURSDMOWDNOQ-UHFFFAOYSA-N 4-methoxy-7h-pyrrolo[2,3-d]pyrimidin-2-amine Chemical compound COC1=NC(N)=NC2=C1C=CN2 CNPURSDMOWDNOQ-UHFFFAOYSA-N 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- AWQSAIIDOMEEOD-UHFFFAOYSA-N 5,5-Dimethyl-4-(3-oxobutyl)dihydro-2(3H)-furanone Chemical compound CC(=O)CCC1CC(=O)OC1(C)C AWQSAIIDOMEEOD-UHFFFAOYSA-N 0.000 description 1
- MGTZCLMLSSAXLD-UHFFFAOYSA-N 5-oxohexanoic acid Chemical compound CC(=O)CCCC(O)=O MGTZCLMLSSAXLD-UHFFFAOYSA-N 0.000 description 1
- 102000055025 Adenosine deaminases Human genes 0.000 description 1
- 235000021357 Behenic acid Nutrition 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 description 1
- IEVNIEKAPWWQDC-LDCVWXEPSA-N CCOC(C[C@@H](CC(C1=CC=CC=C1)S(O)(=O)=O)COC(C)=O)OCC Chemical compound CCOC(C[C@@H](CC(C1=CC=CC=C1)S(O)(=O)=O)COC(C)=O)OCC IEVNIEKAPWWQDC-LDCVWXEPSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-N Caprylic acid Natural products CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 1
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- QSJXEFYPDANLFS-UHFFFAOYSA-N Diacetyl Chemical group CC(=O)C(C)=O QSJXEFYPDANLFS-UHFFFAOYSA-N 0.000 description 1
- 241000222175 Diutina rugosa Species 0.000 description 1
- 206010015108 Epstein-Barr virus infection Diseases 0.000 description 1
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 208000009889 Herpes Simplex Diseases 0.000 description 1
- 206010019972 Herpes viral infections Diseases 0.000 description 1
- 208000029433 Herpesviridae infectious disease Diseases 0.000 description 1
- 241000701074 Human alphaherpesvirus 2 Species 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 125000003412 L-alanyl group Chemical group [H]N([H])[C@@](C([H])([H])[H])(C(=O)[*])[H] 0.000 description 1
- 150000008575 L-amino acids Chemical class 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- 125000003580 L-valyl group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(C([H])([H])[H])(C([H])([H])[H])[H] 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 229920003091 Methocel™ Polymers 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- SGSSKEDGVONRGC-UHFFFAOYSA-N N(2)-methylguanine Chemical compound O=C1NC(NC)=NC2=C1N=CN2 SGSSKEDGVONRGC-UHFFFAOYSA-N 0.000 description 1
- OKIZCWYLBDKLSU-UHFFFAOYSA-M N,N,N-Trimethylmethanaminium chloride Chemical compound [Cl-].C[N+](C)(C)C OKIZCWYLBDKLSU-UHFFFAOYSA-M 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- IPGXWCNRVDQNEM-OFSOJUDTSA-N N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCC=CCCCCCCCCCCC)C=NC2=C1 Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCC=CCCCCCCCCCCC)C=NC2=C1 IPGXWCNRVDQNEM-OFSOJUDTSA-N 0.000 description 1
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 235000021319 Palmitoleic acid Nutrition 0.000 description 1
- 102000019280 Pancreatic lipases Human genes 0.000 description 1
- 108050006759 Pancreatic lipases Proteins 0.000 description 1
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- HDOVUKNUBWVHOX-QMMMGPOBSA-N Valacyclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCOC(=O)[C@@H](N)C(C)C)C=N2 HDOVUKNUBWVHOX-QMMMGPOBSA-N 0.000 description 1
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 108010093894 Xanthine oxidase Proteins 0.000 description 1
- 102100033220 Xanthine oxidase Human genes 0.000 description 1
- IWJJZERAXSFDPY-FJQKOURKSA-N [(2R)-4-[(2S)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] docos-13-enoate Chemical compound NC1=NC=C2N=CN(C2=N1)C[C@@H](CCOC([C@@H](N)C(C)C)=O)COC(CCCCCCCCCCCC=CCCCCCCCC)=O IWJJZERAXSFDPY-FJQKOURKSA-N 0.000 description 1
- AWMUJDHSBADBQB-NAKRPHOHSA-N [(2R)-4-[(2S)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] hexadec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCC=CCCCCCC)C=NC2=C1 AWMUJDHSBADBQB-NAKRPHOHSA-N 0.000 description 1
- AJQSKNMSGMEEED-PPTMTGTBSA-N [(2R)-4-[(2S)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] icos-11-enoate Chemical compound NC1=NC=C2N=CN(C2=N1)C[C@@H](CCOC([C@@H](N)C(C)C)=O)COC(CCCCCCCCCC=CCCCCCCCC)=O AJQSKNMSGMEEED-PPTMTGTBSA-N 0.000 description 1
- MLWPQSDIPXTEIH-OFSOJUDTSA-N [(2R)-4-[(2S)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] octadec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCC=CCCCCCCCC)C=NC2=C1 MLWPQSDIPXTEIH-OFSOJUDTSA-N 0.000 description 1
- SYJGWAIAAHQDPA-BVAGGSTKSA-N [(2R)-4-[(2S)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] tetradec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCC=CCCCC)C=NC2=C1 SYJGWAIAAHQDPA-BVAGGSTKSA-N 0.000 description 1
- XCXSDPVCMWEGML-CJNGLKHVSA-N [(2r)-2-[(2-aminopurin-9-yl)methyl]-4-butanoyloxybutyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCC)C=NC2=C1 XCXSDPVCMWEGML-CJNGLKHVSA-N 0.000 description 1
- IJBNZMZRTGZRQT-PWSUYJOCSA-N [(2r)-2-[(2-aminopurin-9-yl)methyl]-4-hydroxybutyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCO)COC(=O)[C@@H](N)C(C)C)C=NC2=C1 IJBNZMZRTGZRQT-PWSUYJOCSA-N 0.000 description 1
- UFSFOZXONNJSBH-SNVBAGLBSA-N [(2r)-4,4-diethoxy-2-(hydroxymethyl)butyl] acetate Chemical compound CCOC(OCC)C[C@H](CO)COC(C)=O UFSFOZXONNJSBH-SNVBAGLBSA-N 0.000 description 1
- IFKFDWWRFYIIIX-QAPCUYQASA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] 5-oxohexanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)CCCC(C)=O)CCOC(=O)[C@@H](N)C(C)C)C=NC2=C1 IFKFDWWRFYIIIX-QAPCUYQASA-N 0.000 description 1
- PDRZADZPCOGUDA-KNQAVFIVSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] decanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCC)C=NC2=C1 PDRZADZPCOGUDA-KNQAVFIVSA-N 0.000 description 1
- WISSYNKKXRYGCF-FJQKOURKSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] docosanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCCCCCCCCCC)C=NC2=C1 WISSYNKKXRYGCF-FJQKOURKSA-N 0.000 description 1
- LLHURTUHPWCZOS-QPPBQGQZSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] dodecanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCC)C=NC2=C1 LLHURTUHPWCZOS-QPPBQGQZSA-N 0.000 description 1
- JYTUIQPNAWDCFV-NAKRPHOHSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] hexadecanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCCCC)C=NC2=C1 JYTUIQPNAWDCFV-NAKRPHOHSA-N 0.000 description 1
- YXKWTUYXTWERLG-QAPCUYQASA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] hexanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCC)C=NC2=C1 YXKWTUYXTWERLG-QAPCUYQASA-N 0.000 description 1
- GTTSKQBXZFMAHY-PPTMTGTBSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] icosanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCCCCCCCC)C=NC2=C1 GTTSKQBXZFMAHY-PPTMTGTBSA-N 0.000 description 1
- XQAATPRVMCFFDX-XLIONFOSSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] octanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCC)C=NC2=C1 XQAATPRVMCFFDX-XLIONFOSSA-N 0.000 description 1
- RMSUXPCMDFGLHO-BVAGGSTKSA-N [(2r)-4-[(2s)-2-amino-3-methylbutanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] tetradecanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCCCCCCCCCCCC)C=NC2=C1 RMSUXPCMDFGLHO-BVAGGSTKSA-N 0.000 description 1
- SDJJOBYTINBVTQ-QXIHQKPUSA-N [(2r)-4-[(2s,3s)-2-amino-3-methylpentanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] octadec-6-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)[C@@H](C)CC)COC(=O)CCCCC=CCCCCCCCCCCC)C=NC2=C1 SDJJOBYTINBVTQ-QXIHQKPUSA-N 0.000 description 1
- IXCKTUYWRRWQSX-QXIHQKPUSA-N [(2r)-4-[(2s,3s)-2-amino-3-methylpentanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] octadec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)[C@@H](C)CC)COC(=O)CCCCCCCC=CCCCCCCCC)C=NC2=C1 IXCKTUYWRRWQSX-QXIHQKPUSA-N 0.000 description 1
- MNZMFWNCFZPREH-XFAFFCHDSA-N [(2r)-4-[(2s,3s)-2-amino-3-methylpentanoyl]oxy-2-[(2-aminopurin-9-yl)methyl]butyl] tetradec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)[C@@H](C)CC)COC(=O)CCCCCCCC=CCCCC)C=NC2=C1 MNZMFWNCFZPREH-XFAFFCHDSA-N 0.000 description 1
- KXCNUMPXHMIVOF-CJNGLKHVSA-N [(3r)-3-[(2-aminopurin-9-yl)methyl]-4-butanoyloxybutyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)[C@@H](N)C(C)C)COC(=O)CCC)C=NC2=C1 KXCNUMPXHMIVOF-CJNGLKHVSA-N 0.000 description 1
- ATGZURPALDBGHY-PWSUYJOCSA-N [(3r)-3-[(2-aminopurin-9-yl)methyl]-4-hydroxybutyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)N=C2N(C[C@H](CO)CCOC(=O)[C@@H](N)C(C)C)C=NC2=C1 ATGZURPALDBGHY-PWSUYJOCSA-N 0.000 description 1
- FJIWUVILGSEMKW-KBMXLJTQSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] (2s)-2-amino-3-methylbutanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)[C@@H](N)C(C)C)C=NC2=C1 FJIWUVILGSEMKW-KBMXLJTQSA-N 0.000 description 1
- HYMHJVFATZNOLP-QAPCUYQASA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] 5-oxohexanoate Chemical compound N1=C(N)N=C2N(C[C@@H](CCOC(=O)CCCC(C)=O)COC(=O)[C@@H](N)C(C)C)C=NC2=C1 HYMHJVFATZNOLP-QAPCUYQASA-N 0.000 description 1
- KRLSACUKNZDQGR-KNQAVFIVSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] decanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCC)C=NC2=C1 KRLSACUKNZDQGR-KNQAVFIVSA-N 0.000 description 1
- QBKWXZXENZJMOY-FJQKOURKSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] docosanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCCCCCCCCCC)C=NC2=C1 QBKWXZXENZJMOY-FJQKOURKSA-N 0.000 description 1
- SPTXJNIIPIHADK-QPPBQGQZSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] dodecanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCC)C=NC2=C1 SPTXJNIIPIHADK-QPPBQGQZSA-N 0.000 description 1
- KSIZLMOJQHIYKF-NAKRPHOHSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] hexadec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCC=CCCCCCC)C=NC2=C1 KSIZLMOJQHIYKF-NAKRPHOHSA-N 0.000 description 1
- MNMSTGILRMFFIZ-NAKRPHOHSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] hexadecanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCCCC)C=NC2=C1 MNMSTGILRMFFIZ-NAKRPHOHSA-N 0.000 description 1
- GAQMSXAZFSMEQS-QAPCUYQASA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] hexanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCC)C=NC2=C1 GAQMSXAZFSMEQS-QAPCUYQASA-N 0.000 description 1
- ZIJXRKYZOJUTEQ-PPTMTGTBSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] icosanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCCCCCCCC)C=NC2=C1 ZIJXRKYZOJUTEQ-PPTMTGTBSA-N 0.000 description 1
- MIAWPKCOPQMDJN-OFSOJUDTSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] octadec-6-enoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCC=CCCCCCCCCCCC)C=NC2=C1 MIAWPKCOPQMDJN-OFSOJUDTSA-N 0.000 description 1
- DGSKXQREIAGUAP-OFSOJUDTSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] octadec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCC=CCCCCCCCC)C=NC2=C1 DGSKXQREIAGUAP-OFSOJUDTSA-N 0.000 description 1
- OLNSWMQGJWSLGE-OFSOJUDTSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] octadecanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCCCCCC)C=NC2=C1 OLNSWMQGJWSLGE-OFSOJUDTSA-N 0.000 description 1
- WEGSDTZMPJSGHN-XLIONFOSSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] octanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCC)C=NC2=C1 WEGSDTZMPJSGHN-XLIONFOSSA-N 0.000 description 1
- ZWRLPXJBZYBKJU-BVAGGSTKSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] tetradec-9-enoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCC=CCCCC)C=NC2=C1 ZWRLPXJBZYBKJU-BVAGGSTKSA-N 0.000 description 1
- UVFAGDBWYAEPNV-BVAGGSTKSA-N [(3r)-3-[[(2s)-2-amino-3-methylbutanoyl]oxymethyl]-4-(2-aminopurin-9-yl)butyl] tetradecanoate Chemical compound N1=C(N)N=C2N(C[C@H](COC(=O)[C@@H](N)C(C)C)CCOC(=O)CCCCCCCCCCCCC)C=NC2=C1 UVFAGDBWYAEPNV-BVAGGSTKSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 125000005076 adamantyloxycarbonyl group Chemical group C12(CC3CC(CC(C1)C3)C2)OC(=O)* 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940053200 antiepileptics fatty acid derivative Drugs 0.000 description 1
- 125000001124 arachidoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 229940116226 behenic acid Drugs 0.000 description 1
- 125000003910 behenoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- GONOPSZTUGRENK-UHFFFAOYSA-N benzyl(trichloro)silane Chemical compound Cl[Si](Cl)(Cl)CC1=CC=CC=C1 GONOPSZTUGRENK-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical class CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- KWEDUNSJJZVRKR-UHFFFAOYSA-N carbononitridic azide Chemical group [N-]=[N+]=NC#N KWEDUNSJJZVRKR-UHFFFAOYSA-N 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 125000001309 chloro group Chemical class Cl* 0.000 description 1
- 229960000724 cidofovir Drugs 0.000 description 1
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000006482 condensation reaction Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 125000003074 decanoyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(*)=O 0.000 description 1
- WHBIGIKBNXZKFE-UHFFFAOYSA-N delavirdine mesylate Natural products CC(C)NC1=CC=CN=C1N1CCN(C(=O)C=2NC3=CC=C(NS(C)(=O)=O)C=C3C=2)CC1 WHBIGIKBNXZKFE-UHFFFAOYSA-N 0.000 description 1
- 238000006392 deoxygenation reaction Methods 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- KFEVDPWXEVUUMW-UHFFFAOYSA-N docosanoic acid Natural products CCCCCCCCCCCCCCCCCCCCCC(=O)OCCC1=CC=C(O)C=C1 KFEVDPWXEVUUMW-UHFFFAOYSA-N 0.000 description 1
- NQGIJDNPUZEBRU-UHFFFAOYSA-N dodecanoyl chloride Chemical compound CCCCCCCCCCCC(Cl)=O NQGIJDNPUZEBRU-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000009483 enzymatic pathway Effects 0.000 description 1
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 125000005313 fatty acid group Chemical group 0.000 description 1
- 238000001917 fluorescence detection Methods 0.000 description 1
- QEWYKACRFQMRMB-UHFFFAOYSA-N fluoroacetic acid Chemical class OC(=O)CF QEWYKACRFQMRMB-UHFFFAOYSA-N 0.000 description 1
- 229960005102 foscarnet Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- ARBOVOVUTSQWSS-UHFFFAOYSA-N hexadecanoyl chloride Chemical compound CCCCCCCCCCCCCCCC(Cl)=O ARBOVOVUTSQWSS-UHFFFAOYSA-N 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 229910000040 hydrogen fluoride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical class I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- CBVCZFGXHXORBI-PXQQMZJSSA-N indinavir Chemical compound C([C@H](N(CC1)C[C@@H](O)C[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H]2C3=CC=CC=C3C[C@H]2O)C(=O)NC(C)(C)C)N1CC1=CC=CN=C1 CBVCZFGXHXORBI-PXQQMZJSSA-N 0.000 description 1
- 229960001936 indinavir Drugs 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 150000004668 long chain fatty acids Chemical class 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940099690 malic acid Drugs 0.000 description 1
- 150000002690 malonic acid derivatives Chemical class 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-N methyl undecanoic acid Natural products CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 235000021281 monounsaturated fatty acids Nutrition 0.000 description 1
- HOGDNTQCSIKEEV-UHFFFAOYSA-N n'-hydroxybutanediamide Chemical compound NC(=O)CCC(=O)NO HOGDNTQCSIKEEV-UHFFFAOYSA-N 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N n-hexanoic acid Natural products CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- NQHXCOAXSHGTIA-SKXNDZRYSA-N nelfinavir mesylate Chemical compound CS(O)(=O)=O.CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 NQHXCOAXSHGTIA-SKXNDZRYSA-N 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 229940127073 nucleoside analogue Drugs 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 125000002801 octanoyl group Chemical group C(CCCCCCC)(=O)* 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000004043 oxo group Chemical group O=* 0.000 description 1
- 125000003232 p-nitrobenzoyl group Chemical group [N+](=O)([O-])C1=CC=C(C(=O)*)C=C1 0.000 description 1
- 125000001312 palmitoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940116369 pancreatic lipase Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940055726 pantothenic acid Drugs 0.000 description 1
- 229920003175 pectinic acid Chemical class 0.000 description 1
- 229960001179 penciclovir Drugs 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 150000004968 peroxymonosulfuric acids Chemical class 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000006678 phenoxycarbonyl group Chemical group 0.000 description 1
- NMHMNPHRMNGLLB-UHFFFAOYSA-N phloretic acid Chemical compound OC(=O)CCC1=CC=C(O)C=C1 NMHMNPHRMNGLLB-UHFFFAOYSA-N 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 125000001557 phthalyl group Chemical group C(=O)(O)C1=C(C(=O)*)C=CC=C1 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical class OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical class CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- WYVAMUWZEOHJOQ-UHFFFAOYSA-N propionic anhydride Chemical compound CCC(=O)OC(=O)CC WYVAMUWZEOHJOQ-UHFFFAOYSA-N 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical class O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000011946 reduction process Methods 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 description 1
- 229960000311 ritonavir Drugs 0.000 description 1
- 229960001852 saquinavir Drugs 0.000 description 1
- QWAXKHKRTORLEM-UGJKXSETSA-N saquinavir Chemical compound C([C@@H]([C@H](O)CN1C[C@H]2CCCC[C@H]2C[C@H]1C(=O)NC(C)(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)C=1N=C2C=CC=CC2=CC=1)C1=CC=CC=C1 QWAXKHKRTORLEM-UGJKXSETSA-N 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000001593 sorbitan monooleate Substances 0.000 description 1
- 235000011069 sorbitan monooleate Nutrition 0.000 description 1
- 229940035049 sorbitan monooleate Drugs 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 238000012453 sprague-dawley rat model Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000001384 succinic acid Chemical class 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical group [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000007916 tablet composition Substances 0.000 description 1
- TUNFSRHWOTWDNC-HKGQFRNVSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCC[14C](O)=O TUNFSRHWOTWDNC-HKGQFRNVSA-N 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 1
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-M valinate Chemical class CC(C)C(N)C([O-])=O KZSNJWFQEVHDMF-UHFFFAOYSA-M 0.000 description 1
- 150000003679 valine derivatives Chemical class 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/18—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one oxygen and one nitrogen atom, e.g. guanine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Virology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Pharmacology & Pharmacy (AREA)
- Communicable Diseases (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- AIDS & HIV (AREA)
- Tropical Medicine & Parasitology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
本発明は、抗ウイルス剤の分野に関し、とりわけヘルペスおよびレトロウイルス感染に対して有用な非環状ヌクレオシドの誘導体に関する。本発明は、新規化合物、これら化合物を含有する医薬組成物、それを用いたウイルス感染の治療および予防方法、その製造法および新規な中間体を提供する。
発明の背景
多くの非環状ヌクレオシドの実際的な有用性は、それらの薬動力学が比較的穏やかであるために限られている。非環状ヌクレオシドの一般的なバイオアベイラビリティーを改善する努力のなかで多くのプロドラッグアプローチが探求されている。これらアプローチの一つは、非環状側鎖上の1またはそれ以上のヒドロキシ基のエステル誘導体、とりわけ脂肪族エステルを調製することを含む。
ヨーロッパ特許EP165289号には、H2Gとしても知られている有望な抗ヘルペス剤である9−[4−ヒドロキシ−(2−ヒドロキシメチル)ブチル]グアニンが記載されている。ヨーロッパ特許EP186640号は、6−デオキシH2Gを開示している。ヨーロッパ特許EP343133号は、これら化合物、とりわけそのR−(−)エナンシオマーがHIVなどのレトロウイルス感染に対しても活性であることを開示している。H2Gの様々な誘導体、たとえば非環状側鎖上のヒドロキシ基のリン酸エステル、脂肪族エステル(たとえば、ジアセテートおよびジプロピオネート)およびエーテルがEP343133号に開示されている。この特許はまた、典型的に6−ハロゲン化されたプリン残基のN−9位への非環状側鎖の縮合、または別法として、ピリミジンもしくはフラザノ−[3,4−d]ピリミジン残基のイミダゾール環閉環(closure)またはイミダゾール残基のピリミジン環閉環(それぞれ、前駆体ピリミジンまたはイミダゾール残基上にはすでに非環状側鎖が存在している)を含む、これら誘導体の調製法をも開示している。これら各方法の最も広い記載において、非環状側鎖は前以て誘導体化されているが、個々の例はまた無水酢酸および無水プロピオン酸およびDMFを用いたH2Gの1工程ジアシル化をも示している。
ハーンデン(Harnden)ら(J.Med.Chem.32、1738(1989))は、ペンシクロビルとしても知られる非環状ヌクレオシドである9−[4−ヒドロキシ−(3−ヒドロキシメチル)ブチル]グアニンの幾つかの短鎖脂肪族エステルおよびその6−デオキシ類似体を探索した。市場に出回っている抗ウイルス剤であるファンシクロビルは、6−デオキシペンシクロビルのジアセチル誘導体である。
ベンジャミン(Benjamin)ら(Pharm.Res.4、No.2、120(1987))は、ガンシクロビルとしても知られる9−[(1,3−ジヒドロキシ−2−プロポキシ)メチル]グアニンの短鎖脂肪族エステルを開示している。ジプロピオネートエステルは好ましいエステルであると開示されている。
レーク−バカール(Lake−Bakaar)らはAntimicrob.Agents Chemother.33、No.1、110−112(1989)において、H2Gのジアセテートおよびジプロピオネート誘導体および6−デオキシH2Gのモノアセテートおよびジアセテート誘導体を開示している。H2Gのジアセテートおよびジプロピオネート誘導体は、H2Gに比べてバイオアベイラビリティーの穏やかな改善しかもたらさないことが報告されている。
国際特許出願WO94/24134号(1994年10月27日公開)は、ジ−ピバロイルエステル、ジ−バレロイルエステル、モノ−バレロイルエステル、モノ−オレオイルエステルおよびモノ−ステアロイルエステルを含む、ガンシクロビルの6−デオキシN−7類似体の脂肪族エステルプロドラッグを開示している。
国際特許出願WO93/07163号(1993年4月15日公開)および国際特許出願WO94/22887号(1994年10月13日公開)は、ともにモノ−不飽和C18またはC20脂肪酸に由来するヌクレオシド類似体のモノ−エステル誘導体を開示している。米国特許第5,216,142号(1993年6月1日発行)もまたヌクレオシド類似体の長鎖脂肪酸モノ−エステル誘導体を開示している。
非環状ヌクレオシドのプロドラッグを提供する第二のアプローチは、非環状側鎖上の1またはそれ以上のヒドロキシ基のアミノ酸エステルの調製を含む。ヨーロッパ特許EP99493号は一般にアシクロビルのアミノ酸エステルを開示しており、ヨーロッパ特許出願EP308065号(1989年3月22日公開)はアシクロビルのバリンおよびイソロイシンエステルを開示している。
ヨーロッパ特許出願EP375329号(1990年6月27日公開)は、ジ−バリンエステル誘導体、ジ−イソロイシンエステル誘導体、ジ−グリシンエステル誘導体およびジ−アラニンエステル誘導体を含む、ガンシクロビルのアミノ酸エステル誘導体を開示している。国際特許出願WO95/09855号(1995年4月13日公開)は、モノ−バリンエステル誘導体およびジ−バリンエステル誘導体を含む、ペンシクロビルのアミノ酸エステル誘導体を開示している。
DE19526163号(1996年2月1日公開)および米国特許第5,543,414号(1996年8月6日発行)は、ガンシクロビルのアキラルなアミノ酸エステルを開示している。
ヨーロッパ特許出願EP694547号(1996年1月31日公開)は、ガンシクロビルのモノ−L−バリンエステル、およびジ−バリル−ガンシクロビルからのその調製を開示している。
ヨーロッパ特許出願EP654473号(1995年5月24日公開)は、9−[1’,2’−ビスヒドロキシメチル)−シクロプロパン−1’−イル]メチルグアニンの種々のビスアミノ酸エステル誘導体を開示している。
国際特許出願WO95/22330号(1995年8月24日公開)は、非環状ヌクレオシド9−[3,3−ジヒドロキシメチル−4−ヒドロキシ−ブト−1−イル]グアニンの脂肪族エステル、アミノ酸エステルおよび混合アセテート/バリネートエステルを開示している。この文献は、トリバリンエステル誘導体のバリンエステルの一つをアセテートエステルで置き換えたときにバイオアベイラビリティーが低下することを開示している。
発明の簡単な説明
本発明者らは、アミノ酸エステルと脂肪酸エステルとの特定の組み合わせを有するH2Gのジエステル誘導体が親化合物(H2G)と比べて経口バイオアベイラビリティーを有意に改善しうることを見出した。それゆえ、本発明の第一の側面に従い、式I:
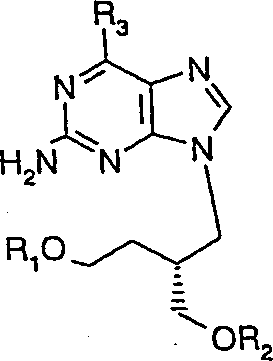
(b)R1が−C(O)C3〜C21の飽和もしくはモノ不飽和の任意に置換されたアルキルであり、R2が−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2であり;
R3はOHまたはHである]
の新規な化合物および薬理学的に許容しうるその塩が提供される。
本発明の混合脂肪酸およびアミノ酸エステルの経口バイオアベイラビリティーに及ぼす有利な効果は、対応する脂肪酸エステルの経口バイオアベイラビリティーと比較して特に予期しないものである。ラットからのH2Gの尿回収アッセイ(表1A)または血漿薬物アッセイ(表1B)を用いた結果に基づき、H2Gのモノ脂肪酸エステルおよびジ−脂肪酸エステルのいずれも親化合物のH2Gに比べて経口バイオアベイラビリティーに改善はみられない。実際、ジ−ステアレート誘導体は親化合物に比べて有意に低いバイオアベイラビリティーとなり、ステアレートエステルがH2Gの経口バイオアベイラビリティーを改善するうえで有害であることが示された。ある種の他の非環状ヌクレオシド類似体において一方または両方のヒドロキシルを対応バリンまたはジ−バリンエステルに変換するとバイオアベイラビリティーが改善されることが報告されている。H2Gを対応モノ−もしくはジ−バリルエステル誘導体に変換しても、親化合物に比べて同様のバイオアベイラビリティーの改善が得られた。H2Gの脂肪酸誘導体がバイオアベイラビリティーを改善するうえで有害であることが示されていることから、それぞれ尿回収アッセイおよび血漿薬物アッセイに基づいてH2Gの混合アミノ酸/脂肪酸ジエステル誘導体がH2Gのバリンジエステル誘導体に比べて改善されたまたは匹敵する経口バイオアベイラビリティーを付与することは予期しないことであった。
本発明の化合物は、とりわけ水痘帯状ヘルペスウイルス、単純ヘルペスウイルス1型および2型、エプスタイン−バーウイルス、ヘルペス6型(HHV−6)および8型(HHV−8)によって引き起こされるものなどのヘルペス感染に対する強力な抗ウイルス剤である。本発明の化合物は、疱疹後の神経痛を含む年寄りにおける帯状疱疹や若年におけるニワトリ水痘などの水痘帯状ヘルペスウイルス感染に対して特に有用であり、該疾患の持続および重篤度を数日間低減することができる。本発明の化合物を用いて治療に供しうるエプスタイン−バーウイルス感染には、以前は治療することができなかったが青年において何ヵ月もの学校不能(scholastic incapacity)を引き起こす感染性単球増殖症/腺熱が含まれる。
本発明の化合物はまた、ある種のレトロウイルス感染、とりわけSIV、HIV−1およびHIV−2に対して、およびトランス作用性(transactivating)ウイルスが示されている感染に対しても活性である。
従って、本発明のさらなる側面は、ヒトまたは動物に有効量の式Iの化合物または薬理学的に許容しうるその塩を投与することからなる、ヒトまたは動物におけるウイルス感染の予防または治療法を提供する。
たとえば側鎖がインビボで開裂される場合に本発明の化合物の塩基部分が天然グアニンとなるように、R3はヒドロキシまたはその互変体=Oであるのが有利である。別の態様として、R3は水素であってよく、それゆえ一般に一層可溶性の6−デオキシ誘導体(インビボでたとえばキサンチンオキシダーゼによりグアニンに酸化されうる)を定めてよい。
式Iの化合物は2R異性体と2S異性体との混合物であるラセミ体の形態で存在していてよい。しかしながら、好ましくは式Iの化合物は少なくとも70%、好ましくは90%のR体、たとえば95%以上のR体を有する。最も好ましくは、式Iの化合物はエナンシオマーとして純粋なR体である。
基R1/R2のアミノ酸はL−アミノ酸に由来するものであるのが好ましい。基R1/R2の脂肪酸は、合計で偶数個の炭素原子を有するものが好ましく、特にデカノイル(C10)、ラウリル(C12)、ミリストイル(C14)、パルミトイル(C16)、ステアロイル(C18)またはエイコサノイル(C20)であるのが好ましい。他の有用な基R1/R2としては、ブチリル、ヘキサノイル、オクタノイルまたはベヘノイル(C22)が挙げられる。さらに有用な基R1/R2としては、ミリストール酸(myristoleic)、ミリステレイド酸(myristelaidic)、パルミトレイン酸、パルミテレイド酸(palmitelaidic)、n6−オクタデセン酸、オレイン酸、エライジン酸、ガンド酸(gandoic)、エルカ酸またはブラシジン酸に由来するものが挙げられる。モノ不飽和脂肪酸エステルは、一般に、その長さに依存して好ましくはω−6、ω−9またはω−11位にトランスの立体配置の二重結合を有する。基R1/R2は、C9〜C17不飽和、またはn:9モノ不飽和アルキルを含む脂肪酸に由来するのが好ましい。
飽和または不飽和脂肪酸またはR1/R2は、ヒドロキシ、C1〜C6アルキル、C1〜C6アルコキシ、C1〜C6アルコキシC1〜C6アルキル、C1〜C6アルカノイル、アミノ、ハロ、シアノ、アジド、オキソ、メルカプトおよびニトロなどよりなる群から独立に選ばれた5つまでの同じかまたは異なる置換基で任意に置換されていてよい。
式Iの最も好ましい化合物は、R1が−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2であり、R2が−C(O)C9〜C17の飽和アルキルであるものである。
本明細書において「低級アルキル」とは1〜7の炭素原子を含む直鎖または分岐鎖アルキルラジカルをいい、メチル、エチル、n−プロピル、イソ−プロピル、n−ブチル、イソ−ブチル、sec−ブチル、t−ブチル、n−ペンチル、1−メチルブチル、2,2−ジメチルブチル、2−メチルペンチル、2,2−ジメチルプロピル、n−ヘキシルなどを含むがこれらに限られるものではない。
本明細書において「N−保護基」または「N−保護」とは、合成手順のあいだの所望でない反応からアミノ酸またはペプチドのN−末端を保護することまたはアミノ基を保護することをいう。よく用いられるN−保護基はグリーン(Greene)の「Protective Groups in Organic Synthesis」(ジョン・ウイリー・アンド・サンズ、ニューヨーク、1981)(参照のため本明細書中に引用する)に記載されている。N−保護基としては、たとえばホルミル、アセチル、プロピオニル、ピバロイル、t−ブチルアセチル、2−クロロアセチル、2−ブロモアセチル、トリフルオロアセチル、トリクロロアセチル、フタリル、o−ニトロフェノキシアセチル、α−クロロブチリル、ベンゾイル、4−クロロベンゾイル、4−ブロモベンゾイル、4−ニトロベンゾイルなどのアセチル基;ベンゼンスルホニル、p−トルエンスルホニルなどのスルホニル基、ベンジルオキシカルボニル、p−クロロベンジルオキシカルボニル、p−メトキシベンジルオキシカルボニル、p−ニトロベンジルオキシカルボニル、2−ニトロベンジルオキシカルボニル、p−ブロモベンジルオキシカルボニル、3,4−ジメトキシベンジルオキシカルボニル、4−メトキシベンジルオモシカルボニル、2−ニトロ−4,5−ジメトキシベンジルオキシカルボニル、3,4,5−トリメトキシベンジルオキシカルボニル、1−(p−ビフェニリル)−1−メチルエトキシカルボニル、α,α−ジメチル−3,5−ジメトキシベンジルオキシカルボニル、ベンズヒドリルオキシカルボニル、t−ブトキシカルボニル、ジイソプロピルメトキシカルボニル、イソプロピルオキシカルボニル、エトキシカルボニル、メトキシカルボニル、アリルオキシカルボニル、2,2,2−トリクロロエトキシカルボニル、フェノキシカルボニル、4−ニトロフェノキシカルボニル、フルオレニル−9−メトキシカルボニル、シクロペンチルオキシカルボニル、アダマンチルオキシカルボニル、シクロヘキシルオキシカルボニル、フェニルチオカルボニルなどのカルバメート形成基;ベンジル、トリフェニルメチル、ベンジルオキシメチルなどのアルキル基;およびトリメチルシリルなどのシリル基が挙げられる。好ましいN−保護基としては、ホルミル、アセチル、ベンゾイル、ピバロイル、t−ブチルアセチル、フェニルスルホニル、ベンジル、t−ブトキシカルボニル(BOC)およびベンジルオキシカルボニル(Cbz)などが挙げられる。
本明細書において「活性エステル誘導体」とは、酸クロライドなどの酸ハライドをいい、ギ酸および酢酸に由来する無水物、イソブチルオキシカルボニルクロライドなどのアルコキシカルボニルハライドからの無水物、N−ヒドロキシスクシンイミド由来のエステル、N−ヒドロキシフタルイミド由来のエステル、N−ヒドロキシベンゾトリアゾール由来のエステル、N−ヒドロキシ−5−ノルボルネン−2,3−ジカルボキサミド由来のエステル、2,4,5−トリクロロフェニル由来のエステルなどが挙げられるがこれらに限られるものではない。
式Iの好ましい化合物として、以下のものが挙げられる:
(R)−9−[2−(ブチリルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[2−(4−アセチルブチリルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[2−(ヘキサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(オクタノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(デカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(ドデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(テトラデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(ヘキサデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(オクタデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[2−(エイコサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[2−(ドコサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−((9−テトラデセノイル)オキシメチル)ブチル]グアニン、
(R)−9−[2−((9−ヘキサデセノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−((6−オクタデセノイル)オキシメチル)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−((9−オクタデセノイル)オキシメチル)ブチル]グアニン、
(R)−9−[2−((11−エイコサノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[2−((13−ドコセノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−2−アミノ−9−[2−(ブチリルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(4−アセチルブチリルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ヘキサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(オクタノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(デカノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(ドデカノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(テトラデカノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(ヘキサデカノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(オクタデカノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(エイコサノイルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[2−(エイコサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ドコサノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−((9−テトラデセノイル)オキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[2−((9−ヘキサデセノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−((6−オクタデセノイル)オキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−((9−オクタデセノイル)オキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[2−((11−エイコサノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、または
(R)−2−アミノ−9−[2−((13−ドコセノイル)オキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
および薬理学的に許容しうるその塩。
さらに好ましい化合物として、以下のものが挙げられる:
(R)−9−[2−(ブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(4−アセチルブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ヘキサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(オクタノイルオキシメチル)−4−(L−バリルオキシ)−ブチル]グアニン、
(R)−9−[2−(デカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ドデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(テトラデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−ヘキサデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(オクタデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(エイコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(エイコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ドコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((9−テトラデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((9−ヘキサデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((6−オクタデセノイル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((9−オクタデセノイル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((11−エイコサノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−((13−ドコセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−2−アミノ−9−[2−(ブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(4−アセチルブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ヘキサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(オクタノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(デカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ドデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(テトラデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ヘキサデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(オクタデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(エイコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−(ドコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−((9−テトラデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−((9−ヘキサデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−((6−オクタデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−((9−オクタデセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−((11−エイコセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、または
(R)−2−アミノ−9−[2−((13−ドコセノイル)オキシメチル)−4−(L−バリルオキシ)ブチル]プリン、
および薬理学的に許容しうるその塩。
式Iの他の好ましい化合物として、以下のものが挙げられる:
(R)−9−[4−(ブチリルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(4−アセチルブチリルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(ヘキサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(オクタノイルオキシ)−2−(L−バリルオキシメチル)−ブチル]グアニン、
(R)−9−[4−(デカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(ドデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(テトラデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(ヘキサデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(オクタデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(エイコサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−(ドコサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((9−テトラデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((9−ヘキサデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((6−オクタデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((9−オクタデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((11−エイコセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−9−[4−((13−ドコセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−2−アミノ−9−[4−(ブチリルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(4−アセチルブチリルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(ヘキサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(オクタノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(デカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(ドデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(テトラデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(ヘキサデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(オクタデカノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(エイコサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−(ドコサノイルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−((9−テトラデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−((9−ヘキサデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−((6−オクタデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−((9−オクタデセノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]プリン、
(R)−2−アミノ−9−[4−((11−エイコサノイル)オキシ)−2−(L−バリルオキシメチル)ブチル]プリン、または
(R)−2−アミノ−9−[4−((13−ドコセノイル)オキシメチル)−2−(L−バリルオキシ)ブチル]プリン、
および薬理学的に許容しうるその塩。
式Iの化合物は塩を形成し、これは本発明の他の側面を形成する。式Iの化合物の適当な薬理学的に許容しうる塩としては、有機酸の塩、とりわけカルボン酸の塩が挙げられ、酢酸、トリフルオロ酢酸、乳酸、グルコン酸、クエン酸、酒石酸、マレイン酸、リンゴ酸、パントテン酸、イセチオン酸、アジピン酸、アルギン酸、アスパラギン酸、安息香酸、酪酸、ジグルコン酸、シクロペンタン酸、グルコヘプタン酸、グリセロリン酸、蓚酸、ヘプタン酸、ヘキサン酸、フマール酸、ニコチン酸、パルミチン酸(palmoate)、ペクチン酸、3−フェニルプロピオン酸、ピクリン酸、ピバル酸、プロピオン酸、酒石酸、ラクトビオン酸、ピボル酸(pivolate)、ショウノウ酸、ウンデカン酸およびコハク酸の塩を含むがこれらに限られるものではない、有機スルホン酸の塩、たとえばメタンスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ショウノウスルホン酸、2−ナフタレンスルホン酸、ベンゼンスルホン酸、p−クロロベンゼンスルホン酸およびp−トルエンスルホン酸の塩;および無機酸の塩、たとえば塩酸、臭化水素酸、ヨウ化水素酸、硫酸、重硫酸、ヘミ硫酸(hemisulfate)、チオシアン酸、過硫酸、リン酸およびスルホン酸の塩が挙げられる。塩酸塩が便利である。
式Iの化合物は水和物として単離できる。本発明の化合物は結晶形、好ましくは均一な結晶形にて単離でき、それゆえ本発明の他の側面は>70%、好ましくは>90%均一な結晶物質、たとえば>95%均一な結晶物質を含む実質的に純粋な結晶形にて式Iの化合物を提供する。
本発明の化合物は経口投与するのに特に適しているが、直腸経由、膣経由、鼻経由、局所的、経皮または非経口的に、たとえば筋肉内、静脈内または硬膜外で投与することもできる。本発明の化合物はたとえばカプセル中にて単独で投与することもできるが、一般に薬理学的に許容しうる担体または希釈剤とともに投与されるであろう。本発明は医薬組成物の調製法にも関し、式Iの化合物または薬理学的に許容しうるその塩を薬理学的に許容しうる担体または希釈剤と混合することからなる。
経口剤型は、通常の担体または結合剤、たとえばステアリン酸マグネシウム、粉乳(chalk)、デンプン、乳糖、ろう、ガムまたはゼラチンを用い、カプセル剤や錠剤などの単位投与剤型として調製する。リポソームまたは合成もしくは天然のポリマー、たとえばHPMCやPVPなどを用い、除放製剤とすることができる。別法として、製剤はまた点鼻または点眼剤、シロップ剤、ゲル剤またはクリーム剤として提供され、任意に香料および/または保存剤および/または乳化剤とともに水、食塩水、エタノール、植物油またはグリセリンなどの通常のビヒクル中の液剤、懸濁剤、エマルジョン、水中油または油中水製剤を含む。
本発明の化合物は、一般に、0.1〜200mg/kg/日の範囲、有利には0.5〜100mg/kg/日、さらに好ましくは10〜50mg/kg/日、たとえば10〜25mg/kg/日の範囲にて毎日の投与量として投与できる。通常の成人に対する典型的な投与割合は、ヘルペス感染の場合、約50〜500mg、たとえば300mgを1日当たり1回または2回であり、HIV感染の場合は該量の2〜10倍である。
抗ウイルス療法において慎重であるように、本発明の化合物は他の抗ウイルス剤、たとえばヘルペス適応症の場合、アシクロビル、バルシクロビル、ペンシクロビル、ファンシクロビル、ガンシクロビルおよびそのプロドラッグ、シドホビル、ホスカーネットなど、およびレトロウイルス適応症の場合、AZT、ddI、ddC、d4T、3TC、ホスカーネット、リトナビル、インジナビル、サキナビル、デラビリジン、ベルテックスVX478、アグロンAG1343などとともに投与できる。
本発明の化合物は新たに、すなわちH2G親化合物のエステル化によって調製でき、該H2G親化合物はヨーロッパ特許出願EP343133号(参照のため本明細書中に引用する)に開示された合成法により調製される。
H2Gの調製のための典型的な反応式を次頁に示す:
R3が水素である本発明の化合物の出発物質は、ヨーロッパ特許EP186640号(その内容を参照のため本明細書中に引用する)に示された方法により調製できる。これら出発物質は、プリンの2−アミノ基を任意に上記通常のN−保護基、とりわけBOC(t−BuO−CO−)、Z(BnO−CO−)またはPh3C−で保護した後、下記H2Gについて記載するようにアシル化することができる。
本発明の化合物は下記反応式AおよびBに記載するようにH2Gから調製できる。
A.直接アシル化
精製後、第一のエステル化工程と同様の手順を用い、R1モノアシル化化合物を側鎖2−CH2OH基上で適当な活性化脂肪酸誘導体でさらにアシル化してジアシル化生成物を得る。ついで、このジエステル生成物を、たとえばトリフルオロ酢酸、HCl(水性)/ジオキサンを用いる通常の脱保護処理または触媒の存在下での水素化に供して式Iの所望の化合物を得る。この化合物は脱保護条件に依存して塩の形態であってよい。
種々のアシル化に用いる活性化R1/R2酸誘導体としては、たとえば、酸ハライド、酸無水物、活性化酸エステル、またはカップリング剤、たとえばジシクロヘキシルカルボジイミドの存在下の酸が挙げられ、この場合、それぞれにおける「酸」は対応するR1/R2アミノ酸またはR1/R2脂肪酸を表す。代表的な活性化酸誘導体としては、酸クロライド、ギ酸および酢酸由来の混合無水物、イソブチルオキシカルボニルクロライドなどのアルコキシカルボニルハライド由来の無水物、N−ヒドロキシスクシンアミド由来のエステル、N−ヒドロキシフタルイミド由来のエステル、N−ヒドロキシ−5−ノルボルネン−2,3−ジカルボキサミド由来のエステル、2,4,5−トリクロロフェノール由来のエステルなどが挙げられる。
B.側鎖4−ヒドロキシ基の保護を経由するアシル化:
反応式BはR1がアミノ酸に由来しR2が脂肪酸に由来する化合物の調製を示すべく例示したが、逆の反応式はR1が脂肪酸に由来しR2がアミノ酸に由来する化合物に適用できるであろう。この反応式は嵩ばった保護基によるH2G側鎖4−ヒドロキシ基の位置選択的な保護に依存している。上記反応式Bにおいて、このことはt−ブチルジフェニルシリルとして示したが、トリチル、9−(9−フェニル)キサンテニル、1,1−ビス(4−メチルフェニル)−1’−ピレニルメチルもまた適している。得られる生成物を上記反応式Aに記載したのと同様の試薬および手順を用いて側鎖2−ヒドロキシメチル基にてアシル化するが、この場合、活性化酸誘導体はR2脂肪酸、たとえばミリスチン酸、ステアリン酸、オレイン酸、エライジン酸クロライドなどである。かくしてモノアシル化した化合物を適当な脱保護処理に供して側鎖4−ヒドロキシ保護基を除去するが、このことは位置選択的保護基に依存したHF/ピリジンなどの試薬、上記反応条件の操作、すなわち試薬濃度、添加速度、温度および溶媒を用いて高度に選択的な仕方で行うことができる。ついで、遊離になった側鎖4−ヒドロキシ基を上記反応式Aに記載したのと同様にして活性化α−アミノ酸でアシル化する。
本明細書におけるたとえば反応式A、B、CまたはDにおいてR1/R2のアミノ酸エステルを導入するための他の方法としては、国際特許出願第WO94/29311号に記載の2−オキサ−4−アザ−シクロアルカン−1,3−ジオン法が挙げられる。
本明細書におけるたとえば反応式A、B、CまたはDにおいてR1/R2の脂肪酸エステルを導入するための他の方法としては、SP435固定化カンジダ・アンタークチクス(Candida antarcticus)(ノボ・ノルディスク)、ブタ膵リパーゼまたはカンジダ・ルゴサ(Candida rugosa)リパーゼなどのリパーゼを用いたプレパラティブ・バイオトランスフォーメーションズ(Preparative Biotransformations)1.11.8(ロバーツ(S.M.Roberts)編、ウイリー・アンド・サンズ、ニューヨーク、1995)に記載の酵素的経路によるものが挙げられる。酵素的なアシル化は、他のアシル基またはプリンの2−アミン上でのN−保護および脱保護工程を避けることが望ましい場合に特に都合がよい。
式IにおいてR3が水素である化合物を得る他の経路は、式Iの対応グアニン化合物(R1/R2のアミノ酸エステル残基はBOCなどの通常のN−保護基で任意に保護してある)をハロなどの活性化剤で6−活性化することである。ついで、かくして活性化された6−プリンをたとえばパラジウム触媒でプリンに還元し、脱保護して所望の6−デオキシH2Gジ−エステルとする。
かくして本発明の他の側面は、
(a)式IにおいてR1およびR2がそれぞれ水素である化合物のプリン2位および/または6位を任意にN−保護し、
(b)(i)任意にN−保護したバリンまたはイソロイシン基、
(ii)任意に置換した飽和またはモノ不飽和のC3〜C21COOH誘導体、または
(iii)位置選択的な保護基
のいずれかで式Iの化合物を側鎖4−ヒドロキシ基にて位置選択的にアシル化し、
(c)(i)任意にN−保護したバリンまたはイソロイシン誘導体、または
(ii)任意に置換した飽和またはモノ不飽和のC3〜C21COOH誘導体
で側鎖2−ヒドロキシメチル基をアシル化し、
(d)R1に位置選択的な保護基が存在する場合には、これを
(i)任意にN−保護したバリンまたはイソロイシン基、または
(ii)任意に置換した飽和またはモノ不飽和のC3〜C21COOH誘導体
で置換し、ついで
(e)得られた化合物を必要なら脱保護する
ことからなる、式Iの化合物の製造方法を提供する。
上記反応式AおよびBではアミノ酸エステルおよび脂肪酸エステルを段階的に加える選択的なアシル化を採用している。式Iの化合物の別の製造方法は、両方のアシル基が同じであるジアシル化H2G誘導体を出発物質とし、これらアシル基の一方を選択的に除去してモノアシル中間体を得、ついで第二の異なるアシル基を上記反応式AおよびBと同様にしてアシル化するものである。
従って、本発明の他の側面は、上記式Iの化合物の製造方法を提供するものであり、該方法は、
(A)式IにおいてR1およびR2がともにバリルまたはイソロイシルエステル(任意にN−保護されている)であるかまたはR1およびR2がともに−C(=O)C3〜C21飽和またはモノ不飽和の任意に置換されたアルキルであるジアシル化合物をモノ脱アシル化し、ついで
(B)かくして遊離になった側鎖4−ヒドロキシまたは側鎖2−ヒドロキシメチル基を対応のバリル、イソロイシルまたは−C(=O)C3〜C21飽和またはモノ不飽和の任意に置換されたアルキルでアシル化し、ついで
(C)必要なら脱保護する
ことからなる。
この別法は、ジアシル化H2G誘導体の調製が容易である点および精製工程が殆どまたは全く必要でない点で有利である。ジアシル化H2G誘導体のアシル基の一方のみの選択的な除去は、反応条件、とりわけ温度、反応物の添加速度および塩基の選択を操作することにより達成できる。
それゆえ、この別法の合成経路に利用できる化合物は、式:
を有するものである。
この別法において合成を容易にするため、R1およびR2はともに最初から同じであるのが好ましく、同じアミノ酸エステルであるのが最も好ましい。かかるジ−アミノ酸エステルは、その調製において一般にN−保護され、この条件にて選択的な脱アシル化工程に直接用いることができる。別のやり方として、そのようなN−保護ジ−アミノアシル化H2G誘導体は、下記に記載するように、脱保護し、必要に応じて再保護することができる。それゆえ、保護されていないジ−アミノアシルH2G誘導体は下記化合物の一つを含む:
(R)−9−[2−(L−イソロイシルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[2−(L−バリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−2−アミノ−9−[4−(L−イソロイシルオキシ)−2−(L−イソロイシルオキシメチル)ブチル]プリン、および
(R)−2−アミノ−9−[4−(L−バリルオキシ)−2−(L−バリルオキシメチル)ブチル]プリン。
これら保護されていないH2Gジアシル化誘導体はアシル基の一方の(典型的には側鎖4−位アシル)選択的な脱アシル化に直接供することができ、ついで遊離した4−ヒドロキシを上記と同様にして酵素的にアシル化する。別のやり方として、保護されていないH2Gジアシル化誘導体を再保護し、ついで選択的な脱アシル化に供し、ついで今度は反応式AおよびBに記載したような脂肪酸エステルによる通常のアシル化を行うことができる。都合のよいことには、かかる再保護工程はその後のアシル化に適した性質を有する異なるN−保護基を用いて行う。たとえば、ジ−アミノ酸H2G誘導体を調製する場合には、保護基の脂肪親和性の性質がアシル化生成物の分離を助けるのでFmocなどの脂肪親和性のN−保護基を用いるのが都合がよい。一方、Fmocの脂肪親和性の性質は脂肪酸でアシル化を行う際には有用性はほとんどなく、それゆえBOCなどの別のN−保護基を用いてジアシル化H2Gを再保護するのが都合がよい。
式Iの化合物の製造を上記本発明の第一の側面の方法における工程(b)(i)、(ii)または(iii)の新規なモノアシル化中間体から開始できることもまた明らかであろう。それゆえ、これら化合物は、式:
(i)−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2、
(ii)−C(=O)C3〜C21飽和またはモノ不飽和の任意に置換されたアルキル、または
(iii)位置選択的な保護基;
R1およびR2の他方は水素;
R3はOHまたはH)
で示されるものである。
それゆえ、有用な化合物としては以下のものが挙げられる:
(R)−9−[2−ヒドロキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(トリチルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(9−(9−フェニル)キサンテニルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(1,1−ビス(4−メチルフェニル)−1’−ピレニルメチルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(デカノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル)−4−(ドデカノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(テトラデカノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル)−4−(ヘキサデカノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(オクタデカノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル)−4−(エイコサノイルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(ドコサノイルオキシ)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(デカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(ドデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(テトラデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(ヘキサデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(オクタデカノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(エイコサノイルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(ドコサノイルオキシメチル)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−ヒドロキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(L−イソロイシルオキシメチル)ブチル]グアニン、
(R)−9−[4−ヒドロキシ−2−(L−バリルオキシメチル)ブチル]グアニン、
(R)−2−アミノ−9−[2−ヒドロキシメチル−4−(L−バリルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[2−ヒドロキシメチル)−4−(L−イソロイシルオキシ)ブチル]プリン、
(R)−2−アミノ−9−[4−ヒドロキシ−2−(L−イソロイシルオキシメチル)ブチル]プリンおよび
(R)−2−アミノ−9−[4−ヒドロキシ−2−(L−バリルオキシメチル)ブチル]プリン。
上記本発明の第一の側面の方法の工程(c)における位置選択的に保護した側鎖4−ヒドロキシ中間体もまた新規な化合物である。
それゆえ、有用な化合物としては以下のものが挙げられる:
(R)−9−[2−デカノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−ドデカノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−テトラデカノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−ヘキサデカノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−オクタデカノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−エイコサノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン、
(R)−9−[2−ドコサノイルオキシメチル−4−(t−ブチルジフェニルシリル)ブチル]グアニン。
式IにおいてR3が−OHである本発明の化合物の他の製造方法を反応式Cに示す。
3を約−20℃〜約100℃の温度、不活性溶媒(たとえば、THFまたはメチルt−ブチルエーテルまたはt−BuOHなど)中にて約0.5〜約4.0モル当量のエステル→アルコール還元剤(たとえば、LiBH4またはCa(BH4)2またはNaBH4またはLiAlH4など)で還元するとジオール4を得る。リパーゼ(たとえば、リパーゼPS−30またはリパーゼPPLまたはリパーゼCCLなど)またはホスホリパーゼ(たとえば、ホスホリパーゼDなど)の存在下での約1.0〜約20.0モル当量のビニルエステル5(R8はC3−C21飽和またはモノ不飽和の任意に置換されたアルキル)との反応による4の酵素的エステル化によりエステル6の所望の立体異性体を得る。この反応は溶媒の不在下でも不活性な溶媒(たとえば、メチルt−ブチルエーテルまたはトルエンまたはヘキサンなど)の存在下でも行うことができる。この反応は約−20℃〜約80℃の温度にて行う。
6を約−25℃〜約100℃の温度にて不活性溶媒(たとえば、メチレンクロライドまたはトルエンまたは酢酸エチルなど)中でハロゲン化剤(たとえば、NBS/P(Ph)3またはNCS/P(Ph)3またはPOCl3またはNCS/P(Ph)3/NaIなど、アセトン中)と反応させるかまたは不活性溶媒(たとえば、メチレンクロライドまたはトルエンまたは酢酸エチルなど)中、約1.0〜約4.0モル当量の塩基(たとえば、トリエチルアミンまたは炭酸カリウムまたはピリジンまたはジメチルアミノピリジンまたはエチルジイソプロピルアミンなど)の存在下で約0.8〜約2.0モル当量のスルホニルハライド(たとえば、ベンゼンスルホニルクロライド、トルエンスルホニルクロライドまたはメタンスルホニルクロライドなど)と反応させることにより、6のアルコール置換基を脱離基(たとえば、ハロゲンまたはスルホネート)に変換してエステル7(X2はハロゲンまたはスルホネート脱離基)を得る。
7を約−25℃〜約140℃にて不活性溶媒(たとえば、DMFまたはTHFまたはアセトニトリルまたはN−メチルピロリドンまたはエタノールなど)中、約1.0〜約6.0モル当量の塩基(たとえば、炭酸カリウムまたはNaHまたはKHまたはNaOHまたはKOHまたはリチウムジイソプロピルアミドなど)の存在下で約0.9〜約2.0モル当量の2−アミノ−4−クロロプリン8と反応させて置換プリン9を得る。
別のやり方として、アルコール6を2−アミノ−4−クロロプリン8とミツノブカップリングして(たとえば、P(Ph)3/ジエチルアジドカルボキシレート)9を得る。
9を約−25℃〜約150℃の温度にて不活性溶媒(たとえば、THFまたはDMFなど)中、約1.0〜約6.0モル当量の塩基(たとえば、カリウムt−ブトキシドまたは炭酸カリウムまたはNaHまたはKHまたはリチウムジイソプロピルアミドなど)の存在下で約2.0〜約20モル当量のアルコールR9OH(R9はベンジルなどのアルコール保護基)と反応させてアルコール10を得る。
10のアルコール保護基R9を除去して(たとえば、Pd/CまたはPd(OH)2などの水素化触媒の存在下、エタノールやベンジルアルコールやメタノールやTHFなどの不活性溶媒中での接触水素化により)置換グアニン11を得る。
11を約−25℃〜約100℃の温度にて(a)不活性溶媒(たとえば、THFまたはDMFなど)中、約0.8〜約2.0モル当量のR10COOHおよびカップリング試薬(たとえば、DCC/DMAPなど)と反応させるか、または(b)不活性溶媒(たとえば、メチレンクロライドまたはTHFまたはピリジンまたはアセトニトリルまたはDMFなど)中、約0〜約3.0モル当量の塩基(たとえば、ピリジンまたはトリエチルアミンまたはエチルジイソプロピルアミンまたはDBUまたは炭酸カリウムなど)の存在下で約0.8〜約2.0モル当量のR10COOHの活性化誘導体(たとえば、酸クロライドまたはN−ヒドロキシスクシンイミドエステルまたはR10C(O)OC(O)R10など)と反応させることによりエステル化してエステル12を得る。
12をまず不活性溶媒(たとえば、THF/H2Oまたはメチレンクロライド/H2Oまたは酢酸エチル/H2Oまたはエタノール/H2Oまたはメタノール/H2Oなど)中、約0.1〜約10.0モル当量の酸(たとえば、トリフルオロメタンスルホン酸またはHClまたは酢酸または硫酸など)と反応させることにより12のアセタール置換基を脱保護し、得られるアルデヒドを還元する。得られた粗製の反応混合物に、約0.1〜約10.0モル当量の塩基(たとえば、重炭酸ナトリウムまたは炭酸カリウムまたはトリエチルアミンまたはピリジンまたはKOHなど)、さらなる不活性溶媒(たとえば、THFおよびまたはメチレンクロライドまたは酢酸エチルまたはメチルt−ブチルエーテルまたはイソプロパノールなど)および約0.3〜約5.0モル当量のアルデヒド還元剤(たとえば、水素化ホウ素ナトリウムまたはRaNi/H2など)を約−25℃〜約100℃の温度にて加えてアルコール13を得る。
13を約25℃〜約100℃の温度にて不活性溶媒(たとえば、THFまたはジオキサンまたはジオキソランまたはDMFまたはメチレンクロライドなど)中、約0.8〜約3.0モル当量のN−保護アミノ酸P1NHCH(R11)COOHまたはその活性化誘導体(P1はN−保護基、R11はイソプロピルまたはイソブチル)と反応させてアルコール14を得る。14をN−脱保護して式IにおいてR3が−OHである本発明の化合物を得る。
別のやり方として、化合物13をP1NHCH(R11)COOH由来の対称無水物(すなわち、P1NHCH(R11)C(O)O−C(O)CH(R11)NHP1)と反応させてR3が−OHである1を得ることができる。
R3が−OHである化合物の他の製造方法を反応式Dに示す。
16を約−20℃〜約100℃の温度、不活性溶媒(たとえば、THFまたはメチルt−ブチルエーテルまたはエタノールまたはt−ブタノールなど)中にて約0.5〜約4.0モル当量のエステル→アルコール還元剤(たとえば、LiBH4またはCa(BH4)2またはNaBH4またはLiAlH4など)で還元するとジオール17を得る。リパーゼ(たとえば、リパーゼPS−30またはリパーゼPPLまたはリパーゼCCLなど)またはホスホリパーゼ(たとえば、ホスホリパーゼDなど)の存在下での約1.0〜約20.0モル当量のビニルエステル5(R8はC3−C21飽和またはモノ不飽和の任意に置換されたアルキル)との反応による17の酵素的エステル化によりエステル18の所望の立体異性体を得る。この反応は溶媒の不在下でも不活性な溶媒(たとえば、メチルt−ブチルエーテルまたはトルエンまたはヘキサンなど)の存在下でも行うことができる。この反応は約−20℃〜約80℃の温度にて行う。
18を約−25℃〜約100℃の温度にて不活性溶媒(たとえば、メチレンクロライドまたはトルエンまたは酢酸エチルなど)中でハロゲン化剤(たとえば、NBS/P(Ph)3またはNCS/P(Ph)3またはPOCl3またはNCS/P(Ph)3/NaIなど、アセトン中)と反応させるかまたは不活性溶媒(たとえば、メチレンクロライドまたはトルエンまたは酢酸エチルまたはピリジンまたはメチルt−ブチルエーテルなど)の存在下、約1.0〜約4.0モル当量の塩基(たとえば、トリエチルアミンまたは炭酸カリウムまたはピリジンまたはジメチルアミノピリジンまたはエチルジイソプロピルアミンなど)の存在下で約0.8〜約2.0モル当量のスルホニルハライド(たとえば、ベンゼンスルホニルクロライド、トルエンスルホニルクロライドまたはメタンスルホニルクロライドなど)と反応させることにより、18のアルコール置換基を脱離基(たとえば、ハロゲンまたはスルホネート)に変換してエステル19(X2はハロゲンまたはスルホネート脱離基)を得る。
19を約−25℃〜約140℃の温度にて不活性溶媒(たとえば、DMFまたはTHFまたはアセトニトリルまたはN−メチルピロリドンまたはエタノールなど)中、約1.0〜約6.0モル当量の塩基(たとえば、炭酸カリウムまたはNaHまたはKHまたはNaOHまたはKOHまたはリチウムジイソプロピルアミドなど)の存在下で約0.9〜約2.0モル当量の2−アミノ−4−クロロプリン8と反応させて置換プリン20を得る。
別のやり方として、アルコール18を2−アミノ−4−クロロプリン8とミツノブカップリングして(たとえば、P(Ph)3/ジエチルアジドカルボキシレート)20を得る。
20を約−25℃〜約150℃の温度にて不活性溶媒(たとえば、THFまたはDMFなど)中、約1.0〜約6.0モル当量の塩基(たとえば、カリウムt−ブトキシドまたは炭酸カリウムまたはNaHまたはKHまたはリチウムジイソプロピルアミドなど)の存在下で約2.0〜約20.0モル当量のアルコールR9OH(R9はベンジルなどのアルコール保護基)と反応させてアルコール21を得る。
21のアルコール保護基R9を除去して(たとえば、Pd/CまたはPd(OH)2などの水素化触媒の存在下、エタノールやベンジルアルコールやメタノールやTHFなどの不活性溶媒中での接触水素化により)置換グアニン22を得る。
23を(a)還元剤(たとえば、HCO2HおよびPd/Cなど)と反応させるか(R12が−CH(Ph)2または−C(Ph)3の場合)、または(b)脱シリル化剤(たとえば、Bu4NFなど)と反応させる(R12が−Si(t−Bu)(Me)2の場合)ことにより23のエーテル置換基を脱保護して13を得る。
アルコール13は反応式Cに示したように1に変換できる。
さらに他のやり方は、アルコール4または17をビニルエステルCH2=CH−OC(O)R10(すなわち、反応式CおよびDにおいてR8=R10)で酵素的にエステル化して6または18中に最終生成物Iの所望のカルボン酸エステルを直接導入することを含む。このことにより、9〜12または20〜23に含まれるエステル加水分解および再エステル化をなしですませることができる。
反応式CおよびDの手順は、非環状側鎖の各ヒドロキシル基が異なるヒドロキシ保護基または前駆体基の使用により識別されるという事実により特徴付けられる。このことにより、アミノ酸基かまたは脂肪酸基のいずれかによる各ヒドロキシ基の選択的なアシル化が可能となる。
反応式CおよびDはR1がアミノ酸由来でありR2が脂肪酸由来である本発明の化合物の態様を参照して説明および記載してある。しかしながら、それぞれの反対の反応式がR1が脂肪酸由来でありR2がアミノ酸由来である化合物に適用しうることが明らかであろう。
発明の詳細な記載
ここで、本発明は下記の限定されない実施例、比較例および添付の図面を参照して例示による説明されるであろう。添付の図面において:
図1は、生物学的実施例3でさらに説明するように、本発明の化合物またはH2Gの他のプロドラッグ誘導体を投与したカニクイザルでの血漿H2Gレベルを時間の関数として表したものである;
図2は、生物学的実施例4でさらに説明するように、種々の投与量の本発明の化合物または従来技術の抗ウイルス剤を投与した単純ヘルペス感染マウスでの生存率を時間の関数として表したものである。
実施例1
(R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
本実施例は、製造反応式Aの適用を説明するものである。
(a)(R)−9−[4−(N−tert−ブトキシカルボニル−L−バリルオキシ)−2−(ヒドロキシメチル)ブチル]グアニン
N−t−BOC−L−バリン(5.58g、25.7ミリモル)、DMAP(0.314g、2.57ミリモル)およびDCC(6.52g、31.6ミリモル)を加える前に、H2G(5g、19.7ミリモル)をDMF(300ml)中に加熱下で溶解し、室温に冷却した。この混合物を室温にて24時間撹拌し、ついで濾過した。生成物をシリカゲル上のクロマトグラフィーにかけ、CH2Cl2/MeOHで溶出して所望の中間体生成物2.4gを得た。
工程(a)の生成物(185mg、0.41ミリモル)をピリジン(5ml)に溶解し、溶液を氷浴中で冷却し、ステアロイルクロライド(179μl、0.531ミリモル)を加えた。溶液を氷浴中にて2時間保持し、ついで室温にて1時間保持した。ついで、これを蒸発させ、シリカゲル上のクロマトグラフィーにかけた。ジクロロメタン/メタノールで溶出して所望の中間体生成物143mgを得た。
(c)(R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)の生成物(138mg、0.192ミリモル)を氷浴中で冷却し、トリフルオロ酢酸(5ml)を加えた。溶液を氷浴中で45分間保持し、ついで蒸発させて油状物を得た。水(0.5〜1ml)を加え、2度蒸発させた。残渣をもう1度水(5ml)に溶解し、濾過し、凍結乾燥して所望の生成物148mgをビストリフルオロ酢酸塩として得た。
(R)−9−[2−(ミリストイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにミリストイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[2−(オレオイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにオレオイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[2−(ブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
(a)(R)−9−[4−(N−tert−ブトキシカルボニル−L−バリルオキシ)−2−(ブチリルオキシメチル)ブチル]グアニン
DCC(110mg、0.53ミリモル)をジクロロメタン(10ml)に溶解し、酪酸(82mg、0.93ミリモル)を加えた。室温で4時間後、混合物を濾過し、濾液を蒸発させた。残渣をピリジン(5ml)に溶解し、(R)−9−[4−(N−tert−ブトキシカルボニル−L−バリルオキシ)−2−ヒドロキシメチル]ブチル]グアニン(200mg、0.44ミリモル)(実施例1、工程(a))を加えた。混合物を室温にて120時間撹拌した。TLCにより反応は不完全であったので上記手順を用いてさらに無水物を調製した。この無水物を加え、混合物をさらに20時間撹拌した。反応混合物を蒸発させ、まずシリカゲル上、ついで酸化アルミニウム上のクロマトグラフィーにかけ、両方の場合にジクロロメタン/メタノールで溶出して中間体生成物79mgを得た。
(b)(R)−9−[2−(ブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(a)の中間体生成物を実施例1工程(c)と同様にして脱保護して所望の生成物84mgをビストリフルオロ酢酸塩として得た。
(R)−9−[2−(デカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにデカノイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[2−(ドコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアリルクロライドと溶媒としてのDMFとジクロロメタンとの混合物との代わりに実施例1工程(a)のDMAP/DCC条件をドコサン酸とともに用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[4−(L−イソロイシルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン
本実施例は、製造反応式Bの適用を説明するものである。
(a)(R)−9−[2−ヒドロキシメチル4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン
H2G(2g、8ミリモル)を乾燥DMFとともに2回共蒸発させ、ついで乾燥DMF(120ml)およびピリジン(1ml)中に懸濁した。懸濁液にジクロロメタン(20ml)中のt−ブチルジフェニルクロロシラン(2.1ml、8.2ミリモル)を0℃にて30分間かけて滴下した。滴下完了時点で反応混合物は透明な溶液となった。反応を0℃で2時間続け、ついで4℃で一夜保持した。反応液にメタノール(5ml)を加えた。室温で20分後、反応混合物を小容量まで蒸発させ、炭酸水素ナトリウム水溶液に注ぎ、ジクロロメタンで2回抽出した。有機相を硫酸ナトリウムで乾燥させ、真空蒸発させた。生成物を、MeOH濃度を段階的に大きくするメタノール/ジクロロメタン系を用いたシリカゲルカラムクロマトグラフィーにより単離した。生成物をCH2Cl2中の7%MeOHで溶出して1.89gを得た。
(b)(R)−9−[2−(ステアロイルオキシメチル)−4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン
(R)−9−[2−ヒドロキシメチル4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン(2.31g、5ミリモル)を乾燥ピリジンとともに2回共蒸発させ、ピリジン(20ml)中に溶解した。溶液にジクロロメタン(2ml)中のステアロイルクロライド(1.86ml、5.5ミリモル、テクニカルグレード)を−5℃にてゆっくりと滴下した。反応液を同温度にて1時間、ついで5℃で2時間保持した。反応をTLCによりモニターした。反応が未完了であったため、−5℃にてステアロイルクロライド(0.29ml)をさらに加えた。5℃にて30分後、メタノール(3ml)を加え、反応混合物を20分間撹拌した。ついで、これを炭酸水素ナトリウム水溶液中に注ぎ、ジクロロメタンで抽出した。有機相を乾燥させ、生成物を、MeOHを段階的に大きくするシリカゲルカラムクロマトグラフィーにより精製し、CH2Cl2中の3.5%MeOHで溶出した(収量2.7g)。
(c)(R)−9−[(4−ヒドロキシ−2−(ステアロイルオキシメチル)ブチル]グアニン
(R)−9−[2−(ステアロイルオキシメチル)−4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン(2.7g、3.56ミリモル)を乾燥THF(30ml)に溶解し、溶液にフッ化水素−ピリジン(1.5ml)を加えた。反応を4℃で一夜保持し、TLCによりモニターした。反応は約80%変換に達した。さらにHF−ピリジン(0.75ml)を加えた。4時間後、TLCは出発物質が消失したことを示した。温度を上げることなく反応混合物を真空濃縮し、さらにピリジン(5ml)を加え、再び蒸発させた。生成物をシリカゲルカラムクロマトグラフィーにより単離した(収量1.26g)。
(d)(R)−9−[4−(N−BOC−L−イソロイシルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン
(R)−9−[4−ヒドロキシ−2−(ステアロイルオキシメチル)ブチル]グアニン(135mg、0.26ミリモル)およびN−BOC−L−イソロイシン(180mg、0.78ミリモル)を乾燥DMFとともに2回共蒸発させ、同溶媒(3.5ml)中に溶解した。溶液に1,3−ジシクロヘキシルカルボジイミド(160mg、0.78ミリモル)および4−ジメチルアミノピリジン(4.8mg、0.039ミリモル)を加えた。18時間反応させた後、反応混合物をセライト濾過し、通常通り処理した。生成物をシリカゲルカラムクロマトグラフィーにより単離し、CH2Cl2中の5%MeOHで溶出した(収量160mg)。
(e)(R)−9−[4−(L−イソロイシルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン
工程(d)の(R)−9−[4−(N−BOC−L−イソロイシルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン(150mg、0.205ミリモル)を0℃にてトリフルオロ酢酸(3ml)で20分間処理した。溶液を真空蒸発させた。残渣をトルエンとともに2回共蒸発させ、真空下に数時間保持した。残渣をMeOH(2ml)中に溶解し、蒸発させてトリフルオロ酢酸塩をガラス状の生成物として得た(収量191mg)。
(R)−9−[2−(デカノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにデカノイルクロライドを用い、実施例7と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[4−(L−イソロイシルオキシ)−2−(ミリストイルオキシメチル)ブチル]グアニン
工程(a)においてN−BOC−バリンの代わりにN−BOC−L−イソロイシンを用い、工程(b)においてステアリルクロライドの代わりにミリストイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロアセチル塩として得た。
(R)−9−[2−(4−アセチルブチリルオキシメチル−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてN−t−Boc−L−バリンの代わりに実施例1工程(a)のDCC/DMAP条件を4−アセチル酪酸とともに用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−9−[2−(ドデカノイルオキシメチル−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにドデカノイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
実施例12
(R)−9−[2−パルミトイルオキシメチル−4−(L−バリルオキシ)ブチル]グアニン
工程(b)においてステアロイルクロライドの代わりにパルミトイルクロライドを用い、実施例1と同様にして標記化合物をビストリフルオロ酢酸塩として得た。
(R)−2−アミノ−9−[2−ステアロイルオキシメチル−4−(L−バリルオキシ)ブチル]プリン
本実施例は、基R1の脱酸素化を示すものである。
(a)(R)−2−アミノ−9−(2−ステアロイルオキシメチル−4−(N−tert−ブトキシカルボニル−L−バリルオキシ)ブチル)−6−クロロプリン:
実施例1工程(b)の(R)−9−(2−ステアロイルオキシメチル−4−(N−tert−ブトキシカルボニル−L−バリルオキシ)ブチル)グアニン(646mg、0.9ミリモルのアセトニトリル中の溶液にテトラメチルアンモニウムクロライド(427mg、2.7ミリモル)、N,N−ジエチルアニリン(0.716ml、4.5ミリモル)およびオキシ塩化リン(0.417ml、4.5ミリモル)を加えた。反応を還流下で保持し、進行をTLCによりモニターした。3時間後、反応混合物を真空蒸発させ、残渣をジクロロメタンに溶解し、ついで冷炭酸水素ナトリウム水溶液に注いだ。有機相を蒸発させ、シリカゲルカラムクロマトグラフィーにより精製した。収量:251mg。
メタノール/酢酸エチル(6ml、3:1V/V)中の(R)−2−アミノ−9−(2−ステアロイルオキシメチル−4−(N−tert−ブトキシカルボニル−L−バリルオキシ)ブチル)−6−クロロプリン(240mg、0.33ミリモル)の溶液にギ酸アンモニウム(105mg、1.65ミリモル)および10%パラジウム/炭素(15mg)を加えた。反応を還流下で1時間保持し、ギ酸アンモニウム(70mg)を再度加えた。さらに1時間後にTLCが反応の完了を示したので、混合物をセライト濾過し、エタノールで充分に洗浄した。濾液を蒸発させ、シリカゲルカラムで精製した。収量:193mg。
(R)−2−アミノ−9−(2−ステアロイルオキシメチル−4−(N−tert−ブトキシカルボニル−L−バリルオキシ)ブチル)プリン(180mg、0.26ミリモル)を0℃にてトリフルオロ酢酸(5ml)で40分間処理した。これを、ついで真空蒸発させ、トルエンおよびメタノールで順番に共蒸発させた。残渣を一夜凍結乾燥して所望の生成物195mgを得た。
(R)−9−[4−ヒドロキシ−2−(ステアロイルオキシメチル)ブチル]グアニンの他の製造法
(a)4,4−ジエトキシ−2−エトキシカルボニル酪酸エチルの製造
(d)(2S)−2−アセトキシメチル−4,4−ジエトキシブチルトルエンスルホネートの製造
50mL容の1首丸底フラスコ中に実施例14工程(d)の生成物(3.88g、10ミリモル)、無水DMF(20mL)、2−アミノ−4−クロロ−プリン(2.125g、12.5ミリモル)およびK2CO3(4.83g)を加えた。得られた懸濁液をN2ブランケット(blanket)下で40℃にて20時間撹拌した。混合物をロータリーエバポレーター上で濃縮して大部分のDMFを除いた。残渣をEtOAc(50mL)およびH2O(50mL)で希釈した。反応混合物を分別漏斗に移し、振盪し、水性層を分離した。水性層をEtOAc(25mL)で抽出した。有機層をコンバインし、5%KH2PO4(75mL)で洗浄した。有機層を分離し、H2O(75mL)、食塩水(75mL)で洗浄し、Na2SO4で乾燥し、濾過し、真空濃縮して粗製の生成物3.95gを得た。粗製の生成物を40mLのメチル−t−ブチルエーテルでスラリー化した。この混合物を4℃で一夜撹拌し、混合物を濾過した。濾液を濃縮して3.35gの生成物を油状物(HPLC分析に基づいて所望の生成物2.6gを含有)として得た。
500mL容の1首丸底フラスコ中にベンジルアルコール(136mL)を加え、0℃に冷却し、ついでKO−t−Bu(36g、321ミリモル)を少しずつ加えた。温度を40℃まで温め、混合物を20分間撹拌した。この混合物に、25mLの無水THFおよびベンジルアルコール(30mL)中に溶解した実施例14工程(e)の粗製生成物(24.7g、64.2ミリモル)を0℃にて加えた。温度を2時間かけてゆっくりと8℃に温めた。反応混合物を500mLの氷に注ぎ、500mLのMTBEで抽出した。有機層を250mLの食塩水で洗浄し、Na2SO4で乾燥し、濾過し、真空濃縮して193gの所望の生成物のベンジルアルコール溶液を得た。HPLC分析は、この溶液が25.96gの所望の生成物を含有することを示した。
100mL容の1首丸底フラスコ中に、無水EtOH(20mL)中に溶解した実施例14工程(f)の粗製の生成物(1.30g、3.13ミリモルの実施例14工程(f)の生成物を含有するベンジルアルコール溶液を9.65g)を加えた。これに5mLの無水EtOH中でスラリー化した0.45gの10%Pd/Cを加えた。反応フラスコを排気し、H2のバルーンでH2を3回加えた。反応フラスコを1気圧H2で与圧し、反応混合物を一夜撹拌した。反応混合物をケイソウ土のパッドで濾過してPd/Cを除いた。揮発成分を真空下で除いた。残渣を25mLの酢酸イソプロピルと混合し、ついで真空濃縮した。残渣をEtOAc(10mL)で希釈し、所望の生成物でシーディングし(seeded)、加熱還流し、ついでCH3CN(2mL)およびMTBE(35ml)を加えた。混合物を30分間撹拌した。沈殿を濾過し、所望の生成物を600mgの一定重量まで乾燥させた。
25mL容の1首丸底フラスコ中に、実施例14工程(g)の生成物(0.650g、2.0ミリモル)、ピリジン(4mL)およびCH2Cl2(2mL)、DMAP(10mg)を加えた。混合物を−5℃に冷却し、CH2Cl2(0.5mL)中に溶解したステアロイルクロライド(790mg、2.6ミリモル)を5分間かけて加えた。得られた混合物を−5℃で16時間撹拌した。無水EtOH(0.138g、3.0ミリモル)を加え、混合物をさらに1時間撹拌した。反応混合物を真空濃縮した。残渣にトルエン(30mL)を加え、ついで混合物を真空濃縮した。再びトルエン(30mL)を残渣に加え、ついで混合物を真空濃縮した。残渣に1%KH2PO4(25mL)を加え、この混合物をCH2Cl2(60mL)で抽出した。有機層を分離し、Na2SO4で乾燥し、濾過し、真空濃縮して1.65gの一定重量とした。粗製の生成物を40gのSiO2上のクロマトグラフィーにかけ、95/5 CH2Cl2−EtOHで溶出して所望の生成物367mgを得た。
25mL容の1首丸底フラスコ中に、THF(1.7mL)に溶解した実施例14工程(h)の生成物(0.234g、0.394ミリモル)を加えた。この溶液にH2O(180mg)中のトリフルオロメタンスルホン酸(0.108g)を加えた。混合物を室温で一夜撹拌した。反応混合物に飽和NaHCO3溶液(10mL)、THF(5mL)、CH2Cl2(2mL)およびNaBH4(0.10g)を加えた。この混合物を30分間撹拌した。反応混合物にKH2PO4の5%溶液(30mL)を加えた。この混合物を2×15mLのCH2Cl2で抽出した。有機層をコンバインし、Na2SO4で乾燥し、濾過し、真空濃縮して207mgの一定重量とした。この物質をEtOAc(8mL)およびCH3CN(0.5mL)から再結晶して所望の生成物173mgを得た。
(R)−9−[4−(N−tert−ブチルオキシカルボニル−L−バリルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニンの他の製造法
(R)−9−[2−(ステアロイルオキシメチル)−4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン(45g)およびTHF(950ml)を2L容フラスコ中でコンバインした。ついで、Boc−L−バリン(3.22g、0.25当量)を加え、ついでテトラブチルアンモニウムフルオライド(THF中に1M、89.05mL)を10分間かけて加えた。透明な反応混合物を室温で2時間50分間撹拌し、反応の進行をTLC(90/10 CH2Cl2/MeOH)によりモニターした。
反応混合物にBoc−L−バリン(35.43g、2.75当量)、DCC(36.67g、2.75当量)およびTHF(25ml)中のジメチルアミノピリジン(1.1g、0.15当量)を加えた。反応混合物を室温で24時間撹拌した。DCCを濾去し、CH2Cl2で洗浄した。濾液を濃縮し、残渣を2リットルのCH2Cl2中に取り、2Lの1/2飽和重炭酸ナトリウム溶液および食塩水溶液で洗浄した。乾燥および蒸発させると約100gの粗製の生成物が得られた。この物質を3%MeOH/CH2Cl2〜5%MeOH/CH2Cl2を用いたシリカクロマトグラフィー(シリカを6000ml)により精製して所望の生成物38.22mgを得た。
実施例16
(R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニンの他の製造法
(a)(R)−9−[2−ヒドロキシメチル)−4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン
H2G(450.0g、1.78モル)およびN,Nジメチルホルムアミド(6.4kg)をブッチ(Bucchi)エバポレーターに入れ、混合物を温めて固体を溶解した。溶液を90℃未満で真空下にて濃縮乾固した。得られた粉末を、スターラー、添加漏斗および温度プローブを備えた22リットル容フラスコに移した。N,N−ジメチルホルムアミド(1.7kg)を加え、ついでピリジン(3.53kg)を加えた。得られた懸濁液を窒素下で−10℃に冷却し、t−ブチルクロロジフェニルシラン(684g、2.49モル)を滴下しながら−5±5℃で撹拌した。得られた混合物を反応が完了するまで(TLC(10:1 メチレンクロライド/メタノール)およびHPLC(4.6×250mmゾルバックス(Zorbax)RxC8(5ミクロン);1.5ml/分の60:40 アセトニトリル−NH4OAC水溶液(0.05M);254nmでUV検出)によりモニター)−5±5℃で撹拌した。水(16kg)を加え、混合物を30分間撹拌して生成物を沈殿させ、ついで混合物を0℃で30分間冷却した。固体を濾過により単離し、生成物のケーキを冷水で洗浄し、空気で吸引乾燥して粗製の生成物をオフホワイトの固体として得た。粗製の固体をピリジン(3kg)中に取り、真空下、60℃で濃縮して水を除去した。乾燥固体残渣をメタノール(10kg)で60℃にて1〜2時間スラリー化し、熱い間に濾過した。濾液を真空濃縮し、固体残渣を酢酸イソプロピル(7kg)とともに30分間還流した。混合物を20℃に冷却し、濾過した。濾過ケーキを50℃で真空乾燥して標記化合物を白色固体(555g)として得た。
(b)(R)−9−[2−(ステアロイルオキシメチル)−4−(t−ブチルジフェニルシリルオキシ)ブチル]グアニン
実施例16工程(a)の生成物(555g、1.113モル)を50リットル容ブッチエバポレーターに入れた。ピリジン(2.7kg)を滴下して固体を溶解し、混合物を真空下、60℃で蒸発乾固した。残渣を新鮮なピリジン(2.7kg)中に取り、スターラー、添加漏斗および温度プローブを備えた22リットル容のフラスコに移した。溶液を窒素下で−5℃に冷却した。メチレンクロライド(1.5kg)中のステアロイルクロライド(440g、1.45モル)の溶液を温度が0℃以下になるように加えた。4−(N,N−ジメチルアミノ)ピリジン(15g、0.12モル)を加え、変換が完了するまで(TLC(10:1 メチレンクロライド/メタノール)およびHPLC(4.6×250mmゾルバックスRxC8(5ミクロン);1.5ml/分の60:40 アセトニトリル−NH4OAC水溶液(0.05M);254nmでUV検出)によりモニター)混合物を−5〜0℃で2〜4時間撹拌した。反応の終了時点でアセトニトリル(8.7kg)を加え、混合物を15分間以上撹拌して生成物を沈殿させた。スラリーを0℃にて2時間冷却し、固体を濾過により単離し、濾過ケーキをアセトニトリル(2kg)で洗浄した。所望の生成物を白色固体(775g)として得た。
(c)(R)−9−[4−ヒドロキシ−2−(ステアロイルオキシメチル)ブチル]グアニン
テトラヒドロフラン(10kg)中の実施例16工程(b)の生成物(765g、0.29モル)の溶液をリアクター中で調製した。テトラヒドロフラン中のテトラ(n−ブチル)アンモニウムフルオライドの溶液(1M溶液を1.7kg、1.7モル)を加え、得られた透明な溶液を20±5℃で4時間撹拌した。水(32kg)を加え、得られたスラリーを1時間撹拌し、ついで0℃に30分間冷却した。沈殿を濾過により単離し、濾過ケーキを水(10kg)およびアセトニトリル(5kg)で順番に洗浄した。真空下、25℃で乾燥した後、702gの粗製の生成物を得た。粗製の生成物を還流THF(4.2kg)および水(160g)中に溶解し、ついで40℃に冷却し、メチレンクロライド(14.5kg)で処理した。混合物を25±5℃に1時間冷却し、ついで5±5℃に1時間冷却して沈殿を完了した。わずかにオフホワイトの粉末を濾過により単離し、真空下、40℃で乾燥させて所望の生成物(416g)を得た。
(d)(R)−9−[4−(N−Cbz−L−バリルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン
THF(750ml)中のN−Cbz−L−バリン(169g、0.67モル)の溶液を、メカニカルスターラー、温度計および添加漏斗を備えた2リットル容フラスコ中に調製した。THF(250ml)中のジシクロヘキシルカルボジイミド(69.3g、0.34モル)の溶液を5分間かけて加え、得られたスラリーを20±5℃で2時間撹拌した。スラリーを濾過し、濾過ケーキをTHF(300ml)で洗浄した。濾液および洗浄液をスターラーおよび温度計を備えた3リットル容フラスコに入れた。実施例16工程(c)の生成物(116g、0.22モル)を固体として加え、THF(250ml)で濯いだ。4−(N,N−ジメチルアミノ)ピリジン(2.73g、0.022モル)を加え、白色スラリーを20±5℃で撹拌した。15分以内に固体はすべて溶解し、反応は1時間以内に完了した(HPLCにより決定:4.6×250mmゾルバックスRxC8カラム;1ml/分の85:15 アセトニトリル−0.2%HClO4水溶液;254nmでUV検出;出発物質は4.1分で溶出し、生成物は5.9分で溶出)。水(5ml)を加えて反応を停止させ、溶液を真空濃縮して薄黄色の半固体を得た。これをメタノール(1.5リットル)中に取り、30分間加熱還流した。溶液を25℃に冷却し、沈殿を濾過により除去した。濾液を真空濃縮して粘性の薄黄色の油状物を得た。アセトニトリル(1L)を加え、得られた白色懸濁液を20±5℃で90分間撹拌した。粗製の固体生成物を濾過により単離し、アセトニトリル(2×100ml)で洗浄し、一夜空気乾燥して所望の生成物をワックス状の粘着性固体(122g)として得た。これをさらに酢酸エチル(500ml)から結晶化し、30℃で真空乾燥することによって精製して所望の生成物を白色のワックス状固体(104g)として得た。
(e)(R)−9−[4−(L−バリルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン
温(40℃)エタノール(2.3L)中の実施例16工程(d)の生成物(77g)の溶液を、5%Pd−C(15.4g)とともに水素化リアクターに入れた。混合物を40psiの水素圧下、40℃で4時間激しく撹拌し、排気し、さらに4〜10時間水素化した。触媒を濾去し、濾液を真空濃縮して白色固体を得た。これをエタノール(385ml)とともに25℃で1時間撹拌し、ついで0℃に冷却し、濾過した。濾過ケーキを空気乾燥し、ついで35℃で真空下で乾燥して標記化合物を白色粉末(46g)として得た。
実施例17
(R)−9−[2−(L−バリルオキシ)−4−(ステアロイルオキシメチル)ブチル]グアニン
(a)(R)−9−[2−ヒドロキシメチル−4−(ステアロイルオキシ)ブチル]グアニン
H2G(506mg;2.0ミリモル)を乾燥N,N−ジメチルホルムアミド(40ml)中にピリジン(400mg;5.06ミリモル)および4−ジメチルアミノピリジン(60mg;0.49ミリモル)とともに溶解した。ステアロイルクロライド(1500mg;4.95ミリモル)を加え、混合物を室温に一夜保った。溶媒のほとんどを真空蒸発させ、残渣を70mlの酢酸エチルおよび70mlの水とともに撹拌し、固体を濾去し、酢酸エチルおよび水で洗浄し、乾燥して680mgの粗製生成物を得た。シリカゲル上のカラムクロマトグラフィー(クロロホルム:メタノール 15:1)により純粋な標記化合物を白色固体として得た。
ジクロロメタン(20ml)中のN−Boc−L−バリン(528mg;2.1ミリモル)およびN,N’−ジシクロヘキシルカルボジイミド(250mg;1.21ミリモル)の混合物を室温で一夜撹拌し、ジシクロヘキシル尿素を濾去し、少量のジクロロメタンで抽出し、濾液を真空蒸発させて小容量とした。(R)−9−[2−ヒドロキシメチル−4−(ステアロイルオキシ)ブチル]グアニン(340mg;0.654ミリモル)、4−ジメチルアミノピリジン(25mg;0.205ミリモル)および乾燥N,N−ジメチルホルムアミド(15ml)を加え、混合物をN2下、50℃で4時間撹拌した。溶媒を真空蒸発させて小容量とした。シリカゲル、ついで酸化アルミニウム上のカラムクロマトグラフィー(溶出液として酢酸エチル:メタノール:水 15:2:1)により185mg(39%)の純粋な標記化合物を白色固体として得た。
(R)−9−[2−(N−Boc−L−バリルオキシメチル)−4−(ステアロイルオキシ)ブチル]グアニン(180mg;0.25ミリモル)に冷トリフルオロ酢酸(2.0g)を加え、溶液を室温で1時間保持し、蒸発して小容量とし、白色の無定形粉末が得られるまでジオキサンで繰り返し凍結乾燥させた。トリフルオロ酢酸塩として得られた標記化合物の収量は定量的なものであった。
(R)−9−[2−ヒドロキシメチル−4−(ステアロイルオキシ)ブチル]グアニンの他の製造法
H2G(7.60g、30ミリモル)を乾燥DMF(200ml)中で加熱溶解した。溶液を濾過して固体不純物を除去し、20℃に冷却し(H2Gは結晶化する)、ピリジン(9.0g、114ミリモル)、4−ジメチルアミノピリジン(0.46g、3.75ミリモル)を添加しついでステアロイルクロライド(20.0g、66ミリモル)をゆっくりと加える間、この温度で撹拌した。撹拌を室温で一夜続けた。ついで、溶媒のほとんどを真空留去し、残渣を200mlの酢酸エチルおよび200mlの水とともに撹拌し、固体を濾去し、酢酸エチルおよび水で洗浄し、乾燥させて粗製の生成物を得た。再結晶の代わりとして、粗製の生成物を100mlの酢酸エチル:メタノール:水(15:2:1)とともにほぼ沸騰するまでしばらく加熱し、懸濁液を30℃にゆっくりと冷却し、濾過して溶液中の2”異性体(2”異性体はより低い温度で結晶化する)のほとんどを得た。抽出手順をもう1回繰り返して真空乾燥した後に、ほとんど異性体を含まない生成物6.57g(42%)を得た。
実施例19
結晶性(R)−9−[2−ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニンの製造
実施例16工程(c)の生成物(20.07g、32.5ミリモル)を無水エタノール(400ml)中に加熱溶解し、濾過し、エタノール(117.5ml)でさらに希釈した。この溶液に水(HPLCグレード、103.5ml)を加え、混合物を35〜40℃に冷却した。混合物を冷却した後、水(HPLCグレード、931.5ml)を充分に撹拌しながら一定速度で16時間かけて加えた。すべての水を加えた後、撹拌を室温で4時間続けた。得られた沈殿を紙で濾過し、室温で真空下にて乾燥させて標記化合物を白色の自由流動性の(free flowing)結晶性粉末(19.43g、97%)として得た(融点169〜170℃)。
実施例20
9−R−(4−ヒドロキシ−2−(L−バリルオキシメチル)ブチル)グアニン
(a)DMF(30ml)中の9−R−(4−(tert−ブチルジフェニルシリルオキシ)−2−(ヒドロキシメチル)ブチル)グアニン(695mg、1.5ミリモル)の溶液に、N−Boc−L−バリン(488mg、2.25ミリモル)、4−ジメチルアミノピリジン(30mg、0.25ミリモル)およびDCC(556mg、2.7ミリモル)を加えた。16時間後、反応液にN−Boc−L−バリン(244mg)およびDCC(278mg)を再度加え、さらに5時間保持した。反応混合物をセライト濾過し、炭酸水素ナトリウム水溶液中に注ぎ、ついでジクロロメタンで抽出した。有機相を蒸発させ、シリカゲルカラムクロマトグラフィーにより精製して950mgのN−保護モノアミノアシル誘導体を得た。
(b)上記中間体(520mg、0.78ミリモル)をTHF(15ml)中に溶解した。溶液にピリジン中のフッ化水素(70%/30%、0.34ml)を加えた。2日後、溶液を蒸発させ、トルエンと共蒸発させた。シリカゲルカラムクロマトグラフィーにより精製して311mgの保護モノアミノアシル化合物を得た。
(R)−9−(2−ヒドロキシメチル−4−(L−イソロイシルオキシ)ブチル)グアニン
(a)DMF(250ml)中の(R)−9−(2−ヒドロキシメチル−4−ヒドロキシブチル)グアニン(2.53g、10ミリモル)の溶液に、N−Boc−L−イソロイシン(2.77g、12ミリモル)、4−ジメチルアミノピリジン(61mg、0.6ミリモル)およびDCC(3.7g、18ミリモル)を加えた。0℃で16時間反応後、N−Boc−L−イソロイシン(1.3g)およびDCC(1.8g)を再度加え、反応液を室温で一夜保持した。反応混合物をセライト濾過し、濾液を蒸発させ、シリカゲルカラムクロマトグラフィーにより精製して1.25gのN−保護モノアミノアシル中間体を得た。
(R)−9−[2−ヒドロキシメチル−4−(L−バリルオキシ)ブチル]グアニン
実施例1工程(a)の生成物を実施例1工程(c)と同様にしてトリフルオロ酢酸で脱保護した。
(R)−9−[2−(L−バリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
(a)(R)−9−[4−(N−Boc−L−バリルオキシ)−2−(N−Boc−L−バリルオキシメチル)ブチル]グアニン
それぞれ2.7当量、0.28当量および3.2当量のN−Boc−バリン、DMAPおよびDCCを用いて実施例1工程(a)に記載の技術を行い、標記化合物を得た。
実施例23工程(a)の中間体を実施例1工程(c)と同様にして脱保護することにより標記化合物をトリフルオロ酢酸塩として得た。
(R)−9−[4−ヒドロキシ−2−(ステアロイルオキシメチル)ブチル]グアニン
実施例7の工程(a)〜(c)に従って標記化合物を調製する。
(R)−9−[2−ヒドロキシメチル−4−(ステアロイルオキシ)ブチル]グアニン
実施例17工程(a)の手順により標記化合物を調製する。
(R)−9−[2−ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニンの他の製造法
(a)(R)−9−[4−N−ベンジルオキシカルボニル−L−バリルオキシ)2−(ヒドロキシメチル)ブチル]グアニン
乾燥H2G(252mg、1ミリモル)、4−ジメチルアミノピリジン(122mg、1ミリモル)およびN−Cbz−L−バリンp−ニトロフェニルエステル(408mg、1.1ミリモル)を乾燥ジメチルホルムアミド(16ml)中に溶解した。23℃で30時間撹拌した後、有機溶媒を除去し、残渣を注意深くクロマトグラフィー(シリカ、2%〜7%メタノール/メチレンクロライド)にかけて所望の生成物を白色固体(151mg、31%)として得た。
(b)(R)−9−[4−N−ベンジルオキシカルボニル−L−バリルオキシ)2−(ステアロイルオキシメチル)ブチル]グアニン
乾燥メチレンクロライド(2ml)中のステアロイルクロライド(394mg、1.3ミリモル)の溶液を、乾燥ピリジン(5ml)中の工程(a)の生成物(243mg、1ミリモル)および4−ジメチルアミノピリジン(20mg)の溶液に、窒素下、−5℃にてゆっくりと滴下した。この温度で反応混合物を12時間撹拌した。メタノール(5ml)を加え、反応液を1時間撹拌した。溶媒を除去した後、残渣をアセトニトリルでトリチュレートし、クロマトグラフィー(シリカ、0〜5%メタノール/メチレンクロライド)にかけて所望の生成物(542mg、72%)を得た。
(c)(R)−9−[2−ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
工程(b)の生成物(490mg、1ミリモル)をメタノール(30ml)中に溶解し、5%Pd/C(100mg)を加えた。水素を充填したバルーンを反応容器の上部に置いた。23℃で6時間後、TLCは出発物質の不在を示した。反応混合物を0.45ミクロンのナイロン膜で濾過して触媒を除去し、溶媒を除いて所望の生成物を白色固体(350mg、99%)として得た。このものは実施例16で得たものと同一であった(スペクトルおよび分析データ)。
実施例27
(R)−9−(4−ヒドロキシ−2−(L−バリルオキシメチル)ブチル)グアニンの他の製造法
実施例23工程(b)の(R)−9−(4−(L−バリルオキシ)−2−(L−バリルオキシメチル)ブチル)グアニン(100mg、0.126ミリモル)を0.1N NaOH水溶液(6.3ml、0.63ミリモル)中に室温にて溶解した。定期的にアリコートを取り、0.5Nトリフルオロ酢酸で中和した。アリコートを蒸発させ、HPLCにより分析して反応の進行をモニターした。4時間後、溶液に0.5Nトリフルオロ酢酸溶液(1.26ml、0.63ミリモル)を加え、反応混合物を蒸発させた。所望の生成物をHPLC(YMC、50×4.6mm、勾配0.1%TFA+0〜50%0.1%TFA(アセトニトリル中)、20分間、254nmにてUV検出)により精製した(収率:13.6%)。
(R)−9−(2−ヒドロキシメチル−4−(L−バリルオキシ)ブチル)グアニンの他の製造法
実施例27の反応溶液をHPLC分離して標記化合物を29.2%の収率で得た。
(R)−9−[2−ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン一塩酸塩
実施例16工程(d)の生成物(360mg、0.479ミリモル)をメタノール(10ml)と酢酸エチル(10ml)との混合物中に溶解した。溶液に10%Pd/C(100mg)および1N HCl(520μl)を加えた。反応混合物を1気圧H2、室温にて2時間撹拌した。反応混合物を濾過し、濾液から溶媒を蒸発させて所望の生成物を結晶性固体(300mg)として得た。
製剤実施例A
錠剤の調合
下記成分を0.15mmふるいでスクリーニングし、乾燥混合する。
10g (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
40g 乳糖
49g 結晶セルロース
1g ステアリン酸マグネシウム
打錠機を用いて混合物を圧錠し、250mgの活性成分を含有する錠剤とする。
製剤実施例B
腸溶錠剤
製剤実施例Aの錠剤に、下記成分を含有する溶液をタブレットコーター(tabletcoater)中でスプレーコーティングする。
120g エチルセルロース
30g プロピレングリコール
10g ソルビタンモノオレエート
1000mlまで 蒸留水
製剤実施例C
除放製剤
50g (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
12g ヒドロキシプロピルメチルセルロース(メトセルK15)
4.5g 乳糖
を乾燥混合し、ポビドンの水性ペーストで顆粒化する。ステアリン酸マグネシウム(0.5g)を加え、混合物を打錠機で圧錠して500mgの活性成分を含有する13mm直径の錠剤とする。
製剤実施例D
ソフトカプセル
250g (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン
100g レシチン
100g ラッカセイ油
本発明の化合物をレシチンおよびラッカセイ油中に分散させ、ソフトゼラチンカプセル中に充填する。
生物学的実施例1
ラットにおけるバイオアベイラビリティー試験
本発明の化合物のバイオアベイラビリティーをラットモデルにおいて親化合物であるH2Gおよび他のH2G誘導体と比較した。本発明の化合物および比較化合物を複数の(multiples of)3匹の個々に体重を計った動物に経口(カテーテルにより胃中へ)で投与して、被験化合物成分の溶解度に依存して水性(実施例4、5、比較例1〜3、5、8)ビヒクル、落花生油(比較例4、9、10)ビヒクルまたはプロピレングリコール(実施例1〜3、6〜12、17、比較例6、7)ビヒクル中に溶解したプロドラッグ0.1ミリモル/kgを与えた。投与後5時間〜投与後約17時間、動物を絶食し、メタボリックケージ中に維持した。投与後24時間の間、採尿し、分析のときまで凍結させた。尿中のH2Gをステーレ
比較例1(H2G)は実施例1〜12の製造に用いたのと同じバッチからのものであった。比較例2(モノVal−H2G)および比較例3(ジVal−H2G)の調製は実施例20および実施例23に示してある。比較例4(ジステアロイルH2G)は、実施例1の工程(b)に匹敵するエステル化条件での非保護H2Gのジエステル化により調製した。比較例5および8(Val/AcH2G)は、無水酢酸および関連モノバリンH2Gを用いて実施例4と同様にして調製した。比較例6(Ala/ステアロイルH2G)は、工程4においてN−t−Boc−L−アラニンを用いて実施例6と同様にして調製した。比較例7(Gly/デカノイル)は、N−t−Boc−L−グリシンで調製した工程(a)中間体を用いて実施例5と同様にして調製した。比較例9および10の調製は、それぞれ実施例24および25に示す。その結果を次頁の表2に示す。
さらに、たとえば比較例5、6および7の結果から、本発明の特定の脂肪酸のみが特定のアミノ酸との組み合わせで薬動力学パラメータにおけるこれら劇的で予測できない増大をもたらすことも明らかである。
生物学的実施例2
ラットにおける血漿濃度
血漿濃度アッセイを雄のスプラーグドーリー由来ラットで行った。投与に先立ち、動物を一夜絶食させたが水分は自由にとらせた。評価すべき各化合物をプロピレングリコール中で10mgH2G/mlに対応する濃度にて溶液/懸濁液として調製し、室温にて8時間振盪した。ラット群(各群少なくとも4匹のラット)に各化合物の10mg/kg(1ml/kg)経口投与量を与えた;この投与量を胃管栄養法(gavage)により投与した。投与後の選択した時点(投与後0.25時間、0.5時間、1時間、1.5時間、2時間、4時間、6時間、9時間、12時間、15時間および24時間)でヘパリン処理血液試料(0.4ml/試料)を各動物の尾静脈から得た。血液試料を直ちに氷浴中で冷却した。採取から2時間以内に血漿を遠心分離により赤血球から分離し、分析のときまで凍結させた。目的成分を血漿タンパク質からアセトニトリル沈殿を用いて分離した。凍結乾燥および再構成後、蛍光検出を用いる逆相HPLCにより血漿濃度を測定した。H2Gおよび他の被験化合物の経口取り込みを、経口投与から得られる曲線下H2G面積を別の群のラットに投与したH2Gの10mg/kg静脈内投与から得られるものと比較することによって決定した。その結果を上記表1Bに示す。
生物学的実施例3
サルにおけるバイオアベイラビリティー
実施例1および比較例3の化合物(上記生物学的実施例1を参照)をカニクイザルに胃管栄養法により経口で投与した。これら溶液は、以下のものを含んでいた:
実施例1 6.0mlのプロピレングリコール中に150mgを溶解、25mg/kgまたは0.0295ミリモル/kgに対応。
比較例3 7.0mlの水中に164mgを溶解、23.4mg/kgまたは0.0295ミリモル/kgに対応。
血液試料を30分、1時間、2時間、3時間、4時間、6時間、10時間および24時間で採取した。血漿を2500rpmで遠心分離により分離し、分析までに凍結乾燥する前に54℃で20分間不活化した。血漿H2Gレベルを上記実施例30のHPLC/UVアッセイによりモニターした。
図1は血漿H2G回収を時間の関数として示す。単一の動物試験から統計的に有意な結論を引き出すことはできないが、本発明の化合物を与えた動物は他のH2Gプロドラッグを与えた動物に比べて若干より速やかで若干より大きなH2Gへの暴露を経験したように思われる。
生物学的実施例4
抗ウイルス活性
単純ヘルペスウイルス−1(HSV−1)感染マウスは、インビボでの抗ウイルス剤の効能を決定するための動物モデルとして用いることができる。LD50の1000倍のHSV−1を腹腔内接種したマウスに、現在市場に出回っている抗ヘルペス剤であるアシクロビルを含む製剤(滅菌水ビヒクル中の2%プロピレングリコール中に21および83mg/kg、毎日3回、経口)かまたは実施例29の化合物(滅菌水ビヒクル中の2%プロピレングリコール中に21および83mg/kg、毎日3回、経口)のいずれかを接種5時間後から開始して5日間連続で投与した。動物の存命を毎日評価した。その結果を図2に示すが、図2は生存率を時間に対して作図したものである。図の説明において、本発明の化合物は実施例29と、アシクロビルはACVと表示してある。生存したHSV−1感染マウスのパーセントは、本発明の化合物を所定量投与した場合に等価量のアシクロビルを投与した場合に比べて有意に大きくなった。
Claims (13)
- 式I
[式中、(a)R1が−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2であり、R2が−C(O)C3〜C21の飽和_アルキルカルボニルであるか;または
(b)R1が−C(O)C3〜C21の飽和_アルキルカルボニルであり、R2が−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2であり;
R3はOHまたはHであり、R3=Oである場合に互変異体を含む_]
の化合物および薬理学的に許容しうるその塩。 - R1が−C(O)CH(CH(CH3)2)NH2または−C(O)CH(CH(CH3)CH2CH3)NH2であり、R2が−C(O)C3〜C21の飽和_アルキルカルボニルであ_る、請求項1に記載の化合物。
- −C(_O)C 3 〜C 21 の飽和アルキルカルボニルが−C(O)C9〜C17の飽和アルキルカルボニルである、請求項1に記載の化合物。
- R3がヒドロキシである請求項1に記載の化合物。
- (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ミリストイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ブチリルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(デカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−(ドコサノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[4−(L−イソロイシルオキシ)−2−(ステアロイルオキシメチル)ブチル]グアニン、
(R)−9−[2−(デカノイルオキシメチル)−4−(L−イソロイシルオキシ)ブチル]グアニン、
R)−9−[4−(L−イソロイシルオキシ)−2−(ミリストイルオキシメチル)ブチル]グアニン、
(R)−9−[2−(ドデカノイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン、
(R)−9−[2−パルミトイルオキシメチル−4−(L−バリルオキシ)ブチル]グアニン、
(R)−2−アミノ−9−[2−ステアロイルオキシメチル−4−(L−バリルオキシ)ブチル]プリン、
(R)−9−[2−(L−バリルオキシメチル)−4−(ステアロイルオキシ)ブチル]グアニン、
または薬理学的に許容しうるその塩
よりなる群れから選ばれる、請求項1に記載の化合物。 - (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニン。
- (R)−9−[2−(ステアロイルオキシメチル)−4−(L−バリルオキシ)ブチル]グアニンまたは薬理学的に許容しうるその塩。
- 薬理学的に許容しうる担体または希釈剤とともに請求項1に記載の化合物を含んでなる医薬組成物。
- 薬理学的に許容しうる担体または希釈剤とともに請求項5に記載の化合物を含んでなる医薬組成物。
- 薬理学的に許容しうる担体または希釈剤とともに請求項6に記載の化合物を含んでなる医薬組成物。
- 請求項1に記載の化合物の有効量を含む、ウイルス感染の治療または予防用剤。
- 水痘帯状ヘルペスウイルス、単純ヘルペスウイルス1型および2型、エプスタイン−バーウイルスまたはヘルペス6型(HHV−6)および8型(HHV−8)を含むヘルペス感染の治療または予防のためのものである請求項11に記載のウイルス感染の治療または予防用剤。
- SIV、HIV−1およびHIV−2を含むレトロウイルス感染の治療または予防のためのものである請求項11に記載のウイルス感染の治療または予防用剤。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SE9600614-3 | 1996-02-16 | ||
| SE9600613-5 | 1996-02-16 | ||
| SE9600614A SE9600614D0 (sv) | 1996-02-16 | 1996-02-16 | Antiviral compounds |
| SE9600613A SE9600613D0 (sv) | 1996-02-16 | 1996-02-16 | Acyclic nucleoside derivatives |
| PCT/SE1997/000241 WO1997030051A1 (en) | 1996-02-16 | 1997-02-14 | Acyclic nucleoside derivatives |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2000504720A JP2000504720A (ja) | 2000-04-18 |
| JP2000504720A5 JP2000504720A5 (ja) | 2004-11-11 |
| JP4171069B2 true JP4171069B2 (ja) | 2008-10-22 |
Family
ID=26662518
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52927997A Expired - Fee Related JP4171069B2 (ja) | 1996-02-16 | 1997-02-14 | 非環状ヌクレオシド誘導体 |
| JP9529280A Ceased JP2000504721A (ja) | 1996-02-16 | 1997-02-14 | 非環状ヌクレオシドの合成 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP9529280A Ceased JP2000504721A (ja) | 1996-02-16 | 1997-02-14 | 非環状ヌクレオシドの合成 |
Country Status (25)
| Country | Link |
|---|---|
| US (5) | US5869493A (ja) |
| EP (2) | EP0888348B1 (ja) |
| JP (2) | JP4171069B2 (ja) |
| KR (1) | KR100464692B1 (ja) |
| CN (1) | CN1067073C (ja) |
| AR (1) | AR005827A1 (ja) |
| AT (2) | ATE231507T1 (ja) |
| AU (1) | AU715062B2 (ja) |
| BG (1) | BG64179B1 (ja) |
| CA (1) | CA2243826C (ja) |
| CZ (1) | CZ292169B6 (ja) |
| DE (2) | DE69718619T2 (ja) |
| DK (1) | DK0888348T3 (ja) |
| EA (2) | EA001404B1 (ja) |
| ES (2) | ES2189941T3 (ja) |
| HU (1) | HU229865B1 (ja) |
| IL (1) | IL124760A (ja) |
| MY (1) | MY123083A (ja) |
| NO (1) | NO322930B1 (ja) |
| NZ (1) | NZ330472A (ja) |
| PL (1) | PL184892B1 (ja) |
| SK (1) | SK284613B6 (ja) |
| TR (1) | TR199801318T2 (ja) |
| TW (1) | TW533213B (ja) |
| WO (2) | WO1997030052A1 (ja) |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6458772B1 (en) | 1909-10-07 | 2002-10-01 | Medivir Ab | Prodrugs |
| US5869493A (en) * | 1996-02-16 | 1999-02-09 | Medivir Ab | Acyclic nucleoside derivatives |
| US6703394B2 (en) | 1996-02-16 | 2004-03-09 | Medivir Ab | Acyclic nucleoside derivatives |
| WO1998021223A1 (en) * | 1996-11-12 | 1998-05-22 | Medivir Ab | Nucleosides |
| US6184376B1 (en) | 1997-02-10 | 2001-02-06 | Mediver Ab | Synthesis of acyclic nucleoside derivatives |
| CA2572577C (en) * | 1997-02-10 | 2011-04-05 | Medivir Ab | Synthesis of acyclic nucleoside derivatives |
| PT988304E (pt) * | 1997-08-15 | 2001-10-31 | Medivir Ab | Analogos de nucleosidos tais como os antivirais que contem inibidores de transcritase reversa retroviral e de adn-polimerase do virus da hepatite b (hbv) |
| AU1711599A (en) * | 1997-12-19 | 1999-07-12 | Bristol-Myers Squibb Company | Prodrugs of lobucavir and methods of use |
| EP1511750A1 (en) * | 2003-04-30 | 2005-03-09 | Teva Pharmaceutical Industries Ltd. | Process for the preparation of famciclovir |
| ES2325088T3 (es) | 2004-12-30 | 2009-08-25 | Medivir Ab | Compuestos utiles en el tratamiento de hiv. |
| US20080233053A1 (en) * | 2005-02-07 | 2008-09-25 | Pharmalight Inc. | Method and Device for Ophthalmic Administration of Active Pharmaceutical Ingredients |
| CN101033238B (zh) * | 2006-03-06 | 2011-02-16 | 中国科学院上海药物研究所 | 嘌呤类化合物双氨基酸酯的制备方法和用途 |
| ZA200702234B (en) * | 2006-03-21 | 2008-07-30 | Cipla Ltd | Preparation of ester of purine derivatives |
| US9296776B2 (en) | 2007-07-09 | 2016-03-29 | Eastern Virginia Medical School | Substituted nucleoside derivatives with antiviral and antimicrobial properties |
| AU2008305666B2 (en) * | 2007-09-21 | 2014-08-28 | Epiphany Biosciences, Inc. | Valomaciclovir polymorphs |
| WO2011022712A1 (en) * | 2009-08-21 | 2011-02-24 | Epiphany Biosciences, Inc. | Method of treating infectious mononucleosis with acylic nucleoside derivatives |
| HUE037408T2 (hu) | 2011-06-10 | 2018-08-28 | Univ Oregon Health & Science | CMV glikoproteinek és rekombináns vektorok |
| KR101763127B1 (ko) | 2015-06-23 | 2017-08-01 | (주)농협아그로 | 끈끈이 엠보싱 트랩 테이프 |
| WO2024118480A1 (en) * | 2022-11-28 | 2024-06-06 | Epiphany Biosciences | Use of antiviral agents, composition of matter, combination preparations/agents to treat chronic diseases associated with epstein-barr virus and other human herpes viruses |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3116282A (en) | 1960-04-27 | 1963-12-31 | Upjohn Co | Pyrimidine nucleosides and process |
| FR2040177A1 (en) | 1969-01-31 | 1971-01-22 | Robugen Gmbh | 2-deoxyribosyl-uracil derivs |
| US3817982A (en) | 1971-12-29 | 1974-06-18 | Syntex Inc | 2{40 ,3{40 -unsaturated nucleosides and method of making |
| US4247544A (en) | 1979-07-02 | 1981-01-27 | The Regents Of The University Of California | C-5 Substituted uracil nucleosides |
| US4267171A (en) | 1979-07-02 | 1981-05-12 | The Regents Of The University Of California | C-5 Substituted cytosine nucleosides |
| JPS57146798A (en) | 1981-03-06 | 1982-09-10 | Yamasa Shoyu Co Ltd | Hexosyl nucleoside derivative |
| NL8202626A (nl) * | 1982-06-29 | 1984-01-16 | Stichting Rega V Z W | Derivaten van 9-(2-hydroxyethoxymethyl)guanine. |
| IL73682A (en) * | 1983-12-20 | 1991-08-16 | Medivir Ab | Antiviral pharmaceutical compositions containing 9-hydroxy aliphatic derivatives of guanine,some new such derivatives and process for their preparation |
| SE8406538D0 (sv) * | 1984-12-21 | 1984-12-21 | Astra Laekemedel Ab | Novel derivatives of purine |
| US4724232A (en) | 1985-03-16 | 1988-02-09 | Burroughs Wellcome Co. | Treatment of human viral infections |
| EP0286825A3 (en) | 1987-03-18 | 1989-04-12 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Use of 3'-fluro-3' deoxythymidine for the manufacture of a medicament for the treatment of virus infections |
| SE8701605D0 (sv) | 1987-04-16 | 1987-04-16 | Astra Ab | Novel medicinal compounds |
| AP160A (en) * | 1987-08-15 | 1991-11-18 | The Wellcome Foundation Ltd | Therapeutic acyclic nucleosides. |
| CA1321994C (en) * | 1987-10-28 | 1993-09-07 | Reid Von Borstel | Acylated uridine and cytidine and uses thereof |
| SE8704298D0 (sv) | 1987-11-03 | 1987-11-03 | Astra Ab | Compounds for use in therapy |
| US5284837A (en) | 1988-05-06 | 1994-02-08 | Medivir Ab | Derivatives of purine, process for their preparation and a pharmaceutical preparation |
| SE8801729D0 (sv) | 1988-05-06 | 1988-05-06 | Astra Ab | Purine derivatives for use in therapy |
| US5216141A (en) | 1988-06-06 | 1993-06-01 | Benner Steven A | Oligonucleotide analogs containing sulfur linkages |
| SE8802173D0 (sv) | 1988-06-10 | 1988-06-10 | Astra Ab | Pyrimidine derivatives |
| DE68922903T2 (de) * | 1988-12-19 | 1995-11-23 | Wellcome Found | Antivirale Pyrimidin- und Purinverbindungen, Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Präparate. |
| FI95384C (fi) | 1989-04-06 | 1996-01-25 | Squibb Bristol Myers Co | Menetelmä 3'-deoksi-3'-substituoitujen metyylinukleosidien valmistamiseksi ja menetelmässä käytettäviä välituotteita |
| IE980216A1 (en) * | 1989-04-17 | 2000-02-23 | Scotia Holdings Plc | Anti-virals |
| US5674869A (en) * | 1990-07-07 | 1997-10-07 | Beecham Group Plc | Pharmaceutical treatment |
| SE9003151D0 (sv) | 1990-10-02 | 1990-10-02 | Medivir Ab | Nucleoside derivatives |
| GB2260319B (en) * | 1991-10-07 | 1995-12-06 | Norsk Hydro As | Acyl derivatives of nucleosides and nucleoside analogues having anti-viral activity |
| US5874578A (en) | 1992-07-13 | 1999-02-23 | Bristol-Myers Squibb | Process for preparing guanine-containing antiviral agents and purinyl salts useful in such process |
| GB9307043D0 (en) * | 1993-04-05 | 1993-05-26 | Norsk Hydro As | Chemical compounds |
| DE4311801A1 (de) * | 1993-04-09 | 1994-10-13 | Hoechst Ag | Neue Carbonsäureester von 2-Amino-7-(1,3-dihydroxy-2-propoxymethyl)purin, deren Herstellung sowie deren Verwendung |
| GB9320316D0 (en) * | 1993-10-01 | 1993-11-17 | Smithkline Beecham Plc | Pharmaceuticals |
| TW282470B (ja) * | 1993-11-18 | 1996-08-01 | Ajinomoto Kk | |
| US5521161A (en) | 1993-12-20 | 1996-05-28 | Compagnie De Developpment Aguettant S.A. | Method of treating HIV in humans by administration of ddI and hydroxycarbamide |
| WO1995022330A1 (en) * | 1994-02-17 | 1995-08-24 | Commonwealth Scientific And Industrial Research Organisation | Antiviral agents |
| US5543414A (en) * | 1994-07-28 | 1996-08-06 | Syntex (Usa) Inc. | Achiral amino acid acyl esters of ganciclovir and its derivatives |
| PE32296A1 (es) * | 1994-07-28 | 1996-08-07 | Hoffmann La Roche | Ester de l-monovalina derivado de 2-(2-amino-1,6-dihidro-6-oxo-purin-9-il) metoxi-1,3-propandiol y sus sales farmaceuticamente aceptables |
| JP3906488B2 (ja) | 1995-02-21 | 2007-04-18 | 味の素株式会社 | プリン誘導体の製造方法 |
| US5700936A (en) * | 1996-01-26 | 1997-12-23 | Syntex (U.S.A.) Inc. | Process for preparing a 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl) methoxy-1,3-propanediol valinate |
| US5756736A (en) * | 1996-01-26 | 1998-05-26 | Syntex (U.S.A.) Inc. | Process for preparing a 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol derivative |
| WO1997027194A1 (en) * | 1996-01-26 | 1997-07-31 | F. Hoffmann-La Roche Ag | Process for preparing purine derivatives |
| US5840890A (en) * | 1996-01-26 | 1998-11-24 | Syntex (U.S.A.) Inc. | Process for preparing a 2-(2-amino-1,6-dihydro-6-oxo-purin-9-yl)methoxy-1,3-propanediol derivative |
| DE69714971T3 (de) * | 1996-01-26 | 2005-12-15 | F. Hoffmann-La Roche Ag | Verfahren zur herstellung von purin-derivaten |
| US5869493A (en) | 1996-02-16 | 1999-02-09 | Medivir Ab | Acyclic nucleoside derivatives |
| US6703394B2 (en) | 1996-02-16 | 2004-03-09 | Medivir Ab | Acyclic nucleoside derivatives |
| EP0827960A1 (en) | 1996-09-10 | 1998-03-11 | Ajinomoto Co., Inc. | Process for producing purine derivatives |
-
1997
- 1997-02-10 US US08/798,216 patent/US5869493A/en not_active Expired - Lifetime
- 1997-02-14 TW TW086101758A patent/TW533213B/zh not_active IP Right Cessation
- 1997-02-14 CZ CZ19982322A patent/CZ292169B6/cs not_active IP Right Cessation
- 1997-02-14 PL PL97328335A patent/PL184892B1/pl unknown
- 1997-02-14 NZ NZ330472A patent/NZ330472A/en not_active IP Right Cessation
- 1997-02-14 WO PCT/SE1997/000242 patent/WO1997030052A1/en not_active Ceased
- 1997-02-14 KR KR10-1998-0706309A patent/KR100464692B1/ko not_active Expired - Fee Related
- 1997-02-14 WO PCT/SE1997/000241 patent/WO1997030051A1/en not_active Ceased
- 1997-02-14 AU AU18182/97A patent/AU715062B2/en not_active Ceased
- 1997-02-14 JP JP52927997A patent/JP4171069B2/ja not_active Expired - Fee Related
- 1997-02-14 HU HU9900680A patent/HU229865B1/hu not_active IP Right Cessation
- 1997-02-14 CN CN97192183A patent/CN1067073C/zh not_active Expired - Fee Related
- 1997-02-14 CA CA002243826A patent/CA2243826C/en not_active Expired - Fee Related
- 1997-02-14 DK DK97903708T patent/DK0888348T3/da active
- 1997-02-14 AR ARP970100594A patent/AR005827A1/es active IP Right Grant
- 1997-02-14 JP JP9529280A patent/JP2000504721A/ja not_active Ceased
- 1997-02-14 MY MYPI97000553A patent/MY123083A/en unknown
- 1997-02-14 TR TR1998/01318T patent/TR199801318T2/xx unknown
- 1997-02-14 EA EA199800729A patent/EA001404B1/ru not_active IP Right Cessation
- 1997-02-14 ES ES97903708T patent/ES2189941T3/es not_active Expired - Lifetime
- 1997-02-14 IL IL12476097A patent/IL124760A/xx not_active IP Right Cessation
- 1997-02-14 DE DE69718619T patent/DE69718619T2/de not_active Expired - Lifetime
- 1997-02-14 DE DE69718626T patent/DE69718626T2/de not_active Expired - Lifetime
- 1997-02-14 EP EP97903708A patent/EP0888348B1/en not_active Expired - Lifetime
- 1997-02-14 AT AT97903709T patent/ATE231507T1/de not_active IP Right Cessation
- 1997-02-14 AT AT97903708T patent/ATE231508T1/de active
- 1997-02-14 SK SK985-98A patent/SK284613B6/sk not_active IP Right Cessation
- 1997-02-14 ES ES97903709T patent/ES2189942T3/es not_active Expired - Lifetime
- 1997-02-14 EP EP97903709A patent/EP0880521B1/en not_active Expired - Lifetime
- 1997-02-14 EA EA200000787A patent/EA002809B1/ru not_active IP Right Cessation
-
1998
- 1998-07-13 NO NO19983216A patent/NO322930B1/no not_active IP Right Cessation
- 1998-07-22 BG BG102647A patent/BG64179B1/bg unknown
- 1998-09-03 US US09/146,194 patent/US6255312B1/en not_active Expired - Lifetime
-
2000
- 2000-04-17 US US09/550,554 patent/US6576763B1/en not_active Expired - Fee Related
-
2008
- 2008-10-03 US US12/245,553 patent/US8124609B2/en not_active Expired - Fee Related
-
2012
- 2012-01-20 US US13/355,231 patent/US20120123119A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8124609B2 (en) | Acyclic nucleoside derivatives | |
| US6878844B2 (en) | Synthesis of acyclic nucleoside derivatives | |
| US6703394B2 (en) | Acyclic nucleoside derivatives | |
| JP4425352B2 (ja) | 非環状ヌクレオシド誘導体の合成 | |
| JP2009298785A (ja) | 非環状ヌクレオシド誘導体の合成 | |
| AU713916C (en) | Synthesis of acyclic nucleosides | |
| CA2238516C (en) | Acyclic nucleoside derivatives | |
| HK1015773B (en) | Acyclic nucleoside derivatives | |
| AU5354799A (en) | Substituted guanine compounds | |
| MXPA98006631A (en) | Synthesis of nucleosid acicli | |
| CA2723085A1 (en) | Synthesis of acyclic nucleoside derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20031219 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20031219 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20071127 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080227 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080414 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080327 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080512 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080425 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080609 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080527 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20080729 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20080808 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110815 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110815 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120815 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130815 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |