JP3983302B2 - ポリアルコールの製造法 - Google Patents
ポリアルコールの製造法 Download PDFInfo
- Publication number
- JP3983302B2 JP3983302B2 JP52829698A JP52829698A JP3983302B2 JP 3983302 B2 JP3983302 B2 JP 3983302B2 JP 52829698 A JP52829698 A JP 52829698A JP 52829698 A JP52829698 A JP 52829698A JP 3983302 B2 JP3983302 B2 JP 3983302B2
- Authority
- JP
- Japan
- Prior art keywords
- reaction
- stage
- formula
- distillation
- formaldehyde
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000004519 manufacturing process Methods 0.000 title claims description 22
- 150000005846 sugar alcohols Polymers 0.000 title description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims description 268
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 claims description 98
- 238000006243 chemical reaction Methods 0.000 claims description 81
- 238000004821 distillation Methods 0.000 claims description 70
- 238000000034 method Methods 0.000 claims description 62
- 239000003054 catalyst Substances 0.000 claims description 49
- -1 methylol group Chemical group 0.000 claims description 48
- 150000001875 compounds Chemical class 0.000 claims description 28
- ZJCCRDAZUWHFQH-UHFFFAOYSA-N Trimethylolpropane Chemical compound CCC(CO)(CO)CO ZJCCRDAZUWHFQH-UHFFFAOYSA-N 0.000 claims description 25
- 238000000926 separation method Methods 0.000 claims description 24
- 150000003512 tertiary amines Chemical class 0.000 claims description 20
- 150000001299 aldehydes Chemical class 0.000 claims description 18
- 125000004432 carbon atom Chemical group C* 0.000 claims description 17
- 238000005191 phase separation Methods 0.000 claims description 15
- 239000007858 starting material Substances 0.000 claims description 15
- 239000011541 reaction mixture Substances 0.000 claims description 13
- 239000012074 organic phase Substances 0.000 claims description 12
- 239000008346 aqueous phase Substances 0.000 claims description 11
- IKHGUXGNUITLKF-UHFFFAOYSA-N Acetaldehyde Chemical compound CC=O IKHGUXGNUITLKF-UHFFFAOYSA-N 0.000 claims description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims description 8
- 238000009833 condensation Methods 0.000 claims description 8
- 230000005494 condensation Effects 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 claims description 6
- WXZMFSXDPGVJKK-UHFFFAOYSA-N pentaerythritol Chemical compound OCC(CO)(CO)CO WXZMFSXDPGVJKK-UHFFFAOYSA-N 0.000 claims description 5
- 239000012071 phase Substances 0.000 claims description 4
- QXJQHYBHAIHNGG-UHFFFAOYSA-N trimethylolethane Chemical compound OCC(C)(CO)CO QXJQHYBHAIHNGG-UHFFFAOYSA-N 0.000 claims description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 claims description 3
- 125000000217 alkyl group Chemical group 0.000 claims description 3
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 3
- 125000003118 aryl group Chemical group 0.000 claims description 3
- 125000004429 atom Chemical group 0.000 claims description 3
- 238000010438 heat treatment Methods 0.000 claims description 2
- 238000002360 preparation method Methods 0.000 claims 1
- 238000009835 boiling Methods 0.000 description 58
- 239000000047 product Substances 0.000 description 42
- 150000001412 amines Chemical class 0.000 description 29
- 238000005984 hydrogenation reaction Methods 0.000 description 27
- JLIDVCMBCGBIEY-UHFFFAOYSA-N vinyl ethyl ketone Natural products CCC(=O)C=C JLIDVCMBCGBIEY-UHFFFAOYSA-N 0.000 description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- 239000000203 mixture Substances 0.000 description 23
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 20
- 239000000243 solution Substances 0.000 description 17
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 230000008878 coupling Effects 0.000 description 11
- 238000010168 coupling process Methods 0.000 description 11
- 238000005859 coupling reaction Methods 0.000 description 11
- HGINCPLSRVDWNT-UHFFFAOYSA-N Acrolein Chemical compound C=CC=O HGINCPLSRVDWNT-UHFFFAOYSA-N 0.000 description 10
- 230000015572 biosynthetic process Effects 0.000 description 9
- 238000003756 stirring Methods 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 7
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 7
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000002904 solvent Substances 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 5
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 5
- 229940044170 formate Drugs 0.000 description 5
- 230000036961 partial effect Effects 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- YIWUKEYIRIRTPP-UHFFFAOYSA-N 2-ethylhexan-1-ol Chemical compound CCCCC(CC)CO YIWUKEYIRIRTPP-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 150000007529 inorganic bases Chemical class 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 4
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 4
- 239000012970 tertiary amine catalyst Substances 0.000 description 4
- SZSSMFVYZRQGIM-UHFFFAOYSA-N 2-(hydroxymethyl)-2-propylpropane-1,3-diol Chemical compound CCCC(CO)(CO)CO SZSSMFVYZRQGIM-UHFFFAOYSA-N 0.000 description 3
- 238000005705 Cannizzaro reaction Methods 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- STNJBCKSHOAVAJ-UHFFFAOYSA-N Methacrolein Chemical compound CC(=C)C=O STNJBCKSHOAVAJ-UHFFFAOYSA-N 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 239000013067 intermediate product Substances 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 230000003068 static effect Effects 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 125000005208 trialkylammonium group Chemical group 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- YYKMQUOJKCKTSD-UHFFFAOYSA-N 2,2-bis(hydroxymethyl)butanal Chemical compound CCC(CO)(CO)C=O YYKMQUOJKCKTSD-UHFFFAOYSA-N 0.000 description 2
- XIKVGYYSAJEFFR-UHFFFAOYSA-N 2-(hydroxymethyl)butanal Chemical compound CCC(CO)C=O XIKVGYYSAJEFFR-UHFFFAOYSA-N 0.000 description 2
- GMLDCZYTIPCVMO-UHFFFAOYSA-N 2-methylidenebutanal Chemical compound CCC(=C)C=O GMLDCZYTIPCVMO-UHFFFAOYSA-N 0.000 description 2
- RTTWLTLNKLTUJR-UHFFFAOYSA-N 2-methylidenepentanal Chemical compound CCCC(=C)C=O RTTWLTLNKLTUJR-UHFFFAOYSA-N 0.000 description 2
- HSJKGGMUJITCBW-UHFFFAOYSA-N 3-hydroxybutanal Chemical compound CC(O)CC=O HSJKGGMUJITCBW-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 150000002373 hemiacetals Chemical class 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 2
- DTUQWGWMVIHBKE-UHFFFAOYSA-N phenylacetaldehyde Chemical compound O=CCC1=CC=CC=C1 DTUQWGWMVIHBKE-UHFFFAOYSA-N 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 235000011121 sodium hydroxide Nutrition 0.000 description 2
- 125000005270 trialkylamine group Chemical group 0.000 description 2
- 150000004072 triols Chemical class 0.000 description 2
- 239000002966 varnish Substances 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- WMYINDVYGQKYMI-UHFFFAOYSA-N 2-[2,2-bis(hydroxymethyl)butoxymethyl]-2-ethylpropane-1,3-diol Chemical compound CCC(CO)(CO)COCC(CC)(CO)CO WMYINDVYGQKYMI-UHFFFAOYSA-N 0.000 description 1
- HWOCOELJTFMTRT-UHFFFAOYSA-N 2-butoxyprop-2-enal Chemical compound CCCCOC(=C)C=O HWOCOELJTFMTRT-UHFFFAOYSA-N 0.000 description 1
- AXRMFIRHWOFRLE-UHFFFAOYSA-N 2-ethoxyprop-2-enal Chemical compound CCOC(=C)C=O AXRMFIRHWOFRLE-UHFFFAOYSA-N 0.000 description 1
- BNBWSNNWJPEFDF-UHFFFAOYSA-N 2-methoxyprop-2-enal Chemical compound COC(=C)C=O BNBWSNNWJPEFDF-UHFFFAOYSA-N 0.000 description 1
- FTUQBXQWSARMAR-UHFFFAOYSA-N 2-methylideneheptadecanal Chemical compound CCCCCCCCCCCCCCCC(=C)C=O FTUQBXQWSARMAR-UHFFFAOYSA-N 0.000 description 1
- IWEWQGKFUYQGSN-UHFFFAOYSA-N 2-methylidenehexanal Chemical compound CCCCC(=C)C=O IWEWQGKFUYQGSN-UHFFFAOYSA-N 0.000 description 1
- LLCLJBFJOXCPLY-UHFFFAOYSA-N 2-methylidenenonanal Chemical compound CCCCCCCC(=C)C=O LLCLJBFJOXCPLY-UHFFFAOYSA-N 0.000 description 1
- FYAKZTPGSCJSJF-UHFFFAOYSA-N 2-methylidenetetradecanal Chemical compound CCCCCCCCCCCCC(=C)C=O FYAKZTPGSCJSJF-UHFFFAOYSA-N 0.000 description 1
- FPBIKMOJDUSGQD-UHFFFAOYSA-N 2-propoxyprop-2-enal Chemical compound CCCOC(=C)C=O FPBIKMOJDUSGQD-UHFFFAOYSA-N 0.000 description 1
- LEBVLOLOVGHEFE-UHFFFAOYSA-N 3,4,4-trimethylpentanal Chemical group CC(C)(C)C(C)CC=O LEBVLOLOVGHEFE-UHFFFAOYSA-N 0.000 description 1
- RUNRFAYALPNCLX-UHFFFAOYSA-N 3-methylheptanal;4-methylpentanal Chemical compound CC(C)CCC=O.CCCCC(C)CC=O RUNRFAYALPNCLX-UHFFFAOYSA-N 0.000 description 1
- NHBINGOOBNESIC-UHFFFAOYSA-N 4,5,5-trimethylhexanal Chemical group CC(C)(C)C(C)CCC=O NHBINGOOBNESIC-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- LRKKUGXSKGITAS-UHFFFAOYSA-N 4-methyl-2-methylidenepentanal Chemical compound CC(C)CC(=C)C=O LRKKUGXSKGITAS-UHFFFAOYSA-N 0.000 description 1
- KQXBFBQRLHMLIS-UHFFFAOYSA-N 4-methylheptanal Chemical compound CCCC(C)CCC=O KQXBFBQRLHMLIS-UHFFFAOYSA-N 0.000 description 1
- JRYJOJWGAMCMTQ-UHFFFAOYSA-N 5,6,6-trimethylheptanal Chemical group CC(C)(C)C(C)CCCC=O JRYJOJWGAMCMTQ-UHFFFAOYSA-N 0.000 description 1
- RRTVBKXBOQHBMV-UHFFFAOYSA-N 5-methylheptanal Chemical compound CCC(C)CCCC=O RRTVBKXBOQHBMV-UHFFFAOYSA-N 0.000 description 1
- GEKRISJWBAIIAA-UHFFFAOYSA-N 5-methylhexanal Chemical compound CC(C)CCCC=O GEKRISJWBAIIAA-UHFFFAOYSA-N 0.000 description 1
- CBOCVOKPQGJKKJ-UHFFFAOYSA-L Calcium formate Chemical compound [Ca+2].[O-]C=O.[O-]C=O CBOCVOKPQGJKKJ-UHFFFAOYSA-L 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 239000004280 Sodium formate Substances 0.000 description 1
- 238000006887 Ullmann reaction Methods 0.000 description 1
- NILLQEAAPZTRRE-UHFFFAOYSA-N [N+](=O)(O)[O-].[N+](=O)(O)[O-].[N+](=O)(O)[O-].[N+](=O)(O)[O-].N(=O)O.N(=O)O.N(=O)O.N(=O)O.N(=O)O Chemical compound [N+](=O)(O)[O-].[N+](=O)(O)[O-].[N+](=O)(O)[O-].[N+](=O)(O)[O-].N(=O)O.N(=O)O.N(=O)O.N(=O)O.N(=O)O NILLQEAAPZTRRE-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 125000002723 alicyclic group Chemical group 0.000 description 1
- 238000005422 blasting Methods 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 235000019255 calcium formate Nutrition 0.000 description 1
- 239000004281 calcium formate Substances 0.000 description 1
- 229940044172 calcium formate Drugs 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000000306 component Substances 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 150000004292 cyclic ethers Chemical class 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 238000005265 energy consumption Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229910052757 nitrogen Inorganic materials 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 229940100595 phenylacetaldehyde Drugs 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 238000003672 processing method Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000004062 sedimentation Methods 0.000 description 1
- HLBBKKJFGFRGMU-UHFFFAOYSA-M sodium formate Chemical compound [Na+].[O-]C=O HLBBKKJFGFRGMU-UHFFFAOYSA-M 0.000 description 1
- 235000019254 sodium formate Nutrition 0.000 description 1
- 210000003813 thumb Anatomy 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- 150000003673 urethanes Chemical class 0.000 description 1
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/68—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
- C07C45/72—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms by reaction of compounds containing >C = O groups with the same or other compounds containing >C = O groups
- C07C45/75—Reactions with formaldehyde
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
- C07C29/136—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH
- C07C29/14—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of a —CHO group
- C07C29/141—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group of >C=O containing groups, e.g. —COOH of a —CHO group with hydrogen or hydrogen-containing gases
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Description
技術水準
カップリング生成物の形成下での方法
トリメチロールプロパン(TMP)は、通常、いわゆる無機カニッツァーロ法により大工業的に製造されている。その際、n−ブチルアルデヒド(n−BA)と過剰のホルムアルデヒド(FA)を、理論量の無機塩基、例えばNaOHまたはCa(OH)2の存在で反応させる。TMPの他に、カニッツァーロ反応により、1当量のギ酸ナトリウムまたはギ酸カルシウムが生じる。カップリング生成物としての無機塩基の生成は、多くの点に関して不利である。第一に、TMPから塩を分離するのは複雑であり、かつ付加的な費用を必要とし、第二に、無機塩−これを有利に使用すべきである場合には−は後処理されなければならず、かつ精製されなければならず、第三にカップリング生成物の生成は、使用される理論量の苛性ソーダ液およびホルムアルデヒドの損失を意味する。その上、この無機カニッツァーロ反応の場合の収率は、ブチルアルデヒドに関連して不十分である、というのも、反応の間に、更に利用されることのできない高沸点成分が形成されるからである。
TMPに説明されるような同様の問題は、他のトリオール、例えばトリメチロールエタン(n−プロパナールおよびホルムアルデヒドから)またはトリメチロールブタン(n−ペンタナールおよびホルムアルデヒドから)の製造の場合に存在する。TMPのように、これらのトリオールは、無機カニッツァーロ法による無機塩のカップリング生成下に製造される。これは同様に、アセトアルデヒドおよびホルムアルデヒドからペンタエリトリットを製造するのに当てはまる。トリメチロールプロパン、トリメチロールエタンおよびトリメチロールブタンは、プラスチック分野におけるワニス、ウレタンおよびポリエステルの製造に多数の使用が見出されているポリオールである。ペンタエリトリットは、ワニスエ業においてしばしば使用される出発物質であり、かつその上、爆破物質(四硝酸ペンタエリトリット)の製造に使用される。
改善された方法によれば、第三アミンの存在でのn−BAとFAとの反応がドイツ連邦共和国特許出願公開第1952738号明細書に記載されている。しかし、約1/6のn−BA/FAの使用された理論量および過剰の理論量のアミンの使用は、TMPの他に理論量のギ酸トリアルキルアンモニウムが生じるという結果となる。アミンおよび形成されたギ酸は、有機塩として留去され、ひいては、この方法を経済的にするために後処理および再回収しなければならない。アミンとしてトリエチルアミンおよびトリメチルアミンが使用された。
有機塩(ギ酸トリアルキルアンモニウム)の生成の可能性を回避することが、欧州特許出願公開第0142090号、同第0289921号明細書およびドイツ連邦共和国特許出願明細書番号P19542035.7およびP19542036.5に記載されている。同様に、これらには、無機塩基の代わりに、塩基として理論量のトリアルキルアミンの使用が記載されている。カップリング生成物として相応するギ酸トリアルキルアンモニウムが、処理中にギ酸メチルに変換される。この有機カニッツァーロ法の利点は、無機の変法に対して、高められた収率にある。ここで、カップリング生成物(ギ酸メチル)が生じ、ひいては1当量のホルムアルデヒドがこの方法において必要とするよりも多く消費されるという不利な事実のままである。
従って、各カップリング生成物なしでTMPを製造する方法は、このために経済的な観点から望ましい。カップリング生成物の形成なしの方法
ドイツ連邦共和国特許出願公開第2507461号明細書の記載によれば、ジメチロールブタナール(DMB)は、n−ブチルアルデヒド(n−BA)とホルムアルデヒド(FA)を触媒量の第三アミンの存在で反応させることにより、中間生成物であるモノメチロールブタナール(MMB)を経て製造されることができる(式1)。引続き、水素添加がTMPに行われる(式2)。
式1:

式3:
該明細書の方法は、工業的な量で使用可能ではない、特別な分枝鎖状第三アミン(例えばジメチルアミノネオペンタノールアミン)が使用されなければならないと言う欠点を有する。工業的に使用可能なアミン、例えばトリエチルアミンは、劣悪な収率が観察される(n−BAに対してTMP 57%)。分枝鎖状アミンは、該明細書に記載されている方法では、方法に返送されず、それにより増大した費用をまねく。同様に、変換されなかったn−BAおよびFAは方法に返送されない。
別の変法は、ドイツ連邦共和国特許出願公開第2702582号明細書に記載されており、その際、ホルムアルデヒドは、アルドール化のために、アルカリ化合物またはアルカリ土類化合物と組合せて第三アミンの存在で、アルデヒドに対して少なくとも8倍過剰で
、−5〜0℃で使用される。形成されたアルドール生成物は、引続き、TMPへと水素添加される。塩基として、線状の第三アミンが使用される。大部分の例において、付加的に、無機塩基が使用される。トリエチルアミンの単独使用の場合(例5、n−BA/FA/NEt3=1/10/0.18)の収率は、水素添加後にTMP 74.6%である。
この方法は、大過剰のホルムアルデヒドおよび低温の使用に基づく。しかしながら、大量のFAは、反応混合物中に存在するアルコール成分のFAアダクトの増大した形成をもたらす(FAとDMBおよびMMBとのアセタールおよびヘミアセタールの形成)。このことは、他方で、使用されるFAに対して望ましくない収率をまねく。その上、アルドール化における無機試薬の使用は、その後の後処理で、TMPの蒸留の際に問題をまねく。低温(−5〜0℃)の遵守は、付加的な技術的費用が必要である。反応に返送されない、第三アミン18モル%が全部で使用される。
ドイツ連邦共和国特許出願公開第2813201号明細書の方法の場合には、ドイツ連邦共和国特許出願公開第2702582号明細書のように、大過剰のホルムアルデヒドが特徴を示しており、かつn−ブチルアルデヒドに対する許容されうる収率は、使用されるホルムアルデヒドの費用のみで達成されるので、この方法は不経済である。
ドイツ連邦共和国特許出願公開第2714516号明細書には、エチルアクロレイン(EA)とホルムアルデヒド(FA)の反応が、塩基性触媒の存在で、1:8〜1:30のEA/FA比で記載されている。DMBの収率は、EAに対して90%まで記載されている。しかしながら、使用されるFAに関連して、単に12%の収率が得られる。
該方法は、大量のFAならびにトリアルキルアミン触媒の多い使用に基づき、不経済である。
従って、本発明の課題は、カップリング生成物を形成せずに、ポリメチロール化合物、例えば、n−ブチルアルデヒドおよびホルムアルデヒドから、n−ブチルアルデヒドおよびホルムアルデヒドに対して高い収率で、トリメチロールプロパンの製造を可能にする方法を発展させることにあった。この方法は、同様に、n−ブチルアルデヒドの高沸点および低沸点の同族のアルカナールからの他のポリアルコールの製造に適しているべきである。
これらの課題は、式I
a)第一(反応)段階で、C原子2個またはそれ以上を有するアルデヒドと、2〜8倍のモル量のホルムアルデヒドを、触媒として第三アミンの存在で反応させ、
b)第二(分離)段階で、反応化合物を、大部分の式Iの化合物を含有する缶出液および第一段階に返送される、大部分の未反応または一部反応した出発物質からなる留出液流に分離するか、または反応混合物を、第一段階から相分離装置を用いて水相および有機相に分離し、かつ有機相を第一段階に返送し、ならびに
c)第三(後反応−蒸留)段階で、第二段階の缶出液または第二段階で相分離により得られた水相を、触媒による処理および/または熱による処理にかけ、その際、不完全にメチロール化された式II
ならびに蒸留の塔底生成物として式Iの化合物を得ることにより特徴付けられる。
本発明による方法の次の記載の範囲内で、「第一(反応)段階」は、「(反応)段階」、「第一段階」または「段階a)」と同義、「第二(分離)段階」は、「(分離)段階」、「第二段階」または「段階b)」と同義、かつ「第三(後反応−蒸留)段階」は、「(後反応−蒸留)段階」、「第三段階」または「段階c)」と同義で呼ばれる。
一般式Iのヒドロキシメチルアルカナールは、引き続いて公知方法において、式IV
(HOCH2)3−C−R IV
[式中、Rは上記の式Iで記載された意味を有する]で示されるポリメチロール化合物に水素化される。
好ましい実施態様に従って、実質的な量の式IIIのメチレン化合物を含有する場合には、返送される留出液または第二段階で相分離により得られ、返送される有機相を、C原子2個またはそれ以上を有する別のアルデヒドを接触させる前に、ホルムアルデヒドおよび第三アミンと前反応させる。
別の好ましい実施態様に従って、留出液または第二段階の留出液流を、熱による後反応にかけ、新たに蒸留し、得られた留出液、または場合によりこの操作を数回繰り返した後の最後の留出液を、段階a)に返送する。
従って、当該方法の好ましい実施態様は、とりわけ、式IIIのメチレン化合物を含有する留出液とホルムアルデヒドとの前反応の使用、および/または場合により、第一段階への返送前に触媒および/または熱による後反応の実施、および/または場合により、蒸留で生じた水性蒸留缶出液の触媒および/または熱による後反応の実施にある。選択的に好ましい実施態様は、分離段階において、相分離により得られた有機相の返送およびホルムアルデヒドとの前反応、および/または場合により、分離工程において、相分離により得られた水相の触媒および/または熱による後反応の実施にある。
本発明による方法は、特に、使用されるアルデヒドの定量的変換および中間に形成される式IIIのメチレン化合物の定量的変換により式Iの化合物に変換することを可能にする反応操作法を含む。こうして、本発明による方法の段階c)での触媒および/または熱による後反応およびその際得られた留出液の段階a)への返送は、使用される出発物質の特に望ましい使用をもたらす。これに関連して、結果として、段階a)において、一方では本来の反応成分であるアルデヒドおよびFAが使用され、他方では、段階b)およびc)に由来し、返送される流れは、前アルデヒド、式IIIのメチレン化合物、ホルムアルデヒド、水およびアミン触媒を含有する。
意外なことに、この流れは、定量的に再び反応へ使用されることができ、その際、望ましくない副成分が新たに生じない。この流れをまず最初に前反応器中で新鮮なFAと前反応させ、引続き段階a)において、こうして得られた混合物と別の出発化合物との反応を実施するようにして反応が実施される場合には、この流れの妨害のない返送は、特にうまく成功する。反応成分の「段階のつけられた添加」のこの挙動は、技術水準に対して、著しく改善された収率をもたらす。
本発明による方法の実質的な特徴は、蒸留缶出液、または分離段階において相分離により生じ、依然としてモノメチロール化されたかもしくはアセトアルデヒドの場合にはモノメチロール化およびジメチロール化された中間化合物を含有する水相の後反応にあり、これにより水素添加前に、当該の完全にメチロール化された化合物および当該の一般式IIIの化合物に変換される。このことは、場合により第三アミンを別に添加しながら、別個の後反応器中で、または反応塔中で、または双方を組み合わせて行われる。
新規方法は、非連続的ならびに連続的に実施されることができる。
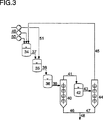
一般的な処理原理を具体的に示すにため、以下に、TMPの製造のための図1に基づいて、略示的に説明される:
単一の反応器または数個の反応器、有利に管状反応器または撹拌釜カスケードから構成されていてよい第一(反応)段階1において、有利に水溶液の形で導入される出発物質であるn−BAおよびFA、ならびに第三アミン触媒は、供給路2、3もしくは4を経て導入され、化合物DMBおよびMMBからなる混合物へ変換され、この混合物は、更に、実質的な成分として、未変換のn−BA、FAおよびEAならびにアミン触媒および場合により水を含有する。管路5を経て、これらの混合物は、(分離)段階に、この場合には、混合物を蒸留により易揮発性成分および難揮発性成分に分離する蒸留装置6、例えば塔または薄層蒸発器に供給される。6において、蒸留条件の適当な調節により、実質的な成分として未反応のn−BAおよびFA、水およびアミン触媒の一部、ならびに既に(反応)段階からの流出液中に含有されており、付加的に蒸留条件下でMMBから水を除去することにより形成されたEAを含有する低沸点分からなる留分を形成する。この低沸点留分は、管路7を経て、冷却器(記載されていない)中でのこれらの縮合後に、再び(反応)段階1へ返送される。実質的にDMB、MMBおよびアミン触媒の一部から構成されている、蒸留6のこの難揮発性塔底生成物は、管路8を経て(後反応−蒸留)段階9へ導入され、所望の場合には管路14および15を経て、付加的に新鮮なFAおよびアミン触媒が導入される。
(後反応−蒸留)段階9は、1個もしくは好ましくは数個の撹拌釜または1個もしくは数個の管状反応器から構成されていてよく、蒸留設備、例えば塔または薄層蒸発器もしくはサムベイ(Sambay)蒸発器がこれに接続されている。段階9において、残りのMMBがDMBへ変換され、生じた反応混合物は、引続き、低沸点分である水、FA、EAおよびアミン触媒の留去により濃縮される。これらの低沸点分は、管路10を経て(反応)段階1中へ返送されるのに対して、難揮発性のDMBは、管路11を経て水素添加反応器12に供給され、その中で、常法で水素と(供給路は記載されていない)接触的に水素添加してTMPに変換され、これは管路13を経て流出される。必要に応じて、水素添加反応器からの流出液を、純蒸留にかけることができる。管路7および10を経て返送された、未反応出発物質、アミン触媒、水および中間生成物EAからの流れは、(反応)段階1において、新鮮に供給される出発物質と一緒に再び前記のように反応される。その際、適当な、常用の調節装置による新鮮な出発物質および場合により新鮮な触媒の供給は、有利に、静平衡
上記の処理方法に対して選択的に、本発明による方法において、有利に、管路5を経て(反応)段階1から流出される反応混合物を蒸留装置6の代わりに相分離装置6aに供給し、かつこの相分離装置中で二相の反応流出液を水相および有機相に分離するようにして進められてもよい。この処理方法は、特に、反応段階1からの反応混合物が2つの液体層を生じる場合に有利であり、これは通常の場合である。これに対して、1からの反応流出液が均質な液状で存在する場合には、従って、例えば、水の添加により二相が新しく形成されてもよい。本発明による方法の結果のための(分離)段階の相分離装置6a中での有機相および水相の定量的な分離は強制的ではないので、使用すべき相分離装置の技術に特に技術的要求に属さない、すなわち、通常、液−液分離に使用される相分離装置、例えばUllmanns Encyclopaedie der technischen Chemie,第4版,第2巻,560〜565頁,Verlag Chemie, Weinheim 1972に記載されているもの、例えば沈降容器、遠心分離機または相分離を改善するための内蔵部材を備えた塔、例えば目皿塔または充填塔、しかし回転する内蔵部材を有する塔も使用することができる。
相分離の際に得られた、実質的な成分としてEA、未反応ブチルアルデヒドおよび少量のアミン触媒、FA、MMB、水およびDMBを含有する有機相は、再び(反応)段階1へ返送され、実質的な成分としてDMB、少量のMMBならびにFAおよびアミン触媒を含有する水相は、(後反応−蒸留)段階9に供給される。蒸留装置6の代わりに、相分離装置6aを使用する場合には、相分離の際に得られた有機相は、蒸留装置6の使用の際の低沸点分と同様の方法で、および水相は、高沸点塔底生成物と同様の方法で、別の処理工程で更に処理されることができる。
個別には、本発明による方法の実施の際に、これは図2に基づく好ましいおよび/または選択的な実施態様のもとで、例えばn−BAおよびFAからのTMPの製造のために、略示的に説明されるようにして有利に行われる。図2における装置が直接に相応する限り、このために同じ符号が使用される。
この出願明細書の目的のために、「前反応器」として単純に特徴付けられている前接続された反応器17は、管路18および19を経て、新鮮なFAは、有利に水溶液の形で、本方法の運転時間の間に、不変の運転条件の調節に必要な、新鮮な第三アミン触媒が計量供給される限り、管路20および/または23を経て図1による(分離)段階6から返送される、未反応n−BAおよびFAならびに形成されたEAを、水およびアミン触媒の他に実質的な成分として含有する低沸点分、ならびに管路10を経て図1による(後反応−蒸留)段階9から返送された、場合により同様に依然としてアミン触媒を含有するFA/水混合物と反応される。
前反応器17中で、エダクトは、例えば次のモル比で存在しうる:n−BA:EAのモル比は、調節されるn−BA変換率または蒸留装置21および28で調節される還流比に強く左右され、1:1000〜1000:1であり;(n−BA+EA)/FAのモル比は、一般に1:0.01〜1:50、好ましくは1:2〜1:20であり、第三アミンの量は、通常、反応混合物のpH値が5〜12、好ましくは6〜11であるように選択される。
水中の有機成分の溶液の全濃度は、前反応器17中で、一般に5〜60重量%、好ましくは10〜45重量%である。反応は、一般に5〜100℃、好ましくは15〜80℃の温度で実施され、かつ滞留時間は、−温度に依存して−一般に0.25〜12時間に調節される。
実質的な成分として未反応FAおよびn−BA、EA、MMBおよびDMBならびにアミン触媒および水を含有する前反応器17からの流出液は、管路63を経て、撹拌釜、管状反応器または有利に撹拌釜カスケードであってよい(反応)段階1に供給される。(反応)段階1において、管路3を経て新鮮なn−BAおよび必要な場合には1における静平衡またはpH値を調節するために、付加的にアミン触媒が管路4を経て計量供給される。撹拌釜カスケードを使用する場合には、カスケードの最初の撹拌釜のみ、一部の撹拌釜または全部の撹拌釜は、n−BAまたは第三アミンで、管路3または4を経て充てんされる。第三アミンの添加は、通常、pH値が再び、5〜12、有利に6〜11であるようにして行われる。管状反応器の使用の際に、反応管の長さに関連して所望のpH範囲に調節するために、管の様々な位置でアミン触媒が後供給されることができる。
1において新鮮に添加されるn−BAと、前反応器17中に後から供給されるFA量とのモル比は、有利に1:2〜1:5、好ましくは1:2〜1:3.5である。依然として前反応器17からの流出液中に過剰に存在するFAが、(反応)段階1においてn−BAと反応して実質的にDMBに変換される。前反応器17および(反応)段階1において導入される第三アミン触媒の量は、(反応)段階1において導入されるn−BAに対して、通常、0.001〜0.2、好ましくは0.01〜0.07当量であり、すなわち、アミンは触媒量で使用される。水性反応混合物中の有機成分の全濃度は、一般に5〜60重量%、好ましくは10〜45重量%である。反応は、一般に5〜100℃、好ましくは15〜80℃の温度で実施される。滞留時間は、−温度に依存して−通常、0.25〜12時間に調節される。
実質的にDMB、少量のMMBおよびEA、過剰のFA、少量の未反応n−BA、水およびアミン触媒を含有する(反応)段階1からの流出液は、管路5を経て(分離)段階6に供給され、これは図1の説明で詳述されるように構成されていてよいし、または選択的に次に詳述されるように前接続されていてよく、その際、図2における符号6を有する括弧により含まれる装置および反応器の全ては、定義によれば図1による(分離)段階6に相当する(分離)段階b)として適しているべきである。
(反応)段階1からの流出液は、(分離)段階6への導入の際に、まず最初に管路5を経て、例えば塔、薄層蒸発器またはサムベイ(Sambay)蒸発器であってよい蒸留装置21に供給され、その中で、1からの流出液は、塔頂を経てまたは蒸留装置21の上部から流出される低沸点分、および塔底生成物として生じるかまたは蒸留装置21の下部から除去される高沸点分に分離される。蒸留は、常法で、一般に50〜200℃、有利に90〜160℃で、一般に0.1〜10バール、有利に0.5〜5バール、殊に大気圧で実施される。塔頂を経て、または蒸留装置21の上部から管路22を経て除去される低沸点分は、実質的な成分として、未反応n−BAおよびFA、EAならびに水およびアミン触媒を含有し、それに対して、塔底からまたは蒸留装置21の下部から流出する高沸点分は、実質的な成分としてDMB、少量のMMBならびに残留量の水、FAおよびアミン触媒を含有する。21から流出した高沸点生成物中のDMBの含有量は、一般に5〜90重量%である。
蒸留装置21中で得られた高沸点混合物は、管路24および8を経て(後反応−蒸留)段階9へ供給され、それに対して、低沸点混合物は、完全にまたは部分流として管路22および23を経て、前反応器17へ返送されてもよいおよび/または管路22および25を経て、縮合後に、例えば撹拌釜、撹拌釜カスケードまたは管状反応器であってよい反応器26に供給されてもよく、そこで、一般に10〜100℃、有利に15〜80℃で、一般に0.25〜12時間の時間で後反応にかけ、その際、留出液中にあるアミン触媒は、留出液中にある成分n−BA、EAおよびFAの反応を生じさせてDMBおよびMMBに変換させる。後処理反応器26からの流出液は、管路27を経て蒸留装置28へ導入され、蒸留装置21中での蒸留に類似して、再び低沸点分および高沸点分への留去が行われ、その際、低沸点分は管路20を経て再び前反応器17へ返送され、高沸点分は管路29および8を経て(後反応−蒸留)段階9中へ導入される。
蒸留装置21からの低沸点留出液の後反応/蒸留から上記の詳述された配列は、図面による(分離)段階6に相当する、(分離)段階b)の範囲で、所望の場合には何度も繰り返されてもよい。(分離)段階6の範囲でのこの後反応/蒸留配列は、蒸留装置21からの低沸点分を、管路22および23を経て前反応器17への直接の還流のために、選択的または補充であってもよく、これは製造すべきポリメチロール化合物の種類に応じて、同等であってもよいし、または直接の還流に関連して有利であってもよい。(分離)段階6から低沸点分の前反応器17への還流は、本発明の有利な実施態様であり、−もちろん、(分離)段階6から返送された低沸点分の流れは、完全に、または部分流として、図面における(反応)段階1に相当する(反応)段階a)へ直接に返送されてもよい。
本発明による方法の上記で詳述された前接続を使用するのに応じて、蒸留装置21から高沸点分は管路24および8を経て、または蒸留装置21および28に由来する、管路24および29を経て1つにまとめられた高沸点分は管路8を経て、図面において(後反応−蒸留)段階9に相当する、(後反応−蒸留)段階c)の後処理反応器30に、高沸点混合物中に含有するMMBとFAとを反応させてDMBに変換させるのを完全にするために供給される。
蒸留装置21の代わりに、図2に基づいて説明された方法の前接続の場合に、層分離装置21aが使用される場合に、従って、その化学組成がほぼ、図1の説明の際に記載されている組成に相当する、層分離の際に得られた有機相で、別の処理段階において、蒸留装置21の使用の際に生じた低沸点分と同じような方法で処理されることができ、21aにおける相分離の際に生じた水相は、別の処理段階において、蒸留装置21の使用の際に生じた高沸点塔底生成物と類似の方法で更に処理されることができる。
例えば撹拌釜または有利に撹拌釜カスケードまたは管状反応器であってよい後処理反応器30において、高沸点混合物は、一般に0.1〜5時間、有利に0.5〜3時間の時間で、一般に30〜200℃、有利に40〜150℃に加熱される。その際、高沸点混合物中に常に依然として触媒的に作用する量で含有されるアミン触媒が、調節された後反応温度で、実施上、MMBと依然として高沸点混合物中に存在するFAまたは場合により管路15を介して付加的に導入されるFAとを完全に反応させてDMBへの変換を生じさせる。部分的に、高沸点混合物中のFAは、MMBおよびDMBに結合されたヘミアセタールの形で存在し、それから、FAはこの触媒による後処理および/または熱による後処理の過程で除去され、それにより、FAはMMBおよびDMBの変換のために再び使用される。
高沸点混合物が管路14を経て、塩基、有利にアルドール化の際に使用される第三アミン触媒の添加下で、5〜12、好ましくは6〜11のpH値にし、ついで上記の温度範囲で処理される場合には、反応器30中の高沸点分の上記の後処理および後反応は、促進されることができ、所望の場合には、温和な条件下で実施されることができる。低分子量第三アミンの代わりに、このためには、ポリマー第三アミン、例えば塩基性イオン交換樹脂が使用されることができる。
後反応反応器30からの流出液は、管路32を経て蒸留装置33、例えば薄層蒸発器またはサムベイ(Sambay)蒸発器へ送り、そこで高沸点DMB、および実質的な成分としてFA、水、アミン触媒および場合により新たに形成されたEAを含有する低沸点分を蒸留により分離し、その際、低沸点分を塔頂を経て、または蒸留装置の上部から除去し、管路10を経て例えば前反応器17および/または所望の場合には直接に(反応)段階1に返送されることができ、その際、塔底からまたは管路11を経て蒸留装置33の下部から高沸点DMBは管路62を経て、水素を予め供給した水素添加反応器12へ導入される。所望の場合には、蒸留は、実質的に水を含有する部分流を濃縮し、除去されるようにして接続されることができる。
今やほぼDMBを除いて、反応生成物として含有し、DMBの含有量が20〜95重量%、好ましくは30〜75重量%である、33からの高沸点生成物は、水素添加反応器12中で接触水素添加される。水素添加触媒としては、特に、銅含有担持触媒、例えば国際特許出願公表番号WO95/32171に記載されているものが適している。同様に、触媒は、例えばこれは欧州特許出願公開第44444号、同第44412号またはドイツ連邦共和国特許出願公開第1957592号明細書に記載されているものが適している。水素添加は、有利に連続的に、例えば触媒積重ね物で充てんされた反応管中で行われ、その際、反応溶液は、触媒積重ね物に関連して、例えば流動運転法または転移流の範囲で導入され、例えばドイツ連邦共和国特許出願公開第1941633号または同第2040501号明細書中に記載されている。反応流出液の部分流が、場合により冷却下に返送され、再び触媒固定層を経て導入されることが有利でありうる。同様に、水素添加を、幾つかの、次々に接続された反応器中で、例えば2〜4個の反応器中で実施することも有利であり、その際、個々の反応器中で、後の反応器の前に、水素添加反応が、例えば50〜98%の部分変換率まで実施されてはじめて後の反応器で水素添加を完全なものにされる。その際、先行する反応器からの水素添加留出液を、後続の反応器への導入前に、例えば冷却装置を用いて、または冷ガス、例えば水素もしくは窒素の噴出により、または冷たい反応溶液の部分流の導入により冷却することも有利である。
水素添加温度は、一般に、50〜180℃、好ましくは90〜140℃にある。水素添加圧として、一般に10〜250バール、好ましくは20〜120バールが使用される。
水素添加は、不活性溶剤の添加下で実施されることができる。溶剤としては、環状エーテル、例えばTHFまたはジオキサンならびに非環式エーテル、同様に低分子アルコール、例えばメダノール、エタノールまたは2−エチルヘキサノールが使用可能である。
その他では、任意の水素添加法を使用することができ、かつ水素添加触媒を使用することができ、例えば、アルデヒドの水素添加には、常用の、標準文献に詳細に記載されている。
こうして得られた粗TMPは、常法で蒸留することにより精製されることができる(記載されていない)。
本発明による方法は、有機溶剤または可溶化剤の添加または未添加で実施されることができる。溶剤または可溶化剤の添加は、特に、出発物質として長鎖アルデヒドの使用の際に、有利なものとして示すことができる。溶剤の使用により、本発明による方法の個々の蒸留の際に形成された低沸点化合物を有する適当な低沸点共沸混合物は、場合によりエネルギー消費をこの蒸留の場合に、低下されることができるおよび/または、高沸点化合物から低沸点分の留去を軽減されることができる。
溶剤として、例えば環式および非環式エーテル、例えばTHF、ジオキサン、メチル−第三ブチルエーテルまたはアルコール、例えばメタノール、エタノールまたは2−エチルヘキサノールが適している。
前反応器、上記の(反応)段階、(分離)段階および(後反応−蒸留)段階において上記の個々に記載された反応は、一般に1〜30バール、有利に1〜15バール、特に好ましくは1〜5バールの圧力で、有利に、当該の反応系の固有圧下で実施されることができる。
新規方法は、実施上、カルボニル基に対してα位にメチレン基を有するすべてのアルカナールが使用可能である。出発物質として、直鎖状もしくは分枝鎖状であってもよいし、または脂環式基を含有していてもよい、2〜24個のC原子を有する脂肪族アルデヒドを使用することができる。同様に、カルボニル基に対してα位にメチレン基を有することを前提として、出発物質としてアル脂肪族(araliphatishe)アルデヒドが使用されることができる。出発物質として、一般にC原子8〜24個を有する、有利にC原子8〜12個を有するアラルキルアルデヒド、例えばフェニルアセトアルデヒドが使用される。好ましくは、C原子2〜12個を有する脂肪族アルデヒド、例えば3−エチル−、3−n−プロピル−、3−イソプロピル−、3−n−ブチル−、3−イソブチル−、3−第二ブチル−、3−第三ブチル−ブタナールならびに相応する−n−ペンタナール、−n−ヘキサナール、−n−ヘプタナール;4−エチル−、4−n−プロピル−、4−イソプロピル−、4−n−ブチル−、4−イソブチル−、4−第二ブチル−、4−第三ブチル−ペンタナール、−n−ヘキサナール、−n−ヘプタナール;5−エチル−、5−n−プロピル−、5−イソプロピル−、5−n−ブチル−、5−イソブチル−、5−第二ブチル−、5−第三ブチル−n−ヘキサナール、−n−ヘプタナール;3−メチルヘキサナール;3−メチルヘプタナール;4−メチルペンタナール、4−メチルヘプタナール、5−メチルヘキサナール、5−メチルヘプタナール;3,3,5−トリメチル−n−ペンチル−、3,3−ジエチル−ペンチル−、4,4−ジエチルペンチル−、3,3−ジメチル−n−ブチル−、3,3−ジメチル−n−ペンチル−、5,5−ジメチルヘプチル−、3,3−ジメチルヘプチル−、3,3,4−トリメチルペンチル−、3,4−ジメチルヘプチル−、3,5−ジメチルヘプチル−、4,4−ジメチルヘプチル−、3,3−ジエチルヘキシル−、4,4−ジメチルヘキシル−、4,5−ジメチルヘキシル−、3,4−ジメチルヘキシル−、3,5−ジメチルヘキシル−、3,3−ジメチルヘキシル−、3,4−ジエチルヘキシル−、3−メチル−4−エチルペンチル−、3−メチル−4−エチルヘキシル−、3,3,4−トリメチルペンチル−、3,4,4−トリメチルペンチル−、3,3,4−トリメチルヘキシル−、3,4,4−トリメチルヘキシル−、3,3,4,4−テトラメチル−ペンチルアルデヒド;殊にC2〜C11−n−アルカナールである。
特に好ましくは、出発化合物として、アセトアルデヒドがペンタエリトリットの製造に、プロピオンアルデヒドがトリメチロールエタンの製造に、n−BAがTMPの製造に、およびn−ペンタナールがトリメチロールブタンの製造に使用される。
アルカナールの代わりに、式IVの化合物、例えばアクロレイン、例えばアクリルアルデヒド(アクロレイン);2−メチルアクリルアルデヒド、2−エチルアクリルアルデヒド、2−プロピルアクリルアルデヒド、2−ブチルアクリルアルデヒド、2−ペンチルアクリルアルデヒド、2−イソプロピルアクリルアルデヒド、2−イソブチルアクリルアルデヒド、2−ヘキシルアルデヒド、2−ヘプチルアクリルアルデヒド、2−ドデシルアクリルアルデヒド、2−ペンタデシルアクリルアルデヒド、2−メトキシアクリルアルデヒド、2−エトキシアクリルアルデヒド、2−プロポキシアクリルアルデヒドまたは2−ブトキシアクリルアルデヒドも使用されることができる。この中では、2−メチルアクロレイン、2−エチルアクロレインおよび2−プロピルアクロレインが好ましい。同様に、アクロレインは、有利に、ペンタエリトリットの生産に使用されることができる。
冒頭に記載された式3から得られるようにして、一般式IVのアクロレインの使用の際に、使用物質として、相応する飽和アルデヒドの使用の場合より少ない1当量のFAが、一般式IIの化合物への完全な反応のために必要とされる。
第三アミンとして、これらの適性に関連してアルデヒドとホルムアルデヒドとの縮合のための公知のアミン、例えばドイツ連邦共和国特許出願公開第2813201号および同第2702582号明細書に記載されているものが当てはまる。特に好ましくは、トリ−n−アルキルアミン、例えばトリエチルアミン、トリ−n−プロピルアミン、トリ−n−ブチルアミンおよび殊にトリメチルアミンである。
本発明による方法は、出発アルデヒドならびにホルムアルデヒドに対する高い収率に優れており、かつアミン触媒の極めて僅かな損失をもたらす。相対的に低いpH値の場合の処理に基づき、カニッツァーロ反応が行われず、それによりカップリング生成物としてのギ酸塩の形成が回避される。
実施例
例1〜4:バッチ式処理方法
例1
n−ブチルアルデヒド288g(4モル)を、10%ホルムアルデヒド3000g(10モル)および45%トリメチルアミン26g(0.2モル)と25℃で5時間撹拌した。その後、反応溶液中に、n−BA0.7モル(n−BAの変換率=82.5%)およびEA0.5モル(EAに対する選択率=15%)が見出された。反応溶液を濃縮し、その際、常圧でサムベイ(Sambay)蒸発器(140℃)により低沸点分10ml/hおよび水の供給で留去した。留出液1195gおよび缶出液2119gが得られ、これを新しくサムベイ(Sambay)蒸発器で濃縮した。これは留出液795gおよび缶出液1324gに相当した。この缶出液を3回サムベイ(Sambay)蒸発器で濃縮した。留出液494gおよび缶出液830gが得られた。集めた留出液(2484g)を30℃で5時間撹拌し、さらにサムベイ(Sambay)蒸発器(140℃)で蒸発させることにより濃縮した。これから、缶出液1435gおよび留出液1049gが得られ、これを例2に記載されているようにして更に処理した。
n−BAまたはEAを含有していない、得られた、集められた缶出液(DMB溶液2265g、pH4.1)を、連続的に水素添加した。このために、反応溶液を水素と一緒に、30バールの圧力で、90℃に加熱した、Cuとして計算して銅25重量%およびSiO275重量%を含有し、国際特許出願公表番号95/32171による方法により製造されたSiO2上の銅触媒100mlを充填された管状反応器中へポンプ供給した。水素添加すべき溶液を、触媒積重ね物に関連する流動法により導入し、圧力下に反応流出液の一部を、反応器中へ返送した(循環運転方法)。供給に相当する量を反応器から連続的に取り出し、放圧し、受け器に集めた。
集めた水素添加流出液を、引続き蒸留した。低沸点分を定量ガスクロマトグラフィー(GC)を用いて集め、オリゴマー、例えばジトリメチロールプロパンまたはTMP−EA−TMP三量体を包含する中間沸点生成物、TMPおよび高沸点縮合生成物を除去した:
例1からの残留している留出液(1049g)に、FA9モルを含有する水溶液1951gおよび45%トリメチルアミン26g(0.2モル)を添加し、混合物を30℃で3時間撹拌した。これにn−ブチルアルデヒド288g(4モル)を添加し、混合物を30℃で4時間撹拌した。その後、例1に記載されているようにして蒸留により後処理した。例3に記載されているようにして再び処理される留出液873g、および水素添加および例1に類似の蒸留後に次の組成の生成物が得られる缶出液2441g(DMB溶液)が得られた:
例2からの留出液873gに、FA9モルを含有する水溶液2127gおよび45%トリメチルアミン26g(0.2モル)を添加し、混合物を30℃で3時間撹拌した。その後、n−ブチルアルデヒド288g(4モル)を添加し、更に、30℃で4時間撹拌し、引続き、例1に記載されているように蒸留により後処理した。留出液1096gが得られ、これを例4に記載されているようにして再び処理し、DMB溶液2218gを、水素添加および蒸留後に次の生成物組成を供給した:
例3からの留出液1096gに、まず最初にFA8.5モルを含有する水溶液1904gおよび45%トリメチルアミン26g(0.2モル)を添加し、混合物を30℃で5時間撹拌した。その後、n−ブチルアルデヒド288g(4モル)を添加し、更に30℃で5時間撹拌した。例1に記載されているような蒸留による後処理後に、留出液1150gが得られ、これは更に処理されてもよい。缶出液として生じたDMB溶液2164gは、水素添加および蒸留後に、次の量の生成物が生じた:
n−BAの使用量 FAの使用量 NMe3の使用量
1152g(16モル) 9558g(36.5モル) 104g(0.79モル)
留出液中に(還流のために)残留する:n−BA 1.75当量およびFA5.1当量
例5〜7:連続的な処理法
例5
図3による装置は、第1表に記載されているようにして、エダクトを充てんし、上記の温度で運転した。MMB、DMBおよびFAの記載は、HPLC分析を用いて測定され、重量%で記入されている。
図3による装置は、第2表に記載されているようにしてエダクトを充てんし、記載された温度で運転した。
こうして得られ、一つにまとめられた塔底生成物を、例1に記載されているようにして水素添加し、流出液を蒸留した:
図3による装置は、第3表に記載されているようにしてエダクトで充てんし、記載された温度で運転した。
こうして得られた塔底生成物を、例1に記載されているようにして水素添加し、流出液を蒸留した。
これらの例の実施のために、例5〜7のために使用された実験室用装置を、図4に略示されているようにして改造した。改造された実験室用装置は、既に記載されている(例5〜7)、撹拌釜34、35および36からなるカスケードおよび溢流管37、38および39およびサムベイ(Sambay)蒸発器40から構成されていた。撹拌釜36の溢流を、直接に溢流管39によりサムベイ(Sambay)蒸発器40の上部に導入し、そこで蒸留により、成分として実質的にn−BA、FA、EAおよび水を含有する低沸点塔頂生成物、および高沸点塔底生成物に分離した。塔頂生成物を、連続的に管路52を経て、縮合後に(記載されていない)撹拌釜34に返送した。サムベイ(Sambay)蒸発器40からの高沸点塔底生成物を、連続的に管路53を経て、新鮮なトリメチルアミン触媒(45%水溶液)の添加後に、管路54を経て後反応器55である加熱され、充てん体(2.5mmラシヒリング)で充てんされた、空容量1000mlを有する管状反応器に供給した。後反応器55の流出液は、連続的に管路56を経て、サムベイ(Sambay)蒸発器44の上部で得られ、そこで蒸留により、実質的にEA、FA、トリメチルアミンおよび水から構成される低沸点塔頂生成物、および高沸点塔底生成物に分離された。低沸点塔頂生成物を、連続的に管路57を経て、縮合後に(記載されていない)撹拌釜34に返送し、これに対して高沸点塔底生成物を、管路58を経て、ストレージタンク59に集め、そこから管路60を経てその後の水素添加に取り出した。外から、管路52および管路57の返送流で、撹拌釜34は、管路49または61を経て新鮮なFA溶液またはn−BAで、および管路50を経て、45%水溶液の形で新鮮なトリメチルアミン触媒で充てんされた。
エダクトおよび触媒の供給された量は、サムベイ(Sambay)蒸発器40および44からの塔底生成物からの流出液の量および第4表の後者の流出液の組成と同様に、まとめられている。MMB、DMBおよびFAの含有量は、HPLCを用いて測定され、重量%で記載されれている。個々の撹拌釜およびサムベイ(Sambay)蒸発器中で使用される温度は、同様に第4表中で見出される。サムベイ(Sambay)蒸発器中での蒸留は、大気圧で実施された。
Claims (15)
- 触媒として第三アミンの使用下で、C原子2〜24個を有するアルデヒドと、ホルムアルデヒドを縮合させることによる、式I
[式中、Rは別のメチロール基またはC原子1〜22個を有するアルキル基またはC原子6〜22個を有するアリール基もしくはアラルキル基を意味する]で示されるメチロールアルカナールの製造方法において、縮合を、
a)第一(反応)段階で、C原子2個またはそれ以上を有するアルデヒドと、2〜8倍のモル量のホルムアルデヒドを、触媒として第三アミンの存在で反応させ、
b)第二(分離)段階で、反応化合物を、大部分の式Iの化合物を含有する缶出液および第一段階に返送される、大部分の未反応または一部反応した出発物質からなる留出液流に分離するか、または反応混合物を、第一段階から相分離装置を用いて水相および有機相に分離し、かつ有機相を第一段階に返送し、ならびに
c)第三(後反応−蒸留)段階で、第二段階の缶出液または第二段階で相分離により得られた水相を、触媒による処理および/または熱による処理にかけ、その際、不完全にメチロール化された式II
で示される化合物を相応する式Iの化合物および相応する式III
[式中、R’は水素であるか、またはRの上記の意味を有する]で示されるメチレン化合物に変換し、こうして得られた反応混合物を蒸留し、式IIIの化合物および未反応ホルムアルデヒドを含有する、この蒸留の塔頂生成物を第一段階に返送し、
ならびに蒸留の塔底生成物として式Iの化合物を得ることにより段階的に実施することを特徴とする、メチロールアルカナールの製造法。 - 返送された留出液または段階b)から返送された有機相を、式IIIのメチレン化合物の実質的な量を含有する場合には、ホルムアルデヒドと第三アミンとの前反応にかけ、これらが段階a)で、C原子2〜24個を含有する相当するアルデヒドと接触させる、請求項1記載の方法。
- 段階b)の留出液を、熱による後反応にかけ、新たに蒸留するか、または場合によりこの操作を数回繰り返した後に、後者の留出液を段階a)に返送する、請求項1記載の方法。
- プロピオンアルデヒドまたはn−ブチルアルデヒドをホルムアルデヒドと反応させる、請求項1記載の方法。
- アセトアルデヒドをホルムアルデヒドと反応させる、請求項1記載の方法。
- 反応を連続的に実施する、請求項1記載の方法。
- 触媒として、第三アミンを、反応混合物中でpH値5〜12に調節するような量で使用する、請求項1記載の方法。
- 触媒として、トリメチルアミンを使用する、請求項1記載の方法。
- 段階c)において、段階a)と同じ触媒を使用する、請求項1記載の方法。
- 段階a)において、C原子2〜24個を有するアルデヒドの代わりに、または付加的に式IIIの相当する化合物を新鮮な供給流として供給する、請求項1記載の方法。
- 第一の段階a)において、管状反応器または撹拌釜カスケードを使用する、請求項1記載の方法。
- 段階c)において、管状反応器または撹拌釜カスケードを使用する、請求項1記載の方法。
- 式IV
(HOCH 2 ) 3 −C−R IV
[式中、Rは別のメチロール基またはC原子1〜22個を有するアルキル基またはC原子6〜22個を有するアリール基もしくはアラルキル基を意味する]で示されるポリメチロール化合物を、式I
[式中、Rは上記の意味を有する]で示され、かつ請求項1から12までのいずれか1項記載の方法により得られるメチロールアルカナールを水素化することにより製造する方法において、公知方法において、式IVのポリメチロール化合物に水素化することを特徴とする、ポリメチロール化合物の製造法。 - トリメチロールエタンまたはトリメチロールプロパンの製造のための、請求項13記載の方法。
- ペンタエリトリトールの製造のための、請求項13記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19653093.8 | 1996-12-20 | ||
| DE1996153093 DE19653093A1 (de) | 1996-12-20 | 1996-12-20 | Verfahren zur Herstellung von Polyalkoholen |
| PCT/EP1997/006776 WO1998028253A1 (de) | 1996-12-20 | 1997-12-04 | Verfahren zur herstellung von polyalkoholen |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2001506658A JP2001506658A (ja) | 2001-05-22 |
| JP2001506658A5 JP2001506658A5 (ja) | 2005-08-11 |
| JP3983302B2 true JP3983302B2 (ja) | 2007-09-26 |
Family
ID=7815413
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP52829698A Expired - Lifetime JP3983302B2 (ja) | 1996-12-20 | 1997-12-04 | ポリアルコールの製造法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6187971B1 (ja) |
| EP (1) | EP0946483B1 (ja) |
| JP (1) | JP3983302B2 (ja) |
| KR (1) | KR100499599B1 (ja) |
| CN (1) | CN1123558C (ja) |
| CA (1) | CA2274083A1 (ja) |
| DE (2) | DE19653093A1 (ja) |
| ES (1) | ES2184146T3 (ja) |
| TW (1) | TW462961B (ja) |
| WO (1) | WO1998028253A1 (ja) |
Families Citing this family (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5994592A (en) * | 1997-02-19 | 1999-11-30 | Nippon Kasei Chemical Company Limited | Process for producing 2,2'-bis(hydroxymethyl)alkanal and 2,2'-bis(hydroxymethyl)alkanoic acid |
| DE19963435A1 (de) | 1999-12-28 | 2001-07-05 | Basf Ag | Verfahren zur Reinigung von durch Hydrierung hergestelltem Trimethylolpropan durch kontinuierliche Destillation |
| DE19963442A1 (de) * | 1999-12-28 | 2001-07-05 | Basf Ag | Farbzahlverbesserung von mehrwertigen Alkoholen durch Hydrierung |
| DE19963438A1 (de) * | 1999-12-28 | 2001-10-31 | Basf Ag | Verfahren zur Herstellung von Polyalkoholen mit Methanolarmem Formaldehyd |
| DE19963437A1 (de) | 1999-12-28 | 2001-07-05 | Basf Ag | Verfahren zur Zersetzung von bei der Synthese mehrwertiger Alkohole gebildeter hochsiedender Nebenprodukte |
| DE10001257A1 (de) * | 2000-01-14 | 2001-07-19 | Bayer Ag | Verfahren zur Herstellung von Trimethylolalkanen |
| DE10041197A1 (de) * | 2000-08-23 | 2002-03-07 | Bayer Ag | TMP/Dampfdruckfiltration |
| DE10050645A1 (de) * | 2000-10-13 | 2002-04-18 | Basf Ag | Farbzahlverbesserung von mehrwertigen Alkoholen durch Hydrierung |
| DE10055180A1 (de) * | 2000-11-08 | 2002-05-29 | Basf Ag | Verfahren zur Hydrierung von Poly- oder Monomethylolalkanalen |
| DE10152525A1 (de) * | 2001-10-24 | 2003-05-08 | Basf Ag | Verfahren zur Zersetzung von Ammoniumformiaten in polyolhaltigen Reaktionsgemischen |
| EP1448502B1 (de) | 2001-11-15 | 2015-10-14 | Basf Se | Verfahren zum enfernen von formaldehydhaltigen acetalen aus mehrwertigen alkoholen durch tempern |
| DE10160180A1 (de) | 2001-12-07 | 2003-06-18 | Basf Ag | Verfahren zur Isolierung von Trimethylolpropan aus einem Reaktionsgemisch |
| DE10223055A1 (de) * | 2002-05-24 | 2003-12-11 | Basf Ag | Verfahren zur Herstellung von Polyesterpolyolen mehrwertiger Alkohole |
| DE10234016A1 (de) | 2002-07-26 | 2004-02-05 | Basf Ag | Verfahren zur Ausbeuteerhöhung bei der Herstellung von mehrwertigen Alkoholen durch Spaltung acetalhaltiger Nebenprodukte |
| SE0301102D0 (sv) | 2003-04-14 | 2003-04-14 | Tetra Laval Holdings & Finance | Method in connection with the production of a apckaging laminate thus produced and a packaging container manufactures from the packaging laminate |
| DE10317543A1 (de) | 2003-04-16 | 2004-11-04 | Basf Ag | Verfahren zur Hydrierung von Methylolalkanalen |
| DE102006009838A1 (de) * | 2006-03-01 | 2007-09-06 | Basf Ag | Verfahren zur Hydrierung von Methylolalkanalen |
| US7388116B2 (en) | 2006-06-06 | 2008-06-17 | Basf Aktiengesellschaft | Hydrogenation of methylolalkanals |
| US20080004475A1 (en) * | 2006-06-28 | 2008-01-03 | Basf Aktiengesellschaft | Process for the production of neopentylglycol using formaldehyde with a low methanol content |
| US7462747B2 (en) * | 2007-01-05 | 2008-12-09 | Basf Aktiengesellschaft | Process for preparing polyalcohols from formaldehyde having a low formic acid content |
| WO2008107333A1 (de) | 2007-03-02 | 2008-09-12 | Basf Se | Verfahren zur herstellung von hydroxypivalinaldehyd und neopentylglykol |
| WO2010066674A2 (de) | 2008-12-09 | 2010-06-17 | Basf Se | Verfahren zur herstellung von polymethylolen |
| JP2012511533A (ja) | 2008-12-09 | 2012-05-24 | ビーエーエスエフ ソシエタス・ヨーロピア | ポリメチロールの製造方法 |
| SG172807A1 (en) | 2009-01-12 | 2011-08-29 | Basf Se | Method for producing polymethylols |
| EP2607341B1 (en) | 2009-12-24 | 2015-11-18 | Oxea Bishop LLC | Trimethylolpropane color improvement |
| US8853465B2 (en) | 2010-05-12 | 2014-10-07 | Basf Se | Process for preparing neopentyl glycol |
| CN102884032A (zh) | 2010-05-12 | 2013-01-16 | 巴斯夫欧洲公司 | 制备新戊二醇的方法 |
| DE102010033844B4 (de) | 2010-08-11 | 2013-01-03 | Oxea Gmbh | Verfahren zur Gewinnung von Di-Trimethylolpropan und mit Trimethylolpropan angereicherten Produktströmen aus den Nebenströmen der Trimethylolpropanherstellung |
| WO2012143309A1 (de) | 2011-04-19 | 2012-10-26 | Basf Se | Verfahren zur herstellung von neopentylglykol |
| CN102304022A (zh) * | 2011-07-05 | 2012-01-04 | 上海华谊(集团)公司 | 一种缩合加氢法制备三羟甲基丙烷反应过程中副产物2-乙基丙烯醛的回收利用方法 |
| CN102391070B (zh) * | 2011-10-19 | 2014-09-03 | 上海华谊(集团)公司 | 利用甲醛生产三羟甲基丙烷的方法 |
| DE102011118956A1 (de) | 2011-11-19 | 2013-05-23 | Oxea Gmbh | Verfahren zur Gewinnung von Di-Trimethylolpropan und mit Trimethylolpropan angereicherten Produktströmen aus den Nebenströmen der Trimethylolpropanherstellung |
| DE102011118993A1 (de) | 2011-11-19 | 2013-05-23 | Oxea Gmbh | Verfahren zur Gewinnung von Di-Trimethylpropan und mit Trimethylpropan angereicherten Produktströmen aus den Nebenströmen der Trimethylolpropanherstellung |
| DE102011118953B4 (de) | 2011-11-19 | 2014-06-05 | Oxea Gmbh | Destillatives Verfahren zur Gewinnung von Di-Trimethylolpropan |
| DE102011122356A1 (de) | 2011-12-23 | 2013-06-27 | Oxea Gmbh | Verfahren zur Gewinnung von mit Trimethylolpropan angereicherten Produktströmen aus den Nebenströmen der Trimethylolpropanherstellung |
| US8710278B1 (en) | 2013-01-31 | 2014-04-29 | Eastman Chemical Company | Process for producing polyols |
| US9056824B2 (en) | 2013-01-31 | 2015-06-16 | Eastman Chemical Company | Preparation of hydroxy aldehydes |
| KR101529828B1 (ko) * | 2013-07-26 | 2015-06-17 | 주식회사 엘지화학 | 메틸올알칸알의 제조방법 |
| KR102283433B1 (ko) * | 2013-08-06 | 2021-07-30 | 옥세아 비숍 엘엘씨 | 메틸올알칸의 개선된 제조 방법 |
| TW201536734A (zh) | 2014-03-12 | 2015-10-01 | Basf Se | 甲酸鹽的分解 |
| KR102224243B1 (ko) * | 2016-10-31 | 2021-03-08 | 주식회사 엘지화학 | 트리메틸올프로판의 제조장치 및 이를 이용한 제조방법 |
| KR102224267B1 (ko) * | 2016-10-31 | 2021-03-08 | 주식회사 엘지화학 | 트리메틸올프로판의 제조장치 및 이를 이용한 제조방법 |
| KR102224268B1 (ko) * | 2016-10-31 | 2021-03-08 | 주식회사 엘지화학 | 트리메틸올프로판의 제조장치 및 이를 이용한 제조방법 |
| KR102245932B1 (ko) * | 2017-10-23 | 2021-04-28 | 주식회사 엘지화학 | 트리메틸올프로판의 제조방법 |
| KR102522282B1 (ko) * | 2018-04-24 | 2023-04-14 | 주식회사 엘지화학 | 포름알데히드의 회수방법 |
| CN113845403B (zh) * | 2021-11-30 | 2023-08-11 | 万华化学集团股份有限公司 | 一种联产2-甲基-1,3-丙二醇和季戊四醇的制备方法 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1952738A1 (de) * | 1968-12-17 | 1970-07-09 | Leuna Werke Veb | Verfahren zur Herstellung von Trimethylolpropan |
| BE758910A (fr) * | 1969-11-15 | 1971-05-13 | Basf Ag | Preparation de dimethyl-2, 2-propanediol-1, 3 |
| DE2507461C3 (de) * | 1975-02-21 | 1986-05-07 | Basf Ag, 6700 Ludwigshafen | Verfahren zur Herstellung von 2,2- Dimethylolalkanalen |
| DE3340791A1 (de) * | 1983-11-11 | 1985-05-23 | Basf Ag, 6700 Ludwigshafen | Verfahren zur herstellung von trimethylolalkanen aus alkanalen und formaldehyd |
| US4740639A (en) * | 1987-06-12 | 1988-04-26 | Eastman Kodak Company | Process for preparation of 2,4-disubstituted-1,5-pentanediols |
| US4851592A (en) * | 1987-10-27 | 1989-07-25 | Eastman Kodak Company | Triethylamine catalyzed neopentyl glycol production utilizing a gas sparged reactor |
| DE3942792A1 (de) * | 1989-12-23 | 1991-06-27 | Hoechst Ag | Verfahren zur herstellung von 2,2-dimethylpropandiol-(1,3) |
| US5532417A (en) * | 1991-04-26 | 1996-07-02 | Aristech Chemical Corporation | Manufacture of neopentyl glycol (IV) |
| US5185478A (en) * | 1991-06-17 | 1993-02-09 | Aristech Chemical Corporation | Manufacture of neopentyl glycol (IIA) |
| US5763690A (en) * | 1992-09-25 | 1998-06-09 | Aristech Chemical Corporation | Manufacture of trimethylolpropane |
-
1996
- 1996-12-20 DE DE1996153093 patent/DE19653093A1/de not_active Withdrawn
-
1997
- 1997-12-04 CN CN97180072A patent/CN1123558C/zh not_active Expired - Fee Related
- 1997-12-04 ES ES97952875T patent/ES2184146T3/es not_active Expired - Lifetime
- 1997-12-04 JP JP52829698A patent/JP3983302B2/ja not_active Expired - Lifetime
- 1997-12-04 US US09/331,370 patent/US6187971B1/en not_active Expired - Fee Related
- 1997-12-04 DE DE59708146T patent/DE59708146D1/de not_active Expired - Lifetime
- 1997-12-04 WO PCT/EP1997/006776 patent/WO1998028253A1/de active IP Right Grant
- 1997-12-04 EP EP19970952875 patent/EP0946483B1/de not_active Expired - Lifetime
- 1997-12-04 CA CA 2274083 patent/CA2274083A1/en not_active Abandoned
- 1997-12-04 KR KR10-1999-7005583A patent/KR100499599B1/ko not_active IP Right Cessation
- 1997-12-20 TW TW086119433A patent/TW462961B/zh not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| EP0946483A1 (de) | 1999-10-06 |
| CA2274083A1 (en) | 1998-07-02 |
| DE59708146D1 (de) | 2002-10-10 |
| DE19653093A1 (de) | 1998-06-25 |
| US6187971B1 (en) | 2001-02-13 |
| ES2184146T3 (es) | 2003-04-01 |
| CN1238753A (zh) | 1999-12-15 |
| CN1123558C (zh) | 2003-10-08 |
| EP0946483B1 (de) | 2002-09-04 |
| TW462961B (en) | 2001-11-11 |
| KR20000062250A (ko) | 2000-10-25 |
| WO1998028253A1 (de) | 1998-07-02 |
| KR100499599B1 (ko) | 2005-07-07 |
| JP2001506658A (ja) | 2001-05-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3983302B2 (ja) | ポリアルコールの製造法 | |
| TWI450883B (zh) | 製備聚羥甲基化物之方法 | |
| KR101584375B1 (ko) | 낮은 포름산 함량을 갖는 포름알데히드로부터 폴리알콜을 제조하는 방법 | |
| JPH027297B2 (ja) | ||
| TWI494294B (zh) | 製備聚羥甲基化物之方法 | |
| US8933277B2 (en) | Process for preparing polymethylols | |
| JP2001507668A (ja) | ポリアルコールの製法 | |
| US6692616B2 (en) | Method for purifying trimethylolpropane, which is produced by hydrogenation, by means of continuous distillation | |
| CA2326779A1 (en) | Process for carrying out aldol condensations | |
| JP2006501206A (ja) | アセタール含有副生物の開裂による多価アルコールを製造する際の収率を増大させる方法 | |
| US20080004475A1 (en) | Process for the production of neopentylglycol using formaldehyde with a low methanol content | |
| JP2005511674A (ja) | 反応混合物からトリメチロールプロパンを単離する方法 | |
| US6586642B2 (en) | Color number improvement in polyhydric alcohols by hydrogenation | |
| CA2114139C (en) | Continuous process for the preparation of 2-ethyl-2- (hydroxymethyl) hexanal and 2-butyl-2-ethyl-1,3-propanediol | |
| US6809224B2 (en) | Method for the separation of formaldehyde from a reaction mixture containing polyols by the addition of solvents before and/or during the distillation | |
| JP2002193854A (ja) | トリメチロール化合物及びギ酸の製造方法 | |
| KR100760776B1 (ko) | 템퍼링 수단에 의해 다가 알콜로부터 포름알데히드-함유아세탈을 제거하는 방법 | |
| CN109790093B (zh) | 在无机碱存在下联合生产多元醇的方法 | |
| JP4599664B2 (ja) | 多価アルコールの製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20041203 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20041203 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20061205 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20061128 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20070305 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20070423 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20070605 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20070704 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100713 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |