JP3790273B2 - シクロペンテノン類、その製造方法及び用途 - Google Patents
シクロペンテノン類、その製造方法及び用途 Download PDFInfo
- Publication number
- JP3790273B2 JP3790273B2 JP51548098A JP51548098A JP3790273B2 JP 3790273 B2 JP3790273 B2 JP 3790273B2 JP 51548098 A JP51548098 A JP 51548098A JP 51548098 A JP51548098 A JP 51548098A JP 3790273 B2 JP3790273 B2 JP 3790273B2
- Authority
- JP
- Japan
- Prior art keywords
- uronic acid
- cyclopentenone
- dihydroxy
- cyclopenten
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images














Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/587—Unsaturated compounds containing a keto groups being part of a ring
- C07C49/703—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups
- C07C49/707—Unsaturated compounds containing a keto groups being part of a ring containing hydroxy groups a keto group being part of a three- to five-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H7/00—Compounds containing non-saccharide radicals linked to saccharide radicals by a carbon-to-carbon bond
- C07H7/02—Acyclic radicals
- C07H7/033—Uronic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/33—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds containing oxygen
- A61K8/35—Ketones, e.g. benzophenone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K8/00—Cosmetics or similar toiletry preparations
- A61K8/18—Cosmetics or similar toiletry preparations characterised by the composition
- A61K8/30—Cosmetics or similar toiletry preparations characterised by the composition containing organic compounds
- A61K8/60—Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q17/00—Barrier preparations; Preparations brought into direct contact with the skin for affording protection against external influences, e.g. sunlight, X-rays or other harmful rays, corrosive materials, bacteria or insect stings
- A61Q17/005—Antimicrobial preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q19/00—Preparations for care of the skin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q11/00—Preparations for care of the teeth, of the oral cavity or of dentures; Dentifrices, e.g. toothpastes; Mouth rinses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q19/00—Preparations for care of the skin
- A61Q19/10—Washing or bathing preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61Q—SPECIFIC USE OF COSMETICS OR SIMILAR TOILETRY PREPARATIONS
- A61Q5/00—Preparations for care of the hair
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Dermatology (AREA)
- Epidemiology (AREA)
- Birds (AREA)
- Emergency Medicine (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Coloring Foods And Improving Nutritive Qualities (AREA)
- Saccharide Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Detergent Compositions (AREA)
- Food Preservation Except Freezing, Refrigeration, And Drying (AREA)
- Cosmetics (AREA)
Description
本発明は、制がん作用等の生理活性を有する安全性の高いシクロペンテノン類に関し、更に詳細には当該化合物の製造方法、該化合物を有効成分とする医薬品に関する。また本発明は食品、飲料等の分野で有用な一連の発明に関する。
従来の技術
従来、臨床上の療法に用いられている薬物はアルキル化剤、代謝阻害剤、植物アルカロイド等の制がん剤、抗生物質、免疫促進剤、免疫調節剤など多岐にわたっているが、これらの薬物療法はいまだ完成したとは言いがたい。
これらのうち、天然物由来であるプロスタグランジンの中で、シクロペンテノン環を有するプロスタグランジンA及びJ類がDNA合成を抑制することにより、安全性の高い制がん剤としての可能性が報告され、それらの各種誘導体が合成されている(特開昭62−96438号公報参照)。
発明が解決しようとする課題
本発明の目的は、制がん作用等の生理作用を有する安全性の高いシクロペンテノン化合物を開発し、該化合物の製造方法及び該化合物を有効成分とする制がん剤等の医薬品、該化合物を含有する食品又は飲料を提供することにある。また本発明の目的は該化合物の使用方法、該化合物の関連化合物等を提供することにある。
課題を解決するための手段
本発明を概説すれば本発明の第1の発明は、下記(a)、(b)、(c)より選択される少なくとも1種の物を加熱処理することを特徴とする下記式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの製造方法に関する。
(a)ウロン酸又はウロン酸誘導体、
(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、
(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物。

(a)ウロン酸又はウロン酸誘導体、
(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、
(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物。
本発明の第3の発明は、下記工程を包含することを特徴とする4,5−ジヒドロキシ−2−シクロペンテン−1−オンの光学活性体の製造方法に関する。
(A):(a)、(b)、(c)より選択される少なくとも1種の物を加熱処理し、4,5−ジヒドロキシ−2−シクロペンテン−1−オンを生成させる工程、
(a)ウロン酸又はウロン酸誘導体、
(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、
(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物。
(B):必要に応じて、得られた加熱処理物より4,5−ジヒドロキシ−2−シクロペンテン−1−オンを単離する工程、
(C):4,5−ジヒドロキシ−2−シクロペンテン−1−オンを光学分割する工程。
本発明の第4の発明は、旋光度が[α]D 20−105°(c0.30,エタノール)である(−)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンに関する。
本発明の第5の発明は、旋光度が[α]D 20+104°(c0.53,エタノール)である(+)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンに関する。
本発明の第6の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とする制がん剤に関する。
本発明の第7の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とするがん細胞分化誘導剤に関する。
本発明の第8の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とするアポトーシス誘発剤に関する。
本発明の第9の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とする抗菌剤に関する。
本発明の第10の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を有効成分として使用することを特徴とするがん細胞分化誘導方法に関する。
本発明の第11の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を有効成分として使用することを特徴とするアポトーシス誘発方法に関する。
本発明の第12の発明は、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有、希釈及び/又は添加してなることを特徴とする食品又は飲料に関する。
本発明の第13の発明は、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物中の、ウロン酸、ウロン酸誘導体、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの生成中間体又は式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンと反応性を有するアミン類、アミノ酸類、ペプチド類又は蛋白質の反応性の少なくとも一部が消失した及び/又は該反応性物質の少なくとも一部が除去されたものであるウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物に関する。
本発明者らは式【1】で表される化合物、4,5−ジヒドロキシ−2−シクロペンテン−1−オン(以下、単にシクロペンテノンと称す)がウロン酸、ウロン酸誘導体、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される少なくとも1種の物の加熱処理物中に生成し、加熱処理物中より単離された当該化合物が制がん作用、アポトーシス誘発作用、抗菌作用等の種々の生理活性を有することを見出し、当該化合物の光学活性体の創製にも成功し、本発明を完成させた。
【図面の簡単な説明】
図1はシクロペンテノンのマススペクトルを示す図である。
図2はシクロペンテノンの1H−NMRスペクトルを示す図である。
図3はアルギン酸加熱処理物より調整したシクロペンテノンのアポトーシス誘発作用を示す図である。
図4はシクロペンテノンのIR吸収スペクトルを示す図である。
図5はシクロペンテノンのUV吸収スペクトルを示す図である。
図6はシクロペンテノンの検量線を示す図である。
図7はグルクロン酸加熱処理物のガスクロマトグラフ結果を示す図である。
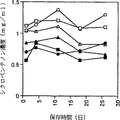
図8は保存時間とシクロペンテノン量の関係を示す図である。
図9は(−)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンの溶出曲線を示す図である。
図10は(+)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンの溶出曲線を示す図である。
図11は(−)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンの1H−NMRスペクトルを示す図である。
図12は(+)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンの1H−NMRスペクトルを示す図である。
図13は培養時間と培養液中の生細胞数の関係を示す図である。
図14は培養時間と培養細胞中において成熟骨髄細胞の占める比率の関係を示す図である。
図15シクロペンテノンの制がん効果を示す図である。
発明の実施の形態
以下、本発明をより具体的に説明する。
本発明において、ウロン酸、ウロン酸誘導体、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物とは、その加熱処理物中にシクロペンテノンが生成されれば特に限定はない。
本発明により、食品又は飲料中に生理活性を有するシクロペンテノン及び/又はその光学活性体の適量を含有させることが可能となった。これらの化合物が有する制がん作用、抗菌作用等によって、本発明の食品又は飲料は制がん性、抗菌性食品又は制がん性、抗菌性飲料として極めて有用である。
また本発明によるシクロペンテノン及び/又はその光学活性体を含有する医薬用組成物が提供され、該医薬用組成物はがんの治療剤又は予防剤として、また防腐剤、抗菌性歯磨剤、抗菌性化粧料、抗菌性浴用剤等の抗菌剤として有用である。
更に本発明によりシクロペンテノン及び/又はその光学活性体を有効成分として使用するがん細胞分化誘導方法、アポトーシス誘発方法が提供され、これらの方法は生化学研究や、がん細胞分化剤、アポトーシス誘発阻害剤等の医薬品のスクリーニングにおいて有用である。
本発明に使用されるシクロペンテノンは、(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物を加熱処理することにより生成される。従って上記(a)、(b)又は(c)を含有しない原料を物理的、化学的、酵素的あるいはその他の手段を用いて生成せしめた(a)、(b)又は(c)を加熱処理することにより、本発明のシクロペンテノンを得ることもできる。
また本発明においてはシクロペンテノンを含有する加熱処理物、該加熱処理物からの部分精製シクロペンテノン及び精製シクロペンテノンを使用することができる。
ウロン酸はグリクロン酸ともいい、アルドースのアルデヒド基はそのままにして他端の第1アルコール基だけをカルボキシル基に酸化したヒドロキシアルデヒド酸の総称であり、天然では動植物の各種の多糖の構成成分として存在する。ウロン酸を含有する多糖としては、ペクチン、ペクチン酸、アルギン酸、ヒアルロン酸、ヘパリン、ヘパラン硫酸、フコイダン、コンドロイチン硫酸、コンドロイチン、デルマタン硫酸等があり、種々の生理機能が知られている。
本発明で使用することができるウロン酸は特に限定されるものでなく、例えばガラクツロン酸、グルクロン酸、グルロン酸、マンヌロン酸、イズロン酸等があり、ウロン酸の誘導体としては、それらのラクトン、それらのエステル、それらのアミド、それらの塩等があり、加熱処理によりシクロペンテノンを生成する物はすべて本発明の誘導体に包含される。ウロン酸のラクトンとしてはグルクロノ−6,3−ラクトン(以下、グルクロノラクトンと略記する)、マンヌロノ−6,3−ラクトン、イズロノ−6,3−ラクトン等が例示される。ウロン酸エステルとしては、例えばメチルエステル、エチルエステル、プロピレングリコールエステル、カルボキシメチルエステル等がありウロン酸より製造することができる。またウロン酸のアミド化によりウロン酸アミドも製造することができる。更にこれらの塩は常法により製造することができる。
次に本明細書において、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物は特に限定されるものでなく、例えばペクチン、ペクチン酸、アルギン酸、ヒアルロン酸、ヘパリン、ヘパラン硫酸、フコイダン、コンドロイチン硫酸、コンドロイチン、デルマタン硫酸、それらの化合物、酵素的、物理的処理物である、その分解物、分解物の誘導体、分解物の塩を使用することができる。
前記の化学的な処理方法としては、原料化合物を例えば室温〜200℃で数秒〜数時間、好ましくは50〜130℃で数秒〜60分処理すれば良く、酸性下この処理を行うとグリコシド結合が加水分解を受け、ペクチンの場合、ガラクツロン酸及び/又はガラクツロン酸エステルを含む分解物が生ずる。また例えばpH6.8、95℃で数分〜数十分処理することによりβ−脱離反応が生じ、235nm付近の吸光度が増大した不飽和ウロン酸及び/又は不飽和ウロン酸エステルを有する糖化合物が得られる。本発明の糖化合物にはウロン酸及び/又はウロン酸エステルを含有する多糖類のβ−脱離反応により生成する非還元末端に不飽和ウロン酸及び/又は不飽和ウロン酸エステルを含有する糖化合物が含まれる。
また前記の酵素学的な処理方法としては、原料糖化合物のウロン酸及び/又はウロン酸エステル含有多糖加水分解酵素、例えばペクチナーゼ、ヒアルロニダーゼ等によるウロン酸及び/又はウロン酸エステル含有多糖の公知の分解が挙げられる。また、ウロン酸及び/又はウロン酸エステル含有多糖リアーゼによるウロン酸及び/又はウロン酸エステル含有多糖の公知の分解が挙げられる。例えばペクチン、ペクチン酸の場合、各々公知のペクチンリアーゼ(EC4.2.2.10)、ペクチン酸リアーゼ(EC4.2.2.2)、エキソポリガラクツロン酸リアーゼ(EC4.2.2.9)で分解することによって、非還元末端に4−デオキシ−L−トレオ−ヘキス−4−エノピラノシル ウロネート(4-deoxy-L-threo-hex-4-enopyranosyl uronate)又はそのメチルエステルを有する糖化合物が得られる。また、ヒアルロン酸の場合はヒアルロン酸リアーゼ(EC4.2.2.1)、アルギン酸の場合はアルギン酸リアーゼ(EC4.2.2.3)が使用される。なお、アルギン酸の場合は非還元末端に4−デオキシ−L−エリトロ−ヘキス−4−エノピラノシル ウロネートを有する糖化合物が得られる。この非還元末端に4−デオキシ−L−トレオ−ヘキス−4−エノピラノシル ウロネート、4−デオキシ−L−エリトロ−ヘキス−4−エノピラノシル ウロネート又はそれらのメチルエステルを有する酵素分解物も本発明の糖化合物に包含される。
更に前記の物理的な処理方法としては、原料糖化合物の近赤外線、赤外線、マイクロ波、超音波処理等が挙げられ、例えばペクチン及び/又はペクチン酸をpH中性又はアルカリ性の溶液中に入れ、温度は適宜、室温以上で、適宜還元下、例えばアスコルビン酸存在下で、時間は1秒以上、好ましくは5秒〜1時間の超音波処理をし、振動エネルギーを与えることが挙げられる。なお超音波以外にもマイクロ波、近赤外線、赤外線等の照射も有効で、これらを組合せ照射しても良い。照射は連続的に行っても良く、断続的に行っても良い。
本発明においてウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物とは、上記のウロン酸及び/又はウロン酸誘導体を含有する糖化合物の含有物であれば特に限定はない。ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物としてはリンゴ、例えばミカン、レモン等の柑橘類、バナナ、白菜、キャベツ、レタス、シソ、カボチャ、セロリ、ゴボウ、エシャロット、ブロッコリー、ピーマン、ほうれん草、タマネギ、人参、人参の葉、大根の葉、茶、ゴマ、マメ、イモ類の双子葉類植物の果実、野菜、葉、種実等、麦、米等の単子葉植物の穀物、褐藻類、例えば昆布、ワカメ等、紅藻類、緑藻類、単細胞緑藻類等の藻類、微生物としてはリオフィラム ウルマリウム、ハタケシメジ、ナメコ、シイタケ、エノキタケ、ヒラタケ、マッシュルーム等の担子菌類、サナギタケ、ノムシタケ等の子のう菌類、酵母、糸状菌、例えば麹菌、細菌、例えば納豆菌、乳酸菌等、動物としては脊椎動物又は無脊椎動物、ブタ皮膚、ウシ皮膚、サメ軟骨、鯨軟骨等が例示され、本発明においては、これらの植物、微生物又は動物由来のウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物を使用することができる。
また本発明においては、ウロン酸及び/又はウロン酸誘導体を含有する酸化合物含有物として、果物果皮、果物搾汁かす、例えばリンゴ搾汁かす、ミカン搾汁かす、野菜搾汁かす、穀類かす、例えば清酒粕、ビールかす、焼酎かす、ウイスキーかす、豆類かす、例えばおから、海藻かす等の農水産・食品加工処理物をそのまま、あるいは乾燥、粉砕して用いても良い。
本発明で使用するウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物はそのまま、若しくは前処理として通常の、煮る、焼く、炒る、焙じる、煎じる、蒸す、炒める、揚げる等の任意の加工方法で処理することができる。
本発明においては、これらのウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物を前記の化合物、酵素的(微生物による発酵を含む)、物理的前処理を行って得られる該含有物の処理物、該処理物よりの精製物を使用しても良い。
ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物には、ウロン酸、ウロン酸誘導体、シクロペンテノンの生成中間体又はシクロペンテノンと反応性を有する物質、例えばアミン類、アミノ酸類、ペプチド類及び/又は蛋白質が含有されており、シクロペンテノンを製造するため好適にはこれらの反応性物質の反応性の少なくとも一部を消失させるための及び/又は該反応性物質の少なくとも一部を除去するための前処理を行うことが好ましい。該前処理方法に特に限定はないが、乾式加熱処理が好適であり、例えば上記のウロン酸及び/又はウロン酸誘導体を含有する糖化合物の含有物の水分を除いたものを60〜400℃で数秒から数日の加熱処理を行うことにより、本発明のシクロペンテノン、その光学活性体の製造に好適な、蛋白質等が不溶化、変性、若しくは不活性化されたウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物を得ることができる。乾式加熱処理の方法としては特開平2−79965号公報に記載の熱風を使用する焙炒処理方法があり、該方法により多量の加熱処理物、即ち焙炒処理物を効率よく調製することができる。焙炒処理物に特に限定はないが、焙炒植物、焙炒動物、焙炒微生物、例えば焙炒野菜、焙炒果実、焙炒穀物、焙炒きのこ、焙炒海藻、焙炒皮質、焙炒軟骨等が例示される。他の例としては該糖化合物含有物を蛋白質分解酵素処理した後、蛋白質分解物を除去した画分が挙げられる。また該糖化合物含有物の粉砕物の水洗物画分、該糖化合物含有物の酸で前処理した画分、該糖化合物含有物のアルカリで前処理した画分等が挙げられる。上記のように、シクロペンテノンの製造に好適であるシクロペンテノン生成性の前処理物は全て本発明で定義したウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物に包含される。
シクロペンテノンの生成中間体とはウロン酸、ウロン酸誘導体、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物の加熱処理中に生成し、更に反応を行えばシクロペンテノンに変化する物質を意味する。シクロペンテンの生成中間体としては、ウロン酸の脱炭酸物、ウロン酸の脱水物、ウロン酸の脱炭酸・脱水物が例示される。
ウロン酸及び/又はウロン酸誘導体を含有する糖化合物である多糖類は公知の化学的、酵素学的、物理的な処理方法により製造することができる。例えばペクチンとしては、例えば柑橘類の果皮及びリンゴの果実より抽出される高分子の多糖類を使用することができる。工業的なペクチン製造の原料はフルーツで、レモン、ライム等の柑橘類のジュースのしぼりかす(主として内果皮)が用いられるほか、リンゴのジュースのしぼりかすも用いられている。ジュースのしぼりかすには主として不溶性のプロトペクチンが含まれており、製造の段階でこれを可溶化(抽出)し、ペクチンを調製する。可溶化は酸性の温水〜熱水で抽出することによって行うことができ、抽出時の温度、pH、時間条件を原料に合せてコントロールすることにより、分子量やエステル化度の一定なペクチンを高収量で製造することができる。抽出液は遠心分離やろ過によって精製し、濃縮後アルコールを添加してペクチンを沈殿させ回収することができる。回収された沈殿を乾燥、粉砕し、所定の乾燥ペクチンを調製することができる。
ペクチンの主構造は、部分的にメチル化されたガラクツロン酸のポリマーである。カルボキシル基はメチルエステル化されたり、遊離の酸のままか、あるいはアンモニウム塩化、カリウム塩化、又はナトリウム塩化されている。ペクチンはメチルエステル化度(DM度:全カルボキシル基に対するメトキシル基の割合)によって、DM度の高いHMペクチン及びDM度の低いLMペクチンに分類され〔吉積智司ほか編、(株)光琳発行、新食品開発用素材便覧、第114〜119頁(1991)〕、本発明においては市販の食品添加物ペクチン〔外山章夫編、食品と科学社発行、天然物便覧、第12版、第138頁(1993)〕、市販のHMペクチン、LMペクチン等(前出の新食品開発用素材便覧)を使用することができる。
合成法により合成されるウロン酸、ウロン酸誘導体、オリゴ糖類も本発明で使用することができる。
本発明に使用する加熱処理物は、(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物を原料として製造することができる。
本発明に使用する加熱処理物の製造における加熱処理方法としては、本発明のシクロペンテノンが生成する条件であれば特に限定は無いが、ウロン酸、ウロン酸誘導体、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、ウロン酸含有物及び/又はウロン酸誘導体を含有する糖化合物含有物を例えば60〜350℃で数秒〜数日、好ましくは80〜150℃で数分〜数日加熱処理すれば良く、ペクチンの場合、例えば80〜150℃で数分〜数日の加熱処理を行うことにより、シクロペンテノンを含有する加熱処理物を得ることができる。またウロン酸、ウロン酸のラクトン、ウロン酸エステルを60〜150℃で数分〜数日加熱処理することによりシクロペンテノンを含有する目的の加熱処理物を得ることができる。これらの加熱処理物には4位と5位のヒドロキシル基がトランスの関係にあるトランス−シクロペンテノンとシスの関係にある微量のシス−シクロペンテノンが含まれている。この加熱処理物から精製したシクロペンテノンを無水ピリジン中で無水酢酸と反応させて得た4,5−ジアセチルシクロペンテノンをシリカゲルカラムクロマトグラフィーで分離し、核磁気共鳴法で各々の画分の構造解析を行うことにより、本加熱処理物中にトランス−シクロペンテノンと微量のシス−シクロペンテノンが含まれていることが確認できる。
加熱処理時のpHは特に限定はないが、中性から酸性下で行うのが好ましく、その原料に応じ加熱処理時のpHを調整すればよいが、通常は酸性下の加熱処理により、シクロペンテノンの生成が加速される。
加熱処理時の原料の濃度はその加熱処理によりシクロペンテノンを生成しうる範囲内であれば特に限定は無く、操作性、収率等の点を考慮し設定すれば良い。
本発明における加熱処理は湿式加熱でも、乾式加熱でも良いが、本発明のシクロペンテノンの生成効率の点からは湿式加熱が好ましい。湿式加熱としては、水蒸気加熱、水蒸気加圧加熱、加圧式加熱等任意の湿式加熱方法を用いることができる。乾式加熱としては、乾燥熱風による直接加熱法、熱源から隔壁を通して加熱する間接加熱法等が使用できる。直接加熱方法としては、気流乾熱法、噴霧乾熱法等があり、間接加熱法としてはドラム乾熱法等が使用できる。
本発明に使用する加熱処理物中のシクロペンテノンは制がん作用、抗菌作用、アポトーシス誘発作用等を指標に採取することができる。採取手段としては、化学的方法、物理的方法等の公知の精製、単離手段を用いれば良く、ゲルろ過法、分子量分画膜による分画法、溶媒抽出法、分留法、イオン交換樹脂、順相、逆相の各種クロマトグラフィー法等の従来公知の精製方法を組合せ、加熱処理物中に生成されたシクロペンテノンを採取することができる。
例えば、ウロン酸としてD−グルクロン酸を使用し、その1%溶液を121℃で4時間加熱処理することにより、加熱処理物中にシクロペンテノンが生成される。この加熱物中のシクロペンテノンを溶媒で抽出し、抽出物を濃縮する。次にこの濃縮物をシリカゲルカラムクロマトグラフィーで分離し、溶出するシクロペンテノン画分を濃縮し、濃縮物からシクロペンテノンをクロロホルムで抽出することにより、加熱処理物中のシクロペンテノンが単離される。
また上記グルクロン酸加熱処理物をイオン交換樹脂カラム、好ましくは陰イオン交換樹脂カラム処理し、非吸着性画分を集めることによりシクロペンテノンが精製される。あるいは上記グルクロン酸加熱物を活性炭カラム処理し、非吸着画分の除去、カラムの洗浄を行った後、親水性有機溶媒、例えばエタノール水溶液、好ましくは40%以上のエタノール水溶液で溶出することにより、精製シクロペンテノンを得ることができる。またこれらの方法を組合わせることにより、高純度のシクロペンテノンを得ることができる。
単離されたシクロペンテノンを光学分割することにより、本発明の(−)−4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び(+)−4,5−ジヒドロキシ−2−シクロペンテン−1−オンを得ることができる。当然、合成方法により得られたシクロペンテノンも本発明により光学分割することができる。
光学活性体の分離はラセミ混合物の機械的分割、優先晶出法、ジアステレオマー塩あるいは包接化合物としての結晶化による分割、酵素・微生物による動力学的分割、クロマトグラフィーによる分割等により行うことができる。
クロマトグラフィーによる分離としては、ガスクロマトグラフィー、液体クロマトグラフィ、薄層クロマトグラフィー等を用いることができ、それぞれに適したキラル固定相を使用すればよい。
液体クロマトグラフィーによる光学分割としてはキラルな固定相を用いる方法、キラルな溶離液を用いる方法、ジアステレオマーとしての分離等を用いることができる。
キラル固定相としてはアミド系固定相、尿素系固定相、配位子交換型固定相、多糖・多糖誘導体固定相、タンパク質固定相、ポリメタクリル酸エステル固定相、ポリメタクリルアミド固定相等が使用できる。
溶離液としてはヘキサン系、アルコール系、水(緩衝液)系等が使用でき、上記固定相との組合せにおいて適宜使用することができる。
本発明で使用するシクロペンテノンの製造方法はいかなる方法でも良く、本発明で開示の方法で製造しても良く、化学合成法〔カーボハイドレートリサーチ(Carbohydrate Res.)、第247巻、第217〜222頁(1993)、ヘルベチカ キミカ アクタ(Helvetica Chimica Acta)、第55巻、第2838〜2844頁(1972)〕で合成しても良く、シクロペンテノンのトランス体、シス体が本発明に使用される。当然、化学合成法で得られたシクロペンテノンの光学活性体も本発明の光学活性体に包含される。またウロン酸、ウロン酸誘導体、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される少なくとも1種の物の加熱処理物中に生成するシクロペンテノン、その精製物、その光学活性体も使用することもできる。
シクロペンテノン及びその光学活性体は、例えばヒト前骨髄性白血病細胞HL−60、ヒト急性リンパ芽球性白血病細胞MOLT−3、肝がん細胞A−549、SV40形質転換肺細胞WI−38VA13、肺がん細胞Hep G2、結腸がん細胞HCT 116、ヒト結腸がん細胞SW480、ヒト結腸がん細胞WiDr、胃がん細胞AGS、ミエローマ細胞等のがん細胞に細胞増殖抑制作用、制がん活性を有し、シクロペンテノン及びその光学活性体は制がん剤の有効成分として使用することができる。また、これらのがん細胞にアポトーシス誘発作用を有する。本発明のシクロペンテノン及びその光学活性体のがん細胞増殖抑制作用機作は本発明をなんら制限するものではないが、例えばがん細胞に対するアポトーシス誘発作用も本発明に包含される。
制がん作用を有するシクロペンテノン及び/又はその光学活性体を有効成分とし、これを公知の医薬用担体と組合せ製剤化すれば制がん剤を製造することができる。制がん剤の製造は一般的には、シクロペンテノン及び/又はその光学活性体を薬学的に許容できる液状又は固体状の担体と配合し、かつ必要に応じて溶剤、分散剤、乳化剤、緩衝剤、安定剤、賦形剤、結合剤、崩壊剤、滑沢剤等を加えて、錠剤、顆粒剤、散剤、粉末剤、カプセル剤等の固形剤、通常液剤、懸濁剤、乳剤等の液剤であることができる。またこれを使用前に適当な担体の添加によって液状となし得る乾燥品とすることができる。
医薬用担体は、上記投与形態及び剤型に応じて選択することができ、経口剤の場合は、例えばデンプン、乳糖、白糖、マンニット、カルボキシメチルセルロース、コーンスターチ、無機塩等が利用される。また経口剤の調製に当っては、更に結合剤、崩壊剤、界面活性剤、潤滑剤、流動性促進剤、矯味剤、着色剤、香料等を配合することもできる。
一方、非経口剤の場合は、常法に従い本発明の有効成分であるシクロペンテノン及び/又はその光学活性体を希釈剤としての注射用蒸留水、生理食塩水、ブドウ糖水溶液、注射用植物油、ゴマ油、ラッカセイ油、ダイズ油、トウモロコシ油、プロピレングリコール、ポリエチレングリコール等に溶解ないし懸濁させ、必要に応じ、殺菌剤、安定剤、等張化剤、無痛化剤等を加えることにより調製される。
本発明の制がん剤は、製剤形態に応じた適当な投与経路で投与される。投与方法も特に限定はなく、内用、外用及び注射によることができる。注射剤は、例えば静脈内、筋肉内、皮下、皮内等に投与し得、外用剤には座剤等も包含される。
制がん剤としての投与量は、その製剤形態、投与方法、使用目的及びこれに適用される患者の年齢、体重、症状によって適宜設定され、一定ではないが一般には製剤中に含有されるシクロペンテノン及び/又はその光学活性体の量が成人1日当り0.1μg〜200mg/kgである。もちろん投与量は、種々の条件によって変動するので、上記投与量より少ない量で十分な場合もあるし、あるいは範囲を超えて必要な場合もある。本発明の薬剤はそのまま経口投与するほか、任意の飲食品に添加して日常的に摂取させることもできる。
シクロペンテノン及び/又はその光学活性体は制がん作用を有するが、低濃度ではがん細胞の分化誘導能を有し、シクロペンテノン及び/又はその光学活性体はがん細胞の分化誘導剤(脱がん剤)としても有用である。シクロペンテノン及び/又はその光学活性体を有効成分とするがん細胞分化誘導剤は、上記制がん剤に準じ、製剤化することができ、制がん剤に準じた方法で投与することができる。
がん細胞分化誘導剤としての投与量は、その製剤形態、投与方法、使用目的及びこれに適用される患者の年齢、体重、症状によって適宜設定され、一定ではないが一般には製剤中に含有されるシクロペンテノン及び/又はその光学活性体の量が成人1日当り0.1μg〜100mg/kgである。もちろん投与量は、種々の条件によって変動するので、上記投与量より少ない量で十分な場合もあるし、あるいは範囲を超えて必要な場合もある。本発明の薬剤はそのまま経口投与するほか、任意の飲食品に添加して日常的に摂取させることもできる。
本発明のがん細胞分化誘導剤はがん細胞分化誘導方法に使用することができる。すなわちシクロペンテノン及び/又はその光学活性体を有効成分として使用することによりがん細胞を分化させることができ、該方法はがん細胞の分化誘導機構の解明、分化誘導剤のスクリーニング等に有用である。
本発明の抗菌剤はシクロペンテノン及び/又はその光学活性体を有効成分とし、これを公知の医薬用担体と組合せ製剤化すれば本発明の抗菌剤を製造することができる。当該製剤の製造は一般的には、シクロペンテノン及び/又はその光学活性体を薬学的に許容できる液状又は固体状の担体と配合し、かつ必要に応じて溶剤、分散剤、乳化剤、緩衝剤、安定剤、賦形剤、結合剤、崩壊剤、滑沢剤等を加えて、錠剤、顆粒剤、散剤、粉末剤、カプセル剤等の固形剤、通常液剤、懸濁剤、乳剤等の液剤とすることができる。またこれを使用前に適当な担体の添加によって液状となし得る乾燥品とすることができる。
医薬用担体は、上記投与形態及び剤型に応じて選択することができ、経口剤の場合は、例えばデンプン、乳糖、白糖、マンニット、カルボキシメチルセルロース、コーンスターチ、無機塩等が利用される。また経口剤の調製に当っては、更に結合剤、崩壊剤、界面活性剤、潤沢剤、流動性促進剤、矯味剤、着色剤、香料等を配合することもできる。
一方、非経口剤の場合は、常法に従い本発明の有効成分であるシクロペンテノン及び/又はそれらの光学活性体を希釈剤としての注射用蒸留水、生理食塩水、ブドウ糖水溶液、注射用植物油、ゴマ油、ラッカセイ油、ダイズ油、トウモロコシ油、プロピレングリコール、ポリエチレングリコール等に溶解ないし懸濁させ、必要に応じ、殺菌剤、安定剤、等張化剤、無痛化剤等を加えることにより調製される。
本発明の抗菌剤は、製剤形態に応じた適当な投与経路で投与される。投与方法も特に限定はなく、内用、外用及び注射によることができる。注射剤は、例えば静脈内、筋肉内、皮下、皮内等に投与し得、外用剤には座剤等も包含される。
抗菌剤としての投与量は、その製剤形態、投与方法、使用目的及びこれに適用される患者の年齢、体重、症状によって適宜設定され、一定ではないが一般には製剤中に含有されるシクロペンテノン及び/又はその光学活性体の量が成人1日当り10μg〜20mg/kgである。もちろん投与量は、種々の条件によって変動するので、上記投与量より少ない量で十分な場合もあるし、あるいは範囲を超えて必要な場合もある。本発明の薬剤はそのまま経口投与するほか、任意の飲食品に添加して日常的に摂取させることもできる。またシクロペンテノン及び/又はその光学活性体含有物を抗菌性飲食品の原料として用いても良い。またエタノール、グリシン、酢酸ナトリウム、アスコルビン酸、グリセリン脂肪酸エステル、食塩、EDTA等の他の抗菌性物質と組合せて使用しても良い。
本発明の抗菌剤はシクロペンテノン及び/又はその光学活性体を有効成分とし、食品又は飲料の保存性を向上させる防腐剤として使用することができる。また、シクロペンテノン及び/又はその光学活性体を食品又は飲料に添加し、食品又は飲料を防腐する方法に使用することができる。
食品又は飲料に添加する場合のシクロペンテノン及び/又はその光学活性体含有抗菌剤の形状は、液状、ペースト状、粉末状、フレーク状、顆粒状等いずれの形状でも良い。取り扱いやすさや、他の添加物と混合物して使用することも考えれば、乾燥して粉末状、フレーク状、顆粒状にすることが好ましい。乾燥方法としては、通常の乾燥方法、例えばスプレー乾燥、ドラム乾燥、棚式乾燥、真空乾燥、凍結乾燥などで行うことができる。
シクロペンテノン及び/又はその光学活性体の食品又は飲料への添加量は食品又は飲料の種類により異なり、その目的に応じた量を添加すれば良い。
本発明の抗菌剤の使用方法として、食品又は飲料に適当な方法で添加する方法が行われる。添加する方法は特に制限はなく、要するに何らかの方法でシクロペンテノン及び/又はその光学活性体が食品又は飲料に含有されれば良い。したがって、本発明の抗菌剤の使用において、添加とはシクロペンテノン及び/又はその光学活性体を食品又は飲料中に含有させるいかなる方法も含む。通常の方法は食品又は飲料の製造工程中に添加するが、シクロペンテノン及び/又はその光学活性体を含有する溶液に食品を一定時間浸漬する方法も用いることができる。更にまた、食品中に添加する方法と浸漬方法を併用することもできる。浸漬方法に適する食品としては、水中でも形くずれしない食品、例えば、蒲鉾、ウインナソーセージ等の魚畜肉練り製品、ゆで麺等の麺類、冷凍前の海老、貝、魚類等の冷凍食品などを挙げることができる。
本発明の抗菌剤を防腐剤として用いることにより、食品又は飲料の保存性を一段と向上させることができる。また冷凍食品や冷菓等においては、冷凍前の加工工程において、汚染した微生物の増殖を抑制することができ、衛生上極めて好ましい結果を得ることができる。本発明の抗菌剤はグラム陽性細菌、グラム陰性細菌の両方に効果を有し、例えばメチシリン耐性黄色ブドウ球菌等の薬剤耐性菌、サルモネラ菌、エンテロトキシン生産性黄色ブドウ球菌、嘔吐型のバシルス セレウス、下痢型のバシルス セレウス、腸出血性大腸菌O−157等の食中毒菌に極めて有効である。更に火落菌にも有効である。また細菌起因性疾病の起因菌、例えば在郷軍人病の起因菌のレジオネラ ニューモフィラ(Legionella pneumophila)、食中毒起因菌のビブリオ パラハエモリチカス(Vibrio parahaemolyticus)、潰瘍起因菌のヘリコバクター ピロリ(Helicobacter pylori)、胃腸炎起因菌のキャンピロバクター ジェジュニ(Campylobacter jejuni)等、例えばLegionella pneumophila(ATCC33153)、Vibrio parahaemolyticus(ATCC17802)、Helicobacter pylori(NCTC11637)、Campylobacter jejuni(ATCC29428)等に抗菌作用を有し、また、酵母、カビ等の微生物にも有効である。特に天然食品由来のシクロペンテノン及び/又はその光学活性体を含有する防腐剤は天然由来の食中毒予防剤、除菌剤として有用性が高い。なお本発明の抗菌剤を用い、衣服、敷布等の殺菌を行うことができ、本発明の抗菌剤を散布すること、本発明の抗菌剤での拭き取り等により、目的物の除菌、殺菌を行うことができる。例えばビルの冷房用水に添加することにより、在郷軍人病の予防を行うことができる。
本発明の抗菌剤は虫歯菌や歯周病菌にも抗菌活性を示し、本発明の抗菌剤を含有する口内用剤を提供することができる。口内用剤の形状は液状、ペースト状等の公知の形状とすることができる。口内用剤としては歯磨剤が例示される。歯磨剤としては液状でもよく、またペースト状、粉末状でもよく、公知の歯磨剤の形状とすることができる。歯磨剤中のシクロペンテノン及び/又はその光学活性体の歯磨剤中の含有量は特に制限されず、虫歯菌や歯周病菌に対する有効濃度が含有されていればよい。歯磨剤中には公知の添加剤、例えば湿潤剤、界面活性剤、結合剤、香料、甘味料等を添加すればよい。本発明の歯磨剤の有効成分としてはシクロペンテノン含有物、例えば野菜、果物等の加熱処理物も使用でき、これらのシクロペンテノン含有の加熱処理物を含有する口内用剤、例えば歯磨剤も本発明に包含される。
本発明の抗菌剤を使用することにより抗菌性化粧料を提供することができる。本発明の化粧料としては有効量のシクロペンテノン及び/又はその光学活性体を含有するクリーム、乳液、ローション、洗顔料、パック等の基礎化粧料、口紅、ファンデーション等のメイクアップ化粧料、ボディソープ、石鹸等の形態に調製することができる。また、頭髪に対しても有効であり、ヘアートニック、ヘアーリキッド、ヘアーセットローション、ヘアーブロー剤、ヘアークリーム、ヘアーコート等のヘアー製品やシャンプー、リンス、ヘアートリートメント等の頭髪用トイレタリー等のヘアーケアー製品の形態にすることができる。化粧料への配合量としては通常化粧料100部(重量部、以下同様)中のシクロペンテノン及び/又はその光学活性体を約10-3〜10部、好ましくは10-2〜1部とすれば良い。化粧料の他の成分は通常化粧料に配合されるものが使用できる。本発明の化粧料はアトピー性皮膚炎の起因菌にも有効に作用し、アトピー性皮膚炎の改善、予防にも著効を有する。
本発明の抗菌剤を使用することにより浴用剤を提供することができる。本発明の浴用剤としては有効量のシクロペンテノン及び/又はその光学活性体を含有する粉末浴用剤、顆粒浴用剤、固形浴用剤、液状浴用剤等の形態に調製することができる。浴用剤への配合量としては通常浴用剤100部(重量部、以下同様)中のシクロペンテノン及び/又はその光学活性体を約10〜100部、好ましくは20〜90部とすれば良く、かくして調製される本発明の浴用剤は、湯200リットルに対し通常5〜25グラム添加すれば良い。浴用剤の他の成分は通常浴用剤に配合されるものが使用できる。本発明の浴用剤はアトピー性皮膚炎の起因菌にも有効に作用し、アトピー性皮膚炎の改善、予防にも著効を有する。また浴場からの病因菌の駆除にも有効である。
以上、本発明により、医薬、化粧料、浴用剤として有用な抗菌剤が提供される。また、シクロペンテノン等を含有する食品又は飲料は食中毒、胃腸炎等の改善及び/又は予防に極めて有用である。
本発明のアポトーシス誘発剤は、アポトーシス誘発性を有するシクロペンテノン及び/又はその光学活性体を有効成分とし、これを上記制がん剤に準じ、製剤化することができ、制がん剤に準じた方法で投与することができる。
アポトーシス誘発剤としての投与量は、その製剤形態、投与方法、使用目的及びこれに適用される患者の年齢、体重、症例によって適宜設定され、一定ではないが一般には製剤中に含有されるシクロペンテノン及び/又はその光学活性体の量が成人1日当り0.1μg〜100mg/kgである。もちろん投与量は、種々の条件によって変動するので、上記投与量より少ない量で十分な場合もあるし、あるいは範囲を超えて必要な場合もある。本発明の薬剤はそのまま経口投与するほか、任意の飲食品に添加して日常的に摂取させることもできる。
なおアポトーシスは、病理的細胞死である壊死と異なり、細胞自身の遺伝子に最初から組込まれている死であると考えられている。すなわち何らかの外部的又は内部的要因が引き金となってアポトーシスをプログラムする遺伝子が活性化され、この遺伝子を基にプログラム死遺伝子タンパク質が生合成され、生成したプログラム死タンパク質により細胞自体が分解され、死に至ると考えられている。
本発明のアポトーシス誘発剤は、このようなアポトーシスを所望の組織、細胞で発現させることができ、不要若しくは病原細胞を自然の形で生体から排除することが可能となり、極めて有用なものである。
本発明のアポトーシス誘発剤はアポトーシス誘発方法に使用することができる。すなわちシクロペンテノン及び/又はその光学活性体を有効成分として使用することによりアポトーシスを誘発させることができ、該方法はアポトーシス誘発機構の解明、アポトーシス誘発剤、アポトーシス誘発阻害剤のスクリーニング等に有用である。
本発明の抗菌性食品又は飲料とは、特に限定はないが、例えば穀物加工品(小麦粉加工品、でんぷん類加工品、プレミックス加工品、麺類、マカロニ類、パン類、あん類、そば類、麩、ビーフン、はるさめ、包装餅等)、油脂加工品(可塑性油脂、てんぷら油、サラダ油、マヨネーズ類、ドレッシング等)、大豆加工品(豆腐類、味噌、納豆等)、食肉加工品(ハム、ベーコン、プレスハム、ソーセージ等)、水産製品(冷凍すりみ、かまぼこ、ちくわ、はんぺん、さつま揚げ、つみれ、すじ、魚肉ハム、ソーセージ、かつお節、魚卵加工品、水産缶詰、つくだ煮等)、乳製品(原料乳、クリーム、ヨーグルト、バター、チーズ、練乳、粉乳、アイスクリーム等)、野菜・果実加工品(ペースト類、ジャム類、漬け物類、果実飲料、野菜飲料、ミックス飲料等)、菓子類(チョコレート、ビスケット類、菓子パン類、ケーキ、餅菓子、米菓類等)、アルコール飲料(日本酒、中国酒、ワイン、ウイスキー、焼酎、ウオッカ、ブランデー、ジン、ラム酒、ビール、清涼アルコール飲料、果実酒、リキュール等)、嗜好飲料(緑茶、紅茶、ウーロン茶、コーヒー、清涼飲料、乳酸飲料等)、調味料(しょうゆ、ソース、酢、みりん等)、缶詰・瓶詰め・袋詰め食品(牛飯、釜飯、赤飯、カレー、その他の各種調理済み食品)、半乾燥又は濃縮食品(レバーペースト、その他のスプレッド、そば・うどんの汁、濃縮スープ類)、乾燥食品(即席麺類、即席カレー、インスタントコーヒー、粉末ジュース、粉末スープ、即席味噌汁、調理済み食品、調理済み飲料、調理済みスープ等)、冷凍食品(すき焼き、茶碗蒸し、うなぎかば焼き、ハンバーグステーキ、シュウマイ、餃子、各種スティック、フルーツカクテル等)、固形食品、液体食品(スープ等)、香辛料類等の農産・林産加工品、畜産加工品、水産加工品等が挙げられる。
本発明の食品又は飲料の製造法は、特に限定はないが、調理、加工及び一般に用いられている食品又は飲料の製造法にある製造を挙げることができ、製造された食品又は飲料にシクロペンテノン及び/又はその光学活性体が含有されていれば良い。
調理及び加工においては、調理、加工後の(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物の加熱処理物中にシクロペンテノン及び/又はその光学活性体が含有されていれば良い。
すなわち調理・加工前、調理・加工時、更には調理・加工後にシクロペンテノン及び/又はその光学活性体を含有する(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物の加熱処理物を添加してもよいし、調理及び加工品やその材料を、シクロペンテノン及び/又はその光学活性体を含有する(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物の加熱処理物へ添加し、該加熱処理物中のシクロペンテノン及び/又はその光学活性体を希釈してもよい。
次に食品又は飲料の製造においては、任意の工程で、加熱処理を行い、加熱処理物中にシクロペンテノン及び/又はその光学活性体を含有させれば良く、シクロペンテノン及び/又はその光学活性体を含有する加熱処理物を添加してもよい。また食品又は飲料やその原料をシクロペンテノン及び/又はその光学活性体を含有する加熱処理物へ添加し、該加熱処理物中のシクロペンテノン及び/又はその光学活性体を希釈してもよい。また、添加は1回又は数回に渡って行ってもよい。したがって、簡便に新規な生理作用を示す食品又は飲料を製造することができる。また製造時において(a)ウロン酸又はウロン酸誘導体、(b)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物、(c)ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物から選択される物を含有せしめ、製造時において生成した加熱処理物中のシクロペンテノン及び/又はその光学活性体を構成成分とする食品又は飲料も本発明に包含される。いずれの工程を経た場合も、シクロペンテノン及び/又はその光学活性体を含有、添加及び/又は希釈してなる食品又は飲料は本発明の食品又は飲料と定義される。
生理作用を有するシクロペンテノン及び/又はその光学活性体の食品中の含有量は特に制限されず、その官能と制がん性、抗菌性等の生理活性の点より適宜選択できるが、例えばシクロペンテノン及び/又はその光学活性体の含有量は食品100部当り5×10-6部以上、食品としての官能、生理作用の面からは好ましくは10-5〜5部、更に好ましくは10-4〜2部である。
また、生理作用を有するシクロペンテノンの飲料中の含有量は特に制限されず、その官能と制がん性、抗菌性等の生理活性の点より適宜選択できるが、例えばシクロペンテノンの含有量は飲料100部当り5×10-6部以上、飲料としての官能、生理作用の面からは好ましくは10-5〜5部、更に好ましくは10-4〜2部である。
本発明の食品又は飲料としては、制がん剤、抗菌性等の生理作用を有するシクロペンテノン及び/又はその光学活性体が含有、添加及び/又は希釈されていれば特にその形状に限定は無く、タブレット状、顆粒状、カプセル状、ゲル状、ゾル状等の形状の経口的に摂取可能な形状物も包含する。
以上、本発明に使用するシクロペンテノン及びその光学活性体は、安価に製造でき、その種々の生理的機能により、食品又は飲料の添加剤として使用することにより、食品又は飲料に簡便に種々の生理的機能、抗菌力、アポトーシス誘発作用、制がん作用等を付与することができ、本発明のシクロペンテノン及びその光学活性体は食品又は飲料への添加剤として極めて有用である。
本発明に使用するシクロペンテノン及びその光学活性体の製造方法はいかなる方法でも良く、本発明で開示の方法で製造しても良く、化学合成方法で合成しても良い。すなわち、シクロペンテノン及び/又はその光学活性体を含有する食品又は飲料、抗菌剤、制がん剤、がん細胞分化誘導剤、アポトーシス誘発剤はすべて本発明に包含される。またシクロペンテノン及び/又はその光学活性体を有効成分として使用するがん細胞分化誘導方法及びアポトーシス誘発方法はすべて本発明に包含される。
本発明のシクロペンテノン及びその光学活性体は100mg/kgの経口投与でマウスに毒性は認められない。
以上、シクロペンテノン及びその光学活性体は、安価に簡便に製造でき、その種々の生理的機能により、医薬、食品等の広い分野において極めて有用な化合物である。
実施例
本発明を以下の実施例により更に具体的に説明するが、本発明はこれらの実施例に何ら限定されるものではない。なお、実施例における%及び部は特記しない限り重量による。
実施例 1
(1)市販のリンゴ製ペクチンを1%になるように水に溶解し、還流冷却器を取り付けたナス型フラスコに入れて110〜120℃に設定した油浴中18時間、42時間、及び66時間加熱した。加熱中のペクチン溶液の温度は100〜102℃であった。
上記ペクチン溶液を遠心して沈殿を除き、その上清を3倍及び10倍に水で希釈した試料を調製した。次に該加熱処理物をNaOHでpH7.0に調整した後、ヒト前骨髄性白血病細胞HL−60細胞(ATCC CCL−240)に対する細胞増殖抑制作用を以下に記載のMTT法で測定した。
すなわち希釈した試料10μlと56℃、30分間処理した牛胎児血清(ギブコ社製)を10%含むRPMI1640培地(日水社製)にて37℃で培養した5000個のHL−60細胞を含むRPMI1640培地100μlを96穴マイクロタイタープレートのウェルに添加し、5%炭酸ガス存在下37℃で48時間培養後、5mg/mlの3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT;シグマ社製)リン酸緩衝食塩水溶液10μlを加えて更に4時間培養を続けた後、顕微鏡で細胞の生育状態を観察した。また、0.04N HCl含有2−プロピルアルコール100μlを加えて良く攪拌し、590nmにおける吸光度を測定してこれを細胞増殖度とした。
その結果、18時間加熱ペクチンの3倍希釈液添加区分、42時間及び66時間加熱ペクチンの3倍、10倍希釈液添加区分において生細胞は観察されず、これらの希釈液濃度において100℃加熱ペクチンは細胞増殖抑制活性を示した。
一方、18時間加熱ペクチンの10倍希釈液添加区分ではほぼすべての細胞が生細胞であったが、対照の水添加区分と比べて590nmにおける吸光度は低かった。
(2)100℃で18〜66時間加熱したペクチン200μlに200μlのメタノールを加えて混合後遠心を行い、上清200μlを減圧下濃縮乾固した。これを10μlの50%メタノール水溶液に溶解し、1μlをシリカゲル60シートF(メルク社製)にスポットした後、展開溶媒(酢酸ブチル:酢酸蒸:蒸留水=3:1:1の上層)で展開した。展開し終えた薄層シリカゲルを乾燥後、AgNO3−NH3溶液(0.1M AgNO3と5N NH3の等量混合物)を噴霧し、加熱することにより、スポットを検出した。その結果、Rf値約0.3付近のスポットが18時間加熱したペクチンで現れ、42時間加熱したペクチンでは18時間加熱したペクチンよりも増加しており、66時間加熱したペクチンでは42時間加熱したペクチンと同程度の量であった。
(3)実施例1−(1)記載の100℃で66時間加熱したペクチン1mlにメタノール1mlを加えて混合し、遠心によって上清を得た。この上清を減圧下濃縮乾固し、100μlのメタノールに懸濁した。この懸濁液を遠心して不溶物を除き、上清をシリカゲル60シートF254にスポットし、実施例1−(2)に記載の溶媒で展開した。薄層の一部を切り取って実施例1−(2)に記載の方法で発色させてRf値約0.3付近のスポットが出現することを確認し、このRf値に相当する部分のシリカゲルを未発色の薄層からかき取った。
かき取ったシリカゲルから1mlのメタノールで3回抽出し、減圧下濃縮乾固してRf値約0.3付近のスポットを単離した。この乾固物を250μlの水に溶解し、更に水で10倍に希釈した液10μlを試料として実施例1−(1)記載のMTT法でHL−60細胞に対する細胞増殖抑制活性を測定した。
その結果、水を添加したウェルではほとんどの細胞が生育していたのに対して本希釈液を添加したウェルでは生細胞が見られなかった。すなわち、このかき取り区分のがん細胞増殖抑制作用を確認した。
(4)実施例1−(3)で単離したRf値約0.3付近のがん細胞増殖抑制物質の質量分析をDX302質量分析計(日本電子社製)を用いて行った。また、重クロロホルム溶媒を用いて核磁気共鳴法(NMR)によって構造を解析した。核磁気共鳴装置はJNM−A500(日本電子社製)を用いた。その結果を以下に示す。
FAB−MS m/z 115〔M+H〕+
マトリックスとしてグリセロールを用いた。
1H−NMR(CDCl3)
δ4.20(1H,d,J=2.4Hz,5−H)、4.83(1H,m,4−H)、6.30(1H,dd,J=1.2,6.1Hz,2−H),7.48(1H,dd,J=2.1,6.1Hz,3−H)
但し、1H−NMRの化学シフト値はCHCl3の化学シフト値を7.26ppmとして表した。
これらの値はアハマド(T.Ahmad)ら〔カーボハイドレート リサーチ(Carbohydrate Res.)第247巻、第217−222頁(1993)〕が報告しているトランス−4,5−ジヒドロキシ−2−シクロペンテン−1−オンの数値と一致した。
図1にそのマスのスペクトルを示し、その縦軸は相対強度(%)、横軸はm/z値を示す。
図2にその1H−NMRスペクトルを示し、その縦軸はシグナルの強度、横軸は化学シフト値(ppm)を示す。
実施例 2
(1)25gのアルギン酸(非膨潤性、和光純薬社製)を475mlの水に懸濁し、121℃で2時間加熱した後、遠心によって得た上清を0.22μmのメンブレンフィルターでろ過し、ろ液を20mlになるまで減圧下濃縮した。これに180mlのエタノールを加えて混合後、−20℃で1時間放置し、遠心によって上清を得た。この上清を減圧下20mlまで濃縮し、180mlのアセトニトリルを加えた後、遠心によって得た上清を減圧下20mlまで濃縮し、再び180mlのアセトニトリルを加えて遠心し、その上清を減圧下15mlまで濃縮した。4mlの上記濃縮液を減圧下約400μlまで濃縮した後、酢酸ブチル:酢酸:水=3:2:2混合液の上層を等量加えて攪拌して遠心上清を集めるという操作を繰り返し、8mlの抽出液を得た。
4mlの上記抽出液をカラムクロマトグラフィー用シリカゲルBW−300SP(2x28cm、富士シリシア化学社製)にアプライし、酢酸ブチル:酢酸:水=3:2:2の上層を溶離液としてコンプレッサーで0.18kg/cmに加圧し、毎分約5mlの流速で分離を行った。1画分当り6〜7mlになるようにフラクショネーションを行い、各画分の一部をとって薄層クロマトグラフィーで分析したところ31番から35番までの画分に高純度のシクロペンテノンが含まれており、35mgのシクロペンテノンを得た。
この画分をパルパックタイプSカラムを用いた順相HPLCで分離し、215nmの紫外線吸収で検出したところ、純度は95.8%であった。
(2)実施例2−(1)で調製したシクロペンテノンを用い、2.86mM、955μM、318μM、106μM、35μM、12μM、及び0.18μMのシクロペンテノン水溶液を調製した。次に該加熱処理物をNaOHでpH7.0に調整した後、HL−60細胞に対するアポトーシス誘発活性を次のように測定した。
56℃、30分間処理した牛胎児血清を10%含むRPMI1640培地にて37℃で培養したHL−60細胞を10%牛胎児血清含有RPMI1640培地にて5.8×104個/1.35mlとなるように懸濁した。
この懸濁液1.35mlに対し、前記シクロペンテノン溶液を0.15ml添加し、37℃、5%二酸化炭素存在下で20時間培養した。
その結果、培養20時間後において106μM以上のシクロペンテノンを添加した区分(終濃度10.6μM)では生細胞数及び細胞生残率の低下が見られ、35μMシクロペンテノンを添加した区分(終濃度3.5μM)ではアポトーシス誘発によるDNAの低分子化と細胞形状の変化(核の凝縮、細胞の縮小、アポトーシス小体の形成)が見られた。
その結果を図3に示す。すなわち図3はHL−60細胞の培養液に様々な濃度のシクロペンテノンを添加したときの培養時間と生細胞数の関係を示す図であり、横軸は培養時間(時間)、縦軸は培養液中の生細胞数(×104個×1.5ml)を示す。
図3中において白四角印(□)は試料無添加(対照)、白ひし形印(◇)は2.86mMシクロペンテノン添加区分、白丸印(○)は955μMシクロペンテノン添加区分、白三角印(△)は318μMシクロペンテノン添加区分、黒四角印(■)は106μMシクロペンテノン添加区分、黒ひし形印(◆)は35μMシクロペンテノン添加区分、黒丸印(●)は12μMシクロペンテノン添加区分をそれぞれ示す。
実施例 3
(1)10gのD-グルクロン酸(シグマ社製G 5269)を1リットルの水に溶解し、121℃で4時間加熱した後約10mlになるまで減圧下濃縮した。これに酢酸ブチル:酢酸:水=3:2:2混合液の上層40mlを加えて混合後、遠心によって得た上清を減圧下約10mlまで濃縮した。
上記抽出液をカラムクロマトグラフィー用シリカゲルBW−300SP(2×28cm、富士シリシア化学社製)にアプライし、酢酸ブチル:酢酸:水=3:2:2の上層を溶離液としてコンプレッサーで0.2kg/cmに加圧し、毎分5mlの流速で分離を行った。1画分当り10mlになるようにフラクショネーションを行い、各画分の一部をとって薄層クロマトグラフィーで分析したところ61番から80番までの画分に高純度のシクロペンテノンが含まれていた。これらの画分を集めて減圧下濃縮した後40mlのクロロホルムで抽出し、抽出液を減圧下濃縮することによって100mgのシクロペンテノンを得た。
この画分をパルパックタイプSカラムを用いた順相HPLCで分離し、215nmの紫外部吸収で検出したところ、純度は98%であった。
(2)上記シクロペンテノンの質量分析をDX302質量分析計を用いて行った。また、重クロロホルム溶媒を用いたNMRによって構造を解析した。核磁気共鳴装置はJNM−A500を用いた。比旋光度はDIP−370型旋光計(日本分光社製)、UV吸収スペクトルはUV−2500分光光度計(島津製作所社製)、赤外吸収スペクトル(IR)はFTIR−8000赤外分光光度計(島津製作所社製)をそれぞれ用いた。質量分析、NMRは実施例1−(4)と同一の結果であった。他の結果を示す。
旋光度:[α]D 20 0°(c1.3,水)
IR(KBr法):3400、1715、1630、1115、1060、1025cm-1に吸収を有する。
UV:λmax215nm(水)
図4にそのIRスペクトルを示し、その横軸は波数(cm-1)、縦軸は透過率を示す。
図5にそのUV吸収スペクトルを示し、その横軸は波長(nm)、縦軸は吸光度を示す。
実施例 4
1%のD-グルクロン酸(シグマ社製G 5269)水溶液を121℃で4時間加熱し、1N NaOHでpHを7に調整した。5mlの本試料を水で平衡化したハイトラップQカラム(5ml;ファルマシア社製)にアプライし、25mlの水でカラムを洗浄した。最初にカラムから溶出してきた5mlをP0画分、2番目に溶出してきた5mlをP1画分、3番目に溶出してきた10mlをP2画分、最後に溶出してきた10mlをP3画分とした。次に25mlの0.5M酢酸で溶出を行った。最初にカラムから溶出してきた5mlをE0画分、2番目に溶出してきた5mlをE1画分、3番目に溶出してきた5mlをE2画分、最後に溶出してきた10mlをE3画分とした。
各画分の一部をとって薄層クロマトグラフィーで分析したところシクロペンテノンはP1画分に含まれており、本画分に定量的に回収されていた。ハイトラップQカラムで精製する前に含まれていた未反応のグルクロン酸と反応生成物であるレダクチン酸はP1画分には見られなかった。
実施例 5
(1)実施例3−(1)記載のシクロペンテノン精製標品を0.67mg/mlとなるように水に溶解した。また、n-オクタコサン(ナカライテスク社製)を1mg/mlとなるようにn-ヘキサン(ナカライテスク社製)に溶解した。チューブ5本に対して、n-オクタコサン溶液は100μl(100μg)ずつ、シクロペンテノン水溶液は各々15μl(10μg)、45μl(30μg)、90μl(60μg)、135μl(90μg)、180μl(120μg)加えた。各々のチューブを減圧下乾燥し、トリメチルシリル化溶液[N,O-ビス(トリメチルシリル)−アセトアミド(ナカライテスク社製):トリメチルクロロシラン(ジーエルサイエンス社製):ピリジン(ピアス社製)=4:1:1の混合液)]を100μlずつ加え、完全に溶解した。
2.1m×3.2mmφのガラスカラム(島津社製)に2%OV17 Uniport HP 60/80 mesh(ジーエルサイエンス社製)を充てんしたものを、ガスクロマトグラフ装置GC-7AG(島津社製)によりキャリアーガスN2で20ml/分の流速で15時間空焼きを行った。
このガスクロマトグラフシステムで、上記各トリメチルシリル化試料を1μlアプライした。分析条件は以下に示すとおりである。
キャリアーガス:N2
流速 :50ml/分
注入口温度 :280℃
初期温度 :80℃、4分
昇温 :8℃/分
最終温度 :270℃
検出 :水素炎イオン化検出装置
この結果、シクロペンテノンは保持時間およそ9.7分のところに、n-オクタコサンは保持時間およそ26.7分のところに単一でピークが検出できた。このときに得られた各ピークの面積は以下に示すとおりである。
すなわちシクロペンテノン量が未知の試料に対してn-オクタコサンを100μg加え減圧下乾燥後、トリメチルシリル化し、上記方法に従いガスクロマトグラフで分析して、シクロペンテノンのピーク面積/n-オクタコサンのピーク面積の値(y)を求めることにより、シクロペンテノン量が定量できる。
(2)1%(重量%)の濃度に調製したグルクロン酸(ナカライテスク社製)水溶液を120℃、4時間オートクレーブで加圧加熱した。これを100μl(グルクロン酸1mg相当)チューブにとりn-オクタコサンを100μg加え減圧下乾燥後、トリメチルシリル化した。これを上記方法に従い、ガスクロマトグラフで分析したところ図7に示すようなパターンが得られた。すなわち図7はグルクロン酸加熱処理物のガスクロマトグラフ結果を示す図であり、縦軸は検出強度を示し、横軸は保持時間(分)を示す。シクロペンテノンは保持時間およそ10.2分〔ピーク(a)〕のところに、n-オクタコサンは保持時間およそ27.8分〔ピーク(b)〕のところで単一でピークが検出される。
この結果、以下に示すとおりとなり、グルクロン酸の加熱処理によるシクロペンテノンへの変換率はモル換算で約15%となった。
シクロペンテノンのピーク面積(a):203,794
n-オクタコサンのピーク面積(b) :305,444
(a)/(b) :0.6672
シクロペンテノン量(μg) :88.4
実施例 6
市販のグルクロノラクトン(メルク社製code No.100282)を1%になるように水に溶解し、121℃で0.5時間、1時間、2時間、4時間、又は16時間加熱した。本加熱処理液を上記実施例5−(1)記載の方法に従ってトリメチルシリル化し、ガスクロマトグラフで分析した。
分析結果を以下に示す。
また実施例3−(1)に記載の方法に準じ、上記グルクロノラクトンの16時間加熱処理液より精製シクロペンテノンを得た。
実施例 7
市販のキャベツ100gに100mlの水を加え、ミキサーで粉砕した。5mlのプロテイナーゼK(20mg/ml;宝酒造社製、9033)を添加して50℃で1時間反応させた後、ろ過してその残渣を1.5リットルの水で洗浄した。洗浄した残渣に100mlの水を加えてpH6.5の懸濁液を得、これを121℃で2時間加熱した後、ろ過することによりpH4.7のろ液を得た。このろ液を減圧下14mlまで濃縮し、36mlのメタノールを添加して混合後、4000×gで10分間遠心して上清を得た。この上清を減圧下濃縮して3.5mlの濃縮液を得た。
この濃縮液100μl中に含まれるシクロペンテノン量を実施例5−(1)に記載の方法で測定したところ、24.9μgであった。ミキサーによる粉砕後、プロテイナーゼK処理と水洗による除タンパク処理を行わず、そのまま加熱した場合にはシクロペンテノンの生成が認められなかった。このことから、100gのキャベツをプロテイナーゼK処理と水洗による除タンパク後に、上記条件で加熱することにより0.87mgのシクロペンテノンが生成していることが明らかになった。
次に実施例3−(1)の方法に準じてこの濃縮液からシクロペンテノンを精製、単離した。
実施例 8
(1)ペクチン(リンゴ製、和光純薬社製)の1%水溶液8mlに1.36単位、0.68単位、又は0.14単位のペクチナーゼ(8.1単位/mg蛋白、シグマ社性、P9932)を加え、25℃に1晩放置した。これらの▲1▼酵素反応物、及び▲2▼酵素反応物の遠心上清、にHClを加えてpH3に調製した後121℃で4時間加熱して加熱処理物を得た。加熱処理物中に含まれるシクロペンテノンを実施例5−(1)記載の方法に従ってトリメチルシリル化し、加熱処理物中のシクロペンテノン量をガスクロマトグラフで測定した。
その結果を表1に示す。
(2)1gのアルギン酸(非膨潤性;和光純薬社製)を100mlの▲1▼0.1N HCl又は▲2▼0.1M Na2CO3に懸濁又は溶解し、95℃で15時間加熱した後、▲1▼には粉末Na2CO3、▲2▼には6N HClを加えてpHを2.7に調整した。一方、▲3▼1gのアルギン酸を100mlの水に懸濁したもの(pH2.7)と▲4▼1gのアルギン酸を100mlの0.1M Na2CO3に溶解した後6N HClでpH2.7に調整したものを調整した。▲1▼〜▲4▼を121℃で4時間加熱したもの(試料▲1▼〜試料▲4▼と呼ぶ)に含まれるシクロペンテノン量を以下の方法で測定した。
試料▲1▼〜試料▲4▼に含まれる不溶物を遠心分離によって除き、上清10μlをTSK gel G2000PWカラム(7.5mm×30cm;東ソー社製)を用いたゲルろ過HPLCによって分析した。なお、移動相は水、流速は1ml/分、カラム温度は40℃とし、215nmにおける吸光度で検出した。標準物質としては実施例3−(1)記載の精製シクロペンテノンを用い、内部標準物質として2−シクロペンテン−1−オン(アルドリッチ社)を用いた。
その結果、試料▲1▼には414μg/ml、試料▲2▼には279μg/ml、試料▲3▼には289μg/ml、試料▲4▼には296μg/mlのシクロペンテノンが含まれていることが明らかになった。すなわち、121℃で4時間加熱した試料▲3▼に比べて、121℃、4時間加熱の前に0.1N HCl中で95℃、15時間加熱した場合に1.43倍のシクロペンテノンが生成した。
各試料より、実施例3−(1)記載の方法で精製シクロペンテノンを調製した。
(3)市販の50gのおからに120mlの水を加えて室温で2時間振とうし、遠心分離とろ過によって固形物を除いた。50mlのろ液に1N H2SO4を加えてpH2に調整し、121℃で4時間加熱して試料▲5▼を得た。
100gのおからに300mlのアセトンを加えて攪拌し、ろ過によって55gの残渣を得た。27.5gの残渣を1リットルの水で洗浄し、水を加えて150mlとした後、1N H2SO4を加えてpH2に調整し、121℃で4時間加熱して試料▲6▼を得た。
アセトン処理の残渣の残り27.5gを200mlの水に懸濁し、プロテイナーゼK(20mg/ml、宝酒造社製)500μlを加えて50℃で2時間反応させた。反応物を1リットルの水で洗浄し、水を加えて180mlとした後、1N H2SO4を加えてpH2に調整し、121℃で4時間加熱て試料▲7▼を得た。
試料▲5▼〜▲7▼をろ過して得たろ液を減圧下約10mlまで濃縮して4倍量のアセトンを加え、遠心して上清を得た。これらの上清を更に減圧下濃縮し、試料▲5▼からは5.5ml、試料▲6▼からは3.8ml、試料▲7▼からは3.0mlの濃縮液を得た。これらの濃縮液に含まれるシクロペンテノン量を実施例8−(2)記載の方法で測定した。
その結果、いずれも50gのおからから出発して、試料▲5▼では0.54mg、試料▲6▼では2.79mg、試料▲7▼では3.98mgのシクロペンテノンが生成していた。また、試料▲5▼〜▲7▼調製途中の121℃、4時間加熱前の試料のいずれにもシクロペンテノンは含まれなかった。従ってプロテイナーゼ処理によって除タンパクすることにより、シクロペンテノン生成量は約40%増加した。
試料▲5▼〜▲7▼の各試料より、実施例3−(1)記載の方法で精製シクロペンテノンを調製した。
50gのおからを200mlの水に懸濁し、6N HClでpH1.5に調整した後、50℃で3時間加熱した。NaOHでpH5.0に調整し、ろ過によってろ液を得た。このろ液を6N HClでpH2.05に調整し、121℃で4時間加熱した。これを試料▲8▼とする。試料▲8▼に含まれるシクロペンテノン量を実施例8−(2)記載の方法で測定した。
その結果、50gのおからから5.0mgのシクロペンテノンが生成していた。なお121℃、4時間加熱前の濾液にはシクロペンテノンは含まれなかった。
(4)市販のアップルファイバー(乾燥粉末、ニチロ社製)に以下の処理を行った。
アップルファイバー0.5gを50mlの水に懸濁し、121℃で4時間加熱した。遠心によって37mlの上清を得た。これを試料▲9▼とする。
アップルファイバー5gを50mlの水に懸濁し、121℃で4時間加熱した。遠心によって20mlの上清を得た。これを試料(10)とする。
アップルファイバー5gを50mlの水に懸濁して室温で2時間振とうし、その遠心上清を121℃で4時間加熱した。遠心によって25mlの上清を得た。これを試料(11)とする。
試料(11)調製時の室温での2時間振とう後の遠心によって得られた沈殿を50mlの水に懸濁し、121℃で4時間加熱した。遠心によって31mlの上清を得た。これを試料(12)とする。
試料▲9▼〜(12)に含まれるシクロペンテノン量を実施例8−(2)記載の方法で測定した。
その結果、試料▲9▼には14.8μg/ml、試料(10)には76.3μg/ml、試料(11)には54.0μg/ml、試料(12)には34.2μg/mlのシクロペンテノンが含まれていた。また、試料▲9▼の121℃、4時間加熱前の試料にはシクロペンテノンは含まれていなかった。したがって、アップルファイバー1gから、試料▲9▼の製造方法では1.09mg、試料(10)の製造方法では0.305mg、試料(11)の製造方法では0.270mg、試料(12)の製造方法では0.212mgのシクロペンテノンが生成した。
試料▲9▼〜(12)の各試料より、実施例3−(1)記載の方法により精製シクロペンテノンを調製した。
実施例 9
市販の煎茶の葉、焙じ茶の葉、烏竜茶の葉、又は紅茶の葉2gに100mlの水を加えてミキサーで粉砕し、1N硫酸でpH3に調整し、121℃で16時間加熱した。これらの加熱処理物に含まれるシクロペンテノン量を実施例8−(2)に記載のゲル濾過HPLC法によって測定した。この結果、煎茶からは0.19mM、焙じ茶からは0.28mM、烏竜茶からは0.34mM、紅茶からは0.12mMのシクロペンテノンが生成していた。これらの原料はその製造過程において乾式加熱処理を受けており、シクロペンテノンが効率よく生成した。
実施例 10
ペクチン(和光純薬工業株式会社製code 167-00542)、アルギン酸(非膨潤性:和光純薬工業株式会社製code 011-13341)、D-α-ガラクツロン酸(ナカライテスク社製code 165-18)、及びD-グルクロン酸(ナカライテスク社製code 169-28)をそれぞれ1%になるように蒸留水に溶解し、各溶液を調製した。更に、ペクチンは、1N 酢酸水溶液に溶解した溶液も調製した。
ペクチンの1%水溶液のpHはpH3.4であった。ペクチンの1%酢酸溶液のpHはpH2.6であった。ガラクツロン酸水溶液の加熱前のpHはpH2.5であった。グルクロン酸水溶液の加熱前のpHはpH2.4であった。アルギン酸水溶液の加熱前のpHはpH3.3であった。
次に、これらの1%溶解液を121℃で2、4、16時間の加熱処理を行い、各々の加熱処理物をNaOHでpH7に調整した後、0.22μmのフィルター滅菌を行い、シクロペンテノン生成量測定試料を調製した。
シクロペンテノンはシリカゲルシート60F254(メルク社製)上にスポットした後、展開溶媒(酢酸ブチル:酢酸:蒸留水=3:1:1の上層)で展開し、展開し終えた薄層シリカゲルを乾燥後、AgNO3−NH3溶液(0.1M AgNO3と5N NH3の等量混合物)を噴霧し、加熱することにより、Rf値約0.3付近のスポットとして検出される。
上記シクロペンテノン生成量測定試料の2倍、5倍、10倍、20倍、50倍、100倍希釈液を調製し、その希釈液のTLCを上記の方法で行った。なお、ペクチンの1%水溶液の2時間加熱処理物の2倍希釈物のシクロペンテノンの生成量を1単位とし、各加熱処理物のシクロペンテノン生成量を測定した。その結果を表2に示す。
各試料において加熱時間の増加とともにシクロペンテノンの生成量の増加が認められ、ガラクツロン酸水溶液の加熱処理物においては30分後に1単位、1時間後には5単位、グルクロン酸水溶液の加熱処理物においては3分後に5単位、1時間後に10単位のシクロペンテノンの生成が認められた。
(1)市販のグルクロノラクトン(メルク社製code No.100282)を1%になるように水に溶解し、121℃で0.5時間、1時間、2時間、4時間、又は16時間加熱した。本加熱処理液を実施例5−(1)記載の方法に従ってトリメチルシリル化し、生成するシクロペンテノン量をガスクロマトグラフで分析した。
測定結果を表3に示す。
まだ上記実施例3−(1)記載の方法に準じ、上記グルクロノラクトンの16時間加熱処理液よりシクロペンテノンを精製、単離した。
(2)上記グルクロノラクトンを1%、2%、5%、10%、又は20%になるように水に溶解し、121℃で4時間加熱した。本加熱処理液を実施例5−(1)記載の方法に従ってトリメチルシリル化し、生成するシクロペンテノン量をガスクロマトグラフで測定した。
測定結果を表4に示す。
(3)上記グルクロノラクトンの1%水溶液のpHをHCl又はNaOHでpH1、2、3、又は4.5に調整し、121℃で4時間加熱した。本加熱処理液を実施例5−(1)記載の方法に従ってトリメチルシリル化し、生成するシクロペンテノン量をガスクロマトグラフで測定した。
測定結果を表5に示す。
(4)1%ガラクツロン酸水溶液のpHをHCl又はNaOHで1、2、3、4、5、6、又は7に調整し、121℃で4時間加熱した。本加熱処理液を実施例5−(1)記載の方法に従ってトリメチルシリル化し、生成するシクロペンテノン量をガスクロマトグラフで測定した。
測定結果を表6に示す。
(5)アルギン酸(非膨潤性)の1%水懸濁液のpHをHCl又はNaOHで1、2、3、又は4に調整した。また、アルギン酸(非膨潤性)を1%濃度で0.1M酢酸緩衝液に溶解してpH5に調整したもの、0.1Mリン酸緩衝液に溶解してpH6又は7に調整したものを作製した。これらを121℃で4時間加熱した。本加熱液に含まれるシクロペンテノン量を以下に示す条件のゲル濾過HPLC法によって測定した。
カラム:TSK gel α−2500(7.8×800mm:東ソー社製)
カラム温度:40℃
移動相:0.01%トリフルオロ酢酸水溶液
流速:1ml/分
検出:215nmにおける吸光度
実施例3−(1)で調製した精製シクロペンテノンを標準物質として、10.0分付近に溶出されてくるシクロペンテノンのピーク面積を測定することによりシクロペンテノン濃度を決定した。
その結果を表7に示す。
実施例 12
ポモシンペクチンタイプLM−13CG(ハーキュリーズ社製)、アルギニックアシッドHFD(大日本製薬社製)、D−グルクロン酸(ナカライテスク社製)及びグルクロノラクトン(メルク社製)を1%となるように水に溶解又は懸濁し、30℃、60℃、95℃、121℃又は132℃で16時間加熱した。この加熱処理物のシクロペンテノンを実施例5−(1)記載の方法に従ってトリメチルシリル化し、加熱処理物中のシクロペンテノン生成量をガスクロマトグラフで測定した。その結果を表8に示す。
(1)コンドロイチン硫酸A(生化学工業社製)、コンドロイチン(ナトリウム塩、生化学工業社製)、デルマタン硫酸(ナトリウム塩、セルビオ社製)、ヘパリン(ナトリウム塩、和光純薬社製)、又はヒアルロン酸(生化学工業社製)を1%になるように水を溶解し、1N HClでpH3に調整した。これらを121℃で4時間又は16時間加熱することによって加熱処理液を調製した。
(2)実施例13−(1)で調製したコンドロイチン硫酸A、コンドロイチン、デルマタン硫酸、またはヘパリンの各加熱処理液100μlをn−オクタコサン(ナカライテスク社製)のn-ヘキサン(ナカライテスク社製)溶液(1mg/ml)100μlと混合し、減圧下乾燥した。これに100μlの上記トリメチルシリル化溶液を加えて完全に溶解し、実施例5−(1)記載の方法で加熱処理物中のシクロペンテノン量を分析した。
その結果を表9に示す。
実施例 14
デルマタン硫酸1gを100mlの水に溶解して1N HClでpH3に調整し、121℃で16時間加熱し加熱処理液を調製した。次に実施例3−(1)の方法に準じて加熱処理物よりシクロペンテノンの精製を行い、30mgの精製シクロペンテノンを得た。
実施例 15
(1)実施例3−(1)記載の精製、単離シクロペンテノンを水、10mMトリス(Tris)−HCl(pH7)、又は10mMトリス(pH10)に25mMになるように溶解し、室温又は4℃で放置した後、実施例13−(3)記載のTLC法で分析した。その結果、水又は10mMトリス−HCl(pH7)に溶解した場合、室温放置、4℃放置ともに1ヵ月経過後において若干の分解物が見られたが大部分は未分解であった。10mMトリス(pH10)に溶解した場合は室温で速やかに分解し、溶解直後にTLC法で分析したところシクロペンテノンのスポットは見られなかった。
水に溶解したシクロペンテノンを121℃で30分間加熱し、TLC法で分析したところ、若干の分解物は見られたが大部分のシクロペンテノンは未分解であった。
(2)1%D−グルクロン酸水溶液を121℃で4時間加熱し、pH未調整の試料とNaOHでpH6.6に調整した試料を調製した。各々を1mlずつ分注し、−20℃、4℃、37℃で保存した後、シクロペンテノンを実施例5−(1)記載の方法に従ってトリメチルシリル化し、試料中のシクロペンテノン量をガスクロマトグラフで測定した。
その結果、25日間保存後では、37℃で保存した場合にシクロペンテノン量がやや減少していたが、4℃、−20℃ではほぼ安定であった。その結果を図8に示す。すなわち図8は保存時間とシクロペンテノン量の関係を示す図であり、横軸は保存時間(日)、縦軸はシクロペンテノン濃度(mg/ml)を示す。図8中において白四角印(□)はpH未調整で−20℃保存、黒四角印(■)はpH6.66で−20℃保存、白丸印(○)はpH未調整で4℃保存、黒丸印(●)はpH6.66で4℃保存、白三角印(△)はpH未調整で37℃保存、黒三角印(▲)はpH6.66で37℃保存を示す。
実施例 16
(1)実施例3−(1)記載の精製、単離シクロペンテノン113.9mgをエタノール2.85mlに溶かした。このエタノール溶液にヘキサン/エタノール(94/6)3.85mlを更に加え、17mg/mlのシクロペンテノン溶液を調製した。この液を0.5μmのフィルターでろ過し、光学分割HPLC試料溶液とした。
この試料溶液を以下の条件で光学分割HPLCを行い、前ピークのシクロペンテノン及び後ピークのシクロペンテノンのフラクションをそれぞれ集め、減圧乾固し、前ピークのシクロペンテノン43.2mg、後ピークのシクロペンテノン43.0mgをそれぞれ得た。
光学分割HPLC条件
カラム:キラールパックAS(ダイセル化学工業)2.0cm×25.0cm
カラム温度:40℃
移動相:ヘキサン/エタノール(94/6)
流速:14.0ml/min
検出:UV 210nm
試料注入量:150μl(2.55mg)
得られた前ピークのシクロペンテノン及び後ピークのシクロペンテノンは両者共に約1%のエナンチオマーを含有していたため再度上記の条件で光学分割した。その結果、前ピークのシクロペンテノン30.0mgから19.7mgのエナンチオマーを含有しないシクロペンテノンを、後ピークのシクロペンテノン37.4mgから27.7mgのエナンチオマーを含有しない後ピークのシクロペンテノンをそれぞれ得た。得られた前ピークのシクロペンテノン及び後ピークのシクロペンテノンの旋光度はそれぞれ[α]D 20−105°(c 0.30、エタノール)及び[α]D 20+104°(c 0.53、エタノール)であり、前ピーク物質が(−)−トランス−4,5−ジヒドロキシ−2−シクロペンテン−1−オン[以下(−)体シクロペンテノンと称す]、後ピーク物質が(+)−トランス−4,5−ジヒドロキシ−2−シクロペンテノン−1−オン[以下(+)体シクロペンテノンと称す]であった。なお旋光度は前記のDIP−370型旋光計(日本分光社製)を用いて測定した。
(−)体シクロペンテノン及び(+)体シクロペンテノンの光学分割HPLCの溶出曲線を図9、図10に示す。すなわち図9は(−)体シクロペンテノンの溶出曲線であり、図9中、縦軸は吸光度、横軸は溶出時間(分)を示す。また、図10は(+)体シクロペンテノンの溶出曲線であり、図10中、縦軸は吸光度、横軸は溶出時間(分)を示す。
次に(−)体シクロペンテノン及び(+)体シクロペンテノンのそれぞれの質量分析、核磁気共鳴法(NMR)による構造解析、UV吸収スペクトルの測定、赤外吸収スペクトルの測定を実施例3−(2)に記載の方法に準じ行った。その結果、両光学活性体は光学分割前のシクロペンテノンと同一の結果を示した。
図11に(−)体シクロペンテノンの1H−NMRスペクトルを示す。図11中その縦軸はシグナルの強度、横軸は化学シフト値(ppm)を示す。
図12に(+)体シクロペンテノンの1H−NMRスペクトルを示す。図12中その縦軸はシグナルの強度、横軸は化学シフト値(ppm)を示す。
(2)前出カーボハイドレートリサーチ記載の方法により、トランス−シクロペンテノンを合成した。また前出ヘルベチカ キミカ アクタ記載の方法に従いシス−シクロペンテノンを合成した。また光学分割法により各々の光学活性体を調製した。
調製した(+)−トランス−シクロペンテノン、(−)−トランス−シクロペンテノン、(+)−シス−シクロペンテノン、(−)−シス−シクロペンテノンにつき各実施例に記載の方法で細胞増殖抑制活性、アポトーシス誘発活性、がん細胞分化誘導活性、抗菌活性を測定したところ、(+)−トランス−シクロペンテノン、(−)−トランス−シクロペンテノン、(+)−シス−シクロペンテノン、(−)−シス−シクロペンテノンは各々細胞増殖抑制活性、アポトーシス誘発活性、がん細胞分化誘導活性、抗菌活性を示した。
実施例 17
実施例16で得た(−)体シクロペンテノンと(+)体シクロペンテノンのそれぞれの1mg/mlエタノール溶液を75%エタノール水溶液で2倍、4倍、8倍、16倍、32倍、64倍、128倍、256倍、512倍、1024倍及び2048倍に希釈し、96穴マイクロタイタープレートの各ウェルに5μlずつ入れて風乾した。5000個のヒト前骨髄性白血病細胞HL−60(ATCC CCL−240)細胞を含む10%牛胎児血清含有RPMI1640培地100μlを各ウェルに加え、5%炭酸ガス存在下37℃で48時間培養した。細胞の形態を光学顕微鏡で観察した後、5mg/mlの3−(4,5−ジメチルチアゾール−2−イル)−2,5−ジフェニルテトラゾリウムブロミド(MTT;シグマ社製)リン酸緩衝食塩水溶液10μlを加えて更に4時間培養を続けた後、顕微鏡で細胞の生育状態を観察した。また、0.04N HCl含有2−プロパノール100μlを加えて良く攪拌し、590nmにおける吸光度を測定してこれを細胞増殖度とした。
その結果、シクロペンテノンの光学活性体の各128倍希釈液添加区分(終濃度0.39μg/ml)においても細胞増殖抑制活性が見られ、またアポトーシス誘発作用が見られた。
実施例 18
(1)1×105/mlのHL−60細胞を含む10%牛胎児血清含有RPMI1640培地に10、1、0.1、又は0.01μg/mlのシクロペンテノンを添加し、5%炭酸ガス存在下37℃で3日間及び6日間培養した後、生細胞数を計数した。その結果、培養6日目においてシクロペンテノンを添加しない対照に比べて10μg/ml添加区分では生細胞は見られず、1μg/ml添加区分では約90%、0.1μg/ml添加区分では約55%、0.01μg/ml添加区分では約40%の細胞増殖阻害が見られた。
その結果を図13に示す。すなわち図13はHL−60細胞の培養液にシクロペンテノンを様々な濃度で添加したときの培養時間と培養液中の生細胞数の関係を示す図であり、横軸は培養時間(日)、縦軸は培養液中の生細胞数(×104/ml)を示す。図中において白四角印(□)は試料無添加(対照)、白菱形印(◇)は10μg/ml、白丸印(○)は1μg/ml、白三角印(△)は0.1μg/ml、黒四角印(■)は0.01μg/mlのシクロペンテノン添加をそれぞれ示す。
(2)HL−60細胞に10-4μg/mlのシクロペンテノンを添加して実施例15−(1)と同様に3日間培養した。細胞の一部をとってスライドガラスに塗抹し、「組織培養の技術」(日本組織培養学会編、朝倉書店、1982年)第191頁に記載のライト−ギムザ染色を行い、光学顕微鏡で観察した。その結果、シクロペンテノン無添加の対照では分化した細胞が10%前後であったのに対してシクロペンテノン添加区分では50%以上の細胞が単球又はマクロファージ様細胞に分化していた。
(3)HL−60細胞に0.5μg/ml又は0.005μg/mlのシクロペンテノンを添加して実施例18−(1)と同様に3日間又は6日間培養した。細胞の一部をとってスライドガラスに塗抹後、ライト−ギムザ染色を行い、光学顕微鏡で観察した。その結果、シクロペンテノン無添加の対照では分化して形態の変化した細胞が約10%であったのに対して、シクロペンテノン0.005μg/ml添加区分では25%以上の細胞が分化して成熟骨髄細胞になっていた。その結果を図14に示す。すなわち図14は培養時間と、培養細胞中において成熟骨髄細胞の占める比率の関係を示す図であり、横軸は培養時間(日)、縦軸は培養細胞中において成熟骨髄細胞の占める比率(%)を示す。図14において白四角印(□)は試料無添加(対照)、白丸印(○)は0.5μg/mlシクロペンテノン添加、白三角印(△)は0.005μg/mlシクロペンテノン添加をそれぞれ示す。
実施例 19
(1)実施例3−(1)記載の精製、単離シクロペンテノンの抗菌作用を以下の菌株で測定した。測定に使用した菌株は被検菌(1):サルモネラ エンテリティディス(Salmonella enteritidis)(食中毒事例株)、被検菌(2):サルモネラ ティフィムリウム(Salmonella typhimurium(食中毒事例株)、被検菌(3):スタフィロコッカス アウレウス(Staphylococcus aureus)FRI 722(A型エンテロトキシン産生株)、被検菌(4):Staphylococcus aureus(メチシリン耐性株)、被検菌(5):バチルス セレウス(Bacillus cereus)(嘔吐型食中毒事例株)及び被検菌(6):Bacillus cereus(下痢型食中毒事例株)である。これらの菌株はいずれも女子栄養大学・衛生学教室保存菌株である。
抗菌作用の測定は、各被検菌に対する増殖抑制効果を指標に行った。すなわち、一定濃度のシクロペンテノンを含む菌体培地に各被検菌を一定量添加した試験菌液を調製し、16hr、48hr培養後の生菌数を比較することにより行った。
まず、感受性測定ブイヨン(日水製)に、一定濃度のシクロペンテノンを加え、2n連続希釈を行った。それに感受性測定ブイヨンで37℃16時間前培養を行った菌液を106/mlになるように接種し、37℃で培養を行った。各菌株の各培養時間ごとの生菌数の測定は、培養液を適宜希釈し表面塗沫培養法により行った。なお、それぞれの菌株の菌数測定に当ってはSalmonellaはDHL(栄研製)、S.aureusはBaird Parker agar(BBL製)、B.cereusはNGKG agar(日水製)を使用した。なお、B.cereusのみは32℃で培養を行った。各培養時間での測定した菌株は、CFU(colony forming units)/mlとして常用対数値で表示した。下記表10に培養16時間後の被検菌株、表11に培養48時間後の被検菌数を表す。シクロペンテノンはいずれの被検菌にも抗菌作用を示した。なお以下、表10〜13において表中の−記号は、被検菌の生育が見られなかったことを意味する。
被検菌(7):エシェリヒア コリ(Escherichia coli)(S−O157:H7、VT1、2産生菌)、被検菌(8):Escherichia coli(Y.3−O157:H7、VT1、2産生株)、被検菌(9):Escherichia coli(Y.1−O157:H7、VT1産生株)、被検菌(10):Escherichia coli(S−O26:HNM、VT1産生株)、被検菌(11):Escherichia coli(S−O111;HNM、VT1、2産生株)及び被検菌(12):Escherichia coli(食品分離株)である。
使用した菌株はいずれも女子栄養大学・衛生学教室保存菌株である。
抗菌作用の測定は、実施例19−(1)と同様に行った。
まず、感受性測定ブイヨン(日水製)に一定濃度のシクロペンテノンを加え、2n連続希釈を行った。それに感受性測定ブイヨンで37℃16時間前培養を行った菌液を106/mlになるように接種し、37℃で培養を行った。なお、菌株の各培養時間ごとの菌株の測定は、培養液を適宜希釈し表面塗抹培養法により行った。なお、菌株の菌数測定にあたってはDHL(栄研製)を使用した。測定した各培養時間での菌数は上記と同様に対数値で表示した。下記表12、表13にその結果を示す。シクロペンテノンはいずれの被検菌にも抗菌作用を示した。
被検菌をL-ブロス(1%トリプトン、0.5%酵母エキス、5%NaCl pH 7.0)で、一晩種培養した。5mlのL-ブロスに25〜200μg/mlのシクロペンテノンを添加した培地及び何も添加していない培地に5μlの種培養液を植菌し、37℃で振とう培養し生育を測定した。培養開始時と8時間後に、富士デジタル濁度計(販売元 富士工業株式会社、製造元 秋山電機製作所)を用い、調製目盛を82.3の条件で培養物の濁度を測定し、8時間後の濁度の値から培養開始時の濁度の値を引いた値(生育濁度)で被検菌の生育を測定した。なお被検菌(18)についてはL−ブロスの代りにブレイン ハート インヒュージョン培地を使用した。
結果を表14に示す。なお表14中の−は未検討を示す。
(4)シクロペンテノンの火落菌に対する抗菌活性を以下の方法で行った。被検菌を10%エタノールを含有するSI培地(日本醸造協会)で5日間静置培養し種菌を得た。10%エタノールを含有するSI培地に最終濃度が0、25、50、75、100μg/mlになるようにシクロペンテノンを添加した培地100mlに被検菌の種菌を0.1%植菌し、5日間静置培養し、濁度を測定した。濁度の測定は、吸光度計を用い、OD600の値を測定した。この値から、被検菌を植菌していない培地のOD600の値を引いた値(生育濁度)で被検菌の生育を測定した。
被検菌は真性火落菌としてラクトバチルス フラクチボランス(Lactobacillus fructivorans)IFO13118(被検菌A)、Lactobacillus fructivorans JCM1198(被検菌B)、ラクトバチルス ホモヒオチイ(Lactobacillus homohiochii)IFO13120(被検菌C)、火落性乳酸菌としてラクトバチルス ラムノサス(Lactobacillus rhamnosus)IFO3532(被検菌D)を用いた。その結果を表15に記載する。
なおシクロペンテノンはサッカロミセス セレビシェ(Saccharomyces cerevisiae)ATCC 9763、カンジダ アルビカンス(Candida albicans)TIMM 0136、アスペルギルス フミガタス(Aspergillus fumigatus)TIMM 1776等の真菌にも高濃度下で抗菌活性を示した。
実施例 20
(1)800、400、200、100、50、25、12.5、6.25、3.13、又は1.56μg/mlのシクロペンテノンを含むトリプトソーヤブイヨン培地(日水社製)にVibrio parahaemolyticus 4387-61又はVibrio parahaemolyticus T4144-1(いずれも国立衛生試験所保存株)を106細胞/mlになるように接種して37℃で24時間静置培養した。その結果、どちらの菌株に対しても12.5μg/ml以上のシクロペンテノン添加区分で菌の生育が見られなかった。
菌の生育が見られなかった区分の培養液50μlを20mlのシクロペンテノンを含まないトリプトソーヤブイヨン寒天培地に塗布し、37℃で24時間培養した。その結果、どちらの菌株でも50μg/ml以上のシクロペンテノンを添加した区分を塗布した寒天培地で菌の生育が見られなかった。
以上より、シクロペンテノンは12.5μg/mlでVibrio parahaemolyticus 4387-61及びVibrio parahaemolyticus T4144-1に対して抗菌作用を、50μg/mlで両菌株に対して殺菌作用を示した。
(2)Campylobacter jejuni A3309(国立衛生試験所保存株)を2%仔ウシ血清(大日本製薬社製)を含む脳心臓滲出液(ディフコ社製)培地に接種し、37℃で16時間、振とう前培養した。この培養液50μlを800、400、200、100、50、25、12.5、6.25、3.13、又は1.56μg/mlのシクロペンテノンを含む20mlの0.5%NaCl含有ミューラー・ヒントン平板培地(BBL社製)に塗布し、37℃で48時間静置培養した。
その結果、12.5μg/ml以上のシクロペンテノンを添加した平板培地上には菌の生育が見られなかった。
以上シクロペンテノンはキャンピロバクターに対して抗菌作用を示した。
(3)被検菌▲1▼Legionella pneumophila(ヒト器官支洗浄液分離菌)、被検菌▲2▼Legionella pneumophila(温泉浴槽水分離菌)(いずれも女子栄養大学・衛生学教室保存菌株)に対するシクロペンテノンの抗菌作用の測定を、各被検菌に対する増殖抑制効果を指標に行った。すなわち、一定濃度のシクロペンテノンを含む液体培地に各被検菌を一定量添加した試験菌液を調製し、16hr、48hr、72hr、96hr培養後の生菌数の検討を行った。
まず、BCYE α ブロス(オキソイド社製)に、一定濃度のシクロペンテノンを加え、2n連続希釈を行った。それにBCYE α ブロスで37℃16時間前培養を行った菌液を106/mlになるように接種し、37℃で培養を行った。各菌株の各培養時間ごとの生菌数の測定は、培養液を適宜希釈し、BCYE α 寒天(オキソイド社製)への表面塗沫培養法により行った。
各培養時間での測定した菌数は、CFU(colony forming units)/mlとして常用対数値で表示した。下記表16にその結果を示す。シクロペンテノンはいずれの被検菌にも抗菌作用を示した。なお表中の−記号は、被検菌の生育が見られなかったことを意味する。
実施例 21
シクロペンテノンの固形がんに対する制がん作用
実施例3−(1)記載のシクロペンテノンを生理的食塩水で所定の濃度に希釈し、以下の試験を行った。
(1)MethA細胞(2×106細胞/マウス)を8週齢の雌性BALB/cマウス(体重約20g)の腹部に皮下注射した。その後、引き続いて同じ箇所に5日間連続してシクロペンテノン(5mg/kg/日)を皮下注射した。一方コントロール群には生理的食塩水を同様に皮下注射した。2週間後にマウス腹部に形成されたがん組織を摘出して、その重量を測定した。結果を表17に示す。
すなわち、コントロール群では平均がん重量は1.41gであったのに対し、シクロペンテノン(5mg/kg/日)投与群では0.0gでがん組織の形成は全く認められず、抑制率は100%であった。
シクロペンテノン投与群には、シクロペンテノンとしての摂取量が約80mg/kg/日となるよう水道水に希釈し、給水瓶にて自由摂取させた。コントロール群には同様に水道水を与えた。餌は両群とも実験期間中、自由摂食とした。
Sarcoma−180皮下移植後40日目の生存個体数はコントロール群で8例中2例、シクロペンテノン投与群で7例で、シクロペンテノン投与による顕著な延命効果が認められた。
(3)マウス白血病P−388(1.1×106cells/マウス)を7週齢の雌性DBA/2マウス(体重約20g)に腹腔内注射した。その後、5日間連続してシクロペンテノン(10mg/kg/日)を腹腔内注射した。一方コントロール群には生理的食塩水を同様に腹腔内注射した。それぞれ8匹ずつの2群において、マウスの生存数、平均生存日数、延命率を算定した。結果を図15に示した。すなわち、コントロール群では平均生存日数が10.3日であったのに対し、シクロペンテノン投与群では31.4日で、その延命率は306.1%を示し、有意な延命効果が認められた。図15はシクロペンテノンの制がん効果を示す図であり、縦軸は生存数、横軸は生存日数を示す。図15中実線がシクロペンテノン投与群、破線は対照群を示す。
同様に、5週齢の雌性ICR系マウス(体重約25g)16匹を用い、Sarcoma−180(5.5×106cells/マウス)を腹腔内注射し、コントロール群8匹及びシクロペンテノン投与群8匹を設定した。
また7週齢の雌性CDF1マウス(体重約20)16匹を用い、IMC(2.0×106cells/マウス)を腹腔内注射し、コントロール群8匹及びシクロペンテノン投与群8匹を設定した。
更に5週齢の雌性DDYマウス(体重約25g)16匹を用い、EAC(1.2×106cells/マウス)を腹腔内注射し、コントロール群8匹及びシクロペンテノン投与群8匹を設定した。
その結果、Sarcoma−180、IMC、EACの各コントロール群の平均生存日数はそれぞれ22.6日、10.8日及び15.8日であったのに対し、シクロペンテノン投与群ではそれぞれ33.4日、20.3日及び40.3日であった。各々の延命率は148%、188%及び255%であり、シクロペンテノン投与によりいずれも顕著な延命効果が見られた。
実施例 22
注射剤
(1)生理食塩液(日本薬局法収載品)にシクロペンテノンを1%濃度で加え注射剤を作製した。
(2)生理食塩水(前記と同じ)にシクロペンテノン及びグリシルリチン酸をそれぞれ0.5%及び0.1%濃度で加え、注射剤を作製した。
実施例 23
錠剤
(1)シクロペンテノン100mgと微結晶性セルロースの適量とを含有する錠剤を調製し、糖衣を施し、錠剤を作製した。
(2)シクロペンテノン0.1mg、グリシルリチン酸ジカリウム10mg及び微結晶セルロースの適量を含有する錠剤を調製し、糖衣を施し、錠剤を作製した。
実施例 24
軟膏
下記組成で軟膏を作成した。
シクロペンテノン 1g
吸水軟膏(日本薬局方収載) 99g
シクロペンテノンをまず少量の吸水軟膏と十分に練り合せ、次いで残った吸水軟膏を徐々に加えて均一になるまで練り合せて軟膏を作製した。
この軟膏は1日4〜5回患部に塗布される。
実施例 25
化粧料
下記組成で抗菌性化粧料としてローションを作製した。
エタノール 10 部
グリセリン 1 部
クエン酸 0.3部
パラオキシ安息香酸メチル 0.2部
シクロペンテノン 0.1部
香料 微量
精製水 全量を100部とする量
実施例 26
浴用剤
下記組成で抗菌性浴用剤を作製した。
無水ボウ硝 20部
炭酸水素ナトリウム 40部
コハク酸 10部
シクロペンテノン 30部
色素 適量
香料 適量
剤形 錠剤
実施例 27
歯磨剤
下記組成で歯磨剤を調製した。
炭酸カルシウム 50.00%
グリセリン 20.00%
カラゲナン 0.50%
カルボキシメチルセルロース 1.00%
ラウリルジエタノールアマイド 1.00%
ショ糖モノラウレート 2.00%
香料 1.00%
サッカリン 0.10%
シクロペンテノン 0.10%
水 残
計 100.00%
実施例 28
下記組成で錠菓を調製した。
砂糖 74.9%
乳糖 20.0%
ショ糖モノラウレート 0.2%
香料 0.5%
精製水 4.3%
シクロペンテノン 0.001%
実施例 29
下記方法で抗菌性飲料を作製した。
(1)ペクチン(ポモシンペクチンLM−13CG:ハーキュリーズ社製)5kgを水道水100リットルに添加し、液温28℃から液温120℃となるまで水蒸気吹込みにより35分間昇温させ、次いで攪拌下で120℃、5時間保温し、次いで冷却し、冷却物135リットルを調製した。次いで冷却物にろ過助剤として、セライト#545(セライト社製)1.35kg、及びシリカ#600−S(中央シリカ社製)1.35kgを添加し、次いでセライト#545の0.1kg、及びシリカ#600−Sの0.1kgでプレコートしたコンパクトフィルター(6インチ16段ろ紙:ADVANTEC#327)でろ過を行った。得られたろ液はプレートヒーター(日阪製作所製)による連続瞬間加熱処理(98℃、60秒)を行った後冷却し、150リットルのシクロペンテノン含有のペクチン加熱処理液を調製した。
シクロペンテノン含有のペクチン加熱処理液のpHは約3.5、酸度は6.2ml、糖度は5.8Brix%であった。なおpHはpHメーターで測定し、酸度は試料10mlをpH7.0に中和するのに要する0.1N NaOH量(ml)で表示した。更に糖度はブリックス糖度計で測定した。
(2)緑茶葉10g、ビタミンC0.2g及びイオン交換水1000mlを用い、常法に従って緑茶を調製した。本発明品1は、上記シクロペンテノン含有ペクチン加熱処理液を用い、製品100ml当り固形物換算で200mg(シクロペンテノン1.6mg含有)を添加した。対照は、無添加のものとした。パネラー20名で、5段階(5良、1悪)の官能評価を行い、その結果の平均値を表18に示した。
実施例 30
下記組成で飲料を調製した。
果糖ブドウ糖液糖 5.00%
砂糖 4.00%
酸味料 1.20%
香料 0.30%
シクロペンテノン含有物 0.5 %
精製水 残
計 100.00
なおシクロペンテノン含有物としては実施例29−(1)記載のシクロペンテノン含有ペクチン加熱処理液を使用し、その固形物換算量を添加した。この飲料100ml中には4mgのシクロペンテノンが含有されている。
実施例 31
新鮮なキャベツ200gを短冊切りし、水(対照)、0.2%のシクロペンテノン水溶液に各100gずつ、5分間浸漬した。その後、軽く水切りし、合成樹脂製袋に入れ室温(20℃)で放置した。24時間後の観察において、シクロペンテノン水溶液に浸漬したものは、水(対照)に浸漬したものに比べ、新鮮度が保持された。
これは日数の増加と共に、差が明確となり、4日後の観察ではシクロペンテノン水溶液に浸漬したものは、水(対照)に浸漬したものに比べ、異臭はなく、新鮮度が保持されていた。
発明の効果
本発明により制がん作用、がん細胞増殖抑制作用、がん細胞分化誘導作用、アポトーシス誘発作用、抗菌作用等の生理活性を有する安全性の高いシクロペンテノン及びその光学活性体が提供され、かつ、該化合物を含有する生理活性機能を有する医薬品、食品及び飲料が提供される。本発明により、シクロペンテノンは天然由来の原料から簡便に、効率良く製造することが可能となり、その光学活性体の安価な提供も可能となった。
本発明により提供されるシクロペンテノン及び/又はその光学活性体は、その生理活性によって、発がん予防、がん抑制効果、抗菌効果を有する医薬品として使用することが可能となり、該医薬品は生体の恒常性の維持、特に胃腸健康保持に有用な医薬品とする。またシクロペンテノン及び/又はその光学活性体を有効成分とする防腐剤、抗菌性化粧料、抗菌性歯磨剤、抗菌性浴用剤が提供される。
本発明により、食品又は飲料中に生理活性を有するシクロペンテノン及び/又はその光学活性体の適量を含有させることが可能となった。このシクロペンテノン及びその光学活性体が有する種々の生理活性によって、本発明の食品又は飲料は発がん予防、制がん効果、抗菌効果、アポトーシス誘発作用等の生体の恒常性(ホメオスタシス)維持機能を有する健康食品又は飲料であり、本発明により、胃腸健康保持に有用な機能性物質入りの食品又は飲料が提供される。また、シクロペンテノンを添加することにより、食品又は飲料の抗菌力を簡便に増強することができ、シクロペンテノン及び/又はその光学活性体を含有する製剤は食品又は飲料の防腐剤としても極めて有用である。
また本発明によりウロン酸、ウロン酸誘導体、シクロペンテノンの生成中間体又はシクロペンテノンと反応性を有するアミン類、アミノ酸類、ペプチド類又は蛋白質の反応性の少なくとも一部が消失した及び/又は該反応性物質の少なくとも一部が除去されたウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物が提供され、該ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物を使用することにより効率よく本発明に使用するシクロペンテノン及び/又はその光学活性体を製造することができる。該ウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物としてはウロン酸及び/又はウロン酸誘導体を含有する糖化合物含有物の乾式加熱処理物がある。乾式加熱処理としては焙炒処理が簡便であり、焙炒植物、焙炒動物、焙炒微生物、例えば焙炒野菜、焙炒果実、焙炒穀物、焙炒きのこ、焙炒海藻、焙炒皮質、焙炒軟骨等は本発明のシクロペンテノン及び/又はその光学活性体の製造において極めて有用である。なお乾式加熱処理の前に被処理物の脱水処理を行うことにより効率よく乾式加熱処理物を調製することができる。
Claims (31)
- 下記(a)、(b)、(c)より選択される少なくとも1種の物を酸性〜中性の条件下で60〜350℃、数秒〜数日加熱処理することを特徴とする下記式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの製造方法。
(a)ウロン酸又はウロン酸ラクトン、
(b)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物、
(c)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- 下記(a)、(b)、(c)より選択される少なくとも1種の物を酸性〜中性の条件下で60〜350℃、数秒〜数日加熱処理し、次いで該加熱処理物より式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンを採取することを特徴とする4,5−ジヒドロキシ−2−シクロペンテン−1−オンの製造方法。
(a)ウロン酸又はウロン酸ラクトン、
(b)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物、
(c)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- ウロン酸がガラクツロン酸、グルクロン酸、グルロン酸、マンヌロン酸及び/又はイズロン酸である請求の範囲1又は2記載の4,5−ジヒドロキシ−2−シクロペンテン−1−オンの製造方法。
- 糖化合物がペクチン、ペクチン酸、アルギン酸、ヒアルロン酸、ヘパリン、ヘパリン硫酸、フコイダン、コンドロイチン硫酸、コンドロイチン、デルマタン硫酸及び/又はその分解物から選択される糖化合物である請求の範囲1又は2記載の4,5−ジヒドロキシ−2−シクロペンテン−1−オンの製造方法。
- 下記工程を包含することを特徴とする4,5−ジヒドロキシ−2−シクロペンテン−1−オンの光学活性体の製造方法。
(A):(a)、(b)、(c)より選択される少なくとも1種の物を酸性〜中性の条件下で60〜350℃、数秒〜数日加熱処理し、4,5−ジヒドロキシ−2−シクロペンテン−1−オンを生成させる工程、
(a)ウロン酸又はウロン酸ラクトン、
(b)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物、
(c)ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物、
(B):必要に応じて、得られた加熱処理物より4,5−ジヒドロキシ−2−シクロペンテン−1−オンを単離する工程、
(C):4,5−ジヒドロキシ−2−シクロペンテン−1−オンを光学分割する工程。 - ウロン酸がガラクツロン酸、グルクロン酸、グルロン酸、マンヌロン酸及び/又はイズロン酸である請求の範囲5記載の4,5−ジヒドロキシ−2−シクロペンテン−1−オンの光学活性体の製造方法。
- 糖化合物がペクチン、ペクチン酸、アルギン酸、ヒアルロン酸、ヘパリン、ヘパラン硫酸、フコイダン、コンドロイチン硫酸、コンドロイチン、デルマタン硫酸及び/又はその分解物から選択される糖化合物である請求の範囲5記載の4,5−ジヒドロキシ−2−シクロペンテン−1−オンの光学活性体の製造方法。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とする制がん剤。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とするがん細胞分化誘導剤。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とするアポトーシス誘発剤。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有することを特徴とする抗菌剤。
- 請求の範囲11記載の抗菌剤を有効成分とする防腐剤。
- 請求の範囲11記載の抗菌剤を有効成分とする歯磨剤。
- 請求の範囲11記載の抗菌剤を有効成分とする化粧料。
- 請求の範囲11記載の抗菌剤を有効成分とする浴用剤。
- 請求の範囲1記載の製造方法で得られる式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンを有効成分として含有することを特徴とする請求の範囲8〜15いずれか1項に記載の剤。
- 請求の範囲1記載の加熱処理物を有効成分として含有することを特徴とする請求の範囲8〜15いずれか1項に記載の剤。
- 請求の範囲2記載の製造方法で採取される式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンを有効成分として含有することを特徴とする請求の範囲8〜15いずれか1項に記載の剤。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を含有、希釈及び/又は添加してなることを特徴とする食品または飲料。
- 請求の範囲1記載の製造方法で得られる式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンを含有、希釈及び/又は添加してなることを特徴とする請求の範囲19記載の食品又は飲料。
- 請求の範囲1記載の加熱処理物を含有、希釈及び/又は添加してなることを特徴とする請求の範囲19記載の食品又は飲料。
- 請求の範囲2記載の製造方法で採取される式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンを含有、希釈及び/又は添加してなることを特徴とする請求の範囲19記載の食品又は飲料。
- 式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オン及び/又はその光学活性体を100重量部当り5×10-6重量部以上含有することを特徴とする請求の範囲19〜22いずれか1項に記載の食品又は飲料。
- 制がん性食品又は制がん性飲料である請求の範囲19〜23いずれか1項に記載の食品又は飲料。
- 抗菌性食品又は抗菌性飲料である請求の範囲19〜23いずれか1項に記載の食品又は飲料。
- ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物中のウロン酸、ウロン酸ラクトン、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの生成中間体、又は式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンと反応性を有するアミン類、アミノ酸類、ペプチド類又は蛋白質の反応性の少なくとも一部が消失した及び/又は該反応性物質の少なくとも一部が除去されたものであるウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- 乾式加熱処理されて、ウロン酸、ウロン酸ラクトン、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの生成中間体、又は式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンと反応性を有するアミン類、アミノ酸類、ペプチド類又は蛋白質の反応性の少なくとも一部が消失した及び/又は該反応性物質の少なくとも一部が除去されたものである請求の範囲26記載のウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- 乾式加熱処理が、ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物を60〜400℃の熱風で数秒から数日の焙炒処理を行うものである請求の範囲27記載のウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物が焙炒植物、培炒動物又は焙炒微生物から選択される請求の範囲28記載のウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- ウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物が焙炒野菜、焙炒果物、焙炒穀物、焙炒きのこ、焙炒海藻、焙炒皮質又は焙炒軟骨から選択される請求の範囲29記載のウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
- 蛋白分解酵素処理されて、ウロン酸、ウロン酸ラクトン、式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンの生成中間体、又は式【1】で表される4,5−ジヒドロキシ−2−シクロペンテン−1−オンと反応性を有するアミン類、アミノ酸類、ペプチド類又は蛋白質の反応性の少なくとも一部が消失した及び/又は該反応性物質の少なくとも一部が除去されたものである請求の範囲26記載のウロン酸及び/又はウロン酸ラクトンを含有する糖化合物含有物。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP27523196 | 1996-09-27 | ||
| JP32590096 | 1996-11-22 | ||
| JP5543497 | 1997-02-25 | ||
| JP9286697 | 1997-03-28 | ||
| JP11604597 | 1997-04-21 | ||
| PCT/JP1997/003052 WO1998013328A1 (fr) | 1996-09-27 | 1997-09-01 | Cyclopentenones, leur procede de preparation et leur utilisation |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP3790273B2 true JP3790273B2 (ja) | 2006-06-28 |
Family
ID=27523222
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP51548098A Expired - Fee Related JP3790273B2 (ja) | 1996-09-27 | 1997-09-01 | シクロペンテノン類、その製造方法及び用途 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6087401A (ja) |
| EP (1) | EP0941981A4 (ja) |
| JP (1) | JP3790273B2 (ja) |
| KR (1) | KR100582159B1 (ja) |
| CN (1) | CN1117057C (ja) |
| AU (1) | AU727370B2 (ja) |
| CA (1) | CA2263563C (ja) |
| EA (2) | EA002969B1 (ja) |
| TW (1) | TW458982B (ja) |
| WO (1) | WO1998013328A1 (ja) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050020687A1 (en) * | 1996-01-14 | 2005-01-27 | Consiglio Nazionale Ricerche | Methods of treating inflammatory and viral disorders by administering cyclopentenone compounds |
| IT1289250B1 (it) | 1996-12-13 | 1998-09-29 | Consiglio Nazionale Ricerche | Impiego di 2 ciclopenten 1-one e suoi derivati come inibitori del fattore nf-kb |
| ES2226096T3 (es) | 1997-03-17 | 2005-03-16 | Takara Bio Inc. | Agentes antivirales. |
| ATE274904T1 (de) * | 1997-03-28 | 2004-09-15 | Takara Bio Inc | 4,5-dihydroxy-2-cyclopenten-1-on für die behandlung von diabetes mellitus |
| US6284801B1 (en) | 1997-04-01 | 2001-09-04 | Takara Shuzo Co., Ltd. | Antirheumatic agents |
| CN1123340C (zh) * | 1997-07-02 | 2003-10-08 | 宝酒造株式会社 | 抗过敏剂 |
| ATE252536T1 (de) | 1997-08-29 | 2003-11-15 | Takara Shuzo Co | Alpha-kristall des cyclopentenons |
| CN1133611C (zh) | 1998-01-19 | 2004-01-07 | 宝酒造株式会社 | 具有细胞凋亡诱发能力的物质 |
| TW526060B (en) | 1998-08-04 | 2003-04-01 | Takara Shuzo Co | Animal feedstuff and its additive |
| ATE281161T1 (de) | 1998-08-18 | 2004-11-15 | Takara Bio Inc | Heilmittel und vorbeugende mittel die cyclopentenonverbindungen als wirksamen stoff enthalten |
| US7326542B2 (en) | 1998-12-02 | 2008-02-05 | Princeton University | Compositions and methods for regulating bacterial pathogenesis |
| US6720415B2 (en) | 1998-12-02 | 2004-04-13 | Princeton University | Compositions and methods for regulating bacterial pathogenesis |
| KR20010102134A (ko) * | 1999-02-19 | 2001-11-15 | 오미야 히사시 | 치료제 |
| EP1175907A4 (en) * | 1999-04-15 | 2004-12-29 | Takara Bio Inc | REMEDIES |
| GB9929702D0 (en) * | 1999-12-16 | 2000-02-09 | Charterhouse Therapeutics Ltd | Chemical compounds and their uses |
| JP2003532698A (ja) | 2000-05-10 | 2003-11-05 | プリンストン ユニバーシティ | 細菌の増殖および病理発生を調節するための化合物ならびに方法 |
| US20020143163A1 (en) * | 2000-08-29 | 2002-10-03 | The University Of Medicine And Dentistry Of New Jersey | Gene conferring resistance to the antibacterial 4,5-dihydroxy-2-cyclopenten-1-one (DHCP), the protein encoded by same, and applications thereof |
| TW200400263A (en) * | 2002-03-13 | 2004-01-01 | Takara Bio Inc | Effect of treatment with 4,5-dihydroxy-2-cyclopenten-1-one (DHCP) on gene expression and quorum-sensing in bacteria |
| JP2007269641A (ja) * | 2006-03-30 | 2007-10-18 | Mie Univ | 修治技術を取り入れたきのこ類の機能性食品およびその生産方法 |
| DK2295538T3 (en) | 2008-07-07 | 2015-12-21 | Takara Bio Inc | Process for producing a pluripotent stem cell |
| AU2012275190B2 (en) | 2011-06-29 | 2016-05-19 | The Penn State Research Foundation | Compositions, methods and kits for treating leukemia |
| JP6053149B2 (ja) * | 2012-03-26 | 2016-12-27 | AdaBio株式会社 | Id遺伝子の発現抑制剤 |
| CN105541927A (zh) * | 2016-02-03 | 2016-05-04 | 广西大学 | 一种源于核糖的稻曲病菌抑制物 |
| CN106518643B (zh) * | 2016-10-14 | 2019-02-15 | 宁波大学 | 一种环戊烯酮类化合物及其制备方法和用途 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2608089B2 (ja) * | 1988-03-09 | 1997-05-07 | 帝人株式会社 | 4−ヒドロキシ−2−シクロペンテノン類およびそれを含有する薬剤組成物 |
| JP2571950B2 (ja) * | 1988-03-11 | 1997-01-16 | 財団法人野口研究所 | シクロペンテノン誘導体及びその製造法 |
| AU615534B2 (en) * | 1988-04-19 | 1991-10-03 | Teijin Limited | 2-substituted-2-cyclopentenones |
| JPH0768163B2 (ja) * | 1989-03-17 | 1995-07-26 | 財団法人野口研究所 | シクロペンテノン誘導体の製法 |
-
1997
- 1997-09-01 AU AU40330/97A patent/AU727370B2/en not_active Ceased
- 1997-09-01 JP JP51548098A patent/JP3790273B2/ja not_active Expired - Fee Related
- 1997-09-01 WO PCT/JP1997/003052 patent/WO1998013328A1/ja not_active Application Discontinuation
- 1997-09-01 US US09/230,868 patent/US6087401A/en not_active Expired - Fee Related
- 1997-09-01 EA EA200100172A patent/EA002969B1/ru not_active IP Right Cessation
- 1997-09-01 KR KR1019997002641A patent/KR100582159B1/ko not_active IP Right Cessation
- 1997-09-01 EP EP97937862A patent/EP0941981A4/en not_active Withdrawn
- 1997-09-01 CA CA002263563A patent/CA2263563C/en not_active Expired - Fee Related
- 1997-09-01 EA EA199900334A patent/EA003923B1/ru not_active IP Right Cessation
- 1997-09-01 CN CN97197822A patent/CN1117057C/zh not_active Expired - Fee Related
- 1997-09-12 TW TW086113265A patent/TW458982B/zh not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| CA2263563C (en) | 2003-12-30 |
| EA200100172A1 (ru) | 2001-08-27 |
| CN1117057C (zh) | 2003-08-06 |
| TW458982B (en) | 2001-10-11 |
| EP0941981A4 (en) | 2003-07-02 |
| KR20000057128A (ko) | 2000-09-15 |
| CN1230165A (zh) | 1999-09-29 |
| EA003923B1 (ru) | 2003-10-30 |
| EP0941981A1 (en) | 1999-09-15 |
| CA2263563A1 (en) | 1998-04-02 |
| EA199900334A1 (ru) | 1999-10-28 |
| AU4033097A (en) | 1998-04-17 |
| WO1998013328A1 (fr) | 1998-04-02 |
| AU727370B2 (en) | 2000-12-14 |
| KR100582159B1 (ko) | 2006-05-24 |
| US6087401A (en) | 2000-07-11 |
| EA002969B1 (ru) | 2002-12-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3790273B2 (ja) | シクロペンテノン類、その製造方法及び用途 | |
| JP4486064B2 (ja) | 藻類由来の生理活性物質を利用した医薬、食品または飲料 | |
| CA2838442C (en) | Long chain glycolipids useful to avoid perishing or microbial contamination of materials | |
| US6482806B1 (en) | Product of heat treatment of uronic acid, food, drink, or drug including the product | |
| AU764582B2 (en) | Therapeutic agents | |
| JP3602854B2 (ja) | ヒドロキシシクロペンタノン | |
| JP3710854B2 (ja) | 水易溶性ヤマモモ科植物抽出物 | |
| JP6888145B2 (ja) | Lps産生抑制剤、及びlps産生抑制用の食品組成物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20050607 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050802 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060328 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060331 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090407 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110407 Year of fee payment: 5 |
|
| LAPS | Cancellation because of no payment of annual fees |