JP2020506916A - 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 - Google Patents
活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 Download PDFInfo
- Publication number
- JP2020506916A JP2020506916A JP2019540538A JP2019540538A JP2020506916A JP 2020506916 A JP2020506916 A JP 2020506916A JP 2019540538 A JP2019540538 A JP 2019540538A JP 2019540538 A JP2019540538 A JP 2019540538A JP 2020506916 A JP2020506916 A JP 2020506916A
- Authority
- JP
- Japan
- Prior art keywords
- antibody
- tnf
- human
- composition
- antibodies
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/24—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against cytokines, lymphokines or interferons
- C07K16/241—Tumor Necrosis Factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/54—Medicinal preparations containing antigens or antibodies characterised by the route of administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Abstract
Description

配列番号1、2、及び3の重鎖可変CDR領域の全て並びに/又は配列番号4、5、及び6の軽鎖可変CDR領域の全てを含む本発明の単離された抗体は、任意の好適なポリヌクレオチドによってコード化される本明細書で開示される抗体のアミノ酸配列、又は任意の単離又は調製された抗体を含む。好ましくは、ヒト抗体又は抗原結合断片は、ヒトTNFに結合し、それにより、タンパク質の少なくとも1つの生物学的活性を部分的又は実質的に中和する。少なくとも1つのTNFタンパク質又は断片の少なくとも1つの生物学的活性を部分的に又は好ましくは実質的に中和する抗体又はその特定された部分若しくは変異体は、タンパク質又は断片に結合し、それによりTNFのTNF受容体への結合を通して、又は他のTNF依存性又は媒介型機序を通して媒介される活性を阻害することができる。本明細書で使用するとき、「中和抗体」という用語は、アッセイに応じて約20〜120%、好ましくは少なくとも約10、20、30、40、50、55、60、65、70、75、80、85、90、91、92、93、94、95、96、97、98、99、100%又はそれ以上、TNF依存性活性を阻害することができる抗体を指す。TNF依存性活性を阻害する抗TNF抗体の能力は、好ましくは、本明細書に記載されかつ/又は当該技術分野において既知の、少なくとも1つの好適なTNFタンパク質又は受容体アッセイによって評価される。本発明のヒト抗体は、任意のクラス(IgG、IgA、IgM、IgE、IgDなど)又はアイソタイプのものであってもよく、κ又はλ軽鎖を含み得る。一実施形態において、ヒト抗体は、IgG重鎖又は規定された断片、例えば、IgG1、IgG2、IgG3又はIgG4のうちの少なくとも1つのアイソタイプを含む。このタイプの抗体は、本明細書に記載され、かつ/又は当該技術分野において既知の、少なくとも1つのヒト軽鎖(例えば、IgG、IgA)及びIgM(例えば、γ1、γ2、γ3、γ4)導入遺伝子を含む、トランスジェニックマウス又は他のトランスジェニック非ヒト哺乳動物を用いることによって調製され得る。別の実施形態において、抗ヒトTNFヒト抗体は、IgG1重鎖及びIgG1軽鎖を含む。
典型的な哺乳類発現ベクターは、mRNAの転写の開始を媒介する少なくとも1つのプロモータ要素、抗体コード配列、並びに転写の終結及び転写物のポリアデニル化に必要なシグナルを含む。付加的な要素には、エンハンサ、コザック配列並びにRNAスプライシングのためのドナー及びアクセプタ部位に隣接している介在配列が含まれる。高効率の転写は、SV40からの初期及び後期プロモータ、レトロウイルス、例えば、RSV、HTLVI、HIVIからの長端末反復(long terminal repeats、LTRS)、及びサイトメガロウイルス(cytomegalovirus、CMV)の初期プロモータで達成することができる。しかしながら、細胞要素を使用することもできる(例えば、ヒトアクチンプロモータ)。本発明の実施において使用するのに好適な発現ベクターとしては、例えば、pIRES1neo、pRetro−Off、pRetro−On、PLXSN若しくはpLNCX(Clonetech Labs,Palo Alto,CA)、pcDNA3.1(+/−)、pcDNA/Zeo(+/−)又はpcDNA3.1/Hygro(+/−)(Invitrogen)、PSVL及びPMSG(Pharmacia,Uppsala,Sweden)、pRSVcat(ATCC37152)、pSV2dhfr(ATCC37146)並びにpBC12MI(ATCC67109)などのベクターが挙げられる。使用することができる哺乳類の宿主細胞には、ヒトHela293、H9及びJurkat細胞、マウスNIH3T3及びC127細胞、Cos1、Cos7及びCV1、ウズラQC1−3細胞、マウスL細胞及びチャイニーズハムスター卵巣(Chinese hamster ovary、CHO)細胞が含まれる。
要約。ヒト重鎖及び軽鎖免疫グロブリン遺伝子を含有するトランスジェニックマウスを使用して、1つ以上のTNF媒介性疾患の処置のための、TNF作用を阻害するために治療的に使用することができる高親和性の完全ヒトモノクローナル抗体を生成する。重鎖及び軽鎖両方のヒト可変及び定常領域抗体導入遺伝子を含有する(CBA/JxC57/BL6/J)F2ハイブリッドマウスをヒト組換えTNFで免疫化する(Taylor et al.,Intl.Immunol.6:579−591(1993)、Lonberg,et al.,Nature368:856−859(1994)、Neuberger,M.,Nature Biotech.14:826(1996)、Fishwild,et al.,Nature Biotechnology14:845−851(1996))。いくつかの融合物が完全ヒトTNF反応性IgGモノクローナル抗体の1つ以上のパネルを生み出した。完全ヒト抗TNF抗体を更に特徴付けする。全てはIgG1κである。かかる抗体は、およそ1×109〜9×1012の親和性定数を有することが分かった。これらの完全ヒトモノクローナル抗体の予期せぬ高親和性により、それらはTNF関連疾患、病態、又は障害における治療用途のための好適な候補となる。
動物。ヒト抗体を発現することができるトランスジェニックマウスは、当該技術分野において既知であり、(例えば、GenPharm International,San Jose,CA、Abgenix,Freemont,CA、及びその他)から市販されており、ヒト免疫グロブリンを発現するが、マウスIgM又はIgκを発現しない。例えば、かかるトランスジェニックマウスは、V(D)J結合、重鎖クラススイッチ及び体細胞突然変異を受けてヒト配列免疫グロブリンのレパートリーを生成する、ヒト配列導入遺伝子を含有する(Lonberg,et al.,Nature368:856−859(1994))。軽鎖導入遺伝子は、例えば、部分的に、生殖系列ヒトVκ領域のほぼ半分を含む酵母人工染色体クローンに由来し得る。加えて、重鎖導入遺伝子は、ヒトμ及びヒトγ1の両方(Fishwild,et al.,Nature Biotechnology14:845−851(1996))並びに/又はγ3定常領域をコード化することができる。適切な遺伝子型系統由来のマウスを免疫化及び融合プロセスにおいて使用して、TNFに対する完全ヒトモノクローナル抗体を生成することができる。
抗ヒトTNFモノクローナル抗体の生成。いくつかの融合を行い、ヒトTNFに特異的な数十の抗体を生み出す各融合物を15のプレート(1440ウェル/融合物)に播種する。これらのうち、いくつかは、ヒト及びマウスIg鎖の組み合わせからなることがわ分かる。残りのハイブリドーマは、ヒト重鎖及び軽鎖のみからなる抗TNF抗体を分泌(secret)する。ヒトハイブリドーマの全てがIgG1κであることが予想される。
いくつかの融合は、ヒトTNFで免疫化されるヒト可変及び定常領域抗体導入遺伝子を含有するハイブリッドマウスからの脾細胞を利用して行われる。IgG1κアイソタイプのいくつかの完全ヒトTNF反応性IgGモノクローナル抗体のセットを生成する。完全ヒト抗TNF抗体を更に特徴付けする。生成された抗体のうちのいくつかは、1×109〜9×1012の親和性定数を有する。これらの完全ヒトモノクローナル抗体の予期せぬ高親和性により、それらは、TNF依存疾患、病態又は関連状態における治療用途に好適なものとなる。
要約。重鎖及び軽鎖両方のヒト可変及び定常領域抗体導入遺伝子を含有する(CBA/JxC57BL/6J)F2ハイブリッドマウス(1〜4)を組換えヒトTNFαで免疫化した。GenTNVと命名された1つの融合が、固定化された組換えヒトTNFαに結合する完全ヒトIgG1κモノクローナル抗体を8つ生み出した。特定直後、8つの細胞株は、更に特徴付けするためにMolecular Biologyに譲渡された。これらMabは配列が完全にヒトであるため、それらはヒトにおけるcA2(Remicade)よりも免疫原性が低いと予想される。
動物ヒト免疫グロブリンを発現するが、マウスIgM又はIgκを発現しないトランスジェニックマウスは、GenPharm Internationalにより開発されてきた。これらのマウスは、V(D)J結合、重鎖クラススイッチ、及び体細胞突然変異を受けて抗原特異的ヒト免疫グロブリン(1)のレパートリーを生成する、機能性ヒト抗体導入遺伝子を含有する。軽鎖導入遺伝子は、部分的に、生殖系列ヒトVκ遺伝子座のほぼ半分を含む酵母人工染色体クローンに由来する。いくつかのVH遺伝子に加えて、重鎖(HC)導入遺伝子は、ヒトμ及びヒトγ1(2)、並びに/又はγ3定常領域の両方をコード化する。本明細書に記載されるモノクローナル抗体を生成するための免疫化及び融合プロセスにおいて、HCo12/KCo5遺伝子型系統由来のマウスを使用した。
GenTNV融合は、Centocorで調製された組換えヒトTNFαで免疫化されたヒト可変及び定常領域抗体導入遺伝子を含有するハイブリッドマウスからの脾細胞を利用して行われた。IgG1κアイソタイプの8つの完全ヒトTNFα反応性IgGモノクローナル抗体を生成した。更なる特徴付け及び開発のために、親細胞株をMolecular Biologyグループに移した。これらの新しいヒト抗体のうちの1つは、Remicadeと比較して、免疫原性及びアレルギー型合併症が減少する潜在的な利益を有して、抗炎症に有用である可能性がある。
要約。TNV表記の8つのヒトモノクローナル抗体(mAb)のパネルは、明らかに高結合活性で固定化されたヒトTNFαに結合することが認められた。8つのmAbのうちの7つは、組換えTNF受容体へのヒトTNFαの結合を効率的に遮断することを示した。7つのmAbをコード化するDNAの配列分析は、全てのmAbがヒトV領域を有していることを確認した。DNA配列は、3対のmAbが互いに同一であり、そのため8つのmAbの元のパネルがTNV14、TNV15、TNV148、及びTNV196で表される4つの別個のmAbのみを含有していることも明らかにした。mAbの推定アミノ酸配列の分析及びインビトロTNFα中和データの結果に基づいて、mAb TNV148及びTNV14を更なる研究のために選択した。
試薬及び細胞。TRIZOL試薬はGibco BRLから購入した。プロテイナーゼKはSigma Chemical Companyから得た。逆転写酵素はLife Sciences,Inc.から得た。Taq DNAポリメラーゼはPerkin Elmer Cetus又はGibco BRLのいずれかから得た。制限酵素はNew England Biolabsから購入した。QIA quick PCR Purification KitはQiagenから得た。QuikChange Site−Directed Mutagenesis KitはStratageneから購入した。Wizardプラスミドミニプレップキット及びRNasinはPromegaからであった。OptiplatesはPackardから得た。125IodineはAmershamから購入した。カスタムオリゴヌクレオチドはKeystone/Biosource Internationalから購入した。この作業で使用したオリゴヌクレオチドの名称、識別番号、及び配列を表2に示す。
オリゴヌクレオチド5’14s及びHuH−J6によりコード化されるアミノ酸を配列の上に示す。「M」アミノ酸残基は翻訳開始コドンを表す。オリゴヌクレオチド5’14s及びHuH−J6の下線付き配列は、それぞれ、BsiWI及びBstBI制限部位を示す。HuH−J6の斜線はエクソン/イントロン境界に対応する。配列がマイナス鎖に対応するオリゴヌクレオチドは、3’−5’配向で書かれていることに留意する。
L28ベクター又はpBCベクターは、初期のAb cDNAクローンを表す。これらのプラスミドのインサートは、中間プラスミドを作製するために不完全な12B75系ベクターに移された。1つの追加の移動工程により、線形化された後に細胞に導入されたか、又は細胞のトランスフェクション前にmAb遺伝子インサートを精製するために使用されたかのいずれかであった最終発現プラスミドがもたらされた。ND=実施せず。
組換え受容体へのTNF結合の阻害。
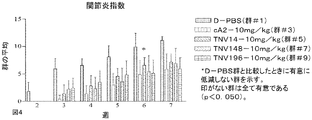
ハイブリドーマ細胞上清に含有される8つのTNV mAbが、受容体へのTNFα結合を阻害することができるかどうかを決定するために、簡単な結合アッセイが行われた。ヒトIgGの標準ELISA分析により、それぞれの細胞上清におけるTNV mAbの濃度を最初に決定した。次に、組換えp55TNF受容体/IgG融合タンパク質p55−sf2をEIAプレート上にコーティングし、様々な量のTNV mAbの存在下で、125I標識TNFαをp55受容体に結合させた。図1に示すように、8つのTNV mAbのうちの1つ(TNV122)を除く全てが、p55受容体へのTNFαの結合を効率的に遮断した。実際、TNV mAbは、陰性対照ハイブリドーマ上清にスパイクされたcA2陽性対照mAbよりもTNFα結合を阻害するのにより有効であるように見えた。これらの結果は、TNV mAbが細胞系アッセイ及びインビボでTNFαの生物活性を遮断するであろう可能性が高く、したがって、追加の分析が必要であることを示すと解釈された。
RNAがヒトmAbをコード化することの確認。
受容体結合アッセイにおいてTNFα遮断活性を示した7つのTNV mAb(TNV14、TNV15、TNV32、TNV86、TNV118、TNV148、及びTNV196)を特徴付ける際の最初の工程として、これらのmAbを生成する7つのハイブリドーマ細胞株から全RNAを単離した。次に、各RNA試料を使用して、各mAbの完全なシグナル配列、完全な可変領域配列、及び定常領域配列の一部を含むヒト抗体重鎖又は軽鎖cDNAを調製した。次に、これらのcDNA生成物をPCR反応で増幅させ、最初に断片をクローニングすることなくPCR増幅DNAを直接配列決定した。配列決定した重鎖cDNAは、マウスに存在する5つのヒト生殖系列遺伝子のうちの1つであるDP−46と>90%同一であった(図2)。同様に、配列決定した軽鎖cDNAは、マウスに存在するヒト生殖系列遺伝子のうちの1つと100%又は98%のいずれかと同一であった(図3)。これらの配列結果は、cDNAに転写され配列決定されたRNA分子がヒト抗体重鎖及びヒト抗体軽鎖をコード化したことを確認した。可変領域がシグナル配列コード配列の5’端にマッピングされるオリゴヌクレオチドを使用してPCR増幅されたため、シグナル配列の最初の数個のアミノ酸は元のTNV翻訳生成物の実際の配列ではない可能性があるが、組換えTNV mAbの実際の配列を表すことに留意するべきである。
各mAbの重鎖及び軽鎖両方の可変領域全体のcDNA配列の分析は、TNV32がTNV15と同一であり、TNV118がTNV14と同一であり、TNV86がTNV148と同一であることを明らかにした。受容体結合アッセイの結果は、DNA配列分析と一致していた、即ち、TNV86及びTNV148の両方が、TNF結合の遮断においてTNV118及びTNV14の両方よりも約4倍良好であった。したがって、後続の作業は、4つの固有のTNV mAbである、TNV14、TNV15、TNV148、及びTNV196にのみ焦点を当てた。
DNA配列結果は、4つのTNV mAbの重鎖をコード化する遺伝子が全て互いに高度に相同であり、全てが同じ生殖系列遺伝子DP−46に由来するように見えることを明らかにした(図2)。加えて、重鎖CDR3配列の各々は非常に類似し、同じ長さのものであるため、そしてそれらが全てJ6エクソンを使用するため、それらは明らかに、単一のVDJ遺伝子再配列事象から生じ、この後に各mAbを固有のものにする体細胞変化が続いた。DNA配列分析は、4つのmAbにおいて2つの別個の軽鎖遺伝子のみが存在したことを明らかにした(図3)。TNV14及びTNV15における軽鎖可変領域コード配列は、互いに同一であり、ヒトκ鎖のVg/38Kファミリーの代表的な生殖系列配列と同一である。TNV148及びTNV196軽鎖コード配列は、互いに同一であるが、2つのヌクレオチド位置での生殖系列配列が異なる(図3)。
rTNV148Bトランスフェクション2からの、生成が最高の653親株のうちの10(使用済24ウェル培養物において5〜10:g/mlを生成)をサブクローニングして、生成がより高い細胞株についてスクリーニングし、より均質な細胞集団を調製した。親株2.320、2.320−17、及び2.320−20のサブクローンのうちの2つは、使用済24ウェル培養物において約50:g/mlを生成し、これは、それらの親株に対して5倍の増加であった。サブクローニングした株2.320−17及び2.320−20の2回目のサブクローニングがもたらした。
rTNV14トランスフェクション3からの生成が最高のSp2/0親株のうちの3つを1回サブクローニングした。サブクローン3.27−1は、生成が19:g/mlであり、使用済の24ウェル培養物において最も高い生産体であることが分かった。この細胞株は、C476Aと表記された(表5)。
元のクローン名の最初の1桁は、細胞株がどのトランスフェクションに由来するかを示す。本明細書に報告されるCコード細胞株の全てが制限酵素で線形化された重鎖及び軽鎖全プラスミドでのトランスフェクションに由来した。
細胞株成長特徴をより慎重に特徴付けし、大規模でmAb生成レベルを決定するために、T75培養物を使用して成長曲線分析を行った。結果は、細胞株の4つのC466シリーズの各々が1.0×106〜1.25×106細胞/mlのピーク細胞密度及び110〜140:g/mlの最大mAb蓄積レベルに達したことを示した(図7)。対照的に、生成が最高のSp2/0サブクローンC467Aは、2.0×106細胞/mlのピーク細胞密度及び25:g/mlの最大mAb蓄積レベルに達した(図7)。成長曲線分析は、rTNV14生成細胞株C476Aに対して行われなかった。
ヒトTNFαに対する8つのヒトmAbの初期パネルから、タンパク質配列及びTNF中和効力を含むいくつかの基準に基づいて、TNV148B並びにTNV14が好ましいものとして選ばれた。100:g/ml超のrTNV148B及び19:g/ml超のrTNV14を生成する細胞株を調製した。
約4週齢のTg197研究マウスを性別及び体重に基づき9つの処置群のうちの1つに割り当て、DulbeccoのPBS(D−PBS)、又は1mg/kg若しくは10mg/kgのいずれかの本発明の抗TNF抗体(TNV14、TNV148、若しくはTNV196)の単回腹腔内ボーラス用量で処置した。
約4週齢のTg197研究マウスを体重に基づき8つの処置群のうちの1つに割り当て、対照品(D−PBS)、又は3mg/kgの抗体(TNV14、TNV148)(0週目)の腹腔内ボーラス投与で処置した。注射は1、2、3及び4週目に全ての動物において繰り返された。群1〜6は、試験品の有効性に関して評価された。群7及び8の動物から得られた血清試料は、2、3及び4週目のTNV14又はTNV148の免疫応答誘導及び薬物動態クリアランスに関して評価された。
約4週齢のTg197研究マウスを性別及び体重に基づき6つの処置群のうちの1つに割り当て、3mg/kg又は5mg/kgのいずれかの抗体(cA2又はTNV148)の単回腹腔内ボーラス投与で処置した。この研究は、D−PBS及び10mg/kgのcA2対照群を利用した。
TNV148(ハイブリドーマ細胞に由来する)及びrTNV148B(トランスフェクトした細胞に由来する)の単回腹腔内投与の有効性を比較するために。約4週齢のTg197研究マウスを性別及び体重に基づき9つの処置群のうちの1つに割り当て、Dulbecco=のPBS(D−PBS)又は1mg/kgの抗体(TNV148、rTNV148B)の単回腹腔内ボーラス投与で処置した。
梗概
活動性乾癬性関節炎(PsA)を有する対象における、静脈内投与された抗TNFαモノクローナル抗体、ゴリムマブの多施設共同、無作為化、二重盲検、プラセボ対照試験
主目的
この研究の主目的は、PsAの徴候及び症状の低減を評価することによって、活動性乾癬性関節炎(PsA)を有する対象におけるゴリムマブ2mg/kgのIV投与の有効性を評価することである。
副次的目的は、IVゴリムマブについて以下を評価することである。
・乾癬皮膚病変、身体機能、健康関連の生活の質、及び他の健康結果の改善に関連する有効性
・構造損傷の進行の阻害
・安全性
・薬物動態(PK)、薬力学(PD)、及び免疫原性
研究の主目的に対処するために、統計的仮説(代替仮説)は、ゴリムマブ2mg/kgが、主要有効性エンドポイントに基づき、活動性PsAを有する対象の徴候及び症状を低減する際に、プラセボよりも統計的に優れていることである。
これは、活動性PsAを有する対象におけるプラセボと比較したIVゴリムマブの有効性及び安全性の3相多施設共同、無作為化、二重盲検、プラセボ対象研究である。約440人の対象が、約90の治験実施機関で無作為化される。対象は、0、4、12、及び20週目にゴリムマブ2mg/kg又はプラセボIV注入を受けるように無作為に割り当てられる。16週目に、早期離脱対象である全ての対象は、治験者によって選択されるような、以下の併用薬介入のうちの1つが認められる。それらのコルチコステロイド用量(最大総用量プレドニゾン10mg/日若しくは同等)、メトトレキサート(MTX)用量(最大総用量25mg/週)、若しくはNSAID用量の増加、又はNSAID、コルチコステロイド(最大用量プレドニゾン10mg/日若しくは同等)、MTX(最大用量25mg/週)、SSZ(最大用量3g/日)、HCQ(最大用量400mg/日)、又はレフルノミド(最大投与量20mg/日)の開始。安定用量のこれらの薬剤への滴定は、24週目の訪問までに早期離脱対象である対象に対して完了されるべきである。24週目に、プラセボ注入を受けた全ての対象は、交差し、ゴリムマブIV注入を受け始める。
研究に適格な対象は、研究薬剤の初回投与前少なくとも6か月間、PsAを有する18歳以上の男性又は女性であり、スクリーニング時にCASPAR基準を満たす。対象は、スクリーニング及びベースラインにおいて活動性疾患の症状(5つ以上の腫脹関節及び5つ以上の圧痛関節)を有し、0.6mg/dL以上のC反応性タンパク質(CRP)レベルを有しなければならない。対象は、生物製剤で処置されていてはならない。対象は、研究中にMTX処置を継続してもよい。
初回スクリーニング訪問では、研究に適格である可能性があると考えられる全ての対象から、研究に登録するために、プロトコル指定の組み入れ及び除外基準に従って、インフォームドコンセントが得られる。無作為化訪問では、対象は再評価され、全ての指定された組み入れ及び除外基準が満たされた場合、対象は、ゴリムマブIV注入又はプラセボIV注入のいずれかを受けるように無作為化される。無作為化は、地理的領域及びベースラインのメトトレキサート(MTX)使用(はい又はいいえ)によって階層化される。
この研究のために選択された有効性評価は、PsAの治療のための治療用生物学的薬剤の以前の試験において確立された。本研究のために選択された患者報告アウトカム(patient reported outcome、PRO)は、PsAにおける他の研究に関する医療文献及び適用可能なUS/EU規制ガイダンス文書で受け入れられている臨床的に関連する測定値と一致する。
・対象の疼痛評価
・対象の疾患の包括的評価
・医師の疾患の包括的評価
・関節評価
・健康評価質問表の障害指標(HAQ−DI)
・乾癬の面積及び重症度指標(Psoriasis Area and Severity Index、PASI)
・手及び足のX線評価
・36項目ショートフォーム健康調査(SF−36)
・指炎評価
・腱付着部炎評価
・Bath強直性脊椎炎疾患活動性指標(BASDAI)
・修正されたNAPSI
・皮膚科学的生活の質指標(DLQI)
・慢性疾患療法の機能評価(FACIT)−疲労
・労働生産性に関する質問票(WLQ)
・生産性VAS
・EuroQol−5D(EQ−5D)質問票
この研究の主エンドポイントは、14週目にACR20応答を達成する対象の割合である。
以下の主要な二次分析エンドポイントは、以下に指定されるような重要度順で列挙される。
・14週目のHAQ−DIスコアにおけるベースラインからの変化。
・14週目にACR50応答を有する患者の割合。
・14週目にPASI75応答を達成する対象の割合(ベースラインの3%以上のBSA乾癬関与を有する)。
・24週目の修正総van der Heijdeシャープスコア(vdH−S)スコアにおけるベースラインからの変化。
血液試料を選択された訪問で収集して、PsAを有する成人対象におけるIVゴリムマブのPKを評価する。研究薬剤がその訪問で投与される場合、薬物動態試料は、IV注入ラインとは異なる腕から採取されるべきである。0、4、12、20、36、及び52週目に、血清ゴリムマブ濃度のための2つの試料が収集され、一方の試料は、注入直前に収集され、他方は、注入の終了の1時間後に収集される。残りの訪問の各々について、血清ゴリムマブ濃度のための1つの試料のみが収集され、これは、研究薬剤の注入がその訪問で投与される場合、注入直前に収集されるべきである。無作為PK試料は、14週目と20週目の訪問の間の集団PK分析のために採取され(14週目又は20週目の訪問時以外)、この無作為試料は、研究薬剤注入の少なくとも24時間前又は後に収集されなければならない。
PsAを有する成人対象におけるゴリムマブの免疫原性を評価するために、ゴリムマブに対する抗体の検出のための血清試料が、時間及びイベントスケジュールに従って収集される。
バイオマーカー試料は、臨床転帰における個人間変動の分子的理解を得るために収集され、これは、薬物に異なる応答を示す集団サブグループを同定するのに役立ち得る。バイオマーカー試料はまた、新たな問題に対処するのに役立つために、かつ将来のより安全な、より効果的な、かつ最終的に個別化された療法の開発を可能にするために使用されてもよい。
ゲノム試験は、疾患又は薬剤に対する反応と特定の遺伝子との関連に関して調査するために行われる。ゴリムマブ又はこの薬物が開発された疾患に関連するDNA研究のみが行われる。ゲノムの幅広い薬理ゲノム学的及び/又はエピジェネティクス試験は、同意が得られた対象において本研究で行われる。この研究のこの部分に参加している対象は、別個のインフォームドコンセントに署名しなければならない。更に、対象は、試験の他の側面への参加、又は試験への今後の参加に影響を与えることなく、随時、そのような同意を撤回することができる。
他の抗TNFα剤の安全プロファイル、並びにこれまでのゴリムマブ安全データに基づき、対象となるいくつかのAEが特定され、この研究において監視及び評価される。これらには、注入反応、検査所見の肝胆汁性異常、TBを含む感染、及び悪性腫瘍が挙げられる。
対象ベースライン、人口統計、及びベースラインの比較可能性を評価するために、疾患特性データは、処置群によって要約される。
有効性及び対象ベースライン分析は、特に明記しない限り、治療企図集団(即ち、無作為化された全ての対象)を利用する。有効性分析に含まれる対象は、割り当てられた処置を受けるかどうかにかかわらず、彼らの割り当てられた処置群に従って要約される。
主エンドポイント分析
主エンドポイントは、14週目にACR20応答を達成する対象の割合である。
以下の主要二次分析は、以下に指定されるような重要度順で実施される。
1.14週目のHAQ−DIスコアにおけるベースラインからの変化が要約され、処置群間で比較される。
2.14週目にACR50応答を有する対象の割合が要約され、処置群間で比較される。
3.14週目にPASI75応答を達成する対象の割合(ベースラインの3%以上の体表面積の乾癬関与を伴う)が要約され、処置群間で比較される。
4.24週目の修正総vdH−Sスコアにおけるベースラインからの変化が要約され、処置群間で比較される。
日常的な安全性評価が実施される。注入反応及びTBを含む感染を含む、AE、SAE、及び適度に関連するAEの発生及びタイプが、処置群によって要約される。NCI CTCAE毒性等級に基づく異常な検査室パラメータ(血液学及び化学)を有する対象の数が、要約される。加えて、ANA及び抗dsDNA抗体を有する対象の数、並びにゴリムマブに対する抗体との注入反応の関係が、要約される。
化学名及び構造
SIMPONI(登録商標)(ゴリムマブ)は、免疫グロブリンG(IgG)1重鎖アイソタイプ(G1m[z]アロタイプ)及びκ軽鎖アイソタイプを有するヒトモノクローナル抗体(mAb)である。ゴリムマブは、配列番号36を含む重鎖(HC)及び配列番号37を含む軽鎖(LC)を有する。ゴリムマブの分子量は、149,802〜151,064ダルトンの範囲である。ゴリムマブは、解剖治療化学(Anatomical Therapeutic Chemical)(ATC)分類法に従い、TNFα阻害剤に分類される(ATCコード:L04AB06)。ゴリムマブは、可溶性及び膜貫通形態の両方の腫瘍壊死因子α(TNFα)に高い親和性で結合し、TNFα生理活性を阻害する。他のTNFスーパーファミリーリガンドに対する結合は観察されなかった。特に、ゴリムマブは、ヒトリンホトキシンに結合しないか、又はそれを中和しない。TNFαは、自己会合して生物活性ホモトリマーを形成し、タンパク質分解によって細胞表面から急速に放出される膜貫通タンパク質として、主に活性化単球、マクロファージ、及びT細胞によって合成される。TNFαのp55又はp75TNF受容体のいずれかへの結合は、受容体細胞質ドメインのクラスター化をもたらし、シグナル伝達を開始する。腫瘍壊死因子は、様々な刺激に応答して産生され、続いて、カスケード依存性アポトーシス経路並びに転写因子核因子(NF)−κB及び活性化因子タンパク質−1(AP−1)の活性化による炎症応答を促進する、主要なセンチネルサイトカインとして同定されている。腫瘍壊死因子はまた、胚中心における免疫細胞の組織におけるその役割を通して免疫応答を調節する。TNFの発現の上昇は、関節リウマチ(RA)などの慢性炎症性疾患、並びに乾癬性関節炎(PsA)及び強直性脊椎炎(AS)などの脊椎関節症に関連しており、これらの疾患に特徴的である関節性炎症及び構造損傷の重要なメディエーターである。
乾癬性関節炎は、乾癬に関連する慢性炎症性、通常はリウマチ因子(RF)陰性関節炎である。一般白人集団における乾癬の有病率は、約2%である。約6%〜39%の乾癬患者がPsAを発症する。
TNFαは、幅広い機能活性を呈する主要な炎症メディエーターと見なされる。2TNFαの過剰産生は、RA及びクローン病患者において実証されるように、炎症に関連する疾患プロセスをもたらす。炎症性サイトカインの主要な供給源である、T細胞と単球/マクロファージとの間の相互作用は、PsAの病因における役割を果たす。7,22TNFαのレベルの増加は、関節流体及び組織において、及びPsA患者における乾癬皮膚病変において検出されている。24,26抗TNFαモノクローナル抗体であるインフリキシマブによる処置は、48時間以内の、活動性PsA患者における乾癬表皮におけるT細胞数並びに滑膜組織におけるT細胞数及びマクロファージ数の有意な低減をもたらすことが報告されている。17インフリキシマブ処置はまた、劇的な臨床的皮膚及び関節応答と並行してPsA患者における滑膜組織における血管新生成長因子も有意に低減した。32
この研究は、活動性PsAの治療において、0及び4週目で、次いで8週間毎(q8w、MTXあり又はなし)に、30分間にわたってIV注入を介して投与された2mg/kgゴリムマブの安全性及び有効性を評価する。
目的
主目的
この研究の主目的は、PsAの徴候及び症状の低減を評価することによって、活動性PsAを有する対象におけるゴリムマブ2mg/kgのIV投与の有効性を評価することである。
副次的目的は、IVゴリムマブについて以下を評価することである。
・乾癬皮膚病変、身体機能、健康関連の生活の質、及び他の健康結果の改善に関連する有効性
・構造損傷の進行の阻害
・安全性
・薬物動態(PK)、薬力学(PD)、及び免疫原性
研究の主目的に対処するために、統計的仮説(代替仮説)は、ゴリムマブ2mg/kgが、主要有効性エンドポイントに基づき、活動性PsAを有する対象の徴候及び症状を低減する際に、プラセボよりも統計的に優れていることである。この研究の一次エンドポイントは、14週目に米国リウマチ学会基準(ACR20と呼ばれる)におけるベースラインからの20%の改善を達成する対象の割合である。このエンドポイントは、規制当局及び臨床的PsAコミュニティによって十分に受け入れられているために選択された。
試験デザインの概要
これは、活動性PsAを有する対象におけるプラセボと比較したIVゴリムマブの有効性及び安全性の3相多施設共同、無作為化、二重盲検、プラセボ対象研究である。約440人の対象が、約90の治験実施機関で無作為化される。対象は、0、4、12、及び20週目にゴリムマブ2mg/kg又はプラセボIV注入を受けるように無作為に割り当てられる。16週目に、早期離脱対象である全ての対象は、治験者によって選択されるような、以下の併用薬介入のうちの1つが認められる。それらのコルチコステロイド用量(最大総用量プレドニゾン10mg/日若しくは同等)、MTX用量(最大総用量25mg/週)、若しくはNSAID用量の増加、又はNSAID、コルチコステロイド(最大用量プレドニゾン10mg/日若しくは同等)、MTX(最大用量25mg/週)、SSZ(最大用量3g/日)、HCQ(最大用量400mg/日)、又はレフルノミド(最大投与量20mg/日)の開始。安定用量のこれらの薬剤への滴定は、24週目の訪問までに早期離脱対象である対象に対して完了されるべきである。
試験母集団
標的研究集団は、スクリーニング時に乾癬性関節炎に関する分類基準(CASPAR)27を満たす、少なくとも6か月間活動性PsAを有する、生物製剤を摂取していない対象である。
対象は、以下のような2つの処置群のうちの1つに、0週目に無作為化される。
・群1(n=220):IVプラセボ注入
・群2(n=220):IVゴリムマブ2mg/kg
この研究では、スクリーニング、二重盲検プラセボ対照、積極的治療、及び安全性フォローアップの4つのフェーズがある。最大6週間のスクリーニングフェーズは、スクリーニング研究評価を実施し、研究適格性を判断するのに十分な時間を可能にする。研究の第2のフェーズは、0週目から24週目までの二重盲検プラセボ対照フェーズである。研究の第3のフェーズは、24週目から52週目までの積極的治療フェーズである。研究の第4のフェーズは、安全性フォローアップフェーズであり、研究薬剤の最後の投与から8週間である。安全性フォローアップは、ゴリムマブの半減期の約5倍に相当する期間にわたって対象を監視することを可能にする。各対象の最初の処置割り当ては、60週間の試験全体にわたって施設及び対象に対して盲検化されている。この期間は、PsAに対する維持療法としてのIVゴリムマブの有効性及び安全性を実証するのに十分な時間を提供する。
無作為化は、処置群に対する対象の評価における偏りを最小限に抑え、既知及び未知の対象属性(例えば、人口統計学的及びベースライン特性)が処置群にわたって均等にバランスがとれるようにする可能性を高め、かつ処置群にわたる統計的比較の妥当性を高めるために使用される。加えて、この研究の2つの群は、地理的領域及びベースラインMTX使用(はい又はいいえ)に基づき階層化される。
この研究のために選択された有効性評価は、PsAの治療のための治療用生物学的薬剤の以前の試験において確立された。このために選択された患者報告アウトカム(PRO)は、PsAにおける他の研究に関する医療文献及び適用可能なUS/EU規制ガイダンス文書で受け入れられている臨床的に関連する測定値と一致する。
・対象の疼痛評価
・対象の疾患の包括的評価
・医師の疾患の包括的評価
・関節評価(腫脹関節数及び圧痛関節数)
・健康評価質問表の障害指標(HAQ−DI)
・乾癬の面積及び重症度指標(Psoriasis Area and Severity Index、PASI)
・手及び足のX線写真
・36項目ショートフォーム健康調査(SF−36)
・指炎評価
・腱付着部炎評価
・Bath強直性脊椎炎疾患活動性指標(BASDAI)
・修正されたNAPSI
・皮膚科学的生活の質指標(DLQI)
・慢性疾患療法の機能評価(FACIT)−疲労
・労働生産性に関する質問票(WLQ)
・生産性VAS
・EuroQol−5D(EQ−5D)質問票
研究に適格な対象は、研究薬剤の初回投与前少なくとも6か月間、PsAと診断された18歳以上の男性又は女性であり、スクリーニング時にCASPAR基準を満たす。適格名対象のためのスクリーニングは、試験薬の投与前6週間以内に実施される。この研究に対象を登録するための組み入れ及び除外基準は、以下の2つのサブセクションに記載される。以下の組み入れ又は除外基準について疑問がある場合、治験責任医師は、この研究に対象を登録する前に適切な治験依頼者担当者に相談しなければならない。
可能性のある対象はそれぞれ、この研究に登録されるために以下の基準の全てを満たさなければならない。
1.対象は、18歳以上の男性又は女性でなければならない。
2.対象は、スクリーニングで実施される身体検査、病歴、バイタルサイン、及び12誘導心電図(ECG)に基づき、医学的に安定でなければならない。この判定は、対象のソースドキュメントに記録され、治験責任医師によって頭文字で略式署名されなければならない。
3.対象は、スクリーニングで実施される臨床検査室試験に基づき、医学的に安定でなければならない。肝臓酵素又は血液学を含む血清化学パネルの結果が正常な基準範囲外である場合、対象は、治験責任医師が異常又は正常からの逸脱が臨床的に有意ではないか、又は研究中の集団に対して適切かつ妥当であると判断した場合にのみ含まれ得る。この判定は、対象のソースドキュメントに記録され、治験責任医師によって頭文字で略式署名されなければならない。組み入れ基準番号5b及び番号18に記載される試験について、結果は、組み入れ基準番号5b及び番号18において許容される適格性範囲内でなければならない。
4.研究薬剤の初回投与の少なくとも6か月前にPsAを有しており、スクリーニング時にCASPAR基準を満たしていること。
5.以下によって定義されるように、活動性PsAと診断されていること。
a.スクリーニング及びベースラインにおいて5つ以上の腫脹関節及び5つ以上の圧痛関節
及び
b.スクリーニング時のC反応性タンパク質(CRP)≧0.6mg/dL。
6.PsAサブセット:DIP関節障害、リウマチ結節が存在しない多発性関節炎、離断性関節炎、非対称性末梢関節炎、又は末梢関節炎を伴う脊椎炎のうちの少なくとも1つを有すること。
7.活動性尋常性乾癬を有するか、又は尋常性乾癬の文書化された履歴を有すること。
8.現在又は以前のDMARD及び/又はNSAID療法にもかかわらず、活動性PsAを有すること。DMARD療法は、DMARDを少なくとも3か月間服用すること、又はDMARD不耐性の証拠として定義される。NSAID療法は、NSAIDを少なくとも4週間服用すること、又はNSAID不耐性の証拠として定義される。
9.無作為化の前に、女性は、以下のいずれかでなければならない。
・妊娠する可能性がないこと;初経前ではないこと;閉経後(少なくとも12か月の無月経を有する45歳超)ではないこと;永久的に不妊ではないこと(例えば、卵管閉塞、子宮摘出、両側卵管切除);又はそうでなければ妊娠不能ではないこと。
・妊娠する可能性があり、かつ臨床研究に参加している対象に対する受胎調節方法の使用に関する地方条例に従う受胎調節の非常に効果的な方法:例えば、避妊の経口、注射、又は埋め込み式ホルモン方法の確立された使用を実施していること;子宮内避妊用具(IUD)又は子宮内システムの配置;バリア法:殺精子フォーム/ゲル/フィルム/クリーム/坐剤付きのコンドーム、又は殺精子フォーム/ゲル/フィルム/クリーム/坐剤付きの閉塞キャップ(ペッサリー若しくは子宮頚部/円蓋キャップ);男性パートナーの断種(精管切除したパートナーは、その対象の唯一のパートナーであるべきである);真の禁欲(これが対象の好ましく、かつ通常の生活習慣と一致する場合)。
10.妊娠する可能性のある女性は、スクリーニング時に陰性血清妊娠試験(β−ヒト絨毛性ゴナドトロピン[β−HCG])、及び無作為化前の0週目に陰性尿妊娠試験を受けなければならない。
11.女性は、研究中、及び研究薬物の最後の投与を受けた後4か月間、妊娠しないか、又は生殖補助の目的で卵子(卵子、卵母細胞)を提供しないと同意しなければならない。
12.妊娠する可能性のある女性との性行為に積極的であり、精管切除を受けていない男性は、研究中、及び研究薬剤の最後の投与を受けた後4か月間、受胎調節のバリア法、例えば、殺精子フォーム/ジェル/フィルム/クリーム/坐剤付きのコンドーム、又は殺精子フォーム/ジェル/フィルム/クリーム/座薬付きの閉塞キャップ(ペッサリー若しくは子宮頚部/円蓋キャップ)を有するパートナーのいずれかを使用することに同意しなければならない。また、男性は全員、研究中及び研究薬剤の最後の投与を受けた後4か月間、精液を提供してはならない。
13.以下の結核(tuberculosis、TB)スクリーニング基準に従って適格であると考えられること:
a.スクリーニング前に、潜在性又は活動性TBの履歴がないこと。潜在性TBの履歴を有し、潜在性TBの治療を現在受けている対象については例外が認められ、研究薬剤の初回投与前に潜在性TBのための治療を開始するか、又は研究薬剤の初回投与前5年以内に潜在性TBのための適切な治療を完了したという文書を有すること。
b.医療履歴及び/又は身体的検査の際に活動性TBを示唆する徴候又は症状を有しないこと。
c.活動性TBを有する人と最近密接な接触がなかったこと、又はそのような接触があった場合は、TB専門医師に紹介して追加評価を受け、保証された場合は、研究薬剤の初回投与前に、潜在性TBのための適切な治療を受けること。
d.研究薬剤の初回投与前6週間以内に、陰性のQuantiFERON(登録商標)−TB Gold試験結果を有するか、又は活動性TBが除外され、潜在性TBのための適切な治療が研究薬剤の初回投与前に開始されている、新たに同定された陽性のQuantiFERON(登録商標)−TB Gold試験結果を有すること。QuantiFERON(登録商標)−TB Gold試験がその国において承認/登録されていないか、又はツベルクリン皮膚検査(TST)が現地の保健機関によって命じられている場合、研究薬剤の初回投与前6週間以内に、陰性TST、又は活動性TBが除外され、潜在性TBのための適切な治療が研究薬剤の初回投与前に開始されている、新たに同定された陽性TSTが更に必要とされる。
i.持続的に不確定なQuantiFERON(登録商標)−TB Gold試験結果を有する対象は、潜在的TBが除外され、彼らの胸部X線写真がTB(活動性又は陳旧性、非活動性TB)を示唆する異常を示さず、対象が治験責任医師によって判断されるようなTBの更なる危険因子を有しない場合、潜在性TBのための治療なしに登録され得る。
ii.QuantiFERON(登録商標)−TB Gold試験及びTSTは、潜在性TBの履歴及び潜在性TBのための進行中の治療、又は上記のように十分な治療を完了したという文書を有する対象に対しては、スクリーニング時に必要とされない。上記のように十分な治療を完了したという文書を有する対象は、潜在性TBのための更なる治療を開始する必要はない。
e.研究薬剤の初回投与前3か月以内に撮影され、適格な有資格放射線科医によって読み取られた胸部X線写真(後−前像)を有し、現在の活動性TB又は陳旧性、非活動性TBの兆候がないこと。
14.MTXを使用する場合、対象は、研究薬剤の初回投与の少なくとも3か月前に25mg/週を超えない用量で治療を開始しなければならず、MTXに起因する重大な毒性副作用を有してはいけない。メトトレキサートの投与経路及び用量は、研究薬剤の初回投与前少なくとも4週間、安定であるべきである。MTXを現在使用していない場合、研究薬剤の初回投与前少なくとも4週間、MTXを受けていてはならない。
15.NSAID又はPsAのための他の鎮痛剤を使用する場合、研究薬剤の初回投与前少なくとも2週間、安定な用量でなければならない。NSAID又はPsAのための他の鎮痛剤を使用していない場合、研究薬剤の初回投与前少なくとも2週間前、NSAID又はPsAのための他の鎮痛剤を受けていてはならない。
16.経口コルチコステロイドを使用する場合、対象は、研究薬剤の初回投与前少なくとも2週間、≦10mgのプレドニゾン/日に相当する安定用量でなければならない。現在、経口コルチコステロイドを使用していない場合、対象は、研究薬剤の初回投与前少なくとも2週間、経口コルチコステロイドを受けていてはならない。
17.研究中に長時間の日光曝露を回避しなければならず、日焼け室又は他の紫外線源を使用してはならない。
18.以下のパラメータ内のスクリーニング検査結果を有すること:
a.ヘモグロビン≧8.5g/dL
b.白血球≧3.5×103/μL
c.好中球≧1.5×103/μL
d.血小板≧100×103/μL
e.血清クレアチニン≦1.5mg/dL
f.AST、ALT、及びアルカリホスファターゼのレベルは、試験を実施する実験室のULN範囲の1.5倍以内でなければならない。
19.対象は、このプロトコルに指定された禁止事項及び制限事項を守る意思があり、それが可能でなければならない。
20.それぞれの対象は、各自が研究の目的とそれに必要な手順を理解し、研究に参加する意思があることを示す、インフォームドコンセントフォーム(ICF)に署名する必要がある。
21.各対象は、研究のために任意選択的なDNA試料を提供することに合意する場合(地域の規制により認められている場合)、別個のインフォームドコンセントフォームに署名しなければならない。任意選択的なDNA研究試料に対して承諾を拒否しても、この研究のへの参加から対象を除外することはない。
22.初回の研究薬剤投与前2週間以内、及び研究期間全体にわたって、アーユルヴェーダ療法医学、漢方薬(複数可)、及び鍼治療を含む補完医療の使用を控える意思があること。
以下の基準のうちのいずれかを満たす任意の潜在的な対象は、この研究の参加から除外される:
1.RA、AS、全身性エリテマトーデス、又はライム病が挙げられるがこれらに限定されない、ゴリムマブ療法の利益の評価を混乱させる可能性がある他の炎症性疾患を有する場合。
2.研究に登録されている間、又は研究薬剤の最後の投与を受けた後4か月以内に、妊娠、授乳、又は妊娠若しくは父親になることを計画している場合。
3.インフリキシマブ、エタネルセプト、アダリムマブ、ゴリムマブ、及びセルトリズマブペゴルが挙げられるがこれらに限定されない、TNFαを低減するために標的とされる任意の生物学的薬剤を使用していた場合。
4.トシリズマブをこれまでに受けていた場合。
5.クロラムブシル、シクロホスファミド、ナイトロジェンマスタード、又は他のアルキル化剤を含む、細胞毒性薬物をこれまでに使用していた場合。
6.ナタリズマブ、エファリズマブ、又はB若しくはT細胞を枯渇させる薬剤(例えば、リツキシマブ、アレムツズマブ、若しくはビシリズマブ)をこれまでに受けていた場合。
7.アレファセプトをこれまでに受けていた場合。
8.アバタセプトをこれまでに受けていた場合。
9.トファシチニブ又は任意の他のヤヌスキナーゼ阻害剤(Janus kinase inhibitor、JAK)の阻害剤をこれまでに受けていた場合。
10.ウステキヌマブをこれまでに受けていた場合。
11.抗IL17療法(例えば、ブロダルマブ、イキセキズマブ、及びセクキヌマブ)をこれまでに受けていた場合。
12.ヒト免疫グロブリン又はゴリムマブ若しくはその賦形剤に対する既知のアレルギー、過敏症、又は不耐性。
13.研究薬剤の初回投与前4週間以内に、MTX以外の任意の全身性免疫抑制薬又はDMARDを受けていた場合。これらの分類の薬剤としては、スルファサラジン(SSZ)、ヒドロキシクロロキン(HCQ)、アザチオプリン、シクロスポリン、ミコフェノール酸モフェチル、金、及びペニシラミンが挙げられるが、これらに限定されない。
14.研究薬剤の初回投与前4週間以内にレフルノミドを受けているか(薬物消失手順を受けることに関係なく)、又は研究薬剤の初回投与前3か月以内にレフルノミドを受けており、薬物排除手順を受けていない場合。
15.研究薬剤の初回投与前4週間以内に、乾癬又は皮膚評価に影響を及ぼし得る任意の全身性薬剤/治療(注射可能なコルチコステロイド、レチノイド、1,25ジヒドロキシビタミンD3及び類似体、プソラレン、スルファタラジン、ヒドロキシ尿素、フマル酸誘導体、又は光線療法が挙げられるが、これらに限定されない)を受けていた場合。
16.任意の研究薬剤の初回投与前2週間以内に、乾癬又は皮膚評価に影響を及ぼし得る局所用薬剤/治療(コルチコステロイド、アントラリン、カルシポトリエン、局所用ビタミンD誘導体、レチノイド、タザロテン、メトキサレン、トリメチルプソラレン、ピメクロリムス、及びタクロリムスが挙げられるが、これらに限定されない)を使用していた場合。
17.研究薬剤の初回投与前4週間の間に、副腎皮質ホルモンを含む、硬膜外、関節内、IM、又はIVコルチコステロイドを受けていた場合。
18.現在リチウムを受けているか、又は研究薬剤の初回投与前4週間以内にリチウムを受けていた場合。
19.研究薬剤の初回投与前3か月以内、研究中、又は研究薬剤の最後の投与後3か月以内に、任意の生ウイルス又は細菌ワクチン接種を受けていたか、又は受けることが予想される場合。
20.慢性腎感染症、慢性胸部感染症(例えば、気管支拡張症)、副鼻腔炎、再発性尿路感染症(例えば、再発性腎盂腎炎)、開放、排膿、若しくは感染した皮膚創傷、又は潰瘍が挙げられるがこれらに限定されない、慢性又は再発性の感染症の履歴があるか又は進行中である場合。
21.感染した関節プロテーゼの履歴を有するか、又はそのプロテーゼが除去若しくは置き換えされていない場合、関節プロテーゼの疑いのある感染のための抗生物質をこれまでに受けていた場合。
22.研究薬剤の初回投与前2か月以内に、深刻な感染症(肝炎、肺炎、敗血症、若しくは腎盂腎炎が挙げられるが、これらに限定されない)を有していたか、又は感染のために入院していたか、又は感染のためのIV抗生物質で治療されていた場合。
23.スクリーニング前に、ヒストプラスマ症又はコクチジオイデス真菌症を含む、活動性肉芽腫性感染症の履歴を有する場合。潜在性TBの履歴を有する適格性に関する情報については組み入れ基準を参照のこと。
24.12か月間のスクリーニングのうちに、Bacille Calmette Guerin(BCG)ワクチン接種を有していた場合。
25.悪性腫瘍又はTBを含む現在の活動性感染症を示唆する異常を示す、研究薬剤の初回投与前3か月以内の胸部X線写真を有する場合。
26.スクリーニング前6か月以内に、非結核性抗酸菌症又は日和見感染症(例えば、サイトメガロウイルス、ニューモシスティス症、アスペルギロシス症)を有していた場合。
27.研究薬剤の初回投与前2か月以内に、帯状ヘルペス感染症を有するか、又は有していた場合。
28.対象は、ヒト免疫不全ウイルス(HIV)抗体陽性の履歴を有するか、又はスクリーニング時にHIV検査結果が陽性である。
29.B型肝炎感染症を有する場合。対象は、B型肝炎ウイルス(HBV)のスクリーニングを受けなければならない。少なくとも、これには、HBsAg(HBV表面抗原)、抗HBs(HBV表面抗体)、及び抗HBC合計(HBVコア抗体合計)のための試験が含まれる。
30.スクリーニング前に6か月の差で、2つの陰性HCV RNA試験結果を有し、かつスクリーニング時に第3の陰性HCV RNA試験結果を有しない限り、C型肝炎ウイルス(HCV)に対する抗体に対して血清陽性である対象。
31.重度、進行性、又は制御されていない腎臓、肝臓、血液、胃腸、内分泌、肺、心臓、神経系、脳、又は精神疾患の現在の徴候又は症状を有する場合。
32.医学的に制御された無症候性CHFを含む、同時のうっ血性心不全(CHF)の履歴を有する場合。
33.移植器官を有する場合(研究薬剤の初回投与前3か月を超えた角膜移植を除く)。
34.リンパ腫を含むリンパ増殖性疾患の既知の履歴、あるいは異常な大きさ若しくは位置のリンパ節腫脹、臨床的に有意な脾腫、又は意義不明の単クローン性ガンマグロブリン血症などの可能性のあるリンパ増殖性疾患を示唆する徴候及び症状既知を有する場合。
35.多発性硬化症又は視神経炎などの既知の脱髄疾患の履歴を有する場合。
36.対象は、スクリーニング前5年以内に悪性腫瘍の履歴を有する場合(例外は、最初の研究薬剤投与前少なくとも3か月間、再発の兆候なしで治療された皮膚の扁平上皮及び基底細胞癌、並びに外科的に治療された子宮頸部の上皮内癌である)。
37.対象は、計画された研究薬剤の初回投与前に、任意の許可されない療法、併用療法を受けていた。
38.対象は、5半減期又は3か月のどちらかより長い方のうちに治験薬(治験ワクチンを含む)を受けているか、又は計画された研究薬剤の初回投与前3か月以内に侵襲的治験医療デバイスを使用していたか、又は治験研究に現在登録されている。
39.対象は、治験責任医師の意見によって、参加が対象の利益を最優先にしていない(例えば、健康状態を損なう)か、又はプロトコル指定の評価を妨げる、制限する、若しくは混乱させ得るような任意の状況を有する。
40.対象は、スクリーニング前1か月以内に大手術(例えば、全身麻酔を必要とする)を受けていたか、又は手術から完全に回復していないか、又は対象が研究に参加することが予想される期間中、若しくは研究薬剤投与の最後の投与後1か月以内に手術を予定している。
41.不十分な忍容性、又は静脈への容易なアクセスが欠いているため、複数の静脈穿刺を受けることができないこと、又は受ける意思がない場合。
42.過去3か月以内に、薬物乱用(薬物又はアルコール)の問題があったことが既知である場合。
43.対象は、治験責任医師又は研究施設の指示に従って提案された研究又は他の研究に直接関与している、治験責任医師又は研究施設の雇用者、並びにこの雇用者又は治験責任医師の家族である。
潜在的な対象は、参加に適格であるために、研究の間に以下の禁止及び制限を順守する意思があり、かつそれが可能でなければならない。
1.妊娠する可能性がある異性との性行為に積極的な女性、及び子供の父親になることが可能な男性の両方が、非常に効果的な避妊方法を使用し、研究期間中、及び研究薬剤の最後の投与後4か月間、避妊の使用を継続することに同意しなければならない。
2.以下の薬物の使用は、IV研究薬剤投与と同時に許可されない。
・TNFαの低減を標的とした生物学的薬剤(インフリキシマブ、SCゴリムマブ、セルトリズマブペゴル、エタネルセプト、yisaipu、CT−P13[Remsima(登録商標)]、及びアダリムマブが挙げられるが、これらに限定されない)。
・IL−1ra(アナキンラ)
・トシリズマブ、又はIL−6若しくはIL−6受容体を標的とする任意の他の生物製剤
・トファシチニブ又は任意の他のJAK阻害剤
・B細胞枯渇剤(例えば、リツキシマブ)
・シクロホスファミド、クロラムブシル、ナイトロジェンマスタードなどの細胞毒性薬物、又は
・他のアルキル化剤
・アバタセプト
・ウステキヌマブ
・抗IL−17剤(例えば、ブロダルマブ、セクキヌマブ、及びイキセキズマブ)
・治験薬
3.以下の薬物の使用は許可されない:SSZ、HCQ、アザチオプリン、経口シクロスポリンA、タクロリムス、ミコフェノール酸モフェチル、レフルノミド、経口又は非経口金を含む、全身性免疫抑制薬又はDMARD(MTX以外)。唯一の例外は、16週目の早期離脱に適格な対象に対するSSZ、HCQ、又はレフルノミドの使用である。
4.研究中に生ウイルス又は生細菌ワクチン接種を受けないことに同意しなければならない。対象はまた、研究薬剤の最後の投与を受けた後3か月間、生ワクチンを受けないことにも同意しなければならない。12か月間のスクリーニングのうちに、Bacille Calmette Guerin(BCG)ワクチン接種を有していてはならない。
5.この研究のための研究薬剤以外の治験医療デバイス又は治験薬を受けないことに同意しなければならない。
6.アスピリン及び選択的シクロオキシゲナーゼ(COX)−2阻害剤を含むNSAID、並びに他の鎮痛剤で治療された対象は、研究が行われている国で承認された通常の市販用量を受けるべきである。NSAID及び他の鎮痛剤の処方は、研究薬剤の初回投与前少なくとも2週間、及び24週目まで調節されるべきではなく、対象が許容できない副作用を生じた場合にのみ変更され得る。24週目の後から52週目まで、単回用量の減少が許可される。そうでなければ、NSAID及び他の鎮痛剤の処方は、対象が許容できない副作用を生じた場合にのみ変更され得る。16週目に、早期離脱に適格な対象は、NSAIDの単回開始、又は彼らのNSAID用量の増加を有し得る。
カプサイシン及びジクロフェナクを含む局所鎮痛剤の使用が許可される。
7.経口コルチコステロイドで治療された対象は、彼らの研究薬剤の初回投与前少なくとも2週間、1日あたり10mg以下のプレドニゾンに相当する安定用量を受け、この用量を24週目まで受け続けるべきである。24週目の後及び52週目まで、経口コルチコステロイドの単回用量の減少が許可される。そうでなければ、経口コルチコステロイドの用量及び種類は、対象が許容できない副作用を生じた場合にのみ、治験責任医師の裁量で変更され得る。16週目に、早期離脱に適格な対象は、彼らの経口コルチコステロイド用量の単回開始又は増加を有し得る(10mg/日のプレドニゾンの最大総用量又は同等)。
コルチコステロイドの硬膜外、IM、又はIV投与は、研究薬剤の初回投与前4週間以内には許可されず、研究全体を通してPsAの治療のために許可されない。PsA以外の兆候のための研究中の硬膜外、IM、及びIVコルチコステロイドの使用を避けるために、あらゆる試みが行われるべきである。PsA以外の兆候のための長期(2週間超)の経口又はIVコルチコステロイドは、研究全体を通して許可されない。PsA以外の兆候に使用される短期(≦2週間)の経口、IV、IM、又は硬膜外コルチコステロイドは、治療医師の意見によって適切な代替物がない状況に限定されるべきである。
関節内ステロイドは、研究薬剤の初回投与前4週間以内に投与されるべきではない。特に研究の最初の24週間の間、関節内コルチコステロイド注射を避ける試みが行われなければならない。しかしながら、必要であれば、対象は、60週間の研究の間、2つ以下の影響を受けた部位に最大2つの関節内、腱鞘、又は嚢のコルチコステロイド注射を受けてもよい。
8.伝統的な医学(例えば、漢方、鍼治療、アーユルヴェーダ療法医学)が挙げられるがこれらに限定されない、PsA疾患活動性又は評価に影響を及ぼし得る補完療法の使用は、60週目まで禁止されている。
適格な対象は、盲検様式で、0週目に一定用量のゴリムマブ2mg/kg又はプラセボを受けるために、自動ウェブ応答システム(IWRS)を使用して無作為に割り当てられる。処置群への対象割り当ては、2つの処置群のうちの1つに1:1の比で、階層化ブロック無作為化法を使用して行われる。層別因子は、地理的領域及びベースラインMTX使用(はい又はいいえ)である。これにより、ベースラインMTX使用と、各地理的領域内の対象の数に対する相対的な処置バランスを確実にする。
投与レジメン及び盲検化
研究薬剤の第1の注入前に、対象は、以下の2つの処置群のうちの1つに1:1の比で無作為に割り当てられる。
群I(n=220):対象は、0、4、12、及び20週目にIVプラセボ注入を受ける。対象は、24週目にIVゴリムマブ2mg/kgに交差し、24、28週目、及びその後q8wに投与を受ける。
群II(n=220):対象は、0、4週目、及びその後q8wにIVゴリムマブ2mg/kgを受ける。対象は、盲検を維持するために、24週目にIVプラセボ注入を受ける。
注記:全ての注入は、30±10分間かけて完了する。
16週目に、腫脹関節数及び圧痛関節数の両方におけるベースラインからの5%未満の改善を有する群I及びIIにおける全ての対象が、二重盲検様式で早期離脱に入る。16週目に、早期離脱対象である全ての対象は、治験者によって選択されるような、以下の併用薬介入のうちの1つが認められる。それらのコルチコステロイド用量(最大総用量プレドニゾン10mg/日若しくは同等)、MTX用量(最大総用量25mg/週)、若しくはNSAID用量の増加、又はNSAID、コルチコステロイド(最大用量プレドニゾン10mg/日若しくは同等)、MTX(最大用量25mg/週)、SSZ(最大用量3g/日)、HCQ(最大用量400mg/日)、又はレフルノミド(最大投与量20mg/日)の開始。安定用量のこれらの薬剤への滴定は、24週目の訪問までに早期離脱対象である対象に対して完了されるべきである。
全てのベースライン後訪問は、指示された週±4日に行われ得る4週目、12週目、14週目、16週目、及び24週目の訪問を除き、研究全体を通して指定された週±7日に行われ得る。推奨される許容可能な時間帯が順守できない場合、治験依頼者には、訪問を予定に入れる前に連絡しなければならない。
24週目まで、又は以下のセクションで指定されるように、対象の併用薬を安定した状態に保持するためにあらゆる努力をするべきである。併用薬用量は、低減され得るか、又は投薬治療は、異常な検査値、副作用、併発症、又は外科処置の性能を理由に一時的に中断され得るが、変化及び変化の理由は、対象の医療記録に明確に文書化されるべきである。
対象は、安定用量のMTXを服用して研究に入ることが許可される。
PsAのために経口コルチコステロイドで治療された対象は、研究薬剤の初回投与前少なくとも2週間、1日あたり≦10mgのプレドニゾンに相当する安定用量を受け、この用量を60週目まで受け続けるべきである。ベースラインにおいて経口コルチコステロイドで治療されていない対象は、研究薬剤の初回投与の少なくとも2週間前に経口コルチコステロイドを中断していなければならず、彼らは、60週目まで経口コルチコステロイドを受けてはならない。
安定用量のNSAID及び他の鎮痛剤の使用が許可される。
MTXを除いて、疾患修飾性抗リウマチ薬/全身性免疫抑制薬は、研究薬剤の初回投与の少なくとも4週間前に中断されなければならず、60週目まで禁止されている。これらのDMARDとしては、SSZ、HCQ、金製剤、ペニシラミン、及びレフルノミドが挙げられるが、これらに限定されない。対象が研究薬剤の初回投与前3か月以内にレフルノミドを受けた場合、対象は、薬物排除手順を受けていなければならない。
生物学的薬剤(例えば、SCゴリムマブ、アナキンラ、エタネルセプト、アダリムマブ、インフリキシマブ、アレファセプト、エファリズマブ、リツキシマブ、ナタリズマブ)、細胞毒性剤(例えば、クロラムブシル、シクロホスファミド、ナイトロジェンマスタード、他のアルキル化剤)、又は治験薬の使用は、60週間の研究中に許可されない。これらの薬剤のいずれかが使用される場合、対象は、更なる研究薬剤の注入から中断される。
アーユルヴェーダ療法医学、漢方薬、又は鍼治療などの非薬物療法を含む補完療法の使用は、60週間の研究中に許可されない。
乾癬に対する局所用薬剤/治療(例えば、コルチコステロイド角質溶解薬[研究全体を通して許可されるサリチル酸シャンプーを除く]、コールタール[研究全体を通して許可されるコールタールシャンプーを除く]、アントラリン、ビタミンD3類似体、又は局所用タロリムス、及びレチノイド)の同時使用は、24週目まで許可されない。
乾癬に対する全身療法の同時使用(例えば、紫外線A[PUVA]とソラレン、全身性レチノイド、シクロスポリン、又はタクロリムス)は、60週目まで許可されない。全身性抗乾癬療法の使用は、研究薬剤の初回投与の少なくとも4週間前に中断されなければならない。
研究手順
概論
妊娠する可能性がある女性に対してのみ、治験責任医師によって必要と判断されるか、又は地方規制によって必要とされる場合、追加の血清又は尿妊娠試験が実施されて、対象の研究への参加中の任意の時点で妊娠がないことを確証することができる。また、追加のTB試験は、治験責任医師によって必要と判断されるか、又は地方規制によって必要とされる場合に実施され得る。
書面によるインフォームドコンセントが得られた後、かつ無作為化前6週間の期間内に、全てのスクリーニング評価が実施される。スクリーニング訪問は、1回を超える訪問に分割され得る。例えば、インフォームドコンセントを得た後、治験責任医師は、最初の訪問において全ての検査室試験を完了する。次いで、対象が中央検査室試験結果によって判定されたように研究に適格である場合にのみ、対象は、スクリーニング手順の残りの部分に戻る。組み入れ基準の全てを満たし、かつ除外基準のいずれも満たさない対象が、研究に登録される。各対象について、研究の時間及びイベントスケジュールに順守するためにあらゆる努力をするべきである。対象は、研究の任意選択的な薬理ゲノム学的研究要素に参加するために、別個の書面の薬理ゲノム学的インフォームドコンセントを提供しなければならない。
異常スクリーニング検査室血液試験及び除外につながるCRPレベルの再試験は、スクリーニング期間中に予定されていない訪問を使用して1回のみ許可される(適格性を再評価するために)。
治療フェーズは、プラセボ対照及び積極的治療フェーズを含む。0週目に、適格な対象は、2つの処置:ゴリムマブIV2mg/kg又はプラセボIVのうちの1つを受けるように無作為に割り当てられる。
乾癬性関節炎応答評価
関節評価
68個の関節の各々が圧痛に関して評価され、66個の関節の各々が腫脹に関して評価される(股関節は、腫脹に関して除外される)。全ての関節は、時間及びイベントスケジュールに示されるように、訪問時に検査される。
関節は、関節を評価することが物理的に不可能である場合にのみ(即ち、ギプスによりアクセス不可能な関節、切断により存在しない関節、アクセスを不可能にするように変形した関節)、IJAによって「評価不可能」と指定されるべきである。他の全ての場合において、IJAは、圧痛及び腫脹に関して各関節を評価するべきである(股関節は、腫脹に関して除外される)。これは、以前の手術を可視的に示すいかなるもの(例えば、瘢痕)、又は彼らが有し得る対象の以前の関節処置/注射の認識(例えば、対象が研究参加前にIJAの患者であった場合)にかかわらず、完了されるべきである。
米国リウマチ学会レスポンスは、複数の疾患評価基準の改善の数値測定として提示される。例えば、ACR20応答10は、以下のように定義される。
1.腫脹関節数(66個の関節)及び圧痛関節数(68個の関節)の両方におけるベースラインからの20%以上の改善、
並びに
2.以下の5つの評価のうちの3つにおけるベースラインからの20%以上の改善:
・患者の疼痛評価(VAS)
・患者の疾患活動性の包括的評価(VAS)
・医師の疾患活動性の包括的評価(VAS)
・患者のHAQ−DIによって測定された身体機能の評価
・CRP
指炎の存在及び重症度は、0〜3のスコアリングシステム(0−指炎なし、1−軽度の指炎、2−中程度の指炎、及び3−重篤な指炎)を使用して両手及び両足において評価される。15,16
腱付着部炎は、Leeds腱付着部炎指標(Leeds Enthesitis Index、LEI)を使用して評価される。18LEIは、PsAを有する対象における腱付着部炎を評価するために開発され、以下の腱付着部に局所的圧力を加えることによって疼痛の有無を評価する。
・肘外側上顆、左及び右
・肘内側上顆、左及び右
・アキレス腱付着部、左及び右
修正総van der Heijdeシャープ(vdH−s)スコアは、手のDIP関節の追加、並びにペンシルインカップ及び全体的な骨溶解変形の評価によって、PsA放射線損傷評価の目的のために修正された元のvdH−Sスコア28である。関節びらんスコアは、手の40個の関節及び足の12個の関節におけるびらん重症度の要約である。各手関節は、関与する表面積に応じて、びらんなしを示す0から関節骨の半分超からの骨の広範な損失を示す5までスコアリングされる。足関節の各側面は、このスケールで等級分けされるため、足関節に対する最大びらんスコアは、10である。したがって、最大びらんスコアは、320である。関節裂隙狭小化(JSN)スコアは、手の40個の関節及び足の12個の関節におけるJSNの重症度を要約する。JSNの評価は、0〜4にスコアリングされ、0は、JSNなしを示し、4は、関節裂隙の完全な損失、骨性強直症、又は完全な脱臼を示す。したがって、最大JSNスコアは、208であり、528は、PsAに対する考え得る最悪の修正総vdH−Sスコアである。
対象の機能状態は、HAQ−DIによって評価される。13この20問の計器は、8つの機能領域(身支度、起床、食事、歩行、衛生、伸展、握力、及び日常生活活動)におけるタスクの達成で人が感じる困難の程度を評価する。各機能領域における応答は、困難なしを示す0からその領域内でタスクを実行することが不可能であることを示す3までスコアリングされる(即ち、より低いスコアは、より良好な機能を示す)。評価の特性は、評価されており、PsAにおけるその妥当性が決定されている。19また、対象の疾患の変化に応答することが示されている。22PsAにおいて、0.30のスコアの減少は、有意義な改善を示すと決定されている。21
PsA最小疾患活動性(MDA)基準は、PsAで使用される7つの結果尺度の複合体である。対象は、7つの結果尺度のうちの5つを満たした場合、MDAを達成することと分類される:圧痛関節数1以下;腫脹関節数1以下;乾癬活動性及び重症度指数1以下又は身体表面積3以下;15以下の患者の疼痛ビジュアルアナログスケール(VAS)スコア;20以下の患者の包括的疾患活動性VASスコア;健康評価質問表(HAQ)スコア0.5以下;及び圧痛腱付着部点1以下。6
医学的アウトカム研究健康指標SF−36質問票は、Rand Health Insurance Experimentの一部として開発され、8つの多項目スケールからなる。
・健康問題による身体機能の制限;
・身体健康問題による日常的役割の活動の制限;
・身体の痛み;
・全般的な精神的健康状態(心理的苦痛及び健康);
・個人的又は感情的問題による日常的役割の活動の制限;
・身体的又は精神的健康問題による社会的機能の制限;
・活力(エネルギー及び疲労);
・全般的な健康状態の認識。
乾癬の面積及び重症度指標
PASIは、乾癬病変の重症度及び療法に対するそれらの応答を評価及び等級分けするために使用されるシステムである。12PASIは、0〜72の範囲であり得る数値スコアを生成する。PASI50応答は、ベースラインからのPASIスコアの50%以上の改善として定義され、PASI75及びPASI90は、同様に定義される。
主エンドポイント
この研究の主エンドポイントは、14週目にACR20応答を達成する対象の割合である。
以下の主要セカンダリーエンドポイントは、以下に指定されるような重要度順で列挙される。
1.14週目のHAQ−DIスコアにおけるベースラインからの変化。
2.14週目にACR50応答を達成する患者の割合。
3.14週目にPASI75応答を達成する対象の割合(ベースラインの3%以上のBSA乾癬関与を有する)。
4.24週目の修正総vdH−Sスコアにおけるベースラインからの変化。
制御されたセカンダリーエンドポイント(多様性に対するタイプIエラー率の制御を伴う)。
以下の制御されたセカンダリーエンドポイントは、主エンドポイント及び主要セカンダリーエンドポイントに加えて分析され、以下に指定されるような重要度順で列挙される。
1.ベースラインにおいて腱付着部炎を有する対象における、14週目の腱付着部炎スコアにおけるベースラインからの変化。
2.ベースラインにおいて指炎を有する対象における、14週目の指炎スコアにおけるベースラインからの変化。
3.14週目のSF−36PCSにおけるベースラインからの変化。
4.24週目にACR50応答を達成する患者の割合。
5.14週目にACR70応答を達成する患者の割合。
6.14週目のSF−36MCSにおけるベースラインからの変化。
主エンドポイント、主要セカンダリーエンドポイント、及び制御されたセカンダリーエンドポイントに加えて、以下のエンドポイントが評価される。
1.2週目にACR20応答を達成する患者の割合。
2.経時的に、ACR20、ACR50、ACR70、及びACR90応答を達成する対象の割合。
3.経時的なACR応答の成分におけるベースラインからの変化。
4.経時的に、ACR応答の各成分において≧20%、≧50%、≧70%、及び≧90%の改善を達成する対象の割合。
5.経時的なHAQ−DIスコアにおけるベースラインからの変化。
6.経時的に、HAQ−DIスコアにおいてPsA対象に対する臨床的に有意義な改善(0.3以上の改善)を達成する対象の割合。
7.経時的な、ベースラインにおいて指炎を有する対象における指炎のベースラインからの変化、及び指炎がある指を有する対象の割合。
8.経時的な、ベースラインにおいて腱付着部炎を有する対象における腱付着部炎スコアにおけるベースラインからの変化、及び腱付着部炎を有する対象の割合。
9.24週目にACR20応答を達成した対象における、52週目にACR応答を達成する対象の割合。ACR50、70、及び90応答者に対する同様のエンドポイントも評価される。
10.24週目にHAQ−DI応答を達成した対象における、52週目にHAQ−DI応答を達成する対象の割合(HAQ−DIスコアの0.3以上の改善を達成する対象)。
11.経時的なMDAを達成する対象の割合。
1.ベースラインにおいて3%以上のBSA乾癬皮膚関与を有する対象については、全体的に、かつベースラインMTX使用によって、経時的にベースラインからのPASIの50%以上、75%以上、90%以上、及び100%の改善を達成する対象の割合。
2.ベースラインにおいて3%以上のBSA乾癬皮膚関与を有する対象については、経時的なPASIのベースラインからの改善。
3.ベースラインにおいて3%以上のBSA乾癬皮膚関与を有する対象については、経時的にPASI75及びACR20応答の両方を達成する対象の割合。
4.ベースラインにおいて3%以上のBSA乾癬皮膚関与を有する対象については、経時的にPASI50及び5以上のDLQIの改善の両方を達成する対象の割合。
5.ベースラインにおいて3%以上のBSA乾癬皮膚関与を有する対象については、経時的にPASI75及び修正されたPsARC応答の両方を達成する対象の割合。
構造損傷エンドポイントについては、2つの読み取りキャンペーンが存在し、読み取りキャンペーン1は、24週目における分析に寄与し、読み取りキャンペーン2は、52週目における分析に寄与する。
1.24週目に0以下の修正総vdH−Sスコアにおけるベースラインからの変化を有する対象の割合。
2.24週目及び52週目の修正総vdH−Sスコアにおけるベースラインからの変化。
3.0週目から24週目、24週目から52週目の修正総vdH−Sスコアにおける変化。24週目及び52週目の領域(手、足)による修正総vdH−Sスコアにおけるベースラインからの変化。
4.24週目及び52週目の損傷の種類(びらん及びJSN)による修正vdH−Sスコアにおけるベースラインからの変化。
5.ベースライン、24週目、及び52週目に関節損傷のない状態(0の修正総vdH−Sスコア、0のびらんスコア、又は0のJSNスコア)を維持している対象の数。
6.24週目及び52週目に0以下又は0.5以下の修正総vdH−Sスコアにおけるベースラインからの変化を有する対象の数。
1.経時的なSF−36のPCSスコア及びMCSスコアにおけるベースラインからの変化。
2.経時的なSF−36スケールにおけるベースラインからの変化。
3.経時的に、5以上のSF−36PCSスコア改善を達成する対象の割合。
4.経時的に、5以上のSF−36MCSスコア改善を達成する対象の割合。
完了
対象は、研究の60週目に評価を完了した場合、研究を完了したと見なされる。いかなる理由でも研究処置を早期に中断する対象は、研究を完了したとは見なされない。
対象の研究処置が治療レジメンの終了前に中断されなければならない場合、これは、研究からの対象の自動脱退をもたらさない。
・研究期間内、又は最後の研究薬剤投与後4か月以内の妊娠及び計画された妊娠。
・人工呼吸器補助を必要とする喘鳴及び/若しくは呼吸困難を伴う気管支けいれんをもたらす反応、又は研究薬剤投与後に生じる症候性低血圧。
・研究薬剤の注入の1〜14日後に生じる、発熱及び/又は発疹を伴う筋肉痛及び/又は関節痛をもたらす反応(血清病を示唆し、他の認識された臨床的症候群の徴候及び症状を表すものではない)。これらは、掻痒、顔、手、又は唇の浮腫、嚥下障害、じんま疹、咽頭炎、及び/又は頭痛を含む他の事象を伴う場合がある。
・日和見感染症。
・非黒色腫皮膚癌を除く悪性腫瘍。
・CHF。
・脱髄疾患。
・対象は、以下のTBスクリーニング基準に従って不適格であると見なされる。
−活動性TBの診断が行われる。
−潜在性TBのための治療を受けている対象は、この治療を早期に中断するか、又は治療に不適合である。
−対象は、フォローアップ評価質問及び/若しくは身体検査に基づき活動性TBを示唆する症状を有するか、又は活動性TBを有する人と最近密接に接触していて、追加の評価を受け続けることができないか、若しくは続けない。
−継続評価を受ける対象は、活動性TBが除外され得、潜在性TBのための適切な治療が、研究薬剤の次の投与前に開始され、完了まで継続され得ない限り、現在の活動性TBの兆候を有する胸部X線写真、及び/又は陽性のQuantiFERON(登録商標)−TB Gold試験結果(及び/又はQuantiFERON(登録商標)−TB Gold試験が承認/登録されていないか、若しくはTSTが現地の保健機関によって命じられている国における陽性のTST結果)を有する。持続的に不確定なQuantiFERON(登録商標)−TB Gold試験結果を有する対象は、潜在的TBが除外され、彼らの胸部X線写真がTB(活動性又は陳旧性、非活動性TB)を示唆する異常を示さず、対象が治験責任医師によって判断されるようなTBの更なる危険因子を有しない場合、潜在性TBのための治療なしに継続し得る。この判定は、治験依頼者のメディカルモニターに速やかに報告され、対象のソースドキュメントに記録され、治験責任医師によって頭文字で略式署名されなければならない。−潜在性TBのための治療を受けている対象は、この治療を早期に中断するか、又は治療に不適合である。
・プロトコル禁止薬剤の開始
・治験責任医師又は治験依頼者のメディカルモニターは、安全性の理由から、それが対象の最善の利益であると確信している。
以下の理由のいずれかで、対象は、研究から脱退させられる。
・追跡不能
・同意の撤回
・死亡
対象は、研究にとどまる一方で、任意選択的な研究試料について同意を撤回し得る。そのような場合、任意選択的な研究試料は破壊される。試料破壊プロセスは、上記のように進む。
対象は、研究のための試料の使用に対する同意を撤回し得る。そのような場合、試料は、それらがもはや臨床研究に必要とされなくなった後に破壊される。研究のための試料保持の詳細は、任意選択的な研究試料についての主要ICF及び別個のICFに提示される。
連続変数、並びに別個の変数の計数及び割合に対するn、平均、SD、中央値、IQ範囲、最小値、及び最大値などの単純な記述要約統計は、大部分のデータを要約するために使用される。
対象の人口統計学的データ(例えば、年齢、人種、性別、身長、体重)及びベースライン疾患特性(例えば、疾患の持続時間、関節数、及びCRP)は、処置群によって要約される。
試料サイズ推定値は、生物製剤のウステキヌマブ(治験依頼者によって開発された抗IL12/23モノクローナル抗体)を用いた治験依頼者の直近のPsA研究からのデータに基づく。活動性PsAを有する対象におけるウステキヌマブの第3相研究(CNTO1275PSA3001)は、最小CRP基準を含み、より現在に近いPsA集団を表す。CNTO1275PSA3001研究のACR20応答速度は、プラセボ、ウステキヌマブ45mg、及び90mgの処置群のそれぞれに対して24週目に22.8%、42.4%、及び49.5%であった。各処置群に220人で合計440人の対象は、カイ二乗検定を使用して、0.05の両側有意水準で、ゴリムマブ2mg/kg群における40%のACR20応答及びプラセボ群における20%の応答を仮定すると、14週目の処置群間の応答者の割合の有意差を検出するために99%の検出力を確実にする(表6)。
暫定分析は計画されていない。しかしながら、独立したデータ監視委員会(DMC)が安全性データを定期的にレビューして、対象の安全性を監視する。
主エンドポイント分析
主エンドポイントは、14週目にACR20応答を達成する対象の割合である。
以下の主要な二次分析は、以下に指定されるような重要度順で実施される。
1.14週目のHAQ−DIスコアにおけるベースラインからの変化が要約され、処置群間で比較される。
2.14週目にACR50応答を達成する対象の割合が要約され、処置群間で比較される。
3.14週目にPASI75応答を達成する対象の割合(ベースラインの3%以上のBSA乾癬関与を伴う)が要約され、処置群間で比較される。
4.24週目の修正総vdH−Sスコアにおけるベースラインからの変化が要約され、処置群間で比較される。
制御されたセカンダリーエンドポイント分析(多様性に対するタイプIエラー率の制御を伴う)
以下の有効性分析は、一次及び主要な二次分析に加えて実施される。
1.ベースラインにおいて腱付着部炎を有する対象における、14週目の腱付着部炎スコアにおけるベースラインからの変化が要約され、処置群間で比較される。
2.ベースラインにおいて指炎を有する対象における、14週目の指炎スコアにおけるベースラインからの変化が要約され、処置群間で比較される。
3.14週目のSF−36PCSにおけるベースラインからの変化が要約され、処置群間で比較される。
4.24週目にACR50応答を有する対象の割合が要約され、処置群間で比較される。
5.14週目にACR70応答を達成する対象の割合が要約され、処置群間で比較される。
6.14週目のSF−36MCSにおけるベースラインからの変化が要約され、処置群間で比較される。
徴候及び症状の低減並びに身体機能に関連した分析
以下のエンドポイントは、処置群によって要約される。要約は、エンドポイントの訪問が指定されていない場合、52週目まで経時的である。処置群間の比較は、24週目の前及び24週目の訪問時に行われる。
1.2週目にACR20応答を達成する対象の割合が、処置群によって要約され、群間で比較される。
2.24週目に、ACR20、ACR50、ACR70、及びACR90応答を達成した対象の割合。要約は、ベースラインMTX使用及び全体によって行われる。加えて、これらのエンドポイントはまた、帰属なしで観察されたデータを使用して要約される。
3.ACR応答の成分におけるベースラインからの変化率が、処置群間で14週目及び24週目に比較され、経時的に要約される。
4.HAQ−DIスコアにおけるベースラインからの変化が、経時的に各処置群について要約され、24週目に処置群間で比較される。
5.HAQ−DI応答者の割合(HAQ−DIスコアの≧0.3の改善を達成する対象)が、経時的に各処置群について要約され、14及び24週目に処置群間で比較される。
6.ベースラインにおいて指炎を有する対象における指炎のベースラインからの変化率、及び指炎がある指を有する対象の割合が、経時的に各処置群について要約され、24週目に処置群間で比較される。
7.ベースラインにおいて腱付着部炎を有する対象における腱付着部炎スコアにおけるベースラインからの変化率、及び腱付着部炎を有する対象の割合が、経時的に各処置群について要約され、24週目に処置群間で比較される。
8.24週目に応答者である対象における、52週目にACR20応答者である対象の割合が、処置群によって要約される。同様の要約が、ACR50、70、及び90応答者について実施される。
9.24週目に応答者である対象における、52週目にHAQ−DI応答者である対象(HAQ−DIスコアの≧0.3の改善を達成する対象)の割合が、処置群によって要約される。
10.MDAを達成する対象の割合が、経時的に各処置群について要約され、14及び24週目に処置群間で比較される。
以下の分析が実施される。
1.ベースラインにおける≧3%のBSA乾癬皮膚関与を有する対象について、ベースラインからのPASIにおける≧50%、≧75%、≧90%、及び100%の改善を達成する対象の割合が、全体的に、かつベースラインMTX使用によって、経時的に各処置群について経時的に要約され、14及び24週目に処置群間で比較される。
2.ベースラインにおける≧3%のBSA乾癬皮膚関与を有する対象について、PASIにおけるベースラインからの改善率が、経時的に各処置群について要約され、14及び24週目に処置群間で比較される。
3.ベースラインにおける3%以上のBSA乾癬皮膚関与を有する対象について、PASI75及びACR20応答の両方を達成する対象の割合が、経時的に各処置群について要約され、14及び24週目に処置群間で比較される。
24週目の分析は、読み取りキャンペーン1からのデータに対して実施され、52週目の分析は、読み取りキャンペーン2からのデータに対して実施される。
1.24週目に0以下の修正総vdH−Sスコアにおけるベースラインからの変化を有した対象の割合が要約され、処置群間で比較される。
2.24週目及び52週目の修正総vdH−Sスコアにおけるベースラインからの変化が、処置群によって、かつ早期離脱状態によって要約される。
3.24週目及び52週目の修正総vdH−Sスコアにおけるベースラインからの変化が、処置群間で比較される。
4.0週目〜24週目及び24週目〜52週目の修正総vdH−Sスコアにおける変化が、処置群によって、かつ早期離脱状態によって要約される。
5.領域(手、足)による修正総vdH−Sスコアにおけるベースラインからの変化は、処置群によって要約され、24週目及び52週目に処置群間で比較される。
6.損傷の種類(びらん及びJSN)による修正vdH−Sスコアにおけるベースラインからの変化が、処置群によって要約され、24週目及び52週目に処置群間で比較される。
7.関節損傷のない状態(0の修正総vdH−Sスコア、0のびらんスコア、又は0のJSNスコア)を維持している対象の数が、処置群によって要約され、24週目及び52週目に処置群間で比較される。
8.0以下又は0.5以下の修正総vdH−Sスコアにおけるベースラインからの変化を有する対象の数が、処置群によって要約され、24週目及び52週目に処置群間で比較される。
9.24週目及び52週目における修正総vdH−Sスコアにおけるベースラインからの変化の累積経験分布関数が提示される。
10.24週目及び52週目における読み取り者による修正総vdH−Sスコア、びらんスコア、及びJSNスコアにおけるベースラインからの変化が、処置群によって要約される。
以下の分析が実施される。
1.24週目のSF−36のPCSスコア及びMCSスコアにおけるベースラインからの変化が、処置群間で比較される。
2.SF−36のPCSスコア及びMCSスコアにおけるベースラインからの変化が、経時的に各処置群について要約される。
3.SF−36スケールにおけるベースラインからの変化が、経時的に各処置群について要約され、14及び24週目に処置群間で比較される。
4.≧5のSF−36PCSスコアの改善を達成する対象の割合が、経時的に要約され、14及び24週目に処置群間で比較される。
5.≧5のSF−36MCSスコアの改善を達成する対象の割合が、経時的に要約され、14及び24週目に処置群間で比較される。
研究は、14週目のACR20を有する対象の割合が、プラセボ群と比較してゴリムマブ群において統計的に有意により大きいことが実証された場合、陽性であると考えられる。
研究薬物の物理的記述
ゴリムマブ
IV投与のための50mgのゴリムマブ最終バイアル化製品(Final Vialed Product、FVP)は、4mLのI型ガラスバイアル瓶内でCNTO148IgGを含有する単回使用の滅菌溶液として供給される。各バイアル瓶は、pH5.5のヒスチジン、ソルビトール、及びポリソルベート80の水性媒体中に12.5mg/mLゴリムマブの4mL溶液を含有する。防腐剤は存在しない。
生理食塩水は、単回使用注入バッグ内でIV注入用の滅菌液体として供給される。防腐剤は存在しない。
メトトレキサート(経口又は注射可能)は、治験依頼者によって供給されず、むしろ商業薬剤から取得されなければならない。
メトトレキサート、NSAID、コルチコステロイド、スルファラジン、ヒドロキシクロロキン、及びレフルノミドは、治験依頼者によって供給されず、むしろ商業薬剤から取得されなければならない。
研究施設では、ゴリムマブ溶液のバイアル瓶は、凍結されず、2℃〜8℃(35.6°F〜46.4°F)において固定された冷蔵庫に保存され、光から保護されなければならない。製品の激しい振とうは、回避されるべきである。投与前に、製品は、粒子状物質及び変色に関して視覚的に検査されるべきである。変色、可視粒子、又は他の異質粒子が溶液中に観察される場合、製品は、使用されるべきではない。
活動性乾癬性関節炎を有する成人患者における静脈内ゴリムマブに関する24週目までの有効性及び安全性
導入:GO−VIBRANT研究は、活動性PsAを有する成人患者(生物製剤を摂取していない)における静脈内(IV)ゴリムマブの安全性及び有効性を評価するようにデザインされた3相、多施設共同、無作為化、二重盲検、プラセボ対照試験である。生物製剤を摂取していない活動性PsA患者を、0、4週目(wk)、及びその後8週間毎にIVゴリムマブ2mg/kg、又はwk0、4、12、及び20にプラセボでwk24にゴリムマブに交差に無作為化した(1:1)。主エンドポイントは、wk14におけるACR20応答であった。多様性が制御されたエンドポイントは、ACR50、ACR70、PASI75、HAQ−DIにおけるベースラインからの変化、腱付着部炎、指炎、wk14のSF−36PCS/MCSスコア、ACR50、及びwk24の修正総vdH−S(構造損傷)スコアにおけるベースラインからの変化を含んだ。有効性分析は、無作為化処置に基づき、wk24までの有害事象(AE)が報告される。治験責任医師は、wk60まで盲検化されている。
1.Basra MKA,Fenech R,Gatt RM,Salek MS,Finlay AY.The Dermatology Life Quality Index 1994−2007:a comprehensive review of validation data and clinical results.Br J Rheumatol.2008;159:997−1035.
2.Beutler B,Greenwald D,Hulmes JD,et al.Identity of tumour necrosis factor and the macrophage−secreted factor cachectin.Nature.1985;316(6028):552−554.
3.Cassell SE,Bieber JD,Rich P,Tutuncu ZN,Lee SJ,Kalunian KC,Wu CW,Kavanaugh A.The modified Nail Psoriasis Severity Index:validation of an instrument to assess psoriatic nail involvement in patients with psoriatic arthritis.J Rheumatol.2007;34(1):123−129.
4.Cella D,Yount S,Sorensen M,Chartash E,Sengupta N,Grober J.Validation of the Functional Assessment of Chronic Illness Therapy Fatigue Scale relative to other instrumentation in patients with rheumatoid arthritis.J Rheumatol.2005;32(5):811−819.
5.Chandran V,Bhella S,Schentag C,Gladman D.Functional Assessment of Chronic Illness Therapy−Fatigue Scale is valid in patients with psoriatic arthritis.Ann Rheum Dis.2007;66:936−939.
6.Coates LC,Helliwell PS.Validation of minimal disease activity criteria for psoriatic arthritis using interventional trial data.Arthritis Care Res(Hoboken).2010;62(7):965−969.
7.Costello P,FitzGerald O.Disease mechanisms in psoriasis and psoriatic arthritis.Curr Rheumatol Rep.2001;3(5):419−427.
8.EuroQol Group.EuroQol−a new facility for the measurement of health−related quality of life.Health Policy.1990;16:199−208.
9.Feldman SR,Gordon KB,Bala M,et al.Infliximab treatment results in significant improvement in the quality of life of patients with severe psoriasis:a double−blind placebo−controlled trial.British Journal of Dermatology.2005;152:954−960.
10.Felson DT,Anderson JJ,Boers M,et al.American College of Rheumatology.Preliminary definition of improvement in rheumatoid arthritis.Arthritis Rheum.1995;38(6):727−735.
11.Finlay AY,Khan GK.Dermatology Life Quality Index(DLQI)−−a simple practical measure for routine clinical use.Clin Exp Dermatol.1994;19(3):210−216.
12.Fredriksson T,Pettersson U.Severe psoriasis−oral therapy with a new retinoid.Dermatologica.1978;157(4):238−244.
13.Fries JF,Spitz P,Kraines RG,Holman HR.Measurement of patient outcomes in arthritis.Arthritis Rheum.1980;23(2):137−145.
14.Garrett S,Jenkinson T,Kennedy LG,Whitelock H,Gaisford P,Calin A.A new approach to defining disease status in ankylosing spondylitis:the Bath Ankylosing Spondylitis Disease Activity Index.J Rheumatol.1994;21(12):2286−2291.
15.Gladman DD,Inman RD,Cook RJ,et al.International spondyloarthritis interobserver reliability exercise−−the INSPIRE study:II.Assessment of peripheral joints,enthesitis,and dactylitis.J Rheumatol.2007;34(8):1740−1745.
16.Gladman DD,Ziouzina O,Thavaneswaran A,Chandran V.Dactylitis in psoriatic arthritis:prevalence and response to therapy in the biologic era.J Rheumatol.2013;40(8):1357−1359.
17.Goedkoop AY,Kraan MC,Teunissen MB,et al.Early effects of tumour necrosis factor alpha blockade on skin and synovial tissue in patients with active psoriasis and psoriatic arthritis.Ann Rheum Dis.2004;63(7):769−773.
18.Healy PJ,Helliwell PS.Measuring clinical enthesitis in psoriatic arthritis:assessment of existing measures and development of an instrument specific to psoriatic arthritis.Arthritis Rheum.2008;59(5):686−691
19.Husted JA,Gladman DD,Cook RJ,Farewell VT.Responsiveness of health status instruments to changes in articular status and perceived health in patients with psoriatic arthritis.J Rheumatol.1998;25(11):2146−2155.
20.Khilji FA,Gonzalez M,Finlay AY.Clinical meaning of change in dermatology life quality index scores.Br J Dermatol.2002;147(suppl 62):25−54.SIMPONI IV(golimumab)Clinical Protocol CNTO148PSA3001 Amendment 2 98 Approved,29 June 2016
21.Mease PJ,Antoni CE,Gladman DD,Taylor WJ.Psoriatic arthritis assessment tools in clinical trials.Ann Rheum Dis.2005;64(Suppl II):ii49−ii54.
22.Mease PJ,Kivitz AJ,Burch FX,et al.Etanercept treatment of psoriatic arthritis:safety,efficacy,and effect on disease progression.Arthritis Rheum.2004;50(7):2264−2272
23.Mease PJ.Measures of psoriatic arthritis:Tender and Swollen Joint Assessment,Psoriasis Area and Severity Index(PASI),Nail Psoriasis Severity Index(NAPSI),Modified Nail Psoriasis Severity Index(mNAPSI),Mander/Newcastle Enthesitis Index(MEI),Leeds Enthesitis Index(LEI),Spondyloarthritis Research Consortium of Canada(SPARCC),Maastricht Ankylosing Spondylitis Enthesis Score(MASES),Leeds Dactylitis Index(LDI),Patient Global for Psoriatic Arthritis,Dermatology Life Quality Index(DLQI),Psoriatic Arthritis Quality of Life(PsAQOL),Functional Assessment of Chronic Illness Therapy−Fatigue(FACIT−F),Psoriatic Arthritis Response Criteria(PsARC),Psoriatic Arthritis Joint Activity Index(PsAJAI),Disease Activity in Psoriatic Arthritis(DAPSA),and Composite Psoriatic Disease Activity Index(CPDAI).Arthritis Care Res(Hoboken).2011;63Suppl11:S64−S85.
24.Partsch G,Steiner G,Leeb BF,Dunky A,Broll H,Smolen JS.Highly increased levels of tumor necrosis factor−alpha and other proinflammatory cytokines in psoriatic arthritis synovial fluid.J Rheumatol.1997;24(3):518−523.
25.Pham T,Guillemin F,Claudepierre P,et al.TNFα antagonist therapy in ankylosing spondylitis and psoriatic arthritis:recommendations of the French Society for Rheumatology.Joint Bone Spine.2006;73:547−553.
26.Ritchlin C,Haas−Smith SA,Hicks D,Cappuccio J,Osterland CK,Looney RJ.Patterns of cytokine production in psoriatic synovium.J Rheumatol.1998;25(8):1544−1552.
27.Taylor W,Gladman D,Helliwell P,Marchesoni A,Mease P,Mielants H;CASPAR Study Group.Classification criteria for psoriatic arthritis:development of new criteria from a large international study.Arthritis Rheum.2006;54(8):2665−2673.
28.van der Heijde DM,van Leeuwen MA,van Riel PL,et al.Biannual radiographic assessments of hands and feet in a three−year prospective followup of patients with early rheumatoid arthritis.Arthritis Rheum.1992;35(1):26−34.
29.van der Linden MA.Disease Activity Scores using C−reactive protein.April5,2004.以下で入手可能:www.umcn.nl/scientist/afdelingen/reumatologie/the_das/the_das_crp.2005年8月19日にアクセス。
30.van Riel P,Fransen J,Scott DL.EULAR handbook of clinical assessments in rheumatoid arthritis.Third ed.Alphen Aan Den Rijn,The Netherlands:Van Zuiden Communications B.V.2004.請求に応じて入手可能。
31.van Riel PL,van Gestel AM,Scott DL,for the EULAR Standing Committee for International Clinical Studies Including Therapeutic Trials−ESCISIT.EULAR Handbook of Clinical Assessments in Rheumatoid Arthritis.Alphen Aan Den Rijn,The Netherlands:Van Zuiden Communications B.V.;2000:40.
32.Veale DJ,Markham T,Fearon U,et al.St Vincents University Hospital,Dublin,Ireland.Therapy in psoriasis and psoriatic arthritis:anti−TNF−alpha clinical and angiogenic responses.Arthritis Rheum.2003;48(suppl):S168−S169.
33.Ware JE Jr,Sherbourne CD.The MOS 36−item short−form health survey (SF−36),I:conceptual framework and item selection.Med Care.1992;30(6):473−483.
34.Ware JE,Kosinski M,Keller SD.Interpretation:Norm−Based.In:SF−36Physical and Mental Health Summary Scales:A User’s Manual.Boston,MA:The Health Institute;1994:8:1−8:42.
35.Zochling J,van der Heijde D,Burgos−Vargas R,et al.ASAS/EULAR recommendations for the management of ankylosing spondylitis.Ann Rheum Dis.2006;65(4):442−452.
Claims (10)
- 活動性乾癬性関節炎の安全かつ有効な治療において使用するための、配列番号36を含む重鎖(HC)及び配列番号37を含む軽鎖(LC)を有する、少なくとも1つの単離された哺乳類抗TNF抗体と、少なくとも1つの薬学的に許容される担体又は希釈剤と、を含む、組成物であって、前記組成物が、IV注入を介して投与され、処置の14週目に、前記抗TNF抗体で処置された患者が、HAQ−DI=−0.60±0.53SD、腱付着部炎=−1.87±1.75SD、指炎=−7.8±8.57SD、SF−36 PCS=8.65±7.60SD、及びSF−36 MCS=5.33±9.95SDからなる群から選択される1つ又は2つ以上の基準におけるベースラインからの平均変化を達成する、組成物。
- 前記抗TNF抗体が、0及び4週目に、次いでその後8週間毎(q8w)に、30±10分にわたって2mg/kgの用量で投与されるように、前記組成物が投与される、請求項1に記載の組成物。
- 前記組成物をメトトレキサート(MTX)と共に、又はメトトレキサートなしで投与することを更に含む、請求項1〜2に記載の組成物。
- 活動性乾癬性関節炎の安全かつ有効な治療において使用するための、配列番号36を含む重鎖(HC)及び配列番号37を含む軽鎖(LC)を有する、少なくとも1つの単離された哺乳類抗TNF抗体と、少なくとも1つの薬学的に許容される担体又は希釈剤と、を含む、組成物であって、前記組成物が、IV注入を介して投与され、処置の24週目に、前記抗TNF抗体で処置された患者が、vdH−S=−0.36±0.144SEにおけるベースラインからの平均変化を達成する、組成物。
- 前記抗TNF抗体が、0及び4週目に、次いでその後8週間毎(q8w)に、30±10分にわたって2mg/kgの用量で投与されるように、前記組成物が投与される、請求項4に記載の組成物。
- 前記組成物をメトトレキサート(MTX)と共に、又はメトトレキサートなしで投与することを更に含む、請求項4〜5に記載の組成物。
- 活動性乾癬性関節炎の安全かつ有効な治療において使用するための、配列番号36を含む重鎖(HC)及び配列番号37を含む軽鎖(LC)を有する、少なくとも1つの単離された哺乳類抗TNF抗体であって、前記抗TNF抗体が、静脈内(IV)注入を介して投与され、前記処置を受けている患者の65%以上が、処置の14週目にACR20を達成する、抗TNF抗体。
- 前記患者の65%以上が、50%以上の処置差(プラセボと比較して改善)で、処置の14週目にACR20を達成する、請求項7に記載の抗TNF抗体。
- 前記抗体が、0及び4週目に、次いでその後8週間毎(q8w)に、30±10分にわたって2mg/kgの用量で投与される、請求項7〜8に記載の抗TNF抗体。
- 前記抗体が、メトトレキサート(MTX)と共に、又はメトトレキサートなしで投与される、請求項7〜8に記載の抗TNF抗体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022110247A JP2022137167A (ja) | 2017-01-30 | 2022-07-08 | 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762452079P | 2017-01-30 | 2017-01-30 | |
| US62/452,079 | 2017-01-30 | ||
| PCT/US2017/061949 WO2018140121A1 (en) | 2017-01-30 | 2017-11-16 | Anti-tnf antibodies, compositions, and methods for the treatment of active psoriatic arthritis |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022110247A Division JP2022137167A (ja) | 2017-01-30 | 2022-07-08 | 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2020506916A true JP2020506916A (ja) | 2020-03-05 |
Family
ID=62977170
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019540538A Pending JP2020506916A (ja) | 2017-01-30 | 2017-11-16 | 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 |
| JP2022110247A Pending JP2022137167A (ja) | 2017-01-30 | 2022-07-08 | 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022110247A Pending JP2022137167A (ja) | 2017-01-30 | 2022-07-08 | 活動性乾癬性関節炎の治療のための抗tnf抗体、組成物、及び方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US20180215819A1 (ja) |
| EP (1) | EP3573658A4 (ja) |
| JP (2) | JP2020506916A (ja) |
| KR (2) | KR20190113858A (ja) |
| CN (1) | CN110234351A (ja) |
| AU (1) | AU2017396503A1 (ja) |
| CA (1) | CA3052095A1 (ja) |
| IL (1) | IL268007A (ja) |
| MA (1) | MA47362A (ja) |
| MX (1) | MX2019008989A (ja) |
| WO (1) | WO2018140121A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210116540A (ko) * | 2019-01-15 | 2021-09-27 | 얀센 바이오테크 인코포레이티드 | 소아 특발성 관절염의 치료를 위한 항-tnf 항체 조성물 및 방법 |
| CN113330031A (zh) * | 2019-01-23 | 2021-08-31 | 詹森生物科技公司 | 用于在治疗银屑病关节炎的方法中使用的抗tnf抗体组合物 |
| EP3976648A1 (en) * | 2019-06-03 | 2022-04-06 | Janssen Biotech, Inc. | Anti-tnf antibody compositions, and methods for the treatment of psoriatic arthritis |
| CN113274349A (zh) * | 2020-02-20 | 2021-08-20 | 百奥泰生物制药股份有限公司 | 抗TNF-α的抗体制剂及其制备方法和用途 |
| KR102394378B1 (ko) | 2020-10-14 | 2022-05-03 | 한림대학교 산학협력단 | 피부 각질세포에서의 아릴탄화수소 수용체 억제자(AhRR) 과발현 방법 |
| WO2023281463A1 (en) * | 2021-07-09 | 2023-01-12 | Janssen Biotech, Inc. | Manufacturing methods for producing anti-tnf antibody compositions |
Family Cites Families (201)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US4309989A (en) | 1976-02-09 | 1982-01-12 | The Curators Of The University Of Missouri | Topical application of medication by ultrasound with coupling agent |
| FR2374910A1 (fr) | 1976-10-23 | 1978-07-21 | Choay Sa | Preparation a base d'heparine, comprenant des liposomes, procede pour l'obtenir et medicaments contenant de telles preparations |
| US4399216A (en) | 1980-02-25 | 1983-08-16 | The Trustees Of Columbia University | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4634665A (en) | 1980-02-25 | 1987-01-06 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US5179017A (en) | 1980-02-25 | 1993-01-12 | The Trustees Of Columbia University In The City Of New York | Processes for inserting DNA into eucaryotic cells and for producing proteinaceous materials |
| US4603106A (en) | 1982-02-22 | 1986-07-29 | The Rockefeller University | Lipoprotein lipase suppression by endotoxin-induced mediator (shock assay) |
| US4822776A (en) | 1981-09-08 | 1989-04-18 | The Rockefeller University | Lipoprotein lipase suppression by endotoxin-induced mediator (shock assay) |
| US5700466A (en) | 1981-09-08 | 1997-12-23 | The Rockefeller University | Method of ameliorating or preventing septic shock using a monoclonal antibody specific to cachectin/tumor necrosis factor |
| US4656134A (en) | 1982-01-11 | 1987-04-07 | Board Of Trustees Of Leland Stanford Jr. University | Gene amplification in eukaryotic cells |
| US5149636A (en) | 1982-03-15 | 1992-09-22 | Trustees Of Columbia University In The City Of New York | Method for introducing cloned, amplifiable genes into eucaryotic cells and for producing proteinaceous products |
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US5168062A (en) | 1985-01-30 | 1992-12-01 | University Of Iowa Research Foundation | Transfer vectors and microorganisms containing human cytomegalovirus immediate-early promoter-regulatory DNA sequence |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4965188A (en) | 1986-08-22 | 1990-10-23 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences using a thermostable enzyme |
| GB2183661B (en) | 1985-03-30 | 1989-06-28 | Marc Ballivet | Method for obtaining dna, rna, peptides, polypeptides or proteins by means of a dna recombinant technique |
| US6492107B1 (en) | 1986-11-20 | 2002-12-10 | Stuart Kauffman | Process for obtaining DNA, RNA, peptides, polypeptides, or protein, by recombinant DNA technique |
| SE448277B (sv) | 1985-04-12 | 1987-02-09 | Draco Ab | Indikeringsanordning vid en doseringsanordning for lekemedel |
| US4766067A (en) | 1985-05-31 | 1988-08-23 | President And Fellows Of Harvard College | Gene amplification |
| EP0212489B1 (en) | 1985-08-16 | 1994-11-30 | The Rockefeller University | Anabolic activity modulator and uses thereof |
| US4870163A (en) | 1985-08-29 | 1989-09-26 | New York Blood Center, Inc. | Preparation of pure human tumor necrosis factor and hybridomas producing monoclonal antibodies to human tumor necrosis factor |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| US5576195A (en) | 1985-11-01 | 1996-11-19 | Xoma Corporation | Vectors with pectate lyase signal sequence |
| US5618920A (en) | 1985-11-01 | 1997-04-08 | Xoma Corporation | Modular assembly of antibody genes, antibodies prepared thereby and use |
| DE3600905A1 (de) | 1986-01-15 | 1987-07-16 | Ant Nachrichtentech | Verfahren zum dekodieren von binaersignalen sowie viterbi-dekoder und anwendungen |
| GB8601597D0 (en) | 1986-01-23 | 1986-02-26 | Wilson R H | Nucleotide sequences |
| US4800159A (en) | 1986-02-07 | 1989-01-24 | Cetus Corporation | Process for amplifying, detecting, and/or cloning nucleic acid sequences |
| SE453566B (sv) | 1986-03-07 | 1988-02-15 | Draco Ab | Anordning vid pulverinhalatorer |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US4767402A (en) | 1986-07-08 | 1988-08-30 | Massachusetts Institute Of Technology | Ultrasound enhancement of transdermal drug delivery |
| US4925673A (en) | 1986-08-18 | 1990-05-15 | Clinical Technologies Associates, Inc. | Delivery systems for pharmacological agents encapsulated with proteinoids |
| US4889818A (en) | 1986-08-22 | 1989-12-26 | Cetus Corporation | Purified thermostable enzyme |
| US5260203A (en) | 1986-09-02 | 1993-11-09 | Enzon, Inc. | Single polypeptide chain binding molecules |
| US4946778A (en) | 1987-09-21 | 1990-08-07 | Genex Corporation | Single polypeptide chain binding molecules |
| US4704692A (en) | 1986-09-02 | 1987-11-03 | Ladner Robert C | Computer based system and method for determining and displaying possible chemical structures for converting double- or multiple-chain polypeptides to single-chain polypeptides |
| DE3631229A1 (de) | 1986-09-13 | 1988-03-24 | Basf Ag | Monoklonale antikoerper gegen humanen tumornekrosefaktor (tnf) und deren verwendung |
| US4921794A (en) | 1987-01-14 | 1990-05-01 | President And Fellows Of Harvard College | T7 DNA polymerase |
| US4795699A (en) | 1987-01-14 | 1989-01-03 | President And Fellows Of Harvard College | T7 DNA polymerase |
| EP0279582A3 (en) | 1987-02-17 | 1989-10-18 | Pharming B.V. | Dna sequences to target proteins to the mammary gland for efficient secretion |
| ATE114723T1 (de) | 1987-03-02 | 1994-12-15 | Enzon Lab Inc | Organismus als träger für ''single chain antibody domain (scad)''. |
| EP0288088B1 (en) | 1987-04-24 | 1994-03-09 | Teijin Limited | Detection of tumor necrosis factor; monoclonal antibody and kit |
| US4873316A (en) | 1987-06-23 | 1989-10-10 | Biogen, Inc. | Isolation of exogenous recombinant proteins from the milk of transgenic mammals |
| CA1341235C (en) | 1987-07-24 | 2001-05-22 | Randy R. Robinson | Modular assembly of antibody genes, antibodies prepared thereby and use |
| US4939666A (en) | 1987-09-02 | 1990-07-03 | Genex Corporation | Incremental macromolecule construction methods |
| IL83878A (en) | 1987-09-13 | 1995-07-31 | Yeda Res & Dev | Soluble protein corresponding to tnf inhibitory protein its preparation and pharmaceutical compositions containing it |
| EP0396612B1 (en) | 1988-01-11 | 1996-07-24 | Xoma Corporation | Novel plasmid vector with pectate lyase signal sequence |
| US6010902A (en) | 1988-04-04 | 2000-01-04 | Bristol-Meyers Squibb Company | Antibody heteroconjugates and bispecific antibodies for use in regulation of lymphocyte activity |
| US4956288A (en) | 1988-04-22 | 1990-09-11 | Biogen, Inc. | Method for producing cells containing stably integrated foreign DNA at a high copy number, the cells produced by this method, and the use of these cells to produce the polypeptides coded for by the foreign DNA |
| US5770198A (en) | 1988-05-18 | 1998-06-23 | The Research Foundation Of The State Of New York | Platelet-specific chimeric 7E3 immunoglobulin |
| US5130238A (en) | 1988-06-24 | 1992-07-14 | Cangene Corporation | Enhanced nucleic acid amplification process |
| DE3823804A1 (de) | 1988-07-14 | 1990-01-18 | Basf Ag | Neutralisation der in vitro und in vivo toxischen eigenschaften von tnf-(alpha) durch monoklonale antikoerper und den davon abgeleiteten fragmenten |
| NZ229922A (en) | 1988-07-18 | 1992-04-28 | Chiron Corp | Monoclonal antibodies specifically binding cachectin (tumor necrosis factor) and compositions |
| US5601819A (en) | 1988-08-11 | 1997-02-11 | The General Hospital Corporation | Bispecific antibodies for selective immune regulation and for selective immune cell binding |
| US5223409A (en) | 1988-09-02 | 1993-06-29 | Protein Engineering Corp. | Directed evolution of novel binding proteins |
| IL94039A (en) | 1989-08-06 | 2006-09-05 | Yeda Res & Dev | Antibodies to tbp - 1 and their use |
| US5142033A (en) | 1988-09-23 | 1992-08-25 | Hoffmann-La Roche Inc. | Structure-independent DNA amplification by the polymerase chain reaction |
| US5091310A (en) | 1988-09-23 | 1992-02-25 | Cetus Corporation | Structure-independent dna amplification by the polymerase chain reaction |
| US5066584A (en) | 1988-09-23 | 1991-11-19 | Cetus Corporation | Methods for generating single stranded dna by the polymerase chain reaction |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US4987893A (en) | 1988-10-12 | 1991-01-29 | Rochal Industries, Inc. | Conformable bandage and coating material |
| JP2919890B2 (ja) | 1988-11-11 | 1999-07-19 | メディカル リサーチ カウンスル | 単一ドメインリガンド、そのリガンドからなる受容体、その製造方法、ならびにそのリガンドおよび受容体の使用 |
| GB8826530D0 (en) | 1988-11-12 | 1988-12-14 | Ped Capacitors Ltd | Electrical capacitors |
| US5342613A (en) | 1988-12-27 | 1994-08-30 | Health Research Inc. | Pharmaceutical compositions and use thereof in the treatment of psoriasis |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US4994370A (en) | 1989-01-03 | 1991-02-19 | The United States Of America As Represented By The Department Of Health And Human Services | DNA amplification technique |
| PT92900A (pt) | 1989-01-24 | 1990-07-31 | Sistema de vectores de expressao para a producao de anticorpos monoclonais quimericos | |
| US5225538A (en) | 1989-02-23 | 1993-07-06 | Genentech, Inc. | Lymphocyte homing receptor/immunoglobulin fusion proteins |
| US5116964A (en) | 1989-02-23 | 1992-05-26 | Genentech, Inc. | Hybrid immunoglobulins |
| US5266491A (en) | 1989-03-14 | 1993-11-30 | Mochida Pharmaceutical Co., Ltd. | DNA fragment and expression plasmid containing the DNA fragment |
| DE3909708A1 (de) | 1989-03-23 | 1990-09-27 | Boehringer Mannheim Gmbh | Verfahren zur herstellung bispezifischer antikoerper |
| ES2238070T3 (es) | 1989-04-21 | 2005-08-16 | Amgen Inc. | Receptor del tnf, proteina ligante del tnf y adn codante para estos. |
| CA2016841C (en) | 1989-05-16 | 1999-09-21 | William D. Huse | A method for producing polymers having a preselected activity |
| AU652539B2 (en) | 1989-05-16 | 1994-09-01 | Medical Research Council | Co-expression of heteromeric receptors |
| CA2016842A1 (en) | 1989-05-16 | 1990-11-16 | Richard A. Lerner | Method for tapping the immunological repertoire |
| CA2017025C (en) | 1989-05-18 | 2008-07-22 | David Wallach | Tumor necrosis factor binding protein ii, its purification and antibodies thereto |
| EP0739904A1 (en) | 1989-06-29 | 1996-10-30 | Medarex, Inc. | Bispecific reagents for aids therapy |
| AU640400B2 (en) | 1989-08-07 | 1993-08-26 | Peptide Technology Ltd. | Tumour necrosis factor binding ligands |
| AU638762B2 (en) | 1989-10-05 | 1993-07-08 | Optein Inc | Cell-free synthesis and isolation of novel genes and polypeptides |
| EP0433900B1 (en) | 1989-12-13 | 1995-09-20 | Yeda Research And Development Company Limited | Expression of the recombinant tumor necrosis factor binding protein I (TBP-I) |
| GB8928874D0 (en) | 1989-12-21 | 1990-02-28 | Celltech Ltd | Humanised antibodies |
| US5580575A (en) | 1989-12-22 | 1996-12-03 | Imarx Pharmaceutical Corp. | Therapeutic drug delivery systems |
| DE69133566T2 (de) | 1990-01-12 | 2007-12-06 | Amgen Fremont Inc. | Bildung von xenogenen Antikörpern |
| TW212184B (ja) | 1990-04-02 | 1993-09-01 | Takeda Pharm Industry Co Ltd | |
| US5427908A (en) | 1990-05-01 | 1995-06-27 | Affymax Technologies N.V. | Recombinant library screening methods |
| DK0600866T3 (da) | 1990-06-01 | 1998-03-09 | Chiron Corp | Præparater og fremgangsmåder til identifikation af biologisk aktive molekyler |
| US5723286A (en) | 1990-06-20 | 1998-03-03 | Affymax Technologies N.V. | Peptide library and screening systems |
| EP0547065B1 (en) | 1990-06-29 | 2001-08-29 | Large Scale Biology Corporation | Melanin production by transformed microorganisms |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| ATE185601T1 (de) | 1990-07-10 | 1999-10-15 | Cambridge Antibody Tech | Verfahren zur herstellung von spezifischen bindungspaargliedern |
| US5580734A (en) | 1990-07-13 | 1996-12-03 | Transkaryotic Therapies, Inc. | Method of producing a physical map contigous DNA sequences |
| CA2089362C (en) | 1990-08-24 | 2000-11-21 | William D. Huse | Methods of synthesizing oligonucleotides with random codons |
| DK0814159T3 (da) | 1990-08-29 | 2005-10-24 | Genpharm Int | Transgene, ikke-humane dyr, der er i stand til at danne heterologe antistoffer |
| US5545806A (en) | 1990-08-29 | 1996-08-13 | Genpharm International, Inc. | Ransgenic non-human animals for producing heterologous antibodies |
| US5633425A (en) | 1990-08-29 | 1997-05-27 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5789650A (en) | 1990-08-29 | 1998-08-04 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US5770429A (en) | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5625126A (en) | 1990-08-29 | 1997-04-29 | Genpharm International, Inc. | Transgenic non-human animals for producing heterologous antibodies |
| US6300129B1 (en) | 1990-08-29 | 2001-10-09 | Genpharm International | Transgenic non-human animals for producing heterologous antibodies |
| US5661016A (en) | 1990-08-29 | 1997-08-26 | Genpharm International Inc. | Transgenic non-human animals capable of producing heterologous antibodies of various isotypes |
| WO1992005258A1 (en) | 1990-09-20 | 1992-04-02 | La Trobe University | Gene encoding barley enzyme |
| IL99552A0 (en) | 1990-09-28 | 1992-08-18 | Ixsys Inc | Compositions containing procaryotic cells,a kit for the preparation of vectors useful for the coexpression of two or more dna sequences and methods for the use thereof |
| GB9022648D0 (en) | 1990-10-18 | 1990-11-28 | Charing Cross Sunley Research | Polypeptide and its use |
| AU8727291A (en) | 1990-10-29 | 1992-06-11 | Cetus Oncology Corporation | Bispecific antibodies, method of production, and uses thereof |
| US5958413A (en) | 1990-11-01 | 1999-09-28 | Celltech Limited | Use of antibodies to TNF or fragments derived thereof and xanthine derivatives for combination therapy and compositions therefor |
| WO1992009690A2 (en) | 1990-12-03 | 1992-06-11 | Genentech, Inc. | Enrichment method for variant proteins with altered binding properties |
| US5582996A (en) | 1990-12-04 | 1996-12-10 | The Wistar Institute Of Anatomy & Biology | Bifunctional antibodies and method of preparing same |
| NZ241119A (en) | 1990-12-20 | 1993-06-25 | Ixsys Inc | Manipulating nucleic acid to optimize the binding characteristics of the encoded binding protein |
| GB9109645D0 (en) | 1991-05-03 | 1991-06-26 | Celltech Ltd | Recombinant antibodies |
| KR100234520B1 (ko) | 1991-01-18 | 1999-12-15 | 그레고리 아보트 | 종양괴사인자 매개병 치료용 의약품 조성물 |
| CA2104698A1 (en) | 1991-02-21 | 1992-08-22 | John J. Toole | Aptamers specific for biomolecules and methods of making |
| US5404871A (en) | 1991-03-05 | 1995-04-11 | Aradigm | Delivery of aerosol medications for inspiration |
| EP0575545B1 (en) | 1991-03-15 | 2003-05-21 | Amgen Inc. | Pegylation of polypeptides |
| EP1857554A1 (en) | 1991-03-18 | 2007-11-21 | New York University | Monoclonal and chimeric antibodies specific for human tumor necrosis factor |
| US5919452A (en) | 1991-03-18 | 1999-07-06 | New York University | Methods of treating TNFα-mediated disease using chimeric anti-TNF antibodies |
| US6277969B1 (en) | 1991-03-18 | 2001-08-21 | New York University | Anti-TNF antibodies and peptides of human tumor necrosis factor |
| US5656272A (en) | 1991-03-18 | 1997-08-12 | New York University Medical Center | Methods of treating TNF-α-mediated Crohn's disease using chimeric anti-TNF antibodies |
| US5698195A (en) | 1991-03-18 | 1997-12-16 | New York University Medical Center | Methods of treating rheumatoid arthritis using chimeric anti-TNF antibodies |
| US6284471B1 (en) | 1991-03-18 | 2001-09-04 | New York University Medical Center | Anti-TNFa antibodies and assays employing anti-TNFa antibodies |
| DK1471142T3 (da) | 1991-04-10 | 2009-03-09 | Scripps Research Inst | Heterodimere receptor-biblioteker under anvendelse af fagemider |
| DE4118120A1 (de) | 1991-06-03 | 1992-12-10 | Behringwerke Ag | Tetravalente bispezifische rezeptoren, ihre herstellung und verwendung |
| US5637481A (en) | 1993-02-01 | 1997-06-10 | Bristol-Myers Squibb Company | Expression vectors encoding bispecific fusion proteins and methods of producing biologically active bispecific fusion proteins in a mammalian cell |
| ATE359842T1 (de) | 1991-07-02 | 2007-05-15 | Nektar Therapeutics | Abgabevorrichtung für nebelförmige medikamente |
| DE69233011T2 (de) | 1991-07-25 | 2003-11-06 | Idec Pharma Corp | Rekombinante antikörper zur humanen therapie |
| MX9204374A (es) | 1991-07-25 | 1993-03-01 | Idec Pharma Corp | Anticuerpo recombinante y metodo para su produccion. |
| EP0525570A3 (en) | 1991-07-31 | 1993-10-06 | Miles Inc. | Anti-idiotypic antibodies that mimic tnf |
| IL99120A0 (en) | 1991-08-07 | 1992-07-15 | Yeda Res & Dev | Multimers of the soluble forms of tnf receptors,their preparation and pharmaceutical compositions containing them |
| DK0605522T3 (da) | 1991-09-23 | 2000-01-17 | Medical Res Council | Fremgangsmåde til fremstilling af humaniserede antistoffer |
| US5270170A (en) | 1991-10-16 | 1993-12-14 | Affymax Technologies N.V. | Peptide library and screening method |
| WO1993008829A1 (en) | 1991-11-04 | 1993-05-13 | The Regents Of The University Of California | Compositions that mediate killing of hiv-infected cells |
| US5733761A (en) | 1991-11-05 | 1998-03-31 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| US5641670A (en) | 1991-11-05 | 1997-06-24 | Transkaryotic Therapies, Inc. | Protein production and protein delivery |
| NO178839C (no) | 1991-11-25 | 1996-06-12 | Ottestad Nils T | Strömningsregulator for opprettholdelse av en stabil strömningsmengde av et fluidum |
| US5932448A (en) | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| ATE463573T1 (de) | 1991-12-02 | 2010-04-15 | Medimmune Ltd | Herstellung von autoantikörpern auf phagenoberflächen ausgehend von antikörpersegmentbibliotheken |
| CA2103887C (en) | 1991-12-13 | 2005-08-30 | Gary M. Studnicka | Methods and materials for preparation of modified antibody variable domains and therapeutic uses thereof |
| US5667988A (en) | 1992-01-27 | 1997-09-16 | The Scripps Research Institute | Methods for producing antibody libraries using universal or randomized immunoglobulin light chains |
| DE4207475A1 (de) | 1992-03-10 | 1993-09-16 | Goldwell Ag | Mittel zum blondieren von menschlichen haaren und verfahren zu dessen herstellung |
| CA2131151A1 (en) | 1992-03-24 | 1994-09-30 | Kevin S. Johnson | Methods for producing members of specific binding pairs |
| US5447851B1 (en) | 1992-04-02 | 1999-07-06 | Univ Texas System Board Of | Dna encoding a chimeric polypeptide comprising the extracellular domain of tnf receptor fused to igg vectors and host cells |
| AU4829593A (en) | 1992-09-23 | 1994-04-12 | Fisons Plc | Inhalation device |
| WO1994008038A1 (en) | 1992-10-02 | 1994-04-14 | Trustees Of Dartmouth College | Bispecific reagents for redirected targeting of low density lipoprotein |
| ATE174804T1 (de) | 1992-10-19 | 1999-01-15 | Dura Pharma Inc | Trockenpulverinhalator |
| US5643252A (en) | 1992-10-28 | 1997-07-01 | Venisect, Inc. | Laser perforator |
| AU5670194A (en) | 1992-11-20 | 1994-06-22 | Enzon, Inc. | Linker for linked fusion polypeptides |
| US5849695A (en) | 1993-01-13 | 1998-12-15 | The Regents Of The University Of California | Parathyroid hormone analogues useful for treatment of osteoporosis and disorders of calcium meatabolism in mammals |
| DK0680451T3 (da) | 1993-01-19 | 1999-07-19 | Glaxo Group Ltd | Aerosoldispenser samt fremgangsmåde ved dens fremstilling |
| AU6132994A (en) | 1993-02-02 | 1994-08-29 | Scripps Research Institute, The | Methods for producing antibody libraries using universal or randomized immunoglobulin light chains |
| CA2155728C (en) | 1993-02-12 | 2000-04-25 | Gerald R. Crabtree | Regulated transcription of targeted genes and other biological events |
| US5770428A (en) | 1993-02-17 | 1998-06-23 | Wisconsin Alumni Research Foundation | Chimeric retrovial expression vectors and particles containing a simple retroviral long terminal repeat, BLV or HIV coding regions and cis-acting regulatory sequences, and an RNA translational enhancer with internal ribsome entry site |
| EP0614989A1 (en) | 1993-02-17 | 1994-09-14 | MorphoSys AG | A method for in vivo selection of ligand-binding proteins |
| US5888511A (en) | 1993-02-26 | 1999-03-30 | Advanced Biotherapy Concepts, Inc. | Treatment of autoimmune diseases, including AIDS |
| ES2159529T5 (es) | 1993-03-05 | 2011-03-09 | Bayer Corporation | Anticuerpos monoclonales humanos anti-tnf alfa. |
| EP0754225A4 (en) | 1993-04-26 | 2001-01-31 | Genpharm Int | HETEROLOGIC ANTIBODY-PRODUCING TRANSGENIC NON-HUMAN ANIMALS |
| GB9313509D0 (en) | 1993-06-30 | 1993-08-11 | Medical Res Council | Chemisynthetic libraries |
| US5444987A (en) | 1993-07-02 | 1995-08-29 | Alsenz; Richard H. | Refrigeration system utilizing a jet enthalpy compressor for elevating the suction line pressure |
| US5514670A (en) | 1993-08-13 | 1996-05-07 | Pharmos Corporation | Submicron emulsions for delivery of peptides |
| US5625825A (en) | 1993-10-21 | 1997-04-29 | Lsi Logic Corporation | Random number generating apparatus for an interface unit of a carrier sense with multiple access and collision detect (CSMA/CD) ethernet data network |
| DE4337197C1 (de) | 1993-10-30 | 1994-08-25 | Biotest Pharma Gmbh | Verfahren zur selektiven Herstellung von Hybridomazellinien, die monoklonale Antikörper mit hoher Zytotoxizität gegen humanes CD16-Antigen produzieren, sowie Herstellung bispezifischer monoklonaler Antikörper unter Verwendung derartiger monoklonaler Antikörper und des CD30-HRS-3-Antikörpers zur Therapie menschlicher Tumore |
| US5814599A (en) | 1995-08-04 | 1998-09-29 | Massachusetts Insitiute Of Technology | Transdermal delivery of encapsulated drugs |
| SE9304060D0 (sv) | 1993-12-06 | 1993-12-06 | Bioinvent Int Ab | Sätt att selektera specifika bakteriofager |
| US5827690A (en) | 1993-12-20 | 1998-10-27 | Genzyme Transgenics Corporatiion | Transgenic production of antibodies in milk |
| JPH09510201A (ja) | 1994-03-07 | 1997-10-14 | メダレツクス・インコーポレーテツド | 臨床的効用を有する二重特異性分子 |
| US6190691B1 (en) | 1994-04-12 | 2001-02-20 | Adolor Corporation | Methods for treating inflammatory conditions |
| US5763733A (en) | 1994-10-13 | 1998-06-09 | Enzon, Inc. | Antigen-binding fusion proteins |
| US5549551A (en) | 1994-12-22 | 1996-08-27 | Advanced Cardiovascular Systems, Inc. | Adjustable length balloon catheter |
| JPH08178054A (ja) | 1994-12-26 | 1996-07-12 | Honda Motor Co Ltd | 自動車の制御装置 |
| FR2728793A1 (fr) | 1994-12-28 | 1996-07-05 | Oreal | Utilisation d'un antagoniste d'histamine, d'un antagoniste d'interleukine 1 et/ou d'un antagoniste de tnf-alpha dans une composition cosmetique, pharmaceutique ou dermatologique et composition obtenue |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US6037453A (en) | 1995-03-15 | 2000-03-14 | Genentech, Inc. | Immunoglobulin variants |
| US5656730A (en) | 1995-04-07 | 1997-08-12 | Enzon, Inc. | Stabilized monomeric protein compositions |
| EP0822830B1 (en) | 1995-04-27 | 2008-04-02 | Amgen Fremont Inc. | Human anti-IL-8 antibodies, derived from immunized xenomice |
| AU2466895A (en) | 1995-04-28 | 1996-11-18 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5730723A (en) | 1995-10-10 | 1998-03-24 | Visionary Medical Products Corporation, Inc. | Gas pressured needle-less injection device and method |
| US6331431B1 (en) | 1995-11-28 | 2001-12-18 | Ixsys, Inc. | Vacuum device and method for isolating periplasmic fraction from cells |
| GB9526100D0 (en) | 1995-12-20 | 1996-02-21 | Intersurgical Ltd | Nebulizer |
| EA001016B1 (ru) | 1996-01-03 | 2000-08-28 | Глаксо Груп Лимитед | Ингаляционное устройство |
| WO1997029131A1 (en) | 1996-02-09 | 1997-08-14 | Basf Aktiengesellschaft | HUMAN ANTIBODIES THAT BIND HUMAN TNF$g(a) |
| US5714352A (en) | 1996-03-20 | 1998-02-03 | Xenotech Incorporated | Directed switch-mediated DNA recombination |
| GB9712818D0 (en) | 1996-07-08 | 1997-08-20 | Cambridge Antibody Tech | Labelling and selection of specific binding molecules |
| ATE210682T1 (de) | 1996-09-03 | 2001-12-15 | Gsf Forschungszentrum Umwelt | Zerstörung von kontaminierenden tumorzellen in stammzelltransplantaten mit bispezifischen antikörpern |
| CA2722378C (en) | 1996-12-03 | 2015-02-03 | Amgen Fremont Inc. | Human antibodies that bind tnf.alpha. |
| US5879681A (en) | 1997-02-07 | 1999-03-09 | Emisphere Technolgies Inc. | Compounds and compositions for delivering active agents |
| US5921447A (en) | 1997-02-13 | 1999-07-13 | Glaxo Wellcome Inc. | Flow-through metered aerosol dispensing apparatus and method of use thereof |
| US6235883B1 (en) | 1997-05-05 | 2001-05-22 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| IL120943A (en) | 1997-05-29 | 2004-03-28 | Univ Ben Gurion | A system for administering drugs through the skin |
| WO1999006834A2 (en) | 1997-08-04 | 1999-02-11 | Ixsys, Incorporated | Methods for identifying ligand specific binding molecules |
| JPH11127855A (ja) | 1997-10-27 | 1999-05-18 | Japan Energy Corp | 組換え型抗ヒトTNF−αヒトモノクローナル抗体 |
| US6902734B2 (en) | 2000-08-07 | 2005-06-07 | Centocor, Inc. | Anti-IL-12 antibodies and compositions thereof |
| UA81743C2 (uk) | 2000-08-07 | 2008-02-11 | Центокор, Инк. | МОНОКЛОНАЛЬНЕ АНТИТІЛО ЛЮДИНИ, ЩО СПЕЦИФІЧНО ЗВ'ЯЗУЄТЬСЯ З ФАКТОРОМ НЕКРОЗУ ПУХЛИН АЛЬФА (ФНПα), ФАРМАЦЕВТИЧНА КОМПОЗИЦІЯ, ЩО ЙОГО МІСТИТЬ, ТА СПОСІБ ЛІКУВАННЯ РЕВМАТОЇДНОГО АРТРИТУ |
| JP2004536786A (ja) * | 2001-03-02 | 2004-12-09 | メディミューン,インコーポレイテッド | インテグリンαvβ3拮抗薬を他の予防薬もしくは治療薬と組み合わせて投与することによる炎症性疾患または自己免疫疾患の予防または治療方法 |
| TWI327597B (en) | 2001-08-01 | 2010-07-21 | Centocor Inc | Anti-tnf antibodies, compositions, methods and uses |
| CA2903138A1 (en) * | 2005-05-16 | 2006-11-23 | Abbvie Biotechnology Ltd. | Use of tnfa inhibitor for treatment of erosive polyarthritis |
| EP1937313A4 (en) * | 2005-08-31 | 2010-03-24 | Centocor Ortho Biotech Inc | HOST CELL LINES USEFUL IN THE PRODUCTION OF A CONSTANT ANTIBODY REGION HAVING IMPROVED FUNCTIONING FUNCTION |
| EP2666472A3 (en) * | 2006-04-10 | 2014-04-02 | Abbott Biotechnology Ltd | Uses and compositions for treatment of psoriatic arthritis |
| US20080311043A1 (en) * | 2006-06-08 | 2008-12-18 | Hoffman Rebecca S | Uses and compositions for treatment of psoriatic arthritis |
| WO2008016633A2 (en) * | 2006-08-02 | 2008-02-07 | Ariad Gene Therapeutics, Inc. | Combination therapy |
| WO2010056399A1 (en) * | 2008-11-17 | 2010-05-20 | Incode Biopharmaceutics, Inc. | Method and composition for modulating the immune system and various inflammatory conditions comprising complement depletors |
| GB201212081D0 (en) | 2012-07-06 | 2012-08-22 | Kalvista Pharmaceuticals Ltd | New polymorph |
| US20160061812A1 (en) * | 2013-03-15 | 2016-03-03 | The University Of Birmingham | Diagnosis and Treatment of Arthritic Conditions |
| US20160130340A1 (en) * | 2013-10-07 | 2016-05-12 | Alderbio Holdings Llc | Dosing regimens using anti-il-6 antibodies for the treatment of rheumatoid and psoriatic arthritis |
-
2017
- 2017-11-16 JP JP2019540538A patent/JP2020506916A/ja active Pending
- 2017-11-16 WO PCT/US2017/061949 patent/WO2018140121A1/en unknown
- 2017-11-16 KR KR1020197025050A patent/KR20190113858A/ko active Application Filing
- 2017-11-16 AU AU2017396503A patent/AU2017396503A1/en active Pending
- 2017-11-16 KR KR1020247008465A patent/KR20240038146A/ko active Application Filing
- 2017-11-16 CA CA3052095A patent/CA3052095A1/en active Pending
- 2017-11-16 EP EP17893875.9A patent/EP3573658A4/en active Pending
- 2017-11-16 MA MA047362A patent/MA47362A/fr unknown
- 2017-11-16 MX MX2019008989A patent/MX2019008989A/es unknown
- 2017-11-16 CN CN201780085136.9A patent/CN110234351A/zh active Pending
- 2017-11-20 US US15/818,063 patent/US20180215819A1/en not_active Abandoned
-
2019
- 2019-07-11 IL IL268007A patent/IL268007A/en unknown
- 2019-07-20 US US16/517,594 patent/US11041020B2/en active Active
-
2021
- 2021-05-14 US US17/320,490 patent/US20210277104A1/en active Pending
-
2022
- 2022-07-08 JP JP2022110247A patent/JP2022137167A/ja active Pending
Non-Patent Citations (3)
| Title |
|---|
| ANN RHEUM DIS, vol. Vol. 73, Issue 9, JPN6021029807, 2014, pages 1689 - 1694, ISSN: 0004564846 * |
| CLINICAL PHARMACOLOGY & THERAPEUTICS, vol. Vol. 87, SUPPLEMENT 1, Number: PI-80, JPN6021029806, 2010, pages 34 - 35, ISSN: 0004564847 * |
| THE JOURNAL OF RHEUMATOLOGY, vol. Vol. 43, No. 6, Number: 87, JPN6021029805, 2016, pages 1186, ISSN: 0004564848 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20190113858A (ko) | 2019-10-08 |
| EP3573658A1 (en) | 2019-12-04 |
| WO2018140121A9 (en) | 2018-12-27 |
| CN110234351A (zh) | 2019-09-13 |
| US20190338022A1 (en) | 2019-11-07 |
| WO2018140121A1 (en) | 2018-08-02 |
| IL268007A (en) | 2019-09-26 |
| JP2022137167A (ja) | 2022-09-21 |
| AU2017396503A1 (en) | 2019-07-11 |
| MA47362A (fr) | 2019-12-04 |
| CA3052095A1 (en) | 2018-08-02 |
| US20210277104A1 (en) | 2021-09-09 |
| US11041020B2 (en) | 2021-06-22 |
| MX2019008989A (es) | 2019-10-09 |
| US20180215819A1 (en) | 2018-08-02 |
| KR20240038146A (ko) | 2024-03-22 |
| EP3573658A4 (en) | 2021-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11014982B2 (en) | Anti-TNF antibodies, compositions, and methods for the treatment of active ankylosing spondylitis | |
| US11041020B2 (en) | Methods for the treatment of active Psoriatic Arthritis | |
| JP2022535534A (ja) | 活動性強直性脊椎炎を治療するための抗tnf抗体、組成物、及び方法 | |
| US20220064278A1 (en) | Anti-TNF Antibody Compositions for Use in Methods for the Treatment of Psoriatic Arthritis | |
| JP2023553702A (ja) | 活動性強直性脊椎炎を処置するための抗tnf抗体、組成物及び方法 | |
| KR20220029593A (ko) | 건선성 관절염의 치료를 위한 항-tnf 항체 조성물, 및 방법 | |
| JP2022518208A (ja) | 若年性特発性関節炎の治療のための抗tnf抗体、組成物、及び方法 | |
| KR20230005284A (ko) | 소아 특발성 관절염을 치료하기 위한 재료 및 방법 | |
| NZ794358A (en) | Anti-tnf antibodies, compositions, and methods for the treatment of active ankylosing spondylitis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20200917 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210803 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20211102 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20211227 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220202 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20220308 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220708 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20220708 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20220721 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20220726 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20220812 |
|
| C211 | Notice of termination of reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C211 Effective date: 20220816 |