JP2017514988A - ナノ銅製剤 - Google Patents
ナノ銅製剤 Download PDFInfo
- Publication number
- JP2017514988A JP2017514988A JP2016554873A JP2016554873A JP2017514988A JP 2017514988 A JP2017514988 A JP 2017514988A JP 2016554873 A JP2016554873 A JP 2016554873A JP 2016554873 A JP2016554873 A JP 2016554873A JP 2017514988 A JP2017514988 A JP 2017514988A
- Authority
- JP
- Japan
- Prior art keywords
- dispersion
- particles
- copper
- dispersant
- formulation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images







Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/05—Metallic powder characterised by the size or surface area of the particles
- B22F1/054—Nanosized particles
- B22F1/0545—Dispersions or suspensions of nanosized particles
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/10—Metallic powder containing lubricating or binding agents; Metallic powder containing organic material
- B22F1/102—Metallic powder coated with organic material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F1/00—Metallic powder; Treatment of metallic powder, e.g. to facilitate working or to improve properties
- B22F1/16—Metallic particles coated with a non-metal
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B22—CASTING; POWDER METALLURGY
- B22F—WORKING METALLIC POWDER; MANUFACTURE OF ARTICLES FROM METALLIC POWDER; MAKING METALLIC POWDER; APPARATUS OR DEVICES SPECIALLY ADAPTED FOR METALLIC POWDER
- B22F9/00—Making metallic powder or suspensions thereof
- B22F9/16—Making metallic powder or suspensions thereof using chemical processes
- B22F9/18—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds
- B22F9/24—Making metallic powder or suspensions thereof using chemical processes with reduction of metal compounds starting from liquid metal compounds, e.g. solutions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82B—NANOSTRUCTURES FORMED BY MANIPULATION OF INDIVIDUAL ATOMS, MOLECULES, OR LIMITED COLLECTIONS OF ATOMS OR MOLECULES AS DISCRETE UNITS; MANUFACTURE OR TREATMENT THEREOF
- B82B1/00—Nanostructures formed by manipulation of individual atoms or molecules, or limited collections of atoms or molecules as discrete units
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B29/00—Single crystals or homogeneous polycrystalline material with defined structure characterised by the material or by their shape
- C30B29/02—Elements
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B29/00—Single crystals or homogeneous polycrystalline material with defined structure characterised by the material or by their shape
- C30B29/60—Single crystals or homogeneous polycrystalline material with defined structure characterised by the material or by their shape characterised by shape
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B7/00—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions
- C30B7/14—Single-crystal growth from solutions using solvents which are liquid at normal temperature, e.g. aqueous solutions the crystallising materials being formed by chemical reactions in the solution
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05K—PRINTED CIRCUITS; CASINGS OR CONSTRUCTIONAL DETAILS OF ELECTRIC APPARATUS; MANUFACTURE OF ASSEMBLAGES OF ELECTRICAL COMPONENTS
- H05K1/00—Printed circuits
- H05K1/02—Details
- H05K1/09—Use of materials for the conductive, e.g. metallic pattern
- H05K1/092—Dispersed materials, e.g. conductive pastes or inks
- H05K1/097—Inks comprising nanoparticles and specially adapted for being sintered at low temperature
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nanotechnology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Metallurgy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Manufacture Of Metal Powder And Suspensions Thereof (AREA)
- Powder Metallurgy (AREA)
Abstract
Description
V=100・(Cu2O濃度(t)−Cu2O濃度(i)/(全Cu%)
で規定され、Cu2O濃度(t)は初期からの経過時間(t)で評価される結晶Cu2Oの濃度であり、価(V)は多くとも3%であり、経過時間(t)は少なくとも2ヵ月である。
1.平均二次粒径(d50)、
2.分散液中の銅粒子の濃度、
3.銅粒子中の単結晶銅粒子の重量百分率、
4.結晶酸化第一銅対結晶ナノ金属銅の濃度比
5.粒子状物質の加熱焼結後の粒子状物質の電気抵抗、および
6.分散液の粘度。
本発明の濃縮分散液において、ナノ銅粒子は20〜200nmの範囲内の平均二次粒径(d50)を有する。これらの分散液中の粒子状物質は、10%超の単結晶金属銅粒子、より典型的には、大部分がまたは主に単結晶の金属銅粒子を含有することができる。
本発明の濃縮分散液中のナノ銅粒子の濃度は、典型的には5重量%〜90重量%の範囲内である。ナノ粒子の生成は、それによって相対的に希釈された分散液が得られる条件で典型的に行われる。洗浄、溶媒添加および/または置換などを含み得る、希釈した分散液の処置は、銅ナノ粒子の凝集の主な一因となり得る。以下に記載の本発明のプロセスは、反応段階で高い割合の単結晶銅ナノ粒子を生成し、および銅ナノ粒子の凝集をかなりの程度まで防ぐことによってその高い割合を主に保持する。
本発明の濃縮分散液において、ナノ銅粒子は、重量ベースで、粒子数ベースで、または断面ベースで、部分的に、大部分が、または主に単結晶の銅粒子であり得る。単結晶粒子の存在は、電子後方散乱回析(EBSD)によって、定性的に証明された。以下にさらに詳細に記載のように、ランダムに選択した点で複数の走査を行うことによって結果の定量を得た。本発明の分散液において、ナノ銅粒子の少なくとも10%が単結晶金属銅粒子であり、より一般的にはナノ銅粒子の少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも40%、少なくとも50%、少なくとも60%、少なくとも70%または少なくとも80%が単結晶金属銅粒子であることを本発明者らは見出した。
本発明の濃縮分散液は、銅ナノ粒子と酸素との反応の結果、少ない割合の酸化第一銅(Cu2O)を含有し得る。酸化第一銅の存在は、粉末X線回析(粉末XRD)によって定性的に証明された。本発明の分散液において、結晶酸化第一銅対ナノ結晶金属銅の濃度比は、多くとも0.4、多くとも0.35、多くとも0.30、および典型的には、多くとも0.25、多くとも0.20、多くとも0.15、多くとも0.12、多くとも0.10、多くとも0.08、多くとも0.06または多くとも0.03であることを本発明者らは見出した。
本発明の濃縮分散液において、加熱焼結後の粒子状物質、詳細には、銅粒子の粒子状物質は、特定の範囲の電気抵抗を特徴とし得る。粒子状物質の電気抵抗は、四点プローブ(4PP)方法を用いて測定した。以下にさらに詳細に記載のように、粒子状物質を300℃で30分間の標準的な加熱焼結後、本発明の製剤中の電気抵抗が、多くとも5×10−3Ω・cm、より典型的には多くとも2×10−3Ω・cm、多くとも1×10−3Ω・cm、多くとも8×10−4Ω・cm、多くとも5×10−4Ω・cm、多くとも2×10−4Ω・cmまたは多くとも1×10−4Ω・cmであることを本発明者らは見出した。焼結した粒子状物質の比電気抵抗は、5×10−5〜5×10−3Ω・cmの範囲内、8×10−5〜2×10−3Ω・cmの範囲内または1×10−4〜1×10−3Ω・cmの範囲内であり得る。
本発明の濃縮分散液において、分散液の粘度は、分散液が使用され得る潜在的応用に大いに影響を及ぼし得る。例えば、インクジェット式印刷に好適な分散液は、噴射温度で、約5〜60cP、より典型的には5〜40cPの粘度を必要とし得る。
少なくとも1種の可溶性銅化合物を水性溶媒に溶解して、溶解した第二銅イオンを含有する第1の水溶液を生成する。次いで、還元剤、アルカリ性水素化ホウ素(例えば、水素化ホウ素ナトリウムまたは水素化ホウ素カリウム)をこの第1の溶液に、好ましくは勢いよく撹拌しながら徐々に、例えば、半連続的に加える。
2CuCl2+NaBH4+3H2O=2Cu+H3BO3+2H2+3HCl+NaCl。
しかし、さまざまな副反応または競合反応が起こることがある。
典型的に、水または水性溶媒を用いて、好適な精製システムで、ステップ1で得られた分散液を精製することができる。不都合な凝集が確実に起こらないようにまたは明らかに減少するように、粒子状物質(または銅粒子)の濃度を常に予め設定した値より下(90重量%未満、好ましくは80重量%未満、70重量%未満または60重量%未満)に維持しながら、使用済みの水性液を交換するための水または水性溶媒の精製システムへの導入を制御することができる。その結果、銅粒子の形状を有害に変化させることなく、または銅粒子を凝集させることなく、水性液中の実質的にすべての塩および大部分の分散剤が除去される。
少なくとも1種の有機溶媒を、ステップ2で利用した方法と同様の方法で、ステップ2で得られた精製分散液の水性液の大部分と置換することができる。同じ精製システムを用いてもよい。水性液を置換する際に、銅粒子のさらなる精製が得られ、これは、種々の生成物および用途に必須であり得る。
ステップ1に記載のようにナノ銅粒子を調製することによって、およびステップ2および3に記載のように処置手順を実施することによって、本発明のナノ銅分散液は格別の安定性(少なくとも6ヵ月、より一般的には少なくとも12ヵ月、少なくとも18ヵ月または少なくとも24ヵ月の有効期間が保証される)を達成することができる。
ここで、以下の実施例を参照する。実施例は上記の説明と共に本発明を非限定的な様式で例示する。
CuCl2−2H2O、99%−Sigma−Aldrich
NaBH2−Acros Organics
ポリビニルピロリドン(PVP)、MW=55,000−Sigma−Aldrich
消泡剤Contraspum1012−Zschimmer&Schwarz(Germany)
エチレングリコール(EG)−Sigma−Aldrich
プロピレングリコール(PG)−Sigma−Aldrich
ヘキシレングリコール(HG)−Sigma−Aldrich
トリプロピレングリコールモノメチルエーテル(TPM)−Sigma−Aldrich
ジエチレングリコールモノメチルエーテル(DGME)−Sigma−Aldrich
エタノール−Sigma−Aldrich
ベンジルアルコール(BA)−Sigma−Aldrich
E=20kV、作動距離−15mm、ビームの分解能は約30〜40nm、スポットサイズは4.5、プローブ電流は約0.5nA、EBSDパターンの収集時間−300msec,積算−50。EBSD画像は、背面電子を使用して撮った。
以降の処理の間に起こり得る酸化を防止するために、得られた粉末をアピエゾンMグリースと混合した。
CuCl2−2H2O(0.6モル)102gとPVP(MW=55,000)18.5gを脱イオン水900mLに溶解した(溶液A)。消泡剤(FOAMASTER(登録商標)NXZ)10滴も同様に加えた。この手順全体を通して、3L容器中の液相にアルゴンを送風した。NaBH4 45gを脱イオン水300mLに溶解した(溶液B)。勢いよく機械撹拌(500〜600rpm)しながら、溶液Bを溶液Aにかなりゆっくりと加えていった(約7cc/分)。最初の温度は約25℃、最終温度は70℃より下であった。金属銅ナノ粒子を生成し、反応混合物のpHが5.1になると溶液Bの添加を終了した。分散液をさらに10分間撹拌し、次いで貯蔵タンクに移し、さらなる処理まで静置した。得られた分散液の色は、銅ナノ粒子分散液特有の暗赤色であった。
実施例1で生成した分散液を使用した。この分散液に追加量の溶液Bをゆっくりと加えた。分散溶液のpHは10.0に達した。このpHで、金属銅粒子は、非常に凝集していた。
PVP 4.62gを使用して、実施例1を繰り返した。これらの条件下で、生成された金属銅粒子のかなりの部分は凝集した。
PVP 9.25gを使用して、実施例1を繰り返した。これらの条件下で、生成された金属銅粒子の一部は凝集した。
PVP 55.5gを使用して、実施例1を繰り返した。これらの条件下で、生成された金属銅粒子は単結晶であることが判明したが、実施例1で得られたものよりはかなり小さかった。
PVP 37.0gを使用して、実施例1を繰り返した。これらの条件下で、生成された金属銅粒子は単結晶であることが判明したが、実施例1で得られたものよりは小さかった。
約37gのナノ銅粒子を含有する実施例1の反応混合物からの分散液1300mLを、脱イオン水約50Lを使用して、クロスフロー精密濾過システムで精製(洗浄)した。分散液中の塩がほとんど除去されるまで、洗浄プロセスを続け、分散剤を予め設定した銅粒子の重量の3%の濃度まで減少させた。得られた洗浄した分散液試料の最終の色は、銅ナノ粒子分散液特有の色の暗赤色であった。
実施例3と同様の方法で調製した銅粒子を約30g含有する、銅粒子の水分散液の一部600mLを、アルゴンの一定送風下で2Lフラスコに導入し、次いで約60gのエチレングリコールを同フラスコに加えた。次いで、フラスコをR−215 Rotavapor(登録商標)エバポレーターに連結し、約10分間加熱した後、水が留出し始めるまで、160ミリバールで徐々に真空度を高めた。留出が完了するまで、真空条件を維持した。生じた60mLの銅分散液は、ナノ銅を約24.4g含有し、全体として約84gの重さであった。生成したエチレングリコール分散液の金属量は29%(金属銅)であり、粘度は34cP(25℃で)であった。
単結晶銅ナノ粒子の存在を、電子後方散乱回析(EBSD)によって、定性的に証明した。
1.集束電子ビームを使用して試料を走査し、回析パターンを得る。
2.回析画像の解釈は、機器ソフトウェア(基本的な銅の結晶データを比較する)を使用して行う。
3.回折の「解」に一致させ、結晶の方位を描写し、すべての菊池線を格子内のその適合する結晶面に相関させる。菊池線と結晶面との間で完全に一致する(理論データによって)場合、その回析は単結晶の方位を決定する。
実施例5の基本的な手順に従って、ナノ銅の試料内の単結晶金属銅ナノ粒子の存在を定量した。定量は、ランダムに選択したポイントで複数回の走査(少なくとも5回の走査、好ましくは少なくとも10回の走査)を行うことによって達成した。
図2は、本発明によって生成されたナノ銅粒子を含有する試料のSEM画像である。試料中のランダムに選択した位置を6箇所で走査した。上述のEBSDを使用して、銅単結晶の完全な一致を6箇所のうち5箇所で得た。
以下の比抵抗試験のために、本発明の銅粒子の分散液を堆積させ、熱処理して調製した。
比抵抗試験のために、本発明の銅粒子のエチレングリコール(EG)分散液を実施例8に示した手順に従って調製した。試料は、約25〜29%の金属量を有した。
本発明の銅ナノ粒子製剤の中の酸化第一銅の存在は、実施例4で生成した銅分散液を使用して、粉末X線回析(XRD)によって定性的および定量的に証明された。

分散液中の粒子状物質および特に分散液中の銅ナノ粒子中の結晶の平均粒径の推定は、XRD走査から得ることができる。
nmでの平均粒径は、表1に示すデータと、デバイ−シェラー式
D=0.9・λ/FWHM(照射)・cos(θ)
とを用いて算出することができる。
(式中、λは、使用した特定の機器におけるX線照射の波長であり、0.1541nmであった。
FWHMは、ピーク(照射での)の半値全幅であり、等しい。
FWHM(°)×3.14:180=0.24×3.14:180[照射)、
「θ」は、回析角であり、「θ」=43.702:2。)
結晶形状係数は0.9と想定されている。したがって、Cuナノ粒子の粒径D(nm)は以下のように算出される。
D=0.9×0.154/[(0.24×3.14:180)・cos(43.702:2)]=約36nm。
実施例3に記載のように調製した銅粒子を約42g含有する、銅の水分散液約900mLを一定のアルゴン送風下で2Lフラスコに導入し、次いで約25gのエチレングリコールを同フラスコに加えた。次いで同フラスコをRotavaporエバポレーターに連結し、実施例4でのように水を留出させた。生成した52gの銅分散液(約20mL)は、31gのナノ銅を含有し、約60%の金属量(金属銅)に相当した。粘度は、25℃で約1400cPであった。
約37gのナノ銅粒子を含有する、実施例1の反応混合物からの分散液1300mLを、分散液中の塩がほとんど除去されるまで、クロスフロー精密濾過システムで精製(洗浄)した。
ナノ銅粒子約37gを含有する、実施例1の反応混合物の分散液1300mLをクロスフロー精密濾過システムで精製(洗浄)した。8回の洗浄サイクルを適用し、これは脱イオン水約32Lに相当した。実施例12Aと同じように、各洗浄サイクルでは、4Lの「新しい」水を濃縮分散液1.3Lに加えた。
通常、PVP/Cu w/w%として示される、分散剤(PVP)/Cu重量/重量比は、熱重量分析(TGA)を用いて、測定することができる。
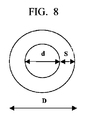
異なるPVP/Cu比率に対して分散剤(PVP)層の厚さの近似値を示すために、銅ナノ粒子は、図8に示すように、外部を有機ポリマーシェルで覆われた銅金属コアを有する球状粒子としてしばしば表される。
(1)PVP/Cu重量比を、異なる試料に関して実施例12(C)に示す手順に従って測定し、K(%PVP/Cu=100K)と表す。
K=WPVP/WCu
(2)二つの層の密度が実質的に文献値に等しい、すなわちCu密度はρCu=8.96g/mLであり、PVP密度はρPVP=1.207g/mLであると仮定すると、密度比は、
ρCu/ρPVP=8.96/1.207=7.4
(3)VCuを銅コアの体積として、VPVPをPVP外部シェルの体積として定義すると、
Wpvp/WCu=K=VPVP×ρPVP/VCu×ρCu
(4)(3)から
VCu/VPVP=(ρPVP/ρCu)/K=1/7.4Kまたは
VPVP=7.4KVCu
(5)粒子全体の体積はVD=VCu+VPVPであり、または(4)と組み合わせて
VD=VCu+7.4KVCu=(1+7.4K)VCu
(6)(5)から
VD/VCu=1+7.4Kとなる。
(7)2つの球の直径比D/dは(VD/VCu)1/3と等しいことから、(6)を用いて、
D/d=(1+7.4K)1/3またはD/d=3√(1+7.4K)
(8)したがって、任意に選択したCuコア径dと、測定したPVP/Cu比Kは、
D=3√(1+7.4K)d、およびS=(D−d)/2である。
このモデルにおいて比率S/dはKの値に依存し、および与えられたKに対して、S/dは異なるコア径を有する粒子について同じであることを強調しなければならない。
実施例3で調製した銅粒子を約28g含有する、銅の水分散液600mLに、アルゴン送風下でプロピレングリコール60gを加えた。次いで、このフラスコをRotavaporエバポレーターに連結し、実施例4と同様に水を留出させた。生成した80gの銅分散液(約60mL)はナノ銅21gを含有し、これは約26%の金属量(金属銅)に相当した。粘度は、25℃で約40cPであった。
実施例3で調製した銅粒子を約28g含有する、銅の水分散液600mLに、アルゴン送風下でヘキシレングリコール60gを加えた。次いで、このフラスコをRotavaporエバポレーターに連結し、実施例4と同様に水を留出させた。生成した82gの銅分散液(約60mL)はナノ銅24gを含有し、これは約29%の金属量(金属銅)に相当した。粘度は、25℃で約180cPであった。
第1のスッテプでは、水−エタノール溶媒交換は、クロスフロー膜システムを使用して行った。このために、実施例3と同様に調製し、約30gのナノ銅粒子を含有する銅の水分散液600mLを2.5Lのエタノールを用いて「洗浄」した。
実施例15Aの手順を繰り返したが、第2のステップではジエチレングリコールモノメチルエーテル(DGME)を使用した。銅粒子を約24g含有する、銅のエタノール分散液400mLに、アルゴン送風下でDGME16gを加えた。得られた銅分散液(38g)はナノ銅23gを含有し、これは約60.5%の金属量(金属銅)に相当した。粘度は、25℃で約40cPであった。
実施例3で調製した銅粒子を約5g含有する、銅の水分散液100mLに、アルゴン送風下でベンジルアルコール10gを加え、二相混合物を生成した。次いで、実施例4と同様に、Rotavaporエバポレーター内で水を留出させた。得られた最終分散液(12g)は、(Cuナノ分散液特有の)暗赤色から紫色であり、(目視検査および光学顕微鏡観察を含む)安定なナノ分散液であることがわかった。
実施例4、11、12および15に記載の例示的な分散液を約20℃で、酸化安定性(「エージング」)試験に供した。結果を表3に示す。種々の試料では、結晶酸化第一銅の濃度(または「酸化第一銅濃度」−粉末XRDを使用した、ピーク1の面積対ピーク1とピーク2の全面積)は、少なくとも1ヵ月、少なくとも2ヵ月、少なくとも3ヵ月または少なくとも12ヵ月にわたって実質的に維持された。
V=100・(Cu2O濃度(t)−Cu2O濃度(i))/(%全Cu)
によって定義される値V内で維持されるように、粒子状物質を適応させ、分散剤と溶媒を選択した。
(式中、
Cu2O濃度(i)はCu2Oの初期濃度または以前に測定した濃度であり、および
Cu2O濃度(t)は時間tで評価したCu2Oの濃度である。)
V=100・(8.6%−8.5%)/(29%)=0.34%
であった。
(式中、29%は分散液中の銅の総濃度(%全Cu)であり、分散液中の%Cu0として表す。)
実施例1で使用したのと同じ比率の試薬を使用したが、この場合、CuCl2溶液をNaBH4溶液に加えた。
(1)粒子状物質の代表的な試料を採取する。
(2)それぞれの液体中の粒子状物質の分散液について分析を行う。
(3)分析を室温で行う。
(4)散乱角は90度である。
Claims (29)
- ナノ金属銅粒子を含有する粒子状物質を含む製剤であって、前記粒子状物質の少なくとも10%が単結晶金属銅粒子であり、前記粒子状物質が20〜200ナノメートル(nm)の範囲内の平均二次粒径(d50)を有し、前記ナノ金属銅粒子が少なくとも1種の分散剤によって少なくとも部分的に覆われており、
前記粒子状物質中の結晶酸化第一銅対前記ナノ金属銅粒子の濃度比が多くとも0.4である、製剤。 - 層を形成している前記分散剤が0.4〜4nm、0.4〜3.5nm、0.4〜3nm、0.4〜2.5nm、0.4〜2.2nmまたは0.4〜2nmの範囲内の計算上の厚さ(S)を有する、請求項1に記載の製剤。
- 前記厚さが少なくとも0.6nm、少なくとも0.8nm、少なくとも1.0nm、少なくとも1.2nmまたは少なくとも1.5nmである、請求項2に記載の製剤。
- 前記分散剤がポリマー分散剤である、請求項1〜3のいずれか一項に記載の製剤。
- 前記粒子状物質の少なくとも2%、少なくとも3%、少なくとも5%または少なくとも7%が、少なくとも50個の粒子を有する高分解能走査型電子顕微鏡(HRSEM)視野において粒子の手動計数によって決定された、三角形の形態を有する単結晶金属銅粒子である、請求項1〜4のいずれか一項に記載の製剤。
- 前記粒子状物質の少なくとも5%、少なくとも8%、少なくとも10%または少なくとも15%が、少なくとも10個の粒子を有する高分解能透過型電子顕微鏡(HRTEM)画像視野において粒子の手動計数によって決定された、三角形の形態を有する単結晶金属銅粒子である、請求項1〜5のいずれか一項に記載の製剤。
- 前記粒子状物質の少なくとも8%、少なくとも10%、少なくとも12%または少なくとも15%、少なくとも18%または少なくとも20%が、少なくとも10個の粒子を有する高分解能透過型電子顕微鏡(HRTEM)視野において粒子の手動計数によって決定された、三角形または六角形の形態を有する単結晶金属銅粒子である、請求項1〜6のいずれか一項に記載の製剤。
- 前記濃度比が多くとも0.35、多くとも0.30、多くとも0.25、多くとも0.20、多くとも0.15、多くとも0.12、多くとも0.10、多くとも0.08、多くとも0.06、多くとも0.05、多くとも0.04または多くとも0.035である、請求項1〜7のいずれか一項に記載の製剤。
- 前記平均二次粒径が大きくとも180nm、大きくとも150nm、大きくとも120nm、大きくとも100nm、大きくとも90nm、大きくとも85nm、大きくとも80nmまたは大きくとも75nmである、請求項1〜8のいずれか一項に記載の製剤。
- 前記平均二次粒径が少なくとも25nm、少なくとも30nm、少なくとも35nm、少なくとも40nm、少なくとも45nmまたは少なくとも50nmである、請求項1〜9のいずれか一項に記載の製剤。
- 前記粒子状物質の少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも40%、少なくとも50%、少なくとも60%、少なくとも70%または少なくとも80%が前記単結晶金属銅粒子である、請求項1〜10のいずれか一項に記載の製剤。
- 前記分散剤がポリビニルピロリドン(PVP)を含む、または主に含む、または本質的にそれからなる、請求項1〜11のいずれか一項に記載の製剤。
- 前記分散剤の重量平均分子量が15,000〜150,000の範囲内である、請求項1〜12のいずれか一項に記載の製剤。
- 少なくとも第1の溶媒をさらに含み、前記粒子状物質および前記溶媒が分散液を生成し、前記分散液中の前記粒子状物質が15重量%〜70重量%、15重量%〜65重量%、20重量%〜75重量%、25重量%〜75重量%、30重量%〜75重量%、20重量%〜65重量%、25重量%〜65重量%または30重量%〜65重量%の範囲内である、請求項1〜13のいずれか一項に記載の製剤。
- 前記第1の溶媒がメタノール、エタノール、イソプロパノール、ベンジルアルコールおよびテルピネオールからなる群から任意選択で選択されるアルコールを含む、または主に含む、請求項14に記載の製剤。
- 前記第1の溶媒がグリコールおよびグリコールエーテルからなる群から選択される少なくとも1種の溶媒を含む、または主に含む、請求項14に記載の製剤。
- 前記分散剤がアラビアゴム、ポリビニルアルコール(PVA)、ポリアクリル酸(PAA)、ポリアリルアミン(PAAM)、ポリスチレンスルホン酸ナトリウム(PSS)、3−(アミノプロピル)トリメトキシシラン(APS)、脂肪酸、ラウリルアミン、セチルトリメチルアンモニウムブロミド(CTAB)、およびテトラオクチルアンモニウムブロミド(TOAB)からなる群から選択される少なくとも1種の分散剤を含む、または主に含む、請求項1〜16のいずれか一項に記載の製剤。
- 前記分散剤対前記粒子状物質の重量比は、多くとも0.04、多くとも0.03、多くとも0.025、多くとも0.022または多くとも0.020、および少なくとも0.015、少なくとも0.016、少なくとも0.017、少なくとも0.018、少なくとも0.019または少なくとも0.020である、請求項1〜17のいずれか一項に記載の製剤。
- 前記分散液中の結晶酸化第一銅の濃度が少なくとも2ヵ月、少なくとも3ヵ月、少なくとも4ヵ月、少なくとも6ヵ月または少なくとも12ヵ月にわたって実質的に維持されるように、前記粒子状物質を適応させ、および前記分散剤と前記溶媒が選択される、請求項15〜18のいずれか一項に記載の製剤。
- 前記分散液中の結晶酸化第一銅の濃度が少なくとも2ヵ月、少なくとも3ヵ月、少なくとも4ヵ月、少なくとも6ヵ月または少なくとも12ヵ月にわたって、絶対百分率ベースで2パーセント以内、1.5パーセント以内または1パーセント以内に維持されるように、前記粒子状物質を適応させ、および前記分散剤と前記溶媒が選択される、請求項15〜19のいずれか一項に記載の製剤。
- (a)第1の分散剤の存在中、酸性水性媒質中の第二銅イオンに水素化ホウ素を添加して、前記第二銅イオンを還元し、第1の分散液中に前記ナノ金属銅粒子を生成するステップと;
(b)生成物分散液中に前記ナノ金属銅粒子を供給するステップと
を含むプロセスによって生成される、請求項1〜20のいずれか一項に記載の製剤。 - 前記水素化ホウ素を添加することが増加分の添加として行われる、請求項21に記載の製剤。
- 前記プロセスは、前記水性媒質のpHが多くとも7であるとき、増加分の添加を省略することをさらに含む、請求項22に記載の製剤。
- 前記プロセスは、前記水性媒質のpHが2.5〜7の範囲内、2.5〜6.5の範囲内、または2.5〜6の範囲内にあるとき、前記増加分の添加を省略することをさらに含む、請求項22に記載の製剤。
- ステップ(a)の間、前記水性媒質が1.5未満、1.0未満または0.5未満のpHに達する、請求項21〜24のいずれか一項に記載の製剤。
- 前記プロセスが、前記粒子状物質の少なくとも10%を単結晶金属銅粒子として維持しながら、および生成物分散液中の前記粒子状物質の前記平均二次粒径を20〜200nmの範囲内に維持しながら、分散液を少なくとも6ヵ月間エージングさせることをさらに含む、請求項21〜25のいずれか一項に記載の製剤。
- 前記平均二次粒径が40〜90nmの範囲内であり、層を生成している前記分散剤が0.6〜3nmの範囲内の計算上の厚さを有し、前記粒子状物質の少なくとも40%が前記単結晶金属銅粒子であり、前記分散剤がPVPを含む、請求項1〜26のいずれか一項に記載の製剤。
- 前記粒子状物質が分散液中に配置されており、前記分散液中の前記粒子状物質の濃度が20%〜65%の範囲内または20%〜60%の範囲内であり、前記分散剤の量が前記銅粒子の重量の多くとも4重量%であり、前記分散液の粘度が25℃で多くとも70cP、多くとも60cP、多くとも50cPまたは多くとも45cPである、請求項27に記載の製剤。
- 前記分散液中の前記粒子状物質の前記濃度が少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、少なくとも45%または少なくとも50%である、請求項28に記載の製剤。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1403731.1A GB201403731D0 (en) | 2014-03-03 | 2014-03-03 | Nanometric copper formulations |
| GB1403731.1 | 2014-03-03 | ||
| PCT/IB2015/051536 WO2015132719A1 (en) | 2014-03-03 | 2015-03-03 | Nanometric copper formulations |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017514988A true JP2017514988A (ja) | 2017-06-08 |
| JP2017514988A5 JP2017514988A5 (ja) | 2018-04-12 |
Family
ID=50490712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016554873A Pending JP2017514988A (ja) | 2014-03-03 | 2015-03-03 | ナノ銅製剤 |
Country Status (10)
| Country | Link |
|---|---|
| US (3) | US10166602B2 (ja) |
| EP (1) | EP3113897B1 (ja) |
| JP (1) | JP2017514988A (ja) |
| KR (1) | KR20160128370A (ja) |
| CN (1) | CN106102970A (ja) |
| BR (1) | BR112016020056B1 (ja) |
| GB (1) | GB201403731D0 (ja) |
| IL (1) | IL247528B (ja) |
| RU (1) | RU2730285C2 (ja) |
| WO (1) | WO2015132719A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20170120135A (ko) * | 2015-03-05 | 2017-10-30 | 오사카 유니버시티 | 구리입자의 제조방법, 구리입자 및 구리페이스트 |
| CN111868977A (zh) * | 2018-05-25 | 2020-10-30 | 本田技研工业株式会社 | 用于氟离子电化学电池的复合电极材料 |
| JPWO2019098196A1 (ja) * | 2017-11-14 | 2020-11-19 | 昭和電工マテリアルズ株式会社 | 組成物、導体及びその製造方法、並びに構造体 |
| JP2021501339A (ja) * | 2017-10-31 | 2021-01-14 | エヌゲイジアイティー・デジタル・ヘルス・インコーポレイテッド | 息の中の化合物を検出し識別するためのポータブルデバイスおよび方法 |
| JP2022163005A (ja) * | 2018-03-29 | 2022-10-25 | 東邦チタニウム株式会社 | ニッケル粉体の製造方法 |
| US11881581B2 (en) | 2016-12-15 | 2024-01-23 | Honda Motor Co., Ltd. | Composite electrode materials for fluoride-ion electrochemical cells |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101886263B1 (ko) * | 2014-02-27 | 2018-08-07 | 더 스쿨 코포레이션 칸사이 유니버시티 | 구리 나노 입자 및 그 제조 방법, 구리 나노 입자 분산액, 구리 나노 잉크, 구리 나노 입자의 저장 방법 및 구리 나노 입자의 소결 방법 |
| GB201403731D0 (en) * | 2014-03-03 | 2014-04-16 | P V Nano Cell Ltd | Nanometric copper formulations |
| CL2015003794A1 (es) * | 2015-12-30 | 2016-07-29 | Univ Chile | Método de obtención de nano partículas de cobre y uso de dichas partículas |
| EP3287499B1 (en) | 2016-08-26 | 2021-04-07 | Agfa-Gevaert Nv | A metallic nanoparticle dispersion |
| WO2018226199A1 (en) * | 2017-06-05 | 2018-12-13 | Nano-Dimension Technologies, Ltd. | Flocculates of metallic, geometrically discrete nanoparticles compositions and methods of forming the same |
| CN115297978A (zh) * | 2020-03-27 | 2022-11-04 | 三井金属矿业株式会社 | 接合用组合物的制造方法 |
| CN112658245B (zh) * | 2020-12-07 | 2022-08-16 | 河南科技大学 | 一种铜纳米颗粒及其制备方法 |
| CN114054746B (zh) * | 2021-10-14 | 2022-08-16 | 华南理工大学 | 粒径呈纳米至微米三峰分布铜粉末及其一次性合成方法与应用 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003342605A (ja) * | 2002-05-21 | 2003-12-03 | Akio Komatsu | 超微粒子、超微粒子結晶膜及び超微粒子結晶の製造方法 |
| WO2013035366A1 (ja) * | 2011-09-08 | 2013-03-14 | 学校法人関西大学 | 分散安定性の高い銅ナノ粒子の製造方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7683098B2 (en) | 1996-09-03 | 2010-03-23 | Ppg Industries Ohio, Inc. | Manufacturing methods for nanomaterial dispersions and products thereof |
| US20050097987A1 (en) * | 1998-02-24 | 2005-05-12 | Cabot Corporation | Coated copper-containing powders, methods and apparatus for producing such powders, and copper-containing devices fabricated from same |
| AU5248600A (en) * | 1999-06-15 | 2001-01-02 | Kimoto, Masaaki | Ultrafine composite metal powder and method for producing the same |
| US6887297B2 (en) * | 2002-11-08 | 2005-05-03 | Wayne State University | Copper nanocrystals and methods of producing same |
| CA2728987C (en) * | 2008-05-16 | 2018-12-04 | Verutek Technologies, Inc. | Green synthesis of nanometals using plant extracts and use thereof |
| WO2010035258A2 (en) * | 2008-09-25 | 2010-04-01 | Nanoready Ltd. | Discrete metallic copper nanoparticles |
| JP5727766B2 (ja) * | 2009-12-10 | 2015-06-03 | 理想科学工業株式会社 | 導電性エマルジョンインク及びそれを用いた導電性薄膜の形成方法 |
| US20110218304A1 (en) * | 2010-03-03 | 2011-09-08 | Tecona Technologies, Inc. | Low cost and high yield method of making large quantity and homogenous metal nanoparticles and controlling their solubility |
| KR101310094B1 (ko) * | 2010-10-26 | 2013-09-24 | 한국과학기술연구원 | 구리 입자를 포함하는 탄소나노섬유, 나노입자, 분산용액 및 그 제조방법 |
| GB2486190A (en) * | 2010-12-06 | 2012-06-13 | P V Nano Cell Ltd | Concentrated dispersion of nanometric silver particles |
| KR101963801B1 (ko) * | 2011-03-29 | 2019-08-01 | 한국과학기술연구원 | 전도성 잉크 조성물, 이의 제조 방법 및 이를 이용한 전도성 박막 제조 방법 |
| US20130230717A1 (en) * | 2011-09-02 | 2013-09-05 | Washington University In St. Louis | Copper nanostructures and methods for their preparation |
| KR101886263B1 (ko) * | 2014-02-27 | 2018-08-07 | 더 스쿨 코포레이션 칸사이 유니버시티 | 구리 나노 입자 및 그 제조 방법, 구리 나노 입자 분산액, 구리 나노 잉크, 구리 나노 입자의 저장 방법 및 구리 나노 입자의 소결 방법 |
| GB201403731D0 (en) * | 2014-03-03 | 2014-04-16 | P V Nano Cell Ltd | Nanometric copper formulations |
| KR101689679B1 (ko) * | 2014-05-30 | 2016-12-26 | 한국화학연구원 | 금속 나노입자 및 이의 제조방법 |
| US20220049277A1 (en) * | 2020-08-17 | 2022-02-17 | The University of the Sciences | Metallic Nanoparticles and Methods of Making and Using the Same |
-
2014
- 2014-03-03 GB GBGB1403731.1A patent/GB201403731D0/en not_active Ceased
-
2015
- 2015-03-03 CN CN201580014550.1A patent/CN106102970A/zh active Pending
- 2015-03-03 BR BR112016020056-0A patent/BR112016020056B1/pt active IP Right Grant
- 2015-03-03 JP JP2016554873A patent/JP2017514988A/ja active Pending
- 2015-03-03 RU RU2016137018A patent/RU2730285C2/ru active
- 2015-03-03 US US15/122,185 patent/US10166602B2/en active Active
- 2015-03-03 IL IL247528A patent/IL247528B/en unknown
- 2015-03-03 EP EP15758302.2A patent/EP3113897B1/en active Active
- 2015-03-03 KR KR1020167026792A patent/KR20160128370A/ko unknown
- 2015-03-03 WO PCT/IB2015/051536 patent/WO2015132719A1/en active Application Filing
-
2018
- 2018-11-28 US US16/202,135 patent/US11590567B2/en active Active
-
2023
- 2023-02-23 US US18/113,096 patent/US20230191486A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003342605A (ja) * | 2002-05-21 | 2003-12-03 | Akio Komatsu | 超微粒子、超微粒子結晶膜及び超微粒子結晶の製造方法 |
| WO2013035366A1 (ja) * | 2011-09-08 | 2013-03-14 | 学校法人関西大学 | 分散安定性の高い銅ナノ粒子の製造方法 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20170120135A (ko) * | 2015-03-05 | 2017-10-30 | 오사카 유니버시티 | 구리입자의 제조방법, 구리입자 및 구리페이스트 |
| KR101990719B1 (ko) | 2015-03-05 | 2019-06-18 | 오사카 유니버시티 | 구리입자의 제조방법 |
| US11881581B2 (en) | 2016-12-15 | 2024-01-23 | Honda Motor Co., Ltd. | Composite electrode materials for fluoride-ion electrochemical cells |
| JP2021501339A (ja) * | 2017-10-31 | 2021-01-14 | エヌゲイジアイティー・デジタル・ヘルス・インコーポレイテッド | 息の中の化合物を検出し識別するためのポータブルデバイスおよび方法 |
| JPWO2019098196A1 (ja) * | 2017-11-14 | 2020-11-19 | 昭和電工マテリアルズ株式会社 | 組成物、導体及びその製造方法、並びに構造体 |
| US11512214B2 (en) | 2017-11-14 | 2022-11-29 | Showa Denko Materials Co., Ltd. | Composition containing organic solvents with different vapor pressures, conductor made from composition, method for manufacturing conductor, and structure comprising conductor |
| JP7276140B2 (ja) | 2017-11-14 | 2023-05-18 | 株式会社レゾナック | 組成物、導体及びその製造方法、並びに構造体 |
| JP2022163005A (ja) * | 2018-03-29 | 2022-10-25 | 東邦チタニウム株式会社 | ニッケル粉体の製造方法 |
| CN111868977A (zh) * | 2018-05-25 | 2020-10-30 | 本田技研工业株式会社 | 用于氟离子电化学电池的复合电极材料 |
| JP2021525941A (ja) * | 2018-05-25 | 2021-09-27 | 本田技研工業株式会社 | フッ化物イオン電気化学セル用バリウムドープ複合電極材料 |
| JP7458996B2 (ja) | 2018-05-25 | 2024-04-01 | 本田技研工業株式会社 | フッ化物イオン電気化学セル用バリウムドープ複合電極材料 |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112016020056B1 (pt) | 2021-06-08 |
| IL247528B (en) | 2022-09-01 |
| RU2016137018A (ru) | 2018-03-20 |
| EP3113897B1 (en) | 2019-10-09 |
| IL247528A0 (en) | 2016-11-30 |
| US11590567B2 (en) | 2023-02-28 |
| US20230191486A1 (en) | 2023-06-22 |
| US10166602B2 (en) | 2019-01-01 |
| US20190291180A1 (en) | 2019-09-26 |
| RU2730285C2 (ru) | 2020-08-21 |
| KR20160128370A (ko) | 2016-11-07 |
| EP3113897A1 (en) | 2017-01-11 |
| CN106102970A (zh) | 2016-11-09 |
| WO2015132719A1 (en) | 2015-09-11 |
| US20160368048A1 (en) | 2016-12-22 |
| RU2016137018A3 (ja) | 2018-11-06 |
| EP3113897A4 (en) | 2017-11-08 |
| GB201403731D0 (en) | 2014-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2017514988A (ja) | ナノ銅製剤 | |
| US11958116B2 (en) | Stable dispersions of monocrystalline nanometric silver particles | |
| TWI389751B (zh) | 銀微粉、銀印墨與銀塗料及該等之製造方法 | |
| JP5377483B2 (ja) | 微小金属粒子含有組成物及びその製造方法 | |
| WO2013047332A1 (ja) | 亜酸化銅粉末およびその製造方法 | |
| JPWO2009060803A1 (ja) | 銅微粒子とその製造方法及び銅微粒子分散液 | |
| JP2006022394A (ja) | 金属銅微粒子の製造方法 | |
| JP2012507114A (ja) | Iva族小粒子の組成物および関連する方法 | |
| CN108025358B (zh) | 导电材料用粉末、导电材料用油墨、导电糊剂以及导电材料用粉末的制造方法 | |
| JP6363138B2 (ja) | 単結晶ナノ銀粒子の安定な分散系 | |
| WO2023013572A1 (ja) | 金属インク、金属インクの製造方法、及び金属層の製造方法 | |
| JP7406047B2 (ja) | ニッケル粉及びニッケル粒子の製造方法 | |
| Kang et al. | Facile and scaleable transformation of Cu nanoparticles into high aspect ratio Cu oxide nanowires in methanol | |
| KR20140106376A (ko) | Pva를 이용한 용액합성법에 의한 나노 산화물 분산강화 합금 분말의 제조방법 | |
| HyunPark et al. | Synthesis of Cu nanoparticles by electron beam irradiation for oxidation stability in ethylene glycol matrix | |
| JP5139846B2 (ja) | ケトンとの親和性に優れた銀微粉および銀インク | |
| JP2008110885A (ja) | アルカリ土類金属炭酸塩粒子の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180301 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180301 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20190116 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190129 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20190418 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20190624 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190729 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20191119 |