JP2005536498A - n−ブタンからのブタジエンの製造方法 - Google Patents
n−ブタンからのブタジエンの製造方法 Download PDFInfo
- Publication number
- JP2005536498A JP2005536498A JP2004520592A JP2004520592A JP2005536498A JP 2005536498 A JP2005536498 A JP 2005536498A JP 2004520592 A JP2004520592 A JP 2004520592A JP 2004520592 A JP2004520592 A JP 2004520592A JP 2005536498 A JP2005536498 A JP 2005536498A
- Authority
- JP
- Japan
- Prior art keywords
- butane
- butene
- butadiene
- stream
- optionally
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/32—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with formation of free hydrogen
- C07C5/327—Formation of non-aromatic carbon-to-carbon double bonds only
- C07C5/333—Catalytic processes
- C07C5/3335—Catalytic processes with metals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/12—Alkadienes
- C07C11/16—Alkadienes with four carbon atoms
- C07C11/167—1, 3-Butadiene
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/32—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with formation of free hydrogen
- C07C5/327—Formation of non-aromatic carbon-to-carbon double bonds only
- C07C5/333—Catalytic processes
- C07C5/3335—Catalytic processes with metals
- C07C5/3337—Catalytic processes with metals of the platinum group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C7/00—Purification; Separation; Use of additives
- C07C7/04—Purification; Separation; Use of additives by distillation
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Water Supply & Treatment (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
本発明は、A)n−ブタン含有気体供給流を供給し、(B)n−ブタン含有気体供給流を第1脱水素化領域へと供給し、n−ブタンを1−ブテン、2−ブテン及び随意にブタジエンへと非酸化的に触媒的に脱水素化して、n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分とを含む第1生成気体流を得、(C)n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分を含む第1生成気体流を、第2脱水素化領域へと供給し、1−ブテン及び2−ブテンをブタジエンへと酸化的に脱水素化して、ブタジエン、n−ブタン、及び蒸気を、場合により第2成分を含む第2生成気体流を得、(D)第2生成気体流からブタジエンを回収する、n−ブタンからのブタジエンの製造方法に関する。
Description
本発明は、n−ブタンからのブタジエンの製造方法に関する。
ブタジエンは、通常は原材料としてナフサから出発して、飽和炭化水素の熱分解(クラッキング)することにより、主に製造されている。ナフサのクラッキングは、メタン、エタン、エテン、アセチレン、プロパン、プロペン、プロピン、アレン、ブテン、ブタジエン、ブチン、メチルアレン、C5及びより高分子量の炭化水素の炭化水素混合物を生じる。クラッキング気体中のアセチレン性不飽和炭化水素、例えばアセチレン、プロピン、1−ブチン、2−ブチン、ブテニン、及びジアセチレン等は、例えば、ディールスアルダー反応でビニルシクロヘキサンへのブタジエンの二量化が後に起こり、そのためにこれらの化合物の痕跡量であっても、銅二量体化触媒の触媒毒となる。ブチン及びアレンは、同様にブタジエンとディールスアルダー反応で反応して、副生成物の形成に至る。三重不飽和C4炭化水素は、一般に、ブタジエンの他の用途に問題となる。
特にブチンは、ブタジエンから蒸留法又は抽出法でのみ取り出し可能であるが、この場合には非常に困難を伴う。そのため、クラッキング装置からのブタジエンを使用する場合には、ブタジエンが対応するブテンへと選択的に部分水素化される水素化段階を、ブタジエンの二量化に前置することが必要である。
別な不利益としては、ナフサ又はその他の炭化水素混合物をクラッキングする場合に、複合炭化水素混合物が得られることがある。例えば、ブタジエンがクラッキング処理で得られる場合に、比較的に大量のエテン又はプロペンが不可避的に副生成物として得られる。
あるいは、ブタジエンは、n−ブタンから出発して、触媒的脱水素化によって製造可能である。しかし、この方法の不利益は、低いブタジエン収率であり、そのために、n−ブタンの触媒的脱水素化は、主として1−ブテン又は2−ブテンを生じる。
本発明の目的は、n−ブタンからブタジエンを製造する方法であって、先行技術の不利益を有さず、且つ高いブタジエン収率が得られる製造方法を提供することにある。
本発明者等は、この目的が、
(A)n−ブタン含有気体供給流を供給し、
(B)n−ブタン含有気体供給流を第1脱水素化領域へと供給し、
非酸化的に触媒的にn−ブタンを1−ブテン、2−ブテン及び随意にブタジエンへと脱水素化して、
n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分とを含む第1生成気体流を得、
(C)n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分を含む第1生成気体流を、第2脱水素化領域へと供給し、
1−ブテン及び2−ブテンをブタジエンへと酸化的に脱水素化して、
ブタジエン、n−ブタン、及び蒸気を、場合により第2成分を含む第2生成気体流を得、
(D)第2生成気体流からブタジエンを回収し、
を含む、n−ブタンからのブタジエンの製造方法によって達成されることを見いだした。
(A)n−ブタン含有気体供給流を供給し、
(B)n−ブタン含有気体供給流を第1脱水素化領域へと供給し、
非酸化的に触媒的にn−ブタンを1−ブテン、2−ブテン及び随意にブタジエンへと脱水素化して、
n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分とを含む第1生成気体流を得、
(C)n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分を含む第1生成気体流を、第2脱水素化領域へと供給し、
1−ブテン及び2−ブテンをブタジエンへと酸化的に脱水素化して、
ブタジエン、n−ブタン、及び蒸気を、場合により第2成分を含む第2生成気体流を得、
(D)第2生成気体流からブタジエンを回収し、
を含む、n−ブタンからのブタジエンの製造方法によって達成されることを見いだした。
第1処理部Aにおいて、n−ブタン含有供給気体流が供給される。通常、原材料はn−ブタン豊富な気体混合物、例えば、液化石油ガス(LPG)である。LPGは、主としてC2〜C5−炭化水素を含む。LPGの組成は非常に多様である。好適には、使用されるLPGは10質量%以上のブタンを含む。
本発明の製造方法のひとつの変形において、n−ブタン含有脱水素化供給気体流の供給は、以下の:
(A1)液化石油ガス(LPG)流を供給し、
(A2)プロパン、及び随意にメタン、エタン、及びペンタンを、LPG流から取り出して、ブタン(n−ブタン、及びイソブタン)含有流を得、
(A3)ブタン含有流からイソブタンを取り出して、n−ブタン含有供給気体流を得て、取り出したイソブタンをn−ブタン/イソブタン混合物へと随意に異性化し、n−ブタン/イソブタン混合物をイソブタン取り出し過程へと再循環する、
工程を含む。
(A1)液化石油ガス(LPG)流を供給し、
(A2)プロパン、及び随意にメタン、エタン、及びペンタンを、LPG流から取り出して、ブタン(n−ブタン、及びイソブタン)含有流を得、
(A3)ブタン含有流からイソブタンを取り出して、n−ブタン含有供給気体流を得て、取り出したイソブタンをn−ブタン/イソブタン混合物へと随意に異性化し、n−ブタン/イソブタン混合物をイソブタン取り出し過程へと再循環する、
工程を含む。
プロパン、及びメタン、エタン、及びペンタンは、1以上の通常の精留塔において取り出される。例えば、低沸点物質(メタン、エタン、プロパン)は、第1塔のオーバーヘッドで取り出すことができ、高沸点物質(ペンタン)は、第2塔の塔底部で取り出すことができる。ブタン(n−ブタン及びイソブタン)を含む流れは、例えば通常の精留塔中でイソブタンが取り出される箇所から、取り出される。残余のn−ブタン含有流は、後のブタン脱水素化のための供給気体流として使用される。
取り出されたイソブタン流を異性化処理することが好適である。この末端で、イソブタン含有流は、異性化反応装置へと供給される。n−ブタンへのイソブタンの異性化は、GB−A2018815に記載されているように行うことができる。得られたn−ブタン/イソブタン混合物は、n−ブタン/イソブタン分離塔へと供給される。
処理部Bにおいて、n−ブタン含有供給気体流は、第1脱水素化領域へと供給され、非酸化的触媒的脱水素化処理される。n−ブタンは、脱水素化反応装置中で、脱水素化触媒を通過して、1−ブタン及び2−ブタンへと部分的に脱水素化され、少量のブタジエンもまた形成される。さらに、水素及び少量のメタン、エタン、エテン、プロパン、及びプロペンが形成される。脱水素化方法に依存して、炭素酸化物(CO、CO2)、水、及び窒素もまた、非酸化的触媒的n−ブタン脱水素化の生成気体混合物中に存在する。さらに、未転化のn−ブタンが生成気体混合物中に存在する。
非酸化的触媒的n−脱水素化は、同時供給物として、場合により酸素含有気体を含んで、行うことができる。
酸化的方法と比較した非酸化的方法の特徴は、流出気体中の水素の存在である。酸化的脱水素化において、遊離の水素は実質的に形成されない。
原理的に、非酸化的触媒的n−ブタン脱水素化は、先行技術から知られるあらゆる種類の反応装置及び方法において行うことができる。本発明に適した脱水素化方法の比較的包括的記載は、「Catalytica(登録商標) Studies Division,Oxidative Dehydrogenation and Alternative Dehydrogenation Processes」(Study Number 4192OD、1993年、430 Ferguson Drive、Mountain View、California、94043−5272、USA)に見ることができる。
好適な反応装置の形態は、固定床管状又は集合管式反応装置である。これらの反応装置において、触媒(脱水素化触媒、及び、同時供給物として酸素と共に作用する場合には、随意に特別な酸化触媒)は、反応管中で又は反応集合管中で固定床として配置される。反応管は、通常は、反応管を取り囲む空間での気体、例えばメタン等の炭化水素の燃焼によって間接的に加熱される。この間接的形態の加熱は固定床の全長の最初の20〜30%にのみ施され、床の残りの長さ部分は、間接的加熱により放出される熱によって必要な反応温度にまで加熱されることが好ましい。通常の反応管の内径は、約10〜15cmである。典型的な脱水素化集合管反応装置は、約300〜1000個の反応管を含む。反応管の内部温度は、通常は、300〜1200℃の範囲、好ましくは500〜1000℃の範囲にある。処理圧力は、通常は、0.5〜8bar、少量の蒸気希釈が使用される場合には(プロパン脱水素化のリンデ過程と同様に)、しばしば1〜2barであり、あるいは高度の蒸気希釈が使用される場合には(Phillips Petroleum Co.のプロパン又はブタンの脱水素化のための蒸気活性改質法(STAR法)と同様に、US4902849、US4996387、及びUS5389342参照)、3〜8barである。典型的な気体時間空間速度(GSHV)は、使用される炭化水素に対して、500〜2000h-1である。触媒の幾何形状は、例えば、球状又はシリンダー状(中空又は非中空状)にすることができる。
非酸化的触媒的n−ブタン脱水素化は、Chem.Eng.Sci.1992年b、47(9−11)2313に記載されているように、流動床中の不均一系触媒の下でも行うことができる。好適には、2つの流動床が、並行して使用され、その1つは一般に再生処理過程にあるものである。処理圧力は、典型的には1〜2barであり、脱水素化温度は一般に550〜600℃である。脱水素化に必要な熱は、脱水素化触媒を反応温度まで予熱することにより反応系へと導入される。酸素含有同時供給物の混合は、予熱装置を不用にし、必要な熱は酸素の存在下での水素の燃焼により反応系中で直接に生成可能である。随意に、水素含有同時供給物を付加的に混合してもよい。
非酸化的触媒的n−ブタン脱水素化は、同時供給物として酸素含有気体を場合により含んで、トレイ(段)反応装置中で行うことができる。この反応装置は、1個以上の連続した触媒床を含む。触媒床の数は、1〜20個、好適には1〜6個、好ましくは1〜4個、特に1〜3個である。触媒床には好ましくは放射状に又は軸状に反応気体が通過する。一般に、そのようなトレイ反応装置は、固定触媒床を使用して操作される。最も簡易な場合には、固定触媒床は、高炉反応装置中で、又は同軸シリンダー状グリッドの環状ギャップ中で、軸状に配置されている。高炉反応装置は、1個のトレイ(段)に対応する。単一の高炉反応装置中で脱水素化を行うことは好ましい実施態様に対応し、その場合には酸素含有同時供給物が使用される。別な好ましい実施の態様において、脱水素化は、3個の触媒床を有するトレイ反応装置中で行われる。同時供給物として酸素含有気体を使用しない方法において、反応気体混合物は、トレイ反応装置中で、ひとつの触媒床から次の触媒床への途中で、例えば高温気体により加熱された熱交換板を通過することによって、又は高温燃焼気体により加熱された管を通過することによって、一定程度の加熱がなされる。
本発明の方法の好ましい実施の態様において、非酸化的触媒的n−ブタン脱水素化は、自熱式に行われる。この末端で、n−ブタン脱水素化の反応気体混合物は、さらに、1個以上の反応領域中で酸素と混合され、反応気体混合物中に存在する水素及び/又は炭化水素は、少なくとも部分的に燃焼され、これにより、1個以上の反応領域中で脱水素化に必要とされる熱の少なくとも一部が、反応気体混合物中で直接に生成される。
一般に、反応気体混合物に添加される酸素含有気体の量は、n−ブタンの脱水素化に必要とされる熱の量が、反応気体混合物中に存在する水素、及び反応気体混合物中に存在する炭化水素、及び/又はコークスの形態で存在する炭素の燃焼によって生成するように選択される。一般に、供給される酸素の総量は、ブタンの総量に対して、0.001〜0.5モル/モル、好ましくは0.005〜0.2モル/モル、より好ましくは0.05〜0.2モル/モルである。酸素は、純粋な酸素としても、あるいは不活性な気体との混合物、例えば空気の形態での酸素含有気体としても、いずれでも使用可能である。不活性な気体及び燃焼により生成した気体は、通常は、さらに希釈をもたらして、それによって不均一系触媒された脱水素化を維持する。
燃焼して熱を生成する水素は、触媒的n−ブタン脱水素化において形成された水素、及び水素含有気体として反応気体混合物へ付加的に添加されたあらゆる水素である。存在する水素の量は、H2/O2のモル比が、反応気体混合物中で、酸素が供給された直後に、1〜10モル/モル、好ましくは2〜5モル/モルとなるように好ましくは添加しなければならない。多段式反応装置においては、酸素含有及びあらゆる水素含有気体のあらゆる中間供給にこのことがあてはまる。
水素は触媒的に燃焼する。使用される脱水素化触媒は、一般に、炭化水素の及び水素の酸素による燃焼をも触媒し、そのために原理的には、特別な酸化触媒の添加は必要ない。一の実施の態様において、操作は、炭化水素の存在下で水素の酸素による燃焼を選択的に触媒する、1以上の酸化触媒の存在下で行われる。CO、CO2、及び水を生成する、これらの炭化水素の酸素による燃焼は、そのためにごくわずかな程度しか進行しない。脱水素化触媒及び酸化触媒は、好ましくは別な反応領域中に存在する。
反応が1以上の段階で行われる場合には、酸化触媒は1の、1以上の、又は全ての反応領域に存在させることができる。
水素の酸化を選択的に触媒する触媒を、反応装置中の他の位置よりも高い酸素分圧である場所、特に酸素含有気体の供給位置付近に、配置することが好適である。酸素含有気体及び/又は水素含有気体は、反応装置中の1以上の位置で供給することもできる。
本発明の方法の一の実施の態様において、酸素含有気体の、及び水素含有気体の中間供給は、トレイ反応装置の各トレイ(段)の上流にある。本発明の別な実施の態様において、酸素含有気体及び水素含有気体は、第1トレイを除く各トレイの上流で供給される。一の実施の態様において、特別な酸化触媒の層が、各供給位置の下流に位置し、脱水素化触媒の層がその後ろにある。別な実施の態様において、特別な酸化触媒は存在しない。脱水素化温度は、一般に400〜1100℃であり、トレイ反応装置の最後の触媒床中の圧力は、一般に0.2〜5bar、好ましくは1〜3barである。GSHVは、一般に500〜2000h-1であり、高負荷操作では、100000h-1にまで上げられ、好ましくは4000〜16000h-1である。
水素の燃焼を選択的に触媒する好適な触媒は、ゲルマニウム、スズ、鉛、ヒ素、アンチモン、及びビスマスの酸化物及び/又はリン酸化物からなる群より選択された酸化物及び/又はリン酸化物を含む。水素の燃焼を触媒する別な好ましい触媒は、周期表のI族及び/又はVIII族遷移元素の貴金属を含む。
使用される脱水素化触媒は、一般に、支持体及び活性成分を含む。支持体は、一般に、熱耐性酸化物又は混合酸化物からなる。脱水素化触媒は、好ましくは、酸化ジルコニウム、酸化亜鉛、酸化アルミニウム、二酸化ケイ素、二酸化チタン、酸化マグネシウム、酸化ランタン、酸化セリウム、及びこれらの混合物からなる群より選択された金属酸化物を、支持体として含む。混合物は、物理的混合物であってもよく、あるいはマグネシウムアルミニウム酸化物又は亜鉛アルミニウム酸化物混合酸化物の化学的混合相であってもよい。好ましい支持体は、二酸化ジルコニウム及び/又は二酸化ケイ素であり、特に好ましいものは、二酸化ジルコニウム及び二酸化ケイ素の混合物である。
脱水素化触媒の活性成分は、一般に、周期表のVIII族遷移元素の金属を1種以上、好ましくはプラチナ及び/又はパラジウム、より好ましくはプラチナを含む。さらに脱水素化触媒は、周期表のI族及び/又はII族の主族元素を1種以上、好ましくはカリウム及び/又はセシウムを含む。脱水素化触媒は、さらにランタノイド及びアクチノイドを含む周期表のIII族の遷移元素を1種以上、好ましくはランタン及び/又はセリウムを含む。最後に、脱水素化触媒は、周期表のIII族及び/又はIV族の主族元素を1種以上、好ましくはホウ素、ガリウム、ケイ素、ゲルマニウム、スズ及び鉛からなる群より選択された1種以上の元素、より好ましくはスズを含む。
好ましい実施の態様において、脱水素化触媒は、周期表のVIII族の遷移元素の1種以上、I及び/又はII族の主族元素の1種以上、III族及び/又はIV族の主族元素の1種以上、及びランタノイド及びアクチノイドを含むIII族の遷移元素の1種以上を含む。
例えば、WO99/46039、US4788371、EP−A705136、WO99/29420、US5220091、US5430220、US5877369、EP0117146、DE−A19937106、DE−A19937105、及びDE−A19937107に記載されているあらゆる脱水素化触媒は、本発明に使用できる。自熱式n−ブタン脱水素化の上述の変形に特に好ましい触媒は、DE−A19937107の実施例1、2、3及び4による触媒である。
蒸気の存在下でn−ブタン脱水素化を行うことが好適である。添加する蒸気は、熱キャリアとして作用し、触媒上の有機沈積物の気化を助け、これにより触媒の炭化を妨げ、触媒のオンストリーム時間(実働時間)を増大する。有機沈積物は、一酸化炭素、二酸化炭素、及び場合により水に転化される。
脱水素化触媒は、本質的に公知の方法で再生することができる。例えば、蒸気を、反応混合物に添加してもよく、又は酸素含有気体を高温で触媒に時折通過させて、沈積した炭素を焼却してもよい。蒸気での希釈は、脱水素化の生成物側へ平衡を移動する。蒸気での再生の後に、触媒は水素含有気体で随意に還元される。
n−ブタン脱水素化は、ブタジエン、1−ブテン、2−ブテン、及び未転化のn−ブタンに加えて、第2成分を含む気体混合物を提供する。通常の第2成分は、水素、蒸気、窒素、CO及びCO2、メタン、エタン、エテン、プロパン、及びプロペンを含有する。第1脱水素化領域から放出される気体混合物の組成は、脱水素化方法に依存して非常に変動しうる。例えば、水素に加えて酸素の供給を伴う好ましい自熱式脱水素化において、生成気体混合物は、比較的に高含量の蒸気及び炭素酸化物を含む。酸素の供給を伴わない方法においては、非酸化的脱水素化の生成気体混合物は、比較的高含量の水素を有する。
非酸化的自熱式n−ブタン脱水素化の生成気体流は、典型的には、0.1〜15体積%のブタジエン、1〜15体積%の1−ブテン、1〜20体積パーセントの2−ブテン、20〜70体積%のn−ブタン、5〜70体積%の蒸気、0〜5体積%の低沸点炭化水素(メタン、エタン、エテン、プロパン、及びプロペン)、0〜30体積%の水素、0〜30体積%の窒素、及び0〜5体積%の炭素酸化物を含む。
本発明によれば、非酸化的触媒的脱水素化に続けて、酸化的脱水素化(オキシデヒドロ化)が処理部Cとして行われる。
原理的には、これはあらゆる種類の反応装置と先行技術から公知の方法において、例えば流動床、トレイ炉、又は固定床管式又は集合管式反応装置において、行うことができる。本発明の方法には後者を使用することが好適である。オキシデヒドロ化を行うために、気体混合物は、酸素:n−ブテンのモル比が0.5以上であることが必要である。好適には、酸素:n−ブテンのモル比が0.55〜50である。この比に調節するために、一般に触媒的脱水素化により生じる生成気体混合物は、酸素又は酸素含有気体、例えば空気と混合される。そして、酸素含有気体混合物は、オキシデヒドロ化へと供給される。
1,3−ブタジエンへとn−ブテンをオキシデヒドロ化するのに特に適した触媒は、概して付加的に鉄を含むMo−Bi−O多金属酸化物系に基づくものである。一般に、触媒系は、周期表の1〜15族の成分、例えばカリウム、マグネシウム、ジルコニウム、クロム、ニッケル、コバルト、カドミウム、スズ、鉛、ゲルマニウム、ランタン、マンガン、タングステン、リン、セリウム、アルミニウム、又はケイ素をも付加的に含む。
有用な触媒及びこれらの調製は、例えばUS4423281(Mo12BiNi8Pb0.5Cr3K0.2Ox、及びMo12BibNi7Al3Cr0.5K0.5Ox)、US4336409((Mo12BiNi6Cd2Cr3P0.5Ox)、DE−A2600128(Mo12BiNi0.5Cr3P0.5Mg7.5K0.1Ox + SiO2)、及びDE−A2440329(Mo12BiCo4.5Ni2.5Cr3P0.5K0.1Ox)に記載されており、これらは参照文献としてここで明確に組み込まれている。
1,3−ブタジエンへとn−ブテンをオキシヒドロ化するために適した種々の多金属酸化物触媒の活性成分の化学量論は、以下の一般式(I)に包含される:
Mo12BiaFebCocNidCreX1 fKgOx (I)
(但し、上記の変数は、
X1 = W、Sn、Mn、La、Ce、Ge、Ti、Zr、Hf、Nb、P、Si、Sb、Al、Cd、及び/又はMg、
a = 0.5〜5、好ましくは0.5〜2、
b = 0〜5、好ましくは2〜4、
c = 0〜10、好ましくは3〜10、
d = 0〜10
e = 0〜10、好ましくは0.1〜4、
f = 0〜5、好ましくは0.1〜2、
g = 0〜2、好ましくは0.01〜1、且つ、
x = (I)中の酸素以外の元素の価数及び/又は出現数によって決定される数値である)。
Mo12BiaFebCocNidCreX1 fKgOx (I)
(但し、上記の変数は、
X1 = W、Sn、Mn、La、Ce、Ge、Ti、Zr、Hf、Nb、P、Si、Sb、Al、Cd、及び/又はMg、
a = 0.5〜5、好ましくは0.5〜2、
b = 0〜5、好ましくは2〜4、
c = 0〜10、好ましくは3〜10、
d = 0〜10
e = 0〜10、好ましくは0.1〜4、
f = 0〜5、好ましくは0.1〜2、
g = 0〜2、好ましくは0.01〜1、且つ、
x = (I)中の酸素以外の元素の価数及び/又は出現数によって決定される数値である)。
本発明の方法においては、オキシヒドロ化にMo−Bi−Fe−O多金属酸化物系を使用することが好ましく、Mo−Bi−Fe−Cr−O、又はMo−Bi−Fe−Zr−O金属酸化物系を使用することが特に好ましい。好ましい系が、例えば、US4547615(Mo12BiFe0.1Ni8ZrCr3K0.2Ox、及びMo12BiFe0.1Ni8AlCr3K0.2Ox)、US4424141(Mo12BiFe3Co4.5Ni2.5P0.5K0.1Ox + SiO2)、DE−A2530959(Mo12BiFe3Co4.5Ni2.5Cr0.5K0.1Ox、Mo13.75BiFe3Co4.5Ni2.5Ge0.5K0.8Ox、Mo12BiFe3Co4.5Ni2.5Mn0.5K0.1Ox、及びMo12BiFe3Co4.5Ni2.5La0.5K0.1Ox)、US3911039(Mo12BiFe3Co4.5Ni2.5Sn0.5K0.1Ox)、DE−A2530959、及びDE−A2447825(Mo12BiFe3Co4.5Ni2.5W0.5K0.1Ox)に記載されている。言及した触媒の調製と特定は、ここで明確に参照されて引用されている文献に包括的に記載されている。
オキシヒドロ化触媒は、一般に、2mmを超える平均粒径を有する成形体として使用される。処理を行うときに観察される圧力降下のために、これより小さな成形体は一般には適さない。有用な成形体には、タブレット、シリンダー、中空シリンダー、環、球、ストランド、ワゴンホイール、又は押出物が含まれる。特別な形状、例えば「三葉状(trilobes)」及び「トライスター状(tristars)」(EP−A−0593646参照)、又は1以上のノッチを外面に有する成形体(US5168090参照)も同様に可能である。
一般に、使用される触媒は、支持体のない触媒として使用される。この場合に、触媒成形体全体は、活性成分からなり、これには助剤、例えばグラファイト又は細孔形成剤、及びさらに別な成分もまた含まれている。特に、支持体のない触媒としてn−ブテンからブタジエンへのオキシヒドロ化に好適に使用されるMo−Bi−Fe−O触媒を使用することが有利であることがわかっている。さらに、触媒の活性成分を支持体、例えば無機、酸化物成形体へと、施すことが可能である。そのような触媒は、一般に被覆触媒と呼ばれる。
ブタジエンへのn−ブテンのオキシヒドロ化は、一般に220〜490℃、好ましくは250〜450℃の温度で行われる。実用上の理由から、反応装置の入口圧力は、一般に、そのプラントとそれに続く後処理の流れ抵抗を克服するのに十分であるように選択される。この反応装置の入口圧力は、一般に0.005〜1MPaで大気圧より大きく、好ましくは0.01〜0.5MPaで大気圧より大きい圧力である。その性質により、反応装置の入口領域で与えられる気体の圧力は、触媒床全体及び不活性部分にわたって実質的に減少する。
形成されるn−ブテンの酸化的脱水素化を伴った、非酸化的触媒的、好ましくは自熱式脱水素化の連結は、使用されるn−ブタンに対しての非常に高いブタジエンの収率をもたらす。非酸化的脱水素化は、穏和な方法で操作することができる。もっぱら非酸化的脱水素化と同等の収率は、選択率の明確な減少という犠牲によってのみ達成される。
ブタジエンと未転化のn−ブタンに加えて、オキシデヒドロ化から放出された第2生成気体流は、蒸気を含む。第2成分としては、一酸化炭素、二酸化炭素、酸素、窒素、メタン、エタン、エテン、プロパン、及びプロペンが、一般に含まれ、場合により水素及びさらに酸素含有炭化水素(酸素化合物として知られる)が含まれる。一般に、1−ブテン及び2−ブテンは非常に小さな割合でのみ含まれる。
例えば、オキシデヒドロ化から放出された生成気体流は、1〜20体積%のブタジエン、0〜1体積%の1−ブテン、0〜1体積%の2−ブテン、0〜50体積%のブタン、2〜50体積%の蒸気、0〜5体積%の低沸点炭化水素(メタン、エタン、エテン、プロパン、及びプロペン)、0〜20体積%の水素、0〜90体積%の窒素、0〜5体積%の炭素酸化物、0〜3体積%酸素化合物を含む。
ブタジエンは、処理部Dにおいて、オキシデヒドロ化で得られた第2生成気体流から回収される。
第2生成気体流からのブタジエンの回収は、以下の:
(D1)生成気体流を水で冷却して、蒸気及び高沸点有機第2成分を凝縮して取り出し、
(D2)水素、一酸化炭素、二酸化炭素、窒素、メタン、エタン、エテン、プロパン、及びプロペンからなる群より選択された、第2生成気体流中の低沸点第2成分を除去して、
ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを、さらに場合により別な第2成分として酸素化合物を含む流れを得、
(D3)酸素化合物を随意に取り出して、ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを得、
(D4)ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れと、ブタジエンを含む流れとに分離し、
(D5)n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、
非酸化的触媒的脱水素化(B)へと、随意に再循環する、
工程を含むことができる。
(D1)生成気体流を水で冷却して、蒸気及び高沸点有機第2成分を凝縮して取り出し、
(D2)水素、一酸化炭素、二酸化炭素、窒素、メタン、エタン、エテン、プロパン、及びプロペンからなる群より選択された、第2生成気体流中の低沸点第2成分を除去して、
ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを、さらに場合により別な第2成分として酸素化合物を含む流れを得、
(D3)酸素化合物を随意に取り出して、ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを得、
(D4)ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れと、ブタジエンを含む流れとに分離し、
(D5)n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、
非酸化的触媒的脱水素化(B)へと、随意に再循環する、
工程を含むことができる。
脱水素化段階から放出された後に、その温度が一般に220〜490℃である高温気体混合物は、通常は水で冷却される。これにより、蒸気及びあらゆる高沸点有機第2成分が凝縮される。
ブタジエン、n−ブタン、及び1−ブテン及び2−ブテンに加えて、脱水素化気体混合物中に存在している低沸点第2成分、例えば水素、一酸化炭素、二酸化炭素、窒素、メタン、エタン、エテン、プロパン、及びプロペンは、その後にC4炭化水素から取り出される。
低沸点第2成分は、通常の精留によって除去することができる。
低沸点第2成分は、高沸点吸着剤を使用した吸着/脱着サイクルで取り出すこともできる。この方法では、実質的に全ての低沸点第2成分(窒素、水素、メタン、エタン、エテン、プロパン、プロペン、炭素酸化物、酸素)が、n−ブタン脱水素化生成気体流から取り出される。
この末端で、C4−炭化水素が、吸着段階の不活性吸着剤に吸着されて、C4−炭化水素を含む吸着剤と、残余の第2成分を含む排出気体とが得られる。脱着段階では、C4−炭化水素及び第2成分の痕跡量が、再び吸着剤から放出される。
吸着段階で使用される不活性吸着剤は、一般に、取り出される炭化水素が生成気体混合物の残余の成分よりも明確に高い溶解度を有している、高沸点の非極性溶媒である。吸着は、吸着剤を通じた生成気体混合物を単に通過することによって行うことができる。しかし、塔(カラム)中で又は回転式吸着装置中で作用させることもできる。操作は、並流、向流、又は逆流で行うことができる。有用な吸着塔の例には、バブルトレイ、遠心トレイ、及び/又は網目板(シーブトレイ)を有するトレイ塔(段塔)、構造充填物、例えばMellapak(登録商標)250Yのように比表面積100〜1000m2/m3を有するシート金属充填物を有する塔、及びランダム充填塔が含まれる。しかし、有用な吸着塔には、細流塔及び噴霧塔、グラファイトブロック吸着装置、表面吸着装置、例えば厚膜及び薄膜吸着装置、及びロータリーカラム、プレートスクラバー、クロススペーススクラバー、ロータリースクラバーも含まれる。
有用な吸着剤は、比較的に非極性の有機溶媒、例えば脂肪族C8〜C18−アルケン、又は芳香族炭化水素、例えばパラフィン蒸留の中油画分、又はバルキーな基を有するエーテル、又はそれらの溶媒の混合物であり、1,2−ジメチルフタレートのような極性溶媒をそれに添加してもよい。別な有用な吸着剤には、ベンゼン酸及びフタル酸のエステルであって、直鎖C1〜C8−アルカノールとのもの、例えばn−ブチルベンゾアート、メチルベンゾアート、エーテルベンゾアート、ジメチルフタレート、ジエチルフタレート、及びさらに熱キャリア油、例えばビフェニル及びジフェニルエーテル、これらの塩素誘導体、及びさらにトリアリールアルケンが含まれる。有用な吸着剤は、ビフェニル及びジフェニルエーテルの混合物、好ましくは共沸混合物組成物、例えば市販のDiphyl(登録商標)である。しばしば、この溶媒混合物は、0.1〜25質量%のジメチルフタレートを含む。別な有用な吸着剤は、オクタン、ノナン、デカン、ウンデカン、ドデカン、トリデカン、テトラデカン、ペンタデカン、ヘキサデカン、ヘプタデカン、及びオクタデカン、又は主成分として上述した直鎖アルカンを有する製油所流から得られる画分である。
脱着のために、吸着状態の吸着剤を、加熱して及び/又は減圧して低圧にする。あるいは、脱着は、ストリッピングによって、あるいは減圧と組み合わせて、1以上の処理工程での加熱及びストリッピングによって、行うこともできる。脱着段階で再生された吸着剤は、吸着段階へと再循環される。
実質的にブタジエン及びn−ブタンからなる流れは、1−ブテン及び2−ブテン、及び別な第2成分としての酸素化合物をも含んだままであってもよい。そのような酸素化合物は、例えばフラン、マレイン酸無水物を含む。その酸素化合物は、別な分離段階でC4炭化水素から取り除くことができ、その分離段階は、同様に、吸着/脱着段階として、又は精留として、形成されていてもよい。
残余の流れは、通常は主としてブタジエン及びn−ブタンを含み、及び付加的に少量の1−ブテン及び2−ブテンを含んでいてもよく、この流れは、別な分離段階で、n−ブタン及び1−ブテン及び2−ブテンを含む流れ、及びブタジエンを含む流れに分離することができる。分離は、例えば、ブタジエンのスクラビングによって、行うことができる。ブタジエンスクラビングは、Weissermehl/Arpe、Industrielle Organische Chemie、第五版、1998年、120/121頁、又はHydrocarbon Processing、2002年三月、頁50Bに記載されているように行うことができる。
n−ブタン及び1−ブテン及び2−ブテンを含む流れは、少なくとも一部は非酸化的触媒的脱水素化(B)へと再循環することができる。
処理部(D)は、好ましくは、少なくとも工程(D1)、(D2)及び(D4)を含む。より好ましくは、工程(D1)〜(D5)を含む。
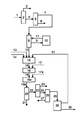
本発明を以下に図を参照して詳細に説明する。
図は、本発明の処理方法の好ましい実施の態様の処理方法フローダイアグラムを示している。実質的にプロパン、n−ブタン及びイソブタン(加えてメタン、エタン、ペンタンを含んでいてもよい)からなる液化石油ガス(LPG)の供給流1を、精留塔2へと供給し、実質的にプロパン及びメタン及びエタンから構成される流れ3と、実質的にn−ブタン及びイソブタン及びペンタンから構成される流れ4とに分離される。精留塔5において、ペンタン6が取り出される。ブタン混合物7は、精留塔8中で、イソブタン9とn−ブタン12とに分離され、イソブタンは、異性化反応装置10中で、n−ブタン/イソブタン混合物11へと異性化され、これは精留塔8へと戻して供給される。n−ブタンは、供給気体流12として、第1脱水素化段階15へと供給され、そこではブタンが1−ブテン、2−ブテン、及びブタジエンとなる非酸化的触媒的脱水素化が行われる。これは好ましくは、酸素又は空気を同時供給13として、随意に水素を同時供給14として供給しながら、自熱式条件下で行われる。第1脱水素化段階は、流動床中で逆混合しながら、又は部分的気体再循環をしながら、例えばドイツ国特許出願P10211275.4(この出願は本発明の優先日において公開されていない)に記載されているように、行うことが好ましい。第1脱水素化段階から放出された生成気体流16は、ブタジエン、1−ブテン、2−ブテン及び未転化のn−ブタンに加えて、蒸気及び通常の第2成分、例えば水素、炭素酸化物、窒素、水素、メタン、エタン、エテン、プロパン及びペンタンを含んでおり、第2脱水素化段階18へと供給され、ここでは酸素又は空気を同時供給17として供給しながら、1−ブテン及び2−ブテンからブタジエンへのオキシデヒドロ化が行われる。第2脱水素化段階は、好ましくは集合管式反応装置で行われる。第2脱水素化段階は好ましくは1以上の段階で、例えば2段階で行われる。オキシデヒドロ化の2段階構成において、第2脱水素化段階は、第1オキシデヒドロ化段階18及び第2オキシデヒドロ化段階18aからなり、空気又は酸素がそれぞれ同時供給17又は17aとして供給される。第2脱水素化段階から放出された生成気体流19a(1段階構成オキシデヒドロ化においては、これは生成気体流19である)は、ブタジエン及び未転化のn−ブタンに加えて、蒸気及び第2成分、例えば水素、炭素酸化物、窒素、メタン、エタン、エテン、プロパン、及び/又はプロペンを、場合により1−ブテン及び2−ブテンの少量の残余を、場合により酸素及び酸素含有炭化水素(酸素化合物)を、含んでいる。生成気体流19aは、随意に熱交換器で予冷した後に、冷却凝縮ユニット20で冷却され、このユニットは、水と高沸点有機副生成物、例えば高沸点炭化水素及び酸素化合物が、凝縮され、流れ21として処理経路から排出されるように、例えば水流動床として、又は流下薄膜式凝縮装置として構成することができる。未凝縮の生成気体成分は、分離段階23へ、流れ22として供給され、ここで低沸点物質及び未凝縮の第2成分24(生成気体流19aに存在する場合: 水素、炭素酸化物、窒素、エタン、エタン、エテン、プロパン、プロペン、及び酸素)が取り出される。分離段階23は、精留塔又は吸着/脱着ユニットとして構成することができる。流れ25は、脱水素化のC4生成物、未転化のn−ブタン、及びあらゆる酸素化合物、例えばフラン及びマレイン酸無水物を含み、精留塔又は吸着/脱着ユニットとして構成できる別な分離段階26へと、随意に供給される。分離段階26において、酸素化合物及び残余の水の痕跡量が取り除かれ、流れ27として処理経路から排出される。流れ28は、ブタジエン及びn−ブタンからなり、少量の1−ブテン及び2−ブテンを含んでいてもよく、これは別な分離段階29、例えばブタジエンスクラビングへと供給され、そこで、n−ブタン及び1−ブテン及び2−ブテンからなる流れ31と、ブタジエンからなる流れ31へと分離される。流れ31は、少なくとも部分的に、非酸化的触媒的脱水素化段階15へと再循環することができる。
本発明を以下の実施例によって詳細に説明する。
[実施例1]
脱水素化触媒前駆体の調製
600mlのエタノール中に11.993gのSnCl2・2H2O及び7.886gのH2PtCl6・6H2Oを含む溶液を、1000gの粉砕したZrO2/SiO2混合酸化物(米国Norton製、ZrO2/SiO2の質量比が95:5)に注ぎかけた。
脱水素化触媒前駆体の調製
600mlのエタノール中に11.993gのSnCl2・2H2O及び7.886gのH2PtCl6・6H2Oを含む溶液を、1000gの粉砕したZrO2/SiO2混合酸化物(米国Norton製、ZrO2/SiO2の質量比が95:5)に注ぎかけた。
混合酸化物は、以下の特性を有する:
篩画分1.6〜2mm; BET表面積:86m2/g; 孔体積:0.28ml/g(水銀多孔度測定法による)。
篩画分1.6〜2mm; BET表面積:86m2/g; 孔体積:0.28ml/g(水銀多孔度測定法による)。
上清のエタノールは、水流ポンプの真空(20mbar)を用いた回転式蒸発装置(ロータリーエバポレーター)で取り除いた。いずれも空気中で、次に100℃で15時間、乾燥処理した後に、560℃で3時間、か焼した。次に、2500mlのH2O中に7.71gのCsNO3、13.559gのKNO3、及び98.33gのLa(NO3)3・6H2Oを含む溶液を、乾燥固体に注いだ。上清の水は、水流ポンプの真空(20mbar)を用いた回転式蒸発装置(ロータリーエバポレーター)で取り除いた。いずれも空気中で、次に100℃で15時間、乾燥処理した後に、560℃で3時間、か焼した。
得られた触媒前駆体は、Pt0.3Sn0.6Cs0.5K0.5La3.0(指数は質量比を表す)の組成物を、キャリアとして(ZrO2)95(SiO2)5(指数は質量比を表す)上に有していた。
[実施例2]
脱水素化領域A反応装置の充填及び触媒前駆体の活性化
実施例1から得られた20mlの触媒前駆体を、垂直管式反応装置(反応装置長:800mm; 壁厚:2mm; 内径:20mm; 反応装置材料:内部アロナイズ処理、即ちアルミニウム酸化物被覆処理、スチール管; 加熱:HTM Reetz製オーブン(LOBA 1100−28−650−2、縦方向中央部長さ650mm)を使用した電気的加熱)に充填して使用した。触媒床の長さは、75mmである。触媒床は、管式反応装置の縦方向中央部に設置した。その上下に残った反応装置内容積は、不活性材料としてステアタイト球(直径4〜5mm)で充填され、これは触媒ベースの上で下から支持されている。
脱水素化領域A反応装置の充填及び触媒前駆体の活性化
実施例1から得られた20mlの触媒前駆体を、垂直管式反応装置(反応装置長:800mm; 壁厚:2mm; 内径:20mm; 反応装置材料:内部アロナイズ処理、即ちアルミニウム酸化物被覆処理、スチール管; 加熱:HTM Reetz製オーブン(LOBA 1100−28−650−2、縦方向中央部長さ650mm)を使用した電気的加熱)に充填して使用した。触媒床の長さは、75mmである。触媒床は、管式反応装置の縦方向中央部に設置した。その上下に残った反応装置内容積は、不活性材料としてステアタイト球(直径4〜5mm)で充填され、これは触媒ベースの上で下から支持されている。
反応管は、次に、500℃の加熱領域に沿った外壁温度で、30分間にわたり、9.3l/h(stp)の水素で満たした。同じ壁温で、水素流を、80体積%の窒素と20体積%の空気の流れにより23l/h(STP)で30分間かけて最初に置換し、次に純粋な空気の同じ流れにより30分間をかけて置換した。壁温を保持しつつ、N2の同じ流れで15分間かけてパージ(浄化)を行い、最後に9.3l/h(STP)の水素で30分間かけて再び還元を行った。触媒前駆体の活性化はこれで完了した。
[実施例3]
オキシデヒドロ化触媒の調製
最初に、0.2質量%の遊離のHNO3含量及び12.5質量%のCo含量(=Co 3.71モル)を有する1750.9gの硝酸コバルト水溶液を、熱耐性ガラス製10L固体反応装置に充填した。14.2質量%のFe含量(=Fe 1.59モル)を有する626.25gの固体Fe(NO3)3・9H2Oを、最初に充填した硝酸コバルト溶液中で、室温で撹拌しながら溶解した。3質量%の遊離のHNO3含量及び11.1質量%のBi含量(=Bi 0.32モル)を有する599.5gの硝酸ビスマス溶液を、蒸気得られた溶液に室温で添加した。106.23gの固体Cr(NO3)3・9H2O(=Cr 0.27モル)を次に添加した。60℃に加熱してさらに撹拌した後に、赤色溶液(溶液A)が得られた。
オキシデヒドロ化触媒の調製
最初に、0.2質量%の遊離のHNO3含量及び12.5質量%のCo含量(=Co 3.71モル)を有する1750.9gの硝酸コバルト水溶液を、熱耐性ガラス製10L固体反応装置に充填した。14.2質量%のFe含量(=Fe 1.59モル)を有する626.25gの固体Fe(NO3)3・9H2Oを、最初に充填した硝酸コバルト溶液中で、室温で撹拌しながら溶解した。3質量%の遊離のHNO3含量及び11.1質量%のBi含量(=Bi 0.32モル)を有する599.5gの硝酸ビスマス溶液を、蒸気得られた溶液に室温で添加した。106.23gの固体Cr(NO3)3・9H2O(=Cr 0.27モル)を次に添加した。60℃に加熱してさらに撹拌した後に、赤色溶液(溶液A)が得られた。
最初に、耐熱性3l撹拌ガラス容器中に2000mlの水を満たした。次に、2.38gのKOH(=K 0.042モル)及び1124.86gの(NH4)6Mo7O24・4H2O(=Mo 6.37モル)を添加して、60℃で溶解した。得られた溶液はやや濁りを示した。(溶液B)
次に、溶液Aを撹拌しながら、溶液Bを溶液Aへとポンプ注入した。102.05gの50質量%のSiO2含量を有するSiO2ゾル(「Ludox TM」、デュポン製、=Si 0.85モル)を、60℃で得られた暗黄色懸濁液へと添加した。
次に、溶液Aを撹拌しながら、溶液Bを溶液Aへとポンプ注入した。102.05gの50質量%のSiO2含量を有するSiO2ゾル(「Ludox TM」、デュポン製、=Si 0.85モル)を、60℃で得られた暗黄色懸濁液へと添加した。
得られた懸濁液を60℃で30分間撹拌し、次に噴霧乾燥した(入口温度370℃、出口温度110〜112℃)。得られた噴霧粉末を、4質量%のグラファイトと混合し、次に直径5mm高さ3mmの固体タブレットへと成錠した。固体タブレット(錠)を、マッフル炉中で、網(ワイアシーブ)(メッシュの大きさ3.5mm)上で、480℃で6時間、熱処理して、100l/hの速度の空気流によって、空気を通過させた。か焼したタブレットは、網を通じて細分して、平均顆粒径2〜3mmを有する触媒細片(スパル)を得た。
オキシデヒドロ化触媒は、公称組成Mo12Bi0.6Fe3Co7Cr0.5Si1.6K0.08Ox(指数は原子比を表す)を有していた。
[実施例4]
脱水素化領域B反応装置の充填
実施例3で得られた95mlの触媒前駆体を、垂直管式反応装置(反応装置長:100cm; 壁厚:2mm; 内径:13mm; 反応装置材料:内部アロナイズ処理されたスチール管、その内部にはサーモウェルが設置され、外径2mmで可動式温度部材(moveable thermal element)を含む; 加熱:電気的加熱、100cmの反応装置長にわたる3個の独立した加熱領域を備え、加熱カラーを使用、Winkler(Heidelberg)製、82cmの最大等温長を反応装置中央部領域にかけて達成)に充填して使用した。触媒床の長さは82cmであった。触媒床は、管式反応装置の等温領域にある。上下に残った反応装置内容積は、不活性材料としてステアタイト球(直径2〜3mm)を充填して、反応管の充填全体は、高さ5cmの触媒ベースの上で下から支持されている。
脱水素化領域B反応装置の充填
実施例3で得られた95mlの触媒前駆体を、垂直管式反応装置(反応装置長:100cm; 壁厚:2mm; 内径:13mm; 反応装置材料:内部アロナイズ処理されたスチール管、その内部にはサーモウェルが設置され、外径2mmで可動式温度部材(moveable thermal element)を含む; 加熱:電気的加熱、100cmの反応装置長にわたる3個の独立した加熱領域を備え、加熱カラーを使用、Winkler(Heidelberg)製、82cmの最大等温長を反応装置中央部領域にかけて達成)に充填して使用した。触媒床の長さは82cmであった。触媒床は、管式反応装置の等温領域にある。上下に残った反応装置内容積は、不活性材料としてステアタイト球(直径2〜3mm)を充填して、反応管の充填全体は、高さ5cmの触媒ベースの上で下から支持されている。
[実施例5]
脱水素化領域A反応装置中のn−ブタンの脱水素化
実施例2の脱水素化領域A反応装置を、500℃の加熱領域に沿った外壁温度で、反応気体混合物として、20l/h(stp)のn−ブタン、3.5l/h(stp)の空気、1.4l/h(stp)の水素、及び10l/h(stp)の蒸気の混合物で、満たした。
脱水素化領域A反応装置中のn−ブタンの脱水素化
実施例2の脱水素化領域A反応装置を、500℃の加熱領域に沿った外壁温度で、反応気体混合物として、20l/h(stp)のn−ブタン、3.5l/h(stp)の空気、1.4l/h(stp)の水素、及び10l/h(stp)の蒸気の混合物で、満たした。
n−ブタン、空気、及び水素は、Brooks製の質量流量調節装置を使用して最初に計量導入し、一方、水はKontron HPLCポンプ420を使用して液体の形態でエバポレーター(蒸発装置)へと最初に計量導入し、その中で蒸発させ、次にn−ブタン及び空気に混合した。充填された充填気体混合物の温度は150℃であった。REKO圧力調節装置を反応装置の出口で使用して、管式反応装置の出口圧力を1.5barに設定した。
得られた生成気体混合物Aの分析量を、大気圧へと減圧し、冷却して存在する蒸気を凝縮して取り出した。残存気体は、GC(HP6890 Chem−Station附属、検出部:FID; TCD: 分離カラム:Al2O3/KCl(Chromopack)、Carboxen1010(Supelco))を使用して分析した。対応する方法で、充填気体混合物も分析した。
3日間の操作時間の後、表1に報告する分析結果が得られた。
1段階あたり32モル%のn−ブタン転化と94モル%のn−ブタン形成の選択率とが、これらの値に相当する。ブタジエン形成の選択率は3.3%に相当する。
[実施例6]
脱水素化領域A反応装置でのn−ブタンの脱水素化と、これに続く脱水素化領域B反応装置でのオキシデヒドロ化。
脱水素化領域A反応装置でのn−ブタンの脱水素化と、これに続く脱水素化領域B反応装置でのオキシデヒドロ化。
実施例4の脱水素化領域B反応装置は、反応気体混合物の単一スループットでのn−ブテン転化が99モル%を超えるような温度に加熱され、反応装置の内部温度は、内部サーモウェル中に設置された温度部材を使用して調節される。
充填は、150l/h(stp)の空気(=20℃)と、実施例5の34.4l/h(stp)の生成気体混合物A(=500℃)との混合物からなる。空気は、Brooks製の質量流量調節装置を使用して計量導入した。気体混合物の充填の温度は、反応装置外壁温度にされた。反応管の出口の圧力調節装置を使用して、反応装置の出口圧力は1.3barに設定された。
圧力調節装置の下流では、得られた生成気体混合物流B(温度=330℃)は、大気圧に減圧され、GC(HP6890 Chem−Station附属、検出部:FID; TCD: 分離カラム:Poraplot Q(Chromopack)、Carboxen1010(Supelco))を使用して分析された。充填気体混合物は、同じ方法で分析した。
3日間の操作時間の後、表2に報告する分析結果が得られた。
1段階あたり99モル%のn−ブタン転化と80モル%のn−ブタン形成の選択率とが、これらの値に相当する。
脱水素化領域AとBの両方にわたるブタジエンの全体収率は、使用したn−ブタンに対して、25%であった。
[比較例]
実施例5に記載の充填気体混合物を、脱水素化領域B反応装置へと直接に通した。同じ反応条件の下で、n−ブタンからブテン又はブタジエンへの転化は生じなかった。
実施例5に記載の充填気体混合物を、脱水素化領域B反応装置へと直接に通した。同じ反応条件の下で、n−ブタンからブテン又はブタジエンへの転化は生じなかった。
Claims (5)
- (A)n−ブタン含有気体供給流を供給し、
(B)n−ブタン含有気体供給流を第1脱水素化領域へと供給し、
n−ブタンを1−ブテン、2−ブテン及び随意にブタジエンへと非酸化的に触媒的に脱水素化して、
n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分を含む第1生成気体流を得、
(C)n−ブタン、1−ブテン及び2−ブテンを、場合によりブタジエンと第2成分を含む第1生成気体流を、第2脱水素化領域へと供給し、
1−ブテン及び2−ブテンをブタジエンへと酸化的に脱水素化して、
ブタジエン、n−ブタン、及び蒸気と、場合により第2成分を含む第2生成気体流を得、
(D)第2生成気体流からブタジエンを回収する、
n−ブタンからのブタジエンの製造方法。 - n−ブタン含有供給気体流の供給が、以下:
(A1)液化石油ガス(LPG)流を供給し、
(A2)プロパン、及び随意にメタン、エタン、及びペンタンを、LPG流から取り出して、ブタン含有流を得、
(A3)ブタン含有流からイソブタンを取り出して、n−ブタン含有供給気体流を得て、取り出したイソブタンをn−ブタン/イソブタン混合物へと異性化し、n−ブタン/イソブタン混合物をイソブタン取り出し過程へと随意に再循環する、
ことを特徴とする、請求項1に記載の製造方法。 - n−ブタンの非酸化的触媒的脱水素化(B)が、自熱式触媒的脱水素化として行われることを特徴とする、請求項1又は請求項2に記載の製造方法。
- 酸化的脱水素化(C)が、1を超える段階で行われることを特徴とする請求項1〜3のいずれかに記載の製造方法。
- 第2生成気体流からのブタジエンの回収(D)が、以下:
(D1)生成気体流を水で冷却して、蒸気及び高沸点有機第2成分を凝縮して取り出し、
(D2)水素、一酸化炭素、二酸化炭素、窒素、メタン、エタン、エテン、プロパン、及びプロペンからなる群より選択された、第2生成気体流中の低沸点第2成分を除去して、
ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを、さらに場合により別な第2成分として酸素化合物を含む流れを得、
(D3)酸素化合物を随意に取り出して、ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを得、
(D4)ブタジエン及びn−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れと、ブタジエンを含む流れとに分離し、
(D5)n−ブタンを、場合により1−ブテン及び2−ブテンを含む流れを、
非酸化的触媒的脱水素化(B)へと、随意に再循環し、
を含むことを特徴とする、請求項1〜4のいずれかに記載の製造方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10231632 | 2002-07-12 | ||
| PCT/EP2003/007523 WO2004007408A1 (de) | 2002-07-12 | 2003-07-10 | Verfahren zur herstellung von butadien aus n-butan |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2005536498A true JP2005536498A (ja) | 2005-12-02 |
Family
ID=30009935
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004520592A Withdrawn JP2005536498A (ja) | 2002-07-12 | 2003-07-10 | n−ブタンからのブタジエンの製造方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US7034195B2 (ja) |
| EP (1) | EP1523462B1 (ja) |
| JP (1) | JP2005536498A (ja) |
| KR (1) | KR100974260B1 (ja) |
| CN (1) | CN100494130C (ja) |
| AT (1) | ATE461165T1 (ja) |
| AU (1) | AU2003254335A1 (ja) |
| BR (1) | BR0311949A (ja) |
| CA (1) | CA2493133A1 (ja) |
| DE (1) | DE50312525D1 (ja) |
| EA (1) | EA007394B1 (ja) |
| MX (1) | MXPA04012234A (ja) |
| MY (1) | MY135793A (ja) |
| NO (1) | NO20045324L (ja) |
| TW (1) | TWI324591B (ja) |
| WO (1) | WO2004007408A1 (ja) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010120933A (ja) * | 2008-10-24 | 2010-06-03 | Asahi Kasei Chemicals Corp | 共役ジオレフィンの製造方法 |
| JP2010534553A (ja) * | 2007-05-10 | 2010-11-11 | エスケー エナジー 株式会社 | 亜鉛フェライト触媒、その製造方法、およびそれを使用する1,3−ブタジエンの製造方法 |
| JP2012067048A (ja) * | 2010-09-27 | 2012-04-05 | Asahi Kasei Chemicals Corp | ブタジエンの製造方法 |
| JP2012512181A (ja) * | 2009-03-25 | 2012-05-31 | ルマス テクノロジー インコーポレイテッド | ブタンからのプロピレンの生成 |
| JP2013502414A (ja) * | 2009-08-17 | 2013-01-24 | ルマス テクノロジー インコーポレイテッド | ブタジエンの製造法 |
| JP2013100244A (ja) * | 2011-11-08 | 2013-05-23 | Mitsui Chemicals Inc | 共役ジオレフィンの製造方法 |
| JP2013189440A (ja) * | 2007-05-30 | 2013-09-26 | Sk Innovation Co Ltd | 多成分系モリブデン酸ビスマス触媒を使用した1,3−ブタジエンの製造方法。 |
| JP2014073462A (ja) * | 2012-10-04 | 2014-04-24 | Cosmo Oil Co Ltd | 金属酸化物触媒、その製造方法及びアルカジエンの製造方法 |
| JP2014519478A (ja) * | 2011-04-04 | 2014-08-14 | エボニック デグサ ゲーエムベーハー | 1−ブテンおよび1,3−ブタジエン誘導体の製造方法 |
| JP2015509108A (ja) * | 2012-01-30 | 2015-03-26 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | n−ブタンからのブタジエンおよび/またはブテン類の製造方法 |
| WO2015186915A1 (ko) * | 2014-06-03 | 2015-12-10 | 주식회사 엘지화학 | 산화탈수소화 반응을 통한 부타디엔의 제조방법 |
| JP2017509483A (ja) * | 2014-12-05 | 2017-04-06 | エルジー・ケム・リミテッド | ブタジエン製造用複合酸化物触媒及びその製造方法 |
| US9738574B2 (en) | 2014-06-03 | 2017-08-22 | Lg Chem, Ltd. | Method for producing butadiene through oxidative dehydrogenation reaction |
| JP2018197215A (ja) * | 2017-05-24 | 2018-12-13 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
| JP2019137664A (ja) * | 2018-02-15 | 2019-08-22 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
| JP2019137663A (ja) * | 2018-02-15 | 2019-08-22 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
Families Citing this family (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10350044A1 (de) * | 2003-10-27 | 2005-05-25 | Basf Ag | Verfahren zur Herstellung von 1-Buten |
| DE10350045A1 (de) * | 2003-10-27 | 2005-05-25 | Basf Ag | Verfahren zur Herstellung von 1-Buten |
| DE10361823A1 (de) * | 2003-12-30 | 2005-08-11 | Basf Ag | Verfahren zur Herstellung von Butadien und 1-Buten |
| DE10361824A1 (de) * | 2003-12-30 | 2005-07-28 | Basf Ag | Verfahren zur Herstellung von Butadien |
| DE10361822A1 (de) * | 2003-12-30 | 2005-08-11 | Basf Ag | Verfahren zur Herstellung von Butadien |
| RU2269504C1 (ru) * | 2004-10-25 | 2006-02-10 | Открытое акционерное общество Научно-исследовательский институт "Ярсинтез" (ОАО НИИ "Ярсинтез") | Способ получения бутадиена-1,3 |
| DE102004054766A1 (de) * | 2004-11-12 | 2006-05-18 | Basf Ag | Verfahren zur Herstellung von Butadien aus n-Butan |
| DE102004059356A1 (de) | 2004-12-09 | 2006-06-14 | Basf Ag | Verfahren zur Herstellung von Butadien aus n-Butan |
| DE102004061514A1 (de) * | 2004-12-21 | 2006-07-06 | Basf Ag | Verfahren zur Herstellung von Butadien aus n-Butan |
| DE102005002127A1 (de) | 2005-01-17 | 2006-07-20 | Basf Ag | Verfahren zur Herstellung von Butadien aus n-Butan |
| US7943038B2 (en) * | 2008-01-29 | 2011-05-17 | Kellogg Brown & Root Llc | Method for producing olefins using a doped catalyst |
| US7910784B2 (en) * | 2008-06-14 | 2011-03-22 | Lummus Technology Inc. | Process for the production of styrene monomer by improving energy efficiency and injecting a recycle gas into the EB vaporizer |
| CN102639227B (zh) | 2009-12-01 | 2014-10-01 | 巴斯夫欧洲公司 | 用于进行自热气相脱氢的反应器 |
| KR20130130791A (ko) | 2010-12-21 | 2013-12-02 | 바스프 에스이 | 자열 기상 탈수소화를 수행하기 위한 반응기 |
| US8802019B2 (en) | 2010-12-21 | 2014-08-12 | Basf Se | Reactor for carrying out an autothermal gas-phase dehydrogenation |
| CA2832887A1 (en) | 2011-04-11 | 2012-10-18 | ADA-ES, Inc. | Fluidized bed method and system for gas component capture |
| CN107254492A (zh) * | 2011-06-17 | 2017-10-17 | 英威达技术有限责任公司 | 产生1,3‑丁二烯的方法 |
| EA025292B1 (ru) | 2011-08-02 | 2016-12-30 | Басф Се | Реактор для проведения автотермического газофазного дегидрирования |
| US8852538B2 (en) | 2011-08-02 | 2014-10-07 | Basf Se | Reactor for carrying out an autothermal gas-phase dehydrogenation |
| EA201490316A1 (ru) | 2011-08-02 | 2014-11-28 | Басф Се | Непрерывный способ проведения автотермического газофазного дегидрирования |
| US9012707B2 (en) | 2011-08-02 | 2015-04-21 | Basf Se | Continuous process for carrying out autothermal gas-phase dehydrogenations |
| KR101338637B1 (ko) * | 2011-10-06 | 2013-12-06 | 삼성토탈 주식회사 | 노르말-부탄의 산화적 탈수소화 반응 촉매용 마그네시아-지르코니아 복합담체의 단일 단계 침전법에 의한 제조방법, 그에 의해 제조된 마그네시아-지르코니아 복합담체에 담지된 마그네슘 오르소바나데이트 촉매 및 상기 촉매를 이용한 노르말-부텐과 1,3-부타디엔의 제조방법 |
| CN102417432A (zh) * | 2011-12-16 | 2012-04-18 | 天津市泰亨气体有限公司 | 采用丁烷催化脱氢制备1,3-丁二烯的方法 |
| US9193647B2 (en) | 2012-01-30 | 2015-11-24 | Basf Se | Process for preparing butadiene and/or butenes from n-butane |
| CN103483123B (zh) * | 2012-06-08 | 2015-05-13 | 中国石油化工股份有限公司 | 一种由正构烷烃制备丁二烯的方法 |
| RU2488440C1 (ru) * | 2012-07-18 | 2013-07-27 | ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ УЧРЕЖДЕНИЕ НАУКИ ИНСТИТУТ ОРГАНИЧЕСКОЙ ХИМИИ им. Н.Д. ЗЕЛИНСКОГО РОССИЙСКОЙ АКАДЕМИИ НАУК (ИОХ РАН) | Катализатор для непрерывного окислительного дегидрирования этана и способ непрерывного окислительного дегидрирования этана с его использованием |
| WO2014047354A1 (en) | 2012-09-20 | 2014-03-27 | ADA-ES, Inc. | Method and system to reclaim functional sites on a sorbent contaminated by heat stable salts |
| US9611191B2 (en) | 2012-12-12 | 2017-04-04 | Basf Se | Reactor for carrying out an autothermal gas-phase dehydrogenation |
| CN103962058A (zh) | 2013-01-30 | 2014-08-06 | 中国石油化工股份有限公司 | 预混合器、径向固定床反应器和丁烯氧化脱氢反应系统 |
| CN103086829B (zh) * | 2013-01-30 | 2016-01-13 | 中国石油化工股份有限公司 | 丁烯氧化脱氢废水回用方法 |
| CN103965005B (zh) * | 2013-01-30 | 2016-04-13 | 中国石油化工股份有限公司 | 丁烯氧化脱氢产物的脱酸方法 |
| CN103965007B (zh) * | 2013-01-30 | 2015-11-18 | 中国石油化工股份有限公司 | 混合碳四增产丁二烯的方法 |
| CN103086830B (zh) * | 2013-01-30 | 2015-04-08 | 中国石油化工股份有限公司 | 生产丁二烯的方法 |
| US10035740B2 (en) | 2013-03-07 | 2018-07-31 | Tpc Group Llc | Multi-stage oxidative dehydrogenation process with inter-stage cooling |
| US10035741B2 (en) | 2013-03-07 | 2018-07-31 | Tpc Group Llc | High throughput oxidative dehydrogenation process |
| CN104250184B (zh) * | 2013-06-28 | 2016-01-13 | 中国石油化工股份有限公司 | 一种低碳烯烃的制备方法 |
| CN104250193B (zh) * | 2013-06-28 | 2016-12-28 | 中国石油化工股份有限公司 | 一种丙烯和丁二烯的制备方法 |
| EP2832716A1 (de) * | 2013-07-29 | 2015-02-04 | LANXESS Deutschland GmbH | 1,3-Butadien-Synthese |
| CN103483130B (zh) * | 2013-09-23 | 2015-05-20 | 浙江大学 | 用Bi/Mo/La/Fe四组分复合氧化物催化剂合成1,3-丁二烯的方法 |
| US20150166439A1 (en) * | 2013-12-16 | 2015-06-18 | Uop Llc | Integration of mto with on purpose butadiene |
| DE102013226370A1 (de) | 2013-12-18 | 2015-06-18 | Evonik Industries Ag | Herstellung von Butadien durch oxidative Dehydrierung von n-Buten nach vorhergehender Isomerisierung |
| US9611192B2 (en) | 2014-06-30 | 2017-04-04 | Uop Llc | Integration of N-C4/N-C4=/BD separation system for on-purpose butadiene synthesis |
| DE102015200702A1 (de) | 2015-01-19 | 2016-07-21 | Evonik Degussa Gmbh | Herstellung von Butadien aus Ethen |
| CN107250326A (zh) | 2015-02-19 | 2017-10-13 | 赛贝克环球科技公司 | 与生产聚乙烯有关的系统和方法 |
| WO2016150940A1 (de) * | 2015-03-26 | 2016-09-29 | Basf Se | Verfahren zur herstellung von 1,3-butadien aus n-butenen durch oxidative dehydrierung |
| WO2016151074A1 (de) | 2015-03-26 | 2016-09-29 | Basf Se | Verfahren zur herstellung von 1,3-butadien aus n-butenen durch oxidative dehydrierung |
| US10927058B2 (en) | 2015-05-15 | 2021-02-23 | Sabic Global Technologies B.V. | Systems and methods related to the syngas to olefin process |
| EP3294837A1 (en) | 2015-05-15 | 2018-03-21 | SABIC Global Technologies B.V. | Systems and methods related to the syngas to olefin process |
| CN106867578B (zh) * | 2015-12-14 | 2018-09-04 | 中国石油天然气股份有限公司 | 一种低碳烃的转化利用工艺 |
| CN106867563B (zh) * | 2015-12-14 | 2018-10-16 | 中国石油天然气股份有限公司 | 一种转化拔头油类轻烃的方法 |
| RU2696137C1 (ru) * | 2016-06-30 | 2019-07-31 | Юоп Ллк | Способ получения бутадиена путем окислительного дегидрирования с последующим прямым дегидрированием |
| WO2018024740A1 (de) | 2016-08-02 | 2018-02-08 | Basf Se | Reaktor zur durchführung der autothermen gasphasendehydrierung eines kohlenwasserstoffhaltigen gasstromes |
| US10391477B2 (en) * | 2016-09-30 | 2019-08-27 | Uchicago Argonne, Llc | Multimetallic catalysts |
| DE102016224063A1 (de) | 2016-12-02 | 2018-06-07 | Thyssenkrupp Ag | Verfahren zur Herstellung von Butadien |
| US10828621B2 (en) | 2017-08-28 | 2020-11-10 | Uchicago Argonne, Llc | Supported multimetallic catalysts for oxidative dehydrogenation of alkanes |
| JP7554754B2 (ja) * | 2019-09-02 | 2024-09-20 | 株式会社Eneosマテリアル | 1,3-ブタジエンの製造方法 |
| JP7699937B2 (ja) * | 2021-03-09 | 2025-06-30 | 千代田化工建設株式会社 | ブタジエンの製造方法 |
| CN113754510B (zh) * | 2021-10-08 | 2022-10-28 | 河北工业大学 | 一种制备1,3-丁二烯的方法 |
| WO2023201244A1 (en) * | 2022-04-12 | 2023-10-19 | Bridgestone Americas Tire Operations, Llc | Butadiene production from used tires |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB508764A (en) * | 1937-10-20 | 1939-07-05 | Ig Farbenindustrie Ag | Improvements in the manufacture and production of butadiene from butane |
| US2376323A (en) * | 1942-01-02 | 1945-05-22 | Phillips Petroleum Co | Process of two-stage catalytic dehydrogenation of paraffin hydrocarbons to diolefin |
| GB628686A (en) * | 1943-10-29 | 1949-09-02 | Universal Oil Prod Co | Production of butadiene |
| US3161670A (en) * | 1960-12-12 | 1964-12-15 | Shell Oil Co | Preparation of olefinic compounds |
| GB1107432A (en) * | 1964-09-01 | 1968-03-27 | Lummus Co | Dehydrogenation process |
| US3718575A (en) * | 1971-07-12 | 1973-02-27 | Universal Oil Prod Co | Hydrocracking for lpg production |
| US3907919A (en) * | 1973-04-09 | 1975-09-23 | El Paso Products Co | Dehydrogenation of hydrocarbons |
| JPS59167525A (ja) * | 1983-03-14 | 1984-09-21 | Japan Synthetic Rubber Co Ltd | 1,3−ブタジエンの製造方法 |
-
2003
- 2003-07-07 MY MYPI20032539A patent/MY135793A/en unknown
- 2003-07-10 AU AU2003254335A patent/AU2003254335A1/en not_active Abandoned
- 2003-07-10 AT AT03763815T patent/ATE461165T1/de not_active IP Right Cessation
- 2003-07-10 WO PCT/EP2003/007523 patent/WO2004007408A1/de not_active Ceased
- 2003-07-10 US US10/516,920 patent/US7034195B2/en not_active Expired - Fee Related
- 2003-07-10 EA EA200500176A patent/EA007394B1/ru not_active IP Right Cessation
- 2003-07-10 KR KR1020057000290A patent/KR100974260B1/ko not_active Expired - Fee Related
- 2003-07-10 BR BR0311949-1A patent/BR0311949A/pt not_active IP Right Cessation
- 2003-07-10 CN CNB038165791A patent/CN100494130C/zh not_active Expired - Fee Related
- 2003-07-10 JP JP2004520592A patent/JP2005536498A/ja not_active Withdrawn
- 2003-07-10 MX MXPA04012234A patent/MXPA04012234A/es unknown
- 2003-07-10 EP EP03763815A patent/EP1523462B1/de not_active Expired - Lifetime
- 2003-07-10 CA CA002493133A patent/CA2493133A1/en not_active Abandoned
- 2003-07-10 DE DE50312525T patent/DE50312525D1/de not_active Expired - Lifetime
- 2003-07-11 TW TW092119023A patent/TWI324591B/zh not_active IP Right Cessation
-
2004
- 2004-12-03 NO NO20045324A patent/NO20045324L/no not_active Application Discontinuation
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010534553A (ja) * | 2007-05-10 | 2010-11-11 | エスケー エナジー 株式会社 | 亜鉛フェライト触媒、その製造方法、およびそれを使用する1,3−ブタジエンの製造方法 |
| JP2013189440A (ja) * | 2007-05-30 | 2013-09-26 | Sk Innovation Co Ltd | 多成分系モリブデン酸ビスマス触媒を使用した1,3−ブタジエンの製造方法。 |
| JP2010120933A (ja) * | 2008-10-24 | 2010-06-03 | Asahi Kasei Chemicals Corp | 共役ジオレフィンの製造方法 |
| JP2012512181A (ja) * | 2009-03-25 | 2012-05-31 | ルマス テクノロジー インコーポレイテッド | ブタンからのプロピレンの生成 |
| JP2013502414A (ja) * | 2009-08-17 | 2013-01-24 | ルマス テクノロジー インコーポレイテッド | ブタジエンの製造法 |
| JP2015061859A (ja) * | 2009-08-17 | 2015-04-02 | ルマス テクノロジー インコーポレイテッド | ブタジエンの製造法 |
| JP2012067048A (ja) * | 2010-09-27 | 2012-04-05 | Asahi Kasei Chemicals Corp | ブタジエンの製造方法 |
| JP2014519478A (ja) * | 2011-04-04 | 2014-08-14 | エボニック デグサ ゲーエムベーハー | 1−ブテンおよび1,3−ブタジエン誘導体の製造方法 |
| JP2013100244A (ja) * | 2011-11-08 | 2013-05-23 | Mitsui Chemicals Inc | 共役ジオレフィンの製造方法 |
| JP2015509108A (ja) * | 2012-01-30 | 2015-03-26 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | n−ブタンからのブタジエンおよび/またはブテン類の製造方法 |
| JP2014073462A (ja) * | 2012-10-04 | 2014-04-24 | Cosmo Oil Co Ltd | 金属酸化物触媒、その製造方法及びアルカジエンの製造方法 |
| WO2015186915A1 (ko) * | 2014-06-03 | 2015-12-10 | 주식회사 엘지화학 | 산화탈수소화 반응을 통한 부타디엔의 제조방법 |
| US9738574B2 (en) | 2014-06-03 | 2017-08-22 | Lg Chem, Ltd. | Method for producing butadiene through oxidative dehydrogenation reaction |
| JP2017509483A (ja) * | 2014-12-05 | 2017-04-06 | エルジー・ケム・リミテッド | ブタジエン製造用複合酸化物触媒及びその製造方法 |
| JP2018197215A (ja) * | 2017-05-24 | 2018-12-13 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
| JP2019137664A (ja) * | 2018-02-15 | 2019-08-22 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
| JP2019137663A (ja) * | 2018-02-15 | 2019-08-22 | Jxtgエネルギー株式会社 | 不飽和炭化水素の製造方法 |
| JP7064896B2 (ja) | 2018-02-15 | 2022-05-11 | Eneos株式会社 | 不飽和炭化水素の製造方法 |
| JP7064897B2 (ja) | 2018-02-15 | 2022-05-11 | Eneos株式会社 | 不飽和炭化水素の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1523462B1 (de) | 2010-03-17 |
| BR0311949A (pt) | 2005-03-22 |
| EA200500176A1 (ru) | 2005-06-30 |
| US20050171311A1 (en) | 2005-08-04 |
| CN1668556A (zh) | 2005-09-14 |
| US7034195B2 (en) | 2006-04-25 |
| WO2004007408A1 (de) | 2004-01-22 |
| MY135793A (en) | 2008-06-30 |
| DE50312525D1 (de) | 2010-04-29 |
| CN100494130C (zh) | 2009-06-03 |
| AU2003254335A1 (en) | 2004-02-02 |
| CA2493133A1 (en) | 2004-01-22 |
| ATE461165T1 (de) | 2010-04-15 |
| TW200413304A (en) | 2004-08-01 |
| EP1523462A1 (de) | 2005-04-20 |
| KR20050018960A (ko) | 2005-02-28 |
| EA007394B1 (ru) | 2006-10-27 |
| TWI324591B (en) | 2010-05-11 |
| MXPA04012234A (es) | 2005-02-25 |
| KR100974260B1 (ko) | 2010-08-06 |
| NO20045324L (no) | 2005-02-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100974260B1 (ko) | N-부탄으로부터 부타디엔을 제조하는 방법 | |
| CN100447117C (zh) | 生产丁二烯的方法 | |
| US8088962B2 (en) | Method for producing butadiene from n-butane | |
| CN100387557C (zh) | 生产丁二烯和1-丁烯的方法 | |
| US7238827B2 (en) | Preparation of at least one partial oxidation and/or ammoxidation product of propylene | |
| CN101084174B (zh) | 由正丁烷制备丁二烯的方法 | |
| KR101168456B1 (ko) | 부타디엔의 제법 | |
| KR20070110080A (ko) | 프로판으로부터의 프로펜 제조 방법 | |
| KR20060136422A (ko) | 부타디엔의 제법 | |
| US6933407B2 (en) | Method for producing methacrylic acid from isobutane | |
| JP4368201B2 (ja) | 4−ビニルシクロヘキセン、エチルベンゼン及びスチレンの製造 | |
| JP2004530723A (ja) | イソブタンからメタクロレインを製造する方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20061006 |