ES2955717T3 - Amidas heterocíclicas de 5 a 7 elementos como inhibidores de JAK - Google Patents
Amidas heterocíclicas de 5 a 7 elementos como inhibidores de JAK Download PDFInfo
- Publication number
- ES2955717T3 ES2955717T3 ES19773584T ES19773584T ES2955717T3 ES 2955717 T3 ES2955717 T3 ES 2955717T3 ES 19773584 T ES19773584 T ES 19773584T ES 19773584 T ES19773584 T ES 19773584T ES 2955717 T3 ES2955717 T3 ES 2955717T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- pharmaceutically acceptable
- acceptable salt
- mmol
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- -1 7-membered heterocyclic amides Chemical class 0.000 title claims description 77
- 229940122245 Janus kinase inhibitor Drugs 0.000 title description 18
- 150000001875 compounds Chemical class 0.000 claims abstract description 278
- 150000003839 salts Chemical class 0.000 claims abstract description 75
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 47
- 208000023504 respiratory system disease Diseases 0.000 claims abstract description 24
- 210000004072 lung Anatomy 0.000 claims description 64
- 125000000217 alkyl group Chemical group 0.000 claims description 49
- 239000003112 inhibitor Substances 0.000 claims description 41
- 208000006673 asthma Diseases 0.000 claims description 33
- 238000011282 treatment Methods 0.000 claims description 25
- 241000124008 Mammalia Species 0.000 claims description 20
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 19
- 206010051604 Lung transplant rejection Diseases 0.000 claims description 19
- 208000024908 graft versus host disease Diseases 0.000 claims description 19
- 206010035664 Pneumonia Diseases 0.000 claims description 17
- 206010035742 Pneumonitis Diseases 0.000 claims description 16
- 229910052757 nitrogen Inorganic materials 0.000 claims description 14
- 230000001684 chronic effect Effects 0.000 claims description 13
- 206010029888 Obliterative bronchiolitis Diseases 0.000 claims description 12
- 201000003848 bronchiolitis obliterans Diseases 0.000 claims description 12
- 208000023367 bronchiolitis obliterans with obstructive pulmonary disease Diseases 0.000 claims description 12
- 239000003937 drug carrier Substances 0.000 claims description 12
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 12
- 230000002685 pulmonary effect Effects 0.000 claims description 11
- 206010067472 Organising pneumonia Diseases 0.000 claims description 10
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 10
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 9
- 208000019693 Lung disease Diseases 0.000 claims description 9
- 230000001154 acute effect Effects 0.000 claims description 9
- 230000004064 dysfunction Effects 0.000 claims description 9
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 9
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 9
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 9
- 201000003883 Cystic fibrosis Diseases 0.000 claims description 8
- 206010006448 Bronchiolitis Diseases 0.000 claims description 7
- 208000034706 Graft dysfunction Diseases 0.000 claims description 7
- 208000004530 Primary Graft Dysfunction Diseases 0.000 claims description 7
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 claims description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 7
- 201000000306 sarcoidosis Diseases 0.000 claims description 7
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 6
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 6
- 206010069351 acute lung injury Diseases 0.000 claims description 6
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 6
- 201000009805 cryptogenic organizing pneumonia Diseases 0.000 claims description 6
- 208000013397 idiopathic acute eosinophilic pneumonia Diseases 0.000 claims description 6
- 206010014561 Emphysema Diseases 0.000 claims description 5
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 5
- 206010006451 bronchitis Diseases 0.000 claims description 5
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 5
- 230000003448 neutrophilic effect Effects 0.000 claims description 5
- 208000018428 Eosinophilic granulomatosis with polyangiitis Diseases 0.000 claims description 4
- 230000000527 lymphocytic effect Effects 0.000 claims description 4
- 208000027004 Eosinophilic disease Diseases 0.000 claims description 3
- 206010061201 Helminthic infection Diseases 0.000 claims description 3
- 206010048643 Hypereosinophilic syndrome Diseases 0.000 claims description 3
- 208000016300 Idiopathic chronic eosinophilic pneumonia Diseases 0.000 claims description 3
- 206010049459 Lymphangioleiomyomatosis Diseases 0.000 claims description 3
- 230000002538 fungal effect Effects 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 230000000172 allergic effect Effects 0.000 claims description 2
- 208000010668 atopic eczema Diseases 0.000 claims description 2
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 claims description 2
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 claims description 2
- 201000002909 Aspergillosis Diseases 0.000 claims 1
- 208000036641 Aspergillus infections Diseases 0.000 claims 1
- 208000027775 Bronchopulmonary disease Diseases 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 30
- 102000042838 JAK family Human genes 0.000 abstract description 5
- 108091082332 JAK family Proteins 0.000 abstract description 5
- 229940043355 kinase inhibitor Drugs 0.000 abstract description 4
- 239000003757 phosphotransferase inhibitor Substances 0.000 abstract description 4
- 238000013519 translation Methods 0.000 abstract description 2
- 239000011541 reaction mixture Substances 0.000 description 94
- 239000000243 solution Substances 0.000 description 94
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 72
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 63
- 210000004027 cell Anatomy 0.000 description 60
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 60
- 238000006243 chemical reaction Methods 0.000 description 54
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 51
- 238000012360 testing method Methods 0.000 description 50
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 47
- 239000000203 mixture Substances 0.000 description 45
- 230000005764 inhibitory process Effects 0.000 description 44
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 42
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 36
- 238000005516 engineering process Methods 0.000 description 36
- 102000004127 Cytokines Human genes 0.000 description 35
- 108090000695 Cytokines Proteins 0.000 description 35
- 238000003556 assay Methods 0.000 description 35
- 235000019439 ethyl acetate Nutrition 0.000 description 35
- 230000036515 potency Effects 0.000 description 35
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 34
- 239000002253 acid Substances 0.000 description 33
- 239000012043 crude product Substances 0.000 description 32
- 239000007787 solid Substances 0.000 description 30
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 29
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 28
- 239000000047 product Substances 0.000 description 28
- 210000001744 T-lymphocyte Anatomy 0.000 description 26
- 239000000872 buffer Substances 0.000 description 26
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 25
- 239000002609 medium Substances 0.000 description 24
- 229910002091 carbon monoxide Inorganic materials 0.000 description 23
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 23
- 238000005481 NMR spectroscopy Methods 0.000 description 22
- 230000002829 reductive effect Effects 0.000 description 22
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 21
- 108010002350 Interleukin-2 Proteins 0.000 description 21
- 102000000588 Interleukin-2 Human genes 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 21
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 21
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 20
- 239000005557 antagonist Substances 0.000 description 20
- 210000004369 blood Anatomy 0.000 description 20
- 239000008280 blood Substances 0.000 description 20
- 239000003814 drug Substances 0.000 description 19
- 238000003756 stirring Methods 0.000 description 19
- 201000010099 disease Diseases 0.000 description 18
- 239000012044 organic layer Substances 0.000 description 18
- 238000002953 preparative HPLC Methods 0.000 description 18
- 206010061218 Inflammation Diseases 0.000 description 17
- 108090000176 Interleukin-13 Proteins 0.000 description 17
- 102000003816 Interleukin-13 Human genes 0.000 description 17
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 17
- 230000004054 inflammatory process Effects 0.000 description 17
- 230000026731 phosphorylation Effects 0.000 description 17
- 238000006366 phosphorylation reaction Methods 0.000 description 17
- 239000000843 powder Substances 0.000 description 17
- 238000002360 preparation method Methods 0.000 description 17
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 17
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- 229940124597 therapeutic agent Drugs 0.000 description 16
- 239000007821 HATU Substances 0.000 description 15
- 230000000694 effects Effects 0.000 description 15
- 239000012091 fetal bovine serum Substances 0.000 description 15
- 239000000543 intermediate Substances 0.000 description 15
- 239000003446 ligand Substances 0.000 description 15
- 210000001519 tissue Anatomy 0.000 description 15
- 102100031294 Thymic stromal lymphopoietin Human genes 0.000 description 14
- 238000004458 analytical method Methods 0.000 description 14
- 239000000725 suspension Substances 0.000 description 14
- 238000004809 thin layer chromatography Methods 0.000 description 14
- 108010029307 thymic stromal lymphopoietin Proteins 0.000 description 14
- 239000003981 vehicle Substances 0.000 description 14
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 13
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- 239000003795 chemical substances by application Substances 0.000 description 13
- 238000010790 dilution Methods 0.000 description 13
- 239000012895 dilution Substances 0.000 description 13
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 108010002616 Interleukin-5 Proteins 0.000 description 12
- 102100039897 Interleukin-5 Human genes 0.000 description 12
- 241001465754 Metazoa Species 0.000 description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 12
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 12
- 210000003979 eosinophil Anatomy 0.000 description 12
- 230000004044 response Effects 0.000 description 12
- 230000000638 stimulation Effects 0.000 description 12
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 11
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 239000007995 HEPES buffer Substances 0.000 description 11
- 108090001005 Interleukin-6 Proteins 0.000 description 11
- 238000012544 monitoring process Methods 0.000 description 11
- 229910052938 sodium sulfate Inorganic materials 0.000 description 11
- 235000011152 sodium sulphate Nutrition 0.000 description 11
- 230000009885 systemic effect Effects 0.000 description 11
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 10
- 239000012114 Alexa Fluor 647 Substances 0.000 description 10
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 10
- 102000004388 Interleukin-4 Human genes 0.000 description 10
- 108090000978 Interleukin-4 Proteins 0.000 description 10
- 102000004889 Interleukin-6 Human genes 0.000 description 10
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 10
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 10
- 239000012911 assay medium Substances 0.000 description 10
- 239000012267 brine Substances 0.000 description 10
- 230000001413 cellular effect Effects 0.000 description 10
- 238000004440 column chromatography Methods 0.000 description 10
- 238000009472 formulation Methods 0.000 description 10
- 230000002401 inhibitory effect Effects 0.000 description 10
- 229940028885 interleukin-4 Drugs 0.000 description 10
- 229940100601 interleukin-6 Drugs 0.000 description 10
- 229940071648 metered dose inhaler Drugs 0.000 description 10
- 239000006199 nebulizer Substances 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 239000011734 sodium Substances 0.000 description 10
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 10
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 9
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 9
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 206010009887 colitis Diseases 0.000 description 9
- 239000003480 eluent Substances 0.000 description 9
- WMWTYOKRWGGJOA-CENSZEJFSA-N fluticasone propionate Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(OC(=O)CC)[C@@]2(C)C[C@@H]1O WMWTYOKRWGGJOA-CENSZEJFSA-N 0.000 description 9
- 238000011534 incubation Methods 0.000 description 9
- 239000000546 pharmaceutical excipient Substances 0.000 description 9
- 230000000770 proinflammatory effect Effects 0.000 description 9
- GLWKVDXAQHCAIO-REYDXQAISA-N 3-[(2z,5z)-2-[[3-(2-carboxyethyl)-5-[[(2r)-4-ethenyl-3-methyl-5-oxo-1,2-dihydropyrrol-2-yl]methyl]-4-methyl-1h-pyrrol-2-yl]methylidene]-5-[[(3z,4r)-3-ethylidene-4-methyl-5-oxopyrrol-2-yl]methylidene]-4-methylpyrrol-3-yl]propanoic acid Chemical compound C\C=C1\[C@@H](C)C(=O)N=C1\C=C(/N\1)C(C)=C(CCC(O)=O)C/1=C/C1=C(CCC(O)=O)C(C)=C(C[C@@H]2C(=C(C=C)C(=O)N2)C)N1 GLWKVDXAQHCAIO-REYDXQAISA-N 0.000 description 8
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 8
- 108060003951 Immunoglobulin Proteins 0.000 description 8
- IGJXAXFFKKRFKU-UHFFFAOYSA-N Phycoerythrobilin Natural products CC=C/1C(NC(C1C)=O)=Cc2[nH]c(C=C3/N=C(CC4NC(=O)C(=C4C)C=C)C(=C3CCC(=O)O)C)c(CCC(=O)O)c2C IGJXAXFFKKRFKU-UHFFFAOYSA-N 0.000 description 8
- 239000006227 byproduct Substances 0.000 description 8
- HEDRZPFGACZZDS-UHFFFAOYSA-N chloroform Substances ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- 230000001419 dependent effect Effects 0.000 description 8
- 229960000289 fluticasone propionate Drugs 0.000 description 8
- 102000018358 immunoglobulin Human genes 0.000 description 8
- 108010012759 phycoerythrobilin Proteins 0.000 description 8
- 229940075993 receptor modulator Drugs 0.000 description 8
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 7
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 7
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 7
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 7
- 230000004913 activation Effects 0.000 description 7
- 239000013543 active substance Substances 0.000 description 7
- 239000000556 agonist Substances 0.000 description 7
- 238000000423 cell based assay Methods 0.000 description 7
- 230000002327 eosinophilic effect Effects 0.000 description 7
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 7
- 125000000623 heterocyclic group Chemical group 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 229910052739 hydrogen Inorganic materials 0.000 description 7
- 239000008101 lactose Substances 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 230000003389 potentiating effect Effects 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 230000011664 signaling Effects 0.000 description 7
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- 238000005160 1H NMR spectroscopy Methods 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- 102100025248 C-X-C motif chemokine 10 Human genes 0.000 description 6
- 102100036170 C-X-C motif chemokine 9 Human genes 0.000 description 6
- 102000019034 Chemokines Human genes 0.000 description 6
- 108010012236 Chemokines Proteins 0.000 description 6
- 108010074328 Interferon-gamma Proteins 0.000 description 6
- 102000008070 Interferon-gamma Human genes 0.000 description 6
- 108010065805 Interleukin-12 Proteins 0.000 description 6
- 102000013462 Interleukin-12 Human genes 0.000 description 6
- 108010066979 Interleukin-27 Proteins 0.000 description 6
- 108091000080 Phosphotransferase Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 108010011005 STAT6 Transcription Factor Proteins 0.000 description 6
- 102100023980 Signal transducer and activator of transcription 6 Human genes 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 6
- 125000004429 atom Chemical group 0.000 description 6
- 125000004432 carbon atom Chemical group C* 0.000 description 6
- 229940112141 dry powder inhaler Drugs 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 150000005828 hydrofluoroalkanes Chemical class 0.000 description 6
- 229960003130 interferon gamma Drugs 0.000 description 6
- 229940117681 interleukin-12 Drugs 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 210000001616 monocyte Anatomy 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- 235000019198 oils Nutrition 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 102000020233 phosphotransferase Human genes 0.000 description 6
- 239000003380 propellant Substances 0.000 description 6
- 229960002052 salbutamol Drugs 0.000 description 6
- 239000012279 sodium borohydride Substances 0.000 description 6
- 229910000033 sodium borohydride Inorganic materials 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- PCFIABOQFAFDAU-UHFFFAOYSA-N 4-bromo-2-fluorobenzoyl chloride Chemical compound FC1=CC(Br)=CC=C1C(Cl)=O PCFIABOQFAFDAU-UHFFFAOYSA-N 0.000 description 5
- 241000223600 Alternaria Species 0.000 description 5
- 206010012438 Dermatitis atopic Diseases 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 101000858088 Homo sapiens C-X-C motif chemokine 10 Proteins 0.000 description 5
- 101000947172 Homo sapiens C-X-C motif chemokine 9 Proteins 0.000 description 5
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 5
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 5
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 5
- 230000004163 JAK-STAT signaling pathway Effects 0.000 description 5
- 208000022873 Ocular disease Diseases 0.000 description 5
- 206010042971 T-cell lymphoma Diseases 0.000 description 5
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 5
- 201000008937 atopic dermatitis Diseases 0.000 description 5
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- BPZSYCZIITTYBL-UHFFFAOYSA-N formoterol Chemical compound C1=CC(OC)=CC=C1CC(C)NCC(O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-UHFFFAOYSA-N 0.000 description 5
- 229960002848 formoterol Drugs 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 5
- 230000002757 inflammatory effect Effects 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 230000004060 metabolic process Effects 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 5
- 229960005018 salmeterol xinafoate Drugs 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000006228 supernatant Substances 0.000 description 5
- OBRNDARFFFHCGE-PERKLWIXSA-N (S,S)-formoterol fumarate Chemical compound OC(=O)\C=C\C(O)=O.C1=CC(OC)=CC=C1C[C@H](C)NC[C@@H](O)C1=CC=C(O)C(NC=O)=C1.C1=CC(OC)=CC=C1C[C@H](C)NC[C@@H](O)C1=CC=C(O)C(NC=O)=C1 OBRNDARFFFHCGE-PERKLWIXSA-N 0.000 description 4
- JSKGYDVSMNCSRH-SSDOTTSWSA-N 2-[(3r)-3-methylpiperazin-1-yl]ethanol Chemical compound C[C@@H]1CN(CCO)CCN1 JSKGYDVSMNCSRH-SSDOTTSWSA-N 0.000 description 4
- 206010002091 Anaesthesia Diseases 0.000 description 4
- KUVIULQEHSCUHY-XYWKZLDCSA-N Beclometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)COC(=O)CC)(OC(=O)CC)[C@@]1(C)C[C@@H]2O KUVIULQEHSCUHY-XYWKZLDCSA-N 0.000 description 4
- 201000004624 Dermatitis Diseases 0.000 description 4
- 229920001917 Ficoll Polymers 0.000 description 4
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 102000015696 Interleukins Human genes 0.000 description 4
- 108010063738 Interleukins Proteins 0.000 description 4
- 208000029523 Interstitial Lung disease Diseases 0.000 description 4
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 4
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 4
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 4
- 241000699666 Mus <mouse, genus> Species 0.000 description 4
- NBBJYMSMWIIQGU-UHFFFAOYSA-N Propionic aldehyde Chemical compound CCC=O NBBJYMSMWIIQGU-UHFFFAOYSA-N 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 239000000443 aerosol Substances 0.000 description 4
- 230000037005 anaesthesia Effects 0.000 description 4
- 229950000210 beclometasone dipropionate Drugs 0.000 description 4
- 229940098773 bovine serum albumin Drugs 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 230000000875 corresponding effect Effects 0.000 description 4
- 231100000673 dose–response relationship Toxicity 0.000 description 4
- 235000019441 ethanol Nutrition 0.000 description 4
- 229960000193 formoterol fumarate Drugs 0.000 description 4
- 210000002865 immune cell Anatomy 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 230000014759 maintenance of location Effects 0.000 description 4
- 201000005962 mycosis fungoides Diseases 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 229940044551 receptor antagonist Drugs 0.000 description 4
- 239000002464 receptor antagonist Substances 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 210000002345 respiratory system Anatomy 0.000 description 4
- 229960000215 ruxolitinib Drugs 0.000 description 4
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 4
- 229960004017 salmeterol Drugs 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 229940083542 sodium Drugs 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 238000002054 transplantation Methods 0.000 description 4
- VQLBLCOABKEJKO-XRIGFGBMSA-N (6s)-4,5,6,7-tetrahydro-3h-imidazo[4,5-c]pyridine-6-carboxylic acid;dihydrochloride Chemical compound Cl.Cl.C1N[C@H](C(=O)O)CC2=C1NC=N2 VQLBLCOABKEJKO-XRIGFGBMSA-N 0.000 description 3
- LVGUZGTVOIAKKC-UHFFFAOYSA-N 1,1,1,2-tetrafluoroethane Chemical compound FCC(F)(F)F LVGUZGTVOIAKKC-UHFFFAOYSA-N 0.000 description 3
- GGGMBSBQEAELRI-UHFFFAOYSA-N 2-(2-ethyl-5-fluoro-4-phenylmethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1F)B1OC(C(O1)(C)C)(C)C)CC GGGMBSBQEAELRI-UHFFFAOYSA-N 0.000 description 3
- YREYLAVBNPACJM-UHFFFAOYSA-N 2-(tert-butylamino)-1-(2-chlorophenyl)ethanol Chemical compound CC(C)(C)NCC(O)C1=CC=CC=C1Cl YREYLAVBNPACJM-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- LERNTVKEWCAPOY-VOGVJGKGSA-N C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 Chemical compound C[N+]1(C)[C@H]2C[C@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 LERNTVKEWCAPOY-VOGVJGKGSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 206010009900 Colitis ulcerative Diseases 0.000 description 3
- 208000011231 Crohn disease Diseases 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 description 3
- 208000003556 Dry Eye Syndromes Diseases 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 3
- 102000013264 Interleukin-23 Human genes 0.000 description 3
- 108010065637 Interleukin-23 Proteins 0.000 description 3
- 229930195725 Mannitol Natural products 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 3
- 206010034277 Pemphigoid Diseases 0.000 description 3
- 208000003251 Pruritus Diseases 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 108010044012 STAT1 Transcription Factor Proteins 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 108010010057 TYK2 Kinase Proteins 0.000 description 3
- 102100040247 Tumor necrosis factor Human genes 0.000 description 3
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 3
- 229940127174 UCHT1 Drugs 0.000 description 3
- 201000006704 Ulcerative Colitis Diseases 0.000 description 3
- 206010046851 Uveitis Diseases 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- TVWAEQRFKRTYIG-JIDHJSLPSA-N acetic acid;4-[(1r)-2-[6-[2-[(2,6-dichlorophenyl)methoxy]ethoxy]hexylamino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol Chemical compound CC(O)=O.C1=C(O)C(CO)=CC([C@@H](O)CNCCCCCCOCCOCC=2C(=CC=CC=2Cl)Cl)=C1 TVWAEQRFKRTYIG-JIDHJSLPSA-N 0.000 description 3
- 206010064930 age-related macular degeneration Diseases 0.000 description 3
- 230000000735 allogeneic effect Effects 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000011324 bead Substances 0.000 description 3
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- 230000002051 biphasic effect Effects 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000002559 chemokine receptor antagonist Substances 0.000 description 3
- 238000002648 combination therapy Methods 0.000 description 3
- 229940125904 compound 1 Drugs 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 102000003675 cytokine receptors Human genes 0.000 description 3
- 108010057085 cytokine receptors Proteins 0.000 description 3
- 201000011190 diabetic macular edema Diseases 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 230000002500 effect on skin Effects 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 230000004761 fibrosis Effects 0.000 description 3
- 238000013100 final test Methods 0.000 description 3
- 238000000684 flow cytometry Methods 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 229960001469 fluticasone furoate Drugs 0.000 description 3
- XTULMSXFIHGYFS-VLSRWLAYSA-N fluticasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(F)[C@@]4(C)C=CC(=O)C=C4[C@@H](F)C[C@H]3[C@@H]2C[C@H]1C)C(=O)SCF)C(=O)C1=CC=CO1 XTULMSXFIHGYFS-VLSRWLAYSA-N 0.000 description 3
- 239000008098 formaldehyde solution Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- 150000003840 hydrochlorides Chemical class 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 229960004078 indacaterol Drugs 0.000 description 3
- QZZUEBNBZAPZLX-QFIPXVFZSA-N indacaterol Chemical compound N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)CNC1CC(C=C(C(=C2)CC)CC)=C2C1 QZZUEBNBZAPZLX-QFIPXVFZSA-N 0.000 description 3
- 230000006698 induction Effects 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 229940047122 interleukins Drugs 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- 239000003199 leukotriene receptor blocking agent Substances 0.000 description 3
- 201000011486 lichen planus Diseases 0.000 description 3
- 208000002780 macular degeneration Diseases 0.000 description 3
- 235000010355 mannitol Nutrition 0.000 description 3
- 239000000594 mannitol Substances 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 3
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 3
- 229960004583 pranlukast Drugs 0.000 description 3
- UAJUXJSXCLUTNU-UHFFFAOYSA-N pranlukast Chemical compound C=1C=C(OCCCCC=2C=CC=CC=2)C=CC=1C(=O)NC(C=1)=CC=C(C(C=2)=O)C=1OC=2C=1N=NNN=1 UAJUXJSXCLUTNU-UHFFFAOYSA-N 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 230000019635 sulfation Effects 0.000 description 3
- 238000005670 sulfation reaction Methods 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 229960000257 tiotropium bromide Drugs 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- UORVGPXVDQYIDP-UHFFFAOYSA-N trihydridoboron Substances B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 3
- 229960000859 tulobuterol Drugs 0.000 description 3
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 3
- 229960004026 vilanterol Drugs 0.000 description 3
- AVAWMINJNRAQFS-LURJTMIESA-N (3s)-n,n-dimethylpyrrolidin-3-amine Chemical compound CN(C)[C@H]1CCNC1 AVAWMINJNRAQFS-LURJTMIESA-N 0.000 description 2
- HECRLGCTGPNABR-LURJTMIESA-N (6S)-3,4,6,7-tetrahydroimidazo[4,5-c]pyridine-5,6-dicarboxylic acid Chemical compound OC(=O)[C@@H]1Cc2nc[nH]c2CN1C(O)=O HECRLGCTGPNABR-LURJTMIESA-N 0.000 description 2
- CCGHCCTXMYYHQC-NSHDSACASA-N (6S)-3,5-bis[(2-methylpropan-2-yl)oxycarbonyl]-6,7-dihydro-4H-imidazo[4,5-c]pyridine-6-carboxylic acid Chemical compound C(C)(C)(C)OC(=O)N1C=NC2=C1CN([C@@H](C2)C(=O)O)C(=O)OC(C)(C)C CCGHCCTXMYYHQC-NSHDSACASA-N 0.000 description 2
- NDAUXUAQIAJITI-LBPRGKRZSA-N (R)-salbutamol Chemical compound CC(C)(C)NC[C@H](O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-LBPRGKRZSA-N 0.000 description 2
- YFMFNYKEUDLDTL-UHFFFAOYSA-N 1,1,1,2,3,3,3-heptafluoropropane Chemical compound FC(F)(F)C(F)C(F)(F)F YFMFNYKEUDLDTL-UHFFFAOYSA-N 0.000 description 2
- PXRYYARWCNIUKT-UHFFFAOYSA-M 1,1-diethyl-4-phenyl-1,4-diazepan-1-ium 4-methylbenzenesulfonate Chemical compound Cc1ccc(cc1)S([O-])(=O)=O.CC[N+]1(CC)CCCN(CC1)c1ccccc1 PXRYYARWCNIUKT-UHFFFAOYSA-M 0.000 description 2
- BIIDVVFATFVXTC-UHFFFAOYSA-N 1-bromo-2-ethyl-4-phenylmethoxybenzene Chemical compound C1=C(Br)C(CC)=CC(OCC=2C=CC=CC=2)=C1 BIIDVVFATFVXTC-UHFFFAOYSA-N 0.000 description 2
- PGMBVOVVYOIMBB-UHFFFAOYSA-N 1-ethyl-3-phenylmethoxybenzene Chemical compound CCC1=CC=CC(OCC=2C=CC=CC=2)=C1 PGMBVOVVYOIMBB-UHFFFAOYSA-N 0.000 description 2
- FXHRAKUEZPSMLJ-UHFFFAOYSA-N 1-methyl-1,4-diazepane Chemical compound CN1CCCNCC1 FXHRAKUEZPSMLJ-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- SVPDDTOIPKRVQR-UHFFFAOYSA-N 2-(2-ethyl-4-phenylmethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1)B1OC(C(O1)(C)C)(C)C)CC SVPDDTOIPKRVQR-UHFFFAOYSA-N 0.000 description 2
- WLUAJCSMROZCDP-UHFFFAOYSA-N 2-methyl-2,5-diazabicyclo[2.2.1]heptane;dihydrobromide Chemical compound Br.Br.C1C2N(C)CC1NC2 WLUAJCSMROZCDP-UHFFFAOYSA-N 0.000 description 2
- WFCSWCVEJLETKA-UHFFFAOYSA-N 2-piperazin-1-ylethanol Chemical compound OCCN1CCNCC1 WFCSWCVEJLETKA-UHFFFAOYSA-N 0.000 description 2
- HMNKTRSOROOSPP-UHFFFAOYSA-N 3-Ethylphenol Chemical compound CCC1=CC=CC(O)=C1 HMNKTRSOROOSPP-UHFFFAOYSA-N 0.000 description 2
- ZZWJKLGCDHYVMB-BWGXUDETSA-N 3-[5-[(1r,2s)-2-(2,2-difluoropropanoylamino)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)propoxy]indazol-1-yl]-n-[(3r)-oxolan-3-yl]benzamide Chemical compound O([C@@H]([C@@H](NC(=O)C(C)(F)F)C)C=1C=C2OCCOC2=CC=1)C(C=C1C=N2)=CC=C1N2C(C=1)=CC=CC=1C(=O)N[C@@H]1CCOC1 ZZWJKLGCDHYVMB-BWGXUDETSA-N 0.000 description 2
- AIAJYBLPGVBVSS-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-imidazo[4,5-b]pyridine Chemical compound C1CCNC2=C1NC=N2 AIAJYBLPGVBVSS-UHFFFAOYSA-N 0.000 description 2
- DWOZNANUEDYIOF-UHFFFAOYSA-L 4-ditert-butylphosphanyl-n,n-dimethylaniline;dichloropalladium Chemical compound Cl[Pd]Cl.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1 DWOZNANUEDYIOF-UHFFFAOYSA-L 0.000 description 2
- VERCIFBQMIUSOI-UHFFFAOYSA-N 6-(2-ethyl-5-fluoro-4-phenylmethoxyphenyl)-1H-indazole Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1F)C1=CC=C2C=NNC2=C1)CC VERCIFBQMIUSOI-UHFFFAOYSA-N 0.000 description 2
- XSMJTRACFNYDFT-UHFFFAOYSA-N 6-(2-ethyl-5-fluoro-4-phenylmethoxyphenyl)-3-iodo-1-(oxan-2-yl)indazole Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1F)C1=CC=C2C(=NN(C2=C1)C1OCCCC1)I)CC XSMJTRACFNYDFT-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 238000012815 AlphaLISA Methods 0.000 description 2
- 241000223602 Alternaria alternata Species 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 208000009137 Behcet syndrome Diseases 0.000 description 2
- 206010006474 Bronchopulmonary aspergillosis allergic Diseases 0.000 description 2
- VOVIALXJUBGFJZ-KWVAZRHASA-N Budesonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(CCC)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O VOVIALXJUBGFJZ-KWVAZRHASA-N 0.000 description 2
- FULGHTIHNXKJCZ-NRFANRHFSA-N C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)C(C)C)N1 Chemical compound C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)C(C)C)N1 FULGHTIHNXKJCZ-NRFANRHFSA-N 0.000 description 2
- 102100024167 C-C chemokine receptor type 3 Human genes 0.000 description 2
- 101710149862 C-C chemokine receptor type 3 Proteins 0.000 description 2
- 102100028990 C-X-C chemokine receptor type 3 Human genes 0.000 description 2
- 102000009660 Cholinergic Receptors Human genes 0.000 description 2
- 108010009685 Cholinergic Receptors Proteins 0.000 description 2
- 208000017667 Chronic Disease Diseases 0.000 description 2
- 208000015943 Coeliac disease Diseases 0.000 description 2
- 206010056979 Colitis microscopic Diseases 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- 102000002045 Endothelin Human genes 0.000 description 2
- 108050009340 Endothelin Proteins 0.000 description 2
- 206010058838 Enterocolitis infectious Diseases 0.000 description 2
- 206010064212 Eosinophilic oesophagitis Diseases 0.000 description 2
- 102000009109 Fc receptors Human genes 0.000 description 2
- 108010087819 Fc receptors Proteins 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 101000916050 Homo sapiens C-X-C chemokine receptor type 3 Proteins 0.000 description 2
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 description 2
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 2
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 206010062016 Immunosuppression Diseases 0.000 description 2
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 2
- 102000003996 Interferon-beta Human genes 0.000 description 2
- 108090000467 Interferon-beta Proteins 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 108090000177 Interleukin-11 Proteins 0.000 description 2
- 102000003815 Interleukin-11 Human genes 0.000 description 2
- 102000013691 Interleukin-17 Human genes 0.000 description 2
- 108050003558 Interleukin-17 Proteins 0.000 description 2
- 108010002386 Interleukin-3 Proteins 0.000 description 2
- 102000000646 Interleukin-3 Human genes 0.000 description 2
- 102100021596 Interleukin-31 Human genes 0.000 description 2
- 101710181613 Interleukin-31 Proteins 0.000 description 2
- 108010002335 Interleukin-9 Proteins 0.000 description 2
- 102000000585 Interleukin-9 Human genes 0.000 description 2
- 108090000862 Ion Channels Proteins 0.000 description 2
- 102000004310 Ion Channels Human genes 0.000 description 2
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 2
- 102000008986 Janus Human genes 0.000 description 2
- 108050000950 Janus Proteins 0.000 description 2
- 206010023439 Kidney transplant rejection Diseases 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 102000012750 Membrane Glycoproteins Human genes 0.000 description 2
- 108010090054 Membrane Glycoproteins Proteins 0.000 description 2
- 102000005741 Metalloproteases Human genes 0.000 description 2
- 108010006035 Metalloproteases Proteins 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- 108010057466 NF-kappa B Proteins 0.000 description 2
- 102000003945 NF-kappa B Human genes 0.000 description 2
- 102000010648 Natural Killer Cell Receptors Human genes 0.000 description 2
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 2
- GFLMRBCCZUOJSI-LBPRGKRZSA-N OC([C@H](CC1=C(C2)N(CC3=CC=CC=C3)C=N1)N2C(O)=O)=O Chemical compound OC([C@H](CC1=C(C2)N(CC3=CC=CC=C3)C=N1)N2C(O)=O)=O GFLMRBCCZUOJSI-LBPRGKRZSA-N 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 201000007902 Primary cutaneous amyloidosis Diseases 0.000 description 2
- 108050000258 Prostaglandin D receptors Proteins 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- 206010037083 Prurigo Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 102000005886 STAT4 Transcription Factor Human genes 0.000 description 2
- 108010019992 STAT4 Transcription Factor Proteins 0.000 description 2
- 206010039491 Sarcoma Diseases 0.000 description 2
- ZBVKEHDGYSLCCC-UHFFFAOYSA-N Seratrodast Chemical compound O=C1C(C)=C(C)C(=O)C(C(CCCCCC(O)=O)C=2C=CC=CC=2)=C1C ZBVKEHDGYSLCCC-UHFFFAOYSA-N 0.000 description 2
- 102100029904 Signal transducer and activator of transcription 1-alpha/beta Human genes 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 description 2
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 2
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 2
- 206010047642 Vitiligo Diseases 0.000 description 2
- URWYQGVSPQJGGB-DHUJRADRSA-N [1-[3-[2-chloro-4-[[[(2r)-2-hydroxy-2-(8-hydroxy-2-oxo-1h-quinolin-5-yl)ethyl]amino]methyl]-5-methoxyanilino]-3-oxopropyl]piperidin-4-yl] n-(2-phenylphenyl)carbamate Chemical compound ClC=1C=C(CNC[C@H](O)C=2C=3C=CC(=O)NC=3C(O)=CC=2)C(OC)=CC=1NC(=O)CCN(CC1)CCC1OC(=O)NC1=CC=CC=C1C1=CC=CC=C1 URWYQGVSPQJGGB-DHUJRADRSA-N 0.000 description 2
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 2
- SFYAXIFVXBKRPK-QFIPXVFZSA-N abediterol Chemical compound C([C@H](O)C=1C=2C=CC(=O)NC=2C(O)=CC=1)NCCCCCCOCC(F)(F)C1=CC=CC=C1 SFYAXIFVXBKRPK-QFIPXVFZSA-N 0.000 description 2
- 229950000192 abediterol Drugs 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- ASMXXROZKSBQIH-VITNCHFBSA-N aclidinium Chemical compound C([C@@H](C(CC1)CC2)OC(=O)C(O)(C=3SC=CC=3)C=3SC=CC=3)[N+]21CCCOC1=CC=CC=C1 ASMXXROZKSBQIH-VITNCHFBSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 239000008186 active pharmaceutical agent Substances 0.000 description 2
- 239000000048 adrenergic agonist Substances 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- BNPSSFBOAGDEEL-UHFFFAOYSA-N albuterol sulfate Chemical compound OS(O)(=O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 BNPSSFBOAGDEEL-UHFFFAOYSA-N 0.000 description 2
- 239000013566 allergen Substances 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 208000004631 alopecia areata Diseases 0.000 description 2
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 description 2
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 description 2
- 238000010976 amide bond formation reaction Methods 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 230000003092 anti-cytokine Effects 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 206010069664 atopic keratoconjunctivitis Diseases 0.000 description 2
- 229950009687 batefenterol Drugs 0.000 description 2
- NBMKJKDGKREAPL-DVTGEIKXSA-N beclomethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O NBMKJKDGKREAPL-DVTGEIKXSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 102000015005 beta-adrenergic receptor activity proteins Human genes 0.000 description 2
- 108040006818 beta-adrenergic receptor activity proteins Proteins 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 238000010256 biochemical assay Methods 0.000 description 2
- 210000000424 bronchial epithelial cell Anatomy 0.000 description 2
- 229960004436 budesonide Drugs 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- 208000000594 bullous pemphigoid Diseases 0.000 description 2
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N butyric aldehyde Natural products CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000004113 cell culture Methods 0.000 description 2
- 230000003915 cell function Effects 0.000 description 2
- 229940125400 channel inhibitor Drugs 0.000 description 2
- 239000000460 chlorine Chemical group 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 208000008609 collagenous colitis Diseases 0.000 description 2
- 239000007822 coupling agent Substances 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- 238000004163 cytometry Methods 0.000 description 2
- 229950008135 dectrekumab Drugs 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 229960004483 doxofylline Drugs 0.000 description 2
- HWXIGFIVGWUZAO-UHFFFAOYSA-N doxofylline Chemical compound C1=2C(=O)N(C)C(=O)N(C)C=2N=CN1CC1OCCO1 HWXIGFIVGWUZAO-UHFFFAOYSA-N 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 2
- 201000000708 eosinophilic esophagitis Diseases 0.000 description 2
- QGFORSXNKQLDNO-UHFFFAOYSA-N erdosteine Chemical compound OC(=O)CSCC(=O)NC1CCSC1=O QGFORSXNKQLDNO-UHFFFAOYSA-N 0.000 description 2
- 229960003262 erdosteine Drugs 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 230000000763 evoking effect Effects 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 229960002714 fluticasone Drugs 0.000 description 2
- MGNNYOODZCAHBA-GQKYHHCASA-N fluticasone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@]1(F)[C@@H]2[C@@H]2C[C@@H](C)[C@@](C(=O)SCF)(O)[C@@]2(C)C[C@@H]1O MGNNYOODZCAHBA-GQKYHHCASA-N 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 238000011134 hematopoietic stem cell transplantation Methods 0.000 description 2
- 229960002885 histidine Drugs 0.000 description 2
- 208000009326 ileitis Diseases 0.000 description 2
- 229940027941 immunoglobulin g Drugs 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 208000027139 infectious colitis Diseases 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 229940125369 inhaled corticosteroids Drugs 0.000 description 2
- 229960001388 interferon-beta Drugs 0.000 description 2
- VBUWHHLIZKOSMS-RIWXPGAOSA-N invicorp Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)C(C)C)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=C(O)C=C1 VBUWHHLIZKOSMS-RIWXPGAOSA-N 0.000 description 2
- 239000011630 iodine Chemical group 0.000 description 2
- 229910052740 iodine Chemical group 0.000 description 2
- 229960001361 ipratropium bromide Drugs 0.000 description 2
- LHLMOSXCXGLMMN-VVQPYUEFSA-M ipratropium bromide Chemical compound [Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 LHLMOSXCXGLMMN-VVQPYUEFSA-M 0.000 description 2
- 229960002725 isoflurane Drugs 0.000 description 2
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 229950008204 levosalbutamol Drugs 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 208000004341 lymphocytic colitis Diseases 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 238000007726 management method Methods 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 229960001664 mometasone Drugs 0.000 description 2
- QLIIKPVHVRXHRI-CXSFZGCWSA-N mometasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(Cl)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CCl)(O)[C@@]1(C)C[C@@H]2O QLIIKPVHVRXHRI-CXSFZGCWSA-N 0.000 description 2
- 229960002744 mometasone furoate Drugs 0.000 description 2
- WOFMFGQZHJDGCX-ZULDAHANSA-N mometasone furoate Chemical compound O([C@]1([C@@]2(C)C[C@H](O)[C@]3(Cl)[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)C(=O)CCl)C(=O)C1=CC=CO1 WOFMFGQZHJDGCX-ZULDAHANSA-N 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 229960004286 olodaterol Drugs 0.000 description 2
- COUYJEVMBVSIHV-SFHVURJKSA-N olodaterol Chemical compound C1=CC(OC)=CC=C1CC(C)(C)NC[C@H](O)C1=CC(O)=CC2=C1OCC(=O)N2 COUYJEVMBVSIHV-SFHVURJKSA-N 0.000 description 2
- 229960000470 omalizumab Drugs 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 230000007119 pathological manifestation Effects 0.000 description 2
- 230000007310 pathophysiology Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- MIXMJCQRHVAJIO-TZHJZOAOSA-N qk4dys664x Chemical compound O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O.C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]2(C)C[C@@H]1O MIXMJCQRHVAJIO-TZHJZOAOSA-N 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 229960003254 reslizumab Drugs 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 229960003090 seratrodast Drugs 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 241000894007 species Species 0.000 description 2
- YCFJXOFFQLPCHD-YFKPBYRVSA-N spinacine Chemical compound C1N[C@H](C(=O)O)CC2=C1NC=N2 YCFJXOFFQLPCHD-YFKPBYRVSA-N 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- SUVMJBTUFCVSAD-UHFFFAOYSA-N sulforaphane Chemical compound CS(=O)CCCCN=C=S SUVMJBTUFCVSAD-UHFFFAOYSA-N 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- VBEKMNXJHBOWCT-SNVBAGLBSA-N tert-butyl (2r)-4-(2-hydroxyethyl)-2-methylpiperazine-1-carboxylate Chemical compound C[C@@H]1CN(CCO)CCN1C(=O)OC(C)(C)C VBEKMNXJHBOWCT-SNVBAGLBSA-N 0.000 description 2
- FMLPQHJYUZTHQS-QMMMGPOBSA-N tert-butyl (3s)-3-methylpiperazine-1-carboxylate Chemical compound C[C@H]1CN(C(=O)OC(C)(C)C)CCN1 FMLPQHJYUZTHQS-QMMMGPOBSA-N 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 231100001274 therapeutic index Toxicity 0.000 description 2
- 230000002992 thymic effect Effects 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 2
- 150000004917 tyrosine kinase inhibitor derivatives Chemical class 0.000 description 2
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 229960004541 umeclidinium bromide Drugs 0.000 description 2
- PEJHHXHHNGORMP-AVADPIKZSA-M umeclidinium bromide Chemical compound [Br-].C=1C=CC=CC=1C([C@@]12CC[N@@+](CCOCC=3C=CC=CC=3)(CC1)CC2)(O)C1=CC=CC=C1 PEJHHXHHNGORMP-AVADPIKZSA-M 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- MWLSOWXNZPKENC-SSDOTTSWSA-N zileuton Chemical compound C1=CC=C2SC([C@H](N(O)C(N)=O)C)=CC2=C1 MWLSOWXNZPKENC-SSDOTTSWSA-N 0.000 description 2
- 229960005332 zileuton Drugs 0.000 description 2
- RVMWIVNHZMXEHS-UWVGGRQHSA-N (1S,2S)-2-[5-(5-chloro-2,4-difluoro-N-(2-fluoro-2-methylpropyl)anilino)-3-methoxypyrazine-2-carbonyl]cyclopropane-1-carboxylic acid Chemical compound ClC=1C(=CC(=C(C=1)N(C=1N=C(C(=NC=1)C(=O)[C@@H]1[C@H](C1)C(=O)O)OC)CC(C)(C)F)F)F RVMWIVNHZMXEHS-UWVGGRQHSA-N 0.000 description 1
- IXXFZUPTQVDPPK-ZAWHAJPISA-N (1r,2r,4r,6r,7r,8r,10s,13r,14s)-17-[4-[4-(3-aminophenyl)triazol-1-yl]butyl]-7-[(2s,3r,4s,6r)-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-13-ethyl-10-fluoro-6-methoxy-2,4,6,8,10,14-hexamethyl-12,15-dioxa-17-azabicyclo[12.3.0]heptadecane-3,9,11,16-tet Chemical compound O([C@@H]1[C@@H](C)C(=O)[C@](C)(F)C(=O)O[C@@H]([C@]2(OC(=O)N(CCCCN3N=NC(=C3)C=3C=C(N)C=CC=3)[C@@H]2[C@@H](C)C(=O)[C@H](C)C[C@@]1(C)OC)C)CC)[C@@H]1O[C@H](C)C[C@H](N(C)C)[C@H]1O IXXFZUPTQVDPPK-ZAWHAJPISA-N 0.000 description 1
- LQHDJQIMETZMPH-ZBFHGGJFSA-N (1r,3s)-n-[[4-cyano-2-(trifluoromethyl)phenyl]methyl]-3-[[4-methyl-6-(methylamino)-1,3,5-triazin-2-yl]amino]cyclohexane-1-carboxamide Chemical compound CNC1=NC(C)=NC(N[C@@H]2C[C@@H](CCC2)C(=O)NCC=2C(=CC(=CC=2)C#N)C(F)(F)F)=N1 LQHDJQIMETZMPH-ZBFHGGJFSA-N 0.000 description 1
- CQIBMEIJDDUKHP-BAHZVNHDSA-N (1s,3s,4r)-4-[(3as,4r,5s,7as)-4-(aminomethyl)-7a-methyl-1-methylidene-3,3a,4,5,6,7-hexahydro-2h-inden-5-yl]-3-(hydroxymethyl)-4-methylcyclohexan-1-ol;acetic acid Chemical compound CC(O)=O.C[C@]1([C@H]2CC[C@]3([C@H]([C@@H]2CN)CCC3=C)C)CC[C@H](O)C[C@@H]1CO CQIBMEIJDDUKHP-BAHZVNHDSA-N 0.000 description 1
- KCSNGWMKFYVMKF-HGYIIGRXSA-N (2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[(2-acetamidoacetyl)amino]propanoyl]amino]-5-amino-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxypropanoyl]amino]-6-aminohexanoyl]amino]-3-hydroxybutanoyl]amino]propanoyl]amino]propanoyl]amino]-6-aminohexanoic acid Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H]([C@H](O)C)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)CNC(C)=O)CC1=CC=CC=C1 KCSNGWMKFYVMKF-HGYIIGRXSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- VXBNTHRZPJLRSS-PTCSXESPSA-N (2s)-2-[(2r,3r,4s,5s,6r)-3-benzoyloxy-2-[(1r,2r,3s,5r)-3-[(2,4-dioxo-1h-pyrimidine-6-carbonyl)amino]-5-[2-[[2-[2-[2-oxo-2-[(3,6,8-trisulfonaphthalen-1-yl)amino]ethoxy]ethoxy]acetyl]amino]ethylcarbamoyl]-2-[(2s,3s,4r,5s,6s)-3,4,5-trihydroxy-6-methyloxan-2- Chemical compound O[C@H]1[C@H](O)[C@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@H]2[C@@H]([C@@H](O[C@@H](CC3CCCCC3)C(O)=O)[C@@H](O)[C@@H](CO)O2)OC(=O)C=2C=CC=CC=2)C[C@H](C(=O)NCCNC(=O)COCCOCC(=O)NC=2C3=C(C=C(C=C3C=C(C=2)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C[C@@H]1NC(=O)C1=CC(=O)NC(=O)N1 VXBNTHRZPJLRSS-PTCSXESPSA-N 0.000 description 1
- NSPHQWLKCGGCQR-DLJDZFDSSA-N (2s)-2-[[(1r,4s,7s,10s,13s,16r,21r,27s,34r,37s,40s)-10-(2-amino-2-oxoethyl)-34-[[(2s)-4-carboxy-2-[[(2s)-3-carboxy-2-[[(2s)-2,4-diamino-4-oxobutanoyl]amino]propanoyl]amino]butanoyl]amino]-37-(2-carboxyethyl)-27-[(1r)-1-hydroxyethyl]-4-methyl-40-(2-methylp Chemical compound N1C(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CC(N)=O)CSSC[C@@H]2NC(=O)[C@H](C)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H]1CSSC[C@@H](C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)CNC(=O)[C@H]([C@@H](C)O)NC2=O NSPHQWLKCGGCQR-DLJDZFDSSA-N 0.000 description 1
- JNGVJMBLXIUVRD-SFHVURJKSA-N (2s)-3-(4-cyanophenoxy)-n-[4-cyano-3-(trifluoromethyl)phenyl]-2-hydroxy-2-methylpropanamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)OC1=CC=C(C#N)C=C1 JNGVJMBLXIUVRD-SFHVURJKSA-N 0.000 description 1
- LOGFVTREOLYCPF-KXNHARMFSA-N (2s,3r)-2-[[(2r)-1-[(2s)-2,6-diaminohexanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxybutanoic acid Chemical compound C[C@@H](O)[C@@H](C(O)=O)NC(=O)[C@H]1CCCN1C(=O)[C@@H](N)CCCCN LOGFVTREOLYCPF-KXNHARMFSA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- HMLGSIZOMSVISS-ONJSNURVSA-N (7r)-7-[[(2z)-2-(2-amino-1,3-thiazol-4-yl)-2-(2,2-dimethylpropanoyloxymethoxyimino)acetyl]amino]-3-ethenyl-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid Chemical compound N([C@@H]1C(N2C(=C(C=C)CSC21)C(O)=O)=O)C(=O)\C(=N/OCOC(=O)C(C)(C)C)C1=CSC(N)=N1 HMLGSIZOMSVISS-ONJSNURVSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- SZNJINHODJLULD-SDNWHVSQSA-N (e)-3-(4-hydroxy-3,5-dimethoxyphenyl)-n-(4-hydroxy-1-methyl-3-octoxy-2-oxoquinolin-7-yl)prop-2-enamide Chemical compound C1=C2N(C)C(=O)C(OCCCCCCCC)=C(O)C2=CC=C1NC(=O)\C=C\C1=CC(OC)=C(O)C(OC)=C1 SZNJINHODJLULD-SDNWHVSQSA-N 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical compound CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 1
- HLGVZNIODPNAME-UHFFFAOYSA-N 1-(1H-indazol-3-yl)-4,5,6,7-tetrahydroimidazo[4,5-c]pyridine Chemical compound N1N=C(C2=CC=CC=C12)N1C=NC=2CNCCC=21 HLGVZNIODPNAME-UHFFFAOYSA-N 0.000 description 1
- NGYNBSHYFOFVLS-LBPRGKRZSA-N 1-[4-chloro-2-hydroxy-3-[(3s)-piperidin-3-yl]sulfonylphenyl]-3-(3-fluoro-2-methylphenyl)urea Chemical compound CC1=C(F)C=CC=C1NC(=O)NC1=CC=C(Cl)C(S(=O)(=O)[C@@H]2CNCCC2)=C1O NGYNBSHYFOFVLS-LBPRGKRZSA-N 0.000 description 1
- RKSHRDUFWGVQSL-UHFFFAOYSA-N 1-bromo-2-ethyl-5-fluoro-4-phenylmethoxybenzene Chemical compound C(C1=CC=CC=C1)OC1=C(C=C(C(=C1)CC)Br)F RKSHRDUFWGVQSL-UHFFFAOYSA-N 0.000 description 1
- XXMFJKNOJSDQBM-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;hydrate Chemical compound [OH3+].[O-]C(=O)C(F)(F)F XXMFJKNOJSDQBM-UHFFFAOYSA-N 0.000 description 1
- 125000003562 2,2-dimethylpentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- FOCKIYZLSZKVDH-UHFFFAOYSA-N 2,3,3a,4-tetrahydro-1h-imidazo[4,5-b]pyridine Chemical group N1C=CC=C2NCNC21 FOCKIYZLSZKVDH-UHFFFAOYSA-N 0.000 description 1
- ZIIUUSVHCHPIQD-UHFFFAOYSA-N 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound CC1=CC(C)=CC(C)=C1S(=O)(=O)NC1=CC=CC(C(F)(F)F)=C1 ZIIUUSVHCHPIQD-UHFFFAOYSA-N 0.000 description 1
- BPXKZEMBEZGUAH-UHFFFAOYSA-N 2-(chloromethoxy)ethyl-trimethylsilane Chemical compound C[Si](C)(C)CCOCCl BPXKZEMBEZGUAH-UHFFFAOYSA-N 0.000 description 1
- RVLCUCVJZVRNDC-IMJSIDKUSA-N 2-[(2s,5s)-5-methyl-3,6-dioxopiperazin-2-yl]acetic acid Chemical compound C[C@@H]1NC(=O)[C@H](CC(O)=O)NC1=O RVLCUCVJZVRNDC-IMJSIDKUSA-N 0.000 description 1
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 1
- ZKLPARSLTMPFCP-OAQYLSRUSA-N 2-[2-[4-[(R)-(4-chlorophenyl)-phenylmethyl]-1-piperazinyl]ethoxy]acetic acid Chemical compound C1CN(CCOCC(=O)O)CCN1[C@@H](C=1C=CC(Cl)=CC=1)C1=CC=CC=C1 ZKLPARSLTMPFCP-OAQYLSRUSA-N 0.000 description 1
- GFPPXZDRVCSVNR-UHFFFAOYSA-N 2-[2-methyl-1-[[4-methylsulfonyl-2-(trifluoromethyl)phenyl]methyl]pyrrolo[2,3-b]pyridin-3-yl]acetic acid Chemical compound CC1=C(CC(O)=O)C2=CC=CN=C2N1CC1=CC=C(S(C)(=O)=O)C=C1C(F)(F)F GFPPXZDRVCSVNR-UHFFFAOYSA-N 0.000 description 1
- UBBJJBFUHAYINW-UHFFFAOYSA-N 2-[4-amino-3-chloro-5-(trifluoromethyl)phenyl]-2-(tert-butylamino)ethanol;hydrochloride Chemical compound Cl.CC(C)(C)NC(CO)C1=CC(Cl)=C(N)C(C(F)(F)F)=C1 UBBJJBFUHAYINW-UHFFFAOYSA-N 0.000 description 1
- FATGTHLOZSXOBC-UHFFFAOYSA-N 2-[5-fluoro-2-methyl-3-(quinolin-2-ylmethyl)indol-1-yl]acetic acid Chemical compound C12=CC(F)=CC=C2N(CC(O)=O)C(C)=C1CC1=CC=C(C=CC=C2)C2=N1 FATGTHLOZSXOBC-UHFFFAOYSA-N 0.000 description 1
- MCIDWGZGWVSZMK-UHFFFAOYSA-N 2-[6-(1h-indol-4-yl)-1h-indazol-4-yl]-5-[(4-propan-2-ylpiperazin-1-yl)methyl]-1,3-oxazole Chemical compound C1CN(C(C)C)CCN1CC1=CN=C(C=2C=3C=NNC=3C=C(C=2)C=2C=3C=CNC=3C=CC=2)O1 MCIDWGZGWVSZMK-UHFFFAOYSA-N 0.000 description 1
- OANCEOSLKSTLTA-REWPJTCUSA-N 2-[[(7s)-7-[[(2r)-2-hydroxy-2-[4-hydroxy-3-(2-hydroxyethyl)phenyl]ethyl]amino]-5,6,7,8-tetrahydronaphthalen-2-yl]oxy]-n,n-dimethylacetamide Chemical compound C1([C@@H](O)CN[C@H]2CCC3=CC=C(C=C3C2)OCC(=O)N(C)C)=CC=C(O)C(CCO)=C1 OANCEOSLKSTLTA-REWPJTCUSA-N 0.000 description 1
- JJBCTCGUOQYZHK-UHFFFAOYSA-N 2-acetyloxybenzoate;(5-amino-1-carboxypentyl)azanium Chemical compound OC(=O)C(N)CCCC[NH3+].CC(=O)OC1=CC=CC=C1C([O-])=O JJBCTCGUOQYZHK-UHFFFAOYSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- HBZAZSCNDMDWEU-WREZULKGSA-N 3,5-diamino-6-chloro-n-[n'-[4-[4-[2-[hexyl-[(2s,3r,4r,5r)-2,3,4,5,6-pentahydroxyhexyl]amino]ethoxy]phenyl]butyl]carbamimidoyl]pyrazine-2-carboxamide Chemical compound C1=CC(OCCN(CCCCCC)C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO)=CC=C1CCCCNC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N HBZAZSCNDMDWEU-WREZULKGSA-N 0.000 description 1
- ASZZHBXPMOVHCU-UHFFFAOYSA-N 3,9-diazaspiro[5.5]undecane-2,4-dione Chemical compound C1C(=O)NC(=O)CC11CCNCC1 ASZZHBXPMOVHCU-UHFFFAOYSA-N 0.000 description 1
- JMWYNUHGKFJVIB-QGFXQWJDSA-N 3-[(3R,9S,12S,15S,18S,24S,30S,33S,36S,39S,42S,45S,48S)-12-(4-aminobutyl)-15-(3-amino-3-oxopropyl)-30-[(2S)-butan-2-yl]-39-[(1R)-1-hydroxyethyl]-33-(hydroxymethyl)-9-[(4-hydroxyphenyl)methyl]-36-methyl-45-octyl-2,8,11,14,17,23,29,32,35,38,41,44,47-tridecaoxo-1,7,10,13,16,22,28,31,34,37,40,43,46-tridecazapentacyclo[46.3.0.03,7.018,22.024,28]henpentacontan-42-yl]propanoic acid Chemical compound C([C@H]1C(=O)N2CCC[C@@H]2C(=O)N2CCC[C@H]2C(=O)N[C@H](C(N[C@@H](CCC(O)=O)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N2CCC[C@H]2C(=O)N2CCC[C@H]2C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N1)[C@@H](C)CC)[C@@H](C)O)=O)CCCCCCCC)C1=CC=C(O)C=C1 JMWYNUHGKFJVIB-QGFXQWJDSA-N 0.000 description 1
- YVPGZQLRPAGKLA-UHFFFAOYSA-N 3-[1-(4-carbamoyl-2-methylphenyl)-5-(4-imidazol-1-ylphenyl)pyrrol-2-yl]propanoic acid Chemical compound CC1=CC(C(N)=O)=CC=C1N1C(C=2C=CC(=CC=2)N2C=NC=C2)=CC=C1CCC(O)=O YVPGZQLRPAGKLA-UHFFFAOYSA-N 0.000 description 1
- STWVLEKJQQRGMO-GUYCJALGSA-N 3-[4-[(2S,5S)-5-[(4-chlorophenyl)methyl]-2-methylmorpholin-4-yl]piperidin-1-yl]-1H-1,2,4-triazol-5-amine Chemical compound ClC1=CC=C(C[C@@H]2N(C[C@@H](OC2)C)C2CCN(CC2)C=2NC(=NN=2)N)C=C1 STWVLEKJQQRGMO-GUYCJALGSA-N 0.000 description 1
- HWNIPKHEEVNHMN-UHFFFAOYSA-N 3-[4-[2-[1-benzhydryl-5-chloro-2-[2-[[2-(trifluoromethyl)phenyl]methylsulfonylamino]ethyl]indol-3-yl]ethylsulfonyl]phenyl]propanoic acid Chemical compound C1=CC(CCC(=O)O)=CC=C1S(=O)(=O)CCC(C1=CC(Cl)=CC=C1N1C(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1CCNS(=O)(=O)CC1=CC=CC=C1C(F)(F)F HWNIPKHEEVNHMN-UHFFFAOYSA-N 0.000 description 1
- VGUSQKZDZHAAEE-UHFFFAOYSA-N 3-[5-amino-4-(3-cyanobenzoyl)pyrazol-1-yl]-n-cyclopropyl-4-methylbenzamide Chemical compound CC1=CC=C(C(=O)NC2CC2)C=C1N(C=1N)N=CC=1C(=O)C1=CC=CC(C#N)=C1 VGUSQKZDZHAAEE-UHFFFAOYSA-N 0.000 description 1
- ULMFXAMQUGLVGA-LJQANCHMSA-N 3-[[2-methoxy-4-[(2-methylphenyl)sulfonylcarbamoyl]phenyl]methyl]-1-methyl-n-[(2r)-4,4,4-trifluoro-2-methylbutyl]indole-5-carboxamide Chemical compound C=1C=C(CC=2C3=CC(=CC=C3N(C)C=2)C(=O)NC[C@H](C)CC(F)(F)F)C(OC)=CC=1C(=O)NS(=O)(=O)C1=CC=CC=C1C ULMFXAMQUGLVGA-LJQANCHMSA-N 0.000 description 1
- USHQRIKZLHNPQR-JTQLQIEISA-N 3-amino-6-methoxy-n-[(2s)-3,3,3-trifluoro-2-hydroxy-2-methylpropyl]-5-(trifluoromethyl)pyridine-2-carboxamide Chemical compound COC1=NC(C(=O)NC[C@](C)(O)C(F)(F)F)=C(N)C=C1C(F)(F)F USHQRIKZLHNPQR-JTQLQIEISA-N 0.000 description 1
- SUVMJBTUFCVSAD-JTQLQIEISA-N 4-Methylsulfinylbutyl isothiocyanate Natural products C[S@](=O)CCCCN=C=S SUVMJBTUFCVSAD-JTQLQIEISA-N 0.000 description 1
- MDHKCIIEVIPVLU-JERHFGHZSA-M 4-[(1r)-2-[6-[2-[(2,6-dichlorophenyl)methoxy]ethoxy]hexylamino]-1-hydroxyethyl]-2-(hydroxymethyl)phenol;diphenyl-[1-(2-phenylmethoxyethyl)-1-azoniabicyclo[2.2.2]octan-4-yl]methanol;bromide Chemical compound [Br-].C1=C(O)C(CO)=CC([C@@H](O)CNCCCCCCOCCOCC=2C(=CC=CC=2Cl)Cl)=C1.C=1C=CC=CC=1C(C12CC[N+](CCOCC=3C=CC=CC=3)(CC1)CC2)(O)C1=CC=CC=C1 MDHKCIIEVIPVLU-JERHFGHZSA-M 0.000 description 1
- SILHYVDKGHXGBL-UHFFFAOYSA-N 4-[1-(3-carboxypropyl)-4-fluoro-7-[2-[4-[4-(3-fluoro-2-methylphenyl)butoxy]phenyl]ethynyl]-2-methylindol-3-yl]butanoic acid Chemical compound C=12N(CCCC(O)=O)C(C)=C(CCCC(O)=O)C2=C(F)C=CC=1C#CC(C=C1)=CC=C1OCCCCC1=CC=CC(F)=C1C SILHYVDKGHXGBL-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- ZQQSRVPOAHYHEL-UHFFFAOYSA-N 4-bromo-2-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(Br)C=C1F ZQQSRVPOAHYHEL-UHFFFAOYSA-N 0.000 description 1
- AGPCZZAYJRAENV-UHFFFAOYSA-N 4-bromo-5-ethyl-2-fluorophenol Chemical compound CCC1=CC(O)=C(F)C=C1Br AGPCZZAYJRAENV-UHFFFAOYSA-N 0.000 description 1
- HLWURFKMDLAKOD-UHFFFAOYSA-N 5-(2,4-diaminopyrimidin-5-yl)oxy-2-methoxy-4-propan-2-ylbenzenesulfonamide Chemical compound C1=C(S(N)(=O)=O)C(OC)=CC(C(C)C)=C1OC1=CN=C(N)N=C1N HLWURFKMDLAKOD-UHFFFAOYSA-N 0.000 description 1
- DKBNZFVZTNDLJJ-XFQAGIBXSA-M 5-[1-hydroxy-2-[1-(4-hydroxyphenyl)propan-2-ylamino]ethyl]benzene-1,3-diol;[(1r,5s)-8-methyl-8-propan-2-yl-8-azoniabicyclo[3.2.1]octan-3-yl] 3-hydroxy-2-phenylpropanoate;bromide Chemical compound [Br-].C=1C(O)=CC(O)=CC=1C(O)CNC(C)CC1=CC=C(O)C=C1.C([C@H]1CC[C@@H](C2)[N+]1(C)C(C)C)C2OC(=O)C(CO)C1=CC=CC=C1 DKBNZFVZTNDLJJ-XFQAGIBXSA-M 0.000 description 1
- YJMYSLFFZJUXOA-UHFFFAOYSA-N 5-bromo-3-[3-(4-chlorophenyl)propoxy]-4-(pyridin-3-ylmethylamino)-1h-pyridazin-6-one Chemical compound C1=CC(Cl)=CC=C1CCCOC1=NNC(=O)C(Br)=C1NCC1=CC=CN=C1 YJMYSLFFZJUXOA-UHFFFAOYSA-N 0.000 description 1
- RFAYDBLGPUAICX-UHFFFAOYSA-N 6-(2-ethyl-5-fluoro-4-phenylmethoxyphenyl)-1-(oxan-2-yl)indazole Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1F)C1=CC=C2C=NN(C2=C1)C1OCCCC1)CC RFAYDBLGPUAICX-UHFFFAOYSA-N 0.000 description 1
- XLLRREYHAULADB-UHFFFAOYSA-N 6-(2-ethyl-5-fluoro-4-phenylmethoxyphenyl)-3-iodo-2H-indazole Chemical compound C(C1=CC=CC=C1)OC1=CC(=C(C=C1F)C1=CC=C2C(=NNC2=C1)I)CC XLLRREYHAULADB-UHFFFAOYSA-N 0.000 description 1
- UFMHRNIBPKQDJY-SFHVURJKSA-N 6-O-benzyl 3-O,5-O-ditert-butyl (6S)-6,7-dihydro-4H-imidazo[4,5-c]pyridine-3,5,6-tricarboxylate Chemical compound N1=CN(C=2CN([C@@H](CC=21)C(=O)OCC1=CC=CC=C1)C(=O)OC(C)(C)C)C(=O)OC(C)(C)C UFMHRNIBPKQDJY-SFHVURJKSA-N 0.000 description 1
- UBLOHCIYTDRGJH-UHFFFAOYSA-N 6-[2-[[4-amino-3-(3-hydroxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]methyl]-3-[(2-chlorophenyl)methyl]-4-oxoquinazolin-5-yl]-n,n-bis(2-methoxyethyl)hex-5-ynamide Chemical compound C=1C=CC=C(Cl)C=1CN1C(=O)C=2C(C#CCCCC(=O)N(CCOC)CCOC)=CC=CC=2N=C1CN(C1=NC=NC(N)=C11)N=C1C1=CC=CC(O)=C1 UBLOHCIYTDRGJH-UHFFFAOYSA-N 0.000 description 1
- QRBVUFXEMHNIDB-UHFFFAOYSA-N 6-[3-(methylamino)azetidin-1-yl]-2-(2-methylpropyl)pyrimidin-4-amine Chemical compound C1C(NC)CN1C1=CC(N)=NC(CC(C)C)=N1 QRBVUFXEMHNIDB-UHFFFAOYSA-N 0.000 description 1
- LFMPVTVPXHNXOT-HNNXBMFYSA-N 6-amino-2-[(2s)-pentan-2-yl]oxy-9-(5-piperidin-1-ylpentyl)-7h-purin-8-one Chemical compound C12=NC(O[C@@H](C)CCC)=NC(N)=C2NC(=O)N1CCCCCN1CCCCC1 LFMPVTVPXHNXOT-HNNXBMFYSA-N 0.000 description 1
- XUSDKDVQICYOIP-UHFFFAOYSA-N 6-bromo-1-(oxan-2-yl)indazole Chemical compound C12=CC(Br)=CC=C2C=NN1C1CCCCO1 XUSDKDVQICYOIP-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- SJVQHLPISAIATJ-ZDUSSCGKSA-N 8-chloro-2-phenyl-3-[(1S)-1-(7H-purin-6-ylamino)ethyl]-1-isoquinolinone Chemical compound C1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)=CC2=CC=CC(Cl)=C2C(=O)N1C1=CC=CC=C1 SJVQHLPISAIATJ-ZDUSSCGKSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 239000010158 AG NPP709 Substances 0.000 description 1
- 229940121819 ATPase inhibitor Drugs 0.000 description 1
- 229940122578 Acetylcholine receptor agonist Drugs 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 108010085238 Actins Proteins 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 229940124258 Adenosine A1 receptor antagonist Drugs 0.000 description 1
- 229940122869 Adenylate cyclase stimulant Drugs 0.000 description 1
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 1
- 102100022455 Adrenocorticotropic hormone receptor Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 208000036065 Airway Remodeling Diseases 0.000 description 1
- 229940123470 Alcohol dehydrogenase 5 inhibitor Drugs 0.000 description 1
- 208000032671 Allergic granulomatous angiitis Diseases 0.000 description 1
- 206010001764 Alopecia scarring Diseases 0.000 description 1
- 208000035939 Alveolitis allergic Diseases 0.000 description 1
- 108010028700 Amine Oxidase (Copper-Containing) Proteins 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 102000013455 Amyloid beta-Peptides Human genes 0.000 description 1
- 108010090849 Amyloid beta-Peptides Proteins 0.000 description 1
- 206010002412 Angiocentric lymphomas Diseases 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 102000001381 Arachidonate 5-Lipoxygenase Human genes 0.000 description 1
- 108010093579 Arachidonate 5-lipoxygenase Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000269837 Artemisia dubia Species 0.000 description 1
- 235000003261 Artemisia vulgaris Nutrition 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 238000011725 BALB/c mouse Methods 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 101150082778 BMP10 gene Proteins 0.000 description 1
- 101150038684 BMP15 gene Proteins 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- RPHFGBIUXJLOEI-IBGZPJMESA-N C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)C)N1 Chemical compound C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)C)N1 RPHFGBIUXJLOEI-IBGZPJMESA-N 0.000 description 1
- RCGGYNUURROHKZ-NRFANRHFSA-N C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)CCC)N1 Chemical compound C(C)C1=C(C=C(C(=C1)O)F)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)O)CCC)N1 RCGGYNUURROHKZ-NRFANRHFSA-N 0.000 description 1
- 102100037853 C-C chemokine receptor type 4 Human genes 0.000 description 1
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 description 1
- 102100028989 C-X-C chemokine receptor type 2 Human genes 0.000 description 1
- 101710098275 C-X-C motif chemokine 10 Proteins 0.000 description 1
- 101710085500 C-X-C motif chemokine 9 Proteins 0.000 description 1
- 101150112860 CCL26 gene Proteins 0.000 description 1
- RYACCWZJZAKABF-ATJOBZSNSA-N CCc1cc(O)c(F)cc1C1CCC2C(C1)NNC2c1nc2C[C@H](N(Cc2[nH]1)C(C)C)C(=O)N1CC(C1)N(C)C Chemical compound CCc1cc(O)c(F)cc1C1CCC2C(C1)NNC2c1nc2C[C@H](N(Cc2[nH]1)C(C)C)C(=O)N1CC(C1)N(C)C RYACCWZJZAKABF-ATJOBZSNSA-N 0.000 description 1
- 229940044663 CMP-001 Drugs 0.000 description 1
- ZNKWRAKPQQZLNX-ZDQHWEPJSA-N CN(CCCn1nnc2cc(CNC[C@H](O)c3ccc(O)c4[nH]c(=O)ccc34)ccc12)[C@H]1CC[C@@H](CC1)OC(=O)C(O)(c1cccs1)c1cccs1 Chemical compound CN(CCCn1nnc2cc(CNC[C@H](O)c3ccc(O)c4[nH]c(=O)ccc34)ccc12)[C@H]1CC[C@@H](CC1)OC(=O)C(O)(c1cccs1)c1cccs1 ZNKWRAKPQQZLNX-ZDQHWEPJSA-N 0.000 description 1
- 229940124003 CRTH2 antagonist Drugs 0.000 description 1
- 239000012275 CTLA-4 inhibitor Substances 0.000 description 1
- 229940045513 CTLA4 antagonist Drugs 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- 101100268548 Caenorhabditis elegans apl-1 gene Proteins 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 102000003902 Cathepsin C Human genes 0.000 description 1
- 108090000267 Cathepsin C Proteins 0.000 description 1
- 229940122079 Cathepsin G inhibitor Drugs 0.000 description 1
- 108010067225 Cell Adhesion Molecules Proteins 0.000 description 1
- 102000016289 Cell Adhesion Molecules Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 108050001186 Chaperonin Cpn60 Proteins 0.000 description 1
- 102000052603 Chaperonins Human genes 0.000 description 1
- 229940125842 Chitinase inhibitor Drugs 0.000 description 1
- 102000012286 Chitinases Human genes 0.000 description 1
- 108010022172 Chitinases Proteins 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- 208000006344 Churg-Strauss Syndrome Diseases 0.000 description 1
- LUKZNWIVRBCLON-GXOBDPJESA-N Ciclesonide Chemical compound C1([C@H]2O[C@@]3([C@H](O2)C[C@@H]2[C@@]3(C[C@H](O)[C@@H]3[C@@]4(C)C=CC(=O)C=C4CC[C@H]32)C)C(=O)COC(=O)C(C)C)CCCCC1 LUKZNWIVRBCLON-GXOBDPJESA-N 0.000 description 1
- BLMQUENFEUQJRY-FERBBOLQSA-N Cl.C(C)C1=C(C=CC(=C1)O)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN[C@@H](C2)C(=O)O)N1 Chemical compound Cl.C(C)C1=C(C=CC(=C1)O)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN[C@@H](C2)C(=O)O)N1 BLMQUENFEUQJRY-FERBBOLQSA-N 0.000 description 1
- 102000012422 Collagen Type I Human genes 0.000 description 1
- 108010022452 Collagen Type I Proteins 0.000 description 1
- 102000008954 Copper amine oxidases Human genes 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- 108010074311 Corticotropin Receptors Proteins 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 102100038496 Cysteinyl leukotriene receptor 1 Human genes 0.000 description 1
- 102100038497 Cytokine receptor-like factor 2 Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 101710137619 DNA gyrase inhibitor Proteins 0.000 description 1
- 229940087106 DNA topoisomerase IV inhibitor Drugs 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical compound C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 229940122858 Elastase inhibitor Drugs 0.000 description 1
- 102000016942 Elastin Human genes 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 206010014950 Eosinophilia Diseases 0.000 description 1
- 102100023688 Eotaxin Human genes 0.000 description 1
- 101710139422 Eotaxin Proteins 0.000 description 1
- 229940122183 Epoxide hydrolase inhibitor Drugs 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 102100028043 Fibroblast growth factor 3 Human genes 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- 229940123570 Fyn tyrosine kinase inhibitor Drugs 0.000 description 1
- 108010003338 GATA3 Transcription Factor Proteins 0.000 description 1
- 102000004610 GATA3 Transcription Factor Human genes 0.000 description 1
- 208000017189 Gastrointestinal inflammatory disease Diseases 0.000 description 1
- 229940123127 Glucocorticoid agonist Drugs 0.000 description 1
- 102000004547 Glucosylceramidase Human genes 0.000 description 1
- 108010017544 Glucosylceramidase Proteins 0.000 description 1
- 102000018899 Glutamate Receptors Human genes 0.000 description 1
- 108010027915 Glutamate Receptors Proteins 0.000 description 1
- 229940122247 Glutathione reductase inhibitor Drugs 0.000 description 1
- VPNYRYCIDCJBOM-UHFFFAOYSA-M Glycopyrronium bromide Chemical compound [Br-].C1[N+](C)(C)CCC1OC(=O)C(O)(C=1C=CC=CC=1)C1CCCC1 VPNYRYCIDCJBOM-UHFFFAOYSA-M 0.000 description 1
- 201000005708 Granuloma Annulare Diseases 0.000 description 1
- 229940118547 Guanylate cyclase stimulant Drugs 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 102100029100 Hematopoietic prostaglandin D synthase Human genes 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 102100035108 High affinity nerve growth factor receptor Human genes 0.000 description 1
- 229940119240 Histamine H4 receptor antagonist Drugs 0.000 description 1
- 229940122236 Histamine receptor antagonist Drugs 0.000 description 1
- 108010023981 Histone Deacetylase 2 Proteins 0.000 description 1
- 102100039999 Histone deacetylase 2 Human genes 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000978362 Homo sapiens C-C motif chemokine 17 Proteins 0.000 description 1
- 101000882759 Homo sapiens Cysteinyl leukotriene receptor 1 Proteins 0.000 description 1
- 101000956427 Homo sapiens Cytokine receptor-like factor 2 Proteins 0.000 description 1
- 101001060280 Homo sapiens Fibroblast growth factor 3 Proteins 0.000 description 1
- 101000988802 Homo sapiens Hematopoietic prostaglandin D synthase Proteins 0.000 description 1
- 101000596894 Homo sapiens High affinity nerve growth factor receptor Proteins 0.000 description 1
- 101001019591 Homo sapiens Interleukin-18-binding protein Proteins 0.000 description 1
- 101000780028 Homo sapiens Natriuretic peptides A Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000823316 Homo sapiens Tyrosine-protein kinase ABL1 Proteins 0.000 description 1
- ZQDGGHCLCKUJQC-VIFPVBQESA-N IC1=NC2=C(CN([C@@H](C2)C(=O)OC)C(=O)OC(C)(C)C)N1 Chemical compound IC1=NC2=C(CN([C@@H](C2)C(=O)OC)C(=O)OC(C)(C)C)N1 ZQDGGHCLCKUJQC-VIFPVBQESA-N 0.000 description 1
- 101150050263 ICAM1 gene Proteins 0.000 description 1
- 101150083678 IL2 gene Proteins 0.000 description 1
- 102000009490 IgG Receptors Human genes 0.000 description 1
- 108010073807 IgG Receptors Proteins 0.000 description 1
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 1
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100021854 Inhibitor of nuclear factor kappa-B kinase subunit beta Human genes 0.000 description 1
- 101710205525 Inhibitor of nuclear factor kappa-B kinase subunit beta Proteins 0.000 description 1
- 229940122355 Insulin sensitizer Drugs 0.000 description 1
- 102000000589 Interleukin-1 Human genes 0.000 description 1
- 108010002352 Interleukin-1 Proteins 0.000 description 1
- 102000003777 Interleukin-1 beta Human genes 0.000 description 1
- 108090000193 Interleukin-1 beta Proteins 0.000 description 1
- 102000051628 Interleukin-1 receptor antagonist Human genes 0.000 description 1
- 108700021006 Interleukin-1 receptor antagonist Proteins 0.000 description 1
- 102000004554 Interleukin-17 Receptors Human genes 0.000 description 1
- 108010017525 Interleukin-17 Receptors Proteins 0.000 description 1
- 102000003810 Interleukin-18 Human genes 0.000 description 1
- 108090000171 Interleukin-18 Proteins 0.000 description 1
- 102100035017 Interleukin-18-binding protein Human genes 0.000 description 1
- 108010067003 Interleukin-33 Proteins 0.000 description 1
- 102000017761 Interleukin-33 Human genes 0.000 description 1
- 102000010787 Interleukin-4 Receptors Human genes 0.000 description 1
- 108010038486 Interleukin-4 Receptors Proteins 0.000 description 1
- 102000010781 Interleukin-6 Receptors Human genes 0.000 description 1
- 108010038501 Interleukin-6 Receptors Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010018951 Interleukin-8B Receptors Proteins 0.000 description 1
- 208000003456 Juvenile Arthritis Diseases 0.000 description 1
- 206010059176 Juvenile idiopathic arthritis Diseases 0.000 description 1
- 102100020880 Kit ligand Human genes 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- 229940121930 L-selectin antagonist Drugs 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002139 L01XE22 - Masitinib Substances 0.000 description 1
- 101150043981 LOXL2 gene Proteins 0.000 description 1
- 229940122145 LYN tyrosine kinase inhibitor Drugs 0.000 description 1
- 102100030635 Leukocyte elastase inhibitor Human genes 0.000 description 1
- 101710091916 Leukocyte elastase inhibitor Proteins 0.000 description 1
- GWNVDXQDILPJIG-SHSCPDMUSA-N Leukotriene C4 Natural products CCCCCC=C/CC=C/C=C/C=C/C(SCC(NC(=O)CCC(N)C(=O)O)C(=O)NCC(=O)O)C(O)CCCC(=O)O GWNVDXQDILPJIG-SHSCPDMUSA-N 0.000 description 1
- 102100023758 Leukotriene C4 synthase Human genes 0.000 description 1
- OTZRAYGBFWZKMX-SHSCPDMUSA-N Leukotriene E4 Natural products CCCCCC=C/CC=C/C=C/C=C/C(SCC(N)C(=O)O)C(O)CCCC(=O)O OTZRAYGBFWZKMX-SHSCPDMUSA-N 0.000 description 1
- 206010024434 Lichen sclerosus Diseases 0.000 description 1
- 208000024078 Localized pagetoid reticulosis Diseases 0.000 description 1
- 208000032376 Lung infection Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 102000008072 Lymphokines Human genes 0.000 description 1
- 108010074338 Lymphokines Proteins 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 229940122696 MAP kinase inhibitor Drugs 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- YFGBQHOOROIVKG-FKBYEOEOSA-N Met-enkephalin Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 YFGBQHOOROIVKG-FKBYEOEOSA-N 0.000 description 1
- 101800001325 Met-enkephalin Proteins 0.000 description 1
- 102400000988 Met-enkephalin Human genes 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102100023482 Mitogen-activated protein kinase 14 Human genes 0.000 description 1
- PVLJETXTTWAYEW-UHFFFAOYSA-N Mizolastine Chemical compound N=1C=CC(=O)NC=1N(C)C(CC1)CCN1C1=NC2=CC=CC=C2N1CC1=CC=C(F)C=C1 PVLJETXTTWAYEW-UHFFFAOYSA-N 0.000 description 1
- UCHDWCPVSPXUMX-TZIWLTJVSA-N Montelukast Chemical compound CC(C)(O)C1=CC=CC=C1CC[C@H](C=1C=C(\C=C\C=2N=C3C=C(Cl)C=CC3=CC=2)C=CC=1)SCC1(CC(O)=O)CC1 UCHDWCPVSPXUMX-TZIWLTJVSA-N 0.000 description 1
- 208000026072 Motor neurone disease Diseases 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 101001065566 Mus musculus Lymphocyte antigen 6A-2/6E-1 Proteins 0.000 description 1
- 229940082332 Muscarinic M1 receptor antagonist Drugs 0.000 description 1
- 229940123467 Muscarinic M2 receptor antagonist Drugs 0.000 description 1
- 229940117926 Muscarinic M4 receptor antagonist Drugs 0.000 description 1
- 108010008409 Muscarinic M5 Receptor Proteins 0.000 description 1
- 102000014415 Muscarinic acetylcholine receptor Human genes 0.000 description 1
- 108050003473 Muscarinic acetylcholine receptor Proteins 0.000 description 1
- 229940121948 Muscarinic receptor antagonist Drugs 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- 102100031789 Myeloid-derived growth factor Human genes 0.000 description 1
- 108010063737 Myristoylated Alanine-Rich C Kinase Substrate Proteins 0.000 description 1
- 102000015695 Myristoylated Alanine-Rich C Kinase Substrate Human genes 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- KSIJIGHGXBEURD-UHFFFAOYSA-N N1(CCCCC1)CCOC1=CC=C(C=C1)C1=NN(C(=C1)C1=CC=C(C=C1)O)C1=CC=C(C=C1)O Chemical compound N1(CCCCC1)CCOC1=CC=C(C=C1)C1=NN(C(=C1)C1=CC=C(C=C1)O)C1=CC=C(C=C1)O KSIJIGHGXBEURD-UHFFFAOYSA-N 0.000 description 1
- XIKVJZOLNXTOFN-UHFFFAOYSA-N N1C=NC2=C1CCCCN2 Chemical class N1C=NC2=C1CCCCN2 XIKVJZOLNXTOFN-UHFFFAOYSA-N 0.000 description 1
- CONJUINEIXHPID-UHFFFAOYSA-N N1N=C(C2=CC=CC=C12)C=1NC2=C(CNCC2)N=1 Chemical compound N1N=C(C2=CC=CC=C12)C=1NC2=C(CNCC2)N=1 CONJUINEIXHPID-UHFFFAOYSA-N 0.000 description 1
- MQXWPWOCXGARRK-HJGJAMNPSA-N NC(=O)C1=CN(C=C1)C1=CN=C(NC[C@@]2(C[C@H](F)C2)C2=NC=CC=C2F)N=C1 Chemical compound NC(=O)C1=CN(C=C1)C1=CN=C(NC[C@@]2(C[C@H](F)C2)C2=NC=CC=C2F)N=C1 MQXWPWOCXGARRK-HJGJAMNPSA-N 0.000 description 1
- 108091008877 NK cell receptors Proteins 0.000 description 1
- 102100034296 Natriuretic peptides A Human genes 0.000 description 1
- 108010077854 Natural Killer Cell Receptors Proteins 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 201000009053 Neurodermatitis Diseases 0.000 description 1
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 1
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 229940125840 OATD-01 Drugs 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229940121825 P-selectin antagonist Drugs 0.000 description 1
- 102100040460 P2X purinoceptor 3 Human genes 0.000 description 1
- 101710189970 P2X purinoceptor 3 Proteins 0.000 description 1
- 102100032341 PCNA-interacting partner Human genes 0.000 description 1
- 101710196737 PCNA-interacting partner Proteins 0.000 description 1
- 239000012270 PD-1 inhibitor Substances 0.000 description 1
- 239000012668 PD-1-inhibitor Substances 0.000 description 1
- 239000012271 PD-L1 inhibitor Substances 0.000 description 1
- 208000002541 Pagetoid Reticulosis Diseases 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 101150034459 Parpbp gene Proteins 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 101710167374 Peptidase 1 Proteins 0.000 description 1
- 229940080774 Peroxisome proliferator-activated receptor gamma agonist Drugs 0.000 description 1
- 229940049937 Pgp inhibitor Drugs 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- 229940123263 Phosphodiesterase 3 inhibitor Drugs 0.000 description 1
- 229940123932 Phosphodiesterase 4 inhibitor Drugs 0.000 description 1
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 description 1
- 229940124034 Phospholipase C inhibitor Drugs 0.000 description 1
- 102000015439 Phospholipases Human genes 0.000 description 1
- 108010064785 Phospholipases Proteins 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- QSXMZJGGEWYVCN-UHFFFAOYSA-N Pirbuterol acetate Chemical compound CC(O)=O.CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=N1 QSXMZJGGEWYVCN-UHFFFAOYSA-N 0.000 description 1
- 208000000766 Pityriasis Lichenoides Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 108010022233 Plasminogen Activator Inhibitor 1 Proteins 0.000 description 1
- 102100039418 Plasminogen activator inhibitor 1 Human genes 0.000 description 1
- 229940124090 Platelet-derived growth factor (PDGF) receptor antagonist Drugs 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 102100027467 Pro-opiomelanocortin Human genes 0.000 description 1
- 206010036774 Proctitis Diseases 0.000 description 1
- 206010036783 Proctitis ulcerative Diseases 0.000 description 1
- 102100024212 Prostaglandin D2 receptor Human genes 0.000 description 1
- 102100024218 Prostaglandin D2 receptor 2 Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- BPZSYCZIITTYBL-YJYMSZOUSA-N R-Formoterol Chemical compound C1=CC(OC)=CC=C1C[C@@H](C)NC[C@H](O)C1=CC=C(O)C(NC=O)=C1 BPZSYCZIITTYBL-YJYMSZOUSA-N 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- LDXDSHIEDAPSSA-OAHLLOKOSA-N Ramatroban Chemical compound N([C@@H]1CCC=2N(C3=CC=CC=C3C=2C1)CCC(=O)O)S(=O)(=O)C1=CC=C(F)C=C1 LDXDSHIEDAPSSA-OAHLLOKOSA-N 0.000 description 1
- NFQIAEMCQGTTIR-UHFFFAOYSA-N Repirinast Chemical compound C12=CC=C(C)C(C)=C2NC(=O)C2=C1OC(C(=O)OCCC(C)C)=CC2=O NFQIAEMCQGTTIR-UHFFFAOYSA-N 0.000 description 1
- 208000018569 Respiratory Tract disease Diseases 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 229940123578 Selectin antagonist Drugs 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- IHAXLPDVOWLUOS-UHFFFAOYSA-N Setipiprant Chemical compound C1=CC=C2C(C(=O)N3CCC=4N(C5=CC=C(F)C=C5C=4C3)CC(=O)O)=CC=CC2=C1 IHAXLPDVOWLUOS-UHFFFAOYSA-N 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 1
- 206010041660 Splenomegaly Diseases 0.000 description 1
- 108010039445 Stem Cell Factor Proteins 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- DYZJXZOQQRXDLE-UHFFFAOYSA-O Suplatast tosilate Chemical compound CCOCC(O)COC1=CC=C(NC(=O)CC[S+](C)C)C=C1 DYZJXZOQQRXDLE-UHFFFAOYSA-O 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 229940124615 TLR 7 agonist Drugs 0.000 description 1
- 229940124614 TLR 8 agonist Drugs 0.000 description 1
- 101150038509 TLR9 gene Proteins 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- 206010043391 Thalassaemia beta Diseases 0.000 description 1
- 108010069102 Thromboxane-A synthase Proteins 0.000 description 1
- 108010046075 Thymosin Proteins 0.000 description 1
- 102000007501 Thymosin Human genes 0.000 description 1
- UGPMCIBIHRSCBV-XNBOLLIBSA-N Thymosin beta 4 Chemical compound N([C@@H](CC(O)=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(O)=O)C(=O)[C@@H]1CCCN1C(=O)[C@H](CCCCN)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(C)=O UGPMCIBIHRSCBV-XNBOLLIBSA-N 0.000 description 1
- 102100035000 Thymosin beta-4 Human genes 0.000 description 1
- KPWYNAGOBXLMSE-UHFFFAOYSA-N Tipelukast Chemical compound CCCC1=C(O)C(C(C)=O)=CC=C1SCCCOC1=CC=C(C(C)=O)C(OCCCC(O)=O)=C1CCC KPWYNAGOBXLMSE-UHFFFAOYSA-N 0.000 description 1
- 239000004012 Tofacitinib Substances 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 102000000887 Transcription factor STAT Human genes 0.000 description 1
- 108050007918 Transcription factor STAT Proteins 0.000 description 1
- 229940122954 Transcription factor inhibitor Drugs 0.000 description 1
- 206010052779 Transplant rejections Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102000013394 Troponin I Human genes 0.000 description 1
- 108010065729 Troponin I Proteins 0.000 description 1
- 102000004987 Troponin T Human genes 0.000 description 1
- 108090001108 Troponin T Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102100022596 Tyrosine-protein kinase ABL1 Human genes 0.000 description 1
- 102000003848 Uteroglobin Human genes 0.000 description 1
- 108090000203 Uteroglobin Proteins 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 108010003205 Vasoactive Intestinal Peptide Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- YEEZWCHGZNKEEK-UHFFFAOYSA-N Zafirlukast Chemical compound COC1=CC(C(=O)NS(=O)(=O)C=2C(=CC=CC=2)C)=CC=C1CC(C1=C2)=CN(C)C1=CC=C2NC(=O)OC1CCCC1 YEEZWCHGZNKEEK-UHFFFAOYSA-N 0.000 description 1
- VCFBPAOSTLMYIV-SANMLTNESA-N [(1s)-1-[3-(cyclopropylmethoxy)-4-(difluoromethoxy)phenyl]-2-(3,5-dichloro-1-oxidopyridin-1-ium-4-yl)ethyl] 3-(cyclopropylmethoxy)-4-(methanesulfonamido)benzoate Chemical compound CS(=O)(=O)NC1=CC=C(C(=O)O[C@@H](CC=2C(=C[N+]([O-])=CC=2Cl)Cl)C=2C=C(OCC3CC3)C(OC(F)F)=CC=2)C=C1OCC1CC1 VCFBPAOSTLMYIV-SANMLTNESA-N 0.000 description 1
- FTQBYQRDFCFCFN-NDEPHWFRSA-N [(6S)-2-[6-(2-ethyl-4-hydroxyphenyl)-1H-indazol-3-yl]-5-propyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-6-yl]-[4-(2-hydroxyethyl)piperazin-1-yl]methanone Chemical compound C(C)C1=C(C=CC(=C1)O)C1=CC=C2C(=NNC2=C1)C1=NC2=C(CN([C@@H](C2)C(=O)N2CCN(CC2)CCO)CCC)N1 FTQBYQRDFCFCFN-NDEPHWFRSA-N 0.000 description 1
- XWHWXAVEQLLHAS-VNGZUUOUSA-N [(6S)-2-[6-(2-ethyl-5-fluoro-4-hydroxyphenyl)-2,3,3a,4,5,6,7,7a-octahydro-1H-indazol-3-yl]-5-methyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-6-yl]-[(2R)-4-(2-hydroxyethyl)-2-methylpiperazin-1-yl]methanone Chemical compound CCc1cc(O)c(F)cc1C1CCC2C(C1)NNC2c1nc2C[C@H](N(C)Cc2[nH]1)C(=O)N1CCN(CCO)C[C@H]1C XWHWXAVEQLLHAS-VNGZUUOUSA-N 0.000 description 1
- DDRLPKKDPQEFGJ-PTKUJLJCSA-N [(6S)-2-[6-(2-ethyl-5-fluoro-4-hydroxyphenyl)-2,3,3a,4,5,6,7,7a-octahydro-1H-indazol-3-yl]-5-propan-2-yl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-6-yl]-(4-methyl-1,4-diazepan-1-yl)methanone Chemical compound CCc1cc(O)c(F)cc1C1CCC2C(C1)NNC2c1nc2C[C@H](N(Cc2[nH]1)C(C)C)C(=O)N1CCCN(C)CC1 DDRLPKKDPQEFGJ-PTKUJLJCSA-N 0.000 description 1
- XDMRJFDJTWYBPV-DZDUBCNZSA-N [(6S)-2-[6-(2-ethyl-5-fluoro-4-hydroxyphenyl)-2,3,3a,4,5,6,7,7a-octahydro-1H-indazol-3-yl]-5-propyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-6-yl]-[(1S,4S)-5-methyl-2,5-diazabicyclo[2.2.1]heptan-2-yl]methanone Chemical compound CCCN1Cc2[nH]c(nc2C[C@H]1C(=O)N1C[C@@H]2C[C@H]1CN2C)C1NNC2CC(CCC12)c1cc(F)c(O)cc1CC XDMRJFDJTWYBPV-DZDUBCNZSA-N 0.000 description 1
- NPSHRQCNIWLYRT-PTKUJLJCSA-N [(6S)-2-[6-(2-ethyl-5-fluoro-4-hydroxyphenyl)-2,3,3a,4,5,6,7,7a-octahydro-1H-indazol-3-yl]-5-propyl-3,4,6,7-tetrahydroimidazo[4,5-c]pyridin-6-yl]-[4-(2-hydroxyethyl)piperazin-1-yl]methanone Chemical compound CCCN1Cc2[nH]c(nc2C[C@H]1C(=O)N1CCN(CCO)CC1)C1NNC2CC(CCC12)c1cc(F)c(O)cc1CC NPSHRQCNIWLYRT-PTKUJLJCSA-N 0.000 description 1
- FYDWDCIFZSGNBU-UHFFFAOYSA-N [1-[2-[[4-[(4-carbamoylpiperidin-1-yl)methyl]benzoyl]-methylamino]ethyl]piperidin-4-yl] n-(2-phenylphenyl)carbamate Chemical compound C=1C=C(CN2CCC(CC2)C(N)=O)C=CC=1C(=O)N(C)CCN(CC1)CCC1OC(=O)NC1=CC=CC=C1C1=CC=CC=C1 FYDWDCIFZSGNBU-UHFFFAOYSA-N 0.000 description 1
- AGQFLEFSRLZALV-KQYNXXCUSA-N [9-[(2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-6-iminopurin-3-yl]phosphonic acid Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(N(C=NC2=N)P(O)(O)=O)=C2N=C1 AGQFLEFSRLZALV-KQYNXXCUSA-N 0.000 description 1
- 229960003697 abatacept Drugs 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- PMZXXNPJQYDFJX-UHFFFAOYSA-N acetonitrile;2,2,2-trifluoroacetic acid Chemical group CC#N.OC(=O)C(F)(F)F PMZXXNPJQYDFJX-UHFFFAOYSA-N 0.000 description 1
- XBJFCYDKBDVADW-UHFFFAOYSA-N acetonitrile;formic acid Chemical compound CC#N.OC=O XBJFCYDKBDVADW-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 229940019903 aclidinium Drugs 0.000 description 1
- 229960005012 aclidinium bromide Drugs 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- 229940099550 actimmune Drugs 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- YAJCHEVQCOHZDC-QMMNLEPNSA-N actrapid Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3N=CNC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@H](C)CC)[C@H](C)CC)[C@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C(N)=O)C1=CNC=N1 YAJCHEVQCOHZDC-QMMNLEPNSA-N 0.000 description 1
- 229950006577 acumapimod Drugs 0.000 description 1
- 229960002964 adalimumab Drugs 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 239000002598 adenosine A1 receptor antagonist Substances 0.000 description 1
- 239000000362 adenosine triphosphatase inhibitor Substances 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000000674 adrenergic antagonist Substances 0.000 description 1
- ISBHYKVAFKTATD-SNVBAGLBSA-N adriforant Chemical compound C1[C@H](NC)CCN1C1=CC(NCC2CC2)=NC(N)=N1 ISBHYKVAFKTATD-SNVBAGLBSA-N 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 239000013576 airborne fungal allergen Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229940083712 aldosterone antagonist Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229950011466 alicaforsen Drugs 0.000 description 1
- ZMJWRJKGPUDEOX-LMXUULCNSA-A alicaforsen Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C=C2)=O)COP([O-])(=S)O[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)CO)[C@@H](OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([S-])(=O)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(NC(=O)C(C)=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(NC(=O)C(C)=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C(N=C(N)C=C2)=O)OP([O-])(=S)OC[C@@H]2[C@H](C[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)C1 ZMJWRJKGPUDEOX-LMXUULCNSA-A 0.000 description 1
- 208000006778 allergic bronchopulmonary aspergillosis Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 229940024142 alpha 1-antitrypsin Drugs 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 108700038111 alunacedase alfa Proteins 0.000 description 1
- JBDGDEWWOUBZPM-XYPYZODXSA-N ambroxol Chemical compound NC1=C(Br)C=C(Br)C=C1CN[C@@H]1CC[C@@H](O)CC1 JBDGDEWWOUBZPM-XYPYZODXSA-N 0.000 description 1
- 229960005174 ambroxol Drugs 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 229960004238 anakinra Drugs 0.000 description 1
- 239000002269 analeptic agent Substances 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 229950004189 andecaliximab Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000006909 anti-apoptosis Effects 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000011230 antibody-based therapy Methods 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 229960001164 apremilast Drugs 0.000 description 1
- IMOZEMNVLZVGJZ-QGZVFWFLSA-N apremilast Chemical compound C1=C(OC)C(OCC)=CC([C@@H](CS(C)(=O)=O)N2C(C3=C(NC(C)=O)C=CC=C3C2=O)=O)=C1 IMOZEMNVLZVGJZ-QGZVFWFLSA-N 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 229960001692 arformoterol Drugs 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 108010038239 aspartyl-alanyl-diketopiperazine Proteins 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229940098165 atrovent Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229950000586 aviptadil Drugs 0.000 description 1
- 108010006060 aviptadil Proteins 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- 229960003060 bambuterol Drugs 0.000 description 1
- ANZXOIAKUNOVQU-UHFFFAOYSA-N bambuterol Chemical compound CN(C)C(=O)OC1=CC(OC(=O)N(C)C)=CC(C(O)CNC(C)(C)C)=C1 ANZXOIAKUNOVQU-UHFFFAOYSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- JUTBAVRYDAKVGQ-UHFFFAOYSA-N bdth2 Chemical compound SCCNC(=O)C1=CC=CC(C(=O)NCCS)=C1 JUTBAVRYDAKVGQ-UHFFFAOYSA-N 0.000 description 1
- 229940092705 beclomethasone Drugs 0.000 description 1
- 229950003452 bedoradrine Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 229950000321 benralizumab Drugs 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 229950010015 bertilimumab Drugs 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 102000014974 beta2-adrenergic receptor activity proteins Human genes 0.000 description 1
- 108040006828 beta2-adrenergic receptor activity proteins Proteins 0.000 description 1
- AXKJGGRSAVLXTE-UHFFFAOYSA-M bevonium methyl sulfate Chemical compound COS([O-])(=O)=O.C[N+]1(C)CCCCC1COC(=O)C(O)(C=1C=CC=CC=1)C1=CC=CC=C1 AXKJGGRSAVLXTE-UHFFFAOYSA-M 0.000 description 1
- ACCMWZWAEFYUGZ-UHFFFAOYSA-N bilastine Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1C(CC1)CCN1CCC1=CC=C(C(C)(C)C(O)=O)C=C1 ACCMWZWAEFYUGZ-UHFFFAOYSA-N 0.000 description 1
- 229960004314 bilastine Drugs 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- GJPICJJJRGTNOD-UHFFFAOYSA-N bosentan Chemical compound COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 GJPICJJJRGTNOD-UHFFFAOYSA-N 0.000 description 1
- 229960003065 bosentan Drugs 0.000 description 1
- AEXFXNFMSAAELR-RXVVDRJESA-N brensocatib Chemical compound C(#N)[C@H](CC1=CC=C(C=C1)C=1C=CC2=C(N(C(O2)=O)C)C1)NC(=O)[C@H]1OCCCNC1 AEXFXNFMSAAELR-RXVVDRJESA-N 0.000 description 1
- 229960003735 brodalumab Drugs 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 201000009267 bronchiectasis Diseases 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229960001838 canakinumab Drugs 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000012292 cell migration Effects 0.000 description 1
- 210000003855 cell nucleus Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- KEWHKYJURDBRMN-XSAPEOHZSA-M chembl2134724 Chemical compound O.[Br-].O([C@H]1C[C@H]2CC[C@@H](C1)[N+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 KEWHKYJURDBRMN-XSAPEOHZSA-M 0.000 description 1
- 239000013000 chemical inhibitor Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000002975 chemoattractant Substances 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000019069 chronic childhood arthritis Diseases 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 229960003728 ciclesonide Drugs 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- RSOZZQTUMVBTMR-XGUNBQNXSA-N colforsin daropate Chemical compound O[C@H]([C@@]12C)CCC(C)(C)[C@@H]1[C@H](OC(=O)CCN(C)C)[C@H](OC(C)=O)[C@]1(C)[C@]2(O)C(=O)C[C@](C)(C=C)O1 RSOZZQTUMVBTMR-XGUNBQNXSA-N 0.000 description 1
- 229950005198 colforsin daropate Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 230000024203 complement activation Effects 0.000 description 1
- 229940121388 complement c3 inhibitor Drugs 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 229940109248 cromoglycate Drugs 0.000 description 1
- IMZMKUWMOSJXDT-UHFFFAOYSA-N cromoglycic acid Chemical compound O1C(C(O)=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C(O)=O)O2 IMZMKUWMOSJXDT-UHFFFAOYSA-N 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 1
- GORAHSAIYZMTHZ-LBFSFEBVSA-N dalazatide Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H]2CSSCC3C(=O)NCC(=O)N[C@H](C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@H](C(N[C@@H](CC(O)=O)C(=O)N[C@H](C(=O)N[C@H](C(=O)N4CCC[C@H]4C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC=4C=CC(O)=CC=4)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCSC)NC(=O)[C@H](CO)NC(=O)[C@H](CC=4N=CNC=4)NC(=O)[C@H](CCCCN)NC2=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(=O)N3)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N1)[C@@H](C)O)[C@@H](C)CC)[C@@H](C)O)=O)[C@@H](C)CC)NC(=O)[C@H](CO)NC(=O)[C@@](CCCNC(N)=N)(OCCOCCN)N(C(C)=O)C(=O)[C@@H](N)CC=1C=CC(OP(O)(O)=O)=CC=1)C(N)=O)[C@@H](C)O)C1=CC=CC=C1 GORAHSAIYZMTHZ-LBFSFEBVSA-N 0.000 description 1
- 229950001360 dalazatide Drugs 0.000 description 1
- 229950003518 danirixin Drugs 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 229960001145 deflazacort Drugs 0.000 description 1
- FBHSPRKOSMHSIF-GRMWVWQJSA-N deflazacort Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@@H]2[C@@H]1[C@@H]1C[C@H]3OC(C)=N[C@@]3(C(=O)COC(=O)C)[C@@]1(C)C[C@@H]2O FBHSPRKOSMHSIF-GRMWVWQJSA-N 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- VGBVAARMQYYITG-DESRROFGSA-N des-acetyl msh Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 VGBVAARMQYYITG-DESRROFGSA-N 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000011903 deuterated solvents Substances 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960002344 dexamethasone sodium phosphate Drugs 0.000 description 1
- PLCQGRYPOISRTQ-FCJDYXGNSA-L dexamethasone sodium phosphate Chemical compound [Na+].[Na+].C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)COP([O-])([O-])=O)(O)[C@@]1(C)C[C@@H]2O PLCQGRYPOISRTQ-FCJDYXGNSA-L 0.000 description 1
- FASDKYOPVNHBLU-SSDOTTSWSA-N dexpramipexole Chemical compound C1[C@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-SSDOTTSWSA-N 0.000 description 1
- 229950004920 dexpramipexole Drugs 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000013024 dilution buffer Substances 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 1
- FVTWTVQXNAJTQP-UHFFFAOYSA-N diphenyl-[1-(2-phenylmethoxyethyl)-1-azoniabicyclo[2.2.2]octan-4-yl]methanol Chemical compound C=1C=CC=CC=1C(C12CC[N+](CCOCC=3C=CC=CC=3)(CC1)CC2)(O)C1=CC=CC=C1 FVTWTVQXNAJTQP-UHFFFAOYSA-N 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000007580 dry-mixing Methods 0.000 description 1
- 229950003468 dupilumab Drugs 0.000 description 1
- 239000000428 dust Substances 0.000 description 1
- 229950004949 duvelisib Drugs 0.000 description 1
- 229960001971 ebastine Drugs 0.000 description 1
- MJJALKDDGIKVBE-UHFFFAOYSA-N ebastine Chemical compound C1=CC(C(C)(C)C)=CC=C1C(=O)CCCN1CCC(OC(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 MJJALKDDGIKVBE-UHFFFAOYSA-N 0.000 description 1
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 1
- JAZBEHYOTPTENJ-UHFFFAOYSA-N eicosapentaenoic acid Natural products CCC=CCC=CCC=CCC=CCC=CCCCC(O)=O JAZBEHYOTPTENJ-UHFFFAOYSA-N 0.000 description 1
- 235000020673 eicosapentaenoic acid Nutrition 0.000 description 1
- 239000003602 elastase inhibitor Substances 0.000 description 1
- 229920002549 elastin Polymers 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 229960000325 emedastine Drugs 0.000 description 1
- KBUZBQVCBVDWKX-UHFFFAOYSA-N emedastine Chemical compound N=1C2=CC=CC=C2N(CCOCC)C=1N1CCCN(C)CC1 KBUZBQVCBVDWKX-UHFFFAOYSA-N 0.000 description 1
- 229950005680 emeramide Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 229950001115 enobosarm Drugs 0.000 description 1
- CSOBIBXVIYAXFM-BYNJWEBRSA-N ensifentrine Chemical compound c-12cc(OC)c(OC)cc2CCn(c(n2CCNC(N)=O)=O)c-1c\c2=N/c1c(C)cc(C)cc1C CSOBIBXVIYAXFM-BYNJWEBRSA-N 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 229960003449 epinastine Drugs 0.000 description 1
- WHWZLSFABNNENI-UHFFFAOYSA-N epinastine Chemical compound C1C2=CC=CC=C2C2CN=C(N)N2C2=CC=CC=C21 WHWZLSFABNNENI-UHFFFAOYSA-N 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229960001123 epoprostenol Drugs 0.000 description 1
- KAQKFAOMNZTLHT-VVUHWYTRSA-N epoprostenol Chemical compound O1C(=CCCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)CCCCC)[C@H](O)C[C@@H]21 KAQKFAOMNZTLHT-VVUHWYTRSA-N 0.000 description 1
- 229960004770 esomeprazole Drugs 0.000 description 1
- SUBDBMMJDZJVOS-DEOSSOPVSA-N esomeprazole Chemical compound C([S@](=O)C1=NC2=CC=C(C=C2N1)OC)C1=NC=C(C)C(OC)=C1C SUBDBMMJDZJVOS-DEOSSOPVSA-N 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229950003562 fevipiprant Drugs 0.000 description 1
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 1
- 229960003592 fexofenadine Drugs 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 239000012997 ficoll-paque Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 229940085861 flovent Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 229960000676 flunisolide Drugs 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000005755 formation reaction Methods 0.000 description 1
- HQVFCQRVQFYGRJ-UHFFFAOYSA-N formic acid;hydrate Chemical compound O.OC=O HQVFCQRVQFYGRJ-UHFFFAOYSA-N 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229940069042 gamunex Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229940121285 gefapixant Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- 229950001864 gemilukast Drugs 0.000 description 1
- 238000009650 gentamicin protection assay Methods 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000002748 glycoprotein P inhibitor Substances 0.000 description 1
- 229960001743 golimumab Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 239000002271 gyrase inhibitor Substances 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 229940125926 hemoglobin modulator Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 239000003396 histamine H4 receptor antagonist Substances 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 102000044064 human CCL17 Human genes 0.000 description 1
- 102000049918 human JAK1 Human genes 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 150000002430 hydrocarbons Chemical group 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 208000024364 idiopathic hypereosinophilic syndrome Diseases 0.000 description 1
- 229960002240 iloprost Drugs 0.000 description 1
- HIFJCPQKFCZDDL-ACWOEMLNSA-N iloprost Chemical compound C1\C(=C/CCCC(O)=O)C[C@@H]2[C@@H](/C=C/[C@@H](O)C(C)CC#CC)[C@H](O)C[C@@H]21 HIFJCPQKFCZDDL-ACWOEMLNSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- SQKXYSGRELMAAU-UHFFFAOYSA-N imidafenacin Chemical compound CC1=NC=CN1CCC(C(N)=O)(C=1C=CC=CC=1)C1=CC=CC=C1 SQKXYSGRELMAAU-UHFFFAOYSA-N 0.000 description 1
- 229950005396 imidafenacin Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000007365 immunoregulation Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- IREJFXIHXRZFER-PCBAQXHCSA-N indacaterol maleate Chemical compound OC(=O)\C=C/C(O)=O.N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)CNC1CC(C=C(C(=C2)CC)CC)=C2C1 IREJFXIHXRZFER-PCBAQXHCSA-N 0.000 description 1
- 229960004735 indacaterol maleate Drugs 0.000 description 1
- 229960000598 infliximab Drugs 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 210000004964 innate lymphoid cell Anatomy 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N inositol Chemical compound OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- 102000006029 inositol monophosphatase Human genes 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000035990 intercellular signaling Effects 0.000 description 1
- 108010042414 interferon gamma-1b Proteins 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940126797 interleukin-17A receptor antagonist Drugs 0.000 description 1
- 229940100602 interleukin-5 Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 229960004508 ivacaftor Drugs 0.000 description 1
- PURKAOJPTOLRMP-UHFFFAOYSA-N ivacaftor Chemical compound C1=C(O)C(C(C)(C)C)=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-UHFFFAOYSA-N 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000008274 jelly Substances 0.000 description 1
- 201000002215 juvenile rheumatoid arthritis Diseases 0.000 description 1
- 108010028309 kalinin Proteins 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 229950002183 lebrikizumab Drugs 0.000 description 1
- 229950007439 lenzilumab Drugs 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 239000003591 leukocyte elastase inhibitor Substances 0.000 description 1
- GWNVDXQDILPJIG-NXOLIXFESA-N leukotriene C4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@@H](C(=O)NCC(O)=O)NC(=O)CC[C@H](N)C(O)=O GWNVDXQDILPJIG-NXOLIXFESA-N 0.000 description 1
- OTZRAYGBFWZKMX-JUDRUQEKSA-N leukotriene E4 Chemical compound CCCCCC=CCC=C\C=C\C=C\[C@@H](SC[C@H](N)C(O)=O)[C@@H](O)CCCC(O)=O OTZRAYGBFWZKMX-JUDRUQEKSA-N 0.000 description 1
- 108010087711 leukotriene-C4 synthase Proteins 0.000 description 1
- 150000002617 leukotrienes Chemical class 0.000 description 1
- 229960001508 levocetirizine Drugs 0.000 description 1
- 229950009923 ligelizumab Drugs 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000008297 liquid dosage form Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 229940125389 long-acting beta agonist Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 108010015964 lucinactant Proteins 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- HWYHZTIRURJOHG-UHFFFAOYSA-N luminol Chemical compound O=C1NNC(=O)C2=C1C(N)=CC=C2 HWYHZTIRURJOHG-UHFFFAOYSA-N 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 208000007282 lymphomatoid papulosis Diseases 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 150000002689 maleic acids Chemical class 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 229960001855 mannitol Drugs 0.000 description 1
- 229950001257 masilukast Drugs 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229960004655 masitinib Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000013160 medical therapy Methods 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229960005108 mepolizumab Drugs 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- RFEAQJBHALKQQR-LURJTMIESA-N methyl (6s)-4,5,6,7-tetrahydro-3h-imidazo[4,5-c]pyridine-6-carboxylate Chemical compound C1N[C@H](C(=O)OC)CC2=C1N=CN2 RFEAQJBHALKQQR-LURJTMIESA-N 0.000 description 1
- FEFIBEHSXLKJGI-UHFFFAOYSA-N methyl 2-[3-[[3-(6-amino-2-butoxy-8-oxo-7h-purin-9-yl)propyl-(3-morpholin-4-ylpropyl)amino]methyl]phenyl]acetate Chemical compound C12=NC(OCCCC)=NC(N)=C2NC(=O)N1CCCN(CC=1C=C(CC(=O)OC)C=CC=1)CCCN1CCOCC1 FEFIBEHSXLKJGI-UHFFFAOYSA-N 0.000 description 1
- 229950010796 methylprednisolone suleptanate Drugs 0.000 description 1
- CDMLLMOLWUKNEK-AOHDELFNSA-M methylprednisolone suleptanate Chemical compound [Na+].C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)COC(=O)CCCCCCC(=O)N(C)CCS([O-])(=O)=O)CC[C@H]21 CDMLLMOLWUKNEK-AOHDELFNSA-M 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 239000003595 mist Substances 0.000 description 1
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229960001144 mizolastine Drugs 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 229950007699 mogamulizumab Drugs 0.000 description 1
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 229960005127 montelukast Drugs 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 208000005264 motor neuron disease Diseases 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 239000000234 muscarinic M1 receptor antagonist Substances 0.000 description 1
- 239000003683 muscarinic M2 receptor antagonist Substances 0.000 description 1
- 239000003149 muscarinic antagonist Substances 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 108700003805 myo-inositol-1 (or 4)-monophosphatase Proteins 0.000 description 1
- IRQWCOFOSMREBQ-UHFFFAOYSA-N n,n-dimethylazetidin-3-amine Chemical compound CN(C)C1CNC1 IRQWCOFOSMREBQ-UHFFFAOYSA-N 0.000 description 1
- PURKAOJPTOLRMP-ASMGOKTBSA-N n-[2-tert-butyl-4-[1,1,1,3,3,3-hexadeuterio-2-(trideuteriomethyl)propan-2-yl]-5-hydroxyphenyl]-4-oxo-1h-quinoline-3-carboxamide Chemical compound C1=C(O)C(C(C([2H])([2H])[2H])(C([2H])([2H])[2H])C([2H])([2H])[2H])=CC(C(C)(C)C)=C1NC(=O)C1=CNC2=CC=CC=C2C1=O PURKAOJPTOLRMP-ASMGOKTBSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- NLUPPCTVKHDVIQ-GASCZTMLSA-N n-[5-[4-[5-[[(2r,6s)-2,6-dimethylmorpholin-4-yl]methyl]-1,3-oxazol-2-yl]-1h-indazol-6-yl]-2-methoxypyridin-3-yl]methanesulfonamide Chemical compound C1=C(NS(C)(=O)=O)C(OC)=NC=C1C1=CC(C=2OC(CN3C[C@@H](C)O[C@@H](C)C3)=CN=2)=C(C=NN2)C2=C1 NLUPPCTVKHDVIQ-GASCZTMLSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229960004398 nedocromil Drugs 0.000 description 1
- RQTOOFIXOKYGAN-UHFFFAOYSA-N nedocromil Chemical compound CCN1C(C(O)=O)=CC(=O)C2=C1C(CCC)=C1OC(C(O)=O)=CC(=O)C1=C2 RQTOOFIXOKYGAN-UHFFFAOYSA-N 0.000 description 1
- 229940069759 nemiralisib Drugs 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 235000021313 oleic acid Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229960004114 olopatadine Drugs 0.000 description 1
- JBIMVDZLSHOPLA-LSCVHKIXSA-N olopatadine Chemical compound C1OC2=CC=C(CC(O)=O)C=C2C(=C/CCN(C)C)\C2=CC=CC=C21 JBIMVDZLSHOPLA-LSCVHKIXSA-N 0.000 description 1
- 150000002892 organic cations Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000001151 other effect Effects 0.000 description 1
- 201000003707 ovarian clear cell carcinoma Diseases 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Chemical group 0.000 description 1
- SHZKQBHERIJWAO-AATRIKPKSA-N ozagrel Chemical compound C1=CC(/C=C/C(=O)O)=CC=C1CN1C=NC=C1 SHZKQBHERIJWAO-AATRIKPKSA-N 0.000 description 1
- 229950003837 ozagrel Drugs 0.000 description 1
- 108010068338 p38 Mitogen-Activated Protein Kinases Proteins 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 208000014965 pancolitis Diseases 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 206010033898 parapsoriasis Diseases 0.000 description 1
- 229950010649 parogrelil Drugs 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229940121655 pd-1 inhibitor Drugs 0.000 description 1
- 229940121656 pd-l1 inhibitor Drugs 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- HIANJWSAHKJQTH-UHFFFAOYSA-N pemirolast Chemical compound CC1=CC=CN(C2=O)C1=NC=C2C=1N=NNN=1 HIANJWSAHKJQTH-UHFFFAOYSA-N 0.000 description 1
- 229960004439 pemirolast Drugs 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- 229960002036 phenytoin Drugs 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 239000002570 phosphodiesterase III inhibitor Substances 0.000 description 1
- 239000002587 phosphodiesterase IV inhibitor Substances 0.000 description 1
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 1
- 239000003371 phospholipase C inhibitor Substances 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229950008515 plecanatide Drugs 0.000 description 1
- 108010018859 plecanatide Proteins 0.000 description 1
- 239000013573 pollen allergen Substances 0.000 description 1
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 238000002600 positron emission tomography Methods 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 230000002206 pro-fibrotic effect Effects 0.000 description 1
- 230000004647 pro-inflammatory pathway Effects 0.000 description 1
- 229960002288 procaterol Drugs 0.000 description 1
- FKNXQNWAXFXVNW-BLLLJJGKSA-N procaterol Chemical compound N1C(=O)C=CC2=C1C(O)=CC=C2[C@@H](O)[C@@H](NC(C)C)CC FKNXQNWAXFXVNW-BLLLJJGKSA-N 0.000 description 1
- 206010036784 proctocolitis Diseases 0.000 description 1
- 208000017048 proctosigmoiditis Diseases 0.000 description 1
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 1
- BHMBVRSPMRCCGG-OUTUXVNYSA-N prostaglandin D2 Chemical compound CCCCC[C@H](O)\C=C\[C@@H]1[C@@H](C\C=C/CCCC(O)=O)[C@@H](O)CC1=O BHMBVRSPMRCCGG-OUTUXVNYSA-N 0.000 description 1
- BHMBVRSPMRCCGG-UHFFFAOYSA-N prostaglandine D2 Natural products CCCCCC(O)C=CC1C(CC=CCCCC(O)=O)C(O)CC1=O BHMBVRSPMRCCGG-UHFFFAOYSA-N 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000001823 pruritic effect Effects 0.000 description 1
- GJAWHXHKYYXBSV-UHFFFAOYSA-N quinolinic acid Chemical compound OC(=O)C1=CC=CN=C1C(O)=O GJAWHXHKYYXBSV-UHFFFAOYSA-N 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 229950004496 ramatroban Drugs 0.000 description 1
- 238000011552 rat model Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 229940124617 receptor tyrosine kinase inhibitor Drugs 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000000611 regression analysis Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 229950009147 repirinast Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- 201000006845 reticulosarcoma Diseases 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 229950000150 revefenacin Drugs 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229950007943 risankizumab Drugs 0.000 description 1
- 229950002845 rivipansel Drugs 0.000 description 1
- 229960002586 roflumilast Drugs 0.000 description 1
- MNDBXUUTURYVHR-UHFFFAOYSA-N roflumilast Chemical compound FC(F)OC1=CC=C(C(=O)NC=2C(=CN=CC=2Cl)Cl)C=C1OCC1CC1 MNDBXUUTURYVHR-UHFFFAOYSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960005328 rupatadine Drugs 0.000 description 1
- WUZYKBABMWJHDL-UHFFFAOYSA-N rupatadine Chemical compound CC1=CN=CC(CN2CCC(CC2)=C2C3=NC=CC=C3CCC3=CC(Cl)=CC=C32)=C1 WUZYKBABMWJHDL-UHFFFAOYSA-N 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 208000001076 sarcopenia Diseases 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 229960004540 secukinumab Drugs 0.000 description 1
- 239000002412 selectin antagonist Substances 0.000 description 1
- 239000000849 selective androgen receptor modulator Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 229940090585 serevent Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 229950007091 setipiprant Drugs 0.000 description 1
- 229950006094 sirukumab Drugs 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- KYIZZXKWRKKFSQ-UHFFFAOYSA-N sodium 9-methyl-3-(1,2,3-triaza-4-azanidacyclopenta-2,5-dien-5-yl)pyrido[1,2-a]pyrimidin-4-one Chemical compound [Na+].CC1=CC=CN(C2=O)C1=NC=C2C1=NN=N[N-]1 KYIZZXKWRKKFSQ-UHFFFAOYSA-N 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 229940124818 soft mist inhaler Drugs 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 229950008588 solithromycin Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229960005559 sulforaphane Drugs 0.000 description 1
- 235000015487 sulforaphane Nutrition 0.000 description 1
- 229910052717 sulfur Chemical group 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000013595 supernatant sample Substances 0.000 description 1
- 229950001956 suplatast Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 229950008041 tadekinig alfa Drugs 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- DATRVIMZZZVHMP-MRVPVSSYSA-N tert-butyl (2r)-2-methylpiperazine-1-carboxylate Chemical compound C[C@@H]1CNCCN1C(=O)OC(C)(C)C DATRVIMZZZVHMP-MRVPVSSYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 229950008998 tezepelumab Drugs 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 239000003848 thrombocyte activating factor antagonist Substances 0.000 description 1
- 239000003769 thromboxane A2 receptor blocking agent Substances 0.000 description 1
- LCJVIYPJPCBWKS-NXPQJCNCSA-N thymosin Chemical compound SC[C@@H](N)C(=O)N[C@H](CO)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CO)C(=O)N[C@H](CO)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@H]([C@H](C)O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCCCN)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](C(C)C)C(=O)N[C@H](C(C)C)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@H](CCC(O)=O)C(O)=O LCJVIYPJPCBWKS-NXPQJCNCSA-N 0.000 description 1
- 108010079996 thymosin beta(4) Proteins 0.000 description 1
- 229940070124 timapiprant Drugs 0.000 description 1
- 238000002877 time resolved fluorescence resonance energy transfer Methods 0.000 description 1
- MQLXPRBEAHBZTK-SEINRUQRSA-M tiotropium bromide hydrate Chemical compound O.[Br-].C[N+]1(C)[C@H]2C[C@@H](C[C@@H]1[C@H]1O[C@@H]21)OC(=O)C(O)(c1cccs1)c1cccs1 MQLXPRBEAHBZTK-SEINRUQRSA-M 0.000 description 1
- 229950004996 tipelukast Drugs 0.000 description 1
- 229960001350 tofacitinib Drugs 0.000 description 1
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 210000003437 trachea Anatomy 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229950000835 tralokinumab Drugs 0.000 description 1
- 229960005342 tranilast Drugs 0.000 description 1
- NZHGWWWHIYHZNX-CSKARUKUSA-N tranilast Chemical compound C1=C(OC)C(OC)=CC=C1\C=C\C(=O)NC1=CC=CC=C1C(O)=O NZHGWWWHIYHZNX-CSKARUKUSA-N 0.000 description 1
- 229950010086 tregalizumab Drugs 0.000 description 1
- 229960002117 triamcinolone acetonide Drugs 0.000 description 1
- YNDXUCZADRHECN-JNQJZLCISA-N triamcinolone acetonide Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H]3OC(C)(C)O[C@@]3(C(=O)CO)[C@@]1(C)C[C@@H]2O YNDXUCZADRHECN-JNQJZLCISA-N 0.000 description 1
- 229950009163 tridecactide Drugs 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 108010036927 trypsin-like serine protease Proteins 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- IYFNEFQTYQPVOC-UHFFFAOYSA-N udenafil Chemical compound C1=C(C=2NC=3C(CCC)=NN(C)C=3C(=O)N=2)C(OCCC)=CC=C1S(=O)(=O)NCCC1CCCN1C IYFNEFQTYQPVOC-UHFFFAOYSA-N 0.000 description 1
- 229960000438 udenafil Drugs 0.000 description 1
- 229960004258 umeclidinium Drugs 0.000 description 1
- 229950011597 vamorolone Drugs 0.000 description 1
- ZYTXTXAMMDTYDQ-DGEXFFLYSA-N vamorolone Chemical compound C([C@H]12)CC3=CC(=O)C=C[C@]3(C)C1=CC[C@@]1(C)[C@H]2C[C@@H](C)[C@]1(O)C(=O)CO ZYTXTXAMMDTYDQ-DGEXFFLYSA-N 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 208000005494 xerophthalmia Diseases 0.000 description 1
- 229960004764 zafirlukast Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Abstract
La invención proporciona compuestos de fórmula (I) donde las variables se definen en la especificación, o una sal farmacéuticamente aceptable de los mismos, que son útiles como inhibidores de la quinasa JAK. La invención también proporciona composiciones farmacéuticas que comprenden dichos compuestos y métodos para usar dichos compuestos para tratar enfermedades respiratorias. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Amidas heterocíclicas de 5 a 7 elementos como inhibidores de JAK
Antecedentes de la invención
Campo de la invención
La invención se refiere a compuestos que resultan útiles como inhibidores de la quinasa JAK. La invención se refiere, además, a composiciones farmacéuticas que comprenden dichos compuestos y a métodos de utilización de dichos compuestos para el tratamiento de enfermedades respiratorias.
Estado de la técnica
El asma es una enfermedad crónica de las vías respiratorias para la que no hay prevención ni cura. La enfermedad se caracteriza por inflamación, fibrosis, hipersensibilidad y remodelado de las vías respiratorias, la totalidad de los cuales contribuyen a la limitación del flujo de aire. Se estima que 300 millones de personas sufren de asma en todo el mundo y que este número crecerá en más de 100 millones para el 2025. En los Estados Unidos, el asma afecta a aproximadamente 6 % a 8 % de la población, convirtiéndolo en una de las enfermedades crónicas más comunes en el país. Aunque la mayoría de pacientes puede conseguir un control de los síntomas del asma mediante el uso de corticoesteroides inhalados que pueden combinarse con un modificador del leucotrieno y/o un agonista beta de acción prolongada, sigue existiendo un subgrupo de pacientes con asma grave cuya enfermedad no se puede controlar con las terapias convencionales. El asma persistente grave se define como una enfermedad que sigue incontrolada bajo dosis elevadas de corticoesteroides inhalados. Aunque los/las asmáticos/as con la enfermedad grave constituyen aproximadamente el 5 % de todos los afectados por asma, presentan un riesgo alto de morbilidad y mortalidad y son responsables de una parte desproporcionada de uso de recursos sanitarios respecto al resto de asmáticos. Sigue existiendo una necesidad de nuevas terapias para tratar estos pacientes.
Las citoquinas son moléculas de señalización intercelular entre las que se incluyen quimioquinas, interferones, interleuquinas, linfoquinas y factores de necrosis tumoral. Las citoquinas resultan cruciales para el crecimiento celular y la inmunorregulación normales, aunque también están detrás de las enfermedades inmunomediadas y contribuyen al crecimiento de células malignas. Se han relacionado los niveles elevados de muchas citoquinas con la patología inflamatoria del asma. Por ejemplo, se ha demostrado que las terapias basadas en anticuerpos con diana en las interleuquinas (IL)-5 y 13 proporcionan un beneficio clínico en subgrupos de pacientes de asma grave. Entre las citoquinas implicadas en la inflamación del asma, muchas actúan a través de rutas de señalización que dependen de la familia Janus de tirosina quinasas (JAK, por sus siglas en inglés), que señaliza mediante la familia de transductores de señales y activadores de transcripción (STAT, por sus siglas en inglés) de factores de transcripción. Entre las citoquinas implicadas en la inflamación del asma que señalizan a través de la ruta de JAK-STAT se incluyen IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-11, IL-13, IL-23, IL-31, IL-27, linfopoyetina estromal tímica (TSLP, por sus siglas en inglés), interferón-γ (IFNγ) y factor estimulante de colonias de granulocitos y macrófagos (GM-CSF).
La familia JAK comprende cuatro miembros: JAK1, JAK2, JAK3 y la tirosina quinasa 2 (TYK2). La unión de las citoquinas a un receptor de citoquina dependiente de JAK induce la dimerización del receptor, lo que resulta en la fosforilación de los residuos de tirosina en la JA0K quinasa, produciendo la activación de la JAK. Las JAK fosforiladas, a su vez, se unen y fosforilan diversas proteínas STAT, que se dimerizan, se internalizan en el núcleo celular y modulan directamente la transcripción génica, conduciendo, entre otros efectos, a efectos cadena abajo asociados a enfermedad inflamatoria. Las JAK habitualmente se asocian con receptores de citoquina en pares, en forma de homodímeros o heterodímeros. Se asocian citoquinas específicas en emparejamientos de JAK específicos. Cada uno de los cuatro miembros de la familia JAK participa en la señalización de por lo menos una de las citoquinas asociadas a la inflamación del asma. En consecuencia, un inhibidor químico con pan-actividad contra todos los miembros de la familia JAK podría modular un amplio abanico de rutas proinflamatorias que contribuyen al asma grave.
Sin embargo, el amplio efecto antiinflamatorio de dichos inhibidores podría suprimir la función normal de las células inmunitarias, posiblemente conduciendo a un riesgo incrementado de infección. Se han observado pruebas de un riesgo de infección incrementado en el caso del inhibidor de JAK tofacitinib, que se administra por vía oral para el tratamiento de la artritis reumatoide. En el asma, la inflamación se localiza en el tracto respiratorio. La inflamación de las vías respiratorias es característica de otras enfermedades respiratorias además del asma. Enfermedad pulmonar obstructiva crónica (EPOC), fibrosis quística (FQ), neumonitis, enfermedades pulmonares intersticiales (incluyendo la fibrosis pulmonar idiopática), lesión pulmonar aguda, síndrome del distrés respiratorio agudo, bronquitis, enfisema y sarcoidosis, y también enfermedades del tracto respiratorio en las que se cree que la fisiopatología está relacionada con citoquinas señalizadoras de JAK. La administración local de un inhibidor de JAK en los pulmones mediante inhalación ofrece el potencial de resultar terapéuticamente eficaz al administrar un potente agente anticitoquina directamente en el sitio de acción, limitando la exposición sistémica y, por lo tanto, limitando el potencial de una inmunosupresión sistémica adversa. Sigue existiendo la necesidad de un inhibidor de JAK potente que resulte adecuado para la administración local en los pulmones para el tratamiento de enfermedades respiratorias.
Las citoquinas de señalización de JAK también desempeñan un papel importante en la activación de los linfocitos T, un subtipo de células inmunitarias que es crucial para muchos procesos inmunitarios. La activación patológica de los linfocitos T es crítica en la etiología de múltiples enfermedades respiratorias. Las células T autorreactivas desempeñan una función en la bronquiolitis obliterante con neumonía organizada (también denominada BONO). De manera similar la BONO, la etiología de los rechazos del trasplante de pulmón se relaciona con una activación aberrante de los linfocitos T del receptor inducida por el pulmón trasplantado del/de la donante. Los rechazos del trasplante pulmonar pueden producirse precozmente, en forma de disfunción primaria del injerto (DPI), neumonía organizada (NO), rechazo agudo (RA), bronquiolitis linfocítica (BL) o pueden producirse años después del trasplante pulmonar, en forma de disfunción crónica del aloinjerto pulmonar (DCIP). La DCIP se conocía anteriormente como bronquiolitis obliterante (BO), aunque actualmente se considera un síndrome que puede presentar diferentes manifestaciones patológicas, incluyendo la BO, DCIP restrictiva (DCIPr o RAS) y disfunción neutrofílica del alotrasplante. La disfunción crónica del aloinjerto pulmonar (DCIP) es un reto importante en la gestión a largo plazo de los receptores de trasplante pulmonar, ya que causa que el pulmón trasplantado pierda funcionalidad progresivamente (Gauthier et al., Curr. Transplant. Rep., 2016, 3(3), 185-191). La DCIP responde mal al tratamiento y, por lo tanto, sigue existiendo una necesidad de compuestos eficaces capaces de prevenir o tratar esta afección. Varias citoquinas dependientes de JAK, tales como IFNγ e IL-5 están reguladas positivamente en la DCIP y el rechazo del trasplante pulmonar (Berastegui et al., Clin. Transplant.2017, 31, e12898). Además, se asocian niveles pulmonares elevados de quimioquinas CXCR3, tales como CXCL9 y CXCL10, que están cadena abajo de la señalización de IFN dependiente de JAK, a resultados peores en pacientes de trasplante pulmonar (Shino et al, PLOS One, 2017, 12 (7), e0180281). Se ha demostrado que la inhibición sistémica de JAK resulta eficaz en el rechazo del trasplante renal (Vicenti et al., American Journal of Transplantation, 2012, 12, 2446-56). Por lo tanto, los inhibidores de JAK presentan el potencial de resultar eficaces en el tratamiento o prevención del rechazo del trasplante pulmonar y la DCIP. Varios eventos similares de activación de los linfocitos T descritos como la base para el rechazo del trasplante pulmonar también se consideran el inductor principal de la enfermedad del injerto pulmonar contra el huésped (EICH), que puede ocurrir posteriormente al trasplante de células madre hematopoyéticas. De manera similar a la DCIP, la EICH pulmonar es una afección progresiva crónica con resultados extremadamente malos y actualmente no se dispone de ningún tratamiento aprobado. Un estudio multicéntrico retrospectivo mediante cuestionarios de 95 pacientes con EICH aguda o crónica refractaria a esteroides que habían recibido el inhibidor de JAK sistémico ruxolitinib como terapia de último recurso mostraron una respuesta completa o parcial al ruxolitinib en la mayoría de pacientes, incluyendo aquellos/as con EICH pulmonar (Zeiser et al., Leukemia, 2015, 29, 10, 2062-68). Debido a que la inhibición sistémica de JAK se asocia a acontecimientos adversos graves y un índice terapéutico pequeño, sigue existiendo una necesidad de un inhibidor de JAK no sistémico dirigido al pulmón e inhalado para prevenir y/o tratar el rechazo del trasplante pulmonar o la EICH pulmonar.
Las solicitudes de patente internacional n.º WO 2017/077288 y n.º WO 2017/077283 describe derivados 4,5,6,7-tetrahidroimidazopiridina y 1,4,5,6,7,8-hexahidroimidazoazepina como inhibidores de quinasa Janus. La solicitud de patente internacional n.º WO 2018/165395 describe derivados 4,5,6,7-tetrahidroimidazopiridina como inhibidores de quinasa Janus.
Sumario de la invención
En un aspecto, la invención proporciona nuevos compuestos que presentan actividad como inhibidores de quinasa JAK.
De acuerdo con lo anterior, la invención proporciona un compuesto de fórmula (I):

en la que:
A es un anillo heterociclilo monocíclico de 6 a 7 elementos que contiene dos átomos de nitrógeno, en el que A está conectada al grupo carbonilo en (I) mediante un átomo de nitrógeno; A está sustituida con 1 a 3 grupos R2 y, opcionalmente, dos grupos R2 forman un anillo puenteado con -(CH2) con A, o
A es pirrolidinilo, en el que el pirrolidinilo está conectado al grupo carbonilo en (I) mediante un átomo de nitrógeno y en el que el pirrolidinilo está sustituido con NR3R4;
R1 es alquilo C1-3,
cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH,
R3 es alquilo C1-3,
y R4 es alquilo C1-3,
o una sal farmacéuticamente aceptable del mismo.
La invención proporciona, además, una composición farmacéutica que comprende un compuesto de la invención y un portador farmacéuticamente aceptable.
La invención proporciona, además, un compuesto de la invención tal como se indica en la presente memoria para la utilización en un método de tratamiento de enfermedad respiratoria, en particular, asma o DCIP, en un mamífero, en el que el método comprende administrar en el mamífero una cantidad terapéuticamente eficaz de un compuesto, o de una composición farmacéutica de la invención.
La invención proporciona, además, un compuesto de la invención tal como se indica en la presente memoria para la utilización en terapia médica, así como la utilización de un compuesto de la invención en la preparación de una formulación o medicamento para el tratamiento de enfermedades respiratorias en un mamífero.
Descripción detallada de la invención
En una realización, la invención proporciona nuevos compuestos que presentan actividad como inhibidores de quinasa JAK. De acuerdo con lo anterior, la invención proporciona un compuesto de fórmula (I):

en la que:
A es un anillo heterociclilo monocíclico de 6 a 7 elementos que contiene dos átomos de nitrógeno, en el que A está conectada al grupo carbonilo en (I) mediante un átomo de nitrógeno; A está sustituida con 1 a 3 grupos R2 y, opcionalmente, dos grupos R2 forman un anillo puenteado con -(CH2) con A, o
A es pirrolidinilo, en el que el pirrolidinilo está conectado al grupo carbonilo en (I) mediante un átomo de nitrógeno y en el que el pirrolidinilo está sustituido con NR3R4;
R1 es alquilo C1-3,
cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH,
R3 es alquilo C1-3,
y R4 es alquilo C1-3,
o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto (I) presenta la fórmula (I'):
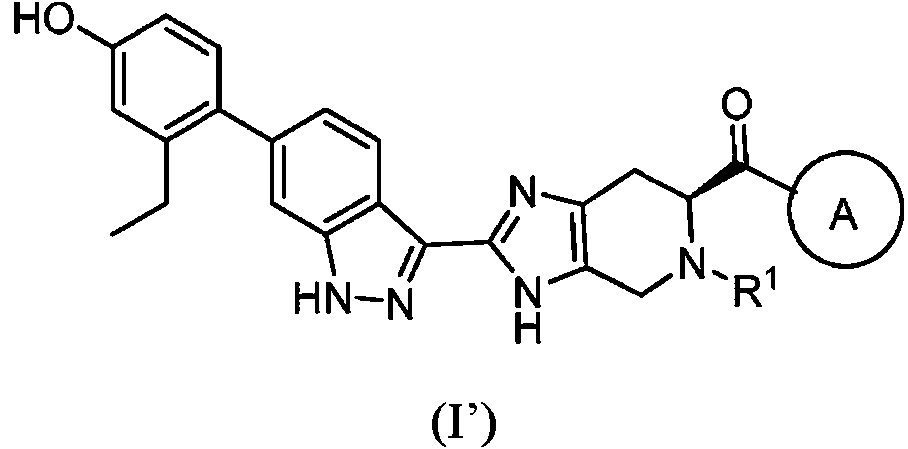
En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo, propilo e isopropilo.
En algunas realizaciones, A se selecciona del grupo que consiste en piperazinilo, 2,5-diazabiciclo[2.2.1]heptanilo y 1,4-diazacicloheptanilo, cada uno de los cuales está sustituido con 1 o 2 grupos R2, en el que cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH o A es pirrolidinilo sustituido con NR3R4, en el que R3 es alquilo C1-3 y R4 es alquilo C1-3.
En algunas realizaciones, A se selecciona del grupo que consiste en:

en la que R2 es alquilo C1-3 sustituido opcionalmente con -OH, R3 es alquilo C1-3 y R4 es alquilo C1-3. En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la invención proporciona un compuesto de fórmula:

o una sal farmacéuticamente aceptable del mismo.
En otra realización, la exposición proporciona un compuesto de fórmula (II):

en la que:
R1 es alquilo C1-3,
A1 se selecciona del grupo que consiste en:
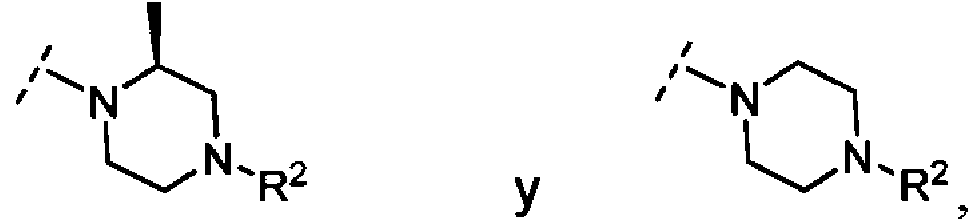
en el que R2 es alquilo C1-3 sustituido opcionalmente con -OH,
o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de fórmula (II) presenta la fórmula (II'):

En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo y propilo.
En algunas realizaciones, A1 se selecciona del grupo que consiste en:

En algunas realizaciones, A1 se selecciona del grupo que consiste en:

y R1 se selecciona del grupo que consiste en metilo y propilo.
En otra realización, la exposición proporciona un compuesto de fórmula (III):
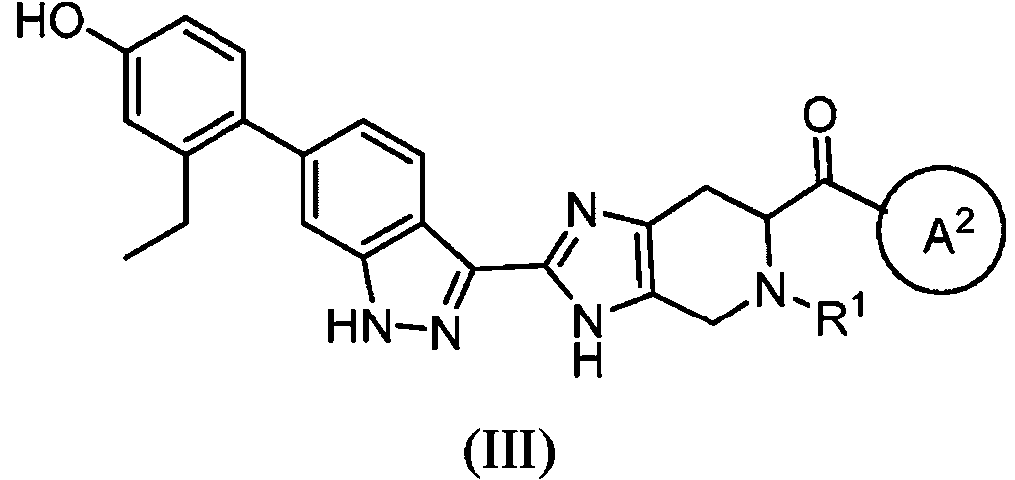
en la que:
R1 es alquilo C1-3,
A2 se selecciona del grupo que consiste en:

en la que R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH, R3 es alquilo C1-3 y R4 es alquilo C1-3,
o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de fórmula (III) presenta la fórmula (III'):

En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo, isopropilo y propilo.
En algunas realizaciones, A2 se selecciona del grupo que consiste en:

En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo, isopropilo y propilo, y A2 se selecciona del grupo que consiste en:

En otra realización, la exposición proporciona un compuesto de fórmula (IV):

en la que:
R1 es alquilo C1-3,
A3 se selecciona del grupo que consiste en:

en la que R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH, R3 es alquilo C1-3 y R4 es alquilo C1-3,
o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, el compuesto de fórmula (IV) presenta la fórmula (IV'):

En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo, isopropilo y propilo.
En algunas realizaciones, A3 se selecciona del grupo que consiste en:

En algunas realizaciones, R1 se selecciona del grupo que consiste en metilo, isopropilo y propilo, y A3 se selecciona del grupo que consiste en:

Además, la parte imidazo de la fracción tetrahidroimidazopiridina existe en formas tautoméricas, ilustradas posteriormente para un fragmento de los compuestos de la exposición.

Según la convención de la IUPAC, dichas representaciones dan lugar a una numeración diferente de los átomos de la parte imidazol: (1H-indazol-3-il)-4,5,6,7-tetrahidro-1H-imidazo[4,5-c]piridina (estructura A) vs. (1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridina (estructura B). Se entenderá que, aunque se muestran las estructuras, o se denominan, en una forma particular, la invención también incluye el tautómero de las mismas.
Los compuestos de la exposición pueden contener uno o más centros quirales y, por lo tanto, dichos compuestos (e intermediarios de los mismos) pueden existir en forma de mezclas racémicas, estereoisómeros puros (es decir, enantiómeros o diastereómeros), mezclas enriquecidas en esteroisómeros y similares. Los compuestos quirales mostrados o denominados en la presente memoria sin una estereoquímica definida en un centro quiral pretenden incluir todos y cada una de las posibles variaciones de estereoisómero en el estereocentro no definido, a menos que se indique de otro modo. La representación o denominación de un estereoisómero particular significa que el estereocentro indicado presenta la estereoquímica designada en el entendido que también pueden encontrarse presentes cantidades menores de otros estereoisómeros, a menos que se indique lo contrario, con la condición de que la utilidad del compuesto representado o denominado no resulte eliminada por la presencia de otro estereoisómero.
Los compuestos de la exposición pueden contener, además, varios grupos básicos (p. ej., grupos amino) y, por lo tanto, dichos compuestos pueden existir en forma de base libre o en diversas formas salinas, tales como una forma de sal monoprotonada, una forma de sal diprotonada, una forma de sal triprotonada, o mezclas de las mismas. La totalidad de dichas formas está comprendida dentro del alcance de la presente invención, a menos que se indique lo contrario.
La presente invención incluye, además, compuestos marcados isotópicamente de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV'), es decir, compuestos de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV') en donde uno o más átomos han sido sustituidos o enriquecidos con un átomo que presenta el mismo número atómico pero una masa atómica diferente de la masa atómica que predominan naturalmente. Entre los ejemplos de isótopos que pueden incorporarse en un compuesto de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV') se incluyen, aunque sin limitarse a ellos, 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O y 18O. Resultan de particular interés los compuestos de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV') enriquecidos en tritio o carbono-14, los cuales pueden utilizarse, por ejemplo, en estudios de distribución en los tejidos. También resultan de particular interés los compuestos de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV') enriquecidos en deuterio, especialmente en un sitio del metabolismo en el que se espera que los compuestos presenten una mayor estabilidad metabólica. Adicionalmente resultan de particular interés los compuestos de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) y (IV') enriquecidos en un isótopo emisor de positrones, tal como 11C, 15O y 13N, los cuales pueden utilizarse en, por ejemplo, estudios de tomografía de emisión de positrones.
Definiciones
Al describir la presente invención, incluyendo sus diversos aspectos y realizaciones, los términos siguientes presentan los significados siguientes, a menos que se indique lo contrario.
El término "alquilo" se refiere a un grupo hidrocarburo saturado monovalente que puede ser lineal o ramificado, o una combinación de los mismos. A menos que se defina lo contrario, dichos grupos alquilo habitualmente contienen entre 1 y 10 átomos de carbono. Entre los grupos alquilo representativos se incluyen, a título de ejemplo, metilo (Me), etilo (Et), n-propilo (n-Pr) o (nPr), isopropilo (i-Pr) o (iPr), n-butilo (n-Bu) o (nBu), sec-butilo, isobutilo, terc-butilo (t-Bu) o (tBu), n-pentilo, n-hexilo, 2,2-dimetilpropilo, 2-metilbutilo, 3-metilbutilo, 2-etilbutilo, 2,2-dimetilpentilo, 2-propilpentilo y similares.
En el caso de que un número específico de átomos de carbono está destinado a un término particular, se muestra el número de átomos de carbono precediendo al término. Por ejemplo, el término "alquilo C1-3" se refiere a un grupo alquilo con 1 a 3 átomos de carbono, en el que los átomos de carbono se encuentran en cualquier configuración químicamente aceptable, incluyendo configuraciones lineales o ramificadas.
El término "cicloalquilo" se refiere a un grupo carbocíclico saturado monovalente que puede ser monocíclico o multíciclico. A menos que se defina lo contrario, dichos grupos cicloalquilo habitualmente contienen entre 3 y 10 átomos de carbono. Entre los grupos cicloalquilo representativos se incluyen, a título de ejemplo, ciclopropilo (cPr), ciclobutilo (cBu), ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, adamantilo y similares.
El término "cpropilo" significa ciclopropilo.
El término "heterociclilo", "heterociclo", "heterocíclico" o "anillo heterocíclico" significa un grupo no aromático cíclico saturado o parcialmente insaturado monovalente, con 3 a 10 átomos anulares en total, en el que el anillo contiene entre 2 y 9 átomos anulares de carbono y entre 1 y 4 heteroátomos anulares seleccionados de entre nitrógeno, oxígeno y azufre. Los grupos heterocíclicos pueden ser monocíclicos o multicíclicos (es decir, fusionados o puenteados). Entre los grupos heterociclilo representativos se incluyen, a título de ejemplo, pirrolidinilo, piperidinilo, piperazinilo, imidazolidinilo, morfolinilo, tiomorfolilo, indolín-3-ilo, 2-imidazolinilo, tetrahidropiranilo, 1,2,3,4-tetrahidroisoquinolín-2-ilo, quinuclidinilo, 7-azanorbornanilo, nortropanilo y similares, en los que el punto de unión se encuentra en cualquier átomo anular de carbono o nitrógeno disponible. En donde el contexto hace que el punto de unión del grupo heterocíclico resulte evidente, dichos grupos alternativamente pueden denominarse especies no valentes, es decir, pirrolidina, piperidina, piperazina, imidazol, tetrahidropirano, etc.
El término "halo" se refiere a flúor, cloro, bromo o yodo.
La expresión "cantidad terapéuticamente eficaz" se refiere a una cantidad suficiente para llevar a cabo el tratamiento al administrarla en un paciente que requiere tratamiento.
El término "tratar" o "tratamiento" significa prevenir, mejorar o suprimir la afección médica, enfermedad o trastorno bajo tratamiento (p. ej., una enfermedad respiratoria) en un paciente (particularmente un ser humano), o aliviar los síntomas de la afección médica, enfermedad o trastorno.
La expresión "sal farmacéuticamente aceptable" se refiere a una sal que resulta aceptable para la administración en un/a paciente o en un mamífero, tal como un ser humano (p. ej., sales que presentan seguridad de mamífero para un régimen posológico dado). Entre las sales farmacéuticamente aceptables representativas se incluyen sales de ácido acético, ascórbico, bencenosulfónico, benzoico, canforsulfónico, cítrico, etanosulfónico, edisílico, fumárico, gentísico, glucónico, glucurónico, glutámico, hipúrico, bromhídrico, clorhídrico, isetiónico, láctico, lactobiónico, maleico, málico, mandélico, metanosulfónico, múcico, naftalenosulfónico, naftaleno-1,5-disulfónico, naftaleno-2,6-disulfónico, nicotínico, nítrico, orótico, pamoico, pantoténico, fosfórico, succínico, sulfúrico, tartárico, p-toluenosulfónico y xinafoico, y similares.
La expresión "sal de los mismos" se refiere a un compuesto formado en el caso de que el hidrógeno de un ácido se sustituya por un catión, tal como un catión metálico o un catión orgánico, y similar. Por ejemplo, el catión puede ser una forma protonada de un compuesto de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) o (IV'), es decir, una forma en que uno o más grupos amino han sido protonados por un ácido. Habitualmente, la sal es una sal farmacéuticamente aceptable, aunque ello no resulta necesario para sales de compuestos intermediarios que no están destinados a la administración en un/a paciente.
Procedimientos sintéticos generales.
Los compuestos de la presente invención, e intermediarios de los mismos, pueden prepararse según los métodos y procedimientos generales siguientes utilizando materiales de partida y reactivos disponibles comercialmente o preparados rutinariamente. Los sustituyentes y variables (p. ej., A, R1, R2, R3, etc.) utilizados en los esquemas siguientes presentan los mismos significados que los definidos en otros sitios de la presente memoria, a menos que se indique lo contrario. Además, pueden utilizarse compuestos que presentan un átomo o grupo funcional ácido o básico, o pueden producirse en forma de una sal, a menos que se indique lo contrario (en algunos casos, el uso de una sal en una reacción particular requerirá la conversión de la sal en una forma no salina, p. ej., una base libre, utilizando procedimientos rutinarios antes de llevar a cabo la reacción).
Aunque puede mostrarse o describirse una realización particular de la presente invención en los procedimientos siguientes, el experto en la materia reconocerá que también pueden prepararse otras realizaciones o aspectos de la presente invención utilizando dichos procedimientos o mediante la utilización de otros métodos, reactivos y materiales de partida conocidos por el experto en la materia. En particular, se apreciará que los compuestos de la invención pueden prepararse mediante una diversidad de rutas de procedimiento en las que se combinan los reactivos en diferentes órdenes para proporcionar intermediarios diferentes en el camino de producción de los productos finales.
En el Esquema a continuación se ilustra un método general de preparación de compuestos finales de la invención.

Se hizo reaccionar el compuesto K-18 con una cetona o aldehído en presencia de un agente reductor, proporcionando el compuesto de fórmula K-19. A continuación, se hizo reaccionar el compuesto K-19 con amina A bajo condiciones normales de formación de enlace amida, proporcionando el compuesto (I). Habitualmente, se pone en contacto ácido carboxílico con aproximadamente 1 a aproximadamente 4 equivalentes de amina A en presencia de un exceso de base. La reacción de formación de enlace amida puede utilizar agentes de acoplamiento, tales como hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio (HATU, por sus siglas en inglés) u otros agentes de acoplamiento de amida conocidos de la técnica. Puede añadirse hidrazina para escindir productos secundarios no deseados. La reacción se lleva a cabo normalmente a temperatura ambiente durante aproximadamente 5 minutos a aproximadamente 24 horas, o hasta que la reacción se haya completado sustancialmente.
Composiciones farmacéuticas.
Los compuestos de la invención y sales farmacéuticamente aceptables de los mismos se utilizan normalmente en la forma de una composición o formulación farmacéutica. Dichas composiciones farmacéuticas. pueden administrarse ventajosamente en el paciente mediante inhalación. Además, las composiciones farmacéuticas pueden administrarse mediante cualquier vía de administración aceptable, incluyendo, aunque sin limitación, los modos de administración oral, rectal, nasal, tópica (incluyendo transdérmica) y parenteral.
De acuerdo con lo anterior, en uno de sus aspectos de composición, la invención se refiere a una composición farmacéutica que comprende un portador o excipiente farmacéuticamente aceptable y un compuesto de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) o (IV'), en el que, tal como se ha definido anteriormente, "compuesto de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) o (IV')" se refiere a un compuesto de fórmula (I), (I'), (II), (II'), (III), (III'), (IV) o (IV') o una sal farmacéuticamente aceptable del mismo. Opcionalmente, dichas composiciones farmacéuticas pueden contener otros agentes terapéuticos y/o de formulación, si se desea. Al comentar las composiciones y usos de las mismas, el "compuesto de la invención" también puede denominarse en la presente memoria como "agente activo". Tal como se utiliza en la presente memoria, la expresión "compuesto de la invención" pretende incluir todos los compuestos comprendidos en la fórmula (I), así como las especies realizadas en la fórmula (I) y sales farmacéuticamente aceptables de las mismas.
Las composiciones farmacéuticas de la invención normalmente contienen una cantidad terapéuticamente eficaz de un compuesto de la presente invención. Sin embargo, el experto en la materia reconocerá que una composición farmacéutica puede contener más de una cantidad terapéuticamente eficaz, es decir, composiciones en masa, o menos de una cantidad terapéuticamente eficaz, es decir, dosis unitarias individuales diseñadas para la administración múltiple a fin de conseguir una cantidad terapéuticamente eficaz.
Habitualmente, dichas composiciones farmacéuticas contendrán entre aproximadamente 0,01 % y aproximadamente 95 % en peso del agente activo, incluyendo, por ejemplo, entre aproximadamente 0,05 % y aproximadamente 30 % en peso, y entre aproximadamente 0,1 % y aproximadamente 10 % en peso del agente activo.
Puede utilizarse cualquier portador o excipiente convencional en las composiciones farmacéuticas. de la invención. La elección de un portador o excipiente particular, o combinaciones de portadores o excipientes, dependerán del modo de administración que se utilice para tratar un paciente particular o tipo de afección médica o estado de enfermedad. A este respecto, la preparación de una composición farmacéutica adecuada para un modo particular de administración se encuentra perfectamente comprendida dentro del alcance de los conocimientos del/de la experto/a en las técnicas farmacéuticas. Además, los portadores o excipientes utilizados en las composiciones farmacéuticas. de la presente invención se encuentran disponibles comercialmente. A título de ilustrativo adicional, las técnicas de formulación convencionales se describen en Remington: The Science y Practice of Pharmacy, 20a edición, Lippincott Williams & White, Baltimore, Maryland (2000) y H.C. Ansel et al., Pharmaceutical Dosage Forms y Drug Delivery Systems, 7a edición, Lippincott Williams & White, Baltimore, Maryland (1999).
Entre los ejemplos representativos de materiales que pueden servir de portadores farmacéuticamente aceptables se incluyen, aunque sin limitarse a ellos, azúcares, tales como lactosa, glucosa y sucrosa; almidones, tales como almidón de maíz y almidón de patata; celulosa, tal como celulosa microcristalina y sus derivados, tales como
carboximetilcelulosa sódica, etilcelulosa y acetato de celulosa; tragacanto en polvo; malta; gelatina; talco; excipientes, tales como manteca de cacao y ceras supositorias; aceites, tales como aceite de cacahuete, aceite de semilla de algodón, aceite de cártamo, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja; glicoles, tales como propilenglicol; polioles, tales como glicerina, sorbitol, manitol y polietilenglicol; ésteres, tales como oleato de etilo y laurato de etilo; agar; agentes tamponadores, tales como hidróxido de magnesio e hidróxido de aluminio; ácido algínico; agua libre de pirógenos; solución salina isotónica; solución de Ringer; alcohol etílico; soluciones tampón de fosfato; y otras sustancias compatibles no tóxicas utilizadas en composiciones farmacéuticas.
Se preparan habitualmente composiciones farmacéuticas mediante la mezcla a fondo e íntima o la combinación del agente activo con un portador farmacéuticamente aceptable, y uno o más ingredientes opcionales. La mezcla uniformemente mezclada resultante a continuación puede conformarse o cargarse en comprimidos, cápsulas, píldoras y similares, utilizando procedimientos y equipos convencionales.
En un aspecto, la composición farmacéutica resulta adecuada para la administración inhalada. Las composiciones farmacéuticas para la administración inhalada normalmente están en la forma de un aerosol o unos polvos. Dichas composiciones se administran generalmente utilizando dispositivos inhaladores de administración, tales como un inhalador de polvos secos (IPS), un inhalador de dosis medidas (IDM), un inhalador-nebulizador o un dispositivo de administración similar.
En una realización particular, la composición farmacéutica se administra mediante inhalación utilizando un inhalador de polvos secos. Dichos inhaladores de polvos secos normalmente administran la composición farmacéutica en forma de unos polvos de flujo libre que se dispersan en el flujo de aire de un/a paciente durante la inspiración. Con el fin de conseguir una composición de polvos de flujo libre, el agente terapéutico normalmente se formula con un excipiente adecuado, tal como lactosa, almidón, manitol, dextrosa, ácido poliláctico (PLA), poliláctido-co-glicólido (PLGA) o combinaciones de los mismos. Habitualmente, el agente terapéutico se microniza y se combina con un portador adecuado para formar una composición adecuada para la inhalación.
Una composición farmacéutica representativa para la utilización en un inhalador de polvos secos comprende lactosa y un compuesto de la invención en forma micronizada. Dicha composición de polvos secos puede prepararse, por ejemplo, mediante combinación de lactosa molida seca con el agente terapéutico y mezclando en seco seguidamente los componentes. A continuación, la composición se carga habitualmente en un dispensador de polvos secos o en cartuchos o cápsulas para inhalación para la utilización con un dispositivo de administración de polvos secos.
Los dispositivos inhaladores de administración de polvos secos adecuados para administrar agentes terapéuticos mediante inhalación se describen en la técnica y se encuentran disponibles comercialmente ejemplos de dichos dispositivos. Por ejemplo, entre los dispositivos o productos inhaladores de administración de polvos secos representativos se incluyen Aeolizer (Novartis); Airmax (IVAX); ClickHaler (Innovata Biomed); Diskhaler (GlaxoSmithKline); Diskus/Accuhaler (GlaxoSmithKline); Ellipta (GlaxoSmithKline); Easyhaler (Orion Pharma); Eclipse (Aventis); FlowCaps (Hovione); Handihaler (Boehringer Ingelheim); Pulvinal (Chiesi); Rotahaler (GlaxoSmithKline); SkyeHaler/Certihaler (SkyePharma); Twisthaler (Schering-Plough); Turbuhaler (AstraZeneca); Ultrahaler (Aventis); y similares.
En otra realización particular, la composición farmacéutica se administra mediante inhalación utilizando un inhalador de dosis medidas. Dichos inhaladores de dosis medida normalmente descargan una cantidad medida de un agente terapéutico utilizando un gas propelente comprimido. De acuerdo con lo anterior, las composiciones farmacéuticas administradas utilizando un inhalador de dosis medidas habitualmente comprenden una solución o suspensión del agente terapéutico en un propelente licuado. Puede utilizarse cualquier propelente licuado adecuado, incluyendo hidrofluoroalcanos (HFA), tal como 1,1,1,2-tetrafluoroetano (HFA 134a) y 1,1,1,2,3,3,3-heptafluoro-n-propano (HFA 227) y clorofluorocarburos, tales como CCl3F. En una realización particular, el propelente es un hidrofluoroalcano. En algunas realizaciones, la formulación de hidrofluoroalcano contiene un cosolvente, tal como etanol o pentano, y/o un surfactante, tal como trioletato de sorbitán, ácido oleico, lecitina y glicerina.
Una composición farmacéutica representativa para la utilización en un inhalador de dosis medida comprende entre aproximadamente 0,01 % y aproximadamente 5 % en peso de un compuesto de la invención; entre aproximadamente 0 % y aproximadamente 20 % en peso de etanol, y entre aproximadamente 0 % y aproximadamente 5 % en peso de surfactante, en el que el resto es un propelente de HFA. Dichas composiciones se preparan habitualmente mediante la adición de hidrofluoroalcano frío o presurizado en un recipiente adecuado que contiene el agente terapéutico, etanol (en caso de estar presente) y el surfactante (en caso de estar presente). Para preparar una suspensión, el agente terapéutico se microniza y después se combina con el propelente. A continuación, la composición se carga en un cartucho de aerosol, que normalmente forma una parte de un dispositivo inhalador de dosis medida.
Los dispositivos inhaladores de dosis medida para la administración de agentes terapéuticos mediante inhalación se describen en la técnica y los ejemplos de dichos dispositivos se encuentran disponibles comercialmente. Por ejemplo, entre los dispositivos inhaladores o productos de dosis medida representativos se incluyen AeroBid Inhaler System (Forest Pharmaceuticals); Atrovent Inhalation Aerosol (Boehringer Ingelheim); Flovent (GlaxoSmithKline); Maxair Inhaler (3M); Proventil Inhaler (Schering); Serevent Inhalation Aerosol (GlaxoSmithKline); y similares.
En otro aspecto particular, la composición farmacéutica se administra mediante inhalación utilizando un inhaladornebulizador. Dichos dispositivos nebulizadores habitualmente producen una corriente de aire a alta velocidad que causa que la composición farmacéutica se pulverice en forma de una niebla que resulta arrastrada hasta el interior del tracto respiratorio del/de la paciente. De acuerdo con lo anterior, al formularlo para el uso en un inhalador-nebulizador, el agente terapéutico puede disolverse en un portador adecuado para formar una solución. Alternativamente, el agente terapéutico puede micronizarse o nanomolerse y combinarse con un portador adecuado para formar una suspensión. Una composición farmacéutica representativa para la utilización en un inhalador-nebulizador comprende una solución o suspensión que comprende entre aproximadamente 0,05 µg/ml y aproximadamente 20 mg/ml de un compuesto de la invención y excipientes compatibles con las formulaciones nebulizadas. En una realización, la solución presenta un pH de entre aproximadamente 3 y aproximadamente 8.
Los dispositivos nebulizadores para la administración de agentes terapéuticos mediante inhalación se describen en la técnica y los ejemplos de dichos dispositivos se encuentran disponibles comercialmente. Por ejemplo, entre los dispositivos nebulizadores o productos representativos se incluyen el inhalador Respimat Softmist (Boehringer Ingelheim); el sistema de administración pulmonar AERx (Aradigm Corp.); el nebulizador reutilizable PARI LC Plus (Pari GmbH) y similares.
En todavía otro aspecto, las composiciones farmacéuticas de la invención pueden prepararse alternativamente en una forma de administración destinada a la administración oral. Las composiciones farmacéuticas adecuadas para la administración oral pueden encontrarse en la forma de cápsulas, comprimidos, píldoras, pastillas, cápsulas, grageas, polvos y gránulos; o en forma de una solución o suspensión en un líquido acuoso o no acuoso; o en forma de una emulsión líquida de aceite en agua o de agua en aceite; o en forma de un elixir o jarabe, cada uno de los cuales contiene una cantidad predeterminada de un compuesto de la presente invención como ingrediente activo.
En el caso de que esté destinada a la administración oral en una forma de administración sólida, la composición farmacéutica de la invención normalmente comprende el agente activo y uno o más portadores farmacéuticamente aceptables, tales como citrato sódico o fosfato dicálcico. Opcional o alternativamente, dichas formas de administración sólidas pueden comprender, además, agentes de carga o extensores, ligantes, humectantes, agentes retardantes de solución, aceleradores de absorción, agentes humectantes, absorbentes, lubricantes, agentes colorantes y agentes tamponadores. También pueden encontrarse presentes en las composiciones farmacéuticas. de la invención, agentes de liberación, agentes humectantes, agentes de recubrimiento, agentes edulcorantes, saborizantes y perfumantes, conservantes y antioxidantes.
Entre las formulaciones alternativas se pueden incluir, además, formulaciones de liberación controlada, formas de administración líquidas para la administración oral, parches transdérmicos y formulaciones parenterales. Los excipientes y métodos convencionales de preparación de dichas formulaciones alternativas se describen en, por ejemplo, la referencia de Remington, supra.
Los ejemplos no limitativos a continuación ilustran composiciones farmacéuticas representantes de la presente invención.
Composición de polvos secos.
Un compuesto micronizado de fórmula (I) (1 g) se combina con lactosa molida (25 g). A continuación, la mezcla combinada se carga en blísteres individuales de un paquete blíster pelable en una cantidad suficiente para proporcionar entre aproximadamente 0,1 mg y aproximadamente 4 mg del compuesto de fórmula I en cada dosis. El contenido de los blísteres se administra mediante la utilización de un inhalador de polvos secos.
Composición de polvos secos.
Se combina un compuesto micronizado de fórmula (I) (1 g) con lactosa molida (20 g) para formar una composición en masa que presenta una proporción en peso de compuesto a lactosa molida de 1:20. La composición combinada se empaqueta en un dispositivo de inhalación de polvos secos capaz de administrar entre aproximadamente 0,1 mg y aproximadamente 4 mg del compuesto de fórmula I en cada dosis.
Composición de inhalador de dosis medida.
Se dispersó un compuesto micronizado de fórmula (I) (10 g) en una solución preparada mediante disolución de lecitina (0,2 g) en agua desmineralizada (200 ml). La suspensión resultante se secó mediante pulverización y después se micronizó para formar una composición micronizada que comprendía partículas con un diámetro medio inferior a aproximadamente 1,5 µm. A continuación, se cargó la composición micronizada en cartuchos de inhalador de dosis medida que contenían 1,1,1,2-tetrafluoroetano presurizado en una cantidad suficiente para proporcionar entre aproximadamente 0,1 mg y aproximadamente 4 mg del compuesto de fórmula I por dosis al administrarlo mediante el inhalador de dosis medida.
Composición para nebulizador.
Se disolvió un compuesto de fórmula (I) (25 mg) en una solución que contenía 1,5 a 2,5 equivalentes de ácido clorhídrico, seguido de la adición de hidróxido sódico para ajustar el pH a un valor entre 3,5 y 5,5 y 3 % en peso de glicerol. La solución se sometió a agitación hasta que todos los componentes estuviesen disueltos. La solución se administró mediante la utilización de un dispositivo nebulizador que proporciona entre aproximadamente 0,1 mg y aproximadamente 4 mg del compuesto de fórmula (I) en cada dosis.
Utilidad.
Los inhibidores de JAK de la invención se han diseñado para el tratamiento de enfermedades inflamatorias y fibróticas del tracto respiratorio. En particular, los compuestos se han diseñado para permitir la administración de un potente agente anticitoquina directamente en el sitio de acción de la enfermedad respiratoria en el pulmón, limitando simultáneamente la exposición sistémica.
Se ha mostrado que los compuestos de la invención son inhibidores potentes de la familia JAK de enzimas: JAK1, JAK2, JAK3 y TYK2. Además, los compuestos han demostrado una potente inhibición de las citoquinas proinflamatorias y profibróticas. Se ha reconocido que el amplio efecto antiinflamatorio de los inhibidores de JAK podría suprimir la función normal de las células inmunitarias, posiblemente conduciendo a un riesgo incrementado de infección. Por lo tanto, los presentes compuestos se han optimizado para limitar la absorción por el pulmón hacia el plasma, minimizando de esta manera el riesgo de inmunosupresión.
Tal como se explica en la sección experimental, posteriormente, la absorción y distribución de los compuestos habituales se ha perfilado en ensayos preclínicos. Se sometieron a ensayo los compuestos 1 a 6 en ratones y mostraron a las 5 horas de la administración, una concentración elevada en el tejido pulmonar y una baja absorción hasta el plasma. Se ha mostrado que los compuestos de la invención inhiben el efecto de la citoquina proinflamatoria IL-13 en tejido pulmonar de ratón. Específicamente, los compuestos han demostrado inhibir la fosforilación inducida por IL-13 de STAT6 en tejido pulmonar, lo que proporciona pruebas del acoplamiento in vivo con la diana JAK localmente en el pulmón. Se ha observado dicho efecto al administrar la citoquina proinflamatoria IL-13 cuatro horas después de la administración del compuesto de ensayo, proporcionando pruebas adicionales de retención significativa en el pulmón.
Se ha demostrado que los compuestos sometidos a ensayo muestran tanto una potente actividad inhibidora al nivel celular como una retención significativa en el tejido pulmonar. Una amplia investigación por los presentes inventores ha determinado que, aunque resulta posible identificar compuestos que son potentes al nivel celular o compuestos que muestran una retención significativa en el pulmón, resulta mucho más difícil encontrar compuestos que muestren ambas características deseables simultáneamente.
La actividad antiinflamatoria de los inhibidores de JAK se ha demostrado robustamente en modelos preclínicos de asma (Malaviya et al., Int. Immunopharmacol., 2010, 10, 829,-836; Matsunaga et al., Biochem. y Biophys. Res. Commun., 2011, 404, 261-267; Kudlacz et al., Eur. J. Pharmacol., 2008, 582, 154-161). Entre las citoquinas implicadas en la inflamación asmática que señalizan a través de la ruta JAK-STAT se incluyen IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-11, IL-13, IL-23, IL-31, IL-27, linfopoyetina estromal tímica (TSLP, por sus siglas en inglés), interferón-γ (IFNγ) y factor estimulador de colonias de granulocitos y macrófagos (GM-CSF). De acuerdo con lo anterior, se espera que los compuestos de la invención resulten útiles para el tratamiento de los trastornos respiratorios inflamatorios, en particular, el asma. La inflamación y fibrosis del pulmón es característica de otras enfermedades respiratorias además del asma, tales como la enfermedad pulmonar obstructiva crónica (EPOC), la fibrosis quística ((FQ), la neumonitis, las enfermedades pulmonares intersticiales (incluyendo la fibrosis pulmonar idiopática), la lesión pulmonar aguda, el síndrome del distrés respiratorio agudo, la bronquitis, el enfisema, la bronquiolitis obliterante y la sarcoidosis. Por lo tanto, los presentes compuestos también se espera que resulten útiles para el tratamiento de la enfermedad pulmonar obstructiva crónica, la fibrosis quística, la neumonitis, las enfermedades pulmonares intersticiales (incluyendo la fibrosis pulmonar idiopática), la lesión pulmonar aguda, el síndrome del distrés respiratorio agudo, la bronquitis, el enfisema, la bronquiolitis obliterante y la sarcoidosis.
En comparación con los análogos fluoro correspondientes (compuestos C-1, C-2, C-3, C-4, C-5 y C-6), se ha mostrado que los compuestos 1 a 6 presentan una actividad de JAK similar. Sin embargo, presentan la ventaja de dar lugar a un metabolismo de sulfatación significativamente inferior, tal como se demuestra en el Ensayo 5. Lo anterior resulta significativo, ya que el metabolismo de sulfatación ocurre en los pulmones, lo que podría conducir a una rápida reducción de la exposición al compuesto parental activo.
Los compuestos de la exposición han demostrado inhibir las citoquinas asociadas a la inflamación. Por lo tanto, los compuestos de la exposición es probable que resulten útiles para el tratamiento de determinadas enfermedades respiratorias específicas, tal como se detalla posteriormente.
La inflamación eosinofílica de las vías respiratorias es un elemento característico de las enfermedades denominadas colectivamente enfermedades pulmonares eosinofílicas (Cottin et al., Clin. Chest. Med., 2016, 37(3), 535-56). Las enfermedades eosinofílicas se han asociado a la señalización de IL-4, IL-13 e IL-5. Entre las enfermedades pulmonares eosinofílicas se incluyen infecciones (especialmente infecciones helmínticas), neumonitis inducida farmacológicamente (inducida, por ejemplo, por fármacos terapéuticos, tales como antibióticos, fenitoína o I-triptófano), neumonitis inducida por hongos (p. ej., aspergilosis broncopulmonar alérgica), neumonitis por hipersensibilidad y granulomatosis eosinofílica con poliangitis (anteriormente conocida como síndrome de Churg-Strauss). Entre las enfermedades pulmonares eosinofílicas de etiología desconocida se incluyen la neumonía eosinofílica aguda idiopática, la neumonía eosinofílica crónica idiopática, el síndrome hipereosinofílico y el síndrome de Löffler.
Se ha asociado un polimorfismo en el gen IL-6 a niveles elevados de IL-6 y a un riesgo incrementado de desarrollar hipertensión arterial pulmonar (HAP) (Fang et al., J. Am. Soc. Hypertens., 2017, 11(3), 171-177). En confirmación del papel de IL-6 en la HAP, la inhibición de la cadena gp130 del receptor de IL-6 mejoró la enfermedad en un modelo de rata de HAP (Huang et al., Can. J. Cardiol., 2016, 32(11), 1356.e1-1356.e10).
Se han implicado algunas citoquinas, tales como IFNγ, IL-12 e IL-6 en un abanico de enfermedades pulmonares no alérgicas, tales como la sarcoidosis y la linfangioleiomiomatosis (El-Hashemite et al., Am. J. Respir. Cell. Mol. Biol., 2005, 33, 227-230, y El-Hashemite et al., Cancer Res., 2004, 64, 3436-3443). También se ha mostrado que los compuestos de la invención inhiben la señalización de IFNγ.
La bronquiectasia y las enfermedades pulmonares infiltrativas son enfermedades asociadas a la inflamación neutrofílica crónica.
La activación patológica de los linfocitos T es crítica en la etiología de múltiples enfermedades respiratorias. Las células T autorreactivas desempeñan una función en la bronquiolitis obliterante con neumonía organizada (también denominada BONO). De manera similar a la BONO, la etiología de los rechazos del trasplante de pulmón se relaciona con una activación aberrante de los linfocitos T del receptor inducida por el pulmón trasplantado del/de la donante. Los rechazos del trasplante pulmonar pueden producirse precozmente, en forma de disfunción primaria del injerto (DPI), neumonía organizada (NO), rechazo agudo (RA), bronquiolitis linfocítica (BL) o pueden producirse años después del trasplante pulmonar, en forma de disfunción crónica del aloinjerto pulmonar (DCIP). La DCIP se conocía anteriormente como bronquiolitis obliterante (BO), aunque actualmente se considera un síndrome que puede presentar diferentes manifestaciones patológicas, incluyendo la BO, DCIP restrictiva (DCIPr o RAS) y disfunción neutrofílica del alotrasplante. La disfunción crónica del aloinjerto pulmonar (DCIP) es un reto importante en la gestión a largo plazo de los receptores de trasplante pulmonar, ya que causa que el pulmón trasplantado pierda funcionalidad progresivamente (Gauthier et al., Curr Transplant Rep., 2016, 3(3), 185-191). La DCIP responde mal al tratamiento y, por lo tanto, sigue existiendo una necesidad de compuestos eficaces capaces de prevenir o tratar esta afección. Varias citoquinas dependientes de JAK, tales como IFNγ e IL-5 están reguladas positivamente en la DCIP y el rechazo del trasplante pulmonar (Berastegui et al., Clin. Transplant.2017, 31, e12898). Además, se asocian niveles pulmonares elevados de quimioquinas CXCR3, tales como CXCL9 y CXCL10, que están cadena abajo de la señalización de IFN dependiente de JAK, a resultados peores en pacientes de trasplante pulmonar (Shino et al, PLOS One, 2017, 12 (7), e0180281). Se ha demostrado que la inhibición sistémica de JAK resulta eficaz en el rechazo del trasplante renal (Vicenti et al., American Journal of Transplantation, 2012, 12, 2446-56). Por lo tanto, los inhibidores de JAK presentan el potencial de resultar eficaces en el tratamiento o prevención del rechazo del trasplante pulmonar y la DCIP. Varios eventos similares de activación de los linfocitos T descritos como la base para el rechazo del trasplante pulmonar también se consideran el inductor principal de la enfermedad del injerto pulmonar contra el huésped (EICH), que puede ocurrir posteriormente al trasplante de células madre hematopoyéticas. De manera similar a la DCIP, la EICH pulmonar es una afección progresiva crónica con resultados extremadamente malos y actualmente no se dispone de ningún tratamiento aprobado. Un estudio multicéntrico retrospectivo mediante cuestionarios de 95 pacientes con EICH aguda o crónica refractaria a esteroides que habían recibido el inhibidor de JAK sistémico ruxolitinib como terapia de último recurso mostraron una respuesta completa o parcial al ruxolitinib en la mayoría de pacientes, incluyendo aquellos/as con EICH pulmonar (Zeiser et al., Leukemia, 2015, 29, 10, 2062-68). Debido a que la inhibición sistémica de JAK se asocia a acontecimientos adversos graves y un índice terapéutico pequeño, sigue existiendo una necesidad de un inhibidor de JAK no sistémico dirigido al pulmón e inhalado para prevenir y/o tratar el rechazo del trasplante pulmonar o la EICH pulmonar. Los compuestos de la exposición presentan las características requeridas para satisfacer dicha necesidad. Más recientemente, la neumonitis inducida por inhibidor de punto de control inmunitario, otra enfermedad pulmonar mediada por linfocitos T, ha surgido con el mayor uso de los inhibidores de punto de control inmunitario. En los pacientes de cáncer tratados con estos agentes estimulantes de linfocitos T, puede desarrollarse neumonitis mortal.
Por lo tanto, en un aspecto, la invención proporciona un compuesto de la invención tal como se indica en la presente memoria para la utilización en un método de tratamiento de una enfermedad respiratoria en un mamífero (p. ej., un ser humano), en el que el método comprende administrar en el mamífero una cantidad terapéuticamente eficaz de un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo, o de una composición farmacéutica que comprende un portador farmacéuticamente aceptable y un compuesto de la invención, o una sal farmacéuticamente aceptable del mismo.
En un aspecto, la enfermedad respiratoria es asma, enfermedad pulmonar obstructiva crónica, fibrosis quística, neumonitis, enfermedad pulmonar obstructiva crónica (EPOC), fibrosis quística (FQ), neumonitis, enfermedades pulmonares intersticiales (incluyendo la fibrosis pulmonar idiopática), lesión pulmonar aguda, síndrome del distrés respiratorio agudo, bronquitis, enfisema, bronquiolitis obliterante o sarcoidosis. En otro aspecto, la enfermedad respiratoria es asma o enfermedad pulmonar obstructiva crónica.
En un aspecto, la enfermedad respiratoria es una infección pulmonar, una enfermedad eosinofílica, una infección helmíntica, hipertensión arterial pulmonar, sarcoidosis, linfangioleiomiomatosis, broncoectasia, una enfermedad pulmonar infiltrante, neumonitis inducida farmacológicamente, neumonitis inducida por hongo, aspergilosis broncopulmonar alérgica, neumonitis por hipersensibilidad, granulomatosis eosinofílica con poliangiitis, neumonía eosinofílica aguda idiopática, neumonía eosinofílica crónica idiopática, síndrome hipereosinofílico, síndrome de Löffler, bronquiolitis obliterante con neumonía organizada, rechazos de trasplante pulmonar agudos y crónicos (incluyendo disfunción primaria del injerto (DPI), neumonía organizada (NO), bronquiolitis linfocítica (BL), rechazo agudo (RA) y DCIP, DCIP restrictiva y disfunción neutrofílica del aloinjerto), bronquiolitis obliterante con neumonía organizada por enfermedad del injerto pulmonar contra el huésped, hipertensión arterial pulmonar, broncoectasia o neumonitis inducida por inhibidor de punto de control inmunitario.
La invención proporciona, además, un compuesto de la invención tal como se indica en la presente memoria para la utilización en un método de tratamiento del asma en un mamífero, en el que el método comprende administrar en el mamífero una cantidad terapéuticamente eficaz de un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo, o de una composición farmacéutica que comprende un portador farmacéuticamente aceptable y un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo.
Al utilizarlos para tratar el asma, los compuestos de la exposición habitualmente se administran en una sola dosis diaria o en múltiples dosis cada día, aunque pueden utilizarse otras formas de administración. La cantidad de agente activo administrada por dosis o la cantidad total administrada al día normalmente serán determinadas por un/a médico/a, a la luz de las circunstancias pertinentes, incluyendo la afección que debe tratarse, la vía de administración seleccionada, el compuesto final administrado y su actividad relativa, la edad, el peso y la respuesta del/de la paciente individual, la gravedad de los síntomas del/de la paciente, y similares.
La invención proporciona, además, un compuesto de la invención tal como se indica en la presente memoria para la utilización en un método de tratamiento de una enfermedad respiratoria (incluyendo, aunque sin limitación, la enfermedad indicada en la presente memoria) en un mamífero, en el que el método comprende administrar en el mamífero una cantidad terapéuticamente eficaz de un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo, o de una composición farmacéutica que comprende un portador farmacéuticamente aceptable y un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo.
Al utilizarlos para tratar una enfermedad respiratoria (incluyendo, aunque sin limitación, la enfermedad indicada en la presente memoria), los compuestos de la exposición habitualmente se administran en una sola dosis diaria o en múltiples dosis cada día, aunque pueden utilizarse otras formas de administración. La cantidad de agente activo administrada por dosis o la cantidad total administrada al día normalmente serán determinadas por un/a médico/a, a la luz de las circunstancias pertinentes, incluyendo la afección que debe tratarse, la vía de administración seleccionada, el compuesto final administrado y su actividad relativa, la edad, el peso y la respuesta del/de la paciente individual, la gravedad de los síntomas del/de la paciente, y similares.
Como inhibidores de JAK, los compuestos de la exposición también pueden resultar útiles para una diversidad de otras enfermedades. Los compuestos de la exposición pueden resultar útiles para una diversidad de indicaciones inflamatorias gastrointestinales entre las que se incluyen, aunque sin limitarse a ellas, enfermedad intestinal inflamatoria, colitis ulcerosa (proctosigmoiditis, pancolitis, proctitis ulcerosa y colitis del lado izquierdo), enfermedad de Crohn, colitis colagenosa, colitis linfocítica, enfermedad de Behcet, enfermedad celíaca, colitis inducida por inhibidor de punto de control inmunitario, ileítis, esofagitis eosinofílica, colitis relacionada con enfermedad del injerto contra el huésped, y colitis infecciosa. La colitis ulcerosa (Reimund et al., J. Clin. Immunology, 1996, 16, 144-150), la enfermedad de Crohn (Woywodt et al., Eur. J. Gastroenterology Hepatology, 1999, 11, 267-276), la colitis colagenosa (Kumawat et al., Mol. Immunology, 2013, 55, 355-364), la colitis linfocítica (Kumawat et al., 2013), la esofagitis eosinofílica (Weinbrand-Goichberg et al., Immunol. Res., 2013, 56, 249-260), la colitis relacionada con la enfermedad del injerto contra el huésped (Coghill et al., Blood, 2001, 117, 3268-3276), la colitis infecciosa (Stallmach et al., Int. J. Colorectal Dis., 2004, 19, 308-315), la enfermedad de Behcet (Zhou et al., Autoimmun. Rev., 2012, 11, 699-704), la celiaquía (de Nitto et al., World J. Gastroenterol., 2009, 15, 4609-4614), la colitis inducida por inhibidor de punto de control inmunitario (p. ej., colitis inducida por inhibidor de CTLA-4 (Yano et al., J. Translation Med., 2014, 12, 191), o la colitis inducida por inhibidor de PD-1- o PD-L1) y la ileítis (Yamamoto et al., Dig. Liver Dis., 2008, 40, 253-259) se caracterizan por la elevación de los niveles de determinadas citoquinas proinflamatorias. Debido a que muchas citoquinas proinflamatorias señalizan mediante la activación de JAK, los compuestos indicados en la presente solicitud podrían ser capaces de aliviar la inflamación y proporcionar un alivio de los síntomas. En particular, los compuestos de la exposición podrían resultar útiles para la inducción y mantenimiento de la remisión de la colitis ulcerosa y para el tratamiento de la enfermedad de Crohn, la colitis inducida por inhibidor de punto de control inmunitario y los efectos gastrointestinales adversos en la enfermedad del injerto contra el huésped. No es parte de la invención un método de
tratamiento de una enfermedad inflamatoria gastrointestinal en un mamífero (p. ej., un ser humano), en el que el método comprende administrar en el mamífero un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica que comprende un portador farmacéuticamente aceptable y un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo.
La dermatitis atópica y otras enfermedades inflamatorias de la piel se han asociado a una elevación de las citoquinas proinflamatorias que se basan en la ruta de JAK-STAT. Por lo tanto, los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, podrían resultar beneficiosos en varias afecciones inflamatorias dérmicas o pruríticas, entre las que se incluyen, aunque sin limitarse a ellas, dermatitis atópica, alopecia areata, vitíligo, soriasis, dermatomiositis, linfoma de células T cutáneo (Netchiporouk et al., Cell Cycle, 2014; 13, 3331-3335) y subtipos (síndrome de Sézary, micosis fungoide, reticulosis pagetoide, piel fláccida granulomatosa, papulosis linfomatoide, pitiriasis liquenoide y varioliforme aguda, linfoma cutáneo de células T CD30+, linfoma de células grandes CD30+ cutáneo secundario, linfoma cutáneo de células T grandes no micosis fungoide, linfoma de células T pleomórficas, linfoma de Lennert, linfoma subcutáneo de células T, linfoma angiocéntrico, linfoma de células NK blásticas, prurigo nodular, liquen plano, amiloidosis cutánea localizada primaria, penfigoide bulloso, manifestaciones cutáneas de la enfermedad del injerto contra el huésped, penfigoide, lupus discoide, granuloma anular, liquen simple crónico, prurito vulvar/escrotal/perianal, liquen escleroso, picor por neuralgia postherpética, liquen plano pilar y foliculitis decalvante. En particular, la dermatitis atópica (Bao et al., JAK-STAT, 2013, 2, e24137), la alopecia areata (Xing et al., Nat. Med. 2014, 20, 1043-1049), el vitíligo (Craiglow et al, JAMA Dermatol. 2015, 151, 1110-1112), el prurigo nodular (Sonkoly et al., J. Allergy Clin. Immunol.2006, 117, 411-417), el liquen plano (Welz-Kubiak et al., J. Immunol. Res.2015, ID:854747), la amiloidosis cutánea localizada primaria (Tanaka et al., Br. J. Dermatol.2009, 161, 1217-1224), el penfigoide bulloso (Feliciani et al., Int. J. Immunopathol. Pharmacol. 1999, 12, 55-61) y las manifestaciones dérmicas de la enfermedad del injerto contra el huésped (Okiyama et al., J. Invest. Dermatol.2014, 134, 992-1000) se caracterizan por la elevación de determinadas citoquinas que señalizan mediante la activación de JAK. De acuerdo con lo anterior, los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, podrían aliviar la inflamación dérmica asociada o prurito inducido por estas citoquinas. En particular, los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, podría preverse que resulte útil para el tratamiento de la dermatitis atópica y otras enfermedades cutáneas inflamatorias. No es parte de la invención un método de tratamiento de una enfermedad inflamatoria de la piel en un mamífero (p. ej., un ser humano), en el que el método comprende aplicar una composición farmacéutica que comprende un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo y un portador farmacéutico en la piel del mamífero. En un aspecto, la enfermedad cutánea inflamatoria es la dermatitis atópica.
Se ha demostrado que muchas enfermedades oculares están asociadas a elevaciones de las citoquinas proinflamatorias que se basan en la ruta de JAK-STAT. Por lo tanto, los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, podrían resultar útiles para el tratamiento de varias enfermedades oculares, entre las que se incluyen, aunque sin limitarse a ellas, uveítis, retinopatía diabética, edema macular diabético, enfermedad del ojo seco, degeneración macular relacionada con la edad y queratoconjuntivitis atópica. En particular, la uveítis (Horai y Caspi, J. Interferon Cytokine Res., 2011, 31, 733-744), la retinopatía diabética (Abcouwer, J. Clin. Cell. Immunol., 2013, Supl.1, 1-12), el edema macular diabético (Sohn et al., American Journal of Opthamology, 2011, 152, 686-694), la enfermedad del ojo seco (Stevenson et al, Arch. Ophthalmol., 2012, 130, 90-100) y la degeneración macular relacionada con la edad (Knickelbein et al, Int. Ophthalmol. Clin., 2015, 55(3), 63-78) se caracterizan por la elevación de determinadas citoquinas proinflamatorias que señalizan mediante la ruta de JAK-STAT. De acuerdo con lo anterior, los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, podrían aliviar la inflamación ocular asociada y revertir la progresión de la enfermedad, o proporcionar un alivio de los síntomas. No es parte de la invención un método de tratamiento de una enfermedad ocular en un mamífero, en el que el método comprende aplicar una composición farmacéutica que comprende un compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo y un portador farmacéutico en el ojo del mamífero. En un aspecto, la enfermedad ocular es uveítis, retinopatía diabética, edema macular diabético, enfermedad del ojo seco, degeneración macular relacionada con la edad o queratoconjuntivitis atópica. En un aspecto, el método comprende administrar el compuesto de la exposición, o una sal farmacéuticamente aceptable del mismo, mediante inyección intravítrea. También pueden utilizarse compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, en combinación con uno o más compuestos útiles para enfermedades oculares.
Los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, también pueden resultar útiles para tratar otras enfermedades, tales como otras enfermedades inflamatorias, enfermedades autoinmunitarias o cánceres. Los compuestos de la exposición, o una sal farmacéuticamente aceptable de los mismos, pueden resultar útiles para tratar uno o más de entre artritis, artritis reumatoide, artritis reumatoide juvenil, rechazo del trasplante, xeroftalmia, artritis soriásica, diabetes, diabetes dependiente de insulina, enfermedad de las neuronas motoras, síndrome mielodisplásico, dolor, sarcopenia, caquexia, choque séptico, lupus eritematoso sistémico, leucemia, leucemia linfocítica crónica, leucemia mielocítica crónica, leucemia linfoblástica aguda, leucemia mielógena aguda, espondilitis anquilosante, mielofibrosis, linfoma de células B, carcinoma hepatocelular, enfermedad de Hodgkin, cáncer de mama, mieloma múltiple, melanoma, linfoma no de Hodgkin, cáncer pulmonar no microcítico, carcinoma de células claras ováricas, tumor ovárico, tumor pancreático, policitemia vera, síndrome de Sjögren, sarcoma de tejido blando, sarcoma, esplenomegalia, linfoma de células T y talasemia mayor.
Terapia de combinación.
Los compuestos de la exposición o una sal farmacéuticamente aceptable de los mismos pueden utilizarse en combinación con uno o más agentes que actúen mediante el mismo mecanismo o mediante diferentes mecanismos para tratar una enfermedad. Los diferentes agentes pueden administrarse secuencial o simultáneamente, en composiciones diferentes o en la misma composición. Entre las clases útiles de agentes para la terapia de combinación se incluyen, aunque sin limitarse a ellos, un agonista de adrenoceptor beta-2, un antagonista de receptor muscarínico, un agonista de glucocorticoide, un antagonista de receptor 44 acoplado con proteína G, un antagonista de inmunoglobulina E, un inhibidor de PDE-4, un antagonista de IL-4, un antagonista de receptor muscarínico M1, un antagonista de receptor de histamina, un antagonista de IL-13, un antagonista de IL-5, un inhibidor de 5-lipooxigenasa, un agonista de adrenoceptor beta, un antagonista de quimioquina CCR3, un estimulante de CFTR, un modulador de inmunoglobulina, un inhibidor de ligando de interleuquina 33, un inhibidor de PDE-3, un inhibidor de fosfoinositida-3 quinasa delta, un antagonista de tromboxano A2, un inhibidor de elastasa, un inhibidor de tirosina quinasa Kit, un antagonista de leucotrieno E4, un antagonista de leucotrieno, un antagonista de PGD2, un inhibidor de ligando de TNF-alfa, un agente ligante de TNF, un inhibidor de cascada del complemento, un inhibidor de ligando de eotaxina, un inhibidor de glutatión reductasa, un antagonista de receptor de histamina H4, un antagonista de IL-6, un estimulante de gen de IL2, un modulador de receptor IIB de Fc de inmunoglobulina gamma, un ligando de interferón gamma, un inhibidor de ligando de interleuquina 13, un inhibidor de ligando de interleuquina 17, un antagonista de L-selectina, un inhibidor de elastasa de leucocito, un antagonista de leucotrieno C4, un inhibidor de leucotrieno C4 sintasa, un inhibidor membranal de cobre amina oxidasa, un inhibidor de metaloproteasa 12, un inhibidor de metaloproteasa 9, un modulador de alérgeno de ácaro, un modulador de receptor muscarínico, un agonista de receptor nicotínico de acetilcolina, un inhibidor de factor nuclear kappa B, un antagonista de selectina P, un inhibidor de PDE-5, un antagonista de receptor de PDGF, un inhibidor de fosfoinositida-3 quinasa gamma, un agonista de TLR-7, un antagonista de TNF, un inhibidor de Abl tirosina quinasa, un antagonista de receptor de acetilcolina, un inhibidor de quitinasa de mamífero ácida, un agonista de receptor de ACTH, un modulador de polimerización de la actina, un antagonista de receptor A1 de adenosina, un estimulante de adenilato ciclasa, un antagonista de adrenoceptor, un ligando de hormona adrenocorticotrópica, un inhibidor de alcohol-deshidrogenasa 5, un estimulador de antitripsina alfa-1, un inhibidor de proteinasa alfa-1, un modulador de receptor de andrógeno, un estimulador de enzima conversor de la angiotensina II, un modulador de adrenoceptor beta-2, un modulador de amiloide beta, un inhibidor de gen BMP10, un inhibidor de gen BMP15, un inhibidor de canales de calcio, un inhibidor de catepsina G, un inhibidor de gen de CCL26, un modulador de quimioquina CCR3, un antagonista de quimioquina CCR4, un inhibidor de molécula de adhesión celular, un estimulante de chaperonina, un inhibidor de quitinasa, un antagonista de colágeno I, un inhibidor de complemento C3, un antagonista de CSF-1, un antagonista de quimioquina CXCR2, un modulador de cadena beta común de receptor de citoquina, un estimulante de proteína 4 de linfocito T citotóxico, un estimulador de desoxirribonucleasa I, un estimulador de desoxirribonucleasa, un inhibidor de dipeptidilpeptidasa I, un inhibidor de ADN girasa, un modulador de receptor prostanoide DP, un antagonista de selectina E, un inhibidor de receptor de tirosina quinasa de la familia de EGFR, un modulador de elastina, un antagonista de endotelina ET-A, un antagonista de endotelina ET-B, un inhibidor de epóxido hidrolasa, un antagonista de receptor de FGF3, un inhibidor de tirosina quinasa Fyn, un inhibidor de factor de transcripción GATA3, un modulador de glucosilceramidasa, un modulador de receptor de glutamato, un inhibidor de ligando de GM-CSF, un estimulante de guanilato ciclasa, un inhibidor de ATPasa H+K+, un modulador de hemoglobina, un agonista de heparina, un inhibidor de histona desacetilasa, un estimulador de histona desacetilasa-2, un inhibidor de HMG CoA reductasa, un inhibidor de I-kappa B quinasa beta, un inhibidor de gen ICAM1, un antagonista de IL-17, un modulador de receptor de IL-17, un antagonista de IL-23, un modulador de receptor de IL-4, un modulador de inmunoglobulina G, un agonista de inmunoglobulina G1, un modulador de inmunoglobulina G, un antagonista de receptor IA de Fc de inmunoglobulina epsilón, un antagonista de receptor IIB de Fc de inmunoglobulina gamma, un modulador de inmunoglobulina kappa, un sensibilizador de insulina, un ligando de interferón beta, un antagonista de receptor similar a interleuquina 1, un inhibidor de ligando de interleuquina 18, un antagonista de receptor 17A de interleuquina, un inhibidor de ligando de interleuquina-1 beta, un inhibidor de ligando de interleuquina-5, un inhibidor de ligando de interleuquina-6, un inhibidor de canal-3 de potasio activado por voltaje KCNA, un inhibidor de ligando Kit, un agonista de laminina-5, un antagonista de receptor de leucotrieno CysLT1, un antagonista de receptor de leucotrieno CysLT2, un inhibidor de gen LOXL2, un inhibidor de tirosina quinasa Lyn, un inhibidor de proteína MARCKS, un inhibidor de proteína 4 asociada a MDR, un modulador de metaloproteasa-2, un modulador de metaloproteasa-9, un antagonista de receptor mineralocorticoide, un antagonista de receptor muscarínico M2, un antagonista de receptor muscarínico M4, un antagonista de receptor muscarínico M5, un agonista de receptor A de péptido natriurético, un modulador de receptor de células asesinas naturales, un estimulador de subunidad alfa-7 de receptor nicotínico ACH, un modulador de receptor de células NK, un modulador de factor nuclear kappa B, un agonista de receptor de factor de crecimiento opioide, un inhibidor de glucoproteína P, un antagonista de purinoceptor P2X3, un inhibidor de MAP quinasa p38, un modulador de peptidasa 1, un inhibidor de fosfolipasa A, un inhibidor de fosfolipasa C, un inhibidor 1 de activador del plasminógeno, un antagonista de receptor de factor activador de plaquetas, un agonista de PPAR gamma, un agonista de prostaciclina, un inhibidor de proteína tirosina quinasa, un estimulador de dominio SH2 de inositol fosfatasa 1, un inhibidor de transducción de señales, un inhibidor de canales de sodio, un modulador de STAT-3, un inhibidor de antígeno-1 de células madre, un modulador de superóxido dismutasa, un inhibidor de glucoproteína CD28 de superficie de células T, un inhibidor de glucoproteína CD8 de superficie de células T, un agonista de TGF-beta, un antagonista de TGF-beta, un inhibidor de tromboxano sintetasa, un inhibidor de ligando de linfoproteína estromal tímica, un agonista de timosina, un ligando de timosina beta-4, un agonista de TLR-8, un agonista de TLR-9, un estimulante de gen TLR9, un inhibidor de topoisomerasa IV, un
estimulador de músculo esquelético rápido troponina I, un estimulador de troponina T de músculo esquelético rápido, un antagonista de receptor de IL-1 de tipo I, un modulador de receptor de TNF de tipo II, un modulador de canales iónicos, un estimulador de uteroglobina y un agonista de VIP.
Entre los agentes específicos que pueden utilizarse en combinación con los presentes compuestos inhibidores de JAK se incluyen, aunque sin limitarse a ellos, acetato de rosiptor, bromuro de umeclidinio, secukinumab, acetato de metenkefalina, acetato de tridecáctide, propionato de fluticasona, sulforafano estabilizado por ciclodextrina alfa, tezepelumab, furoato de mometasona, BI-1467335, dupilumab, aclidinium, formoterol, AZD-1419, HI-1640V, rivipansel, CMP-001, manitol, ANB-020, omalizumab, tregalizumab, Mitizax, benralizumab, golimumab, roflumilast, imatinib, REGN-3500, masitinib, apremilast, RPL-554, Actimmune, adalimumab, rupatadina, parogrelil, MK-1029, dipropionato de beclometasona, formoterol fumarato, mogamulizumab, seratrodast, UCB-4144, nemiralisib, CK-2127107, fevipiprant, danirixina, bosentán, abatacept, EC-18, duvelisib, dociparstat, ciprofloxacino, salbutamol HFA, erdosteína, PrEP-001, nedocromilo, CDX-0158, salbutamol, enobosarm, R-TPR-022, lenzilumab, furoato de fluticasona, trifenatato de vilanterol, propionato de fluticasona, salmeterol, PT-007, PRS-060, remestemcel-L, citrulina, RPC-4046, óxido nítrico, DS-102, gerilimzumab, Actair, furoato de fluticasona, umeclidinio, vilanterol, AG-NPP709, Gamunex, infliximab, Ampion, acumapimod, canakinumab, INS-1007, CYP-001, sirukumab, propionato de fluticasona, mepolizumab, pitavastatina, solitromicina, etanercept, ivacaftor, anakinra, MPC-300-IV, bromuro de glucopirronio, bromuro de aclidinio, FP-025, risankizumab, glucopirronio, fumarato de formoterol, Adipocell, YPL-001, bromuro de tiotropio, bromuro de glucopirronio, maleato de indacaterol, andecaliximab, olodaterol, esomeprazol, vacuna contra los ácaros del polvo, Vacuna contra el alérgeno del polen de la artemisa, vamorolona, gefapixant, revefenacina, gefitinib, ReJoin, tipelukast, bedoradrina, SCM-CGH, SHP-652, RNS-60, brodalumab, BIO-11006, bromuro de umeclidinio, trifenatato de vilanterol, bromuro de ipratropio, tralokinumab, PUR-1800, VX-561, VX-371, olopatadina, tulobuterol, fumarato de formoterol, acetónido de triamcinolona, reslizumab, xinafoato de salmeterol, propionato de fluticasona, dipropionato de beclometasona, fumarato de formoterol, bromuro de tiotropio, ligelizumab, RUTI, bertilimumab, omalizumab, bromuro de glucopirronio, SENS-111, dipropionato de beclometasona, CHF-5992, LT-4001, indacaterol, bromuro de glucopirronio, furoato de mometasona, fexofenadina, bromuro de glucopirronio, azitromicina, AZD-7594, formoterol, CHF-6001, batefenterol, OATD-01, olodaterol, CJM-112, rosiglitazona, salmeterol, setipiprant, interferónbeta inhalado, AZD-8871, plecanátide, fluticasona, salmeterol, monoglicéridos de ácido eicosapentaenoico, lebrikizumab, RG-6149, QBKPN, mometasona, indacaterol, AZD-9898, piruvato sódico, zileuton, CG-201, imidafenacina, CNTO-6785, CLBS-03, mometasona, RGN-137, procaterol, formoterol, CCI-15106, POL-6014, indacaterol, beclometasona, MV-130, GC-1112, Allergovac depot, MEDI-3506, QBW-251, ZPL-389, udenafilo, GSK-3772847, levocetirizina, AXP-1275, ADC-3680, timapiprant, abediterol, AZD-7594, bromuro de ipratropio, sulfato de salbutamol, tadekinig alfa, ACT-774312, domasa alfa, iloprost, batefenterol, furoato de fluticasona, alicaforsen, ciclesonida, emeramida, arformoterol, SB-010, Ozagrel, BTT-1023, Dectrekumab, levalbuterol, pranlukast, ácido hialurónico, GSK-2292767, Formoterol, NOV-14, Lucinactant, salbutamol, prednisolona, ebastina, cipecilato de dexametasona, GSK-2586881, BI-443651, GSK-2256294, VR-179, VR-096, hdm-ASIT+, budesónide, GSK-2245035, VTX-1463, emedastina, dexpramipexol, levalbuterol, N-6022, fosfato de dexametasona sódica, PIN-201104, OPK-0018, TEV-48107, suplatast, BI-1060469, Gemilukast, interferón gamma, dalazatide, bilastina, propionato de fluticasona, xinafoato de salmeterol, RP-3128, bromuro de bencicloquidio, reslizumab, PBF-680, antagonista de CRTH2, pranlukast, xinafoato de salmeterol, propionato de fluticasona, monohidrato de bromuro de tiotropio, masilukast, RG-7990, doxofilina, abediterol, bromuro de glucopirronio, TEV-46017, ASM-024, propionato de fluticasona, bromuro de glucopirronio, xinafoato de salmeterol, salbutamol, TA-270, Flunisólide, cromoglicato sódico, Epsi-gam, ZPL-521, salbutamol, aviptadilo, TRN-157, Zafirlukast, Stempeucel, pemirolast sodio, nadolol, propionato de fluticasona xinafoato de salmeterol, RV-1729, sulfato de salbutamol, dióxido de carbono bromuro de perfluorooctilo, APL-1, dectrekumab VAK-694, acetilsalicilato de lisina, zileuton, TR-4, terapia de células progenitoras mesenquimales derivadas de tejido adiposo alogénico humano, MEDI-9314, PL-3994, HMP-301, TD-5471, NKTT-120, pemirolast, dipropionato de beclometasona, trantinterol, monosodio alfa luminol, IMD-1041, AM-211, TBS-5, ARRY-502, seratrodast, midismasa recombinante, ASM-8, deflazacort, bambuterol, RBx-10017609, ipratropio fenoterol, fluticasona formoterol, epinastina, WIN-901X, VALERGEN-DS,OligoG-COPD-5/20, tulobuterol, Oxis Turbuhaler, DSP-3025, ASM-024, mizolastina, budesónide salmeterol, LH-011, AXP-E, inmunoglobulina humana de histaminas, YHD-001, teofilina, ambroxol erdosteína, ramatrobán, montelukast, pranlukast, AG-1321001, tulobuterol, ipratropio salbutamol, tranilast, suleptanato de metilprednisolona, daropato de colforsín, repirinast y doxofilina.
No es parte de la invención una composición farmacéutica que comprende un compuesto de la exposición o una sal farmacéuticamente aceptable del mismo y otro u otros agentes terapéuticos. El agente terapéutico puede seleccionarse de la clase de agentes especificada anteriormente y de la lista de agentes específicos indicados anteriormente. En algunas realizaciones, la composición farmacéutica resulta adecuada para la administración en los pulmones. En algunas realizaciones, la composición farmacéutica resulta adecuada para la administración inhalada o nebulizada. En algunas realizaciones, la composición farmacéutica son unos polvos secos o una composición líquida.
No es parte de la invención un método de tratamiento de una enfermedad o trastorno en un mamífero que comprende administrar en el mamífero un compuesto de la exposición o una sal farmacéuticamente aceptable del mismo y otro u otros agentes terapéuticos.
Al utilizarlos en terapia de combinación, los agentes pueden formularse en una única composición farmacéutica, o los agentes pueden proporcionarse en composiciones separadas que se administran simultáneamente o en tiempos
separados, por la misma vía de administración o por vías de administración diferentes. Dichas composiciones pueden envasarse por separado o pueden envasarse juntas en forma de kit. Los dos o más agentes terapéuticos en el kit pueden administrarse por la misma vía de administración o por vías de administración diferentes.
Ejemplos
Los ejemplos sintéticos y biológicos a continuación se proporcionan a título ilustrativo de la invención y no deben interpretarse en modo alguno como limitativos del alcance de la invención. En los ejemplos, posteriormente, las abreviaturas siguientes presentan los significados siguientes, a menos que se indique lo contrario. Las abreviaturas no definidas a continuación presentan sus significados generalmente aceptados.
ACN = acetonitrilo
DCM = diclorometano
DIPEA= N,N-diisopropiletilamina
DMF = N,N-dimetilformamida
EtOAc = acetato de etilo
h = hora(s)
HATU= hexafluorofosfato de N,N,N',N'-tetrametil-O-(7-azabenzotriazol-1-il)uronio
IPA = alcohol isopropílico
IPAc = acetato de isopropilo
MeOH= metanol
min = minuto(s)
Pd(PPh3)4 = tetrakis(trifenilfosfina)paladio(0)
TA = temperatura ambiente
TFA = ácido trifluoroacético
THF = tetrahidrofurano
bis(pinacolato)diboron = 4,4,5,5,4',4',5',5'-octametil-[2,2']bi[[1,3,2]dioxaborolanilo]
Los reactivos y solventes se adquirieron de proveedores comerciales (Aldrich, Fluka, Sigma, etc.) y se utilizaron sin purificación posterior. Se realizó un seguimiento de las mezclas de reacción mediante cromatografía de capa fina (CCF), cromatografía líquida de alto rendimiento analítica (HPLC anal.) y espectrometría de masas. Las mezclas de reacción se trataron tal como se ha indicado específicamente en cada reacción; habitualmente se purificaron mediante extracción y otros métodos de purificación, tales como la cristalización dependiente de la temperatura y del solvente, y la precipitación. Además, las mezclas de reacción se purificaron rutinariamente mediante cromatografía de columna o mediante HPLC preparativa, habitualmente utilizando rellenos de columna C18 o BDS y eluyentes convencionales. Las condiciones de HPLC preparativas normales se indican posteriormente.
La caracterización de los productos de reacción se llevó a cabo rutinariamente mediante espectrometría de masas y de RMN-1H. Para el análisis de RMN, las muestras se disolvieron en solvente deuterado (tal como CD3OD, CDCl3 o d6-DMSO) y se adquirieron los espectros de RMN-1H con un instrumento Varian Gemini 2000 (400 MHz) bajo condiciones de observación estándares. La identificación mediante espectrometría de masas de los compuestos se llevó a cabo mediante un método de ionización por electropulverización (ESMS, por sus siglas en inglés) con un instrumento modelo API 150 EX de Applied Biosystems (Foster City, CA) o un instrumento Waters (Milford, MA), acoplado con sistema de autopurificación.
Condiciones de las HPLC preparativas.
Columna: C18, 5 µm.21,2 × 150 mm o C18, 5 µm 21 × 250 o C14, 5 µm 21×150 mm Temperatura de la columna Temperatura ambiente
Caudal: 20,0 ml/min
Fases móviles: A = Agua TFA al 0,05 %
B = ACN TFA al 0,05 %
Volumen de inyección: (100 a 1500 µl)
Longitud de onda del detector: 214 nm
Los compuestos en bruto se disolvieron en agua:ácido acético 1:1 a una concentración aproximada de 50 mg/ml. Se llevó a cabo una tanda de ensayo a escala analítica de 4 minutos utilizando una columna C18 de 2,1x50 mm, seguido de una tanda a escala preparativa de 15 o 20 minutos utilizando una inyección de 100 µl con el gradiente basado en la retención en % de B de la tanda de ensayo a escala analítica. Los gradientes exactos eran dependientes de la muestra. Las muestras con impurezas que eluían próximas se comprobaron con una columna C18 de 21x250 mm y/o con una columna C14 de 21x150 mm, para mejorar la separación. Las fracciones que contenían el producto deseado se identificaron mediante análisis de espectrometría de masas.
Preparación 1: 4-(benciloxi)-2-etilfenil)trifluoro-λ4-borano, sal potásica I-5

(a) 1-(Benciloxi)-3-etilbenceno (I-2)
A una solución bajo agitación de 3-etilfenol (I-1) (25,0 g, 204,0 mmoles) en ACN (250 ml, 10 vol.) se añadió carbonato potásico (42,0 g, 306 mmoles) a temperatura ambiente. La masa de reacción resultante se sometió a agitación a temperatura ambiente durante 15 minutos, seguido de la adición de bromuro de bencilo (24,0 ml, 204 mmoles) gota a gota. La mezcla de reacción resultante se sometió a agitación durante 6 horas a temperatura ambiente. Tras completarse la reacción (seguimiento mediante CCF), la masa de reacción resultante se vertió en agua (1,0 l), seguido de la extracción del compuesto con EtOAc (2x2 l). Las capas orgánicas agrupadas se lavaron con agua fría y con solución hipersalina, y se secaron sobre sulfato sódico, se filtraron y se evaporaron bajo presión reducida. A continuación, el producto en bruto se purificó mediante cromatografía de columna sobre gel de sílice (100 a 200 M) mediante la utilización de los eluyentes EtOAc al 2 % en hexano, obteniendo el producto deseado (I-2) en forma de compuesto aceitoso ligeramente amarillento (35,0 g, 81 %). RMN 1H (400 MHz, cloroformo-d) δ 7,46-7,44 (m, 2H), 7,39 (t, J = 7,6 Hz, 2H), 7,34-7,31 (m, 1H), 7,21 (t, J = 7,6 Hz), 6,86-6,80 (m, 3H), 5,07 (s, 2H), 2,64 (q, J = 7,6 Hz, 2H), 1,24 (t, J = 7,6 Hz, 3H).
(b) 4-(Benciloxi)-1-bromo-2-etilbenceno (I-3)
A una solución bajo agitación helada de 1-(benciloxi)-3-etilbenceno (I-2) (35,0 g, 164 mmoles) en ACN (525 ml, 15 vol.) se añadió N-bromosuccinimida (32,0 g, 181 mmoles) en partes a lo largo de un periodo de 15 minutos. La mezcla de reacción resultante se sometió a agitación durante la hora siguiente a temperatura ambiente. Tras completarse la reacción (seguimiento mediante CCF), la masa de reacción resultante se vertió en agua helada (1,50 l), seguido de la extracción del compuesto con EtOAc (2x1 l). Las capas orgánicas agrupadas se lavaron con agua y se secaron sobre sulfato sódico, se filtraron y se evaporaron bajo presión reducida, obteniendo el producto en bruto. Se añadió n-hexano (250 ml) al material en bruto, resultando en una suspensión, seguido de la filtración a través de un embudo sinterizado. Se evaporó el licor madre bajo presión reducida, obteniendo el producto deseado, I-3, en forma de compuesto aceitoso amarillo pálido (42,0 g, 87 %). RMN 1H (400 MHz, cloroformo-d) δ 7,52-7,29 (m, 7H), 6,88 (s, 1H), 6,68 (d, J = 6,0 Hz, 1H), 5,04 (s, 2H), 2,69 (q, J = 7,6 Hz, 2H), 1,20 (t, J = 7,5 Hz, 3H).
(c) 2-(4-(Benciloxi)-2-etilfenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (I-4)
Una solución bajo agitación of 4-(benciloxi)-1-bromo-2-etilbenceno (1-3) (42,0 g, 144 mmoles), bis(pinacolato)diboro (44,0 g, 173 mmoles) y acetato potásico (28 g, 288 mmoles) en dioxano (440 ml) se desgasificó mediante purga con N2 (g) durante 15 min, seguido de la adición de complejo de PdCl2(dppf)·DCM (11,0 g, 15 mmoles). La mezcla de reacción resultante se calentó a 80 °C durante las siguientes 16 h. Tras completar la reacción (seguimiento mediante CCF), la masa de reacción se filtró a través de un lecho de Celite y el licor madre se evaporó bajo presión reducida, obteniendo el producto en bruto. El producto en bruto se purificó mediante cromatografía de columna sobre gel de sílice (100 a 200 M) mediante la utilización de los eluyentes EtOAc al 1% en hexano, obteniendo el producto deseado (I-4) en forma de compuesto aceitoso ligeramente amarillento (32,0 g, 66 %). RMN 1H (400 MHz, cloroformo-d) δ 7.74 (d, J = 8.4 Hz, 1H), 7.45-7.36 (m, 5H), 6.84-6.78 (m, 2H), 5.08 (s, 2H), 2.91 (q, J = 7.6 Hz), 1.33 (s, 12H), 1.19 (t, J = 7.6 Hz, 3H).
(d) 4-(Benciloxi)-2-etilfenil)trifluoro-λ4-borano, sal potásica (I-5)
A una solución bajo agitación de compuesto 2-(4-(benciloxi)-2-etilfenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (I-4) (20 g, 59,0 mmoles), en acetona:metanol (200 ml, proporción 1:1, 10 vol.), se añadió una solución 3 M de hidrogenofluoruro de potasio (23,0 g, 295 mmoles, disueltos en 98,0 ml de agua). La mezcla de reacción resultante se sometió a agitación a temperatura ambiente durante 16 horas. Tras completarse la reacción (seguimiento mediante CCF), la masa de reacción resultante se evaporó bajo presión reducida. El sólido obtenido de esta manera se introdujo en agua (100 ml) y se sometió a agitación a temperatura ambiente durante 30 min. La masa de reacción resultante se filtró a través de
un embudo sinterizado, se lavó con n-hexano y se secó bajo presión reducida, proporcionando el producto deseado (I-5) en forma de un sólido blanco (14,0 g, 74 %). RMN 1H (400 MHz, cloroformo-d) δ 7,43 (d, J = 7,2 Hz, 2H), 7,37 (t, J = 7,5 Hz, 2H), 7,30 (t, J = 7,1 Hz, 1H), 7,22 (d, J = 8,0 Hz), 6,58 (s, 1H), 6,53 (d, J = 7,9 Hz, 1H), 5,00 (s, 2H), 2,65 (q, J = 7,5 Hz, 2H), 1,07 (t, J = 7,4 Hz, 3H).
Preparación 2: (A)-3-bencil-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(tercbutilo) (I-11)

(a) Ácido (S)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, sal hidrocloruro (I-7)
A una suspensión bajo agitación helada de L-histidina (I-6) (5,0 kg, 32,14 moles) en agua (40 l, 8 vol.) se añadió ácido clorhídrico concentrado (3,93 l, 33,75 moles), seguido de la adición de formaldehído (5,50 l, 67,5 moles, solución acuosa al 37 %) gota a gota. La solución resultante se sometió a agitación durante 30 minutos a la misma temperatura y después se calentó a 80 °C durante 8 horas. Se realizó el seguimiento del progreso de la reacción mediante CL-EM. Se eliminó el agua bajo presión reducida, obteniendo el producto en bruto, y el producto en bruto resultante se sometió a agitación durante 2 horas en tolueno (20 l). Se separaron las capas orgánicas bajo presión reducida para eliminar el exceso de agua y se secó azeotrópicamente el compuesto. A continuación, el material resultante se introdujo en éter dietílico (20 l) y se sometió a agitación durante 2 horas. A continuación, se filtró el material sólido y se secó al aire, obteniendo el producto deseado (I-7) en forma de un sólido blanquecino (6,50 Kg, 85 %). RMN 1H (400 MHz, D2O) δ 8,69 (s, 1H), 4,56 (d, J = 15,4 Hz, 1H), 4,42 (d, J = 15,5 Hz, 1H), 4,20 (dd, J = 5,5, 5,2 Hz, 1H), 3,42 (dd, J = 5,0, 17,0 Hz, 1H), 3,11 (dd, J = 10,2, 16,8 Hz, 1H).
(b) Ácido (S)-3,5-bis(terc-butoxicarbonil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (I-8)
A una solución helada bajo agitación de dihidrocloruro de ácido (S)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (I-7) (6,10 Kg, 25,40 moles) en 1,4-dioxano (48 l, 8 vol.) y agua (48 l, 8 vol.) se añadió trietilamina (12,36 l, 89 moles) gota a gota, seguido de la adición de dicarbonato de di-terc-butilo (18,07 l, 78,74 moles, disueltos en 5 l de 1,4-dioxano) a lo largo de un periodo de 30 min. La mezcla de reacción resultante se sometió a agitación a temperatura ambiente durante las siguientes 16 horas. Tras completar la reacción (seguimiento mediante CCF y CL-EM), la mezcla de reacción amarillenta se diluyó con agua (10 l) y se lavó sucesivamente con éter dietílico (2×10 l) y EtOAc (2×7,50 l). Se descartó la fase orgánica. La capa acuosa se enfrió y se llevó a pH ~3 con solución de HCl 6 N; la fase acuosa se extrajo con EtOAc (3×10 l). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre sulfato sódico y se concentraron bajo presión reducida. El residuo aceitoso se cristalizó a partir de EtOAc al 30 %:hexanos, proporcionando el producto deseado (I-8) en forma de sólido blanquecino (5,1 Kg, 55 %). (m/z): [M+H]+ calc. para C17H25N3O6: 368,18; observado: 368,21.
(c) (S)-6,7-Dihidro-3H-imidazo[4,5-c]piridín-3,5,6(4H)-tricarboxilato de 6-bencil-3,5-di-terc-butilo (I-9)
A una solución helada de ácido (S)-3,5-bis(terc-butoxicarbonil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (I-8) (5,1 Kg, 13,88 moles) en DCM (51 l, 10 vol.) se añadió secuencialmente solución acuosa saturada de bicarbonato sódico (41,0 l, 8 vol.), yoduro de tetrabutilamonio (5,13 Kg, 13,88 moles) y bromuro de bencilo (2,47 l, 20,82 moles). La mezcla de reacción resultante se sometió a agitación a temperatura ambiente durante las siguientes 16 horas. Tras completar la reacción (seguimiento mediante CCF y CL-EM), se separó la solución bifásica. La capa acuosa se extrajo con DCM (3×10 l). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre sulfato sódico, se filtraron y se concentraron bajo presión reducida, obteniendo el producto en bruto, que se purificó mediante cromatografía de columna a través de gel de sílice (100-200 M) mediante la utilización de los eluyentes EtOAc al 40 % en hexano, obteniendo el producto deseado (I-9) en forma de aceite viscoso (4,50 Kg, 72 %). (m/z): [M+H]+ calc. para C24H31N3O6: 458,22; observado: 458,60.
(d) (S)-3,4,6,7-Tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-10)
A una solución helada de 3,5-di-terc-butil (S)-6,7-dihidro-3H-imidazo[4,5-c]piridín-3,5,6(4H)-tricarboxilato de 6-bencilo (I-9) (4,50 Kg, 9,84 moles) en IPA (45 l, 10 vol.) se añadió hidróxido amónico (36 l, 8 vol.) gota a gota. La mezcla de reacción resultante se sometió a agitación adicional a temperatura ambiente durante las siguientes 16 horas. Tras completar la reacción (seguimiento mediante CCF y CL-EM), la mezcla resultante se diluyó con agua (25 l), seguido de extracción con EtOAc (3×20 l). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre sulfato sódico, se filtraron y se concentraron bajo presión reducida, rindiendo el producto en bruto, que se purificó mediante cromatografía de columna a través de gel de sílice (100-200 M) mediante la utilización de los eluyentes MeOH al 2 % en DCM, obteniendo el producto deseado (I-10) en forma de un aceite viscoso espeso (2,70 Kg, 77 %). (m/z): [M+H]+ calc. para C19H23N3O4: 358,17; observado: 358,33.
(e) (S)-3-Bencil-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-11) A una solución helada de (S)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-10) (2,70 kg, 7,55 moles) en DCM (32,4 l, 12 vol.) se añadió hidróxido sódico 1 N acuoso (24,3 l, 9 vol.), seguido de la adición secuencial de yoduro de tetrabutilamonio (2,80 Kg, 7,55 moles) y bromuro de bencilo (0,99 l, 8,31 moles). La mezcla de reacción resultante se sometió a agitación a temperatura ambiente durante las siguientes 2 horas. Tras completar la reacción (seguimiento mediante CCF y CL-EM), se separó la solución bifásica. La capa acuosa se extrajo con DCM (3×10 l). Las capas orgánicas agrupadas se lavaron con solución hipersalina, se secaron sobre sulfato sódico, se filtraron y se concentraron bajo presión reducida, rindiendo el producto en bruto, que se purificó mediante cromatografía de columna sobre gel de sílice (100-200 M) mediante la utilización de los eluyentes EtOAc al 40 % en hexano, obteniendo el producto deseado (I-11) en forma de un aceite viscoso (1,70 Kg, 63 %). (m/z):
[M+H]+ calc. para C26H29N3O4: 448,22; observado: 448,20.
Preparación 3: (A)-3-bencil-2-(6-(4-(benciloxi)-2-etilfenil)-1H-indazol-3-il)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (1-16)

(a) Cloruro de 4-bromo-2-fluorobenzoilo (I-13)
A una solución bajo agitación helada de ácido 4-bromo-2-fluorobenzoico (I-12) (1,25 Kg, 5,71 moles) en DCM (12,5 l, 15 vol.) se añadió cloruro de oxalilo (0,98 l, 11,42 moles) gota a gota. La mezcla de reacción resultante se sometió a agitación durante 10 min a la misma temperatura. A continuación, se añadió DMF (150 ml) gota a gota a la mezcla de reacción. La masa de reacción resultante se dejó que se calentase hasta la temperatura ambiente y se sometió a agitación durante 2 horas. Tras completar la reacción (seguimiento mediante CCF), se eliminó el exceso de cloruro de oxalilo bajo presión reducida bajo una atmósfera de nitrógeno para obtener el producto en bruto (I-13) (1,08 Kg, 80 %), que se utilizó en la etapa siguiente sin purificación adicional.
(b) (S)-3-Bencil-2-(4-bromo-2-fluorobenzoil)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-14)
A una solución bajo agitación de (S)-3-bencil-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-11) (1,70 Kg, 3,80 moles) en ACN (13,6 l, 8 vol.) se añadió trietilamina (2,11 l, 15,2 moles) seguido de la adición de cloruro de 4-bromo-2-fluorobenzoilo (I-13) (1,08 Kg, 4,56 moles en 3,4 l de ACN, 2 vol.) a temperatura ambiente. Tras completar la adición, el color de la mezcla de reacción resultante cambió de marrón a amarillo pálido. La mezcla de reacción resultante se sometió a agitación a la misma temperatura durante 30 min, y se realizó un seguimiento mediante CCF del progreso de la reacción. La mezcla de reacción resultante se desactivó con agua helada (10 l), seguido de la extracción con EtOAc (3×5 l) y las capas orgánicas agrupadas se lavaron con solución
hipersalina. Las capas orgánicas se secaron sobre sulfato sódico, se filtraron y se concentraron bajo presión reducida, rindiendo el producto en bruto, que se purificó mediante cromatografía de columna sobre gel de sílice (100-200M) mediante la utilización de los eluyentes EtOAc al 20 % en hexano para obtener el producto deseado (I-14) (1,74 Kg, 71 %). %). (m/z): [M+H]+ calc. para C33H31BrFN3O5: 648,14; observado: 648,20.
(c) (S)-3-bencil-2-(6-bromo-1H-indazol-3-il)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-15)
A una solución bajo agitación de (S)-3-bencil-2-(4-bromo-2-fluorobenzoyl)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-14) (1,74 kg, 2,68 moles) en THF (26,0 l, 15 vol.) se añadió hidrato de hidrazina (0,705 l, 13,4 moles) a temperatura ambiente. La mezcla de reacción resultante se calentó a 60 °C durante 3 horas. Tras completarse la reacción (seguimiento mediante CCF), la masa de reacción resultante se vertió en agua helada (10 l), seguido de la extracción del compuesto con EtOAc (3x10 l). Las capas orgánicas agrupadas se lavaron con solución hipersalina y se secaron sobre sulfato sódico, se filtraron y se evaporaron bajo presión reducida, rindiendo el producto en bruto, que se purificó mediante cromatografía de columna sobre gel de sílice (100-200M) mediante la utilización de los eluyentes EtOAc al 20 % en hexano para obtener el producto deseado (I-15) en forma de un sólido blanquecino (1,12 Kg, 65 %). (m/z): [M+H]+ calc. para C33H32BrN5O4: 642,16; observado: 642,21.
(d) (S)-3-Bencil-2-(6-(4-(benciloxi)-2-etilfenil)-1H-indazol-3-il)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-16)
Se cargó bis(pinacolato)diboro (250 g, 984 mmoles) en un matraz de pared única de 3 cuellos de 5 l previamente esmerilado utilizando química de fluoruro, junto con propán-2-ol (1,882 l, 2,46E+04 mmoles) y la mezcla se sometió a agitación hasta la disolución total. La disolución era endotérmica (-4 °C). En un matraz de Erlenmeyer de 4 l, previamente grabado mediante reacción con flúor, hidroxifluoruro de fluoruro potásico (538 g, 6891 mmoles) se disolvió en agua (2,306 l, 1,28E+05 mmoles), formando una solución de 3M. La disolución era endotérmica (-12 °C). A continuación, la solución se filtró para eliminar la pequeña cantidad de materias insolubles respecto del hidroxifluoruro de fluoruro potásico. Una vez ambas soluciones se habían disuelto por completo, se cargó el contenido del matraz de Erlenmeyer en un matraz de una sola pared por partes a lo largo de 15 minutos. Se observó una exoterma moderada (+10 °C). La solución se tornó una suspensión gris espesa semiopaca y translúcida durante la adición y se incrementó la agitación para mantener el contenido bien mezclado. La mezcla se sometió a agitación durante 1,5 h, y después se filtró a través de un embudo fritado de vidrio grueso (4 l, previamente esmerilado). La filtración requirió 30 a 45 minutos para completarse. Se descartó el filtrado bifásico transparente. Se secaron los sólidos blancos durante 10 minutos en el filtro (se observó agrietamiento de la torta). Se transfirieron los sólidos de vuelta a un matraz de una sola pared de 3 cuellos de 5 litros limpio y se mezclaron nuevamente con agua formando una suspensión (1,33 l, 7,38E+04 mmoles). La suspensión se sometió a agitación durante 2 h, después de las cuales se formó un hidrogel homogéneo transparente. La solución se sometió a agitación durante otra hora, momento en que se separaron los sólidos/gel utilizando un embudo de vidrio grueso de 4 l (previamente esmerilado). Se dejó que los sólidos se secasen sobre el filtro durante 30 minutos. Se transfirieron los sólidos de vuelta a un matraz de una sola pared de 3 cuellos de 5 l y se mezclaron nuevamente en acetona formando una suspensión (1,084 l, 1,48E+04 mmoles). La suspensión blanca/gris se sometió a agitación durante 1 h y a continuación se filtró a través de un embudo de vidrio grueso de 4 l (previamente esmerilado). La filtración requirió 20 minutos para completarse y después se secaron sobre el embudo durante otra hora. Durante este tiempo, los sólidos se sometieron a agitación ocasional para garantizar un secado homogéneo. Después del secado sobre el filtro quedaron unos polvos blancos ligeros. Se secaron los sólidos durante 20 h a 55 °C bajo vacío con una purga lenta de nitrógeno, proporcionando un sólido blanco esponjoso (se recogieron 200,3 g).
A una solución bajo agitación de (S)-3-bencil-2-(6-bromo-1H-indazol-3-il)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-15) (10,0 g, 16,0 mmoles) en 2-metiltetrahidrofurano (100 ml, 10 vol.) se añadió 4-(benciloxi)-2-etilfenil)trifluoro-λ4-borano, sal potásica (I-5) (8,0 g, 20 mmoles) y el sólido blanco esponjoso obtenido anteriormente (0,20 g). La mezcla de reacción resultante se desgasificó con gas nitrógeno durante 30 minutos. A esta solución, se añadió una solución acuosa preparada de carbonato de cesio (20,0 g, 62,0 mmoles en 60 ml de agua, 6 vol.). La mezcla de reacción resultante se desgasificó adicionalmente durante 15 minutos, seguido de la adición de bis(di-terc-butil(4-dimetilaminofenil)fosfina)dicloropaladio (II) (0,66 g, 0,93 mmoles), y la mezcla de reacción se evacuó bajo vacío y se enjuagó con nitrógeno. La mezcla de reacción resultante se calentó a 110 °C durante 20 horas. Tras completar la reacción (seguimiento mediante CCF y CL-EM), la mezcla de reacción resultante se enfrió hasta la temperatura ambiente y se filtró a través de Celite; después se lavó adicionalmente con EtOAc (3×0,5 l). Las capas orgánicas agrupadas se lavaron con solución de hidróxido sódico 1 N (3×0,5 l). A continuación, se lavaron las capas orgánicas agrupadas con solución hipersalina y se secaron sobre sulfato sódico, se filtraron y se evaporaron bajo presión reducida, rindiendo el producto en bruto, que se purificó mediante cromatografía de columna de gel de sílice (100-200 M) mediante la utilización de los eluyentes EtOAc al 20 % en hexano, obteniendo el producto deseado (I-16) (en forma de mezcla de regioisómeros N-bencilo) en forma de sólido amarillo pálido (8,0 g, 66 %). (m/z): [M+H]+ calc. para C48H47N5O5: 774,36; observado: 774,59.
6-carboxílico, hidrocloruro (I-18)

(a) (S)-3-Bencil-2-(6-(4-(benciloxi)-2-etilfenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxilato de bencilo, hidrocloruro (I-17)
Se disolvió (S)-3-bencil-2-(6-(4-(benciloxi)-2-etilfenil)-1H-indazol-3-il)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) (I-16) (1,0 g, 1,292 mmoles) en dioxano (8 ml) y agua (1,5 ml), seguido de la adición de solución de ácido clorhídrico 4 M en dioxano (7 ml, 28,0 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se siguió mediante CL-EM). A continuación, la mezcla de reacción se congeló y se liofilizó, y el producto en bruto (I-17) se utilizó directamente en la reacción siguiente (se partió de que el rendimiento era cuantitativo). (m/z): [M+H]+ calc. para C43H39N5O3: 674,31; observado: 674,3.
(b) Ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, hidrocloruro (I-18)
Se disolvió (S)-3-bencil-2-(6-(4-(benciloxi)-2-etilfenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxilato de bencilo, hidrocloruro (I-17) (0,918 g, 1,292 mmoles) en 2-propanol (15 ml), solución de ácido clorhídrico 5 M en agua (0,258 ml, 1,292 mmoles) y agua (0,25 ml) a 50 °C; después se añadió paladio, al 10 % en peso sobre carbono, al 50 % en agua (0,138 g, 0,065 mmoles). A continuación, el matraz de reacción se purgó con nitrógeno, se unió un balón de hidrógeno y la mezcla de reacción se sometió a agitación a 50 °C durante 4 días, rellenando el balón de hidrógeno según se necesitase (el progreso de la reacción se siguió mediante CL-EM). A continuación, se separaron todos los sólidos mediante filtración y se concentró la solución resultante. El residuo se disolvió en ACN/agua 1:1, se congeló y se liofilizó. Los polvos resultantes (I-18) se utilizaron sin purificación adicional (se partió de que el rendimiento era cuantitativo). (m/z): [M+H]+ calc. para C22H21N5O3: 404,17; observado: 404,2.
Preparación 5: ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (I-19)

Se suspendió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (I-18) (0,25 g, 0,568 mmoles) en DMF (2,5 ml) y acetona (2,5 ml); después se añadió ácido acético (0,098 ml, 1,705 mmoles) y cianoborohidruro sódico (0,179 g, 2,84 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 24 horas (el progreso de la reacción se siguió mediante CL-EM). Se concentró la mezcla de reacción y a continuación el producto en bruto se purificó mediante cromatografía de fase inversa (gradiente de 50-70 % de ACN/agua, columna C18aq de 50 g), proporcionando la sal de TFA del compuesto del título (149 mg, rendimiento: 47 %). (m/z): [M+H]+ calc. para C25H27N5O3: 446,21; observado: 446,3.
- - - - - - - - - - - - - , , , - - - , -c]piridín-6-carboxílico (I-20)

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (I-18) (0,160 g, 0,364 mmoles) y propionaldehído (0,039 ml, 0,546 mmoles) en metanol (3,0 ml); a continuación, se añadió cianoborohidruro sódico (0,069 g, 1,091 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 24 horas (el progreso de la reacción se siguió mediante CL-EM). Se concentró la mezcla de reacción y el producto en bruto se purificó mediante cromatografía de fase inversa (gradiente de 5-70 % de ACN/agua, columna C18 de 50 g), proporcionando la sal de TFA del compuesto del título (78 mg, rendimiento: 38 %). (m/z): [M+H]+ calc. para C25H27N5O3: 446,21; observado: 446,3.
Preparación 7: ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (I-21)

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (I-18) (0,160 g, 0,364 mmoles) y solución de formaldehído, al 37 % en peso en agua (0,032 ml, 0,436 mmoles) en metanol (3,0 ml); después, se añadió cianoborohidruro sódico (0,069 g, 1,091 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 4 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió borohidruro sódico (14 mg, 0,364 mmoles) para desactivar el exceso de formaldehído; a continuación, se concentró la mezcla de reacción. El producto en bruto se purificó mediante cromatografía de fase inversa (gradiente de 5-70 % de ACN/agua, columna C18 de 50 g), proporcionando la sal de TFA del compuesto del título (110 mg, rendimiento: 57 %). (m/z):
[M+H]+ calc. para C23H23N5O3: 418,18; observado: 418,2.
Preparación 8: (R)-2-(3-metilpiperazín-1-il)etán-1-ol, dihidrocloruro (I-24)

(a) (R)-4-(2-hidroxietil)-2-metilpiperazín-1-carboxilato de terc-butilo (I-23)
Se disolvió (R)-1-boc-2-metil-piperazina (0,2 g, 0,999 mmoles), DIPEA (0,698 ml, 3,99 mmoles) y 2-bromoetanol (0,085 ml, 1,198 mmoles) en DMF (5 ml) y la mezcla de reacción se sometió a agitación a 60 °C durante 16 horas (el progreso de la reacción se siguió mediante CL-EM). Se concentró la mezcla de reacción; a continuación, se añadieron 10 ml de éter dietílico al residuo. La solución se sometió a sonicación hasta que había desaparecido el gel residual y se había formado un sólido. A continuación, se separó la solución de éter respecto del sólido mediante decantación. Seguidamente, se utilizó el sólido directamente en la reacción siguiente (se partió de que el rendimiento era cuantitativo). (m/z):
[M+H]+ calc. para C12H24N2O3: 245,18; observado: 245,4.
(b) (R)-2-(3-metilpiperazín-1-il)etán-1-ol, dihidrocloruro (I-24)
Se disolvió (R)-4-(2-hidroxietil)-2-metilpiperazín-1-carboxilato de terc-butilo (0,244 g, 0,999 mmoles) en dioxano (3,0 ml) y agua (0,5 ml); a continuación, se añadió solución de ácido clorhídrico 4 M en dioxano (2,0 ml, 65,8 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se
siguió mediante CL-EM). La mezcla de reacción se congeló y se liofilizó y el producto resultante se utilizó sin purificación (se partió de que el rendimiento era cuantitativo). (m/z): [M+H]+ calc. para C7H16N2O: 145,13; observado: 145,4.
Ejemplo 1: (S)-(2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)(4-(2-hidroxietil)piperazín-1-il)metanona

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-20) (40 mg, 0,071 mmoles), n-(2-hidroxietil)piperazina (0,018 ml, 0,143 mmoles) y DIPEA (0,025 ml, 0,143 mmoles) en DMF (1,5 ml); a continuación, se añadió HATU (40,8 mg, 0,107 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,011 ml, 0,357 mmoles) para cortar los productos secundarios no deseados; a continuación, la solución se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante HPLC preparativa (gradiente de 5-70 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (31 mg, rendimiento: 56 %). (m/z):
[M+H]+ calc. para C31H39N7O3: 558,31; observado: 558,3. RMN 1H (400 MHz, DMSO-d6) δ 13,10 (s, 1H), 12,27 (d, J = 48,92 Hz, 1H), 9,40 (s, 1H), 8,28 (t, J = 8,10 Hz, 1H), 7,30 (s, 1H), 7,04 (m, 2H), 6,71 (d, J = 2,46 Hz, 1H), 6,64 (dd, J = 2,50, 8,23 Hz, 1H), 4,44 (t, J = 5,31 Hz, 1H), 4,11 (q, J = 5,26, 2H), 3,96 (m, 1H), 3,86-3,52 (m, 6H), 3,49 (q, J = 6,01 Hz, 2H), 2,95 (m, 2H), 2,48 (q, J = 7,48 Hz, 2H), 2,42-2,21 (m, 4H), 2,37 (t, J = 6,20 Hz, 2H), 1,42 (m, 2H), 1,00 (t, J = 7,52 Hz, 3H), 0,81 (m, 3H).
Ejemplo 2: ((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((1S,4S)-5-metil-2,5-diazabiciclo[2.2.1]heptán-2-il)metanona

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-20) (40 mg, 0,071 mmoles), dihidrobromuro de (1S,4S)-5-metil-2,5-diazabiciclo[2.2.1]heptano (29,4 mg, 0,107 mmoles) y DIPEA (0,062 ml, 0,357 mmoles) en DMF (1,5 ml); después se añadió HATU (40,8 mg, 0,107 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,011 ml, 0,357 mmoles) para cortar los productos secundarios no deseados; a continuación, la solución se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, se concentró la mezcla de reacción y el producto en bruto se purificó mediante HPLC preparativa (gradiente de 5-70 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (32 mg, rendimiento: 59 %). (m/z): [M+H]+ calc. para C31H37N7O2: 540,30; observado: 540,3.
Ejemplo 3: ((S)-2,4-dimetilpiperazín-1-il)((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona

(a) (S)-4-((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carbonil)-3-metilpiperazín-1-carboxilato de terc-butilo (I-25)
Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-21) (55 mg, 0,103 mmoles), (S)-4-n-boc-2-metilpiperazina (41,5 mg, 0,207 mmoles) y DIPEA (0,036 ml, 0,207 mmoles) en DMF (1,5 ml); a continuación, se añadió HATU (59,0 mg, 0,155 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 16 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,016 ml, 0,517 mmoles) para cortar los productos secundarios no deseados; a continuación, la mezcla de reacción se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante cromatografía de fase inversa (gradiente de 5-70 % de ACN/agua, columna C18 de 50 g), proporcionando la sal de TFA del compuesto del título (54 mg, rendimiento: 72 %). (m/z):
[M+H]+ calc. para C33H41N7O4: 600,33; observado: 600,3.
(b) ((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((S)-2-metilpiperazín-1-il)metanona (I-26)
Se disolvió (S)-4-((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carbonil)-3-metilpiperazín-1-carboxilato de terc-butilo, TFA (I-25) (0,126 g, 0,177 mmoles) en dioxano (1,5 ml) y agua (0,3 ml); a continuación, se añadió solución de ácido clorhídrico 4 M en dioxano (1,5 ml, 6,00 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 2 horas (el progreso de la reacción se siguió mediante CL-EM). La mezcla de reacción se congeló y se liofilizó, y los polvos resultantes se utilizaron directamente en la reacción siguiente (se partió de que el rendimiento era cuantitativo). (m/z): [M+H]+ calc. para C28H33N7O2: 500,27; observado: 500,3.
(c) ((S)-2,4-dimetilpiperazín-1-il)((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona
Se disolvió ((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((S)-2-metilpiperazín-1-il)metanona, dihidrocloruro (0,101 g, 0,176 mmoles) y solución de formaldehído, al 37 % en peso en agua (0,016 ml, 0,212 mmoles) en metanol (3,0 ml); a continuación, se añadió cianoborohidruro sódico (0,055 g, 0,882 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 16 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió borohidruro sódico (7 mg, 0,176 mmoles) para desactivar cualquier formaldehído remanente. Se concentró la mezcla de reacción; a continuación, se purificó el producto en bruto mediante HPLC preparativa (gradiente de 5-60 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (28 mg, rendimiento: 21 %). (m/z): [M+H]+ calc. para C29H35N7O2: 514,29; observado: 514,3.
Ejemplo 4: (S)-(2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)(4-metil-1,4-diazepán-1-il)metanona

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-19) (50 mg, 0,089 mmoles), 1-metilhomopiperazina (0,022 ml, 0,179 mmoles) y DIPEA (0,031 ml, 0,179 mmoles) en DMF (1,5 ml); a continuación, se añadió HATU (51,0 mg, 0,134 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,014 ml, 0,447 mmoles) para cortar los productos secundarios no deseados y la solución se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante HPLC preparativa (gradiente de 2-70 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (29 mg, rendimiento: 42 %). (m/z): [M+H]+ calc. para C31H39N7O2: 542,32; observado: 542,3. RMN 1H (400 MHz, DMSO-d6) δ 13,08 (s, 1H), 12,21 (d, J = 29,9 Hz, 1H), 9,40 (s, 1H), 8,27 (d, J = 8,33 Hz, 1H), 7,30 (s, 1H), 7,04 (t, J = 8,05, 2H), 6,71 (d, J = 2,53 Hz, 1H), 6,64 (dd, J = 2,54, 8,23 Hz, 1H), 4,11 (m, 3H), 3,91-3,52 (m, 6H), 2,97 (m, 1H), 2,91-2,53 (m, 4H), 2,49 (q, J = 7,46, 2H), 2,23 (d, J = 13,9 Hz, 3H), 1,76 (m, 2H), 1,0 (m, 9H).
- - - - - - - - - - - - - , , , - - - , - - -il)((R)-4-(2-hidroxietil)-2-metilpiperazín-1-il)metanona

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-21) (55 mg, 0,103 mmoles), (R)-2-(3-metilpiperazín-1-il)etán-1-ol, dihidrocloruro (I-24) (33,7 mg, 0,155 mmoles) y DIPEA (0,090 ml, 0,517 mmoles) en DMF (1,5 ml); a continuación, se añadió HATU (59,0 mg, 0,155 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 16 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,016 ml, 0,517 mmoles) para cortar los productos secundarios no deseados; a continuación, la mezcla de reacción se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante HPLC preparativa (gradiente de 5-70 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (22 mg, rendimiento: 28 %). (m/z): [M+H]+ calc. para C30H37N7O3: 544,30; observado: 544,3.
Ejemplo 6: ((S)-3-(dimetilamino)pirrolidín-1-il)((S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona

Se disolvió ácido (S)-2-(6-(2-etil-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (I-19) (50 mg, 0,089 mmoles), (S)-(-)-3-(dimetilamino)pirrolidina (0,023 ml, 0,179 mmoles) y DIPEA (0,031 ml, 0,179 mmoles) en DMF (1,5 ml); a continuación, se añadió HATU (51,0 mg, 0,134 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 3 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió hidrazina (0,014 ml, 0,447 mmoles) para cortar los productos secundarios no deseados, y la solución se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante HPLC preparativa (gradiente de 5-70 % de ACN/agua, columna C18), proporcionando la sal de TFA del compuesto del título (37 mg, rendimiento: 53 %). (m/z): [M+H]+ calc. para C31H39N7O2: 542,32; observado: 542,3.
Preparación 9: 2-(4-(benciloxi)-2-etil-5-fluorofenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano

(a) 1-(Benciloxi)-4-bromo-5-etil-2-fluorobenceno
A una solución of 4-bromo-5-etil-2-fluorophenol (20 g, 910,32 mmoles) en ACN (250 ml) se añadió K2CO3 (31,55 g, 228,3 mmoles) seguido de bromuro de bencilo (13,10 ml, 109,58 mmoles) gota a gota. La mezcla de reacción resultante se sometió a agitación a 80 °C durante 2 h. La capa acuosa se extrajo con EtOAc (tres veces), se agrupó y se lavó con solución hipersalina. La capa orgánica se secó sobre Na2SO4 y se evaporó bajo presión reducida, proporcionando el intermediario del título en forma de un líquido aceitoso amarillo pálido (25 g, rendimiento: 89 %). RMN 1H (400 MHz, cloroformo-d) δ 7,48 - 7,30 (m, 5H), 7,27 (d, J = 10,5 Hz, 1H), 6,87 (d, J = 8,7 Hz, 1H), 5,12 (s, 2H), 2,66 (q, J = 7,5 Hz, 2H), 1,16 (t, J = 7,5 Hz, 3H).
(b) 2-(4-(Benciloxi)-2-etil-5-fluorofenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano
A una solución del producto de la etapa anterior (12,5 g, 40,45 mmoles) en dioxano (100 ml) se añadió bis(pinacolato)diboro (15,40 g, 60,67 mmoles) y KOAc (11,9 g, 121,35 mmoles). La mezcla de reacción se purgó con nitrógeno durante 15 min seguido de la adición de [1,1'-bis(difenilfosfino)ferroceno]dicloropaladio (II), complejo con
diclorometano (1,65 g, 2,023 mmoles). La mezcla de reacción resultante se sometió a agitación y se calentó a 110 °C durante 3 h, se filtró a través de Celite y el residuo se lavó con EtOAc. El filtrado se diluyó con un exceso de EtOAc (200 ml) y se lavó con agua (100 ml), seguido de solución hipersalina (100 ml), se secó sobre sulfato sódico y se concentró al vacío, obteniendo el producto en bruto, que se purificó mediante cromatografía de columna sobre gel de sílice (100-200), eluyendo con EtOAc al 3-5 %: hexano, proporcionando el producto deseado en forma de un sólido blanquecino (9,50 g, rendimiento: 66 %). RMN 1H (400 MHz, cloroformo-d) δ 7,54 - 7,27 (m, 6H), 6,81 (d, J = 7,9 Hz, 1H), 5,16 (s, 2H), 2,84 (q, J = 7,5 Hz, 2H), 1,32 (s, 12H), 1,14 (t, J = 7,5 Hz, 3H).
Preparación 10: 6-(4-(benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-3-(trimetilestanil)-1H-indazol

(a) 6-(4-(Benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol
A una solución of 6-bromo-1-(tetrahidro-2H-pirán-2-il)-1H-indazol (50 g, 178,57 mmoles) y 2-(4-(benciloxi)-2-etil-5-fluorofenil)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (76,3 g, 214,29 mmoles) en DMF:H2O (480:120 ml) se añadió K3PO4 (94,64 g, 446,86 mmoles). La mezcla de reacción se desgasificó con nitrógeno durante 15 min, seguido de la adición de catalizador Pd(PPh3)2Cl2 (6,26 g, 8,93 mmoles) y la mezcla se desgasificó nuevamente con nitrógeno durante 5 min bajo agitación y se calentó a 100-110 °C durante 5 h. La mezcla de reacción se filtró a través de Celite y el residuo se lavó con EtOAc. El filtrado se diluyó con EtOAc, se lavó con agua fría y solución hipersalina, se secó sobre sulfato sódico y se concentró al vacío, proporcionando el producto en bruto, que se purificó mediante cromatografía de columna flash, proporcionando el intermediario del título en forma de un sólido blanco (65 g, rendimiento: 86 %). (m/z): [M+H]+ calc. para C27H27FN2O2: 431,21; observado: 431,46. RMN 1H (400 MHz, cloroformo-d) δ 8,06 - 7,98 (m, 2H), 7,70 (d, J = 8,2 Hz, 1H), 7,51 - 7,32 (m, 5H), 7,08 (dd, J = 809,6, 8,3 Hz, 1H), 7,03 (d, J = 11,9 Hz, 1H), 6,95 (d, J = 8,5 Hz, 1H), 5,76 - 5,64 (m, 1H), 5,20 (s, 2H), 4,04 (d, J = 10,1 Hz, 1H), 3,72 (t, J = 9,7 Hz, 1H), 2,52 (q, J = 7,5 Hz, 2H), 2,22 - 2,02 (m, 3H), 1,80 - 1,71 (m, 3H), 1,06 (t, J = 7,5 Hz, 3H).
(b) 6-(4-(Benciloxi)-2-etil-5-fluorofenil)-1H-indazol
A una solución del producto de la etapa anterior (65 g, 151,16 mmoles) en metanol (700 ml) se añadió HCl conc. (120 ml) y la solución resultante se calentó a 60-65 °C durante 3 h, se enfrió a TA y se concentró al vacío. El residuo se disolvió en EtOAc y se lavó con solución acuosa saturada de NaHCO3 y agua. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró al vacío, proporcionando el intermediario del título en forma de un sólido blanco (52 g, 99 % (en bruto)). RMN 1H (400 MHz, Cloroformo-d) δ 8.13 (s, 1H), 7,77 (d, J = 8,3 Hz, 1H), 7,59 - 7,30 (m, 6H), 7,10 (d, J = 8,3 Hz, 1H), 7,01 (d, J = 11,8 Hz, 1H), 6,96 (d, J = 8,4 Hz, 1H), 5,21 (s, 2H), 2,53 (q, J = 7,5 Hz, 2H), 1,05 (t, J = 7,5 Hz, 3H).
(c) 6-(4-(Benciloxi)-2-etil-5-fluorofenil)-3-yodo-1H-indazol
[0166] A una solución of 6-(4-(benciloxi)-2-etil-5-fluorofenil)-1H-indazol (56 g, 161,18 mmoles) en DMF (400 ml) se añadió KOH (36,2 g, 647,39 mmoles) y la mezcla se sometió a agitación durante 5 min. Se añadió una solución de yodo (82,2 g, 323,69 mmoles) en DMF (100 ml) lentamente a 0 °C y se sometió a agitación a TA durante 30 min, se diluyó con agua (3×150 ml) y se extrajo con EtOAc (3×200 ml). La capa orgánica se lavó con solución acuosa saturada de metabisulfito sódico (3×200 ml) y agua (400 ml), se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida, obteniendo producto en bruto que se purificó mediante cromatografía de columna, proporcionando el
intermediario del título en forma de un semisólido parduzco (64 g, rendimiento: 84 %). RMN 1H (400 MHz, cloroformod) δ 10,49 (s, 1H), 7,57 - 7,32 (m, 7H), 7,16 (d, J = 8,3 Hz, 1H), 7,04 - 6,91 (m, 2H), 5,20 (s, 2H), 2,51 (q, J = 7,4 Hz, 2H), 1,04 (t, J = 7,5 Hz, 3H).
(d) 6-(4-(Benciloxi)-2-etil-5-fluorofenil)-3-yodo-1-(tetrahidro-2H-pirán-2-il)-1H-indazol
A una solución helada del producto de la etapa anterior (60 g, 127,12 mmoles) en DCM (700 ml) se añadió ácido ptoluenosulfónico (4,84 g, 25,423 mmoles) seguido de 3,4-dihidro-2H-pirano (17,43 ml, 190,68 mmoles) gota a gota. La mezcla de reacción se sometió a agitación a TA durante la noche, se diluyó con DCM y se lavó con solución acuosa saturada de NaHCO3 y solución hipersalina. La capa orgánica se secó sobre Na2SO4 anhidro y se concentró bajo presión reducida, proporcionando el producto en bruto, que se purificó mediante cromatografía flash (gel de sílice), proporcionando el intermediario del título en forma de un sólido blanquecino (64 g, rendimiento: 91 %).
(m/z): [M+H]+ calc. para C27H26FIN2O2: 557,10; observado: 557,30. RMN 1H (400 MHz, cloroformo-d) δ 7,56 - 7,31 (m, 7H), 7,14 (d, J = 8,3 Hz, 1H), 7,01 (d, J = 11,8 Hz, 1H), 6,95 (d, J = 8,5 Hz, 1H), 5,68 (d, J = 9,3 Hz, 1H), 5,20 (s, 2H), 4,08 - 3,99 (m, 1H), 3,77 - 3,64 (m, 1H), 2,50 (q, J= 7,2 Hz, 2H), 2,23 - 1,97 (m, 3H), 1,81 - 1,68 (m, 3H), 1,06 (t, J = 7,4 Hz, 3H).
(e) 6-(4-(benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-3-(trimetilestanil)-1H-indazol
A una solución of 6-(4-(benciloxi)-2-etil-5-fluorofenil)-3-yodo-1-(tetrahidro-2H-pirán-2-il)-1H-indazol (20 g, 35,97 mmoles) en tolueno (150 ml) se añadió hexametildiestaño (9,2 ml, 43,17 mmoles). La mezcla de reacción se desgasificó con nitrógeno durante 20 min, seguido de la adición de tetrakis (2,0 g, 1,80 mmoles) y después se sometió a agitación a 100 °C durante 2 h, se enfrió hasta la TA, se filtró a través de Celite y el residuo se lavó con EtOAc. El filtrado se concentró y se purificó mediante cromatografía de columna (sobre alúmina neutra), eluido con EtOAc al 2-5 %:hexano, proporcionando el compuesto del título (17,50 g, rendimiento: 82 %).
(m/z): [M+H]+ calc. para C30H35FN2O2Sn 595,17, 593,17; observado: 595,49, 593,55. RMN 1H (400 MHz, cloroformod) δ 7,68 (d, J = 8,0 Hz, 1H), 7,57 - 7,29 (m, 6H), 7,13 - 7,00 (m, 2H), 6,96 (d, J = 8,4 Hz, 1H), 5,81 - 5,68 (m, 1H), 5,21 (s, 2H), 4,13 - 4,00 (m, 1H), 3,81 - 3,66 (m, 1H), 2,54 (q, J = 7,3 Hz, 2H), 2,23 - 2,00 (m, 2H), 1,87 - 1,59 (m, 4H), 1,08 (t, J = 7,5 Hz, 3H), 0,47 (s, 9H).
Preparación 11: (S)-2-yodo-3-((2-trimetilsilil)etoxi)metil)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5- terc-butil-6-metilo

(a) Ácido (S)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico
A una suspensión bajo agitación de L-histidina (50 g, 322,24 mmoles) en agua (420 ml) se añadió HCl conc. (29 ml) gota a gota a 0 °C seguido de formaldehído (55 ml, 676,72 mmoles) de una vez a 0 °C. La mezcla de reacción resultante se sometió a agitación durante 30 min y después se calentó a 75 °C durante 6 h y se concentró. El producto en bruto resultante se sometió a agitación durante 2 h con éter dietílico, se filtró y se lavó con IPA:THF (100:300 ml), proporcionando la sal HCl del intermediario del título en forma de un sólido blanquecino (75 g, rendimiento: 99 % (en bruto)). (m/z): [M+H]+ calc. para C7H9N3O2: 168,07; observado: 168,17.
(b) (S)-4,5,6,7-Tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxilato de metilo
A una solución bajo agitación del producto de la etapa anterior (75,0 g, 312,5 mmoles) en metanol (1500 ml) se añadió SOCl2 (45,6 ml, 625 mmoles) gota a gota a 0 °C y se sometió a agitación a TA durante 16 h, después se calentó a reflujo (70 °C) durante 1 h. El solvente se eliminó mediante destilación y el producto en bruto se trituró con metanol, seguido de éter dietílico, proporcionando la sal HCl en bruto del intermediario del título en forma de un sólido blanquecino (80 g en bruto).
RMN 1H (400 MHz, DMSO-d6) δ 9,05 (s, 1H), 4,71 (dd, J = 9,4, 5,2 Hz, 1H), 4,36 (d, J = 15,5 Hz, 1H), 4,30 (d, J = 15,6 Hz, 1H), 3,82 (s, 3H), 3,44 - 3,21 (m, 2H).
(c) (S)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(terc-butil)-6-metilo
A una solución bajo agitación del producto de la etapa anterior (80,0 g, 314,96 mmoles) en metanol (1000 ml) se añadió DIPEA (282 ml, 1574 mmoles) seguido de dicarbonato de di-terc-butilo (172 ml, 787,48 mmoles) a 0 °C. La mezcla de reacción se sometió a agitación a TA durante 16 h y después se añadió NH3 líquido (150 ml, al 25 % en agua) y la mezcla de reacción se sometió a agitación nuevamente durante 16 h a TA, se eliminó el metanol mediante destilación y el residuo se extrajo en DCM (3×200 ml). Los extractos orgánicos agrupados se secaron sobre Na2SO4 anhidro, se concentraron y se purificaron mediante cromatografía flash (gel de sílice de malla 100-200), se eluyeron con MeOH al 5 %:DCM, proporcionando el intermediario del título (41 g, rendimiento: 46 %). (m/z): [M+H]+ calc. para C13H19N3O4: 282,14; observado: 282,21. RMN 1H (400 MHz, DMSO-d6) δ 11,85 (s, 1H), 7,50 (s, 1H), 5,18 (dd, J = 49,3, 5,1 Hz, 1H), 4,51 (t, J = 14,2 Hz, 1H), 4,09 (dd, J = 43,9, 16,1 Hz, 1H), 3,59 (s, 3H), 3,08 (d, J = 15,5 Hz, 1H), 2,94 (d, J = 15,1 Hz, 1H), 1,45 (s, 9H).
(d) (S)-2-Yodo-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(terc-butil)-6-metilo
A una solución del producto de la etapa anterior (41,0 g, 145,9 mmoles) en THF (500 ml) se añadió A-yodosuccinimida (66,0 g, 291,8 mmoles) a 0 °C y la solución resultante se sometió a agitación a TA durante 4 h, se diluyó con agua y se extrajo con acetato de etilo. La parte orgánica se lavó con solución al 10 % de tiosulfato sódico (3×200 ml). La capa orgánica agrupada se secó sobre sulfato sódico anhidro y se concentró, proporcionando el compuesto del título, 60 g (en bruto), que se utilizó en la etapa siguiente sin purificación adicional. (m/z): [M+H]+ calc. para C13H18IN3O4: 408,03; observado: 408,31. RMN 1H (400 MHz, DMSO-d6) δ 12,48 (s, 1H), 5,34 - 4,97 (m, 1H), 4,67 - 4,35 (m, 1H), 4,12 - 3,95 (m, 1H), 3,60 (s, 3H), 3,14 - 2,82 (m, 2H), 1,44 (s, 9H).
(e) (S)-2-Yodo-3-((2-trimetilsilil)etoxi)metil)-3,4,6,7-tetrahidro -5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(tercbutil)-6-metilo.
A una solución bajo agitación de (S)-2-yodo-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(tercbutil)-6-metilo (40 g, 0,098 moles) en DMF (150 ml) se añadió DIPEA (35,1 ml, 0,19 moles) a 0°C. La mezcla de reacción se sometió a agitación durante 10 min y a continuación se añadió cloruro de 2-(trimetilsilil)-etoximetilo (19,1 ml, 0,10 moles) gota a gota a 0 °C. La mezcla de reacción resultante se sometió a agitación durante 3 h a TA. Tras 4 h, se añadió agua fría y la mezcla de reacción se extrajo con EtOAc (2×200 ml). La capa orgánica se secó sobre sulfato sódico anhidro, se concentró y se purificó mediante cromatografía flash, se eluyó con EtOAc al 20-35 %:hexano, proporcionando el producto del título en forma de un líquido viscoso amarillo pálido (27 g). (m/z): [M+H]+ calc. para C19H32IN3O5Si: 538,12; observado: 538,42. RMN 1H (400 MHz, DMSO-d6) δ 5,33 - 5,04 (m, 3H), 4,79 - 4,56 (m, 1H), 4,54 - 4,14 (m, 1H), 3,60 (s, 3H), 3,47 (t, J = 7,8 Hz, 2H), 3,31 - 3,16 (m, 1H), 2,97 (t, J = 18,9 Hz, 1H), 1,44 (s, 9H), 0,92 - 0,74 (m, 2H), -0,03 (s, 9H).
Preparación 12: ácido (6S)-5-(terc-butoxicarbonil)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol-3-il)-3-((2-(trimetilsilil)etoxi)metil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico

(a) (6S)-2-(6-(4-(Benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol-3-il)-3-((2-(trimetilsilil)etoxi) metil)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(terc-butil)-6-metilo
A una solución bajo agitación de (S)-2-yodo-3-((2-trimetilsilil)etoxi)metil)-3,4,6,7-tetrahidro -5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 5-(terc-butil)-6-metilo (17,0 g, 31,65 mmoles) en tolueno (500 ml) se añadió 6-(4-(benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-3-(trimetilestanil)-1H-indazol (20 g, 34,82 mmoles). La mezcla de reacción se purgó con argón durante 15 min, se añadió Pd(PPh3)4 (3,6 g, 3,16 mmoles) y yoduro de cobre (1,20 g, 6,33 mmoles) y la mezcla de reacción se sometió a agitación a 120 °C durante 16 h. La mezcla de reacción se filtró a través de Celite; el filtrado se concentró bajo presión reducida y se purificó mediante cromatografía de columna en gel de sílice (columna Redisep de 80 g, eluido con DCM durante 10 min y después EtOAc al 15-20 % en hexano, proporcionando el intermediario del título en forma de un sólido amarillo (15,10 g, rendimiento de 58 %).
(m/z): [M+H]+ calc. para C46H58FN5O7Si: 840,41; observado: 840,54. RMN 1H (400 MHz, cloroformo-d) δ 8,43 (s, 1H), 7,54 - 7,33 (m, 6H), 7,20 (s, 1H), 7,05 (d, J = 11,4 Hz, 1H), 6,95 (d, J = 8,5 Hz, 1H), 6,09 - 5,69 (m, 3H), 5,59 - 5,36 (m, 1H), 5,20 (s, 2H), 4,97 - 4,80 (m, 1H), 4,12 - 3,90 (m, 1H), 3,68 (s, 3H), 3,57 - 3,47 (m, 2H), 3,40 (d, 1H), 3,21 -3,05 (m, 1H), 2,74 - 2,34 (m, 4H), 2,25 - 2,07 (m, 2H), 1,94 - 1,65 (m, 4H), 1,54 (s, 9H), 1,12 - 0,99 (m, 3H), 0,91 - 0,75 (m, 2H), -0,12 (s, 9H).
(b) (6S)-2-(6-(4-(Benciloxi)-2-etil-5-fluorofenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol-3-il)-3-((2-(trimetilsilil)etoxi)metil)-3,4,6,7-tetrahidro-5H-imidazo[4,5-c]piridín-5,6-dicarboxilato de 6-bencil-5-(terc-butilo) A un matraz de fondo redondo se añadió el producto de la etapa anterior (15,0 g, 17,85 mmoles) en tolueno (400 ml), alcohol bencílico (46,3 ml) y Ti(OEt)4 (7,15 ml, 35,70 mmoles) y la mezcla de reacción se sometió a reflujo vigorosamente (140 °C) durante 48 h, se diluyó con agua y se extrajo con DCM. La suspensión se filtró, el filtrado se secó sobre Na2SO4, se concentró bajo presión reducida y se purificó mediante cromatografía de columna en gel de sílice (columna Redisep de 80 g, EtOAc al 0-5 % en hexanos) durante 20 min para eliminar el exceso de alcohol bencílico; después se eluyó con EtOAc al 10-15 % en hexano), proporcionando el intermediario del título. RMN 1H consistente con la estructura. (m/z): [M+H]+ calc. para C52H62FN5O7Si: 916,44; observado: 916,86.
(c) Ácido (6S)-5-(terc-butoxicarbonil)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol-3-il)-3-((2-(trimetilsilil)etoxi)metil)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico
A una solución bajo agitación del producto de la etapa anterior (21,0 g, 22,92 mmoles) en 1:1 IPA:THF (400 ml) se añadió Pd(OH)2 (5,0 g). La mezcla de reacción se sometió a agitación a TA durante 16 h bajo un balón de hidrógeno, se filtró a través de Celite, se concentró bajo presión reducida y se purificó mediante cromatografía de columna de gel de sílice (columna Redisep de 80 g, se eluyó con EtOAc al 25-40 % en hexano), proporcionando el compuesto del título (6,1 g, 8,29 mmoles) en forma de un sólido blanquecino). (m/z): [M+H]+ calc. para C38H50FN5O7Si: 736,35; observado: 736,5. RMN 1H consistente con la estructura. (m/z): [M+H]+ calc. para C38H50FN5O7Si: 736,35; observado: 736,5. RMN 1H (400 MHz, DMSO-d6) δ 12,94 (s, 1H), 9,86 (s, 1H), 8,34 (t, J = 7,6 Hz, 1H), 7,66 (s, 1H), 7,20 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 11,8 Hz, 1H), 6,93 (d, J = 9,1 Hz, 1H), 6,11 - 5,77 (m, 3H), 5,33 - 5,06 (m, 1H), 4,87 - 4,56 (m, 1H), 4,52 - 4,14 (m, 1H), 3,97 - 3,69 (m, 2H), 3,53 - 3,40 (m, 2H), 3,23 - 3,11 (m, 1H), 3,11 - 2,93 (m, 1H), 2,47 - 2,44 (m, 2H), 2,13 - 1,96 (m, 2H), 1,68 (d, J = 70,9 Hz, 4H), 1,48 (s, 9H), 1,02 (t, J = 7,5 Hz, 3H), 0,86 - 0,68 (m, 2H), -0,17 (s, 9H).
Preparación 13: ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico

A una solución bajo agitación de ácido (6S)-5-(terc-butoxicarbonil)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1-(tetrahidro-2H-pirán-2-il)-1H-indazol-3-il)-3-((2-(trimetilsilil)etoxi)-metil)-4,5,6,7-tetrahidro-3/7-imidazo[4,5-c]piridín-6-carboxílico (5,7 g, 7,75 mmoles) en dioxano:agua 5:1 (60 ml) se añadió HCl conc. (20 ml) gota a gota a 0 °C. La mezcla de reacción se calentó y se sometió a agitación a 90 °C durante 16 h y se destiló bajo vacío, proporcionando el residuo en bruto, que se trituró secuencialmente con éter dietílico frío y acetonitrilo, proporcionando la sal HCl del compuesto del título (3,6 g, rendimiento: 95 %) en forma de un sólido marrón pálido. (m/z): [M+H]+ calc. para C22H20FN5O3: 422,16; observado: 422,24. RMN 1H (400 MHz, D2O/DMSO-d6) δ 8,22 (d, J = 8,4 Hz, 1H), 7,49 (s, 1H), 7,19 (d, J = 8,1 Hz, 1 H), 6,99 (d, J = 11,9 Hz, 1 H), 6,91 (d, J = 9,0 Hz, 1H), 4,56-4,51 (m, 1H), 4,36 (d, J = 15,5 Hz, 1H), 4,30 (d, J = 15,5 Hz, 1H), 3,35 - 3,25 (m, 1H), 3,15 - 3,05 (m, 1H), 2,4 - 2,55 (m, 2H), 0,97 (t, J = 7,5 Hz, 3H).
Preparación 14: ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (400 mg, 0,874 mmoles) y propionaldehído (0,095 ml, 1,310 mmoles) en DMF (7 ml) se añadió cianoborohidruro sódico (165 mg, 2,62 mmoles) y la mezcla de reacción se sometió a agitación a la TA durante la noche. Se añadió borohidruro sódico (33 mg, 0,874 mmoles), se concentró la solución, y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (179 mg, rendimiento: 37 %). (m/z): [M+H]+ calc. para C25H26FN5O3: 464,20; observado: 464,5.
Preparación 15: ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H imidazo[4,5-c]piridín-6-carboxílico

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (400 mg, 0,874 mmoles), acetona (0,192 ml, 2,62 mmoles) y ácido acético (0,150 ml, 2,62 mmoles) en DMF (7 ml); se añadió cianoborohidruro sódico (274 mg, 4,37 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió borohidruro sódico (33 mg, 0,874 mmoles), se concentró la solución y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (115 mg, rendimiento: 23 %). (m/z): [M+H]+ calc. para C25H26FN5O3: 464,20; observado: 464,5.
Preparación 16: ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, HCl (8') (300 mg, 0,655 mmoles) y formaldehído al 37 % en peso en agua (0,059 ml, 0,786 mmoles) DMF (5 ml), se añadió cianoborohidruro sódico (165 mg, 2,62 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió borohidruro sódico (25 mg, 0,655 mmoles), se concentró la solución y se purificó mediante cromatografía flash (columna de 100 g, 5-75 % de ACN/agua), proporcionando la sal de TFA del compuesto del título (85 mg, rendimiento: 24 %).
(m/z): [M+H]+ calc. para C23H22FN5O3: 436,17; observado: 436,45.
Ejemplo 7: (S)-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)(4-(2-hidroxietil)piperazín-1-il)metanona C-1

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (30 mg, 0,052 mmoles), 2-(piperazín-1-il)etanol, 2HCl (0,19 ml, 0,156 mmoles) y DIPEA (0,027 mL, 0,156 mmoles) en DMF (1,5 ml) se añadió HATU (29,6 mg, 0,078 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió hidrazina (5 eq.), se concentró la mezcla de reacción y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (15,4 mg, rendimiento: 37 %). (m/z): [M+H]+ calc. para C31H38FN7O3: 576,30; observado: 576,2.
Ejemplo 8: ((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((1S,4S)-5-metil-2,5-diazabiciclo-[2.2.1]heptán-2-il)metanona C-2
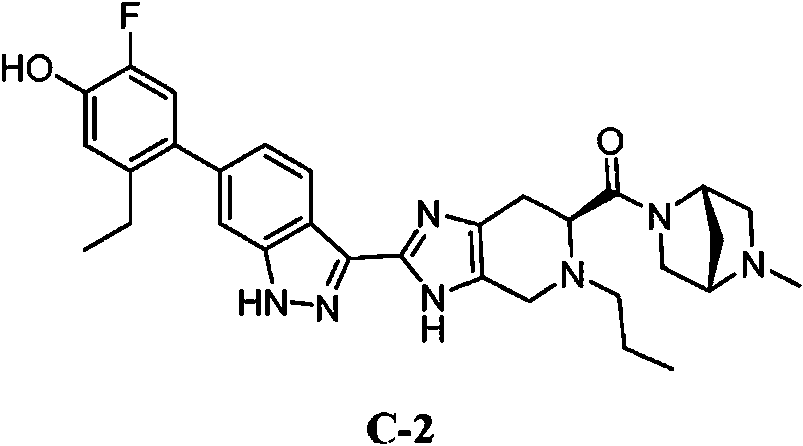
A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-propil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (30 mg, 0,052 mmoles), dihidrobromuro de (1S,4S)-5-metil-2,5-diazabiciclo[2.2.1]heptano (42,7 mg, 0,156 mmoles) y DIPEA (0,064 ml, 0,364 mmoles) en DMF (1,5 ml) se añadió HATU (29,6 mg, 0,078 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió hidrazina (5 eq.), se concentró la mezcla de reacción y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (27 mg, rendimiento: 66 %). (m/z): [M+H]+ calc. para C31H36FN7O2: 558,29; observado: 558,3. RMN 1H (400 MHz, metanol-d4) δ 8,17 (dt, 1H), 7,59 - 7,50 (m, 1H), 7,32 (dd, 1H), 6,95 (d, 1H), 6,90 (d, 1H), 5,03 - 4,91 (m, 2H), 4,56 - 4,34 (m, 2H), 4,30 - 3,88 (m, 4H), 3,76 - 3,55 (m, 1H), 3,28 - 3,10 (m, 1H), 3,10 - 2,96 (m, 4H), 2,81 - 2,62 (m, 2H), 2,53 (q, 2H), 2,47 - 2,33 (m, 1H), 2,31 - 2,14 (m, 1H), 1,79 - 1,57 (m, 2H), 1,07 (t, 3H), 0,97 (td, 3H).
Ejemplo 9: ((S)-2,4-dimetilpiperazín-1-il)((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona C-3

(a) (S)-4-((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carbonil)-3-metilpiperazín-1-carboxilato de terc-butilo
Se disolvió ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico (0,120 g, 0,276 mmoles), (S)-4-n-boc-2-metilpiperazina (0,110 g, 0,551 mmoles) y DIPEA (0,096 ml, 0,551 mmoles) en DMF (3,0 ml); a continuación, se añadió HATU (0,157 g, 0,413 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 16 horas (el progreso de la reacción se siguió mediante CL
EM). Se añadió hidrazina (0,043 ml, 1,378 mmoles) para cortar los productos secundarios no deseados; a continuación, la mezcla de reacción se sometió a agitación a temperatura ambiente durante 10 minutos. A continuación, la mezcla de reacción se concentró y el producto en bruto se purificó mediante cromatografía de fase inversa (gradiente de 5-70 % de ACN/agua, columna C18 de 50 g), proporcionando la sal de TFA del compuesto del título (126 mg, rendimiento: 62 %). (m/z): [M+H]+ calc. para C33H40FN7O4: 618,32; observado: 618,3.
(b) ((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((S)-2-metilpiperazín-1-il)metanona
Se disolvió (S)-4-((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carbonil)-3-metilpiperazín-1-carboxilato de terc-butilo, TFA (0,126 g, 0,172 mmoles) en dioxano (1,5 ml) y agua (0,3 ml); a continuación, se añadió ácido clorhídrico 4 M en dioxano (1,5 ml, 49,4 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 1 hora (el progreso de la reacción se siguió mediante CL-EM). La mezcla de reacción se congeló y se liofilizó, y los polvos resultantes se utilizaron directamente en la reacción siguiente (se partió de que el rendimiento era cuantitativo). (m/z):
[M+H]+ calc. para C28H32FN7O2: 518,26; observado: 518,3.
(c) ((S)-2,4-dimetilpiperazín-1-il)((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona
Se disolvió ((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((S)-2-metilpiperazín-1-il)metanona, dihidrocloruro (0,102 g, 0,173 mmoles) y solución de formaldehído, al 37 % en peso en agua (0,015 ml, 0,207 mmoles) en metanol (3,0 ml); a continuación, se añadió cianoborohidruro sódico (0,054 g, 0,864 mmoles) y la mezcla de reacción se sometió a agitación a temperatura ambiente durante 20 horas (el progreso de la reacción se siguió mediante CL-EM). Se añadió borohidruro sódico (7 mg, 0,173 mmoles) para desactivar cualquier formaldehído remanente. Se concentró la mezcla de reacción; a continuación, el producto en bruto se purificó mediante HPLC preparativa (gradiente de 5-60 % de ACN/agua, columna C18), proporcionando el compuesto del título en forma de sal TFA (59 mg, rendimiento: 45 %). (m/z): [M+H]+ calc. para C29H34FN7O2: 532,28; observado: 532,3.
Ejemplo 10: (S)-(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)(4-metil-1,4-diazepan-1-il)metanona C-4

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (30 mg, 0,052 mmoles), 1-metilhomopiperazina (0,019 ml, 0,156 mmoles) y DIPEA (0,036 ml, 0,208 mmoles) en DMF (1 ml) se añadió HATU (29,6 mg, 0,078 mmoles) y la mezcla de reacción se sometió a agitación a TA durante 3 h. Se añadió hidrazina (5 eq.) y la mezcla de reacción se sometió a agitación a TA durante 10 min, se concentró y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (26,9 mg, rendimiento: 66 %). (m/z): [M+H]+ calc. para C31H38FN7O2: 560,31; observado: 560,2.
Ejemplo 11: ((S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)((R)-4-(2-hidroxietil)-2-metil-piperazín-1-il)metanona C-5

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-metil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (30 mg, 0,052 mmoles), (R)-2-(3-metilpiperazín-1-il)etanol, 2 HCl (35,6 mg,
0,164 mmoles) y DIPEA (0,057 ml, 0,328 mmoles) en DMF (1 ml) se añadió HATU (31,1 mg, 0,082 mmoles) y la mezcla de reacción se sometió a agitación a la TA durante la noche. Se añadió hidrazina (8,57 µl, 0,273 mmoles) y se concentró la mezcla de reacción y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (15,6 mg, rendimiento: 36 %).
(m/z): [M+H]+ calc. para C30H36FN7O3: 562,29; observado: 562,2.
Ejemplo 12: ((S)-3-(dimetilamino)pirrolidín-1-il)((S)-5-etil-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona C-6

A una solución de ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (179 mg, 0,310 mmoles), (S)-N,N-dimetilpirrolidín-3-amina (0,079 ml, 0,620 mmoles) y DIPEA (0,162 ml, 0,930 mmoles) en DMF (4 ml) se añadió HATU (177 mg, 0,465 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió hidrazina (5 eq.), se concentró la mezcla de reacción y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (107 mg, rendimiento: 44 %). (m/z): [M+H]+ calc. para C31H38FN7O2: 560,31; observado: 560,2. RMN 1H (400 MHz, metanol-d4) δ 8,21 (d, 1H), 7,50 (s, 1H), 7,26 (d, 1H), 6,94 (d, 1H), 6,90 (d, 1H), 4,83 - 4,66 (m, 1H), 4,48 - 4,25 (m, 2H), 4,23 - 4,12 (m, 1H), 4,12 - 3,93 (m, 2H), 3,93 - 3,63 (m, 3H), 3,62 - 3,48 (m, 1H), 3,26 - 3,09 (m, 1H), 2,98 (d, 6H), 2,67 - 2,57 (m, 1H), 2,53 (q, 2H), 2,44 - 2,12 (m, 1H), 1,41 (t, 3H), 1,31 (d, 3H), 1,05 (t, 3H).
Ejemplo 13: (S)-(3-(dimetilamino)azetidín-1-il)(2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-il)metanona D-1

A una solución of ácido (S)-2-(6-(2-etil-5-fluoro-4-hidroxifenil)-1H-indazol-3-il)-5-isopropil-4,5,6,7-tetrahidro-3H-imidazo[4,5-c]piridín-6-carboxílico, TFA (179 mg, 0,310 mmoles), N,N-dimetilazetidin-3-amina, 2 HCl (107 mg, 0,465 mmoles) y DIPEA (0,162 ml, 0,930 mmoles) en DMF (4 ml), se añadió HATU (177 mg, 0,465 mmoles) y la mezcla de reacción se sometió a agitación a TA durante la noche. Se añadió hidrazina (5 eq.), se concentró la mezcla de reacción y se purificó mediante HPLC preparativa, proporcionando la sal de TFA del compuesto del título (63 mg, rendimiento: 26 %). (m/z): [M+H]+ calc. para C30H36FN7O2: 546,29; observado: 546,7. RMN 1H (400 MHz, DMSO-d6) δ 9,90 (s, 1H), 8,29 (dd, 1H), 7,34 (s, 1H), 7,07 (d, 1H), 7,01 (d, 1H), 6,89 (d, 1H), 4,35 - 4,18 (m, 1H), 4,11 - 3,94 (m, 1H), 3,94 - 3,73 (m, 3H), 3,70 - 3,57 (m, 2H), 3,06 - 2,94 (m, 2H), 2,87 - 2,66 (m, 2H), 2,48 - 2,40 (m, 2H), 2,13 - 2,00 (m, 6H), 1,07 (t, 3H), 1,03 - 0,93 (m, 6H).
Ensayos biológicos
Los compuestos de la invención se caracterizaron en uno o más de los ensayos biológicos siguientes.
Ensayo 1: ensayos bioquímicos de quinasa JAK.
Se llevó a cabo un panel de cuatro ensayos bioquímicos LanthaScreen de JAK (JAK 1, 2, 3 y Tyk2) en un tampón de reacción de quinasa habitual (HEPES 50 mM, pH 7,5, Brij-35 al 0,01 %, MgCl210 mM y EGTA 1 mM). Los enzimas JAK etiquetados con GST recombinantes y un sustrato peptídico STAT1 etiquetado con GFP se obtuvieron de Life Technologies.
Se preincubaron compuestos diluidos en serie con cada uno de los cuatro enzimas JAK y el sustrato en microplacas blancas de 384 pocillos (Corning) a temperatura ambiente durante 1 h. A continuación, se añadió ATP para iniciar las reacciones de quinasa en un volumen total de 10 µl, con DMSO al 1 %. Las concentraciones de enzima finales para JAK 1, 2, 3 y Tyk2 eran de 4,2 nM, 0,1 nM, 1 nM y 0,25 nM, respectivamente; las concentraciones Km de ATP correspondientes utilizadas fueron de 25 µM, 3 µM, 1.6 µM y 10 µM, mientras que la concentración de sustrato era de 200 nM para la totalidad de los cuatro ensayos. Se dejó que continuasen las reacciones de quinasa durante 1 hora a temperatura ambiente antes de añadir una preparación de 10 µl de EDTA (concentración final: 10 mM) y Tb-anticuerpo anti-pSTAT1 (pTyr701) (Life Technologies, concentración final: 2 nM) en tampón de dilución TR-FRET (Life Technologies). Las placas se dejaron bajo incubación a temperatura ambiente durante 1 h antes de leerlas en el lector EnVision (Perkin Elmer). Se registraron las señales de relación de emisiones (520 nm/495 nm) y se utilizaron para calcular los valores en porcentaje de inhibición basados en los controles de DMSO y de fondo.
Para el análisis de dosis-respuesta, los datos porcentuales de inhibición se representaron frente a las concentraciones de compuesto y se determinaron los valores de IC50 a partir de un modelo de ajuste robusto de 4 parámetros con el software Prism (GraphPad Software). Los resultados se expresaron como pIC50 (logaritmo negativo de IC50) y posteriormente se convirtieron en pKi (logaritmo negativo de la constante de disociación, Ki) utilizando la ecuación de Cheng-Prusoff.
Los compuestos de ensayo con un valor de Ki más bajo o un valor de pKi más alto en cada uno de los cuatro ensayos de JAK mostraron una mayor inhibición de la actividad de JAK.
Ensayo 2: inhibición de pSTAT5 estimulada por IL-2 en células T Tall-1.
Se midió la potencia de los compuestos de ensayo para la inhibición de la fosforilación de STATS estimulada por interleuquina-2 (IL-2) en la línea celular de linfocitos T humanos Tall-1 (DSMZ) utilizando AlphaLisa. Debido a que IL-2 señaliza a través de JAK1/3, el presente ensayo proporciona una medida de la potencia celular de JAK1/3.
Se midió STATS fosforilado mediante el kit AlphaLISA SureFire Ultra pSTAT5 (Tyr694/699) (Perkin Elmer).
Se cultivaron linfocitos T humanos de la línea celular Tall-1 en un incubador a 37 ºC, humidificado y con 5 % de CO2, en RPMI (Life Technologies) complementado con suero de feto bovino inactivado por calor al 15 % (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies y 1X Pen/Strep (Life Technologies). Los compuestos se diluyeron en serie en DMSO y se dispensaron acústicamente en pocillos vacíos. Los medios de ensayo (DMEM sin rojo fenol (Life Technologies) complementados con FBS al 10 % (ATCC)) se dispensaron (4 µl/pocillo) y las placas se sometieron a agitación a 900 rpm durante 10 min. Las células se sembraron a razón de 45.000 células/pocillo en medio de ensayo (4 µl/pocillo) y se incubaron a 37 ºC, 5 % de CO2 durante 1 hora, seguido de la adición de IL-2 (R&D Systems; concentración final: 300 ng/ml) en medio de ensayo precalentado (4 µl) durante 30 minutos. Tras la estimulación con citoquina, las células se lisaron con 6 µl de 3x tampón de lisis AlphaLisa (Perkin Elmer) que contenía 1x tabletas PhosStop y cOmplete (Roche). El lisado se sometió a agitación a 900 rpm durante 10 minutos a temperatura ambiente (TA). Se midieron los STAT fosforilados mediante el kit pSTAT5 AlphaLisa (Perkin Elmer). Se dispensó mezcla de perlas aceptoras recién preparada sobre el lisado (5 µl) bajo luz verde filtrada de <100 lux. Las placas se sometieron a agitación a 900 rpm durante 2 min, se centrifugaron brevemente y se incubaron durante 2 h a TA en la oscuridad. Se dispensaron las perlas donadoras (5 µl) bajo luz verde filtrada de <100 lux. Las placas se sometieron a agitación a 900 rpm durante 2 minutos, se centrifugaron brevemente y se incubaron durante la noche a TA en la oscuridad. Se midió la luminiscencia bajo excitación a 689 nm y emisión a 570 nm utilizando un lector de placas EnVision (Perkin Elmer) bajo luz verde filtrada de <100 lux.
Para determinar la potencia de inhibición de los compuestos de ensayo en respuesta a IL-2, se midió la intensidad media de emisión de las perlas unidas a pSTAT5 en una línea celular de linfocitos T humanos. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de intensidad de señal frente a concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50) (medias ± desviaciones estándar).
Resultados de ensayo in vitro.
Tabla 1

(continuación)

Ensayo 3: modelo murino (ratón) de inducción de pSTAT6 inducida por IL-13 en tejido pulmonar.
La IL-13 es una citoquina importante que subyace a la fisiopatología del asma (Kudlacz et al., Eur. J. Pharmacol, 2008, 582,154-161). IL-13 se une a receptores de superficie celular que activan miembros de la familia Janus de quinasas (JAK), que a continuación fosforilan STAT6 y en consecuencia activan rutas posteriores de transcripción. En el modelo explicado, se administró una dosis de IL-13 localmente en los pulmones de ratones para inducir la fosforilación de STAT6 (pSTAT6), que a continuación se midió como criterio de valoración.
En el ensayo se utilizaron ratones Balb/c adultos de Harlan. El día del estudio, los animales fueron anestesiados levemente con isoflurano y se les administró vehículo o compuesto de ensayo (1 mg/ml, 50 µl de volumen total a lo largo de varias respiraciones) mediante aspiración oral. Los animales se tumbaron de costado después de la dosis y se les realizó un seguimiento hasta la recuperación total de la anestesia antes de devolverlos a su jaula de origen. Cuatro horas después se anestesiaron brevemente de nuevo y se les retó con vehículo o IL-13 (dosis total administrada de 0,03 µg, volumen total: 50 µl) mediante aspiración oral antes de realizarse un seguimiento para la recuperación de la anestesia y devolverlos a su jaula de origen. Una hora después de la administración de vehículo o IL-13, se recolectó sangre completa y los pulmones tanto para la detección de pSTAT6 en homogenados de pulmón utilizando un kit de ensayo AlphaLISA® SureFire® Ultra™ HV p-STAT6 (Tyr641) de Perkin Elmer, como un análisis de concentración total de fármaco en pulmón y plasma. Se centrifugaron muestras de sangre (centrífuga Eppendorf, 5804R) durante 4 minutos a aproximadamente 12.000 rpm a 4 ºC para recoger plasma. Se enjuagaron los pulmones en DPBS (solución salina tamponada con fosfato de Dulbecco), se secaron superficialmente con papel, se congelaron instantáneamente, se pesaron y se homogeneizaron a una dilución de 1:3 en ácido fórmico al 0,1 % en agua para HPLC. Se determinaron los niveles plasmático y pulmonar de compuesto de ensayo mediante análisis de CL-EM frente a patrones analíticos construidos en una curva patrón en la matriz de ensayo. Se determinó una relación de pulmón a plasma como la proporción entre la concentración en pulmón, en ng/g, y la concentración en plasma, en ng/ml, a las 5 horas.
La actividad en el modelo se puso de manifiesto por una reducción del nivel de pSTAT6 presente en los pulmones de los animales tratados a las 5 horas en comparación con animales de control tratados con vehículo y retados con IL-13. La diferente entre los animales de control que habían sido tratados con vehículo, retados con IL-13 y los animales de control que habían sido tratados con vehículo y retados con vehículo marcó los efectos de inhibición de 0 % y 100 %, respectivamente, en cualquier experimento dado. Los compuestos analizados en el ensayo mostraron una inhibición de la fosforilación de STAT6 a las 5 horas después del reto con IL-13, tal como se documenta posteriormente.
Tabla 2: inhibición de pSTAT6 y exposición en plasma/pulmón observada.

La observación de una concentración significativa en los compuestos sometidos a ensayo en pulmón de ratón confirmó que la inhibición observada de inducción de pSTAT6 inducida por IL-13 fue un resultado de la actividad del compuesto de ensayo. La relación de pulmón a plasma a las 5 horas mostró que los compuestos 1 a 6 mostraban significativamente más exposición en el pulmón que exposición en plasma en los ratones.
Ensayo 4: inhibición de la liberación de TARC evocada por TSLP en células mononucleares de sangre periférica humana.
La linfopoyetina estromal tímica (TSLP, por sus siglas en inglés) y la quimioquina regulada y activada del timo (TARC, por sus siglas en inglés) se sobreexpresaban en las vías respiratorias asmáticas y se correlacionaban con la gravedad
de la enfermedad. En los pulmones, la TSLP podría ser liberada por las células epiteliales bronquiales en respuesta a alérgenos e infecciones víricas. La TSLP señaliza a través de un heterodímero IL-7Rα/TSLPR que se encuentra en un amplio abanico de tejidos y tipos celulares, incluyendo células epiteliales, células endoteliales, neutrófilos, macrófagos y mastocitos. La unión de TSLP a su receptor induce un cambio conformacional que activa JAK1 y JAK2 para fosforilar diversos factores de transcripción, incluyendo STATS y STATS. En las células inmunitarias, lo anterior desencadena una cascada de sucesos intracelulares que resulta en proliferación celular, antiapoptosis, migración de células dendríticas y producción de citoquinas Th2 y quimioquinas. En las células mononucleares de sangre periférica (PBMC), la TSLP posee un efecto proinflamatorio mediante la activación de las células dendríticas mieloides, atrayendo y estimulando los linfocitos T, un proceso mediado por la TARC quimioatrayente.
En el presente ensayo se mostró que la estimulación de TSLP induce la liberación de TARC a partir de las PBMC, y que esta respuesta resulta atenuada de una manera dependiente de la dosis con el tratamiento con compuesto. Se midieron las potencias de los compuestos de ensayo en la inhibición de la liberación de TARC.
Se descongelaron alícuotas de PBMC (previamente aisladas de sangre completa y congeladas en alícuotas a -80 ºC) procedentes de 3 a 5 donantes, a 37 ºC, y se añadieron gota a gota a 40 ml de medio RPMI completo y filtrado a esterilidad, precalentado, en tubos Falcon de 50 ml. Se peletizaron las células y se resuspendieron en medio completo a razón de 2,24x106 células/ml. Las células se sembraron a razón de 85 µl (190.000 células) por pocillo en una microplaca de fondo plano de 96 pocillos tratada para el cultivo de tejidos. Las células se dejaron en reposo durante 1 hora a 37 ºC con 5 % de CO2.
Los compuestos se recibieron como soluciones patrón 10 mM en DMSO. Se llevaron a cabo diluciones en serie de 3,7 veces, generando 9 concentraciones de compuesto de ensayo en DMSO a 300X la concentración final de ensayo. Se llevaron a cabo diluciones intermedias de 150 veces en medio completo, generando compuesto a 2X la concentración de ensayo final utilizando DMSO al 0,2 %. Tras el periodo de reposo de 1 hora, se añadieron 95 µl de compuesto 2X a cada pocillo de PBMC para un abanico de concentraciones finales del ensayo de 33,3 µM a 0,95 µM. Se añadieron 95 µl de DMSO al 0,2 % en medio completo a los pocillos de control no tratados. Las células se pretrataron con compuesto durante 1 hora a 37 ºC con 5 % de CO2 antes de la estimulación.
Se reconstituyó proteína TLSP humana recombinante a 10 µg/ml en DPBS estéril con BSA al 0,1 % y se almacenó en alícuotas a -20 ºC. Inmediatamente antes del uso, se descongeló una alícuota y se preparó a 20X la concentración de ensayo final en medio completo. Se añadieron 10 µl de 20X TSLP a cada pocillo de PBMC para una concentración de ensayo final de 10 ng/ml. Se añadieron 10 µl de medio completo a los pocillos de control no estimulados. Las células se estimularon en la presencia de compuesto durante 48 horas a 37 ºC con 5 % de CO2.
Tras la estimulación, se recolectaron los sobrenadantes de cultivo celular y se detectaron los niveles de TARC mediante ensayo de inmunosorción ligada a enzima (ELISA, por sus siglas en inglés) utilizando el kit ELISA Quantikine CCL17/TARC humano (R&D Systems, n.º DDN00) siguiendo las instrucciones del fabricante.
Para el análisis de respuesta a dosis, se representó gráficamente el log [compuesto de ensayo (M)] frente a los valores de respuesta en porcentaje para cada donante, y se determinaron los valores de IC50 utilizando un análisis de regresión no lineal con el software GraphPad Prism utilizando el algoritmo de dosis-respuesta sigmoidal de 4 parámetros con pendiente variable. Los datos se expresaron como valores medios de pIC50 (logaritmo decadal negativo de IC50) calculados a partir de los valores de pIC50 de donantes individuales y redondeados un decimal. Los valores de potencia de inhibición por los compuestos originales y sus análogos desfluoro modificados se resumen en la Tabla 3.
Tabla 3: valores de potencia de inhibición de los compuestos de ensayo de liberación de TARC evocada por TSLP en células mononucleares de sangre periférica humana.

Ensayo 5: metabolismo de S9 pulmonar.
Se evaluó la estabilidad metabólica in vitro de los compuestos 1 a 6 y de C-1 a C-6 en la fracción S9 pulmonar humana (compuesto 1 µM; proteína S9, 1 mg/ml). Las muestras de 0, 15, 30 y 60 minutos se analizaron para compuesto parental mediante CL-EM/EM de alta resolución. Las fracciones S9 pulmonares de origen humano (lote n.º 1410245) se adquirieron de XenoTech LLC (Lenexa, KS). Se adquirió NADPH (Sigma Aldrich, N1630) y 5-fosfosulfato de 3-fosfoadenosina (PAPS, por sus siglas en inglés) (Sigma Aldrich, A1651) de Sigma Aldrich (St. Louis, MO). Se obtuvo acetonitrilo y agua de VWR (Radnor, PA) y eran de grado HPLC o superior. El raloxifeno y el ácido fórmico se obtuvieron de Sigma Aldrich (St. Louis, MO). Las incubaciones de S9 pulmonar se llevaron a cabo en un baño de agua a 37 ºC en una placa de polipropileno de 96 pocillos. Las soluciones de S9 pulmonar consistían en fosfato potásico 100 mM tamponado a pH 7,4 (BD Biosciences, Woburn, MA) complementado con NADPH 1 mM (Sigma-Aldrich, St. Louis, MO), cloruro de magnesio 3 mM (Sigma Aldrich, M1028) y en presencia de cofactor PAPS 100 µM (Sigma-Aldrich, St. Louis, MO), con concentraciones de proteína en la incubación final de 1 mg/ml. Se diluyeron soluciones madre en DMSO 10 mM de raloxifeno (n=1) y de compuesto (n=1) en tampón y se añadieron a las incubaciones, rindiendo concentraciones de sustrato 1 µM (DMSO al 0,001 % v/v). Los volúmenes de incubación de 400 µl y puntos temporales se obtuvieron a los 0, 15, 30 y 60 minutos mediante la extracción de una alícuota de 70 µl y la dilución en 140 µl de acetonitrilo (ácido fórmico al 0 %). Todas las muestras se centrifugaron a 2250 g durante 10 minutos a 5 ºC. Se extrajo el sobrenadante (50 µl) de las muestras centrifugadas y se diluyó en 100 µl de agua para HPLC que contenía el patrón interno. Las muestras se analizaron en un automuestreador Dionex Ultimate 3000 y se analizaron utilizando un espectrómetro de masas de alta resolución Thermo Q-Exactive (Thermo, Waltham, MA) en modo de escaneo completo junto con una columna Atlantis T33 µM - 2,1x50 mm (Waters Inc., 186003717). La fase móvil A consistía en agua ácido fórmico al 0,2 % y la fase móvil B consistía en acetonitrilo ácido fórmico al 0,2 %. Se consiguió la integración del pico utilizando el software Gubbs GMSU (Gubbs Inc., Alpharetta, GA). Para cada muestra, se calcularon las relaciones de superficie de los picos mediante la división de la superficie del pico del analito por la superficie del pico del patrón interno. Para cada incubación, se fijaron las relaciones de superficie de pico de los analitos en cada t0 a 100 % y las relaciones de superficies de pico de las muestras del minuto 60 se convirtieron a porcentajes remanentes respecto al t0 correspondiente. La determinación de la formación de metabolito sulfato se realizó cualitativamente mediante observación del pico de elución temprana en el canal del ion parental que, basándose en datos internos históricos, correspondía al metabolito O-sulfato de cada compuesto parental. Los resultados del ensayo se resumen en la Tabla 4 (n=2 réplicas).
Tabla 4: estabilidad metabólica en la fracción S9 pulmonar humana.

En comparación con sus análogos fluoro correspondientes (compuestos C-1 a C-6), los compuestos 1 a 6 dieron lugar a un metabolismo de sulfatación significativamente inferior en la fracción S9 pulmonar.
Ensayo 6: farmacocinética en plasma y pulmón de ratón.
Se determinaron las concentraciones en plasma y en pulmón de los compuestos de ensayo y las relaciones de los mismos de la manera siguiente. En el ensayo se utilizaron ratones BALB/c de Charles River Laboratories. Los compuestos de ensayo se formularon individualmente en propilenglicol al 20 % en tampón citrato de pH 4 a una concentración de 0,2 mg/ml y se introdujeron 50 µl de la solución de administración en las tráqueas de los ratones mediante aspiración oral. En diversos puntos temporales (normalmente 0,167, 2, 6 y 24 h) después de la administración, se extrajeron muestras de sangre mediante punción cardíaca y se extirparon los pulmones intactos de los ratones. Se centrifugaron muestras de sangre (centrífuga Eppendorf, 5804R) durante 4 minutos a aproximadamente 12.000 rpm a 4 ºC para recoger plasma. Los pulmones se secaron superficialmente con papel, se pesaron y se homogeneizaron a una dilución 1:3 en agua estéril. Se determinaron las concentraciones plasmática y pulmonar de compuesto de ensayo mediante análisis de CL-EM frente a patrones analíticos construidos en una curva patrón en la matriz de ensayo. Se determinó la relación de pulmón a plasma como la proporción de superficie bajo la curva (AUC, por sus siglas en inglés) pulmonar, en µg/h, a AUC plasmática, en µg·h/ml, en donde "AUC" se define convencionalmente como la superficie bajo la curva de concentración de compuesto de ensayo frente al tiempo.
Tabla 5: exposición en plasma y tejido pulmonar tras una única administración mediante aspiración oral de compuestos de ensayo.

Ensayo 7: ensayo de supervivencia de eosinófilos mediada por IL-5.
La potencia de los compuestos de ensayo para la supervivencia de los eosinófilos mediada por IL-5 puede medirse en eosinófilos humanos aislados a partir de sangre completa humana ("TodasLasCélulas"). Debido a que IL-5 señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se aislaron eosinófilos humanos a partir de sangre completa humana recién extraída (TodasLasCélulas) de donantes sanos. Se mezcló la sangre con dextrano al 4,5 % (Sigma-Aldrich) en una solución de cloruro sódico al 0,9 % (Sigma-Aldrich). Se dejó que los glóbulos rojos se sedimentasen durante 35 minutos. Se separó la capa superior rica en leucocitos y se aplicó en una capa sobre Ficoll-Paque (GE Healthcare) y se centrifugó a 600g durante 30 minutos. Se eliminaron las capas de plasma y células mononucleares antes de lisaron la capa de granulocitos para eliminar cualquier glóbulo rojo contaminante. Se purificaron adicionalmente los eosinófilos utilizando un kit de aislamiento de eosinófilos humanos (Miltenyi Biotec). Se incubó una fracción de los eosinófilos purificados con anti-CD16 FITC (Miltenyi Biotec) durante 10 minutos a 4 ºC en la oscuridad. Se analizó la pureza utilizando un citómetro de flujo LSRII (BD Biosciences).
Las células se cultivaron en un incubador a 37 ºC, humidificado y con 5 % de CO2, en RPMI 1640 (Life Technologies) complementado con suero de feto bovino inactivado por calor al 10% (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies y 1X Pen/Strep (Life Technologies). Las células se sembraron a razón de 10.000 células/pocillo en medio (50 µl). Se centrifugó la placa a 300g durante 5 minutos y se separaron los sobrenadantes. Los compuestos se diluyeron en serie en DMSO y, a continuación, se diluyeron 500 veces adicionales hasta una concentración de ensayo final 2x en medio. Los compuestos de ensayo (50 µl/pocillo) se añadieron a las células y se incubaron a 37 ºC, 5 % de CO2 durante 1 hora, seguido de la adición de IL-5 (R&D Systems; concentraciones finales: 1 ng/ml y 10 pg/ml) en medio de ensayo precalentado (50 µl) durante 72 horas.
Tras la estimulación de citoquinas, las células se centrifugaron a 300 g durante 5 minutos y se lavaron dos veces con DPBS frío (Life Technologies). Para determinar la viabilidad y la apoptosis, las células fueron incubadas con yoduro de propidio (Thermo Fisher Scientific) y APC-anexina V (BD Biosciences) y se analizaron utilizando un citómetro de flujo LSRII (BD Biosciences). Se determinaron los valores de IC50 a partir del análisis de las curvas de viabilidad de porcentaje de viabilidad celular frente a concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50).
Ensayo 8: inhibición de quimioquinas CXCL9 y CXCL10 inducidas por IFNγ e IL-27 en cultivos 3D de vías respiratorias humanas.
Pueden obtenerse cultivos de tejidos EpiAirway de Mattek (AIR-100). Los cultivos se derivan de donantes asmáticos. En un inserto de cultivo celular, se cultivaron células epiteliales traqueales/bronquiales de origen humano y se diferenciaron sobre un soporte de membrana porosa, dejando una interfaz aire-líquido con medio de cultivo caliente bajo las células y una atmósfera de ensayo gaseoso en la parte superior. Los tejidos se cultivaron en medio de mantenimiento (Mattek, AIR-100-MM) en un incubador humidificado a 37 ºC con 5 % de CO2. Pueden someterse a ensayo cuatro donantes. El día 0, los cultivos de tejidos se trataron con compuestos de ensayo a concentraciones de 10 µM, 1 µM y/o 0,1 µM. Los compuestos se diluyeron en dimetilsulfóxido (DMSO, Sigma) hasta una concentración final de 0,1 %. El DMSO a 0,1 % puede utilizarse como control de vehículo. Los compuestos de ensayo se incubaron con cultivos durante 1 hora a 37 ºC y 5 % de CO2 seguido de la adición de medio precalentado que contenía IFNγ (R&D Systems) o IL-27 (R&D Systems) a una concentración final de 100 ng/ml. Los cultivos de tejidos se mantuvieron durante 8 días. Se sustituyó el medio cada 2 días con medio fresco que contenía compuestos e IFNγ o IL-27. El día 8, se recogieron cultivos de tejidos y sobrenadantes para el análisis. Se sometieron a ensayo muestras de sobrenadante para CXCL10 (IP-10) y CXCL9 (MIG) utilizando análisis de Luminex (EMD Millipore). Los datos se expresan como % de inhibición /- desviación estándar (±STDV). Se determinó el porcentaje de inhibición como la potencia inhibidora del compuesto frente a la secreción de CXCL10 o CXCL9 inducida por IFNγ o IL-27 en comparación con células tratadas con vehículo. Los datos son de medias de 3 o 4 donantes.
Ensayo 9: ensayo celular de potencia de JAK: inhibición de IFNγ estimulada por IL-2/anti-CD3 en PBMC humanas.
La potencia de los compuestos de ensayo en la inhibición del interferón gamma (IFNγ) estimulado por interleuquina-2 (IL-2)/anti-D3 puede medirse en células mononucleares de sangre periférica (PBMC) humana aisladas a partir de
sangre completa humana (Stanford Blood Center). Debido a que IL-2 señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
(1) Se aislaron células mononucleares de sangre periférica (PBMC) humanas a partir de sangre completa humana de donantes sanos utilizando un gradiente de Ficoll. Las células se cultivaron en un incubador a 37 ºC, humidificado y con 5 % de CO2, en RPMI (Life Technologies) complementado con suero de feto bovino inactivado por calor al 10 % (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies y 1X Pen/Strep (Life Technologies). Las células se sembraron a razón de 200.000 células/pocillo en medio (50 µl) y se cultivaron durante 1 h. Los compuestos se diluyeron en serie en DMSO y, a continuación, se diluyeron 500 veces adicionales (hasta una concentración de ensayo final de 2x) en medio. Los compuestos de ensayo (100 µl/pocillo) se añadieron a las células y se incubaron a 37 ºC, 5 % de CO2 durante 1 hora, seguido de la adición de IL-2 (R&D Systems; concentraciones finales: 100 ng/ml) y anti-CD3 (BD Biosciences; concentración final: 1 µg/ml) en medio de ensayo precalentado (50 µl) durante 24 h.
(2) Tras la estimulación con citoquina, las células se centrifugaron a 500 g durante 5 min y se separaron los sobrenadantes y se congelaron a -80ºC. Para determinar la potencia de inhibición del compuesto de ensayo en respuesta a IL2-/anti-CD3, se midieron las concentraciones de IFNγ en el sobrenadante mediante ELISA (R&D Systems). Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de la concentración de IFNγ frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50).
Ensayo 10: ensayo celular de potencia de JAK: inhibición de pSTATS estimulada por IL-2 en linfocitos T CD4+.
La potencia de los compuestos de ensayo en la inhibición de la fosforilación de STATS estimulada por interleuquina-2 (IL-2)/anti-CD3 puede medirse en linfocitos T positivos para CD4 (CD4+) en células mononucleares de sangre periférica (PBMC) humana aisladas a partir de sangre completa humana (Stanford Blood Center) mediante citometría de flujo. Debido a que IL-2 señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se identificaron linfocitos T CD4+ utilizando un anticuerpo anti-CD4 conjugado con ficoeritrobilina (PE) (clon RPA-T4, BD Biosciences), mientras que se utilizó un anticuerpo p-STAT5 conjugado con Alexa Fluor 647 (pY694, clon 47, BD Biosciences) para detectar la fosforilación de STATS.
(1) Se siguió el protocolo del ensayo 9, párrafo (1), con la excepción de que la estimulación de citoquina con anti-CD3 se llevó a cabo durante 30 min en lugar de 24 h.
(2) Tras la estimulación de citoquina, las células se fijaron con solución de fijación precalentada (200 µl, BD Biosciences) durante 10 min a 37 ºC, 5 % de CO2, se lavaron dos veces con tampón DPBS (1 ml, Life Technologies) y se resuspendieron en tampón Perm III helado (1000 µl, BD Biosciences) durante 30 min a 4 ºC. Las células se lavaron dos veces con FBS al 2 % en DPBS (tampón FACS) y después se resuspendieron en tampón de FACS (100 µl) que contenía anti-CD4 PE (dilución 1:50) y anti-CD3 anti-CD3Alexa Fluor 647 (dilución 1:5) durante 60 min a temperatura ambiente en la oscuridad. Tras la incubación, las células se lavaron dos veces en tampón FACS antes de analizarlas utilizando un citómetro de flujo LSRII (BD Biosciences). Para determinar la potencia de inhibición de los compuestos de ensayo en respuesta a IL-2/anti-CD3, se midió la mediana de la intensidad fluorescencia (MIF) de pSTAT5 en los linfocitos T CD4+. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de MIF frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50).
Ensayo 11: ensayo celular de potencia de JAK: inhibición de pSTAT6 estimulada por IL-4 en linfocitos T CD3+.
La potencia de los compuestos de ensayo en la inhibición de la fosforilación de STAT6 estimulada por interleuquina-4 (IL-4) puede medirse en linfocitos T positivos para CD3 (CD3+) en células mononucleares de sangre periférica (PBMC) humana aisladas a partir de sangre completa humana (Stanford Blood Center) mediante citometría de flujo. Debido a que IL-4 señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se identificaron linfocitos T CD3+ utilizando un anticuerpo anti-CD3 conjugado con ficoeritrobilina (PE) (clon UCHT1, BD Biosciences), mientras que se utilizó un anticuerpo de p-STAT6 conjugado con Alexa Fluor 647 (pY641, clon 18/P, BD Biosciences) para detectar la fosforilación de STAT6.
Se aislaron células mononucleares de sangre periférica (PBMC) humanas a partir de sangre completa humana de donantes sanos tal como en los ensayos 9 y 10. Las células se sembraron a razón de 250.000 células/pocillo en medio (200 µl), se cultivaron durante 1 h y después se resuspendieron en medio de ensayo (50 µl) (RPMI complementado con albúmina de suero bovino al 0,1 % (Sigma), Glutamax 2 mM, HEPES 25 mM y 1X Penstrep) que contenía diversas concentraciones de compuestos de ensayo. Los compuestos se diluyeron en serie en DMSO y, a continuación, se diluyeron 500 veces (hasta una concentración de ensayo final 2x) en medio de ensayo. Los compuestos de ensayo (50 µl) se incubaron con células a 37 ºC, 5% de CO2, durante 1 h, seguido de la adición de IL-4 (50 µl) (R&D Systems; concentración final: 20 ng/ml) en medio de ensayo precalentado durante 30 min. Tras la estimulación con citoquinas, las células se fijaron con solución de fijación precalentada (100 µl) (BD Biosciences) durante 10 min a 37 ºC, con 5 %
de CO2, se lavaron dos veces con tampón de FACS (1 ml) (FBS al 2 % en DPBS) y se resuspendieron en tampón Perm III helado (1000 µl) (BD Biosciences) durante 30 min a 4 ºC. Las células se lavaron dos veces con tampón de FACS y después se resuspendieron en tampón de FACS (100 µl) que contenía anti-CD3 PE (dilución 1:50) y antipSTAT6 Alexa Fluor 647 (dilución 1:5) durante 60 min a temperatura ambiente en la oscuridad. Tras la incubación, las células se lavaron dos veces en tampón FACS antes de analizarlas utilizando un citómetro de flujo LSRII (BD Biosciences).
Para determinar la potencia de inhibición del compuesto de ensayo en respuesta a IL-4, se midió la mediana de la intensidad de fluorescencia (MIF) de pSTAT6 en los linfocitos T CD3+. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de MIF frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50).
Ensayo 12: ensayo celular de potencia de JAK: inhibición de pSTAT3 estimulada por IL-6 en linfocitos T CD3+.
Puede utilizarse un protocolo análogo al del Ensayo 11 para determinar la potencia del compuesto de ensayo para la inhibición de la fosforilación de STATS estimulada por interleuquina-6 (IL-6). Se utilizó un anticuerpo anti-pSTAT3 conjugado con Alexa Fluor 647 (pY705, clon 4/P, BD Biosciences) para detectar la fosforilación de STATS.
Ensayo 13: modelo murino de inflamación eosinofílica inducida por Alternaria alternata del pulmón.
La eosinofilia de las vías respiratorias es una característica distintiva del asma humano. Alternaria alternata es un alérgeno aéreo fúngico que puede exacerbar el asma en el ser humano y que induce la inflamación eosinofílica en los pulmones de ratones (Havaux et al., Clin Exp Immunol.2005 Feb;139(2): 179-88). En ratones se ha demostrado que Alternaria activa indirectamente las células linfoides innatas de tipo 2 residentes en el tejido del pulmón, que responde (p. ej., IL-2 e IL-7) y libera citoquinas dependientes de JAK (p. ej., IL-5 e IL-13) y coordina la inflamación eosinofílica (Bartemes et al., J Immunol.2012 Feb 1;188(3):1503-13).
En el estudio se utilizaron ratones C57 macho de siete a nueve semanas de edad de Taconic. El día del estudio, los animales fueron anestesiados levemente con isoflurano y se les administró vehículo o compuesto de ensayo (0,03 a 1,0 mg/ml, 50 µl de volumen total a lo largo de varias respiraciones) mediante aspiración orofaríngea. Los animales se tumbaron de costado después de la dosis y se les realizó un seguimiento hasta la recuperación total de la anestesia antes de devolverlos a su jaula de origen. Una hora después se anestesiaron brevemente de nuevo y se les retó con vehículo o extracto de Alternaria (200 µg de extracto administrado en total; volumen total: 50 µl) mediante aspiración orofaríngea antes de realizarse un seguimiento para la recuperación de la anestesia y devolverlos a su jaula de origen. Cuarenta y ocho horas después de la administración de Alternaria, se recogió líquido de lavado broncoalveolar (LLBA) y se realizó un recuento de los eosinófilos en el LLBA utilizando el sistema de hematología Advia 120 (Siemens). La actividad en el modelo se puso de manifiesto por una reducción del nivel de eosinófilos presente en el LLBA de los animales tratados a las cuarenta y ocho horas, en comparación con animales de control retados con Alternaria y tratados con vehículo. Los datos se expresan como porcentaje de inhibición de la respuesta de eosinófilos en el LLBA de animales retados con Alternaria y tratados con vehículo. Para calcular el porcentaje de inhibición, el número de eosinófilos del LLBA para cada condición se convirtió en porcentaje respecto al nivel medio de eosinófilos del LLAB de animales retados con Alternaria y tratados con vehículo y se restó del porcentaje cien.
Ensayo 14: ensayo celular de potencia de JAK: inhibición de pSTAT1 inducida por IFNγ.
Se midió la potencia de los compuestos de ensayo en la inhibición de la fosforilación de STAT1 estimulada por interferón gamma (IFNγ) en monocitos positivos para CD14 (CD14+) derivados de sangre completa humana (Stanford Blood Center) mediante citometría de flujo. Debido a que IFNγ señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se identificaron los monocitos utilizando un anticuerpo anti-CD14 conjugado con isotiocianato de fluoresceína (FITC, por sus siglas en inglés) (clon RM052, Beckman Coulter) y se utilizó un anticuerpo anti-pSTAT1 conjugado con Alexa Fluor 647 (pY701, clon 4a, BD Biosciences) para detectar la fosforilación de STAT1.
Se aislaron células mononucleares de sangre periférica (PBMC) humanas a partir de sangre completa humana de donantes sanos utilizando un gradiente de Ficoll. Las células se cultivaron en un incubador a 37 ºC, humidificado y con 5 % de CO2, en RPMI (Life Technologies) complementado con suero de feto bovino inactivado por calor al 10 % (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies y 1X Pen/Strep (Life Technologies). Las células se sembraron a razón de 250.000 células/pocillo en medio (200 µl), se cultivaron durante 2 h y después se resuspendieron en medio de ensayo (50 µl) (RPMI complementado con albúmina de suero bovino al 0,1 % (Sigma), Glutamax 2 mM, HEPES 25 mM y 1X Penstrep) que contenía diversas concentraciones de compuesto de ensayo. El compuesto se diluyó en serie en DMSO y, a continuación, se diluyeron 1000 veces adicionales en medio para llevar la concentración de DMSO final a 0,1 %. Se incubaron las diluciones de compuesto de ensayo con células a 37 ºC y 5 % de CO2 durante 1 h, seguido de la adición de IFNγ precalentado (R&D Systems) en medio (50 µl) a una concentración final de 0,6 ng/ml durante 30 min. Tras la estimulación de citoquinas, se fijaron las células con solución de fijación precalentada (100 µl) (BD Biosciences) durante 10 min a 37 ºC, 5 % de CO2; se lavaron dos veces con
tampón de FACS (1 ml) (BSA al 1 % en PBS), se resuspendieron en anti-CD14 FITC:tampón de FACS 1:10 (100 µl) y se incubaron a 4 ºC durante 15 min. Las células se lavaron una vez y después se resuspendieron en tampón Perm Buffer III helado (BD Biosciences) (100 µl) durante 30 min a 4 ºC. Las células se lavaron dos veces con tampón de FACS y después se resuspendieron en anti-pSTAT1 Alexa Fluor 647:tampón de FACS 1:10 (100 µl) durante 30 min a TA en la oscuridad, se lavaron dos veces en tampón de FACS y se analizaron utilizando un citómetro de flujo MACSQuant (Miltenyi).
Para determinar la potencia de inhibición del compuesto de ensayo, se midió la mediana de la intensidad de fluorescencia (MIF) de pSTAT1 en los monocitos CD14+. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de MIF frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50). El compuesto 1 mostró un valor de pIC50 de 7,5 en este ensayo. El compuesto 4 mostró un valor de pIC50 de 7,3 en este ensayo.
Ensayo 15: ensayo celular de potencia de JAK: inhibición de pSTATS inducida por GM-CSF.
Se midió la potencia de los compuestos de ensayo en la inhibición de la inhibición de la fosforilación de STATS estimulada por factor estimulador de colonias de macrófagos-granulocitos (GM-CSF) en monocitos derivados de sangre completa humana (Stanford Blood Center) mediante citometría de flujo. Debido a que GM-CSF señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se identificaron los monocitos utilizando un anticuerpo anti-CD14 conjugado con isotiocianato de fluoresceína (FITC) (clon RM052, Beckman Coulter) y se utilizó un anticuerpo anti-pSTAT5 conjugado con Alexa Fluor 647 (pY694, BD Biosciences) para detectar la fosforilación de STATS.
Se aislaron células mononucleares de sangre periférica (PBMC) humanas a partir de sangre completa humana de donantes sanos utilizando un gradiente de Ficoll. Las células se cultivaron en un incubador a 37 ºC, humidificado y con 5 % de CO2, en RPMI (Life Technologies) complementado con suero de feto bovino inactivado por calor al 10 % (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies y 1X Pen/Strep (Life Technologies). Las células se sembraron a razón de 250.000 células/pocillo en medio (200 µl), se cultivaron durante 2 h y después se resuspendieron en medio de ensayo (50 µl) (RPMI complementado con albúmina de suero bovino al 0,1 % (Sigma), Glutamax 2 mM, HEPES 25 mM y 1X Penstrep) que contenía diversas concentraciones de compuesto de ensayo. El compuesto se diluyó en serie en DMSO y, a continuación, se diluyeron 1000 veces adicionales en medio para llevar la concentración de DMSO final a 0,1 %. Se incubaron las diluciones de compuesto de ensayo con células a 37 ºC y 5 % de CO2 durante 1 h, seguido de la adición de IFNγ precalentado (R&D Systems) en medio (50 µl) a una concentración final de 0,3 ng/ml durante 15 min. Tras la estimulación de citoquinas, se fijaron las células con solución de fijación precalentada (100 µl) (BD Biosciences) durante 10 min a 37 ºC, 5 % de CO2; se lavaron dos veces con tampón de FACS (1 ml) (BSA al 1 % en PBS), se resuspendieron en anti-CD14 FITC:tampón de FACS 1:10 (100 µl) y se incubaron a 4 ºC durante 15 min. Las células se lavaron una vez y después se resuspendieron en tampón Perm Buffer III helado (BD Biosciences) (100 µl) durante 30 min a 4 ºC. Las células se lavaron dos veces con tampón de FACS y después se resuspendieron en anti-pSTAT1 Alexa Fluor 647:tampón de FACS 1:10 (100 µl) durante 30 min a TA en la oscuridad, se lavaron dos veces en tampón de FACS y se analizaron utilizando un citómetro de flujo MACSQuant (Miltenyi).
Para determinar la potencia de inhibición del compuesto de ensayo, se midió la mediana de la intensidad de fluorescencia (MIF) de pSTAT5 en los monocitos CD14+. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de MIF frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50). El compuesto 1 mostró un valor de pIC50 de 6,7 en este ensayo. El compuesto 4 mostró un valor de pIC50 de 6,7 en este ensayo.
Ensayo 16: ensayo celular de potencia de JAK: inhibición de pSTAT4 inducida por IL-12.
Se midió la potencia de los compuestos de ensayo en la inhibición de la fosforilación de STAT4 estimulada por interleuquina-12 (IL-12) en linfocitos T positivos para CD3 (CD3+) derivados de sangre completa humana (Stanford Blood Center) mediante citometría de flujo. Debido a que IL-12 señaliza a través de JAK, el presente ensayo proporciona una medida de la potencia celular de JAK.
Se identificaron linfocitos T CD3+ utilizando un anticuerpo anti-CD3 conjugado con ficoeritrina (PE) (clon UCHT1, BD Biosciences), mientras que se utilizó un anticuerpo anti-STAT4 conjugado con Alexa Fluor 647 (clon 38/p-Stat4, BD Biosciences) para detectar la fosforilación de STAT4.
Se aislaron células mononucleares de sangre periférica (PBMC) humanas a partir de sangre completa humana de donantes sanos utilizando un gradiente de Ficoll. Las células se cultivaron en RPMI (Life Technologies) complementado con suero de feto bovino al 10 % (FBS, Life Technologies), Glutamax 2 mM (Life Technologies), HEPES 25 mM (Life Technologies), 1X Pen/Strep (Life Technologies), anticuerpo anti-CD3 purificado unido a placa (5 µg/ml, clon UCHT1, BD Biosciences) y anticuerpo anti-CD28 soluble (1 µg/ml, clon CD28.2, BD Biosciences) durante 3 días en un incubador humidificado a 37 ºC con 5 % de CO2. Se recolectaron las células, se lavaron con medio y
después se resuspendieron en medio que contenía interleuquina-2 (IL-2, 10 ng/ml, R&D Systems). Las células se cultivaron durante 3 días en un incubador humidificado a 37 ºC con 5 % de CO2. Se recolectaron las células, se lavaron con RPMI y se sembraron a razón de 250.000 células/pocillo en medio (200 µl), se cultivaron durante 2 h y se resuspendieron en medio de ensayo (50 µl) (RPMI complementado con albúmina de suero bovino al 0,1 % (Sigma), Glutamax 2 mM, HEPES 25 mM y 1X Penstrep) que contenía diversas concentraciones de los compuestos de ensayo. El compuesto se diluyó en serie en DMSO y, a continuación, se diluyeron 1000 veces adicionales en medio para llevar la concentración de DMSO final a 0,1 %. Las diluciones de los compuestos de ensayo se incubaron con células a 37 ºC, 5% de CO2, durante 1 h, seguido de la adición de IL-12 (R&D Systems) en medio (50 µl) a una concentración final de 10 ng/ml durante 30 min. Tras la estimulación con citoquinas, las células se fijaron con solución de fijación precalentada (100 µl) (BD Biosciences) durante 10 min a 37 ºC, con 5 % de CO2; se lavaron dos veces con tampón de FACS (1 ml) (BSA al 1% en PBS) y se resuspendieron en tampón Perm III helado (1000 µl) (BD Biosciences) durante 30 min a 4 ºC. Las células se lavaron dos veces con tampón de FACS y después se resuspendieron en tampón de FACS (100 µl) que contenía anti-CD3 PE (dilución 1:50) y anti-pSTAT4 Alexa Fluor 647 (dilución 1: 10) durante 45 min a temperatura ambiente en la oscuridad. Tras la incubación, las células se lavaron dos veces en tampón de FACS antes de analizarlas utilizando un citómetro de flujo MACSQuant (Miltenyi). Para determinar la potencia de inhibición del compuesto de ensayo, se midió la mediana de la intensidad de fluorescencia (MIF) de pSTAT4 en los linfocitos CD3+. Se determinaron los valores de IC50 a partir del análisis de las curvas de inhibición de MIF frente a la concentración de compuesto. Los datos se expresan como valores de pIC50 (logaritmo decadal negativo de IC50). El compuesto 1 mostró un valor de pIC50 de 6,2 en este ensayo. El compuesto 4 mostró un valor de pIC50 de 6,0 en este ensayo.
Estructura cristalina.
Se obtuvo una estructura cocristalina del compuesto D-1 unido a JAK1 humano a una resolución de 2,28 Å. Se observó que el ligando se unía al sitio de unión de ATP. Se identificaron siete interacciones de enlace de hidrógeno específicas basándose en la distancia de 3,5 Å o menos entre los átomos donante y aceptor. Cabe destacar que se identificó una interacción de enlace de hidrógeno entre el carbonilo de la amida exocíclica del compuesto D-1 y la cadena lateral de Arg879 de la JAK1. Puede esperarse una interacción similar para los compuestos de la invención. En estudios de modelado anteriores, dicha interacción se ha propuesto como una manera de proporcionar selectividad a JAK1 sobre otras tirosina-quinasas, ya que quinasas de otro modo estrechamente relacionadas (p. ej., TRKA, VEGFR y ABL1) no poseen un residuo de arginina en la posición equivalente. Los resultados observados de interacción de enlace de hidrógeno en la estructura cristalina y la selectividad mejorada del quinoma en comparación con la serie que no posee la amida exocíclica valida esta hipótesis de diseño.
Claims (22)
1. Un compuesto de fórmula (I):

en la que:
A es un anillo heterociclilo monocíclico de 6 a 7 elementos que contiene dos átomos de nitrógeno, en el que A está conectada al grupo carbonilo en (I) mediante un átomo de nitrógeno; A está sustituida con 1 a 3 grupos R2 y, opcionalmente, dos grupos R2 forman un anillo puenteado con -(CH2)- con A; o A es pirrolidinilo, en el que el pirrolidinilo está conectado al grupo carbonilo en (I) mediante un átomo de nitrógeno y en el que el pirrolidinilo está sustituido con NR3R4;
R1 es alquilo C1-3;
cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH;
R3 es alquilo C1-3;
y R4 es alquilo C1-3;
o una sal farmacéuticamente aceptable del mismo.
2. El compuesto según la reivindicación 1, o una sal farmacéuticamente aceptable del mismo, que presenta la fórmula (I'):

o un compuesto de fórmula (II):

en la que:
R1 es alquilo C1-3;
A1 se selecciona del grupo que consiste en:

en el que R2 es alquilo C1-3 sustituido opcionalmente con -OH,
o
un compuesto de fórmula (III):

en la que:
R1 es alquilo C1-3,
A2 se selecciona del grupo que consiste en:

en la que cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH; R3 es alquilo C1-3 y R4 es alquilo C1-3,
o
un compuesto de fórmula (IV):

en la que:
R1 es alquilo C1-3,
A3 se selecciona del grupo que consiste en:

en la que cada R2 es, independientemente, alquilo C1-3 sustituido opcionalmente con -OH, R3 es alquilo C1-3 y R4 es alquilo C1-3.
3. El compuesto según la reivindicación 1 o 2, o una sal farmacéuticamente aceptable del mismo, en el que R1 se selecciona del grupo que consiste en metilo, propilo e isopropilo, por ejemplo, metilo y propilo.
4. El compuesto según cualquiera de las reivindicaciones 1 a 3, o una sal farmacéuticamente aceptable del mismo, en el que A se selecciona del grupo que consiste en piperazinilo, 2,5-diazabiciclo[2.2.1]heptanilo y 1,4-diazacicloheptanilo, cada uno de los cuales está sustituido con 1 o 2 grupos R2, en el que cada R2 es,
independientemente, alquilo C1-3 sustituido opcionalmente con -OH o A es pirrolidinilo sustituido con NR3R4, en el que R3 es alquilo C1-3 y R4 es alquilo C1-3.
5. El compuesto según cualquiera de las reivindicaciones 1 a 4, o una sal farmacéuticamente aceptable del mismo, en el que A se selecciona del grupo que consiste en:

en la que R2 es alquilo C1-3 sustituido opcionalmente con -OH, R3 es alquilo C1-3 y R4 es alquilo C1-3.
6. Compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
7. El compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
8. El compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
9. El compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
10. El compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
11. El compuesto según la reivindicación 1 que presenta la fórmula:

o una sal farmacéuticamente aceptable del mismo.
12. Una composición farmacéutica que comprende un compuesto según cualquiera de las reivindicaciones 1 a 11 o una sal farmacéuticamente aceptable del mismo, y un portador farmacéuticamente aceptable del mismo.
13. Un compuesto, o sal farmacéuticamente aceptable del mismo, según cualquiera de las reivindicaciones 1 a 11, para uso en el tratamiento de una enfermedad respiratoria en un mamífero.
14. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 13, en el que la enfermedad respiratoria se selecciona del grupo que consiste en asma, enfermedad pulmonar obstructiva crónica, fibrosis quística, neumonitis, fibrosis pulmonar idiopática, lesión pulmonar aguda, síndrome de distrés respiratorio agudo, bronquitis, enfisema, bronquiolitis obliterante, sarcoidosis, una enfermedad eosinofílica, una infección helmíntica, hipertensión arterial pulmonar, linfangioleiomiomatosis, broncoectasia, una enfermedad pulmonar infiltrante, neumonitis inducida farmacológicamente, neumonitis inducida por hongo, aspergilosis broncopulmonar alérgica, neumonitis por hipersensibilidad, granulomatosis eosinofílica con poliangiitis, neumonía eosinofílica aguda idiopática, neumonía eosinofílica crónica idiopática, síndrome hipereosinofílico, síndrome de Löffler, bronquiolitis obliterante con neumonía organizada, enfermedad del injerto pulmonar contra el huésped y neumonitis inducida por inhibidor de punto de control inmunitario.
15. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 13, en el que la enfermedad respiratoria es asma o enfermedad pulmonar obstructiva crónica.
16. Un compuesto, o una sal farmacéuticamente aceptable del mismo, según cualquiera de las reivindicaciones 1 a 11, para la utilización en el tratamiento del rechazo del trasplante pulmonar en un mamífero.
17. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 16, en el que el rechazo del trasplante pulmonar se selecciona del grupo que consiste en disfunción primaria del injerto, neumonía organizada, rechazo agudo, bronquiolitis linfocítica y disfunción crónica del aloinjerto pulmonar.
18. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 16, en el que el rechazo del trasplante pulmonar es rechazo agudo del trasplante pulmonar.
19. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 16, en el que el rechazo del trasplante pulmonar es disfunción crónica del aloinjerto pulmonar.
20. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 16, en el que el rechazo del trasplante pulmonar se selecciona del grupo que consiste en bronquiolitis obliterante, disfunción crónica restrictiva del aloinjerto pulmonar y disfunción neutrofílica del aloinjerto.
21. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 14, en el que la enfermedad respiratoria es lesión pulmonar aguda.
22. El compuesto, o una sal farmacéuticamente aceptable del mismo, para la utilización según la reivindicación 14, en el que la enfermedad respiratoria es el síndrome agudo de distrés respiratorio.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862726583P | 2018-09-04 | 2018-09-04 | |
| PCT/US2019/049342 WO2020051139A1 (en) | 2018-09-04 | 2019-09-03 | 5 to 7 membered heterocyclic amides as jak inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2955717T3 true ES2955717T3 (es) | 2023-12-05 |
Family
ID=68051893
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19773584T Active ES2955717T3 (es) | 2018-09-04 | 2019-09-03 | Amidas heterocíclicas de 5 a 7 elementos como inhibidores de JAK |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US10836763B2 (es) |
| EP (1) | EP3837010B1 (es) |
| JP (1) | JP7383696B2 (es) |
| KR (1) | KR20210056380A (es) |
| CN (1) | CN112703037B (es) |
| AR (1) | AR116114A1 (es) |
| AU (1) | AU2019335200A1 (es) |
| BR (1) | BR112021004063A2 (es) |
| CL (1) | CL2021000515A1 (es) |
| CO (1) | CO2021002976A2 (es) |
| EA (1) | EA202190686A1 (es) |
| ES (1) | ES2955717T3 (es) |
| IL (1) | IL281150B2 (es) |
| MX (1) | MX2021002484A (es) |
| PH (1) | PH12021550324A1 (es) |
| SG (1) | SG11202101751XA (es) |
| TW (1) | TWI793365B (es) |
| UA (1) | UA127328C2 (es) |
| WO (1) | WO2020051139A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016004079A1 (en) | 2014-06-30 | 2016-01-07 | Black & Decker Inc. | Battery pack for a cordless power tools |
| CN108349972B (zh) | 2015-11-03 | 2021-06-08 | 施万生物制药研发Ip有限责任公司 | 用于治疗呼吸疾病的jak激酶抑制剂化合物 |
| CA2997772A1 (en) | 2017-03-09 | 2018-09-09 | Theravance Biopharma R&D Ip, Llc | Fused imidazo-piperidine jak inhibitors |
| JP2021535176A (ja) | 2018-09-04 | 2021-12-16 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | Jak阻害剤およびその中間体を調製するためのプロセス |
| IL281146B2 (en) | 2018-09-04 | 2023-12-01 | Theravance Biopharma R& D Ip Llc | Dimethyl amino aztidine amides as JAK inhibitors |
| AU2019335200A1 (en) * | 2018-09-04 | 2021-03-11 | Theravance Biopharma R&D Ip, Llc | 5 to 7 membered heterocyclic amides as JAK inhibitors |
| TW202144343A (zh) | 2020-03-02 | 2021-12-01 | 美商施萬生物製藥研發 Ip有限責任公司 | Jak抑制劑化合物之結晶水合物 |
| WO2022081872A1 (en) * | 2020-10-16 | 2022-04-21 | Gb008, Inc. | Janus kinase inhibitors |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI262914B (en) | 1999-07-02 | 2006-10-01 | Agouron Pharma | Compounds and pharmaceutical compositions for inhibiting protein kinases |
| CA2532800C (en) | 2003-07-23 | 2013-06-18 | Exelixis, Inc. | Anaplastic lymphoma kinase modulators and methods of use |
| US20050090529A1 (en) | 2003-07-31 | 2005-04-28 | Pfizer Inc | 3,5 Disubstituted indazole compounds with nitrogen-bearing 5-membered heterocycles, pharmaceutical compositions, and methods for mediating or inhibiting cell proliferation |
| US7884109B2 (en) | 2005-04-05 | 2011-02-08 | Wyeth Llc | Purine and imidazopyridine derivatives for immunosuppression |
| US8648069B2 (en) | 2007-06-08 | 2014-02-11 | Abbvie Inc. | 5-substituted indazoles as kinase inhibitors |
| EP3495369B1 (en) | 2007-06-13 | 2021-10-27 | Incyte Holdings Corporation | Use of salts of the janus kinase inhibitor (r)-3-(4-(7h-pyrrolo[2,3-d]pyrimidin-4-yl)-1h- pyrazol-1-yl)-3- cyclopentylpropanenitrile |
| RU2561104C2 (ru) | 2008-06-20 | 2015-08-20 | Дженентек, Инк. | Триазолопиридиновые соединения-ингибиторы jak и способы |
| JP2010111624A (ja) | 2008-11-06 | 2010-05-20 | Shionogi & Co Ltd | Ttk阻害作用を有するインダゾール誘導体 |
| JP5651681B2 (ja) | 2009-04-03 | 2015-01-14 | 大日本住友製薬株式会社 | 代謝型グルタミン酸受容体5介在障害の治療のための化合物、およびその使用方法 |
| DK3001903T3 (en) | 2009-12-21 | 2017-12-18 | Samumed Llc | 1H-PYRAZOLO [3,4 -?] PYRIDINES AND THERAPEUTIC APPLICATIONS THEREOF |
| US8575336B2 (en) | 2011-07-27 | 2013-11-05 | Pfizer Limited | Indazoles |
| SI3318565T1 (sl) | 2013-12-05 | 2021-07-30 | Pfizer Inc. | Pirolo(2,3-D)pirimidinil, pirolo(2,3-B)pirazinil in pirolo(2,3-D)piridinil akrilamidi |
| CN106459048A (zh) | 2014-05-14 | 2017-02-22 | 辉瑞公司 | 吡唑并吡啶类和吡唑并嘧啶类 |
| WO2016026078A1 (en) | 2014-08-19 | 2016-02-25 | Changzhou Jiekai Pharmatech Co., Ltd. | Heterocyclic compounds as erk inhibitors |
| KR101663277B1 (ko) | 2015-03-30 | 2016-10-06 | 주식회사 녹십자 | TNIK, IKKε 및 TBK1 억제제로서의 피라졸계 유도체 및 이를 포함하는 약학적 조성물 |
| EP3712152B1 (en) * | 2015-11-03 | 2021-01-13 | Topivert Pharma Limited | 4,5,6,7-tetrahydro-1h-imidazo[4,5-c]pyridine and 1,4,5,6,7,8-hexahydroimidazo[4,5-d]azepine derivatives as janus kinase inhibitors |
| DK3371185T3 (da) * | 2015-11-03 | 2020-11-30 | Topivert Pharma Ltd | 4,5,6,7-tetrahydro-1h-imidazo[4,5-c]pyridin og 1,4,5,6,7,8-hexahydroimidazo[4,5-d]azepinderivater som janus-kinase-inhibitorer |
| CN108349972B (zh) | 2015-11-03 | 2021-06-08 | 施万生物制药研发Ip有限责任公司 | 用于治疗呼吸疾病的jak激酶抑制剂化合物 |
| JP2019524716A (ja) * | 2016-07-14 | 2019-09-05 | ファイザー・インク | バニン1酵素の阻害薬としての新規のピリミジンカルボキサミド |
| CA2997772A1 (en) * | 2017-03-09 | 2018-09-09 | Theravance Biopharma R&D Ip, Llc | Fused imidazo-piperidine jak inhibitors |
| TWI808083B (zh) | 2017-05-01 | 2023-07-11 | 美商施萬生物製藥研發 Ip有限責任公司 | Jak抑制劑化合物之結晶型式 |
| AR111495A1 (es) | 2017-05-01 | 2019-07-17 | Theravance Biopharma R&D Ip Llc | Compuestos de imidazo-piperidina fusionada como inhibidores de jak |
| EP3609498A1 (en) | 2017-05-01 | 2020-02-19 | Theravance Biopharma R&D IP, LLC | Methods of treatment using a jak inhibitor compound |
| IL281146B2 (en) | 2018-09-04 | 2023-12-01 | Theravance Biopharma R& D Ip Llc | Dimethyl amino aztidine amides as JAK inhibitors |
| AU2019335200A1 (en) * | 2018-09-04 | 2021-03-11 | Theravance Biopharma R&D Ip, Llc | 5 to 7 membered heterocyclic amides as JAK inhibitors |
| JP2021535176A (ja) | 2018-09-04 | 2021-12-16 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | Jak阻害剤およびその中間体を調製するためのプロセス |
| JP2022506111A (ja) | 2018-10-29 | 2022-01-17 | セラヴァンス バイオファーマ アール&ディー アイピー, エルエルシー | Jak阻害剤としての2-アザビシクロヘキサン化合物 |
| KR20210132666A (ko) | 2019-02-25 | 2021-11-04 | 허난 메디노 파마슈티컬 테크놀로지 컴퍼니 리미티드 | Jak 억제제 화합물 및 그 사용 |
| KR20210137087A (ko) | 2019-03-05 | 2021-11-17 | 인사이트 코포레이션 | 만성 폐 동종이식 기능장애의 치료를 위한 jak1 경로 억제제 |
| CN112279848A (zh) | 2019-07-25 | 2021-01-29 | 四川海思科制药有限公司 | 一种泛JAKs抑制剂及其用途 |
-
2019
- 2019-09-03 AU AU2019335200A patent/AU2019335200A1/en not_active Abandoned
- 2019-09-03 MX MX2021002484A patent/MX2021002484A/es unknown
- 2019-09-03 UA UAA202101706A patent/UA127328C2/uk unknown
- 2019-09-03 IL IL281150A patent/IL281150B2/en unknown
- 2019-09-03 ES ES19773584T patent/ES2955717T3/es active Active
- 2019-09-03 SG SG11202101751XA patent/SG11202101751XA/en unknown
- 2019-09-03 WO PCT/US2019/049342 patent/WO2020051139A1/en active Application Filing
- 2019-09-03 JP JP2021512242A patent/JP7383696B2/ja active Active
- 2019-09-03 US US16/559,138 patent/US10836763B2/en active Active
- 2019-09-03 BR BR112021004063-3A patent/BR112021004063A2/pt unknown
- 2019-09-03 CN CN201980057601.7A patent/CN112703037B/zh active Active
- 2019-09-03 TW TW108131710A patent/TWI793365B/zh active
- 2019-09-03 AR ARP190102519A patent/AR116114A1/es unknown
- 2019-09-03 KR KR1020217009933A patent/KR20210056380A/ko not_active Application Discontinuation
- 2019-09-03 EP EP19773584.8A patent/EP3837010B1/en active Active
- 2019-09-03 EA EA202190686A patent/EA202190686A1/ru unknown
-
2020
- 2020-10-13 US US16/949,067 patent/US11713315B2/en active Active
-
2021
- 2021-02-15 PH PH12021550324A patent/PH12021550324A1/en unknown
- 2021-03-02 CL CL2021000515A patent/CL2021000515A1/es unknown
- 2021-03-04 CO CONC2021/0002976A patent/CO2021002976A2/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| US10836763B2 (en) | 2020-11-17 |
| SG11202101751XA (en) | 2021-03-30 |
| AU2019335200A1 (en) | 2021-03-11 |
| EA202190686A1 (ru) | 2021-07-23 |
| CN112703037A (zh) | 2021-04-23 |
| MX2021002484A (es) | 2021-05-12 |
| PH12021550324A1 (en) | 2021-10-04 |
| EP3837010C0 (en) | 2023-07-05 |
| CL2021000515A1 (es) | 2021-07-23 |
| IL281150A (en) | 2021-04-29 |
| US20200071325A1 (en) | 2020-03-05 |
| JP2021535174A (ja) | 2021-12-16 |
| WO2020051139A1 (en) | 2020-03-12 |
| TW202024075A (zh) | 2020-07-01 |
| IL281150B1 (en) | 2023-11-01 |
| BR112021004063A2 (pt) | 2021-05-25 |
| CO2021002976A2 (es) | 2021-05-31 |
| US11713315B2 (en) | 2023-08-01 |
| UA127328C2 (uk) | 2023-07-19 |
| JP7383696B2 (ja) | 2023-11-20 |
| EP3837010B1 (en) | 2023-07-05 |
| IL281150B2 (en) | 2024-03-01 |
| AR116114A1 (es) | 2021-03-31 |
| EP3837010A1 (en) | 2021-06-23 |
| TWI793365B (zh) | 2023-02-21 |
| CN112703037B (zh) | 2023-10-20 |
| US20210024517A1 (en) | 2021-01-28 |
| KR20210056380A (ko) | 2021-05-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2882186T3 (es) | Inhibidores de JAK que contienen una amida heterocíclica de 4 miembros | |
| ES2955717T3 (es) | Amidas heterocíclicas de 5 a 7 elementos como inhibidores de JAK | |
| US11634419B2 (en) | Dimethyl amino azetidine amides as JAK inhibitors | |
| CA3053853C (en) | Dimethyl amino azetidine amides and 5 to 7 membered heterocyclic amides as jak inhibitors | |
| EA040902B1 (ru) | 5-7-членные гетероциклические амиды в качестве ингибиторов jak |