ES2910049T3 - Moduladores heterocíclicos de la síntesis de lípidos - Google Patents
Moduladores heterocíclicos de la síntesis de lípidos Download PDFInfo
- Publication number
- ES2910049T3 ES2910049T3 ES16712623T ES16712623T ES2910049T3 ES 2910049 T3 ES2910049 T3 ES 2910049T3 ES 16712623 T ES16712623 T ES 16712623T ES 16712623 T ES16712623 T ES 16712623T ES 2910049 T3 ES2910049 T3 ES 2910049T3
- Authority
- ES
- Spain
- Prior art keywords
- agents
- therapeutic agent
- alkyl
- cancer
- fasn
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/451—Non condensed piperidines, e.g. piperocaine having a carbocyclic group directly attached to the heterocyclic ring, e.g. glutethimide, meperidine, loperamide, phencyclidine, piminodine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4525—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Abstract
Un compuesto seleccionado entre **(Ver fórmula)** y **fórmula)** o una sal farmacéuticamente aceptable de los mismos, para su uso en el tratamiento de un cáncer, en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de célula no pequeña (NSCLC) y tumores colorrectales con mutación KRAS.
Description
DESCRIPCIÓN
Moduladores heterocíclicos de la síntesis de lípidos
Campo
La presente divulgación se refiere en general a moduladores heterocíclicos de la síntesis de lípidos y procedimientos de uso de los mismos. Los presentes moduladores heterocíclicos de la síntesis de lípidos pueden utilizarse para el tratamiento del carcinoma de pulmón de células no pequeñas y de los tumores colorrectales con mutación KRAS.
Antecedentes
Los enfoques terapéuticos dominantes que se emplean actualmente para tratar el cáncer incluyen la extirpación quirúrgica de los tumores primarios, la irradiación del tumor y la aplicación parenteral de agentes citotóxicos antimitóticos. Por desgracia, sólo una parte relativamente pequeña de los pacientes con cáncer tiene tumores que son "adictos" a una vía específica y, por tanto, pueden ser tratados con los últimos agentes dirigidos. El continuo predominio de estas terapias establecidas desde hace tiempo se refleja en la falta de mejora de las tasas de supervivencia de la mayoría de los cánceres. Además del limitado éxito clínico, los efectos secundarios devastadores acompañan a las terapias clásicas. Tanto las terapias basadas en la radiación como las citotóxicas provocan la destrucción de las células hematopoyéticas y epiteliales intestinales que se dividen rápidamente, lo que conlleva el compromiso de la función inmunitaria, la anemia y el deterioro de la absorción de nutrientes. La intervención quirúrgica suele provocar la liberación de células tumorales a la circulación o a los sistemas linfáticos, a partir de los cuales pueden establecerse posteriormente tumores metastásicos. Es necesario mejorar los procedimientos de tratamiento del cáncer.
Los documentos WO 2012/122391 A1 y WO 2014/008197 A1 divulgan moduladores de la síntesis de ácidos grasos y su uso en el tratamiento de infecciones virales, cánceres y trastornos metabólicos.
Sumario
La invención está definida por las reivindicaciones adjuntas.
La presente divulgación aborda las deficiencias de los tratamientos contra el cáncer proporcionando novedosos moduladores heterocíclicos de la síntesis de lípidos que tienen actividades anticancerígenas mejoradas.
En diferentes aspectos, la presente invención proporciona compuestos que tienen una Fórmula seleccionada del grupo que consiste en:
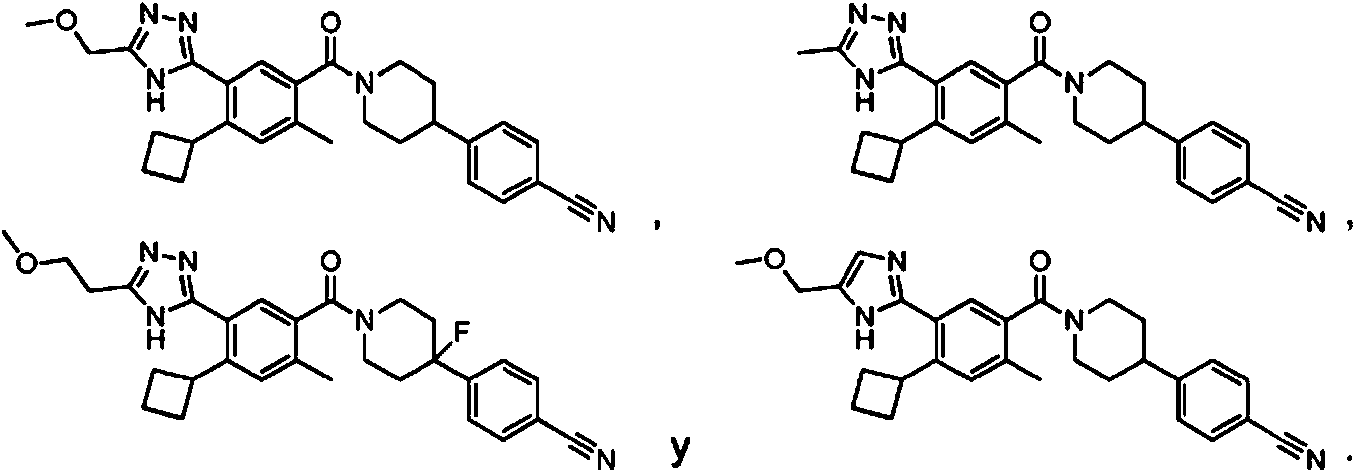
o una sal farmacéuticamente aceptable del mismo para su uso en el tratamiento del cáncer, en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de células no pequeñas (NSCLC) y tumores colorrectales con mutación KRAS.
En otros aspectos, la invención proporciona un compuesto que tiene una Fórmula seleccionada del grupo que consiste en:

o una sal farmacéuticamente aceptable de los mismos, para su uso en el tratamiento del cáncer, en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de células no pequeñas (NSCLC) y tumores colorrectales con mutación KRAS.
En diferentes aspectos, la presente divulgación proporciona
(i) un primer agente terapéutico como el descrito anteriormente; y
(ii) un segundo agente terapéutico;
para su uso en el tratamiento del cáncer, en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de células no pequeñas (NSCLC) y tumores colorrectales con mutación KRAS.
Breve descripción de los dibujos
La FIG. 1 ilustra una correlación entre la inhibición del FASN y la inhibición del VHC.
Descripción detallada
La presente divulgación aborda las deficiencias en el tratamiento de condiciones caracterizadas por la desregulación de la función FASN en un sujeto, como el cáncer, proporcionando novedosos moduladores heterocíclicos de la síntesis de lípidos.
En ciertos aspectos, la presente divulgación proporciona composiciones y procedimientos para el tratamiento del cáncer, en los que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de células no pequeñas (NSCLC) y tumores colorrectales con mutación KRAS. La sintetasa de ácidos grasos es responsable de la conversión de malonil-CoA en ácidos grasos de cadena larga, que es una reacción temprana en la biosíntesis de ácidos grasos. La sintetasa de ácidos grasos se sobreexpresa en muchas células cancerosas. Sin pretender estar ligado a ninguna teoría en particular, se plantea la hipótesis de que la inhibición de la expresión de la sintetasa de ácidos grasos o la selectividad de la actividad de la sintetasa de ácidos grasos suprime la proliferación e induce la muerte celular de las células cancerosas, con poca toxicidad hacia las células normales.
Definiciones
Las fracciones químicas denominadas fracciones químicas univalentes (es decir, alquilo, arilo, etc.) también abarcan fracciones multivalentes estructuralmente admisibles, como entienden los expertos en la técnica. Por ejemplo, mientras que una fracción de "alquilo" generalmente se refiere a un radical monovalente (es decir, CH3CH2-), en circunstancias apropiadas una fracción de "alquilo" también puede referirse a un radical divalente(es decir, -CH2CH2-, que es equivalente a un grupo "alquileno"). Del mismo modo, en circunstancias en las que se requiere una fracción divalente, los expertos en la técnica entenderán que el término "arilo" se refiere al correspondiente grupo arileno divalente.
Se entiende que todos los átomos tienen su número normal de valencias para la formación de enlaces (es decir, 4 para el carbono, 3 para el N, 2 para el O y 2, 4 o 6 para el S, dependiendo del estado de oxidación del átomo). En
ocasiones, una fracción puede definirse, por ejemplo, como (A)aB, en la que a es 0 o 1. En estos casos, cuando a es
0 la fracción es B y cuando a es 1 la fracción es AB.
Cuando un sustituyente puede variar en el número de átomos o grupos del mismo tipo (es decir, los grupos alquilo pueden ser C1, C2, C3, etc.), el número de átomos o grupos repetidos puede representarse mediante un intervalo (es decir, alquilo C1-C6 ) que incluye todos y cada uno de los números del intervalo y todos y cada uno de los subintervalos.
Por ejemplo, los alquilos C1-C3 incluyen los alquilos C1, C2 , C3 , C1-2, C1-3 y C2-3.
"Alcanoilo" se refiere a un grupo carbonilo con un grupo alquilo inferior como sustituyente.
"Alquilamino" se refiere a un grupo amino sustituido por un grupo alquilo.
"Alcoxi" se refiere a un átomo de O sustituido por un grupo alquilo como se define en el presente documento, por ejemplo, metoxi [-OCH3, un alcoxi C1]. El término "alcoxi C1-6" abarca alcoxi C1, alcoxi C2, alcoxi C3 , alc C6 y cualquier subintervalo de los mismos.
"Alcoxicarbonilo" se refiere a un grupo carbonilo con un grupo alcoxi como sustituyente.
"Alquilo", "alquenilo" y "alquinilo" se refieren a grupos alifáticos de cadena recta y ramificada opcionalmente sustituidos, que tienen de 1 a 30 átomos de carbono, o preferentemente de 1 a 15 átomos de carbono, o más preferentemente de
1 a 6 átomos de carbono. Los ejemplos de grupos alquilo incluyen, sin limitación, metilo, etilo, propilo, isopropilo, butilo, tert-butilo, isobutilo, pentilo, hexilo, vinilo, alilo, isobutenilo, etinilo y propinilo. El término "heteroalquilo", tal como se utiliza en el presente documento, contempla un alquilo con uno o más heteroátomos.
"Alquileno" se refiere a un radical divalente opcionalmente sustituido que es un fragmento de hidrocarburo ramificado o no ramificado, que contiene el número especificado de átomos de carbono y que tiene dos puntos de unión. Un ejemplo es el propileno [-CH2CH2CH2-, un alquileno C3].
"Amino" se refiere al grupo -NH2.
"Arilo" se refiere a los grupos aromáticos opcionalmente sustituidos, que tienen al menos un anillo con un sistema de electrones pi conjugado e incluye grupos arilo carbocíclicos y biarilos, todos los cuales pueden estar opcionalmente sustituidos. Los grupos fenilo y naftilo son grupos arilo carbocíclicos preferidos.
"Aralquilo" o "arilalquilo" se refieren a grupos arilo sustituidos con alquilo. Los ejemplos de grupos aralquilo incluyen butilfenilo, propilfenilo, etilfenilo, metilfenilo, 3,5-dimetilfenilo, tert-butilfenilo.
El "carbamoil", tal como se utiliza en el presente documento, contempla un grupo de la estructura
o
--------C------ NRn2
donde en RN se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C1 a C12, heteroalquilo de C1 a C12, alquenilo, alquinilo, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoilo, carbamoilo, sulfonilo, sulfonato y sulfonamida.
"Carbonilo" se refiere a un grupo de la estructura

"Cicloalquilo" se refiere a un anillo opcionalmente sustituido, que puede ser saturado o insaturado y monocíclico, bicíclico o tricíclico formado totalmente por átomos de carbono. Un ejemplo de grupo cicloalquilo es el grupo ciclopentenilo (C5H7-), que es un grupo cicloalquilo insaturado de cinco carbonos (C5).
"Heterociclo" se refiere a un sistema de anillos de cicloalquilo de 5 a 7 miembros opcionalmente sustituido, que contiene 1, 2 o 3 heteroátomos, que pueden ser iguales o diferentes, seleccionados entre N, O o S, y que opcionalmente contiene un doble enlace.
"Halógeno" se refiere a un radical de átomo de cloro, bromo, flúor o yodo. El término "halógeno" también contempla los términos "halo" o "haluro"
"Heteroátomo" se refiere a un átomo que no es de carbono, donde boro, nitrógeno, oxígeno, azufre y fósforo son heteroátomos preferidos, siendo nitrógeno, oxígeno y azufre heteroátomos particularmente preferidos en los compuestos de la presente divulgación.
"HeteroarNo" se refiere a grupos arilo opcionalmente sustituidos que tienen de 1 a 9 átomos de carbono y el resto de los átomos son heteroátomos, e incluye aquellos sistemas heterocíclicos descritos en "Handbook of Chemistry and Physics", 49a edición, 1968, R. C. Weast, editor; The Chemical Rubber Co., Cleveland, Ohio. Véase en particular la sección C, Rules for Naming Organic Compounds, B. Fundamental Heterocyclic Systems. Los heteroarilos adecuados incluyen tienilo, pirrilo, furilo, piridilo, pirimidilo, pirazinilo, pirazolilo, oxazolilo, isoxazolilo, imidazolilo, tiazolilo, piranilo, tetrazolilo, pirrolilo, pirrolinilo, piridazinilo, triazolilo, indolilo, isoindolilo, indolizinilo, benzimidazolilo, quinolilo, isoquinolilo, indazolilo, benzotriazolilo, tetrazolopiridazinilo, oxadiazolilo, benzoxazolilo, benzoxadiazolilo, tiadiazolilo, benzotiazolilo, benzotiadiazolilo, y similares.
Una fracción "opcionalmente sustituida" puede estar sustituida con uno a cuatro, o preferentemente con uno a tres, o más preferentemente con uno o dos sustituyentes que no son hidrógeno. A menos que se especifique de otro modo, cuando el sustituyente está en un carbono, se selecciona del grupo formado por -OH, -CN, -NO2, halógeno, alquilo C1 a C12, heteroalquilo C1 a C12, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoilo, carbamoilo, sulfonilo sustituido, sulfonato, sulfonamida y amino, ninguno de los cuales está sustituido adicionalmente. A menos que se especifique de otro modo, cuando el sustituyente está en un nitrógeno, se selecciona del grupo que consiste en alquilo C1 a C12, heteroalquilo de C1 a C12, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoilo, carbamoilo, sulfonilo, sulfonato y sulfonamida, ninguno de los cuales está sustituido adicionalmente.
El término "sulfonamida", tal como se utiliza en el presente documento, contempla un grupo que tiene la estructura
o
---s— nrn2
o
en la que RN se selecciona del grupo que consiste en hidrógeno, -OH, alquilo C1 a C12, heteroalquilo de C1 a C12, alquenilo, alquinilo, cicloalquilo, heterociclo, arilo, heteroarilo, aralquilo, alcoxi, alcoxicarbonilo, alcanoilo, carbamoilo, sulfonilo sustituido, sulfonato y sulfonamida.
El término "sulfonato", tal y como se utiliza en el presente documento, contempla un grupo que tiene la estructura
o
--- s— o Rs
o
en la que Rs se selecciona del grupo que consiste en hidrógeno, alquilo C1-C10, alquenilo C2-C10, alquinilo C2-C10, alcanoilo C1-C10 o alcoxicarbonilo C1-C10.
"Sulfonilo", tal como se utiliza en el presente documento, solo o como parte de otro grupo, se refiere a un grupo SO2. La fracción SO2 está opcionalmente sustituida.
Los compuestos de la presente divulgación pueden existir como estereoisómeros, en los que están presentes centros asimétricos o quirales. Los estereoisómeros se designan como (R) o (S), dependiendo de la configuración de los sustituyentes alrededor del átomo de carbono quiral. Los términos (R) y (S) utilizados en el presente documento son configuraciones tal como se definen en las Recomendaciones de la IUpAC de 1974 para la Sección E, Fundamental Stereochemistry, Pure Appl. Chem. (1976), 45: 13-30. La presente divulgación contempla diferentes estereoisómeros y mezclas de los mismos y se incluyen específicamente dentro del alcance de la presente divulgación. Los estereoisómeros incluyen enantiómeros, diastereoisómeros y mezclas de enantiómeros o diastereoisómeros. Los estereoisómeros individuales de los compuestos de la presente divulgación pueden prepararse sintéticamente a partir de materiales de partida comercialmente disponibles que contengan centros asimétricos o quirales, o mediante la preparación de mezclas racémicas seguidas de una resolución bien conocida por aquellos con conocimientos ordinarios en la técnica. Estos procedimientos de resolución se ejemplifican mediante (1) la unión de una mezcla de enantiómeros a un auxiliar quiral, la separación de la mezcla resultante de diastereoisómeros por recristalización o cromatografía y la liberación del producto ópticamente puro del auxiliar o (2) la separación directa de la mezcla de enantiómeros ópticos en columnas cromatográficas quirales.
Además, las fracciones divulgadas en el presente documento que existen en múltiples formas tautoméricas incluyen todas tales formas comprendidas en una estructura tautomérica determinada.
Los átomos individuales en los compuestos divulgados pueden ser cualquier isótopo de ese elemento. Por ejemplo, el hidrógeno puede estar en forma de deuterio.
"Farmacéuticamente aceptable" significa que ha sido aprobado o puede ser aprobado por una agencia reguladora del
gobierno federal o estatal o que figura en la Farmacopea de los Estados Unidos o en otra farmacopea generalmente reconocida para su uso en animales, y más particularmente en humanos. Puede ser un material que no sea biológicamente o de otro modo indeseable, es decir, el material puede ser administrado a un individuo sin causar ningún efecto biológico indeseable o interactuar de manera perjudicial con cualquiera de los componentes de la composición en la que está contenido.
El término "sal farmacéuticamente aceptable" de un compuesto significa una sal que es farmacéuticamente aceptable y que posee la actividad farmacológica deseada del compuesto original. Dichas sales incluyen, por ejemplo, sales de adición de ácidos y sales de adición de bases.
Las "sales de adición de ácido" de acuerdo con la presente divulgación, se forman con ácidos inorgánicos como el ácido clorhídrico, el ácido bromhídrico, el ácido sulfúrico, el ácido nítrico, el ácido fosfórico y similares; o formados con ácidos orgánicos como el ácido acético, el ácido propiónico, el ácido hexanoico, el ácido ciclopentanopropiónico, el ácido glicólico, el ácido pirúvico, el ácido láctico, el ácido malónico, el ácido succínico, el ácido málico, el ácido fumárico, el ácido tartárico, el ácido cítrico, ácido benzoico, ácido 3(4-hidroxibenzoil)benzoico, ácido cinámico, ácido mandélico, ácido metanosulfónico, ácido etanosulfónico, ácido 1,2-etanodisulfónico, ácido 2-hidroxietanosulfónico, ácido bencenosulfónico, ácido 2-naftalenosulfónico, ácido 4-metilbiciclo-[2.2.2]oct-2-eno-1-carboxílico, ácido glucoheptónico, ácido 4,4'-metilenbis-(3-hidroxi-2-eno-1-carboxílico), ácido 3-fenilpropiónico, ácido trimetilacético, ácido butilacético terciario, ácido lauril sulfúrico, ácido glucónico, ácido glutámico, ácido hidroxinaftóico, ácido salicílico, ácido esteárico, ácido mucónico y similares.
Las "sales de adición de base" de acuerdo con la presente divulgación se forman cuando un protón ácido presente en el compuesto original se sustituye por un ion metálico, es decir, un ion de metal alcalino, un ion alcalinotérreo o un ion de aluminio, o se coordina con una base orgánica. Las bases orgánicas aceptables incluyen la etanolamina, dietanolamina, trietanolamina, trometamina, N-metilglucamina y similares. Las bases inorgánicas aceptables incluyen hidróxido de aluminio, hidróxido de calcio, hidróxido de potasio, carbonato de sodio, hidróxido de sodio y similares. Debe entenderse que una referencia a una sal farmacéuticamente aceptable incluye las formas de adición de disolvente o las formas cristalinas de las mismas, en particular los solvatos o polimorfos. Los solvatos contienen cantidades estequiométricas o no estequiométricas de un disolvente, y suelen formarse durante el proceso de cristalización. Los hidratos se forman cuando el disolvente es el agua, o los alcoholatos cuando el disolvente es el alcohol. Los polimorfos incluyen las diferentes disposiciones de empaquetamiento cristalino de la misma composición elemental de un compuesto. Los polimorfos suelen tener diferentes patrones de difracción de rayos X, espectros infrarrojos, puntos de fusión, densidad, dureza, forma del cristal, propiedades ópticas y eléctricas, estabilidad y solubilidad. Varios factores, como el disolvente de recristalización, la velocidad de cristalización y la temperatura de almacenamiento, pueden causar que predomine una forma de cristal individual.
El término "tratar" incluye la administración de los compuestos o agentes de la presente invención a un sujeto para prevenir o retrasar, aliviar o detener o inhibir el desarrollo de los síntomas o condiciones asociadas con los trastornos asociados a la sintetasa de ácidos grasos, es decir, el crecimiento del tumor asociado al cáncer. Un médico experto sabrá cómo utilizar procedimientos estándar para determinar si un paciente padece una enfermedad asociada a la actividad de la sintetasa de ácidos grasos, es decir examinando al paciente y determinando si padece una enfermedad de la que se sabe que está asociada a la actividad de la sintetasa de ácidos grasos o analizando los niveles de sintetasa de ácidos grasos en el plasma sanguíneo o en el tejido del individuo del que se sospecha que padece una enfermedad asociada a la sintetasa de ácidos grasos, y comparando los niveles de sintetasa de ácidos grasos en el plasma sanguíneo o en el tejido del individuo del que se sospecha que padece una enfermedad asociada a la sintetasa de ácidos grasos en el plasma sanguíneo o en el tejido de un individuo sano. El aumento de los niveles de securina es indicativo de enfermedad. En consecuencia, la presente invención proporciona, entre otras cosas, procedimientos para administrar un compuesto de la presente invención a un sujeto, y determinar la actividad de la sintetasa de ácidos grasos en el sujeto. La actividad de la sintetasa de ácidos grasos en el sujeto puede determinarse antes y/o después de la administración del compuesto.
Una "cantidad terapéuticamente eficaz" o "cantidad farmacéuticamente eficaz" significa la cantidad que, cuando se administra a un sujeto, produce los efectos para los que se administra. Por ejemplo, una "cantidad terapéuticamente eficaz", cuando se administra a un sujeto para inhibir la actividad de la sintetasa de ácidos grasos, es suficiente para inhibir la actividad de la sintetasa de ácidos grasos. Una "cantidad terapéuticamente eficaz", cuando se administra a un sujeto para tratar una enfermedad, es suficiente para efectuar el tratamiento de dicha enfermedad.
Excepto cuando se indique, los términos "sujeto" o "paciente" se utilizan indistintamente y se refieren a mamíferos como pacientes humanos y primates no humanos, así como a animales de experimentación como conejos, ratas y ratones, y otros animales. Por consiguiente, el término "sujeto" o "paciente", tal como se utiliza en el presente documento, significa cualquier paciente o sujeto mamífero al que puedan administrarse los compuestos de la invención. En un aspecto ejemplar de la presente invención, para identificar a los pacientes sujetos para el tratamiento de acuerdo con los procedimientos de la invención, se emplean procedimientos de discriminación aceptados para determinar los factores de riesgo asociados con una enfermedad o condición objetivo o sospechosa, o para determinar el estado de una enfermedad o condición existente en un sujeto. Estos procedimientos de discriminación incluyen, por ejemplo, exámenes convencionales para determinar los factores de riesgo asociados a la enfermedad o afección
objetivo o que se sospecha. Estos y otros procedimientos rutinarios permiten al médico clínico seleccionar a los pacientes que necesitan una terapia, usando los procedimientos y formulaciones de la presente invención.
Los detalles de la invención se exponen en la descripción que se acompaña a continuación. Aunque en la práctica o en las pruebas de la presente invención pueden utilizarse procedimientos y materiales similares o equivalentes a los descritos en el presente documento, a continuación se describen procedimientos y materiales ilustrativos. Otras características, objetos y ventajas de la invención serán evidentes a partir de la descripción y de las reivindicaciones. En la especificación y en las reivindicaciones adjuntas, las formas singulares también incluyen el plural, a menos que el contexto lo dicte claramente de otro modo. A menos que se definan de otro modo, todos los términos técnicos y científicos utilizados en el presente documento tienen el mismo significado que se entiende comúnmente por una persona con conocimientos ordinarios en la técnica a la que pertenece esta invención.
Por conveniencia, algunos términos empleados en la especificación, los ejemplos y las reivindicaciones se recogen aquí. A menos que se definan de otra manera, todos los términos técnicos y científicos utilizados en esta divulgación tienen el mismo significado que comúnmente entiende una persona con destreza ordinaria en la técnica a la que pertenece esta divulgación. La definición inicial proporcionada para un grupo o término proporcionado en esta divulgación, aplica a ese grupo o término a lo largo de la presente divulgación individualmente, o como parte de otro grupo, a menos que se indique de otro modo.
Moduladores de la vía FASN
Un aspecto de la presente divulgación incluye un procedimiento de tratamiento del cáncer mediante el contacto de una célula con un agente que modula la vía de síntesis de los ácidos grasos. Este procedimiento de tratamiento del cáncer puede realizarse in vitro, poniendo en contacto las células cancerosas con un agente que modula la vía de síntesis de los ácidos grasos, o in vivo administrando un agente que modula la vía de síntesis de los ácidos grasos, a un sujeto que tiene cáncer. En un aspecto, un agente puede ser un inhibidor de la vía de síntesis de los ácidos grasos.
A continuación se describen ejemplos de inhibidores de la vía de síntesis de ácidos grasos que pueden utilizarse en los procedimientos y composiciones de la presente divulgación.
En algunos aspectos, los compuestos tienen una Fórmula seleccionada del grupo que consiste en


Síntesis de compuestos
También se describen en el presente documento procedimientos para sintetizar los compuestos de la presente divulgación. Los compuestos de la presente divulgación pueden sintetizarse como se indica en
Esquemas 1-13 sintéticos a continuación.

R" es hidrógeno o alquilo;
Ri es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(R13)(R14),-(CH2)qC(=O)N(R13)(R14), CF3 , -OCF3 , o -S(=O)2R20;
q es 0, 1,2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6, o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6, o alquilo C1-6 ;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son cada uno independientemente hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3 , -OCF3, o -S(=O)2R20;
R13 y R14 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16), o -S(=O)2R20;
R15 y R16 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino; y
R17 es hidrógeno o alquilo.
Esquema 2

en el que:
R1 es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(Ri3)(Ri4),-(CH2}qC(=O)N(Ri3)(Ri4), CF3 , -OCF3 , o -S(=O)2R20i
q es 0, 1,2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6, o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6, o alquilo C1-6 ;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son cada uno independientemente hidrógeno, halo, ciano, alquilo C|_6, alcoxi C1-6, CF3 , -OCF3, o -S(=O)2R20;
R13 y R14 son cada uno independientemente hidrógeno, alquilo C|_6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16) o -S(=O)2R20; R15 y R16 son cada uno independientemente hidrógeno, alquilo C1.6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino;
R23 es hidrógeno, -N(R13)(R14), alquilo C1.6, alcoxi C1.6, está ausente si L1 es N, o R23 y R24 tomados junto con los átomos a los que están unidos, se unen para formar un heterociclilo, heteroarilo o cicloalquilo y R24 es hidrógeno, -N(R13)(R14), alquilo C1-6, alcoxi C1-6, -(alcoxi C1-6)(heterociclilo), heterociclilo, o R23 y R24 tomados junto con los átomos a los que están unidos se unen para formar un heterociclilo, heteroarilo o cicloalquilo.
Esquema 3

en el que:
LG es un grupo saliente;
Nu es un nucleófilo;
L2 , L3 , L4 y L4 son cada uno independientemente CH o N;
R1 es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(R13)(R14),-(CH2)qC(=O)N(R13)(R14), CF3 , -OCF3 ,
o -S(=O)2R20;
q es 0, 1,2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6, o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6, o alquilo C1-6 ;
R3 es hidrógeno, hidroxilo, halo, alquilo C1-6 o alcoxi C1.6;
R21 y R22 son cada uno independientemente hidrógeno, halo, ciano, alquilo C|.6, alcoxi C1-6, CF3 , -OCF3, o -S(=O)2R20;
R13 y R14 son cada uno independientemente hidrógeno, alquilo C|.6, cicloalquilo, arilo, heterociclilo,
heteroarilo, hidroxialquilo, alquilamino, -N(R15R16), o -S(=O)2R20;
R15 y R16 son cada uno independientemente hidrógeno, alquilo C|.6, cicloalquilo, arilo, heterociclilo,
heteroarilo, hidroxialquilo o alquilamino; y
R17 es hidrógeno o alquilo.

en el que:
Ri es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(Ri3)(Ri4), -(CH2)qC(=O)N(Ri3)(Ri4), CF3 , -OCF3, o -S(=O)2R20Í
q es 0, 1,2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6, o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6, o alquilo C1-6 ;
R3 es hidrógeno, hidroxilo, halo, alquilo C1.6 o alcoxi C1-6;
R21 y R22 son cada uno independientemente hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1.6, CF3 , -OCF3, o -S(=O)2R20;
R13 y R14 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16), o -S(=O)2R20;
R15 y R16 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino;
R17 es hidrógeno o alquilo; y
R24 es hidrógeno, -N(R13)(R14), alquilo C1.6, alcoxi C1.6, -(alcoxi C1-6)(heterociclilo), o heterociclilo.
Esquema 5

en el que:
Ri es hidrógeno, ciano, halo, alquilo C1-6, alcoxi C1-6, -C(=O)N(Ri3)(Ri4), -(CH2)qC(=O)N(Ri3)(Ri4), CF3 , -OCF3, o -S(=O)2R20Í
q es 0, 1,2, 3 o 4;
R20 es hidrógeno o alquilo C1-6, alcoxi C1.6, o -N(R13)(R14);
R2 es hidrógeno, halo, alcoxi C1-6, o alquilo C1-6 ;
R3 es hidrógeno, hidroxilo, halo, alquilo C1-6 o alcoxi C1-6;
R21 y R22 son cada uno independientemente hidrógeno, halo, ciano, alquilo C1-6, alcoxi C1-6, CF3 , -OCF3, o -S(=O)2R20;
R13 y R14 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo, alquilamino, -N(R15R16), o -S(=O)2R20;
R15 y R16 son cada uno independientemente hidrógeno, alquilo C1-6, cicloalquilo, arilo, heterociclilo, heteroarilo, hidroxialquilo o alquilamino;
R17 es hidrógeno o alquilo;
R24 es hidrógeno, -N(R13)(R14), alquilo C1-6, alcoxi C1-6, -(alcoxi C1-6)(heterociclilo), o heterociclilo;
R29 es hidrógeno, alquilo C1-6, alcoxi C1.6, hidroxialquilo, heteroarilo, heterociclilo, -N(R15R16), -C(=O)R46, o -R48C(=O)R47;
R34 es hidrógeno, alquilo C1.6, alcoxi C1.6, cicloalquilo, hidroxilo, hidroxialquilo, arilo, heterociclilo, heteroarilo, alquilamino, CF3 , -OCF3, -S(=O)2R20, o -N(R15R16); y m 0, 1, o 2.
Los esquemas 6-13 proporcionan una síntesis para compuestos ejemplares de fórmula IX en la que:
R1 es H, -CN, halógeno, alquilo C1-C4 recto o ramificado, -O-(cicloalquilo C3-C5), -O-(alquilo C1-C4 recto o ramificado), en la que:
El cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno; y
cuando R1 no es H, -CN o halógeno, está opcionalmente sustituido con uno o más halógenos;
cada R2 es independientemente hidrógeno, halógeno o alquilo C1-C4 recto o ramificado;
R3 es H, -OH, o halógeno;
R21 es H, halógeno, alquilo C1-C4 recto o ramificado, cicloalquilo C3-C5, en el que el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o de nitrógeno;
R22 es H, halógeno o alquilo C1-C2 ;
R23 es H o alquilo C1-C4 recto o ramificado; y
R24 es H, alquilo C1-C4 recto o ramificado, -(alquilo C1-C4VOH, -(alquilo C1-C4)t-ot-(cicloalquilo C3-C5), o -(alquilo C1-C4)t-O-(C1-C4 alquilo recto o ramificado ) en la que: t es 0 o 1; y
el cicloalquilo C3-C5 incluye opcionalmente un heteroátomo de oxígeno o nitrógeno.








En los Ejemplos se proporcionan procedimientos adicionales para producir compuestos particulares de acuerdo con la presente divulgación.
Muchas de estas técnicas son bien conocidas en la técnica. Sin embargo, muchas de las técnicas conocidas están elaboradas en Compendium of Organic Synthetic Methods (Vol. 1, 1971 Vol. 2, 1974 Vol. 3, 1977 Vol. 4, 1980 Vol. 5, 1984y Vol. 6, así como March en Advanced Organic Chemistry (1985); Comprehensive Organic Synthesis. Selectivity, Strategy & Efficiency in Modern Organic Chemistry. En 9 volúmenes (1993) Advanced Organic Chemistry Parte B: Reactions and Synthesis, segunda edición (1983) Advanced Organic Chemistry, Reactions, Mechanisms, and Structure, segunda edición (1977) Protecting Groups in Organic Synthesis, segunda edición; y Comprehensive Organic Transformations (1999).
Vías de infección viral
Los objetivos de las células huésped inhibidas por los presentes compuestos y procedimientos desempeñan un papel en las vías de replicación y/o infección virales. La focalización de estos objetivos de las células huésped modula las vías de replicación y/o infección de los virus. En aspectos preferidos, los objetivos identificados de células huésped con modulados directa o indirectamente utilizando las composiciones de la presente divulgación. La modulación de
dichos objetivos de las células huésped también puede realizarse focalizándose en entidades de las vías de señalización ascendentes o descendentes de los objetivos de las células huésped.
De acuerdo con la presente divulgación, la infección viral puede ser tratada focalizándose en la vía de síntesis de ácidos grasos, y en particular a la sintetasa de ácidos grasos. El HRV es representativo de los virus que pueden ser tratados de acuerdo con la presente divulgación. Al igual que otros virus, la replicación de1HRV implica seis fases: transmisión, entrada, replicación, biosíntesis, ensamblaje y salida. La entrada ocurre por endocitosis, la replicación y el ensamblaje del vRNP tienen lugar en el núcleo, y el virus brota de la membrana plasmática. En el paciente infectado, el virus se focaliza en las células epiteliales de las vías respiratorias. Los presentes compuestos y procedimientos se focalizan en y modulan al menos uno de los objetivos de las células huésped implicadas en dichas vías.
En el caso de algunos virus se ha avanzado mucho en la elucidación de los pasos que intervienen durante la infección de las células huésped. Por ejemplo, los experimentos iniciados a principios de la década de 1980 demostraron que el virus de la gripe sigue un programa de entrada endocítica por pasos, con elementos compartidos con otros virus como los alfa- y los rabdovirus (Marsh y Helenius 1989; Whittaker 2006). Los pasos incluyen: 1) Unión inicial a los receptores de glicoconjugados que contienen ácido siálico en la superficie celular; 2) señalización inducida por la partícula del virus; 3) endocitosis por mecanismos celulares dependientes e independientes de la clatrina; 4) penetración mediada por la hemaglutinina (HA) inducida por el ácido desde los endosomas tardíos; 5) descobertura de la cápside dependiente de la proteína M2 y de la matriz (M1) activada por el ácido; y, 6) transporte intracitosólico e importación nuclear de vRNPs. Estos pasos dependen de la ayuda de la célula huésped en forma de receptores de clasificación, maquinaria de formación de vesículas, regulación mediada por quinasa, acidificación de los organelos y, muy probablemente, actividades del citoesqueleto.
La adhesión de la gripe a la superficie de las células ocurre a través de la unión de la subunidad HA1 a las glicoproteínas de la superficie celular y a los glicolípidos que llevan fracciones de oligosacáridos con residuos terminales de ácido siálico (Skehel y Wiley 2000). El enlace por el que el ácido siálico está conectado al siguiente sacárido contribuye a la especificidad de la especie. Las cepas aviares, incluida la H5N1, prefieren un enlace a-(2,3) y las cepas humanas un enlace a-(2,6) (Matrosovich 2006). En las células epiteliales, la unión ocurre preferentemente a las microvellosidades de la superficie apical, y la endocitosis ocurre en base a estas extensiones (Matlin 1982). Todavía no se sabe si la unión del receptor induce señales que preparan a la célula para la invasión, pero es probable porque la activación de la proteína quinasa C y la síntesis de fosfatidilinositol-3-fosfato (PI3P) son necesarias para una entrada eficiente (Sieczkarski et al. 2003; Whittaker 2006).
La internalización endocítica ocurre unos minutos después de la unión (Matlin 1982; Yoshimura y Ohnishi 1984). En las células de cultivo de tejidos, el virus de la gripe utiliza tres tipos diferentes de procesos celulares: 1) fosas preexistentes recubiertas de clatrina, 2) fosas recubiertas de clatrina inducidas por el virus y 3) endocitosis en vesículas sin capa visible (Matlin 1982; Sieczkarski y Whittaker 2002; Rust et al. 2004). La videomicroscopía con virus fluorescentes mostró que las partículas del virus experimentaban un rápido movimiento mediado por la actina en la periferia de la célula, seguido de un transporte mediado por microtúbulos y dirigido por el extremo negativo hacia la zona perinuclear de la célula. Las imágenes de células vivas indicaron que las partículas del virus entraron primero en una subpoblación de endosomas tempranos móviles y periféricos, que las transportan hacia el interior del citoplasma antes de que tenga lugar la penetración (Lakadamyali et al. 2003; Rust et al. 2004). El proceso endocítico está regulado por quinasas de proteínas y lípidos, el proteasoma, así como por factores de clasificación dependientes de Rabs y la ubiquitina (Khor et al. 2003; Whittaker 2006).
El paso de penetración en la membrana está mediado por la activación mediada por un pH bajo de la HA trimérica y metaestable, y la conversión de esta proteína de fusión viral de tipo I a una conformación competente para la fusión de membranas (Maeda et al. 1981; White et al. 1982). Esto ocurre aproximadamente 16 minutos después de la internalización, y el umbral de pH varía entre las cepas, en el intervalo de 5,0-5,6. La membrana objetivo es la membrana limitante de los endosomas intermedios o tardíos. El mecanismo de fusión ha sido ampliamente estudiado (Kielian y Rey 2006). Además, se observó que la fusión en sí misma no parece requerir ningún componente de la célula huésped, excepto una membrana de bicapa lipídica y un sistema de acidificación funcional (Maeda et al. 1981; White et al. 1982). El paso de penetración es inhibido por agentes como las bases débiles lisosomotrópicas, los ionóforos carboxílicos y los inhibidores de la bomba de protones (Matlin 1982; Whittaker 2006).
Para permitir la importación nuclear de las vRNPs entrantes, la cápside tiene que ser desmontada. Este paso implica la acidificación del interior del virus a través de los canales M2 sensibles a la amantadina, lo que provoca la disociación de Ml de los vRNPs (Bukrinskaya et al. 1982; Martin y Helenius 1991; Pinto et al. 1992). El transporte de las vRNPs individuales a los complejos de poros nucleares y su transferencia al núcleo depende de los receptores de transporte nuclear celulares (O'Neill et al. 1995; Cros et al. 2005). La replicación de los ARN virales (síntesis de las cadenas positiva y negativa) y la transcripción ocurren en complejos estrechamente asociados a la cromatina del núcleo. Es evidente que, aunque muchos de los pasos son catalizados por la polimerasa viral, hay factores celulares implicados, incluyendo los factores activadores de la ARN polimerasa, una chaperona HSP90, hCLE y un factor UAP56 de empalme humano. La expresión de los genes virales está sujeta a un complejo control celular a nivel transcripcional, un sistema de control dependiente de las quinasas celulares (Whittaker 2006).
El ensamblaje final de una partícula de gripe ocurre durante un proceso de gemación en la membrana plasmática. En las células epiteliales, la gemación ocurre únicamente en el dominio de la membrana apical (Rodríguez-Boulan 1983). En primer lugar, las vRNPs progenitoras son transportadas dentro del nucleoplasma a la envoltura nuclear, después del núcleo al citoplasma y, finalmente, se acumulan en la periferia celular. La salida del núcleo depende de la proteína NEP y M1 viral, y de una variedad de proteínas celulares que incluyen CRM1 (un receptor de exportación nuclear), caspasas y posiblemente algunas chaperonas de proteína nuclear. La fosforilación juega un papel en la exportación nuclear regulando la síntesis de M1 y NEP, y también a través del sistema MAPK/ERK (Bui et al. 1996; Ludwig 2006). La señalización de la proteína G y de la proteína quinasa está implicada en la gemación del virus de la gripe de las células huésped infectadas (Hui E. y Nayak D, 2002).
Las tres proteínas de membrana del virus son sintetizadas, plegadas y ensambladas en oligómeros en el RE (Doms et al. 1993). Pasan a través del complejo de Golgi; soportan una maduración a través de la modificación de sus fracciones de carbohidratos y la escisión proteolítica. Tras alcanzar la membrana plasmática, se asocian con M1 y las vRNPs en un proceso de gemación que da lugar a la inclusión de las ocho vRNPs y a la exclusión de la mayoría de los componentes de la célula huésped, excepto los lípidos.
La infección de la gripe está asociada con la activación de varias cascadas de señalización, incluyendo la vía MAPK (ERK, JNK, p38 y b Mk -1/ERK5), el módulo de señalización IkB/NF-kB, la cascada Raf/MEK/ERK y la muerte celular programada (Ludwig 2006). Esto da lugar a una serie de efectos que limitan el progreso de la infección, como la activación transcripcional del IFNb, la muerte celular apoptótica y el bloqueo del escape del virus desde los endosomas tardíos (Ludwig 2006).
La mayoría de los estudios anteriores sobre las interacciones entre virus y células se realizaron en cultivos de tejidos utilizando cepas de virus adaptadas al cultivo de tejidos o a huevo. Los virus de estos ejemplos se adaptaron de tal manera que se indujeron cambios que afectaron a la unión del receptor y al tropismo (Matrosovich 2006). La infección con cepas patógenas de tipo silvestre proporciona una imagen más natural de la interacción viral con las proteínas del huésped. Se sabe que en las vías respiratorias humanas, la gripe A y B infectan principalmente a las células epiteliales no ciliadas del tracto respiratorio superior que llevan NeuSAc a-(2,6)-Gal, mientras que las cepas aviares infectan a las células epiteliales ciliadas con ácidos siálicos ligados a a-(2,3) más profundamente en las vías respiratorias (Matrosovich et al. 2004a).
Además, se ha avanzado en la elucidación de los pasos implicados durante la infección por e1HRV de las células del huésped. Se puede considerar que eventos seleccionados de la infección por rinovirus de las vías respiratorias humanas normales ocurren de forma secuencial. Se cree que los pasos iniciales de la patogénesis del rinovirus incluyen la entrada del virus a través de la nariz, el transporte mucociliar del virus a la faringe posterior y el inicio de la infección en las células epiteliales ciliadas y no ciliadas de las vías respiratorias superiores. La replicación viral alcanza su punto máximo por término medio a las 48 horas de iniciada la infección y persiste hasta 3 semanas. A la infección sigue la activación de varios mecanismos inflamatorios, que pueden incluir la liberación o generación de interleuquinas, bradiquininas, prostaglandinas y, posiblemente, histamina y la estimulación de los reflejos parasimpáticos. Se inician procesos fisiopatológicos que incluyen la vasodilatación de los vasos sanguíneos nasales, la exudación de plasma, la secreción glandular y la estimulación de las fibras nerviosas, lo que provoca dolor y desencadena los reflejos del estornudo y la tos. La enfermedad clínica resultante es una rinosinusitis, una faringitis y una bronquitis que, por término medio, dura una semana.
Se han identificado cambios en los perfiles de expresión génica durante las infecciones por rinovirus in vivo (Proud, D., et al., Am. J. Respir. Crit. Care Med. Vol 178. pp 962-968, 2008). Se obtuvieron raspados epiteliales nasales antes y durante la infección experimental por rinovirus, y se evaluó la expresión génica mediante microarreglo. La viperina se identifica como una proteína antiviral inducida por el interferón (IFN), las infecciones virales y las moléculas asociadas a los patógenos. Para evaluar adicionalmente el papel de la viperina en las infecciones por rinovirus, se utilizaron infecciones por rinovirus adquiridas de forma natural, células epiteliales humanas cultivadas y la eliminación del ARN de interferencia corta. Se midieron las puntuaciones de los síntomas y los títulos virales en los sujetos inoculados con rinovirus o con un control simulado, y se evaluaron los cambios en la expresión génica 8 y 48 horas después de la inoculación. No se observaron cambios en la expresión génica inducidos por el rinovirus, 8 horas después de la infección viral, pero 11.887 transcripciones génicas se alteraron significativamente en los raspados obtenidos 2 días después de la inoculación. Los principales grupos de genes sobrerregulados incluyen quimioquinas, moléculas de señalización, genes que responden al interferón y antivirales. La infección por rinovirus altera significativamente la expresión de muchos genes asociados a la respuesta inmunitaria, incluidas las quimioquinas y los antivirales. Algunos de los genes notablemente inducidos por la infección de1HRV-16 incluyen, pero no se limitan a, CCL2, CCL8, CXCL11, CXCL10, CXCL13, CXCL9, CCL20, IFIT2, GBP1, IFIT1, GIP2, IFIT4, IL28B, IRF7, CIG5, NOS2A, OAS3, OASL, OAS2, OAS1, MX2, MX1, PLSCR1, SOCS1, SOCS2, MDA5, RIGI, SOCS3, ICAM-1, HAPLN3, MMP12, EPSTI1, y TNC.
Vía de síntesis de los ácidos grasos
Varios aspectos de la presente divulgación se refieren a composiciones y procedimientos que modulan la actividad de la vía de síntesis de ácidos grasos, para tratar una infección viral o tratar el cáncer. La vía de síntesis de los ácidos grasos en los seres humanos puede utilizar cuatro enzimas: 1) la acetil-CoA carboxilasa (ACC), que puede sintetizar malonil-CoA; 2) la enzima málica, que puede producir NADPH; 3) la citrato liasa, que puede sintetizar acetil-CoA; y 4) la sintetasa de ácidos grasos, que puede catalizar la síntesis dependiente de NADPH de ácidos grasos a partir de acetil-CoA y malonil-CoA. En varios aspectos, la presente divulgación se refiere al tratamiento de infecciones virales y del cáncer, mediante la modulación de la actividad de la proteína sintetasa de ácidos grasos.
Los productos finales de la sintetasa de ácidos grasos son ácidos grasos libres que pueden utilizar una formación enzimática de derivados separada, con coenzima-A para su incorporación a otros productos. En el ser humano, la síntesis de ácidos grasos puede ocurrir en dos sitios: el hígado, donde se puede fabricar ácido palmítico (Roncari, (1974) Can. J. Biochem, 52:221-230) y la glándula mamaria lactante, donde se pueden producir ácidos grasos C10-C14 (Thompson, et al., (1985) Pediatr. Res., 19:139-143).
Los ácidos grasos pueden ser sintetizados en el citoplasma a partir de acetil-CoA. El acetil-CoA puede ser generado a partir del piruvato por la piruvato deshidrogenasa (PDH) y por la p-oxidación de los ácidos grasos en la mitocondria. Una "lanzadera de citrato" puede transportar acetil-CoA desde la mitocondria al citoplasma. El acetil-CoA puede reaccionar con el oxaloacetato para producir citrato, y una translocasa de tricarboxilato puede transportar el citrato de la mitocondria al citosol. En el citoplasma, el citrato puede volver a escindirse en oxaloacetato y acetil-CoA, una reacción que puede ser catalizada por la ATP-citrato liasa. El oxaloacetato puede convertirse de nuevo en piruvato para entrar nuevamente en la mitocondria.
El acetil-CoA puede convertirse en malonil-CoA. La acetil-CoA carboxilasa (ACC) es un sistema enzimático multifuncional complejo que contiene biotina, y que puede catalizar la carboxilación de acetil-CoA a malonil-CoA. Esta conversión es un paso irreversible limitante de la velocidad en la síntesis de ácidos grasos. La ACC puede desempeñar tres funciones: proteína transportadora de carboxil biotina, carboxilasa de biotina y carboxiltransferasa. La carboxilación de la biotina dependiente de ATP, un grupo prostético (cofactor), puede ser seguida por la transferencia del grupo carboxilo a la acetil-CoA.
HCO3' ATP acetil-CoA -> ADP Pi malonil-CoA
Existen dos formas de ACC, alfa y beta, codificadas por dos genes diferentes ACC-alfa (también conocido como ACC, ACAC, ACC1, ACCA y ACACA) que pueden codificar proteínas altamente enriquecidas en los tejidos lipogénicos. Se han encontrado múltiples variantes de transcripción empalmadas alternativamente, divergentes en la secuencia y que codifican distintas isoformas para este gen. La ACC-beta (también conocida como ACC2, ACCb , HACC275 y ACACB) puede codificar una proteína que se cree que controla la oxidación de los ácidos grasos mediante la capacidad de la malonil-CoA de inhibir la carnitina-palmitoil-CoA transferasa I, el paso que limita la tasa de captación y oxidación de los ácidos grasos por las mitocondrias. El ACC-beta puede estar implicado en la regulación de la oxidación de los ácidos grasos, más que en la biosíntesis de los mismos. Hay pruebas de la presencia de dos isoformas ACC-beta.
El ACC puede ser regulado por la fosforilación/desfosforilación de los residuos de serina seleccionados. Por ejemplo, la quinasa activada por AMP (AMPK) puede fosforilar el ACC, y esta fosforilación puede inhibir la capacidad del ACC para producir malonil-CoA. En ACACA, la AMPK puede fosforilar Ser79, Ser1200 y Ser1215 (Park, S.H., et al., (2002) J. Appl. Physiol. 92:2475-82). La AMPK puede fosforilar Ser218 en la ACACB (Hardie, D.G. (1992) Biochim. Biophys. Acta 1123:231-8). Además, la proteína quinasa dependiente del AMPc (proteína quinasa A, o PKA) puede fosforilar el ACC.
El ACC puede ser regulado por transformación alostérica por citrato o palmitoil-CoA. Por ejemplo, el citrato puede ser un efector positivo (es decir, el citrato puede activar alostéricamente el ACC). La concentración de citrato puede ser alta cuando hay una entrada adecuada de acetil-CoA en el ciclo de Krebs. El exceso de acetil-CoA puede entonces convertirse, a través del malonil-CoA, en ácidos grasos. La palmitoil-CoA puede ser un efector negativo. El palmitoil-CoA, que es el producto de la sintetasa de ácidos grasos (FASN), puede promover la conformación inactiva del ACC, lo que puede reducir la producción de malonil-CoA (un proceso de inhibición por retroalimentación). El AMP puede regular la síntesis de ácidos grasos mediante la regulación de la disponibilidad de malonil-CoA. El receptor a de unión de insulina puede activar una fosfatasa para desfosforilar el ACC, lo que puede eliminar el efecto inhibidor.
El gen de la sintetasa de ácidos grasos (también conocido como FAS, OA-519, SDR27X1; MGC14367; MGC15706; FASN) está implicado en la síntesis de ácidos grasos. La enzima codificada por este gen es una proteína multifuncional de aproximadamente 272 kDa con múltiples dominios, cada uno con actividades enzimáticas distintivas que pueden desempeñar un papel en la biosíntesis de los ácidos grasos. La FASN puede catalizar la síntesis de palmitato a partir de acetil-CoA y malonil-CoA, en presencia de NADPH, en ácidos grasos saturados de cadena larga. En algunas líneas celulares de cáncer, se ha encontrado que la proteína FASN está fusionada con el receptor alfa de estrógeno (ER-alfa), en el que el extremo N de FASN está fusionado en el marco con el extremo C de ER-alfa.
La proteína FASN puede existir en el citosol como un dímero de subunidades idénticas. La FASN consta de tres dominios catalíticos en la sección N-terminal (-cetoacil sintetasa (KS), malonil/acetiltransferasa (MAT) y deshidrasa (DH)). La sección N-terminal está separada por una región central de unos 600 aminoácidos de cuatro dominios C-terminales (enoil reductasa (ER), -cetoacil reductasa (KR), proteína transportadora de acilo (ACP) y tioesterasa (TE)). Se ha reportado la estructura cristalina de una sintetasa de ácidos grasos de mamífero (Maier, T., et al., (2008) Science 321: 1315-1322). Cada uno de los dominios catalíticos de FASN puede ser el objetivo de los procedimientos de tratamiento de la infección viral de la invención proporcionada.
Los pasos enzimáticos de la síntesis de ácidos grasos pueden implicar la condensación descarboxilante, la reducción, la deshidratación y otra reducción y pueden dar lugar a una fracción de acilo saturado. El NADPH puede ser un donador de electrones en las reacciones reductoras.
Actividad anticancerígena
En varios aspectos, la presente divulgación proporciona procedimientos para tratar el cáncer en un sujeto, comprendiendo el procedimiento la administración a un sujeto que necesita dicho tratamiento, de una cantidad eficaz de un compuesto como el descrito anteriormente.
El cáncer se selecciona del grupo que consiste en cáncer de pulmón de células no pequeñas; y tumores colorrectales con mutación KRAS.
Las células cancerosas de rápida proliferación activan la vía de síntesis de ácidos grasos para suministrar los altos niveles de lípidos necesarios para el ensamblaje de la membrana y el metabolismo oxidativo. (Flavin, R. et al. (2010) Future Oncology. 6(4):551-562) Los inhibidores de la síntesis de ácidos grasos han demostrado actividad in vivo en modelos preclínicos de cáncer. (Orita, H. et al. (2007) Clinical Cancer Research. 13(23):7139-7145 y Puig, T. et al. (2011) Breast Cancer Research, 13(6):R131) Además, la síntesis de ácidos grasos favorece la formación de nuevos vasos sanguíneos y los inhibidores de esta vía tienen actividad en modelos in vitro de angiogénesis. (Browne, C.D., et al. (2006) The FASEB Journal, 20(12):2027-2035). Los compuestos actualmente divulgados demostraron la capacidad de inducir selectivamente la detención del ciclo celular en las células HUVEC, sin causar la muerte celular general por apoptosis. Ver Ejemplos.
El tratamiento del cáncer de la presente invención incluye un efecto antitumoral que puede evaluarse por medios convencionales, como la tasa de respuesta, el tiempo de progresión de la enfermedad y/o la tasa de supervivencia. Los efectos antitumorales de la presente invención incluyen, pero no se limitan a, la inhibición del crecimiento tumoral, el retraso del crecimiento tumoral, la regresión del tumor, el encogimiento del tumor, el aumento del tiempo hasta el rebrote del tumor al cesar el tratamiento, y la ralentización de la progresión de la enfermedad. Por ejemplo, se espera que cuando la combinación de la presente invención se administre a un animal de sangre caliente, como un ser humano, que necesite un tratamiento para el cáncer que implique un tumor sólido, dicho uso en el tratamiento produzca un efecto, medido por, por ejemplo, uno o más de los siguientes: el alcance del efecto antitumoral, la tasa de respuesta, el tiempo hasta la progresión de la enfermedad y la tasa de supervivencia.
Tratamiento
También se proporcionan en el presente documento composiciones farmacéuticas que comprenden los compuestos de la presente divulgación. Las presentes composiciones y procedimientos tienen actividad anticancerígena.
En diferentes aspectos, la presente divulgación proporciona composiciones farmacéuticas que comprenden uno cualquiera de los compuestos descritos anteriormente y un portador, excipiente o diluyente farmacéuticamente aceptable.
Ciertos aspectos de la presente divulgación se refieren a procedimientos de uso de composiciones farmacéuticas y kits que comprenden uno o más agentes que inhiben la vía de síntesis de ácidos grasos para el tratamiento del cáncer. Ciertos aspectos de la presente divulgación se refieren a procedimientos de uso de composiciones farmacéuticas y kits que comprenden uno o más agentes que inhiben la sintetasa de ácidos grasos para el tratamiento del cáncer. Otro aspecto de la presente invención proporciona composiciones farmacéuticas, y kits para su uso en el tratamiento de sujetos animales que tienen cáncer o están en riesgo de desarrollarlo. El término "sujeto", tal como se utiliza en el presente documento, incluye a los seres humanos y a otros mamíferos. El término "tratar'1, tal como se utiliza en el presente documento, incluye la obtención de un beneficio terapéutico y/o un beneficio profiláctico.
La composición farmacéutica puede modular la vía de síntesis de los ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN. En el que, el término modular incluye la inhibición de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN o, alternativamente, la
activación de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN.
La reducción de la actividad de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN, también se denomina "inhibición" de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN. El término "inhibe" y sus conjugaciones gramaticales, como "inhibidor", no requieren una inhibición completa, sino que se refieren a una reducción de la actividad de síntesis de ácidos grasos, es decir, de la expresión del gen FASN o de la actividad de la proteína FASN. En otro aspecto, dicha reducción es de al menos el 50 %, al menos el 75 %, al menos el 90 %, y puede ser de al menos el 95 % de la actividad de la enzima en ausencia del efecto inhibidor, es decir, en ausencia de un inhibidor. Por el contrario, la frase "no inhibe" y sus conjugaciones gramaticales se refieren a situaciones en las que hay menos del 20 %, menos del 10 %, y puede ser menos del 5 %, de reducción de la actividad enzimática en presencia del agente. Además, la frase "no inhibe sustancialmente" y sus conjugaciones gramaticales se refieren a situaciones en las que hay menos del 30 %, menos del 20 % y, en algunos aspectos, menos del 10 % de reducción de la actividad enzimática en presencia del agente.
El aumento de la actividad de la vía de síntesis de ácidos grasos, es decir, la expresión del gen F ASN o la actividad de la proteína FASN, también se denomina "activación" de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN. El término "activado" y sus conjugaciones gramaticales, como "activando", no requieren una activación completa, sino que se refieren a un aumento de la actividad de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN. En otro aspecto, dicho aumento es de al menos el 50 %, al menos el 75 %, al menos el 90 %, y puede ser de al menos el 95 % de la actividad de la enzima en ausencia del efecto de activación, es decir, en ausencia de un activador. Por el contrario, la frase "no se activa" y sus conjugaciones gramaticales se refieren a situaciones en las que hay menos del 20 %, menos del 10 %, y puede ser menos del 5 %, de aumento de la actividad enzimática en presencia del agente. Además, la frase "no activa sustancialmente" y sus conjugaciones gramaticales se refieren a situaciones en las que hay menos del 30 %, menos del 20 % y, en otro aspecto, menos del 10 % de aumento de la actividad enzimática en presencia del agente.
La capacidad de reducir la actividad enzimática es una medida de la potencia o la actividad de un agente, o de una combinación de agentes, hacia o contra la enzima. La potencia puede medirse mediante ensayos libres de células, de células completas y/o in vivo en términos de valores IC50, Ki y/o ED50. Un valor IC50 representa la concentración necesaria de un agente para inhibir la actividad enzimática a la mitad (50 %), en un conjunto dado de condiciones. Un valor Ki representa la constante de afinidad de equilibrio para la unión de un agente inhibidor a la enzima. Un valor ED50 representa la dosificación necesaria de un agente para obtener una respuesta semimáxima en un ensayo biológico. Los detalles de estas medidas serán apreciados por aquellos con destreza ordinaria en la técnica, y pueden encontrarse en textos estándar sobre bioquímica, enzimología y similares.
Formulaciones, vías de administración y dosificaciones efectivas
Otro aspecto de la presente invención se refiere a las formulaciones, las vías de administración y las dosificaciones eficaces para las composiciones farmacéuticas que comprenden un agente o una combinación de agentes de la presente invención.
Los compuestos de la invención pueden administrarse en forma de formulaciones farmacéuticas, incluidas las adecuadas para la administración oral (incluida la bucal y la sublingual), rectal, nasal, tópica, en parches transdérmicos, pulmonar, vaginal, en supositorios o parenteral (incluida la intramuscular, intraarterial, intratecal, intradérmica, intraperitoneal, subcutánea e intravenosa) o en una forma adecuada para la administración mediante aerosol, por inhalación o por insuflación. Se puede encontrar información general sobre los sistemas de administración de fármacos en Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems (Lippencott Williams & Wilkins, Baltimore Md. (1999).
En diferentes aspectos, la composición farmacéutica incluye portadores y excipientes (incluidos, pero no limitados, a amortiguadores, carbohidratos, manitol, proteínas, polipéptidos o aminoácidos como la glicina, antioxidantes, bacteriostáticos, agentes quelantes, agentes de suspensión, agentes espesantes y/o conservantes), agua, aceites, incluidos los de origen de petróleo, animal, vegetal o sintético, como el aceite de cacahuete, aceite de soja, aceite mineral, aceite de sésamo y similares, soluciones salinas, soluciones acuosas de dextrosa y glicerina, agentes aromatizantes, agentes colorantes, agentes de separación y otros aditivos, adyuvantes o aglutinantes aceptables, otras sustancias auxiliares farmacéuticamente aceptables que se requieran para aproximarse a las condiciones fisiológicas, como agentes amortiguadores del pH, agentes para ajuste de la tonicidad, agentes emulsificantes, agentes humectantes y similares. Algunos ejemplos de excipientes son almidón, glucosa, lactosa, sacarosa, gelatina, malta, arroz, harina, tiza, gel de sílice, estearato de sodio, monoestearato de glicerol, talco, cloruro de sodio, leche desnatada en polvo, glicerol, propileno, glicol, agua, etanol y similares. En otro aspecto, la preparación farmacéutica está sustancialmente libre de conservantes. En otro aspecto, La preparación farmacéutica puede contener al menos un conservante. La metodología general sobre formas farmacéuticas de dosificación se encuentra en Ansel et al., Pharmaceutical Dosage Forms and Drug Delivery Systems (Lippencott Williams & Wilkins, Baltimore Md. (1999)). Se
reconocerá que, mientras que cualquier portador adecuado conocido por aquellos de habilidad ordinaria en la técnica puede ser empleado para administrar las composiciones de esta invención, el tipo de portador variará dependiendo del modo de administración.
Los compuestos también pueden ser encapsulados dentro de liposomas utilizando una tecnología bien conocida. Las microesferas biodegradables también pueden emplearse como portadoras de las composiciones farmacéuticas de esta invención. Las microesferas biodegradables adecuadas se divulgan, por ejemplo, en los documentos de EE.UU. N° 4,897,2685,075,1095,928,647 5,811,1285,820,8835,853,7635,814,344 y 5,942,252.
El compuesto puede administrarse en liposomas o microesferas (o micropartículas). Los procedimientos para preparar liposomas y microesferas para su administración a un paciente son bien conocidos por los expertos en la técnica. El documento de EE.UU. N° 4,789,734 describe procedimientos para encapsular materiales biológicos en liposomas. Esencialmente, el material se disuelve en una solución acuosa, se añaden los fosfolípidos y lípidos adecuados, junto con los tensioactivos si es necesario, y al material se aplica diálisis o ultrasonido, según sea necesario. Una revisión de los procedimientos conocidos es proporcionada porG. Gregoriadis, Capítulo 14, "Liposomes", Drug Carriers in Biology and Medicine, pp. 2.sup.87-341 (Academic Press, 1979).
Las microesferas formadas por polímeros o proteínas son bien conocidas por los expertos en la técnica, y pueden adaptarse a la medida para que pasen por el tracto gastrointestinal directamente al torrente sanguíneo. Alternativamente, el compuesto puede incorporarse y las microesferas, o el compuesto de microesferas, implantarse para una liberación lenta durante un período de tiempo que varía de días a meses. Véanse, por ejemplo, los documentos de EE.UU. Números 4,906,474, 4,925,673 y 3,625,214y Jein, TIPS 19:155-157 (1998).
La concentración del fármaco puede ser ajustada, el pH de la solución amortiguado y la isotonicidad ajustada, para ser compatibles con la inyección intravenosa, como es bien conocido en la técnica.
Los compuestos de la invención pueden formularse como una solución o suspensión estéril, en vehículos adecuados, bien conocidos en la técnica. Las composiciones farmacéuticas pueden ser esterilizadas mediante técnicas de esterilización convencionales y conocidas, o pueden ser filtradas en condiciones de esterilidad. Las soluciones acuosas resultantes pueden ser envasadas para su uso como están, o liofilizadas, combinándose la preparación liofilizada con una solución estéril antes de su administración. Las formulaciones y los soportes adicionales adecuados se describen en Remington "The Science and Practice of Pharmacy" (20a ed., Lippincott Williams & Wilkins, Baltimore MD).
Los agentes o sus sales farmacéuticamente aceptables pueden proporcionarse solos o en combinación con uno o más agentes o con una o más otras formas. Por ejemplo, una formulación puede comprender uno o más agentes en proporciones particulares, dependiendo de las potencias relativas de cada agente y de la indicación prevista. Por ejemplo, en las composiciones para focalizarse en dos objetivos diferentes del huésped, y cuando las potencias son similares, se puede utilizar una proporción de agentes de aproximadamente 1:1. Las dos formas pueden formularse juntas, en la misma unidad de dosificación, es decir, en una crema, un supositorio, un comprimido, una cápsula, un atomizado de aerosol o un paquete de polvo para disolver en una bebida; o cada forma puede formularse en una unidad separada, es decir, dos cremas, dos supositorios, dos comprimidos, dos cápsulas, un comprimido y un líquido para disolver el comprimido, dos atomizados en aerosol, o un paquete de polvo y un líquido para disolver el polvo, etc.
El término "sal farmacéuticamente aceptable" significa aquellas sales que conservan la eficacia y las propiedades biológicas de los agentes utilizados en la presente invención, y que no son biológicamente o de otro modo indeseables. Por ejemplo, una sal farmacéuticamente aceptable no interfiere con el efecto beneficioso de un agente de la invención, en la inhibición de la vía de síntesis de ácidos grasos, es decir, la inhibición de la expresión del gen FASN o la actividad de la proteína FASN.
Las sales típicas son las de los iones inorgánicos, como, por ejemplo, los iones de sodio, potasio, calcio, magnesio y similares. Dichas sales incluyen sales con ácidos inorgánicos u orgánicos, como ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido nítrico, ácido sulfúrico, ácido metanosulfónico, ácido p-toluenosulfónico, ácido acético, ácido fumárico, ácido succínico, ácido láctico, ácido mandélico, ácido málico, ácido cítrico, ácido tartárico o ácido maleico. Además, si el o los agente(s) contiene(n) un grupo carboxi u otro grupo ácido, pueden convertirse en una sal de adición farmacéuticamente aceptable con bases inorgánicas u orgánicas. Ejemplos de bases adecuadas incluyen hidróxido de sodio, hidróxido de potasio, amoníaco, ciclohexilamina, diciclohexilamina, etanolamina, dietanolamina, trietanolamina y similares.
Un éster o amida farmacéuticamente aceptable se refiere a aquellos que conservan la eficacia propiedades biológicas y de los agentes utilizados en la presente invención, y que no son biológicamente o de otro modo indeseables. Por ejemplo, el éster o la amida no interfiere con el efecto beneficioso de un agente de la invención, en la inhibición de la vía de síntesis de ácidos grasos, es decir, la inhibición de la expresión del gen FASN o la actividad de la proteína FASN. Los ésteres típicos incluyen etilo, metilo, isobutilo, etilenglicol y similares. Las amidas típicas incluyen amidas no sustituidas, alquilamidas, dialquilamidas y similares.
En otro aspecto, un agente puede administrarse en combinación con uno o más otros compuestos, formas y/o
agentes, es decir, como se ha descrito anteriormente. Las composiciones farmacéuticas que comprenden combinaciones de un inhibidor de la vía de síntesis de los ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, con uno o varios otros agentes activos, pueden formularse para que comprendan determinadas proporciones molares. Por ejemplo, pueden utilizarse proporciones molares de aproximadamente 99:1 a aproximadamente 1:99 de un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, respecto al otro agente activo. En algunos subconjuntos de los aspectos, el intervalo de relaciones molares del inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN:otro agente activo se selecciona de aproximadamente 80:20 a aproximadamente 20:80; de aproximadamente 75:25 a aproximadamente 25:75, de aproximadamente 70:30 a aproximadamente 30:70, de aproximadamente 66:33 a aproximadamente 33:66, de aproximadamente 60:40 a aproximadamente 40:60; de aproximadamente 50:50; y de aproximadamente 90:10 a aproximadamente 10:90. La relación molar de un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN:otro agente activo puede ser de aproximadamente 1:9, y en otro aspecto puede ser de aproximadamente 1:1. Los dos agentes, formas y/o compuestos pueden formularse juntos, en la misma unidad de dosificación, es decir, en una crema, supositorio, comprimido, cápsula o paquete de polvo para disolver en una bebida; o cada agente, forma y/o compuesto puede formularse en unidades separadas, es decir, dos cremas, supositorios, comprimidos, dos cápsulas, un comprimido y un líquido para disolver el comprimido, un atomizado en aerosol, un paquete de polvo y un líquido para disolver el polvo, etc.
Si es necesario o deseable, los agentes y/o combinaciones de agentes pueden administrarse con otros agentes más. La elección de los agentes que pueden ser coadministrados con los agentes y/o combinaciones de agentes de la presente invención puede depender, al menos en parte, de la condición que se está tratando. Las formulaciones pueden contener además uno o más suplementos, como vitamina C, E u otros antioxidantes.
En ciertos aspectos, los compuestos de la presente divulgación pueden administrarse en combinación con un terapéutico conocido contra el cáncer. Por ejemplo, los compuestos pueden administrarse en combinación con paclitaxel (disponible comercialmente como Taxol, Bristol-Myers Squibb), doxorrubicina (también conocida bajo el nombre comercial Adriamicina), vincristina (conocida bajo los nombres comerciales Oncovin, Vincasar PES y Vincrex), actinomicina D, altretamina, asparaginasa, bleomicina, busulfán, cabazitaxel, capecitabina, carboplatino, carmustina, clorambucil, cisplatino, ciclofosfamida, citarabina, dacarbazina, daunorubicina, docetaxel, epirubicina, etopósido, fludarabina, fluorouracilo, gemcitabina, hidroxiurea, idarubicina, ifosfamida, irinotecán, lomustina, melfalán, mercaptopurina, metotrexato, mitomicina, mitozantrona, oxaliplatino, procarbazina, esteroides, estreptozocina, taxotere, tamozolomida, tioguanina, tiotepa, tomudex, topotecán, treosulfán, UFT (uracil-tegufur), vinblastina, vindesina, agentes dirigidos a moduladores inmunitarios como PD-1, PDL-1 e IDOl, es decir, nivolumab, pembrolizumab, MPDL3280A y MEDI4736; agentes dirigidos a la deficiencia en la reparación del ADN, por ejemplo, olaparib; agentes dirigidos a los receptores de tirosina quinasa, como EGFR, ERBB2, c-MET, VEGFR2 e IGFR1, por ejemplo, erlotinib, necitumumab, traztuzamab, pertuzamab, lapatinib, crizotinib, cabozantinib, onartuamab, ramucirumab o bevacizumab; agentes dirigidos a la MAPK/e Rk , la MAPK quinasa, MEK1 y/o MEK2, es decir, selumetinib o trametinib; agentes dirigidos a los receptores hormonales, como los receptores de andrógenos y estrógenos, es decir, enzalutamida, abiraterona o tamoxifeno; agentes dirigidos a las vías de la MAP quinasa o PI3K-AKT, es decir, cobimetinib, vemurafenib y everolimus; bloqueadores de la vía Her2 (ErbB2), como lapatinib, trastuzumab y Kadyzla; bloqueadores de mTOR, como ralapogs (es decir sirolimus); inhibidores de mTORC1/mTORC1; bloqueadores de la vía de la angiogénesis o del VEGFR, como avastin, nexavar o sutent; moduladores de la aromatasa, como exemtesane o femora; moduladores de la señalización androgénica, como enzalutamida, bicalutamida; y bloqueadores de B-RAF, como Tafinlar o Zelboraf, o similares.
El (los) agente(s) (o las sales, ésteres o amidas farmacéuticamente aceptables de los mismos) puede(n) ser administrado(s) per se o en forma de una composición farmacéutica en la que el (los) agente(s) activo(s) se encuentra(n) en una adición o mezcla con uno o más portadores farmacéuticamente aceptables. Una composición farmacéutica, tal como se utiliza en el presente documento, puede ser cualquier composición preparada para su administración a un sujeto. Las composiciones farmacéuticas para su uso de acuerdo con la presente invención pueden formularse de manera convencional utilizando uno o más portadores fisiológicamente aceptables, que comprenden excipientes, diluyentes y/o auxiliares, es decir, que facilitan la transformación de los agentes activos en preparaciones que pueden administrarse. La formulación adecuada puede depender, al menos en parte, de la vía de administración elegida. El/los agente/s útil/es en la presente invención, o las sales, ésteres o amidas farmacéuticamente aceptables de los mismos, pueden administrarse a un paciente utilizando una serie de rutas o modos de administración, incluyendo aplicaciones orales, bucales, tópicas, rectales, transdérmicas, transmucosales, subcutáneas, intravenosas e intramusculares, así como por inhalación.
Para la administración oral, los agentes pueden formularse fácilmente combinando el/los agente(s) activo(s) con portadores farmacéuticamente aceptables bien conocidos en la técnica. Dichos portadores permiten que los agentes de la invención se formulen en forma de comprimidos, incluidos los comprimidos masticables, píldoras, grageas, cápsulas, pastillas, caramelos duros, líquidos, geles, jarabes, pastas líquidas, polvos, suspensiones, elixires, obleas y similares, para su ingestión oral por parte del paciente que se va a tratar. Dichas formulaciones pueden comprender portadores farmacéuticamente aceptables, incluyendo diluyentes o rellenos sólidos, medios acuosos estériles y diversos disolventes orgánicos no tóxicos. Un portador sólido puede ser una o más sustancias que también pueden
actuar como diluyentes, agentes aromatizantes, agentes de solubilidad, lubricantes, agentes de suspensión, aglutinantes, conservantes, agentes de desintegración de comprimidos o un material de encapsulación. En los polvos, el portador suele ser un sólido finamente dividido que es una mezcla con el componente activo finamente dividido. En los comprimidos, el componente activo se mezcla generalmente con el soporte que tiene la capacidad de unión necesaria, en proporciones adecuadas y se compacta en la forma y tamaño deseados. Los polvos y los comprimidos contienen preferentemente entre el uno (1) y el setenta (70) por ciento del compuesto activo. Los portadores adecuados incluyen, pero no se limitan a, carbonato de magnesio, estearato de magnesio, talco, azúcar, lactosa, pectina, dextrina, almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio, una cera de baja fusión, manteca de cacao y similares. Generalmente, los agentes de la invención se incluirán en niveles de concentración que varían desde aproximadamente el 0,5 %, aproximadamente el 5 %, aproximadamente el 10 %, aproximadamente el 20 % o aproximadamente el 30 % hasta aproximadamente el 50 %, aproximadamente el 60 %, aproximadamente el 70 %, aproximadamente el 80 % o aproximadamente el 90 % en peso de la composición total de las formas de dosificación oral, en una cantidad suficiente para proporcionar una unidad de dosificación deseada.
Las suspensiones acuosas para uso oral pueden contener agente(s) de esta invención con excipientes farmacéuticamente aceptables, como un agente de suspensión (es decir, metilcelulosa), un agente humectante (es decir, lecitina, lisolecitina y/o un alcohol graso de cadena larga), así como agentes colorantes, conservantes, agentes aromatizantes y similares.
En otro aspecto, pueden requerirse aceites o disolventes no acuosos para poner los agentes en solución, debido, por ejemplo, a la presencia de grandes fracciones lipofílicas. Como alternativa, pueden utilizarse emulsiones, suspensiones u otras preparaciones, por ejemplo, preparaciones liposomales. Respecto a las preparaciones liposomales, se puede utilizar cualquier procedimiento conocido para preparar liposomas para el tratamiento de una enfermedad. Véase, por ejemplo, Bangham, et al., J. Mol. Biol. 23: 238-252 (1965) y Szoka, et al., Proc. Acad. Nat. Sci. USA 75: 4194-4198 (1978). También se pueden unir ligandos a los liposomas para dirigir estas composiciones a sitios particulares de acción. Los agentes de esta invención también pueden integrarse en productos alimenticios, es decir, en queso crema, mantequilla, aderezo para ensaladas o helado, para facilitar la solubilización, la administración y/o el cumplimiento en ciertas poblaciones de pacientes.
Las preparaciones farmacéuticas para uso oral pueden obtenerse como excipiente sólido, opcionalmente moliendo una mezcla resultante, y procesando la mezcla de gránulos, después de añadir auxiliares adecuados, si se desea, para obtener comprimidos o núcleos de grageas. Los excipientes adecuados son, en particular, rellenos como azúcares, incluyendo lactosa, sacarosa, manitol o sorbitol; elementos aromatizantes, preparados de celulosa como, por ejemplo, almidón de maíz, almidón de trigo, almidón de arroz, almidón de patata, gelatina, goma tragacanto, metilcelulosa, hidroxipropilmetilcelulosa, carboximetilcelulosa de sodio y/o polivinilpirrolidona (PVP). Si se desea, pueden añadirse agentes de desintegración, como la polivinilpirrolidona entrecruzada, el agar o el ácido algínico o una sal de éste, como el alginato de sodio. Los agentes también pueden formularse como una preparación de liberación sostenida.
Los núcleos de grageas pueden estar provistos de recubrimientos adecuados. Para ello, pueden utilizarse soluciones concentradas de azúcar, que pueden contener opcionalmente goma arábiga, talco, polivinilpirrolidona, gel de carbopol, polietilenglicol y/o dióxido de titanio, soluciones de laca y disolventes orgánicos o mezclas de disolventes adecuados. Pueden añadirse colorantes o pigmentos a los comprimidos o recubrimientos de grageas, para su identificación o para caracterizar diferentes combinaciones de agentes activos.
Las preparaciones farmacéuticas que pueden utilizarse por vía oral incluyen cápsulas de gelatina con cierre a presión, así como cápsulas blandas y selladas hechas de gelatina y un plastificante, como glicerol o sorbitol. Las cápsulas con cierre a presión pueden contener los ingredientes activos en mezcla con rellenos como la lactosa, aglutinantes como los almidones, y/o lubricantes como el talco o el estearato de magnesio y, opcionalmente, estabilizantes. En las cápsulas blandas, los agentes activos pueden estar disueltos o suspendidos en líquidos adecuados, como aceites grasos, parafina líquida o polietilenglicoles líquidos. Además, se pueden añadir estabilizantes. Todas las formulaciones para la administración oral deberían estar en dosificaciones adecuadas para la administración.
Otras formas adecuadas para la administración oral incluyen preparaciones en forma líquida, incluyendo emulsiones, jarabes, elixires, soluciones acuosas, suspensiones acuosas, o preparaciones en forma sólida que están destinadas a ser convertidas poco antes de su uso en preparaciones en forma líquida. Las emulsiones pueden prepararse en soluciones, por ejemplo, en soluciones acuosas de propilenglicol o pueden contener agentes emulsificantes, por ejemplo, como lecitina, monooleato de sorbitán o acacia. Las soluciones acuosas pueden prepararse disolviendo el componente activo en agua y añadiendo colorantes, aromas, estabilizantes y agentes espesantes adecuados. Las suspensiones acuosas pueden prepararse dispersando el componente activo finamente dividido en agua con material viscoso, como gomas naturales o sintéticas, resinas, metilcelulosa, carboximetilcelulosa sódica y otros agentes de suspensión bien conocidos. Entre los rellenos o portadores adecuados con los que se pueden administrar las composiciones se encuentran agar, alcohol, grasas, lactosa, almidón, derivados de celulosa, polisacáridos, polivinilpirrolidona, sílice, solución salina estéril y similares, o las mezclas de los mismos utilizadas en cantidades adecuadas. Las preparaciones en forma sólida incluyen soluciones, suspensiones y emulsiones, y pueden contener,
además del componente activo, colorantes, aromas, estabilizantes, amortiguadores, edulcorantes artificiales y naturales, agentes de dispersión, espesantes, agentes de solubilidad y similares.
Se puede preparar un jarabe o una suspensión, añadiendo el compuesto activo a una solución acuosa concentrada de un azúcar, es decir, sacarosa, a la que también se puede añadir cualquier ingrediente accesorio. Dichos ingredientes accesorios pueden incluir aromatizantes, un agente para retardar la cristalización del azúcar o un agente para aumentar la solubilidad de cualquier otro ingrediente, es decir, como un alcohol polihídrico, por ejemplo, glicerol o sorbitol.
Cuando se formulan compuestos de la invención para la administración oral, puede ser deseable utilizar formulaciones gastrorretentivas para mejorar la absorción desde el tracto gastrointestinal (GI). Una formulación que se retiene en el estómago durante varias horas puede liberar compuestos de la invención lentamente y proporcionar una liberación sostenida que puede utilizarse en los procedimientos de la invención. La divulgación de tales formulaciones gastrorretentivas se encuentra en Klausner, E.A., Lavy, E., Barta, M., Cserepes, E., Friedman, M., Hoffman, A., 2003 "Novel gastroretentive dosage forms: evaluation of gastroretentivity and its effect on levodopa in humans" Pharm. Res.
20, 1466-73 Hoffman, A., Stepensky, D., Lavy, E., Eyal, S., Klausner, E., Friedman, M. 2004 "Pharmacokinetic and pharmacodynamic aspects of gastroretentive dosage forms" Int. J. Pharm. 11, 141-53 Streubel, A., Siepmann, J., Bodmeier, R., 2006 "Gastroretentive drug delivery systems" Expert Opin. Drug Deliver. 3, 217-3y Chavanpatil, M.D., Jain, P., Chaudhari, S., Shear, R., Vavia, P.R., "Novel sustained release, swellable and bioadhesive gastroretentive drug delivery system for olfoxacin" Int. J. Pharm. 2006 epub 24 de marzo. Se pueden utilizar técnicas expandibles, flotantes y bioadhesivas para maximizar la absorción de los compuestos de la invención.
Los compuestos de la invención pueden formularse para la administración parenteral (es decir, por inyección, por ejemplo, inyección en bolo o infusión continua) y pueden presentarse en forma de dosis unitaria en ampollas, jeringas precargadas, infusión de pequeño volumen o en envases multidosis con un conservante añadido. Las composiciones pueden adoptar formas tales como suspensiones, soluciones o emulsiones en vehículos oleosos o acuosos, por ejemplo, soluciones en polietilenglicol acuoso.
Para las formulaciones inyectables, el vehículo puede elegirse entre los que se conocen en la técnica como adecuados, incluyendo soluciones acuosas o suspensiones de aceite, o emulsiones, con aceite de sésamo, aceite de maíz, aceite de semilla de algodón o aceite de cacahuete, así como elixires, manitol, dextrosa o una solución acuosa estéril, y vehículos farmacéuticos similares. La formulación también puede comprender composiciones poliméricas que sean biocompatibles y biodegradables, como el ácido poli(láctico-co-glicólico). Estos materiales se pueden convertir en micro o nanoesferas, cargadas con el fármaco y recubiertas o derivadas, para proporcionar un rendimiento superior de liberación sostenida. Los vehículos adecuados para la inyección periocular o intraocular incluyen, por ejemplo, suspensiones del agente terapéutico en agua de calidad para inyección, liposomas y vehículos adecuados para sustancias lipofílicas. Otros vehículos para la inyección periocular o intraocular son bien conocidos en la técnica.
En un aspecto preferido, la composición se formula de acuerdo con los procedimientos de rutina, como una composición farmacéutica adaptada para la administración intravenosa a seres humanos. Normalmente, las composiciones para la administración intravenosa son soluciones en amortiguador acuoso isotónico estéril. Cuando sea necesario, la composición también puede incluir un agente de solubilizad y un anestésico local como la lidocaína, para aliviar el dolor en el lugar de la inyección. Por lo general, los ingredientes se suministran por separado o mezclados en forma de dosificación unitaria, por ejemplo, como polvo seco liofilizado o concentrado sin agua en un recipiente sellado herméticamente como una ampolla o una bolsita que indica la cantidad de agente activo. Cuando la composición vaya a administrarse por infusión, puede dispensarse con un frasco de infusión que contenga agua o solución salina estériles de calidad farmacéutica. Cuando la composición se administra por inyección, se puede proporcionar una ampolla de agua para inyección o solución salina estériles, para que los ingredientes se puedan mezclar antes de la administración.
Cuando la administración es por inyección, el compuesto activo puede formularse en soluciones acuosas, específicamente en amortiguadores fisiológicamente compatibles como la solución de Hanks, la solución de Ringer o el amortiguador salino fisiológico. La solución puede contener agentes de formulación tales como agentes de suspensión, de estabilización y/o de dispersión. Alternativamente, el compuesto activo puede estar en forma de polvo para su constitución con un vehículo adecuado, es decir, agua estéril libre de pirógenos, antes de su uso. En otro aspecto, la composición farmacéutica no comprende un adyuvante ni ninguna otra sustancia añadida para mejorar la respuesta inmunitaria estimulada por el péptido. En otro aspecto, la composición farmacéutica comprende una sustancia que inhibe una respuesta inmunitaria al péptido. Los procedimientos de formulación son conocidos en la técnica, por ejemplo, como se divulga en Remington's Pharmaceutical Sciences, última edición, Mack Publishing Co., Easton P.
Adicionalmente a las formulaciones descritas anteriormente, los agentes también pueden ser formulados como una preparación de depósito. Estas formulaciones de acción prolongada pueden administrarse por implante o administración transcutánea (por ejemplo, por vía subcutánea o intramuscular), inyección intramuscular o uso de un parche transdérmico. Así, por ejemplo, los agentes pueden formularse con materiales poliméricos o hidrófobos
adecuados (por ejemplo, como una emulsión en un aceite aceptable) o resinas de intercambio iónico, o como derivados poco solubles, por ejemplo, como una sal poco soluble.
En otro aspecto, las composiciones farmacéuticas que comprenden uno o más agentes de la presente invención ejercen efectos locales y regionales cuando se administran tópicamente o se inyectan en o cerca de sitios particulares de infección. La aplicación tópica directa, es decir, de un líquido viscoso, solución, suspensión, soluciones en base a dimetilsulfóxido (DMSO), formulaciones liposomales, gel, jalea, crema, loción, ungüento, supositorio, espuma o atomizado en aerosol, puede utilizarse para la administración local, para producir, por ejemplo, efectos locales y/o regionales. Los vehículos farmacéuticamente apropiados para dicha formulación incluyen, por ejemplo, alcoholes alifáticos inferiores, poliglicoles (es decir, glicerol o polietilenglicol), ésteres de ácidos grasos, aceites, grasas, siliconas y similares. Dichas preparaciones también pueden incluir conservantes (es decir, ésteres de ácido p-hidroxibenzoico) y/o antioxidantes (es decir, ácido ascórbico y tocoferol). Ver también Dermatological Formulations: Percutaneous absorption, Barry (Ed.), Marcel Dekker Incl, 1983. En otro aspecto, las formulaciones locales/tópicas que comprenden un inhibidor de la vía de síntesis de los ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, se utilizan para tratar las infecciones virales epidérmicas o de las mucosas.
Las composiciones farmacéuticas de la presente invención pueden contener un portador cosmética o dermatológicamente aceptable. Dichos portadores son compatibles con la piel, las uñas, las membranas mucosas, los tejidos y/o el cabello, y pueden incluir cualquier portador cosmético o dermatológico utilizado convencionalmente que cumpla estos requisitos. Dichos portadores pueden ser fácilmente seleccionados por alguien de destreza ordinaria en la técnica. En la formulación de ungüentos para la piel, un agente o una combinación de agentes de la presente invención puede formularse en una base de hidrocarburo oleaginoso, una base de absorción anhidra, una base de absorción de agua en aceite, una base agua en aceite removible con agua y/o una base soluble en agua. Entre los ejemplos de dichos portadores y excipientes se incluyen, pero no se limitan a, humectantes (por ejemplo, urea), glicoles (por ejemplo, propilenglicol), alcoholes ( por ejemplo, etanol), ácidos grasos (por ejemplo, ácido oleico), tensioactivos ( por ejemplo, miristato de isopropilo y lauril sulfato de sodio), pirrolidonas, monolaurato de glicerol, sulfóxidos, terpenos (por ejemplo, mentol), aminas, amidas, alcanos, alcanoles, agua, carbonato de calcio, fosfato de calcio, diversos azúcares, almidones, derivados de la celulosa, gelatina y polímeros como los polietilenglicoles.
Las ungüentos y cremas pueden, por ejemplo, formularse con una base acuosa u oleosa con la adición de agentes espesantes y/o gelificantes adecuados. Las lociones pueden estar formuladas con una base acuosa u oleosa y, en general, también contendrán uno o más agentes emulsificantes, agentes estabilizantes, agentes dispersantes, agentes de suspensión, agentes espesantes o agentes colorantes. La construcción y el uso de parches transdérmicos para la administración de agentes farmacéuticos es bien conocida en la técnica. Véase, por ejemplo los documentos de EE.UU. N° 5,023,252, 4,992,445 y 5,001,139. Dichos parches pueden construirse para la administración continua, pulsátil o a demanda de agentes farmacéuticos.
Los lubricantes que pueden utilizarse para formar las composiciones farmacéuticas y las formas de dosificación de la invención incluyen, pero no se limitan a, estearato de calcio, estearato de magnesio, aceite mineral, aceite mineral ligero, glicerina, sorbitol, manitol, polietilenglicol, otros glicoles, ácido esteárico, lauril sulfato de sodio, talco, aceite vegetal hidrogenado (es decir, aceite de cacahuete, aceite de semilla de algodón, aceite de girasol, aceite de sésamo, aceite de oliva, aceite de maíz y aceite de soja), estearato de zinc, oleato de etilo, laurato de etilo, agar o mezclas de los mismos. Los lubricantes adicionales incluyen, por ejemplo, un gel de sílice siloide, un aerosol coagulado de sílice sintética, o mezclas de los mismos. Opcionalmente, se puede añadir un lubricante, en una cantidad inferior a aproximadamente el 1 por ciento en peso de la composición farmacéutica.
Las composiciones de acuerdo con la presente invención pueden presentarse en cualquier forma adecuada para su aplicación tópica, incluyendo soluciones acuosas, acuoso-alcohólicas u oleosas, dispersiones en loción o suero, geles acuosos, anhidros u oleosos, emulsiones obtenidas por dispersión de una fase grasa en una fase acuosa (O/W o aceite en agua) o, a la inversa, (W/O o agua en aceite), microemulsiones o alternativamente microcápsulas, dispersiones de tipo iónico y/o no iónico de micropartículas o de vesículas lipídicas. Estas composiciones pueden prepararse de acuerdo con los procedimientos convencionales. Aparte de los agentes de la invención, las cantidades de los diversos constituyentes de las composiciones de acuerdo con la invención son las utilizadas convencionalmente en la técnica. Estas composiciones constituyen, en particular, cremas, leches, lociones, geles o espumas de protección, tratamiento o cuidado para la cara, las manos, el cuerpo y/o las membranas mucosas, o para la limpieza de la piel. Las composiciones también pueden consistir en preparaciones sólidas que constituyen jabones o barras limpiadoras.
Las composiciones de la presente invención también pueden contener adyuvantes comunes en los campos cosmético y dermatológico, como agentes gelificantes hidrofílicos o lipofílicos, agentes activos hidrofílicos o lipofílicos, agentes conservantes, antioxidantes, disolventes, fragancias, rellenos, protectores solares, absorbentes de olores y colorantes. Las cantidades de estos diversos adyuvantes son las que se utilizan convencionalmente en los campos considerados y, por ejemplo, son de aproximadamente 0,01 % a aproximadamente 20 % del peso total de la composición. Dependiendo de su naturaleza, estos adyuvantes pueden introducirse en la fase grasa, en la fase acuosa y/o en las vesículas lipídicas.
En otro aspecto, las infecciones virales oculares pueden ser tratadas eficazmente con soluciones oftálmicas, suspensiones, ungüentos o insertos que comprenden un agente o una combinación de agentes de la presente invención. Los colirios pueden prepararse disolviendo el ingrediente activo en una solución acuosa estéril, como una solución salina fisiológica, una solución amortiguadora, etc., o combinando composiciones en polvo que se disuelven antes de su uso. Pueden elegirse otros vehículos, como se conoce en la técnica, incluyendo pero no limitándose a: solución salina de equilibrio, solución salina, poliéteres solubles en agua como el polietilenglicol, polivinilos, como el alcohol polivinílico y la povidona, derivados de la celulosa como metilcelulosa e hidroxipropilmetilcelulosa, derivados del petróleo, como aceite mineral y vaselina blanca, grasas animales, como lanolina, polímeros de ácido acrílico, como gel de carboxipolimetileno, grasas vegetales, como aceite de cacahuete, y polisacáridos, como dextranos, y glicosaminoglicanos, como hialuronato sódico. Si se desea, se pueden añadir los aditivos que se utilizan habitualmente en los colirios. Dichos aditivos incluyen agentes isotonizantes (es decir, cloruro de sodio, etc.), agentes amortiguadores (es decir, ácido bórico, monohidrogenofosfato de sodio, dihidrógeno fosfato de sodio, etc.), conservantes(es decir, cloruro de benzalconio, cloruro de bencetonio, clorobutanol, etc.), espesantes ( es decir, sacáridos como lactosa, manitol, maltosa, etc.; es decir, ácido hialurónico o su sal como el hialuronato de sodio, el hialuronato de potasio, etc. ; es decir mucopolisacárido como el condroitín sulfato, etc.; es decir, poliacrilato de sodio, polímero de carboxivinilo, poliacrilato entrecruzado, alcohol polivinílico, polipirrolidona, metilcelulosa, hidroxipropilmetilcelulosa, hidroxietilcelulosa, carboximetilcelulosa, hidroxipropilcelulosa u otros agentes conocidos por los expertos en la técnica).
La solubilidad de los componentes de las presentes composiciones puede ser mejorada por un tensioactivo u otro co solvente apropiado en la composición. Tales cosolventes incluyen polisorbato 20, 60 y 80, Pluronic F68, F-84 y P-103, ciclodextrina u otros agentes conocidos por los expertos en la técnica. Dichos codisolventes pueden emplearse a un nivel de aproximadamente 0,01 % a 2 % en peso.
Las composiciones de la invención pueden envasarse en forma de multidosis. Se pueden preferir los conservantes para evitar la contaminación microbiana durante el uso. Los conservantes adecuados incluyen: cloruro de benzalconio, timerosal, clorobutanol, metilparabeno, propilparabeno, alcohol feniletílico, edetato disódico, ácido sórbico, Onamer M, u otros agentes conocidos por los expertos en la técnica. En los productos oftálmicos del estado de la técnica anterior, dichos conservantes pueden emplearse a un nivel del 0,004 % al 0,02 %. En las composiciones de la presente solicitud, el conservante, preferentemente el cloruro de benzalconio, puede emplearse a un nivel del 0,001 % a menos del 0,01 %, es decir, del 0,001 % al 0,008 %, preferentemente alrededor del 0,005 % en peso. Se ha descubierto que una concentración de cloruro de benzalconio del 0,005 % puede ser suficiente para preservar las composiciones de la presente invención del ataque microbiano.
En otro aspecto, las infecciones virales del oído pueden tratarse eficazmente con soluciones, suspensiones, ungüentos o insertos óticos que comprenden un agente o una combinación de agentes de la presente invención.
En otro aspecto, los agentes de la presente invención se entregan en forma soluble más que en forma de suspensión, lo que permite una absorción más rápida y cuantitativa a los sitios de acción. En general, las formulaciones como jaleas, cremas, lociones, supositorios y ungüentos pueden proporcionar una zona con una exposición más prolongada a los agentes de la presente invención, mientras que las formulaciones en solución, es decir, los aerosoles, proporcionan una exposición más inmediata y de corto plazo.
En otro aspecto relacionado con la aplicación tópica/local, las composiciones farmacéuticas pueden incluir uno o más potenciadores de la penetración. Por ejemplo, las formulaciones pueden comprender portadores o excipientes adecuados en fase sólida o de gel que aumenten la penetración o ayuden al suministro de agentes o combinaciones de agentes de la invención a través de una barrera de permeabilidad, es decir, la piel. Muchos de estos compuestos que mejoran la penetración son conocidos en la técnica de la formulación tópica, e incluyen, por ejemplo, agua, alcoholes (por ejemplo, terpenos como metanol, etanol, 2-propanol), sulfóxidos (por ejemplo, dimetilsulfóxido, decilmetilsulfóxido, tetradecilmetilsulfóxido), pirrolidonas (por ejemplo, 2-pirrolidona, N-metil-2-pirrolidona, N-(2-hidroxietil)pirrolidona), laurocapram, acetona, dimetilacetamida, dimetilformamida, alcohol tetrahidrofurfurílico, L-aaminoácidos, tensioactivos aniónicos, catiónicos, anfóteros o no iónicos ( por ejemplo, miristato de isopropilo y lauril sulfato de sodio), ácidos grasos, alcoholes grasos (por ejemplo, ácido oleico), aminas, amidas, amidas de ácido clofíbrico, lauramida de hexametileno, enzimas proteolíticas, a-bisabolol, d-limoneno, urea y N,N-dietil-m-toluamida, y similares. Otros ejemplos incluyen humectantes (por ejemplo, la urea), glicoles (por ejemplo, propilenglicol y polietilenglicol), monolaurato de glicerol, alcanos, alcanoles, ORGELASE, carbonato de calcio, fosfato de calcio, diversos azúcares, almidones, derivados de celulosa, gelatina y/u otros polímeros. En otro aspecto, las composiciones farmacéuticas incluirán uno o más de estos potenciadores de la penetración.
En otro aspecto, las composiciones farmacéuticas para aplicación local/tópica pueden incluir uno o más conservantes antimicrobianos, como compuestos de amonio cuaternario, mercuriales orgánicos, p-hidroxi-benzoatos, alcoholes aromáticos, clorobutanol y similares.
Las infecciones virales gastrointestinales pueden tratarse eficazmente con soluciones, suspensiones, ungüentos,
enemas y/o supositorios administrados por vía oral o rectal, que comprenden un agente o una combinación de agentes de la presente invención.
Las infecciones virales respiratorias pueden tratarse eficazmente con soluciones en aerosol, suspensiones o polvos secos que comprenden un agente o una combinación de agentes de la presente invención. La administración por inhalación es especialmente útil en el tratamiento de infecciones virales del pulmón, como una infección por HRV. El aerosol puede administrarse a través del sistema respiratorio o pasos nasales. Por ejemplo, un experto en la técnica reconocerá que una composición de la presente invención puede suspenderse o disolverse en un portador apropiado, es decir, un propelente farmacéuticamente aceptable, y administrarse directamente en los pulmones mediante un aerosol nasal o un inhalante. Por ejemplo, una formulación en aerosol que comprende un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, puede disolverse, suspenderse o emulsionarse en un propelente o en una mezcla de disolvente y propelente, es decir, para su administración como aerosol nasal o inhalante. Las formulaciones en aerosol pueden contener cualquier propelente aceptable bajo presión, como un propelente cosmética o dermatológica o farmacéuticamente aceptable, como se utiliza convencionalmente en la técnica.
Una formulación en aerosol para la administración nasal es generalmente una solución acuosa diseñada para ser administrada a los conductos nasales en gotas o aerosoles. Las soluciones nasales pueden ser similares a las secreciones nasales en el sentido de que generalmente son isotónicas y están ligeramente amortiguadas para mantener un pH de aproximadamente 5,5 a aproximadamente 6,5, aunque también pueden utilizarse valores de pH fuera de este intervalo. También pueden incluirse en la formulación agentes antimicrobianos o conservantes.
Una formulación en aerosol para inhalaciones e inhalantes puede diseñarse de manera que el agente o la combinación de agentes de la presente invención sea transportada al árbol respiratorio del sujeto, cuando se administra por la vía respiratoria nasal u oral. Las soluciones de inhalación pueden administrarse, por ejemplo, mediante un nebulizador. Las inhalaciones o insuflaciones, que comprenden fármacos finamente pulverizados o líquidos, pueden administrarse al sistema respiratorio como un aerosol farmacéutico de una solución o suspensión del agente o combinación de agentes en un propelente, es decir, para ayudar a su salida. Los propelentes pueden ser gases licuados, incluidos los halocarbonos, por ejemplo, los fluorocarbonos, como los hidrocarburos clorados fluorados, los hidroclorofluorocarbonos y los hidroclorocarbonos, así como los hidrocarburos y los éteres de hidrocarburos.
Los propelentes de halocarbono útiles en la presente invención incluyen propelentes de fluorocarbono, en los que todos los hidrógenos se sustituyen por flúor, propelentes de clorofluorocarbono en los que todos los hidrógenos se sustituyen por cloro y al menos un flúor, propelentes de fluorocarbono que contienen hidrógeno y propelentes de clorofluorocarbono que contienen hidrógeno. Los propelentes de halocarbono se describen en Johnson, en el documento No. 5,376,359, emitido el 27 de diciembre de 1994 Byron et al., en el documento No. 5,190,029, emitido el 2 de marzo de 1993y Purewal et al., en el documento N° 5,776,434, emitido el 7 de julio de 1998. Los propelentes de hidrocarburos útiles en la invención incluyen, por ejemplo, propano, isobutano, n-butano, pentano, isopentano y neopentano. También se puede utilizar una mezcla de hidrocarburos como propelente. Los propelentes de éter incluyen, por ejemplo, el éter dimetílico, así como los éteres. Una formulación de aerosol de la invención también puede comprender más de un propelente. Por ejemplo, la formulación del aerosol puede comprender más de un propelente de la misma clase, como dos o más fluorocarbonos; o más de uno, más de dos, más de tres propelentes de diferentes clases, como un fluorohidrocarburo y un hidrocarburo. Las composiciones farmacéuticas de la presente invención también pueden dispensarse con un gas comprimido, es decir, un gas inerte como dióxido de carbono, óxido nitroso o nitrógeno.
Las formulaciones en aerosol también pueden incluir otros componentes, por ejemplo, etanol, isopropanol, propilenglicol, así como tensioactivos u otros componentes como aceites y detergentes. Estos componentes pueden servir para estabilizar la formulación y/o lubricar los componentes de la válvula.
La formulación en aerosol puede envasarse a presión y puede formularse como aerosol utilizando soluciones, suspensiones, emulsiones, polvos y preparaciones semisólidas. Por ejemplo, una formulación de aerosol en solución puede comprender una solución de un agente de la invención como un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, en un propelente (sustancialmente) puro o como una mezcla de propelente y disolvente. El disolvente puede utilizarse para disolver el agente y/o retardar la evaporación del propelente. Los disolventes útiles en la invención incluyen, por ejemplo, agua, etanol y glicoles. Se puede utilizar cualquier combinación de disolventes adecuados, opcionalmente combinados con conservantes, antioxidantes y/u otros componentes del aerosol.
Una formulación en aerosol también puede ser una dispersión o una suspensión. Una formulación de aerosol en suspensión puede comprender una suspensión de un agente o combinación de agentes de la presente invención, es decir, un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, y un agente dispersante. Los agentes dispersantes útiles en la invención incluyen, por ejemplo, el trioleato de sorbitán, el alcohol oleico, el ácido oleico, la lecitina y el aceite de maíz. Una formulación de aerosol de suspensión también puede incluir lubricantes, conservantes, antioxidantes y/u otros componentes de aerosol.
Una formulación en aerosol puede formularse de forma similar como una emulsión. Una formulación de aerosol en emulsión puede incluir, por ejemplo, un alcohol como etanol, un tensioactivo, agua y un propelente, así como un agente o combinación de agentes de la invención, es decir, una vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN. El tensioactivo utilizado puede ser no iónico, aniónico o catiónico. Un ejemplo de formulación de aerosol en emulsión comprende, por ejemplo, etanol, tensioactivo, agua y propelente. Otro ejemplo de formulación de aerosol en emulsión comprende, por ejemplo, aceite vegetal, monoestearato de glicerilo y propano.
Los compuestos de la invención pueden formularse para su administración como supositorios. Primero se funde una cera de baja fusión, como una mezcla de triglicéridos, glicéridos de ácidos grasos, Witepsol S55 (marca comercial de Dynamite Nobel Chemical, Alemania) o manteca de cacao, y se dispersa el componente activo de forma homogénea, por ejemplo, mediante agitación. A continuación, la mezcla homogénea fundida se vierte en moldes de tamaño adecuado, se deja enfriar y se solidifica.
Los compuestos de la invención pueden formularse para su administración vaginal. Pesarios, amortiguadores, cremas, geles, pastas, espumas o aerosoles que contengan los portadores, además del ingrediente activo, son conocidos en la técnica como adecuados.
Se prevé, además, que los compuestos de la invención puedan unirse de forma que puedan liberarse, a polímeros biocompatibles para su uso en formulaciones de liberación sostenida en, dentro de o unidos a insertos para la administración tópica, intraocular, periocular o sistémica. La liberación controlada de un polímero biocompatible puede utilizarse también con un polímero soluble en agua, para formar una formulación instilable. La liberación controlada de un polímero biocompatible, como por ejemplo, las microesferas o nanoesferas de PLGA, puede utilizarse también en una formulación adecuada para la implantación o inyección intraocular para la administración de liberación sostenida. Puede utilizarse cualquier polímero biodegradable y biocompatible adecuado.
Las composiciones farmacéuticas adecuadas para su uso en la presente invención incluyen composiciones en las que los ingredientes activos están presentes en una cantidad efectiva, es decir, en una cantidad efectiva para lograr un beneficio terapéutico y/o profiláctico en un huésped con al menos una infección viral o en un sujeto con cáncer. La cantidad real efectiva para una aplicación particular dependerá de la condición o condiciones que se traten, de la condición del sujeto, de la formulación y de la vía de administración, así como de otros factores conocidos por los expertos en la técnica. La determinación de una cantidad efectiva de un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen FASN o de la actividad de la proteína FASN, está bien dentro de las capacidades de los expertos en la técnica, a la luz de la divulgación del presente documento, y se determinará utilizando técnicas de optimización de rutina.
La cantidad efectiva para su uso en humanos puede determinarse a partir de modelos animales. Por ejemplo, una dosis para los seres humanos puede formularse para lograr concentraciones circulantes, hepáticas, tópicas y/o gastrointestinales que se han encontrado eficaces en los animales. Un experto en la técnica puede determinar la cantidad efectiva para el uso humano, especialmente a la luz de los datos experimentales de los modelos animales descritos en el presente documento. Basándose en los datos de animales, y en otros tipos de datos similares, los expertos en la técnica pueden determinar las cantidades eficaces de las composiciones de la presente invención, apropiadas para los seres humanos.
La cantidad efectiva, cuando se refiere a un agente o combinación de agentes de la invención, significará generalmente los intervalos de dosis, modos de administración, formulaciones, etc., que han sido recomendados o aprobados por cualquiera de las diversas organizaciones reguladoras o asesoras en las técnicas médicas o farmacéuticas (es decir, FDA, AMA) o por el fabricante o proveedor.
Además, las dosis apropiadas para un inhibidor de la vía de síntesis de ácidos grasos, es decir, un inhibidor de la expresión del gen fAs N o de la actividad de la proteína FASN, pueden determinarse basándose en los resultados experimentales in vitro. Por ejemplo, la potencia in vitro de un agente en la inhibición de un componente de la vía de síntesis de ácidos grasos, es decir, la expresión del gen FASN o la actividad de la proteína FASN, proporciona información útil en el desarrollo de dosis eficaces in vivo para lograr efectos biológicos similares.
En otro aspecto, la administración de los agentes de la presente invención puede ser intermitente, por ejemplo la administración una vez cada dos días, cada tres días, cada cinco días, una vez a la semana, una o dos veces al mes, y similares. En otro aspecto, la cantidad, las formas y/o las cantidades de las diferentes formas pueden variar en diferentes momentos de la administración.
Un experto en la técnica sería capaz de vigilar en un paciente el efecto de la administración de un agente particular.
Habiendo descrito ahora de forma general diversos aspectos y facetas de la invención, la misma se entenderá más fácilmente a través de la referencia a los siguientes ejemplos, que se proporcionan a modo de ilustración, y no pretenden ser limitantes, a menos que se especifique.
Ejemplos
La divulgación se ilustra adicionalmente con los siguientes ejemplos. Los ejemplos que no están comprendidos en el alcance de las reivindicaciones son únicamente ilustrativos.
Ejemplo 1 - Inhibición de FASN por los compuestos de la presente divulgación
Determinación de la actividad bioquímica de FASN: La enzima FASN se aisló de células SKBr3. SKBr3 es una línea celular de cáncer de mama humano con altos niveles de expresión de FASN. Se estima que FASN comprende aproximadamente 25 % de las proteínas citosólicas en esta línea celular. Las células SKBr3 se homogeneizaron en un homogeneizador de rebote y después se centrifugaron durante 15 minutos a 4 °C para eliminar las partículas. A continuación, se analizó el contenido proteico del sobrenadante, se diluyó hasta la concentración adecuada y se utilizó para medir la actividad de FASN. La presencia de FASN se confirmó mediante un análisis de mancha occidental. Un procedimiento similar para el aislamiento de FASN a partir de células SKBr3 se describe en Teresa, P. et al. (Clin. Cancer Res. 2009; 15(24), 7608-7615).
La actividad de FASN del extracto celular SKBr3 se determinó midiendo la oxidación del NADPH o la cantidad de coenzima A (CoA) que contiene tiol liberada durante la reacción de la sintetasa de ácidos grasos. El tinte CPM (7-dietilamino-3-(4'-maleimidil-fenil)-4-metilcumarina) contiene un grupo reactivo tiol que aumenta su emisión de fluorescencia al reaccionar con el grupo sulfhidrilo de la CoA. Las actividades bioquímicas se determinaron utilizando la medición de fluorescencia de la liberación de CoA mediante un procedimiento descrito en Chung C.C. et al. (Assay and Drug Development Technologies, 2008, 6(3), 361-374).
Ejemplo 2 de referencia - Actividad antiviral
La actividad antiviral de la Fórmula (I-Z) se evaluó utilizando el sistema de replicón del VHClb

El replicón se construyó utilizando la línea celular ET (luc-ubi-neo/ET), una línea celular de hepatoma humano Huh7 que alberga un replicón del VHC con un reportero de luciferasa (Luc) estable y tres mutaciones adaptadas al cultivo celular (Pietschmann, et al (2002) J. Virol. 76:4008-4021). El ensayo de evaluación antiviral del replicón del VHC examinó los efectos de los compuestos a seis concentraciones de semilog. El interferón humano alfa-2b se incluyó en cada paso como compuesto de control positivo. Los cultivos subconfluyentes de la línea ET se sembraron en placas de 96 pocillos dedicadas al análisis del número de células (citotoxicidad) o de la actividad antiviral, y al día siguiente se añadieron los fármacos en los pocillos correspondientes. Las células se procesaron 72 horas más tarde, cuando las células aún eran subconfluyentes. Se determinaron los valores EC50 (concentraciones que inhiben el replicón en un 50 % y 90 %, respectivamente), IC50 (concentración que disminuye la viabilidad celular en un 50 %) y Si (índice selectivo: IC50/EC50). Los niveles de replicón del ARN del VHC se evaluaron como actividad Luc derivada del ARN del VHC o como ARN del VHC mediante la RT-PCR de TaqMan. Se utilizaron dos procedimientos para estimar el recuento de células (citotoxicidad). Cuando se empleó el sistema de ensayo Luc, se utilizó el ensayo (Promega) colorimétrico de proliferación celular CytoTox-1 para estimar el número de células, mientras que los niveles de ARN ribosómico (ARNr) determinados mediante RT-PCR de TaqMan se utilizaron como indicación del número de células en el ensayo basado en ARN. En el cuadro 2 se resumen los resultados.
Tabla 2.

Ejemplo 3 de referencia - Inhibición de FASN se correlaciona con la inhibición del VHC
Las actividades antivirales de 15 compuestos de la presente divulgación se midieron utilizando el sistema de replicón del VHC. La línea 1b celular de Replicon (HCV 1b/Luc-Neo replicon (1b Con1 con el gen Firefly integrado)) se estableció siguiendo procedimientos publicados (Lohmann et al. (1999) Science 285(5424):110-113, Lohmann et al. (2001) J. Virol. 75(3): 1437-1449 y Qi et al. (2009) Antiviral Res. 81(2): 166-173) utilizando Huh7 por selección G418. El replicón se ensambló utilizando fragmentos de genes sintéticos. La línea GTlb tiene PV-EKT y alberga 3 mutaciones adaptativas E1202G(NS3), T1280I(NS3), K1846T(NS4B) y el armazón es Con1. El medio de cultivo era:
a) Suplemento DMEM con 10 % FBS, G418 (250 pg/ml), estreptomicina (100 pg/ml)/penicilina (100 U/ml), L-glutamina (100*), NEAA (100*)
b) Medios preparados de la siguiente manera:
i) 500 ml de medio DMEM (Gibco, Cat#1 1960-077)
ii) 57 ml de suero fetal bovino (Gibco, Cat# 16140-071)
iii) 5,7 ml de penicilina-estreptomicina (Gibco, Cat#15140-122)
iv) 5,7 ml de MEM aminoácidos no esenciales (Gibco, Cat#1 11140-050)
v) 5,7 ml de L-glutamina (Gibco, Cat#125030-081)
vi) 574,1 ml de medio 2,87 ml de G41850 mg/ml [0,25 mg/ml final] (Gibco, Cat# 10131-027)
Los compuestos se disolvieron en DMSO para suministrar una reserva de 10 mM o se utilizaron a partir de soluciones madre de DMSO. Los compuestos se diluyeron para generar diluciones seriadas de 10 puntos de semilogaritmo (3,16 veces) para el ensayo en placas de 384 pocillos (Echo qualified PP de 384 pocillos (Labcyte Cat#P-05525)) más DMSO por duplicado. Este experimento se repitió tres veces en tres días diferentes.
Las células se cosecharon cuando la confluencia alcanzó el 90 %-100 %. Las concentraciones celulares se ajustaron a 8*104 células/ml y se añadieron a microplacas de ensayo blancas de 384 pocillos (con tratamiento de cultivo de tejidos - Greiner Cat#781080) para alcanzar una densidad celular final de 2.000 células/pocillo. Las placas se incubaron a 5 % de CO2 y 37 °C durante 72 horas.
Después de 72 horas de incubación se prepararon el reactivo Bright-Glo Luciferase (Promega, cat# E2650) y Cell Titer Flo (Promega, cat#G6080/1/2) y se almacenaron en la oscuridad mientras se equilibraba a temperatura ambiente. Las células tratadas se equilibraron igualmente a temperatura ambiente. Se añadieron 10 pl de Cell Titer Flo a cada pocillo de células tratadas con el compuesto y se incubaron en placas de microtitulación durante aproximadamente 0,5 horas. La viabilidad de las células se midió con un lector Envision (disponible en Perkin Elmer) para estimar la citotoxicidad. Se añadieron 30 pl de sustrato de luciferasa de luciérnaga a cada pocillo y se midió la quimioluminiscencia como indicador del grado de replicación del VHC.
La actividad antirreplicón (% de inhibición) se calcula mediante la ecuación:

La citotoxicidad se calcula mediante la ecuación:
Se determinó que había una correlación entre la potencia de la inhibición de FASN y la actividad antiviral como se ilustra en la Tabla 3 a continuación y en la FIG. 1. Se observa que ninguno de los compuestos causó una citotoxicidad significativa.
Tabla 3

(Continuación)

Ejemplo 4 de referencia - Los inhibidores de FASN conservan su actividad contra los mutantes del VHC que confieren resistencia a los agentes antivirales de acción directa
Uno de los principales retos en el tratamiento de la hepatitis C es la rápida aparición de resistencia en respuesta a los agentes antivirales de acción directa. La resistencia suele producirse cuando el virus genera un mutante puntual que soporta las funciones virales esenciales pero impide la unión de los agentes antivirales. Se probaron tres inhibidores de FASN (compuestos 55, 20 y 70) respecto a su capacidad de inhibir mutantes del VHC que confieren resistencia a agentes antivirales representativos. Cada uno de estos mutantes se introdujo en una construcción GTlb en base a un armazón de Con1 que contenía el gen PVIRES-Luciferase Ubi-Neo y que albergaba una mutación adaptativa (S2204I). (Lohmann et al. (1999) Science 285(5424): 110-113, Lohmann et al. (2001) J. Virol. 75(3): 1437-1449 y Qi et al. (2009) Antiviral Res. 81(2): 166-173). Las actividades antivirales se midieron por el procedimiento descrito en el Ejemplo 3.
Las mutaciones estudiadas se muestran en la Tabla 4 a continuación.
Tabla 4. Mutaciones estudiadas

Se probaron un inhibidor alostérico conocido de NS4B (Compuesto A), un inhibidor conocido de NS5A (Compuesto B), un inhibidor no nucleósido conocido de NS5B (Compuesto C), un inhibidor conocido de la proteasa NS3/NS4A (Compuesto D) y un inhibidor nucleósido conocido de NS5B (Compuesto E) en paralelo con los inhibidores de FASN de la presente divulgación, para confirmar el rendimiento de las mutaciones de resistencia.
A continuación se muestran las EC50 antivirales de los distintos compuestos contra el panel de mutantes, junto con el cambio relativo en la EC50 en relación con el replicón de tipo silvestre de GTlb. La variación normal del ensayo es de ± 3-4 veces. Los cambios de la EC50 fuera de este intervalo implican resistencia y se indican en negrilla. Los 3 inhibidores de FASN retienen su actividad en todo el panel de mutantes, mientras que los agentes antivirales de acción directa despliegan resistencia frente a las mutaciones en sus respectivos sitios de unión.
Tabla 5. EC50 antivirales.

Tabla 6. Veces de cambio de la EC50 en relación con el tipo silvestre
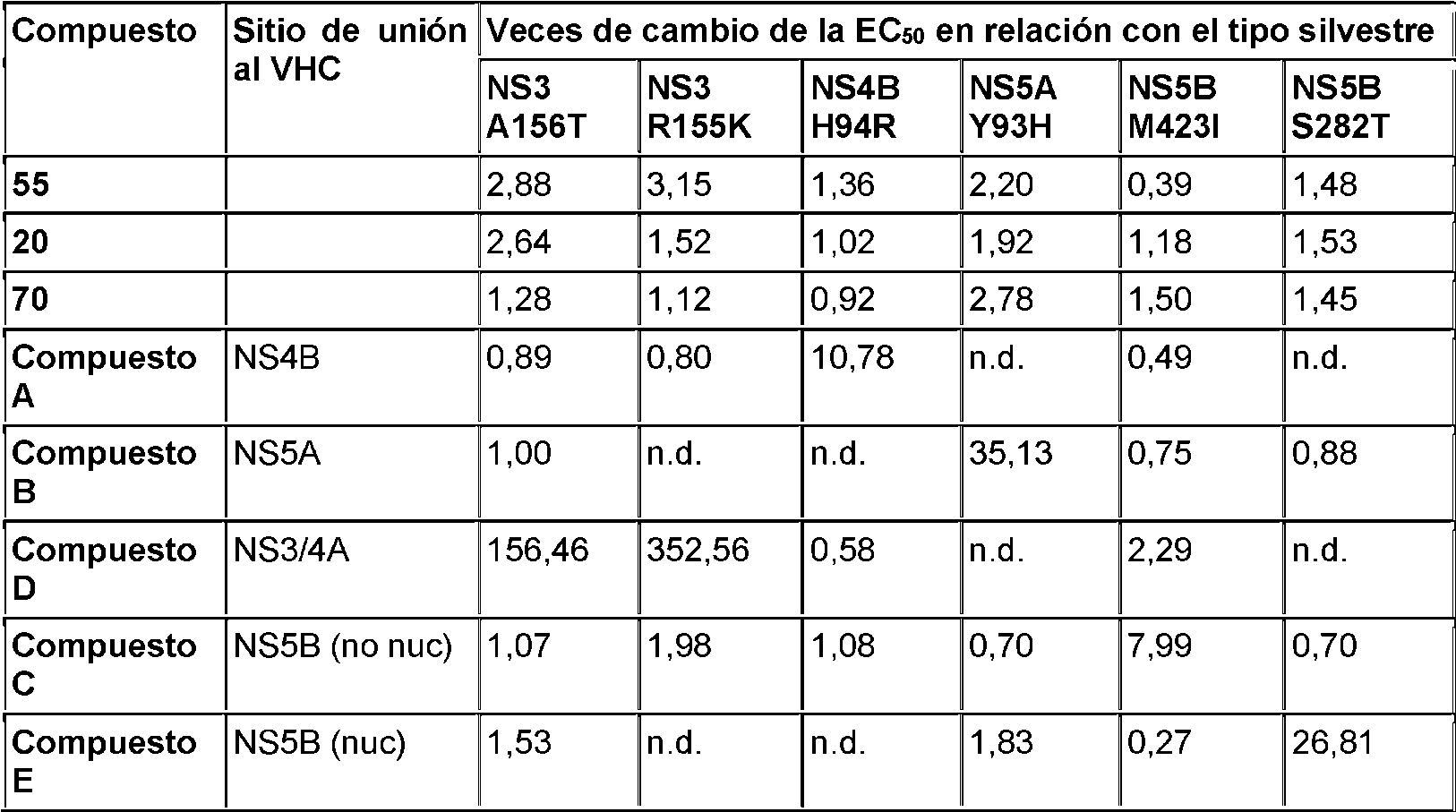
Ejemplo 5 de referencia - Inhibidores de FASN útiles en terapias de combinación
Este ejemplo describe la actividad y la citotoxicidad antiviral in vitro del compuesto de Fórmula (V-K) en combinación con IFN-a, Ribavirina, Compuestos B, C, D y E contra una línea celular de replicón de VHC GTlb
Materiales:
Virus: El plásmido replicón GTlb se ensambló utilizando fragmentos de genes sintéticos. El genoma del replicón contiene segmentos del gen PVIRES-Luciferasa Ubi-Neo y alberga 1 mutación adaptativa (S22041), y el armazón es Con1. La línea celular GTlb de replicón se estableció mediante los siguientes procedimientos publicados.
Medio y reactivos: En la Tabla 7 se detallan los reactivos del medio de cultivo utilizados en este ejemplo.
Tabla 7. Lista de reactivos de medios de cultivo

Instrumentos analíticos: Para realizar los ensayos de este ejemplo se utilizaron los siguientes instrumentos analíticos:
• POD-810
• Topcount (PE)
• Envision (PE)
• Multidrop (Thermo)
Procedimientos:
Preparación de las placas de compuestos para el ensayo de compuesto individual: Los compuestos se suministraron como polvos secos y se reconstituyeron en DMSO para generar soluciones madre. El sistema POD-810 se utilizó para generar diluciones seriadas de 10 puntos de semilog (3,16 veces) para el ensayo en placas de 96 pocillos. Las concentraciones más altas de la prueba se detallan para cada compuesto en la Tabla 8.
Protocolo de ensayo (compuestos individuales): Cada compuesto se ensayó con diluciones seriadas de 3,16 veces (semilogaritmo) para 10 concentraciones más DMSO por duplicado. Las células GTlb del replicón del VHC se cosecharon y se ajustaron a una concentración celular de 8E+04 células/ml. Se utilizó un Multidrop para colocar 100 pl/pocillo en microplacas de 96 ensayos para alcanzar una densidad celular final de 8.000 células/pocillo. Las placas se incubaron al 5 % de CO2 a 37 °C durante 72 horas.
Al final de la incubación de 72 horas, se midieron la actividad antiviral y la citotoxicidad. El reactivo Bright-Glo Luciferase y el Cell Titer Flo se prepararon y almacenaron en la oscuridad mientras se equilibraban a temperatura ambiente. También se dejó que las placas de células se equilibraran a temperatura ambiente. Se utilizó un Multidrop para añadir 20 pL de Cell Titer Flo a cada pocillo de células tratadas con compuestos y sin ellos. Las placas se incuban durante 1 hora y se mide la viabilidad celular en un lector Envision para calcular la citotoxicidad. Se añaden 50 microlitros de sustrato de luciferasa de luciérnaga a cada pocillo, se incuba durante 2 minutos y se mide la quimioluminiscencia para calcular EC50.
La actividad antirreplicón (% de inhibición) se calculó mediante la siguiente ecuación:
% d e inh ib ición = [ !-( ( Compuesto - Fondo )/(DMSO- Fondo)) x 100]
Compuestos de prueba y configuración del ensayo para los estudios de combinación de dos compuestos: En este análisis también se utilizaron las soluciones madre de DMSO de los compuestos utilizados en el ensayo de compuesto individual. Las matrices de dilución de combinación fueron generadas por POD-810 en microplacas de ensayo de 96 pocillos. El sistema POD-810 se utilizó para generar diluciones seriadas de 7 puntos y 2 veces en formato de matriz. A continuación se detalla la concentración máxima probada para cada compuesto.
Tabla 8. Actividades y concentraciones máximas previstas de los compuestos probadosen los estudios de agente individual y de combinación

El compuesto de Fórmula (V-K) se probó solo y en combinación con los compuestos detallados en la Tabla 9. También se probó cada compuesto por sí solo como agente individual.
Tabla 9. Combinaciones de compuestos para la evaluación in vitro.
Configuración del ensayo (combinaciones de dos fármacos): Cada compuesto se ensayó con diluciones seriadas de dos veces para 7 concentraciones en formato de matriz más cada fármaco solo. Las células GTlb del replicón del VHC se cosecharon y se ajustaron a una concentración celular de 8E+04 células/ml. Se utilizó un Multidrop para colocar 100 pl en microplacas de ensayo de 96 para alcanzar una densidad celular final de 8.000 células/pocillo. Las placas se incubaron al 5 % de CO2 a 37 °C durante 72 horas.
Al final de la incubación de 72 horas, se midieron la actividad antiviral y la citotoxicidad. El reactivo Bright-Glo Luciferase y el Cell Titer Flo se prepararon y almacenaron en la oscuridad mientras se permitía que alcanzaran el equilibrio a temperatura ambiente. También se dejó que las placas de células se equilibraran a temperatura ambiente. Se utilizó un Multidrop para añadir 20 pl de Cell Titer Flo a cada pocillo de células tratadas con compuestos y sin ellos. Las placas se incubaron durante 1 hora y se midió la viabilidad celular en un lector Envision para calcular la citotoxicidad. A continuación, se retiró el líquido de las placas, tras lo cual se añadieron 50 pl de PBS y 50 pl de solución de sustrato de luciferasa de luciérnaga en cada pocillo; tras un periodo de incubación de 2 minutos, se midió la quimioluminiscencia (para el cálculo de la replicación del VHC). Los datos seanalizaron con MacSynergyMR II.
Resultados del ensayo:
Actividad y citotoxicidad de los compuestos. Los valores EC50 y CC50 se resumen en la Tabla 10.
T a b la 10. EC50 y CC50 d e c a d a c o m p u e s to d e p ru eb a

E fec to d e la c o m b in a c ió n . El efecto de la combinación de los pares de compuestos se calculó utilizando MacSynergyMR II y esos resultados se resumen en la Tabla 11 a continuación.
T a b la 11. S u m a rio d e los e fe c to s d e la c o m b in a c ió n d e los pares d e c o m p u e s to s

C o n c lu s io n e s
Los factores Z de los pares de compuestos resumidos en la Tabla 12 indican que la calidad del ensayo es mejor que el estándar QC.
T a b la 12. S u m a rio d e l fa c to r Z d e los pares d e c o m p u e s to

Los valores de EC50 de los compuestos individuales en la matriz de combinación (resumidos en la Tabla 13) son consistentes con los datos de EC50 obtenidos para la inhibición de compuesto individual de la Tabla 10.
Tabla 13. Sumario de la EC50 de la dosis individual en la combinación de compuestos

Se demostró que el compuesto de Fórmula (V-K) tiene una actividad antiviral aditiva, sin aumentar la citotoxicidad en combinación con agentes que representan una variedad de mecanismos. Estos resultados se resumen en el cuadro 14.
Tabla 14. Sumario de los mecanismos antivirales que son aditivos con el compuesto de Fórmula (V-K). El término "antiviral de acción directa" ("AAD") se refiere a un compuesto que se une a una proteína viral y la inhibe, más que a una proteína del huésped.

El IFN-a y el RBV representan el estándar de cuidado actual para el tratamiento de la infección por hepatitis C, y recientemente se han aprobado los inhibidores de la proteasa del VHC Telaprevir y Boceprivir. La actividad antiviral aditiva y la falta de citotoxicidad mejorada en combinación con IFN-a y RBV sugieren además que los compuestos de esta invención no interferirán con procesos críticos del huésped, como la defensa celular (IFN-a) o la biosíntesis de nucleótidos de guanidina (RBV). Los compuestos de esta invención, como el compuesto de Fórmula (V-K), deberían por tanto ser terapéuticamente útiles, si se administran en regímenes de combinación con el estándar de cuidado actual. Además, las actividades antivirales aditivas observadas con el compuesto B, el compuesto C y el compuesto E sugieren que las moléculas de esta invención, como el compuesto de fórmula (V-K), pueden combinarse productivamente con agentes actualmente en desarrollo que se focalizan en mecanismos más nuevos (es decir, inhibidores de NS5A y NS5B).
Ejemplo 6 - Actividad antitumoral - Ensayo de citotoxicidad múltiple
Las células se cultivaron en RPMI1640, FBS 10 %, L-alanil-L-glutamina 2 mM, ImM Na piruvato o un medio especial en una atmósfera humidificada de 5 % de CO2 a 37 °C. Las células se sembraron en placas de 384 pocillos y se incubaron en una atmósfera humidificada de 5 % de CO2 a 37 °C. Los compuestos se añadieron 24 horas después de la siembra de las células. Al mismo tiempo, se generó una placa de células sin tratar de tiempo cero.
Después de un período de incubación de 72 horas, las células fueron fijadas y teñidas con anticuerpos marcados con fluorescencia y tinte nuclear, para permitir la visualización de los núcleos, las células apoptóticas y las células mitóticas. Las células apoptóticas se detectaron utilizando un anticuerpo anti-caspasa-3 activa. Las células mitóticas se detectaron utilizando un anticuerpo anti-fosfohistona-3.
Los compuestos se diluyeron en serie en incrementos de semilogaritmo (3,16 veces) y se ensayaron en 10
concentraciones en una concentración final de ensayo de 0,1 % de DMSO a partir de la concentración de ensayo más alta especificada en el capítulo de información de la muestra. La microscopía de fluorescencia automatizada se llevó a cabo utilizando un GE Healthcare IN Cell Analyzer 1000, y las imágenes se recogieron con un objetivo 4X.
Se adquirieron imágenes tiff de doce bits utilizando el InCell Analyzer 1000 3.2 y se analizaron con el software Developer Toolbox 1.6. Los valores de EC50 e IC50 se calcularon utilizando una regresión no lineal para ajustar los datos a un modelo de respuesta a la dosis sigmoidal de Un Sitio, de 4 puntos y 4 parámetros, donde: y (ajuste) = A [(B - A)/(1 ((C/x) A D)]. El ajuste de curvas, los cálculos de EC50/IC50 y la generación de informes se realizan mediante un motor de reducción de datos personalizado, basado en el software MathlQ (AIM).
El ensayo de citotoxicidad múltiple utiliza una técnica de análisis basada en la imagen celular, en la que las células se fijan y se tiñen con anticuerpos marcados con fluorescencia y tinte nuclear, para visualizar los núcleos y las células apoptóticas y mitóticas. Las células apoptóticas se detectan utilizando un anticuerpo anti-caspasa-3 activo. Las células mitóticas se detectan utilizando un anticuerpo anti-fodfohistona-3.
La proliferación celular se mide por la intensidad de la señal del tinte nuclear incorporado. El resultado del ensayo de proliferación celular se denomina recuento celular relativo. Para determinar el punto final de la proliferación celular, los datos de salida de proliferación celular se transforman en porcentaje de control (POC) utilizando la siguiente fórmula:
POC = recuento relativo de células (pocilios de compuesto) / recuento relativo de células (pocilios de vehículo) x 100
La placa de tiempo cero no tratada se utiliza para determinar el número de duplicaciones en un período de ensayo de 72 horas: Número de duplicaciones en 72 horas = LN[Número de células (punto final de 72 horas) 'Número de células (momento cero)]/LN(2). El resultado de cada biomarcador es el aumento en veces sobre el fondo del vehículo normalizado al recuento celular relativo en cada pocillo.
El marcador de la caspasa-3 activada etiqueta las células desde la etapa temprana hasta la tardía de la apoptosis. El resultado se muestra como un aumento de veces de las células apoptóticas sobre el fondo del vehículo normalizado al recuento celular relativo en cada pocillo. Las concentraciones del compuesto de prueba que causan una inducción de 5 veces en la señal de la caspasa-3 indican una inducción significativa de la apoptosis. Los pocillos con concentraciones superiores al recuento celular relativo IC95 se eliminan del análisis de inducción de caspasa-3.
El marcador fosfo-histona-3 etiqueta las células mitóticas. El resultado se muestra como una inducción en veces de células mitóticas sobre el fondo del vehículo normalizado al recuento celular relativo en cada pocillo. Cuando la inducción en veces de la señal de las células mitóticas sobre el fondo es ~1, no hay "efecto" en el ciclo celular. Un aumento de dos o más veces en la señal de fosfo-histona-3 sobre el fondo del vehículo indica una inducción significativa del bloqueo mitótico por parte del compuesto de ensayo.
Una disminución de dos o más veces en la señal de fosfo-histona-3 puede indicar el bloqueo G1/S, sólo cuando los niveles de citotoxicidad están por debajo del recuento IC95 celular relativo medido. Cuando se observan disminuciones de 2 o más veces en la señal de fosfo-histona-3 a concentraciones superiores al recuento IC95 celular relativo, la disminución del recuento de células mitóticas se debe con máxima probabilidad a un efecto citotóxico más general, más que a un verdadero bloqueo de la fase G1/S. Los pocillos con concentraciones superiores al recuento IC95 celular relativo se eliminan del análisis de fosfo-histona-3.
La proliferación celular medida por recuentos relativos de células fue el criterio para la respuesta positiva.
Apoptosis:
un aumento de >5 veces en la señal de la caspasa-3 activada indica una respuesta apoptótica Mitosis:
un aumento de >2 veces en la fosfo-histona-3 indica un bloqueo mitótico
una disminución de <2 veces en la fosfo-histona-3 indica un bloqueo G1/S
Tabla 15. Resultados

(Continuación)
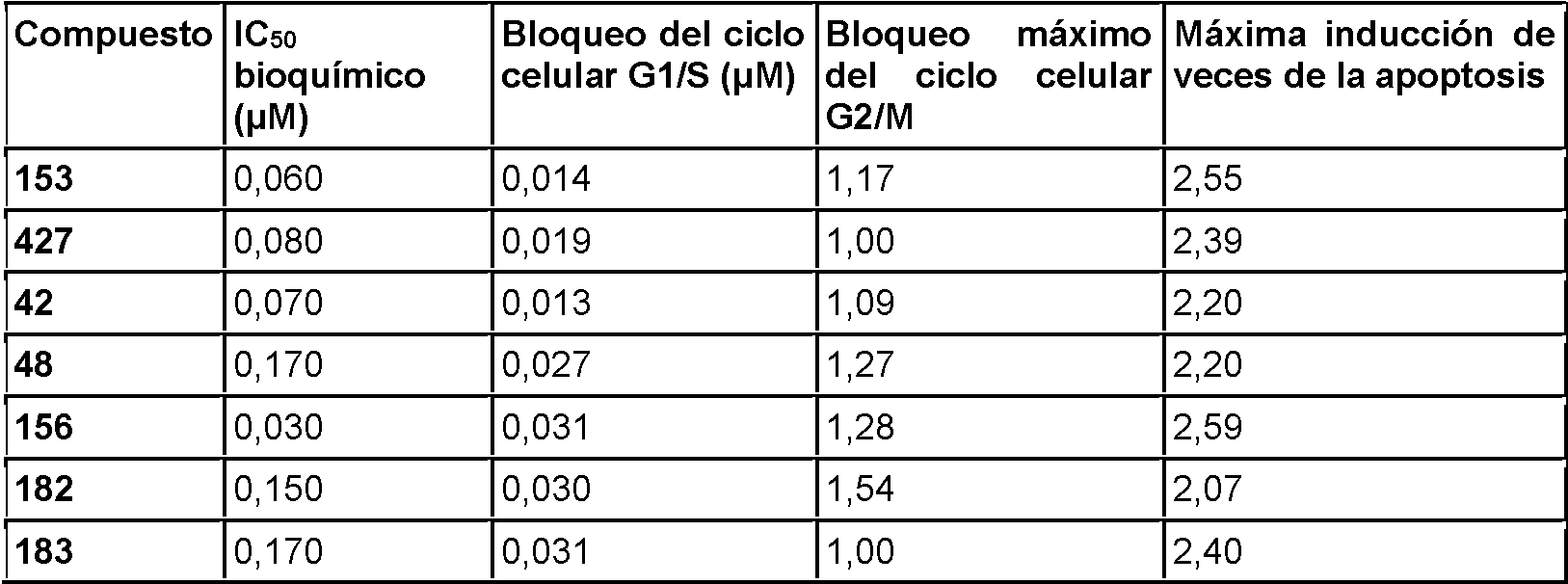
Claims (5)
1. Un compuesto seleccionado entre

o una sal farmacéuticamente aceptable de los mismos, para su uso en el tratamiento de un cáncer,
en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de célula no pequeña (NSCLC) y tumores colorrectales con mutación KRAS.
2. Un primer agente terapéutico y un segundo agente terapéutico para su uso en el tratamiento de un cáncer en un sujeto, en el que el primer agente terapéutico se selecciona del grupo que consiste en

o una sal farmacéuticamente aceptable de los mismos,
en el que el cáncer se selecciona del grupo que consiste en carcinoma de pulmón de célula no pequeña (NSCLC) y tumores colorrectales con mutación KRAS.
3. El primer agente terapéutico y el segundo agente terapéutico para el uso de la reivindicación 2, en el que el segundo agente terapéutico es un agente terapéutico contra el cáncer seleccionado entre paclitaxel, doxorrubicina, vincristina, actinomicina D, altretamina, asparaginasa, bleomicina, busulfán, cabazitaxel, capecitabina carboplatino, carmustina, clorambucil, cisplatino, ciclofosfamida, citarabina, dacarbazina, daunorubicina, docetaxel, epirubicina, etopósido, fludarabina, fluorouracilo, gemcitabina, hidroxiurea, idarubicina, ifosfamida, irinotecán, lomustina, melfalán, mercaptopurina, metotrexato, mitomicina, mitozantrona, oxaliplatino, procarbazina, esteroides, estreptozocina, taxotere, tamozolomida, tioguanina, tiotepa, tomudex, topotecán, treosulfán, uracil-tegufur, vinblastina, vindesina, nivolumab, pembrolizumab, MPDL3280A, MED14736, olaparib, erlotinib, necitumumab, traztuzamab, pertuzamab, lapatinib, crizotinib, cabozantinib, onartuamab, ramucirumab, bevacizumab, enzalutamida, abiraterona, tamoxifeno, cobimetinib, vemurafenib, everolimus, Kadyzla, sirolimus, avastin, nexavar, sutent, exemtesane, femora, enzalutamida, bicalutamida, Tafinlar y Zelboraf.
4. El primer agente terapéutico y el segundo agente terapéutico para el uso de las reivindicaciones 2 o 3, en el que el primer y el segundo agentes terapéuticos deben administrarse en la misma unidad de dosificación.
5. El primer agente terapéutico y el segundo agente terapéutico para el uso de la reivindicación 2 o 3, en el que el primer y el segundo agentes terapéuticos deben administrarse en unidades de dosificación separadas.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562135631P | 2015-03-19 | 2015-03-19 | |
| PCT/US2016/022471 WO2016149271A1 (en) | 2015-03-19 | 2016-03-15 | Heterocyclic modulators of lipid synthesis |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2910049T3 true ES2910049T3 (es) | 2022-05-11 |
Family
ID=55640910
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16712623T Active ES2910049T3 (es) | 2015-03-19 | 2016-03-15 | Moduladores heterocíclicos de la síntesis de lípidos |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US10189822B2 (es) |
| EP (1) | EP3277675B1 (es) |
| JP (1) | JP6758314B2 (es) |
| KR (1) | KR20170129151A (es) |
| CN (1) | CN107428728A (es) |
| CA (1) | CA2979696A1 (es) |
| ES (1) | ES2910049T3 (es) |
| WO (1) | WO2016149271A1 (es) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20170119786A1 (en) | 2011-03-08 | 2017-05-04 | 3-V Biosciences, Inc. | Heterocyclic modulators of lipid synthesis |
| AU2013286894B2 (en) | 2012-07-03 | 2017-12-07 | Sagimet Biosciences Inc. | Heterocyclic modulators of lipid synthesis |
| ES2833454T3 (es) | 2014-08-15 | 2021-06-15 | Sagimet Biosciences Inc | Inhibidor de la ácido graso sintasa para su uso en el tratamiento de un cáncer farmacorresistente |
| KR20240036703A (ko) * | 2016-11-11 | 2024-03-20 | 새지메트 바이오사이언시스, 인코포레이티드 | 지질 합성의 헤테로사이클릭 조절인자 |
| NZ755835A (en) | 2017-01-17 | 2023-12-22 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| CN109320594B (zh) * | 2018-11-13 | 2021-09-28 | 四川大学 | 一种禽传染性支气管炎和新城疫的病毒样颗粒、制备方法及应用 |
| WO2023220223A1 (en) * | 2022-05-13 | 2023-11-16 | Merck Sharp & Dohme Llc | Inhibitors of human respiratory syncytial virus and metapneumovirus |
| WO2023220225A1 (en) * | 2022-05-13 | 2023-11-16 | Merck Sharp & Dohme Llc | Inhibitors of human respiratory syncytial virus and metapneumovirus |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3625214A (en) | 1970-05-18 | 1971-12-07 | Alza Corp | Drug-delivery device |
| US4906474A (en) | 1983-03-22 | 1990-03-06 | Massachusetts Institute Of Technology | Bioerodible polyanhydrides for controlled drug delivery |
| US4789734A (en) | 1985-08-06 | 1988-12-06 | La Jolla Cancer Research Foundation | Vitronectin specific cell receptor derived from mammalian mesenchymal tissue |
| US5023252A (en) | 1985-12-04 | 1991-06-11 | Conrex Pharmaceutical Corporation | Transdermal and trans-membrane delivery of drugs |
| NL8720442A (nl) | 1986-08-18 | 1989-04-03 | Clinical Technologies Ass | Afgeefsystemen voor farmacologische agentia. |
| US5075109A (en) | 1986-10-24 | 1991-12-24 | Southern Research Institute | Method of potentiating an immune response |
| US5811128A (en) | 1986-10-24 | 1998-09-22 | Southern Research Institute | Method for oral or rectal delivery of microencapsulated vaccines and compositions therefor |
| US5001139A (en) | 1987-06-12 | 1991-03-19 | American Cyanamid Company | Enchancers for the transdermal flux of nivadipine |
| US4992445A (en) | 1987-06-12 | 1991-02-12 | American Cyanamid Co. | Transdermal delivery of pharmaceuticals |
| US4897268A (en) | 1987-08-03 | 1990-01-30 | Southern Research Institute | Drug delivery system and method of making the same |
| US5766573A (en) | 1988-12-06 | 1998-06-16 | Riker Laboratories, Inc. | Medicinal aerosol formulations |
| US5190029A (en) | 1991-02-14 | 1993-03-02 | Virginia Commonwealth University | Formulation for delivery of drugs by metered dose inhalers with reduced or no chlorofluorocarbon content |
| US5376359A (en) | 1992-07-07 | 1994-12-27 | Glaxo, Inc. | Method of stabilizing aerosol formulations |
| WO1994015635A1 (en) | 1993-01-11 | 1994-07-21 | Dana-Farber Cancer Institute | Inducing cytotoxic t lymphocyte responses |
| CA2630460C (en) | 2005-12-01 | 2013-01-08 | F. Hoffmann-La Roche Ag | Heteroaryl substituted piperidine derivatives as l-cpt1 inhibitors |
| US7994222B2 (en) | 2006-09-05 | 2011-08-09 | Bipar Sciences, Inc. | Monitoring of the inhibition of fatty acid synthesis by iodo-nitrobenzamide compounds |
| WO2008059214A1 (en) | 2006-11-13 | 2008-05-22 | Astrazeneca Ab | Bisamlde derivatives and use thereof as fatty acid synthase inhibitors |
| TW200833663A (en) | 2006-12-21 | 2008-08-16 | Astrazeneca Ab | Therapeutic agents |
| TW200831092A (en) | 2006-12-21 | 2008-08-01 | Astrazeneca Ab | Therapeutic agents |
| WO2008075077A1 (en) | 2006-12-21 | 2008-06-26 | Astrazeneca Ab | Piperidine derivatives for the treatment of obesity |
| AR086837A1 (es) | 2011-03-08 | 2014-01-29 | 3 V Biosciences Inc | Moduladores heterociclicos de sintesis lipidica |
| ES2624131T3 (es) * | 2011-03-08 | 2017-07-13 | 3-V Biosciences, Inc. | Moduladores heterocíclicos de la síntesis de lípidos |
| US9624173B2 (en) | 2011-03-08 | 2017-04-18 | 3-V Biosciences, Inc. | Heterocyclic modulators of lipid synthesis |
| AU2013286894B2 (en) * | 2012-07-03 | 2017-12-07 | Sagimet Biosciences Inc. | Heterocyclic modulators of lipid synthesis |
| KR20160100329A (ko) * | 2013-12-20 | 2016-08-23 | 3-브이 바이오사이언시스, 인코포레이티드 | 지질 합성의 헤테로사이클릭 조절물질 및 이들의 조합물 |
| ES2833454T3 (es) * | 2014-08-15 | 2021-06-15 | Sagimet Biosciences Inc | Inhibidor de la ácido graso sintasa para su uso en el tratamiento de un cáncer farmacorresistente |
-
2016
- 2016-03-15 CN CN201680015585.1A patent/CN107428728A/zh active Pending
- 2016-03-15 ES ES16712623T patent/ES2910049T3/es active Active
- 2016-03-15 KR KR1020177026446A patent/KR20170129151A/ko not_active Application Discontinuation
- 2016-03-15 JP JP2017549031A patent/JP6758314B2/ja active Active
- 2016-03-15 US US15/558,813 patent/US10189822B2/en active Active
- 2016-03-15 WO PCT/US2016/022471 patent/WO2016149271A1/en active Application Filing
- 2016-03-15 CA CA2979696A patent/CA2979696A1/en active Pending
- 2016-03-15 EP EP16712623.4A patent/EP3277675B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP6758314B2 (ja) | 2020-09-23 |
| JP2018508547A (ja) | 2018-03-29 |
| EP3277675B1 (en) | 2022-01-19 |
| US20180079746A1 (en) | 2018-03-22 |
| RU2017132497A (ru) | 2019-04-19 |
| RU2017132497A3 (es) | 2019-07-17 |
| US10189822B2 (en) | 2019-01-29 |
| CA2979696A1 (en) | 2016-09-22 |
| WO2016149271A1 (en) | 2016-09-22 |
| KR20170129151A (ko) | 2017-11-24 |
| CN107428728A (zh) | 2017-12-01 |
| EP3277675A1 (en) | 2018-02-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2910049T3 (es) | Moduladores heterocíclicos de la síntesis de lípidos | |
| ES2883274T3 (es) | Moduladores heterocíclicos de la síntesis de lípidos | |
| CN103702561B (zh) | 阿片样物质受体配体以及使用和制备其的方法 | |
| KR102027598B1 (ko) | 타이로신 키나아제 저해제 | |
| ES2470673T3 (es) | Composiciones y métodos para el tratamiento de la isquemia y de la lesión por isquemia y reperfusi�n | |
| EP3251694A1 (en) | Drug combinations for the treatment of duchenne muscular dystrophy | |
| TW201300385A (zh) | 用於治療cns病症和癌症的8-乙基-6-(芳基)吡啶并[2,3-d]嘧啶-7(8h)-酮 | |
| PT2201840E (pt) | Inibidores da tirosina quinase de bruton | |
| ES2733092T3 (es) | Derivados de benzodioxol como inhibidores de la fosfodiesterasa | |
| CN104093717A (zh) | 用于治疗细胞增殖性障碍的pak抑制剂 | |
| ES2886935T3 (es) | Inhibidores de FASN para su uso en el tratamiento de esteatohepatitis no alcohólica | |
| CA3017028C (en) | Fibrotic treatment | |
| US10413583B2 (en) | Synthetic substrates for enzymes that catalyze reactions of modified cysteines and related methods | |
| US20150259292A1 (en) | Heterocyclic modulators of lipid synthesis | |
| WO2021141969A1 (en) | Nanomaterials | |
| CN106715421A (zh) | 用于治疗hiv相关病症的方法和组合物 | |
| RU2549441C2 (ru) | Способы и фармацевтические композиции для лечения синдрома дауна | |
| JP2022501390A (ja) | 化合物及びその用途 | |
| CN105377838A (zh) | 新抗疟剂 | |
| EP4101468A1 (en) | Anticancer agent composition | |
| US9797882B2 (en) | Method of screening for a compound for inhibitory activity of FN14-tweak interaction | |
| RU2774758C2 (ru) | Гетероциклические модуляторы синтеза липидов | |
| ES2694202T3 (es) | Idebenona deuterada | |
| US11197854B1 (en) | Inhibitors for targeting flaviviruses | |
| US20200330453A1 (en) | Therapeutic agent for solid cancers, which comprises axl inhibitor as active ingredient |