ES2906305T3 - Profármacos de derivados de triazol sustituido y sus usos - Google Patents
Profármacos de derivados de triazol sustituido y sus usos Download PDFInfo
- Publication number
- ES2906305T3 ES2906305T3 ES18785394T ES18785394T ES2906305T3 ES 2906305 T3 ES2906305 T3 ES 2906305T3 ES 18785394 T ES18785394 T ES 18785394T ES 18785394 T ES18785394 T ES 18785394T ES 2906305 T3 ES2906305 T3 ES 2906305T3
- Authority
- ES
- Spain
- Prior art keywords
- compounds
- compound
- triazol
- general formula
- receptor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Abstract
Un compuesto de fórmula general (I): **(Ver fórmula)** en la que R1 representa un grupo de fórmula **(Ver fórmula)** en la que representa el punto de unión con el anillo 1,2,4-triazolilo, o una de las sales farmacéuticamente aceptables de estos, solvatos de estos o solvatos de las sales de estos.
Description
DESCRIPCIÓN
Profármacos de derivados de triazol sustituido y sus usos
La presente invención se refiere a profármacos de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxamida, 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxamida y 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxamida, a procedimientos para la preparación de dichos compuestos, a composiciones farmacéuticas que contienen dichos compuestos y a dichos compuestos o composiciones para su uso en el tratamiento y/o prevención de enfermedades, en particular para el tratamiento y/o prevención de enfermedades renales y cardiovasculares.
Los profármacos son derivados de un principio activo que se somete in vivo a una biotransformación enzimática y/o química en una o más etapas antes de liberarse el principio activo real. Normalmente se usa un residuo de profármaco para mejorar el perfil de propiedades del principio activo subyacente [P. Ettmayer et ál, J. Med. Chem.
47, 2393 (2004)]. Para lograr un perfil óptimo de efectos es necesario con respecto al diseño del residuo de profármaco, así como el mecanismo de liberación deseado ajustarse de manera muy precisa al principio activo individual, la indicación, el sitio de acción y la vía de administración. Una gran cantidad de medicamentos se administran como profármacos que muestran una biodisponibilidad mejorada en comparación con el principio activo subyacente, que se logra, por ejemplo, mejorando el perfil fisicoquímico, específicamente la solubilidad, las propiedades de absorción activa y pasiva o la distribución específica del tejido. Un ejemplo que se puede mencionar de la literatura de amplio espectro sobre profármacos es: H. Bundgaard (Ed.), Design of Prodrugs: Bioreversible derivatives for various functional groups and chemical entities, Elsevier Science Publishers B.V., 1985. 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxamida (Ejemplo 4A), 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxamida (Ejemplo 6A) y 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxamida (Ejemplo 8A) son antagonistas altamente potentes y selectivos del receptor V1a, como se desvela en el documento WO 2017/191102-A1 (ejemplos 1 y 2) y el documento WO 2017/191107-A1 (ejemplo 1).

La vasopresina es una neurohormona que básicamente regula la homeostasis del agua y el tono vascular. Se produce en neuronas endócrinas especializadas en el Nucleus supraopticus y N. paraventricularis en la pared del tercer ventrículo (hipotálamo) y es transportada desde allí a través de los procedimientos neurales hacia los lóbulos posteriores de la hipófisis (neurohipófisis). Allí la hormona es liberada en el torrente sanguíneo en respuesta a diferentes estímulos fisiológicos y patofisiológicos. Una regulación neurohormonal alterada se manifiesta esencialmente en una elevación del tono simpático y una activación inapropiada del sistema renina-angiotensinaaldosterona (RAAS). Mientras la inhibición de estos componentes por bloqueadores del receptor beta por un lado y los inhibidores ACE o bloqueadores del receptor de angiotensina por el otro es ahora una parte inherente del tratamiento farmacológico de las enfermedades cardiovasculares, la elevación inapropiada de la secreción de vasopresina actualmente no se puede tratar adecuadamente.
La vasopresina ejerce su acción principalmente uniéndose a tres receptores, que se clasifican como receptores V1a, V1b y V2 y que pertenecen a la familia de los receptores acoplados a la proteína G.
Los receptores V2 se ubican en el epitelio tubular distal y el epitelio de los túbulos recolectores del riñón. Su activación hace que estos epitelios sean permeables al agua. Este fenómeno se debe a la incorporación de acuaporinas (canales especiales de agua) en la membrana luminal de las células epiteliales. Por consiguiente, la inhibición farmacológica de la acción de la vasopresina sobre el receptor V2 da como resultado una mayor excreción de orina. Por lo tanto, los fármacos con actividad antagonista de V2 parecen ser particularmente adecuados para el tratamiento de todas las patologías asociadas con una sobrecarga del cuerpo con agua.
Los receptores V1b (también denominaos receptores V3) son detectables principalmente en el sistema nervioso central. Junto con la hormona liberadora de corticotropina (CRH), la vasopresina regula la secreción basal e inducida por estrés de la hormona adrenocorticotropa (ACTH) mediante el receptor V1b.
Los receptores V1a se ubican principalmente en células de músculo liso vascular (VSMC) pero también en
cardiomiocitos, fibroblastos y células renales especializadas como células mesangiales glomerulares o células de la mácula densa que controlan la liberación de renina [Wasilewski MA, Myers VD, Recchia FA, Feldman AM, Tilley DG, Cell Signal., 28(3), 224-233, (2016)]. La activación del receptor VSMC V1a por vasopresina provoca la liberación de calcio intracelular y vasoconstricción acorde. Entonces, la estimulación de los receptores VSMC V1a provoca resistencia vascular aumentada y mejor poscarga cardíaca. El gasto cardiaco se ve perjudicado por la vasoconstricción mediada por V1a. El aumento en la poscarga y la estimulación directa de los receptores V1a en cardiomiocitos puede llevar a hipertrofia cardíaca y remodelación que incluye fibrosis. Los ratones con sobreexpresión específica cardíaca del receptor V1a desarrollan hiperetrofia cardíaca que lleva a dilatación y disfunción del ventrículo izquierdo, lo que sugiere un rol esencial para el receptor V1a en el desarrollo de insuficiencia cardíaca [Li X, Chan TO, Myers V, Chowdhury I, Zhang XQ, Song J, Zhang J, Andrel J, Funakoshi H, Robbins J, Koch WJ, Hyslop T, Cheung JY, Feldman AM, Circulation.; 124, 572-581 (2011)].
El receptor V1a también se expresa en la vasculatura medular y cortical renal en la que media la vasoconstricción de vasos renales y afecta al flujo sanguíneo renal global. Por lo tanto, la activación del receptor V1a puede reducir el flujo sanguíneo medular lo que induce otros procedimientos patológicos como hipoxia tisular, oxígeno reducido y suministro de energía acorde para procedimientos de transporte, así como daños directos de las células mesangiales y de la mácula densa. Se ha demostrado que la activación del receptor V1a mesangial media la señalización de TGFp y provoca un aumento en la producción de colágeno IV. Mientras esta señalización contribuye a la acumulación de matriz extracelular y remodelación en el riñón, se cree que ocurren rutas de señalización similares en las células cardíacas, especialmente después de un infarto del miocardio, lo cual enfatiza el rol principal del receptor V1a en el desarrollo de procedimientos hipertróficos y fibróticos como respuesta a niveles de vasopresina patofiosiológicos elevados [Wasilewski MA, Myers VD, Recchia FA, Feldman AM, Tilley DG. Señalización del receptor de arginina vasopresina y resultados funcionales en insuficiencia cardíaca. Cell Signal., 28(3), 224-233 (2016)].
Dado que los receptores V1a se expresan principalmente en VSMC y por tanto participan en la función vascular, es posible un enlace con enfermedades vasculares como enfermedad arterial periférica (PAD) que incluye claudicación e isquemia aguda de las extremidades, así como disfunción microvascular coronaria (CMD).
Aparte de esto, los receptores V1a también se expresan en plaquetas humanas y el hígado. El significado de receptores V1a de plaquetas no se entiende totalmente, aunque la vasopresina induce la agregación de plaquetas humanas mediante el receptor V1a a altas concentraciones ex vivo. Por lo tanto, la inhibición de agregación plaquetaria inducida por vasopresina por antagonistas del receptor V1a es un ensayo farmacológico ex vivo útil que usa tejido humano que expresa de forma endógena el receptor V1a [Thibonnier M, Roberts JM, J Clin Invest.; 76:1857-1864, (1985)].
La vasopresina estimula la gluconeogénesis y glucogenólisis mediante activación del receptor V1a hepático. Estudios animales han demostrado que la vasopresina perjudica la tolerancia a la glucosa que podría inhibirse por un antagonista del receptor V1a proporcionando así un enlace del receptor de vasopresina V1a con la diabetes mellitus. [Taveau C, Chollet C, Waeckel L, Desposito D, Bichet DG, Arthus MF, Magnan C, Philippe E, Paradis V, Foufelle F, Hainault I, Enhorning S, Velho G, Roussel R, Bankir L, Melander O, Bouby N. Vasopressin and hydration play a major role in the development of glucose intolerance and hepatic steatosis in obese rats. Diabetologia, 58(5), 1081-1090, (2015)]. Se ha demostrado que la vasopresina contribuye al desarrollo de albuminuria y nefropatía inducida por diabetes en modelos animales, lo que es coherente con los descubrimientos epidemiológicos en seres humanos.
Se descubrió recientemente que la vasopresina también parece cumplir una función causal en el desarrollo de la preeclampsia. La infusión crónica de vasopresina durante el embarazo en ratones es suficiente para inducir todos los fenotipos fetales y maternales principales asociados con la preeclampsia humana, que incluye hipertensión específica del embarazo [Santillan Mk , Santillan DA, Scroggins s M, Min JY, Sandgren JA, Pearson NA, Leslie KK, Hunter SK, Zamba GK, Gibson-Corley KN, Grobe JL. Vasopressin in preeclampsia: a novel very early human pregnancy biomarker and clinically relevant mouse model. Hypertension. 64(4), 852-859, (2014)].
Los niveles de vasopresina pueden estar elevados en mujeres con dismenorrea (trastorno ginecológico caracterizado por dolor pélvico de calambres cíclicos) durante la menstruación, que parece aumentar la contracción del músculo liso miometrial. Se descubrió recientemente que un antagonista del receptor V1a de vasopresina selectivo (relcovaptan/SR-49059) puede reducir las contracciones intrauterinas provocadas por la vasopresina. Por estas razones, los agentes que inhiben la acción de la vasopresina sobre el receptor V1a parecen ser adecuados para el tratamiento de varias enfermedades cardíacas. En particular, los agentes que inhiben la acción de vasopresina selectivamente en el receptor V1a ofrecen un perfil especialmente ideal para el tratamiento de pacientes de otra manera normovolémicos, es decir, aquellos que no son elegibles para descongestión mediante, por ejemplo, altas dosis de diuréticos de bucle o antagonistas de V2 y en los que la acuaresis inducida mediante inhibición de V2 puede ser no deseada.
Determinados derivados de 4-fenil-1,2,4-triazol-3-ilo se han descrito en el documento WO 2005/063754-A1 y el documento WO 2005/105779-A1 por actuar como antagonistas del receptor V1a de vasopresina que son útiles para
el tratamiento de trastornos ginecológicos, particularmente trastornos menstruales tales como dismenorrea.
En el documento WO 2011/104322-A1, un grupo particular de 1,2,4-triazol-3-onas unidas por bis-arilo, que incluyen 5-fenil-1,2,4-triazol-3-ilo y derivados de 1-fenil-1,2,3-triazol-4-ilo de este, se han descrito como antagonistas de los receptores V2 y/o V1a de vasopresina útiles para el tratamiento y/o prevención de enfermedades cardiovasculares. Sin embargo, los compuestos descritos no muestran selectividad suficiente hacia el receptor V1a y en su mayoría muestran actividad combinada en ambos receptores de vasopresina V1a y V2. Con todo, como se describe anteriormente, una alta afinidad, así como una alta selectividad para el receptor V1a es una condición previa deseada para el tratamiento de patologías en las que no se desea una descongestión y puede llevar a una homeostasis de fluido corporal desregulado que incluye reducción de osmolalidad en plasma sanguíneo en individuos de otra manera normovolémicos.
En el documento WO 2016/071212-A1 determinados derivados de 5-(hidroxialquil)-1-fenil-1,2,4-triazol han sido descritos, que actúan como antagonistas potentes de receptores de vasopresina V1a y V2 y, además, muestran una potencia acuarética considerablemente potenciada in vivo después de la aplicación oral. Los compuestos se describen como útiles para el tratamiento y/o la prevención de enfermedades cardiovasculares y renales. Con todo, como se describe anteriormente, una alta afinidad, así como una alta selectividad para el receptor V1a es una condición previa deseada para el tratamiento de patologías en las que no se desea una descongestión y puede llevar a una homeostasis de fluido corporal desregulado que incluye reducción de osmolalidad en plasma sanguíneo en individuos de otra manera normovolémicos.
En el documento WO 2017/191107-A1 y el documento WO 2017/191102-A1 se han descrito determinados derivados de 5-(carboxamida)-1-fenil-1,2,4-triazol así como en el documento WO 2017/191114-A1 se han descrito derivados específicos de 5-(hidroxialquil)-1-heteroaril-1,2,4-triazol, que representan antagonistas altamente selectivos y potentes del receptor V1a y son particularmente útiles para el tratamiento y/o prevención de enfermedades cardiovasculares y renales en sujetos que no sufren de sobrecarga de fluidos y que, por lo tanto, no deberían descongestionarse.
Otros derivados novedosos de 1,2,4-triazol 5-(carboxamida)-sustituidos, 5-(fluoroalquil)-sustituidos y 3-(hidroxialquil)-sustituidos han sido descritos como antagonistas de receptores de vasopresina V2 y/o V1a en los documentos WO 2017/191105-A1, WO 2017/191112-A1, WO 2017/191115-A1 y WO 2018/073144-A1.
Un perfil de actividad con una alta selectividad para el receptor V1a tiene un bajo potencial para provocar efectos secundarios fuera del objetivo no deseados y también ayudaría a reducir la cantidad de sustancia que será necesaria para lograr y mantener el efecto terapéutico deseado, limitando así la posibilidad de efectos secundarios no aceptables y/o interacciones fármaco-fármaco no deseadas durante el tratamiento de pacientes que ya podrían estar en alto riesgo, tales como, por ejemplo, en enfermedades cardíacas o renales agudas o crónicas.
Un problema técnico a resolver de acuerdo con la presente invención por lo tanto puede ser la identificación y suministro de nuevos compuestos que actúen como potentes antagonistas del receptor V1A de vasopresina. Otro objeto de la invención es identificar y proporcionar nuevos compuestos con una alta afinidad y selectividad vis-a-vis el receptor de vasopresina V1a. Los compuestos pretenden evitar inducir la acuaresis mediante inhibición de V2. Además, se pretende que los compuestos tengan un perfil terapéutico similar o mejorado en comparación con los compuestos conocidos de la técnica anterior, por ejemplo, con respecto a sus propiedades in vivo, por ejemplo, sus características farmacocinéticas y farmacodinámicas y/o su perfil metabólico y/o su relación dosis-actividad.
Sin embargo, los compuestos del Ejemplo 4A, Ejemplo 6A y Ejemplo 8A que son antagonistas altamente potentes y selectivos del receptor de V1a, tienen una solubilidad limitada en agua y medio fisiológico, haciendo difícil, por ejemplo, la administración intravenosa de los compuestos del Ejemplo 4A, Ejemplo 6A y Ejemplo 8A. Además, la biodisponibilidad de los compuestos después de la administración oral de los compuestos debería mejorar. Por lo tanto, era otro objeto de la presente invención identificar derivados o profármacos de los compuestos del Ejemplo 4A, Ejemplo 6A y Ejemplo 8A que tienen una solubilidad mejorada en el medio mencionado y, al mismo tiempo, permiten la liberación controlada de los compuestos del Ejemplo 4A, Ejemplo 6A y Ejemplo 8a en el cuerpo del paciente después de la administración y/o que tienen una buena biodisponibilidad después de la administración oral. Sorprendentemente, se ha descubierto ahora que determinados profármacos de los compuestos del Ejemplo 4A, Ejemplo 6A y Ejemplo 8A tienen este perfil específico y hace que los compuestos de la invención sean útiles para el tratamiento y/o prevención de enfermedades, que están asociadas con la activación del receptor de V1a. Los compuestos de la presente invención son particularmente útiles para el tratamiento y/o prevención de enfermedades renales y cardiovasculares en sujetos que no sufren sobrecarga de fluidos y que por lo tanto no deberían descongestionarse.
La invención proporciona compuestos de fórmula general (I)

en la que
R1 representa un grupo de fórmula

en la que
# representa el punto de unión con el anillo 1,2,4-triazolilo
y sales farmacéuticamente aceptables de estos, sus solvatos y solvatos de las sales de estos.
Los términos como se mencionan en el presente texto tienen los significados a continuación:
La expresión “que comprende”, cuando se usa en la memoria descriptiva, incluye “que consiste en”.
En las fórmulas del grupo que representa R1, el punto final de la línea marcada por # no representa un átomo de carbono o un grupo CH2, pero es parte del enlace con el átomo con que R1 está unido.
Es posible que los compuestos de fórmula general (I) existan como variantes isotópicas.
La expresión “variante isotópica” de un compuesto o un reactivo se define como un compuesto que muestra una proporción no natural de uno o más de los isótopos que constituyen dicho compuesto.
La expresión “variante isotópica de un compuesto de fórmula general (I)” se define como un compuesto de fórmula general (I) que muestra una proporción poco natural de uno o más de los isótopos que constituyen dicho compuesto. La expresión “proporción poco natural” significa una proporción de dicho isótopo que es mayor que su abundancia natural. Las abundancias naturales de isótopos a ser aplicadas en este contexto se describen en “Isotopic Compositions of the Elements 1997”, Pure Appl. Chem., 70(1), 217-235, 1998. Ejemplos de dichos isótopos incluyen isótopos estables y radioactivos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, azufre, flúor, cloro, bromo y yodo, tales como 2H (deuterio), 3H (tritio), 11C, 13C, 14C, 15N, 17O, 18O, 32P, 33P, 33S, 34S, 35S, 36S, 18F, 36Cl, 82Br, 123I, 124I, 125i, 129i y 131i, respectivamente.
Con respecto al tratamiento y/o prevención de los trastornos especificados en el presente documento las una o más variantes isotópicas de los compuestos de fórmula general (I) contienen preferentemente deuterio (“compuestos que contienen deuterio de fórmula general (I)”). Las variantes isotópicas de los compuestos de fórmula general (I) en las que se incorporan uno o más isótopos radioactivos, tales como 3H o 14C, son útiles, por ejemplo, en los estudios de distribución de tejido de sustrato y/o fármaco. Estos isótopos se prefieren particularmente por la facilidad de su incorporación y detectabilidad. Los isótopos que emiten positrones, tales como 18F o 11C, se pueden incorporar en un compuesto de fórmula general (I). Estas variantes isotópicas de los compuestos de fórmula general (I) son útiles para aplicaciones de imagenología in vivo. Los compuestos que contienen deuterio y que contienen 13C de fórmula general (I) se pueden usar en análisis de espectrometría de masas (H. J. Leis et ál., Curr. Org. Chem., 1998, 2, 131) en el contexto de estudios preclínicos o clínicos.
Las variantes isotópicas de los compuestos de fórmula general (I) pueden prepararse generalmente por procedimientos conocidos por los expertos en la técnica, tales como los descritos en los esquemas y/o ejemplos en el presente documento, sustituyendo un reactivo por una variante isotópica de dicho reactivo, preferentemente por un reactivo que contiene deuterio. Dependiendo de los sitios deseados de deuteración, en algunos casos el deuterio
de D2O se puede incorporar directamente en los compuestos o en reactivos que son útiles para sintetizar dichos compuestos (Esaki et ál., Tetrahedron, 2006, 62, 10954; Esaki et ál., Chem. Eur. J., 2007, 13, 4052). El gas de deuterio también es un reactivo útil para incorporar deuterio en moléculas. La deuteración catalítica de enlaces olefínicos (H. J. Leis et ál., Curr. Org. Chem., 1998, 2, 131; J. R. Morandi et ál., J. Org. Chem., 1969, 34 (6), 1889) y enlaces acetilénicos (N. H. Khan, J. Am. Chem. Soc., 1952, 74 (12), 3018; S. Chandrasekhar et ál., Tetrahedron Letters, 2011, 52, 3865) es una vía directa para la incorporación de deuterio. Los catalizadores metálicos (es decir Pd, Pt, y Rh) en presencia de gas de deuterio se pueden usar para intercambiar directamente deuterio por hidrógeno en grupos funcionales que contienen hidrocarburos (J. G. Atkinson et ál., patente estadounidense 3966781). Una variedad de reactivos deuterados y bloques de construcción sintéticos están comercialmente disponibles de compañías tales como, por ejemplo, C/D/N Isotopes, Quebec, Canadá; Cambridge Isotope Laboratories Inc., Andover, MA, EUA y CombiPhos Catalysts, Inc., Princeton, NJ, EUA. Se proporciona más información acerca del estado de la técnica con respecto al intercambio de deuterio-hidrógeno, por ejemplo, en Hanzlik et ál., J. Org. Chem.
55, 3992-3997, 1990; R. P. Hanzlik et ál., Biochem. Biophys. Res. Commun. 160, 844, 1989; P. J. Reider et ál., J. Org. Chem. 52, 3326-3334, 1987; M. Jarman et ál., Carcinogenesis 16(4), 683-688, 1995; J. Atzrodt et ál., Angew. Chem., Int. Ed. 2007, 46, 7744; K. Matoishi et ál., Chem. Commun. 2000, 1519-1520; K. Kassahun et ál., WO2012/112363.
La expresión “compuesto que contiene deuterio de fórmula general (I)” se define como un compuesto de fórmula general (I), en el que uno o más átomos de hidrógeno se remplazan por uno o más átomos de deuterio y en el que la abundancia de deuterio en cada posición deuterada del compuesto de fórmula general (I) es mayor que la abundancia natural de deuterio, que es de alrededor del 0,015 %. Particularmente, en un compuesto que contiene deuterio de fórmula general (I) la abundancia de deuterio en cada posición deuterada del compuesto de fórmula general (I) es mayor que el 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 % u 80 %, preferentemente mayor que el 90 %, 95 %, 96 % o 97 %, aún más preferentemente mayor que el 98 % o 99 % en dicha o dichas posiciones. Se entiende que la abundancia de deuterio en cada posición deuterada es independiente de la abundancia de deuterio en otra u otras posiciones deuteradas.
La incorporación selectiva de uno o más átomos de deuterio en un compuesto de fórmula general (I) puede alterar las propiedades fisicoquímicas (tales como por ejemplo acidez [C. L. Perrin, et ál., J. Am. Chem. Soc., 2007, 129, 4490; A. Streitwieser et ál., J. Am. Chem. Soc., 1963, 85, 2759;], basicidad [C. L. Perrin et ál., J. Am. Chem. Soc., 2005, 127, 9641; C. L. Perrin, et ál., J. Am. Chem. Soc., 2003, 125, 15008; C. L. Perrin in Advances in Physical Organic Chemistry, 44, 144], lipofilicidad [B. Testa et ál., Int. J. Pharm., 1984, 19(3), 271]) y/o el perfil metabólico de la molécula y puede dar como resultado cambios en la relación del compuesto original con los metabolitos o en las cantidades de metabolitos formados. Dichos cambios pueden dar como resultado determinadas ventajas terapéuticas y, por lo tanto, se pueden preferir en algunas circunstancias. Se han reportado relaciones reducidas de metabolismo y cambio metabólico, cuando la relación de metabolitos cambia (A. E. Mutlib et ál., Toxicol. Appl. Pharmacol., 2000, 169, 102; D. J. Kushner et ál., Can. J. Physiol. Pharmacol., 1999, 77, 79). Estos cambios en la exposición al fármaco original y metabolitos pueden tener consecuencias importantes con respecto a la farmacodinámica, tolerabilidad y eficacia de un compuesto que contiene deuterio de fórmula general (I). En algunos casos la sustitución de deuterio reduce o elimina la formación de un metabolito no deseado o tóxico y potencia la formación de un metabolito deseado (por ejemplo, nevirapina: A. M. Sharma et ál., Chem. Res. Toxicol., 2013, 26, 410; Efavirenz: A. E. Mutlib et ál., Toxicol. Appl. Pharmacol., 2000, 169, 102). En otros casos el efecto principal de la deuteración es reducir el índice de la depuración sistémica. Como resultado, la semivida biológica del compuesto aumenta. Los beneficios clínicos potenciales incluirían la capacidad de mantener exposición sistémica similar con niveles máximos reducidos y niveles de depresión elevados. Esto puede dar como resultado efectos secundarios menores y eficacia potenciada, dependiendo de la relación farmacocinética/farmacodinámica particular del compuesto. ML-337 (C. J. Wenthur et ál., J. Med. Chem., 2013, 56, 5208) y odanacatib (K. Kassahun et ál., WO2012/112363) son ejemplos para este efecto del deuterio. Se han reportado aun otros casos en los cuales los índices reducidos de metabolismo dan como resultado un aumento de la exposición del fármaco sin cambiar el índice de depuración sistémica (por ejemplo, Rofecoxib: F. Schneider et ál., Arzneim. Forsch. / Drug. Res., 2006, 56, 295; Telaprevir: F. Maltais et ál., J. Med. Chem., 2009, 52, 7993). Los fármacos deuterados que muestran este efecto pueden tener requisitos de dosificación reducidos (por ejemplo, menor cantidad de dosis o menor dosificación para alcanzar el efecto deseado) y/o pueden producir menores cargas de metabolito.
Un compuesto de fórmula general (I) puede tener múltiples sitios potenciales de ataque para metabolismo. Para optimizar los efectos antes descritos sobre las propiedades fisicoquímicas y el perfil metabólico, se pueden seleccionar compuestos que contienen deuterio de fórmula general (I) con determinado patrón de uno o más intercambios de deuterio-hidrógeno. Particularmente, el o los átomos de deuterio del compuesto o compuestos que contienen deuterio de fórmula general (I) se unen con un átomo de carbono y/o se ubican en esas posiciones del compuesto de fórmula general (I), que son sitios de ataque para metabolizar enzimas tales como, por ejemplo, citocromo P450.
Cuando en el presente documento se utilice la forma plural para la palabras compuestos, sales, polimorfos, hidratos, solvatos y similares, esto también significa un único compuesto, sal, polimorfo, isómero, hidrato, solvato o similar.
Un “compuesto estable” o “estructura estable”, se refiere a un compuesto que es lo suficientemente potente para sobrevivir el aislamiento a un nivel útil de pureza de una mezcla de reacción y su formulación en un agente
terapéutico eficaz.
Los profármacos son derivados de un principio activo. Las expresiones “principio activo subyacente”, “fármaco correspondiente subyacente” y “fármaco correspondiente” se usan como sinónimos en la presente invención.
Los compuestos de la presente invención opcionalmente contienen un centro asimétrico, dependiendo de la ubicación y naturaleza de los diferentes sustituyentes deseados. Es posible que un átomo de carbono asimétrico esté presente en la configuración (R) o (S), lo que puede dar como resultado mezclas racémicas. En determinados casos, es posible que la asimetría también esté presente debido a rotación limitada alrededor de un enlace dado, por ejemplo, el enlace central unido a dos anillos aromáticos sustituidos de los compuestos especificados. Los compuestos preferidos son aquellos que producen la actividad biológica más deseable. Los isómeros y estereoisómeros separados, puros o parcialmente purificados o mezclas racémicas de los compuestos de la presente invención también se incluyen en el alcance de la presente invención. La purificación y la separación de dichos materiales se pueden lograr por técnicas estándar conocidas en la técnica.
Los isómeros ópticos se pueden obtener mediante resolución de las mezclas racémicas de acuerdo con procedimientos convencionales, por ejemplo, por la formación de sales diastereoméricas usando un ácido o base ópticamente activa o formación de diastereómeros covalentes. Ejemplos de ácidos apropiados son ácido tartárico, diacetiltartárico, ditoluoiltartárico y canforsulfónico. Las mezclas diastereoméricas pueden ser separadas en sus diastereómeros individuales en función de sus diferencias físicas y/o químicas mediante procedimientos conocidos en la técnica, por ejemplo, por cromatografía o cristalización fraccionada. Las bases o ácidos ópticamente activos luego se liberan de las sales diastereoméricas separadas. Un procedimiento diferente para la separación de isómeros ópticos implica el uso de cromatografía quiral (por ejemplo, columnas de CLAR usando una fase quiral) con o sin derivatización convencional, óptimamente elegidas para maximizar la separación de los enantiómeros. Las columnas de CLAR adecuadas que usan una fase quiral están comercialmente disponibles, tales como las fabricadas por Daicel, por ejemplo, Chiracel OD y Chiracel OJ, por ejemplo, entre muchas otras, que se pueden seleccionar de forma rutinaria. Las separaciones enzimáticas con o sin derivatización, también son útiles. Los compuestos ópticamente activos de la presente invención asimismo se pueden obtener por síntesis quiral usando materiales de partida ópticamente activos. Para distinguir los diferentes tipos de isómeros entre sí se hace referencia a las normas IUPAC sección E (Pure Appl Chem 45, 11-30, 1976).
La presente invención incluye todos los estereoisómeros posibles de los compuestos de la presente invención como estereoisómeros simples o como cualquier mezcla de dichos estereoisómeros, por ejemplo, isómeros (R)- o (S)-, en cualquier proporción. El aislamiento de un estereoisómero individual, por ejemplo, un enantiómero individual o un diastereómero individual, de un compuesto de la presente invención se alcanza por medio de cualquier procedimiento adecuado del estado de técnica, tal como cromatografía, especialmente cromatografía quiral, por ejemplo.
En el contexto de la presente invención, la expresión "enantioméricamente puro" se entiende como que significa que el compuesto en cuestión con respecto a la configuración absoluta del centro quiral está presente en un exceso enantiomérico de más del 95 %, preferentemente más del 97 %. El exceso enantiomérico, ee, se calcula evaluando el cromatograma correspondiente CLAR en una fase quiral usando la fórmula a continuación:
ee = [EA (% de área) - EB (% de área)] x 100 % / [EA (% de área) EB (% de área)] (EA: enantiómero principal, EB: enantiómero secundario)
La presente invención también cubre formas útiles de los compuestos de la presente invención, tales como hidratos, solvatos, sales, en particular sales farmacéuticamente aceptables y/o coprecipitados.
Los compuestos de la presente invención pueden existir como un hidrato o como un solvato en el que los compuestos de la presente invención contienen disolventes polares, en particular agua, metanol o etanol, por ejemplo, como elemento estructural de la estructura cristalina de los compuestos. Es posible que la cantidad de disolventes polares, en particular agua, exista en una relación estequiométrica o no estequiométrica. En el caso de solvatos estequiométricos, por ejemplo, un hidrato, hemi-, (semi-), mono-, sesqui-, di-, tri-, tetra-, penta- etc. solvatos o hidratos, respectivamente, son posibles. La presente invención incluye todos dichos hidratos o solvatos. Los hidratos, en particular hemihidratos (semihidratos) son solvatos preferidos en el contexto de la presente invención. Además, es posible que los compuestos de la presente invención existan en forma libre, por ejemplo, como una base libre o como un ácido libre o como un zwitterion o que existan en forma de una sal. Dicha sal puede ser cualquier sal, ya sea sal de adición orgánica o inorgánica, particularmente cualquier sal de adición inorgánica u orgánica farmacéuticamente aceptable, que se usa habitualmente en farmacia, o que se usa, por ejemplo, para aislar o purificar los compuestos de la presente invención.
La expresión “sal farmacéuticamente aceptable” se refiere a sales de adición inorgánicas u orgánicas de un compuesto de la presente invención. Por ejemplo, véase S. M. Berge, et ál. “Pharmaceutical Salts,” J. Pharm. Sci.
1977, 66, 1-19.
Una sal farmacéuticamente aceptable adecuada de un compuesto de la presente invención que es suficientemente ácida, es una sal de metal alcalino, por ejemplo una sal de sodio, sal de potasio o sal de litio, una sal de metal alcalinotérreo, por ejemplo una sal de calcio, sal de magnesio o sal de estroncio o una sal de aluminio o una sal de zinc o una sal de amonio derivada de amoníaco o de una amina orgánica primaria, secundaria o terciaria con de 1 a 20 átomos de carbono, tales como etilamina, dietilamina, trietilamina, etildiisopropilamina, monoetanolamina, dietanolamina, trietanolamina, diciclohexilamina, dimetilaminoetanol, dietilaminoetanol, tris(hidroximetil)aminometano, procaína, dibencilamina, N-metilmorfolino, arginina, lisina, 1,2-etilendiamina, N-metilpiperidina, N-metil-glucamina, N;N-dimetil-glucamina, N-etil-glucamina, 1,6-hexanodiamina, glucosamina, sarcosina, serinol, 2-amino-1,3-propanodiol, 3-amino-1,2-propanodiol, 4-amino-1,2,3-butanotriol o una sal con un ion de amonio cuaternario con de 1 a 20 átomos de carbono, tales como tetrametilamonio, tetraetilamonio, tetra(npropil)amonio, tetra(n-butil)amonio, N-bencil-N,N,N-trimetilamonio, colina o benzalconio. Se prefiere una sal de sodio, sal de potasio, sal de litio, sal de calcio o sal de magnesio, la más preferida es una sal de potasio.
Las sales de metal alcalino y alcalinotérreo de compuestos ácidos de la presente invención se preparan haciendo reaccionar los compuestos de la presente invención con la base apropiada mediante una variedad de procedimientos conocidos.
La presente invención incluye todas las sales posibles de los compuestos de la presente invención como sales simples o como cualquier mezcla de dichas sales, en cualquier proporción.
En el presente texto, en particular en la Sección Experimental, para la síntesis de productos intermedios y de ejemplos de la presente invención, cuando un compuesto se menciona como una forma salina con la base o el ácido correspondiente, la composición estequiométrica exacta de dicha forma salina, como se obtiene por la preparación y/o procedimiento de purificación correspondientes es, en la mayoría de los casos, desconocida.
Salvo que se especifique lo contrario, los sufijos de nombres químicos o fórmulas estructurales con relación a sales, tales como "sal de sodio”, “sal de potasio”, o ”x Na+”, “x K+”, por ejemplo, se refieren a una forma salina, cuya estequiometría no se especifica.
Esto se aplica de forma análoga a casos en los cuales se obtuvieron productos intermedios de síntesis o compuestos de ejemplos o sales de estos por los procedimientos de preparación y/o purificación descritos, como solvatos, tales como hidratos, con (si se define) composición estequiométrica desconocida.
Además, la presente invención incluye todas las formas cristalinas posibles o polimorfos de los compuestos de la presente invención ya sea como polimorfo simple o como mezcla de más de un polimorfo, en cualquier proporción. Se prefieren compuestos de fórmula general (I) en la que
R1 representa un grupo de fórmula

en la que
# representa el punto de unión con el anillo 1,2,4-triazolilo.
También se prefiere dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo que tiene la fórmula a continuación
o sales farmacéuticamente aceptables de este, sus solvatos y solvatos de las sales de este.
También se prefiere dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo que tiene la fórmula a continuación

La presente invención cubre los compuestos de fórmula general (I) que se describen en la Sección Ejemplos de este texto.
La invención proporciona además un procedimiento para preparar los compuestos de fórmula general (I) o las sales farmacéuticamente aceptables de estos, sus solvatos o solvatos de las sales de estos, en los que
[A] los compuestos de fórmula

en la que
R1 tiene el significado como se define para los compuestos de fórmula general (I) anteriormente,
se hacen reaccionar en la primera etapa con oxicloruro de fósforo y en la segunda etapa se hidrolizan para dar compuestos de fórmula general (I)
o
[B] los compuestos de fórmula

en los que
R1 tiene el significado como se define para los compuestos de fórmula general (I) anteriormente,
se hacen reaccionar en la primera etapa con difosfato de tetrabencilo y en la segunda etapa los grupos bencilo se eliminan en condiciones de reducción para dar compuestos de fórmula general (I), opcionalmente seguido,
cuando fuera apropiado, de conversión de los compuestos de fórmula general (I) en sus correspondientes sales farmacéuticamente aceptables de estos, sus solvatos o los solvatos de las sales de estos por tratamiento con los disolventes y/o bases correspondientes.
La primera etapa en la reacción [A] se realiza generalmente haciendo reaccionar un compuesto de fórmula (II) con oxicloruro de fósforo en un disolvente inerte en presencia de una base, opcionalmente en presencia de un aditivo, preferentemente en un intervalo de temperaturas de -10 °C a 50 °C, más preferentemente de 0 ° C a 30 °C. Las reacciones se pueden realizar a presión atmosférica, elevada o reducida (por ejemplo, de 0,5 a 5 bar); en general, las reacciones se realizan a presión atmosférica.
Los disolventes inertes son, por ejemplo, hidrocarburos halogenados tales como diclorometano o triclorometano, éter tal como dietil éter o metil ferc-butil éter, hidrocarburos tales como benceno o tolueno, u otros disolventes tales como dioxano, dimetilformamida o tetrahidrofurano. Además, es posible usar mezclas de los disolventes. Se prefiere tetrahidrofurano.
Bases adecuadas son, por ejemplo, bases orgánicas tales como trialquilaminas, por ejemplo, trietilamina o diisopropiletilamina, o piridina. Se prefiere trietilamina.
Aditivos adecuados son, por ejemplo, 4-N,N-dimetilaminopiridina.
La segunda etapa en la reacción [A] se realiza generalmente agregando una base o agua, preferentemente en un intervalo de temperaturas de -10 °C a 50 °C, más preferentemente de 0 °C a 30 °C. Las reacciones se pueden realizar a presión atmosférica, elevado o reducida (por ejemplo, de 50 a 500 kPa ((0,5 a 5 bar)); en general, las reacciones se realizan a presión atmosférica.
Son bases adecuadas, por ejemplo, soluciones de hidróxidos de metal alcalino tales como hidróxido de sodio acuoso, hidróxido de litio acuoso o hidróxido de potasio acuoso, o hidrogenocarbonatos de metal alcalino acuosos tales como hidrogenocarbonato de sodio acuoso o hidrogenocarbonato de potasio acuoso o, carbonatos de metal alcalino acuoso tales como carbonato de sodio acuoso o carbonato de potasio acuoso. Se prefiere solución de hidrogenocarbonato de sodio acuoso.
La primera etapa en la reacción [B] se realiza generalmente haciendo reaccionar un compuesto de fórmula (II) con difosfato de tetrabencilo en un disolvente inerte en presencia de una base, preferentemente en un intervalo de temperaturas de -10 °C a 50 °C, más preferentemente de 0 °C a 30 °C. Las reacciones se pueden realizar a presión atmosférica, elevada o reducida (por ejemplo, de 50 a 500 kPa ((0,5 a 5 bar)); en general, las reacciones se realizan a presión atmosférica.
Los disolventes inertes son, por ejemplo, hidrocarburos halogenados tales como diclorometano o triclorometano, éteres tales como dietil éter o metil ferc-butil éter u otros disolventes tales como dioxano, dimetilformamida o tetrahidrofurano. Además, es posible usar mezclas de los disolventes. Se prefiere tetrahidrofurano.
Son bases adecuadas, por ejemplo, ferc-butóxido de potasio o ferc-butóxido de sodio, hidruro de sodio, N-butillitio, diisopropilamida de litio, bis(trimetilsilil)amida de sodio o bis(trimetilsilil)amida de litio, se prefiere bis(trimetilsilil)amida de litio.
La segunda etapa en la reacción [B] se realiza generalmente con un agente de reducción en un disolvente inerte, preferentemente en un intervalo de temperaturas de -10 °C a 50 °C, más preferentemente de 0 °C a 30 °C. Las reacciones se pueden realizar a presión atmosférica, elevada o reducida (por ejemplo, de 50 a 500 kPa ((0,5 a 5 bar)); en general, las reacciones se realizan a presión atmosférica.
Los disolventes inertes son, por ejemplo, etanol o mezclas de dioxano y agua o tetrahidrofurano y agua. Se prefiere etanol.
Los agentes de reducción son, por ejemplo, paladio sobre carbono e hidrógeno, dihidróxido de paladio, dicloruro de estaño, tricloruro de titanio o formiato de amonio. Se prefiere paladio sobre carbono e hidrógeno.
Los compuestos de fórmula (II) o bien están comercialmente disponibles, son conocidos en la literatura o se pueden preparar a partir de materiales de partida fácilmente disponibles por adaptación de procedimientos estándar descritos en la literatura. Los procedimientos detallados y las referencias de la literatura para preparar los materiales de partida también se pueden encontrar en la Parte Experimental, en la sección sobre la preparación de los materiales de partida y los productos intermedios.
La Presente invención cubre procedimientos para preparar compuestos de la presente invención de fórmula general (I), dichos procedimientos comprenden las etapas como se describe en la Sección Experimental en el presente documento.
Los esquemas y procedimientos descritos a continuación ilustran rutas sintéticas para los compuestos de fórmula general (I) de la invención y no pretenden ser taxativos.
La preparación de los compuestos de la invención se puede ilustrar por medio del siguiente esquema sintético: Esquema 1

Los compuestos de fórmula general (I) de la presente invención se pueden convertir en cualquier sal, preferentemente sales farmacéuticamente aceptables, como se describe en el presente documento, por cualquier procedimiento que sea conocido por el experto en la técnica. De manera similar, cualquier sal de un compuesto de fórmula general (I) de la presente invención se puede convertir en el compuesto libre, por cualquier procedimiento que sea conocido por el experto en la técnica.
Los compuestos de la presente invención tienen propiedades farmacológicas valiosas y se pueden usar para la prevención y/o tratamiento de varias enfermedades y estados inducidos por enfermedades en humanos y otros mamíferos. Los compuestos de fórmula general (I) de la presente invención demuestran un espectro farmacológico de acción y perfil farmacocinético valiosos. Se ha descubierto, de forma sorprendente, que los compuestos de la presente invención inhiben de forma eficaz el receptor de vasopresina V1a y por lo tanto es posible que dichos compuestos se usen para el tratamiento y/o prevención de enfermedades, preferentemente enfermedades renales y cardiovasculares en seres humanos y animales.
En el contexto de la presente invención, el término “tratamiento” o “tratar” incluye inhibir, demorar, aliviar, mitigar, detener, reducir o provocar la regresión de una enfermedad, trastorno, afección o estado, el desarrollo y/o evolución de estos y/o los síntomas de estos. El término “prevención” o “prevenir” incluye reducir el riesgo de tener, contraer o experimentar una enfermedad, trastorno, afección o estado, el desarrollo y/o evolución de estos y/o los síntomas de estos. El término prevención incluye profilaxis. El tratamiento o prevención de una enfermedad, trastorno, afección o estado puede ser parcial o completo.
Los compuestos de la presente invención son antagonistas altamente potentes y en particular selectivos del receptor de vasopresina V1a. Los compuestos de la invención por lo tanto se espera que sean altamente valiosos como agentes terapéuticos para el tratamiento y/o prevención de enfermedades, en particular para el tratamiento y/o prevención de enfermedades renales y cardiovasculares.
Tal como se usa en el presente documento, la expresión “antagonista del receptor de vasopresina V1a” se refiere a un compuesto que funciona inhibiendo (parcial o completamente) o bloqueando el receptor de vasopresina V1a, evitando así la activación del receptor por vasopresina.
En una realización, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son activos en el receptor V1a. En otra realización los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento muestran inhibición del receptor V1a de acuerdo con el estudio en B-4 con una CI50 < 100 nM. En otra realización los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento muestran inhibición del receptor V1a de acuerdo con el estudio en B-4 con una CI50 < 20 nM. En otra realización los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento muestran inhibición del receptor V1a de acuerdo con el estudio en B-4 con una CI50 < 10 nM. En otra realización los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento muestran inhibición del receptor V1a de acuerdo con el estudio en B-4 con una CI50 < 5 nM.
En una realización más, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son selectivamente activos en el receptor V1a y son menos activos, sustancialmente menos activos y/o inactivos en otros receptores de vasopresina, tales como los subtipos V1b y/o V2. En otra realización, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son al menos 10 veces selectivos para el receptor V1a en comparación con el receptor V2 como se determina de acuerdo con el estudio en B-4. En otra realización, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son al menos 15 veces selectivos para el receptor V1a en comparación con el receptor V2 como se determina de acuerdo con el estudio en B-4. En otra realización, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son al menos 20 veces selectivos para el receptor V1a en comparación con el receptor V2 como se determina de acuerdo con el estudio en B-4. En otra realización, los fármacos correspondientes subyacentes de los compuestos descritos en el presente documento son al menos 30
veces selectivos para el receptor V1a en comparación con el receptor V2 como se determina de acuerdo con el estudio en B-4.
Los compuestos de acuerdo con la invención son adecuados para el tratamiento y/o prevención de enfermedades renales, en particular de enfermedades renales agudas y crónicas, enfermedades renales diabéticas y de insuficiencia renal aguda y crónica. Las expresiones generales “enfermedad renal” o “enfermedad del riñón” describen una clase de afecciones en las que los riñones fallan al filtrar y eliminar los desperdicios de la sangre. Hay dos formas principales de enfermedad renal: enfermedad renal aguda (lesión renal aguda, LRA) y enfermedad renal crónica (ERC). Los compuestos de acuerdo con la invención además se pueden usar para el tratamiento y/o prevención de secuelas de lesión renal aguda que surgen de múltiples lesiones tales como lesión por reperfusiónisquemia, administración por radiocontraste, cirugía de bypass cardiopulmonar, choque y sepsis. En el sentido de la presente invención, el término fallo renal o insuficiencia renal comprende tanto las manifestaciones agudas como las crónicas de insuficiencia renal, así como enfermedades renales subyacentes o relacionadas tales como hipoperfusión renal, hipotensión intradialítica, uropatía obstructiva, glomerulopatías, nefropatía IgA, glomerulonefritis, glomerulonefritis aguda, glomeruloesclerosis, enfermedades tubulointersticiales, enfermedades nefropáticas tales como enfermedad renal primaria y congénita, nefritis, síndrome de Alport, inflamación renal, enfermedades renales inmunológicas tales como rechazo a trasplante renal, enfermedades renales inducidas por complejo inmunitario, nefropatía inducida por sustancias tóxicas, nefropatía inducida por medio de contraste; glomerulonefritis de cambio mínimo (lipoide); glomerulonefritis membranosa; glomerulosclerosis segmentaria focal (GESF); síndrome urémico hemolítico (SUH), amiloidosis, síndrome de Goodpasture, granulomatosis de Wegener, púrpura Schonlein-Henoch, nefropatía diabética y no diabética, pielonefritis, quistes renales, nefroesclerosis, nefroesclerosis hipertensiva y síndrome nefrótico, que se pueden caracterizar de forma diagnóstica, por ejemplo, por creatitina reducida anormalmente y/o excreción de agua, concentraciones de urea, nitrógeno, potasio y/o creatinina en sangre aumentadas anormalmente, actividad alterada de enzimas renales tales como, por ejemplo, glutamil sintetasa, osmolalidad alterada en la orina o volumen de orina, microalbuminuria elevada, macroalbuminuria, lesiones de glomérulos y arteriolas, dilatación tubular, hiperfosfataemia y/o necesidad de diálisis. La presente invención también comprende el uso de los compuestos de acuerdo con la invención para el tratamiento y/o prevención de secuelas de insuficiencia renal, por ejemplo, edema pulmonar, insuficiencia cardíaca, uremia, anemia, alteraciones de electrolitos (por ejemplo, hiperpotasemia, hiponatremia) y alteraciones en huesos y el metabolismo de carbohidratos. Los compuestos de acuerdo con la invención también son adecuados para el tratamiento y/o prevención de enfermedad renal poliquística (ERP) y del síndrome de secreción inadecuada de HAD (SSIHA).
Las enfermedades cardiovasculares en este contexto que se pueden tratar y/o prevenir con los compuestos de la invención incluyen, pero no se limitan a, las siguientes: insuficiencia cardíaca aguda y crónica que incluye insuficiencia cardíaca crónica empeorada (u hospitalización por insuficiencia cardíaca) y que incluye insuficiencia cardíaca congestiva, hipertensión arterial, hipertensión resistente, hipertensión pulmonar arterial, enfermedad cardíaca coronaria, angina pectoris estable e inestable, arritmias auriculares y ventriculares, alteraciones del ritmo auricular y ventricular y arritmias de conducción, por ejemplo bloques auriculoventriculares de grado I-III (AVB I-III), de taquiarritmia supraventricular, fibrilación auricular, palpitación auricular, fibrilación ventricular, palpitación ventricular, taquiarritmia ventricular, taquicardia torsade-de-pointes, extrasístole auricular y ventricular, extrasístole de unión AV, síndrome del seno enfermo, síncopes, taquicardia de reentrada del nodo AV y síndrome Wolff-Parkinson-White, síndrome coronario agudo (SCA), enfermedades cardíacas autoinmunes (pericarditis, endocarditis, valvulitis, aortitis, cardiomiopatías), choque tal como choque cardiogénico, choque séptico y choque anafiláctico, aneurismas, cardiomiopatía de Boxer (contracción ventricular prematura), otras enfermedades tromboembólicas e isquemias tales como alteraciones de perfusión periférica, lesión por reperfusión, trombosis arterial y venosa, insuficiencia del miocardio, disfunción endotelial, daño micro y macrovascular (vasculitis) y para prevenir reestenosis tal como terapias post trombosis, angioplastia transluminal percutánea (ATP), angioplastia coronaria transluminal percutánea (ACTP), trasplante cardiaco y operaciones de bypass, aterosclerosis, alteraciones del metabolismo de lípidos, hipolipoproteinemias, dislipidemias, hipertrigliceridemias, hiperlipidemias e hiperlipidemias combinadas, hipercolesterolemias, abetalipoproteinemia, sitosterolemia, xantomatosis, enfermedad de Tangier, adipositas, obesidad, síndrome metabólico, ataques transitorios e isquémicos, apoplejía, enfermedades cardiovasculares inflamatorias, enfermedades vasculares periféricas y cardíacas, trastornos de circulación periférica, espasmos de las arterias coronarias y arterias periféricas y edema tal como, por ejemplo, edema pulmonar, edema cerebral, edema renal y edema relacionado con insuficiencia cardíaca.
En el sentido de la presente invención, la expresión insuficiencia cardíaca también incluye formas de enfermedad más específicas o relacionadas tales como insuficiencia cardíaca derecha, insuficiencia cardíaca izquierda, insuficiencia global, cardiomiopatía isquémica, cardiomiopatía dilatativa, defectos cardiacos congénitos, defectos de la válvula cardíaca, insuficiencia cardíaca con defectos de la válvula cardíaca, estenosis de la válvula mitral, insuficiencia de la válvula mitral, estenosis de la válvula aórtica, insuficiencia de la válvula aórtica, estenosis tricúspide, insuficiencia tricúspide, estenosis de la válvula pulmonar, insuficiencia de la válvula pulmonar, defectos combinados de la válvula cardíaca, inflamación del músculo cardiaco (miocarditis), miocarditis crónica, miocarditis aguda, miocarditis vírica, insuficiencia cardíaca diabética, cardiomiopatía toxica por alcohol, enfermedades de almacenamiento cardiaco, insuficiencia cardíaca con fracción de eyección conservada (ICFEc o insuficiencia cardíaca diastólica), e insuficiencia cardíaca con fracción de eyección reducida (ICFEr o insuficiencia cardíaca sistólica).
Los compuestos de la presente invención pueden ser particularmente útiles para el tratamiento y/o prevención del síndrome cardiorrenal (SCR) y sus diferentes subtipos. La expresión comprende determinados trastornos del corazón y los riñones por medio de los cuales la disfunción aguda o crónica en un órgano puede inducir disfunción aguda o crónica en el otro.
Además, los compuestos de acuerdo con la invención se pueden usar para el tratamiento y/o prevención de enfermedad arterial periférica (EAP) que incluye claudicación e incluye isquemia de extremidad crítica así como disfunción microvascular coronaria (DMC) que incluye DMC tipo 1-4, fenómeno primario y secundario de Raynaud, alteraciones en la microcirculación, claudicación, neuropatías periféricas y autonómicas, microangiopatías diabéticas, retinopatía diabética, úlceras en extremidad diabética, gangrena, síndrome de CREST, trastornos eritematosos, enfermedades reumáticas y para promover la cicatrización.
Además, los compuestos de la invención son adecuados para tratar enfermedades urológicas y enfermedades del sistema urogenital masculino y femenino tales como, por ejemplo, síndrome prostático benigno (SPB), hiperplasia prostática benigna (HPB), agrandamiento prostático benigno (APB), obstrucción de la salida de la vejiga (OSV), síndromes del tracto urinario inferior (STUI), vejiga hiperactiva neurogénica (VHN), cistitis intersticial (CI), incontinencia urinaria (IU) tales como por ejemplo, incontinencia mixta, de urgencia, de esfuerzo y continua (IUM, IUE, IUE, IUC), dolores pélvicos, disfunción eréctil, dismenorrea y endometriosis.
Los compuestos de acuerdo con la invención también se pueden usar para el tratamiento y/o prevención de enfermedades inflamatorias, enfermedades asmáticas, enfermedad pulmonar obstructiva crónica (EPOC), síndrome de dificultad respiratoria aguda (SDRA), lesión pulmonar aguda (LPA), déficit de alfa-1-antitripsina (DAAT), fibrosis pulmonar, enfisema pulmonar (por ejemplo, enfisema pulmonar inducido por tabaco) y fibrosis quística FQ). Además, los compuestos de la invención se pueden usar para el tratamiento y/o prevención de hipertensión arterial pulmonar (HPA) y otras formas de hipertensión pulmonar (HP), que incluye hipertensión pulmonar asociada con enfermedad ventricular izquierda, infección por VIH, anemia de células falciformes, tromboembolismo (HPTEC), sarcoidosis, enfermedad pulmonar obstructiva crónica (EPOC) o fibrosis pulmonar.
Además, los compuestos de acuerdo con la invención pueden ser útiles para el tratamiento y/o prevención de cirrosis hepática, ascitas, diabetes mellitus y complicaciones diabéticas tales como, por ejemplo, neuropatía y nefropatía.
Además, los compuestos de la invención se pueden usar para el tratamiento y/o prevención de trastornos nerviosos centrales tales como estados de ansiedad, depresión, glaucoma, cáncer tal como en particular tumores pulmonares, y desregulación del ritmo circadiano tal como jet lag y trabajo por turnos.
Además, los compuestos de acuerdo con la invención pueden ser útiles para el tratamiento y/o prevención de afecciones de dolor, enfermedades de los adrenales tales como, feocromocitoma y apoplejía adrenal, enfermedades del intestino tales como, por ejemplo, enfermedad de Crohn y diarrea, trastornos menstruales tales como, por ejemplo, dismenorrea, endometriosis, parto prematuro y tocólisis.
Debido a su perfil de selectividad y actividad se cree que los compuestos de la presente invención son particularmente adecuados para el tratamiento y/o prevención de enfermedades renales agudas y crónicas que incluyen nefropatía diabética, insuficiencia cardíaca crónica y aguda, preeclampsia, enfermedad arterial periférica (EAP), disfunción microvascular coronaria (DMC), síndrome de Raynaud y dismenorrea.
Las enfermedades mencionadas anteriormente han sido bien caracterizadas en seres humanos, pero también existen con una etiología comparable en otros mamíferos y se pueden tratar en estos con los compuestos y procedimientos de la presente invención.
La presente invención se refiere además al uso de los compuestos de acuerdo con la invención para preparar una composición farmacéutica para el tratamiento y/o prevención de enfermedades, especialmente las enfermedades antes mencionadas.
La presente invención se refiere además al uso de los compuestos de acuerdo con la invención en un procedimiento para el tratamiento y/o la prevención de enfermedades, en especial las enfermedades antes mencionadas.
De acuerdo con otro aspecto, la presente invención cubre combinaciones farmacéuticas, en particular medicamentos, que comprenden al menos un compuesto de fórmula general (I) de la presente invención y al menos uno o más de otros principios activos, en particular para el tratamiento y/o prevención de enfermedades, especialmente las enfermedades antes mencionadas.
Particularmente, la presente invención cubre una combinación farmacéutica que comprende:
• uno o más primeros ingredientes activos, en particular compuestos de fórmula general (I) como se definen anteriormente y
• uno o más ingredientes activos adicionales, en particular para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades antes mencionadas.
El término “combinación” en la presente invención se usa como lo conocen los expertos en la técnica, siendo posible que dicha combinación sea una combinación fija, una combinación no fija o un kit de partes.
Una “combinación fija” en la presente invención se usa como lo conocen los expertos en la técnica y se define como una combinación en la que, por ejemplo, un primer principio activo, tal como uno o más compuestos de fórmula general (I) de la presente invención y un principio activo adicional se presentan juntos en una dosificación unitaria o en una sola entidad. Un ejemplo de una “combinación fija” es una composición fija en la que un primer principio activo y otro principio activo adicional están presentes mezclados para la administración simultánea, tal como en una formulación. Otro ejemplo de una “combinación fija” es una combinación farmacéutica en la que un primer principio activo y otro principio activo adicional están presentes en una unidad sin mezclarse.
Una combinación no fija o “kit de partes” en la presente invención se usa como lo conocen los expertos en la técnica y se define como una combinación en la que un primer principio activo y otro principio activo están presentes en más de una unidad. Un ejemplo de una combinación no fija o kit de partes es una combinación en la que el primer principio activo y el otro principio activo están presentes por separado. Es posible que los componentes de la combinación no fija o kit de partes se administren por separado, de manera secuencial, de manera simultánea, de manera concurrente o de manera escalonada cronológicamente.
Los compuestos de la presente invención se pueden administrar como el agente farmacéutico solo o en combinación con uno o más ingredientes farmacéuticamente activos en los que la combinación causa efectos adversos no aceptables. La presente invención también cubre dichas combinaciones farmacéuticas. Por ejemplo, los compuestos de la presente invención se pueden combinar con agentes conocidos para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades antes mencionadas.
En particular los compuestos de la presente invención se pueden usar en combinación fija o separada con
• agentes antitrombóticos, por ejemplo y preferentemente del grupo de los inhibidores de agregación plaquetaria, anticoagulantes y sustancias profibrinolíticos;
• agentes reductores de la presión arterial, por ejemplo y preferentemente del grupo de antagonistas de calcio, antagonistas de angiotensina AII, inhibidores de ACE, inhibidores de NEP, inhibidores de vasopeptidasa, antagonistas de endotelina, inhibidores de renina, bloqueadores alfa, bloqueadores beta, antagonistas del receptor de mineralocorticoides y diuréticos;
• agentes antidiabéticos (agentes hipoglucémicos o antihiperglucémicos), tales como, por ejemplo y preferentemente, insulina y derivados, sulfonilureas, biguanidas, tiazolidindionas, acarbosa, inhibidores de DPP4, análogos de GLP-1, o inhibidores de SGLT (gliflozinas);
• nitratos orgánicos y donantes de NO, por ejemplo, nitroprusiato de sodio, nitroglicerina, mononitrato de isosorbida, dinitrato de isosorbida, molsidomina o SIN-1 y NO por inhalación;
• compuestos que inhiben la degradación de guanosín monofosfato cíclico (GMPc), por ejemplo, inhibidores de fosfodiesterasas (PDE) 1, 2, 5 y/o 9, en particular inhibidores de PDE-5 tales como sildenafilo, vardenafilo, tadalafilo, udenafilo, dasantafilo, avanafilo, mirodenafilo, lodenafilo, CTP-499 o PF-00489791;
• agentes inotrópicos positivos, tales como, por ejemplo, glucósidos cardiacos (digoxina) y agonistas betaadrenérgicos y dopaminérgicos tales como isoproterenol, adrenalina, noradrenalina, dopamina o dobutamina;
• péptidos natriuréticos, tales como, por ejemplo, péptido natriurético auricular (PNA, anaritida), péptido natriurético de tipo B o péptido natriurético cerebral (PNB, nesiritida), péptido natriurético de tipo C (PNC) o urodilatina;
• sensibilizadores del calcio, tales como, por ejemplo y preferentemente, levosimendán;
• los activadores hemo- y NO independientes de guanilato ciclasa soluble (GCs), por ejemplo y preferentemente, los compuestos descritos en los documentos WO 01/19355, WO 01/19776, WO 01/19778, WO 01/19780, WO 02/070462 y WO 02/070510;
• los estimulantes NO-independientes, pero hemodependientes, de guanilato ciclasa soluble (GCs), por ejemplo y preferentemente, los compuestos descritos en los documentos 00/06568, WO 00/06569, WO 02/42301, w O 03/095451, WO 2011/147809, WO 2012/004258, WO 2012/028647 y WO 2012/059549;
• agentes que estimulan la síntesis de GMPc, por ejemplo y preferentemente, moduladores GCs, por ejemplo y preferentemente, riociguat, cinaciguat, vericiguat o BAY 1101042;
• inhibidores de elastasa de neutrófilo humana (ENH), tales como por ejemplo sivelestat o DX-890 (reltran);
• compuestos que inhiben la cascada de transducción de señal, en particular inhibidores de tirosina y/o serina/treonina cinasa, tales como, por ejemplo, nintedanib, dasatinib, nilotinib, bosutinib, regorafenib, sorafenib,
sunitinib, cediranib, axitinib, telatinib, imatinib, brivanib, pazopanib, vatalanib, gefitinib, erlotinib, lapatinib, canertinib, lestaurtinib, pelitinib, semaxanib o tandutinib;
• compuestos que influyen en el metabolismo enérgico del corazón, tales como, por ejemplo y preferentemente, etomoxir, dicloroacetato, ranolazina o trimetazidina o agonistas del receptor de adenosina A1 parcial o completo tales como GS-9667 (antes conocido como CVT-3619), bialanato de capadenosona y neladenosona (BAY 1067197);
• compuestos que influyen en la frecuencia cardíaca, tales como, por ejemplo y preferentemente, ivabradina; • activadores de miosina cardíaca, tales como, por ejemplo y preferentemente, omecamtiv mecarbil (CK-1827452);
• fármacos antiinflamatorios tales como fármacos antiinflamatorios no esteroideos (AINE) que incluyen ácido acetilsalicílico (aspirina), ibuprofeno y naproxeno, glucocorticoides tales como, por ejemplo y preferentemente, prednisona, prednisolona, metilprednisolona, triamcinolona, dexametasona, beclometasona, betametasona, flunisolida, budesonida o fluticasona o derivados del ácido 5-aminosalicílico, antagonistas de leucotrieno, inhibidores de TNF-alfa y antagonistas del receptor de quimiocina tales como inhibidores de CCR1,2 y/o 5; • agentes que alteran el metabolismo de la grasa, por ejemplo y preferentemente, del grupo de agonistas del receptor tiroideo, inhibidores de la síntesis de colesterol, tales como, por ejemplo y preferentemente, inhibidores de la síntesis de HMG-CoA-reductasa o escualeno, inhibidores de ACAT, inhibidores de CETP, inhibidores de MTP, agonistas PPAR-alfa, PPAR-gamma y/o PPAR-delta, inhibidores de la absorción de colesterol, inhibidores de lipasa, adsorbentes de ácido biliar polimérico, inhibidores de la reabsorción del ácido biliar y antagonistas de lipoproteína (a).
Se prefiere que se entienda que los agentes antitrombóticos son compuestos del grupo de inhibidores de agregación plaquetaria, anticoagulantes y sustancias profibrinolíticas.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de agregación plaquetaria, por ejemplo y preferentemente, aspirina, clopidogrel, ticlopidina o dipiridamol.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de trombina, por ejemplo y preferentemente, ximelagatrán, dabigatrán, melagatrán, bivalirudina o enoxaparina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de GPIIb/IIIa, por ejemplo y preferentemente, tirofibán o abciximab.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor del factor Xa, por ejemplo y preferentemente, rivaroxabán, apixabán, otamixabán, fidexabán, razaxabán, fondaparinux, idraparinux, DU-176b, p Md-3112, YM-150, KFA-1982, e Md-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 o SSR-128428.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con heparina o un derivado de heparina de bajo peso molecular (BPM).
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de vitamina K, por ejemplo y preferentemente coumarina.
Los agentes reductores de la presión arterial son preferentemente compuestos del grupo de antagonistas de calcio, antagonistas de angiotensina AII, inhibidores de ACE, inhibidores de NEP, inhibidores de vasopeptidasa, antagonistas de endotelina, inhibidores de renina, bloqueadores alfa, bloqueadores beta, antagonistas del receptor de mineralocorticoides y diuréticos.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de calcio, por ejemplo y preferentemente, nifedipina, amlodipina, verapamil o diltiazem.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un bloqueador del receptor alfa-1, por ejemplo y preferentemente, prazosina o tamsulosina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un bloqueador beta, por ejemplo y preferentemente, propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazolol, sotalol, metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol o bucindolol.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en
combinación con un antagonista del receptor de angiotensina AII, por ejemplo y preferentemente, losartán, candesartán, valsartán, telmisartán, irbesartán, olmesartán, eprosartán, embursartán o azilsartán.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de vasopeptidasa o inhibidor de endopeptidasa neutra (NEP), tal como por ejemplo y preferentemente, sacubitril, omapatrilat o AVE-7688.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista del receptor de angiotensina doble AII/inhibidor de NEP (ARNI), por ejemplo y preferentemente, LCZ696.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de ACE, por ejemplo y preferentemente, enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril, benazepril o trandopril.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de endotelina, por ejemplo y preferentemente, bosentán, darusentán, ambrisentán, tezosentán, sitaxsentán, avosentán, macitentán o atrasentán.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de renina, por ejemplo y preferentemente, aliskiren, SPP-600 o SPP-800.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista del receptor de mineralocorticoides, por ejemplo y preferentemente finerenona, espironolactona, canrenona, canrenoato de potasio, eplerenona, esaxerenona (CS-3150) o apararenona (MT-3995), CS-3150 o MT-3995.
En una realización preferida de la invención los compuestos de acuerdo con la invención se administran en combinación con un diurético tal como, por ejemplo y preferentemente, furosemida, bumetanida, piretanida, torsemida, bendroflumetiazida, clorotiazida, hidroclorotiazida, xipamida, indapamida, hidroflumetiazida, meticlotiazida, politiazida, triclorometiazida, clorotalidona, metolazona, quinetazona, acetazolamida, diclorofenamida, metazolamida, glicerina, isosorbida, manitol, amilorida o triamtereno.
Los agentes que alteran el metabolismo de la grasa se entiende que son preferentemente compuestos del grupo de inhibidores de CETP, agonistas del receptor tiroideo, inhibidores de la síntesis de colesterol tales como inhibidores de la síntesis de HMG-CoA-reductasa o escualeno, inhibidores de ACAT, inhibidores de MTP, agonistas PPAR-alfa, PPAR-gamma y/o PPAR-delta, inhibidores de la absorción de colesterol, adsorbentes del ácido biliar polimérico, inhibidores de la reabsorción del ácido biliar, inhibidores de lipasa y antagonistas de lipoproteína (a).
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de CETP, por ejemplo y preferentemente, dalcetrapib, anacetrapib, BAY 60-5521 o vacuna CETP (Avant).
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista del receptor tiroideo, por ejemplo y preferentemente D-tiroxina, 3,5,3-triiodotironina (T3), CGS 23425 o axitirom (CGS 26214).
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de HMG-CoA-reductasa de la case de estatinas, por ejemplo y preferentemente, lovastatina, simvastatina, pravastatina, fluvastatina, atorvastatina, rosuvastatina o pitavastatina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor para la síntesis de escualeno, por ejemplo y preferentemente, BMS-188494 o TAK-475.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de ACAT, por ejemplo y preferentemente, avasimiba, melinamida, pactimiba, eflucimiba o SMP-797.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de MTP, por ejemplo y preferentemente, implitapida, R-103757, BMS-201038 o JTT-130.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista de PPAR-gamma, por ejemplo y preferentemente, pioglitazona o rosiglitazona.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista de PPAR-delta, por ejemplo y preferentemente, GW 501516 o BAY 68-5042.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor para la absorción de colesterol, por ejemplo y preferentemente, ezetimiba, tiquesida o pamaquesida.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de lipasa, por ejemplo y preferentemente, orlistat.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un adsorbente del ácido biliar polimérico, por ejemplo y preferentemente, colestiramina, colestipol, colesolvam, CholestaGel o colestimida.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de la reabsorción de ácido biliar, por ejemplo y preferentemente, inhibidores ASBT (= IBAT) tales como AZD-7806, S-8921, AK-105, BARI-1741, SC-435 o SC-635.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista de lipoproteína (a), por ejemplo y preferentemente, gemcabeno calcio (CI-1027) o ácido nicotínico.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista TGFbeta, a modo de ejemplo y con preferencia, pirfenidona o fresolimumab.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con inhibidores de HIF-PH, a modo de ejemplo y con preferencia, molidustat o roxadustat.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista CCR2, a modo de ejemplo y con preferencia, CCX-140.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un antagonista TNFalfa, a modo de ejemplo y con preferencia, adalimumab.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de galectina-3, a modo de ejemplo y con preferencia, GCS-100.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agonista BMP-7, a modo de ejemplo y con preferencia, THR-184.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un modulador p53, a modo de ejemplo y con preferencia, QPI-1002.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de NOX1/4, a modo de ejemplo y con preferencia, GKT-137831.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un medicamento que afecta el metabolismo de la vitamina D, a modo de ejemplo y con preferencia, colecalciferol o paracalcitol.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agente citostático, a modo de ejemplo y con preferencia, ciclofosfamida.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un agente inmunosuporesor, a modo de ejemplo y con preferencia, ciclosporina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un aglutinante de fosfato, a modo de ejemplo y con preferencia, sevelamer o carbonato de lantano. En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un calcimimético para terapia de hiperparatiroidismo.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con agentes para terapia para falta de hierro, a modo de ejemplo y con preferencia, productos de hierro.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con agentes para la terapia de hiperuricemia, a modo de ejemplo y con preferencia, alopurinol o rasburicasa.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con hormona de glicoproteína para la terapia de anemia, a modo de ejemplo y con preferencia, eritropoyetina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con productos biológicos para terapia inmunitaria, a modo de ejemplo y con preferencia, abatacept, rituximab, eculizumab o belimumab.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con inhibidores de Jak, a modo de ejemplo y con preferencia, ruxolitinib, tofacitinib, baricitinib, CYT387, GSK2586184, lestaurtinib, pacritinib (SB1518) o TG101348.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con análogos de prostaciclina para terapia de microtrombos.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con una terapia alcalina, a modo de ejemplo y con preferencia, bicarbonato de sodio.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de mTOR, a modo de ejemplo y con preferencia, everolimus o rapamicina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de NHE3, a modo de ejemplo y con preferencia, AZD1722.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un modulador eNOS, a modo de ejemplo y con preferencia, sapropterina.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con un inhibidor de CTGF, a modo de ejemplo y con preferencia, FG-3019.
En una realización preferida de la invención, los compuestos de acuerdo con la invención se administran en combinación con antidiabéticos (agentes hipoglucémicos o antihiperglucémicos), tales como por ejemplo y preferentemente, insulina y derivados, sulfonilureas tales como tolbutamida, carbutamida, acetohexamida, clorpropamida, glipizida, gliclazida, glibenclamida, gliburida, glibornurida, gliquidona, glisoxepida, gliclopiramida, glimepirida, JB253 y JB558, meglitinidas tales como repaglinida y nateglinida, biguanidas tales como metformina y buformina, tiazolidindionas tales como rosiglitazona y pioglitazona, inhibidores de alfa-glucosidasa tales como miglitol, acarbosa y voglibosa, inhibidores de DPP4 tales como vildagliptina, sitagliptina, saxagliptina, linagliptina, alogliptina, septagliptina y teneligliptina, análogos de GLP-1 tales como exenatida (también exendina-4, liraglutida, lixisenatida y taspoglutida, o inhibidores de SGLT (gliflozinas) tales como canagliflozina, dapagliflozina y empagliflozina.
En una realización particularmente preferida, los compuestos de acuerdo con la invención se administran en combinación con uno o más de los agentes terapéuticos adicionales seleccionados del grupo que consiste en diuréticos, antagonistas de angiotensina AII, inhibidores de ACE, bloqueantes del receptor beta, antagonistas del receptor de mineralocorticoides, antidiabéticos, nitratos orgánicos y donantes de NO, activadores y estimulantes de la guanilato ciclasa soluble (sGC) y agentes inotrópicos positivos.
En otra realización particularmente preferida, los compuestos de acuerdo con la invención se administran en combinación con uno o más de los agentes terapéuticos adicionales seleccionados del grupo que consiste en diuréticos, antagonistas de angiotensina AII, inhibidores de ACE, bloqueantes del receptor beta, antagonistas del receptor de mineralocorticoides, antidiabéticos, nitratos orgánicos y donantes NO, activadores y estimulantes de la guanilato ciclasa soluble (sGC),agentes inotrópicos positivos, agentes antiinflamatorios, agentes inmunosupresores, aglutinantes de fosfato y/o compuestos que modulan el metabolismo de la vitamina D.
Entonces, en una realización adicional, la presente invención se refiere a composiciones farmacéuticas que comprenden al menos uno de los compuestos de acuerdo con la invención y uno o más agentes terapéuticos adicionales para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades antes mencionadas.
Además, los compuestos de la presente invención se pueden usar tal cual o en composiciones, en investigaciones y diagnósticos o como estándares de referencia analítica y similares, que son bien conocidos en la técnica.
Cuando los compuestos de la presente invención se administran como productos farmacéuticos a seres humanos y otros mamíferos, se puede administrar solos o como una composición farmacéutica que contiene, por ejemplo, del 0,1 al 99,5 % (más preferentemente, del 0,5 % al 90 %) de principio activo junto con uno o más excipientes farmacéuticamente aceptables.
Entonces, en otro aspecto, la presente invención se refiere a composiciones farmacéuticas que comprenden al menos uno de los compuestos de acuerdo con la invención, convencionalmente juntos con uno o más de los excipientes inertes, no tóxicos, farmacéuticamente aceptables y al uso de estos para el tratamiento y/o prevención de enfermedades, especialmente de las enfermedades antes mencionadas.
Es posible que los compuestos de acuerdo con la invención puedan tener actividad sistémica y/o local. Con este fin,
se pueden administrar de forma adecuada, tal como, por ejemplo, mediante vía oral, parenteral, pulmonar, nasal, sublingual, lingual, bucal, rectal, vaginal, dérmica, transdérmica, conjuntival, ótica o como implante o stent.
Para estas vías de administración, es posible que los compuestos de acuerdo con la invención se puedan administrar en formas de administración adecuadas.
Para administración oral, es posible formular los compuestos de acuerdo con la invención en formas de dosificación conocidas en la técnica que suministran los compuestos de la invención rápidamente y/o de forma modificada, tal como, por ejemplo, comprimidos (comprimidos revestidos o no revestidos, por ejemplo con revestimiento de liberación controlada o entérico que se disuelven con una demora o son insolubles), comprimidos de desintegración oral, películas/obleas, películas/liofilizados, cápsulas (por ejemplo cápsulas de gelatina dura o blanda), comprimidos revestidos con azúcar, gránulos, píldoras, polvos, emulsiones, suspensiones, aerosoles o soluciones. Es posible incorporar los compuestos de acuerdo con la invención en forma cristalina y/o amorfa y/o disuelta en dichas formas de dosificación.
La administración parenteral puede tener lugar evitando una etapa de absorción (por ejemplo, intravenosa, intraarterial, intracardíaca, intraespinal o intralumbar) o incluyendo una absorción (por ejemplo, intramuscular, subcutánea, percutánea o intraperitoneal). Las formas de administración adecuadas para administración parenteral son, entre otras, preparaciones para inyección e infusión en forma de soluciones, suspensiones, emulsiones, liofilizados o polvos estériles.
Ejemplos que son adecuados para otras vías de administración son formas farmacéuticas para inhalación (entre otros, inhaladores en polvo, nebulizadores), gotas nasales, soluciones nasales, aerosoles nasales; comprimidos/películas/obleas/cápsulas para administración lingual, sublingual o bucal; supositorios; gotas para los ojos, ungüentos para los ojos, baños para los ojos, insertos oculares, gotas para los oídos, aerosoles para los oídos, polvos para los oídos, enjuagues para los oídos, tapones para los oídos; cápsulas vaginales, suspensiones acuosas (lociones, mixturae agitandae), suspensiones lipofílicas, emulsiones, ungüentos, cremas, sistemas terapéuticos transdérmicos (tales como por ejemplo, parches), leche, pastas, espumas, polvos, implantes o stents.
Los compuestos de acuerdo con la invención se pueden incorporar en las formas de administración establecidas. Esto puede tener lugar de forma conocida en sí misma mezclando con excipientes farmacéuticamente aceptables. Los excipientes farmacéuticamente aceptables incluyen, entre otros,
• rellenos y vehículos (por ejemplo, celulosa, celulosa microcristalina (tales como, por ejemplo, Avicel®), lactosa, manitol, almidón, fosfato de calcio (tal como, por ejemplo, Di-Cafos®)),
• bases para ungüentos (por ejemplo, vaselina, parafinas, triglicéridos, ceras, cera de lana, alcoholes de cera de lana, lanolina, ungüento hidrófilo, polietilenglicoles),
• bases para supositorios (por ejemplo, polietilenglicoles, manteca de cacao, grasa dura),
• disolventes (por ejemplo, agua, etanol, iso-propanol, glicerol, propilenglicol, aceites grasos triglicéridos de longitud de cadena media, polietilenglicoles líquidos, parafinas),
• tensioactivos, emulsionantes, dispersantes o humectantes (por ejemplo, dodecil sulfato de sodio), lecitina, fosfolípidos, alcoholes grasos (tales como, por ejemplo, Lanette®), ésteres de ácido graso de sorbitán (tales como, por ejemplo, Span®), ésteres de ácido graso de polioxietilen sorbitán (tales como, por ejemplo, Tween®), glicéridos de ácido graso de polioxietileno (tales como, por ejemplo, Cremophor®), ésteres de ácido graso de polioxietileno, éteres de alcohol graso de polioxietileno, ésteres de ácido graso de glicerol, poloxámeros (tales como, por ejemplo, Pluronic®),
• amortiguadores, ácidos y bases (por ejemplo, fosfatos, carbonatos, ácido cítrico, ácido acético, ácido clorhídrico, solución de hidróxido de sodio, carbonato de amonio, trometamol, trietanolamina),
• agentes de isotonicidad (por ejemplo, glucosa, cloruro de sodio),
• adsorbentes (por ejemplo, sílices altamente dispersas),
• agentes que aumentan la viscosidad, formadores de gel, espesantes y/o aglutinantes (por ejemplo, polivinilpirrolidona, metilcelulosa, hidroxipropilmetilcelulosa, hidroxipropilcelulosa, carboximetilcelulosa sódica, almidón, carbómeros, ácidos poliacrílicos (tales como, por ejemplo, Carbopol®); alginatos, gelatina),
• desintegrantes (por ejemplo, almidón modificado, carboximetilcelulosa sódica, glicolato de almidón de sodio (tales como, por ejemplo, Explotab®), polivinilpirrolidona reticulada, croscarmelosa sódica (tal como, por ejemplo, AcDiSol®)),
• reguladores de flujo, lubricantes, deslizantes y agentes liberadores de moho (por ejemplo, estearato de magnesio, ácido esteárico, talco, sílices altamente dispersos (tales como, por ejemplo, Aerosil®)),
• materiales de revestimiento (por ejemplo, azúcar, laca) y formadores de película o membranas de difusión que se disuelven rápidamente o de forma modificada (por ejemplo, polivinilpirrolidonas (tales como, por ejemplo, Kollidon®), alcohol polivinílico, hidroxipropilmetilcelulosa, hidroxipropilcelulosa, etilcelulosa, ftalato de hidroxipropilmetilcelulosa, acetato de celulosa, acetato ftalato de celulosa, poliacrilatos, polimetacrilatos tales como, por ejemplo, Eudragit®)),
materiales para cápsulas (por ejemplo, gelatina, hidroxipropilmetilcelulosa),
polímeros sintéticos (por ejemplo, poliláctidos, poliglicólidos, poliacrilatos, polimetacrilatos (tales como, por ejemplo, Eudragit®), polivinilpirrolidonas (tales como, por ejemplo, Kollidon®), alcoholes polivinílicos, acetatos de polivinilo, óxidos de polietileno, polietilenglicoles y sus copolímeros y copolímeros de bloque),
plastificantes (por ejemplo, polietilenglicoles, propilenglicol, glicerol, triacetina, citrato de triacetilo, ftalato de dibutilo),
potenciadores de penetración,
estabilizadores (por ejemplo, antioxidantes tales como, por ejemplo, ácido ascórbico, palmitato de ascorbilo, ascorbato de sodio, butilhidroxianisol, butilhidroxitolueno, galato de propilo),
conservantes (por ejemplo, parabenos, ácido sórbico, tiomersal, cloruro de benzalconio, acetato de clorhexidina, benzoato de sodio),
colorantes (por ejemplo, pigmentos inorgánicos tales como, por ejemplo, óxidos de hierro, dióxido de titanio), saborizantes, edulcorantes, agentes para el enmascaramiento del olor y/o sabor.
La presente invención se refiere además a una composición farmacéutica que comprende al menos un compuesto de acuerdo con la invención, convencionalmente junto con uno o más excipientes farmacéuticamente adecuados y a su uso de acuerdo con la presente invención.
En función de técnicas de laboratorio estándar conocidas para evaluar compuestos útiles para el tratamiento de trastornos cardiovasculares y renales, mediante pruebas de toxicidad estándar y mediante ensayos farmacológicos estándar para la determinación del tratamiento de las afecciones antes identificadas en mamíferos y, mediante comparación de estos resultados con los resultados de principios activos o medicamentos conocidos que se usan para tratar estas afecciones, la dosificación eficaz de los compuestos de la presente invención se puede determinar fácilmente para el tratamiento de cada indicación deseada. La cantidad del principio activo a administrar en el tratamiento de una de estas afecciones puede variar ampliamente de acuerdo con dichas consideraciones, como el compuesto particular y la unidad de dosificación empleada, el modo de administración, el período de tratamiento, la edad y sexo del paciente tratado y la naturaleza y el alcance de la afección tratada.
La cantidad total del principio activo a ser administrado generalmente varía de alrededor de 0,001 mg/kg a alrededor de 200 mg/kg de peso corporal por día y, preferentemente, de alrededor de 0,01 mg/kg a alrededor de 20 mg/kg de peso corporal por día. Los programas de dosificación clínicamente útiles varían de una a dosificación de tres veces al día a una vez cada cuatro semanas. Además, es posible “días libres de fármacos” en los cuales al paciente no se le dosifica un fármaco durante un período de tiempo, lo que es beneficioso en el balance global entre la tolerabilidad y el efecto farmacológico. Es posible que una dosificación unitaria contenga de alrededor de 0,5 mg a alrededor de 1500 mg de principio activo y se puede administrar una o más veces por día o menos de una vez al día. La dosificación diaria promedio para la administración por inyección, que incluye inyecciones intravenosas, intramusculares, subcutáneas y parenterales y el uso de técnicas de infusión, será preferentemente de 0,01 a 200 mg/kg del peso corporal total. De manera ilustrativa, el compuesto de la presente invención se puede administrar parenteralmente con una dosis de alrededor de 0,001 mg/kg a alrededor de 10 mg/kg, preferentemente de alrededor de 0,01 mg/kg a alrededor de 1 mg/kg de peso corporal. En una administración oral, un ejemplo de intervalo de dosis es alrededor de 0,01 a 100 mg/kg, preferentemente alrededor de 0,01 a 20 mg/kg y más preferentemente, alrededor de 0,1 a 10 mg/kg de peso corporal. Los valores de intervalos intermedios a los antes mencionados también pretenden ser parte de la invención.
Por supuesto, el régimen de dosificación continuo e inicial específico para cada paciente varía de acuerdo con la naturaleza y gravedad de la afección según se determine por el médico tratante, la actividad del compuesto específico empleado, la edad y estado general del paciente, el tiempo de administración, la vía de administración, la velocidad de excreción del fármaco, las combinaciones de fármacos y similares. El modo de tratamiento deseado y la cantidad de dosis de un compuesto de la presente invención o una sal o éster o composición farmacéuticamente aceptable de este puede ser determinado por los expertos en la técnica usando pruebas de tratamiento convencionales.
Los siguientes ejemplos de realizaciones ilustran la invención. La invención no se limita a los ejemplos.
Los porcentajes en las siguientes pruebas y ejemplos son, salvo que se especifique lo contrario, en peso; las partes
son en peso. Las relaciones de disolvente, las relaciones de dilución y las concentraciones informadas para líquidos/soluciones líquidas se basan cada una en el volumen.
SECCIÓN EXPERIMENTAL SECCIÓN EXPERIMENTAL - PARTE GENERAL
Los picos de RMN formados se indican como aparecen en los espectros, no se han considerado posibles efectos de orden mayor.
Los nombres químicos se generaron usando el software ACD/Name de ACD/Labs. En algunos casos los nombres generalmente aceptados de reactivos comercialmente disponibles se usaron en lugar de los nombres generados por ACD/Name.
La siguiente Tabla 1 enumera las abreviaturas usadas en este párrafo y en la sección de Ejemplos siempre que no se expliquen en el cuerpo del texto. Otras abreviaturas tienen sus significados habituales propiamente dichos para un experto en la técnica.
Tabla 1: Abreviaturas
La siguiente lista enumera las abreviaturas usadas en el presente documento.
Abreviatura Significado
abs absoluto
a amplio (señal de RMN 1H)
conc. concentrado
CI ionización química
d doblete (señal de RMN 1H)
d día(s)
DAD detector de matriz de diodos
DCM diclorometano
dd Doble doblete
peryodinano Dess-Martin 1,1,1-triacetoxi-1,1-dihidro-1,2-benziodoxol-3-(1H)-ona
DMSO dimetilsulfóxido
ESI ionización por electropulverización (IE)
h hora(s)
HATU 3-óxido hexafluorofosfato de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio
CLAR cromatografía líquida de alto rendimiento
CL-EM Cromatografía líquida - espectrometría de masa
m multiplete (señal de RMN 1H)
min minuto(s)
EM Espectrometría de masa
MTBE metil-ferc-butiléter
RMN espectroscopía de resonancia magnética nuclear: los desplazamientos químicos (8) se proporcionan en ppm. Los desplazamientos químicos se corrigieron estableciendo la señal DMSO en 2,50 ppm salvo que se especifique lo contrario.
de t. de teoría
PDA matriz de foto diodos
Tr tiempo de retención (según se mide con CLAR o CLUR) en minutos s singulete (señal de RMN1H)
CFS Cromatografía de fluidos supercríticos
DQS detector de cuadropolo simple
t triplete (señal de RMN 1H)
td Triple doblete (señal de RMN 1H)
TFA ácido trifluoroacético
THF tetrahidrofurano
CLUR cromatografía líquida de ultra rendimiento
Los diferentes aspectos de la invención descritos en esta solicitud se ilustran con los siguientes ejemplos que no pretenden limitar la invención.
Los ejemplos de experimentos de prueba descritos en el presente documento sirven para ilustrar la presente invención y la invención no se limita a los ejemplos dados.
Todos los reactivos, para los cuales la síntesis no se describe en la parte experimental, o bien están comercialmente disponibles o son compuestos conocidos o se pueden formar a partir de compuestos conocidos por procedimientos conocidos por un experto en la técnica.
Los compuestos e intermedios producidos de acuerdo con los procedimientos de la invención pueden requerir purificación. La purificación de compuestos orgánicos es conocida por los expertos en la técnica y hay varias formas de purificar el mismo compuesto. En algunos casos, puede no ser necesario purificar. En algunos casos, los compuestos se pueden purificar por cristalización. En algunos casos, las impurezas se pueden eliminar por agitación usando un disolvente adecuado. En algunos casos, los compuestos se pueden purificar por cromatografía, particularmente cromatografía ultrarrápida de columna, usando, por ejemplo, cartuchos de gel de sílice preempaquetados, por ejemplo, cartuchos Biotage SNAP KP-Sil® o KP-NH® en combinación con un sistema de autopurificación Biotage (SP4® o Isolera Four®) y eluyentes tales como gradientes de acetato de hexano/etilo o DCM/metanol. En algunos casos, los compuestos se pueden purificar por CLAR preparativa usando por ejemplo un autopurificador Waters equipado con un detector de matriz de diodos y/o espectrómetro de masa de ionización por electropulverización en línea en combinación con una columna de fase inversa preempaquetada adecuada y eluyentes tales como gradientes de agua y acetonitrilo que pueden contener aditivos tales como ácido trifluoroacético, ácido fórmico o amoníaco acuoso.
En algunos casos, los procedimientos de purificación como se describen anteriormente pueden proporcionar esos compuestos de la presente invención que poseen una funcionalidad suficientemente ácida o básica en la forma de una sal, tal como, en el caso de un compuesto de la presente invención que es suficientemente básico, por ejemplo, una sal formiato o trifluoroacetato o, en el caso de un compuesto de la presente invención que es suficientemente ácida, por ejemplo, una sal de amonio. Una sal de este tipo se puede transformar tanto en su forma de base libre como de ácido libre, respectivamente, por varios procedimientos conocidos por el experto en la técnica o se pueden usar como sales en posteriores ensayos biológicos. Se entenderá que la forma específica (por ejemplo, sal, base libre, etc.) de un compuesto de la presente invención como se aísla y como se describe en el presente documento no es necesariamente la única forma en la que dicho compuesto se puede aplicar a un ensayo biológico para cuantificar la actividad biológica específica.
En el caso de los productos intermedios de síntesis y ejemplos de trabajo de la invención descritos a continuación, cualquier compuesto especificado en la forma de una sal de la base o el ácido correspondiente es generalmente una sal de composición estequiométrica exacta desconocida, como se obtiene por la preparación y/o procedimiento de purificación correspondiente. Salvo que se especifique en más detalle, las adiciones a nombres y fórmulas estructurales, tales como “clorhidrato”, “trifluoroacetato”, “sal de sodio”, “sal de potasio”, "sal de dipotasio", "fosfato de hidrógeno", "fosfato" o “x HCl”, “x CF3COOH”, “x Na+”, “x K+”, “x Ca2+”, “x Mg2+” por lo tanto no se entenderán en un sentido estequiométrico en el caso de dichas sales, sino que tienen meramente carácter descriptivo en relación con los componentes formadores de sales presentes allí.
Esto se aplica correspondientemente si los intermedios de síntesis o ejemplos de trabajo o sales de estos se obtuvieron en la forma de solvatos, por ejemplo, hidratos, de composición estequiométrica desconocida (si son de un tipo definido) por los procedimientos de preparación y/o purificación descritos.
Procedimientos CLAR y CL-EM:
Procedimiento 1 (CL/EM)
Instrumento: sistema Waters Acquity SQD UPLC; columna: Waters Acquity UPLC HSS T3 1,8 |j, 50 x 1 mm; eluyente A: 1 l de agua+ 0,25 ml de ácido fórmico al 99 %, eluyente B: 1 l de acetonitrilo 0,25 ml de ácido fórmico al 99 %, gradiente: 0,0 min al 90 % de A 1,2 min al 5 % de A 2,0 min al 5 % de A; horno: 50 °C; velocidad de flujo: 0,40 ml/min; detección UV: 208-400 nm.
Procedimiento 2 (CL/EM):
Instrumento MS: Thermo Scientific FT-MS; tipo de instrumento UCLAR+: Thermo Scientific UltiMate 3000; Columna: Waters, HSST3, 2,1 x 75 mm, C18 1,8 jm; eluyente A: 1 l de agua ácido fórmico al 0,01 %, eluyente B: 1 l de acetonitrilo ácido fórmico al 0,01 %, gradiente: 0,0 min al 10 % de B ^ 2,5 min al 95 % de B ^ 3,5 min al 95 % de B; horno: 50 °C; velocidad de flujo: 0,90 ml/min; detección UV: 210 nm/ senda de integración óptima 210-300 nm. Procedimiento 3 (CL-EM):
Instrumento: Agilent MS Quad 6150; CLAR: Agilent 1290; Columna: Waters Acquity UPLC HSS T3 1,8 j 50 x 2,1 mm; eluyente A: 1 l de agua 0,25 ml de 99 % ácido fórmico, eluyente B: 1 l de acetonitrilo 0,25 ml de 99 % ácido fórmico; gradiente: 0,0 min al 90 % de A ^ 0,3 min al 90 % de A ^ 1,7 min al 5 % de A ^ 3,0 min al 5 % de A; horno: 50 °C; flujo: 1,20 ml/min; detección UV: 205 - 305 nm.
Procedimiento 4 (CLAR preparativa):
Columna: Chromatorex o Reprosil C1810 jm , 125 x 30 mm; eluyente A: agua ácido fórmico al 0,1 %, eluyente B: acetonitrilo ácido fórmico al 0,1 %; gradiente: 3 min al 10 % de B, 17,5 min al 95 % de B, 19,5 min al 100 % de B, 20 min al 10 % de B; flujo: 75 ml/min; tiempo de ejecución: 20 min; detección a 210 nm.
Procedimiento 5 (CL-EM):
Instrumento: Waters Single Quad MS System; Instrument Waters UPLC Acquity; Columna: Waters BEH C181,7 jm 50 x 2,1 mm; eluyente A: 1 l de agua 1,0 ml (solución acuosa de amoníaco, 25 %)/l, eluyente B: 1 l de acetonitrilo; gradiente: 0,0 min al 92 % de A ^ 0,1 min 9al 2 % de A ^ 1,8 min al 5 % de A ^ 3,5 min al 5 % de A; horno: 50 °C; flujo: 0,45 ml/min; detección UV: 210 nm (208-400 nm).
Microondas
El reactor de microondas usado fue un sistema de microondas Initiator+ con robot sixty de Biotage®.
Difractometría de rayos X
Difractómetro de transmisión PANalytical X'Pert PRO con contador PIXcel (multicanal):
radiación: cobre, K alfa
monocromador primario: espejo de rayos X
enfocado longitud de onda (K1): 1,5406 A
longitud de onda (K2): 1,5444 A
parámetros de generador: 40 kV, 40 mA
rango de medición: 2-38°
condiciones ambiente: 25 °C, 40 -60 % de hr
Termogravimetría
Analizador termogravimétrico TGA 7; fabricante: Perkin-Elmer; tasa de calentamiento: 10 Kmin_1; gas de purga: nitrógeno, 20-30 ml/min; crisol: crisol de aluminio abierto; preparación de muestra: ninguna.
Espectros infrarrojos
Espectrómetro Bruker Tensor 37; resolución espectral 2 cm-1; cantidad de mediciones individuales 64; rango de cantidad de ondas 4000-550 cm-1; preparación de muestra: ninguna.
SECCIÓN EXPERIMENTAL- MATERIALES DE PARTIDA E INTERMEDIOS
Ejemplo 1A
{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetonitrilo

En un recipiente de reacción de 2 l, 100 g (273 mmol) de ácido {3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acético (síntesis descrita como Ejemplo 8A en el documento WO 2010/105770-A1), 43,3 g (547 mmol) de piridina y 33 mg (0,3 mmol) de 4-dimetilaminopiridina se disolvieron en 300 ml de THF. La solución resultante se trató a 5 °C con 52,8 g (438 mmol) de cloruro de 2,2-dimetilpropanoílo durante 15 minutos y la mezcla resultante se agitó a temperatura ambiente durante 2,5 horas. Después de enfriarse hasta 0 °C, se agregaron 183 ml de solución de amoníaco acuosa al 28 % durante 1 h mientras la temperatura de la solución se mantuvo entre 10 °C y 20 °C y la mezcla resultante luego se agitó a 5 °C durante un período de tiempo adicional de 1 h. Luego se agregaron 500 ml de metil ferc-butiléter y 300 ml de ácido cítrico acuoso al 20 % mientras se mantuvo la temperatura interna entre 10 °C y 20 °C. Las fases se separaron y la fase orgánica se lavó con 300 ml de ácido cítrico acuoso al 20 % seguido de 300 ml solución de hidrogenocarbonato de sodio acuoso saturado y finalmente con 300 ml de solución de cloruro de sodio acuosa al 10 %. La fase orgánica se evaporó a 60 °C a presión reducida hasta que se obtuvo un residuo oleoso. Luego se agregaron 300 ml THF y la solución se evaporó nuevamente hasta que se obtuvo una solución oleosa. Esta operación se repitió una segunda vez. El residuo oleoso se recuperó en 360 ml de THF y se trató con 172 g (820 mmol) de ácido trifluoroacético anhídrido durante 20 min a una temperatura de entre 10 °C y 20 °C. La solución resultante luego se agitó a temperatura ambiente durante 1 h. Se agregaron 720 ml de 4-metil-2-pentanona y 650 ml de solución de hidróxido de sodio acuosa al 7,5 % a una temperatura de entre 10 °C y 20 °C. Finalmente el valor del pH se ajustó hasta pH = 9,5 usando solución de hidróxido de sodio acuosa al 7,5 %. Después de la fase de separación, la fase orgánica se lavó dos veces con 450 ml de solución acuosa de cloruro de sodio al 10 %. La fase orgánica se evaporó a una temperatura de 80 °C a presión reducida mientras se agregaban 1200 ml de n-heptano. La suspensión formada se enfrió hasta 20 °C y se formó un sólido que se eliminó por filtración y se lavó con 200 ml de n-heptano y luego se secó a presión reducida (50 °C, 3 kPa (30 mbar)) proporcionando 88 g (93 % teórico) de {3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}acetonitrilo en forma de un sólido.
RMN 1H (400 MHz, DMSO-da): 8 [ppm] = 7,78 (d, 2H), 7,55 (d, 2H), 6,91 (d, 1H), 5,17 (s, 2 H), 4,34-4,23 (m, 1 H), 3,98 (dd, 1H), 3,81 (dd, 1H).
Ejemplo 2A
2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}etanimidato de metilo

En un recipiente de reacción de 4 l, de 200 g (576,9 mmol) de {3-(4-clorofenil)-5-oxo-4-[(2Sj-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-7H-1,2,4-triazol-1-il}acetonitrilo (Ejemplo 1A) en 1600 ml de metanol se trataron con 5,2 g (28 mmol) de metanolato de sodio (30 % en metanol) y la mezcla resultante se agitó a 50 °C durante 2,5 horas. La solución luego se evaporó a 50 °C a presión reducida hasta que se obtuvo una solución oleosa. Se agregaron
2000 ml de metil ferc-butiléter y la solución se concentró hasta alcanzar un volumen de 800 ml. Luego se agregaron 3000 ml de n-heptano y se formó una suspensión. Después de enfriar hasta 20 °C, el sólido se filtró y lavó con 500 ml de n-heptano y luego se secó a presión reducida (50 °C, 3 kPa (30 mbar)) proporcionando 175 g (80 % teórico) de 2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}etanimidato de metilo en forma de un sólido.
RMN 1H (400 MHz, DMSO-da): 8 [ppm] = 8,01 (s, 1H), 7,78 (d, 2H), 7,62 (d, 2H), 6,93 (s a, 1H), 4,50 (s, 2 H), 4,35 4,23 (m, 1 H), 3,96 (dd, 1H), 3,81 (dd, 1H), 3,67 (s, 3H).
Ejemplo 3A
3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxilato de metilo

Una solución de 1,0 g de 2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluor-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-ilo}etanimidato de metilo (Ejemplo 2A, 2,64 mmol) en 20 ml de 1,4-dioxano se enfrió hasta 10 °C y luego se trató con 388 mg (3,17 mmol) de clorooxoacetato de metilo y 0,55 ml (3,18 mmol) de N,N-diisopropiletilamina. La mezcla resultante luego se agitó durante 30 min. Una solución preagitada de 1,10 g (3,17 mmol) de sal de 4-metilbencenosulfonato 2-hidrazin-3-(trifluorometil)piridina (1:1), 0,65 ml (3,72 mmol) N,N-diisopropiletilamina y 506 mg (3,19 mmol) de sulfato de cobre (II) anhidro en 10 ml de 1,4-dioxano se agregó a la mezcla de reacción y la mezcla resultante luego se agitó durante la noche a temperatura ambiente. Luego se agregó agua y la fase acuosa se extrajo con acetato de etilo, las fases orgánicas combinadas se lavaron con solución acuosa de cloruro de sodio, se secaron sobre sulfato de magnesio y se evaporaron al vacío proporcionando 777 mg (50 % teórico) del compuesto del título en forma de un sólido.
CL-EM (Procedimiento 1): Tr= 1,00 min; MS (ESI pos): m/z = 592,6 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,93 (d, 1H), 8,60 (dd, 1H), 7,98 (dd, 1H), 7,75 (d, 2H), 7,67-7,57 (m, 2H), 6,91 (d, 1H), 5,22 (s, 2H), 4,37-4,22 (m, 1H), 4,10-3,97 (m, 1H), 3,85 (dd, 1H), 3,77 (s, 3H).
Ejemplo 4A
3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxamida

1,80 g de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxilato de metilo (Ejemplo 3A, 3,04 mmol) se disolvió en 10,0 ml de una solución de amoníaco (7 N en metanol, 70,0 mmol). La mezcla resultante se agitó durante 1 h a temperatura ambiente. El disolvente se eliminó al vacío y el producto bruto se purificó por CLAR preparativa (Procedimiento 4). La liofilización del producto que contiene las fracciones proporcionó 1,49 g (85 % teórico) del compuesto del título en forma de un sólido.
CL-EM (Procedimiento 3): Tr = 1,20 min; MS (ESI pos): m/z = 577 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,87 (d, 1H), 8,51 (d, 1H), 8,39 (s, 1H), 7,99 (s, 1H), 7,90 (dd, 1H), 7,827,68 (m, 2H), 7,63 (d, 2H), 6,90 (s, 1H), 5,22-5,07 (m, 2H), 4,29 (s a, 1H), 4,16-3,94 (m, 1H), 3,85 (dd, 1H).
Ejemplo 5A
3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxilato de metilo

En argón, una solución de 150 mg (0,40 mmol) de metil-2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}etanimidato (Ejemplo 2A) en 3 ml de THF anhidro se trató a 0 °C con 75 pl (0,44 mmol) de A/,W-diisopropiletilamina y 40 pl (0,44 mmol) de clorooxoacetato de metilo y la mezcla se agitó luego a 0 °C durante 30 min. 93 mg (0,44 mmol) de [2-(trifluorometil)fenil]hidrazina se agregaron luego, seguido por 145 pl (0,83 mmol) de W,A/-diisopropiletilamina. La mezcla resultante se agitó durante 2 h a temperatura ambiente, seguido por 1 h a 120 °C en el microondas y luego se evaporó. El residuo obtenido se purificó por CLAR preparativa (Procedimiento 4) para proporcionar 75 mg (32 % teórico) del compuesto del título.
CL-EM (Procedimiento 2): Tr = 2,01 min; MS (ESI pos): m/z = 591,1 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,07-7,56 (m, 8H), 6,91 (d, 1H), 5,17 (d, 2H), 4,37-4,21 (m, 1H), 4,09-3,80 (m, 2H), 3,74 (s, 3H).
Ejemplo 6A
3-({3-(4-dorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxamida

Una mezcla de 55 mg (0,093 mmol) de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxilato de metilo (Ejemplo 5A) en una solución de amoníaco (7 N en metanol, 0,8 ml, 5,6 mmol) se agitó durante la noche a temperatura ambiente y la mezcla se evaporó luego. El residuo obtenido se purificó por CLAR preparativa (Procedimiento 4) para proporcionar 55 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 2): Tr= 1,77 min; MS (ESI pos): m/z = 576,1 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,23 (s, 1H), 8,00-7,55 (m, 9H), 6,90 (d, 1H), 5,22-5,01 (m, 2H), 4,40-4,18 (m, 1H), 4,10-3,76 (m, 2H).
Ejemplo 7A
3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxilato de metilo

Una solución de 150 mg de metil-2-{3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluor-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-ilo}etanimidato (Ejemplo 2A, 26,4 mmol) en 3 ml de THF se enfrió hasta 0 ° C y luego se trató con 58,2 mg (0,48 mmol) de clorooxoacetato de metilo y 275 j l (1,58 mmol) de N,N-diisopropiletilamina. La mezcla resultante se calentó hasta temperatura ambiente y se agitó durante 1 h y se enfrió nuevamente hasta 0 °C. Luego se agregaron 62,6 mg (0,436 mmol) de 3-cloro-2-hidrazinopiridina y la mezcla de reacción se calentó hasta temperatura ambiente y luego se agitó durante 1 h, seguido de 1 h a 120 °C en un recipiente sellado en irradiación de microondas. El producto en bruto se purificó por CLAR preparativa (Procedimiento 4). La liofilización del producto que contiene fracciones proporcionó 25,3 mg (11 % teórico) del compuesto del título.
CL-EM (Procedimiento 2): Tr= 1,82 min; MS (ESI pos): m/z = 558,1 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,70-8,24 (m, 2H), 7,89-7,56 (m, 5H), 6,92 (d, 1H), 5,22 (s, 2H), 4,46-4,20 (m, 1H), 3,79 (s, 5H).
Ejemplo 8A
3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxamida

5,1 g de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxilato de metilo (Ejemplo 7A, 9,134 mmol) se disolvió en 42,5 ml de una solución de amoníaco (7 N en metanol, 297 mmol). La mezcla resultante se agitó durante 2 h a temperatura ambiente. La solución luego se vertió en hielo y la mezcla se agitó durante 10 min. El precipitado se filtró y lavó con agua, lo cual proporcionó 3,5 g de producto bruto. La fase acuosa se extrajo con acetato de etilo. La fase orgánica se secó con sulfato de magnesio, se filtró y el disolvente se eliminó al vacío. El producto en bruto se purificó por cromatografía ultrarrápida (gel de sílice, diclorometano/metanol, 97/3) proporcionando 4,00 g (81 % teórico) del compuesto del título en forma de un sólido.
CL-EM (Procedimiento 2): Tr= 1,62 min; MS (ESI pos): m/z = 543,1 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 8 [ppm] = 8,55 (dd, 1H), 8,39 (s, 1H), 8,25 (dd, 1H), 8,00 (s, 1H), 7,76 (d, 2H), 7,69 (dd, 1H), 7,62 (d, 2H), 6,90 (d, 1H), 5,18 (d, 2H), 4,36-4,23 (m, 1H), 4,06-3,97 (m, 1H), 3,85 (dd, 1H).
SECCIÓN EXPERIMENTAL - EJEMPLOS
Ejemplo 1
Dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo

A 0 °C, una solución de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-tnfluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-tnazol-1-il}metil)-1 -[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-5-carboxamida (Ejemplo 4A, 2,00 g, 3,47 mmol) en tetrahidrofurano (40 ml, 490 mmol) se trató con 4-W,A/-dimetilaminopindina (635 mg, 5,20 mmol) y trietilamina (720 pl, 5,2 mmol). Luego se agregó gota a gota oxicloruro de fósforo (480 pl, 5,2 mmol). La mezcla resultante se agitó durante 40 min a 0 °C y luego durante 50 min a temperatura ambiente. Luego se agregó agua (4,0 ml) y una solución acuosa saturada de hidrogenocarbonato de sodio (24 ml) y la mezcla resultante se agitó durante la noche a temperatura ambiente. Luego se evaporó el tetrahidrofurano a temperatura ambiente. La solución resultante se diluyó con agua y se extrajo con acetato de etilo. La fase acuosa se llevó a pH = 1 con una solución de ácido clorhídrico (1 N), se diluyó con una solución acuosa saturada de cloruro de sodio y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con solución acuosa saturada de cloruro de sodio, se secaron sobre sulfato de magnesio y se concentraron. El residuo se purificó por CLAR preparativa (Procedimiento 4) para proporcionar 1,32 g (58 % teórico) del compuesto del título.
CL-EM (Procedimiento 3): Tr = 0,94 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN 1H (400 MHz, DMSO-da): 8 [ppm] = 8,86 (d, 1H), 8,58-8,29 (m, 2H), 8,07-7,82 (m, 2H), 7,79-7,45 (m, 4H), 5,33 5,00 (m, 2H), 4,95 - 4,77 (m a, 1 H), 4,29-3,89 (m, 2H), 3,36 (s a, 2h , superposición con pico de HDO).
Ejemplo 1-1
Dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo hemihidrato

Una suspensión de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)pindin-2-il]-1H-1,2,4-tnazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (4,46 g, 6,79 mmol) en 446 ml de tolueno se agitó a 25 °C durante siete días. El sólido se eliminó por filtración y el filtrado se evaporó. El sólido y el residuo obtenidos del filtrado se mezclaron juntos y se suspendieron en una mezcla de diclorometano/n-heptano (200 ml, 55:45). La mezcla resultante se agitó a 40 °C durante ocho días. El material sólido se filtró y se examinó por difractometría de rayos X y se corresponde con el compuesto del título en forma de material cristalino (forma de hemihidrato).
Tabla 2: Difractometría de rayos X en polvo del compuesto obtenido en forma de material cristalino en el ejemplo 1-1

(continuación)

Tabla 3: Espectro infrarrojo del compuesto obtenido como material cristalino en el ejemplo 1-1

(continuación)

Las bandas marcadas con * son específicas del ácido libre.
Ejemplo 2
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo potásico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 1, 104 mg, 158 pmol) en agua (3,0 ml) se trató con hidrogenocarbonato de potasio (31,6 mg, 316 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 124 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,67 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 8,87-8,80 (m, 1H), 8,52 (d, 1H), 7,95 (dd, 1H), 7,72-7,57 (m, 4H), 5,45-5,36 (m, 1H), 5,35-5,26 (m, 1H), 4,99-4,55 (m, 1H, superposición con pico HDO), 4,20-4,07 (m, 2H).
Ejemplo 3
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo potásico
Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-dorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con hidrogenocarbonato de potasio (5,33 mg, 53,3 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 39,5 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,67 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN 1H (500 MHz, D2O): 5 [ppm] = 8,87-8,80 (m, 1H), 8,54-8,48 (m, 1H), 7,97-7,91 (m, 1H), 7,71-7,57 (m, 4H), 5,43 5,37 (m, 1H), 5,35-5,28 (m, 1H), 4,90-4,64 (m, 1H, superposición con pico de HDO), 4,23-4,10 (m, 2H).
Ejemplo 4
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con carbonato disodio (5,65 mg, 53,3 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 41,2 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,68 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN iH (500 MHz, D2O): 5 [ppm] = 8,86-8,82 (m, 1H), 8,54-8,49 (m, 1H), 7,98-7,92 (m, 1H), 7,72-7,58 (m, 4H), 5,43 5,37 (m, 1H), 5,34-5,28 (m, 1H), 4,88-4,65 (m, 1H, superposición con pico de HDO), 4,20-4,06 (m, 2H).
Ejemplo 5
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con hidrogenocarbonato de sodio (4,48 mg, 53,3 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 38,6 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,68 min; MS (ESI pos): m/z = 657 [M+H]+
RMN iH (500 MHz, D2O): 5 [ppm] = 8,86-8,81 (m, 1H), 8,53-8,48 (m, 1H), 7,97-7,92 (m, 1H), 7,71-7,58 (m, 4H), 5,42 5,36 (m, 1H), 5,34-5,27 (m, 1H), 4,68 (s, 1H, superposición con pico de HDO), 4,22-4,10 (m, 2H).
Ejemplo 6
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo de litio

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trmuorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con hidróxido de litio hidrato (1:1) (4,47 mg, 107 pmol), luego se agitó durante 15 min a temperatura ambiente y se Mofilizó proporcionando 38,7 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,67 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 8,86-8,82 (m, 1H), 8,54-8,49 (m, 1H), 7,98-7,91 (m, 1H), 7,72-7,58 (m, 4H), 5,43 5,37 (m, 1H), 5,34-5,28 (m, 1H), 4,90-4,68 (m, 1H, superposición con pico de HDO), 4,21-4,06 (m, 2H).
Ejemplo 7
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo de litio

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trmuorometil)piridin-2-il]-1H-1,2,4-triazol-3-M}metM)-3-(4-dorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-M]-1,1,1-trifluoropropan-2-Mo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con hidrato de hidróxido de litio (1:1) (2,24 mg, 53,3 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 38,3 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,67 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN iH (500 MHz, D2O): 6 [ppm] = 8,83 (d, 1H), 8,51 (d, 1H), 7,94 (dd, 1H), 7,71-7,58 (m, 4H), 5,42-5,37 (m, 1H), 5,34-5,28 (m, 1H), 4,89-4,65 (m, 1H, superposición con pico de HDO), 4,22-4,10 (m, 2H).
Ejemplo 8
Fosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo cálcico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trmuorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 1, 100 mg, 152 pmol) en agua (1 l) se trató con carbonato de calcio (15,2 mg, 152 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 93,9 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,69 min; MS (ESI pos): m/z = 657,1 [M+H]+
RMN 1H (500 MHz, D2O): 5 [ppm] = 8,86-8,82 (m, 1H), 8,54-8,49 (m, 1H), 7,98-7,92 (m, 1H), 7,72-7,57 (m, 4H), 5,45 5,37 (m, 1H), 5,35-5,28 (m, 1H), 4,91-4,66 (m, 1H, superposición con pico HDO), 4,21-4,09 (m, 2H).
Ejemplo 9
Fosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo de magnesio

Una solución de (dihidrogenofosfato de 2S)-3-[1-({5-carbamoil-1-[3-(trmuorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo (Ejemplo 1, 35,0 mg, 53,3 pmol) en agua (1,0 ml) se trató con carbonato de magnesio (4,49 mg, 53,3 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 40,1 mg (cuant.) del compuesto del título.
CL-e M (Procedimiento 1): Tr = 0,68 min; MS (ESI pos): m/z = 657,0 [M+H]+
RMN iH (500 MHz, D2O): 5 [ppm] = 8,87-8,81 (m, 1H), 8,54-8,48 (m, 1H), 7,98-7,91 (m, 1H), 7,73-7,58 (m, 4H), 5,43 5,37 (m, 1H), 5,35-5,27 (m, 1H), 4,92-4,60 (m, 1H, superposición con pico HDO), 4,20-4,06 (m, 2H).
Ejemplo 10
Dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo

A 0 °C, una solución de 3-({3-(4-dorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-5-carboxamida (Ejemplo 6A, 1000 mg, 1,74 mmol) en tetrahidrofurano (20 ml, 250 mmol) se trató con 4-N,N-dimetilaminopiridina (318 mg, 2,60 mmol) y trietilamina (360 pl, 2,6 mmol). Se agregó gota a gota oxicloruro de fósforo (240 pl, 2,6 mmol). La mezcla resultante se agitó durante 40 min a 0 °C y luego durante 50 min a temperatura ambiente. Luego se agregó agua (2,0 ml) y una solución acuosa saturada de hidrogenocarbonato de sodio (12 ml) y la mezcla resultante se agitó durante la noche a temperatura ambiente. Luego se evaporó el tetrahidrofurano a temperatura ambiente. La solución resultante se diluyó con agua y se extrajo con acetato de etilo. La fase acuosa se llevó a pH = 1 con una solución de ácido clorhídrico (1 N), se diluyó con una solución de cloruro de sodio acuosa saturada y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con solución saturada de cloruro de sodio, se secaron sobre sulfato de magnesio y se concentraron. El residuo se purificó por CLAR preparativa (Procedimiento 4) para proporcionar 876 mg (77 % teórico) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,74 min; MS (ESI pos): m/z = 656,0 [M+H]+
RMN 1H (400 MHz, DMSO-d6): 6 [ppm] = 8,30 (s a, 1H), 7,96-7,50 (m, 8H), 5,25-4,96 (m, 2H), 4,97 -4 ,74 (m a, 1H), 4,26-3,91 (m, 2H), 3,57 (s a, 2H, superposición con pico de HDO).
Ejemplo 11
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo potásico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (1,0 ml) se trató con hidrogenocarbonato de potasio (10,7 mg, 107 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 40,4 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,73 min; MS (ESI pos): m/z = 656,0 [M+H]+
RMN iH (500 MHz, D2O): 6 [ppm] = 7,97-7,93 (m, 1H), 7,87-7,79 (m, 2H), 7,73-7,59 (m, 5H), 5,38-5,33 (m, 1H), 5,30 5,24 (m, 1H), 4,88-4,66 (m, 1H, superposición con pico de HDO), 4,20-4,06 (m, 2H).
Ejemplo 12
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo potásico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (1,0 ml) se trató con hidrogenocarbonato de potasio (5,34 mg, 53,4 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 36,3 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,73 min; MS (ESI pos): m/z = 656,0 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 7,97-7,93 (m, 1H), 7,88-7,79 (m, 2H), 7,71-7,58 (m, 5H), 5,39-5,24 (m, 2H), 4,90 4,67 (m, 1H, superposición con pico de HDO), 4,19-4,09 (m, 2H).
Ejemplo 13
(Hidrogenofosfato de 2S)-3-[1-({5-carbamoiM-[2-(trifluorometil)fenN]-1H-1,2,4-triazol-3-N}metN)-3-(4-dorofenN)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-M]-1,1,1 -trifluoropropan-2-ilo sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (1,0 ml) se trató con carbonato disodio (5,66 mg, 53,4 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 37,9 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,74 min; MS (ESI pos): m/z = 656,1 [M+H]+
RMN iH (500 MHz, D2O): 6 [ppm] = 7,97-7,93 (m, 1H), 7,89-7,78 (m, 2H), 7,74-7,57 (m, 5H), 5,40-5,22 (m, 2H), 4,92 4,64 (m, 1H, superposición con pico HdO), 4,22-4,04 (m, 2H).
Ejemplo 14
Hidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (1,0 ml) se trató con hidrogenocarbonato de sodio (4,48 mg, 53,4 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 36,2 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,47 min; MS (ESI pos): m/z = 656,0 [M+H]+
RMN 1H (500 MHz, D2O): 5[ppm] = 7,97-7,93 (m, 1H), 7,87-7,79 (m, 2H), 7,72-7,59 (m, 5H), 5,39-5,24 (m, 2H), 4,90 4,65 (m, 1H, superposición con pico HdO), 4,20-4,09 (m, 2H).
Ejemplo 15
Fosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo de magnesio

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (50 ml) se trató con carbonato de magnesio (4,50 mg, 53,4 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 37,3 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 2): Tr= 1,34 min; MS (ESI pos): m/z = 656,1 [M+H]+
RMN iH (500 MHz, D2O): 5[ppm] = 7,97-7,93 (m, 1H), 7,88-7,79 (m, 2H), 7,72-7,58 (m, 5H), 5,39-5,24 (m, 2H), 4,92 4,63 (m, 1H, superposición con pico de HDO), 4,20-4,09 (m, 2H).
Ejemplo 16
Fosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo de calcio

Una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[2-(trifluorometil)fenil]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 10, 35,0 mg, 53,4 pmol) en agua (15 ml) se trató con acetato de calcio monohidrato (9,40 mg, 53,4 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 37,9 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 2): Tr = 1,33 min; MS (ESI pos): m/z = 656,1 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 8,00-7,91 (m, 1H), 7,89-7,77 (m, 2H), 7,73-7,55 (m, 5H), 5,42-5,22 (m, 2H), 4,99 4,50 (m, 1H, superposición con pico de HDO), 4,27-3,99 (m, 2H).
Ejemplo 17
Dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-cloro-fenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo

A 0 °C, una solución de 3-({3-(4-clorofenil)-5-oxo-4-[(2S)-3,3,3-trifluoro-2-hidroxipropil]-4,5-dihidro-1H-1,2,4-triazol-1-il}metil)-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-5-carboxamida (Ejemplo 8A, 200 mg, 368 pmol) en tetrahidrofurano (4,2 ml, 52 mmol) se trató con 4-N,N-dimetilaminopiridina (67,5 mg, 552 pmol) y trietilamina (77 pl, 550 pmol). Luego se agregó gota a gota oxicloruro de fósforo (52 pl, 0,55 mmol). La mezcla resultante se agitó durante 40 min a 0 °Cy luego durante 50 min a temperatura ambiente. Luego se agregó agua (420 pl) y una solución acuosa saturada de hidrogenocarbonato de sodio (2,5 ml) y la mezcla resultante se agitó durante la noche a temperatura ambiente. Luego se evaporó el tetrahidrofurano a temperatura ambiente. La solución resultante se diluyó con agua y se extrajo con acetato de etilo. La fase acuosa se llevó a pH = 1 con una solución de ácido clorhídrico (1 N), se diluyó con una solución de cloruro de sodio acuosa saturada y se extrajo con acetato de etilo. Las capas orgánicas combinadas se lavaron con solución saturada de cloruro de sodio, se secaron sobre sulfato de magnesio y se concentraron. El residuo se purificó por CLAR preparativa (Procedimiento 4) para proporcionar 126 mg (55 % teórico) del compuesto del título.
CL-EM (Procedimiento 2): Tr = 1,22 min; MS (ESI neg): m/z = 621,0 [M-H]-RMN iH (400 MHz, DMSO-d6): 6 [ppm] = 8,64-8,34 (m, 2H), 8,24 (dd, 1H), 7,96 (s, 1H), 7,80-7,48 (m, 5H), 5,25-5,00 (m, 2H), 4,93 - 4,76 (m a, 1H), 4,20-3,87 (m, 2H), 3,34 (s a, 2H, superposición con pico de HDO).
Ejemplo 18
Hidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo potásico

Una solución de (dihidrogenofosfato de 2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-dorofenN)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-N]-1,1,1-trifluoropropan-2-No (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (2,0 ml) se trató con hidrogenocarbonato de potasio (11,2 mg, 112 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 40,2 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,64 min; MS (ESI pos): m/z = 623,0 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 8,54 (dd, 1H), 8,22 (dd, 1H), 7,76-7,58 (m, 5H), 5,42 - 5,29 (m, 2H), 4,92-4,63 (m, 1H, superposición con pico de HDO), 4,20-4,07 (m, 2 H).
Ejemplo 19
Hidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1 -(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-M]-1,1,1 -trifluoropropan-2-ilo potásico

Una solución de dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-dorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-N]-1,1,1-trifluoropropan-2-ilo (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (2,0 ml) se trató con hidrogenocarbonato de potasio (5,62 mg, 56,2 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 37,1 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,65 min; MS (ESI pos): m/z = 623,0 [M+H]+
RMN iH (500 MHz, D2O): 6 [ppm] = 8,55-8,51 (m, 1H), 8,23-8,18 (m, 1H), 7,75-7,57 (m, 5H), 5,35 (q, 2H), 4,90-4,63 (m, 1H, superposición con pico de HDO), 4,22-4,10 (m, 2H).
Ejemplo 20
Hidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-dorofenN)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-N]-1,1,1-trifluoropropan-2-No (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (2,0 ml) se trató con carbonato disódico (5,95 mg, 56,2 pmol), luego se agitó durante 15 min a temperatura ambiente y se MofiMzó proporcionando 38,9 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,65 min; MS (ESI pos): m/z = 623,0 [M+H]+
RMN 1H (500 MHz, D2O): 6 [ppm] = 8,56-8,53 (m, 1H), 8,25-8,20 (m, 1H), 7,75-7,58 (m, 5H), 5,42-5,28 (m, 2H), 4,90 4,64 (m, 1H, superposición con pico HdO), 4,21-4,06 (m, 2H).
Ejemplo 21
Hidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1 -(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-il sódico

Una solución de dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (2,0 ml) se trató con dihidrogenofosfato de (4,72 mg, 56,2 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 36,4 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,64 min; MS (ESI pos): m/z = 623,1 [M+H]+
RMN iH (500 MHz, D2O): 6 [ppm] = 8,56-8,51 (m, 1H), 8,24-8,18 (m, 1H), 7,75-7,56 (m, 5H), 5,43-5,28 (m, 2H), 4,92 4,59 (m, 1H, superposición con pico de HDO), 4,25-4,09 (m, 2H).
Ejemplo 22
Fosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo de magnesio

Una solución de dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (53 ml) se trató con carbonato de magnesio (4,74 mg, 56,2 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 37,8 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 2): Tr= 1,13 min; MS (ESI pos): m/z = 623,0 [M+H]+
RMN 1H (500 MHz, D2O): 5 [ppm] = 8,57-8,52 (m, 1H), 8,26-8,19 (m, 1H), 7,77-7,58 (m, 5H), 5,43-5,28 (m, 2H), 4,95 4,58 (m, 1H, superposición con pico de HDO), 4,07 (s, 2 H).
Ejemplo 23
Fosfato de (2S)-3-[1-{[5-carbamoiM-(3-doropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-dorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo cálcico

Una solución de dihidrogenofosfato de (2S)-3-[1-{[5-carbamoil-1-(3-cloropiridin-2-il)-1H-1,2,4-triazol-3-il]metil}-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo (Ejemplo 17, 35,0 mg, 56,2 pmol) en agua (2,0 ml) se trató con de acetato de calcio monohidrato (1:1) (9,89 mg, 56,2 pmol), luego se agitó durante 15 min a temperatura ambiente y se liofilizó proporcionando 38,5 mg (cuant.) del compuesto del título.
CL-EM (Procedimiento 1): Tr = 0,65 min; MS (ESI pos): m/z = 623,0 [M+H]+
RMN iH (500 MHz, D2O): 5 [ppm] = 8,56-8,52 (m, 1H), 8,25-8,19 (m, 1H), 7,76-7,58 (m, 5H), 5,42-5,29 (m, 2H), 4,68 (s, 1H, superposición con pico de HDO), 4,23-4,10 (m, 2H).
Ejemplo 24
Fosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-il}-metil)-3-(4-dorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1 -trifluoropropan-2-ilo dipotásico

A 0 °C , una solución de dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometil)piridin-2-il]-1H-1,2,4-triazol-3-N}metN)-3-(4-dorofenN)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-N]-1,1,1-trifluoropropan-2-ilo (Ejemplo 1, 400 mg, 0,609mmol) en acetonitrilo (12 ml) se trató con una solución de hidrogenocarbonato de potasio (1,2 ml, 1 M, 1,2 mmol). La mezcla resultante se agitó durante 5 min a 0 °C, 5 min a temperatura ambiente y se liofilizó.
50 mg del compuesto liofilizado se disolvieron en 2-propanol (3 ml). La solución resultante se concentró lentamente en agitación a temperatura ambiente. Después de 3 semanas, el disolvente se evaporó completamente. El sólido obtenido se examinó por difractometría de rayos X y correspondía al compuesto del título en forma de material mesomorfo. Una estequiometría de potasio de 1,9±0,2 se calculó según el análisis TGA (9,9 % del disolvente residual en masa) y la cromatografía de iones (9,1 % de potasio en masa) de acuerdo con la siguiente fórmula:
Estequiometria = [( %potasio) / (1 - %disolvente residual - %potasio)] x [(Mácido libre) / (Mpotasio)]
CL-EM (Procedimiento 5): Tr = 0,67 min; MS (ESI pos): m/z = 657,1 [M+H]+
RMN 1H (500 MHz, D2O): 5 [ppm] = 8,84 (d, 1H), 8,51 (d, 1H), 7,95 (dd, 1H), 7,74-7,56 (m, 4H), 5,47-5,23 (m, 2H), 4,23-4,03 (m, 2H).
Tabla 4: Difractometría de rayos X en polvo del compuesto obtenido en el ejemplo 24

Tabla 5: Espectro infrarrojo del compuesto obtenido en el ejemplo 24

(continuación)

Las bandas marcadas con * son específicas de la sal de potasio.
SECCIÓN EXPERIMENTAL - ENSAYOS BIOLÓGCIOS
Abreviaturas y acrónimos:
Acc. n.° número de acceso
AP fosfatasa alcalina
AVP arginina vasopresina
Bmáx máxima capacidad de unión al ligando
BSA albúmina en suero bovino
cAMP adenosina monofosfato cíclica
Cat. n.° número de catálogo
ADNc ácido desoxirribonucleico complementario
CHO ovario de hámster chino
CRE elemento de respuesta a cAMP
Ct umbral de ciclo
DMEM/F12 medio de Eagle modificado por Dulbecco/ medio Ham's F12 (1:1) ADN ácido desoxirribonucleico
DMSO dimetilsulfóxido
DTT ditiotreitol
concentración eficaz máxima media
EDTA ácido etilendiamina-tetraacético
FAM carboxifluoresceína succinimidil éster
c.f. concentración final
FCS suero de ternero fetal
HEPES ácido 4-(2-hidroxietil)piperazin-1-etanosulfónico
concentración inhibidora media máxima
Kd constante de disociación
Ki constante de disociación de un inhibidor
mRNA ácido ribonucleico mensajero
PBS solución salina amortiguada con fosfato
PEG polietilenglicol
p.o. per os, peroral
ARN ácido ribonucleico
RTPCR reacción en cadena de la polimerasa en tiempo real
SPA ensayo de proximidad de centelleo
TAMRA carboxitetrametilrodamina
TRIS; Tris 2-amino-2-hidroximetilpropan-1,3-diol
v/v volumen/volumen; concentración, porcentaje de solución
La demostración de la actividad de los compuestos de la presente invención se puede lograr a través de ensayos in vitro, ex vivo, e in vivo que son conocidos en la técnica. Por ejemplo, para demostrar la actividad de los compuestos de la presente invención, se pueden usar los siguientes ensayos.
B-1. Ensayo celular in vitro para determinar actividad del receptor de vasopresina
La identificación de agonistas y antagonistas de los receptores de vasopresina V1a y V2 de seres humanos, ratas y perros, así como la cuantificación de la actividad de los compuestos de la invención, se realizan usando líneas celulares recombinantes. Estas líneas celulares derivan originalmente de célula epitelial de ovario de hámster (ovario de hámster chino, CHO K1, ATCC: American Type Culture Collection, Manassas, VA 20108, EUA). Estas líneas celulares de prueba expresan constitutivamente los receptores V1a y V2 de ser humano, rata o perro. En el caso de receptores V1a acoplados a G«q, las células también se transfectan de forma estable con una forma modificada de fotoproteínas sensibles a calcio aequorina (V1a de ser humano y rata) u obelina (V1a de perro), que, después de la reconstitución con el cofactor coelenterazina, emiten luz cuando hay aumentos en las concentraciones de calcio libre [Rizzuto R, Simpson AW, Brini M, Pozzan T, Nature 358, 325-327 (1992); Illarionov BA, Bondar VS, Illarionova VA, Vysotski ES, Gene 153 (2), 273-274 (1995)]. Las células del receptor de vasopresina resultantes reaccionan con estimulación de receptores V1a expresados recombinantemente por liberación intracelular de iones de calcio, que se pueden cuantificar por la luminiscencia de fotoproteína resultante. Los receptores V2 acoplados con Gs se transfectan de forma estable en líneas celulares que expresan el gen para luciferasa de luciérnaga bajo el control de un promotor responsable de CRE. La activación de receptores V2 induce la activación del promotor de respuesta a CRE mediante aumento de cAMP, induciendo así la expresión de luciferasa de luciérnaga. La luz emitida por fotoproteínas de líneas celulares V1a, así como la luz emitida por luciferasa de luciérnaga de líneas celulares V2 corresponden a la activación o inhibición del receptor de vasopresina correspondiente. La bioluminiscencia de las líneas celulares se detecta usando un luminómetro adecuado [Milligan G, Marshall F, Rees S, Trends in Pharmacological Sciences 17, 235-237 (1996)].
Procedimiento de prueba:
Líneas celulares del receptor de vasopresina V1a:
El día previo al ensayo, las células se colocan en medio de cultivo (DMEM/F12, FCS al 2 %, glutamina 2 mM, HEPES 10 mM, 5 pg/ml de coelenterazina) en placas de microtitulación de 384 pocillos y se mantienen en una incubadora celular (96 % de humedad, 5 % en v/v de CO2, 37 °C). El día del ensayo, los compuestos de prueba en varias concentraciones se colocan durante 10 minutos en los pocillos de la placa de microtitulación antes de agregar el agonista [Arg8]-vasopresina en una concentración CE50. La señal de luz resultante se mide inmediatamente en un luminómetro.
Líneas celulares del receptor de vasopresina V2:
El día previo al ensayo, las células se colocan en medio de cultivo (DMEM/F12, FCS al 2 %, glutamina 2 mM, HEPES 10 mM) en placas de microtitulación de 384 pocillos y se mantienen en una incubadora celular (96 % de humedad, 5 % en v/v de CO2, 37 °C). El día del ensayo, los compuestos de prueba en varias concentraciones y el agonista [Arg8]-vasopresina en una concentración CE50 se agregan juntos a los pocillos y las placas se incuban durante 3 horas en una incubadora celular. Tras la adición del reactivo de lisis celular Triton™ y el sustrato de luciferina, se mide la luminiscencia de luciferasa de luciérnaga en un luminómetro.
La Tabla 1A a continuación enumera valores individuales de CI50 para los compuestos de la invención (que incluyen mezclas racémicas, así como enantiómeros separados) que se obtuvieron de líneas celulares transfectadas con el receptor V1a o V2 humano: Esto significa que estos valores CI50 son los valores para el profármaco y no para el fármaco respectivo subyacente ya que el profármaco es principalmente estable en condiciones de ensayo. Los datos para el fármaco respectivo subyacente se muestran en los Experimentos mencionados a continuación.
Tabla 1A:

(continuación)

B-2. Ensayo de unión radioactivo
Los valores de CI50 y K se pueden determinar en ensayos de unión radioactivos usando fracciones de membrana de línea celular de riñón embrionario humano 293 (HEK293) o líneas celulares CHO-K1 que expresan los receptores de vasopresina humana V1a y V2 correspondientes.
Los receptores de vasopresina recombinante humana V1a expresados en células HEK293 se usan en 50 mM de amortiguador Tris-HCl, pH 7,4, MgCh5 mM, 0,1 % de BSA usando técnicas estándar. Alícuotas de membranas preparadas se incuban con compuestos de prueba en varias concentraciones en duplicados y 0,03 nM [125I]fenilacetil-D-Tyr(Me)-Phe-Gln-Asn-Arg-Pro-Arg-Tyr-NH2 durante 120 minutos a 25 °C. La unión no específica se estima en presencia de 1 pM [Arg8]Vasopresina. Los receptores se filtran y lavan, los filtros luego se cuentan para determinar el [125I]fenilacetil-D-Tyr(Me)-Phe-Gln-Asn-Arg-Pro-Arg-Tyr-NH2 específicamente unido.
Las células CHO-K1 transfectadas de forma estable con un plásmido que codifica el receptor de vasopresina recombinante humana V2 se usan para preparar membranas en 50 mM de amortiguador Tris-HCl, pH 7,4, MgCh 10 mM, 0,1 % de BSA usando técnicas estándar. Alícuotas de membranas preparadas se incuban con compuestos de prueba en varias concentraciones en duplicados y 4 nM [3H](Arg8)-Vasopresina durante 120 minutos a 25 °C. La unión no específica se estima en presencia de (Arg8)-Vasopresina1 mM. Las membranas se filtran y se lavan 3 veces y los filtros se cuentan para determinar [3H](Arg8)-Vasopresina específicamente unida.
Los valores de CI50 se determinan por análisis de regresión de mínimos cuadrados no lineales usando MathlQTM (ID Business Solutions Ltd., Reino Unido). La constante de inhibición Ki se calcula usando la ecuación de Cheng y Prusoff (Cheng, Y., Prusoff, W.H., Biochem. Pharmacol. 22:3099-3108, 1973).
Para verificar la conversión de profármacos en fármacos respectivos, los profármacos se incuban con y sin fosfatasa alcalina. La escisión del resto de fosfato por fosfatasa alcalina convierte el profármaco en el fármaco respectivo subyacente [Coleman J. E., Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441-483]. Como el profármaco y el fármaco respectivo subyacente muestran diferentes valores CI50, el valor del profármaco después del tratamiento con fosfatasa alcalina se asemeja al CI50 del fármaco respectivo.
B-3. Ensayo celular in vitro para detectar la acción de antagonistas del receptor de vasopresina V1a en la regulación de genes profibróticos
La línea celular H9C2 (American Type Culture Collection No. ATCC CRL-1446), descrita como un tipo de cardiomiocito aislado de tejido cardíaco de rata, expresa de forma endógena el receptor de vasopresina V1a AVPR1A en gran cantidad de copias, mientras la expresión de AVPR2 no se puede detectar. Asimismo, la línea celular NRK49F (ATCC No. CRL1570) aislada de tejido de riñón de rata, muestra un patrón de expresión similar de expresión de AVPR1A mRNA alto y reducción de expresión de AVPR2. Para ensayos celulares que detectan la inhibición de la regulación dependiente del receptor AVPR1A de expresión génica por antagonistas del receptor, el procedimiento es el siguiente:
Las células H9C2 o células NRK49F se siembran en placas de microtitulación de 6 pocillos para cultivo celular a una densidad celular de 50000 células/pocillo en 2,0 ml de medio Opti-MEM (Invitrogen Corp., Carlsbad, CA, EUA, n.° de catálogo 11058-021) y se mantienen en una incubadora celular (96 % de humedad, 8 % en v/v de CO2, 37 °C).
Después de 24 horas, se cargan conjuntos de tres pocilios (triplicado) con solución vehicular (control negativo) y solución de vasopresina ([Arg8]-vasopresina acetato, Sigma, n.° de catálogo V9879) o compuesto de prueba (disuelto en vehículo: agua con 20 % en v/v de etanol) y solución de vasopresina. En el cultivo celular, la concentración de vasopresina final es 1 nM. La solución del compuesto de prueba se agrega al cultivo celular en pequeños volúmenes, de modo que no se exceda una concentración final de 0,03 % de etanol en el ensayo celular. Después de un tiempo de incubación de 5 horas, el sobrenadante de cultivo se quita con succión, las células adherentes se lisan en 350 pl de amortiguador RLT (Qiagen, n.° de catálogo 79216) y el ARN se aísla del lisado usando el kit RNeasy (Qiagen, n.° 74104). A esto le sigue digestión de ADNsa (Invitrogen, n.° de catálogo 18068 015), síntesis de ADNc (Promaga, ImProm-II Reverse Transcription System, n.° de catálogo A3800) y reacción en cadena de polimerasa por transcripción inversa (RCPTI) (pPCR MasterMix RT-QP2X-03-075, Eurogentec, Seraing, Bélgica). Todos los procedimientos se realizan de acuerdo con los protocolos de trabajo de los fabricantes del reactivo de prueba. Los conjuntos de cebadores para el RCPTI se seleccionan según las secuencias génicas de ARNm (NCBI GenBank Entrez Nucleotide Data Base) usando el programa Primer3Plus con sondas etiquetadas con 6-FAM TAMRA. El RCPTI para determinar la expresión de ARNm relativa de células de varios lotes de ensayo se realiza usando el detector de secuencia Applied Biosystems ABI Prism 7700 en formato de placa de microtitulación de 384 pocillos de acuerdo con las instrucciones operativas del instrumento. La expresión génica relativa se representa por el valor delta-delta Ct [Applied Biosystems, User Bulletin No. 2 ABI Prism 7700 SDS, 11 de diciembre de 1997 (actualizado 10/2001)] con referencia al nivel de expresión del gen L-32 de proteína ribosómica (n.° de acceso GenBank NM_013226) y el valor Ct umbral de Ct = 35.
Para verificar la conversión de los profármacos en los fármacos respectivos, los profármacos se incuban con y sin fosfatasa alcalina. La escisión del resto de fosfato por fosfatasa alcalina convierte el profármaco en el fármaco respectivo subyacente [Coleman J. E., Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441-483]. Como el profármaco y el fármaco respectivo subyacente muestran diferentes valores CI50, el valor del profármaco después del tratamiento con fosfatasa alcalina se asemeja al CI50 del fármaco respectivo.
B-4. Ensayo celular in vitro para verificación de conversión del profármaco en fármaco
Para verificar la conversión de los profármacos en los fármacos respectivos, los profármacos se incuban con y sin fosfatasa alcalina. La escisión del resto de fosfato por fosfatasa alcalina convierte el profármaco en el fármaco respectivo subyacente [Coleman J. E., Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441-483]. Como el profármaco y el fármaco respectivo subyacente muestran diferentes valores CI50, el valor del profármaco después del tratamiento con fosfatasa alcalina se asemeja al CI50 del fármaco respectivo. La determinación de los valores CI50 de los compuestos de la invención en el receptor V1a humano se lleva a cabo usando una línea celular recombinante. La línea celular deriva originalmente de una célula epitelial de ovario de hámster (ovario de hámster chino, CHO K1, ATCC: American Type Culture Collection, Manassas, VA 20108, EUA). Esta línea celular de prueba expresa constitutivamente el receptor V1a humano. Las células también se transfectan de forma estable con una forma modificada de fotoproteína sensible a calcio aequorina que, después de la reconstitución con el cofactor coelenterazina, emite luz tras aumentos en la concentración de calcio libre [Rizzuto R, Simpson AW, Brini M, Pozzan T, Nature 358, 325-327 (1992); Illarionov BA, Bondar VS, Illarionova VA, Vysotski ES, Gene 153 (2), 273-274 (1995)]. La célula del receptor de vasopresina resultante reacciona con la estimulación del receptor V1a expresado recombinantemente por liberación intracelular de iones de calcio, que se puede cuantificar por la luminiscencia de la fotoproteína resultante. La bioluminiscencia de la línea celular se detecta usando un luminómetro adecuado [Milligan G, Marshall F, Rees S, Trends in Pharmacological Sciences 17, 235-237 (1996)].
Procedimiento de prueba:
El día previo al ensayo, las células se siembran en medio de cultivo (DMEM/F12, FCS al 2 %, glutamina 2 mM, HEPES 10 mM, 5 pg/ml de coelenterazina) en placas de microtitulación de 384 pocillos y se mantienen en una incubadora celular (96 % de humedad, 5 % en v/v de CO2, 37 °C). El día del ensayo, 100 pl de compuestos de prueba a una concentración de 100 pM se incuban durante 15 minutos con 500 unidades de fosfatasa alcalina a 37 °C en amortiguador tyrode (HEPES 20 mM, NaCl 130 mM, KCl 5 mM, NaHCO35 mM, MgCh2 mM, pH 7,4). Después de la incubación los compuestos se diluyen inmediatamente y se colocan durante 10 minutos a 37 °C en las células V1a sembradas. Para la detección de la actividad antagonista de los compuestos se agrega [Arg8]-vasopresina a concentración CE50 y la señal de luz resultante se mide inmediatamente en un luminómetro.
Se realizó la CL-EM de las muestras de compuesto (Ejemplo 4A y Ejemplo 1) con y sin fosfatasa alcalina (AP). Procedimiento 6 (CL-EM):
Instrumento MS: Instrumento Waters TOF; UPLC tipo Instrumento: Waters Acquity I-CLASS; Columna: Waters, HSST3, 2,1 x 50 mm, C181,8 pm; eluyente A: 1 l de agua ácido fórmico al 0,01 %; eluyente B: 1 l de acetonitrilo ácido fórmico al 0,01 %; gradiente: 0,0 min al 2 % de B ^ 0,5 min al 2 % de B ^ 7,5 min al 95 % de B ^ 10,0 min al 95 % de B; Horno: 50 °C; Flujo: 1,00 ml/min; detección UV: 210 nm.
Ejemplo 4A con AP
CL-EM (Procedimiento 6): Tr = 3,83 min; MS (ESI pos): m/z = 577,1 [M+H]+
Ejemplo 4A sin AP
CL-EM (Procedimiento 6): Tr = 3,84 min; MS (ESI pos): m/z = 577,1 [M+H]+
Ejemplo 1 con AP
CL-EM (Procedimiento 6): Tr = 3,83 min; MS (ESI pos): m/z = 577,1 [M+H]+
Ejemplo 1 sin AP
CL-EM (Procedimiento 6): Tr = 3,19 min; MS (ESI pos): m/z = 657,1 [M+H]+
Tabla 2A: valores CI50 de fármacos y sus respectivos profármacos en presencia y ausencia de fosfatasa alcalina (AP)

B-5. La inhibición de vasopresina indujo agregación de plaquetas humanas para la verificación de la conversión del profármaco en fármaco
Las plaquetas humanas expresan el receptor de V1a de forma endógena. Se descubrió que las concentraciones de vasopresina de arginina (AVP) relativamente altas (aprox. 50-100 nM) estimulan la agregación de plaquetas ex vivo. Por lo tanto, las plaquetas enriquecidas con sangre humana pueden servir como tejido que expresa V1a para estudios farmacológicos con antagonistas de vasopresina.
Para verificar la conversión de los profármacos en los fármacos respectivos, los profármacos se incuban con y sin fosfatasa alcalina. La escisión del resto de fosfato por la fosfatasa alcalina convierte el profármaco en el fármaco. Como el profármaco y el fármaco respectivo subyacente muestran diferentes valores CI50, el valor del profármaco después del tratamiento con fosfatasa alcalina se asemeja al CI50 del fármaco respectivo.
100|jl del compuesto de prueba a una concentración de 100 jM se incuban durante 15 min con 500 unidades de fosfatasa alcalina a 37 °C en amortiguador Tyrode modificado (NaCl 134 mM, NaH2PO4*H2O 12 mM, NaH2PO4*H2O 0,34 mM, KCl 2,9 mM, HEPES 5 mM, glucosa 5 mM) y se almacenan a 4 °C hasta su uso adicional en ensayo de agregación plaquetaria.
La sangre humana se recoge en tubos de plástico que contienen un volumen 1/10 de citrato de trisodio 0,106 M por punción venosa en voluntarios sanos no fumadores (n = 4/grupo) que no tomaron fármacos durante al menos 1 semana. El plasma rico en plaquetas (PRP) se obtiene por centrifugación de la muestra de sangre a 140 g durante 20 min a temperatura ambiente. El sedimento resultante se descarta y el PRP se centrifuga adicionalmente (11.000 rpm, 1 min) para producir plasma pobre en plaquetas (PPP). La agregación de plaquetas se mide de forma turbidimétrica usando un agregómetro (APACT 4). La reacción se sigue monitoreando los cambios en la transmisión de luz en alícuotas de 178 j l de PRP, con agitación continua a 37 °C, en comparación con control de PPP. Varias concentraciones de antagonistas de vasopresina (en 2 jl) se agregan a PRP 5 min antes de la adición de 20 j l de AVP (concentración final 100 nM). Los efectos inhibidores de los compuestos se determinan midiendo la amplitud máxima de la curva de agregación en comparación con la respuesta de control. Los valores CI50 se calculan según una curva de inhibición de respuesta a la concentración por un programa de regresión no lineal iterativo. Todos los valores se expresan como valores promedio (Tabla 3A).
Tabla 3A: Efectos de compuestos de prueba en agregación en plasma rico en plaquetas humano.

(continuación)

B-6. Efectos sobre la contracción de anillos de vasos de ratas aislados
Aorta aislada
Los compuestos de prueba se pueden investigar en anillos aórticos aislados de ratas macho Wistar que expresan el receptor V1a. Las ratas macho Wistar se sometieron a eutanasia usando dióxido de carbono. La aorta se remueve y coloca en amortiguador helado Krebs-Henseleit de la siguiente composición (en mmol/l): NaCl 112, KCl 5,9, CaCh 2,0, MgCl21,2, NaH2PO41,2, NaHCO325, glucosa 11,5. La aorta se corta en anillos de 3 mm y se transfiere a 20 ml de baños de órganos que contienen solución Krebs-Henseleit equilibrada con 95 % de O2, 5 % de CO2 a 37 °C. Para registrar la tensión isométrica los anillos se montan entre dos ganchos. La tensión restante se ajusta hasta 3 g. Después de un período de equilibrio, cada experimento se inicia exponiendo la preparación a K+ (50 mM) de solución Krebs-Henseleit. Los anillos aórticos luego se precontraen usando 1 nmol/l de Arg-vasopresina. Después de establecida una contracción estable, se construye una curva de respuesta a dosis cumulativa del compuesto de prueba. La contracción estabilizada inducida por Arg-vasopresina se define como el 100 % de tensión. La relajación se expresa como in porcentaje de tensión.
Arteria renal aislada
Las ratas macho Wistar (200-250 g) se sometieron a eutanasia usando dióxido de carbono. La arteria renal se remueve y coloca en amortiguador helado Krebs-Henseleit de la siguiente composición (en mmol/l): NaCl 112, KCl 5,9, CaCl22,0, MgCh 1,2, NaH2PO41,2, NaHCO325, glucosa 11,5. Para medir la tensión isométrica, los segmentos de anillo, de 2 mm de longitud, se montan en un miógrafo de cámara de vasos pequeños (Danish Myo Technology A/S, Dinamarca) usando dos cables de tungsteno sujetos a montajes de mandíbula. Un montaje de mandíbula se une a un micrómetro permitiendo el control de la circunferencia del vaso. El otro montaje de mandíbula se une con un transductor de fuerza para medir el desarrollo de tensión. La preparación total se mantiene en una cámara con solución de sal fisiológica a 37 °C burbujeada con oxígeno. Después de 30 min de período de equilibro, los vasos se extienden a su diámetro óptimo de lumen para desarrollo de tensión activo que se determina en función de la relación de tensión de circunferencia interna-pared. La circunferencia interna se establece en un 90 % de la que sería la de los vasos si se hubieran expuesto a una tensión pasiva equivalente a la producida por una presión transmural de 100 mmHg.
Después, los vasos se lavan tres veces con amortiguador Krebs-Henseleit y se dejan equilibrar durante 30 min. La contractilidad luego se analiza por una exposición de dos veces a una solución de K+ alto (50 mmol/l de KCl). Después de lavar con amortiguador Krebs-Henseleit los vasos luego se precontraen luego usando 1 nmol/l de Argvasopresina. Después de establecida una contracción estable, se construye una curva de respuesta a dosis cumulativa del compuesto de prueba. La contracción estabilizada inducida por Arg-vasopresina se define como el 100 % de tensión. La relajación se expresa como un porcentaje de tensión.
Para verificar la conversión de los profármacos en los fármacos respectivos, los profármacos se incuban con y sin fosfatasa alcalina. La escisión del resto de fosfato por fosfatasa alcalina convierte el profármaco en el fármaco respectivo subyacente [Coleman J. E., Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 441-483]. Como el profármaco y el fármaco respectivo subyacente muestran diferentes valores CI50, el valor del profármaco después del tratamiento con fosfatasa alcalina se asemeja al CI50 del fármaco respectivo.
B-7. Ensayo in vivo para detectar efectos cardiovasculares: medición de la presión sanguínea en ratas anestesiadas (modelo de 'desafío' de vasopresina)
Se usan ratas macho Sprague-Dawley (250-350 g de peso corporal) con anestesia de inyección de ketamina/xilazina/pentobarbital. Tubos de polietileno (PE-50, Intramedic®), que se precargan con solución de cloruro de sodio isotónica que contiene heparina (500 IU/ml), se introducen en la vena yugular y la vena femoral y luego se relacionan. Mediante un acceso venoso, con la ayuda de una jeringa, se inyecta Arg-vasopresina (SIGMA); la sustancia de prueba se administra mediante el segundo acceso venoso. Para la determinación de la presión sanguínea sistólica, se relaciona un catéter de presión (Millar SPR-3202F) en la arteria carótida. El catéter arterial está conectado a un transductor de presión que alimenta sus señales a una computadora de registro equipada con software de registro adecuado. En un experimento típico, al animal experimental se le administran 3-4 inyecciones de bolo sucesivas en intervalos de 10-15 min con una cantidad definida de Arg-vasopresina (30 ng/kg) en solución de cloruro de sodio isotónica. Cuando la presión sanguínea ha alcanzado nuevamente los niveles iniciales, la sustancia de prueba se administra como un bolo con infusión continua posterior en un disolvente adecuado. Después de esto, en intervalos definidos (10-15 min), se administra nuevamente la misma cantidad de Argvasopresina que al comienzo. En función de los valores de presión sanguínea, se hace una determinación de la
medida con la que la sustancia de prueba contrarresta el efecto hipertenso de la Arg-vasopresina. Los animales de control solo reciben disolvente en vez de la sustancia de prueba.
Después de la administración intravenosa, los compuestos de la invención en comparación con los controles del disolvente ocasionan una inhibición del aumento de presión sanguínea por Arg-vasopresina.
B-8. Ensayo in vivo para detectar los efectos mediados por el receptor V2 de vasopresina: investigaciones de diuresis en ratas conscientes mantenidas en jaulas de metabolismo
Se mantienen ratas Wistar (400-500 g de peso corporal) con acceso libre a alimento (Altromin) y agua potable. Durante el experimento, los animales se mantienen con acceso libre a agua potable durante 7 horas individualmente en jaulas de metabolismo adecuadas para ratas de esta clase de peso (Tecniplast Deutschland GmbH, D-82383 HohenpeilJenberg). Al comienzo del experimento, se les administra a los animales la sustancia de prueba en un volumen de 1 ml/kg de peso corporal de un disolvente adecuado (2-hidroxipropil-beta-ciclodextrina) mediante sonda en el estómago. Los animales de control solo recibieron disolvente. Las pruebas de control y sustancia se realizan en paralelo el mismo día. Los grupos de control y los grupos de dosis de sustancia consisten en de 6 a 8 animales. Durante el experimento, la orina excretada por los animales se recoge continuamente en un receptor en la base de la jaula. El volumen de orina se determina por separado para cada animal. Antes de comenzar el experimento, el peso corporal de los animales individuales se determina.
Después de la administración intravenosa, en comparación con las aplicaciones de control de disolvente, un compuesto de bloqueo del receptor V2 de vasopresina ocasionaría una excreción aumentada de orina, que se basa esencialmente en una excreción aumentada de agua (acuaresis).
La Tabla 4A a continuación muestra los cambios observados en la excreción urinaria con relación al control de disolvente (= 100 %) para los ejemplos de compuestos de la invención y los compuestos de comparación a tres dosificaciones diferentes:
Tabla 4A:

Los resultados mostrados en Tabla 4A demuestran que los compuestos de la presente invención no poseen ninguna actividad de bloqueo de V2 dependiente de la dosis significativa a las dosis indicadas in vivo. Esto es contrario al Ejemplo 82 del documento WO2016/071212 que dio surgimiento a un incremento dependiente de la dosis, de hasta catorce veces, en el volumen urinario versus el grupo de control del vehículo a una dosis de 3 mg/kg i.v.
B-9. Ensayo in vivo para detectar efectos renales protectores: Modelo de lesión de isquemia aguda/reperfusión en roedores
Se obtienen ratones macho C57Bl/6J criados en laboratorio de 6-8 semanas de edad en Taconic Biosciences y se obtienen ratas Sprague Dawley® de 6-8 semanas de edad en Charles River. Tanto las ratas como los ratones se mantienen en condiciones de laboratorio estándar, ciclos de 12 horas de luz-oscuridad con acceso a alimentación normal y tomando agua ad líbitum. Para el modelo de lesión por reperfusión isquémica se usa un total de 10-12 ratas o ratones en cada grupo de control y experimental.
Los animales se anestesian con isoflurano inhalado continuamente. Se realiza una nefrectomía derecha a través de una incisión en el flanco derecho 7 días antes de los procedimientos isquémicos en los riñones contralaterales. Para la isquemia renal se realiza una incisión en el flanco izquierdo. Los vasos renales se exponen por disección del pedículo renal izquierdo. Se usan grapas vasculares no traumáticas para detener el flujo de sangre (arteria y vena) durante 45 min (ratas) o 25 min (ratones) de isquemia. La reperfusión se establece removiendo las grapas. La pared abdominal (capa muscular y piel) se cierra con suturas de polipropileno 5.0. Se aplica Temgesic® (buprenorfina, 0,025 mg/kg s.c.) como analgésico.
Se recoge orina de cada animal en jaulas metabólicas durante la noche antes de sacrificarlos 24 h después de la isquemia. Después de sacrificarlos, se obtienen las muestras de sangre bajo anestesia terminal. Después de la centrifugación de las muestras de sangre, se aísla el suero. Se mide la creatinina en suero y la urea en suero mediante analizador de bioquímica clínica (Pentra 400). Para la evaluación de biomarcadores de lesión en riñones
en suero y urinarios (lipocalina asociada con neutrófilo gelatinasa [NGAL], ELISA de molécula de lesión renal - 1 [KIM-1] y Osteopontina) se realizan de acuerdo con el protocolo de los fabricantes. Se mide la creatinina y albúmina urinarias para determinar la relación albúmina/creatinina.
El ARN total se aísla de los riñones. Los riñones izquierdos se congelaron instantáneamente en nitrógeno líquido en el sacrificio. El tejido de los riñones luego se homogeniza y se obtiene el ARN. El ARN total se transcribe en ADNc. Usando NGAL renal PCR en tiempo real TaqMan, la expresión de ARNm de Osteopontina, KIM-1, Nefrina y Podocina se analiza en tejidos de riñones completos.
Las diferencias entre grupos se analizaron por ANOVA unidireccional con correcciones de Dunnett para múltiples comparaciones. La importancia estadística se define como < 0,05. Todos los análisis estadísticos se realizan usando GraphPad Prism 7.
B-10. Ensayo in vivo para detectar efectos cardiovasculares: investigaciones hemodinámicas en perros anestesiados
Se anestesian perros beagle machos (Beagle, Marshall BioResources, EUA) con un peso de entre 10 y 15 kg con pentobarbital (30 mg/kg i.v., Narcoren®, Merial, Alemania) para las intervenciones quirúrgicas y los exámenes hemodinámicos y funcionales. Pancuroniumbromide (Pancuronium Inresa, Inresa, Alemania, 2-4 mg/animal i.v.) sirve además como un relajante muscular. Los perros se intuban y ventilan con una mezcla de aire ambiente/oxígeno (30/70 %), alrededor de 2,5-4 l/min. La ventilación ocurre usando un ventilador de GE Healthcare (Avance, Alemania) y se monitorea usando un analizador de dióxido de carbono (-Datex Ohmeda). Se mantiene la anestesia por infusión continua de pentobarbital (50 pg/kg/min); se usa fentanilo como un analgésico (10 pg/kg/h).
En intervenciones preparatorias, los perros se equipan con un marcapasos cardíaco. Al comienzo del experimento, se implanta un marcapasos cardíaco de Biotronik (Logos®, Alemania) en un bolsillo de piel subcutáneo y se pone en contacto con el corazón mediante un electrodo de marcapasos (Siello S60®, Biotronik, Alemania) que se avanza a través de la vena yugular externa con iluminación en el ventrículo derecho.
Entonces los accesos se remueven y los perros se despiertan espontáneamente de la anestesia. Después de 7 días adicionales, se activa el marcapasos descrito anteriormente y el corazón se estimula a una frecuencia de 220 latidos por minuto.
Los experimentos de prueba de fármaco real se realizan 28 días después del comienzo de la estimulación del marcapasos usando la siguiente instrumentación:
• Introducción de un catéter en la vejiga para liberación de la vejiga y para medir el flujo de orina
• Unión de electrocardiografía (ECG) lleva a las extremidades para la medición de ECG
• Introducción de un introductor de vaina relleno con solución de cloruro de sodio en la arteria femoral. Este tubo está conectado con un sensor de presión (Braun Melsungen, Melsungen, Alemania) para medir la presión arterial sistémica
• Introducción de un catéter Millar Tip (tipo 350 PC, Millar Instruments, Houston, EUA) a través de un puerto asegurado en la arteria carótida, para medir la hemodinámica cardíaca
• Introducción de un catéter Swan-Ganz (CCOmbo 7.5F, Edwards, Irvine, EUA) por la vena yugular en la arteria pulmonar, para medir la salida cardíaca, saturación de oxígeno, presiones arteriales pulmonares y presión venosa central
• Colocación de un catéter venoso en la vena cefálica para infundir pentobarbital para remplazo líquido y para muestreo de sangre (determinación de niveles de plasma de la sustancia o de otros valores sanguíneos clínicos) • Colocación de un catéter venoso en la vena safena para infundir fentanilo y para la administración de la sustancia
• Infusión de vasopresina (Sigma) en dosificación en aumento hasta una dosis de 4 mU/kg/min. Las sustancias farmacológicas luego se analizan con esta dosificación.
Las señales primarias se amplían si es necesario (ACQ7700, Data Sciences International, EUA o Edwards-Vigilance-Monitor, Edwards, Irvine, EUA) y se introducen posteriormente en el sistema Ponemah (Data Sciences International, EUA) para evaluación. Las señales se registran continuamente a lo largo del período experimental y se procesan además de forma digital por dicho software y se promedian durante 30 segundos.
B-11. Efectos agudos en cambios mediados por vasopresina de hemodinámica sistémica, flujo sanguíneo renal y oxigenación en ratas anestesiadas
Los experimentos se realizan en ratas macho Sprague Dawley (Charles River, Alemania; 350-450 g peso corporal).
En la preparación de los procedimientos quirúrgicos las ratas se anestesian con isoflurano y se colocan en una tabla calentada para mantener la temperatura corporal interna a 37 °C, se evalúan usando un termómetro rectal insertado. La anestesia por inhalación con isoflurano se aplica usando un vaporizador calibrado para inducir y mantener la anestesia con isoflurano 5 % en vol. y 2 % en vol., respectivamente. Una cánula PE50 se coloca en la arteria femoral derecha para monitorear la presión arterial promedio (PAM). Otro catéter PE50 se inserta en la vena femoral para infusión intravenosa. El riñón izquierdo se expone por una incisión en el flanco izquierdo y se separa de las uniones perirrenales. La cápsula del riñón permanece intacta. Durante la cirugía y posteriores períodos de equilibrio y control, las ratas reciben una infusión intravenosa de solución salina (NaCl al 0,9 %) a una velocidad de 100 pl/min. Las mediciones renales de flujo sanguíneo renal (FSR) se registran usando una sonda de flujo sanguíneo Doppler láser (Oxford Optronix, Reino Unido) que se une y estabiliza a la superficie del riñón. La sonda se inserta en la corteza renal a una profundidad de 2 mm y otra en la médula renal a 4 mm. El flujo sanguíneo renal, el oxígeno y la temperatura se miden por sonda de sensor combinada (Oxford Optronix, Reino Unido). La PAM, la frecuencia cardíaca (FC), FSR, temperatura y oxígeno se registran continuamente. Después de la cirugía y equilibrio, las mediciones iniciales se determinan durante 20 minutos. A las ratas luego se les suministra una infusión intravenosa con vasopresina a una velocidad de 50 ng/kg/min en 100 pl/kg durante 20 minutos. En el tercer período, se determina el efecto de infusión combinada de vasopresina y un antagonista del receptor de vasopresina dual (Ejemplo 82 del documento WO 2016/071212) o un antagonista de V1a selectivo (Ejemplo 1) o vehículo que se aplica como bolo en diferente concentración.
Tabla 5A: Efectos del antagonista del receptor de vasopresina dual Ejemplo 82 del documento WO2016/071212 en cambios mediados por vasopresina (AVP) de hemodinámica sistémica y renal en ratas.

Tabla 6A: Efectos del antagonista del receptor V1a de vasopresina selectivo Ejemplo 4A en cambios mediados por vasopresina (AVP) de hemodinámica sistémica y renal en ratas.

Tabla 7A: Efectos del antagonista del receptor V1a de vasopresina Ejemplo 1 en cambios mediados por vasopresina (AVP) de hemodinámica sistémica y renal en ratas.

B-12. Estabilidad de pH
0,15 mg del compuesto de prueba se disolvieron en 0,1 ml de dimetilsulfóxido y 0,4 ml de acetonitrilo. Para la disolución completa el vial de CLAR con la solución de muestra se agita y se trata con irradiación ultrasónica. Luego se agrega 1,0 ml de la respectiva solución amortiguadora y la muestra se centrifuga en vórtice. La solución de muestra se analiza por CLAr para determinar la cantidad del compuesto de prueba en un momento particular durante un período de tiempo de 24 h a 37 °C. Las áreas pico en porcentaje se usan para la cuantificación.
Amortiguadores
pH 4,0: amortiguador Fluka, Orden n.° 33643 (11,76 g de ácido cítrico, 2,57 g de cloruro de sodio y 2,72 g de hidróxido de sodio).
pH 7,4: 90 g de cloruro de sodio, 13,61 g de dihidrogenofosfato de potasio y 83,35 g de solución de hidróxido de sodio 1 M se componen hasta 1 litro con agua Millipore y luego se diluyen 1:10. Ajuste con ácido fosfórico a pH = 7,4.
pH 10: amortiguador Fluka, orden n.° 33649 (4,77 g de Borax, 0,73 g de hidróxido de sodio).
Porciones de 15 pl de la solución de muestra se analizan por CLAR en diferentes momentos (0 h, 1 h, 2 h, 4 h y 24 h) a 37 °C. Las áreas pico en porcentaje se usan para cuantificación.
Procedimiento CLAR
Eluyente: A = 1 ml de ácido trifluoroacético /l en agua; B = 1 ml ácido trifluoroacético/l en acetonitrilo
Columna: Nucleodur 100 C18ec, 3pm, 50 x 2 mm
Temperatura: 37 °C
Detección: 214 nm
Inyección: 15 pl
Gradiente: Tiempo (min) A ( %) B ( %) Flujo (ml/min)
0,0 98 2 0,75
1,0 98 2 0,75
15,0 5 95 0,75
17,5 5 95 0,75
17,7 98 2 1,50
18,2 98 2 1,50
18,5 98 2 1,00
19,0 98 2 0,75
Las relaciones del área pico (F) en diferentes momentos con relación al área pico en el punto de partida se muestran en la Tabla 8A para los ejemplos representativos:
Tabla 8A:

B-13. Solubilidad
Procedimiento experimental
Para cada sustancia se pesan 0,5 - 0,6 mg exactamente. En cada caso, se agrega medio suficiente a la muestra de modo tal que se obtiene una concentración de c = 500 pg/ml. La solución de muestra se agita durante 24 h a temperatura ambiente y 1400 rpm.
Otro peso de muestra de 0,5 - 0,6 mg es necesario para la solución de calibración de DMSO. Esta muestra se llena con DMSO hasta una concentración de c = 600 pg/ml. Dos soluciones de calibración se preparan de esta solución madre. En un vial CLAR de 2 ml, 1000 pl de DMSO se introducen inicialmente y 34,4 pl de la solución madre se colocan con pipeta (c = 20 pg/ml). 71,4 pl de esta solución (c = 20 pg/ml) se colocan en otro vial CLAR de 2 ml que contiene 500 pl de DMSO (c = 2,5 pg/ml).
Después de agitar las soluciones de prueba, 230 pl del sobrenadante se transfieren a un tubo de centrifugación y se centrifugan a 42000 rpm (223000 g) durante 30 min. 180 pl del sobrenadante se toman luego y cada uno se diluye con DMSO (1:5 muestra 1; 1:100 muestra 2) y se transfieren a viales de CLAR. Las dos soluciones de calibración y las soluciones de muestra diluidas se analizan por CLAR. La cuantificación se realiza en las correspondientes áreas pico.
Disolvente
Agua destilada; ácido trifluoroacético (Merck; 1.08262.0100); acetonitrilo (grado de CLAR); DMSO (Merck; 8.02912.2500).
Medio
Amortiguador de citrato pH 4: amortiguador Fluka, Orden n.° 33643 (11,76 g de ácido cítrico, 2,57 g de cloruro de sodio y 2,72 g de hidróxido de sodio).
Amortiguador pH 7: amortiguador Fluka, Orden n.° 33646 (3,52 g de dihidrogenofosfato de potasio, 7,26 g de fosfato de hidrógeno de potasio).
Amortiguador PBS pH 7,4: Solución de 6,18 g de cloruro de sodio y 3,96 g de dihidrogenofosfato de sodio en 1 l de agua destilada, ajustada con solución de hidróxido de sodio acuosa 1 M a pH 7,4.
Amortiguador-Tris pH 8,5: 0,6057 g de TRIS se disolvieron en 95 ml de agua, se ajustó a pH 8,5 con ácido clorhídrico acuoso, se llenó con agua hasta alcanzar 100 ml de volumen.
Equipo CLAR
Agilent 1100 o aparato comparable con detección UV, longitud de onda variable (z.B. haz de diodos); baño ultrasónico; Vibromix von Janke & Kunkel; Thermomixer de Eppendorf
Procedimiento CLAR
Eluyente A: 1 ml de ácido trifluoroacético/l agua; Eluyente B: 1 ml ácido trifluoroacético/l acetonitrilo Gradiente: Tiempo (min) A ( %) B ( %) Flujo (ml/min)
0,0 98 2 1,5
0,2 98 2 1,5
3,3 10 90 1,5
4,0 10 90 1,5
4,1 98 2 2,5
4,7 98 2 2,5
5,0 98 2 1,5
Columna: Zorbax Extend-c18, 50 x 3,0 mm, 3,5 |jm; temperatura de columna: 30 °C; Flujo: 1,5 ml/min; detector: 214/254 nm; volumen de inyección: 20 jl.
La solubilidad de los ejemplos representativos se muestra en la Tabla 9A.
Tabla 9A:
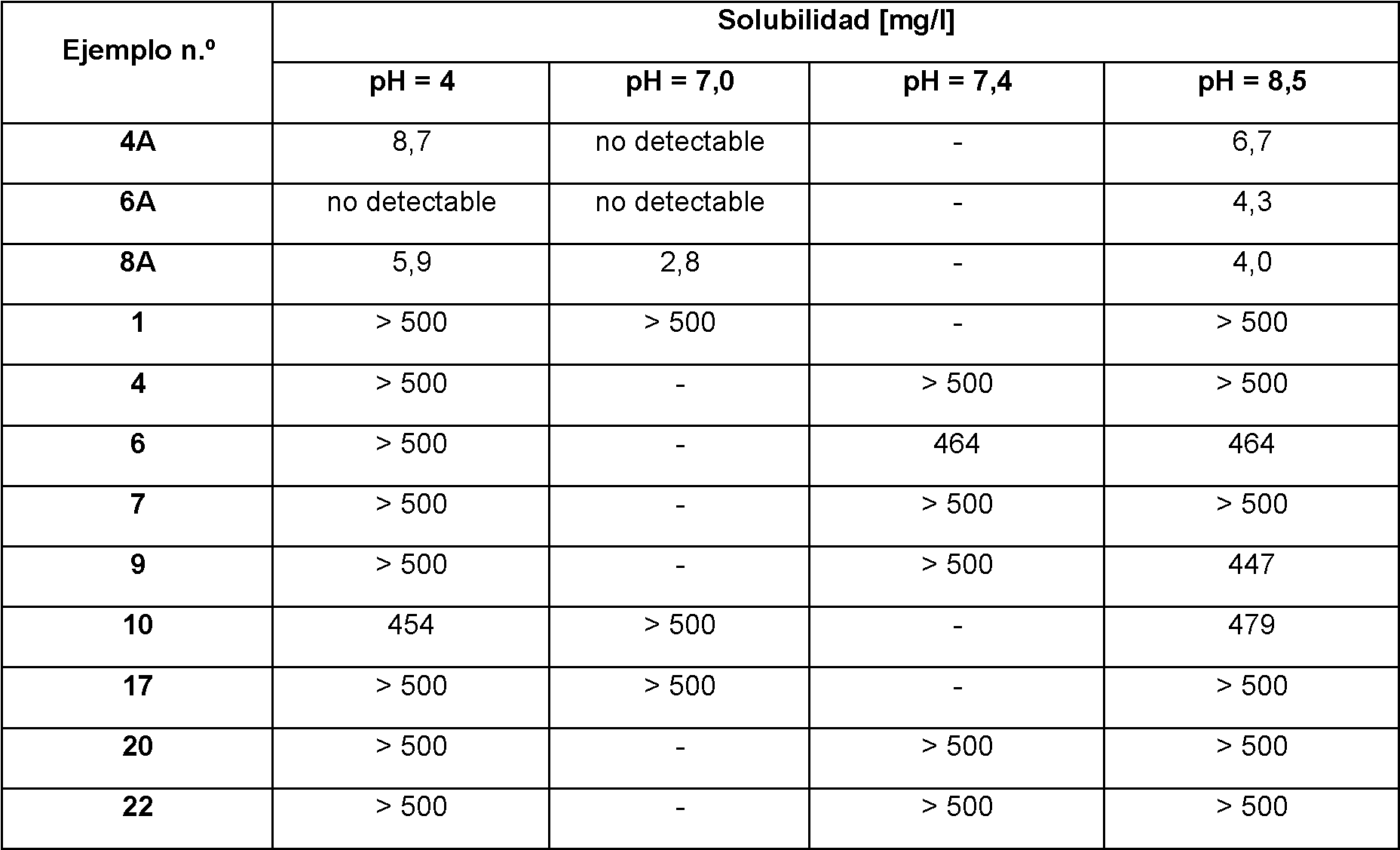
B-14. Determinación de parámetros farmacocinéticos después de administración intravenosa y oral Los parámetros farmacocinéticos de los compuestos de acuerdo con la invención se determinan en ratas macho Wistar, perros hembra Beagle y monos hembra Cynomolgus. La administración intravenosa se lleva a cabo por un vehículo de formulación adecuado para la respectiva especie, como una formulación de plasma/DMSO o solución salina fisiológica (pH 4 o más) para ratas o una formulación de agua/PEG400/etanol o solución salina fisiológica (pH 4 o más) para perros y monos. En todas las especies, la administración oral de la sustancia disuelta se realiza por alimentación forzada, usando un vehículo de formulación adecuado, como formulación de agua/PEG400/etanol o solución salina fisiológica. La toma de muestra de sangre de ratas se simplifica insertando un catéter adecuado en la vena yugular externa derecha antes de la administración de la sustancia. La operación se realiza al menos un día antes del experimento con anestesia de isoflurano y de la administración de un analgésico (atropina/Rimadyl (3/1) 0,1 ml s.c.). La sangre se toma (generalmente al menos 6 puntos de tiempo) en un período de tiempo que incluye puntos de tiempo terminales de al menos 7 horas hasta un máximo de 72 horas después de la administración de la sustancia. Cuando se toma la sangre, se pasa a tubos que contienen un anticoagulante adecuado, preferentemente
K-EDTA. Luego el plasma sanguíneo se obtiene por centrifugación y opcionalmente se almacena a -20 °C hasta el procesamiento adicional.
Un estándar interno (que también puede ser una sustancia químicamente no relacionada) se agrega a las muestras de los compuestos de acuerdo con la invención, las muestras de calibración y clasificadores y le sigue la precipitación de proteínas por medio de exceso de acetonitrilo. De manera alternativa, el estándar interno se agrega al acetonitrilo y la mezcla luego se agrega en exceso a muestras de los compuestos de acuerdo con la invención, muestras de calibración y calificadores para precipitación de proteínas. La adición de una solución amortiguadora coincidió con las condiciones de CL y posterior mezcla en vórtice, es seguida por la centrifugación a 2800 g. El sobrenadante se analiza por CL-EM/EM usando C18 o columnas de fase inversa de bifenilo y mezclas de fase móvil variable. Las sustancias se cuantifican mediante las alturas o áreas máximas de los cromatogramas de iones extraídos de experimentos de monitoreo de iones seleccionados específicos.
La concentración de plasma/gráficas de tiempo determinados se usan para calcular los parámetros farmacocinéticos como ABC (área bajo la curva), Cmáx (máxima concentración), t1/2 (semivida terminal), F (biodisponibilidad), TPM (tiempo de permanencia medio) y CL (depuración), usando un programa de cálculo farmacocinético validado.
Dado que la cuantificación de sustancia se realiza en plasma, es necesario determinar la distribución de sangre/plasma de la sustancia para ser capaz de ajustarse a los parámetros farmacocinéticos correspondientes. Con este fin, una cantidad definida de sustancia se incuba en sangre entera de la especie en cuestión en una mezcla oscilante durante 20 min. Después de la centrifugación a 2800 g, la concentración de plasma se midió (por medio de CL-EM/EM usando C18 o columnas de fase inversa de bifenilo y mezclas de fase móvil variable) y se determinó calculando la relación de la concentración de sangre entera contra la concentración de plasma (valor Csangre/Cplasma).
C) Ejemplos de trabajo de composiciones farmacéuticas
Las sustancias de acuerdo con la invención pueden convertirse en preparaciones farmacéuticas de la siguiente forma:
Comprimido:
Composición:
100 mg del compuesto del Ejemplo 1, 50 mg de lactosa (monohidrato), 50 mg de almidón de maíz, 10 mg de polivinilpirrolidona (PVP 25) (de BASF, Alemania) y 2 mg de estearato de magnesio.
Peso del comprimido 212 mg. Diámetro 8 mm, radio de curvatura 12 mm.
Producción:
La mezcla del compuesto del Ejemplo 1, lactosa y almidón se granula con una solución de fuerza al 5 % (m/m) del PVP en agua. Después de secarse, los gránulos se mezclan con el estearato de magnesio durante 5 min. Esta mezcla se comprime en una prensa de formación de comprimidos convencional (véase anteriormente formato del comprimido).
Suspensión oral
Composición:
1000 mg del compuesto del Ejemplo 1, 1000 mg de etanol (96 %), 400 mg de Rhodigel (goma xantana de FMC, EUA) y 99 g de agua.
10 ml de suspensión oral corresponde a una sola dosis de 100 mg del compuesto de la invención.
Producción:
El Rhodigel se suspende en etanol y el compuesto del Ejemplo 1 se agrega a la suspensión. El agua se agrega con agitación. La mezcla se agita durante alrededor de 6 h hasta que la hinchazón del Rhodigel esté completa.
Solución i.v. estéril:
El compuesto de acuerdo con la invención se disuelve en una concentración por debajo de la solubilidad de saturación en un disolvente fisiológicamente aceptable (por ejemplo, solución de cloruro de sodio isotónica, solución de glucosa al 5 % y/o solución de PEG 400 al 30 %). La solución se esteriliza por filtración y se rellena en recipientes de inyección estériles y libres de pirógenos.
Claims (11)
1. Un compuesto de fórmula general (I):

en la que
R1 representa un grupo de fórmula

en la que
# representa el punto de unión con el anillo 1,2,4-triazolilo,
o una de las sales farmacéuticamente aceptables de estos, solvatos de estos o solvatos de las sales de estos. 2. Un compuesto de fórmula general (I) de acuerdo con la Reivindicación 1, caracterizado porque
R1 representa un grupo de fórmula

en la que
# representa el punto de unión con el anillo 1,
2,4-triazolilo.
3. Dihidrogenofosfato de (2S)-3-[1-({5-carbamoil-1-[3-(trifluorometM)piridin-2-il]-1H-1,2,4-triazol-3-il}metil)-3-(4-clorofenil)-5-oxo-1,5-dihidro-4H-1,2,4-triazol-4-il]-1,1,1-trifluoropropan-2-ilo de acuerdo con la Reivindicación 1 de fórmula a continuación

o una de las sales farmacéuticamente aceptables de estos, solvatos de estos o solvatos de las sales de estos. 4. Dihidrogenofosfato de (2S)-3-[1-({5-CarbamolM-[3-(trlfluorometll)plrldln-2-ll]-1H-1,2,4-trlazol-3-ll}metll)-3-(4-clorofenll)-5-oxo-1,5-dlhldro-4H-1,2,
4-trlazol-4-ll]-1,1,1-trlfluoropropan-2-llo de acuerdo con la Reivindicación 1 de fórmula a contlnuaclón

5. Procedimiento para preparar un compuesto de fórmula general (I) o una de las sales farmacéuticamente aceptables de estos, solvatos de estos o solvatos de las sales de estos de acuerdo con la Relvlndlcaclón 1, caracterizado porque
[A] un compuesto de fórmula

en la que
R1 tiene el significado como se define para los compuestos de fórmula general (I) dado en la reivindicación 1, se hace reaccionar en la primera etapa con oxlcloruro de fósforo y en la segunda etapa se hidroliza para proporcionar un compuesto de fórmula general (I),
o
[B] un compuesto de fórmula

en la que
R1 tiene el significado como se define para los compuestos de fórmula general (I) dado en la reivindicación 1, se hace reaccionar en la primera etapa con difosfato de tetrabencilo y en la segunda etapa los grupos bencilo se eliminan en condiciones de reducción para proporcionar un compuesto de fórmula general (I),
opcionalmente le sigue, cuando fuera apropiado, convertir el compuesto de fórmula general (I) en sus correspondientes sales farmacéuticamente aceptables de estos, solvatos de estos o los solvatos de las sales de estos por tratamiento con los disolventes y/o bases correspondientes.
6. Compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para su uso en el tratamiento y/o prevención de enfermedades.
7. Compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para su uso en el tratamiento y/o prevención de enfermedades renales agudas y crónicas que incluyen nefropatía diabética, insuficiencia cardíaca aguda y crónica, preeclampsia, enfermedad arterial periférica (EAP), disfunción microvascular coronaria (DMC), síndrome de Raynaud, dismenorrea, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de HAD (SSIHA).
8. Uso de un compuesto como se define en cualquiera de las reivindicaciones 1 a 4 para la fabricación de una composición farmacéutica para el tratamiento y/o prevención de enfermedades renales agudas y crónicas que incluyen nefropatía diabética, insuficiencia cardíaca aguda y crónica, preeclampsia, enfermedad arterial periférica (EAP), disfunción microvascular coronaria (DMC), síndrome de Raynaud, dismenorrea, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de HAD (SSIHA).
9. Composición farmacéutica que comprende un compuesto como se define en cualesquiera de las reivindicaciones 1 a 4 y uno o más excipientes farmacéuticamente aceptables.
10. Composición farmacéutica de la reivindicación 9 que comprende uno o más primeros ingredientes activos, en particular compuestos de fórmula general (I) de acuerdo con una cualquiera de las reivindicaciones 1 a 4 y uno o más ingredientes activos adicionales, en particular uno o más agentes terapéuticos adicionales que se seleccionan del grupo que consiste en diuréticos, antagonistas de angiotensina AII, inhibidores de ACE, bloqueadores del receptor beta, antagonistas del receptor mineralocorticoide, nitratos orgánicos, donantes de NO, activadores y estimuladores de la guanilato ciclasa soluble y agentes inotrópicos positivos, agentes antiinflamatorios, agentes inmunosupresores, aglutinantes de fosfato y/o compuestos que modulan el metabolismo de vitamina D.
11. La composición farmacéutica como se define en la reivindicación 9 o 10 para su uso en el tratamiento y/o prevención de enfermedades renales agudas y crónicas que incluyen nefropatía diabética, insuficiencia cardíaca aguda y crónica, preeclampsia, enfermedad arterial periférica (EAP), disfunción microvascular coronaria (DMC), síndrome de Raynaud, dismenorrea, síndrome cardiorrenal, hiponatremia hipervolémica y euvolémica, cirrosis hepática, ascitis, edema y el síndrome de secreción inadecuada de HAD (SSIHA).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17197935 | 2017-10-24 | ||
| PCT/EP2018/078364 WO2019081292A1 (en) | 2017-10-24 | 2018-10-17 | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2906305T3 true ES2906305T3 (es) | 2022-04-18 |
Family
ID=60162099
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES18785394T Active ES2906305T3 (es) | 2017-10-24 | 2018-10-17 | Profármacos de derivados de triazol sustituido y sus usos |
Country Status (38)
| Country | Link |
|---|---|
| US (1) | US11298367B2 (es) |
| EP (1) | EP3700913B1 (es) |
| JP (1) | JP2021500365A (es) |
| KR (1) | KR20200069361A (es) |
| CN (1) | CN111225921A (es) |
| AR (1) | AR113791A1 (es) |
| AU (1) | AU2018356352A1 (es) |
| BR (1) | BR112020007983A2 (es) |
| CA (1) | CA3079767A1 (es) |
| CL (1) | CL2020001076A1 (es) |
| CO (1) | CO2020004945A2 (es) |
| CR (1) | CR20200176A (es) |
| CU (1) | CU24604B1 (es) |
| CY (1) | CY1124938T1 (es) |
| DK (1) | DK3700913T3 (es) |
| DO (1) | DOP2020000076A (es) |
| EA (1) | EA202091025A1 (es) |
| EC (1) | ECSP20023056A (es) |
| ES (1) | ES2906305T3 (es) |
| HR (1) | HRP20220060T1 (es) |
| HU (1) | HUE057278T2 (es) |
| IL (1) | IL273949A (es) |
| JO (1) | JOP20200099A1 (es) |
| LT (1) | LT3700913T (es) |
| MA (1) | MA50438B1 (es) |
| MX (1) | MX2020004191A (es) |
| NI (1) | NI202000030A (es) |
| PE (1) | PE20210394A1 (es) |
| PH (1) | PH12020550473A1 (es) |
| PL (1) | PL3700913T3 (es) |
| PT (1) | PT3700913T (es) |
| RS (1) | RS62984B1 (es) |
| SG (1) | SG11202003049WA (es) |
| SI (1) | SI3700913T1 (es) |
| TW (1) | TW201932117A (es) |
| UA (1) | UA126456C2 (es) |
| UY (1) | UY37948A (es) |
| WO (1) | WO2019081292A1 (es) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021259852A1 (en) | 2020-06-25 | 2021-12-30 | Bayer Aktiengesellschaft | Process for preparing 5-(alkoxycarbonyl)-and 5-(carboxamide)-1-aryl-1,2,4-triazole derivatives |
| WO2022112213A1 (en) | 2020-11-30 | 2022-06-02 | Bayer Aktiengesellschaft | Crystalline forms of 3-[[3-(4-chlorophenyl)-5-oxo-4-((2s)-3,3,3-trifluoro- 2-hydroxypropyl)-4,5-dihydro-1h-1,2,4-triazol-1-yl]methyl]-1-[3- (trifluoromethyl)pyridin-2-yl]-1h-1,2,4-triazole-5-carboxamide |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3966781A (en) | 1970-12-17 | 1976-06-29 | Merck Sharp & Dohme (I.A.) Corporation | Deuteration of functional group-containing hydrocarbons |
| DE19834044A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Neue substituierte Pyrazolderivate |
| DE19834047A1 (de) | 1998-07-29 | 2000-02-03 | Bayer Ag | Substituierte Pyrazolderivate |
| DE19943635A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943639A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Dicarbonsäurederivate mit neuartigen pharmazeutischen Eigenschaften |
| DE19943636A1 (de) | 1999-09-13 | 2001-03-15 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE19943634A1 (de) | 1999-09-13 | 2001-04-12 | Bayer Ag | Neuartige Dicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| AR031176A1 (es) | 2000-11-22 | 2003-09-10 | Bayer Ag | Nuevos derivados de pirazolpiridina sustituidos con piridina |
| DE10110750A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Neuartige Aminodicarbonsäurederivate mit pharmazeutischen Eigenschaften |
| DE10110749A1 (de) | 2001-03-07 | 2002-09-12 | Bayer Ag | Substituierte Aminodicarbonsäurederivate |
| DE10220570A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Ag | Carbamat-substituierte Pyrazolopyridine |
| KR100854872B1 (ko) | 2003-12-22 | 2008-08-28 | 화이자 인코포레이티드 | 바소프레신 길항제로서의 트리아졸 유도체 |
| WO2005105779A1 (en) | 2004-04-28 | 2005-11-10 | Pfizer Limited | 3-heterocyclyl-4-phenyl-triazole derivatives as inhibitors of the vasopressin v1a receptor |
| JP2011502982A (ja) * | 2007-11-06 | 2011-01-27 | アストラゼネカ・アクチエボラーグ | 好中球エラスターゼの阻害剤としてのある種の2−ピラジノン誘導体およびその使用 |
| DE102010001064A1 (de) | 2009-03-18 | 2010-09-23 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 2-Acetamido-5-Aryl-1,2,4-triazolone und deren Verwendung |
| AR076542A1 (es) * | 2009-05-22 | 2011-06-22 | Merck Sharp & Dohme | Antagonistas del receptor de orexina de isonicotinamida |
| AU2011219746B2 (en) | 2010-02-27 | 2015-04-23 | Bayer Intellectual Property Gmbh | Bisaryl-bonded aryltriazolones and use thereof |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| CN106977530A (zh) | 2010-07-09 | 2017-07-25 | 拜耳知识产权有限责任公司 | 环稠合的嘧啶和三嗪以及其用于治疗和/或预防心血管疾病的用途 |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| DE102010043379A1 (de) | 2010-11-04 | 2012-05-10 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 6-Fluor-1H-Pyrazolo[4,3-b]pyridine und ihre Verwendung |
| US8791162B2 (en) | 2011-02-14 | 2014-07-29 | Merck Sharp & Dohme Corp. | Cathepsin cysteine protease inhibitors |
| MX2016004532A (es) * | 2013-10-16 | 2016-07-22 | Bayer Cropscience Ag | Combinaciones de compuestos activos que comprenden un derivado de (tio)carboxamida y un compuesto fungicida. |
| CN107074783B (zh) | 2014-11-03 | 2020-06-05 | 拜耳制药股份公司 | 羟烷基取代的苯基三唑衍生物及其用途 |
| UY36571A (es) * | 2015-03-05 | 2016-09-30 | Bayer Cropscience Ag | Combinaciones de compuestos activos |
| AR108265A1 (es) | 2016-05-03 | 2018-08-01 | Bayer Pharma AG | Derivados de feniltriazol sustituidos con amida y usos de estos |
| US9988367B2 (en) | 2016-05-03 | 2018-06-05 | Bayer Pharma Aktiengesellschaft | Amide-substituted pyridinyltriazole derivatives and uses thereof |
| WO2017191114A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Aktiengesellschaft | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof |
| WO2017191105A1 (en) | 2016-05-03 | 2017-11-09 | Bayer Pharma Aktiengesellschaft | Amide-substituted aryltriazole derivatives and uses thereof |
| EP3452457B1 (en) | 2016-05-03 | 2020-03-18 | Bayer Pharma Aktiengesellschaft | Oxoalkyl-substituted phenyltriazole derivatives and uses thereof |
| US10525041B2 (en) | 2016-05-03 | 2020-01-07 | Bayer Pharma Aktiengesellschaft | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof |
| WO2018073144A1 (en) | 2016-10-20 | 2018-04-26 | Bayer Pharma Aktiengesellschaft | Hydroxyalkyl-substituted triazole derivatives and uses thereof |
-
2018
- 2018-10-17 PL PL18785394T patent/PL3700913T3/pl unknown
- 2018-10-17 ES ES18785394T patent/ES2906305T3/es active Active
- 2018-10-17 AU AU2018356352A patent/AU2018356352A1/en not_active Abandoned
- 2018-10-17 CR CR20200176A patent/CR20200176A/es unknown
- 2018-10-17 CU CU2020000040A patent/CU24604B1/es unknown
- 2018-10-17 CA CA3079767A patent/CA3079767A1/en active Pending
- 2018-10-17 SG SG11202003049WA patent/SG11202003049WA/en unknown
- 2018-10-17 PE PE2020000813A patent/PE20210394A1/es unknown
- 2018-10-17 CN CN201880068846.5A patent/CN111225921A/zh active Pending
- 2018-10-17 WO PCT/EP2018/078364 patent/WO2019081292A1/en unknown
- 2018-10-17 PT PT187853940T patent/PT3700913T/pt unknown
- 2018-10-17 US US16/758,742 patent/US11298367B2/en active Active
- 2018-10-17 MX MX2020004191A patent/MX2020004191A/es unknown
- 2018-10-17 UA UAA202003139A patent/UA126456C2/uk unknown
- 2018-10-17 HR HRP20220060TT patent/HRP20220060T1/hr unknown
- 2018-10-17 SI SI201830540T patent/SI3700913T1/sl unknown
- 2018-10-17 BR BR112020007983-9A patent/BR112020007983A2/pt not_active Application Discontinuation
- 2018-10-17 JO JOP/2020/0099A patent/JOP20200099A1/ar unknown
- 2018-10-17 DK DK18785394.0T patent/DK3700913T3/da active
- 2018-10-17 EP EP18785394.0A patent/EP3700913B1/en active Active
- 2018-10-17 HU HUE18785394A patent/HUE057278T2/hu unknown
- 2018-10-17 JP JP2020522855A patent/JP2021500365A/ja active Pending
- 2018-10-17 MA MA50438A patent/MA50438B1/fr unknown
- 2018-10-17 EA EA202091025A patent/EA202091025A1/ru unknown
- 2018-10-17 LT LTEPPCT/EP2018/078364T patent/LT3700913T/lt unknown
- 2018-10-17 RS RS20220036A patent/RS62984B1/sr unknown
- 2018-10-17 KR KR1020207014412A patent/KR20200069361A/ko not_active Application Discontinuation
- 2018-10-22 TW TW107137148A patent/TW201932117A/zh unknown
- 2018-10-24 AR ARP180103099A patent/AR113791A1/es unknown
- 2018-10-24 UY UY0001037948A patent/UY37948A/es not_active Application Discontinuation
-
2020
- 2020-04-13 IL IL273949A patent/IL273949A/en unknown
- 2020-04-21 EC ECSENADI202023056A patent/ECSP20023056A/es unknown
- 2020-04-22 CL CL2020001076A patent/CL2020001076A1/es unknown
- 2020-04-22 CO CONC2020/0004945A patent/CO2020004945A2/es unknown
- 2020-04-22 PH PH12020550473A patent/PH12020550473A1/en unknown
- 2020-04-23 NI NI202000030A patent/NI202000030A/es unknown
- 2020-05-06 DO DO2020000076A patent/DOP2020000076A/es unknown
-
2022
- 2022-02-01 CY CY20221100084T patent/CY1124938T1/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2866109T3 (es) | Derivados de piridiniltriazol sustituidos con amida y usos de estos | |
| US10526314B2 (en) | Hydroxyalkyl-substituted heteroaryltriazole derivatives and uses thereof | |
| US10525041B2 (en) | Fluoroalkyl-substituted aryltriazole derivatives and uses thereof | |
| EP3452470B1 (en) | Amide-substituted phenyltriazole derivatives and uses thereof | |
| US20210253557A1 (en) | Substituted triazole derivatives and uses thereof | |
| ES2906305T3 (es) | Profármacos de derivados de triazol sustituido y sus usos | |
| US11149023B2 (en) | Substituted triazole derivatives and uses thereof | |
| WO2019081291A1 (en) | PRODRUGS OF SUBSTITUTED TRIAZOLE DERIVATIVES AND USES THEREOF | |
| EP3700899A1 (en) | Substituted triazole derivatives and uses thereof |