ES2864223T3 - Compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida para el tratamiento y profilaxis de infección vírica - Google Patents
Compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida para el tratamiento y profilaxis de infección vírica Download PDFInfo
- Publication number
- ES2864223T3 ES2864223T3 ES15804128T ES15804128T ES2864223T3 ES 2864223 T3 ES2864223 T3 ES 2864223T3 ES 15804128 T ES15804128 T ES 15804128T ES 15804128 T ES15804128 T ES 15804128T ES 2864223 T3 ES2864223 T3 ES 2864223T3
- Authority
- ES
- Spain
- Prior art keywords
- amino
- tetrahydrofuran
- thiazolo
- hydroxy
- dione
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 *N(c1nc(N)nc(O)c1S1)C1=O Chemical compound *N(c1nc(N)nc(O)c1S1)C1=O 0.000 description 13
- MMTLSQQNKWIWQR-HUDPQJTASA-N C=CC([C@H](CC1)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O Chemical compound C=CC([C@H](CC1)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O MMTLSQQNKWIWQR-HUDPQJTASA-N 0.000 description 1
- MOYIRJVGPKHXDN-HTAVTVPLSA-N CCC(C(CC1)O[C@@H]1OC(C)(C)O)OC(C(C)C)=O Chemical compound CCC(C(CC1)O[C@@H]1OC(C)(C)O)OC(C(C)C)=O MOYIRJVGPKHXDN-HTAVTVPLSA-N 0.000 description 1
- FWOIAGYUWKTWMH-YXMLORGKSA-N CCC(C(C[C@H]1OC(C)=O)=[O]C1OC(C)=O)OC(C)=O Chemical compound CCC(C(C[C@H]1OC(C)=O)=[O]C1OC(C)=O)OC(C)=O FWOIAGYUWKTWMH-YXMLORGKSA-N 0.000 description 1
- ZCJZGIGROVUOPX-IZZREWJQSA-N CCC(C(C[C@H]1OC(C)=O)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O Chemical compound CCC(C(C[C@H]1OC(C)=O)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O ZCJZGIGROVUOPX-IZZREWJQSA-N 0.000 description 1
- ITEJEZHUERWUHO-DAJGMKRESA-N CCC(C(C[C@H]1SC)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O Chemical compound CCC(C(C[C@H]1SC)OC1N(C(N=C(N)NC1=O)=C1S1)C1=O)O ITEJEZHUERWUHO-DAJGMKRESA-N 0.000 description 1
- HDHGQPWZDXVBJA-GSLILNRNSA-N CCC([C@H]1O[C@@H]2OC(C)(C)O[C@@H]2C1)O Chemical compound CCC([C@H]1O[C@@H]2OC(C)(C)O[C@@H]2C1)O HDHGQPWZDXVBJA-GSLILNRNSA-N 0.000 description 1
- HZVZHHILVMJQKX-PKVWOUKASA-N CS(N[C@H]1[C@H](N(C(N=C(N)NC2O)=C2S2)C2=O)O[C@H](COBr)C1)(=O)=O Chemical compound CS(N[C@H]1[C@H](N(C(N=C(N)NC2O)=C2S2)C2=O)O[C@H](COBr)C1)(=O)=O HZVZHHILVMJQKX-PKVWOUKASA-N 0.000 description 1
- SQCDIQSUYXCFJD-ULHQUTTKSA-N NC(NC1=O)=NC(N2[C@@]3(CO[C@H](CO)C3)C#N)=C1SC2=O Chemical compound NC(NC1=O)=NC(N2[C@@]3(CO[C@H](CO)C3)C#N)=C1SC2=O SQCDIQSUYXCFJD-ULHQUTTKSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/24—Heterocyclic radicals containing oxygen or sulfur as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Epidemiology (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de fórmula (I), **(Ver fórmula)** en la que R1 es hidroxi, alquilo C1-6, alquilcarbonil C1-6-O-, alquil C1-6-S-, acido o alquenilo C2-6; R2 es alquilo C1-6, alcoxi C1-6alquilo C1-6, cicloalquilo C3-7, alquinilo C2-6, alquenilo C2-6, bencilo y tiofenilo; R3 es hidrógeno o alquilcarbonilo C1-6; o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Description
DESCRIPCIÓN
Compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida para el tratamiento y profilaxis de infección vírica
La presente invención se refiere a compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida novedosos, que tienen actividad agonista del receptor de tipo Toll y sus profármacos de los mismos, así como su fabricación, composiciones farmacéuticas que los contienen y su uso potencial como medicamentos.
CAMPO DE LA INVENCIÓN
La presente invención se refiere a compuestos de fórmula (I) y (Ia),

y sus profármacos, fórmula (II) y (IIa),
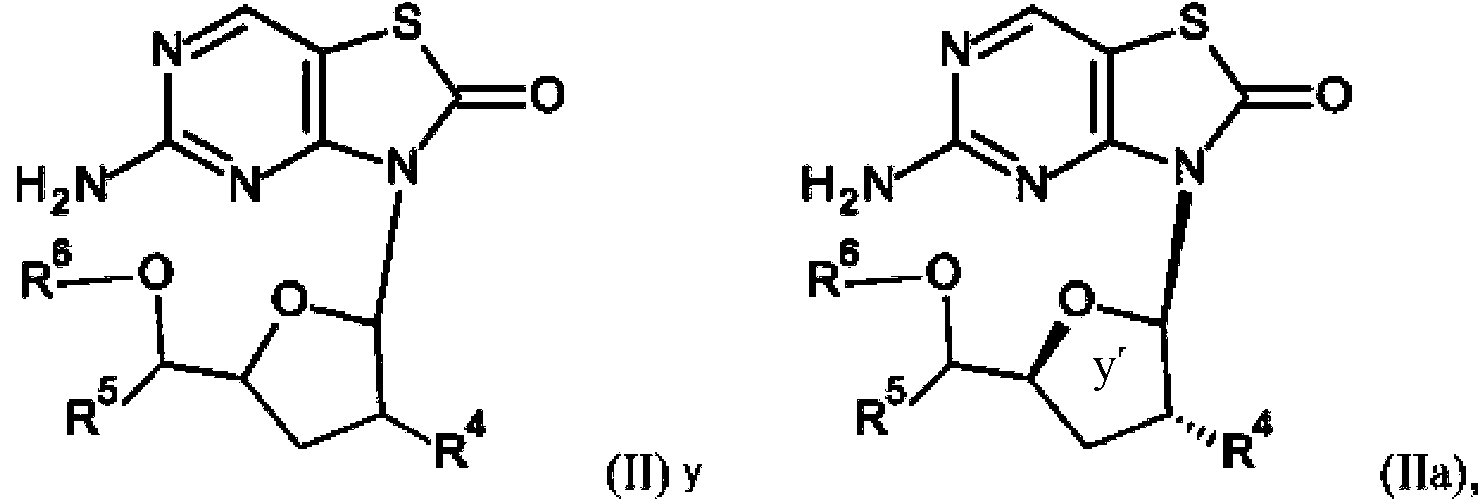
en las que R1 a R6 se describen a continuación, o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos.
Los receptores de tipo Toll (TLR) detectan una amplia gama de patrones moleculares asociados a patógenos (PAMP) conservados. Desempeñan un papel importante de detección de patógenos invasores y posterior inicio de respuestas inmunitarias innatas. Existen 10 miembros conocidos de la familia de TLR en seres humanos, que son proteínas transmembranarias de tipo I que presentan un dominio rico en leucina extracelular y una cola citoplásmica que contiene un dominio de receptor Toll/interleucina (IL)-1 (TIR) conservado. Dentro de esta familia, TLR3, TLR7 TLR8 y TLR9 se localizan dentro de los endosomas. TLR7 se puede activar por unión a un ligando de molécula pequeña específico (es decir, agonista de TLR7) o su ligando natural (es decir, ARN monocatenario, ARNmc). Después de la unión de ARNmc a TLR7, se cree que el receptor en su forma dimerizada experimenta un cambio estructural que da lugar al posterior reclutamiento de proteínas adaptadoras en su dominio citoplásmico, incluyendo el gen de respuesta principal de diferenciación mieloide 88 (MyD88). Después del inicio de la cascada de señalización del receptor por medio de la vía MyD88, se activan factores de transcripción citoplásmicos tales como el factor regulador de interferones 7 (IRF-7) y el factor nuclear kappa B (NF-kB). A continuación, estos factores de transcripción se translocan al núcleo e inician la transcripción de diversos genes, por ejemplo, IFN-a y otros genes de atocinas antivíricas. TLR7 se expresa predominantemente en células plasmocitoides, y también en linfocitos B. La alteración en la reactividad de células inmunitarias podría contribuir a la reducción en las respuestas inmunitarias innatas durante infecciones víricas crónicas. Por lo tanto, la activación inducida por agonista de TLR7 podría representar un enfoque novedoso para el tratamiento de infecciones víricas crónicas. (D. J Connolly y L. AJ O'Neill, Current Opinion in Pharmacology 2012,
12:510-518, P. A. Roethle et al, J. Med. Chem. 2013, 56, 7324-7333).
El tratamiento actual de infección por VHB crónica se basa en dos tipos diferentes de fármacos: los análogos nucleos(t)ídicos antivíricos tradicionales y el IFN-a pegilado (PEG-IFN-a) más reciente. Los análogos nucleos(t)ídicos orales actúan inhibiendo la replicación del VHB. Esta es una serie de tratamiento de por vida durante el que a menudo se produce resistencia farmacológica. Como opción alternativa, se ha usado IFN-a pegilado (PEG-IFN-a) para tratar a algunos pacientes con infección por VHB crónica dentro de una duración de tratamiento limitada. Aunque ha logrado seroconversión en HBeAg al menos en un pequeño porcentaje de pacientes con VHB, el efecto adverso lo hace poco tolerable. De forma notable, la cura funcional definida como seroconversión de HBsAg es muy rara con los dos tratamientos actuales. Por lo tanto, es una necesidad urgente obtener una opción terapéutica de nueva generación para tratar a pacientes con VHB para una cura funcional. El tratamiento con un agonista de TLR7 oral representa una solución prometedora para proporcionar mayor eficacia con mejor tolerabilidad. Actualmente se usa IFN-a pegilado (PEG-IFN-a) para tratar VHB crónica y es una alternativa al tratamiento potencialmente de por vida con análogos nucleos(t)ídicos antivíricos. En un subconjunto de pacientes con VHB crónica, el tratamiento con PEG-IFN-a puede inducir control inmunológico mantenido del virus después de una duración limitada de tratamiento. Sin embargo, el porcentaje de pacientes con VHB que logran seroconversión con tratamiento con interferón es bajo (hasta un 27 % para pacientes con HBeAg positivo) y típicamente el tratamiento es poco tolerado. Además, la cura funcional (definida como pérdida de HBsAg y seroconversión) también es muy infrecuente tanto con tratamiento con PEG-IFN-a como nucleos(t)ídico. Dadas estas limitaciones, existe una necesidad urgente de obtener una mejora en las opciones terapéuticas para tratar e inducir una cura funcional para VHB crónica. El tratamiento con un agonista de TLR7 de molécula pequeña oral es un enfoque prometedor que tiene el potencial para proporcionar una mayor eficacia y tolerabilidad (T. Asselah et al, Clin Liver Dis 2007, 11, 839-849).
De hecho, se han considerado varios agonistas de TLR7 identificados para propósitos terapéuticos. Hasta la fecha, imiquimod (ALDARA™) es un fármaco agonista de TLR7 aprobado por la FDA estadounidense para su uso tópico para tratar lesiones cutáneas por papilomavirus humano. Se han evaluado el agonista doble de TLR7/8 resiquimod (R-848) y el agonista de TLR7 852a para tratar herpes genital humano y melanoma metastásico resistente a quimioterapia, respectivamente. ANA773 es un agonista de TLR7 profármaco oral, desarrollado para el tratamiento de pacientes con infección por virus de la hepatitis C (VHC) crónica e infección por hepatitis B crónica. GS-9620 es un agonista de TLR7 disponible por vía oral. Un estudio de fase Ib demostró que el tratamiento con GS-9620 era seguro, bien tolerado y daba como resultado la inducción de ARNm de ISG15 dependiente de la dosis en pacientes con hepatitis B crónica (E. J. Gane et al, Annu Meet Am Assoc Study Liver Dis (1-5 noviembre, Washington, D.C.) 2013, Abst 946). Por lo tanto, existe una gran necesidad clínica no cubierta de desarrollar agonistas de TLR7 potentes y seguros como nuevo tratamiento del VHB para ofrecer más soluciones terapéuticas o reemplazar el tratamiento parcialmente eficaz existente. El documento WO2006/066080 divulga 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona con actividad antivírica.
El documento WO2009026292 divulga 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona como agonistas de TLR7. El documento US5041426 divulga nucleósidos y nucleótidos de sistemas de anillo tiazolo[4,5-d]pirimidina con actividad antivírica.
SUMARIO DE LA INVENCIÓN
La presente invención proporciona una serie de compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida novedosos, que tienen actividad agonista del receptor de tipo Toll y sus profármacos. La invención también proporciona la bioactividad de dichos compuestos para inducir un incremento en el nivel de SEAP activando receptores de tipo Toll, tales como receptor TLR7, la conversión metabólica de profármacos en compuestos originales en presencia de hepatocitos humanos, y el uso terapéutico o profiláctico de dichos compuestos y sus composiciones farmacéuticas que comprenden estos compuestos y sus profármacos para tratar o prevenir enfermedades infecciosas como VHB o VHC. La presente invención también proporciona compuestos con actividad superior. Además, los compuestos de fórmula (I) y (Ia) también mostraron perfiles de solubilidad y FC buenos.
La presente invención se refiere a compuestos novedosos de fórmula (I) y (Ia),

en las que
R1 es hidroxi, alquilo C i - 6 , haloalquilo Ci - 6 , alquilcarbonil C1 -6 -O-, alquil C1 - 6 -S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1 -6 -NH-, (alquil Ci -6)2N-, alquilcarbonil C1 - 6 -NH- o amino heterocíclico;
R2 es hidrógeno, alquilo C1 - 6 , alcoxi C i - 6 alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 , alquenilo C2 -6 , bencilo y tiofenilo; R3 es hidrógeno o alquilcarbonilo C1-6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos; con la condición de que se excluyan 5-amino-7-hidroxi-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
La presente invención también se refiere a los profármacos de fórmula (II) y (IIa),

en las que
R4 es hidroxi, alquilo C1 - 6 , haloalquilo C1 - 6 , alquilcarbonil C1 -6 -O-, alquil C1 - 6 -S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1 -6 -NH-, (alquil Ci -6)2N-, alquilcarbonil C1 - 6 -NH- o amino heterocíclico;
R5 es hidrógeno, alquilo C1 - 6 , alcoxi C i - 6 alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 , alquenilo C2 -6 , bencilo y tiofenilo; R6 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos; con la condición de que se excluyan 5-amino-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-2-oxotiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
La invención también se refiere a su fabricación, medicamentos a base de un compuesto de acuerdo con la invención y su producción así como al uso de compuestos de fórmula (I) o (Ia) o sus profármacos, fórmula (II) o (IIa), de los mismos como agonista de TLR7. En consecuencia, los compuestos de fórmula (I) y (Ia) o sus profármacos de fórmula (II) y (IIa) son útiles para el tratamiento o profilaxis de VHB y/o VHC con agonistas de receptores de tipo Toll.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
A menos que se defina de otro modo, todos los términos técnicos y científicos usados en el presente documento tienen el mismo significado que se entiende comúnmente por un experto en la técnica a la que pertenece la presente invención. Además, las siguientes definiciones se exponen para ilustrar y definir el significado y alcance de los diversos términos usados para describir la invención.
DEFINICIONES
Como se usa en el presente documento, el término "alquilo C i - a " indica un grupo alquilo de cadena lineal o ramificada saturado que contiene de 1 a 6, en particular de 1 a 4 átomos de carbono, por ejemplo metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, ferc-butilo y similares. Los grupos "alquilo C i - a " particulares son metilo, etilo y n-propilo.
El término "halógeno" y "halo" se usan de manera intercambiable en el presente documento e indican flúor, cloro, bromo o yodo.
El término "haloalquilo Ci - a " se refiere a un grupo alquilo en el que al menos uno de los átomos de hidrógeno del grupo alquilo se ha reemplazado por átomos de halógeno iguales o diferentes, en particular átomos de flúor. Los ejemplos de haloalquilo C i - a incluyen monofluoro-, difluoro- o trifluoro-metilo, -etilo o -propilo, por ejemplo 3,3,3-trifluoropropilo, 2-fluoroetilo, 2,2,2-trifluoroetilo, fluorometilo, difluorometilo y trifluorometilo.
El término anillo "heterocíclico" indica un sistema de anillo mono o bicíclico saturado o parcialmente insaturado monovalente de 3 a 10 átomos de anillo, que comprende de 1 a 5 heteroátomos de anillo seleccionados de N, O y S, siendo los restantes átomos de anillo carbono. En modos de realización particulares, anillo heterocíclico es un sistema de anillo monocíclico saturado monovalente de 4 a 7 átomos de anillo, que comprende 1,2, o 3 heteroátomos de anillo seleccionados de N, O y S, siendo los restantes átomos de anillo carbono. Los ejemplos para anillo heterocíclico saturado monocíclico son aciridinilo, oxiranilo, acetidinilo, oxetanilo, pirrolidinilo, tetrahidrofuranilo, tetrahidrotienilo, pirazolidinilo, imidazolidinilo, oxazolidinilo, isoxazolidinilo, tiazolidinilo, piperidinilo, tetrahidropiranilo, tetrahidrotiopiranilo, piperacinilo, morfolinilo, tiomorfolinilo, 1,1-dioxo-tiomorfolin-4-ilo, acepanilo, diacepanilo, homopiperacinilo y oxacepanilo. Los ejemplos para anillo heterocíclico saturado bicíclico son 8-aza-biciclo[3.2.1]octilo, quinuclidinilo, 8-oxa-3-aza-biciclo[3.2.1]octilo, 9-aza-biciclo[3.3.1]nonilo, 3-oxa-9-aza-biciclo[3.3.1]nonilo, y 3-tia-9-aza-biciclo[3.3.1]nonilo. Los ejemplos para anillo heterocíclico parcialmente insaturado son dihidrofurilo, imidazolinilo, dihidrooxazolilo, tetrahidropiridinilo y dihidropiranilo.
El término "amino heterocíclico" indica un grupo amino con el átomo de nitrógeno en el anillo heterocíclico.
El término "alquenilo C2 -a " indica un grupo alquenilo de cadena lineal o ramificada insaturado que contiene de 2 a 6, en particular de 2 a 4 átomos de carbono, por ejemplo vinilo, propenilo, alilo, butenilo y similares. Los grupos "alquenilo C2 -a " particulares son alilo y vinilo.
El término "alquinilo C2 -a " indica un grupo alquinilo de cadena lineal o ramificada insaturado que contiene de 2 a 6, en particular de 2 a 4 átomos de carbono, por ejemplo etinilo, 1 -propinilo, propargilo, butinilo y similares. Los grupos "alquinilo C2 -a " particulares son etinilo y 1 -propinilo.
El término "cicloalquilo C3 -7 ", solo o en combinación, se refiere a un anillo de carbono saturado que contiene de 3 a 7 átomos de carbono, en particular de 3 a 6 átomos de carbono, por ejemplo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo y similares. El grupo "cicloalquilo C3 -7 " particular es ciclopropilo.
El término "carbonilo" solo o en combinación se refiere al grupo -C(O)-.
El término "alquilcarbonilo C1 - a " se refiere a un grupo alquil C1 - a -C(O)-, en el que "alquilo C1 - a " es como se define anteriormente. El grupo "alquilcarbonilo C i - a " particular es acetilo.
El término "enantiómero" indica dos estereoisómeros de un compuesto que son imágenes especulares no superponibles entre sí.
El término "diastereómero" indica un estereoisómero con dos o más centros de quiralidad y con moléculas que no son imágenes especulares entre sí. Los diastereómeros tienen diferentes propiedades físicas, por ejemplo, puntos de fusión, puntos de ebullición, propiedades espectrales y reactividades.
El término "sales farmacéuticamente aceptables" indica sales que no son biológicamente o de otro modo indeseables. Las sales farmacéuticamente aceptables incluyen sales de adición tanto de ácido como de base.
El término "sal de adición de ácido farmacéuticamente aceptable" indica las sales farmacéuticamente aceptables formadas con ácidos inorgánicos tales como ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido carbónico, ácido fosfórico, y ácidos orgánicos seleccionados de las clases alifática, cicloalifática, aromática, aralifática,
heterocíclica, carboxílica y sulfónica de ácidos orgánicos tales como ácido fórmico, ácido acético, ácido propiónico, ácido glicólico, ácido glucónico, ácido láctico, ácido pirúvico, ácido oxálico, ácido málico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido aspártico, ácido ascórbico, ácido glutámico, ácido antranílico, ácido benzoico, ácido cinámico, ácido mandélico, ácido embónico, ácido fenilacético, ácido metanosulfónico, ácido etanosulfónico, ácido p-toluenosulfónico y ácido salicíclico.
El término "sal de adición de base farmacéuticamente aceptable" indica las sales farmacéuticamente aceptables formadas con una base orgánica o inorgánica. Los ejemplos de bases inorgánicas aceptables incluyen sales de sodio, potasio, amonio, calcio, magnesio, hierro, cinc, cobre, manganeso y aluminio. Las sales derivadas de bases no tóxicas orgánicas farmacéuticamente aceptables incluyen sales de aminas primarias, secundarias y terciarias, aminas sustituidas incluyendo aminas sustituidas naturales, aminas cíclicas y resinas de intercambio iónico básicas, tales como isopropilamina, trimetilamina, dietilamina, trietilamina, tripropilamina, etanolamina, 2-dietilaminoetanol, trimetamina, diciclohexilamina, lisina, arginina, histidina, cafeína, procaína, hidrabamina, colina, betaína, etilendiamina, glucosamina, metilglucamina, teobromina, purinas, pipericina, piperidina, N-etilpiperidina y resinas de poliamina.
Los compuestos de la fórmula general (I) o (Ia) y sus profármacos que contienen uno o varios centros quirales pueden estar presentes como racematos, mezclas diastereoméricas o bien isómeros individuales ópticamente activos. Los racematos se pueden separar de acuerdo con procedimientos conocidos en los enantiómeros. En particular, las sales diastereoméricas que se pueden separar por cristalización se forman a partir de mezclas racémicas por reacción con un ácido ópticamente activo tal como ácido D- o L-tartárico, ácido mandélico, ácido málico, ácido láctico o ácido alcanforsulfónico.
Los compuestos de la invención pueden presentar el fenómeno de tautomería. Aunque los dibujos de fórmulas no pueden representar expresamente todas las formas tautoméricas posibles, se debe entender que pretenden representar cualquier forma tautomérica del compuesto representado y no se deben limitar simplemente a una forma de compuesto específica representada por los dibujos de fórmulas. Por ejemplo, se entiende para la fórmula (III) que independientemente de si los sustituyentes se muestran o no en su forma enol o ceto, representan el mismo compuesto (como se muestra en el ejemplo a continuación).

Rx se refiere a cualquier sustituyente factible.
Algunos de los compuestos de la invención pueden existir como estereoisómeros individuales (es decir, esencialmente libres de otros estereoisómeros), racematos y/o mezclas de enantiómeros y/o diastereómeros. Se pretende que todos dichos estereoisómeros individuales, racematos y mezclas de los mismos estén dentro del alcance de la presente invención. Preferentemente, los compuestos de la invención que son ópticamente activos se usan en forma ópticamente pura. Como se entiende en general por los expertos en la técnica, un compuesto ópticamente puro que tiene un centro quiral (es decir, un átomo de carbono asimétrico) es uno que consiste esencialmente en uno de los dos posibles enantiómeros (es decir, es enantioméricamente puro), y un compuesto ópticamente puro que tiene más de un centro quiral es uno que es tanto diastereoméricamente puro como enantioméricamente puro. Preferentemente, los compuestos de la presente invención se usan en una forma que es al menos un 90 % ópticamente pura, es decir, una forma que contiene al menos un 90 % de un isómero individual (80 % de exceso enantiomérico ("e.e.") o exceso diastereomérico ("e.d.")), más preferentemente al menos un 95 % (90 % e.e. o e.d.), incluso más preferentemente al menos un 97,5 % (95 % e.e. o e.d.), y lo más preferentemente al menos un 99 % (98 % e.e. o e.d.). Adicionalmente, los compuestos de fórmula (I) y (Ia) y sus profármacos, fórmula (II) y (IIa), y otros compuestos de la invención pretenden cubrir formas solvatadas así como no solvatadas de las estructuras identificadas. Por ejemplo, la fórmula (I) o (Ia) incluye compuestos de la estructura indicada en formas tanto hidratada como no hidratada. Otros ejemplos de solvatos incluyen las estructuras en combinación con isopropanol, etanol, metanol, DMSO, acetato de etilo, ácido acético o etanolamina.
El término "profármaco" indica una forma o derivado de un compuesto que se metaboliza in vivo, por ejemplo, por líquidos biológicos o enzimas por un sujeto después de la administración, en una forma farmacológicamente activa del compuesto para producir el efecto farmacológico deseado. Los profármacos se describen, por ejemplo, en "The
Organic Chemistry of Drug Design and Drug Action", por Richard B. Silverman, Academic Press, San Diego, 2004, capítulo 8 Prodrugs and Drug Delivery Systems, pp. 497-558.
Un "metabolito farmacéuticamente activo" pretende querer decir un producto farmacológicamente activo producido a través del metabolismo en el cuerpo de un compuesto especificado o sal del mismo. Después de entrar en el cuerpo, la mayoría de los fármacos son sustratos para reacciones químicas que pueden cambiar sus propiedades físicas y efectos biológicos. Estas conversiones metabólicas, que normalmente afectan a la polaridad de los compuestos de la invención, alteran la forma en que los fármacos se distribuyen en y se excretan del cuerpo. Sin embargo, en algunos casos, se requiere el metabolismo de un fármaco para el efecto terapéutico.
El término "cantidad terapéuticamente eficaz" indica una cantidad de un compuesto o molécula de la presente invención que, cuando se administra a un sujeto, (i) trata o evita la enfermedad, afección o trastorno particular, (ii) atenúa, mejora o elimina uno o más síntomas de la enfermedad, afección o trastorno particular, o (iii) evita o retrasa la aparición de uno o más síntomas de la enfermedad, afección o trastorno particular descrito en el presente documento. La cantidad terapéuticamente eficaz variará dependiendo del compuesto, el estado de enfermedad que se está tratando, la gravedad de la enfermedad tratada, la edad y salud relativa del sujeto, la vía y forma de administración, el juicio del médico especialista o veterinario, y otros factores.
El término "composición farmacéutica" indica una mezcla o solución que comprende una cantidad terapéuticamente eficaz de un ingrediente farmacéutico activo conjuntamente con excipientes farmacéuticamente aceptables que se va a administrar a un mamífero, por ejemplo, un ser humano que lo necesite.
AGONISTA DE TLR7 Y PROFÁRMACO
La presente invención se refiere a un compuesto de fórmula (I),

en la que
R1 es hidroxi, alquilo C i - 6 , haloalquilo Ci - 6 , alquilcarbonil C1 -6 -O-, alquil C1 - 6 -S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1 -6 -NH-, (alquil Ci -6)2N-, alquilcarbonil C1 - 6 -NH- o amino heterocíclico;
R2 es hidrógeno, alquilo C1 - 6 , alcoxi C i - 6 alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 , alquenilo C2 -6 , bencilo y tiofenilo;
R3 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo;
con la condición de que se excluyan 5-amino-7-hidroxi-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
Otro modo de realización de la presente invención es (ii) un compuesto de fórmula (I), en la que
R1 es hidroxi, metilo, propilo, fluoroisopropilo, acetiloxi, metilsulfanilo, acido, ciano, alilo, 2-metilalilo, metilsulfonilamino, dimetilamino, acetilamino, pirrolidinilo, morfolinilo o piperidinilo;
R2 es hidrógeno, metilo, etilo, propilo, butilo, ciclopropilo, ciclopentilo, vinilo, alilo, bencilo, etinilo, 1 -propinilo, metoximetilo o tiofenilo;
R3 es hidrógeno, acetilo o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (iii) un compuesto de fórmula (Ia),

en la que
R1 es hidroxi, alquilo C i - 6 , haloalquilo Ci - 6 , alquilcarbonil C1 -6 -O-, alquil C1 - 6 -S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1 -6 -NH-, (alquil Ci -6)2N-, alquilcarbonil C1 - 6 -NH- o amino heterocíclico;
R2 es hidrógeno, alquilo C1 - 6 , alcoxi Ci - 6 alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 - 6 , alquenilo C2 - 6 , bencilo o tiofenilo; R3 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo;
con la condición de que se excluyan 5-amino-7-hidroxi-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-7-hidroxi-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
Otro modo de realización de la presente invención es (iv) un compuesto de fórmula (Ia), en la que
R1 es hidroxi, metilo, propilo, fluoroisopropilo, acetiloxi, metilsulfanilo, acido, ciano, alilo, 2-metilalilo, metilsulfonilamino, dimetilamino, acetilamino, pirrolidinilo, morfolinilo o piperidinilo;
R2 es hidrógeno, metilo, etilo, propilo, butilo, ciclopropilo, ciclopentilo, vinilo, alilo, bencilo, etinilo, 1-propinilo, metoximetilo o tiofenilo;
R3 es hidrógeno, acetilo o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (v) un compuesto de fórmula (I) o (Ia), en la que
R1 es hidroxi, alquilo C1 - 6 , alquilcarbonil C1 -6 -O-, alquil C1 -6 -S-, acido o alquenilo C2 -6 ;
R2 es alquilo C1 - 6 , alcoxi Ci - 6 alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 - 6 , alquenilo C2 -6 , bencilo y tiofenilo;
R3 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (vi) un compuesto de fórmula (I) o (Ia), en la que
R1 es hidroxi, metilo, propilo, acetiloxi, metilsulfanilo, acido o alilo;
R2 es metilo, etilo, propilo, butilo, ciclopropilo, ciclopentilo, vinilo, alilo, bencilo, etinilo, 1-propinilo, metoximetilo o
tiofenilo;
R3 es hidrógeno, acetilo o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (vii) un compuesto de fórmula (I) o (Ia), en la que R1 es hidroxi, alquilo Ci - 6 , alquil C1 - 6 -S-, acido o alquenilo C2 -6.
Otro modo de realización de la presente invención es (viii) un compuesto de fórmula (I) o (Ia), en la que R1 es hidroxi, metilo, n-propilo, metilsulfanilo, acido o alilo.
Otro modo de realización de la presente invención es (ix) un compuesto de fórmula (I) o (Ia), en la que R2 es alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 o alquenilo C2 -6.
Otro modo de realización de la presente invención es (x) un compuesto de fórmula (I) o (Ia), en la que R2 es metilo, etilo, n-propilo, ciclopropilo, vinilo, etinilo o 1 -propinilo.
Otro modo de realización de la presente invención es (xi) un compuesto de fórmula (I) o (Ia), en la que R3 es hidrógeno o alquilcarbonilo C 1 - 6.
Otro modo de realización de la presente invención es (xii) un compuesto de fórmula (I) o (Ia), en la que R3 es hidrógeno o isobutirilo.
Otro modo de realización de la presente invención es (xiii) un compuesto de fórmula (I) o (Ia), en la que
R1 es hidroxi, alquilo C1 - 6 , alquil C1 - 6 -S-, acido o alquenilo C2 - 6 ;
R2 es alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 o alquenilo C2 - 6 ;
R3 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xiv) un compuesto de fórmula (I) o (Ia), en la que
R1 es hidroxi, metilo, propilo, metilsulfanilo, acido o alilo;
R2 es metilo, etilo, propilo, ciclopropilo, vinilo, etinilo o 1 -propinilo;
R3 es hidrógeno o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xv) un compuesto de fórmula (I) o (Ia), en la que
R1 es haloalquilo C1 - 6 , alquil C1 - 6 -S-, ciano, alquenilo C2 - 6 , alquilsulfonil C1 - 6 -NH-, (alquil Ci -6)2N-, alquilcarbonil C1 - 6 -NH- o amino heterocíclico;
R2 es hidrógeno;
R3 es hidrógeno;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xvi) un compuesto de fórmula (I) o (Ia), en la que
R1 es fluoroisopropilo, metilsulfanilo, ciano, 2-metilalilo, metilsulfonilamino, dimetilamino, acetilamino, pirrolidinilo, morfolinilo o piperidinilo;
R2 es hidrógeno;
R3 es hidrógeno;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xvii) un compuesto de fórmula (I) o (la), en la que R1 es alquil C1-6-S- o amino heterocíclico.
Otro modo de realización de la presente invención es (xviii) un compuesto de fórmula (I) o (Ia), en la que R1 es metilsulfanilo o pirrolidinilo.
Otro modo de realización de la presente invención es que (xix) compuestos particulares de fórmula (I) o (Ia) son los siguientes:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; acetato de [(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]; acetato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo];
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxietil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-3-enil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipentil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibutil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[ciclopentil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-fenil-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-3-metil-butil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; acetato de [[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropilmetilo];
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxiprop-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-[hidroxi(2-tienil)metil]tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-metoxi-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-rnetilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxialil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-((2R,3R,5S)-3-acido-5-((S)-1-hidroxietil)tetrahidrofuran-2-il)tiazolo[4,5-d]pirimidin-2,7(3H,6H)-diona;
3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1R)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]metanosulfonamida;
W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]acetamida; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-morfolino-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(1-piperidil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-(dimetilamino)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; (2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-carbonitrilo;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-(1-fluoro-1-metil-etil)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(2-metilalil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona y 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofu ran-2-il]propilo];
o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos.
Otro modo de realización de la presente invención es que (xx) compuestos más particulares de fórmula (I) o (Ia) son los siguientes:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibutil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxiprop-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxialil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1R)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; y 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofu ran-2-il]propilo];
o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos.
Otro modo de realización de la presente invención es (xxi) un compuesto de fórmula (II),

en la que
R4 es hidroxi, alquilo Ci-6, haloalquilo Ci-6, alquilcarbonil C1-6-O-, alquil C1 -6-S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1-6-NH-, (alquil Ci -6)2N-, alquilcarbonil C1 -6-NH- o amino heterocíclico;
R5 es hidrógeno, alquilo C1 -6 , alcoxi Ci-6alquilo C1 -6 , cicloalquilo C3 -7 , alquinilo C2 -6 , alquenilo C2 -6 , bencilo y tiofenilo; R6 es hidrógeno o alquilcarbonilo C1 -6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo;
con la condición de que se excluyan 5-amino-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
Otro modo de realización de la presente invención es (xxii) un compuesto de fórmula (II), en la que
R4 es hidroxi, metilo, n-propilo, fluoroisopropilo, acetiloxi, metilsulfanilo, acido, ciano, alilo, 2-metilalilo, metilsulfonilamino, dimetilamino, acetilamino, pirrolidinilo, morfolinilo o piperidinilo;
R5 es hidrógeno, metilo, etilo, propilo, butilo, ciclopropilo, ciclopentilo, vinilo, alilo, bencilo, etinilo, 1-propinilo, metoximetilo o tiofenilo;
R6 es hidrógeno, acetilo o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xxiii) un compuesto de fórmula (IIa),

en la que
R4 es hidroxi, alquilo C1 -6 , haloalquilo C1 -6 , alquilcarbonil C1-6-O-, alquil C1 -6-S-, acido, ciano, alquenilo C2 -6 , alquilsulfonil C1-6-NH-, (alquil C1 -6 )2N-, alquilcarbonil C1 -6-NH- o amino heterocíclico;
R5 es hidrógeno, alquilo C i - 6 , alcoxi C i - 6 alquilo Ci - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 , alquenilo C2 -6 , bencilo y tiofenilo; R6 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo;
con la condición de que se excluyan 5-amino-3-[3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; acetato de [2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-ilo]; acetato de [4-acetoxi-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo y sus diastereómeros.
Otro modo de realización de la presente invención es (xiv) un compuesto de fórmula (IIa), en la que
R4 es hidroxi, metilo, n-propilo, fluoroisopropilo, acetiloxi, metilsulfanilo, acido, ciano, alilo, 2-metilalilo, metilsulfonilamino, dimetilamino, acetilamino, pirrolidinilo, morfolinilo o piperidinilo;
R5 es hidrógeno, metilo, etilo, propilo, butilo, ciclopropilo, ciclopentilo, vinilo, alilo, bencilo, etinilo, 1 -propinilo, metoximetilo o tiofenilo;
R6 es hidrógeno, acetilo o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xxv) un compuesto de fórmula (II) o (IIa), en la que
R4 es hidroxi, alquilo C1 - 6 , alquil C1 - 6 -S-, acido o alquenilo C2 - 6 ;
R5 es alquilo C1 - 6 , cicloalquilo C3 - 7 , alquinilo C2 -6 o alquenilo C2 - 6 ;
R6 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xxvi) un compuesto de fórmula (II) o (IIa), en la que
R4 es hidroxi, metilo, propilo, metilsulfanilo, acido o alilo;
R5 es metilo, etilo, propilo, ciclopropilo, vinilo, etinilo o 1 -propinilo;
R6 es hidrógeno o isobutirilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xxvii) un compuesto de fórmula (II) o (IIa), en la que
R4 es hidroxi o alquilcarbonil C1 -6 -O-;
R5 es alquilo C1 -6 o cicloalquilo C3 - 7 ;
R6 es hidrógeno o alquilcarbonilo C 1 - 6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es (xxviii) un compuesto de fórmula (II) o (IIa), en la que
R4 es hidroxi o acetiloxi;
R5 es etilo o ciclopropilo;
R6 es hidrógeno o acetilo;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
Otro modo de realización de la presente invención es que (xxix) compuestos particulares de fórmula (II) o (IIa) son los siguientes:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona;
acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]; acetato de 1-[(2S,4R,5S)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo; acetato de [(S)-[(2S,4R,5S)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropilmetilo];
y 5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)rnetil]-3-hidroxi-tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona; o sal, enantiómero o diastereómero farmacéuticamente aceptable de los mismos.
SÍNTESIS
Los compuestos de la presente invención se pueden preparar por cualquier medio convencional. Los procedimientos adecuados para sintetizar estos compuestos así como sus materiales de partida se proporcionan en los esquemas a continuación y en los ejemplos. Todos los sustituyentes, en particular, R1 a R11 son como se define anteriormente a menos que se indique de otro modo. Además, y a menos que se establezca explícitamente de otro modo, todas las reacciones, condiciones de reacción, abreviaturas y símbolos tienen los significados bien conocidos para un experto en química orgánica.
Esquema 1
Ċ

Ra es R2 o R5; Rb es acilo, benzoílo, ferc-butil(dimetil)sililo o ferc-butil(difenil)sililo; Rc es hidrógeno o hidroxi; Re es R1 o R4.
El tratamiento del compuesto V con disulfuro de carbono y yodometano en presencia de una base apropiada, tal como NaH, da metanotioato de metilsulfanilo 1a. La desoxilación de metanotioato de metilsulfanilo 1a con un reactivo apropiado, tal como hidruro de tri-n-butilestaño, da 1b. La desprotección de 1b con un ácido apropiado, tal como ácido acético, da el diol 1c. La oxidación de 1c con oxidante, tal como metaperyodato de sodio, da el aldehido 1d. El tratamiento del aldehido 1d con un reactivo nucleófilo apropiado, tal como reactivo de Grignard, da el alcohol VI. La protección del alcohol VI con un anhídrido de ácido o cloruro de ácido apropiado, tal como cloruro acético o anhídrido de benzoílo, da el éster VII. El tratamiento del éster VII con ácido acético y anhídrido acético en presencia de un ácido apropiado, tal como ácido sulfúrico condensado, da el éster VIII. El acoplamiento del éster VIII y IX en presencia de un agente de eterificación de silicio apropiado, tal como W,O-bis(trimetilsilil)acetamida, y ácido de Lewis, tal como TMSOTf, da el intermedio X . La desprotección del intermedio X con un reactivo apropiado, tal como K2CO3 o TBAF, y la purificación por HPLC preparativa da los compuestos deseados P1, P2 o P3. La protección del compuesto P1 con un anhídrido de ácido o cloruro de ácido apropiado, tal como cloruro acético o anhídrido acético, da el compuesto P3. La oxidación de P2 con un oxidante apropiado, tal como peryodinano de Dess-Martin, da la cetona XI. La reducción de la cetona XI con un reductor apropiado, tal como hidruro de ferc-butoxialuminio, da el alcohol XII. El tratamiento del alcohol XII con anhídrido sulfónico o cloruro de sulfonilo da el intermedio XIII. El tratamiento del intermedio XIII con un reactivo nucleófilo apropiado, tal como acida de sodio, da el compuesto XIV. La desprotección de XIV con una base apropiada, tal como K2CO3 , o un reactivo de fluoruro apropiado, tal como TBAF, y la purificación por HPLC preparativa da el compuesto P4.
Esquema 2:
Ra es R2 o R5; Rc es hidrógeno o hidroxi; Re es R1 o R4.
El ácido 28a se convierte en cloruro de acilo 28b con un reactivo apropiado, tal como dicloruro de oxalilo. El cloruro de acilo 28b se trata con un reactivo nucleófilo apropiado, tal como reactivo de Grignard, para dar la cetona XV. La reducción de la cetona XV con un reductor apropiado, tal como L-selectrida, da el alcohol XVI. La protección de XVI con terc-butilclorodifenilsilano da el intermedio XVII. La alquilación de XVII con un reactivo alquilante, tal como yodometano, en presencia de una base apropiada, tal como diisopropilamida de litio, da el intermedio XVIII. La desprotección de XVIII con un reactivo apropiado, tal como TBAF, da el alcohol XIX. La protección del alcohol XIX con cloruro de benzoílo da el éster XX. La reducción del éster XX con un reductor apropiado, tal como hidruro de diisobutilaluminio, da el alcohol XXI. La esterificación del alcohol XXI con cloruro acético o anhídrido acético da XXII. El acoplamiento de XXII y IX en presencia de un agente de eterificación de silicio apropiado tal como N,O-bis(trimetilsilil)acetamida y ácido de Lewis da XXIII. La desprotección de XXIII con una base apropiada, tal como K2CO3 , y la purificación por HPLC preparativa da el compuesto P5.
Esquema 3

La protección del alcohol 25e con cloruro de benzoílo da el intermedio 32a. La desprotección de 32a con un ácido apropiado, tal como clorhidrato, en presencia de un disolvente apropiado, tal como metanol, da el intermedio 32b. El tratamiento de 32b con anhídrido sulfónico o cloruro de sulfonilo da el intermedio XXIV. El tratamiento de XXIV con un reactivo nucleófilo, tal como acida de sodio y aminas, da el intermedio XXV. El acoplamiento de XXV y IX en presencia de un agente de eterificación de silicio apropiado tal como N,O-bis(trimetilsilil)acetamida y ácido de Lewis da XXVI. La desprotección de XXVI con una base apropiada, tal como K2CO3 , y la purificación por HPLC preparativa da el compuesto XXVII.
Esquema 4:

El tratamiento de XXIX con un reactivo apropiado, tal como clorometanotioato de O-fenilo, en presencia de una base apropiada, tal como DMAP, da el intermedio XXX. El tratamiento del intermedio XXX con un reactivo de organoestannano, tal como alil(tri-n-butil)estannano, da el intermedio XXXI. La desprotección de XXXI con una base apropiada, tal como K2CO3 , y la purificación por HPLC preparativa da el compuesto P6.
La presente invención también se refiere a un procedimiento para la preparación de un compuesto de fórmula (I), (la), (II) o (lla) que comprende la reacción de:
(a) la reacción de un compuesto de fórmula (X),

con una base o reactivo de fluoruro, en la que Ra es R2 o R5; Rb es acilo, benzoílo, terc-butil(dimetil)sililo o terc-butil(difenil)sililo; Rc es hidrógeno o hidroxi;
(b) la reacción de un compuesto de fórmula (P1),

con un anhídrido de ácido o cloruro de ácido, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi;
(c) la reacción de un compuesto de fórmula (XIV),

con una base o un reactivo de fluoruro, en la que Ra es R2 o R5; Rb es acilo, benzoílo, terc-butil(difenil)sililo; Rc es hidrógeno o hidroxi;
Re es R1 o R4;
(d) la reacción de un compuesto de fórmula (XXIII),
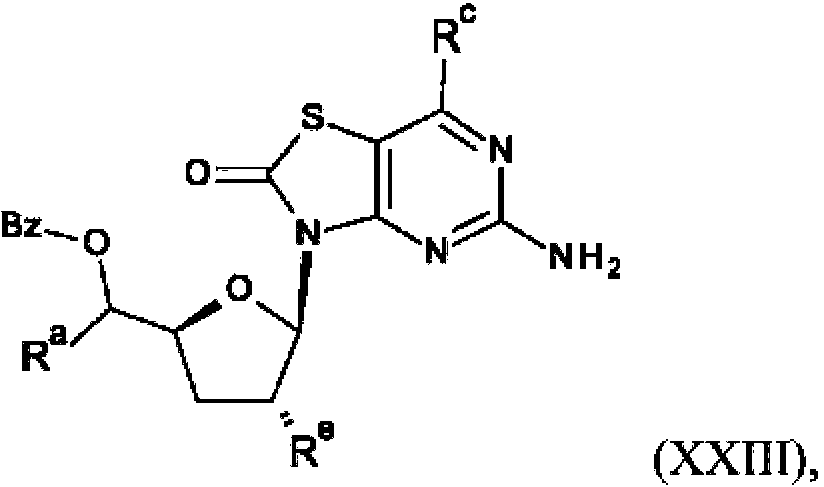
con una base, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi; Re es R1 o R4;
(e) la reacción de un compuesto de fórmula (XXVI),

con una base, en la que Rc es hidrógeno o hidroxi; Re es R1 o R4;
(f) la reacción de un compuesto de fórmula (XXXI),
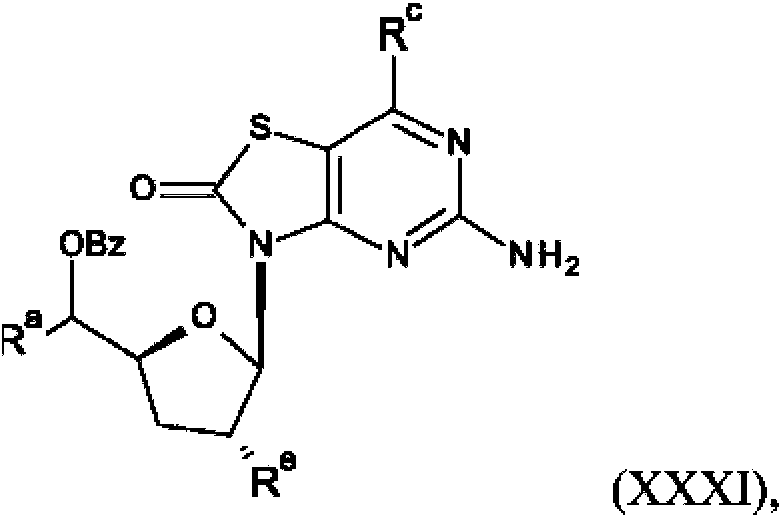
con una base, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi; Re es R1 o R4; o
en la que Ra, Rb, Rc, Re, R1, R2, R3, R4, R5 y R6 se definen anteriormente.
En la etapa (a), la base puede ser por ejemplo K2CO3 , el reactivo de fluoruro puede ser por ejemplo TBAF.
En la etapa (b), el anhídrido de ácido o cloruro de ácido puede ser por ejemplo cloruro acético o anhídrido acético.
En la etapa (c), (d), (e) y (f), la base puede ser por ejemplo K2CO3 , el reactivo de fluoruro puede ser por ejemplo TBAF.
Un compuesto de fórmula (I), (Ia), (II) y (IIa) cuando se fabrica de acuerdo con el procedimiento anterior también es un objetivo de la invención.
COMPOSICIONES FARMACÉUTICAS Y ADMINISTRACIÓN
Otro modo de realización proporciona composiciones farmacéuticas o medicamentos que contienen los compuestos de la invención y un vehículo, diluyente o excipiente terapéuticamente inerte, así como procedimientos de uso de los compuestos de la invención para preparar dichas composiciones y medicamentos. En un ejemplo, se pueden formular los compuestos de fórmula (I) o (Ia) o sus profármacos mezclando a temperatura ambiente al pH apropiado, y al grado deseado de pureza, con vehículos fisiológicamente aceptables, es decir, vehículos que no son tóxicos para los receptores a las dosificaciones y concentraciones empleadas en una forma de administración galénica. El pH de la formulación depende principalmente del uso particular y la concentración de compuesto, pero preferentemente varía en cualquier valor de aproximadamente 3 a aproximadamente 8. En un ejemplo, un compuesto de fórmula (I) o (Ia) o sus profármacos se formulan en un tampón acetato, a pH 5. En otro modo de realización, los compuestos de fórmula (I) o (Ia) o sus profármacos son estériles. El compuesto se puede almacenar, por ejemplo, como composición sólida o amorfa, como formulación liofilizada o como solución acuosa.
Las composiciones se formulan, dosifican y administran de forma consecuente con la buena práctica médica. Los factores que se deben tener en consideración en este contexto incluyen el trastorno particular que se está tratando, el mamífero particular que se está tratando, el estado clínico del paciente individual, la causa del trastorno, el sitio de administración del agente, el procedimiento de administración, la programación de la administración y otros factores conocidos por los médicos. La "cantidad eficaz" del compuesto que se va a administrar se regirá por dichas consideraciones, y es la cantidad mínima necesaria para activar el receptor TLR7 y dar lugar a producir INF-a y otras citocinas, que se pueden usar, pero no están limitadas, para el tratamiento o prevención de pacientes con infección vírica por hepatitis B y/o C.
En un ejemplo, la cantidad farmacéuticamente eficaz del compuesto de la invención administrado por vía parenteral
por dosis estará en el intervalo de aproximadamente 0,1 a 50 mg/kg, de forma alternativa de aproximadamente 0,1 a 30 mg/kg de peso corporal del paciente por día, siendo el intervalo inicial típico del compuesto usado de 0,3 a 15 mg/kg/día. En otro modo de realización, las formas de dosificación unitaria orales, tales como comprimidos y cápsulas, contienen preferentemente de aproximadamente 20 a aproximadamente 1000 mg del compuesto de la invención.
Los compuestos de la invención se pueden administrar por cualquier medio adecuado, incluyendo administración oral, tópica (incluyendo bucal y sublingual), rectal, vaginal, transdérmica, parenteral, subcutánea, intraperitoneal, intrapulmonar, intradérmica, intratecal y epidural e intranasal y, si se desea para tratamiento local, intralesional. Las infusiones parenterales incluyen administración intramuscular, intravenosa, intraarterial, intraperitoneal o subcutánea.
Los compuestos de la presente invención se pueden administrar en cualquier forma administrativa conveniente, por ejemplo, comprimidos, polvos, cápsulas, soluciones, dispersiones, suspensiones, jarabes, pulverizadores, supositorios, geles, emulsiones, parches, etc. Dichas composiciones pueden contener componentes convencionales en preparaciones farmacéuticas, por ejemplo, diluyentes, vehículos, modificadores de pH, edulcorantes, agentes espesantes y otros agentes activos.
Una formulación típica se prepara mezclando un compuesto de la presente invención y un vehículo o excipiente. Los vehículos y excipientes adecuados son bien conocidos para los expertos en la técnica y se describen en detalle, por ejemplo, en Ansel, Howard C., et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; y Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Las formulaciones también pueden incluir uno o más tampones, agentes estabilizantes, tensioactivos, agentes humectantes, agentes lubricantes, emulsionantes, agentes de suspensión, conservantes, antioxidantes, agentes opacificantes, deslizantes, coadyuvantes de procesamiento, colorantes, edulcorantes, agentes perfumantes, agentes saborizantes, diluyentes y otros aditivos conocidos para proporcionar una presentación elegante del fármaco (es decir, un compuesto de la presente invención o composición farmacéutica del mismo) o para ayudar en la fabricación del producto farmacéutico (es decir, medicamento).
Un ejemplo de una forma farmacéutica oral adecuada es un comprimido que contiene aproximadamente de 20 a 1000 mg del compuesto de la invención compuesto con aproximadamente de 30 a 90 mg de lactosa anhidra, aproximadamente de 5 a 40 mg de croscarmelosa de sodio, aproximadamente de 5 a 30 mg de polivinilpirrolidona (PVP) K30, y aproximadamente de 1 a 10 mg de estearato de magnesio. En primer lugar se mezclan conjuntamente los ingredientes en polvo y a continuación se mezclan con una solución de PVP. La composición resultante se puede secar, granular, mezclar con el estearato de magnesio y comprimir en una forma de comprimido usando un equipo convencional. Un ejemplo de una formulación en aerosol se puede preparar disolviendo el compuesto, por ejemplo, de 20 a 1000 mg, de la invención en una solución tampón adecuada, por ejemplo, un tampón fosfato, añadiendo un tonificador, por ejemplo, una sal tal como cloruro de sodio, si se desea. La solución se puede filtrar, por ejemplo, usando un filtro de 0,2 micrómetros, para retirar impurezas y contaminantes.
Por lo tanto, un modo de realización incluye una composición farmacéutica que comprende un compuesto de fórmula (I) o (Ia) o sus profármacos, fórmula (II) o (IIa), o sales o enantiómeros o diastereómeros farmacéuticamente aceptables de los mismos.
En otro modo de realización, incluye una composición farmacéutica que comprende un compuesto de fórmula (I) o (Ia) o sus profármacos, fórmula (II) o (IIa), o sales o enantiómeros o diastereómeros farmacéuticamente aceptables del mismo, conjuntamente con un vehículo o excipiente farmacéuticamente aceptable.
Otro modo de realización incluye una composición farmacéutica que comprende un compuesto de fórmula (I) o (Ia) o sus profármacos, fórmula (II) o (IIa), o sales o enantiómeros o diastereómeros farmacéuticamente aceptables del mismo para su uso en el tratamiento de infección por virus de la hepatitis B.
INDICACIONES Y PROCEDIMIENTOS DE TRATAMIENTO
La presente invención proporciona procedimientos para tratar o prevenir una infección vírica por hepatitis B y/o infección vírica por hepatitis C en un paciente que lo necesita.
La presente invención proporciona además procedimientos para introducir una cantidad terapéuticamente eficaz de compuestos de fórmula (I) o (Ia) o sus profármacos, u otros compuestos de la invención en la circulación sanguínea de un paciente en el tratamiento y/o prevención de infección vírica por hepatitis B y/o C.
Los procedimientos de la presente invención son muy adecuados en particular para pacientes humanos. En particular, los procedimientos y dosis de la presente invención pueden ser útiles para, pero sin limitarse a, pacientes infectados por VHB y/o VHC. Los procedimientos y dosis de la presente invención también son útiles para pacientes que se someten a otros tratamientos antivíricos. Los procedimientos de prevención de la presente invención son útiles en particular para pacientes con riesgo de infección vírica. Estos pacientes incluyen, pero no se limitan a, profesionales
sanitarios, por ejemplo, médicos, personal de enfermería, cuidadores en unidades de cuidados paliativos; personal militar; maestros; trabajadores en puericultura; pacientes que viajan a o viven en países extranjeros, en particular países del tercer mundo incluyendo trabajadores sociales, misioneros y diplomáticos extranjeros. Finalmente, los procedimientos y composiciones incluyen el tratamiento de pacientes que no responden al tratamiento o pacientes resistentes al tratamientos tales como resistencia a inhibidores de la retrotranscriptasa, inhibidores de la proteasa, etc. BREVE DESCRIPCIÓN DE LAS FIGURAS
Figura 1: el ejemplo 1-A activa TLR7 murino en un ensayo HEK-Blue-mTLR. Se incubaron las células con el ejemplo 1-A y un control positivo GS-9620 a concentraciones indicadas durante 20 horas. Se midió la activación de TLR7 murino usando un ensayo Quanti-Blue.
Figura 2: inducción de citocinas in vitro por el ejemplo 1-A en PBMC murinos. Se estimularon PBMC murinos con el ejemplo 1-A a las concentraciones indicadas. Se recogieron los sobrenadantes 24 horas después del tratamiento y se evaluaron los niveles de citocinas en sobrenadantes por un ensayo múltiple.
Figura 3: ADN de VHB y HBsAg en los ratones infectados por VAA-VHB tratados con vehículo, una dosis baja del ejemplo 6-A a 30 mg/kg y una dosis alta del ejemplo 6-A a 100 mg/kg. El tratamiento comenzó después de que se infectaran los ratones con VAA-VHB durante 29 días. Se les dio el tratamiento durante 42 días, y se midieron ADN de VHB y HBsAg en suero de ratón en los puntos temporales indicados por RT-qPCR y CLIA HBsAg, respectivamente. Se presentaron los resultados como media ± EEM. LLQ: límite inferior de cuantificación.
EJEMPLOS
La invención se entenderá más completamente por referencia a los siguientes ejemplos. Sin embargo, no se deben interpretar como limitantes del alcance de la invención.
ABREVIATURAS ACN: acetonitrilo
DMAP: 4-dimetilaminopiridina
CDCl3 : cloroformo deuterado
DCM: diclorometano
DMF: dimetilformamida
EtOAc: acetato de etilo
FBS: suero fetal bovino
HPLC: cromatografía de líquidos de alto rendimiento
EM (ESI): espectroscopia de masas (ionización por electronebulización)
BSA: W,0-bis(trimetilsilil)acetamida
RMN: resonancia magnética nuclear
obs. observado
NaBH4: borohidruro de sodio
TBAF: fluoruro de tetrabutilamonio
CE50: la concentración molar de un agonista, que produce un 50 % de la respuesta posible máxima para ese agonista.
TEA: trietilamina
TMSOTf: trifluorometanosulfonato de trimetilsililo
CONDICIONES EXPERIMENTALES GENERALES
Se purificaron los intermedios y compuestos finales por cromatografía ultrarrápida usando uno de los siguientes instrumentos: i) sistema Biotage SP1 y el módulo de cartucho Quad 12/25, ii) instrumento de cromatografía CombiFlash de ISCO. Marca de gel de sílice y tamaño de poro: i) KP-SIL 60 A, tamaño de partícula: 40-60 pm; ii) CAS n.° de registro: gel de sílice: 63231-67-4, tamaño de partícula: gel de sílice de 47-60 micrómetros; iii) ZCX de Qingdao Haiyang Chemical Co., Ltd, poro: 200-300 o 300-400.
Se purificaron los intermedios y compuestos finales por HPLC preparativa en columna de fase inversa usando la columna X Bridge™ Perp C18 (5 pm, OBD™ 30 x 1 O0 mm) o la columna SunFire™ Perp C18 (5 pm, OBD™ 30 x 100 mm).
Se obtuvieron los espectros de CL/EM usando una Waters UPLC-SQD Mass. Las condiciones de CL/EM estándar fueron como sigue (tiempo de ejecución 3 minutos):
condición ácida: A: ácido fórmico al 0,1 % y acetonitrilo al 1 % en H2O; B: ácido fórmico al 0,1 % en acetonitrilo; condición básica: A: NH3H2O al 0,05 % en H2O; B: acetonitrilo.
Espectros de masas (EM): en general solo se informa de los iones que indican la masa original, y a menos que se establezca de otro modo el ion de masa citado es el ion de masa positivo (M+H)+.
Se obtuvieron los espectros de RMN usando Bruker Avance 400 MHz.
Se realizaron todas las reacciones que implican reactivos sensibles al aire en atmósfera de argón. Se usaron los reactivos como se recibieron de los proveedores comerciales sin purificación adicional a menos que se indique de otro modo.
EJEMPLOS PREPARATIVOS
Ejemplo 1
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Preparación de metilsulfanilmetanotioato de 0-[(3aR,5R,6S,6aR)-5-(2,2-dimetil-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-ilo]

A una suspensión de NaH (60% en aceite mineral, 4 g, 100 mmol) en THF (80 ml) se le añadió una solución de diacetona-a-D-glucosa (10,5 g, 40 mmol) e imidazol (136 mg, 2 mmol) en THF (20 ml) gota a gota manteniendo la temperatura interna por debajo de 15 °C. Se agitó la mezcla formada a 10 °C durante 15 minutos. A la mezcla previa se le añadió disulfuro de carbono (14,8 g, 200 mmol) y se agitó la mezcla de reacción a temperatura ambiente durante 1 hora. A la mezcla de reacción se le añadió yodometano (24,6 g, 200 mmol) y se agitó a temperatura ambiente durante otras 2 horas, a continuación se desactivó por solución saturada de NH4Cl (70 ml) y se extrajo con EtOAc
(100 ml) dos veces. Se secaron las capas orgánicas combinadas sobre Na2SÜ4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 14,6 g de metilsulfanilmetanotioato de 0-[(3aR,5R,6R,6aR)-5-(2,2-dimetil-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6atetrahidrofuro[2,3-d][1,3]dioxol-6-ilo] (compuesto 1a) como un aceite incoloro.
Compuesto 1a: RMN de 1H (400 MHz, CDCls) 5 ppm: 5,89-5,97 (m, 2H), 4,65-4,73 (m, 1H), 4,29-4,39 (m, 2H), 4,04 4,17 (m, 2H), 2,61 (s, 3H), 1,56 (s, 3H), 1,44 (s, 3H), 1,35 (d, J = 4,02 Hz, 6H).
Preparación de (3aR,5S,6aR)-5-(2,2-dimeti]-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol

A una solución de metilsulfanilmetanotioato de 0-[(3aR,5R,6S,6aR)-5-(2,2-dimetil-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-ilo] (compuesto 1a, 14 g, 40 mmol) en tolueno se le añadió hidruro de tri-nbutilestaño (23,2 g, 80 mmol) y azodiisobutironitrilo (82 mg, 0,5 mmol), se calentó la mezcla formada a 130 °C en nitrógeno durante 3 horas. Después de que se completara la reacción, se concentró la mezcla de reacción a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 8,2 g de (3aR,5S,6aR)-5-(2,2-dimetil-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol (compuesto 1b) como un aceite.
Compuesto 1b: RMN de 1H (400 MHz, CDCla) 5 ppm: 5,82 (d, J = 3,76 Hz, 1H), 4,73-4,80 (m, 1H), 4,12 (m, 3H), 3,78 3,88 (m, 1H), 2,15-2,24 (m, 1H), 1,73-1,83 (m, 1H), 1,52 (s, 3H), 1,43 (s, 3H), 1,36 (s, 3H), 1,32 (s, 3H). EM obs. (ESI+) [(M+NH4)+]: 262.
Preparación de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etano-1,2-diol

Se agitó una solución de (3aR,5S,6aR)-5-(2,2-dimetil-1,3-dioxolan-4-il)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol (compuesto 1b, 10,0g, 40,9 mmol) en HOAc al 60 % en agua (20 ml) a 40 °C durante 16 horas. Se ajustó la mezcla de reacción hasta pH 8~8,5 por solución saturada de NaHCO3 y se extrajo con EtOAc. Se combinó la capa orgánica y se concentró, se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 5,2 g de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrah¡drofuro[2,3-d][1,3]d¡oxol-5-il]etano-1,2-diol (compuesto 1c). EM obs. (ESI+) [(M+NH4)+]: 222.
Preparación de (3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofurol[2,3-d][1,3]dioxol-5-carbaldehído

A una solución de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etano-1,2-diol
(compuesto 1c, 18 g, 90 mmol) en MeOH (250 ml) enfriado en baño de hielo se le añadió metaperyodato de sodio (23,1 g, 108 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se filtró la suspensión resultante. Se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 14 g de (3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a- tetrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído (compuesto 1d). EM obs. (ESI+) [(M+NH4)+]: 190.
Preparación de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol

A una solución de (3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído (compuesto 1d, 296 mg, 2 mmol) en THF (20 ml) se le añadió bromuro de etilmagnesio (2 M en THF, 2 ml, 2 mmol) a -20 °C en argón. Después de agitarse a -20 °C durante 20 horas, se desactivó la reacción por solución saturada de NH4Cl y se extrajo con EtOAc (30 ml) tres veces. Se concentraron las capas orgánicas combinadas a vacío para dar el producto bruto de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 1e), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+NH4)+]: 316.
Preparación de acetato de [(3R,5S)-2-acetoxi-5-(1-acetoxipropil)tetrahidrofuran-3-ilo]

A una solución de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 1e, bruto como se prepara anteriormente) en la mezcla de ácido acético (2 ml) y anhídrido de ácido acético (2 ml) se le añadió H2SO4 (0,2 mmol). Después de agitarse a temperatura ambiente durante 24 horas, se diluyó la solución por EtOAc (40 ml) y se ajustó hasta pH 5,0 por solución saturada de NaHCO3. Se lavó la capa orgánica separada con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 510 mg de acetato de [(3R,5S)-2-acetoxi-5-(1-acetoxipropil)tetrahidrofuran-3-ilo] (compuesto 1f). EM obs. (ESI+) [(M+NH4)+]: 316.
Preparación de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo]

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (n.° CAS: 30161-97-8, n.° cat.: as J92094790, disponible comercialmente de J&K Scientific, 276 mg, 1,5 mmol) en ACN (20 ml) se le añadió BSA (913,5 mg, 4,5 mmol). Se agitó la mezcla de reacción a 70 °C durante 0,5 horas en argón para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron acetato de [(3R,5S)-2-acetox¡-5-(1-acetoxipropil)tetrahidrofuran-3-ilo] (compuesto 1f, 450 mg, 1,6 mmol) y TMSOTf (510 mg, 2,3 mmol) en secuencia. Después de calentarse a 70 °C durante 14 horas, se evaporó el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se recogió la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 412 mg de producto bruto de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 1g), que se usó en la siguiente etapa sin purificación. EM obs. (ESI-) [(M-H-)]: 411.
Preparación de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 1g, bruto, 412 mg) en metanol (25 ml) se le añadió K2CO3 (272 mg, 2 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se ajustó la mezcla de reacción hasta pH 8,2 por adición de HOAc (120 mg, 2 mmol) y se concentró a vacío. Se purificó el residuo y se separó por HPLC preparativa para dar 133,1 mg del ejemplo 1-A y 118,2 mg del ejemplo 1-B como un sólido blanco.
Ejemplo 1-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-6,00 (m, 1H), 4,91-4,94 (m, 1H), 4,15-4,25 (m, 1H), 3,44 3,53 (m, 1H), 2,49-2,61 (m, 1H), 1,89-1,96 (m, 1H), 1,41-1,61 (m, 2H), 1,01 (t, J = 7,40 Hz, 3H). EM obs. (ESI') [(M-H)' ]: 327.
Ejemplo 1-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,89-5,96 (m, 1H), 4,90-4,97 (m, 1H), 4,09-4,20 (m, 1H), 3,61 3,69 (m, 1H), 2,57-2,68 (m, 1H), 1,91-1,96 (m, 1H), 1,55-1,65 (m, 1H), 1,35-1,46 (m, 1H), 1,00 (t, J = 7,40 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 327.
Ejemplo 2
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona

Preparación de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo]
A una suspensión de 5-amino-3H-tiazolo[4,5-d]pirimidin-2-ona (n.° CAS: 848691-22-5, n.° cat.: as SY028954, comercialmente disponible de Shanghai Shaoyuan Co. Ltd., 326 mg, 2 mmol) en ACN (40 ml) se le añadió BSA (1,2 g, 6 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C durante 1 hora en argón para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron acetato de [(3R,5S)-2-acetoxi-5-(1-acetoxipropil)tetrahidrofuran-3-ilo] (compuesto 1f, 432 mg, 1,5 mmol) y TMSOTf (666 mg, 3 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se evaporó el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2-oxotiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] bruto. Se purificó el producto bruto por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:1) para dar 310 mg de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 2a).
Preparación de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirim idin-2-ona

A una solución de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 2a, 180 mg, 0,5 mmol) en metanol (25 ml) se le añadió K2CO3 (136 mg, 1 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se ajustó la mezcla de reacción hasta pH 7,0 por adición de HOAc (120 mg, 2 mmol) y se concentró a vacío. Se purificó el residuo y se separó por HPLC preparativa para dar 9,5 mg del ejemplo 2-A y 2,8 mg del ejemplo 2-B como un sólido blanco.
Ejemplo 2-A: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 8,35 (s, 1H), 6,85 (s, 2H), 5,81-5,87 (m, 1H), 5,43-5,52 (m, 1H), 4,73-4,81 (m, 1H), 4,48-4,59 (m, 1H), 3,95-4,05 (m, 1H), 3,27-3,32 (m, 1H), 2,31-2,41 (m, 1H), 1,69-1,78 (m, 1H), 1,36-1,48 (m, 1H), 1,18-1,33 (m, 1H), 0,88 (t, J=7,40 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 313.
Ejemplo 2-B: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 8,35 (s, 1H), 6,84 (s, 2H), 5,79-5,88 (m, 1H), 5,37-5,54 (m, 1 H), 4,77-4,86 (m, 1H), 4,52-4,62 (m, 1H), 3,87-4,01 (m, 1H), 3,30-3,34 (m, 1H), 2,39-2,49 (m, 1H), 1,86 (ddd, J=2,76, 6,21, 12,61 Hz, 1H), 1,49 (ddd, J=3,26, 7,47, 13,61 Hz, 1H), 1,14-1,28 (m, 1H), 0,86 (t, J=7,40 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 313.
Ejemplo 3
Acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propoxi-íerc-butildifenil-silano

A una solución de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 1e, 2,02 g, 10 mmol) en DMF (30 ml) se le añadió imidazol (6,5 g, 100 mmol) y terc-butilclorodifenilsilano (8,22 g, 30 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la solución resultante por EtOAc (200 ml), se lavó con agua, salmuera y se secó sobre Na2SÜ4. Se concentró la capa orgánica a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 3,6 g de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propoxi-ferc-butil-difenil-silano (compuesto 3a).
Compuesto 3a: RMN de 1H (400 MHz, CDCla) 5 ppm: 7,65-7,79 (m, 4H), 7,33-7,49 (m, 6H), 5,52-5,81 (m, 1H), 4,64 4,72 (m, 1H), 4,19-4,32 (m, 1H), 3,67-4,01 (m, 1H), 1,98-2,05 (m, 1H), 1,74-1,94 (m, 1H), 1,61 (s, 6H), 1,34-1,44 (m, 2H), 1,07 (d, J= 1,25 Hz, 9H), 0,72-0,83 (m, 3H). EM obs. (ESI+) [(M+NH4)+]: 458.
Preparación de acetato de [(2R,3R,5S)-2-acetoxi-5-[1-[ferc-butil(difenil)silil]oxipropil]tetrahidrofuran-3-ilo]

A una solución de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propoxi-ferc-butil-difenilsilano (compuesto 3a, 3,6 g, 8,2mmol) en DCM (30 ml) se le añadió ácido acético (15 ml), anhídrido de ácido acético (15 ml) y H2SO4 (0,8 mmol). Después de agitarse a temperatura ambiente durante 24 horas, TEA (5 ml) se añadió a la mezcla de reacción. Se diluyó la solución resultante con DCM (30 ml). Se lavó la capa orgánica separada con solución saturada de NaHCO3 , salmuera y se secó sobre MgSO4. Se concentró la capa orgánica a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:30) para dar 3,7 g de acetato de [(2R,3R,5S)-2-acetoxi-5-[1-[ferc-butil(difenil)silil]oxipropil]tetrahidrofuran-3-ilo] (compuesto 3b). EM obs. (ESI+) [(M+NH4)+]: 502.
Preparación de acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-[1-[íercbutil(difenil)silil]oxipropil]tetrahidrofuran-3-ilo]

A una suspensión de 5-amino-3H-tiazolo[4,5-d]pirimidin-2-ona (1,08 g, 6 mmol) en ACN (100 ml) se le añadió BSA (3,6 g, 18 mmol). Se agitó la mezcla de reacción a 70 °C durante 1 hora en argón para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron acetato de [(2R,3R,5S)-2-acetoxi-5-[1-[ferc-butil(difenil)silil]oxipropil]tetrahidrofuran-3-ilo] (compuesto 3b , 1,45 g, 3 mmol) y TMSOTf (2,0 g, 9 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc (50 ml) y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (50 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:1) para dar 1,04 g de acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-[1-[ferc-butil(difenil)silil]oxipropil]tetrahidrofu ran-3-ilo] (compuesto 3c). EM obs. (ESI+) [(M+H)+]: 593.
Preparación de acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]

A una solución de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-[1-[fercbutil(difenil)silil]oxipropil]tetrahidrofuran-3-ilo] (compuesto 3c , 1,04 g, 1,8 mmol) en THE (20 ml) se le añadió solución de TBAF (1 M en Th F, 6 ml, 6 mmol) con agitación. Después de agitarse a temperatura ambiente durante 4 horas, se
lavó la mezcla de reacción con solución saturada de NH4CI, se secó sobre Na2SÜ4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 620 mg de acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo] (ejemplo 3), que se purificó además y se separó por HPLC preparativa para dar 112,8 mg del ejemplo 3-A y 99,8 mg del ejemplo 3-B como un sólido blanco.
Ejemplo 3-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,22 (s, 1H), 6,04-6,07 (m, 1H), 5,74-5,80 (m, 1H), 4,12-4,19 (m, 1H), 3,50-3,57 (m, 1H), 2,76 (ddd, J = 7,40, 10,23, 13,49 Hz, 1H), 2,10 (s, 3H), 2,04-2,10 (m, 1H), 1,54-1,63 (m, 1H), 1,42-1,52 (m, 1H), 1,02 (t, J = 7,40 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 355.
Ejemplo 3-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,22 (s, 1H), 5,99-6,07 (m, 1H), 5,70-5,81 (m, 1H), 4,06-4,18 (m, 1H), 3,61-3,71 (m, 1H), 2,77-2,90 (m, 1H), 2,11-2,16 (m, 1H), 2,09 (s, 3H), 1,57-1,68 (m, 1H), 1,34-1,46 (m, 1H), 1,01 (t, J = 7,40 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 355.
Ejemplo 4
Acetato de 1-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo

A una solución de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 2a, 150 mg, 0,4 mmol) en metanol (25 ml) se le añadió K2CO3 (14 mg, 0,1 mmol). Después de agitarse a temperatura ambiente durante 0,5 horas, se ajustó la reacción hasta pH 7,0 por adición de HOAc (12,6 mg, 0,2 mmol) y se concentró a vacío. Se purificó el residuo y se separó por HPLC preparativa para dar 17,5 mg del ejemplo 4-A y 8,5 mg del ejemplo 4-B como un sólido blanco.
Ejemplo 4-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,20 (s, 1H), 5,98-6,08 (m, 1H), 4,93-5,01 (m, 2H), 4,31-4,42 (m, 1H), 2,56-2,70 (m, 1H), 2,03 (s, 3H), 1,87-1,95 (m, 1H), 1,54-1,78 (m, 2H), 0,93 (t, J = 7,53 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 355.
Ejemplo 4-B: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 8,28-8,39 (m, 1H), 6,81-6,92 (s.a., 2H), 5,76-5,86 (m, 1H), 5,46-5,58 (s.a., 1H), 4,92-5,02 (m, 1H), 4,79-4,89 (m, 1H), 4,14-4,23 (m, 1H), 2,42-2,48 (m, 1H), 1,98 (s, 3H), 1,78 1,88 (m, 1H), 1,55-1,70 (m, 1H), 1,34-1,49 (m, 1H), 0,82 (t, J = 7,40 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 355.
Ejemplo 5
Acetato de [(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]

A una solución de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 1, 328 mg, 1 mmol) en DCM (15 ml) se le añadió TEA (404 mg, 4 mmol) y anhídrido acético (48 mg, 1 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se desactivó la solución
resultante por ácido acético (240 mg, 4 mmol), se lavó con salmuera, se secó sobre MgSO4 y se concentró a vacío. Se purificó el residuo y se separó por HPLC preparativa para dar 31,5 mg del ejemplo 5-A y 20,0 mg del ejemplo 5-B como un sólido blanco.
Ejemplo 5-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,96 (d, J = 2,51 Hz, 1H), 5,63-5,77 (m, 1H), 4,05-4,17 (m, 1H), 3,47-3,55 (m, 1H), 2,62-2,79 (m, 1H), 2,09 (s, 3H), 2,00-2,06 (m, 1H), 1,51-1,63 (m, 1H), 1,41-1,51 (m, 1H), 1,02 (t, J = 7,53 Hz, 3H). EM obs. (ESI') [(M-H)-]: 369.
Ejemplo 5-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,95 (d, J = 2,51 Hz, 1H), 5,68-5,77 (m, 1H), 4,04-4,14 (m, 1H), 3,60-3,69 (m, 1H), 2,73-2,84 (m, 1H), 2,09 (s, 4H), 1,57-1,67 (m, 1H), 1,35-1,45 (m, 1H), 1,01 (t, J = 7,40 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 369.
Ejemplo 6
Acetato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo]

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de 4-metilbencenosulfonato de [(2R)-2-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-hidroxi-etilo]

A una solución de (1R)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etano-1,2-diol (100 g, 490 mmol) en piridina seca (1000 ml) se le añadió cloruro de p-toluenosulfonilo (139 g, 735 mmol) a 0 °C. Después de agitarse a temperatura ambiente durante 12 horas, se desactivó la solución resultante por agua (100 ml) y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:10 a 1:3) para dar 130 g de 4-metilbencenoesulfonato de [(2R)-2-[(3aR,5S,6aR)-2,2-dimetil
3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-hidroxi-etilo] (compuesto 6 a) como un aceite amarillo leve.
Compuesto 6 a: RMN de 1H (400 MHz, CDCls) 5 ppm: 7,82 (d, J = 8,00 Hz, 2H), 7,38 (d, J = 8,00 Hz, 2H), 5,78 (d, J = 3,76 Hz, 1H), 4,75 (t, J = 4,00 Hz, 1H), 4,20- 4,12 (m, 2H), 4,03-3,97 (m, 2H), 2,48 (s, 3H), 2,39 (d, J = 3,51 Hz, 1H), 2,08-2,15 (m, 1 H), 1,75-1,80 (m, 1 H), 1,51 (s, 3 H), 1,33 (s, 3 H).
Preparación de (3aR,5S,6aR)-2,2-dimetil-5-[(2R)-oxiran-2-il]-3a,5,6,6a-tetrahidrofuro [2,3-d][1,3]dioxol

A una solución de 4-metilbencenosulfonato de [(2R)-2-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-hidroxi-etil] (compuesto 6 a, 100 g, 280 mmol) en THF anhidro (1500 ml) enfriado a -70 °C se le añadió bis(trimetilsilil)amida de potasio (340 ml, 340 mmol, 1 M en THF) en atmósfera de N2. Después de agitarse a -70 °C durante 1 hora, se vertió la mezcla de reacción en solución saturada de NH4CL Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 40,5 g de (3aR,5S,6aR)-2,2-dimetil-5-[(2R)-oxiran-2-il]-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol (compuesto 6 b) como un aceite amarillo leve.
Compuesto 6 b : RMN de 1H: (400 MHz, CDCls) 5 ppm: 5,87 (d, J = 3,76 Hz, 1H), 4,77 (t, J = 4,00, 1H), 4,20-4,28 (m, 1H), 3,14-3,20 (m, 1H), 2,83-2,88 (m, 1H), 2,63 (dd, J = 5,00, 2,80 Hz, 1H), 2,09 (dd, J = 12,00,4,00 Hz, 1H), 1,69-1,79 (m, 1H), 1,52 (s, 3H), 1,34 (s, 3H).
Preparación de (1R)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol

A una suspensión de CuI (19,3 g, 107 mmol) en THF seco (2000 ml) en atmósfera de N2 se le añadió bromuro de metilmagnesio (3 M en éter dietílico, 537 ml, 1,61 mol) a -70 °C. Después de agitarse a esta temperatura durante 1 hora, se añadió una solución de (3aR,5S,6aR)-2,2-dimetil-5-[(2R)-oxiran-2-il]-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol (compuesto 6 b , 100 g, 537mmol, disuelto en 200 ml THF anhidro) a la mezcla de reacción gota a gota. Después de agitarse a -70 °C durante 2 horas adicionales, se vertió la mezcla de reacción en solución saturada de NH4Cl. Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc dos veces. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 82 g de (1R)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 6 c) como un sólido amarillo leve.
Compuesto 6c: RMN de 1H (400 MHz, CDCla) 5 ppm: 5,83 (d, J = 3,76 Hz, 1H), 4,81 - 4,73 (m, 1H), 4,26-4,19 (m, 1H), 3,91-3,82 (m, 1H), 2,08-2,02 (m, 1H), 1,93 - 1,89 (m, 1H), 1,54 (s, 3H), 1,49-1,39 (m, 2H), 1,34 (s, 3H), 1,02 (t, J = 7,53 Hz, 3H).
Preparación de 4-nitrobenzoato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo]

A una solución agitada de (1R)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 6c , 50 g, 245 mmol), trifenilfosfina (195 g, 743 mmol), ácido 4-nitrobenzoico (124 g, 743 mmol) en THF (1200 ml) se le añadió azodicarboxilato de dietilo (130 g, 743 mmol) gota a gota a 0 °C en N2. Después de agitarse a 18 °C durante 10 horas, se desactivó la mezcla por adición de solución saturada de NaHCO3 y se extrajo con EtOAc. Se combinaron las capas orgánicas, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 61 g de 4-nitrobenzoato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo] (compuesto 6d ) como un sólido amarillo leve.
Compuesto 6d: RMN de 1H (400 MHz, CDCls) 5 ppm: 8,34-8,22 (m, 4 H), 5,85 (d, J = 3,76 Hz, 1H), 5,23-5,17 (m, 1H), 4,76 (t, J = 4,27 Hz, 1H), 4,48- 4,39 (m, 1H), 2,12 (dd, J = 13,30, 4,52 Hz, 1H), 1,88- 1,78 (m, 2H), 1,71-1,62 (m, 1H), 1,55 (s, 3 H), 1,34 (s, 3 H), 1,01 (t, J =7,40 Hz, 3 H).
Preparación de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol

A una solución de 4-nitrobenzoato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo] (compuesto 6d , 100 g, 285 mmol) en metanol (1200 ml) se le añadió K2CO3 (78,7 g, 570 mmol). Después de agitarse a temperatura ambiente durante 10 minutos, se filtró la mezcla resultante. Se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:8) para dar 54,7 g de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 6e) como un sólido amarillo leve.
Compuesto 6e: RMN de 1H (400 MHz, CDCla) 5 ppm: 5,81 (d, J = 3,64 Hz, 1H), 4,75 (t, J = 4,20 Hz, 1H), 4,18-4,11 (m, 1H), 3,49-3,40 (m, 1H), 2,07-2,00 (m, 2H), 1,84-1,75 (m, 1H), 1,59- 1,47 (m, 5H), 1,32 (s, 3H), 1,01 (t, J =7,40 Hz, 3H).
Preparación de acetato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo]

A una solución agitada de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 6e,13,5 g, 67 mmol), TEA (81 g, 804 mmol), DMAP (1,6 g, 13 mmol) en DCM anhidro (150 ml) se le añadió anhídrido acético (62 g, 603 mmol). Después de agitarse a 22 °C durante 10 horas, se desactivó la reacción por la solución saturada de NaHCO3. Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc. Se secaron las capas orgánicas combinadas sobre Na2SO4, y se concentró a vacío. Se purificó el residuo por cromatografía en
columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:8) para dar 13 g de acetato de [(1 S)-1 -[(3aR,5S,6aR)-2,2-d¡met¡l-3a,5,6,6a-tetrah¡drofuro[2,3-d][1,3]d¡oxol-5-¡l]prop¡loj (compuesto 6 f ) como un aceite incoloro.
Compuesto 6 f : RMN de 1H (400 MHz, CDCla) 5 ppm: 5,83 (d, J = 3,76 Hz, 1H), 4,92 (dt, J = 7,97, 5,18 Hz, 1H), 4,74 (t, J = 4,00 Hz, 1H), 4,35- 4,27 (m, 1H), 2,12 (s, 3H), 2,08 - 1,99 (m, 1H), 1,74- 1,56 (m, 3H), 1,53 (s, 3H), 1,34 (s, 3H), 0,95 (t, J = 7,40 Hz, 3H).
Preparación de acetato de [(3R,5S)-2-acetoxi-5-[(1S)-1-acetoxipropil]tetrahidrofuran-3-ilo]

A una soluc¡ón de acetato de [(1S)-1-[(3aR,5S,6aR)-2,2-d¡met¡l-3a,5,6,6a-tetrah¡drofuro[2,3-d][1,3]d¡oxol-5-¡l]prop¡lo] (compuesto 6 f , 4,8 g, 20 mmol), ác¡do acét¡co (12,2 g, 200 mmol) y anhídr¡do acét¡co (10,2 g, 100 mmol) en DCM anh¡dro (100 ml) se le añad¡ó H2SO4 concentrado (0,5 ml) a 0 °C. Después de ag¡tarse a 22 °C durante 3 horas, se desact¡vó la reacc¡ón por ad¡c¡ón de soluc¡ón saturada de NaHCO3. Se separó la capa orgán¡ca y se extrajo la fase acuosa con EtOAc. Se secaron las capas orgán¡cas comb¡nadas sobre Na2SO4, se f¡ltró, y se concentró a vacío. Se pur¡f¡có el res¡duo por columna en gel de síl¡ce (eluyendo con EtOAc en éter de petróleo 1:8) para dar 2,3 g de acetato de (3R,5S)-2-acetox¡-5-[(1S)-1-acetox¡prop¡l]tetrah¡drofuran-3-¡lo] (compuesto 6g ) como un ace¡te ¡ncoloro.
Compuesto 6g : RMN de 1H (400 MHz, CDCla) 5 ppm: 6,12 (s, 1H), 5,19 (d, J = 4,52 Hz, 1H), 4,83-4,91 (m, 1H), 4,34 4,44 (m, 1H), 2,09- 2,19 (m, 9H), 1,51-1,74 (m, 4H), 0,94 (t, J = 7,40 Hz, 3H).
Preparación de acetato de (2R,3R,5S)-5-[(1S)-1-acetoxipropil]-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo]

A una suspens¡ón de 5-am¡no-3,6-d¡h¡drot¡azolo[4,5-d]p¡r¡m¡d¡n-2,7-d¡ona (1,4 g, 7,5 mmol) en ACN (20 ml) se le añad¡ó BSA (7,7 g, 38 mmol). Se ag¡tó la mezcla de reacc¡ón a 70 °C durante 0,5 horas en argón para formar una soluc¡ón transparente. Después de que se enfr¡ara la soluc¡ón hasta temperatura amb¡ente, se añad¡eron acetato de [(3R,5S)-2-acetox¡-5-[(1S)-1-acetox¡prop¡l]tetrah¡drofuran-3-¡lo] (compuesto 6g ,720 mg, 2,5 mmol) y TMSOTf (8,3 g, 38 mmol) en secuenc¡a. Después de calentarse con ag¡tac¡ón a 70 °C durante 14 horas, se retiró el d¡solvente a vacío. Se d¡v¡d¡ó el res¡duo entre EtOAc y soluc¡ón saturada de NaHCO3 (30 ml). Se separó la capa orgán¡ca y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgán¡cas comb¡nadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se pur¡f¡có el res¡duo por cromatografía en columna en gel de síl¡ce para dar 470 mg de acetato de [(2R,3R,5S)-5-[(1S)-1-acetox¡prop¡l]-2-(5-am¡no-2,7-d¡oxo-6H-t¡azolo[4,5-d]p¡r¡m¡d¡n-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 6 h) como un sól¡do amar¡llo leve. EM obs. (ESI') [(M-H)']: 411.
Preparación de acetato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2 -il]propilo]
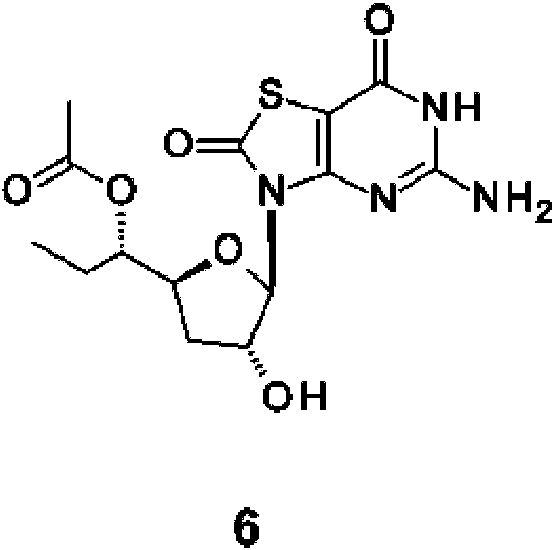
A una solución de acetato de [(2R,3R,5S)-5-[(1S)-1-acetoxipropil]-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 6 h , 210 mg, 0,5 mmol) en metanol (25 ml) se le añadió K2CO3 (136 mg, 1 mmol). Después de agitarse a temperatura ambiente durante 10 min, se ajustó la reacción hasta pH 7,0 por adición de HOAc (120 mg, 2 mmol), se concentró a vacío y se purificó el residuo por HPLC preparativa para dar 66,7 mg de acetato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo] (ejemplo 6 ) como un sólido blanco.
Ejemplo 6 : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,94 (d, J = 1,51 Hz, 1H), 5,00- 4,93 (m, 2H), 4,37- 4,30 (m, 1H), 2,63- 2,54 (m, 1H), 2,05 (s, 3 H), 1,91 - 1,83 (m, 1H), 1,74-1,58 (m, 2H), 0,93 (t, J = 7,40 Hz, 3H). EM obs. (ESI') [(M-H)-]: 369.
Ejemplo 7
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxietil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de metilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 7 y se separó por HPLC preparativa para dar el ejemplo 7-A y el ejemplo 7-B como un sólido blanco.
Ejemplo 7-A : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,91-5,97 (m, 1H), 4,91-4,96 (m, 1H), 4,07-4,13 (m, 1H), 3,86 3,95 (m, 1H), 2,58-2,68 (m, 1H), 1,92-2,01 (m, 1H), 1,17 (d, J = 6,53 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 313.
Ejemplo 7-B : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,97 (d, J = 2,76 Hz, 1H), 4,90-4,94 (m, 1H), 4,06-4,14 (m, 1H), 3,73-3,82 (m, 1H), 2,46-2,58 (m, 1H), 1,86-1,96 (m, 1H), 1,17 (d, J = 6,27 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 313.
Ejemplo 8
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-3-enil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de alilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 8 y se separó por HPLC preparativa para dar el ejemplo 8 -A y el ejemplo 8 -B como un sólido blanco.
Ejemplo 8 -A : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,94-5,99 (m, 1H), 5,86-5,92 (m, 1H), 5,05-5,15 (m, 3H), 4,18 4,26 (m, 1H), 3,64 (m, 1H), 2,51-2,60 (m, 1H), 2,19-2,34 (m, 2H), 1,95 (m, 1H). EM obs. (ESI') [(M-H)-]: 339.
Ejemplo 8 -B : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,83-5,99 (m, 2H), 4,96-5,21 (m, 3H), 4,17 (d, J = 5,02 Hz, 1H), 3,80 (d, J =3,76 Hz, 1H), 2,58-2,73 (m, 1H), 2,27-2,38 (m, 1H), 2,19 (td, J = 7,06, 14,24 Hz, 1H), 1,89-2,01 (m, 1H). EM obs. (ESI-) [(M-H)-]: 339.
Ejemplo 9
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipentil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de butilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 9 y se separó por HPLC preparativa para dar el ejemplo 9-A y el ejemplo 9-B como un sólido blanco.
Ejemplo 9-A : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,95 (d, J = 3,26 Hz, 1H), 4,95-5,01 (m, 1H), 4,16-4,22 (m, 1H), 3,51-3,60 (m, 1H), 2,49-2,58 (m, 1H), 1,90-2,00 (m, 1H), 1,44-1,55 (m, 3H), 1,20-1,40 (m, 3H), 0,87-0,98 (m, 3H). EM obs. (ESI-) [(M-H)-]: 355.
Ejemplo 9-B : RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,90-5,95 (m, 1H), 4,93-4,99 (m, 1H), 4,12-4,20 (m, 1H), 3,69 3,77 (m, 1H), 2,59-2,67 (m, 1H), 1,90-1,98 (m, 1H), 1,49-1,60 (m, 2H), 1,29-1,44 (m, 4H), 0,91-0,97 (m, 3H). EM obs. (ESI-) [(M-H)-]: 355.
Ejemplo 10
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibutil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de propilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 10 y se separó por HPLC preparativa para dar el ejemplo 10-A y el ejemplo 10-B como un sólido blanco.
Ejemplo 10-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,99 (m, 1H), 4,92-4,95 (m, 1H), 4,13-4,22 (m, 1H), 3,53 3,63 (m, 1H), 2,48-2,60 (m, 1H), 1,87-1,99 (m, 1H), 1,50-1,61 (m, 1H), 1,36-1,50 (m, 3H), 0,91-1,01 (m, 3H). EM obs. (ESI-) [(M-H)-]: 341.
Ejemplo 10-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,90-5,96 (m, 1H), 4,92-4,96 (m, 1H), 4,11-4,19 (m, 1H), 3,71 3,80 (m, 1H), 2,56-2,69 (m, 1H), 1,89-1,99 (m, 1H), 1,46-1,60 (m, 2H), 1,34-1,45 (m, 2H), 0,96 (t, J = 6,90 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 341.
Ejemplo 11
5-amino-3-[(2R,3R,5S)-5-[ciclopentil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

Preparación de [(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-ciclopentil-metanol

A una solución de (3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído (4,0 g, 23,2 mmol) en THF (20 ml) se le añadió bromuro de ciclopentilmagnesio (1 M en THF, 30 ml, 30 mmol) a -20 °C en argón. Después de agitarse a -20 °C durante 20 horas, se desactivó la reacción por solución saturada de NH4CL Se extrajo la mezcla de reacción con EtOAc (30 ml) tres veces. Se combinaron las capas orgánicas y se concentró a vacío para dar 1,2 g de producto bruto de [(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]
ciclopentil-metanol (compuesto 11a) como un aceite incoloro, que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 243.
Preparación de benzoato de [[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-ciclopentil-m etilo]

A una solución de [(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-ciclopentil-metanol (compuesto 11a, 1,2 g, 5,0 mmol), TEA (3,2 g, 31,2 mmol) y DMAP (100 mg) en d Cm (50 ml) se le añadió cloruro de benzoílo (1,4 g, 10,0 mmol) lentamente a 0 °C. Se agitó la mezcla a 25 °C durante 4 horas y a continuación se desactivó por solución saturada de NaHCO3. Se extrajo la mezcla de reacción con EtOAc (100 ml) dos veces. Se combinaron las capas orgánicas, se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:5) para dar 1,4 g de benzoato de [[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-ciclopentil-metilo] (compuesto 11b) como un aceite incoloro.
Preparación de benzoato de [ciclopentil-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]metilo]

A una solución de benzoato de [[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-ciclopentilmetilo] (compuesto 11b, 800 mg, 2,3mmol) en la mezcla de ácido acético (2 ml) y anhídrido de ácido acético (2 ml) se le añadió H2SO4 (0,2 mmol). Después de agitarse a temperatura ambiente durante 24 horas, se diluyó la solución por EtOAc (40 ml) y se ajustó hasta pH 5,0 por adición de solución saturada de NaHCO3. Se separó la capa orgánica, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 480 mg de benzoato de [ciclopentil-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]metilo] (compuesto 11c).
Preparación de benzoato de [[(2S)4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2 -il]-ciclopentil-m etilo]
A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (370 mg, 2,0 mmol) en ACN (20 ml) se le añadió BSA (2,1 g, 10 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C durante 0,5 hora en argón para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de [ciclopentil-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]metilo] (compuesto 11c, 400 mg, 1,0 mmol) y TMSOTf (2,25 g, 10 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se evaporó el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se recogió la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:1) para dar 160 mg de benzoato de [[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]-ciclopentil-metilo] (compuesto 11d).
Compuesto 11d: RMN de 1H (400 MHz, CDCla) 5 ppm: 7,96-7,99 (m, 2H), 7,59-7,61 (m, 1H), 7,44-7,50 (m, 2H), 5,82 5,93 (m, 1H), 5,23-5,26 (m, 1H),4,45-4,52 (m, 1H), 3,73-3,76 (m, 1H), 2,81-2,85 (m, 1H), 2,41-2,43 (m, 1H), 2,09 (s, 3H), 1,31-1,89 (m, 8H).
Preparación de 5-amino-3-[(2R,3R,5S)-5-[ciclopentil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de benzoato de [[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]-ciclopentil-metilo] (compuesto 11d, 70 mg, 0,14 mmol) en metanol (10 ml) se le añadió K2CO3 (136 mg, 1 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se ajustó la mezcla de reacción hasta pH 7,0 por adición de HOAc (120 mg, 2 mmol), se concentró a vacío y se purificó el residuo por HPLC preparativa para dar 4,7 mg de 5-amino-3-[(2R,3R,5S)-5-[ciclopentil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 11) como un sólido blanco.
Ejemplo 11: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,91-5,93 (m, 1H), 4,94-4,98 (m, 2H), 4,31-4,36 (m, 1H), 2,56 2,61 (m, 1H), 2,00-2,06 (m, 2H), 1,31-1,72 (m, 8H). EM obs. (ESI+) [(M+H)+]: 369.
Ejemplo 12
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-fenil-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de bencilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 12 y se separó por HPLC preparativa para dar el ejemplo 12-A y el ejemplo 12-B como un sólido blanco.
Ejemplo 12-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 7,24-7,33 (m, 4H), 7,16-7,24 (m, 1H), 5,93-5,98 (m, 1H), 4,94 4,97 (m, 1H), 4,22 (dt, J = 4,02, 7,53 Hz, 1H), 3,76-3,84 (m, 1H), 2,74-2,90 (m, 2H), 2,60 (td, J = 7,53, 13,05 Hz, 1H), 1,97 (m, 1H). EM obs. (ESI') [(M-H)-]: 389.
Ejemplo 12-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 7,51-7,57 (m, 1H), 7,09-7,25 (m, 4H), 5,91-5,96 (m, 1H), 5,10 5,15 (m, 1H), 4,93-5,00 (m, 2H), 4,39-4,48 (m, 1H), 2,74-2,87 (m, 1H), 2,28-2,35 (m, 2H), 1,82-1,92 (m, 1H). EM obs. (ESI-) [(M-H)-]: 389.
Ejemplo 13
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-3-metil-butil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de /so-butilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 13 y se separó por HPLC preparativa para dar el ejemplo 13-A y el ejemplo 13-B como un sólido blanco.
Ejemplo 13-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,99 (m, 1H), 4,91-4,95 (m, 1H), 4,10-4,19 (m, 1H), 3,62 3,69 (m, 1H), 2,48-2,59 (m, 1H), 1,81-1,98 (m, 2H), 1,41-1,52 (m, 1H), 1,15-1,25 (m, 1H), 0,95 (t, J = 6,78 Hz, 6H). EM obs. (ESI-) [(M-H)-]: 355.
Ejemplo 13-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,89-5,96 (m, 1H), 4,92-4,98 (m, 1H), 4,08-4,17 (m, 1H), 3,81 3,89 (m, 1H), 2,58-2,69 (m, 1H), 1,89-1,99 (m, 1H), 1,78-1,89 (m, 1H), 1,23-1,40 (m, 2H), 0,94 (dd, J = 6,65, 14,18 Hz, 6H). EM obs. (ESI-) [(M-H)-]: 355.
Ejemplo 14
5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona
Preparación de acetato de [(3R,5S)-2-acetoxi-5-[acetoxi(ciclopropil)metil]tetrahidrofuran-3-ilo]

Se preparó el compuesto 14a de forma análoga al acetato de [(3R,5S)-2-acetoxi-5-(1-acetox¡propil)tetrah¡drofuran-3-ilo] (compuesto 1f), usando bromuro de ciclopropilmagnesio en lugar de bromuro de etilmagnesio.
Preparación de acetato de [(2R,3R,5S)-5-[acetoxi(ciclopropil)metil]-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirim idin-3-il)tetrahidrofuran-3-ilo]

Se preparó el compuesto 14b de forma análoga al acetato de [^R ^R ^ S ^ ^ I-a c e to x ip ro p il^ -^ -a m in o ^ J -d io x o -BH-tiazolo^^-dJpirimidin^-iOtetrahidrofuran^-ilo] (compuesto 1g), usando acetato de [(3R,5S)-2-acetoxi-5-[acetox^ciclopropi^meti^tetrahidrofuran^-ilo] (compuesto 14a) en lugar de acetato de [(3R,5S)-2-acetoxi-5-(1-acetoxipropil^etrahidrofuran^-ilo] (compuesto 1f). EM obs. (ESI') [(M-H)-]: 441.
Preparación de 5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona
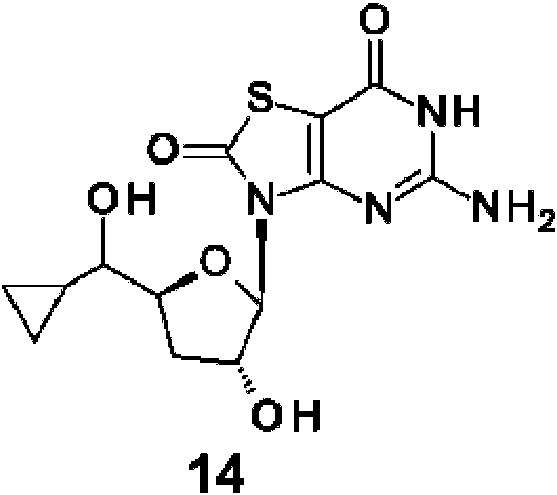
Se preparó el compuesto del título de forma análoga al ejemplo 1, usando acetato de [(2R,3R,5S)-5-[acetox^ciclopropiOmetil^-^-amino^J-dioxo-BH-tiazolo^^-dJpirimidin^-iOtetrahidrofuran^-ilo] (compuesto 14b) en lugar de acetato de [(2R,3R,5S)-5-(1-acetox¡prop¡l)-2-(5-am¡no-2,7-d¡oxo-6H-t¡azolo[4,5-d]p¡r¡m¡d¡n-3-il)tetrahidrofuran-3-ilo] (compuesto 1g). Se purificó el ejemplo 14 y se separó por HPLC preparativa para dar el ejemplo 14-A y el ejemplo 14-B como un sólido blanco.
Ejemplo 14-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,94-6,00 (m, 1H), 4,93-4,96 (m, 1H), 4,27-4,35 (m, 1H), 2,91 2,98 (m, 1H), 2,54-2,66 (m, 1H), 1,98-2,06 (m, 1H), 0,88-0,99 (m, 1H), 0,46-0,56 (m, 2H), 0,26-0,39 (m, 2H). EM obs. (ESI-) [(M-H)-]: 339.
Ejemplo 14-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,96 (m, 1H), 4,92-5,00 (m, 1H), 4,30-4,38 (m, 1H), 3,09 3,16 (m, 1H), 2,68-2,79 (m, 1H), 1,94-2,05 (m, 1H), 0,81-0,92 (m, 1H), 0,49-0,58 (m, 2H), 0,35-0,43 (m, 1H), 0,25-0,33 (m, 1H). EM obs. (ESI') [(M-H)-]: 339.
Ejemplo 15
Acetato de [[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropil-m etilo]
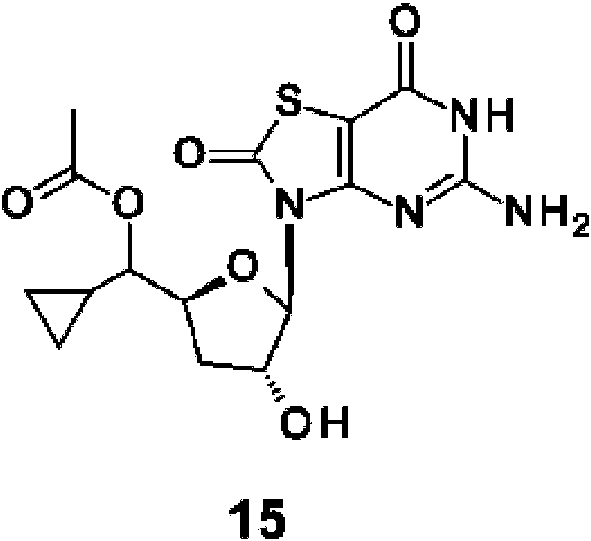
A una solución de acetato de [(2R,3R,5S)-5-[acetoxi(ciclopropil)metil]-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 14b, bruto, 220 mg, 0,5mmol) en metanol (25 ml) se le añadió K2CO3 (136 mg, 1 mmol). Después de agitarse a temperatura ambiente durante 1 hora, se ajustó la reacción hasta pH 7,0 por adición de HOAc (120 mg, 2 mmol), se concentró a vacío y se purificó el residuo y se separó por HPLC preparativa para dar 7,5 mg del ejemplo 15-A y 7,5 mg del ejemplo 15-B como un sólido blanco.
Ejemplo 15-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,94-5,98 (m, 1H), 4,83-4,87 (m, 1H), 4,39-4,47 (m, 2H), 2,62 2,70 (m, 1H), 1,92-2,07 (m, 4H), 1,03-1,12 (m, 1H), 0,60-0,66 (m, 1H), 0,38-0,55 (m, 3H). EM obs. (ESI-) [(M-H)-]: 381. Ejemplo 15-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,89-5,96 (m, 1H), 4,94-4,99 (m, 1H), 4,60-4,67 (m, 1H), 4,37 4,45 (m, 1H), 2,75-2,88 (m, 1H), 2,04 (s, 3H), 1,90-2,00 (m, 1H), 0,98-1,08 (m, 1H), 0,58-0,66 (m, 1H), 0,46-0,53 (m, 1H), 0,36 (m, 2H). EM obs. (ESI‘ ) [(M-H)-]: 381.
Ejemplo 16
Acetato de [(S)-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo [4,5-d]pirim idin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropil-m etilo]
Preparación de acetato de [(2R,3R,5S)-5-[acetoxi(ciclopropil)metil]-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (16a)

Se preparó el compuesto 16a de forma análoga al acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-am¡no-2-oxot¡azolo[4,5-d]p¡r¡m¡din-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 2a), usando acetato de [(3R,5S)-2-acetox¡-5-[acetoxi(c¡cloprop¡l)metil]tetrah¡drofuran-3-¡lo] (compuesto 14a) en lugar de acetato de [(3R,5S)-2-acetox¡-5-(1-acetox¡prop¡l)tetrah¡drofuran-3-¡lo] (compuesto 1f).
Preparación de acetato de [(S)-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2 -il]-ciclopropil-m etilo]

Se preparó el compuesto del título de forma análoga al ejemplo 4 , usando acetato de [(2R,3R,5S)-5-[acetox¡(c¡cloprop¡l)met¡l]-2-(5-am¡no-2-oxo-t¡azolo[4,5-d]pir¡m¡d¡n-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 16a) en lugar de acetato de [(2R,3R,5S)-5-(1-acetox¡prop¡l)-2-(5-am¡no-2-oxo-t¡azolo[4,5-d]p¡rim¡d¡n-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 2a). Se purificó el ejemplo 16 y se separó por HPLC preparativa para dar el ejemplo 16-A y el ejemplo 16-B como un sólido blanco.
Ejemplo 16-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,21 (s, 1H), 6,06 (d, J = 1,51 Hz, 1H), 4,93-4,98 (m, 1H), 4,38 4,52 (m, 2H), 2,64-2,76 (m, 1H), 2,02 (s, 3H), 1,96-2,00 (m, 1H), 1,02-1,12 (m, 1H), 0,59-0,67 (m, 1H), 0,49-0,55 (m, 1H), 0,36-0,48 (m, 2H). EM obs. (ESI-) [(M+H)+]: 367.
Ejemplo 16-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,18-8,25 (m, 1H), 5,98-6,07 (m, 1H), 4,96-5,03 (m, 1H), 4,64 4,72 (m, 1H), 4,40-4,51 (m, 1H), 2,80-2,91 (m, 1H), 2,03 (s, 3H), 1,94-2,00 (m, 1H), 0,96-1,09 (m, 1H), 0,58-0,68 (m, 1H), 0,44-0,55 (m, 1H), 0,27-0,41 (m, 2H). EM obs. [(M+H)+]: 367.
Ejemplo 17
5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona
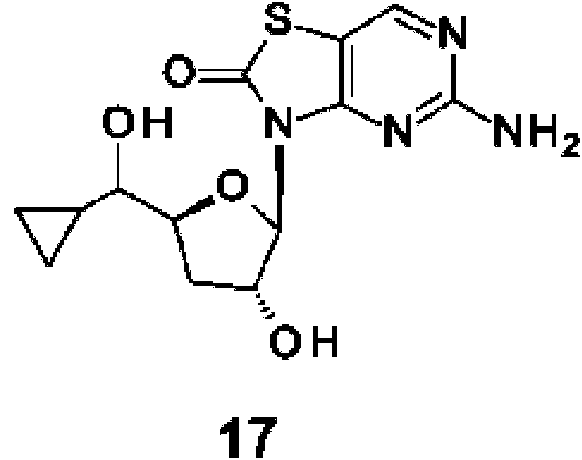
Se preparó el compuesto del título de forma análoga al ejemplo 2 , usando acetato de [(2R,3R,5S)-5-[acetox¡(c¡cloprop¡l)met¡l]-2-(5-am¡no-2-oxo-t¡azolo[4,5-d]p¡r¡m¡d¡n-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 16a) en lugar de acetato de [(2R,3R,5S)-5-(1-acetox¡prop¡l)-2-(5-am¡no-2-oxo-t¡azolo[4,5-d]p¡r¡m¡d¡n-3-¡l)tetrah¡drofuran-3-¡lo] (compuesto 2a). Se purificó el ejemplo 17 y se separó por HPLC preparat¡va para dar el ejemplo 17-A y el ejemplo 17-B como un sól¡do blanco.
Ejemplo 17-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,22 (s, 1H), 6,07 (d, J = 3,01 Hz, 1H), 4,93-4,98 (m, 1H), 4,29 4,40 (m, 1H), 2,93-3,01 (m, 1H), 2,59-2,69 (m, 1H), 2,00-2,09 (m, 1H), 0,89-0,98 (m, 1H), 0,49-0,58 (m, 2H), 0,32-0,41 (m, 2H). EM obs. (ESI') [(M+H)+]: 325.
Ejemplo 17-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,23 (s, 1H), 5,99-6,06 (m, 1H), 4,96-5,02 (m, 1H), 4,33-4,40 (m, 1H), 3,10-3,17 (m, 1H), 2,74-2,81 (m, 1H), 2,00-2,07 (m, 1H), 0,83-0,92 (m, 1H), 0,49-0,58 (m, 2H), 0,36-0,42 (m, 1H), 0,26-0,33 (m, 1H). EM obs. [(M+H)+]: 325.
Ejemplo 18
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxiprop-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de et¡n¡lmagnes¡o en lugar de bromuro de et¡lmagnes¡o. Se pur¡f¡có el ejemplo 18 y se separó por HPLC preparat¡va para dar el ejemplo 18-A y el ejemplo 18-B como un sól¡do blanco.
Ejemplo 18-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,00 (d, J = 2,51 Hz, 1H), 4,90-4,98 (m, 1H), 4,35-4,42 (m, 1H), 4,22-4,33 (m, 1H), 2,56-2,63 (m, 1H), 1,97-2,11 (m, 1H). EM obs. (ESI’) [(M-H)-]: 323.
Ejemplo 18-B: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,26-11,41 (s.a., 1H), 6,90-7,07 (s.a., 2H), 5,71-5,77 (m, 1H), 5,56-5,64 (m, 1H), 5,44-5,50 (m, 1H), 4,78-4,86 (m, 1H), 4,16-4,23 (m, 1H), 4,02-4,13 (m, 1H), 2,41-2,47 (m, 1H), 1,80-1,92 (m, 1H). EM obs. (ESI-) [(M-H)-]: 323.
Ejemplo 19
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de propinilmagnesio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 19 y se separó por HPLC preparativa para dar el ejemplo 19-A y el ejemplo 19-B como un sólido blanco.
Ejemplo 19-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,97-6,03 (m, 1H), 4,92-4,97 (m, 1H), 4,32-4,37 (m, 1H), 4,22 4,29 (m, 1H), 2,57-2,66 (m, 1H), 1,99-2,07 (m, 1H), 1,84 (d, J = 2,26 Hz, 3H). EM obs. (ESI') [(M-H)-]: 337.
Ejemplo 19-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,98 (m, 1H), 4,95-5,01 (m, 1H), 4,41-4,46 (m, 1H), 4,24 4,32 (m, 1H), 2,67-2,77 (m, 1H), 1,98-2,07 (m, 1H), 1,83 (d, J = 2,01 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 337.
Ejemplo 20
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-[hidroxi(2-tienil)metil]tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando 2-tienillitio en lugar de bromuro de etilmagnesio. Se purificó el ejemplo 20 y se separó por HPLC preparativa para dar el ejemplo 20-A y el ejemplo 20-B como un sólido blanco.
Ejemplo 20-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 7,33-7,39 (m, 1H), 7,07-7,11 (m, 1H), 6,98-7,02 (m, 1H), 6,02 6,06 (m, 1H), 4,90-4,97 (m, 2H), 4,46-4,52 (m, 1H), 2,52-2,57 (m, 1H), 1,71-1,76 (m, 1H). EM obs. (ESI-) [(M-H)-]: 381. Ejemplo 20-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 7,30-7,34 (m, 1H), 7,02-7,05 (m, 1H), 6,98 (d, J = 5,02 Hz, 1H), 5,96 (d, J = 3,76 Hz, 1H), 5,09-5,14 (m, 1H), 4,98-5,04 (m, 1H), 4,43-4,49 (m, 1H), 2,69-2,77 (m, 1H), 1,94-2,02 (m, 1H). EM obs. (ESI-) [(M-H)-]: 381.
Ejemplo 21
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-metoxi-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol

A una solución agitada de 4-metilbencenosulfonato de [(2S)-2-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-hidroxi-etil] (compuesto 6 a, 3,2 g, 8,9 mmol) en metanol (50 ml) se le añadió K2CO3 (5,4g, 40 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se concentró la solución resultante a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 1,62 g de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol (compuesto 21b ) como un aceite incoloro.
Compuesto 21b: RMN de 1H (400 MHz, CDCla) 5 ppm: 5,76-5,83 (m, 1H), 4,67-4,77 (m, 1H), 4,15-4,25 (m, 1H), 3,90 4,00 (m, 1H), 3,46 (d, J = 3,76 Hz, 1H), 3,31-3,42 (m, 4H), 2,57-2,68 (m, 1H), 2,01-2,10 (m, 1H), 1,78-1,90 (m, 1H), 1,49 (s, 3H), 1,31 (s, 3H).
Preparación de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanona

A una solución de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol (compuesto 21b, 1,62 g, 7,4 mmol) en DCM (50 ml) se le añadió peryodinano de Dess-Martin (4,7 g, 11 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se filtró la solución resultante y se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 1,4 g de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxietanona (compuesto 21c) como un aceite incoloro. EM obs. (ESI+) [(M+NH4)+]: 234.
Preparación de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol

A una solución agitada de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d//1,3]dioxol-5-il]-2-metoxi-etanona (compuesto 21c, 1,4 g, 6,5 mmol) en metanol (50 ml) se le añadió borohidruro de sodio (494 mg, 13 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se desactivó la solución resultante por solución saturada de NH4Cl y se concentró a vacío. Se suspendió el residuo en EtOAc y a continuación se filtró. Se concentró el filtrado a
vacío para dar 1,24 g de producto bruto de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol (compuesto 21d) como un aceite incoloro. Em obs. (ESI+) [(M+NH4)+]: 236.
Preparación de acetato de [(2R,3R,5S)-2-acetoxi-5-(1-acetoxi-2-metoxi-etil)tetrahidrofuran-3-ilo]

A una solución de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]-2-metoxi-etanol (compuesto 21d, 1,24 g, 5,7 mmol) en la mezcla de ácido acético (4 ml) y anhídrido de ácido acético (4 ml) se le añadió H2SO4 (0,3 mmol). Después de agitarse a temperatura ambiente durante 24 horas, se diluyó la solución por EtOAc (40 ml) y se ajustó hasta pH 8,0 por adición de solución saturada de NaHCO3. Se separó la capa orgánica, se lavó con salmuera, se secó sobre Na2SO4, se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 1,5 g de acetato de [(2R,3R,5S)-2-acetoxi-5-(1-acetoxi-2-metoxi-etil)tetrahidrofuran-3-ilo] (compuesto 21e) como un aceite incoloro. EM obs. (ESI+) [(M+NH4)+]: 322.
Preparación de acetato de [(2R,3R,5S)-5-(1-acetoxi-2-metoxi-etil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirim idin-3-il)tetrahidrofuran-3-ilo]

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (368 mg, 2,0 mmol) en ACN (20 ml) se le añadió BSA (1,2 mg, 6,0 mmol). Se agitó la mezcla de reacción a 70 °C durante 0,5 horas en argón para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron acetato de [(2R,3R,5S)-2-acetoxi-5-(1-acetoxi-2-metoxi-etil)tetrahidrofuran-3-ilo] (compuesto 21e, 304 mg, 1,0 mmol) y TMSOTf (666 mg, 3,0 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 320 mg de producto bruto de acetato de [(2R,3R,5S)-5-(1-acetoxi-2-metoxi-etil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 21f), que se usó en la siguiente etapa sin purificación. EM obs. (ESI') [(M-H)-]: 427.
Preparación de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-metoxi-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de acetato de [(2R,3R,5S)-5-(1-acetoxi-2-metoxi-etil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 21 f , preparado anteriormente) en metanol (25 ml) se le añadió K2CO3 (272 mg, 2 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se ajustó la mezcla de reacción hasta pH 7 8 por adición de HOAc (240 mg, 4 mmol), se concentró a vacío y se purificó el residuo y se separó por HPLC preparativa para dar 22,6 mg del ejemplo 21-A y 22,3 mg del ejemplo 21-B como un sólido blanco.
Ejemplo 21-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,92-5,98 (m, 1H), 4,92-4,95 (m, 1H), 4,29-4,37 (m, 1H), 3,72 3,79 (m, 1H), 3,42-3,51 (m, 2H), 3,38 (s, 3H), 2,56-2,68 (m, 1H), 1,91-2,01 (m, 1H). EM obs. (ESI') [(M-H)-]: 343. Ejemplo 21-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,91-5,96 (m, 1H), 4,92-4,95 (m, 1H), 4,20-4,28 (m, 1H), 3,85 3,91 (m, 1H), 3,49-3,56 (m, 1H), 3,39-3,45 (m, 1H), 3,37 (s, 3H), 2,63-2,73 (m, 1H), 1,95-2,03 (m, 1H). EM obs. (ESI-) [(M-H)-]: 343.
Ejemplo 22
5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2 -il]propilo

A una solución de acetato de [(2R,3R,5S)-5-(1-acetoxipropil)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-3-ilo] (compuesto 1g, 7,0 g, 16,9 mmol) en metanol (200 ml) se le añadió K2CO3 (1,18 g, 8,5 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se ajustó la mezcla de reacción hasta pH 6,0 por adición de HOAc (1,2 g, 17 mmol), se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 2,8 g de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo (compuesto 22a) como un sólido amarillo. EM obs. (ESI-) [(M-H)-]: 369.
Preparación de acetato de 1-[(2S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-oxotetrahidrofuran-2 -il]propilo

A una solución agitada de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo (compuesto 22a, 2,8 g, 7,6 mmol) en THF (100 ml) se le añadió peryodinano de Dess-Martin (4,8 g, 11,3 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se filtró la solución resultante y se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM 1:10) para dar 2,8 g de producto bruto de acetato de 1-[(2S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-oxo-tetrahidrofuran-2-il]propilo (compuesto 22b). EM obs. (ESI') [(M-H)-]: 367.
Preparación de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2 -il]propilo

A una solución agitada de acetato de 1-[(2S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-oxotetrahidrofuran-2-il]propilo (compuesto 22b, 2,8 g, 7,6 mmol) en THF (50 ml) se le añadió hidruro de litio y tri-tercbutoxialuminio (1 M en THF, 15 ml, 15 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se desactivó la solución resultante por solución saturada de NH4Cl y se filtró. Se concentró el filtrado a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM 1:10) para dar 1,76 g de
producto bruto de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo (compuesto 22c). EM obs. (ESI') [(M-H)']: 369. (Refiérase a Tetrahedron 1984, 40, 125 135).
Preparación de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(trifluorom etilsulfoniloxi)tetrahidrofuran-2 -il]propilo

A una solución agitada de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo (compuesto 22c, 1,76 g, 0,46 mmol) en DCM (30 ml) se le añadió piridina (154 mg, 1,9 mmol) y anhídrido trifluorometanosulfónico (197 mg, 0,7 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se lavó la solución resultante con agua, salmuera, se secó sobre Na2SÜ4. Se concentró la capa orgánica a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 420 mg de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]propilo (compuesto 22d). EM obs. (ESI-) [(M-H)-]: 502.
Preparación de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metilsulfaniltetrahidrofuran-2 -il]propilo

A una solución agitada de acetato de 1-[(2S,4S,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]propilo (compuesto 22d, 420 mg, 0,83 mmol) en DMF (7 ml) se le añadió tiometóxido de sodio (84 mg, 1,2 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la solución resultante con EtOAc, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar producto bruto de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metilsulfanil-tetrahidrofuran-2-il]propilo (compuesto 22e), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI-) [(M-H)-]: 399.
Preparación de 5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona
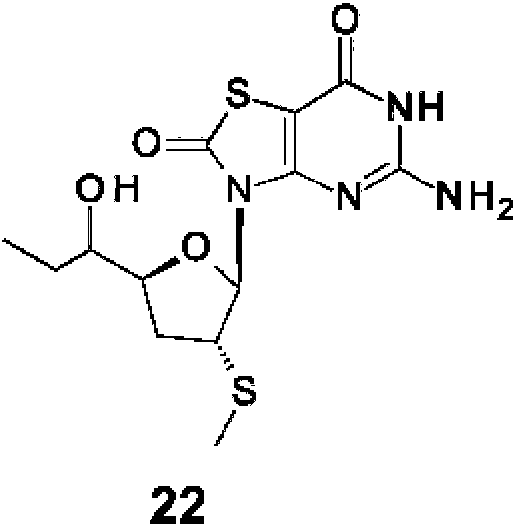
A una solución de acetato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metilsulfaniltetrahidrofuran-2-il]propilo (compuesto 22e, 200 mg, 0,5 mmol) en metanol (25 ml) se le añadió K2CO3 (272 mg, 2 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se ajustó la mezcla de reacción hasta pH 7,0 por adición de HOAc (120 mg, 2 mmol), se concentró a vacío y se purificó el residuo y se separó por HPLC preparativa para dar 4,7 mg del ejemplo 22 -A y 1,8 mg del ejemplo 22 -B como un sólido blanco.
Ejemplo 22-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,09-6,16 (m, 1H), 4,09-4,16 (m, 1H), 3,97-4,06 (m, 1H), 3,47 3,57 (m, 1H), 2,61-2,72 (m, 1H), 2,13 (s, 3H), 1,95-2,06 (m, 1H), 1,41-1,61 (m, 2H), 1,01 (t, J = 7,2Hz, 3H). EM obs. (ESI-) [(M-H)-]: 357.
Ejemplo 22-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,05-6,12 (m, 1H), 4,01-4,11 (m, 2H), 3,65-3,74 (m, 1H), 2,67 2,78 (m, 1H), 2,12 (s, 3H), 1,98-2,05 (m, 1H), 1,52-1,65 (m, 1H), 1,31-1,47 (m, 1H), 1,01 (t, J = 7,2Hz, 3H). EM obs. (ESI-) [(M-H)-]: 357.
Ejemplo 23
5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona
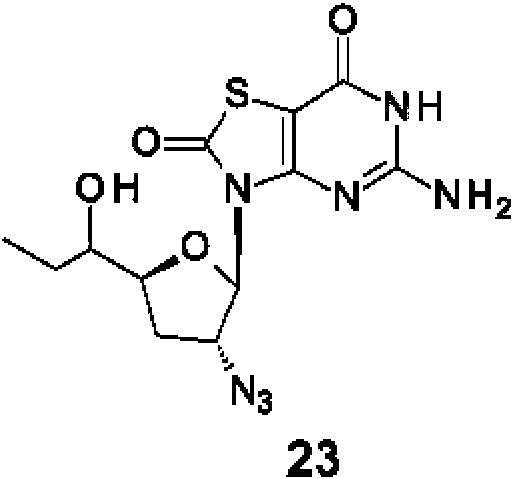
Se preparó el compuesto del título de forma análoga al ejemplo 22 , usando acida de sodio en lugar de tiometóxido de sodio. Se purificó el ejemplo 23 y se separó por HPLC preparativa para dar el ejemplo 23-A y el ejemplo 23-B como un sólido blanco.
Ejemplo 23-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,99 (m, 1H), 4,95-5,00 (m, 1H), 4,06-4,14 (m, 1H), 3,47 3,55 (m, 1H), 2,63-2,75 (m, 1H), 2,03-2,12 (m, 1H), 1,51-1,61 (m, 1H), 1,43-1,51 (m, 1H), 1,01 (t, J = 7,40 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 352.
Ejemplo 23-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,92-5,97 (m, 1H), 4,94-4,98 (m, 1H), 4,02-4,10 (m, 1H), 3,62 3,68 (m, 1H), 2,72-2,80 (m, 1H), 2,06-2,15 (m, 1H), 1,53-1,68 (m, 1H), 1,33-1,45 (m, 1H), 1,00 (t, J = 7,40 Hz, 3H). EM obs. (ESI-) [(M-H)-]: 352.
Ejemplo 24
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxialil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 1, usando bromuro de etilenmagnesio en lugar de etilmagnesio. Se purificó el ejemplo 24 y se separó por HPLC preparativa para dar el ejemplo 24-A y el ejemplo 24-B como un sólido blanco.
Ejemplo 24-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,96-6,01 (m, 1H), 5,86-5,96 (m, 1H), 5,33-5,43 (m, 1H), 5,16 5,22 (m, 1H), 4,91-4,95 (m, 1H), 4,16-4,23 (m, 1H), 4,09-4,16 (m, 1H), 2,53-2,63 (m, 1H), 1,87-1,95 (m, 1H). EM obs. (ESI-) [(M-H)-]: 357.
Ejemplo 24-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,93-5,95 (m, 1H), 5,84-5,92 (m, 1H), 5,37 (td, J = 1,76, 17,32 Hz, 1H), 5,17-5,23 (m, 1H), 4,93-4,99 (m, 1H), 4,29 (s.a., 1H), 4,22 (d, J = 4,52 Hz, 1H), 2,57-2,68 (m, 1H), 1,88 1,98 (m, 1H). EM obs. (ESI') [(M-H)']: 357.
Ejemplo 25
5-amino-3-((2R,3R,5S)-3-acido-5-((S)-1-hidroxietil)tetrahidrofuran-2-il)tiazolo[4,5-d]pirimidin-2,7(3H,6H)-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de (2R,3S,5R)-5-[[ferc-butil(difenil)silil]oximetil]tetrahidrofuran-2,3,4-triol

A una solución agitada de D-arabinosa (50 g, 0,33mol) en DMF (500 ml) se le añadió imidazol (45 g, 0,66 mol) y terc-butilclorodifenilsilano (109 g, 0,4 mol). Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la solución resultante con EtOAc (2000 ml), se lavó con agua, salmuera y se secó sobre Na2SO4. Se concentró la capa orgánica a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:11) para dar 33 g de (2R,3S,5R)-5-[[terc-butil(difenil)silil]oximetil]tetrahidrofuran-2,3,4-triol (compuesto
25a).
Compuesto 25a: RMN de 1H (400 MHz, CDCI3) 5 ppm: 7,66-7,75 (m, 4H), 7,38-7,52 (m, 6H), 5,44-5,50 (m, 1H), 4,30 (d, J = 1,76 Hz, 1H), 4,25 (m, 1H), 4,09 (m, 1H), 3,93-4,00 (m, 1H), 3,84-3,89 (m, 1H), 3,74-3,78 (m, 1H), 1,02-1,09 (m, 9H).
Preparación de (3aS,5R,6aS)-5-[[ferc-butil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3] dioxol-6 -ol

A una solución agitada de (2R,3S,5R)-5-[[ferc-butil(difenil)silil]oximetil]tetrahidrofuran-2,3,4-triol (compuesto 25a, 33 g, 85 mmol) en acetona (250 ml) se le añadió 2,2-dimetoxipropano (13,2 g, 127 mmol) y ácido p-toluensulfónico (1 g, 5,8 mmol). Después de agitarse a 60 °C durante 2 horas, se ajustó la solución resultante hasta pH 7,0 por adición de solución saturada de NaHCO3 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:11) para dar 20 g de (3aS,5R,6aS)-5-[[ferc-butil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-ol (compuesto 25b).
Compuesto 25b: RMN de 1H (400 MHz, CDCla) 5 ppm: 7,66-7,71 (m, 4H), 7,41 (d, J = 7,78 Hz, 6H), 5,87-5,93 (m, 1H), 4,55-4,60 (m, 1H), 4,42-4,49 (m, 1H), 4,04-4,10 (m, 1H), 3,80-3,89 (m, 2H), 1,35 (s, 3H), 1,31 (s, 3H), 1,09 (s, 9H).
Preparación de [(3aS,5R,6aS)-5-[[ferc-butil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-il]oxi-fenoxi-metanotiona

A una solución de (3aS,5R,6aS)-5-[[ferc-butil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-ol (compuesto 25b, 23 g, 50 mmol) en DCM (200 ml) se le añadió clorometanotioato de O-fenilo (10,3 g, 60 mmol) y piridina (7,9 g, 100 mmol) en DCM. Después de agitarse a temperatura ambiente durante la noche, se lavó la mezcla resultante con salmuera, se secó sobre Na2SO4, y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 20 g de [(3aS,5R,6aS)-5-[[fercbutil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-il]oxi-fenoxi-metanotiona (compuesto 25c). EM obs. (ESI+) [(M+NH4)+]: 582.
Preparación de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metoxi-íerc-butildifenil-silano
A una solución de [(3aS,5R,6aS)-5-[[ferc-butil(difenil)silil]oximetil]-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-6-il]oxi-fenoxi-metanotiona (compuesto 25c, 17 g, 30 mmol) en tolueno (150 ml) se le añadió tri(trimetilsilil)silano(16,4 g, 66 mmol) y azodiisobutironitrilo (98 mg, 0,6 mmol), se calentó la mezcla a 130 °C en nitrógeno durante 3 horas. Después de que se completara la reacción, se concentró la reacción a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:10) para dar 11 g de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metoxi-terc-butil-difenil-silano (compuesto 25d).
Compuesto 25d: RMN de 1H (400 MHz, CDCb) 5 ppm: 7,70 (qd, J = 1,89, 5,87 Hz, 4H), 7,37-7,46 (m, 6H), 5,78-5,83 (m, 1H), 4,72-4,78 (m, 1H), 4,27-4,35 (m, 1H), 3,84 (d, J = 6,78 Hz, 2H), 2,25-2,33 (m, 1H), 2,13-2,19 (m, 1H), 1,35 (s, 3H), 1,30 (s, 3H), 1,08 (s, 9H).
Preparación de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metanol

A una solución de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metoxi-terc-butil-difenilsilano (compuesto 25d, 11 g, 26,6 mmol) en THF (100 ml) se le añadió solución TBAf (1 M en THF, 6 ml, 6 mmol) con agitación. Después de agitarse a temperatura ambiente durante 4 horas, se lavó la solución de reacción con solución saturada de NH4Cl, se secó sobre Na2SO4 , se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:2) para dar 5,8 g de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6atetrahidrofuro[2,3-d][1,3]dioxol-5-il]metanol (compuesto 25e).
Compuesto 25e: RMN de 1H (400 MHz, CDCla) 5 ppm: 5,81-5,89 (m, 1H), 4,75-4,83 (m, 1H), 4,32-4,41 (m, 1H), 3,81 3,91 (m, 1H), 3,60-3,70 (m, 1H), 2,21-2,28 (m, 1H), 1,97-2,09 (m, 1H), 1,57-1,59 (s, 6H).
Preparación de (3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído
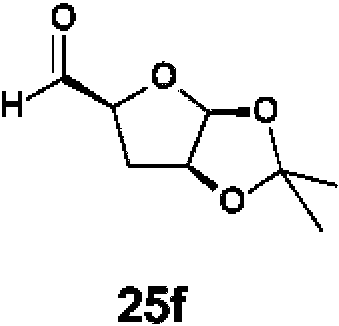
A una solución agitada de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metanol (compuesto 25e, 2 g, 11,5 mmol) en THF (20 ml) se le añadió peryodinano de Dess-Martin (7,2 g, 17,2 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se filtró la solución resultante y se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 1,2 g de (3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído (compuesto 25f). EM obs. (ESI+) [(M+NH4)+]: 190.
Preparación de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etanol
A una solución de (3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tctrahidrofuro[2,3-d][1,3]dioxol-5-carbaldehído (compuesto 25f, 800 mg, 1 mmol) en THF (20 ml) se le añadió bromuro de metilmagnesio (2 M en THF, 0,7 ml, 1,4 mmol) a -20 °C en argón. Después de agitarse a -20 °C durante 20 horas, se desactivó la reacción por solución saturada de NH4Cl, se extrajo con EtOAc (30 ml) tres veces. Se combinaron las capas orgánicas y se concentró a vacío para dar 400 mg de producto bruto de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etanol (compuesto 25g), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+n H4)+]: 206.
Preparación de benzoato de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etilo

A una solución enfriada de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etanol (compuesto 25g, 200 mg, 1,06 mmol) en DCM ( 8 ml) se le añadió cloruro de benzoílo (178 mg, 1,28 mmol) y DMAP (259 mg, 2 mmol). Después de la adición, se calentó la mezcla de forma natural hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se diluyó la mezcla resultante con EtOAc y se lavó con una solución acuosa saturada de NH4CL Se secó la capa orgánica sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 170 mg de benzoato de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etilo (compuesto 25h).
Compuesto 25h: RMN de 1H (400 MHz, CDCla) 5 ppm: 8,05 (s, 2H), 7,56-7,63 (m, 1H), 7,46 (s, 2H), 5,85 (d, J = 4,02 Hz, 1H), 5,38-5,52 (m, 1H), 4,73-4,83 (m, 1H), 4,13-4,25 (m, 1H), 2,22 (d, J = 4,77 Hz, 2H), 1,62 (s, 3H), 1,46 (d, J = 6,27 Hz, 3H), 1,34 (s, 3H).
Preparación de benzoato de 1-[(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]etilo

Se agitó una solución de benzoato de 1-[(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]etilo (compuesto 25h, 170 mg, 18 mmol) en la solución de HCl (0,1 N en MeOH, 3 ml) a temperatura ambiente durante la noche. Se neutralizó la mezcla resultante por hidróxido de amonio y se concentró a vacío. Se suspendió el residuo en EtOAc y a continuación se filtró, se concentró el filtrado a vacío para dar 148 mg de producto bruto de benzoato de 1-[(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]etilo (compuesto 25i), que se usó en la siguiente etapa directamente. EM obs. (ESI+) [(M+H)+]: 267.
Preparación de benzoato de 1-[(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]etilo
A una solución de benzoato de 1-[(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]etilo (compuesto 25i, 483 mg) y DMAP (885 mg, 7,3 mmol) en DCM (20 ml) se le añadió anhídrido trifluorometanosulfónico (665 mg, 2,36 mmol) a 0 °C. Después de agitarse a temperatura ambiente durante 0,5 h, se desactivó la reacción por solución saturada de NaHCO3 y se extrajo con DCM tres veces. Se combinaron las capas orgánicas, se secó sobre Na2SO4 y se concentró a vacío para dar 740 mg de producto bruto de benzoato de 1-[(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]etilo (compuesto 25j) como un aceite, que se usó en la siguiente etapa directamente sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 400.
Preparación de benzoato de 1-[(2S,4R)-4-acido-5-metoxi-tetrahidrofuran-2-il]etilo

A una solución de benzoato de 1-[(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]etilo (compuesto 25j, 400 mg, 1 mmol) en DMF (2 ml) se le añadió acida de sodio (65 mg, 1,05 mmol) a temperatura ambiente y se agitó la mezcla a temperatura ambiente durante 16 horas. Se dividió la mezcla de reacción entre EtOAc y H2O, se separó la capa orgánica y se extrajo la capa acuosa con EtOAc dos veces. Se combinaron las capas orgánicas, se lavó con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 600 mg de producto bruto de benzoato de 1-[(2S,4R)-4-acido-5-metoxi-tetrahidrofuran-2-il]etilo (compuesto 25k), que se usó en la siguiente etapa sin purificación adicional.
Preparación de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo [4,5-d]pirimidin-3-il)-4-acidotetrahidrofuran-2 -il]etilo

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (186 mg, 1 mmol) en ACN (10 ml) se le añadió BSA (630 mg, 3 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de 1-[(2S,4R)-4-acido-5-metoxi-tetrahidrofuran-2-il]etilo (compuesto 25k, 300 mg, 1,0 mmol) y TMSOTf (1,15 g, 5 mmol) en secuencia. Después de calentarse a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 600mg de producto bruto de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-acido-tetrahidrofuran-2-il]etilo (compuesto 25I), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 442.
Preparación de 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxietil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

A una solución de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-acidotetrahidrofuran-2-il]etilo (compuesto 25I, 600 mg, bruto) en MeOH (8 ml) se le añadió K2CO3 (187 mg, 1,4 mmol). Después de agitarse a temperatura ambiente durante 5 horas, se concentró la mezcla de reacción a vacío y se purificó el residuo por HPLC preparativa para dar 30 mg de 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxietil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 25) como un sólido blanco.
Ejemplo 25: RMN de 1H (400 MHz, cfó-DMSO) 5 ppm: 11,26 (s, 1H), 6,96 (s.a., 2H), 5,76 (d, J = 3,3 Hz, 1H), 5,03 (dt, J = 7,4, 2,8 Hz, 1H), 4,71 (d, J =5,0 Hz, 1H), 3,80 (dt, J = 9,2, 6,1 Hz, 1H), 3,57-3,67 (m, 1H), 2,53-2,68 (m, 1H), 2,04 (ddd, J = 13,2, 6,0, 2,6 Hz, 1H), 1,05 (d, J = 6,3 Hz, 3H). EM obs. (ESI') [(M-H)-]: 338.
Ejemplo 26
3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de benzoato de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo

A una solución agitada de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol bruto (compuesto 1e, 8,08 g, 40,0 mmol) y piridina (16,1 ml, 200 mmol) en DCM se le añadió cloruro de benzoílo (5,0 ml, 43,0 mmol) gota a gota a 0 °C. Después de la adición, se calentó la mezcla hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se lavó la mezcla resultante con ácido clorhídrico 1 N, salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 6,86 g de benzoato de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6atetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo (compuesto 26a).
Preparación de benzoato de 1-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]propilo

A una solución agitada de benzoato de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-iljpropilo (compuesto 26a, 6,73 g, 22,0 mmol) y anhídrido acético (11 ml) en ácido acético (44 ml) y cloroformo (11 ml) se le añadió ácido sulfúrico concentrado (200 ul) gota a gota. Después de agitarse a temperatura ambiente durante la noche, se diluyó la mezcla resultante con EtOAc (100 ml) y se lavó con una solución acuosa saturada de NaHCO3 (100 ml) tres veces. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:10) para dar benzoato de 5,1 g 1-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]propilo (compuesto 26b) como un aceite viscoso.
Preparación de benzoato de 1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d] pirim idin-3-il)tetrahidrofuran-2 -il]propilo

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (2,82 g, 16,8 mmol) en ACN (100 ml) se le añadió BSA (10,4 ml, 42 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de 1-[(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]propilo (compuesto 26b, 4,9 g, 14,0 mmol) y TMSOTf (4,7 ml, 2,321mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM 1:200) para dar 5,27 g de producto bruto de benzoato de 1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo (compuesto 26c) como un sólido amarillo claro. EM obs. (ESI+) [(M+H)+]: 475.
Preparación de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2 -il]propilo
A una solución de benzoato de 1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo (compuesto 26c, 4,98 g, 10,5 mmol) en metanol (105 ml) se le añadió K2CO3 (1,38 g, 10,0 mmol). Después de agitarse a temperatura ambiente durante 1 hora, se ajustó la reacción hasta pH 7,0 con HOAc (1,2 g, 20,0 mmol), se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM 1:200) para dar 4,5 g de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo (compuesto 26d) como un sólido marrón claro. EM obs. (ESI+) [(M+H)+]: 433.
Preparación de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2 -il]propilo

A una solución de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo (compuesto 26d, 4,32 g, 10,0 mmol) en DCM (60 ml) se le añadió DMAP (2,44 g, 20 mmol) y clorometanotioato de O-fenilo (1,6 ml, 12,0 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se concentró la solución resultante a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:10 a 1:1) para dar 1,9 g de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2-il]propilo (compuesto 26e). EM obs. (ESI+) [(M+H)+]: 569.
Preparación de benzoato de 1-[(2S,4R,5R)-4-alil-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2 -il]propilo

Se desgasificó una mezcla de benzoato de 1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2-il]propilo (compuesto 26e, 1,14 g, 2,0 mmol), 2,2'-azobisisobutironitrilo (168 mg, 1 mmol) y alil(tributil)stannano (3,08 ml, 10 mmol) en tolueno anhidro (15 ml) con argón y a continuación se calentó con agitación a 80 °C durante 4 horas. Se agitó la mezcla resultante con solución saturada de NH4F a temperatura ambiente durante 2 horas y se extrajo con DCM dos veces. Se secaron las capas orgánicas combinadas sobre Na2SO4, se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:3) para dar 820 mg de benzoato de 1-[(2S,4R,5R)-4-alil-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo (compuesto 26f) como un sólido marrón. EM obs. (ESI+) [(M+H)+]: 457.
Preparación de 3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona

A una solución de benzoato de 1-[(2S,4R,5R)-4-alil-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo (compuesto 26f, 800 mg, 1,75 mmol) en metanol (25 ml) se le añadió K2CO3 (483 mg, 3,5 mmol). Después de agitarse a temperatura ambiente durante 12 horas, se diluyó la reacción por solución saturada de NH4Cl y se extrajo con DCM. Se combinaron las capas orgánicas y se concentró a vacío. Se purificó el residuo por HPLC preparativa para dar 200 mg de 3-[(2R,3^,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6wtiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 26). Se separaron además 100 mg del ejemplo 26 por cromatografía de fluidos supercríticos (SFC) para dar 32,0 mg del ejemplo 26-A y 30,8 mg del ejemplo 26 -B como un sólido blanco.
Ejemplo 26: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,32 (s.a., 1H), 6,89 (s.a., 2H), 5,64-5,81 (m, 2H), 4,92-5,10 (m, 2H), 4,45-4,63 (m, 1H), 3,74-3,93 (m, 1H), 3,37-3,48 (m, 1H), 2,97-3,14 (m, 1H), 2,12-2,39 (m, 3H), 1,61-1,79 (m, 1H), 1,36-1,52 (m, 1H), 1,14-1,29 (m, 1H), 0,88 (q, J = 7,36 Hz, 3H) EM obs. (ESI+) [(M+H)+]: 353.
Ejemplo 26-A: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,24 (s.a., 1H), 6,87 (s.a., 2H), 5,66-5,80 (m, 2H), 4,95-5,11 (m, 2H), 4,45 (d, J = 6,53 Hz, 1H), 3,82-3,93 (m, 1H), 3,39-3,49 (m, 1H), 2,95-3,06 (m, 1H), 2,14-2,30 (m, 3H), 1,66 (ddd, J = 4,89, 7,09, 12,11 Hz, 1H), 1,34-1,49 (m, 1H), 1,18-1,33 (m, 1H), 0,88 (t, J = 7,28 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 353.
Ejemplo 26-B: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,28 (s.a., 1H), 6,87 (s.a., 2H), 5,65-5,79 (m, 2H), 4,92-5,10 (m, 2H), 4,57 (d, J = 4,77 Hz, 1H), 3,78 (q, J = 6,78 Hz, 1H), 3,39-3,50 (m, 1H), 3,03-3,15 (m, 1H), 2,35 (ddd, J = 5,77, 8,66, 12,42 Hz, 1H), 2,16 (t, J = 7,15 Hz, 2H), 1,73 (td, J = 7,34, 12,42 Hz, 1H), 1,42-1,55 (m, 1H), 1,14-1,28 (m, 1H), 0,87 (t, J = 7,28 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 353.
Ejemplo 27
Ejemplo 27-A y ejemplo 27-B: 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona y 5-amino-3-[(2R,3R,5S)-5-[(1R)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona
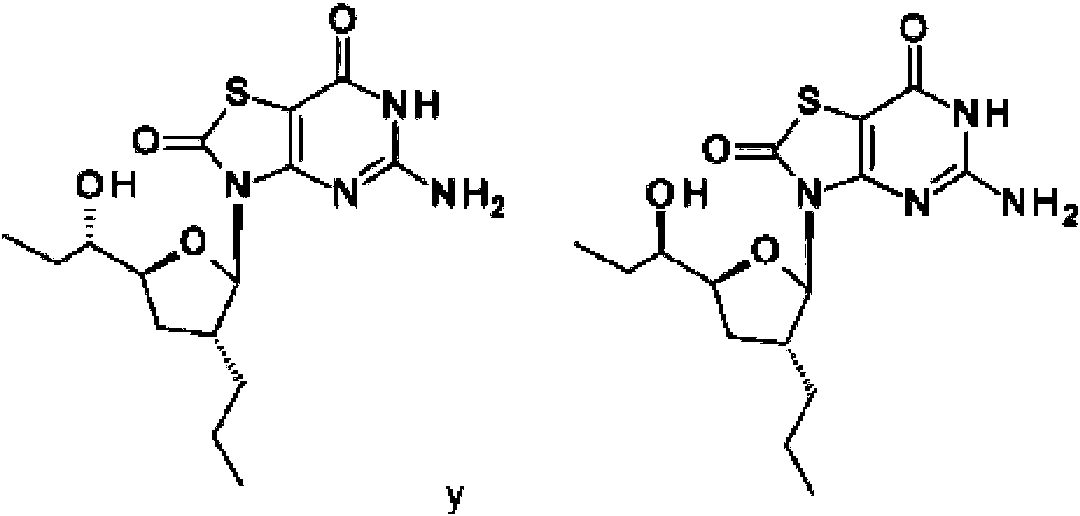
Se agitó una solución del ejemplo 26-A (50 mg, 0,15 mmol) en metanol con PtO2 (10 mg) en atmósfera de hidrógeno durante 4 horas a temperatura ambiente. Se filtró la mezcla de reacción para retirar PtO2. Se concentró el filtrado a vacío y se purificó el residuo y se separó por HPLC preparativa para dar 32,1 mg del ejemplo 27-A como un sólido blanco.
Ejemplo 27-A: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 10,86-11,27 (s.a., 1H), 6,81-7,00 (s.a., 2H), 5,64-5,72 (m, 1H), 4,41-4,49 (m, 1H), 3,82-3,91 (m, 1H), 2,81-2,95 (m, 1H), 2,16-2,30 (m, 1H), 1,57-1,69 (m, 1H), 1,34-1,50 (m, 4H), 1,22-1,34 (m, 4H), 0,79-0,94 (m, 6H). EM obs. (ESI+) [(M+H)+]: 355.
Se preparó el ejemplo 27-B de forma análoga al ejemplo 27-A, usando el ejemplo 26-B en lugar del ejemplo 26-A. Ejemplo 27-B: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,18-11,47 (s.a., 1H), 6,79-7,02 (s.a., 2H), 5,61-5,75 (m, 1H), 4,57 (d, J = 4,27 Hz, 1H), 3,69-3,85 (m, 1H), 3,45 (m, 1H), 2,94-3,08 (m, 1H), 2,35 (m, 1H), 1,71 (m, 1H), 1,42 1,55 (m, 1H), 1,31-1,42 (m, 2H), 1,12-1,31 (m, 3H), 0,69-0,95 (m, 6H). EM obs. (ESI+) [(M+H)+]: 355.
Ejemplo 28
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

A una solución de ácido (S)-2-aminopentanodioico (100 g, 680 mmol) en H2O (500 ml) se le añadió HCl (140 ml, 1,6 mol) y a continuación NaNO2 (70,4 g, 1,02 mol) en H2O (100 ml) se le añadió lentamente a -5 °C- 0 °C, y se agitó la mezcla de reacción a temperatura ambiente durante 24 horas. Se concentró el disolvente a vacío por debajo de 50 °C. Se suspendió el residuo en EtOAc (500 ml) y se filtró. Se secó el filtrado sobre Na2SO4 y se concentró a vacío para dar 56 g de producto bruto de ácido (2S)-5-oxotetrahidrofuran-2-carboxílico (compuesto 28a) como un aceite amarillo, que se usó en la siguiente etapa sin purificación adicional.
Preparación de cloruro de (2S)-5-oxotetrahidrofuran-2-carbonilo

A una solución de ácido (2S)-5-oxotetrahidrofuran-2-carboxílico (compuesto 28a, 70 g, 539 mmol) y una gota de DMF en DCM anhidro (500 ml) se le añadió dicloruro de oxalilo (137 g, 1,07 mol) gota a gota. Se agitó la mezcla de reacción a temperatura ambiente durante 3 horas. A continuación se concentró la reacción a vacío para dar 70 g de producto bruto de cloruro de (2S)-5-oxotetrahidrofuran-2-carbonilo (compuesto 28b), que se usó en la siguiente etapa sin purificación adicional.
Preparación de (5S)-5-propanoiltetrahidrofuran-2-ona

A una solución de cloruro de (25)-5-oxotetrahidrofuran-2-carbonilo (compuesto 28b, 70 g, 473 mmol) en THF seco (400 ml) se le añadió bromuro de etilmagnesio (173 ml, 520 mmol, 3M en THF) lentamente a -78 °C en N2. Después de la adición, se agitó la mezcla de reacción a -78 °C durante otras 2 horas. A continuación se desactivó la mezcla con solución saturada de NH4Cl y se extrajo con EtOAc (500 ml) dos veces. Se concentraron las capas orgánicas combinadas a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:7 a 1:3) para dar 35 g de (5S)-5-propanoiltetrahidrofuran-2-ona (compuesto 28c) como un aceite amarillo claro.
Preparación de (5S)-5-(1-hidroxipropil)tetrahidrofuran-2-ona

A una solución de (5S)-5-propanoiltetrahidrofuran-2-ona (compuesto 28c, 35 g, 246,5 mmol) se le añadió L-selectrida (320 ml, 320 mmol, 1 M en t Hf ) a -78 °C en N2. Después de la adición, se agitó la mezcla de reacción a -78 °C durante 2 horas. A continuación se desactivó la mezcla de reacción con HCl 2 N (200 ml) y se extrajo con EtOAc (400 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera (100 ml), se concentró a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:7 e 1:3) para dar 20 g de (5S)-5-(1-hidroxipropil)tetrahidrofuran-2-ona (compuesto 28d) como un aceite amarillo. (Refiérase a Eur. J. Med. Chem. 1997, 32, 617-623).
Preparación de (5S)-5-[(1S)-1-[ferc-butil(difenil)silil]oxipropil]tetrahidrofuran-2-ona y (5S)-5-[(1R)-1-[íercbutil(difenil)silil]oxipropil]tetrahidrofuran-2-ona

A una solución de (5S)-5-(1-hidroxipropil)tetrahidrofuran-2-ona (compuesto 28d, 9 g, 62,5 mmol) en DMF (100 ml) se le añadió terc-butilclorodifenilsilano (42,8 g, 156 mmol) e imidazol (10,6 g, 156 mmol) en N2. Después de agitarse a 50 °C durante 12 horas, se diluyó la mezcla con agua y se extrajo con EtOAc. Se lavaron las capas orgánicas combinadas con salmuera (100 ml) y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:3) para dar 18 g de (5S)-5-[1-[tercbutil(difenil)silil]oxipropil]tetrahidrofuran-2-ona. Se purificaron además 10 g de la mezcla y se separó por SFC para dar 5,6 g de (5S)-5-[(1S)-1-[terc-butil(difenil)silil]oxipropil]tetrahidrofuran-2-ona (compuesto 28e-S) y 2 g de (5S)-5-[(1R)-1-[terc-butil(difenil)silil]oxipropil]tetrahidrofuran-2-ona (compuesto 28e-R). (Refiérase a Tetrahedron. 1997, 53, 6281 6294).
Compuesto 28e-S: RMN de 1H (CDCb 400 MHz) 5 ppm: 7,73-7,69 (m, 4H), 7,46- 7,39 (m, 6H), 4,56 (m, 1H), 3,66 (m, 1H), 2,64-2,47 (m, 2H), 2,20-2,15 (m, 1H), 1,72-1,67 (m, 1H), 1,47-1,42 (m, 1H), 1,15-1,05 (m, 9H), 0,82-0,73 (t, 3H).
Compuesto 28e-R: RMN de 1H (CDCb 400 MHz) 5 ppm: 7,72-7,69 (m, 4H), 7,48- 7,39 (m, 6H), 4,54 (m, 1H), 3,92 (m, 1H), 2,60-2,47 (m, 2H), 2,38-2,31 (m, 1H), 2,19-2,12 (m, 1H), 1,50-1,41 (m, 1H), 1,05 (s, 9H), 0,74-0,72 (t, 3H).
Preparación de (3R,5S)-5-[(1S)-1-[íerc-butil(difenil)silil]oxipropil]-3-metil-tetrahidrofuran-2-ona

A una solución de (5S)-5-[(1S)-1-[terc-butil(difenil)silil]oxipropil]tetrahidrofuran-2-ona (compuesto 28e-S, 3,0 g, 7,8 mmol) en THF (60 ml) a -78 °C se le añadió diisopropilamida de litio (2 M en THF, 5,9 ml, 11,8 mmol) gota a gota. Después de la adición, se agitó la reacción a -78 °C durante 1 hora. A la mezcla se le añadió yodometano (5,5 g, 39 mmol) y se agitó la mezcla a -78 °C durante 1 hora adicional. Se desactivó la mezcla con solución saturada de NH4Cl (40 ml), se extrajo con EtOAc (100 ml) dos veces. Se combinaron las capas orgánicas, se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:10) para dar 2,7 g de (3R,5S)-5-[(1S)-1-[tercbutil(difenil)silil]oxipropil]-3-metil-tetrahidrofuran-2-ona (compuesto 28f) como un aceite incoloro. (Refiérase a Tetrahedron. 1997, 53, 6281-6294).
Preparación de (3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-ona
A una solución de (3R,5S)-5-[(1S)-1-[terc-butil(difenil)silil]oxipropil]-3-metil-tetrahidrofuran-2-ona (compuesto 28f, 2,7 g, 6,8 mmol) en Th F (10 ml) se le añadió TBAF (1 M en THF, 13,6 ml, 13,6 mmol) y se agitó la mezcla a temperatura ambiente durante 12 horas. A continuación se concentró la mezcla a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:30 a 1:20) para dar 1,02 g de (3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-ona (compuesto 28g) como un aceite incoloro.
Preparación de benzoato de [(1S)-1-[(2S,4R)-4-metil-5-oxo-tetrahidrofuran-2-il]propilo]

A una solución de (3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-ona (compuesto 28g, 1,0 g, 6,3 mmol), TEA (3,2 g, 31,2 mmol) y Dm A p (100 mg) en DCM (50 ml) se le añadió cloruro de benzoílo (1,8 g, 12,6 mmol) lentamente a 0 °C. Se agitó la mezcla a 25 °C durante 4 horas y a continuación se desactivó por solución saturada de NaHCO3, se extrajo con EtOAc (100 ml) dos veces. Se combinaron las capas orgánicas, se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:5) para dar 1,4 g de benzoato de [(1 S)-1-[(2S,4R)-4-metil-5-oxotetrahidrofuran-2-il]propilo] (compuesto 28h) como un aceite incoloro.
Preparación de benzoato de [(1S)-1-[(2S,4R)-5-hidroxi-4-metil-tetrahidrofuran-2-il]propilo]

A una solución de benzoato de [(1 S)-1-[(2S,4R)-4-metil-5-oxo-tetrahidrofuran-2-il]propilo] (compuesto 28h, 1,3 g, 5,0 mmol) en THF (100 ml) se le añadió hidruro de diisobutilaluminio (11 ml, 11 mmol) gota a gota a -78 °C y se agitó la mezcla a -78 °C durante 2 horas. Se desactivó la mezcla por solución saturada de NH4Cl (5 ml) y se extrajo con EtOAc (100mL) dos veces. Se combinaron las capas orgánicas, se lavó con salmuera (50ml), se secó sobre Na2SO4 y se concentró a vacío para dar 1,2 g de producto bruto de benzoato de [(1 S)]-1-[(2S,4R)-5-hidroxi-4-metiltetrahidrofuran2-il]propilo] (compuesto 28i), que se usó en la siguiente etapa sin purificación adicional.
Preparación de benzoato de [(1S)-1-[(2S,4R)-5-acetoxi-4-metil-tetrahidrofuran-2-il]propilo]
A una solución de benzoato de [(1 S)-1-[(2S,4R)-5-hidroxi-4-metil-tetrahidrofuran-2-il]propilo] (compuesto 28i, bruto, 1,2 g, 4,5 mmol) en piridina (60 ml) se le añadió anhídrido de ácido acético (0,918 g, 9 mmol) y d Ma P (200 mg) con agitación. Después de agitarse 25 °C durante 2 horas, se desactivó la mezcla con solución saturada de NaHCO3 y se extrajo con EtOAc (40 ml). Se combinaron las capas orgánicas, se lavó con salmuera (50 ml), se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo de 1:20 a 1:5) para dar 1,0 g de benzoato de [(1 S)-1-[(2S,4R)-5-acetoxi-4-metil-tetrahidrofuran-2-il]propilo] (compuesto 28j) como un aceite incoloro.
Preparación de benzoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metiltetrahidrofuran-2-il]propilo] (compuesto 28k-A) y benzoato de [(1R)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirim idin-3-il)-4-metil-tetrahidrofuran-2-il]propilo] (compuesto 28k-B)

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (872 mg, 4,7 mmol) en ACN (20 ml) se le añadió BSA (913,5 mg, 4,5 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 2 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de [(1 S)-1-[(2S,4R)-5-acetoxi-4-metil-tetrahidrofuran-2-il]propilo] (compuesto 28j, 294 mg, 0,95 mmol) y TMSOTf (1,44 g, 6,6 mmol) en secuencia. Después de agitarse con agitación a 20 °C durante 14 h, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo y se separó por HPLC preparativa para dar 9,7 mg de benzoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metiltetrahidrofuran-2-il]propilo] (compuesto 28k-A) y 8,4 mg de benzoato de [(1R)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-metil-tetrahidrofuran-2-il]propilo] (compuesto 28k-B) como un sólido blanco.
Compuesto 28k-A: RMN de 1H (400 MHz, CD3OD) 5 ppm: 7,97-8,05 (m, 2H), 7,55-7,62 (m, 1H), 7,41-7,48 (m, 2H), 5,77 (d, J = 4,14 Hz, 1H), 5,19 -5,27 (m, 1H), 4,35 (dt, J = 8,38, 6,48Hz, 1H), 3,16 (dd, J = 7,40, 4,27 Hz, 1H), 2,57 (dt, J = 12,33,8,77 Hz, 1 H), 1,67-1,87 (m, 3H), 1,16 (d, J = 7,28 Hz, 3H), 0,95 (t, J = 7,47 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 431.
Compuesto 28k-B: RMN de 1H (400 MHz, CD3OD) 5 ppm: 8,03- 8,08 (m, 2H), 7,61-7,66 (m, 1H), 7,48 -7,55 (m, 2H), 6,41 (d, J = 7,65 Hz, 1H), 5,06 (td, J = 6,65, 4,27 Hz, 1H), 4,75 (d, J = 7,15 Hz, 1H), 2,72-2,84 (m, 1H), 2,25-2,42 (m, 1H), 1,97-2,06 (m, 1H), 1,75-1,85 (m, 2H), 0,97 (t, J = 7,40 Hz, 3H), 0,89 (d, J = 6,90 Hz, 3 H). EM obs. (ESI+) [(M+H)+]: 431.
Preparación de 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de compuesto benzoato de [(1 S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4
metil-tetrahidrofuran-2-il]propilo] (compuesto 28k-A, 120 mg, 0,27 mmol) en MeOH (2 ml) se le añadió K2CO3 (58 mg, 0,42 mmol). Después de agitarse a temperatura ambiente durante 5 horas, se ajustó la mezcla de reacción hasta pH 7 burbujeando CO2 y a continuación y se concentró a vacío. Se purificó el residuo por HPLC preparativa para dar 24 mg de 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 28) como un sólido blanco.
Ejemplo 28: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,81 (d, J = 7,03 Hz, 1 H), 4,03-4,18 (m, 1 H), 3,45-3,48 (m, 1 H), 3,05-3,11 (m, 1 H), 2,37-2,42 (m, 1 H), 1,80-1,85 (m, 1 H), 1,40 - 1,62 (m, 2 H), 1,11 (d, J = 6,78 Hz, 3 H), 1,00 (t, J = 7,40 Hz, 3 H). EM obs. (ESI+) [(M+H)+]: 327.
Ejemplo 29
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona
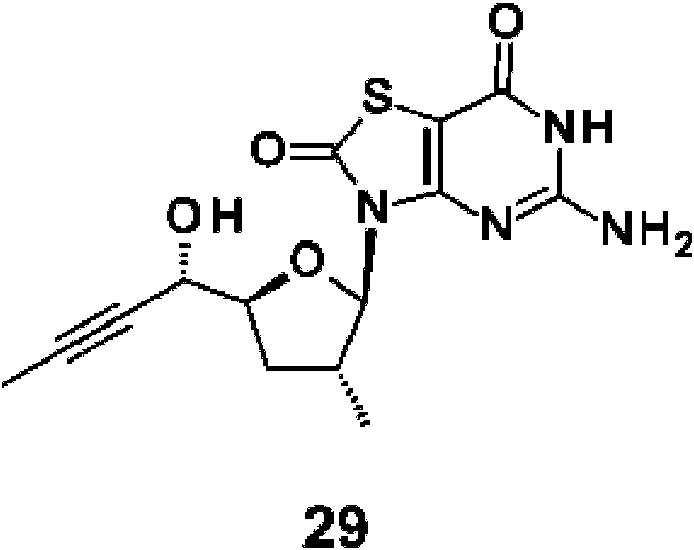
Se preparó el compuesto del título de forma análoga al ejemplo 28 usando bromuro de 1-propinilmagnesio en lugar de bromuro de etilmagnesio. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 29) como un sólido blanco.
Ejemplo 29: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,83-5,85 (m, 1H), 4,74-4,78 (m, 1H), 4,42-4,44 (m, 1H), 4,12 4,13 (m, 1H), 3,15-3,17 (m, 1H), 1,87-1,89 (m, 1H), 1,83-1,84 (m, 3H), 1,10-1,12 (m, 3H). EM obs. (ESI+) [(M+H)+]: 337.
Ejemplo 30
5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d|pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 28 usando bromuro de ciclopropilmagnesio en lugar de bromuro de etilmagnesio. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 30) como un sólido blanco.
Ejemplo 30: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,82 (d, J = 6,78 Hz, 1H), 4,20 (dt, J = 7,87, 5,22 Hz, 1H), 3,05 (d, J = 7,78 Hz, 1H), 2,92 (dd, J = 8,34, 4,58 Hz, 1H), 2,38-2,47 (m, 1H), 1,83 (dt, J = 12,39, 7,73 Hz, 1H), 1,10 (d, J = 6,90 Hz, 3 H), 0,87-0,98 (m, 1H), 0,46-0,52 (m, 2 H), 0,26-0,39 (m, 2 H). EM obs. (ESI+) [(M+H)+]: 339.
Ejemplo 31
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona
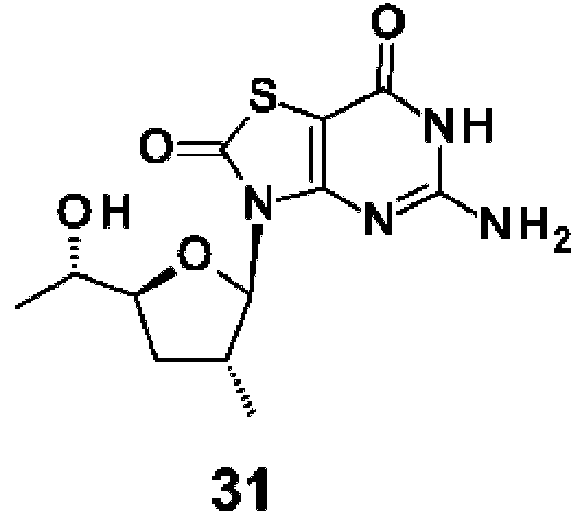
Se preparó el compuesto del título de forma análoga al ejemplo 29 usando bromuro de metilmagnesio en lugar de bromuro de etilmagnesio. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 31) como un sólido blanco. Ejemplo 31: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,81 (d, J = 6,52 Hz, 1H), 3,90-4,06 (m, 1H), 3,74-3,86 (m, 1H), 2,97-3,16 (m, 1H), 2,25-2,41 (m, 1H), 1,68-1,83 (m, 1H), 1,15 (d, J = 6,40 Hz, 3H,), 1,10 (d, J = 6,90 Hz, 3H). EM obs. (ESI+) [(M+H)+]: 313.
Ejemplo 32
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de benzoato de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metilo
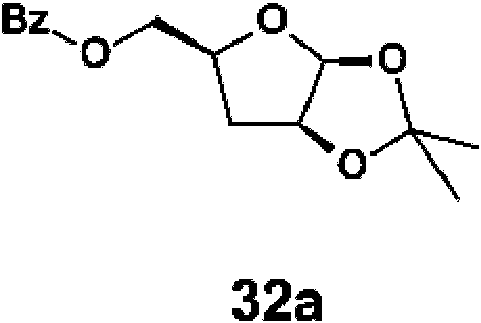
A una solución enfriada de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metanol bruto (compuesto 25e, 7,6 g, 43,6 mmol) y TEA (8,6 g, 109 mmol) en DCM se le añadió cloruro de benzoílo (9,1 g, 65,5 mmol) gota a gota a 0 °C con agitación. Después de la adición, se calentó la mezcla de forma natural hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se lavó la mezcla resultante con ácido clorhídrico 1 N, salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 5,8 g de benzoato de 1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo (compuesto 32a).
Compuesto 32a: RMN de 1H (400 MHz, CDCla) 5 ppm: 8,06-8,16 (m, 2H), 7,54-7,62 (m, 1H), 7,42-7,52 (m, 2H), 5,88 (d, J = 3,76 Hz, 1H), 4,81 (d, J = 1,00 Hz, 1H), 4,51-4,65 (m, 2H), 4,46 (dd, J = 4,64, 10,16 Hz, 1H), 2,27-2,34 (m, 1H), 2,19 (s, 1H), 1,63 (s, 3H), 1,36 (s, 3H).
Preparación de benzoato de [(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]metilo

A una solución de benzoato de [(3aS,5S,6aS)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]metilo (compuesto 32a, 5,1 g, 18 mmol) en MeOH (50 ml) se le añadió H2SO4 (0,5 ml) a temperatura ambiente. Después de agitarse a 80 °C durante 0,5 horas, se enfrió la mezcla de reacción hasta temperatura ambiente, se neutralizó por
NaHCO3 sólido y se concentró a vacío. Se redisolvió el residuo en EtOAc, se lavó con agua dos veces. Se secó la capa orgánica separada sobre Na2SO4 y se concentró a vacío para dar 4,1 g de producto bruto de benzoato de [(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]metilo (compuesto 32b) como un aceite, que se usó en la siguiente etapa directamente. EM obs. (ESI+) [(M+NH4)+]: 270.
Preparación de benzoato de [(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]metilo

A una solución de benzoato de [(2S,4S)-4-hidroxi-5-metoxi-tetrahidrofuran-2-il]metilo (compuesto 32b, 4,1 g, 17 mmol), piridina (4,8 g, 60 mmol) y DMAP (300 mg, 2,5 mmol) en DCM (50 ml) se le añadió anhídrido trifluorometanosulfónico (8,5 g, 30 mmol) a -30 °C. Después de agitarse a -30 °C- 0 °C durante 2 horas, se desactivó la reacción por solución saturada de NaHCO3 y se extrajo con DCM tres veces. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró para dar 6,6 g de producto bruto de benzoato de [(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]metilo (compuesto 32c) como un aceite, que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 385.
Preparación de benzoato de [(2S,4R)-5-metoxi-4-pirrolidin-1-il-tetrahidrofuran-2-il]metilo

A una solución de benzoato de [(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]metilo (compuesto 32c, 400 mg, 1,04 mmol) en DMF (3 ml) se le añadió pirrolidina (142 mg, 2,0 mmol) a temperatura ambiente y se agitó la mezcla a temperatura ambiente durante 16 horas. Se diluyó la mezcla de reacción con agua y se extrajo con EtOAc, se combinaron las capas orgánicas y se concentró a vacío para dar 230 mg de producto bruto de benzoato de [(2S,4R)-5-metoxi-4-pirrolidin-1-il-tetrahidrofuran-2-il]metilo (compuesto 32d) como un aceite, que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 306.
Preparación de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-pirrolidin-1-iltetrahidrofuran-2-il]metilo

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (138 mg, 0,75 mmol) en ACN (5 ml) se le añadió BSA (535 mg, 2,6 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de [(2S,4R)-5-metoxi-4-pirrolidin-1-il-tetrahidrofuran-2-il]metilo (compuesto 32d, 230 mg, 0,75 mmol) y TMSOTf (832 mg, 3,75 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 250 mg de producto bruto de
benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-pirrolidin-1-il-tetrahidrofuran-2-iljmetilo (compuesto 32e) como un sólido amarillo. EM obs. (ESI+) [(M+H)+]: 458.
Preparación de 5-amino-3-[(2S,4R,5R)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de compuesto benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-pirrolidin-1 -il-tetrahidrofuran-2-il]metilo (compuesto 32e, 250 mg, 0,54 mmol) en MeOH (8 ml) se le añadió K2CO3 (138 mg, 1,0 mmol). Después de agitarse a temperatura ambiente durante 5 horas, se concentró la mezcla de reacción a vacío y se purificó el residuo por HPLC preparativa para dar 10,0 mg de 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1 -il-tetrahidrofuran-2-il]-6W-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 32) como un sólido blanco.
Ejemplo 32: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,08 (d, J = 6,8 Hz, 1H), 4,30-4,37 (m, 1H), 3,78-3,89 (m, 2H), 3,54-3,61 (m, 1H), 2,57-2,64 (m, 2H), 2,50-2,56 (m, 2H), 2,42-2,49 (m, 1H), 2,20-2,30 (m, 1H), 1,79 (m, 4H). EM obs. (ESI+) [(M+H)+]: 354.
Ejemplo 33
W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]metanosulfonamida
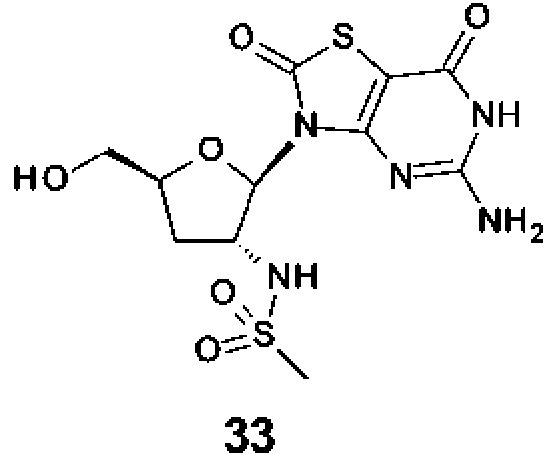
Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de benzoato de [(2S,4R,5R)-4-acido-5-metoxi-tetrahidrofuran-2-il]metilo

A una solución de benzoato de [(2S,4S)-5-metoxi-4-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]metilo (compuesto 32c, 1,1 g, 2,8 mmol) en DMF (5 ml) se le añadió acida de sodio (372 mg, 5,7 mmol) a temperatura ambiente y se agitó la mezcla a temperatura ambiente durante 16 horas. Se diluyó la mezcla de reacción con agua y se extrajo con EtOAc, se combinaron las capas orgánicas y se concentró a vacío para dar 1,1 g de producto bruto de benzoato de [(2S,4R,5R)-4-acido-5-metoxi-tetrahidrofuran-2-il]metilo (compuesto 33a) como un aceite, que se usó en la siguiente etapa sin purificación adicional.
Preparación de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-acidotetrahidrofuran-2-il]metilo
A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (730 mg, 3,97 mmol) en ACN (15 ml) se le añadió BSA (2,8 mg, 13,8 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de [(2S,4R,5R)-4-acido-5-metoxi-tetrahidrofuran-2-il]metilo (compuesto 33a, bruto, 1,1 g, 3,97 mmol) y TMSOTf (4,4 g, 19,5 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (30 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 500 mg de producto bruto de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-acido-tetrahidrofuran-2-il]metilo (compuesto 33b) como un sólido amarillo. EM obs. (ESI+) [(M+H)+]: 430.
Preparación de benzoato de [(2S,4R,5R)-4-amino-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo

A una solución de compuesto benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-acidotetrahidrofuran-2-il]metilo (compuesto 33b, 200 mg, 0,466 mmol) y trifenilfosfina (364 mg, 1,39 mmol) en THF (10 ml) se le añadió agua (0,5 ml) a temperatura ambiente. Después de agitarse a 80 °C durante 1 hora, se filtró la reacción y se concentró el filtrado a vacío para dar 80 mg de producto bruto de benzoato de [(2S,4R,5R)-4-amino-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo (compuesto 33c), que se usó en la siguiente etapa directamente. EM obs. (ESI+) [(M+H)+]: 404.
Preparación de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(metanosulfonamido)tetrahidrofuran-2-il]metilo

A una solución de compuesto benzoato de [(2S,4R,5R)-4-amino-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo (compuesto 33c, bruto, 80 mg, 0,198 mmol) en DCM (10 ml) y THF (2 ml) se le añadieron TEA (44 mg, 0,436 mmol) y cloruro de metanosulfonilo (27 mg, 0,237 mmol) a 0 °C. Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la mezcla de reacción con DCM, se lavó con agua, salmuera, se secó sobre Na2SO4 y se concentró a vacío para dar 80 mg de producto bruto de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(metanosulfonamido)tetrahidrofuran-2-il]metilo (compuesto 33d), que se usó en la siguiente etapa directamente. e M obs. (ESI') [(M-H)']: 480.
Preparación de W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]metanosulfonamida

A una solución de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(metanosulfonamido)tetrahidrofuran-2-il]metilo (compuesto 33d, bruto, 80 mg, 0,54 mmol) en MeOH (5 ml) se le añadió K2CO3 (80 mg, 0,6 mmol). Después de agitarse a temperatura ambiente durante 5 horas, se concentró la mezcla de reacción a vacío y se purificó el residuo por HPLC preparativa para dar 8,0 mg de N-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]metanosulfonamida (ejemplo 33) como un sólido blanco.
Ejemplo 33: 8 mg, RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,45-11,78 (s.a., 1H), 7,82 (d, J = 8,5 Hz, 1H), 6,99 (s.a.,
2H), 5,79 (d, J =4,8 Hz, 1H), 4,68-4,84 (m, 2H), 4,15 (dt, J = 12,7, 6,3 Hz, 1H), 3,46 (m, 2H), 2,89 (s, 3H), 2,37-2,44 (m, 1H), 1,90-2,00 (m, 1H). EM obs. (ESI+) [(M+H)+]: 378.
Ejemplo 34
W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]acetamida

Se preparó el compuesto del título de forma análoga al ejemplo 33 usando cloruro de acilo en lugar de cloruro de metilsulfonilo. Después de purificarse por HPLC preparativa, se dio N-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]acetamida (ejemplo 34) como un sólido blanco.
Ejemplo 34: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 6,09 (dd, J = 9,0, 5,8 Hz, 1H), 5,08 (t, J =5,6Hz, 1H), 3,92-4,01 (m, 1H), 3,66 (dd, J = 11,4, 6,1 Hz, 1H), 3,50-3,53 (m, 1H), 3,10-3,23 (m, 2H), 1,56-1,61 (m, 1H), 1,23-1,38 (m, 1H), 0,93 (t, J = 7,3 Hz, 1H). EM obs. (ESI+) [(M+H)+]: 342.
Ejemplo 35
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-morfolino-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 32 usando morfolina en lugar de pirrolidina. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-morfolino-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 35) como un sólido blanco.
Ejemplo 35: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,17 (d, J = 5,3 Hz, 1H), 4,26 (tt, J = 7,5, 4,8 Hz, 1H), 3,88-3,97 (m, 1H), 3,65-3,75 (m, 6 H), 2,52-2,64 (m, 4H), 2,37 (ddd, J = 13,2, 8 ,8 , 7,4 Hz, 1H), 2,21 (ddd, J = 13,1, 7,8, 5,0 Hz, 1H). EM obs. (ESI+) [(M+H)+]: 370.
Ejemplo 36
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(1-piperidil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 32 usando piperidina en lugar de pirrolidina. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(1-piperidil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 36) como un sólido blanco.
Ejemplo 36: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,16 (d, J = 6,0 Hz, 1H), 4,28 (d, J = 2,8 Hz, 1H), 4,01 (t, J = 4,8 Hz, 1H),, 3,74 (dd, J =3,6, 11,6 Hz, 1H), 3,63 (dd, J = 4,8, 12,0 Hz, 1H), 2,54-2,21 (m, 6H), 1,63-1,48 (m, 6H). EM obs. (ESI+) [(M+H)+]: 368.
Ejemplo 37
5-amino-3-[(2R,3R,5S)-3-(dimetilamino)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 32 usando dimetilamina en lugar de pirrolidina. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-3-(dimetilamino)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 37) como un sólido blanco.
Ejemplo 37: RMN de 1H (400 MHz, CD3OD) 5 ppm: 6,08 (d, J = 6,52 Hz, 1H), 4,26-4,32 (m, 1H), 3,87-3,95 (m, 1H), 3,77 (dd, J = 11,92, 2,89 Hz, 1H), 3,57 (dd, J = 11,92, 3,64 Hz, 1H), 2,41 (ddd, J = 13,11, 8,47, 5,02 Hz, 1H), 2,27 (s, 6H), 2,19 (ddd, J = 12,89, 8,63, 7,59 Hz, 1H). EM obs. (ESI+) [(M+H)+]: 328.
Ejemplo 38
(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-carbonitrilo

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

Preparación de benzoato de [(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (4,42 g, 24,0 mmol) en ACN (20 ml) se le añadió BSA (14,8 ml, 60,0 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 h para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron benzoato de [(2S,4R)-4,5-diacetoxitetrahidrofuran-2-il]metilo (n.° CAS: 4613-71-2, n.° cat.: MD04725, comercialmente disponible de Carbosynth Limited, 6,45 g, 20,0 mmol) y TMSOTf (5,5 ml, 30,0 mmol) en secuencia.
Después de calentarse con agitación a 70 °C durante 14 horas, se retiró el disolvente a vacío. Se dividió el residuo entre EtOAc y solución saturada de NaHCO3 (30 ml). Se separó la capa orgánica y se extrajo la fase acuosa con EtOAc (100 ml) dos veces. Se lavaron las capas orgánicas combinadas con salmuera, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice para dar 5,7 g de benzoato de [(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo (compuesto 38a) como un sólido amarillo claro.
Preparación de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38b) y benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]metilo (compuesto 38c)

Se agitó una mezcla de benzoato de [(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]metilo (compuesto 38a, 4,72 g,10,6 mmol) y carbonato de potasio (1,46 g, 10,6 mmol) en metanol (106 ml) a temperatura ambiente durante 3,5 horas. Se desactivó la reacción por adición de ácido acético (1,5 ml). Se concentró la mezcla resultante a vacío para retirar el disolvente y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol al 0-5 % en DCM) para dar 1,0 g de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38b) y 2,8 g de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]metilo (compuesto 38c) como un sólido pálido.
Preparación de 5-amino-3-[(2R,3R,5S)-5-[[ferc-butil(dimetil)silil]oximetil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38b, 3,8 g, 12,7 mmol) en DMF (30 ml) se le añadió imidazol (2,6 g, 38 mmol) y terc-butilclorodimetilsilano (4,2 g, 28 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la solución resultante con EtOAc (200 ml), se lavó con agua, salmuera, se secó sobre Na2SO4. Se concentró la capa orgánica a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc en éter de petróleo 1:5) para dar 3,3 g de 5-amino-3-[(2R,3R,5S)-5-[[terc-butil(dimetil)silil]oximetil]-3-hidroxitetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38d).
Compuesto 38d: RMN de 1H (400 MHz, CDCla) 5 ppm: 11,21 (s, 1H), 6,94 (s.a., 2H), 5,70-5,80 (m, 1H), 5,44 (d, J =4,5 Hz, 1H), 4,79 (ddt, J = 6,6, 4,4, 2,2 Hz, 1H), 4,11-4,24 (m, 1H), 3,58-3,70 (m, 2H), 2,32 (ddd, J = 12,7, 9,4, 6,8 Hz, 1H), 1,80 (ddd, J = 12,7, 6,1,2,0 Hz, 1H), 0,79-0,92 (s, 9H), 0 (s, 6H).
Preparación de 5-amino-3-[(2R,5S)-5-[[íerc-butil(dimetil)silil]oximetil]-3-oxo-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución de 5-amino-3-[(2R,3R,5S)-5-[[terc-butil(dimetil)silil]oximetil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38d, 3,3 g, 7,8 mmol) en THF (100 ml) se le añadió peryodinano de Dess-Martin (3,68 g, 8,76 mmol) con agitación. Después de agitarse a temperatura ambiente durante 2 horas, se filtró la solución resultante y se concentró el filtrado a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM) 1:10 para dar 2,4 g de producto bruto de 5-amino-3-[(2R,5S)-5-[[tercbutil(dimetil)silil]oximetil]-3-oxo-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38e). EM obs. (ESI+) [(M+H)+]: 413.
Preparación de 5-amino-3-[(2R,3S,5S)-5-[[íerc-butil(dimetil)silil]oximetil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

A una solución agitada de 5-amino-3-[(2R,5S)-5-[[terc-butil(dimetil)silil]oximetil]-3-oxo-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38e, 1,0 g, 2,43 mmol) en THF (5 ml) se le añadió solución de hidruro de litio y tri-terc-butoxialuminio (1 M en THF, 2,7 ml, 2,7 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se desactivó la solución resultante por solución saturada de NH4Cl y se filtró. Se concentró el filtrado a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol en DCM 1:10) para dar 750 mg de producto bruto de 5-amino-3-[(2R,3S,5S)-5-[[terc-butil(dimetil)silil]oximetil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38f). (Refiérase a Tetrahedron 1984, 40, 125-135).
Compuesto 38f: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 7,00 (s.a., 2H), 6,08 (d, J = 7,5 Hz, 1H), 5,35 (d, J = 5,5 Hz, 1H), 4,53-4,65 (m, 1H), 3,91-3,99 (m, 1H), 3,82-3,89 (m, 1H), 3,70 (dd, J = 10,5, 4,3 Hz, 1H), 2,11 (t, J = 8,7 Hz, 2H), 0,84-0,86 (m, 9H), 0,84-0,88 (m, 10H), 0,84- 0,86 (m, 9H), 0,01 (d, J = 2,3 Hz, 6H).
Preparación de trifluorom etanosulfonato de [5-amino-3-[(2R,3S,5S)-5-[[íerc-butil(dimetil)silil]oximetil]-3-(trifluorom etilsulfoniloxi)tetrahidrofuran-2-il]-2-oxo-tiazolo[4,5-d]pirim idin-7-ilo]
A una solución agitada de 5-amino-3-[(2R,3S,5S)-5-[[ferc-butil(dimetil)silil]oximetil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 38f, 100 mg, 0,24mmol) en DCM (15 ml) se le añadió DMAP (147 mg, 1,2 mmol) y cloruro de trifluorometanosulfonilo (122 mg, 0,7 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se lavó la solución resultante con agua, salmuera, se secó sobre Na2SO4. Se concentró la capa orgánica a vacío para dar 120 mg de producto bruto de trifluorometanosulfonato de [5-amino-3-[(2R,3R,5S)-5-[[tercbutil(dimetil)silil]oximetil]-3-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]-2-oxo-tiazolo[4,5-d]pirimidin-7-ilo] (compuesto 38g), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI') [(M-H)-]: 679.
Preparación de (2R,3S,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-[[íercbutil(dim etil)silil]oxim etil]tetrahidrofuran-3-carbonitrilo
A una solución agitada de trifluorometanosulfonato de [5-amino-3-[(2R,3S,5S)-5-[[ferc-butil(dimetil)silil]oximetil]-3-(trifluorometilsulfoniloxi)tetrahidrofuran-2-il]-2-oxo-tiazolo[4,5-d]pirimidin-7-ilo] (compuesto 38g, bruto, 120 mg, 0,2 mmol) en DMF (2 ml) se le añadió cianuro de sodio (100 mg, 2,3 mmol). Después de agitarse a temperatura ambiente durante 2 horas, se diluyó la solución resultante con EtOAc, se lavó con salmuera, se secó sobre Na2SÜ4 y se concentró a vacío para dar 100 mg de producto bruto de (2R,3S,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-[[íerc-butil(dimetil)silil]oximetil]tetrahidrofuran-3-carbonitrilo (compuesto 38h), que se usó en la siguiente etapa sin purificación adicional. EM obs. (ESI+) [(M+H)+]: 424.
Preparación de (2R,3S,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-carbonitrilo

A una solución agitada de (2R,3S,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-[[fercbutil(dimetil)silil]oximetil]tetrahidrofuran-3-carbonitrilo (compuesto 38h, bruto, 100 mg) en THF (5 ml) se le añadió solución de TBAf (1 M en THF, 6 ml, 6 mmol) a 0 °C. Después de agitarse a temperatura ambiente durante 4 horas, se lavó la solución de reacción con solución saturada de NH4Cl, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por HPLC preparativa para dar 9 mg de (2R,3S,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-carbonitrilo (ejemplo 38) como un sólido blanco.
Ejemplo 38: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,11-11,99 (s.a., 1H), 7,10 (s.a., 2H), 6,15 (d, J = 4,8 Hz, 1H), 4,86 (s.a., 1H), 4,33 (dt, J = 9,5, 4,7 Hz, 1H), 4,07-4,20 (m, 1H), 3,50 (d, J = 4,8 Hz, 2H), 2,44-2,48 (m, 1H), 2,23-2,36 (m, 1H). EM obs. (ESI+) [(M+H)+]: 310.
Ejemplo 39
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de forma análoga al ejemplo 38 usando metilsulfuro de sodio en lugar de cianuro de sodio. Después de purificarse por HPLC preparativa, se dio 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfaniltetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 39) como un sólido blanco.
Ejemplo 39: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,37-11,55 (s.a., 1H), 6,91-7,10 (s.a., 2H), 5,94 (d, J = 4,8 Hz, 1H), 4,75 (t, J = 5,9 Hz, 1H), 4,03-4,15 (m, 2H), 3,45-3,55 (m, 3H), 2,09 (s, 3H), 1,92 (ddd, J = 12,8, 6,8, 4,8 Hz, 1H). EM obs. (ESI+) [(M+H)+]: 331
Ejemplo 40
5-amino-3-[(2R,3R,5S)-3-(1-fluoro-1-metil-etil)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema:

A una solución de (Z)-3-[(4S)-2,2-dimetil-1,3-dioxolan-4-il]prop-2-enoato de etilo (n.° CAS: 91926-90-8, n.° cat.: PB1131897, comercialmente disponible de Pharma Block (Nanjing) R&D Co., Ltd, 4,0 g, 20,0 mmol) en metanol se le añadió una cantidad catalítica de ácido sulfúrico concentrado (25 pl de ácido sulfúrico concentrado al 10 % en metanol). Se agitó la mezcla a temperatura ambiente durante 2 horas. Se concentró la mezcla resultante a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol al 0-10 % en DCM) para dar 2,25 g de (2S)-2-(hidroximetil)-2H-furan-5-ona (compuesto 40b) como un aceite viscoso.
Preparación de (2S)-2-[[ferc-butil(difenil)silil]oximetil]-2H-furan-5-ona

A una solución de 2,25 g de (2S)-2-(hidroximetil)-2H-furan-5-ona (compuesto 40b, 2,11 g, 16,0 mmol) e imidazol (1,63 g, 24,0 mmol) en DCM se le añadió ferc-butil(cloro)difenilsilano (5,2 ml, 20,0 mmol) gota a gota. Se agitó la mezcla resultante a temperatura ambiente durante 2 horas. Se lavó la mezcla resultante con salmuera. Se extrajo la capa acuosa con DCM. Se combinaron las capas orgánicas, se lavó con ácido clorhídrico 1 N, se secó sobre Na2SÜ4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 4,6 g de (2S)-2-[[ferc-butil(difenil)silil]oximetil]-2H-furan-5-ona (compuesto 40c) como un sólido blanco. EM obs. (ESI+) [(M+H)+]: 353.
Preparación de (5S)-5-[[ferc-butil(difenil)silil]oximetil]tetrahidrofuran-2-ona

Se agitó una solución de (2S)-2-[[terc-butil(difenil)silil]oximetil]-2H-furan-5-ona (compuesto 40c, 2,8 g, 8,0 mmol) en EtOAc (40 ml) con paladio al 10 % sobre carbono (280 mg) en atmósfera de hidrógeno durante la noche. Se filtró la mezcla resultante y se concentró el filtrado a vacío para dar 2,7 g de (5S)-5-[[tercbutil(difenil)silil]oximetil]tetrahidrofuran-2-ona (compuesto 40d) como un aceite viscoso. EM obs. (ESI+) [(M+H)+]: 355.
Preparación de (3S,5S)-5-[ferc-butil(difenil)silil]oximetil]-3-(1-hidroxi-1-metil-etil)tetrahidrofuran-2-ona

A una solución enfriada de (5S)-5-[[terc-butil(difenil)silil]oximetil]tetrahidrofuran-2-ona (compuesto 40d, 5,00 g, 14,0 mmol) en tetrahidrofurano seco (28 ml) se le añadió a solución de bis(trimetilsilil)azanida de litio (1,3 M en THF, 11,8 ml, 15,4 mmol) gota a gota a -78 °C en argón. Después de la adición, se agitó la mezcla a -78 °C durante 1 hora. A continuación se añadió acetona destilada (1,23 ml, 15,4 mmol) gota a gota a la mezcla y se agitó la mezcla resultante a -78 °C durante otras 2 horas. Se desactivó la reacción por solución saturada de NH4Cl y se extrajo con EtOAc (30 ml) tres veces. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 5,7 g de (3S,5S)-5-[[terc-butil(difenil)silil]oximetil]-3-(1-hidroxi-1-metil-etil)tetrahidrofuran-2-ona (compuesto 40e) como un aceite amarillo claro. EM obs. (ESI+) [(M+NH4)+]: 430. (Para la síntesis, refiérase a: Tetrahedron 1997,53, 6281-6294).
Preparación de (3R,5S)-5-[[íerc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-ona

A una solución de trifluoruro de dietilaminoazufre (414 pl, 3,0 mmol) en DCM (10 ml) a -78 °C se le añadió una solución de (3S,5S)-5-[[terc-butil(difenil)silil]oximetil]-3-(1-hidroxi-1-metil-etil)tetrahidrofuran-2-ona (compuesto 40e, 1,03 g, 2,5 mmol) en DCM (10 ml) gota a gota. Se calentó la mezcla resultante hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se concentró la mezcla resultante a vacío y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-20 % en éter de petróleo) para dar 820 mg de (3R,5S)-5-[[terc-butil(difenil)silil]oximetil]-3-(1 -fluoro-1 -metil-etiijtetrahidrofuran-2-ona (compuesto 40f) como un aceite viscoso. EM obs. (ESI+) [(M+H)+]: 415.
Preparación de acetato de [(3R,5S)-5-[[íerc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-ilo]
A una solución enfriada de (3R,5S)-5-[[terc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-ona
(compuesto 40f, 820 mg, 1,92 mmol) en DCM seco (10 ml) a -78 °C se le añadió hidruro de diisobutilaluminio (1,0 M en tolueno, 6,0 ml, 6,0 mmol) gota a gota. Se agitó la mezcla resultante a -78 °C durante 1 hora. A continuación a la mezcla se le añadió piridina (790 mg, 10 mmol), anhídrido acético (0,93 ml, 10,0 mmol) y DMAP (732 mg, 6,0 mmol). Se dejó que la mezcla de reacción se calentara hasta -20 °C lentamente y se agitó a -20 °C durante varias horas hasta que se completó la reacción. Se desactivó la mezcla resultante por salmuera y se extrajo con EtOAc (30 ml) tres veces. Se combinaron las capas orgánicas, se secó sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-20 % en éter de petróleo) para dar 360 mg de acetato de [(3R,5S)-5-[[ferc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-ilo] (compuesto 40g) como un aceite viscoso. Em obs. (ESI+) [(M+H)+]: 459.
Preparación de 5-amino-3-[(2R,3R,5S)-5-[[ferc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metiletil)tetrahidrofuran-2-il]-6H-tiazolo [4,5-d]pirimidin-2,7-diona

Se calentó una mezcla de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (162 mg, 0,87 mmol) y bis(trimetilsilil)acetamida (527 mg, 2,61 mmol) con agitación a 75 °C en argón hasta que la mezcla se volvió transparente. Se enfrió la mezcla hasta temperatura ambiente. A la mezcla de reacción previa, se le introdujeron acetato de [(3R,5S)-5-[[ferc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-ilo] (compuesto 40g, 280 mg, 1,02 mmol) y trifluorometanosulfonato de trimetilsililo (290 mg, 1,31 mmol). Se calentó la mezcla resultante a 75 °C en argón durante 3 horas. A continuación se concentró la mezcla resultante a vacío para retirar el disolvente y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol al 0-5 % en DCM) para dar 225 mg de 5-amino-3-[(2R,3R,5S)-5-[[ferc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metil-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 40h) como un sólido marrón. EM obs. (ESI+) [(M+H)+]: 583.
Preparación de 5-amino-3-[(2R,3R,5S)-3-(1-fluoro-1-metil-etil)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

Se calentó una mezcla de 5-amino-3-[(2R,3R,5S)-5-[[ferc-butil(difenil)silil]oximetil]-3-(1-fluoro-1-metiletil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (compuesto 40h, 66 mg, 0,12 mmol) y NH4F (133 mg, 3,6 mmol) en metanol a reflujo durante 1,5 h. Se concentró la mezcla resultante a vacío y se purificó el residuo por HPLC preparativa para dar 10 mg de 5-amino-3-[(2R,3R,5S)-3-(1-fluoro-1-metil-etil)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 40) como un sólido blanco.
Ejemplo 40: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,27-11,40 (s.a., 1H), 6,90-7,08 (s.a., 2H), 6,03-6,10 (m, 1H), 4,65-4,74 (m, 1H), 3,92-4,03 (m, 1H), 3,42-3,53 (m, 2H), 3,18-3,30 (m, 1H), 2,10-2,23 (m, 1H), 1,96-2,06 (m, 1H), 1,19 1,42 (m, 6H). EM obs. (ESI+) [(M+H)+]: 345.
Ejemplo 41:
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(2-metilalil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona

Se preparó el compuesto del título de acuerdo con el siguiente esquema.

Preparación de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2-il]m etilo

Se agitó una mezcla de benzoato de [(2S,4R,5S)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]metilo (compuesto 38c, 2,08 g, 5,0 mmol), clorometanot¡oato de 0-fen¡lo (0,80 ml) y DMAP (1,22 g, 10,0 mmol) en DCM (50 ml) a temperatura ambiente durante la noche. Se lavó la mezcla resultante con salmuera, se
secó sobre Na2SÜ4, y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 2,20 g de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2-il]metilo (compuesto 41a) como un sólido pálido. EM obs. (ESI-) [(M-H)-]: 539.
Preparación de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(2-metilalil)tetrahidrofuran-2-il]metilo

Se desgasificó una mezcla de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-fenoxicarbotioiloxi-tetrahidrofuran-2-il]metilo (compuesto 41a, 324 mg, 0,60 mmol), 2,2'-azobis(2-metilpropionitrilo) (50 mg, 0,30 mmol) y tributil(2-metilalil)stannano (0,70 ml, 3,0 mmol) en tolueno anhidro (10 ml) con argón y a continuación se calentó con agitación a 80 °C durante 4 horas. Se agitó la mezcla resultante con NH4F acuoso saturado a temperatura ambiente durante 2 horas, y a continuación se extrajo con DCM dos veces. Se secaron las capas orgánicas combinadas sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 190 mg de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(2-metilalil)tetrahidrofuran-2-il]metilo (compuesto 41b) como un sólido marrón. EM obs. (ESI-) [(M-H)-]: 441.
Preparación de 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(2-metilalil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirim idin-2,7-diona

Se agitó una solución de benzoato de [(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-(2-metilalil)tetrahidrofuran-2-il]metilo (compuesto 41b, 180 mg, 0,41 mmol) en metanol con K2CO3 (150 mg, 1,09 mmol) a temperatura ambiente durante 4 horas. Se desactivó la reacción por adición de ácido acético y se concentró la mezcla resultante a vacío. Se purificó el residuo por HPLC preparativa para dar 41 mg de 3-[(2R,3R,5S)-3-alil-5-(hidroximetil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona (ejemplo 41) como un polvo blanco.
Ejemplo 41: RMN de 1H (400 MHz, de-DMSO) 5 ppm: 11,03-11,29 (m, 1H), 6,76-7,04 (m, 2H), 5,68 (d, J = 6,02 Hz, 1H), 4,62-4,70 (m, 3H), 3,96-4,10 (m, 1H), 3,47 (t, J = 5,27 Hz, 2H), 3,24-3,30 (m, 1H), 2,16-2,25 (m, 1H), 2,13 (d, J = 7,53 Hz, 2H), 1,67-1,76 (m, 1H), 1,62 (s, 3H). EM obs. (ESI+) [(M+H)+]: 339.
Ejemplo 42
2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo]

Se preparó el compuesto del título de acuerdo con el siguiente esquema.
Preparación de 2-metilpropanoato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo]

A una solución enfriada de (1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propan-1-ol (compuesto 6e, 505,6 mg, 2,5 mmol) en piridina se le añadió cloruro de isobutirilo (0,39 ml, 3,72 mmol) gota a gota mientras se enfriaba con un baño de agua helada. Después de la adición, se calentó la mezcla hasta temperatura ambiente y se agitó a temperatura ambiente durante la noche. Se diluyó la mezcla resultante con EtOAc y se lavó con una solución saturada de NhUCl. Se secó la capa orgánica sobre Na2SO4 y se concentró a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-30 % en éter de petróleo) para dar 470 mg de 2-metilpropanoato de [(1 S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo] (compuesto 42a).
Preparación de 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-4,5-diacetoxitetrahidrofuran-2-il]propilo]

A una solución agitada de 2-metilpropanoato de [(1S)-1-[(3aR,5S,6aR)-2,2-dimetil-3a,5,6,6a-tetrahidrofuro[2,3-d][1,3]dioxol-5-il]propilo] (compuesto 42a, 470 mg, 1,73 mmol), anhídrido acético (0,81 ml, 8,64 mmol) y ácido acético (0,51 ml, 8,64 mmol) en DCM (10 ml) se le añadió ácido sulfúrico concentrado (18,4 pl, 0,17 mmol). Se agitó la mezcla resultante a temperatura ambiente durante la noche. Se concentró la mezcla resultante a vacío. Se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con EtOAc al 0-20 % en éter de petróleo) para dar 105 mg de 2-metilpropanoato de [(1 S)-1-[(2S,4R,5R)-4,5-diacetoxitetrahidrofuran-2-il]propilo] (compuesto 42b).
Preparación de 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirim idin-3-il)tetrahidrofuran-2-il]propilo]

A una suspensión de 5-amino-3,6-dihidrotiazolo[4,5-d]pirimidin-2,7-diona (63 mg, 0,34 mmol) en ACN (5 ml) se le añadió BSA (252 pl, 1,02 mmol). A continuación se agitó la mezcla de reacción resultante a 70 °C en argón durante 0,5 horas para formar una solución transparente. Después de que se enfriara la solución hasta temperatura ambiente, se añadieron 2-metilpropanoato de [(1s)-1-[(2S,4R,5R)-4,5-diacetoxitetrahidrofuran-2-il]propilo] (compuesto 42b, 105 mg, 0,34 mmol) y TMSOTf (113 pl, 0,51 mmol) en secuencia. Después de calentarse con agitación a 70 °C durante 14 horas, se concentró la mezcla resultante a vacío para retirar el disolvente y se purificó el residuo por cromatografía en columna en gel de sílice (eluyendo con metanol al 0-5 % en DCM) para dar 75 mg de 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo] (compuesto 42c) como un sólido amarillo claro.
Preparación de 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo]

Se agitó una mezcla de 2-metilpropanoato de [(1S)-1-[(2S,4R,5R)-4-acetoxi-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)tetrahidrofuran-2-il]propilo] (compuesto 42c, 70 mg,0,16 mmol) y K2CO3 (13,2 mg, 0,096 mmol) en metanol (0,5 ml) y tetrahidrofurano (2 ml) a temperatura ambiente durante la noche. Se desactivó la reacción por adición de ácido acético (1 gota). Se concentró la mezcla resultante a vacío para retirar los disolventes y se purificó el residuo por HPLC preparativa para dar 18,2 mg de 2-metilpropanoato de [(1 S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo] (ejemplo 42) como un sólido pálido.
Ejemplo 42: RMN de 1H (400 MHz, CD3OD) 5 ppm: 5,89-5,95 (m, 1H), 4,93-5,01 (m, 2H), 4,29-4,38 (m, 1H), 2,51 2,63 (m, 2H), 1,83-1,93 (m, 1H), 1,58-1,76 (m, 2H), 1,15 (dd, J = 4,02, 7,03 Hz, 6H), 0,88-0,95 (m, 3H). EM obs. (ESI+)
[(M+H)+j: 399.
Ejemplo 43
Ensayo de células HEK293-Blue-hTLR-7:
Se adquirió una línea celular de HEK293-Blue-hTLR-7 estable de InvivoGen (n.° cat: hkb-htlr7, San Diego, California, EE. u U.). Se diseñaron estas células para estudiar la estimulación de TLR7 humano realizando un seguimiento de la activación de NF-kB. Se dispuso un gen indicador de SEAP (fosfatasa alcalina embrionaria secretada) bajo el control del promotor mínimo de IFN-Srfusionado a cinco sitios de unión a NF-kB y AP-1. Se indujo la SEAP activando NF-kB y AP-1 por medio de estimulación de células HEK-Blue-hTLR7 con ligandos de TLR7. Por lo tanto, se reguló la expresión indicadora por el promotor de NF-kB tras la estimulación de TFL7 humano durante 20 horas. Se determinó la actividad indicadora de SEAP de sobrenadante del cultivo celular usando el kit QUANTI-Blue™ (n.° cat.: rep-qb1, Invivogen, San Diego, Ca, EE. UU.) a una longitud de onda de 640 nm, un medio de detección que se vuelve púrpura o azul en presencia de la fosfatasa alcalina.
Se incubaron células HEK293-Blue-hTLR7 a una densidad de 250.000~450.000 células/ml en un volumen de 180 pl en una placa de 96 pocillos en medio Eagle modificado de Dulbecco (DMEM) que contenía 4,5 g/l de glucosa, 50 U/ml de penicilina, 50 mg/ml de estreptomicina, 100 mg/ml de normocina, L-glutamina 2 mM, suero fetal bovino termoinactivado al 10 % (v/v) durante 24 h. A continuación se incubaron las células HEK293-Blue-hTLR-7 con adición de 20 pl de compuesto de prueba en una dilución en serie en presencia de DMSO final al 1 % y se realiza la incubación a 37 °C en una estufa con CO2 durante 20 horas. A continuación se incubaron 20 pl del sobrenadante de cada pocillo con 180 pl de solución de sustrato Quanti-blue a 37 °C durante 2 horas y se leyó la absorbancia a 620~655 nm usando un espectrofotómetro. La vía de señalización por la que la activación de TLR7 da lugar a la activación de NF-kB posterior se ha aceptado ampliamente y, por lo tanto, también se usó un ensayo indicador similar para evaluar el agonista de TLR7 (Tsuneyasu Kaisho y Takashi Tanaka, Trends in Immunology, volumen 29, número 7, julio 2008, páginas 329,sci; Hiroaki Hemmi et al, Nature Immunology 3, 196-200 (2002)).
La actividad agonista de TLR7 en el ensayo HEK293-hTLR-7 de los compuestos de la presente invención se enumera en la tabla 1. Se sometieron a prueba los ejemplos en el ensayo anterior y se descubrió que tenían una CE50 de aproximadamente 10 pM a aproximadamente 90 pM.
Tabl 1 A i i m n l n HEK2 -hTLR-7
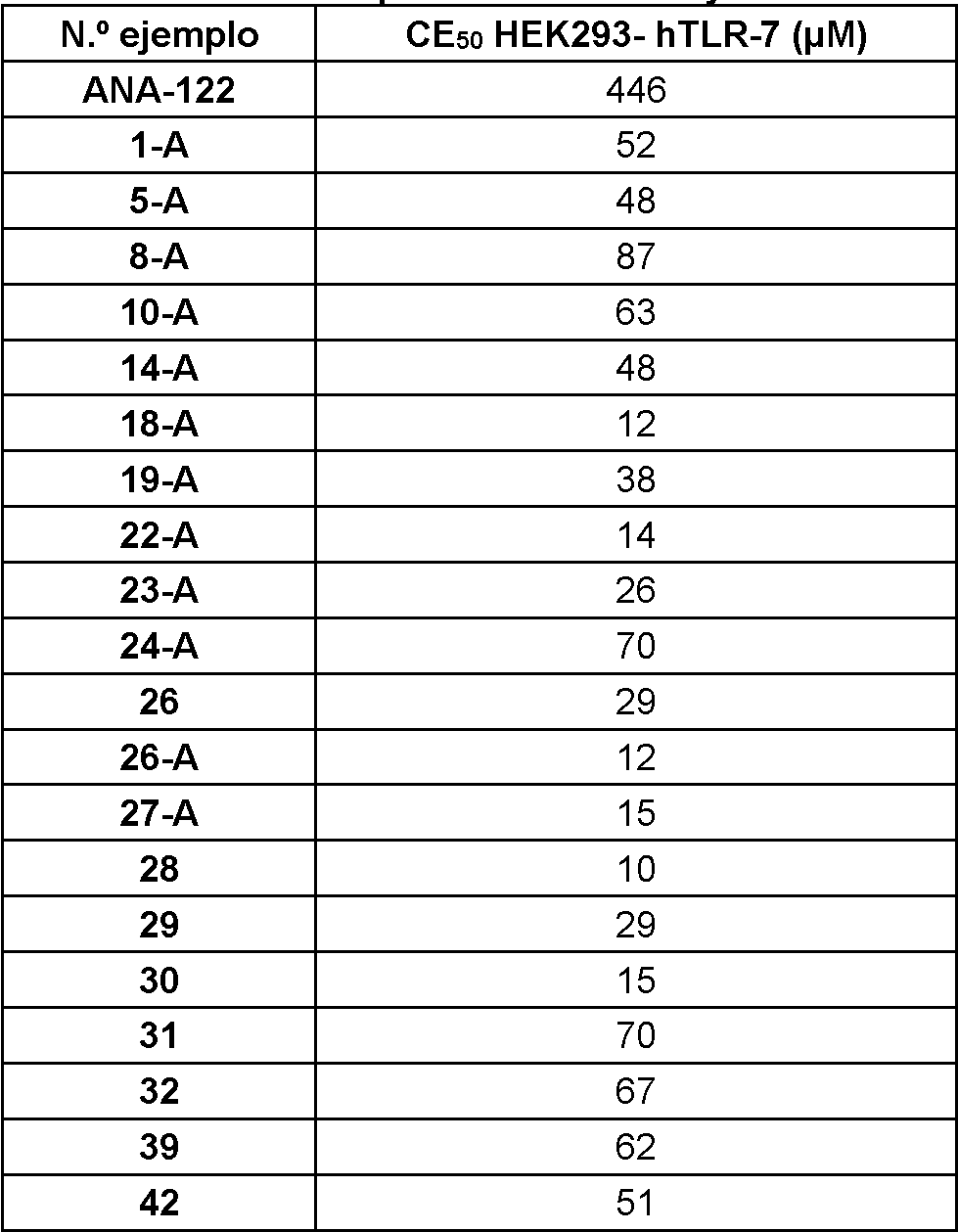
También se sometió a prueba un compuesto de referencia divulgado en la patente W02006066080(A1) como compuesto 122 (en el presente documento denominado ANA-122) para determinar la actividad agonista de TLR7 en el ensayo HEK293-hTLR-7 mencionado anteriormente, se descubrió que la CE50 de ANA-122 era de 446 pM.

Ejemplo 44
Metabolismo de profármacos: fórm ula (II) o fórmula (Ila)
Se llevó a cabo un estudio para evaluar la conversión metabólica de profármacos, fórmula (II) o fórmula (Ila), a en compuestos de fórmula (I) o fórmula (Ia) de la presente invención. Los profármacos, fórmula (II) o fórmula (IIa), se pueden metabolizar al compuesto activo de fórmula (I) o fórmula (Ia) y otros compuestos de la invención en el cuerpo si se sirven como profármacos. A menudo se usan hepatocitos para evaluar el grado de conversión metabólica de profármacos en el cuerpo de animales o seres humanos.
Se llevó a cabo un estudio para evaluar la conversión metabólica de los profármacos, ejemplo 2-A, ejemplo 3-A, ejemplo 4-A, ejemplo 16-A y ejemplo 17-A, en las correspondientes formas activas, ejemplo 1-A y ejemplo 14-A, en presencia de hepatocitos humanos. Se realizó un seguimiento de la formación de las formas activas, ejemplo 1-A y ejemplo 14-A, en el estudio. Por comparación, también se evaluó la conversión metabólica de famciclovir a penciclovir.
Suspensión de hepatocitos
Se adquirió medio de placa de hepatocitos crioconservados (n.° cat.: PY-HMD-01) del RILD Research Institute for Liver Diseases (Shanghai) Co.Ltd. Se adquirió hepatocito humano crioconservado (n.° cat.: X008005, n.° lote: VRR) de In Vitro Technologies (Baltimore, MD).
Se preparó la suspensión de hepatocitos madre a partir de hepatocitos crioconservados en medio de placa a la concentración de 1,8x10® células/ml.
Soluciones de trabajo de compuestos
Se disolvieron compuestos en DMSO para preparar soluciones madre 50 mM. Se diluyeron 10 pl de la solución madre en 5 ml de medio de placa para obtener una solución de trabajo 100 pM.
Incubaciones
Se prepararon suspensiones de reacción en una placa de cultivo celular de 24 pocillos mezclando 200 pl de suspensión de hepatocitos (de cyno o humanos) y 200 pl de solución de trabajo. La incubación final contenía 0,9x10® células/ml y compuesto 50 pM. Se incubaron las mezclas anteriores a 37 °C en una atmósfera de CO2 al 5 % humidificada, con agitación a 150 rpm.
Preparación de muestras para análisis
Después de 180 min de incubación, se transfirieron 200 pl de la mezcla de incubación a un tubo de 1,5 ml y se desactivó con 400 pl de solución de detención (acetonitrilo helado con tolbutamida 0,2 pM como patrón interno). Se centrifugaron las muestras a 12000 rpm durante 10 minutos y los sobrenadantes resultantes se sometieron a análisis CL-EM/EM.
Se prepararon las curvas de calibración de la siguiente forma. A 200 pl de suspensión celular (densidad celular de 1,8 millones de células/ml), se le añadieron 198 pl de medio de placa de hepatocitos y 2 pl de la concentración apropiada del compuesto en DMSO. Se mezclaron exhaustivamente las muestras y se transfirieron 200 pl de la mezcla a 400 pl de la solución de detención (véase anteriormente). El intervalo de curva estándar es de 1 pM a 25 pM.
Bioanálisis
Se cuantificaron los compuestos en un instrumento de CL-EM/EM API5500 en el modo MRM ESl-positivo. Se resumen los resultados de conversión de profármacos y generación de metabolitos en la tabla 2.
Tabla 2: Concentración de los metabolitos formados en hepatocitos humanos después de una incubación de 3 horas de 50 pM r f rm

En hepatocitos humanos, se metabolizaron los compuestos de ejemplo 2-A, ejemplo 3-A, ejemplo 4-A, ejemplo 16-A y ejemplo 17-A así como famciclovir para proporcionar los correspondientes metabolitos activos de ejemplo 1-A, ejemplo 14-A y penciclovir, respectivamente.
Ejemplo 45
El agonista de TLR7 ejemplo 1-A activa TLR7 murino
Se evaluó la potencia del agonista de TLR7 ejemplo 1-A que activa TLR7 murino usando una línea celular HEK293-Blue-mTLR7 estable disponible de InvivoGen (n.° cat.: Hkb-mtlr7, San Diego, California, EE. UU.). De forma similar a HEK293-Blue-hTLR7 como se describe en el ejemplo 43, se diseña HEK293-Blue-mTLR7 para estudiar la estimulación de TLR7 murino realizando un seguimiento de la activación de NF-kB. Se dispuso un gen indicador de SEAP bajo el control del promotor mínimo de IFN-p^ fusionado a cinco sitios de unión a NF-kB y AP-1. Se indujo la expresión de SEAP por la activación de NF-kB y AP-1 tras estimulación de TLR7 murino con ligandos de TLR7. Se determinó la expresión de SEAP en el sobrenadante del cultivo celular usando un kit QUANTI-Blue™ (n.° cat.: Rep-qb1, Invivogen, San Diego, Ca, EE. UU.), un medio de detección que se vuelve púrpura/azul en presencia de la fosfatasa alcalina, a una longitud de onda de 655 nm.
Se incubaron células HEK293-Blue-mTLR7 a una densidad de 250.000~450.000 células/ml en un volumen de 180 pl en una placa de 96 pocillos en medio Eagle modificado de Dulbecco (DMEM) que contenía 4,5 g/l de glucosa, 50 U/ml de penicilina, 50 mg/ml de estreptomicina, 100 mg/ml de normocina, L-glutamina 2 mM, suero fetal bovino termoinactivado al 10 % (v/v) durante 24 horas. A continuación se incubaron las células HEK293 con 20 pl del compuesto de prueba en una dilución en serie en presencia de DMSO al 1 % final a 37 °C en una estufa con CO2 durante 20 horas. Se incubaron 20 pl del sobrenadante de cada pocillo con 180 pl de solución de sustrato Quanti-blue a 37 °C durante 2 horas y se midió la absorbancia a 655 nm usando un espectrofotómetro.
Como se muestra en la figura 1, el ejemplo 1-A activa TLR7 murino de manera dependiente de la dosis con una CE50 de 71,8 pM.
Ejemplo 46
El agonista de TLR7 ejemplo 1-A induce citocinas antivíricas en leucocitos monomorfonucleares en la sangre periférica (PBMC) murinos in vitro
Para demostrar la activación de TLR7 en leucocitos por el compuesto de la presente invención, se sometieron a estimulación PBMC murinos (disponibles de ALLCells, LLC.) por el ejemplo 1-A. Se sembraron 70 millones de PBMC murinos (C57BL/6 cepa) en cada pocillo de una placa de 24 pocillos a la concentración de 2x106/ml en 1,5 ml de suero fetal bovino al 10 % que contiene medio RPMI-1640. Se incubaron los PBMC sembrados con el ejemplo 1-A en un intervalo de concentración de 2 a 10 pM durante 24 horas. Se recogieron 50 pl de medio de cultivo celular y se analizaron con un kit 36plex Procarta Multiplex (ebioscience EPX360-26092-901, eBioscience), que midió los niveles de 15 analitos de citocinas, siguiendo las instrucciones del fabricante.
Como se muestra en la figura 2, se descubrió que se indujeron niveles altos de IFNa, IP10, TNFa e IL-6 por el ejemplo 1-A de manera dependiente de la dosis. El incremento en los niveles de estas citocinas en los PBMC estimulados demuestra que el agonista de TLR7 ejemplo 1-A induce activación inmunitaria y tiene el potencial para tratar enfermedades infecciosas.
Ejemplo 47
El ejemplo 4-A reduce ADN de VHB y HBsAg en el modelo VAA-VHB
Se evaluó el ejemplo 4-A para determinar su eficacia antivírica in vivo usando un modelo de ratón VAA-VHB. Se generó este modelo de ratón para infección por VHB inyectando a ratones C57BL/6 un virus adenoasociado (VAA) recombinante que porta un genoma de VHB (virus de la hepatitis B) replicable (VAA-VHB). En 2-3 semanas después de la infección, se detectaron niveles altos de marcadores víricos de VHB, tales como ADN genómico de VHB y HBsAg (antígeno de superficie del VHB) en el suero de ratones infectados. Con viremia del VHB persistente y un sistema inmunológico completamente competente, el modelo VAA-VHB es adecuado para investigar la eficacia in vivo del ejemplo 4-A.
Se realizaron dos estudios in vivo independientes para evaluar la potencia antivírica del ejemplo 4-A a 100 mg/kg y 300 mg/kg respectivamente. Para cada estudio, estuvieron disponibles diez ratones C57BL/6 machos de 4-5 semanas de edad, sin patógenos específicos, del Shanghai Laboratory Animal Center of Chinese Academy of Sciences (SLAC) y se alojaron en un animalario en jaulas ventiladas individualmente en condiciones de temperatura y luz controladas siguiendo las pautas de cuidado animal institucional. Se adquirió la reserva de virus VAA-VHB del Beijing FivePlus Molecular Medicine Institute (Beijing, China). Se inyectó a ratones C57BL/6 200 pl de virus recombinante en tampón de solución salina a través de inyección en la vena de la cola. Se extrajo sangre a los ratones el día 14 después de la inyección para el seguimiento de los niveles de HBsAg, HBeAg y ADN genómico de VHB en suero, y se agruparon aleatoriamente en base a estos niveles de biomarcadores de VHB. A continuación se trataron los ratones agrupados siguiendo el diseño de estudio que se muestra en la tabla 3.
Tabla 3. Estudio in vivo en modelo de ratón AAV-HBV

Se trataron los ratones en los grupos 1 y 3 con vehículo placebo (Klucel LF al 2 %, polisorbato 80 al 0,1 % y parabenos al 0,1 % en agua); se administró por vía oral a los ratones en los grupos 2 y 4 el ejemplo 4-A a 100 mg/kg y 300 mg/kg respectivamente, cada dos días (q.o.d.). Se trataron todos los ratones durante un total de 6 semanas. Se obtuvieron muestras de suero dos veces por semana para el seguimiento de los niveles de biomarcadores de VHB. Se midió HBsAg en suero usando los kits CLIA (Autobio Diagnostics Co., Ltd, Zhengzhou, China) de acuerdo con las instrucciones del fabricante. El límite inferior de cuantificación (LLQ) para HBsAg fue de 0,1 ng/ml. Se usó una dilución de suero de 500 veces (para HBsAg) para obtener valores dentro del intervalo lineal de la curva estándar. Se extrajo ADN de VHB en suero usando un kit MagNA Pure 96 DNA and Viral NA Small Volume Kit (Roche) siguiendo las instrucciones del fabricante. Se analizaron las muestras de ADN por PCR cuantitativa ultrarrápida (qPCR) usando un conjunto de cebador y sonda específicos de VHB para amplificación y detección específicas de una región del genoma del VHB de 128 pb del nucleótido 2969 a 3096. El LLQ para el ADN de VHB fue de 20 copias/pl.
Como se muestra en la figura 3, después del tratamiento de 6 semanas, el ejemplo 4-A a 100 mg/kg indujo una reducción de más de 2 log en el ADN de VHB y una reducción de 1,5 log en HBsAg. A una dosis mayor de 300 mg/kg, el ejemplo 4-A redujo el ADN de VHB en más de 3 log y HBsAg en 2,7 log al final del tratamiento. Los resultados de este estudio demuestran claramente la eficacia antivírica in vivo del ejemplo 4-A y subrayan el potencial de los compuestos de la presente invención para desarrollar un tratamiento novedoso para enfermedades infecciosas.
Claims (18)
1. Un compuesto de fórmula (I),

en la que
R1 es hidroxi, alquilo Ci-6, alquilcarbonil C1-6-O-, alquil C1-6-S-, acido o alquenilo C2-6 ;
R2 es alquilo C1-6 , alcoxi Ci-6alquilo C1-6 , cicloalquilo C3-7 , alquinilo C2-6 , alquenilo C2-6 , bencilo y tiofenilo;
R3 es hidrógeno o alquilcarbonilo C1-6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
2. Un compuesto de acuerdo con la reivindicación 1, en el que R1 es hidroxi, alquilo C1-6 , alquil C1-6-S-, acido o alquenilo C2-6.
3. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 2, en el que R2 es alquilo C1-6, cicloalquilo C3-7, alquinilo C2-6 o alquenilo C2-6.
4. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3, en el que R3 es hidrógeno o alquilcarbonilo C1-6.
5. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en el que
R1 es haloalquilo C1-6 , alquil C1-6-S-, ciano, alquenilo C2-6 , alquilsulfonil C1-6-NH-, (alquil Ci -6)2N-, alquilcarbonil C1-6-NH- o amino heterocíclico;
R2 es hidrógeno;
R3 es hidrógeno;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
6. Un compuesto de acuerdo con la reivindicación 5, en el que R1 es alquil C1-6-S- o amino heterocíclico.
7. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 6 seleccionado de:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; acetato de [(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]; acetato de [(1S)-1-[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo];
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxietil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-3-enil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipentil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibutil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[ciclopentil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-fenil-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-3-metil-butil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)meti1]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; acetato de [[(2S,4R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropilmetilo];
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxiprop-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-[hidroxi(2-tienil)metil]tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxi-2-metoxi-etil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxialil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-((2R,3R,5S)-3-acido-5-((S)-1-hidroxietil)tetrahidrofuran-2-il)tiazolo[4,5-d]pirimidin-2,7(3H,6H)-diona;
3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1R)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]metanosulfonamida;
W-[(2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-il]acetamida; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-morfolino-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(1-piperidil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-(dimetilamino)-5-(hidroximetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; (2R,3R,5S)-2-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-5-(hidroximetil)tetrahidrofuran-3-carbonitrilo;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-(1-fluoro-1-metil-etil)-5-(hidroxirnetil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-(2-metilalil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona y 2-metilpropanoato de [(1S)-1-[(2R,4R,5S)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofuran-2-il]propilo];
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
8. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7 seleccionado de:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibutil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]piri midinc-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxiprop-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxibut-2-inil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(1-hidroxipropil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-3-acido-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxialil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
3-[(2R,3R,5S)-3-alil-5-(1-hidroxipropil)tetrahidrofuran-2-il]-5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1R)-1-hidroxipropil]-3-propil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxipropil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxibut-2-inil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-[(S)-ciclopropil(hidroxi)metil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-[(1S)-1-hidroxietil]-3-metil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona;
5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-pirrolidin-1-il-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; 5-amino-3-[(2R,3R,5S)-5-(hidroximetil)-3-metilsulfanil-tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona; y 2-metilpropanoato de [(1S)-1-[(2R,3R,5R)-5-(5-amino-2,7-dioxo-6H-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxitetrahidrofu ran-2-il]propilo];
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
9. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 8, en el que el compuesto es
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]-6H-tiazolo[4,5-d]pirimidin-2,7-diona.
10. Un compuesto de fórmula (II),

en la que
R4 es hidroxi, alquilo Ci-6, alquil C1-6-S-, acido o alquenilo C2-6 ;
R5 es alquilo C1-6 , cicloalquilo C3-7 , alquinilo C2 -6 o alquenilo C2-6 ;
R6 es hidrógeno o alquilcarbonilo C1-6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
11. Un compuesto de acuerdo con la reivindicación 10, en el que
R4 es hidroxi o alquilcarbonil C1-6-O-;
R5 es alquilo C1-6 o cicloalquilo C3-7;
R6 es hidrógeno o alquilcarbonilo C1-6 ;
o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
12. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 10 a 11 seleccionado de:
5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona;
acetato de [(2R,3R,5S)-2-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-5-(1-hidroxipropil)tetrahidrofuran-3-ilo]; propilacetato de 1-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-ilo]; acetato de [(S)-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]-ciclopropilmetilo];
y 5-amino-3-[(2R,3R,5S)-5-[ciclopropil(hidroxi)metil]-3-hidroxi-tetrahidrofuran-2-il]tiazo1o[4,5-d]pirimidin-2-ona; o sal, enantiómero o diastereómero farmacéuticamente aceptable del mismo.
13. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 10 a 12, en el que el compuesto es 5-amino-3-[(2R,3R,5S)-3-hidroxi-5-(1-hidroxipropil)tetrahidrofuran-2-il]tiazolo[4,5-d]pirimidin-2-ona.
14. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 10 a 12, en el que el compuesto es acetato de 1-[(2S,4R,5R)-5-(5-amino-2-oxo-tiazolo[4,5-d]pirimidin-3-il)-4-hidroxi-tetrahidrofuran-2-il]propilo.
15. Un procedimiento para la preparación de un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 14 que comprende las siguientes etapas:
(a) la reacción de un compuesto de fórmula (X),

con una base o reactivo de fluoruro, en la que Ra es R2 o R5; Rb es acilo, benzoílo, terc-butil(dimetil)sililo o terc-butil(difenil)sililo; Rc es hidrógeno o hidroxi;
(b) la reacción de un compuesto de fórmula (P1),

con un anhídrido de ácido o cloruro de ácido, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi;
(c) la reacción de un compuesto de fórmula (XIV),

con una base o un reactivo de fluoruro, en la que Ra es R2 o R5; Rb es acilo, benzoílo o terc-butil(difenil)sililo; Rc es hidrógeno o hidroxi; Re es R1 o R4;
(d) la reacción de un compuesto de fórmula (XXIII),

con una base, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi; Re es R1 o R4;
(e) la reacción de un compuesto de fórmula (XXVI),

con una base, en la que Rc es hidrógeno o hidroxi; Re es R1 o R4;
(f) la reacción de un compuesto de fórmula (XXXI),

con una base, en la que Ra es R2 o R5; Rc es hidrógeno o hidroxi; Re es R1 o R4; o
en la que Ra, Rb, Rd, Re, R1, R2, R3, R4, R5 y R6 se definen como en una cualquiera de las reivindicaciones 1 a 6, 10 y 11.
16. Un compuesto o sales, enantiómeros o diastereómeros farmacéuticamente aceptables de acuerdo con una cualquiera de las reivindicaciones 1 a 14 para su uso como sustancia terapéuticamente activa.
17. Una composición farmacéutica que comprende un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 14 y un vehículo terapéuticamente inerte.
18. Un compuesto o sales, enantiómeros o diastereómeros farmacéuticamente aceptables de acuerdo con una cualquiera de las reivindicaciones 1 a 14 para su uso en el tratamiento o profilaxis de infección por el virus de la hepatitis B.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2014093224 | 2014-12-08 | ||
| CN2015086987 | 2015-08-14 | ||
| PCT/EP2015/078439 WO2016091698A1 (en) | 2014-12-08 | 2015-12-03 | 3-substituted 5-amino-6h-thiazolo[4,5-d]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2864223T3 true ES2864223T3 (es) | 2021-10-13 |
Family
ID=54771116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15804128T Active ES2864223T3 (es) | 2014-12-08 | 2015-12-03 | Compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida para el tratamiento y profilaxis de infección vírica |
Country Status (30)
| Country | Link |
|---|---|
| US (4) | US9441008B2 (es) |
| EP (1) | EP3230298B1 (es) |
| JP (1) | JP6817217B2 (es) |
| KR (2) | KR102557560B1 (es) |
| CN (2) | CN106164079B (es) |
| AU (1) | AU2015359692B2 (es) |
| BR (1) | BR112017011941B1 (es) |
| CA (1) | CA2963717C (es) |
| CL (1) | CL2017001426A1 (es) |
| CR (1) | CR20170237A (es) |
| DK (1) | DK3230298T3 (es) |
| EA (1) | EA038744B1 (es) |
| ES (1) | ES2864223T3 (es) |
| HR (1) | HRP20210454T1 (es) |
| HU (1) | HUE053704T2 (es) |
| IL (1) | IL251285B (es) |
| LT (1) | LT3230298T (es) |
| MA (1) | MA41134B1 (es) |
| MX (1) | MX379474B (es) |
| MY (1) | MY194002A (es) |
| PE (1) | PE20170679A1 (es) |
| PH (1) | PH12017501061A1 (es) |
| PL (1) | PL3230298T3 (es) |
| PT (1) | PT3230298T (es) |
| RS (1) | RS61612B1 (es) |
| SG (1) | SG11201704512PA (es) |
| SI (1) | SI3230298T1 (es) |
| TW (1) | TWI733656B (es) |
| UA (1) | UA124184C2 (es) |
| WO (1) | WO2016091698A1 (es) |
Families Citing this family (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102557560B1 (ko) * | 2014-12-08 | 2023-07-20 | 에프. 호프만-라 로슈 아게 | 바이러스 감염의 치료 또는 예방을 위한 3-치환된 5-아미노-6H-티아졸로[4,5-d]피리미딘-2,7-다이온 화합물 |
| MA52701A (fr) | 2015-03-16 | 2021-03-31 | Hoffmann La Roche | Traitement combiné avec un agoniste tlr7 et un inhibiteur d'assemblage de capside du virus de l'hépatite b |
| EP4001283A1 (en) * | 2015-05-12 | 2022-05-25 | F. Hoffmann-La Roche AG | Substituted aminothiazolopyrimidinedione derivatives for the treatment and prophylaxis of virus infection |
| EP3317289B1 (en) * | 2015-06-30 | 2021-10-27 | F. Hoffmann-La Roche AG | Novel substituted aminothiazolopyrimidinedione for the treatment and prophylaxis of virus infection |
| CN108430515B (zh) | 2015-10-29 | 2021-11-12 | 诺华股份有限公司 | 包含toll-样受体激动剂的抗体缀合物 |
| MA46535A (fr) | 2016-10-14 | 2019-08-21 | Prec Biosciences Inc | Méganucléases modifiées spécifiques de séquences de reconnaissance dans le génome du virus de l'hépatite b |
| CN110167947B (zh) * | 2017-01-06 | 2022-05-03 | 豪夫迈·罗氏有限公司 | 乙酸[(1S)-1-[(2S,4R,5R)-5-(5-氨基-2-氧代-噻唑并[4,5-d]嘧啶-3-基)-4-羟基-四氢呋喃-2-基]丙基]酯的固体形式 |
| CA3048768A1 (en) | 2017-01-06 | 2018-07-12 | F. Hoffmann-La Roche Ag | Process for the preparation of 3-substituted 5-amino-6h-thiazolo[4,5-d]pyrimidine-2,7-dione compounds |
| US12012421B2 (en) | 2017-07-07 | 2024-06-18 | Hoffmann-La Roche Inc. | Solid forms of [(1S)-1-[(2S,4R,5R)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-tetrahydrofuran-2-yl]propyl] acetate |
| US12012420B2 (en) | 2017-11-21 | 2024-06-18 | Hoffmann-La Roche, Inc. | Solid forms of [(1S)-1-[(2S,4R,5R)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-tetrahydrofuran-2-yl]propyl] acetate |
| KR20200100113A (ko) | 2017-12-15 | 2020-08-25 | 실버백 테라퓨틱스, 인크. | 간염 치료용 항체 구조물-약물 접합체 |
| CN111511754B (zh) | 2017-12-20 | 2023-09-12 | 捷克共和国有机化学与生物化学研究所 | 活化sting转接蛋白的具有膦酸酯键的2’3’环状二核苷酸 |
| EP3728283B1 (en) | 2017-12-20 | 2023-11-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3' cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| KR102526964B1 (ko) | 2018-02-26 | 2023-04-28 | 길리애드 사이언시즈, 인코포레이티드 | Hbv 복제 억제제로서의 치환된 피롤리진 화합물 |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| TWI833744B (zh) | 2018-04-06 | 2024-03-01 | 捷克科學院有機化學與生物化學研究所 | 3'3'-環二核苷酸 |
| TWI818007B (zh) | 2018-04-06 | 2023-10-11 | 捷克科學院有機化學與生物化學研究所 | 2'3'-環二核苷酸 |
| TW202005654A (zh) | 2018-04-06 | 2020-02-01 | 捷克科學院有機化學與生物化學研究所 | 2,2,─環二核苷酸 |
| TW201945388A (zh) | 2018-04-12 | 2019-12-01 | 美商精密生物科學公司 | 對b型肝炎病毒基因體中之識別序列具有特異性之最佳化之經工程化巨核酸酶 |
| US20190359645A1 (en) | 2018-05-03 | 2019-11-28 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides comprising carbocyclic nucleotide |
| US11059824B2 (en) | 2018-05-25 | 2021-07-13 | Primmune Therapeutics, Inc. | Substituted purines as TLR7 agonists |
| BR112021000538A2 (pt) | 2018-07-13 | 2021-04-06 | F. Hoffmann-La Roche Ag | Oligonucleotídeos para modular a expressão de rtel1 |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| FI3873903T3 (fi) | 2018-10-31 | 2024-03-26 | Gilead Sciences Inc | Substituoituja 6-azabentsiimidatsoliyhdisteitä HPK1-inhibiittoreina |
| KR102658602B1 (ko) | 2018-10-31 | 2024-04-19 | 길리애드 사이언시즈, 인코포레이티드 | Hpk1 억제 활성을 갖는 치환된 6-아자벤즈이미다졸 화합물 |
| WO2020162705A1 (ko) | 2019-02-08 | 2020-08-13 | 성균관대학교산학협력단 | 톨-유사 수용체 7 또는 8 작용자와 콜레스테롤의 결합체 및 그 용도 |
| EP3934757B1 (en) | 2019-03-07 | 2023-02-22 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| CA3129011C (en) | 2019-03-07 | 2023-12-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| EP3935065A1 (en) | 2019-03-07 | 2022-01-12 | Institute of Organic Chemistry and Biochemistry ASCR, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| TW202210480A (zh) | 2019-04-17 | 2022-03-16 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TWI751517B (zh) | 2019-04-17 | 2022-01-01 | 美商基利科學股份有限公司 | 類鐸受體調節劑之固體形式 |
| TWI826690B (zh) | 2019-05-23 | 2023-12-21 | 美商基利科學股份有限公司 | 經取代之烯吲哚酮化物及其用途 |
| WO2020257407A1 (en) | 2019-06-19 | 2020-12-24 | Silverback Therapeutics, Inc. | Anti-mesothelin antibodies and immunoconjugates thereof |
| US11339159B2 (en) | 2019-07-17 | 2022-05-24 | Pfizer Inc. | Toll-like receptor agonists |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| KR20220074917A (ko) | 2019-09-30 | 2022-06-03 | 길리애드 사이언시즈, 인코포레이티드 | Hbv 백신 및 hbv를 치료하는 방법 |
| IL293293A (en) | 2019-11-26 | 2022-07-01 | Primmune Therapeutics Inc | Tlr7 agonists |
| DK4069729T3 (da) | 2019-12-06 | 2025-04-07 | Prec Biosciences Inc | Optimerede, modificerede meganukleaser med specificitet for en genkendelsessekvens i hepatitis b-virusgenomet |
| EP4081639A1 (en) | 2019-12-24 | 2022-11-02 | F. Hoffmann-La Roche AG | Pharmaceutical combination of a therapeutic oligonucleotide targeting hbv and a tlr7 agonist for treatment of hbv |
| AU2020415307A1 (en) * | 2019-12-24 | 2022-06-09 | F. Hoffmann-La Roche Ag | Method of treating virus infection using a TLR7 agonist |
| EP4081217A1 (en) | 2019-12-24 | 2022-11-02 | F. Hoffmann-La Roche AG | Pharmaceutical combination of antiviral agents targeting hbv and/or an immune modulator for treatment of hbv |
| CA3170551A1 (en) | 2020-03-02 | 2021-09-10 | Yong Taik Lim | Live-pathogen-mimetic nanoparticles based on pathogen cell wall skeleton, and production method thereof |
| AR121620A1 (es) | 2020-03-20 | 2022-06-22 | Gilead Sciences Inc | Profármacos de nucleósidos 4-c-sustituidos-2-halo-2-deoxiadenosina y métodos de preparación y uso de los mismos |
| US20230346924A1 (en) | 2020-08-04 | 2023-11-02 | Progeneer Inc. | Mrna vaccine comprising adjuvant capable of kinetic control |
| JP2023536954A (ja) | 2020-08-04 | 2023-08-30 | プロジェニア インコーポレイテッド | 活性化部位が一時的に不活性化したトール様受容体7または8作用薬と機能性薬物の結合体およびその用途 |
| WO2022031011A1 (ko) | 2020-08-04 | 2022-02-10 | 성균관대학교산학협력단 | 동력학적으로 작용하는 아주번트 앙상블 |
| JP2023536999A (ja) | 2020-08-05 | 2023-08-30 | エフ. ホフマン-ラ ロシュ アーゲー | B型肝炎患者のオリゴヌクレオチド処置 |
| CA3215049A1 (en) | 2021-04-10 | 2022-10-13 | Baiteng ZHAO | Folr1 binding agents, conjugates thereof and methods of using the same |
| WO2022226317A1 (en) | 2021-04-23 | 2022-10-27 | Profoundbio Us Co. | Anti-cd70 antibodies, conjugates thereof and methods of using the same |
| EP4337223A1 (en) | 2021-05-13 | 2024-03-20 | Gilead Sciences, Inc. | Combination of a tlr8 modulating compound and anti-hbv sirna therapeutics |
| AU2022299051B2 (en) | 2021-06-23 | 2025-03-13 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| AU2022298639C1 (en) | 2021-06-23 | 2025-07-17 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| KR20240025616A (ko) | 2021-06-23 | 2024-02-27 | 길리애드 사이언시즈, 인코포레이티드 | 다이아실글리세롤 키나제 조절 화합물 |
| KR20240005901A (ko) | 2021-06-23 | 2024-01-12 | 길리애드 사이언시즈, 인코포레이티드 | 디아실글리세롤 키나제 조절 화합물 |
| TW202320857A (zh) | 2021-07-06 | 2023-06-01 | 美商普方生物製藥美國公司 | 連接子、藥物連接子及其結合物及其使用方法 |
| IL312635A (en) | 2021-11-11 | 2024-07-01 | Hoffmann La Roche | Pharmaceutical combinations for treatment of hbv |
| CN121620516A (zh) * | 2023-08-08 | 2026-03-06 | 豪夫迈·罗氏有限公司 | 用于制备3-取代的5-氨基-6H-噻唑并[4,5-d]嘧啶-2,7-二酮化合物的方法 |
| AR133955A1 (es) | 2023-09-26 | 2025-11-19 | Profoundbio Us Co | Agentes de unión a ptk7, conjugados de éstos y métodos de uso de los mismos |
| TW202547866A (zh) | 2024-01-10 | 2025-12-16 | 丹麥商珍美寶股份有限公司 | Slitrk6結合劑、其共軛物及使用彼之方法 |
| US20250381289A1 (en) | 2024-02-29 | 2025-12-18 | Genmab A/S | Egfr and c-met bispecific binding agents, conjugates thereof and methods of using the same |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| US20250345389A1 (en) | 2024-05-13 | 2025-11-13 | Gilead Sciences, Inc. | Combination therapies |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5476659A (en) | 1982-11-09 | 1995-12-19 | The Scripps Research Institute | Cancerous B cell treatment using substituted nucleoside derivatives |
| US5166141A (en) | 1983-11-01 | 1992-11-24 | Scripps Clinic And Research Foundation | Immunostimulating 7-deaza-7-oxa- and 7-deaza-7-oxo-analogs of 8-substituted-guanine-9-(1'-beta-D-aldoglycosidyl) derivatives and methods of treating test animals |
| US5041426A (en) | 1987-12-21 | 1991-08-20 | Brigham Young University | Immune system enhancing 3-β-d-ribofuranosylthiazolo[4,5-d]pyridimine nucleosides and nucleotides |
| HUP0001186A3 (en) | 1996-10-16 | 2002-04-29 | Icn Pharmaceuticals Inc Costa | Purine l-nucleosides,analogs and pharmaceutical compositions thereof |
| EP1072607A3 (en) | 1996-10-16 | 2001-09-12 | ICN Pharmaceuticals, Inc. | Purine L-nucleosides, analogs and uses therof |
| WO2005016235A2 (en) | 2003-04-14 | 2005-02-24 | The Regents Of The University Of California | Combined use of impdh inhibitors with toll-like receptor agonists |
| ES2342069T4 (es) | 2003-09-05 | 2011-01-26 | Anadys Pharmaceuticals, Inc. | Ligandos de tlr7 para el tratamiento de la hepatitis c. |
| CN101212968B (zh) * | 2004-12-17 | 2011-11-16 | 安那迪斯药品股份有限公司 | 3,5-二取代的和3,5,7-三取代的-3H-噁唑并以及3H-噻唑并[4,5-d]嘧啶-2-酮化合物及其前药 |
| PL1824482T3 (pl) * | 2004-12-17 | 2014-07-31 | Anadys Pharmaceuticals Inc | 3,5-Dipodstawione oraz 3,5,7-tripodstawione związki 3H-oksazolo- oraz 3H-tiazolo[4,5-d]pirymidyn-2-onowe i ich proleki |
| SG152296A1 (en) | 2005-11-21 | 2009-05-29 | Anadys Pharmaceuticals Inc | Novel process for the preparation of 5-amino-3h-thiazolo[4,5-d]pyrimidin-2- one |
| AR061024A1 (es) | 2006-05-22 | 2008-07-30 | Novartis Ag | Maleato de 5-amino-3- (2'3'-di-o-acetil-beta-d-ribofuranosil)-3h-tiazolo[4,5-d] pirimidin-2-ona. |
| WO2007150002A2 (en) * | 2006-06-22 | 2007-12-27 | Anadys Pharmaceuticals, Inc. | PRODRUGS OF 5-AMINO-3-(3'-DEOXY-β-D-RIBOFURANOSYL)-THIAZOLO[4,5-d] PYRIMIDIN-2,7-DIONE |
| BRPI0714831A2 (pt) | 2006-07-18 | 2013-04-02 | Anadys Pharmaceuticals Inc | composto, composiÇço farmacÊutica e mÉtodos de modulaÇço de atividades imuno da citoquina em paciente, de tratamento de infecÇço de vÍrus c da hepatite em paciente e de desordem relacionada com proliferaÇço em mamÍfero necessitado do mesmo |
| BRPI0717741A2 (pt) | 2006-10-17 | 2014-04-08 | Anadys Pharmaceuticals Inc | Compostos e respectivos método de preparação, método de redução de composto de sulfonil substituído, composição farmacêutica método de tratamento ou prevenção de doença |
| WO2009026292A1 (en) * | 2007-08-20 | 2009-02-26 | Anadys Pharmaceuticals, Inc. | Dosing methods for treating disease |
| KR102557560B1 (ko) | 2014-12-08 | 2023-07-20 | 에프. 호프만-라 로슈 아게 | 바이러스 감염의 치료 또는 예방을 위한 3-치환된 5-아미노-6H-티아졸로[4,5-d]피리미딘-2,7-다이온 화합물 |
| MA52701A (fr) | 2015-03-16 | 2021-03-31 | Hoffmann La Roche | Traitement combiné avec un agoniste tlr7 et un inhibiteur d'assemblage de capside du virus de l'hépatite b |
| EP4001283A1 (en) | 2015-05-12 | 2022-05-25 | F. Hoffmann-La Roche AG | Substituted aminothiazolopyrimidinedione derivatives for the treatment and prophylaxis of virus infection |
| EP3317289B1 (en) | 2015-06-30 | 2021-10-27 | F. Hoffmann-La Roche AG | Novel substituted aminothiazolopyrimidinedione for the treatment and prophylaxis of virus infection |
-
2015
- 2015-12-03 KR KR1020177015128A patent/KR102557560B1/ko active Active
- 2015-12-03 KR KR1020237024143A patent/KR20230113824A/ko not_active Ceased
- 2015-12-03 ES ES15804128T patent/ES2864223T3/es active Active
- 2015-12-03 AU AU2015359692A patent/AU2015359692B2/en active Active
- 2015-12-03 MX MX2017007044A patent/MX379474B/es unknown
- 2015-12-03 EA EA201791011A patent/EA038744B1/ru unknown
- 2015-12-03 BR BR112017011941-2A patent/BR112017011941B1/pt active IP Right Grant
- 2015-12-03 JP JP2017548325A patent/JP6817217B2/ja active Active
- 2015-12-03 CN CN201580018026.1A patent/CN106164079B/zh active Active
- 2015-12-03 RS RS20210297A patent/RS61612B1/sr unknown
- 2015-12-03 CN CN201710845723.5A patent/CN107445980B/zh active Active
- 2015-12-03 CR CR20170237A patent/CR20170237A/es unknown
- 2015-12-03 SG SG11201704512PA patent/SG11201704512PA/en unknown
- 2015-12-03 PL PL15804128T patent/PL3230298T3/pl unknown
- 2015-12-03 MY MYPI2017702051A patent/MY194002A/en unknown
- 2015-12-03 LT LTEP15804128.5T patent/LT3230298T/lt unknown
- 2015-12-03 WO PCT/EP2015/078439 patent/WO2016091698A1/en not_active Ceased
- 2015-12-03 EP EP15804128.5A patent/EP3230298B1/en active Active
- 2015-12-03 MA MA41134A patent/MA41134B1/fr unknown
- 2015-12-03 PT PT158041285T patent/PT3230298T/pt unknown
- 2015-12-03 UA UAA201706823A patent/UA124184C2/uk unknown
- 2015-12-03 PE PE2017000551A patent/PE20170679A1/es unknown
- 2015-12-03 CA CA2963717A patent/CA2963717C/en active Active
- 2015-12-03 HU HUE15804128A patent/HUE053704T2/hu unknown
- 2015-12-03 US US14/958,619 patent/US9441008B2/en active Active
- 2015-12-03 DK DK15804128.5T patent/DK3230298T3/da active
- 2015-12-03 HR HRP20210454TT patent/HRP20210454T1/hr unknown
- 2015-12-03 SI SI201531553T patent/SI3230298T1/sl unknown
- 2015-12-07 TW TW104140978A patent/TWI733656B/zh active
-
2016
- 2016-07-11 US US15/207,338 patent/US10040815B2/en active Active
-
2017
- 2017-03-20 IL IL251285A patent/IL251285B/en unknown
- 2017-06-05 CL CL2017001426A patent/CL2017001426A1/es unknown
- 2017-06-07 PH PH12017501061A patent/PH12017501061A1/en unknown
-
2018
- 2018-07-02 US US16/025,170 patent/US10618929B2/en active Active
-
2020
- 2020-04-13 US US16/847,552 patent/US11180525B2/en active Active
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2864223T3 (es) | Compuestos de 5-amino-6H-tiazolo[4,5-d]pirimidin-2,7-diona 3-sustituida para el tratamiento y profilaxis de infección vírica | |
| US10000512B2 (en) | Compounds for the treatment of infectious diseases | |
| US10065973B2 (en) | Substituted aminothiazolopyrimidinedione for the treatment and prophylaxis of virus infection | |
| WO2016180691A1 (en) | Novel oxathiolane carboxylic acids and derivatives for the treatment and prophylaxis of virus infection | |
| HK1245764B (zh) | 3-取代的5-氨基-6h-噻唑并[4,5-d]嘧啶-2,7-二酮化合物 | |
| HK1229793B (en) | 3-substituted 5-amino-6h-thiazolo[4,5-d]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection | |
| HK1229793A1 (en) | 3-substituted 5-amino-6h-thiazolo[4,5-d]pyrimidine-2,7-dione compounds for the treatment and prophylaxis of virus infection | |
| HK1248711B (zh) | 用於治疗和预防病毒感染的新的取代的氨基噻唑并嘧啶二酮 |