ES2790640T3 - Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante - Google Patents
Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante Download PDFInfo
- Publication number
- ES2790640T3 ES2790640T3 ES18198969T ES18198969T ES2790640T3 ES 2790640 T3 ES2790640 T3 ES 2790640T3 ES 18198969 T ES18198969 T ES 18198969T ES 18198969 T ES18198969 T ES 18198969T ES 2790640 T3 ES2790640 T3 ES 2790640T3
- Authority
- ES
- Spain
- Prior art keywords
- mmol
- chloro
- oxo
- mixture
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 C[C@](C1=Cc2cc(*)ccc2NC1=O)N Chemical compound C[C@](C1=Cc2cc(*)ccc2NC1=O)N 0.000 description 2
- SFOFUPMHMIGQMT-UHFFFAOYSA-N CC(C)(C)OC(Nc(c(F)c1)ccc1Cl)=O Chemical compound CC(C)(C)OC(Nc(c(F)c1)ccc1Cl)=O SFOFUPMHMIGQMT-UHFFFAOYSA-N 0.000 description 1
- IUZJFCDYXXLQGV-REZTVBANSA-N CC(C)(C)S(/N=C/C(C(Nc1c2)=O)=Cc1cc(Cl)c2F)=O Chemical compound CC(C)(C)S(/N=C/C(C(Nc1c2)=O)=Cc1cc(Cl)c2F)=O IUZJFCDYXXLQGV-REZTVBANSA-N 0.000 description 1
- QRZBEHUXMWTNTE-UHFFFAOYSA-N CC(C)Oc(cc(cc1)N)c1Cl Chemical compound CC(C)Oc(cc(cc1)N)c1Cl QRZBEHUXMWTNTE-UHFFFAOYSA-N 0.000 description 1
- HUUXACSGKZKEEJ-UHFFFAOYSA-N CC(c(cc(cc(c(OC)c1)Cl)c1n1)c1OC)O Chemical compound CC(c(cc(cc(c(OC)c1)Cl)c1n1)c1OC)O HUUXACSGKZKEEJ-UHFFFAOYSA-N 0.000 description 1
- HMOJUEVHEFCRHO-UHFFFAOYSA-N CCC(C(Nc1c2)=O)=Cc1cc(Cl)c2F Chemical compound CCC(C(Nc1c2)=O)=Cc1cc(Cl)c2F HMOJUEVHEFCRHO-UHFFFAOYSA-N 0.000 description 1
- VMUOXNQVBSAYQR-UHFFFAOYSA-N CN(C(C#N)=CC=C1F)C1=O Chemical compound CN(C(C#N)=CC=C1F)C1=O VMUOXNQVBSAYQR-UHFFFAOYSA-N 0.000 description 1
- SLCDOTDJNCIYCJ-VIFPVBQESA-N C[C@@H](C1=Nc2cc(Cl)ccc2NC1=O)NC1=CC=C(C#N)N(C)C1=O Chemical compound C[C@@H](C1=Nc2cc(Cl)ccc2NC1=O)NC1=CC=C(C#N)N(C)C1=O SLCDOTDJNCIYCJ-VIFPVBQESA-N 0.000 description 1
- NNSNVNVXUVDZMJ-UHFFFAOYSA-N Nc(c(CO)c1)cc(F)c1Cl Chemical compound Nc(c(CO)c1)cc(F)c1Cl NNSNVNVXUVDZMJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Una composición farmacéutica que comprende el compuesto de Fórmula I-13 o una sal farmacéuticamente aceptable del mismo y un portador farmacéuticamente aceptable: **(Ver fórmula)** en donde la composición contiene de aproximadamente el 0,1% a aproximadamente el 99% del compuesto de Fórmula I-13 en peso o volumen.
Description
DESCRIPCIÓN
Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante
Campo de la invención
La presente invención está dirigida a inhibidores de proteínas de deshidrogenasa isocitrato mutante (mt-IDH) con actividad neomórfica útil en el tratamiento de enfermedades o trastornos asociados con tales proteínas IDH mutantes que incluyen trastornos de proliferación celular y cánceres. Específicamente, la invención se refiere a compuestos y composiciones que inhiben mt-IDH, métodos para tratar las enfermedades o trastornos asociados con mt-IDH, y métodos de síntesis de estos compuestos.
Antecedentes de la invención
Las deshidrogenasas de isocitrato (IDH) son enzimas que participan en el ciclo del ácido cítrico (metabolismo celular). Catizan la descarboxilación oxidativa de isocitrato a 2-oxoglutarato (es decir, a-cetoglutarato, a-KG). Hay tres isoformas dentro de la familia IDH. IDH-1, expresada en el citoplasma y el peroxisoma, IDH-2, localizada en las mitocondrias, ambas utilizan NADP+ como cofactor y existen como homodímeros. IDH-3 se localiza en la matriz mitocondrial y utiliza NAD+ como cofactor y existe como tetrámero. Se han identificado mutaciones en IDH-1 (citosólica) e IDH-2 (mitocondrial) en diversas enfermedades o trastornos que incluyen glioma, glioblastoma multiforme, paraganglioma, tumores primigeniales neuroectodérmicos primordiales supratentoriales, leucemia mieloide aguda (AML), cáncer de próstata, cáncer de tiroides, cáncer de colon, condrosarcoma, colangiocarcinoma, linfoma periférico de células T y melanoma (L. Deng y otros, Trends Mol. Med., 2010, 16, 387; T. Shibata y otros, Am. J. Pathol., 201 1, 178 (3), 1395; Gaal et al., J. Clin. Endocrinol. Metab. 2010; Hayden et al., Cell Cycle, 2009; Balss et al., Acta Neuropathol., 2008). Las mutaciones se han encontrado en o cerca de los residuos clave en el sitio activo: G97D, R100, R132, H133Q y A134D para IDH1, y R140 y R172 para IDH2. (Véase L. Deng et al., Nature, 2009, 462, 739; L. Sellner et al., Eur. J. Haematol., 2011, 85, 457).
Se ha demostrado que las formas mutantes de IDH-1 e IDH-2 pierden actividad de tipo salvaje, y en cambio exhiben una actividad neomorfa (también conocida como actividad de ganancia de función), de reducir el alfacetoglutarato a 2-hidroxiglutarato (2 -HG). (véase PS Ward et al., Cancer Cell, 2010, 17, 225; Zhao et al., Science 324, 261 (2009); Dang et.al Nature 462, 739 (2009)). En general, la producción de 2-HG es enantioespecífica, lo que resulta en la generación del enantiómero D (también conocido como el enantiómero R o R-2-HG). Las células normales tienen niveles basales bajos de 2-HG, mientras que las células que albergan mutaciones en IDH1 o IDH2 muestran niveles significativamente elevados de 2-HG. También se han detectado altos niveles de 2-HG en tumores que albergan las mutaciones. Por ejemplo, se han detectado niveles altos de 2-HG en el plasma de pacientes con IDH mutante que contienen AML. (véase S. Gross et al., J. Exp. Med., 2010, 207 (2), 339). Se ha demostrado que los altos niveles de 2-HG bloquean el ADN dependiente de a-KG y las desmetilasas de histonas, y en última instancia, resultan en una desdiferenciación inadecuada de células progenitoras hematopoyéticas en pacientes con AML (Wang et al., Science 340, 622 (2013) ; Losman et al., Science 339, 1621 (2013)).
Además, los pacientes con enfermedad de Oilier y síndrome de Mafucci (dos trastornos raros que predisponen a los tumores cartilaginosos) han demostrado ser un mosaico somático para las mutaciones de IDH1 y 2 y exhiben altos niveles de D-2-HG. (véase Amary et al., Nature Genetics, 2011 y Pansuriya et al., Nature Genetics, 2011).
La inhibición de mt-IDHs y su actividad neomórfica con inhibidores de moléculas pequeñas, por lo tanto, tiene el potencial de ser un tratamiento para los cánceres y otros trastornos de la proliferación celular.
Sumario de la invención
La presente invención proporciona composiciones farmacéuticas que comprenden el compuesto de Fórmula I-13 o una sal farmacéuticamente aceptable del mismo y un portador farmacéuticamente aceptable:

en donde la composición contiene de aproximadamente el 0,1% a aproximadamente el 99% del compuesto de Fórmula 1-13 en peso o volumen.
La presente invención también proporciona mezclas que comprenden el compuesto de Fórmula I-13 de la invención y un compuesto de Fórmula II-1 y un compuesto de Fórmula III:

Hal = Cl o F.
La presente invención también proporciona un proceso para preparar un compuesto de Fórmula 1-13 o una sal farmacéuticamente aceptable del mismo:

en donde dicho proceso comprende el paso de hacer reaccionar un compuesto de Fórmula II-1 con un compuesto de Fórmula III:

Hal = Cl o F.
Un primer aspecto de la divulgación se refiere a compuestos de Fórmula I:

y sales farmacéuticas, enantiómeros, hidratos, solvatos y tautómeros de los mismos,
en donde:
cada W1 y W2 es independientemente CH, CF o N;
W3 es independientemente CR2 o N;
U es N o CR6;
A se selecciona del grupo que consiste en H, D, halógeno, CN, -CHO, -COOH, - COOR, -CONH2, -CONHR, R'S(O)2-, -O(CH2)nC(O)R', R'SO-, heteroarilo, -SOMe, -SO2Me,

en donde X e Y son independientemente en cada aparición C, N, NR', S y O, siempre que el anillo que contiene
X e Y no pueda tener más de 4 N o átomos de NH o más de un átomo S o O, y en donde S y O no son contiguos;
R y R' cada vez que aparecen se seleccionan independientemente del grupo que consiste en H, OH, CN, -CH2CN, halógeno, -NR7R8, CHCF2, CF3, alquilo C1-C6, R7S(O)2-, alcoxi C1-C6, alquenilo C2-C6, a cicloalquilo C3-C8, cicloalquilalquilo C3-C8, heterociclilo de 3 a 8 miembros, arilo y heteroarilo, en donde cada R está opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en OH, halógeno, alcoxi C1-C6, NH2, R7S(O)2-, CN, cicloalquilo C3-C8, heterociclilo de 3 a 8 miembros, arilo, heteroarilo y R7S(O)-;
R1 es independientemente OH, CN, halógeno, CHCF2, CF3, alquilo C1-C6, alcoxi C1-C6, alquenilo C2-C6, alquenilo
C2-C6, cicloalquilo C3-C8, heterociclilo, arilo o heteroarilo de 3 a 8 miembros, en donde cada alquilo C1-C6, alquenilo C2-C6, alquinilo C2-C6, cicloalquilo C3-C8, heterociclilo, arilo o heteroarilo de 3 a 8 miembros está opcionalmente sustituido una o más veces con sustituyentes seleccionados del grupo que consiste en halógeno,
OH, NH2, CN, alquilo C1-C6 y alcoxi C1-C6;
cada R2 es independientemente H, OH, CN, halógeno, CF3, CHF2, bencilo, alquilo C1-C6, alcoxi C1-C6, NH2, -O(CH2)nR', -O(CH2)nC(O)NHR', -O(CH2)nC(O)R', NHR7, -N(R7)(R8), NHC(O)R7, NHS(O)R7, NHS(O)2R7, NHC(O)OR7, NHC(O)NHR7, -S(O)2NHR7, NHC(O)N(R8)R7, OCH2R7, CHRR' u OCHR'R7, en donde alquilo C1-C6, alcoxi C1-C6 está opcionalmente sustituido con uno o más sustituyentes seleccionados del grupo que consiste en alquilo C1-C6, alcoxi C1-C6, alquenilo C2-C6, alquinilo C2-C6, cicloalquilo C3-C8, cicloalquilo C3-C8 s uno o más halógenos, heterociclilo de 3 a 8 miembros, arilo, -heteroaril-C(O)NH2, y heteroarilo;
o R1 y R2 pueden combinarse para formar un cicloalquilo C4-C6 o un heterociclilo de 3 a 8 miembros que contiene al menos un átomo seleccionado del grupo que consiste en N, O y S;
R3 es H, D, alquilo C1-C6, o; -OH;
R4 y R5 son independientemente H, D, halógeno, CH2OH, alquilo C1-C3 o alquilo C1-C3 sustituido con halógeno, o
R4 y R5 cuando se combinan pueden formar un cicloalquilo C3-C6 o heterociclilo C3-C6;
cada R6 es H, halógeno, alquilo C1-C6, alquilo C1-C6 sustituido con halógeno, alcoxi C1-C6, alcoxi C1-C6 sustituido con uno o más halógenos, alquenilo C2-C6, alquinilo C2-C6, cicloalquilo C3-C8, heterociclilo de 8 miembros, arilo o heteroarilo;
R7 y R8 son independientemente H, alquilo C1-C6, alcoxi C1-C6, alquenilo C2-C6, alquinilo C2-C6, cicloalquilo C8, heterociclilo, arilo y heteroarilo de 3 a 8 miembros; o cuando se combinan, R7 y R8 pueden formar un anillo heterociclilo o heteroarilo de 3 a 8 miembros;
Rg es independientemente H, D, CD3, CF3, alquilo C1-C6, alquenilo C2-6, alquinilo C3-6, cicloalquilo C3-C8, en donde el alquilo, alquenilo, alquinilo y cicloalquilo está opcionalmente sustituido con amino, OH, halo o alcoxi;
n es 0, 1 o 2; y
r es 0, 1 o 2;
con la condición de que cuando A es H, entonces R1 no es alquilo C1-C6 o alcoxi C1-C6 y R1 y R2 no pueden combinarse para formar un heterociclilo de 3 a 8 miembros.
Otro aspecto de la divulgación se refiere a un método para tratar una enfermedad o trastorno asociado con la deshidrogenasa isocitrato mutante. El método implica administrar a un paciente que necesita un tratamiento para enfermedades o trastornos asociados con la deshidrogenasa isocitrato mutante, una cantidad eficaz de un compuesto de Fórmula I.
Otro aspecto de la divulgación se refiere a un método que inhibe la deshidrogenasa isocitrato mutante. El método implica administrar a un paciente que lo necesite una cantidad eficaz del compuesto de Fórmula I.
Otro aspecto de la divulgación se refiere a un método para reducir el 2-hidroxiglutarato. El método comprende administrar a un paciente que lo necesite una cantidad eficaz del compuesto de Fórmula I.
Otro aspecto de la divulgación se refiere a composiciones farmacéuticas que comprenden un compuesto de Fórmula I y un vehículo farmacéuticamente aceptable. El vehículo farmacéuticamente aceptable puede incluir además un excipiente, diluyente o tensioactivo.
La presente divulgación proporciona además métodos de tratamiento de enfermedades y cánceres de proliferación celular que incluyen, sin limitación, glioma, glioblastoma multiforme, paraganglioma, tumores neuroectodérmicos prematiales supratentoriales, leucemia mieloide aguda (AML) cáncer de próstata, cáncer de tiroides, cáncer de colon, cáncer de colon, colangiocarcinoma, linfoma periférico de células T, melanoma, colangiocarcinoma intrahepático (IHCC), síndrome mielodisplásico (MDS), enfermedad mieloproliferativa (MPD) y
otros tumores sólidos.
La presente invención también proporciona potentes inhibidores de mt-IDH con excelentes propiedades de tipo farmacológico para los cánceres y otros trastornos de la proliferación celular. Los inhibidores de la presente invención pueden dirigirse a IDH1 o IDH2 mutados.
La presente invención proporciona además el desarrollo de inhibidores de IDH potentes, oralmente activos y selectivos como agentes terapéuticos para diversas enfermedades o trastornos que incluyen cánceres. La divulgación también proporciona dichos agentes para su uso en un tratamiento para cánceres sólidos y hematológicos para los cuales actualmente no hay terapias dirigidas disponibles para pacientes que padecen estas afecciones o trastornos.
Breve descripción de los dibujos de la invención.
La Figura 1 ilustra un gráfico que muestra la potencia de los inhibidores de IDH1 en el ensayo enzimático IDH1-R132H utilizando los compuestos 1-1, 1-5 y 1-20.
Descripción detallada de la invención
Las mutaciones IDH1 o IDH2 son un objetivo validado genéticamente en muchos cánceres sólidos y hematológicos, pero actualmente no hay terapias dirigidas disponibles para pacientes que necesitan tratamiento para afecciones específicas asociadas con la actividad mt-IDH. La IDH no mutante (por ejemplo, de tipo salvaje) cataliza la descarboxilación oxidativa de isocitrato a a-cetoglutarato, reduciendo así NAD+ (NADP+) a NADh (NADPH) (documento WO 2013/102431 a Cianchetta et al.). Las mutaciones de IDH presentes en ciertas células cancerosas dan como resultado una nueva capacidad de la enzima para catalizar la reducción dependiente de NADPH de a-cetoglutarato R(-)-2-hidroxiglutarato (2HG). 2HG no está formado por IDH de tipo salvaje. La producción de 2HG contribuye a la formación y progresión del cáncer (Dang, L et al., Nature, 2009, 462: 739-44, que se incorpora aquí como referencia en su totalidad). La presente divulgación proporciona inhibidores de mt-IDH y medidas profilácticas para reducir la formación y progresión de 2HG en las células.
En un primer aspecto de la divulgación, se describen los compuestos de Fórmula I:

y sales farmacéuticamente aceptables, enantiómeros, hidratos, solvatos y tautómeros de los mismos, en los que A, U, W1, W2, W3, R1-R6 y R9 son como se describió anteriormente.
Los detalles de la invención se exponen en la siguiente descripción adjunta. Aunque los métodos y materiales similares o equivalentes a los descritos en el presente documento se pueden usar en la práctica o el ensayo de la presente invención, ahora se describen métodos y materiales ilustrativos. Otras características, objetos y ventajas de la invención serán evidentes a partir de la descripción y de las reivindicaciones. En la especificación y las reivindicaciones adjuntas, las formas singulares también incluyen el plural a menos que el contexto indique claramente lo contrario. A menos que se defina lo contrario, todos los términos técnicos y científicos utilizados en este documento tienen el mismo significado que entiende comúnmente un experto en la técnica a la que pertenece esta invención.
Definiciones
Los artículos "un" y "una" se utilizan en esta divulgación para referirse a uno o más de uno (es decir, al menos uno) del objeto gramatical del artículo. A modo de ejemplo, "un elemento" significa un elemento o más de un elemento.
El término "y/o" se usa en esta descripción para significar "y" u "o" a menos que se indique lo contrario. Se entiende que el término "opcionalmente sustituido" significa que un resto químico dado (por ejemplo, un grupo alquilo) puede (pero no se requiere que) estar unido a otros sustituyentes (por ejemplo, heteroátomos). Por ejemplo, un grupo alquilo que está opcionalmente sustituido puede ser una cadena alquilo completamente saturada
(es decir, un hidrocarburo puro). Alternativamente, el mismo grupo alquilo opcionalmente sustituido puede tener sustituyentes diferentes de hidrógeno. Por ejemplo, puede, en cualquier punto a lo largo de la cadena, estar unido a un átomo de halógeno, un grupo hidroxilo o cualquier otro sustituyente descrito en el presente documento. Por lo tanto, el término "opcionalmente sustituido" significa que un resto químico dado tiene el potencial de contener otros grupos funcionales, pero no tiene necesariamente otros grupos funcionales. Los sustituyentes adecuados utilizados en la sustitución opcional de los grupos descritos incluyen, sin limitación, halógeno, oxo, CN, -COOH, -CH2CN, -O alquilo C1-C6, alquilo C1-C6, alquenilo -OC1-C6, alquinilo -CO1-C6, alquenilo C1-C6, alquinilo C1-C6, -OH, -OP(O)(OH)2, -OC(O)alquilo C1-C6, -C(O)alquilo C1-C6, -OC(O)alquilo OC1-C6, NH2, NH (alquilo C1-C6), N(alquilo C1-Ca)2, -NHC(O)alquilo Ci-Ca,
-C(o)NHC1alquilo, -S(O)2-C1-Caalquilo, -S(O)NHC1-Caalquilo, y S(O)N (C1-Caalquilo)2
A menos que se defina específicamente de otro modo, el término "arilo" se refiere a grupos hidrocarbonados aromáticos cíclicos que tienen de 1 a 2 anillos aromáticos, incluyendo grupos monocíclicos o bicíclicos tales como fenilo, bifenilo o naftilo. Cuando contienen dos anillos aromáticos (bicíclicos, etc.), los anillos aromáticos del grupo arilo pueden unirse en un solo punto (por ejemplo, bifenilo) o fusionarse (por ejemplo, naftilo).
El grupo arilo puede estar opcionalmente sustituido con uno o más sustituyentes, por ejemplo, 1 a 5 sustituyentes, en cualquier punto de unión. Los ejemplos de sustituyentes incluyen, pero no se limitan a, -H, -halógeno, -O-alquilo
C1-Ca, alquilo C1-Ca, alquenilo -oC^Ca, alquinilo -OC1-Ca, alquileno -C1-Ca, -alquinilo C1-Ca, -OH, -OP(O)(OH)2, OC(O)alquilo C1-Ca, -C(O)alquilo C1-Ca, -OC(O)alquilo C1-Ca, NH2, NH(alquilo C1-Ca), N(alquilo C1-Ca)2, -S(O)2-alquilo C1-Ca, -S(O)NH-alquilo C1-Ca, y S(O)N(alquilo C1-Ca)2. Los sustituyentes pueden ser sustituidos opcionalmente. Además, cuando contienen dos anillos fusionados, los grupos arilo aquí definidos pueden tener un anillo insaturado o parcialmente saturado fusionado con un anillo completamente saturado. Los sistemas de anillos ejemplares de estos grupos arilo incluyen indanilo, indenilo, tetrahidronaftalenilo y tetrahidrobenzoantenulenilo.
A menos que se defina específicamente, heteroarilo significa un radical aromático monocíclico monovalente de 5 a 10 átomos del anillo o un radical aromático policíclico, que contiene uno o más heteroátomos del anillo seleccionados de N, O o S, siendo los átomos restantes del anillo C. Heteroarilo como se define aquí también significa un grupo heteroaromático bicíclico en el que el heteroátomo se selecciona entre N, O o S. El radical aromático está opcionalmente sustituido de manera independiente con uno o más sustituyentes descritos en este documento. Los ejemplos incluyen, pero no se limitan a, furilo, tienilo, pirrolilo, piridilo, pirazolilo, pirimidinilo, imidazolilo, pirazinilo, indolilo, tiofen-2-ilo, quinolilo, benzopiranilo, tiazolilo y derivados de los mismos. Además, cuando contienen dos anillos fusionados, los grupos arilo aquí definidos pueden tener un anillo insaturado o parcialmente saturado fusionado con un anillo completamente saturado. Los sistemas de anillos ejemplares de estos grupos heteroarilo incluyen indolinilo, indolinonilo, dihidrobenzotiofenilo, dihidrobenzofurano, cromanilo, tiocromanilo, tetrahidroquinolinilo, dihidrobenzotiazina y dihidrobenzoxanilo.
Halógeno o "halo" se refiere a flúor, cloro, bromo y yodo.
Alquilo se refiere a un hidrocarburo saturado de cadena lineal o ramificada que contiene 1-12 átomos de carbono. Ejemplos de un grupo alquilo C1-Ca incluye, pero no se limita a, metilo, etilo, propilo, butilo, pentilo, hexilo, isopropilo, isobutilo, sec-butilo, terc-butilo, isopentilo, neopentilo e isohexilo.
"Alcoxi" se refiere a un hidrocarburo saturado de cadena lineal o ramificada que contiene 1-12 átomos de carbono que contiene un "O" terminal en la cadena. Los ejemplos de grupos alcoxi incluyen, sin limitación, grupos metoxi, etoxi, propoxi, butoxi, t-butoxi o pentoxi.
"Alquenilo" se refiere a un hidrocarburo insaturado de cadena lineal o ramificada que contiene 2-12 átomos de carbono. El grupo "alquenilo" contiene al menos un doble enlace en la cadena. Los ejemplos de grupos alquenilo incluyen etenilo, propenilo, n-butenilo, iso-butenilo, pentenilo o hexenilo.
"Alquinilo" se refiere a un hidrocarburo insaturado de cadena lineal o ramificada que contiene 2-12 átomos de carbono. El grupo "alquinilo" contiene al menos un triple enlace en la cadena. Los ejemplos de grupos alquenilo incluyen etinilo, propargilo, n-butinilo, iso-butinilo, pentinilo o hexinilo.
"Cicloalquilo" significa anillos de carbono saturados monocíclicos que contienen 3-18 átomos de carbono.
Ejemplos de grupos cicloalquilo incluyen, sin limitaciones, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptanilo, ciclooctanilo, norborano, norborenilo, biciclo[2.2.2]octanilo, o biciclo[2.2.2]octenilo.
"Cicloalquilalquilo" significa anillos de carbono saturados monocíclicos que contienen 3-18 átomos de carbono sustituidos adicionalmente con grupos alquilo C1-Ca. En general, los grupos cicloalquilalquilo descritos en este documento muestran la siguiente fórmula
a

donde m es un número entero de 1 a 6 y n es un número entero de 1 a 16.
Anillos monocíclicos "heterocidilo" o "heterocicloalquilo" que contienen carbono y heteroátomos tomados de oxígeno, nitrógeno o azufre y en los que no hay electrones n deslocalizados (aromaticidad) compartidos entre el anillo de carbono o los heteroátomos; anillos de heterociclilo incluyen, pero no se limitan a, oxetanilo, azetadinilo, tetrahidrofuranilo, pirrolidinilo, oxazolinilo, oxazolidinilo, tiazolinilo, tiazolidinilo, piranilo, tiopiranilo, tetrahidropiranilo, dioxalinilo, piperidinilo, morfolinilo, tiomorfolinilo, S-óxido, tiomorfolinilo S-dióxido, piperazinilo, azepinilo, oxepinilo, diazepinilo, tropanilo y homotropanilo. De acuerdo con la presente invención, heterociclilo de 3 a 8 miembros se refiere a estructuras de anillos no aromáticos saturados o parcialmente saturados que contienen entre 3 y 8 átomos en los cuales hay al menos un grupo de átomos seleccionados del grupo N, O o S.
El término "solvato" se refiere a un complejo de estequiometría variable formado por un soluto y un disolvente. Dichos disolventes para los fines de la invención no pueden interferir con la actividad biológica del soluto. Los ejemplos de disolventes adecuados incluyen, pero no se limitan a, agua, MeOH, EtOH y AcOH. Los solvatos en los que el agua es la molécula solvente se denominan típicamente hidratos. Los hidratos incluyen composiciones que contienen cantidades estequiométricas de agua, así como composiciones que contienen cantidades variables de agua.
El término "isómero" se refiere a compuestos que tienen la misma composición y peso molecular pero que difieren en sus propiedades físicas y/o químicas. La diferencia estructural puede estar en la constitución (isómeros geométricos) o en la capacidad de rotar el plano de la luz polarizada (estereoisómeros). Con respecto a los estereoisómeros, los compuestos de Fórmula (I) pueden tener uno o más átomos de carbono asimétricos y pueden aparecer como racematos, mezclas racémicas y como enantiómeros o diastereómeros individuales.
La descripción también incluye composiciones farmacéuticas que comprenden una cantidad eficaz de un compuesto descrito y un vehículo farmacéuticamente aceptable. Las "sales farmacéuticamente aceptables" representativas incluyen, por ejemplo, sales solubles en agua e insolubles en agua, como el acetato, amsonato (4,4-diaminostilbeno-2,2-disulfonato), bencenosulfonato, benzonato, bicarbonato, bisulfato, bitartrato, borato, bromuro, butirato, calcio, edetato de calcio, camsilato, carbonato, cloruro, citrato, clavularia, dihidrocloruro, edetato, edisilato, estoilato, fiunarato, gluconato, fiunato, gluconato, glutamato, glicolilarsanilato, hexafluorofosfato, hexilresorcinato, hidrabamina, hidrobromuro, hidrocloruro, hidroxinaftoato, yoduro, isotionato, lactato, lactobionato, laurato, magnesio, malato, maleato, mandelato, mesilato, metilbromuro, metilnitrato, metilsulfato, mucato, napsilato, nitrato, sal de amonio de N-metilglucamina, 3-hidroxi-2-naftoato, oleato, oxalato, palmitato, pamoato (1,1-meteno-bis-2-hidroxi-3-naftoato, einbonato), pantotato, fosfato/difosfato, picrato, poligalacturonato, propionato, p-toluensulfonato, salicilato, estearato, subacetato, succinato, sulfato, sulfosalicilato, suramato, tanato, tartrato, teoclato, tosilato, trietiodido y sales de valerato.
Un "paciente" o "sujeto" es un mamífero, por ejemplo, un humano, ratón, rata, cobaya, perro, gato, caballo, vaca, cerdo o primate no humano, como un mono, chimpancé, babuino o rhesus.
Una "cantidad efectiva" cuando se usa en relación con un compuesto es una cantidad efectiva para tratar o prevenir una enfermedad en un sujeto como se describe en el presente documento.
El término "portador", como se usa en esta descripción, abarca portadores, excipientes y diluyentes y significa un material, composición o vehículo, tal como una carga líquida o sólida, diluyente, excipiente, solvente o material de encapsulación, involucrado en transportar un agente farmacéutico de un órgano, o parte del cuerpo, a otro órgano, o parte del cuerpo de un sujeto.
El término "tratar" con respecto a un sujeto, se refiere a mejorar al menos un síntoma del trastorno del sujeto. El tratamiento incluye curar, mejorar, o al menos parcialmente mejorar el trastorno.
El término "trastorno" se usa en esta descripción para significar, y se usa indistintamente con, los términos afección o enfermedad, a menos que se indique lo contrario.
El término "administrar", "administrado" o "administración" tal como se usa en esta descripción se refiere a la administración directa de un compuesto descrito o una sal farmacéuticamente aceptable del compuesto descrito o
una composición a un sujeto, o administrar un derivado profármaco o análogo del compuesto o sal farmacéuticamente aceptable del compuesto o composición para el sujeto, que puede formar una cantidad equivalente de compuesto activo dentro del cuerpo del sujeto.
El término "profármaco", como se usa en esta descripción, significa un compuesto que se puede convertir in vivo por medios metabólicos (por ejemplo, por hidrólisis) a un compuesto descrito.
En un aspecto de la invención, A es CN. En esta realización, Rg puede ser además H, alquilo C1-C6 o cicloalquilo C3-C6. En otro aspecto, Rg también puede ser metilo o etilo.
En otro aspecto de los compuestos de Fórmula I, U es N. En esta realización, A puede ser además CN. En otros aspectos de la invención, se describen los compuestos de Fórmula I en los que A es H o F.
En otros aspectos de la invención, se describen los compuestos de Fórmula I en los que A es

Otro aspecto de la invención se refiere a compuestos de Fórmula I en la que R4 y R5 son H.
En otro aspecto de la invención, R3 es H, metilo o etilo.
En otro aspecto de los compuestos de Fórmula I, R4 es H y R5 es metilo.
En otro aspecto más de la invención, R4 es H y R5 es (S)-metilo.
En otro aspecto, R4 y R5 son halógenos.
En otro aspecto de los compuestos de Fórmula I, R4 es F y R5 es metilo.
En otro aspecto, R4 y R5 pueden combinarse para formar un cicloalquilo C3-C6.
En un aspecto de los compuestos de Fórmula I, Wi, W2 y W3 son todos CH.
En un aspecto de los compuestos de Fórmula I, Wi, W2 o W3 son CF.
En un aspecto, Wi o W3 es CH o N.
En un aspecto, W3 es CR2.
En otro aspecto de la invención, Ri puede ser halógeno. En otro aspecto, Ri es cloro.
En un aspecto de la invención, R2 puede ser H, halógeno o alcoxi Ci-C6. En otro aspecto, R2 puede también puede ser alcoxi Ci-C6 sustituido con heteroarilo o heterociclilo de 3 a 8 miembros.
En otro aspecto, los compuestos ilustrativos de Fórmula I son:
5- {[(6-cloro-2-oxo-i,2-dihidroquinolin-3-il)metil]amino}-i-metil-6-oxo-i,6-dihidropiridin-2-carbonitrilo;
6- cloro-3-{[(i-etil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-i,2-dihidroquinolin-2-ona;
6-cloro-3-{[(i-metil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-i,2-dihidroquinolin-2-ona;
5- {[(6-cloro-2-oxo-i,2-dihidroquinolin-3-il)metil]amino}-6-oxo-i,6-dihidropiridin-2-carbonitrilo;
6- cloro-3-{[(i-ciclopropil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-i,2-dihidroquinolin-2-ona;
6-cloro-3-{[(i,6-dimetil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-i,2-dihidroquinolin-2-ona;
3-{[(6-bromo-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-6-cloro-i,2-dihidroquinolin-2-ona;
6-cloro-3-({[2-oxo-6-(trifluorometil)-i,2-dihidropiridin-3-il]amino}metil)-i,2-dihidroquinolin-2-ona;
6-cloro-3-({[i-metil-2-oxo-6-(trifluorometil)-i,2-dihidropiridin-3-il]amino}metil)-i,2-dihidroquinolin-2-ona;
5- {[(6-cloro-2-oxo-i,2-dihidroquinolin-3-il)metil]amino}-6-oxo-i,6-dihidropiridin-3-carboxilato de metilo;
6- cloro-7-metoxi-3-{[(i-metil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-i,2-dihidroquinolin-2-ona;
6-cloro-3-{[(i-metil-2-oxo-i,2-dihidropiridin-3-il)amino]metil}-7-(piridin-2-ilmetoxi)-i,2-dihidroquinolina-2- uno;
5-{[(1S)-1-(6-doro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1R)-1-(6-doro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1s)-1-(6-doro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-(6-doro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropirazina-2-carbonitrilo;
5-{[(1R)-1-(6-doro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[1-(6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo; 5-{[(1S)-1-(6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1R)-1-(6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[1-(6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo; 5-{[(1S)-1 -[6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1R)-1-[6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-({1-[6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il]etil}amino)-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-{6-cloro-2-oxo-7-[(1R)-1-(piridin-2-il)etoxi]-1,2-dihidroquinolin-3-il}etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1S)-1-[6-cloro-7-(ciclopropilmetoxi)-2-oxo-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-[(1-{6-cloro-7-[(3,3-difluorociclobutil)metoxi]-2-oxo-1,2-dihidroquinolin-3-il}etil)amino]-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1S)-1-[6-cloro-2-oxo-7-(propan-2-iloxi)-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-(6-cloro-8-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-(6-cloro-2-oxo-1,2-dihidro-1,8-naftiridin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo; 5-{[(1R)-1-(7-cloro-3-oxo-3,4-dihidroquinoxalin-2-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo; y
5-{[(1S)-1-(7-cloro-3-oxo-3,4-dihidroquinoxalin-2-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo.
En otro aspecto, los compuestos ilustrativos de Fórmula I incluyen:
5-{[(1S)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-6-oxo-1-(trifluorometil)-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-[6-cloro-7-(2-hidroxipropan-2-il)-2-oxo-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1S)-1-(6-cloro-7-ciclopropil-2-oxo-1,2-dihidro-1,8-naftiridin-3-il)etil]amino-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-(6-cloro-7-metil-2-oxo-1,2-dihidro-1,8-naftiridin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo;
5-{[(1S)-1-{6-cloro-7-[(2-hidroxi-2-metilpropil)amino]-2-oxo-1,2-dihidroquinolin-3-il}etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1S)-1-[7-(azetidin-1-il)-6-cloro-2-oxo-1,2-dihidro-1,8-naftiridin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5-{[(1S)-1-[7-(azetidin-1-il)-6-cloro-2-oxo-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
5- {[(1S)-1-[6-cloro-7-(3,3-difluoroazetidin-1-il)-2-oxo-1,2-dihidroquinolin-3-il]etil]amino}-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo;
6- cloro-3-[(1S)-1-{[1-metil-2-oxo-6-(1H-1,2,3,4-tetrazol-1-il)-1,2-dihidropiridin-3 -il]amino}etil]-1,2-dihidro-quinolin-2-ona; y
5-{[(1S)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil]amino}-1-metil-6-oxo-1,6-dihidropiridina-2-carboxamida.
En un aspecto, los compuestos de la invención tienen la fórmula la:

En otro aspecto, los compuestos de la invención tienen la Fórmula Ia-1:

En otro aspecto de la invención, los compuestos de Fórmula I son enantiómeros. En algunas realizaciones Los compuestos son (S)-enantiómero. En otras realizaciones, los compuestos también pueden ser (R)-enantiómero. En otras realizaciones más, los compuestos de Fórmula I pueden ser enantiómeros (+) o (-).
En otro aspecto de la invención, los compuestos de Fórmula I contienen isótopos de átomos que forman la estructura de Fórmula I. Isótopos aquí significa, cada uno de dos o más formas del mismo elemento (por ejemplo, H y D; 12C y 13C) que contienen números iguales de protones pero diferentes números de neutrones en sus núcleos, y por lo tanto difieren en masa atómica relativa.
Debe entenderse que en la presente invención están incluidas todas las formas isoméricas, incluyendo mezclas de las mismas. Si el compuesto contiene un doble enlace, el sustituyente puede estar en la configuración E o Z. Si el compuesto contiene un cicloalquilo disustituido, el sustituyente de cicloalquilo puede tener una configuración cis o trans. Todas las formas tautoméricas también están destinadas a ser incluidas.
Métodos de uso de los compuestos descritos
Otro aspecto de la divulgación se refiere a un método para tratar una enfermedad o trastorno asociado con la deshidrogenasa isocitrato mutante. El método implica administrar a un paciente que necesite un tratamiento para enfermedades o trastornos asociados con la deshidrogenasa isocitrato mutante, una cantidad eficaz de las composiciones y compuestos de Fórmula I.
Otro aspecto de la divulgación está dirigido a un método que inhibe la deshidrogenasa isocitrato mutante. El método implica administrar a un paciente que lo necesite una cantidad eficaz de las composiciones o compuestos de Fórmula I.
Los ejemplos de una proteína IDH mutante que tiene una actividad neomórfica son la IDH1 mutante y la IDH2 mutante. Una actividad neomórfica asociada con el mutante IDH1 y el mutante IDH2 es la capacidad de producir 2-hidroxiglutarato (actividad neomórfica 2-HG), específicamente R-2-HG (actividad neomórfica R-2-HG). Las mutaciones en IDH 1 asociadas con la actividad neomórfica 2-HG, específicamente la actividad neomórfica R-2-HG, incluyen mutaciones en los residuos 97, 100 y 132, por ejemplo, g 97D, R100Q, R132H, R132C, R132S, R132G, R132l y R132V. Las mutaciones en IDH2 asociadas con la neoactividad 2-HG, específicamente la actividad neomórfica R-2-HG, incluyen mutaciones en los residuos 140 y 172, por ejemplo, R140Q, R140G, R172K, R172M, R172S, R172G y R172W.
Otro aspecto de la divulgación se refiere a un método para reducir el 2-hidroxiglutarato. El método comprende administrar a un paciente que lo necesite una cantidad eficaz de las composiciones o compuestos de Fórmula I.
Un uso terapéutico de los compuestos o composiciones de la presente invención que inhiben el mt-IDH es proporcionar tratamiento a pacientes o sujetos que padecen enfermedades de proliferación celular y cánceres que incluyen, sin limitación, glioma, glioblastoma multiforme, paraganglioma, tumores neuroectodérmicos primordiales supratentoriales, leucemia mieloide aguda (LMA), cáncer de próstata, cáncer de tiroides, cáncer de colon, condrosarcoma, colangiocarcinoma, linfoma linfático de células T periféricas, melanoma, colangiocarcinoma intrahepático (IHCC), síndrome mielodisplásico (MDS), enfermedad mieloproliferativa (MPD), y otros tumores sólidos. Los tratamientos dirigidos para estos cánceres y las enfermedades de proliferación celular no están disponibles actualmente para los pacientes que padecen estas afecciones. Por lo tanto, existe la necesidad de nuevos agentes terapéuticos selectivos para estas condiciones.
Los compuestos descritos de la invención pueden administrarse en cantidades eficaces para tratar o prevenir un trastorno y/o prevenir el desarrollo de los mismos en sujetos.
La administración de los compuestos descritos se puede realizar a través de cualquier modo de administración para agentes terapéuticos. Estos modos incluyen la administración sistémica o local, como los modos de administración oral, nasal, parenteral, transdérmica, subcutánea, vaginal, bucal, rectal o tópica.
Según el modo de administración previsto, las composiciones descritas pueden estar en forma de dosificación sólida, semisólida o líquida, tal como, por ejemplo, inyectables, comprimidos, supositorios, píldoras, cápsulas de liberación prolongada, elixires, tinturas, emulsiones, jarabes, polvos, líquidos, suspensiones o similares, a veces en dosis unitarias y compatibles con las prácticas farmacéuticas convencionales. Del mismo modo, también se pueden administrar en forma intravenosa (tanto en bolo como en infusión), intraperitoneal, subcutánea o intramuscular, y todas ellas usan formas bien conocidas por los expertos en la técnica farmacéutica.
Las composiciones farmacéuticas ilustrativas son comprimidos y cápsulas de gelatina que comprenden un compuesto de la invención y un vehículo farmacéuticamente aceptable, tal como a) un diluyente, por ejemplo, agua purificada, aceites triglicéridos, tales como aceite vegetal hidrogenado o parcialmente hidrogenado, o mezclas de los mismos, aceite de maíz, aceite de oliva, aceite de girasol, aceite de cártamo, aceites de pescado, como EPA o DHA, o sus ésteres o triglicéridos o mezclas de los mismos, ácidos grasos omega-3 o sus derivados, lactosa, dextrosa, sacarosa, manitol sorbitol, celulosa, sodio, sacarina, glucosa y/o glicina; b) un lubricante, por ejemplo, sílice, talco, ácido esteárico, su sal de magnesio o calcio, oleato de sodio, estearato de sodio, estearato de magnesio, benzoato de sodio, acetato de sodio, cloruro de sodio y/o polietilenglicol; también para tabletas; c) un aglutinante, por ejemplo, silicato de aluminio y magnesio, pasta de almidón, gelatina, tragacanto, metilcelulosa, carboximetilcelulosa de sodio, carbonato de magnesio, azúcares naturales como la glucosa o beta-lactosa, edulcorantes de maíz, gomas naturales y sintéticas tales como acacia, tragacanto o alginato de sodio, ceras y/o polivinilpirrolidona, si se desea; d) un
desintegrante, por ejemplo, almidones, agar, metilcelulosa, bentonita, goma xantana, ácido algiico o su sal de sodio, o mezclas efervescentes; e) absorbente, colorante, saborizante y edulcorante; f) un emulsionante o agente dispersante, como Tween 80, Labrasol, HPMC, DOSS, caproilo 909, labrafac, labrafil, peceol, transcutol, capmul MCM, capmul PG-12, captex 355, gelucire, vitamina E TGPS u otro emulsionante aceptable ; y/o g) un agente que mejora la absorción del compuesto tal como ciclodextrina, hidroxipropil-ciclodextrina, PEG400, PEG200.
Las composiciones líquidas, particularmente inyectables, pueden prepararse, por ejemplo, por disolución, dispersión, etc. Por ejemplo, el compuesto descrito se disuelve o se mezcla con un disolvente farmacéuticamente aceptable tal como, por ejemplo, agua, solución salina, acuosa, dextrosa, glicerol, etanol, y similares, para formar así una solución o suspensión isotónica inyectable. Se pueden usar proteínas como la albúmina, partículas de quilomicrones o proteínas séricas para solubilizar los compuestos.
Los compuestos descritos también pueden formularse como un supositorio que puede prepararse a partir de emulsiones o suspensiones grasas; utilizando polialquilenglicoles tales como propilenglicol, como vehículo.
Los compuestos descritos también pueden administrarse en forma de sistemas de administración de liposomas, tales como pequeñas vesículas unilamelares, grandes vesículas unilamelares y vesículas multilamelares. Los liposomas se pueden formar a partir de una variedad de fosfolípidos, que contienen colesterol, estearilamina o fosfatidilcolinas. En algunas realizaciones, una película de componentes lipídicos se hidrata con una solución acuosa de medicamento en una forma de capa lipídica que encapsula el medicamento, como se describe en la Patente de EE.UU. N° 5.262.564.
Los compuestos descritos también pueden administrarse mediante el uso de anticuerpos monoclonales como vehículos individuales a los que se acoplan los compuestos descritos. Los compuestos descritos también pueden acoplarse con polímeros solubles como portadores de fármacos dirigibles. Dichos polímeros pueden incluir polivinilpirrolidona, copolímero de pirano, polihidroxipropilmetacrilamidafenol, polihidroxietilaspanamidefenol o polietilenoxidoplicilina sustituida con residuos de palmitoilo. Además, los compuestos divulgados pueden acoplarse a una clase de polímeros biodegradables útiles para lograr la liberación controlada de un fármaco, por ejemplo, ácido poliláctico, polepsilon caprolactona, ácido polihidroxibutírico, poliortoésteres, poliacetales, polihidropiranos, policianoacrilatos y reticulados. Polímeros de bloques anfipáticos de hidrogeles. En una realización, los compuestos descritos no se unen covalentemente a un polímero, por ejemplo, un polímero de ácido policarboxílico, o un poliacrilato.
La administración parenteral inyectable se usa generalmente para inyecciones subcutáneas, intramusculares o intravenosas e infusiones. Los inyectables pueden prepararse en formas convencionales, ya sea como soluciones o suspensiones líquidas o formas sólidas adecuadas para disolverse en líquido antes de la inyección.
Otro aspecto de la divulgación está dirigido a composiciones farmacéuticas que comprenden un compuesto de Fórmula I y un vehículo farmacéuticamente aceptable. El vehículo farmacéuticamente aceptable puede incluir además un excipiente, diluyente o tensioactivo.
Las composiciones pueden prepararse de acuerdo con métodos convencionales de mezcla, granulación o recubrimiento, respectivamente, y las presentes composiciones farmacéuticas pueden contener de aproximadamente 0,1% a aproximadamente 99%, de aproximadamente 5% a aproximadamente 90%, o de aproximadamente 1% a aproximadamente el 20% del compuesto descrito en peso o volumen.
El régimen de dosificación que utiliza el compuesto descrito se selecciona de acuerdo con una variedad de factores que incluyen el tipo, especie, edad, peso, sexo y condición médica del paciente; la severidad de la condición a tratar; la vía de administración; la función renal o hepática del paciente; y el compuesto particular descrito empleado. Un médico o veterinario con experiencia ordinaria en la técnica puede determinar y prescribir fácilmente la cantidad efectiva del medicamento requerido para prevenir, contrarrestar o detener el progreso de la enfermedad.
Las cantidades de dosificación efectiva de los compuestos descritos, cuando se usan para los efectos indicados, varían de aproximadamente 0,5 mg a aproximadamente 5000 mg del compuesto descrito según sea necesario para tratar la afección. Las composiciones para uso in vivo o in vitro pueden contener aproximadamente 0,5, 5, 20, 50, 75, 100, 150, 250, 500, 750, 1000, 1250, 2500, 3500 o 5000 mg del compuesto descrito, o, en un intervalo de una cantidad a otra cantidad en la lista de dosis. En una realización, las composiciones están en forma de una tableta que se puede puntuar.
Método de síntesis de los compuestos.
Los compuestos de la presente invención se pueden preparar mediante una variedad de métodos, que incluyen química estándar. Las rutas sintéticas adecuadas se representan en los esquemas que se dan a continuación.
Los compuestos de Fórmula (I) se pueden preparar por métodos conocidos en la técnica de la síntesis orgánica tal como se expone en parte mediante los siguientes esquemas sintéticos. En los esquemas descritos a continuación, se entiende que los grupos protectores para grupos sensibles o reactivos se emplean cuando es necesario de acuerdo con los principios generales o la química. Los grupos protectores se manipulan de acuerdo con los métodos estándar de síntesis orgánica (TW Greene y PGM Wuts, "Protective Groups in Organic Synthesis", tercera edición, Wiley, Nueva York, 1999). Estos grupos se eliminan en una etapa conveniente de la síntesis de compuestos utilizando métodos que son fácilmente evidentes para los expertos en la técnica. Los procesos de selección, así como las condiciones de reacción y el orden de su ejecución, serán consistentes con la preparación de los compuestos de Fórmula (I).
Los expertos en la técnica reconocerán si existe un estereocentro en los compuestos de Fórmula (I). En consecuencia, la presente invención incluye ambos estereoisómeros posibles (a menos que se especifique en la síntesis) e incluye no solo compuestos racémicos sino también los enantiómeros y/o diastereómeros individuales. Cuando se desea un compuesto como un único enantiómero o diastereómero, se puede obtener por síntesis estereoespecífica o por resolución del producto final o cualquier intermedio conveniente. La resolución del producto final, un producto intermedio o un material de partida puede verse afectado por cualquier método adecuado conocido en la técnica. Véase, por ejemplo, "Stereochemistry of Organic Compounds" por EL Eliel, SH Wilen y LN Mander (Wiley-lnterscience, 1994).
Los compuestos descritos en el presente documento pueden prepararse a partir de materiales de partida disponibles comercialmente o sintetizarse usando procesos orgánicos, inorgánicos y/o enzimáticos conocidos. Preparación de compuestos
Los compuestos de la presente invención se pueden preparar de varias maneras bien conocidas por los expertos en la técnica de la síntesis orgánica. A modo de ejemplo, los compuestos de la presente invención se pueden sintetizar usando los métodos descritos a continuación, junto con los métodos sintéticos conocidos en la técnica de la química orgánica sintética, o variaciones en los mismos, como lo aprecian los expertos en la materia. Los métodos preferidos incluyen, pero no se limitan a los métodos descritos a continuación. Compuestos de la presente invención La fórmula (I) se puede sintetizar siguiendo los pasos descritos en los Esquemas 1-2, que comprenden diferentes secuencias de compuestos intermedios de ensamblaje II, III, IV y V. Los materiales de partida están disponibles comercialmente o se fabrican mediante procedimientos conocidos en la literatura reportada o como se ilustra.
Esquema 1
en donde A, U, Wi, W2, W3, R1-R9 se definen en la Fórmula (I).
Las formas generales de preparación de moléculas diana de Fórmula I mediante el uso de intermedios II, III, IV y V se describen en el Esquema 1 y 2. Desplazamiento de haluros de arilo (III) con intermedios amina (II) en condiciones estándar de sustitución nucleófila utilizando una base tal como N,N-diisopropiletilamina y/o carbonato de potasio, el carbonato de cesio en el disolvente DMSO o DMF proporciona los compuestos de Fórmula I. La aminación reductora del aldehído (IV) con amina (V) se realiza bajo un procedimiento estándar (AcOH y NaBH(OAc)3) para preparar el compuesto de Fórmula I (donde R4 = R5 = H). Una mezcla de enantiómeros, diastereoisómeros, isómeros cis/trans resultante del proceso se puede separar en sus componentes individuales mediante una técnica de sal quiral, cromatografía usando fase normal, fase inversa o columna quiral, dependiendo de la naturaleza de la separación.
Debe entenderse que en la descripción y fórmulas mostradas anteriormente, los diversos grupos A, U, W1, W2, W3, R1-R6 y R9 y otras variables son como se definieron anteriormente, excepto que se indique lo contrario. Además, para fines sintéticos, los compuestos de los esquemas 1 y 2 son meros representantes con radicales elegidos para ilustrar la metodología sintética general del compuesto de Fórmula I como se define aquí.
Ejemplos
La divulgación se ilustra adicionalmente mediante los siguientes ejemplos y esquemas de síntesis, que no deben interpretarse como limitantes de esta divulgación en alcance o espíritu a los procedimientos específicos descritos en este documento. Debe entenderse que los ejemplos se proporcionan para ilustrar ciertas realizaciones y que no se pretende con ello ninguna limitación del alcance de la divulgación.
La Tabla 6 proporciona la actividad de compuestos ilustrativos de Fórmula I en ensayos IDH1-R132H, IDH1-R132C, IDH1-MS-HTC116-R132H, e IDH1-MS-HTC116-R132C.
Métodos analíticos, materiales e instrumentación.
A menos que se indique lo contrario, los reactivos y disolventes se utilizan como recibidos de proveedores comerciales. Los espectros de resonancia magnética nuclear de protones (RMN) se obtuvieron en los espectrómetros Bruker o Varian a 300 MHz. Los espectros se dan en ppm (8) y las constantes de acoplamiento, J, se informan en Hertz. Tetrametilsilano (TMS) se utilizó como estándar interno. Los espectros de masas se recolectaron utilizando un espectrómetro de masas Waters ZQ Single Quad (ionización por electropulverización de trampa iónica (ESI)). Los análisis de cromatografía líquida de alto rendimiento (HPLC) se obtuvieron utilizando una columna XBridge Fenyl o C18 (5 mm, 50x4,6 mm, 150x4,6 mm o 25034,6 mm) con detección UV (Waters 996 PDA) a 254 nm o 223 nm usando una programa de gradiente de solvente estándar (Método 1-4).
Método 1 de LCMS (ESI, método de 4 min):
Instrumentos:
HPLC: Waters HT2790 Alliance MS: Waters ZQ Single Quad Espectrómetro de masas
UV: Waters 996 PDA 95% de agua/5% de metanol con 0.1% de ácido fórmico
Condiciones:
Fase móvil A 95% de agua/5% de metanol con 0,1% de ácido fórmico
Fase Móvil B (B) 95% de metanol/5% de agua con 0,1% de ácido fórmico
Columna XBridge Fenyl o C18, 5 pm 4,6 x 50 mm
Temperatura de la columna Ambiente
Gradiente LC Lineal 5-95% B en 2.5 min, mantenga 95% B a 3,5 min
Caudal de LC 3 mL/min
Longitud de onda UV 220 nm y 254 nm
Modo de ionización Ionización por electropulverización; positivo/negativo
Método 2 de LCMS (ESI, método de 10 min):
Instrumentos:
HPLC: Waters HT2790 Alliance MS: Espectrómetro de masas de un solo patio Waters ZQ
UV: Waters 996 PDA
Condiciones:
Fase móvil A (A) 95% de agua/5% de metanol con 0,1% de ácido fórmico
Fase Móvil B (b ) 95% de metanol/5% de agua con 0,1% de ácido fórmico
Columna XBridge C18, 5 pm 4,6 x 150 mm
Temperatura de columna Ambiente
Gradiente LC Lineal 5-95% B en 5,5 min, mantenga 95% B a 7,5 min
Caudal LC 1,2 mL/min
Longitud de onda UV 220 nm y 254 nm
Modo de ionización Ionización por electropulverización; positivo/negativo
Método 3 de LCMS: (APCI, 20 min )
Instrumentos y condiciones:
Serie HPLC-Agilent 1100.
Columna: Agela Technologies Durashell C18, 3 |jm, 4,6 x 50 mm,).
Fase Móvil A: ACN 0,1% TFA.
Fase móvil B: Agua 0,1% TFA.
Gradiente: 15
Tiempo (min) %B
00 95
15 05
18 05
20 95 20
Velocidad de flujo: 1 mL/min.
Temperatura de la columna:
Detector: 254 nm.
Método 4 de LCMS (ESI, método de 2,5 min):
Instrumentos y condiciones:
HPLC: Disolvente binario
Waters Acquity Gerente MS: Detector de masa Waters ZQ
UV: Waters Acquity PDA
Fase móvil A (A) 95% de agua/5% de acetonitrilo con 0,1% de ácido fórmico en formato de amonio 10 mM. Fase Móvil B (b ) 95% de acetonitrilo/5% de agua con 0,09% de ácido fórmico.
Columna Water Acquity UPLC BEH C18, 1,7 jm, 2,1 x 50 mm
Temperatura de columna 35°C
Gradiente LC 5-100% de B en 2,0 min, mantenga el 100% de B en 2,2 min
Velocidad de flujo LC 0,6 mL/min
Longitud de onda UV 220 nm y 254 nm
Modo de ionización Ionización por electropulverización; positivo/negativo
Las abreviaturas utilizadas en los siguientes ejemplos y en otras partes del presente documento son:
AC2O Anhídrido acético
ACN Acetonitrilo
BOP amoníaco 4-(3-(piridin-3-ilmetil)ureido)bencenosulfinato
CDCla clorhidrato deuterado
Cs2CO3 carbonato de cesio CuSO4 sulfato de cobre
8 cambio químico
DCM diclorometano o cloruro de metileno
DCE 1,2-dicloroetano
DEAD azodicarboxilato de dietilo
DIAD diisopropil azodicarboxilato
DIEA W,W-diisopropiletilamina
DMA W,W-dimetilacetamida
DME dimetoxietano
DMF W,W-dimetilformamida
DMP Dess-Martin Periodinane
DMSO dimetilsulfóxido
DMSO-de dimetilsulfóxido deuterado
dppf 1,1'-Bis(difenilfosfino)ferroceno
EDCI W-(3-dimetilaminopropil)-W-etilcarbodiimida clorhidrato
EDTA ácido etilendiaminotetraacético
ee exceso enantiomérico
EtOAc acetato de etilo
EtOH etanol
1H RMN resonancia magnética nuclear de protones
HOAc ácido acético
HATU 2-(3H-[1,2,3]triazolo[4,5-b]piridin-3-il)-1,1,3,3-tetrametilisouronio hexafluorofosfato
HCl Ácido clorhídrico
HOBT 1H-benzo[d][1,2,3]triazol-1-ol hidrato
HPLC cromatografía líquida de alta presión
Hz hertz
IPA alcohol isopropílico
KOAc acetato de potasio
K2CO3 carbonato de potasio
LAH hidruro de litio y aluminio
LCMS cromatografía líquida/espectrometría de masa
(M+1) masa 1
m-CPBA ácido m-cloroperbenzoico
MeOH metanol
MeMgBr bromuro de metil magnesio
MS espectrometría de masas
NaBH4 borohidruro de sodio
Na2SO4 sulfato de sodio
Pd(dppf)Cl2 [1,1'-Bis(difenilfosfino)ferroceno]dicloropaladio(II)
Paladio tetrakis Tetrakis(trifenilfosfina)paladio(0)
Rt tiempo de retención
TBDMS-Cl Cloruro de terc-butil dimetilsililo
TEA trietilamina
THF tetrahidrofurano
TLC cromatografía de capa fina
Xantphos 4,5-Bis(difenilfosfino)-9,9-dimetilxanteno
Ejemplo 1 - Intermedio N-1:(s)-3-(1-ammoetM)-6-cloroqumoMn-2(1H)-ona clorhidrato

Paso 1: (R,E)-N-((2,6-dicloroquinolm-3-il)metíleno)-2-metílpropano-2-sulfínamida.

A una mezcla de 2,6-dicloroquinolina-3-carbaldehído (15,0 g, 66,37 mmol) y (R)-2-metilpropano-2
sulfinamida (8,85 g, 73,14 mmol) en 1,2-dicloroetano. (150 ml) se añadió CuSO4 (16.0 g, 100,25 mmol). La mezcla resultante se calentó a 55°C y se agitó a 55°C durante la noche. Después de que TLC y MS mostraron una desaparición completa de los materiales de partida, la mezcla se enfrió a temperatura ambiente y se filtró a través de una capa de Celite®. La almohadilla de celite se enjuagó luego con CH2Ch. El filtrado se evaporó a sequedad al vacío y se purificó por cromatografía en columna de SO2 (0 a 25% de hexanos/EtOAc) para proporcionar el compuesto del título, (R,E)-N-((2,6-dicloroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida, como un sólido amarillo (17,7 g, 81% de rendimiento).
Paso 2: (R)-W-((S)-1-(2,6-d¡cloroqumolm-3-M)etM)-2-metilpropano-2-sulfmam¡da.

A una solución de (R,E)-N-((2,6-dicloroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (8,85 g, 26,88 mmol) en CH2Ch anhidro (200 ml)) a -60°C se añadió gota a gota MeMgBr (solución 3M en éter dietílico, 13,5 ml, 40,54 mmol). La mezcla de reacción resultante se agitó a aproximadamente -60 a -50°C durante 3 horas y luego se agitó a -20°C durante la noche bajo una atmósfera de N2. Después de que la TLC y la MS mostraron una desaparición completa de los materiales de partida, se añadió NH4Cl saturado (163 ml) a -20°C y la mezcla resultante se agitó durante 10 minutos. La fase acuosa se extrajo con CH2Ch (100 ml x 3), se secó sobre Na2SO4 anhidro, se filtró y se evaporó. El residuo se purificó por cromatografía en columna en un sistema de cromatografía ISCO® (SO2: columna de oro; gradiente; hexanos a 100% de EtOAc) para proporcionar el compuesto del título, (R)-N-((S)-1-(2,6)-dicloroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida, como un sólido amarillo (5,8 g, 63% de rendimiento).
Paso 3: (S)-3-(1-ammoetil)-6-cloroqumolm-2(1H)-ona clorhidrato (II-1).

Se calentó una mezcla de (R)-N-((S)-1-(2,6-dicloroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (6,6 g, 19,13 mmol) en 1,4-dioxano (41 ml) y IN HCl (41 ml) a reflujo durante la noche. Los disolventes se evaporaron al vacío y el residuo resultante se disolvió en agua caliente y se liofilizó. El producto bruto se trituró con éter dietílico para proporcionar el compuesto del título II-1 en forma de un sólido amarillo (9,0 g, ee: 98,4%). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,4 (br s, 1 H), 8,32 (br s, 2 H), 8,07 (s, 1 H), 7,85 (d, J = 2,2 Hz, 1 H), 7,63 (dd, Ji = 8,8 Hz, J 2 = 2,5 Hz, 1 H), 7,40 (d, J = 8,8 Hz, 1 H), 4,40-4,45 (m, 1 H), 1,53 (d, J = 8,5 Hz, 3 H). LCMS (Método 3): Rt 3,42 min, m/z 223,1 [M+H]+.
Ejemplo 2- Intermedio 11-2: (R)-3-(1-ammoetil)-6-cloroqumolm-2(1H)-ona clorhidrato.
Paso 1: (R)-W-((2,6-d¡cloroqumolm-3-¡l)met¡leno)-2-met¡lpropano-2-sulfmam¡da.
A una mezcla de 2,6-didoroquinolina-3-carbaldehído (500 mg, 2,21 mmol) y (R)-2-metilpropano-2-sulfinamida (295 g, 2,43 mmol) en 1,2-dicloroetano. (15 ml) se añadió CuSO4 (530 mg, 3,31 mmol). La mezcla resultante se calentó a 55°C y se agitó a 55°C durante 18 horas. Una vez que TLC y MS mostraron la desaparición completa de los materiales de partida, la mezcla de reacción se enfrió a temperatura ambiente y se filtró a través de una almohadilla de Celite®. La almohadilla de celite se enjuagó luego con CH2Ch. El filtrado se evaporó a sequedad al vacío y se purificó por cromatografía en columna en un sistema de cromatografía ISCO® (SO2; hexanos a 60% de EtOAc/hexanos) para proporcionar el compuesto del título, (R)-W-((2,6-dicloroquinolina-3-il)metileno)-2-metilpropano-2-sulfinamida, como un sólido amarillo (510 mg, 70% de rendimiento).
Paso 2: (R)-W-((R)-1-(2,6-d¡cloroqumolm-3-¡l)et¡l)-2-met¡lpropano-2-sulfmam¡da.
A una solución de (R)-W-((2,6-dicloroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (505 mg, 1,534 mmol) en THF anhidro (8 ml) a a 0°C se añadió gota a gota MeMgBr (solución 3M en éter dietílico, 0,56 ml, 1,687 mmol). La mezcla se agitó a 0°C durante 3 horas bajo una atmósfera de N2. Después de que la CCF y la MS mostraron una desaparición completa de los materiales de partida, se añadió NH4Cl saturado (5 ml) a 0°C y la mezcla resultante se agitó durante 10 minutos. La fase acuosa se extrajo con EtOAc (10 ml x 3), se secó sobre Na2SO4 anhidro, se filtró y se evaporó. El residuo se purificó por cromatografía en columna en un sistema de cromatografía ISCO® (SO2; hexanos a 80% de EtOAc/hexanos) para proporcionar el compuesto del título como el isómero R,R como un sólido amarillo pálido (200 mg, 38%) y el isómero de R,S como un sólido amarillo pálido (93 mg, 18% de rendimiento).
Paso 3: (R)-3-(1-ammoet¡l)-6-cloroqumolm-2(1H)-ona clorh¡drato (11-2).

Una mezcla de (R)-W-((R)-1-(2,6-dicloroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (190 mg, 0,55 mmol) en 1,4-dioxano (2 ml) y IN HCl (1,1 ml, 1,1 mmol) se calentó a 150°C durante 30 minutos en un reactor de microondas. Los disolventes se evaporaron y el residuo se disolvió en agua caliente y se liofilizó para proporcionar el compuesto del título II-2 en forma de un sólido amarillo (148 mg, rendimiento cuantitativo). 1H RMN (300 MHz, DMSO-de): 8 ppm 12,35 (br s, 1 H), 8,28 (br s, 2 H), 8,05 (s, 1 H), 7,86 (d, J = 2,2 Hz, 1 H), 7,63 (dd, Ji = 8,8 Hz, J 2 = 2,5 Hz, 1 H), 7,40 (d, J = 8,8 Hz, 1 H), 4,40-4,45 (m, 1 H), 1,53 (d, J = 8,5 Hz, 3 H). LCMS (Método 3): Ta 3,40 min, m/z 223,1 [M+H]+.
Ejemplo 3 - Un enfoque alternativo al Intermed¡o II-1

Paso 1: 3-acet¡l-6-cloroqumolm-2(1W)-ona.

Una mezcla de 2-amino-5-clorobenzaldehído (0,5 g, 3,21 mmol) y 2,2,6-trimetil-4H-1,3-dioxin-4-ona (0,594 g, 4,18 mmol) en xilenos (10 ml) bajo una atmósfera de nitrógeno se calentó a reflujo durante 3 horas y luego se enfrió a temperatura ambiente. La mezcla de reacción se filtró y se lavó con xilenos dos veces para proporcionar el compuesto del título, 3-acetil-6-cloroquinolin-2(1H)-ona (330 mg, 46.3 %). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,22 (br, 1 H), 8,41 (s, 2 H), 8,00 (s, 1 H), 7,63 (d, J = 8,8 Hz, 1 H), 7,32 (dd, Ji = 8,8 Hz, J 2 = 2,5 Hz, 1 H), 2.58 (s, 3 H). LCMS (Método 1): m/z 222,94 [M+H]+.
Paso 2: ((S)-W-((S)-1-(6-cloro-2-oxo-1,2-dihidroqummolm -3-il)etil)-2-metilpropano-2-sulfinamida.

Una mezcla de tetraetoxititanio (144 mg, 0,632 mmol),(s)-2-metilpropano-2-sulfinamida (38,3 mg, 0,316 mmol) y 3-acetil-6-cloroquinolin-2(1H)-ona (70 mg, 0,316 mmol) en THF (20 ml) se calentaron a 80°C durante la noche y luego se enfriaron a temperatura ambiente. A esta mezcla se le añadió NaBH4 (59,7 mg, 1,579 mmol) a -50°C. La mezcla se calentó lentamente a temperatura ambiente durante la noche. Se añadió MeOH (2 ml) para apagar el exceso de NaBH4 y se siguió con la adición de agua. La mezcla resultante se filtró para eliminar los sólidos y la fase acuosa se extrajo con EtOAc dos veces, se secó sobre Na2SO4 y se concentró. El residuo se purificó en un sistema de cromatografía Biotage® utilizando una columna de SO2 de 25 g con gradiente de elución (20% a 100% de AcOEt/Hexanos, luego 0-5% de MeOH/DCM) para proporcionar (S)-A/-((S)-1-(2,6-dicloroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (39 mg, 38% de rendimiento). 1H r Mn (300 m Hz , DMsO-d6): 8 ppm 12,05 (br, 1 H), 7,95 (s, 1 H), 7,84 (s, 1 H), 7,38 (d, J = 8,8 Hz, 1 H), 5,76 (d, J = 8,06 Hz, 1 H), 5,37 (m, 1 H), 4,55 (m, 1 H), 1,44 (d, J = 6,82 Hz, 3 H), 1,18 (s, 9 H). LCMS (Método 1): Ta 2,22 min; m/z 327,96 [M+H]+.
Paso 3:
(S)-3-(1-ammoetN)-6-cloroqumolm-2(1H)-ona clorhidrato (II-1).

En una solución de ((S)-W-((S)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (150 mg, 0,459 mmol) en MeOH (5 ml) se añadió HCl (2 ml, 8,0 mmol, 4M en 1,4-dioxano). La mezcla se agitó a temperatura ambiente durante la noche. A esta mezcla se agregaron 6 ml de éter etílico y el precipitado resultante se recogió por filtración, se lavó con éter etílico (2 x) y luego se secó para proporcionar clorhidrato de (S)-3-(1-aminoetil)-6-cloro quinolin-2(1H)-ona (50 mg, 42% de rendimiento). 1H RMN (300 Mhz, DMSO-d6): 8 ppm 12,4 (br s, 1 H), 8,32 (br s, 2 H), 8,07 (s, 1 H), 7,85 (d, J = 2,2 Hz, 1 H), 7,63 (dd, Ji = 8,8 Hz, J 2 = 2,5 Hz, 1 H), 7,40 (d, J = 8,8 Hz, 1 H), 4,40-4,45 (m, 1 H), 1,53 (d, J = 8,5 Hz, 3 H). LCMS (Método 1): Ta 1,22 min, m/z 223,1 [M+H]+. La pureza del enantiómero (ee%) de II-1 (> 98 %) se determinó por análisis de HPLC quiral.
Ejemplo 4 - Método alternativo (R)-3-(1-ammoetN)-6-cloroqumolm-2(1H)-ona clorhidrato (II-2).

Paso 1: ((R)-W-((R)-1-(6-cloro-2-oxo-1,2-dihidroqumoMn-3-N)etM)-2-metNpropano-2-sulfmamida

Una mezcla de tetraetoxititanio (412 mg, 1.805 mmol)(R)-2-metilpropano-2-sulfinamida (131 mg, 1.083 mmol) y 3-acetil-6-cloroquinolin-2(1H)-ona (160 mg, 0,722 mmol) en THF (20 ml) se calentó a 80°C durante la noche, luego se enfrió a temperatura ambiente. A esta mezcla se le añadió NaBH4 (137 mg, 3.61 mmol)-50°C. La mezcla se calentó lentamente a temperatura ambiente durante la noche. Se añadió MeOH (2 ml) para apagar el exceso de NaBH4 y se siguió con la adición de agua. La mezcla resultante se filtró para eliminar los sólidos y la fase acuosa se extrajo con EtOAc dos veces, se secó sobre Na2SO4 y se concentró. El residuo se purificó en un sistema de cromatografía Biotage® utilizando una columna de 25 g de SO2 con elución en gradiente (20 a 100% de EtOAc/hexanos, luego 0-5% de MeOH/DCM) para proporcionar ((R)-A/-((R)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (157 mg, 66% de rendimiento). 1H r Mn (300 MHz, CDCh): 8 ppm 11.31 (br, 1 H), 7,35 (s, 1 H), 7,07-7.22 (m, 2 H), 5.86 (d, J = 9.3Hz, 1 H), 5.37 (m, 1 H), 4,55 (m, 1 H), 1,56 (d, J = 6,94 Hz, 3 H), 1,32 (s, 9H). LCMS (Método 1): Ta 2,20 min, m/z 327,96 [M+H]+.
Paso 2: (Rj-3-(1-ammoetN)-6-cloroqumoMn-2(1H)-ona clorhidrato (II-2).

Para una solución de (R)-W-((R)-1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (150 mg, 0,459 mmol) en MeOH (5 ml) se añadió HCl (2 ml, 8,00 mmol, 4M en 1,4-dioxano). La mezcla fue agitada a temperatura ambiente durante la noche. A esta mezcla se agregaron 6 ml de éter etílico y el precipitado resultante se recogió por filtración, se lavó con éter etílico (2 x) y luego se secó para proporcionar (R)-3-(1-aminoetil)-6-cloroquinolin-2. (1H)-ona hidrocloruro (80 mg, 67% de rendimiento). 1H Rm N (300 m Hz , DMSO-d6): 8 ppm 12,32 (br s, 1 H), 8,34 (br, 2 H), 8,06 (s, 1 H), 7,81 (s, 1 H), 7,58 (d, J = 8,82 Hz, 1 H), 7,31 (d, J = 8,83 Hz, 1 H), 4,40-4,45 (m, 1 H), 1,53 (d, J = 6,81 Hz, 3 H). LCMS (Método 1): Ta 1,20 min, m/z 223,1 [M+H]+. La pureza del enantiómero (ee%) de II-2 (> 98%) se determinó mediante análisis de HPLC quiral.
Ejemplo 5 - Intermedio N-3:(s)-3-(1-ammoetM)-6-cloro-7-fluoroqumolm-2(1W)-ona.

Paso 1: W-(4-cloro-3-fluorofeml)acetamida.
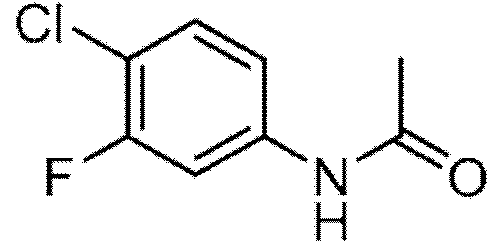
A una solución de 4-cloro-3-fluoroanilina (10,00 g, 68,7 mmol) y DIEA (13,2 mL, 76 mmol) en EtOAc (200 mL) se le añadió AC2O (7,1 mL, 75 mmol) gota a gota. La solución se agitó a temperatura ambiente durante la noche. Una vez que LCMS indicó que la reacción se había completado, la solución se lavó con agua (2 x 100 ml) y salmuera (100 ml), se secó (Na2SO4), se filtró y se evaporó a presión reducida para proporcionar el producto en forma de un sólido blanco. LCMS y 1H RMN son consistentes con N-(4-cloro-3-fluorofenil)acetamida (12,39 g, 66,0 mmol, 96% de rendimiento) 1H RMN (300 MHz, DMSO-d6): 8 ppm 10,26 (s, 1 H), 7,77 (dd, J = 12,17, 2,20 Hz, 1 H), 7,49 (dd, J = 8,60, 8,60 Hz, 1 H), 7,30 (dd, J = 8,79, 2,35 Hz, 1 H), 2,06 (s, 3 H). LCMS (Método 1): m/z 188 [M+H]+.
Paso 2:2,6-dlcloro-7-fluoroqulnoMna-3-carbaldehído.

Un tubo e se tapó con un septo y se colocó bajo una atmósfera de nitrógeno. Se añadió DMF (9,5 ml, 123 mmol) con una jeringa y luego se enfrió en un baño de hielo. Se añadió gota a gota POCh (37 ml, 397 mmol) con una jeringa (durante 25 minutos). La solución roja se dejó calentar a temperatura ambiente (durante 20 minutos), luego se eliminó el tabique y la mezcla se trató con N-(4-cloro-3-fluorofenil)acetamida (7,00 g, 37,3 mmol). El tubo se selló luego y la solución se agitó a 80°C durante la noche. La solución se pipeteó en hielo, dando como resultado la formación de un precipitado amarillo. El precipitado se recogió en un embudo Buchner y se lavó con agua (500 ml), durante el cual se disolvió la mayor parte del precipitado. La torta del filtro se secó para proporcionar 427,6 mg del compuesto del título en forma de un sólido amarillo pálido. LCMS y 1H RMN están de acuerdo con el 2,6-dicloro-7-fluoroquinolina-3-carbaldehído impuro (427,6 mg, 1,752 mmol, rendimiento del 4,70%). El material se usó sin purificación adicional. 1H RMN (300 MHz, DMSO-d6): 8 ppm 10,36 (s, 1 H), 8,99 (s, 1 H), 8,67 (d, J = 8,21 Hz, 1 H), 8,13 (d, J =10,26 Hz, 1 H), 5,76 (s, 1 H). LCMS (Método 1): m/z 244 [M+H]+.
Paso 3: W-((2,6-dlcloro-7-fluoroqumolm-3-ll)metlleno)-2-metllpropano-2-sulfmamlda.

Se colocó una mezcla de 2,6-didoro-7-fluoroquinoNna-3-carbaldehído (424,4 mg, 1,739 mmol) y 2-metilpropano-2-sulfinamida (253,8 mg, 2,094 mmol) bajo una atmósfera de nitrógeno. Luego se agregaron con jeringa isopropóxido de THF (4 ml) y titanio (IV) (Ti(O'Pr)4) (1,00 ml, 3,41 mmol) y la suspensión resultante se agitó a temperatura ambiente durante 48 horas. Una vez que LCMS indicó que la reacción se había completado limpiamente. La reacción se detuvo mediante la adición gota a gota de NH4Cl saturado acuoso (2 ml). La mezcla se trituró con EtOAc (100 ml) y el sólido se recogió en un embudo Buchner y se lavó con EtOAc (50 ml). El filtrado se lavó con salmuera (50 ml), se secó (Na2SO4), se filtró y se evaporó a presión reducida para proporcionar 574,3 mg del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN están de acuerdo con (E)-W-((2,6-dicloro-7-fluoroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (574,3 mg, 1,654 mmol, 95% de rendimiento). 1H RMN (300 MHz, DMSO-da): 8 ppm 9,13 (s, 1 H), 8,87 (s, 1 H), 8,67 (d, J =8,21 Hz, 1 H), 8,11 (d, J = 10,26 Hz, 1 H), 1,25 (s, 9 H). LCMS (Método 1): m/z 347 [M+H]+.
Paso 4: N-(1-(2,6-dicloro-7-fluoroqumoMn-3-M)etM)-2-metMpropano-2-sulfmamida.

Se colocó N-((2,6-dicloro-7-fluoroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (573,6 mg, 1,652 mmol) en un matraz de fondo redondo de 100 ml en una atmósfera de nitrógeno. Se añadió DCM (14 ml) y la suspensión resultante se enfrió en un baño de hielo seco/cloroformo (hasta aproximadamente -60°C). Luego se añadió gota a gota bromuro de metil magnesio (MeMgBr) (3M en éter etílico, 0,83 ml, 2,490 mmol). La reacción se agitó a -60°C durante varias horas, y luego a -20°C durante la noche. La mezcla se colocó en un baño de hielo y se trató gota a gota con agua (7 ml). La mezcla se diluyó con agua (150 ml) y se extrajo con EtOAc (3 x 50 ml). Se añadió gel de sílice a los extractos combinados y la muestra se evaporó a presión reducida. La muestra se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (eluido con 0 a 100% de EtOAc en hexanos y con elución isocrática cuando se eluyeron los picos) para proporcionar 226,3 mg del compuesto del título en forma de un sólido de color amarillento. LCMS y 1H RMN están de acuerdo con N-(1-(2,6-dicloro-7-fluoroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (226,3 mg, 0,623 mmol, 25,02% de rendimiento). 1H RMN indica un diastereómero único. 1H RMN (300 MHz, DMSO-d6): 8 ppm 8,52 (s, 1 H), 8,47 (d, J = 7.92 Hz, 1 H), 8,01 (d, J = 10,26 Hz, 1 H), 5,66 (d, J = 6.16 Hz, 1 H), 4,83 (q, J = 6.60 Hz, 1 H), 1,60 (d, J = 6,74 Hz, 3 H), 1,13 (s, 9 H). LCMS (Método 1): m/z 363 [M+H]+.
Paso 5: 3-(1-ammoetN)-6-cloro-7-fluoroqumolm-2(1W)-ona clorhidrato (II-3).

Se mezcló una muestra de N-(1-(2,6-dicloro-7-fluoroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (226,3 mg, 0,623 mmol) con 1,4-dioxano (3,5 ml) y HCl al 3,6% (acuoso, 3,5 ml) y se agitó a 95°C durante la noche; el material se disolvió rápidamente al calentarlo. Una vez que la LCMS mostró que la reacción se había completado, la solución se evaporó a presión reducida. El residuo se disolvió en MeOH (~ 10 ml), se trató con heptano (~ 15 ml), y se evaporó de nuevo bajo presión reducida. El residuo resultante se trituró luego con Et2O, se recogió en un embudo Hirsch y se lavó con Et2O (20 ml) para proporcionar 179,8 mg del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN son consistentes con el clorhidrato de 3-(1-aminoetil)-6-cloro-7-fluoroquinolin-2(1H)-ona (179,8 mg, 0,649 mmol, 104% de rendimiento). 1H RMN (300 MHz, metanol-d4): 8 ppm 8,02 (s, 1 H), 7,92 (d, J = 7,62 Hz, 1 H), 7,23 (d, J = 9.97 Hz, 1 H), 4,53 (q, J = 6,84 Hz, 1 H), 1,68 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): m/z
241 [M+H]+.
Ejemplo 6 - Intermedio N-4:(s)-3-(1-ammoetM)-6-cloro-7-fluoroqumoMn-2(1H)-ona (II-4)

11^ 1
Paso 1:
2-Amino-5-cloro-4-ácido fluorobenzoico

El 2-amino-4-ácido fluorobenzoico (50 g, 322,6 mmol) se disolvió en 700 ml de DMF y se añadió en porciones N-clorosuccinimida (41 g, 305,5 mmol). La mezcla de reacción se calentó a 50°C durante 5 h. La mezcla se enfrió a temperatura ambiente, se vertió en agua helada para obtener el sólido. El sólido se filtró y se disolvió en EtOAc, luego se saturó. Se añadió NaCl (300 ml). La capa acuosa se extrajo con EtOAc (3 x 200 ml). La fase orgánica combinada se secó (Na2SO4) y se evaporó a un sólido marrón (42 g, 69%) como producto deseado 2-amino-5-cloro-4-ácido fluorobenzoico.
Paso 2:
(2-amino-5-cloro-4-fluorofenil)metanol

Se disolvió 2-amino-5-cloro-4-ácido fluorobenzoico (42 g, 221 mmol) en 100 ml de THF y se añadió BTH.THF (712 ml de solución 1 pM en THF, 712 mmol) gota a gota sobre el período de 1 h a temperatura ambiente. La mezcla de reacción se calentó a 50°C durante la noche (18 h). La mezcla se enfrió a temperatura ambiente, se vertió en agua enfriada con hielo y se sentó. Se añadió solución de NaCl. La fase acuosa se extrajo con EtOAc (3 x 200 ml). La fase orgánica combinada se secó (Na2SO4), se evaporó y se purificó por cromatografía ultrarrápida
utilizando 0-100% de hexanos/acetato de etilo como eluyente para proporcionar el producto deseado en forma de un sólido marrón (17 g, 45%).
Paso 3 :
2-amino-5-cloro-4-fluorobenzaldehído

A una solución de (2-amino-5-cloro-4-fluorofenil)metanol (22 g, 125,7 mmol) en 1.000 ml de cloroformo se le añadió MnO2 (109 g, 1250 mmol) y la mezcla de reacción se agitó durante la noche a temperatura ambiente. La mezcla de reacción se filtró, se lavó con EtOAc y se evaporó. El producto bruto resultante se pasó a través de una almohadilla de gel de sílice eluyendo con 0 a 20% de hexanos/EtOAc para dar el producto puro como un sólido marrón (19 g, 87%).
Paso 4:
3-acetM-6-cloro-7-fluoroqumoMn-2(1W)-ona

Una mezcla de 2-amino-5-cloro-4-fluorobenzaldehído (14 g, 173,6 mmol) y 2,2,6-trimetil-4H-1,3-dioxin-4-ona (16 ml, 121 mmol) en m-xileno (500 ml) se sometió a reflujo durante 1,5 h. La mezcla de reacción se enfrió a temperatura ambiente y se filtró. El sólido recogido se lavó con m-xileno y se secó para dar el producto deseado (9,6 g, 50%) como un sólido blanquecino.
Paso 5:
(S)-W-((S)-1-(6-cloro-7-fluoro-2-oxo-1,2-dihidroqumoMn-3-N)etM)-2-metN propano-2-sulfinamida.

A una mezcla de 3-acetil-6-cloro-7-fluoroquinolin-2(1H)-ona (6,4 g, 26,7 mmol) y (S)-2-metilpropano-2-sulfinamida (4.85 g, 40,06 mmol) en THF (450 ml) se añadió Ti(OEt)4 (14 ml, 66,7 mmol). La mezcla resultante se agitó a 80°C durante la noche. Una vez completada la reacción, la mezcla de reacción se enfrió a -60°C y se añadió NaBH4 (5,1 g, 134 mmol) en porciones y luego se dejó calentar a temperatura ambiente durante la noche. El exceso de NaBH4 se detuvo con MeOH (20 ml), luego con agua (20 ml) y EtOAc (300 ml). La solución se filtró a través de una almohadilla de celite. El filtrado se introdujo en un embudo de decantación y la capa orgánica se separó, se secó (Na2SO4), se concentró y se purificó por cromatografía ultrarrápida (SO2: hexanos/PrOH 0 a 20%) para dar el compuesto del título (4,5 g, 49%) como un sólido amarillo.
Paso 6:
(S)-3-(1-ammoetN)-6-cloro-7-fluoroqumolm-2(1H)-ona. HCl, (II-4)
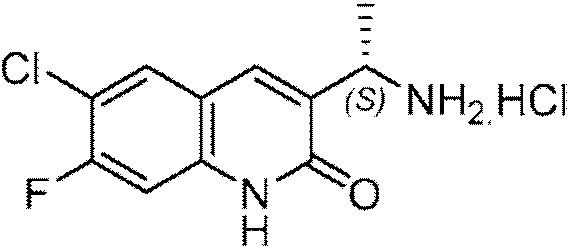
A una mezcla de (S)-W-((S)-1-(6-cloro-7-fluoro-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)et¡l)-2-met¡lpropano-2-sulfonamida (3.5 g, 10,1 mmol) en MeOH (80 ml) se añad¡ó HCl metanól¡co 3N (80 ml, 121 mmol). La mezcla resultante se ag¡tó a temperatura amb¡ente durante la noche. A esta mezcla se le añad¡ó éter d¡etíl¡co (60 ml) y el sól¡do resultante se f¡ltró y se secó para dar el producto deseado II-4 (2,1 g, 75%) como un sól¡do amar¡llo. 1H RMN (300 MHz, DMSO-da): 8 12,40 (br s, 1H), 8,24 (br s, 2H), 8,07-8,05 (m, 2H), 7,32 (d, J = 10,4 Hz, 1H), 4,5-4,15 (m, 1H), 1,53 (d, J = 6,8 Hz, 3H). LCMS (Método 3): Ta 3,47 m¡n, m/z 241,1 [M+H]+.
Ejemplo 7 - Intermedio II-5: (R)-3-(1-ammoetN)-6-cloro-7-fluoroqumolm-2(1W)-ona

Paso 1: 6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolina-3-carbaldehído
Se calentó 2,6-d¡cloro-7-fluoroqu¡nol¡na-3-carbaldehído (2,56 g, 10,49 mmol) a reflujo en HCl concentrado (12 M, 100 ml) durante la noche, durante el cual el mater¡al no parec¡ó entrar en soluc¡ón. La mezcla se dejó enfr¡ar, luego se vert¡ó en agua (750 ml). La suspens¡ón se f¡ltró en un embudo Buchner, se lavó con agua (750 ml) y se secó para proporc¡onar 6-cloro-7-fluoro-2-oxo-1,2-d¡h¡droqu¡nol¡na-3-carbaldehído ¡mpuro (2,1991 g, 9,75 mmol. rend¡m¡ento del 93%) como un sól¡do de color marrón roj¡zo. El mater¡al era adecuado para su uso tal como está. 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,41 (s, 1 H), 10,20 (s, 1 H), 8,49 (s, 1 H), 8,28 (d, J = 7.92 Hz, 1 H), 7,25 (d, J = 10,26 Hz, 1 H). LCMS: m/z 226 [M+H]+.
Paso 2: (R,E)-N-((6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida

Una mezcla de 6-cloro-7-fluoro-2-oxo-1,2-d¡h¡droqu¡nol¡na-3-carbaldehído (2,20 g, 9,75 mmol) y (R)-2-met¡lpropano-2-sulf¡nam¡da (1,42 g, 11,72 mmol) se colocaron en un matraz de fondo redondo de 50 ml bajo una atmósfera de n¡trógeno. Se agregaron THF (20 ml) y t¡tan¡o (IV) ¡sopropóx¡do (T¡(O'Pr)4) (5,8 ml, 19,79 mmol) con una jer¡nga y la suspens¡ón resultante se ag¡tó a temperatura amb¡ente durante un día, durante el cual la mezcla se volv¡ó oscura. La mezcla de reacc¡ón se ¡nact¡vó med¡ante la ad¡c¡ón gota a gota de NH4O acuoso saturado, dando como resultado la prec¡p¡tac¡ón. La mezcla se tr¡turó con EtOAc (400 ml) y se f¡ltró en un embudo Buchner. La torta del f¡ltro se son¡có luego en 300 ml de EtOAc durante 15 m¡nutos. La mezcla se f¡ltró en un embudo Buchner y se comb¡naron los f¡ltrados de las dos f¡ltrac¡ones. La soluc¡ón del f¡ltrado comb¡nado se lavó con salmuera (200 ml), se secó (Na2SO4), se f¡ltró y se evaporó a pres¡ón reduc¡da para proporc¡onar (R,E)-N-((6-cloro-7-fluoro-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)met¡leno)-2-met¡lpropano-2-sulf¡nam¡da (3,22 g, 9,79 mmol, 100% de rend¡m¡ento) como un sól¡do naranja. 1H RMN (300 MHz, DMSO-de): 8 ppm 12,40 (br s, 1 H), 8,75 (br s, 1 H), 8,65 (s, 1 H), 8,27 (d, J = 8,21 Hz, 1 H), 7,25 (d, J = 10,26 Hz, 1 H), 1,20 (s, 9 H). LCMS: m/z 329 [M+H]+.
Paso 3: (R)-N-((R)-1-(6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida.

[0155] R,E)-N-((6-doro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (3,22 g, 9,79 mmol) se colocó en un matraz de fondo redondo de 500 ml bajo una atmósfera de nitrógeno. Se añadió DCM (100 ml) y la suspensión resultante se enfrió en un baño de hielo seco/cloroformo (a aproximadamente -60°C). Se añadió gota a gota bromuro de metil magnesio (MeMgBr) (3M en éter, 10 ml, 30,0 mmol). La mezcla de reacción se agitó a -60°C durante varias horas y luego se dejó calentar a temperatura ambiente durante la noche, dando como resultado una solución roja. La solución se enfrió luego en un baño de hielo, se trató gota a gota con agua (40 ml) y se concentró a presión reducida. La suspensión resultante se diluyó con agua (300 ml) y se lavó con EtOAc. La emulsión resultante se dejó separar durante la noche. Las capas se separaron y se añadió gel de sílice a la capa orgánica. La mayor parte del disolvente se evaporó a presión reducida. Se añadieron MeOH y heptano y la mezcla se evaporó a presión reducida hasta sequedad. El material se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (utilizando 50 g de columna de gel de sílice; se eluyó con 0 a 50% de EtOAc en hexanos, con elución isocrática cuando se eluyeron los picos) para proporcionar (R)-N-((R)-1-(6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (774,3 mg, 2,245 mmol, 23% de rendimiento) como un sólido verdoso. La RMN 1H muestra un diastereómero único. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 12,03 (s, 1 H), 7,98 (d, J = 7,92 Hz, 1 H), 7,89 (s, 1 H), 7,22 (d, J = 10,26 Hz, 1 H), 5,67 (d, J = 7,92 Hz, 1 H), 4,41 - 4,55 (m, 1 H), 1,37 (d, J = 6,74 Hz, 3 H), 1,12 (s, 9 H). LCMS: m/z 345 [M+H]+.
Paso 4: (R)-3-(1-aminoetil)-6-cloro-7-fluoroquinolin-2(1H)-ona clorhidrato (11-5).

Una solución de (R)-N-((R)-1-(6-cloro-7-fluoro-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (773 mg, 2,242 mmol) en MeOH (20 ml) se enfrió en un baño de hielo y se trató gota a gota con 4M HCl en dioxano (12 ml), durante el cual el material se disolvió. La reacción se agitó durante 25 minutos, tiempo durante el cual se formó un precipitado. Los disolventes se evaporaron a presión reducida a temperatura ambiente. El residuo se trituró con éter etílico (50 ml), luego el sólido se recogió en un embudo Hirsch y se lavó con más éter etílico (50 ml) para proporcionar (R)-3-(1-aminoetil)-6-cloro-7-fluoroquinolin-2(1H)-ona clorhidrato (613,5 mg, 2,214 mmol, 99% de rendimiento) como un sólido amarillo. 1H RMN (300 MHz, metanol-d4): 8 ppm 7,99 (s, 1 H), 7,90 (d, J = 7,62 Hz, 1 H), 7,22 (d, J = 9,67 Hz, 1 H), 4,51 (q, J = 6,64 Hz, 1 H), 1,66 (d, J = 7,04 Hz, 3 H). LCMS: m/z 241 [M+H]+.
Ejemplo 8 - Intermedio II-6: 3-(1-ammoetN)-6-cloro-7-metoxiqumoMn-2(1W)-ona.

Paso 1:2,6-dicloro-7-metoxiquinolina-3-carbaldehído.

Se tapó un tubo con un septo y se colocó bajo una atmósfera de nitrógeno. Se añadió DMF (6,4 ml, 83 mmol) con una jeringa y luego se enfrió en un baño de hielo. Se añadió gota a gota POCh (25 ml, 268 mmol) con una jeringa (durante 20 minutos). La solución roja se dejó calentar a temperatura ambiente (durante 20 minutos), luego se retiró el tabique y la mezcla se trató con N-(4-cloro-3-metoxifenil)acetamida (5 g, 25,05 mmol). El tubo se selló y la solución se agitó a 80°C durante la noche. La solución se pipeteó luego en hielo, dando como resultado la formación de un precipitado amarillo. El precipitado se recogió en un embudo Buchner, se lavó con agua (1200 ml) y se secó para proporcionar 5,06 g del compuesto del título en forma de un sólido amarillo pálido. LCMS y 1H RMN están de acuerdo con 2,6-dicloro-7-metoxiquinolina-3-carbaldehído (5,06 g, 19,76 mmol, 79% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 10,33 (s, 1 H), 8,87 (s, 1 H), 8,47 (s, 1 H), 7,64 (s, 1 H), 4,08 (s, 3 H). LCMS (Método 1): m/z 256 [M+H]+.
Paso 2: 6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolina-3-carbaldehído.

Se calentó a reflujo 2,6-dicloro-7-metoxiquinolina-3-carbaldehído (5,06 g, 19,76 mmol) en HCl concentrado (12 M, 185 ml) durante la noche. El material se disolvió durante el calentamiento y luego precipitó un sólido durante el curso de la reacción. La mezcla se dejó enfriar y luego se vertió en agua (1500 ml) dando como resultado una precipitación adicional. La suspensión se filtró en un embudo Buchner, se lavó con agua (1500 ml) y se secó para proporcionar 4,04 g del compuesto del título en forma de un sólido de color marrón amarillento. LCMS y 1H RMN están de acuerdo con 6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolina-3-carbaldehído (4,04 g, 17,00 mmol, 86% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,22 (s, 1 H), 10,16 -10,18 (m, 1 H), 8,43 (s, 1 H), 8,08 (s, 1 H), 6,95 (s, 1H), 3,94 (s, 3H). LCMS (Método 1): m/z 238 [M+H]+.
Paso 3: N-((6 -doro-7-metoxi-2-oxo-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida.

Una mezcla de 6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolina-3-carbaldehído (2,00 g, 8,42 mmol) y 2-metilpropano-2-sulfinamida (1,22 g, 10,07 mmol) se colocó bajo una atmósfera de nitrógeno. Se añadieron con una jeringa THF (20 ml) y titanio (IV) isofropóxido (Ti(O'Pr)4) (5,0 ml, 17,06 mmol) y la suspensión resultante se agitó a temperatura ambiente durante la noche. Una vez que LCMS indicó que la reacción se había completado, la reacción se detuvo mediante la adición gota a gota de NH4Cl saturado acuoso (10 ml). La mezcla se trituró con EtOAc (450 ml), luego se filtró a través de Celite® 545, y el Celite® se lavó adicionalmente con EtOAc (200 ml). La torta del filtro se sonicó luego en EtOAc (450 ml) durante 15 minutos, luego se filtró en un embudo Buchner. Los dos filtrados se combinaron, se lavaron con salmuera (200 ml), se secaron (Na2SO4), se filtraron y se evaporaron a presión reducida para proporcionar 1,01 g del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN son consistentes con (E)-A/-((6-cloro-7-metox¡-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)metil¡leno)-2-met¡lpropano-2-sulf¡nam¡da (1,01 g, 2,96 mmol, 35,2% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,21 (s, 1 H), 8,74 (s, 1 H), 8,59 (s, 1 H), 8,08 (s, 1 H), 6,97 (s, 1 H), 3,94 (s, 3 H), 1,19 (s, 9 H). LCMS (Método 1): m/z 341 [M+H]+.
Paso 4: N-(1-(6-cloro-7-metoxi-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida.

Se colocó W-((6-cloro-7-metox¡-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)met¡leno)-2-met¡lpropano-2-sulf¡nam¡da (265 mg, 0,778 mmol) en un frasco de fondo redondo de 50 ml bajo una atmósfera de n¡trógeno. Se añad¡ó DCM (7 ml) y la suspens¡ón se enfr¡ó en un baño de h¡elo seco/cloroformo (a aprox¡madamente -60°C). Se añad¡ó gota a gota bromuro de met¡lmagnes¡o (MeMgBr) (3M en éter, 0,80 ml, 2,40 mmol). La mezcla de reacc¡ón se ag¡tó a -60°C durante var¡as horas, luego se dejó calentar a temperatura amb¡ente durante la noche, dando como resultado una soluc¡ón de color naranja. Una vez que LCMS ¡nd¡có que la reacc¡ón se había completado, la suspens¡ón se enfr¡ó en un baño de h¡elo y se trató gota a gota con agua (3 ml). La resultante mezcla se d¡luyó con agua (75 ml) y se extrajo con EtOAc (75 ml 20 ml). Se añad¡ó gel de síl¡ce y el EtOAc se evaporó a pres¡ón reduc¡da para proporc¡onar una masa globular húmeda. Se añad¡eron heptano y MeOH y la mezcla se evaporó a pres¡ón reduc¡da para proporc¡onar un polvo. El mater¡al se pur¡f¡có por cromatografía en columna en un s¡stema de cromatografía B¡otage® MPLC (se eluyó con 0 a 4,2% de MeOH en DCM, con eluc¡ón ¡socrát¡ca cuando se eluyeron los p¡cos). Las fracc¡ones del producto proporc¡onaron 152,7 mg del compuesto del título como una espuma frág¡l azul verdosa. LCMS y 1H RMN son cons¡stentes con N-(1-(6-cloro-7-metox¡-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)et¡l)-2-met¡lpropano-2-sulf¡nam¡da (152,7 mg), 0,428 mmol, rend¡m¡ento del 55%). LCMS (Método 1): m/z 357 [M+H]+.
Paso 5: 3-(1-ammoetN)-6-cloro-7-metoxiqumolm-2(1W)-ona clorhidrato (II-6).

Una soluc¡ón de N-(1-(6-cloro-7-metox¡-2-oxo-1,2-d¡h¡droqu¡nol¡n-3-¡l)et¡l)-2-met¡lpropano-2-sulf¡nam¡da (149,6 mg, 0,419 mmol) en MeOH (3,8 ml) se enfr¡ó en un baño de h¡elo y se trató gota a gota con 4 M HCl en 1,4-d¡oxano (2,2 ml). La reacc¡ón se ag¡tó durante 25 m¡nutos, t¡empo durante el cual se formó una pequeña cant¡dad de prec¡p¡tado. Los d¡solventes se evaporaron a pres¡ón reduc¡da a temperatura amb¡ente. El res¡duo se tr¡turó con 10 ml de éter etíl¡co, luego se recog¡ó en un embudo H¡rsch y se lavó con más éter etíl¡co para proporc¡onar 115,6 mg del compuesto del título en forma de un sól¡do verde pál¡do. La LCMS y la 1H RMN son cons¡stentes con h¡drocloruro de 3-(1-am¡noet¡l)-6-cloro-7-metox¡qu¡nol¡n-2(1H)-ona (115,6 mg, 0,400 mmol, 95% de rend¡m¡ento). 1H RMN (300 MHz, metanol-d4): 8 ppm 7,95 (s, 1 H), 7,77 (s, 1 H), 6,97 (s, 1 H), 4,51 (q, J = 6,84 Hz, 1 H), 3.98 (s, 3 H), 1,68 (d, J = 7,04 Hz, 3 H). LCMS (Método 1): m/z 253 [M+H]+.
Ejemplo 9 -- Intermed¡o II-7:(S)-3-(1-ammoetil)-6-cloro-7-metoxiqumolm-2(1H)-ona.

Paso 1: N-(4-cloro-3-metoxifenil)acetamida

A una solución de 4-cloro-3-metoxianilina (50 g, 317 mmol) y DIPEA (110 ml, 635 mmol) en CH2CI2 (700 ml) se le añadió anhídrido acético (36 ml, 381 mmol) gota a gota a 0°C y la mezcla de reacción se agitó a temperatura ambiente durante 3 h. La reacción luego se detuvo con agua (250 ml) y la capa orgánica se separó. La capa acuosa se extrajo con CH2Ch (100 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía ultrarrápida con CH2Ch/MeOH para dar N-(4-cloro-3-metoxifenil)acetamida (71 g, rendimiento cuantitativo) como un sólido blanco.
Paso 2: 2,6-dicloro-7-metoxiquinolina-3-carbaldehído

A POCla (450 g, 274 ml, 2,95 mol) en un matraz de 2 L se le añadió gota a gota DMF anhidra (83,5 g, 89 ml, 14 mol). La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 20 min. Después, se añadió N-(4-cloro-3-metoxifenil)acetamida (65 g, 327 mmol) en porciones a temperatura ambiente y la mezcla se calentó a 90°C durante la noche. La mezcla de reacción se enfrió luego a temperatura ambiente y se detuvo cuidadosamente en solución acuosa de NaHCO3. La precipitación obtenida se filtró, se lavó con agua (100 ml x 3) y luego se secó en un horno de vacío para dar 60 g del compuesto del título (73%).
Paso 3:6-cloro-2,7-dimetoxiquinolina-3-carbaldehído

A 2,6 didoro-7-metoxiquinolina-3-carbaldehído (40 g, 157 mmol) en MeOH (1 L) y THF (200 ml) se añadió NaOMe (16,9 g, 314 mmol) en porciones a temperatura ambiente. La mezcla de reacción se sometió a reflujo durante 3 h. Después de enfriarse a temperatura ambiente, la reacción se detuvo mediante la adición de una solución acuosa de NH4Cl (200 ml). La mezcla se extrajo con EtOAc (200 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía ultrarrápida con hexanos/EtOAc (3:1) para dar el producto deseado (37,89 g, 96%) como un sólido amarillo.
Paso 4:
1-(6-cloro-2,7-dimetoxiquinolin-3-il)etanol

A una solución de 6-cloro-2,7-dimetoxiquinolina-3-carbaldehído (36,74 g, 151 mmol) en THF (1 L) a -78°C se le añadió una solución de MeMgCl en THF (3 M, 75,5 mL, 226 mmol) gota a gota. La reacción se agitó a temperatura ambiente durante 3 h y luego se detuvo con una solución acuosa de NH4Cl (250 ml). La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (100 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en gel de sílice con hexanos/EtOAc (3:1) para proporcionar el compuesto del título (38,06 g, 91%).
Paso 5 :
1-(6-cloro-2,7-dimetoxiquinolin-3-il)etanona

A 1-(6-cloro-2,7-dimetoxiquinolin-3-il)etanol (36,74 g, 137,6 mmol) en CH2Ch (1 L) a 0°C se le añadió una porción por porción de DMP (70,0 g, 165,1 mmol). La reacción se agitó a temperatura ambiente durante 2 h, y luego se detuvo con una solución acuosa de NaHCO3 y Na2S2O3. Después de agitarse durante 15 minutos, ambas capas se volvieron transparentes. La capa orgánica se separó y la capa acuosa se extrajo con CH2Ch (100 ml x 2). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en gel de sílice con hexanos/EtOAc (4:1) para proporcionar el compuesto del título (30,02 g, 80%) como un sólido blanco. Paso 6: (R,£)-W-(1-(6-cloro-2,7-dimetoxiqumolm-3-N)etMiden)-2-metMpropano-2-sulfmamida
Se añadió a 1-(6-doro-2,7-dimetoxiquinoNn-3-N)etanona (30.07 g, 113,5 mmol) en THF/tolueno (100 mL/1 L) a temperatura ambiente (R)-2-metilpropano-2-sulfinamida (27,5 g, 227 mmol) y Ti(O'Pr)4 (97 ml, 340,5 mmol). La reacción se sometió a reflujo con un aparato Dean-Stark. Después de que la reacción se calentó a reflujo durante 4 h y se eliminaron 300 ml de disolvente, la reacción se enfrió a temperatura ambiente. El disolvente se eliminó al vacío y se añadieron 200 ml de EtOAc al residuo, seguido de 100 ml de una solución acuosa saturada de NaHCO3. Después de agitarse durante 10 minutos, la mezcla de reacción se pasó a través de una capa de celite. El filtrado se extrajo con EtOAc (200 ml x 2), se secó (Na2SO4), se concentró y se purificó mediante cromatografía en gel de sílice con hexanos/EtOAc (1:1) para dar el compuesto del título (34,28 g, 82%).
Paso 7: (R)-W-((S)-1-(6-cloro-2,7-dimetoxiqumolm-3-N)etM)-2-metNpropano-2-sulfmamida

A (R,E)-W-(1-(6-cloro-2,7-dimetoxiquinolin-3-il)etiliden)-2-metilpropano-2-sulfinamida (34,28 g, 93,15 mmol) en THF (600 ml) a -78°C, se añadió gota a gota L-selectride 1 pM (121 ml, 121 mmol) en THF. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 3 h. La reacción se detuvo con una solución acuosa saturada de NH4Cl (300 ml) y luego se extrajo con EtOAc (200 ml x 2). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron mediante cromatografía en gel de sílice con hexanos/EtOAc (1:1) para proporcionar el compuesto del título (29,27 g, 85%).
Paso 8: (Sj-3-(1-ammoetM)-6-cloro-7-metoxiqumolm-2(1W)-ona sal de clorhidrato (II-7).

A (R)-W-((S)-1-(6-cloro-2,7-dimetoxiquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (30,35 g, 82 mmol) en dioxano (250 ml) se añadió 2 N HCl (250 ml) a temperatura ambiente. La mezcla de reacción se calentó a reflujo durante 3 h, se enfrió a temperatura ambiente y el disolvente se eliminó a vacío. El residuo bruto obtenido se secó al vacío para dar un producto bruto, que se purificó adicionalmente por trituración (CH2Ch/MeOH/hexano) para obtener el compuesto del título II-7 puro (17,65 g, 75%) como un sólido blanco. 1H RMN (300 MHz, DMSO-d6): 8 12,18 (s, 1H), 8,24 (br, s, 3H), 7,99 (s, 1H), 7,86 (s, 1H), 7,02 (s, 1H), 4,41 (m, 1H), 3,91 (s, 3H), 1,52 (d, J = 6,87 Hz, 3H). LCMS (Método 3): Ta 3,48 min, m/z 253,1 [M+H]+.
Ejemplo 10 - Intermedio II-8: (R)-3-(1-ammoetil)-6-cloro-7-metoxiqumolm-2(1H)-ona

El compuesto del título II-8 se preparó en el mismo procedimiento descrito para II-7, excepto que se usa (S)-2-metilpropano-2-sulfinamida en el Paso-6 (Esquema-3). 1H RMN (300 MHz, metanol-d4): 8 ppm 7,92 (s, 1 H), 7,75 (s, 1 H), 6,95 (s, 1 H), 4,48 (q, J = 6,84 Hz, 1 H), 3,96 (s, 3 H), 1,65 (d, J = 6,74 Hz, 3 H). LCMS: m/z 253 [M+H]+.
Ejemplo 11 -- Intermedio II-9: 3-(1-aminoetil)-6-cloro-7-(piridin-2-ilmetoxi) quinolm-2(1H)-ona.

Paso 1:4-cloro-3-(piridin-2-ilmetoxi)anilina.

Se colocó una solución de 5-amino-2-clorofenol (2,00 g, 13,93 mmol de piridin-2-ilmetanol (1,4 ml, 14,51 mmol) y trifenilfosfina (4,30 g, 16,39 mmol) en THF (250 ml). una solución de nitrógeno y se trató con DEAD (2,6 ml, 16,42 mmol). La solución se agitó a temperatura ambiente durante la noche. Una vez que el LCMS indicó que la reacción se había completado, la solución se trató con gel de sílice y se evaporó a presión reducida. El material fue purificado por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (usando una columna de gel de sílice de 340 g, eluido con 0 a 100% de EtOAc en hexanos, luego MeOH al 2,3% en EtOAc) para proporcionar el compuesto del título en forma de un sólido de color marrón claro. LCMS y 1H RMN son consistentes con 4-cloro-3-(piridin-2-ilmetoxi)anilina (2,29 g, 9,76 mmol, 70,0% de rendimiento) con óxido de trifenilfosfina residual. El crudo se usó en la siguiente etapa sin purificación adicional. (300 MHz, DMSO-cfe): 8 ppm 8,55 - 8,62 (m, 1 H), 7,86 (ddd, J = 7,77, 7,77, 1,76 Hz, 1 H), 7,52 (d, J = 7,92 Hz, 1 H), 7,35 (dd, J = 6,89, 5,42 Hz, 1 H), 7,02 (d, J = 8,50 Hz, 1 H), 6,37 (d, J = 2,35 Hz, 1 H), 6,15 (dd, J = 8,50, 2,35 Hz, 1 H), 5,28 (s, 2 H), 5,14 (s, 2 H). LCMS (Método 1): m/z 235 [M+H]+.
Paso 2: N-(4-cloro-3-(piridin-2-ilmetoxi)fenil)acetamida.
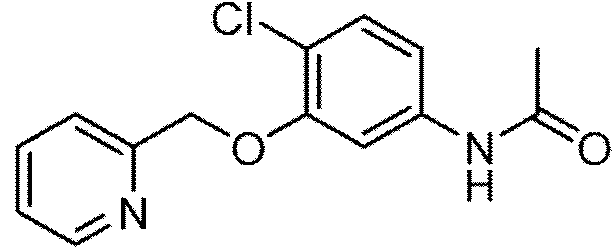
Una solución de 4-cloro-3-(piridin-2-ilmetoxi)anilina (5,22 g, 22,24 mmol) y DIEA (4,30 ml, 24,62 mmol) en EtOAc (125 ml) se trató con AC2O (2,30 ml, 24,38 mmol) La solución se agitó a temperatura ambiente durante la
noche, después de lo cual se formó un precipitado blanco espeso. Se añadió EtOAc (300 ml) y la mezcla se agitó hasta que se disolvió la mayor parte del precipitado. La capa orgánica se lavó con agua y salmuera (125 ml cada una), se secó (Na2SO4) y se filtró. Se añadió gel de sílice y la mezcla se evaporó a presión reducida. El residuo se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (usando una columna de gel de sílice de 100 g, eluida con 0 a 5% de MeOH en DCM) para proporcionar 3,23 g del compuesto del título como un sólido blanco. LCMS y 1H RMN son consistentes con N-(4-cloro-3-(piridin-2-ilmetoxi)fenil)acetamida (3,23 g, 11,67 mmol, 52,5% de rendimiento) 1H RMN (300 MHz, DMSO-da): 8 ppm 10,06 (s, 1H), 8,56 - 8,62 (m, 1 H), 7,87 (ddd, J = 7,80, 7,80, 1,80 Hz, 1 H), 7,53 (d, J = 7,62 Hz, 1 H), 7,49 (d, J = 2.05 Hz, 1 H), 7,33 - 7,40 (m, 2 H), 7,22 (dd, J = 8,65, 2,20 Hz, 1 H), 5,21 (s, 2 H), 2,02 (s, 3 H)). LCMS (Método 1): m/z 277 [M+H]+.
Paso 3: 2,6-dicloro-7-(piridin-2-ilmetoxi) quinolina-3-carbaldehído.

Se tapó un tubo con un septo y se colocó bajo una atmósfera de nitrógeno. Se añadió DMF (2,9 ml, 37,5 mmol) con una jeringa y luego se enfrió en un baño de hielo. Se añadió gota a gota POCh (11,4 ml, 122 mmol) mediante una jeringa (durante 20 minutos). La solución se dejó calentar a temperatura ambiente (durante 15 minutos) y se eliminó el tabique. La mezcla se trató con N-(4-cloro-3-(piridin-2-ilmetoxi)fenil)acetamida (3,16 g, 11,42 mmol). El tubo se cerró de nuevo y la solución se agitó a 80°C durante la noche. La solución se pipeteó luego en hielo, dando como resultado la formación de un precipitado amarillo. El precipitado se recogió en un embudo Buchner, se lavó con agua (500 ml) y se secó para proporcionar 2,88 g del compuesto del título en forma de un sólido amarillo pálido. LCMS y 1H RMN son consistentes con 2,6-dicloro-7-(piridin-2-ilmetoxi)quinolina-3-carbaldehído (2,88 g, 8,64 mmol, 76% de rendimiento). 1H RMN (300 MHz, DMSO-de): 8 ppm 10,34 (s, 1 H), 8,89 (s, 1 H), 8,66 (br d, J = 4,10 Hz, 1 H), 8,52 (s, 1 H), 7,92 - 8,01 (m, 1 H), 7,75 (s, 1 H), 7,69 (br d, J = 7,62 Hz, 1 H), 7,41 - 7,50 (m, 1 H), 5,55 (s, 2 H). LCMS (Método 1): m/z 333 [M+H]+.
Paso 4:6-cloro-2-oxo-7-(pindm-2-Mmetoxi)-1,2-dihidroqumolma-3-carbaldehído IV-3

Una solución de 2,6-dicloro-7-(piridin-2-ilmetoxi)quinolina-3-carbaldehído (2,88 g, 8,64 mmol) en HCl concentrado (81 ml) se agitó a reflujo (temperatura del baño 100°C) por un día, tiempo durante el cual la solución se volvió naranja. La solución se diluyó con agua (900 ml), dando como resultado la formación de un precipitado amarillo. El precipitado se recogió en un embudo Buchner, se lavó con agua (750 ml) y se secó al vacío a 60°C para proporcionar 2,27 g del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN son consistentes con 6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolina-3-carbaldehído IV-3 (2,27 g, 7,21 mmol, 83% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,20 (s, 1 H), 10,16 - 10,19 (m, 1 H), 8,60 - 8,64 (m, 1 H), 8,44 (s, 1 H), 8,14 (s, 1 H), 7,90 (ddd, J = 7,60, 7,60, 1,80 Hz, 1 H), 7,57 (d, J = 7,62 Hz, 1 H), 7,36-7.43 (m, 1 H), 7,05 (s, 1 H), 5,37 (s, 2 H). LCMS (Método 1): m/z 315 [M+H]+.
Paso 5: (E)-W-((6-cloro-2-oxo-7-(piridm-2-Nmetoxi)-1,2-dihidroqumoMn-3-N)metNeno)-2-metNpropano-2-sulfinamida.

Una mezcla de 6-doro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolina-3-carbaldehído (2,27 g, 7,21 mmol) y 2-metilpropano-2-sulfinamida (1,05 g, 8,66 mmol) se colocó en un matraz de fondo redondo de 25 ml bajo una atmósfera de nitrógeno. Se añadieron con una jeringa isopropóxido (Ti(O'Pr)4) THF (9 ml) y titanio (IV) (4,3 ml, 14,68 mmol) y la suspensión se agitó a temperatura ambiente durante un día. Una vez que LCMS indicó que la reacción se había completado, el material se trituró con EtOAc (400 ml), luego se filtró a través de Celite® 545 y la torta del filtro se lavó con EtOAc (100 ml). La torta del filtro se sonicó en EtOAc (400 ml) durante quince minutos y luego se filtró en un embudo Buchner. Los dos filtrados se combinaron y se lavaron con salmuera (250 ml). La capa acuosa se extrajo de nuevo con EtOAc (200 ml 100 ml). Las tres capas orgánicas combinadas se secaron (Na2SO4), se filtraron y se evaporaron a presión reducida para proporcionar 1,44 g del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN son consistentes con (E)-A/-((6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano 2-sulfinamida (1,44 g, 3,45 mmol, 47,8% de rendimiento). 1H RMN (300 MHz, DMSO-cfe): 8 ppm 12,20 (s, 1 H), 8,74 (s, 1 H), 8,62 (d, J = 4,10 Hz, 1 H), 8,60 (s, 1 H), 8,13 (s, 1 H), 7,90 (ddd, J = 7,80, 7,80, 1,80 Hz, 1 H), 7,58 (d, J = 7,92 Hz, 1 H), 7,40 (dd, J = 7,18, 4,54 Hz, 1 H), 7,06 (s, 1 H), 5,36 (s, 2 H), 1,19 (s, 9 H). LCMS (Método 1): m/z 418 [M+H]+.
Paso 6: N-(1-(6-cloro-2-oxo-7-(piridm-2-Mmetoxi)-1,2-dihidroqumoMn-3-M)etM)-2-metMpropano-2-sulfmamida.

(E)-W-((6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (1,44 g, 3,45 mmol) se colocó en un matraz de fondo redondo de 250 mL bajo una atmósfera de nitrógeno. Se añadió DCM (27 ml) y la suspensión se enfrió en un baño de hielo seco/cloroformo (a aproximadamente -60°C). Se añadió gota a gota bromuro de metilmagnesio (MeMgBr) (3M en éter, 3,50 ml, 10,50 mmol). El baño frío se dejó calentar a temperatura ambiente durante la noche dando como resultado una suspensión naranja. Una vez que LCMS indicó que la reacción se había completado, la suspensión se enfrió en un baño de hielo y se trató gota a gota con agua (10 ml) dando como resultado la emulsificación. La emulsión se diluyó con EtOAc (400 ml) y se lavó con agua (400 ml). Se añadió gel de sílice a la capa orgánica y el disolvente se evaporó a presión reducida. El material se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (se eluyó con MeOH del 0 al 6% en DCM con elución ¡socrática cuando se eluyeron los picos) para proporcionar 1,17 g del compuesto del título como una espuma amarilla quebradiza. LCMS y 1H RMN son consistentes con N-(1-(6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (1,17 g, 2,70 mmol, 78% de rendimiento). La RMN indicó una mezcla de diastereómeros. LCMS (Método 1): m/z 434 [M+H]+.
Paso 7: 3-(1-ammoetN)-6-cloro-7-(piridm-2-Nmetoxi)qumoMn-2(1H)-ona clorhidrato (II-9).
Una solución de N-(1-(6-cloro-2-oxo-7-(piridin-2-ilmetoxi)-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida. (167,3 mg, 0,386 mmol) en MeOH (3,5 ml) se enfrió en un baño de hielo y se trató gota a gota con 4 M HCl en 1,4-dioxano (2 ml). La reacción se agitó durante 20 minutos y en cinco minutos comenzó a formarse un precipitado. Los disolventes se evaporaron a presión reducida a temperatura ambiente. El residuo se trituró con 10 ml de éter etílico, se recogió en un embudo Hirsch y se lavó con más éter etílico para proporcionar 145,8 mg del compuesto del título en forma de un sólido amarillo pálido. LCMS y 1H RMN están de acuerdo con el clorhidrato de 3-(1-aminoetil)-6-cloro-7-(piridin-2-ilmetoxi)quinolin-2(1H)-ona (145,8 mg, 0,398 mmol, 103% de rendimiento). 1H RMN (300 MHz, metanol-d^: 8 ppm 8,91-8,95 (m, 1 H), 8,68 (ddd, J = 7,90, 7,90, 1,50 Hz, 1 H), 8,29 (d, J = 7,62 Hz, 1 H), 8,04-8,11 (m, 1 H), 8,00 (s, 1 H), 7,90 (s, 1 H), 7,17 (s, 1 H), 5,66 (s, 2 H), 4,53 (q, J) = 6,84 Hz, 1 H), 1,69 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): m/z 352 [M+Na]+.
Ejemplo 12 - Intermedio 11-10: (S)-3-(1-ammoetil)-6-cloro-7-(pmdm-2-Nmetoxi)qumolm-2(1W)-ona.

Paso 1:1-(2,6-Dicloro-7-(pirídm-2-ilmetoxi)qumolm-3-N)etanona.

A una solución de 2,6-dicloro-7-(piridin-2-ilmetoxi)quinolina-3-carbaldehído (1,0 g, 3,0 mmol) (preparada en el mismo procedimiento descrito para el paso 1-3 mostrado en el Esquema-4) en CH2Ch (40 ml) se añadió gota a gota bromuro de metilo y magnesio (MeMgBr) (solución 3M en éter dietílico, 1,5 ml, 4,50 mmol) a 0°C. La mezcla resultante entonces se agitó a temperatura ambiente durante 1,5 horas. Una vez completada la reacción, la mezcla se inactivó lentamente con agua (3 ml) y se extrajo con CH2Ch (50 ml). La capa orgánica se separó y se secó sobre Na2SO4 anhidro. Los disolventes se evaporaron a sequedad. El residuo resultante se disolvió en CH2Ch (25 ml) y se trató con Periodinato de Dess-Martin (2,54 g, 6,00 mmol). La mezcla se agitó a temperatura ambiente durante la noche. A continuación, la mezcla se inactivó con una co-solución acuosa de NaHCO3 al 20% y Na2S2O3 al 20% (10 ml) y se agitó durante 5 minutos a temperatura ambiente. La solución se extrajo con CH2Ch (40 ml), se secó sobre Na2SO4 anhidro, se filtró y se evaporó. El residuo resultante se purificó por cromatografía en columna en un sistema de cromatografía ISCO® (columna de SO2: se eluyó con C^Ch/MeOH 0 a 10%) para proporcionar el compuesto del título (800 mg, 79%).
Paso 2: (R,EJ-W-(1-(2,6-d¡cloro-7-(pmdm-2-¡lmetox¡)qumolm-3-¡l)et¡Mden)-2-met¡lpropano-2-sulfmamida.

A una mezcla de 1-(2,6-didoro-7-(piridin-2-ilmetoxi)quinolin-3-il)etanona (2,18 g, 6,56 mmol) y (R)-2-metilpropano-2-sulfinamida (1,19 g, 9,84 mmol) en THF: Tolueno (40 mL: 180 mL), se agregó isopropóxido de titanio (IV) (Ti(O'Pr)4) (3,96 mL, 13,30 mmol). La mezcla resultante se sometió a reflujo con un aparato Dean-Stark durante 7 horas. La mezcla se enfrió luego a temperatura ambiente, se inactivó con agua y se diluyó con EtOAc (300 ml). La capa orgánica se lavó con agua (100 ml), se secó sobre Na2SO4 anhidro, se filtró y se evaporó a sequedad. El residuo resultante se purificó por cromatografía en columna en un sistema de cromatografía ISCO® (columna de SiO2: se eluyó con Hex/EtOAc del 0 al 100%) para proporcionar el compuesto del título en forma de un sólido amarillo (1,4 g, 50% de rendimiento). El material de partida cetona también se recuperó (250 mg, 11% de rendimiento).
Paso 3: (RJ-W-((S)-1-(2,6-d¡cloro-7-(p¡r¡dm-2-¡lmetox¡)qumolm-3-¡l)etM)-2-met¡lpropano-2-sulfmam¡da.

Para una solución de (R,E)-W-(1-(2,6-dicloro-7-(piridin-2-ilmetoxi)quinolin-3-il)etiliden)-2-metilpropano-2-sulfinamida (900 mg, 1,99 mmol) en THF (25 ml) a -40 a -50°C se añadió L-selectride (1M en THF, 1,98 ml, 2,59 mmol) gota a gota. La mezcla resultante se agitó a una temperatura de -40 a -50°C durante 2 horas. Una vez completada la reacción, la mezcla se inactivó con hielo a -50°C, se extrajo con EtOAc (100 ml), se secó y se evaporó. El residuo resultante se purificó por cromatografía en columna en un sistema de cromatografía ISCo® (columna SO2: Hex/EtOAc 0 a 100%) seguido de trituración con hexanos-cloruro de metileno para proporcionar el compuesto del título (266 mg, 30% de rendimiento).
Paso 4: (S)-3-(1-ammoet¡l)-6-cloro-7-(p¡ndm-2-¡lmetox¡)qumolm-2(1H)-ona una sal de TFA (II-10).

A una mezcla de (R)-A/-((S)-1-(2,6-dicloro-7-(piridin-2-ilmetoxi)quinolin-3-il)etil)-2-metilpropano-2-sulfinamida (1,1 g, 2,43 mmol) en 1,4-dioxano (6,6 ml), se añadió IN HCl acuoso (6,6 ml) a temperatura ambiente. La mezcla resultante se calentó a 120°C durante la noche. Después de que la TLC y la MS mostraron que la reacción se había completado, los disolventes se eliminaron en un evaporador rotatorio y se liofilizaron para proporcionar un sólido amarillo. El sólido bruto se purificó por cromatografía de fase inversa en un sistema de cromatografía ISCO® (columna C18: se eluyó con H2O/MeCN/(0,1% CF3CO2H 0 a 100%) y las fracciones se controlaron mediante LCMS. Las fracciones puras se combinaron y se liofilizaron para proporcionar el compuesto del título II-10 (920 mg, 86% de rendimiento) como la sal de TFA. 1H RMN (300 MHz, DMSO-cfe): 812,17 (br s, 1 H), 8,62 (d, J = 4,95 Hz, 1 H), 8,09 (br s, 2 H), 7,96-7.85 (m, 3 H), 7,59 (d, J = 7,9 Hz, 1 H), 7,42-7.37 (m, 1 H), 7,08 (d, J = 2,5 Hz, 1 H), 5,33 (s, 2 H), 4,39-4,38 (m, 1 H), 1,51 (d, J = 6,8 Hz, 3 H). LCMS (método 3): Ta 3,3 min, m/z 329,1 [M+H]+.
Ejemplo 13 - Intermed¡o 11-11:(s)-3-(1-ammoet¡l)-6-doro-1,8-naft¡r¡dm-2(1W)-ona.

Paso 1: 3-acet¡l-6-cloro-1,8-naft¡ridm-2(1W)-ona.

Una mezcla de 2-amino-5-cloronicotinaldehído (1 g, 6,39 mmol) y 2,2,6-trimetil-4H-1,3-dioxin-4-ona (1,362 g, 9,58 mmol) en xilenos (10 ml) se calentó a reflujo durante 3 horas, luego se enfrió a temperatura ambiente, se filtró y se lavó con xilenos dos veces para proporcionar 914 mg de 3-acetil-6-cloro-1,8-naftiridin-2(1H)-ona (64,3% de rendimiento). 1H RMN (300 MHz, DMSO-da): 812,68 (br, 1 H), 8,63 (s, 1 H), 8,49 (s, 1 H), 8,39 (s, 1 H), 2,48 (s, 3 H)). LCMS (Método 1): Ta 1,60 min, m/z 223,03 [M+H]+.
Paso 2: (S)-W-((S)-1-(2,6-d¡cloroqumolm-3-¡l)etM)-2-metilpropano-2-sulfmam¡da.

Una mezcla de tetraetoxititanio (512 mg, 2,25 mmol), (R)-2-metilpropano-2-sulfinamida (163 mg, 1,35 mmol) y 3-acetil-6-cloro-1,8-naftiridin-2(1H)-ona (200 mg, 0,898 mmol) en THF (15 ml) se calentó a 80°C durante la noche, luego se enfrió a temperatura ambiente. A esta mezcla se le añadió NaBH4 (170 mg, 4,49 mmol) y la mezcla se calentó lentamente a temperatura ambiente durante la noche. Luego se añadió MeOH para apagar cualquier exceso de NaBH4, seguido de la adición de agua. La mezcla se filtró para eliminar los sólidos, luego se extrajo con EtOAc dos veces, se secó sobre Na2SO4 y se concentró. El residuo se purificó en un sistema de cromatografía Biotage® utilizando una columna de SO2 de 25 g eluida en un gradiente (primero 20% a 100% de EtOAc/hexanos, luego 0-5% de MeOH/DCM) para proporcionar (S)-W-((S)-1-(2,6-dicloroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (123 mg, 42% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 88,40 (s, 1 H), 7,74 (s, 1 H), 7,75 (s, 1 H), 7,24 (s, 1 H), 5,24 (d, J = 9,45 Hz, 1 H), 4,42 (m, 3 H), 1,54 (d, J = 6,93Hz, 3H), 1,20 (s, 9H). LCMS (Método 1): Ta 2,07 min, m/z 328,98 [M+H]+.
Paso 3: (S)-3-(1-ammoetil)-6-cloro-1,8-naftiridm-2(1W)-ona (II-11).
A una solución de ((S)-N-((S)-1-(6-doro-2-oxo-1,2-dihidro-1,8-naftiridin-3-il)etil)-2. Se añadió HCl (2 ml, 8,00 mmol, 4M en 1,4-dioxano) a la mezcla de metilpropano-2-sulfinamida (123 mg, 0,375 mmol) en MeOH (5 ml), luego la mezcla se agitó a temperatura ambiente durante la noche. A esta mezcla se agregaron 6 ml de éter etílico y el precipitado resultante se filtró, se lavó con éter etílico (2 x), se secó y se concentró para proporcionar (S)-3-(1-aminoetil)-6-cloro-1,8-naftiridin-2(1H)-ona, HCl (96 mg, 98% de rendimiento). 1H RMN (300 MHz, DMSO-da): 812,75 (br s, 1 H), 8,60-8,35 (s, 1 H), 8,26 (br, 1 H) 8,07 (s, 1 H), 4,40-4,50 (m, 1 H), 1,51 (d, J = 6,78 Hz, 3 H). LCMS (Método 1): Ta 0,87 min, m/z 224,99 [M+H]+.
Ejemplo 14 - Intermedio II-12: (R)-3-(1-ammoetN)-6-doroqumoxaMn-2(1H)-ona
[0219]

Paso 1: Etil 3-((4-cloro-2-nitrofenil)amino)-3-oxopropanoato.

A una solución de 4-cloro-2-nitroanilina (42.3 g, 245 mmol) en CH2Ch (1 L) se le añadió etil-3-cloro-3-oxopropanoato (48 g, 319 mmol) gota a gota y la mezcla de reacción se agitó a temperatura ambiente durante la noche. El disolvente se eliminó al vacío y el residuo resultante se disolvió en una cantidad mínima de MTBE (200 ml) y hexanos (800 ml) que se añadieron lentamente. Cualquier producto que precipitó de la solución se filtró y el filtrado se concentró y se purificó por cromatografía en columna. El sistema de cromatografía con gradiente de hexanos/acetato de etilo se diluyó para proporcionar el producto deseado adicional. El compuesto del título se obtuvo con un rendimiento del 98% (69,85 g).
Paso 2: 7-cloro-2-(etoxicarbonil)-3-oxo-3,4-dihidroquinoxalina 1-óxido (A) y 7-cloro-2-(metoxicarboninil)-3-
oxo-3,4-dihidroquinoxalina 1-óxido (B).

A una solución de 3-((4-cloro-2-nitrofenil)amino)-3-oxopropanoato de etilo (68 g, 238 mmol) y benzoato de metilo (150 ml) en DMF anhidro (500 ml) a 0°C se añadió gota a gota KOtBu (solución 1 pM en Th F, 500 ml, 500 mmol). La mezcla de reacción se agitó a 0°C durante 4 horas y luego se detuvo con una solución acuosa saturada de NH4CL La mezcla se extrajo con CH2Ch (300 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron mediante cromatografía ultrarrápida de SO2 y se eluyeron con CH2Ch/MeOH para proporcionar una mezcla de A/B (42,54 g, 67% de rendimiento, relación A/B 1:2) como sólido. Esto se usó en el siguiente paso sin purificación adicional.
Paso 3:
7-cloro-3-oxo-3,4-dihidroquinoxalino-2-carboxilato de etilo (D) y 7-cloro-3-oxo-3,4-dihidro-quinoxino-2-carboxilato de metilo (C).

A una mezcla de compuestos A y B (42,54 g, 159 mmol) en DMF (200 ml) se añadió PBr3 (85,9 g, 318 mmol) gota a gota a temperatura ambiente. La mezcla de reacción se agitó a temperatura ambiente durante 3 horas y luego se detuvo con agua con hielo y se extrajo con CH2Ch (200 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía ultrarrápida usando CH2Ch/MeOH (9: 1) como eluyente para proporcionar C/D (36,6 g, 91% de rendimiento) como un sólido. Esto se usó en el siguiente paso sin purificación adicional.
Paso 4:
Etil 3.7-dicloroquinoxalina-2-carboxilato (E) y metil 3,7-dicloro quinoxalina-2-carboxilato (F).

A una mezcla de compuestos C/D (36,6 g, 145 mmol) en un matraz de 1 L, se añadió POCh (150 ml) en una porción y la mezcla resultante se calentó a reflujo durante 3 horas. La mezcla se enfrió luego a temperatura ambiente y se detuvo cuidadosamente con una solución acuosa de NaHCO3. La mezcla se extrajo con CH2Ch (200 ml x 3). La capa orgánica combinada se secó (Na2SO4), se concentró y se purificó por cromatografía ultrarrápida con SO2 usando hexano/acetato de etilo (9:1) como eluyente para proporcionar E/F (23,7 g, 61% de rendimiento) como un sólido. Esta mezcla se usó en la siguiente etapa sin purificación adicional.
Paso 5:
Metil 7-cloro-3-metoxiquinoxalina-2-carboxilato.
A una mezcla de compuestos E/F (22,11 g, 81,9 mmol) en THF/MeOH (9:1, 300 ml) se añadió NaOMe (0,5 M, 360 ml) gota a gota a 0°C. La mezcla resultante se agitó a temperatura ambiente durante 3 horas y se inactivó con NH4Cl sólido (20 g). El disolvente se eliminó a vacío y se añadió agua (200 ml). La mezcla se extrajo con CH2Ch (150 ml x 3) y las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía ultrarrápida con SiO2 usando hexanos/acetato de etilo (9:1) como eluyente para proporcionar el compuesto del título (19.1). g, 88% de rendimiento) como un sólido.
Paso 6:
7-cloro-3-metoxiquinoxalina-2-carbaldehído (G) y oxibis((7-doro-3-metoxiquinoxalin-2-il)metanol) (H).

A metil 7-cloro-3-metoxiquinoxalina-2-carboxilato (5,3 g, 20 mmol) en CH2Ch (250 ml) se le añadió hidruro de diisobutilaluminio (1 M, 30 ml) gota a gota a -78°C. La mezcla resultante se agitó a -78°C durante 3 horas y luego se detuvo con MeOH (a -78°C, 20 ml). Después de agitarse durante 0,5 horas, la mezcla se calentó a temperatura ambiente y se añadió una solución acuosa de potasio sódico L-tartrato (100 ml). Luego se separó la capa orgánica y la capa acuosa se extrajo con CH2Ch (50 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía ultrarrápida con SO2 usando hexanos/acetato de etilo (1:1) como eluyente para proporcionar G (1,02 g, rendimiento del 23%) y H (2,24 g, rendimiento del 50%). La estructura de H se asignó en base a MS y 1H RMN.
Paso 7:
(R,Ej-W-((7-cloro-3-metoxiqumoxalm-2-N)metNeno)-2-metNpropano-2-sulfmamida.

Al compuesto H (2,24 g, 5,1 mmol) en DCE (300 ml) a temperatura ambiente se añadió (R)-2-metilpropano-2-sulfinamida (2,44 g, 20,1 mmol) y CuSO4 (4,85 g, 30,3 mmol). La reacción se calentó a 60°C y se agitó durante 4 horas. La mezcla de reacción se enfrió luego a temperatura ambiente y se detuvo con 50 ml de una solución acuosa saturada de NaHCO3. Después de agitarse durante 10 minutos, la mezcla de reacción se filtró a través de una capa de Celite®. El filtrado se extrajo con CH2Ch (50 ml x 3), se secó (Na2SO4), se concentró y se purificó por cromatografía en columna en un sistema de cromatografía ISCO® usando hexanos/acetato de etilo como eluyente para proporcionar el compuesto del título (2,21 g, 67% de rendimiento).
Paso 8:
(R)-N-((R)-1-(7-cloro-3-metoxiquinoxalin-2-il)etil)-2-metilpropano-2-sulfinamida.

A (R,E)-A/-((7-cloro-3-metoxiquinoxalin-2-il)metileno)-2-metilpropano-2-sulfinamida (2,21 g, 6,8 mmol) en CH2Cl2 (150 mL) se añadió cloruro de metil magnesio (MeMgCl) (3M en THF, 3,4 ml) gota a gota a -78°C. La mezcla resultante se agitó a -78°C durante 2 horas y luego se detuvo con una solución acuosa de NH4Cl (20 ml). Después de agitarse durante 10 minutos, la capa orgánica se separó y la capa acuosa se extrajo con CH2Cl2 (25 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en columna en un sistema de cromatografía ISCO® usando hexanos/acetato de etilo como eluyente para proporcionar el
compuesto del título (1,18 g, rendimiento del 51%).
Paso 9:
(R)-3-(1-ammoet¡l)-6-cloroqumoxalm-2(1H)-ona (II-12).

En el compuesto (R)-N-((R)-1-(7-cloro-3-metox¡qu¡noxal¡n-2-¡l)et¡l)-2-met¡lpropano-2-sulf¡nam¡da (1,29 g, 3,46 mmol) en CH3CN (100 ml) se añad¡ó yodotr¡met¡ls¡lano (3,46 g, 17,3 mmol) gota a gota a 0°C. La mezcla se calentó a reflujo durante 2 horas, se enfr¡ó a temperatura amb¡ente y se detuvo con MeOH (10 ml). El d¡solvente se el¡m¡nó al vacío, y el res¡duo se pur¡f¡có por cromatografía ¡nversa C-18 en un s¡stema de cromatografía ISCO® usando agua (TFA al 0,1%)/CH3c N (TFA al 0,1%) como eluyente para proporc¡onar el compuesto II-12 (1,22 g), 95% de rend¡m¡ento) como una sal de TFA.
Ejemplo 15 - Intermedio 11-13: (S)-3-(1-ammoet¡l)-6-cloroqumoxalm-2(1H)-ona

Paso 1:
(S,E)-W-((7-cloro-3-metox¡qumoxalm-2-¡l)met¡leno)-2-met¡lpropano-2-sulfmam¡da.

Al compuesto H (2,31 g, 5,2 mmol) en DCE (300 ml) a temperatura amb¡ente se añad¡ó (S)-2-met¡lpropano-2-sulf¡nam¡da (2,52 g, 20,8 mmol) y CuSO4 (5,0 g, 31,2 mmol). La mezcla de reacc¡ón resultante se calentó a 60°C y se ag¡tó durante 4 horas. La mezcla de reacc¡ón se enfr¡ó luego a temperatura amb¡ente y se detuvo con 50 ml de una soluc¡ón acuosa saturada de NaHCO3. Después de ag¡tarse durante 10 m¡nutos, la mezcla se f¡ltró a través de una capa de Cel¡te®. El f¡ltrado se extrajo con CH2Ch (50 ml x 3), se secó (Na2SO4), se concentró y se pur¡f¡có por cromatografía en columna en un s¡stema de cromatografía ISCO® usando hexanos/acetato de et¡lo como eluyente para proporc¡onar el compuesto del título (2,62 g, 78% de rend¡m¡ento).
Paso 2:
(S)-W-((S)-1-(7-cloro-3-metox¡qumoxalm-2-¡l)et¡l)-2-met¡lpropano-2-sulfmam¡da.

Al compuesto (S,E)-W-((7-cloro-3-metox¡qu¡noxal¡n-2-¡l)met¡leno)-2-met¡lpropano-2-sulf¡nam¡da (2,62 g, 8,0 mmol) en CH2Ch (150 mL) se añadió cloruro de metil magnesio (MeMgCl) (3M en THF, 4,0 ml) gota a gota a -78°C. La mezcla resultante se ag¡tó a -78°C durante 2 horas y luego se detuvo con una soluc¡ón acuosa de NH4Cl (20 ml). Después de ag¡tarse durante 10 m¡nutos, la capa orgán¡ca se separó y la capa acuosa se extrajo con CH2Ch (25 ml x 3). Las capas orgán¡cas comb¡nadas se secaron (Na2SO4), se concentraron y se pur¡f¡caron por cromatografía en columna en un s¡stema de cromatografía ISCO® usando hexanos/acetato de et¡lo como eluyente para proporc¡onar el compuesto del título (1,69 g, 62%).
Paso 14:
(S)-3-(1-ammoetN)-6-doroqumoxalm-2(1W)-ona (11-13).

Al compuesto (S)-W-((S)-1-(7-cloro-3-metox¡qu¡noxal¡n-2-¡l)et¡l)-2-met¡lpropano-2-sulf¡nam¡da (350 mg, 1,03 mmol) en CH3CN (40 ml) se añad¡ó yodotr¡met¡ls¡lano (1,03 g, 5,15 mmol) gota a gota a 0°C. La mezcla se somet¡ó luego a reflujo durante 2 horas. Después de enfr¡arse a temperatura amb¡ente, la reacc¡ón se detuvo con MeOH (2 ml). El d¡solvente se el¡m¡nó al vacío y el res¡duo se pur¡f¡có por cromatografía ¡nversa C-18 en un s¡stema de cromatografía ISCO® usando agua (0,1% de TFA)/c HsCN (0,1% de TFA) como eluyente para proporc¡onar el compuesto del título (267 mg, 79). % de rend¡m¡ento) como una sal de TFA.
Ejemplo 16 - Intermedio 11-14: (3-((S)-1-ammoetN)-6-doro-7-((R)-1-(pindm-2-N)etoxi)qumoMn-2(1H)-ona 1

Paso 1: tere-butilo (3-((terc-butNdimetNsNM) oxi)-4-clorofenilo)carbamato.

Una solución de 5-amino-2-clorofenol (10,00 g, 69,7 mmol) en THF (350 ml) se trató con dicarbonato de diferc-butilo (20 ml, 86 mmol) y se agitó a reflujo durante la noche. El disolvente se evaporó a presión reducida para proporcionar un aceite marrón. El aceite se disolvió luego en EtOAc (300 ml), se lavó con agua, NaHCO3 acuoso saturado, y salmuera (300 ml cada una), se secan (Na2SO4), se filtran y se evaporan a presión reducida para proporcionar 21,01 g de carbamato de terc-butil (4-cloro-3-hidroxifenil) impuro como un aceite marrón (LCMS: m/z 244 [M+H]+). Este material se disolvió en DMF (130 ml) y se enfrió en un baño de hielo. Luego se añadió lentamente imidazol (11,74 g, 172 mmol) (durante aproximadamente 10 minutos). Se añadió una solución de TBDMS-Cl (14,98 g, 99 mmol) en DMF (45 ml) (durante aproximadamente 2 minutos). El baño de hielo se retiró y la solución se agitó a temperatura ambiente durante la noche. Una vez que LCMS indicó que la reacción se había completado, la solución se diluyó con EtOAc (1 L) y se lavó con agua (2 x 600 ml), NaHCO3 acuoso semisaturado (600 ml), NH4O acuoso semisaturado (600 ml), NaHCO3 saturado (600 ml) y salmuera (600 ml). La capa orgánica se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar 28,00 g de un sólido marrón. La muestra se disolvió en EtOAc, se añadió gel de sílice (33 g) y el disolvente se evaporó a presión reducida. El material se dividió en dos lotes, cada uno de los cuales se purificó mediante cromatografía en columna en un sistema de cromatografía Biotage® MPLC usando una columna de gel de sílice de 330 g eluido con 0 a 5% de EtOAc en hexanos y con elución isocrática a 4,5% o 5% de EtOAc cuando el producto eluyó. Las fracciones del producto se recogieron y proporcionaron 21,76 g de carbamato de terc-butilo (3-((tercbutildildimetil-silil)oxi)-4-clorofenil) (21,76 g, 60,8 mmol, 88% de rendimiento) como un sólido de color melocotón. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 9,43 (s, 1 H), 7,23-7,28 (m, 1 H), 7,22 (d, J = 2,35 Hz, 1 H), 7,09-7.16 (m, 1 H), 1,46 (s, 9 H), 0,99 (s, 9 H), 0,21 (s, 6 H). LCMS (Método 1): m/z 358 [M+H]+.
Paso 2: tere-butilo (4-cloro-2-formil-5-hidroxifenilo)carbamato (J).

Se cargó un matraz de fondo redondo de 500 ml con 3 bocas y secado con terc-butilo (3-((tercbutildimetilsilil)oxi)-4-clorofenil)carbamato (10 g, 27.9 mmol). Se unió un embudo de adición secado al horno y el sistema se lavó con nitrógeno. Se añadió éter etílico (113 ml) con una jeringa. La solución amarilla resultante se enfrió en un baño de acetonitrilo/hielo seco (hasta aproximadamente -40°C). Luego se añadió t-BuLi (1,7 pM en pentano, 40 ml, 68,0 mmol) al embudo de adición con una cánula. La solución de t-BuLi se añadió gota a gota a la solución de éter (durante aproximadamente 10 minutos), tiempo durante el cual la solución de éter se volvió turbia gradualmente con un precipitado. La mezcla se agitó a aproximadamente -40°C durante 2,5 horas, luego se añadió gota a gota DMF (11 ml) con una jeringa (durante ~ 10 minutos), durante el tiempo en que los sólidos volvieron a la solución. El baño de acetonitrilo/hielo seco se reemplazó con un baño de hielo y la solución amarilla se agitó a 0°C durante 1,75 horas. La reacción se detuvo luego mediante la adición gota a gota de agua (25 ml), dando como resultado la formación de un precipitado de color naranja. Se retiró el baño de hielo y la muestra se diluyó con agua (125 ml), dando como resultado la disolución del precipitado. La mezcla se agitó, y las capas se separaron. La capa acuosa se acidificó a pH ~ 4-5 con AcOH. El precipitado resultante se extrajo con EtOAc (200 ml), se lavó con agua (2 x 100 ml), se secó (Na2SO4), se filtró y se evaporó a presión reducida para proporcionar ferc-butilo (4-cloro-2-formil-5-hidroxifenil)carbamato como una solución amarilla (4,79 g, 17,63 mmol, 63% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 11,72 (s, 1 H), 10,50 (s, 1 H), 9,68 (br s, 1 H), 7,99 (s, 1 H), 7,88 - 7,91 (m, 1 H), 1,48 (s, 9 H). LCMS (Método 1): m/z 216 (M-56, pérdida de f-Bu).
Paso 3: (R)-tert-butil (4-cloro-2-formil-5-(1-(piridin-2-il)etoxi)fenil)carbamato.

Una mezcla de (S)-1-(piridin-2-il)etanol (454,3 mg, 3,69 mmol), terc-butil (4-cloro-2-formil-5-hidroxifenil)carbamato (1 g), 3,68 mmol) y trifenilfosfina (1,158 g, 4,42 mmol) se colocaron en un matraz de fondo redondo de 100 ml bajo una atmósfera de nitrógeno. Se añadió THF (40 ml) con una jeringa. La solución amarilla resultante se enfrió en un baño de hielo y luego se añadió gota a gota DIAD (0,86 ml, 4,42 mmol). El baño de hielo se retiró y la solución se agitó a temperatura ambiente durante la noche. Una vez que LCMS indicó que la reacción se había completado, se añadió gel de sílice y el disolvente se evaporó a presión reducida. La muestra se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (usando una columna de gel de sílice de 50 g eluida con 0 a 13% de EtOAc en hexanos) para proporcionar 473,7 mg de un sólido blanco. LCMS y RMN son consistentes con (R)-terc-butil (4-cloro-2-formil-5-(1-(piridin-2-il)etoxi)fenil)carbamato contaminado con material de partida fenólico (~5:1 producto a material de partida por RMN). El material se usó para la siguiente etapa sin purificación adicional. 1H RMN (300 MHz, DMSO-da): 8 ppm 10,42 (s, 1 H), 9,73 (s, 1 H), 8,54-8,60 (m, 1 H), 7,98 (s, 1 H), 7,92 (s, 1 H), 7,82 (ddd, J = 7,80, 7,80, 1,80 Hz, 1 H), 7,44 (br d, J = 7,90 Hz, 1 H), 7,30-7,36 (m, 1 H), 5,64 (q, J = 6,35 Hz, 1 H), 1,67 (d, J = 6,45 Hz, 3 H), 1,46 (s, 9 H). LCMS (Método 1): m/z 377 [M+H]+.
Paso 4: (S)-etil 3-((terc-butoxicarbonil)amino)butanoato (K).

Una suspensión de (S)-3-ácido aminobutanoico (6,25 g, 60,6 mmol) en EtOH (27,5 ml) se enfrió en un baño de hielo. Luego se añadió gota a gota cloruro de tionilo (7,5 ml, 103 mmol) durante 40 minutos, tiempo durante el cual el aminoácido se disolvió. El baño de hielo se dejó fundir y la solución se agitó a temperatura ambiente durante la noche. La mezcla se evaporó a presión reducida y el residuo se mezcló con más EtOH (60 ml) y se evaporó nuevamente a presión reducida para proporcionar un aceite. El aceite se disolvió en DCM (55 ml) y se enfrió en un baño de hielo. Se añadió gota a gota TEA (25 ml, 179 mmol) durante 15 minutos con agitación, dando como resultado una mezcla lechosa. Luego se añadió dicarbonato de di-terc-butilo (17 ml, 73,2 mmol). El baño de hielo se dejó fundir y la mezcla se agitó a temperatura ambiente durante cinco días. La mezcla resultante se filtró a través de Celite® 545 en un embudo Buchner y la torta del filtro se lavó con DCM (50 ml). El filtrado se lavó con ácido cítrico acuoso saturado (20 ml) y agua (2 x 100 ml), se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar el compuesto del título en forma de un aceite transparente. 1H RMN es consistente con (S)-etil 3-((terc- butoxicarbonil)amino)butanoato (13,47 g, 58,2 mmol, rendimiento del 96%). 1H RMN (300 MHz, CDCh): 8 ppm 4,95 (br s, 1 H), 4,15 (q, J = 7,13, 2 H), 3,98-4.10 (m, 1 H), 2,40-2,57 (m, 2) H), 1,44 (s, 9 H), 1,27 (t, J = 7,18, 3 H), 1,22 (d,J = 6,74, Hz, 3 H).
Paso 5 y 6: 3-((S)-1-ammoetM)-6-cloro-7-((R)-1-(pmdm-2-N)etoxi)qumoMn-2(1H)-ona clorhidrato (11-14).
Se colocaron un matraz de fondo redondo de 25 ml secado en horno y una barra de agitación bajo una atmósfera de nitrógeno. Luego se añadieron con una jeringa THF (2,25 ml) y diisopropilamina (0,27 ml, 1,894 mmol). La solución se enfrió utilizando un baño de hielo seco/acetona (-78°C) y se añadió gota a gota n-BuLi (1,6 pM en hexano, 1,15 ml, 1,84 mmol) durante 5 minutos. Después de agitarse durante 10 minutos, se añadió gota a gota (durante 5 minutos) una solución de (S)-etil 3-((terc-butoxicarbonil)amino)butanoato K (115,3 mg, 0,499 mmol) en THF (0,5 ml). La solución se agitó durante 75 minutos a -78°C y luego una solución de (R)-ferc-butil (4-cloro-2-formil-5-(1-(piridin-2-il)etoxi)fenil)carbamato (188,7 mg, 0,501 mmol) en THF (1,0 mL) se añadió gota a gota con una jeringa. La solución de reacción se volvió amarilla cuando se añadió el aldehido. La reacción se agitó a -78°C durante 13 minutos y luego se detuvo mediante la adición de una solución acuosa saturada de NH4Cl (2,5 ml). La mezcla se repartió entre EtOAc y agua (10 ml cada uno). La capa orgánica se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar una mezcla impura de isómeros de (3S)-etil 3-((terc-butoxicarbonil)amino)-2-((2-((terc-butoxicarbonil)amino)-5-cloro-4-((P)-1-(piridin-2-il)etoxi)fenil)(hidroxi)metil)butanoato como un aceite amarillo (344,8 mg; LCMS: m/z +608 [M+H]+). El material bruto (334 mg) se disolvió en 1,4-dioxano (5 ml), se trató con HCl acuoso 12 pM (0,125 ml) y se agitó a 110°C durante 90 minutos, tiempo durante el cual se precipitó un material rojo. La mezcla se dejó enfriar y el sobrenadante se decantó y se desechó. Se añadió heptano (~ 4 ml) al precipitado rojo que quedaba en el fondo redondo y luego se evaporó a presión reducida para proporcionar 161,8 mg de un sólido rojo. El material se trituró con PrOH (5 ml) y el precipitado resultante se recogió en un embudo Hirsch y se lavó con PrOH (1 ml) y éter etílico (~ 20 ml) para proporcionar 3-((S)-1-aminoetilo)-6-cloro-7-((P)-1-(piridin-2-il)etoxi)quinolin-2(1H)-ona clorhidrato (104,2 mg, 0,274 mmol, 55% de rendimiento) como un sólido rojo, impuro pero adecuado para su uso tal como es. 1H RMN (300 MHz, metanol^): 8 ppm 8,81-8,87 (m, 1 H), 8,55-8,64 (m, 1 H), 8,18 (d, J = 7,92 Hz, 1 H), 7,96-8,04 (m, 1 H), 7,95 (s, 1 H), 7,85 (s, 1 H), 6,99 (s, 1 H), 5,98 (q, J = 6,84 Hz, 1 H), 4,48 (q, J = 6,84 Hz, 1 H), 1,86 (d,J = 6,45 Hz, 3 H), 1,64 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): m/z 344 [M+H]+.
Ejemplo 17 - Intermedio 11-15: (S)-3-(1-ammoetN)-6-cloro-7-(ciclopropNmetoxi)qumoMn-2(1H)ona

Paso 1: tere-butilo (4-doro-5-(ddopropMmetoxi)-2-formMfenMo)carbamato.

Una mezcla de ciclopropilmetanol (0,145 ml, 1,838 mmol), terc-butil (4-cloro-2-formil-5-hidroxifenil)carbamato J (499,4 mg, 1,838 mmol) y trifenilfosfina (579,4 mg, 2,209 mmol) se colocó en un matraz de fondo redondo de 100 ml bajo una atmósfera de nitrógeno y luego se añadió THF (20 ml) con una jeringa. La solución naranja resultante se enfrió en un baño de hielo y se añadió gota a gota DIAD (0,43 ml, 2,184 mmol). El baño de hielo se retiró y la solución se agitó a temperatura ambiente durante 48 horas. Una vez que LCMS indicó que la reacción se había completado, se añadió gel de sílice y el disolvente se evaporó a presión reducida. La muestra se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC usando una columna de gel de sílice de 25 g eluida con 0 a 3% de EtOAc en hexanos para proporcionar terc-butilo (4-cloro-5-(ciclopropilmetoxi)-2-formilfenilo)carbamato (410,6 mg, 1,260 mmol, 68.6% de rendimiento) como un sólido amarillento. 1H RMN (300 MHz, DMSO-d6): 8 ppm 10,57 (s, 1 H), 9,75 (s, 1 H), 7,95-8,00 (m, 2 H), 4,02 (d, J = 7,04 Hz, 2 H), 1,49 (s, 9 H), 1,23-1,31 (m, 1 H), 0,57-0,66 (m, 2 H), 0,38-0,46 (m, 2 H). LCMS (Método 1): m/z 270 (pérdida de f-Bu).
Paso 2 y 3: (S)-3-(1-ammoetM)-6-cloro-7-(ciclopropNmetoxi)qumoMn-2(1W)-ona clorhidrato (11-15).

Se colocaron un matraz de fondo redondo de 25 ml secado al horno y una barra de agitación bajo una atmósfera de nitrógeno y se añadieron con jeringa THF (5,6 ml) y diisopropilamina (0,53 ml, 3,72 mmol). La solución se enfrió en un baño de hielo seco/acetona (a -78°C) y se añadió gota a gota n-BuLi (1,6 pM en hexano, 2,35 ml, 3,76 mmol) durante un período de 5 minutos. Después de agitarse durante 15 minutos, se añadió gota a gota (durante 5 minutos) una solución de (S)-etil 3-((terc-butoxicarbonil)amino)butanoato K (286 mg, 1,238 mmol) en THF (1,25 ml). La solución se agitó durante 80 minutos a -78°C y se añadió una solución de ferc-butil (4-cloro-5-(ciclopropilmetoxi)-2-formilfenil)carbamato (403,2 mg, 1,238 mmol) en THF (2,5 ml) gota a gota con jeringa. La solución de reacción se volvió amarilla cuando se añadió el aldehido. La reacción se agitó a -78°C durante 12 minutos y luego se detuvo mediante la adición de una solución acuosa saturada de NH4Cl (6 ml). La mezcla se repartió entre EtOAc y agua (25 ml cada una) y la capa orgánica se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar 724,5 g de un aceite amarillento. El material se disolvió en 1,4-dioxano (12,5 ml), se trató con HCl 12 pM (acuoso; 0,32 ml) y se agitó a 110°C durante 70 minutos, tiempo durante el cual la solución se espesó con un precipitado rosa. La muestra se dejó enfriar y el disolvente se evaporó a presión reducida para proporcionar 1,13 g de un sólido rojo fibroso. El material se trituró con /-PrOH (15 ml) y el precipitado resultante se recogió en un embudo Buchner y se lavó con /-PrOH (20 ml) y éter etílico (~ 60 ml) para proporcionar (S)-3-(1-aminoetil)-6-cloro-7-(ciclopropilmetoxi)quinolin-2(1H)-onocloruro (146,1 mg, 0,444 mmol, 36% de rendimiento) como un sólido blanco como el papel. 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,13 (br s, 1 H), 8,21 (br s, 3 H), 7,98 (s, 1 H), 7,86 (s, 1 H), 6,98 (s, 1 H), 4,32-4,46 (m, 1 H), 3,96 (d, J = 6,40 Hz, 2 H), 1,51 (d, J= 6,70 Hz, 3 H), 1,21-1,35 (m, 1 H), 0,55-0,68 (m, 2 H), 0,35-0,46 (m, 2 H). LCMS (Método 1): m/z 293 [M+H]+.
Ejemplo 18 - Intermedio 11-16: 3-(1-ammoetil)-6-cloro-7-((3,3-difluorocidobutil)metoxi)qumolm-2(1H)-ona

Paso 1: W-(4-cloro-3-((3,3-dmuorociclobutM)metoxi)feml)acetamida.

Se colocó una solución de 5-amino-2-clorofenol (3 g, 20,90 mmol) (3,3-difluorociclobutil)metanol (2,66 g, 21,78 mmol) en THF (375 ml) bajo una atmósfera de nitrógeno y se trató con DEAD (3,90 ml, 24,63 mmol). La
solución se agitó a temperatura ambiente durante 48 horas. Una vez que el LCMS indicó una progresión adecuada de la reacción, el gel de sílice se añadió a la solución y se evaporó a presión reducida. El material se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MpLC (utilizando 340 g de columna de gel de sílice eluida con 0 a 100% de EtOAc en hexanos con elución isocrática cuando se eluyeron los picos) para proporcionar 3,89 g del compuesto del título como un líquido marrón. La LCMS fue consistente con 4-cloro-3-((3,3-difluorociclobutil)metoxi)anilina impura (m/z 248 [M+H]+). La muestra se disolvió en EtOAc (80 ml) y se trató con DIEA (3,00 ml, 17,18 mmol) y AC2O (1,60 ml, 16,96 mmol). La solución se agitó a temperatura ambiente durante la noche. La solución se lavó con agua y salmuera (50 ml cada una), se secó (Na2SO4), se filtró y se evaporó a presión reducida. El residuo se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (usando una columna de gel de sílice de 50 g, se eluyó con 0 a 50% de EtOAc en hexanos con elución isocrática cuando se eluyeron los picos) para proporcionar 3,16 g del compuesto del título como una luz. Aceite pardo, que lentamente se cristaliza en reposo. LCMS y 1H RMN son consistentes con N-(4-cloro-3-((3,3-difluorociclobutil)metoxi)fenil)acetamida (3,16 g, 10,91 mmol, 52% de rendimiento). En la RMN un protón está oscurecido por la señal del solvente. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 11,91 (s, 1 H), 8,54-8,67 (m, 1 H), 7,80 7,95 (m, 2 H), 7,68 (s, 1 H), 7,56 (d, J = 7,30 Hz, 1 H), 7,34-7,44 (m, 1 H), 7,29 (d, J = 9,10 Hz, 1 H), 7,13-7,22 (m, 1 H), 7,03 (s, 1 H), 6,31 (br s, 1 H), 6,22 (d, J = 7,90 Hz, 1 H), 5,30 (s, 2 H), 4,10-4,26 (m, 2 H), 3,78 (s, 3 H). LCMS (Método 1): m/z 290 [M+H]+.
Paso 2: 2,6-didoro-7-((3,3-difluoroddobutM)metoxi)qumolma-3-carbaldehído.

Se tapó un tubo con un septo y se colocó bajo una atmósfera de nitrógeno. Luego se añadió DMF (2,15 ml, 27,8 mmol) con una jeringa y la mezcla de reacción resultante se enfrió en un baño de hielo. Se añadió gota a gota POCla (8,40 ml, 90 mmol) con una jeringa (10 minutos), tiempo durante el cual se precipitó un material blanco. A continuación, la solución se dejó calentar a temperatura ambiente durante 10 minutos y la mezcla se trató con N-(4-cloro-3-((3,3-difluorociclobutil)metoxi)fenil)acetamida (2,44 g, 8,42 mmol). La mezcla se agitó a 80°C durante dos días. La solución roja espesa resultante se pipeteó en hielo, dando como resultado un precipitado amarillo. El precipitado se recogió en un embudo Buchner, se lavó con agua (~ 500 ml) y se secó para proporcionar 2,38 g del compuesto del título en forma de un sólido amarillo pálido. LCMS y 1H RMN son consistentes con 2,6-dicloro-7-((3,3-difluorociclobutil)metoxi)quinolina-3-carbaldehído (2,38 g, 6,88 mmol, 82% de rendimiento). 1H RMN (300 Mhz , DMSO-d6): 8 ppm 10,31-10,36 (m, 1 H), 8,88 (s, 1 H), 8,48 (s, 1 H), 7,65 (s, 1 H), 4,37 (d, J = 4,69 Hz, 2 H), 2,53 2,84 (m, 5 H). LCMS (Método 1): m/z 346 [M+H]+.
Paso 3: 6-doro-7-((3,3-difluoroddobutM)metoxi)-2-oxo-1,2-dihidroqumoMna-3-carbaldehído.

Se agitó una solución de 2,6-dicloro-7-((3,3-difluorociclobutil)metoxi)quinolina-3-carbaldehído (2,66 g, 7,68 mmol) en HCl concentrado (75 ml) a 100°C para un día durante el cual se formó una corteza roja en la superficie del matraz. La mezcla se diluyó con agua (800 ml), dando como resultado la formación de un precipitado rojo. La mezcla se dejó reposar a temperatura ambiente durante 4 días. El precipitado se recogió luego en un embudo Buchner, se lavó con agua (1 L) y se secó al vacío a 50°C para proporcionar 2,16 g del compuesto del título en forma de un sólido rojo. LCMS y 1H RMN son consistentes con 6-cloro-7-((3,3-difluorociclobutil)metoxi)-2-oxo-1,2-dihidroquinolina-3-carbaldehído (2,16 g, 6,59 mmol, 86% de rendimiento). 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,21 (s, 1 H), 10,16-10,18 (m, 1 H), 8,43 (s, 1 H), 8,09 (s, 1 H), 6,94 (s, 1 H), 4,20 (d, J = 4,10 Hz, 2 H), 2,54-2,80 (m, 5 H). LCMS (Método 1): m/z +328 [M+H]+.
Paso 4: (E)-W-((6-doro-7-((3,3-difluoroddobutN)metoxi)-2-oxo-1,2-dihidroqumolm-3-N)metNeno)-2-metilpropano-2-sulfinamida.

Una mezcla de 6-doro-7-((3,3-difluorocidobutil)metoxi)-2-oxo-1,2-dihidroquinolina-3-carbaldehído (499,6 mg, 1,525 mmol) y 2-metilpropano-2-sulfinamida (222,1 mg, 1,832 mmol) se colocó en un matraz de fondo redondo de 25 ml bajo una atmósfera de nitrógeno. THF (3,0 ml) y titanio (IV) isopropóxido (Ti(O'Pr)4) (0,90 ml, 3,07 mmol) se añadieron con una jeringa, y la suspensión se agitó a temperatura ambiente durante la noche. Una vez que LCMS indicó que la reacción estaba casi completa, la reacción se detuvo mediante la adición gota a gota de una solución acuosa saturada de NH4Cl (2 ml). El material se trituró luego con EtOAc (100 ml) y el precipitado resultante se filtró a través de Celite®. La torta del filtro se lavó con EtOAc (50 ml), se trató con ultrasonidos en EtOAc durante 15 minutos y se filtró usando un embudo Buchner. Los filtrados se combinaron y se lavaron con salmuera (100 ml), se secaron (Na2SO4), se filtraron y se evaporaron a presión reducida para proporcionar 413 mg del compuesto del título en forma de un sólido amarillo. LCMS y 1H RMN son consistentes con (E)-W-((6-cloro-7-((3,3-difluorociclobutil)metoxi)-2-oxo-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (413 mg, 0,958 mmol, 62,9% de rendimiento). 1H RMN (300 MHz, DMSO-da): 8 ppm 12,21 (s, 1 H), 8,74 (s, 1 H), 8,59 (s, 1 H), 8,09 (s, 1 H), 6,95 (s, 1 H), 4,19 (d, J = 4,40 Hz, 2 H), 2,55-2,79 (m, 5 H), 1,19 (s, 9 H). LCMS (Método 1): m/z 431 [M+H]+.
Paso 5: N-(1-(6-doro-7-((3,3-difluoroddobutN)metoxi)-2-oxo-1,2-dihidroqumoMn-3-N)etM)-2-metNpropano-2-sulfinamida.

(E)-W-((6-cloro-7-((3,3-difluorociclobutil)metoxi)-2-oxo-1,2-dihidroquinolin-3-il)metileno)-2-metilpropano-2-sulfinamida (411,3 mg, 0,955 mmol) se colocó en un matraz de fondo redondo de 100 ml bajo una atmósfera de nitrógeno. Se añadió DCM (7,6 ml) y la suspensión se enfrió en un baño de hielo seco/cloroformo (a aproximadamente -60°C). Se añadió gota a gota bromuro de metilmagnesio (MeMgBr, 3M en éter) (0,95 ml, 2,85 mmol). El baño frío se dejó calentar a temperatura ambiente durante la noche, dando como resultado una solución de color naranja. Una vez que el LCMS indicó la finalización de la reacción, la solución se enfrió en un baño de hielo y se trató gota a gota con agua (5 ml), dando como resultado la precipitación. La mezcla se diluyó con EtOAc (100 ml) y se lavó con agua (100 ml). Se añadió gel de sílice a la capa orgánica y el disolvente se evaporó a presión reducida. El material se purificó por cromatografía en columna en un sistema de cromatografía Biotage® MPLC (eluido con 0 a 5% de MeOH en DCM con elución isocrática a 3,2% de MeOH) para proporcionar 345,5 mg del compuesto del título como una espuma frágil marrón. LCMS y 1H RMN son consistentes con N-(1-(6-cloro-7-((3,3-difluorociclobutil)metoxi)-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (345,5 mg, 0,773 mmol, rendimiento del 81%). RMN muestra una mezcla ~1:1 de diastereómeros. LCMS (Método 1): m/z 447 [M+H]+.
Paso 6: 3-(1-AmmoetN)-6-doro-7-((3,3-difluoroddobutM)metoxi)qumolm-2(1W)-ona clorhidrato (II-16).

Una solución de W-(1-(6-cloro-7-((3,3-difluorociclobutil)metoxi)-2-oxo-1,2-dihidroquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (342,7 mg, 0,767 mmol) en MeOH (7,0 mL) se enfrió en un baño de hielo y se trató gota a gota con 4M HCl en 1,4-dioxano (4 mL). La solución se agitó luego durante 25 minutos. Los disolventes se
evaporaron a presión reducida a temperatura ambiente. El residuo se trituró con 20 ml de éter etílico y el precipitado resultante se recogió en un embudo Hirsch y se lavó con más éter etílico para proporcionar 271,4 mg de un sólido rosa. LCMS y 1H RMN son consistentes con el clorhidrato de 3-(1-aminoetil)-6-cloro-7-((3,3-difluorociclobutil)metoxi)quinolin-2(1H)-ona (271,4 mg, 0,716 mmol, 93% de rendimiento)). 1H RMN (300 Mhz , metanol-d4): 8 ppm 7,95 (s, 1 H), 7,79 (s, 1 H), 6,96 (s, 1 H), 4,48-4,55 (m, 1 H), 4,20 (d, J = 4,10 Hz, 2 H), 2,56 -2,79 (m, 5 H), 1,68 (d, J = 7,04 Hz, 3 H). LCMS (Método 1): m/z 343 [M+H]+.
Ejemplo 19 - Intermedio 11-17: (S)-3-(1-ammoetN)-6-cloro-8-fluoroqumolm-2(1H)-ona

Paso 1:
terc-Butilo
(4-cloro-2-fluorofenilo)carbamato.

Una solución de 4-cloro-2-fluoroanilina (2 g, 13,74 mmol) y dicarbonato de di-ferc-butilo (6,4 ml, 27,6 mmol) en 1,4-dioxano (50 ml) se agitó a reflujo durante 2 dias. Entonces se evaporó el disolvente. El aceite resultante se diluyó con MeOH, agua y solución acuosa de hidróxido de amonio (10 ml cada uno) y se agitó vigorosamente durante 45 minutos. Se separó la capa inferior orgánica. El material orgánico se diluyó con EtOAc (50 ml) y se lavó con agua (50 ml), solución acuosa de HCl al 3,6% (2 x 50 ml), solución acuosa saturada de NaHCO3 (50 ml) y luego nuevamente con agua (2 x 50 ml). La capa orgánica se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar carbamato de ferc-butil (4-cloro-2-fluorofenilo) (3,0011 g, 12,22 mmol, 89% de rendimiento) como un líquido rojizo que solidificó en reposo. 1H RMN (300 MHz, DMsO-d6): 8 ppm 9,12 (s, 1 H), 7,63 (t, J = 8,65 Hz, 1 H), 7,42 (dd, J = 10,85, 2,35 Hz, 1 H), 7,18 -7,24 (m, 1 H), 1,45 (s, 9 H). LCMS (Método 1): m/z 246 [M+H]+.
Paso 2: terc-Butilo (4-cloro-2-fluoro-6-formilfenilo)carbamato.

Un matraz de fondo redondo de 500 ml de 3 bocas secado en horno se ajustó con un embudo de adición secado en horno y se colocó bajo una atmósfera de nitrógeno. Se añadieron mediante una jeringa carbamato de ferc-butilo (4-cloro-2-fluorofenilo) (5,44 g, 22,14 mmol) y éter etílico (91 ml). La solución transparente se enfrió en un
baño de acetonitrilo/hielo seco (a aproximadamente -40°C). Se añadió ferc-butillitio (1,7 pM en pentano, 33 ml, 22,14 mmol) al embudo de adición con una cánula. La solución de f-BuLi se añadió gota a gota a la solución de éter (durante ~ 10 minutos), tiempo durante el cual la solución de éter comenzó a volverse naranja. La solución se agitó a aproximadamente -40°C durante 2 horas, tiempo durante el cual progresivamente se volvió más naranja. Se añadió gota a gota DMF (8,7 ml, 112 mmol) (durante ~ 10 minutos), dando como resultado la precipitación de un sólido amarillo. El baño de hielo seco/MeCN se reemplazó con un baño de hielo y la mezcla se agitó durante 2 horas adicionales. La reacción se detuvo luego mediante la adición gota a gota de agua (20 ml), dando como resultado una mezcla marrón y se retiró el baño de hielo. La mezcla se diluyó con EtOAc (100 ml), se lavó con agua (2 x 100 ml), se secó (Na2SO4), se filtró y se evaporó a presión reducida para proporcionar 5,45 g de un sólido negro aceitoso. El material se trituró con hexanos (50 ml), se recogió en un embudo Buchner y se lavó con más hexanos para proporcionar 2,73 g de carbamato de ferc-butilo (4-cloro-2-fluoro-6-formilfenilo) como un polvo amarillo. El filtrado se evaporó a presión reducida, el residuo se trituró en hexanos (~ 15 ml) y el sólido amarillo resultante se recogió en un embudo Hirsch para proporcionar una segunda cosecha del compuesto del título (0,66 g). Se recuperó un total de 3.39 g (12,4 mmol, 56 % de rendimiento) de carbamato de ferc-butil (4-cloro-2-fluoro-6-formilfenilo). 1H RMN (300 MHz, DMSO-cfe): 8 ppm 9,93 (d, J = 0,88 Hz, 1 H), 9,47 (s, 1 H), 7,81-7,90 (m, 1 H), 7,55-7,61 (m, 1 H), 1,44 (s, 9 H). LCMS (Método 1): m/z 296 [M+Na].
Pasos 3 y 4: (S)-3-(1-ammoetil)-6-cloro-8-fluoroqumolm-2(1H)-ona clorhidrato (II-17).

Se colocaron un matraz de fondo redondo de 200 ml secado al horno y una barra de agitación bajo una atmósfera de nitrógeno. THF (17 ml) y diisopropilamina (1,59 ml, 11,16 mmol) se agregaron con una jeringa. La solución resultante se enfrió en un baño de hielo seco/acetona (a aproximadamente -78°C) y luego se añadió gota a gota n-butillitio (1,6 pM en hexano, 7,1 ml, 11,36 mmol) durante un período de 5 minutos. Después de agitarse durante 15 minutos, se añadió gota a gota una solución de (S)-etil 3-((terc-butoxicarbonil)amino)butanoato K (860,7 mg, 3,72 mmol) en THF (3,75 ml) durante 5 minutos. La solución se agitó durante 80 minutos a -78°C y luego se añadió una solución de carbamato de ferc-butilo (4-cloro-2-fluoro-6-formilfenilo) (1016,4 mg, 3,71 mmol) en THF (7,5 ml). gota a gota con jeringa. La reacción se agitó a -78°C durante otros 22 minutos y luego se detuvo mediante la adición de una solución acuosa saturada de NH4Cl (17 ml). La mezcla se repartió entre EtOAc y agua (100 ml cada uno). La capa orgánica se secó (MgSO4), se filtró y se evaporó a presión reducida para proporcionar 1,88 g del compuesto del título como una goma naranja. El material se disolvió en 1,4-dioxano (38 ml), se trató con HCl acuoso 12 pM (0,96 ml) y se agitó a 110°C durante 50 minutos. La muestra se dejó enfriar. El disolvente se evaporó a presión reducida para proporcionar 1,24 g de un sólido rojo. El material se trituró en IPA (25 ml), se recogió en un embudo Hirsch y se lavó secuencialmente con IPA (5 ml) y éter etílico (~ 20 ml) para proporcionar (S)-3-(1-aminoetilo)-6-cloro-8-fluoroquinolin-2(1H)-ona clorhidrato (370,4 mg, 1,337 mmol, 36% de rendimiento) como un sólido rojo. 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,41 (s, 1 H), 8,33 (br s, 3 H), 8,10 (s, 1 H), 7,67-7,76 (m, 2 H), 4,38-4,53 (m, 1 H), 1,52 (d, J = 7,04 Hz, 3 H). LCMS (Método 1): m/z 241 [M+H]+.
Ejemplo 20 - Intermedio 11-18: (S)-3-(1-aminoetil)-6-cloro-7-isopropoxi quinolm-2(1H)-ona

-18
Paso 1:
4-Cloro-3-isopropoxianilina

Se sometió a reflujo una mezcla de 5-amino-2-clorofenol (20 g, 139 mmol) y 2-bromopropano (26 mL, 278 mmol) y K2CO3 (38,4 g, 278 mmol) en CH3CN (300 mL) durante 24 h. La mezcla de reacción se enfrió a temperatura ambiente, se filtró y el sólido se lavó con acetato de etilo (150 ml). El filtrado se concentró y el residuo se purificó por ISCO (SO2: Hex/EtOAc 0 a 40%) para dar el compuesto del título, 4-cloro-3-isopropoxianilina (22,6 g, 87%).
Paso 2:
W-(4-cloro-3-isopropoxifeml)acetam¡da

A una mezcla de 4-cloro-3-isopropoxianilina (22,5 g, 121 mmol) en CH2Ch (200 ml) se le añadió DIPEA (42 ml, 242 mmol) seguido de anhídrido acético (17 ml, 181 mmol). La mezcla resultante se agitó a temperatura ambiente durante 3 h. Una vez completada la reacción, se añadió agua (100 ml) y se agitó durante 10 minutos. La capa organica se separó, se lavó con IN HCl (ac., 200 ml), salmuera (150 ml) y se secó sobre Na2SO4 anhidro. La solución se filtró y se concentró. El residuo crudo se recristalizó en CH2Ch/hexanos para dar el compuesto deseado N-(4-Cloro-3-isopropoxifenil)acetamida (19,6 g, 71%).
Paso 3:
2,6-d¡cloro-7-¡sopropox¡qu¡nol¡na-3-carbaldehído
Se añadió DMF (15 ml, 193,6 mmol) a un tubo de sellado de 350 ml y se enfrió a 0°C. A esta solución se le añadió oxicloruro de fósforo (60,1 ml, 645,6 mmol) gota a gota durante 40-50 min. La mezcla resultante se llevó a temperatura ambiente, seguido de la adición de N-(4-cloro-3-isopropoxifenil)acetamida (14,7 g, 64,5 mmol) en porciones y se calentó a 80°C durante la noche. La mezcla se enfrió a temperatura ambiente y se vertió cuidadosamente sobre hielo triturado. El precipitado amarillo se filtró, se lavó con agua y se secó sobre P2O5 durante la noche para proporcionar 2,6-dicloro-7-isopropoxiquinolina-3-carbaldehído como un sólido amarillo (17,5 g, 95%). Paso 4: 6-cloro-7-isopropoxi-2-metoxiquinolina-3-carbaldehído

A 2,6-dicloro-7-isopropoxiquinolina-3-carbaldehído (5,8 g, 20,4 mmol) en un co-disolvente de MeOH: THF (1 :1 , 100 ml) se añadió NaOMe (2,2 g, 40,8 mmol) en lo que respecta a ta. La mezcla de reacción se sometió a reflujo durante 3 h. Después de enfriarse a temperatura ambiente, la reacción se detuvo con una solución acuosa de NH4'Cl (20 ml). La mezcla se extrajo con EtOAc (25 ml x 3). La capa orgánica combinada se secó (Na2SO4), se concentró y se purificó por cromatografía ultrarrápida con hexano/EA (3:1) para dar 6-cloro-7-isopropoxi-2-metoxiquinolina-3-carbaldehído (5,07 g, 89%) como un sólido amarillo.
Paso 5:
1-(6-Cloro-7-isopropoxi-2-metoxiquinolin-3-il)etanol

A 6-cloro-7-isopropoxi-2-metoxiquinolina-3-carbaldehído (5,07 g, 18,17 mmol) en THF (100 ml) a -78°C se le añadió una solución de MeMgCl en THF (3 M, 9,1 mL, 27,2 mmol) gota a gota. La reacción se agitó a temperatura ambiente (temperatura ambiente) durante 3 h y luego se detuvo con una solución acuosa de NH4O (50 ml). La capa orgánica se separó y la capa acuosa se extrajo con EtOAc (25 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en gel de sílice con hexano/EA (3:1) para dar el compuesto 1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-ilo)etanol (4,06 g, 76%).
Paso 6:
1-(6-Cloro-7-isopropoxi-2-metoxiquinolin-3-il)etanona

A 1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-il)etanol (4,06 g, 13,8 mmol) en CH2Ch (50 mL) a temperatura ambiente se agregó porción tras porción DMP (7,0 g, 16,5 mmol). La reacción se agitó a temperatura ambiente durante 2 h, y luego se detuvo con una solución acuosa de NaHCO3 y Na2S2O3. Después de agitarse durante 15 minutos, ambas capas se volvieron transparentes. La capa orgánica se separó y la capa acuosa se extrajo con CH2Cl2 (30 ml x 2). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en gel de sílice con hexano/EA (4:1) para dar 1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-il)etanona (3,67 g, 72%) como un sólido blanco.
Paso 7: (R,Ej-W-(1-(6-cloro-7-isopropoxi-2-metoxiqumolm-3-N)etMiden)-2-metNpropano-2-sulfmamida

A 1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-il)etanona (3,67 g, 12,5 mmol) en THF/tolueno (20 mL: 400 mL) a la temperatura ambiente se agregó (R)-2-metilpropano-2-sulfinamida (3,03 g, 25 mmol) y Ti(O'Pr)4 (11 ml, 37,5 mmol). La reacción se sometió a reflujo con un aparato Dean-Stark. Después de que la reacción se calentó a reflujo durante 4 h y se eliminaron 150 ml de disolvente, la reacción se enfrió a temperatura ambiente. El disolvente se eliminó al vacío y se añadieron 50 ml de EtOAc al residuo, seguido de la adición de 20 ml de una solución acuosa saturada de NaHCO3. Después de agitarse durante 10 minutos, el sólido se eliminó a través de una capa de celite. El filtrado se extrajo con EtOAc (200 ml x 2), se secó (Na2SO4), se concentró y se purificó por cromatografía en gel de sílice con hexano/EA (1:1) para dar el compuesto del título (4,32 g, 87%).
Paso 8: (R)-W-((S)-1-(6-cloro-7-isopropoxi-2-metoxiqumolm-3-N)etM)-2-metNpropano-2-sulfmamida

A (R,E)-A/-(1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-il)etiliden)-2-metilpropano-2-sulfinamida (4,32 g, 10,9 mmol) en THF (100 ml) a -78°C, se añadió 1 pM L-selectride (14,2 ml, 14,2 mmol) en THF gota a gota. La mezcla de reacción se calentó a temperatura ambiente y se agitó durante 3 h. La reacción se detuvo con una solución acuosa saturada de NH4Cl (30 ml) y luego se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron por cromatografía en gel de sílice con hexano/EA (1:1) para dar el compuesto deseado (3,58 g, 82%).
Paso 9: (S)-3-(1-ammoetN)-6-cloro-7-isopropoxiqumolm-2(1H)-ona sal clorhidrato (11-18).

A (R)-W-((S)-1-(6-cloro-7-isopropoxi-2-metoxiquinolin-3-il)etil)-2-metilpropano-2-sulfinamida (3,58 g, 8,99 mmol) en dioxano (50 ml) se añadió 2 N HCl (50 ml) a temperatura ambiente. La reacción se sometió a reflujo durante 3 h. El disolvente se eliminó a vacío y el residuo se secó a vacío para proporcionar el II-18 crudo, que se purificó adicionalmente por trituración (CH2Ch/MeOH/hexano) para dar el compuesto II-18 puro (2,44 g, 86%) como blanco sólido. 1H RMN (300 MHz, DMSO-cfe): 812,10 (s, 1H), 8,29 (br, s, 3H), 7,98 (s, 1H), 7,83 (s, 1H), 7,08 (s, 1H), 4,66 (m, 1H), 4,38 (m, 1H), 3,91 (s, 3H), 1,52 (d, J = 6,87 Hz, 3H), 1,37 (d, J = 6,03 Hz, 6H).
LCMS (Método 3, APCI): TR = 8,06 min, m/z = 281,1 [M+H]+.
Ejemplo 21 - Intermedio III-1: 5-fluoro-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo

Paso 1: 1-óxido de 2-ciano-5-fluoropiridina.

Se añadió gota a gota una solución de 5-fluoropicolinonitrilo (7,27 g, 59,5 mmol) en CHCI3 (60 mL) mediante un embudo de adición a una solución de m-CPBA (<77%, 22,00 g, 98 mmol) en CHCh 160 mL). La solución se agitó a reflujo durante 4 días, momento en el que la LCMS mostró una conversión de aproximadamente el 85%. La muestra se dejó enfriar, luego se añadió sulfito de sodio (12,4 g, 98 mmol) y la muestra se agitó a temperatura ambiente durante tres horas, tiempo durante el cual la solución se espesó con un precipitado blanco. La muestra se diluyó con DCM (300 ml) y se filtró en un embudo Buchner, y la torta del filtro se lavó con DCM (~ 400 ml). Un material blanco precipitó en el filtrado. La mezcla del filtrado se lavó con NaHCO3 acuoso saturado (400 ml), durante lo cual los sólidos se fueron a la solución. La capa orgánica se lavó con agua (300 ml), luego se secó (MgSO4) y se filtró. Se añadió gel de sílice y la mezcla se evaporó a presión reducida. El material se cromatografió por Biotage MPLC (340 g en columna de gel de sílice) con 0 a 100% de EtOAc en hexanos, con elución isocrática cuando los picos se desprendieron para proporcionar 1-óxido de 2-ciano-5-fluoropiridina (4,28 g, 31,0 mmol, 52 % de rendimiento) como un sólido blanco. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 8,85 - 8,93 (m, 1 H), 8,23 (dd, J = 9,09, 6,74 Hz, 1 H), 7,53 - 7,64 (m, 1 H). LCMS (Método 1): Ta 0,57 min., m/z 138,9 [M+H]+.
Paso 2: Acetato de 6-ciano-3-fluoropiridin-2-ilo

Se calentó una solución de 1-óxido de 2-ciano-5-fluoropiridina (4,28 g, 31,0 mmol) en anhídrido acético (40 ml, 424 mmol) a reflujo (baño a 150°C) durante tres días, durante los cuales la solución clara se volvió oscura. La muestra se concentró a presión reducida. El residuo se disolvió en MeOH (30 ml) y se agitó durante 1 hora. Se añadió gel de sílice y el disolvente se evaporó a presión reducida. El material se cromatografió por Biotage MPLC (100 g de columna de gel de sílice) con 0 a 23% de EtOAc en hexanos para proporcionar acetato de 6-ciano-3-fluoropiridin-2-ilo (3,32 g, 18,43 mmol, 60% ydl) como líquido transparente que solidificó al enfriar. RMN 1H (300 MHz, CLOROFORM-d): 8 ppm 7,65 - 7,75 (m, 2 H), 2,42 (s, 3 H). LCMS (Método 1): Ta 1,54 min., m/z 138,8 (pérdida de acetato).
Paso 3: 5-fluoro-6-oxo-1,6-dihidropiridin-2-carbonitrilo.

Una solución de acetato de 6-ciano-3-fluoropiridin-2-ilo (3,32 g, 18,43 mmol) en MeOH (40 ml) se trató con carbonato de potasio (5,10 g, 36,9 mmol) y se agitó a temperatura ambiente cuatro horas. La LCMS a las 2 horas mostró que la reacción se había completado. El disolvente se evaporó a presión reducida. El residuo se disolvió en agua (l0o ml) y se acidificó a pH <1 con 1M HCl. La solución se extrajo con EtOAc (3x100 ml). Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se evaporaron a presión reducida para proporcionar 5-fluoro-6-oxo-1,6-dihidropiridin-2-carbonitrilo (2,34 g, 16,94 mmol, rendimiento al 92%) como blanco sólido. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 12,92 (br s, 1 H), 7,73 (br s, 1 H), 7,43 (br s, 1 H). LCMS (Método 1): Ta 0,70 min., m/z 138,9 [M+H]+.
Paso 4: 5-fluoro-1-metil-6-oxo-1,6-dihidropiridina-2-carbonitrilo (III-1)

Se trató una mezcla de 5-fluoro-6-oxo-1,6-dihidropiridin-2-carbonitrilo (2,31 g, 16,73 mmol) y carbonato de potasio (4,86 g, 35,2 mmol) en un matraz de fondo redondo de 200 ml. Con DMF (46 ml) y se agitó durante 15 minutos. Se añadió Mel (1,2 ml, 19,19 mmol) y la mezcla se agitó a temperatura ambiente durante 45 minutos. El disolvente se evaporó a presión reducida. El residuo se mezcló con agua (150 ml) y se extrajo con DCM (2x150 ml). Los extractos orgánicos combinados se secaron (MgSO4), se filtraron, se trataron con gel de sílice y se evaporaron a presión reducida, luego se evaporaron más a 60°C en alto vacío. El material se cromatografió por Biotage MPLC con 0 a 35% de EtOAc en hexanos, con elución isocrática a 16% de EtOAc y 35% de EtOAc mientras que los picos se desprendieron. El pico que se desprendió con un 16% de EtOAc era material metilado en O y se descartó. El pico que se desprendió con un 35% de EtOAc proporcionó el compuesto del título III-I (1,70 g, 11,17 mmol, 67% de rendimiento) en forma de un sólido. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 7,53 (dd, J = 9,38, 7,62 Hz, 1 H), 7,18 (dd, J = 7,77, 4,84 Hz, 1 H), 3,60 (s, 3 H). LCMS (Método 1): Ta 0,94 min., m/z 152,9 [M+H]+.
Ejemplo 22 - Intermedio V-2: 5-amino-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo

Una solución de 5-aminopicolinonitrilo (5,50 g, 46 mmol, 1 eq.) En 300 ml de DCM se enfrió a 0°C y luego se trató con TEA (20 ml, 144 mmol, 3,1 eq.) seguido gota a gota por la adición de anhídrido trifluoroacético (20 ml, 144 mmol, 3,1 eq.). Después de agitarse durante la noche a temperatura ambiente, la mezcla de reacción se vertió en hielo y se extrajo con DCM. La purificación pasando por un tapón de gel de sílice (hexano/EtOAc, 75/25)
proporcionó N-(6-cianopiridin-3-il)-2,2,2-trifluoroacetamida (7,24 g, 73%) como un sólido blanco. TLC: Hexano/EtOAc, 8/2.
Paso 2:
N-(6-cianopiridin-3-il)-2,2,2-trifluoroacetamida-N-óxido.
Se enfrió una solución de N-(6-cianopiridin-3-il)-2,2,2-trifluoroacetamida (7,24 g, 33,7 mmol, 1 eq.) en 270 ml de CHCl3 en un baño de hielo, luego se trató gota a gota. con una solución de mCPBA (7,68 g, 39 mmol, 1,15 eq.) en 65 ml de CHCh. La mezcla de reacción se sometió a reflujo durante 24 horas y luego se vertió en H2O. Después de agitarse con NaHSO3 acuoso al 10% y NaHCO3, el sólido se recogió y se enjuagó con H2O, luego con CHCl3. Esto proporcionó 1,86 g (24%) del compuesto del título como un sólido blanco. N-(6-cianopiridin-3-il)-2,2,2-trifluoroacetamida sin reaccionar (4,70 g, 65%) se recuperó por extracción del filtrado y purificación por cromatografía en gel de sílice (hexano/EtOAc, 75/25).
Paso 3:
5-amino-6-oxo-1,6-dihidropiridin-2-carbonitrilo.
Una suspensión de N-(6-cianopiridin-3-il)-2,2,2-trifluoroacetamida-N-óxido (0,81 g, 3,5 mmol, 1 eq.) en 10,5 ml de THF se trató con TEA (0,75 mL, 5,3 mmol, 1,5 eq.) seguido de la adición gota a gota de anhídrido trifluoroacético (1,74 mL, 12,5 mmol, 3,5 eq.). Después de agitarse durante la noche a temperatura ambiente, se agregaron trozos de hielo y 12 ml de NaOH al 10%. Después de agitarse a temperatura ambiente durante 1 hora, la mezcla de reacción se acidificó a pH ~ 4 con HOAc y se recogió el sólido precipitado, proporcionando 0,31 g 5-Amino-6-oxo-1,6-dihidropiridin-2-carbonitrilo (64%) como un sólido de color beige. TLC: DCM/Me-OH, 97/3.
Paso 4:
5-amino-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo (V-2).
Una solución de 5-amino-6-oxo-1,6-dihidropiridin-2-carbonitrilo (500 mg, 3,7 mmol, 1 eq.) en 18 ml de DMF se trató con K2CO3 anhidro (1,0 g, 7,26 mmol, 2 eq.) y CH3I (0,175 ml, 4,0 mmol, 1,1 eq) y se agitó a temperatura ambiente durante 1,5 h. A la mezcla de reacción se le añadió agua, seguido de extracción con EtOAc (2x), los extractos se secaron (Na2SO4) y se evaporaron para proporcionar un sólido de color marrón. El análisis del producto bruto por RMN indicó una relación de ~ 8/2 del producto deseado vs el isómero O-metilado. La trituración del sólido con Et2O proporcionó 160 mg del producto deseado (29%). La purificación de los lavados con Et2O mediante cromatografía preparativa C18 ISCO proporcionó 82 mg adicionales del compuesto V-1 del título como la sal de TFA (15%).
TLC: Hexano/EtOAc, 1/1. 1H-RMN (300 MHz, d6DMSO): 86,94 (d, J = 7,68), 6,42 (ancho s, 2H), 6,33 (d, J = 7,68), 3,55 (s, 3H). LC/MS (Métodos 3): Ta 3,0 min., m/z 150 [M+H]+.
Tabla 1: Los intermedios listados en la Tabla 1 se prepararon utilizando los métodos descritos anteriormente o se obtuvieron de fuentes comerciales.

(continuado)

(continuado)

Ejemplo 23 - 5-(((6-cloro-2-oxo-1,2-dihidroquinolin-3-il)metil)amino)-1 -metN-6-oxo-1,6-dihidropindm-2-carbonitrilo (I -1)

A un matraz de botella redonda de 100 ml se le añadió 6-cloro-2-oxo-1,2-dihidroquinolina-3-carbaldehído IV-1 (69,6 mg, 0,335 mmol), 5-amino-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo V-2 (50 mg, 0,335 mmol) y ácido acético (0,096 ml, 1,676 mmol) en DCM (10 ml). Finalmente, se cargó triacetoxiborohidmro de sodio (107 mg, 0,503 mmol) y se agitó vigorosamente a temperatura ambiente bajo un flujo de N2 durante la noche. La mezcla de reacción se diluyó con EtOAc (60 ml), luego se lavó con NaHCO3 saturado, agua (x2) y salmuera. El extracto orgánico se secó sobre Na2SO4, se filtró y se concentró para producir un crudo, que se purificó mediante HPLC preparativa en fase inversa en Gilson para producir una mezcla de producto y subproducto desconocido (~ 32 mg, 28% de rendimiento, 81% Pureza por HPLC). La mezcla se sometió a una segunda purificación por HPLC para proporcionar un producto puro deseado (4 mg, rendimiento del 3,5%). 1H RMN (300 MHz, CDCh) 8 ppm 7,97 (s, 1 H), 7,56 (br s, 1 H), 7,45 (br d, J = 11,43 Hz, 2 H), 7,36 (br d, J = 8,79 Hz, 1 H), 7,12 -7,20 (m, 1 H), 6,66 -6,78 (m, 1 H), 6,00 (br d, J = 7,92 Hz, 1 H), 3,68 (s, 2 H), 3,31 (br s, 3 H). LCMS (Método 1): Ta 2,37 min, m/z 340,97 [M+H]+.
Ejemplo 24 - 6-cloro-3-((1-etN-2-oxo-1,2-dihidropmdm-3-Nammo)metN)qumoMn-2(1H)-ona (1-2)

Paso 1: 1-etil-3-mtropmdm-2(1H)-ona.

Se trató una mezcla de 3-nitropiridin-2(1H)-ona (1,00 g, 7,14 mmol) y K2CO3 (3,00 g, 21,71 mmol) en DMF (30 ml) con yoduro de etilo (0,60 ml, 7,42 mmol) y se agita a 50°C durante la noche. LCMS indicó una mezcla 4:1 de producto y material de partida. Se añadió más yoduro de etilo (0,25 ml) y la reacción se agitó a 60°C durante cinco horas. La mezcla amarilla se diluyó con agua (100 ml) y se extrajo con EtOAc (3 x 100 ml). Los extractos orgánicos combinados se secaron (MgSO4), se filtraron y se evaporaron a presión reducida para proporcionar 1,08 g de un
sólido amarillo. El material se disolvió en unos pocos ml de DCM y se cromatografió por Biotage MPLC (25 g de columna de gel de sílice, 0 a 10% de MeOH en DCM, con elución isocrática a 3% de MeOH) para proporcionar 1-etil-3-nitropiridin-2(1H)-ona (898,9 mg, 5,35 mmol, rendimiento del 74,9%) como un sólido amarillo. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 8,38 (dd, J = 7,92, 2,05 Hz, 1 H), 8,24 (dd, J = 6,60, 2,20 Hz, 1 H), 6,44 (dd, J = 7,62, 6,45 Hz, 1 H), 4,05 (q, J = 7,04 Hz, 2 H), 1,26 (t, J = 7,18 Hz, 3 H). LCMS (Método 1): Ta 0,96 min., m/z 169,0 [M+H]+.
Paso 2: 3-ammo-1-etMpiridm-2(1H)-ona

Una solución de 1-etil-3-nitropiridin-2(1H)-ona (891,2 mg, 5,30 mmol) y cloruro de estaño (II) dihidrato (5,03 g, 22,29 mmol) en EtOAc (30 ml) en un matraz de fondo redondo de 200 mL se agitó a 80°C durante dos horas; la LCMS a las 1,5 horas mostró que la reacción se había completado limpiamente. La solución se dejó enfriar y se diluyó con EtOAc (50 ml), luego se añadió NaHCO3 (8 g) en pequeñas porciones y la mezcla se agitó durante 20 minutos, momento en el que se había producido poca efervescencia y la mezcla todavía era fuertemente ácida (pH ~ 1). Se añadió agua (50 ml) en porciones con agitación completa, primero magnéticamente y luego a mano como se formó un precipitado, dando como resultado una mezcla azul oscuroade pH ~ 8, La mezcla se filtró en un embudo Buchner y la torta del filtro se lavó con varias porciones de EtOAc (~ 100 ml en total). Las capas de filtrado se separaron. La fase acuosa se extrajo con EtOAc (3 x 50 ml) y todos los compuestos orgánicos se combinaron y se secaron (Na2SO4), se filtraron y se evaporaron a presión reducida. El sólido azulado resultante (0,64 g) se disolvió en unos pocos ml de DCM y se cromatografió por Biotage MPLC (25 g de columna de gel de sílice, 0 a 9% de MeOH en DCM, con elución isocrática a 3,8% de MeOH). El sólido azul así obtenido se disolvió en DCM, se trató con gel de sílice y se evaporó a presión reducida. El material se recromatografió por Biotage MPLC (25 g de columna de gel de sílice, 0 a 100% de EtOAc en hexanos, con elución isocrática a 67% de EtOAc) para proporcionar 3-amino-1-etilpiridin-2(1H)-ona (517,7 mg, 3,75 mmol, 70,7% de rendimiento) como un sólido ligeramente azul. 1H RMN (300 MHz, DMSO-d6): 8 ppm 6,88 (dd, J = 6,89, 1,91 Hz, 1 H), 6,41 (d d, J = 7,04, 1,76 Hz, 1 H), 6,03 (dd, J = 6,90, 6,90 Hz, 1 H), 5,06 (s, 2 H), 3,89 (q, J = 7,13 Hz, 2 H), 1,19 (t, J = 7,18 Hz, 3 H). LCMS (Método 1): Ta 0,76 min., m/z 139,0 [M+H]+.
Paso 3: 6-cloro-3-((1-etN-2-oxo-1,2-dihidropmdm-3-Nammo)metN)qumolm-2(1W)-ona (1-2).

Una suspensión de 6-cloro-2-oxo-1,2-dihidroquinolina-3-carbaldehído (100,1 mg, 0,482 mmol) y 3-amino-1-etilpiridin-2(1H)-ona (67,1 mg, 0,486). mmol) en MeOH (1,5 ml) y el tolueno (1,5 ml) se trató con AcOH (27,6 ml) y se agitó a 50°C durante 5,5 horas, durante las cuales se descargó el color azul del material de partida de piridinona. Los disolventes se evaporaron a presión reducida. El residuo rojo se trató sucesivamente con dos alícuotas de tolueno (3 ml cada una) y se evaporó a presión reducida. El residuo se suspendió en DCM (3 ml) y se trató con AcOH (135,4 ml) y triacetoxiborohidruro de sodio (164,3 mg, 0,775 mmol), luego se colocó bajo nitrógeno y se agitó a temperatura ambiente durante la noche; en unos pocos minutos, el material se disolvió y en una hora precipitó un material. La muestra se diluyó con DCM/MeOH/EtOAc, se trató con gel de sílice y se evaporó a presión reducida. El material se cromatografió por Biotage MPLC (0 a 100% de EtOAc en hexanos) para proporcionar el compuesto del título (I-2) (25,7 mg, 0,078 mmol, 16,16% de rendimiento, HPLC de pureza del 100% a 220 nm) como un sólido verdoso. 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,02 (s, 1 H), 7,79 (d, J = 2,05 Hz, 1 H), 7,65 (s, 1 H), 7,49 (dd, J = 8,65, 2,20 Hz, 1 H), 7,30 (d, J = 8,79 Hz, 1 H), 6,90 (dd, J = 4,30, 4,30 Hz, 1 H), 5,95 -6,11 (m, 3 H), 4,16 (d, J) = 5,90 Hz, 2 H), 3,93 (q, J = 6,84 Hz, 2 H), 1,22 (t, J = 7,04 Hz, 3 H). LCMS (Método 4): Ta 1,15 min., m/z 330,0 [M+H]+.
Tabla 2: Los co m p u e s to s e n u m e ra d o s en la T a b la 2 se p re pa ra ron u tilizan do m é tod os s im ila re s a los d e sc rito s para la p re p a ra c ió n de I-1 & I-2.


Tabla 3. Señal de LCMS y cambios químicos de RMN de cada compuesto enumerado en la Tabla 2.
-1,2-

(continuado)

Ejemplo 25 -- (S)-5-((1-(6-cloro-2-oxo-1,2-dihidroqumoMn-3-N)etM)ammo)-1-metN-6-oxo-1,6-dihidropmdm-2-carbonitrilo (I-13)

Una mezcla de 5-fluoro-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo III-1 (1,23 g, 8,09 mmol),(s)-3-(1-aminoetilo)-6-doroquinolin-2(1H)-ona clorhidrato II-1 (1,91 g, 7,37 mmol) y W,W-diisopropiletNamina (3,8 ml, 21,8
mmol) en dimetilsulfóxido anhidro (57 ml) bajo N2 se calentó a 110°C y se agitó durante 6 horas. Después de enfriarse a temperatura ambiente, la mezcla se repartió entre EtOAc/H2O (750 ml/750 ml). La capa orgánica se separó, se secó (Na2SO4) y se concentró al vacío. El residuo se purificó en ISCO dos veces (40 g de columna de gel de sílice, EtOAc/hexano 0 ~ 100%; 80 g de columna de gel de sílice, MeOH/diclorometano 0 ~ 5%). Las fracciones incoloras se combinaron y el diclorometano se eliminó a presión reducida en el rotavapor hasta que precipitó una gran cantidad de sólido blanco. El sólido blanco se recogió por filtración y se lavó con MeOH frío. Luego se mezcló con MeCN/H2O (10 ml/25 ml) y se liofilizó para proporcionar el compuesto del título 1-13 en forma de un sólido blanco (790 mg). pf 262-264°C. 1H RMN (300 MHz, DMSO-cfe): 12,07 (s, 1H), 7,75 (s, 1H), 7,73 (d, J = 2,2 Hz, 1H), 7,51 (d d,J = 8,6, 2,3 Hz, 1H), 7,31 (d, J = 8,8 Hz, 1H), 6,97 (d, J = 8,0 Hz, 1H), 6,93 (d, J = 7,7 Hz, 1H), 5,95 (d, J = 8,0 Hz, 1H), 4,68 (m, 1H), 3,58 (s, 3H), 1,50 (d, J = 6,6 Hz, 3H). LCMS (Método 3): 100% puro a 254 nm, Ta 10,78 min, m/z 355, 357 [M+H]+. El filtrado y las fracciones coloreadas (TLC puro) del segundo ISCO se combinaron y trataron con carbón activado y se filtraron (hasta que el filtrado sea incoloro). El filtrado se concentró luego a presión reducida en rotavapor para eliminar el diclorometano hasta que precipitó una gran cantidad de sólido blanco. El sólido blanco se recogió por filtración y se lavó con MeOH frío. Luego se mezcló con MeCN/H2O (10 ml/25 ml) y se liofilizó para proporcionar el compuesto del título 1-13 en forma de un sólido blanco (970 mg). pf 262-264°C. RMN 1H (300 MHz, DMSO-d6): 12,06 (s, 1H), 7,75 (s, 1H), 7,73 (d, J = 2,5 Hz, 1H), 7,51 (dd, J = 8,6, 2,3 Hz, 1H), 7,31 (d, J = 8,8 Hz, 1H), 6,97 (d, J = 8,0 Hz, 1H), 6,92 (d, J = 8,0 Hz, 1H), 5,95 (d, J = 8,0 Hz, 1H), 4,68 (m, 1H), 3,58 (s, 3H), 1,50 (d, J = 6,9 Hz, 3H). LCMS (Método 3): 100% puro a 254 nm, m/z 355, 357 [M+H]+. El rendimiento total para dos lotes combinados es del 67%.
Ejemplo 26 --(S)-5-((1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)amino)-6-oxo-1,6-dihidropiridina-2-bonitrilo (I-14)

Una mezcla de DIEA (0,165 ml, 0,943 mmol), (S)-3-(1-aminoetil)-6-cloroquinolin-2(1H)-ona II-1 (70 mg, 0,314 mmol), y 5-Fluoro-6-oxo-1,6-dihidropiridin-2-carbonitrilo (52,1 mg, 0,377 mmol) en DMSO (1 ml) se calentó a 110°C durante 2 horas. La mezcla de reacción se enfrió a temperatura ambiente, luego se trató con EtOAc, se lavó con agua dos veces, se secó y se concentró. La purificación de biotipos con 0 a 10% de MeOH/DCM en una columna de 10 g produjo (S)-5-((1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)amino)-6-oxo-1,6-dihidro piridina-2-carbonitrilo (12,1 mg, 11,3%). 1H RMN (300 MHz, DMSO-d6) 8 ppm 12,03 (s, 1 H), 7,72 (s, 2 H), 7,47 (m, 1 H), 7,28 (m, 1H), 6,84 (m, 1 H), 6,68 (m, 1H), 5,93 (m, 1H), 4,66 (m, 1H), 1,45 (d, J = 6,74Hz, 3H). LCMS (Método 3): Ta 2,35 min, m/z 361,05 [M+Na]+.
Ejemplo 27 -- (S)-5-((1-(6-doro-7-fluoro-2-oxo-1,2-dihidroqumolm-3-N)etM)ammo)-1-metN-6-oxo-1,6-dihidropiridin-2-carbonitrilo (I-16)

Una mezcla de (S)-3-(1-aminoetil)-6-cloro-7-fluoroquinolin-2(1H)-ona clorhidrato II-4 (1,00 g, 3,61 mmol), 5-fluoro-1 -El metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo 111-1 (604 mg, 3,97 mmol), N,N-diisopropiletilamina (1,9 ml, 10,8 mmol) en DMSO (15 ml) se calentó a 110°C En un tubo de sellado durante 16 h. MS y TLC mostraron conversión limpia. La mezcla de reacción se vertió en agua (300 ml) con agitación vigorosa. El sólido se filtró y se lavó con agua, y luego se disolvió en EtOAc y se secó sobre sulfato de sodio. Después de la filtración, la solución se concentró con gel de sílice y se purificó por cromatografía ultrarrápida en columna (SO2: diclorometano/EtOAc 0 a 50%) para proporcionar el compuesto objetivo 1-16 en forma de un sólido amarillo pálido (1,20 g, 89%). 1H RMN (300 MHz, DMSO-d6) 812,12 (s, 1H), 7,95 (d, J = 7,9 Hz, 1H), 7,74 (s, 1H), 7,21 (d, J = 10,4 Hz, 1H), 6,94 (d, J = 7,9 Hz, 1H), 6,92 (d, J = 7,4 Hz, 1H), 5,94 (d, J = 8,2 Hz, 1H), 4,69-4,62 (m, 1H), 3,58 (s, 3H), 1,49 (d, J = 6,6 Hz, 3H); LCMS (Método 3): Ta 5,00 min, m/z 373.1, 375,1 [M+H]+.
Ejemplo 28-(s)-5-((1-(6-cloro-2-oxo-1,2-dihidroquinolin-3-il)etil)amino)-1-metil-6-oxo-1,6-dihidropirazina-2-carbonitrilo (1-17)

Paso 1: 6-bromo-3-doro-1-metNpirazm-2(1H)-ona.

Una mezcla de 6-bromo-3-cloropiraz¡n-2(1H)-ona (2 g, 9,55 mmol) y carbonato de potasio (2,77 g, 20,04 mmol) en un matraz de fondo redondo de 200 ml se trató con DMF (25 ml) y se agitó durante 15 minutos. Se añadió MeI (0,69 ml, 11,04 mmol) y la mezcla se agitó a temperatura ambiente durante 45 minutos. El disolvente se evaporó a presión reducida. El residuo se mezcló con agua (75 ml) y se extrajo con DCM (2 x 75 ml). Los extractos orgánicos combinados se secaron (MgSO4), se filtraron, se trataron con gel de sílice y se evaporaron a presión reducida, luego se evaporaron más a 60°C en alto vacío. El material se cromatografió por Biotage MPLC (gel de sílice, 0 a 35% de EtOAc en hexanos), con elución isocrática a 16% de EtOAc y 30% de EtOAc, mientras que desaparecieron los picos de la masa deseada. El pico que se desprendió con 30% de EtOAc proporcionó 6-bromo-3-cloro-1-metilpirazin-2(1H)-ona (1,30 g, 5,82 mmol, 61% de rendimiento) como un sólido blanco. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 7,50 (s, 1 H), 3,63 (s, 3 H). LCMS (Método 1): Ta 1,44 min., m/z 222,9, 224,9 [M+H]+.
Paso 2: (S)-3-(1-((5-bromo-4-metil-3-oxo-3,4-dihidropirazin-2-il)amino)etil)-6-cloroquinolin-2(1H)-ona
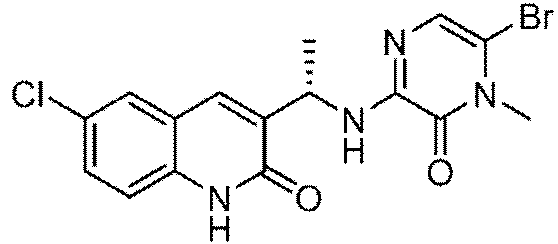
Una mezcla de (S)-3-(1-am¡noet¡l)-6-cloroqu¡nol¡n-2(1H)-ona clorhidrato II-1 (200 mg, 0,772 mmol) y 6-bromo-3-cloro-1-metilpirazin-2(1H)-ona (189,2 mg, 0,847 mmol) en DMSO (5 ml) se trató con DIEA (400 ml, 2,290 mmol) y se agitó a 110°C durante cinco horas. La muestra se mezcló con agua (75 ml) y se extrajo con DCM (2 x 50 ml). Las capas orgánicas combinadas se secaron (Na2SO4) y se filtraron, se añadió gel de sílice y el disolvente se evaporó a presión reducida. La muestra se cromatografió por Biotage MPLC (25 g de columna de gel de sílice, 0 a 100% de EtOAc en hexanos, con elución isocrática cuando se desprendieron los picos) para proporcionar (S)-3-(1-(^-bromo^-metilo^-oxo^^-dihidropirazin^-i^amino^til^-cloro quinolin-2 (1 H)-ona (32,9 mg, 0,080 mmol, 10% de rendimiento) como un sólido naranja. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 11,99 (s, 1 H), 7,70 - 7,75 (m, 2 H), 7,56 ( d, J = 7,92 Hz, 1 H), 7,46 - 7,52 (m, 1 H), 7,30 ( d, J = 8,79 Hz, 1 H), 6,88 - 6,96 (m, 1 H), 5,02 - 5,17 (m, 1 H), 3,50 -3,60 (m, 3 H), 1,44 ( d, J = 6,74 Hz, 3 H). LCMS (Método 1): Ta 2,55 min., m/z 410,8 [M+H]+.
P a s o 3 :
(S)-5-((1-(6-doro-2-oxo-1,2-dihidroqumolm-3-N)etN)ammo)-1-metN-6-oxo-1,6-dihidropirazma-2-carbonitrilo (1-17).

Una mezcla de (S)-3-(1-((5-bromo-4-met¡l-3-oxo-3,4-d¡h¡drop¡raz¡n-2-¡l)am¡no)et¡l)-6-cloroqu¡nol¡n-2(1H)-ona (31,0 mg, 0,076 mmol), Pd2(dba)3 (7,4 mg, 8,08 jmol), 1,1'-b¡s(d¡fen¡lfosf¡no)ferroceno (8,7 mg, 0,016 mmol) y d¡c¡anoz¡nc (18,1 mg, 0,154 mmol) se colocó bajo n¡trógeno en un v¡al de 2-dram. Se añad¡ó DMF (1,4 ml) con una jer¡nga. La atmósfera fue evacuada y reemplazada con n¡trógeno tres veces. La mezcla fue ag¡tada a temperatura amb¡ente durante la noche. LCMS ¡nd¡có que la reacc¡ón se había completado l¡mp¡amente. El d¡solvente se evaporó a pres¡ón reduc¡da. El res¡duo se repart¡ó entre agua (15 ml) y DCM (2x15 ml). Los extractos orgán¡cos comb¡nados se secaron (Na2SO4) y se f¡ltraron, se añad¡ó gel de síl¡ce y el d¡solvente se evaporó a pres¡ón reduc¡da. El mater¡al se cromatograf¡ó por B¡otage MPLC (0 a 65% de EtOAc en hexanos, con eluc¡ón ¡socrát¡ca cuando se el¡m¡naron los p¡cos) para proporc¡onar el compuesto del título 1-17 (20,1 mg, 0,055 mmol, 72,0% de rend¡m¡ento, HPLC pureza 96,5% a 220°c ). nm) como un sól¡do de color naranja. 1H RMN (300 MHz, DMSO-cfe): 8 ppm 12,03 (s, 1 H), 8,59 (d, J = 8,50 Hz, 1 H), 7,77 (s, 1 H), 7,72 (d, J = 2,35 Hz, 1 H), 7,47 - 7,55 (m, 2 H), 7,31 (d, J = 8,79 Hz, 1 H), 5,18 - 5,31 (m, 1 H), 3,48 (s, 3 H), 1,48 (d, J = 6,74 Hz, 3 H). LCMS (Método 4): Ta 1,25 m¡n., m/z 356,1 [M+H]+.
Ejemplo 29-(s)-5-((1-(6-doro-7-metoxi-2-oxo-1,2-dihidroqumolm-3-N)etM)ammo)-1-metN-6-oxo-1,6-dihidropiridina-2-carbonitrilo (1-20)

Una mezcla de 5-fluoro-1-met¡l-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡tr¡lo III-1 (58 mg, 0,38 mmol), (S)-3-(1-am¡noet¡lo)-6-cloro-7-metox¡qu¡nol¡n-2(1H)-ona clorh¡drato II-7 (100 mg, 0,35 mmol) y N,N-d¡¡soprop¡let¡lam¡na (180 |jl, 1,04 mmol) en n-BuOH (3 ml) se calentó a 110°C en un tubo sellado bajo N2 y se ag¡tó durante la noche. La mezcla se concentró luego a pres¡ón reduc¡da y el res¡duo se pur¡f¡có en ISCo (20 g de columna de gel de síl¡ce, EtOAc/hexanos 0 ~ 100%). El sól¡do blanquec¡no obten¡do se tr¡turó con EtOAc/hexanos, se f¡ltró, se d¡solv¡ó en MeCN/H2O cal¡ente (10 ml/10 ml) y luego se l¡of¡l¡zó para proporc¡onar el compuesto del título 1-20 en forma de un sól¡do blanco (78 mg, 58%). 1H RMN (300 MHz, DMSO-d6) 8: 11,90 (s, 1H), 7,74 (s, 1H), 7,68 (s, 1H), 6,98 (d, J = 7,7 Hz, 1H), 6,95 (s, 1H)), 6,90 (d, J = 7,9 Hz, 1H), 5,95 (d, J = 7,9 Hz, 1H), 4,65 (m, 1H), 3,88 (s, 3H), 3,58 (s, 3H), 1,48 (d, J = 6,9 Hz, 3H). LCMS (Método 3): Ta 4,98 m¡n, m/z 385 [M+H]+.
Ejemplo 30 - 5-(((S)-1-(6-cloro-2-oxo-7-((R)-1-(piridin-2-il)etoxi)-1,2-dihidroquinolina-3-yl)etil)amino)-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo (1 -26 )

Una mezcla de 5-fluoro-1-met¡l-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡tr¡lo III-1 (35,2 mg, 0,231 mmol) y 3-((S)-1
am¡noet¡lo)-6-cloro-7-((R)-1-(p¡rid¡n-2-¡l)etox¡)qu¡nol¡n-2(1H)-ona clorhidrato 11-14 (80 mg, 0,210 mmol) II-8 se trató con DMSO (1,5 ml) y DIEA (111 ml, 0,636 mmol). La solución se agitó a 110°C durante cinco horas. La muestra se mezcló con agua (20 ml) y se extrajo con DCM (2 x 15 ml). Los extractos se lavaron con agua (2 x 20 ml), se secaron (Na2SO4) y se filtraron, se añadió gel de sílice y el disolvente se evaporó a presión reducida. El material se cromatografió por Biotage MPLC (10 g columna de gel de sílice) con 0 a 3,4% de MeOH en hexanos. El material así obtenido se disolvió en MeCN (2 ml), se trató con agua (1 ml), se congeló en un baño de hielo seco/acetona y se liofilizó para proporcionar el compuesto del título (I-26) (32,7 mg, 0,069 mmol, 33% de rendimiento, HPLC pureza 100% a 220 nm) como un sólido blanco. 1H RMN (300 MHz, DMSO-d6): 8 ppm 11,75 (s, 1 H), 8,55 - 8,62 (m, 1 H), 7,80 (dd, J = 7,50, 7,50 Hz, 1 H), 7,74 (s, 1 H), 7,64 (s, 1 H), 7,39 (d, J = 7,62 Hz, 1 H), 7,32 (dd, J = 7,48, 4,84 Hz, 1 H), 6,96 (d, J = 7,62 Hz, 1 H), 6,82 -6,89 (m, 2 H), 5,93 (d, J = 7,92 Hz, 1H), 5,50 (q, J = 6,16 H z, 1 H), 4,61 (s, 1 H), 3,57 (s, 3 H), 1,66 (d, J = 6,16 Hz, 3 H), 1,44 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): Ta 2,61 min., m/z 475,9 [M+H]+.
Ejemplo 31 -- (S)-5-((1-(6-doro-7-(ddopropMmetoxi)-2-oxo-1,2-dihidroqumoMn-3-N)etM)ammo)-1-metM-6-oxo-1,6-dihidropiridin-2-carbonitrilo (1-27)

Una solución de 5-fluoro-1-metil-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡trilo 111-1 (18,3 mg, 0,120 mmol) y (S)- 3-(1-am¡noet¡lo)-6-cloro-7-(cicloprop¡lmetox¡)qu¡nol¡n-2(1H)-ona clorhidrato II-15 (35 mg, 0,106 mmol) se trató con DMSO (0,8 ml) y DIEA (57 ml, 0,326 mmol). La solución se agitó a 110°C durante 3,5 horas. La muestra se mezcló con agua (20 ml) y se extrajo con DCM (2 x 10 ml). Los extractos combinados se lavaron con agua (2 x 20 ml), se secaron (Na2SO4) y se filtraron, se añadió gel de sílice y el disolvente se evaporó a presión reducida. El material se cromatografió por Biotage MPLC (10 g de columna de gel de sílice) con 0 a 70% de EtOAc en hexanos. El material así obtenido se disolvió en MeCN (0,8 ml), se trató con agua (0,4 ml), se congeló en un baño de hielo seco/acetona y se liofilizó para proporcionar el compuesto del título (I-27) (23,9 mg, 0,056 mmol). Rendimiento del 52,9%, pureza por HPLC >99% a 220 nm) como un sólido blanco. 1H RMN (300 MHz, DMSO-d6): 8 ppm 11,83 (s, 1 H), 7,73 (s, 1 H), 7,67 (s, 1 H), 6,97 (d, J = 7,92 Hz, 1 H), 6,92 (s, 1 H), 6,89 (d, J = 7,92 Hz, 1 H), 5,95 (d, J = 7,92 Hz, 1 H), 4,61 -4,70 (m, 1 H), 3,92 (d, J = 6,74 Hz, 2 H), 3,58 (s, 3 H), 1,48 (d, J = 6,74 Hz, 3 H), 1,21 - 1,33 (m, 1 H), 0,56 - 0,65 (m, 2 H), 0,34 - 0,44 (m, 2 H). LCMS (Método 1): Ta 2,61 min., m/z 424,9 [M+H]+.
Ejemplo 32 - 5-((1-(6-doro-7-((3,3-difluoroddobutN)metoxi)-2-oxo-1,2-dihidroqumoMn-3-N)etN)ammo)-1-metN-6-oxo-1,6-dihidropiridin-2-carbonitrilo (1-28)
Una mezcla de 5-fluoro-1-metil-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡trilo III-1 (26,7 mg, 0,176 mmol) y 3-(1-am¡noet¡l)-6-cloro-7-((3,3-difluoroc¡clobut¡l)metox¡)qu¡nol¡n-2(1H)-ona clorhidrato II-16 (59,7 mg, 0,157 mmol) se trató con DMSO (1 ml) y DlEA (84 pl, 0,481 mmol). La solución se agitó a 110°C durante ocho horas. LCMS indicó que la reacción se había completado. La muestra se mezcló con agua (15 ml) y se extrajo con DCM (3 x 10 ml). Los extractos se secaron (Na2SO4), se filtraron, se trataron con gel de sílice y se evaporaron a presión reducida. El material se cromatografió por Biotage MPLC (10 g columna de gel de sílice, 0 a 75% en EtOAc en hexanos) para proporcionar el compuesto del título 1-28 (40,5 mg, 0,085 mmol, 54.2% de rendimiento, HPLC pureza 100% a 220 nm) como un sólido blanquecino. 1H RMN (300 MHz, DMSO-d6): 8 ppm 11,90 (s, 1 H), 7,76 (s, 1 H), 7,68 (s, 1 H), 6,97 (d, J = 7,62 Hz, 1 H), 6,94 (s, 1 H), 6,91 (d, J = 7,62 Hz, 1 H), 5,95 (d, J = 7,62 Hz, 1 H), 4,65 (quin, J = 6,82 Hz, 1 H), 4,12 (d,J = 4,10 Hz, 2 H), 3,58 (s, 3 H), 2,52 - 2,80 (m, 5 H), 1,48 (d, J = 6,74 Hz, 3 H). LCMS (Método 4): Ta
1,51 min., m/z 475,1 [M+H]+.
Ejemplo 33 - (S)-5-((1-(6-doro-7-isopropoxi-2-oxo-1,2-dihidroqumolm-3-N)etM)ammo)-1-metN-6-oxo-1,6-dihidropiridina-2-carbonitrilo (1-29)

Una mezcla de (S)-3-(1-am¡noet¡l)-6-cloro-7-¡sopropox¡qumolm-2(1H)-ona clorhidrato II-18 (128 mg, 0,4 mmol, 1 eq.), 5-fluoro-1-metil-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡trilo (67 mg, 0,44 mmol, 1,1 eq.) y DIPEA (148 mg, 1,2 mmol, 3 eq.) en 4 ml de DMSO se calentó a 130-135°C durante 80 m¡nutos. La mezcla de reacc¡ón se vert¡ó luego en agua y el sól¡do resultante se recog¡ó y se enjuagó con agua. La cromatografía en 3,5 g de gel de síl¡ce usando un grad¡ente de DCM a DCM/EtOH (98/2) segu¡do de tr¡turac¡ón con H2O/MeOH proporc¡onó I-29 (93 mg, 56%) como un sól¡do blanquec¡no. 1H RMN (300 MHz, DMSO-cfe) 8: 11,80 (ancho s, 0,7H), 7,72 (s, 1H), 7,66 (s, 1H), 6,98 (s, 1H), 6,96 (s, 1H), 6,89 (d, J = 7,41, 1H), 5,93 (d, J = 7,68, 1H), 4,62 (m, 2H), 3,57 (s, 3H), 1,47 (d, J = 7,41, 3H), 1,33 (d,J = 6,03, 6H). LC/MS (Método 3), Ta 5,5 m¡n, m/z 413 [M+H]+.
Ejemplo 34 -- (S)-5-((1-(6-doro-8-fluoro-2-oxo-1,2-dihidroqumolm-3-N)etM)ammo)-1-metN-6-oxo-1,6-dihidropiridina-2-carbonitrilo (1-30)

Una soluc¡ón de (S)-3-(1-am¡noet¡l)-6-cloro-8-fluoroqu¡nol¡n-2(1H)-ona clorh¡drato II-17 (91,7 mg, 0,331 mmol) y 5-fluoro-1-met¡l-6-oxo-1,6-d¡h¡drop¡r¡d¡n-2-carbon¡tr¡lo III-1 (56,8 mg, 0,373 mmol) en DMSO (2,0 ml) se trató con DlEA (172 ml, 0,985 mmol)) y se ag¡tó a 110°C durante cuatro horas. La muestra se añad¡ó a agua (30 ml) y el prec¡p¡tado resultante se extrajo con DCM (2 x 20 ml) y EtOAc (10 ml). Los extractos orgán¡cos comb¡nados se secaron (Na2SO4), se f¡ltraron, se trataron con gel de síl¡ce y se evaporaron a pres¡ón reduc¡da. El mater¡al se cromatograf¡ó por B¡otage MPLC (10 g de columna de gel de síl¡ce) con 0 a 45% de EtOAc en hexanos, con eluc¡ón ¡socrát¡ca cuando los p¡cos se desprend¡eron. Las fracc¡ones del producto se comb¡naron, se lavaron con agua (2 x 30 ml) y se evaporaron a pres¡ón reduc¡da. El res¡duo se d¡solv¡ó en MeCN (4 ml) y agua (2 ml), se congeló (h¡elo seco y baño de acetona) y se l¡of¡l¡zó para proporc¡onar el compuesto del título I-30 (62,0 mg, 0,166 mmol, 50,3% de rend¡m¡ento, pureza por Hp LC 100 % a 220 nm) como un sól¡do amar¡llo gr¡sáceo. 1H RMN (300 MHz, DMSO-d6): 8 ppm 12,15 (s, 1 H), 7,77 (s, 1 H), 7,56 - 7,65 (m, 2 H), 6,97 (d, J = 7,92 Hz, 1 H), 6,93 (d, J = 7,62 Hz, 1 H), 5,94 (d, J = 7,92 Hz, 1 H), 4,61 - 4,75 (m, 1 H), 3,58 (s, 3 H), 1,50 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): Ta 2,39 m¡n., m/z 373,0 [M+H]+.
Ejemplo 35 --(S)-5-((1-(6-cloro-2-oxo-1,2-dihidro-1,8-naftiridin-3-il)etil)amino)-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo (I-31)

La mezcla de (S)-3-(1-am¡noet¡l)-6-cloro-1,8-naft¡r¡d¡n-2(1H)-ona II-11 (100 mg, 0,447 mmol), 5-fluoro-1metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo III-1 (82 mg, 0,537 mmol) y DIEA (0,234 ml, 1,341 mmol) en DMSO (1 ml) se calentó a 110°C durante dos horas. LC-MS mostró la formación del producto. La mezcla de reacción se enfrió luego a temperatura ambiente, seguido de la adición de agua y filtración. La purificación de biotipos del producto bruto con 0-10% de MeOH/DCM en una columna de 25 g produjo (S)-5-((1-(6-cloro-2-oxo-1,2-dihidro-1,8-naftiridina)-3-il)etil)amino)-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo I-31 (53,8 mg, 33.8%). 1H RMN (300 MHz, DMSO- efe) 8 ppm 12,52 (s, 1 H), 8,49 (d, J = 2,64 Hz, 1 H), 8,24 (d, J = 2,64 Hz, 1 H), 7,72 (s, 1 H), 6,71 - 7,07 (m, 2 H), 5,91 (d, J = 8,21 Hz, 1 H), 4,52 - 4,85 (m, 1 H), 3,46 - 3.74 (s, 3 H), 1,48 (d, J = 6,74 Hz, 3 H). LCMS (Método 1): Ta 2,22 min, m/z 356,01 [M+H]+.
Ejemplo 36-(s)-5-((1-(7-cloro-3-oxo-3,4-dihidroquinoxalin-2-il)etil)amino)-1-metil-6-oxo-1,6-dihidropiradina-2-carbonitrilo (1-33)

Al compuesto II-13 (59 mg, 0,175 mmol) en DMSO (5 ml) en un tubo sellado se le añadió 5-fluoro-1-metil-6-oxo-1,6-dihidropiridin-2-carbonitrilo III-1 (35 mg, 0,23 mmol) y DIEA (0,5 ml). La mezcla de reacción se calentó hasta 110°C y se agitó durante 3 h. La mezcla de reacción se enfrió luego a ta, se diluyó con agua (30 ml) y se extrajo con EtOAc (50 ml x 4). Las capas orgánicas combinadas se secaron (Na2SO4), se concentraron y se purificaron mediante C-18 ISc O inverso con agua (TFA al 0,1%) a CH3CN (TFA al 0,1%) para dar el compuesto del título (I-33) (22 mg, 34%) Como un sólido blanco. 1H RMN (300 MHz, DMSO-d6): 812,71 (s, 1H), 7,82 (d, J = 6,57 Hz, 1H), 7,90 (s, 1H), 7,81 (s, 1 H), 7,59 (d, J = 2,19 Hz, 1H), 7,59 (dd, J = 9,06 Hz, 2,19 Hz, 1H), 7,32 (d, J = 8,79 Hz, 1H), 7,05 (d, J = 7,71 Hz, 1H), 6,93 (d, J = 7,98 Hz, 1H), 6,31 (d, J = 7,98 Hz, 1H), 5,00 (m, 1H), 3,59 (s, 3H), 1,49 (d, J = 6,60 Hz, 3H). LCMS (Método 3): Ta 5,30 min, m/z 357,1 [M+H]+.


Tabla 5. Señal de LCMS y cambios químicos de RMN de cada compuesto enumerado en la Tabla 4.
-2--2--2-

(continuado)

(continuado)

Ejemplo 37 - Ensayo enzimático IDH1-R132H e IDH1-R132C
Los ensayos se realizaron en una placa negra de 384 pocilios. Se incubó una parte alícuota de 250 nL de compuesto con l0 ml de IDH1-R132H 30 nM o proteína recombinante IDH1-R132C l0 nM en tampón de ensayo (Tris 50 mM pH = 7,5, NaCl 150 mM, MgCh 5 mM, 0,1% (p/v) albúmina de suero bovino y 0,01% de Triton X-100) en cada pocillo a 25°C durante 15 minutos. Después de que la placa se centrifugó brevemente, se añadió a cada pocillo una alícuota de 10 pl de a-cetoglutarato 2 mM y una solución de NADPH 20 mM preparada en tampón de ensayo y la reacción se mantuvo a 25°C durante 45 minutos. Se añadió a cada pocillo una alícuota de 10 pl de solución de diaforasa (0,15 U/ml de diaforasa y 30 pM de resazurina en tampón de ensayo). La placa se mantuvo a 25°C durante 15 minutos y luego se leyó en un lector de placas con longitudes de onda de excitación y emisión a 535 nm y 590 nm, respectivamente. La CI50 de un compuesto dado se calculó ajustando la curva de respuesta a la dosis de inhibición del consumo de NADPH a una concentración dada con la ecuación logística de cuatro parámetros.
Ejemplo 38 - Ensayo celular de 2-HG utilizando células mutantes IDH1 de HCT116
Se cultivaron células mutantes IDH1-R132H e IDH1-R132C de HCT116 en medios de crecimiento (5A de McCoy, suero bovino fetal al 10%, solución antibiótica-antimicótica 1X y 0,3 mg/ml de G418) en 5% de CO2 en un incubador a 37°C. Para preparar el ensayo, las células se tripsinizaron y se resuspendieron en medios de ensayo (5A de McCoy sin L-glutamina, suero bovino fetal al 10%, solución antibiótica antimicótica 1X y 0,3 mg/ml de G418). Se transfirió una alícuota de 10.000 células/100 ml a cada pocillo de una placa de cultivo de tejidos transparente de 96 pocillos. Las células se incubaron en CO2 al 5% a 37°C en una incubadora durante la noche para permitir una correcta unión celular. Luego se agregó una parte alícuota de 50 pl de compuesto que contiene medios de ensayo a cada pocillo y la placa de ensayo se mantuvo en CO2 al 5% a 37°C en una incubadora durante 24 horas. Luego se retiró el medio de cada pocillo y se agregó a cada pocillo 150 pl de una mezcla de metanol/agua (80/20 v/v). Las placas se mantuvieron a -80°C en el congelador durante la noche para permitir la lisis celular completa. Se analizó una alícuota de 125 ml de sobrenadante extraído mediante espectrometría de alta masa de RapidFire (Agilent) para determinar el nivel de 2-HG celular. La CI50 de un compuesto dado se calculó ajustando la curva de respuesta a la dosis de la inhibición celular 2-HG a una concentración dada con la ecuación logística de cuatro parámetros.
La Tabla 6 a continuación proporciona la actividad de cada compuesto según la leyenda de que "++++" indica una inhibición a una concentración <0,01 pM; "+++" indica la inhibición a una concentración entre 0,01 pM y 0,1 pM del compuesto descrito; "++" indica la inhibición a una concentración de 0,1 pM a 1 pM del compuesto descrito; y "+" indica la inhibición a una concentración > 1 pM para la enzima IDH1 R132H, HCT116 IDH1 R132H y HCT116 IDH1 R132C. Para la enzima IDH1 R132C, "++++" indica una inhibición a una concentración <0,1 mM; "+++" indica la inhibición a una concentración entre 0,1 mM y 1 mM del compuesto descrito; "++" indica la inhibición a una concentración de 1 mM a 10 mM del compuesto descrito; y "+" indica inhibición a una concentración> 10 mM.
Tabla 6 Resultados de los compuestos ilustrativos de Fórmula I en los ensayos IDH1-R132H, IDH1-R132C, IDH1-MS-HTC116-R132H, y IDH1-MS-HTC116-R132C.

(continuado)

Claims (11)
1. Una composición farmacéutica que comprende el compuesto de Fórmula I-13 o una sal farmacéuticamente aceptable del mismo y un portador farmacéuticamente aceptable:

en donde la composición contiene de aproximadamente el 0,1% a aproximadamente el 99% del compuesto de Fórmula I-13 en peso o volumen.
2. La composición farmacéutica de acuerdo con la reivindicación 1, en donde la composición contiene de aproximadamente el 5% a aproximadamente el 90% del compuesto de Fórmula I-13 en peso o volumen.
3. La composición farmacéutica de acuerdo con la reivindicación 1 o la reivindicación 2, en donde el porcentaje del compuesto de Fórmula I-13 presente en la composición se mide en peso.
4. La composición farmacéutica de acuerdo con la reivindicación 1, en donde el compuesto de Fórmula I-13 está presente en una pureza enantiomérica (% ee) mayor del 98% como se determina por análisis de HPLC quiral.
5. La composición farmacéutica de acuerdo con la reivindicación 1, en donde la composición es un comprimido o cápsula de gelatina.
6. Una mezcla que comprende el compuesto de Fórmula I-13 como se define en la reivindicación 1 y un compuesto de Fórmula II-1 y un compuesto de Fórmula III:

Hal = Cl o F.
7. La mezcla como se reivindica en la reivindicación 6, en donde el compuesto de Fórmula III es un compuesto de Fórmula III-1:

8. Un proceso para preparar un compuesto de Fórmula I-13 o una sal farmacéuticamente aceptable del mismo:

en donde dicho proceso comprende el paso de hacer reaccionar un compuesto de Fórmula II-1 con un compuesto de Fórmula III:

Hal = Cl o F.
9. El proceso de la reivindicación 8, en donde el compuesto de Fórmula II-1 tiene un exceso enantiomérico (% ee) mayor del 98%.
10. El proceso de la reivindicación 8 o la reivindicación 9 en donde el compuesto de Fórmula III es un compuesto de Fórmula III-1:

11. El proceso de cualquiera de las reivindicaciones 8-10, en donde la reacción del compuesto de Fórmula II-1 con el compuesto de Fórmula III se lleva a cabo en condiciones adecuadas para la sustitución nucleófila.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462053006P | 2014-09-19 | 2014-09-19 | |
| US201562128089P | 2015-03-04 | 2015-03-04 | |
| US201562150812P | 2015-04-21 | 2015-04-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2790640T3 true ES2790640T3 (es) | 2020-10-28 |
Family
ID=54291606
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20157755T Active ES2953347T3 (es) | 2014-09-19 | 2015-09-18 | Derivados de piridin-2(1H)-ona quinolinona como inhibidores de isocitrato deshidrogenasa mutante |
| ES18198969T Active ES2790640T3 (es) | 2014-09-19 | 2015-09-18 | Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante |
| ES15778433T Active ES2704897T3 (es) | 2014-09-19 | 2015-09-18 | Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20157755T Active ES2953347T3 (es) | 2014-09-19 | 2015-09-18 | Derivados de piridin-2(1H)-ona quinolinona como inhibidores de isocitrato deshidrogenasa mutante |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15778433T Active ES2704897T3 (es) | 2014-09-19 | 2015-09-18 | Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante |
Country Status (36)
| Country | Link |
|---|---|
| US (8) | US9834539B2 (es) |
| EP (4) | EP3194376B1 (es) |
| JP (1) | JP6648115B2 (es) |
| KR (1) | KR102209667B1 (es) |
| CN (3) | CN117695280A (es) |
| AU (3) | AU2015317329B2 (es) |
| BR (1) | BR112017005238B1 (es) |
| CA (1) | CA2961817C (es) |
| CL (1) | CL2017000658A1 (es) |
| CO (1) | CO2017003241A2 (es) |
| CY (2) | CY1121149T1 (es) |
| DK (2) | DK3447050T3 (es) |
| EA (1) | EA034336B1 (es) |
| EC (1) | ECSP17022933A (es) |
| ES (3) | ES2953347T3 (es) |
| FI (1) | FI3733662T3 (es) |
| HR (1) | HRP20200666T1 (es) |
| HU (2) | HUE062424T2 (es) |
| IL (3) | IL292608A (es) |
| LT (2) | LT3194376T (es) |
| MA (2) | MA40481A (es) |
| ME (1) | ME03776B (es) |
| MX (2) | MX2017003404A (es) |
| MY (2) | MY176250A (es) |
| NZ (2) | NZ758641A (es) |
| PE (1) | PE20171056A1 (es) |
| PH (1) | PH12017500517A1 (es) |
| PL (3) | PL3194376T3 (es) |
| PT (3) | PT3447050T (es) |
| RS (2) | RS60140B1 (es) |
| SA (1) | SA517381129B1 (es) |
| SG (1) | SG11201702194SA (es) |
| SI (2) | SI3194376T1 (es) |
| TW (1) | TWI686390B (es) |
| WO (1) | WO2016044789A1 (es) |
| ZA (2) | ZA201702127B (es) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102985557B (zh) | 2009-03-13 | 2018-06-15 | 安吉奥斯医药品有限公司 | 用于细胞增殖相关病症的方法和组合物 |
| CA2793835C (en) | 2009-10-21 | 2021-07-20 | Agios Pharmaceuticals, Inc. | Methods and compositions for cell-proliferation-related disorders |
| RS57843B1 (sr) | 2014-02-06 | 2018-12-31 | Heptares Therapeutics Ltd | Biciklična aza jedinjenja kao agonisti muskarinskog receptora m1 |
| BR112017005444A2 (pt) | 2014-09-19 | 2018-04-24 | Bayer Pharma AG | indazoles substituídos por benzil como inibidores bub1. |
| JP6648116B2 (ja) * | 2014-09-19 | 2020-02-14 | フォーマ セラピューティクス,インコーポレイテッド | 変異イソクエン酸デヒドロゲナーゼ阻害剤としてのキノリノンピリミジン組成物 |
| US9771349B2 (en) | 2014-09-19 | 2017-09-26 | Forma Therapeutics, Inc. | Pyridinyl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| CA2961807A1 (en) | 2014-09-19 | 2016-03-24 | Forma Therapeutics, Inc. | Phenyl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| JP6648115B2 (ja) | 2014-09-19 | 2020-02-14 | フォーマ セラピューティクス,インコーポレイテッド | 変異イソクエン酸デヒドロゲナーゼ阻害剤としてのピリジン−2(1h)−オンキノリノン誘導体 |
| CA2971872C (en) | 2014-12-22 | 2023-10-10 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Mutant idh1 inhibitors useful for treating cancer |
| US9624175B2 (en) | 2015-04-21 | 2017-04-18 | Forma Therapeutics, Inc. | Fused-bicyclic aryl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| WO2016171756A1 (en) | 2015-04-21 | 2016-10-27 | Forma Therapeutics, Inc. | Quinolinone five-membered heterocyclic compounds as mutant-isocitrate dehydrogenase inhibitors |
| ES2919280T3 (es) | 2017-05-04 | 2022-07-22 | Bayer Cropscience Ag | Derivados de 2-{[2-(feniloximetil)piridin-5-il]oxi}-etanamina y compuestos relacionados como pesticidas, por ejemplo para la protección de plantas |
| US11311527B2 (en) | 2018-05-16 | 2022-04-26 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH-1) |
| US11013734B2 (en) | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Treating patients harboring an isocitrate dehydrogenase-1 (IDH-1) mutation |
| EP3720442B1 (en) | 2018-05-16 | 2022-12-28 | Forma Therapeutics, Inc. | Inhibiting mutant idh-1 |
| FI3720442T3 (fi) | 2018-05-16 | 2023-03-17 | Forma Therapeutics Inc | Mutantti idh-1:n inhibitio |
| US11013733B2 (en) * | 2018-05-16 | 2021-05-25 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mlDH-1) |
| WO2020232381A1 (en) | 2019-05-16 | 2020-11-19 | Forma Therapeutics, Inc. | INHIBITING MUTANT ISOCITRATE DEHYDROGENASE 1 (mIDH-1) |
| WO2019222551A1 (en) | 2018-05-16 | 2019-11-21 | Forma Therapeutics, Inc. | Solid forms of ((s)-5-((1-(6-chloro-2-oxo-1,2-dihydroquinolin-3-yl)ethyl)amino)-1-methyl-6-oxo-1,6-dihydropyridine-2-carbonitrile |
| TW202104207A (zh) * | 2019-04-17 | 2021-02-01 | 美商健生生物科技公司 | 二氫乳清酸脫氫酶抑制劑 |
| US11566013B1 (en) * | 2019-11-20 | 2023-01-31 | Forma Therapeutics, Inc. | Inhibiting mutant isocitrate dehydrogenase 1 (mIDH1) |
| CN112321571B (zh) * | 2020-10-27 | 2022-06-03 | 浙江工业大学 | 2-呋喃-喹啉-4-甲酰胺类化合物及其应用 |
| CN112341389B (zh) * | 2020-10-27 | 2022-07-29 | 浙江工业大学 | 一种含氮芳杂环取代的喹啉甲酰胺类衍生物及其应用 |
| CN115850240B (zh) * | 2022-12-28 | 2023-09-19 | 北京康立生医药技术开发有限公司 | 一种治疗急性髓系白血病药物奥卢他西尼的合成方法 |
Family Cites Families (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL99731A0 (en) | 1990-10-18 | 1992-08-18 | Merck & Co Inc | Hydroxylated pyridine derivatives,their preparation and pharmaceutical compositions containing them |
| US5262564A (en) | 1992-10-30 | 1993-11-16 | Octamer, Inc. | Sulfinic acid adducts of organo nitroso compounds useful as retroviral inactivating agents anti-retroviral agents and anti-tumor agents |
| US20030105124A1 (en) | 2000-04-27 | 2003-06-05 | Susan Beth Sobolov-Jaynes | Substituted benzolactam compounds |
| CA2488635C (en) | 2002-06-12 | 2012-10-23 | Abbott Laboratories | Antagonists of melanin concentrating hormone receptor |
| JPWO2004043936A1 (ja) | 2002-11-14 | 2006-03-09 | 協和醗酵工業株式会社 | Plk阻害剤 |
| RU2284325C2 (ru) | 2003-12-17 | 2006-09-27 | Общество С Ограниченной Ответственностью "Асинэкс Медхим" | Производные фенил-3-аминометил-хинолона-2 в качестве ингибиторов no-синтетазы, способ их получения, биологически активные соединения и фармацевтическая композиция на их основе |
| JPWO2005095382A1 (ja) | 2004-03-30 | 2007-08-16 | 協和醗酵工業株式会社 | 抗腫瘍剤 |
| WO2006054912A1 (fr) | 2004-11-18 | 2006-05-26 | Obchestvo S Ogranichennoy Otvetstvennost'u 'asineks Medhim' | Derives d'aryl(hetaryl)-3-aminomethylchinolone-2 utilises en tant qu'inhibiteurs de no-synthetase et de cyclo-oxygenase-2, procedes de leur fabrication et compositions pharmaceutiques sur leur base |
| TW200803855A (en) | 2006-02-24 | 2008-01-16 | Kalypsys Inc | Quinolones useful as inducible nitric oxide synthase inhibitors |
| CA2657287A1 (en) * | 2006-07-17 | 2008-01-24 | Merck & Co., Inc. | 1-hydroxy naphthyridine compounds as anti-hiv agents |
| JP2010043004A (ja) | 2006-12-06 | 2010-02-25 | Dainippon Sumitomo Pharma Co Ltd | 新規2環性複素環化合物 |
| CN101679321B (zh) | 2007-04-30 | 2012-10-03 | 普罗米蒂克生物科学公司 | 三嗪衍生物、含所述衍生物的组合物以及所述衍生物在制备用于治疗癌症和自身免疫性疾病药物中的应用 |
| JP2012529511A (ja) | 2009-06-08 | 2012-11-22 | アブラクシス バイオサイエンス リミテッド ライアビリティー カンパニー | トリアジン誘導体類及びそれらの治療応用 |
| EP2509600B1 (en) * | 2009-12-09 | 2017-08-02 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds for use in the treatment of cancer characterized as having an idh mutation |
| US9073941B2 (en) | 2010-06-28 | 2015-07-07 | Academia Sinica | Compounds and methods for treating tuberculosis infection |
| CN102558049B (zh) * | 2010-12-17 | 2015-02-04 | 中国科学院上海药物研究所 | 一类双香豆素类化合物及其制备方法和用途 |
| US20120184562A1 (en) | 2011-01-19 | 2012-07-19 | Kin-Chun Luk | 1,6- and 1,8-naphthyridines |
| US20120184548A1 (en) | 2011-01-19 | 2012-07-19 | Romyr Dominique | Carboxylic acid aryl amides |
| US9464065B2 (en) | 2011-03-24 | 2016-10-11 | The Scripps Research Institute | Compounds and methods for inducing chondrogenesis |
| CN102827170A (zh) | 2011-06-17 | 2012-12-19 | 安吉奥斯医药品有限公司 | 治疗活性组合物和它们的使用方法 |
| CN103814020B (zh) * | 2011-06-17 | 2017-07-14 | 安吉奥斯医药品有限公司 | 治疗活性组合物和它们的使用方法 |
| EA025183B1 (ru) | 2011-09-27 | 2016-11-30 | Новартис Аг | 3-пиримидин-4-ил-оксазолидин-2-оны в качестве ингибиторов мутантной idh |
| AU2012358317B2 (en) | 2011-12-21 | 2017-12-14 | Indiana University Research And Technology Corporation | Anti-cancer compounds targeting Ral GTPases and methods of using the same |
| DK2800743T3 (en) * | 2012-01-06 | 2018-06-14 | Agios Pharmaceuticals Inc | THERAPEUTIC ACTIVE RELATIONS AND PROCEDURES FOR USE THEREOF |
| BR112015022483A2 (pt) * | 2013-03-14 | 2017-07-18 | Novartis Ag | 3-pirimidin-4-il-oxazolidin-2-onas como inibidores de idh mutante |
| US10478445B2 (en) | 2013-07-03 | 2019-11-19 | Georgetown University | Boronic acid derivatives of resveratrol for activating deacetylase enzymes |
| US9957235B2 (en) | 2014-02-11 | 2018-05-01 | Bayer Pharma Aktiengesellschaft | Benzimidazol-2-amines as mIDH1 inhibitors |
| US9771349B2 (en) | 2014-09-19 | 2017-09-26 | Forma Therapeutics, Inc. | Pyridinyl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| CA2961807A1 (en) | 2014-09-19 | 2016-03-24 | Forma Therapeutics, Inc. | Phenyl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| JP6648116B2 (ja) * | 2014-09-19 | 2020-02-14 | フォーマ セラピューティクス,インコーポレイテッド | 変異イソクエン酸デヒドロゲナーゼ阻害剤としてのキノリノンピリミジン組成物 |
| JP6648115B2 (ja) * | 2014-09-19 | 2020-02-14 | フォーマ セラピューティクス,インコーポレイテッド | 変異イソクエン酸デヒドロゲナーゼ阻害剤としてのピリジン−2(1h)−オンキノリノン誘導体 |
| CA2971872C (en) | 2014-12-22 | 2023-10-10 | The United States Of America As Represented By The Secretary, Department Of Health And Human Services | Mutant idh1 inhibitors useful for treating cancer |
| GB2533925A (en) | 2014-12-31 | 2016-07-13 | Univ Bath | Antimicrobial compounds, compositions and methods |
| US9624175B2 (en) | 2015-04-21 | 2017-04-18 | Forma Therapeutics, Inc. | Fused-bicyclic aryl quinolinone derivatives as mutant-isocitrate dehydrogenase inhibitors |
| WO2016171756A1 (en) | 2015-04-21 | 2016-10-27 | Forma Therapeutics, Inc. | Quinolinone five-membered heterocyclic compounds as mutant-isocitrate dehydrogenase inhibitors |
| WO2017019429A1 (en) | 2015-07-27 | 2017-02-02 | Eli Lilly And Company | 7-phenylethylamino-4h-pyrimido[4,5-d][1,3]oxazin-2-one compounds and theit use as mutant idh1 inhibitors |
| US10137130B2 (en) | 2016-02-26 | 2018-11-27 | Celgene Corporation | Methods of treatment of malignancies |
| JP6682043B2 (ja) | 2016-06-06 | 2020-04-15 | イーライ リリー アンド カンパニー | 変異体idh1阻害剤 |
| WO2017223202A1 (en) | 2016-06-22 | 2017-12-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Thiazole derivatives useful as mutant idh1 inhibitors for treating cancer |
| UA123640C2 (uk) | 2016-12-16 | 2021-05-05 | Елі Ліллі Енд Компані | Сполука 7-фенілетиламіно-4н-піримідо[4,5-d][1,3]оксазин-2-ону (варіанти) як інгібітор мутантів idh1 та idh2 |
-
2015
- 2015-09-18 JP JP2017515059A patent/JP6648115B2/ja active Active
- 2015-09-18 US US14/858,167 patent/US9834539B2/en active Active
- 2015-09-18 FI FIEP20157755.8T patent/FI3733662T3/fi active
- 2015-09-18 DK DK18198969.0T patent/DK3447050T3/da active
- 2015-09-18 EP EP15778433.1A patent/EP3194376B1/en active Active
- 2015-09-18 ES ES20157755T patent/ES2953347T3/es active Active
- 2015-09-18 SG SG11201702194SA patent/SG11201702194SA/en unknown
- 2015-09-18 MY MYPI2017700895A patent/MY176250A/en unknown
- 2015-09-18 MX MX2017003404A patent/MX2017003404A/es active IP Right Grant
- 2015-09-18 EP EP20157755.8A patent/EP3733662B1/en active Active
- 2015-09-18 RS RS20200418A patent/RS60140B1/sr unknown
- 2015-09-18 PE PE2017000471A patent/PE20171056A1/es unknown
- 2015-09-18 ES ES18198969T patent/ES2790640T3/es active Active
- 2015-09-18 MA MA040481A patent/MA40481A/fr unknown
- 2015-09-18 PT PT181989690T patent/PT3447050T/pt unknown
- 2015-09-18 LT LTEP15778433.1T patent/LT3194376T/lt unknown
- 2015-09-18 MA MA053352A patent/MA53352A/fr unknown
- 2015-09-18 ME MEP-2020-78A patent/ME03776B/me unknown
- 2015-09-18 SI SI201530537T patent/SI3194376T1/sl unknown
- 2015-09-18 TW TW104131044A patent/TWI686390B/zh active
- 2015-09-18 AU AU2015317329A patent/AU2015317329B2/en active Active
- 2015-09-18 CN CN202311265288.0A patent/CN117695280A/zh active Pending
- 2015-09-18 CA CA2961817A patent/CA2961817C/en active Active
- 2015-09-18 EP EP23176702.1A patent/EP4257131A3/en active Pending
- 2015-09-18 IL IL292608A patent/IL292608A/en unknown
- 2015-09-18 LT LTEP18198969.0T patent/LT3447050T/lt unknown
- 2015-09-18 NZ NZ758641A patent/NZ758641A/en unknown
- 2015-09-18 CN CN202010391529.6A patent/CN111909130B/zh active Active
- 2015-09-18 CN CN201580050441.5A patent/CN107001328B/zh active Active
- 2015-09-18 NZ NZ73037315A patent/NZ730373A/en unknown
- 2015-09-18 DK DK15778433.1T patent/DK3194376T3/en active
- 2015-09-18 WO PCT/US2015/051055 patent/WO2016044789A1/en active Application Filing
- 2015-09-18 RS RS20181586A patent/RS58184B1/sr unknown
- 2015-09-18 PL PL15778433T patent/PL3194376T3/pl unknown
- 2015-09-18 HU HUE20157755A patent/HUE062424T2/hu unknown
- 2015-09-18 ES ES15778433T patent/ES2704897T3/es active Active
- 2015-09-18 PL PL20157755.8T patent/PL3733662T3/pl unknown
- 2015-09-18 BR BR112017005238-5A patent/BR112017005238B1/pt active IP Right Grant
- 2015-09-18 EP EP18198969.0A patent/EP3447050B1/en active Active
- 2015-09-18 PT PT201577558T patent/PT3733662T/pt unknown
- 2015-09-18 PT PT15778433T patent/PT3194376T/pt unknown
- 2015-09-18 KR KR1020177010585A patent/KR102209667B1/ko active IP Right Grant
- 2015-09-18 EA EA201790657A patent/EA034336B1/ru unknown
- 2015-09-18 PL PL18198969T patent/PL3447050T3/pl unknown
- 2015-09-18 SI SI201531117T patent/SI3447050T1/sl unknown
- 2015-09-18 HU HUE15778433A patent/HUE041460T2/hu unknown
- 2015-09-18 MY MYPI2020001463A patent/MY197533A/en unknown
-
2017
- 2017-03-07 US US15/452,256 patent/US20170174658A1/en not_active Abandoned
- 2017-03-14 IL IL251163A patent/IL251163B/en active IP Right Grant
- 2017-03-15 MX MX2019013203A patent/MX2019013203A/es unknown
- 2017-03-17 CL CL2017000658A patent/CL2017000658A1/es unknown
- 2017-03-17 PH PH12017500517A patent/PH12017500517A1/en unknown
- 2017-03-19 SA SA517381129A patent/SA517381129B1/ar unknown
- 2017-03-27 ZA ZA2017/02127A patent/ZA201702127B/en unknown
- 2017-04-03 CO CONC2017/0003241A patent/CO2017003241A2/es unknown
- 2017-04-13 EC ECIEPI201722933A patent/ECSP17022933A/es unknown
-
2018
- 2018-04-27 US US15/964,844 patent/US10414752B2/en active Active
-
2019
- 2019-01-21 CY CY20191100075T patent/CY1121149T1/el unknown
- 2019-03-01 US US16/290,240 patent/US10550098B2/en active Active
- 2019-04-17 ZA ZA2019/02446A patent/ZA201902446B/en unknown
- 2019-12-12 US US16/712,951 patent/US10889567B2/en active Active
- 2019-12-16 AU AU2019283765A patent/AU2019283765B2/en active Active
-
2020
- 2020-04-24 HR HRP20200666TT patent/HRP20200666T1/hr unknown
- 2020-05-04 CY CY20201100407T patent/CY1122865T1/el unknown
- 2020-11-23 US US17/101,018 patent/US11498913B2/en active Active
-
2021
- 2021-04-18 IL IL282363A patent/IL282363B/en unknown
- 2021-08-10 AU AU2021215141A patent/AU2021215141B2/en active Active
-
2022
- 2022-11-09 US US17/984,069 patent/US20230125739A1/en not_active Abandoned
-
2023
- 2023-11-09 US US18/506,060 patent/US20240150319A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2790640T3 (es) | Derivados de piridin-2-(1H)-ona-quinolinona como inhibidores de isocitrato dehidrogenasa mutante | |
| ES2706525T3 (es) | Derivados de piridinilquinolinona como inhibidores de isocitrato deshidrogenasa mutante | |
| ES2706888T3 (es) | Referencia cruzada a aplicaciones relacionadas |