ES2787225T3 - Métodos para ajustar los niveles de producción de carotenoides y composiciones en géneros de Rhodosporidium y Rhodotorula - Google Patents
Métodos para ajustar los niveles de producción de carotenoides y composiciones en géneros de Rhodosporidium y Rhodotorula Download PDFInfo
- Publication number
- ES2787225T3 ES2787225T3 ES15870471T ES15870471T ES2787225T3 ES 2787225 T3 ES2787225 T3 ES 2787225T3 ES 15870471 T ES15870471 T ES 15870471T ES 15870471 T ES15870471 T ES 15870471T ES 2787225 T3 ES2787225 T3 ES 2787225T3
- Authority
- ES
- Spain
- Prior art keywords
- polynucleotides
- fungal host
- genetic manipulation
- carotenoids
- gene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 235000021466 carotenoid Nutrition 0.000 title claims abstract description 93
- 150000001747 carotenoids Chemical class 0.000 title claims abstract description 93
- 238000000034 method Methods 0.000 title claims abstract description 54
- 241000223252 Rhodotorula Species 0.000 title claims abstract description 34
- 238000004519 manufacturing process Methods 0.000 title claims abstract description 26
- 239000000203 mixture Substances 0.000 title claims abstract description 17
- 102000040430 polynucleotide Human genes 0.000 claims abstract description 87
- 108091033319 polynucleotide Proteins 0.000 claims abstract description 87
- 239000002157 polynucleotide Substances 0.000 claims abstract description 87
- 230000002538 fungal effect Effects 0.000 claims abstract description 64
- 108090000765 processed proteins & peptides Proteins 0.000 claims abstract description 30
- 229920001184 polypeptide Polymers 0.000 claims abstract description 29
- 102000004196 processed proteins & peptides Human genes 0.000 claims abstract description 29
- 230000015572 biosynthetic process Effects 0.000 claims abstract description 20
- HRQKOYFGHJYEFS-RZWPOVEWSA-N gamma-carotene Natural products C(=C\C=C\C(=C/C=C/C=C(\C=C\C=C(/C=C/C=1C(C)(C)CCCC=1C)\C)/C)\C)(\C=C\C=C(/CC/C=C(\C)/C)\C)/C HRQKOYFGHJYEFS-RZWPOVEWSA-N 0.000 claims abstract description 15
- OAIJSZIZWZSQBC-GYZMGTAESA-N lycopene Chemical compound CC(C)=CCC\C(C)=C\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C=C(/C)CCC=C(C)C OAIJSZIZWZSQBC-GYZMGTAESA-N 0.000 claims abstract description 12
- UPYKUZBSLRQECL-UKMVMLAPSA-N Lycopene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1C(=C)CCCC1(C)C)C=CC=C(/C)C=CC2C(=C)CCCC2(C)C UPYKUZBSLRQECL-UKMVMLAPSA-N 0.000 claims abstract description 11
- AIBOHNYYKWYQMM-MXBSLTGDSA-N torulene Chemical compound CC(C)=C\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C AIBOHNYYKWYQMM-MXBSLTGDSA-N 0.000 claims abstract description 10
- JEVVKJMRZMXFBT-XWDZUXABSA-N Lycophyll Natural products OC/C(=C/CC/C(=C\C=C\C(=C/C=C/C(=C\C=C\C=C(/C=C/C=C(\C=C\C=C(/CC/C=C(/CO)\C)\C)/C)\C)/C)\C)/C)/C JEVVKJMRZMXFBT-XWDZUXABSA-N 0.000 claims abstract description 9
- 235000012661 lycopene Nutrition 0.000 claims abstract description 9
- 239000001751 lycopene Substances 0.000 claims abstract description 9
- 229960004999 lycopene Drugs 0.000 claims abstract description 9
- ZCIHMQAPACOQHT-ZGMPDRQDSA-N trans-isorenieratene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/c1c(C)ccc(C)c1C)C=CC=C(/C)C=Cc2c(C)ccc(C)c2C ZCIHMQAPACOQHT-ZGMPDRQDSA-N 0.000 claims abstract description 9
- OENHQHLEOONYIE-UKMVMLAPSA-N all-trans beta-carotene Natural products CC=1CCCC(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C OENHQHLEOONYIE-UKMVMLAPSA-N 0.000 claims abstract description 6
- 235000013734 beta-carotene Nutrition 0.000 claims abstract description 6
- 239000011648 beta-carotene Substances 0.000 claims abstract description 6
- TUPZEYHYWIEDIH-WAIFQNFQSA-N beta-carotene Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2=CCCCC2(C)C TUPZEYHYWIEDIH-WAIFQNFQSA-N 0.000 claims abstract description 6
- 229960002747 betacarotene Drugs 0.000 claims abstract description 6
- OENHQHLEOONYIE-JLTXGRSLSA-N β-Carotene Chemical compound CC=1CCCC(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C OENHQHLEOONYIE-JLTXGRSLSA-N 0.000 claims abstract description 6
- HRQKOYFGHJYEFS-UHFFFAOYSA-N Beta psi-carotene Chemical compound CC(C)=CCCC(C)=CC=CC(C)=CC=CC(C)=CC=CC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C HRQKOYFGHJYEFS-UHFFFAOYSA-N 0.000 claims abstract description 5
- 239000011663 gamma-carotene Substances 0.000 claims abstract description 5
- 235000000633 gamma-carotene Nutrition 0.000 claims abstract description 5
- 238000010353 genetic engineering Methods 0.000 claims description 46
- 108020004414 DNA Proteins 0.000 claims description 36
- 238000012217 deletion Methods 0.000 claims description 26
- 230000037430 deletion Effects 0.000 claims description 26
- 230000002018 overexpression Effects 0.000 claims description 24
- 230000003828 downregulation Effects 0.000 claims description 22
- 230000009368 gene silencing by RNA Effects 0.000 claims description 21
- 230000009088 enzymatic function Effects 0.000 claims description 20
- 230000002779 inactivation Effects 0.000 claims description 16
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 13
- 239000008103 glucose Substances 0.000 claims description 13
- 230000001105 regulatory effect Effects 0.000 claims description 11
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 claims description 9
- 230000002103 transcriptional effect Effects 0.000 claims description 9
- 229910052799 carbon Inorganic materials 0.000 claims description 8
- 238000002744 homologous recombination Methods 0.000 claims description 8
- 230000006801 homologous recombination Effects 0.000 claims description 8
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 7
- 108010017070 Zinc Finger Nucleases Proteins 0.000 claims description 7
- 229940041514 candida albicans extract Drugs 0.000 claims description 7
- 239000012138 yeast extract Substances 0.000 claims description 7
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 claims description 6
- 102100025169 Max-binding protein MNT Human genes 0.000 claims description 6
- 101710163270 Nuclease Proteins 0.000 claims description 6
- 239000011780 sodium chloride Substances 0.000 claims description 6
- 108091006107 transcriptional repressors Proteins 0.000 claims description 6
- 238000005286 illumination Methods 0.000 claims description 4
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 claims description 3
- 102000053602 DNA Human genes 0.000 claims description 3
- 229930006000 Sucrose Natural products 0.000 claims description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 claims description 3
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 claims description 3
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 claims description 3
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 claims description 3
- 239000005720 sucrose Substances 0.000 claims description 3
- 108091030071 RNAI Proteins 0.000 claims 2
- 108090000623 proteins and genes Proteins 0.000 description 70
- 239000002773 nucleotide Substances 0.000 description 49
- 125000003729 nucleotide group Chemical group 0.000 description 49
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 32
- 230000014509 gene expression Effects 0.000 description 28
- 210000004027 cell Anatomy 0.000 description 26
- 239000012634 fragment Substances 0.000 description 24
- 241000221523 Rhodotorula toruloides Species 0.000 description 23
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 19
- 239000002609 medium Substances 0.000 description 18
- 150000007523 nucleic acids Chemical class 0.000 description 18
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 17
- 108020004999 messenger RNA Proteins 0.000 description 17
- 102000039446 nucleic acids Human genes 0.000 description 16
- 108020004707 nucleic acids Proteins 0.000 description 16
- 102100024633 Carbonic anhydrase 2 Human genes 0.000 description 15
- 101000760643 Homo sapiens Carbonic anhydrase 2 Proteins 0.000 description 15
- 239000013598 vector Substances 0.000 description 14
- 108091028043 Nucleic acid sequence Proteins 0.000 description 13
- 102000004169 proteins and genes Human genes 0.000 description 13
- 102100024650 Carbonic anhydrase 3 Human genes 0.000 description 12
- 241000233866 Fungi Species 0.000 description 12
- 101000760630 Homo sapiens Carbonic anhydrase 3 Proteins 0.000 description 12
- 239000000047 product Substances 0.000 description 11
- 108091026890 Coding region Proteins 0.000 description 10
- 101150063578 ald1 gene Proteins 0.000 description 10
- NUHSROFQTUXZQQ-UHFFFAOYSA-N isopentenyl diphosphate Chemical compound CC(=C)CCO[P@](O)(=O)OP(O)(O)=O NUHSROFQTUXZQQ-UHFFFAOYSA-N 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 108020004705 Codon Proteins 0.000 description 9
- 102100037957 Dixin Human genes 0.000 description 9
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 9
- 101100321983 Homo sapiens ABCD2 gene Proteins 0.000 description 9
- 101000857682 Homo sapiens Runt-related transcription factor 2 Proteins 0.000 description 9
- 101710178747 Phosphatidate cytidylyltransferase 1 Proteins 0.000 description 9
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 9
- 239000013612 plasmid Substances 0.000 description 9
- NCYCYZXNIZJOKI-UHFFFAOYSA-N vitamin A aldehyde Natural products O=CC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-UHFFFAOYSA-N 0.000 description 9
- 102100020970 ATP-binding cassette sub-family D member 2 Human genes 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- 108091034117 Oligonucleotide Proteins 0.000 description 8
- 238000002105 Southern blotting Methods 0.000 description 8
- 239000002299 complementary DNA Substances 0.000 description 8
- 230000000694 effects Effects 0.000 description 8
- 238000012224 gene deletion Methods 0.000 description 8
- 102100031075 Serine/threonine-protein kinase Chk2 Human genes 0.000 description 7
- 150000001413 amino acids Chemical class 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 238000005516 engineering process Methods 0.000 description 7
- 230000002068 genetic effect Effects 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 235000020945 retinal Nutrition 0.000 description 7
- 239000011604 retinal Substances 0.000 description 7
- 108700028369 Alleles Proteins 0.000 description 6
- 241000196324 Embryophyta Species 0.000 description 6
- 241000221778 Fusarium fujikuroi Species 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 230000006696 biosynthetic metabolic pathway Effects 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- 238000003209 gene knockout Methods 0.000 description 6
- 239000002679 microRNA Substances 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 108010001545 phytoene dehydrogenase Proteins 0.000 description 6
- CBIDRCWHNCKSTO-UHFFFAOYSA-N prenyl diphosphate Chemical compound CC(C)=CCO[P@](O)(=O)OP(O)(O)=O CBIDRCWHNCKSTO-UHFFFAOYSA-N 0.000 description 6
- 238000003762 quantitative reverse transcription PCR Methods 0.000 description 6
- 238000013519 translation Methods 0.000 description 6
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 5
- 241000589155 Agrobacterium tumefaciens Species 0.000 description 5
- 241000235553 Blakeslea trispora Species 0.000 description 5
- 101150065050 carS gene Proteins 0.000 description 5
- 239000003086 colorant Substances 0.000 description 5
- 102000006602 glyceraldehyde-3-phosphate dehydrogenase Human genes 0.000 description 5
- 108020004445 glyceraldehyde-3-phosphate dehydrogenase Proteins 0.000 description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N methanol Natural products OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000001965 potato dextrose agar Substances 0.000 description 5
- 230000002207 retinal effect Effects 0.000 description 5
- 238000003757 reverse transcription PCR Methods 0.000 description 5
- YVLPJIGOMTXXLP-UHFFFAOYSA-N 15-cis-phytoene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC(C)=CC=CC=C(C)CCC=C(C)CCC=C(C)CCC=C(C)C YVLPJIGOMTXXLP-UHFFFAOYSA-N 0.000 description 4
- 102000000452 Acetyl-CoA carboxylase Human genes 0.000 description 4
- 108010016219 Acetyl-CoA carboxylase Proteins 0.000 description 4
- 108010018763 Biotin carboxylase Proteins 0.000 description 4
- 108700011259 MicroRNAs Proteins 0.000 description 4
- 102000009658 Peptidylprolyl Isomerase Human genes 0.000 description 4
- 108010020062 Peptidylprolyl Isomerase Proteins 0.000 description 4
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 4
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 4
- 241000235401 Phycomyces blakesleeanus Species 0.000 description 4
- 101710173432 Phytoene synthase Proteins 0.000 description 4
- 108010011939 Pyruvate Decarboxylase Proteins 0.000 description 4
- 238000007792 addition Methods 0.000 description 4
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 4
- 230000000692 anti-sense effect Effects 0.000 description 4
- 238000000065 atmospheric pressure chemical ionisation Methods 0.000 description 4
- 230000001851 biosynthetic effect Effects 0.000 description 4
- 150000001746 carotenes Chemical class 0.000 description 4
- 235000005473 carotenes Nutrition 0.000 description 4
- 238000005119 centrifugation Methods 0.000 description 4
- 238000012512 characterization method Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 238000009396 hybridization Methods 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 238000010369 molecular cloning Methods 0.000 description 4
- 230000008520 organization Effects 0.000 description 4
- 229930002330 retinoic acid Natural products 0.000 description 4
- 229960003471 retinol Drugs 0.000 description 4
- 235000020944 retinol Nutrition 0.000 description 4
- 239000011607 retinol Substances 0.000 description 4
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 229960001727 tretinoin Drugs 0.000 description 4
- VWFJDQUYCIWHTN-YFVJMOTDSA-N 2-trans,6-trans-farnesyl diphosphate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CO[P@](O)(=O)OP(O)(O)=O VWFJDQUYCIWHTN-YFVJMOTDSA-N 0.000 description 3
- 241000238876 Acari Species 0.000 description 3
- 108020004635 Complementary DNA Proteins 0.000 description 3
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 101150022635 Dixdc1 gene Proteins 0.000 description 3
- 108700024394 Exon Proteins 0.000 description 3
- VWFJDQUYCIWHTN-FBXUGWQNSA-N Farnesyl diphosphate Natural products CC(C)=CCC\C(C)=C/CC\C(C)=C/COP(O)(=O)OP(O)(O)=O VWFJDQUYCIWHTN-FBXUGWQNSA-N 0.000 description 3
- 102100035111 Farnesyl pyrophosphate synthase Human genes 0.000 description 3
- NCYCYZXNIZJOKI-OVSJKPMPSA-N Retinaldehyde Chemical compound O=C\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-OVSJKPMPSA-N 0.000 description 3
- 241000223253 Rhodotorula glutinis Species 0.000 description 3
- 108020004459 Small interfering RNA Proteins 0.000 description 3
- 241000123675 Sporobolomyces roseus Species 0.000 description 3
- 244000301083 Ustilago maydis Species 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 229960004261 cefotaxime Drugs 0.000 description 3
- GPRBEKHLDVQUJE-VINNURBNSA-N cefotaxime Chemical compound N([C@@H]1C(N2C(=C(COC(C)=O)CS[C@@H]21)C(O)=O)=O)C(=O)/C(=N/OC)C1=CSC(N)=N1 GPRBEKHLDVQUJE-VINNURBNSA-N 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000009833 condensation Methods 0.000 description 3
- 230000005494 condensation Effects 0.000 description 3
- 101150038198 crgA gene Proteins 0.000 description 3
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 3
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- YYJNOYZRYGDPNH-MFKUBSTISA-N fenpyroximate Chemical compound C=1C=C(C(=O)OC(C)(C)C)C=CC=1CO/N=C/C=1C(C)=NN(C)C=1OC1=CC=CC=C1 YYJNOYZRYGDPNH-MFKUBSTISA-N 0.000 description 3
- 230000010354 integration Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000011144 upstream manufacturing Methods 0.000 description 3
- AJPADPZSRRUGHI-RFZPGFLSSA-N 1-deoxy-D-xylulose 5-phosphate Chemical compound CC(=O)[C@@H](O)[C@H](O)COP(O)(O)=O AJPADPZSRRUGHI-RFZPGFLSSA-N 0.000 description 2
- YVLPJIGOMTXXLP-UUKUAVTLSA-N 15,15'-cis-Phytoene Natural products C(=C\C=C/C=C(\CC/C=C(\CC/C=C(\CC/C=C(\C)/C)/C)/C)/C)(\CC/C=C(\CC/C=C(\CC/C=C(\C)/C)/C)/C)/C YVLPJIGOMTXXLP-UUKUAVTLSA-N 0.000 description 2
- YVLPJIGOMTXXLP-BAHRDPFUSA-N 15Z-phytoene Natural products CC(=CCCC(=CCCC(=CCCC(=CC=C/C=C(C)/CCC=C(/C)CCC=C(/C)CCC=C(C)C)C)C)C)C YVLPJIGOMTXXLP-BAHRDPFUSA-N 0.000 description 2
- OINNEUNVOZHBOX-QIRCYJPOSA-N 2-trans,6-trans,10-trans-geranylgeranyl diphosphate Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\CC\C(C)=C\COP(O)(=O)OP(O)(O)=O OINNEUNVOZHBOX-QIRCYJPOSA-N 0.000 description 2
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 2
- 101150073246 AGL1 gene Proteins 0.000 description 2
- 108010085238 Actins Proteins 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 241000589158 Agrobacterium Species 0.000 description 2
- 102100026608 Aldehyde dehydrogenase family 3 member A2 Human genes 0.000 description 2
- 101100096931 Arabidopsis thaliana SEP1 gene Proteins 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 239000002028 Biomass Substances 0.000 description 2
- 101150095530 CDS1 gene Proteins 0.000 description 2
- 108091033409 CRISPR Proteins 0.000 description 2
- 108010078791 Carrier Proteins Proteins 0.000 description 2
- 241000450599 DNA viruses Species 0.000 description 2
- 101710140859 E3 ubiquitin ligase TRAF3IP2 Proteins 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 108010007508 Farnesyltranstransferase Proteins 0.000 description 2
- 108010087894 Fatty acid desaturases Proteins 0.000 description 2
- 102000009114 Fatty acid desaturases Human genes 0.000 description 2
- 102100039291 Geranylgeranyl pyrophosphate synthase Human genes 0.000 description 2
- 108010026318 Geranyltranstransferase Proteins 0.000 description 2
- 101100378577 Hypocrea jecorina agl2 gene Proteins 0.000 description 2
- 108010065958 Isopentenyl-diphosphate Delta-isomerase Proteins 0.000 description 2
- 102100027665 Isopentenyl-diphosphate Delta-isomerase 1 Human genes 0.000 description 2
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 2
- 108010058996 Long-chain-aldehyde dehydrogenase Proteins 0.000 description 2
- 238000010222 PCR analysis Methods 0.000 description 2
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 2
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 2
- 101150017020 ROC1 gene Proteins 0.000 description 2
- 108091081024 Start codon Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108091036066 Three prime untranslated region Proteins 0.000 description 2
- 102100028601 Transaldolase Human genes 0.000 description 2
- 108020004530 Transaldolase Proteins 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 206010064930 age-related macular degeneration Diseases 0.000 description 2
- 229930002945 all-trans-retinaldehyde Natural products 0.000 description 2
- 230000008468 bone growth Effects 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 108091008053 gene clusters Proteins 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 208000002780 macular degeneration Diseases 0.000 description 2
- 108091070501 miRNA Proteins 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 230000000243 photosynthetic effect Effects 0.000 description 2
- 235000011765 phytoene Nutrition 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000002028 premature Effects 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 230000006798 recombination Effects 0.000 description 2
- 238000005215 recombination Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 230000036559 skin health Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 229960005322 streptomycin Drugs 0.000 description 2
- 230000008471 teeth growth Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- KBPHJBAIARWVSC-XQIHNALSSA-N trans-lutein Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2C(=CC(O)CC2(C)C)C KBPHJBAIARWVSC-XQIHNALSSA-N 0.000 description 2
- 108091006106 transcriptional activators Proteins 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 101710165761 (2E,6E)-farnesyl diphosphate synthase Proteins 0.000 description 1
- JKQXZKUSFCKOGQ-JLGXGRJMSA-N (3R,3'R)-beta,beta-carotene-3,3'-diol Chemical compound C([C@H](O)CC=1C)C(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)C[C@@H](O)CC1(C)C JKQXZKUSFCKOGQ-JLGXGRJMSA-N 0.000 description 1
- LXJXRIRHZLFYRP-VKHMYHEASA-L (R)-2-Hydroxy-3-(phosphonooxy)-propanal Natural products O=C[C@H](O)COP([O-])([O-])=O LXJXRIRHZLFYRP-VKHMYHEASA-L 0.000 description 1
- VMSLCPKYRPDHLN-UHFFFAOYSA-N (R)-Humulone Chemical compound CC(C)CC(=O)C1=C(O)C(CC=C(C)C)=C(O)C(O)(CC=C(C)C)C1=O VMSLCPKYRPDHLN-UHFFFAOYSA-N 0.000 description 1
- KJTLQQUUPVSXIM-ZCFIWIBFSA-M (R)-mevalonate Chemical compound OCC[C@](O)(C)CC([O-])=O KJTLQQUUPVSXIM-ZCFIWIBFSA-M 0.000 description 1
- PKAUICCNAWQPAU-UHFFFAOYSA-N 2-(4-chloro-2-methylphenoxy)acetic acid;n-methylmethanamine Chemical compound CNC.CC1=CC(Cl)=CC=C1OCC(O)=O PKAUICCNAWQPAU-UHFFFAOYSA-N 0.000 description 1
- 101710158485 3-hydroxy-3-methylglutaryl-coenzyme A reductase Proteins 0.000 description 1
- 108020003589 5' Untranslated Regions Proteins 0.000 description 1
- 102000007469 Actins Human genes 0.000 description 1
- 102000007698 Alcohol dehydrogenase Human genes 0.000 description 1
- 108010021809 Alcohol dehydrogenase Proteins 0.000 description 1
- 102000005369 Aldehyde Dehydrogenase Human genes 0.000 description 1
- 108020002663 Aldehyde Dehydrogenase Proteins 0.000 description 1
- 241001225321 Aspergillus fumigatus Species 0.000 description 1
- JEBFVOLFMLUKLF-IFPLVEIFSA-N Astaxanthin Natural products CC(=C/C=C/C(=C/C=C/C1=C(C)C(=O)C(O)CC1(C)C)/C)C=CC=C(/C)C=CC=C(/C)C=CC2=C(C)C(=O)C(O)CC2(C)C JEBFVOLFMLUKLF-IFPLVEIFSA-N 0.000 description 1
- 101150026586 CAR1 gene Proteins 0.000 description 1
- 101150083136 CAR2 gene Proteins 0.000 description 1
- 241000701489 Cauliflower mosaic virus Species 0.000 description 1
- 108700010070 Codon Usage Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 108010051219 Cre recombinase Proteins 0.000 description 1
- 241000195493 Cryptophyta Species 0.000 description 1
- 241000192700 Cyanobacteria Species 0.000 description 1
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 1
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 1
- 108050008072 Cytochrome c oxidase subunit IV Proteins 0.000 description 1
- 102000000634 Cytochrome c oxidase subunit IV Human genes 0.000 description 1
- LXJXRIRHZLFYRP-VKHMYHEASA-N D-glyceraldehyde 3-phosphate Chemical compound O=C[C@H](O)COP(O)(O)=O LXJXRIRHZLFYRP-VKHMYHEASA-N 0.000 description 1
- KJTLQQUUPVSXIM-UHFFFAOYSA-N DL-mevalonic acid Natural products OCCC(O)(C)CC(O)=O KJTLQQUUPVSXIM-UHFFFAOYSA-N 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 101100273207 Dictyostelium discoideum carC gene Proteins 0.000 description 1
- 102000016680 Dioxygenases Human genes 0.000 description 1
- 108010028143 Dioxygenases Proteins 0.000 description 1
- 102100026620 E3 ubiquitin ligase TRAF3IP2 Human genes 0.000 description 1
- 102100034545 FAD synthase region Human genes 0.000 description 1
- 101710156207 Farnesyl diphosphate synthase Proteins 0.000 description 1
- 101710125754 Farnesyl pyrophosphate synthase Proteins 0.000 description 1
- 101710089428 Farnesyl pyrophosphate synthase erg20 Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 1
- OINNEUNVOZHBOX-XBQSVVNOSA-N Geranylgeranyl diphosphate Natural products [P@](=O)(OP(=O)(O)O)(OC/C=C(\CC/C=C(\CC/C=C(\CC/C=C(\C)/C)/C)/C)/C)O OINNEUNVOZHBOX-XBQSVVNOSA-N 0.000 description 1
- 102100036669 Glycerol-3-phosphate dehydrogenase [NAD(+)], cytoplasmic Human genes 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000934858 Homo sapiens Breast cancer type 2 susceptibility protein Proteins 0.000 description 1
- 101000848289 Homo sapiens FAD synthase region Proteins 0.000 description 1
- 101001072574 Homo sapiens Glycerol-3-phosphate dehydrogenase [NAD(+)], cytoplasmic Proteins 0.000 description 1
- 108010020056 Hydrogenase Proteins 0.000 description 1
- 101710115465 Lon protease Proteins 0.000 description 1
- 239000006142 Luria-Bertani Agar Substances 0.000 description 1
- 239000006137 Luria-Bertani broth Substances 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- 102000005455 Monosaccharide Transport Proteins Human genes 0.000 description 1
- 108010006769 Monosaccharide Transport Proteins Proteins 0.000 description 1
- 241000306281 Mucor ambiguus Species 0.000 description 1
- 101000997933 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) (2E,6E)-farnesyl diphosphate synthase Proteins 0.000 description 1
- 101001015102 Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Dimethylallyltranstransferase Proteins 0.000 description 1
- 229910018890 NaMoO4 Inorganic materials 0.000 description 1
- 229930182559 Natural dye Natural products 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 102000005877 Peptide Initiation Factors Human genes 0.000 description 1
- 108010044843 Peptide Initiation Factors Proteins 0.000 description 1
- 239000001888 Peptone Substances 0.000 description 1
- 108010080698 Peptones Proteins 0.000 description 1
- 101710150389 Probable farnesyl diphosphate synthase Proteins 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 241001157811 Pucciniomycotina Species 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 241001502496 Rhodotorula toruloides ATCC 204091 Species 0.000 description 1
- 102000004431 Riboflavin transporter Human genes 0.000 description 1
- 101100010928 Saccharolobus solfataricus (strain ATCC 35092 / DSM 1617 / JCM 11322 / P2) tuf gene Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 102000009105 Short Chain Dehydrogenase-Reductases Human genes 0.000 description 1
- 108010048287 Short Chain Dehydrogenase-Reductases Proteins 0.000 description 1
- 101000848282 Siganus canaliculatus Acyl-CoA Delta-6 desaturase Proteins 0.000 description 1
- 108091027967 Small hairpin RNA Proteins 0.000 description 1
- 244000144916 Streptopus roseus Species 0.000 description 1
- 102000006467 TATA-Box Binding Protein Human genes 0.000 description 1
- 108010044281 TATA-Box Binding Protein Proteins 0.000 description 1
- 101150001810 TEAD1 gene Proteins 0.000 description 1
- 101150074253 TEF1 gene Proteins 0.000 description 1
- 102100029898 Transcriptional enhancer factor TEF-1 Human genes 0.000 description 1
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 1
- 208000010011 Vitamin A Deficiency Diseases 0.000 description 1
- JKQXZKUSFCKOGQ-LQFQNGICSA-N Z-zeaxanthin Natural products C([C@H](O)CC=1C)C(C)(C)C=1C=CC(C)=CC=CC(C)=CC=CC=C(C)C=CC=C(C)C=CC1=C(C)C[C@@H](O)CC1(C)C JKQXZKUSFCKOGQ-LQFQNGICSA-N 0.000 description 1
- QOPRSMDTRDMBNK-RNUUUQFGSA-N Zeaxanthin Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCC(O)C1(C)C)C=CC=C(/C)C=CC2=C(C)CC(O)CC2(C)C QOPRSMDTRDMBNK-RNUUUQFGSA-N 0.000 description 1
- XMWHRVNVKDKBRG-CRCLSJGQSA-N [(2s,3r)-2,3,4-trihydroxy-3-methylbutyl] dihydrogen phosphate Chemical compound OC[C@](O)(C)[C@@H](O)COP(O)(O)=O XMWHRVNVKDKBRG-CRCLSJGQSA-N 0.000 description 1
- RRLHMJHRFMHVNM-BQVXCWBNSA-N [(2s,3r,6r)-6-[5-[5-hydroxy-3-(4-hydroxyphenyl)-4-oxochromen-7-yl]oxypentoxy]-2-methyl-3,6-dihydro-2h-pyran-3-yl] acetate Chemical compound C1=C[C@@H](OC(C)=O)[C@H](C)O[C@H]1OCCCCCOC1=CC(O)=C2C(=O)C(C=3C=CC(O)=CC=3)=COC2=C1 RRLHMJHRFMHVNM-BQVXCWBNSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 238000000862 absorption spectrum Methods 0.000 description 1
- 238000000246 agarose gel electrophoresis Methods 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- JKQXZKUSFCKOGQ-LOFNIBRQSA-N all-trans-Zeaxanthin Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2=C(C)CC(O)CC2(C)C JKQXZKUSFCKOGQ-LOFNIBRQSA-N 0.000 description 1
- 125000003275 alpha amino acid group Chemical group 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000003674 animal food additive Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 150000001491 aromatic compounds Chemical class 0.000 description 1
- 235000013793 astaxanthin Nutrition 0.000 description 1
- 239000001168 astaxanthin Substances 0.000 description 1
- MQZIGYBFDRPAKN-ZWAPEEGVSA-N astaxanthin Chemical compound C([C@H](O)C(=O)C=1C)C(C)(C)C=1/C=C/C(/C)=C/C=C/C(/C)=C/C=C/C=C(C)C=CC=C(C)C=CC1=C(C)C(=O)[C@@H](O)CC1(C)C MQZIGYBFDRPAKN-ZWAPEEGVSA-N 0.000 description 1
- 229940022405 astaxanthin Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000013611 chromosomal DNA Substances 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000002856 computational phylogenetic analysis Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 210000001840 diploid cell Anatomy 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000009144 enzymatic modification Effects 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 208000036234 familial restrictive 6 cardiomyopathy Diseases 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 238000000855 fermentation Methods 0.000 description 1
- 230000004151 fermentation Effects 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 231100000221 frame shift mutation induction Toxicity 0.000 description 1
- 230000037433 frameshift Effects 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000003208 gene overexpression Methods 0.000 description 1
- GVVPGTZRZFNKDS-JXMROGBWSA-N geranyl diphosphate Chemical compound CC(C)=CCC\C(C)=C\CO[P@](O)(=O)OP(O)(O)=O GVVPGTZRZFNKDS-JXMROGBWSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000036074 healthy skin Effects 0.000 description 1
- 108010002685 hygromycin-B kinase Proteins 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 230000000984 immunochemical effect Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 230000006372 lipid accumulation Effects 0.000 description 1
- 235000012680 lutein Nutrition 0.000 description 1
- 239000001656 lutein Substances 0.000 description 1
- 229960005375 lutein Drugs 0.000 description 1
- KBPHJBAIARWVSC-RGZFRNHPSA-N lutein Chemical compound C([C@H](O)CC=1C)C(C)(C)C=1\C=C\C(\C)=C\C=C\C(\C)=C\C=C\C=C(/C)\C=C\C=C(/C)\C=C\[C@H]1C(C)=C[C@H](O)CC1(C)C KBPHJBAIARWVSC-RGZFRNHPSA-N 0.000 description 1
- ORAKUVXRZWMARG-WZLJTJAWSA-N lutein Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CCCC1(C)C)C=CC=C(/C)C=CC2C(=CC(O)CC2(C)C)C ORAKUVXRZWMARG-WZLJTJAWSA-N 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 239000000978 natural dye Substances 0.000 description 1
- 108010058731 nopaline synthase Proteins 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 239000002417 nutraceutical Substances 0.000 description 1
- 235000021436 nutraceutical agent Nutrition 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 235000019319 peptone Nutrition 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 235000020777 polyunsaturated fatty acids Nutrition 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000010839 reverse transcription Methods 0.000 description 1
- 108020001053 riboflavin transporter Proteins 0.000 description 1
- 229920002477 rna polymer Polymers 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 238000002864 sequence alignment Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 108010027126 short chain trans-2-enoyl-CoA reductase Proteins 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229960000268 spectinomycin Drugs 0.000 description 1
- UNFWWIHTNXNPBV-WXKVUWSESA-N spectinomycin Chemical compound O([C@@H]1[C@@H](NC)[C@@H](O)[C@H]([C@@H]([C@H]1O1)O)NC)[C@]2(O)[C@H]1O[C@H](C)CC2=O UNFWWIHTNXNPBV-WXKVUWSESA-N 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 235000019529 tetraterpenoid Nutrition 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 230000014621 translational initiation Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 239000012137 tryptone Substances 0.000 description 1
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 239000006200 vaporizer Substances 0.000 description 1
- 238000012795 verification Methods 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 235000019155 vitamin A Nutrition 0.000 description 1
- 239000011719 vitamin A Substances 0.000 description 1
- 229940045997 vitamin a Drugs 0.000 description 1
- 238000003260 vortexing Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- FJHBOVDFOQMZRV-XQIHNALSSA-N xanthophyll Natural products CC(=C/C=C/C=C(C)/C=C/C=C(C)/C=C/C1=C(C)CC(O)CC1(C)C)C=CC=C(/C)C=CC2C=C(C)C(O)CC2(C)C FJHBOVDFOQMZRV-XQIHNALSSA-N 0.000 description 1
- -1 y-carotene Chemical class 0.000 description 1
- 239000007222 ypd medium Substances 0.000 description 1
- 235000010930 zeaxanthin Nutrition 0.000 description 1
- 239000001775 zeaxanthin Substances 0.000 description 1
- 229940043269 zeaxanthin Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/37—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from fungi
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/0008—Oxidoreductases (1.) acting on the aldehyde or oxo group of donors (1.2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P23/00—Preparation of compounds containing a cyclohexene ring having an unsaturated side chain containing at least ten carbon atoms bound by conjugated double bonds, e.g. carotenes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Biophysics (AREA)
- Medicinal Chemistry (AREA)
- Mycology (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Gastroenterology & Hepatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Un método para ajustar el nivel de producción y la composición de carotenoides en un huésped fúngico que comprende: (a) manipular genéticamente uno o más polinucleótidos en la biosíntesis de carotenoides en un huésped fúngico, en donde uno o más polinucleótidos se seleccionan de (i) los polinucleótidos expuestos en las SEQ ID NOs: 1, 3, 5, 7, 8, 10, 11, 13, 14, 16, 17, 19 y 20 o una secuencia homóloga que comparte al menos el 75 % de identidad con los mismos o (ii) uno o más polinucleótidos que codifican uno o más polipéptidos expuestos en las SEQ ID NOs: 2, 4, 6, 12, 15, 18 y 21 o una secuencia homóloga que comparte al menos el 75 % de identidad con los mismos, y en donde el huésped fúngico es Rhodosporidium o Rhodotorula,y (b) cultivar el huésped fúngico para producir carotenoides, en donde los carotenoides se seleccionan del grupo que consiste en licopeno, beta-caroteno, gamma-caroteno, toruleno y torularodina o derivados de estos, mediante lo cual se ajusta el nivel de producción o la composición de los carotenoides.
Description
DESCRIPCIÓN
Métodos para ajustar los niveles de producción de carotenoides y composiciones en géneros de Rhodosporidium y Rhodotorula
Referencia cruzada a solicitudes relacionadas
La presente solicitud se relaciona con y reivindica la prioridad a la solicitud de patente provisional de los Estados Unidos núm. de serie 62/091,913, presentada el 15 de diciembre de 2014. Esta solicitud se incorpora en la presente descripción como referencia en su totalidad.
Envío de secuencia
La presente solicitud se presenta junto con una lista de secuencias en formato electrónico. El Listado de secuencias se titula 2577244PCTSequenceListing.txt, creado el 03 de noviembre de 2015 y tiene un tamaño de 184 kb. La información de la Lista de Secuencias en formato electrónico se incorpora en la presente descripción como referencia en su totalidad.
Antecedentes de la invención
La presente invención se refiere al campo de la biotecnología fúngica, más particularmente a métodos de ingeniería genética para la producción de carotenoides en huéspedes fúngicos seleccionados de los géneros Rhodosporidium y Rhodotorula.
Las publicaciones y otros materiales usados en la presente descripción para ilustrar los antecedentes de la invención, y en particular, los casos que proporcionan detalles adicionales con respecto a la práctica, se incorporan como referencia, y por conveniencia se referencian en el texto siguiente por el autor y la fecha y se enumeran alfabéticamente por el autor en la bibliografía anexa.
Está bien documentado que la producción de carotenoides se inicia con la biosíntesis de geranilgeranil difosfato (GGPP) catalizada por la GGPP sintasa para la condensación de C15 farnesil difosfato (FPP) y C5 isopentenil difosfato (IPP). Posteriormente, dos moléculas de GGPP se condensan adicionalmente para formar el precursor incoloro fitoeno, que es catalizado por la fitoeno sintasa. En hongos y eubacterias, la fitoeno desaturasa cataliza las 4 etapas de desaturación del fitoeno para producir el licopeno de color rojo, mientras que esto se realiza por fitoeno desaturasa y Y-caroteno o turasa y desaturasa separadas en plantas, algas y cianobacterias. El licopeno se cicla por la caroteno ciclasa para formar ycaroteno y 8-caroteno monocíclicos, y a-caroteno y p-caroteno dicíclicos [1, 2]. Aguas arriba de la vía biosintética, el FPP se produce por la condensación catalizada por farnesil difosfato sintasa (FPS), de IPP y C10 geranil difosfato (GPP), este último producido por la condensación catalizada por la GPP sintasa, de IPP y dimetilalil difosfato (DMAPP), el producto de la isomerasa IPP (IPI). El IPP y el DMAPP pueden sintetizarse a través de la vía del mevalonato (MVP) o de la vía de 2-C-metil-D-eritritol 4-fosfato/1-desoxi-D-xilulosa 5-fosfato (MEP/DOXP) [3, 4].
Los carotenoides son tetraterpenoides de 40 carbonos (C40) [5]. Los carotenoides no oxigenados, tales como el ycaroteno, el p-caroteno y el licopeno se conocen como carotenos. Las modificaciones enzimáticas adicionales de los carotenos producen moléculas que contienen oxígeno, tales como luteína, retinol (vitamina A), zeaxantina y astaxantina [6, 7]. La biosíntesis de carotenoides se produce en todos los organismos fotosintéticos [8] y en muchos microorganismos no fotosintéticos, tales como bacterias y hongos [1, 5, 9, 10] y algunos insectos [11].
Los carotenoides desempeñan un papel importante en la salud y el desarrollo humano y animal [12-15]. Por ejemplo, una mayor ingestión de carotenoides en la dieta se asoció con un riesgo menor de degeneración macular relacionada con la edad (ADM) [13]; la deficiencia de vitamina A se asocia con un crecimiento anormal del esqueleto y los dientes y la infertilidad en ratas [14]; el retinal (retinaldehído) es esencial para la visión, mientras que el ácido retinoico es esencial para la salud de la piel, la remineralización de los dientes y el crecimiento óseo [16]; la ingestión de licopeno se relaciona con riesgo menor de cáncer de próstata [17]. Los carotenoides son colorantes naturales con muchos colores disponibles [18-20]. Los carotenoides son precursores para la producción de compuestos aromáticos valiosos [21]. El p-caroteno puede escindirse por la citocromo oxidasa P450 para producir retinal (retinaldehído) [16], que es esencial para la visión y cuando se convierte en ácido retinoico, es esencial para la salud cutánea, la remineralización de los dientes y el crecimiento óseo. Por lo tanto, los carotenoides son valiosos aditivos de alimentos y piensos, nutracéuticos y cosmecéuticos.
El retinol, el retinal y el ácido retinoico se conocen como retinoides, que se derivan del fallo de la salud cutánea, la remineralización de los dientes y el crecimiento óseo. El retinol y el retinal son interconvertibles y se catalizan por la alcohol deshidrogenasa y las deshidrogenasa/reductasas de cadena corta, mientras que las familias de enzimas aldehído deshidrogenasa y citocromo P450 catalizan la oxidación irreversible de retinal a ácido retinoico. La identificación de enzimas que catalizan la oxidación del retinol in vivo ha sido controversial, lo que se debe en parte a la dificultad por la naturaleza reversible de esta reacción [22].
Rhodosporidium y Rhodotorula son dos géneros fúngicos que pertenecen al subfilum Pucciniomycotina. Pueden cultivarse en forma de células individuales en una densidad celular muy alta en fermentadores a una velocidad de crecimiento rápida y acumular niveles altos de triacilglicéridos [23-26]. Rhodosporidium y Rhodotorula son capaces de producir niveles altos de carotenoides [27-30], con beta-caroteno, gamma-caroteno, torularodina y toruleno como componentes principales [31]. Torularodina y toruleno son colorantes potenciales e inductores de la expresión génica. Aparte de la identificación de un supuesto homólogo de CAR2 [32], no hay ningún informe sobre la vía biosintética de carotenoides en Rhodosporidium y Rhodotorula. Cualquier método que mejore la productividad y la pureza del producto de los carotenoides y sus derivados tiene valor y significado comercial.
Resumen de la invención
La presente invención se refiere al campo de la biotecnología fúngica, más particularmente a métodos de ingeniería genética para la producción de carotenoides en huéspedes fúngicos seleccionados de los géneros Rhodosporidium y Rhodotorula.
Así, en un aspecto, la presente invención proporciona un método para ajustar el nivel de producción y composición de carotenoides en un huésped fúngico. De acuerdo con este aspecto, el método comprende la manipulación genética de uno o más de los polinucleótidos implicados en la biosíntesis de carotenoides. En una modalidad, los carotenoides son licopeno, beta-caroteno, gamma-caroteno, toruleno, torularodina o derivados de estos. En algunas modalidades, un derivado es un derivado hidroxilado, un derivado glicosilado o un derivado oxidado. En otra modalidad, el huésped fúngico es Rhodosporidium o Rhodotorula. En algunas modalidades, el método comprende manipular genéticamente uno o más polinucleótidos implicados en la biosíntesis de carotenoides en un huésped fúngico y hacer crecer el huésped fúngico para producir los carotenoides, por lo que se ajusta el nivel de producción o la composición de los carotenoides.
En una modalidad, la manipulación genética comprende la regulación negativa de uno o más polinucleótidos seleccionados de SEQ ID NOs: 1, 3, 5, 7, 8, 9, 10, 11, 13, 14, 16, 17, 19 y 20, o un homólogo que comparte al menos el 75 % de identidad de nucleótidos con los mismos en un huésped fúngico. En otra modalidad, la manipulación genética comprende la regulación negativa de uno o más polinucleótidos que codifican polipéptidos seleccionados de SEQ ID NOs: 2, 4, 6, 12, 15, 18 y 21 o un homólogo que comparte al menos el 75 % de identidad con los mismos en un huésped fúngico. En algunas modalidades, la regulación negativa se compara con un huésped fúngico sin la manipulación genética. En otras modalidades, uno o más polinucleótidos se regulan negativamente por ARNi, un represor transcripcional artificial o un promotor débil.
En una modalidad, la manipulación genética es la inactivación total de la función enzimática en un huésped fúngico. En algunas modalidades, la inactivación se logra mediante la deleción de todo o una parte de uno o más polinucleótidos seleccionados de las SEQ ID NOs: 10, 11, 16, 17, 19 y 20 o un homólogo que comparte al menos el 75 % de identidad de nucleótidos con los mismos. En otras modalidades, la inactivación se logra mediante la deleción de todo o una parte de uno o más polinucleótidos que codifican polipéptidos seleccionados entre las SEQ ID NOs: 12, 18 y 21 o un homólogo que comparte al menos el 75 % de identidad de nucleótidos con los mismos. En una modalidad, la deleción se realiza mediante el uso de una técnica de recombinación homóloga. En otra modalidad, la deleción se ayuda mediante el uso de una nucleasa artificial. En una modalidad adicional, la nucleasa artificial es una nucleasa de dedos de zinc (ZFN) o un complejo Cas9-ARNg.
En una modalidad, la manipulación genética implica la sobreexpresión de uno o más polinucleótidos seleccionados de SEQ ID NOs: 1, 3, 5, 7, 8, 9, 10, 11, 13, 14, 16, 17, 19 y 20, o un homólogo que comparte al menos el 75 % de identidad de nucleótidos con los mismos. En otra modalidad, la manipulación genética implica la sobreexpresión de uno o más polinucleótidos que codifican polipéptidos seleccionados de SEQ ID NOs: 2, 4, 6, 12, 15, 18 y 21; o un homólogo que comparte al menos el 75 % de identidad con los mismos. En algunas modalidades, la sobreexpresión está mediada por la introducción de un casete genético sintético en una célula fúngica huésped. En otra modalidad, el casete comprende un promotor heterólogo unido operativamente al polinucleótido, opcionalmente unido operativamente a un terminador transcripcional. En algunas modalidades, la sobreexpresión se compara con un huésped fúngico sin manipulación genética.
En algunas modalidades, la manipulación genética implica una combinación de las manipulaciones genéticas descritas previamente. En una modalidad, la manipulación genética comprende la regulación negativa de uno o más de los polinucleótidos y la sobreexpresión de uno o más polinucleótidos diferentes. En otra modalidad, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos y la sobreexpresión de uno o más de diferentes polinucleótidos. En una modalidad adicional, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos y la regulación negativa de uno o más polinucleótidos diferentes. En una modalidad adicional, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos, la sobreexpresión de uno o más de diferentes polinucleótidos y la regulación negativa de uno o más polinucleótidos diferentes.
En algunas modalidades, los polinucleótidos descritos en la presente descripción que se han incorporado de manera estable en el genoma fúngico están unidos operativamente a un promotor que permite la expresión eficiente en especies
del género Rhodosporidium y el género Rhodotorula. Los promotores para cada polinucleótido incorporado pueden ser iguales o diferentes. En algunas modalidades, los promotores son promotores encontrados en especies del género Rhodosporidium y el género Rhodotorula. Los ejemplos de promotores adecuados incluyen, pero no se limitan a, promotores de los genes siguientes que codifican las proteínas siguientes: gliceraldehído 3-fosfato deshidrogenasa (GPD), proteína transportadora de acil-CoA (ACP), ácido graso desaturasa, factor de elongación de traducción (TEF), piruvato descarboxilasa (PDC), enolasa (2-fosfoglicerato deshidratasa) (ENO), peptidilprolil isomerasa (PPI), acetil-CoA carboxilasa (ACC) o transaldolasa. En otras modalidades, los genes descritos en la presente descripción también incluyen un terminador transcripcional de ARNm que puede encontrarse en cualquier especie eucariota y sus virus de ADN.
En otra modalidad, la presente invención proporciona un método para producir carotenoides que comprende cultivar una célula fúngica huésped descrita en la presente descripción en condiciones adecuadas para producir carotenoides. Puede usarse cualquier medio con al menos el 5 % de fuente de carbono. En algunas modalidades, la fuente de carbono es glucosa, manosa, glicerol, sacarosa, xilosa o sus combinaciones. En una modalidad, el medio es MinCAR que contiene 30-100 g de glucosa, 1,5 g de extracto de levadura, 0,5 g de (N H ^S O 4 , 2,05 g de K2HPO4 , 1,45 g de KH2PO4 , 0,6 g de MgSO4 , 0,3 g de NaCl, 10 mg de CaCh, 1 mg de FeSO4 , 0,5 mg de ZnSO4, 0,5 mg de CuSO4 , 0,5 mg de H3BO4 , 0,5 mg de MnSO4, 0,5 mg de NaMoO4 (por litro). El medio se ajusta preferentemente a pH 5-7. En algunas modalidades, el cultivo celular se realiza preferentemente a 25 °-35 °C. En otras modalidades, el cultivo se realiza preferentemente en una condición con iluminación.
Breve descripción de las figuras
La figura 1 muestra la organización de pRH201. LB: borde izquierdo de T-DNA; RB: borde derecho de T-DNA; Pgpd: Promotor de 595 pb de Umgpdi; Pgpdi: Promotor de 795 pb de RtGPDI; hpt-3: gen de resistencia a la higromicina optimizado por codones que se basa en el sesgo de uso de codones en R. toruloides; Tnos: terminador del gen de la nopalina sintasa de A. tumefaciens. Los sitios de corte de enzimas de restricción únicos se muestran en rojo. loxP-RE y loxP-LE y los sitios de reconocimiento de mutantes para Cre recombinasa. Sp/Str son genes de resistencia para espectinomicina y estreptomicina; eGFP-His6 es un gen de codón adaptado que codifica la proteína de fusión de etiqueta eGFP-histidina de R. toruloides; 35S: terminador del gen 35S del virus del mosaico de la coliflor.
Las Figuras 2A-2E muestran la identificación de mutantes RCM y la caracterización del gen CAR1. Figura 2A: Fenotipos de color de colonias de mutantes RCM. Todas las cepas se inocularon en estrías en placas PDA y se incubaron a 28 °C durante 2 días. Figura 2B: Diagrama esquemático de CAR1 y su estrategia de deleción. Figura 2C: Análisis de Southern blot del mutante nulo del candidato CARI (A cari). Las secuencias homólogas usadas para la deleción de CARI fueron de 1036 pb (CAR1L) y de 830 pb (CAR1R) de longitud, que oscilan de -89 a 947 y de 2098 a 2928 del codón de inicio traduccional. El fragmento de ADN marcado con digoxigenina CAR1R se usó como sonda para la detección de ADN total HindIII. Figura 2D: Colores de colonias de WT, mutante nulo (A cari) y cepa de complementación (A carIC) cultivadas en placa PDA. Figura 2E: Perfiles de carotenoides en la cepa de tipo salvaje de R. toruloides, Acari y AcarIC. Se analizó el contenido de cuatro componentes principales de carotenoides.
Las Figuras 3A-3D muestran el grupo de genes biosintéticos de carotenoides en R. toruloides. Figura 3A: Organización genómica de grupos de genes biosintéticos de carotenoides en 5 hongos carotenogénicos, Blakeslea trispora, Fusarium fujikuroi, Phycomyces blakesleeanus y Sporobolomyces roseus. Figura 3B: Deleción de CAR3, CCD1 y CDS1. El panel superior muestra los esquemas de deleción y el panel inferior muestra el análisis de Southern blot de mutantes de desactivación génica. Las barras negras indican las sondas usadas para la hibridación mediante Southern blot. Figura 3C: Fenotipo de color de colonias de mutantes nulos implicados en la vía biosintética de carotenoides en R. toruloides. Figura 3D: Perfiles de carotenoides en cepa de tipo salvaje y mutantes nulos de R. toruloides de genes CCD1 y CDS1.
La figura 4 muestra los perfiles de carotenoides en cepa de tipo salvaje de R. toruloides, mutante nulo Ald1 (ald1) y sobreexpresión de Ald1OE.
La figura 5 muestra los niveles relativos de ARNm de genes biosintéticos de carotenoides después de cambiar a iluminación. El nivel de expresión de cada gen se realizó mediante qRT-PCR y se normalizó para el gen Actin (ACT1).
Las Figuras 6A-6C muestran la caracterización de Rocl. Figura 6A: Estructura esquemática de ROC1 y estrategia de deleción de genes. Figura 6B: Análisis de árbol filogenético de reguladores negativos de la biosíntesis de carotenoides fúngicos. El número de registro de NCBI GenBank se siguió del nombre del gen. Figura 6C: Comparación de dominios de dedo RING y dominios LON. Las secuencias de consenso se indican en la línea inferior de cada una. Los números de registro de GenBank (con secuencias en la Figura 6C establecidas en las secuencias que se indican): B. trispora crgA: CAE51310.1 (SEQ ID NO:26); M circinelloides crgA: CAB61339.2 (SEQ ID NO:28); A. fumigatus crgA: XP_755380.1 (SEQ ID NO:25); F. fujikurol carS: CCP50075.1 (SEQ ID NO:27); P. blakesleeanus carS: ADU04395.1 (SEQ ID NO:29); U. maydis: EAK85777.1 (SEQ ID NO:31); R toruloides: (SEQ ID NO:30)
Las Figuras 7A-7E muestra la deleción de ROC1. Figura 7A: Diferencias de color de colonias entre recombinación ectópica y homóloga de ROC1 de desactivación génica. Figura 7B: Verificación mediante Southern blot de mutantes de deleción génica. Se usó Digoxigenin-labeled DAN Molecular Weight Marker VII (Roche Diagnosis, Estados Unidos) como marcador. Figura 7C: Morfología celular del mutante nulo y tipo salvaje de rocl. La barra representa 10 pm. Figura 7D: transcritos de ARNm de genes de carotenoides en la cepa mutante y de tipo salvaje de roc i en condiciones de iluminación.
Figura 7E: Comparación de la producción de carotenoides en la cepa de tipo salvaje, mutante de desactivación génica (Arocl) y su cepa de complementación (AroclC).
Descripción detallada de la invención
La presente invención se refiere al campo de la biotecnología fúngica, más particularmente a métodos de ingeniería genética para la producción de carotenoides en huéspedes fúngicos seleccionados de los géneros Rhodosporidium y Rhodotorula.
A menos que se defina de cualquier otra manera, todos los términos técnicos y científicos usados en la presente descripción tienen el mismo significado que el que se conoce comúnmente por el experto en la técnica a la que pertenece la invención.
El término "aproximadamente" significa dentro de un intervalo de un valor estadísticamente significativo. Dicho intervalo puede estar dentro de un orden de magnitud, preferentemente dentro del 50 %, más preferentemente dentro del 20 %, aún más preferentemente dentro del 10 %, y aún más preferentemente dentro del 5 % de un valor o intervalo dado. La variación permisible abarcada por el término "aproximadamente" depende del sistema particular en estudio, y puede apreciarse fácilmente por un experto en la técnica.
Como se usa en la presente descripción, "alelo" se refiere a cualquiera de una o más formas alternativas de un locus génico, todos los alelos se relacionan con un rasgo o característica. En una célula u organismo diploide, los dos alelos de un gen dado ocupan los loci correspondientes en un par de cromosomas homólogos.
El término "regulación negativa" se refiere a la disminución del nivel de expresión de un polinucleótido, como un gen, en comparación con un control que usa cualquier método conocido en la técnica, como ARNi, un represor transcripcional artificial para atacar específicamente el gen si es de interés, como ZFN y Cas9 que se fusionan a un dominio represor transcripcional y se unen a una secuencia de ADN específica en un promotor de un gen o secuencia codificante para lograr la regulación negativa; o el uso de un promotor más débil para impulsar la expresión del gen de interés. El término "regulado negativamente" se usa en la presente descripción para indicar que la expresión del gen objetivo se reduce en 1-100 %. Por ejemplo, la expresión puede reducirse en aproximadamente 5 %, 10 %, 15 %, 20 %, 25 %, 30 %, 35 %, 40 %, 45 %, 50 %, 55 %, 60 %, 65 %, 70 %, 75 %, 80 %, 85 %, 90 %, 95 % o 99 %.
Como se usa en la presente descripción, una "ventana de comparación" se refiere a un segmento conceptual de al menos 6 posiciones contiguas, generalmente de aproximadamente 50 a aproximadamente 100, más generalmente de aproximadamente 100 a aproximadamente 150, en el que una secuencia se compara con una secuencia de referencia del mismo número de posiciones contiguas después que las dos secuencias se alinean de manera óptima. La ventana de comparación puede comprender adiciones o deleciones (es decir, brechas) de aproximadament el 20 % o menos en comparación con la secuencia de referencia (que no comprende adiciones o deleciones) para la alineación óptima de las dos secuencias. Los expertos en la técnica deben hacer referencia a los métodos detallados usados para la alineación de secuencias, como en Wisconsin Genetics Software Package Release 7.0 (Genetics Computer Group, 575 Science Drive Madison, Wisconsin, Estados Unidos).
Una molécula "dsRNA" or "ARNi", como se usa en la presente descripción en el contexto de ARNi, se refiere a un compuesto, el cual es capaz de regular negativamente o reducir la expresión de un gen o la actividad del producto de dicho gen en una medida suficiente para lograr un efecto biológico o fisiológico deseado. El término "dsRNA" o "molécula de ARNi", como se usa en la presente descripción, se refiere a uno o más de un dsRNA, siRNA, miRNA, hpRNA, ihpRNA.
El término "expresión" con respecto a una secuencia génica se refiere a la transcripción del gen y, según sea apropiado, a la traducción del transcrito de ARNm resultante a una proteína. Por lo tanto, como quedará claro por el contexto, la expresión de una secuencia codificante de proteínas es el resultado de la transcripción y traducción de la secuencia codificante.
Como se usa en la presente descripción, "genotipo" se refiere a la constitución genética de una célula u organismo.
El término "homólogo" como se usa en la presente descripción se refiere a un gen relacionado con un segundo gen por descendencia de una secuencia de ADN ancestral común. El término, homólogo, puede aplicarse a la relación entre genes separados por el evento de especiación (ortólogo) o a la relación entre genes separados por el evento de duplicación genética (parálogo). El término homólogo se usa genéricamente para referirse a todas las especies.
"Enlace operable" o "unido operativamente" como se usa en la presente descripción, se entiende que significa, por ejemplo, la disposición secuencial de un promotor y el ácido nucleico a expresarse y, si es apropiado, elementos reguladores adicionales tales como, para ejemplo, un terminador, de tal manera que cada uno de los elementos reguladores pueda cumplir su función en la expresión recombinante del ácido nucleico para formar dsRNA. Esto no requiere necesariamente un enlace directo en el sentido químico. Las secuencias de control genético como, por ejemplo, las secuencias potenciadoras, también pueden ejercer su función en la secuencia objetivo desde posiciones que están algo distantes, o incluso desde otras moléculas de ADN (localización cis o trans). Las disposiciones preferidas son
aquellas en las que la secuencia de ácido nucleico que va a expresarse de forma recombinante se coloca aguas abajo de la secuencia que actúa como promotor, de modo que las dos secuencias se unen covalentemente entre sí. Las secuencias reguladoras o de control pueden colocarse en el lado 5' de la secuencia de nucleótidos o en el lado 3' de la secuencia de nucleótidos como se conoce bien en la técnica.
El término "sobreexpresión" se refiere al aumento en el nivel de expresión de un polinucleótido, como un gen, en comparación con un control que usa cualquier método conocido en la técnica, como un activador transcripcional artificial para atacar específicamente el gen si es de interés, tales como ZFN y Cas9 que se fusionan a un dominio activador transcripcional y se unen a una secuencia de ADN específica en un promotor de un gen; o el uso de un promotor más fuerte para impulsar la expresión del gen de interés. El término "sobreexpresión" se usa en la presente descripción para indicar que la expresión del gen objetivo aumenta en un 25 % o más en comparación con un control. Por ejemplo, la expresión puede aumentarse en aproximadament el 25 %, 50 %, 100 %, 200 %, 500 %, 1000 % y así sucesivamente.
Como se usa en la presente descripción, el "fenotipo" se refiere a las características detectables de una célula u organismo, cuyas características son la manifestación de la expresión génica.
Los términos "polinucleótido", "ácido nucleico" y "molécula de ácido nucleico" se usan indistintamente en la presente descripción para referirse a un polímero de nucleótidos que puede ser un arreglo lineal natural o sintético y secuencial de nucleótidos y/o nucleósidos, que incluyen el ácido desoxirribonucleico, el ácido ribonucleico y derivados de estos. Incluye ADN cromosómico, plásmidos autorreplicantes, polímeros infecciosos de ADN o ARN y ADN o ARN que desempeñan un papel primordialmente estructural. A menos que se indique lo contrario, los ácidos nucleicos o el polinucleótido se escriben de izquierda a derecha en una orientación de 5'a 3', los nucleótidos se denominan por sus códigos de una sola letra comúnmente aceptados. Los intervalos numéricos incluyen los números que definen el intervalo.
Los términos “polipéptido”, “péptido” y “proteína” se usan indistintamente en la presente descripción para referirse a un polímero de residuos de aminoácidos. Los términos se aplican a polímeros de aminoácidos en los que uno o más residuos de aminoácidos es un análogo químico artificial de un aminoácido de origen natural correspondiente, así como a polímeros de aminoácidos de origen natural. Los aminoácidos pueden denominarse por sus símbolos de tres letras o una letra comúnmente conocidos. Las secuencias de aminoácidos se escriben de izquierda a derecha en orientación de amino a carboxilo, respectivamente. Los intervalos numéricos incluyen los números que definen el intervalo.
Como se usa en la presente descripción, el término "identidad de secuencia", "similitud de secuencia" u "homología" se usa para describir relaciones de secuencia entre dos o más secuencias de nucleótidos. El porcentaje de "identidad de secuencia" entre dos secuencias se determina mediante la comparación de dos secuencias alineadas óptimamente sobre una ventana de comparación como la longitud total de una SEQ ID NO: referenciada, en donde la porción de la secuencia en la ventana de comparación puede comprender adiciones o deleciones (es decir, brechas) en comparación con la secuencia de referencia (que no comprende adiciones o deleciones) para la alineación óptima de las dos secuencias. El porcentaje se calcula mediante la determinación del número de posiciones en las que el residuo de aminoácido o de base de ácido nucleico idéntico aparece en ambas secuencias para producir el número de posiciones coincidentes, tras dividir el número de posiciones coincidentes por el número total de posiciones en la ventana de comparación, y multiplicar el resultado por 100 para obtener el porcentaje de identidad de secuencia. Se plantea que una secuencia que es idéntica en cada posición en comparación con una secuencia de referencia es idéntica a la secuencia de referencia y viceversa. Se plantea que una primera secuencia de nucleótidos cuando se observa en la dirección 5' a 3' es un "complemento" de, o es complementaria a, una segunda secuencia o de referencia de nucleótidos observada en la dirección 3' a 5' si la primera secuencia de nucleótidos muestra complementariedad completa con la segunda secuencia o de referencia. Tal como se usa en la presente descripción, se plantea que las moléculas de secuencias de ácido nucleico presentan "complementariedad completa" cuando cada nucleótido de una de las secuencias que se leen en la dirección 5' a 3' es complementaria a cada nucleótido de la otra secuencia cuando se lee en la dirección 3' a 5'. Una secuencia de nucleótidos que es complementaria a una secuencia de nucleótidos de referencia presentará una secuencia idéntica a la secuencia complementaria inversa de la secuencia de nucleótidos de referencia. Estos términos y descripciones están bien definidos en la técnica y se conocen fácilmente por los expertos en la técnica.
El término "inactivación total de la función enzimática", como se usa en la presente descripción, se refiere a la pérdida completa de la función o actividad enzimática para una enzima dada o un polipéptido dado que tiene función o actividad enzimática en comparación con un control en el que la enzima no ha sido inactivada. La función enzimática puede inactivarse totalmente mediante la deleción de todo o parte de un polinucleótido que codifica el polipéptido que tiene función enzimática. "Parte de un polinucleótido", como se usa en la presente descripción con respecto a la inactivación de la función enzimática, se refiere a una porción del polinucleótido, cuya deleción es suficiente para causar la inactivación total de la función enzimática. Una parte del polinucleótido podría ser un solo nucleótido o múltiples nucleótidos siempre que la deleción de como resultado la terminación prematura del polipéptido o una mutación de desplazamiento de marco, cada uno de los cuales resultaría en un polipéptido inactivo. Una parte del polinucleótido podría ser una parte que codifica al menos el 10 % pero menos del 90 % del polipéptido, lo que también da como resultado la pérdida de la función enzimática.
El término "ajustar", como se usa en una modalidad de la presente descripción, se refiere a aumentar o disminuir el rendimiento de un carotenoide en al menos el 25 %, en al menos el 30 %, en al menos el 35 %, en al menos el 40 %, en
al menos el 45 %, o aproximadamente! 50 % o más en un huésped fúngico manipulado genéticamente en comparación con un huésped fúngico control no manipulado genéticamente. El término "ajustado", como se usa en una modalidad de la presente descripción, se refiere a un rendimiento aumentado o disminuido de un carotenoide en al menos el 25 %, en al menos el 30 %, en al menos el 35 %, en al menos el 40 %, en al menos el 45 %, o aproximadamente el 50 % o más en un huésped fúngico manipulado genéticamente en comparación con un huésped fúngico control no manipulado genéticamente.
El término "ajustar", como se usa en otra modalidad de la presente descripción, se refiere a un cambio en la composición de carotenoides en un huésped fúngico manipulado genéticamente en comparación con un huésped fúngico control no manipulado genéticamente. El término "ajustado", como se usa en otra modalidad de la presente descripción, se refiere a una composición cambiada de carotenoides en un huésped fúngico manipulado genéticamente en comparación con un huésped fúngico control no manipulado genéticamente.
Así, en un aspecto, la presente invención proporciona un método para ajustar el nivel de producción y composición de carotenoides en un huésped fúngico. De acuerdo con este aspecto, el método comprende la manipulación genética de uno o más de los polinucleótidos implicados en la biosíntesis de carotenoides. En una modalidad, los carotenoides son licopeno, beta-caroteno, gamma-caroteno, toruleno, torularodina o derivados de estos. En algunas modalidades, un derivado es un derivado hidroxilado, un derivado glicosilado o un derivado oxidado. En otra modalidad, el huésped fúngico es Rhodosporidium o Rhodotorula. En algunas modalidades, el método comprende manipular genéticamente uno o más polinucleótidos implicados en la biosíntesis de carotenoides en un huésped fúngico y hacer crecer el huésped fúngico para producir los carotenoides, por lo que se ajusta el nivel de producción o la composición de los carotenoides.
En una modalidad, la manipulación genética comprende la regulación negativa de uno o más polinucleótidos seleccionados de SEQ ID NOs:1, 3, 5, 7, 8, 9, 10, 11, 13, 14, 16, 17, 19 y 20, o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de nucleótidos con los mismos en un huésped fúngico. En otra modalidad, la manipulación genética comprende la regulación negativa de uno o más polinucleótidos que codifican polipéptidos seleccionados de SEQ ID NO:2, 4, 6, 12, 15, 18 y 21, o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de aminoácidos con los mismos en un huésped fúngico. En algunas modalidades, la regulación negativa se compara con un huésped fúngico sin manipulación genética. En otras modalidades, uno o más polinucleótidos se regulan negativamente por ARNi, un represor transcripcional artificial o un promotor débil.
La regulación negativa de un polinucleótido de la presente invención puede lograrse mediante el uso de técnicas bien conocidas, que incluyen, pero no se limitan a, técnicas de ARNi, como dsRNA, miRNA, siRNA, smRNA, hpRNA o ihpRNA (denominadas colectivamente moléculas de ARNi), supresión de sentido (co-supresión), antisentido, y similares. Dichas técnicas se describen en la Patente de los Estados Unidos núm. 7,312,323 y las referencias citadas allí. Por ejemplo, la reducción podría lograrse, por ejemplo, con la transformación de una célula fúngica huésped para comprender un promotor y otras regiones reguladoras 5' y/o 3' descritas en la presente descripción unidas a una secuencia de nucleótidos antisentido, horquilla, molécula interferente de ARN, ARN bicatenario, microARN u otra molécula de ácido nucleico, de manera que la expresión de la molécula preferida por los tejidos interfiera con la traducción del ARNm de la secuencia de ADN nativa o inhiba la expresión de la secuencia de ADN nativa en las células vegetales. Para una descripción más detallada de las técnicas de ARNi o técnicas de microARN, véase, por ejemplo, las Patentes de los Estados Unidos núms.
5,034,323; 6,326,527; 6,452,067; 6,573,099; 6,753,139; y 6,777,588. Véase también las publicaciones internacionales núms. WO 97/01952, WO 98/36083, WO 98/53083, WO 99/32619 y WO 01/75164; y la Publicación de la Solicitud de Patente de los Estados Unidos núms. 2003/0175965, 2003/0175783, 2003/0180945, 2004/0214330, 2005/0244858, 2005/0277610, 2006/0130176, 2007/0265220, 2008/0313773, 2009/0094711, 2009/0215860, 2009/0308041, 2010/0058498 y 2011/0091975. Las moléculas de ARNi o las moléculas de microARN (denominadas colectivamente en la presente descripción como moléculas de ARNi) pueden prepararse por un experto en la técnica mediante el uso de técnicas bien conocidas en la técnica, que incluyen técnicas para la selección y prueba de moléculas de ARNi y moléculas de microARN que son útiles para la regulación negativa de un polinucleótido de la presente invención. Véase, por ejemplo, Wesley y otros (2001), Mysara y otros (2011) y Yan y otros (2012).
Típicamente se ha encontrado que el dsRNA de 200-700 pb es particularmente adecuado para inducir ARNi en vegetales. También se ha encontrado que los ARN de horquilla que contienen un intrón, por ejemplo, un constructo que comprende una secuencia codificante de ARN en una dirección de sentido operativamente unida a un intrón operativamente unido a una secuencia codificante de ARN en una dirección antisentido o viceversa que es capaz de formar un ARN de intrónhorquilla (ihpRNA), es adecuado para inducir ARNi en vegetales. Véase, por ejemplo, Wang y otros (2000), Fuentes y otros (2006), Bonfim y otros (2007) Vanderschuren y otros (2007a, 2007b), Zrachya y otros (2007). Por ejemplo, puede prepararse un constructo de ácido nucleico que incluye un ácido nucleico que se transcribe en un a Rn que puede hibridarse consigo mismo, por ejemplo, un ARN bicatenario que tiene una estructura de horquilla. Además, las estructuras de horquilla pueden prepararse como se describe por Guo y otros. (2003).
Por ejemplo, puede prepararse un constructo de ácido nucleico que incluye un nucleico que se transcribe en un ARN que puede hibridarse consigo mismo, por ejemplo, un ARN bicatenario que tiene una estructura de horquilla. En algunas modalidades, una hebra de la porción del tallo de un ARN bicatenario comprende una secuencia que es similar o idéntica
a la secuencia codificante de sentido, o un fragmento de esta, de un polinucleótido como se describe en la presente descripción, y que es de aproximadamente 10 nucleótidos a aproximadamente 1800 nucleótidos de longitud. La longitud de la secuencia que es similar o idéntica a la secuencia codificante de sentido puede ser de 10 nucleótidos a 1000 nucleótidos, de 15 nucleótidos a 600 nucleótidos, de 20 nucleótidos a 500 nucleótidos, o de 25 nucleótidos a 100 nucleótidos, o cualquier longitud dentro de los 10 nucleótidos a 2500 nucleótidos. La otra hebra de la porción del tallo de un ARN bicatenario comprende una secuencia que es similar o idéntica a la hebra antisentido, o un fragmento de esta, de la secuencia codificante del polipéptido de interés, y puede tener una longitud que es más corta, igual o mayor que la longitud correspondiente de la secuencia de sentido. En algunos casos, una hebra de la porción del tallo de un ARN bicatenario comprende una secuencia que es similar o idéntica a la región no traducida 3' o 5', o un fragmento de esta, del ARNm que codifica un polipéptido descrito en la presente descripción, y la otra la hebra de la porción del tallo del ARN bicatenario comprende una secuencia que es similar o idéntica a la secuencia que es complementaria a la región no traducida 3' o 5', respectivamente, o un fragmento de esta, del ARNm que codifica un polipéptido descrito en la presente descripción. En otras modalidades, una hebra de la porción del tallo de un ARN bicatenario comprende una secuencia que es similar o idéntica a la secuencia de un intrón o un fragmento de este en el pre-ARNm transcrito a partir de un polinucleótido descrito en la presente descripción, y la otra hebra de la porción del tallo comprende una secuencia que es similar o idéntica a la secuencia que es complementaria a la secuencia del intrón o fragmento de este en el pre-ARNm.
La porción del bucle de un ARN bicatenario puede ser de 3 nucleótidos a 5000 nucleótidos, por ejemplo, de 3 nucleótidos a 2500 nucleótidos, de 15 nucleótidos a 1000 nucleótidos, de 20 nucleótidos a 500 nucleótidos, o de 25 nucleótidos a 200 nucleótidos, o cualquier longitud dentro de los 3 nucleótidos a 5000 nucleótidos. La porción de bucle del ARN puede incluir un intrón o un fragmento de este. Un ARN bicatenario puede tener cero, uno, dos, tres, cuatro, cinco, seis, siete, ocho, nueve, diez, o más estructuras de horquilla.
En una modalidad, la manipulación genética es la inactivación total de la función enzimática en un huésped fúngico. En algunas modalidades, la inactivación se logra mediante la deleción de todo o parte de uno o más polinucleótidos seleccionados de SEQ ID NOs:10, 11, 16, 17, 19 y 20, o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de nucleótidos con los mismos. En otras modalidades, la inactivación se logra mediante la deleción de todo o parte de uno o más polinucleótidos que codifican polipéptidos seleccionados de SEQ ID NO:12, 18 y 21 o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de aminoácidos con los mismos. En una modalidad, la deleción se realiza mediante el uso de técnicas de recombinación homóloga. Las técnicas de recombinación homóloga se conocen bien por el experto en la técnica. En otra modalidad, la deleción se ayuda mediante el uso de una nucleasa artificial. En una modalidad adicional, la nucleasa artificial es una nucleasa de dedos de zinc (ZFN) o un complejo Cas9-ARNg. Las tecnologías de nucleasa artificial se conocen bien por el experto en la técnica. Véase, por ejemplo, Durai y otros (2005), Makarova y otros (2011) y Mali y otros (2013).
En una modalidad, la manipulación genética implica la sobreexpresión de uno o más polinucleótidos seleccionados de SEQ ID NOs:1, 3, 5, 7, 8, 9, 10, 11, 13, 14, 16, 17, 19 y 20, o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de nucleótidos con los mismos. En otra modalidad, la manipulación genética implica la sobreexpresión de uno o más polinucleótidos que codifican polipéptidos seleccionados de SEQ ID NOs:2, 4, 6, 12, 15, 18 y 21, o un homólogo que comparte al menos el 75 %, al menos el 80 %, al menos el 85 %, al menos el 90 %, al menos el 95 %, al menos el 98 % o al menos el 99 % de identidad de aminoácidos con los mismos. En algunas modalidades, la sobreexpresión está mediada por la introducción de un casete genético sintético en una célula fúngica huésped. En otra modalidad, el casete comprende un promotor heterólogo unido operativamente al polinucleótido, opcionalmente unido operativamente a un terminador transcripcional. En algunas modalidades, la sobreexpresión se compara con un huésped fúngico sin manipulación genética.
En algunas modalidades, la manipulación genética implica una combinación de las manipulaciones genéticas descritas previamente. En una modalidad, la manipulación genética comprende la regulación negativa de uno o más de los polinucleótidos y la sobreexpresión de uno o más polinucleótidos diferentes. En otra modalidad, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos y la sobreexpresión de uno o más polinucleótidos diferentes. En una modalidad adicional, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos y la regulación negativa de uno o más polinucleótidos diferentes. En una modalidad adicional, la manipulación genética comprende la inactivación total de la función enzimática de uno o más polipéptidos codificados por uno o más polinucleótidos, la sobreexpresión de uno o más polinucleótidos diferentes y la regulación negativa de uno o más polinucleótidos diferentes.
La Tabla 1 muestra ejemplos representativos de manipulación genética en huéspedes fúngicos de la presente invención y el efecto con respecto al ajuste de la producción o composición de carotenoides.
Tabla 1

En algunas modalidades, los polinucleótidos descritos en la presente descripción se han incorporado de manera estable en el genoma fúngico. En estas modalidades, los polinucleótidos están unidos operativamente a un promotor que permite la expresión eficiente en especies del género Rhodosporidium y el género Rhodotorula. Los promotores para cada polinucleótido incorporado pueden ser iguales o diferentes. En algunas modalidades, los promotores son promotores encontrados en especies del género Rhodosporidium y el género Rhodotorula. En otras modalidades, los promotores son promotores que se encuentran en otras especies fúngicas. Los ejemplos de promotores adecuados incluyen, pero no se limitan a, promotores de los genes siguientes que codifican las proteínas siguientes: gliceraldehído 3-fosfato deshidrogenasa (GPD), proteína transportadora de acil-CoA (ACP), ácido graso desaturasa, factor de elongación de traducción (TEF), piruvato descarboxilasa (PDC), enolasa (2-fosfoglicerato deshidratasa) (ENO), peptidilprolil isomerasa (PPI), acetil-CoA carboxilasa (ACC) o transaldolasa. En otras modalidades, los genes descritos en la presente descripción también incluyen un terminador transcripcional de ARNm que puede encontrarse en cualquier especie eucariota y sus virus de ADN.
En algunas modalidades, un promotor adecuado se describe en la Publicación de Solicitud de Patente Internacional núm. WO 2012/169969, incorporado por referencia en la presente descripción en su totalidad. Esta solicitud publicada describe varias secuencias de polinucleótidos derivadas de la región aguas arriba del gen de la gliceraldehído fosfato deshidrogenasa (GPD1), el gen del factor de iniciación de la traducción (TEF1) y el gen de la estearoil-CoA-delta 9-desaturasa (FAD1) que funcionan como promotores en hongos. Los promotores descritos en esta solicitud publicada se establecen en las SEQ ID NOs:94-101. En otras modalidades, se describen promotores adicionales en la Publicación de Solicitud de Patente Internacional núm. WO 2014/142747, incorporado por referencia en la presente descripción en su totalidad. Los promotores descritos en esta solicitud publicada se establecen en las SEQ ID NOs:102-118.
Además, los fragmentos operables de las secuencias promotoras descritas en la presente descripción pueden aislarse mediante el uso de ensayos convencionales de tamizaje de promotores y pueden tamizarse para la selección eficiente de las células fúngicas transformadas mediante el uso de las técnicas descritas en la presente descripción. En una modalidad, un fragmento operable, también denominado una porción promotora en la presente descripción, tiene aproximadamente 400 pares de bases hasta aproximadamente 1100 pares de bases de longitud comenzando desde la posición -1 del codón ATG. Como se usa en la presente descripción "hasta" se refiere a la longitud de la porción promotora de los promotores expuestos en las SEQ ID NOs descritas. Por lo tanto, "hasta" se refiere a la longitud máxima de la secuencia promotora si hay menos de 1100 nucleótidos de los promotores de las SEQ ID NOs descritas.
En una modalidad, se proporciona una secuencia promotora que tiene al menos el 60 % de identidad con cualquiera de estas secuencias promotoras. En otra modalidad, se proporciona una secuencia promotora que tiene al menos el 70 % de identidad con cualquiera de estas secuencias promotoras. En una modalidad adicional, se proporciona una secuencia promotora que tiene al menos el 80 % de identidad con cualquiera de estas secuencias promotoras. En una modalidad adicional, se proporciona una secuencia promotora que tiene al menos el 90 % de identidad con cualquiera de estas secuencias promotoras. En otra modalidad, se proporciona una secuencia promotora que tiene al menos el 95 % de identidad con cualquiera de estas secuencias promotoras. En otra modalidad, se proporciona una secuencia promotora que tiene al menos el 98 % de identidad con cualquiera de estas secuencias promotoras.
Los genes que se incorporarán de manera estable en el genoma fúngico están típicamente en forma de un constructo de ADN o polinucleótido que comprende las secuencias promotoras descritas en la presente descripción, una secuencia codificante de polipéptidos unidos operativamente descrita en la presente descripción y una secuencia terminadora transcripcional de ARN unida operativamente. En una modalidad, puede usarse cualquier terminador transcripcional operable en especies de hongos. Los terminadores se ubican típicamente aguas abajo (3') del gen, después del codón de parada (TGA, TAG o TAA). Los terminadores desempeñan un papel importante en el procesamiento y la estabilidad del ARN así como también en la traducción.
Un constructo de ADN o ácido nucleico que comprende un promotor operable en hongos, una secuencia de ADN que codifica proteínas y un terminador operable en hongos también puede denominarse en la presente descripción como un casete de expresión. El casete de expresión puede incluir otras regiones reguladoras transcripcionales como se conocen
bien en la técnica. En otras modalidades, el constructo de ADN o ácido nucleico o casete de expresión comprende además un marcador de selección. Los marcadores de selección se conocen bien por los expertos en la técnica como casetes de expresión que incorporan dichos marcadores y promotores seleccionables para impulsar su expresión, tal como se describe en la Publicación de Solicitud de Patente Internacional núm. WO 2012/169969. Cualquier promotor adecuado unido operativamente a cualquier marcador de selección adecuado puede usarse en la presente invención. En algunas modalidades, pueden usarse una o más moléculas de ADN en las que cada molécula de ADN tiene uno o más constructos de ácido nucleico.
En otra modalidad, la presente invención proporciona un método para producir carotenoides que comprende cultivar una célula fúngica huésped descrita en la presente descripción en condiciones adecuadas para producir carotenoides. Puede usarse cualquier medio con al menos el 5 % de fuente de carbono. En algunas modalidades, la fuente de carbono es glucosa, manosa, glicerol, sacarosa, xilosa o sus combinaciones. En una modalidad, el medio es MinCAR que contiene 30-100 g de glucosa, 1,5 g de extracto de levadura, 0,5 g de (N H ^S O 4 , 2,05 g de K2HPO4 , 1,45 g de KH2PO4 , 0,6 g de MgSO4 , 0,3 g de NaCl, 10 mg de CaCh, 1 mg de FeSO4 , 0,5 mg de ZnSO4, 0,5 mg de CuSO4 , 0,5 mg de H3BO4 , 0,5 mg de MnSO4, 0,5 mg de NaMoO4 (por litro). El medio se ajusta preferentemente a pH 5-7. En algunas modalidades, el cultivo celular se realiza preferentemente a 25 ° - 35 °C. En otras modalidades, el cultivo se realiza preferentemente en una condición con iluminación.
En la preparación del constructo de ácido nucleico o un casete de expresión, los diversos fragmentos de ADN pueden manipularse, para proporcionar las secuencias de ADN en la orientación adecuada y, según sea apropiado, en el marco de lectura apropiado. Con este fin, pueden emplearse adaptadores o enlazadores para unir los fragmentos de ADN u otras manipulaciones pueden estar implicadas para proporcionar sitios de restricción convenientes, eliminación de ADN superfluo, eliminación de sitios de restricción, o similares. Para este propósito, pueden estar involucradas las mutagénesis in vitro, reparación de cebadores, restricción, hibridación, resustituciones, por ejemplo, transiciones y transversiones.
Los ácidos nucleicos de la presente invención también pueden sintetizarse, ya sea totalmente o en parte, especialmente cuando se desea proporcionar secuencias preferidas en vegetales, mediante métodos que se conocen en la técnica. Por lo tanto, todos o una porción de los ácidos nucleicos de la presente invención pueden sintetizarse mediante el uso de codones preferidos por un huésped seleccionado. Los codones preferidos por especie pueden determinarse, por ejemplo, a partir de los codones usados con mayor frecuencia en las proteínas que se expresan en una especie huésped particular. Otras modificaciones de las secuencias de nucleótidos pueden dar como resultado mutantes con actividad ligeramente alterada.
Puede ser útil generar un número de hongos individuales transformados con cualquier constructo recombinante para recuperar hongos libres de cualquier efecto posicional. También puede ser preferible seleccionar los hongos que contienen más de una copia del constructo de polinucleótido introducido de manera que se obtengan niveles altos de expresión de la molécula recombinante.
La práctica de la presente invención emplea, a menos que se indique lo contrario, técnicas convencionales de química, biología molecular, microbiología, ADN recombinante, genética, inmunología, biología celular, cultivo celular y biología transgénica, que están dentro del estado de la técnica. Véase, por ejemplo, Maniatis y otros, 1982, Molecular Cloning (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, Nueva York); Sambrook y otros, 1989, Molecular Cloning, 2da Ed. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, Nueva York); Sambrook y Russell, 2001, Molecular Cloning, 3ra Ed. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, Nueva York); Ausubel y otros, 1992), Current Protocols in Molecular Biology (John Wiley & Sons, incluidas las actualizaciones periódicas); Glover, 1985, ADN Cloning (IRL Press, Oxford); Russell, 1984, Molecular biology of plants: a laboratory course manual (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.); Anand, Techniques for the Analysis of Complex Genomes, (Academic Press, Nueva York, 1992); Guthrie y Fink, Guide to Yeast Genetics and Molecular Biology (Academic Press, Nueva York, 1991); Harlow y Lane, 1988, Antibodies, (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, Nueva York); Nucleic Acid Hybridization (B. D. Hames & S. J. Higgins eds. 1984); Transcription And Translation (B. D. Hames & S. J. Higgins eds.
1984); Culture Of Animal Cells (R. I. Freshney, Alan R. Liss, Inc., 1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical Guide To Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press, Inc., N.Y.); Methods In Enzymology, Vols. 154 y 155 (Wu y otros eds.), Immunochemical Methods In Cell And Molecular Biology (Mayer y Walker, eds., Academic Press, London, 1987); Handbook Of Experimental Immunology, Volúmenes I-IV (D. M. Weir y C. C. Blackwell, eds., 1986); Riott, Essential Immunology, 6ta Edición, Blackwell Scientific Publications, Oxford, 1988; Fire y otros, ARN Interference Technology: From Basic Science to Drug Development, Cambridge University Press, Cambridge, 2005; Schepers, ARN Interference in Practice, Wiley-VCH, 2005; Engelke, RNA Interference (ARNi): The Nuts & Bolts of siRNA Technology, DNA Press, 2003; Gott, RNA Interference, Editing, and Modification: Methods and Protocols (Methods in Molecular Biology), Human Press, Totowa, NJ, 2004; Sohail, Gene Silencing by RNA Interference: Technology and Application, CRC, 2004.
EJEMPLOS
La presente invención se describe con referencia a los siguientes Ejemplos, que se ofrecen a modo de ilustración y no pretenden limitar la invención de ninguna manera. Se utilizaron técnicas estándar bien conocidas en la técnica o técnicas específicamente descritas a continuación.
Ejemplo 1
Cepas, productos químicos, medios y condiciones de cultivo:
Las cepas ATCC 10657, ATCC 10788, ATCC 204091 de Rhodosporidium toruloides (anteriormente conocido como Rhodotorula glutinis), la cepa ATCC 90781 de Rhodotorula glutinis se adquirieron de ATCC (Estados Unidos). Las cepas WP1 de Rhodotorula glutinis graminis y FGSC 10293 (IAM13481) de Sporobolomyces roseus se obtuvieron de Fungal Genetics Stock Center (Universidad de Missouri, Estados Unidos). Las cepas AGL1 [33] y AGL2 [34] de A. tumefaciens se usaron para ATMT.
Las cepas de Rhodosporidium se cultivaron a 28 °-30 °C en YPD Broth (1 % de extracto de levadura, 2 % de peptona, 2 % de glucosa) o en agar patata-dextrosa sólido (PDA). A. tumefaciens se cultivó a 28 °C en medio 2YT líquido o sólido (1,6 % de triptona, 1 % de extracto de levadura, 0,5 % de NaCl). Escherichia coli XL1-Blue se cultivó en caldo Luria-Bertani (LB) o en agar LB y se usó para la manipulación rutinaria del ADN. Para acumular la producción de carotenoides y lípidos, R. toruloides se cultivó en medio productor de carotenoides (MinCAR) y medio de acumulación de lípidos (MinRL2), respectivamente. MinCAR y MinRL2 se modificaron del medio de carotenoides [35] y el medio lipídico [36]. El medio MinCAR contiene (por litro) 70 g de glucosa, 1,5 g de extracto de levadura, 0,5 g de (NH4)2SO4 , 2,05 g de K2HPO4, 1,45 g de KH2PO4, 0,6 g de MgSO4, 0,3 g de NaCl, 10 mg de CaCh, 1 mg de FeSO4, 0,5 mg de ZnSO4 , 0,5 mg de CuSO4 , 0,5 mg de H3BO4, 0,5 mg de MnSO4, 0,5 mg de NaMoO4 (pH 6,1). El medio MinRL2 contiene (por litro) 100 g de glucosa, 1,5 g de extracto de levadura, 0,5 g de (NH4)2SO4, 2,05 g de K2HPO4, 1,45 g de KH2PO4, 0,6 g de MgSO4 , 0,3 g de NaCl, 10 mg de CaCh, 1 mg de FeSO4 , 0,5 mg de ZnSO4 , 0,5 mg de CuSO4 , 0,5 mg de H3BO4, 0,5 mg de MnSO4 , 0,5 mg de NaMoO4 (pH 6,1).
Ejemplo 2
Aislamiento de ADN genómico y ARN total
El ADN genómico y el ARN de R. toruloides se extrajeron como se describió anteriormente [37]. El ADN extraído se analizó cualitativamente mediante electroforesis en gel de agarosa y se cuantificó con el Espectrofotómetro NanoDrop® ND-1000 (Nanodrop Technologies, Estados Unidos).
Ejemplo 3
Transformación mediada por Agrobacterium tumefaciens(ATMT):
La transformación de los hongos a través de ATMT se realizó como se describió anteriormente, a menos que se indique lo contrario [37]. Los vectores binarios se transformaron mediante electroporación en A. tumefaciens AGL1 o AGL2 mediante el uso de una cubeta al 0,2 mM acoplada a un electroporador BioRad que se fijó a 2,5 kV, 25 pF, 400 O. Los transformantes se seleccionaron en placas de agar 2YT suplementadas con estreptomicina (100 pg/ml).
Ejemplo 4
Constructos de ADN
Los oligonucleótidos usados se enumeran en la Tabla 2. Todas las enzimas de restricción y modificación se adquirieron de New England Biolabs (NEB, Massachusetts, Estados Unidos).
Tabla 2



Diversos promotores, como el promotor de U. maydis gpdl (Pgpd, 595 pb de longitud) [38, 39] y RtGPDI (795 pb) [37], se han descrito previamente y se amplificaron mediante el uso de la plantilla del plásmido pEX1 [42] y el ADN genómico de R. toruloides ATCC 10657, mediante el uso de pares de cebadores Pgpd-Sf/Pgpd-Nr y Rt011S/Rt012N, respectivamente. Los productos resultantes de PCR se digirieron con SpeI y NcoI y se clonaron mediante ligación de 3 fragmentos con el fragmento de ADN sintético Bsp de 1030 pbHI/SmaI del gen hpt-3 [37] y el fragmento de ADN de pEC3GPD-GUS de 8855 pb SpeI/SacI (extremo romo), que crea pEC3GPD-HPT3 y pEC3GPDR-HPT3, respectivamente.
Para crear constructos de deleción para CARI, CAR3 y ALD1, el fragmento de ADN que cubre las regiones codificantes completas del gen (3,0 kb, 2,8 kb y 3,0 kb, respectivamente) se amplificó mediante el uso de ADN genómico de R. toruloides ATCC 10657 como plantilla y el par de oligos Rt127-2/Rt128-2, CAR3Lf/CAR3Rr2 y ALD1Lf/ALD1Rr como pares de cebadores, respectivamente. El producto de PCR de extremos romos se ligó al vector pEX2 PmeI/SacI (extremos romos) para crear el vector intermedio pEX2CAR1, pEX2CAR3 y pEX2ALD1, respectivamente. Posteriormente, el casete de resistencia a la higromicina (RtGPDi::hpt-3::Tnos con la versión de 795 pb del promotor RtGPD1 que impulsa la expresión de hpt-3) amplificado a partir del plásmido pRH2034 se ligó al pEX2CAR1 cortado con SmaI/MfeI, pEX2CAR3 cortado con PvuM/BglII, y pEX2ALD1 cortado con XhoI/BspHI (extremo romo) para crear el plásmido específico del gen, pKOCAR1, pKOCAR3 y pKOALD1, respectivamente.
Para la deleción de CCD1, el brazo derecho e izquierdo (0,9 kb cada uno) para recombinación homóloga se amplificó mediante el uso de ADN genómico de R. toruloides ATCC 10657 como plantilla y el par de oligos específico CCD1L-Kf/CCD1L-Hry CCD1R-Bf/CCD1R-Str, respectivamente. El plásmido de deleción génica pKOCCD1 se creó mediante la ligación de cuatro fragmentos que consisten en brazo derecho digerido por KpnI/HindIII, brazo izquierdo digerido por BamHI/StuI, casete de resistencia a la higromicina HindIII /BamHI de pDXP795hptR [32] y el vector pEX2 digerido por KpnI/SacI. Las cepas de E. coli recombinante con los fragmentos correctos se identificaron mediante PCR de colonias seguido de secuenciación del ADN de todo el casete de recombinación. Se aplicó una estrategia similar para la deleción de CDS1. Los pares de oligos CDS1L-Stf/CDS1L-Hr y CDS1R-Bf/CDS1R-Sr se usaron para amplificar los brazos de homología derecha e izquierda de CDS1 (0,5 kb cada uno), respectivamente. Los fragmentos de ADN se digirieron mediante el uso de StuI/HindIII para el brazo izquierdo y BamHI/SacI para el brazo derecho, que se ligaron con el casete de resistencia a la higromicina HindIII/BamHI y el vector pEX2 digerido con SmaI/SacI.
Para la expresión in vitro de ALD1, las secuencias de ADNc se amplificaron mediante RT-PCR con la plantilla de ARN total y los oligos Rt203Nf y Rt204Br. Los productos de PCR de doble digestión con NcoI-BamHI se clonaron en el vector pRH2034 en los mismos sitios que se crea el plásmido pRHALD1, en el que una fusión Ald1-eGFP se impulsó por el promotor RtGPD1.
Para la deleción de ROC1, se usaron pares de los oligos CARSL-Stf/CARSL-Hr y CARSR-Bf/CARSR-Sr para amplificar los fragmentos flanqueantes homólogos 5' y 3' (0,9 kb cada uno). Se realizó una ligación de cuatro fragmentos con el vector binario pEX2 SacI-PmeI, los lados 5' de StuI-HindIII, el casete resistente a la higromicina optimizado con el codón BamHI-HindIII de pDXP795hptR (P GPDi::hpt-3::T nos) y los lados 3' de BamHI-SacI para generar el plásmido de deleción génica pKOROC1, donde Pgpd1 es el promotor de gliceraldehído 3-fosfato de R. toruloides ATCC 10657 con el número de registro de GenBank de JN208861, hpt-3 es el gen optimizado con el codón que codifica la higromicina fosfotransferasa (JQ806387), y Tnos es el terminador de agrobacterium (Liu, Koh y otros 2013).
Para estudios de complementación de Acarl, el locus del genoma de CAR1 que oscila de 389556 a 393649 nt del andamio WGS # 18 se amplificó mediante el uso de la plantilla de R. toruloides y los oligos Rt127-2 y CDSL1. Los productos de PCR de 4,1 kb fueron de extremos romos y se ligaron con el vector pRH2034 linealizado con SpeI (extremo romo)-PmeI para crear el plásmido de complementación pRHCAR1. Para estudios de complementación de Arocl, el locus del genoma
de ROC1 que oscila entre 622910 y 627480 nt del andamio WGS # 9 se amplificó mediante el uso de la plantilla de R. toruloides y los oligos Rt303Sf y Rt304Pmr. Los productos de PCR de 4,6 kb se digirieron doblemente con SpeI y PmeI, y se ligaron con el vector pRH2034 linealizado con SpeI-PmeI para crear el plásmido de complementación pRHROC1.
Ejemplo 5
PCR de colonias y análisis de Southern blot
El análisis por PCR de colonias fúngicas se usó para la detección de candidatos de mutantes de deleción génica. Brevemente, se cultivaron colonias individuales de transformantes en 150 pl de YPD Broth suplementado con cefotaxima (300 mg/ml) e higromicina (150 mg/ml) durante varias horas. Se usó un microlitro de cultivo celular para el análisis de PCR de colonias con un par de oligos apropiado dentro de la región de selección génica (Tabla 2). El PCR se realizó mediante el uso de i-Taq polimerasa (í-DnA Biotech, Singapur) y el programa siguiente: iniciación a 95 °C durante 5 minutos, seguido de 35 ciclos a 94 °C durante 30 segundos, a 58 °C durante 30 segundos y a 72 °C durante 45 segundos, y una mayor extensión a 72 °C durante 5 minutos. Se usó una electroforesis para la identificación de mutantes candidatos que carecen de fragmentos de ADN que podrían amplificarse mediante el uso de la plantilla de ADN genómico de WT.
Para verificar los verdaderos mutantes de deleción génica sin ninguna integración ectópica, los ADN genómicos se digirieron con HindlII, PstI, Pvul, Pvul, HinclI y Pvul para los supuestos mutantes de desactivación génica Acarl, Acar3, Accdl, Acdsl, Aald l y Arocl, respectivamente. Los fragmentos de ADN que contienen los brazos derechos de CARI, CAR3, ALD1, ROC1 y los brazos izquierdos de CCD1, CDS1 se marcaron con digoxigenina mediante el uso de DIG-High prime DNA labeling and detection starter Kit II (Roche Diagnostics, Estados Unidos). La hibridación Southern blot se realizó de acuerdo con las instrucciones del fabricante.
Ejemplo 6
PCR de transcripción inversa cuantitativa
El ARN total se extrajo como se describió anteriormente [37]. Antes del PCR de transcripción inversa cuantitativa (qRT-PCR), el ARN total se trató con DNasa I (Roche Diagnostics) para eliminar las trazas de ADN y se recuperó mediante precipitación con etanol. El ADNc se sintetizó mediante el uso de iScript™ Reverse Transcription Supermix (Bio-Rad, Estados Unidos) y el q-PCR se realizó en el Sistema de detección de secuencia ABI PRISM 7900HT mediante el uso de ABI SYBR® Select Master Mix (Life Technologies, Estados Unidos). Las condiciones del qRT-PCR fueron las siguientes: una etapa inicial de desnaturalización a 50 °C durante 2 min y a 95 °C durante 10 min, seguida de 40 ciclos de desnaturalización a 95 °C durante 15 segundos, hibridación a 60 °C durante 1 minuto. Las muestras se analizaron por triplicado y los datos se obtuvieron mediante el uso del programa SDS 2.4 (Life Technologies, Estados Unidos). Los niveles relativos de expresión génica se calcularon contra el gen de referencia ACT1 (SEQ ID NOs: 22, 23, 24 para genómica, ADNc y proteína, respectivamente) mediante el uso del método 2-AACt con el programa RQ Manager v1.2.1 (Applied Biosystems, Estados Unidos).
Ejemplo 7
Detección de genes involucrados en la biosíntesis de carotenoides
La cepa haploide de R. toruloides ATCC 10657 se mutagenizó mediante inserciones aleatorias de T-DNA mediante la transformación del vector binario de T-DNA pRH201 mediada por Agrobacterium tumefaciens (Figura 1). Los transformantes se seleccionaron en medio de agar YPD suplementado con 300 pg/ml de cefotaxima y 150 pg/ml de higromicina. Después de incubarse a 28 °C durante 5 días, los transformantes que muestran colores albinos o pálidos se transfirieron a medio YPD líquido (300 pg/ml de cefotaxima, 150 pg/ml de higromicina) para la propagación. Después de rayar en las placas de PDA suplementadas con los antibióticos anteriores, las colonias individuales que mostraron un fenotipo de color estable se nombraron Rhodosporidium Carotenoide Mutant (RCM).
Ejemplo 8
Identificación de posiciones de etiquetado de T-DNA
Las posiciones de la etiqueta T-DNA en el genoma se identificaron mediante PCR termal de entrelazado asimétrico de alta eficacia (hiTAIL-PCR) [43, 44]. Se usaron cebadores específicos (HRSP1, HRSP2 y HRSP3) y el cebador arbitrario LAD1-4 para las secuencias de flanqueo del borde izquierdo (LB) de la T-DNA mientras que los cebadores específicos (HRRSP1, HRRSP2 y HRRSP3) y el cebador arbitrario LAD1-4 para las secuencias flanqueantes del borde derecho (RB). Las reacciones de PCR se llevaron a cabo con i-Taq DNA polymerase (i-DNA, Singapur) en un controlador térmico programable PTC-200™ (Bio-Rad, Estados Unidos). Los productos del PCR se purificaron mediante el uso del kit de extracción de gel (Qiagen, Estados Unidos) y se secuenciaron directamente mediante el uso del kit terminador BigDye (Applied Biosystems, Estados Unidos) con oligos HRRSP3 (para secuencias flanqueantes RB) o HRSP3 (secuencias flanqueantes LB). Para las muestras que dieron resultados de secuenciación pobres, los productos del PCR se clonaron en el vector pGTM-T easy (Promega, Estados Unidos) y se secuenciaron mediante el uso de los oligos M13FP y M13RP.
Ejemplo 9
Extracción de carotenoides
Las células se cultivaron en 50 ml de medio MinCAR en matraces de agitación a 30 °C y se sedimentaron por centrifugación. Después de lavar dos veces con agua, se determinó la masa celular húmeda mediante el pesaje y mezclado con la misma masa de perlas de vidrio lavadas con ácido (0,4-0,6 mm de diámetro, Sigma-Aldrich) y 5 ml de DMSO. Las células se lisaron mediante agitación vigorosa en vórtex durante 10 minutos, 1 hora de incubación a 65 °C seguida de congelación a -20 °C. Después de descongelar, la suspensión se centrifugó a 10000 g, y el sobrenadante que contenía carotenoides solubles en DMSO se transfirió a un tubo nuevo mientras que el residuo celular insoluble se volvió a extraer con 30 ml de éter de petróleo ligero - acetato de etilo (36:19) durante 10 minutos a temperatura ambiente. Los contenidos de las dos extracciones se combinaron y se extrajeron con 2 ml de NaCl saturado. La fase de disolvente se recogió después de la centrifugación y se secó bajo un flujo de gas nitrógeno. Las muestras se redisolvieron en hexano y se almacenaron a -20 °C antes de un análisis posterior.
Ejemplo 10
Métodos de cuantificación
La biomasa celular (peso seco de la célula) se determinó mediante la liofilización del sedimento celular recogido por centrifugación hasta que se alcanzó un peso constante.
La concentración de glucosa en los medios se cuantificó mediante HPLC. El medio se separó de las células mediante centrifugación y se filtró a través de una membrana de nylon de 0,2 pm. Se inyectaron 10 pl de la muestra y se pasaron a través de una columna Aminex 87H de 300 x 7,0 mm (Bio-Rad) a una tasa de flujo constante de 0,7 ml/min mediante el uso de ácido sulfúrico 5 mM como fase móvil. La columna se mantuvo a 50 °C y se detectó la glucosa con un detector de índice de refracción (RID, Shimadzu, Japón). La concentración de glucosa en el cultivo celular se determinó mediante el uso de una curva de calibración construida con la solución acuosa de glucosa estándar.
Los picos principales se determinaron mediante los espectros de absorción en hexano y espectrometría de masas. La técnica de ionización química a presión atmosférica (APCI) es como se describió anteriormente [46, 47] con algunas modificaciones. Brevemente, las muestras (x pl) se analizaron en un sistema Shimadzu UPLC-MS (APCI) (Shimadzu, Japón) equipado con una columna de YMC-carotenoide (fase inversa C30, 03 pm, 150 mm x 3,0 mm I.D., YMC, Japón) a una tasa de flujo de 0,3 ml/min en un gradiente lineal en 3 minutos, desde la mezcla A al 100 % (MeOH/tercbutilmetiléter/agua, 30:1:10, v/v/v) hasta la mezcla B al 50 % (MeOH/terc-butilmetiléter, 1:1, v/v) seguido de la mezcla B al 100 % durante 0,5 min y después la columna se mantuvo en las condiciones durante 15 min a una tasa de flujo de 0,6 ml/min. Se usó APCI en modo positivo para la identificación de componentes de carotenoides con 15 L/s de gas nitrógeno como envoltura y gas auxiliar. La temperatura del vaporizador y el capilar se fijó a 350 °C y 150 °C, respectivamente, y los voltajes de las lentes de los capilares y los tubos se fijaron a 50 V y 125 V, respectivamente.
Ejemplo 11
Caracterización de mutantes biosintéticos de carotenoides
La detección de aproximadamente 20 000 transformantes de T-DNA mediante la identificación visual de los colores de las colonias conduce a la identificación de seis mutantes de carotenoides, que se denominan RAM1-5 (mutantes de carotenoides de R. toruloides) (Figs. 2a-2E, Tabla 3). La búsqueda mediante hiTAIL-PCR y BLAST de las etiquetas de secuencia reveló que los T-DNA se insertaron en genes que codifican un supuesto transportador de riboflavina, aldehído hidrogenasa, transportador de hexosa, factor asociado a la proteína de unión a TATA, fitoeno desaturasa y aldehído deshidrogenasa grasa, respectivamente (Tabla 3). Se conoce bien el papel de la fitoeno desaturasa, o Carl en hongos (EC 1.3.99.30) en la producción de licopeno [48].
Tabla 3

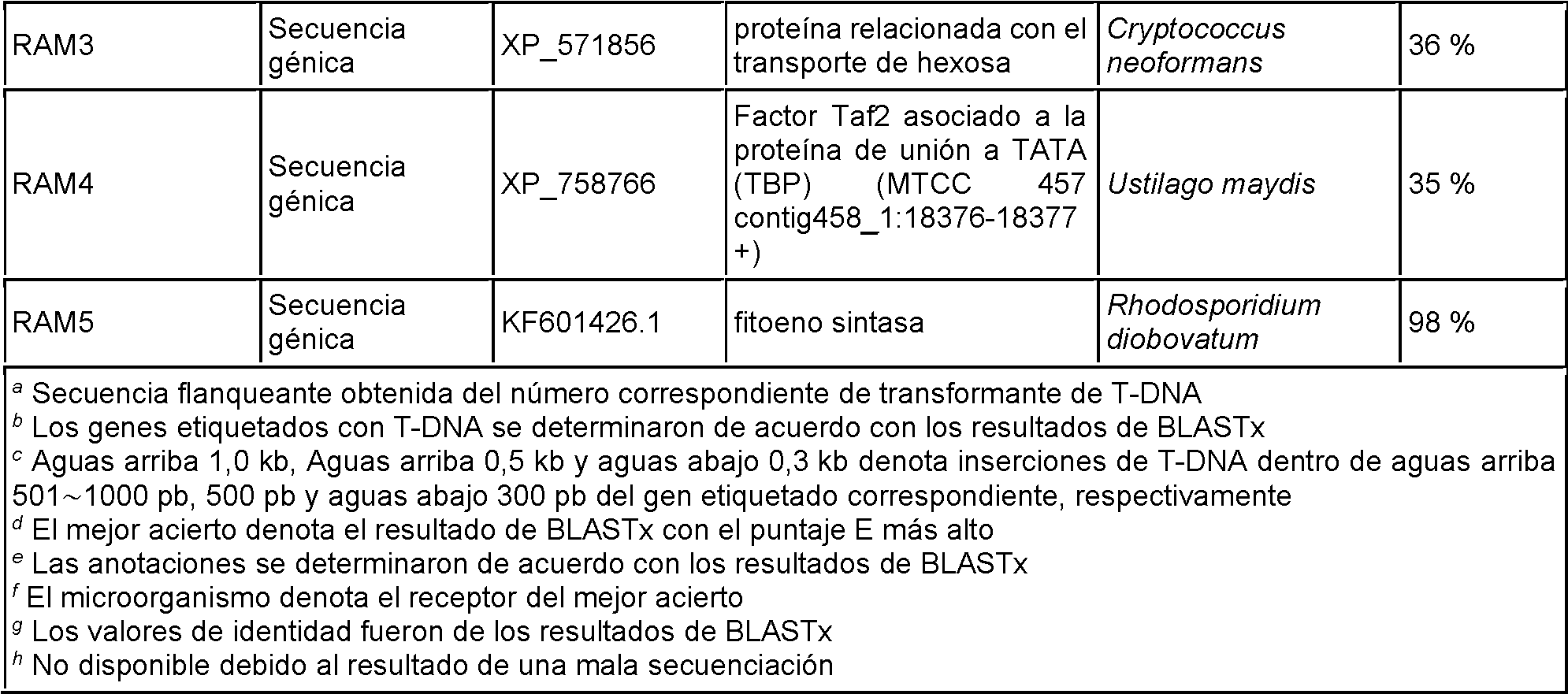
Ejemplo 12
Análisis del grupo de genes de biosíntesis de carotenoides en R. toruloides mediante Genética inversa
El hiTAIL-PCR reveló que el fenotipo albino RCM5 es el resultado de un T-DNA insertado entre 391802 nt y 391803 nt en el andamio genómico # 18 (AEVR02000018) de R. glutenis ATCC 204091, ubicado en el 3er exón del supuesto gen de la fitoeno desaturasa (CARI, locus del genoma RTG_00274) (Figura 2B), una enzima implicada en la biosíntesis de carotenoides. Para confirmar la función de este gen que se interrumpió por la inserción de T-DNA, el supuesto CARI CDS se eliminó por desactivación génica específica mediante el uso de ATMT de pKOCARI, en el que la secuencia de nucleótidos que oscila entre 948 y 2097 del CDS se reemplazó por el casete resistente a la higromicina (PGPDi::hpt-3:.Tnos, Figura 2B). El mutante nulo correcto (úcarl) se verificó mediante análisis de Southern blot (Figura 2C). Como se esperaba, la colonia Acar1 mostró un color cremoso en lugar del color naranja observado en WT. El color cremoso de A cari podría restaurarse a naranja mediante la integración ectópica del alelo del gen CARI en el genoma (cepa AcarIC en la Figura 2D). Los análisis de los perfiles de carotenoides confirmaron la pérdida de picos de p-caroteno, Y-caroteno, toruleno y torularodina en Acari y todos los picos perdidos se restauraron en la cepa complementada, AcarIC, con la integración del T-DNA del vector binario pRHCAR1, donde el alelo completo de CARI que oscila desde -1166 aguas arriba hasta 517 aguas abajo de los codones transcripcionales de inicio y parada, respectivamente (Figura 2E). Los resultados respaldan fuertemente que CARI codifica una de las enzimas clave involucradas en la vía biosintética de carotenoides. Para identificar más genes en la vía biosintética de carotenoides, se realizaron búsquedas de tBLASTn mediante el uso de la GGPP sintasa de U. maydis (CAR3) (XP_760606, GenBank) y fitoeno sintasa/caroteno ciclasa (CAR2) (XP_762434) y las secuencias ortólogas correspondientes en R. toruloides se identificaron exitosamente. CAR3 CDS se encontró ubicado en nt 849806-851310 en el andamio # 13 (locus del genoma RTG_00457, AVER02000013) mientras CAR2 en nt 396838-399094 en el andamio # 18.
Nosotros hemos informado que la desactivación génica CAR2 conduce al fenotipo albino [32]. Sin embargo, los detalles sobre su estructura génica continuan siendo desconocidos. Mediante el uso de la amplificación rápida de los extremos de ADNc (RACE) y las técnicas de PCR de transcripción inversa (RT-PCR), se obtuvieron las secuencias de ADNc para CAR1 (SEQ ID NO:1), CAR2 (SEQ ID NO:3), y CAR3 (SEQ ID NO:5). Los ADNc deCAR1, CAR2 y CAR3 abarcan 2430, 2334, y 1546 nt genómico de longitud, que contienen 10, 8 y 6 exones y codifican proteínas de 554 (Car1, SEQ ID NO:2), 608 (Car2, SEQ ID NO:4) y 359 (Car3, SEQ ID NO:6) aa, con 19, 77 y 41 nt 5'u Tr en los ADNc, respectivamente. Las secuencias genómicas correspondientes se enumeran en las SEQ ID NOs: 7, 8 y 9, respectivamente. El empalme de los 3 ARNm sigue estrictamente la regla canónica GU-AG. Los resultados también revelaron que el T-DNA de RAM5 se integró entre 681 y 682 desde el codón de inicio de CAR1, lo que da como resultado la terminación prematura de la traducción del ARNm de CAR1 después del 158vo aa (Figura 2B).
CAR1 y CAR2 se ubican en el mismo andamio # 18, separados por una secuencia de ADN de 4354 pb. Esta organización es análoga a varios otros hongos carotenogénicos, tales como Blakeslea trispora, Fusarium fujikuroi, Phycomyces blakesleeanus y Sporobolomyces roseus [49-52] (Figura 3A). Una búsqueda homóloga (BLASTx, NCBI) de la secuencia de ADN entre el locus genómico CAR1 y CAR2 descubrió dos genes supuestos que codifican una dioxigenasa de escisión de carotenoides (Ccd1) y una carotenoide desaturasa (Cds1) (Figura 3a ). Las RACE 5' y 3' y el RT-PCR revelaron que los ADNc de CCD1 (SEQ ID NO:11) y CDS1 (SEQ ID NO:14) abarcan 2079 y 793 nt genómico en longitud, que contienen 4 y 3 exones con 41 y 19 nt 5'Ut R que codifican proteínas con 636 (Ccd1, SEQ ID NO:12) y 224 aa (Cds1, SEQ ID n O:14), respectivamente. Nuevamente, el empalme sigue estrictamente la regla canónica GU-AG. Las secuencias genómicas correspondientes se enumeran en SEQ ID NOs: 10 y 13, respectivamente.
La organización divergente de CARI y CAR2 son análogas a Mucor circinelloides (andamio # 1), Phycomyces blakesleeanus (andamio # 5), Blakeslea trispora y F. fujikuroi (cromosoma #11) excepto que los genes CCD1 y CDS1 se encuentran solo en R. toruloides. El genoma de S. roseus parece que sufrió una recombinación entre CAR1 y CAR2, que dio como resultado la pérdida de CDS1 y la translocación de CAR1 (Figura 5A). F. fujikuroi CarXse ubica fuera del CarRA (CAR2 ortólogo) y CarB (CAR1 ortólogo) y se considera como el ortólogo de CCD1 porque su producto proteico exhibe una secuencia de aa altamente homóloga a Ccd1 (43 % de identidad). La sintenia genética compartida entre estos hongos carotenogénicos sugiere un origen evolutivo común de los grupos de genes carotenoides.
Para confirmar sus funciones en la biosíntesis de carotenoides, se crearon mutantes nulos. Similar a A cari y A car2, las colonias de A car3 exhibieron un fenotipo de color cremoso mientras que A ccdl y A cdsl mostraron también colores diferentes significativamente a WT (Figura 3C). El análisis de sus carotenoides mediante HPLC reveló que la producción de carotenoides se abolió totalmente en Acar3, Acar2 y Acari (datos no mostrados). Los niveles de producción total de carotenoides aumentaron ligeramente en A ccdl en un 18 %, pero disminuyeron drásticamente en Acdsi en un 69 % (Figura 3D). Excepto la ligera disminución de p-caroteno (6 %), con respecto a los componentes de los carotenoides acumulados, la deleción de CCD1 podría dar como resultado un aumento del 86 %, el 14 % y el 65 % en torularodina, toruleno y ycaroteno, respectivamente (Figura 3D). En el mutante cdsl, la cuantificación de todos los componentes de carotenoides disminuyó, especialmente más de la mitad disminuyó en torularodina y toruleno (Figura 3D).
Ejemplo 13
Efecto de la deleción de ALD1 sobre la producción de carotenoides
El mutante RCM6 se identificó inicialmente en un examen de detección de genes que afectan la biosíntesis de ácidos grasos y se encontró que el gen representativo ALD1, gen que codifica la aldehido deshidrogenasa grasa, está involucrado en la degradación de un ácido graso poliinsaturado (ácido alfa-linolénico, C18:3n=9) (Solicitud de patente provisional de Estados Unidos núm. 62/047,300 presentada el 8 de septiembre de 2014, incorporada como referencia en la presente descripción). En cuanto a la diferencia significativa en el color de la célula frente a WT, también se estudió el papel de ALD1 en la biosíntesis de carotenoides. La deleción génica mutante de A a ld l se generó por recombinación homóloga a través de ATMT mediante el uso del vector binario pKOALD1. Para obtener la sobreexpresión génica mutante, la proteína de fusión Ald1-RtGFP se fusionó con el promotor RtGPDI y se integró ectópicamente en Aaldl a través de ATMT (mutante OEALD1). Después de una fermentación de 5 días en el medio productor de carotenoides MinCAR, el rendimiento total de carotenoides en Aaldl fue similar a WT, sin embargo, el nivel de beta-caroteno aumentó en aproximadamente el 36 %. Por el contrario, los niveles de toruleno y de torularodina aumentaron significativamente en la cepa de sobreexpresión de ALD1 (Figura 4). Una secuencia genómica, secuencia de ADNc y secuencia de proteína para a LD1 se establecen en las SEQ ID NOs: 16, 17 y 18, respectivamente.
Ejemplo 14
Perfiles de ARNm de CARI, CAR2, CAR3, ALD1, CCD1 y CDS1
Se inocularon colonias individuales de R. toruloides ATCC 10657 y mutantes de carotenoides en 50 ml de YPD Broth en matraces cónicos de 250 ml y se cultivaron a 28 °C y 250 rpm hasta la saturación. Los cultivos celulares se separaron en mitades y se continuó el cultivo para cada uno en una plataforma de agitación durante 5 horas más, donde la iluminación se trasmitió mediante el uso de una luz fluorescente (Philips TLD 36 W/865, luz blanca de 4 W/m2, a 75 cm de distancia) o se evitó tras cubrirlo con papel de aluminio. Se tomaron muestras de las células en varios puntos de tiempo y se extrajeron los ARN totales. El qRT-PCR se realizó por triplicado mediante el uso de pares de oligos qCAR3f/qCAR3r, qCAR2f/qCAR2r, qCAR1f/qCAR1r, qCCD1f/qCCD1r, qCDS1f/qCDS1r y qALD1f/qALD1r para CAR3, CAR2, CARI, CCD1, CDS1, ALD1, respectivamente (Tabla 2). Los niveles relativos de ARNm se calcularon frente al gen de referencia de actina (par de oligos qACT1f/qACT1r, Tabla 2) mediante el uso del método 2-AACt. Como se muestra en la Figura 5, los niveles de ARNm de CARI y CAR2fueron mucho más bajos que los de CAR3 y probablemente fueron el cuello de botella para la redirección del flujo de carbono hacia la producción de carotenoides. El ARNm de CARI, CAR2, CAR3 y ALD1 se indujo mediante la luz.
Ejemplo 15
Caracterización de ROC1
En Fusarium fujikuroi, la interrupción de carS mostró una mejor biosíntesis de carotenoides independientemente de la iluminación con la luz (Avalos y Cerda-Olmedo 1987). Sin embargo, los genes involucrados en las etapas anteriores de la vía biosintética de terpenoides como FPP sintasa y HMGR, no se vieron afectadas por la deleción de carS (Rodríguez-Ortiz, Limón y otros 2009).
Mediante el uso de la secuencia de proteínas carS de Fusarium fujikuroi (NCBI GenBank número de registro CCP50075.1) como consulta para buscar las secuencias de escopeta de genoma completo de R. glutinis ATCC 204091 a través del programa en línea tBLASTn (NCBI, Estados Unidos), se encontró un ortólogo supuesto en el andamio de secuencia # 9
(valor E y cobertura de consulta de 4E-37 y 53 %, respectivamente). Para ser consistente con la nomenclatura génica del gen supuesto de R. toruloides se denominó ROC1 (Regulador de la biosíntesis de carotenoides).
La RACE 5' y 3' produjo un par de fragmentos de ADNc de aproximadamente 0,9 y 0,7 kb para ROC1 (datos no mostrados). Mediante el uso del par de oligos Rt301Nf y Rt302Evr (Tabla 2), el ADNc de longitud completa de ROC1 podría amplificarse exitosamente mediante RT-PCR (datos no mostrados). La comparación entre el ADNc y las secuencias genómicas reveló 5 exones para ROC1 (Figura 6A), donde las uniones de empalme se atienen estrictamente a la regla cónica GU-AG.
ROC1 abarca una región del genoma de 3136 nt (SEQ ID NO:19) que codifica un ARNm de 2805 nt con un 5'UTR de 84 nt (SEQ ID NO:20). ROC1 codifica una proteína de 934-aa (SEQ ID NO:21) que contiene un dominio de dedo RING (tipo C3HC4, dominio conservado NCBI cd00162) y un dominio La (LON) de proteasa dependiente de ATP (pfam02190). La secuencia muestra el 96 % y el 97 % de identidad de aa con un homólogo en R. toruloides cepa NP11 (EMS19961.1) y CECT1137 (CDR44527.1, respectivamente).
Rocl comparte menos del 20 % de identidad con los ortólogos de varios hongos carotenogénicos, excepto los de dos zigomicetos, B. trispora y M. cricinelloides (Figura 6B). Además, las secuencias de proteínas dentro del dominio de dedo RING central también muestran una homología muy baja, algunas incluso carecen del motivo C3HC4 (Figura 6C).
Ejemplo 16
Efectos de la deleción de ROC1
Para comprender genéticamente el papel de ROC1 en la biosíntesis de carotenoides, el gen ROC1 se eliminó mediante recombinación homóloga (Figura 7A). La transformación con el vector de deleción génica (pKOROC1) mostró dos tipos de transformantes con diferencias de color obvias, naranja claro y fuerte (Figura 7A), y se encontró que los transformantes naranja fuerte son los verdaderos mutantes de desactivación génica mediante el análisis de Southern blot (Figura 7B). Los mutantes nulos roc l mostraron una morfología y crecimiento celular similar a WT (Figuras 7C y 7D). Sin embargo, pudo observarse una mejora significativa de la biosíntesis de caroteno en mutantes roc l y esto podría restaurarse completamente mediante la introducción de un alelo génico WT ROC1 en un mutante roc l (Figura 7D). Sorprendentemente, la producción de biomasa celular disminuyó significativamente en el mutante de complementación (Figura 7D). El análisis cuantitativo reveló que el cultivo de 5 días en medio MinCAR produjo aproximadamente 1,5 veces más carotenoides en el mutanteroc1 en comparación con los de WT (Figura 7E).
El uso de los términos " un", " una" y "el/la" y referentes similares en el contexto de la descripción de la invención (especialmente en el contexto de las siguientes reivindicaciones) se debe interpretar que abarca tanto el singular como el plural, a menos que se indique de cualquier otra manera en la presente descripción o se contradiga evidentemente por el contexto. Los términos "que comprende", "que tiene", "que incluye" y "que contiene" deben interpretarse como términos abiertos (es decir, que significan "que incluye pero sin limitarse a") a menos que se indique lo contrario. La mención de intervalos de valores en la presente descripción tiene meramente la intención de servir como un método abreviado para referirse individualmente a cada valor por separado que cae dentro del intervalo, a menos que se indique de cualquier otra forma en la presente, y cada valor por separado se incorpora en la descripción como si se mencionara individualmente en la presente. Todos los métodos que se describen en la presente descripción pueden llevarse a cabo en cualquier orden adecuado a menos que se indique de otra manera en la presente descripción o que el contexto lo contradiga claramente de otra manera. El uso de cualquiera y todos los ejemplos, o lenguaje ilustrativo (por ejemplo, “tal como”) proporcionados en la presente descripción simplemente tiene el propósito de ilustrar mejor la invención y no representa una limitación en el alcance de la invención al menos que se reivindique de cualquier otra forma. Ningún lenguaje en la descripción debe interpretarse como que indica cualquier elemento no reivindicado como esencial para la práctica de la invención.
Bibliografía
1. Hirschberg J: Carotenoid biosynthesis in flowering plants. Current opinion in plant biology 2001, 4:210-218.
2. Armstrong GA, Hearst JE: Carotenoids 2: Genetics and molecular biology of carotenoid pigment biosynthesis. FASEB J 1996,10:228-237.
3. Eisenreich W, Bacher A, Arigoni D, Rohdich F: Biosynthesis of isoprenoids via the non-mevalonate pathway. Cellular and Molecular Life Sciences CMLS 2004, 61:1401-1426.
4. Martin VJ, Pitera DJ, Withers ST, Newman JD, Keasling JD: Engineering a mevalonate pathway in Escherichia coli for production of terpenoids. Nature biotechnology 2003, 21:796-802.
5. Armstrong GA, Hearst JE: Carotenoids 2: Genetics and molecular biology of carotenoid pigment biosynthesis. The FASEB Journal 1996, 10:228-237.
6. Kajiwara S, Kakizono T, Saito T, Kondo K, Ohtani T, Nishio N, Nagai S, Misawa N: Isolation and functional identification of a novel cDNA for astaxanthin biosynthesis from Haematococcus pluvialis, and astaxanthin synthesis in Escherichia coli. Plant molecular biology 1995, 29:343-352.
7. Fraser PD, Miura Y, Misawa N: In vitro characterization of astaxanthin biosynthetic enzymes. Journal of Biological Chemistry 1997, 272:6128-6135.
8. Cunningham FX, Gantt E: Genes and Enzymes of Carotenoid Biosynthesis in Plants. Annu Rev Plant Physiol Plant Mol Biol 1998, 49:557-583.
9. Mapari SA, Nielsen KF, Larsen TO, Frisvad JC, Meyer AS, Thrane U: Exploring fungal biodiversity for the production of water-soluble pigments as potential natural food colorants. Current Opinion in Biotechnology 2005, 16:231-238.
10. Takaichi S: Carotenoids and carotenogenesis in anoxygenic photosynthetic bacteria. In The photochemistry of carotenoids. Springer; 1999: 39-69
11. Cobbs C, Heath J, Stireman III JO, Abbot P: Carotenoids in unexpected places: Gall midges, lateral gene transfer, and carotenoid biosynthesis in animals. Molecular phylogenetics and evolution 2013, 68:221-228.
12. Sporn M, Dunlop N, Newton D, Smith J: Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids). In Federation proceedings. 1976: 1332-1338.
13. Seddon JM, Ajani UA, Sperduto RD, Hiller R, Blair N, Burton TC, Farber MD, Gragoudas ES, Haller J, Miller DT: Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Jama 1994, 272:1413-1420.
14. Wolbach SB, Howe PR: Tissue changes following deprivation of fat-soluble A vitamin. The Journal of experimental medicine 1925, 42:753-777.
15. Miki W: Biological functions and activities of animal carotenoids. Pure and Applied Chemistry 1991, 63:141-146. 16. Estrada AF, Brefort T, Mengel C, Díaz-Sánchez V, Alder A, Al-Babili S, Avalos J: Ustilago maydis accumulates isefort T, Mengel C, Díaz-Sánchez V, Alder A, Al-Babili S, Avalos J: J: io Fungal Genetics and Biology 2009, 46:803-813.
17. Giovannucci E, Ascherio A, Rimm EB, Stampfer MJ, Colditz GA, Willett WC: Intake of carotenoids and retino in relation to risk of prostate cancer. Journal of the national cancer institute 1995, 87:1767-1776.
18. Spencer KG: Pigmentation supplements for animal feed compositions. Google Patents; 1989.
19. Herring P: The carotenoid pigments of< i> Daphnia magna</i> straus-I. The pigments of animals feed< i> Chlorella pyrenoidosa</i> and pure carotenoids. Comparative biochemistry and physiology 1968, 24:187-203.
20. Mortensen A: Carotenoids and other pigments as natural colorants. Pure and Applied Chemistry 2006, 78:1477-1491.
21. Peter F, Andrea L-R, Peter W, Naoharu W: Carotenoid Cleavage Enzymes in Animals and Plants. In Carotenoid-Derived Aroma Compounds. Volume 802: American Chemical Society; 2001: 76-88: ACS Symposium Series],
22. Duester G: Involvement of alcohol dehydrogenase, short-chain dehydrogenase/reductase, aldehyde dehydrogenase, and cytochrome P450 in the control of retinoid signaling by activation of retinoic acid synthesis. Biochemistry 1996, 35:12221-12227.
23. Li Y, Zhao ZK, Bai F: High-density cultivation of oleaginous yeast Rhodosporidium toruloides Y4 in fed-batch culture. Enzyme and Microbial Technology 2007, 41:312-317.
24. Zhao X, Hu C, Wu S, Shen H, Zhao ZK: Lipid production by Rhodosporidium toruloides Y4 using different substrate feeding strategies. J Ind Microbiol Biotechnol 2011, 38:627-632.
25. Pan JG, Kwak MY, Rhee JS: High density cell culture of Rhodotorula glutinis using oxygen-enriched air. Biotechnology letters 1986, 8:715-718.
26. Frengova GI, Beshkova DM: Carotenoids from Rhodotorula and Phaffia: yeasts of biotechnological importance. J Ind Microbiol Biotechnol 2009, 36:163-180.
27. Cerdá-Olmedo E: Production of carotenoids with fungi. In Biotechnology of vitamins, pigments and growth factors. Springer; 1989: 27-42
28. Buzzini P, Martini A: Production of carotenoids by strains of< i> Rhodotorula glutinis</i> cultured in raw materials of agro-industrial origin. Bioresource technology 2000, 71:41-44.
29. Frengova GI, Beshkova DM: Carotenoids from Rhodotorula and Phaffia: yeasts of biotechnological importance. Journal of industrial microbiology & biotechnology 2009, 36:163-180.
30. Bhosale P, Gadre R: pBhosale P production in sugarcane molasses by a Rhodotorula glutinis mutant. Journal of Industrial Microbiology and Biotechnology 2001,26:327-332.
31. Sperstad S, Lutn^s B, Stormo S, Liaaen-Jensen S, Landfald B: Torularhodin and torulene are the major contributors to the carotenoid pool of marine Rhodosporidium babjevae (Golubev). Journal of Industrial Microbiology and Biotechnology 2006, 33:269-273.
32. Koh CM, Liu Y, Du M, Ji L: Molecular characterization of KU70 and KU80 homologues and exploitation of a KU70-deficient mutant for improving gene deletion frequency in Rhodosporidium toruloides. BMC Microbiology 2014, 14:50.
33. Lazo GR, Stein PA, Ludwig RA: A DNA transformation-competent Arabidopsis genomic library in Agrobacterium. Biotechnology (N Y) 1991, 9:963-967.
34. Cai L, Sun L, Fu L, Ji L: media compositions, selection methods and agrobacterium strains for transformation of plants. Google Patents; 2009.
35. Buzzini P, Martini A: Production of carotenoids by strains of Rhodotorula glutinis cultured in raw materials of agroindustrial origin. Bioresource Technology 1999, 71:41-44.
36. Wu S, Hu C, Jin G, Zhao X, Zhao ZK: Phosphate-limitation mediated lipid production by Rhodosporidium toruloides. Bioresour Technol 2010, 101:6124-6129.
37. Liu Y, Koh CM, Sun L, Hlaing MM, Du M, Peng N, Ji L: Characterization of glyceraldehyde-3-phosphate dehydrogenase gene RtGPD1 and development of genetic transformation method by dominant selection in oleaginous yeast Rhodosporidium toruloides. Appl Microbiol Biotechnol 2013, 97:719-729.
38. Smith TL, Leong SA: Isolation and characterization of a Ustilago maydis glyceraldehyde-3-phosphate dehydrogenaseencoding gene. Gene 1990, 93:111-117.
39. Ji L, Jiang ZD, Liu Y, Koh CM, Zhang LH: A Simplified and efficient method for transformation and gene tagging of Ustilago maydis using frozen cells. Fungal Genet Biol 2010, 47:279-287.
40. Punt PJ, Dingemanse MA, Kuyvenhoven A, Soede RD, Pouwels PH, van den Hondel CA: Functional elements in the promoter region of the Aspergillus nidulans gpdA gene encoding glyceraldehyde-3-phosphate dehydrogenase. Gene 1990, 93:101-109.
41. Steiner S, Philippsen P: Sequence and promoter analysis of the highly expressed TEF gene of the filamentous fungus Ashbya gossypii. Mol Gen Genet 1994, 242:263-271.
42. Liu Y, Koh CMJ, Sun L, Hlaing MM, Du M, Peng N, Ji L: Characterization of glyceraldehyde-3-phosphate dehydrogenase gene RtGPD1 and development of genetic transformation method by dominant selection in oleaginous yeast Rhodosporidium toruloides. Applied microbiology and biotechnology 2013, 97:719-729.
43. Liu YG, Whittier RF: Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics 1995, 25:674-681.
44. Liu YG, Chen Y: High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 2007, 43:649-650, 652, 654 passim.
45. Saelices L, Youssar L, Holdermann I, Al-Babili S, Avalos J: Identification of the gene responsible for torulene cleavage in the Neurospora carotenoid pathway. Mol Genet Genomics 2007, 278:527-537.
46. Bijttebier S, D'Hondt E, Noten B, Hermans N, Apers S, Voorspoels S: Ultra high performance liquid chromatography versus high performance liquid chromatography: stationary phase selectivity for generic carotenoid screening. J Chromatogr A 2014, 1332:46-56.
47. Bijttebier SK, D'Hondt E, Hermans N, Apers S, Voorspoels S: Unravelling ionization and fragmentation pathways of carotenoids using orbitrap technology: a first step towards identification of unknowns. J Mass Spectrom 2013, 48:740-754.
48. Hausmann A, Sandmann G: A single five-step desaturase is involved in the carotenoid biosynthesis pathway to betacarotene and torulene in Neurospora crassa. Fungal Genet Biol 2000, 30:147-153.
49. Keller NP, Turner G, Bennett JW: Fungal secondary metabolism-from biochemistry to genomics. Nature Reviews Microbiology 2005, 3:937-947.
50. Linnemannstons P, Prado MM, Fernandez-Martin R, Tudzynski B, Avalos J: A carotenoid biosynthesis gene cluster in Fusarium fujikuroi: the genes carB and carRA. Mol Genet Genomics 2002, 267:593-602.
51. Thewes S, Prado-Cabrero A, Prado MM, Tudzynski B, Avalos J: Characterization of a gene in the car cluster of Fusarium fujikuroi that codes for a protein of the carotenoid oxygenase family. Mol Genet Genomics 2005, 274:217-228.
52. Jin JM, Lee J, Lee YW: Characterization of carotenoid biosynthetic genes in the ascomycete Gibberella zeae. FEMS Microbiol Lett 2010, 302:197-202.
53. Moline M, Flores MR, Libkind D, Dieguez Mdel C, Farias ME, van Broock M: Photoprotection by carotenoid pigments in the yeast Rhodotorula mucilaginosa: the role of torularhodin. Photochemical & photobiological sciences: Official journal of the European Photochemistry Association and the European Society for Photobiology 2010, 9:1145-1151.
54. Moline M, Libkind D, van Broock M: Production of torularhodin, torulene, and beta-carotene by Rhodotorula yeasts. Methods Mol Biol 2012, 898:275-283.
55. Buzzini P, Innocenti M, Turchetti B, Libkind D, van Broock M, Mulinacci N: Carotenoid profiles of yeasts belonging to the genera Rhodotorula, Rhodosporidium, Sporobolomyces, and Sporidiobolus. Can J Microbiol 2007, 53:1024-1031.
56. Liu ZJ, Sun YJ, Rose J, Chung YJ, Hsiao CD, Chang WR, Kuo I, Perozich J, Lindahl R, Hempel J, Wang BC: The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat Struct Biol 1997, 4:317-326.
57. Lee LY, Gelvin SB: T-DNA binary vectors and systems. Plant Physiol 2008, 146:325-332.
Bonfim, K. y otros: RNAi-mediated resistance to Bean golden mosaic virus in genetically engineered common bean (Phaseolus vulgaris). Mol Plant Microbe Interact 2007, 20:717-726.
Durai, S. y otros: Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucl Acids Res 2005, 33:5978-5990.
Fuentes, A. y otros: Intron-hairpin RNA derived from replication associated protein C1 gene confers immunity to tomato yellow leaf curl virus infection in transgenic tomato plants. Transgenic Res 2006, 15:291-304.
Guo, H.S. y otros: A chemical-regulated inducible RNAi system in plants. Plant J 2003, 34:383-392.
Makarova, K.S. y otros: Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 2011, 9:467-477.
Mali, P. y otros: RNA-guided human genome engineering via Cas9. Science 2013, 339:823-826.
Mysara, M. y otros: MysiRNA-designer: a workflow for efficient siRNA design. PLOS one 2011, 6(10):e25642.
Vanderschuren, H. y otros: Transgenic cassava resistance to African cassava mosaic virus is enhanced by viral DNA-A bidirectional promoter-derived siRNAs. Plant Mol Biol 2007a, 64:549-557.
Vanderschuren, H. y otros: Engineering resistance to geminiviruses--review and perspectives. Plant Biotechnology Journal 2007b, 5:207-220.
Wang, M.B. y otros: A single copy of a virus-derived transgene encoding hairpin RNA gives immunity to barley yellow dwarf virus. Mol Plant Pathol 2000, 1:347-356.
Wesley, S.V. y otros: Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 2001, 27:581-590.
Yan, P. y otros: High-throughput construction of intron-containing hairpin RNA vectors for RNAi in plants. PLOS one 2012, 7(5):e38186.
Zrachya, A. y otros: Production of siRNA targeted against TYLCV coat protein transcripts leads to silencing of its expression and resistance to the virus. Transgenic Res 2007, 16:385-398.
Claims (15)
1. Un método para ajustar el nivel de producción y la composición de carotenoides en un huésped fúngico que comprende:
(a) manipular genéticamente uno o más polinucleótidos en la biosíntesis de carotenoides en un huésped fúngico, en donde uno o más polinucleótidos se seleccionan de (i) los polinucleótidos expuestos en las SEQ ID NOs: 1, 3, 5, 7, 8, 10, 11, 13, 14, 16, 17, 19 y 20 o una secuencia homóloga que comparte al menos el 75 % de identidad con los mismos o (ii) uno o más polinucleótidos que codifican uno o más polipéptidos expuestos en las SEQ ID NOs: 2, 4, 6, 12, 15, 18 y 21 o una secuencia homóloga que comparte al menos el 75 % de identidad con los mismos, y en donde el huésped fúngico es Rhodosporidium o Rhodotorula,y
(b) cultivar el huésped fúngico para producir carotenoides, en donde los carotenoides se seleccionan del grupo que consiste en licopeno, beta-caroteno, gamma-caroteno, toruleno y torularodina o derivados de estos, mediante lo cual se ajusta el nivel de producción o la composición de los carotenoides.
2. El método de la reivindicación 1, en donde la manipulación genética es la regulación negativa del uno o más polinucleótidos, en donde la regulación negativa se compara con un huésped fúngico que no tiene la manipulación genética.
3. El método de la reivindicación 2, en donde uno o más polinucleótidos se regulan negativamente por ARNi, un represor transcripcional artificial o un promotor débil.
4. El método de la reivindicación 1, en donde la manipulación genética es la sobreexpresión de uno o más polinucleótidos, en donde la sobreexpresión se compara con un huésped fúngico que no tiene la manipulación genética.
5. El método de la reivindicación 1, en donde la manipulación genética es la inactivación total de una o más funciones enzimáticas de uno o más polipéptidos codificados por uno o más polinucleótidos.
6. El método de la reivindicación 5, en donde la manipulación genética es la deleción de todo o una parte de uno o más de los polinucleótidos, en donde el uno o más polinucleótidos se seleccionan de (i) los polinucleótidos expuestos en las SEQ ID NOs:10, 11, 16, 17, 19 y 20 o (ii) uno o más polinucleótidos codifican uno o más polipéptidos expuestos en las SEQ ID NOs:12, 18 y 21.
7. El método de la reivindicación 6, en donde la deleción se realiza mediante recombinación homóloga, preferentemente en donde la deleción se realiza mediante el uso de una nucleasa artificial seleccionada de una nucleasa de dedos de zinc o un complejo Cas9-ARNg.
8. El método de la reivindicación 2 o 3, en donde la manipulación genética incluye además la sobreexpresión de uno o más polinucleótidos diferentes al uno o más polinucleótidos regulados negativamente, en donde la sobreexpresión se compara con un huésped fúngico que no tiene la manipulación genética.
9. El método de cualquiera de las reivindicaciones 5 a 7, en donde la manipulación genética incluye además la sobreexpresión de uno o más polinucleótidos diferentes al uno o más polinucleótidos que codifican uno o más polipéptidos que tienen pérdida de función enzimática, en donde la sobreexpresión se compara con un huésped fúngico que no tiene la manipulación genética.
10. El método de cualquiera de las reivindicaciones 4, 8 o 9, en donde el uno o más polinucleótidos se sobreexpresan mediante la introducción de una o más moléculas de ADN en el huésped fúngico, en donde cada molécula de ADN comprende uno o más constructos y en donde cada constructo comprende un promotor heterólogo unido operativamente a uno de los polinucleótidos unidos operativamente a un terminador transcripcional.
11. El método de cualquiera de las reivindicaciones 5 a 7, en donde la manipulación genética incluye además la regulación negativa de uno o más polinucleótidos diferentes al uno o más polinucleótidos que codifican uno o más polipéptidos que tienen pérdida de función enzimática, en donde la regulación negativa se compara con un huésped fúngico que no tiene la manipulación genética.
12. El método de la reivindicación 9 o 10, en donde la manipulación genética incluye además la regulación negativa de uno o más polinucleótidos diferentes al uno o más polinucleótidos que codifican uno o más polipéptidos que tienen pérdida de la función enzimática o uno o más polinucleótidos que se sobreexpresan, en donde la regulación negativa se compara con un huésped fúngico que no tiene la manipulación genética.
13. El método de la reivindicación 11 o 12, en donde el uno o más polinucleótidos son regulados negativamente por ARNi, un represor transcripcional artificial o un promotor débil.
14. El método de cualquiera de las reivindicaciones 1 a 13, en donde el huésped fúngico se cultiva en un medio que comprende al menos el 5 % de una fuente de carbono a una temperatura de aproximadamente 25 °C a
aproximadamente 35 °C, preferentemente bajo iluminación, preferentemente en donde la fuente de carbono es glucosa, manosa, glicerol, sacarosa, xilosa o sus combinaciones.
15. El método de la reivindicación 18 o 19, en donde el medio comprende 30-100 g/l de glucosa, 1,5 g/l de extracto de levadura, 0,5 g/l de (N H ^S O 4 , 2,05 g/l de K2HPO4 , 1,45 g/l de KH2PO4, 0,6 g/l de MgSO4, 0,3 g/l de NaCl, 10 mg de CaCl2 , 1 mg/l de FeSO4 , 0,5 mg/l de ZnSO4, 0,5 mg/l de CuSO4, 0,5 mg/l de H3BO4 , 0,5 mg/l de MnSO4 , 0,5 mg/l de NaMoO4 , en donde el pH del medio es de aproximadamente 5 a aproximadamente 7.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462091913P | 2014-12-15 | 2014-12-15 | |
| PCT/SG2015/050491 WO2016099401A1 (en) | 2014-12-15 | 2015-12-14 | Methods for tuning carotenoid production levels and compositions in rhodosporidium and rhodotorula genera |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2787225T3 true ES2787225T3 (es) | 2020-10-15 |
Family
ID=56127088
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15870471T Active ES2787225T3 (es) | 2014-12-15 | 2015-12-14 | Métodos para ajustar los niveles de producción de carotenoides y composiciones en géneros de Rhodosporidium y Rhodotorula |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US10308691B2 (es) |
| EP (1) | EP3234105B1 (es) |
| ES (1) | ES2787225T3 (es) |
| SG (1) | SG11201704890UA (es) |
| WO (1) | WO2016099401A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108220329A (zh) * | 2017-12-29 | 2018-06-29 | 四川省农业科学院生物技术核技术研究所 | 一种敲除转基因植物中潮霉素抗性基因的方法 |
| KR102167388B1 (ko) * | 2020-01-15 | 2020-10-19 | 코스맥스 주식회사 | 신규 로도스포리디움 토룰로이데스 균주 및 그 균주 배양액을 포함하는 화장료 조성물 |
| KR20250021610A (ko) * | 2022-02-24 | 2025-02-13 | 머쉬랩스 게엠베하 | 유색 진균 균사체의 생산 |
| CN116855523B (zh) * | 2023-07-14 | 2024-03-22 | 合曜生物科技(南京)有限公司 | 一种高产依克多因的圆红冬孢酵母工程菌及其构建方法与应用 |
| CN120099065B (zh) * | 2025-05-09 | 2025-08-29 | 浙江工业大学 | 基于rna干扰动态调控酿酒酵母中质粒拷贝数的方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1866428A2 (en) * | 2005-03-18 | 2007-12-19 | Microbia, Inc. | Production of carotenoids in oleaginous yeast and fungi |
| BR112013031723B1 (pt) | 2011-06-10 | 2020-10-13 | Temasek Life Sciences Laboratory Limited | construção de polinucleotídeo, método para transformação de uma célula fúngica e promotor isolado |
| CN105408484B (zh) | 2013-03-14 | 2020-05-19 | 淡马锡生命科学研究院有限公司 | 来自红冬孢属和红酵母属的多核苷酸序列及其用途 |
-
2015
- 2015-12-14 ES ES15870471T patent/ES2787225T3/es active Active
- 2015-12-14 EP EP15870471.8A patent/EP3234105B1/en active Active
- 2015-12-14 WO PCT/SG2015/050491 patent/WO2016099401A1/en not_active Ceased
- 2015-12-14 US US15/535,466 patent/US10308691B2/en active Active
- 2015-12-14 SG SG11201704890UA patent/SG11201704890UA/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| EP3234105A4 (en) | 2018-05-16 |
| EP3234105A1 (en) | 2017-10-25 |
| WO2016099401A1 (en) | 2016-06-23 |
| US20170362285A1 (en) | 2017-12-21 |
| EP3234105B1 (en) | 2020-04-15 |
| US10308691B2 (en) | 2019-06-04 |
| SG11201704890UA (en) | 2017-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Galarza et al. | Over-accumulation of astaxanthin in Haematococcus pluvialis through chloroplast genetic engineering | |
| ES2787225T3 (es) | Métodos para ajustar los niveles de producción de carotenoides y composiciones en géneros de Rhodosporidium y Rhodotorula | |
| Cunningham Jr et al. | A portfolio of plasmids for identification and analysis of carotenoid pathway enzymes: Adonis aestivalis as a case study | |
| KR102205236B1 (ko) | 아세틸 전달효소 및 이의 카로티노이드 생산에 있어서의 용도 | |
| EP2718442B1 (en) | Genetic manipulation and expression systems for pucciniomycotina and ustilaginomycotina subphyla | |
| CN115197947B (zh) | 一种三角褐指藻热激转录因子PtHSF1基因及其编码蛋白和应用 | |
| ES2724998T3 (es) | Métodos para la producción eficiente de ácidos grasos poliinsaturados (PUFA) en especies de Rhodosporidium y Rhodotorula | |
| Csernetics et al. | Expression of three isoprenoid biosynthesis genes and their effects on the carotenoid production of the zygomycete Mucor circinelloides | |
| KR20190079575A (ko) | 세포 소기관이 변이된 재조합 효모 및 이를 이용한 아이소프레노이드 생산 방법 | |
| CN118813433A (zh) | 一种生产β-胡萝卜素的基因工程菌及其制备方法和应用 | |
| TW201544593A (zh) | 用以製備蝦紅素之重組多核苷酸序列及其用途 | |
| CN118401672A (zh) | 能够产生维生素a的耶氏酵母属的遗传修饰酵母 | |
| CN109477120B (zh) | 香根草 | |
| CN103154250A (zh) | 具有改变的类胡萝卜素含量的生物体以及制备其的方法 | |
| WO2015199043A1 (ja) | カロテノイド合成酵素およびその利用 | |
| JP2018510647A (ja) | 芳香性化合物の製造 | |
| Bhatia et al. | Algal Biorefineries and the Circular Bioeconomy: Industrial Applications and Future Prospects | |
| CN106414744B (zh) | 适于酵母中的异源基因表达的启动子 | |
| Sharma et al. | Genetic engineering in improving the output of algal biorefinery | |
| JP6632976B2 (ja) | 藻類及びその製造方法、並びに該藻類を用いたバイオマスの製造方法 | |
| Sui et al. | Isolation and characterization of a neoxanthin synthase gene functioning in fucoxanthin biosynthesis of Phaeodactyum tricornutum | |
| JP6829483B2 (ja) | 形質転換体およびその利用 | |
| US20210230652A1 (en) | Systems, methods and compositions for the generation novel high yielding waxes from microalgae | |
| Nogueira | Optimisation of high-value isoprenoid production in plants | |
| JP2015226469A (ja) | 新規dna、およびそれを利用したカロテノイドの製造方法 |