ES2766474T3 - Método para producir flutemetamol - Google Patents
Método para producir flutemetamol Download PDFInfo
- Publication number
- ES2766474T3 ES2766474T3 ES16782221T ES16782221T ES2766474T3 ES 2766474 T3 ES2766474 T3 ES 2766474T3 ES 16782221 T ES16782221 T ES 16782221T ES 16782221 T ES16782221 T ES 16782221T ES 2766474 T3 ES2766474 T3 ES 2766474T3
- Authority
- ES
- Spain
- Prior art keywords
- solid phase
- extraction cartridge
- general formula
- compound represented
- phase extraction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0446—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/041—Heterocyclic compounds
- A61K51/044—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins
- A61K51/0453—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine, rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
- C07D277/66—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2 with aromatic rings or ring systems directly attached in position 2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Physics & Mathematics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Optics & Photonics (AREA)
- Psychiatry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Hospice & Palliative Care (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Treatment Of Liquids With Adsorbents In General (AREA)
- Ink Jet (AREA)
- Saccharide Compounds (AREA)
Abstract
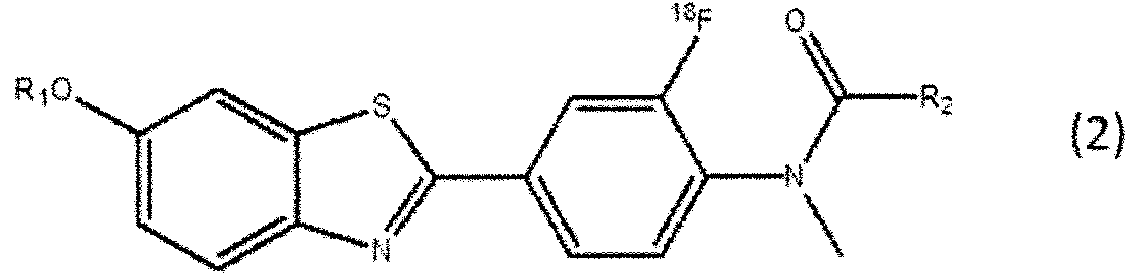
Un método para producir [18F]flutemetamol, que comprende las etapas de: (a) hacer reaccionar un compuesto representado por la siguiente fórmula general (1) con un fluoruro radiactivo para obtener un compuesto representado por la siguiente fórmula general (2); (b) permitir que una base fuerte actúe sobre la mezcla de reacción de la etapa (a) que contiene el compuesto representado por la fórmula general (1) y el compuesto representado por la fórmula general (2); (c) después de la etapa (b), purificar el compuesto representado por la siguiente fórmula general (2) utilizando un cartucho de extracción en fase sólida de fase inversa; y (d) separar ambos grupos protectores (hidroxi y amina) por hidrólisis ácida en presencia de agua, un disolvente orgánico o una mezcla de los mismos, para obtener [18F]flutemetamol.**Fórmula** en donde R1 es un grupo protector de hidroxi, y C(O)R2 representa un grupo protector de amino.**Fórmula** en donde R1 y R2 tienen el mismo significado que en el compuesto representado por la fórmula general (1).
Description
DESCRIPCIÓN
Método para producir flutemetamol
Campo de la invención
La presente invención se refiere a un método para producir [18F] flutemetamol.
Antecedentes de la invención
La inyección de [18F]flutemetamol es un agente utilizado para visualizar placas p-amiloides en el cerebro mediante tomografía por emisión de positrones, y es útil en el diagnóstico de la demencia de tipo Alzheimer.
Como un método para producir [18F]flutemetamol, por ejemplo, utilizando un sintetizador radiofarmacéutico "FASTlab", un método para hacer reaccionar AH111907 (6-etoximetoxi-2-(4'-(N-formil-N-metil)amino-3'-nitro)fenilbenzotiazol) con un fluoruro radiactivo para reemplazar el grupo nitro de AH111907 por 18F, luego convertir el residuo AH111907 en una sustancia menos liposoluble por una base fuerte, separando grupos protectores del grupo hidroxi y el grupo amino del producto de sustitución 18F de AH111907 (6-etoximetoxi-2-(4'-(N-formil-N-metil)amino-3'-[18F]fluoro)fenilbenzotiazol), luego realizar la purificación utilizando un cartucho de extracción en fase sólida (documento WO2011/044406).
Sin embargo, el rendimiento de [18F]flutemetamol es bajo en el método descrito en documento WO2011/044406, por lo tanto, el suministro a una amplia gama de producción en masa ha sido difícil. Por lo tanto, con el fin de suministrar una formulación de [18F]flutemetamol a más pacientes, se ha requerido mejorar la productividad.
Sumario de la invención
La presente invención se ha realizado teniendo en cuenta la situación anterior, y un objeto de la presente invención es mejorar la productividad de [18F]flutemetamol.
De acuerdo con un aspecto de la presente invención, se proporciona un método para producir [18F]flutemetamol, que incluye las etapas de:
(a) hacer reaccionar un compuesto representado por la siguiente fórmula general (1) con un fluoruro radiactivo para obtener un compuesto representado por la siguiente fórmula general (2);
(b) permitir que una base fuerte actúe sobre la mezcla de reacción de la etapa (a) que contiene el compuesto representado por la siguiente fórmula general (1) y el compuesto representado por la siguiente fórmula general (2); (c) después de la etapa (b), purificar el compuesto representado por la siguiente fórmula general (2) utilizando un cartucho de extracción en fase sólida de fase inversa; y
(d) separar ambos grupos protectores (hidroxi y amina) por hidrólisis ácida en presencia de agua, un disolvente orgánico o una mezcla de los mismos, para obtener [18F]flutemetamol.

en donde R1 es un grupo protector de hidroxi, y C(O)R2 representa un grupo protector de amino.

en donde R1 y R2 tienen el mismo significado que en el compuesto representado por la fórmula general (1).
De acuerdo con la presente invención, se puede mejorar la productividad de [18F]flutemetamol.
Descripción detallada
El término "alquilo" en esta memoria, utilizado solo o como parte del otro grupo, designa un grupo hidrocarbonado saturado o saturado ramificado tal como metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc.-butilo, n-pentilo o nhexilo.
Además, el término "haloalquilo" utilizado en esta memoria, solo o como parte del otro grupo, designa uno en el que uno o más hidrógenos de alquilo se reemplaza por flúor, cloro, bromo o yodo.
Además, el término "alcoxi" en esta memoria, utilizado solo o como parte del otro grupo, designa un grupo hidrocarbonado saturado o saturado ramificado tal como metoxi, etoxi, propoxi, isopropoxi, butoxi, isobutoxi, terc.-butoxi, n-pentoxi o n-hexiloxi.
Además, el término "arilo" en esta memoria. utilizado solo o como parte del otro grupo, designa un hidrocarburo aromático monocíclico o de anillo condensado tal como fenilo o naftilo.
(a) Etapa de marcaje con 18F
En la etapa de marcaje con 18F, el compuesto representado por la fórmula general (1) (al que se alude en lo sucesivo también como "compuesto precursor de marcaje") se hace reaccionar con un fluoruro radiactivo para obtener un compuesto representado por la siguiente fórmula general (2) (al que se alude en lo sucesivo también como "compuesto intermedio de marcaje con 18F").
Como el grupo protector de hidroxi de R1 se pueden utilizar los descritos en Protective Groups in Organic Synthesis de Greene (publicado por Wiley-Interscience, 4a edición, emitido el 30 de octubre de 2006). El grupo representado por OR1 es preferiblemente un grupo alcoximetoxi que tiene de 1 a 6 átomos de carbono, y ejemplos incluyen un grupo etoximetoxi y un grupo metoximetoxi.
R2 se selecciona de hidrógeno, alquilos que tienen de 1 a 10 átomos de carbono, haloalquilos que tienen de 1 a 10 átomos de carbono, arilos que tienen de 6 a 14 átomos de carbono, arilalquilos que tienen de 6 a 14 átomos de carbono y -(CH2CH2O)p-CH3, en donde p es un número entero de 1 a 10. R2 es preferiblemente hidrógeno o un alquilo que tiene 1 a 10 átomos de carbono, más preferiblemente hidrógeno o metilo, y aún más preferiblemente hidrógeno.
El compuesto precursor de marcaje puede sintetizarse, por ejemplo, utilizando un método descrito en el documento W02007/020400. Un ejemplo preferido del compuesto precursor de marcaje es 6-etoximetoxi-2-(4'-(N-formil-N-metil)amino-3'-nitro)fenilbenzotiazol (AH111907), y se describe un ejemplo del método de síntesis del mismo en el Ejemplo 1 del documento WO2007/020400.
El fluoruro radioactivo se puede obtener mediante la adición de un contraión catiónico a una solución acuosa que contiene un ion fluoruro [18F] obtenido de [18O]agua por reacción nuclear 18O(p,n)18F para separar el agua. El contraión catiónico es preferiblemente uno que tiene suficiente solubilidad en un disolvente de reacción anhidro, de modo que se puede mantener la solubilidad del ion fluoruro [18F]. Ejemplos incluyen iones tetraalquilamonio y de metales alcalinos (iones sodio, iones potasio, iones cesio, iones rubidio) que forman un complejo con un catalizador de transferencia de fase (por ejemplo, 4,7,13,16,21,24-hexaoxa-1,10-diazabiciclo[8.8.8]hexacosano (nombre comercial: Kryptofix 2.2.2)), y se prefiere tetrabutilamonio. El [18F]fluoruro de tetrabutilamonio se puede preparar, por ejemplo, haciendo pasar el [18O]agua que contiene iones [18F]fluoruro obtenida por la reacción nuclear de 18O(p,n)18F a través de una resina de intercambio aniónico para adsorber el ion [18F]fluoruro a la resina de intercambio aniónico, eluyéndola con una solución acuosa de hidrógeno-carbonato de tetrabutilamonio, y formando una mezcla azeotrópica con acetonitrilo.
La etapa de marcaje con 18F puede llevarse a cabo en un disolvente apropiado. Como disolvente se puede utilizar acetonitrilo, dimetilformamida, dimetilsulfóxido, dimetilacetamida, tetrahidrofurano, dioxano, 1,2-dimetoxietano, sulfolano, N-metilpirrolidona, un derivado de imidazolio, tal como hexafluorofosfato de 1 -butil-3-metilimidazolio, un derivado de piridinio, tal como hexafluoroborato de 1 -butil-4-metilpiridinio, un compuesto de fosfonio o un líquido iónico tal como un compuesto de tetraalquilamonio, y se prefiere dimetilsulfóxido.
La etapa de marcaje con 18F puede llevarse a cabo, por ejemplo, en el intervalo de 15 a 180°C, preferiblemente de 80 a 150°C, y más preferiblemente de 120 a 140°C, y puede llevarse a cabo más preferiblemente a aproximadamente 130°C.
(b) Etapa de descomposición del precursor
En la etapa de descomposición del precursor se deja que una base fuerte actúe sobre la mezcla de reacción en la etapa de marcaje con 18F que contiene el compuesto precursor de marcaje y el compuesto intermedio de marcaje con 18F. De este modo, el residuo del compuesto precursor de marcaje contenido en la mezcla de reacción en la etapa de marcaje con 18F se convierte en un compuesto altamente polar. Como compuesto altamente polar se pueden considerar los que se muestran en la Fig. 1 del documento WO2011/044406. En la etapa de descomposición del precursor, el compuesto intermedio de marcaje con 18F permanece sin reaccionar con una base fuerte.
La base fuerte incluye alcóxidos de metales alcalinos, hidróxidos de metales alcalinos y similares, y se utiliza preferiblemente metóxido de sodio, etóxido de sodio, hidróxido de sodio, hidróxido de potasio, hidruro de sodio o metilmercaptano sódico. La base fuerte es más preferiblemente metóxido de sodio o etóxido de sodio, y más preferiblemente metóxido de sodio.
La etapa de descomposición del precursor se lleva a cabo preferiblemente en presencia de un disolvente. El disolvente incluye alcoholes alquílicos, y se prefiere metanol.
La etapa de descomposición del precursor puede llevarse a cabo, por ejemplo, en el intervalo de 15 a 180°C, preferiblemente de 80 a 150°C, y más preferiblemente de 120 a 140°C, y puede llevarse a cabo más preferiblemente a aproximadamente 130°C.
(c) Primera etapa de purificación
En la primera etapa de purificación, después de la etapa de descomposición del precursor, el compuesto intermedio de marcaje con 18F se purifica utilizando un cartucho de extracción en fase sólida de fase inversa. De este modo, el compuesto intermedio de marcaje con 18F y el compuesto altamente polar obtenido en la etapa de descomposición del precursor se separan.
Como cartucho de extracción en fase sólida de fase inversa se utiliza uno que usa una carga en el que se modifica un grupo sililo con alquilo que tiene preferiblemente 8 o más átomos de carbono y más preferiblemente 18 o más átomos de carbono, y se utiliza además, preferiblemente, un cartucho de extracción en fase sólida empaquetado con gel de sílice triacontilo sililado, en que un grupo sililo está modificado con 30 átomos de carbono. Un cartucho de extracción en fase sólida de fase inversa de este tipo está disponible comercialmente, por ejemplo, de Macherey-Nagel. El cartucho de extracción en fase sólida de fase inversa se acondiciona preferiblemente con acetonitrilo y agua antes de su uso.
La purificación del compuesto intermedio de marcaje con 18F utilizando un cartucho de extracción en fase sólida de fase inversa no está particularmente limitado, siempre que se lleve a cabo utilizando una técnica del método de extracción en fase sólida normal. Un ejemplo se explicará más adelante.
Primero, el compuesto intermedio de marcaje con 18F a través de la etapa de descomposición del precursor se mantiene en un cartucho de extracción en fase sólida de fase inversa [etapa de retención (c-1)]. Preferiblemente, después de la etapa de descomposición del precursor, la mezcla de reacción que contiene el compuesto intermedio de marcaje con 18F y el compuesto altamente polar arriba descrito se diluye añadiendo agua, y se carga en un cartucho de extracción en fase sólida de fase inversa.
Posteriormente, el cartucho de extracción en fase sólida de fase inversa se lava con una mezcla líquida de agua y uno o más disolventes orgánicos seleccionados de un grupo que consiste en acetonitrilo, tetrahidrofurano y alcoholes alquílicos que tienen 1 a 3 átomos de carbono [etapa de lavado (c-2)]. El disolvente utilizado para el lavado es preferiblemente una mezcla líquida de agua y acetonitrilo, y como relación de mezcla de los mismos, por ejemplo, el contenido de acetonitrilo puede ser del 35 al 45% en volumen, y preferiblemente del 39,5 al 40,5% en volumen del líquido de mezcla completa. La temperatura del cartucho de extracción en fase sólida de fase inversa está preferiblemente en el intervalo de 19 a 34°C y más preferiblemente en el intervalo de 20 a 30°C. Esta etapa de lavado se puede repetir una pluralidad de veces. De este modo, el compuesto altamente polar arriba descrito se puede eluir del cartucho de extracción en fase sólida de fase inversa, al tiempo que el compuesto intermedio de marcaje con 18F se mantiene en un cartucho de extracción en fase sólida de fase inversa.
Posteriormente, el compuesto intermedio de marcaje con 18F se eluye con un alcohol alquílico que tiene de 1 a 3 átomos de carbono a partir del cartucho de extracción en fase sólida de fase inversa [etapa de elución (c-3)]. El alcohol alquílico que tiene de 1 a 3 átomos de carbono incluye metanol, etanol, 1-propanol y 2-propanol, y el etanol es más preferible desde el punto de vista de la seguridad. En este momento, el gas nitrógeno puede fluir desde la lumbrera de entrada del cartucho de extracción en fase sólida de fase inversa o puede ser aspirado desde la lumbrera de descarga. El material eluido obtenido puede utilizarse en la siguiente etapa tal como está o después de concentrar el disolvente bajo calentamiento o presión reducida.
(d) Etapa de desprotección
En la etapa de desprotección, los grupos protectores del grupo hidroxi y el grupo amino se separan cada uno para obtener [18F]flutemetamol.
La etapa de desprotección puede llevarse a cabo de acuerdo con la descripción de Protective Groups in Organic Synthesis de Greene (publicado por Wiley-Interscience, 4a edición, emitido el 30 de octubre de 2006), y se prefiere llevar a cabo la hidrólisis ácida utilizando un ácido orgánico o ácido inorgánico. Como ácido se utiliza preferiblemente un ácido inorgánico tal como ácido sulfúrico, ácido clorhídrico, ácido fosfórico o ácido bromhídrico, y se utiliza más preferiblemente ácido clorhídrico.
La etapa de desprotección puede llevarse a cabo en presencia de agua, un disolvente orgánico tal como un alcohol alquílico que tiene 1 a 4 átomos de carbono o acetonitrilo o una mezcla líquida de los mismos, y se prefiere añadir un ácido a un material eluido de etanol obtenido por la etapa de elución de la primera etapa de purificación y luego llevar a cabo la etapa de desprotección.
La etapa de desprotección se lleva a cabo preferiblemente a 100°C o más.
(e) Segunda etapa de purificación
En la segunda etapa de purificación, después de la etapa de desprotección, el [18F]flutemetamol se purifica utilizando un cartucho de extracción en fase sólida de fase inversa.
Como tipo de cartucho de extracción en fase sólida de fase inversa utilizado en la segunda etapa de purificación, puede utilizarse uno que se puede utilizar en la primera etapa de purificación, y se utiliza preferiblemente un cartucho de extracción en fase sólida empaquetado con gel de sílice sililada con triacontilo en el que un grupo sililo está modificado con 30 átomos de carbono.
La purificación del [18F]flutemetamol utilizando un cartucho de extracción en fase sólida de fase inversa no está particularmente limitada, siempre que se lleve a cabo utilizando una técnica del método de extracción en fase sólida normal. Un ejemplo se explicará a continuación.
Primero, el [18F]flutemetamol a través de la etapa de desprotección se mantiene en un cartucho de extracción en fase sólida de fase inversa [etapa de retención (e-1)]. Preferiblemente, después de la etapa de desprotección, el producto bruto de [18F]flutemetamol se diluye añadiendo agua, de modo que el contenido del disolvente orgánico tomado de la etapa previa (por ejemplo, etanol tomado de la primera etapa de purificación) es del 50% en volumen o menos, y se carga en un cartucho de extracción en fase sólida de fase inversa.
Posteriormente, el cartucho de extracción en fase sólida de fase inversa se lava con agua o una mezcla líquida de agua y uno o más disolventes orgánicos seleccionados de un grupo que consiste en acetonitrilo, tetrahidrofurano y alcoholes alquílicos con 1 a 3 átomos de carbono [etapa de lavado (e-2)]. El disolvente utilizado para el lavado es preferiblemente una mezcla líquida de agua y acetonitrilo, y como relación de mezcla de los mismos, por ejemplo, el contenido de acetonitrilo puede ser del 35 al 45% en volumen, y preferiblemente del 39,5 al 40,5% en volumen de la mezcla líquida completa. La temperatura del cartucho de extracción en fase sólida de fase inversa está preferiblemente en el intervalo de 19 a 34°C y más preferiblemente en el intervalo de 20 a 30°C. Esta etapa de lavado se puede repetir una pluralidad de veces y, en este momento, el cartucho de extracción en fase sólida de fase inversa se lava preferiblemente con agua. Con ello se puede separar disolvente innecesario y el reactivo de desprotección, al tiempo que el [18F]flutemetamol se mantiene en el cartucho de extracción en fase sólida de fase inversa.
Posteriormente, el [18F]flutemetamol se eluye con etanol del cartucho de extracción en fase sólida de fase inversa [etapa de elución (e-3)]. Posteriormente, el agua puede pasar más y combinarse con el material eluido. Además, el nitrógeno gaseoso puede fluir desde la lumbrera de entrada del cartucho de extracción en fase sólida de fase inversa o puede ser aspirado desde la lumbrera de descarga.
(f) Tercera etapa de purificación
En la tercera etapa de purificación, después de la segunda etapa de purificación, el [18F]flutemetamol se purifica utilizando un cartucho de extracción en fase sólida de interacción hidrófila (HILIC).
Como cartucho de extracción en fase sólida HILIC, por ejemplo, se puede utilizar uno empaquetado con gel de sílice o gel de sílice en la que se introduce un grupo funcional altamente polar, tal como amino, amida, ciano, diol, un derivado de polisuccinimida, ion híbrido o ciclodextrina. Aquí, es preferible una fase de amino sólida a base de gel de sílice, y es más preferible una empaquetada con gel de sílice aminopropilado. De este modo, las impurezas se pueden capturar con el cartucho de extracción en fase sólida HILIC, mientras que se permite que pase el [18F]flutemetamol. Un cartucho de extracción en fase sólida HILIC de este tipo está disponible comercialmente, por ejemplo, de Waters, Agilent Technologies y similares. El cartucho de extracción en fase sólida HILIC se acondiciona preferiblemente haciendo pasar acetonitrilo o etanol antes de su uso, seguido de flujo de nitrógeno a secar.
Posteriormente, el material eluido obtenido en la segunda etapa de purificación se deja pasar directamente a través del cartucho de extracción en fase sólida HILIC. Posteriormente, se puede hacer pasar agua y se puede combinar el material eluido. Además, el gas nitrógeno puede fluir desde la lumbrera de entrada del cartucho de extracción en fase sólida HILIC o ser aspirado de la lumbrera de descarga.
El material eluido obtenido puede contener un soporte, diluyente, emulsión, excipiente, extensor, dispersante, tampón, conservante, solubilizante, antiséptico, colorante, estabilizador y similares, farmacéuticamente aceptable, con el fin de que tenga una forma adecuada para la administración de [18F]flutemetamol a un cuerpo vivo, preferiblemente una forma de inyección. La solución que contiene [18F]flutemetamol obtenida se filtra deseablemente con un filtro de membrana.
El ejemplo de formulación de [18F]flutemetamol se describe, por ejemplo, en el documento WO2009/027452.
De acuerdo con el método de la presente invención arriba descrito, la primera etapa de purificación llevada a cabo convencionalmente después de la etapa de desprotección se lleva a cabo después de la etapa de marcaje con 18F y antes de la etapa de desprotección. Cuando la primera etapa de purificación se lleva a cabo después de la etapa de desprotección, están presentes muchas impurezas que contienen el compuesto altamente polar derivado del compuesto precursor de marcaje, por lo que la pérdida de [18F]flutemetamol aumenta con la separación de impurezas. Por otro lado, la primera etapa de purificación se lleva a cabo antes de la etapa de desprotección, por lo que el compuesto altamente polar derivado del compuesto precursor de marcaje se puede separar antes de la etapa de desprotección. De esta manera, el compuesto intermedio de marcaje con 18F se puede purificar, al tiempo que se evita la pérdida del compuesto intermedio de marcaje con 18F, que es un compuesto intermedio del [18F]flutemetamol, por lo que se puede obtener [18F]flutemetamol con un rendimiento mayor que uno convencional y con una calidad equivalente a una convencional. Por lo tanto, de acuerdo con la presente invención, se puede mejorar la productividad de [18F]flutemetamol.
EJEMPLOS
En lo que sigue se describirá adicionalmente la presente invención preferiblemente en detalle con referencia a los ejemplos, pero la presente invención no se limita al contenido de los ejemplos. Aquí, como reactivo y miembro de la columna utilizado en los ejemplos, se utilizó un componente de un sintetizador radiofarmacéutico FASTlab (para la síntesis de [18F]flutemetamol) fabricado por GE Healthcare o un componente equivalente al mismo.
Ejemplos 1 a 3
(a) Etapa de marcaje con18F
[18O]agua que contiene ion fluoruro [18F], obtenida por irradiación de protones de [18O]agua utilizando un ciclotrón se hizo pasar a través de una columna de intercambio aniónico, y el ion fluoruro [18F] se adsorbió y recogió. Posteriormente, la columna se lavó con agua (3 mL), luego se eluyó utilizando una solución acuosa 0,15 mol/L de hidrogenó-carbonato de tetrabutilamonio (0,35 mL) y acetonitrilo (1 mL), y el material eluido obtenido se evaporó. A esto se añadió una solución de dimetilsulfóxido (1 mL) de 6-etoximetoxi-2-(4-(N-formil-N-metil)amino-3-nitro)fenilbenzotiazol (AH111907) (75 pmol), y la mezcla se calentó a 130°C durante 15 minutos y luego se enfrió. (b) Etapa de descomposición del precursor
Se añadió una solución de metanol (11% (p/p), 1 mL) de metóxido de sodio al líquido de reacción después de enfriar en la etapa (a), y la mezcla se calentó a 130°C durante 5 minutos y se enfrió.
(c) Primera etapa de purificación
Se añadió agua (2 mL) al líquido de reacción después de enfriar en la etapa (b), y la mezcla se hizo pasar a través de una columna de gel de sílice sililada con triacontilo (C30) para contener el compuesto intermedio de marcaje con 18F. Además, el lavado se realizó haciendo pasar una solución acuosa de acetonitrilo al 40% (v/v) (6 mL) a través de la columna C30 a través de un recipiente de reacción, luego se realizó nuevamente un lavado, haciendo pasar directamente una solución acuosa al 40% (v/v) de acetonitrilo (6 mL) a través de la columna C30. Se hizo pasar etanol (2 mL) a través de esta columna C30 para recoger un material eluido.
(d) Etapa de desprotección
Se añadieron 4 mol/L (2,0 mL) de ácido clorhídrico al material eluido recogido en la etapa (c), y la mezcla se calentó a 125°C durante 5 minutos para obtener una solución de [18F]flutemetamol no purificada.
(e) Segunda etapa de purificación
La solución de [18F]flutemetamol no purificada, obtenida en la etapa (d), se enfrió, luego se añadió agua (10 mL) a la misma, y la mezcla se hizo pasar a través de una columna C30 no utilizada diferente de la utilizada en la etapa (b) para contener el [18F]flutemetamol en la columna C30. El lavado se realizó haciendo pasar una solución acuosa de acetonitrilo al 40% (v/v) (6 mL) a través de la columna C30, luego el lavado se realizó haciendo pasar agua (6 mL). El [18F]flutemetamol se eluyó con etanol (3,5 mL) de la columna C30.
(f) Tercera etapa de purificación
El material eluido en la etapa (e) se hizo pasar a través de una columna (columna de NH2) empaquetada con gel de sílice aminopropilado. El lavado se llevó a cabo haciendo pasar agua (9,3 mL) a través de la columna C30 utilizada en la etapa (e) y la columna de NH2, en este orden, y se recogió cada uno de los materiales eluidos en un recipiente al que se añadió un tampón 18,8 mmol/L de ácido fosfórico (37,2 mL) que contenía 0,7% (p/v) de polisorbato 80 y 1,2% (p/v) de cloruro de sodio.
Ejemplos Comparativos 1 y 2
Se llevaron a cabo las etapas (a) y (b) de los Ejemplos 1 a 3, y se llevaron a cabo las siguientes etapas.
(c') Etapa de desprotección
Se añadió ácido clorhídrico 4 mol/L (0,6 mL) al líquido de reacción obtenido en la etapa (b), y la mezcla se calentó a 125°C durante 5 minutos para obtener una solución de [18F]flutemetamol no purificada.
(d') Primera etapa de purificación
La solución de [18F]flutemetamol no purificada obtenida en la etapa (c') se enfrió, luego se añadió agua (2 mL) a la misma, y la mezcla se hizo pasar a través de una columna C30 para contener el [18F]flutemetamol. Además, el lavado se realizó haciendo pasar una solución acuosa de acetonitrilo al 40% (v/v) (12 mL) a través de la columna C30 a través de un recipiente de reacción, luego el lavado se realizó haciendo pasar agua (5 mL) a través de la columna C30. Se hizo pasar acetonitrilo (2 mL) a través de esta columna C30 para recoger un material eluido.
(e') Segunda etapa de purificación
El material eluido obtenido en la etapa (d') se purificó haciéndolo pasar a través de una columna de NH2, luego la solución de acetonitrilo (1 mL) se hizo pasar adicionalmente a través de la columna de NH2, y estas soluciones se mezclaron.
(f') Tercera etapa de purificación
Se añadió agua (5 mL) a la solución obtenida en la etapa (e'), y se hizo pasar a través de una columna C30 no utilizada, diferente de la utilizada en la etapa (d') para mantener el [18F]flutemetamol en la columna C30, luego se realizó el lavado haciendo pasar agua (4 mL) tres veces a través de la columna C30. Se hizo pasar etanol (3,5 mL) a través de la columna C30, y se hizo pasar agua (9,3 mL) a través de la columna C30. Cada uno de los materiales eluidos se recogió en un recipiente al que se le añadió un tampón ácido fosfórico 18,8 mmol/L (37,2 mL) que contenía 0,7% (p/v) de polisorbato 80 y 1,2% (p/v) de cloruro de sodio.
Los resultados de los Ejemplos 1 a 3 y los Ejemplos Comparativos 1 y 2 se muestran en la Tabla 1. En la Tabla 1, la "cantidad de radiactividad (MBq)" es una cantidad de radiactividad de ion fluoruro [18F] al comienzo de la síntesis, utilizada en cada uno de los ejemplos y ejemplos comparativos, el "tiempo de síntesis (minuto)" es un tiempo requerido para realizar cada uno de los ejemplos y ejemplos comparativos, y el "rendimiento radioquímico (%)" es un rendimiento radioquímico de [18F]flutemetamol basado en la cantidad de radiactividad de ion [18F]fluoruro después de la corrección de la atenuación al comienzo de la síntesis, la "pureza radioquímica (%)" es una pureza radioquímica de [18F]flutemetamol, y la "cantidad total de impurezas no radiactivas (pg/mL)" es una concentración de impurezas no radioactivas en la solución de [18F]flutemetamol obtenida.
La pureza radioquímica de [18F]flutemetamol y la concentración de impurezas no radiactivas se analizaron mediante los métodos que se muestran a continuación.
1. Análisis de la pureza radioquímica de [18F]flutemetamol
El análisis se realizó por TLC. Las condiciones son las siguientes.
Placa TLC: gel de sílice 60 F254 (fabricada por Merck)
Fase móvil: acetato de etilo / dietilamina = 100/1
Dispositivo de medición: Rita Star (fabricado por raytest)
2. Análisis de la concentración de impurezas no radiactivas en la solución de [18F]flutemetamol
El análisis se realizó por HPLC equipada con un detector UV. Las condiciones son las siguientes.
Columna: Luna C18 (2) (fabricada por Phenomenex, tamaño: 4.6 x 150 mm, 3 pm)
Fase móvil: un tampón acetato de amonio de 20 mmoL (pH 6,0) / acetonitrilo = 62/38 ^ 40/10 (0 ^ 9 minutos), 40/10 ^ 10/90 (9 ^ 10 minutos), 10/90 (10 ^ 20 minutos), 10/90 ^ 62/38 (20 ^ 20,5 minutos), 62/38 (20,5 ^ 30 minutos)
Caudal: 1.0 mL/minuto
Detector: fotómetro de absorción de luz visible ultravioleta (longitud de onda de detección: 330 nm)
Tabla 1

Como se muestra en la Tabla 1, el rendimiento radioquímico de [18F]flutemetamol se mejoró mediante los métodos de los Ejemplos 1 a 3, y el tiempo de síntesis podría acortarse. Además, se mejoró la pureza radioquímica y no se encontró el marcado aumento en la cantidad total de impurezas. Por lo tanto, se demostró que, de acuerdo con la presente invención, se puede obtener [18F]flutemetamol que tiene la misma calidad, al tiempo que se mejora la productividad más que una convencional.
Claims (8)
1. Un método para producir [18F]flutemetamol, que comprende las etapas de:
(a) hacer reaccionar un compuesto representado por la siguiente fórmula general (1) con un fluoruro radiactivo para obtener un compuesto representado por la siguiente fórmula general (2);
(b) permitir que una base fuerte actúe sobre la mezcla de reacción de la etapa (a) que contiene el compuesto representado por la fórmula general (1) y el compuesto representado por la fórmula general (2);
(c) después de la etapa (b), purificar el compuesto representado por la siguiente fórmula general (2) utilizando un cartucho de extracción en fase sólida de fase inversa; y
(d) separar ambos grupos protectores (hidroxi y amina) por hidrólisis ácida en presencia de agua, un disolvente orgánico o una mezcla de los mismos, para obtener [18F]flutemetamol.

en donde R1 es un grupo protector de hidroxi, y C(O)R2 representa un grupo protector de amino.
en donde R1 y R2 tienen el mismo significado que en el compuesto representado por la fórmula general (1).
2. El método para producir [18F]flutemetamol de acuerdo con la reivindicación 1, en el que la etapa (c) incluye las etapas de:
(c-1) mantener el compuesto representado por la fórmula general (2) en un cartucho de extracción en fase sólida de fase inversa;
(c-2) lavar el un cartucho de extracción en fase sólida de fase inversa con una mezcla líquida de uno o más disolventes orgánicos seleccionados de agua y un grupo que consiste en acetonitrilo, tetrahidrofurano y alcoholes alquílicos con 1 a 3 átomos de carbono; y
(c-3) eluir el compuesto representado por la fórmula general (2) con un alcohol alquílico con 1 a 3 átomos de carbono del cartucho de extracción en fase sólida de fase inversa.
3. El método para producir [18F]flutemetamol de acuerdo con la reivindicación 2, en el que en la etapa (c-2) el cartucho de extracción en fase sólida de fase inversa se lava con una mezcla líquida de agua y acetonitrilo.
4. El método para producir [18F]flutemetamol de acuerdo con la reivindicación 2 o 3, en el que en la etapa (c-3) el alcohol alquílico con 1 a 3 átomos de carbono es etanol.
5. El método para producir [18F]flutemetamol de acuerdo con una cualquiera de las reivindicaciones 1 a 4, en el que el cartucho de extracción en fase sólida de fase inversa en la etapa (c) es uno empaquetado con gel de sílice triacontilo sililado.
6. El método para producir [18F]flutemetamol de acuerdo con una cualquiera de las reivindicaciones 1 a 5, que comprende, además, las etapas de:
(e) después de la etapa (d), purificar el [18F]flutemetamol utilizando un cartucho de extracción en fase sólida de fase inversa; y
(f) después de la etapa (e), purificar el [18F]flutemetamol utilizando un cartucho de extracción en fase sólida de interacción hidrófila.
7. El método para producir [18F]flutemetamol de acuerdo con la reivindicación 6, en el que el cartucho de extracción en fase sólida de fase inversa en la etapa (e) es uno empaquetado con gel de sílice triacontilo sililado.
8. El método para producir [18F]flutemetamol de acuerdo con la reivindicación 6 o 7, en el que el cartucho de extracción en fase sólida de interacción hidrófila en la etapa (f) es uno empaquetado con gel de sílice aminopropilado.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015211413A JP6485914B2 (ja) | 2015-10-28 | 2015-10-28 | フルテメタモルの製造方法 |
| PCT/EP2016/074840 WO2017071980A1 (en) | 2015-10-28 | 2016-10-17 | Method for producing flutemetamol |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2766474T3 true ES2766474T3 (es) | 2020-06-12 |
Family
ID=57144967
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16782221T Active ES2766474T3 (es) | 2015-10-28 | 2016-10-17 | Método para producir flutemetamol |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10472338B2 (es) |
| EP (1) | EP3368518B1 (es) |
| JP (1) | JP6485914B2 (es) |
| KR (1) | KR20180073581A (es) |
| CN (1) | CN108137520B (es) |
| AU (1) | AU2016344537B2 (es) |
| CA (1) | CA2998985C (es) |
| ES (1) | ES2766474T3 (es) |
| HK (1) | HK1249098A1 (es) |
| IL (1) | IL258148B (es) |
| MX (1) | MX2018005046A (es) |
| RU (1) | RU2733398C2 (es) |
| WO (1) | WO2017071980A1 (es) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6485914B2 (ja) | 2015-10-28 | 2019-03-20 | 日本メジフィジックス株式会社 | フルテメタモルの製造方法 |
| JP6770837B2 (ja) * | 2016-06-28 | 2020-10-21 | 日本メジフィジックス株式会社 | 放射性フッ素標識有機化合物を製造する方法 |
| KR20200016863A (ko) * | 2017-06-23 | 2020-02-17 | 니혼 메디피직스 가부시키가이샤 | 방사성 할로겐 표지 화합물의 제조 방법 및 방사성 의약의 제조 방법 |
| GB201805253D0 (en) * | 2018-03-29 | 2018-05-16 | Ge Healthcare Ltd Ip | Solid phase extraction |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE1010280A3 (fr) * | 1996-05-02 | 1998-05-05 | Coincidence S A | Procede et dispositif de synthese de 2-[18f] fluoro-2-deoxy-d-glucose. |
| GB0229686D0 (en) * | 2002-12-20 | 2003-01-29 | Amersham Plc | Solid-phase fluorination of benzothiazoles |
| CA2438032C (en) | 2003-03-14 | 2013-05-07 | University Of Pittsburgh | Benzothiazole derivative compounds, compositions and uses |
| WO2006133732A1 (en) * | 2005-06-17 | 2006-12-21 | Advanced Accelerator Applications | Process for synthesizing labelled compounds |
| GB0516564D0 (en) * | 2005-08-12 | 2005-09-21 | Ge Healthcare Ltd | Fluorination process |
| US7897811B2 (en) * | 2006-05-11 | 2011-03-01 | Nihon Medi-Physics Co., Ltd. | Process for production of radioactive fluorine-labeled organic compound |
| WO2009027452A2 (en) * | 2007-08-30 | 2009-03-05 | Ge Healthcare Limited | Radiopharmaceutical composition |
| WO2011044410A2 (en) * | 2009-10-08 | 2011-04-14 | Ge Healthcare Limited | Solid phase extraction purification method |
| JP6485914B2 (ja) | 2015-10-28 | 2019-03-20 | 日本メジフィジックス株式会社 | フルテメタモルの製造方法 |
-
2015
- 2015-10-28 JP JP2015211413A patent/JP6485914B2/ja active Active
-
2016
- 2016-10-17 ES ES16782221T patent/ES2766474T3/es active Active
- 2016-10-17 CA CA2998985A patent/CA2998985C/en active Active
- 2016-10-17 EP EP16782221.2A patent/EP3368518B1/en active Active
- 2016-10-17 KR KR1020187011715A patent/KR20180073581A/ko active IP Right Grant
- 2016-10-17 CN CN201680062917.1A patent/CN108137520B/zh active Active
- 2016-10-17 AU AU2016344537A patent/AU2016344537B2/en active Active
- 2016-10-17 RU RU2018113741A patent/RU2733398C2/ru active
- 2016-10-17 WO PCT/EP2016/074840 patent/WO2017071980A1/en active Application Filing
- 2016-10-17 US US15/764,309 patent/US10472338B2/en active Active
- 2016-10-17 MX MX2018005046A patent/MX2018005046A/es active IP Right Grant
-
2018
- 2018-03-15 IL IL258148A patent/IL258148B/en active IP Right Grant
- 2018-07-06 HK HK18108745.1A patent/HK1249098A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| RU2018113741A3 (es) | 2020-02-20 |
| AU2016344537A1 (en) | 2018-04-12 |
| CA2998985C (en) | 2023-10-31 |
| CN108137520A (zh) | 2018-06-08 |
| CN108137520B (zh) | 2022-06-21 |
| MX2018005046A (es) | 2018-06-13 |
| WO2017071980A1 (en) | 2017-05-04 |
| IL258148B (en) | 2020-08-31 |
| US20190055207A1 (en) | 2019-02-21 |
| US10472338B2 (en) | 2019-11-12 |
| EP3368518A1 (en) | 2018-09-05 |
| JP2017081847A (ja) | 2017-05-18 |
| BR112018007545A2 (pt) | 2018-10-23 |
| HK1249098A1 (zh) | 2018-10-26 |
| KR20180073581A (ko) | 2018-07-02 |
| IL258148A (en) | 2018-05-31 |
| JP6485914B2 (ja) | 2019-03-20 |
| RU2733398C2 (ru) | 2020-10-01 |
| AU2016344537B2 (en) | 2020-09-17 |
| RU2018113741A (ru) | 2019-11-28 |
| EP3368518B1 (en) | 2019-12-04 |
| CA2998985A1 (en) | 2017-05-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2766474T3 (es) | Método para producir flutemetamol | |
| ES2917875T3 (es) | Método automatizado para la preparación de composiciones de 18F-fluciclovina | |
| ES2642086T3 (es) | Procedimiento para la producción de ligandos de beta amiloide marcados con F-18 | |
| ES2610574T3 (es) | Procedimiento para producir un compuesto orgánico marcado con flúor radiactivo | |
| ES2710073T3 (es) | Nueva formulación y método de síntesis | |
| ES2654528T3 (es) | Procedimiento de preparación de un derivado de purina marcado, dicho derivado y sus usos | |
| ES2841109T3 (es) | Composiciones de 18F-fluciclovina en tampones citrato | |
| ES2884934T3 (es) | Un proceso de producción de flutemetamol | |
| ES2838752T3 (es) | Método para preparar un compuesto alifático de fluoruro orgánico y método para purificar un compuesto alifático de fluoruro orgánico | |
| BR112018007545B1 (pt) | Método para produzir [18f] flutemetamol | |
| Okumura et al. | Method for producing [18 F] flutemetamol | |
| JP7159157B2 (ja) | 放射性フッ素標識化合物の製造方法および放射性医薬の製造方法 | |
| KR101592291B1 (ko) | 고순도 및 고비방사능의 [18f]플루오로-l-도파의 제조 방법 | |
| CA3034905A1 (en) | Method for producing radiopharmaceutical composition | |
| JP2022500482A (ja) | 選択的アジド置換反応および前駆体除去を用いたフッ素−18標識フルオロメチル置換放射性医薬品の製造方法 | |
| BR112013015003B1 (pt) | Método e método radiossintético para obter derivados de aminoácidos protegidos |