ES2753407T3 - Conjugado de péptido-resina y su utilización - Google Patents
Conjugado de péptido-resina y su utilización Download PDFInfo
- Publication number
- ES2753407T3 ES2753407T3 ES14742356T ES14742356T ES2753407T3 ES 2753407 T3 ES2753407 T3 ES 2753407T3 ES 14742356 T ES14742356 T ES 14742356T ES 14742356 T ES14742356 T ES 14742356T ES 2753407 T3 ES2753407 T3 ES 2753407T3
- Authority
- ES
- Spain
- Prior art keywords
- peptide
- tir
- arg
- pro
- resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/06—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents
- C07K1/061—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups
- C07K1/063—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length using protecting groups or activating agents using protecting groups for alpha-amino functions
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/04—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers
- C07K1/042—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers characterised by the nature of the carrier
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/04—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers
- C07K1/045—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length on carriers using devices to improve synthesis, e.g. reactors, special vessels
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/001—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof by chemical synthesis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Gastroenterology & Hepatology (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Un procedimiento para preparar un péptido, o una sal del mismo, de un conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f),**Fórmula** en el que la resina es una resina polimérica escindible de TFA seleccionada de entre resinas de tritilo, 2-cloro-tritlo, 4- metil-tritilo y 4-metoxi-tritilo; y Pr1 es un grupo protector seleccionado de entre el grupo que consiste en: Fmoc, Boc, Cbz, Npys y Alloc; dicho procedimiento comprendiendo las etapas de: (a) desproteger la función α-amino del extremo N-terminal de dicho conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f) para eliminar el grupo protector Pr1; (b) acoplar al menos un aminoácido o péptido protegido en el extremo N-terminal que presente una función de ácido carboxílico activado o libre con la función α-amino desprotegida de la etapa (a), alargando así el compuesto de las fórmulas (2b), (2c), (2d), (2e) o (2f), (c) repetir, opcionalmente, las etapas (a) y (b) una o más veces, en las que el al menos un aminoácido o péptido protegido en el extremo N-terminal es idéntico o diferente de aquel de la etapa anterior (b); (d) escindir el péptido resultante de la resina; (e) guanilar el péptido obtenido en la etapa (d); (f) eliminar, opcionalmente, todos los grupos protectores que queden después de la etapa (d); (g) aislar y opcionalmente purificar el péptido así obtenido.
Description
DESCRIPCIÓN
Conjugado de péptido-resina y su utilización
La presente invención se refiere a conjugados de péptido-resina adecuados para su utilización en la síntesis de péptidos. Más específicamente, la invención se refiere a péptidos cortos que contienen diaminoácidos en su secuencia y a su utilización en la síntesis de amidas de péptidos.
ANTECEDENTES DE LA INVENCIÓN
La utilización de resinas de tipo tritilo ácido-lábil en la síntesis de fase sólida de péptidos y péptidos protegidos se conoce bien en la técnica (véase, por ejemplo, Barlos K, Chatzi O, Gatos D, Stavropoulos G., Int J Pept Protein Res. junio de 1991;37(6):513-20). Los conjugados de péptido-resina se enlazan, por lo general, a la resina por medio de un enlace éster conjugado con carboxilo terminal (véase, por ejemplo, el documento de EE. UU. 7.939.629). Los conjugados de péptido-resina enlazados a la resina por medio de la cadena lateral de un residuo de aminoácido de lisina terminal también son conocidos (véase, por ejemplo, el documento de EE. UU. 2009/0292106).
Las resinas de Rinck-amida comúnmente utilizadas resultan en amidas de péptidos que contienen, en muchos casos, varios subproductos, que pueden tener su origen en la escisión parcial del enlazante.
Giraud y col. en el documento de EE. UU. 2010/0197891 describen un procedimiento para anclar una cadena de péptidos creciente durante la síntesis química a un soporte de fase sólida por medio del grupo amino de una cadena lateral de aminoácidos. Sin embargo, el documento de EE. UU. 2010/0197891 no describe la unión de una amida de péptido a la resina.
La presente invención busca proporcionar nuevos conjugados de péptido-resina para su utilización en la síntesis de péptidos. Más particularmente, en una realización, la invención busca proporcionar nuevos conjugados de péptidoresina y procedimientos relacionados con los mismos para permitir la preparación de péptidos que exhiben uno o más de los siguientes: rendimientos mejorados, una pureza más alta, menos reacciones secundarias y condiciones de reacción más leve.
Los aspectos de la invención se establecen en las reivindicaciones adjuntas y se describen con más detalles a continuación.
DECLARACIÓN DE LA INVENCIÓN
Un primer aspecto de la invención se refiere a un procedimiento para preparar un péptido, o una sal del mismo, de un conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f),
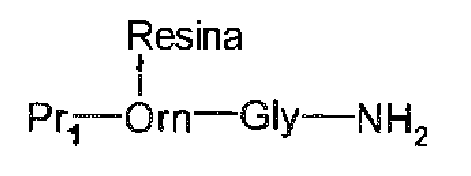
(2b)

(2c)

(2d)

Resina
I
Pr1— Orn T yr(tB u)—NH2
(2f)
en el que la resina es una resina polimérica escindible de TFA seleccionada de entre resinas de tritilo, 2-cloro-tritlo, 4-metil-tritilo y 4-metoxi-tritilo; y
Pri es un grupo protector seleccionado de entre el grupo que consiste en: Fmoc, Boc, Cbz, Npys y Alloc; dicho procedimiento comprendiendo las etapas de:
(a) desproteger la función a-amino del extremo N-terminal de dicho conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f) para eliminar el grupo protector Pri;
(b) acoplar al menos un aminoácido o péptido protegido en el extremo N-terminal que presente una función de ácido carboxílico activado o libre con la función a-amino desprotegida de la etapa (a), alargando así el compuesto de las fórmulas (2b), (2c), (2d), (2e) o (2f),
(c) repetir, opcionalmente, las etapas (a) y (b) una o más veces, en las que el al menos un aminoácido o péptido protegido en el extremo N-terminal es idéntico o diferente de aquel de la etapa anterior (b);
(d) escindir el péptido resultante de la resina;
(e) guanilar el péptido obtenido en la etapa (d);
(f) eliminar, opcionalmente, todos los grupos protectores que queden después de la etapa (d);
(g) aislar y opcionalmente purificar el péptido así obtenido.
De manera ventajosa, la utilización de los conjugados de amida de péptidos reivindicados en este documento permite la preparación de péptidos en mejores rendimientos y/o una pureza más alta y/o con menos reacciones secundarias. Además, las condiciones de reacción leve requeridas para eliminar las amidas de péptidos de la resina permiten que las amidas de péptidos se obtengan de manera protegida o parcialmente protegida, es decir, que el procedimiento permite la escisión de la resina de amidas de péptidos parcialmente protegidos. Esto permite que los péptidos obtenidos se conviertan selectivamente en los péptidos guanilados correspondientes. Los conjugados reivindicados en este documento son particularmente útiles en la preparación de un número de péptidos específicos descritos en esta invención.
DESCRIPCIÓN DETALLADA
Como se utiliza en esta invención, el término «alquilo» incluye grupos alquilo de cadena ramificada y recta saturada que pueden sustituirse (mono o poli) o no. Preferentemente, el grupo alquilo es un grupo alquilo C1-20, más preferentemente un C1-15, incluso más preferentemente un grupo alquilo C1-12, incluso más preferentemente un grupo alquilo C1-6 , y más preferentemente un grupo alquilo C1-3. En particular, los grupos alquilo preferidos incluyen, por ejemplo, metil, etil, propil, isopropil, butil, isobutil, tert-butil, pentil y hexil. Los sustituyentes incluyen, por ejemplo, uno o más grupos seleccionados de entre OH, O-alquilo, halógeno, NH2 , NH-alquilo, N-(alquilo)2 , CF3 , NO2 , CN, COO-alquilo, c Oo H, CONH2 , CO-NH-alquilo, CO-N(alquilo)2 , SO2-alquilo, SO2NH2 y SO2-NH-alquilo.
Como se utiliza en esta invención, el término «arilo» se refiere a un grupo aromático C6-12 que puede sustituirse (mono o poli) o no. Los ejemplos típicos incluyen fenil y naftenil, etc. Los sustituyentes incluyen, por ejemplo, uno o más grupos seleccionados de entre OH, O-alquilo, halógeno, NH2 , NH-alquilo, N-(alquilo)2 , CF3 , NO2 , CN, COO-alquilo, COOH, CONH2 , CO-NH-alquilo, CO-N(alquilo)2, SO2-alquilo, SO2NH2 y SO2-NH-alquilo.
El término «aralquilo» se utiliza como una conjunción de los términos alquilo y arilo, como se indicó anteriormente. Como se utiliza en esta invención, el término «aroil» se refiere a un «Ar-CO» radical, donde Ar es un grupo arilo, como se definió anteriormente. Los ejemplos de los grupos aroil incluyen benzoil y naftoil.
Como se utiliza en esta invención, el término «acilo» se refiere a un «Alquil-CO» radical, donde alquilo es tal como se definió anteriormente.
Las sales farmacéuticamente aceptables de los compuestos de la invención incluyen la adición de ácidos adecuados o sales básicas de los mismos. En Berge y col. J Pharm Sci, 66, 1-19 (1977) se puede encontrar una reseña de las sales farmacéuticas adecuadas. Las sales se forman, por ejemplo, con ácidos inorgánicos fuertes, como ácidos minerales, por ejemplo, ácido sulfúrico, ácido fosfórico o ácidos hidrogenados; con ácidos carboxílicos orgánicos fuertes, como los ácidos alcanocarboxílicos de 1 a 4 átomos de carbono sustituidos o no (por ejemplo, por un halógeno), como el ácido acético; con ácidos dicarboxílicos saturados o insaturados, por ejemplo los ácidos oxálico, malónico, succínico, maleico, fumárico, ftálico o tetraftálico; con ácidos hidroxicarboxílicos, por ejemplo, el ácido ascórbico, el ácido glicólico, el ácido láctico, el ácido tartárico o el ácido cítrico; con aminoácidos, por ejemplo, el ácido
aspártico o el ácido glutámico; o con ácidos sulfónicos orgánicos, como los ácidos (Ci-C4)-alquil- o aril-sulfónicos, sustituidos o no (por ejemplo, por un halógeno) como el ácido metanosulfónico o p-toluenosulfónico. Particularmente, se prefieren las sales de acetato.
En todos los aspectos de la presente invención anteriormente analizada, la invención incluye, donde corresponda, todos los enantiómeros y tautómeros de los compuestos de la invención. El experto en la materia reconocerá los compuestos que poseen propiedades ópticas (uno o más átomos de carbono quiral) o características tautoméricas. Los enantiómeros y/o tautómeros correspondientes pueden aislarse/prepararse mediante los procedimientos conocidos en la técnica.
Algunos de los compuestos de la invención pueden existir como estereoisómeros y/o isómeros geométricos, por ejemplo, pueden poseer uno o más centros asimétricos y/o geométricos y, por tanto, pueden existir en dos o más formas estereoisoméricas y/o geométricas. La presente invención contempla la utilización de todos los estereoisómeros individuales e isómeros geométricos de esos compuestos, y sus mezclas. Los términos utilizados en las reivindicaciones abarcan estas formas.
La presente invención también incluye todas las variaciones isotópicas de los compuestos o sales farmacéuticamente aceptables de los mismos. Una variación isotópica de un agente de la presente invención o una sal farmacéuticamente aceptable del mismo se define como uno en el que al menos un átomo se reemplaza con un átomo que presenta el mismo número atómico, pero una masa atómica diferente de la masa atómica que se encuentra generalmente en la naturaleza. Los ejemplos de isótopos que pueden incorporarse al agente y sus sales farmacéuticamente aceptables incluyen los isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, azufre, flúor y cloro, como 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F y 36Cl, respectivamente. Ciertas variaciones isotópicas del agente y sus sales farmacéuticamente aceptables, por ejemplo, aquellas en las que se incorpora el isótopo radioactivo, como 3H o 14C, son útiles en los estudios de la distribución de fármacos y/o sustratos en el tejido. Los isótopos tritiados, es decir, 3H, y carbono-14, es decir, 14C, son particularmente preferidos por su facilidad de preparación y detectabilidad. Además, la sustitución con isótopos tales como el deuterio, es decir, 2H, puede permitir ciertas ventajas terapéuticas que resultan de una mayor estabilidad metabólica, por ejemplo, el aumento de la vida media in vivo o requerimientos de dosis reducidos y, por tanto, puede preferirse en algunas circunstancias. Las variaciones isotópicas del agente de la presente invención y las sales farmacéuticamente aceptables del mismo de esta invención, en general, se pueden preparar mediante procedimientos convencionales utilizando variaciones isotópicas de reactivos adecuados.
Los aminoácidos naturales incluyen alanina, arginina, asparagina, ácido aspártico, cisteína, ácido glutámico, glutamina, glicina, histidina, isoleucina, leucina, lisina, metionina, fenilalanina, prolina, serina, treonina, triptófano, tirosina y valina.
Como se utiliza en esta invención, el término «aminoácido no natural» incluye los aminoácidos alfa y alfa-disustituido, los aminoácidos N-alquilo, el ácido láctico, los derivados de haluro de aminoácidos naturales como la trifluorotirosina, la p-Cl-fenilalanina, la p-F-fenilalanina, la p-Br-fenilalanina, la p-NO2-fenilalanina, la fenilglicina, la azaglicina, la sarcosina, la penicilamina, el D-2-metil triptófano, la fosfoserina, la fosfotreonina, la fosfotirosina, la p-I-fenilalanina, la L-alil-glicina, la p-alanina, el ácido p-aspártico, la p-ciclohexilalanina, la citrulina, la homoserina, la homocisteína, el ácido piroglutámico, el ácido L-a-aminobutírico, el ácido L-Y-aminobutírico, el ácido L-a-aminoisobutírico, la aciclohexilglicina, el ácido diaminobutírico, el ácido diaminopimélico, la N-e-dinitrofenil-lisina, la L-1-naftilalanina, la L-2-naftilalanina, la 3-(2-piridil)-L-alanina, la 3-(3-piridil)-L-alanina, la 3-(4-piridil)-L-alanina, la N-£-metil-lisina, la N,N-edimetil-lisina, la N,N,N-e-trimetil-lisina, el ácido 3-mercaptopropiónico, el ácido L-e-aminocaproico, el ácido 7-aminoheptanoico, el ácido 6-aminohexanoico la L-metionina sulfona, la ornitina, la L-norleucina, la L-norvalina, la pnitro-L-fenilalanina, la L-hidroxiprolina, el ácido Y-glutámico, el ácido Y-aminobutírico, la L-tioprolina, los derivados de metil de la fenilalanina (Fe) como la 4-metil-Fe, la pentametil-Fe, la (4-amino)L-Fe, la (metil)L-Tir, la (4-isopropil)L-Fe, el L-Tic (1,2,3,4-tetrahidroisoquinolina-3-carboxil ácido), el ácido L-diaminopropiónico y la (4-bencil)L-Fe.
El péptido de la presente invención puede comprender aminoácidos en forma de L o D, es decir, uno o más residuos, preferentemente todos los residuos pueden estar en forma de L o D.
Como se utiliza en esta invención, el término «péptido sintético» se refiere a un péptido que se sintetiza químicamente. Los péptidos sintéticos pueden prepararse a partir de aminoácidos naturales o no, o una combinación de los mismos.
Como se utiliza en esta invención, el término «péptido natural» se refiere a un péptido que se encuentra en la naturaleza.
Los presentes inventores han demostrado que los diaminoácidos que contienen amidas de péptidos cortos unidos a resinas sensibles altamente ácidas adecuadas desde su cadena lateral pueden proporcionar amidas de péptidos y amidas de péptidos parcialmente protegidos más grandes con una función amino de cadena lateral de diaminoácido selectivamente liberada. Estas pueden modificarse selectiva y adicionalmente en la cadena lateral de diaminoácido. Una modificación importante de la cadena lateral de diaminoácido es, por ejemplo, la guanidilación selectiva de Orn a Arg para dar péptidos que contengan Arg. Esto es ventajoso sobre la síntesis de péptidos que contienen Arg mediante la utilización de derivados de Arg protegidos en su cadena lateral porque la desprotección de la cadena lateral de Arg,
utilizando los grupos protectores guanidino habituales generalmente da origen a la formación de varios subproductos y, además, en muchos casos, resulta incompleta.
También se forman varios productos secundarios durante la eliminación de amidas de péptidos de resinas que proporcionan amidas de péptidos como las resinas de Rink-amida.
Los presentes inventores han descubierto que la unión de péptidos que contienen diaminoácidos cortos, en resinas del tipo tritilo, procede con alto rendimiento y que los péptidos y amidas de péptidos parcialmente protegidos que se obtienen son de alta pureza, mayor a la obtenida mediante la utilización del procedimiento de síntesis de amida de péptido correspondiente, la cual utiliza una resina de amida. Las amidas de péptidos y amidas de péptidos parcialmente protegidos escindidos de la resina pueden transformarse en un alto rendimiento para péptidos que contienen aminoácidos guanilados en su secuencia, como Arg, D-Arg y homo-Arg. Las amidas de péptidos obtenidas presentan una pureza más alta que los péptidos que contienen Arg correspondientes, sintetizados utilizando derivados de Arg protegidos con cadenas laterales, por ejemplo, Fmoc-Arg(Pbf)-OH.
Las amidas de péptidos cortos adecuadas requeridas (1) se obtienen mediante procedimientos conocidos en la técnica, por ejemplo, mediante la desprotección de la función amino de cadena lateral del péptido corto. Las amidas de péptidos protegidos se conjugan posteriormente con resinas sensibles muy ácidas del tipo tritilo a través de la función amino de cadena lateral del diaminoácido contenido en la cadena peptídica, según el esquema a continuación.
Por ejemplo, el diaminoácido que está contenido en la parte del extremo C-terminal de la amida de péptido se hace reaccionar con un haluro de resina, A-CI, en la presencia de una base para dar un conjugado de péptido-resina (2) en alto rendimiento:
Pr1-Y-D¡a-X-NR1R2 ------------------Prr Y-D ia-X
DI PEA
Fórmula (1) Fórmula (2)
en la que Pn se selecciona de entre H, alquilo, arilo, aralquilo, acilo, aroil y un grupo protector;
Y es un enlace directo; o un residuo de aminoácido natural o no natural opcionalmente protegido; o un péptido natural o sintético que comprende de 2 a 200 residuos de aminoácidos naturales o no naturales, cada uno de los cuales está opcionalmente protegido;
Dia es un diaminoácido natural o no natural;
A es una resina polimérica conjugada para la función amino de cadena lateral del diaminoácido;
X es un residuo de aminoácido natural o no natural opcionalmente protegido; o un péptido que comprende de 2 a 15 residuos de aminoácidos naturales o no naturales, cada uno de los cuales está opcionalmente protegido; R1 y R2 , cada uno, se seleccionan independientemente de H, alquilo, arilo, aralquilo, NH2 , NH-CO-NH2.
En una realización preferida, la base es una base de trialquilamina, más preferentemente DIPEA.
En una realización preferida, A es una resina polimérica escindible de TFA conjugada en la función amino de cadena lateral del Dia.
Más preferentemente, A es una resina escindible de TFA del tipo tritilo.
Incluso más preferentemente, A se selecciona de entre resinas de tritilo, de 2-cloro-tritilo, de 4-metil-tritilo y de 4-metoxi-tritilo como se muestra a continuación, en las que Q puede estar ausente o es un enlazador entre el grupo tritilo y la matriz polimérica P, por ejemplo, un grupo carboxilo.

Las amidas de péptidos con uniones de resina también pueden obtenerse por medio de la unión del diaminoácido a la resina a través de la función amino de cadena lateral y el acoplamiento en la resina con la amida de aminoácido o amida de péptido, como se muestra en el esquema a continuación:
A A
C l-A I H-X-NR-i R2
Fmoc-Dia-OH ---------------- Fmoc-Dia-OH Fmoc-Dia-X-NR.] R2
DIPEA DIC/HOBt
(3)
Las amidas de péptidos (3) también se pueden obtener mediante la amidación de péptidos con unión de resina, como se muestra a continuación:
A A
Fmoc-Dia-X-OH  -X-NR1 R2
-X-NR1 R2
Los péptidos con unión de resina de las fórmulas (2) y (3) se pueden utilizar en la síntesis peptídica de fase sólida. Después de completar el ensamble de la cadena, las amidas de péptidos obtenidas de la fórmula general (2) se pueden escindir de las resinas extremadamente sensibles al ácido en la forma de péptido parcialmente protegido de la fórmula general (1) o en la forma completamente desprotegida, como se muestra en el esquema a continuación con la fórmula general (4):

Fm oc-D ia-X-NR iR2 P r1-Y-D¡a-X-NR1R2

H-Y-Dia-X-NR-iR? Pr1-Y-D ia-X-NR1R2

Desproteccion
H-Y-Gua-X-NR-iRo Pr-1-X-G ua-Y-NR1 R2
SBS = Procedimiento etapa por etapa
(SBS, del ingles step-by-step)
Los péptidos desprotegidos selectivamente en la función de cadena lateral de diaminoácido (1) pueden modificarse adicionalmente en la cadena lateral del diaminoácido, como se muestra en el esquema anterior, mediante la guanidilación. La etapa de guanidilación puede efectuarse mediante cualquier procedimiento conocido en la técnica para, por ejemplo, utilizar los reactivos de guanidilación, como la S-metiltiourea, la 1-H-1,2,4-triazol-carboxamidina, etc.
En una realización preferida, el reactivo de guanlidilación se selecciona de entre 1H-pirazol-1-carboxamidina 2, 1-H-1,2,4-triazol-carboxamidinas, triflil guanidina y tosilato de benzotriazol-1-carboxamidinio.
Los péptidos parcialmente protegidos guanidilados obtenidos de la fórmula general (5) entonces pueden desprotegerse totalmente para dar la función de cadena lateral de guanidil que contiene péptidos de la fórmula general (6), en el que Gua es un grupo guanidilo de cadena lateral que contiene aminoácidos como la Arg.
Los grupos protectores para los aminoácidos resultarán familiares para la persona experta en la técnica (véase, por ejemplo, Chem. Rev. 2009, 109, 2455-2504). Estos grupos protectores pueden separarse en tres grupos, como se indica a continuación:
• grupos protectores del extremo N-terminal
• grupos protectores del extremo C-terminal
• grupos protectores de cadena lateral
Los ejemplos de grupos protectores de extremo N-terminal altamente preferidos para los aminoácidos incluyen, entre otros, el t-Boc (ferf-butiloxicarbonil) y Fmoc (9-fluorenilmetiloxicarbonil). Su labilidad es causada por el grupo carbamato que libera CO2 de inmediato para una etapa de desacoplamiento irreversible. Otro grupo adecuado basado en carbamato es el grupo benciloxi-carbonilo (Z o Cbz); este se elimina en condiciones más duras.
Otro ejemplo preferido es el grupo protector aliloxicarbonilo (alloc), que a menudo se utiliza para proteger un ácido carboxílico, un grupo hidroxilo o amino, cuando se requiere un esquema de desprotección ortogonal.
Otro ejemplo preferido es el grupo nitro-2-piridinasulfenilo (Npys), que es útil para la protección y activación de grupos amino y hidroxilo en la síntesis peptídica. El grupo Npys se introduce de inmediato mediante el tratamiento de aminoácidos con cloruro de 3-nitro-2-piridinasulfenilo. El grupo Npys se elimina fácilmente mediante el tratamiento con HCl muy diluido, por ejemplo, 0,1-0,2 N HCI en dioxano, pero es resistente al ácido trifluoroacético y 88 % ácido fórmico. Npys también se elimina selectivamente bajo condiciones neutrales, utilizando trifenilfosfina o 2-piridinatiol 1-óxido sin afectar a los grupos protectores de benciloxicarbonilo (Z), tert-butiloxicarbonilo (Boc), 2-(4-bifenilil)propil(2)oxicarbonilo (Bpoc), 9-fluorenilmetiloxicarbonilo (Fmoc), bencilo (Bzl) o tert-butilo (tBu).
Las cadenas laterales de aminoácidos representan una amplia gama de grupos funcionales y son sitios de reactividad no específica durante la síntesis peptídica. Debido a esto, se requieren muchos grupos protectores que, por lo general, están basados en el grupo bencilo (Bzl) o tert-butilo (tBu). Los grupos protectores específicos utilizados durante la
síntesis de un cierto péptido varían según la secuencia de péptidos y el tipo de protección de extremo N-terminal utilizada. Generalmente, a los grupos protectores de cadena lateral se los conoce como grupos protectores permanentes o semipermanentes, porque pueden resistir múltiples ciclos de tratamiento químico durante la síntesis y solo se eliminan durante el tratamiento con ácidos fuertes, después de completar la síntesis peptídica.
Los aminoácidos individuales y purificados se hacen reaccionar con esos grupos protectores antes de la síntesis y después se eliminan selectivamente durante etapas específicas de la síntesis peptídica.
En una realización preferida, Pri es un grupo protector Fmoc.
Más preferentemente, la resina es una resina de tritilo o de 4-metoxitritilo.
Los conjugados de péptido-resina se pueden preparar mediante un proceso que comprende hacer reaccionar una amida de péptido con un haluro de resina adecuado en un solvente adecuado, en la presencia de una base.
Preferentemente, el haluro se selecciona de entre cloruro, bromuro y yoduro.
Preferentemente, el solvente se selecciona de entre DCM, DCE, DMF, NMP, THF, DME y sus mezclas.
Preferentemente, la base se selecciona de entre DIPEA, NMM, DBU, piridina, DMAP y TEA.
En una realización preferida, los aminoácidos o péptidos protegidos en su extremo N-terminal de las etapas (b) y (c) están protegidos con Fmoc.
En una realización preferida, el al menos un aminoácido o péptido protegido en su extremo N-terminal de la etapa (c) repetida en último lugar está protegido por un grupo protector que es ortogonal respecto de Fmoc.
En una realización preferida, el grupo protector ortogonal es Boc.
En una realización preferida, el paso (d) comprende escindir el péptido de la resina mediante el tratamiento con un ácido.
En una realización preferida, la etapa (e) comprende tratar el péptido con un reactivo de guanilado seleccionado de entre 1H-pirazol-1-carboxamidina 2, 1-H-1,2,4-triazol-carboxamidina, triflil guanidina y tosilato de benzotriazol-1-carboxamidinio.
En una realización preferida, el conjugado de péptido-resina se selecciona de entre los siguientes:
Resina

Resina
I
F m o c— O rn — P ro— N H 2
(2h)
Resina

(2 i)
Resina
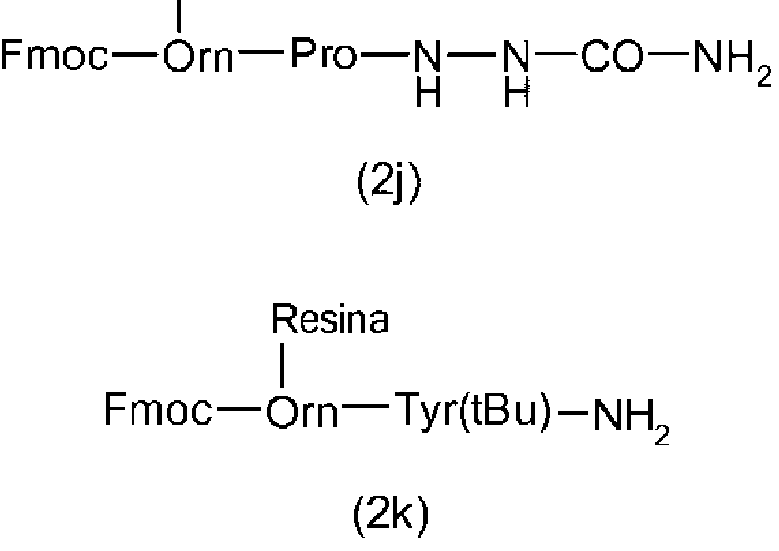
en el que la resina es una resina de tritilo o una resina de 4-metoxitritilo.
En una realización preferida, el procedimiento de la invención se utiliza para preparar un péptido seleccionado de entre los siguientes:
[2] Arg8-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Arg-Gli-NH2 ;
[5 ] 3-Mercaptopropionil-Tir-Fe-Gln-Asn-Cis-Pro-D-Arg-Gli-NH2;
[8] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2 ;
[9 ] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt sal de acetato;
[10] Pir-His-T rp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt;
[1 1 ] Pir-His-Trp-Ser-Tir-D-His(Bzl)-Leu-Arg-Pro-NHEt sal de acetato;
[1 2 ] Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-NHEt sal de acetato;
[20]
H-Tir-Pro-Ser-Lis-Pro-Asp-Asn-Pro-Gli-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met- Ala-Arg-Tir-Tir-Ser-Ala-Leu-Arg-His-Tir-lle-Asn-Leu-lle-Tr-Arg-Gln-Arg-Tir- NH2 ; y
[21]
H-Tir-Pro-lle-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tir-Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-GIn-Arg-Tir-NH2.
Los péptidos específicos preparados mediante el procedimiento de la invención incluyen los siguientes:
* denota que está fuera del alcance de la invención
*Acetato de atosiban,
3-Mercaptopropionil-D-Tir(Et)-Ile-Tr-Asn-Cis-Pro-Orn-Gli-NH2 sal de acetato
Acetato de vasopresina,
Arg-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Arg-Gli-NH2 sal de acetato
*Acetato de ornipresina,
H-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Orn-Gli-NH2 sal de acetato
*Acetato de terlipresina,
H-Gli-Gli-Gli-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Lis-Gli-NH2 sal de acetato
Acetato de desmopresina,
3-Mercaptopropionil-Tir-Fe-Gln-Asn-Cis-Pro-D-Arg-Gli-NH2 sal de acetato
*Acetato de triptorelina,
Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-Gli-NH2 sal de acetato
*Acetato de nafarelina
Pir-His-Trp-Ser-Tir-D-2-Nal-Leu-Arg-Pro-Gli-NH2 sal de acetato
Goserelina
Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2
Acetato de buserelina
Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt sal de acetato
Leuprolida (Leuprolin)
Pir-His-Trp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt
Acetato de histrelina
Pir-His-Trp-Ser-Tir-D-His(Bzl)-Leu-Arg-Pro-NHEt sal de acetato
Acetato de deslorelina alto
Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-NHEt sal de acetato
*Cetrorelix
Ac-D-2-Nal-4-cloro-D-Fe-p-(3-piridil)-D-Ala-Ser-Tir-D-Cit-Leu-Arg-Pro-D-Ala-NH2 sal de acetato
*Ozarelix
Ac-D-2-Nal-D-4-Cpa-D-3-Pal-Ser-N-Me-Tir-D-Hci-Nle-Arg-Pro-D-Ala-NH2 sal de acetato
*Acetato de ziconotida
H-Cis-Lis-Gli-Lis-Gli-Ala-Lis-Cis-Ser-Arg-Leu-Met-Tir-Asp-Cis-Cis-Tr-Gli-Ser- Cis-Arg-Ser-Gli-Lis-Cis-NH2 sal de acetato
*Afamelanotida
Ac-Ser-Tir-Ser-Nle-Glu-His-D-Fe-Arg-Trp-Gli-Lis-Pro-Val-NH2 sal de acetato
*Acetato de GRF (humana)
H-Tir-Ala-Asp-Ala-lle-Fe-Tr-Asn-Ser-Tir-Arg-Lis-Val-Leu-Gli-Gln-Leu-Ser-Ala-Arg- Lis-Leu-Leu-Gln-Asp-lle-Met-Ser-Arg-Gln-Gln-Gli-Glu-Ser-Asn-Gln-Glu-Arg-Gli-Ala- Arg-Ala-Arg-Leu-NH2 sal de acetato
*Amida de GRF (1-29) (humana)
H-Tir-Ala-Asp-Ala-lle-Fe-Tr-Asn-Ser-Ti-Arg-Lis-Val-Leu-Gli-Gln-Leu-Ser-Ala-Arg-Lis-Leu-Leu-Gln-Asp-lle-Met-Ser-Arg-NH2
‘ Acetato de a-melanotropina (humana)
Ac-Ser-Tir-Ser-Met-Glu-His-Fe-Arg-Trp-Gli-Lis-Pro-Val-NH2 sal de acetato
Acetato de neuropéptido Y (humano, rata)
H-Tir-Pro-Ser-Lis-Pro-Asp-Asn-Pro-Gli-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met-Ala-Arg-Tir-Tir-Ser-Ala-Leu-Arg-His-Tir-lle-Asn-Leu-lle-Tr-Arg-Gln-Arg-Tir-NH2 sal de acetato
Acetato del péptido YY (humano)
H-Tir-Pro-lle-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tir- Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-Gln-Arg-Tir-NH2 sal de acetato
El procedimiento de la invención es adecuado para preparar una variedad de diferentes péptidos, incluyendo, entre otros, los siguientes:
[2] Arg-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Arg-Gli-NH2 ;
[5] 3-Mercaptopropionil-Tir-Fe-Gln-Asn-Cis-Pro-D-Arg-Gli-NH2 sal de acetato;
[8] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2 ;
[9 ] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt sal de acetato;
[10] Pir-His-T rp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt;
[11] Pir-His-Trp-Ser-Tir-D-His(Bzl)-Leu-Arg-Pro-NHEt sal de acetato;
[1 2 ] Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-NHEt sal de acetato;
[20]
H-Tir-Pro-Ser-Lis-Pro-Asp-Asn-Pro-Gli-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met- Ala-Arg-Tir-Tir-Ser-Ala-Leu-Arg-His-Tir-lle-Asn-Leu-lle-Tr-Arg-Gln-Arg-Tir- NH2 sal de acetato; y
[21]
H-Tir-Pro-lle-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn- Arg-Tir-Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-GIn-Arg-Tir- NH2 sal de acetato.
De manera ventajosa, el procedimiento reivindicado en este documento permite amidas de péptidos y amidas de péptidos parcialmente protegidos escindidos de la resina para transformarse en un alto rendimiento para péptidos que contienen aminoácidos guanilados en su secuencia, como Arg, D-Arg y homo-Arg. Las amidas de péptidos obtenidas presentan una pureza más alta que los péptidos que contienen Arg correspondientes, sintetizados utilizando derivados de Arg protegidos con cadenas laterales, por ejemplo, Fmoc-Arg(Pbf)-OH.
La presente invención se describe adicionalmente por medio de los siguientes ejemplos no limitantes.
EJEMPLOS
Síntesis de fase sólida de péptidos y sus segmentos protegidos.
Procedimiento general.
Preparación de resinas de tritilo cargadas con péptidos unidos a través de la cadena lateral de un diaminoácido, procedimiento general.
La resina de cloruro de tritilo (100 g; carga de 0,9-1,6 mmol/g) de CBL-Patras se colocó en un reactor de síntesis peptídica de 2 L y se hinchó con 700 ml de diclorometano (DCM) durante 30 min a 25 °C. La resina se filtró y una solución de 100 mmol de
Ácido de péptido Fmoc y diisopropiletilamina (DIEA) en DCM se adicionó, de modo tal que la proporción de mmol del péptido Fmoc/DIPEA se volvió de 0,80. La mezcla se agitó bajo nitrógeno durante 4 horas a 25 °C. Después, los sitios activos restantes de la resina se neutralizaron mediante la adición de 10 ml de metanol (MeOH) y se procedió a la reacción durante 1 hora a temperatura ambiente. La resina se filtró y se lavó 4X con 400 ml de DMF, y se deshinchó con 3 lavados de 500 ml de isopropanol (IPA) y 4X con 400 ml de DEE. La resina se secó a un peso constante. Del 60 al 80 % del mmol del péptido utilizado se unió a la resina.
Todas las resinas del tipo tritilo en el paciente se encuentran comercialmente disponibles (CBL-Patras y otros).
A3. Amidación de ácidos peptídicos unidos en una fase sólida a través de una cadena lateral de aminoácido de un diaminoácido, procedimiento general.
Un péptido con unión de resina (5,0 g = 0,2-1,2 mmol/g) se colocó en el reactor de fase sólida y se trató con 0,25 1,4 mmol de HOBt y DIC. La mezcla se agitó durante 1 hora a temperatura ambiente y después se adicionó una solución de amoniaco o alquilamina en DMF o HOBt.sal de NH3 o una amida de péptido en DMF en un exceso molar de 1,00-1,5. A continuación, la mezcla se agitó durante 2 horas a temperatura ambiente y el procedimiento se repitió una vez. Después, la resina se filtró y se lavó 5X con DMF, 3X con IPA y 3X con DEE, y se secó al vacío a un peso constante.
B. Síntesis de fase sólida, un protocolo general
B1. Hinchamiento de la resina
La resina se colocó en un reactor de 15 ml y se trató dos veces con 7 ml de NMP, y después se filtró.
B2. Activación del aminoácido
El aminoácido (equiv. a 3,0) y 1-hidroxibenzotriazol (equiv. a 4,0) se pesó y se disolvió en un reactor con 2,5 de su volumen en NMP. Después se enfrió a hasta 0 °C. A continuación, se añadió DIC (equiv. a 3,0) y la mezcla se agitó durante 15 min.
B3. Acoplamiento
La solución que se preparó en B2 después se adicionó al reactor B1. El reactor se lavó una vez con un volumen de DCM y se adicionó al reactor que se agitó por 1-3 h a 25 a 30 °C. En una muestra, se efectuó la prueba de Kaiser para determinar si la reacción se había completado. Si no se había completado la reacción de acoplamiento al cabo de 3 horas (prueba de Kaiser positiva), la mezcla de reacción se filtraba y reacoplaba con una solución fresca de aminoácido activado. Después de completar la reacción de acoplamiento, la mezcla de reacción se filtró y se lavó 4 veces con NMP (5 volúmenes por lavado).
B4. Eliminación del grupo Fmoc
La resina resultante en B3 se filtró y después se trató durante 30 min con 5 ml de una solución que contenía el 25 % del volumen de piperidina. Después, se lavó la resina tres veces con 5 ml de NMP.
B5. Alargamiento de la cadena de péptidos
Después de la incorporación de cada aminoácido, las etapas B1-B5 se repitieron hasta completar la cadena de péptidos.
Para la introducción de cada aminoácido individual, se utilizaron los siguientes aminoácidos Fmoc: Fmoc-Gli-OH, Fmoc-Ala-OH, Fmoc-Val-OH, Fmoc-Ile-OH, Fmoc-Leu-OH, Fmoc-Met-OH, Fmoc-Fe-OH, Fmoc-Pro-OH, Fmoc-Asp(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Lis(Boc)-OH, Fmoc-Orn(Mtt)-OH, Fmoc-Orn(Mmt)-OH, Fmoc-Orn(Boc)-OH, Fmoc-Lis(Mmt)-OH, Fmoc-Lis(Mtt)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Ser(Trt)-OH, Fmoc-Tr(tBu)-OH, Fmoc- Tr(Trt)-OH, Fmoc-Tir(tBu)-OH, Fmoc-Tir(Clt)-OH, Fmoc-Asn-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln-OH, Fmoc- Gln(Trt)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-His(Trt)-OH, Fmoc-Cis(Trt)-OH, Fmoc-Cis(Mmt)-OH and Fmoc-Cis(Acm)-OH y los siguientes aminoácidos Boc: Boc-Fe-OH y Boc-Gli-OH.
C. Procedimiento general para la escisión de resinas del tipo tritilo de péptidos parcialmente protegidos y de sus segmentos protegidos que contienen grupos Fmoc o Boc u otro grupo protector adecuado en sus extremos N-terminales y que se desprotegen selectivamente a una ornitina, una lisina individual o cualquier otra cadena lateral de diaminoácido.
El péptido o segmento de péptido con unión de resina que se produjo como se describió anteriormente en B1-B5 y se unió a una Lis, Orn o cualquier otra cadena lateral de diaminoácido específica con una resina de 2-clorotritilo (Clt), de Trt-Mmt o de Mtt se lavó 4 veces con 5 ml de NMP, 3 veces con 5 ml de IPA y finalmente 5 veces con 7 ml de DCM para eliminar completamente cualquier NMP residual u otros componentes básicos. A continuación, la resina se enfrió a 0 °C, se filtró de DCM y se trató seis veces con una solución de 10 ml del 1,0-1,5 % de TFA en DCM/TES(95:5) a 5 °C. La resina se filtró y se lavó tres veces con 10 ml de DCM. Después se adicionó piridina a los filtrados (equiv. a 1,3 en relación con el TFA) para neutralizar el TFA. La solución de escisión en DCM después se mezcló con un volumen igual de agua.
La mezcla resultante se destiló a presión reducida, a fin de eliminar el DCM (350 torr a 28 °C). El péptido o segmento de péptido se precipitó después de la eliminación del DCM. El péptido resultante se lavó con agua y éter y se secó a 30-35 °C bajo 15 Torr de vacío. De manera alternativa, el DCM puede eliminarse al vacío y el péptido parcialmente protegido se puede precipitar mediante la adición de DEE o éter de diisopropilo (DIE).
D. Procedimiento general para la guanilación del péptido parcialmente protegido que se desprotege selectivamente en la cadena lateral de uno o más diaminoácidos.
El péptido parcialmente desprotegido en la cadena lateral de diaminoácido (1,0 mmol) se disuelve en 10-15 ml de DMF o en una mezcla adecuada de DMF/agua y después se neutraliza la solución mediante la adición de DIPEA o 1N-NaOH. A continuación, se adiciona un exceso molar de 1,0-1,5 del reactivo de guanilación, por ejemplo, 1-H-1,2,4-triazol-carboxamidina hidrocloruro, y la mezcla se agita hasta determinar que la guanilación ha sido completada mediante HPLC, TLC o la prueba de Kaiser. Después, se diluye la mezcla con salmuera y el producto se extrae en la fase orgánica con EtAc o DCM, lo cual es seguido de una extracción de ácido-base estándar. La solución obtenida del péptido guanilado posteriormente se concentra en el precipitado de RE con la adición de DEE o DIE y se desprotege según nuestro procedimiento general que se indica a continuación.
E. Desprotección del péptido - Procedimiento general.
El péptido parcialmente protegido, como se describió anteriormente (0,01-0,005 mmol) se trató con 10 ml de TFA/TES o TIPS/tioanisol/agua (85:5:5:5) o TFA/DTT/agua (90:5:5) durante 3 horas a 5 °C y durante 1 hora a 15 °C. La solución resultante se concentró al vacío y, después, el péptido desprotegido se precipitó mediante la adición de DEE o DIE y se lavó tres veces con 10 ml de DEE o DIE. El sólido resultante se secó al vacío (25 °C, 1-10 Torr) hasta alcanzar un peso constante.
Ejemplo 1. Síntesis de Atosiban a partir de un Fmoc-Orn-Gli-NH2 unido a través de la cadena lateral de ornitina en la resina de tritilo.

La resina de cloruro de tritilo (20,0 g; carga de 24,4 mmol) de la resina de cloruro de tritilo se colocó en un reactor de síntesis peptídica y se hinchó con 200 ml de DCM/DMF (1:1) durante 30 min a 25 °C. A continuación, se adicionaron 4,63 g (10 mmol) de Fmoc-Orn-Gli-NH2.HCl, producido mediante procedimientos conocidos en la técnica de Fmoc-Orn(Boc)-Gli-NH2 y HCI en dioxano. La mezcla se agitó durante la noche a temperatura ambiente. Después, los sitios activos restantes de la resina se neutralizaron mediante la adición de 10 ml de metanol (MeOH) y se hicieron reaccionar durante 2 horas adicionales a temperatura ambiente. Después, la resina se filtró y se lavó 4X con 400 ml de DMF, y se deshinchó con 3 lavados de 500 ml de isopropanol (IPA) y 4X con 400 ml de DEE. Posteriormente, se hinchó nuevamente en DMF. A continuación, se efectuó el alargamiento de la cadena peptídica según los procedimientos estándares, utilizando Fmoc-Pro-OH, Fmoc-Cis(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Tr(Trt)-OH, Fmoc-Ile-OH y Fmoc-D-Tir(Et)-OH. Finalmente, se introdujo el ácido S-Trt-mercaptopropiónico de extremo N-terminal mediante un procedimiento similar utilizado para la introducción de los aminoácidos Fmoc. Después, la resina se lavó 6X con DMF y 6X con DCM. A continuación, la resina se trató 6X con 100 ml del 1 % de TFA en DCM y, a los filtrados combinados, se adicionaron 2,54 g (100 mmol) de iodo. La solución se agitó hasta completar la reacción de oxidación (20 min). Después, el exceso de iodo se neutralizó por extracción con una solución del 3 % de Na2S2O3 y se eliminó el DCM en el RE y se precipitó con DEE. A continuación, el péptido obtenido se desprotegió utilizando procedimientos estándares, se lo purificó mediante HPLC y se procedió a su liofilización. Rendimiento: 7,14 g de sal de TFA, 78 % del contenido del péptido (44,8 %).
Ejemplo 2. Síntesis de Atosiban a partir de un Fmoc-Orn-OH unido a través de la cadena lateral de ornitina en la resina de 4-metoxi.
Se disolvió 1 mmol de Fmoc-Orn-OH en 15 ml de DCM. A continuación, se adicionaron 1,5 mmol de DIPEA y 1 g de resina de 4-metoxitritilo (1,2 mmol/g) y la mezcla se agitó durante la noche. Se adicionó 1 ml de metanol y la mezcla se agitó durante 4 horas adicionales a temperatura ambiente. Después, la resina se filtró, se lavó 3X con d Cm , 3X con DMF, 3X con iPrOH y 3X con hexano, y se secó al vacío a un peso constante para dar el Fmoc-Orn(resina de 4-metoxitritilo)-OH.
Se suspendieron 2,0 g (1,0 mmol) de Fmoc-Orn(resina de 4-metoxitritilo)-OH en 10,0 ml de DMF, se enfriaron a 5 °C y se hicieron reaccionar con 0,18 g (1,3 mmol) de HOBt y 0,13 g (1,0 mmol) de DIC. La mezcla se agitó durante 5 minutos a 5 °C y después se calentó hasta alcanzar 15 °C. A continuación, se adicionaron 0,22 g (2 mmol) de hidrocloruro de amida de glicina y 0,39 g (3,0 mmol) de DIPEA y la mezcla se agitó durante 90 min a temperatura ambiente. El procedimiento se repitió y el Fmoc-Orn(resina de 4-metoxitritilo)-Gli-NH2 obtenido se utilizó para la síntesis de Atosiban, como se describió anteriormente. Rendimiento: 0,79 g de sal de TFA, 76 % del contenido del péptido (48,6 %).
Ejemplo 3. Síntesis de Fmoc-Orn-Pro-NH2, Fmoc-Orn-Pro-NHEt y Fmoc-Orn-Pro-NH-NH-CO- NH2 con unión de resina, procedimiento general.
El Fmoc-Orn(Boc)-OH, el Fmoc-Orn(Mtt)-OH o el Fmoc-Orn(Mmt)-OH se acopló mediante procedimientos conocidos en la técnica con H-Pro-NH2 , H-Pro-NHEt o H-Pro-NH-NH-CO-NH2. Después, se desprotegió la cadena lateral del producto obtenido con TFA en DCM. El dipéptido de ornitina obtenido bien secado de 10,0 mmol después se disolvió en 100 ml de DCM/DMF (1:1). A continuación, esta solución se adicionó a 10,0 g de resina de cloruro de 2-clorotritilo o tritilo, de 4-metiltritilo o de 4-metoxitritilo (carga = 0,9-1,6 mmol/g) y, a la mezcla obtenida, se adicionaron 30,0 mmol de DIPEA. Después, la mezcla se agitó durante 12 horas a temperatura ambiente. A continuación, se adicionaron 5.0 ml de MeOH y la mezcla se agitó durante 2 horas adicionales a temperatura ambiente. Después, la resina se filtró y se lavó 6X con DMF, 3X con IPA y 3X con DEE, y se secó al vacío a un peso constante. Rendimiento de 12,5-14,5 g con una carga total de 7,9-8,7 mmol.
Ejemplo 4. Síntesis de Fmoc-Orn(resina de 4-metoxitritilo)-Tir(tBu)-NH2
El Fmoc-Orn(Mtt)-OH o el Fmoc-Orn(Mmt)-OH se acopló mediante procedimientos conocidos en la técnica con H-Tir(tBu)-NH2. Después, se desprotegió la cadena lateral del producto obtenido con un 1 % de TFA en DCM/TES (97:3) durante 2 h a temperatura ambiente. El dipéptido de ornitina obtenido bien secado de 1,0 mmol después se disolvió en 10 ml de DCM/DMF (1:1). A continuación, esta solución se adicionó a 2,0 g de resina de cloruro de 4-metoxitritilo (carga = 1,42 mmol/g) y, a la mezcla obtenida, se adicionaron 3,0 mmol de DIPEA. Después, la mezcla se agitó durante 12 horas a temperatura ambiente. A continuación, se adicionaron 0,5 ml de MeOH y la mezcla se agitó durante 2 horas adicionales a temperatura ambiente. Después, la resina se filtró y se lavó 6X con DMF, 3X con IPA y 3X con hexano, y se secó al vacío a un peso constante. Rendimiento de 3,02 g con una carga total de 0,86 mmol (0,28 mmol de dipéptido/g).
Ejemplo 5. Síntesis de Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2 (Goserelina).
1.0 g (0,64 mmol) de Fmoc-Orn(resina de 4-metoxitritilo)-Pro-NH-NH-CO-NH2 se acoplaron secuencialmente con Fmoc-Leu-OH, Fmoc-D-Ser(tBu)-OH, Fmoc-Tir(Clt)-OH, Fmoc-Ser(Trt)-OH, Fmoc-Trp-OH, Fmoc-His(Mmt)-OH y ácido piroglutámico. La resina obtenida después se lavó 6X con DMF, 3X con IPA y 3X con DEE, y se secó al vacío. A continuación, se adicionaron 20 ml de 1 % de TFA en DCM/TES (97:3) y la resina se filtró y se lavó con un 1 % de TFA en DCM/TES (97:3). Los filtrados combinados se concentraron al vacío, se precipitaron mediante la adición de DEE y se secaron al aire. Después, la resina restante se lavó 3X con DMF y 4X con DMF/agua (1:1). A los filtrados combinados se adicionó el sólido obtenido mediante el tratamiento de la resina con TFA y la solución se neutralizó mediante la adición de 1N-NaOH. A continuación, se adicionaron 145,5 mg (1,0 mmol) 1-H-1,2,4-triazol-carboxamida hidrocloruro y 258 mg (2,0 mmol) de DIPEA y la mezcla se agitó durante la noche. Después, la mezcla se acidificó hasta alcanzar un pH=2,5, se concentró y se purificó mediante RP-HPLC y se liofilizó. Rendimiento: 555,3 mg (67,6 %).
Ejemplo 6. Síntesis de Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt (Buserelina)
1.0 g (0,55 mmol) de Fmoc-Orn-Pro-NHEt se acoplaron secuencialmente con Fmoc-Leu-OH, Fmoc-D-Ser(tBu)-OH, Fmoc-Tir(Clt)-OH, Fmoc-Ser(Trt)-OH, Fmoc-Trp-OH, Fmoc-His(Mmt)-OH y ácido piroglutámico. Según el procedimiento descrito anteriormente, obtuvimos 477,0 mg de péptido (61,8 %).
Ejemplo 7. Síntesis de Pir-His-Trp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt (Leuprolida, Leuprolin)
1.0 g (0,55 mmol) de Fmoc-Orn-Pro-NHEt se acoplaron secuencialmente con Fmoc-Leu-OH, Fmoc-D-Leu-OH, Fmoc-Tir(Clt)-OH, Fmoc-Ser(Trt)-OH, Fmoc-Trp-OH, Fmoc-His(Mmt)-OH y ácido piroglutámico. Según el procedimiento descrito anteriormente, obtuvimos 488,.1 mg (61,5 %).
Ejemplo 8. Síntesis de H-Tir-Pro-Ile-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tir- Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-Gln-Arg-Tir-NH2 (péptido YY).
1.0 g (0,28 mmol) de Fmoc-Orn(resina de 4-metoxitritilo)-Tir(tBu)-NH2 se acoplaron secuencialmente con Fmoc-Gln(Trt)-OH, Fmoc-Orn(Mmt)-OH, Fmoc-Tr(tBu)-OH, Fmoc-Val-OH, Fmoc-Leu-OH, Fmoc-Asn(Trt)-OH, Fmoc-Leu-OH, Fmoc-Tir(tBu)-OH, Fmoc-His(Trt)-OH, Fmoc-Orn(Mmt)-OH, Fmoc-Leu-OH, Fmoc-Ser(tBu)-OH, Fmoc-Ala-OH, Fmoc-Tir(tBu)-OH, Fmoc-Tir(tBu)-OH, Fmoc-Orn(Mmt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Leu-OH, Fmoc-Glu(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Pro-OH, Fmoc-Ser(tBu)-OH, Fmoc-Ala-OH, Fmoc-Asp(tBu)-OH, Fmoc-Glu(tBu)-OH, Fmoc-Gli-OH, Fmoc-Pro-OH, Fmoc-Ala-OH, Fmoc-Glu(tBu)-OH, Fmoc-Pro-OH, Fmoc-Lis(Boc)-OH, Fmoc-Ile-OH, Fmoc-Pro-OH y Fmoc-Tir(tBu)-OH. Después, el péptido se escindió de la resina y se desprotegió parcialmente en la cadena lateral de ornitina y se guaniló posteriormente con 442,5 mg (3 mmol) de 1-H-1,2,4-triazol-carboxamidina hidrocloruro en DMF y DIPEA. A continuación, el producto se precipitó mediante la adición de agua, se filtró, se lavó con agua y DEE, se desprotegió, se purificó mediante RP-HPLc y se liofilizó. Rendimiento 289,9 mg (24,4 %).
Varias modificaciones y variaciones de los aspectos descritos de la invención se volverán evidentes para los expertos en la materia. Si bien la invención ha sido descrita en conexión con realizaciones preferidas específicas, debe
entenderse que la invención, como se la reivindica, no debe limitarse indebidamente a dichas realizaciones específicas. En efecto, se pretende que varias modificaciones a los modos descritos de realizar la invención, que resultan obvias para los expertos en la materia en los campos relevantes, estén dentro del alcance de las siguientes reivindicaciones.
Claims (1)
- REIVINDICACIONES1. Un procedimiento para preparar un péptido, o una sal del mismo, de un conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f),
 (2b)Resina
(2b)Resina
 (2d)Resina
(2d)Resina (26 )
(26 ) (2f)en el que la resina es una resina polimérica escindible de TFA seleccionada de entre resinas de tritilo, 2-cloro-tritlo, 4-metil-tritilo y 4-metoxi-tritilo; yPn es un grupo protector seleccionado de entre el grupo que consiste en: Fmoc, Boc, Cbz, Npys y Alloc; dicho procedimiento comprendiendo las etapas de:(a) desproteger la función a-amino del extremo N-terminal de dicho conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f) para eliminar el grupo protector Pri;(b) acoplar al menos un aminoácido o péptido protegido en el extremo N-terminal que presente una función de ácido carboxílico activado o libre con la función a-amino desprotegida de la etapa (a), alargando así el compuesto de las fórmulas (2b), (2c), (2d), (2e) o (2f),(c) repetir, opcionalmente, las etapas (a) y (b) una o más veces, en las que el al menos un aminoácido o péptido protegido en el extremo N-terminal es idéntico o diferente de aquel de la etapa anterior (b);(d) escindir el péptido resultante de la resina;(e) guanilar el péptido obtenido en la etapa (d);(f) eliminar, opcionalmente, todos los grupos protectores que queden después de la etapa (d);(g) aislar y opcionalmente purificar el péptido así obtenido.2. El procedimiento de la reivindicación 1, en el que los aminoácidos o péptidos protegidos en su extremo N-terminal de las etapas (b) y (c) están protegidos con Fmoc.3. El procedimiento de la reivindicación 2, en el que el al menos un aminoácido o péptido protegido en su extremo N-terminal de la etapa (c) repetida en último lugar está protegido por un grupo protector que es ortogonal respecto de Fmoc.4. El procedimiento de cualquiera de las reivindicaciones 1 a 3, en el que la etapa (d) comprende la escisión del péptido de la resina mediante el tratamiento con un ácido.5. El procedimiento de cualquiera de las reivindicaciones 1 a 4, en el que la etapa (e) comprende tratar el péptido con un reactivo de guanilado seleccionado de entre 1H-pirazol-1-carboxamida 2, 1-H-1,2,4-triazol-carboxamidina, triflil guanidina y tosilato de benzotriazol-1-carboxamidinio.6. El procedimiento de cualquiera de las reivindicaciones 1 a 5, en el que el conjugado de péptido-resina se selecciona de entre los siguientes:
(2f)en el que la resina es una resina polimérica escindible de TFA seleccionada de entre resinas de tritilo, 2-cloro-tritlo, 4-metil-tritilo y 4-metoxi-tritilo; yPn es un grupo protector seleccionado de entre el grupo que consiste en: Fmoc, Boc, Cbz, Npys y Alloc; dicho procedimiento comprendiendo las etapas de:(a) desproteger la función a-amino del extremo N-terminal de dicho conjugado de péptido-resina de las Fórmulas (2b), (2c), (2d), (2e) o (2f) para eliminar el grupo protector Pri;(b) acoplar al menos un aminoácido o péptido protegido en el extremo N-terminal que presente una función de ácido carboxílico activado o libre con la función a-amino desprotegida de la etapa (a), alargando así el compuesto de las fórmulas (2b), (2c), (2d), (2e) o (2f),(c) repetir, opcionalmente, las etapas (a) y (b) una o más veces, en las que el al menos un aminoácido o péptido protegido en el extremo N-terminal es idéntico o diferente de aquel de la etapa anterior (b);(d) escindir el péptido resultante de la resina;(e) guanilar el péptido obtenido en la etapa (d);(f) eliminar, opcionalmente, todos los grupos protectores que queden después de la etapa (d);(g) aislar y opcionalmente purificar el péptido así obtenido.2. El procedimiento de la reivindicación 1, en el que los aminoácidos o péptidos protegidos en su extremo N-terminal de las etapas (b) y (c) están protegidos con Fmoc.3. El procedimiento de la reivindicación 2, en el que el al menos un aminoácido o péptido protegido en su extremo N-terminal de la etapa (c) repetida en último lugar está protegido por un grupo protector que es ortogonal respecto de Fmoc.4. El procedimiento de cualquiera de las reivindicaciones 1 a 3, en el que la etapa (d) comprende la escisión del péptido de la resina mediante el tratamiento con un ácido.5. El procedimiento de cualquiera de las reivindicaciones 1 a 4, en el que la etapa (e) comprende tratar el péptido con un reactivo de guanilado seleccionado de entre 1H-pirazol-1-carboxamida 2, 1-H-1,2,4-triazol-carboxamidina, triflil guanidina y tosilato de benzotriazol-1-carboxamidinio.6. El procedimiento de cualquiera de las reivindicaciones 1 a 5, en el que el conjugado de péptido-resina se selecciona de entre los siguientes: en el que la resina es una resina de tritilo o una resina de 4-metoxitritilo.7. El procedimiento de la reivindicación 1 para preparar un péptido seleccionado de entre los siguientes:[2] Arg-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Arg-Gli-NH2 ;[5 ] 3-Mercaptopropionil-Tir-Fe-Gln-Asn-Cis-Pro-D-Arg-Gli-NH2;[8] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2 ;[9 ] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt sal de acetato;[10] Pir-His-Trp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt;[1 1 ] Pir-His-Trp-Ser-Tir-D-His(Bzl)-Leu-Arg-Pro-NHEt sal de acetato;[1 2 ] Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-NHEt sal de acetato;[20]H-Tir-Pro-Ser-Lis-Pro-Asp-Asn-Pro-Gli-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met-Ala-Arg- Tir-Tir-Ser-Ala-Leu-Arg-His-Tir-lle-Asn-Leu-lle-T r-Arg-GIn-Arg-Tir-NH2; y[21]H-Tir-Pro-lle-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tir-Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-Gln-Arg-Tir-NH2.7
en el que la resina es una resina de tritilo o una resina de 4-metoxitritilo.7. El procedimiento de la reivindicación 1 para preparar un péptido seleccionado de entre los siguientes:[2] Arg-Cis-Tir-Fe-Gln-Asn-Cis-Pro-Arg-Gli-NH2 ;[5 ] 3-Mercaptopropionil-Tir-Fe-Gln-Asn-Cis-Pro-D-Arg-Gli-NH2;[8] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-Azagli-NH2 ;[9 ] Pir-His-Trp-Ser-Tir-D-Ser(tBu)-Leu-Arg-Pro-NHEt sal de acetato;[10] Pir-His-Trp-Ser-Tir-D-Leu-Leu-Arg-Pro-NHEt;[1 1 ] Pir-His-Trp-Ser-Tir-D-His(Bzl)-Leu-Arg-Pro-NHEt sal de acetato;[1 2 ] Pir-His-Trp-Ser-Tir-D-Trp-Leu-Arg-Pro-NHEt sal de acetato;[20]H-Tir-Pro-Ser-Lis-Pro-Asp-Asn-Pro-Gli-Glu-Asp-Ala-Pro-Ala-Glu-Asp-Met-Ala-Arg- Tir-Tir-Ser-Ala-Leu-Arg-His-Tir-lle-Asn-Leu-lle-T r-Arg-GIn-Arg-Tir-NH2; y[21]H-Tir-Pro-lle-Lis-Pro-Glu-Ala-Pro-Gli-Glu-Asp-Ala-Ser-Pro-Glu-Glu-Leu-Asn-Arg-Tir-Tir-Ala-Ser-Leu-Arg-His-Tir-Leu-Asn-Leu-Val-Tr-Arg-Gln-Arg-Tir-NH2.7
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1310921.0A GB201310921D0 (en) | 2013-06-19 | 2013-06-19 | Peptide-resin conjugate and use thereof |
| PCT/IB2014/062430 WO2014203193A1 (en) | 2013-06-19 | 2014-06-19 | Peptide-resin conjugate and use thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2753407T3 true ES2753407T3 (es) | 2020-04-08 |
Family
ID=48914802
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14742356T Active ES2753407T3 (es) | 2013-06-19 | 2014-06-19 | Conjugado de péptido-resina y su utilización |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20160137689A1 (es) |
| EP (1) | EP3010930B1 (es) |
| CN (1) | CN105408344B (es) |
| AU (1) | AU2014282839B9 (es) |
| BR (1) | BR112015031679B1 (es) |
| CA (1) | CA2915439C (es) |
| ES (1) | ES2753407T3 (es) |
| GB (1) | GB201310921D0 (es) |
| HK (1) | HK1217022A1 (es) |
| WO (1) | WO2014203193A1 (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3088418A1 (en) * | 2015-04-28 | 2016-11-02 | Vallaurix Pte. Ltd. | Pharmaceutical compound |
| WO2019234108A1 (en) * | 2018-06-05 | 2019-12-12 | Dsm Ip Assets B.V. | Methods for the synthesis of arginine-containing peptides |
| CN110041407B (zh) * | 2019-02-26 | 2022-10-28 | 南京肽业生物科技有限公司 | 一种基于Fmoc二肽的合成醋酸德舍瑞林的方法 |
| JP2020169141A (ja) * | 2019-04-04 | 2020-10-15 | 株式会社日立製作所 | 樹脂との密着性に優れたペプチドならびにそれを用いた生体適合機能材料 |
| CN115038711B (zh) * | 2021-01-04 | 2023-05-02 | 湖北健翔生物制药有限公司 | 一种阿托西班的合成方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7691968B2 (en) * | 2002-05-03 | 2010-04-06 | Avecia Biologics Limited | Process for the synthesis of peptides amides by side-chain attachment to a solid phase |
| EP1737889B1 (en) | 2004-10-19 | 2010-09-08 | Lonza AG | Method for solid phase peptide synthesis |
| CN101379075B (zh) | 2006-02-08 | 2013-05-15 | 隆萨股份公司 | 类胰高血糖素肽的合成 |
| CN101522704A (zh) * | 2006-10-05 | 2009-09-02 | 隆萨股份公司 | 肽合成的方法 |
| KR101046846B1 (ko) * | 2006-10-12 | 2011-07-06 | 동국제약 주식회사 | 고체상 합성법을 이용한 펩타이드의 제조방법 |
| CN102127146B (zh) * | 2010-12-24 | 2013-04-24 | 深圳翰宇药业股份有限公司 | 一种制备醋酸阿托西班的方法 |
| WO2013098802A2 (en) * | 2011-12-29 | 2013-07-04 | Chemical & Biopharmaceutical Laboratories Of Patras S.A. | Solid phase peptide synthesis via side chain attachment |
| CN102653555B (zh) * | 2012-05-18 | 2015-04-22 | 深圳翰宇药业股份有限公司 | 一种固相制备戈舍瑞林的方法 |
-
2013
- 2013-06-19 GB GBGB1310921.0A patent/GB201310921D0/en not_active Ceased
-
2014
- 2014-06-19 US US14/899,270 patent/US20160137689A1/en not_active Abandoned
- 2014-06-19 EP EP14742356.0A patent/EP3010930B1/en active Active
- 2014-06-19 BR BR112015031679-4A patent/BR112015031679B1/pt active IP Right Grant
- 2014-06-19 WO PCT/IB2014/062430 patent/WO2014203193A1/en active Application Filing
- 2014-06-19 AU AU2014282839A patent/AU2014282839B9/en active Active
- 2014-06-19 CN CN201480042295.7A patent/CN105408344B/zh active Active
- 2014-06-19 CA CA2915439A patent/CA2915439C/en active Active
- 2014-06-19 ES ES14742356T patent/ES2753407T3/es active Active
-
2016
- 2016-05-03 HK HK16105020.5A patent/HK1217022A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| US20160137689A1 (en) | 2016-05-19 |
| AU2014282839B2 (en) | 2019-12-12 |
| BR112015031679B1 (pt) | 2024-03-05 |
| AU2014282839B9 (en) | 2019-12-19 |
| CA2915439C (en) | 2021-11-09 |
| CN105408344A (zh) | 2016-03-16 |
| GB201310921D0 (en) | 2013-07-31 |
| BR112015031679A8 (pt) | 2021-06-29 |
| AU2014282839A1 (en) | 2016-01-21 |
| HK1217022A1 (zh) | 2016-12-16 |
| EP3010930A1 (en) | 2016-04-27 |
| CA2915439A1 (en) | 2014-12-24 |
| BR112015031679A2 (pt) | 2017-07-25 |
| EP3010930B1 (en) | 2019-09-18 |
| CN105408344B (zh) | 2020-08-11 |
| WO2014203193A1 (en) | 2014-12-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2957399T3 (es) | Método de síntesis de péptidos | |
| ES2239364T3 (es) | Sintesis de peptidos en fase solida. | |
| ES2786225T3 (es) | Método para preparar AMG 416 | |
| ES2753407T3 (es) | Conjugado de péptido-resina y su utilización | |
| US8828938B2 (en) | Method for the manufacture of degarelix | |
| ES2352204T3 (es) | Método de síntesis peptídica en fase sólida. | |
| CN111479819B (zh) | 制备肽的方法和处理碱的方法 | |
| ES2308539T3 (es) | Ciclacion de peptidos. | |
| TW201915009A (zh) | 合成依特卡肽(Etelcalcetide)或其鹽類之方法 | |
| ES2885869T3 (es) | Procedimiento para la fabricación de degarelix y sus productos intermedios | |
| BRPI0718996B1 (pt) | método para a preparação de um peptídio da fórmula h-d-?-nal-[cys-tyr-d-trp-lys-val-cys]-thr-nh2 ou h-d-?-nal-cys(acm)-tyr-d-trp-lys-val-cys(acm)-thr | |
| TW201024316A (en) | Process for the preparation of Pramlintide | |
| WO2005087794A1 (en) | Process for octreotide synthesis | |
| KR101046846B1 (ko) | 고체상 합성법을 이용한 펩타이드의 제조방법 | |
| Ruczyński et al. | Problem of aspartimide formation in Fmoc‐based solid‐phase peptide synthesis using Dmab group to protect side chain of aspartic acid | |
| US9150615B2 (en) | Process for the preparation of leuprolide and its pharmaceutically acceptable salts | |
| JP5445456B2 (ja) | ジベンゾフルベンの除去方法 | |
| JP4615221B2 (ja) | ヘプタペプチドオキシトシンアナログを製造するための中間体及び方法 | |
| FI102380B (fi) | Menetelmä peptidien valmistamiseksi kiinteäfaasisynteesin avulla | |
| CN116507763A (zh) | 抑制由二酮哌嗪形成导致的缺损的肽合成方法 | |
| WO2006105199A2 (en) | Compositions and methods for synthesis of peptide and related conjugate | |
| CN108148113A (zh) | 一种nmda受体调控剂四肽衍生物的固相合成方法 | |
| CA3238634A1 (en) | Synthetic process for production of modified gcc receptor agonists | |
| AU6183099A (en) | Synthesis of cyclic peptides |