ES2394870T5 - Procedimiento para la fabricación de olefinas fluoradas - Google Patents
Procedimiento para la fabricación de olefinas fluoradas Download PDFInfo
- Publication number
- ES2394870T5 ES2394870T5 ES07863530T ES07863530T ES2394870T5 ES 2394870 T5 ES2394870 T5 ES 2394870T5 ES 07863530 T ES07863530 T ES 07863530T ES 07863530 T ES07863530 T ES 07863530T ES 2394870 T5 ES2394870 T5 ES 2394870T5
- Authority
- ES
- Spain
- Prior art keywords
- reaction
- catalyst
- conversion
- stream
- fluorinated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 41
- 150000001336 alkenes Chemical class 0.000 title claims description 33
- 238000004519 manufacturing process Methods 0.000 title claims description 8
- 238000006243 chemical reaction Methods 0.000 claims description 187
- 239000003054 catalyst Substances 0.000 claims description 135
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 45
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 40
- 238000005796 dehydrofluorination reaction Methods 0.000 claims description 31
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 claims description 25
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 21
- 238000006704 dehydrohalogenation reaction Methods 0.000 claims description 18
- 229910052799 carbon Inorganic materials 0.000 claims description 17
- 125000001153 fluoro group Chemical group F* 0.000 claims description 15
- 229910052739 hydrogen Inorganic materials 0.000 claims description 14
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 13
- 239000000376 reactant Substances 0.000 claims description 12
- VJGCZWVJDRIHNC-UHFFFAOYSA-N 1-fluoroprop-1-ene Chemical compound CC=CF VJGCZWVJDRIHNC-UHFFFAOYSA-N 0.000 claims description 9
- 150000005828 hydrofluoroalkanes Chemical class 0.000 claims description 9
- 238000000605 extraction Methods 0.000 claims description 7
- 229910001512 metal fluoride Inorganic materials 0.000 claims description 7
- MSSNHSVIGIHOJA-UHFFFAOYSA-N pentafluoropropane Chemical compound FC(F)CC(F)(F)F MSSNHSVIGIHOJA-UHFFFAOYSA-N 0.000 claims description 6
- 229910044991 metal oxide Inorganic materials 0.000 claims description 4
- 150000004706 metal oxides Chemical class 0.000 claims description 4
- 229910052723 transition metal Inorganic materials 0.000 claims description 3
- 150000003624 transition metals Chemical class 0.000 claims description 3
- 238000004064 recycling Methods 0.000 claims description 2
- 150000001875 compounds Chemical class 0.000 description 24
- FYIRUPZTYPILDH-UHFFFAOYSA-N 1,1,1,2,3,3-hexafluoropropane Chemical compound FC(F)C(F)C(F)(F)F FYIRUPZTYPILDH-UHFFFAOYSA-N 0.000 description 19
- 239000000047 product Substances 0.000 description 17
- 150000001335 aliphatic alkanes Chemical class 0.000 description 16
- 238000006722 reduction reaction Methods 0.000 description 16
- ZDCWZRQSHBQRGN-UHFFFAOYSA-N 1,1,1,2,3-pentafluoropropane Chemical compound FCC(F)C(F)(F)F ZDCWZRQSHBQRGN-UHFFFAOYSA-N 0.000 description 15
- 229910052731 fluorine Inorganic materials 0.000 description 13
- FXRLMCRCYDHQFW-UHFFFAOYSA-N 2,3,3,3-tetrafluoropropene Chemical compound FC(=C)C(F)(F)F FXRLMCRCYDHQFW-UHFFFAOYSA-N 0.000 description 12
- 238000006467 substitution reaction Methods 0.000 description 11
- 238000005984 hydrogenation reaction Methods 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 239000011737 fluorine Substances 0.000 description 9
- HCDGVLDPFQMKDK-UHFFFAOYSA-N hexafluoropropylene Chemical compound FC(F)=C(F)C(F)(F)F HCDGVLDPFQMKDK-UHFFFAOYSA-N 0.000 description 9
- 239000007789 gas Substances 0.000 description 8
- 229910052751 metal Inorganic materials 0.000 description 8
- 239000002184 metal Substances 0.000 description 8
- 239000000203 mixture Substances 0.000 description 8
- 125000005843 halogen group Chemical group 0.000 description 7
- PXHVJJICTQNCMI-UHFFFAOYSA-N nickel Substances [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 7
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 6
- 230000003197 catalytic effect Effects 0.000 description 6
- 239000007795 chemical reaction product Substances 0.000 description 6
- 230000001276 controlling effect Effects 0.000 description 6
- 238000001816 cooling Methods 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 238000007033 dehydrochlorination reaction Methods 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 238000012856 packing Methods 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- DMUPYMORYHFFCT-UHFFFAOYSA-N 1,2,3,3,3-pentafluoroprop-1-ene Chemical compound FC=C(F)C(F)(F)F DMUPYMORYHFFCT-UHFFFAOYSA-N 0.000 description 4
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- 239000003085 diluting agent Substances 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 229910052763 palladium Inorganic materials 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- CDOOAUSHHFGWSA-OWOJBTEDSA-N (e)-1,3,3,3-tetrafluoroprop-1-ene Chemical compound F\C=C\C(F)(F)F CDOOAUSHHFGWSA-OWOJBTEDSA-N 0.000 description 3
- 229910000792 Monel Inorganic materials 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- -1 unsaturated alkyl radical Chemical class 0.000 description 3
- 239000012808 vapor phase Substances 0.000 description 3
- NSGXIBWMJZWTPY-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropane Chemical class FC(F)(F)CC(F)(F)F NSGXIBWMJZWTPY-UHFFFAOYSA-N 0.000 description 2
- PFFGXVGPSGJOBV-UHFFFAOYSA-N 1,1,1,3-tetrafluoropropane Chemical compound FCCC(F)(F)F PFFGXVGPSGJOBV-UHFFFAOYSA-N 0.000 description 2
- NDMMKOCNFSTXRU-UHFFFAOYSA-N 1,1,2,3,3-pentafluoroprop-1-ene Chemical class FC(F)C(F)=C(F)F NDMMKOCNFSTXRU-UHFFFAOYSA-N 0.000 description 2
- CDOOAUSHHFGWSA-UHFFFAOYSA-N 1,3,3,3-tetrafluoropropene Chemical compound FC=CC(F)(F)F CDOOAUSHHFGWSA-UHFFFAOYSA-N 0.000 description 2
- RRUCZSCEAWFDBM-UHFFFAOYSA-N 1-bromo-2,3,3,3-tetrafluoroprop-1-ene Chemical compound BrC=C(F)C(F)(F)F RRUCZSCEAWFDBM-UHFFFAOYSA-N 0.000 description 2
- IRPGOXJVTQTAAN-UHFFFAOYSA-N 2,2,3,3,3-pentafluoropropanal Chemical compound FC(F)(F)C(F)(F)C=O IRPGOXJVTQTAAN-UHFFFAOYSA-N 0.000 description 2
- FDMFUZHCIRHGRG-UHFFFAOYSA-N 3,3,3-trifluoroprop-1-ene Chemical compound FC(F)(F)C=C FDMFUZHCIRHGRG-UHFFFAOYSA-N 0.000 description 2
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 2
- KLZUFWVZNOTSEM-UHFFFAOYSA-K Aluminum fluoride Inorganic materials F[Al](F)F KLZUFWVZNOTSEM-UHFFFAOYSA-K 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000011651 chromium Substances 0.000 description 2
- QCMJBECJXQJLIL-UHFFFAOYSA-L chromium(6+);oxygen(2-);difluoride Chemical compound [O-2].[O-2].[F-].[F-].[Cr+6] QCMJBECJXQJLIL-UHFFFAOYSA-L 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- QDOXWKRWXJOMAK-UHFFFAOYSA-N dichromium trioxide Chemical class O=[Cr]O[Cr]=O QDOXWKRWXJOMAK-UHFFFAOYSA-N 0.000 description 2
- QUPDWYMUPZLYJZ-UHFFFAOYSA-N ethyl Chemical compound C[CH2] QUPDWYMUPZLYJZ-UHFFFAOYSA-N 0.000 description 2
- 238000007701 flash-distillation Methods 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 238000003682 fluorination reaction Methods 0.000 description 2
- 229910000856 hastalloy Inorganic materials 0.000 description 2
- 239000011261 inert gas Substances 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 150000002739 metals Chemical class 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 238000003541 multi-stage reaction Methods 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- SMCNZLDHTZESTK-UHFFFAOYSA-N 2-chloro-1,1,1,2-tetrafluoropropane Chemical compound CC(F)(Cl)C(F)(F)F SMCNZLDHTZESTK-UHFFFAOYSA-N 0.000 description 1
- VRVIDSRWPUGFBU-UHFFFAOYSA-N 2-chloro-1,1,1-trifluoropropane Chemical compound CC(Cl)C(F)(F)F VRVIDSRWPUGFBU-UHFFFAOYSA-N 0.000 description 1
- ZGOMEYREADWKLC-UHFFFAOYSA-N 3-chloro-1,1,1,3-tetrafluoropropane Chemical compound FC(Cl)CC(F)(F)F ZGOMEYREADWKLC-UHFFFAOYSA-N 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- WGLPBDUCMAPZCE-UHFFFAOYSA-N Trioxochromium Chemical compound O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000009903 catalytic hydrogenation reaction Methods 0.000 description 1
- 239000003518 caustics Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 229910000423 chromium oxide Inorganic materials 0.000 description 1
- UOUJSJZBMCDAEU-UHFFFAOYSA-N chromium(3+);oxygen(2-) Chemical class [O-2].[O-2].[O-2].[Cr+3].[Cr+3] UOUJSJZBMCDAEU-UHFFFAOYSA-N 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 208000012839 conversion disease Diseases 0.000 description 1
- 239000002826 coolant Substances 0.000 description 1
- 238000005260 corrosion Methods 0.000 description 1
- 230000007797 corrosion Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 239000003546 flue gas Substances 0.000 description 1
- 239000004446 fluoropolymer coating Substances 0.000 description 1
- 238000004817 gas chromatography Methods 0.000 description 1
- 238000010574 gas phase reaction Methods 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 229910001026 inconel Inorganic materials 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- SHXXPRJOPFJRHA-UHFFFAOYSA-K iron(iii) fluoride Chemical compound F[Fe](F)F SHXXPRJOPFJRHA-UHFFFAOYSA-K 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- ORUIBWPALBXDOA-UHFFFAOYSA-L magnesium fluoride Chemical compound [F-].[F-].[Mg+2] ORUIBWPALBXDOA-UHFFFAOYSA-L 0.000 description 1
- 229910001635 magnesium fluoride Inorganic materials 0.000 description 1
- YFKIWUQBRSMPMZ-UHFFFAOYSA-N methane;nickel Chemical compound C.[Ni] YFKIWUQBRSMPMZ-UHFFFAOYSA-N 0.000 description 1
- DOTMOQHOJINYBL-UHFFFAOYSA-N molecular nitrogen;molecular oxygen Chemical compound N#N.O=O DOTMOQHOJINYBL-UHFFFAOYSA-N 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 229910001120 nichrome Inorganic materials 0.000 description 1
- 150000002815 nickel Chemical class 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 239000010970 precious metal Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 125000004805 propylene group Chemical class [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 239000010948 rhodium Substances 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 1
- 229910052707 ruthenium Inorganic materials 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 238000007039 two-step reaction Methods 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/25—Preparation of halogenated hydrocarbons by splitting-off hydrogen halides from halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/35—Preparation of halogenated hydrocarbons by reactions not affecting the number of carbon or of halogen atoms in the reaction
- C07C17/354—Preparation of halogenated hydrocarbons by reactions not affecting the number of carbon or of halogen atoms in the reaction by hydrogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/38—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C21/00—Acyclic unsaturated compounds containing halogen atoms
- C07C21/02—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds
- C07C21/18—Acyclic unsaturated compounds containing halogen atoms containing carbon-to-carbon double bonds containing fluorine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2527/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- C07C2527/02—Sulfur, selenium or tellurium; Compounds thereof
- C07C2527/053—Sulfates or other compounds comprising the anion (SnO3n+1)2-
- C07C2527/054—Sulfuric acid or other acids with the formula H2Sn03n+1
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
DESCRIPCIÓN
Procedimiento para la fabricación de olefinas fluoradas
Antecedentes de la invención
Las olefinas fluoradas, como una clase, presentan muchos y variados usos, incluyendo como compuestos intermedios químicos y monómeros.
Se conocen diversos métodos para preparar olefinas fluoradas. Por ejemplo, la Patente de EE.UU. N° 5.679.875 desvela métodos para fabricar 1,1,1,2,3-pentafluoropropeno y 1,1,1,2,3-pentafluoropropano; la Patente de EE.UU. N° 6.031.141 desvela un procedimiento catalítico usando catalizadores que contienen cromo para la deshidrofluoración de hidrofluorocarbonos a fluoroolefinas; la Patente de EE.UU. N° 5.396.000 desvela un procedimiento para producir CF3CHFCH2F usando deshidrohalogenación catalítica en fase de vapor para producir CF3CF=CHF y HF, seguido por hidrogenación catalítica en fase de vapor de CF3CF=CHF en presencia de HF; la Patente de EE.UU. N° 6.548.719 desvela un procedimiento para producir fluoroolefinas por deshidrohalogenación de un hidrofluorocarbono en presencia de un catalizador de transferencia de fases; la Publicación de Patente de EE.UU. N° 2006/0106263 desvela la producción y purificación de compuestos hidrofluoroolefínicos y la Patente Internacional WO 98/33755 desvela un procedimiento catalítico para la deshidrofluoración de hexafluoropropanos a pentafluoropropenos.
La Patente de EE.UU. N° 5.986.151 desvela un procedimiento para preparar 1,1,1-trifluoropropeno por deshidrofluoración de 1,1,1,3,3-pentafluoropropano en presencia de un catalizador de paladio sobre carbono para formar 1,1,1,3-tetrafluoropropeno, reducción de 1,1,1,3-tetrafluoropropeno para formar 1,1,1,3-tetrafluoropropano y después deshidrofluoración, 1,1,3-tetrafluoropropano para producir 1,1,1-trifluoropropeno. La Patente de EE.UU. N° 6.194.619 se refiere a un procedimiento para producir hidrocarburos sustituidos con flúor de las fórmulas RCH2CF2R y RFC=CHR. Se preparan compuestos de fórmula RFC=CHR por hidrogenación de una correspondiente perfluoroolefina y después deshidrogenación del dihidrocompuesto resultante. Knunyants I L et al (Bulletin of the Academy of Sciences of the USSR, 1.960, 1.312-1.317) describen la hidrogenación catalítica y la posterior deshidrofluoración de diversas perfluoroolefinas. La Patente de EE.UU. N° 5.672.787 desvela un procedimiento para la hidrogenación de 1,1,1,4,4-hexafluorobuteno para proporcionar 1,1,1,4,4-hexafluorobutano. Los solicitantes han descubierto que los procedimientos del tipo descrito anteriormente presentan desventajas y/o no son tan eficaces y/o económicos como sería necesario en la práctica para producción comercial a gran escala. Por ejemplo, los solicitantes han apreciado que no es posible en general, siguiendo las explicaciones de las publicaciones anteriores sólo, conseguir un procedimiento que tenga a la vez un alto grado de conversión última y un alto grado de selectividad para la olefina fluorada deseada.
Sumario de la invención
Los solicitantes han apreciado desde el punto de vista de la invención, que ninguna de las publicaciones indicadas anteriormente desvela un procedimiento integrado que comprenda las etapas de hidrogenar de manera selectiva una o más olefinas altamente fluoradas para producir uno o más hidrofluorocarbonos, seguido por la etapa de deshidrofluoración del hidrofluorocarbono o de los hidrofluorocarbonos resultantes para producir una o más olefinas fluoradas deseadas, en particular propeno y HF, tampoco desvelan dicho procedimiento combinado con una etapa de recuperación de HF muy eficaz.
La presente invención proporciona un procedimiento para la producción de un producto olefínico fluorado que comprende las etapas de:
(a) poner en contacto una corriente de alimentación que comprende un fluoropropeno que tiene de tres a seis sustituyentes flúor y agentes reaccionantes de hidrógeno con una primera cantidad de catalizador para convertir dichos agentes reaccionantes en hidrofluoroalcano a una primera velocidad de conversión y para producir una primera corriente de salida que comprende dicho hidrofluoroalcano, fluoropropeno no reaccionado e hidrógeno; (b) poner en contacto dicha primera corriente de salida con una segunda cantidad de un catalizador para convertir dicho fluoropropeno no reaccionado en un hidrofluoroalcano a una segunda velocidad de conversión, en la que dicha segunda cantidad de catalizador es mayor que dicha primera cantidad de catalizador y en la que dicha segunda velocidad de conversión es mayor que dicha primera velocidad de conversión;
(c) deshidrohalogenar al menos una porción de dicho hidrofluoroalcano de dicha etapa de contacto (b) para producir una corriente de producto que comprende una olefina fluorada y producto HF y
(d) opcionalmente, separar dicho HF de dicha corriente de producto.
La presente divulgación se refiere a procedimientos para la síntesis de alquenos fluorados y olefinas fluoradas preferibles que tienen de tres a seis átomos de carbono y un grado de sustitución de flúor de N. En algunas realizaciones muy preferidas, las olefinas fluoradas deseadas comprenden uno o más fluoroalquenos C2 a C6, preferiblemente compuestos con una fórmula como sigue:
X1CFzR3-z
donde X1 es un radical alquilo, sustituido o no sustituido, insaturado C1, C2, C3, C4 o C5, cada R es independientemente CI, F, Br, I o H y z es 1 a 3. Son muy preferidos entre dichos compuestos los propenos y butenos que tienen de 3 a 5 sustituyentes de flúor y entre éstos se prefieren especialmente los tetrafluoropropenos (HFO-1234).
Los procedimientos preferidos comprenden hacer reaccionar un material de partida olefínico fluorado con un grado de sustitución con halógeno de N+1 teniendo sustancialmente el mismo número de átomos de carbono que la olefina u olefinas fluoradas que se tienen que sintetizar con un grado de sustitución con halógeno de N. Preferiblemente, el material de partida olefínico fluorado con un grado de sustitución de flúor de N 1 se desvela para condiciones de reacción eficaces para producir un producto de reacción que contiene uno o más alcanos fluorados con el mismo número de átomos de carbono que la olefina. En un aspecto preferido, esta etapa de conversión de la olefina comprende una reacción que se refiere a veces en la presente memoria por conveniencia, pero no necesariamente como limitación, como una etapa de reducción o hidrogenación. El alcano fluorado se convierte después preferiblemente en una olefina fluorada con un grado de sustitución de flúor de N. En un aspecto preferido, esta etapa de conversión de alcano comprende una reacción que se refiere a veces en la presente memoria por conveniencia, pero no necesariamente como limitación, como una reacción de deshidrohalogenación o más en particular, en ciertas realizaciones como reacción de deshidrofluoración o deshidrocloración.
Los presentes procedimientos comprenden preferiblemente las etapas de:
(a) hidrogenar en una reacción multietapa, un compuesto de fórmula (I)
(CXnY3-n)(CR1a R2b)zCX=CHmX2-m (I)
en condiciones eficaces para formar al menos un alcano fluorado de fórmula (II)
(CXnY3-n) (CR1a R2b)zCHXCHm+1X2-m (II)
donde:
cada X es independientemente CI, F, I o Br;
cada Y es independientemente H, CI, F, I o Br;
cada R1 es independientemente H, CI, F, l, Br o radical metilo o etilo no sustituido o sustituido con halógeno; cada R2 es independientemente H, CI, F, I, Br o radical metilo o etilo no sustituido o sustituido con halógeno;
n es 1, 2 ó 3;
a y b son cada uno 1 ó 2, siempre que a+b = 2;
m es 0, 1 ó 2 y
Z es 0, 1, 2 ó 3 y
(b) deshidrohalogenar el compuesto de fórmula (II) en condiciones eficaces para producir una fluoroolefina con un grado menor de sustitución con flúor que el compuesto de fórmula (I), preferiblemente para producir un compuesto de fórmula (III):
(CXnY3-n)(CR1a R2b)zCX=CHmX2-m (III)
donde cada n tiene el mismo valor que en la fórmula (I) y m es 0 ó 1.
En algunas realizaciones preferidas, el agente reaccionante de fórmula (I) comprende una olefina de tres carbonos de fórmula (I A) en la que z es 0, es decir
CXnY3-nCX=CHmX2-m (IA)
para producir un alcano de tres carbonos de fórmula (IIA) como sigue:
(CXnY3-n)CHXCHm+ l X2-m (IIA)
donde X, Y, n y m son todos como se indicó anteriormente, compuesto que se deshidrohalogena después para formar un compuesto de fórmula (IIIA)
(CXnY3-n)CHXCHm+1X2-m (IIIA)
donde n tiene el mismo valor que en la fórmula (IA) y m es 0 ó 1.
En algunos aspectos muy preferidos de tales realizaciones, un carbono terminal saturado de los compuestos de las fórmulas (I) o (IA) está completamente sustituido con flúor (por ejemplo, n en el carbono terminal saturado es 3 y cada X en ese carbono es F) e incluso más preferiblemente n es 3 y cada X en el compuesto es F.
En la presente invención, el compuesto de Fórmula (IA) es un fluoropropeno que tiene de tres a seis sustituyentes flúor y potencialmente otros sustituyentes halógeno, incluyendo por ejemplo hexafluoropropeno (esto es, Z es 0, n es 3, m es 0 y todos los X son F) o pentafluoropropeno (esto es, Z es 0, n=3, m es 1 y todos los X son F) y el compuesto de fórmula (IIA) preferiblemente comprende y más preferiblemente se selecciona del grupo que consiste en, uno o más de los siguientes alcanos fluorados: clorotrifluoropropano (HCFC-244) y pentafluoropropano (HFC-245) y hexafluoropropano (HFC-236), incluyendo todos los isómeros de cada uno de éstos, pero preferiblemente 1-cloro,1,3,3,3-tetrafluoropropano (HCFC-244fa), 1,1,1,3,3-pentafluoropropano (HFC-245fa), 1,1,1,2,3-pentafluoropropano (HFC-245eb) y 1,1,1,2,3,3-hexafluoropropano (HFC-236ea). En algunas realizaciones preferidas el alcano fluorado producido por la etapa de conversión presenta un grado de sustitución con flúor de N+1.
En realizaciones preferidas, la etapa (a) de conversión en la que la olefina se convierte en un alcano se realiza en condiciones eficaces para proporcionar una conversión de la fórmula (I) de al menos aproximadamente 40%, más preferiblemente al menos aproximadamente 55% e incluso más preferiblemente al menos aproximadamente 70%. En algunas realizaciones preferidas la conversión es al menos aproximadamente 90% y más preferiblemente aproximadamente 99%. Además, en algunas realizaciones preferidas, la conversión del compuesto de fórmula (I) para producir un compuesto de fórmula (II) se realiza en condiciones eficaces para proporcionar una selectividad de fórmula (II) de al menos aproximadamente 60%, más preferiblemente al menos aproximadamente 80% y más preferiblemente al menos aproximadamente 90% e incluso más preferiblemente aproximadamente 100%.
En realizaciones preferidas, la etapa (b) de conversión en la que el alcano se convierte en una olefina fluorada con un grado de fluoración de N se realiza en condiciones eficaces para proporcionar una conversión de la fórmula (II) de al menos aproximadamente 40%, más preferiblemente al menos aproximadamente 55% e incluso más preferiblemente al menos aproximadamente 70%. En algunas realizaciones preferidas la conversión es al menos aproximadamente 90% y más preferiblemente aproximadamente 95%. Además, en algunas realizaciones preferidas, la conversión del compuesto de fórmula (II) para producir un compuesto de fórmula (III) se realiza en condiciones eficaces para proporcionar una selectividad de la fórmula (III) de al menos aproximadamente 60%, más preferiblemente al menos aproximadamente 80% y más preferiblemente al menos aproximadamente 90% e incluso más preferiblemente aproximadamente 98%.
Breve descripción del dibujo
La FIG. 1 es un diagrama de flujo del proceso esquemático según una realización de la presente invención.
La FIG. 2 es un diagrama de flujo del proceso semiesquemático según una realización de la etapa de hidrogenación de la presente invención.
Descripción detallada
Ahora se describirá una realización de la presente invención en relación con la Figura 1 en que una corriente 1 de alimentación que comprende al menos una olefina fluorada con un grado de sustitución de halógeno, y preferiblemente un grado de sustitución con flúor, de N+1 se somete a una primera etapa 50 de conversión. Esta olefina fluorada es un fluoropropeno que tiene de tres a seis sustituyentes flúor. La etapa 50 de conversión implica preferiblemente también una corriente 2 de alimentación que comprende un agente reductor. La etapa 50 de conversión incluye preferiblemente proporcionar uno o más recipientes de reacción, al menos uno de los cuales contiene preferiblemente un catalizador de reducción o hidrogenación e introducir las corrientes/ y 2 en el recipiente o los recipientes en condiciones eficaces para conseguir la conversión deseada.
Aunque las corrientes 1 y 2 en la figura se muestran por conveniencia como corrientes separadas, esto se hace por conveniencia y la presente invención no está así limitada. Por ejemplo, se podían combinar las corrientes en ciertas
realizaciones fuera del recipiente e introducir después en el recipiente juntas o en otras realizaciones la corriente 1 y la corriente 2 podían comprender cada una diversas corrientes separadas, cada una de las cuales se introduce en el recipiente o los recipientes en momentos diferentes y/o en posiciones diferentes. Este mismo convenio se ha usado y se ejerce en la presente memoria durante todo el uso de la terminología “corriente”, tanto en la descripción como en las figuras, a menos que se indique específicamente de otro modo.
La etapa 50 de conversión preferida produce al menos una corriente 3 de producto de reacción que contiene un alcano fluorado según la presente invención. La corriente 3 se usa como agente reaccionante en la etapa 51 de conversión, opcionalmente se purifica además la corriente 3 antes de que se alimente a la etapa 51, en la que el alcano fluorado en la corriente 3 se convierte en una olefina fluorada con un grado de sustitución con halógeno y en algunas realizaciones preferidas sustitución con flúor, de N según la presente invención. La etapa 51 de conversión incluye preferiblemente proporcionar uno o más recipientes de reacción, al menos uno de los cuales contiene preferiblemente un catalizador de deshidrohalogenación e introducir al menos la corriente 3 en el recipiente o los recipientes en condiciones eficaces para producir la fluoroolefina deseada.
En realizaciones preferidas, la etapa 51 de conversión produce un producto de reacción que incluye no sólo la fluoroolefina deseada, sino también HF. En tales realizaciones se prefiere en general introducir la corriente 4 en una etapa 52 de separación en que se separa al menos una porción del HF de la corriente para producir al menos una primera corriente 6 relativamente rica (en comparación con la corriente 4 de alimentación) en la olefina fluorada y al menos una segunda corriente relativamente rica (en comparación con la corriente 4 de alimentación) en HF.
Aspectos preferidos de cada una de las etapas 50, 51 y 52 se describen a continuación.
La etapa de reducción
Aunque se considera que la etapa de reducción se puede realizar en operación discontinua, se prefiere que la reacción de reducción se realice como una operación sustancialmente continua. Además, aunque es posible que la reacción de reducción pueda implicar en ciertas realizaciones una reacción de fase líquida, se considera que en realizaciones preferidas la reacción de reducción comprende, e incluso más preferiblemente consiste en, al menos dos etapas de reacción en fase vapor.
Con respecto al número de etapas de reacción, los solicitantes han encontrado sorprendentemente e inesperadamente que se puede conseguir conversión de la reacción total y selectividad a niveles relativamente altos por el uso de al menos dos etapas de reacción en la que la primera etapa de reacción se realiza en condiciones eficaces para conseguir una primera velocidad de conversión relativamente baja para producir un primer efluente de la reacción en etapas y al menos una segunda etapa de reacción que se alimenta por al menos una porción de dicho efluente de la primera etapa y que se realiza en condiciones eficaces para conseguir una segunda velocidad de conversión mayor que dicha primera velocidad. La cantidad de catalizador presente en la segunda etapa de la reacción es mayor que la cantidad de catalizador presente en la primera etapa de la reacción. Preferiblemente, las condiciones de reacción distintas de la cantidad de catalizador se controlan en cada una de las etapas primera y segunda para conseguir la conversión deseada según la presente invención. Como se usa en la presente memoria, el término "condiciones de reacción" se desea que incluya el singular y significa control de uno o más cualesquiera parámetros del procedimiento que se puede modificar por el operador de la reacción para producir la conversión del material de alimentación según las explicaciones contenidas en la misma. Como ejemplo, pero no como limitación de ningún modo, la conversión del material de alimentación se puede controlar o regular por control o regulación de uno o más cualesquiera de lo siguiente: la temperatura de la reacción, el caudal de los agentes reaccionantes, la presencia de diluyente, la forma y el tamaño del recipiente de reacción, la presión de la reacción y una cualquiera de las combinaciones de éstos y otros parámetros del procedimiento que estarán disponibles y serán conocidos para los expertos en la materia a la vista de la descripción contenida en presente memoria.
Los solicitantes han encontrado que en realizaciones preferidas la etapa de controlar la conversión en la primera etapa de la reacción de hidrogenación se consigue por selección juiciosa y control de la cantidad de catalizador presente en la primera etapa de reacción en relación con la velocidad de alimentación de uno o más de los agentes reaccionantes y/o por selección juiciosa y control de la temperatura de reacción y preferiblemente por selección juiciosa y control de ambos de estos parámetros del procedimiento. La etapa de seleccionar de manera juiciosa la cantidad de catalizador que se tiene que usar en la primera etapa de reacción incluye la etapa de estimar la cantidad de catalizador necesaria teóricamente para convertir el 100% del material de alimentación. Dicho cálculo se puede obtener por cualquiera y todos los métodos conocidos para hacer tal cálculo, que debería ser evidente para los expertos en la materia a la vista de las explicaciones contenidas en la presente memoria. Además, la etapa de seleccionar de manera juiciosa la cantidad de catalizador también puede implicar realizar estudios escalonados, piloto o similares para determinar la cantidad del catalizador particular que se esté usando que sea necesaria para convertir el 100% del material de alimentación en la velocidad de alimentación en otros parámetros del procedimiento que se ha elegido de otro modo. Basándose en este cálculo, las realizaciones preferidas de la presente invención incluyen después el paso de proporcionar en la primera etapa de reacción una cantidad de catalizador que está sustancialmente por debajo de la cantidad requerida para conversión del 100% e incluso más
preferiblemente es suficientemente baja para que de cómo resultado una conversión de la olefina de alimentación de desde aproximadamente 10% a aproximadamente 60%, más preferiblemente de aproximadamente 10% a aproximadamente 40% e incluso más preferiblemente de aproximadamente 10% a 25%. Una vez más, los expertos en la materia apreciarán que el paso de elegir de manera juiciosa la cantidad de catalizador puede incluir además efectuar estudios escalonados, piloto u otros estudios adicionales con la cantidad reducida de catalizador y ajustar la cantidad de catalizador de acuerdo con esto. Se considera que todos esos estudios y cálculos se pueden conseguir sin experimentación excesiva a la vista de las explicaciones contenidas en la presente memoria.
En realizaciones preferidas, por lo tanto, la etapa de controlar la conversión en la primera etapa del reactor comprende alimentar el agente reaccionante olefínico en la primera etapa de reacción a una velocidad que está sustancialmente por encima de, y al menos 60% aproximadamente 90% por encima de la productividad del catalizador presente en la primera etapa de reacción. Los solicitantes han encontrado, sin estar limitados por o a ninguna teoría particular, que el uso de tal exceso de agente reaccionante en la primera etapa de reacción permite que los materiales de alimentación sirvan como medio de eliminación de calor. Puesto que la reacción de reducción o de hidrogenación de la presente invención es en general exotérmica y normalmente sustancialmente exotérmica, el uso de tal material de alimentación en exceso presenta el efecto en realizaciones preferidas de mantener la temperatura del reactor por debajo de la que existiría si no se tuviera que usar un exceso el material de alimentación, asumiendo que se mantuvieran igual todas las demás condiciones del procedimiento.
Los solicitantes han encontrado que el paso de mantener una conversión muy baja de agente reaccionante según la presente invención en una primera etapa de reacción presenta un efecto ventajoso sobre la selectividad de la reacción para el alcano deseado. En otras palabras, aunque la cantidad de conversión que tiene lugar en la primera etapa de reacción se controla que esté muy por debajo de la que se desea para la etapa de reducción total, los solicitantes han encontrado que un porcentaje mayor, mejorado, del material de alimentación se convierte en el alcano deseado en la primera etapa de reacción (esto es, se consigue selectividad mejorada) por control de la conversión como se describe en la presente memoria. Más específicamente, se prefiere en muchas realizaciones que la selectividad para el alcano deseado en la primera etapa de reacción es al menos aproximadamente 80%, más preferiblemente al menos aproximadamente 90% e incluso más preferiblemente al menos aproximadamente 95% y en muchas realizaciones preferidas aproximadamente 97% o mayor.
En algunas realizaciones preferidas, el paso de controlar la conversión en la primera etapa de reacción incluye además retirar calor de la reacción enfriando al menos una porción de la mezcla de reacción. Se considera que los expertos en la materia podrán idear sin experimentación excesiva y muchos medios y mecanismos para lograr tal enfriamiento a la vista de las explicaciones contenidas en la presente memoria y todos esos medios y mecanismos están con el alcance de la presente invención.
En realizaciones preferidas, al menos una porción del efluente de la primera etapa de reacción se alimenta directamente, u opcionalmente después de algún tratamiento adicional, a una segunda etapa de reacción en que la olefina fluorada no reaccionada que queda en el efluente después de la primera etapa de reacción se convierte en el alcano fluorado según la presente invención. La segunda etapa de reacción o etapas de reacción posteriores si hay, se realiza en condiciones eficaces para convertir la olefina fluorada contenida en la corriente de alimentación en la segunda etapa del reactor a una velocidad de conversión que es mayor que el porcentaje de conversión en la primera etapa de reacción. En algunas realizaciones preferidas, por ejemplo, el porcentaje de conversión en la segunda etapa de reacción es de aproximadamente 20% a aproximadamente 99%, dependiendo en gran parte del número total de etapas de agentes reaccionantes usado para efectuar la etapa de conversión total. Por ejemplo, en realizaciones que consisten en un sistema de reacción de dos etapas, se considera que la conversión en la segunda etapa de reacción es preferiblemente mayor que 95% e incluso más preferiblemente aproximadamente 100%. Sin embargo, como los expertos en la materia apreciarán de las explicaciones contenidas en la presente memoria, tal reacción en dos etapas puede no ser suficiente para producir la selectividad deseada para el alcano fluorado. En tales casos, está dentro del alcance de la presente invención que la etapa de conversión pueda comprender más de dos etapas de reacción, incluyendo en algunas realizaciones tantas como 10 o más etapas de reacción.
En realizaciones preferidas, el paso de conversión de olefina fluorada de la presente invención comprende aproximadamente cuatro etapas de reacción. Aunque se entiende que los parámetros particulares usados en cada etapa de reacción pueden variar extensamente dentro del alcance de la presente invención, dependiendo de muchos factores, incluyendo la olefina fluorada deseada que se tiene que producir, la materia prima disponible y otras restricciones del procedimiento específicas, la siguiente Tabla proporciona intervalos preferidos y mas preferidos de ciertos parámetros del procedimiento aplicables a algunas realizaciones preferidas de la presente invención (se entiende que todos los valores numéricos en la tabla están precedidos por la palabra "aproximadamente.")
TABLA 1

Para los fines de ilustración, pero no necesariamente como limitación, una disposición de múltiples etapas de reacción según un paso de reducción preferido según la presente invención se ilustra en Figura 2. Aunque se considera que el paso 50 de reducción se puede realizar en una operación en modo discontinuo o semicontinuo, se prefiere que el paso 50 de reducción sea una operación continua del tipo general mostrado en la Figura 2. En la Figura 2, se proporciona un reactor 50A de primera etapa con catalizador en una cantidad eficaz para proporcionar una conversión de la olefina fluorada contenida en la corriente 1 de desde aproximadamente 10% a aproximadamente 60% según la presente invención. En realizaciones preferidas, la primera etapa de reacción de la presente invención, por ejemplo el recipiente 50A, se proporciona con un medio para enfriar la mezcla de reacción (no mostrado). Por ejemplo, el recipiente 50A de reacción puede ser un recipiente de reacción con camisa en que el espacio entre el recipiente y la camisa contiene un medio de enfriamiento para retirar calor del reactor. Por supuesto, se puede emplear otro medio para enfriar la mezcla de reacción, tal como el uso de un serpentín de enfriamiento interno, la introducción de diluyente de enfriamiento adicional en la mezcla de reacción y similares.
El efluente 3A de la primera etapa de reacción es preferiblemente, pero no necesariamente, enfriado además previamente a su introducción en la segunda etapa de reacción, tal como el recipiente 50B de reacción, por ejemplo por uso de un intercambiador 60A de calor. De hecho, en ciertas realizaciones, puede ser deseable que el intercambiador 60A de calor y/o uno o más de los intercambiadores 60B y 60C de calor aguas abajo, proporcionen la capacidad de añadir calor al efluente previamente a su introducción en la siguiente etapa de reacción. Esta capacidad es deseable en algunas realizaciones debido a que se prefiere en general, aunque no exclusivamente, que el nivel de conversión en cada etapa de reacción posterior sea mayor que en la etapa previa inmediatamente precedente. Para conseguir este resultado, puede ser deseable y/o necesariamente elevar la temperatura de una o más de las corrientes de alimentación como un medio para contribuir a una mayor temperatura de reacción en la siguiente etapa de reacción. Por supuesto, los expertos en la materia apreciarán que están disponibles muchos medios y mecanismos para controlar la temperatura en todas las etapas de reacción posteriores y todos esos medios y mecanismos están dentro del alcance de la presente invención y se pueden usar para controlar la conversión de la etapa de reacción según la presente invención.
El tamaño y la forma y otras características del propio recipiente de reacción pueden variar ampliamente con el alcance de la presente invención y se considera que el recipiente asociado con cada etapa puede ser diferente de o igual que el recipiente asociado con las etapas de la reacción aguas arriba y aguas abajo. Además, se considera que todas las etapas de la reacción pueden tener lugar en el interior de un solo recipiente, siempre que se proporcionen los medios y mecanismos necesarios para controlar la conversión. Por ejemplo, puede ser deseable en ciertas realizaciones utilizar un único reactor tubular para cada etapa de reacción, proporcionando control de la conversión por la selección juiciosa de la cantidad y/o distribución de catalizador por todo el reactor tubular. En tal caso, es posible controlar además la conversión en diferentes secciones del mismo reactor tubular por control de la cantidad de calor retirada de o añadida a diferentes secciones del reactor tubular.
Los expertos en la materia podrán seleccionar fácilmente el tipo de catalizador o de catalizadores usados para el paso de hidrogenación de la presente invención a la vista de las explicaciones contenidas en la presente memoria. Por ejemplo, se prefiere en ciertas realizaciones que al menos una, pero preferiblemente todas, las etapas de
reacción utilice catalizador de paladio, solo o junto con otros catalizadores. Con respecto a esto, se puede usar uno o más del catalizador de hidrogenación descrito en la Patente de EE.UU. 5.679.875, para una o más de las etapas de reacción según la presente invención. En algunas realizaciones preferidas, el catalizador comprende preferiblemente paladio soportado sobre carbono, tal como una malla de carbono.
Así, algunas realizaciones de los métodos presentes comprenden poner en contacto una olefina fluorada según la fórmula I, específicamente, un fluoropropeno que tiene de tres a seis sustituyentes flúor, y un agente de hidrogenación, tal como H2 , con una primera cantidad de catalizador en una primera etapa de reacción para producir una corriente de reacción que comprende hidrofluorocarbono(s), olefina fluorada no reaccionada e hidrógeno; poner en contacto al menos una porción de esta primera corriente de efluente con una segunda cantidad de catalizador en una segunda etapa de reacción para producir un hidrofluorocarbono, en la que la segunda cantidad de catalizador es mayor que la primera cantidad de catalizador y en la que la conversión en la olefina fluorada es mayor en la segunda etapa de reacción. Preferiblemente, al menos una porción del efluente de las etapas segunda y/o cualquiera posterior de reacción se pone en contacto después con un catalizador para la deshidrofluoración para producir una corriente de producto que comprende el propeno fluorado deseado y HF. En algunas realizaciones preferidas, la etapa de deshidrofluoración va seguida por un paso de separación preferido que comprende alimentar al menos una porción de la corriente de producto por una operación de extracción usando un agente de extracción, tal como una corriente de ácido sulfúrico, a HF extracto de la corriente de producto. La etapa de deshidrohalogenación y la etapa de separación según aspectos preferidos de la presente invención se discuten con detalle a continuación.
Deshidrohalogenación
Se considera que la etapa de reacción de deshidrohalogenación se puede formar previamente usando una amplia variedad de parámetros del procedimiento y condiciones del procedimiento a la vista de todas las explicaciones contenidas en la presente memoria, tal como por ejemplo se considera que el paso de deshidrohalogenación puede comprender, en ciertas realizaciones no preferidas, una reacción en fase líquida. Sin embargo, se prefiere en muchas realizaciones de la presente invención que este paso de la reacción comprenda una reacción en fase gas, preferiblemente en presencia de catalizador, preferiblemente un catalizador de metal, e incluso más preferiblemente uno o más catalizadores basados en metales de transición (incluyendo en algunas realizaciones preferidas catalizadores de haluro de metal de transición), tales como FeCh, oxifluoruro de cromo, Ni (incluyendo malla de Ni), NiCl2, CrF3 y mezclas de los mismos, soportados o en masa. Otros catalizadores incluyen catalizadores soportados sobre carbono, catalizadores a base de antimonio (tal como SbCIs), catalizador a base de aluminio (tal como AIF3, Al2O3 y Al2O3 fluorado). Se espera que se puedan usar muchos otros catalizadores dependiendo de los requerimientos de realizaciones particulares, incluyendo por ejemplo catalizador a base de paladio, catalizadores a base de platino, catalizadores a base de rodio y catalizadores a base de rutenio. Por supuesto, se pueden usar en asociación dos o más cualesquiera de estos catalizadores u otros catalizadores no mencionados en la presente memoria.
En general se prefiere que los catalizadores estén fluorados. En realizaciones preferidas, la fluoración de los catalizadores comprende exponer el catalizador a una corriente de HF a aproximadamente la temperatura y presión de la reacción. La reacción de deshidrohalogenación en fase gas se puede realizar, por ejemplo, introduciendo una forma gaseosa de un compuesto de fórmula (II) en un recipiente de reacción o reactor adecuado. Preferiblemente, el recipiente está formado por materiales que son resistentes a la corrosión como revestimientos de Hastelloy, Inconel, Monel y/o fluoropolímeros. Preferiblemente, el recipiente contiene catalizador, por ejemplo un lecho catalítico fijo o fluido, empaquetado con un catalizador de deshidrohalogenación adecuado, con medios adecuados para calentar la mezcla de reacción a la temperatura de reacción deseada.
Aunque se considera que se puede usar una amplia variedad de temperaturas de reacción, dependiendo de factores relevantes tales como el catalizador que se esté usando y el producto de reacción más deseado, en general se prefiere que la temperatura de reacción para el paso de deshidrohalogenación sea de aproximadamente 150°C a aproximadamente 600°C, preferiblemente aproximadamente de 200°C a aproximadamente 550°C e incluso más preferiblemente de aproximadamente 220°C a aproximadamente 450°C.
En general también se considera que se puede usar una amplia variedad de presiones de reacción, dependiendo de nuevo de factores relevantes tales como el catalizador específico que se esté usando y el producto de reacción más deseado. La presión de la reacción puede ser, por ejemplo, superatmosférica, atmosférica o a vacío.
En ciertas realizaciones, se puede usar un gas diluyente inerte y/o un agente oxidante, tal como nitrógeno, oxígeno y mezcla de nitrógeno y oxígeno, en asociación con el compuesto de fórmula (II) como alimentación al paso de deshidrohalogenación. Cuando se usa tal diluyente y/o agente oxidante, se prefiere en general que la salida de la alimentación comprenda compuesto de fórmula (II) en una cantidad de desde aproximadamente 5% a mayor que 95% en peso basado en el peso combinado de diluyente y compuesto de fórmula (II).
Se considera que la cantidad de catalizador usado variará dependiendo de los parámetros particulares presentes en
cada realización. En realizaciones preferidas, el tiempo de contacto, que se expresa como la relación del volumen del catalizador (ml) al flujo total de alimentación (ml/s) es de aproximadamente 0,1 segundos a aproximadamente 1.000 segundos y preferiblemente de aproximadamente 2 segundos a aproximadamente 120 segundos.
Una reacción de deshidrohalogenación preferida comprende una reacción de deshidrofluoración. Por ejemplo, para realizaciones en que el producto de fórmula (III) deseado es HFO-1234yf, se prefiere en ciertas realizaciones que el compuesto de fórmula (II) comprenda 1,1,1,2,3 pentafluoropropano. Los solicitantes han encontrado que en tales realizaciones se prefiere usar como catalizador un catalizador de óxido de cromo fluorado y catalizador de fluoruro de aluminio, un catalizador de fluoruro férrico y una mezcla de catalizadores que comprende fluoruro de magnesio y fluoruro de aluminio. En algunas realizaciones, se puede usar catalizador a base de níquel, un catalizador a base de carbono o una combinación de éstos.
Además, se prefiere en general realizar al menos una porción sustancial de la reacción a una temperatura de desde aproximadamente 200°C a aproximadamente 600°C, eligiéndose la temperatura particular dependiendo de muchos factores, incluyendo en particular el tipo de catalizador que se esté usando. Por ejemplo, se prefiere que cuando tiene lugar la reacción de deshidrofluoración en presencia de un catalizador fluorado o de metal fluorado, entonces las temperaturas de la reacción son preferiblemente de aproximadamente 200°C a aproximadamente 550°C, más preferiblemente de aproximadamente 220°C a aproximadamente 450°C e incluso más preferiblemente de aproximadamente 250°C a aproximadamente 375°C. Para realizaciones en que el catalizador comprende carbono activado, metales sobre carbono activado, especialmente sales de hierro, cobalto y níquel sobre carbono activado y paladio sobre carbono, la temperatura de la reacción es preferiblemente de aproximadamente 200°C a aproximadamente 600°C, e incluso más preferiblemente de aproximadamente 300°C a aproximadamente 500°C. En realizaciones preferidas, el tiempo de contacto es de aproximadamente 0,1 s a aproximadamente 1.000 segundos y preferiblemente de aproximadamente 2 s a aproximadamente 120 segundos.
Preferiblemente, en tales realizaciones de deshidrofluoración, la conversión es al menos aproximadamente 50%, más preferiblemente al menos aproximadamente 65% e incluso más preferiblemente al menos aproximadamente 90%. Preferiblemente, la selectividad para HFO-1234yf es al menos aproximadamente 70%, más preferiblemente al menos aproximadamente 80% y más preferiblemente al menos aproximadamente 90%.
Otra reacción de deshidrohalogenación preferida comprende una reacción de deshidrocloración. Por ejemplo, para realizaciones en que el producto de fórmula (III) deseado es HFO-1234ze, se prefiere que el compuesto de fórmula (II) comprenda 1,1,1,3-tetrafluoro-3-cloropropano. Los solicitantes han encontrado que en ciertas realizaciones se prefiere usar para esta reacción un catalizador a base de níquel a una temperatura de reacción de desde aproximadamente 200°C a aproximadamente 550°C, más preferiblemente de aproximadamente 250°C a aproximadamente 500°C, e incluso más preferiblemente de aproximadamente 300 a 400°C. En algunas otras realizaciones se prefiere usar para esta reacción un catalizador de carbono activado a una temperatura de reacción de desde aproximadamente 250°C a aproximadamente 550°C, más preferiblemente de aproximadamente 300°C a aproximadamente 550°C, e incluso más preferiblemente aproximadamente 400°C. En otras realizaciones se prefiere usar para esta reacción un catalizador que comprende 3% de paladio sobre carbono a una temperatura de reacción de desde aproximadamente 200°C a aproximadamente 500°C, más preferiblemente de aproximadamente 225°C a aproximadamente 475°C, e incluso más preferiblemente aproximadamente 400°C. En otras realizaciones más se prefiere usar para esta reacción un catalizador que comprende 2% de níquel sobre carbono a una temperatura de reacción de desde aproximadamente 400°C a aproximadamente 500°C, más preferiblemente de aproximadamente 400°C a aproximadamente 500°C, e incluso más preferiblemente aproximadamente 450°C. En otras realizaciones, se prefiere usar para esta reacción un catalizador que comprende oxifluoruro de cromo a una temperatura de reacción de desde aproximadamente 200°C a aproximadamente 500°C, más preferiblemente de aproximadamente 250°C a aproximadamente 450°C, e incluso más preferiblemente aproximadamente 300°C.
En tales realizaciones de deshidrocloración es una opción introducir al reactor gas inerte y/o un gas oxidante, tal como nitrógeno y/u oxígeno o mezcla de nitrógeno y oxígeno, en una relación en volumen de fórmula (ll):inerte (u oxígeno) de desde aproximadamente 100:0,5 a aproximadamente 100:75, siendo incluso más preferida una relación de aproximadamente 100:2.
En aspectos preferidos de las realizaciones de deshidrocloración, el tiempo de contacto es de aproximadamente 0,1 a aproximadamente 1.000 s y preferiblemente de aproximadamente 3 a aproximadamente 120 s.
Preferiblemente, en tales realizaciones de deshidrocloración, la conversión es al menos aproximadamente 50%, más preferiblemente al menos aproximadamente 65%, e incluso más preferiblemente al menos aproximadamente 90%. Preferiblemente, la selectividad para HFO-1234ze, e incluso más preferiblemente para trans-HFO-1234ze, es al menos aproximadamente 70%, más preferiblemente al menos aproximadamente 80% y más preferiblemente al menos aproximadamente 90%.
En general la dirección de flujo de los componentes gaseosos en el paso de deshidrohalogenación no es crítica,
pero en algunas realizaciones preferidas el flujo del procedimiento es en la dirección hacia abajo por un lecho del catalizador.
Preferiblemente, antes de cada ciclo de uso, se seca el catalizador de deshidrohalogenación, se pretrata y se activa. Puede ser ventajoso también en algunas realizaciones regenerar periódicamente el catalizador después de uso prolongado aunque en su lugar en el reactor. El pre-tratamiento puede incluir calentar el catalizador a aproximadamente 250°C a aproximadamente 430°C con una corriente de nitrógeno u otro gas inerte. El catalizador puede ser activado después tratándolo con una corriente de HF diluida con un gran exceso de gas nitrógeno para obtener alta actividad catalítica. La regeneración del catalizador se puede realizar por cualquier medio conocido en la técnica tal como, por ejemplo, haciendo pasar aire u oxígeno por el catalizador a temperaturas de desde aproximadamente 100°C a aproximadamente 400°C durante desde aproximadamente 1 hora a aproximadamente 3 días dependiendo del tamaño del reactor.
Separación
Como se mencionó anteriormente, además de producir una olefina fluorada, es decir propeno fluorado, la reacción de deshidrofluoración también produce HF. En una realización preferida, se retira HF de la corriente de producto de deshidrofluoración por extracción a contracorriente con ácido sulfúrico. En esta realización, la corriente de producto que contiene el HF se alimenta en una dirección a una columna, preferiblemente una columna empaquetada. Al mismo tiempo, se alimenta una corriente de ácido sulfúrico, preferiblemente de manera en contracorriente, a la misma columna empaquetada. El empaquetamiento de la columna apropiado se puede determinar fácilmente por un experto en la materia. Los materiales de empaquetamiento de columna adecuados incluyen los fabricados de materiales poliméricos no metálicos, metales y aleaciones que no son reactivos en presencia de HF o ácido sulfúrico, tal como PTFE, PFA, hastelloy, monel y metales nobles. Preferiblemente, la corriente de ácido sulfúrico contiene de aproximadamente 50% a aproximadamente 100% de ácido sulfúrico y más preferiblemente aproximadamente 80% de ácido sulfúrico. En una realización, la corriente de ácido sulfúrico se alimenta de manera continua a la parte superior de la columna empaquetada a una velocidad de alimentación de aproximadamente dos veces la velocidad de alimentación de la corriente de producto, que en realizaciones preferidas se alimenta desde el fondo de la columna empaquetada y se mueve en una dirección hacia arriba en general sustancialmente de manera en contracorriente a la corriente que fluye sustancialmente hacia abajo que contiene el ácido sulfúrico. En algunas realizaciones, una corriente que comprende ácido sulfúrico y HF se retira de la cola de la columna y preferiblemente al menos una porción de la corriente y lo más preferiblemente sustancialmente todo de la corriente, se vuelve a reciclar a la torre de extracción. La etapa de reciclado se repite preferiblemente hasta que la concentración de HF en la cola de la columna es mayor que aproximadamente 10% en peso de HF.
En una realización, la mezcla de ácido sulfúrico y HF que contiene más de aproximadamente 10% en peso de HF se carga en un recipiente separado. La mezcla se calienta después a una temperatura suficiente para vaporizar y vaporizar de manera instantánea HF, que se recoge.
Otra realización incluye purificar el HF recogido de la destilación instantánea.
Opcionalmente, el HF o HCI generado de la reacción de deshidrohalogenación se lava de la corriente de producto usando agua o disoluciones caústicas.
Los siguientes ejemplos se proporcionan como ilustraciones específicas de la invención. Se debería observar, sin embargo, que la invención no está limitada a los detalles específicos explicados en los ejemplos.
Ejemplos
Ejemplo Comparativo C-1: Reacción de hexafluoropropeno e hidrógeno
Un reactor de una sola etapa, con camisa pequeño, con una camisa exterior unida a un baño de enfriamiento de circulación de 31°C se carga con 0,2 g de Pd/C al 1% (malla 4-6) mezclado con malla de nicromo de un tamaño similar para proporcionar un volumen de lecho catalítico total de 1 cc. Previamente a la introducción de los gases hidrógeno y hexafluoropropeno, la temperatura del lecho es inicialmente aproximadamente 21°C. Sin embargo, cuando se introducen hidrógeno (0,37 mol/h) y hexafluoropropeno (0,26 mol/h), la temperatura del lecho se eleva a casi 70°C en aproximadamente 1 minuto.
Ejemplo Comparativo C-2: Reacción de hexafluoropropeno e hidrógeno
El mismo reactor de una sola etapa, con camisa, pequeño, que en el Ejemplo C-1, se carga con una pequeña cantidad de Pd al 1% sobre carbono. Se mezclan previamente hidrógeno y hexafluoropropeno y después se introducen en el reactor. Se deja que la temperatura del lecho catalítico se estabilice a 69°C. Se analizan los gases de salida para determinar la conversión y la selectividad para CF3CHFCF2H. La conversión promedio es 93,2% mientras que la selectividad promedio es 95,7%.
Ejemplo Comparativo C-3: Reacción de hexafluoropropeno e hidrógeno
Se repite el Ejemplo Comparativo C-2 usando el mismo reactor de una sola etapa, excepto que la temperatura del fluido de circulación se reduce a 21°C. La temperatura del lecho se estabiliza a 61,5°C. En estas condiciones, la conversión se reduce a 88,6% mientras que la selectividad aumenta a 97,0%.
Ejemplos 1 y 2: Reacciones de Reducción Multi-Etapa
Los reactores usados en los siguientes ejemplos son reactores multi-etapa construidos de secciones de tubería 316 SS, plan 40 de 3,81 cm (1,5").
La cantidad de catalizador usada para cargar cada etapa de la reacción se calcula por estimación primero de la productividad del catalizador (gramos de alimentación convertidos por gramo de catalizador por hora). La productividad se estima a partir de estudios de alcance usando un pequeño reactor. Después, la velocidad de producción deseada se fija a aproximadamente 4,5 kg (10 lb) por hora, permitiendo que se estime la cantidad total de catalizador necesaria para conversión del 100%. Usando esta información, se calcula una cantidad estimada de catalizador necesaria para convertir 10-15% de la olefina en el primer reactor.
La carga de catalizador en los siguientes ejemplos es como sigue:
Sección 1 (3,81 cm (1,5") x 30,48 cm (1 pie)): 10 g de catalizador (Pd al 1% en peso en carbono de malla 4-8) con el resto cargado con empaquetamiento saliente SS de 0,63 cm (W), catalizador igualmente distribuido por todo.
Sección 2 (3,81 cm (1,5") x 60,96 cm (2 pie)): 25 g de catalizador distribuido como en la Sección 1.
Sección 3 (3,81 cm (1,5") x 91,44 (3 pie)): 73,4 g de catalizador con 1.200 cc de empaquetamiento distribuido como en la Sección 1.
Sección 4 (3,81 cm (1,5") x 121,92 (4 pie)): 158 g de catalizador distribuido con 1.400 cc de empaquetamiento. Catalizador total = 267 g.
Ejemplo 1: Reducción multi-etapa de hexafluoropropeno
Se introduce hexafluoropropeno en el reactor multi-etapa y se reduce de manera continua durante un periodo de 58 horas durante lo cual la velocidad de alimentación promedio es 6,5 kg/h (14,5 Ib/h) (o aproximadamente 16,4 l por minuto). La velocidad de alimentación de hidrógeno promedio es 25 l por minuto. Las muestras se toman en diversos puntos a lo largo de la serie de reactores para seguir el porcentaje de conversión y la selectividad. Después de la segunda etapa de la reacción, la conversión es aproximadamente 40%; después de la cuarta etapa de reacción, la conversión es 99,5% con selectividad para CF3CHFCF2H de 99%. La temperatura de los gases que salen inmediatamente de las etapas de la reacción es 66°C para la primera etapa, 104°C para la segunda etapa, 173°C para la tercera etapa y 100C para la cuarta etapa. La temperatura máxima en cualquier etapa de reacción es aproximadamente 230°C. El primer baño se mantiene a 55°C mientras que el segundo baño se mantiene a 111°C.
Ejemplo 2: Reducción multi-etapa de 1,2,3,3,3-Pentafluoropropeno-1
Se hidrogena 1,2,3,3,3-pentafluoropropeno-1 usando el mismo reactor que en el Ejemplo 1 usando una velocidad de alimentación de 6,6 kg/h (14,6 Ib/h) para un total de 64 horas. La velocidad de alimentación promedio de hidrógeno es 25 litros por minuto. Se toman muestras en diversos puntos a lo largo de la serie de reactores para seguir el porcentaje de conversión y la selectividad. Después del segundo reactor, la conversión es aproximadamente 54%. Aunque después del cuarto reactor, la conversión es del 100% con la selectividad para CF3CHFCH2F de 98%. La temperatura de los gases que salen inmediatamente de los reactores es 99°C para el primer reactor, 95°C para el segundo reactor, 173°C para el tercer reactor y 104°C para el cuarto reactor. La temperatura máxima en cualquier reactor es aproximadamente 240°C. El primer baño se mantiene a 59°C y el segundo baño se mantiene a 116°C. Ejemplos 3-8
Las siguientes reacciones de deshidrofluoración se realizan en un reactor Monel cilindrico. El calentamiento se proporciona insertando el reactor en un horno eléctrico. Se registran las temperaturas del proceso usando un termopar multipunto colocado en el interior del reactor y dentro del lecho catalítico. Se alimenta el hidrofluorocarbono en el fondo del reactor montado de manera vertical y se vaporiza antes de alcanzar el lecho catalítico. Se hacen pasar los gases del efluente por una válvula de muestreo de gases para controlar el progreso de la reacción usando análisis GC.
Ejemplo 3: Deshidrofluoración de 1,1,1,2,3,3-hexafluoropropano (HFC-236ea) sobre catalizador de Cr2O3 fluorado.
Se hace pasar HFC-236ea por 20 cc de Cr2Ü3 fluorado a una velocidad de 12 g/h en un intervalo de temperatura de desde 250 a 350°C a 1 atm. Se generan dos isómeros de 1,1,1,2,3-pentafluoropropeno, específicamente, 1225yeZ y 1225yeE, durante la reacción. Como se muestra en la Tabla Ej 3 a continuación, a medida que aumenta la temperatura de reacción de 250 a 350°C, la conversión de HFC-236ea aumenta de 65,2% a 96,0%, al tiempo que la selectividad para 1225yeZ disminuye ligeramente de 97,0% a 94,6%. A 250°C, 1225 isómeros parecen ser los únicos productos. Estos resultados indican que el catalizador de CrzO fluorado es muy activo y selectivo para convertir HFC-236ea en 1225yeZ.
Tabla Ej 3. Efecto de la temperatura de reacción sobre la realización de Cr2O3 fluorado durante la deshidrofluoración de HFC-236ea

Ejemplo 4: Deshidrofluoración de HFC-236ea por catalizadores de fluoruro de metal
Los catalizadores para uso en este ejemplo incluyen AIF3 , FeF3 y MgF2 al 10% - AIF3 al 90%. Se hace pasar HFC-236ea por 20 cc de cada catalizador a una velocidad de 12 g/h a 350°C a 1 atm. Como se muestra en la Tabla Ej 4 a continuación, tanto AIF3 como MgF2 al 10% - AIF3 al 90% proporcionan alta actividad (conversión > 95% de HFC-236ea) para deshidrofluoración de HFC-236ea, mientras que FeF3 presenta actividad mucho menor (conversión < 60% de HFC-236ea). La selectividad para 1225yeZ sobre los catalizadores de AIF3 y MgF2 al 10% - AIF3 al 90% es aproximadamente 92% a 350°C.
Tabla Ej 4. Deshidrofluoración de HFC-236ea sobre catalizadores de fluoruro de metal

Ejemplo 5 - Deshidrofluoración de HFC-236ea sobre catalizadores de metal soportados sobre carbono activado
Los catalizadores para uso en este ejemplo incluyen tres catalizadores de metal soportados sobre carbono activado, específicamente, Fe/CA al 0,5% en peso, Ni/CA al 0,5% en peso y Co/CA al 0,5% en peso. Se pasa HFC-236ea sobre 20 cc de cada catalizador a una velocidad de 12 g/h a 350°C a 1 atm. Como se muestra en la Tabla Ej 5, entre los catalizadores de metal no precioso soportados sobre carbono activado, el hierro presenta la actividad más alta. A una temperatura de reacción de 350°C y 1 atm, el catalizador de Fe/CA al 0,5% en peso proporciona una selectividad de 1225yeZ de aproximadamente 90% y una conversión de HFC-236ea de aproximadamente 80%. Tabla Ej 5: Deshidrofluoración de HFC-236ea sobre catalizadores de metal soportados sobre carbono activado a 350°C


Ejemplo 6: Deshidrofluoración de CF
3
CHFCH
2
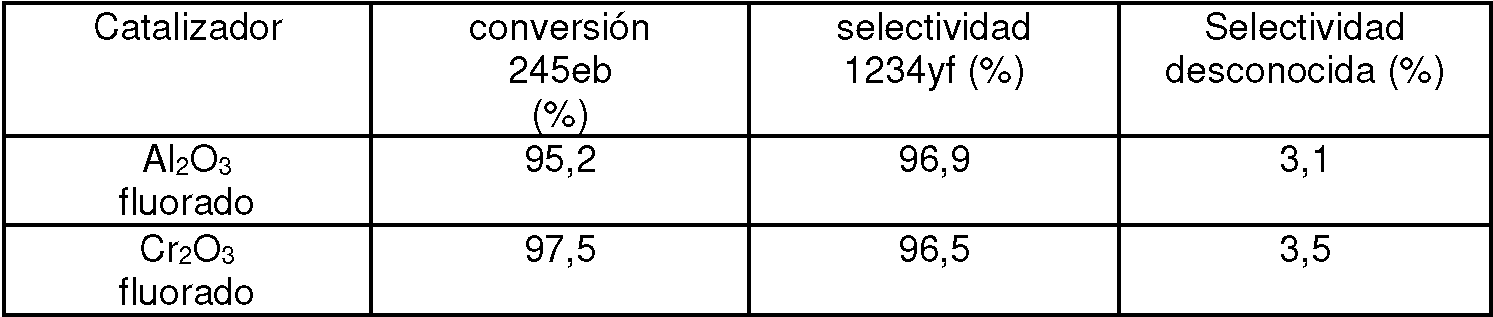
F (HFC-245eb) sobre catalizadores de alúmina fluorada y óxido de cromo
Los catalizadores para uso en este ejemplo incluyen dos catalizadores de óxido de metal fluorados, específicamente, AI2O3 y Cr2Ü3 fluorados. Se hace pasar HFC-245eb sobre 20 cc de cada catalizador a una velocidad de 12 g/h a 300 °C a 1 atm. Se genera 2,3,3,3-tetrafluoropropeno (HFC-1234yf) como producto de reacción. Como se muestra en la Tabla Ej 6, ambos catalizadores de óxido de metal fluorados proporcionan una conversión de HFC-245eb por encima de 95% y selectividad de 1234yf por encima de 96%. Estos resultados indican que los catalizadores de AI2O3 y C 2O3 fluorados son muy activos y selectivos para convertir 245eb en 1234yf.
Tabla Ej 6. Deshidrofluoración de HFC-245eb sobre catalizadores de óxido de metal fluorados
Ejemplo 7: Deshidrofluoración de HFC-245eb sobre catalizadores de fluoruro de metal
Los catalizadores para uso en este ejemplo incluyen tres catalizadores de fluoruro de metal, específicamente, MgF2 , AIF3 y CeF4. Se hace pasar HFC-245eb sobre 20 cc de cada catalizador a una velocidad de 12 g/h a 350°C a 1 atm. Como se muestra en la Tabla Ej 7, entre los fluoruros de metal investigados, AIF3 presenta la actividad más alta con alta selectividad. Este catalizador proporciona una conversión de HFC-245eb de aproximadamente 97% y a una selectividad de 1234yf de aproximadamente 97%.
Tabla Ej 7. Deshidrofluoración de HFC-245eb sobre catalizadores de fluoruro de metal

Ejemplo 8: Deshidrofluoración de HFC-245eb sobre carbono activado y catalizadores de Pd soportado sobre carbono activado
Los catalizadores para uso en este ejemplo incluyen carbono activado y catalizadores de Pd soportado sobre carbono activado. Se hace pasar HFC-245eb por 20 cc de cada catalizador a una velocidad de 12 g/h a 1 atm. Como se muestra en la Tabla Ej 8, el carbono activado muestra muy poca actividad para convertir 245eb en 1234yf incluso a 525°C, mientras que el catalizador de Pd soportado sobre carbono activado al 0,5% en peso proporciona una conversión de 245eb de aproximadamente 92% y una selectividad de 1234yf de aproximadamente 98% a 450°C. Estos resultados indican que los catalizadores de metal de valencia cero pueden ser activos para la deshidrofluoración de 245eb en 1234yf.
Tabla Ej 8. Deshidrofluoración de HFC-245eb sobre catalizadores de carbono activado (CA) y Pd soportado sobre CA

Ejemplo 9: Extracción en ácido sulfúrico de HF generado a partir de reacción de deshidrofluoración
La corriente de producto del Ejemplo 3 (a 350°C), que contiene 4,0% en peso de HFC-236ea, 78,9% en peso de 1225yeZ, 3,3% en peso de 1225yeE, 12,6% en peso de HF y 1,2% de desconocido es alimentada a la cola de una columna empaquetada a una velocidad de alimentación de aproximadamente 1,3 kg (2,9 lb) por hora durante aproximadamente 4 horas.
Una corriente de aproximadamente 80% en peso de ácido sulfúrico (H2SO4/H2O 80/20) se alimenta de manera continua a la parte superior de la misma columna empaquetada a una velocidad de alimentación de aproximadamente 2,5 kg (5,6 lb) por hora durante la misma base de tiempo. Una corriente gaseosa que sale de la parte superior de la columna incluye una mayoría de los compuestos orgánicos con menos de 0,5% en peso de HF. La concentración de HF en el ácido sulfúrico en los fondos de la columna es aproximadamente 5,8% en peso.
Las colas de la columna que contienen ácido sulfúrico y HF se vuelven a reciclar a la torre de extracción hasta que la concentración de HF es mayor que 10% en peso de Hf. Con posterioridad, se carga la mezcla de ácido sulfúrico y HF en un recipiente de Teflon® de 7,6 l (2 galones). Se calienta la mezcla a aproximadamente 140°C para vaporizar y vaporizar de manera instantánea HF, que se recoge. El producto HF recogido contiene 6.000 ppm de agua y 217 ppm de azufre. El ácido sulfúrico contiene aproximadamente 500 ppm de carbono orgánico total.
El HF recogido de la destilación instantánea se destila en una columna de destilación y se recupera HF anhidro. El HF anhidro recuperado contiene aproximadamente 37 ppm de impurezas de azufre.
Ejemplo 10: Conversión de 1-bromo, 2,3,3,3-tetrafluoropropeno en 2,3,3,3-tetrafluoropropeno
Se introduce 1-bromo, 2,3,3,3-tetrafluoropropeno en el reactor multi-etapa del Ejemplo 1 y se reduce de manera continua. Se toman muestras en diversos puntos a lo largo de la serie de reactores para seguir el porcentaje de conversión y la selectividad. Después de la segunda etapa de reacción, la conversión es de aproximadamente 40%; después de la cuarta etapa de reacción, la conversión es 99,5% con selectividad para CF3CHFCH2Br de 99%.
El alcano producto producido en el reactor multietapa se introduce en un reactor catalítico donde se deshidroclora a 2,3,3,3-tetrafluoropropeno a un alto nivel de conversión y selectividad.
Claims (7)
1. Un procedimiento para la producción de un producto olefínico fluorado que comprende las etapas de:
(a) poner en contacto una corriente de alimentación que comprende un fluoropropeno que tiene de tres a seis sustituyentes flúor y agentes reaccionantes de hidrógeno con una primera cantidad de un catalizador para convertir dichos agentes reaccionantes en un hidrofluoroalcano a una primera velocidad de conversión y para producir una primera corriente de salida que comprende dicho hidrofluoroalcano, fluoropropeno no reaccionado e hidrógeno; (b) poner en contacto dicha primera corriente de salida con una segunda cantidad de un catalizador para convertir dicho fluoropropeno no reaccionado en un hidrofluoroalcano a una segunda velocidad de conversión, en la que dicha segunda cantidad de catalizador es mayor que dicha primera cantidad de catalizador y en la que dicha segunda velocidad de conversión es mayor que dicha primera velocidad de conversión;
(c) deshidrohalogenar al menos una porción de dicho hidrofluoroalcano de dicha etapa (b) de contacto para producir una corriente de producto que comprende una olefina fluorada y HF producto y
(d) opcionalmente, separar dicho HF de dicha corriente de producto.
2. El procedimiento según la reivindicación 1, en el que la etapa (d) comprende además poner en contacto al menos una porción de dicha corriente de producto con ácido sulfúrico para extraer HF de dicha corriente de producto.
3. El procedimiento según la reivindicación 2, en el que dicha puesta en contacto en la etapa (d) produce una corriente de ácido sulfúrico de efluente que contiene al menos una porción de1HF de la corriente de producto y en el que dicho procedimiento comprende además el paso de reciclar al menos una porción de dicha corriente de ácido sulfúrico de efluente por una torre de extracción.
4. El procedimiento según la reivindicación 1, en el que dicho hidrofluoroalcano comprende al menos uno de pentafluoropropano (HFC-245) o (HFC-236).
5. El procedimiento según la reivindicación 1, en el que dicho producto comprende CFH=CFCF3.
6. El procedimiento según la reivindicación 1, en el que dichas etapas (a) y (b) de conversión comprenden cada una el uso de paladio soportado sobre carbono.
7. El procedimiento según la reivindicación 1, en el que dicha etapa de deshidrohalogenación comprende una deshidrofluoración catalizada y dicho catalizador se selecciona del grupo que consiste en uno o más óxidos de metal fluorados, fluoruros de metal, metales de transición soportados sobre carbono y combinaciones de éstos.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US588464 | 1984-03-12 | ||
| US11/588,464 US7560602B2 (en) | 2005-11-03 | 2006-10-27 | Process for manufacture of fluorinated olefins |
| PCT/US2007/082601 WO2008057794A1 (en) | 2006-10-27 | 2007-10-26 | Process for the manufacture of fluorinated olefins |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2394870T3 ES2394870T3 (es) | 2013-02-06 |
| ES2394870T5 true ES2394870T5 (es) | 2021-07-27 |
Family
ID=39201461
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES07863530T Active ES2394870T5 (es) | 2006-10-27 | 2007-10-26 | Procedimiento para la fabricación de olefinas fluoradas |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US7560602B2 (es) |
| EP (1) | EP2076478B2 (es) |
| JP (1) | JP5588174B2 (es) |
| KR (2) | KR20140090702A (es) |
| CN (1) | CN101553453B (es) |
| ES (1) | ES2394870T5 (es) |
| RU (1) | RU2457195C2 (es) |
| WO (1) | WO2008057794A1 (es) |
Families Citing this family (61)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7880040B2 (en) * | 2004-04-29 | 2011-02-01 | Honeywell International Inc. | Method for producing fluorinated organic compounds |
| US8067649B2 (en) * | 2004-04-29 | 2011-11-29 | Honeywell International Inc. | Method for producing fluorinated organic compounds |
| US8766020B2 (en) | 2008-07-31 | 2014-07-01 | Honeywell International Inc. | Process for producing 2,3,3,3-tetrafluoropropene |
| US7833434B2 (en) * | 2006-06-27 | 2010-11-16 | E. I. Du Pont De Nemours And Company | Tetrafluoropropene production processes |
| US8377327B2 (en) | 2006-06-27 | 2013-02-19 | E I Du Pont De Nemours And Company | Tetrafluoropropene production processes |
| US8263816B2 (en) | 2006-06-27 | 2012-09-11 | E I Du Pont De Nemours And Company | 1,2,3,3,3-Pentafluoropropene production processes |
| RU2445302C2 (ru) | 2006-06-27 | 2012-03-20 | Е.И. Дюпон Де Немур Энд Компани | Способы получения тетрафторпропена |
| EP4328213A3 (en) | 2006-10-27 | 2024-04-03 | Honeywell International Inc. | A high purity 2,3,3,3-tetrafluoropropene |
| US8013194B2 (en) † | 2008-03-14 | 2011-09-06 | Honeywell International Inc. | Process for the manufacture of fluorinated olefins |
| GB0625214D0 (en) * | 2006-12-19 | 2007-01-24 | Ineos Fluor Holdings Ltd | Process |
| US9040759B2 (en) * | 2007-07-06 | 2015-05-26 | Honeywell International Inc. | Preparation of fluorinated olefins via catalytic dehydrohalogenation of halogenated hydrocarbons |
| US7884254B2 (en) | 2007-08-08 | 2011-02-08 | Honeywell International Inc. | Dehydrochlorination of hydrochlorofluorocarbons using pre-treated activated carbon catalysts |
| US8044250B2 (en) * | 2007-11-16 | 2011-10-25 | Honeywell International Inc. | Manufacture of 1,1,1,2,3,3-hexafluoropropane and 1,1,1,2-tetrafluoropropane via catalytic hydrogenation |
| JP4849058B2 (ja) | 2007-11-21 | 2011-12-28 | ダイキン工業株式会社 | 含フッ素オレフィンの製造方法 |
| EP2292574B1 (en) | 2007-12-27 | 2015-05-06 | Arkema France | Process for producing 2,3,3,3-tetrafluoropropene |
| GB0801209D0 (en) | 2008-01-23 | 2008-02-27 | Ineos Fluor Holdings Ltd | Process |
| JP5136111B2 (ja) * | 2008-02-19 | 2013-02-06 | セントラル硝子株式会社 | フルオロオレフィンとフッ化水素の分離方法 |
| US8710282B2 (en) | 2008-03-14 | 2014-04-29 | Honeywell International Inc. | Integrated process for the manufacture of fluorinated olefins |
| FR2929272B1 (fr) | 2008-03-28 | 2010-04-09 | Arkema France | Procede pour la preparation du 2,3,3,3-tetrafluoro-1-propene |
| CA3093427A1 (en) | 2008-05-07 | 2009-11-12 | The Chemours Company Fc, Llc | Compositions comprising 1,1,1,2,3-pentafluoropropane or 2,3,3,3-tetrafluoropropene |
| GB0808836D0 (en) * | 2008-05-15 | 2008-06-18 | Ineos Fluor Ltd | Process |
| FR2935703B1 (fr) * | 2008-09-11 | 2010-09-03 | Arkema France | Procede de preparation de composes fluores. |
| US8609909B2 (en) * | 2009-01-30 | 2013-12-17 | Honeywell International Inc. | Process for the purification of hydrofluoroolefins |
| US8487145B2 (en) | 2009-06-03 | 2013-07-16 | E I De Pont De Nemours And Company | Catalysts and process to manufacture 2,3,3,3-tetrafluoropropene |
| CN102448921B (zh) * | 2009-06-03 | 2014-04-23 | 纳幕尔杜邦公司 | 制备2,3,3,3-四氟丙烯的催化剂和方法 |
| FR2948362B1 (fr) * | 2009-07-23 | 2012-03-23 | Arkema France | Procede de preparation de composes fluores |
| FR2948361B1 (fr) | 2009-07-23 | 2011-08-05 | Arkema France | Procede de preparation de composes fluores |
| CN101628850B (zh) * | 2009-08-14 | 2012-09-19 | 西安近代化学研究所 | Z-1,2,3,3,3-五氟丙烯的制备方法 |
| CN101628849B (zh) * | 2009-08-14 | 2012-10-10 | 西安近代化学研究所 | Z-1,2,3,3,3-五氟丙烯的制备方法 |
| US8129574B2 (en) * | 2009-08-31 | 2012-03-06 | Honeywell International Inc. | Hydrogenation process for fluorocarbons |
| US8158549B2 (en) | 2009-09-04 | 2012-04-17 | Honeywell International Inc. | Catalysts for fluoroolefins hydrogenation |
| EP2558544B2 (en) | 2010-04-16 | 2024-09-04 | The Chemours Company FC, LLC | Chillers containing a composition comprising 2,3,3,3-tetrafluoropropene and 1,1,1,2-tetrafluoroethane |
| US20110269999A1 (en) | 2010-04-29 | 2011-11-03 | Honeywell International Inc. | Process for dehydrohalogenation of halogenated alkanes |
| US8436218B2 (en) * | 2010-05-27 | 2013-05-07 | Honeywell International Inc. | Azeotrope-like composition of hexafluoropropane, hexafluoropropene and hydrogen fluoride |
| US8513474B2 (en) * | 2010-06-24 | 2013-08-20 | Honeywell International Inc. | Process for the manufacture of fluorinated olefins |
| FR2962130B1 (fr) * | 2010-06-30 | 2012-07-20 | Arkema France | Composition a base de 2,3,3,3-tetrafluoropropene |
| FR2962442B1 (fr) | 2010-07-09 | 2016-02-26 | Arkema France | Composition stable de 2,3,3,3-tetrafluoropropene |
| JP5434970B2 (ja) * | 2010-07-12 | 2014-03-05 | セントラル硝子株式会社 | ドライエッチング剤 |
| US8680345B2 (en) | 2011-01-07 | 2014-03-25 | Honeywell International Inc. | Low temperature production of 2-chloro-3,3,3-trifluoropropene |
| CN103328424A (zh) | 2011-02-04 | 2013-09-25 | 旭硝子株式会社 | 2,3,3,3-四氟丙烯的纯化方法 |
| TW201247315A (en) | 2011-05-16 | 2012-12-01 | Du Pont | Catalytic hydrogenation of fluoroolefins, alpha-alumina supported palladium compositions and their use as hydrogenation catalysts |
| JP6034653B2 (ja) * | 2012-10-22 | 2016-11-30 | 住友精化株式会社 | パラフィンの製造方法 |
| CN103073386B (zh) * | 2012-12-31 | 2014-08-20 | 浙江衢化氟化学有限公司 | 一种2,3,3,3-四氟丙烯的制备方法 |
| US9272968B2 (en) | 2013-03-14 | 2016-03-01 | Honeywell International Inc. | Process to suppress the formation of 3,3,3-trifluoropropyne in fluorocarbon manufacture |
| US9290424B2 (en) * | 2013-03-14 | 2016-03-22 | Honeywell International Inc. | Processes for the hydrogenation of halogenated alkenes and the manufacture of fluorinated olefins |
| KR101500655B1 (ko) | 2013-09-17 | 2015-03-12 | 한국과학기술연구원 | 금속불소화물 촉매 제조방법 및 이를 이용한 탈불산화 방법 |
| CN110845295A (zh) | 2014-08-14 | 2020-02-28 | 科慕埃弗西有限公司 | 通过脱氟化氢来制备E-1,3,3,3-四氟丙烯(HFC-1234ze)的方法 |
| FR3033791B1 (fr) | 2015-03-18 | 2017-04-14 | Arkema France | Stabilisation du 1-chloro-3,3,3-trifluoropropene |
| JP6623561B2 (ja) * | 2015-05-29 | 2019-12-25 | ダイキン工業株式会社 | 含フッ素オレフィンの製造方法 |
| KR102580755B1 (ko) | 2015-06-05 | 2023-09-19 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | 하이드로플루오로올레핀 및 이의 사용 방법 |
| GB2540427B (en) | 2015-07-17 | 2017-07-19 | Mexichem Fluor Sa De Cv | Process for the preparation of 2,3,3,3-tetrafluoropropene (1234yf) |
| GB201512510D0 (en) | 2015-07-17 | 2015-08-19 | Mexichem Fluor Sa De Cv | Process |
| KR20160017661A (ko) | 2016-01-29 | 2016-02-16 | 한국과학기술연구원 | 헥사플루오로프로필렌으로부터 2,3,3,3-테트라플루오로프로펜과 1,3,3,3-테트라플루오로프로펜의 제조방법 |
| KR101914236B1 (ko) | 2017-01-11 | 2018-12-28 | 한국과학기술연구원 | 1,2,3,3,3-펜타플루오로프로펜 제조방법 |
| JP6624126B2 (ja) * | 2017-03-13 | 2019-12-25 | ダイキン工業株式会社 | 含フッ素オレフィンの製造方法 |
| CN119118781A (zh) | 2018-06-06 | 2024-12-13 | 霍尼韦尔国际公司 | 用于HCFC-244bb的脱氯化氢以制备HFO-1234yf的方法 |
| CN113195443A (zh) * | 2018-12-19 | 2021-07-30 | 大金工业株式会社 | 氟代乙烷的制造方法和氟代烯烃的制造方法 |
| JP6835060B2 (ja) * | 2018-12-27 | 2021-02-24 | ダイキン工業株式会社 | シクロブテンの製造方法 |
| CN111558386B (zh) * | 2020-04-10 | 2023-05-12 | 西安近代化学研究所 | 气相氟化合成氢氟烯烃用氟化铁基催化剂及其制备方法和应用 |
| ES3063120T3 (en) | 2021-04-15 | 2026-04-15 | Zhejiang Res Institute Of Chemical Industry Co Ltd | Method for preparing 2,3,3,3-tetrafluoropropene |
| CN116874345B (zh) * | 2023-09-06 | 2024-08-06 | 北京宇极科技发展有限公司 | 气相连续制备e-1,1,1,4,5,5,5-七氟-4-(三氟甲基)戊-2-烯的方法 |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4033899A (en) * | 1974-12-30 | 1977-07-05 | Texaco Inc. | Alkylation fluosulfonic-sulfuric acid catalyst recovery process with silica-alumina removal of fluoride compounds |
| US4138355A (en) * | 1976-08-10 | 1979-02-06 | Halocarbon Products Corporation | Fluorine substitution in 1,1,1-trihalomethanes |
| US5334783A (en) * | 1988-03-14 | 1994-08-02 | Hoechst Aktiengesellschaft | Process for the preparation of hexafluoropropene |
| RU2026279C1 (ru) * | 1989-02-03 | 1995-01-09 | Е.И.Дюпон Де Немур Энд Компани | Способ гидрогенолиза и/или дегидрогалогенирования фторгалоуглеродов и/или фторгалоуглеводородов |
| US5180860A (en) * | 1990-06-28 | 1993-01-19 | E. I. Du Pont De Nemours And Company | Dehydrohalogenation process |
| US5171902A (en) * | 1990-10-11 | 1992-12-15 | E. I. Du Pont De Nemours And Company | Saturated linear polyfluorohydrocarbons, processes for their production, and their use in cleaning compositions |
| US5268122A (en) | 1991-08-28 | 1993-12-07 | E. I. Du Pont De Nemours And Company | Gem-dihydropolyfluoroalkanes and monohydropolyfluoroalkenes, processes for their production, and use of gem-dihydropolyfluoroalkanes in cleaning compositions |
| US5459275A (en) * | 1991-12-28 | 1995-10-17 | Ricoh Company, Ltd. | Pyrenylamine derivatives |
| US5679875A (en) | 1992-06-05 | 1997-10-21 | Daikin Industries, Ltd. | Method for manufacturing 1,1,1,2,3-pentafluoropropene 1,1,1,2,3-pentafluoropropane |
| US5396000A (en) | 1993-05-24 | 1995-03-07 | E. I. Du Pont De Nemours And Company | Process for the manufacture of 1,1,1,2,3,-pentafluoropropane |
| US5488189A (en) * | 1993-12-14 | 1996-01-30 | E. I. Du Pont De Nemours And Company | Process for fluorinated propanes and pentanes |
| DE4421702A1 (de) * | 1994-06-21 | 1996-01-04 | Bayer Ag | Verfahren zur Herstellung von 1,1,1,4,4,4-Hexafluorbutan in flüssiger Phase |
| JP3543863B2 (ja) * | 1994-12-16 | 2004-07-21 | ダイキン工業株式会社 | 1,1,1,2,3,3−ヘキサフルオロプロパンの製造方法 |
| US5895639A (en) * | 1996-07-03 | 1999-04-20 | Alliedsignal Inc. | Separation of hydrogen fluoride from a fluorocarbon/hydrogen fluoride azeotropic mixture by sulfuric acid |
| US5710352A (en) * | 1996-09-19 | 1998-01-20 | Alliedsignal Inc. | Vapor phase process for making 1,1,1,3,3-pentafluoropropane and 1-chloro-3,3,3-trifluoropropene |
| US6369284B1 (en) * | 1997-01-31 | 2002-04-09 | E. I. Du Pont De Nemours And Company | Catalytic manufacture of pentafluoropropenes |
| US5945573A (en) * | 1997-01-31 | 1999-08-31 | E. I. Du Pont De Nemours And Company | Process for the manufacture of 1,1,1,3,3-pentafluoropropane |
| US5986151A (en) * | 1997-02-05 | 1999-11-16 | Alliedsignal Inc. | Fluorinated propenes from pentafluoropropane |
| US6031141A (en) * | 1997-08-25 | 2000-02-29 | E. I. Du Pont De Nemours And Company | Fluoroolefin manufacturing process |
| EP1043297B8 (en) * | 1997-12-26 | 2005-09-07 | Zeon Corporation | Process for the preparation of compounds having -ch2-chf- groups |
| JP2001240569A (ja) * | 2000-02-29 | 2001-09-04 | Nippon Zeon Co Ltd | −ch2−chf−基を有する化合物の製造方法 |
| US6548719B1 (en) | 2001-09-25 | 2003-04-15 | Honeywell International | Process for producing fluoroolefins |
| US7230146B2 (en) * | 2003-10-27 | 2007-06-12 | Honeywell International Inc. | Process for producing fluoropropenes |
| US7405334B2 (en) * | 2003-05-23 | 2008-07-29 | E. I. Du Pont De Nemours And Company | Process for the reduction of acidic contaminates in fluorinated hydrocarbons |
| US7371363B2 (en) * | 2003-07-15 | 2008-05-13 | Honeywell International Inc. | Methods of purifying hydrogen fluoride |
| WO2005037743A1 (en) * | 2003-10-14 | 2005-04-28 | E.I. Dupont De Nemours And Company | Process for the preparation of 1,1,1,3,3-pentafluoropropane and 1,1,1,2,3-pentafluoropropane |
| KR101125467B1 (ko) * | 2003-10-14 | 2012-03-27 | 이 아이 듀폰 디 네모아 앤드 캄파니 | 1,1,1,3,3-펜타플루오로프로판 및1,1,1,3,3,3-헥사플루오로프로판의 제조 방법 |
| MXPA06012466A (es) * | 2004-04-29 | 2007-01-31 | Honeywell Int Inc | Procesos para la sintesis de 1,3,3,3-tetrafluoropropeno y 2,3,3,3-tetrafluoropropeno. |
| CN1972887B (zh) * | 2004-04-29 | 2010-10-13 | 霍尼韦尔国际公司 | 1,3,3,3-四氟丙烯的合成方法 |
| US7371904B2 (en) * | 2004-04-29 | 2008-05-13 | Honeywell International Inc. | Processes for synthesis of 1,3,3,3-tetrafluoropropene |
| US7897823B2 (en) * | 2004-10-29 | 2011-03-01 | E. I. Du Pont De Nemours And Company | Process for production of azeotrope compositions comprising hydrofluoroolefin and hydrogen fluoride and uses of said azeotrope compositions in separation processes |
| US20060106262A1 (en) * | 2004-11-18 | 2006-05-18 | Optimer Photonics, Inc. | Electrooptic chromophores with large optical birefringence for applications at high speed and short wavelengths |
| US8044250B2 (en) † | 2007-11-16 | 2011-10-25 | Honeywell International Inc. | Manufacture of 1,1,1,2,3,3-hexafluoropropane and 1,1,1,2-tetrafluoropropane via catalytic hydrogenation |
-
2006
- 2006-10-27 US US11/588,464 patent/US7560602B2/en active Active
-
2007
- 2007-10-26 RU RU2008122213/04A patent/RU2457195C2/ru not_active IP Right Cessation
- 2007-10-26 EP EP07863530.7A patent/EP2076478B2/en active Active
- 2007-10-26 KR KR1020147017940A patent/KR20140090702A/ko not_active Ceased
- 2007-10-26 CN CN2007800400996A patent/CN101553453B/zh active Active
- 2007-10-26 WO PCT/US2007/082601 patent/WO2008057794A1/en not_active Ceased
- 2007-10-26 ES ES07863530T patent/ES2394870T5/es active Active
- 2007-10-26 KR KR1020087012977A patent/KR20090071510A/ko not_active Ceased
- 2007-10-26 JP JP2009534879A patent/JP5588174B2/ja active Active
-
2009
- 2009-02-19 US US12/389,110 patent/US7786333B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2076478B2 (en) | 2020-11-25 |
| ES2394870T3 (es) | 2013-02-06 |
| JP5588174B2 (ja) | 2014-09-10 |
| KR20090071510A (ko) | 2009-07-01 |
| WO2008057794A1 (en) | 2008-05-15 |
| CN101553453A (zh) | 2009-10-07 |
| EP2076478A1 (en) | 2009-07-08 |
| RU2457195C2 (ru) | 2012-07-27 |
| RU2008122213A (ru) | 2010-04-27 |
| KR20140090702A (ko) | 2014-07-17 |
| US20090209791A1 (en) | 2009-08-20 |
| US7786333B2 (en) | 2010-08-31 |
| EP2076478B1 (en) | 2012-10-03 |
| CN101553453B (zh) | 2013-05-29 |
| JP2010508294A (ja) | 2010-03-18 |
| US7560602B2 (en) | 2009-07-14 |
| US20070179324A1 (en) | 2007-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2394870T5 (es) | Procedimiento para la fabricación de olefinas fluoradas | |
| ES2355541T5 (es) | Proceso de co-producción de HFC-1225ye y HFC-1234yf por reacciones de 4 pasos | |
| JP5732062B2 (ja) | フルオロオレフィンを製造するための統合方法 | |
| JP6013353B2 (ja) | 2,3,3,3−テトラフルオロプロペンの製造法 | |
| ES2705051T3 (es) | Procedimiento para la fabricación de olefinas fluoradas |