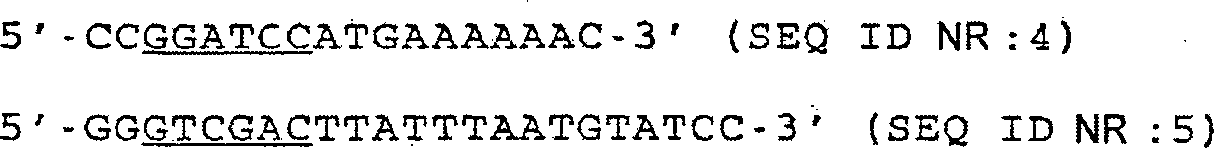-
GEBIET DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft ein Verfahren zum Spalten von DNA,
umfassend das Reagieren der DNA mit einer ortsspezifischen Endonuclease,
die eine spezifische Nucleotidsequenz erkennt.
-
HINTERGRUND DER ERFINDUNG
-
Die
Endonuclease ist eine Nuclease (Nucleinsäure abbauendes Enzym), das
die Phosphodiesterbindung einer Polynucleotidkette hydrolysiert.
Die Endonuclease erkennt und bindet an eine spezifische Nucleotidsequenz
entlang von DNA-Molekülen,
wobei die Moleküle
innerhalb der Erkennungssequenz gespalten werden.
-
Die
Endonuclease ist ein notwendiges Enzym für die heutigen fortschrittlichen
gentechnischen Verfahren zur Clonierung und Analyse von Genen.
-
Von
einer ortsspezifischen Endonuclease Endo. SceI (hier nachstehend
auch als „Scel" bezeichnet) aus
einem eukaryontischen Mikroorganismus (z.B. Hefe) ist bekannt, dass
sie ein Heterodimer ist, die Untereinheiten mit 75 kDa und 50 kDa
besitzt. Die Untereinheiten von Scel sowie Gene, die die Untereinheiten
codieren, wurden cloniert, und deren Nucleotidsequenzen wurden bestimmt
(für die
75 kDa-Untereinheit vgl. Morishima, N. et al., J. Biol. Chem. 265,
15189–15197
(1990) und für
die 50 kDA-Untereinheit vgl. JP-B-7-77556).
-
Um
die vorstehend beschriebene Endonuclease zur künstlichen Modifikation eines
biochemischen Mittels, eines Gens oder dergleichen in großem Maßstab zu
nutzen, muss eine Massenproduktion der Endonuclease mit einem Genexpressionssystem
erfolgen. Die Endonuclease funktioniert erst, wenn sie eine spezifische Nucleotidsequenz
erkennt, d.h. die Endonuclease muss für die zu erkennende Nucleotidsequenz
spezifisch sein.
-
Die
50 kDa-Untereinheit der vorstehend beschriebenen Endonuclease Scel
wird von den Mitochondriumgenomen der Hefe (Saccharomyces cerevisiae)
codiert. Ein Gen eines Hefe-Mitochondriumgenoms enthält für Mitochondrien
einzigartige Codons, die sich von den Aminosäurecodons (Universalcodons)
unterscheiden, die in Genexpressionssystemen aus Organismen verwendet
werden, die im Allgemeinen zur Proteinexpression (E. coli, Baculovirus,
Hefe etc.) im großem
Maßstab
verwendet werden. Falls dieses Gen des Mitochondriumgenoms direkt
verwendet wird, produziert das Proteinexpressionssystem kaum ein
Protein einer ursprünglichen
Aminosäuresequenz.
Während
beispielsweise TGA als Universalcodon ein Stoppcodon ist, ist es in
Mitochondrien ein unterschiedliches Codon, das eine andere Aminosäure (Trp)
codiert. Ein Gen kann in Mitochondrien normal exprimiert werden,
aber die Expression desselben Gens kann aufgrund einer vom Stoppcodon
verursachten unvollständigen Übersetzung
in einem allgemeinen Expressionssystem wie E. coli zu einem unvollständigen Protein
führen.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Die
vorliegende Erfindung zielt auf die Bereitstellung eines Verfahrens
zum Spalten von DNA ab, umfassend das Reagieren der DNA mit einer
ortsspezifischen Endonuclease, die eine spezifische Nucleotidsequenz
erkennt.
-
Die
hier genannten Erfinder haben intensive Studien betrieben, um die
vorstehend beschriebenen Probleme zu lösen. Sie waren folglich bei
der Bereitstellung eines Verfahrens zum Spalten von DNA erfolgreich, das
das Reagieren der DNA mit einer ortsspezifischen Endonuclease umfasst,
die eine spezifische Nucleotidsequenz erkennt, wobei die vorliegende
Erfindung vollendet wurde.
-
Demzufolge
betrifft die vorliegende Erfindung ein Verfahren zum Spalten von
DNA, die die Sequenz aus 26 Nucleotiden: GCCCAGACATATCCCTGAATGATACC
umfasst, umfassend das Reagieren der DNA mit:
- (i)
einem Endonucleaseprotein, das die in SEQ ID NR: 3 dargestellte
Aminosäuresequenz
umfasst; oder
- (ii) einem Endonucleaseprotein, das eine von SEQ ID NR: 3 durch
Substitution von Gly mit Lys bei Aminosäure 217 und Asn mit Asp bei
Aminosäure
346 abgeleitete Aminosäuresequenz
umfasst,
wobei die DNA innerhalb der Sequenz aus 26 Nucleotiden:
GCCCAGACATATCCCTGAATGATACC gespalten wird, um gestufte Enden zu
erzeugen. Die Beschreibung der vorliegenden Erfindung verwendet
eine Endonuclease, die die Nucleotidsequenz: GCCCAGACATATCCCTGAATGATACC
erkennen kann.
-
Ferner
wird in der Beschreibung auch ein rekombinantes Protein von entweder
(a) oder (b) beschrieben:
- (a) ein Protein,
das die in SEQ ID NR: 3 dargestellte Aminosäuresequenz umfasst; oder
- (b) ein Protein mit einer Endonucleaseaktivität zur Erkennung
der Nucleotidsequenz: GCCCAGACATATCCCTGAATGATACC, wobei das Protein
mindestens eine Deletion, Substitution oder Addition der Aminosäure in der
in SEQ ID NR: 3 dargestellten Aminosäuresequenz umfasst.
-
Außerdem wird
in der Beschreibung auch ein Gen beschrieben, das das rekombinante
Protein von entweder a oder b codiert:
- (a)
ein Protein, das die in SEQ ID NR: 3 dargestellte Aminosäuresequenz
umfasst; oder
- (b) ein Protein mit einer Endonucleaseaktivität zur Erkennung
der Nucleotidsequenz: GCCCAGACATATCCCTGAATGATACC, wobei das Protein
mindestens eine Deletion, Substitution oder Addition der Aminosäure in der
in SEQ ID NR: 3 dargestellten Aminosäuresequenz umfasst.
-
Zudem
wird in der Beschreibung ein Gen beschrieben, das DNA von entweder
(c) oder (d) enthält:
- (c) DNA, das eine in SEQ ID NR: 2 dargestellte
Nucleotidsequenz umfasst; oder
- (d) DNA, die ein Protein mit einer Endonucleaseaktivität zur Erkennung
der Nucleotidsequenz: GCCCAGACATATCCCTGAATGATACC codiert, wobei
die DNA unter stringenten Bedingungen mit DNA hybridisieren kann,
die die in SEQ ID NR: 2 dargestellte Nucleotidsequenz umfasst.
-
Außerdem wird
in der Beschreibung ein rekombinanter Vektor beschrieben, der das
vorstehend beschriebene Gen umfasst.
-
Zusätzlich wird
in der Beschreibung eine Transformante beschrieben, die den vorstehend
beschriebenen rekombinanten Vektor umfasst.
-
Außerdem wird
in der Beschreibung ein Verfahren zur Herstellung der Endonuclease
beschrieben, umfassend die Schritte der Züchtung der vorstehend beschriebenen
Transformante; und das Gewinnen einer Endonuclease aus einer Kultur,
die die Nucleotidsequenz: GCCCAGACATATCCCTGAATGATACC erkennen kann.
-
In
der Beschreibung wird auch eine Endonuclease beschrieben, die durch
das Verfahren hergestellt wird.
-
Schließlich wird
in der Beschreibung ein Kit beschrieben, der die Endonuclease und/oder
das rekombinante Protein und/oder das Gen und/oder den rekombinanten
Vektor und/oder die vorstehend erwähnte Transformante umfasst.
-
Die
Bestandteile des Kits können
in Behältern
wie Phiolen, fakultativ in Puffern und/oder Lösungen, verpackt werden. Gegebenenfalls
können
ein oder mehrere Bestandteile in ein und denselben Behälter verpackt
werden.
-
Es
sollte angemerkt werden, dass ausschließlich das Verfahren, wie in
Patentanspruch 1 definiert, Teil der Erfindung ist.
-
KURZE BESCHREIBUNG
DER ZEICHNUNGEN
-
1 stellt
die Aminosäuresequenz
einer Endonuclease vor und nach der Modifikation dar;
-
2 stellt
die Schritte zur Konstruktion von Plasmid pY673L dar;
-
3 stellt
die Schritte zur Konstruktion der Plasmide pEN1.7 und pEN0.5 dar;
-
4 stellt
die Nucleotidsequenz des Gens der 50 kDa-Untereinheit von Scel dar,
das modifiziert wurde, so dass sie mit dem Universalcode übereinstimmt;
-
5A und 5B sind
Photos der Elektrophorese, die die sequenzspezifischen Endonucleaseaktivitäten der
50 kDa-Untereinheit der modifizierten Scel zeigen;
-
6A und 6B stellen
den Substitutionsort der 50 kDa-Untereinheit aus Saccharomyces uvarum und
die für
die Substitution verwendeten Oligonucleotide dar; und
-
7A und 7B sind
Photos der Elektrophorese, die die sequenzspezifischen Endonucleaseaktivitäten der
50 kDa-Untereinheit aus Saccharomyces uvarum zeigen.
-
DETAILLIERTE
BESCHREIBUNG
-
Die
DNA des Mitochondriumgenoms, das die kleinere Untereinheit (50 kDa)
einer Endonuclease aus Hefe codiert, kann in großem Maßstab exprimiert werden, indem
ein Proteinexpressionssytem wie E. coli oder Hefe verwendet wird.
Bei der Vollendung dieses Ziels können die für Mitochondrien einzigartigen
Codons in Universalcodons in einem Gen, das eine Aminosäuresequenz
der kleineren Untereinheit codiert, modifiziert werden. In der Beschreibung
wird eine derartige modifizierte, kleinere Untereinheit erwähnt, die
26 Basenpaare der spezifischen Nucleotidsequenz erkennen und spalten
kann.
-
Eine
Endonuclease, wie hier beschrieben (d.h. die 50 kDa-Untereinheit
einer Endonuclease aus Hefe, hier nachstehend auch als „Endo.
SceI-50 kDa" bezeichnet),
wird wie folgt hergestellt.
-
(1) Konstruktion der mutierten
Aminosäure
und Einführung
der Mutation
-
Die
kleinere Untereinheit der Endonuclease Scel aus Saccharomyces cerevisiae
oder die kleinere Untereinheit der Endonuclease SuvI aus Saccharomyces
uvarum wird als Ziel zur Einführung
einer Mutation verwendet. Die kleineren Untereinheiten sowohl von
Scel als auch von SuvI besitzen ein Molekulargewicht von 50 kDa.
Jedoch unterscheiden sie sich dadurch voneinander, dass sie zwei
unterschiedliche Aminosäuren
besitzen (6A).
-
Das
Gen, das die Untereinheit (50 kDa) von Scel (hier nachstehend als „ENS2" bezeichnet) codiert, wird
von einem Mitochondriumgenom codiert und enthält daher genetische Codes,
die für
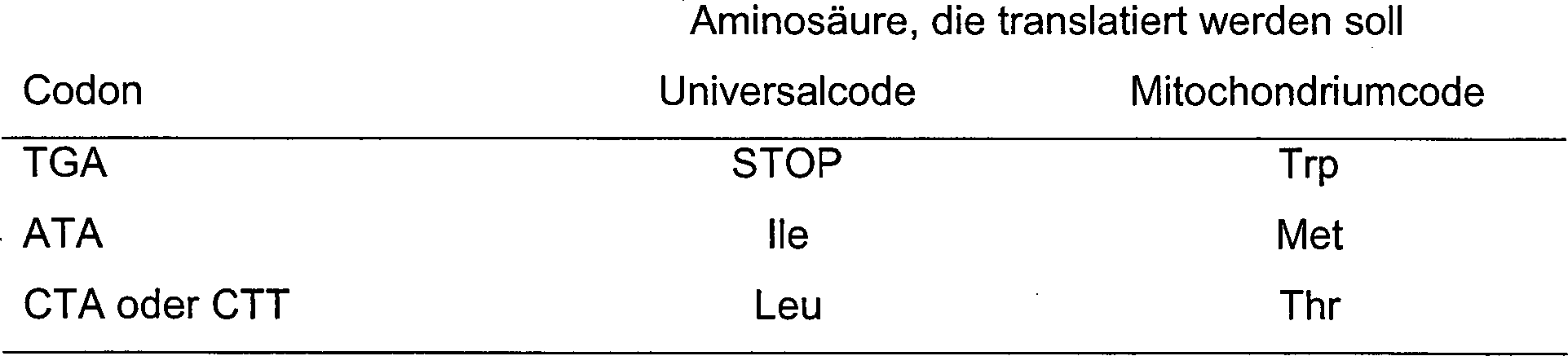
Mitochondrien einzigartig sind (Tabelle 1).
-
Tabelle
1 Unterschied
zwischen dem Mitochondriumcode und Universalcode
-
Die
Nucleotidsequenz von ENS2 ist bekannt (JP-B-7-77556; Nakagawa, K.,
Morishima, N. und Shibata, T., J. Biol. Chem. 266, 1977–1984 (1991)).
Wenn ENS2 in einem allgemeinen Expressionssystem wie E. coli nach
dem Universalcode exprimiert wird, wird die Translation bei TGA
unterbrochen, das als Stoppcodon gelesen wird, wie aus Tabelle 1
ersichtlich ist (beispielsweise schließt die in JP-B-7-77556 beschriebene
ENS2 ein TGA-Stoppcodon an den Nucleotiden 97–99 ein). Während ATA nach dem Universalcode
als Ile gelesen wird, wird es nach dem Mitochondriumcode als Met
gelesen.
-
Um
ein Expressionssystem im normalen Maßstab für ENS2 zu konstruieren, ist
es notwendig, den genetischen Code von ENS2 zu modifizieren, so
dass die Aminosäuresequenz,
die bei der Expression in einem allgemeinen Expressionssystem (z.B.
E. coli) erhalten wird, mit einer Aminosäuresequenz identisch ist, wie
sie in einem Mitochondrium-Expressionssystem exprimiert wird. Daher
wird ein Codon für
Trp (TGA) nach dem Mitochondriumcode (Tabelle 1) durch ein Codon
(TGG) substituiert, das nach dem Universalcode in Trp translatiert
wird. Eine derartige Substitution wird auch bei ATA sowie CTA und
CTT angewandt, die nach dem Universalcode in Ile bzw. Leu translatiert
werden (Tabelle 1). Es gibt keinen Bedarf, andere degenerierende
Codons zu substituieren, die nach dem Universalcode Ile oder Leu
codieren.
-
Grundsätzlich gibt
es innerhalb der Aminosäuresequenz
der kleineren Untereinheit von Scel (476 Aminosäuren) 37 Aminosäuren als
Kandidaten für
Modifikationen. Ihre Positionen sind in 1 und Tabelle
2 dargestellt. Was die Endonuclease aus Saccharomyces uvarum (SuvI)
anbelangt, sind Gly bei 217 und Asn bei 346 (1) zusätzlich durch
Lys bzw. Asp substituiert, so dass insgesamt 39 Aminosäuren modifiziert
sind. Es ist nicht notwendig, alle der vorstehenden 37 oder 39 Aminosäuren zu
substituieren. Die Anzahl der Substitutionen kann 36 oder 35 oder
weniger betragen. Translationen in Codons, die für Mitochondrien einzigartig
sind, können
unvollständig
sein, solange die nachstehend erwähnten 26 Nucleotide (SEQ ID
NR: 1) von der Nuclease erkannt werden. Die Positionen der Substitution
sind nachstehend in Tabelle 2 zusammengefasst.
-
-
Die
Substitution der Aminosäuren
erfolgt durch Substitution der Nucleotidsequenz des Gens, das die Aminosäuren codiert,
durch eine andere Nucleotidsequenz (ortsgerichtete Mutagenese).
Beispiele der Mutagenese schließen
ein, sind aber nicht eingeschränkt
auf das Verfahren der ortsgerichteten Mutagenese von T. Kunkel (Kunkel,
T. A., Proc. Natl. Acad. Sci. USA. 82, 488–492 (1985)) und das Gapped-Duplex-Verfahren.
Es gibt auch eine modifizierte Version des Kunkel-Verfahrens, bei dem
gleichzeitig maximal 16 Oligonucleotide zur Modifikation verwendet
werden (anstatt der Verwendung von 1 oder 2 Oligonucleotiden wie
beim herkömmlichen
Kunkel-Verfahren), um eine Vielzahl von Positionen effizient zu
substituieren. Eine Mutation kann unter Verwendung eines Kits zur
Einführung
von Mutationen (beispielsweise Mutant-K (Takara Shuzo, Co., Ltd.) oder
Mutant-G (Takara Shuzo, Co., Ltd.)), der die ortsgerichtete Mutagenese
nutzt, oder unter Verwendung eines in vitro-LA-PCR-Mutagenesereihen-Kits
(Takara Shuzo, Co., Ltd.) eingeführt
werden.
-
Die
Oligonucleotide werden unter Verwendung der Nucleotidsequenz von
ENS2 als Matrize (1431 Basenpaare: Nakagawa, K. et al., J. Biol.
Chem. 266, 1977–1984
(1991); JP-B-7-77556) konstruiert und synthetisiert, so dass mindestens
eine Base, die mit der Mutation eingeführt werden soll, von etwa 8
bis 30 Basen flankiert wird (wobei jedes Oligonucleotid insgesamt
etwa 18 bis 60 Basen besitzt). Die Oligonucleotide können durch
chemische Synthese unter Verwendung eines herkömmlichen Synthesegeräts erhalten
werden.
-
(2) Herstellung des Endonucleasegens,
das mit der Mutation eingeführt
wurde
-
Jedes
der, wie vorstehend in (1) beschrieben, erhaltenen Oligonucleotide
ist am 5'-Ende phosphoryliert,
wird unter Verwendung von ENS2 als Matrize synthetisiert und Ligierungsreaktionen
unterzogen. Diese Reaktionen können
unter Verwendung von T4-Polynucleotidkinase (Takara Shuzo, Co.,
Ltd.), T4 DNA-Polymerase (Takara Shuzo, Co., Ltd.), T4-DNA-Ligase
(Takara Shuzo, Co., Ltd.) oder dergleichen erfolgen.
-
Die
Nucleotidsequenz der so erhaltenen DNA wird bestimmt. Die Bestimmung
der Nucleotidsequenz kann nach einem bekannten Verfahren wie dem
chemischen Maxam-Gilbert-Modifikationsverfahren oder einem Didesoxykettenabbruchverfahren
unter Verwendung des M13-Phagen durchgeführt werden. Im Allgemeinen
wird die Sequenz unter Verwendung eines automatischen Nucleotidsequenziergeräts (z.B.
ALF (Pharmacia), 373A-DNA-Sequenziergerät (Perkin-Elmer) etc.) bestimmt.
-
Die
SEQ ID NR: 2 und 3 geben die Nucleotidsequenz eines Gens, das eine
Endonuclease codiert, und die Aminosäuresequenz einer Endonuclease
beispielhaft an, die im Verfahren der vorliegenden Erfindung verwendet
werden. Die Endonuclease erlangt die wesentliche Funktion der Endonuclease
Scel oder SuvI, d.h. die Funktion, die Consensussequenz „CANRYNNANNCYYGTTW" und eine dazu ähnliche
Sequenz zu erkennen, indem sie an die größere Untereinheit der natürlichen
Endonuclease bindet. Die Endonuclease übt die Funktion der kleineren
Einheit der natürlichen
Endonuclease aus und kann die 26 Basen spezifisch erkennen, die durch
GCCCAGACATATCCCTGAATGATACC (SEQ ID NR: 1) dargestellt werden.
-
Die
Endonuclease kann die Mutationen Gly217Lys und Asn346Asp einschließen und
immer noch die vorstehenden 26 Basen (SEQ ID NR: 1) erkennen.
-
Der
Ausdruck „kann
erkennen", wie hier
verwendet, bezieht sich auf die Funktion der Endonuclease, an eine
Stelle der 26 Basen innerhalb des Gens zu binden und das Gen zu
spalten, so dass die 26 Basenpaare in zwei Fragmente mit gestuften
Enden getrennt werden.
-
Die
DNA, die mit dem vorstehenden Gen (SEQ ID NR: 2) unter stringenten
Bedingungen hybridisieren kann, kann auch in das Gen eingeschlossen
werden, das die Endonuclease codiert. Die stringenten Bedingungen
sind beispielsweise eine Natriumkonzentration von 15 bis 900 mM
und eine Temperatur von 37 bis 70°C, vorzugsweise
68°C.
-
(3) Herstellung und Transformation
des rekombinanten Vektors
-
(i) Herstellung des rekombinanten
Vektors
-
Ein
rekombinanter Vektor kann erhalten werden, indem das Gen, das die
Endonuclease codiert, in einen geeigneten Vektor ligiert (eingebaut)
wird. Der Vektor zum Einbau des Gens ist nicht auf einen Spezifischen
begrenzt, solange er in einer Wirtszelle replizierbar ist. Beispiele
eines derartigen Vektors schließen
ein, sind aber nicht auf Plasmid-DNA und Phagen-DNA begrenzt.
-
Die
Plasmid-DNA ist beispielsweise ein Plasmid aus E. coli (z.B. pRSET,
pTZ19R, pBR322, pBR325, pUC118, pUC119 etc.), ein Plasmid aus Bacillus
(z.B. pUB110, pTP5 etc.) oder ein Plasmid aus Hefe (z.B. YEp13,
YEp24, YCp50 etc.). Die Phagen-DNA ist beispielsweise der λ-Phage oder
dergleichen. Auf ähnliche Weise
kann ebenfalls ein tierischer Virusvektor wie ein Retrovirus- oder Vacciniavirus-Vektor
oder ein Insektenvirusvektor wie ein Baculovirusvektor verwendet
werden.
-
Um
das die Endonuclease codierende Gen in den Vektor einzubauen, wird
die gereinigte DNA mit einem geeigneten Restriktionsenzym gespalten.
Dann wird das gespaltene Fragment in die Restriktionsstelle oder
eine Multiclonierungsstelle der geeigneten Vektor-DNA eingebaut.
-
Das
Gen sollte in den Vektor eingebaut werden, so dass das Gen funktionieren
kann. Falls gewünscht, kann
der Vektor andere Gene als das Gen und den Promotor einschließen, beispielsweise
ein cis-Element (z.B. ein Enhancerelement), ein Spleiß-Signal,
ein Poly(A)-Schwanz-Signal, einen Selektionsmarker und eine Ribosomen-bindende
Sequenz (SD-Sequenz). Beispiele des Selektionsmarkers schließen das
Dihydrofolatreduktasegen, das Ampicillin-Resistenzgen und das Neomycin-Resistenzgen
ein.
-
(ii) Herstellung der Transformante
-
Eine
Transformante kann erhalten werden, indem der rekombinante Vektor
in eine Wirtszelle so eingeführt
wird, dass das Gen von Interesse exprimiert werden kann. Die Wirtszelle
ist nicht auf eine Spezifische eingeschränkt, solange sie die Endonuclease
codierende DNA exprimieren kann. Als Beispiele sind Bakterien wie
die Gattung Escherichia (z.B. Escherichia coli), die Gattung Bacillus
(z.B. Bacillus subtilis), die Gattung Pseudomonas (z.B. Pseudomonas
putida), die Gattung Rhizobium (z.B. Rhizobium meliloti), Hefe wie
Schizosaccharomyces pombe, tierische Zellen (z.B. COS- und CHO-Zellen)
und Insektenzellen (z.B. Sf9 und Sf21) angegeben.
-
Wenn
ein Bakterium wie E. coli als Wirt verwendet wird, wird bevorzugt,
dass sich der rekombinante Vektor autonom replizieren kann und einen
Promotor, eine ribosomale Bindungsstelle, das Gen und eine Transkriptionsterminationssequenz
einschließt.
Der rekombinante Vektor kann auch ein Gen zur Kontrolle des Promotors
einschließen.
-
Für E. coli
sind E. coli K12 und DH1 als Beispiele angegeben und für Bacillus
sind Bacillus subtilis MI 114 und 207-21 als Beispiele angegeben.
-
Als
Promotor kann jeder Promotor verwendet werden, solange er in der
Wirtszelle wie E. coli exprimiert werden kann. Beispielsweise kann
ein Promotor, der von E. coli oder einem Phagen stammt, z.B. der
trp-Promotor, lac-Promotor,
pL-Promotor oder pR-Promotor,
verwendet werden. Künstlich
konstruierte und modifizierte Promotoren wie der tac-Promotor können ebenfalls
verwendet werden.
-
Der
rekombinante Vektor kann in das Bakterium nach jedem Verfahren zur
Einführung
von DNA in ein Bakterium eingeführt
werden. Beispielsweise können
das Calciumionen-Verfahren (Cohen, S. N. et al., Proc. Natl. Acad.
Sci., USA, 69: 2110–2114
(1972)) und ein Elektroporationsverfahren verwendet werden.
-
Eine
Hefe wie Saccharomyces cerevisiae, Saccharomyces uvarum, Schizosaccharomyces
pombe oder Pichia pastoris kann ebenfalls als Wirt verwendet werden.
In diesem Fall kann der Promotor jeder Promotor sein, der in der
Hefe exprimiert werden kann. Beispiele eines derartigen Promotors
schließen
ein, sind jedoch nicht begrenzt auf, den gal1-Promotor, gal10-Promotor,
Hitzeschockprotein-Promotor, MF 1-Promotor, PHO5-Promotor, PGK-Promotor,
GAP-Promotor, ADH-Promotor und AOX1-Promotor.
-
Der
rekombinante Vektor kann in die Hefe durch jedes Verfahren zur Einführung von
DNA in eine Hefe eingeführt
werden. Beispielsweise können
das Elektroporationsverfahren (Becker, D. M. et al., Methods Enzymol.,
194, 182–187
(1990)), das Sphäroblastenverfahren
(Hinnen, A. et al., Proc. Natl. Acad. Sci., USA, 75, 1929–1933 (1978))
oder das Lithiumacetatverfahren (Itoh, N., J. Bacteriol., 153, 163–168 (1983))
verwendet werden.
-
Eine
tierische Zelle wie die Affenzelle COS-7, Vero, die chinesische
Hamsterovarienzelle (CHO-Zelle), die Maus-L-Zelle, die Ratten-GH3-
oder menschliche FL-Zelle können
ebenfalls als Wirt verwendet werden. Als Promotor kann beispielsweise
der SR-Promotor, SV40-Promotor, LTR-Promotor oder CMV-Promotor verwendet
werden. Andere Promotoren als diese, beispielsweise ein früher Gen-Promotor
des menschlichen Cytomegalievirus können ebenfalls verwendet werden.
-
Der
rekombinante Vektor kann in die tierische Zelle beispielsweise durch
ein Elektroporationsverfahren, ein Calciumphosphatverfahren oder
ein Lipofektionsverfahren eingeführt
werden.
-
Eine
Insektenzelle wie die Sf9-Zelle, Sf21-Zelle oder dergleichen kann
ebenfalls als Wirt verwendet werden. Der rekombinante Vektor kann
in die Insektenzelle beispielsweise durch ein Calciumphosphatverfahren,
ein Lipofektionsverfahren oder ein Elektroporationsverfahren eingeführt werden.
-
(5) Herstellung der Endonuclease
-
Die
Endonuclease kann durch Züchtung
der vorstehend beschriebenen Transformante und durch Gewinnen der
Endonuclease aus der Kultur davon erhalten werden. Der Begriff „Kultur", wie hier verwendet,
bezieht sich auf einen Kulturüberstand,
eine gezüchtete
Zelle oder mikrobielle Zelle oder eine Zelle oder mikrobielle Zelltrümmer.
-
Die
Transformante wird nach einem allgemeinen Verfahren gezüchtet, das
zur Züchtung
des Wirts verwendet wird.
-
Ein
Medium zur Züchtung
der Transformante, die von einem Wirtsmikroorganismus wie E. coli
oder Hefe erhalten wird, kann entweder ein natürliches oder ein synthetisches
Medium sein, solange es Kohlenstoffquellen, Stickstoffquellen, anorganische
Salze und dergleichen enthält,
die vom Mikroorganismus assimilierbar sind, und solange damit eine
effiziente Züchtung
der Transformante erfolgen kann.
-
Als
Kohlenstoffquellen können
Kohlenwasserstoffe wie Glucose, Fructose, Saccharose, Stärke; organische
Säuren
wie Essigsäure,
Propionsäure;
und Alkohole wie Ethanol und Propanol verwendet werden.
-
Als
Stickstoffquellen können
Ammoniak, Ammoniumsalze anorganischer oder organischer Säuren wie Ammoniumchlorid,
Ammoniumsulfat, Ammoniumacetat, Ammoniumphosphat, andere stickstoffhaltige
Verbindungen; Pepton, Fleischextrakt, Maisquellwasser und dergleichen
verwendet werden.
-
Als
anorganische Substanzen können
Kaliumdihydrogenphosphat, Dikaliumhydrogenphosphat, Magnesiumphosphat,
Magnesiumsulfat, Natrium chlorid, Eisen(II)-Sulfat, Mangansulfat,
Kupfersulfat, Calciumcarbonat und dergleichen verwendet werden.
-
Die
Züchtung
erfolgt im Allgemeinen 12 bis 18 Stunden bei 37°C unter aeroben Bedingungen
wie Schütteln
oder Rühren
mit Belüftung.
Während
der Züchtung
wird der pH-Wert bei 6,5 bis 7,5, vorzugsweise bei 7,0, gehalten.
Der pH-Wert wird mit anorganischer oder organischer Säure, einer
alkalischen Lösung
oder dergleichen reguliert.
-
Während der
Züchtung
können
dem Medium gegebenenfalls Antibiotika wie Ampicillin, Tertacyclin oder
dergleichen zugegeben werden.
-
Bei
der Züchtung
eines Mikroorganismus, der mit einem Expressionsvektor unter Verwendung
eines induzierbaren Promotors transformiert wurde, kann dem Medium
bei Bedarf ein Induktor zugesetzt werden. Wenn beispielsweise ein
Mikroorganismus, der mit einem Expressionsvektor unter Verwendung
des lac-Promotors oder trp-Promotors transformiert wurde, gezüchtet wird,
kann dem Medium Isopropyl-1-thio-β-D-galactosid
(IPTG) bzw. Indolessigsäure
(IES) zugegeben werden.
-
Eine
Transformante, die unter Verwendung von tierischen Wirtszellen erhalten
wurde, kann in einem allgemein verwendeten Medium wie RPM11640-Medium oder DMEM-Medium
oder einem Medium gezüchtet werden,
das durch Ergänzen
des allgemein verwendeten Mediums mit fötalem Rinderserum und dergleichen erhalten
wird.
-
Die
Züchtung
erfolgt im Allgemeinen 1 bis 3 Tage bei 37°C unter 5% CO2.
Während
der Züchtung
kann dem Medium ein Antibiotikum wie Kanamycin, Penicillin oder
dergleichen zugegeben werden.
-
Nach
der Züchtung,
bei der eine mikrobielle Zelle oder eine andere Zelle intrazellulär Endonuclease produziert,
wird die Endonuclease durch Aufschluss der mikrobiellen Zelle oder
der anderen Zelle extrahiert. Wenn eine mikrobielle Zelle oder eine
andere Zelle Endonuclease extrazellulär produziert, wird die Kulturlösung direkt
verwendet. Alternativ wird die mikrobielle Zelle oder die andere
Zelle über
Zentrifugation oder dergleichen entfernt, bevor die Endonuclease
aus der Kultur über
ein allgemeines biochemisches Verfahren zur Proteinisolierung und
-reinigung wie Ammoniumsulfatpräzipitation,
Gelchromatographie, Ionenaustauschchromatographie, Affinitätschromatographie
oder eine Kombination davon isoliert und gereinigt wird.
-
BEISPIELE
-
Beispiel 1: Herstellung
der einzelsträngigen
Matrizen-DNA, die die Untereinheit von Scel codiert und Desoxyuracil
enthält
-
Das
Gen der Endo. SceI-50 kDa-Untereinheit ENS2 (1431 Basenpaare; Nakagawa,
K., Morishima, N. und Shibata, T., J. Biol. Chem. 266, 1977–1984 (1991))
wurde gleichzeitig in zwei Genbereichen modifiziert, d.h. innerhalb
der stromaufwärtsliegenden
Einheit mit 1,0 Kilobasenpaaren und der stromabwärtsliegenden Einheit mit 0,4
Kilobasenpaaren.
-
Für diesen
Zweck wurden das EcoRI/EcoRI-Fragment (1671 Basenpaare), enthaltend
das Gen der 50 kDa-Untereinheit mit der gesamten Länge (Nakagawa,
K., Morishima, N. und Shibata, T. J., Biol. Chem. 266, 1977–1984 (1991)),
und das PstI/EcoRI-Fragment (534 Basenpaare), enthaltend die stromabwärtsliegende Einheit
des Gens der 50 kDa-Untereinheit, getrennt in das Phagmid pUC118
(Takara Shuzo, Co., Ltd.) cloniert und als Plasmide pEN1.7 bzw.
pEN0.5 bezeichnet (3). Diese Phagmide wurden zur
Transformation in die E. coli-Stämme
CJ236 (Takara Shuzo, Co., Ltd.) eingeführt. Die E. coli-Transformanten-Stämme wurden
12 Stunden oder länger
bei 37°C
einer Schüttelkultur
unterzogen, um Vorkulturlösungen
herzustellen. 20 μl
jeder Vorkulturlösung
wurden 2 ml 2 × YT-Kulturmedium
zugegeben, enthaltend Ampicillin (100 μg/ml) (Sambrook, J., Fritsch,
E. F. und Maniatis, T., Molecular Cloning: a laboratory Manual,
Zweite Ausgabe, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, New York (1989)) und 1 Stunde bei 37°C gezüchtet. Jedem Medium wurde der
Helferphage M13KO7 (2,0 × 1012 plaquebildende Einheit (PBE); Takara Shuzo,
Co., Ltd.) zugegeben, so dass er 0,4 Vol.-% des Mediums ausmachte,
und das Ergebnis wurde 1 Stunde bei 37°C gezüchtet. Danach wurde Kanamycin
(100 μg/ml)
zugegeben und das Ergebnis wurde 14 Stunden bei 37°C gezüchtet. Die
während
der Züchtung
aus E. coli in das Medium freigesetzten Phagenpartikel wurden gewonnen.
Genau gesagt wurden 1,5 ml jeder Kulturlösung in einer Mikrozentrifuge
zentrifugiert (14,000 UpM, 5 min). 1,2 ml Überstand wurden gesammelt und
unter denselben Bedingungen zentrifugiert, um die Zellen vollständig zu entfernen,
so dass 1,0 ml Überstand
erhalten wurde. Das anschließende
Verfahren zur Herstellung der DNA erfolgte nach der in der Versuchsanleitung
von J. Sambrook et al. (Sambrook, J., Fritsch, E. F. und Maniatis, T.,
Molecular Cloning: a laboratory Manual, Zweite Ausgabe, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, New York (1989)) zusammengefassten
DNA-Reinigung des Bakteriophagen M13.
-
Beispiel 2: Synthese der
Oligonucleotide zur Einführung
der ortsgerichteten Mutation
-
(i) Synthese des Einzelstanges
-
Das
Gen der 50 kDa-Untereinheit ENS2 wird vom Mitochondriumgenom codiert
und schließt
daher den genetischen Code ein, der für Mitochondrien einzigartig
ist (Tabelle 1). Um ENS2 einem allgemeinen Expressionssystem in
großem
Maßstab
zu unterziehen, müssen
diese einzigartigen Codons substituiert werden, damit sie mit dem
Universalcode übereinstimmen.
Nach Beispiel 2 werden die Basen unter Verwendung eines modifizierten
Verfahrens von T. Kunkel (Kunkel, T. A., Proc. Natl. Acad. Sci.
USA 82, 488–492
(1985)) substituiert. Während
beim allgemeinen Kunkel-Verfahren nur ein oder zwei Oligonucleotide
zur Modifikation verwendet werden, werden beim vorliegenden Verfahren
maximal 16 Oligonucleotide zur effizienten Substitution an multiplen
Positionen verwendet.
-
Es
wurden 33 Oligonucleotide konstruiert, die jeweils die zu substituierende
Base enthielten, die von etwa 10 bis 15 Basen flankiert wurde (Tabelle
3).
-
-
-
In
Tabelle 3 stellt/stellen die Base(n) in Klammern unter jeder Sequenz
die ursprüngliche(n)
Base(n) dar, die durch die unterstrichene(n) Base(n) substituiert
wurde(n). Die in Kleinbuchstaben dargestellten Basen stellen diejenigen
dar, bei denen bereits das ursprüngliche
Oligonucleotid substituiert wurde.
-
Die
Längen
der Oligonucleotide variieren innerhalb des Bereiches von 18 bis
52 Basen, und sie schließen
die Mutation von einem bis maximal 4 Resten ein. Diese Oligonucleotide
wurden zur Substitution von 50 Basenpaaren des 1431 bp großen Gens
der 50 kDa-Untereinheit verwendet, um 37 Codons zu modifizieren. Das
5'-Ende jedes Oligonucleotids
wurde phosphoryliert, so dass die später beschriebene DNA-Ligase-Reaktion
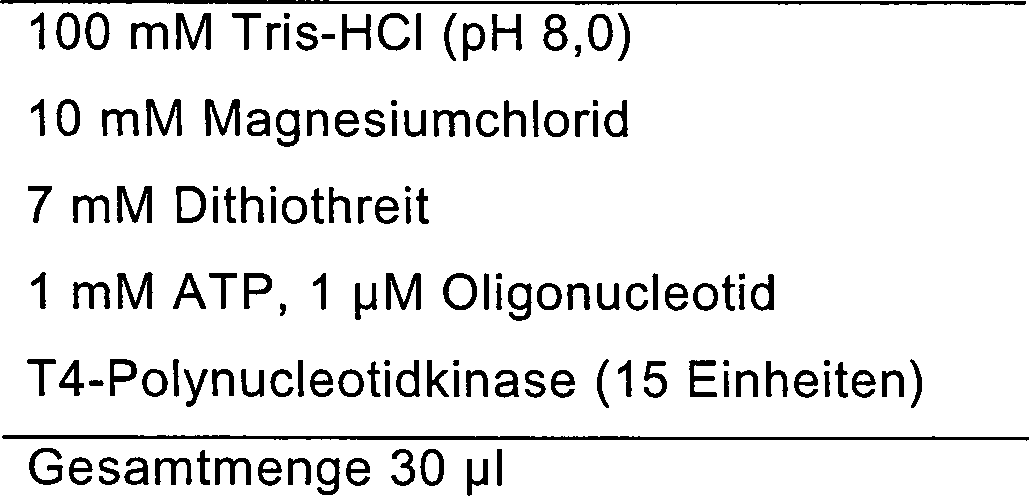
erfolgen konnte. Die Zusammensetzung des Reaktionsgemisches für die Phosphorylierung
ist nachstehend dargestellt:
-
-
Das
Reaktionsgemisch wurde einer 15-minütigen Phosphorylierungsreaktion
bei 37°C
unterzogen, und dann wurde das Enzym durch eine 10-minütige Behandlung
bei 70°C
inaktiviert.
-
(ii) Synthese des Komplementärstranges
-
Die
in (i) erhaltenen Oligonucleotide wurden wie folgt behandelt, um
Doppelstränge
zu erhalten. Die Zusammensetzungen des Anlagerungspuffers und des
Verlängerungsreaktionspuffers
sind nachstehend dargestellt.
-
-
Verlängerungsreaktionspuffer
-
Zu
1 μl Anlagerungspuffer
und 0,2 pMol der einzelsträngigen
Matrizen-DNA wurde
destilliertes Wasser gegeben, was zu einer Gesamtmenge von 10 μl führte. 1 μl der Lösung wurde
dispergiert, so dass sie mit 1 μl der
phosphorylierten Oligonucleotidlösung
gemischt wurde. Das sich ergebende Gemisch wurde 15 min bei 65°C und dann
15 min bei 37°C
stehengelassen, wobei sich das Oligonucleotid an die einzelsträngige DNA anlagerte.
Der Lösung
wurden 25 μl
Verlängerungspuffer,
60 Einheiten E. coli-DNA-Ligase und 1 Einheit T4-DNA-Polymerase zugegeben und 2 Stunden
bei 25°C
stehengelassen, so dass ein Komplementärstrang synthetisiert wurde.
3 μl 0,2
M Ethylendiamintetraessigsäuretetranatriumsalz
(pH 8,0) wurden zugegeben, um die Enzymreaktion zu beenden, nach
der das Enzym durch 5-minütige
Behandlung bei 65°C
inaktiviert wurde. Diese Reaktionslösung wurde direkt für die nachstehende
Transformation verwendet.
-
Beispiel 3: Transformation
-
E.
coli BMH71-18-mutS (Takara Shuzo, Co., Ltd.) wurde so verwendet,
dass die Nucleotidsequenz des Wildtyp-DNA-Stranges (die einzelsträngige DNA,
die mit CJ236 hergestellt wurde) in der doppelsträngigen Plasmid-DNA,
die durch Komplementärstrangsynthese
erhalten wurde, durch einen Mutantentyp substituiert wurde. In diesem
E. coli-Stamm wurde Desoxyuracil, das in der mit CJ236 hergestellten
einzelsträngigen
DNA enthalten war, durch das Enzym Uracil-DNA-Glycosylase hydrolysiert
und dann unter Verwendung des DNA-Stranges erneut synthetisiert,
der die substituierte Base als Matrize enthielt (Lindahl, T., Ann.
Rev. Biochem. 51, 61–87
(1982)).
-
Das
gesamte Reaktionsgemisch mit dem synthetisierten Komplementärstrang
wurde der 100 μl-Lösung zugegeben,
die die kompetenten Zellen von BMH71-18 mutS enthielt. Kompetente
E. coli-Zellen wurden nach dem Verfahren von H. Inoue et al. (Inoue,
H., Nojima, H. und Okayama, H., Gene 96, 23–28 (1990)) hergestellt.
-
Der
Lösung
wurde Medium zugegeben und 1 Stunde bei 37°C stehengelassen. Dann wurden
30 μl des
Helferphagen (a. a. O.) zugegeben und weitere 30 min bei 37°C stehengelassen,
damit die Infektion stattfand. 40 μl der BMH71-18-Kulturlösung, die
intrazellulär
sowohl den Helferpagen als auch das Plasmid enthielt, wurde fraktioniert
und 2 ml des 2 × YT-Mediums,
enthaltend Ampicillin (100 μg/ml)
und Kanamycin (100 μg/ml), zugegeben.
Das sich ergebende Medium wurde 16 bis 20 Stunden bei 37°C einer Schüttelkultur
unterzogen, um einen Phagen herzustellen.
-
Die
mikrobielle Zelle wurde durch Zentrifugation (14.000 UpM, 5 min)
entfernt. Der Überstand,
der den Phagenpartikel enthielt, der die einzelsträngige DNA
des Plasmids mit der substituierten Base einbaut, wurde entfernt.
Mit 20 μl Überstand
wurden 80 μl
des Stammes MV1184 (Takara Shuzo, Co., Ltd.) gemischt, der 12 Stunden
oder länger
gezüchtet
worden war, und man ließ ihn
10 min bei 37°C
stehen, damit die einzelsträngige DNA
des Phagen in die Zelle injiziert wurde.
-
Der
Stamm MV1184, der das sich aus der Replikation der integrierten
einzelsträngigen
DNA ergebende Plasmid enthielt, wurde zur Selektion auf einem LB-Agarmedium,
enthaltend 100 μg/ml
Ampicillin, angeimpft (Sambrook, J., Fritsch, E. F. und Maniatis,
T., Molecular Cloning: a laboratory Manual, Zweite Ausgabe, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor New York (1989)).
Die Nucleotidsequenzen der Gene der 50 kDa-Untereinheit wurden bei
einigen Clonen mit einem automatischen Sequenziergerät ALF (Pharmacia)
analysiert, um den Einbau der vorbestimmten Substitutionen zu bestätigen. Der
Fluoreszenzprimer für
die Analyse der Nucleotidsequenz wurde von Pharmacia (Uppsala, Schweden)
erworben. Die DNA-Sequenzierreaktion basierte auf dem Sanger-Verfahren
(Sanger, F. et al., Proc. Natl. Acad. Sci., 74, 5463–5467 (1977))
nach der Vorschrift von Pharmacia.
-
Für das Ausgangsmaterial,
Plasmid pEN1.7, wurden 40 Nucleotidsubstitutionen mittels 9 Zyklen
des Mutageneseverfahrens durchgeführt, während 10 Nucleotidsubstitutionen
für pEN0.5
mittels 4 Zyklen eines derartigen Verfahrens durchgeführt wurden.
-
Waren
einmal alle Substitutionen bestätigt,
wurden die stromaufwärtsliegenden
und stromabwärtsliegenden
Einheiten an die PstI-Schnittstelle
gebunden, so dass ein Gen erhalten wurde, das die 50 kDa-Untereinheit
mit vollständigen
Substitutionen codiert (4, SEQ ID NR: 2).
-
Beispiel 4: Konstruktion
des Expressionsplasmids für
die 50 kDa-Untereinheit
-
Um
die Bindung zwischen dem modifizierten Gen der 50 kDa-Untereinheit
und dem Vektor zur Induktion ihrer Expression zu ermöglichen,
wurden die Restriktionsschnittstellen in die 5'- und 3'-Enden des modifizierten Gens über die
Polymerasekettenreaktion (PCR) eingeführt. Die Reaktion erfolgte
unter Ver wendung von Taq-DNA-Polymerase (Takara Shuzo, Co., Ltd.)
nach der Vorschrift des Herstellers. Die Sequenzen der verwendeten
Primer sind nachstehend dargestellt.
-
-
Die
unterstrichenen Teile sind die neu eingeführten BamHI- und SalI-Erkennungssequenzen.
Die Reaktion erfolgte mit 25 Zyklen: 1 min bei 94°C; 2 min
bei 45°C;
und 3 min bei 72°C.
-
Das
durch PCR amplifizierte DNA-Fragment (1447 Basenpaare) wurde über (0,8%)
Agarose-Gelelektrophorese (Sambrook, J., Fritsch, E. F. und Maniatis,
T. Molecular Cloning: a laboratory Manual, Zweite Ausgabe, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New York (1989))
aufgetrennt und zur Bestätigung
mit Ethidiumbromid gefärbt.
Das Fragment wurde aus dem Agarosegel unter Verwendung des Geneclean-Kits
(BIO101, Kalifornien, USA) gewonnen.
-
Das
gewonnene DNA-Fragment wurde mit BamHI und SalI behandelt und in
pRSET (Invitrogen Corp.) und pTZ19R (Pharmacia) subcloniert, so
dass pSC50 bzw. pTZSC50 erhalten wurde. Das Plasmid pSC50 wurde
zur Induktion der Expression verwendet. Plasmid pTZSC50 wurde einer
DNA-Sequenzierung unter Verwendung des Fluoreszenzprimers (a. a.
O.) unterzogen, so dass bestätigt
wurde, dass keine zusätzliche
Mutation während
der PCR eingeführt
worden war.
-
Beispiel 5: Einführung der
Expression der 50 kDa-Untereinheit
-
Das
Expressionsplasmid pSC50 wurde in kompetente E. coli BL21 (DE3)
pLysS-Zellen (Invitrogen Corp.) eingeführt. Die Transformantenzelle
wurde über
Nacht bei 37°C über eine
Schüttelkultur
in einem flüssigen
LB-Medium, enthaltend Ampicillin (150 μg/ml) und Chloramphenicol (34 μg/ml) vorgezüchtet (Sambrook, J.,
Fritsch, E. F. und Maniatis, T., Molecular Cloning: a laboratory Manual,
Zweite Ausgabe, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, New York (1989)). Die vorgezüchtete Lösung (4 Vol.-% einer sich ergebenden
Kultur) wurde zentrifugiert (2.500 × g, 10 min), um die mikrobielle
Zelle zu gewinnen. Dieses Präzipitat wurde
in einer kleinen Menge frischem Medium suspendiert, das dann einem
flüssigen
Medium zugegeben wurde. Die Schüttelkultur
(37°C) erfolgte,
bis der Suspensionsspiegel von etwa 0,5 bei 600 Nanometer (nm) (OD600)
erhalten wurde. Anschließend
wurde die Schüttelkultur
bei 18°C
fortgesetzt. Wenn die OD600 etwa 0,8 erreichte, wurde Isopropyl-1-thio-β-D-galactosid
(IPTG) bis zur Endkonzentration von 0,4 mM zugegeben, um die Induktion
der Expression der 50 kDa-Untereinheit zu initiieren. Nachdem E.
coli weiteren 12 Stunden einer Schüttelkultur bei 18°C unterzogen
wurde, wurde er durch Zentrifugation rückgewonnen, schnell mit flüssigem Stickstoff
eingefroren und bei –80°C gelagert.
-
Beispiel 6: Reinigung
der 50 kDa-Unterinheit aus E. coli.
-
Die
bei –80°C gelagerte
mikrobielle Zelle wurde bei Raumtemperatur aufgetaut. Die anschließenden Behandlungen
wurden bei 4°C
oder auf Eis durchgeführt.
Die mikrobielle Zelle wurde in Puffer A (20 mM Tris-HCl-Puffer (pH
8,0), 500 mM Natriumchlorid, 5 mM Imidazol, 1 mM Phenylmethylsulfonylfluorid
(PMSF; Sigma Aldrich Japan K. K., Tokio, Japan), 0,1% NP-40 (Nacalai
Tesque, Inc., Kyoto, Japan)) suspendiert. Die sich ergebende Suspension
wurde schnell mit flüssigem
Stickstoff eingefroren und unter laufendem Wasser aufgetaut, um
E, coli aufzuschließen.
Die Suspension wurde 5-mal 30 sec mit einem Ultraschallgerät (UR-200P,
Tomy Seiko Co., Ltd., Tokio, Japan) bei maximaler Leistung behandelt.
-
Die
behandelte Lösung
wurde bei 4°C
(39.000 × g,
20 min) zentrifugiert. Der erhaltene Überstand wurde durch einen
0,45 μm-Mylex-Filter
(Millipore, Massachusetts, USA) filtriert. Die Probe wurde auf eine
Säule (ø10 mm,
2,0 ml) aufgetragen, die mit Probond Nickelchelate Resin (Invitrogen
Corp.) beladen war, das mit Puffer A äquilibriert worden war. Dann
wurde die Probe mit 20 ml Puffer A (10-faches Volumen des Harzes) gewaschen.
Nach einem weiteren Waschschritt mit 12 ml Puffer A, enthaltend
60 mM Imidazol (6-faches Volumen des Harzes), wurde das Ergebnis
einer Gradientenelution mit 60–50
mM Imidazol enthaltendem Puffer A (Gesamtmenge 80 ml) unterzogen.
Die eluierte Fraktion wurde einer Natriumdodecylsulfat-Polyacrylamidgelelektrophorese
(Laemmli, U. K. Nature, 227, 680–685 (1970)) unterzogen und
mit Coomasie Brilliantblau gefärbt,
um das Vorliegen der 50 kDa-Untereinheit zu bestätigen. Die 50 kDa-Untereinheit
enthaltende Fraktion wurde gegen Puffer B (20 mM Tris-HCl-Puffer
(pH 7,5), 300 mM Natriumchlorid, 1 mM Ethylendiamintetraessigsäuretetranatriumsalz,
1 mM Dithiothreit) dialysiert. Das gereinigte Protein wurde unter
Verwendung eines Mittels zum Proteintest (Bio-Rad, Kalifornien,
USA) nach dem Mikrotestverfahren des Herstellers quantifiziert.
Als Standardprotein wurde Rinderserumalbuminlösung (Sigma Aldrich Japan K.
K.) verwendet. Infolgedessen wurden 300 μg gereinigtes Protein aus 25
g (Nassgewicht) der mikrobiellen Zelle erhalten.
-
Beispiel 7: Messung der
Endonucleaseaktivität
-
Ein
Substrat zur Messung einer Endonucleaseaktivität der 50 kDa-Untereinheit wurde
wie folgt hergestellt. Ein EcoRI/EcoRI-Fragment, das den oli2-Bereich
auf Mitochondrium-DNA enthielt, von dem bekannt ist, dass Endo.
SceI darin spaltet (1671 Basenpaare; Nakagawa, K. et al., EMBO J.
11, 2707–2715
(1992)), wurde in das Phagemid pUC119 (Takara Shuzo, Co., Ltd.)
subcloniert, wobei das Ergebnis als pY673L (2) bezeichnet
wurde. Plasmid pBR322 (Takara Shuzo, Co., Ltd.) wurde als Kontroll-DNA-Substrat
verwendet. Die Plasmide pY673L und pBR322 wurden verwendet, um E.
coli zu transformieren. Dann wurden die Plasmide aus E. coli extrahiert
und unter Verwendung einer Qiagen-Säule gutgereinigt (Qiagen Japan,
Tokio, Japan).
-
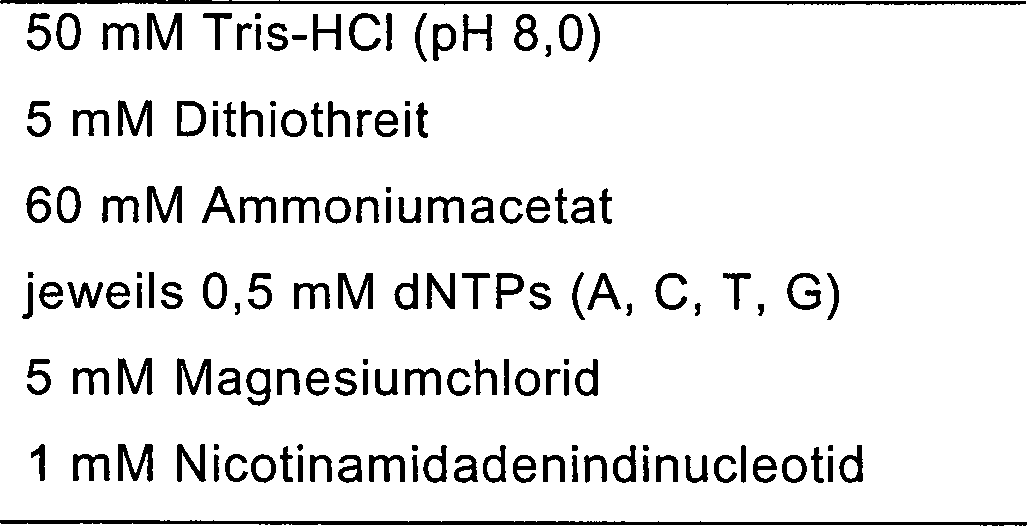
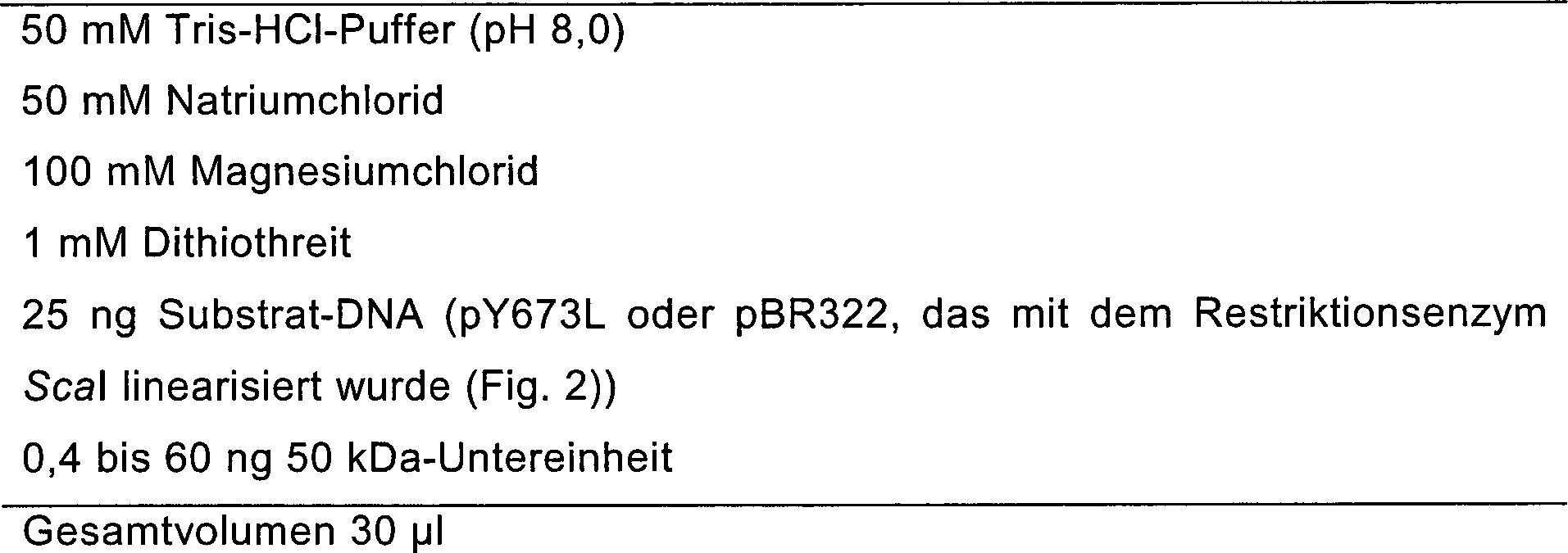
Die
Zusammensetzung der für
die Messung der Endonucleaseaktivität der 50 kDa-Untereinheit verwendeten
Reaktionslösung
ist nachstehend dargestellt.
-
-
Nach
Durchführung
der 30-minütigen
DNA-Spaltungsreaktion bei 37°C
wurden Ethylendiamintetraessigsäuretetranatriumsalz
und Natriumdodecylsulfat bis zu Endkonzentrationen von 10 mM bzw.
0,3% zugegeben, um die Reaktion zu beenden. Die gespaltene DNA wurde
einer 0,8%-igen Agaroseelektrophorese entweder direkt oder nach
der Konzentrierung der DNA durch Phenolextraktion und Ethanolpräzipitation
unterzogen.
-
Nach
der Elektrophorese wurde das Gel mit Ethidiumbromid (Sigma Aldrich
Japan K. K.) oder SYBR-Green (Takara Shuzo, Co., Ltd.) gefärbt, um
die Spaltung der DNA zu bestätigen.
Die DNA wurde unter Verwendung eines FMBIO Imaging-Gerätes (Takara
Shuzo, Co., Ltd.) nachgewiesen, um die DNA-Spaltung zu bestimmen.
-
Beispiel 8: Nachweis der
sequenzspezifischen Endonuclease
-
Die
dimere Endo. SceI erkennt und spaltet in vivo und in vitro 26 Basenpaare,
die der Consensussequenz innerhalb des oli2-Genbereichs auf der
Mitochondrium DNA ähneln
(Nakagawa, K., Morishima, N. und Shibata, T., EMBO J. 11, 2707–2715 (1992))
(2). Die gereinigte 50 kDa-Untereinheit von Endo.
SceI spaltete eine spezifische Sequenz selbst (SEQ ID NR: 1).
-
Die
spezifische Spaltung von oli2 mit der 50 kDa-Untereinheit unter
Verwendung von Plasmid pY673L, das oli2 als Substrat enthält, wurde
bestätigt
(5A). Bezugnehmend auf 5A sind
die Spuren 1 bis 8 die Ergebnisse, die mit 0,5, 1,0, 2,0, 4,0, 8,0,
16,0, 32,0 bzw. 64,0 ng der 50 kDa-Untereinheiten erhalten wurden. Mit
64 ng der 50 kDa-Untereinheit wurden 60% von pY673L (25 ng) in der
Reaktionslösung
sequenzspezifisch bei 37°C
innerhalb von 30 min gespalten, wobei DNA-Fragmente mit 3,4 und
1,4 Kilobasen nachgewiesen wurden.
-
Die
DNA wurde nicht mit der 50 kDa-Untereinheit unter Verwendung von
Plasmid pBR322 als Substrat gespalten, und es wurde kein Spaltungsfragment
nachgewiesen. Bezugnehmend auf 5B sind
die Spuren 1 bis 7 die Ergebnisse, die mit 2,3, 4,5, 9,0, 18,0,
36,0, 72,0 bzw. 144 ng der 50 kDa-Untereinheiten erhalten wurden.
Bei Verwendung einer Überschussmenge
(200 ng) der 50 kDa-Untereinheit fand man keine Spaltung bei pBR322
oder anderen DNAs (Mitochondrium-DNA (80 Kilobasenpaare) aus einem
knospenden Hefestamm, DNA des E. coli-Phagen λ (47 Kilobasenpaare) und DNA
des Bacillus-Phagen ø105
(38 Kilobasenpaare), die keine spezifische Sequenz (26 Basenpaare)
innerhalb des oli2-Genbereichs enthielten.
-
Beispiel 9: Massenproduktion
der 50 kDa-Untereinheit aus Saccharomyces uvarum und Nachweis ihrer
Aktivität
-
Die
Endo. SuvI-50 kDa-Untereinheit, ein homologes Protein der Endo.
SceI-50 kDa-Untereinheit aus Saccharomyces cerevisiae liegt in Saccharomyces
uvarum vor (Nakagawa, K., Morishima, N. und Shibata, T., J. Bio.
Chem. 266, 1977–1984
(1991)). Beide Untereinheiten besitzen 476 Aminosäurereste,
aber auf der Aminosäureebene
gibt es zwei Unterschiede zwischen ihnen. Die Unterschiede bei den
Aminosäuren
zwischen der Endo. SceI- und der Endo. SuvI-50 kDa-Untereinheit
sind in 6A dargestellt.
-
Für die Endo.
SuvI-50 kDa-Untereinheit wurde ein Gen zur Expression in großem Maßstab durch
Einführung
zweier zusätzlicher
Modifikationen in das modifizierte Gen für die Endo. SceI-50 kDa-Untereinheit
hergestellt. Die für
die Substitutionen verwendeten Oligonucleotide der Aminosäuren für die Endo.
SuvI-50 kDa-Untereinheit sind in 6B dargestellt.
Bezugnehmend auf 6B entsprechen die Basen in
Klammern der Nucleotidsequenz der Endo. SceI-50kDa-Untereinheit.
Für diesen
Zweck wurden zwei Oligonucleotide neu synthetisiert, um Mutationen
nach dem vorstehend beschriebenen Genmodifikationsverfahren einzuführen. Die
Mutation wurde durch DNA-Sequenzierung
bestätigt.
Das modifizierte Gen wurde in den pRSET-Vektor (Invitrogen Corp.)
subcloniert, der dann durch das Transformationsverfahren in E. coli
BL21 (DE3) pLys eingeführt
wurde.
-
Die
Endo. SuvI-50 kDa-Untereinheit wurde nach dem vorstehend beschriebenen
Verfahren exprimiert und gereinigt, das für die Endo. SceI-50 kDa-Untereinheit
angewandt wurde. Die gereinigte Endo. SuvI-50 kDa-Untereinheit wurde
verwendet, um Plasmid pY673L spezifisch zu spalten.
-
Folglich
war die Endo. SuvI-50 kDa-Untereinheit bei der sequenzspezifischen
Spaltung der Schnittstelle für
die Endo. SceI-50 kDa-Untereinheit auf Plasmid pY673L ähnlich wirksam
(7A).
-
Dagegen
spaltete die Endo. SuvI-50 kDa-Untereinheit das Plasmid pBR322 überhaupt
nicht (7B). Auch andere DNAs (Mitochondrium-DNA
von knospender Hefe, DNA des Bacillus-Phagen ø105 und DNA des E. coli-Phagen λ, die den
oli2-Genbereich nicht enthielten) wurden von der Endo. SuvI-50 kDa-Untereinheit nicht
gespalten.
-
In
den 7A und 7B sind
die Spuren 1 bis 7 die Ergebnisse, die mit 2,3, 4,5, 9,0, 18,0,
36,0, 72,0 bzw. 144 ng der 50 kDa-Untereinheiten erhalten wurden.
-
Dementsprechend
werden eine ortsspezifische Endonuclease, die eine spezifische Nucleotidsequenz erkennen
kann, ein die Nucleotidsequenz codierendes Gen, ein rekombinanter,
das Gen enthaltender Vektor, eine den Vektor enthaltende Transformante
und ein Verfahren zur Herstellung der Endonuclease bereitgestellt. Da
die Endonuclease eine spezifische Sequenz aus 26 Basen erkennen
kann, ist sie bei der Modifikation und Kartierung von DNA in einem
großen
Anwendungsbereich, d.h. Plasmid bis Genom, auf dem Gebiet der Gentechnik
und Biochemie nützlich.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-