DE69821019T2 - 3-carboalkoxy-2,3-dihydro-1h-phenothiazin-4(10h)-one derivate - Google Patents
3-carboalkoxy-2,3-dihydro-1h-phenothiazin-4(10h)-one derivate Download PDFInfo
- Publication number
- DE69821019T2 DE69821019T2 DE69821019T DE69821019T DE69821019T2 DE 69821019 T2 DE69821019 T2 DE 69821019T2 DE 69821019 T DE69821019 T DE 69821019T DE 69821019 T DE69821019 T DE 69821019T DE 69821019 T2 DE69821019 T2 DE 69821019T2
- Authority
- DE
- Germany
- Prior art keywords
- methyl
- dihydro
- use according
- coor
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 11
- 150000003839 salts Chemical class 0.000 claims abstract description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 7
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 6
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 5
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims abstract description 3
- 125000001309 chloro group Chemical group Cl* 0.000 claims abstract 3
- 206010010904 Convulsion Diseases 0.000 claims description 8
- -1 phenothiazine compound Chemical class 0.000 claims description 8
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 7
- 229950000688 phenothiazine Drugs 0.000 claims description 5
- 239000003814 drug Substances 0.000 claims description 4
- 241000124008 Mammalia Species 0.000 claims description 3
- 229910052801 chlorine Inorganic materials 0.000 claims description 3
- 208000034308 Grand mal convulsion Diseases 0.000 claims description 2
- 206010061334 Partial seizures Diseases 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 claims description 2
- 125000005843 halogen group Chemical group 0.000 claims 1
- 150000002990 phenothiazines Chemical class 0.000 abstract description 9
- 229910052739 hydrogen Inorganic materials 0.000 abstract description 4
- 125000000217 alkyl group Chemical group 0.000 abstract description 3
- 229910052736 halogen Inorganic materials 0.000 abstract description 3
- 150000002367 halogens Chemical class 0.000 abstract description 3
- 239000001257 hydrogen Substances 0.000 abstract description 3
- 125000001246 bromo group Chemical group Br* 0.000 abstract 1
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 53
- 239000000203 mixture Substances 0.000 description 45
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 239000013078 crystal Substances 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 27
- 238000002844 melting Methods 0.000 description 25
- 230000008018 melting Effects 0.000 description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 23
- 238000012360 testing method Methods 0.000 description 21
- 239000004480 active ingredient Substances 0.000 description 20
- 238000009472 formulation Methods 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 17
- 241001465754 Metazoa Species 0.000 description 17
- 239000007787 solid Substances 0.000 description 17
- 238000010992 reflux Methods 0.000 description 16
- 239000000460 chlorine Chemical group 0.000 description 15
- 150000001875 compounds Chemical class 0.000 description 15
- 238000001816 cooling Methods 0.000 description 15
- 238000010438 heat treatment Methods 0.000 description 14
- 239000012452 mother liquor Substances 0.000 description 14
- 239000000538 analytical sample Substances 0.000 description 13
- 239000001961 anticonvulsive agent Substances 0.000 description 11
- 238000001556 precipitation Methods 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 10
- 229960003965 antiepileptics Drugs 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 230000001773 anti-convulsant effect Effects 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- 238000000034 method Methods 0.000 description 9
- 239000000243 solution Substances 0.000 description 9
- 241000699670 Mus sp. Species 0.000 description 8
- 239000012071 phase Substances 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 7
- TZJGKEPNZDAOES-UHFFFAOYSA-N ethyl 2-methyl-4,6-dioxocyclohexane-1-carboxylate Chemical compound CCOC(=O)C1C(C)CC(=O)CC1=O TZJGKEPNZDAOES-UHFFFAOYSA-N 0.000 description 7
- YOELNWPMMURSQH-UHFFFAOYSA-N methyl 2-methyl-4,6-dioxocyclohexane-1-carboxylate Chemical compound COC(=O)C1C(C)CC(=O)CC1=O YOELNWPMMURSQH-UHFFFAOYSA-N 0.000 description 7
- LDGHLZFFKMEAOE-UHFFFAOYSA-N 2-amino-5-bromobenzenethiol Chemical compound NC1=CC=C(Br)C=C1S LDGHLZFFKMEAOE-UHFFFAOYSA-N 0.000 description 6
- TYRZAGMAVZESQX-UHFFFAOYSA-N 2-amino-5-chlorobenzenethiol Chemical compound NC1=CC=C(Cl)C=C1S TYRZAGMAVZESQX-UHFFFAOYSA-N 0.000 description 6
- VRVRGVPWCUEOGV-UHFFFAOYSA-N 2-aminothiophenol Chemical class NC1=CC=CC=C1S VRVRGVPWCUEOGV-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 238000006243 chemical reaction Methods 0.000 description 6
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- VUMZNLOQJGKGNE-UHFFFAOYSA-N 2-amino-5-methylbenzenethiol Chemical compound CC1=CC=C(N)C(S)=C1 VUMZNLOQJGKGNE-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 239000008187 granular material Substances 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- KOBHMYVVCCJHNM-UHFFFAOYSA-N 1,2,3,10-tetrahydrophenothiazin-4-one Chemical class S1C2=CC=CC=C2NC2=C1C(=O)CCC2 KOBHMYVVCCJHNM-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 239000006071 cream Substances 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- RZXMPPFPUUCRFN-UHFFFAOYSA-N p-toluidine Chemical compound CC1=CC=C(N)C=C1 RZXMPPFPUUCRFN-UHFFFAOYSA-N 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 238000005481 NMR spectroscopy Methods 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000007135 neurotoxicity Effects 0.000 description 3
- 231100000228 neurotoxicity Toxicity 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 239000006072 paste Substances 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- NARLFVSETWHKRP-UHFFFAOYSA-N 3,4-dihydro-2h-1,4-thiazine Chemical class C1CSC=CN1 NARLFVSETWHKRP-UHFFFAOYSA-N 0.000 description 2
- KGTJEVRYDFNQKA-UHFFFAOYSA-N 3,5-dihydro-2h-furo[3,4-b][1,4]thiazine Chemical class S1CCN=C2COC=C21 KGTJEVRYDFNQKA-UHFFFAOYSA-N 0.000 description 2
- QSNSCYSYFYORTR-UHFFFAOYSA-N 4-chloroaniline Chemical compound NC1=CC=C(Cl)C=C1 QSNSCYSYFYORTR-UHFFFAOYSA-N 0.000 description 2
- ZLILRRGWBOKBIG-UHFFFAOYSA-N 4h-1,4-benzothiazine Chemical class C1=CC=C2NC=CSC2=C1 ZLILRRGWBOKBIG-UHFFFAOYSA-N 0.000 description 2
- VMNXKIDUTPOHPO-UHFFFAOYSA-N 6-chloro-1,3-benzothiazol-2-amine Chemical compound C1=C(Cl)C=C2SC(N)=NC2=C1 VMNXKIDUTPOHPO-UHFFFAOYSA-N 0.000 description 2
- 241000220479 Acacia Species 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- ZFDIRQKJPRINOQ-HWKANZROSA-N Ethyl crotonate Chemical compound CCOC(=O)\C=C\C ZFDIRQKJPRINOQ-HWKANZROSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 2
- AFBPFSWMIHJQDM-UHFFFAOYSA-N N-methyl-N-phenylamine Natural products CNC1=CC=CC=C1 AFBPFSWMIHJQDM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 201000010099 disease Diseases 0.000 description 2
- 150000002081 enamines Chemical class 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000011835 investigation Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 235000010603 pastilles Nutrition 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- ZNNZYHKDIALBAK-UHFFFAOYSA-M potassium thiocyanate Chemical compound [K+].[S-]C#N ZNNZYHKDIALBAK-UHFFFAOYSA-M 0.000 description 2
- 229940116357 potassium thiocyanate Drugs 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M sodium chloride Inorganic materials [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- JKUYRAMKJLMYLO-UHFFFAOYSA-N tert-butyl 3-oxobutanoate Chemical compound CC(=O)CC(=O)OC(C)(C)C JKUYRAMKJLMYLO-UHFFFAOYSA-N 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000001256 tonic effect Effects 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- ZFDIRQKJPRINOQ-UHFFFAOYSA-N transbutenic acid ethyl ester Natural products CCOC(=O)C=CC ZFDIRQKJPRINOQ-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- WDFQBORIUYODSI-UHFFFAOYSA-N 4-bromoaniline Chemical compound NC1=CC=C(Br)C=C1 WDFQBORIUYODSI-UHFFFAOYSA-N 0.000 description 1
- YTUXGQUUYDSWGY-UHFFFAOYSA-N 5,7-dichloro-3-methyl-4h-1,4-benzothiazine Chemical class ClC1=CC(Cl)=C2NC(C)=CSC2=C1 YTUXGQUUYDSWGY-UHFFFAOYSA-N 0.000 description 1
- VZEBSJIOUMDNLY-UHFFFAOYSA-N 6-bromo-1,3-benzothiazol-2-amine Chemical compound C1=C(Br)C=C2SC(N)=NC2=C1 VZEBSJIOUMDNLY-UHFFFAOYSA-N 0.000 description 1
- DZWTXWPRWRLHIL-UHFFFAOYSA-N 6-methyl-1,3-benzothiazol-2-amine Chemical compound CC1=CC=C2N=C(N)SC2=C1 DZWTXWPRWRLHIL-UHFFFAOYSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- REXJVSQUIVJFGR-UHFFFAOYSA-N BrC1=CC=C2NC(CC(C(C3=O)C(=O)OC(C)(C)C)C)=C3SC2=C1 Chemical compound BrC1=CC=C2NC(CC(C(C3=O)C(=O)OC(C)(C)C)C)=C3SC2=C1 REXJVSQUIVJFGR-UHFFFAOYSA-N 0.000 description 1
- 239000004358 Butane-1, 3-diol Substances 0.000 description 1
- 101100165177 Caenorhabditis elegans bath-15 gene Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 206010009346 Clonus Diseases 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 206010048768 Dermatosis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102000004300 GABA-A Receptors Human genes 0.000 description 1
- 108090000839 GABA-A Receptors Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical class Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101100353526 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pca-2 gene Proteins 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000027089 Parkinsonian disease Diseases 0.000 description 1
- 206010034010 Parkinsonism Diseases 0.000 description 1
- CWRVKFFCRWGWCS-UHFFFAOYSA-N Pentrazole Chemical compound C1CCCCC2=NN=NN21 CWRVKFFCRWGWCS-UHFFFAOYSA-N 0.000 description 1
- 241000269799 Perca fluviatilis Species 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229940125907 SJ995973 Drugs 0.000 description 1
- 238000007351 Smiles rearrangement reaction Methods 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- PFRUBEOIWWEFOL-UHFFFAOYSA-N [N].[S] Chemical compound [N].[S] PFRUBEOIWWEFOL-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000002421 anti-septic effect Effects 0.000 description 1
- 229940125715 antihistaminic agent Drugs 0.000 description 1
- 239000000739 antihistaminic agent Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 229940064004 antiseptic throat preparations Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- FBOSKQVOIHEWAX-UHFFFAOYSA-N benzothiazine Chemical class C1=CC=C2N=CCSC2=C1 FBOSKQVOIHEWAX-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000005056 compaction Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002447 crystallographic data Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 231100000223 dermal penetration Toxicity 0.000 description 1
- VILAVOFMIJHSJA-UHFFFAOYSA-N dicarbon monoxide Chemical group [C]=C=O VILAVOFMIJHSJA-UHFFFAOYSA-N 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 238000007877 drug screening Methods 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 206010015037 epilepsy Diseases 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 229960002767 ethosuximide Drugs 0.000 description 1
- HAPOVYFOVVWLRS-UHFFFAOYSA-N ethosuximide Chemical compound CCC1(C)CC(=O)NC1=O HAPOVYFOVVWLRS-UHFFFAOYSA-N 0.000 description 1
- MLINRHYVNRGQIA-UHFFFAOYSA-N ethyl 7-chloro-2-methyl-4-oxo-1,2,3,10-tetrahydrophenothiazine-3-carboxylate Chemical compound ClC1=CC=C2NC(CC(C)C(C3=O)C(=O)OCC)=C3SC2=C1 MLINRHYVNRGQIA-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000006052 feed supplement Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000003966 growth inhibitor Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 239000008309 hydrophilic cream Substances 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 238000013095 identification testing Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000012073 inactive phase Substances 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 230000002045 lasting effect Effects 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 125000001434 methanylylidene group Chemical group [H]C#[*] 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 239000003883 ointment base Substances 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 238000007243 oxidative cyclization reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 229960005152 pentetrazol Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 229960002036 phenytoin Drugs 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- AEQFSUDEHCCHBT-UHFFFAOYSA-M sodium valproate Chemical compound [Na+].CCCC(C([O-])=O)CCC AEQFSUDEHCCHBT-UHFFFAOYSA-M 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000013222 sprague-dawley male rat Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 150000005846 sugar alcohols Polymers 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- UYFCCIULQCUZJI-UHFFFAOYSA-N tert-butyl 10H-phenothiazine-3-carboxylate Chemical compound C(=O)(OC(C)(C)C)C=1C=CC=2NC3=CC=CC=C3SC=2C=1 UYFCCIULQCUZJI-UHFFFAOYSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 150000004897 thiazines Chemical class 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- DTMHTVJOHYTUHE-UHFFFAOYSA-N thiocyanogen Chemical compound N#CSSC#N DTMHTVJOHYTUHE-UHFFFAOYSA-N 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 229940102566 valproate Drugs 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 238000005550 wet granulation Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D279/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one sulfur atom as the only ring hetero atoms
- C07D279/10—1,4-Thiazines; Hydrogenated 1,4-thiazines
- C07D279/14—1,4-Thiazines; Hydrogenated 1,4-thiazines condensed with carbocyclic rings or ring systems
- C07D279/18—[b, e]-condensed with two six-membered rings
- C07D279/20—[b, e]-condensed with two six-membered rings with hydrogen atoms directly attached to the ring nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
- A61P25/12—Antiepileptics; Anticonvulsants for grand-mal
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pain & Pain Management (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Description
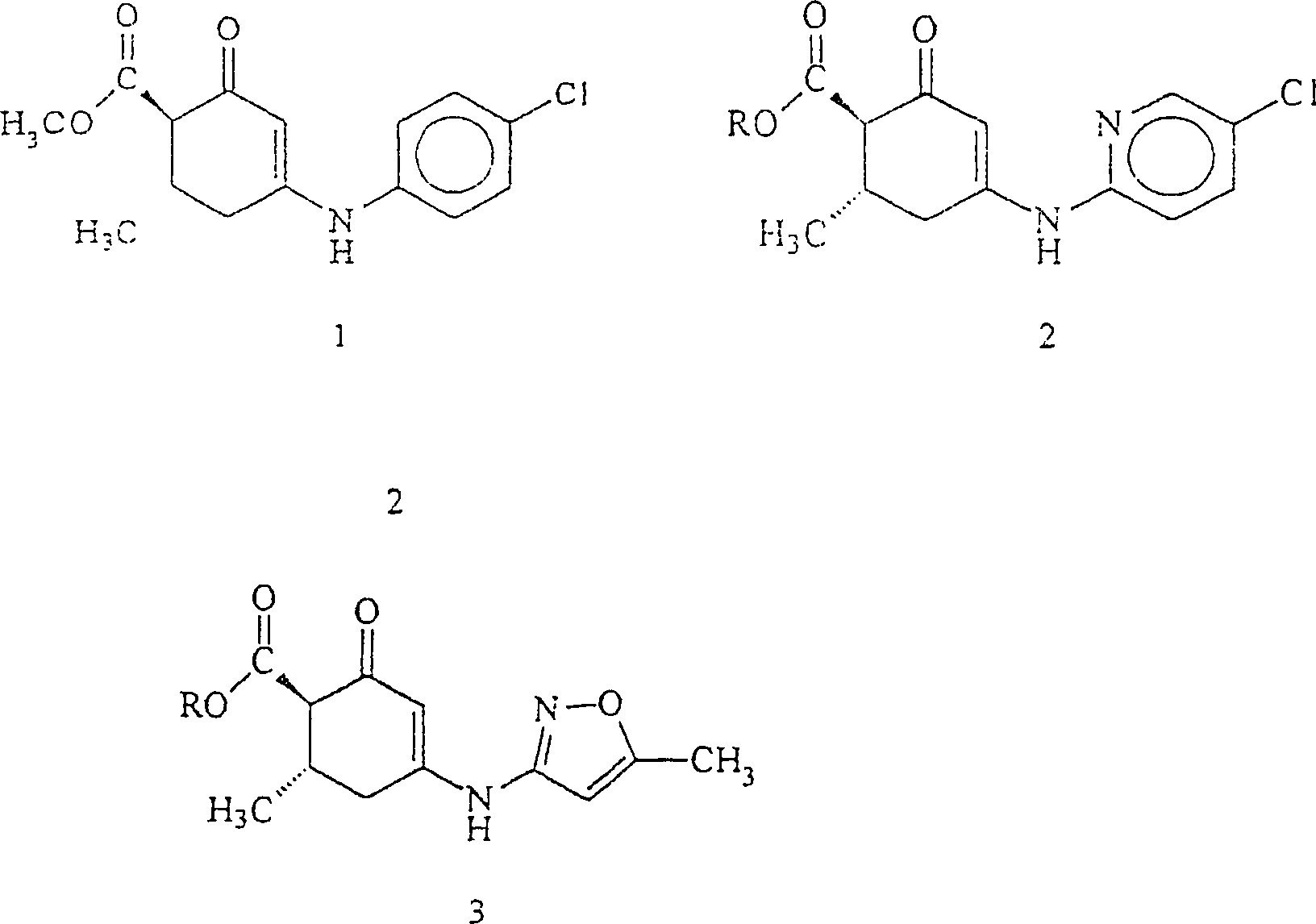
- Bei einem Versuch, Analoga des Prototyp-Antikonvulsivums zu entdecken, wurden Methyl-4-[(4'-chlorphenyl)-amino]-6-methyl-2-oxo-3-cyclohexen-1-carboxylat, 1, Alkyl-4-[(5'-chlor-2'-pyridinyl)amino]-6-methyl-2-oxo-3-cyclohexen-1-carboxylate, 2 [R=CH3 (2a); R=C2H5 (2b)] und Alkyl-4-[(5'-methyl)-3'-isoxazolylamino]-6-methyl-2-oxo-3-cyclohexen-1-carboxylate, 3 [R=CH3 (3a); R=C2H5 (3b); R=C(CH3)3 (3c)] ins Auge gefaßt. Edafiogho, I. O.; Hinko, C. N.; Chang, H.; Moore, J. A.; Mulzac, D.; Nicholson, J. M.; Scott, K. R.: Synthesis and anticonvulsant activity of enaminones. J. Med. Chem. 1992, 35, 2798–2805. Scott, K. R.; Edafiogho, I. O.; Richardson, E. L.; Farrar, V. A.; Moore, J. A.; Tietz, E. I.; Hinko, C. N.; Chang, H.; El-Assadi, A; Nicholson, J. M.: Synthesis and anticonvulsant activity of enaminones. 2. Further structure activity correlations. J. Med. Chem. 1993, 36, 1947–1955. US-Patent Nr. 5,580,894.
-
- Des weiteren wurde gezeigt, daß das Phenothiazin-Grundringsystem eine wesentliche pharmakologisch aktive Gruppe für Beruhigungsmittel, Antikrebsmittel, entzündungshemmende Mittel, Antihistaminika, Wurmmittel, Lokalanästhetika, Antiseptika, Wachstumshemmer und bei der Behandlung von neuropsychiatrischen Störungen ist. (a) Gupta, R. R. Herausgeber: Phenothiazines and 1,4-benzothiazines. Chemical and biomedical aspects. Elsevier, Amsterdam, The Netherlands, 1988. (b) Benz, G.; Fengler, G.; Meyer, H.; Niemers, E.; Fiedler, V.; Mardin, M.; Mayer, D.; Perzborn, E.; Seuter, F. et al.: Use of 4H-1,4-benzothiazines in the prevention and therapy of respiratory diseases, inflammations/rheumatism, thromboembolic diseases, ischemias and infarctions, heart rhythm disturbances, arteriosclerosis, and dermatosis, drugs for this purpose, and active substances contained in these drug. Chem. Abstr. 1986, 105, 60620y; Deutsche Offenlegungsschrift
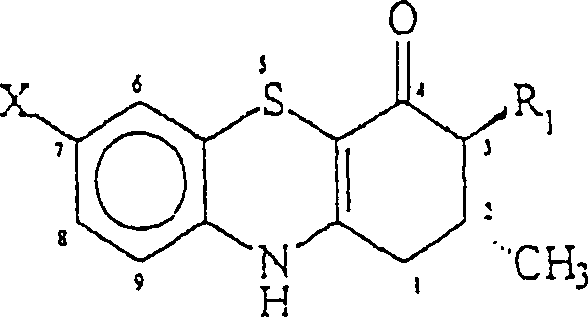
DE 3,426,564 . (c) Niemers, E.; Gruetzmann, R.; Mardin, M.; Busse, W. D.; Meyer, H.: Annellated 4H-1,4-benzothiazine lipoxygenase inhibitors. Chem. Abstr. 1984, 100, l85787k; Deutsche OffenlegungsschriftDE 3,229,122 . Es wird angenommen, daß der Mechanismus für die Vielzahl von therapeutischen Aktivitäten auf das Vorhandensein einer Falte entlang der Stickstoff-Schwefel-Achse zurückzuführen ist. Gupta, R. R.; Saraswat, V.; Gupta, V.; Rajoria, C. M.; Gupta, A.; Jain, M.: Synthesis of 5,6- and 5,7-dichloro-3-methyl-4H-1,4-benzothiazines and their conversion into sulfones. J. Heterocyclic Chem. 1993, 30, 803–806. Eine antikonvulsive Aktivität wurde für eine verwandte Reihe von 2,3-Dihydro-3-oxo-5H-pyrido[3,4-b][1,4]benzothiazin-4-carbonitrilen berichtet. Chorvat, R. J.; Desai, B. N.; Radak, S. E.; Bloss, J.; Hirsch, J.; Tenen, S.: Synthesis, benzodiazepine receptor binding, and anticonvulsant activity of 2,3-dihydro-3-oxo-5H-pyrido-[3,4-b][1,4]benzothiazine-4-carbonitriles. J. Med. Chem. 1983, 26, 845–850. - Die vorliegende Erfindung betrifft die Verwendung einer Reihe von Phenothiazinen. Insbesondere betrifft die vorliegende Erfindung die Verwendung einer Phenothiazinverbindung der Formel:wobei R1 H oder -COOR ist, wobei R ausgewählt ist aus der Gruppe, bestehend aus verzweigten oder unverzweigten Alkylresten, die 1 bis 4 Kohlenstoffatome enthalten, und wobei X ausgewählt ist aus H, verzweigten oder unverzweigten Alkylresten, die 1 bis 4 Kohlenstoffatome enthalten, und Halogen, und pharmazeutisch verträgliche Salze davon zur Herstellung einer pharmazeutischen Zusammensetzung zur Behandlung von Grand mal und partiellen Anfällen bei einem Säuger. Besonders bevorzugte Phenothiazine sind diejenigen, bei denen X Wasserstoff, Brom, Chlor oder Methyl ist, R1 COOR ist und R Methyl, Ethyl oder t-Butyl ist.
- Die vorstehend erwähnten Phenothiazine sind vorteilhaft, da sie Mittel für das Zentralnervensystem mit einer antikonvulsiven Aktivität sind, die eine besonders außergewöhnliche Wirksamkeit gegen Elektroschockanfälle besitzen.
- Aufgrund ihrer biologischen Reaktion sind die erfindungsgemäßen Verbindungen brauchbar beim Verhindern, Lindern, Bekämpfen oder Untersuchen einer Vielzahl von Krankheiten und unerwünschten psychologischen Zustanden bei Säugern, einschließlich Menschen, Haustieren, zoologischen Gattungen, Stalltieren und Labortieren wie Affen, Kaninchen, Ratten und Mäuse. Solche Krankheiten und Zustände umfassen Epilepsie, Parkinsonismus, Huntington-Chorea und Alzheimer-Krankheit.
- KURZE BESCHREIBUNG DER ZEICHNUNGEN
-
1 zeigt die Röntgen-Struktur der Verbindung 4b (R=C2N5; X=H). -
2 zeigt die Röntgen-Struktur der Verbindung 4b in einer Einheitszelle. - In Erwägung der konformationell eingeschränkten Analoga von 1, wurden Analoga mit den zwei Stellungen b- zu dem enaminischen Stickstoff, der in einer tricyclischen Struktur verbunden ist, durch die vorliegenden Erfinder untersucht. Die vorliegende Erfindung betrifft eine Bindung durch ein Schwefelatom. Folglich wurde eine neue Reihe von 3-Carboalkoxy-2,3-dihydro-1H-phenothiazin-4[10H]-onen (4a–c und 4e–m) sowie das unsubstituierte Analogon, 4d, synthetisiert. Diese Verbindungen sind in der Tabelle I angegeben.
-
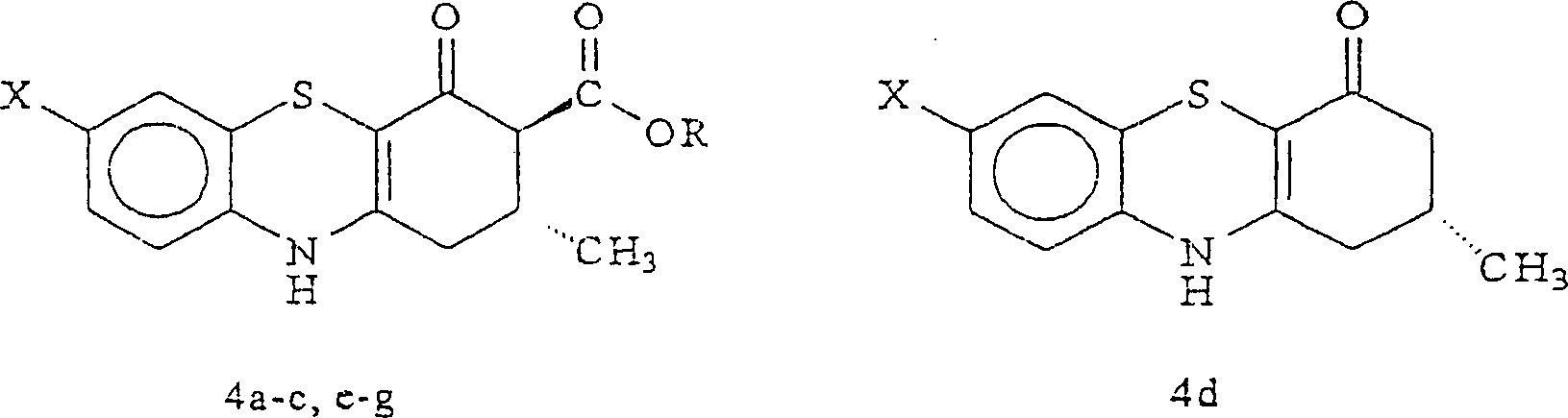
- Tabelle I. Physikalische Eigenschaften der Phenothiazine
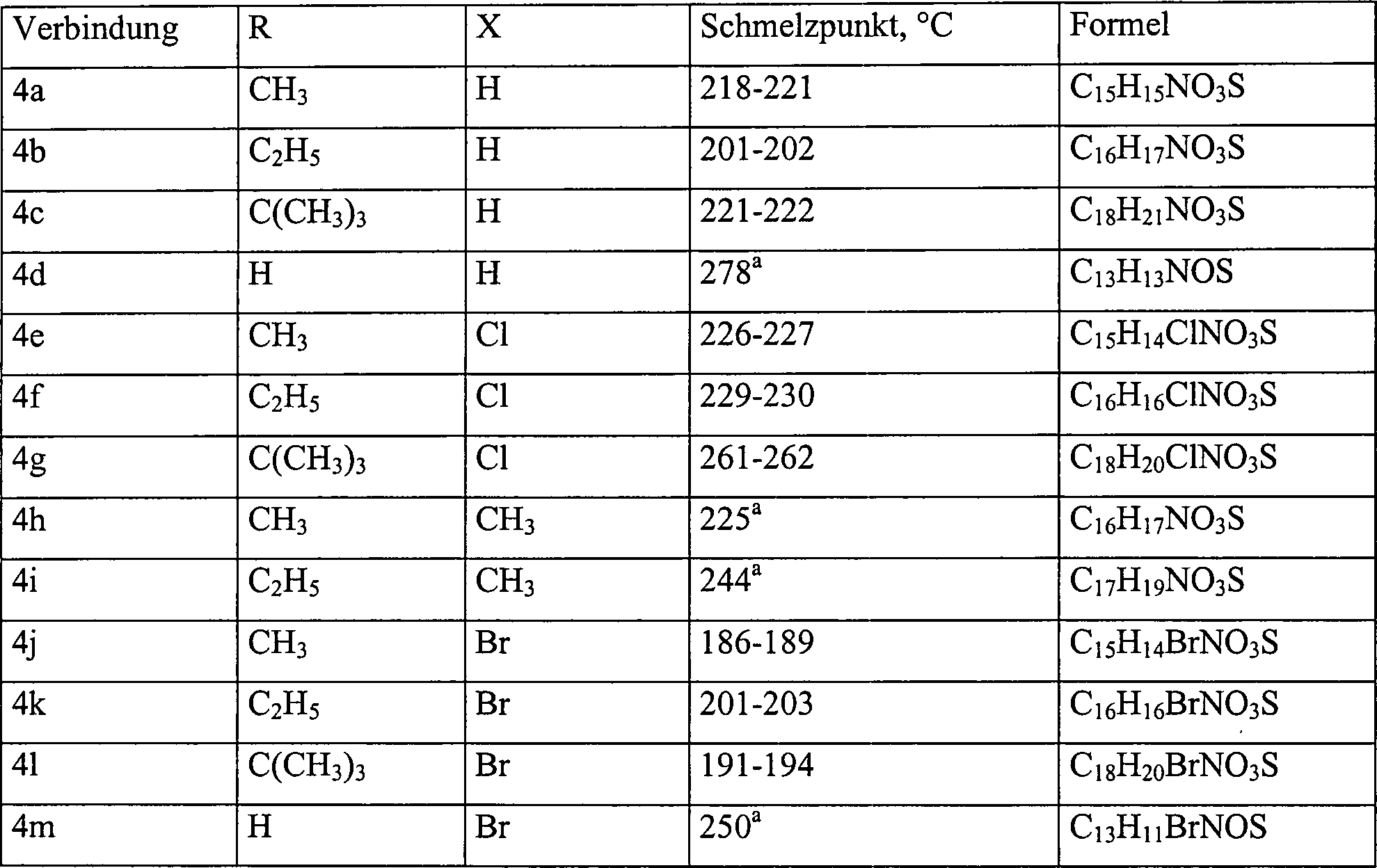
- Syntheseschema
- Thiazine wurden früher unter Verwendung von Enaminen (abgeleitet von acetylenischen Nitrilen und Estern) synthetisiert. Roberts, R. R.; Landor, S. R.: 2,3-Dihydro-4H-1,4-thiazines and 5,6-dihydro-3H-furo[3,4-b]-1,4-thiazines from 4-tetrahydropyranyloxyalk-2-ynenitriles. Tetrahedron Lett. 1993, 34, 5681–5684. Ein erstes Schema für die Durchführung der Erfindung ist wie folgt.
-

- Es wird angenommen, daß dieses Schema einer klassischen Einstufenreaktion von Miyano und Mitarbeitern (Miyano, S.; Abe, N.; Sumoto, K.: Synthesis of 2,3-dihydro-1H-phenothiazin-4(10H)-ones. J. Chem. Soc. Chem. Comm. 1975, 760) folgt, die die Kondensierung und oxidative Cyclisierung der in geeigneter Weise substituierten 2-Aminobenzolthiole, 6, mit dem b-Dicarbonylester, 7 [R=CH3 (7a); R=C2H5 (7b); R=C(CH3)3 (7c)] in unter Rückfluß erhitztem DMSO beinhaltet, um 4a–c und 4e–l in angemessen reiner Form bereitzustellen. Die Vorläufer-Thiolverbindungen 6 sind abgeleitet von der basenkatalysierten, hydrolytischen Spaltung der 6-substituierten 2-Aminobenzothiazole, 5, hergestellt durch die Wirkung von Kalium- (oder Ammonium-) Thiocyanat und Brom (das in situ Thiocyanogen, [(SCN)2] erzeugt) an p-substituierten Anilinen, wie in der Literatur beschrieben. Mital, R.; Jain, S. K.: Synthesis of some 5-substituted 2-aminobenzenethiols and the conversion into phenothiazines via Smiles rearrangement. J. Chem. Soc. (C) 1969, 2148–2150.
- Ein alternatives Schema für die Durchführung der vorliegenden Erfindung ist nachstehend angegeben.
-
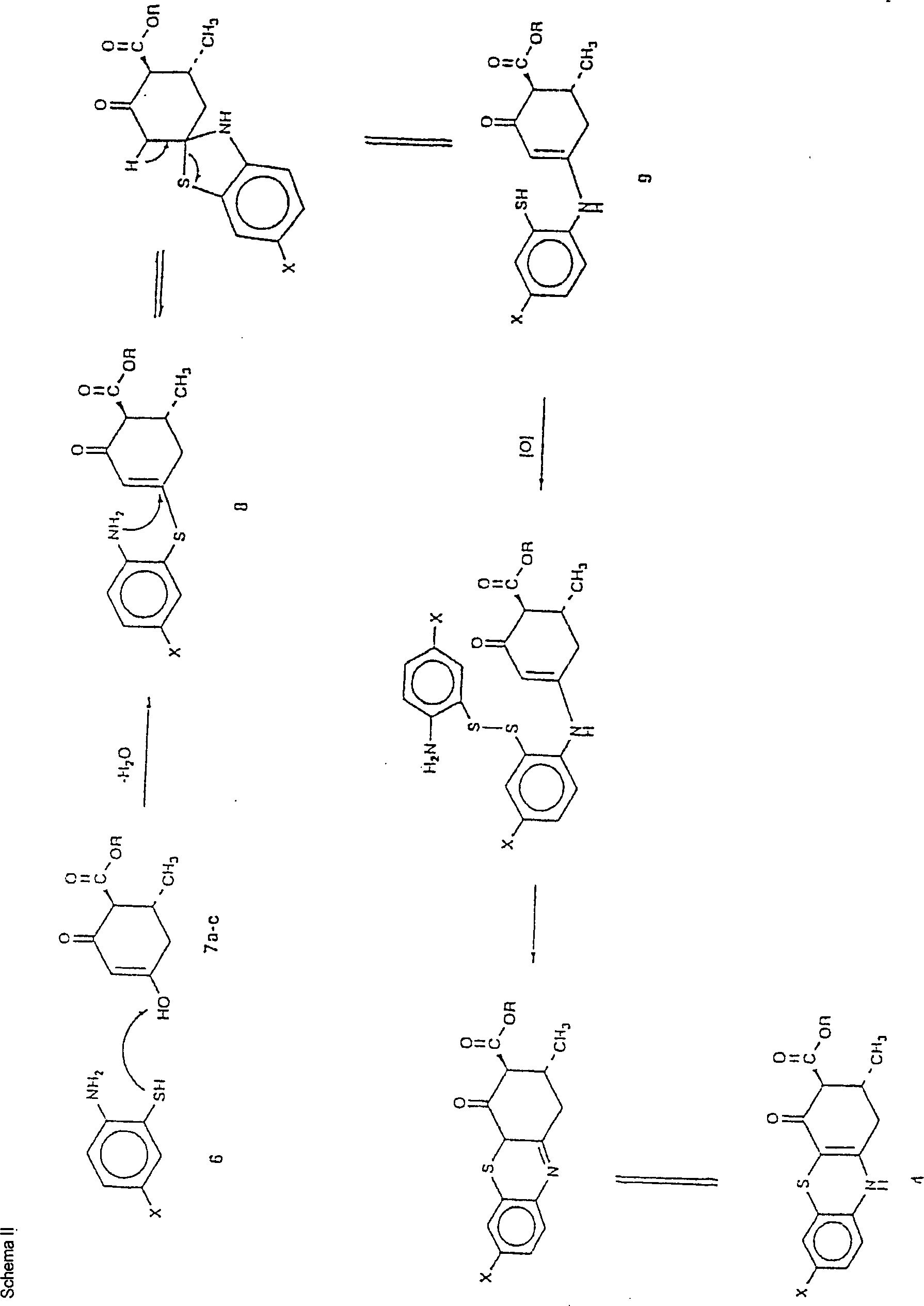
- Bei diesem alternativen Schema beinhaltet ein herausragendes mechanistisches Merkmal für die Konstruktion von 4a–c und 4e–l die Umwandlung des Ensulfids 8 zu enaminischem Keton 9, gefolgt vom oxidativen Ringschluß der letzteren Verbindung. Dieser Mechanismustyp wurde vorgeschlagen. Roberts, R. R.; Landor, S. R.: 2,3-Dihydro-4H-1,4-thiazines and 5,6-dihydro-3H-furo[3,4-b]-1,4-thiazines from 4-tetrahydropyranyloxyalk-2-ynenitriles. Tetrahedron Lett. 1993, 34, 5681–5684.
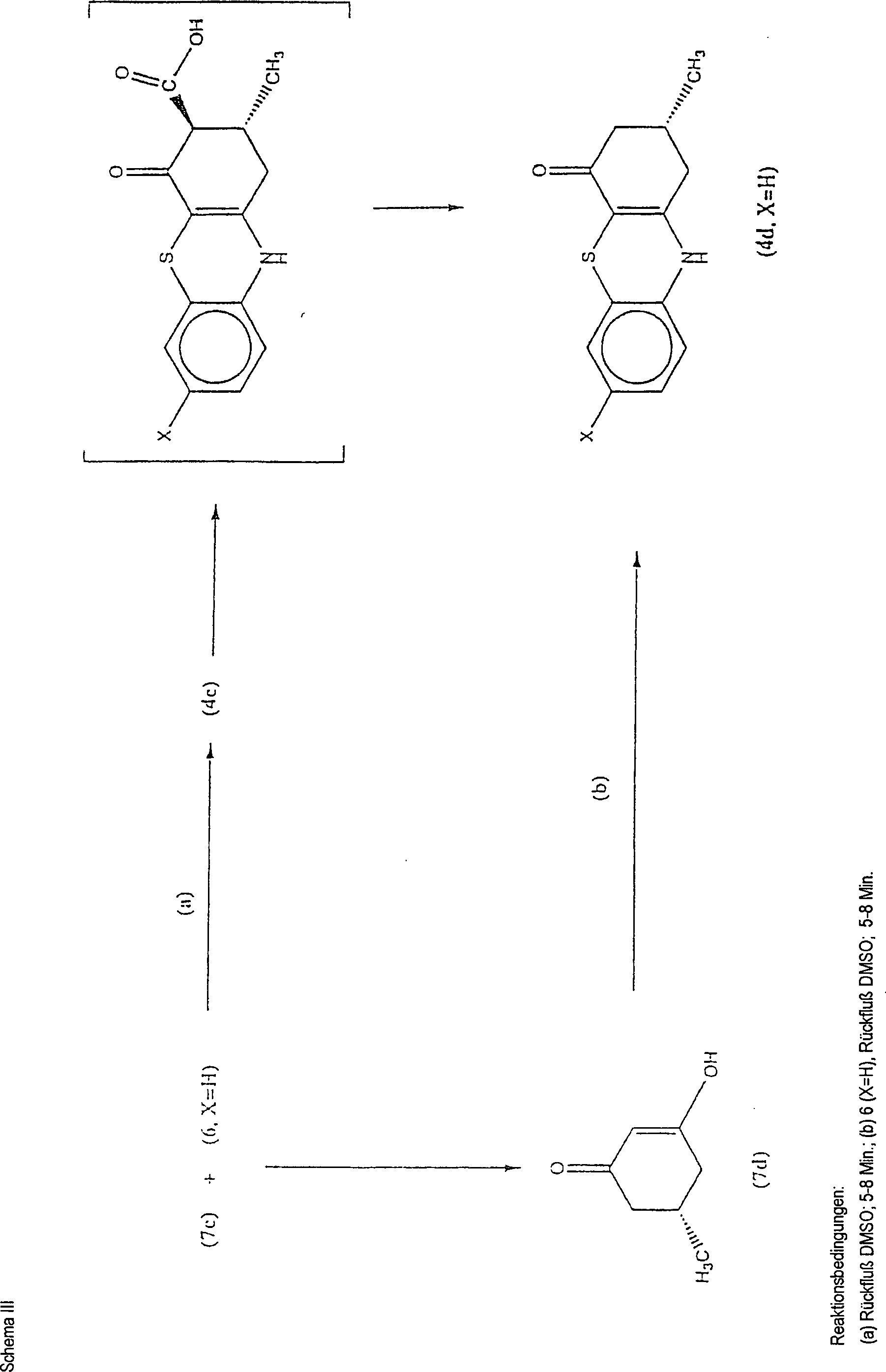
- Ohne durch eine bestimmte Theorie gebunden sein zu wollen, wird angenommen, daß, wenn die experimentellen Bedingungen für den letzten Schritt für jeden Ester identisch sind (d. h. 40 Minuten Rückfluß in DMSO), die ter-Butylverbindung 4c (X=H) einer thermischen Zersetzung unterzogen wird, um 2,3-Dihydro-1H-phenothiazin-4[10H]-on, 4d (X=H) zu ergeben. Die thermische Zersetzung des gewünschten Phenothiazins 4c (X=H) zu Butylen und die b-Ketosäure (4, R=X=H) wird vermutet, gefolgt von der Decarboxylierung der letzteren Verbindung zu 4d (X=H). Dies wird durch die Bildung der 4c Reihe unter Verwendung von kürzeren Rückflußzeiten in DMSO bewiesen. Stabilitätsuntersuchungen des 3-Carbo-ter-butoxy-phenothiazins (41, X=Br) bestätigen die Instabilität des 3-Carbo-ter-butoxy-Substituenten mit unter Rückfluß erhitztem DMSO für Zeiträume bis zu 40 Minuten, wobei 4m (X=Br) gebildet wird. Friary und Mitarbeiter haben gezeigt, daß 7c (R=C(CH3)3) unter säurekatalysierten Bedingungen leicht decarboxyliert wird, um 7d (siehe das nachstehende Schema III) zu bilden. Friary, R. J.; Gilligan, J. M.; Szajewski, R. P.; Falci, K. J.; Franck, R. W.: Heterocyclic syntheses via the intramolecular acylation of enamines derived from animo acids. J. Org. Chem. 1973, 38, 3487–3490.
-
- Die erfindungsgemäßen Verbindungen können leicht in der Form pharmazeutisch verträglicher Salze bereitgestellt werden. Geeignete pharmazeutisch verträgliche Salze umfassen Salze, die von anorganischen oder organischen Säuren abgeleitet sind. Typische Beispiele pharmazeutisch verträglicher Salze umfassen Salze, die von anorganischen Säuren abgeleitet sind, wie Salzsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Salpetersäure und dergleichen, und organische Säuren wie Essigsäure, Propionsäure, Glycolsäure, Oxasäure, Brenztraubensäure, Äpfelsäure, Bernsteinsäure, Malonsäure, Maleinsäure, Fumarsäure, Zitronensäure, Weinsäure, Mandelsäure, Zimtsäure, Benzoesäure, p-Toluolsulfonsäure, Salicylsäure, Ethansulfonsäure, Methansulfonsäure und so weiter. Die Bildung solcher Salze liegt innerhalb der Fähigkeiten eines Durchschnittsfachmanns.
- Verabreichung
- Für jeden Zweck und jede Indikation hängt die Menge an erforderlichem Bestandteil von einer Anzahl von Faktoren ab, einschließlich der Schwere des zu behandelnden Zustands und der Identität des Empfängers ab und liegt letztendlich im Ermessen des behandelnden Arztes oder Tierarztes. Im allgemeinen liegt jedoch für jeden dieser Zwecke und jede dieser Indikationen eine geeignete Wirkdosis vorzugsweise im Bereich von 0,1 bis 250 mg pro Kilogramm Körpergewicht des Empfängers pro Tag, stärker bevorzugt im Bereich von 0,1 bis 10 mg pro Kilogramm Körpergewicht pro Tag.
- Während es möglich ist, die Wirkbestandteile allein zu verabreichen, wird bevorzugt, sie als pharmazeutische Formulierungen darzureichen. Die Formulierungen der vorliegenden Erfindung sowohl für den Gebrauch bei Tieren als auch beim Menschen umfassen mindestens einen Wirkbestandteil wie vorstehend definiert, zusammen mit einem oder mehreren verträglichen Trägern davon und gegebenenfalls andere therapeutische Bestandteile. Der bzw. die Träger muß bzw. müssen in dem Sinn "verträglich" sein, daß er bzw. sie mit den anderen Bestandteilen der Formulierung kompatibel sind und für den Empfänger derselben nicht schädlich sind.
- Die Formulierungen umfassen diejenigen, die für eine orale, rektale, nasale, topische (einschließlich bukkale und sublinguale), vaginale oder parenterale (einschließlich subkutane, intramuskuläre, intravenöse, intradermale, intrathekale und epidurale) Verabreichung geeignet sind. Die Formulierungen können geeigneterweise in Einheitsdosierungsform dargereicht werden und können mittels irgendeines der Verfahren, die in der Technik der Pharmazie wohlbekannt sind, hergestellt werden. Solche Verfahren schließen eines Schritt des Inverbindungbringens des Wirkbestandteils mit dem Träger, der einen oder mehrere zusätzliche Bestandteile bildet ein. Im allgemeinen werden die Formulierungen durch gleichmäßiges und inniges Inverbindungbringen des Wirkbestandteils mit flüssigen Trägern oder feinverteiaten festen Trägern oder beidem und dann, falls notwendig, Formen des Produkts zu beispielsweise einer Tablettenform hergestellt.
- Erfindungsgemäße Formulierungen, die für die orale Verabreichung geeignet sind, können als getrennte Einheiten wie Kapseln, Gelatinekapseln oder Tabletten, die jeweils eine vorbestimmte Menge des Wirkbestandteils enthalten, als Pulver oder Körnchen, als Lösung oder Suspension in einer wässerigen Flüssigkeit oder einer nichtwässerigen Flüssigkeit, als Bolus, als Elektuarium oder Paste dargereicht werden.
- Eine Tablette kann durch Komprimieren oder Formen, gegebenenfalls mit einem oder mehreren pharmazeutisch verträglichen Exzipientien hergestellt werden. Komprimierte Tabletten können durch Komprimieren des Wirkbestandteils in einer freifließenden Form wie als Pulver oder Körnchen, gegebenenfalls im Gemisch mit einem Bindemittel, Gleitmittel, inertem Verdünnungsmittel, Konservierungsmittel, oberflächenaktiven Mittel oder Dispersionsmittel in einer geeigneten Maschine hergestellt werden. Geformte Tabletten können durch Formen einer Mischung der pulverisierten Verbindung, die mit einem inerten flüssigen Verdünnungsmittel angefeuchtet ist, in einer geeigneten Maschine hergestellt werden. Die Tabletten können gegebenenfalls mit einem Überzug versehen oder eingekerbt werden und können formuliert werden, um für eine langsame oder gesteuerte Freisetzung des darin enthaltenen Wirkbestandteils zu sorgen.
- Für topische Anwendungen werden die Formulierungen vorzugsweise als Salbe oder Creme aufgetragen. Bei einer Formulierung in einer Salbe können die Wirkbestandteile entweder mit einer paraffinischen oder wassermischbaren Salbenbasis verwendet werden. Alternativ können die Wirkbestandteile in einer Creme mit einer Öl-in-Wasser-Cremebasis formuliert werden.
- Falls gewünscht, kann die wässerige Phase der Cremebasis beispielsweise mindestens 30% Gew./Gew. eines mehrwertigen Alkohols, d. h. eines Alkohols mit zwei oder mehr Hydroxylgruppen, wie Propylenglycol, Butan-1,3-diol, Mannitol, Sorbitol, Glycerol und Polyethylenglycol und Mischungen davon, umfassen. Die topischen Formulierungen können wünschenswerterweise eine Verbindung einschließen, die die Absorption oder Durchdringung des Wirkbe standteils durch die Haut oder andere betroffene Bereiche verbessert. Beispiele solcher dermaler Durchdringungsverstärker umfassen Dimethylsulfoxid und verwandte Analoga.
- Formulierungen, die für die topische Verabreichung im Auge geeignet sind, umfassen auch Augentropfen, bei denen der Wirkbestandteil in einem geeigneten Träger, insbesondere einem wässerigen Lösungsmittels für den Wirkbestandteil gelöst (oder suspendiert) ist.
- Formulierungen, die für die topische Verabreichung im Mund geeignet sind, schließen Pastillen ein, die den Wirkbestandteil enthalten, vorzugsweise in einem mit Aromastoffen versetzten Grundstoff, üblicherweise Saccharose und Akazie oder Tragant; Pastillen, die den Wirkbestandteil in einer inerten Basis wie Gelatine und Glycerin oder Saccharose und Akazie umfassen, und Mundwässer, die den Wirkbestandteil in einem geeigneten flüssigen Träger umfassen.
- Formulierungen für die rektale Verabreichung können als Täpfchen mit einer geeigneten Basis dargereicht werden, die beispielsweise Kakaobutter oder ein Stearat enthält.
- Formulierungen, die für die nasale Verabreichung geeignet sind, bei denen der Träger ein Feststoff ist, schließen ein grobes Pulver mit einer Teilchengröße von beispielsweise im Bereich von etwa 20 bis etwa 500 Mikron, die durch schnelle Inhalierung durch die Nasenpassage verabreicht werden, ein. Geeignete Formulierungen, bei denen der Träger eine Flüssigkeit ist, für die Verabreichung als beispielsweise Nasenspray oder Nasentropfen, schließen wässerige oder ölige Lösungen des Wirkbestandteils ein.
- Formulierungen, die für die vaginale Verabreichung geeignet sind, können als Pessare, Tampons, Cremes, Gele, Pasten, Schäume oder Sprühformulierungen dargereicht werden, die zusätzlich zum Wirkbestandteil solche Träger enthalten, deren Eignung in der Technik bekannt ist.
- Formulierungen, die für die parenterale Verabreichung geeignet sind, schließen wässerige und nichtwässerige, sterile Injektionslösungen ein, die Antioxidantien, Puffer, Bakteriostatika und gelöste Stoffe, die die Formulierung mit dem Blut des beabsichtigten Empfängers isotonisch machen, und wässerige und nichtwässerige, sterile Suspensionen, die Suspensionsmittel und Dickungsmittel einschließen können, enthalten können. Die Formulierungen können in Einheitsdosis- oder Mehrfachdosis-Behältern, beispielsweise versiegelten Ampullen und Phiolen, dargereicht werden und können in gefriergetrocknetem (lyophilisierten) Zustand gelagert werden, der nur die Zugabe des sterilen flüssigen Trägers, beispielsweise Wasser, für die Injektion, unmittelbar vor der Verwendung erfordert. Unvorbereitete Injektionslösungen und -suspensionen können aus sterilen Pulvern, Körnchen und Tabletten der vorstehend beschriebenen Art hergestellt werden. Formulierungen für die intramuskuläre Verabreichung sind besonders bevorzugt.
- Bevorzugte Einheitsdosierungsformulierungen sind diejenigen, die eine Tagesdosis oder eine Tagesteildosis des Wirkbestandteils, wie hier vorstehend angegeben, oder einen angemessenen Bruchteil davon enthalten.
- Es ist ersichtlich, dass, zusätzlich zu den vorstehend besonders erwähnten Bestandteilen, die erfindungsgemäßen Formulierungen andere Mittel, die in der Technik mit Bezug auf den Typ der fraglichen Formulierungen üblich sind, enthalten können. Beispielsweise können diejenigen, die für die orale Verabreichung geeignet sind, Geschmacksmittel enthalten.
- Die vorliegende Erfindung stellt des weiteren veterinäre Zusammensetzungen bereit, die mindestens einen Wirkstoff, wie vorstehend definiert, zusammen mit einem veterinären Träger davon enthalten.
- Veterinäre Träger sind Materialien, die für den Zweck der Verabreichung der Zusammensetzung brauchbar sind, und sie können feste, flüssige oder gasförmige Materialien sein, die für den Zweck der Verabreichung der Zusammensetzung brauchbar sind, und sie sind anderweitig in der veterinären Technik inert oder verträglich und mit dem Wirkbestandteil kompatibel. Diese veterinären Zusammensetzungen können oral, parenteral oder mittels eines anderen gewünschten Wegs verabreicht werden.
- Für die orale Verabreichung können die Zusammensetzungen in der Form einer Tablette, eines Körnchenarzneitranks, einer Paste, einer Gelatinekapsel, einer Kapsel oder eines Futterergänzungsmittel vorliegen. Körnchen können durch wohlbekannte Techniken der Naßgranulierung, Vorverdichtung oder Stoßen hergestellt werden. Sie können Tieren über einen inerten flüssigen Träger zur Bildung eines Arzneitranks oder in einer Suspension mit Wasser oder einer Ölbasis verabreicht werden. Vorzugsweise sind weitere zusätzliche Bestandteile wie ein Dispensiermittel enthalten.
- Weitere Formulierungsinformationen sind beispielsweise im US-Patent Nr. 5,079,252 zu finden.
- Experimenteller Teil
- Schmelzpunkte werden auf einer Thomas-Hoover Kapillar-Schmelzpunktvorrichtung bestimmt und sind unberichtigt. Die Ergebnisse sind in der vorstehenden Tabelle I gezeigt. IR-Spektren sind auf Proben in KBr als verdünnte Chloroformlösungen in angepaßten Natriumchloridzellen oder sorgfältig mit einem Perkin-Elmer 1330 Spektrophotometer aufgezeichnet und sind in Tabelle 2 angegeben.
- Tabelle II. IR-Daten in KBr-Pellets

- 1H NMR-Spekten sind auf einem General Electric QE 300 MHz Spectrometer in deuterierten Lösungsmitteln unter Verwendung von Tetramethylsilan als internem Standard aufgezeichnet. Die Ergebnisse sind in Tabelle III angegeben.
- Tabelle III. 1H NMR (DMSO-d6) Korrelationstabelle für 2,3-Dihydro-1H-phenothiazin-4(10H)-one

- Tabelle III (Forts.)

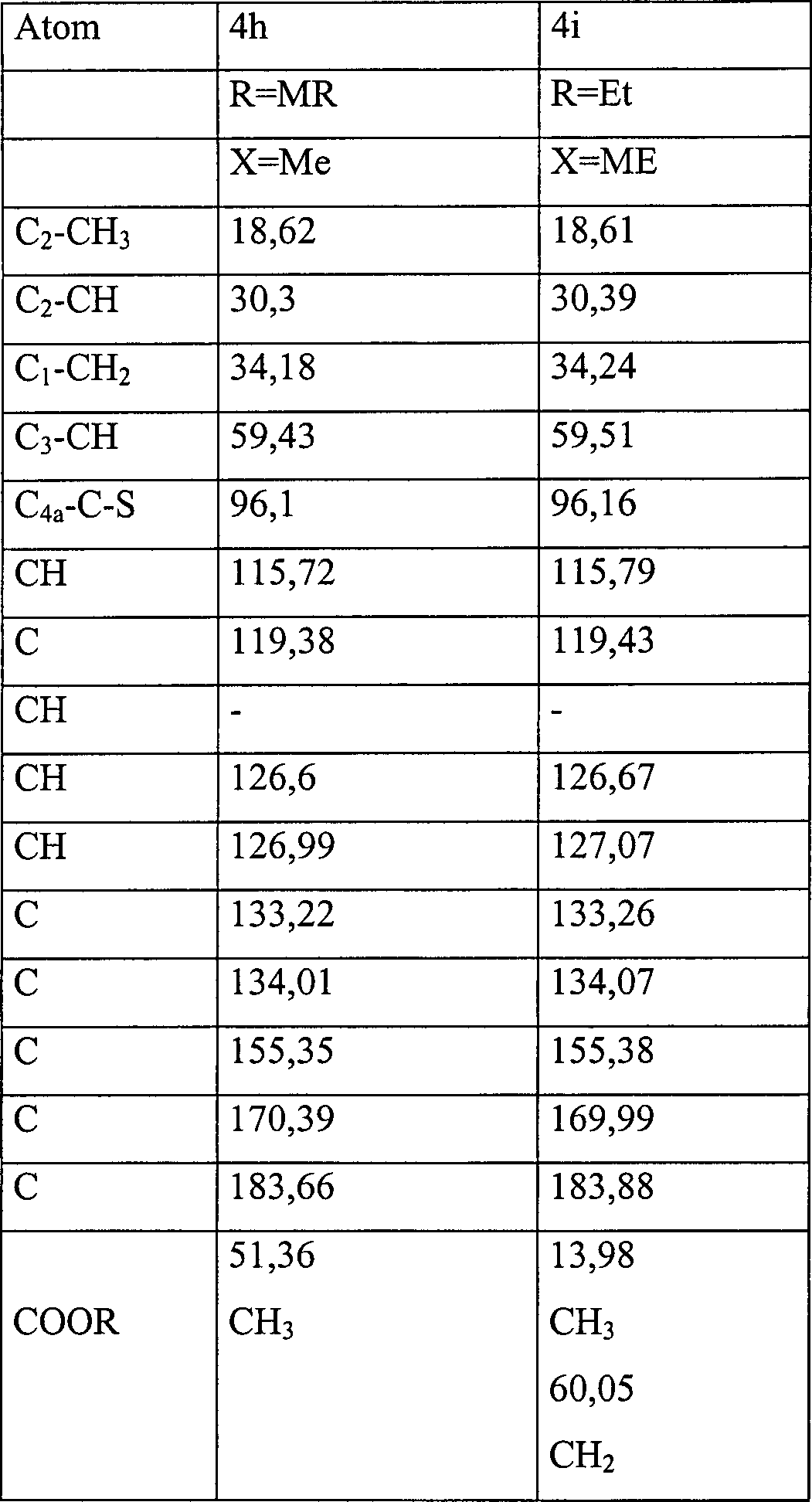
- Die Stereochemie über die C2-C3-Bindung der Phenothiazine 4a–c und 4e–i wird von der 1H-NMR-Spektroskopie bestätigt, die das Vorhandensein eines Methin-Dubletts bei etwa d 3,1 mit JH-H 11 Hz angibt. Wasserstoffatome bei C1,2 fallen als 3H Multiplett bei d 2,3 zusammen. Zuweisungen zu den H8,9 aromatischen Wasserstoffen können durch Feststellen der schwachen meta-Kopplung (J = 3,0 Hz) von H6-8 eindeutig gemacht werden. Um zwischen den isomeren Formen 4 und 5a zu unterscheiden, wird ein Nuclear Overhauser Differenzexperiment durchgeführt. Die Konfiguration des Schwefelatoms mit Bezug auf die Carbonylkohlenstoffatome wird bestätigt. Des weiteren ermöglicht die 2D-Korrelation dieser und anderer Signale 13C Zuweisungen.
-
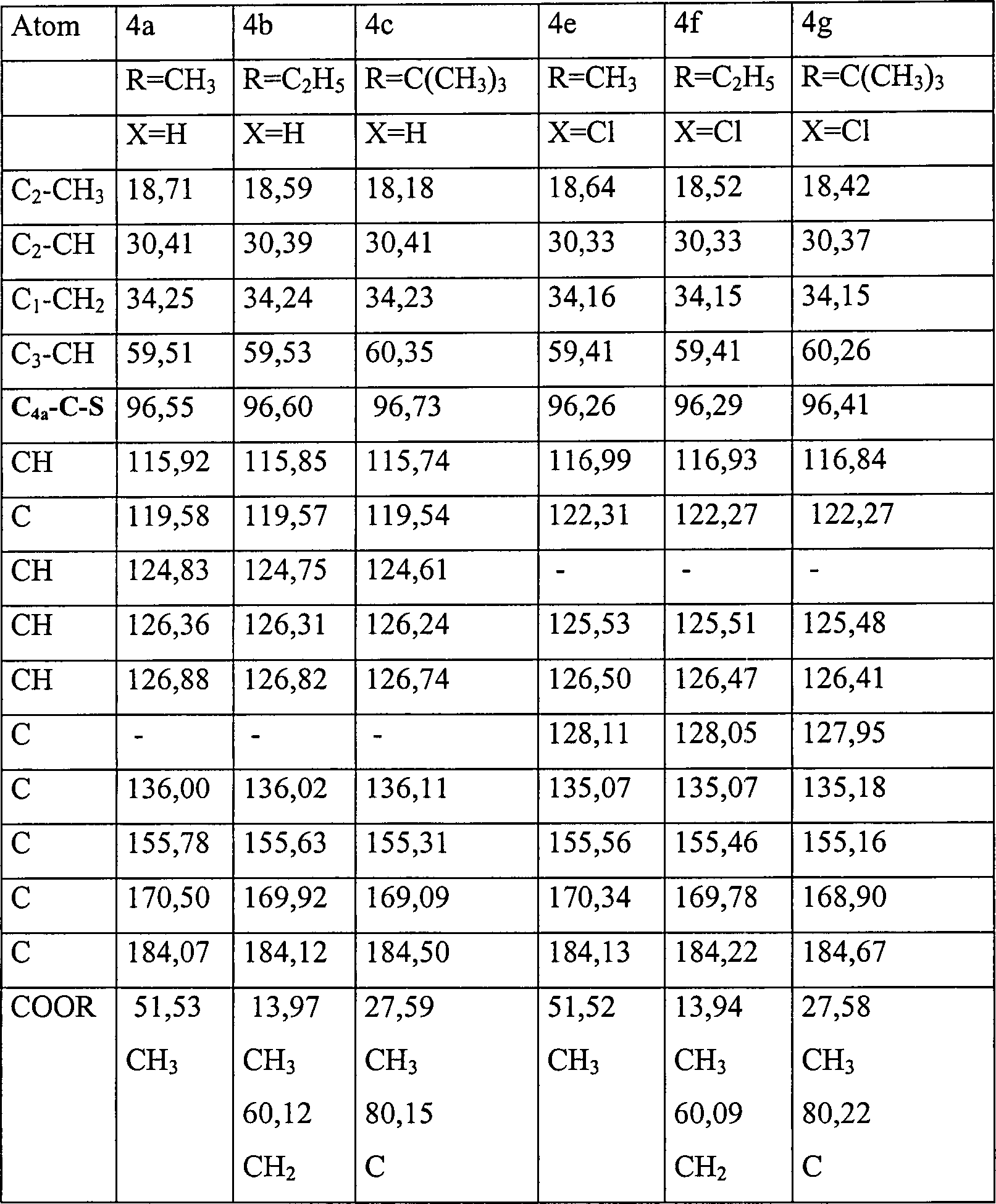
- 13C NMR-Daten werden auf einem General Electric QE 300-mHz Spektrometer in deuterierten Lösungsmitteln unter Verwendung von Tetramethylsilan als internem Standard aufgezeichnet und sind in Tabelle N angegeben.
- Tabelle IV. 13C NMR (DMSO-d6) Korrelationstabelle für 2,3-Dihydro-1H-phenothiazin-4(10H)-one
- Tabelle IV (Forts.)
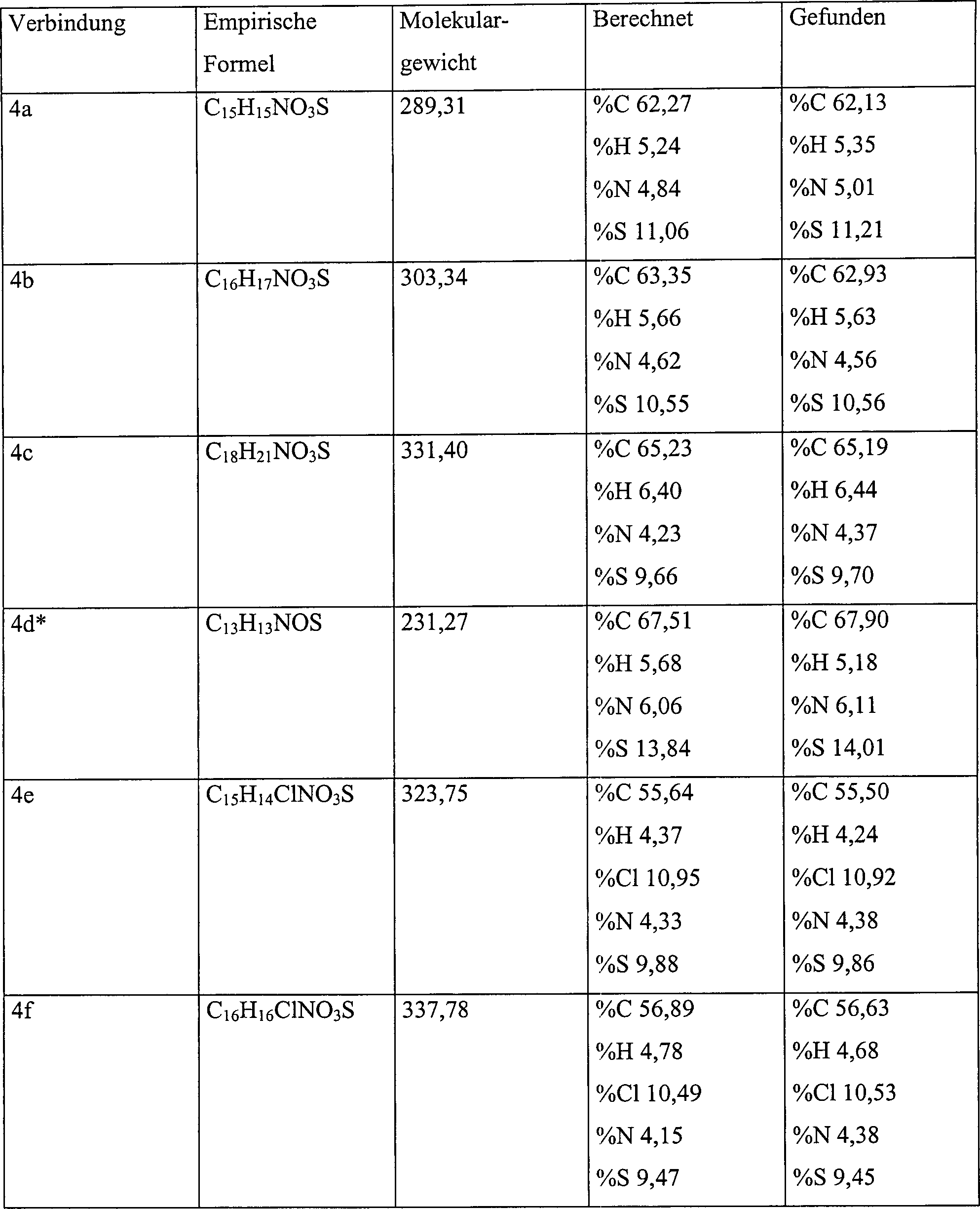
- Elementaranalysen (C, H, N, S und Halogen) werden durchgeführt. Die Ergebnisse sind in Tabelle V gezeigt, wo die Analysen nur durch die Symbole der Elemente angegeben sind.
- Tabelle V. Analytische Ergebnisse
-

-
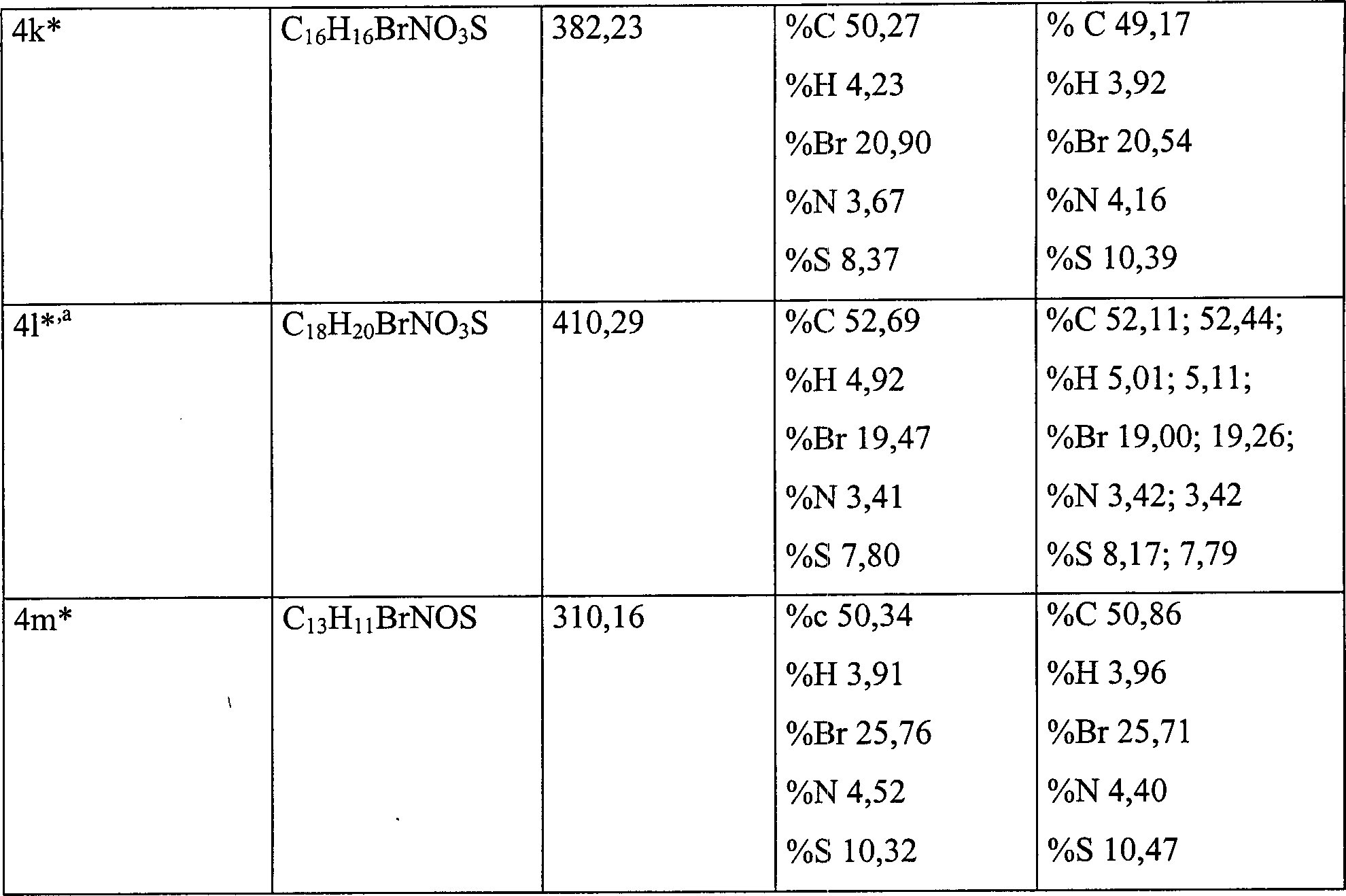
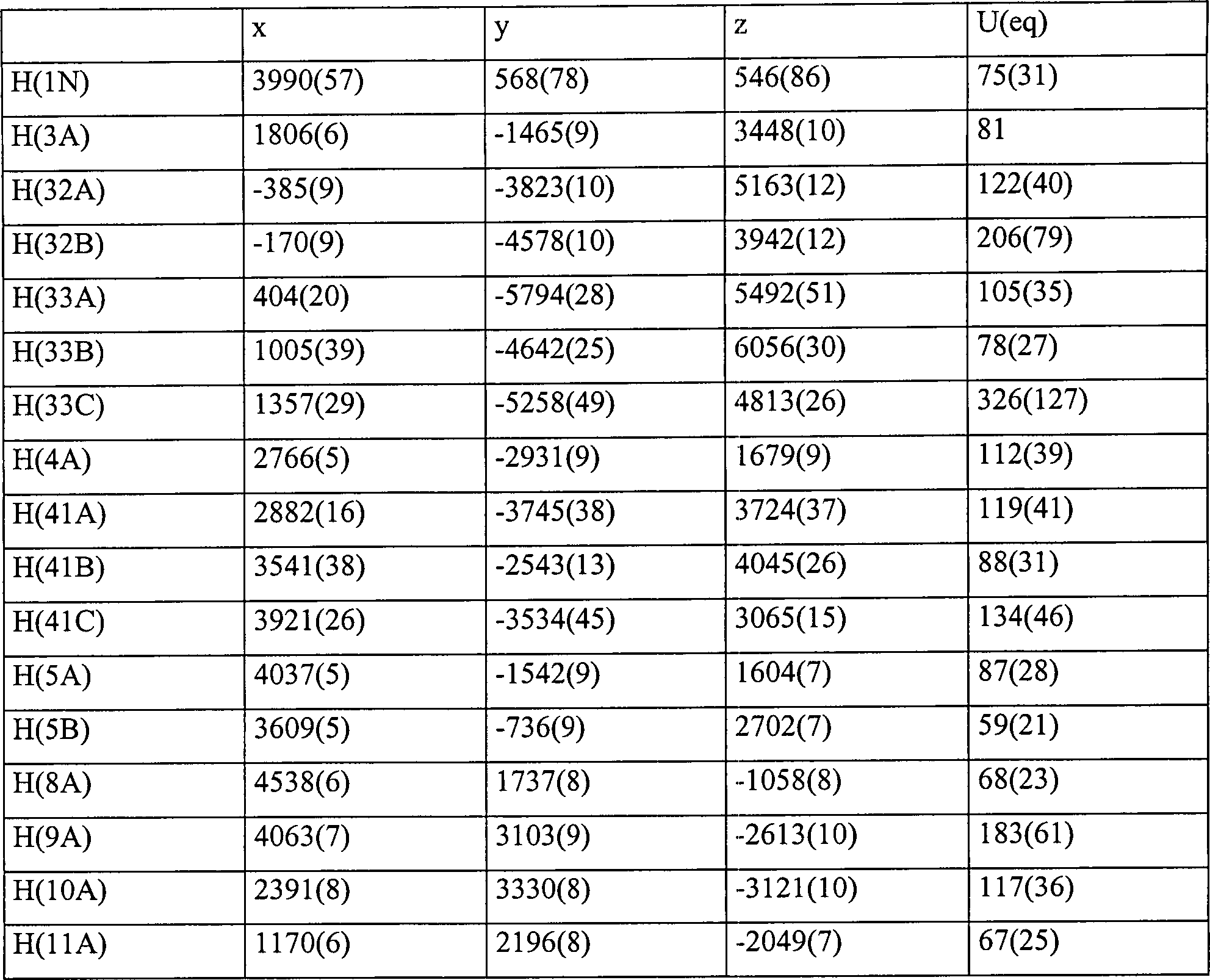
- Kristallographische Daten wurden auch für die Verbindung 4b erhalten. Diese Daten zusammen mit einer Strukturverfeinerung sind in der nachstehenden Tabelle VIa angegeben. Verbindungslängen (Å) und Winkel (°) für die Verbindung 4b sind in der nachstehenden Tabelle VIb angegeben. Atomkoordinaten [× 104] und äquivalente isotrope Verschiebungsparameter [Å2 × 103] sind in Tabelle VIc angegeben, in der U(eq) als ein Drittel des orthogonalisierten Uij Tensors definiert ist. Anisotrope Verschiebungsparameter [Å × 103] für die Verbindung 4b sind in der nachstehenden Tabelle IVd angegeben. Der anisotrope Verschiebungsfaktorexponent nimmt die Form: –2n2 [(ha*)2 U11 + .... + 2hka*b*U12] an. Wasserstoffkoordinaten (× 104) und isotrope Verschiebungsparameter (Å2 × 103) für die Verbindung 4b sind in der Tabelle VIe gezeigt.
- Die Röntgenkristallstrukturen, die für die Verbindung 4b bestimmt wurden, sind allein und innerhalb einer Elementarzelle in
1 bzw. 2 gezeigt. TABELLE VIaIdentifikationscode mll Empirische Formel CH16H17NO3S Formelgewicht 303,37 Temperatur 293(2) K Wellenlänge 0,71073 Å Kristallsystem Orthorhombisch Raumgruppe Pca21 Elementarzellenabmessungen a = 13,257(4) Å α = 90° b = 10,439(3) Å β = 90° c = 10,875(3) Å γ = 90° Volumen 1505,0 (8) Å3 Z 4 Dichte (berechnet) 1,339 Mg/m3 Absorptionskoeffizient 0,224 mm–1 F(000) 640 Kristallgröße 0,13 × 0,52 × 0,26 mm θ Bereich für Datensammlung 3,07 bis 24,99° Indexbereiche 0 ≤ h ≤ 15, –12 ≤ k ≤ 0, 0 ≤ M ≤ 12 Gesammelte Reflektionen 1399 Unabhängige Reflektion 1399 (Rint = 0,0000) (fortgesetzt) Verfeinerungsverfahren Vollmatrix der kleinsten Quadrate an F2 Daten/Beschränkungen/Parameter 1399/1/211 Anpassungsgüte an F2 1,067 Endgültige R Indices [I > 2σ(I)] R1 = 0,0576, wR2 = 0,1177 R Indices (alle Daten) R1 = 0,1026, wR2 = 0,1419 Absolute Strukturparameter – größte Differenz zwischen 0,1 (2) Peak und Minimum 0,284 und –0,208 eÅ–3 - Tabelle VIb
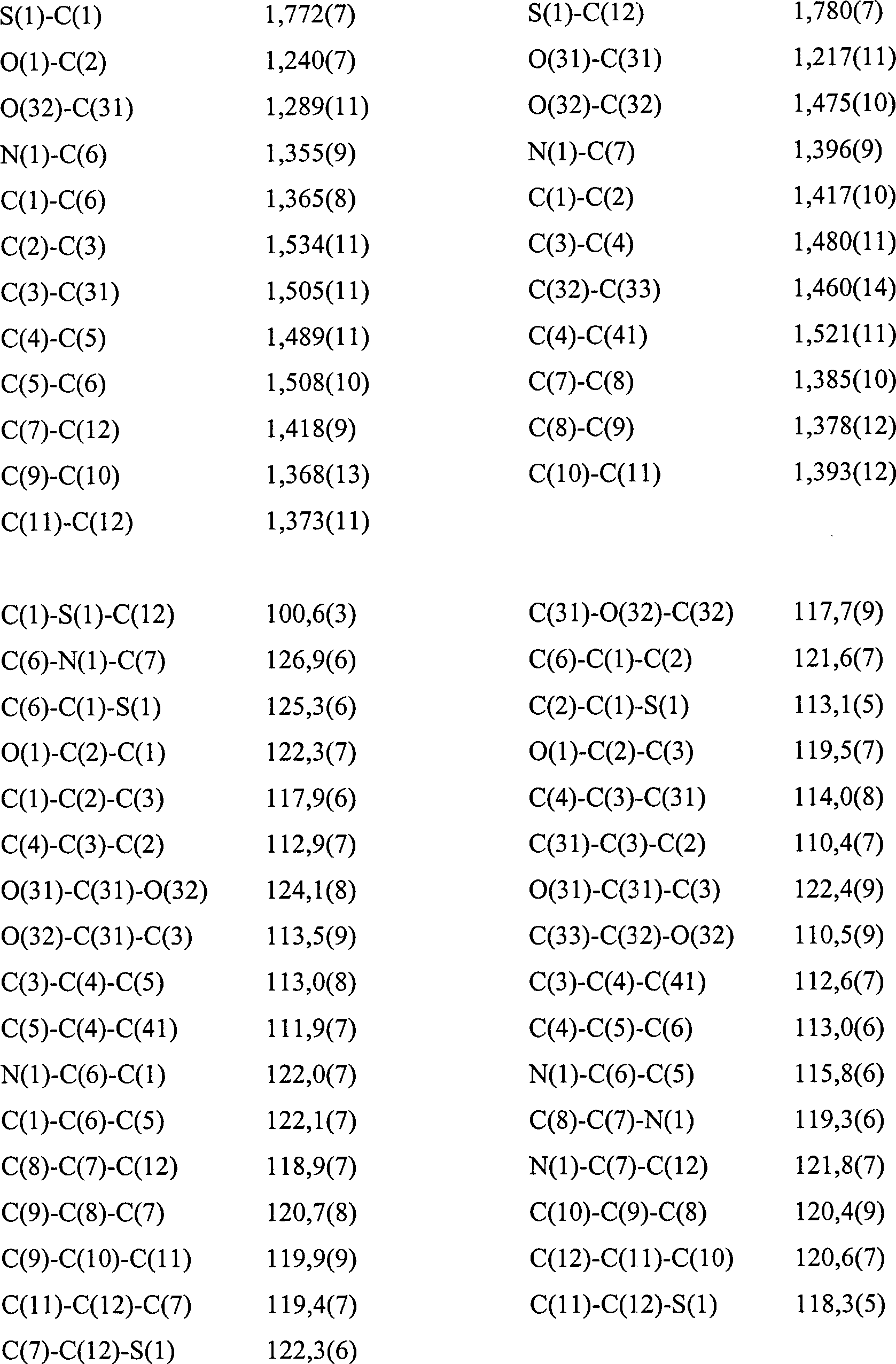
- Zur Erzeugung äquivalenter Atome verwendete Symmetrietransformationen:
- Tabelle VIc
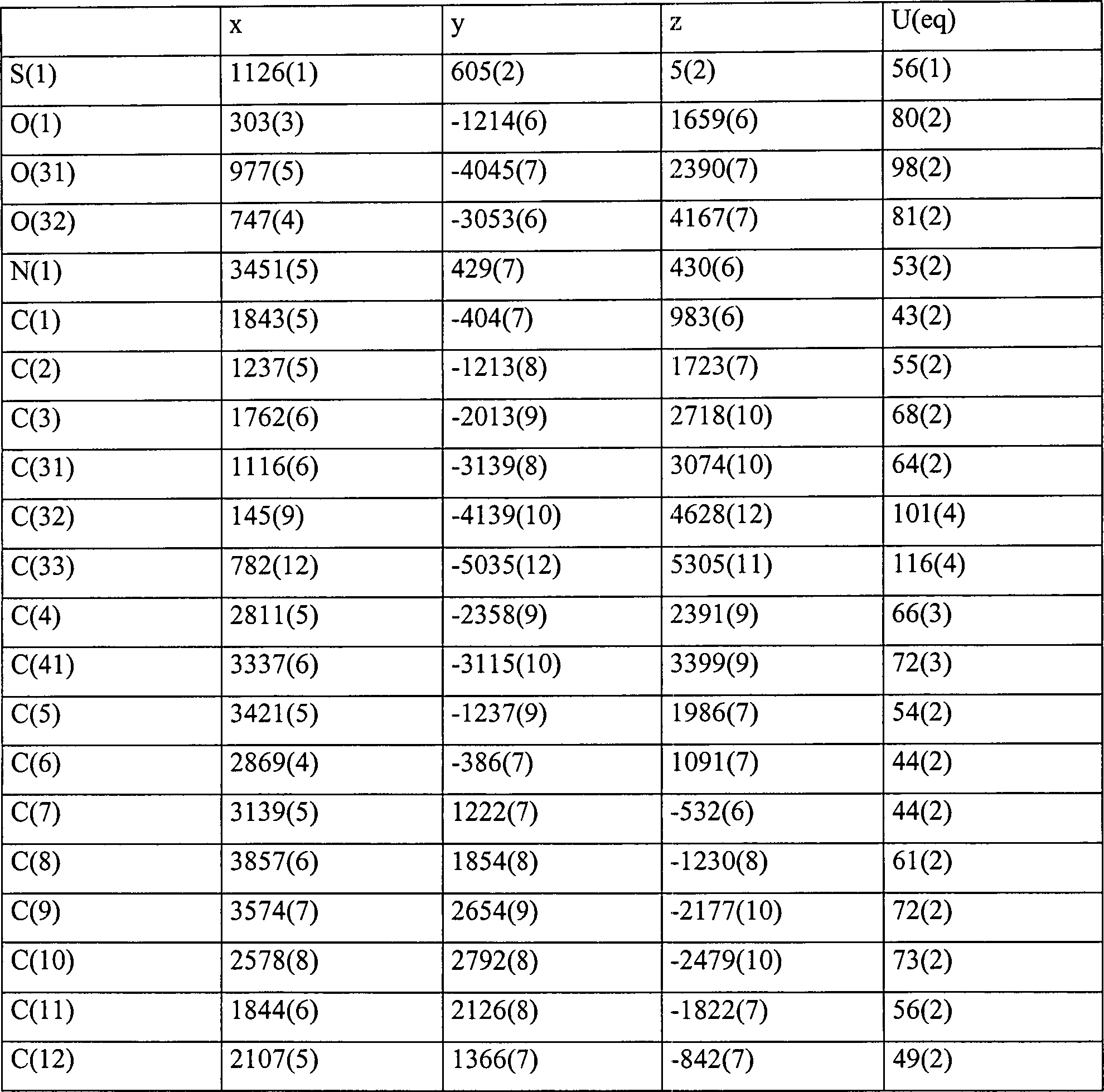
- Tabelle VId

- Tabelle VIe
- Synthese
- ter-Butyl-6-methyl-2,4-dioxocyclohexancarboxylat, Methyl-6-methyl-2,4-dioxo-cyclohexancarboxylat, Ethyl-6-methyl-2,4-dioxocyclohexancarboxylat, 5-Chlor-2-aminobenzolthiol, 5-Methyl-2-aminobenzolthiol und 5-Brom-2-aminobenzolthiol werden mittels Verfahren der Literatur hergestellt. Friary, R. J.; Gilligan, J. M.; Szajewski, R. P.; Falci, K. J.; Franck, R. W.: Heterocyclic syntheses via the intramolecular acylation of enamines derived from animo acids. J. Org. Chem. 1973, 38, 3487–3490. Edafiogho, I. 0.; Hinko, C. N.; Chang, H.; Moore, J. A.; Mulzac, D.; Nicholson, J. M.; Scott, K. R.: Synthesis and anticonvulsant activity of enaminones. J. Med. Chem. 1992, 35, 2798–2805. Spencer, T. A.; Newton, M. D.; Baldwin, S. W.: Condensation of diethyl malonate with methyl vinyl ketone. J. Org. Chem. 1964, 29, 787–789. Mital, R.; Jain, S. K.: Synthesis of some 5-substituted 2-aminobenzenethiols and the conversion into phenothiazines via Smiles rearrangement. J. Chem. Soc. (C) 1969, 2148–2150. 2-Aminothiophenol, Natrium, Ethylcrotonat, Methylacetoacetat, Ethylacetoacetat, tert.-Butylacetoacetat, Kaliumthiocyanat, 4-Chloranilin, p-Toluidin und Brom werden von der Aldrich Chemical Company erhalten und ohne Reinigung verwendet.
- Methyl-6-methyl-2,4-dioxo-cyclohexancarboxylat (7a)
- Einer frisch hergestellten Lösung aus Natrium (17,78 g, 0,77 Grammatom) in Methanol (220 ml) wird Methylacetoacetat 89,66 g (0,77 Mol) zugegeben, und die Mischung wird auf einem Eisbad 15 Minuten nach der Zugabe gerührt. Ethylcrotonat (100 ml eines 96%igen Produkts ≈ 88,13 g (100%; 0,77 Mol) wird tropfenweise zugegeben, und die Mischung wird bei Raumtemperatur weitere 30 Minuten gerührt. Nach dem Erhitzen unter Rückfluß (2 Std.) wird Methyl-6-methyl-2,4-dioxo-cyclohexancarboxylatenolat, das sich abtrennt, gefiltert, und der feste Rückstand wird in einer minimalen Menge kalten Wassers gelöst. Die wässerige Lösung wird mit Schwefelsäure (500 ml einer 2 M Lösung) angesäuert, der Niederschlag wird mit Dichlormethan (4 × 300 ml) extrahiert und die organische Phase über Natriumsulfat getrocknet. Der Rückstand wird eingedampft und der Rückstand aus Toluol umkristallisiert, um die Titelverbindung zu ergeben: Ausbeute 37 g, 40%, Schmelzpunkt 122–123°C. Die Mutterlauge der Reaktion wird zur Trockene eingedampft, in kaltem Wasser gelöst und mit Dichlormethan (nach Ansäuern mit den gleichen 2 M Schwefelsäure wie zuvor verwendet) extrahiert. Nach Eindampfen und Umkristallisieren aus Toluol ist der Schmelzpunkt identisch demjenigen der ursprünglichen Ausbeute. Gesamtausbeute: 47 g; 51%.
- Ethyl-6-methyl-2,4-dioxocyclohexancarboxylat (7b)
- Das gleiche Verfahren wie vorstehend verwendet wird unter Verwendung einer äquivalenten Menge wasserfreien Ethanols als Lösungsmittel und Ethylacetoacetat modifiziert; Ausbeute 93,4 g (47%); Schmelzpunkt 89–91°C (aus Ethylacetat: Petrolether, Siedepunkt 54°C).
- ter-Butyl-6-methyl-2,4-dioxocyclohexancarboxylat (7c)
- Das gleiche Verfahren wie vorstehend verwendet wird unter Verwendung einer äquivalenten Menge wasserfreien Ethanols als Lösungsmittel und tert.-Butylacetoacetat modifiziert; Ausbeute 90,2 g (44%); Schmelzpunkt 145–146°C (lit. Schmelzpunkt 130–131,5°) aus Aceton : Wasser.
- 5-Chlor-2-aminobenzolthiol (6, X=Cl)
- Kaliumthiocyanat (38,8 g, 0,40 Mol) und 4-Chloranilin (50,9 g, 0,40 Mol) werden in 300 ml Eisessig gelöst. Die Lösung wird auf unterhalb 10°C in einem Eisbad gekühlt und unter Rühren wird Brom (10 ml, 0,40 Mol) in Eisessig (50 ml) tropfenweise während einer Stunde zugegeben, wobei die Temperatur unterhalb 10°C mit externer Kühlung gehalten wird. Rühren wird für weitere 30 Minuten durchgeführt. Die Reaktionsmischung wird bei Raumtemperatur 16 Stunden gerührt. Das Hydrobromidsalz aus 2-Amino-6-chlorbenzothiazol 5 (X=Cl) wird mit Hexan (2 × 25 ml) gewaschen. Das Salz wird in warmem Wasser (300 ml) gelöst, und das Produkt wird durch Zugabe von verdünntem (10%igen) Natriumhydroxid ausgefällt. Das rohe Produkt wird gefiltert und aus Ethanol umkristallisiert (Schmelzpunkt 198–200°C, Ausbeute 16,47 g, 63,3%). Eine Mischung aus 2-Amino-6-chlorbenzothiazol 5 (15 g, 0,08 Mol), Kaliumhydroxid (75 g, 1,34 Mol) und Wasser (150 ml) wird unter Rühren und unter Rückfluß über Stickstoff 8 Stunden erhitzt. Die heftige Reaktion läßt langsam nach. Die Mischung wird dann mit Wasser (20 ml) verdünnt und gefiltert. Dem Filtrat werden 5 N Essigsäure unter kräftigem Rühren und Kühlen zugegeben, bis es gerade zu pH Papier sauer ist. Die gelben Kristalle werden gefiltert, mit kaltem Wasser gewaschen und aus absolutem Ethanol umkristallisiert und mit neutralem Norit entfärbt. Ausbeute von 5-Chlor-2-aminobenzolthiol (6, X=Cl); 40%; Schmelzpunkt 108–110°C (lit. 110°C).
- 5-Methyl-2-aminobenzolthiol (6, X=CH3)
- Das gleiche Verfahren wird mit p-Toluidin wiederholt, um 2-Amino-6-methylbenzothiazol 5 (X=CH3) 12,47 g (87,4%); Schmelzpunkt 128–130°C aus Methanol zu ergeben. 5-Methyl-2-aminobenzolthiol (6, X=CH3) wird in 38%iger Ausbeute mittels der durch Kaliumhydroxid katalysierten Hydrolyse während 8 Stunden hergestellt; Schmelzpunkt 112–113°C (lit. 113–115°C) aus absolutem Ethanol.
- 5-Brom-2-aminobenzolthiol (6, X=Br)
- Das gleiche Verfahren wurde mit p-Bromanilin wiederholt, um 2-Amino-6-brombenzothiazol 5 (X=Br) 21,47 g (94,4%); Schmelzpunkt 213–214°C aus Methanol zu ergeben. 5-Brom-2-aminobenzolthiol (6, X=Br) wurde in 38%iger Ausbeute mittels der durch Kaliumhydroxid katalysierten Hydrolyse während 8 Stunden hergestellt; Schmelzpunkt 112–113°C (lit. 113–115°C) aus absolutem Ethanol.
- 3-Carbomethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4a, R=H)
- Eine Mischung aus Methyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7a (5,52 g, 30 mMol) und 2-Aminothiophenol, 6 (X=H, 3,75 g, 30 mMol) in DMSO (10 ml) wird in einen vorerhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4a, (R=H), Schmelzpunkt 218–221°C ergibt sich als hellorangene Kristalle; Ausbeute: 1,47 g (55,0%).
- 3-Carbethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4b, R=H)
- Eine Mischung aus Ethyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 76 (5,94 g, 30 mMol) und 2-Aminothiophenol, 6 (X=H, 3,75 g, 30 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus EtOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4b, (R=H), Schmelzpunkt 201–202°C ergibt sich als hellorangene Kristalle; Ausbeute: 1,80 g (30,8%).
- 3-Carbo-ter-butoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4c, R=H)
- Eine Mischung aus ter-Butyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7c (3,10 g, 14 mMol) und 2-Aminothiophenol, 6 (X=H, 1,72 g, 14 mMol) in DMSO (10 ml) wird in einen vorerhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 10 Minuten unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4c, (R=H), Schmelzpunkt 221–222°C ergibt sich als hellorangene Kristalle; Ausbeute: 2,10 g (48,6%).
- 7-Chlor-3-Carbomethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4e, R=Cl)
- Eine Mischung aus Methyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7a (2,53 g, 13,7 mMol) und 5-Chlor-2-aminobenzolthiol, 6, (X=Cl, 2,20 g, 13,7 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4e, (R=Cl), Schmelzpunkt 226–227°C ergibt sich als hellorangene Kristalle; Ausbeute: 1,70 g (36,7%).
- 7-Chlor-3-carbethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4f, R=Cl)
- Eine Mischung aus Ethyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7b (2,6 g, 13,1 mMol) und 5-Chlor-2-aminobenzolthiol, 6 (X=Cl, 2,0 g, 13,1 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag, wird getrennt zweimal aus EtOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4f, (R=Cl), Schmelzpunkt 229–230°C ergibt sich als hellorangene Kristalle; Ausbeute: 2,05 g (49,4%).
- 7-Chlor-3-carbo-ter-butoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4g, R=Cl)
- Eine Mischung aus ter-Butyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7c (1,42 g, 6,3 mMol) und 5-Chlor-2-aminobenzolthiol, 6 (X=Cl, 1,00 g, 6,3 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 10 Minuten unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4g, (R=Cl), Schmelzpunkt 261–262°C ergibt sich als hellorangene Kristalle; Ausbeute: 1,12 g (50,9%).
- 3-Carbomethoxy-2,7-dimethyl-2,3-dihydro-1H-phenothiazin-410H)-on (4 h, R=Me)
- Eine Mischung aus Methyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7a (0,83 g, 4,5 mMol) und 5-Methyl-2-aminobenzolthiol, 6 (X=Me, 0,83 g, 4,5 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4 h, (R=Me), Schmelzpunkt 225°C ergibt sich als hellorangene Kristalle; Ausbeute: 0,39 g (21,7%).
- 3-Carbethoxy-2,7-dimethyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4i, R=Me)
- Eine Mischung aus Ethyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7b (1,43 g, 7,2 mMol) und 5-Methyl-2-aminobenzolthiol, 6 (X=Me, 1,0 g, 7,2 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird 0,5 Std. gerührt und unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus EtOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4i, (R=Me), Schmelzpunkt 224°C ergibt sich als hellorangene Kristalle; Ausbeute: 0,50 (21,9%).
- 2-Methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4d, FC=H)
- Eine Mischung aus ter-Butyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7c (3,09 g, 13,7 mMol) und 2-Aminothiophenol, 6 (X=H, 1,71 g, 13,7 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4d, (R=H), Schmelzpunkt 278°C ergibt sich als hellorangene Kristalle; Ausbeute: 0,46 g (15,4%).
- 7-Brom-3-carbomethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4j, R=Br)
- Eine Mischung aus Methyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7a (1,43 g, 7,7 mMol) und 5-Brom-2-aminobenzolthiol, 6 (X=Br, 1,44 g, 7,7 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4j, (R=Br), Schmelzpunkt 186–189°C ergibt sich als hellorangene Kristalle; 0,14 g (13,9%).
- 7-Brom-3-carbethoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4k, R=Br)
- Eine Mischung aus Ethyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7b (5,94 g, 30 mMol) und 5-Brom-2-aminobenzolthiol, 6 (X=Br, 5,8 g, 30 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 0,5 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4k, (R=Br), Schmelzpunkt 201–203°C ergibt sich als hellorangene Kristalle; 2,24 g (56,7%).
- 7-Brom-3-carbo-ter-butoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (41, R=Br)
- Eine Mischung aus ter-Butyl-6-methyl-2,4-dioxocyclohexan-1-carboxylat, 7c (1,42 g, 6,3 mMol) und 5-Brom-2-aminobenzolthiol, 6 (X=Br, 1,0 g, 4,5 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 10 Minuten bei 155°C erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 41, R=Br, Schmelzpunkt 191–194°C ergibt sich als hellorangene Kristalle; 1,99 g (77%).
- 7-Brom-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (4m, R=Br)
- Eine Mischung aus 7-Brom-3-carbo-ter-butoxy-2-methyl-2,3-dihydro-1H-phenothiazin-4[10H]-on (41 R=Br, 3,09 g, 13,7 mMol) in DMSO (10 ml) wird in einen vorherhitzten Heizmantel verbracht. Die Reaktionsmischung wird gerührt und 3 Std. unter Rückfluß erhitzt. Beim Abkühlen bildet die Reaktionsmischung einen Feststoff. Die Kristalle werden gefiltert und die verbleibende Mutterlauge wird in kaltes Wasser gegossen, woraufhin eine weitere Ausfällung stattfindet. Jeder Niederschlag wird getrennt zweimal aus MeOH umkristallisiert und erweist sich als identisch. Eine analytische Probe von 4m, (R=Br), Schmelzpunkt 250°C (dec.) ergibt sich als hellorangene Kristalle; 2,67 g (63,2%).
- Pharmakologie
- Die anfänglichen Bewertungen bezüglich einer antikonvulsiven Aktivität umfassen die Testverfahren der Phasen I, VIA und VIB. Diese Tests werden intraperitoneal bei männlichen Carworth Farms Nr. 1 (CF1) Mäusen, die 18 bis 25 g wiegen (Phase n, oder oral bei männlichen Sprague-Dawley Ratten, die 100 bis 150 g wiegen (Phasen VIA und VIB), durchgeführt. Die Bewertung basiert auf drei Tests: maximaler Elektroschock (MES), subkutan (scMet) und dem Rotorod-Test mit Bezug auf neurologische Toxizität (Tox). (a) Anticonvulsant Screening Project, Antiepileptic Drug Development Program, National Institutes of Health, DHEW Publ (NIH)(U.S.) 1978, NIH 78–1093. (b) Porter, R. J.; Cereghino, J. J.; Gladding, G. D.; Hessie, B. J.; Kupferberg, H. J.; Scoville, B.; White, B. G.: Antiepileptic drug development program. Cleveland Clin. Q. 1984, 51, 293–305. (c) Krall, R. L.; Penry, J. K.; White, B. G.; Kupferberg, H. J.; Swinyard, E. A.: Antiepileptic drug development: II. Anticonvulsant drug screening. Epilepsia 1978, 19, 400–428. Beim MES-Test werden maximale Elektroschock-Anfälle mit einem 60 Perioden Wechselstrom einer Stärke von 50 mA (5 bis 7 mal derjenigen, die notwendig ist, um minimale Elektroschock-Anfälle auszulösen), ausgelöst, der über Hornhautelektroden 0,2 Sekunden zugeführt wird. Ein Tropfen einer 0,9%igen physiologischen Kochsalzlösung wird auf das Auge vor der Anlegung der Elektroden verbracht, um den Tod des Tiers zu verhindern. Das Abschaffen der tonischen Extensionskomponente des Hinterbeins beim Anfall wird als Schutz definiert und die Ergebnisse sind als Anzahl der geschützten Tiere/Anzahl der getesteten Tiere ausgedrückt.
- Bei dem sc MET-Test werden 85 mg/kg Pentylentetrazol, das bei mehr als 95% der Mäuse Anfälle verursacht, als 0,5%ige Lösung subkutan in der hinteren Mittellinie verabreicht. Das Tier wird 30 Minuten beobachtet. Das Nichtbeobachten von selbst einem Schwellenanfall (einer einzigen Episode von klonischen Spasmen einer Dauer von mindestens 5 Sekunden) wird als Schutz definiert, und die Ergebnisse werden als Anzahl der geschützten Tiere/Anzahl der getesteten Tiere ausgedrückt.
- Der Rotorod-Test wird zur Bewertung der Neurotoxizität verwendet. Das Tier wird auf eine gerändelte Kunststoffstange mit einem Durchmesser von 1 Zoll verbracht, der mit 6 UpM gedreht wird. Normale Mäuse können unbegrenzt auf einer Stange verbleiben, die sich bei dieser Geschwindigkeit dreht. Neurologische Toxizität wird definiert als das Nichtverbleiben des Tiers auf der Stange während 1 Minute, und die Ergebnisse sind als Anzahl der Tiere, die Toxizität aufweisen/Anzahl der getesteten Tiere ausgedrückt.
- Bei der Phase I werden die Verbindungen in 0,5%iger wässeriger Methylcellulose suspendiert und intraperitoneal mit drei Dosierungsstärken (30, 100 und 300 mg/kg) verabreicht, wobei die antikonvulsive Aktivität und die motorische Beeinträchtigung 30 Minuten und 4 Stunden (und in einigen Fällen 2 Stunden und 6 Stunden) nach der Verabreichung festgestellt wurden. Die Ergebnisse des Testens der Phase I sind in der Tabelle VII gezeigt. Tabelle VII. Antikonvulsives Untersuchungsprojekt (ASP) – Testergebnisse der Phase I bei Mäusen (ip)Tabelle VII (Forts.)
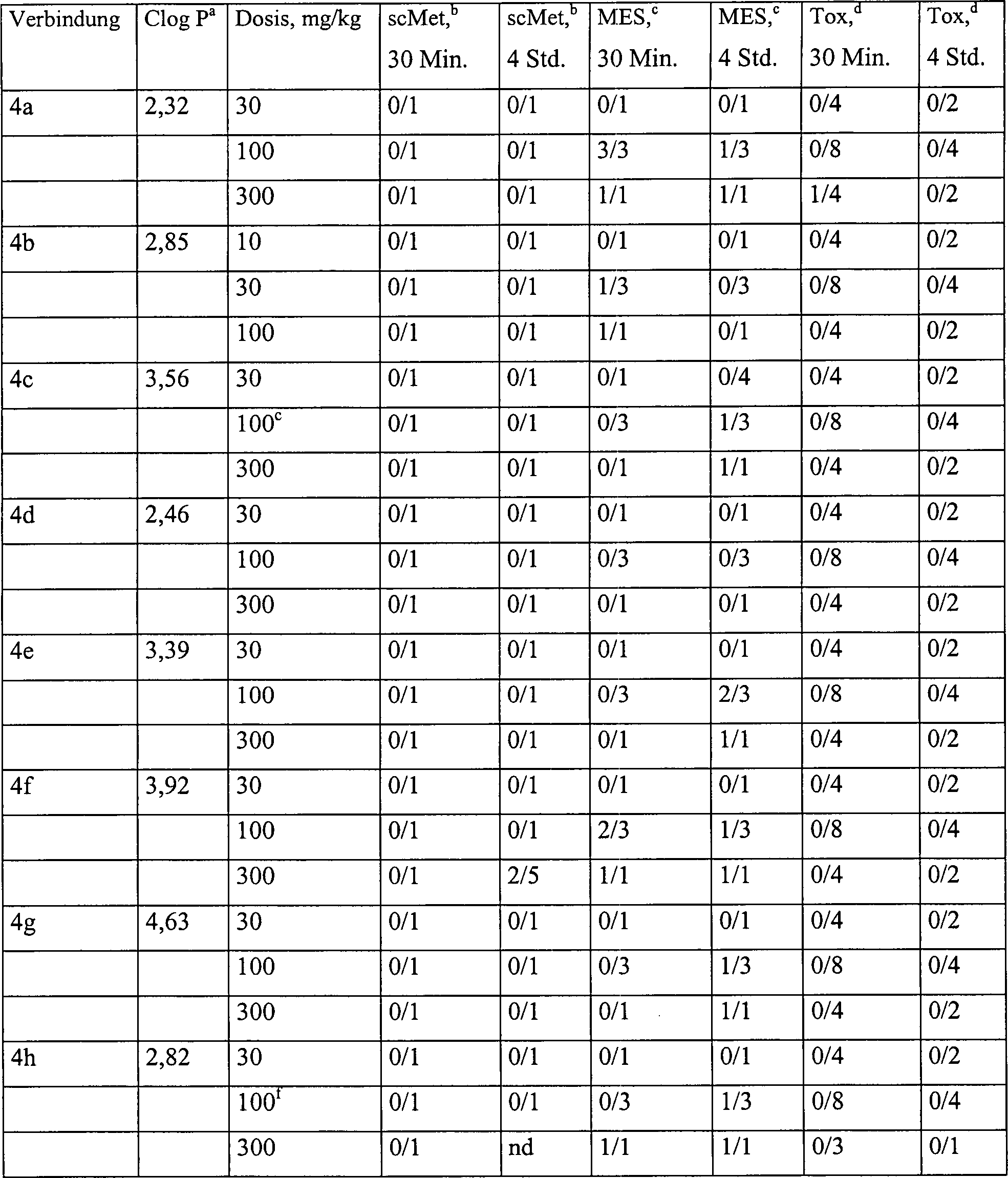

- nd
- nicht bestimmt.
- Um die Ergebnisse zwischen den Nagerspezien zu differenzieren, werden 4b (X=H) sowie 4a (R=H), 4e (R=Cl), 4f (R=Cl) und 4i (R=CH3) anschließend mit Bezug auf die orale (po) Aktivität bei Ratten (Phase VIA oder VIB) bewertet. Die Daten sind in Tabelle VIII angegeben. Tabelle VIII. Orale Daten bei RattenTabelle VIII (Forts.)

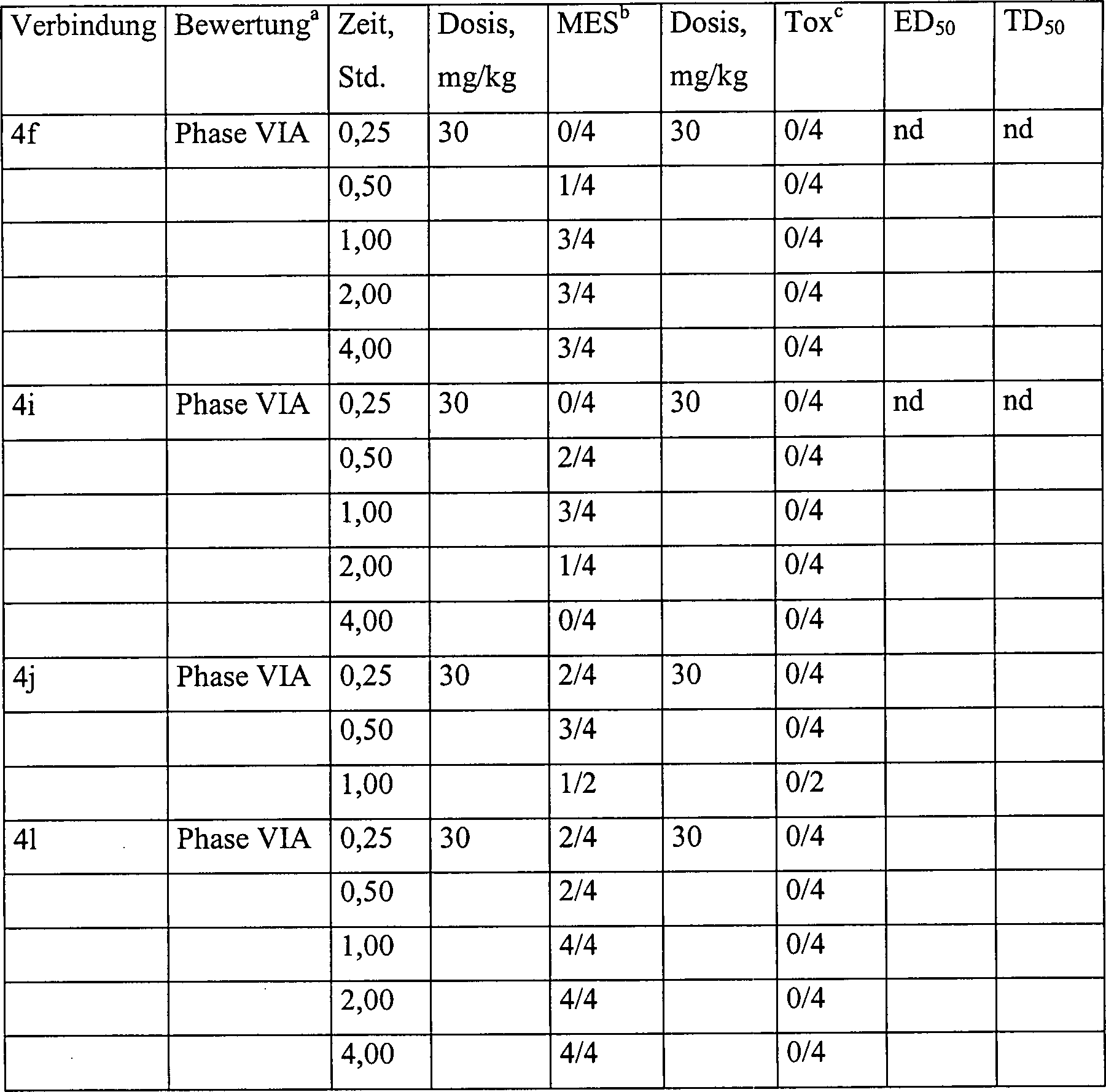
- nd
- nicht bestimmt
- Es ist zu beachten, daß sich die Quantifizierung der Wirksamkeit von 4b als schwierig erweist. Ein erster gemessener ED50 von 13,9 mg/kg wird mit einem zweiten gemessenen ED50 von 47,2 mg/kg und einem TD50 > 500 mg/kg verglichen, was einen Schutzindex (TD50/ED50) > 10,6 angibt. Dieser Unterschied bei den ED50-Ergebnissen ist sehr wahrscheinlich auf Löslichkeitschwierigkeiten zurückzuführen, da diese Verbindungen sehr wasserlöslich sind.
- Ein TTE-Test wird mit dem inaktiven Phenothiazin 4d (R=H) der Phase I durchgeführt. Piredda, S. G.; Woodhead, J. H.; Swinyard, E. A.: Effect of stimulus intensity on the profile of anticonvulsant activity of phenytoin, ethosuximide and valproate. J. Pharmacol. Exp. Ther. 1985, 232, 741–745. Der TTE-Test ist ein klinisch nichtselektives, elektrokonvulsives Anfallsmodell, das Verbindungen, die die Anfallsschwelle erhöhen, sowie diejenigen identifiziert, die eine Ausbreitung des Anfalls verhindern. Des weiteren kann dieser Test bestimmte Verbindungen identifizieren, die sowohl bei dem MES- als auch dem scMet-Test inaktiv sind. Der TTE-Test ist ähnlich der MES-Untersuchung, jedoch wird bei ihm ein geringeres Niveau des elektrischen Stroms verwendet. Der geringere Strom macht den TTE-Test empfindlicher, jedoch weniger unterscheidungskräftig als die MES-Untersuchung. Diese Fähigkeit macht das Modell attraktiv, weil es die Identifizierung von Verbindungen gestattet, die durch die Standardidentifizierungsuntersuchung weggelassen worden wären. Falls bei dem TTE-Test gefunden wird, daß eine Verbindung eine beträchtliche Aktivität hat, während sie bei der erneuten MES-Untersuchung inaktiv bleibt, wird sie ein Kandidat für ein fortgeschrittenes Testen. Diese TTE-aktiven Verbindungen können Verbindungen darstellen, die durch einen neuen Mechanismus wirken.
- 20 Mäuse werden mit 100 mg/kg der Testverbindung vorbehandelt. Bei mehreren Zeiträumen (15 Min., 30 Min., 1 Std., 2 Std. und 4 Std.) nach der Behandlung mit der Testverbindung werden vier Mäuse zu jedem Zeitpunkt einem elektrischen Strom von 12,5 mA 0,2 Sekunden über Hornhautelektroden ausgesetzt. Diese Stimulierung ruft einen TTE-Anfall bei den Tieren hervor. Für jeden Zeitraum werden die Ergebnisse als Verhältnis der Anzahl der geschützten Tiere zu der Anzahl der getesteten Tiere ausgedrückt. Die Ergebnisse für 4d sind in der Tabelle IX angegeben. Tabelle IX. Tonischer Schwellenextensions- (TTE)a-Test und MES-Bestätigung: Mäuse ip für 4d
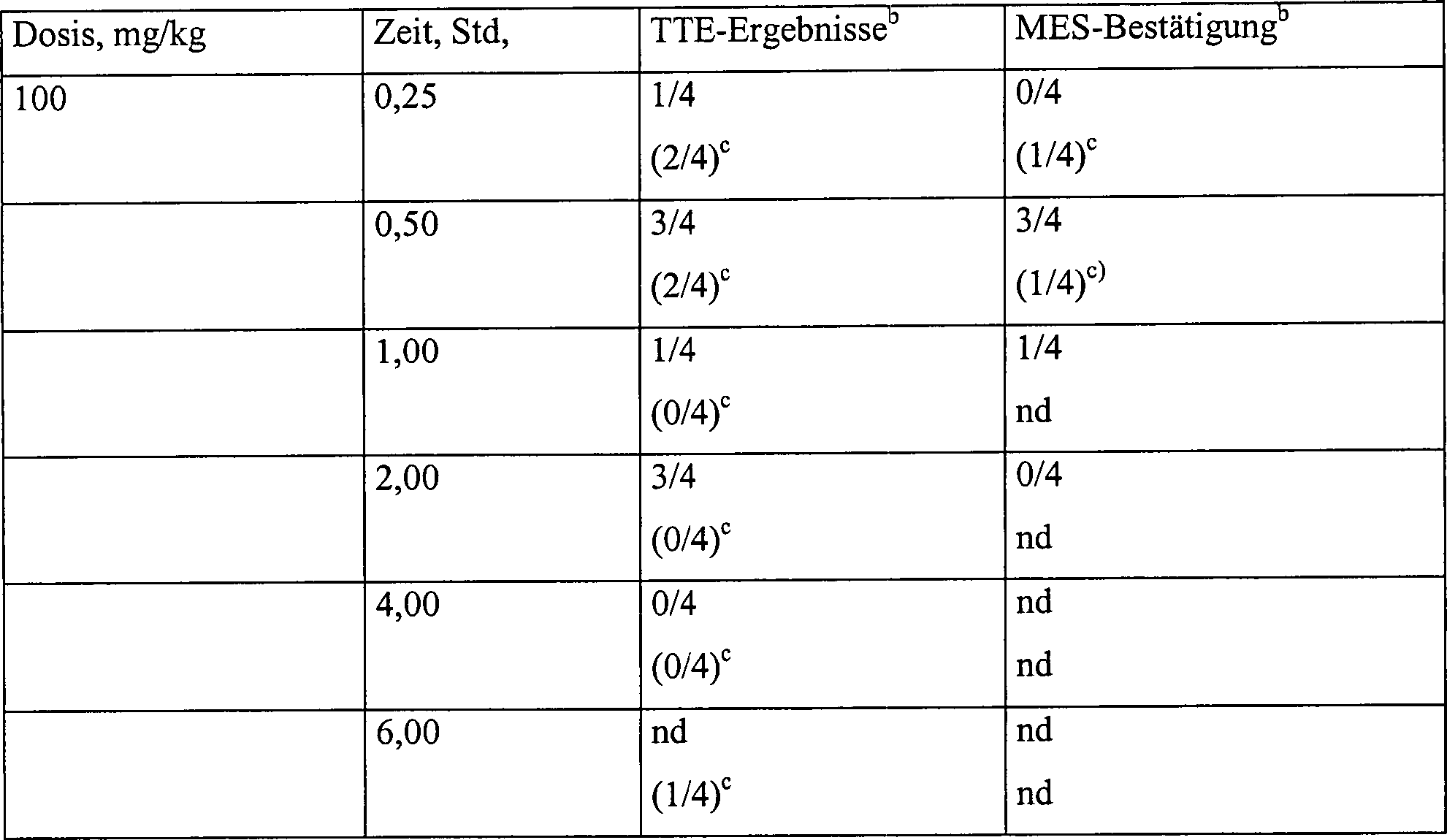
- nd
- nicht bestimmt.
Claims (9)
- Verwendung einer Phenothiazin-Verbindung der Formel:und pharmazeutisch verträglicher Salze davon, wobei R1 ein Wasserstoffatom oder -COOR ist, wobei R aus verzweigten oder unverzweigten Allcylresten ausgewählt ist, die 1 bis 4 Kohlenstoffatome enthalten, und wobei X aus einem Wasserstoffatom, verzweigten oder unverzweigten Alkylresten, die 1 bis 4 Kohlenstoffatome enthalten, und einem Halogenatom ausgewählt ist, zur Herstellung eines Arzneimittels zur Behandlung von Grand mal und partiellen Anfällen bei einem Säuger.

- Verwendung nach Anspruch 1, wobei X ein Wasserstoffatom ist, R1 ein Rest COOR ist und R eine Methylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Wasserstoffatom ist, R1 ein Rest COOR ist und R eine Ethylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Chloratom ist, R1 ein Rest COOR ist und R eine Methylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Chloratom ist, R1 ein Rest COOR ist und R eine Ethylgruppe ist.
- Verwendung nach Anspruch 1, wobei X eine Methylgruppe ist, R1 ein Rest COOR ist und R eine Ethylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Bromatom ist, R1 ein Rest COOR ist und R eine Methylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Bromatom ist, R1 ein Rest COOR ist und R eine Ethylgruppe ist.
- Verwendung nach Anspruch 1, wobei X ein Bromatom ist, R1 ein Rest COOR ist und R eine tert-Butylgruppe ist.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/958,320 US5994349A (en) | 1997-10-27 | 1997-10-27 | 3-carboalkoxy-2, 3-dihydro-1H-phenothiazin-4[10H]-one derivatives |
| US958320 | 1997-10-27 | ||
| PCT/US1998/022693 WO1999021560A1 (en) | 1997-10-27 | 1998-10-27 | 3-carboalkoxy-2,3-dihydro-1h-phenothiazin-4[10h]-one derivatives |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69821019D1 DE69821019D1 (en) | 2004-02-12 |
| DE69821019T2 true DE69821019T2 (de) | 2004-10-28 |
Family
ID=25500854
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69821019T Expired - Fee Related DE69821019T2 (de) | 1997-10-27 | 1998-10-27 | 3-carboalkoxy-2,3-dihydro-1h-phenothiazin-4(10h)-one derivate |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US5994349A (de) |
| EP (1) | EP1027055B1 (de) |
| AT (1) | ATE257382T1 (de) |
| AU (1) | AU748596B2 (de) |
| CA (1) | CA2308281C (de) |
| DE (1) | DE69821019T2 (de) |
| DK (1) | DK1027055T3 (de) |
| ES (1) | ES2212370T3 (de) |
| IL (1) | IL135842A (de) |
| PT (1) | PT1027055E (de) |
| WO (1) | WO1999021560A1 (de) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201536795A (zh) * | 2013-05-31 | 2015-10-01 | Medeia Therapeutics Ltd | 作為細胞保護劑用之芳基噻嗪化合物 |
| CN106117247B (zh) * | 2016-06-29 | 2018-02-16 | 东华大学 | 一种2‑甲基‑1,2,3,9‑四氢苯并[b]吡咯[1,4]‑噻嗪‑1,3‑二酮类化合物的制备方法 |
| KR102829709B1 (ko) | 2018-09-03 | 2025-07-04 | 에프. 호프만-라 로슈 아게 | 바이사이클릭 헤테로아릴 유도체 |
| EP3894411B1 (de) | 2018-12-13 | 2024-06-19 | F. Hoffmann-La Roche AG | 7-phenoxy-n-(3-azabicyclo[3.2.1]octan-8-yl)-6,7-dihydro-5h-pyrrolo[1,2-b][1,2,4]triazol-2-amin-derivate und ähnliche verbindungen als gamma-secretase-modulatoren zur behandlung von alzheimer's erkrankung |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3229122A1 (de) * | 1982-08-04 | 1984-02-09 | Bayer Ag, 5090 Leverkusen | Anellierte 4h-1,4-benzothiazine, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| DE3426564A1 (de) * | 1984-07-19 | 1986-01-30 | Bayer Ag, 5090 Leverkusen | Verwendung von 4h-1,4-benzothiazinen bei der prophylaxe und therapie von erkrankungen der atemwege, entzuendungen/rheuma, thromboembolischen erkrankungen, ischaemien und infarkten, herzrhythmusstoerungen, arteriosklerose und dermatosen, arzneimittel zu diesem zweck sowie wirkstoffe, die in diesen arzneimitteln enthalten sind |
| US5580894A (en) * | 1996-01-11 | 1996-12-03 | Howard University | Isoxazolyl enaminones |
-
1997
- 1997-10-27 US US08/958,320 patent/US5994349A/en not_active Expired - Fee Related
-
1998
- 1998-10-27 CA CA002308281A patent/CA2308281C/en not_active Expired - Fee Related
- 1998-10-27 PT PT98955131T patent/PT1027055E/pt unknown
- 1998-10-27 WO PCT/US1998/022693 patent/WO1999021560A1/en not_active Ceased
- 1998-10-27 DK DK98955131T patent/DK1027055T3/da active
- 1998-10-27 AT AT98955131T patent/ATE257382T1/de not_active IP Right Cessation
- 1998-10-27 DE DE69821019T patent/DE69821019T2/de not_active Expired - Fee Related
- 1998-10-27 AU AU12013/99A patent/AU748596B2/en not_active Ceased
- 1998-10-27 ES ES98955131T patent/ES2212370T3/es not_active Expired - Lifetime
- 1998-10-27 EP EP98955131A patent/EP1027055B1/de not_active Expired - Lifetime
- 1998-10-27 IL IL13584298A patent/IL135842A/en not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| WO1999021560A1 (en) | 1999-05-06 |
| DK1027055T3 (da) | 2004-04-26 |
| EP1027055B1 (de) | 2004-01-07 |
| IL135842A0 (en) | 2001-05-20 |
| IL135842A (en) | 2004-06-01 |
| US5994349A (en) | 1999-11-30 |
| AU748596B2 (en) | 2002-06-06 |
| DE69821019D1 (en) | 2004-02-12 |
| CA2308281A1 (en) | 1999-05-06 |
| EP1027055A1 (de) | 2000-08-16 |
| AU1201399A (en) | 1999-05-17 |
| PT1027055E (pt) | 2004-05-31 |
| CA2308281C (en) | 2004-08-24 |
| ATE257382T1 (de) | 2004-01-15 |
| EP1027055A4 (de) | 2001-12-05 |
| ES2212370T3 (es) | 2004-07-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE3852555T2 (de) | Substituierte 2-Aminobenzothiazole und Derivate verwendbar als cerebrovasculare Mittel. | |
| EP0002482B1 (de) | 4-Hydroxy-2H-1,2-benzothiazin-3-carboxamid-1,1-dioxide, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| EP0013376B1 (de) | Ein Isoxazolderivat, Verfahren zu seiner Herstellung und diese Verbindung enthaltende Mittel | |
| DE69613240T2 (de) | N-aminoalkylfluorencarboxamide, neue klasse von dopaminerezeptor subtype spezifische liganden | |
| CH667268A5 (de) | 1,2-benzisothiazol-3-ylpiperazine. | |
| CH650782A5 (de) | Carbostyrilverbindungen, verfahren zu deren herstellung und arzneimittel, welche diese enthalten. | |
| DE2915318A1 (de) | Cycloalkyltriazole, verfahren zu ihrer herstellung und diese enthaltende arzneimittel | |
| DE69005557T2 (de) | Substituierte 4-Amino-2-butinyl-Harnstoffe und -thioharnstoffe sowie deren Derivate als zentral wirkende muscarinische Mittel. | |
| DD281596A5 (de) | Verfahren zur hersteung antiarrhytmischer mittel | |
| DE69821019T2 (de) | 3-carboalkoxy-2,3-dihydro-1h-phenothiazin-4(10h)-one derivate | |
| DE2931418A1 (de) | Neue imidazochinoxaline und deren salze, verfahren zu deren herstellung, deren verwendung als arzneimittel und die sie enthaltenden pharmazeutischen zusammensetzungen | |
| DE3112984A1 (de) | 2-(3-(4-(3-chlor-4-fluorphenyl)-1-piperazinyl) propyl)-1,2,4-triazolo(4,3-a)pyridin-3(2h)-on dessen pharmazeutische anwendung und herstellung | |
| EP0061056A1 (de) | 5,6-Dimethyl-pyrrolo(2,3-b)pyridine, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| DE2535599C2 (de) | Substituierte Zimtsäureamide, Verfahren zu ihrer Herstellung und pharmazeutische Zubereitungen, welche diese Verbindungen enthalten | |
| DE2854727A1 (de) | Polyfluorhydroxyisopropyl-substituierte bicyclische und tricyclische carbostyrile, verfahren zu ihrer herstellung und sie enthaltende arzneimittel | |
| DE2912414A1 (de) | 2-(4-aethyl-1-piperazinyl)-4-phenylchinolin und dessen salze mit saeuren, verfahren zu ihrer herstellung und ihre verwendung als antidepressive wirkstoffe | |
| DD213922A5 (de) | Verfahren zur herstellung von phenothiazinverbindungen | |
| DE2448389A1 (de) | Derivate von 4-hydroxy-3-nitro-alpha- pyridonen enthaltende arzneimittel, neue 4-hydroxy-3-nitro-alpha-pyridone und verfahren zu ihrer herstellung | |
| DE3242442A1 (de) | Benzylidenderivate, verfahren zu ihrer herstellung und sie enthaltende arzneimittel | |
| DE68921470T2 (de) | Diazabicycloalkan-derivate. | |
| DE1095836B (de) | Verfahren zur Herstellung von in 10-Stellung basisch substituierten Phenthiazinderivaten | |
| DD251982A5 (de) | Verfahren zur herstellung von neuen in 11-stellung substituierten 5,11-dihydro-6h-pyride-(2,3-b)(1,4) benzodiazipin-6-onen | |
| DE3031161C2 (de) | ||
| DE2435381C2 (de) | 8-(2-Dimethylaminoäthyl)-3-oxo-4-phenyl-1-thia-4,8-diazaspiro[4.5]decan und seine Salze mit Säuren, Verfahren zu ihrer Herstellung und deren Verwendung in Arzneipräparaten | |
| DE3245950A1 (de) | Verfahren zur herstellung substituierter pyridine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8339 | Ceased/non-payment of the annual fee |