-
HINTERGRUND DER ERFINDUNG
-
Die
als Streptococcus pneumoniae (Pneumokokken, Pn) klassifizierten
pathogenen Bakterien wurden in 84 antigene Serotypen unterteilt,
basierend auf dem Kapselpolysaccharid (Pn-Ps) des Organismus. Krankheitszustände, die
diesen Organismen zugeschrieben werden können, umfassen Lungenentzündung, Meningitis,
Mittelohrentzündung,
Bakteriämie
und akute Verschlimmerungen von chronischer Bronchitis, Sinusitis, Arthritis
und Konjunktivitis. Der Großteil
dieser Erkrankungen wird jedoch von einer begrenzten Untergruppe der
84 bekannten Isolate verursacht Somit kann ein polyvalentes Vakzin,
welches die Pn-Ps der am häufigsten vorkommenden
und am stärksten
pathogenen Isolate des Organismus enthält, Schutz gegen einen sehr
hohen Prozentsatz der am häufigsten
genannten Pathogene dieser Klasse bieten.
-
Es
wurden polyvalente Vakzine hergestellt, welche zur Induktion schützender
Immunantworten gegen die Pneumokokken bei Erwachsenen wirksam sind. „PNEUMOVAX® 23” (Pneumokokkenvakzin,
polyvalentes, MSD; siehe PDR, Ausgabe von 1990, Seite 1431) ist
beispielsweise eine flüssige
Zusammensetzung, die 50 μg/ml
von jedem der 23 verschiedenen unkonjugierten Pneumokokken-Polysaccharide
enthält,
von denen alle bei der ATCC hinterlegt sind und eine mögliche Quelle
von Ausgangsmaterial für
diese Erfindung bieten. „PNEUMOVAX® 23” umfaßt jedes
der folgenden freien, d. h. unkonjugierten, Polysaccharide: 1, 2,
3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19F,
19A, 20, 22F, 23F und 33F, die etwa 90% der Pneumokokken-Blutisolate
ausmachen. Derartige Vakzine sind jedoch am wenigsten wirksam bei
dem Teil der Bevölkerung,
der das höchste
Risiko für
Pneumokokken-Infektionen
aufweist: Personen mit geschwächter B-Zell-Immunabwehr, ältere Personen
und Kleinkinder, die jünger
als zwei Jahre sind, welche für
den Immunschutz von T-Zell-Antworten abhängig sind. Nachdem unkonjugierte
Polysaccharide schlechte Induktoren von T-Zell-Immunantworten sind,
ist die Überführung der
Pn-Ps in Immunogene, welche zur Induktion von T-Zell-Antworten imstande
sind, der Schlüssel,
um bei dieser Zielpopulation einen adäquaten Schutz zu erzielen.
Die Verwendung ist jedoch nicht auf diese Gruppe von Individuen
beschränkt.
Beispielsweise induziert die Verabreichung eines Vakzins, das eines
oder mehrere der neuen Konjugate umfaßt, an einen weiblichen Säuger vor
oder während
der Schwangerschaft Antikörper
bei der Mutter, welche einen sich entwickelnden Fötus und
einen Säugling
passiv schützen
können,
selbst wenn das Vakzin dem Fötus
oder Kleinkind nicht direkt verabreicht wird. Solche Konjugat-Vakzine
sollten sich auch als geeignet erweisen zur Induktion von Antikörpern für einen
maximalen passiven Schutz von Risikogruppen wie Neugeborene oder
Geschwister von infizierten Individuen.
-
Polysaccharide,
die allein als wenig immunogen befunden worden waren, zeigten sich
als ziemlich gute Immunogene, sobald sie mit einem immunogenen Protein,
PRO, konjugiert sind [Marburg et al.,
US-Patent-Nr. 4,695,624 ;
Schneerson et al., New Dev. with Hum. & Vet. Vaccines, 77–94 (1980); Schneerson
et al., J. Exptl. Med., 152, 361 (1980); Anderson, Infection and
Immunity, 39, 233 (1983)]. Ein Hauptproblem bei der Herstellung
solcher Konjugate ist jedoch die viskose und inhomogene Natur des
Polysaccharid-Ausgangsmaterials und somit die Schwierigkeit bei
der chemischen Definition des Konjugatprodukts. Somit ist ein Verfahren erforderlich,
bei dem die Ausgangsmaterialien so gut wie möglich definiert sind und jeder
Schritt auf dem Syntheseweg hinsichtlich des gebildeten Zwischenprodukts überprüfbar ist.
Das hier beschriebene Verfahren genügt dieser Anforderung, indem
hochgradig immunogene Konjugat-Immunogene gegen die zugehörigen Pathogene,
von denen das Pn-Ps stammt, bereitgestellt werden. Die Konjugate
eignen sich für
Kleinkinder, die jünger
als zwei Jahre alt sind.
-
Marburg
et al., [J. Am. Chem. Soc., 108, 5282 (1986), und
US-Patent 4,695,624 ;
4,830,852 ;
4,882,317 ] offenbarten ein Mittel
zur Konjugation von Polysacchariden und immunogenen Proteinen durch
bigenerische Abstandhalter. Das PRO wurde derivatisiert, um nukleophile
oder elektrophile Seitengruppen zu zeigen (PRO*), während ein
Partner-Ps funktionalisiert wurde, um Seitengruppen der entgegengesetzten
Reaktivität
zu zeigen (Ps*). Nach der Kombination von Ps* mit PRO* wurden bigenerische
Abstandshalter gebildet, die Ps kovalent mit PRO verbanden (Ps-PRO).
Nach saurer Hydrolyse wird der bigenerische Abstandhalter als ungewöhnliche
Aminosäure
freigesetzt, durch Aminosäureanalyse
quantitativ bestimmt und dadurch ein Mittel zum Nachweis der Kovalenz
geboten.
-
EP-A-097407 offenbart
Vakzine gegen pathogene Bakterien mit Kapselpolysacchariden und
ein Verfahren zu deren Herstellung.
-
EP-A-208375 offenbart
Glykoprotein-Konjugate mit trivalenter immunogener Aktivität, die als
Vakzine eingesetzt wurden.
-
WO-A-8706267 offenbart
Antigene mit immunogenen Determinanten von Gruppe B-Streptococcus-Bakterien und Vakzine
gegen solche Bakterien.
-
Obwohl
die drei obigen Referenzen Techniken zur partiellen Hydrolyse und
Fraktionierung nach Größe und Reinheit
von Streptococcus pneumoniae-Polysaccharid-Antigenen beschreiben,
offenbaren oder nahelegen sie nirgends das verbesserte Verfahren,
das durch die vorliegende Erfindung bereitgestellt wird.
-
Diese
Erfindung offenbart ein Verfahren, welches gegenüber dem im
US-Patent 4,695,624 ;
4,830,852 ; und
4,882,317 offenbarten verbessert ist.
Die Verbesserungen umfassen die Präparation des Pn-Ps-Ausgangsmaterials,
welche spezifischere, reproduzierbarere und steuerbarere physikalische
Eigenschaften aufweist, als sie von rohen Pn-Ps-Präparationen
geboten werden, einschließlich:
erhöhte
Löslichkeit,
erhöhte
Filtrierbarkeit, erhöhte
Reinheit (Verringerung der Verunreinigung mit gruppenspezifischem
C-Polysaccharid (C-Ps)) und verringertes Molekulargewicht, verringerte
Polydispersität und
Viskosität.
Die resultierenden, hier offenbarten Konjugate sind hinsichtlich
Steigerungen in der Einheitlichkeit und Bequemlichkeit der Herstellung, besserer
Antigenizität
und besserer Reinheit des Endprodukts gegenüber denjenigen verbessert,
welche durch das Verfahren von
4,695,624 bereitgestellt
werden. Besonders signifikant ist die 3–20fache Verringerung von gruppenspezifischem
C-Polysaccharid und Peptidoglykan in den Pn-Ps vor der Konjugation
gegenüber den
Ps-Präparationen
des Patents
4,695,624 vor
der Konjugation. Obwohl die Anwesenheit der C-Polysaccharid-Verunreinigung
die Immunantworten gegen die typenspezifischen Antigene nicht stört, könnte C-Ps
an der Konjugationsreaktion teilnehmen und diese für das typenspezische
Ps weniger spezifisch und kontrolliert machen. Ferner kann die Produktion
von Anti-C-Polysaccharid-Antikörpern
mit der Gewebezerstörung
korrelieren, die bei einigen ungeklärten Pneumokokken-Infektionen
beobachtet wurde.
-
Diese
Erfindung offenbart neben dem neuen Konjugatprodukt ein neues Verfahren
zur Herstellung partiell hydrolysierter, hochgereinigter Pneumokokken-Polysaccharid-Zwischenprodukte,
neue Zusammensetzungen, die ein bis zehn verschiedene Konjugate
umfassen, und Verfahren zum Einsatz der Erfindung. Von besonderem
Interesse sind die in „PNEUMO-VAX® 23” (Pneumokokkenvakzin,
polyvalentes, MSD; siehe PDR, Ausgabe von 1990, Seite 1431) eingeschlossenen
Kapselpolysaccharide. Eine am meisten bevorzugte Untergruppe sind
die Kapselpolysaccharide der Streptococcus pneumoniae-Subtypen 6B,
23F, 19F, 14, 18C, 4 und 9V, da geschätzt wird, daß diese
kleine Gruppe von Pneumokokken-Subtypen für zwischen 75–85% der Pneumokokken-Infektionen
bei Kleinkindern und Kindern verantwortlich ist. Die hier bereitgestellten
Verfahren sind auf ein breites Spektrum von Pneumokokken- und anderen
bakteriellen Polysacchariden anwendbar.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Chemisch
gut definiertes Pneumokokken-Polysacharid (Pn-Ps) wird hergestellt
durch partielle Hydrolyse einer rohen Präparation von Pn-Ps auf einen
Endpunkt, der so vorherbestimmt ist, daß die antigene Integrität des Pn-Ps
aufrechterhalten wird. Das partiell hydrolysierte Pn-Ps ist im wesentlichen
gereinigt und geeignet zur Herstellung von Konjugaten von Pn-Ps
und immunogenem Protein (PRO), (Pn-Ps-PRO).
-
Neue
und hochgradig antigene Pn-Ps-PRO-Konjugate der Erfindung, welche
den Komplex des Proteins der äußeren Membran
(OMPC) von Neisseria meningitidis b oder rekombinante oder gereinigte
Untereinheiten davon, z. B. MIEP, oder andere immunogene Trägerproteine
in kovalenter Verknüpfung
mit partiell hydrolysierten und hochgereinigten Pn-Ps-Zwischenprodukten
von den am häufigsten
vorkommenden Pneumokokken-Isolaten umfassen, sind geeignet zur Verhinderung
von Pneumokokken-Infektionen bei Säugern. Die Konjugate sind von
besonderem Nutzen in Vakzin-Zusammensetzungen zur Stimulierung von
Anti-Pneumokokken-Immmunantworten bei Säugern, insbesondere bei Individuen
mit geschwächter
B-Zell-Immunabwehr, älteren
Individuen und menschlichen Kleinkindern, die jünger als zwei Jahre alt sind,
da die Konjugate T-Zell-Antworten hervorrufen. Pn-Ps-OMPC- und Pn-Ps-MIEP-Konjugate
werden hergestellt durch ein Verfahren, welches umfaßt die Schritte
der Isolierung von Kapsel-Ps aus Kulturen von Streptococcus pneumoniae (Pneumokokken,
Pn), partiellen Hydrolyse oder physikalischen Scherung der Pn-Ps,
Fraktionierung der Pn-Ps, um ein Pn-Ps-Produkt mit verringerter
Molekülgröße, Polydispersität, Vskosität zu ergeben,
und anschließenden
kovalenten Konjugation des Pn-Ps mit OMPC oder MIEF.
-
AUFGABEN DER ERFINDUNG
-
Eine
Aufgabe dieser Erfindung ist die Bereitstellung neuer, partiell
hydrolysierter und hochgereinigter, in antigener Hinsicht typenspezifischer
Pneumokokken-Kapselpolysaccharide (Pn-Ps), die als Zwischenprodukte
bei der Herstellung von T-Zell-abhängigen Konjugaten der Pn-Ps
und immunogenen Proteine geeignet sind. Eine weitere Aufgabe ist
die Bereitstellung von T-Zell-abhängigen Konjugaten der Pn-Ps
und immunogenen Proteine, geeignet für Vakzin-Zusammensetzungen,
um Pneumokokken-Infektionen zu verhindern, insbesondere bei Kleinkindern,
die jünger
als zwei Jahre alt sind, und bei Personen mit geschwächter B-Zell-Immunabwehr.
Eine weitere Aufgabe ist die Bereitstellung eines Verfahrens, welches
gegenüber
dem im
US-Patent Nr. 4,695,624 bereitgestellten
Verfahren verbessert ist, zur Herstellung von kovalenten Konjugaten
von Pneumokokken-Polysaccharid und immunogenem Protein (Pn-Ps-PRO),
worin die Verbesserung in der besseren chemischen Definiertheit
und Reinheit der Ausgangs-Pn-Ps, der Zwischenprodukte und des Endprodukts
und erhöhten
Einheitlichkeit und Leichtigkeit der Durchführung des Verfahrens selbst
besteht. Eine noch weitere Aufgabe ist die Bereitstellung von chemisch
definierten Pn-Ps-PRO-Konjugaten nach dem verbesserten Verfahren,
welche T-Zell-Abhängigkeit
zeigen, geeignet zur Induktion von schützendem Serum-Antikörper gegen pathogene
Pneumokokken, insbesondere bei Kleinkindern, die jünger als
zwei Jahre alt sind, und bei Individuen mit geschwächter Immunabwehr.
Eine weitere Aufgabe ist die Bereitstellung eines Behandlungsverfahrens unter
Einsatz dieser Konjugate in immunologisch wirksamen Mengen in Vakzin-Formulierungen,
um pneumokokken-induzierte Krankheiten wie Mittelohrentzündung, Meningitis,
Lungenentzündung,
Bakteriämie
und die akuten Verschlimmerungen von chronischer Arthritis, Sinusitis,
Bronchitis und Konjunktivitis zu verhindern.
-
DETAILLIERTE BESCHREIBUNG DER ERFINDUNG
-
A. Das neue Pn-Ps-PRO-Konjugat und Polysaccharid-Zwischenprodukte:
-
Das
Pn-Ps-PRO-Konjugatprodukt dieser Erfindung weist ein Verhältnis von
Pn-Ps zu PRO zwischen 0,05 und 0,5 mg Polysaccharid/mg Protein,
ein hohes Maß an
Kovalenz, eine minimale Verunreinigung des Konjugats durch freies
Pn-Ps und einzigartige pysikalische und chemische Eigenschaften
auf, die von den neuen Polysaccharid-Komponenten verliehen werden.
-
Das
Konjugat umfaßt
ein immunogenes Protein (PRO), das über einen Abstandhalter kovalent
an ein neues, partiell hydrolysiertes und hochgereinigtes Pneumokokken-Kapselpolysaccharid
(Pn-Ps) gekoppelt ist. Das immunogene Protein ist vorzugsweise der
Komplex des Proteins der äußeren Membran
(OMPC), der von einer Kultur von Neisseria meningitidis b stammt.
Ein Verfahren zur Präparation
von OMPC ist im wesentlichen wie im
US-Patent
4,271,147 und in Beispiel 1 angegeben. Als Alternative
ist eine Untereinheit von OMPC, z. B. MIEP, die durch Dissoziation
des OMPC oder durch ihre rekombinante Expression gebildet wird,
ebenfalls bevorzugt. Eine Methode zur Erreichung dieses Ziels wird
in der Patentanmeldung USSN 555,978; 555,329 und 555,204 (entsprechend
EP-A-0467714 ) und
in Beispiel 2, 16–23,
angegeben.
-
Das
neue, partiell hydrolysierte und hochgereinigte Pneumokokken-Kapselpolysaccharid
(Pn-Ps) ist eine Präparation
eines antigenen Polysaccharids, das von einer Kultur eines der Pneumokokken-Subtypen (wie
im folgenden in dem Abschnitt, der ein neues Konjugationsverfahren
beschreibt, und in den Beispielen 3–10 beschrieben) stammt. Das
Pn-Ps besitzt ein mittleres Molekulargewicht zwischen etwa 1 × 105 und 1 × 106 Dalton, im Mittel weniger als etwa 1000
sich wiederholende Einheiten pro Molekül, einen Grad der Verunreinigung
durch C-Polysaccharid von weniger als etwa 3% und einen Antigenizitätsindex
zwischen 0,4 und 1,1, und vorzugsweise zwischen 0,7 und 1,1. Dieser
letzte Parameter ist das relative Ausmaß der typenspezifischen Anti-Pneumokokken-Antikörperbindung,
welche pro Masseeinheit des neuen Pn-Ps gezeigt wird, im Vergleich
zu rohem Pn-Ps, das bei der ATCC hinterlegt ist. Darüber hinaus
ist das neue Pn-Ps einer Konjugation mit immunogenem Protein zugänglich,
um das Pn-Ps-PRO-Produkt
der Erfindung zu bilden. Einige physikalische und chemische Eigenschaften
von zwei verschiedenen Pn6B-Ps- und zwei verschiedenen Pn23F-Ps-Präparationen
sind in Tabelle I unten angegeben, während die folgende Beschreibung
zeigt, wie diese Eigenschaften gemessen werden. Das im folgenden
offenbarte Verfahren stellt ein Verfahren bereit zur Herstellung
von Pn-Ps-Zwischenprodukten und Pn-Ps-PRO-Konjugaten mit einer großen Vielfalt
von Pneumokokken-Subtypen, einschließlich, aber nicht beschränkt auf,
diejenigen, welche aus den Subtypen 1, 2, 3, 4, 5, 68, 7F, 8, 9N,
9V, 10A, 11A, 12F, 14, 158, 17F, 18C, 19F, 19A, 20, 22F, 23F und
33F ausgewählt
sind. Wie oben erwähnt,
sind eine bevorzugte Untergruppe von Pneumokokken-Polysacchariden
diejenigen, welche von den Pneumokokken-Subtypen 4, 6B, 9V, 14,
18C, 19F und 23F stammen. Für
Fachleute auf diesem Gebiet wird offensichtlich sein, daß neben
irgendwelchen dieser Polysaccharide oder anstelle irgendwelcher
dieser Polysaccharide ersatzweise andere eingesetzt werden können, wenn
es in der Riskopopulation erforderlich wird. So können Pn1-Ps
und Pn5-Ps wie Pn4-Ps oder Pn9V-Ps behandelt werden, ebenso wie
Neisseria Meningitidis B-, C- oder Gruppe B-Streptokokken-Polysaccharide,
während
Pn7F-Ps wie Pn14-Ps behandelt werden kann, wie im folgenden näher beschrieben,
und in ein multivalentes Vakzin eingeschlossen werden. Für Fachleute
wird auch ersichtlich sein, daß mit
dem Einschluß von
Pn6B-Ps durch kreuzreaktive Antikörper Schutz gegen Pn6A geboten
wird. Dies gilt auch für
eine Reihe anderer Pneumokokken-Subtypen.
-
Multivalente
Vakzine, sind diejenigen, welche Mischungen von verschiedenen Pn-Ps-PRO-Konjugaten
umfassen, die jeweils separat mit einem gegebenen Pn-Ps-Subtyp hergestellt
wurden. Darüber
hinaus sind multivalente Vakzine diejenigen, bei denen mehrere verschiedene
Pn-Ps-Subtypen alle gleichzeitig oder nacheinander mit einem gegebenen
PRO konjugiert werden.
-
1. Charakterisierung der neuen Polysaccharid-Zwischenprodukte:
-
Die
physikalischen und chemischen Eigenschaften der partiell hydrolysierten,
gereinigten Pneumokokken-Kapselpolysaccharid-Zwischenprodukte hängen von
dem Pneumokokken-Subtyp ab, von dem sie stammen, und den Manipulationen,
denen sie nach dem hier offenbarten Verfahren unterworfen werden.
Im allgemeinen weisen die Pn-Ps Zwischenprodukte im Vergleich zu
rohem, von einer Bakterienkultur stammendem Polysaccharid eine 2-
bis 10fach geringere Molekülgröße und Polydispersität auf. Die
reduzierte Größe erlaubt
eine bessere Handhabung des Polysaccharids während der Konjugation und bei
der Entfernung von freiem Pn-Ps nach der Konjugation, höhere Pn-Ps-Reinheit/-Homogenität, niedrigere
Polydispersität
der Molekülgröße des Pn-Ps
und im wesentlichen unveränderte
Antigenizität.
Diese Eigenschaften des neuen Pn-Ps tragen wesentlich zu der entsprechenden
Bildung eines gut definierten, hochgradig typenspezifischen antigenen
Pn-Ps-PRO-Produkts bei.
-
i. Pn-Ps-Molekulargewicht und -Polydispersität:
-
Durch
Messung des Gewichtsmittel-Molekulargewichts MW mittels
Diffusion, Sedimentation oder Chromatographie und des Zahlenmittel-Molekulargewichts
MN durch eine kolligative Eigenschaft wie
Viskosität,
Gefrierpunktserniedrigung oder Siedepunktserhöhung wird die Polydispersität der Pn-Ps-Präparation
als das Verhältnis
MW/MN erhalten.
Je näher
diese Zahl bei 1 liegt, desto homogener ist die Polysaccharid-Präparation. Die
Polydispersität
einer Anzahl von Pn-Ps-Präparationen
ist hier angegeben und ein bevorzugtes Verfahren zur Erzielung dieser
erhöhten
Homogenität
ist ebenfalls offenbart
-
Der
Verteilungskoeffizient
- Vo
- = Hohlraumvolumen
der Säule
- Vi
- = Gesamtpermeationsvolumen
- Ve
- = Elutionsvolumen
der Probe
- Kd
- = Verteilungskoeffizient
der Probe
jeder rohen und partiell hydrolysierten Pn-Ps-Präparation
wird durch Größenausschlußchromatographie (SEC)
oder Hochleistungs-Größenausschlußchromatographie
(HPSEC) eines Aliquots des Polysaccharids nach im Stand der Technik
bekannten Verfahren gemessen. Der so erhaltene Kd ist
ein Maß des
mittleren hydrodynamischen Volumens der Polysaccharid-Präparation.
Mit abnehmender Molekülgröße des Pn-Ps
durch physikalische Scherung oder thermische Hydrolyse oder Beschallungshydrolyse
gemäß dem offenbarten
Verfahren nimmt das Elutionsvolumen Ve des Pn-Ps zu und somit auch
der Kd.
-
Eine
bevorzugte Säulenmatrix
für diesen
Zweck ist SEPHAROSE CL2B-Gel (Pharmacia Nr. 17-0120-01). Das Hohlraumvolumen
der Säule
(Vo) wird mit Blue Dextran 2000 (Pharmacia Nr. 17-0360-01) und das
Gesamtpermeationsvolumen (Vi) aus einem Natriumchloridsalz-Peak
bestimmt. Gemäß einem
Verfahren wird die Pn-Ps-Probe mit 2,5 mg/ml in destilliertem Wasser
hergestellt und ein Injektionsvolumen von 1 ml eingesetzt Das Verhältnis Vo/Vi
sollte im Bereich von 0,32–0,37
liegen. Der Kd für Dextran T500 (Pharmacia Nr.
17-0320-01) sollte zwischen 0,37–0,49 liegen. Ein bevorzugtes
HPSEC-System umfaßt eine
7,5 × 600
mm TSK G6000 PW-Säule,
die auf 50°C
erwärmt
ist.
-
In
einem sehr bevorzugten Verfahren wird SEC oder HPSEC mit einem Differentialrefraktometer,
welcher die relative Analytenkonzentration als Funktion des Elutionsvolumens überwacht,
und einem Differentialviskometer, welcher die spezifische Viskosität des Analyten
als Funktion des Elutionsvolumens überwacht, kombiniert. Eine
Universalkalibrierungskurve [log (Grenzviskosität mal Molekulargewicht) gegen
Retentionsvolumen] wird anhand der Analyse einer Reihe von monodispersen
Polyethylenoxid-Standards konstruiert. Die Diagramme der Konzentration
und spezifischen Viskosität
können
verwendet werden, um für
die Probe ein Diagramm von Molekulargewicht gegen Elutionsvolumen
zu berechnen, welches wiederum eingesetzt wird, um die Werte für MN und MW zu berechnen,
aus denen der Polydispersitätsindex
(MW/MN) berechnet
wird [Yau, W. W., und Rementer, S. W., J. Lig. Chromatog., 13, 627–675 (1990);
Nagy, J. Lig. Chrom., 13, 677–691
(1990); Benoit et al., J. Ch. Phys. Tome., 63, 1507–1514 (1966)].
Bei der vorliegenden Erfindung wurde die Grenzviskosität in 0,1
M Natriumphosphatpuffer, pH 7,2, gemessen.
-
Nachdem
das mittlere Molekulargewicht einer Pn-Ps-Präparation bestimmt wurde, ist
die mittlere Anzahl sich wiederholender Einheiten pro Molekül leicht
zu bestimmen durch Teilung des Polymer-Molekulargewichts durch das
Molekulargewicht der sich wiederholenden Einheit (siehe Tabelle
II).
-
ii. Retention der typenspezifischen Pn-Ps-Antigenizität:
-
Es
ist wichtig, daß für jedes
rohe Pn-Ps, das einer physikalischen Scherung oder einer chemischen, thermischen,
Beschallungs- oder enzymatischen Hydrolyse unterworfen wird, ein
Endpunkt bestimmt wird, an dem sich die antigene Integrität zu verlieren
beginnt. Dieser Endpunkt wird bequem bestimmt durch Korrelation der
Viskosität
mit irgendeinem aus einer Anzahl von immunologischen Tests, die
im Stand der Technik bekannt sind. In einem bevorzugten Verfahren
wird ein Aliquot der Polysaccharidlösung gemessen durch den Ouchterlony-Doppelimmundiffusions-Assay
unter Verwendung von Pneumokokken-Subtyp-spezifischem Antikörper. Das
Auftreten einer weißen
Präzipitinbande
im Agar, welche neben der Präzipitinbande
einer Probe von rohem Pn-Ps liegt, die in eine benachbarte Vertiefung
eingebracht wurde, nach einer Diffusionsperiode bietet eine qualitative
Identifizierung der Reaktanten und einen Hinweis darauf, daß die antigene
Integrtät
des Polysaccharids intakt bleibt. Ein mehr quantitativer immunologischer
Assay wird durch Geschwindigkeitsnephelometrie-Analyse oder einen
RIA erreicht.
-
Geschwindigkeitsnephelometrie
mißt die Änderung
oder die Geschwindigkeit der Änderung
in der Intensität
von Streulicht während
der Bildung von Antigen-Antikörper-Komplexen.
Die Reaktion läuft
in einer Reaktionszelle ab, durch die ein Lichtstrahl geleitet wird.
Im vorliegenden Fall werden die Komplexe durch eine Immunpräzipitinreaktion
gebildet, welche in Lösung
stattfindet, wenn ein spezifischer Antikörper (Ab) mit seinem spezifischen
Antigen (Ag), d. h., Pn-Ps, reagiert. Da die Bildung eines Ag-Ab-Komplexes von der
Anwesenheit von Ag- und Ab-Molekülen
in optimalen Verhältnissen
abhängt,
nimmt der Grad der Komplexbildung für eine konstante Menge von
Ab mit der Menge von Ag bis zu einem maximalen Niveau zu; größere Mengen an
Ag führen
zur Bildung von weniger Komplex. So wird durch Aufrechterhaltung
eines konstanten Niveaus an Ab und Messung der Lichtstreuung mit
zunehmenden Konzentrationen an Ag eine Standardkurve gebildet. Es ist
möglich,
die Ag-Konzentration für
eine Ps(oder derivatisiertes Ps)-Präparation zu berechnen, wenn
Proben mit ihren spezifischen Ab unter den gleichen Bedingungen,
die zur Entwicklung der Standardkurve verwendet wurden, umgesetzt
werden.
-
Ein
Vergleich der immunologisch durch Geschwindigkeitsnephelometrie
berechneten Konzentration mit der Konzentration, die auf chemische
oder physikalische Weise (durch Kolorimetrie, Refraktionsindex oder durch
Gesamthydrolyse und quantitative Bestimmung von Monosacchariden – siehe
unten) erhalten wurde, ergibt einen Index der Antigenizität für die Ps-Proben.
Eine Trockengewichtsanalyse von Polysacchariden ist nur angebracht,
wenn der Gehalt an flüchtigen
Stoffen der Pulverpräparation
bekannt ist. Polysaccharide sind notorisch hygroskopisch und können schätzungsweise
von 5 bis 30 Gewichtsprozent flüchtige
Stoffe enthalten. Somit sind Trockengewichtsmessungen für sich allein
nicht besonders verläßlich. Ein
Verfahren, das zur Bestimmung der Polysaccharidkonzentration mit
vernünftiger
Genauigkeit eingesetzt wurde, ist ein kolorimetrischer Assay, wenn
der Assay mit einer Standardlösung
des Polysaccharids von Interesse geeicht ist. Beispielsweise können Pn6B-Ps,
Pn18C-Ps, Pn19F-Ps und Pn23F-Ps alle quantitativ bestimmt werden
durch den Methylpentose-Assay von Dische und Shettles (J. Biol.
Chem., 175, 595–603
(1948)]. Pn4-Ps, Pn9V-Ps, Pn14-Ps und Pn19F-Ps können durch den Hexosamingehalt
quantitativ bestimmt werden und Pn9V kann auch durch den Uronsäuregehalt
quantitativ bestimmt werden. Der Phenol-Schwefelsäure-Assay
[Dubois et al., Anal. Chem., 28, 350–356 (1956)] ist geeignet zur
quantitativen Bestimmung aller dieser Pn-Ps-Präparationen als Teil einer verfahrensbegleitenden Überprüfung während der
Konjugatherstellung. Das andere eingesetzte Verfahren ist die Verwendung
eines Refraktionsindexsignals als Maß für die Masse des Analyten, ebenfalls
geeicht mit einer Standardlösung
des interessierenden Polysaccharids. Obwohl der kolorimetrische
Assay zur Überprüfung des
Polysaccharidgehalts der Proben während der Derivatisierung und
des Konjugationsprozesses eingesetzt wird, wird das letztere Verfahren
während
der physikalischen Charakterisierung der Polysaccharid-Präparation
durch HPSEC-Universalkalibrierungsanalyse und zur Berechnung des
Antigenizitätsindexes
eingesetzt. Dem rohen Ausgangs-Pn-Ps wird ein Antigenizitätsindexwert
von 1,0 zugeordnet. Ein Index der relativen Antigenizität wird für Versuchsproben
berechnet und ein Wert von 0,4–1,1
wird als zufriedenstellend betrachtet. Es ist möglich, einen Antigenizitätsindex
von größer als
1,0 zu bekommen, wenn das Polysaccharid während der Hydrolyse und dem
Fraktionierungsschritt erheblich gereinigt wird. Es ist auch theoretisch möglich, daß die Größenreduktion
allein den Antigenizitätsindex
einer Präparation
erhöhen
könnte,
indem die Flexibilität
der Polysaccharidmoleküle
erhöht
und somit die sterische Hinderung um die antigenen Epitope verringert
wird. Diese Bestimmungen erfolgen als verfahrensbegleitende Prüfung auf
hydrolysierte, fraktionierte und derivatisierte Ps-Proben. Proben,
die relative Antigenizitäten
von < 0,4 aufweisen,
werden verworfen, d. h. nicht konjugiert. Es stehen Anti-Pn-Ps-Antikörper-Präparationen
zur Verfügung,
welche zur Charakterisierung von Pneumokokken-Polysacchariden geeignet
sind. Bezugsquellen von Anti-Pn-Ps-Antikärpern schließen die
Health Research Inc., Albany, N. Y., und das Staten Serum Institute
ein. Alternativ können
typenspezifische Anti-Pn-Ps-Antikörper für diesen Zweck nach im Stand
der Technik bekannten Verfahren hergestellt werden unter Verwendung
von im Handel erhältlichen
rohen Pn-Ps als Immunogen [Baker et al., Immunology 20, 469 (1971);
Brooke, M. S., J. Immunol., 95, 358 (1966); Kearney, R., und Halladay,
W. J., Aust. J. Exp. Biol. Med. Sci., 48, 227(1970); Schneerson,
R., et al., Prag. Allergy, 33, 144 (1983); Robbins, J. B., Infect.
Immun., 26, 1116 (1979)].
-
Ein
weiteres Indiz für
erhalten gebliebene Antigene Integrität ist die Beibehaltung der
korrekten chemischen Zusammensetzung der Pn-Ps-Präparation.
Beispielsweise weist Pn6B-Ps eine sich wiederholende Einheit von
[α-Gal(1-3)-α-Glu(1-3)-α-L-Rhap(1-4)-D-Ribit-5-(PO4(2)] auf, so daß das Molverhältnis der
Kohlenhydratkomponenten Ribit:Rhamnose:Galactose:Glucose etwa 1:1:1:1
beträgt
Dieses Verhältnis
kann beispielsweise nach der Hydrolyse des Polysaccharids mit 36%iger
Fluorwasserstoffsäure
für etwa
2 Stunden bei 45–65°C, gefolgt
von 2 M Trifluoressigsäure
für 10–20 Stunden
bei 100°C
und Hochleistungs-Anionenaustauschchromatographie mit gepulstem
amperometrischem Nachweis bestimmt werden. Vier Peaks, die etwa gleiche
Molmengen der Kohlenhydratkomponenten repräsentieren, sind somit ein Indiz
für die
erhalten gebliebene Integrität.
Im wesentlichen theoretische Verhältnisse der Kohlenhydratkomponenten
bleiben für
alle neuen Pn-Ps-Verbindungen dieser Erfindung innerhalb von etwa
20% erhalten. Die Abweichungen von den theoretischen Werten sind
in erster Linie auf methodische Grenzen zurückzuführen. Somit hat nach einer
Gesamthydrolyse Pn23F-Ps ein Verhältnis von etwa Glycerin:Rhamnose:Galactose:Glucose
= 1:2:1:1;
Pn14-Ps ein Verhältnis
von etwa N-Acetylglucosamin:Galactose:Glucose = 1:2:1;
Pn19F-Ps
ein Verhältnis
von etwa Rhamnose:N-Acetylmannosamin:Glucose = 1:1:1;
Pn18C-Ps
ein Verhältnis
von etwa Glucose:Galactose:Rhamnose:Glycerin:Acetat = 3:1:1:1:1;
Pn9V-Ps
ein Verhältnis
von etwa Glucose:Galactose:N-Acetylmannosamin:Glucuronsäure: Galacturonsäure:Acetat
= 2:1:1:1:1:1,7; und
Pn4-Ps ein Verhältnis von etwa N-Acetylmannosamin:N-Acetylfucosamin:Galactosamin:
Galactose:Pyruvat = 1:1:1:1:1.
-
Ferner
wurde kürzlich
festgestellt, daß Pn4-Ps
eine weitere Komponente enthält,
identifiziert durch HPLC-Analyse, welche 2-Aminopneumosamin (2-Amino-2,6-didesoxytalose)
zu sein scheint, ebenso Pn5-Ps [Barker et al., Carbohydrate Res.,
224–233
(1966)]. Pn19F-Ps besitzt ebenfalls eine weitere Komponente, wahrscheinlich
ein Hexosamin, das in der Literatur noch nicht erwähnt wurde
und wofür
eine definitive Identifizierung noch aussteht. Diese und weitere
theoretische Zusammensetzungen von Polysaccharid-Repeats werden
in den folgenden Referenzen angegeben: J. E. G. van Dam et al.,
Carbohyd. Res. 187, 267 (1988); H. J. Jennings, Adv. Carbohyd. Chem.
41, 155 (1983) und die dort angegebenen Referenzen; J. C. Richards
und M. Perry, Biochem. Cell. Biol. 66, 758 (1988). Neben den Kohlenhydratkomponenten
gibt es bei mehreren der interessierenden Pn-Ps Phosphat-, Acetat-
und Pyruvat-Seitengruppen, wobei einige davon immunodominante Merkmale
sind. Als solche können
diese Komponenten auch nachgewiesen werden (siehe Beispiel 30).
Die quantitative Bestimmung von Monosacchariden ist auch ein brauchbares
Mittel zur quantitativen Bestimmung der Polysaccharidkonzentration
einer Probe.
-
Ein
weiteres Element bei der Antigenizität der vorliegenden Polysaccharide
ist die Erhaltung dessen, was als ”Konformationsepitop” in dem
Polysaccharid bezeichnet worden ist [siehe z. B. Wessels, M. R.,
und Kasper, D. L., J. Exp. Med., 169, 2121–2131 (1989)]. Dieses Niveau
der Antigenizität
scheint nur in hochmolekularen Formen des Saccharids Ausdruck zu
finden, und die hier beschriebenen Verfahren sind auch auf die Erhaltung
dieses Niveaus der Polysaccharid-Immogenizität gerichtet
-
iii. Minimale Verunreinigung durch C-Polysaccharid:
-
Ein
weiterer kritischer Parameter ist der Grad der Verunreinigung durch
C-Polysaccharid. Dieser Wert kann bestimmt werden durch vollständige Säurehydrolyse
einer Polysaccharid-Präparation,
Chromatographie des Hydrolysats und konduktometrischen Nachweis
von Cholin [Hermans et al., Red. Trav. Chim. Pays-Bas, 107, 600
(1988)]. Alternativ kann das unhydrolysierte Polysaccharid mittels
NMR hinsichtlich Cholin analysiert werden. Die NMR Technik setzt
das Verhältnis
des Cholinsignals zum Rhamnosemethylsignal (für Pn-Ps, die eine Rhamnose
enthalten; ein anderes Signal für
andere Pn-Ps) zur Berechnung des C-Ps-Gehalts ein. Das chromatographische
Verfahren setzt entweder das Verhältnis des Cholinsignals zum
Polysaccharidgehalt, bestimmt durch konduktometrischen Assay, oder
das Verhältnis
des Cholinsignals zu einem der Pn-Ps-Komponenten-Peaks ein, um den
C-Ps-Gehalt zu berechnen. Bei jedem Verfahren erlauben Standards
bekannter Konzentrationen an Cholin die direkte Berechnung des in
einer Polysaccharid-Präparation
vorhandenen Cholinniveaus, unter Heranziehung der theoretischen
Wiederholungsstruktur von C-Ps [Hermans et al., vollst. Referenz
siehe oben] wird die Konzentration von C-Ps in einer Polysaccharid-Präparation
bestimmt Polysaccharidkonzentrationen von Pn-Ps-Proben werden nach
im Stand der Technik bekannten Verfahren gemessen. Beispielsweise
kann durch vollständige
Hydrolyse des Polysaccharids und Messung der Konzentration eines spezifischen
Monosaccharids die Gesamtpolysaccharid-Konzentration bestimmt werden.
Durch Vergleich der C-Ps-Konzentration
mit der Gesamtpolysaccharid-Konzentration wird der Grad der Verunreinigung
durch C-Polysaccharid
(Gew./Gew.) bestimmt. C-Polysaccharid-Niveaus von weniger als 3%
(Gew./Gew.) des Gesamtpolysaccharids werden als annehmbar betrachtet,
jedoch noch bevorzugter sind Niveaus von weniger als 1%.
-
Die
chemischen und physikalischen Eigenschaften von zwei Pn6B-Ps-Chargen
und zwei Pn23F-Ps-Chargen
sind in Tabelle I unten zusammengefaßt. Diese Daten demonstrieren
die Reproduzierbarkeit der Parameter von einer Charge zur anderen,
welche aus dem neuen, hier beschriebenen Verfahren resultiert TABELLE I Charakteristische Eigenschaften von hydrolysierten
und fraktionierten Pn-Ps
| Pn-Ps-Präparation | 6B-1 | 6B-2 | 23F-1 | 23F-2 |
| Endviskosität | 1,094 | 1,147 | 1,350 | 1,376 |
| Kd
(HPSEC) | 0,62 | 0,62 | 0,49 | 0,49 |
| Kd
(CL-2B) | 0,64 | 0,60 | 0,41 | n.
b. |
| Monosaccharid | S | S | S | S |
| Antigenizität | | | | |
| Ouchterlony | S | S | S | S |
| Nephelose | S | S | S | S |
| (Phenol:Schwefelsäure) | S | S | S | S |
| S: Zufriedenstellend |
-
In
der folgenden Tabelle II sind chemische und physikalische Parameter
verschiedener roher Pneumokokken-Polysaccharide und der korrespondierenden
neuen hydrolysierten und fraktionierten (hyd. + frak.) Verbindungen
dieser Erfindung angegeben. Die angegebenen Zahlen sind Näherungswerte
innerhalb des Versuchsfehlers und der Nachweisgrenzen für die hergestellten
komplexen Polysaccharidverbindungen.
-
-
2. Charakterisierung des neuen Pn-Ps-PRO-Konjugats:
-
i. Analyse der Identität und Quantität von Pn-Ps:
-
Es
ist sehr nützlich,
die Quantität
und chemische Integrität
von Pn-Ps in einem Endkonjugat durch eine unabhängige Technik zu verifizieren,
um die Antigenizität
(Integrität)
sicherzustellen und das Pn-Ps/PRO-Verhältnis zu
berechnen. Ein Verfahren beinhaltet die vollständige Hydrolyse unter Einsatz
von 2 M TFA bei 100°C für 5–16 h, je
nach der optimalen Hydrolysezeit für jedes spezielle Pn-Ps. Gleiche
Mengen von Pn-Ps-PRO-Konjugat und PRO-Hydrolysat (auf der Basis
von Lowry-Protein) werden durch Hochleistungs-Anionenaustauschchromatographie
mit gepulstem amperometrischem Nachweis analysiert, um Monosaccharid-Profile
zu erhalten. Zur Korrektur bezüglich
der von PRO beigesteuerten Monosaccharide, z. B. aus in OMPC vorhandenem
Lipopolysaccharid (LPS), wird das Profil von PRO von dem Profil
des Pn-Ps-PRO-Konjugats unter Einsatz geeigneter Computer-Software wie dem
Nelson-Programm „subtrahiert”. Die Menge
des Pn-Ps im Konjugat wird dann berechnet durch Vergleich des Profils
der bekannten Menge des derivatisierten Pn-Ps-Hydrolysats und der
korrigierten Pn-Ps-PRO-Konjugate. Das Pn-Ps/PRO-Verhältnis wird
ebenfalls auf diese Weise gemessen.
-
ii. Analyse hinsichtlich neuer Aminosäuren, welche
Konjugation und Versehung mit Endgruppen anzeigen
-
Nach
der Konjugation von Pn-Ps mit PRO werden Proben des Konjugats in
6 N HCl hydrolysiert und einer Aminosäureanalyse unterworfen. Dieses
Verfahren weist die Anwesenheit und die Menge einmaliger Aminosäuren wie
S-Carboxymethylhomocystein (S-CMHC) und S-Carboxymethylcysteamin
(S-CMCA) nach. Die erstere Aminosäure wird als Teil der chemischen
Verknüpfung
durch die chemische Reaktion zwischen derivatisiertem Pn-Ps und
PRO, wie im folgenden Verfahrensabschnitt beschrieben, gebildet
und wird als definitiver Beweis der kovalenten Verknüpfung von
derivatisiertem Pn-Ps mit PRO betrachtet. Die Bildung einer solchen
kovalenten Verknüpfung
ist für
die T-Zell-Immunogenizität
des Konjugat-Vakzins essentiell. Unmittelbar nach Beendigung der
Konjugat-Reaktion werden nicht umgesetzte Bromacetamidgruppierungen
mit N-Acetylcysteamin-Endgruppen
versehen. Die Hydrolyse dieser Verknüpfung resultiert in der Freisetzung
von S-Carboxymethylcysteamin
(S-CMCA), das auch bei der Aminosäureanalyse nachgewiesen wird.
Der Nachweis dieser Aminosäure
bestätigt
das erfolgreiche Versehen der reaktiven Bromacetamidgruppen mit
Endgruppen, was sie somit für
etwaige unerwünschte
chemische Reaktionen nicht mehr verfügbar macht. Akzeptable Niveaus
an Kovalenz und Endgruppen liegen zwischen etwa 1–15% für S-CMHC/Lys
und etwa 0–5%
für S-CMCA/Lys.
-
iii. Analyse des aluminiumhydroxid-adsorbierten
Vakzins:
-
In
einer bevorzugten Ausführungsform
ist das Pn-Ps-PRO-Konjugat an Aluminiumhydroxid (Aluminiumoxid,
Al(OH)3-Gel (siehe Abschnitt C. unten))
adsorbiert, was zu einer Potenzierung der Immunantwort auf das Vakzin
führt.
Andere mögliche
Vakzin-Formulierungen umfassen die Formulierung in physiologisch
annehmbaren Verdünnungsmitteln
und die Verwendung anderer Adjuvanzien, Immunmodulatoren oder inerter Trägerstoffe
als das Aluminiumhydroxidgel. Die Analyse dieses aluminiumoxid-adsorbierten
Materials wird wie folgt durchgeführt.
-
Das
aluminiumoxid-adsorbierte Pn-Ps-PRO kann hinsichtlich Zusammensetzung
und Stabilität
nach der Desorption des Konjugats von Alaun analysiert werden. Dies
wird erreicht durch Dialyse des aluminiumoxid-adsorbierten Pn-Ps-PRO
gegen eine 3%ige Natriumcitratlösung
für 16
h bei Raumtemperatur. Das resultierende lösliche Aluminiumcitratsalz
wandert aus der Dialysemembran und läßt das Pn-Ps-PRO zurück. Dieses
Verfahren ist von Bedeutung, um zu bestätigen, daß sich die korrekte Menge an
Pn-Ps-PRO in der aluminiumoxid-adsorbierten Formulierung befindet.
Jedoch enthalten einige Formulierungen wie Pn6B-Ps-OMPC und Pn23F-Ps-OMPC
10, 5, 2 und 1 μg
Pn-Ps/ml (siehe
Abschnitt C. unten), Konzentrationen deutlich unterhalb chemischer
Nachweisverfahren. Um deshalb Analysen der Kohlenhydratzusammensetzung
durchzuführen,
werden die adsorbierten Pn-Ps-OMPC-Vakzine zuerst pelletiert, um
die wäßrige Flüssigkeit
zu entfernen, und das Pellet wird vor der Citratlösung in
einem Fünftel
des ursprünglichen
Volumens resuspendiert. Nach der Dialyse liegt das solubilisierte
Pn-Ps-OMPC mit 50, 25, 10 und 5 μg
Pn-Ps/ml vor. Diese Konzentrationen sind dann sowohl einer Pn-Ps-
als auch Proteinanalyse zugänglich,
um die Dosierungsniveaus zu bestätigen.
-
Citrat-desorbierte
Proben werden auch hinsichtlich der möglichen Anwesenheit von freiem
Pn-Ps analysiert, ein Gesichtspunkt, der für die Immunogenizität und Einheitlichkeit
der Produktion bedeutsam ist. Diese Analyse wird durchgeführt mittels
Chromatographie auf einer SEPHAROSE CL-2B- oder SEPHACRYL S1000SF-Größenfraktionierungssäule, worin
Pn-Ps-OMPC von Pn-Ps abgetrennt werden kann. Die Anwesenheit und
Menge des freien Pn-Ps wird als Antigen durch Geschwindigkeitsnephelometrie
gemessen. Der Grad der Verunreinigung des Pn-Ps-OMPC durch freies
Pn-Ps beträgt
weniger als 15% des insgesamt vorhandenen Pn-Ps.
-
iv. Pyrogen-Test: Abwesenheit von signifikanter
Pyrogenizität:
-
Das
Konjugatprodukt dieser Erfindung wird hinsichtlich der Abwesenheit
nachteiliger Temperaturerhöhungseffekte
getestet. Konjugatprodukte wurden nach dem hier offenbarten Verfahren
hergestellt und befunden, annehmbare Niveaus an Pyrogenizität aufzuweisen.
-
Methode (IV)
-
Das
Pn-Ps-Konjugat-Vakzin wird getestet wie in 21 CFR, Abschnitt 610.13(b)
beschrieben.
-
1) Methode (IM)
-
Ein
zweites Maß für die Pyrogenizität ist der
Kaninchen-IM-Test. Dieser Test ahmt den Einsatz des Produkts in
der Klinik besser nach und reflektiert unserer Meinung nach genauer
die scheinbare Endotoxinbelastung des Produkts.
-
Jedes
Kaninchen erhält
1,0 ml Vakzin durch intramuskuläre
Injektion. Der Test wird mit mindestens drei Kaninchen durchgeführt. Die
Temperatur wird 5 h lang nach der Injektion überwacht. Andere Testmethoden
sind wie in 21 CFR Abschnitt 610.13(b) beschrieben (Die Testdosis
basiert auf der Polysaccharidkonzentration).
-
v. Art der kovalenten Verknüpfung:
-
Die
Pn-Ps-PRO-Konjugate, wie z. B. Pn-Ps-OMPC oder Pn-Ps-MIEP, können durch
bigenerische Abstandhalter gekoppelt werden, die eine Thioethergruppe
und ein primäres
Amin enthalten, welche hydrolytisch labile kovalente Bindungen mit
dem Polysaccharid und dem PRO, wie z. B. OMPC oder MIEP, bilden.
Bevorzugte Konjugate gemäß dieser
Erfindung sind diejenigen, welche durch die Formeln Pn-Ps-A-E-S-B-PRO
oder Pn-Ps-A'-S-E'-B'-PRO dargestellt
werden können.
A-E-S-B und A'-S-E'-B' stellen bigenerische
Abstandhalter dar, welche hydrolytisch stabile kovalente Thioetherbindungen
enthalten und welche kovalente Bindungen (wie hydrolytisch labile
Ester- oder Amidbindungen) mit den Makromolekülen PRO und Pn-Ps bilden. In
dem Abstandhalter A-E-S-B ist S Schwefel; E ist das Transformationsprodukt
einer thiophilen Gruppe, die mit einer Thiolgruppe umgesetzt wurde,
und wird repräsentiert
durch
worin R H oder CH
3 darstellt und p 1 bis 3 ist; A ist
worin W O oder NH ist, m
0 bis 4 ist, n 0 bis 3 ist und Y CH
2, O,
S, NR' oder CHCO
2H darstellt, wobei R' H oder C
1-
oder C
2-Alkyl ist, so daß, falls Y CH
2 ist,
m und n nicht beide gleich Null sein können und falls Y O oder S ist,
m größer als
1 und n größer als
1 ist; B ist
worin q 0 bis 2 ist, Z NH
2,
COOH oder H darstellt, wobei
R' und p wie oben
definiert sind und D
oder
ist.
-
In
dem Abstandhalter A'-S-E'-B' ist S Schwefel;
A' ist
worin a 1 bis 4 ist und R'' CH
2 oder
darstellt, wobei Y' NH
2 oder
NHCOR' ist, und
W, p und R' wie
oben definiert sind, und E' ist
das Transformationsprodukt einer thiophilen Gruppe, die mit einer
Thiolgruppe umgesetzt wurde, und wird repräsentiert durch
worin R wie oben definiert
ist, und B' ist
oder E' ist
und B' ist
worin p 1 bis 3 ist. Ferner
sind von den bigenerischen Abstandhaltern A-E-S-B und A'-S-E'-B' die E-S-B- und A'-S-E'-Komponenten bestimmbar
und quantifizierbar, wobei diese Identifizierung die Kovalenz der
Konjugatbindung reflektiert, welche die Seite des Thioetherschwefels,
welche von dem kovalent modifizierten Polysaccharid stammt, mit
der Seite des Abstandshalters, welche von dem funktionalisierten
Protein stammt, verknüpft.
-
Die
Konjugate Pn-Ps-A-E-S-B-PRO gemäß dieser
Erfindung können
Abstandhalter enthalten, deren Komponenten u. a. Derivate einschließen von:
Kohlendioxid,
1,4-Butandiamin und S-Carboxymethyl-N-acetylhomocystein; Kohlendioxid,
1,5-Pentandiamin und
S-Carboxymethyl-N-acetylhomocystein; Kohlendioxid, 3-Oxa-1,5-pentandiamin
und S-Carboxymethyl-N-acetylhomocystein; Kohlendioxid, 1,4-Butandiamin
und S-Carboxymethyl-N-acetycystein;
Kohlendioxid, 1,3-Propandiamin und S-Carboxymethyl-N-benzoylhomocystein;
Kohlendioxid, 3-Aza-1,5-pentandiamin und S-Carboxymethyl-N-acetylcystein;
Kohlendioxid, 1,2-Ethandiamin,
Glycin und S-(Succin-2-yl)-N-acetylhomocystein. Die Konjugate Pn-Ps-A'-S-E'-B'-PRO gemäß dieser
Erfindung können
Abstandhalter enthalten, deren Komponenten u. a. Derivate einschließen von:
Kohlendioxid
und S-Carboxymethylcysteamin; Kohlendioxid und S-(α-Carboxyethyl)cysteamin;
Kohlendioxid und S-Carboxymethylhomocysteamin; Kohlendioxid, S-(Succin-2-yl)cysteamin
und Glycin; Kohlendioxid und S-Carboxymethylcystein.
-
B. Verfahren zur Herstellung neuer Pn-Ps-Zwischenprodukte
und Pn-Ps-PRO-Konjugate:
-
Bei
der Offenbarung dieses Verfahrens werden mehrere Stufen gesondert
beschrieben:
-
I. Polysaccharid-Herstellung:
-
- a) Isolierung von rohem Pneumokokken-Polysaccharid,
Pn-Ps;
- b) partielle Hydrolyse oder mechanische Scherung des rohen Pn-Ps;
- c) Fraktionierung des partiell hydrolysierten Pn-Ps nach Größe und Reinheit;
-
II. Konjugation:
-
- a) Funktionalisierung des fraktionierten Pn-Ps,
um den elektrophilen oder nukleophilen Reaktanten Pn-Ps* zu bilden,
vorzugsweise um etwa 21 reaktive Bromacetylgruppen pro 100 sich
wiederholender Pn-Ps-Oligosaccharideinheiten aufzuweisen;
- b) Isolierung des immunogenen Proteins (PRO), vorzugsweise des
Neisseria meningitidis B-OMPC
oder einer Untereinheit davon;
- c) Funktionalisierung des PRO, um den nukleophilen oder elektrophilen
Reaktanten PRO* zu bilden, vorzugsweise OMPC oder eine Untereinheit
davon, wie z. B. MIEP, um reaktive Sulfhydrylgruppierungen aufzuweisen;
- d) Konjugation des Polysaccharids (Pn-Ps*) von Schritt (a) mit
dem Protein (PRO*) von Schritt (c);
- e) Versehen des Pn-Ps-PRO-Konjugats mit Endgruppen, um restliche
funktionelle Gruppen zu eliminieren;
- f) Isolierung des Konjugatprodukts.
-
I.a.) Isolierung von rohem Pneumokokken-Polysaccharid,
Pn-Ps:
-
Pneumokokken-Kapselpolysaccharide
unterscheiden sich in chemischer und antigener Hinsicht aufgrund
der Zusammensetzung und der Verknüpfung der sich wiederholenden
Oligosaccharideinheit des gegebenen Kapselpolysaccharid-Serotyps.
Die Isolierung der Polysaccharide muß je nach den charakteristischen physikalischen
Eigenschaften des gegebenen Polysaccharids auf etwas verschiedene
Weise ablaufen. Im allgemeinen werden jedoch nach bekannten Verfahren
[Beispiel 3 und Williams, C. A., und Chase, M. W., Methods in Immunology
and Immunochemistry, Bd. I, Academic Press (1967)] die Bakterien
gezüchtet
und das Pn-Ps gewonnen, während
die Pathogene selbst von der ATCC erhältlich sind. Kurz gesagt, im
Anschluß an eine
Großkultur
der Bakterien in geeigneten Nährmedien,
die bekannterweise das Pneumokokken-Wachstum unterstützen, wird
ein Bakterizid wie Phenol oder Toluol zur Abtötung der Organismen zugegeben
(Beispiel 3).
-
Anschließend wird
eine Alkoholfraktionierung des Polysaccharids in zwei Stufen durchgeführt. In
der ersten Stufe wird eine niedrige Alkoholkonzentration eingesetzt,
um Zelltrümmer
und andere unerwünschte Verunreinigungen
auszufällen,
während
das rohe Pn-Ps in Lösung
verbleibt. Eine nachfolgende Zugabe von mit Wasser mischbarem Alkohol
auf eine vorherbestimmte Konzentration fällt die Kapselpolysaccharide
aus, während
weitere Verunreinigungen in der Überstandsflüssigkeit
zurückgelassen
werden. Einer erneuten Suspension in einem wäßrigen Medium folgt die Entfernung
kontaminierender Proteine und Nukleinsäuren nach bekannten Verfahren
wie Nukleaseverdauung oder proteolytische Verdauung oder Lösungsmittelextraktion. Das
rohe Polysaccharid wird durch Alkoholfällung gewonnen und getrocknet,
um ein Pulver des rohen Pn-Ps zu bilden (Beispiel 3).
-
I.b) Partielle Hydrolyse oder mechanische
Scherung des rohen Pn-Ps:
-
Rohes
Polysaccharid, im wesentlichen wie oben beschrieben hergestellt
[siehe auch Beispiel 3 unten], wurde in unkonjugiertem Zustand eingesetzt,
um Pneumokokken-Vakzine zu formulieren, die zur Verwendung bei Erwachsenen
und Kindern im Alter von über
zwei Jahren bestimmt sind. Die folgenden Verfahrensschritte ergeben
ein neues, partiell hydrolysiertes, gereinigtes Pn-Ps-Produkt mit
einmaligen und definierten chemischen und physikalischen Eigenschaften
(siehe Tabelle II), das zur Herstellung von Konjugat-Vakzinen geeignet
ist. Die Größenreduktion
des rohen Pn-Ps trägt
zum Erfolg der anschließenden
Reinigungsschritte bei, um ein hochgereinigtes Pn-Ps-Produkt zu
ergeben. Bei Verwendung zur Herstellung von Konjugaten ist darüber hinaus
die Konjugation effizienter, wenn das neue Pn-Ps dieser Erfindung
eingesetzt wird. Dies liegt daran, daß wäßrige Lösungen des rohen Polysaccharidmaterials
hochviskos und schlecht löslich
sind und dessen Konjugate weitgehend unlöslich und unfiltrierbar sind.
Der Konjugationsprozeß selbst
ist schwierig durchzuführen,
was zu einer niedrigen Ausbeute an Konjugat führt Ferner wird die Abtrennung
von unkonjugiertem Pn-Ps von dem Endkonjugat erleichtert, wenn das
Pn-Ps vor der Konjugation eine verringerte Größe und Viskosität und verbesserte
Löslichkeit
aufweist. Dies ist insofern erheblich, als es die Anwesenheit von
freiem Pn-Ps in Konjugat-Präparationen
schwierig macht, die tatsächliche
Dosis an verabreichtem Konjugat-Pn-Ps abzuschätzen, und nachdem es das konjugierte
Pn-Ps ist, welches die signifikante T-Zell-stimulierende Wirkung
besitzt, bedeutet die Anwesenheit von unkonjugiertem Pn-Ps eine
Verminderung des immunologisch ”relevanten” Pn-Ps.
-
Das
wie oben hergestellte, trockene, rohe Kapselpolysaccharid kann auch
vor oder nach der partiellen Hydrolyse gereinigt werden, z. B. durch
Anionenaustauschchromatographie oder ein anderes chromatographisches
Verfahren, wie in Beispiel 6 für
Pn14-Ps gezeigt. Die chromatographische Adsorption-Desorption kann entweder
positiv oder negativ angewandt werden. Im positiven Modus wird das
Pn-Ps an das Harz adsorbiert, wobei Verunreinigungen in der Lösung zurückgelassen
werden, welche vor der Pn-Ps-Desorption weggewaschen werden. Im
negativen Modus werden Verunreinigungen aus der Pn-Ps-Lösung adsorbiert
und verworfen, was das Pn-Ps in der Lösung in gereinigtem Zustand
zurückläßt. Alternativ
kann das Pn-Ps direkt einer partiellen thermischen Hydrolyse, wie
in Beispiel 4 für
Pn6B-Ps gezeigt, oder einer Beschallungshydrolyse, wie in Beispiel
6 für Pn14-Ps
gezeigt, unterworfen werden. Andere Hydrolysemittel, z. B. chemische,
enzymatische oder physikalische (z. B. eine Hochdruckzelle), sind
ebenfalls bekannt.
-
Die
partielle Hydrolyse erfolgt durch eine limitierte thermische Behandlung
in einem wäßrigen Medium, vorzugsweise
bei 50 bis 110°C
für etwa
1 Stunde bis etwa 48 Stunden. Alternativ wird eine limitierte Schallbehandlung
mit hoher Energie von 5 Sekunden bis 5 Minuten so oft wie erforderlich,
mit Abkühlperioden,
wiederholt, um die gewünschte
Viskosität
oder den gewünschten
Kd-Endpunkt zu erreichen. Das Verfahren
der Beschallungshydrolyse ist der thermischen Hydrolyse für Polysaccharide
mit komplexen Strukturen (siehe unten) vorzuziehen. Andere im Stand
der Technik bekannte geeignete Mittel, um eine partielle Hydrolyse
von Polysacchariden zu bewirken, sind ebenfalls anwendbar. Beispielsweise
kann eine limitierte chemische Hydrolyse mit Säure, endolytische Enzymbehandlung
oder physikalische Scherung in einem Mischer oder einer Mühle ebenfalls
zur Verringung der mittleren Kettengröße des Pn-Ps eingesetzt werden.
-
In
einer bevorzugten Ausführungsform
wird das Pn-Ps mittels Passage durch einen Homogenisator bei einer
Temperatur zwischen etwa 0°C
und 30°C
und Drücken
zwischen etwa 13,8 MPa und 103 MPa (2000 psi und 15000 psi) einer
physikalischen Scherung unterworfen, so vorherbestimmt, daß sich ein
Pn-Ps-Produkt mit den erwünschten
Eigenschaften hinsichtlich Größe, Polydispersität und Antigenizität (siehe
Beispiel 10) ergibt.
-
Ein
Ziel-Endpunkt der Hydrolyse, günstig
durch Lösungsviskosität oder durch
Hochleistungs-Größenausschlußchromatographie
gemessen, wird für
jedes Polysaccharid in einem Pilotmaßstab so vorherbestimmt, daß die Antigenizität des Polysaccharids
nicht eliminiert wird. Wie oben erörtert, wird eine nominale Fähigkeit zur
Bindung von typenspezifischem Anti-Pneumokokken-Antikörper, die
nicht weniger als 70% der gezeigten Bindung für eine gleiche Konzentration
des rohen Pn-Ps-Ausgangsmaterials beträgt, als zufriedenstellend betrachtet.
Dies bedeutet nicht, daß nicht
Pn-Ps mit wesentlich niedrigerem MN, MW oder niedrigerer Anzahl sich wiederholender
Einheiten pro Molekül
(Tabelle II) nach diesem Verfahren hergestellt werden könnten und
solche Pn-Ps, welche nicht in der Lage sein mögen, oberhalb der 70%-Ausschlußgrenze
zu reagieren, welche oben für
den Geschwindigkeitsnephelometrie-Assay festgelegt wurde, nach einer
Konjugation in Tieren immunogen sein könnten. Dies bedeutet, daß Pn-Ps
mit niedrigem Molekulargewicht trotz der Abwesenheit einer nenneswerten
Fähigkeit
zur Bindung von typenspezifischem Anti-Pn-Ps-Antikörper in
konjugiertem Zustand von dem Immunsystem des Säugers erkannt werden könnten und
eine gute typenspezifische Anti-Pneumokokken-Antwort hervorgerufen
werden könnte.
In diesem Fall sollte der Begriff durch den Begriff ”immunogen” als operatives
Kriterium zur Akzeptanz oder Verwerfung einer gegebenen Pn-Ps-Präparation
ersetzt werden. In der Praxis ist es jedoch am bequemsten, statt
der in vivo-Immunogenitätsparameter
die in vitro-Antigenizitätsparameter
zur Verfahrenskontrolle zu verwenden.
-
Im
allgemeinen ist dasselbe Größenreduktionsverfahren
auf die meisten Polysaccharide anwendbar. Während das Pn6B-Ps jedoch nach
längerer
thermischer Größenreduktion
seine Antigenizität
behält,
kann Pn23F-Ps seine strukturelle Integrität verlieren (Entfernung der
Glycerin-Phosphat-Seitenketten) und erfordert die sanftere Größenreduktion,
welche mittels Schall oder physikalischer Scherung erzielt werden
kann. Physikalische Scherung, z. B. in einem Gaulin-Homogenisator,
ist aus mehreren Gründen
ein bevorzugtes Verfahren. Erstens ist das Verfahren einer Anwendung
in größerem Maßstab zugänglich.
Zweitens erfordern die Verfahren der Beschallungs- und thermischen
Hydrolyse im allgemeinen eine nachfolgende Fraktionierung des hydrolysierten
Pn-Ps, um Polydispersitäten
im Bereich zwischen 1,0 und 1,5 zu erzielen. Das Verfahren der physikalischen
Scherung ergibt jedoch im allgemeinen ein Pn-Ps-Produkt mit einer
Polydispersität,
die ohne weitere Fraktionierung in diesen Bereich fällt, obwohl
erforderlichenfalls eine Fraktionierung eingesetzt werden kann,
um zusätzliche
Reinheitssteigerungen und Verminderungen der C-Ps-Verunreinigung
zu erzielen. Drittens kann das Verfahren der physikalischen Scherung
im Vergleich zu den Mitteln der thermischen Hydrolyse oder Beschallungshydrolyse
den Vorteil einer größeren Reproduzierbarkeit
für jedes
gegebene Pn-Ps aufweisen. Viertens scheint das Verfahren der physikalischen
Scherung einen gewissen Vorteil bei der Herstellung des Pn-Ps-Produkts
zu bieten, welches für
eine gegebene Größe mehr
Antigenizität
behält
als ein Pn-Ps derselben Größe, welches
durch Beschallungshydrolyse oder thermische Hydrolyse erzeugt wurde.
-
Die
Viskosität,
die zum mittleren Pn-Ps-Molekulargewicht in Beziehung steht, ist
ein bequemer Prozeßparameter
zur Überwachung
und kann während
der Hydrolyse leicht verfolgt werden, um den Grad der Größenreduktion
zu limitieren und zu kontrollieren. Chemisch und physikalisch nicht
unterscheidbare Chargen von Pn6B-Ps und Pn23F-Ps wurden einfach
hergestellt durch Größenreduktion
des Polysaccharids auf eine einheitliche Ziel-Endviskosität (siehe
Tabelle I oben). Ein solcher Einsatz von verfahrensbegleitenden
Vskositätsmessungen
ist auf ein breites Spektrum von rohen Polysacchariden anwendbar,
was deren hydrolytische Größenreduktion
ohne Änderung
der antigenen Eigenschaften der resultierenden Pn-Ps erlaubt Wie
oben geschildert, wird die Retention der Antigenizität leicht
festgestellt, z. B. durch einen Ouchterlony-Doppelimmundiffusions-Assay,
Geschwindigkeitsnephelometrie oder andere im Stand der Technik bekannte
Verfahren.
-
Ziel-Endviskositäten für Lösungen von
1 mg/ml verschiedener Pn-Ps-Präparationen
in 0,9% Natriumchlorid (Salzlösung)
sind in Tabelle III unten angegeben. Ähnliche Werte sind für Pn-Ps,
die von anderen Pneumokokken-Subtypen stammen, geeignet TABELLE
III Lösungsviskosität für rohe und
hydrolysierte Pn-Ps:
| Pn-Ps-Subtyp | Viskosität von rohem
Pn-Ps | Ziel-Endviskosität |
| | (Zentistokes) | (Zentistokes) |
| Pn4-Ps | 1,8 | 1,5–1,00 |
| Pn6-Ps | 1,4 | 1,3–1,00 |
| Pn9V-Ps | 1,4 | 1,3–1,00 |
| Pn14-Ps | 1,2 | 1,1–0,95 |
| Pn18C-Ps | 2,0 | 1,5–1,00 |
| Pn19F-Ps | 1,4 | 1,3–1,00 |
| Pn23F-Ps | 1,6 | 1,5–1,00 |
-
Bei
einigen Pneumokokken-Polysacchariden ist es vorteilhaft, einen zusätzlichen
Reinigungsschritt wie einen Ionenaustauschschritt vor oder nach
der partiellen Hydrolyse einzuschließen. Im Falle von Pn14-Ps erfolgt
dieser Schritt durch eine diskontinuierliche Adsorption von anionischen
Verunreinigungen an Whatman DE52-Harz vor der partiellen Beschallungshydrolyse.
Das Polysaccharid, das bei dem leicht sauren ph-Wert der Behandlung
neutral ist, wird als Überstandsfraktion
bereit zur Hydrolyse gewonnen.
-
Die
Molekulargewichtswerte für
Pn6B-Ps-Präparationen
betragen etwa 900 Kilodalton (kD) vor und etwa 300 kD nach der Größenreduktion
und Fraktionierung. Für
Pn23F-Ps betragen die jeweiligen Werte etwa 1000 kD oder mehr davor
und etwa 400–500
kD danach. Somit ist eine Reduktion der Pn-Ps-Größe auf etwa 500 plus-minus
etwa 300 Kilodalton ein angemessenes Ziel für diese Phase des Verfahrens
für jeden Pn-Ps-Subtyp.
-
Eine
erneute Fällung
des partiell hydrolysierten Materials mit vorherbestimmten Alkoholkonzentrationen
erlaubt die Gewinnung und weitere Reinigung des partiell hydrolysierten
Pn-Ps wie im Unterabschnitt (c) unten beschrieben.
-
I.c) Fraktionierung der partiell hydrolysierten
Pn-Ps nach Größe und Reinheit:
-
Die
Polydispersität
einer Pn-Ps-Präparation
weist nicht nur auf die Varianz der Kettenlänge des subtypenspezifischen
Pn-Ps hin, sondern zeigt auch an, daß gruppenspezifisches C-Polysaccharid
sowie andere Verunreinigungen in der Pn-Ps-Präparation verbleiben können. Wie
oben festgestellt, ist eine Verunreinigung durch restliches C-Polysaccharid
nicht nützlich
und kann sogar mit negativen Immunantworten in Zusammenhang stehen.
-
Die
Auswahl eines engen Bereichs der mittleren Polysaccharid-Molekülgröße (verringerte
Polydispersität)
wird günstig
erzielt durch differentielle Alkohollöslichkeit, z. B. Ethanol- und
vorzugsweise Isopropanol(IPA)-Löslichkeit,
nach der Größenreduktion.
Die Grundlage dieser Selektion ist, daß für eine gegebene Pn-Ps-Präparation
die Alkohollöslichkeit
umgekehrt proportional zur Kettenlänge ist, welche wiederum proportional
zum Molekulargewicht ist. So wurde das Verfahren erfolgreich angewandt,
um quantitativ Molekülpopulationen
mit einheitlicher Größe und mit
signifikant verbesserter Homogenität gegenüber den größenreduzierten Ausgangs-Pn-Ps
zu isolieren. Eine verfahrensbegleitende Kontrolle von IPA-Fraktionierungen
wird ermöglicht
durch die Durchführung
eines Pilotexperiments, um den IPA-Bereich, in dem das Pn-Ps ausfällt, vorauszusagen.
Ein antikörper-gesteuerter
Nephelose-Assay wird zur Überwachung
der Fraktionierung eingesetzt, um eine quantitative Pn-Ps-Gewinnung
sicherzustellen. Durch diese Verbesserung wird die Verunreinigung durch
C-Polysaccharid, das gruppenspezifische Polysaccharid, welches vielen
verschiedenen Pneumokokken-Isolaten gemeinsam ist, gegenüber dem
bei rohen Pn-Ps-Präparationen
gefundenen Niveau um etwa das 3- bis 20fache verringert. Ferner
wird die Polydispersität
der Molekülgröße der Pn-Ps-Präparation
gleichzeitig auf zwischen etwa 1,0 und 1,4 verringert.
-
Eine
zur IPA-Fraktionierung der größenreduzierten
Pn-Ps alternative Vorgehensweise ist die Chromatographie der wäßrigen,
größenreduzierten
Pn-Ps durch ein geeignetes Größenausschlußharz, z.
B. CL-2B-Harz oder irgendein anderes Harz, welches imstande ist,
Polysaccharid im Molekulargewichtsbereich von 200–1000 Kilodalton
einzuschließen
und zu fraktionieren. HPSEC unter Einsatz einer starren Größenausschluß-Matrix
ist in dieser Hinsicht günstig,
um die Laufzeit zu verringern und die Auflösung zu erhöhen. Die Selektion von Fraktionen,
die von der Säule
mit einer vorherbestimmten Viskosität oder Retentionszeit eluieren,
oder durch On-Line-Nachweis ergibt eine Population von Pn-Ps-Molekülen mit
den erwünschten,
oben offenbarten Eigenschaften hinsichtlich Größe, Viskosität und Reinheit.
-
Präparationen
von Pn-Ps, die den zusätzlichen
Schritten von IPA- oder chromatographischer Fraktionierung unterzogen
wurden, verhalten sich während
der chemischen Kopplungsschritte einheitlicher und ergeben deshalb
Konjugate mit reproduzierbaren Eigenschaften. Es werden auch signifikante
gleichzeitige Erhöhungen
der Pn-Ps-Reinheit erzielt, insbesondere werden die C-Ps-Niveaus
stark verringert.
-
Als
Ergebnis der oben beschriebenen Manipulationen und Messungen sind
bevorzugte Eigenschaften für
die Pn-Ps-Zwischenprodukte so wie in Tabelle II oben zusammengefaßt.
-
II.a) Funktionalisierung des fraktionierten
Pn-Ps, um den elektrophilen oder nukleophilen Reaktanten Pn-Ps* zu
bilden, vorzugsweise um etwa 10–40
reaktive Bromacetylgruppen pro 100 Pn-Ps-Monomer-Einheiten aufzuweisen:
-
Das
Pn-Ps aus Schritt I. (c) oben ist ausreichend homogen und besitzt
Eigenschaften wie verbesserte Löslichkeit
und verringerte Viskosität,
um das Pn-Ps einer Konjugation zugänglich zu machen. Für Fachleute sind
viele unterschiedliche Schemata verfügbar, um Konjugate von Polysacchariden
und anderen Gruppierungen herzustellen. Das hier offenbarte Verfahren
ist nur ein möglicher
Weg zur Nutzbarmachung des neuen, partiell hydrolysierten und fraktionierten
Pn-Ps-Zwischenprodukts dieser Erfindung, um Konjugate zu bilden, und
sollte nicht als der ausschließliche
Modus zur Verwendung des Pn-Ps-Zwischenprodukts verstanden werden.
-
Das
von Marburg, S., et al., [
US-Patent
4,695,624 ; J. Am. Chem. Soc. 108, 5282 (1986)] offenbarte Verfahren
mit einem bigenerischen Abstandhalter ist ein bevorzugtes Verfahren
zur Konjugation des fraktionierten und größenreduzierten Pn-Ps mit einem
immunogenen Protein. Das Pn-Ps wird funktionalisiert, um elektrophile
oder nukleophile Gruppen zu zeigen. Das resultierende Pn-Ps* ist
dann imstande, mit einem umgekehrt funktionalisierten Protein, PRO*,
zu reagieren. Das Verfahren dieser Erfindung umfaßt auch
die Wahl eines Nukleophils oder Bisnukleophils, welches mit dem
aktivierten Polysaccharid reagieren wird, um ein kovalent modifiziertes
Polysaccharid mit elektrophilen Seitenpositionen oder Thiolseitengruppen
zu bilden, wodurch das Bedürfnis
zur weiteren Funktionalisierung des bis-nukleophil modifizierten
Polysaccharids vor der Umsetzung des kovalent modifizierten Polysaccharids
mit dem kovalent modifizierten Protein entfällt. Die Funktionalisierung
des Proteins mit jeder Gruppierungsform kann auch in mehr als einem
Schritt durchgeführt werden,
je nach der Wahl der Reaktanten in diesen Schritten.
-
Unabhängig von
der erwünschten
Funktionalität
des Pn-Ps* muß das
größenreduzierte
fraktionierte Pn-Ps zuerst in einem Lösungsmittel solubilisiert werden,
welches den Funktionalisierungsprozeß nicht stören wird. Nachdem es die Hydroxylgruppen
des Pn-Ps sind, welche einer Funktionalisierung am ehesten zugänglich sind,
ist die Trennung des Pn-Ps von Wasser, um die erste Funktionalisierung
zu bewirken, kritisch. Der Ersatz saurer Pn-Ps-Wasserstoffe durch
ein hydrophobes Kation wie Tetra- oder
Tributylammonium ermöglicht dem
Pn-Ps, in nicht-wäßrigen Lösungsmitteln
wie DMSO oder DMF löslich
zu werden. Natürlich
besteht keine Notwendigkeit, diesen Ersatz bei Pn-Ps durchzuführen, welche
neutral sind (z. B. Pn14-Ps oder Pn7F-Ps). Sobald sich das Pn-Ps
in einer nicht-wäßrigen Lösung befindet,
kann es mit einem Biselektrophil wie Carbonyldiimidazol umgesetzt
werden, um ein Imidazoyldiurethan zu bilden. Die Anzahl der funktionellen
Gruppen pro 100 Pn-Ps-Monomereinheiten
wird zu diesem Zeitpunkt kontrolliert durch Zugabe einer limitierten
Menge von etwa 1/5 des Carbonyldiimidazol-Reagens im Vergleich zu
den gesamten Pn-Ps-Monomeren auf molarer Basis, so daß im Mittel
nur etwa 10–40
von 100 Pn-Ps-Monomereinheiten derivatisiert werden. Diese Spezies
ist einer nukleophilen Substitution durch Reagenzien wie i) Cystamin-Dihydrochlorid,
welche die Bildung von nukleophilen Pn-Ps*-Derivaten erlauben, oder
ii) 1,4-Butandiamin, welche die Bildung von elektrophilen Pn-Ps*-Derivaten
erlauben, zugänglich,
wie offenbart im
US-Patent 4,695,624 und
Marburg et al., J. Am. Chem. Soc., 108, 5282 (1986).
-
Es
wurde kürzlich
festgestellt, daß aufgrund
der Tatsache, daß einige
saure Pneumokokken-Polysaccharide,
wie Pn9V-Ps, Pn4-Ps, Pn1-Ps, Pn5-Ps und Neisseria meningitidis B-
oder C-Polysaccharide,
freie Carbonsäuregruppen
sowie freie Hydroxylgruppen aufweisen, die Konjugationschemie dieser
Polysaccharide im Vergleich zu neutralen Polysacchariden oder Polysacchariden,
welche aufgrund der Anwesenheit von Phosphodiesterbindungen wie
in Polyribosylribitphosphat anionisch sind, in leicht unterschiedlicher
Weise vonstatten geht. Im allgemeinen geht die Konjugationschemie
von carboxylat-freien Polysacchariden durch Überführung freier Polysaccharidhydroxyle
in Urethanverknüpfungen
vonstatten, d. h. von
worin R
a den
Rest der Atomkette, die das Polysaccharid mit Protein verknüpft, darstellt.
-
Bei
den Carbonsäure
enthaltenden Polysacchariden wie Pn-9V-Ps, welches Glucuronat enthält, oder Pn4-Ps,
welches Pyruvatgruppen enthält,
geht die Chemie jedoch von
worin R
a wiederum
den Rest der Atomkette, die das Polysaccharid mit Protein verknüpft, darstellt.
Beobachtet wird deshalb, daß die
Esterfunktionalität
des Urethans bei dem Carbonsäure
enthaltenden Polysaccharid entweder nicht gebildet wird oder zusätzlich an
diesen Stellen eine einfache Amidbindung gebildet wird. Die Konjugationschemie
geht aufgrund der Anwesenheit der Carbonsäurefunktionalitäten mit
höherer
Geschwindigkeit vonstatten.
-
Da
angenommen wird, daß die
Carbonsäurefunktionalitäten einen
wichtigen Beitrag zur Antigenizität und Immunogenizität dieser
Klasse von anionischen Polysacchariden darstellen, muß die Konjugationschemie für diese
Polysaccharide sorgfältig
gesteuert werden. Das Bedürfnis
nach hohen Verhältnissen
von Polysaccharid zu Protein im Endkonjugat muß gegen die Notwendigkeit zur
Aufrechterhaltung der antigenen Integrität ausbalanciert werden. Dieses
Ziel wird erreicht durch Begrenzung des Ausmaßes der anfänglichen, durch Carbonyldiimidazol
vermittelten Aktivierung. Sobald das Amid gebildet ist, kann der
nächste
Schritt unter Beteiligung von Cystamin-Dihydrochlorid oder 1,4-Butandiamin
wie oben angegeben und im folgenden weiter erläutert vonstatten gehen.
-
Alternativ
können
die Carboxylgruppen durch Verwendung von Trimethylsilyl oder ähnlichen
Schutzgruppen, welche später
durch milde alkalische Bedingungen entfernt werden können, oder
durch Verwendung von 2,4-Dimethoxybenzylestern, welche säurelabil
wären,
reversibel geschützt
und dann von den Schutzgruppen befreit werden. In diesem Fall kann
die Konjugationschemie auf übliche
Weise über
die Polysaccharid-Hydroxylgruppen vonstatten gehen.
-
1. Herstellung von nukleophilem Pn-Ps*:
-
Das
oben erhaltene, durch Carbonyldiimidazol aktivierte, größenreduzierte
und fraktionierte Ps kann in wäßrigen oder
anderen Lösungsmitteln
mit Reagenzien, wie z. B. im
US-Patent
4,695,624 offenbart, umgesetzt werden. Ein bevorzugtes
Reagens ist Cystamin-Dihydrochlorid. Die anschließende Entfernung
von überschüssigem Cystamin
und die Reduktion mit Dithiothreit oder Dithioerythreit ergibt das
nukleophile sulfhydryl-funktionalisierte Pn-Ps. Dieses Pn-Ps*-Derivat
ist zur Reaktion mit einem elektrophilen PRO* in der Lage, z. B.
dort wo das Protein modifiziert wurde, um Bromacetylseitengruppen
aufzuweisen.
-
2. Herstellung von elektrophilem Pn-Ps*:
-
Das
oben erhaltene, durch Carbonyldiimidazol aktivierte, größenreduzierte
und fraktionierte Pn-Ps kann in wäßrigen oder anderen Lösungsmitteln
mit Reagenzien wie im Patent
4,695,624 offenbart,
vorzugsweise mit 1,4-Butandiamin (BuA
2),
umgesetzt werden. Eine nachfolgende Acylierung des Pn-Ps-BuA
2 mit
p-Nitrophenylbromacetat oder einem ähnlichen Reagens erzeugt das
elektrophile Pn-Ps-BuA
2-BrAc-Derivat, welches zur Reaktion mit
einem nukleophilen PRO*, z. B. einem sulfhydryl- modifizierten Protein, in der Lage ist. Der
Grad der Derivatisierung wird zu diesem Zeitpunkt durch NMR und
Vergleich des 1,4-Butandiamin-Integrals mit einem geeigneten Monosaccharidsignal
wie demjenigen des Methyls von Rhamnose gemessen. Bevorzugte Derivatisierungsgrade
betragen zwischen 10 bis 40% und am meisten bevorzugt etwa 20%.
-
II.B.) Isolierung des immunogenen Proteins
(PRO), vorzugsweise des Neisseria meningitidis B-OMPC oder einer Untereinheit davon:
-
Die
Proteingruppierung sollte sich als Immunverstärker verhalten. Es ist wünschenswert,
bei der Wahl des Proteins diejenigen zu vermeiden, welche zu einer
unspezifischen Aktivierung der Immunantwort des Empfängers führen (Reaktogenizität). Im
US-Patent 4,695,624 verwendeten
Marburg et al. den Komplex des Proteins der äußeren Membran (OMPC), abgeleitet
von Neisseria meningitidis, um Polysaccharid-Protein-Konjugate herzustellen.
OMPC hat sich für
diese Erfindung als geeignet erwiesen, obwohl andere immunogene
Proteine wie Tetanus- oder Diphterie-Toxoid oder Pertussinogen eingesetzt
werden können.
-
Es
wurden verschiedene Verfahren zur Reinigung von OMPC aus den gramnegativen
Bakterien entwickelt [Frasch et al., J. Exp. Med. 140, 87 (1974);
Frasch et al., J. Exp. Med. 147, 629 (1978); Zollinger et al.,
US-Patent 4,707,543 (1987);
Helling et al., Acta Path. Microbiol. Scand. Abschn. C 89, 69 (1981);
Helting et el.,
US-Patent 4,271,147 ).
Der hier verwendete OMPC wurde präpariert wie im Beispiel 1 beschrieben.
Ferner ist eine Proteinuntereinheit, isoliert durch Dissoziation
von OMPC oder durch rekombinante Expression von Material, das für ein OMPC-Komponentenprotein
kodiert, insbesondere das Hauptmembranprotein (auch als das mitogene
Induktionsprotein MIP oder als das hauptsächlich immunverstärkende Protein,
MIEP, bezeichnet), ebenfalls bevorzugt. Ein Verfahren zur Gewinnung
der Untereinheitsproteine ist offenbart in Beispiel 2, 16–23 und
in der US-Patentanmeldung USSN 555,978; 555,329; 555,204; und 639,457
(entsprechend
EP-A-0467714 ):
-
II.c) Funktionalisierung des PRO zur Erzeugung
des nukleophilen oder elektrophilen Reaktanten PRO*, vorzugsweise
OMPC oder eine Untereinheit davon, um reaktive Sulfhydrylgruppierungen
zu zeigen:
-
Das
wie in II.(b) oben isolierte PRO wird anschließend funktionalisiert, um entweder
elektrophile oder nukleophile Gruppen zu zeigen. Das resultierende
PRO* ist dann imstande, mit einem gegenteilig funktionalisierten
Pn-Ps*, wie in II.(a) oben hergestellt, zu reagieren.
-
1. Bildung von elektrophilen PRO*-Derivaten:
-
Das
isolierte PRO, z. B. der OMPC von Neisseria oder MIEP von OMPC,
wird vorzugsweise umgesetzt mit einem Reagens wie N-(Bromacetyl)-6-aminocapronsäure-p-nitrophenylester,
das imstande ist, mit den ε-Aminogruppen
von Lysin auf PRO zu reagieren. Das resultierende bromacetylierte
PRO* ist imstande, mit den nukleophilen Derivaten von Pn-Ps*, wie
in II.(a)1. oben hergestellt, zu reagieren.
-
2. Bildung von nukleophilen PRO*-Derivaten:
-
Das
isolierte PRO, z. B. der OMPC von Neisseria oder MIEP, wird mit
einem Reagens wie N-Acetylhomocysteinthiolacton umgesetzt, um das
Sulfhydryl-Derivat des Proteins zu erzeugen. Dieses nukleophile Derivat
ist zur Reaktion mit einem elektrophilen Pn-Ps*, hergestellt wie
in II.(a)2. oben, in der Lage. Typische Ergebnisse dieser Prozeßphase ergeben
Sulfhydryl-Titer von zwischen etwa 0,1 und 0,3 μMol/mg Protein.
-
II.d) Konjugation des Polysaccharids (Pn-Ps*)
von Schritt II.(a) mit dem Protein (PRO*) von Schritt II(c):
-
Nach
Bildung der reaktiven Spezies Pn-Ps* und PRO* nach den obigen Schritten
II.(a) und II.(c) werden die gegenteilig aktivierten Reaktionspartner
miteinander in einem Masseverhältnis
von etwa 1:1 in Kontakt gebracht. Die Reaktionsmischung sollte durch
Spülen
mit Stickstoff von Luft befreit werden, verschlossen und bei Raumtemperatur
etwa 4 Tage lang bei zwischen 17° und
40°C reagieren
gelassen werden. Beispiele solcher Reaktionen umfassen:
worin
ein aktiviertes Polysaccharid, das mit 4-Bromacetamidobutylamin
umgesetzt wurde, mit einem Protein, das mit N-Acetylhomocysteinthiolacton
umgesetzt wurde, zur Bildung eines Konjugats umgesetzt wird, und:
(worin
Y'' ein C
2-C
8-Alkylrest ist), worin ein amino-derivatisiertes
Polysaccharid, das mit aktivierter Maleimidosäure umgesetzt wurde, mit einem
carboxy-aktivierten Protein, das mit einem Aminothiol aminiert wurde,
zur Bildung eines Konjugats umgesetzt wird.
-
Gleichermaßen können beliebige
der kovalent modifizierten Polysaccharide mit Thiolseitengruppen mit
dem bakteriellen Protein OMPC oder MIEP, welches elektrophile Zentren
als Seitengruppen aufweist, umgesetzt werden, um ein kovalentes
Konjugat zu ergeben. Ein Beispiel einer solchen Reaktion ist:
worin
ein aktiviertes Polysaccharid, das mit einem Aminothiol umgesetzt
wurde, mit einem carboxy-aktivierten Protein,
das mit Monohalogenacetyl-Derivaten eines Diamins umgesetzt wurde,
zur Bildung eines Konjugats umgesetzt wird. Eine sehr bevorzugte
Verknüpfung
gemäß dieser
Erfindung ist der Abstandhalter der Formel:
für Verknüpfungen über die Polysaccharid-Hydroxylgruppen,
oder
im Falle von Polysacchariden,
welche Carbonsäuregruppen
tragen.
-
Sollte
es erforderlich sein, die elektrophile Aktivität eines Überschusses an Halogenacetylgruppen
zu eliminieren, wird die Reaktion des Konjugats mit einem niedermolekularen
Thiol wie N-Acetylcysteamin diesen Zweck erfüllen. Die Verwendung dieses
Reagenzes, N-Acetylcysteamin, erlaubt auch die Bestätigung hinsichtlich
der eingesetzten Halogenacetylgruppierungen, da das gebildete S-Carboxymethylcysteamin
eindeutig mit dem Verfahren von Spackman, Moore und Stein nachgewiesen
werden kann.
-
II.e) Versehen des Pn-Ps-PRO-Konjugats
mit Endgruppen, um restliche funktionelle Gruppen zu eliminieren:
-
Restliche
elektrophile Gruppen entweder auf dem PRO* oder dem Pn-Ps* werden
am Ende der Reaktion gequencht durch Zugabe eines etwa 2–10fachen
molaren Überschusses
gegenüber restlichen
reaktiven Gruppen auf dem Konjugat an einem niedermolekularen Nukleophil,
z. B. N-Ethylmaleimid
(NEM), um restliche freie Sulfhydryle mit Endgruppen zu versehen,
oder einem Elektrophil, z. B. N-Acetylcysteamin, um restliche Bromacetylgruppierungen
mit Endgruppen zu versehen.
-
II.f) Isolierung des Konjugatprodukts:
-
Das
mit Endgruppen versehene Produkt-Konjugat wird von unkonjugiertem
PRO, Pn-Ps und anderen Reaktanten durch Ultrazentrifugation oder
Diafiltration abgetrennt. Das Konjugat-Pellet wird in einer wäßrigen Pufferwaschlösung resuspendiert,
welche eine Detergensbehandlung, z. B. 0,5% Desoxycholin und Salz
wie 0,1 M Tris, pH 7–9,
und etwa 10 mM EDTA einschließen
kann, um restliche Pyrogene zu entfernen, und wird zwischen 1 und
25 h lang bei Raumtemperatur stehengelassen. Das Konjugat wird erneut
pelletiert, In wäßrigem Puffer
ohne Detergens resuspendiert und dann durch Ultrazentrifugation
bei etwa 100.000 × g
unter Verwendung eines Festwinkelrotors etwa 2 h lang bei etwa 1° bis 20°C erneut
pelletiert oder irgendeinem aus einer Vielfalt von anderen Reinigungsverfahren
unterworfen, einschließlich
Difiltrations-Gelpermeation, Ionenaustauschchromatographie, Gradientenzentrifugation
oder einer anderen Differentialadsorptionschromatographie, um nicht-kovalent gebundene
Polysaccharide und Proteine zu eliminieren, unter Verwendung von
Ps-, PRO- und Geschwindigkeitsnephelometrie-Assays
sowie des Kovalenz-Assays für
den bigenerischen Abstandhalter (siehe unten) als in vitro-Verfahren
zur Verfolgung der erwünschten
biologischen Aktivität.
-
Die
weitere Abtrennung von Reagenzien kann durch Größenausschlußchromatographie in einer Säule erfolgen
oder die Abtrennung kann im Falle sehr großer, unlöslicher Proteine durch Ultrazentrifugation
erfolgen. Der Resuspension des Konjugats in sterilem Wasser und
dem Stehenlassen für
etwa einen Tag bei 4°C, um
eine vollständige
Solubilisierung zu erlauben, folgt eine Zentrifugation bei niedrigerer
Geschwindigkeit, um unlösliche
Teilchen zu entfernen. Die Überstandslösung enthält das Endprodukt,
welches steril filtriert zu Vakzin-Zusammensetzungen in immunologisch
wirksamen Dosierungsniveaus formuliert und steril in Flaschen abgefüllt werden
kann.
-
Eine
Analyse des Konjugats, um die Kovalenz und somit die Stabilität des Konjugats
zu bestätigen, wird
durchgeführt
mittels Hydrolyse (vorzugsweise mit 6 N HCl bei 100°C für 20 h)
des Konjugats und anschließender
quantitativer Analyse hinsichtlich der einmaligen Aminosäure des
hydrolysestabilen Abstandhalters, welcher die Thioetherbindung enthält, und
der Aminosäurebestandteile
des Proteins. Der Beitrag der Aminosäuren des Proteins kann erforderlichenfalls
eliminiert werden durch Vergleich mit dem entsprechenden Aminosäurestandard
für das
betreffende Protein, wobei der verbleibende Aminosäurewert
die Kovalenz des Konjugats reflektiert, oder die Aminosäure des
Abstandhalters kann so ausgesucht werden, daß sie außerhalb des Aminosäurestandards
des Proteins in der Analyse erscheint. Der Kovalenz-Assay ist auch
geeignet zur Überwachung
von Reinigungsverfahren, um die Konzentrationserhöhung der
biologisch aktiven Komponenten zu kennzeichnen. In den obigen Beispielen
resultiert die Hydrolyse von

in der Freisetzung von S-Carboxymethylhomocystein,
die Hydrolyse von
resultiert in der Freisetzung
der Aminodicarbonsäure,
und die Hydrolyse von
resultiert in der Freisetzung
von S-Carboxymethylcysteamin, H
2NCH
2CH
2SCH
2CO
2H, durch Spaltung des Ps-A-E-S-B-PRO-Moleküls an Peptidverknüpfungen
und anderen hydrolytisch instabilen Bindungen. Chromatographieverfahren,
z. B. diejenigen von Spackman, Moore und Stein, können dann
günstig
angewandt und das Verhältnis
der Aminosäurebestandteile
bestimmt werden.
-
Die
optimale Produktion von IgG-Antikörper erfordert das Zusammenwirken
von B- und T-Lymphozyten
mit Spezifität
für das
Antigen von Interesse. T-Lymphozyten sind nicht imstande, Polysaccharide
zu erkennen, können
jedoch zu Anti-Polysaccharid-IgG-Antikörper-Antworten beitragen, falls
das Polysaccharid kovalent mit einem Protein verknüpft ist,
welches die T-Zelle erkennen kann.
-
C. Vakzin-Formulierungen und Anwendbarkeit:
-
In
einer bevorzugten Ausführungsform
wird das Konjugatprodukt an Aluminiumhydroxidgel adsorbiert. Dies
erfolgt beispielsweise durch Präparation
einer Konjugat-Stammlösung,
die einer Konzentration von 20 μg/ml
Pn-Ps äquivalent
ist. Portionen können
1:1, 1:5 und 1:10 mit sterilem Wasser verdünnt werden. Portionen jeder
dieser Proben, einschließlich
einer Portion der 20 μg/ml-Stammlösung, werden
mit einem Aluminiumhydroxid-Verdünnungsmittel,
das 0,85 mg/ml Al+3, 1,7% NaCl (Gew./Vol.)
und 100 μg/ml
Thimerosol enthält, 1:1
verdünnt.
Der pH-Wert der Lösung
wird mit 1 N NaOH auf etwa 7,5 eingestellt, was Lösungen mit
einer Pn-Ps-Konzentration von 10, 5, 2 und 1 μg/ml ergibt Dosen von zwischen
etwa 0,1 und 0,75 ml jeder dieser Formulierungen eignen sich zur
Verabreichung an Empfänger
unterschiedlichen Alters und Gewichtsbereichs. Es wurde festgestellt,
daß das
wie beschrieben formulierte Vakzin bei 2–3 Monate alten Affenbabys
signifikante subtypenspezifische Anti-Pneumokokken-Polysaccharid-Immunantworten
auf Pn6B-Ps-OMPC, Pn14-Ps-OMPC, Pn19F-Ps-OMPC und Pn23F-Ps-OMPC induzierte.
Das Pn-Ps-OMPC-Konjugat-Vakzin wurde ferner befunden, bei athymischen
Mäusen
T-Zell-abhängig
zu sein.
-
Aus
dieser Offenbarung sollte deutlich geworden sein, daß andere
Polysaccharide, welche Eigenschaften wie hier definiert aufweisen,
und Verfahren zur Herstellung der Ps mit diesen Eigenschaften zur
Herstellung von anderen Konjugaten als solchen, welche partiell
hydrolysierte und fraktionierte Pneumokokken-Polysaccharide umfassen,
günstig
einsetzbar sein werden. Diese Konjugate könnten dann eingesetzt werden
zur Verhinderung von Krankheiten, die von anderen pathogenen Organismen
verursacht werden. Beispielsweise könnten die Gruppe-B-Streptokokken,
eine Ursache von Neugeborenenmeningitis, Neisseria meningitidis
B oder C, eine Ursache von Meningitis bei Kleinkindern, oder E.
coli, eine wichtige Ursache von Harntraktsinfektionen und anderen
opportunistischen Infektionen, als Polysaccharid-Quellen verwendet
werden. Diese Polysaccharide sowie die Pn-Ps und deren kovalente
Konjugate können
auch wichtige Komponenten für
Kombinationsvakzin-Formulierungen bereitstellen. Solche Kombinationen
können
beispielsweise immunologisch wirksame Mengen an Adjuvans, z. B.
Freunds oder Ribi, oder immunmodulatorische Verbindungen wie die
Interleukine, Interferone (siehe z. B. die Verbindungen, welche
aufgelistet sind in: Market Letter, 30. Nov., 1987, S. 26–27; Genetic
Engineering News, Jan. 1988, Bd. 8, S. 23) oder weitere Immunogene
einschließen. In
einer bevorzugten Ausführungsform
ist eine Zusammensetzung, die immunologisch wirksame Mengen des Konjugats
dieser Erfindung umfaßt,
mit einem oder mehreren der Vakzine gegen Hepatitis B, Hepatitis
A, Non-A-Non-B-Hepatitis,
AIDS, Diphterie, Keuchhusten, Tetanus, Masern, Mumps, Röteln, Varicella,
Polio oder Haemophilus influenzae b eingeschlossen. Bevorzugte weitere
Vakzine, ausgewählt
aus den soeben genannten, sind aus PevaxHIB®, Recombivax
HB®,
M-M-R® und
einem trivalenten DTP-Vakzin ausgewählt.
-
Die
folgenden Beispiele dienen zur weiteren Offenbarung der Erfindung.
-
BEISPIEL 1
-
Herstellung von Neisseria meningitidis
B11 Serotyp 2 OMPC
-
A. Fermentation
-
1. Neisseria meningitidis Gruppe B11
-
Ein
Röhrchen,
enthaltend die lyophilisierte Kultur von Neisseria meningitidis
[erhalten von Dr. M. Artenstein, Walter Reed Army Institute of Research
(WRAIR), Washington, D. C.], wurde geöffnet und Eugonbouillon (BBL)
zugegeben. Die Kultur wurde auf Mueller-Hinton-Agarschrägen ausgestrichen
und bei 37°C
mit 5% CO2 36 h lang inkubiert, wonach das
Wachstum in 10%iges Magermilchmedium (Difco) geerntet wurde und Aliquots
bei –70°C eingefroren
wurden. Die Identität
des Organismus wurde bestätigt
durch Agglutination mit spezifischem Antiserum, bezogen von WRAIR,
und Typisierungsserum, bezogen von Difco.
-
Ein
Gefäß der Kultur
aus der zweiten Passage wurde aufgetaut und auf 10 Columbia Sheep Blood-Agarplatten (CBAB-BBL)
ausgestrichen. Die Platten wurden bei 37°C mit 5% CO2 18
h lang inkubiert, wonach das Wachstum in 100 ml 10%iges Magermilchmedium
geerntet wurde, Aliquots in 0,5-ml-Mengen entnommen und bei –70°C eingefroren
wurden. Der Organismus wurde durch Agglutination mit spezifischem
Antiserum, Zuckerfermentation und Gram-Färbung positiv identifiziert.
-
Ein
Gefäß der Kultur
aus dieser Passage wurde aufgetaut, mit Mueller-Hinton-Bouillon
verdünnt
und auf 40 Mueller-Hinton-Agarplatten ausgestrichen. Die Platten
wurden bei 37° C
mit 6% CO2 18 h lang inkubiert, wonach das
Wachstum in 17 ml 10%iges Magermilchmedium geerntet wurde, in Aliquots
von 0,3-ml-Mengen aufgeteilt und bei –70°C eingefroren wurde. Der Organismus
wurde durch Gram-Färbung, Agglutination
mit spezifischem Antiserum und Oxidasetest positiv identifiziert.
-
2. Fermentation und Gewinnung von Zellpaste
-
- a. Impfgutentwicklung – Das Impfgut wurde gezüchtet aus
einem eingefrorenen Gefäß von Neisseria
meningitidis Gruppe B, B-11 von oben (Passage 4). 10 Mueller-Hinton-Agarschrägen wurden
angeimpft und 6 etwa 18 h später
geerntet und als Impfgut für
drei 250-ml-Kolben von Gotschlichs Hefedialysatmedium bei pH 6,35
eingesetzt. Die OD660 wurde auf 0,18 eingestellt
und es wurde inkubiert, bis die OD660 zwischen
1 und 1,8 lag. 1 ml dieser Kultur wurde verwendet, um jeden von
fünf 2-l-Erlenmeyerkolben
(jeweils 1 l Medium enthaltend; siehe unten) anzuimpfen, und bei
37°C in
einem Schüttler
bei 200 UpM inkubiert. Die OD wurde in stündlichen Intervallen nach der
Animpfung überwacht.
4 l Bouillonkultur bei einer OD660 von 1,28 waren
das Ergebnis.
-
70-l-Impffermenter – Etwa 4
l Impfkultur wurden dazu verwendet, einen sterilen 70-l-Fermenter
anzuimpfen, der etwa 40 l vollständiges
Produktionsmedium enthielt (siehe unten). Die Bedingungen für die 70-l-Fermentation
umfaßten
37° C, 185
UpM bei 10 l/Mm. Lufteintrag und konstante pH-Kontrolle bei etwa
pH 7,0 für
etwa 2 h. Für
diesen Ansatz betrug die endgültige
OD660 nach 2 h 0,732.
-
800-l-Produktionsfermenter – Etwa 40
l Impfkultur wurden verwendet, um einen sterilen 800-l-Fermenter
anzuimpfen, der 568,2 l vollständiges
Produktionsmedium (siehe unten) enthielt. Der Ansatz wurde bei 37°C, 100 UpM
mit 60 l/Min. Lufteintrag und konstanter pH-Kontrolle bei pH 7,0
inkubiert Für
diesen Ansatz betrug die endgültige
OD 13 h nach der Animpfung 5,58. 3. Vollständiges Medium für Erlenmeyerkolben
und 70- und 800-l-Fermenter
| Fraktion
A |
| L-Glutaminsäure | 1,5
g/l |
| NaCl | 6,0
g/l |
| Na2HPO4·wasserfrei | 2,5
g/l |
| NH4Cl | 1,25
g/l |
| KCl | 0,09
g/l |
| L-Cystein-HCl | 0,02
g/l |
-
Fraktion B (Gutschlichs Hefedialysat):
-
1280
g Difco-Hefeextrakt wurden in 6,4 l destilliertem Wasser gelöst. Die
Lösung
wurde in zwei Amicon DC-30-Hohlfaserdialyseeinheiten mit drei H10SM-Kartuschen
dialysiert. 384 g MgSO4·7-H2O
und 3200 g Dextrose wurden in dem Dialysat gelöst und das Gesamtvolumen mit
destilliertem Wasser auf 15 l gebracht. Der pH wurde mit NaOH auf
7,4 eingestellt, es erfolgte eine Sterilisierung mittels Passage
durch ein 0,22-μm-Filter und
die Überführung in
den Fermenter, der Fraktion A enthielt.
-
Für die Erlenmeyerkolben:
1 l Fraktion A und 25 ml Fraktion B wurden zugegeben und der pH
mit NaOH auf 7,0–7,2
eingestellt.
-
Für den 70-l-Fermenter:
41,8 l Fraktion A und 900 ml Fraktion B wurden zugegeben und der
pH mit NaOH auf 7,0–7,2
eingestellt.
-
Für den 800-l-Fermenter.
553 l Fraktion A und 15,0 l Fraktion B wurden zugegeben und der
pH mit NaOH auf 7,1–7,2
eingestellt.
- b. Ernte und Inaktivierung
Nachdem
die Fermentation beendet war, wurde Phenol in ein separates Gefäß gegeben,
in das anschließend
die Zellbouillon überführt wurde,
was eine Phenolendkonzentration von etwa 0,5% ergab. Das Material
wurde unter leichtem Rühren
bei Raumtemperatur gehalten, bis die Kultur nicht mehr lebensfähig war (etwa
24 h).
- c. Zentrifugation
Nach etwa 24 h bei 4°C wurden die 614,4 l inaktivierter
Kulturflüssigkeit
durch Sharples-Durchlaufzentrifugen zentrifugiert. Das Gewicht der
Zellpaste nach der Phenol behandlung betrug 3,875 kg.
-
Alternativ
wurde die durch Phenol abgetötete
Fermentationsbouillon durch Diafiltration wie unten beschrieben
geerntet.
-
B. OMPC-Isolierung
-
Schritt 1. Konzentration und Diafiltration
-
Die
durch Phenol inaktivierte Kultur wurde auf etwa 30 l konzentriert
und in sterilem destilliertem Wasser unter Verwendung von 0,2-μm-Hohlfaserfiltern
(ENKA) diafiltriert.
-
Schritt 2. Extraktion
-
Ein
gleiches Volumen an 2 × TED-Puffer
[0,1 M TRIS-, 0,01 M EDTA-Puffer, pH 8,5, mit 0,5% Natriumdesoxycholat]
wurde zu den konzentrierten diafiltrierten Zellen zugegeben. Die
Suspension wurde in einen temperaturregulierten Tank überführt zur
OMPC-Extraktion bei 56°C
unter Rühren
für 30
Minuten.
-
Der
Extrakt wurde mit etwa 18.000 UpM in einer Sharples-Durchlaufzentrifuge
bei einer Durchflußrate von
etwa 80 ml/Min. bei etwa 4°C
zentrifugiert. Die viskose Überstandsflüssigkeit
wurde dann gewonnen und bei 4°C
gelagert. Die extrahierten Zellpellets wurden erneut in TED-Puffer
extrahiert wie oben beschrieben. Die Überstandsflüssigkeiten wurden vereinigt
und bei 4°C
gelagert.
-
Schritt 3. Konzentration durch Ultrafiltration
-
Der
vereinigte Extrakt wurde in ein temperaturreguliertes Gefäß überführt, das
mit AG-Tech-0,1-μm-Polysulfonfiltern
verbunden war. Die Temperatur des Extrakts wurde in dem Gefäß während des
Konzentrationsprozesses bei 25°C
gehalten. Die Probe wurde bei einem durchschnittlichen Transmembrandruck
von zwischen 75,9 und 165,6 kPa (11 und 24 psi) 10fach konzentriert.
-
Schritt 4. Gewinnung und Waschen des OMPC
-
Das
Retentat aus Schritt 3 wurde bei etwa 160.000 × g (35.000 UpM) bei etwa 70°C in einer
Durchlaufzentrifuge mit einer Durchflußrate zwischen 300 bis 500
ml/Min. zentrifugiert und die Überstandsflüssigkeit verworfen.
-
Das
OMPC-Pellet wurde in TED-Puffer (190 ml Puffer; 20 ml/g Pellet)
suspendiert. Schritt 2 und Schritt 4 wurden zweimal wiederholt (unter
Auslassung von Schritt 3).
-
Schritt 5. Gewinnung des OMPC-Produkts
-
Die
gewaschenen Pellets von Schritt 4 wurden in 100 ml destilliertem
Wasser mit einem Glasstab und einem Dounce-Homogenisator suspendiert,
um eine vollständige
Suspension sicherzustellen. Die wäßrige OMPC-Suspension wurde
dann mittels Passage durch ein 0,22-μm-Filter filtersterilisiert
und der TED-Puffer durch Wasser ersetzt mittels Diafiltration gegen
steriles destilliertes Wasser unter Verwendung eines 0,1-μm-Hohlfaserfilters.
-
BEISPIEL 2
-
Reinigung einer Untereinheit von OMPC:
-
Präparation
von gereinigtem MIEP aus OMPC oder aus rekombinanten Zellen durch
Polyacrylamidgelelektrophorese
-
Acrylamid/BIS(37,5:1)-Gele,
18 × 14
cm, 3 mm dick, wurden verwendet. Das Stützgel war 4%iges Polyacrylamid
und das Trennungsgel war 12%iges Polyacrylamid. Etwa 5 μg OMPC-Protein
oder rekombinantes Wirtszellprotein wurden pro Gel eingesetzt. Zu
1 ml OMPC wurden 0,5 ml Probenpuffer (4% Glycerin, 300 mM DTT, 100
mM TRIS, 0,001% Bromphenolblau, pH 7,0) zugegeben. Die Mischung
wurde 20 Minuten lang auf 105°C
erwärmt
und ihr gestattet, auf Raumtemperatur abzukühlen, bevor sie auf das Gel
aufgetragen wurde. Das Gel wurde bei 200–400 mA unter Kühlen laufengelassen,
bis das Bromphenolblau den unteren Rand des Gels erreichte. Ein
vertikaler Streifen des Gels wurde ausgeschnitten (etwa 1–2 cm breit)
und mit Coomassie/Kupfer(II)-acetat (0,1%) angefärbt. Der Streifen wurde entfärbt, bis
die MIEP-Bande (etwa 38 KD) sichtbar wurde. Der Streifen wurde dann
an seine ursprüngliche
Gelposition gebracht und die MIEP-Fläche aus dem verbleibenden Gel
mit Hilfe eines Skalpells ausgeschnitten.
-
Die
ausgeschnittene Fläche
wurde in Würfel
geschnitten (etwa 5 mm) und mit 0,01 M TRIS-Puffer, pH 8,1, eluiert.
Nach zwei Elutionszyklen wurde das Eluat hinsichtlich der Reinheit
mittels SDS-PAGE bewertet. Das Eluat wurde mit einem gemeinsamen
Pool von Eluaten kombiniert und 48 h lang gegen 60 mM Ammoniak-Ameisensäure, pH
10, dialysiert Alternativ kann das eluierte Protein gegen 50%ige
Essigsäure
in Wasser dialysiert werden. Nach der Dialyse wurde das eluierte
Protein bis zur Trockne eingedampft Das Material wurde weiter gereinigt
mittels Passage durch eine PD10-Größenfraktionierungssäule (Pharmacia,
Piscataway, NJ), und bei Raumtemperatur gelagert.
-
BEISPIEL 3
-
Kultivierung von Streptococcus pneumoniae-Subtypen
und Isolierung von rohem Pn-Ps:
-
I. Kultivierung von Pneumokokken:
-
Verfahren
zur Kultivierung von Pneumokokken sind im Stand der Technik wohlbekannt
[Chase, M. W., Methods of Immunology and Immunochemistry 1, 52 (1967)].
Isolate von Pneumokokken-Subtypen sind von der ATCC erhältlich.
Die Bakterien sind identifiziert als verkapselte, unbewegliche,
grampositive, lanzettenförmige
Diplokokken, welche auf Blutagar alpha-hämolytisch sind. Die Subtypen
werden auf der Basis der Quelling-Reaktion unter Verwendung spezifischer
Antiseren differenziert. Haupt- und Stamm-Impfkulturen werden vorzugsweise lyophilisiert
oder unterhalb von 8°C
gehalten. In einem bevorzugten Kulturverfahren werden Stammkulturen
mit Heart Infusion Broth rekonstituiert, auf Heart Infusion Agar, enthaltend
10% fibrinfreies Kaninchenblut, ausplattiert und etwa 18 Stunden
lang bei 37°C ± 2°C inkubiert.
-
Das
Wachstum auf der Platte wird in Heart Infusion Broth resuspendiert
und ein Aliquot des resuspendierten Wachstums wird zur Animpfung
von 100 ml Heart Infusion Broth, enthaltend 10% fibrinfreies Kaninchenblut,
verwendet, welche als stationäre
Kultur etwa 18 Stunden lang bei 37°C ± 2°C inkubiert wird. Diese 100
ml flüssiger
Kultur (Arbeitsimpfgut) werden durch mikroskopische Überprüfung eines
gramgefärbten
Ausstrichs und durch Wachstum auf Heart Infusion-Blutagarplatten
auf Reinheit überprüft. Das
Arbeitsimpfgut kann bis zu 14 Tage bei 2–8°C gelagert oder sofort eingesetzt
werden. Zwei-Liter-Erlenmeyerkolben oder andere geeignete Gefäße, die
Pneumococcus Inoculum Medium (YUF) mit Dextrose (25 g/l) enthalten,
werden mit Arbeitsimpfgut angeimpft und etwa 8–24 Stunden lang bei 37°C ± 2°C stationär inkubiert.
Der Inkubationszeitraum variiert je nach dem gezüchteten Streptococcus pneumoniae-Typ
wie angegeben. Der pH-Wert der Fermentation wird durch die periodische
Zugabe von 12%iger Natriumbicarbonatlösung so eingestellt, daß ein Ziel-pH-Bereich
von 6,0 bis 7,2 aufrechterhalten wird, bis eine optische Dichte
von 1,5 bis 4,0 erreicht ist. Die optische Dichte wird bei 660 Nanometer überprüft. Eine
Probe des Wachstums wird mikroskopisch untersucht und eine serologische
Agglutinationsreaktion durchgeführt,
um die Reinheit zu überprüfen. Das
Wachstum dieser Stufe wird in einen Impffermenter überführt, der
40 l Pneumococcus Fermenter Medium, zusammengesetzt aus destilliertem
Wasser, einer trockenen Charge der Komponenten für Pneumococcus-Impfmedium (YUF), Yeast
Extract Ultrafiltrate, UCON und Dextrose (etwa 25 g/l) enthält. Die
Kultur wird bei 37°C ± 2°C unter leichtem
Rühren
etwa 2–12
Stunden lang inkubiert. Der pH wird durch die periodische Zugabe
von Natriumhydroxidlösung
auf 6,0 bis 7,2 eingestellt. Ein Fermenter, der 525 l Pneumococcus
Fermenter Medium, zusammengesetzt aus destilliertem Wasser, einer
trockenen Charge der Komponenten für Pneumococcus Production Medium
(YUF), Yeast Extract Ultrafiltrate, UCON, und Dextrose (etwa 25
g/l) enthält,
wird mit etwa 50 l einer 2–12stündigen Impfkultur
angeimpft. Die Kultur wird bei 37°C ± 2°C unter leichtem
Rühren
6–30 Stunden
lang inkubiert, je nachdem Typ, der gezüchtet wird. Der pH-Wert wird
durch periodische Zugaben von Natriumhydroxidlösung auf 6,0 bis 7,2 eingestellt.
Der Fermentation folgt eine Bestimmung der optischen Dichte, und
die Fermentation wird beendet, wenn die Dextrose vollständig verbraucht
ist, wie durch keine weiteren pH-Veränderungen angezeigt.
-
Die
pathogenen Organismen werden unmittelbar nach Beendigung der Fermentation
abgetötet.
Dies wird erreicht, indem Phenol bis zu einer Konzentration von
etwa 1% zugegeben und die Fortsetzung der Abtötung für 2–12 Stunden bei Umgebungstemperatur
gestattet wird.
-
II) Isolierung von rohem Pn-Ps:
-
Denaturierter
Alkohol wird der abgetöteten
Kultur in einer ausreichenden Menge zugegeben, um Zelltrümmer und
Nukleinsäuren
auszufällen,
welche durch Zentrifugation entfernt werden. Das rohe Polysaccharid wird
dann aus der Überstandsflüssigkeit
durch Zugabe von weiterem denaturiertem Ethanol gefällt. Die
Feststoffe werden durch Zentrifugation gewonnen und die Überstandsflüssigkeit
verworfen.
-
Die
Nukleinsäureverunreinigung
wird verringert durch Solubilisierung des Polysaccharids in einer
neutralen wäßrigen Lösung wie
1–5%igem
Natriumacetat oder 0,05 M Phosphatpuffer, zu der Nuklease und etwa 0,01
M Magnesiumchlorid zugegeben wird. Nach etwa 60–120 Minuten bei etwa 36°C wird der
pH-Wert auf etwa 8,0 eingestellt und eine Protease wie Trypsin zugegeben,
um proteinhaltige Verunreinigungen zu verdauen.
-
Weitere
Verunreinigungen können
durch erneute Fällung
des Polysaccharids in Natriumacetat mit denaturiertem Alkohol oder
Isopropanol, gefolgt von erneuter Solubilisierung in destilliertem
Wasser, eliminiert werden. Die Zugabe von Cetrimoniumbromid bei
etwa 8°C
fällt Verunreinigungen
aus, welche durch Zentrifugation entfernt werden. Die Zugabe von
Natriumacetat und eines Aliquots an denaturiertem Alkohol oder Isopropanol
erlaubt die Entfernung weiterer Verunreinigungen. Das Polysaccharid
wird durch Zugabe von weiterem Alkohol und Zentrifugation gewonnen.
Der Niederschlag wird mit absolutem Ethanol gewaschen, bis ein weißes Pulver
erhalten wird. Das Polysaccharid wird durch Filtration gewonnen,
mit absolutem Ethanol und Aceton gewaschen und im Vakuum getrocknet,
um das rohe Pn-Ps als Pulver zu ergeben.
-
BEISPIEL 4
-
Herstellung von partiell hydrolysiertem,
gereinigtem Pn6B-Ps:
-
- (1) Thermische Hydrolyse: Eine 3,0-g-Portion
von rohem Pn6B-Ps-Pulverwurde in 1200 ml Salzlösung (0,9% NaCl) unter Rühren bei
Raumtemperatur für
etwa 4 Stunden solubilisiert und über Nacht bei 4°C gelagert.
Die Lösung
wurde dann in einer Kältefinger-Rückflußkühler-Apparatur
24 Stunden lang bei 100°C hydrolysiert
und auf Raumtemperatur abgekühlt.
Natriumacetat-Reagens (59,7 g) wurde bis zu einer Endkonzentration
von 3% (Gew./Vol.) zugegeben.
- (2) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungspilotstudie
und ein antikörpergesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das Pn6B-Ps
bei 40–50%
IPA ausfallen wurde.
- (3) Erste IPA-Zugabe: Die hydrolysierte Probe (Volumen – 1210 ml,
aus Schritt 1 oben) wurde durch die Zugabe von 932 ml IPA (tropfenweise
unter Rühren
bei Raumtemperaturzugegeben) auf 43,5% IPA gebracht. Die Probe wurde
15–30
Minuten lang rühren
gelassen und dann 30 Minuten lang bei 11.000 × g zentrifugiert (Beckman
JA-10-Rotor, 8.000 UpM; 20°C).
Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter gesammelt. Der Niederschlag
wurde direkt auf dem Trichter mit absolutem EtOH, dann Aceton gewaschen
und im Vakuum über CaCl2 bei Raumtemperatur in Vorbereitung auf
die Analyse getrocknet.
- (4) Zweite IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 43,5% IPA (Volumen 2020 ml, aus Schritt 3 oben] wurde durch
tropfenweise Zugabe von 93,5 ml IPA unter Rühren bei Raumtemperatur auf
46,0% IPA gebracht. Die Probe wurde stehengelassen und zentrifugiert
wie in Schritt 3 oben. Das Pellet wurde trituriert, gesammelt, gewaschen
und getrocknet wie in Schritt 3 oben. Das Pn6B-Ps-Produkt wog 1650
mg und besaß einen
Kd von 0,62 und einen Phosphorgehalt von
3,3%.
-
BEISPIEL 5
-
S. pneumoniae 6B-OMPC-Konjugat Pn6B-Ps-OMPC:
-
A. Herstellung von Dowex 50 × 2 Tetrabutylammoniumharz
[Dowex 50 (Bu4N+)]:
-
Dowex
50 × 2
(200–400
Mesh), H+-Form (72 g), wurde in Wasser aufgeschlämmt, auf
eine Säule
aufgetragen und nacheinander mit Wasser, 6 N HCl und dann Wasser
gewaschen, bis der Ausfluß beim
Test mit pH-Papier neutral war. Eine 10%ige wäßrige Lösung von Tetrabutylammoniumhydroxid
wurde dann durch die Säule
laufengelassen, bis der Ausfluß beim
Test stark alkalisch war. Schließlich wurde Wasser durch die
Säule laufengelassen,
bis der Ausfluß beim
Test wieder neutral war.
-
B. Pn6B(Bu4N+):
-
Pn68-Ps
(600 mg), größenreduziert
und fraktioniert (hinsichtlich der physikalischen Eigenschaften
siehe Tabelle I, Pn6B-Ps-Charge 1), wurde in sterilem destilliertem
Wasser (60 ml) gelöst
und die Lösung
magnetisch gerührt,
bis alle Feststoffe in Lösung
gingen (1,5 h). Die Polysaccharidlösung wurde auf das gespülte Harz
aufgetragen und ihr gestattet, durch Schwerkraft das Bett zu passieren
(4,5 h). Die Säule
wurde mit Wasser (10–12
ml) gewaschen und die vereinigten Ausflüsse lyophilisiert, was 640
mg trockenes Pn6B-Ps-Tetra-n-butylammoniumsalz, Pn6B(n-Bu4N+), ergab.
-
C. Pn6B-BuA2:
-
Pn6B(n-Bu4N+) (640 mg) wurde
in Dimethylsulfoxid (DMSO) (24 ml) gelöst und 30 Minutenlang magnetisch
gerührt,
nach welcher Zeit alle Feststoffe in Lösung zu sein schienen. Zu dieser
Mischung wurde 1,1'-Carbonyldiimidazol
(44,2 mg) zugegeben und die Reaktion bei Raumtemperatur (60 Min.)
gerührt.
In einem separaten Kolben wurde eine Lösung von Butandiamin-Dihydrochlorid
(BuA2·2HCl,
1,022 g) in Wasser (16 ml) durch die Zugabe von 10 N NaOH basisch
gemacht (pH 10,2). Die Lösung
wurde durch ein steriles 0,2-μm-Filter
filtriert und in einem Eisbad gekühlt. Die gealterte DMSO-Mischung, enthaltend
das aktivierte Polysaccharid, wurde zu der kalten BuA2·2HCl-Lösung in
einem langsamen, stetigen Strom zugegeben und die resultierende
Lösung
bei 0°C
gerührt
(15 Min.). Die Reaktionsmischung wurde auf Raumtemperatur aufwärmen gelassen
und 1 weitere Stunde gerührt,
wonach sie in einen Dialyseschlauch überführt und gegen folgendes dialysiert
wurde (4°C):
1] 15 l 0,1 M Natriumphosphatpuffer, pH 7,0, für 6 h; 2] 15 l 0,01 M Natriumphosphatpuffer,
pH 7,0, 12 h; 3] 15 l 0,01 M Natriumphosphatpuffer, pH 7,0, 9 h;
4] 15 l destilliertes H2O, 17,5 h. Der Inhalt
des Dialyseschlauchs wurde lyophilisiert, was 222 mg Pn6B-1,4-Butandiamin
(Pn6B-BuA2) ergab. Das NMR (300 MHz, D2O) von etwa 5 mg dieses Materials zeigte
durch Vergleich der Integrale der Resonanzen der Butandiamin- Methylene und der
Rhamnose-Methylprotonen von Pn6B-Ps eine Beladung von 22 Diaminresten
pro 100 sich wiederholender Pn6B-Ps-Monomereinheiten.
-
Pn6BuA2-BrAc:
-
Pn6B-BuA2 (210 mg) wurde in 0,1 M Kolthoff-Borat-Phosphat-Puffer,
pH 9,04, (21 ml) gelöst
und die Mischung 30 Minuten lang magnetisch gerührt, um die Lösung zu
bewirken. Zu dieser wäßrigen Lösung wurde eine
Mischung zugegeben, die aus p-Nitrophenylbromacetat (210 mg) in
Acetonitril (2,6 ml) bestand und die Reaktion über Nacht gerührt (20
h, 4°C).
Die Lösung
wurde in einen Dialyseschlauch überführt und
gegen folgendes dialysiert (4°C):
- 1] 15 l steriles destilliertes H2O,
12,3 h; 2] 15 l steriles destilliertes H2O,
8,25 h; 3] 15 l steriles destilliertes Wasser, 5,5 h. Von dem Inhalt
des Beutels wurden 1,7 ml für
Assays entnommen (NMR und HPSEC-Universalkalibrierung
oder Molekülgrößenanalyse)
und dann 0,449 g getrocknetes Salz von Phosphatpuffer, pH 8, (hergestellt
durch Lyophilisierung einer 0,1 M Natriumphosphatlösung von
pH 8), zugegeben. Nach vollständiger
Lösung
(30 Min.) wurde die Lösung
durch ein steriles 0,2-μm-Filter
filtriert, was eine Lösung von
Pn6B-BuA2-BrAe bei pH 8 ergab.
-
Pn6B-OMPC:
-
Steriler
OMPC (40 ml, 4,5 mg/ml) wurde durch Ultrazentrifugation (4°C, 43 K UpM,
2 h) in vier 10-ml-Zentrifugenrhrchen pelletiert. Jedes Pellet wurde
in 3 ml einer steril filtrierten (0,22 μm) Thiolierungsmischung resuspendiert,
welche aus dem folgenden bestand:
N-Acetylhomocysteinthiolacton-Hydrochlorid
(164 mg), Ethylendiamintetraessigsäure-Dinatriumsalz (255 mg) und
Dithiothreit (53 mg) in Na2B4O7-Puffer, pH 11,09 (30 ml). Die resuspendierten
Pellets wurden homogenisiert (Dounce), vereinigt, das Gefäß entgast
und mit Stickstoff als Schutzgas versehen und über Nacht (19 h) bei Raumtemperatur
stehengelassen. Die Lösung
wurde auf drei Ultrazentrifugenröhrchen
aufgeteilt, mit 1 M KH2PO4 überschichtet
und das Protein pelletiert (4°C,
43 K UpM, 2 h). Die Pellets wurden in 0,1 M Natriumphosphatpuffer,
pH 8, (30 ml) resuspendiert, homogenisiert (Dounce) und erneut pelletiert
(4°C, 43
K UpM, 2 h). Das sterile Proteinpellet wurde in der filtrierten
Pn6B-BuA2-BrAc-Lösung resuspendiert verwendet.
Ein Ellman's-Test
wurde sofort durchgeführt
und zeigte einen SH-Titer von 34 μMol.
Die Reaktionsmischung wurde entgast, mit Stickstoff als Schutzgas
versehen und 91 h lang bei Raumtemperatur stehengelassen.
-
Das
Protein wurde durch Zugabe von 1 ml einer (steriles 0,22 μm-Filter)
Lösung,
bestehend aus dem folgenden: N-Ethylmaleimid (75 mg) in 5 ml 0,1
M Natriumphosphatpuffer, pH 8,0, mit Endgruppen versehen. Diese
Mischung wurde 4 h lang bei Raumtemperatur stehengelassen, wonach
300 μl N-Acetylcysteamin (bei 0,22 μm steril
filtriert) zugegeben und die Lösung
weitere 19,5 h lang stehengelassen wurde.
-
Das
sterile, mit Endgruppen versehene Konjugat wurde auf vier Zentrifugenröhrchen aufgeteilt,
mit 0,1 M Natriumphosphatpuffer, pH 7, überschichtet und durch Ultrazentrifugation
(4°C, 43
K UpM, 2 h) pelletiert, dann in sterilem 0,1 M NaPO
4-Puffer,
pH 7, (42 ml) resuspendiert und homogenisiert (Dounce). Nach erneuter Zentrifugation
wie zuvor wurden die Pellets in einem Dounce-Homogenisator in einer
Gesamtmenge von 50 ml sterilem destilliertem Wasser resuspendiert
Nach Stehenlassen für
17 h bei 4°C
wurde die Konjugat-Präparation
3,5 Minuten lang bei 1000 UpM in einem TH-4-Rotor in einer TJ-6-Zentrifuge zentrifugiert
und eine kleine Menge Sediment entfernt Die endgültige Konjugatprodukt-Suspension wurde
auf Protein (Lowry), Pn6B-Polysaccharid (Phenol/Schwefelsäure), unkonjugiertes
Polysaccharid (Größenausschlußchromatographie-Geschwindigkeitsnephelometrie)
und Aminosäuren
(Aminosäureanalyse)
untersucht. Die Ergebnisse waren wie folgt:
| Pn6B-Polysaccharid | 0,33
mg/ml |
| Protein | 2,2
mg/ml |
| Pn6B-Ps/OMPC | 0,15 |
| freies
Pn6B-Ps | < 5 Flächen-% |
| S-Carboxymethylhomocystein/Lysin | 7,7% |
| S-Carboxymethylcysteamin/Lysin | 1,6% |
-
BEISPIEL 6
-
Herstellung von partiell hydrolysiertem,
gereinigtem Pn14-Ps:
-
- (1) Behandlung mit Anionenaustauscherharz:
Eine Portion von 2,81 g Pn14-Ps-Pulverwurde in 1124 ml destilliertem
H2O unter Rühren bei Raumtemperatur etwa
4 Stunden lang solubilisiert und dann bei 4°C über Nacht gelagert. Die Lösung wurde
zu 60 g DE52 (Whatman, Diethylaminoethylcellulose) zugegeben, die etwa
15 h lang in destilliertem H2O bei einem
pH von etwa 5–6
vorgequollen worden war. Die Aufschlämmung wurde auf einer Schüttelplattform
bei Raumtemperatur etwa 15 h lang sanft geschüttelt, wonach sie in einem
Beckman JA-10-Rotor 15 Minuten lang bei 5000 UpM und 20°C zentrifugiert
wurde. Die Überstandsflüssigkeit
wurde durch einen Sinterglastrichter (150 ml, mittlere Porosität) weiter
geklärt
und in einem 2-Liter-Seitenhalskolben gesammelt.
- (2) Beschallungshydrolyse: Das DE52-behandelte Pn14-Ps (Volumen – 1100 ml,
aus Schritt 1 oben) wurde in einem Kunststoffbecherglas auf einem
Eisbad mit einem Branson Sonifier (Halbzoll-Sonde, Einstellung 8)
2 Minuten lang beschallt. Die Probe wurde etwa 15 Minuten lang abkühlen gelassen,
während
die Viskosität
bestimmt wurde, und dann für
weitere einminütige
Intervalle beschallt. Eine Endviskosität von 1,096 Zentistokes wurde
nach der letzten Schallbehandlung erzielt. Die hydrolysierte Probe
wurde auf Raumtemperatur gebracht und Natriumacetat-Reagens (18,0
g) wurde bis zu einer Endkonzentration von 1% (Gew./Vol.) zugegeben.
- (3) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungspilotstudie
und ein antikörpergesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das Pn14-Ps
zwischen 35–45%
IPA ausfallen würde.
- (4) Erste IPA-Zugabe: Die hydrolysierte Probe [Volumen – 1090 ml,
aus Schritt 2 oben] wurde durch die Zugabe von 706 ml IPA (tropfenweise
unter Rühren
bei Raumtemperatur zugegeben) auf 39,3% IPA gebracht. Die Probe
wurde 15–30
Minuten lang rühren
gelassen und dann 30 Minuten lang bei 11.000 × g zentrifugiert (Beckman
JA-10-Rotor; 8.000 UpM; 20°C)
und die Überstandsflüssigkeit
dekantiert. Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter
gesammelt. Der Niederschlag wurde direkt auf dem Trichter mit absolutem
EtOH, dann Aceton gewaschen und im Vakuum über CaCl2 bei
Raumtemperatur getrocknet in Vorbereitung auf die Analyse.
- (5) Zweite IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 39,3% IPA [Volumen – 1712 ml,
aus Schritt 4 oben] wurde durch tropfenweise Zugabe von 73,5 ml
IPA unter Rühren
bei Raumtemperatur auf 41,8% IPA gebracht. Die Probe wurde stehengelassen
und zentrifugiert wie in Schritt 4 oben. Das Pellet wurde trituriert,
gesammelt, gewaschen und getrocknet wie in Schritt 4 oben. Das Pn14-Ps-Produkt wog
1399 mg.
- (6) Dialyse und Lyophilisierung: Eine Portion (1385,6 mg) der
Probe aus Schritt 5 oben wurde in 554 ml destilliertem H2O bei Raumtemperatur 2–3 Stunden lang solubilisiert.
Die Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch (MW-Ausschluß 12.000;
45 mm) überführt und
unter 2 weiteren Austauschen von destilliertem H2O
27 Stunden lang gegen destilliertes H2O
dialysiert. Dann wurde die dialysierte Probe in Lyophilisierungskolben überführt, in
einem Trockeneis:Methanol-Bad rollschichtgefroren und mit einem
Virtis(Freezemobile)-Lyophilisator 2–1/2 Tage bis zur Trockne lyophilisiert.
Die Ausbeute an Pn14-Ps-Endprodukt,
welches einen Kd von 0,56 besaß, betrug
1326,8 mg.
-
Für Fachleute
sollte aus dieser Offenbarung offensichtlich sein, daß andere
neutrale Pn-Ps-Subtypen wie Pn7F-Ps nach dem hier offenbarten Verfahren
hergestellt werden und wie Pn14-Ps, das ebenfalls ein neutrales
Polysaccharid ist, konjugiert werden könnten.
-
BEISPIEL 7
-
Konjugation des Komplexes des Proteins
der äußeren Membran
mit Pneumokokken-14-Polysaccharid, Pn14-Ps-OMPC:
-
a. Herstellung des 1,4-Butandiamin-Derivats
von Pn14-Ps (Pn14-BuA2):
-
Eine
410-mg-Portion von Pn14-Ps nach einer 3-ständigen Lagerung im Vakuum über P205
wurde mit 26 ml Dimethylsulfoxid (DMSO) bedeckt und zur Lösung 0,75
h lang gerührt
Dazu wurden 62 mg Carbonyldiimidazol zugegeben und die resultierende
Lösung
80 Minuten lang bei Raumtemperatur (RT) gerührt.
-
Eine
Lösung,
enthaltend 1,067 g 1,4-Butandiamin-Dihydrochlorid (BuA2·2HCl)
in 38,5 ml H2O wurde hergestellt und deren
pH mit 2,5 N NaOH auf 10,20 eingestellt. Diese Lösung wurde durch ein 0,2-μm-GV-Filter von
Millex filtriert und in einem Eisbad gekühlt.
-
Die
gealterte DMSO-Lösung
wurde zu der kalten BuA2-Lösung zugegeben
und weitere 10 Minuten lang in dem Eisbad gerührt. Sie wurde dann 50 Minuten
lang bei RT stehengelassen, wonach die Lösung in zwei 12-Inch-Längen von
Spectrapor-2-Dialyseschlauch eingebracht wurde, 1 cm vom oberen
Rand der Flüssigkeit
abgeklemmt und dialysiert wurde gegen: 1) 15 l 0,1 M Natriumphosphatpuffer,
pH 7,0, für
16,5 h; 2) 15 l 0,1 M Natriumphosphatpuffer, pH 7,0, für 8 h; 3)
15 l 0,1 M Natriumphosphatpuffer, pH 7,0, für 8 h; 4) 15 l H2O für 17,5 h.
Sie wurde dann lyophilisiert, um 210 mg des 1,4-Butandiamin-Derivats
von Pn14-Ps(Pn14-BuA2) zu ergeben.
-
Ein
NMR-Spektrum einer Probe von etwa 5 mg zeigte eine ”Beladung” von etwa
31 Butandiaminresten pro 100 sich wiederholender Polysaccharideinheiten,
festgestellt durch Vergleich der Integrale der Butandiamin-Methylene
und der N-Acetylmethyl(von Pn14-Ps)-Resonanzen.
-
b. Herstellung des bromacetylierten Butandiamin-Derivats
von Pn14-Ps(Pn14-BuA2-BrAc):
-
Pn14-BuA2 (210 mg) wurde mit 36 ml eines 0,1 M Borat-Phosphat-Puffers,
pH 9,0, bedeckt und 2,5 h lang gerührt, um die Lösung zu
bewirken. Dann wurden 195 mg p-Nitrophenylbromacetat, gelöst in 4
ml Acetonitril, zugegeben. Die resultierende Mischung wurde 21 h
bei 4°C
gerührt
Sie wurde dann in einem Spectrapor-2-Schlauch dialysiertgegen: 1)
15 l destilliertes H2O für 6 h, 2) 15 l destilliertes
H2O für
14,5 h und 3) 15 l destilliertes H2O für 6 h. Von
dem dialysierten Inhalt des Beutels wurden 2,0 ml für Assays
entnommen und dann 492 mg getrocknetes Salz von Phosphatpuffer,
pH 8,0, (hergestellt durch Lyophilisierung einer 0,1 M Natriumphosphatlösung, pH
8,0) zugegeben. Die Lösung
wurde durch zwei 0,2-μm-Corning-Filter
filtriert, was in einer wäßrigen Lösung, pH
8,0, von Pn14-BuA2-BrAc (43 ml) resultierte.
-
c. Konjugation von OMPC mit Pn14-BuA2-BrAc-Ps:
-
50
ml OPMC (Konzentration 3,2 mg/ml) wurden in fünf 10-ml-Zentrifugenröhrchen eingebracht
und in einem Beckman 80-Ti-Rotor mit 43.000 UpM (43 K) 2 h lang
bei 4°C
zentrifugiert. Eine Thiolierungsmischung wurde hergestellt durch
Lösen von
350 mg EDTA (Ethylendiamintetraessigsäure-Dinatriumsalz) und 64 mg Dithiothreit
(DTT) in 30 ml Na264O7-Puffer, pH 11,0. 346 mg N-Acetylhomocysteinthiolacton
wurden zugegeben und die Lösung
durch ein 0,2-μm-Corning-Filter (Bechertyp)
filtriert.
-
Die
Pellets aus der obigen Zentrifugation wurden jeweils mit 3 ml der
filtrierten Thiolierungsmischung (insgesamt 15 ml) disloziert, in
einen Dounce-Homogenisator überführt und
resuspendiert. Die Röhrchen
wurden durch reihenweises Umfüllen
von weiteren 5 ml der Thiolierungslösung gespült Der Spülprozeß wurde mit weiteren 5 ml Thiolierungslösung wiederholt.
Die kombinierten Spüllösungen wurden
im Dounce homogenisiert und das gesamte resuspendierte Material
(25 ml) in einen 100-ml-Rundkolben überführt.
-
Nach
Verschließen
mit einem Septum und Ersetzen der Luft durch N2 unter
Verwendung eines Firestone-Ventils wurde die Reaktionsmischung 21
h lang stehengelassen. Die 25-ml-Reaktionsmischung
wurde dann auf drei Zentrifugenröhrchen
aufgeteilt, wovon jedes mit 1 M KH2PO4 (wäßrig) aufgefüllt und
dann 2 h lang bei 43 K UpM und 4°C
zentrifugiert wurde. Die Überstandsflüssigkeiten
wurden entfernt und die Pellets in 0,1 M Natriumphosphatpuffer,
pH 8,0, resuspendiert (eine Gesamtmenge von 30 ml war das endgültige Resuspensionsvolumen).
-
Anschließend wurde
eine zweite Ultrazentrifugation (2 h, 4°C, 43 K UpM) durchgeführt. Nach
Entfernung der Überstandsflüssigkeit
wurden die Pellets mit Hilfe des Dounce-Verfahrens in der oben hergestellten, filtrierten
Pn14-BiA2-BrAc-Lösung resuspendiert. Ein Ellman-Assay
zu diesem Zeitpunkt zeigte eine Gesamtmenge von etwa 23 μMol Thiol
an.
-
Es
sollte beachtet werden, daß die
Filtration der Pn14-BuA2-BrAc-Lösung gerade
vor der Resuspension des thiolierten Proteins geschieht. Die resultierende
Reaktion (d. h. Pn14-BuA2-BrAc mit thioliertem OMPC)
wurde unter Stickstoff (unter Entgasen) in einer N2-Box
bei RT 114 h lang stehengelassen.
-
Die
Reaktion wurde dann mit Endgruppen versehen (d. h., die reaktiven
Gruppierungen auf dem Pn14-Ps und OMPC werden deaktiviert) wie folgt:
Eine Lösung,
enthaltend 75 mg N-Ethylmaleimid (NEM) in 5 ml 0,1 M Natriumphosphatpuffer,
pH 8,0, wurde zugegeben und die Mischung weitere 22,5 h lang stehengelassen.
-
Die
mit Endgruppen versehene Reaktionsmischung (35 ml) wurde auf 4 Zentrifugenröhrchen aufgeteilt und
zentrifugiert (43 K, 2 h, 4°C).
Die Pellets wurden in 40 ml TED-Puffer (0,1 M Tris, 0,01 M EDTA,
0,5% 000, pH 8,5) resuspendiert und 19 h lang bei Raumtemperatur
stehengelassen. Die Lösung
wurde dann zentrifugiert (43 K, 2 h, 4°C). Die Pellets wurden in 40
ml 0,1 M Natriumphosphatpuffer, pH 8, resuspendiert und dann erneut
zentrifugiert (43 K, 2 h, 4°C).
Diese Pellets wurden in 44 ml destilliertem H
2O
resuspendiert und 17 h lang bei 4°C
stehengelassen. Eine Zentrifugation bei niedriger Geschwindigkeit
(1000 UpM, 3,5 Min.) ergab ein kleines Pellet, welches verworfen
wurde. Die Überstandsflüssigkeit
wurde entnommen, was 43 ml rohes Konjugat mit den folgenden analytischen
Eigenschaften ergab:
| Test | Ergebnisse |
| a.
Ps-Gehalt | 387 μg/ml |
| b.
Protein | 1300 μg/ml |
| Ps/Protein-Verhältnis (berechnet) | 0,30 |
| c.
Freies Ps | < 5 Flächen-% |
| d.
Aminosäureanalyse | |
| SCMHC/Lysin | 9,8% |
| SCMC/Lysin | 3,5% |
-
BEISPIEL 8
-
Herstellung des Pn23F-Ps-Zwischenprodukts:
-
- (1) Beschallungshydrolyse: Eine 3,0-g-Portion
von Pn23F-Ps-Pulver wurde in 1200 ml Salzlösung (0,9% NaCl) unter Rühren bei
Raumtemperatur für
etwa 4 h solubilisiert. Die Lösung
wurde dann in einem Kunststoffbecherglas in einem Eisbad mit einem
Branson Sonifier (Halbzoll-Sonde, Einstellung 8) für Intervalle von
3 Minuten bis zu insgesamt 15 Minuten beschallt. Die Viskosität wurde
nach jedem Intervall überprüft. Nach
15 Minuten wurde eine weitere Beschallung von 5 Minuten durchgeführt, um
eine Endviskosität
von 1,206 Zentistokes zu erhalten. Die hydrolysierte Probe wurde
auf Raumtemperatur gebracht und Natriumacetat-Reagens (58,4 g) bis
zu einer Endkonzentration von 3% (Gew./Vol.) zugegeben.
- (2) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungspilotstudie
und ein antikörpergesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das Pn23F-Ps
zwischen 35–45%
IPA ausfallen würde.
- (3) Erste IPA-Zugabe: Die hydrolysierte Probe [Volumen = 1165
ml, aus Schritt 1 oben] wurde durch die Zugabe von 810 ml IPA (tropfenweise
unter Rühren
bei Raumtemperaturzugegeben) auf 41,0% IPA gebracht. Die Probe wurde
15–30
Minuten lang röhrengelassen
und dann 30 Minuten lang bei 11.000 × g zentrifugiert (Beckman
JA-10-Rotor, 8.000 UpM; 20°C).
Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter gesammelt. Der Niederschlag wurde
direkt auf dem Trichter mit absolutem EtOH, dann Aceton gewaschen
und im Vakuum über
CaCl2 bei Raumtemperatur in Vorbereitung
auf die Analyse getrocknet.
- (4) Zweite IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 41,0% IPA [Volumen = 1925 ml, aus Schritt 3 oben] wurde durch
die tropfenweise Zugabe von 85,0 ml IPA unter Rühren bei Raumtemperatur auf
43,5% IPA gebracht. Die Probe wurde stehengelassen und zentrifugiert
wie in Schritt 3 oben. Das Pellet wurde trituriert, gesammelt, gewaschen
und getrocknet wie in Schritt 3 oben. Das Pn23F-Ps-Produkt wog 1795
mg.
- (5) Dialyse und Lyophilisierung: Eine Portion (1779 mg) der
Pn-Ps-Probe aus Schritt 4 oben wurde in 712 ml destilliertem H2O bei Raumtemperatur 3–4 h lang solubilisiert. Die
Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch überführt (MG-Ausschluß 12.000;
45 mm) und unter zwei weiteren Austauschen von destilliertem H2O 27 h lang bei 4°C gegen destilliertes Wasser
dialysiert. Dann wurde die Probe in Lyophilisierungskolben überführt, in
einem Trockeneis:Methanol-Bad rollschichtgefroren und mit einem
Virtis(Freezemobile)-Lyophilisator 2–3 Tage lang lyophilisiert.
Die Ausbeute des Ps-Endprodukts betrug 1703 mg. Das Endprodukt besaß einen
Kd = 0,60.
-
BEISPIEL 9
-
Konjugation des Komplexes des Proteins
der äußeren Membran
mit dem Pn23F-Ps-Zwischenprodukt:
-
a. Herstellung von Dowex 50 × 2 (200–400 Mesh)
Tetrabutylammoniumform-Harz [Dowex 50 (Bu4N+)]
-
Dowex
50 × 2
(200–400
Mesh), H+-Form (72 g), wurde in H2O aufgeschlämmt (das bei diesen Verfahren
verwendete Wasser war pyrogenfreies, steriles, destilliertes Wasser),
auf eine Säule
aufgetragen und nacheinander gewaschen mit 1] 800 ml H2O;
2] 400 ml 6 N HCl; 3] 300 ml H2O, bis der
Ausfluß für pH-Papier neutral ist;
4] 250 g einer 10%igen wäßrigen Tetrabutylammoniumhydroxidlösung, bis
der Ausfluß für pH-Papier
stark alkalisch ist; 5] 750 ml H2O.
-
b. Herstellung von Pn23F-Ps Tetrabutylammoniumform
[Pn23F(Bu4N+)]:
-
Eine
34-ml-Säule
von Dowex 50 × 2
(Bu4N+) wurde mit
70 ml H2O gewaschen. Eine 450-mg-Portion von
größensortiertem
Pn23F-Ps wurde mit 50 ml H2O bedeckt und
0,5 h lang gerührt.
Diese Lösung
wurde auf die Säule
aufgetragen und durch Schwerkraft perkolieren gelassen (ca. 2 h).
Zu diesem Zeitpunkt wurde an das untere Ende der Säule Vakuum
angelegt und die Elution (unter Vakuum) für eine weitere Stunde fortgesetzt.
Die Säule
wurde mit 25 ml H2O gewaschen und die vereinigten
Ausflüsse
lyophilisiert, was 0,5 g des Pn23F(Bu4N+)-Salzes ergab. Dieses wurde in einem Vakuumexsikkator über P2O5 für etwa 17
h gelagert.
-
c. Herstellung des 1,4-Butandiamin-Derivats
von Pn23F-Ps (Pn23F-BuA2):
-
Die
0,5 g des Pn23F (Bu4N+)
von Schritt b oben wurden mit 25 ml Dimethylsulfoxid (DMSO) bedeckt und
zur Lösung
15 Minuten lang gerührt.
Dazu wurden 22 mg Carbonyldiimidazol (CDI) zugegeben und die resultierende
Lösung
bei Raumtemperatur (RT) 0,5 h lang gerührt.
-
Eine
Lösung,
enthaltend 507 mg 1,4-Butandiamin-Dihydrochlorid (BuA2·2HCl)
in 32 ml H2O, wurde hergestellt und deren
pH mit 2,5 N NaOH auf 10,23 eingestellt. Diese Lösung wurde durch ein 0,2-μm-GV-Filter von Millex
filtriert und in einem Eisbad gekühlt.
-
Die
gealterte DMSO-Lösung
wurde zu der kalten BuA2-Lösung zugegeben
und eine weitere Stunde in dem Eisbad gerührt. Sie wurde dann 1 h lang
bei RT stehengelassen, wonach die Lösung in 2 × 12 Inch eines Spectrapor-Dialyseschlauchs
eingebracht wurde, 1 cm vom oberen Rand der Flüssigkeit abgeklemmt und dialysiert
wurde gegen: 1) 15 l 0,1 M Natriumphosphatpuffer, pH 7,0, für 16 h;
2) 15 l 0,01 M Natriumphosphatpuffer, pH 7,0, für 10,5 h; 3) 15 l 0,01 M Natriumphosphatpuffer,
pH 7,0, für
12,5 h; 4) 15 l H2O für 10,5 h. Sie wurde dann lyophi-lisiert,
um 220 mg des 1,4-Butandiamin-Derivats von Pn23F-Ps (Pn23F-BuA2) zu ergeben.
-
Ein
NMR-Spektrum von etwa 6,9 mg zeigte eine ”Beladung” von etwa 23,5 Butandiaminresten
pro 100 sich wiederholender Polysaccharitdeinheiten, festgestellt
durch Vergleich der Integrale der Butandiamin-Methylene und der Rhamnosemethyl(von
Pn23F)-Resonanzen.
-
d. Herstellung des bromacetylierten Butandiamin-Derivats
von Pn23F-Ps (Pn23F-BuA-BrAc):
-
Pn23F-BuA2 (214 mg) wurde mit 23 ml eines 0,1 M Borat-Phosphat-Puffers,
pH 9,0, bedeckt und 30 Minuten lang gerührt, um die Lösung zu
bewirken. Dann wurden 230 mg p-Nitrophenylbromacetat in 6 ml Acetonitril
zugegeben. Die resultierende Mischung wurde 23 h lang bei 4°C gerührt. Sie
wurde dann in einem Spectrapor 2-Schlauch dialysiert gegen: 1) 15
l H2O für
8 h, 2) 15 l H2O für 12 h und 3) 15 l H2O für
6 h. Von dem dialysierten Inhalt des Beutels wurden 1,5 ml für Assays
entnommen und dann 490 mg getrocknetes Salz von Phosphatpuffer,
pH 8,0, (hergestellt durch Lyophilisierung einer 0,1 M Natriumphosphatlösung, pH
8,0) zugegeben. Das Lösen
erfordert ungefähr
15 Minuten, nach welcher Zeit durch ein 0,2-μm-Corning-Filter filtriert wird,
was eine wäßrige Lösung, pH
8,0, von Pn23F-BuA2-BrAc ergibt.
-
e. Konjugation von OMPC mit Pn23F-BuA2-BrAc-Ps:
-
60
ml OMPC (3,1 mg/ml) wurden in 6 10-ml-Zentrifugenrährchen eingebracht
und in einem Beckman 80-Ti-Rotor
bei 43.000 UpM (43 K) 2 h lang bei 4°C zentrifugiert. Eine Thiolierungsmischung
wurde hergestellt durch Lösen
von 260 mg EDTA (Ethylendiamintetraessigsäure-Dinatriumsalz) und 52 mg
Dithiothreit (DTT) in 30 ml Na2B4O7. Thiolacton wurde
zugegeben und die Lösung
durch ein 0,2-μm-Corning-Filter (Bechertyp)
filtriert.
-
Die
Pellets aus der obigen Zentrifugation wurden jeweils mit 3 ml der
filtrierten Thiolierungsmischung (insgesamt 20 ml) disloziert und
in einen Dounce-Homogenisator überführt und
resuspendiert Die Röhrchen wurden
durch reihenweises Umfüllen
von weiteren 6 ml der Thiolisierungslösung gespült. Das Spülverfahren wurde mit weiteren
4 ml Thiolisierungslösung
wiederholt. Die vereinigten Spüllösungen wurden
in dem Dounce homogenisiert und das gesamte resuspendierte Material
(28 ml) in einen 100-ml-Rundkolben überführt. Nach Verschließen mit
einem Septum und Ersetzen der Luft durch N2 unter
Verwendung eines Firestone-Ventils wurde die Reaktionsmischung 19
h lang stehengelassen. Die 28-ml-Reaktionsmischung wurde dann auf
drei Zentrifugenröhrchen
aufgeteilt, von denen jedes mit 1 M Kaliumphosphat (wäßrig) aufgefüllt und
dann 2 h lang bei 43 K UpM und 4°C
in einem Beckmann 80 Ti-Rotor
zentrifugiert wurde. Die Überstandsflüssigkeiten
wurden entfernt und die Pellets in 0,1 M Natriumphosphatpuffer,
pH 8,0, resuspendiert (eine Gesamtmenge von 30 ml war das endgültige Resuspensionsvolumen).
-
Anschließend wurde
eine zweite Ultrazentrifugation (2 h, 4°C, 43 K UpM) durchgeführt Nach
Entfernung der Überstandsflüssigkeit
wurden die Pellets nach dem Dounce-Verfahren in der filtrierten Pn23F-BuA2-BrAc-Lösung resuspendiert,
die in Abschnitt 7.I.C.3d hergestellt worden war. Ein Ellman-Assay zu
diesem Zeitpunkt zeigte eine Gesamtmenge von etwa 28 μMol Thiol
in der resultierenden Lösung
an.
-
Es
sollte beachtet werden, daß die
Filtration der Pn23F-BuA2-BrAc-Lösung gerade
vor der Resuspension des thiolierten Proteins geschieht. Die resultierende
Reaktion (d. h. Pn23F-BuA2-BrAc mit thioliertem OMPC)
wurde unter N2 (unter Entgasen) in einer
N2-Box bei RT 117 h lang stehengelassen.
-
Die
Reaktion wurde dann mit Endgruppen versehen (d. h. die reaktiven
Gruppierungen auf dem Pn23F-Ps
und OMPC werden deaktiviert) wie folgt Eine Lösung, enthaltend 75 mg N-Ethylmaleimid
(NEM) in 5 ml 0,1 M Natriumphosphatpuffer, pH 8,0, wurde durch ein
0,22-μm-Filter
filtriert, der Reaktion zugegeben und 18 h lang stehengelassen.
-
Das
Gesamtvolumen der mit Endgruppen versehenen Konjugationsmischung
betrug 38,5 ml und 1,5 ml 0,1 M Natriumphosphatpuffer, pH 8,0, wurden
zugegeben, um das Gesamtvolumen auf 40 ml zu bringen. Mit 35 ml
dieser Lösung
wurden zu gleichen Teilen vier 10-ml-Zentrifugenröhrchen beschickt
und jedes davon mit 0,1 M Natriumphosphatpuffer, pH 8,0, aufgefüllt Diese
wurden bei 43 K UpM, 2 h, 4°C,
zentrifugiert. Die Überstandsflüssigkeiten
wurden entfernt und jedes der Pellets mit 8 ml TED-Puffer (0,1 M Tris,
pH 8,5, 0,01 M EDTA, 0,5% Na-Desoxycholat) disloziert und in einen
Dounce-Homogenisator überführt. Die
Zentrifugenröhrchen
wurden der Reihe nach mit weiteren 8 ml TED-Puffer gespült und die
Pellets resuspendiert (insgesamt 40 ml) und bei Raumtemperatur 20
h lang stehengelassen. Das gealterte Material wurde zentrifugiert
(wie oben beschrieben) in vier 10-ml-Röhrchen
bei 43 K, 2 h, 4°C.
Jedes der Pellets wurde mit 8 ml TED-Puffer disloziert, die Röhrchen der
Reihe nach mit 8 ml TED-Puffer gespült, resuspendiert und zentrifugiert
wie oben beschrieben. Diese Pellets wurden dann in insgesamt 40
ml 0,1 M Natriumphosphatpuffer, pH 7, resuspendiert und wie oben
beschrieben erneut zentrifugiert. Die Pellets wurden in insgesamt
44 ml Wasser resuspendiert und 17 h lang bei 4°C stehengelassen. Eine kleine
Menge unlöslicher
Stoffe wurde durch eine Zentrifugation bei geringer Geschwindigkeit
(1000 UpM, 3,5 Min.) entfernt, was das Produkt in der Überstandsflüssigkeit
ergab.
-
Die
resultierende Überstandsflüssigkeit
ist die Wirkstoffsubstanz, rohes Konjugat-Vakzin. Das Konjugat besaß die folgenden
analytischen Eigenschaften:
| Test | Ergebnisse |
| a.
Ps-Gehalt | 284 μg/ml |
| b.
Protein | 2025 μg/ml |
| Ps/Protein-Verhältnis (berechn.) | 0,14 |
| c.
Freies PS | < 5 Flächen-% |
| d.
Aminosäureanalyse | |
| SCMHC/Lysin | 6,7% |
| SCMC/Lysin | 1,6% |
-
BEISPIEL 10
-
Herstellung von Pn-Ps durch Gaulin-Homogenisierung:
-
Rohes
Pneumokokken-Pulver wurde in einer Konzentration von 1,5 mg/ml in
Wasser durch Mischen über
Nacht bei 4°C
solubilisiert. Es wurde auch eine stärker konzentrierte Lösung von
Pn-Ps mit 10 mg/ml hergestellt Die Zugabe von 50 mM CaCl
2 war erfolgreich bei der Verringerung der
Viskosität
der 10 mg/ml-Lösung auf
die Viskosität
der 1,5 mg/ml-Lösung.
Das solubilisierte Pn-Ps wurde dann durch einen Gaulin-Homogenisator
geleitet, der auf eine von vier Druckeinstellungen eingestellt war:
13,8, 34,5, 69,0 oder 103 MPa (2000, 5000, 10000 oder 15000 psi).
Das gescherte Pn-Ps wurde dann durch Zugabe von 60%igem Isopropanol,
der mit einer 2 M Stammlösung
auf 50 mM CaCl
2 gebracht worden war, gewonnen.
Das Pellet wurde mit 100%igem Ethanol in einem Omnimixer gewaschen
und filtriert, um das gefällte
Pn-Ps zu gewinnen. Das Pn-Ps wird auf dem Filter mit Aceton gewaschen
und dann über
CaSO
4 (Drierit) getrocknet und bis zur Analyse bei –70°C gelagert
Aliquots des gescherten Pn-Ps werden mit etwa 1 mg/ml resuspendiert
und hinsichtlich der Molekulargröße und Polydispersität durch
HPSEC-Universalkalibrierung und hinsichtlich des Antigenizitätsindexes
durch Geschwindigkeitsnephelometrie analysiert.
| Pn-Ps-Subtyp | MG,
bei dem die Antigenizität
abzunehmen beginnt | Polydispersität |
| 68 | 500.000 | 1,19 |
| 14 | 300.000 | 1,15 |
| 19
F | 250.000 | 1,09 |
| 23
F | 250.000 | 1,15 |
-
BEISPIEL 11
-
Maus-T-Zell-Stimulierung:
-
Dieser
Test wird durchgeführt,
um die T-Zell-Abhängigkeit/Immunogenizität von Pn-Ps-Konjugat-Vakzinen in Mäusen festzustellen.
Dieses Modell wurde verwendet, da Kinder im Alter von weniger als
zwei Jahren normalerweise gut auf T-abhängige Antigene ansprechen.
Athymische Mäuse
besitzen ein abnormes Thymusepithel und deshalb ist ihre Antwort
auf T-abhängige
Antigene signifikant geringer als bei ihren normalen kongenen Wurfgenossen.
-
Eine
Einzelverdünnung
von Vakzin, um eine Dosierung von 0,5 μg Polysaccharid zu ergeben,
wurde intraperitoneal erwachsenen athymischen Mäusen (nu/nu) und deren kongenen
Kontroll-Wurfgenossen (nu/+) am Tag 0,7 und 28 injiziert. Den Mäusen wurde
eine Woche später
Blut entnommen und deren individuelle Seren auf Antikörperantwort
mittels Radioimmunoassay (RIA) getestet.
-
Bei
dem RIA wurde jedes Mausserum mit C14-markiertem
Pn-Ps kombiniert Ein etwaiger gebildeter Antigen-Antikörper-Komplex
wurde dann durch Zugabe von gesättigtem
Ammoniumsulfat gefällt
Jede behandelte Probe wurde in einem Betazähler 1 Minute lang gezählt. Die
Pn6B-Ps-OqMPC-, Pn14-Ps-OMPC-, Pn19F-Ps-OMPC-,
Pn23F-Ps-OMPC-, Pn18C-Ps- und Pn4-Ps-Konjugate dieser Erfindung
wurden auf diese Weise getestet und befunden, bei den Nu/+-Mäusen eine
gute T-Zell-Stimulierung hervorzurufen.
-
BEISPIEL 12
-
Immunogenizität von Pn-Ps-Konjugaten bei
Rhesusaffenbabys:
-
Dieser
Test wird durchgeführt,
um die Immunogenizität
von entweder rohem Konjugat oder gefüllten Behältern von Pn-Ps-OMPC- oder
Pn-Ps-MIEP-Konjugat-Vakzin bei Affenbabys festzustellen. Das Affenbaby-Modell
hat sich als ausgezeichneter klinischer Indikator für das PedvaxHIBTM-Konjugat-Vakzin erwiesen [Vella et al.,
Pediatrics, 5. April, Erg., S. 668–675 (1990)] und wurde deshalb
als Modell für
die Pn-Ps-Konjugat-Vakzin-Beurteilung ausgewählt.
-
Eine
Dosis Vakzin wird intramuskulär
(0,25 ml in jede von zwei Stellen) zwei bis drei Monate alten Affenbabys
am Tag 0 und 28 injiziert. Den Affen wird am Tag 0, 28 und 42 Blut
entnommen und die individuellen Seren auf Antikörperantwort mittels Radioimmunoassay
(RIA) getestet.
-
Bei
dem RIA wird jedes Affenserum mit C14-markiertem
Pn-Ps kombiniert. Etwaiger gebildeter Antigen-Antikörper-Komplex wird dann durch
die Zugabe von gesättigtem
Ammoniumsulfat gefällt.
Jede behandelte Probe wird 1 Minute lang in einem Betazähler gezählt. Die
immunogene Antwort auf das Vakzin ist zufriedenstellend, wenn mindestens
50% der Versuchstiere eine Antikörperantwort
von mindestens 1 μg
nach Erhalt von zwei Dosen Vakzin aufweisen.
-
Pn6B-Ps-OMPC,
Pn23F-Ps-OMPC, Pn19F-Ps-OMPC, Pn18C-Ps-OMPC, Pn4-Ps-OMPC und Pn14-Ps-OMPC
zeigten, daß sie
starke anti-typenspezifische Antikörperantworten hervorrufen.
Ferner zeigte eine tetravalente Zusammensetzung, umfassend Pn6B-Ps-OMPC,
Pn23F-Ps-OMPC, Pn19F-Ps-OMPC und Pn14-Ps-OMPC, gute Anti-Pn-Ps-Antikörperantworten
auf alle vier Serotypen.
-
BEISPIEL 13
-
Schutzwirkung von Pneumokokken-Konjugaten
bei Chinchillas:
-
Jedem
Chinchilla wurde subkutan oder intramuskulär 0, 0,25, 1,0 oder 4,0 μg Pn6B-Ps-OMPC,
adsorbiert an Al(OH)3, injiziert. Den Chinchillas
wurde nach 0, 2, 4, 6 und 8 Wochen Blut entnommen.
-
Die
Tiere wurden mit Streptococcus pneumoniae 6B 8 Wochen nach der Injektion
herausgefordert und alle 1–3
Tage durch Otoskopie und Tympanometrie überwacht. Flüssigkeitsansammlungen
im Mittelohr wurden für
die Kultur abgesaugt und die Tiere zwei Wochen nach der Herausforderung
getötet
Die getöteten
Tiere wurden hinsichtlich einer Mittelohr-Histopathologie analysiert.
Es gab eine 60%ige Sterblichkeit bei Tieren, die kein Konjugat erhielten,
während
sogar die niedrigste Dosis zu 0% Sterblichkeit führte. Es gab keinen Schutz gegen
eitrige Mittelohrentzündung
bei den Tieren, welche kein Konjugat erhielten, während diejenigen,
welche Konjugat enthielten, auf Niveaus zwischen 60 und 100% über alle
Dosierungsbereiche hinweg geschützt
waren.
-
BEISPIEL 14
-
Anti-Pneumokokken-Immunantworten bei 2–5 Jahre
alten Kindern:
-
2–5 Jahre
alte Kinder, die zwei Dosen von jeweils 0,5 oder 5 μg Pn6B-Ps
enthielten, wurden auf die Produktion von Anti-Pn6B-Ps-Antikörpern durch
RIA und ELISA untersucht. Es wurden signifikante Erhöhungen der
Anti-Pn6B-Ps-Antikörper
beobachtet.
-
BEISPIEL 15
-
Geschwindigkeitsnephelometrie von Pneumokokken-Polysacchariden:
-
Der
Zweck dieses Assays ist die Bestimmung des Polysaccharidgehalts
und Antigenizitätsindexes
von freiem Pn-Ps und Konjugat-Präparationen
unter Anwendung von Geschwindigkeitsnephelometrie.
-
Der
Bereich der Standardkurve für
die Geschwindigkeitsnephelometrie differiert für die verschiedenen Pn-Ps wie
die Antwort pro Pn-Ps-Masseeinheit und der lineare Abschnitt des
Antwort-gegen-Pn-Ps-Antigenkonzentration-Diagramms
differiert für
jedes Pn-Ps. Das hier angegebene Verfahrensbeispiel ist spezifisch
für Pn6B-Konjugat
und trifft nicht notwendigerweise auf Konjugate anderer Pn-Ps-Typen
zu. Ferner werden alaun-adsorbierte Proben und deren jeweilige Standards
in 3% Natriumcitrat statt 0,9% NaCl verdünnt, wie im folgenden beschrieben.
Wäßrige Konjugat-Proben
und -Standards (d. h. diejenigen, welche nicht auf Alaun vorliegen)
werden in 0,9% NaCl verdünnt
und wiederum auf Nominalkonzentrationen verdünnt, von denen angenommen wird,
daß sie
innerhalb der Grenzen der Standardkurve liegen.
-
a) Reagenzien
-
- Salzlösung:
0,9% wäßriges NaCl
- Anti-Pn-Ps-Seren: Antiseren (Health Research, Inc., Albany,
NY) werden 30fach mit Salzlösung
verdünnt
- Standards: Herstellung von Standards von 1,0, 1,5, 2,0, 2,5,
3,0 und 4,0 μg/ml
Pn-Ps-Konjugat aus einer Stammlösung
von 387 μg/ml,
deren Konzentration durch den Phenol-Schwefelsäure-Assay für Polysaccharid bestimmt wurde.
- Testproben: Vorbereitung in Natriumcitrat-Stammlösung, um
eine Endkonzentration von 3% Natriumcitrat zu haben, und Reihenverdünnungen
der Testproben auf theoretische Konzentrationen von 1,0, 2,0 und
3,0 μg Pn-Ps/ml.
-
b) Verfahren
-
Assay
aller Proben und Standards unter Verwendung des Beckman ICS-Geschwindigkeitsnephelometers
und Anwendung von Doppelbestimmungen. Bestimmung der Konzentration
in den Proben aus der Standardkurve. Multiplikation der Probenkonzentration
mit dem Ver-dünnungsfaktor
und Mittelung der Werte für
jede Testprobe.
-
Wie
zuvor erwähnt,
werden die Proben, die bei diesem Verfahren befunden werden, Antigenizitätsindizes
unterhalb von 70% aufzuweisen, für
die Konjugation verworfen, um sicherzustellen, daß das verwendete Pn-Ps
die gewünschten
immunologischen Eigenschaften besitzt.
-
BEISPIEL 16
-
Klonierung von genomischer DNA, die für MIEP kodiert:
-
Etwa
0,1 g der durch Phenol inaktivierten N. meningitidis-Zellen (siehe
Beispiel 1) wurden in ein frisches Röhrchen eingebracht. Die durch
Phenol inaktivierten Zellen wurden in 567 μl TE-Puffer [10 mM TRIS-HCl, 1 mM EDTA, pH
8,0] resuspendiert. Zu den resuspendierten Zellen wurden 30 μl 10%iges
SDS und 3 μl
20 mg/ml Proteinase K (Sigma) zugegeben. Die Zellen wurden gemischt
und etwa 1 h lang bei 37°C
inkubiert, wonach 100 μl
5 M NaCl zugegeben und sorgfältig
gemischt wurde. 80 μl
1%iges Cetyltrimethylammoniumbromid (CTAB) in 0,7 M NaCl wurden
dann zugegeben, es wurde sorgfältig
gemischt, und 10 Minuten lang bei 65°C inkubiert. Ein gleiches Volumen
(etwa 0,7 bis 0,8 ml) Chloroform/Isoamylalkohol (bei einem Verhältnis von
24:1) wurde zugegeben, sorgfältig
gemischt und etwa 5 Minuten lang bei etwa 10.000 × g zentrifugiert.
Die wäßrige (obere)
Phase wurde in ein neues Röhrchen überführt und
die organische Phase verworfen. Ein gleiches Volumen an Phenol/Chloroform/-Isoamylalkohol (bei
einem Verhältnis
von 25:24:1) wurde der wäßrigen Phase
zugegeben, sorgfältig
gemischt und etwa 5 Minuten lang bei 10.000 × g zentrifugiert. Die wäßrige Phase (oben)
wurde in ein neues Röhrchen überführt und
0,6 Volumina (etwa 420 μl)
Isopropylalkohol zugegeben, sorgfältig gemischt und die ausgefällte DNA
10 Minuten lang bei 10.000 × g
zentrifugiert Die Überstandsflüssigkeit
wurde verworfen und das Pellet mit 70%igem Ethanol gewaschen. Das
DNA-Pellet wurde getrocknet und in 100 μl TE-Puffer resuspendiert und
repräsentiert
genomische N. meningitidis-DNA.
-
Es
wurden zwei DNA-Oligonukleotide synthetisiert, welche dem 5'-Ende des MIEP-Gens
und dem 3'-Ende des MIEP-Gens
entsprechen (Murakami, E. C., et al., (1989), Infection and Immunity,
57, S. 2318–23]. Die
Sequenz des für
das 5'-Ende des
MIEP-Gens spezifischen DNA-Oligonukleotids war: 5' ACTAGTTGCAATGAAAAAATCCCTG-3'; und die für das 3'-Ende des MIEP-Gens
war: 5'-GAATTCAGATTAGGAATTTGTT-3'. Diese DNA-Oligonukleotide
wurden als Primer für
eine Polymerasekettenreaktions(PCR)-Amplifizierung des MIEP-Gens
unter Verwendung von 10 ng genomischer N. meningitidis-DNA eingesetzt.
Der PCR-Amphfizierungsschritt erfolgte nach den vom Hersteller (Perkin
Elmer) angegebenen Verfahren.
-
Die
amplifizierte MIEP-DNA wurde dann mit den Restriktionsendonukleasen
SpeI und EcoRI verdaut. Das 1,3-Kilobasen(kb)-DNA-Fragment, enthaltend
den vollständigen
kodierenden Bereich von MIEP, wurde durch Elektrophorese auf einem
1,5%igen Agarosegel isoliert und aus dem Gel durch Elektroelution
gewonnen [Current Protocols in Molecular Biology, (1987), Ausubel,
R. M., Brent, R., Kingston, R. E., Moore, D. D., Smith, J. A., Seidman,
J. G., und Struhl, K., Hrsg., Greene Publishing Assoc.].
-
Der
Plasmidvektor pUC-19 wurde mit SpeI und EcoRI verdaut. Die gelgereinigte
SpeI-EcoRI-MIEP-DNA wurde in den SpeI-EcoRI-pUC-19-Vektor ligiert
und zur Transformation des E. coli-Stammes DH5 eingesetzt. Transformanten,
welche den pUC-19-Vektor mit der 1,3-kbp-MIEP-DNA enthielten, wurden durch
Restriktionsendonukleasekartierung identifiziert und die MIEP-DNA
sequenziert, um deren Identität
sicherzustellen.
-
BEISPIEL 17
-
Konstruktion des pC1/1.Gal10p(B)ADH1t-Vektors:
-
Der
Gal10-Promotor wurde aus dem Plasmid YEp52 [Broach et al., (1983)
in Experimental Manipulation of Gene Expression, Inouye, M. (Hrsg.),
Academic Press, S. 83–117]
isoliert durch Gelreinigung des 0,5-Kilobasenpaar(kbp)-Fragments, das nach
Spaltung mit Sau3A und HindIII erhalten worden war. Der ADH1-Terminator
wurde aus dem Vektor pGAP.tADH2 [Kniskern et el., (1986), Gene,
46, S. 135–141]
isoliert durch Gelreinigung des 0,35-kbp-Fragments, das durch Spaltung
mit HindIII und SpeI erhalten worden war. Die beiden Fragmente wurden
mit T4-DNA-Ligase mit dem gelgereinigten pUC18ΔHindIII-Vektor ligiert (die HindIII-Stelle wurde
eliminiert durch Verdauung von pUC18 mit HindIII, Erzeugung stumpfer
Enden mit dem Klenow-Fragment von E. coli-DNA-Polymerase I und Ligierung
mit T4-DNA-Ligase),
welcher mit BamHI und SphI verdaut worden war; um den Stammvektor
pGal10.tADH1 zu bilden. Dieser besitzt eine nur einmal vorkommende
HindIII-Klonierungsstelle an der Gal10p.ADH1,-Verbindungsstelle.
-
Die
nur einmal vorkommende HindIII-Klonierungsstelle von pGal10.tADH1
wurde zu einer nur einmal vorkommenden BamHI-Klonierungsstelle verändert durch
Verdauung von pGal10.tADH1 mit HindIII, Gelreinigung der geschnittenen
DNA und Ligierung unter Verwendung von T4-DNA-Ligase mit dem folgenden
HindIII-BamHl-Linker:
-
Bei
dem resultierenden Plasmid, pGal10(B)tADH1, ist die HindIII-Stelle
deletiert und eine nur einmal vorkommende BamHI-Klonierungsstelle
geschaffen worden.
-
Das
Gal10p.tADH1-Fragment wurde aus pGal10(B)tADH1 durch Verdauung mit
SmaI und SphI isoliert, mit T4-DNA-Polymerase stumpfendig gemacht
und gelgereinigt Der Hefe-Shuttle-Vektor pC1/1 (Brake et al., (1984),
Proc. Natl. Acad. Sci. USA, 81, S. 4642–4646] wurde mit SphI verdaut,
mit T4-DNA-Polymerase stumpfendig
gemacht und gereinigt Dieses Fragment wurde mittels T4-DNA-Ligase
in den Vektor ligiert. Die Ligierungsreaktionsmischung wurde dann
verwendet, um E. coli-HB101-Zellen zu Ampicillinresistenz zu transformieren
und Transformanten wurden gescreent durch Hybridisierung mit einem
Einzelstrang des 32P-markierten HindIII-BamHI-Linkers.
Die neue Vektorkonstruktion pC1/1.Gal10p(B)ADH1t wurde
durch Verdauung mit HindIII und BamHI bestätigt.
-
BEISPIEL 18
-
Konstruktion
eines Hefe-MIEP-Expressionsvektors mit MIEP- + Leader-DNA-Sequenzen
Ein DNA-Fragment, enthaltend den vollständigen kodierenden Bereich
von MIEP, wurde durch Verdauung von pUC19.MIEP#7 mit SpeI und EcoRI,
Gelreinigung der MIEP-DNA erzeugt und mit T4-DNA-Polymerase stumpfendig gemacht.
-
Der
interne Hefe-Expressionsvektor pC1/1.Gal10p(B)ADH1t wurde
mit BamHI verdaut, mit alkalischer Kalbsdarmphosphatase dephosphoryliert
und mit T4-DNA-Polymerase stumpfendig gemacht. Die DNA wurde gelgereinigt,
um ungeschnittenen Vektor zu entfernen.
-
Das
stumpfendige 1,1-kbp-Fragment von MIEP wurde mit dem stumpfendigen pC1/1.Gal10p(B)ADH1t-Vektor
ligiert und die Ligierungsreaktionsmischung eingesetzt, um kompetente
E. coli-DH5-Zellen zu Ampicillinresistenz zu transformieren. Transformanten
wurden gescreent durch Hybridisierung mit einem 32P-markierten
DNA-Oligonukleotid: 5'...
AAGCTCGGATCCTAGTTGCAATG...3',
welches konstruiert wurde, um Homologie zu Sequenzen aufzuweisen,
welche die MIEP-Vektor-Verbindungsstelle überlappen. DNA-Präparationen
wurden aus hybridisierungspositiven Transformanten hergestellt und
mit KpnI und SelI verdaut, um zu verifizieren, daß das MIEP-Fragment
in der korrekten Orientierung zur Expression durch den Gal10-Promotor
vorlag. Eine weitere Bestätigung
der DNA-Konstruktion wurde erhalten durch Didesoxysequenzierung
vom Gal10-Promotor in den für
MIEP kodierenden Bereich.
-
Expression
von MIEP durch die Transformanten wurde durch Western-Blot-Analyse
nachgewiesen. Rekombinantes MIEP, das in den Transformanten produziert
worden war, wanderte auf Polyacrylamidgelen zusammen mit MIEP, das
aus OMPC-Vesikeln gereinigt worden war, und konnte immunologisch
mit MIEP-spezifischen Antikärpern
reagieren.
-
BEISPIEL 19
-
Konstruktion des Hefe-MIEP-Expressionsvektors
mit einer 5'-modifizierten
MIEP-DNA-Sequenz
-
Ein
DNA-Oligonukleotid, enthaltend eine HindIII-Stelle, einen konservierten
nicht-translatierten 5'-Hefe-Leader (NTL), ein
Methionin-Startcodon (ATG), die ersten 89 Codons des reifen MIEP
(beginnend mit Asp an Position +20) und eine KpnI-Stelle (an Position
+89), wurde unter Einsatz der Polymerasekettenreaktionstechnik (PCR)
erzeugt. Die PCR wurde durchgeführt
wie vom Hersteller angegeben (Perkin Elmer Cetus) unter Verwendung
des Piasmids pUC19MIEP42#7 als Matrize und der folgenden DNA-Oligomeren
als Primer:
-
Zur
Entfernung des 5'-Bereichs
des MIEP-Klons wurde das Plasmid pUC19MIEP42#7 mit KpnI und HindIII
verdaut und das 3,4-kbp-Vektorfragment agarosegelgereinigt. Das
280-bp-PCR-Fragment wurde mit KpnI und HindIII verdaut, agarosegelgereinigt
und mit dem 3,4-kbp-Vektorfragment ligiert Transformanten von E.
coli HB101 (BRL) wurden durch DNA-Oligonukleotid-Hybridisierung
gescreent und die DNA aus positiven Transformanten durch Restriktionsenzymverdauung
analysiert Um sicherzustellen, daß keine Mutationen während des
PCR-Schritts eingeführt
wurden, wurde die 280 bp lange, durch PCR erzeugte DNA der positiven Transformanten
sequenziert. Das resultierende Plasmid enthält ein HindIII-EcoRI-Insert,
das aus einem Hefe-NTL, ATG-Codon und dem gesamten offenen Leserahmen
(ORF) von MIEP, beginnend am Asp-Codon (Aminosäure +20), besteht.
-
Die
Hefe-MIEP-Expressionsvektoren wurden wie folgt konstruiert. Die
Vektoren pGal10/pC1/1 und pGAP/pC1/1 [Vlasuk, G. P., et al., (1989)
J. B. C., 264, S. 12, 106–12,
112] wurden mit BamHI verdaut, mit dem Klenow-Fragment von DNA-Polymerase
I glattendig gemacht und mit alkalischer Kalbsdarmphosphatase dephosphoryliert.
Diese linearen Vektoren wurden mit dem oben beschriebenen, mit Klenow
behandelten und gelgereinigten HindIII-EcoRI-Fragment, welches den
Hefe-NTL, das ATG und den ORF von MIEP enthält, ligiert unter Bildung von
pGal10/pC1/1-MIEP und pGAP/pC1/1-MIEP.
-
Der
Saccharomyces cerevisiae-Stamm U9 (gal10pgal4-) wurde mit dem Plasmid
pGal10/pC1/1-MIEP transformiert. Rekombinante Klone wurden isoliert
und auf Expression von MIEP überprüft. Die
Klone wurden bei 37°C
unter Schütteln
in synthetischem Medium (leu-), das 2% Glucose (Gew./Vol.) enthielt,
bis zu einer OD660 von etwa 6,0 gezüchtet. Anschließend wurde
Galactose bis 2% (Gew./Vol.) zugegeben, um Expression von MIEP durch
den Gal10-Promotor zu induzieren. Die Zellen wurden nach der Galactose-Induktion
weitere 45 h lang auf eine OD660 von etwa
9,0 gezüchtet.
Die Zellen wurden dann durch Zentrifugation geerntet. Das Zellpellet
wurde mit destilliertem Wasser gewaschen und eingefroren.
-
Western-Blot für rekombinantes MIEP:
-
Zur
Feststellung, ob die Hefe MIEP exprimierte, wurde eine Western-Blot-Analyse
durch-geführt. 12%ige
Novex-Laemmli-Gele, 1 mm, 10 bis 15 Vertiefungen, wurden eingesetzt
Die Hefezellen wurden in H2O mit Hilfe von
Glasperlen aufgebrochen (Natriumdodecylsulfat (SDS) kann während des
Aufbruchprozesses mit 2% eingesetzt werden). Die Zelltrümmer wurden
durch einminütige
Zentrifugation bei 10.000 × g
entfernt.
-
Der Überstand
wurde mit Probenlaufpuffer gemischt, wie für die Polyacrylamidgelreinigung
von MIEP beschrieben. Die Proben wurden bei 35 mA laufengelassen,
unter Verwendung von OMPC als Referenzkontrolle, bis der Bromphenolblau-Farbstoffmarker
aus dem Gel läuft.
-
Die
Proteine wurden unter Verwendung einer NOVEX-Transferapparatur auf
Nitrocellulosepapier mit 0,45 μm
Porengröße überführt Nach
dem Transfer wurde das Nitrocellulosepapier mit 5%igem Rinderserumalbumin
in phosphatgepufferter Salzlösung
1 h lang blockiert, wonach 15 ml einer 1:1000-Verdünnung
von Anti-MIEP-Kaninchen-Antiserum (erzeugt durch Immunisierung mit
gelgereinigtem MIEP nach Standardverfahren) zugegeben wurden. Nach
einer Übernachtinkubation
bei Raumtemperatur wurden 15 ml einer 1:1000-Verdünnung von
mit alkalischer Phosphatase konjugiertem Anti-Kaninchen-Ziegen-IgG zugegeben. Nach
2 h Inkubation wurde der Blot unter Verwendung von FAST RED TR SALT
(Sigma) und Naphthol-AS-MX-Phosphat (Sigma) entwickelt.
-
BEISPIEL 20
-
bakterielle Expression von rekombinantem
MIEP
-
Das
Plasmid pUC19-MIEP, enthaltend das 1,3-Kilobasenpaar-MIEP-Gen-Insert,
wurde mit den Restriktionsendonukleasen SpeI und EcoRI verdaut Das
1,1-kbp-Fragment wurde isoliert und auf einem Agarosegel nach im
Stand der Technik bekannten Standardverfahren gereinigt. Das Plasmid
pTACSD, enthaltend den Zwei-Cistron-TAC-Promotor und eine nur einmal
vorkommende EcoRI-Stelle, wurde mit EcoRI verdaut. Stumpfe Enden
wurden sowohl bei der 1,3-kbp-MIEP-DNA als auch dem pTACSD-Vektor unter Verwendung von
T4-DNA-Polymerase (Boehringer Mannheim) nach den Anweisungen des
Herstellers erzeugt. Die stumpfendige 1,3-kbp-MIEP-DNA wurde in
den stumpfendigen Vektor unter Verwendung von T4-DNA-Ligase (Boehringer
Mannheim) nach den Anweisungen des Herstellers ligiert. Die ligierte
DNA wurde eingesetzt, um den E. coli-Stamm DH5alQMAX (BRL) nach
den Anweisungen des Herstellers zu transformieren. Die transformierten
Zellen wurden auf Agarplatten ausplattiert, die 25 μg Kanamycin/ml
und 50 μg
Penicillin/ml enthielten, und etwa 15 h lang bei 37°C inkubiert.
Ein DNA-Oligonukleotid
mit einer zu MIEP homologen Sequenz wurde mit 32P
markiert und zum Screenen von Nitrocellulosefiltern, die lysierte
denaturierte Kolonien von den Platten von Transformanten enthielten,
unter Anwendung von Standard-DNA-Hybridisierungstechniken eingesetzt.
Kolonien, die sich bei der Hybridisierung als positiv erwiesen,
wurden mit Hilfe von Restriktionsendonukleasen kartiert, um die
Orientierung des MIEP-Gens zu bestimmen.
-
Die
Expression des MIEP durch die Transformanten wurde durch Western-Blot-Analyse
nachgewiesen. Rekombinantes MIEP, das in den Transformanten produziert
worden war, wanderte auf Polyacrylamidgelen zusammen mit MIEP, das
aus OMPC-Vesikeln gereinigt worden war, und konnte immunologisch
mit MIEP-spezifischen Antikörpern
reagieren.
-
BEISPIEL 21
-
Konjugation von Pn-Ps an N. meningitidis-MIEP:
-
-
10
mg MIEP in 3 ml 0,1 M Boratpuffer, pH 11,5, wurden mit 10 mg Ethylendiamintetraessigsäure-Dinatriumsalz (EDTA,
Sigma Chemicals) und 4 mg Dithiothreit (Sigma Chemicals) gemischt.
Die Proteinlösung wird
gründlich
mit N2 gespült. 125 mg N-Acetylhomocysteinthiolacton
(Aldrich Chemicals) werden der MIEP-Lösung zugegeben und die Mischung
16 h lang bei Raumtemperatur inkubiert. Sie wird dann zweimal unter
N2 gegen 2 l 0,1 M Boratpuffer, pH 9,5,
der 4 mM EDTA enthält,
24 h lang bei Raumtemperatur dialysiert. Das thiolierte Protein
wird dann hinsichtlich des Thiolgehalts durch Ellmans Reagens (Sigma
Chemicals) untersucht und die Proteinkonzentration durch Bradford-Reagens
(Pierce Chemicals) bestimmt Für
die Konjugation von MIEP an Pn-Ps wird ein 1,5facher Überschuß (Gew./Gew.)
von bromacetyliertem Pn-Ps der MIEP-Lösung zugegeben und der pH mit
1 N NaOH auf 9–9,5
eingestellt. Die Mischung wird unter N2 6–8 h lang
bei Raumtemperatur inkubieren gelassen. Am Ende der Reaktionszeit
werden 25 μl
N-Acetylcysteamin (Chemical Dynamics) der Mischung zugegeben und
18 h lang unter N2 bei Raumtemperatur stehengelassen.
Die Konjugat-Lösung
wird mit 1 N HCl auf zwischen pH 3 bis 4 angesäuert und eine Zentrifugation
10 Minuten lang bei 10.000 × g
durchgeführt.
1 ml der Überstandsflüssigkeit
wird direkt auf eine Säule
von FPLC-Superose 6B (1,6 × 50
cm, Pharmacia) aufgebracht und das Konjugat mit PBS eluiert. Der
Peak des Hohlraumvolumens, welcher das Polysaccharid-Protein-Konjugat
(Pn-Ps-MIEP) enthält,
wird gepoolt. Die Konjugat-Lösung
wird dann zur Sterilisation durch ein 0,22-μm-Filter filtriert.
-
BEISPIEL 22
-
Demonstration der Immunogenizität von Pn-Ps-MIEP-Konjugaten:
-
Immunisierungen:
Männliche
Balb/c-Mäuse
(Charles River, Wilmington, MA) werden mit kovalent an MIEP konjugiertem
Pn-Ps unter Einsatz von 2,5 μg
Pn-Ps in 0,5 ml vorgebildeten Alauns IP immunisiert. Kontrollmäuse werden
mit äquivalenten
Mengen an Pn-Ps immunisiert, die als Pn-Ps-CRM (Anderson, M. E.,
et al., (1985), J. Pediatrics, 107, S. 346–351] (2,5 μg Pn-Ps/6,25 μg CRM; 1/4
der Human-Dosis), Pn-Ps-DT (2,5 μg
Pn-Ps/1,8 μg
DT; 1/10 der Human-Dosis, so daß konstante
Mengen von Pn-Ps eingesetzt werden) und Pn-Ps-OMPC (2,5 μg Pn-Ps/35 μg OMPC; 1/4
der Human-Dosis) gegeben werden.
-
6–13,5 Wochen
alte Rhesusaffenbabys werden mit Pn-Ps-MIEP-Konjugaten, adsorbiert
an Alaun, immunisiert. Jeder Affe enthält 0,25 ml Konjugat an zwei
verschiedenen Injektionsstellen für eine Gesamtdosis von 0,5
ml. Die Affen werden am Tag 0,28 und 56 immunisiert und es werden
alle 2 bis 4 Wochen Blutproben entnommen.
-
Die
Antikörperantworten
werden durch den ELISA gemessen, welcher die Klasse und Subklasse
der Immunglobulinantwort unterscheidet. Ein RIA, welcher die Gesamtmenge
an Anti-Pn-Ps-Antikörper
quantitativ bestimmt, wird ebenfalls eingesetzt, um die Affen-Antwort
zu bewerten.
-
Pn-Ps-MIEP-Konjugate
sind imstande, bei Mäusen
eine Immunantwort hervorzurufen, bestehend aus Anti-Pn-Ps-IgG-Antikörper und
einer Erinnerungsantwort. Dies steht im Gegensatz zu den Pn-Ps-CRM
und Pn-Ps-DT, welche keinen meßbaren
Anti-Pn-Ps-Antikörper
hervorrufen. Somit wirkt MIEP als immunologisches Trägerprotein
für Pn-Ps
und ist imstande, bei kovalenter Konjugation mit dem Pn-Ps-Antigen eine Anti-Pn-Ps-Antikörperantwort
zu veranlassen. Gereinigtes MIEP ist deshalb ein effektives immunologisches
Trägerprotein,
welches das heterogene OMPC bei der Konstruktion bakterieller Polysaccharid-Konjugat-Vakzine ersetzt.
-
BEISPIEL 23
-
Quantitative Bestimmung des C-Polysaccharid-Gehalts
in Pn-Ps-Präparationen:
-
Es
wurden Systeme zur quantitativen Bestimmung von C-Polysaccharid
entwickelt, die auf NMR, enzymatischen oder chromatographischen
Methoden beruhen. Im vorliegenden Fall wurde die chromatographische
Abtrennung von Cholin (eine Komponente von C-Ps) aus Proben von
hydrolysierten Pn-Ps angewandt und mit diesen anderen Methoden verglichen.
Das Cholin wurde abgetrennt auf einer Kationenaustauschersäule, gekoppelt
mit einem Nachweis unterdrückter
Leitfähigkeit.
-
Proben
von Pn-Ps wurden durch Behandlung mit 36%iger Fluorwasserstoffsäure für 2 h bei
45–65°C, gefolgt
von 2 M Trifluoressigsäure
für 16
h bei 100°C,
vollständig
hydrolysiert Nach der Hydrolyse wurden 200–300 μg der Probe injiziert in ein
Dionex BioLC-Chromatographiesystem mit einer Omnipac PCX-500-Analyse-
und Überwachungssäule, einer
Ion Pac CTC-1-Kationeneinfangsäule,
einem CMMS-2-Micromembransuppressor,
regeneriert mit 50 mM Tetrabutylammoniumhydroxid (10 ml/Mmn.), und
einer Leitfähigkeitsdetektoreinstellung
auf eine Empfindlichkeit von 1 μSiemens.
Die Probe wurde unter Verwendung von 5% 200 mM HCl, 5% 20%iges Acetonitril,
85% MilliQ-Wasser, 5% 20 mM Diaminopropionsäure isokratisch eluiert. Cholin eluierte
als scharfer Peak nach etwa 10 Minuten.
-
Gereinigtes
C-Ps (erhalten vom Statens Serum Institut) wurde mit diesem Verfahren
hinsichtlich des Cholingehalts analysiert und ein Wert von 5,4 Gew.-%
Cholin erhalten. Dieser Wert stimmt mit veröffentlichten Angaben des Cholingehalts
von C-Ps überein.
-
Dieser
Faktor wurde zur Berechnung der C-Ps-Konzentration in verschiedenen
Proben von Pn-Ps-Präparationen
verwendet, indem die Nanomolmengen des durch HPLC erhaltenen Cholins
in Massenwerte umgerechnet wurden. Durch Vornahme der Umrechnung
von 5,4 Gew.-% Cholin wurde die C-Ps-Menge nach Gewicht berechnet.
Proben mit C-Ps-Konzentrationen über
3% wurden als für
die Konjugation unakzeptabel verworfen. Die folgende Tabelle zeigt
die Korrelation dieser Methode mit den NMR- und enzymatischen Methoden
und zeigt typische C-Ps-Verunreinigungsniveaus in Präparationen
von Pn-Ps verschiedener Reinheitsgrade:
| Probe | NMR | Enzymatisch | HPLC |
| Pn6B-Ps | 20% | n.
b. | 18,4% |
| Pn6B-Ps | 1,6% | 0,3–1,0% | 1,2% |
| Pn23F-Ps | n.
b. | 2,8% | 3,7% |
| Pn14-PS | 2,9% | 2,4% | 3,2% |
| Pn19F-Ps | 2,7% | 2,6% | 2,6% |
-
BEISPIEL 24
-
Herstellung von partiell hydrolysiertem,
gereinigtem Pn18C-Ps:
-
- (1) Beschallungshydrolyse: Eine 3,0-g-Portion
von Pn18C-Ps-Pulver wurde in 1200 ml Salzlösung (0,9% NaCl) unter Rühren bei
Raumtemperatur für
etwa 3–4
h solubilisiert, dann bedeckt und bei 4°C über Nacht gelagert Die Lösung wurde
dann in einem Kunststoffbecherglas im Eisbad mit einem Branson Sonifier (Halbzoll-Sonde,
Einstellung 8) für
Intervalle von 20 Minuten (in 5-minütigen Aktivitätsintervallen)
bis insgesamt 40 Minuten beschallt. Die Viskosität wurde nach jedem Intervall überprüft.
- Nach 40 Minuten wurde eine weitere 10-minütige Beschallung durchgeführt, um
eine Endviskosität
von 1,218 Zentistokes zu erhalten. Die hydrolysierte Probe (Volumen – 1188 ml)
wurde auf Raumtemperatur gebracht und Natriumacetat-Reagens (59,2
g) wurde bis zu einer Endkonzentration von 3% (Gew./Vol.) zugegeben.
- (2) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungsuntersuchung
und ein antikörper-gesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das
Pn18C-Ps zwischen 40–50%
IPA ausfallen würde.
- (3) Erste IPA-Zugabe: Die hydrolysierte Probe [Volumen = 1200
ml, aus Schritt 1 oben] wurde durch die Zugabe von 894 ml IPA (tropfenweise
unter Rühren
bei Raumtemperatur zugegeben) auf 42,7% IPA gebracht Die Probe wurde
15–30
Minuten rühren
gelassen und dann 30 Minuten lang bei 11.000 × g zentrifugiert (Beckman
JA-10-Rotor, 8.000 UpM; 20°C).
Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter gesammelt Der Niederschlag wurde
direkt auf dem Trichter mit absolutem EtOH, dann Aceton gewaschen
und im Vakuum über
CaSO4 (Drierit) bei Raumtemperatur in Vorbereitung
auf die Analyse getrocknet.
- (4) Zweite IPA-Zugabe und Gewinnung des Zwischenprodukts: Die Überstandsflüssigkeit
von 42,7% IPA [Volumen = 2016 ml, aus Schritt 3 oben] wurde durch
tropfenweise Zugabe von 92,0 ml IPA unter Rühren bei Raumtemperatur auf
45,2% IPA gebracht Die Probe wurde stehengelassen und zentrifugiert wie
in Schritt 3 oben. Das Pellet wurde trituriert, gesammelt, gewaschen
und getrocknet wie in Schritt 3 oben. Das Pn18C-Ps-Zwischenprodukt
wog 1609 mg.
- (5) Dialyse und Lyophilisierung: Eine Portion (1612,5 mg) der
Probe aus Schritt 4 oben wurde in 645 ml destilliertem H2O bei Raumtemperatur etwa 2 h lang solubilisiert.
Die Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch überführt (MG-Ausschluß 12.000;
45 mm) und unter zwei weiteren Austauschen von destilliertem H2O 30 h lang gegen destilliertes H2O bei 4°C
dialysiert. Dann wurde die Probe in Lyophilisierungskolben überführt, in
einem Trockeneis:Methanol-Bad rollschichtgefroren und mit einem
Virtis(Freezemobile)-Lyophilisator 2–3 Tage lang lyophilisiert
Die Ausbeute des Ps-Endprodukts betrug 1487 mg.
-
BEISPIEL 25
-
S. Pneumoniae 18C-OMPC-Konjugat, Pn18C-Ps-OMPC:
-
- A. Herstellung von Dowex 50 × 2 (200–400 Mesh)
Tetrabutylammoniumform-Harz [Dowex 50 (Bu4N+)]: Dowex 50 × 2 (200–400 Mesh), H+-Form,
(500 g) wurde in H2O aufgeschlämmt, auf
eine Säule
aufgebracht und nacheinander gewaschen mit 1] 600 ml H2O;
2] 1000 ml 6 N HCl; 3] 400 ml H2O, bis der
Ausfluß für pH-Papier
neutral war; 4] 72 g einer 10%igen wäßrigen Tetrabutylammoniumhydroxidlösung, bis
der Ausfluß für pH-Papier
stark alkalisch war; 5] 1000 ml H2O bis
zur Neutralität.
- B. Herstellung von S. Pneumoniae Typ 18C-Polysaccharid, Tetrabutylammonium-form
[Pn18C(Bu4N+)]: Eine
60-ml-Säule
von Dowex 50 × 2
(Bu4N+) wurde mit
250 ml H2O gewaschen. Pn18C-Polysaccharid (MG-reduziert
(650 mg)) wurde mit 65 ml H2O bedeckt und
1 h lang gerührt,
wonach alles in Lösung
zu sein schien. Diese Lösung
wurde auf die Säule
aufgetragen und durch Schwerkraft perkolieren gelassen (2 h lang
und dann 1 h lang unter Vakuum). Die Säule wurde mit 150 ml H2O gewaschen und die vereinigten Ausflüsse lyophilisiert,
was 655 mg des 18C(Bu4N+)-Salzes ergab. 25
mg wurden für
eine NMR-Analyse entnommen und Material zurückbehalten.
- C. Herstellung des 1,4-Butandiamin-Derivats von 18C(18C-BuA2):
18C(Bu4N+) (630 mg) wurde mit 143 ml DMSO (Dimethylsulfoxid)
bedeckt und 3,25 h lang gerührt.
Nach dieser Zeit war der gesamte Feststoff gelöst und 1 ml wurde für eine Karl-Fischer-Titration hinsichtlich
des Wassergehalts entnommen. Es wurde ein Wert von 28,2 μMol H2O/ml festgestellt (insgesamt 4 mMol). Zu dieser
Lösung
wurden 165,1 mg Carbonyldiimidazol (CDI) zugegeben und die resultierende
Lösung
2,0 h lang bei Raumtemperatur (RT) gerührt. Eine Lösung, enthaltend 1,260 g 1,4-Butandiamin-Dihydrochlorid (BuA2·2HCl)
in 40 ml H2O, wurde hergestellt und deren
pH mit 2,5 N NaOH auf 10,20 eingestellt. Diese Lösung wurde in einem Eisbad
gekühlt.
Die gealterte DMSO-Lösung
wurde langsam der kalten BuA2-Lösung zugegeben
und weitere 10 Minuten im Eisbad gerührt. Sie wurde dann 50 Minuten
lang bei RT gerührt, wonach
die Lösung
in einen SPECTRAPOR 2-Dialyseschlauch eingebracht wurde, 1,27 cm
(1/2 Inch) vom oberen Rand der Flüssigkeit abgeklemmt und wie
folgt dialysiert wurde gegen: 1] 15 l 0,1 M NaPO4-Puffer, pH
7,0, für
13,0 h; 2] 15 l 0,01 M NaPO4-Puffer, pH
7,0, für
11 h; 3] 15 l 0,01 M NaPO4-Puffer, pH 7,0,
für 10,8
h; 4] 15 l H2O für 9,5 h. Das Volumen betrug
zu diesem Zeitpunkt 190 ml. Ein 7,5-ml-Aliquot wurde entnommen und
separat für
einen NMR-Assay lyophilisiert. Die verbleibenden 182,5 ml wurden
auf 416 mg des 1,4-Butandiamin-Derivats von 18C(Pn18C-BuA2) lyophilisiert. Ein NMR-Spektrum von etwa
5 mg zeigte eine ”Beladung” von 10
Butandiaminresten pro 100 Polysaccharidmonomereinheiten, festgestellt
durch Vergleich der Integrale der Resonanzen der internen Butandiamin-Methylene
und der Rhamnose-Methyle (von 18C).
- D. Herstellung des bromacetylierten Butandiamin-Derivats von
18C(Pn18C-BuA2-BrAc): 18C-BuA2 (416 mg)
wurde mit 36 ml eines 0,1 M Puffers (Kolthoff-Borat-Phosphat), pH
9,04, bedeckt und gerührt,
um die Lösung
zu bewirken. Dann wurden 256 mg p-Nitrophenylbromacetat in 4,48
ml Acetonitril zugegeben. Die resultierende Mischung wurde 20 h
lang bei 4°C
gerührt.
Sie wurde dann in einem SPECTRAPOR 2-Schlauch wie folgt dialysiert
1] gegen 15 l H2O für 6 h; 2] gegen 15 l H2O für
6 h; 3] gegen 15 l H2O für 6 h. Zu diesem Zeitpunkt
lag ein Volumen von 60 ml vor, von dem 1,7 ml für Assays (NMR, Ouchterlony
und Viscotek) entnommen und dann 2,42 g getrocknetes Salz von Phosphatpuffer,
pH 8 (hergestellt durch Lyophilisierung einer 0,1 M NaPO4-Lösung von
pH 8) zugegeben wurden. Nach der Lösung wurde durch ein 0,2-Micron-CORNING-Filter filtriert,
was eine wäßrige Lösung, pH
8, von 18C-BuA2-BrAc ergab. Die Filtration
war langsam und erforderte vier Becherfilter.
- E. Konjugation von OMPC (N. meningitidis) mit Pn18C-BuA2-BrAc: Der Komplex des Proteins der äußeren Membran
(N. meningitidis), OMPC, 3,2 mg/ml, 80 ml, wurde in vier 25-ml-Zentrifugenröhrchen eingebracht und
in einem 60 Ti-Rotor bei 43.000 UpM (43 K) 2 h lang bei 4°C zentrifugiert.
Eine Thiolierungsmischung wurde hergestellt durch Lösen von
680 mg EDTA (Ethylendiamintetraessigsäure-Dinatriumsalz) und 120 mg
Dithiothreit (DTT) in 40 ml eines Na2B4O7-Puffers, pH 11,09.
320 mg N-Acetylhomocysteinthiolacton wurden hinzugegeben und dann
die Lösung
durch ein 0,2-μm-Corning-Filter
(Bechertyp) filtriert. Die Pellets aus der obigen Zentrifugation
wurden mit 5 ml der filtrierten Thiolierungsmischung (insgesamt
20 ml) disloziert und in einen DOUNCE-Homogenisator überführt und
resuspendiert. Die Röhrchen
wurden durch reihenweises Umfüllen
von weiteren 2/10 ml der Thiolierungslösung gespült Die vereinigten Lösungen wurden im
DOUNCE homogenisiert und das gesamte resuspendierte Material (40
ml) in einen 100-ml-Rundkolben überführt Die
Glasgeräte
wurde mit weiteren 20 ml der Thiolisierungslösung gespült und diese dem Reaktionskolben
zugegeben. Nach dem Verschließen
des Kolbens mit einem Septum und Ersetzen der Luft durch N2 unter Verwendung eines FIRESTONE-Ventils wurde die
Reaktionsmischung 18,5 h lang stehengelassen. Die 60 ml wurden dann
auf vier Zentrifugenröhrchen
aufgeteilt, von denen jedes mit 1 M KH2PO4 (wäßrig) aufgefüllt und
dann 2 h lang bei 43 K und 4°C
zentrifugiert wurde. Die Überstände wurden
entfernt und die Pellets in 0,1 M NaPO4-Puffer,
pH 8, resuspendiert (eine Gesamtmenge von 40 ml war das endgültige Resuspensionsvolumen).
Diese Lösung
wurde zu gleichen Teilen in zwei 25-ml-Zentrifugenröhrchen (Polycarbonat) überführt und
die Glasgeräte
(DOUNCE etc.) wurden mit etwa 10 ml Phosphatpuffer, pH 8, gespült und diese
zum Auffüllen
der Zentrifugenröhrchen
verwendet Dann wurde eine zweite Ultrazentrifugation (2 h, 4°C, 43 K)
durchgeführt.
Die Pellets wurden in 30 ml 0,1 M PO4-Puffer,
pH 8, resuspendiert Ein Ellman-Assay zeigte eine Gesamtmenge von
24 μMol
SH oder etwa 100 nMol/mg OMPC an. Das thiolierte Protein wurde in
einen 100-ml-Rundkolben überführt und
die filtrierte 18C-BuA2-BrAc-Lösung zugegeben. Die
resultierende Reaktion (d. h. 18C-BuA2-BrAc
mit thioliertem OMPC) wurde unter N2 (unter
Entgasen) in der N2-Box 89 h lang bei Raumtemperatur
stehengelassen.
Die Reaktion wurde dann wie folgt mit Endgruppen
versehen: Eine Lösung,
enthaltend 75 mg N-Ethylmaleimid
(NEM) in 5 ml 0,1 M NaPO4-Puffer, pH 8,
wurde durch ein 0,22-μm-Filterfiltriert
und 2 ml der obigen Reaktionsmischung zugegeben und 4 h lang stehengelassen.
Dann
wurden 0,5 ml N-Acetylcysteamin in 2,5 ml 0,1 M PO4-Puffer,
pH 8, durch ein 0,22-μm-Filter
filtriert und 1,0 ml dieser Lösung
der Reaktion zugegeben und 22,5 h lang stehengelassen.
Das
mit Endgruppen versehene Produkt wurde dann zu gleichen Teilen in
vier 25-ml-Zentrifugenröhrchen eingebracht
und mit insgesamt 8 ml 0,1 M PO4-Puffer,
pH 8, überschichtet
und bei 43 K, 2 h, 4°C
zentrifugiert. Nach Entfernung der Überstände wurden die Pellets in einem
DOUNCE-Homogenisator
in einer Gesamtmenge von 40 ml TED-Puffer resuspendiert, die Glasgeräte mit weiteren
10 ml TED-Puffer gespült
und die Lösung
in zwei 25-ml-Röhrchen überführt. Diese
Röhrchen
wurden 15,25 h lang bei Raumtemperatur gelagert und dann 2 h lang
bei 43 K UpM und 24°C
zentrifugiert Die resultierenden Pellets wurden in einem DOUNCE-Homogenisator
in einer Gesamtmenge von 30 ml TED-Puffer resuspendiert, in zwei
25-ml-Zentrifugenröhrchen überführt, die
Glasgeräte
mit weiteren 20 ml TED-Puffer gespült und 2 h lang bei 43 K und 4°C erneut
zentrifugiert. Die Pellets wurden in 50 ml Phosphatpuffer, pH 7,
resuspendiert und einer dritten Zentrifugation bei 43 K für 2 h bei
4°C unterworfen.
Die Pellets wurden in 82 ml Wasser resuspendiert und in 20,5-ml-Portionen
in zwei sterile 50-ml-Kunststoffzentrifugenröhrchen (FALCON) überführt. Nach
dem Stehenlassen bei 4°C
für 18
h wurde die Konjugat-Präparation
3,5 Minuten lang bei 1000 UpM in einem TH-Rotor in einer TJ-6-Zentrifuge
zentrifugiert. Die endgültige
Konjugatprodukt-Suspension wurde auf Protein (Lowry), 18C-Polysaccharid
(Phenol/Schwefelsäure),
unkonjugiertes Polysaccharid (Größenausschlußchromatographie-Geschwindigkeitsnephelometrie)
und Aminosäuren
(SPINCO) untersucht:
Polysaccharid = 339 μg/ml;
Protein = 2,57 mg/ml;
freies
Polysaccharid: < 5%
(Grenze des Versuchsfehlers);
S-Carboxymethylhomocystein/Lysin
= 0,025;
S-Carboxymethylcysteamin/Lysin = 0,005.
-
BEISPIEL 26
-
Herstellung des Pn4-Ps-Zwischenprodukts:
-
- (1) Beschallungshydrolyse: Eine 1,0-g-Portion
von Pn4-Ps-Pulver wurde in 400 ml Salzlösung (0,9% NaCl) unter Rühren bei
Raumtemperatur etwa 4 h lang solubilisiert Die Lösung wurde dann in einem Kunststoffbecherglas
in einem Eisbad mit einem Branson Sonifier (Halbzoll-Sonde, Einsteilung
8) für
Intervalle von 10 Minuten bis zu insgesamt 20 Minuten beschallt
Die Viskosität
wurde nach jedem Intervall überprüft. Nach 20
Minuten wurde eine Endviskosität
von 1267 Zentistokes erhalten. Die hydrolysierte Probe wurde auf Raumtemperatur
gebracht und Natriumacetat-Reagens (18,7 g) wurde bis zu einer Endkonzentration
von 3% (Gew./Vol.) zugegeben.
- (2) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungspilotstudie
und ein antikörpergesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das Pn4-Ps
zwischen 45–55%
IPA ausfallen würde.
- (3) Erste IPA-Zugabe: Die hydrolysierte Probe [Volumen = 385
ml, aus Schritt 1 oben] wurde durch die Zugabe von 379 ml IPA (tropfenweise
unter Rühren
bei Raumtemperatur zugegeben) auf 49,7% IPA gebracht. Die Probe
wurde 15–30
Minuten lang röhrengelassen
und dann 30 Minuten lang bei 11.000 × g (Beckman JA-10-Rotor; 8.000
UpM; 20°C)
zentrifugiert. Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter gesammelt. Der Niederschlag wurde
direkt auf dem Trichter mit absolutem EtOH, dann Aceton gewaschen
und im Vakuum über
CaCl2 bei Raumtemperatur in Vorbereitung
auf die Analyse getrocknet.
- (4) Zweite IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 49,7% IPA [Volumen 727 ml, aus Schritt 3 oben] wurde durch tropfenweise
Zugabe von 38 ml IPA unter Rühren
bei Raumtemperatur auf 52,2% IPA gebracht. Die Probe wurde stehengelassen
und zentrifugiert wie in Schritt 3 oben. Das Pellet wurde trituriert,
gesammelt, gewaschen und getrocknet wie in Schritt 3 oben. Das Pn4-Ps-Produkt
wog 516 mg.
- (5) Dialyse und Lyophilisierung: Eine Portion (500 mg) der Pn-Ps-Probe
aus Schritt 4 oben wurde in 200 ml destilliertem H2O
bei Raumtemperatur 2–3
h lang solubilisiert. Die Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch (MG-Ausschluß 12.000;
45 mm) überführt und
unter zwei weiteren Austauschen von destilliertem H2O
C 27 h lang bei 4°C
gegen destilliertes H2O dialysiert. Dann
wurde die Probe in Lyophilisierungskolben überführt, in einem Trockeneis:-Methanol-Bad
rollschichtgefroren und mit einem Virtis(Freezemobile)-Lyophilisator
2–3 Tage
lang lyophilisiert. Die Ausbeute des Ps-Endprodukts betrug 491 mg.
Das Endprodukt besaß einen
Kd = 0,69.
-
Für Fachleute
sollte aus dieser Offenbarung ersichtlich sein, daß andere
carboxyl-enthaltende Pn-Ps-Subtypen wie Pn1-Ps oder Pn5-Ps nach
dem hier offenbarten Verfahren hergestellt werden könnten und
wie Pn4-Ps oder Pn9V-Ps, die ebenfalls saure Polysaccharide sind,
konjugiert werden könnten.
-
BEISPIEL 27
-
S. Pneumoniae Typ 4-OMPC-Konjugat, Pn4-Ps-OMPC:
-
- A. Herstellung von Dowex 50 × 2 (200–400 Mesh)
Tetrabutylammoniumform-Harz [Dowex 50 (Bu4N+)]: Dowex 50 × 2 (200–400 Mesh), H+-Form,
(500 g) wurde in H2O aufgeschlämmt (CM-66),
auf eine Säule
aufgebracht und nacheinander gewaschen mit 1) 600 ml H2O;
2] 1000 ml 6 N HCl; 3] 400 ml H2O, bis der
Ausfluß für pH-Papier
neutral war; 4] 72 g einer 10%igen wäßrigen Tetrabutylammoniumhydroxidlösung, bis
der Ausfluß für pH-Papier
stark alkalisch war; 5] 1000 ml H2O bis
zur Neutralität.
- B. Herstellung von S. Pneumoniae Typ 4-Polysaccharid, Tetrabutylammoniumform
[Pn4(Bu4N+)]: Eine 65-ml-Säule von
Dowex 50 × 2
(Bu4N+) wurde mit
520 ml H2O gewaschen. Pn4-Polysaccharid
(MG-reduziert) (400 mg) wurde mit 35 ml H2O
bedeckt und 20 Minuten lang gerührt,
wonach alles in Lösung
zu sein schien (das Rühren
wurde über
Nacht fortgesetzt). Diese Lösung
wurde auf die Säule
aufgetragen und durch Schwerkraft perkolieren gelassen und die Säule wurde
mit 150 ml H2O gewaschen und die vereinigten
Ausflüsse
lyophilisiert, was 504 mg des Pn4(Bu4N+)-Salzes ergab.
- C. Herstellung des 1,4-Butandiamin-Derivats von Pn4(Pn4-BuA2): Pn4(Bu4N+) (97 mg) wurde mit 16 ml DMSO (Dimethylsulfoxid)
bedeckt und im Verlauf eines Zeitraums von 15 Minuten bei 52°C in die
Lösung gerührt. Nach
dieser Zeit war der gesamte Feststoff gelöst und die Lösung wurde
auf Raumtemperatur abgekühlt.
Zu
dieser Lösung
wurden 2 mg Carbonyldiimidazol (CDI), gelöst in 160 μl DMSO, zugegeben und die resultierende
Lösung
1,0 h lang bei Raumtemperatur (RT) gerührt. Eine Lösung, enthaltend 0,500 g 1,4-Butandiamin-Dihydrochlorid
(BuA2·2HCl)
in 5 ml H2O, wurde hergestellt und deren
pH mit 5,0 N NaOH auf 10,20 eingestellt. Diese Lösung wurde in einem Eisbad
gekühlt.
Die gealterte DMSO-Lösung
wurde langsam der kalten BuA2-Lösung zugegeben
und weitere 5 Minuten in dem Eisbad gerührt. Sie wurde dann 1 h lang
bei RT gerührt,
wonach die Lösung
in einen SPECTRAPOR-2-Dialyseschlauch eingebracht wurde, 1,27 cm
(1/2 Inch) vom oberen Rand der Flüssigkeit abgeklemmt und wie
folgt dialysiert wurde gegen: 1] 4 l 10,1 M NaPO4-Puffer,
pH 7,0, für
15,0 h; 2] 4 l 0,01 M NaPO4-Puffer, pH 7,0,
für 9 h;
3] 4 l 0,01 M NaPO4-Puffer, pH 7,0, für 21 h;
4] 4 l H2O für 20 h. Die Lösung wurde
auf 70 mg des 1,4-Butandiamin-Derivats von Pn4 (Pn4-BuA2)
lyophilisiert. Ein NMR-Spektrum von etwa 5 mg zeigte eine ”Beladung” von 22 Butandiaminresten
pro 100 Polysaccharidmonomereinheiten, festgestellt durch Vergleich
der Integrale der Resonanzen der internen Butandiamin-Methylene
und der N-Acetylmethyle (von Pn4).
- D. Herstellung des bromacetylierten Butandiamin-Derivats von
Pn4 (Pn4-BuA2-BrAc): Pn4-BuA2 (54 mg) wurde
mit 5,5 ml eines 0,1 M Puffers (Kolthoff-Borat-Phosphat), pH 9,04,
bedeckt und gerührt,
um die Lösung
zu bewirken. Dann wurden 55 mg p-Nitrophenylbromacetat in 1,0 ml
Acetonitril zugegeben. Die resultierende Mischung wurde 17 h lang
bei 4°C
gerührt.
Sie wurde dann in einem SPECTRAPOR-2-Schlauch wie folgt dialysiert:
1] gegen 16 l H2O für 24 h; 2] gegen 16 l H2O für
8 h; 3] gegen 16 l H2O für 23 h. Zu diesem Zeitpunkt
lag ein Volumen von 12,5 ml vor, von dem 1,0 ml für Assays
(NMR, Ouchterlony und Viscotek) entnommen wurde und dann 275 mg
eines getrockneten Salzes von Phosphatpuffer, pH 8 (hergestellt
durch Lyophilisierung einer 0,1 M NaPO4-Lösung, pH
8), zugegeben wurde. Nach der Lösung
wurde durch ein 0,2-Mikron-CORNING-Filter
filtriert, was eine wäßrige Lösung, pH
8, von Pn4-BuA2-BrAc ergab.
- E. Konjugation von OMPC (N. meningitidis) mit Pn4-BuA2-BrAc: Der Komplex des Proteins der äußeren Membran
(N. meningitidis, OMPC, 4,34 mg/ml) (5 ml) wurde in einem 80-Ti-Rotor
bei 43.000 UpM (43 K) 2 h lang bei 4°C zentrifugiert. Eine Thiolierungsmischung
wurde hergestellt durch Lösen
von 85 mg EDTA (Ethylendiamintetraessigsäure-Dinatriumsalz) und 15 mg
Dithiothreit (DTT) in 10 ml eines Na2B4O7-Puffers, pH 11,09.
50 mg N-Acetylhomocysteinthiolacton wurden zugegeben und dann die
Lösung
durch ein 0,2-pm-Filter filtriert Die Pellets aus der obigen Zentrifugation
wurden mit 5 ml der filtrierten Thiolierungsmischung disloziert
und in einen DOUNCE-Homogenisator überführt und resuspendiert Die resuspendierte Lösung wurde
in ein Zentrifugenröhrchen überführt, mit
einem Septum verschlossen und die Luft durch N2 unter
Verwendung eines FIRESTONE-Ventils ersetzt. Die Reaktionsmischung
wurde 19 h lang stehengelassen und dann in ein Zentrifugenröhrchen überführt, das
mit 1 M KH2PO4 (wäßrig) aufgefüllt und
dann 2 h lang bei 43 K und 4°C
zentrifugiert wurde. Die Überstände wurden
entfernt und die Pellets in 10 ml 0,1 M NaPO4-Puffer,
pH 8, resuspendiert. Diese Lösung
wurde in ein Zentrifugenröhrchen überführt und
dann eine zweite Ultrazentrifugation (2 h, 4°C, 43 K) durchgeführt. Die
Pellets wurden in 11,5 ml der in Abschnitt D hergestellten Pn4-BuA2-BrAc-Lösung
resuspendiert. Ein Ellman-Assay zeigte eine Gesamtmenge von 3,44 μMol SH oder
etwa 158 nMol SH/mg OMPC an. Die resultierende Reaktion (d. h. Pn4-BuA2-BrAc mit thioliertem OMPC) wurde unter
N2 (unter Entgasen) in der N2-Box
66 h lang bei Raumtemperatur stehengelassen.
Die Reaktion wurde
dann wie folgt mit Endgruppen versehen: Eine Lösung, enthaltend 5 mg N-Ethylmaleimid
(NEM) in 1 ml 0,1 M NaPO4-Puffer, pH 8,
wurde durch ein 0,22-μm-Filter
filtriert und der obigen Reaktionsmischung zugegeben und die Lösung 5 h
lang stehengelassen. Dann wurden 0,1 ml N-Acetylcysteamin in 0,4
ml 0,1 M PO4-Puffer, pH 8, durch ein 0,22-μm-Filter
filtriert und diese Lösung
der Reaktion zugegeben und 14,5 h lang stehengelassen.
Die
Reaktionsmischung wurde dann 2 h lang bei 43 K, 4°C zentrifugiert
und das Pellet in 8 ml 1 × TED-Puffer
resuspendiert. Diese Lösung
wurde über
Nacht bei Raumtemperatur stehengelassen und dann 2 h lang bei 43
K, 4°C zentrifugiert.
Das Pellet wurde in 8 ml TED-Puffer resuspendiert und sofort 2 h
lang bei 43 Kund 4°C
erneut zentrifugiert. Das Pellet wurde dann in 10 ml 0,1 M PO4-Puffer, pH 7,0, resuspendiert und 2 h lang
bei 43 K und 4°C
rezentrifugiert. Dieses endgültige
Pellet wurde in 7,5 ml H2O resuspendiert.
Nach dem Stehenlassen über
Nacht bei 4°C
wurde die Suspension 3 Minuten lang bei 1000 UpM zentrifugiert und
der Überstand
als des endgültige
Konjugat entnommen.
-
| Assays: |
Lowry-Protein:
0,920 mg/ml; |
| |
Phenol-Schwefelsäure-Assay:
0,212 mg/ml; |
| |
Ps/PRO
= 0,23; |
| |
SCMHC/Lys
= 0,031; |
| |
SCMC/Lys
= 0,022. |
-
Nach
Verabreichung dieses Konjugats an Mäuse oder afrikanische Meerkatzen
wurden hohe Titer von Anti-Pn4-Ps-Antikörper induziert, wie gemessen
durch einen Pn4-Ps-spezifischen ELISA-Assay.
-
BEISPIEL 28
-
Herstellung des Pn9V-Ps-Zwischenprodukts:
-
- (1) Beschallungshydrolyse: Eine 1,0-g-Portion
Pn9V-Ps-Pulverwunde in 400 ml Satzlösung (0,9% NaCl) unter Rühren bei
Raumtemperatur etwa 4 h lang solubilisiert. Die Lösung wurde
dann in einem Kunststoffbecherglas in einem Eisbad mit einem Branson
Sonifier (Halbzoll-Sonde, Einstellung 8) für ein Intervall von 3 Minuten
beschallt. Die Viskosität
wurde nach diesem Intervall überprüft. Nach
13 Minuten wurde eine weitere einminütige Beschallung durchgeführt, um
eine Endviskosität
von 1,117 Zentistokes zu erhalten. Die hydrolysierte Probe wurde
auf Raumtemperatur gebracht und Natriumacetat-Reagens (19,5 g) bis
zu einer Endkonzentration von 3% (Gew./Vol.) zugegeben.
- (2) Serologische Untersuchung: Eine Isopropanol(IPA)-Fraktionierungspilotstudie
und ein antikörpergesteuerter
Endpunkt-Nephelose-Assay, durchgeführt mit einer 10-ml-Portion
der Probe, zeigten, daß das Pn9V-Ps
zwischen 40–45%
IPA ausfallen würde.
- (3) Erste IPA-Zugabe: Die hydrolysierte Probe [Volumen = 391
ml, aus Schritt 1 oben] wurde durch die Zugabe von 281 ml IPA (tropfenweise
unter Rühren
bei Raumtemperatur zugegeben) auf 41,8% IPA gebracht. Die Probe
wurde 15–30
Minuten lang röhrengelassen
und dann 30 Minuten lang bei 11.000 × g (Beckman JA-10-Rotor; 8.000
UpM; 20°C)
zentrifugiert. Das Abfallpellet wurde mit absolutem EtOH in einem 250-ml-Omnimix-Gefäß trituriert,
dann auf einem 60-ml-Sinterglastrichter gesammelt. Der Niederschlag wurde
direkt auf dem Trichter mit absolutem EtOH, dann Aceton gewaschen
und im Vakuum über
CaCl2 bei Raumtemperatur in Vorbereitung
auf die Analyse getrocknet.
- (4) Zweite IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 41,8% IPA (Volumen = 637 ml, aus Schritt 3 oben] wurde durch
tropfenweise Zugabe von 28,6 ml IPA unter Rühren bei Raumtemperatur auf
44,3% IPA gebracht. Die Probe wurde stehengelassen und zentrifugiert
wie in Schritt 3 oben. Das Pellet wurde trituriert, gesammelt, gewaschen
und getrocknet wie in Schritt 3 oben. Das Pn9V-Ps-Produkt wog 342,2
mg.
- (5) Dialyse und Lyophilisierung: Eine Portion (347 mg) der Pn-Ps-Probe
aus Schritt 4 oben wurde in 139 ml destilliertem H2O
bei Raumtemperatur 4–5
h lang solubilisiert. Die Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch (MG-Ausschluß 12.000;
45 mm) überführt und
unter zwei weiteren Austauschen von destilliertem H2O
25 h lang bei 4°C
gegen destilliertes Wasser dialysiert. Dann wurde die Probe in Lyophilisierungskolben überführt, in
einem Trockeneis:Methanol-Bad rollschichtgefroren und mit einem
Virtis(Freezemobile)-Lyophilisator 2–3 Tage lang lyophilisiert.
Die Ausbeute des Ps-Endprodukts betrug 303,5 mg. Das Endprodukt
besaß einen
Kd = 0,60.
- (6) Dritte IPA-Zugabe und Produktgewinnung: Die Überstandsflüssigkeit
von 44,3% IPA [Volumen = 655 ml, aus Schritt 4 oben] wurde durch
tropfenweise Zugabe von 30,8 ml IPA unter Rühren bei Raumtemperatur auf
46,8% IPA gebracht. Die Probe wurde stehengelassen und zentrifugiert
wie in Schritt 3 oben. Das Pellet wurde trituriert, gesammelt, gewaschen
und getrocknet wie in Schritt 3 oben. Das Pn9V-Ps-Produkt wog 410,8
mg.
- (7) Dialyse und Lyophilisierung: Eine Portion (420,4 mg) der
Pn-Ps-Probe aus Schritt 6 oben wurde in 168 ml destilliertem H2O bei Raumtemperatur 4–5 h lang solubilisiert. Die
Lösung
(2,5 mg/ml) wurde in einen Dialyseschlauch (MG-Ausschluß 12.000;
45 mm) überführt und
unter zwei weiteren Austauschen von destilliertem H2O
25 h lang bei 4°C
gegen destilliertes H2O dialysiert. Dann
wurde die Probe in Lyophilisierungskolben überführt, in einem Trockeneis:Methanol-Bad
rollschichtgefroren und mit einem Virtis(Freezemobile)-Lyophilisator
2–3 Tage
lang lyophilisiert. Die Ausbeute des Ps-Endprodukts betrug 342,5
mg. Das Endprodukt besaß einen
Kd = 0,65.
- (8) Für
Fachleute ist offensichtlich, daß die Produkte in den Schritten
4 und 6 mit einer größeren Zugabe von
IPA hätten
gemeinsam gewonnen werden können,
dann dialysiert und lyophilisiert als ein einziges Produkt mit den
analytischen Eigenschaften des gewichteten Mittels der Eigenschaften
der individuellen Subfraktionen. Für Fachleute sollte auch offensichtlich
sein, daß Pn1-Ps
oder Pn5-Ps auf dieselbe Weise wie Pn9V-Ps oder Pn4-Ps, wie hier
offenbart, behandelt werden könnten.
-
BEISPIEL 29
-
Konjugation von Pn9V-Ps mit OMPC:
-
Pn9V-Ps,
hergestellt nach Beispiel 28, wird auf dieselbe Weise konjugiert
wie Pn4-Ps, wie in Beispiel 27 gezeigt.
-
BEISPIEL 30
-
Quantitative Bestimmung von Acetat in
Pn9V/18C-Ps und Pyruvat in Pn4-Ps:
-
Es
wurde ein Verfahren entwickelt, um die Retention von O-Pyruvatgruppen
in Pn4-Ps und O-Acetatgruppen in Pn9V-Ps und Pn18C-Ps während der
Behandlung der Pneumokokken(Pn)-Kapselpolysaccharide (Ps) quantitativ
zu bestimmen. Die O-Acetyl- oder O-Pyruvatgruppen werden zuerst
durch Hydrolyse freigesetzt, dann wird das Acetat und Pyruvat in
dem PnPs-Hydrolysat identifiziert und unter Verwendung von Hochleistungs-Anionenaustauschchromatographie,
gekoppelt mit unterdrückter
Leitfähigkeit,
quantitativ bestimmt.
-
Nach
diesem Verfahren wurden Proben von behandelten und unbehandelten
Pn4, Pn9V und Pn18C analysiert. Die vorläufigen Ergebnisse zeigten ein
Molverhältnis
von etwa 1:1 und 0,8:1 von Pyruvat zu jeder sich wiederholenden
Ps-Einheit für
unbehandeltes bzw. größensortiertes
Pn4. Die Molverhältnisse
von Acetat zu jeder sich wiederholenden Ps-Einheit in Pn18C-Ps wurden
als 1:1 und 0,8:1 für
unbehandelte bzw. größensortierte
Proben; und 1,7:1 und 1,5:1 für
unbehandeltes bzw. größensortiertes
Pn9V befunden. Eine Probe des wäßrigen rohen
Pn18C-Ps-OMPC-Konjugats wurde ebenfalls hinsichtlich des Molverhältnisses
von O-Acetat zu jeder sich wiederholenden Ps-Einheit analysiert
und dieses als etwa 0,5:1 befunden.
-
In
der Literatur wurde berichtet, daß die Pyruvatgruppe eine wirkungsvolle
Immundeterminante bei Kapselpolysacchariden vom Typ 4 ist und daß deren
Entfernung zu deutlichen Änderungen
der immunologischen Spezifität
führt [Heidelberger,
M., Dudman, W. F., und Nimmich, W., 'Immunochemical relationships of certain
capsular polysaccharides of Klebsiella, pneumococci, and Rhizobia. 'J. Immunol., 104:
1321–1328, (1970);
Higginbotham, J. D., Heidelberger, M., und Gutschlich, E., 'Degradation of a
pneumococcal type-specific polysaccharide with exposure of group-specificity. 'Proc. Natl. Acad.
Sci. USA, 67: 138–142,
(1970)). Gleichermaßen
eliminierte die Entfernung der O-Acetatgruppe bei dem Typ Pn18C-Ps-Polysaccharid
dessen immunologische Spezifität.
[Estrada-Parra, S., und Heidelberger, M., 'The specific polysaccharide of type
XVIII pneumococcus. 'Biochemistry,
2: 1288–1294,
(1963)). Es war deshalb essentiell, eine quantitative Methode zur Bestimmung
des Acetats und Pyruvats in Pn18C-Ps und Pn4 zu entwickeln. O-Acetylgruppen
in Pn9V könnten auch
eine wichtige Rolle bei der immunologischen Struktur von Pn9V spielen,
da de-O-acetyliertes Pn9V keine antigene Reaktivität aufwies,
wie bestimmt durch Geschwindigkeitsnephelometriemessung.
-
Wir
fanden heraus, daß O-Acetat
aus Pn9V- und Pn18C-Ps unter alkalischen Bedingungen (pH 11) bei
4°C leicht
freigesetzt wird und O-Pyruvat aus Pn4 nach Erwärmung auf 65°C leicht
freigesetzt wird. Wir stellten fest, daß Acetat und Pyruvat aus einer
hydrolysierten PnPs-Probe unter Verwendung einer OmniPac PAX500-Säule bei
einer Durchflußrate
von 1 mVMin. mit 0,98 mM NaOH und 2% MeOH als mobiler Phase abgetrennt
werden können.
Der Nach-weis erfolgte durch Nachweis unterdrückter Leitfähigkeit unter Verwendung von
25 mM H2SO4 als
Regenerationsmittel bei einer Durchflußrate von 10 ml/Min. Optimale
Hydrolysebedingungen für
die quantitative HPLC-Analyse von O-Acetat und O-Pyruvat aus Pn18C-Ps, 9V bzw. Pn4 werden
in diesem Beispiel offenbart.
-
Meßausrüstung
-
Ein
Dionex BioLC war mit einer OmniPac PAX-500-Überwachungs- und Analysesäule (4,6 × 250 mm) ausgerüstet. Der
Nachweis unterdrückter
Leitfähigkeit
erfolgte unter Verwendung von 25 mN Schwefelsäure als Regenerationsmittel.
Die Durchflußrate
wurde mit einer Dionex-Autoregenerationseinheit auf 10 ml/Min. eingestellt.
Die mobile Phase und das Gradientenprogramm für die Abtrennung von Acetat
und Pyruvat aus einer Probe von hydrolysiertem PnPs sind in der
folgenden Tabelle aufgelistet:
- Puffer 1 – 1 mM Natriumhydroxid
- Puffer 2 – 100%
Methanal
- Puffer 3 – 200
mM Natriumhydroxid
- Puffer 4 – Wasser
-
| Zeit |
%
Puffer 1 |
%
Puffer 2 |
%
Puffer 3 |
%
Puffer 4 |
Durchfluß (ml/Min.) |
| 0 |
98 |
2 |
0 |
0 |
1 |
| 12,5 |
98 |
2 |
0 |
0 |
1 |
| 12,6 |
58 |
2 |
0 |
40 |
1,5 |
| 20,0 |
58 |
2 |
0 |
40 |
1,5 |
| 20,1 |
98 |
2 |
0 |
0 |
1,5 |
| 30,0 |
98 |
2 |
0 |
0 |
1,5 |
| 30,1 |
98 |
2 |
0 |
0 |
1 |
| 50,0 |
98 |
2 |
0 |
0 |
1 |
-
Unter
Einsatz dieser Bedingungen und einer Detektorempfindlichkeit von
3 μSiemens
können
jeweils 4 nMol Acetat und Pyruvat, die bei Retentionszeiten von
etwa 5,2 bzw. 9,5 Minuten eluieren, ohne weiteres nachgewiesen werden.
-
Probenvorbereitung
-
Gereinigte
PnPs-Proben von Merck Manufacturing Division wurden einer Karl-Fisher-Titration
unter Verwendung eines volumetrischen Aquastar V3000-Feuchtigkeitstitrators
unterworfen, um den Restgehalt an H2O zu
bestimmen, und dann in Milli-Q-H2O in einer
Konzentration von 1,0 mg Trockengewicht pro ml gelöst. Proben
(100 μg/ml)
wurden in 2 mM NaOH 16 h lang bei Raumtemperatur behandelt, um O-Acetat
aus Pn9V-Ps- und Pn18C-Ps-Proben zu entfernen. Die Pn4-Ps-Proben
wurden in 1 mM HCl bei 65°C
16 h lang zur Entfernung von O-Pyruvat aus Pn4 hydrolysiert. Die
Proben von größensortiertem
Pn9V- und Pn18C-Ps und des wäßrigen rohen
Pn18C-Ps-OMPC-Konjugats wurden ebenfalls Analysen hinsichtlich der
Monosaccharidzusammensetzung unterworfen durch Hoch-pH-Anionaustauschchromatographie
und gepulsten amperometrischen Nachweis. Die Analyse der Monosaccharidzusammensetzung
erfolgte, um die korrekte Konzentration von PnPs in den größensortierten
Proben und den Proben des wäßrigen rohen
Konjugats zu erhalten. Acetat-, Pyruvat- und N-Acetylmannosamin-Standards
wurden in Milli-Q-H2O in der Konzentration
von 200 nMol/ml gelöst.
-
Hydrolyse von Proben und Standards
-
Die
De-O-acetylierung von Pn18C-Ps wurde durch Behandlung von Pn18C-Ps
bei vier NaOH-Konzentrationen
(1, 2, 5 und 50 mM) bei verschiedenen Temperaturen (4, 25, 45 und
65°C) und
verschiedenen Zeiten (3, 5 und 16 h) untersucht. Standardlösungen von
Acetat, Pyruvat und N-Acetylmannosamin
waren ebenfalls in der Studie eingeschlossen, um festzustellen,
ob die für
die De-O-acetylierung erforderlichen Bedingungen auch in einem Abbau
von Acetat/Pyruvat oder dem Verlust von N-Acetylgruppen resultieren
würden.
-
Die
Entfernung von Pyruvat aus Pn4 wurde nach der Behandlung mit entweder
Natriumhydroxid (50 mM/100°C/16
h) oder Salzsäure
bei verschiedenen Konzentrationen (1, 10, 100 mM), Zeiten (3, 5
und 16 h) und Temperaturen (65, 85 und 100°C) untersucht.
-
Geschwindigkeitsnephelometrie
-
Die
geschwindigkeitsnephelometrische Aktivität von Pn9V-Ps, Pn18C-Ps und
Pn4-Ps vor und nach der De-O-acetylierung oder De-O-pyruvylierung
wurde gemessen. Die Proben wurden auf 1, 1,5, 2 und 2,5 μg/ml verdünnt.
-
Hochleistungs-Größenausschlußchromatographie (HPSEC)
-
Die
HPSEC von Pn9V-Ps, Pn18C-Ps und Pn4 vor und nach De-O-acetylierung
oder De-O-pyruvylierung wurde gemessen. Eine 7,5 × 600 mm
TSK G6000PW-Säule,
ausgerüstet
mit einem Durchflußbegrenzer, wurde
auf 50°C
bei 5,5 MPa–6,9
MPa (800–1000
psi) erwärmt
und mit 0,2 M Natriumacetat mit 0,3 ml/Min. äquilibriert. Eine 60-μg-Probe (1
mg/ml) wurde in die Säule
injiziert und mit der mobilen Phase mit 0,3 ml/Min. eluiert. Das
Säuleneluat
wurde mit einer nach der Säule
erfolgenden Zugabe von 0,5 M NaOH bei einer Flußrate von 0,5 ml/Min. gemischt
und mit einem gepulsten amperometrischen Dionex-Monitor überwacht und der Kd gemessen.
-
Assay-Empfindlichkeit und Linearität
-
Die
Linearität
und Empfindlichkeit des Detektors wurden bei 3 μSiemens sowohl für Pyruvat
als auch für
Acetat bestimmt. Pyruvat und Acetat waren bei einer unteren Grenze
von 0,125 nMol nachweisbar. Die Detektorantwort für beide
Komponenten war bis 2 nMol linear mit Korrelationskoeffizienten
von 0,9999 und 0,9992 für
Pyruvat bzw. Acetat.
-
Optimierung der Entfernung von O-Acetat
aus Pn18C-Ps
-
Vorstudien
des Zeitverlaufs der Hydrolyse von Pn18C-Ps zeigten die Labilität der O-Acetylgruppe
gegenüber
alkalischer Hydrolyse bei niedrigen Temperaturen. 2 mM Natriumhydroxid
war ausreichend, um Pn18C-Ps nach einer 16-ständigen Inkubation bei 4°C vollständig zu
de-O-acetylieren. Behandlungen bei höheren Temperaturen (> 25°C) wurden befunden, N-Acetat
aus N-Acetylmannosamin freizusetzen, was die Messung des Pn9V-O-Acetats
stören
würde.
Die optimale Hydrolysebedingung zur Entfernung von O-Acetat aus
PnPs wurde als 16 h bei 4°C
befunden. Es wurde festgestellt, daß weniger als 1% Acetat aus
einem Standard von N-Acetylmannosamin entfernt wurden, der 16 h
lang bei Raumtemperatur mit 2 mM NaOH behandelt worden war.
-
Optimierung der Entfernung von O-Pyruvat
aus Pn4
-
Hydrolysestudien
von Pn4 wurden anfangs unter Anwendung von Natriumhydroxid-Hydrolyse
unternommen. Es wurde schnell festgestellt, daß bei Verwendung von Natriumhydroxid
sehr wenig Pyruvat gewonnen wurde. Anfängliche Kontrollstudien demonstrierten,
daß Pyruvat
von Pn4 in H2O bei 100°C abgespalten wurde. Mit dieser
Information wurde beschlossen, die Optimierungsstudien für die O-Pyruvatfreisetzung
aus Pn4 unter Anwendung von HCl-Hydrolyse bei erhöhten Temperaturen
durchzuführen.
Es fand ein gewisser Abbau des Pyruvats bei höheren Temperaturen statt. Dies
kann mit einer Probe eines Pyruvatstandards demonstriert werden,
der unter denselben Bedingungen wie Pn4 hydrolysiert wurde. Eine
maximale Gewinnung von Pyruvat fand statt, wenn die Hydrolyse in
1 mM HCl für
16 h bei 65°C
durchgeführt
wurde.
-
Analyse von O-Acetat und O-Pyruvat in
PnPs-Proben
-
Verschiedene
Proben, die Ausgangs-PnPs, größensortierte
PnPs und ein Pn18C-Ps-OMPC-Konjugat repräsentierten, wurden auf O-Acetat/Pyruvat
analysiert durch das oben beschriebene HPLC-Verfahren nach einer
Hydrolyse in 2 mM NaOH bei Raumtemperatur zur Freisetzung von O-Acetat
in Pn9V-Ps/18C oder in 1 mM HCl bei 65°C zur Freisetzung von O-Pyruvat
in Pn4-Ps. Die Ergebnisse dieser Untersuchung sind im folgenden
dargestellt:
| Probe | Verhältnis von
Pyruvat/Acetat zu jeder sich wiederholenden Ps-Einheit |
| Pn4,
Probe 1 | 1,0 |
| Pn4,
Probe 2 | 0,8 |
| Pn9V
Probe 1 | 1,7 |
| Pn9V,
Probe 2 | 1,5 |
| Pn18C-Ps,
Probe 1 | 1,0 |
| Pn18C-Ps,
Probe 2 | 0,8 |
| Pn18C-Ps-OMPC,
wäßriges Rohmaterial | 0,5 |
-
Die
Ergebnisse zeigen, daß die
Retention der Seitengruppen in den größensortierten PnPs etwa 90% für Pn9V-Ps
und 80% für
Pn4 und 18C betrug. Die Retention von O-Acetat im wäßrigen rohen Pn18C-Ps-OMPC-Konjugat wurde
als etwa 50% befunden. Die theoretischen Werte für Pn18C-Ps und Pn4 sind 1 Mol
Acetat oder Pyruvat pro Mol sich wiederholender Ps-Einheit und für Pn9V beträgt das Verhältnis 2:1.
[Jansson, P.-E., Lindberg, B., und Lindquist, U. 'Structural studies
of the capsular polysaccharides from Streptococcus pneumoniae Type
4. 'Carbohyd. Res.,
95: 73–80,
(1981). Lugrowski, C., und Jennings, H. J. 'Structural determination of the capsular
polysaccharide of Streptococcus pneumoniae Type 18C. 'Carbohyd. Res. 131:
119–129,
(1984). Perry, M. B., Daoust, V., und Carlos, D. J. 'The specific capsular
polysaccharide of Streptococcus pneumoniae Type 9V. 'Can. J. Biochem.
59: 524–533,
(1981)]. Die geringere Retention von O-Acetat, die bei dem Pn18C-Ps-OMPC-Konjugat
festgestellt wurde, ist aufgrund der Hydrolyseempfindlichkeit von
O-Acetylgruppen unter alkalischen Bedingungen bei niedrigen Temperaturen
zu erwarten.
-
Die
geschwindigkeitsnephelometrische Aktivität von Proben von Pn4-, Pn9V-
und Pn18C-Ps und de-O-pyruvylierter
und de-O-acetylierter Proben wurde gemessen. Die Ergebnisse zeigten,
daß die
Nephelose-Aktivität nach der
Entfernung dieser Seitengruppen vollständig verlorenging, trotz des
Kd der unbehandelten Proben. Die Kd-Werte wurden durch das oben
beschriebene HPSEC-Verfahren erhalten. Der Kd von Pn4 nach De-O-pyruvylierung
durch milde Säurehydrolyse
wurde jedoch von 0,60 auf 0,68 erhöht und das Erscheinen in der
Nähe des
Salzvolumens. Die Antigenizltätsdaten
für Pn4-
und Pn18C-Ps stützen
die Arbeit anderer Forscher bezüglich
der Bedeutung dieser Seitengruppen bei der immunologischen Reaktivität von Pneumokokken-Polysaccharid.
Die Ergebnisse für
Pn9V legen nahe, daß neben
Glucuronsäure
die O-Acetylgruppen dieses Moleküls
ebenfalls wichtige immunologische Determinanten sind.
-
So
wurde nach dieser Methode ein schnelles, empfindliches Verfahren
zur quantitativen Analyse von O-Pyruvylketal
in Pn4 und O-Acetat in Pn9V- und Pn18C-Ps entwickelt. Dieses Verfahren
ist wertvoll bei der Festlegung des richtigen Verfahrens zur Größensortierung
und Konjugation von Pn4-, Pn9V- und Pn18C-Ps, um die antigene Struktur
dieser Polysaccharide aufrechtzuerhalten.
-
BEISPIEL 31
-
Isolierung von Pn6B-Ps-MIEP-Konjugat:
-
- 1. Zwei Konjugat-Reaktionsmischungsproben (wobei
eine eine gegen H2O dialysierte Probe der
anderen darstellt) wurden bis zur Verwendung bei 3–8°C gelagert.
- 2. 0,2 M MOPS-Puffer, pH 7,2, wurde den Proben zugegeben, um
eine Endkonzentration von etwa 7 mM zu erhalten. Festes GuHCl wurde
der Probe zugegeben, um eine Endkonzentration von 4,2 M zu erreichen (Anmerkung:
1,42 g GuHCl/ml Probe sollten zugegeben werden, um die Volumenerhöhung aufgrund
der Zugabe des festen GuHCl zu kompensieren. Gleichermaßen sollte
die Pufferzugabe eingestellt werden, um die Volumenerhöhung auszugleichen,
so daß die
Probenzusammensetzung der Zusammensetzung des Säulenelutionsmittels näher kommt.
Alternativ könnte
die Probe vor der Chromatographie gegen das Säulenelutionsmittel dialysiert
werden.)
- 3. 2,8 ml Probe (enthaltend etwa 1 mg Protein auf der Basis
eines Lowry-Protein-Assays) wurden in eine 2,6 × 96 cm-Säule von Sephacryl S-1000, äquilibriert
in 10 mM MOPS, pH 7,2, 6 M GuHCl, bei einer Durchflußrate von
0,6 ml/Min. injiziert. Der Säulenausfluß wunde
kontinuierlich bei 280 nm (Perkin Elmer LC 235-Diodenanordnungsdetektor) überwacht
und 3-ml-Fraktionen gesammelt.
- 4. Die Proteinverteilung basierte auf A280 (sowie Spektren)
und die Pn6B-Ps-Verteilung auf einem für Pn6B-Ps spezifischen RIA-Assay.
Auf der Basis der Elutionspositionen von Pn6B-Ps-BrAc allein und
in physikalischen Mischungen mit unaktiviertem MIEP wurden Pools
von Fraktionen, die sowohl Ps als auch Protein enthielten, hergestellt
und diejenigen, welche abweichend von den Positionen eluierten,
die für
das Pn6B-Ps-BrAc beobachtet wurden, wurden als mutmaßliches
Pn6B-Ps-MIEP-Konjugat bezeichnet
- 5. Pools wurden durch Ultrafiltration unter Verwendung einer
YM-30-Membran konzentriert und unter Verwendung von Milli-Q-H2O diafiltriert. Protein- und Pn6B-Ps-Gehalt
wurden aus quantitativen Untersuchungen der Zusammensetzung bestimmt
Die Anwesenheit von SCMHC wurde durch Aminosäureanalyse nachgewiesen.
-
BEISPIEL 32
-
Direkter RIA-Assay auf Pneumokokken-Polysaccharid
Pn18C-Ps
-
Dieser
Assay wird zur quantitativen Bestimmung des Pneumokokken-Polysaccharids
vom Typ 18C eingesetzt. Es ist ein Mehrschichten-Sandwich-RIA-Assay.
Polystyrolperlen werden mit Kaninchen-Anti-Pn18C-Ps beschichtet Die Perlen werden
in einer Probenlösung,
die Pn18C-Ps enthält,
inkubiert. Nach der Inkubation werden die Perlen gewaschen und in
einer zweiten Lösung
erneut inkubiert, die einen Maus-Antikörper gegen Pn18C-OMPC enthält. Nach
dieser Inkubation werden die Perlen gewaschen und ein drittes Mal in
einer Lösung
inkubiert, die ein 125I-Anti-Maus-Ziegen-IgG
enthält
Die Platten werden einmal mehr gewaschen, wonach die Perlen zur
Zählung
in Kunststoffröhrchen überführt werden.
Unbekannte Proben von Pn18C-Ps werden zur quantitativen Bestimmung
mit einer Standardkurve verglichen.
-
Ausrüstung
-
- 1. RIA-Kit: Abbott Labs, Diagnostic Div., Katalog-Nr.
6171-10.
- 2. Qwik Wash System, Abbott Labs, Diagnostic Div.
- 3. Einstellbare Pipetten und Einmal-Pipettenspitzen (Ausführung Eppendorf
digital)
- 4. Gamma-Zähler
(Ausführung
Abbott Autologic).
- 5. 1/4-Inch-Polystyrolperlen mit spiegelnder Oberfläche: Precision
Plastic Ball Co., 3000 N. Cicero Ave., Chicago, Illinois 60641.
-
Reagenzien
-
- 1. New York State Health Services Anti-Pn18C-Ps-Antikörper, Los
R18-44 oder ein Äquivalent.
- 2. Anti-Pn18C-Ps-OMPC-Mausantiseren (Pool 11260-235 oder ein Äquivalent).
- 3. 125I-markierte Anti-Maus-Ziegen-IgG-Antiseren:
NEX 159, New England Nuclear, 549 Albany Street, Boston, MA 02118.
- 4. Inkubationspuffer: RCM8 enthaltend
-
| 1,0%
BSA |
Sigma
A2153 |
| 0,1%
Azid |
Sigma
S2002 |
-
| 8 Teile
fötales
Kalbsserum |
Sigma
F3885 |
| 1 Teil
Ziegenserum |
Sigma
G6767 |
| 1 Teil
Kaninchenserum |
Sigma
R4505 |
| 0,05%
TWEEN 20 |
Sigma
P1379 |
| 0,1%
Azid |
Sigma
S2002 |
- 3. Zugabe einer mit Anti-Pn18C-Ps
beschichteten Perle zu jeder Vertiefung der Platte, die eine Probe
oder einen Standard enthält,
und sanftes Bewegen der Platte, um sicherzustellen, daß alle Perlen
vollständig
mit Puffer bedeckt sind.
- 4. Bedeckung der Platte mit einer haftenden Schutzfolie, die
mit dem RIA-Kit geliefert wurde, und Inkubation der Platte für 6 h bei
Raumtemperatur.
- 5. Waschen der Platte unter Verwendung der Qwik Wash-Apparatur
und entionisierten Wassers.
- 6. Verdünnung
des Anti-18C-Mausantikörpers
1:1000 in Verdünnungsmittel.
- 7. Zugabe von 200 μl
dieser Lösung
zu jeder Vertiefung, die eine Perle enthält.
- 8. Bedeckung der Platte und Inkubation über Nacht bei Raumtemperatur.
- 9. Waschen der Platte unter Verwendung der Qwik Wash-Apparatur
und entionisierten Wassers.
- 10. Verdünnung
von 125I-markiertem Anti-Maus-Ziegenantikörper auf
15000 CpM/10 μl
in Verdünnungsmittel
(≈ 1:160-Verdünnung).
- 11. Zugabe von 20 μl
dieser Lösung
zu jeder Vertiefung, die eine Perle enthält.
- 12. Bedeckung der Platte und Inkubation für 2 h bei 37°C.
- 13. Waschen der Platten unter Verwendung der Qwik Wash-Apparatur
und entionisierten Wassers.
- 14. Überführung der
Perlen in die Kunststoffröhrchen,
die mit dem RIA-Kit geliefert wurden, und Zählung unter Verwendung eines
geeigneten Gamma-Zählers.
-
Berechnungen
-
- 1. Kombination der Doppelmessungen, um ein
Mittel für
jede Probe, jeden Standard und die Inkubationspufferkontrolle zu
erhalten. Subtraktion der Inkubationspufferkontrolle von alten Standards
und Proben.
- 2. Verwendung eines für
statistische Berechnungen ausgerüsteten
Rechners, Eingabe der Daten für
die Standardkurve und Berechnung des Korrelationskoeffizienten und
der Steigung der Linie.
- 3. Verwendung der geeigneten Standardkurve (frei für frei,
Konjugat für
Konjugat), Berechnung der Antwort der Proben und Korrektur hinsichtlich
der Verdünnungen.
-
Dasselbe
oben beschriebene Verfahren ist durch Ersatz der typenspezifischen
Reagenzien auf jede der anderen Pn-Ps-Spezies anwendbar.

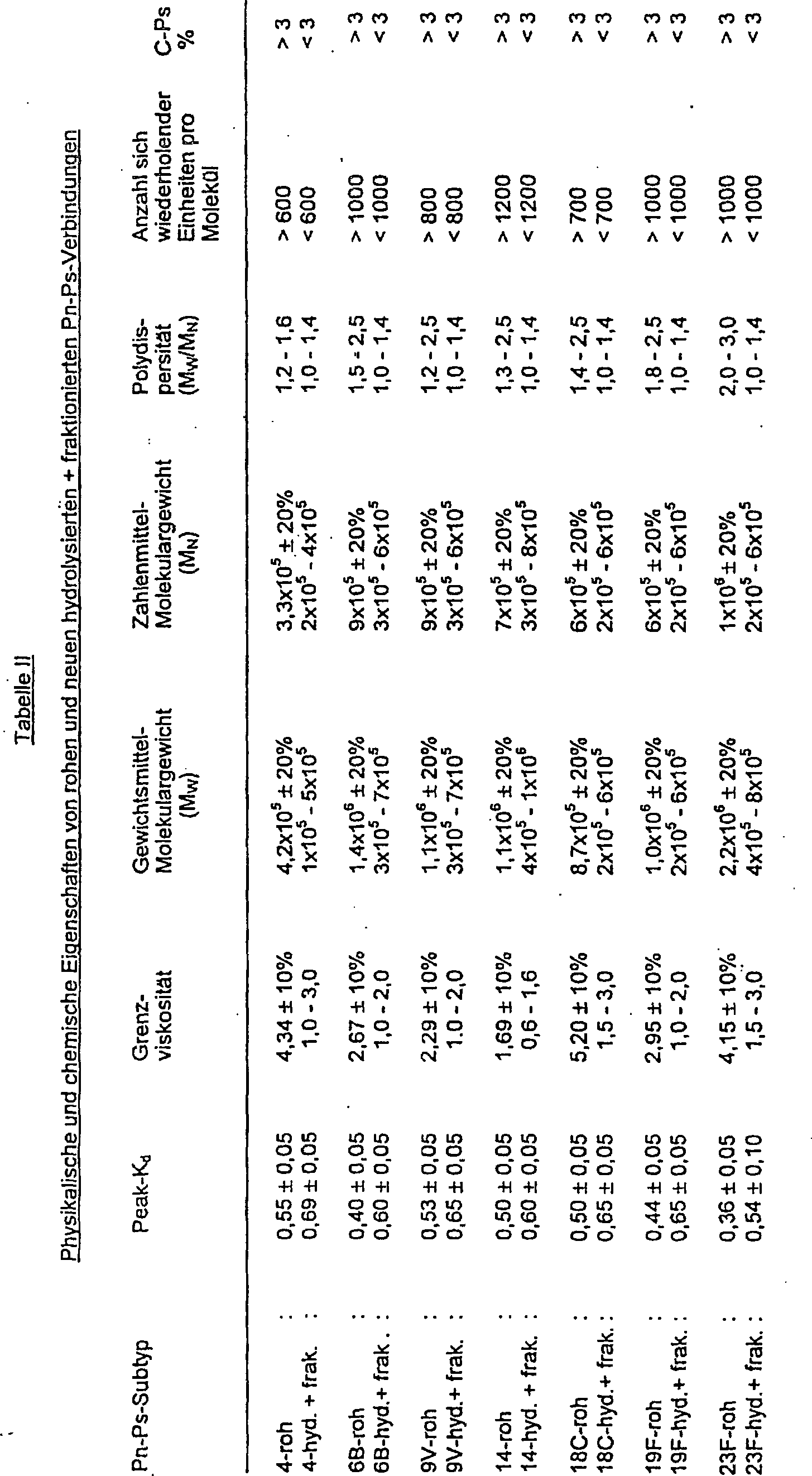
 worin W O oder NH ist, m 0 bis 4 ist, n 0 bis 3 ist und Y CH2, O, S, NR' oder CHCO2H darstellt, wobei R' H oder C1- oder C2-Alkyl ist, so daß, falls Y CH2 ist, m und n nicht beide gleich Null sein können und falls Y O oder S ist, m größer als 1 und n größer als 1 ist; B ist
worin W O oder NH ist, m 0 bis 4 ist, n 0 bis 3 ist und Y CH2, O, S, NR' oder CHCO2H darstellt, wobei R' H oder C1- oder C2-Alkyl ist, so daß, falls Y CH2 ist, m und n nicht beide gleich Null sein können und falls Y O oder S ist, m größer als 1 und n größer als 1 ist; B ist worin q 0 bis 2 ist, Z NH2,
worin q 0 bis 2 ist, Z NH2, COOH oder H darstellt, wobei R' und p wie oben definiert sind und D
COOH oder H darstellt, wobei R' und p wie oben definiert sind und D oder
oder ist.
ist.
 darstellt, wobei Y' NH2 oder NHCOR' ist, und W, p und R' wie oben definiert sind, und E' ist das Transformationsprodukt einer thiophilen Gruppe, die mit einer Thiolgruppe umgesetzt wurde, und wird repräsentiert durch
darstellt, wobei Y' NH2 oder NHCOR' ist, und W, p und R' wie oben definiert sind, und E' ist das Transformationsprodukt einer thiophilen Gruppe, die mit einer Thiolgruppe umgesetzt wurde, und wird repräsentiert durch worin R wie oben definiert ist, und B' ist
worin R wie oben definiert ist, und B' ist oder E' ist
oder E' ist und B' ist
und B' ist worin p 1 bis 3 ist. Ferner sind von den bigenerischen Abstandhaltern A-E-S-B und A'-S-E'-B' die E-S-B- und A'-S-E'-Komponenten bestimmbar und quantifizierbar, wobei diese Identifizierung die Kovalenz der Konjugatbindung reflektiert, welche die Seite des Thioetherschwefels, welche von dem kovalent modifizierten Polysaccharid stammt, mit der Seite des Abstandshalters, welche von dem funktionalisierten Protein stammt, verknüpft.
worin p 1 bis 3 ist. Ferner sind von den bigenerischen Abstandhaltern A-E-S-B und A'-S-E'-B' die E-S-B- und A'-S-E'-Komponenten bestimmbar und quantifizierbar, wobei diese Identifizierung die Kovalenz der Konjugatbindung reflektiert, welche die Seite des Thioetherschwefels, welche von dem kovalent modifizierten Polysaccharid stammt, mit der Seite des Abstandshalters, welche von dem funktionalisierten Protein stammt, verknüpft.


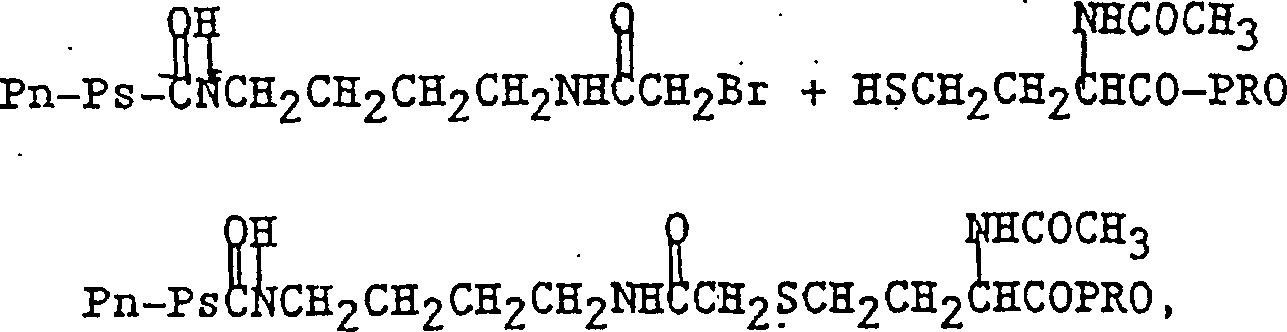 (worin Y'' ein C2-C8-Alkylrest ist), worin ein amino-derivatisiertes Polysaccharid, das mit aktivierter Maleimidosäure umgesetzt wurde, mit einem carboxy-aktivierten Protein, das mit einem Aminothiol aminiert wurde, zur Bildung eines Konjugats umgesetzt wird.
(worin Y'' ein C2-C8-Alkylrest ist), worin ein amino-derivatisiertes Polysaccharid, das mit aktivierter Maleimidosäure umgesetzt wurde, mit einem carboxy-aktivierten Protein, das mit einem Aminothiol aminiert wurde, zur Bildung eines Konjugats umgesetzt wird.
 für Verknüpfungen über die Polysaccharid-Hydroxylgruppen, oder
für Verknüpfungen über die Polysaccharid-Hydroxylgruppen, oder im Falle von Polysacchariden, welche Carbonsäuregruppen tragen.
im Falle von Polysacchariden, welche Carbonsäuregruppen tragen.
 resultiert in der Freisetzung der Aminodicarbonsäure,
resultiert in der Freisetzung der Aminodicarbonsäure, und die Hydrolyse von
und die Hydrolyse von resultiert in der Freisetzung von S-Carboxymethylcysteamin, H2NCH2CH2SCH2CO2H, durch Spaltung des Ps-A-E-S-B-PRO-Moleküls an Peptidverknüpfungen und anderen hydrolytisch instabilen Bindungen. Chromatographieverfahren, z. B. diejenigen von Spackman, Moore und Stein, können dann günstig angewandt und das Verhältnis der Aminosäurebestandteile bestimmt werden.
resultiert in der Freisetzung von S-Carboxymethylcysteamin, H2NCH2CH2SCH2CO2H, durch Spaltung des Ps-A-E-S-B-PRO-Moleküls an Peptidverknüpfungen und anderen hydrolytisch instabilen Bindungen. Chromatographieverfahren, z. B. diejenigen von Spackman, Moore und Stein, können dann günstig angewandt und das Verhältnis der Aminosäurebestandteile bestimmt werden.

