-
Hintergrund
der Erfindung
-
Die
vorliegende Erfindung betrifft ein neues Verfahren für die Synthese
von Amphetamin, Methamphetamin und verwandten Verbindungen aus Derivaten
von Phenylpropanolamin-Säureadditionssalzen.
Dieses neue Verfahren, das angewendet wird, um d-Amphetamin zu produzieren,
weist gegenüber
d-Amphetamin-Produktionswegen
des Standes der Technik mehrere Vorteile auf: kürzere Zykluszeiten, weniger
arbeitsaufwändige
Schritte und eine bessere chemische Hygiene. Gewisse Kombinationen
von pharmazeutisch annehmbaren Salzen von d,l-Amphetamin und d-Amphetamin
sind bei der Behandlung von Aufmerksamkeitsdefizit-Störungen nützlich.
-
Es
sind viele Verfahren zur Herstellung von Amphetamin und verwandten
Verbindungen im Stand der Technik bekannt, einschließlich der
kommerziell verwendeten Leukart-Wallach-Reaktion für die Produktion
von racemischem Amphetamin aus Phenylaceton. Zum Beispiel wird in
einem kommerziellen Verfahren Phenylaceton mit Formamid und Ameisensäure unter
Bildung von (±)-N-Formylamphetamin
(racemischem N-Formylamphetamin) umgesetzt. Das racemische N-Formylamphetamin
wird dann mit Schwefelsäure
hydrolysiert, die Lösung
wird basisch gemacht, und das resultierende d,l-Amphetamin ((±)-Amphetamin; racemische
Amphetamin) wird mit einer Gesamtausbeute von etwa 60% destilliert.
-
Bei
den illegalen Synthesen von Amphetamin und verwandten Verbindungen,
wie jenen, die man bei Internet-Suchen findet, werden Phenylpropanolamin
und Pseudoephedrin, die aus rezeptfreien Husten- und Schnupfenprodukten
isoliert werden, in Amphetamin bzw. Methamphetamin überführt (siehe
zum Beispiel Otto Snow, Amphetamine Synthesis (Thoth Press: Spring
Hill, Florida, 1998); http://www.hyperreal.org/drugs/synthesis/meth.synth.;
oder http://hive.lycaeum.org/book-store.htm/). Unter Befolgen eines
der Verfahren, die bei der illegalen Herstellung von Amphetamin
und verwandten Verbindungen verwendet werden, wurde d,l-Norephedrin
mit Iodwasserstoffsäure
und rotem Phosphor refluxiert, wodurch man eine Mischung von Amphetamin und
einer Verbindung, von der angenommen wird, dass sie eine Bis-Verbindung,
1-Phenyl-2-(phenylisopropyl)aminopropan,
ist, zu gleichen Teilen erhielt. Mittels eines weiteren Verfahrens
ergab das Erwärmen
von Norephedrin mit Thionylchlorid bei Rückflusstemperatur, gefolgt
von katalytischer Hydrierung des resultierenden 2-Amino-1-chlor-1-phenylpropan-Hydrochlorids
Amphetamin. Um die Gefahren des Arbeitens mit Thionylchlorid, Iodwasserstoffsäure und
rotem Phosphor zu vermeiden, war ein anderer Weg wünschenswert.
Die Überführung der
Hydroxylgruppe von Phenylpropanolamin in einen benzylischen Acyloxyester,
gefolgt von der Entfernung durch Hydrogenolyse, das Verfahren der
vorliegenden Erfindung, wurde untersucht, und es wurde gefunden,
dass es ein guter Weg war. Diese drei getrennten Synthesewege sind
in den Beispielen von Schema 1 zusammengefasst, wobei ein Verfahren
der Erfindung als unterer Reaktionsweg veranschaulicht ist. In diesem
Schema wird Amphetamin nur zur Erläuterung verwendet, diese Synthesewege
sind auf verwandte Verbindungen mit für den Fachmann offensichtlichen
Substitutionsmustern anwendbar.
-
-
Derzeit
wird Dextroamphetamin durch ein langwieriges, arbeitsaufwändiges Verfahren
aus racemischem Amphetamin erhalten. Es wird über eine Tartratsalz-Auftrennung, gefolgt
von einer Alkalisierung und Destillation, mit 23% Ausbeute aus racemischem
Amphetamin erhalten. In dem Tartratsalz-Auftrennungsschritt wird
eine heiße
Lösung
von 37%-iger Salzsäure,
Methanol, Weinsäure
und dem racemischen Amphetamin aus einem Reaktor in Edelstahlgefäße abgelassen,
und man lässt
die heiße
Mischung ungestört
16 Stunden abkühlen,
während
hauptsächlich
das d-Amphetamintartrat-Salz kristallisiert. Das Lösungsmittel
wird dann von jedem der Edelstahlgefäße abdekantiert, und das gewonnene
d-Amphetamintartrat-Salz wird von Hand in eine Zentrifuge übergeführt, wo
das Salz trockengeschleudert, wieder mit Methanol aufgeschlämmt und
wieder trockengeschleudert wird. Der Tartrat-Auftrennungsschritt
wird dann wiederholt, bis das erhaltene Salz die gewünschten
Spezifikationen von Schmelzpunkt und optischer Drehung erfüllt.
-
Unter
Verwendung des Verfahrens der Erfindung kann Dextroamphetamin (S-(+)-Amphetamin) stereospezifisch
aus einem Phenylpropanolamin mit der S-Konfiguration am Kohlenstoff, der die
Aminogruppe trägt,
z.B. 1R,2S-(–)-Norephedrin oder
1S,2S-(+)-Norpseudoephedrin, hergestellt werden (die erythro-Form von Phenylpropanolamin
ist Norephedrin, und die threo-Form ist Norpseudoephedrin). Im Verfahren
der Erfindung werden die ansonsten höheren Kosten der geeigneten
Phenylpropanolamin-Diastereomere, die für die Herstellung von Dextroamphetamin
nützlich
sind, durch die kürzeren
Zykluszeiten, ein weniger arbeitsaufwändiges Verfahren und eine bessere
chemische Hygiene aufgewogen.
-
Zusammenfassung
der Erfindung
-
Das
Verfahren umfasst die Esterbildung und dann Entfernung der benzylischen
Acyloxygruppe durch katalytische Hydrierung oder katalytische Transferhydrierung.
Wie oben angemerkt, weist das Verfahren, wenn es auf die Produktion
von d-Amphetamin angewendet wird, mehrere Vorteile gegenüber derzeitigen
d-Amphetamin-Produktionswegen
auf: kürzere
Zykluszeiten, weniger arbeitsaufwändige Schritte und eine bessere
chemische Hygiene. Eine weitere Optimierung von Ausbeuten und Betriebszykluszeiten
unter Verwendung von dem Fachmann bekannten Optimierungsverfahren
würden
diese Vorteile nur steigern.
-
Das
allgemeine Verfahren ist im nachstehenden Schema 2 gezeigt.
-
-
In
Schema 2 ist R1 Wasserstoff oder eine Niederalkylgruppe;
ist
jedes R2 unabhängig ein Wasserstoff, ein Halogen,
eine Niederalkylgruppe, eine Niederalkoxygruppe, eine mit 1 bis
5 Halogen substituierte Niederalkylgruppe, eine mit 1–5 Halogen
substituierte Niederalkoxygruppe oder stellen beide R2 zusammen,
wenn sie an benachbarten Kohlenstoffen vorliegen, eine -O(CH2)xO-Gruppe dar,
worin x für
1 bis 4 steht, wodurch sie eine Ringstruktur bilden, die mit der
Phenylgruppe kondensiert ist;
ist R3 eine
(C1-C8)-Alkylgruppe,
eine (C1-C12)-Aralkylgruppe,
eine (C1-C12)-Alkarylgruppe oder
eine Phenylgruppe, jede gegebenenfalls mit 1 bis 5 Substituenten
substituiert, die aus Halogen, Hydroxy oder (C1-C6)-Alkyl ausgewählt sind; und
ist HX ein Äquivalent
einer organischen oder anorganischen Säure; bevorzugte Säuren umfassen
Fluorwasserstoffsäure,
Chlorwasserstoffsäure,
Bromwasserstoffsäure,
Iodwasserstoffsäure,
Schwefelsäure,
Phosphorsäure,
Salpetersäure,
Ameisensäure,
Essigsäure,
Propionsäure
und andere Carbonsäuren,
wie Benzoesäure, Weinsäure, Bernsteinsäure, Asparaginsäure, Zuckersäure, Oxalsäure, Äpfelsäure und
dergleichen.
-
In
Schritt A wird das Phenylpropanolaminsalz-Ausgangsmaterial der Formel
II mit einem Acylierungsmittel, in diesem Beispiel (R3CO)2O in R3CO2H, in einem Lösungsmittel bei erhöhter Temperatur
acyliert, um das entsprechende acylierte Phenylpropanolaminsalz
der Formel III zu bilden. In Schritt B wird das acylierte Phenylpropanolaminsalz
der Formel III unter Verwendung von katalytischer Hydrierung oder
katalytischer Transferhydrierung hydriert, wodurch man eine Verbindung
der Formel I erhält.
-
Für einen
direkten Weg zu Dextroamphetamin weisen sowohl 1R,2S-(–)-Norephedrin als auch 1S,2S-(+)-Norpseudoephedrin
die richtige sterische Konfiguration am Kohlenstoff auf, der die
Aminogruppe trägt,
die erforderlich ist, um d-Amphetamin [S-(+)-Amphetamin] zu erzeugen,
wie in Schema 3 gezeigt. 1R,2S-(–)-Norephedrin ist allgemein
im Handel erhältlich.
Dieses gleiche Verfahren erzeugt d-Methamphetamin ausgehend von
entweder 1R,2S-(–)-Ephedrin
oder 1S,2S-(+)-Pseudoephedrin.
-
-
Detaillierte
Beschreibung der Erfindung
-
Definitionen
der verwendeten Ausdrücke
und Konventionen
-
Den
Ausdrücken,
die nicht speziell hierin definiert sind, sollten die Bedeutungen
zugewiesen werden, die ihnen der Fachmann im Licht der Offenbarung
und des Zusammenhangs zuweisen würde.
Wie in der Beschreibung und den beigefügten Ansprüchen verwendet, weisen jedoch,
falls nichts Gegenteiliges angeführt ist,
die folgenden Ausdrücke
die angegebene Bedeutung auf, und man hält sich an die folgenden Konventionen.
-
In
den nachstehend definierten Gruppen, Resten oder Einheiten wird
die Zahl der Kohlenstoffatome häufig
vor der Gruppe angegeben, zum Beispiel bedeutet (C1-C10)-Alkyl eine Alkylgruppe oder einen Alkylrest mit
1 bis 10 Kohlenstoffatomen. Der Ausdruck "Nieder", wenn er auf irgendeine Kohlenstoff-haltige
Gruppe angewendet wird, bedeutet eine Gruppe, die 1 bis 8 Kohlenstoffatome
enthält,
wie es für
die Gruppe angemessen ist (d.h., eine cyclische Gruppe muss mindestens
3 Atome aufweisen, um einen Ring aufzubauen). Allgemein ist bei
Gruppen, die zwei oder mehr Untergruppen umfassen, die zuletzt genannte
Gruppe der Anbringungspunkt des Restes, zum Beispiel bedeutet "Alkylaryl" einen einwertigen
Rest der Formel Alk-Ar-, während "Arylalkyl" einen einwertigen
Rest der Formel Ar-Alk- bedeutet (worin Alk eine Alkylgruppe ist
und Ar eine Arylgruppe ist). Weiter soll die Verwendung eines Ausdrucks,
der einen einwertigen Rest bezeichnet, so betrachtet werden, dass
er, wenn ein zweiwertiger Rest angemessen ist, den zweiwertigen
Rest bezeichnet und umgekehrt. Falls nicht anders angegeben, werden
die herkömmlichen
Definitionen der Steuerung der Energieniveaus und herkömmliche
stabile Atomvalenzen angenommen und in allen Formeln und Gruppen
erreicht.
-
Die
Ausdrücke "Alkyl" oder "Alkylgruppe" bedeuten einen verzweigten
oder geradkettigen gesättigten aliphatischen
einwertigen Kohlenwasserstoffrest mit 1–10 Kohlenstoffen. Beispiele
für diesen
Ausdruck sind Gruppen wie Methyl, Ethyl, n-Propyl, 1-Methylethyl
(Isopropyl), n-Butyl, n-Pentyl, 1,1-Dimethylethyl (tert-Butyl) und
dergleichen. Er kann als "Alk" abgekürzt werden.
-
Die
Ausdrücke "Alkenyl" oder "Alkenylgruppe" bedeuten einen verzweigten
oder geradkettigen aliphatischen einwertigen Kohlenwasserstoffrest
mit 2–10
Kohlenstoffen, der mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung
enthält.
Beispiele für
diesen Ausdruck sind Gruppen wie Ethenyl, Propenyl, n-Butenyl, Isobutenyl,
3-Methylbut-2-enyl, n-Pentenyl, Heptenyl, Octenyl, Cyclohexylbutenyl,
Decenyl und dergleichen.
-
Die
Ausdrücke "Alkinyl" oder Alkinylgruppe" bedeuten einen verzweigten
oder geradkettigen aliphatischen einwertigen Kohlenwasserstoffrest
mit 2–10
Kohlenstoffen, der mindestens eine Kohlenstoff-Kohlenstoff-Dreifachbindung
enthält.
Beispiele für
diesen Ausdruck sind Gruppen wie Ethinyl, Propinyl, n-Butinyl, 2-Butinyl,
3-Methylbutinyl, n-Pentinyl, Heptinyl, Octinyl, Decinyl und dergleichen.
-
Die
Ausdrücke "Alkoxy" oder "Alkoxygruppe" bedeuten einen einwertigen
Rest der Formel AlkO-, worin Alk eine Alkylgruppe ist. Beispiele
für diesen
Ausdruck sind Gruppen wie Methoxy, Ethoxy, Propoxy, Isopropoxy,
Butoxy, sek-Butoxy, tert-Butoxy,
Pentoxy und dergleichen.
-
Die
Ausdrücke "Aryloxy" oder "Aryloxygruppe" bedeuten einen einwertigen
Rest der Formel ArO-, worin Ar Aryl ist. Beispiele für diesen
Ausdruck sind Gruppen wie Phenoxy, Naphthoxy und dergleichen.
-
Die
Ausdrücke "Alkylcarbonyl", "Alkylcarbonylgruppe", "Alkanoyl" oder "Alkanoylgruppe" bedeuten einen einwertigen
Rest der Formel -C(O)Alk, worin Alk Alkyl oder Wasserstoff ist.
-
Die
Ausdrücke "Aryl" oder "Arylgruppe" bedeuten einen substituierten
oder unsubstituierten carbocyclischen aromatischen einwertigen oder
zweiwertigen Rest mit 6 bis 14 Kohlenstoffatomen mit einem einzigen Ring
(z.B. Phenyl oder Phenylen) oder mehreren kondensierten Ringen (z.B.
Naphthyl oder Anthryl). Falls nicht anders angegeben, kann der Arylring
an jedem geeigneten Kohlenstoffatom angebracht sein, das eine stabile
Struktur zum Ergebnis hat, und kann, falls substituiert, an jedem
geeigneten Kohlenstoffatom mit einem oder mehreren Substituenten
substituiert sein, die aus Halogen, Alkyl, Alkoxy, Aryl, Acyl, Nitro,
Cyano und dergleichen ausgewählt
sind, das eine stabile Struktur zum Ergebnis hat. Beispielhafte
Arylgruppen umfassen Phenyl, Naphthyl, Anthryl, Phenanthryl, Indenyl,
Heptalenyl, Biphenyl, Biphenylenyl, Azulenyl, Pentalenyl und dergleichen.
Sie kann als "Ar" abgekürzt werden.
-
Die
Ausdrücke "Arylcarbonyl", "Arylcarbonylgruppe", "Aroyl" oder "Aroylgruppe" bedeuten einen einwertigen
Rest der Formel -C(O)Ar, worin Ar Aryl ist, wie oben definiert.
-
Die
Ausdrücke "Acyl" oder "Acylgruppe" bedeuten einen einwertigen
Rest der Formel -C(O)R, worin R ein Substituent ist, der aus Wasserstoff,
Alkyl, Alkenyl, Alkinyl, Aryl, Arylalkyl, Cycloalkyl und dergleichen
ausgewählt
ist, wobei jeder gegebenenfalls mit einer oder mehreren Gruppen
substituiert sein kann, die aus Halogenen, Alkoxy, Hydroxy, Nitro,
Cyano, Alkylaryl und dergleichen ausgewählt sind. Bevorzugt ist R Niederalkyl oder
Phenyl, jeweils gegebenenfalls substituiert. Als solche umfassen
die Ausdrücke
Alkylcarbonylgruppen und Arylcarbonylgruppen.
-
Der
Ausdruck "Acylierungsmittel" bedeutet einen Reaktanten,
der, wenn er mit einer Verbindung umgesetzt wird, welche eine nukleophile
Stelle aufweist, die mit dem Acylierungsmittel reagieren kann, bewirkt, dass
eine Acylgruppe kovalent an eine oder mehrere Stellen der Verbindung
gebunden wird. Acylierungsmittel umfassen, ohne jedoch darauf beschränkt zu sein,
Reagenzien mit der Formel RC(O)X, in der X ein Halogen, eine Acyloxygruppe
der Formel R'C(O)O-
ist, worin R' die
gleiche Bedeutung der im vorstehenden Absatz definierten Gruppe
R aufweist. Als solcher umfasst der Ausdruck Carbonsäuren, Carbonsäureanhydride,
Niederester von Carbonsäuren
und Säurehalogenide.
Bevorzugt sind die Acylierungsmittel Säurehalogenide oder Säureanhydride.
Derartige Acylie rungsmittel können
mono-, di-, tricarboxylische oder polycarboxylische Acylierungsmittel
sein. Die Säureanhydride
können
symmetrische, asymmetrische oder gemischte Anhydride sein. Zusätzlich umfassen
Acylierungsmittel in situ erzeugte o-Acylisoharnstoffe, Verbindungen,
die aus der Umsetzung einer Säure
(z.B. Bromessigsäure)
und eines Carbodiimids (z.B. DIC und DCC) und isolierter o-Acylisoharnstoffe
erzeugt werden. Beispielhafte und bevorzugte Acylierungsmittel umfassen
Acetylchlorid, Acetylbromid, Propionylchlorid, Benzoylchlorid, Acetanhydrid,
Propionsäureanhydrid,
Buttersäureanhydrid, Valeriansäureanhydrid,
Hexansäureanhydrid,
Ameisensäureanhydrid,
Benzoesäureanhydrid,
Trifluoracetylchlorid, Methyltrifluoracetat, Ethyltrifluoracetat
und dergleichen.
-
Der
Ausdruck "Acetylierungsmittel" bedeutet ein Acylierungsmittel,
in dem die Acylgruppe Acetyl ist.
-
Die
Ausdrücke "Acylierung", "Acylieren" und dergleichen
beziehen sich auf eine chemische Reaktion, durch welche eine Acylgruppe
unter Verwendung eines Acylierungsmittels an eine andere Verbindung
oder Einheit addiert wird.
-
Die
Ausdrücke "Acetylierung", "Acetylieren" und dergleichen
bezeichnen eine chemische Reaktion, durch welche eine Acetylgruppe
unter Verwendung eines Acetylierungsmittels an eine andere Verbindung
oder Einheit addiert wird.
-
Der
Ausdruck "aliphatische
Gruppe" bedeutet
eine nicht-aromatische gerad- oder verzweigtkettige Kohlenwasserstoffgruppe.
Als solcher umfasst der Ausdruck Alkyl-, Alkenyl- und Alkinylgruppen.
-
Der
Ausdruck "alicyclische
Gruppe" bedeutet
eine nicht aromatische cyclische Kohlenwasserstoffgruppe. Als solcher
umfasst der Ausdruck Cycloalkyl-, Cycloalkenyl- und Cycloalkinylgruppen.
-
Der
Ausdruck "Carbonsäure" bedeutet eine organische
Säure der
Formel RC(O)OH, in der R ein Substituent, der aus Wasserstoff ausgewählt ist,
oder ein Substituent ist, der aus einer niederaliphatischen Gruppe, niederalicyclischen Gruppe,
einer Arylgruppe, einer Arylalkylgruppe oder einer Alkylarylgruppe
ausgewählt
ist, die gegebenenfalls mit Halogen, Alkyl, Alkoxy, Aryl usw. substituiert
ist. Beispiele für
diesen Ausdruck sind Ameisensäure,
Essigsäure,
Propansäure,
Butansäure,
2-Methylpropansäure,
Pentansäure,
Propensäure, 2-Methylpropensäure, 2-Butensäure, Zimtsäure, Benzoesäure, Cyclobutancarbonsäure, Salicylsäure und
dergleichen.
-
Der
Ausdruck "Dicarbonsäure" bedeutet eine organische
Säure der
Formel R(C(O)OH)2, in der R entweder eine
Bindung (d.h. Oxalsäure)
oder eine zweiwertige Kohlenwasserstoffgruppe ist, die aus einer (C1-C6)-Alkylengruppe,
die gegebenenfalls mit Hydroxy, Halogen, Alkoxy und dergleichen
substituiert ist, niederalicyclischen Gruppe, einer Arylgruppe,
einer Arylalkylgruppe oder einer Alkylarylgruppe ausgewählt ist. Beispiele
für diesen
Ausdruck sind Oxalsäure,
Malonsäure,
Bernsteinsäure,
Glutarsäure,
Adipinsäure,
Maleinsäure,
Fumarsäure,
Phthalsäure,
Isophthalsäure,
Terephthalsäure,
Zuckersäure
und dergleichen.
-
Der
Ausdruck "Tricarbonsäure" bedeutet eine organische
Säure der
Formel R(C(O)OH)3, in der R entweder eine
Bindung (d.h. Oxalsäure)
oder ein Substituent ist, der aus einer niederaliphatischen Gruppe,
niederalicyclischen Gruppe, einer Arylgruppe, einer Arylalkylgruppe
oder einer Alkylarylgruppe ausgewählt ist, die gegebenenfalls
mit Hydroxy, Halogen, Alkoxy und dergleichen substituiert ist. Ein
Beispiel für
diesen Ausdruck ist Citronensäure.
-
Die
Ausdrücke "Alkylcarbonyloxy" oder "Alkylcarbonyloxygruppe" bedeuten einen einwertigen
Rest der Formel -OC(O)Alk, in der Alk Alkyl ist, das gegebenenfalls
mit Hydroxy, Halogen, Alkoxy und dergleichen substituiert ist.
-
Die
Ausdrücke "Arylcarbonyloxy" oder "Arylcarbonyloxygruppe" bedeuten einen einwertigen
Rest der Formel -OC(O)Ar, in der Ar Aryl ist, das gegebenenfalls
mit Hydroxy, Halogen, Alkoxy und dergleichen substituiert ist.
-
Der
Ausdruck "Edelmetallkatalysator" bedeutet einen festen
Metallkatalysator in irgendeiner Form, die für das Erzielen der Hydrierungsreaktionen
der vorliegenden Erfindung geeignet und wirksam ist. Beispielhafte und
bevorzugte Edelmetallkatalysatoren umfassen Platin, Palladium, Ruthenium,
Osmium, Iridium, Rhodium und dergleichen oder deren Mischungen,
wobei das Metall oder die Legierung in Form: (a) eines feinen zerteilten
Metalls oder einer fein zerteilten Legierung (z.B. Pulver, Granalien
usw.) oder eines Metalls oder einer Legierung mit hoher Oberfläche (z.B.
porös,
Schwamm, Netzplatin oder Palladiummohr), (b) einer Vorstufen-Verbindung
(z.B. des Oxids), das vor oder während
der Hydrierung in den aktiven Katalysator überführt wird, oder (c) verteilt
auf einem anorganischen Träger,
im Allgemeinen mit hoher Oberfläche,
wie Kohlenstoff, aktiviertem Kohlenstoff, Siliciumdioxid, Aluminiumoxid
oder andere Metalloxiden (z.B. Calciumoxid), Metallcarbonaten (z.B.
Calciumcarbonat), Metallsulfaten (z.B. Bariumsulfat) oder dergleichen
bereitgestellt ist, wobei das geträgerte Edelmetall bevorzugt
zu 0,5 Gew.-% bis 10 Gew.-%, bevorzugter 1 Gew.-% bis 5 Gew.-% vorliegt.
-
Experimentelle
Ergebnisse
-
In
den folgenden Experimenten schlossen die verwendeten analytischen
Methoden quantitative und qualitative Analysen ein, die mittels
Hochleistungsflüssigkeitschromatographie-(HPLC-)
und Gas-Flüssigkeits-Chromatographie-(GLC-)Verfahren durchgeführt wurden.
-
I. SYNTHESEVERFAHREN
DES STANDES DER TECHNIK
-
Für Vergleichszwecke
wurden die zwei oben erwähnten
Syntheseverfahren des Standes der Technik getestet und die Ergebnisse
erhalten.
-
1.
Iodierung von Norephedrin-Hydrochlorid und Reduktion zu Amphetamin
-
Ein
100 ml-Rundkolben mit einem Magnetrührer wurde mit 20 ml 57%-iger
HI-Lösung beschickt. (1R,2S)-(–)-Norephedrin
(10,0 g, 0,066 Mol) wurde dann unter Rühren zu dem Kolben gegeben.
Roter Phosphor (1,0 g) wurde zu der gerührten Mischung gegeben, und
die Reaktionsmischungstemperatur wurde dann innerhalb weniger Minuten
auf 40°C
erhöht.
Die Reaktionsmischung wurde dann auf 100°C erhöht, und Proben wurden in Zeitabständen für eine HPLC-Analyse
entnommen. Die Ergebnisse waren wie folgt:
- a.
2 Stunden (58% Norephedrin, 6,8% Amphetamin, 6% Bis-Verbindung);
- b. 4 Stunden (50% Norephedrin, 10,2% Amphetamin, 14,7% Bis-Verbindung);
- c. 6 Stunden (41% Norephedrin, 15,6% Amphetamin, 17,7% Bis-Verbindung);
und
- d. 22 Stunden (48,95% Amphetamin, 48,5% Bis-Verbindung).
-
Nach
22 Stunden bei 100°C
wurde die Reaktionsmischung abgekühlt und filtriert, um den Phosphor zu
entfernen. Es trennte sich eine ölige
Schicht ab (1,3 g, mittels GLC als Bis-Verbindung identifiziert).
Die wässrige
Schicht wurde mit 50%-iger Natriumhydroxid-Lösung alkalisch gemacht und
mit Ether extrahiert. Der Ether-Extrakt wurde über wasserfreiem Magnesiumsulfat
getrocknet und konzentriert, wodurch man 4,1 g gelbes Öl erhielt,
von dem mittels HPLC identifiziert wurde, dass es aus 83,23% Amphetamin
und 16,27% Bis-Verbindung bestand. Die berechnete Ausbeute an Amphetamin
war deshalb 3,41 g (38,3%).
-
2.
Herstellung von 2-Amino-1-chlor-1-phenylpropan-Hydrochlorid aus
Norephedrin-Hydrochlorid und Reduktion zu Amphetamin
-
Ein
100 ml-Rundkolben mit einem Magnetrührer wurde mit Thionylchlorid
(24,47 g, 15 ml) und Norephedrin-Hydrochlorid (5,38 g) beschickt.
Die Mischung wurde gerührt
und etwa 1 Stunde lang am Rückfluss erwärmt und
auf Umgebungstemperatur abkühlen
gelassen. Das überschüssige Thionylchlorid
wurde durch Verdampfen auf einem Rotationsverdampfer entfernt. Der
Rückstand
im Kolben wurde mit Ether (50 ml) digeriert, und der Festkörper wurde
gesammelt. Das rohe feste Produkt wurde aus Methanol-Isopropylether
umkristallisiert, und der gereinigte Festkörper wurde gesammelt (2,83
g, 48%). Das gereinigte 2-Amino-1-chlor-1-phenylpropan-Hydrochlorid wurde
dann wie folgt einer Hydrogenolyse unterzogen. Eine Lösung von
2-Amino-1-chlor-1-phenylpropan-Hydrochlorid (2,38 g, 0,0137 Mol)
in einer Mischung aus 50 ml Ethanol/16 ml Wasser wurde hergestellt
und in eine Parr-Flasche überführt. Unter
Stickstoffatmosphäre
wurde eine Portion von 10% Palladium auf Kohlenstoff zu der Lösung in
der Parr-Flasche
gegeben. Die Parr-Flasche wurde dann auf einer Parr-Schüttelapparatur
installiert, und eine Inertatmosphäre wurde in der Flasche erzeugt und
aufrechterhalten. Die Parr-Flasche wurde dann mit Wasserstoff auf
einen Druck von etwa 50 psi gebracht und geschüttelt, bis die Wasserstoffaufnahme
aufgehört
hatte. Der Inhalt der Parr-Flasche wurde filtriert, um den Katalysator
zu entfernen, und das Filtrat wurde unter verringertem Druck eingedampft,
um das Ethanol zu entfernen. Die verbleibende wässrige Lösung wurde mit 50%-iger Natriumhydroxid-Lösung basisch
gemacht. Die ölige
obere Schicht wurde mit Ether extrahiert, und die Etherschicht wurde
von der wässrigen
Schicht abgetrennt. Der Ether-Extrakt
wurde mit Trocknungsmittel getrocknet und filtriert, um das Trocknungsmittel
zu entfernen. Das Filtrat wurde konzentriert, wodurch man rohes
Amphetamin als Öl
erhielt (1,70 g, 72,3%).
-
II. DAS VERFAHREN DER
ERFINDUNG
-
Das
Verfahren der Erfindung und die experimentellen Ergebnisse unter
Verwendung des Verfahrens der Erfindung werden nachstehend erörtert. Norephedrin-Hydrochlorid
ist im Handel erhältlich
und wurde verwendet, um die Reaktionen zu erläutern, die in diesem Verfahren
nützlich
sind. Diese Bedingungen können auch
auf Ephedrin-Hydrochlorid oder Pseudoephedrin-Hydrochlorid angewendet werden, um d-Methamphetamin
zu erzeugen. Weiter sollte der Fachmann wissen, wie dieses Verfahren
auf die Herstellung von vielen mit Amphetamin verwandten Verbindungen
anzuwenden ist, wie sie im Schema 2 abgedeckt werden. Literaturverfahren
zur Herstellung von Amphetamin und Methamphetamin berichten über die
Retention der Konfiguration am Kohlenstoff, der die Aminogruppe
trägt (siehe
z.B. Noggle, DeRuiter und Clark, J. Chrom. Sci. 25, 38–42 (1987);
Allen und Kiser, J. Forensic Sciences 32(4), 953–962 (1987)). Deshalb wird
erwartet, dass d,l-Norephedrin und d,l-Norpseudoephedrin das gleiche
Produkt, d,l-Amphetamin ergeben, und es wird erwartet, dass 1R,2S-(–)-Norephedrin und 1S,2S-(+)-Norpseudoephedrin
Dextroamphetamin [d-Amphetamin,
S-(+)-Amphetamin] ergeben. Es wird auch erwartet, dass d,l-Ephedrin und d,l-Pseudoephedrin
das gleiche Produkt, d,l-Methamphetamin ergeben, und es wird erwartet,
dass 1R,2S-(–)-Ephedrin
und 1S,2S-(+)-Pseudoephedrin Dextromethamphetamin
[d-Methamphetamin, S-(+)-Methamphetamin]
ergeben. Die in den folgenden Tabellen verwendeten Abkürzungen
sind NE für
Norephedrin, O-AcNE für
O-Acetylnorephedrin, N-AcNE für
N-Acetylnorephedrin, O,N-DiAcNE für O,N-Diacetylnorephedrin,
Amp für
Amphetamin und N-AcAmp für
N-Acetylamphetamin. Diese Verbindungen wurden synthetisiert und
als Bezugssubstanzen für
HPLC-Analysen verwendet.
-
A.
Herstellung von 2-Amino-1-acetoxy-1-phenylpropan-Hydrochlorid (O-Acetylnorephedrin)
aus Norephedrin-Hydrochlorid
-
Ein
100 ml-Dreihalsrundkolben, der mit einer Wärmesteuerung, einem Rührer, einem
Kühler
und einem Gasgluckertopf ausgestattet war, wurde mit 18,77 g (0,10
Mol) d,l-Norephedrin-Hydrochlorid, Essigsäure (18 ml) und Acetanyhdrid
(12,24 g, 0,12 Mol) beschickt. Die Reaktionsmischung wurde mit einem
Heizmantel erwärmt,
wobei die Wärmesteuerung
auf 80°C
eingestellt war. Nachdem sich die Reaktionsmischung geklärt hatte,
wurde sie 2 Stunden bei 80°C
gehalten. Heptan (36 ml) wurde unter raschem Rühren langsam zu der Reaktionsmischung
bei 80°C
gegeben, dann ließ man
die Mischung auf Raumtemperatur abkühlen. Die Aufschlämmung wurde über Nacht
bei Umgebungstemperatur gerührt,
und der Festkörper
wurde durch Filtration gesammelt. Der granuläre Festkörper wurde unter Umgebungsbedingungen über Nacht
getrocknet, wodurch man 18,13 g Produkt (89,99% Ausbeute) erhielt.
-
B.
Herstellung von 2-Amino-1-acetoxy-1-phenylpropan-Hydrochlorid (O-Acetylnorephedrin)
aus Norephedrin-Hydrochlorid und Reduktion zu Amphetamin ohne Isolierung
des Zwischenprodukts
-
Das
folgende Verfahren ist ein Beispiel, in dem die Reduktion des Zwischenprodukts
O-Acetylnorephedrin ohne Isolierung des O-Acetylnorephedrins durchgeführt wird.
-
Ein
100 ml-Dreihalsrundkolben, der mit einem Magnetrührer, einem Kühler und
einer Wärmesteuerung ausgestattet
war, wurde mit d,l-Norephedrin-Hydrochlorid (9,39 g, 0,050 Mol),
Acetanhydrid (6,12 g, 0,060 Mol) und Essigsäure (18 ml) beschickt. Die
Reaktionsmischung wird mit einem Heizmantel erwärmt, wobei eine Wärmesteuerung
bei 80°C
eingestellt ist. Als die Temperatur etwa 60°C erreichte, wurde die Reaktionsmischung
exotherm, und die Temperatur stieg auf 84°C. Die Reaktionsmischung wurde
auf 80°C
abgekühlt
und 2 Stunden bei 80°C
gehalten. Die HPLC-Analyse der Reaktionsmischung zu diesem Zeitpunkt
zeigte 90,69% O-Acetylnorephedrin
und 6,88% O,N-Diacetylnorephedrin. Die Reaktionsmischung wurde über Nacht
bei Umgebungstemperatur gerührt
und dann mit 20 ml Ethanol verdünnt.
Die resultierende Lösung
wurde in eine Parr-Flasche überführt, welche
mit Stickstoffgas gespült
wurde, und etwa 1 g 10% Palladium-Kohlenstoff-Katalysator (50% Wasser) wurde dazugegeben.
Die Parr-Flasche wurde auf einer Parr-Schüttelapparatur installiert und
mit Stickstoff unter Inertatmosphäre gesetzt. Die Parr-Flasche
wurde dann mit Wasserstoff auf einen Druck von 45 psi gebracht und
dann unter Wasserstoffdruck bei Umgebungstemperatur geschüttelt. In
10 Minuten wurde ein Druckabfall von 3 psi beobachtet. Die Parr-Flaschen-Temperatursteuerung
wurde auf 55°C eingestellt,
und der Druck erhöhte
sich von 42 psi auf 47 psi. Die Druckänderungen über die nächsten 5 bis 6 Stunden wurden
aufgezeichnet. Die Wärme
wurde dann abgeschaltet, und man ließ die Reaktionsmischung auf
Umgebungstemperatur abkühlen.
Der Enddruck wurde aufgezeichnet, und die Parr-Flasche wurde entspannt
und mit Stickstoffgas gespült.
Die Mischung wurde filtriert, um den Katalysator zu entfernen, und
das Filtrat wurde analysiert, und es wurde gefunden, dass es aus
2,45% Norephedrin, 57,59% Amphetamin, 20,71% O-Acetylnorephedrin,
10% N-Acetylamphetamin und 3,75% O,N-Diacetylnorephedrin bestand.
Die berechnete Ausbeute betrug 3,89 g (57%).
-
C. Weitere Beispiele für die Acetylierung
von Norephedrin-Hydrochlorid
-
Wie
oben erläutert,
kann O-Acetylnorephedrin-Hydrochlorid in guter Ausbeute aus Norephedrin-Hydrochlorid
durch 2-stündiges
Erwärmen
mit Acetanhydrid (bevorzugt 1,2 bis 2,0 Äquivalenten) in Essigsäure (bevorzugt
1 bis 2 ml/g Norephedrin-Hydrochlorid) in einer mild exothermen
Reaktion bei 50–80°C hergestellt werden,
obwohl die exothermen Reaktionstemperaturen 80°C überschreiten könnten. Im
Allgemeinen verbleibt, wenn weniger als 20% Überschuss an Acetanhydrid verwendet
werden, etwas unumgesetztes Norephedrin, zum Beispiel reagierte
es, wenn ein 10%-iger Überschuss
von Acetanhydrid verwendet wurde, mit nur etwa 80% des Norephedrin-Hydrochlorids.
Es wurde gefunden, dass die verwendete Essigsäure-Menge nicht kritisch zu
sein scheint, da die Reaktion gut mit entweder 1 oder 2 ml/g Norephedrin-Hydrochlorid
vonstatten ging und ohne Schwierigkeit bei 1 ml/g gerührt werden
konnte. Als jedoch keine Essigsäure
verwendet wurde, bestand das erhaltene Produkt aus einer Mischung
von 16,71% Norephedrin, 67,08% O-Acetylnorephedrin und
14,43% O,N-Diacetylnorephedrin. Die Ergebnisse von anderen Labor-Herstellungen
sind in Tabelle 1 zusammengefasst.
-
-
(i). Nebenprodukte
-
Tabelle
1 führt
die Nebenprodukte auf, die in verschiedenen Acetylierungsexperimenten
gefunden wurden. Es wurden geringe Mengen an unumgesetztem Norephedrin
und O,N-Diacetylnorephedrin als Nebenprodukte in den isolierten
Produkten gefunden. Das Signal für
Amphetamin im Experiment L kann ein Artefakt sein.
-
(ii). Kristallisationslösungsmittel
-
Um
die Kristallisationsgeschwindigkeit des O-Acetylnorephedrin-Hydrochlorid-Salzes zu erhöhen und die
Entfernung von Essigsäure
und jeglichen überschüssigen Acetanhydrids,
das vorliegen kann, zu unterstützen,
wurde die Reaktionsmischung mit Heptan (Experiment I), Methyl-tert-butylether
(Experimente F und H), Ethanol (Experiment G), Isopropanol, Methylisobutylketon,
Acetonitril, Ethylacetat, Tetrahydrofuran oder einem anderen Lösungsmittel
behandelt, in dem das Produkt wenig Löslichkeit aufweist. Das feste
Produkt, das sich bildete, wurde durch Filtration gesammelt. Heptan
ist ein bevorzugtes Lösungsmittel,
da der erzeugte Festkörper
dicht war und nicht so sehr solvatisiert zu sein schien wie bei
anderen versuchten Lösungsmitteln.
-
(iii). Geschwindigkeit
der Acetylierungsreaktion
-
Die
Geschwindigkeit der Acetylierungsreaktion wird durch die Ergebnisse
der Hochleistungsflüssigkeitschromatographie-(HPLC-)Analysen
einer Reaktion in verschiedenen Zeitabständen demonstriert (Experiment
A). Diese Ergebnisse sind in Tabelle 2 angegeben und zeigen, dass
die Reaktion innerhalb von 20 Minuten, nachdem die Reaktionsmischung
klar geworden ist, nahezu vollständig
ist.
-
-
(iv). Chemische Reaktivitäten von
freier Norephedrin-Base und Salzen
-
Die
im Handel erhältliche
Vorstufe zu Dextroamphetamin ist 1R,2S-(–)-Norephedrin. Es wurden Versuche unternommen,
die freie Norephedrin-Base zu O-acetylieren, aber gemäß einer
Analyse der Carbonylregion des Infrarot-Spektrums erzeugte dies hauptsächlich N-Acetylamphetamin.
In einem Experiment wurde die freie Norephedrin-Base in Essigsäure mit
0,5 Äquivalenten
Schwefelsäure
behandelt und dann mit 1,1 Äquivalenten
Acetanhydrid unter Erwärmen
auf 63°C
behandelt, wodurch man eine klare Lösung erhielt. Katalytische
Reduktion, gefolgt von einer Alkalisierung, ergab ein Öl, das aus
13,96% Amphetamin und 68% Norephedrin zusammengesetzt war, ein Hinweis,
dass das Acetylierungsverfahren bei einem Sulfatsalz nur teilweise
erfolgreich war. In einem weiteren Experiment wurde eine 0,10 Mol-Portion
Norephedrin-Hydrochlorid mit
einem 20%-igen Überschuss
an Acetanhydrid in 2 ml Essigsäure/g
Salz acetyliert, und man folgte dem Fortschritt der Reaktion über 2 Stunden
(Tabelle 2). Die Reaktionsmischung wurde mit 130 ml Wasser verdünnt und
22 Stunden lang einer katalytischen Hydrierung unterzogen, wodurch
man 10,4 g Öl
erhielt. Die lange Reaktionszeit ist der Anwesenheit von Essigsäure in der
Reduktionsmischung zuzuschreiben. Die Ausbeute an Amphetamin aus
Norephedrin-Hydrochlorid, basierend auf dem Gewicht des Produkts
und auf HPLC-Analyse, beträgt
66% (Experimente A und B). Bei diesen Experimenten kann gesehen
werden, dass die besten Ergebnisse mit den Salzen von Phenylpropylamin
oder Norephedrin, insbesondere dem Hydrochloridsalz, erhalten wurden.
-
D.
Katalytische Transfer-Hydrierung von O-Acetylnorephedrin
-
Ein
500 ml-Dreihalsrundkolben, der mit einem Rührer, Zugabetrichter, Kühler und
einer Wärmesteuerung
ausgestattet war, wurde mit O-Acetylnorephedrin- Hydrochlorid (47,0 g, 0,205 Mol), 131
ml Wasser und 1,0 g 10% Palladium auf Kohlenstoff-Katalysator (50%
Wasser) beschickt. Eine Lösung
von Ammoniumformiat (HCOONH4; 15,50 g, 0,246
Mol) in 20 ml Wasser wurde dann hergestellt. Die Reaktionsmischung
in dem Dreihalsrundkolben wurde dann in einem Wasserbad erwärmt, das
auf 71°C
temperaturgesteuert war. Als die Reaktionsmischungstemperatur 68°C erreichte,
wurden etwa 6 ml der Ammoniumformiat-Lösung
in 2 ml-Inkrementen in 5-minütigen
Zeitabständen
in die Reaktionsmischung gegeben. Als die Gasentwicklung begann
aufzuhören,
wurde der Rest der Ammoniumformiat-Lösung dazugetropft, und die
Reaktionsmischung wurde in etwa 1 Stunde im Wasserbad gerührt. Die
Wasserbad-Temperatursteuerung wurde dann abgeschaltet, und die Reaktionsmischung
wurde unter Rühren über Nacht
auf Umgebungstemperatur abkühlen
gelassen. Die abgekühlte
Reaktionsmischung wurde dann filtriert, um den Katalysator zu entfernen,
und das Filtrat wurde mit 26 ml (0,50 Mol) 50%-iger Natriumhydroxid-Lösung behandelt.
Die alkalisierte Reaktionsmischung wurde dann in einen Scheidetrichter überführt und
0,5 Stunden stehen gelassen. Die untere wässrige Schicht wurde von der oberen öligen Schicht
abgetrennt, und die ölige
Schicht wurde gewonnen (40,83 g). Das Öl wurde analysiert, und die
Analyse hatte die folgenden Ergebnisse: GLC-Gewichtsprozent-Assay:
60,18% Amphetamin; Karl-Fischer-Titration: 23,15% Wasser; HPLC-Analyse:
79,03% Amphetamin, 14,6% N-Acetylnorephedrin, 5,49% N-Acetylamphetamin
und 0,76% Norephedrin; berechnete Ausbeute: 24,6 g Amphetamin (88,8%).
-
E.
Katalytische Hydrierung von O-Acetylnorephedrin-Hydrochlorid
-
Eine
Parr-Flasche wurde mit O-Acetylnorephedrin-Hydrochlorid (11,43 g)
und 75 ml Wasser beschickt. Die Parr-Flasche wurde mit Stickstoff
gespült,
und der Katalysator wurde dazugegeben. Die Flasche wurde dann auf
einer Parr-Apparatur
mit einem anfänglichen
Druck von 55 psi geschüttelt.
Nach 2 Stunden betrug der Druckabfall 4 psi (am Tank). Die Parr-Flasche
wurde eine weitere Stunde geschüttelt,
um die Vollständigkeit
der Hydrogenolyse sicherzustellen, und dann entspannt und mit Stickstoff
gespült.
Der Inhalt der Parr-Flasche wurde dann filtriert, um den Katalysator
zu entfernen, und das Filtrat wurde mit 15 ml 50%-iger Natriumhydroxid-Lösung basisch
gemacht, und man ließ die
Mischung über
Nacht stehen. Die Mischung wurde dann in einen Scheidetrichter überführt, um
die untere wässrige
Schicht von der oberen öligen
Schicht abzutrennen; die ölige
Schicht wurde gewonnen (7,00 g). Das Öl wurde analysiert, und die
Analyse hatte die folgenden Ergebnisse: GLC-Gewichtsprozent-Assay:
63,02% Amphetamin; HPLC-Analyse: 87,22% Amphetamin, 10,8% N-Acetylnorephedrin,
0,37% N-Acetylamphetamin;
Karl-Fischer-Analyse: 23,699% Wasser; berechnete Ausbeute: 4,41
g Amphetamin (65,33%).
-
F. Weitere Beispiele für die Reduktion
von O-Acetylnorephedrin-Hydrochlorid
zu Amphetamin
-
Die
Reduktion von O-Acetylnorephedrin-Hydrochlorid zu Amphetamin wird
entweder durch katalytische Hydrierung oder durch katalytische Transfer-Hydrierung bewerkstelligt.
Die katalytische Hydrierung kann in etwa 4 Stunden bei Raumtemperatur
in Wasser unter Verwendung von 10% Palladium-auf-Kohlenstoff-Katalysator (50% nass mit
Wasser) bei 50–55
psi Wasserstoffdruck auf einer Parr-Schüttelvorrichtung erzielt werden.
Unter Verwendung dieses Verfahrens findet der größte Teil der Wasserstoff-Aufnahme
innerhalb von 2 Stunden statt. Die katalytische Transfer-Hydrierung
unter Verwendung von Ammoniumformiat und 10% Palladium auf Kohlenstoff
(50% nass mit Wasser) in Wasser ist innerhalb von 20–30 Minuten
ab Initiierung der Reaktion vollständig, wenn die Ammoniumformiat-Lösung in
einer Portion zugesetzt wird.
-
1. Katalytische
Hydrierung
-
Die
Ergebnisse von Labor-Hydrierungsexperimenten sind in den Tabellen
3 und 4 gezeigt. In Tabelle 4 wurden die Gewichtsprozent Amphetamin
mittels GLC bestimmt, und die prozentuale Zusammensetzung wurde
mittels HPLC bestimmt.
-
-
Die
Löslichkeit
von O-Acetylnorephedrin-Hydrochlorid in Wasser beträgt 5,5 ml/g
(Experiment X). Hydrierungsreaktionen bei Wasserkonzentrationen
gleich oder größer als
5,5 ml/g scheinen innerhalb von 2 Stunden zu 90–95% vollständig zu sein (Experiment T).
Die Anwesenheit von Essigsäure
scheint die Reduktion zu verlangsamen (Experiment B). Ethanol verringert
die Löslichkeit
des Salzes und kann so ein größeres Lösungsmittelvolumen
für die
Hydrierung erfordern, damit sie leicht vonstatten geht (Experiment
BB). Wenn entweder Essigsäure
oder Ethanol verwendet wird, ist ein weiterer Schritt bei der Aufarbeitung
erforderlich, um das organische Lösungsmittel zu entfernen. Dies
macht Wasser zu einem ausgezeichneten Lösungsmittel für die katalytische
Reduktion, da das Volumen nicht übermäßig ist,
es nicht brennbar ist und es preiswert ist. Ein rigoroses Spülen des
Hydriergefäßes mit
einem Inertgas vor Einführung
des Palladium-Katalysators
ist bei Wasser nicht erforderlich, wie es bei einem brennbaren Lösungsmittel
der Fall wäre.
Das aus den Experimenten T, V und Y erhaltene Amphetamin wurde aus
der wässrigen
Lösung
nach Alkalisierung anstelle einer Extraktion mit einem Lösungsmittel
abgetrennt. Mittels Karl-Fischer-Wasseranalyse wurde gezeigt, dass
das rohe Amphetamin 23–24%
Wasser enthielt. Dies trägt
Ausbeuten von mehr als 100% und niedrigeren Amphetamin-Gewichtsprozent-Werten
Rechnung.
-
2. Katalytische
Transfer-Hydrierung
-
Bei
der katalytischen Transfer-Hydrierung kann eine Mischung von O-Acetylnorephedrin-Hydrochlorid, Wasser
und 10% Palladium auf Kohlenstoff (50% nass mit Wasser) mit 1,2 Äquivalenten
Ammoniumformiat behandelt und erwärmt werden, bis eine exotherme
Reaktion, begleitet von einer Gasentwicklung, stattfindet. Die Reaktion
ist vollständig,
wenn die Gasentwicklung aufhört.
-
Die
Ergebnisse der Experimente unter Verwendung von katalytischer Transfer-Hydrierung sind in
Tabelle 5 gezeigt. Diese Reaktionen wurden bei Temperaturen zwischen
60°C–80°C vorgenommen,
außer
der höheren
Temperatur, die bei der Exothermen erhalten wurde.
-
-
Die
Mengen der Reaktanten bei den katalytischen Transfer-Hydrierungsreaktionen,
die in Tabelle 5 aufgeführt
sind, sind in Tabelle 6 gezeigt.
-
-
-
Wenn
ein großer Überschuss
an Ammoniumformiat verwendet wird, sublimiert ein weißer Festkörper in
den Kühler.
Eine Sublimation wurde bei lediglich 10–20% Überschuss an Ammoniumformiat
nicht beobachtet. Ein 10–20%-iger Überschuss
an Ammoniumformiat scheint für
eine vollständige
Reaktion ausreichend zu sein. Die Exotherme und die Gasentwicklung
wurden im Experiment DD, das mit Methanol durchgeführt wurde, bei
etwa 52°C
ebenfalls gesehen.
-
3. Vergleich
der Hydrogenolyse-Reaktionen
-
Bei
beiden Arten der Hydrogenolyse-Reaktionen besteht die Aufarbeitung
mit einer Ausnahme aus dem Filtrieren der Reaktionsmischung, um
den Palladium-auf-Kohlenstoff-Katalysator
zu entfernen, der Alkalisierung des Filtrats auf pH 14, der Abtrennung
der wässrigen
Schicht von der Amphetamin-Schicht und der Destillation des Amphetamins.
Diese Ausnahme war das Experiment DD, bei dem die filtrierte Reaktionsmischung
konzentriert und der Rückstand
in Wasser aufgenommen und alkalisiert wurde. Die zwei Arten von
Hydrogenolyse-Reaktionen
sind in der Effizienz vergleichbar, wobei die katalytische Transfer-Hydrierung schneller
ist. Jedoch gibt es eine Sicherheitsfrage, da bei der katalytischen
Transfer-Hydrierungsreaktion eine Induktionsperiode auftritt, die
von einer beträchtlichen
Gasentwicklung begleitet ist, wenn die Reaktion beginnt. Die Reaktion
kann teilweise durch anfängliche
Zugabe von nur etwa 25% des Ammoniumformiats als wässrige Lösung und
Erwärmen
der Mischung, bis die Reaktion beginnt, gesteuert werden. Der Rest
der Ammoniumformiat-Lösung
kann dann mit einer geeigneten Geschwindigkeit zugesetzt werden.
-
Ammoniumformiat
scheint die Umlagerung von O-Acetylnorephedrin-Hydrochlorid zu N-Acetylnorephedrin
zu katalysieren. Drei Proben von O-Acetylnorephedrin-Hydrochlorid (Experiment
Z umkristallisiert) wurden 6 Stunden bei Raumtemperatur, bei 60°C bzw. bei
60°C mit
zugesetztem Ammoniumformiat gerührt. Proben
wurden in Zeitabständen
für HPLC-Analysen
entnommen. Bei Raumtemperatur nahm der Prozentsatz an O-Acetylnorephedrin-Hydrochlorid über eine
Zeitspanne von 6 Stunden von 99,30 auf 99,12% ab, und der Prozentsatz
an N-Acetylnorephedrin
nahm von 0,10% auf 0,21% zu. Bei 60°C nahm der Prozentsatz an O-Acetylnorephedrin-Hydrochlorid über eine
6-stündige
Zeitspanne von 99,18 auf 97,79 ab, wobei der Prozentsatz an N-Acetylnorephedrin
von 0,21% auf 1,1% zunahm. Bei der Ammoniumformiat-Mischung bei
60°C fiel
der Prozentsatz an O-Acetylnorephedrin-Hydrochlorid innerhalb von
30 Minuten auf 91,23% und nach 6 Stunden auf 77,20%, während die
Menge von N-Acetylnorephedrin
von 7,51% auf 20,95% zunahm. Der katalytische Hydrierungsweg scheint
deshalb der katalytischen Transfer-Hydrierung im Hinblick auf die
Induktionszeitspanne, die Gasentwicklung und das Umlagerungspotential
vorzuziehen zu sein.
-
Das
Amphetamin, das aus mehreren Hydrogenolyse-Reaktionen beider Arten
erhalten wurde, wurde mittels HPLC und/oder GLC analysiert, und
die Ergebnisse sind in Tabelle 7 zusammengefasst. Die Produkte der
Experimente Experiment P und Experiment Q wurden durch Extraktion
mit Ether und Trocknen des Extrakts zur Entfernung jeglichen Wassers
erhalten. Es wurde durch Karl-Fischer-Analysen gezeigt, dass das
in den Experimenten R, S und U erhaltene Produkt Wasser enthielt,
und dieses wird in den Amphetamin-Gewichtsprozentanalysen kompensiert.
Die prozentuale Ausbeute an Amphetamin ist aus dem tatsächlichen
Gewicht des isolierten Produkts und dem Prozentsatz an Amphetamin
in dem Produkt berechnet, wie durch GLC- oder HPLC-Gewichtsprozent-Analysen
bestimmt. Die Zusammensetzung des Produkts ist die Flächen-Prozent-Analyse.
-
-
-
Anstelle
des Vorliegens von Amphetamin-Gewichtsprozent-Daten wurden die Flächen-Prozent
von Amphetamin in der Produktmischung verwendet, um die erwarteten
Ausbeute von Amphetamin zu berechnen. In anderen Reaktionen war
das erhaltene Produkt nicht trocken.
-
Alle
hierin erwähnten
Veröffentlichungen,
Patentanmeldungen, Patente und anderen Literaturstellen werden in
ihrer Gesamtheit durch Bezugnahme aufgenommen. Weiter haben alle
technischen und wissenschaftlichen Ausdrücke, die hierin verwendet werden,
die gleiche Bedeutung, wie sie üblicherweise
vom Fachmann verstanden wird, an denn sich diese Erfindung richtet,
falls nicht anders definiert.
-
Wie
erforderlich, schließt
die obige Beschreibung die beste Weise ein, die derzeit für die Durchführung der
Erfindung in Betracht gezogen wird. Man wird bemerken, dass die
Erfindung mit Bezug auf zahlreiche spezielle Ausführungsformen
und Beispiele beschrieben worden ist; es wird betont, dass diese
Ausführungsformen
und Beispiele nicht als Beschränkung
der Erfindung angesehen werden sollten, sondern lediglich für den Zweck
der Beschreibung der allgemeinen Prinzipien der Erfindung und der
Erläuterung
der Erfindung angegeben sind. Deshalb können, obwohl geeignete Verfahren,
Vorrichtungen und Materialien für
die Durchführung oder
das Testen der vorliegenden Erfindung vorstehend beschrieben sind,
andere geeignete Verfahren, Vorrichtungen und Materialien, die den
hierin beschriebenen ähnlich
oder äquivalent
und in der Technik wohlbekannt sind oder anschließend entwickelt
werden, ebenfalls verwendet werden, ohne vom Geist oder Bereich der
Erfindung abzuweichen. Da diese verschiedenen Äquivalente und Ersetzungen
vom gewöhnlichen
Fachmann in Anbetracht der vorstehenden Offenbarung erkannt werden,
werden sie als innerhalb des Bereichs der vorliegenden Erfindung
liegend angesehen, wie er durch die beigefügten Ansprüche definiert ist. Nur die
beigefügten
Ansprüche
definieren den Bereich der Erfindung.





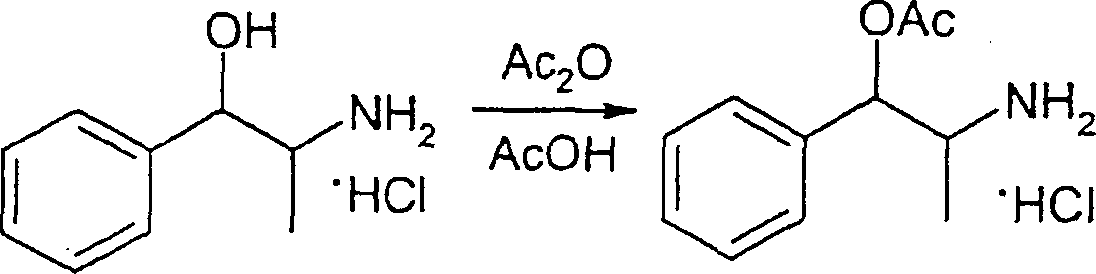











 über die Zwischenprodukt-Verbindung der Formel III
über die Zwischenprodukt-Verbindung der Formel III worin: R1 Wasserstoff oder eine C1-C8-Alkylgruppe ist; jedes R2 unabhängig Wasserstoff, Halogen, eine C1-C8-Alkylgruppe, C1-C8-Alkoxygruppen, eine C1-C8-Alkylgruppe, die mit 1 bis 5 Halogenen substituiert ist, C1-C8-Alkoxygruppen ist, die mit 1 bis 5 Halogenen substituiert sind, oder beide R2 zusammen, wenn sie an benachbarten Kohlenstoffen vorliegen, -O(CH2)xO- darstellen, worin x für 1 bis 4 steht, wodurch eine Ringstruktur gebildet wird, die mit der Phenylgruppe kondensiert ist; R3 eine C1-C8-Alkylgruppe, eine C1-C12-Aralkylgruppe, eine C1-C12-Alkarylgruppe oder eine Phenylgruppe ist, wobei jede gegebenenfalls mit 1 bis 5 Substituenten substituiert ist, die aus Halogen, Hydroxy oder C1-C6-Alkyl ausgewählt sind; und HX ein Äquivalent einer organischen oder anorganischen Säure ist, wobei das Verfahren umfasst: (a) Acylieren des Phenylpropanolaminsalzes der Formel II mit einem Acylierungsmittel in einem Lösungsmittel bei erhöhter Temperatur, um eine Reaktionsmischung herzustellen, die ein O-acyliertes Phenylpropanolaminsalz der Formel III enthält, welches durch die Zugabe eines Kristallisationslösungsmittels isoliert werden kann, oder diese Mischung kann gegebenenfalls im nächsten Schritt verwendet werden; und (b) Hydrieren des O-acylierten Phenylpropanolaminsalzes in Anwesenheit eines Katalysators, um die Verbindung der Formel I herzustellen.
worin: R1 Wasserstoff oder eine C1-C8-Alkylgruppe ist; jedes R2 unabhängig Wasserstoff, Halogen, eine C1-C8-Alkylgruppe, C1-C8-Alkoxygruppen, eine C1-C8-Alkylgruppe, die mit 1 bis 5 Halogenen substituiert ist, C1-C8-Alkoxygruppen ist, die mit 1 bis 5 Halogenen substituiert sind, oder beide R2 zusammen, wenn sie an benachbarten Kohlenstoffen vorliegen, -O(CH2)xO- darstellen, worin x für 1 bis 4 steht, wodurch eine Ringstruktur gebildet wird, die mit der Phenylgruppe kondensiert ist; R3 eine C1-C8-Alkylgruppe, eine C1-C12-Aralkylgruppe, eine C1-C12-Alkarylgruppe oder eine Phenylgruppe ist, wobei jede gegebenenfalls mit 1 bis 5 Substituenten substituiert ist, die aus Halogen, Hydroxy oder C1-C6-Alkyl ausgewählt sind; und HX ein Äquivalent einer organischen oder anorganischen Säure ist, wobei das Verfahren umfasst: (a) Acylieren des Phenylpropanolaminsalzes der Formel II mit einem Acylierungsmittel in einem Lösungsmittel bei erhöhter Temperatur, um eine Reaktionsmischung herzustellen, die ein O-acyliertes Phenylpropanolaminsalz der Formel III enthält, welches durch die Zugabe eines Kristallisationslösungsmittels isoliert werden kann, oder diese Mischung kann gegebenenfalls im nächsten Schritt verwendet werden; und (b) Hydrieren des O-acylierten Phenylpropanolaminsalzes in Anwesenheit eines Katalysators, um die Verbindung der Formel I herzustellen.
 worin: R1 Wasserstoff oder eine C1-C8-Alkylgruppe ist; jedes R2 unabhängig Wasserstoff, Halogen, eine C1-C8-Alkylgruppe, C1-C8-Alkoxygruppen, eine C1-C8-Alkylgruppe, die mit 1 bis 5 Halogenen substituiert ist, C1-C8-Alkoxygruppen ist, die mit 1 bis 5 Halogenen substituiert sind, oder beide R2 zusammen -O(CH2)xO- darstellen, worin x für 1 bis 4 steht, wodurch eine Ringstruktur gebildet wird, die mit der Phenylgruppe kondensiert ist; R3 eine C1-C8-Alkylgruppe, eine C1-C12-Aralkylgruppe, eine C1-C12-Alkarylgruppe oder eine Phenylgruppe ist, wobei jede gegebenenfalls mit 1 bis 5 Substituenten substituiert ist, die aus Halogen, Hydroxy oder C1-C6-Alkyl ausgewählt sind; und HX ein Äquivalent einer organischen oder anorganischen Säure ist, wobei das Verfahren umfasst: Hydrieren des O-acylierten Phenylpropanolaminsalzes, um eine Verbindung der Formel I herzustellen.
worin: R1 Wasserstoff oder eine C1-C8-Alkylgruppe ist; jedes R2 unabhängig Wasserstoff, Halogen, eine C1-C8-Alkylgruppe, C1-C8-Alkoxygruppen, eine C1-C8-Alkylgruppe, die mit 1 bis 5 Halogenen substituiert ist, C1-C8-Alkoxygruppen ist, die mit 1 bis 5 Halogenen substituiert sind, oder beide R2 zusammen -O(CH2)xO- darstellen, worin x für 1 bis 4 steht, wodurch eine Ringstruktur gebildet wird, die mit der Phenylgruppe kondensiert ist; R3 eine C1-C8-Alkylgruppe, eine C1-C12-Aralkylgruppe, eine C1-C12-Alkarylgruppe oder eine Phenylgruppe ist, wobei jede gegebenenfalls mit 1 bis 5 Substituenten substituiert ist, die aus Halogen, Hydroxy oder C1-C6-Alkyl ausgewählt sind; und HX ein Äquivalent einer organischen oder anorganischen Säure ist, wobei das Verfahren umfasst: Hydrieren des O-acylierten Phenylpropanolaminsalzes, um eine Verbindung der Formel I herzustellen.